Embed Size (px)
Citation preview

1
ALINE DOS SANTOS GARCIA GOMES AVALIAÇÃO DO POTENCIAL DOS ÁCIDOS OLEANÓLICO E URSÓLICO, ISOLADOS
DE Lippia lacunosa, EM INIBIR A FUNÇÃO DE TRANSPORTE DA Pdr5p DE Saccharomyces cerevisiae E DE QUIMIOSENSIBILIZAR UMA CEPA DE Candida
albicans RESISTENTE A FLUCONAZOL
Dissertação de Mestrado apresentada ao Programa de Pós Graduação em Ciências (Microbiologia), Instituto de Microbiologia Prof. Paulo de Góes da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciências Biológicas (Microbiologia)
Orientador: Prof. Dr. Antonio Ferreira Pereira
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO INSTITUTO DE MICROBIOLOGIA PROF PAULO DE GÓES
FEVEREIRO DE 2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
FICHA CATALOGRÁFICA
GARCIA-GOMES, Aline dos Santos Avaliação do potencial dos ácidos oleanólico e ursólico, isolados de Lippia lacunosa, em inibir a função de transporte da Pdr5p de Saccharomyces cerevisiae e de quimiosensibilizar uma cepa de Candida albicans resistente a fluconazol / Aline dos Santos Garcia Gomes – Rio de Janeiro. 156, número de folhas. Dissertação (Mestrado em Ciências Biológicas). Universidade Federal do Rio de Janeiro/ Instituto de Microbiologia Prof. Paulo de Góes, 2010. Orientador: Antonio Ferreira Pereira Referencias Bibliográficas f: 121 1. MDR 2. S. cerevisiae 3. Pdr5p 4. Transportadores ABC 5. Lippia lacunosa 6. Ácido oleanólico 7. Ácido ursólico 8. C. albicans 9. Curcumina I. Ferreira-Pereira, Antonio II. UFRJ. Instituto de Microbiologia Prof. Paulo de Góes, Mestrado em Ciências Biológicas III. Avaliação do potencial dos ácidos oleanólico e ursólico, isolados de Lippia lacunosa, em inibir a função de transporte da Pdr5p de Saccharomyces cerevisiae e de quimiosensibilizar uma cepa de Candida albicans resistente a fluconazol.

3
ALINE DOS SANTOS GARCIA GOMES
AVALIAÇÃO DO POTENCIAL DOS ÁCIDOS OLEANÓLICO E URSÓLICO, ISOLADOS DE Lippia lacunosa, EM INIBIR A FUNÇÃO DE TRANSPORTE DA Pdr5p DE Saccharomyces cerevisiae E DE QUIMIOSENSIBILIZAR UMA CEPA DE Candida
albicans RESISTENTE A FLUCONAZOL Rio de Janeiro, 26 de fevereiro de 2010. ______________________________________________________________________
Dr. Antonio Ferreira Pereira, Prof. Associado, UFRJ
______________________________________________________________________
Dra. Ana Paula Vieira Colombo, Prof. Adjunto, UFRJ)
______________________________________________________________________
Dra.Tânia Beatriz Creczynski-Pasa, Prof. Associado, UFSC)
______________________________________________________________________
Dra. Eveline Gomes Vasconcelos, Prof. Associado, UFJF)
______________________________________________________________________
Dra. Rosangela Maria de Araujo Soares, Prof. Associado, UFRJ)

4
O presente trabalho foi realizado no Laboratório de
Bioquímica Microbiana, Departamento de
Microbiologia Geral, Instituto de Microbiologia Prof.
Paulo de Góes, Centro de Ciências da Saúde
(CCS), Universidade Federal do Rio de Janeiro,
sob a orientação do Prof. Dr. Antonio Ferreira
Pereira.

5
AGRADECIMENTOS
Inicialmente gostaria de agradecer a minha família, pelo apoio sempre constante e pela
torcida de que a formação que venho escolhendo para minha vida seja sempre a melhor. Não
teria como deixar de agradecer a meu noivo, José Miguel Machado, pelo carinho, amor, por me
entender, por estar sempre disponível para fazer qualquer figura não só para esta dissertação mas
também pôsteres de congressos e apresentações em geral, sem dúvida é ele que traz um toque de
beleza às minhas apresentações.
Aos meus amigos de laboratório, Antonio, Luciana, Geralda, Fernanda, Júlia e, mais
recentemente, Ana, gostaria de dizer que cada um de vocês contribuiu de uma maneira especial
para este trabalho, seja fazendo meus dias mais felizes e engraçados, seja me dando puxões de
orelha e me ensinando, muitas vezes, coisas que com certeza vou levar para o resto da vida, seja
me auxiliando indiretamente ou diretamente em meus experimentos, deixo a vocês aqui o meu
muito obrigada, vocês sem dúvida foram parte essencial destes anos que passei no Laboratório
de Bioquímica Microbiana e estão marcados em mim.
Obrigada a todos!

6
Como sempre, não teria como não
dedicar este trabalho aos meus pais.
As minhas vitórias são, e sempre serão, de vocês também.

7
RESUMO
Aline dos Santos Garcia Gomes
AVALIAÇÃO DO POTENCIAL DOS ÁCIDOS OLEANÓLICO URSÓLICO, ISOLADOS DE Lippia lacunosa, EM INIBIR A FUNÇÃO DE TRANSPORTE DA Pdr5p DE
Saccharomyces cerevisiae E DE QUIMIOSENSIBILIZAR UMA CEPA DE Candida albicans RESISTENTE A FLUCONAZOL
Orientador: Antonio Ferreira Pereira Resumo da Dissertação de Mestrado submetida ao Programa de Pós-Graduação em Ciências
(Microbiologia), Instituto de Microbiologia Prof. Paulo de Góes da Universidade Federal do Rio
de Janeiro, como parte dos requisitos necessários para a obtenção do título de Mestre em
Ciências Biológicas.
Dentre os diversos mecanismos que contribuem para o fenótipo de resistência a múltiplas drogas (MDR) em células fúngicas, a superexpressão de bombas de efluxo pertencentes às superfamílias de transportadores ABC e MFS, é a causa mais frequente de resistência a antifúngicos em isolados clínicos. Pdr5p, um transportador ABC presente em Saccharomyces cerevisiae, transporta uma grande variedade de substratos distintos, apresentando sobreposição de substratos com outros transportadores de fungos patogênicos, como os de Candida albicans, fazendo deste um interessante modelo de estudo para a identificação de novos compostos que poderiam funcionar como inibidores do MDR. O isolamento do ácido oleanólico e de uma mistura de ácido oleanólico e ácido ursólico de Lippia lacunosa, possibilitou um estudo para analisar a capacidade desses isômeros, diferenciados apenas pela posição de um grupamento metila, em inibir o efluxo de rodamina 6G promovido pela Pdr5p. O ácido oleanólico não foi capaz de restaurar o acúmulo deste composto, enquanto a mistura, quando usada na mesma concentração, mostrou uma capacidade moderada de inibir este efluxo. A utilização do ácido ursólico purificado mostrou que este composto era o responsável pela inibição visualizada para a mistura, apresentando uma forte inibição (~ 72%) da função de transporte da Pdr5p, necessitando de apenas 50 µM para promover a inibição do transporte. A caracterização da resistência de uma cepa de C. albicans hiperesistente a fluconazol isolada de um paciente HIV+ possibilitou uma avaliação do uso do ácido ursólico como um quimiosensibilizador quando utilizado em combinação com fluconazol, permitindo a inibição do crescimento desta cepa. Uma interação sinérgica entre estes dois compostos foi encontrada, permitindo reduzir o MIC80 desta cepa de 256 µg/ml para 4 µg/ml. Com este estudo nosso grupo mostrou uma nova função para o ácido ursólico como um inibidor do fenótipo de MDR apresentado por células fúngicas e um possível uso na clínica para sensibilizar cepas de C. albicans hiperesistentes ao tratamento com fluconazol.

8
ABSTRACT
Aline dos Santos Garcia Gomes
EVALUATION OF THE POTENTIAL OF OLEANOLIC AND URSOLIC ACID,
ISOLATED FROM Lippia lacunosa, TO INHIBIT THE TRANSPORT FUNCTION OF Pdr5p FROM Saccharomyces cerevisiae AND TO CHEMOSENSITIZE A Candida
albicans STRAIN HIPERESISTANT TO FLUCONAZOLE
Orientador: Antonio Ferreira Pereira Resumo da Dissertação de Mestrado submetida ao Programa de Pós-Graduação em Ciências
(Microbiologia), Instituto de Microbiologia Prof. Paulo de Góes da Universidade Federal do Rio
de Janeiro, como parte dos requisitos necessários para a obtenção do título de Mestre em
Ciências Biológicas.
Among several mechanisms contributing to the multiple drug resistance (MDR) phenotype in fungal cells, overexpression of efflux pumps, belonging to the ABC superfamily and to the MFS superfamily, is the most frequent cause of resistance to antifungal in clinical isolates. Pdr5p, an ABC pump present in Saccharomyces cerevisiae, transports a broad array of distinct substrates, with some overlaps with other pumps like those from Candida albicans, making it an interesting model of study useful for screening of new compounds that may function as MDR inhibitors. The isolation of oleanolic acid and a mixture of oleanolic and ursolic acid from Lippia lacunosa, enable a study to analyze the ability of these two isomers, differentiable only in the position of a methyl group, in inhibit rhodamine 6G efflux promoted by Pdr5p. Oleanolic acid was not able to restore rhodamine 6G accumulation, while the mixture, when used in the same concentration, showed a moderate capability to inhibit the efflux. The use of purified ursolic acid showed that this compound was the responsible of the inhibition visualized for the mixture, presenting a strong inhibition (~ 72%) of the transport function mediated by Pdr5p, needing 50 µM to inhibit this transport. A characterization of C. albicans strain hyperesistant to fluconazole isolated from a HIV+- patient enabled an evaluation of the use of ursolic acid as a chemosinsetizer when used in combination with fluconazole, permitting the growth inhibition of this strain. A synergistic interaction was found, permitting reduce the MIC80 from 256 µg/ml to 4 µg/ml. With this study our group showed a new function for ursolic acid as a MDR inhibitor for fungal cells expressing a MDR phenotype and a possible use in clinical practice to sensitize hyperesistant strains of C. albicans to fluconazole treatment.

9
Índice
1. Introdução 1
1.1 O uso de plantas medicinais pela humanidade 1
1.2 Resistência a múltiplas drogas 3
1.3 Os principais transportadores relacionados ao MDR em fungos 6
1.3.1 Transportadores ABC 6
1.3.1.1 Histórico 6
1.3.1.2 Definição e estrutura 6
1.3.1.3 Ciclo catalítico 10
1.3.1.4 Mecanismo de transporte 11
1.3.1.5 Sítio de ligação a drogas 14
1.3.2 Transportadores MFS 15
1.3.2.1 Histórico 15
1.3.2.2 Definição e estrutura 16
1.3.2.3 Mecanismo de transporte 18
1.4 Resistência a múltiplas drogas em fungos 19
1.4.1 Saccharomyces cerevisiae: um modelo de estudo 19
1.4.1.2 As cepas AD1234567 e AD124567 21
1.4.2 Candida albicans: um fungo de interesse clínico 22
1.5 Funções fisiológicas dos transportadores relacionados ao MDR em
fungos
25
1.6 Os terpenóides: ácido oleanólico e ácido ursólico 27
1.7 Revertendo o processo de MDR em fungos 32
2. Justificativa 35
3. Objetivos 36
4. Material e métodos 37
4.1 Produtos químicos e drogas 37
4.2 Isolamento de compostos 37
4.3 Crescimento dos microrganismos 37
4.4 Avaliação da toxicidade dos compostos frente as cepas de S. 38

10
cerevisiae e C. albicans
4.5 Efluxo de rodamina 6G 38
4.5.1 Análise qualitativa – Microscopia ótica de fluorescência 38
4.5.2 Análise quantitativa – Citometria de fluxo 39
4.6 Determinação da concentração mínima inibitória de antifúngicos de
uso clínico para cepas de C. albicans
40
4.7 Avaliação de atividade sinérgica entre ácido ursólico e fluconazol
frente ao isolado clínico de C. albicans
40
4.8 Avaliação da capacidade do isolado clínico de C. albicans em
extruir compostos fluorescentes
41
4.9 Análises estatísticas 41
5. Resultados 42
5.1 Avaliação da toxicidade do AO e da mistura AO/AU frente a cepas
AD1234567 e AD124567 de S. cerevisiae
42
5.2 Avaliação da capacidade do AO e da mistura AO/AU em inibir o
efluxo de rodamina 6G realizado pela Pdr5p
45
5.3 Avaliação da toxicidade do AU frente às cepas AD1234567 e
AD124567 de S. cerevisiae
50
5.4 Avaliação da capacidade do AU em inibir o efluxo de rodamina 6G
realizado pela Pdr5p
52
5.5 Caracterização da resistência apresentada pelo isolado clínico de C.
albicans
54
5.5.1 Avaliação de resitência a antifúngicos de uso clínico 54
5.5.2 Avaliação da capacidade do isolado clínico de C. albicans em
reter fluoróforos substratos de bombas ABC e MFS
56
5.6 Avaliação da toxicidade do AU frente ao isolado clínico de C.
albicans
58
5.7 Avaliação de interação entre AU e fluconazol quando usados em
combinação contra o isolado clínico de C. albicans
60
8. Discussão 63
9. Conclusões 70

11
Anexo I 72
1. Curcumina 73
2. Material e Métodos 76
3. Resultados 76
3.1 Avaliação da toxicidade da curcumina frente às cepas Candida
abicans
76
3.2 Avaliação da interação entre curcumina e fluconazol quando
usados em combinação contra o isolado clínico de C. albicans
81
3.3 Avaliação da capacidade da curcumina em inibir o efluxo de
vermelho do nilo do isolado clínico Pri de C. albicans
84
4. Discussão 86
5. Conclusões 87
10. Referências
88

12
LISTA DE ABREVIATURAS
µg microgramas
µM micromolar
Å Angstron
a.C. antes de Cristo
AAS ácido acetil salicílico
ABC do inglês “ATP binding cassette” (Casete ligador de ATP)
AD1234567 cepa mutante de S. cerevisiae deletada de transportadores de multidrogas
AD124567 cepa mutante de S. cerevisiae que super expressa a Pdr5p
AIDS do inglês “Acquired Immunodeficiency Syndrome” (Síndrome da
Imunodeficiência Adquirida)
AO ácido oleanólico
AU ácido ursólico
AO/AU mistura ácido oleaólico + ácido ursólico
ATP adenosina trifosfato
ATP-Na adenosina trifosfato ligada a sódio
BHI do inglês “Brain Heart Infusion” (infusão coração/cérebro)
CCD cromatografia em camada delgada
CaCDR1p transportador do tipo ABC, produto do gene CDR1 de C. albicans
CaCDR2p transportador do tipo ABC, produto do gene CDR2 de C. albicans
CDR3p produto de gene CDR3 de C. albicans
CaMDR1p transportador do tipo MFS, produto do gene MDR1 de C. albicans
CFDA do inglês “Carboxyfluorescein diacetate” (fluoróforo substrato da MDR1p)
CgCDR1p transportador do tipo ABC produto do gene CDR1 de C. glabrata
CLSI do inglês “Clinical and Laboratory Standards Institute”
COX-2 cicloxigenase-2
d.C depois de Cristo
DMSO dimetilsufóxido
FK506 lactona cíclica, inibidor do transporte promovido pela Pdr5p (Tacrolimus)
g grama

13
HIV do inglês “Human Imuno Deficiency Virus” (Vírus da Imunodeficiência
Humana)
kDa quilodaltons
LacY Lactato permease
M molar
MATE do inglês “multidrug and toxic-compound extrusion family”
MDR do inglês “Multidrug resistance” (Resistência a múltiplas drogas)
MFS do inglês “Major facilitator superfamily (Superfamília dos principais
facilitadores)
MIC concentração mínima inibitória
min minutos
mM milimolar
MRP1 transportador do tipo ABC de mamíferos
MRSA estafilococos resistente a meticilina
NBD do inglês “Nucleotide Binding Domain” (Domínio de ligação ao
nucleotídeo)
NPPN Núcleo de pesquisa de produtos naturais
NF-κB fator nuclear kappa B
nm nanômetro
Nm nanomolar oC graus célsius
OppD transportador de oligopeptídeos de S. typhimurium
PBS tampão salina fosfato
PDR do inglês “Pleiotropic Drug Resistance” (Resistência pleiotrópica a drogas)
PDR5 gene codificador da Pdr5p
Pdr1p fator de transcrição, produto do gene PDR1 de S. cerevisiae
Pdr3p fator de transcrição, produto do gene PDR3 de S. cerevisiae
Pdr5p produto do gene PDR5 de Saccharomyces cerevisiae
P-gp do inglês “P Glycoprotein” (Glicoproteína-P)
pH potencial hidrogeniônico
Pri isoado clínico de C. albicans resistênte a fluconazol

14
RND do inglês “resistance nodulation division”
rpm rotações por minuto
SD meio de cultura Saboroud dextrose
SDS dodecil sulfato de sódio
SMR do inglês “small multidrug resistance family”
SNQ2 gene codificador da Sqn2p
Snq2p produto do gene SNQ2 de S. cerevisiae
TMD do inglês “Transmembrane Domains” (domínios transmembranares)
TMS do inglês “Transmembrane Segment (”segmento transmembranar”)
TNF fator de necrose tumoral
v/v volume/volume
VRE enterococos resistente a vancomicina
YPD meio de crescimento contendo extrato de levedura, peptona e dextrose
YOR1 gene codificador da Yor1p
Yor1p produto do gene YOR1 de S. cerevisiae

15
LISTA DE FIGURAS
Fig 1. Esquema das famílias de transportadores de múltiplas drogas.
Fig 2. Organização estrutural dos Transportadores ABC. Fig 3. Modelos propostos para o arranjo dos domínios dos Transportadores ABC.
Fig. 4. Modelos propostos para o mecanismo de transporte de drogas pela P-gp.
Fig. 5. Organização estrutural dos transportadores da família MFS.
Fig. 6. Exemplar do vegetal Lippia lacunosa utilizado para o isolamento do ácido oleanólico e da
mistura ácido oleanólico/ácido ursólico testados neste trabalho.
Fig. 7: Propostas de drogas alternativas que possam atuar em células que apresentem o fenótipo
de MDR.
Fig. 8: Avaliação da toxicidade do AO e da mistura AO/AU sobre o crescimento da cepa
AD124567.
Fig. 9: Avaliação da toxicidade do AO e da mistura AO/AU sobre o crescimento da cepa
AD1234567.
Fig. 10. Avaliação qualitativa da inibição do efluxo de rodamina 6G após tratamento da cepa
AD124567 com AO ou AO/AU.
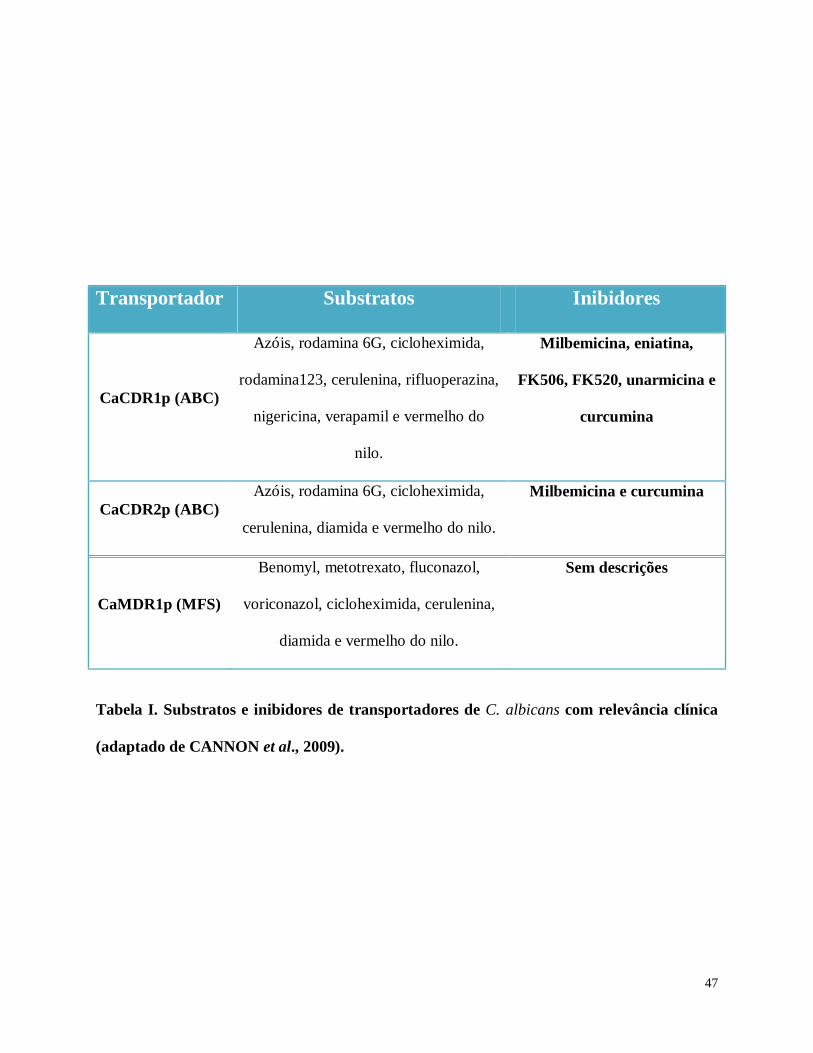
Fig. 11. Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida pelo tratamento
da cepa AD124567 com AO.
Fig. 12: Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida pelo tratamento
da cepa AD124567 com AO/AU.
Fig. 13: Avaliação da toxicidade do AU frente às cepas AD124567 e AD1234567 de S.
cerevisiae.
Fig. 14: Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida pelo tratamento
da cepa AD124567 com AU.
Fig. 15: Avaliação da capacidade da cepa 24433 e do isolado Pri de C. albicans em extruir
compostos fluorescentes.
Fig. 16: Avaliação da toxicidade do AU frente à cepa 24433 e ao isolado clínico Pri de C.
albicans.
Fig. 17: Avaliação da capacidade de quimiosensibilização do AU em associação ao fluconazol.
ANEXO

16
Fig. 1: Curcuma longa e complexo curcuminóide.
Fig. 2: Avaliação da toxicidade da curcumina frente à cepa ATCC24433 de C. albicans.
Fig. 3: Avaliação da toxicidade da curcumina frente ao isolado clínico Pri de C. albicans.
Fig. 4: Avaliação da toxicidade da curcumina frente ao isolado clínico 88 de C. albicans.
Fig. 5: Avaliação da capacidade de quimiosensibilização da curcumina em associação ao
fluconazol.
Fig. 6: Avaliação da capacidade da curcumina em inibir o efluxo do fluoróforo vermelho do nilo
do isolado Pri de C. albicans.

17
LISTA DE TABELAS
Tabela I. Substratos e inibidores de transportadores de C. albicans com relevância clínica
(modificado de CANNON et al. 2009).
Tabela II. Avaliação da concentração mínima inibitória (MIC) de antifúngicos de uso clínico
para o isolado Pri e cepa ACTCC24433. Tabela III. Avalição da interação entre fluconazol e AU apresentada durante o tratameto do
isolado Pri de C. albicans.
ANEXO
Tabela I. Avaliação da interação entre fluconazol e curcumina apresentada durante o tratamento
do isolado Pri de C. albicans.

