Embed Size (px)
Citation preview

UNIVERSIDADE DO EXTREMO SUL CATARINENSE – UNESC
UNIDADE ACADÊMICA DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
ANALISE DA COORDENAÇÃO MOTORA EM CAMUNDONGOS
SUBMETIDOS AO TRAUMATISMO CRÂNIO-ENCEFÁLICO E PRÉ-
CONDICIONADOS COM NMDA
TAYANA COSTA
CRICIÚMA, JANEIRO DE 2009

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
UNIVERSIDADE DO EXTREMO SUL CATARINENSE – UNESC
UNIDADE ACADÊMICA DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
TAYANA COSTA
ANALISE DA COORDENAÇÃO MOTORA EM CAMUNDONGOS
SUBMETIDOS AO TRAUMATISMO CRÂNIO-ENCEFÁLICO E PRÉ-
CONDICIONADOS COM NMDA
Dissertação apresentada ao Programa de Pós-graduação em Ciências da Saúde, como requisito parcial à obtenção do título de mestre em Ciências da Saúde.
Orientadora: Profa. Dra. Carina Rodrigues
Boeck
CRICIÚMA, JANEIRO DE 2009

3
Dados Internacionais de Catalogação na Publicação Bibliotecária: Flávia Caroline Cardoso – CRB 14/840 Biblioteca Central Prof. Eurico Back – UNESC
C837a Costa, Tayana. Análise da coordenação motora em camundongos
submetidos ao traumatismo crânio-encefálico e pré-condicionados com NMDA / Tayana Costa; orientadora: Carina Rodrigues Boeck. – Criciúma: Ed. do autor, 2009.
84f. : il. ; 30 cm.
Dissertação (Mestrado) - Universidade do Extremo Sul
Catarinense, Programa de Pós-Graduação em Ciências da Saúde, 2009.
1. Traumatismo cranioencefálico. 2. Lesão cerebral. 3. N-metil-D-aspartato. 4. Atividade motora. I. Título.
CDD. 21ª ed. 616.81

4

5
“É certo que a confiança do homem em suas
próprias forças o torna capaz de realizar coisas
materiais que não se podem fazer quando se
duvida de si mesmo...”
(Allan Kardec)

6
RESUMO
Tem sido descrito que danos celulares posteriores ao trauma seja mediados
pelos neurotransmissores excitatórios, glutamato e aspartato, através da
excessiva ativação dos receptores N-metil-D-aspartato (NMDA). O pré-
condicionamento com NMDA é uma estratégia de neuroproteção que previne o
dano celular induzido por eventos como a isquemia e convulsões. O objetivo deste
estudo é investigar o efeito do pré-condicionamento com NMDA em camundongos
submetidos a um modelo de traumatismo crânio-encefálico (TCE) através da
avaliação da coordenação motora e do dano celular posterior ao trauma. Para tal,
camundongos CF-1 adultos machos foram pré-tratados com solução salina ou
NMDA (75 mg/kg) 24 horas antes do trauma. Os animais foram anestesiados por
inalação com O2/N2O (33%:66%) e em seguida submetidos ao modelo
experimental de TCE pelo impacto de um peso de 50 g no crânio. A destreza dos
animais foi avaliada pelo teste de Impressão das Pegadas (Footprint), em que os
animais têm as patas dianteiras e traseiras pintadas com tinta não-tóxica nas
cores preta e vermelha, respectivamente. Os resultados indicam que o pré-
condicionamento com NMDA previne os prejuízos motores provocados pelo TCE
nos camundongos. Neste modelo de trauma, não foi verificado dano celular, o que
pode indicar que o trauma tenha sido do tipo leve. Em resultados adicionais,
verifica-se que o pré-condicionamento com NMDA, assim como o TCE, levou a
uma diminuição dos níveis das subunidades do tipo NR1 dos receptores NMDA,
mas induziram um aumento das subunidades NR2B, sem alterar os níveis das
subunidades NR2A. Apesar de que, tanto o mecanismo de proteção (pré-

7
condicionamento) quanto o de dano desencadeado pelo trauma alteram
similarmente os níveis dos receptores NMDA, contudo, o papel neuroprotetor
desempenhado pelo pré-condicionamento é conhecido. Assim, é provável que
outras proteínas intracelulares determinem o destino da célula. Tendo em vista o
papel protetor do pré-condicionamento com NMDA, entende-se que este
mecanismo possa desempenhar funções preventivas ao SNC e/ou de manutenção
contra neuropatologias que envolvam o sistema glutamatérgico no TCE.
Palavras chaves: TCE; pré-condicionamento com NMDA; proteção; coordenação
motora.

8
ABSTRACT
Cell damage after trauma can be mediated by excitatory neurotransmitter,
glutamate and aspartate, through excessive activation of receptor N-methyl-D-
aspartate (NMDA). The NMDA preconditioning is a strategy for neuroprotection
that prevents cellular damage induced by ischemia and seizures. The aim was to
investigate the effect of NMDA preconditioning in mice subjected to a traumatic
brain injury (TBI) by evaluating the motor coordination and cellular damage. Mice
CF-1 adult male were pre-treated with saline or NMDA (75mg/kg) 24 hours before
the brain trauma. The animals ware anesthetized by inhalation with O2/N2O
(33%:66%) and then subjected to experimental model of TBI by the impact of a
weight 50 grams in the skull. The coordination of the animals was evaluated by
Footprint Test in which the hind- and forefeet of the mice were coated with black
and red nontoxic paints, respectively. Results indicate that NMDA preconditioning
prevents the coordination deficit damage caused by TBI. In this trauma model,
cellular damage was not observed, suggesting that trauma was mild. Additionally,
both NMDA preconditioning and trauma induce a decrease of level on NMDA
receptor subunits NR1 and an increase on NR2B without affect NR2A levels.
Besides, even trauma and preconditioning operate at the same way with regards
NMDA receptors subunits content, the preconditioning is a well known strategy of
protection. Thus, probably other intracellular proteins contribute to opposite effect
of trauma and preconditioning. In point of view of the protective role of NMDA
preconditioning, is certain that this mechanism can prevent the central nervous

9
system functions and/or maintenance against neuropathology involving the
glutamatergic system in TBI.
Key words: TBI; NMDA preconditioning; protection; motor coordination.

1
LISTA DE ABREVIATURAS
AMPA - ácido alfa-amino-3-hidroxi-5-metil-4-isoxazol propionato
AVC - acidente vascular cerebral
iGluR - receptores ionotrópicos de glutamato
mGluR - receptores metabotrópicos de glutamato
NMDA - N-metil-D-aspartato
SNC - sistema nervoso central
TCE - traumatismo crânio encefálico

SUMÁRIO
1.Introdução................................................................................................... 14
1.1. Conseqüências fisiológicas e estruturais do TCE................................... 15
1.2. Sistema Glutamatérgico.......................................................................... 17
1.2.1. Receptores NMDA............................................................................... 18
1.2.2. Excitotoxicidade Glutamatérgica no TCE............................................. 19
1.3. Neuroproteção......................................................................................... 21
1.3.1. Pré-condicionamento com NMDA........................................................ 23
2. Objetivos.................................................................................................... 26
3. Métodos e Resultados: Capítulo 1.............................................................
3.1. Artigo: NMDA preconditioning improves short and long-term motor
deficits outcome after diffuse traumatic brain injury………………………….
27
28
3.2. Métodos e Resultados Adicionais: Capítulo 2:
3.2.1. Metodologia.................................................................................
54
3.2.2. Aspectos Éticos............................................................................ 54
3.2.3. Animais Experimentais................................................................ 54
3.2.4. Tratamento com NMDA............................................................... 55
3.2.5. Traumatismo Crânio Encefálico................................................... 55
3.2.6. Separação de Proteínas por Eletroforese (SDS-Page)............... 56
3.2.7. Imunodetecção de proteínas (Western Blot)................................ 57
3.2.8. Dosagem de Proteína.................................................................. 57
3.2.9. Análise Estatística........................................................................ 58
3.2.10. Resultados................................................................................ 58

13
4. Discussão................................................................................................... 63
5. Conclusão.................................................................................................. 73
6. Referências................................................................................................ 73

14
1 INTRODUÇÃO
De acordo com o National Institute of Neurological Disorders and Stroke
(NINDS, USA, 2007), o traumatismo crânio encefálico (TCE) é definido como “um
traumatismo repentino que causa dano ao cérebro”. Esta definição inclui ambas as
lesões abertas e fechadas. Segundo dados do Centro de Pesquisas em Educação
e Prevenção da Rede SARAH (CESPE, Brasília, 2008), o TCE é uma importante
causa de morte e de deficiência física e mental, sendo apenas superado pelo
acidente vascular cerebral (AVC). Na Rede SARAH de Hospitais do Aparelho
Locomotor foram atendidos, em 2001, 344 pacientes com TCE e 1419 pacientes
com AVC. A idade média dos pacientes com TCE foi 28,9 anos e 75,6 por cento
foram do sexo masculino. A idade média dos pacientes com AVC foi 60 anos e a
proporção homem/ mulher foi igual a 1:1.
Após o TCE muitos pacientes mostram uma diversidade de seqüelas
persistentes que podem incluir a dor de cabeça, a fadiga, danos na memória,
déficit de concentração e atenção, mudanças de personalidade, depressão,
irritabilidade, distúrbios do sono e disfunção sexual (Lewine et al., 2007), diversos
déficits motores, déficits na velocidade e no equilíbrio (Brink et al., 1970, Schaaf et
al., 1997), processos desencadeados por injúrias que acometem o sistema
nervoso central (SNC) (Xiong et al., 2007). Os prejuízos motores e a deficiência
cognitiva podem persistir durante anos depois do TCE moderado ou severo (Oddy
et al., 1985; Adelson et al., 1997; Lindner et al., 1998; Piot-Grosjean et al., 2001).
Pacientes podem recuperar a capacidade funcional dos danos motores e
cogitivos, porém essa recuperação dependerá de fatores múltiplos como o local da

