Embed Size (px)
Citation preview


À minha mãe.

AGRADECIMENTOS
Ao Prof. Dr. PAULO CELSO ISOLANI pela dedicação,
amizade e conhecimento científico oferecido durante a sua orien
tação.
A "maninha" Ediane Mansani e aos Profs. José Ma
nuel Riveros e Peter TÍedemann pela amizade, incentivos, alegrias
e tri s tezas compartilhadas.
Ao amigo Walter Kaiser, conselheiro e incentiva-
dor.
Aos colegas Keiko Takashima, Sonia Maria José, -
Sergio Galembeck, Sergio R.A. Leite, Hidetake Imasato, José BartQ
lomeu C. de Melo, Ourides Santini Filho, Paschoal Bronzo, váska
ra Barrilli, Esther Sbampato, Rosana C.L. Pereira, Maria José Ro
drigues de Almeida pelo convívio agradável e amistoso.
Ao Srs. Geraldo Ayrosa, Waldemar Perin e Mário -
Otsuka, pela assistência dedicada e eficiente na manutenção da
aparelhagem do laboratório.
Ao Prof. Osvaldo Sala, pelo uso do espectrômetro
infravermelho e de sua oficina mecânica.
A FAPESP, pela concessão da bolsa de mestrado.
Ao CNPq, como à FAPESP também, pelos equipam~n
tos fornecidos ao laboratório.


62 b) Estudo em função da quantidade
de energia do laser • • •••• _~." ~ ... ~ . ... ,,, .... 41
6 2 c) Dependência do rendimento da dissociação
com relação ao comprimento de
onda da irradiação ~ . _ .... " ...... " ... , • .. .., ....... 41
6 2 d) Cinética de decomposição do éter
dietilico • • ,. .. ft. . v Ci' • ~ .... . ., ~ ~ . • c. o l' • • c; .... 45
6 2 e) Verificação da lei de Beer-Lambert . .. .. ' ...... 11 50
6 . 3 Estudo da variação do rendimento
individual dos produtos •••• ~ • • ••• 5 O
6 . 3 a) Variação do rendimento individual dos
produtos em função da
quantidade de energia
por pulso ~ . ' .-o .. " •• • • ., ••• - o
6 . 3 . b) Variação do rendimento individual
dos produtos em função da
..... o I t ' O"' # ....
pressão de éter dietílico c . ' .. c ,. ...... ., .... .. . .
64 Estudos com adição de gases
52
57
inertes \Ôr •• .., • • tio .... ~ 62
6 4 , 1 Influência de um gás inerte no
rendimento dos produtos de uma reação
fotoquímica o • ,, ' • • • CJ o .... o . - •• 62
642 Experimentos com éter
dietílico e
gases inertes O .~ - •• O •• · CJct.o •••••• _ ••••• 63
6 5 Estudos em misturas de
éter dietílico
com óxido
nítrico. .. ·"·· ........ ,, .... .. -•...... 0. • 68
6 5 A ação de captadores de
radicais livres t'l opc e • ., o~ ,. .~ o ., \J' • •• ' • • . ' • • • • 68
652 Irradiação de amostras éter/NO •••• 11 . ... . " ••• 69
6 6 Estudo do sistema éter/oxigênio • • o ,. •••• 11 ••• 71
6 6 Combustões • • •••••••• .... . .... ~ ... 7 1

66 2
6 6 . 3
Rendimentos dos produtos na
irradiação do
sistema éter/oxigênio ... ., . ., ...... ., ~ .,. .. ... . Estudo cinético do sistema
74
éter/oxigênio .. O.fi..II.« • ••••• Ct~ .. O • •••• ." 78
o fenômeno de quebra dielétrica " ... . ' ........ . 78
A quebra dielétrica no sistema
éter/oxigênio ••.• •• •.••• ' •. ~ •. ~_ •••• • • 83
Considerações sobre a combustão
do éter dietilico ............ .
7 Conclusões, ••.•••••
. . .... " ... 83
87
Referências bibliográficas . .•.. • • ~ •. ~, < . .. ... 97

I
RESUMO
Foi estudada a dissociaçã o multifotônica do éter dietí
l i co i nd uz id a por ab s or ção de luz i nfr avermelha, prov enien te de um
l aser de CO 2 , tipo TEA .
Na irradiação com a linha P(2 0) da banda 00 0 1_0 20 0
(1046 cm- 1) , os produto s observado s por análi se espec t roscópica in
fraver mel ha e cromatografia em fa se gasos a f or am: H2 ' CO, CH 4 , C2H2,
C2H4 , C2H6 , CH3CHO e C2H50H.
A e fi c i ê nc i a de di ssoc i ação f oi proporc i ona l à s pre s -
sões in i cia i s de ét e r a uma potên c i a f i xa e cresc ente em função da
pot ên cia do l a ser a uma pressão fi xa . A eficiência com relação ao
co mpr ime nto de onda do la s er seguiu aprox imad amente o espectro de
absorção I .V. do ét e r, s ugerindo qu e a dis s oc iação seja iniciada
por um pr oces so de ab s orção multifo t ôni c a ressonante .
o estudo da variação do rendimento individual dos prod~
tos em f un ç ão da potência apresentou um comportamento crescente pa
ra to dos os produto s , enquanto que a variação do rendimento indivi
du a l dos mes mos em fun ção da pres s ão inicial de éter para CO e CH 4
fo r am cre scentes e para C2H6 , CH 3CH O e C2H50H foram decrescentes; o
c om po rtament o do C2H4 foi linear e quase constante, o que sugere
que ele se f orme por dissociação unimolecular do éter, enquanto que
a f or maç ão de CO, CH 4 e C2H6 deve envolver processos colisionais.
Na irradiação de mistura s de éter com argônio, neônio,
hidrogênio, hélio e N20, os rendimentos individuais absolutos cai
ram, poss ivelmente porque tais ga s es agem como desativadores de mo
léculas de éter excitadas pelo laser, por transferência de energia
V-To
Adicionando-se ao éter um captador de radicais, no caso

NO, f oi verificado apenas um pequeno aumento no rendimento relativ o
de CH 4 em detrime nto do C2H6 .
Na irradiação do sistema éter/oxigênio foi observada a
quebra di elétrica do sistema, tratando-se este fenô me no de uma ver
dadeira comb ustão , uma vez que o processo todo é desencadeado com
apenas um pulso do lase r para pre ssões aci ma de 40,0 Torr de 02,se~
do CO 2 o produto principal. Entret an to, abaixo do limiar de quebra
dielétrica o rendimento indiv idu al dos produtos foi crescente c om o
aumento do número de pul s os e também em função do aumento da pres são
de 02 ' exce t o o etano l que ne s te último es tudo de c re sc eu. Ne s te ca
so, os prod utos obse rvado s f oram os me s mos qu e no éter pur o , exceto
pequ en a quant idade de CO 2 .
Em s ínt ese , o C2H4 e C2HSOH de vem se form a r da dis soc i~
ção unimole cular do é ter, ao passo que H2 ' CO, CH 4 , C2H2 , C2H6 e
CH 3CHO são obtidos , em su a maior parte, através de rea ções qu~ en
volvem processos colisionais, radicalares ou não.

ABSTRACT
The photodissociation of diethy l et her induced by
infrared multiphoton absorption from focuse d radiation of a
TEA CO 2 laser wa s inve s tigated.
111
After i rra diating with the P(20) line of the 00 0 1_02 0 0
band (1 046 cm - 1) the decomposition products were a nalyzed by IR
s pe ctroscop y and gas chro matography. Hydrogen, carbon monoxi de,
methane, ethylene , acetyl ene, e than e, acetaldehy de and ethanol wer e
detected .
The overall decomposition efficiency wa s proportional
to the init ial pressure for a fixed irradiation energy, and increases
with pulse e ,l ergy at constant sa mpl e pres s ure.
The decomposition efficiency was also obser ved to be
wavelength dependent and followed, roughly , the IR abso rp tion
spectrum pro file suggesting a reson an t multiphoton ab so rpt io n
initiated reaction.
Indi vidual product yi e ld s were found to inc re ase by
increasing pu l se energies.
The var iati on of the initial ether pressure showed
increasing yields for CO and CH 4 , and de creas ing yields for C2H6 ,
CH 3CHO and C2HSOH. The C2H4 yield was almost constant with sa mpl e
pressure, s uggesting that it i s f or me d through unimol ecu la r
decomposition , wh ile the formation of CO, CH 4 and C2H6 must inv olve
collisional processes.
The us e of argon, helium, neon , hydrogen, and nitrous
oxide as buffer gases decreases the absolute product yields,

probably due to the deactivation of excited molecules via collisional
V-T energy transfer.
The use of a free radical scavenger, nitric oxide,
indicated a small increase for CH 4 yield and a decrease for C2H6 .
When irradiating ether/ ox ygen mixtures, a strong
reaction initiated by dielectric breakdown was observed . This
phenomenon occurs with one la se r pulse when the oxygen pressure
is abo ve 40 Torr, and i s practic a lly a true combustion, resulting
in the formation of CO 2 as the maj or pr od uct and traces of co .
However, under the threshold for dielectric breakdown the products
yields in crease when increasing the number of pulses an d 02
pre ss ure. In this case the products are the same as in neat ether,
exc ep t for small quantities of CO 2 .

1. INTRODUÇÃO
A fotoquí mi ca infravermelha realça um novo tipo de
fotoquímica, onde as reações ocorrem no estado eletrônico fund~
mental. Isto oferece elevada especificidade pois a radiação i~
fravermelha proveniente de um la ser é monocromática e pode ser
ajustada a freqüências iguais ou combinações das freqüências de
ab s orção das moléculas gasosas a serem excitadas . Isto requer
o us o de multi freqüências de radiação infravermelha para obten
ção da fotoexcitação até o limite de dissociação, desde que as
séries de transição vibracionais-rotacionais não sejam muito
afastadas. Uma rica estrut ur a de transições vibracionais-rota
cionais nas moléculas poli atômicas permite a abso r ção de um ele
vado número de fóto ns infravermelhos se a radiação do laser for
suficientemente intensa. Tal processo de absorção é tão seleti
vo que po de induzir até separações isotópicas.
Os químicos têm um interesse especial na - química
dos lasers co m potências da ordem de megawatts. A irradiação -
pulsada com estas potências favorece determinadas reações de ele
vadas temperaturas vibracionais ( > 1000K) que são forçados a
ocorrer à temperatura amb iente. A temperatura efetiva é facil
mente variável em um amplo intervalo, modificando a energia ra
diante por área (fluência) . Resulta disto, portanto, a pos~ibl
lidade de um estudo detalhado de reações que eram antes inaces
síveis devido à elevada energia de ativação.
Assim , rea ções induzid as por lasers apresentam
maior "limpeza" do que as r eações realizadas em aparelhagem tér
mica clássica por diversas razões:

2.
a) o reage nte é exposto a elevada temperatura por um curto inter
valo de tempo . A velocidade de aq uec imento é da ordem de 106
- 10 11 K/s; b ) a s ub seqüente velocidade de re s friamento oco rre
num inter valo de 103 - 106 K/s; c) se o feixe do laser for me-
nor que a seção de ch oque da cela de reação, efei tos de parede -
são praticamente desprez ívei s ; e d ) se o feixe do la ser irradiar
não some nte o gás ma s ta mb ém a cela, as reações que ocorrem nas
paredes da ce la são acentuadas.
Na maioria dos ca s os, a e tap a primária das rea ções
induzidas por lasers são reações de decomposição . Contudo,ex i s-
tem exemplos em que oco rr em isomerizaçõe s ou combinações bimole-
culares produzindo novas molécula s [1, 2 J .
Uma vez descoberto o fenô meno de absorçã o de ene ~ia
I.V. o interesse gera l passo u a se r em c omo analisar os div ersos
tip os de reações ind uz i das por la sers .
Dive r sos método s ex pe rimen ta i s têm si do utili za do s
no es tudo das r eaç ões multif otônica s . Entre os métod os emprega-
dos, alé m da anál i se dos produtos por espectrometria infraver me
lha, cromatografia em fase ga s osa e espec tr ometria de massa, são
destacados os segu intes:
a) espectroscopi a de intermediári os [3J; b ) ionização multifotQ
nica seguida de análise de massa [4J ; c ) dis s ociação nã o coli
sional pela intersecção de um feixe molecular com um f ei xe prov~
niente de um laser [5J; expe rim ent os co m pul sos c urtos e a ba i
xas pres sões ; d) análise do s produto s po r exc i tação ressonan te
fluores cent e por mei o de um l ase r de co rant e [6J, onde é verifi
cada a distribuiç ão energé tica populacional vibracional dos frag
mentos ori undos da di sso ciaçã o ; e) estudo de luminescência visí
velou ultravioleta de radica is excitado s a estados eletrôn icos

3.
elevados [7J; f) luminescência infravermelha de produtos de re a
ções bimoleculares entre os frag mentos da dissociação e amostras
de gases inertes [8J; g) distribuição do centro de massa da ener
gia cinética dos fragmentos obtidos em dissociações c om feixes -
moleculares [9J; h) razão is ot ópi ca intramolecular [10J ; e i)
detec ção optoacústica, baseada na medida da intensidade de uma -
onda acústica num gás absorvente onde ocorre a conversão de ener
gia vibracional das moléculas em movimento translacional [11,12J.
Essas reações podem ser estudadas por dupla ou múl
tiplas freqüências e em função do comprimento de onda e in te nsi
dade do l aser , duração do pul so , pre s são da amostra e ret ard amen
to do intervalo de tempo entre os pulsos .
Uma análise geral do fenômeno de di ss oc iação por ra
diação infravermelha é oferecida na referência 13 .
1.1 Objetivo desta dissertação
o objetivo des ta di ssertação foi o de estudar a di s
sociação da molécula de éter di etílico induzida por luz infrave~
melha, proveniente de um laser de CO 2 tipo TEA. O suprimento de
grandes quantidades de energia a freqüências determinadas ou se
letivas, correspondendo a um modo vibracional particular da mol~
cuIa de éter dietílico, proporcionou um estudo à parte da piróll
se e fotólise no ultravioleta da mesma .
Portant o, serão abordados, neste trabalho, al€m do
modelo teórico discutido na literatura e de algumas considerações
gerais , os seguintes tópicos no que concerne à molécula de éter
dietílico:
a) observação do fenômeno de dissociação e análise dos produtos;
b) comportamento da amostra e dos produtos observados com rela-

4.
ção aos segui nt es parâmetros variáveis : qu ant idade de energia
por pulso, comprimento de onda da irr adiação e pre ssão da amos
tra;
c) com portamento do r endimento dos produtos na irradiação da
amostra em mistura co m outr os gases (inertes à irradiação)fo~
necendo s ub sídios para a interpretação do mecanism o de disso -
ciação ;
d) prop osição de um poss ív el mecanismo de formação dos produtos.
2 . Modelo Básico do Processo de Absorção
Multifotônica e Dissociação de
Moléculas Poliatômicas num
Campo Infravermel ho
Os aut or es de a l guns modelos teóricos , qu e tentam -
explicar o processo de absorção e dissociação mul t ifot ônica en-
contram uma relativa dificuldade em explicar como as moléculas -
so brepõem a "barreira anarmôn ica " a intensidades de luz moderada
mente inten s as « 10 12w/ cm 2). A "barreira anarmônica" é exp lic~
da em virt ud e da a nar monicidade eletro - óptica , onde os nív eis vi
braciona i s vão se aproxi mando com o aumento do número quân tic o -
vibracional, o que resulta numa perda paulatina de res so nância -
entre as t ransições e a radi ação do l ase r.
Assi m, foi propos t o inicialmente que num intenso ca~
po resso na nte infr avermelho o alargame nt o por potência da transl
ção devido ao efeito s tark dinâmico compe nsari a a anarmonicidade
[14J. Neste caso a intensidade da ra di ação infrave rm elha deve
ser extremame nte e l evada (acima de 1010W/cm2) . No estudo reali -
zado com a molécula de 8C1 3 [11J foi calcu l ado qu e a intensidade
reque r ida para a dis soc ia ção da mesma ser ia da ordem de 10 11 W/ 2
/cm . Porém, a inten sidade expe riment al ob se rvada para a ocor -