18
1. INTRODUÇÃO
1.1 O uso de plantas medicinais pela humanidade
A tentativa de descobrir compostos com capacidade de curar doenças e aliviar dores
remonta ao aparecimento do homem na Terra. O emprego das plantas no controle de diversas
doenças e pragas é provavelmente tão antigo quanto o próprio aparecimento da humanidade
(FRANÇA, 2003). Os conhecimentos e experiências acumulados pelas civilizações foram
transmitidos, ao longo dos anos, de geração a geração, levando ao descobrimento de alguns dos
medicamentos mais importantes utilizados na medicina moderna (SIMÕES et. al., 1986).
Hipócrates, no final do século cinco a.C., teria mencionado de 300 a 400 plantas
medicinais em sua obra Corpus Hippocraticu (SCHULTES, 1978), enquanto Dioscórides
escreveu De Materia Medica no primeiro século d.C., um catálogo de plantas medicinais que se
tornou um protótipo para as atuais farmacopéias. A própria Bíblia oferece descrições de diversos
tipos de plantas com poderes de “cura” (COWAN, 1999).
De um modo geral a natureza é responsável pela produção da maioria das substâncias
orgânicas conhecidas, sendo o reino vegetal o detentor da maior parcela de diversidade química
conhecida e registrada na literatura (VIEGAS JR, BOLZANI & BARREIRO, 2006). As plantas
possuem várias vias metabólicas secundárias que dão origem a diversos compostos como
alcalóides, flavonóides, taninos, cumarinas, terpenos, dentre outros, cujas funções, até pouco
tempo eram desconhecidas. Hoje já se sabe que tais compostos são utilizados como mecanismo
de defesa contra predação por microrganismos, insetos e animais herbívoros, como é o caso dos

19
terpenóides, que conferem odor característico, dificultando a predação de plantas (COWAN,
1999 LIMA, 2001; NIERO et al., 2003).
Estima-se que existam de 250.000 a 500.000 espécies de plantas na Terra e apesar de
tanta diversidade vegetal, somente cerca de 5% das plantas têm sido estudadas fitoquimicamente
e uma porcentagem ainda menor avaliadas sob os aspectos biológicos medicinais (CECHINEL
FILHO & YUNES, 1998).
No início dos estudos químicos de extratos vegetais buscando o isolamento e
identificação de substâncias ativas, as plantas estudadas eram as de uso popular, que geralmente
já estavam incorporadas às farmacopéias da época (VIEGAS JR, BOLZANI & BARREIRO,
2006). As plantas eram, portanto, modelos moleculares fornecidos pela natureza que
fundamentaram estudos de relação entre estrutura e atividade e inspiraram o desenvolvimento da
síntese orgânica clássica. Dentre as substâncias derivadas de plantas que são utilizadas até hoje
ou que sofreram algum tipo de modificação química podemos citar o ópio, preparado a partir de
bulbos de Papaver somniferum, conhecido e usado há séculos devido às suas propriedades
soporíferas e analgésicas, havendo relatos de seu uso desde 4000 a.C. (VIEGAS JR, BOLZANI
& BARREIRO, 2006); os salicilatos, provenientes do extrato da espécie Salix alba, descobertos
em 1757, e descritos por Edward Stone com propriedades analgésicas e antipiréticas e que mais
de cem anos a frente dariam origem ao ácido acetil salicílico (AAS); forscolina, obtida de Coleus
barbatus, que apresenta promissores efeitos contra hipertensão, glaucoma, asma e contra certos
tumores; a artemisinina, presente em Artemisia annua, que exerce potente atividade antimalárica;
o diterpenóide anticancerígeno taxol, isolado de plantas do gênero Taxus, que após sua síntese
em escala industrial, já se encontra disponível no mercado farmacêutico; e por fim, como um

20
último exemplo, mas não menos importante, a quinina, a qual durante quase 300 anos foi o único
princípio ativo eficaz contra a malária (HEDNER & EVERTS, 1998).
No século XX, o surgimento dos antibióticos produzidos por fermentação microbiana,
aliado ao desenvolvimento de fármacos sintéticos produzidos pela indústria farmacêutica, logo
depois da I Grande Guerra, foram causas marcantes no declínio do uso de plantas medicinais e
conseqüentemente, no investimento em fármacos de origem vegetal (MONTANARI &
BALZANI, 2001). Após a síntese do AAS, ocorreu o fim do período no qual a busca por
substâncias naturais terapeuticamente ativas era feito ao acaso, iniciando-se a era da síntese
química de novos compostos com atividade biológica. Porém, essa metodologia mostrou-se
muito mais complexa e dispendiosa, tendo em vista o pequeno número de novos compostos que
venciam as etapas pré-clínicas e clínicas (VIEGAS Jr, BOLZANI & BARREIRO, 2006).
Neste período, porém, a descoberta de novos compostos naturais com indiscutível
importancia médica, como a vinblastina e vincristina, ambas extraídas da espécie Catharantus
roseus (JOHNSON et al., 1963), bem como de análogos do etoposídeo e do taxol, fizeram
resurgir o interesse da indústria farmacêutica pelos produtos de origem natural.
Devido a todo esse contexto apresentado, os produtos naturais vêm recuperando seu
espaço na pesquisa científica e na indústria farmacêutica seja por si mesmo, ou como fonte de
inspiração para novos modelos moleculares bioativos, o que pode ser claramente observado pelo
aumento de trabalhos publicados nesta área, tanto em congressos como em periódicos nacionais
e internacionais, além do surgimento de periódicos específicos sobre produtos naturais ativos
(CECHINEL FILHO & YUNES, 1998).
No Brasil, diversos grupos étnicos usam emplastros, decocções, infusões e extratos brutos
de plantas sem nenhum conhecimento científico, mas que são passados de geração em geração

21
como tendo propriedades curativas. Sendo assim, essas plantas usadas na medicina popular
durante milhares de anos constituem uma escolha óbvia de estudo, principlamente em nosso país,
favorecido pela grande diversidade da flora com potencial terapêutico, o que estimula os
pesquisadores a estudar suas possíveis ações farmacológicas, bem como ações tóxicas. A fim de
produzir medicamentos eficazes e seguros, com menos efeitos colaterais, muitos compostos
biologicamente ativos já vem sendo isolados de diversas espécies distintas de plantas (GUERRA
& NODARI, 2003).
1.2 Resistência a múltiplas drogas
O processo de resistência a múltiplas drogas, ou MDR (do inglês “Multidrug
Resistance”) foi observado inicialmente por oncologistas clínicos, quando estes perceberam que
cânceres tratados com diferentes tipos de fármacos desenvolviam uma resistência cruzada a
diversos agentes citotóxicos aos quais estes tumores não haviam sido expostos previamente
(revisto em AMBUDKAR et al., 1999). Este processo já havia sido demonstrado na década de
1960, quando surgiram relatos das primeiras bactérias multiresistentes a antibióticos (SHEPS &
LING, 2007).
Este tipo de resistência na quimioterapia antimicrobiana e anticâncer é a maior causa de
falhas destes tratamentos, sendo uma das mais importantes e preocupantes (LAGE, 2003).
O mecanismo de MDR melhor caracterizado, denominado “MDR clássico”, se baseia na
super expressão de uma bomba de efluxo de drogas dependente de energia denominada
Glicoproteína P (do inglês “P-glycoprotein” ou “P-gp”), detectada inicialmente como uma
glicoproteína de superfície superexpressa em cultura de células cancerosas selecionadas por

22
MDR (revisto em AMBUDKAR et al., 1999). Este fenótipo consiste na expulsão, do interior
celular ou ainda da membrana plasmática, de diversos tipos de fármacos, estruturalmente e
funcionalmente distintas, as quais guardam entre si apenas o caráter levemente hidrofóbico em
comum, promovendo uma redução de sua concentração no meio intracelular (GOTTESMAN &
PASTAN, 1993).
Após a descoberta deste processo, um grande número de proteínas transportadoras de
múltiplas drogas homólogas foi identificado em células cancerosas e em microrganismos
patogênicos (LAGE, 2003).
Logo no início da observação deste fenômeno e posterior detecção de uma proteína
visível em gel corado como uma banda de aproximadamente 170 kDa, inexistente em células
sensíveis e proeminentes em células super resistentes, era difícil crer que uma única proteína
fosse responsável por todo o fenótipo de resistência, interagindo com cada um dos muitos
substratos (RIORDAN & LING, 1979). A aceitação de que esta teria efetivamente uma função
como bomba de efluxo de uma incrível gama de substratos veio com o reconhecimento de que a
P-gp era um homólogo de uma família multifuncional de proteínas transportadoras em bactérias,
a qual passou a ser denominada a família dos Transportadores ABC (do inglês “ATP Binding
Cassette”) (revisto em SHEPS & LING, 2007).
Existem cinco famílias de proteínas de efluxo que estão associadas com o fenótipo de
MDR (Figura 1) estando presentes em todos os organismos: a superfamília de transportadores
ABC, a MFS (do inglês “Major Facilitator Superfamily”), a MATE (do inglês “Multidrug and
Toxic-compound Extrusion Family”), a SMR (do inglês “Small Multidrug Resistance Family”) e
a RND (do inglês “Resistance Nodulation Division”) (LAGE, 2003).

23
Estes transportadores podem ser divididos em duas classes baseado em sua fonte de
energia. A primeira classe é composta por proteínas capazes de realizar transporte ativo primário,
utilizando como energia a hidrólise de ATP, enquanto a segunda classe é composta por proteínas
capazes de realizar transporte ativo secundário, usando um gradiente iônico, através de simporte
ou antiporte (PUTMAN, VAN VEEN & KONINGS, 2000; CHANG, 2003).
Os transportadores primários exercem seu papel principalmente em células eucarióticas
enquanto em células procarióticas os transportadores secundários são predominantes (LAGE,
2003).

24
Fig. 1. Esquema das famílias de transportadores de múltiplas drogas. Transportadores
ABC (do inglês “ATP binding cassette”), a MFS (do inglês “major facilitator superfamily”), a
MATE (do inglês “multidrug and toxic-compound extrusion family”), a SMR (do inglês “small
multidrug resistance family”) e a RND (do inglês “resistance nodulation division”) (adaptado de
LAGE, 2003).
Transportadores de Múltiplas Drogas
Transportadores ativos secundários Transportadores ativos primários
MATE
SMR
Transportadores ABC RND MFS

25
1.3 Os principais transportadores relacionados ao MDR em eucariotos
1.3.1 Transportadores ABC
1.3.1.1 Histórico
Os transportadores ABC foram primeiramente identificados e caracterizados em
bactérias. A fisiologia dos sistemas de transportes mediando à captação de nutrientes por
bactérias (especialmente Escherichia coli e Salmonella typhimurium) foi estudada em detalhes
na década de 1970 e em 1974 foi caracterizada uma classe de transportadores sensível ao choque
osmótico e energizado pela hidrólise de ATP, sendo esta devido à perda de sua proteína de
ligação ao substrato, uma proteína periplasmática (revisto em HIGGINS, 2001).
Após o sequenciamento completo do transportador de oligopeptídeos (OppD) de S.
typhimurium, notou-se que essas proteínas possuíam uma região de nucleotídeos bastante
conservada, similar aos previamente identificados em ATP sintetase, miosina e adenilato quinase
(WALKER et al., 1982), sugerindo portanto que a hidrólise de ATP estaria acoplada ao processo
de transporte. Esta hipótese foi posteriormente confirmada (BISHOP et al., 1989, MIMMACK et
al., 1989) ao se observar um domínio de ligação ao ATP nestes transportadores. Em 1985 ficou
reconhecido que as subunidades ligadoras de ATP em transportadores de bactérias definiriam
uma superfamília de proteínas as quais teriam uma organização estrutural formada por uma
unidade básica composta por quatro domínios (HIGGINS et al., 1985).
No ano seguinte, o primeiro transportador pertencente a essa família em eucariotos,
responsável pela resistência a múltiplas drogas em humanos, foi identificado e denominado

26
como Glicoproteína P (CHEN et al., 1986, GERLACH et al., 1986; GROS, CROOP &
HOUSMAN, 1986), e em 1990 o termo Transportador ABC começou a ser usado (HYDE et al.,
1990).
1.3.1. 2 Definição e estrutura
Os transportadores ABC podem ser encontrados na maioria das espécies e exercendo
diversos papéis fisiológicos. Em bactérias possuem papel central na eliminação de compostos e
captação de nutrientes, podendo também atuar como bomba de efluxo de drogas, enquanto que
em fungos, o seu papel principal é o de resistência a antifúngicos (MADIGAN, MARTINKO &
PARKER, 2004). No geral, os transportadores do tipo ABC estão relacionados ao transporte de
substâncias através de membranas, estando localizados com frequência na membrana plasmática,
mas também podendo estar presentes nas membranas de algumas organelas (RUTHERFORD &
WILLINGHAM, 1993; MOLINARI et al., 1994; MARALDI et al., 1999).
Dentre diversos mecanismos que parecem contribuir para o fenômeno de MDR, a
superexpressão de bombas de efluxo pertencentes à superfamília dos transportadores ABC é a
causa mais frequente de desenvolvimento de resistência a antifúngicos, herbicidas e drogas
citotóxicas (PRASAD & PANWAR, 2004).
A estrutura desses transportadores é caracterizada pela presença de uma unidade básica
constituída por quatro domínios, sendo dois domínios transmembranares (TMD) e dois domínios
de ligação ao nucleotídeo (NBD), os quais podem ser produtos de um mesmo gene ou de genes
distintos. Em procariotos cada um dos quatro domínios pode ser codificado como um
polipeptídio separado, porém em alguns transportadores esses domínios podem ser fundidos uns
aos outros de diversas formas (Figura 2). Na maioria das proteínas ABC de eucariotos, é possível

27
encontrar esses domínios sendo codificados como um polipeptídio único, o qual contém todos os
quatro domínios (ALTENBERG, 2003).
Os TMDs são tipicamente formados por seis alfas hélices, também denominados
seguimentos transmembranares (TMS), que atravessam a membrana plasmática formando um
total de 12 por transportador (Figura 3). Estes formam o caminho pelo qual o composto atravessa
a membrana e determina a especificidade do transportador, uma vez que possui sítios de ligação
ao substrato (HIGGINS, 1992). Os NBDs são domínios hidrofílicos e estão perifericamente
associados com a face citoplasmática da membrana (Figura 3). Eles representam uma região
altamente conservada de aproximadamente 215 aminoácidos, podendo ser também denominados
de domínio ABC, pelo qual estes transportadores são definidos, sendo a conservação deste
domínio essencial para definição e delimitação da família ABC (revisto por HIGGINS, 2001).
Como para todas as outras ATPases, o domínio de ligação ao ATP dos transportadores ABC
contém a região conservada walker A (seqüência rica em glicina) e walker B (seqüência
hidrofóbica) (WALKER et al.,1982), além de uma terceira seqüência conservada (“LSGGQ”),
denominada motivo C, exclusiva dos domínios ABC e mais conhecida como “assinatura ABC”
(HYDE et al., 1990).

28
Fig. 2. Organização estrutural dos Transportadores ABC. Domínios de ligação ao ATP
(NBD) (ovais laranja) e Domínios transmembranares (TMD) (quadrados verde). Os domínios
podem se apresentar fundidos em diversas, 2001). combinações: (A) codificados como 4
polipeptídios separados, (B) NBDs fundidos, (C) TMDs fundidos, (D) cada NBD fundido a um
TMD, (E) um único TMD fundindo a um NBD e o outro TMD e NBD codificados em
polipeptídios separados e (F) todos os quatro domínios fundidos em um único polipeptídio
(desenho realizado por JM Machado – designer gráfico, adaptado de ALTENBERG, 2003)

29
Fig. 3. Modelos propostos para o arranjo dos domínios dos Transportadores ABC. A
região amarela representa o domínio de ligação ao nucleotídeo (NBD), contendo os motivos
Walker A, Walker B e assinatura ABC; Domínios transmembranares (TMD) estão representados
como áreas verdes, sendo formados por seis segmentos transmembranares (TMS). (A)
configuração [NBD-TMD], como no ADP1p de Saccharomyces cerevisiae; (B) configuração
[TMD-NBD]; como no LmrA de Lactococcus lactis; (C) configuração [NBD-TMD]2 como na
P-gp de mamíferos (D) configuração [TMD-NBD]2, como no Pdr5p de Saccharomyces
cerevisiae; (E) representação da estrutura de poro adquirida por transportadores ABC. Droga,
substrato do transportador, representada como círculo vermelho (desenho realizado por JM
Machado – designer gráfico, adaptado de LAGE, 2003 e CANNON et al., 2009).

30
Em eucariotos, em geral, os transportadores apresentam uma topologia [TMD-NBD]2 ou
[NBD-TMD]2, sendo a segunda topologia também chamada de configuração invertida (Figura
3).
Com relação a sua estrutura tridimensional, os TMDs se arrumam de modo a formar um
poro central, o qual, provavelmente, atua como o caminho de translocação do substrato (Figura
3). Apesar de estudos de cristalografia terem demonstrado diversas arquiteturas para o poro
central de proteínas ABC distintas, a maioria pode ser classificada em duas estruturas diferentes,
uma estrutura que apresenta uma abertura voltada para o citoplasma (FERREIRA-PEREIRA, et
al., 2003; PIKETT et al., 2007; GERBER et al., 2008; ALLER et al., 2009; KHARE et al.,
2009) e uma outra com a abertura para o meio extracelular (LOCHER, LEE & REES, 2002;
DOWSON & LOCHER, 2006; OLDHAM et al., 2007).
1.3.1.3 Ciclo catalítico
A grande maioria dos estudos visando analisar o processo de catálise realizado por
transportadores dessa família foram realizados com a P-gp, porém devido à homologia que esta
proteína possui com outros transportadores da mesma família acredita-se que esse mesmo ciclo
poderia ser utilizado como um modelo para diversos transportadores da família ABC.
Al-Shawi & Sênior (1993) sugeriram que a ligação do ATP ao NBD não é necessária
para que ocorra a ligação do fármaco à P-gp, sendo assim os eventos de ligação do fármaco e
ligação do ATP são independentes um do outro. Porém, sabemos que para que ocorra o
transporte de uma molécula é essencial que ocorra a ligação e hidrólise do nucleotídeo

31
trifosfatado (TEODORI et al., 2002) e que os dois NBDs podem ligar e hidrolisar ATP
(GOTTESMAN, PASTAN & AMBUDKAR, 1996).
Para que ocorra o transporte de uma única molécula do fármaco são necessários dois
eventos não simultâneos de hidrólise de ATP (SAFA, 1992). No modelo inicialmente descrito
por Senior e colaboradores, em 1995, e complementado por Sauna e Ambudkar (2000), foi
descrito que a ligação do substrato aos TMDs estimula a atividade ATPásica da P-gp, porém
apenas um NBD de cada vez atua como um sítio catalítico, e a conformação deste impede o
segundo NBD de hidrolisar outro ATP. Após a hidrólise do nucleotídeo trifosfatado ocorre uma
mudança conformacional no sítio de ligação à droga que libera o substrato para o exterior
celular. A hidrólise no segundo sítio de ligação ao ATP parece ser necessária para restaurar a
conformação inicial do transportador, para que este possa ligar um novo substrato, completando
um ciclo catalítico (revisto por LINTON, 2007). Modificações conformacionais entre os dois
estados citados previamente, parecem ser essenciais para o transporte (HOLLENSTEIN, FREI &
LOCHER, 2007; MOUSSATOVA et al., 2008; OLDHAM, DAVIDSON & CHEN, 2008). Para
procariotos, geralmente os NBDs adotam um arranjo no qual um fica de frente para o outro
ficando inseridas no meio as duas moléculas de ATP (SMITH et al., 2002; CHEN et al., 2003;
VERDON et al., 2003; ZAITSEVA et al., 2005, WENG, FAN & WANG, 2009). Durante a
hidrólise do ATP a interface do dímero deve ser desfeita e os NBDs voltam a se isolar (LU et al.
2005; ZAITSEVA et al., 2006).

32
1.3.1.4 Mecanismos de transporte
Acredita-se que os transportadores ABC envolvidos na resistência a múltiplas drogas
tenham um mecanismo molecular de transporte em comum. Diversos modelos foram sugeridos
para explicar sua ampla gama de especificidade por compostos quimicamente tão distintos
(VARMA et al., 2003), porém até o momento não se reconheceu nenhum mecanismo de
transporte proposto como sendo o mecanismo de transporte geral dos transportadores ABC.
Atualmente apenas três modelos são aceitos como possíveis mecanismos de transporte
(Figura 4), dentre eles: o modelo de poro (ALTENBERG et al., 1994), flipase (HIGGINS &
GOTTESMAN, 1992) e “aspirador hidrofóbico” (RAVIV et al., 1990). No modelo de poro, o
fármaco seria captada diretamente do citoplasma e translocada para o meio extracelular; no
modelo de flipase o fármaco se acumularia no folheto interno da membrana plasmática enquanto
a proteína transportadora por um movimento de “flip-flop” transportaria o fármaco para o folheto
externo, onde este poderia se difundir no espaço extracelular; e por fim, no modelo de aspirador
hidrofóbico propõe-se que a molécula seria detectada e expulsa diretamente da bicamada
lipídica, da mesma forma como para o modelo de flipase. Neste caso, porém, o fármaco se ligaria
a proteína e seria transportado por um canal central para o meio extracelular (FERREIRA-
PEREIRA et al., 2003; AL-SHAWI & OMOTE, 2005; SIARHEYEVA, LOPEZ, &
GLAUBITZ, 2006). Hoje é observado como duas estruturas conformacionais distintas que se
alternam para promover o transporte de um substrato parece ser uma combinação desses
mecanismos inicialmente propostos.

33
Fig. 4. Modelos propostos para o mecanismo de transporte de drogas pela P-gp. (a) Modelo
de poro, (b) modelo flipase e (c) modelo de aspirador hidrofóbico (desenho refeito por JM
Machado – designer gráfico, extraído de VARMA et al., 2003).

34
1.3.1.5 Sítio de ligação a drogas
Esforços têm sido feitos na tentativa de identificar resíduos na Pdr5p, que estariam
relacionados com sua funcionalidade. Egner e colaboradores (1998) iniciaram esses estudos
utilizando ferramentas que promoviam mutações randomizadas, e posteriormente, em 2000,
estudos de mutação sítio-dirigida (EGNER et al., 2000).
Acredita-se que para o funcionamento da Pdr5p, e outros transportadores ABC,
aminoácidos específicos nos domínios transmembranares funcionariam no reconhecimento e
ligação do substrato (EGNER et al., 1998; MOODY et al., 2002; NEYFAKH, 2002).
Egner e colaboradores (1998) e Wolfger e colaboradores (2001) indicaram que na Pdr5p,
assim como para P-gp, uma região de ligação a drogas estaria localizada nos TMS, uma vez que
mutações pontuais nestes geravam mutantes com especificidade alterada pelo substrato. Em seus
estudos foi mostrado que mutações no TMS 10 promoviam alterações na especificidade do
transportador.
Posteriormente, o grupo de Golin (2003) propôs a existência de pelo menos três sítios de
ligação à droga na Pdr5p, e uma possível interação de diferentes substratos em um mesmo sítio,
significando, portanto, que neste transportador poderia ocorrer a sobreposição de sítios. Neste
mesmo trabalho também foi observada uma dependência do tamanho do fármaco (que deve ser
em torno de 200-225 Å) para que o transportador pudesse transportá-la.
Além disso, Tutulan-Cunita e colaboradores (2005) analisando diversos mutantes,
observaram que mutações nos TMS 4 e 9 e nas alças hidrofílicas que conectam os TMS na
porção extracelular (por ex. EL6) conferiam diferentes graus de alterações na especificidade ao

35
substrato, comprovando assim a idéia que estes transportadores albergariam múltiplos sítios de
interação com drogas.
Características específicas dos substratos, além do tamanho da molécula, também são
levadas em consideração, tanto que moléculas anfipáticas e hidrofóbicas que possam caber no
sítio podem se transformar em substratos. Dentro de seu sítio de ligação, o substrato estabelece
diversas ligações de Van der Waals com resíduos hidrofóbicos encontrados nos TMS e/ou
alterações eletrostáticas com resíduos carregados (TUTULAN-CUNITA et al., 2005).
1.3.2 Transportadores MFS
1.3.2.1 Histórico
A família MFS (Superfamília dos Principais Facilitadores) compreende uma superfamília
de proteínas encontradas em todos os reinos, representando o maior grupo de transportadores que
promovem o transporte acoplado a íons (SAIER et al., 1999). Tais transportadores são capazes
de translocar seus substratos através de membranas, acoplando a este o transporte de um íon (H+
ou Na+) ou a outro soluto, contra seu gradiente (KABACK et al., 2001).
Inicialmente se acreditava que os transportadores MFS estariam envolvidos
exclusivamente na captação de açúcares (MAIDEN et al., 1987; HENDERSON & MAIDEN,
1990), porém estudos posteriores revelaram que sistemas de efluxo de drogas também
pertenciam a essa família (GRIFFITH et al., 1992; PAULSEN & SKURRAY, 1994). Esta
família de transportadores foi primariamente identificada na década de 1990, quando

36
pesquisadores observaram que um grande grupo de transportadores de solutos se relacionava
evolucionariamente (HENDERSON, 1990; GRIFFITH et al., 1992; MARGER & SAIER, 1993).
Em 1993 Marger e Saier, com o auxílio de análises estatísticas, observaram que tais
transportadores compartilhavam uma estrutura em comum, dividindo-os em famílias e dentre
essas, cinco compreenderiam o que hoje conhecemos como a superfamília MFS, nomeclatura
também definida pelo mesmo grupo.
Após a divisão proposta por Marger e Saier (1993), Paulsen e colaboradores (1996)
utilizando estudos filogenéticos, dividiram a família em duas subfamílias de acordo com sua
topologia, inserindo assim mais uma família às cinco propostas inicialmente, uma vez que se
conseguiu identificar membros que apresentavam divergências em suas topologias (PAULSEN
et al., 1996). Pouco tempo depois, em 1998, uma nova divisão foi proposta, sendo baseada
exclusivamente em similaridades de sequências, criando assim uma divisão das proteínas MFS
em 17 famílias, e conseguindo mostrar que as famílias filogenéticas estavam correlacionadas
com a função da proteína, de forma que, cada uma das famílias reconheceria e transportaria uma
classe específica de substratos estruturalmente relacionados (PAO, PAULSEN & SAIER, 1998).
1.3.2.2 Definição e estrutura
Como já descrito anteriormente, nesta família encontramos proteínas que realizam
transporte ativo secundário, não utilizando, portanto, a energia metabólita da célula (ATP). O
gradiente de concentração de uma substância representa um armazenamento de energia química
potencial que pode ser utilizado para realizar trabalho (ROBERT & MATTHEW, 1996) e é este

37
trabalho que impulsionará o transporte de uma segunda substância contra seu gradiente de
concentração.
Até o momento, mais de 300 proteínas, que pertencem a essa superfamília, já foram
identificadas, incluindo a TPO1 de S. cerevisiae (DERISSI et al., 2000; ROGERS et al., 2001),
CaMd1p de Candida albicans (BEN-YAACOV et al., 1994) e LacY de Eschericcia coli,
também denominada lactose permease, o transportador MFS mais bem caracterizado até o
momento (ABRAMSON et al., 2003).
Para praticamente todos os transportadores MFS existe uma topologia básica, composta
por um único domínio transmembranar formado por 12 alfas hélices, conectadas por alças intra e
extracelulares, tendo tanto o seu domínio N-terminal quanto o C-terminal localizados na porção
citoplasmática (PAO, PAULSEN & SAIER, 1998; SAIER, 2003; SAIER et al., 1999). Porém,
algumas exceções podem estar presentes nessa família, onde encontramos membros com 14, 24
ou apenas 6 alfa hélices (Figura 5). As duas hélices adicionais nos membros de 14 alfas hélices
surgiram, provavelmente, a partir da inserção da alça citoplasmática central na membrana,
enquanto que os membros de 24, podem ser oriundos de um evento de fusão de genes (SAIER,
2003) e os de 6, provavelmente funcionam como homodímeros (LAW, MALONEY & DA-
NENGWANG, 2008).

38
(A)
(B)
(C)
Fig. 5. Organização estrutural dos transportadores da família MFS. Os
transportadores MFS podem ser formados por TMDs que possuam: (A) 6 segmentos
transmembranares, (B) 12 segmentos transmembranares ou (C) 14 segmentos transmembranares.
As porções N- e C- terminais estão livres no citoplasma (desenho realizado por JM Machado –
designer gráfico, baseado no modelo proposto por GAUR et al., 2008).

39
No geral, pouca homologia é encontrada quando comparamos as duas metades da
proteína, o que sugere que o surgimento desta molécula ocorreu após um evento de duplicação
e/ou fusão de genes (MAIDEN et al., 1987). Quando comparamos proteínas dentro de uma
mesma família é possível observar altas taxas de similaridade (PAO, PAULSEN & SAIER,
1998), porém, quando a comparação é realizada em nível de superfamília, proteínas individuais
compartilham baixa identidade ou similaridade de sequência, sendo unidos apenas por um par de
sequências conservadas localizadas nas porções N- e C- terminais e em algumas alças (MAIDEN
et al., 1987)
Estudos de cristalografia 3D de alta resolução com as proteínas LacY, GlpT, EmrD, e de
baixa resolução de OxlT, mostram que apesar das diferenças nas sequências dessas proteínas
MFS, todas compartilhavam praticamente a mesma estrutura tridimensional. Essa descoberta
permite propor um modelo estrutural básico compartilhado pelas proteínas dessa família,
funcionando como um esqueleto para todas as proteínas MFS, independente de sua função
(revisto por LAW, MALONEY & WANG, 2008). Até o momento encontramos disponível
detalhes da estrutura 3D para 3 transportadores, sendo todos eles de procariotos (HIRAI et al.,
2002; HUANG et al., 2003; YIN et al., 2006). As estruturas disponíveis indicam não só uma
conservação no dobramento mas também na arquitetura da proteína, consistente com a formação
de dois domínios com um poro central.
Algumas características podem ser consideradas típicas de transportadores MFS, como: a
presença de 12 alfas hélices transmembranares (topologia mais comum) com as porções N- e C-
terminais localizadas no citoplasma; uma sequência composta por 400-600 aminoácidos e uma
assinatura conservada (RXXXR) entre as alças 2-3 (MAIDEN et al., 1987; PAO, PAULSEN &
SAIER, 1998; HIRAI, HEYMANN & SUBRAMANIAM, 2003).