15
lesão (Shelton & Reding, 2001; Morecraft al et., 2002; Wenzelburger et al., 2005;
Morecraft et al., 2007), o grau de dano infligido (Kuhtz-Buschbeck et al., 2003;
Katz et al., 2004), gênero (Niemeier et al., 2007) e a idade dos pacientes (Katz &
Alexander, 1994; Katz et al., 2004; Livingston et al., 2005).
1.1 Conseqüências fisiológicas e estruturais do TCE
Primeiramente, pacientes que sofreram TCE podem ter contusão da
superfície ou dilaceração, focais ou difusas como hemorragia intracranial, e dano
axonal. Além da destruição neuronal local resultante do insulto primário, o
ferimento mecânico induz secundariamente no cérebro uma cascata progressiva
de eventos relacionados, que geralmente induzem a uma injúria mais severa,
incluindo isquemia, o edema cerebral, ferimento difuso axonal, excitotoxidade,
disfunção mitocondrial, e desregulação da homeostase do cálcio (Yi & Ranzell,
2006; Richardson et al., 2007). Os pacientes que sobrevivem ao trauma
inicialmente são suscetíveis a estes insultos cerebrais secundários que são
iniciados pela liberação de mediadores endógenos neurotóxicos e inflamatórios
pelas células no SNC.
Estudos experimentais em animais mostraram as mudanças estruturais que
acontecem depois do TCE no sistema nervoso central (Nudo, 1999; 2006;
Carmichael, 2003; Butefisch, 2006), que envolvem tipicamente uma combinação
de mecanismos de insultos focais e difusos (Gennarelli & Graham, 2005). O TCE
pode resultar em mudanças neuropatológicas que afetam a densidade do tecido, o
fluxo cerebral de sangue e a integridade da massa branca e cinzenta do cérebro

16
(Mcallister et al., 2001). Outro dano comum no caso do TCE é a contusão cortical
focal e um dano axonal difuso (Gentry et al., 1988). Embora a contusão cortical
focal seja uma causa importante de inabilidade relacionada ao TCE, o dano axonal
difuso contribui mais para esta inabilidade (Levine et al., 2006).
O dano axonal difuso é iniciado pelo rompimento da homeostase iônica e
da permeabilidade alterada do axolema e terminado horas mais tarde pela
desconexão do segmento axonal e consequente morte do neurônio (Povlishock &
Christman, 1995; Maxwell et al., 1997). Inicialmente, o dano axonal difuso é a
causa de confusão mental, da perda da consciência, ou do coma devido ao
rompimento das fibras ascendentes envolvidas na atenção (Gennarelli et al.,
1982).
A resposta neuroinflamatória endógena profunda após o TCE,
fisiopatologicamente ocorre para defender o SNC dos patógenos e para reparar o
tecido lesado, mas esta também é responsável pelo desenvolvimento das lesões
ao cérebro e dos resultados adversos. Dentre os potenciais promotores de dano
secundário decorrente do trauma, o neurotransmissor glutamato tem importante
função neste processo danoso (Rao et al., 2001). Após o TCE a concentração
extracelular de glutamato é aumentada imediatamente após o impacto e perdura
por até 24 horas (Faden et al., 1989; Koizumi et al., 1997), ocasionando
excitotoxidade nas células gliais (Nicholls & Attwell, 1990) e morte de neurônios
(Smith et al., 1997).

17
1.2 Sistema Glutamatérgico
O sistema glutamatérgico envolve uma série de receptores que são
ativados pelo aminoácido glutamato (Cotman et al., 1995). O glutamato é o
principal neurotransmissor excitatório e mais abundante no cérebro (Yi & Hazell,
2006; Bressan & Pilowsky, 2003), participando de muitas sinalizações celulares
excitatórias e de fenômenos plásticos vinculados à aprendizagem e memória
(Fonnum, 1984; Izquierdo, 1989), além de participar do desenvolvimento do SNC,
da diferenciação e morte celular (Mcdonald & Johnston, 1990; Meldrum, 2000).
O glutamato exerce suas funções sinalizadoras sinápticas agindo em
proteínas específicas localizadas na membrana celular neural. Os receptores
glutamatérgicos são classificados de acordo com as suas propriedades
farmacológicas e funcionais, e são divididos em receptores ionotrópicos (iGluR) e
metabotrópicos (mGluR). Os receptores ionotrópicos são canais iônicos que
quando ativados tornam-se permeáveis a cátions e são divididos em: N-metil D-
aspartato (NMDA), ácido alfa-amino-3-hidroxi-5-metil-4-isoxazol propionato (AMPA)
e cainato (Mori & Mishima, 1995). A estimulação de qualquer destes receptores
ionotrópicos leva ao influxo de cálcio (Ca2+) e/ou sódio (Na+) resultando na
despolarização da membrana, ativando assim, indiretamente os canais de cálcio
dependentes de voltagem. Os oito tipos de receptores metabotrópicos são
associados a sistemas transmembrana acoplados a proteínas-G divididos em três
grandes famílias de receptores de acordo com as suas seqüências homólogas e
com a modulação de efetores intracelulares. O Grupo I inclui os receptores mGluR

18
1 e 5, estão associados a ativação da fosfolipase C (PLC); o Grupo II (mGluR 2 e
3) e o Grupo III (mGluR 4, 6, 7 e 8) estão associados negativamente a adenilato
ciclase (Conn & Pin, 1997).
A extensão da estimulação dos receptores glutamatérgicos é determinada
pela concentração do glutamato no espaço extracelular. É de grande importância
para as células que o glutamato seja mantido em baixas concentrações, pois a
excessiva liberação e o conseqüente aumento nos seus níveis extracelulares o
tornam tóxico em decorrência da excessiva estimulação de seus receptores (Lipton
& Rosenberg, 1994). O sistema de transporte de glutamato é a via mais rápida de
remoção do glutamato extracelular, interrompendo a sua ação nos seus receptores
(Rosenberg et al., 1992). Os transportadores de glutamato estão presentes na
membrana celular de neurônios e das células da glia. Os transportadores
astrocitários são considerados mais importantes, pois além de manterem baixa a
concentração extracelular do glutamato, degradam o aminoácido à glutamina no
citoplasma, que será liberada e captada pelo neurônio para regeneração de
glutamato (Schousboe, 1981; Yi & Hazell, 2006)
1.2.1 Receptores NMDA
O receptor NMDA quando ativado torna-se altamente permeável a íons
sódio (Na+) e principalmente cálcio (Ca2+) (Kandel et al., 2001) e seu potencial
excitatório são mais duradouros (Dale & Roberts, 1985), tendo um papel
importante na regulação das sinapses no SNC (Carroll & Zukin, 2002).

19
A excessiva ativação dos receptores NMDA, porém, está envolvida na
epilepsia e em danos cerebrais associados à isquemia/hipoglicemia (Olney, 1994)
e também com o TCE (Clausen & Bullock, 2001). Pois a excessiva ativação dos
canais-receptores NMDA desfaz a homeostase celular dos íons Ca2+, levando a
excitotoxicidade e a lesão no neurônio provocado pelo aumento do influxo do íon,
além de danificar as funções mitocondriais (Clausen & Bullock, 2001) e
subseqüente morte por necrose e/ou apoptose que ocorrem como resultado deste
acúmulo intracelular de Ca2+ (Arundine & Tymianski, 2003). O receptor NMDA é
formado pela composição de quatro de subunidades, formando uma proteína
heteromérica constituída por NR1, NR2 (A-D) e NR3. Diferentes combinações
destas subunidades determinam especificidades na capacidade funcional de cada
receptor, como, por exemplo, afinidade do NR1 pela glicina que é um co-agonista
obrigatório (GAGLIARD, 2000) e do NR2 pelo glutamato e limiar para abertura do
canal iônico (SHULER et al., 2008).
1.2.2 Excitotoxicidade Glutamatergica no TCE
O conceito de excitotoxicidade foi introduzido por Olney (1969) para
descrever a neurotoxicidade associada com a administração exógena de altas
concentrações glutamato, ou de compostos que agem como agonistas em
receptores glutamatérgicos. Alterações bioquímicas foram identificadas através
dos modelos experimentais de trauma; estes incluem mudanças na homeostase
iônica (cálcio, potássio, sódio, magnésio), indução de radicais livres, mudanças
imunes inflamatórias e alterações de sistemas múltiplos do neurotransmissores,

20
inclusive no sistema glutamatérgico (Panter & Faden, 1992; Mcintosh, 1994). A
excitotoxicidade glutamatérgica não é o único fator desencadeante de
neuropatologias, mas tanto pode ser um evento primário, como uma conseqüência
de um evento danoso já ocorrido.
Em várias condições neuropatológicas crônicas ou agudas em que o
glutamato participa, pode haver alterações na sua captação e/ou liberação,
exacerbando a injúria desencadeada por doenças como: a isquemia, a
hipoglicemia, o edema cerebral, a lesão por trauma mecânico, a epilepsia, a
demência de Alzheimer, a enfermidade de Huntington e doenças neurológicas
relacionadas à SIDA (Obrenovitch et al., 2000). O aumento no nível de glutamato
extracelular após o TCE (ou demais doenças citadas acima), causa um aumento
na ativação dos receptores de glutamato, desencadeando assim os eventos
secundários do trauma como a morte neural, aumento do tempo da
despolarização da célula, desequilíbrio iônico, diminuição dos níveis intracelulares
de ATP, aumentos nos níveis intracelulares de Ca2+ o que favorece ao edema
cerebral e pressão intracranial elevada (Yi & Ranzell, 006)
Em um modelo de trauma em ratos verificou-se que a injúria celular pós-
TCE é mediada pelos neurotransmissores excitotóxicos endógenos (glutamato e
aspartato) por ativação dos receptores NMDA (Ikonomidou et al., 1999), pois um
tratamento prévio com antagonistas de receptor NMDA foi capaz de diminuir a
morte celular. O glutamato parece ser destrutivo na fase aguda, imediatamente
após o ferimento traumático ou isquêmico, mas após este período, assume suas
funções fisiológicas normais, que incluem a promoção da sobrevivência neuronal
(Ikonomidou & Turski, 2002).