5.
rência da reação foi da ordem de 109W/cm 2 [15J. Além disto, foi
observ ada também uma forte excitação e dissociação de moléculas
de SF6
[11 J a potências relativamente moderadas (de 106 a 107
W/cm 2 ), de maneira que este efeito por alargamento da s transições
não exp lica a facilidade com que as moléculas sobrepõem a "barrei
ra anarmô nica" a intensidades de luz moderadamente intensas.
Outro modelo, o do "qu ase -contínuo vibracional", on
de um intenso cam po I.V. estaria interagindo com o contínuo for-
mado pela sob repo s i ção dos nívei s vibraci onais de modo qu e um
elevado número de vibraçõe s norm a i s se riam excitadas s imultanea-
mente [6,16 ,17J, e xplica em part e po rqu e a energia a bs orvida por
uma molécu la excede consideravelmente a energia de di s sociação -
da ligação molecular em ressonãncia com a radiação I.V.. Porém,
este modelo falha na explicação da excitação isotópica seletiva
dos mais bai xos níveis vibracion a i s di sc retos das moléculas já
estudadas .
Uma combinação dos dois modelos citados, a compens~
ção da anarmonicidad e para as primeir as transições através do
alargamento por potência e o quase-contínuo vibracional, seria -
uma possível interpretação dô ocorrência de dissociação por pro-
c essas nã o colisionais [16J. Entretanto, isto não esclarece a
excitação e dissociação mole cular a baixas int e ns idad es (10 6_10 7
W/ cm 2 ) .
O modelo aceito a t ualm ente é o do mecani smo que com
pensa a anarmonicidade para as primeiras transições pela vari~ão
da energia ro ta cional molecular. Esta teoria surgiu a partir dos
resultados experimentais do rendimento da dissocia ção da molécu-
la de SF 6 em função da freqüência de irradiação [11J. Existem
evidências de que as t r ans i çõ e s vibracionais se dão entre níveis
essencialment e discretos até v=3, enquan to que para v ~ 4 a densi

dade de níveis rotacionais-vibracionais torna-se tão grande que
forma um "quase-contínuo". Esta elevada densidade de níveis a
elevadas energias resulta do fato de qu e existem diversas manei
ras pela qual uma certa quantidade de energia pode ser distribuí
da entre os vários modos vibracionais, combinações e harmônicos
da molécula . A implicação prática deste modelo é que uma vez
efetuada a excitação a v ~ 4, ocorre excitação suplementar no
quase-contínuo, aonde praticamente qualquer freqüência pode ser
absorvida com grande eficiência, já qu e haverá r essonâ ncia qua
se perfeita co m algum nível desta região. O s egredo da obten
ção de uma absorção eficiente é a de ultrapassar o gargalo en
tre v=O e v=4 . É neste contexto que está inserido o modelo PQ~
parte do modelo P/Q/R/Contínuo [18,19J, válido para moléculas -
poliatômicas que apresentam uma banda vibracional com ramos P,
Q e R correspondentes às varia ções rotacionais 6J= -1, O, +1 ,
para transições rotac ionais-vibraci onais. Se o laser estiver
sintonizado no ramo P da transição vibracional O --->1, o deslo
camento da freqüência anarmônicas em etapas progressivas da ex
citação fará com que a freqüência do laser ressoe com as regiões
do ramo Q(2º fóton) e posteriormente R (na 3ª absorção), devido
à estrutura rotacional. Entretanto para a obtenção de melhores
resultados, é sugerido que a freqüência do laser co incida com o
ramo Q da transição vibracional 1 -->2. Isto ocorrerá quando a
freqüência do laser for igual à diferença entre a freqüência do
primeiro harmônico e da freqüência fundamental da vibração ·con
siderada.
Por outro lado, para que ocorra uma eficiente exci
tação de todas as moléculas, é necessário que aquelas que não
se encontram no estado rotacional excitado ressonante com o la
ser, sejam levada s a tal, de modo a absorver a radiaçã o. Isto
6.

7.
pode ser conseguido por repopulação rotacional causada por coli
sões inelá stic as com outras moléculas . Além disso, na ma i or pa~
te dos casos, ess as colisões alargam a estrutura fina das bandas
vibracionais , o que facilita o "casamento" com a freqüênci a emi-
tida pelo laser.
Um outro mecanismo ba se ad o numa divisão anarmônica
( " anharmon i c spl i tti ng") dos nívei s vibracionais excitados dege-
n e r a dos, em p a r t i c u I a r p a r a a m o I é c u I a de S F 6' f o i s u g e r i d o [2 O J.
Nas moléculas em que o limite infer ior do quase-contínuo vibraci~
nal está acima do nível v= 4 ( moléculas pequenas), as tran siçõe s
oco r re m entre v=3 e os nív eis do mesmo. Neste c aso não é neces -
sário que o alar gam ento por potência co mpe nse a fal ta de s i nto
nia da freqüência do laser c om o nível interme di ár io, v=4. A p~
quenos alargamen to s por potência a velocidade de transição de
dois fótons, v=3 - > v=5 , permi t e a existência de um "vazamento"
a partir dos níveis infer iores ( v=0 ,1,2 e 3) para o quase-contí
-7 nuo vibracional ao longo de um pulso de laser da ordem de 10 -
segundos [21 J .
A c ombin ação do quase-contínuo vibracional com a com
pensação anarmôn ica r otac ional e o "vazamen to " nas t r a nsições
subsequentes formam a ba se de interpretação teórica da excitação
multifot ônica e di ssociação de mol éculas po l iatômicas . Com rela
ção aos trabalhos teóricos é dado cert o ênfase ao s seguintes tó-
picos:
a) o mo del o do osci lador anarmônico e da excitação multifotônica
ressonante de s eus níveis; b) o modelo do oscilador anarmônico
rotaciona l com respei to à compensação anarmônica r otacional de
diversas transi çõ es inferiores; e c) a combinação dos níveis vi
bracionais inferiores co m os do qu ase - con tí nuo vibr ac ion al [22-
-25J. Estes modelos básicos são considerados também co m maiores

8.
detalhes na referência 11 .
Deve ser levado em conta ainda no processo de excit~ .
ção e dissociação multifotônica, qu e o requisito fundamental des
te processo não é exi stir somente uma ressonância entre a freqüê~
cia do laser e as transições rota cionais-vibraciona is da moléc ul~
mas que também existe uma dependênc ia da cinética de todo o pro-
cesso com a intensidade e a fluência do laser, notadame nt e na ex-
citação acima do limite de dissociação molecular, assunto este
tratado no capítulo 3.
3. Terminologia e considerações gerais sobre
fotoquímica com lasers
Dentre algumas das de signações referentes aos parâm~
tros do laser , são encontrados frequentemente, na literatura os
termos: intensidade e fluência. No s processos fotoquímicos a ve-
locidade de fotoativação é basicamente proporcional à densidade -
de energia radiante . Entretanto, é exposto simplesmente como se~
do proporcional à intensidade, em c ujo cont exto está s ituada a de
finição de intensidade do feixe do laser,
Intensidade do feixe (W/cm 2 ) = velocidade da
luz (cm/s) x densida-
de de energia radian
te (J/~m 3).
= nº de f ót ons/uni dade
de área x unidade de
tempo .
Quanto à fluência (ou dose), é ca r acter i zada como a
quantidade de energia radiante que atravessa uma área unitária nor
mal à direção do feixe do la ser, cuja unidade é J/c m2 .
Tanto a fluência co mo a intensidade do laser sã o pa-

9.
râmetros importantes no controle do nível final de excitaç ão da mo
lécula, no momento em que a dissociaç ão ocorre [12,26-28J. A exci
tação de moléculas através dos níveis discret os é fun ção da inten
sidade do laser , enquanto a excitação s ubseqüente dentro do quase
-contínuo depende da fluência [28J. A por çã o principal do pulso
do laser (como o laser de CO 2 tipo TEA) deve ser intensa o suficien
te para excitar a molécula através dos níveis discretos até o qua
se - contínuo e o restante do pulso deve conter fluência suficiente
para leva r a mol éc ula no qu ase -contínuo vibracional até o limite
de dissociação. Se a intensidade luminosa r equerida para a excita
ção na regi ão dos níveis discretos for menor do que a intensidade
do pico do pulso do laser, então o rendimento da dissociação será
fun ção somente da fluência do feixe e não da intensidade. No caso
de molécul a s pequenas e leves, o qua s e-contínuo se inicia a nívei s
rel a tivamente mais alto s de energia, de modo que a int e ns idade do
laser é c rítica na determinação do rend i mento da decomposição [28J.
Po r outro lado, a excitação acima do limite de disso
ciação depende ta nto da intensidade como da fluência, pois quanto
maior a fluência , ma i or o nível energético a que as moléculas po
dem ser levadas . Contu do a exci t ação ac ima do limite de diss oci a
çã o com pet e com a velocidade de de com posição, de modo que a inten
s id ade do laser pass a a ser preponderante nesta região, considera~
do- se a i mportâ nci a novamente do aspecto cinético . O nível final
de ex ci ta ção deve se r mais alto quanto maior for a intensidade do
laser, consequentemente um maior nível de excitação leva a uma dis
tribuição de energia translacional aos fragmentos da decomposição,
energia es ta em excesso, pois, normalmente, a energia de excitação
fo rn ec id a é sempre superior à energia de dissocia çã o da amostra em
es tudo [29J .
Outras denominações comun s são os termos "reação tér-


energia do laser e transferem-na ao reagente por meio de colisões
[31J, indicam se um determinado tipo de reação é térmica ou não;
já o efeito da adição de certa quantidade de gás inerte à amostra
irradiada deve reduzir a conversão dos produtos por pulso, caso
se trate de uma reação térmica [32J; e o efeito da pressão do rea
gente [33J onde, no caso do rendimento de uma reação te nd er sem
pre a aumenta r com o aumento da pressão da amostra indica se o
processo em estudo é colisional, são alguns dos métod os expe rime~
tais que têm sido utilizado s na distinção entre processos térm i
cos e fotoquímicos.
As distribuiçõe s dos produtos em amostras que sofre
ram decomposição térmica e decomposi ção fotoquímica verdadeira,po
dem ser comparadas de modo a fornecer es clarecimento s a respeito
do processo de dissociação [34J. Analogamente, a espectroscopia
em função do tempo pode indicar o tem po de surgimento dos produ
tos de uma reação, como também auxiliar na diferenciação entre
reações t érmicas ou radicalares [35J.
o termo "processos colisionais", empregado com rela
tiva freqüência neste trabalho, está relacionado principalmente a
processos de transferência de energia ' intermolecular, nos quais
moléculas excitadas tanto pelo processo térmi co como fotoquímico
são ativadas ou desativadas por colisõe s com outras moléculas, ex
citadas ou não, onde estas adquirem energia s uficien te para se
dissociarem ou iniciar determinada reação, de natu rez a radicalar
ou não. É certo que nestes casos a largura do pul so do laser e a
pressão do sistema influem decisivam ent e na dinâmica destes pro
cessos [36J.
Uma observação deve ser feita ainda quanto à a prese~
tação da energia de ativação ou calor de forma ção, relacionado às
reações indicadas. Na maioria das vezes (salvo indicação dos au-
11.

tores) os valores destacados foram obtidos de reações térmicas pu
blicadas na literatura, facilitando portanto a comparação e inter
pretação da reação em estudo no mecanismo global.
4. A molécula de éter dietílico e outros éteres
4.1 A fotodissociação do Éter dietílico: pirólise,
fotólise no ultravioleta e o presente
trabalho
o interesse particular no estudo da fotodissociação
do éter dietílico por meio da radiação infravermelha, está rela
cionado com os comportamentos peculiares observáveis das reações
químicas induzidas por um laser, já mencionadas anteriormente .
o simples fato de se depositar determinada quantida
de de energia em um modo vibracional particular da molécula, leva
a crer que a dissociação da mesma ao longo de certa via de rea-
ção irá possivelmente diferir da reação de decompo sição térmica.
Freeman, Hinshelwood e Danby [37,38J, combinaram a
análise de espectrometria de ma ssa e de cromatografia de fase de
vapor para observar os produtos formados da decomposição térmica
do éter dietílico, a pre ssões iniciais de 80 a 1000 mmHg e a tem
peratura de 525 0 C, em vário s e s tá gio s da r eaç ão ( co m ou se m inibi
ção ) .
Os produtos observados da decomposição foram: acetal
deído, etano, monóxido de carbono, metano, etileno , hidrogênio e
etanol, sendo a origem dos mesmos explicado pelas seguintes rea
ções :
C2H50C 2H5 ---- > CH3CHO + C2H6
C2H50C 2H5 ---- > CH 3CHO + C2H4 + H2
12.
- - -~--------~--__ .-J

CH 3CHO ---- > CH 4 + CO
C2H50C 2H5 ---- > C2H50H + C2H4
e possivelmente
13 .
Analogamente, Laidler e McKenney [39,40J, estudaram a
pirólise do éter dietílico entre 560 a 620 °c e a pressões varian
do de 15 a 320 mmHg .
Dados analíticos e cin éticos comprovaram uma quebra -
molecular do éter em etanol e etileno, confirmando o trabalho de
Danby e Fre eman , onde a componente de reação de primeira ordem co n
siste na formaçã o de etano 1 e et ileno e em reações envolvendo ra
dic ais livres, o que explicaria a f ormaçã o de acetaldeído e e tano.
Postularam um mecanismo envo lv endo uma iniciação de primeira ord em
e uma etapa de terminação de cadeia entre C2H5 e éH 2CH 20C 2H5 . Es
te mecanismo insere um estado estacionário cuj a equaç ão de veloci
dade apresenta uma cinética de primeira ordem a baixas pressões de
éter e uma cinética de ordem 3/2 a e levad as press ões de éter. ° me
canismo proposto, com alguma simp lifi cação , foi o seguinte :
[ 1 m J C2H5OC 2H5 > C2H50H + C2H4
[1] C2H5OC 2H5 > C2H5O + é 2H5
[2 J C2H5 + C2H5OC 2H5 ---- > C2H6 + CH 2CH 2OC 2H5
[3 J C2H5 + C2H5OC 2H5 - -- > C2H6 + CH 3CHOC 2H5
[4 J C2H5O ---- > CH 3 + CH 20
[5J CH 3 + C2H5OC 2H5 --- > CH 4 + CH 2CH2OC 2H5
[6 ] éH 3 + C2H5OC 2H5 ---> CH 4 + CH 3CHOC 2H5
[7 ] CH 2CH 20C 2H5 + C2H5OC 2H5 --- > CH 3CHOC 2H5 + C2H5OC 2H5

[8 J
[9 J
C· H CH OC H --> C2H4 + C2H50 2 2 2 5
CH3
CHO C2H5 --> CH 3CHO + C2H5
[ 10 J C2H 5 + CH 2CH 20C 2H 5 ---> C 4 H90C 2H 5
14.
Recent emen te , Mikuni e Takahasi publicaram os result~
dos da fotólise do éter dietílico na fase de vapor, irradiado em
184,9nm na região do ultr avi oleta [41J. OS produtos obtidos fo
ram: metano, etano, etileno, propano, n-butan o, etanol, acetal deí-
do, 2-etox ibutano, 2 , 3-di etox ibut a no e formaldeído. Os possíveis
processos apresentados foram os seguintes:
--- >
sendo as duas últimas v ias po uc o prováveis, de acordo com o re ndi -
mento quântico observado pelos autores.
A fotoq uími ca do éter dietílico realizada na região
do infra vermelh o durante o presente estudo, além de c omplementa r -
mais um estudo utilizando uma outra fonte de energ ia, no cas~ um
laser de CO 2 , apres en to u também alguns aspectos diferentes compar~
da aos dois tipos de estudo já de scr i tos . De maneira geral, os
produtos da dissociação do ét er observados em nosso estudo for am
praticamente os me s mos: monó xi do de carbono, met ano, etileno, ace-
tileno, etano , acetaldeído, eta nol e hidrogênio.
° comportamento observado foi semelhante especialmen-