40
1.3.2.3 Mecanismos de transporte
Com relação à cinética de transporte realizada por estes transportadores, dois mecanismos
são observados: simporte, no qual ocorre o transporte de duas ou mais moléculas
simultaneamente na mesma direção energizado pelo gradiente eletroquímico de uma dessas
moléculas, e o antiporte, que transporta duas ou mais moléculas em direções opostas.
Nos transportadores antiportes, relacionados com efluxo de drogas, acredita-se que exista
um único sítio de ligação que funcionaria em um mecanismo de acesso alternado, podendo o
transportador adquirir duas principais conformações que se alternam, com aberturas voltadas
para o citoplasma ou para o espaço extracelular (VIDAVAR, 1966; JENCKS, 1980; TANFORD,
1982; TANFORD, 1983; WEST, 1997), nesse caso o transporte de uma molécula seria realizado
pela interpolação dessas duas conformações.
Alguns modelos para explicar o mecanismo de transporte são propostos: no mecanismo
de “cadeira de balanço” ou “gangorra” (HUANG et al., 2003) sugere-se que um substrato é
fixado no sítio de ligação, em seguida o acesso alternado é realizado a partir de mudanças
conformacionais que poderão gerar acessos para as diferentes superfícies (MALONEY, 1994);
um segundo mecanismo seria o de poro (PATLAK, 1957) e neste, o substrato se moveria pelo
centro do transportador assim como ocorre em um canal de íons, porém com aberturas que
seriam alternadas entre o meio intra e extracelular, sendo que esta visão é mais comumente aceita
para outros transportadores que não são MFS (revisto por LAW, MALONEY & WANG, 2008).

41
1.4 Resistência a múltiplas drogas em fungos
1.4.1 Saccharomyces cerevisiae: um modelo de estudo
O reino dos Fungos compreende uma variabilidade de aproximadamente 1,5 milhões de
espécies, sendo que 200 dessas teriam alguma associação com o homem. Alguns fungos podem
coexistir com humanos de forma comensal, não causando nenhum dano a eles, enquanto outros
podem ser considerados patógenos (Revisto por CANNON et al., 2009). Os fungos e as
leveduras estão presentes em praticamente todos os habitats da Terra, participando em diversos
processos biológicos.
Leveduras, como a S. cerevisiae, participam da alimentação do homem desde épocas em
que o homem vivia apenas da colheita. Com o estabelecimento da prática agrícola, as colheitas
passaram a ser processadas em bebidas e alimentos elaborados, com a colaboração das leveduras.
Em relíquias do antigo Egito é possível encontrar papiros e esculturas que indicam a existência,
já naquela época, de panificadoras e casas de fermentação. Atualmente, as leveduras possuem
aplicações biotecnológicas podendo ser utilizadas na tecnologia ambiental e em pesquisas de
base, onde são avaliados metabolismo de drogas, biologia celular e molecular e na indústria
farmacêutica e de alimentos e bebidas (WALKER, 1998). A espécie S. cerevisiae possui
aproximadamente seis mil genes e o seu genoma foi o primeiro a ser completamente sequenciado
(GOFFEAU et al., 1996).
As leveduras (patogênicas ou não), assim como outras células, são capazes de
desenvolver mecanismos de resistência quando são desafiadas com antifúngicos ou outras drogas
com poder citotóxico (revisto por PRASAD & PANWAR, 2004). Um desses mecanismos

42
envolve a superexpressão de bombas de efluxo relacionadas ao processo de MDR, como
membros das famílias ABC e MFS (LAGE, 2003). O fenótipo apresentado pela S. cerevisiae se
assemelha ao processo de MDR de células de mamíferos, sendo denominado Resistência
Pleiotrópica a Drogas (PDR) (BALZI & GOFFEAU, 1994; 1995).
O primeiro transportador ABC homólogo a P-gp encontrado em sistema diferente de
células de mamífero foi descrito em S. cerevisiae. Esse transportador é formado por 1290
resíduos de aminoácidos e foi denominado Ste6p. Este exibe a configuração [TMD-NBD]2 e está
envolvido não com a resistência a fármacos, mas com o transporte do feromônio fator-a (LAGE,
2003). Após essa constatação, duas diferentes análises do genoma da levedura revelaram
aproximadamente 30 genes (TAGLICHT & MICHAELIS, 1998; DECOTTIGNIES &
GOFFEAU, 1997) que codificam possíveis proteínas ABC, sendo essas subdivididas em cinco
subfamílias: PDR, MDR, MRP/CFTR, RL1, YEF3 e ALDP (MICHAELIS & BERKOWER,
1995; DECOTTIGNIES & GOFFEAU, 1997; ROGERS et al., 2001). A subfamília PDR é a
maior, compreendendo grande parte das proteínas ABC desta levedura, sendo seus membros
também encontrados em outros fungos, como Candida albicans, Candida glabrata e Aspergillus
nidulans (DEL SORBO, SCHOONBEEK & DE WAARD, 2000). Desta análise gênica, foi
possível concluir que cinco dos aproximadamente 30 genes identificados, codificavam
transportadores ABC mediando o fenótipo de MDR quando presentes em múltiplas cópias:
PDR5, PDR11, SNQ2, YCF1 e YOR1 (DECOTTIGNIES et al., 1995). Destes, os produtos
Pdr5p, Pdr11p e Snq2p apresentam a chamada topologia invertida [NBD-TMD]2 (BALZI et al.,
1994; BISSINGER & KUCHLER, 1994), nomeclatura denominada em relação à estrutura
apresentada por outros transportadores ABC como a P-gp de mamíferos.

43
O produto do gene PDR5 é o transportador mais bem caracterizado em leveduras
(GOLIN et al., 2007). A superexpressão desse gene gera uma resistência a centenas de compotos
sem relação química entre si, dentre as quais estão incluídas: cicloheximida, rodamina 6G,
rodamina 123, drogas anticâncer, trifluoperazina, nigericina, hormônios esteróides, como
progesterona, muitas classes de antimicóticos, como os azóis, e fungicidas agrícolas (ex.
anilinopirimidinas, benzimidazois, ditiocarbamatos e análogos de estrobilurina)
(KOLACZKOWSKI et al., 1996).
PDR5, SNQ2 e YOR1 compartilham ativadores transcricionais em comum e esses
ativadores são produtos dos genes regulatórios PDR1 e PDR3 e co-regulam constitutivamente a
rede PDR (BALZI & GOFFEAU, 1995). Mutações de ganho de função, nesses fatores de
transcrição, induzem a superexpressão dessas proteínas em membranas plasmáticas. De uma
forma geral, a expressão dos transportadores ABC em S. cerevisiae está sob o controle de uma
complexa rede de regulação que envolve, além dos fatores de transcrição Pdr1p e Pdr3p, outros
fatores reguladores de resposta ao estresse (KOLACZKOWSKA & GOFFEAU, 1999).
Já foi demonstrado que a Pdr5p compartilha além da hidrólise de nucleotídeos
trifosfatados, substratos e moduladores com outros transportadores de fungos e mamíferos
(revisto por CANNON et al., 2009), o que torna esta proteína um ótimo modelo para estudos na
busca de inibidores para transportadores homólogos em microrganismos patogênicos, como o
transportador CgCdr1p (SANGLARD, 1999) e CaCdr1p de cepas resistentes a azóis de C.
glabrata e C. albicans, respectivamente, que também codificam transportadores de múltiplas
drogas, e são os produtos dos genes mais comumente associados ao efluxo de drogas dependente
de energia em isolados clínicos resistentes a fluconazol (SANGLARD & BILLIE, 2002). Além

44
dos substratos já descritos podemos destacar a cicloheximida, benomil, flusilazol, nuarimal,
fenapromil e sorafen como substratos exclusivos da Pdr5p (ROGERS et al., 2001).
1.4.1.1 As cepas AD1234567 e AD124567 de S. cerevisiae
Os transportadores ABC de leveduras são normalmente expressos em quantidades muito
baixas em cepas selvagens, além de apresentarem sobreposição no seu espectro de ação, o que
gera dificuldades nos estudos bioquímicos e fisiológicos dessas proteínas em cepas selvagens
(ROGERS et al., 2001).
Cepas mutantes foram desenvolvidas por Decottignies e colaboradores (1998) com o
objetivo de se conseguir estudar o transportador de interesse descartando a influência dos outros
transportadores da levedura. Nessas cepas foram deletados diversos tipos de transportadores
como YOR1, SNQ2, PDR5, YCF1, PDR10, PDR11 e PDR3 em diferentes combinações, além
de mutações de ganho de função nos genes dos ativadores transcricionais PDR1 e PDR3,
levando a um aumento da expressão de seus genes alvos.
Com essas mutações foram obtidas diversas cepas, dentre elas a AD1234567 e
AD124567, sendo a primeira uma cepa na qual todos os genes mencionados acima (indicados
por números na nomeclatura da cepa) foram deletados, gerando uma cepa hipersensível a
diversos compostos, os quais serviriam como substratos para os transportadores ABC (ROGERS
et al., 2001), podendo ser utilizada, portanto, como um controle para ensaios com células
intactas; e a segunda uma cepa que expressa ativamente grandes quantidades do transportador
Pdr5p funcional, o que torna esse cepa altamente resistente.

45
1.4.2 Candida albicans: um fungo de interesse clínico
A espécie C. albicans faz parte da flora anfibionte dos homens (PERFECT &
CASADEVALL, 2006), colonizando pele e mucosas de pessoas saudáveis e atuando
comensalmente no trato gastrointestinal, cavidade oral e vagina, podendo, frequentemente,
causar infecções superficiais sem gravidade alguma, mas também sendo capaz de se disseminar e
causar infecções invasivas. Podem ser considerados patógenos oportunistas, uma vez que
ocasionam infecções quando seu hospedeiro está com o sistema imunológico debilitado
(MAVOR, THEWES & HUBE, 2005), como no caso da Síndrome da Imunodeficiência
Adquirida (BACCAGLINI et al., 2007), causando sérias infecções da mucosa oral e podendo,
até mesmo, se disseminar por todo organismo humano.
Infecções fúngicas sistêmicas, como as que podem ser ocasionadas por diversas espécies
de Candida, são de difícil diagnóstico e além disso, o limitado número de antifúngicos
disponíveis no mercado, quando comparado com a amplitude de antibióticos colabora para suas
altas taxas de mortalidade (revisto por CANNON et al., 2009).
Existem apenas cinco classes de antifúngicos disponíveis para o uso clínico: análogos de
5-flucitosina, que interferem na síntese de ácidos nucléicos; os poliênicos, que atuam pela
formação de poros na porção ergosterol da membrana plasmática; as equinocandinas, a mais
nova classe de antifúngicos, que inibem a síntese da β1-3 glucana sintase; as alilaminas, que
atuam na síntese de ergosterol, e os azóis, que também atuam na síntese de ergosterol, sendo o
fluconazol, o representante desta classe de antifúngicos mais amplamente utilizado devido a sua
relativa baixa toxicidade e sua melhor solubilidade, quando comparado aos antifúngicos de
outras classes (revisto por CANNON et al., 2009).

46
Na espécie C. albicans podemos observar diversas estratégias para o desenvolvimento de
resistência, como alterações e super expressão dos sítios alvos dos antifúngicos e super expressão
na membrana plasmática, de bombas de efluxo capazes de extruir essas moléculas, impedindo
que sua concentração intracelular ideal seja alcançada (WHITE, MARR & BOWDEN, 1998;
revisto por AKINS 2005; COWEN & STEINBACH, 2008), porém, devemos lembrar que o
processo de resistência é sempre multifatorial, envolvendo, portanto, várias estratégias de
resistência (ROGERS & BARKER, 2003).
A resistência de isolados clínicos de C. albicans a análogos de flucitosina está
relacionada com mutações na enzima alvo; para polienos, a redução de ergosterol de membrana
está relacionada com a resistência; enquanto que para azóis, diversos mecanismos já foram
observados, como aumento da expressão e/ou mutações pontuais na enzima alvo (Erg11p) e
superexpressão de bombas de efluxo pertencentes as famílias MFS e ABC (revisto em
CANNON et al., 2007).
Dentre as bombas de efluxo relacionadas com o processo de MDR, a espécie C. albicans
é capaz de expressar diversos transportadores in vitro (WHITE, 1997; ROGERS & BARKER,
2003), porém não se sabe ao certo qual o nível de expressão desses transportadores é requerido,
in vivo, para se tornarem significativos clinicamente (CANNON et al., 2009). Análises do
genoma identificaram pelo menos 27 proteínas do tipo ABC; dessas proteínas, sete foram
confirmadas como sendo transportadores ABC, mas apenas duas, CaCDR1 (PRASAD et al.,
1995) e CaCDR2 (SANGLARD et al., 1997), estariam envolvidas com o fenótipo de MDR
transportando diversos compostos (Tabela I) (GAUR, CHOUDHURY & PRASAD, 2005) e
apresentando sua expressão aumentada em isolados resistentes a fluconazol e itraconazol
(ROGERS & BARKER, 2003; KARABABA et al., 2004; XU et al., 2006).
47
Transportador Substratos Inibidores
CaCDR1p (ABC)
Azóis, rodamina 6G, cicloheximida,
rodamina123, cerulenina, rifluoperazina,
nigericina, verapamil e vermelho do
nilo.
Milbemicina, eniatina,
FK506, FK520, unarmicina e
curcumina
CaCDR2p (ABC) Azóis, rodamina 6G, cicloheximida,
cerulenina, diamida e vermelho do nilo.
Milbemicina e curcumina
CaMDR1p (MFS)
Benomyl, metotrexato, fluconazol,
voriconazol, cicloheximida, cerulenina,
diamida e vermelho do nilo.
Sem descrições
Tabela I. Substratos e inibidores de transportadores de C. albicans com relevância clínica
(adaptado de CANNON et al., 2009).

48
Mutantes nulos em CaCDR1 se mostram extremamente sensíveis ao fluconazol
(SANGLARD et al., 1996), enquanto mutantes nulos apenas em CaCDR2, apresentam
sensibilidade reduzida, não alcançando níveis parecidos com o mutante para CaCDR1.
Entretanto, mutantes duplamente nulos, se tornam mais sensíveis quando comparados a mutantes
nulos apenas para CaCDR1 (SANGLARD et al., 1997). Estes dados confirmam que o
transportador CaCDR1 seria o principal transportador na expressão do fenótipo de MDR
(HOLMES et al., 2008; TSAO, RAHKHOODAEE & RAYMOND, 2009).
Além dos transportadores ABC, também se encontra transportadores MFS atuando em
espécies de Candida. Até o momento, seis genes foram anotados como possíveis transportadores
MFS, porém apenas um, o CaMDR1, está relacionado com o processo de MDR (FLING et al.,
1991; BEN-YAACOV et al., 1994) (Tabela I). Sua superexpressão está associada a isolados
clínicos que apresentam resistência a fluconazol, e seus mutantes nulos, apresentam resistência
diminuída a este antifúngico (WHITE, 1997; WIRSCHING, MICHEL & MORSCHHAUSER,
2000; PEREA et al., 2001). É interessante notar que, apesar dos transportadores CDR conferirem
resistência a diversos azóis (NAKAMURA et al., 2001), o produto do gene MDR1 confere
resistência apenas ao azol fluconazol (WIRSCHING &MORSCHHAUSER, 2000; KOHLI et al.,
2001), porém ambas as classes de transportadores estão fortemente relacionadas com a
resistência a azóis em isolados clínicos resistentes a fluconazol (SANGLARD & BILLIE, 2002)

49
1.5 Funções fiosiológica dos transportadores relacionados ao MDR em fungos
É possível que uma família tão grande, quanto à família ABC, existisse apenas com o
com o intuito de promover resistência a fármacos? Provavelmente não, e muitas evidências já
surgiram com relação à função fisiológica dos transportadores ABC em células de mamíferos
(revisto por AMBUDKAR et al., 1999), o que levanta suspeita de que nos fungos, assim como
em outros tipos celulares, tais transportadores também possuiriam outras funções fisiológicas
além de extruir fármacos do meio intracelular.
Reforçando esta hipótese está o fato de alguns estudos demonstrarem que proteínas
homólogas a estes transportadores não realizam efluxo de fármacoss, estando diretamente
relacionados com funções fisiológicas nas células. Um exemplo seriam os transportadores
CDR3p e Ste6p, de C. albicans e S. cerevisiae, respectivamente, que guardam grande homologia
com CDR1p, CDR2p e Pdr5p, mas não estão envolvidos com o efluxo de drogas (MCGRATH &
VERSHAVSKY, 1989; BALAN, ALARCO & RAYMOND, 1997; FRANZ, MICHEL &
MORSCHHAUSER, 1998; PRASAD, PANWAR & SMRITI, 2002).
Estudos realizados por Decottignies e colaboradores (1995) mostraram que a
superexpressão de SNQ2 e PDR5 acarreta em redução da fase exponencial de crescimento,
sugerindo, portanto, que ambas as proteínas seriam necessárias para o desenvolvimento celular.
Até o momento, estudos apontam no geral apenas duas funções fisiológicas destes
transportadores em fungos: manutenção da distribuição assimétrica de fosfolipídios na
membrana plasmática e extrusão de catabólitos.
A superexpressão, não apenas de PDR5, mas também de CDR1 está diretamente
relacionada com a fase do crescimento celular, sendo sua expressão maior na fase exponencial e

50
até mesmo na fase estacionária do crescimento (SERVOS, HAASE & BRENDEL, 1993;
SZCZYPCA et al., 1994; DECOTTIGNIES et al., 1995; KATZMANN et al., 1995; PRASAD et
al., 1995; SANGLARD et al., 1997; WEMMIE & MOYE-ROWLEY, 1997; KOLACZKOWSKI
et al., 1998; KRISHNAMUYTHY et al., 1998; LYONS & WHITE, 2000), o que poderia ter
uma ligação com o fato de que nesta fase existe uma grande concentração de catabólitos no
interior celular, sendo assim, esta expressão aumentada permitiria à célula se livrar do acúmulo
de metabólitos tóxicos durante seu desenvolvimento e metabolismo.
A distribuição assimétrica dos fosfolipídios na membrana plasmática dos fungos parece
ser mantida por alguns transportadores do tipo ABC. Na maioria dos tipos celulares
fosfatidiletanolamina e fosfatidilserina estão localizadas no folheto interno da membrana
plasmática, enquanto fosfatidilcolina, esfingomielina e glicolipídios estão localizados no folheto
externo (DIAZ & SCHROIT, 1996; WILLIAMSON & SCHLEGEL, 1997).
Recentemente, observou-se que alguns transportadores de C. albicans e S. cerevisiae
estão relacionados com essa distribuição assimétrica. Cdr1p e Cdr2p parecem atuar em
movimentos de fli-flop, no qual a fosfatidiletanolamina do folheto interno é translocada para o
folheto externo (DOGRA et al., 1999; SMRITI et al., 2002). Enquanto isso, Cdr3p, um
transportador ABC de C. albicans que não tem envolvimento com o processo de resistência a
múltiplas drogas, atua na translocação de fosfolipídios do folheto externo para o folheto interno
(SMRITI et al., 2002). Essa diferença na direção do transporte pode ter relação com a capacidade
de transporte de fármacos, não conferida a Cdr3p (SMRITI et al., 2002).
Assim como para C. albicans, alguns transportadores de S. cerevisiae também estão
envolvidos na translocação de fosfolipídios. Decottignies e colaboradores (1998) mostraram que
tanto Pdr5p quanto Yor1p também estão envolvidos na translocação de fosfatidiletanolamina.

51
Até o momento, atividades secretoras não foram observadas para transportadores de
fungos, a não ser para Ste6p, de S. cerevisiae, que está envolvido na secreção do peptídeo
ferohormônio fator-a, essencial para o processo de brotamento (MCGRATH & VERSHAVSKY,
1989).
1.6. Os terpenóides: ácido ursólico e oleanólico
Os terpenos constituem uma família bem diversificada de substâncias naturais,
produzidos por uma grande variedade de plantas, principalmente as coníferas, estando presentes
nas sementes, folhas, flores, raízes e madeira de plantas superiores, bem como em musgos, algas
e líquens.
Inicialmente considerou-se que os terpenos seriam derivados do 2-metil-butadieno, mais
conhecido como isopreno, no entanto, com o tempo percebeu-se que os terpenos não derivavam
do isopreno, uma vez que este nunca foi encontrado como produto natural, sendo seu verdadeiro
precursor o ácido mevalônico. Apesar disso, a regra do isopreno ainda é usada para classificá-
los. Sendo assim, são classificados de acordo com a quantidade de unidades de isopreno em:
hemiterpenóides (C5), monoterpenóides (C10), sesquiterpenóides (C15), diterpenóides (C20),
triterpenóides (C30) e carotenóides (C40) (SPITZER, 2003).
Essas substâncias estão presentes nos óleos essenciais de muitas plantas e flores, sendo
esses óleos muito utilizados nas indústrias alimentícias, cosmética e farmacêutica (SPITZER,
2003).

52
Dentre diversas ações biológicas, os terpenóides que ocorrem naturalmente, apresentam
propriedades antiinflamatórias, antimicrobianas, antiparasitárias, tripanocida, larvicida e
hipoglicemiante, além de inibir a agregação plaquetária e interferir em passos da transdução de
sinais (CALIXTO et al., 1998; 2000) bem como atuação em bombas de efluxo relacionadas com
o processo de MDR (COREA et al., 2003; BRAGA et al., 2007).
O ácido oleanólico (AO) e o seu isômero, ácido ursólico (AU) (Figura 6), são triterpenos
que se encontram abundantes na natureza, sendo o AO encontrado em mais de 120 espécies
distintas de plantas (WANG & JIANG, 1992), como por exemplo a Lippia lacunosa, encontrada
no Brasil (Figura 6). Ambos podem existir naturalmente como ácidos livres ou aglicanas,
também denominadas saponinas (PRICE, JOHNSON & FENWICK, 1987; MAHATO et al.,
1988; WANG & JIANG, 1992) quando possuem uma aglicana ligada a uma ou mais cadeias de
açúcar. Estruturalmente esses compostos se diferem apenas pela posição de um grupamento
metila (revisto por LIU, 1995).
Durante as últimas três décadas, estudos farmacológicos envolvendo AU e AO indicaram
que estes triterpenóides possuem muitos efeitos benéficos à saúde humana (revisto por LIU,
1995) apresentando baixa toxicidade para células de mamíferos (SCHIMMER et al., 2003).
Em 1969, Gupta e colaboradores, foram os primeiros a identificar uma ação
antinflamatória proporcionada pelo AO. O efeito antinflamatório é uma propriedade comum aos
triterpenóides (PRICE et al., 1987; MAHATO et al., 1988), estando o AO e o AU em posição de
destaque por apresentarem um alto grau de inibição deste processo.
A capacidade hepatoprotetora destes compostos recebe um grande destaque, sendo muito
explorada e estudada sua capacidade em proteger o fígado contra injúrias causadas tanto por
compostos tóxicos quanto contra fibrose e cirrose (LIU,1995). Após resultados terapêuticos

53
satisfatórios em testes clínicos (Hunan Med. Inst., 1975), o AO tem sido usado com sucesso por
médicos chineses com o intuito de tratar doenças do fígado, como hepatite aguda e crônica,
dentre outras doenças (QU, 1981; WU & LI, 1986; CHEN & WANG, 1989). No Brasil, também
encontramos medicamentos indicados para regeneração da pele, antinflamatório, prevenção de
manchas, tratamento de acne e hepatoprotetor, que também possuem AO ou AU em sua
composição, como Colastil®, Tri-Def® e Ursacol®. O efeito benéfico ao fígado pode estar
relacionado não só com sua atividade antinflamatória, mas também antioxidante, bem como
devido a seus efeitos em enzimas que metabolizam drogas (LIU, 1995).
Outras atividades biológicas também são relacionadas a estes compostos, como
propriedades antihipertensiva, antimicrobiana, anti ateriosclerótica e antitumoral (revisto por
LIU, 2005; HORIUCHI et al., 2007), nesta última podendo atuar como inibidores do processo de
angiogênese, invasão tumoral e metástase (OVESNA et al., 2004).

54
(A) (B)
(C)
Fig. 6. Exemplar do vegetal Lippia lacunosa utilizado para o isolamento do ácido
oleanólico e da mistura ácido oleanólico/ácido ursólico testados neste trabalho. (a) Arbusto
em seu local de cultivo e coleta - Horto da Faculdade de Farmácia – Bioquímica da Universidade
Federal de Juíz de Fora (UFJF), (b) detalhe da inflorescência (fotos gentilmente cedidas pela
Profa. Suzana Leitão e a pós-graduanda Aline Castellar – Faculdade de Farmácia/UFRJ), (c)
estrutura química dos compostos isolados, ácido oleanólico (posição I = H e posição II = CH3) e
ácido ursólico (posição I = CH3 e posição II = H).

55
Com relação a sua atividade antitumoral, diversos grupos apontam que ambos compostos
podem atuar em etapas distintas do desenvolvimento tumoral, como a iniciação e promoção, bem
como na inibição de proliferação de células tumorais e na indução de apoptose dessas células
(HUANG et al., 1994; HSU et al., 2004; LIU, 2005; PATHAK et al., 2007; MANU &
KUTTAN, 2008; YAN et al., 2009; SHAN et al., 2009). Dentre muitos relatos de atuação desses
compostos em células cancerosas, podemos destacar: a indução de apoptose em células de
leucemia mielóide aguda e em linhagens de câncer de fígado, proporcionadas, respectivamente,
pela utilização de AO e derivados modificados quimicamente, e pelo AO e AU (KONOPLEVA
et al., 2004; YAN et al., 2009); a capacidade do AU em induzir apoptose e inibir a proliferação
de células de câncer de ovário humano (WANG et al., 2009) bem como induzir apoptose em
células de câncer de próstata, mesmo quando a proteína relacionada à resistência a apoptose
(Bcl2) está sendo constitutivamente expressa (ZHANG et al., 2010); o potencial, apresentado
pelo AU em inibir a ativação do NF-κB, um fator de transcrição que atua de forma crítica na
sobrevivência e proliferação de células tumorais (SHISHODIA et al., 2003) e a capacidade do
AO e seu derivado contendo uma molécula de dextrose ligada (DEX-AO), in vitro e in vivo, de
inibir a atividade de células de osteosarcoma (HUA et al., 2009).
No Japão o uso destes compostos é recomendado para o tratamento de câncer de pele
(MUTO et al., 1990), já tendo sido patenteado por japoneses, formulações cosméticas contendo
AO/AU para uso tópico com o intuito de proteger contra este tipo de câncer (ISHIDA et al.,
1990).
Em estudos de avaliação da capacidade antimicrobiana destes compostos, Horiuchi
(2007) observou esta atividade apenas contra bactérias gram positivas, como Staphylococcus
aureus resistente a meticilina (MRSA), Staphylococcus pneumonie e enterococos resistentes a

56
vancomicina (VRE). Neste trabalho também se concluiu que para esta atividade, o AU
apresentava um potencial superior ao AO. Análises mostram que nenhum dos compostos
apresenta atividade antifúngica contra C. albicans, necessitando de concentrações acima de 128
µg/ml para se impedir a proliferação deste fungo (HORIUCHI et al., 2007). Outros estudos
confirmam esse dado, enfatizando a atuação do AO na membrana desses microrganismos
(SZAKIEL et al., 2008).
Existem na literatura dados contraditórios com relação à atividade microbicida frente a
bactérias gram negativas e estudos mais recentes priorizam que esta atividade não é observada
para nenhum dos dois compostos (WOLDEMICHAEL et al. 2003; HORIUCHI et al., 2007).
Para AO já foram observadas atividades sinérgicas com Rifampicina, Isoniazida e Etambutol, já
foram observadas no tratamento in vitro contra Mycobacterium tuberculosis (GAUR et al.,
2010).
Alguns estudos demonstram a capacidade do AO em inibir transportadores relacionados
ao fenótipo de MDR. Braga e colaboradores (2007) mostraram a capacidade deste composto em
bloquear o efluxo do substrato fluorescente da bomba de efluxo MRP1, o composto CFDA,
porém o mesmo não era capaz de impedir o efluxo de Rodamina 123 da bomba P-gp. Neste
trabalho também foi demonstrada a baixa toxicidade do AO frente a células de mamíferos,
evidenciando sua capacidade citotóxica apenas contra células cancerosas. Para o AU, um estudo
recente evidenciou sua capacidade em inibir o efluxo dos substratos fluorescentes da P-gp a
daunorubucina e rodamina 123, porém o mesmo composto não foi capaz de inibir o efluxo de
calceína, um substrato fluorescente da bomba MRP1 (NABEKURA et al., 2009), o que pode
indicar que tais compostos atuariam em sítios distintos nessas bombas.