21
Estudos demonstraram também uma diminuição nos níveis dos
transportadores gliais para glutamato, GLT-1 e GLAST, do córtex cerebral
ipsilateral e contralateral ao trauma, associado ao aumento nos níveis
extracelulares do glutamato observados 48 horas após o trauma (Van Landeghem
et al., 2001). Esta alteração na quantidade de transportadores gliais para
glutamato também é observada após a isquemia cerebral (Rao et al., 2000).
Enquanto o TCE conduz a uma hipoperfusão sangüínea, ao mesmo tempo
o glutamato e os distúrbios iônicos produzem demanda metabólica aumentada no
tecido afetado produzindo o aumento na taxa metabólica cerebral local para a
glicose (Mendelow & Crawford, 1997) que poderia estar relacionado a
excitotoxidade glutamatérgica.
1.3 Neuroproteção
A neuroproteção é um processo que interrompe e/ou previne uma cascata
patológica que ocorre durante um processo de dano tóxico (Mcintosh, 1993;
Gagliard, 2000). Em muitos modelos de excitotoxicidade em que os níveis
extracelulares de glutamato estão elevados, antagonistas dos receptores
glutamatérgicos são utilizados como neuroprotetores. Os antagonistas dos
receptores NMDA, AP-5 (antagonista do tipo competitivo, bloqueia o sítio de
ligação de NMDA ao receptor) e MK-801 (antagonista do tipo não-competitivo,
bloqueia o canal iônico no receptor) (Monaghan et al., 1989) fazem parte do grupo
de antagonistas utilizados para bloquear respostas neurotóxicas desencadeadas
pela ativação dos canais-receptores NMDA, devido a liberação de aminoácidos

22
durante um dano isquêmico ou convulsivo (Olney et al., 1987; Novelli et al., 1988;
Breukel et al., 1998; Sierra-Paredes et al., 2000; Meloni et al., 2002).
A neuroproteção pode ser induzida por baixas doses de NMDA, que ativam
os receptores sinápticos e extrasinápticos de NMDA (Francesc et al., 2006). A
atividade sináptica do receptor de NMDA promove a neuroproteção pela ativação
pró-sobrevida contra os sinais patológicos, incluindo a ativação do fator nuclear
CREB (proteína de ligação ao elemento de resposta ao AMPc) e expressão gênica
(Lee et al., 2005; Papadia et al., 2005).
Alguns estudos com modelos animais de trauma mostraram efeitos
benéficos dos antagonistas do receptor NMDA como por exemplo MK-801,
fenciclidina, D-CPP-ene, Pfizer PC 101-606 (Hamm et al., 1993) e o íon magnésio
(Mg2+) (Heath & Vink, 1999).
Outra substância que age de modo antagônico nos receptores NMDA está
sendo investigado como um neuroprotetor contra os eventos secundários
resultantes do aumento de glutamato pós-TCE (Alves et al., 2003). Em um modelo
animal de TCE foi demonstrado que após o tratamento com topiramato houve uma
recuperação neurológica mais rápida em relação ao desempenho da memória e
motor (O'Dell & Hamm, 1995).
O TCE induzido por percussão em ratos causou uma elevação na
marcação do glutamato e do aspartato extracelular junto ao local do traumatismo.
Este aumento dos aminoácidos excitatórios é relacionado à severidade do dano e
associado a uma redução no estado bioenergético celular e do Mg2+ livre
intracelular, condições essas recuperadas após tratamento com um antagonista

23
não–competitivo (dextrofan) ou competitivo (CPP) do receptor NMDA, atuando
benéficamente pós-TCE (Faden et al, 1989).
Apesar de evidências indicarem que os antagonistas de NMDA possuem
melhores propriedades neuroprotetoras em processos excitotóxicos provocados
por ferimentos decorrentes do trauma no cérebro de ratos jovens quando
comparados a ratos adultos, pouco ainda se sabe sobre os mecanismos celulares
envolvidos no dano celular pelo TCE (Bernert & Turski, 1995).
1.3.1 Pré-condicionamento com NMDA
O TCE também pode induzi a morte apoptótica assim como a morte
necrótica da célula, e ambos os tipos de morte podem ser modulados
farmacologicamente. Estas modulações farmacológicas conduzem à avaliação de
numerosas estratégias para a neuroproteção cerebral, incluindo inibidores de
canais ionicos, corticosteróides, antioxidantes, antagonistas dos receptores do
glutamato, antagonistas dos receptores opióides, assim como vários moduladores
antiinflamatórios. Recentemente foi demostrado que a apoptose foi reduzida
usando um inibidor da caspase-3, levando assim a uma melhora e prevenção dos
déficits neurológica após o TCE (Yakovlev, et al., 1997), fornecendo bases para o
desenvolvimento de estratégias para neuroproteção no TCE. Há outras estratégias
farmacológicas que contribuem para o entendimento dos mecanismos celulares
envolvidos no dano desencadeado pelo TCE. Algumas destas estratégias de
neuroproteção utilizam antagonistas de receptores NMDA contra os danos
causados pelo TCE (Faden et al., 1989; Temple et al., 2001).

24
Outra linha de evidência demonstra que o uso de agonistas em baixíssimas
doses, em que a toxicidade estaria bem abaixo do limiar de dano, pode induzir
tolerância nas células contra danos glutamatérgicos. O NMDA e o glutamato
podem atuar como agentes de pré-condicionamento químicos para melhorar
significativamente o dano isquêmico (Schurr et al., 2001) em fatias de hipocampo,
como em cultura de células (Xu et al., 2002) e in vivo (Boeck et al., 2004). Assim a
administração de doses sub-convulsivas de NMDA via intraperitoneal tem sido
utilizada como modelo de pré-condicionamento químico in vivo frente a diversos
insultos letais posteriores (Ogita et al., 2003; Boeck et al., 2004).
O pré-condicionamento diminui a prevalência de morte celular decorrente
de alguma lesão (Murry et al., 1990). Estudos estratégicos de pré-
condicionamento como isquemia (Chen & Simon, 1997), hipóxia (Pugliese et al.,
2003) e hipotermia (Nisho et al., 2000) resultaram em proteção contra o dano
isquêmico subseqüente.
Vários dados experimentais demonstram que o aumento extracelular de
glutamato aumenta a atividade do receptor NMDA tornando-o um importante
mediador da excitotoxidade após o trauma. As doenças que acometem o SNC
apresentam em comum uma exacerbação na transmissão mediada pelo
neurotransmissor glutamato, que está associado à gênese de quadros patológicos
agudos (TCE, isquemia cerebral e convulsões) e crônico-degenerativos (doenças
de Alzheimer, Huntington, escleroses). O estudo de vias celulares que possam
modular a transmissão glutamatérgica é de fundamental importância para a
compreensão do desenvolvimento de mecanismos fisiológicos e fisiopatológicos
no SNC. Tendo em vista o papel protetor do pré-condicionamento com NMDA,

25
entende-se que este mecanismo possua funções preventivas ao SNC e/ou de
manutenção contra neuropatologias, neste estudo o TCE, que envolvam o sistema
glutamatérgico.

26
2. OBJETIVOS
2.1. Geral:
Verificar a presença de dano celular no cérebro de camundongos submetidos ao
modelo de traumatismo crânio encefálico com ou sem o pré-tratamento com
NMDA, e os efeitos sobre a atividade locomotora.
2.2. Específicos:
2.2.1 Verificar possíveis alterações de dano celular no córtex frontal e cerebelo
dos animais pré-condicionados com NMDA e submetidos ao TCE.
2.2.2 Verificar a coordenação motora em camundongos pré-condicionados ou não
com NMDA e expostos ao traumatismo crânio encefálico.
2.2.3 Avaliar o conteúdo das subunidades NMDA (NR1, NR2a, NR2b) em cerebelo
de camundongos submetidos ao TCE e pré-condicionados com NMDA.

27
CAPITULO 1
3 METODOS E RESULTADOS
3.1 Artigo
Conforme resolução 01/2007 do PPGCS, parte dos resultados está apresentada
na forma de artigo cientifico, submetido para publicação no periódico Journal of
Neurotrauma.

28
NMDA preconditioning improves short and long-term m otor deficits outcome
after diffuse traumatic brain injury
Tayana Costa, Leandra C. Constantino, Bruna M. Pescador, Bruno Herculano,
Carla I. Tasca, Carina R. Boeck
Laboratório de Neurociências, Programa de Pós-Graduação em Ciências da
Saúde, Unidade Acadêmica de Ciências da Saúde, Universidade do Extremo Sul
Catarinense, 88806-000 Criciúma, SC, Brasil;
Corresponding author: Prof. Carina R. Boeck, M.Sc., Ph. D – Laboratório de
Neurociências, PPGCS, Universidade do Extremo Sul Catarinense, 88806-000
Criciúma, SC, Brasil. Phone: #55 48 3431 2759. E- mail: [email protected]

29
Abstract
Traumatic brain injury (TBI) causes impairment of fine motor functions in humans
and non-human mammalians that often persists for months after the injury occurs.
Neuroprotective strategies for prevention of the sequelae of TBI and understanding
the molecular mechanisms and cellular pathways are related to the glutamatergic
system. It has been suggested that cellular damage subsequent to TBI is mediated
by the excitatory neurotransmitters, glutamate, and aspartate, through the
excessive activation of the N-methyl D-aspartate (NMDA) receptors. Thus,
preconditioning with low dose of NMDA was used as a strategy for protection
against locomotor deficits observed following TBI in mice. Male adult mice CF-1
were preconditioned with NMDA (75 mg/kg) 24 hours before the TBI induction.
Under anesthesia with O2/N2O (33%: 66%) inhalation, the animals were submitted
to the experimental model of trauma that occurs by the impact of a 50g weight on
the skull. Sensorimotor gating was evaluated using fore- and hindlimb footprints
and rotarod at 1.5, 6, or 24 hours following TBI. Cellular damage also was
assessed 24 hours after cortical trauma. Mice preconditioned with NMDA were
protected against all motor deficits revealed by footprint tests, but not those
observed in rotarod tasks. Although mice showed motor deficits after TBI, no
cellular damage was observed after TBI. These data corroborate the hypothesis
that glutamatergic excitotoxicity, especially via NMDA receptors, contributes to
severity of trauma. They also point to a putative neuroprotective mechanism
induced by a sub-lethal dose of NMDA to improve motor behavioral deficits after
TBI.

30
Key-words: NMDA preconditioning; neuroprotection; trauma brain injury; motor
behavior.

31
Introduction
The cellular damage following trauma brain injury (TBI) constitutes a
dynamic pathophysiologic process of the brain because of the combination of
primary (unavoidable damage, occurs at the time of injury) with secondary damage
(avoidable damage, occurs at variable times after injury). The severity of
secondary injury level over time depends on the degree of mechanical impact
(Katz et al., 2004; Ucar et al., 2006) which can be classified as mild, moderate or
severe (Fujimoto et al., 2004; Ucar et al., 2006). The cascade of events
subsequent to the acute brain injury includes glutamatergic excitotoxicity (Bernert
and Turski, 1996) leading to impairment of synaptic plasticity, modulation of N-
methyl-D-aspartate (NMDA) receptor activity and contents (Miller et al., 1990;
Schumann et al., 2008), and persistent cognitive-, sensory-, and motor dysfunction
(Fujimoto et al., 2004). Excitotoxicity induced by glutamates involves at least in part
intracellular Ca2+ level elevation through excessive activation of NMDA receptors
(Choi, 1988). NMDA receptor antagonists have been used as neuroprotective
agents in TBI in clinical trials (Willis et al., 2004) and animal models (Bernert and
Turski, 1996; Pohl et al., 1999; Kuo et al., 2007). However, human trials of NMDA
receptor antagonists have no shown positive effect on clinical outcomes or
intolerance side effects, because a persistent blockade of glutamate receptors
impairs trophic effects of glutamate, which are responsible for the reorganization of
neural cells after occurrence of the brain injury (Muir and Lees, 1995).
Despite the well-known neurotoxic mechanisms blocked by NMDA receptor
antagonists, NMDA agonists can evoke a neuroprotective action (Sei et al., 1998;