15.
te no tocante à dissociação unimolecular dando como produto s eta
nol e etileno, assim c omo na form ação de monóxido de carbono e me
ta no que são produtos de reações de caráter colisi onal, de radi
cais livres ou não . Entretanto, a forma ção de CO , CH 4 e H2 deve
ser decorrente, também, da decomposição do etanol e acetal deído ,
assim como a formação de etano pode se r explicada pela recombina
ção de dois radicais CH 3 .
Assim , o processo primário deve envol ver uma quebra
molecular do éter em etileno e eta nol - ou mesmo em etano e ace
taldeído em menor proporção - co mo uma via predom i na nte , enquanto
que a formação dos outros produtos é decorrente de um processo cQ
lisional . As ev i dências expe ri me ntais que apoiam esta hipótese -
serão apresentadas nos capítulos 6 e seguintes.
Considerados os três diferentes tipos de dissociação
apresenta dos, sucede que do fato de se depositar determinada qua~
tidade de energi a em um modo vi bracional particular da molécula -
por absorção multifotônica origi na-se um proces so global de dissQ
ciação que di fere da decomposi ção pirolítica como da dissociaçã o
provocada no ultravioleta, não obstante o fato de alguns dos pro
duto s majoritários form ados ser em os mesmos .
4.2 O e s pect r o de abso r ç ão do éter dietí l i c o
na r egi ão de in t ere s se
Quanto ao estudo espectral na região do infravermàho
do éter dietílico em fase de vapor [42J, este apresentou a co~xi~
tência de dois confôrm eros, TG e TT, à tem peratu r a ambiente, pre
dominando em tais con dições a forma TT de simetria C2v :

TT (C 2v ) TG (C ) 1
16.
Os 39 modos normais de vibração, co m relação ao con-
fôrmero TT são reduzidos pela teoria de grupo a 12 modo s de s ime-
tri a da espécie Al' 8 da A2' 11 da 81 e 8 da 8r Dessas, SA1 ,4A2'
58 1 e 48 2 (total de 18 ) são vibrações do grupo metila, 3 de c ad a
espécie (total de 12) são vibrações do grupo metileno e 4Al' lA 2'
38 1 e 18 1 (total de 9) correspondem a vi br ações do esqueleto da -
molécula, desde que sej a assumido que todas as vibrações são es-
tritamente independ entes. Com exceção do modo normal de vibração
do grupo COC, todas as vibrações podem ser consideradas como com-
binações em fase ou fora de fase das correspondente s vibrações
dos dois grupos etila separados. As vibrações no plano combinam
em fase originando os modos A1 e as fora de fase originam os mo
dos 81. As combinações da s vibrações fora do plano em fase cor
respondem às espécies simétricas 82 e as fora de fase são combina
ções A2 . Somente as espécies correspondentes a Al' 81 e 82 são
ativas no infravermelho.
Os 39 modos normais do confôrmero TG pertencem à mes
ma classe de simetria e são ativas no infravermelho. As freqüên-
cias de absorção do grupo etila T são essencialmente idêntica s as
do confôrmero TT, enquanto que as freqüências de absorção do gru
po etila G diferem enormemente a bai xas freqüências.
A absorção do éter dietílico na faixa de trabalho do

17.
laser de CO 2 (9,6 ~ m) é devida aos modos vibracionais de deformação
angular (rocking) dos grupos metila e metileno. No entanto, devido
ao elevado grau de interpretação entre as band as de absorção são ob
- 1 ) se rvadas, por exem plo, duas absorções combinadas A1(1153 cm e
8 1(1045 cm- 1) que consistem numa deformação de um grupo metila aco
plado a um estiramento c-c.
Assim , a absorção por parte da amostra de éter dietíli
co não segue um pr ocesso res sonante puro com relação a um determina
do modo vibracional , podendo ser de scrito par a o confôrmero TT como
sendo predominantemente uma deforma ção (rocking) do grupo metila, -
CH 3-C, com alguma participação de balanço(wagging) do metileno,
C-CH 2- O, e estiramento C-C . Em contrapartida, a absorção por parte
do confôrmero TG deve ocorrer por me io de um estiramento c-c
do esqueleto da mo lécu la .
4.3 Resenha de alguns processos fotoquímicos com
laser de CO 2 realizados com outros
éteres
puro
Dentre as amostras de éteres, relacionadas na literatu-
r a, f o r a m e n c o n t r a dos o s e s t u dos d o é t e r e til v i n í 1 i c o (E V E) [43 - 46 J,
o éter n- butil vinílico [47J e do éter dimetílico [48J, irradiados -
co m um laser de CO 2 .
o primeiro despertou um relativo interesse devido à com
petição observada ent re as duas vias de dissociação de menor- energ i~
. . ( 1) E V E --> C H 3 C H O + C 2 H 4' e (2) E V E - > C H 2 C H O + C 2 H 5 . R o s e n f e 1 d
et aI. [43J estudaram a dissociação multifotônica do EVE a pressões
de 5 a 440 Torr, reação esta de natureza colisional. Tra balhand o
com um feixe de laser nã o focalizado, so mente aceta l deído e eti leno
foram observados, mas com o feixe focalizado quantidades compa rá veis
de ceteno, etano e butano também f ora m observadas, indica ção de um
ERRATA: Onde se lê int erpreta~ão l ei a- se interpenetra~ão.

18.
processo competitivo de radicais livres. Brenner, por sua vez, es
tudou a dissociaç ão do EVE usando pulsos do laser de diferentes du
rações (0 . 2 a 2 jJs) mantendo constante a fluência [44]. Com pulsos
de 2 JJ S de duração, somente a reação (1) foi observada, e nquanto
que na irradiação com pulsos de 200 ns o processo radicalar foi
preponderante. Est e re s ultado foi interpretado como se ndo uma po~
s ív el evidência de uma distr ibui ção não estatística da energia na s
molé culas precedendo a decomposição. Finalmente, a deco mpo siç ão -
infravermelha mu lt i fo tônica do EVE roi investigada pel a téc nica de
um feixe molecular cruzados com o do laser. Os produt os das rea-
çõ es (1 ) e (2) s ão f ormado s com a energia translacional de 31 e 5
kc al/mol , r espe c tivamente [46J. Com o aumento da inten s id ade do
l as er e da fluência a via correspondente a maior energia radicalar
foi fa vorecida, de acordo com a predi ção estatística da t eoria da
velocidade de reações unimoleculare s RRKM, na qual é assumida uma
dis tribuição estatística da energia entre todos os gr aus de liber-
da de da molécula [49J.
A irradiação de amostras de éter n-butil vinílico,com
as ba ndas de comprimento de onda de 9,6 a 10,6 um, apresentou uma
distint a diferença na distribui ção dos produt os nestes dois comprl
mentos de onda, destacando-se a produ ção de acetileno somente na
amostra irradiada a 9,6 jJ m [47J. Ou t ros produtos observados foram
aldeídos , ce t enos, cetonas e etileno; as espécies e concentrações
rela tiva s dos produtos da dissociação não apresentaram variações _
em f unçãd d~ pressão na faixa de a 40 Torr.
Nó que diz respeito à decomposição do é ter dimetílico
[48J, fêl v~r ifi ca dá á decomposição preferencial de (13 CH3 )2 0 ou
(CD~'~ê êffi ffi i§ tU f t § ~ffl a ~tet dimetí l ico comum, quando da irradia
~~§ e§ ªfflê~tiá têm é li~h a P(1 6) e cer ca de 0,25 J/pulso na região
dê ~·4 , Monóxido de carbono enriquecido _

19.
com metano, etano e metanol foram alguns dos produtos obser-
vados. Foi observada, também, uma pequena seletividade isotópica
13 - 180 . . b - d t · t para o C em comparaçao com , pOIS a VI r açao e es Iramen o
assimé trico do grupo C-O-C apresent a um largo deslocamento para o
átomo central, ma s pequenos deslo camentos para os átom os termina~
da molécula. Assim a excitação na extremidade de maior comprime~
to de onda da banda de absorção do éter dimetílico favo rec eu sele
tivamente a f orma ção de C180 ao invé s do 13 CO , já que o CO forma -
d t .. t 180 f ~ . .. o apresen ou um enrIquecImen o em de pr e e r enC I a a enr I quec~
mento em 13C.
Durante o transcorrer do presente trabalho foi veri-
ficado ta mbém o comportamento do éter dimetílico sob a ação da ra
diação infravermelha proveniente do laser de CO 2 . Foram detec ta
dos os produtos : CO, CH 4 , C2H6 e também H2 e C2H4 , isto irr ad ian
do a amostra tanto com a linha P(20) emitida pela banda vibracio-
nal centrada em 9,6 ~m como em 10 ,6 ~m. Tais resultados es tão de
acordo com os dados obtidos na referência 48.
5. Parte Experimental
A radi açã o monocr omáti ca in fraver melh a, uti l izada ne~
te trabalho , foi obtida de um laser de CO 2 ti po TE A (excitação
trans versal a pressão atmosféric a) , pul s ado, modelo 215 G da Ta-
chisto I nc., Needham, MA, USA. Este laser em it e c omp ri mentos de
onda discretos correspondendo às tran siçõ es 00 0 1 - 02 00 (ban da
centra da em 9 ,6 ~ m) e 00 0 1 - 1000 (banda centrada em 10,6 ~m ) da
molécula de CO 2 [50J .
° meio ativo deste laser é constituído de um f lu xo -
d 1 ° I . -1 dI· -1 I .-1 e , .mIn e He,0,8 mIn de CO 2 e 0,6 .mIn de N2 atra-
vés de um tubo que contém doi ~ eletrodo s int e rn os, pelos quais é
descarregada a energia armazenada de um banco de cap acitore s , que

por sua vez recebem uma tensão de até 30 kV.
São permitidas, pelas regras de seleção, variações
do nú mero quântico ro t acion al de mai s ou meno s doi s para cada
uma das transições mencionadas, caracterizadas por duas bandas
rotacionais- vi br ac ionais, as quai s são de s ign adas pela s letras
R(6 J = +2) e P( 6 J = - 2) em e s pectro scop ia de ab so r ção [ 5 1J.
As linhas de emissão do laser cor respondem portan-
to às bandas de diferen ç a observada s em um espec tro normal de
absor ção no i nfravermelh o do gás carbônico.
Por parte da molécula de éter dietíl i co, esta ab
s orve os comprimentos de onda das linhas da banda do laser de -
CO2 correspo nden tes à tran s i ç ão 00 0 1 - 02 0 0 (9 , 6 ]J m) . A abs or
ção ocorr e co m o laser sintonizado tanto nas linhas r otacionais
do ra mo R como do ramo P, desta ba nda de emissão do laser [52,
53 J.
o l aser de CO 2 , da Tachisto Inc., é constituído de
um t ubo para os gas es, situado entre dois ref let ores formando a
cavidade óptica . Um destes, 100% r ef letor, é uma rede de difra
ção, en quanto ·qu e o outro, 90% refletor, é um semi-espelho de
germânio .
Uma vez selecionada a linha de emissão pe la posi
ção da rede, a mesma foi identificada por um analisador de es
pectro de CO 2 , mod e lo 16 A, da Optical Engineering, Inc., Santa
Rosa, CA, USA .
Qua nto à quantidade de energia incidente na amos
tra por pulso, esta foi medida com um medidor de potência, mod~
lo 2 10, da Coherent Radiation, PaIo Alto, CA, USA, que possui -
se ns or tipo ca lorímetro de disco. A quantidade máxima de ener
gia por pulso , nas linhas de ma ior intensidade de emissão, foi
20.

21.
de 1,2 J, e a freqüência de irradiação foi fixada em 30 pulsos por
mi uto (0,5 Hz).
Oois tipos de celas de vidro foram util izadas para a
irradiação da amostra de éter dietílico, dotadas de janela s de ma
terial transparente à radiação infravermelha, (NaCI), dispostas pe~
pendicularmente ao eixo cilíndrico. Uma delas, um tubo de vidro
em f orma de T, de 12 cm de comprimento e diâmetro interno de 2 cm,
provida de uma torneira e junta cônica fêmea 14/35, foi utilizada
na anál i se es pectrométrica infr avermelha dos produtos da di ssoc ia
ção e nas experiências acompanhadas por medida de pressão (Fig . 1 ).
A outra cela , com 22 cm de comprimento e diâmetro interno de 2 cm,
comporta duas torneiras de teflon isolando o corpo principal da ce
la por intermédio de um "O ring". Uma dessas torneiras possib!
litou a ligação do tubo central a um reservatório c om a finalidade
de armazenar uma amostra gaso sa , que foi usa da co mo padrão para a
análise cromatog ráfica, resguardada ' do feixe do la s er; a outra fi
ca interposta entre a cela e a linha de vácuo (F ig. 2).
Uma vez irradiad a a s ub s tância, o co nteúd o armazenado
no reservatório lateral era misturado aos produtos da dissociação
por um intervalo de 20 minutos - e em seguida efetuada a análise -
uo s mesm os.
Durante a conexão das celas na linha de vácuo foi uti
lizada graxa de silicone para alto vácuo, inerte em relaçã o à amo~
tra irradiada e aos produtos da dissocia ção , apenas na junta ' côni
ca 14/35.
As ligações entre as celas e a linha de vácuo foram
as mais curtas possíveis, de modo a minimizar a perda do s p~odutos
formados, tanto por diluição como por adsorção dos mesmos na linh~
nos experimen tos em que foi acompanhada a eficiência de dissocia-

2
12 em
I~
FIGURA 1:
Corpo da Cela
2 - Janelas de NaCl selando o tubo com
o auxilio de um "O Ring"
3 - Junta 14/35 para conexão com a linha
de vácuo
22.

\.
2
3
4
FIGURA 2
- Corpo da cela 22 em d e c omp = 2 c~
2 - Janelas de NaCI selando o tub o com o auxilio
de um "O Ring"
3 Torneiras d e te fl on
4 Reservatório para a a mostra do padrão
cromatográfico
5 - Junta 14/35 para conexão co~ a l i nha de vácuo
23.