57
1.7. Revertendo o fenótipo MDR em fungos
Devido ao crescente número de pacientes imunodebilitados, seja pelo aparecimento da
síndrome da imunodeficiência adquirida (do inglês, AIDS), da terapia anticâncer ou pelo grande
número de transplantes realizados, vê-se um aumento significativo na incidência de infecções
fúngicas resistentes.
As infecções fúngicas sistêmicas geralmente são fatais, sendo poucos os compostos
disponíveis para serem usados na terapia antifúngica, além disso, muitas desses fármacos não são
utilizadas por apresentarem alta toxicidade, “resistência” ou alto custo, restando assim, um
pequeno leque de escolhas para compor uma terapia eficaz (revisto por CANNON et al., 2009).
O desenvolvimento de novos compostos antifúngicos continua, porém a maioria dos
compostos desenvolvidos atua em alvos intracelulares, o que torna essas moléculas suscetíveis
aos mecanismos de detoxificação celular ou efluxo, caracterizando sua resistência. Cannon e
colaboradores (2009) propuseram alguns interessantes mecanismos de ação para possíveis novos
fármacos que possam atuar em seu sitio alvo sem sofrer efluxo proporcionado pelos
transportadores MDR, como pode ser observado na Figura 7, onde quatro modelos estão
demonstrados. Destes quatro modelos o que chama mais a atenção seria o de compostos
denominadas quimiosensibilizadoras (Figura 7b), que poderiam atuar inibindo as bombas de
efluxo e permitindo que antifúngicos já disponíveis no mercado possam ser usados sem serem
extruídos das células, permitindo assim, a reversão do fenótipo de MDR.
Dentre os compostos quimiosensibilizadoras já conhecidas estão o FK506 (MAESAKI et
al., 1998) e a ciclosporina A (MARCHETTI et al., 2000), ambos inibidores do processo de MDR
em células de mamíferos, sendo o FK506 também inibidor de transportadores ABC de fungos,

58
porém ambos são imunossupressores, o que impossibilita seu uso para tratamentos de longa
duração na clínica.
Para a quimioterapia antifúngica não encontramos compostos quimiosensibilizadores
sendo utilizados na clínica, porém, alguns compostos já foram mostrados como promissores em
reverter tal fenótipo, como eniatina, fenotiazinas, isonitrila e derivados do ácido gálico
(KOLACZKWSKI, MICHALAK & MOTOHASHI, 2003; HIRAGA et al., 2005;
YAMAMOTO et al., 2005; RANGEL et al., 2010), no entanto, os testes realizados se resumem a
Pdr5p e a transportadores de C. albicans expressos em S. cerevisiae.
Diversos grupos passaram a buscar em fontes naturais como plantas, possíveis inibidores
de transportadores ABC, tendo em vista que muitos dos compostos que podem ser isolados,
como alcalóides, flavonóides e terpenóides já foram testados em células cancerosas que
apresentam o fenótipo de MDR com algum êxito (KUNTZ et al., 1999; IKEGAWA et al., 2000;
COREA et al., 2003; BRAGA et al, 2007; RANGEL et al., 2008).

59
(A) (B) (C) (D)
Fig. 7: Propostas de fármacos alternativos que possam atuar em células que apresentem o
fenótipo de MDR. (A) Célula suscetível a um composto que não é substrato para as bombas de
efluxo, ou que é rapidamente fungicida. (B) o composto é administrada em combinação com
inibidor para a bomba. (C) O composto é um inibidor multifuncional que afeta ao mesmo tempo
seu alvo, a atividade da bomba de efluxo e a transcrição do transportador. (D) a captação do
fármaco é aumentada e supera o efluxo. Fator de transcrição(FT) (Adaptado de CANNON et al.,
2009)..

60
2. JUSTIFICATIVA
A resistência a múltiplas drogas é um processo preocupante. Tendo em vista a variedade
da flora brasileira e as diversas referências, vindas do uso popular e corroboradas por pesquisas
científicas de seu uso no tratamento de enfermidades, torna-se evidente o interesse de nosso
grupo por compostos isolados de plantas que possam ter algum efeito inibitório de
transportadores que conferem o fenótipo MDR.
Em nosso estudo preliminar, em colaboração com o grupo da Prof. Dra. Suzana
Guimarães Leitão, do Departamento de Alimentos e Produtos Naturais da Faculdade de
Farmácia – UFRJ e sua aluna de mestrado Aline Castellar, iniciado em minha monografia,
frações da espécie Lippia lacunosa se mostraram eficazes em promover a inibição do
transportador Pdr5p, sendo isolado inicialmente um triterpeno da fração mais ativa. No início
deste trabalho realizamos a identificação deste triterpeno, denominado ácido oleanólico e um
novo isolamento proporcionou a identificação de uma mistura dos terpenos ácido oleanólico e
ácido ursólico, o que nos estimulou a avaliar a capacidade desses compostos em atuar sobre o
fenótipo de resistência a múltiplas drogas em uma cepa de S.cerevisiae e um isolado clínico de
C. albicans.

61
3. OBJETIVOS
• Avaliar a toxicidade do ácido oleanólico, da mistura ácido oleanólico/ursólico, isolados
da espécie Lippia lacunosa e do ácido ursólico purificado (comercial), frente a cepas
Saccharomyces cerevisiae.
• Avaliar a capacidade do ácido oleanólico, da mistura ácido oleanólico/ursólico e do ácido
ursólico purificado em inibir o transporte do substrato fluorescente, rodamina 6G,
promovido pelo transportador Pdr5p de Saccharomyces cerevisiae.
• Avaliar a toxicidade do ácido ursólico purificado frente às cepas de Candida albicans,
utilizando a cepa padrão ATCC24433 e o isolado clínico hiperesistente a fluconazol,
obtido de mucosa oral de paciente HIV+, procedendo a uma caracterização da resistência
deste isolado.
• Avaliar o potencial do ácido ursólico em reduzir a concentração mínima inibitória (MIC)
do fluconazol sobre o isolado clínico hiper resistente de Candida albicans, avaliando o
tipo de interação que esses dois compostos possuem.

62
4. MATERIAL E MÉTODOS
4.1 Produtos químicos e fármacos
AO e a mistura AO/AU foram isolados da espécie vegetal Lippia lacunosa e
solubilizados em DMSO 100%. O AU foi obtido comercialmente (Sigma Aldrich) e também
solubilizado em DMSO 100%. Fluconazol (Aquafarma®) e rodamina 6G (Sigma®) foram
dissolvidos em água destilada, sendo o fluconazol solubilizado em água destilada estéril.
Itraconazol (Sigma®), FK506 (Tecoland Corporation® USA) e vermelho do Nilo (Sigma®)
foram solubilizados em DMSO 100%. Concentrações de DMSO de no máximo 2% foram
utilizadas nos ensaios, dados anteriores de nosso grupo indicam que nestas concentrações não
ocorre interferencia no crescimento celular.
4.2 Isolamento de compostos
O extrato em diclorometano originado das folhas de Lippia lacunosa foi fracionado em
coluna com suporte de sílica de malha de 70 – 230 . As frações foram recolhidas até o volume de
300 ml. Como sistema de solventes foi utilizado diclorometano / acetato de etila seguido de
acetato de etila / metanol, ambos em proporções decrescentes.
As frações foram analisadas em cromatografia em camada delgada (CCD) em placas de
sílica e reveladas com solução de ácido sulfúrico com posterior aquecimento por dois minutos a
100 oC. As manchas observadas na placa, referentes à triterpenos, foram raspadas e purificadas e

63
os compostos identificados por Ressonância Mangnética Nuclear de H1 e C13 no Núcleo de
Pesquisa de Produtos Naturais (NPPN) da Universidade Federal do Rio de Janeiro.
4.3 Crescimento dos microrganismos
De acordo com o procedimento a ser realizado o cultivo das cepas utilizadas neste
trabalho foi realizado como descrito a seguir: as cepas mutantes de S. cerevisiae AD124567 ou
AD1234567 foram crescidas em meio de cultura YPD (dextrose 2 %, extrato de levedura 1 % e
peptona 2 %) a partir de um inóculo inicial de 2 x 105 células/ml, até se obter aproximadamente
3,0 de densidade ótica, a 600 nm (primeira metade da fase exponencial de crescimento celular).
Para as cepas de C. albicans, o crescimento foi realizado em meio BHI ou Sabouraud Dextrose
(SD) dependendo do experimento a ser realizado. Para solidificar os meios de cultura, quando
necessário, 2% de ágar foi utilizado. O isolado clínico de C. albicans (Pri) foi mantido em
estoque de glicerol (20% v/v de glicerol) e estocado a - 20 oC e sempre que necessário, reativado
em meio líquido antes da inoculação em meio sólido. A cepa ATCC 24433 e as cepas de S.
cerevisisiae foram mantidas em meio sólido a 4 oC.
4.4 Avaliação da toxicidade dos compostos frente às cepas de S. cerevisiae e C.
albicans
O efeito tóxico do AO, da mistura AO/AU e do AU foi avaliado em meio líquido como
descrito no protocolo M27-A2 do CLSI com pequenas modificações. Para cepas de S. cerevisiae,
concentrações crescentes de AO, AO/AU e AU foram utilizadas. A partir de um crescimento em

64
meio líquido, 4,0 x 103 foram incubadas em meio YPD em placas de 9 poços. As células foram
inoculadas em cada poço juntamente com concentrações crescentes dos compostos, sendo o
volume final em cada poço igual a 200 µl. Para S. cerevisiae, a placa foi incubada a 28 oC por
48h. O crescimento celular foi avaliado de acordo com a turbidez do meio de cultura, medida a
600 nm em leitor de microplacas (FLUOstar OPTIMA BMG Labtech). Para as cepas de C.
albicans o mesmo protocolo foi realizado, utilizando-se o meio líquido SD e concentrações
crescentes dobradas apenas do AU, sendo a placa incubada a 37 oC por 48 h. Como controle foi
utilizado o poço contendo apenas meio de cultura e células, sendo este ponto considerado 100%
de crescimento celular.
4.5 Efluxo de Rodamina 6G
4.5.1 Análise Qualitativa - Microscopia óptica de fluorescência
• Avaliação da capacidade do AO e da mistura AO/AU em inibir o efluxo de
rodamina 6G proporcionado pela Pdr5p de S. cerevisiae
Com objetivo de avaliar o potencial dos compostos isolados em promover o acúmulo de
rodamina 6G (um substrato fluorescente da Pdr5p) foi utilizado o protocolo descrito por
Yamamoto e colaboradores (2005), com algumas modificações. As cepas AD124567 e
AD1234567 foram crescidas em meio YPD até se obter aproximadamente 3,0 de densidade ótica
a 600 nm. As células foram lavadas e ressuspendidas em tampão PBS glicosado (2% p/v) pH 7,0
e 2,0 x 106 células/ml foram incubadas por 2 horas com AO ou a mistura AO/AU na

65
concentração final de 100 µM, com posterior adição de rodamina 6G (concentração final de 4
µM) e incubação por 30 minutos. Depois de lavadas 3 vezes com tampão PBS pH 7,0, as células
foram visualizadas em microscópio de fluorescência (Nikon Eclipse E400) .
Como controle positivo de acúmulo de rodamina 6G utilizamos a cepa sensível
AD1234567, bem como uma cepa selvagem de S. cerevisiae (gentilmente cedida pelo Prof. Dr
Allen Hagler – Titular do Departamento de Microbiologia Geral /IMPPG/UFRJ), isolada de
bromélia e que não foi manipulada geneticamente em laboratório. O composto FK506 foi
utilizado como controle positivo de inibição do transporte de rodamina 6G, na concentração final
de 20 µM.
4.5.2 Análise Quantitativa - Citometria de fluxo
• Avaliação da capacidade do AO, da mistura AO/AU e do AU em inibir o efluxo
de rodamina 6G proporcionado pela Pdr5p de S. cerevisiae
Este teste foi realizado como descrito por Cernicka e colaboradores (2007) com algumas
modificações. As cepas AD1234567 e AD124567 foram crescidas até se obter aproximadamente
3,0 de densidade ótica a 600 nm, quando então foram lavadas com tampão PBS pH 7,0. Após as
lavagens, 2,0 x 106 células foram ressuspendidas em PBS glicosado (2% p/v) pH 7,0 para um
volume final de 500 µl. A essa suspensão de células foi adicionado o composto de interesse em
concentrações crescentes. Para testes com AO e a mistura AO/AU foram usadas as
concentrações de 10, 25, 50, 100 e 200 µM e para os testes com AU foram usadas as
concentrações de 10, 25, 50, 100, 125, 150 e 200 µM. Estas células foram posteriormente

66
incubadas por 30 min a 30 ºC com as concentrações de interesse de cada um dos compostos
juntamente com rodamina 6G (concentração final de 4 µM). Após o tempo de incubação, 500 µl
de PBS pH 7,0 gelado foi adicionado, com o objetivo de diminuir o metabolismo celular, sendo
as células então centrifugadas a 5000 rpm em centrífuga refrigerada e posteriormente
ressuspendidas em 500 µl de PBS glicosado pH 7,0 (2% p/v) contendo as mesmas
concentrações anteriores dos compostos, seguindo uma nova incubação por mais 30 min a 30ºC.
Após o tempo de incubação as células foram lavadas e armazenadas em gelo até serem avaliadas
em citômetro de fluxo (BD FACSCalibur). Como controle de acúmulo de rodamina 6G
utilizamos a cepa AD1234567. Como controle de inibição do efluxo de rodamina 6G foi
utilizado o inibidor clássico FK506 a 20 µM. As avaliações em citômetro de fluxo foram
realizadas no Instituto Nacional do Câncer, com a colaboração do Prof. Dr. Marcelo Alex
Carvalho e do doutorando Renato Sampaio Carvalho.
4.6 Determinação da concentração mínima inibitória de antifúngicos de uso clínico
para cepas de C. albicans
Para determinar a concentração mínima inibitória de antifúngicos de uso clinico, foi
utilizado o protocolo M27-A2 do CLSI com pequenas modificações. Para as cepas de C.
albicans utilizamos os antifúngicos fluconazol (2 a 128 µg/ml) e itraconazol (0,1 a 2 µg/ml). A
partir de um crescimento em meio líquido, 4 x 103 células/poço foram incubadas em meio SD em
placas de 96 poços. As células foram inoculadas em cada poço juntamente com concentrações
crescentes dos fármacos, sendo o volume final em cada poço igual a 200 µl. A placa foi incubada
a 37 oC por 48 h. O crescimento celular foi avaliado em cada poço como turbidez, medida em

67
leitor de microplacas (FLUOstar OPTIMA BMG Labtech) a 600 nm. Como controle foi
utilizado o poço contendo apenas meio de cultura e células, sendo este ponto considerado 100%.
4.7 Avaliação de atividade sinérgica entre ácido ursólico e fluconazol frente ao
isolado clínico de C. albicans
Com objetivo de avaliar a existência de um efeito combinatório entre AU e fluconazol,
experimentos de “checkerboard” foram realizados, para o isolado Pri (cepa obtida de coleta de
infecção orofaríngea de paciente HIV+) (PORTELA, 2006), de acordo com o protocolo descrito
pelo Mukherjee e colaboradores (2005). Interações entre as moléculass foram classificadas de
acordo com o índice de inibição fracionária (FICI), um modelo não paramétrico definido pela
equação: FICI = FIC do composto A + FIC do compostoa B (MUKHERJEE et al., 2005). FIC do
composto A é definido como o MIC80 do AU combinado com fluconazol dividido pelo MIC80 do
AU sozinho, e o FIC do composto B é definido como MIC80 do fluconazol combinado com o AU
dividido pelo MIC80 do fluconazol sozinho. Quando o MIC80 para alguma dos compostos não é
alcançado, se admite que o MIC80 passe a equivaler a próxima concentração dobrada. Os valores
obtidos por esta equação definem o tipo de interação entre as moléculass. Valores abaixo ou
igual a 0,5 indicam interação sinérgica, entre 0,5 – 1,0 indicam atividade aditiva, entre 1 – 2,
indicam atividade indiferente e maior de 2,0, efeitos antagônicos.
Para este experimento utilizamos o protocolo de checkerboard como descrito por
Marchetti e colaboradores (2000), seguindo as recomendações do CLSI no qual concentrações
crescentes, dobradas, de AU e de fluconazol foram utilizadas sozinhas ou em combinação.

68
4.8 Avaliação da capacidade do isolado clínico de C. albicans em extruir compostos
fluorescentes
Com objetivo de se caracterizar a capacidade do isolado clínico de C. albicans em extruir
fluoróforos padrões, as cepas ATCC 24433 e Pri, foram crescidas em meio BHI até se obter
aproximadamente 6,0 de densidade ótica (primeira metade da fase fase exponencial de
crescimento celular). As células foram lavadas e ressuspendidas em tampão PBS glicosado (2%
p/v) pH 7,0 e 2 x 106 células/ml foram incubadas por 30 minutos à 30oC com rodamina 6G
(substrato das bombas CaCDR1p e CaCDR2p) na concentração final de 4 µM ou Vermelho do
Nilo (substrato das bombas CaCDR1p, CaCDR2p e CaMDR1p) na concentração final de 7 µM.
Depois de lavadas três vezes com tampão PBS pH 7,0, as células foram visualizadas em
microscópio de fluorescência. Neste caso a cepa ATCC 24433 foi utilizada como controle
positivo para acúmulo de rodamina 6G.
4.9 Análises estatísticas
Quando necessário utilizamos os testes de Anova e Dunnets, utilizando para realização
dos mesmos o programa Excell (Microsoft®), dotado das equações dos referidos testes, para os
cálculos matemáticos.

69
5. RESULTADOS
5.1 Avaliação da toxicidade do AO e da mistura AO/AU frente a cepas AD1234567 e
AD124567 de S. cerevisiae
Nossos experimentos se iniciaram com teste visando avaliar a toxicidade do AO e da
mistura AO/AU frente às cepas AD1234567 e AD124567, a partir de agora denominadas
hipersensível e hiperesistente, respectivamente. Utilizando-se concentrações crescentes de AO e
da mistura AO/AU, que variavam de 10 a 200 µM no meio de cultura, estas células foram
incubadas por 48 horas na presença e ausência dos compostos, possibilitando avaliar diferenças
no crescimento celular em cada uma dessas concentrações. Como demonstrado no resultado
apresentado na figura 8, para a cepa hiperesistente podemos notar que nenhum dos compostos foi
capaz de influenciar negativamente o crescimento celular desta cepa, mesmo na maior
concentração testada (200 µM), apresentando inibição máxima de 3,17 ± 2,1 % e 4,87 ± 3,5 %
do crescimento celular, quando tratada, respectivamente, como AO e AO/AU.
Prosseguindo nossos testes, o mesmo experimento citado previamente foi realizado, desta
vez com a cepa hipersensível (Figura 9). Foi possível observar uma inibição de 36,5 ± 2,5 %do
crescimento celular quando esta cepa foi submetida ao tratamento com AO, e 9,98 ± 5,48 % de
inibição quando tratada com AO/AU, sendo que estes valores são observados apenas quando
ambos foram testados na concentração de 200 µM.

70
Composto (µM)
0 50 100 150 200
Cre
scim
ento
cel
ular
(% re
lativ
a ao
con
trole
)
0
20
40
60
80
100
120
140
Fig. 8: Avaliação da toxicidade do AO e da mistura AO/AU sobre o crescimento da cepa
AD124567. A cepa AD124567 foi crescida em meio YPD por 48 horas na presença de
concentrações crescentes de AO (●) ou AO/AU (○) como descrito em Material e Métodos.
Como controle utilizamos o crescimento das cepas na ausência do composto. Os resultados
refletem a média de três experimentos ± erro padrão.

71
Composto (µM)
0 50 100 150 200
Cre
scim
ent
o ce
lula
r (%
rela
tiva
ao c
ontro
le)
0
20
40
60
80
100
120
Fig. 9: Avaliação da toxicidade do AO e da mistura AO/AU sobre o crescimento da cepa
AD1234567. A cepa AD1234567 foi crescida em meio YPD por 48 horas na presença de
concentrações crescentes de AO (●) ou AO/AU (○) como descrito em Material e Métodos.
Como controle utilizamos o crescimento das cepas na ausência do composto. Os resultados
refletem a média de três experimentos ± erro padrão.

72
5.2 Avaliação da capacidade do AO e da mistura AO/AU em inibir o efluxo de
rodamina 6G realizado pela Pdr5p
Em uma segunda etapa, foi realizada uma avaliação da capacidade destes compostos em
inibir o transporte de rodamina 6G, um substrato fluorescente da bomba de efluxo Pdr5p,
utilizando para isto uma concentração única desses compostos (100 µM final) no tratamento da
cepa hiperesistente.
Uma análise qualitativa, utilizando uma abordagem de microscopia óptica, nos permitiu
observar que na concentração testada o AO parece ser capaz de inibir o transporte do substrato
fluorescente, sendo possível observar a retenção de uma pequena quantidade de fluorescência no
interior da célula (Figura 10). Para a mistura AO/AU, conseguimos observar um acúmulo de
rodamina 6G na célula hiperesistente, visualmente maior que o acúmulo observado para o
tratamento com o AO puro (Figura 10), sugerindo uma maior eficiência da mistura de ambos os
compostos.
Para quantificarmos e analisarmos a diferença nos níveis de fluorescência procedemos a
uma avaliação quantitativa da inibição observada nos experimentos de microscopia, utilizando,
dessa vez, concentrações crescentes do AO, variando de 10 a 200 µM, para avaliar a capacidade
deste composto em inibir o efluxo de rodamina 6G, permitindo assim sua retenção no interior da
célula hiperesistente. Os resultados obtidos se referem a porcentagem de inibição relativa ao
controle, célula hipersensível, que equivale ao nosso máximo de retenção do fluoróforo. Para
este experimento, trabalhamos com a abordagem de citometria de fluxo, em colaboração com o
Prof. Dr. Marcelo Alex Carvalho, do Instituto Federal do Rio de Janeiro (IFRJ) e doutorando
Renato Sampaio Carvalho, utilizando o citômetro do Instituto Nacional do Câncer (INCA). Neste
experimento podemos observar que apenas na maior concentração testada de AO (200 µM), foi

73
possível observar um efeito inibitório significativo, porém, muito inferior ao efeito observado
pelo tratamento com o inibidor clássico FK506, alcançando apenas 16,86 ± 3,01 % de inibição
do efluxo (Figura 11). Um controle realizado neste ensaio permitiu a exclusão da hipótese de
autofluorescencia do AO. Para isto as células foram submetidas a tratamento apenas com este
composto, em sua maior concentração (200 µM) e foram analisadas por citometria (dado não
mostrado).

74
Fig. 10. Avaliação qualitativa da inibição do efluxo de rodamina 6G após tratamento
da cepa AD124567 com AO ou AO/AU. Para avaliar a capacidade dos compostos em inibir o
efluxo de rodamina 6G foi realizado como descrito em Material e Métodos. Para este
experimento utilizamos como controles positivos de acúmulo de rodamina 6G as cepas
AD1234567 (PDR -) e selvagem (WT); para controle positivo de inibição utilizamos o fármaco
FK506 a 20 µM e para controle positivo de extrusão, a cepa AD124567 (PDR +). AO e AO/AU
foram utilizados a 100 µM.
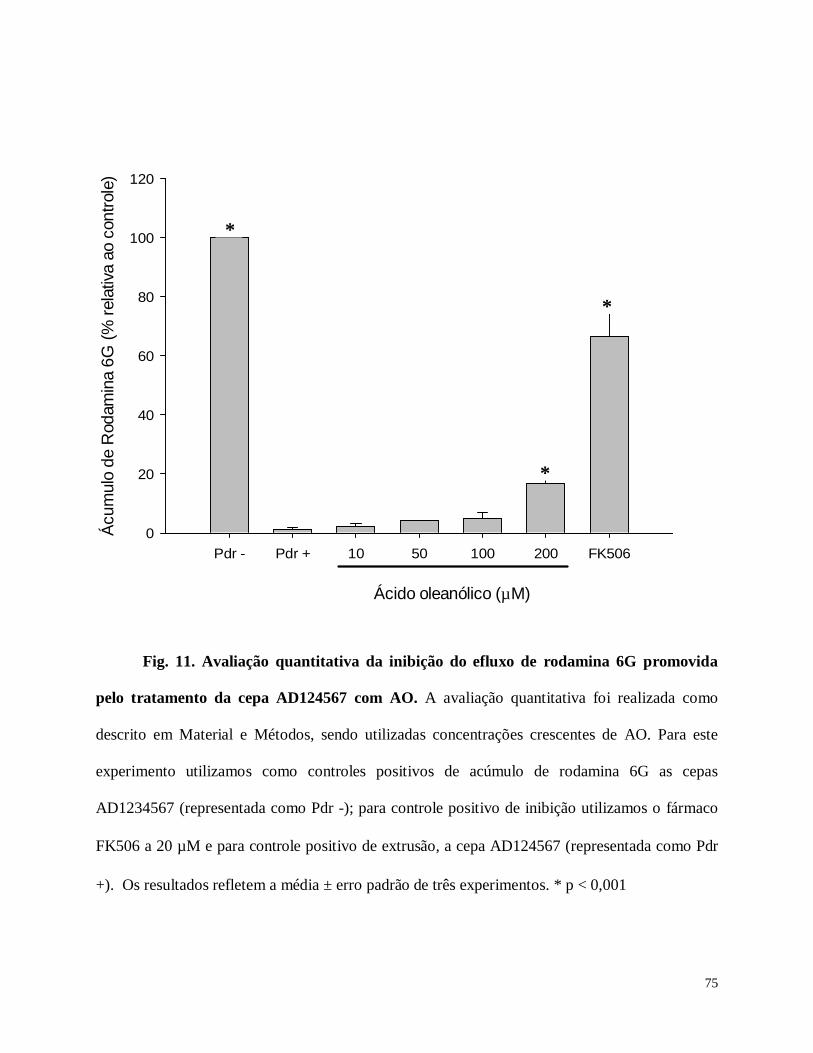
75
Ácido oleanólico (µM)
Pdr - Pdr + 10 50 100 200 FK506
Ácum
ulo
de R
odam
ina
6G (%
rela
tiva
ao c
ontro
le)
0
20
40
60
80
100
120
Fig. 11. Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida
pelo tratamento da cepa AD124567 com AO. A avaliação quantitativa foi realizada como
descrito em Material e Métodos, sendo utilizadas concentrações crescentes de AO. Para este
experimento utilizamos como controles positivos de acúmulo de rodamina 6G as cepas
AD1234567 (representada como Pdr -); para controle positivo de inibição utilizamos o fármaco
FK506 a 20 µM e para controle positivo de extrusão, a cepa AD124567 (representada como Pdr
+). Os resultados refletem a média ± erro padrão de três experimentos. * p < 0,001
*
*
*

76
Realizamos também uma avaliação quantitativa da inibição inicialmente observada por
microscopia quando se tratava a célula hiperesistente com a mistura AO/AU (Figura 12). Para
isto foram utilizadas concentrações crescentes do AO/AU, variando de 10 a 200 µM, utilizando o
mesmo protocolo citado para AO (Figura 11).
Neste experimento podemos constatar um padrão de inibição do efluxo de rodamina 6G
do tipo dose dependente, no qual a maior inibição observada, de 56,76 ± 15,08 % , foi alcançada
com a concentração máxima testada. (Figura 12). Utilizamos o mesmo controle previamente
citado, excluindo assim a hipótese de autofluorescencia da mistura AO/AU (dado não mostrado).