32
Damschroder-Williams et al., 1995; Jonas et al., 2001). Several studies have
shown that preconditioning evokes protection against brain damage induced by
ischemia (Kitagawa et al., 1991; Pagliaro et al., 2001; Ferguson et al., 2008),
seizures (Sasahira et al., 1995), and closed head injury (Shein et al., 2005). NMDA
preconditioning is a “chemical preconditioning” state evoked by sub-toxic
concentrations of NMDA that induces a tolerance state to a subsequent lethal
event (Rejdak et al., 2001). NMDA preconditioning is protective against injury
induced by kainate (Ogita et al., 2003) and seizures (Boeck et al., 2004;
Vandresen-Filho et al., 2007) in vivo; also by glutamates (Chuang et al., 1992;
Marini et al., 1998; Boeck et al., 2005) or oxygen- and glucose-deprivation (Grabb
et al., 1999) or both stimuli (Lin et al., 2008) in vitro. Cellular damage and long-term
behavior deficits associated with TBI can be activated by an excitotoxic mechanism
triggered by secondary hypoxia (Saatman et al., 2008). NMDA receptors
participate in neuroprotection evoked by hypoxia preconditioning (Tauskela et al.,
2003), but only a few studies have analyzed behavioral parameters after
preconditioning (Boeck et al., 2005; Rybnikova et al., 2005). Thus, the aim of the
present study is investigate NMDA preconditioning as a strategy against
sensorimotor deficits after TBI caused by diffuse trauma in an animal model.
Methodology
Animals: Male CF-1 mice (2-3 months, 30-35 g) were obtained from our breeding
colony (UNESC). The animals were housed six to cage with food and water
available ad libitum and were maintained on a 12-h light/dark cycle (lights on at

33
7:00 a.m.). All experimental procedures involving animals were performed in
accordance with the National Institute of Heath’s Guide for the Care and Use of
Laboratory Animals and the recommendations of the Brazilian Society for
Neuroscience and Behavior (SBNeC) for animal care, designed to minimize
suffering and limit the number of animals used. Animals were used only once and
to avoid circadian variations all experiments were carried out between 8:00 a.m.
and 4:00 p.m. This study was approved by the local ethics committee (Comitê de
Ética em Pesquisa da Universidade do Extremo Sul Catarinense, n° 776/2007).
Treatment with NMDA: Animals were pretreated intraperitoneally (i.p.) with NMDA
with a low nonconvulsant dose (75 mg/kg, 10 mL/kg) or vehicle (saline, 0.9% NaCl,
w/v) 24 hours before cortical trauma injury induction (Ferre et al., 1994). NMDA
was dissolved in saline and pH adjusted to 7.4 with NaOH. Mice were housed in
acrylic boxes (25x25x25 cm) and were observed for 30 min immediately after
NMDA administration for occurrence of behavioral alterations (Boeck et al., 2004).
Diffuse Traumatic Brain Injury: A diffuse TBI was produced using the closed
head weight-drop method previously described (Adelson et al., 1996) with minor
modifications. The trauma apparatus consists of a metal tube, 1 m length and 10
mm inner diameter, attached to a ring stand. The mice, anesthetized by inhalation
with a mixture of O2/N2O (33%:66%) led through a mask, were placed on a loosely
fixed foam bed with their heads slightly elevated to avoid spinal cord damage.
A brass weight of 50 g with concentric grooves on its face fell downward freely
from a cm height of 80. Just after impact, the foam bed with the animal was slid out

34
from under the tube to prevent a second impact to it on recoil. After trauma, the
mice received supporting oxygenation with 100% O2 until they were fully awake
(approximately one minute) and were then brought back to their cages. The extent
of posttraumatic sensorimotor impairment was assessed at defined time intervals
after trauma (t = 1.5h, 6h, and 24h), as described below. At corresponding time-
points mice were euthanized for subsequent analysis of extracted brain
hemispheres.
Sensorimotor Evaluation: Two tests of fore- and hind limb coordination, balance,
and sensorimotor gating are reported here in accordance with previous studies; the
performance of the animals was analyzed 1.5h, 6h, and 24h had lapsed after the
TBI occurred (Bristow, et al., 1996). All animals were trained immediately before
NMDA administration, and each mouse was evaluated individually for one period of
analysis after TBI for two tests: first, the animal was submitted to a footprint test
and later to rotarod test. The animals were divided into groups in accordance with
the treatments that were to be administered (5-9 mice per group per time = total of
93 animals): SAL = saline i.p. + anesthesia (control); NMDA = NMDA i.p. +
anesthesia; TBI = saline i.p. + trauma; NMDA + TBI = NMDA i.p. + trauma.
Footprint: To assess the motor coordination and gaiting (instability of posture,
incoordination, tremor, dysmetria, and impairment of fine control of movements) we
used a footprint test as previously described (Carter et al., 1999). To obtain
footprints, the hind- and forefeet of the mice were coated with black and red
nontoxic paints, respectively. The animals were then allowed to walk along a 40-

35
cm-long, 8-cm-wide runway into an enclosed box with 20-cm-high walls on either
side (Dietrich et al., 2005). All mice had three training runs and a fresh sheet of
white paper was placed on the floor of the runway for each run. The footprint
patterns were analyzed for four-step parameters (all measured in centimeters).
Stride length = the average distance measured of forward movement between
each stride; forelimb width = the average distance measured between left- and
right front footprints; hind limb width = the average distance measured between
left- and right hind footprints. These values were determined by measuring the
perpendicular distance from a given step to a line connecting its opposite
preceding- and succeeding steps. Step alternation = the average distance
measured from left- or right front footprint/hind footprint overlap. When the center
of the hind footprint fell on top of the center of the preceding front footprint, a value
of zero was recorded. When the footprints did not overlap, the distance between
the center points of the footprints was recorded. For each step parameter, three
values were measured from each run, excluding footprints made at the beginning-
and end of the run where the animal was initiating- and finishing movement,
respectively. The mean value of each set of three values was used. Total time and
velocity of movements in three trials of maximum 2 min each were recorded by
blind observer 24h after TBI.
Rotarod: In the rotarod apparatus (Insight Equipments, Ribeirão Preto, Brazil) all
mice were habituated to running (3.7 cm in diameter, 12 rpm) until they could
remain there for up to 60 s without falling for 8 trials with intervals of 10 min. In

36
tests during daytime/periods the mouse was provoked to run in a rotarod for up to
60 s without falling for 3 trials (Dunham and Miya, 1957). The mean latency to fall
off the rotarod and the number of falls was recorded.
Cellular damage evaluation: Immediately after sensorimotor performance, mice
were sacrificed by decapitation; their cerebral cortices and cerebellum were
dissected, rapidly frozen, and stored at −80 °C unt il measurement of the DNA
damage, or were immediately prepared for cellular viability assessment by MTT
reduction assay.
Cellular viability: Cell survival following TBI was measured by dehydrogenase
activities to reduce MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide] (Mosmann, 1983; Liu et al., 1997). The tetrazolium ring of MTT was
cleaved by various dehydrogenase enzymes into viable cells, and then precipitated
as a blue formazan product. The cerebral cortex and cerebellum were cut into 400
µm thick transversal slices with a McIlwain tissue chopper, followed by transfer of
the sections to phosphate-buffered saline supplemented with 0.6% glucose (pH
7.4), and were separated into individual slices. Two slices from each cerebral
structure were incubated with MTT (0.5 mg/mL) in PBS buffer for 20 min at 37 °C.
The medium was aspirated, the precipitated formazan solubilized with dimethyl
sulfoxide, and the viable cells quantified spectrophotometrically at a wavelength of
550 nm. Variability due to differences in slice size was found minimal.

37
DNA fragmentation: DNA fragmentations were evaluated 24 h after TBI induction
in mice with- or without preconditioning treatment. Ten to 20 mg of cerebral cortex
or cerebellum tissue was used for DNA extraction. The tissue was homogenized in
a lysis buffer (GenomicPrep™, GE Healthcare) followed by incubation for 1 h at 65
°C. Proteins and RNA were digested with proteinase K (Sigma, 100 µg/ml) and
RNAse (Sigma, 20 µg/ml), at 55 °C for 12 h and at 37 °C for 1 h, resp ectively.
Proteins and RNA were then precipitated by centrifugation of the samples at
16,000g for 15min at 4 °C. DNA was precipitated for 12 h with 100% isopropanol at
4 °C and 70% ethanol. Centrifuged samples (correspo nding to 40 µg DNA) were
then submitted to a 2% agarose gel electrophoresis with ethidium bromide (100
µg/ml) and visualized in UV light (Yang and Paul, 1997).
Statistical analysis : Results were analyzed with STATISTICA version 7.0
software (StatSoft, Inc., USA). All data are presented as mean values ± SEM
(standard error of the mean), and the parameter of the normality was fulfilled by
Shapiro-Wilk test. Each value reflects the mean of 6 to 9 animals per group for
behavioral parameters or the mean of 3 to 6 animals per group for cellular viability
analysis. In all cases, statistical analyses were analyzed employing two-way
analysis of variance (ANOVA) with NMDA pre-treatment and TBI as the main
factors, followed by the Fisher LSD test for behavioral analysis. The level of
significant difference was accepted at P < 0.05.
Results

38
All parameters were analyzed at 1.5h, 6h, or 24h following TBI, with the
exception of cellular viability assays, which were analyzed only after 24h of TBI.
The brain samples from mice submitted to TBI presented no signs of external
damage to the cerebral cortex 24h after trauma, no hemorrhages in the
periventricular white matter below the cortex, and no apparent distortion of the
inner structures. All mice that suffered trauma survived the procedure. In all groups
of animals, the impact did not induce any DNA damage in the cerebral cortex
(impact area) or in the cerebellum (distant area) (Fig. 1). Besides, pretreatment
with NMDA or TBI did not alter the cellular viability as measured by MTT assay 24
h after trauma (Table 1, P>0.05 in all cases).
Assessment of motor coordination was performed to evaluate motor
damage caused by TBI. All mice were trained immediately before pretreatment
with NMDA for analysis of gait performance in the footprint test and were tested at
1.5h, 6h, or 24h after TBI. Sensorimotor behavioral tests revealed uncoordinated
movements in traumatic mice when compared to control mice (SAL); however,
preconditioning with NMDA prevented the distortion of gait for all parameters of
mice showed deficits. The irregular stride length (SL) and hind limb stride (HLS)
observed in mice at 1.5h after TBI were prevented in preconditioned mice
(F(1,27)=1.662, P<0.001, and F(1,28)=3.323, P<0.05; respectively) (Fig. 2A and C).
Qualitatively, the generated patterns clearly differed showing that TBI mice display
irregularly spaced shorter strides as compared with evenly spaced and accurately
positioned footprints of SAL mice (Fig. 2E). The measure of percentage of
movements in relation of SAL mice demonstrated clearly the significant preventive
effect of preconditioning in the coordination deficit displayed by traumatic mice