24.
ção pelo aumento da pressão na cela. No restante do s ex pe r im e nt os,
após a introdução dos reagentes na cela, a mesma era descon ectada -
da linha de vácuo, sendo irradiada e t r a ns ferida, para a análise
dos produtos, a outro sistema de vácuo, l i gad o ao cr omatógr afo de
gás.
A inserção dos reag e nte s na cela de irradiação foi po~
sí vel por i nte r méd io de uma linha de vác uo metálica , de cobre, man-
tida por um a bomba mecânica rotativa e outra de difusão refrigerada
a água, capaz de fornecer pressão mínima de cer ca de 1 x 10- 5 Torr
e com uma velocidade de degaseament o i nf e rior a lmTorr/hora, após
aquecimento . Por out r o l ado, os tubo s contendo as amostras líqui -
das e gasosas foram conectad os à linha de vácuo por mei o de uniões
"Caj on " prov i da s de anéi s de borra c ha ( "O-Ring s " ), t end o si do dega -
seificada a amo s tra líquida, no iníci o de cada ex periên c i a, pelo
c ongelamento da mesma co m ni t rogêni o líq ui do e bomb ea men to su bs eque.!2
te.
Uma fita de aqueciment o envolvendo a lin ha de vác uo as
segu rou maior rapidez e ef iciência na limpeza da mes ma, acelera ndo
a dessorção de ' impurezas das par edes .
As medidas de pressão foram feit as com um manômetro de
capacitância MKS 8aratron, da MKS In st rum ents, Inc., Burlingto n,M A,
USA, empregado no intervalo até 10,000 ~ 0,0 0 1 Torr , e com um manô -
metr o tipo Bo urdon, da Wallace e Tiernan, Belleville, NJ, US A, de
+ 0,0 a 100,0 - 0,2 Torr.
Todas as experiências foram realizadas à te mperatu ra -o ambiente de 24,5 C, sendo o laborató ri o term os tatizado durante o ve
rão em 24,~ ! 0,5 oCo
o feixe do laser ·foi focalizado no centro da cela de
irradiação por meio de uma lente convergente de telureto de c ádmio,

25.
de distância focal de 25,4 cm, sendo o valor da quantidade de ener
gia por pulso incidente na cela medido logo após a lente. Tanto a
lente como o suporte para a ce l a foram ajustados sobre um banco óp-
tico. Por fim , o conjunto terminava com um anteparo de grafite, lo
calizado após a cela, com a finalidade de interceptar o feixe do la
ser (F ig. 3) .
A partir deste arranjo óptico a fluência medida no pon
to fo ca l foi de 10 J/c m2 para uma quantidade de energia por pulso
de 1 Jou le . Tal medida foi obtida pelo registro da forma do fei xe
do l ase r num papel térmico, medindo-se a área do me smo para uma de
terminada quantidade de energia por pulso.
o modo de oscilação transversal do laser, em que as
experiências foram realizadas, foi o TEM 70 , correspondendo o número
7 ao modo vertical de oscilação do laser na cavidade, o qual se re
flete na estrutura do feixe, cuja se ção transversal contêm tantas -
regiões quantos forem os ventres das ondas estacionárias na cavi da
de; o número ze ro corresponde ao modo de oscilação horizontal fund~
mental do laser na cavidade, constituíndo-se portanto em um feixe -
gaussiano nesta di mensão. Tal modo de oscilação foi obtido atr avés
do reg is tr o do f e ixe num papel térmico e posterior contagem do núm e
r o de fai xas horizontais correspondente s às regiões ven trais .
Quanto à identifi cação dos produto s , a análi se espec
trométrica infravermelha foi reali za da num espectro fotômetro infra
vermelho Perkin Elmer modelo IR-180, Norwalk, USA, na região "de
4000 a 600 cm -1
A ident ifica ção e dosagem dos produto s por cromatogra
fia em fase gasosa foi realizada em um cromatógrafo de gás CG-20D ,
com um detector de condutividade térmica , da Instrumentos Científi-
cos CG ktda. , Sã o Paulo, SP, Bras il. Uma válvula de amostragem es-


27.
pecial, com um "loop" permitiu a introdução da amostra, diretamente
da cela de irradiação, nas colunas cromatográficas, através de um
sistema de vácuo mantido por uma bomba me câ ni ca rotativa e de difu
são adaptada ao s istema . Uma fit a de aq uecimento envolvendo o
"loop " gara ntiu a de ssorção rápida e eficiente de substâncias du
rante a limpeza, sen do a pressão do sis tema medida por intermédio -
de um sensor t ipo Pirani.
Na identificação do s produtos por cromatografia em fa
se gasosa foram empre gad a s as seguintes co luna s: Porapak-S , Porapa~
-T, Apiezo n L a 10%, Carbowax - 20M e Peneira Molecular PM-13X . Na
dos age m de CO, CH 4 , C2H4 e C2H6 foi ut iliz ada a coluna Porapak -S,te~
do sido empreg ado CO 2 ( Matheson, 99 , 9% de pure za) co mo padrão crom~
tográfico, enquanto que na dosagem de CH 3CHO e C2HSOH a coluna usa
da foi a Carbo wax- 20 M e o propanol (Merck, grau espectroscópico)co
mo padrão. No que concerne aos estudo s dos sistemas éter/argôn i o e
éter/0 2 a coluna usada foi a Peneira Molecular PM-13X na dosagem de
CO e CH4 , pois nas outras colunas os tempos de retenção do CO e CH 4 coincidiram com os tempos de reten ção do oxigênio e do argônio, res
pectivamente. Tanto o oxigênio como o argônio serviram, nesta dos~
gem, como padrões, por estarem em excesso na ce la. Nestas experiê~
cias, o tempo de mistura entre a amostra e os gases foi fixado em
20 minutos, tempo após o qual as análises cromatográficas qua ntita
tivas indicaram a co mpleta homogeneização da mistura.
o gás de arraste usado em todas as ex pe riê ncias. com o
cromatógrafo foi hélio, motivo pelo qual não fo i possível rea l izar
a dosagem de hidrogênio, po r este apresentar uma d i ferenç a muito p~
quena no valor da condutividade térmica.
As colunas cromatográficas foram condicionadas a 200 0 C,
no início de cada experiência, pelo período de duas horas, tendo si

28.
do assegu r ada a ausência de impureza s que por ventu ra prejudicassem
as dosage ns.
As áreas e as pre ssões das s ubstância s dos ada s e do
padrão fo r am r elaci onados pela seguinte expressão,
P.= P 1 P
A. lC
r : pre ss ão do pGd r Go ; p
ond e ,
P. : pressão da s ubstância i (a se r dosada); 1
A: $rea do pad rão ; p
A. lC
fi :
área da
correção
fator de
substânc ia
( f i ) ;
correção,
i co rrigid a, isto é ,
A dado f .= ~ por x
1 A. 1
A.x fator 1
P . 1 --
P P
de
on de o fator de c or re çã o é a rela ção en tr e as áreas do pad rão uti
lizado e a amostra a se r dosada [ 54J .
As áreas fora m determ inadas às vezes por triangula-
ção, po r ém na maioria da s experiências por um integrador eletrôn i-
co co ns truído na oficina de e l et r ôni ca do In s tituto de Química da
USP.
Os reagentes gasosos foram da Matheson Gas Products,
E. Rutherfo rd, NJ, USA, se m purificação prévia no laboratório, en-
quanto que o éter dietíl ico, proveniente do laboratório de Ou ímic a
Orgâni ca do In s tituto de Qu ími ca da USP , isento de al coó is e a l deí
dos . To do s os re agentes fo ram anali sad os por cromatografia em fa-
se gasosa para as se gurar s ua pureza.

29.
6. Resultados e Discussão
6.1 Identificação dos produtos obtidos da
irradiação do éter dietílico
A amostra de éter diet ílico , 5,00 Torr, foi irradia
da com a linha P(20) da banda 00 0 1-0 20 0 do la ser de CO 2 , a 1046,8
cm- 1 , freqüência esta que correspond e ao centro da band a de absor
ção do éter dietílico na fai xa de trabalho do laser de CO 2 (908 -
-1092 cm- 1)[18 , 53 J. O número de pul sos foi fixado e m 200 e a qua~
tid ade de energia por pulso em 1, 0 J .
Uma vez irr adiada a amostra, fo i realizada a ide nti-
ficação dos produtos por dua s técni cas :
a) Espe c tr osco pia no infravermelho:
A co mp aração do espectro infravermelho da amostra ir
radiada apresen tou três bandas signifi ca tivas como produ tos da
dissociação (Fig. 4), identificado s pelas se guint es característi-
cas :
a . 1 ) - 1 Uma banda pequena a 1745 cm de carbonila
(C=O) , proven i en te de aceta ldeíd o, c uja absor çã o é devid a a vi br a
ções de estirament o axial [55J.
a . 2) Uma banda fina e intensa em 948 cm- 1 (lit. 949
cm - 1) correspon dend o à vibra ç ão de defor mação angular da molécula
de C2H4 (etileno) co mo um todo, designada por v 7 (b 1u) , cujo ~ome~
to de dipolo oscila perpendicularm en t e ao ei xo C=C e ao plan o da
molécu la [ 55 - 57 J. Este modo norm al de vibra çã o v7
da mo lécu la -
pode ser representado como,
v 7 !
'\ 8

/~ ~ ( I I
\ I I
o
H C - C 3
H
9 , ,
El 8 8 u u u u O O LI'\ O O O ..:t O O O ,....
LI'\ M N
FIGURA 4
Em cima: espectro infravermelh o do éter dietilico não irradiado
Emba ixo: espectro da amostra irradiada com os respectivos produtos
de dissociação -1 -1
Resolução: 2 cm (a 4000 cm )
H2
C=CH2
, , 9 6 u u
O 00 O ..:t O 0"1
HC=CH
I..N o
'9 u
00 N ,....

31.
a.3) Uma ba nd a fina e de relativa intensidade em 728
-1 ( . -1 ) d d b d d b - f cm llt . 729, 1 cm correspon en o a uma an a e a sorçao un-
damental perpendicula r de flexã o C-H da molécula de C2H2 (ace tile
no) [55-58J, caracterizada por v5 e representada como,
A presença dos produtos, C2H2 , C2H4 e CH 3CHO foi tam
bém co nfirmada por cromatografia em fase gasosa.
b) Cromatografia em fase gasosa:
Os produtos da dissociação da amostra de éter dietíli
co foram identificados por cromatografia em fase gaso sa pela comp~
ra ção do s tempos de retenção dos mesmos com os tempos de retenção
de cada produto puro ins e rido na co lun a cromatográfic a , mantendo -
as mesmas condições experimentais. Foram utilizada s na identifica
ção de cada produto, pelo meno s , dois tipos de colunas, comprovan-
do os mesmos como produ t os da disso ci ação.
Assim, os produtos: hidrogênio, monóxido de carbono,
metano, etileno, acetileno e etano fora m identificados em colunas
de Porapak-S, Porapak-T e Peneira Molecular PM-13X (figs. 5,6,7)
acetaldeído e etanol foram identificados em colunas de Apiezo n-L -
10% e Carbowax 20M ( Figs. 8,9). A tabela apresenta os tempos de
retenção do s produtos identificados.
Foi observado, também , que a am ostra de éter dietíli -
co ( 4, 90 Torr) quando irradiada com a linha P(20) e R(30) (944,2 e
982,1 cm- 1 , respectivamente) com energia de 0,5 J/pulso e 200 pul
sos, da banda 00 0 1_10 0 0 (a 10,6 wm), apresentou os mesmos pr odutos
de dissociação já detectado s por cromatografia em fase gasosa, em
coluna Porapak-S, excetuando-se some nte o acetileno. Este produto

Tabela 1
Coluna de Po rapak-T (50 °C )
Fluxo do gás de arra s te ( He): 26,0 ml/min
Pr od ut os verificados Tempo de r etenção e m se gundos
(TR
)
39 , 4
48,7
129 ,9
146,2
Colun a de Porapak - S (50°C)
Fluxo de gás de ar ras te (He) : 25 , 5 ml / min
Pr odutos verificados
H2
CO
CH 4
C2H4
C2 H2
C2
H6
TR (seg)
37 , 1
49 , 9
51 , O
126 ,4
145,0
158 , 9
32.

Coluna de 10% Apiezon-L (50 oC)
Flu xo do gá s de arraste ( He ) : 31,3 ml/min
Produto Ve ri fica do
aceta ldeído (C H3CHO)
T R(seg)
91 , 6
Coluna de Carbow ax-20M (50°C)
Fluxo do gás de arraste (He) : 25 , 0 ml/min
Produ tos veri fi cados T R(seg)
143 , 8
4 17, 6
Coluna de Pene ir a Molec ul ar -1 3X (35 0 C)
Fluxo do gás de arraste (H e ) : 30 ml / min.
Produtos verificados T R(seg)
32,5
254
289
33.
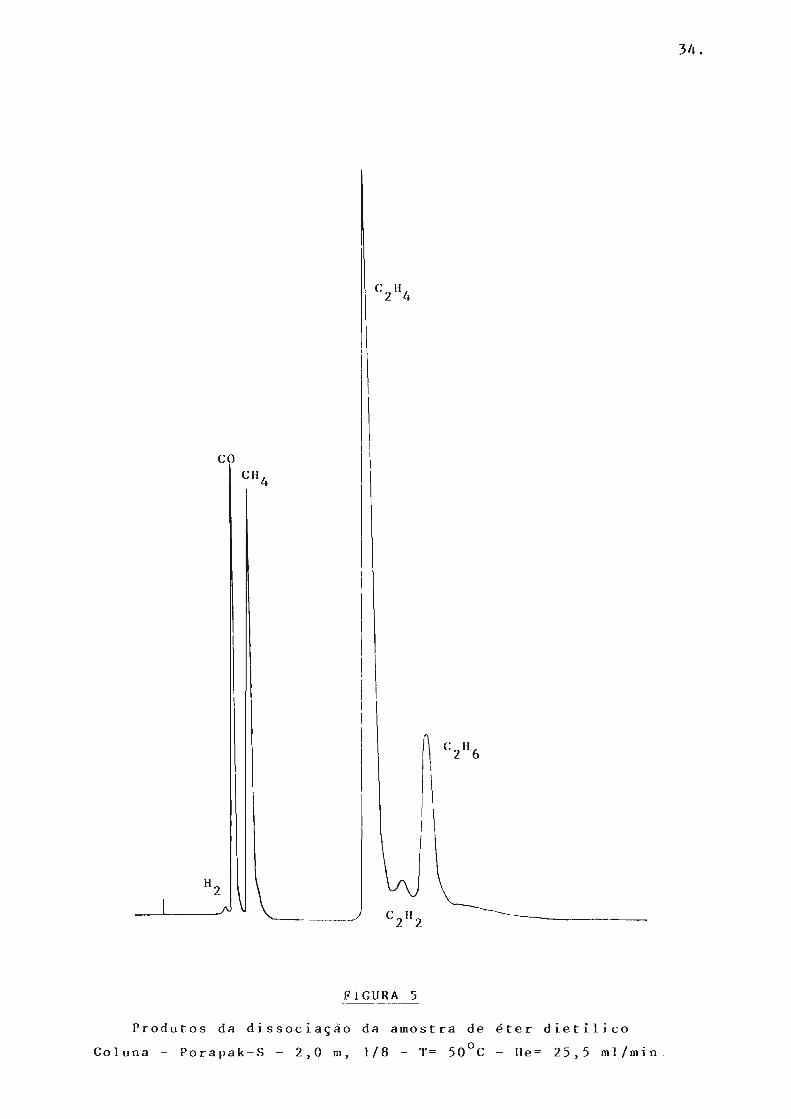
co
FIGURA 5
Produtos da dissociação da amostra d e éter dietilico
Coluna - Porapak-S - 2,0 m, 1/8 - T= 50°C - He= 25,5 ml/min .
34.

-4 __________ ~~ __________________ . _______ __
FIGURA 7
Produtos da dissociação da amostra do éter dietilico
Coluna - Peneira Molecular 13X, T= 3S o C - He= 30 ml/min
Comp _ 2,0 m - 1/8 de diâmetro
co
FIGURA 6
Produtos da dissociação da amostra de éter dietilico
Coluna - Porapak-T - 1,8 m, · 1/8, T= 50 o C, He= 26,0 ml/min



38.
também não foi observado no trabalho de Hofmann, Kl~pffer e Sc h~
fe r [47J, quando da irradiaçã o do éter n-buti l vinílico, apr e se n
tando portanto as amostra s uma dep en dên cia na di s tribui ção dos
produto s de dissociação com o comprime nto de onda emitido pelo la
ser, isto é, o acetileno som ente f oi observado na amost r a irradia
da a 9 , 6 wm. est e r es ult ado não just if ica um pro cesso de disso
cia ção selet ivo, de s de qu e o acetil eno pode ser fo rmado pela dis
sociaçã o s ec undária de alg um produto oriundo da dissociação ini
cial do éte r dietílico. Ta l produt o pode ser estáve l ou instável
ou mesmo ser excitado internament e , porém deve apresenta r uma di
feren ça de absor ção a 9,6 wm e a 10, 6 ~ m em nosso caso, o que ex
pli ca ria o fato observado. Um forte candidato é o etileno, o que
s e rá discutido posteriormente.
6.2 Estudo da dependência do rendimento
da dissociação
Após a id enti f ica ção dos produtos f oi estudada a de
pend ênc ia du ren dime nt o da dissoci aç ão com relação a algumas va
riáveis experi mentais, que fo r am: a) pre ssão ini cial de éter di
et íl i co, b) energi a por pul so do l as er e c) comprimento de onda
da irradiação .
Nesta primeira parte do e s t udo foi observado o rendi
ment o global dos produtos (efic i ência de dissoc i ação) e a medida
experimental usa da para se guir este processo fo i o aumento de
pre ssã o na cela de ir rad iação, por s er o p8râmetro mais fáci l de
se r medido ne s te caso . O tipo de cela usado nestes expe rimento s
está ilustrado na f i gura 1 .