77
Ácido oleanólico/Ácido ursólico (µM)
Pdr - Pdr + 10 50 100 200 FK506
Acú
mul
o de
Rod
amin
a 6G
(% re
lativ
a ao
con
trole
)
0
20
40
60
80
100
120
Fig. 12: Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida
pelo tratamento da cepa AD124567 com AO/AU. A avaliação quantitativa foi realizada como
descrito em Material e Métodos, sendo utilizadas concentrações crescentes de AO/AU. Para este
experimento utilizamos como controles positivos de acúmulo de rodamina 6G as cepas
AD1234567 (representada como Pdr -); para controle positivo de inibição utilizamos o fármaco
FK506 a 20 µM e para controle positivo de extrusão, a cepa AD124567 (representada como Pdr
+). Os resultados refletem a média ± erro padrão de três experimentos. * p < 0,001
*
* *

78
5.3 Avaliação da toxicidade do AU frente às cepas AD1234567 e AD124567 de S. cerevisiae
Com o objetivo de avaliarmos o potencial citotóxico do AU frente às cepas hiperesistente
e hipersensível de S. cerevisae utilizamos concentrações crescentes deste composto, variando de
10 a 200 µM, com tempo de incubação de 48 horas, utilizando o mesmo protocolo descrito para
AO e AO/AU.
De acordo com o resultado apresentado no gráfico da figura 13 podemos observar uma
inibição de 31,2 ± 9,88 % e 32,9 ± 1,9 % do crescimento celular para as cepas hiperesistente e
cepa hipersensível, respectivamente, quando submetidas ao tratamento com a maior
concentração de AU (200 µM).

79
Ácido ursólico (µM)
0 50 100 150 200
Cre
scim
ento
cel
ular
(% re
lativ
a ao
con
trole
)
0
20
40
60
80
100
120
Fig. 13: Avaliação da toxicidade do AU frente às cepas AD124567 e AD1234567 de S.
cerevisiae. Para avaliar toxicidade do AU frente as cepas AD124567 (●) e AD1234567 (○) de S.
cerevisiae foram incubadas com concentrações crescentes de AU como descrito em Material e
Métodos. Como controle utilizamos o crescimento das cepas na ausência do composto. Os
resultados refletem a média de três experimentos ± erro padrão.

80
5.4 Avaliação da capacidade do AU em inibir o efluxo de rodamina 6G realizado
pela Pdr5p
Avaliações da capacidade deste composto em inibir o efluxo de rodamina 6G das células
hiperesistentes de S. cerevisiae foram realizadas por citometria de fluxo.
Para este teste, assim como feito para AO e AO/AU, utilizamos concentrações crescentes
de AU variando de 10 a 200 µM (Figura 14). Neste experimento podemos constatar inibições
significativas a partir de 50 µM de AU (71,37 ± 10,48 %), quando comparamos com o acúmulo
observado para a cepa hipersensível, resultado comparável ao obtido para o tratamento com FK
506 a 20 µM, o qual foi capaz de inibir 77,94 ± 8,44 %. É possível observar um efeito inibitório
do tipo dose dependente, que parece atingir seu potencial máximo de inibição do efluxo de
rodamina 6G com 125 µM de AU, onde alcançamos uma inibição de 91,2± 3,68 %. Neste caso
também foi realizado controle de autofluorescencia do AU, assim como já descrito para AO e
AO/AU (dado não mostrado).

81
Ácido ursólico (µM)
Pdr - Pdr + 10 25 50 100 125 15 200 FK506
Acú
mul
o de
Rod
amin
a 6G
(% re
lativ
a)
0
20
40
60
80
100
0
Fig. 14: Avaliação quantitativa da inibição do efluxo de rodamina 6G promovida
pelo tratamento da cepa AD124567 com AU. A avaliação quantitativa foi realizada como
descrito em Material e Métodos. Concentrações crescentes de AU foram utilizadas. Para este
experimento foi utilizado como controle positivo de acúmulo de rodamina 6G a cepa
AD1234567 (representada como Pdr -); para controle positivo de inibição utilizamos o fármaco
* *
* * *
* *

82
FK506 a 20 µM e para controle positivo de extrusão, a cepa AD124567 (representada como Pdr
+). Resultados refletem a média ± erro padrão de três experimentos. * p < 0,001
5.5 Caracterização da resistência apresentada pelo isolado clínico de C. albicans
5.5.1 Avaliação de resistência a antifúngicos de uso clínico
Estudos visando caracterizar a resistência ao fluconazol apresentado pelo isolado clínico
Pri de C. albicans foram realizados. Inicialmente foi avaliada a menor concentração inibitória
(MIC80) para dois azóis de uso clínico, itraconazol e fluconazol, para o isolado Pri e para a cepa
padrão ATCC24433 (Tabela 2).
Para avaliação do MIC80 utilizamos o protocolo M27-A2, padronizado pelo CLSI. Para
realização deste protocolo utilizamos o itraconazol em concentrações que variavam de 0,1 a 1
µg/ml e o fluconazol em concentrações de 2 a 512 µg/ml. O crescimento celular foi avaliado por
medida de turbidez a 600 nm, sendo considerado como MIC80 a concentração do fármaco na qual
observamos uma redução de no mínimo 80% do crescimento celular.
Para o isolado Pri foi obtido um MIC80 de 256 µg/ml para fluconazol e 0,1 µg/ml para
itraconazol. Para a cepa padrão ATCC24433 observamos um MIC80 de < 2 µg/ml para
fluconazol e 1,6 µg/ml para itraconazol.

83
Concentração Mínima Inibitória (MIC)* Cepa
Fluconazol (µg/ml) Itraconazol (µg/ml)
C. albicans Pri 256 0,1
C. albicans ATCC 24433 < 2 1,6
Tabela II. Avaliação da concentração mínima inibitória (MIC) de antifúngicos de
uso clínico para o isolado Pri e cepa ACTCC24433. Resultados obtidos de acordo com a
metodologia descrita previamente em Material e Métodos, seguindo as normas descritas pelo
CLSI. * Os valores apresentados equivalem ao MIC80 após 48 horas de incubação a 37oC.

84
5.5.2 Avaliação da capacidade do isolado clínico de C. albicans em reter
fluoróforos substratos de bombas ABC e MFS
Dando continuidade a caracterização da resistência do isolado Pri, com relação aos
transportadores relacionados ao processo de MDR, foi avaliada a capacidade desta cepa em
extruir os compostos fluorescentes, rodamina 6G e vermelho do nilo. Para esta avaliação foi
utilizada a abordagem de miscroscopia óptica de fluorescência.
A cepa Pri e ATCC24433 foram submetidas à incubação com rodamina 6G ou vermelho
do nilo, sendo posteriormente observados em microscopia óptica de fluorescência. Nos
resultados mostrados na figura 15 é possível observar a capacidade de ambas as cepas em
acumular rodamina 6G, porém o acúmulo de vermelho do nilo é visualizado apenas para a cepa
padrão ATCC24433. A fluorescência observada no interior das células do isolado clínico Pri,
tratadas com vermelho do nilo, se mostrou tênue, quando comparada à fluorescência acumulada
pela cepa ATCC24433.
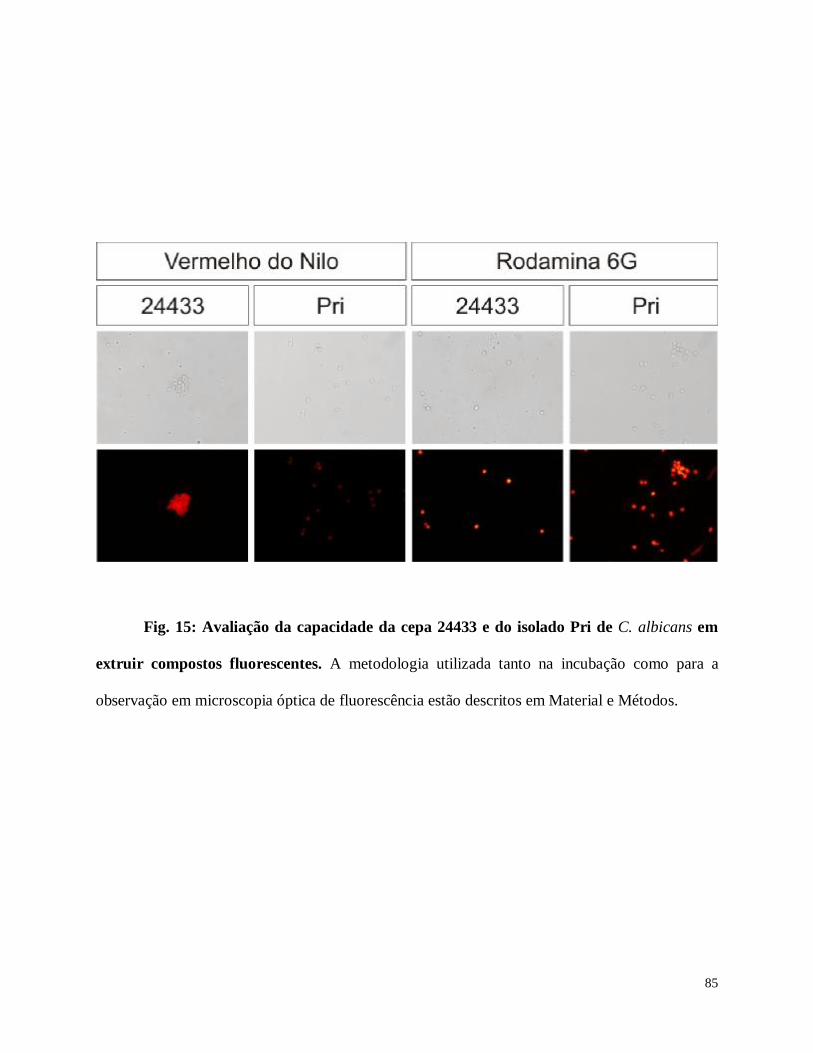
85
Fig. 15: Avaliação da capacidade da cepa 24433 e do isolado Pri de C. albicans em
extruir compostos fluorescentes. A metodologia utilizada tanto na incubação como para a
observação em microscopia óptica de fluorescência estão descritos em Material e Métodos.

86
5.6 Avaliação da toxicidade do AU frente ao isolado clínico de C. albicans
Com o objetivo de avaliar a toxicidade do AU para as cepas de C. albicans foi utilizada a
cepa ATCC24433 e o isolado Pri, aplicando a metodologia descrita pelo protocolo M27-A2 do
CLSI, no qual foram usadas concentrações de AU que variavam de 8 a 256 µM.
No gráfico observado na figura 16 é possível notar que o AU se mostra não tóxico para o
isolado Pri, inibindo apenas 1,12% ± 4,0 do crescimento celular na maior concentração utilizada.
Com relação a cepa ATCC, foi possível observar uma inibição de 43,0% ± 5,7 do crescimento
celular quando utilizada a mesma concentração do AU. Com esses dados podemos observar,
portanto, um MIC80 para o AU de 512 µM para ambas as cepas.

87
Ácido Ursólico (µM)
0 50 100 150 200 250
Cre
scim
ento
cel
ular
(%
rela
tiva
ao c
ontro
le)
0
20
40
60
80
100
120
Fig. 16: Avaliação da toxicidade do AU frente à cepa 24433 e ao isolado clínico Pri de
C. albicans. Para avaliar a toxicidade do AU para a cepa ATCC24433 (○) e isolado Pri (l)
foram incubadas com concentrações crescentes de AU como descrito em Material e Métodos.
Como controle foi utilizado o crescimento das cepas na ausência do composto. Os dados
apresentados refletem a média de três experimentos ± erro padrão.

88
5.7 Avaliação de interação entre AU e fluconazol quando usados em combinação
contra o isolado clínico de C. albicans
Para avaliar a interação do AU com o antifúngico fluconazol utilizamos a metodologia de
“checkerboard” descrita em Materiais e Métodos. Neste ensaio, diferentes combinações de
concentrações de cada um dos compostos foi utilizada, visando obter uma concentração de AU
que seja capaz de sensibilizar a cepa Pri ao tratamento com fluconazol, revertendo assim, o seu
fenótipo de resistência ao fluconazol.
Para este ensaio o AU foi utilizado em concentrações que variavam de 2 a 32 µM e o
fluconazol em concentrações de 2 a 128 µg/ml. Os resultados obtidos indicam que se utilizando
o AU na concentração de 4 µM é possível reduzir o MIC80 do fluconazol de 256 µg/ml para 4
µg/ml (Figura 17).
Com estes resultados foi possível calcular o tipo de interação que estas duas moléculas
possuem, sendo para isto calculado o FICI (Índice de Inibição Fracionária). Na Tabela III são
apresentados os valores de FIC para cada umas das moléculas. Para o fluconazol obtivemos um
FIC de 0,0937 ± 0,078 e para o AU um FIC de 0,0104 ± 0,0026, obtendo-se, com isso, um valor
de FICI de 0,1041 ± 0,0807, indicativo de atividade sinérgica entre os dois compostos.

89
Fig. 17: Avaliação da capacidade de quimiosensibilização do AU em associação ao
fluconazol. O isolado Pri foi incubado na ausência e na presença de 4 µM de AU combinado
com concentrações crescentes de fluconazol como mostrado na figura. Como controle de
crescimento (barra CTR) foi utilizado a mesma cepa na ausência de ambos os compostos. Os
resultados refletem a média de três experimentos ± erro padrão.

90
Cepa FIC AU FIC fluconazol FICI
Isolado Pri 0,0104 ± 0,0026 0,0937 ± 0,078 0,1041 ± 0,0807
Tabela III. Avaliação da interação entre fluconazol e AU apresentada durante o
tratamento do isolado Pri de C. albicans. Os dados mostrados na tabela foram obtidos a partir
do “checkerboard”, utilizando a equação: FICI = FIC do AU + FIC do fluconazol, onde FIC do
AU equivale ao MIC80 do AU combinado com fluconazol dividido pelo MIC80 do AU sozinho,
enquanto o FIC de fluconazol equivale ao MIC80 do fluconazol combinado com AU dividido
pelo MIC80 do fluconazol sozinho. Valores de FICI ≤ 0,5 indicam atividade sinérgica; > 0,5 ≤ 1,
indicam atividade aditiva; > 1 e ≤ 2 indicam atividade indiferente e > 2 indicam atividade
antagônica.

91
8. DISCUSSÃO
Em estudos anteriores, utilizando a partição em diclorometano, obtida a partir do extrato
etanólico bruto de folhas de Lippia lacunosa, foi demonstrado que esta partição era capaz de
inibir a atividade catalítica da enzima Pdr5p de S. cerevisiae e, seus compostos inicialmente
isolados, um flavonóide e um triterpeno, eram capazes de inibir o efluxo de rodamina 6G
(GARCIA-GOMES, 2007). Análises cromatográficas dessa partição em diclorometano
permitiram o isolamento de dois compostos majoritários. Com o auxílio da Ressonância
Mangnética Nuclear de H1 e C13, os terpenos isolados foram identificados como ácido oleanólico
(AO) e ácido ursólico (AU). Destes, o AO foi obtido como composto isolado, em purificações
iniciais, enquanto outra amostra, obtida mais tardiamente, continha AO/AU na proporção de 6:4,
sendo de difícil separação.
Uma vez que não existem dados na literatura que mostrem a atividade de terpenos em
fungos apresentando o processo de MDR, nossa discussão será mais fortemente desenvolvida
com base nos resultados encontrados por nosso grupo.
Em estudos iniciais, avaliamos a toxicidade desses isolados frente às cepas hipersensível e
hiperesistente de S. cerevisiae, confirmando dados da literatura que apontam a baixa toxicidade
destes terpenos para leveduras (HORIUCHI et al., 2007). Nesses experimentos podemos
observar uma maior inibição do crescimento da cepa hipersensível quando tratada com AO, em
comparação com a inibição de crescimento da cepa hiperesistente. Neste caso obtivemos 36,5%
de inibição do crescimento da cepa hipersensível e 9,98% para cepa hiperesistente, mostrando
que o AO é tóxico apenas para a cepa hipersensível, quando utilizado na maior concentração
testada. Estes dados analisados em conjunto sugerem que o AO poderia ser um substrato para o

92
transportador, sendo transportado para o meio extracelular, permitindo assim, que uma cepa que
possua superexpressão de Pdr5p, seja capaz de suportar maiores concentrações de AO em seu
meio de cultura.
Para a mistura AO/AU podemos observar que a mesma não foi capaz de inibir
significativamente o crescimento das cepas hiperesistente e hipersensível, obtendo inibições de
4,87% e 9,98%, respectivamente.
Com o objetivo de analisar a capacidade do AO e do AO/AU em inibir o efluxo do
substrato fluorescente rodamina 6G, proporcionado pela Pdr5p, inicialmente realizamos uma
microscopia óptica de fluorescência, na qual as células hiperesistentes foram tratadas com 100
µM de cada um desses compostos, utilizando como controles as cepas hipersensível e selvagem,
bem como o inibidor clássico FK506 a 20 µM. Este inibidor, pertencente a classe das lactonas, já
foi mostrado como um eficiente inibidor do efluxo de rodamina 6G proporcionado pela Pdr5p,
atuando de modo competitivo para alcançar esta inibição (HENDRYCH et al., 2009). As
imagens obtidas nos permitiram constatar que os compostos apresentavam efeitos distintos com
relação a inibição do transporte mediado pela Pdr5p, sendo, visualmente, observada uma maior
capacidade de inibição mediada pela mistura AO/AU, comparável, visualmente, a inibição
proporcionada pelo FK506. Já com relação ao AO, foi observada uma fraca inibição onde poucas
células apresentavam uma leve fluorescência interna.
Com o objetivo de quantificar a inibição observada por microscopia, seguimos para uma
abordagem em citometria de fluxo. Para ambos os compostos utilizamos concentrações de 10,
25, 50, 100 e 200 µM. Os resultados obtidos para o tratamento com AO nos permitiram
comprovar que este composto era realmente incapaz de promover tal inibição, necessitando de
altas concentrações para se obter apenas 16,86% de inibição do efluxo do fluoróforo. Este

93
resultado parace ser comum para transportadores ABC, tendo em vista que estudos de Braga e
colaboradores (2007) mostraram que esse mesmo composto também não era capaz de inibir o
efluxo de rodamina 123 de uma linhagem de célula de mamífero que superexpressava o
transportador P-gp, um transportador ABC que guarda homologia não só estrutural, mas também
de substratos, com a Pdr5p (KOLACZKOWSKI et al., 1996). Esse resultado pode ser
complementar ao descrito previamente com relação ao crescimento celular (Figura 8 e 9), já que
levantamos a hipótese de que o AO pode ser transportado pela Pdr5p. Neste caso o AO seria
transportado para o meio extracelular sem interferir no transporte de rodamina, provavelmente
por não interagir com o mesmo sítio de ligação de rodamina, o que poderia indicar sítios
distintos para estes compostos. Estes resultados não colaboraria com a idéia de uma inibição do
tipo competitiva, na qual o composto testado competiria pelo sítio de ligação ao fármaco, não
permitindo que esse, mesmo sendo um substrato, fosse capaz de impedir o efluxo de outra
molécula, como foi demonstrado para os derivados de ácido gálico na Pdr5p (RANGEL et al.,
2010).
Os mesmos estudos realizados com a mistura AO/AU mostraram que esta apresentava
uma capacidade bem maior de inibir o efluxo do fluoróforo rodamina 6G, conseguindo inibir
56,76 % quando utilizamos a maior concentração da mistura, mesmo tendo em sua composição
aproximadamente 60% de AO. Com este resultado, duas hipóteses foram levantadas: na primeira
hipótese o AO e o AU poderiam atuar de forma sinérgica. Este tipo de associação é muito
comum em extratos de plantas, em que temos a presença de vários compostos juntos
desenvolvendo uma ação biológica, mas pouco analisada devido a dificuldades de se combinar
concentrações e compostos em variedades tamanha que se torne possível fazer um estudo de
sinergismo. Em nossa segunda hipótese, o composto realmente eficaz em promover a inibição do

94
transporte promovido pela Pdr5p seria o AU mesmo se apresentando em menor quantidade na
mistura (40%).
Uma maneira simples de responder a questão levantada seria repetir os testes, dessa vez
utilizando-se apenas o AU. Como a purificação do AU não foi possível a partir da nossa mistura
AO/AU, tivemos como alternativa a opção de utilizar um AU comercial (Sigma©) com grau de
pureza superior a 98%. Com as análises de efluxo de substrato fluorescente, foi possível observar
que o AU comercial se mostrou mais eficaz em inibir o transporte promovido pela Pdr5p quando
comparado não só com o AO, mas também quando comparado com a mistura, alcançando uma
forte inibição a 50 µM (71,38%) comparável a obtida com 20 µM de FK506 (77,94%),
alcançando um máximo de inibição na concentração de 125 µM (91,2%). Estes dados nos
permitiram concluir que nossa segunda hipótese era a que se encaixava melhor para explicar a
inibição obtida com a mistura AO/AU. Neste caso podemos confirmar a capacidade do AU em
inibir o transporte de rodamina 6G pela Pdr5p, já tendo sido mostrada a capacidade deste
composto em inibir o efluxo de substratos fluorescentes de células que superexpressam P-gp
(NABEKURA et al., 2009).
Analisando os resultados obtidos até o momento, e resultados já presentes na literatura
com relação a atividade diferenciada entre AO e AU (HORIUCHI et al., 2007; BRAGA et al.,
2007; NABEKURA et al., 2009), podemos inferir que o posicionamento de um grupamento
metila é capaz de promover interações distintas das moléculas testadas com transportadores do
tipo ABC. O grupamento metila na posição I, como ocorre para o AU (Figura 6) permite uma
interação com a Pdr5p que impede o transporte de rodamina 6G, o que não ocorre quando o
mesmo grupamento se encontra na posição II, caso do AO (Figura 6).