39
(F(1,27)=1.797, P<0.001 for SL; F(1,28)=2.468, P<0.05 for HLS) (Fig 5A). The
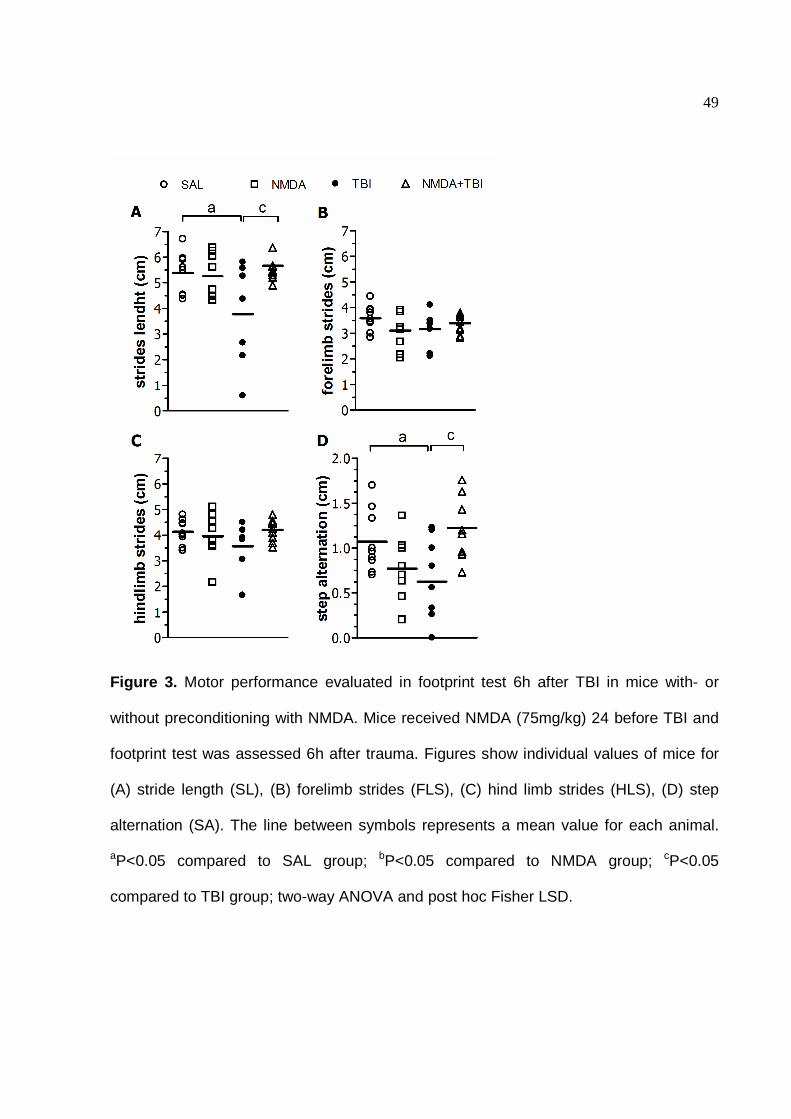
distortion of gait appearing in stride length (SL) defects was observed up to 6h
following traumatic injury to the cortex demonstrated in the mean of individual
measures (F(1,28)=5.698, P<0.05) (Fig. 3A) and in percentage of SAL group (Fig. 5)
with significant amelioration in mice preconditioned with NMDA (F(1,28)=5.986;
P<0.05). In the same period, mice revealed loss of rhythm coordination after TBI in
the form of deficits in step alternation (SA) (Fig. 3C and 5C) that were prevented by
NMDA treatment (F(1,30)=11.643; P<0.001). Long-term sensorimotor coordination
deficits were observed 24h after TBI in mice, who showed defects in forelimb stride
(FLS) (F(1,26)=0.153; P<0.05), hind limb stride (HLS) (F(1,26)=1.173; P<0.05), and
step alternation (SA) (F(1,26)=2.872; P<0.05) (Fig. 4 and 5). Animals preconditioned
with NMDA and submitted to TBI did not display forelimb or hind limb stride
defects. Twenty four hours after TBI all mice spent the same time for run in the
runway and also, the velocity to cover was the same among the groups (Table 2;
P>0.05 in all cases).
The mice were tested on rotarod at 1.5h, 6h, or 24h following traumatic
injury with or without treatment with NMDA (Fig. 6). The TBI mice were not able to
stay on the rotarod as evaluated 1.5h after TBI, independent of treatment with
NMDA when compared to the SAL group (F(3,24)=4.940; P<0.001 for 1.5h) (Fig.
6A). Interestingly, while SAL mice showed an improvement in rotarod performance
after 6h, all traumatic animals entirely failed to show motor learning and falling
more than SAL mice did (F(1,22)=0.113; P<0,05) with a nonsignificant tendency to
stay less time on rotarod (F1,22)=0.084; P=0.057). When 24h had lapsed after TBI,
rotarod performance was found not altered in all groups of animals.

40
Discussion
The present study reports a neuroprotective effect of NMDA preconditioning
against sensorimotor deficits in traumatic injury to the cortex of mice. Some
preliminary conclusions about severity of trauma can be drawn from the lack of
cellular damage and the absence of focal lesions of TBI mice. It is clear that
trauma to the cortex did not induce cellular damage or animal death after lapse of
24h, which could indicate a mild brain trauma, such as previously reported for
diffuse brain injury (Foda and Marmarou, 1994). However, mild trauma motor and
learning deficits, such as those observed in this study, are also observed in
humans (Kashluba et al., 2008) and in animal models (Milman et al., 2005).
Glutamatergic excitotoxicity is involved in several injuries related to the
brain including TBI. NMDA receptor antagonists have been used with significant
success against Alzheimer dementia (Farlow et al., 2008), but against trauma
injury its use is still being discussed (Muir, 2006). NMDA preconditioning is an
experimental strategy to evaluate a mechanism of protection that activates
expressive protective signaling pathways in the cells (Jiang et al., 2003; Soriano et
al., 2006). TBI can induce necrotic- and apoptotic cellular death (Raghupathi, 2004;
Wong et al., 2005) following primary- or secondary damage with participation of
glutamatergic excitotoxicity (Faden et al., 1989), a mechanism of neural death
which can be prevented by NMDA preconditioning (Ogita et al., 2003; Boeck et al.,
2004). Preventive intervention could be an important protective strategy against
unavoidable injury induced by brain surgery (Jadhav et al., 2007), but the present
study also explores a method to contribute to the overall functional improvement

41
over TBI. In ischemia, studies of preconditioning have contributed to knowledge of
the disease and perspectives of treatment (Billinger et al., 1999; Vohra and
Galiñanes, 2006).
The present data showed that the neuroprotection evoked by low activation
of glutamatergic system through NMDA preconditioning was efficient against
sensorimotor deficits displayed by traumatic mice in a model of diffuse trauma.
These data are in accordance with the clinical observations that show motor
deficiencies after moderate TBI (Adelson et al., 1997; Piot-Grosjean et al., 2001).
The TBI in mice evoked a synchronous bilateral contraction of both fore- and hind
limb extensors since the distance measured between left/right and right/left hind
footprints were prevented by NMDA preconditioning. Cerebellar ataxia is
characterized by instability of posture and gait, incoordination, tremor, dysmetria,
muscular hypotonia, and impairment of fine control of movements (Grusser-
Cornehls and Baurle, 2001). The lack of balance and deficits in movements involve
many areas of the brain that may have suffered injury following TBI, such as the
motor association cortex and the cerebellum. This latter observation is particularly
interesting, since the impact on the brain of the animal probably led to functional
cortical- and cerebellar deficits without affecting its viability, which could contribute
to their recovery verified by the rotarod test.
NMDA preconditioning induces pro-survival signaling and provides
protection against cellular damage after brain injuries such as seizures and
ischemia (Boeck et al., 2004; Saleh et al., 2009). Recently, a study showed that
controlled closed head injury (CHI) changes the content of NMDA receptor
subunits in the cortex and hippocampus (Schumann et al., 2008). The authors

42
suggest that the decrease in NR1, NR2A, and NR2B observed in the cortex (local
impact) could be due to neuronal death, since they observed an increased
phosphorylation of NR2B on Y1472, which stabilizes the receptors on the cell
surface (Roche et al., 2001). If our results were due to the same alteration on
NMDA receptors after TBI, probably NMDA preconditioning was preventing it.
However, is important to note that the mechanisms triggered by NMDA
preconditioning involve other intracellular proteins beyond NMDA receptors. In
cultured neurons, NMDA preconditioning has been shown to result in no alteration
in specific [3H]MK-801 binding (Damschroder-Williams et al., 1995), and its
protective effects are blocked in presence of the protein synthesis inhibitor
cyclohexamide (Marini et al., 1992) showing the dependence on new proteins to
the neuroprotective effects of preconditioning.
Altogether, NMDA preconditioning leads to tolerance against functional
sensorimotor deficits induced by TBI in mice. This apart, it is probable that the
trauma subjected was mild because cellular damage was not observed and mice
recovered motor uncoordination when evaluated after 24h in rotarod. NMDA
preconditioning is a chemical mechanism leading to cell resistance that could be
investigated to point to new strategies to protect the brain from trauma injury.
Acknowledgements: This work was supported by funds from the Brazilian
National Council Research (CNPq grants, n° 478249/2 006-3 and FAPESC grants,
n° 12348/2007-0 to CRB) and UNESC.
.