39 .
6.2.a) Estudo do rendimento da dissociação relacio-
nada com a pressão inicial de éter dietílico
Neste estudo foi observada a variação da pressão ini
cial do éter dietílico em função do número de pulsos previamente
determina do numa va r iação de 50 até completar o númer o de 200 . A
quantidade de energia de 0,8 J/pulso e a linha de em i ssão P(20) a
9,6 wm foram os parâmetros fixos utilizados na experiência. A ta
bela 2 contém os dados obtidos .
De imediato é comprovad o um aumento da eficiência de
dissociação em relação a um va lor inicial de pressão do éter cres
cente . Entretanto a porcentagem de di ssociação obtida a uma pre~
são inicial de éter de 1,00 Torr para 50 e 200 pulsos foi, 13% e
27% respectivamente , que comparada à porcentagem ob t ida quando da
irradiação de 3,00 Torr (11,3% e 28%), 5,00 Torr ( 13,6% e 33,2%),
e 8, 00 Torr (14,6 % e 35,8%) de éter dietílico não apresenta uma
variação muito significativa , talvez devido à des a tiv ação das mo -
léculas de éter exci tadas pelo laser, po is com um aument o da pre~
são inicial no sis tema o número de col i sõ es por unidade de tempo
que ocorrem passa a ser bastante significativo, principalmente a
uma pressão acima de 10 , 00 To rr.
Uma estimativa da fr eqüência de colisões neste siste
ma pode ser calculada através da fórmula :
Para um diâmetro de colisão méd io, 7 a = 0 ,7 5nm , R= 8,31 x 10 e r g/
/grau moI, T= 298 K e M= 74, o número de colisões calcu lado foi
- 1 -1 de 0,024 Torr ns Sendo o pul so do laser da ordem de 40 ns
(largura a meia altura) e a pres s ão de éter na maioria das expe
riências de 5 , 00 Torr , o núm~ro de colisões durante o pico inici
al do pulso é de aproximadamente 5. Portanto , duran te cada pulso

Pressão inicia l
de éter (To rr)
0,50
1 ,00
2,00
3,00
4,00
5 , 00
6,00
7,00
8,00
9 ,00
40.
Tabe la 2
Pressão em Torr . (média) após irradiar
50 100 150 200 pulsos
0,57 0,6 3 0,69 0 , 73
1 , 1 3 1 , 1 7 1 ,23 1 , 27
2 ,23 2 , 38 2 ,48 2,56
3,34 3,54 3,70 3 ,84
4 , 44 4 , 7 1 4 , 95 5 , 10
5 , 68 6, 10 6 , 41 6,66
6,92 7,4 3 7,83 8 , 12
8 , 30 8, 66 9, 10 9,43
9 , 17 9,90 10 , 45 10 , 86
10,30 11 ,04 ----

41.
do la s er existe a probabilidade de oco rrerem de zero a cinco coli
sões.
6.2.b) Estudo em função da quantidade de energia do
laser.
Nesta experiência foi verificada a eficiência de dis
sociação, em intervalos de 50 puls os perfazendo um tot al de 200 , a
uma pressão in icial fixa de éter dietílico de 3,00 Torr, variando
a quantidade de energia por pulso. a linha de emissão do laser
foi P(2 0) a 9,6 ~ m. Os resultados são ap r esen tado s na tabela 3.
Neste caso a quantidade de energia incidente na amos
tra tem grande influência no proce sso de dissocia ção da molécula -
de éter dietílico. Tanto a int e nsidade como a fluência foram alte
radas, porém a largura do pulso foi mantida constante durante o
transcorrer da experiência.
6.2 .c ) Dependência do rendimento da dissociação com
re lação ao comprimento de onda da irradiação
Neste estudo , a amostra de éter dietílico (3,00 Torr)
foi irradiada com os comprimentos de onda correspondentes às li
nhas da banda do laser de CO 2 , centrada em 9 , 6 ~ m, tendo sido acom
panhado o número de pul sos necessários pa r a atingir uma vari ação -
de pr es s ão determinada, 0,5 Torr, c om uma quantidade de energia
por pulso constante (1,0 J) .
Tal rendimento seguiu aproximadamente o perfil do es
pectro de absorção infravermelho da amostra de éter não i r rad iada,
indicação de um provável processo de abso r ção ressonante (F ig. 10 ).
Os dados apresentados na tabela 4 representa m a média de quatro ex
perimentos no mínimo .
Uma análise geral de ste resultado indica uma ausência
de seletividade no processo de absorção, pois o rendimento é pratl

Quanti dade de energia
por pulso (J)
1 , O
0 ,9
0 , 8
0 , 7
0,6
0,5
0 ,4
0,3
0,2
42 .
Tabela 3
Pressão em Torr. (média) após irradiar
50 100 150 2 00 pu lso ~
3,47 3,74 3 , 96 4,1 0
3 , 37 3 , 62 3 , 87 3 ,94
3, 40 3 , 65 3 , 84 3,97
3 , 34 3 , 56 3 , 71 3 , 86
3, 2 3 3 , 40 3,51 3,67
3 , 13 3 , 26 3 , 37 3 , 47
3 , 09 3 , 13 3 , 20 3 , 26
3,03 3,07 3 , 10 3, 11
3,01 3 , 01 3 , 03 3 , 05

Linha Rot ac ional
P ( 12)
P ( 14)
P ( 16)
P ( 18)
P(20)
P(22)
P(24)
P(26)
R (8)
R ( 10)
R ( 12)
R ( 14)
R ( 16 )
R ( 18)
R(20)
R(22)
Tabela 4
Comprimento de o nda
( ]J m)
9 ,4 8 8
9,5 0 4
9 , 519
9 ,53 6
9 , 552
9 , 56 9
9 , 58 6
9,603
9 , 3 42
9 , 329
9,3 17
9 ,3 05
9 , 29 3
9 , 28 2
9 , 2 71
9 , 26 0
Mé d ia do nú mero de
pulsos
51
47
44
5 1
55
58
6 3
64
44
37
36
38
4 1
42
42
4 3
43 .

~ • .-1 u ~
(~
,t:J H O CIl
,t:J
-<
o o o
o
o o
L-____ ----L-. __ ---=--'I---,,---, 1080 1070
-.L _ ____ -1 ._,----=-_---=..;,....,.-=--_----=-J".."..~-1060 1050 1040 1030
-1 Número de onda(cm )
FIGURA 10
Rendimento da dissociação com o comprimento de
onda do laser
Em c~ma : "Re ndimento" em função do número de
onda da radiação
Emba ixo: Espectro in fr averme lho do éter
dietil i co
44.
o
10
20
30
40 2: c, S
50 (1)
t1 o
60 o. (t)
"O
70 c ~
CIl O
80 CIl
90
100
1 10

45.
camente o mesmo quer se trabalhe no ramo R ou P da mesma banda de
t ransição do la s er. Contudo é necessário levar em consideração a
elev ada quantidade de graus de liberdade vibracionais que a molé-
cuIa de éter apresenta favorecendo provavelmente este tipo de com
portamento.
Experimentos s emelhant es for am real i zados com a mol~
c uIa de SF 6 [60J , evidenciando que com o aumento da intensidade -
(W/cm 2 ) do lase r o proce sso resson an t e pode ser mascarado devido ao alar
gamento por pot ênc ia [6 1J. Por s ua ve z, o máximo de dis soc iação
depende esse ncia lmente da pressão , sen do de s locado para a região
de compri ment o de onda maior com o aumento da mesma [61J . Certa-
mente o númer o de colisões con tribui para o mascaramento do pro-
cesso de absorção de fótons por parte da amostra , deslocando o má
ximo de cu rv a de dissociação para a região de comprimento de onda
maior, onde a seç ão de choque da ab so rção multifotônica é maior.
En tretanto, mesmo trab alhando com 3,00 Torr de éter
dietílico e s em verifi ca r para qual valor da quantidade de ener-
gi a do laser um proce s so de absorção de fótons ocorreria de for-
ma mais eficiente, foi observada uma razoável se melhan ça entre o
espec t ro de absorção linear e a dep endência do número de fóto ns -
absorv id as pela amo s tra.
6 2.d) Cinética de decomposição do éter dietílico
Co m o intuito de obter mais uma evidência para o prQ
cesso de dis soc i ação do é ter dietílico, foi realizado um tratamen
to c inético da de com posição da amos tra , tendo sido observada a va
riação da pressão do éte r remanescente em função do número de pul
so s. Os valores obtidos e as co nd ições experimentais estão regi~
trados na tabela 5, enquanto que na figura 11 está o gráfico log~
rítmico corre s pond ente. O comportamento observado é linear até -
45 pul s os, exib indo no início da irradiaçã o um pro cesso de primel

nº de pulsos
o
15
30
45
60
75
90
105
120
135
150
165
180
195
210
225
Tabe l a 5
Quantidade de energia por pul so : 1,0 J
Linha P(20) a 9,6 wm
Pressão inicial de éter: 3,04 Torr
Pressão total de éter
e pro du tos
Pressão do éter
remanescente
3,04 3 ,04
3,25 2,83
3,46 2,62
3,60 2,48
3 , 71 2 , 37
3,82 2,26
3,91 2 ,17
4,00 2,08
4 , 06 2,02
4 , 1 1 1 ,97
4,17 1 ,91
4,23 1 ,85
4,29 1 , 79
4,34 1 ,74
4, 38 1 ,7O
4,42 1 ,66
46.

,-... i-I Q)
~
' Q)
1,100
Q) 0 , 900 "O
o 110 til til Q)
i-I p.. '-"
0,700
0,500 O 15
47.
o
o
o
o
o
o
o
o
o
I 1 I I 1 I I 30 45 60 75 90 105 120 135 150 165 180 195
Número de pulsos
FIGURA 1 1
Cinéti ca da d ec ompos ição do éte r dietilico
Logaritmo da pressão residual de éter em
função do número d e pul s os do la ser

48.
ra ordem global.
Um outro tratamento numérico foi tentado através das
su postas vias de decomposição, semelhante ao realizado por Leite
[6 2J em sua tese de doutoramento.
Admitindo as seguinte s via s de decompo siçã o para o
éter di etílico:
1 . (C 2HS)2 0 k 1 --> C2H4 + C2HSOH
P o
- x x x
2. ( C2HS )2 0 k
2 > CH 3CHO + C2H6
P o - Y Y Y
onde P = pressão inicial de éter o
Pt = pr essã o total no tempo t,
segue - se que :
2P + x + y o
com x y- (pa ra P = 3,00 ou P = 5,0 0 Torr), o o 3
x + -
3 = 2P o
4 + - x
3
Portanto a pressão de éter no
P P = ( C2HS)2 0 o - x - y =
p(C H ) O = 3P - Pt 252
o
Diferenciando com rela çã o a t ,
dP (C H ) O 252
kP = dt (C 2HS) 20
x=
tempo t será:
P x 3Po - x - - = o 3
(k = k 1
+ k 2)
- Pt

dP (C H ) O 2 52
In
=
49.
- kdt , que integr ando fornece
In Po - kt .
Uma vez que todos os pul sos têm a mesma duração , o número destes PQ
de substituir o tem po t na fórmula acima. O co mpo rtamento de
In P (C 2H5 )2 0 em função de s(nº de pulsos) calculado desta maneira
apresentou uma linearidade que foi obedecida até os primeiros 45
pul s os do la s er, concordando com o apre se nt ado no tratamento ante-
rior.
Um outro método para a verifica ção da decomposição do
reagente foi apres entado por Kramer [63], e Keefer, Allen Jr. e Per
son [ 6 4J, onde foi proposta uma equa ç ão análoga a uma equação ciné-
tica para a obtenção da possíve l ordem de decompo sição da amostra -
irradiada, partindo da diferencial que relaciona o desaparecime nt o
do reagente em função do número de pulsos ( s),
-d P amostra
ds
Integ r ando resulta:
ex:
[p ]n-l amostra
[ Jn-l Pamostra o
ex: (n - l)s .
O gráfico CP )n - l
em função de s deve se r uma reta; portan -amostra
to por tentativas é determinado n-l, de modo que o gráfico
n-l l/CP t) em função de s seja linear . amos ra'

50.
utilizando os dados da tabela 5, o valor de n calc ulado
para o experimento com a molécula de éter dietílico foi de 0,92. A~
sim, neste caso, para os primeiros 60 pulsos do laser foi verifica
do que a decomposição do éter segue quase uma dissociação de primei
ra orde m global, co rroborand o os re s ultados dos dois modelos cinéti
cos utilizados previamente.
6.2.e) Verificação da lei de Beer-Lambert
Outro experimento realizado foi a verificação da obediência
à lei de Beer-Lambert para este provável pr ocess o de absorção r essQ
nante, mesmo sabendo que a absorção multifotônica apresenta uma fe
nomeno logia comp le xa que não segue geralmente esta lei [65, 66J . Na
figura 12, está representado o comportamento do sistema na absorçã o
da energia do laser, para pres s ões de éter inferiores a 30,0 Torr.
As c ondições exp erimentais com relação à parte óptica foram idênti
cas aos estudos anteriores, com o feixe focalizado, pois foi obser
vado que irradiando a amost ra sem focalizaç ão do me smo não oc orria
a di ssoc iação do éter, mesmo trabalhand o a uma quantid ade de ener
gia de 1 , 2 J/puls o. O valor do coeficiente de absor ção foi idênti
co nas duas janelas de NaCI e a qu an tidade de energia transmitida -
com a presença de amostra na ce l a conti nuava a mesma, apó s 200 pul
sos, dentro de um erro expe riment al de 1%.
6.3 . Estudo da variação do rendimento individual dos
produtos
Uma vez realizadas as experiências acompanhando a efi ci
ência de dissociação (re ndimento global) pela varia ção da pressão -
no s i st ema, foi analisada a variação do rendimento individual dos
produtos provenientes da dissociação da amostra de éter, por análi-

51.
0,900
0,800
0,700
0,600 C'J .... U c:: <ca 0,500 ..o
'"' o t/J 0,400 ..o ~
0,300
0,200
O, 100
o
0,000 5,00 10,00 15,00 20,00 25,00 30,00 35,00 40,00
Pressão de éter dietilico (Torr)
FIGURA 12
Absorbância da amostra de éter díetilíco
em função da pressão

52.
se cromatográfica quantitativa em fase ga sosa .
Est as experiências visavam estudar a vari ação destes -
rendimentos co m os se guintes parâmetros experimentais:
a) quant idad e de energia por pulso e
b) pressão inicial da amostra de éter dietíli co.
6.3.a) Variação do rendimento individual dos produtos
em função da quantidade de energia por pulso
Neste estudo foi observada a influência da intensidade
e da fluência na formação dos produtos, pois ambos estes parâmetros
estão relacionados com a quantidade de energia por pulso provenie~
te do laser . Nas figura s 13 e14 estão representados os comporta-
mentos destes ren dimento s e os dados experimentais na tabela 6 .
Dentre os produtos apresentados, o etano segue um com-
portamento linear crescente somente a energias inferiores a 0 ,6 J/
/pulso, tendendo acima de tal valor a apresentar um rend i mento qu~
se que constante .
Dentre as hipóteses para explicar o processo de disso -
ciação é conhecida a importância da intens id ade, já que ocorre uma
competição entre a velocidade de bombeamento ao quase - contínuo ou
até mesmo ao contínuo com as col i sões moleculares causan do relaxa-
ção rota cional e vibracional [1 2 J, con tribuindo a f l uê ncia pa ra ,
que seja alcançado o li mite de dissociação onde novamente a inten-
sidade é impo rta nt e, já que a quantidade de energia ac ima do limi-
te de dissociação vai depen der da relaç ão entre a cinética de de-
composição e a velocidade de bombeamento [28J. Contudo, não é po~
sível se saber a partir de qu e valor para a intensidade o processo
é favorecido, assim co mo para que valores de fluê ncia a molécula -
passa do quase-contínuo ao limite de dissociação, com os dados ex-

Quant idade de
energia por pulso (J)
0,2
0,4
0,6
0,8
1 , °
Tabela 6
Pressão de éter dietílico: 5 , 00 Torr,
Lin ha P(20) a 9 ,6 -w m,
número de pulsos: 200 .
Pressão dos Produtos/Pressão inicial de éter dietílico
co CH 4 C2
H4
C2H2 C2
H6
CH 3CHO
0,00 97 0,0071 0 ,0217 0,0122
0,414 0,0440 0 ,0912 0 , 0351 0 ,01 43
0,0764 0,0741 0, 1421 0,0512 0,0237
0,1101 0,0978 0, 1764 0,0085 0,0597 0,0285
0,1383 0, 1231 0 ,2114 0 , 017 4 0,0663
C2H5 0H
0, 00 75
0,0142
0,0248
0,07.77
V1 VJ .

o u ,,..
,..., .,.. .oJ
0,220
0,200
0,180
Q) O, 160 ,,.. '1:l
o 1('0 (/)
(/)
0,140
Q) O, 120 I-<
p.; ........
(/)
o .oJ
=' '1:l o ~
p.;
(/)
o '1:l
o 1('0 (/)
(/)
Q)
I-< p.;
O, 100
0,080
0,060
0,040
0,020
0,000
Quantidade de Energia (J)
FIGURA 13
Rendimento dos produtos da dissociação do
éter dietilico em função da quantidade
de energia por pulso
54.