95
Analisando conjuntamente os resultados de toxicidade e inibição de efluxo, podemos
sugerir que, tendo em vista que o AU não interfere negativamente no crescimento das cepas de S.
cereviseae e ao mesmo tempo é capaz de inibir o efluxo do substrato fluoróforo, este composto
parece inibir diretamente o transportador. Esta inibiçãom pode ocorrer, provavelmente, devido a
uma interação com o mesmo sítio de ligação de rodamina 6G ou em algum sítio próximo, que
poderia inclusive ser o NBD, inibindo ou não a atividade ATPásica deste transportador. Em
ambos os casos, poderia ocorrer um impedimento alostérico, não possibilitando o efluxo do
fluoróforo.
De acordo com os resultados de viabilidade, podemos inferir que o AU não é um substrato
para a bomba, até mesmo devido as altas concentrações necessárias para se observar inibições
significativas do crescimento celular, sendo assim, provavelmente ele não atuaria como um
inibidor do tipo competitivo. Esses resultados corroboram a idéia de que neste caso, o AU deve
se ligar a algum sítio próximo ao da rodamina, seja nos TMS ou nos NBDs, e de certa forma
dificultar o acesso da rodamina 6G ao seu sítio de ligação, inibindo, assim, o seu efluxo.
Após a obtenção desses resultados, aumentou nosso interesse em avaliar a atuação deste
composto em um isolado clínico de C. albicans, que apresentava o fenótipo de resistência a
fluconazol, obtida de um paciente HIV+ a partir de um infecção de orofaringe, denominado Pri
(BRAGA-SILVA et al., 2007). Com este isolado também foi possível analisar o processo de
resistência com o objetivo de descobrir se nesta cepa poderia estar ocorrendo uma
superexpressão de uma bomba de efluxo que conferiria o fenótipo MDR.
Na tentativa de analisar esse fenótipo foram utilizadas duas abordagens: uso demoléculas
que fossem substratos e a utilização de fluoróforos que também seriam substratos.
Transportadores do tipo ABC de C. albicans, quando superexpressos, são capazes de conferir

96
resistência à classe dos azóis como um todo (LAMPING et al., 2008), já no caso de
transportadores do tipo MFS, os mesmos conferem resistência a azóis específicos, não sendo
capazes de conferir resistência por exemplo a itraconazol (revisto por CANNON et al., 2009).
Partindo desses dados já descritos na literatura, realizamos avaliações de MIC80 para fluconazol e
itraconazol, utilizando como controle uma cepa padrão sensível, denominada ATCC24433.
Neste caso podemos observar que a cepa Pri apresentava alta resistência para fluconazol,
apresentando um MIC80 de 256 µg/ml, valor bem acima dos pontos de referência propostos pelo
CLSI, no qual para uma cepa ser resistente a fluconazol seu MIC80 deve ser maior ou igual a 64
µg/ml, enquanto para uma cepa ser considerada sensível o MIC80 deve ser menor ou igual a 8
µg/ml; enquanto isso para itraconazol encontramos um MIC80 de 0,1 µg/ml, indicativo de
sensibilidade para este azol, já que de acordo com o mesmo órgão citado acima, para uma cepa
ser considerada resistente a este azol, seu MIC80 deve ser maior que 1 µg/ml, sendo considerado
sensível quando obtemos MIC80 menor ou igual a 0,125 µg/ml. Estes resultados já apresentavam
um indício de que neste isolado clínico provavelmente não deveria ocorrer a superexpressão de
transportadores do tipo ABC, podendo ocorrer uma expressão aumentada, provavelmente, de
uma bomba do tipo MFS, uma vez que observamos um padrão de resistência apenas a fluconazol
e de sensibilidade a itraconazol.
Em nossa segunda metodologia de caracterização avaliamos o potencial dessa cepa em
extruir compostos fluorescentes, como rodamina 6G e vermelho do nilo. Sabemos que a
rodamina 6G é um substrato clássico para bombas do tipo ABC, porém recentemente em um
estudo de Ivnitski-Steele e colaboradores (2009) foi demonstrado que o vermelho do nilo,
corante de regiões ricas em lipídios, é um substrato não só para os transportadores ABC,
CaCDR1p e CaCDR2p, mas também para o transportador MFS CaMDR1p, fazendo com que

97
ambos os compostos possam ser utilizados para se obter informações sobre o tipo de
transportador presente em uma cepa. Utilizando os dois fluoróforos podemos avaliar a presença
de um transportador ABC, sem definir qual deles estaria presente, tendo em vista que a C.
albicans pode expressar tanto CaCDR1p quanto CaCDR2p (GAUR, CHOUDHURY &
PRASAD, 2005). A observação da presença de um transportador MFS, também pode ser
realizada, neste caso, uma vez que o vermelho do nilo pode ser extruído por qualquer um desses
transportadores e a rodamina, apenas pelos transportadores do tipo ABC, sendo o transportador
MFS CaMDR1p mais relacionado com o fenótipo MDR em C. albicans (FLING et al., 1991;
BEN-YAACOV et al., 1994).
Para este experimento utilizamos os dois fluoróforos já citados e as cepas Pri e
ATCC24433 em uma abordagem de microscopia óptica de fluorescência. Neste caso observamos
que ambas as cepas eram capazes de reter a rodamina 6G em seu interior, não ocorrendo,
portanto, extrusão visualmente significativa deste fluoróforo do interior celular. Entretanto,
quando as cepas foram incubadas com vermelho do nilo, foi possível observar que a cepa Pri
acumulava este fluoróforo em quantidade muito inferior a cepa ATCC24433.
Analisando conjuntamente os dados obtidos com as avaliações do MIC80 e a extrusão de
fluoróforos, foi possível coletar mais indícios de que o isolado clínico Pri apresenta um fenótipo
de MDR, devido provavelmente a superexpressão de uma bomba de efluxo do tipo MFS.
Neste momento, tendo em mãos um composto com atividade contra o transporte mediado
pela Pdr5p de S. cerevisiae, uma bomba do tipo ABC, e uma cepa de C. albicans, superesistente
a fluconazol devido a um fenótipo MDR, provavelmente mediado pela superexpressão de um
transportador do tipo MFS e sabendo que existem similaridades entre estas duas famílias, como
por exemplo: estrutura em alfa hélice dos TMS, sobreposição de substratos e o fato de que

98
alguns transportadores ABC mesmo após deleção de seus NBDs permaneceram capazes de
transportar fármacos, inclusive permitindo que sua unidirecionalidade fosse alterada, fazendo
com que compostos possam também ser captados para o interior celular (revisto por VENTER et
al., 2005); aumentou o nosso interesse em avaliar a capacidade do AU, já identificado neste
trabalho como inibidor do transporte de rodamina 6G promovido pela Pdr5p, em atuar
resensibilizando esta cepa ao tratamento com fluconazol.
Para utilizar o AU como possível substância quimiosensibilizadora se torna necessário
analisar primariamente sua toxicidade frente à cepa de referência ATCC24433 e para o isolado
Pri. Para isso, utilizamos a abordagem padronizada pelo CLSI para obtenção do MIC80 deste
composto. Os resultados apresentados na figura 16 mostram que o AU não é tóxico para o
isolado Pri até a concentração máxima testada (256 µM), alcançando apenas 1,12% de inibição
do crescimento celular, fazendo com que seu MIC80 corresponda a 512 µM. Diferentemente, para
a cepa padrão ATCC24433 observamos que o AU se mostrava tóxico dose dependente,
alcançando inibição máxima (43,0%), a 256 µM, e de acordo com as regras propostas pelo CLSI,
seu MIC80 corresponderia também a 512 µM. Apesar dessa diferença de inibição do
crescimento, estes dados estão em concordância com dados da literatura que mostram que para
uma ação antifúngica são necessárias concentrações superiores a 128 µg/ml tanto para AU
quanto para AO (HORIUCHI et al., 2007) Estes resultados se assemelham aos obtidos para o
AO com a Pdr5p, indicando que provavelmente, para o transportador expresso neste isolado
clínico, o AU seja um substrato, diferente do que ocorre quando este composto é utilizado com o
intuito de inibir o efluxo de rodamina 6G proporcionado pela Pdr5p.
Avaliações da ação conjunta do AU com fluconazol permitiram observar que na presença
de 4 µM deste composto, o MIC80 do fluconazol para o isolado Pri, que antes se apresentava

99
como 256 µg/ml, passa a ser 4 µg/ml (Figura 17), permitindo concluir, portanto, que o AU
resensibiliza esta cepa ao tratamento com fluconazol, revertendo seu fenótipo resistente.
Com estes dados foi possível calcular o índice de inibição fracionária (FICI) que nos
permitiu avaliar o tipo de interação que o AU possui quando usado em conjunto com o
fluconazol, sendo encontrado um valor de 0,1041, condizente com sinergismo, o que indica que
ambas as moléculas atuariam em conjunto para inibir o crescimento celular.
Até o momento, os testes com a finalidade de se obter moléculas quimiosensibilizadoras
têm sido realizados em cepas mutadas e até mesmo com expressão heteróloga de transportadores,
o que por fim acaba sendo um modelo de estudo que não reflete a realidade, até mesmo devido
ao fato de que, no caso de Candida spp., por exemplo, não é possível determinar ao certo o nível
de expressão, para os transportadores relacionados a MDR, que conferem relevância clínica a um
isolado (CANNON et al., 2009). A expressão heteróloga de proteínas de membrana pode levar a
modificações celulares como, por exemplo, alterações na estrutura da membrana plasmática, o
que pode resultar em análises equivocadas de substratos e inibidores (WIRSCHING, 2001).
Além disso, a geração de um “background” que favoreça o estudo de um único transportador,
sem outras interferências, acaba sendo artificial, uma vez que sabemos que o processo de
resistência, em fungos, muitas vezes é multifatorial, dependendo de vários mecanismos. Neste
caso torna-se interessante analisar um isolado clínico, no qual se observa o fenótipo de MDR.
Por existir um limitado número de classes de antifúngicos disponíveis, a escolha
terapêutica para o tratamento de infecções fúngicas sistêmicas, acaba sendo extremamente
limitado (revisto por CANNON et al., 2009), o que pode favorecer o desenvolvimento de
resistência, principalmente nos casos em que se observam altas taxas de reincindiva da infecção,
como para pacientes HIV+, o que poderia contribuir para um maior impacto do fenótipo MDR

100
em fungos. Neste contexto, a utilização de substâncias resensibilizadoras, ou
quimiosensibilizadoras, permitiria que este processo fosse sobreposto. Neste trabalho
descrevemos pela primeira vez um estudo de avaliação da atividade inibitória do AO e do AU
frente ao transportador ABC Pdr5p, de S. cerevisiae. Este modelo de estudo se mostrou bastante
útil para identificar o AU como um novo inibidor do processo de MDR apresentado por uma
cepa de C. albicans que apresentava um fenótipo de hiperesistencia a fluconazol, e ao que tudo
indica, devido a expressão aumentada de um transportador MFS, provavelmente o CaMDR1,
conferindo a este trabalho grande relevância clínica.
9. CONCLUSÕES
• O AO e a mistura AO/AU, isolados de Lippia lacunosa não apresentam
toxicidade significativa para a cepa AD124567. Enquanto a mistura AO/AU se
mostrou tóxica quando utilizada na maior concentração testada. no crescimento da
cepa AD1234567. O AO apresenta uma toxicidade dose-dependente para a cepa
AD1234567, mais evidenciada quando utilizado na maior concentração testada. O
AU de origem comercial apresentou toxicidade dose-dependente para ambas as
cepas, alcançando grau significativo de inibição do crescimento celular quando
utilizado na maior concentração testada.
• Com relação à capacidade dos compostos testados em inibir o efluxo de rodamina
6G, promovido pela Pdr5p super expressa na cepa AD124567, o AO não foi

101
capaz de promover tal inibição. Entretanto, a mistura AO/AU mostrou uma
inibição do tipo dose-dependente do efluxo, apesar de necessitar de altas
concentrações para conseguir um nível de inibição comparável ao inibidor
clássico FK506. O AU se mostrou eficaz em promover esta inibição, necessitando
de apenas 50 µM para alcançar uma inibição comparável a promovida pelo
inibidor clássico. Os resultados obtidos indicam que não ocorre um sinergismo
entre AO, sendo a presença do AU na mistura AO/AU, responsável em promover
o acúmulo de rodamina 6G.
• A abordagem utilizada permitiu caracterizar o isolado clínico Pri de C. albicans
como: hiperesistente a fluconazol, sensível a itraconazol, capaz de reter rodamina
6G em seu interior e de extruir vermelho do nilo. Estas características são
indicativas da possível atuação de um transportador MFS, provavelmente
CaMDR1p
• O AU se mostrou atóxico para o isolado Pri de C. albicans, porém tóxico de
forma dose-dependente para a cepa padrão ATCC2443.
• Quando utilizado em combinação com fluconazol, o AU é capaz de reverter o
fenótipo resistente do isolado Pri, sendo necessário baixas concentrações de AU
para reduzir o MIC80 para 4 µg/ml, caracterizando esta atividade como sinérgica.

102
• A utilização de nosso modelo de S. cerevisiae, com superexpressão do
transportador ABC Pdr5p se mostrou eficaz para seleção de inibidores, não
somente para transportadores ABC mas também para transportadores MFS,
devido a similaridades entre estas duas famílias de bomba de efluxo.

103
ANEXO I
Dados recentes demonstram a utilização da curcumina, um composto de origem natural,
como inibidor de transportadores relacionados ao processo de MDR, mostrando inclusive sua
capacidade de inibir transportadores ABC de fungos (SHARMA et al., 2009)
Paralelamente a esta dissertação, após conseguir a caracterização de um isolado clínico
de Candida albicans resistente a fluconazol, e pelos dados sugerirem a presença de uma bomba
de efluxo do tipo MFS, cresceu nosso interesse em analisar se a inibição inicialmente
visualizada, com a utilização da curcumina, para transportadores ABC de C. albicans expressos
em mutantes de Sacharomyces cerevisiae, poderia ocorrer também no isolado clínico
caracterizado nesta dissertação, possibilitando a sensibilização desta cepa ao tratamento com
fluconazol.
Esta parte do trabalho foi desenvolvida em colaboração com a Prof. Dra Rosangela
Soares e o mestrando Alexandre Curvelo, sendo nossos resultados apresentados neste anexo.

104
1. INTRODUÇÃO
1.1 Curcumina
A Curcumina é um polifenol lipofílico natural, de coloração amarela alaranjado,
produzido pelos rizomas da planta Curcuma longa (Figura 1), espécie amplamente cultivada na
Índia e outras partes do sudoeste asiático, fazendo parte do tempero turmérico e compondo
temperos como curry e mostardas, amplamente utilizados na cozinha indiana (revisto por
JURENKA et al. 2009; BENGMARK, MESA & GIL, 2009; PIANTINO et al., 2009).
O turmérico é composto por um grupo de curcuminóides denominados curcumina,
demetoxi-curcumina e bis-dimetoxi-curcumina, além de alguns óleos voláteis, açúcares,
proteínas e resinas. Este complexo curcuminóide (Figura 1) é conhecido como açafrão da índia e
seu componente principal, o qual confere sua cor alaranjada, é a curcumina, inicialmente
identificada por Lampe e Milobedzka (1913).
Na Índia e em muitos países asiáticos, a curcumina é utilizada por séculos pela medicina
Ayurvédica devido a suas propriedades antinflamatórias (AMMON & WAHL, 1991), sendo
utilizada para o tratamento de artrite, colite e hepatite (BENGMARK, MESA & GIL, 2009). A
segurança da curcumina já foi avaliada em diversos estudos com modelos animais (QURESHI et
al., 1992; SHANKAR et al., 1980), ficando claro que este composto não é tóxico mesmo quando
altas doses são utilizadas em animais de laboratório (BRAVANI et al., 1980; CHAINAINI,
2003).
Diversas atividades biológicas já foram atribuídas à curcumina, dentre elas:
antinflamatória, antioxidante, antimicrobiana e anticâncer, atuando em diversos tipos de

105
linhagens cancerosas (revisto por JURENKA, 2009; PIANTINO et al., 2009), e anti-HIV-1, pela
inibição de suas sequências longas de repetição (LTR) (LI et al., 1993).
Com relação a sua atividade antinflamatória, pesquisas apontam que a curcumina é
altamente pleiotrópica, sendo capaz de interagir com diversas moléculas envolvidas no processo
inflamatório. A modulação deste processo está relacionada com a regulação negativa da
ciclooxigenase-2 (COX-2), lipoxigenase e óxido nítrico sintase induzida (iNOS), pela inibição da
produção de citocinas inflamatória, TNF-α, e diversas interleucinas, bem como inibição da
ativação de NF-κB. Sua atuação em gastrintestinal já foi amplamente relatada, atuando em
dispepsia, infecções por Helicobacter pylori, úlcera péptica, doença de Crohn e colite ulcerativa
(revisto por JURENKA, 2009).

106
(A) (B)
(C)
Fig. 1: Curcuma longa e complexo curcuminóide. (A) Foto da espécie Curcuma longa.
(B) Rizoma da espécie C. longa. (C) Estrutura dos componentes do complexo curcuminóide:
curcumina, demetoxi-curcumina e Bis-demetoxi-curcumina.

107
Muitos efeitos antitumorais já foram descritos para este composto em diversos modelos
pré-clínicos de tumores sólidos de pâncreas, cólon e reto, próstata e de mama (revisto por
PIANTINO et al., 2009). Seu efeito anticâncer pode estar diretamente relacionado ao seu efeito
antinflamatório, uma vez que já é sabido que estados proinflamatórios estão relacionados à
promoção de tumores (BENNETT, 1986; QIAO et al., 1995).
Dentre os efeitos antinflamatórios relacionados com seu potencial anticarcinogênico
podemos citar: inibição de NF- κB e COX-2, uma vez que níveis aumentados de COX-2 estão
associados a diversos tipos de câncer (HUANG, LYSZ & FERRARO, 1991; SURH et al., 2001);
inibição do metabolismo de ácido aracdônico; diminuição de expressão de interleucinas pró-
inflamatórias, que resultam na diminuição do crescimento de linhagens de células cancerosas e
regulação negativa de proteínas como Cinase C, que medeiam o processo inflamatório e
proliferação de célula tumoral (LIU, LIN & LIN, 1993).
Além dessas atividades, já foi descrita também sua atividade antimicrobiana, contra
fungos, tendo melhores atividades contra C. albicans (MARTINS et al., 2009; SHARMA et al.,
2009) e Paracoccidioides brasilensis (MARTINS et al., 2009). No estudo de Martins e
colaboradores (2009) também foi avaliada a capacidade da curcumina em inibir não só a
proliferação da espécie C. albicans, mas também sua capacidade em inibir a adesão de Candida
spp. à células de epitélio bucal, demonstrando um maior potencial antifúngico da curcumina
quando comparado ao antifúngico fluconazol. Sharma e colaboradores (2009), também
demonstraram a capacidade deste composto em impedir o desenvolvimento de hifas na mesma
espécie, diminuindo, portanto, sua capacidade invasora.
Estudos recentes demonstram a atuação modulatória da curcumina em células cancerosas
que apresentam o fenótipo de MDR. Neste caso já foi mostrado que este composto natural é

108
capaz de modular a atividade dos transportadores do tipo ABC de humanos como MRP1 e P-gp
(ANUCHAPREEDA et al., 2002; CHEARWAE et al., 2004; CHEARWAE et al., 2006).
É interessante citar que a curcumina já se mostrou capaz de inibir transportadores de
fungos como a Pdr5p e também CaCDR1p e CaCDR2p, de C. albicans super expressos em
mutantes de S. cerevisiae, neste caso, sendo capaz de atuar em sinergia com antifúngicos de uso
clínico para impedir a proliferação celular dessa espécie (SHARMA et al., 2009). Neste
contexto, tendo em mãos o isolado Pri de C. albicans, decidimos analisar a capacidade da
curcumina em promover a reversão do fenótipo de resistência apresentado por esta cepa.
2. MATERIAL E MÉTODOS
Neste anexo utilizamos as mesmas metodologias descritas no tópico Material e Métodos
desta dissertação.
3. RESULTADOS
3.1 Avaliação da toxicidade da curcumina frente às cepas de C. albicans
Experimentos iniciais visando avaliar o potencial tóxico da curcumina frente a C.
albicans foram realizados utilizando a cepa ATCC24433, o isolado clínico Pri (PORTELA,
2006), resistente a fluconazol e um novo isolado clínico, denominado 88 (gentilmente cedida
pela Prof. Dra Rosangela Soares), altamente sensível a fluconazol, sendo utilizadas
concentrações crescentes de curcumina até um máximo de 50 µM

109
Para todas as cepas testadas podemos observar uma inibição do crescimento do tipo dose-
dependente (Figuras 2, 3, 4). No caso da cepa ATCC24433, podemos observar uma inibição que
parece ser estabilizada quando utilizamos 20 µM de curcumina, não ocorrendo grande diferença
de inibição mesmo quando utilizamos 50 µM, obtendo assim uma inibição máxima de 40,78% e
um IC50 de 14,31 µM (Figura 2); para o isolado clínico Pri observamos uma inibição máxima de
63,26% e um IC50 de 17,65 µM (Figura 3); e por fim, para o isolado clínico 88, observamos uma
inibição máxima de 90,0% e um IC50 de 17,01 µM (Figura 4).

110
Curcumina (µM)
0 10 20 30 40 50
Cre
scim
ento
cel
ular
(% d
o co
ntro
le)
0
20
40
60
80
100
120
Fig. 2: Avaliação da toxicidade da curcumina frente à cepa ATCC24433 de C.
albicans. A cepa foi crescida em meio SD por 48 horas na presença de concentrações crescentes
de curcumina como descrito em Material e Métodos. Como controle utilizamos o crescimento da
cepa na ausência do composto. Para cálculos de IC50 utilizamos a equação Ki = f(x)=
Vo/(1+(x/Ki)) +R
IC50 = 14,31 µM ± 5.39

111
Curcumina (µM)
0 10 20 30 40 50
Cre
scim
ento
cel
ular
(% d
o co
ntro
le)
0
20
40
60
80
100
120
Fig. 3: Avaliação da toxicidade da curcumina frente ao isolado clínico Pri de C.
albicans. A cepa foi crescida em meio SD por 48 horas na presença de concentrações crescentes
de curcumina como descrito em Material e Métodos. Como controle utilizamos o crescimento da
cepa na ausência do composto. Para cálculos de IC50 utilizamos a equação Ki = f(x)=
Vo/(1+(x/Ki)) +R
IC50 = 17,65 µM ± 2,28

112
Curcumina (µM)
0 10 20 30 40 50
Cre
scim
ento
cel
ular
(% d
o co
ntro
le)
0
20
40
60
80
100
120
Fig. 4: Avaliação da toxicidade da curcumina frente ao isolado clínico 88 de C.
albicans. A cepa foi crescida em meio SD por 48 horas na presença de concentrações crescentes
de curcumina como descrito em Material e Métodos. Como controle utilizamos o crescimento da
cepa na ausência do composto. Para cálculos de IC50 utilizamos a equação Ki = f(x)=
Vo/(1+(x/Ki))
IC50 = 17,01 µM ± 8,16

113
3.2 Avaliação de interação entre curcumina e fluconazol quando usados em
combinação contra o isolado clínico de C. albicans
Para avaliar a interação da curcumina com o antifúngico fluconazol utilizamos a
metodologia de “checkerboard” (MUKHERJEE et al., 2005). Neste ensaio diferentes
combinações de concentrações de cada um dos compostos foram utilizadas, visando encontrar
uma concentração de curcumina que fosse capaz de sensibilizar a cepa Pri ao tratamento com
fluconazol, revertendo assim o seu fenótipo de resistência a este antifúngico.
Para este ensaio a curcumina foi utilizada em concentrações que variavam de 4 a 32
µg/ml e o fluconazol em concentrações de 2 a 128 µg/ml. Os resultados obtidos indicam que se
utilizando a curcumina na concentração de 4 µg/ml, o que equivale a 11 µM, é possível reduzir o
MIC80 do fluconazol de 256 µg/ml para 2 µg/ml (Figura 4).
Com estes resultados foi possível calcular o tipo de interação que estas duas moléculas
possuem, usando os valores calculados do FICI cuja metodologia e fórmula foram descritos
previamente. Na tabela I são apresentados os valores de FIC para cada umas das moléculas. Para
o fluconazol obtivemos um FIC de 0,0182 e para o curcumina um FIC de 0,0208, obtendo-se,
com isso, um valor de FICI de 0,0391, o qual é indicativo de atividade sinérgica entre os dois
compostos.

114
Fig. 5: Avaliação da capacidade de quimiosensibilização da curcumina em
associação ao fluconazol. O isolado Pri foi incubado na ausência (barra em preto) e na presença
de apenas 11 µM de curcumina (barras em cinza) combinado com concentrações crescentes de
fluconazol como descrito em Material e Métodos. Como controle de crescimento (barra CTR) foi
utilizado a mesma cepa na ausência de ambos os compostos. Os resultados refletem a média de
três experimentos ± erro padrão.

115
Cepa FIC Curcumina FIC fluconazol FICI
Isolado Pri 0,0208 ± 0,0052 0,0182 ± 0,0069 0,0391 ± 0,008
Tabela I. Avaliação da interação entre fluconazol e curcumina apresentada durante o
tratamento do isolado Pri de C. albicans. Os dados mostrados na tabela foram obtidos a partir
do “checkerboard”, utilizando a equação: FICI = FIC do AU + FIC do fluconazol, onde FIC da
curcumina equivale ao MIC80 da curcumina combinada com fluconazol dividido pelo MIC80 do
AU sozinho, enquanto o FIC de fluconazol equivale ao MIC80 do fluconazol combinado com AU
dividido pelo MIC80 do fluconazol sozinho. Valores de FICI ≤ 0,5 indicam atividade sinérgica;
valores > 0,5 ≤ 1, indicam atividade aditiva; valore > 1 e ≤ 2 indicam atividade indiferente e
valores > 2 indicam atividade antagônica.

116
3.3 Avaliação da capacidade da curcumina em inibir o efluxo de vermelho do nilo do
isolado clínico Pri de C. albicans
Para avaliar a capacidade da curcumina em impedir o efluxo de vermelho do nilo do
isolado Pri, utilizamos a metodologia descrita em materiais e métodos para AU, neste caso,
utilizando a curcumina em uma concentração final de 11 µM (Figura 5). Análises em
microscópio óptico de fluorescência nos permitiram observar que com este tratamento, a cepa Pri
perdia sua capacidade de extruir o fluoróforo vermelho do nilo, sendo possível a observação de
fluorescência no interior celular superior à fluorescência visualizada quando as células não eram
tratadas com curcumina, e similar a fluorescência observada para a cepa controle ATCC24433.
Para este teste também utilizamos como controle o fluoróforo rodamina 6G, substrato apenas
para transportadores ABC.

117
Fig. 6: Avaliação da capacidade da curcumina em inibir o efluxo do fluoróforo
vermelho do nilo do isolado Pri de C. albicans. Para avaliar a capacidade da curcumina em
inibir o efluxo de vermelho do nilo foi realizada a metodologia descrita em Material e Métodos,
sendo utilizado 11 µM de curcumina. Para este experimento utilizamos como controles positivos
de acúmulo de rodamina 6G e vermelho do nilo a cepa ATCC24433, sendo ambos os controles
feitos na ausência d curcumina.

118
4. DISCUSSÃO
Nos resultados apresentados nas figuras 2, 3 e 4, podemos observar que a curcumina
apresenta um alto poder inibitório do crescimento celular das cepas de leveduras utilizadas neste
trabalho. Esta atividade parece variar de cepa para cepa dentro de uma mesma espécie, de C.
albicans, obtendo-se neste estudo uma maior inibição para o crescimento do isolado 88 (Figura
4), onde uma inibição de aproximadamente 100% do crescimento celular foi alcançada pelo
tratamento com 50 µM de curcumina.
É interessante notar que a cepa de referência ATCC24433 conseguiu se manter viável
crescendo em concentrações superiores (Figura 2), quando comparada com o isolado Pri (Figura
3). Nesta dissertação constatamos que esta cepa de referência apresenta resistência a itraconazol,
mas não a fluconazol, sendo capaz de reter rodamina 6G e vermelho do nilo em seu interior, o
que exclui a possibilidade de super expressão de um transportador ABC ou MFS. Entretanto,
algum mecanismo de resistência (provavelmente envolvendo um mecanismo de detoxificação
celular) parece estar presente, permitindo que esta cepa sobreviva em altas concentrações deste
antifúngico e neste caso, também, de curcumina.
Os dados de inibição de crescimento celular estão em concordância com dados da
literatura que indicam a atividade antifúngica da curcumina contra Candida spp. (MARTINS et
al., 2009). Alíquotas retiradas do experimento de “checkerboard”, posteriormente semeadas em
ágar BHI e incubadas por 48 horas a 37 oC, nos permitiram constatar que a curcumina não possui
atividade fungicida e sim fungistática (dado não mostrado).

119
Além disso, neste estudo foi observada uma interação sinérgica entre fluconazol e
curcumina, na qual com a utilização de 11 µM deste composto foi possível reverter o fenótipo de
resistência a fluconazol apresentado pelo isolado clínico Pri (Figura 5 e Tabela I). Entretanto a
curcumina não induz ao desenvolvimento de uma atividade fungicida deste fármaco,
prevalecendo a sua característica de composto fungistático. Com a mesma concentração foi
possível observar que a curcumina é capaz de inibir o efluxo de vermelho do nilo do isolado Pri,
permitindo que este fluoróforo seja retido no interior celular de modo comparável a cepa sensível
ATCC24433 (Figura 6).
Os resultados aqui apresentados em combinação com dados da literatura que mostram
que a curcumina é capaz de inibir transportadores ABC de C. albicans (SHARMA et al., 2009),
reiteram o que já foi constatado nesta dissertação, onde podemos perceber que estudos que
apresentam inibidores de bombas do tipo ABC, podem também revelar potenciais inibidores de
bombas do tipo MFS devido a similaridades compartilhadas entre estas bombas (revisto por
VENTER et al., 2005).

120
5. CONCLUSÕES
• A curcumina apresenta atividade antifúngica contra Candida albicans, porém não
apresentando capacidade fungicida e sim fungistática.
• Usada em combinação com fluconazol, a curcumina em baixas concentrações, é
capaz de reduzir o MIC80 do fluconazol do isolado Pri para 4 µg/ml, apresentando
um FICI de 0,0391, indicativo de atividade sinérgica entre estes dois compostos.
• A curcumina é capaz de promover a inibição do efluxo do fluoróforo vermelho do
nilo do isolado Pri, sendo necessário baixas concentrações desse composto para se
obter, de forma qualitativa, uma restauração da fluorescência intracelular.