42
References
Adelson PD, Dixon CE, Robichaud P and Kochanek PM. (1997). Motor and cognitive functional deficits following diffuse traumatic brain injury in the immature rat. J Neurotrauma. 14:99-108.
Adelson PD, Robichaud P, Hamilton RL and Kochanek PM. (1996). A model of diffuse traumatic brain injury in the immature rat. J Neurosurg. 85:877-884.
Bernert H and Turski L. (1996). Traumatic brain damage prevented by the non-N-methyl-D-aspartate antagonist 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f] quinoxaline. Proc Natl Acad Sci U S A. 93:5235-5240.
Billinger M, Fleisch M, Eberli FR, Garachemani A, Meier B and Seiler C. (1999). Is the development of myocardial tolerance to repeated ischemia in humans due to preconditioning or to collateral recruitment? J Am Coll Cardiol. 33:1027-1035.
Boeck CR, Ganzella M, Lottermann A and Vendite D. (2004). NMDA preconditioning protects against seizures and hippocampal neurotoxicity induced by quinolinic acid in mice. Epilepsia. 45:745-750.
Boeck CR, Kroth EH, Bronzatto MJ and Vendite D. (2005). Adenosine receptors co-operate with NMDA preconditioning to protect cerebellar granule cells against glutamate neurotoxicity. Neuropharmacology. 49:17-24.
Bristow LJ, Flatman KL, Hutson PH, Kulagowski JJ, Leeson PD, Young L and Tricklebank MD. (1996). The atypical neuroleptic profile of the glycine/N-methyl-D-aspartate receptor antagonist, L-701,324, in rodents. J Pharmacol Exp Ther. 277:578-585.
Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB and Morton AJ. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 19:3248-3257.
Choi DW. (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1:623-634.
Chuang DM, Gao XM and Paul SM. (1992). N-methyl-D-aspartate exposure blocks glutamate toxicity in cultured cerebellar granule cells. Mol Pharmacol. 42:210-216.
Damschroder-Williams P, Irwin RP, Lin SZ and Paul SM. (1995). Characterization of the excitoprotective actions of N-methyl-D-aspartate in cultured cerebellar granule neurons. J Neurochem. 65:1069-1076.
Dietrich MO, Mantese CE, Anjos G, Souza DO and Farina M. (2005). Motor impairment induced by oral exposure to methylmercury in adult mice. Environ. Toxicol. Pharmacol. 19(1), 169-175.
Dunham NW and Miya TS. (1957). A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Am Pharm Assoc (Baltim). 46:208-209.
Faden AI, Demediuk P, Panter SS and Vink R. (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 244:798-800.
Farlow MR, Graham SM and Alva G. (2008). Memantine for the treatment of Alzheimer's disease: tolerability and safety data from clinical trials. Drug Saf. 31:577-585.

43
Ferguson AL and Stone TW. (2008). Adenosine preconditions against ouabain but not against glutamate on CA1-evoked potentials in rat hippocampal slices. Eur J Neurosci. 28:2084-2098.
Ferre S, Gimenez-Llort L, Artigas F and Martinez E. (1994). Motor activation in short- and long-term reserpinized mice: role of N-methyl-D-aspartate, dopamine D1 and dopamine D2 receptors. Eur J Pharmacol. 255:203-213.
Foda MA and Marmarou A. (1994). A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg. 80:301-313.
Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N and McIntosh TK. (2004). Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 28:365-378.
Grabb MC and Choi DW. (1999). Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci. 19:1657-1662.
Grusser-Cornehls U and Baurle J. (2001). Mutant mice as a model for cerebellar ataxia. Prog Neurobiol. 63:489-540.
Jadhav V, Solaroglu I, Obenaus A and Zhang JH. (2007). Neuroprotection against surgically induced brain injury. Surg Neurol. 67:15-20; discussion 20.
Jiang X, Zhu D, Okagaki P, Lipsky R, Wu X, Banaudha K, Mearow K, Strauss KI and Marini AM. (2003). N-methyl-D-aspartate and TrkB receptor activation in cerebellar granule cells: an in vitro model of preconditioning to stimulate intrinsic survival pathways in neurons. Ann N Y Acad Sci. 993:134-145; discussion 159-160.
Jonas W, Lin Y and Tortella F. (2001). Neuroprotection from glutamate toxicity with ultra-low dose glutamate. Neuroreport. 12:335-339.
Kashluba S, Hanks RA, Casey JE and Millis SR. (2008). Neuropsychologic and functional outcome after complicated mild traumatic brain injury. Arch Phys Med Rehabil. 89:904-911.
Katz DI, White DK, Alexander MP and Klein RB. (2004). Recovery of ambulation after traumatic brain injury. Arch Phys Med Rehabil. 85:865-869.
Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N, Kimura K and Kamada T. (1991). 'Ischemic tolerance' phenomenon detected in various brain regions. Brain Res. 561:203-211.
Kuo JR, Lo CJ, Chio CC, Chang CP and Lin MT. (2007). Resuscitation from experimental traumatic brain injury by agmatine therapy. Resuscitation. 75:506-514.
Lin CH, Chen PS and Gean PW. (2008). Glutamate preconditioning prevents neuronal death induced by combined oxygen-glucose deprivation in cultured cortical neurons. Eur J Pharmacol. 589:85-93.
Liu Y, Peterson DA, Kimura H and Schubert D. (1997). Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem. 69:581-593.
Marini AM and Paul SM. (1992). N-methyl-D-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci U S A. 89:6555-6559.
Marini AM, Rabin SJ, Lipsky RH and Mocchetti I. (1998). Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-D-aspartate. J Biol Chem. 273:29394-29399.

44
Miller LP, Lyeth BG, Jenkins LW, Oleniak L, Panchision D, Hamm RJ, Phillips LL, Dixon CE, Clifton GL and Hayes RL. (1990). Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 526:103-107.
Milman A, Rosenberg A, Weizman R and Pick CG. (2005). Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J Neurotrauma. 22:1003-1010.
Mosmann T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 65:55-63.
Muir KW. (2006). Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 6:53-60.
Muir KW and Lees KR. (1995). Clinical experience with excitatory amino acid antagonist drugs. Stroke. 26:503-513.
Ogita K, Okuda H, Yamamoto Y, Nishiyama N and Yoneda Y. (2003). In vivo neuroprotective role of NMDA receptors against kainate-induced excitotoxicity in murine hippocampal pyramidal neurons. J Neurochem. 85:1336-1346.
Pagliaro P, Gattullo D, Rastaldo R and Losano G. (2001). Ischemic preconditioning: from the first to the second window of protection. Life Sci. 69:1-15.
Piot-Grosjean O, Wahl F, Gobbo O and Stutzmann JM. (2001). Assessment of sensorimotor and cognitive deficits induced by a moderate traumatic injury in the right parietal cortex of the rat. Neurobiol Dis. 8:1082-1093.
Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hubner C, Olney JW, Turski L and Ikonomidou C. (1999). N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci U S A. 96:2508-2513.
Raghupathi R. (2004). Cell death mechanisms following traumatic brain injury. Brain Pathol. 14:215-222.
Rejdak R, Rejdak K, Sieklucka-Dziuba M, Stelmasiak Z and Grieb P. (2001). Brain tolerance and preconditioning. Pol J Pharmacol. 53:73-79.
Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD and Wenthold RJ. (2001). Molecular determinants of NMDA receptor internalization. Nat Neurosci. 4:794-802.
Rybnikova E, Vataeva L, Tyulkova E, Gluschenko T, Otellin V, Pelto-Huikko M and Samoilov MO. (2005). Mild hypoxia preconditioning prevents impairment of passive avoidance learning and suppression of brain NGFI-A expression induced by severe hypoxia. Behav Brain Res. 160:107-114.
Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A and Manley GT. (2008). Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 25:719-738.
Saleh MC, Connell BJ and Saleh TM. (2009). Ischemic tolerance following low dose NMDA involves modulation of cellular stress proteins. Brain Res. 1247:212-220.
Sasahira M, Lowry T, Simon RP and Greenberg DA. (1995). Epileptic tolerance: prior seizures protect against seizure-induced neuronal injury. Neurosci Lett. 185:95-98.
Schumann J, Alexandrovich GA, Biegon A and Yaka R. (2008). Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma. 25:945-957.

45
Sei Y, Fossom L, Goping G, Skolnick P and Basile AS. (1998). Quinolinic acid protects rat cerebellar granule cells from glutamate-induced apoptosis. Neurosci Lett. 241:180-184.
Shein NA, Horowitz M and Shohami E. (2007). Heat acclimation: a unique model of physiologically mediated global preconditioning against traumatic brain injury. Prog Brain Res. 161:353-363.
Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H and Hardingham GE. (2006). Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 26:4509-4518.
Tauskela JS, Brunette E, Monette R, Comas T and Morley P. (2003). Preconditioning of cortical neurons by oxygen-glucose deprivation: tolerance induction through abbreviated neurotoxic signaling. Am J Physiol Cell Physiol. 285:C899-911.
Ucar T, Tanriover G, Gurer I, Onal MZ and Kazan S. (2006). Modified experimental mild traumatic brain injury model. J Trauma. 60:558-565.
Vandresen-Filho S, de Araujo Herculano B, Franco JL, Boeck CR, Dafre AL and Tasca CI. (2007). Evaluation of glutathione metabolism in NMDA preconditioning against quinolinic acid-induced seizures in mice cerebral cortex and hippocampus. Brain Res. 1184:38-45.
Vohra HA and Galinanes M. (2006). Myocardial preconditioning against ischemia-induced apoptosis and necrosis in man. J Surg Res. 134:138-144.
Willis C, Lybrand S and Bellamy N. (2004). Excitatory amino acid inhibitors for traumatic brain injury. Cochrane Database Syst Rev.CD003986.
Wong J, Hoe NW, Zhiwei F and Ng I. (2005). Apoptosis and traumatic brain injury. Neurocrit Care. 3:177-182.
Yang GM, Paul SM. (1997). Cultured cerebellar granule neurons as a model of neuronal apoptosis. In: Neuromethods. Poirier, J. (ed.). Humana Press Inc. 29, pps. 47–66.

46
Figure 1. Evaluation of DNA fragmentation in internucleosomal fractions in brain 24h
after TBI in mice with- or without preconditioning with NMDA. (A) Cerebral cortex and (B)
cerebellum from mice were dissected 24h after TBI and DNA was extracted and DNA
“laddering” visualized in agarose gel electrophoresis with ethidium bromide. The lanes in
the gel are groups of treatments. These gel results are representative of three individual
electrophoresis observations.
Table 1. Percent cellular survival measured 24h after TBI using MTT assay.
SAL NMDA TBI NMDA+TBI P
Cortex 100 ± 25.4 102.6 ± 26 70.1 ± 31.3 69.5 ± 40. 5 > 0.05
Cerebellum 100 ± 22 92.5 ± 49 87.5 ± 12.2 87.9 ± 18 .3 > 0.05
NMDA was administrated 24h before cortical impact and after 24h the cerebral cortex
and cerebellum from mice were prepared to MTT assay accord to Material and Methods.
Data are presented as mean of percent control ± SD.

47

48
Figure 2. Motor performance evaluated in footprint test 1.5h after TBI in mice with- or
without preconditioning with NMDA. Mice received NMDA (75mg/kg) 24 before TBI and
footprint test was assessed 1.5h after trauma. Figures show individual values of mice for
(A) strides length (SL), (B) forelimb strides (FLS), (C) hind limb strides (HLS), (D) step
alternation (SA). (E) The representative walking footprint patterns of mice 1.5h after TBI.
The stride pattern of mice treated with NMDA was omitted because it was similar to that
of the SAL group (control). The line between symbols represents a mean value for each
animal. aP<0.05 compared to SAL group; bP<0.05 compared to NMDA group; cP<0.05
compared to TBI group; two-way ANOVA and post hoc Fisher LSD.
49
Figure 3. Motor performance evaluated in footprint test 6h after TBI in mice with- or
without preconditioning with NMDA. Mice received NMDA (75mg/kg) 24 before TBI and
footprint test was assessed 6h after trauma. Figures show individual values of mice for
(A) stride length (SL), (B) forelimb strides (FLS), (C) hind limb strides (HLS), (D) step
alternation (SA). The line between symbols represents a mean value for each animal.
aP<0.05 compared to SAL group; bP<0.05 compared to NMDA group; cP<0.05
compared to TBI group; two-way ANOVA and post hoc Fisher LSD.