0,030 ~
ra .~
u . ~
c:: .~ 0, 025
1-1 ~ ~
~
~ "O
0,020 o
Ira (/)
(/)
~ 1-1
Il.. 0,015 -(/)
o ~
:l "O o ~
0 , 010 c..
(/)
o "O
o Ira
0,005 (/)
(/)
~ ~
Il..
0,000
6 ~
, 9
~ O CH)CHO
• C2
H5
OH
L- .--I
0,2 0,4 0,6 0,8 1,0
Quantidade de energ1a (J)
FIGURA 14
Rendimento dos produtos da dissociação em função
da quantidade d e energia por pulso .
55 .






61.
dominando sob re o processo de absor ção multifotônica no caso do
etano 1 formado neste estudo, de mod o a se explicar o decrés ci mo -
do rendimento de etanol e o aumento do rendimento de CH 4 e CO no
estudo em função da pressão de éter dietílico irradiad o .
o mesmo deve ocorrer com o acetaldeído, o qual se de
com põe em CO e CH 4 , envo lvendo colisões ou re açõe s de radicais li
vres [69]. Laidler [39] verificou que a temperaturas em tôrno de
600 0 C o acetal deído se decompõe em CH 4 e co. Tal temperatura vi
bracional, em noss as condições experimentais, é seguramente ob ti
da na região focal durante o proces so de irradiação da am ostra, de
forma que uma fração de acetaldeído f orm ado da decompo s iç ão do
éter tem possivelmente energia inter na s uficiente para se di s so-
cia r em co e CH4 , sendo uma provável explicação para o rendimento
crescente obtido no estudo em função da pressão da amostra para
estes produtos. Po r outro lado, foi obser vado que o processo de
dissociação de acetaldeído pela irr adiação da linha P(20) a 9 , 6 -
~ m é ineficiente [70], es perando-se uma mínima di ssociação des
te produto por absorção multifotônica nas no ssas con di ções ex perl
mentais , j á que os e xperimentos e nv olveram irrad iações com 200
pulsos.
o comportamento observad o para o acetileno, crescente
co m o aumento da pressão inicial de é ter suge re, que o mesmo se
or ig ina de um proce sso colisional. ' Neste caso o a umento da pres
são do sistema favorece um maior número de co lisõ e s por un i dade -
de tempo , de modo que o etileno formado da dissociação do éter ad
quire energia para se decompor em H2 e C2H2 , por transferência de
energia vi br acional, colidindo com as moléculas de éter exc itadas
pelo laser. No entanto, deve ser cons id e rado também que o etile
no formado é bombeado durante a cauda do pulso do laser de CO2
[7 1], podendo ser decomposto poster i ormente de forma a contribuir
1 1

62.
para a formação de C2H2 , embora com um rendimento relativamente bai
xo, conforme observado, também, na referência 67.
6.4 Estudos com adição de gases inertes
6.4.1. Influência de um gás inerte no rendimento dos
produtos de uma reação fotoquímica
A adição de um gás inerte a um reagente que absorve luz
infravermelha eleva a capacidade calorífica da fase gasosa de modo
a reduzir a magnitude do salto de temperatura produzido por uma de
terminada ener gia absoluta [18J. Devido à dependência exponencial
da velocidade de reação co m 1/T, tal adição deve acarretar um decrés
cimo na conversão de produtos de qu alq uer reação térmi ca . Uma vez
conhecendo os parâmetros de ativação, a magnitude do decréscimo da
velocidade térmica pode se r prevista.
Se por um lado a adição de um gás inerte acarreta uma
diminuição no rendimento de uma reação fotoquímica em decorrência -
de relaxação V-T/R[30,63,72-81J, por outro lado, a adição de um gás
inerte à amost r a a ser irradiada pode elevar o rendimento da reação
[82,83J.
Os parâmetros controláveis com relação ao laser são : in
tensidade [12J , baixa fluência (supressão [84J) ou elevada fluência
(relaxação rotacional resultando em repopulação dos níveis da molé-
cuIa de modo a prosseguir no processo de absorção [85J) e a forma
do pulso do laser (quanto mais longo o pulso do laser maior a prob~
bilidade de ocorrer desativação por colisões a uma pressão de gás
inerte constante). Todos estes fatores podem influir, ora ele vando
ora diminuindo o rendimento de certas reações fotoquímicas que con
tenham gás inerte no sistema [12,83-87J .
Nestes sistemas o efeito colisional é pr eponderante . Co

63.
lisões entre moléculas e xc itadas com outras não excitadas podem ser
classificadas em dois tipos: transf erência de energia de uma moléc~
la excitada a outra não excitada (ou vice-versa) ou distribuição da
energia de excitação entre os diversos modos internos da molécula -
excitada (transferência de energia V-V ) . Como a energia interna de
uma molécula devido à sua intera ção com o campo de r adiação é acumu
lada num determinado modo para pequ ena energia total absorvida,a re
distribuição desta mesma energia a outros modos deve influenciar a
probabilidade da molécula se dissociar e de promover reações quími
cas [88J. Portanto, aparte o controle do processo de difusão e do
processo térmico, são esperados a partir de colisões entre molécu
las de gases inertes e da espécie absorvente, relaxação ro taci onal,
defasagem do processo de absorção coerente e uma redistribuição ou
supressão da energia vibracional [83J.
6.4.2 - Experimentos com éter dietílico e gases inertes
No que toca ao éter die tílico, foi realizada a irradia
ção da mesma em presença de argônio, hidrogênio, hélio, neônio e
óxido nitroso, verificando-se quanti ta tivamente a variação do rendi
mento dos prod utos por cromatografia em fase gasosa. Um estudo mais
rigoroso foi realizado com argônio, variando a pressão do mesmo pa
ra uma pressão constante de éter, conforme apresentado na tabela 8.
Quando da irradiação da amostra de éter em mistura com
pressões de argônio variando de zero a 3,00 Torr, o rendimento dos
produtos não foi significativamente afetado, porém a pressões de a~
gônio de 5,00 a 60,0 Torr ocorreu um decréscimo marcante nos mesmos
(Fig . 16 ) .
De um modo geral, o argônio agiu como um desativador
das moléculas de éter dietílico excitadas pelo laser, o que explica
os rendimentos absolutos inferiores obtidos em todos os experimen-

-I
-• ...
Tabe la 8
Po tência Pressão de Pressã o dos Produtos/Pressão i nic ial de éter dietílico
(J/ p ) Argônio (To rr) CO CH4
C2H4 C2H6
0 , 5 0,00 (* ) 0,0448 0 , 1005 0,0305
0,5 1 ,0O 0 , 0666 0,1244 0 , 0399
0,5 2,00 0 , 0483 0,0945 0,0309
0 , 5 3,00 0,0575 0, 108 O 0,0329
0,6 5,00 0 , 0260 0,0194 0,1241 0,0466
0,6 20 , 0 0,0505 0 ,0 485 0 , 1084 0 ,04 04
0,6 40 , 0 0,0782 0,0554 0,0954 0 ,030 8
0,6 60,0 0,1118 0,0875 0,0954 0,0293
0,6 0,00(**) 0,0764 0,0741 0,1 42 1 0,0512
(*) ° argôni o apresenta mesmo tempo de reten ç ão (Porapak - S) .
(**) Valores obtidos do estudo em função da potência.
Lin h a P(20) a 9, 6 jJ m.
CH 3 CHO C2H5
OH
0 , 0202 0,0168
0,0192 0,0 16 7
0 ,0 183 0,0 122
0,0163 0 , 011 1
0,0237 0,0 248
0\ +:- .

I-< Q) ~
~
Q)
"O
"""" co
'''" u
'''" c ',,"
o IC'Il CIl CIl Q)
I-< p.. -CIl o ~
::l "O o I-< ~
CIl o
"O
o IC'Il CIl CIl Q)
I-< p..
O, 150
O, 100
0 , 050
0,000
65.
~
/~ C2H6
/~
10,0 20,0 30,0 40,0 50,0 60,0
Pressão de argônio (Torr)
FIGURA 16
Influência de um gás inerte (argônio) no rendimento
dos produtos da dissociação do éter dietílico
T IlI l ll

66.
tos, quando comparados aos da irradiação do éter puro, apesar de os
rendimentos relativos se comportarem de maneira bem diversa. Neste
sistema a transferência de energia V-T, R durante colisões entre as
moléculas de éter excitadas com moléculas de argônio deve predomi-
nar.
° decréscimo do rendimento de etileno, etanol, acetal
deído e etano pode ser explicado pelo que foi mencionado. Isto po
deria sugerir, talvez, a existência de duas vias de dissociação uni
molecular do éter dietílico: a dissociação em etileno e etanol e a
outra, acetaldeído e etano. É certo que a primeira via deve predo
minar, visto que o rendimento do etileno é sig nificativamente maior
em relação aos outros produto s . A se gunda via (formação de eta no)
ocorre quase com certeza por via radicalar (recombinação de 2C H3 ),e
o comportamento apresentado estaria relacionado com a dificuldade -
de colisão entre dois radicais CH 3 , pela diluição, favorecendo a
formação de CH 4 por colisão com qualquer mol éc ula hidrogenad a, con
forme já discutido anteriormente. Laidler[39J verificou que o eta
no se decompõe em etileno e hidrogênio a temperaturas pró ximas de
600 oC o Este processo seria viáve l, caso o re ndiment o de etileno -
não fosse decrescente em função da pressão de arg ônio, pois certa
mente temperaturas vibracionais superiores a esta são obtidas na re
gião focal [18J.
Qua nto ao rendimento crescente do CO e CH 4 com o aumen
to da pressão de argônio, está relacionado, provavelmente, ao com
portamento decrescente do etanol e acetaldeído envolvidos em proce~
sos colisionais de dissociação, originando H2' CO e CH 4 .
Na Tabela 9, por sua vez, estão os dados referentes aos
estudos dos sistema s éter/H 2 , He, Ne e N20.
Foi observado que para o sistema éter/gases nobres,He e
111 '1

:; ..I
Tabela 9
Gá s mi s tura do Potência Pressão dos Produtos/P r essão i nicial de éter dietílico
(T orr ) (J/p) CO CH 4 C2 H4
He
5,00 0,6 0,050 8 0,0474 0,0979
20,00 0,6 0,0333 0,0269 0 , 064 3
Ne
20 ,0 0 0,6 0,0927 0, 06 50 0 , 1078
NO
26 , 5 0 , 6 (**) 0 ,091 8 O, 1003
H2 20,0 0,6 0,0622 0,0427 0 ,0708
N2 0
20,0 0 , 6 0 , 0190 0,0193 0 , 0658
nenhum gás
misturado (*) 0 , 6 0,0764 0,0741 O, 1421
(*) Valor obtid o do estudo em funç ão da potência.
(**) ° tempo de reten ção d o NO é igual ao de CO.
Linha P(20) a 9,6 ~ m.
C2H6 CH3
CHO C2H5 OH
0,0346
0,0263
0,0361
0,0229
0,0254
0 ,0 192 0,0 15 1 0 ,01 35
0 , 0512 0,0237 0 ,0248
0\ -....J

68.
Ne, o rendimento dos produtos foi menor, comparado com os valores
de referência (éter puro), somente o CO na mistura éter/Ne destoa
desta afirmação. ° baixo rendimento dos produto s dev e estar rel~
cionado, ta mbém, com a desativação de moléculas de éter excitadas
pe lo laser, por transferência de energia V-T, do mesmo modo que
no estudo do sistema éter/argônio. ° mesmo foi verificado para
o sistema éter/H 2 , aonde nem mesmo foi observado um aumento no ren
dimento de C2H6 o que seria esperad o , caso a rea ção
ocorresse de maneira significativa.
A tentativa de irradiar o éter misturado com N20 , foi
a de observar um possível processo de combu s tão, poi s o óxido ni
troso é um comburente conhecido [89J. Nada ocorreu a tais pres
sões, a não ser um va lor inferior no re ndim ento dos produtos de
dissociação do éter, como no experimento com os gases nobr es . Nem
mesmo um novo pro duto foi detectado como no estudo da reação en
tre átomos de boro em presença de óxido nitroso [90J.
6.5 - Estudos em misturas de éter dietílico com óxido
nítrico
6.5.1 - A ação de captadores de radicais livres
o estudo da ação de captadores de radicai s livres no
processo de formação dos produtos de uma reação química em fase -
gasosa auxilia a proposição do mecanismo da mesma . Os métodos
utilizados para tais estudos são diversos, inclu i ndo a de tecção -
de radicais livres pelo estudo espectroscópicos dos estados exci
tados dos radicais [3J, assim como análise dos produtos finais ob

69.
servando reação dos radicais formados com o captador ou a inibição
da cinética de formação dos pr odutos detectados [67,91-94J.
Nem sempre a adição de captadores inibe uma reação de
pendendo da reatividade dos radicais formados como é o caso do sis
tema CH 30CH 3/NO ou propileno [95J, ou no estudo da eficiência da
reação de decomposição do etanol no processo de absorção multifotô
nica do sistema CH 3CH 20H/NO, apresentando um aumento da eficiên-
cia global da reação até pressões de 3,00 Torr de NO, decaindo a
pressões mais elevadas deste último [62J.
6.5.2 - Irradiação de amostras éter/NO
No estudo da irradiação do sistema éter/NO, foi verifi
cada um aumento do rendimento de CH 4 em detrimento de um menor va
lor para C2H6 , de acordo com os resultados da tabela 10. As con
dições e xperimentais foram as seguintes:
Pressão de éter dietíli co: 5,00 Torr
Número de pulsos: 200
Quanti dade de energia por pulso: 0,6 J
Lin ha do laser: P(20) a 9,6 ~ m
Coluna cromatográfica: Porapak-S (35 0 C)
Este resultado não permite uma conclusão objetiva,pri~
cipalmente por não ter sido detectado por cromatografia em fase
gasosa a formaçã o de HNO e nem tão pouco CH 3NO, C2HSNO, HCN ou
CH 3CN, produtos observados na piróli se inibida por NO do próprio -
éter dietílico [38,40J. Uma explicação seria a de que na região
focal- onde provavelmente todo o processo acontece- deve ocorrer -
uma rápida recomb inação dos radicais formados, através de reações
com energia de ativação muito próxima de zero, fato este que torna
praticamente ineficiente a ação de captad or es de radicais livres.

Pressão de
NO
(Torr)
26,5
Éter puro
(Torr)
5,00
Tabe l a 10
Pressão dos Produtos/Pressão inicial de éter
dietílico
co
0,0 9 18 0,1003 0 ,0 229
0,0764 0,0741 0,1421 0,0512
(*) Apresent a mesmo tempo de retenção que o NO.
70.