121
10. REFERÊNCIAS
ABRAMSON, J., SMIRNOVA, I., KASHO, V., VERNER, G., KABACK, H. R. & IWATA,
S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science v.
3:610–615.
AKINS, R. A. (2005). An update on antifungal targets and mechanisms of resistance in Candida
albicans. Med. Mycol. 43:285–318.
ALLER, S. G., YU, J., WARD, A., WENG, Y., CHITTABOINA, S., ZHUO, R. P., HARRELL,
P. M., TRINH, Y. T., ZHANG, Q. H., URBATSCH, I. L. & CHANG, G. (2009) Structure of P-
glycoprotein reveals a molecular basis for poly-specific drug binding. Science v. 323, 1718-1722
AL-SHAWI, M. K. & OMOTE, H. 2005. The remarkable transport mechanism of P-
glycoprotein; a Multidrug Transporter. J Bioenerg Biomembr v. 37: 489–496.
AL-SHAWI, M. K. & SENIOR, A. E. 1993. Characterization of the adenosine triphosphatase
activity of Chinese hamster P-glycoprotein. J. Biol. Chem. v. 268:4197-4206.
ALTENBERG, G. A. 2003. The Engine of ABC Proteins. News Physiol Sci v. 18:191-195.
ALTENBERG, G. A., VANOYE, C. G.,HORTON J. K. & REUSS, L. (1994) Unidirectional
fluxes of rhodamine 123 in multidrug-resistant cells: evidence against direct drug extrusion from
the plasma membrane. Proc Natl Acad Sci U S A. v. 91: 4654–4657

122
AMBUDKAR, S. V., DEY, S., HRYCYNA, C. A., RAMACHANDRA, M., PASTAN, I. &
GOTTESMAN, M. M. 1999. Biochemical, cellular, and pharmacological aspects of the
multidrug transporter. Annu. Rev. Pharmacol. Toxicol. v. 39:361–98.
AMMON, H. P. & WAHL, M. A. (1991) Pharmacology of Curcuma longa. Planta Med v.57:1-
7
ANUCHAPREEDA, S., LEECHANACHAI, P., SMITH, M. M., AMBUDKAR, S. V. &
LIMTRAKUL, P. N. (2002) Modulation of P-glycoprotein expression and function by curcumin
in multidrug-resistant human KB cells.Biochem Pharmacol. v.64:573-82.
BACCAGLINI, L., ATKINSON, J. C., PATTON, L. L., GLICK, M., FICARRA, G. &
PETERSON, D. E.. 2007. Management of oral lesions in HIV-positive patients. Oral Surg. Oral
Med. Oral Pathol. Oral Radiol. Endod. 103(Suppl. S50):e1–e23
BALAN, I., ALARCO, A. M. & RAYMOND, M. (1997) The Candida albicans CDR3 gene
codes for an opaque-phase ABC transporter. J. Bacteriol. v. 179, 7210–7218.
BALZI, E. & GOFFEAU, A. 1995. Yeast multidrug resistance: the PDR network. J. Bioenerg.
Biomembr. v. 27:71-76.
BALZI, E., AND A. GOFFEAU. 1994. Genetics and biochemistry of yeast multidrug resistance.
Biochim. Biophys. Acta 1187:152–162.

123
BALZI, E.; WANG, M.; LETERNE, S.; CAPIEAUX, E. & GOFFEAU, A. 1994. PDR5, a novel
yeast multidrug resistance gene conferring transporter controlled by the transcription regulator
PDR1. J. Biol. Chem. v. 269: 2206-2214.
BENGMARK, S., MESA, M. D. & GIL, A. (2009) Plant-derived health - the effects of turmeric
and curcuminoids. Nutr Hosp. v.24:273-281
BENNETT, A. (1986) The production of prostanoids in human cancers, and their implications
for tumor progression. Prog Lipid Res v.25:539-542.
BEN-YAACOV, R., KNOLLER, S., CALDWELL, G. A., BECKER, J. M. & KOLTIN, Y.
(1994) Candida albicans Gene Encoding Resistance to Benomyl and Methotrexate Is a
Multidrug Resistance Gene. Antimicrobial Agents and Chemotherapy, : 648-652
BISHOP L., AGBAYANI R., AMBUDKAR S. V., MALONEY P. C. & AMES G. F. L. (1989).
Reconstitution of a bacterial periplasmic permease in proteoliposomes and demonstration of
ATP hydrolysis concomitant with transport, Proc. Natl. Acad. Sci. USA v. 86:6953–6957.
BISSINGER, P. H. & KUCHLER, K. 1994. Molecular cloning and expression of the
Saccharomyces cerevisiae STS1 gene product. A yeast ABC transporter conferring mycotoxin
resistance. J. Biol. Chem. v. 269:4180-4186.

124
BRAGA, F.; AYRES-SARAIVA, D.; GATTASS, C. R. & CAPELLA, M. A. (2007). Oleanolic
acid inhibits the activity of the multidrug resistance protein ABCC1 (MRP1) but not of the
ABCB1 (P-glycoprotein): possible use in cancer chemotherapy. Cancer Lett. v. 248:147-152.
BRAVANI SHANKAR T. N, SHANTHA, N. V., RAMESH, H. P., MURTHY, I. A. &
MURTHY, V. S. (1980) Toxicity studies on Turmeric (Curcuma longa): acute toxicity studies in
rats, guinea pigs & monkeys. Indina J Exp Biol. v. 18: 73-75.
CALIXTO, J. B., BEIRITH, A., FERREIRA, J., SANTOS, A. R. S., CECHINEL-FILHO, V. &
YUNES, R. A. (2000). Naturally ocuuring antinociceptive substances from plants. Phytother.
Res. v. 14:401-418.
CALIXTO, J. B., SANTOS, A. R. S., CECHINEL-FILHO, V & YUNES, R. A. 1998. A review
of the plants of genus Phyllanthus: their chemistry, pharmacology, and therapeutic potencial.
Med. Chem. Res. v. 18:225-258.
CANNON, R. D., LAMPING, E., HOLMES, A. R., NIIMI, K., BARET, P. V., KENIYA, M.
V., TANABE, K., NIIMI, M., GOFFEAU, A. & MONK, B. C. (2009) Efflux-mediated
antifungal drug resistance. Clin Microbiol Rev. v. 22:291-321
CANNON, R. D., LAMPING, E., HOLMES, A. R., NIIMI, TANABE, K., NIIMI & MONK, B.
C. 2007. Candida albicans drug resistance – another way to cope with stress. Microbiology v.
153: 3211-3217.

125
CECHINEL FILHO, V. & YUNES, R. A. 1998. Estratégias para a obtenção de compostos
farmacologicamente ativos a partir de plantas medicinais. Conceitos sobre modificação estrutural
para otimização da atividade. Química nova. v. 21:99-105.
CERNICKA, J. A. , KOZOVSKA, Z. A, HNATOVA, M. A., VALACHOVIC, M. B.,
HAPALA I. B., RIEDL, Z. C, ORGY, H. A.J., & SUBIK, G. C. (2007) Chemosensitisation of
drug-resistant and drug-sensitive yeast cells to antifungals j. International J. Antimicrobial
agents v. 29: 170–178
CHAINANI, W. N. (2003) Safety and anti-inflammatory activity of curcumin: a component of
turmeric (Curcuma longa). J Alternative and Complementary Medicine v.9: 161-168.
CHANG, G. (2003). Multidrug resistance ABC tansporters. FEBS Letters v. 555:102-105.
CHEARWAE, W., SHUKLA, S., LIMTRAKUL, P. & AMBUDKAR, S. V. (2006) Modulation
of the function of the multidrug resistance-linked ATP-binding cassette transporter ABCG2 by
the cancer chemopreventive agent curcumin.Mol Cancer Ther. v.5:1995-2006.
CHEARWAE, W., ANUCHAPREEDA, S., NANDIGAMA, K., AMBUDKAR, S. V.
& LIMTRAKUL, P. (2004) Biochemical mechanism of modulation of human P-glycoprotein
(ABCB1) by curcumin I, II, and III purified from Turmeric powder. Biochem
Pharmacol. v.68:2043-52.

126
CHEN, C. J., CHIN, J. E., UEDA, K., CLARK, D. P., PASTAN, I., GOTTESMAN, M. M. &
RONINSON, I. B. (1986) Internal duplication and homology with bacterial transport proteins in
the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. v. 47:381-9.
CHEN, C.P. AND WANG, H.Y. (1989) The effects of oleanolic acid and dimethyl
dicarbonate biphenyls on acute liver injury. Zhong Cheng Yao Study 10, 11(S)-14(S)133
CHEN, J., LU, G., LIN, J., DAVIDSON, A. L., & QUIOCHO, F. A. (2003) A tweezers-like
motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell v. 12, 651-661
COREA, G., FATTORUSSO, E., LANZOTTI, V., TAGLIALATELA-SCAFATI, O.,
APPENDINO, G., BALLERO, M., SIMON, P. N., DUMONTET, C. & DI PIETRO, A. (2003).
Jatrophane diterpenes as P-glycoprotein inhibitors. First insights of structure-activity
relationships ans discovery of a new, powerful lead. J. Med. Chem. v. 46:3395-3402
COWAN, M. M. (1999). Plant products as antimicrobial agents. Clinical Microbiol. Rev. v.
12:564–582.
COWEN, L. E., & STEINBACH, W. J. (2008). Stress, drugs, and evolution: the role of cellular
signaling in fungal drug resistance. Eukaryot. Cell 7:747– 764.
DAWSON, R. J. P. & LOCHER, K. P. (2006) Structure of a bacterial multidrug ABC
transporter. Nature 443, 180-185

127
DECOTTIGNIES, A. & GOFFEAU, G, A. (1997) Complete inventory of the yeast ABC
proteins. Nat. Genet. v. 15:137-145.
DECOTTIGNIES, A., LAMBERT, L., CATTY, P., DEGAND, H., EPPING, E. A., MOYE-
ROWLEY, W. S., BALZI, E. & GOFFEAU, A. (1995). Identification and characterization of
SNQ2, a new multidrug ATP binding cassette transporter of the yeast plasma membrane. J Biol
Chem. v. 270(30):18150-7.
DECOTTIGNIES, A.; GRANT, A. M.; NICHOLS, J. W.; DE WET, H.; MCINTOSH, D. B. &
GOFFEAU, A. (1998). ATPase and multidrug transport activities of the overexpressed yeast
ABC protein Yor1p. J. Biol. Chem. v. 27312612-22.
DEL SORBO, G.; SCHOONBEEK, H. & DE WAARD, M. A. (2000). Fungal transporters
involved in efflux of natural toxic compounds and fungicides. Fungal Genet. Biol. v. 30:1-15.
DERISI, J., B. VAN DEN HAZEL, P. MARC, E., BALZI, P. BROWN, J. A. C., C., &
GOFFEAU, A.. (2000). Genome microarray analysis of transcriptional activation in multidrug
resistance yeast mutants. FEBS Lett. 470:156–160
DIAZ, C. & SCHROIT, A. J. (1996) Role of translocases in the generation of phosphatidylserine
asymmetry. J. Membr. Biol., v. 151, 1–9.

128
DOGRA, S., KRISHNAMURTHY, S., GUPTA, V., DIXIT, B. L., GUPTA, C. M.,
SANGLARD, D. & PRASAD, R. (1999) Asymmetric distribution of phosphatidylethanolamine
in C. albicans, possible mediation by cdr1, a multidrug transporter belonging to ATP binding
cassette (abc) superfamily. Yeast v. 15: 111–121.
EGNER, R., ROSENTHAL, F. E., SANGLARD, D. & KUCHLER, K. (1998). Genetic
separation of FK506 susceptibility and drug transport in the yeast Pdr5 ATP-binding cassette
multidrug resistance transporter. Mol. Biol. Cell v. 9:523-543.
EGNER, R., BAUER, B. E. & KUCHLER, K. (2000) The transmembrane domain 10 of the
yeast Pdr5p ABC antifungal efflux pump determines both substrate specificity and inhibitor
susceptibility.
FERREIRA-PEREIRA, A.; MARCO, S.; DECOTTIGNIES, A.; NADER, J.; GOFFEAU, A. &
RIGAUD, J. L. (2003). Three-dimensional reconstruction of the Saccharomyces cerevisiae
multidrug resistance protein Pdr5p. J. Biol. Chem. v. 278:11995-11999.
FLING, M. E., KOPF, J., TAMARKIN, A., GORMAN, J. A., SMITH, H. A. & KOLTIN, Y.
(1991). Analysis of a Candida albicans gene that encodes a novel mechanism for resistance to
benomyl and methotrexate. Mol. Gen. Genet. Vv 227:318–329.

129
FRANÇA, S. C. (2003). Abordagens biotecnológicas para a obtenção de substâncias ativas. Em:
Simões, C.M.O. et al. Farmacognosia: da planta ao medicamento. 5a ed. Florianópolis/Porto
Alegre: UFSC/UFRS, p.123.
FRANZ, R., MICHEL, S. AND MORSCHHAUSER, J. (1998) A fourth gene from the Candida
albicans CDR family of ABC transporters. Gene, v. 220, 91–98.
GARCIA-GOMES (2007) Lippia lacunosa e Lippia rotundifolia: possíveis fontes de compostos
capazes de reverter o fenótipo de resistência a múltiplas drogas? (monografia submetida ao
IMPPG-UFRJ). Universidade Federal do Rio de Janeiro/RJ.
GAUR, M., CHOUDHURY, D., & PRASAD, R. (2005). Complete inventory of ABC proteins
in human pathogenic yeast, Candida albicans. J. Mol. Microbiol. Biotechnol. v 9:3–15.
GAUR, M., PURI, N; MONOHARLAL, R.; RAI, V.; MUKHOPADHAYAY, G;
CHOUDHURY, D & GE, F., ZENG, F., LIU, S., GUO, N., YE, H., SONG, Y., FAN, J., WU,
X., WANG, X., DENG, X., JIN, Q. & YU, L. (2010) In vitro synergistic interactions of oleanolic
acid in combination with isoniazid, rifampicin or ethambutol against Mycobacterium
tuberculosis. J Med Microbiol. [Epub ahead of print]
GERBER, S., COMELLAS-BIGLER, M., GOETZ, B. A., & LOCHER, K. P. (2008) Structural
basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science 321, 246-250.

130
GERLACH, J. H., ENDICOTT, J. A., JURANKA, P. F., HENDERSON, G., SARANGI,
F., DEUCHARS, K. L. & LING, V. (1986). Homology between P-glycoprotein and a bacterial
haemolysin transport protein suggests a model for multidrug resistance. Nature v. 324: 485–489.
GOFFEAU, A., BARRELL, B. G., BUSSEY, H., DAVIS, R. W., DUJON, B., FELDMANN, H.,
GALIBERT, F., HOHEISEL, J. D., JACQ, C., JOHNSTON, M., LOUIS, E. J., MEWES, H. W.,
MURAKAMI, Y., PHILIPPSEN, P., TETTELIN, H. & OLIVER, S. G. (1996). Life with 6000
genes. Science. v. 25;274:546, 563-7.
GOLIN, J., AMBDUKAR, S. V., GOTTESMAN, M. M.; HABIB, A.D., SCZEPANSKI, J.,
ZICCARDI, W. & MAY, L. (2003). Studies with novel Pdr5p substrates demonstrate a strong
size dependence for xenobiotic efflux. J. Biol. Chem. v. 273:5963-5969.
GOLIN, J.; KON, Z. N.; MARTELLO, J.; HANSON, L.; SUPERNAVAGE, S.; AMBUDKAR,
S. V. & SAUNA, Z. E. (2007). Complete inhibition of the Pdr5p multidrug efflux pump ATPase
activity by its transport substrate clotrimazole suggests that GTP as well as ATP may be used as
an energy source. Biochemistry. v. 46:13109-19.
GOTTESMAN, M. M. & PASTAN, I. (1993). Biochemistry of multidrug resistance mediated by
the multidrug transporter. Annu Rev Biochem v. 62: 385-427.
GOTTESMAN, M. M., PASTAN, I. & AMBUDKAR, S. V. (1996). P-glycoprotein and MDR.
Curr Opin Genet Dev. v. 6:610-617.

131
GRIFFITH, J. K., BAKER, M. E., ROUCH, D. A., PAGE, M. G. P., SKURRAY, R. A.,
PAULSEN, I. T., CHATER, K. F., BALDWIN, S. A. & HENDERSON, P. J. F. (1992).
Membrane transport proteins: Implications of sequence comparisons. Curr Opin Cell Biol v.4:
684-695.
GROS, P., CROOP, J. & HOUSMAN, D. (1986) Mammalian multidrug resistance gene:
complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell. v.
47:371-80.
GUERRA, M. P. & NODORI, R. O. (2003). Biodiversidade: aspectos biológicos, geográficos,
legais e éticos. Em: Simões, C.M.O. et al. Farmacognosia: da planta ao medicamento. 5. Ed.
Florianópolis/Porto alegre: UFSC/UFRS, p.13–20.
HEDNER, T. & EVERTS, B. (1998). The Early Clinical History of Salicylates in Rheumatology
and Pain. Clinical Rheumatology. v. 17:1725.
HENDERSON, P. J. F. (1990). Proton-linked sugar transport systems in bacteria. J Bioenerg
Biomembr 22525-569.
HENDRYCH, T., KODEDOVÁ, M., SIGLER, K. & GÁŠKOVÁ, D. (2009). Characterization
of the kinetics and mechanisms of inhibition of drugs interacting with the S. cerevisiae multidrug
resistance pumps Pdr5p and Snq2p. Biochim Biophys Acta. 1788:717-23.

132
HIGGINS, C. F. & GOTTESMAN, M. M. (1992). Is the multidrug transporter a flippase?
Trends Biochem. Sci.v. v. 17: 18-21.
HIGGINS, C. F. (2001). ABC transporters: physiology, structure and mechanism – an overview.
Res. Microbiol. v. 152: 205–210.
HIGGINS, C. F., Hiles, I. D., WHALLEY, K. & JAMIESON, D. J. (1985). Nucleotide binding
by membrane components of bacterial periplasmic binding protein-dependent transport systems.
EMBO J. v. 4:1033–1040.
HIRAGA, K., YAMAMOTO, S., FUKUDA, H., HAMANAKA, N. & ODA, K. (2005) Enniatin
has a new function as an inhibitor of Pdr5p, one of the ABC transporters in Saccharomyces
cerevisiae. Biochem Biophys Res Commun. v. 328:1119-25.
HIRAI, T., HEYMANN, J. A., SHI, D., SARKER, R., MALONEY, P. C. & SUBRAMANIAM,
S. (2002) Three-dimensional structure of a bacterial oxalate transporter. Nat Struct Biol v.
9:597–600.
HIRAI, T., HEYMANN, J.A., MALONEY, P.C. & SUBRAMANIAM, S. (2003). Structural
model for 12-helix transporters belonging to the major facilitator superfamily. J. Bacteriol. 185:
1712–1718

133
HOLLENSTEIN, K., FREI, D. C., & LOCHER, K. P. (2007) Structure of an ABC transporter in
complex with its binding protein. Nature 446, 213-216
HOLMES, A.R., LIN, Y.H., NIIMI, K., LAMPING, E., KENIYA, M., NIIMI, M., TANABE,
K., MONK, B.C. & CANNON, R. D. (2008) ABC transporter Cdr1p contributes more than
Cdr2p does to uconazole efux in uconazole-resistant Candida albicans clinical
isolates,Antimicrob. Agents Chemother. v. 52: 3851–3862.
HORIUCHI, K., SHIOTA, S., HATANO, T., YOSHIDA, T., KURODA, T. U. & TSUCHIYA
T. (2007) Antimicrobial activity of oleanolic acid from Salvia ofcinalis and related compounds
on vancomycin-resistant enterococci (VRE) Biol. Pharm. Bull. v. 30: 1147—1149.
HSU, Y.L., KUO, P.L. & LIN, C.C. (2004). Proliferative inhibition, cell-cycle dysregulation,
and induction of apoptosis by ursolic acid in human non-small cell lung cancer A549 cells. Life
Sci., v. 75:2303-2316.
HUA, Y., ZHANG, Z., LI, J., LI, Q., HU, S., LI, J., SUN, M. & CAI, Z. (2009) Oleanolic acid
derivative Dex-OA has potent anti-tumor and anti-metastatic activity on osteosarcoma cells in
vitro and in vivo. Invest New Drugs. Epub ahead of print.
HUANG, M.T., HO, C.T., WANG, Z.Y., FERRARO, T., LOU, Y.R., STAUBER, K., MA, W.,
GEORGIADIS, C., LASKIN, J.D. & CONNEY, A.H. (1994). Inhibition of skin tumorigenesis
by rosemary and its constituents carnosol and ursolic acid. Cancer Res., v. 54:701-708.

134
HUANG, Y., LEMIEUX, M. J., SONG, J., AUER, M. & WANG, D. N. (2003). Structure and
mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science v. 301:616–
20.
Hunan Med. Inst. (1975) Pharmacological studied of hepatoprotective compounds from
Swertia mileensis. Tradi-tional Medicine (Zhong Chao Yao) v. 6: 47-62.
HYDE, S. C., EMSLEY, P., HASTHORN, M. J., MIMMACK, M. M., GILEADI, U, PEARCE,
S. R., GALLAGHER, M. P., GILL, D. R, HUBBARD, R. E. & HIGGINS, C. F. (1990).
Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance
and bacterial transport. Nature v. 346:362-365.
IKEGAWA, T., USHIGOME, F., KOYABU, N., MORIMOTO, S., SHOYAMA, Y., NAITO,
M., TSURUO, T., OHTANI, H. & SAWADA, Y. (2000). Inhibition of p-glycoprotein by orange
juice components, polymethoxyflavones in adriamycin-resistant human myelogenous leukimia
(K562/ADM) cells. Cancer Lett. v. 160: 21-28.
ISHIDA, M., OKUBO, T., KOSHIMIZU, K., DAITO, H., TOKUDA, H., KIN, T.,
YAMAMOTO, T. & YAMAZAKI, N. (1990) Topical preparations containing ursolic acid
and/or oleanolic acid for prevention of skin cancer. Chemical Abstract v. 113, 12173.
IVNITSKI-STEELE, I., HOLMES, A. R., LAMPING, E., MONK, B. C., CANNON, R. D.
& SKLAR, L. A. (2009) Identification of Nile red as a fluorescent substrate of the Candida

135
albicans ATP-binding cassette transporters Cdr1p and Cdr2p and the major facilitator
superfamily transporter Mdr1p. Anal Biochem. v. 394: 87-91.
JENCKS, W. P. (1980). The utilization of binding energy in coupled vectorial processes. Adv.
Enzymol. Relat.Areas Mol. Biol. v. 51:75–106.
JOHNSON, I. S., ARMSTRONG, J. G., GORMAN, M. & BUERNETT, J. P. (1963). The vinca
alkaloids : a new class of oncolytic agents. Cancer Res. v. 23: 1390-1427.
JURENKA, J. S. M. T.(2009) Anti-infammatory properties of curcumin, a major constituent of
Curcuma longa: a review of preclinical and clinical research. Alternative Medicine Review
v.14: 141-153.
KABACK, H.R., SAHIN-TOTH, M., & WEINGLASS, A.B. (2001). The kamikaze approach to
membrane transport. Nat. Rev. Mol. Cell Biol. v. 2: 610–620.
KARABABA, M., A. T. COSTE, B. ROGNON, J. BILLE, & D. SANGLARD. (2004).
Comparison of gene expression proles of Candida albicans azole-resistant clinical isolates and
laboratory strains exposed to drugs inducing multidrug transporters. Antimicrob. Agents
Chemother. v. 48:3064–3079;
KATZMANN, D. J., HALLSTROM, T. C., VOET, M., WYSOCK, W., GOLIN, J.,
VOLCKERT, G. & MOYE-ROWLEY, W. S. (1995) Expression of an atp-binding cassette

136
transporter-encoding gene (yor1) is required for oligomycin resistance in Saccharomyces
cerevisiae. Mol. Cell. Biol., V. 15, 6875–6883.
KHARE, D., OLDHAM, M. L., ORELLE, C., DAVIDSON, A. L., & CHEN, J. (2009)
Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell 33, 528-
536
KOLACZKOWSKA, A. & GOFFEAU, A. (1999). Regulation of pleiotropic drug resistance in
yeast. Drug Resist. Updates v. 2:403-414.
KOLACZKOWSKI, M., KOLACZKOWSKA, A., LUCZYNSKI, J., WITEK, S. & GOFFEAU,
A. (1998) In vivo characterization of the drug resistance profile of the major abc transporters and
other components of the yeast pleiotropic drug resistance network. Microb. Drug resist., v. 4:
143–158.
KOLACZKOWSKI, M., MICHALAK, K. & MOTOHASHI, N. (2003). Phenothiazines as
potent modulators of yeast multidrug resistance . Int. J. Antimicr. Agents v.22:279-283.
KOLACZKOWSKI, M., VAN DER REST, M., KOLACZKOWSKA, A., SOUMILLION, J. P.,
KONINGS, W. N. & GOFFEAU, A. (1996). Anticancer drug, ionophoric peptides, and steroids
as substrates of the yeast multidrug transporter Pdr5p. J. Biol. Chem. v. 271:31543-31548.

137
KONOPLEVA, M., TSAO, T., ESTROV, Z., LEE, R.M., WANG, R.Y., JACKSON, C.E.,
MCQUEEN, T., MONACO, G., MUNSELL, M., BELMONT, J., KANTARJIAN, H., SPORN,
M.B. & ANDREEFF, M., (2004). The synthetictriterpenoid 2-cyano-3,12-dioxooleana-1 9-dien-
28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous
leukemia. Cancer Research v. 64: 7927–7935.
KUNTZ S., WENZEL, U. & DANIEL, H. (1999). Comparative analysis of the effects of
flavonoids on proliferation, cytotoxicity, and apoptosis in human colon cancer cell lines. Eur. J.
Nutr. v. 38:133-142.
LAGE, H. (2003). ABC-transporters: implications on drug resistance from microorganisms to
human cancers. Int. J. Antimicrob. Agents v. 22:188-199.
LAMPE, V. & MILOBEDZKA J. (1913) Sem título disponível. Ber Dtsch Chem Ges v.46:2235,
citado por JURENKA, 2009.
LAMPING, E., MONK, B. C., NIIMI, K., HOLMES, A. R., TSAO, S., TANABE, K., NIIMI,
M., UEHARA, Y., CANNON, R. D., LAW, C. J., MALONEY, P. C. & DA-NENGWANG, D.
(2008). Ins and Outs of Major Facilitator Superfamily Antiporters. Annu. Rev. Microbiol.
62:289-305.
LAW, C. J., MALONEY, P. C. & WANG, D. N. (2008) Ins and outs of major facilitator
superfamily antiporters. Annu Rev Microbiol v. 62: 289-305.