50
Figure 4. Motor performance evaluated in footprint test 24h after TBI in mice with or
without preconditioning with NMDA. Mice received NMDA (75mg/kg) 24 before TBI and
footprint test was assessed 24h after trauma. Figures show individual values of mice for
(A) strides length (SL), (B) forelimb strides (FLS), (C) hind limb strides (HLS), (D) step
alternation (SA). The line between symbols represents a mean value for each animal.
aP<0.05 compared to SAL group; bP<0.05 compared to NMDA group; cP<0.05
compared to TBI group; two-way ANOVA and post hoc Fisher LSD.

51
Figure 5. Percentage of motor performance evaluated in footprint test after TBI in mice
with- or without preconditioning with NMDA. Mice received NMDA (75mg/kg) 24 before
TBI and footprint test was assessed 1.5h (A), 6h (B), or 24h (C) after trauma. Data are
shown as mean of percent of control ± S.E.M. (7-9 mice/group). aP<0.05 compared to

52
SAL group; bP<0.05 compared to NMDA group; cP<0.05 compared to TBI group; two-
way ANOVA and post hoc Fisher LSD. SL = strides length; FLS = forelimb strides; HLS
= hind limb strides; SA = step alternation.
Table 2. Performance of mice on runway 24h after TBI.
SAL NMDA TBI NMDA+TBI P
trials 5.0 ± 4.2 10.1 ± 9.9 8.7 ± 3 4.5 ± 2.5 Spent time, s
test 4.5 ± 1.8 16.2 ± 11.8 15.3 ± 13.3 8.9 ± 8.6 > 0.05
trials 14.7 ± 10.6 11.0 ± 7.6 11.4 ± 7.7 13.1 ± 5.2 Velocity,
cm/s test 12.0 ± 2.5 7.9 ± 5.8 7.3 ± 5 12.0 ± 9.6 > 0.05
Mice were trained in three subsequent trails before NMDA administration and were
tested 24h after cortical accord to Material and Methods. Data are presented as mean ±
SD of 6-10 mice.

53
Figure 6. Motor- and balance performance evaluated in rotarod task after TBI in mice
with- or without preconditioning with NMDA. Mice received NMDA (75mg/kg) 24 before
TBI and (A) latency for the first fall and (B) on the number of falls from the rotarod was
assessed 1.5h (A), 6h (B), or 24h, (C) after trauma. Data are shown as mean of percent
of control ± S.E.M. (7-9 mice/group). aP<0.05 compared to SAL group; two-way ANOVA
and post hoc Fisher LSD.

54
CAPITULO 2
3.2 METODOS E RESULTADOS ADICIONAIS
3.2.1 Materiais:
Os materiais utilizados incluem o N-metil-D-aspartato (NMDA) adquirido do
laboratório Sigma; anticorpos primários anti-NR1, anti-NR2A e anti-NR2B obtidos do
laboratório Upstate; anticorpos secundários anti-imunoglobulina de camundongo ou
coelho adquiridos do laboratório Sigma. Os demais reagentes utilizados foram obtidos
dos laboratórios Sigma, Amresco e Invitrogen.
3.2.2 Aspéctos Éticos
Todos os procedimentos experimentais foram feitos de acordo com as
recomendações da Sociedade Brasileira de Neurociências e Comportamento
(SBNeC). O Projeto já está aprovado pelo Comitê de Ética em Pesquisa da
Universidade do Extremo Sul Catarinense (UNESC; prot. 776/2007).
3.2.3 Animais Experimentais
Os camundos albinos CF1 machos (30-40g) foram mantidos em ambiente
climatizado com água e comida disponíveis. Os nossos protocolos para experimentos

55
com animais são projetados de maneira que o animal tenha o mínimo de sofrimento
possível e com limite de animais sacrificados.
3.2.4 Tratamento com NMDA
Os animais foram divididos em grupos de pré-tratamento com NMDA (pré-
condicionados) e tratados com solução fisiológica (animais controle de pré-tratamento).
Ainda, os animais que receberam solução fisiológica (NaCl 0,9 g%) ou NMDA
intraperitonialmente (i.p.) foram divididos em grupos de dano cerebral por TCE, como
segue: 1) Salina (salina i.p. + anestesia); 2) NMDA (NMDA i.p. + anestesia); 3) TCE
(salina i.p. + trauma); 4) NMDA + TCE (NMDA i.p. + trauma). Após a administração i.p.
NMDA (75 mg/kg de animal) os animais foram colocados individualmente em caixas de
acrílico (25 x 25 x 25 cm) e foram observados por 30 minutos para a ocorrência de
mudanças comportamentais.
3.2.5 Traumatismo Crânio-Encefálico (TCE)
Vinte e quatro horas após a administração de NMDA os camundongos foram
submetidos ao modelo experimental de TCE em um aparato de queda de peso em
formato de bala, como previamente descrito (Adelson et al., 1996). Os animais foram
anestesiados por inalação com uma mistura de O2/N2O (33%:66%) através de uma
máscara. O animal foi posicionado abaixo de um tubo de acrílico de 80 m fixado a um
suporte de metal com circunferência de 1cm e o dano cerebral pelo impacto foi induzido
pela queda do peso de 50g que corresponde ao trauma moderado. O animal foi fixado a

56
uma espuma para permitir a deflexão da cabeça após o impacto, pois após o impacto
do peso na cabeça do animal, a espuma foi retirada imediatamente para evitar um
segundo impacto no recuo do peso. Após o trauma cada animal recebeu um suporte de
oxigenação com 100% de O2 até estar totalmente desperto. Apesar do elevado risco de
mortalidade decorrente do traumatismo, no presente estudo não houve morte dos
animais pelo TCE. Assim, foram utilizados 24 animais divididos em quatro grupos com
um número de seis animais por grupo.
3.2.6 Separação de Proteínas Por Eletroforese (SDS- PAGE)
Seis horas após o TCE os animais foram mortos por deslocamento cervical para
a retirada do cerebelo. As amostras de cerebelo foram solubilizadas em tampão de
amostra composto por 4% de duodecil sulfato de sódio (SDS), 50 mM de Tris-HCl, 5
mM de EDTA e 8 % de β-mercaptoetanol, pH 6,8; e diluídas em solução de diluição
com 40 % de glicerol, 25 mM de Tris-HCl e azul de bromofenol pH 6,8. As proteínas
foram separadas por SDS-PAGE [eletroforese por gel de poliacrilamida-SDS (sodium
dodecyl sulfate polyacrylamide gel electrophoresis)] usando gel de separação com
gradiente de acrilamida (Laemmli, 1970). A concentração da acrilamida no gel de
separação é de 7,5%. O gel de entrada será de 4 % de acrilamida. A amostras foram
aplicadas na quantidade de 30 µg de proteína/poço. A eletroforese foi realizada com
corrente fixa de 40 mA e voltagem máxima de 140 mV (para 2 géis), por
aproximadamente 2 horas. Após a corrida, os géis foram submetidos à
eletrotransferência em tampão 50 Mm de Tris-HCl, 140 mM de glicina e 10 % de

57
metanol por 1 hora, em corrente de no máximo 100 mA e voltagem de 350 mV a 4 °C,
para transferência das proteínas do gel para uma membrana de nitrocelulose de 0,4 µm
de porosidade (GE Life Science).
3.2.7 Imunodetecção de Proteínas ( WESTERN BLOT)
Após a eletrotransferência, as membranas foram bloqueadas por 1 hora com
albumina bovina, em 5 % de TBS (10 mM de Tris, 150 mM de NaCl, pH 7,5) e em
seguida lavadas com TBS-T (0,05 % de Tween-20, 10 mM de Tris, 150 mM de NaCl,
pH 7,5). As membranas foram incubadas com anticorpos primários contra os receptores
NR1A (1:450), NR2A (1:500) ou NR2B (1:500) por 14-16 horas (overnight). Para a
detecção dos complexos imunes, as membranas foram novamente lavadas com TBS-T
e os anticorpos secundários (conjugado a peroxidase) anti-mouse (para NR1) e anti-
rabbit (para NR2A e NR2B) foram incubados por 1 hora (1:8000). Após lavagens com
TBS-T a imunodetecção foi revelada por emissão de quimiluminescência com
reagentes luminescentes (GE Life Science), seguindo as recomendações do fabricante,
e impressão de imagem em filme auto-radiográfico. As imagens foram visualizadas em
software para quantificação densitométrica das bandas protéicas (Scion Image, Beta
4.0.2; Scion Corporation, Frederick, MD).
3.2.8 Dosagem de Proteína
As proteínas foram dosadas através do método de Peterson, (1977) descrito

58
brevemente a seguir. Sobre alíquotas de 2 µl das amostras foram adicionados 398 mL
de água e 400 mL do reagente de Lowry (0,2 N de NaOH; 2,5% de SDS; 5% de
Na2CO3; 0,2% de CuSO4 e 0,1% de tartarato duplo de Na+/K+). As amostras foram
homogeneizadas imediatamente e deixadas para reagir por dez minutos. Em seguida
foi adicionado 200 mL do reagente de Folin 0,4 N seguido de homogeinização e
incubação por 30 minutos. A leitura do complexo de cor foi realizada em
espectrofotômetro (comprimento de onda de 750 nm) e as concentrações protéicas
foram obtidas a partir uma curva-padrão utilizando albumina de soro bovino (BSA; 0,1
mg/mL).
3.2.9 Análise Estatística
Os resultados foram analisados por Two-way ANOVA usando como fatores o
tratamento e o TCE (software STATISTICA 7.0; StatSoft, Inc., USA). Quando o valor de
P foi menor que 0,05 o teste post-hoc Duncan foi utilizado para avaliação das medidas
bioquímicas e moleculares. Os dados estão apresentados como média ± erro padrão da
média da densidade óptica das bandas protéicas.
3.2.10. Resultados
Os camundongos foram pré-condicionados com NMDA e submetidos ao TCE
para avaliação das subunidades do receptor NMDA em amostras de cerebelo seis

59
horas após o TCE, pois se sabe que o dano secundário já possa estar ocorrendo,
preparadas para identificação das subunidades a partir do Western blot.
Na figura 1 está representada a quantificação da subunidade NR1. Observa-se
uma diminuição do conteúdo da subunidade NR1 no cerebelo dos animais pré-
condicionados com NMDA assim como daqueles submetidos ao TCE, com ou sem pré-
tratamento com NMDA, quando comparado ao grupo controle. Entretanto não houve
nenhuma mudança significativa na subunidade NR2A do receptor NMDA, nos grupos
investigados como representa a figura 2.
Na figura 3 está representada a quantificação da subunidade NR2B. Observa-se
um aumento do conteúdo da subunidade NR2B no cerebelo dos animais pré-
condicionados com NMDA assim como daqueles submetidos ao TCE (com ou sem pré-
tratamento com NMDA) quando comparado ao grupo controle.