71.
Uma solução seria a de irradiar uma pequena quantidade de éter
( < 1 ,00 Torr) em excesso de NO. Contudo, a sensibilidade da apa
relhagem na detecção dos produtos finais foi o fator limitante em
nosso trabalho.
A ineficiência do NO como captador de radicais livres,
foi verificada também na referência 63, com relação ao radical CH~
em que reações deste radical compet em com a rápida reação de recom
binação, levando à formação de C2H6 . Todavia, o rendimento supe
rior de CH 4 , no sis tema éter/NO, deve estar relacionado também com
a eficiência de colisão dos radicai s CH 3 forma dos com uma molécula
hidrogenada qualquer, de mod o a hidr oge nar o radic al ao invés de
permitir recombinação do mesmo, como c itado no c aso acima [63J.
6.6 - Estudo do sistema éter/oxigênio
6.6.1 - Combustões
Os estudos de queima s de co mpostos outro s que não pe
que nos hidrocarbonetos, têm contrib uído muito pouco para o enriqu~
cimento deste tema, provavelmente devido aos comple xos mec an ismo s .
que envo lvem a combustão destes sistemas. O estudo do sis te ma
éter/0 2 não foge à regra. Porém, é de se esperar que ele deva se
comportar de maneira semelhante às reações de oxidação dos hidro
carbonetos maiores que o metano estudados até o momento [96,97J.
A forma çã o de radicais livres, pelo craqueamento térml
co dos compostos orgânicos, certamente contribui para as etapas de
iniciação de uma reação de combustão, gerando material altamente -
reat ivo para sustentar o processo. Co ntudo é aceito que a combus
tã o s e inicia pela reação
---->
cuja end otermicidade está em torno de 40 a 50 kcal / mol [9 8J . Desde

72.
que o oxigênio, por si só, é considerado um di-radical livre e es
tando em abundância nos processos de combustão, ele reage com vá-
rios dos radicais formados, produzindo espécies instáveis que se
decompõem pr omovendo material reativo:
R· + O2 ----> RO . 2
o processo em cadeia pode terminar por recombinação dos vários ra-
dic a i s formados ou desativação na s parede s . Certamente, em alguns
dos pro dutos resultantes que apresentem uma ligação fraca esta po-
derá ser rompida, mesmo a temperaturas relativamente bai xas , como
por exemplo as reações:
ROOH ----> RO· + OH ·
que apresentam energia de ativação próximas a 40 kcal/mol.
Para hidrocarbonetos maiores que o metano, a in ic iação
envolve a remoção de um hidrogênio (secundário ou terciário), de
modo que o radical formado é atacado por uma molécul a de oxigênio :
Ambas as reações são exotérmicas e envolvem uma baixa energia de
ativação, portanto são rápidas .
Pouca informação é encontrada na literatura de siste-
mas de substâncias orgânicas em mistura com oxigênio perfazendo a
combustão induzida por radiação infravermelha. Em muitas das ex-
periências o oxigênio age como um gás inerte estimulador, inibidor,

73.
captador de radicais livres, ocasionador de transfer ência de ener
gia vibracional intramolecular ou mesmo responsável por um enriqu~
cimento isotópico [6 2 ,85,91,94,99-1 06 ] resultados diversos ao de
uma combustão .
Entretanto, Riveros et al e irradiaram amostra de eta
nol em presença de oxigênio com o intuito de verificar o comp orta
mento do mesmo através da irradiação com laser infravermelho. Ao
ser i rradiada a mistura de etanol com oxigênio, foi observada uma
lum i nescência azulada em torno da região focal, sendo observadas -
re açõe s quimi luminescentes que perduram por tempos muito maiores -
do que o pulso do laser [107J.
OS produtos da irradiação do etanol com oxigênio, iden
ti ficados por cromatogra f ia em fase gasosa em c oluna de Porapak-S,
for am: metano, etileno, acetileno, eta no e dió xido de carbono . O
dió xi do de carbono foi formado em quantidade relativamente pequena
Leit e observou que o aumento da pre ssão de oxigênio na mistura, de
5 ,00 para 15,0 Torr, resultou em aumento da produção de dióxido de
car bono, metano e acetileno, mas não modificou a do etileno e do
etano. Explico u este efeito admitindo uma redistribuição int ermo
lecular de energia por meio de colisões com moléculas de oxig êni o ,
mais freqüentes na experiência com ma io r pressão deste; as vias
que levaram à formação de etileno e etan o não ser iam se nsíveis a
tal efeito .
Final mente, aventou a hip ótese para a pequena extensão
em que oc orreu a combustã o : no vol ume de reação a co ncentraçã o de
radicais intermediários seria tão grande que a probabilidade de re
combinação predom inaria, ao invés da colisã o e reaçã o subseqüente
com moléculas de oxigênio. Também devido ao curto t empo de vida
das moléculas de etanol superexcitadas, a probabilid ade de colisões

74.
com moléculas de oxigênio antes de se decomporem ser i a pequena.
6.6.2 - Rendimentos dos produtos na irradiação do
sistema éter/oxigênio
Na irradiação da amostra de éter dietílico (5 ,0 0 Torr )
em mistu ra com oxi gênio (5,00-35,00 Torr), foi detect ada apenas
uma pequena quantidade de CO 2 , juntament e c om os dema is pro dut os -
já observados devendo-se isto, prov a velm ente, a reações rápidas e~
tre radicais formados na região foc al, ao invés de col isões e rea
ções subseqüentes dos mesmos com molé cula s de oxigênio, poi s as
rea çõ e s entre dois radicais apresent am energia de ativação
pró ximas de zero [ 108J .
muito
Em seguida, foi verificad o se por ventura algum dos prQ
duto s da dissoci ação do éter era con su mido priori tari amente, ini
ciando assim o pro ce sso de combu stão, de modo que foi realizado o
estudo do rendimento individual dos produtos em fun ção da pres-
são de O2 misturada ao éter dietíli co . Os dados e as condições ex
perimentais es tão indicados na tabela 11.
A figura 17 ilustra o cresc iment o dos r endiment os de
CO, CH4 , C2H4 , C2H6 e CH 3CHO até pres s ões de 25 , 0 Torr de oxigê ni o,
decaindo com pressões maiores, isto é, próximas ao limite de que
bra dielétrica (fenômeno discutido na secção 6.6.5 ) . este c ompor
tamento crescente deve estar relacion ado com a ativa ç ão de molécu
las de éter por colisões com molécul as de O2 ou repopulação rota
cional . Nes te último caso, deve ocorrer uma redistr i buição inter
mo l ecula r de energia por meio de co l isões com moléculas de oxigê
nio, onde o efeito de repopulação rota c i on al nas moléculas de éter
aumenta a eficiência de absorção de energia, e xplicando o aum ento
do ren di mento de C2H4 . Ao mesmo tempo, deve ocorrer o efeito de -
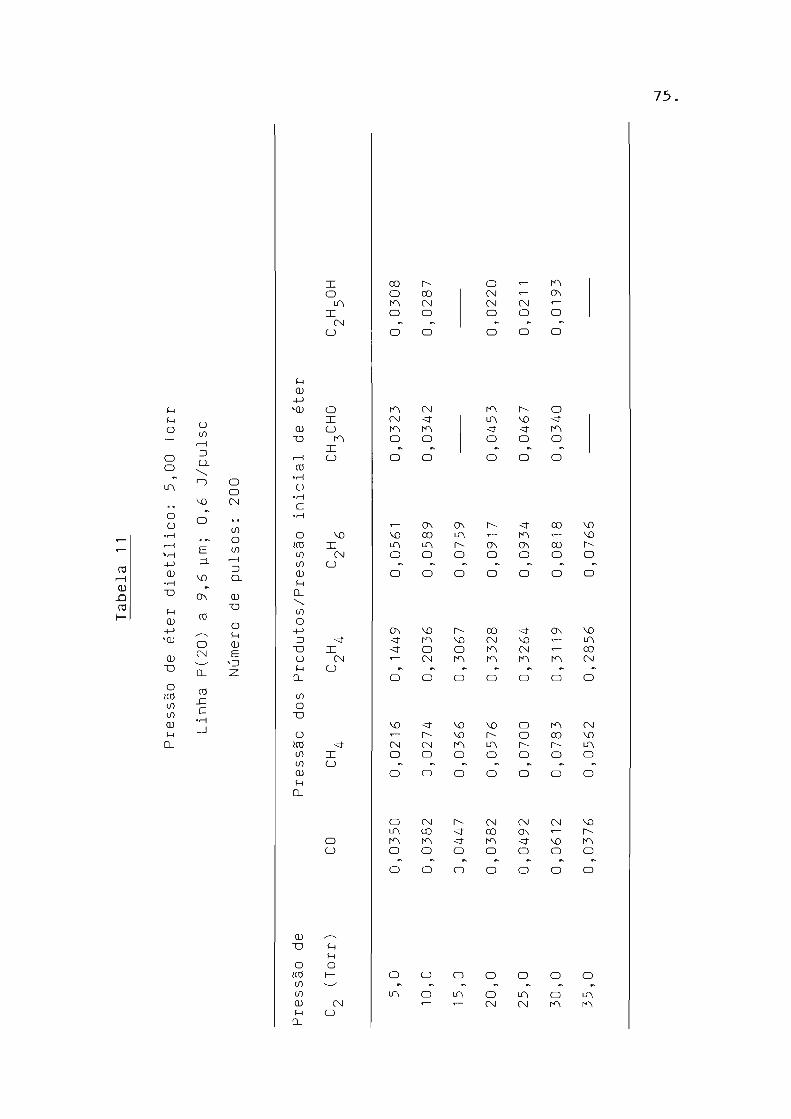


77.
banho térmico, pois durante colisões de moléculas de éter excitadas
pelo laser com moléculas de 02 existe transferência de energia vi
bracional para o volume de reação do sistema. Esta energia cedida
ao sist ema, por sua vez, pode favorecer processos de reações coli
sionais radicalares ou não, o que explica, por exemplo, o aumento -
dos ren dimentos de CO, CH 4 e C2H6 . No entanto, a pressões de 02
acima de 25 ,0 Torr, havendo maior di l uiç ão na região focal, a recom
binação dos radicais formados é em parte dificultada. Em toda a
fai xa estud ada, certamente ocorre um a competi ção entre o pro cesso -
de repopulação rotacional e o de tran sferê ncia de energia. A pres
sões maiores, é possível que o proc esso de de sa tivação semelhante -
ao do sistema éter/gases inertes, pr edomine, diminuindo o rendimen
to dos produtos [86J.
A propósito, a partir de pressões de oxigênio ac i ma de
20 , 0 To rr , o rendimento de CH 4 foi s uperior ao de CO, resultado
opo s to ao ver ifi ca do na irradiação do éte r pur o . I s to se deve , po~
sivelmente, a reações que consomem CO originando CO 2 ' pois pequena
quant idade deste último foi detectada por cromatografia em fase ga
sosa .
Ao mesmo tempo, o C2H50H aprese nto u um rendimento decre~
cente, pois com o aumento da pressão de oxigênio deve a umentar o nú
mero de colisõ es, obtendo o mesmo, através de banho térmico, ener
gia suficiente para se dissociar em H2' CO, CH 4 e CH 3CHO, o que po
de explicar em parte o rendimento crescente destes pr odutos. Uma
supressão da via responsável pela formaçã o de etan ol impli car ia fa
talmente num rendimento decrescente para o etileno (j á que ambos d~
vem ser formados pela dissociação unimolecular do éter), hipótese -
es ta descartada pelos resultados experimentais obtido s .

78.
6.6.3 - Estudo cinético do sistema éter/oxigênio
Outro estudo realizado, abaixo do limite de quebra dielé
trica, foi o estudo cinético do sistema éter (5,00 Torr)/02(10,OTor~
que indicou um rendimento crescente para todos os produtos de disso
ciação do éter em função do aumento do número de pulsos a uma mesma
quantidade de energia. O comportamento dos rendimentos dos produtos
está reproduzido na Figura 18, e os resultados obtidos estão na Tabe
la 12.
O comportamento apresentado, rendimento crescente para -
todos os produtos, foi análogo ao da irradiação da amostra de éter
puro. Contudo, era esperada também uma indicação de que algum dos
produtos formados apresentasse um comportamento decrescente, indican
do uma possivel contribuição para a pequena formação de CO 2 , ou trou
xesse algum esclarecimento quanto às vias de dissociação propostas -
anteriormente. Com relação a este último item, o estudo deste siste
ma permite afirma r que o ox igênio age como um estimulador, pois os
rendimentos abso lut os observados com 200 pulsos nesta mistura são su
periores aos valores obtidos na amostra pura. Entre tanto, simulta
neamente à dissociação unimolecular do éter, devem ocorrer também
processos colisionais, pela própria natureza dos produtos forma do s:
CO, CH 4 , C2H6 e mesmo CH 3CHO. Portanto, não ocorre um estimulo ou
inibição de determinada via de dissociação, mas sim a participação -
de alguns produtos formados, como por exemplo o etanol, favorecendo
a formação de CO, CH 4 e H2 pela própria dissociação em processos co
lisionais.
6 , 6.4 - O fenômeno de quebra di elétrica
Uma proposta para o complexo mecanismo em que ocor re a
quebra dielétrica de um gá s i~duzid a por um laser, seria a de que



81.
uma ionização inicial causada pela absorção de fótons pelo gás, re
sultaria num plasma de íons a uma elevada temperatura (s uperior a
10 4K). A expansão deste plasma, no entanto, resultaria numa onda
de choque . Entretanto, é possível com o auxílio da espectroscopia
por resolução de tempo determinara que intervalo ou a que temperat~
ra do plasma em expansão são formados os produtos estáveis. Além
disto, não é possível com base nos produtos formados estabelecer se
os mesmos s ão formados por combina ções entre reações íon-molécula,
radical-radical ou radical-átomo.
Deve ser considerado ainda que o fenômeno de quebra di
elétrica induzida por lasers não está restrito a uma interação res
sonante entre a radiação e a matéria absorvente, de modo que não re
quer um "c asa mento" entre a linha de absorção da amostra e a freqLE.Q
cia do laser [109J . No entanto, a quebra dielétrica induzida por -
laser de CO 2 envolve um mecanismo de rea ção em ca deia co nhecido co
mo avalanch e de ionização. ° mecanismo requer a presença de uma p~
quena quantida de de elétrons, os quais acelerado s pelo campo do la
ser, ocasionam uma avalanche, culminando num plasma ionizado. Os
elétrons iniciais podem ter origem a partir de partícula s de poeira,
raios cós mi cos ou mesmo das janelas da cela de reação [110J.
Miknis e Biscar [llJ relataram um estudo de reações do
t iacic lobutano e tiaciclopentano em mistura com 02 induzidas por um
laser de r ubi. Os produtos gasosos identificados foram: CO 2 , 50 2 '
H25, H20, C05 , C5 2 e C2H2 . Nem tod os esses produtos f oram observa
dos para cada pressão de 02 adicionada, e nem tão pou co observados
para am bo s os compostos . Nenhuma r eaç ão foi observada em tiociclo
pentano na ausência de 02' ou com pre s são parcial de 02 de 50 Torr.
A uma pre ssão de 02 de 100 Torr, CO 2 , 502 e H20 foram detectados co
mo produtos de reação para ambos os compostos e somente a pressões