138
LI, C. J., ZHANG, L. J., DEZUBE, B. J., CRUMPACKER, C. S. & PARDEE, A. B. (1993)
Three inhibitors of type 1 human immunodeficiency virus long terminal repeat-directed gene
expression and virus replication. Proc. Natl. Acad. Sci. v.90: 1839-184.
LIMA, E. O. (2001). Plantas e suas propriedades antimicrobianas: uma breve análise histórica.
Em: Simões, C.M.O. et al. Farmacognosia: da planta ao medicamento. 5a ed. Florianópolis/Porto
Alegre: UFSC/UFRS.
LINTON, K. J. (2007). Structure and function of ABC transporters. Phisiology v. 22: 122-130
LIU, J. (1995) Pharmacology of oleanolic acid and ursolic acid. Journal of
Ethnopharmacology. v. 49: 57-68
LIU, J. (2005) Oleanolic acid and ursolic acid: Research perspectives. Journal of
Ethnopharmacology v. 100:92–94
LIU, J. Y., LIN, S. J. & LIN, J. K. (1993) Inhibitory efects of curcumin on protein kinase C
activity induced by 12-O-tetradecanoyl-phorbol-13-acetate in NIH 3T3 cells. Carcinogenesis
v.14:857-861

139
LOCHER, K. P., LEE, A. T. & REES, D. C. (2002) The E. coli BtuCD structure: a framework
for ABC transporter architecture and mechanism. Science v. 296, 1091-1098.
LU, G., WESTBROOKS, J. M., DAVIDSON, A. L. & CHEN, J. (2005) Proc. Natl. Acad. Sci.
U. S. A. v. 102, 17969-17974.
LYONS, C. N. & WHITE, T. C. (2000) Transcriptional analyses of antifungal drug resistance in
Candida albicans. Antimicrob. Agents chemother., v. 44, 2296–2303.
MAESAKI, S., MARICHAL, P., HOSSAIN, M. A., SANGLARD, D., VANDEN BOSSCHE,
H. & KOHNO, S. (1998). Synergic effects of tacrolimus and azole antifungal agents against
azole-resistant Candida albicans strains. J. Antimicrob. Chemother. v. 42:747-753.
MAHATO, S. B., SARKAR, S. K. & PODDAR, G. 1988) Triterpenoid saponins.
Phytochemistty v. 27, 3037-3067.
MAIDEN, M. C. J., DAVIS, E. O., BALDWIN, S. A., MOORE, D. C. M. & HENDERSON, P.
J. F. (1987) Mammalian and bacterial sugar-transport proteins are homologous. Nature v.
325:641–43

140
MANU, K.A. & KUTTAN, G., (2008). Ursolic acid induces apoptosis by activating p53 and
caspase-3 gene expressions and suppressing NF-kappaB mediated activation of Bcl-2 in B16F-
10 melanoma cells. Int Immunopharmacol. v. 8:974-81.
MARALDI, N.M., ZINI, N., SANTI, S., SCOTLANDI, K., SERRA, M. & BALDINI, N. (1999)
P-glycoprotein subcellular localization and cell morphotype in MDR1 gene-transfected human
osteosarcoma cells. Biol. Cell v.91: 17–28.
MARCHETTI, O.; MOREILLON, P.; GLAUSER, M. P.; BILLE, J. & SANGLARD, D. (2000).
Potent synergism of the combination of fluconazole and cyclosporine in Candida albicans.
Antimicrobial Agents and Chemotherapy v.44: 2373-2381.
MARGER, M. D., & SAIER, J. R. (1993). A major superfamily of transmembrane facilitators
that catalyse uniport, symport and antiport. Trends Biochem. Sci. 18:13–20.
MARTINS, C. V. B., DA SILVA, D. L., NERES, A. T. M., MAGALHÃES, T. F. F.,
WATANABE, G. A., MODOLO, L. SABINO, V., DEFÁTIMA, A. A. & DE RESENDE, M. A.
(2009) Curcumin as a promising antifungal of clinical interest. J Antimicrobial Chemotherapy v.
63, 337–339.
MAVOR, A. L.; THEWES, S. & HUBE, B. (2005). Systemic fungal infections caused by
Candida species: epidemiology, infection process and virulence attributes. Curr Drug Targets v.
6: 863–874.

141
MCGRATH, J. P. & VERSHAVSKY, A. (1989) The yeast ste6 gene encodes a homologue of
the mammalian multidrug resistance p-glycoprotein. Nature v. 340, 400–404.
MICHAELIS, S. & BERKOWER, C. (1995) Sequence comparison of yeast atp-binding cassette
proteins. Cold spring harbor symp. Quant. Biol. 291–307.
MIMMACK, M. L., GALLAGHER, M. P., HYDE, S. C., PEARCE, S. R., BOOTH, I. R. &
HIGGINS, C. F. (1989). Energy-coupling to periplasmic binding protein-dependent transport
systems: stoichiometry of ATP hydrolysis during transport, Proc. Natl. Acad. Sci. USA v.
86:8257–8261.
MOLINARI, M., CIANFRIGLIA, S., MESCHINI, A., CALCABRINI, G. & ARANCIA (1994)
P-glycoprotein expression in the Golgi apparatus of multidrug-resistant cells. Int. J. Cancer v. 59:
789–795.
MONTANARI, C. A. & BOLZANI, V. S. 2001. Planejamento racional de fármacos baseado em
produtos naturais. Quim. Nova. v.24:105-111.
MOODY, J.E., MILLEN, L., BINNS, D., HUNT, J.F. & THOMAS, P.J. (2002) Cooperative,
ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-
binding transporters. J. Biol. Chem. v. 277, 21111–21114.

142
MOUSSATOVA, A., KANDT, C., O'MARA, M. L., & TIELEMAN, D. P. (2008) ATP-binding
cassette transporters in Escherichia coli. Biochim. Biophys. Acta. .v 1778, 1757-1771
NABEKURA, T., YAMAKI, T., HIROI, T., UENO, K. & KITAGAWA, S. (2009) Inhibition of
anticancer drug efflux transporter P-glycoprotein by rosemary phytochemicals. Pharmacol
Res. Epub ahead of print
NAKAMURA, K., M. NIIMI, K. NIIMI, A. R. HOLMES, J. E. YATES, A. DECOTTIGNIES,
B. C. MONK, A. GOFFEAU, & R. D. CANNON. (2001). Functional expression of Candida
albicans drug efux pump Cdr1p in a Saccharomyces cerevisiae strain decient in membrane
transporters. Antimicrob. Agents Chemother. v. 45:3366–3374
NEYFAKH, A. A. (2002) Mystery of the drug transporters: the answer can be simple. Mol.
Microbiol. v. 44, 1123–1130.
NIERO, R.; MALHEIROS, A.; BITTENCOURT, C. M. S.; BIAVATTI, M. W.; LEITE, S. N. &
CECHINEL-FILHO, V. (2003) Aspectos químicos e biológicos de plantas medicinais e
considerações sobre fitoterápicos. Em. BRESOLIN, T. M. B.; CECHINEL-FILHO, V. Ciências
Farmacêuticas: Contribuição ao desenvolvimento de novos fármacos e medicamentos. UNIVALI
(ed.), p. 239.
OLDHAM, M. L., DAVIDSON, A. L., & CHEN, J. (2008) Structural insights into ABC
transporter mechanism. Curr. Opin. Struct. Biol. v. 18, 726–733.

143
OLDHAM, M. L., KHARE, D., QUIOCHO, F. A., DAVIDSON, A. L., & CHEN, J. (2007)
Crystal structure of a catalytic intermediate of the maltose transporter. Nature v. 450, 515-521.
OVESNA, Z., VACHALKOVA, A., HORVATHOVA, K. & TOTHOVA, D. (2004).
Pentacyclic triterpenoic acids: new chemoprotective compounds. Minireview Neoplasma v. 51,
327–333
PAO, S. S., PAULSEN, I. T. & SAIER, M. H. (1998). Major facilitator superfamily. Microbiol.
Mol. Biol. Rev. v. 62:1–34.
PATHAK, A.K., BHUTANI, M., NAIR, A.S., AHN, K.S., CHAKRABORTY, A., KADARA,
H., GUHA, S., SETHI, G. & AGGARWAL, B.B. (2007). Ursolic acid inhibits STAT3 activation
pathway leading to suppression of proliferation and chemosensitization of human multiple
myeloma cells. Mol. Cancer Res. v. 5: 943-955.
PATLAK C. 1957. Contributions to the theory of active transport. II. The gate type noncarrier
mechanism and generalizations concerning tracer ow, efciency, andmeasurement of energy
expenditure. Bull. Math. Biophys. v. 19:209–35.
PAULSEN, I. T. & SKURRAY, R. A. (1994). The POT family of transport proteins. Trends
Biochem. Sci. v. 19:404.

144
PEREA S.; LOPEZ-RIBOT J.L.; KIRKPATRICK W.R.; MCATEE R.K.; SANTILLAN R.A.;
MARTINEZ M.; CALABRESE D.; SANGLARD D. & PATTERSON T. F. (2001) Prevalence
of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains
displaying high-level uconazole resistance isolated from human immunodeciency virus-
infected patients, Antimicrob. Agents Chemother. v. 45: 2676–2684.
PERFECT, J. R. & CASADEVALL A. (2006) Fungal molecular pathogenesis: what can it do
and why do we need it? In Molecular principles of fungal pathogenesis. Heitman J, Filler SG,
Edwards JE Jr, Mitchell AP, eds, pp 3–11. ASM Press, Washington DC.
PIANTINO, C. B.; SALVADORI, F. A.; AYRES, P. P.; KATO, R. B.; SROUGI, V.; LEITE, K.
R. & SROUGI, M. (2009) An Evaluation of the Anti-neoplastic Activity of Curcumin in Prostate
Cancer Cell Lines. Internatioal Braz J Urol . v. 35: 354-361.
PORTELA, M. B (2006) Estudo de proteínas funcionais de Candida spp. isoladas de cavidade
bucal de crianças infectadas pelo vírus da Imunodeficiência humana. Dissertação (Mestrado em
Ciências). Universidade Federal do Rio de Janeiro/RJ.
PRASAD R.; WORGIFOSSE, P. D.; GOFFEAU, A. & BALZI, E. (1995) Molecular cloning and
characterisation of a novel gene of C.albicans, CDR1,conferring multiple resistance to drugs and
antifungals. Curr. Genet. v. 27:320–329.

145
PRASAD, R. (2005). Complete inventory of ABC proteins in human pathogenic yeast, Candida
albicans. J. Mol. Microbiol. Biotechnol. .v 9:3–15.
PRASAD, R. & PANWAR, S. 2004. Physiological functions of multidrug transporters in yeast.
Cur.Sci. v. 86: 62-73.
PRASAD, R.; PANWAR, S. L. & SMRITI, 2002. Drug resistance in yeast – an emerging
scenario. In Advances in Microbial Physiology (ed. Poole, R. K.), Academic Press v. 46: 156–
201. Press, Washington, DC, USA.
PRICE, K. R.; JOHNSON, L. T.; &FENWICK, G. R. (1987) The chemistry and biological
significance of saponins in foods and feedingstuffs. CRC Critical Review Food Science
and Nutrition v. 26, 27-135.
PUTMAN, M.; VAN VEEN, H. W. & KONINGS, W. N. (2000). Molecular properties of
bacterial multidrug transporters. Microbiol. Mol. Rev. v. 64:672-693.
QIAO, L. ; KOZONI, V. ; TSIOULIAS, G. J., et al. (1995) Selected eicosanoids increase the
proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim
Biophys Acta v.1258:215-223.
QU, R.Y. (1981) Oleanolic acid in treating virus hepatitis. Guangzhou Med. v. 3, 41-43.

146
RANGEL, L. P.; ABREU, L. F.; ANDRADE, A. R.; LEITÃO, S. G.; LEITÃO, G. G. &
FERREIRA-PEREIRA, A. (2008). Effect of different extracts from the Brazilian Atlantic Forest
on the Pdr5p ATPase activity. Rev. Bras. Farmacogn. v.18:30-36.
RANGEL, L. P.; FRITZEN, M.; YUNES, R. A.; LEAL, P. C.; CRECZYNSKI-PASA, T. B. &
FERREIRA-PEREIRA, A. (2010) Inhibitory effects of gallic acid ester derivatives on
Saccharomyces cerevisiae multidrug resistance protein Pdr5p. Accepted Article Online: Dec 24
2009 6:10AM DOI: 10.1111/j.1567-1364.2009.00603.x
RAVIV, Y.; POLLARD, H. B.; BRUGGEMANN, E. P.; PASTAN, I. & GOTTESMAN, M. M.
(1990) Photosensitized labeling of a functional multidrug transporter in living drug-resistant
tumor cells. J. Biol. Chem. 265, 3975-3980.
RIORDAN, J. R. & LING, V. (1979). Purification of P-glycoprotein from plasma membrane
vesicles of Chinese hamster ovary cell mutants with reduced colchicine permeability. J Biol
Chem v. 254:12701–12705.
ROBERT, M., MATTHEW, N. Fisiologia 3ª Ed. Rio de Janeiro, Editora Guanabara Koogan,
1996.
ROGERS, B.; A. DECOTTIGNIES, M.; KOLACZKOWSKI, E.; CARVAJAL, E.; BALZI &
GOFFEAU, A. (2001). The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J.
Mol. Microbiol. Biotechnol. 3:207–214.

147
ROGERS, P. D. & BARKER, K. S. (2003). Genome-wide expression prole analysis reveals
coordinately regulated genes associated with stepwise acquisition of azole resistance in Candida
albicans clinical isolates. Antimicrob. Agents Chemother. v. 47:1220–1227.
RUTHERFORD, A. V. & WILLINGHAM, M. C. (1993) Ultrastructural localization of
daunomycin in multidrug-resistant cultured cells with modulation of the multidrug transporter, J.
Histochem. Cytochem. v. 41 1573–1577.
SAFA, A. R. (1992). Photoaffinity labeling of P-glycoprotein in multidrug resistant cells. 1992.
Cancer Invest. v. 10:295-305.
SAIER, M. H. 2003. Tracing pathways of transport protein evolution. Mol. Microbiol. v.
48:1145–56.
SAIER, M. H.; BEATTY, J. T.; GOFFEAU, A.; HARLEY, K. T. & HEIJNE,W. H. (1999). The
major facilitator superfamily. J. Mol. Microbiol. Biotechnol. v. 1:257–79.
SANGLARD, D. (1999). The ATP Binding Cassette transporter gene CgCDR1 from Candida
glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrobial
Agents and Chemotherapy. v. 43:2753-2765.

148
SANGLARD, D.; ISCHER, F.; MONOD, M. & BILLE, J. (1997). Cloning of Candida albicans
genes conferring resistance to azole antifungal agents: characterization of CDR2, a new
multidrug ABC transporter gene. Microbiology v. 143:405–416.
SANGLARD, D.; ISCHER, F.; MONOD, M. & BILLE, J. (1996). Susceptibilities of Candida
albicans multidrug transporter mutants to various antifungal agents and other metabolic
inhibitors. Antimicrob. Agents Chemother. v. 40:2300–2305.
SAUNA, Z. E. & AMBUDKAR, S. V. (2000). Evidence for a requirement for ATP hydrolysis at
two distinct steps during single turnover of the catalytic cycle of human P-glycoprotein. Proc.
Natl. Acad. Sci. USA v. 97:2515-2520.
SCHIMMER, A. D.; PEDERSEN, I. M.; KITADA, S. et al (2003). Functional blocks in caspase
activation pathways are common in leukemia and predict patient response to induction
chemotherapy. Cancer Res v. 63:1242– 8.
SCHULTES, R. E. (1978). The kingdom of plants. In Medicines from the Earth. W. A. R.
Thomson (ed.), McGraw-Hill Book Co., New York, p. 208.
SENIOR, A. E.; AL-SHAWI, M.K. & URBATSCH, I. L. (1995) The catalytic cycle of P-
glycoprotein, FEBS Lett. v. 377:285-289.

149
SERVOS, J.; HAASE, E. & BRENDEL, M. (1993) Gene snq2 of Saccharomyces cerevisiae,
which confers resistance to 4-nitroquino- line-n-oxide and other chemicals, encodes a 169 kda
protein homologous to atp-dependent permeases. Mol. Gen. Genet. v. 236, 214–218.
SHAN, J.; XUAN, Y.; ZHENG, S.; DONG, Q. &ZHANG, S. (2009) Ursolic acid inhibits
proliferation and induces apoptosis of HT-29 colon cancer cells by inhibiting the EGFR/MAPK
pathway . J Zhejiang Univ Sci B v. 10: 668-674.
SHANKAR, T. N.; SHANTHA, N. V.; RAMESH, H. P.; MURTHY, I. A. & MURTHY, V. S.
(1980). Toxicity studies on Turmeric: acute toxicity studies in rats, guinea pigs and monkeys.
Indian J Exp. Biol. v. 18:73-75.
SHARMA, M.; MANOHARLAL, R.; PURI, N. & PRASAD, R. (2009) Antifungal curcumin
induces reactive oxygen species and triggers an early 1 apoptosis but prevents hyphae
development by targeting the global 2 repressor TUP1 in Candida albicans. Bioscience Reports
Immediate publication doi:10.1042/BSR20090151
SHEPS, J. A. & LING, V. (2007). Preface: the concept and consequences. Pflugers Arch. v.
453:545-53.
SHISHODIA, S.; MAJUMDAR, S.; BANERJEE, S. & AGGARWAL, B. B. (2003) Ursolic acid
inhibit nuclear factor-nB activation induced by carcinogenic agents through suppression of InBa

150
kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix
metalloproteinase 9, and cyclin D1. Cancer Res v. 63:4375 – 83.
SIARHEYEVA, A.; LOPEZ, J. J. & GLAUBITZ, C. (2006). Localization of Multidrug
Transporter Substrates within Model Membranes. J. Bioche. v. 45:6203-6211.
SIMÕES, C. M. O.; MENTZ, L. A.; SCHENKEL, E. O.; IRGANG, B. E. & STEHMAN, J. R.
(1986). Plantas da medicina popular do Rio Grande do Sul. 5a ed., Porto Alegre: Editora da
UFRS, p.176.
SMITH, P. C.; KARPOWICH, N.; MILLEN, L.; MOODY, J. E.; ROSEN, J.; THOMAS, P. J. &
HUNT, J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation
of a nucleotide sandwich dimer. Mol. Cell 10, 139-149.
SMRITI, K. S.; DIXIT, B. L.; GUPTA, C. M.; MILEWSKI, S. & PRASAD, R. (2002) ABC
transporters Cdr1p, Cdr2p and Cdr3p of a human pathogen Candida albicans are general
phospholipid translocators. Yeast v. 19, 1–16.
SPITZER, C. M. O. S. V. (2003). Óleos Voláteis. Em: Simões, C.M.O. et al. Farmacognosia: da
planta ao medicamento. 5a Ed. Florianópolis/Porto alegre: UFSC/UFRS, p.467–496.
SURH, Y. J.; CHUN, K. S.; CHA, H. H.; HAN, S. S.; KEUM, Y. S.; PARK, K. K. & LEE, S. S.
(2001) Molecular mechanisms underlying chemopreventive activities of anti-inflammatory

151
phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B
activation. Mutat Res. v.1: 480-481.
SZAKIEL, A.; RUSZKOWSKI, D.; GRUDNIAK, A.; KUREK, A.; WOLSKA, K. I.;
DOLIGALSKA, M. & JANISZOWSKA, W. (2008) Antibacterial and antiparasitic activity of
oleanolic acid and its glycosides isolated from marigold (Calendula officinalis). Planta Med. v.
74: 1709-15.
SZCZYPCA, M. S.; WEMMIE, J. A.; MOYE-ROWLEY, W. S. & THIELE, D. J. (1994) A
yeast metal resistance protein similar to human cystic fibrosis transmembrane conductance
regulator (cftr) and multidrug resistance-associated protein. J. Biol. Chem. v. 269: 22853–
22857.
TAGLICHT, D. & MICHAELIS, S. (1998). Saccharomyces cerevisiae ABC proteins and their
relevance to human health and disease. Methds. Enzymol. v. 292:130-162.
TANFORD C. (1983).Mechanism of free energy coupling in active transport. Annu. Rev.
Biochem. v. 52: 379–409.
TANFORD, C. (1982). Simple model for the chemical potential change of a transported ion in
active transport. Proc. Natl. Acad. Sci. USA v. 79: 2882–84.

152
TEODORI, E., DEI, S.; SCAPECCHI, S. & GUALTIERI, F. (2002). The medicinal chemistry of
multidrug resistance (MDR) reversing drugs. Il Farmaco v. 57: 385-415.
TSAO, S.; RAHKHOODAEE, F. & RAYMOND, M. (2009) Relative contributions of the
Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance, Antimicrob.
Agents Chemother. v. 53: 1344–1352.
TUTULAN-CUNITA, A. C.; MIKOSHI, M.; MIZUNUMA, M.; HIRATA, D. & MIYAKAWA,
T. (2005) Mutational analysis of the yeast multidrug resistance ABC transporter Pdr5p with
altered drug specificity. Genes Cells. v. 10: 409-20.
VARMA, M. V. S.; ASHOKRAJ, Y.; DEY, C. S. & PANCHAGNULA, R. (2003). P-
glycoprotein inhibitors and their screening: a perspective from biovailability enhancement.
Pharmacol. Res. v. 48: 347-359.
VERDON, G.; ALBERS, S. V.; VAN OOSTERWIJK, N.; DIJKSTRA, B. W.; DRIESSEN, A.
J. M. & THUNNISSEN, A. (2003) Formation of the productive ATP-Mg2+-bound dimer of
GlcV, an ABC-ATPase from Sulfolobus solfataricus. J. Mol. Biol. v. 334, 255-267
VIDAVAR, G. A. (1966). Inhibition of parallel ux and augmentation of counter ux shown by
transport models not involving a mobile carrier. J. Theor. Biol. v. 10: 301–6;

153
VIEGAS JR, C.; BOLZANI, V. S. & BARREIRO, E. J. (2006). Os Produtos Naturais e a
Química Moderna. Química Nova. v.29: 326-337.
WALKER, G. M (1998) Yeast physiology and Biotechnology. John Wiley and Snos Ltda.
Englad pp 350.
WALKER, J. E.; SARASTE, M.; RUNSWICK, M. J. & GAY, N. J. (1982). Distantly related
sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring
enzymes and a common nucleotide-binding fold, EMBO J. v. 1: 945–951.
WANG, X.; LI, L.; WANG, B. & XIANG, J. (2009) Effects of ursolic acid on the proliferation
and apoptosis of human ovarian cancer cells. J Huazhong Univ Sci Technolog Med Sci. v. 29:
761-4.
WANG, B. & JIANG, Z. H. (1992) Studies on oleanolic acid. Chinese Pharmaceutical
Journal 27, 393-397.J Huazhong Univ Sci Technolog Med Sci. 2009 Dec;29(6):761-4. Epub
2009 Dec 29.
WEMMIE, J. A. & MOYE-ROWLEY, W. S. (1997) Mutational analysis of the Saccharomyces
cerevisiae ATP-binding cassette transporter protein ycf1p. mol. microbiol. v. 25: 683–694.
WEST, I. C. 1997. Ligand conduction and the gated-pore mechanism of transmembrane
transport. Biochim. Biophys. Acta 1331: :213–34.

154
WHITE, T. C. (1997). Increased mRNA levels of ERG16, CDR, and MDR1 correlate with
increases in azole resistance in Candida albicans isolates from a patient infected with human
immunodeciency virus. Antimicrob. Agents Chemother. v. 41:1482–1487.
WHITE, T. C.; MARR, K. A.; & BOWDEN, R. A. (1998). Clinical, cellular, and molecular
factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. v. 11:382–402
WILLIAMSON, P. & SCHLEGEL, R. A. (1997) Back and forth: the regulation and function of
transbilayer phopholipid movement in eukaryotic cells. Mol. Membr. Biol. v. 11, 199–216.
WIRSCHING, S.; MICHEL, S. & MORSCHHAUSER, J. (2000). Targeted gene disruption in
Candida albicans wild-type strains: the role of the MDR1 gene in uconazole resistance of
clinical Candida albicans isolates. Mol. Microbiol. v. 36: 856–865.
WOLDEMICHAEL, G. M.; SINGH, M. P.; MAIESE, W. M. & TIMMERMANN, B. N. (2003)
Constituents of antibacterial extract of Caesalpinia paraguariensis Burk. Z Naturforsch C. v. 58:
70-5.
WOLFGER, H.; MAMNUN, Y. M. & KUCHLER, K. (2001). Fungal ABC proteins: pleiotropic
drug resistance, stress response and cellular detoxification. Res. Microbiol. v. 152: 375-389.
WU, K.H. & LI, Z.S. (1986) Oleanohc acid, a new drug for hepatitis. In: Oleanoli acid.
Guiyang Pharmaceut. Co., pp. l-5.

155
XU, Z., L. X.; ZHANG, J. D.; ZHANG, Y. B.; CAO, Y. Y.; YU, D. J.; WANG, K.; YING, W.
CHEN, S. & JIANG, Y. Y. (2006). cDNA microarray analysis of differential gene expression
and regulation in clinically drug-resistant isolates of Candida albicans from bone marrow
transplanted patients. Int. J. Med. Microbiol. v. 296: 421–434.
YAMAMOTO, S.; HIRAGA, K.; ABIKO, A.; HAMANAKA, N. & ODA, K. (2005). A new
function for isonitrile as an inhibitor of the Pdr5p multidrug ABC transporter in Saccharomyces
cerevisiae.Biochem. Biophys. Res. Commun. v. 330: 622:8.
YAN, S. L. ; HUANG, C. Y. ; WU, S. T. & YIN, M. C. (2009) Oleanolic acid and ursolic acid
induce apoptosis in four human liver cancer cell lines. Toxicol In Vitro. Epub ahead of print.
YIN, Y.; HE, X.; SZEWCZYK P; NGUYEN, T. & CHANG, G. (2006). Structure of the
multidrug transporter emrd from Escherichia coli. Science V. 312: 741–44.
YUNES, R. A.; CALIXTO, J. B. Plantas medicinais sob a ótica da química medicinal moderna.
Argos, Chapecó, p. 479-499.
ZAITSEVA, J.; JENEWEIN, S.; JUMPERTZ, T.; HOLLAND, I. B. & SCHMITT, L. (2005)
H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter
HlyB. Embo J. v. 24: 1901-1910.

156
ZAITSEVA, J.; OSWALD, C.; JUMPERTZ, T.; JENEWEIN, S.; WIEDENMANN, A.;
HOLLAND, I. B. & SCHMITT, L. (2006). A structural analysis of asymmetry required for
catalytic activity of an ABC-ATPase domain dimer. Embo J. v. 25: 3432-3443.
ZHANG, Y. X.; KONG, C. Z.; WANG, L. H.; LI, J. Y.; LIU, X. K.; XU, B.; XU, C. L. & SUN,
Y. H. (2010) Ursolic acid overcomes Bcl-2-mediated resistance to apoptosis in prostate cancer
cells involving activation of JNK-induced Bcl-2 phosphorylation and degradation. J. Cell
Biochem. Epub ahead of print.

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo





