82.
de O2
acima de 150 Torr os demais produtos foram detectados. Por ou
tro lado, observaram que a pressões de cerca de 1500 Torr de O2 a
reação foi suprimi da. Sugeriram, também, que a distribuição dos pr~
dutos é provavelmente afetada pela temperatura do plasma, assim como
as reações químicas que ocorrem durante a quebra dielétrica do gás
dependem da potência do laser, a qual por sua vez pode ser relaciona
da c om a temperatura máxima do plasma. Opinaram ainda que em suas -
experiências, devido à elevada temperatura obtida, tiociclobutano e
tiociclopentano alcançam uma "nã o existência" e somente íons, átomos
e radicais livres de elevada energia são formados, os quais reagem -
ao longo da frente de choque produzindo produtos estáveis.
Aldeman [112J observou a emissão de C2 como a única emis
são poliatômica na quebra dielétrica induzida por laser em tetraclo
reto de carbono, clorofórmio, acetona, meta nol e hex a no. Outras
emissões observa das foram de espécies atômicas ionizadas incluindo -
C2+ . Analoga mente, outros autore s estudaram os diver s os radicais li
vres originados pela fotólise atravé s de um laser infravermelho
[113-116J, responsáveis ou não por emissão na região do visível.
Ronn e Langsam [109J, por sua ve z, estudaram a quebra di
elétrica de metais carbonílicos, de SF 6 e OCS. Observaram que o fe
nômeno da quebra di elétrica depende da pressão da amostra gasosa em
estudo, da pressão do captador utilizado no experimento e da potên
cia e duração do pulso, parâmetros estes que variam de acordo com a
amostra em estudo . Quanto à pressão da amostra, para este fe nômeno,
está intimamente relacionada à sua polarizabilidade e momento de di
polo, de tal forma que um decréscimo da polarizabilidade da amostra
gasosa está condicio nada a um aumento do momento de dipolo da mesma.
Verificaram que moléculas de elevad o momento de dipolo apresentam
quebra di elétrica a pressões relativamente baixas, por exemplo, de

83.
22 Torr para OCS.
6.6.5 - A quebra dielétrica no sistema éter/oxigênio
No que concerne à amostra de éter dietílic o , ocorreu um a
forte luminescênci a em todo o volume da ce la logo no s primeiros pul-
sos de irradiação. O si s tema constituído de 5,00 Torr de é ter e
pressões entre 35 , 0 e 50,0 Torr de ox igênio fo i irradiado co m a li-
nha P(20) a 9,6 ~ m, com uma quantid ade de energia fi xa da em 0 ,6 J/ pul
so . A anál ise dos produtos em coluna Porapak-S (3S oC), por cromato-
grafia em fase gasosa, revelou traços de hidrogênio e uma área gran-
de correspo ndente a dió xi do de carbono. Nesta coluna o oxigênio
ap re sentou mesmo tempo de retenção qu e o monóxido de carbono, sendo
necessário anal isar os produtos da co mbu s tão em coluna PM-13X (3S oC),
quando foi observado também CO com o produto da combu s tão. Os rendi-
mento s obti dos e s tão na tabela 13. Es ta é uma indica çã o de que não
se trata de uma c ombustão completa, ma s si m de um fenômeno explosivo,
conforme mencio nado por Pilcher et aI [117J, que obtiveram a combus-
tão co mp leta do éter obtendo o va l or de + 6 H= - 657,52 - 0,18 kcal/mol
para o pro ces so , mostrando trat a r- se de um proce ss o altamente exotér
mico.
Outro resultado relev ante é o de que a pressões de oxig~
nio acima de 40,0 To rr a combustão semp re ocor r eu apenas com o pri-
meiro pulso prove niente do la ser de CO 2 , Es te fato está com certez a
re la cionado com o próprio processo de quebra dielétrica do gás: nes-
tas condições, forma-s e um plasma na região focal e os radicais ou
íons, formado s em grande qu antidad e, podem propagar a reação por to-
da a ma ssa gasosa [99,111, 118-1 20J.
6.6.6 - Considerações sobre a combustão do éter dietíli-
co
De acordo com alguns do s resultados divul gad os na litera

Pressão de
°2(Torr)
40,0
84.
Tabela 13
Pressão do s produtos/Pressão de éter dietíli co
H2 CO CO 2
0 , 0900 0,6200

85.
tura, pode ser aventada, em parte, que a combustão do éter dietíli-
co, observada neste trabalho, deva ocorrer de maneira semelhante ao
observado para o n-heptano. Coats e Williams [121] relataram um es
tudo da combustão do n-heptano pela técnica de tubo de choque onde
verificaram o desaparecimento do hep tano mesmo antes da ignição de
modo a dividir o processo em três r egiões: degradação do n-heptano
produzindo C2H4 e CO; desaparecimento de C2H4 e produ ção de C2H2 ; e
consumo de C2H2 e O2 com formação de CO 2 .
Assim, no si s tema éter/0 2 é possível que o éter se de
componha inicialmente nos produtos detectados, os quais reagiriam -
numa rápida seqüência formando CO e H2' que por sua vez pode r iam
reagir com um excesso de oxigênio presente originando CO 2 e H20.
Com base nos resultados e modelos apresentados para as
etapas de oxidação de pequenos hidrocarbonetos [96,122], é aventado
o seguinte esquema simplificado para a combustão do éter dietílico :
I I I I CO CH 4 C2~ C2H4 C2H2
!l~l~ 1 1 .
êH30/r~r C3H3 C2H
1 ~ CH20 ~CH3CHO /
1 t CHO CH3CO CH 2CO 1 ~ .v ~ co co CH 2
H·, O·, OH·, H2' H20 e C2H50H foram omitidos por que stões de simpll
ficação, assim como etapas que envolvem C2H3 · , C2H· , :CH 2 , CH·, C2 e
co

86.
outras espécies intermediários .
Com relação ao mecanismo da oxidação do CO, este pode
envolver átomos de oxigênio, poré m a mane ir a exata da reação ocor
rer é duvidosa [97J . Le wi s e von Elbe [1 23J suge riram qu e a rami
ficação da cade ia ocorre através da oz onaj a ss im,
-->
03 + CO --> CO 2 + 20
Contudo, 03 apresenta pouca reativid ade com CO abaixo de 523 K.Ou
tro es qu ema proposto seria :
O + CO ---> CO 2*
--- > CO 2 + 20
um pr ocesso exotérm ic o e possível de oco rrer. Certament e, em sis
temas contendo hidr ogênio, o pr opagado r de cadeia mais eficiente
é o radical OH · , co mo no caso da combu s tão de CO contendo traç os
de água [124J :
CO + OH· ---> CO 2 + H·
H· + O2 ---> OH· + O·
( E = 0.6 kcal/mol), a
( E = 16 . 8 kcal/mol ) a
mais outras reações envolvendo oxigênio atômi co [125- 127J .
Outra hipóte se , porém remota, seria a de que estando
o é ter dietílico misturado com oxigênio e sendo irradiado c om uma
quantid a de suficiente de energia, seja convertido ini c ialmente em
peró xi dos in st áveis, os quais propag a riam o processo por toda a -
fase gasosa .
Não estendendo em demasi a o as s unto, é apresentada a
seguir uma lista de algumas rea ções possíve i s no sistema éterl 02
[92,94 ,1 24,128, 129-132J.

8 7 .
Reações O Ho E a
( kca l/mol) (kcal/mol)
H· + O2 --> OH' + O 16,8 17
CO + OH · - > CO 2 + H· -24,4 0,60
H' + OH· --> H2 + ° - 1 , ° 7,0
CH· + O· -->CO + H· - 177 6,5
CH· + °2 - > CO + OH' -159
CH · + O2 - > CO
2 + H· -1 84
OH· + H2 - > H· + H20 -1 5 ,0 5,3
OH' + C2H6 - > H20 + C2H5 - 21 ,2
H· + CO 2 --> OH' + CO 24,9
OH · + CH4 - > H2 0 + CH 3 -15,2 6,5
°3 + C2H6 --> (CO, H2O,H 2 ,C0 2 , CH 4 ) 13,9 a 14,7
7 . Conc lusões
De uma ma neira gera l a absorção de um fóton infra v erm~
lho nã o ind uz uma drástica transforma çã o nas propriedades químicas
dos reage nte s , como os fótons visíveis e ultravioleta s . Contudo,
um a intensa radiação i nfravermelha de determinada freqüência pode
seletivamente e eficientemente depositar vários fótons em molécu
las isoladas causando a dissociação das mesmas. Neste conte xto es
tá in s erido este trabalho com a amostra de éter di etílico.
De acordo com os resultados obtidos, foram notadas cer
tas particularidades no pr ocesso de dissocia çã o infravermelha do
éter dietílico no que di z r espeito aos pro dutos f or mados e ao com
po rtamento dos mesmos . Por exe mplo: a formação de acetile no e o
rendimento decrescente de e tano, álcool etílico e acetaldeído com

88.
o aum ento da pressão da amostra irradiada a presentaram um comport~
mento diferente do observado na pirólise do éter dietílico [37J, -
não obst ante o fato de os produto s majorit á rios serem os mesmos.
Quanto à fotólise do éter no ultravioleta [ 41J, a formação por
via molecular do etileno é a via que menos contribui para a forma-
ção do mesmo, predominando a via radicalar como um todo na origem
dos prod uto s formado s .
Entretanto, a decomposição do é ter dietíli c o pela ab-
sorção multifotônica no infravermelho, observada nes te trabalho,e~
tá relacio nada, muito pr ovavelemente, a um proces s o de rearranjo -
molecula r c ujos pr odutos são obtido s a partir de um estado de tran
sição de quatro centros, semelhante ao que apresentou Freeman [133J
par a a sua pirólise:
* H H I I
-----> H - C C - H ( I ) I I H----O - C2H5
Tal processo foi sugerido para explicar a diferença da dependência
de pressão nos dois modos de decompo s ição do éter. O modo que fo~
ma acetaldeído envolve energia de ativação provavelmente superior
à da reação de formação de etanol. A formação de etano 1 (I), en-
volve uma nova ligação no oxigênio e a clivagem simultânea da lig~
ç ão SC-H, sendo uma reação de primeira ordem. Além disso, levando
-se em conta o elevado rendimento de etileno em relação aos demais
produt os (34,6%, irradiando a amostra com 200 pulsos a 1 J), prati

89.
camente constante com o aumento da pressão de éter irradiado, assim
como a linearidade apresentada pela soma dos rendimentos do etileno
e etano 1 em função da pressão da amo st ra, é muito provável que a
formação destes produtos ocorra quase que exclusivamente pela dissQ
ciação uni molecular já citada como processo (I), envolvendo uma
energia de ativação de aproximadamente 84 kcal/mol [39J, e o .6. H =16 , 6
kcal/mol.
Não é de sc art ada a po ssibi lidade de ocorrência da rea-
ção (11) ; es ta provavelmente deve ocorre r na di ssociaçã o da amostra
de éter irrad i ada devido à s altas energ i as int er nas que podem ser
atingidas (t alvez por transferênci a colisio na l de energia vibracio-
nal de molécu las de éter excitada s for ne cen do mai s energia a moléc~
las de éter j á excitadas internamen te; de modo que a f or mação de
etano não deve ocorrer so mente de vido à r eco mbina çã o de dois radi
cais livres CH 3 . Não foi encontrado, na literatura, um valor da
energia de ativação para a rea ção (1 1 ) , que entretanto dev e ser su-
perio r à do processo ( I) .
Analogamente, a soma do s rendimentos de acetaldeído e
etano apresentou uma constância em f unção do aumento da pressão da
amost ra irradiada, podendo ser isto um indício de se originarem de
uma mesma vi a de dissociação a partir do éter dietílico.
Por outro lado, o rendimento decrescente apresentado p~
lo etanol e acetaldeído em função do aumento da pressão de éter po-
de ser interpretado como provindo da dissociação dos mesmos, atra-
vés de col isões, de modo a contribuírem para o rendimento crescente
de CO e CH 4 , fato este observado também no si s tema éter/argônio. No
entanto, seria esperado um rendimento crescente, também, para o eta
no em função da pressão da amostra irra diada, conforme ocorreu com
o CO e CH 4 , caso seja considerado que estes produtos se originam a

90.
partir de reações de colisões entre as moléculas do sistema. Uma
possível explicação para o rendimento decrescente do etano com o
aumento da pressão de éter, seria a de que o mesmo (ou parte do
mesmo) é orioinado a partir da recombinação de radicais CH 3 ; as
sim, com o aumento da pressão de éter au menta o número de colisões
por unidade de tempo, favorecendo reações radicalares outras -com
qualquer espécie hidrogenada - inclusive o próprio éter -, que
contribuam para a formação de CH 4 , o que pode ajudar a explicar o
seu rendimento crescente.
Outra possibilidade a ser considerada seria a reação
de dissociação do etano a qual Rice e Her zfe ld [1 34J , Lin e Back
[135J e Loucks e Laidler [136J, mostraram ser de elevada endot er-
micidade;
----- > 2CH 3 e ----- > C2H5 + H
E ~ 80 a 86 kcal/mol a o 6 H ( 00 ) = 98,1 kcal/mol.
Apesar de a temperatura vibracional em nosso sistema ser muito al
ta, nem propano nem butano foram observados como produtos, de mo-
do que esta disso ciação não deve ocorrer de maneira significativa •
neste sistema.
Por conseguinte, a formaç ão de CO, CH 4 e H2 deve es
tar relacionada com o rendimento decrescente do acetaldeído, en-
volvido em colisõ es ou reações de radicais livres [69J. Laidler
[39J, verificou que a temperaturas em torno de 600 °c o acetaldeí
do se decompõe em CH 4 e CO. Tal temperatura vibracional é segur~
mente obtida na região focal durante o processo de irradiação da
amostra, de sorte que uma fração do acetaldeído formado da decom
posição do éter contém, possivelmente, energia interna suficiente
para se dissociar em CO e CH 4", sendo isto uma provável explicação
para o rendimento crescente destes produtos no estudo em função -


92.
de iniciação da decomposição do éter podem ser:
(1) e
. . ( 2 ) ----- > C2H5 + OC 2H5
Rebbert e Laidler [139J, assim como McKenney [39J deram preferência
à quebra da ligação C-O, para explicar o mecanismo da pirólise do
é t er dietílico, justificando apresentar uma menor energia de ativa-
çã o , em vez do mecanismo proposto por Rice e Herzfeld, e Smith e
Hinshelwood [140J, isto é, a quebra da ligação C-C . Todavia, Gowen
l ock [ 141J mostrou que os argumentos de Rebbert e Laidler não eram
conclusivos e que a energia de ativação para ambos os processos é
praticamente idêntica (77,5 a 78,0 kcal/mol). Portanto, o mecanis-
mo proposto para essas duas possíveis vias de dissociação do éter ,
podem ser apresentadas como :
1 ) (C 2H5 )20 + nhv ----> OH 3 + CH 2OC 2H5
* CH 2OC 2H5 ---> CH 3 + CH 3CHO
2CH 3 ---> C2H6
* 6 HO= CH 3CHO ----> CH 4 + CO -4,6 kcal/mol
ou ainda
CH 3 + parede ----> remoção (término de cadeia).
o Ó H = 78,0 kcal/mol
6 HO= 14,5 kcal/mol . * > CH 3 + HCHO
E = 10 kcal/mol a
* HCHO -----> 6 HO= 1 ,6 kcal/mol
6 HO= -88,2 kcal/mol




96.
condições em que foi irradiada a amostra de éter dietílico apresen-
tou alguma indicação [70J, é provável que a formação de acetileno -
seja mesmo decorrente de reações de dissocia ç ão por proces sos coli-
sionais dos produtos primários provenientes do éter dietílico.
Em síntese, as prováv eis via s mais importantes para a
dissociação do éter dietílico indu zid a pelo laser de CO 2 são as se
guintes:
L (C 2HS) 20 + nh v > C2H4 + C2HSOH
* * C2HSOH > CH 3CHO + H2
* * C2HSOH > CH 4 + HC HO
* HCHO :> H2 + CO
* CH 3CHO > CO + CH4
* C2H4 > C2H2 + H2
2. (C 2HS)20 + nh v > CH 3CHO + C2H6
* CH 3CHO > CO + CH 4
3. (C 2HS)20 + nh v " CH 3 + CH 2OC 2HS
CH 2OC 2HS > CH 3 + CH 3CHO
. 2CH 3 > C2H6
CH 3CHO > CO + CH 4
CH 3 + ( C2HS)2 0 > C2H6 + CH 2OC 2HS
4 . (C 2HS)20 + nh v > 2C 2H4 + H2O, (possivelmente).




