60
Figura 1 : Efeito do pré-condicionamento com NMDA e do TCE no conteúdo da
subunidade NR1 do receptor NMDA. As amostras de cerebelo dos camundongos foram
dissecadas 6 horas após o TCE. As proteínas foram separadas por SDS-PAGE e a
imunodetecção dos complexos foi realizada pela técnica de Western blot. Painel
superior: autoradiograma representativo da detecção da subunidade NR1. Painel
inferior: quantificação da imunoreatividade como valores arbitrários de densidade óptica
pelo software Scion Image e estatisticamente comparados com two-way ANOVA, post
hoc Duncan. Os dados expressos como média ± erro padrão da média (n = 5). *P <
0,05, comparado ao grupo controle. CRL = salina i.p. + anestesia; NMDA = NMDA i.p. +
anestesia; TCE = salina i.p. + trauma; NMDA+TCE = NMDA i.p. + trauma.

61
Figura 2 : Efeito do pré-condicionamento com NMDA e do TCE no conteúdo da
subunidade NR2A do receptor NMDA. As amostras de cerebelo dos camundongos
foram dissecadas 6 horas após o TCE. As proteínas foram separadas por SDS-PAGE e
a imunodetecção dos complexos foi realizada pela técnica de Western blot. Painel
superior: autoradiograma representativo da detecção da subunidade NR2A. Painel
inferior: quantificação da imunoreatividade como valores arbitrários de densidade óptica
pelo software Scion Image e estatisticamente comparados com two-way ANOVA, post
hoc Duncan. Os dados expressos como média ± erro padrão da média (n = 5). *P <
0,05, comparado ao grupo controle. CRL = salina i.p. + anestesia; NMDA = NMDA i.p. +
anestesia; TCE = salina i.p. + trauma; NMDA+TCE = NMDA i.p. + trauma.

62
Figura 3: Efeito do pré-condicionamento com NMDA e do TCE no conteúdo da
subunidade NR2B do receptor NMDA. As amostras de cerebelo dos camundongos
foram dissecadas 6 horas após o TCE. As proteínas foram separadas por SDS-PAGE e
a imunodetecção dos complexos foi realizada pela técnica de Western blot. Painel
superior: autoradiograma representativo da detecção da subunidade NR2B. Painel
inferior: quantificação da imunoreatividade como valores arbitrários de densidade óptica
pelo software Scion Image e estatisticamente comparados com two-way ANOVA, post
hoc Duncan. Os dados expressos como média ± erro padrão da média (n = 5). *P <
0,05, comparado ao grupo controle. CRL = salina i.p. + anestesia; NMDA = NMDA i.p. +
anestesia; TCE = salina i.p. + trauma; NMDA+TCE = NMDA i.p. + trauma.

63
4 DISCUSSÃO
O glutamato tem um importante papel no mecanismo de excitotoxicidade,
responsável por danos ao cérebro. A carga intracelular de cálcio na excitotoxidade é
ocasionada pela ativação excessiva de receptores NMDA pelo glutamato (Clausen &
Bullock, 2001). Diversos estudos clínicos frente ao monitoramento desses eventos vêm
sendo realizados e poucos com sucesso em relação à neuroproteção e TCE.
De acordo com pesquisas de Gagliard, (2000) doses subtóxicas de NMDA
fornecem um pré-condicionamento químico ao cérebro, que conduz a tolerância celular
a doses tóxicas subseqüentes de agonistas de receptores glutamatérgicos; e que o
objetivo da neuroproteção é fazer com que a cascata patológica se interrompa.
Neste estudo com relação a análise de DNA e MTT não se observou diferenças
significativa entre os grupos Sal; TCE+Sal; NMDA; TCE+NMDA. Talvez esses
resultados possam ser devido ao peso de 50g utilizado refira-se ao trauma moderado a
leve quando comparado com outros modelos de trauma moderado (Leinhase et al.,
2006). Ou devido a indução de neurogenese cerebral após o TCE (Richardson et al.,
2007) ou porque a morte pode ser necrótica e talvez nenhuma das técnicas permita a
avaliação exata.
Nos resultados adicionais deste estudo se observa que o pré-condicionamento
com NMDA 24h antes do trauma proporcionou modificações nas subunidades protéicas
do receptor NMDA. Apesar de a maioria dos estudos sobre essas subunidades NR1,
NR2A e NR2B ainda estarem em andamento sabe-se que essas proteínas estão
envolvidas em processos de neuroproteção por sua especificidade de agonistas e
antagonistas que bloqueiam tais subunidades (LIU et al., 2007).

64
Neste trabalho observa-se uma diminuição do conteúdo da subunidade NR1 no
cerebelo dos grupos de animais pré-condicionados com NMDA sem trauma, pré-
condicionados com NMDA+TCE e submetidos ao TCE sem pré-condicionamento
quando comparados ao grupo controle. Giza (2006) não observou no córtex, após o
trauma, mudanças nos níveis de NR1 e NR2B. Kreutz et al. (1998) descobriram que a
sobrevivência seguida do trauma axonal do nervo ópico é altamente dependente do
aumento da expressão do NR1. Baseado nestas observações sugere-se que a
subunidade NR1 pode levar a uma diferente composição do receptor NMDA nativo e
diferente resposta na ativação ao glutamato dependendo da sua localização. O NR1 é
uma proteína essencial para a atividade do canal do receptor de NMDA incluindo
agonistas e antagonista, permeabilidade de Ca2+, bloqueiam Mg2+ , modulam a glicina,
ativam a poliamina e inibem Zn2+. E são expressas em quase todas as células neurais
(Nakanishi, 1992).
Com relação ao NR2A observa-se neste estudo que não houve nenhuma
mudança significativa nesta subunidade do receptor NMDA no cerebelo 6h após o TCE.
BESSHO et al., (1994) mostraram que a as células cerebelares tornam-se muito
vulneráveis à toxicidade mediada pelo NMDA quando há o aumento dos níveis do
NR2A, reforçada pelo influxo de Ca2+ mediada pelo receptor. Em contrapartida Lio et
al., (2007) refere que a ativação dos receptores sinápticos ou extrasinápticos de NR2A
promove a sobrevivência neuronal e exerce uma ação neuroprotetora e de encontro a
dano neuronal mediado pelo receptor NMDA observadas igualmente in vivo e in vitro.
Os dados da pesquisa de Giza et al., (2006) sugerem que a redução na expressão de
NR2A no trauma induzido seja um mecanismo provável para a neuroplasticidade.
Estudos de Horak et al., (2008) mostram que a expressão de NR2A é a resonsável pelo

65
aumento da toxicidade, aumentando a entrada de cálcio então reduzir NR1 e não
alterar NR2A possa estar relacionado com neuroproteção.
Para finalizar, os achados adicionais deste estudo, observa-se nesta pesquisa
que a subunidade NR2B teve um aumento no seu conteúdo em todos os grupos
tratados quando comparado ao grupo controle. Schumann (2008) observou que após o
trauma em ratos a subunidade NR2B estava aumentada no hipocampo, mas no córtex
estava diminuída, associando isto a um aumento na fosforilação da tirosina de NR2B.
Mas as causas do aumento do NR2B ou a diminuição do NR1 no cerebelo ainda não
são conhecidas. A mudança desenvolvida nas subunidades NR2A e NR2B, na
superfície sináptica, foi correlacionada com a mudança no tempo passado por essas
subunidades dentro da superfície sináptica (Grog et al., 2006). Nosso tempo estudado
foi de 6h após o TCE e suas diferentes quantificações de proteínas nesse tempo,
localização cerebral e funções no cerebelo ainda não são sabidas com exatidão.
Apesar das limitações em relação ao tempo da pesquisa e disponibilidades de animais
não se verificou córtex frontal. Mas devido aos resultados encontrados estima-se que
no córtex frontal também possa ter alguma altercação nessas subunidades
considerando que o fármaco NMDA foi sistêmico e que o TCE não foi focal,
necessitando de estudos mais específicos e detalhados sobre o tema complementar.
Para que assim se possam compreender melhor os mecanismos envolvidos nos
prejuízos motores em camundongos expostos ao TCE.
O cerebelo é uma estrutura que esta ligada ao aprendizado motor (Boyden et al.,
2004), regula o sincronismo dos movimentos instruídos (Buonomano & Mauk, 2004) e
equilíbrio dinâmico. Lesão pós trauma no córtex cerebelar interrompe primeiramente a
resposta aos movimentos instruídos (Perrett et al., 1993), podendo levar ao

66
desequilíbrio e prejuízo motor em diversos tempos. No aprendizado, de seres humanos,
para alcançar a memória motora e executar os movimentos corretos em um campo de
força, deve-se realizar o movimento diversas vezes e em tempos diversos. Tudo isso
antes de se alterar o campo de força para que a memória motora não se torne
resistente à supressão pela aprendizagem de um campo de força novo (Brashers-Krug
et al., 1996; Shadmehr & Brashers-Krug, 1997) ou no caso desta pesquisa o campo
novo estudado seria a avaliação motora após o TCE no rotarod e footprint.
Para esta tarefa, a execução dos movimentos imediatamente depois da
aprendizagem conduz à ativação do córtex frontal, mas nas horas depois que o
treinamento está completo, o cerebelo torna-se ativo durante o desempenho da tarefa
(Shadmehr & Holcomb 1997). Assim o papel do cerebelo pode mudar com o tempo
após a aquisição da aprendizagem, o que é consistente com a noção geral que a
consolidação pode envolver a redistribuição da informação entre partes diferentes do
cérebro, das áreas a curto prazo às áreas a longo prazo (Marr, 1971, Junque, 1999).
Como neste estudo os tempos de verificação da coordenação motora foram de
1h e 30 min e 6h podendo estar estimulando áreas de curto prazo, como o córtex frontal
e 24h ativando o cerebelo, mas essa hipótese não descarta o fato de ambas as
estruturas estarem em constante e incessante atividade.
O córtex frontal agrupa funções motoras, de expressão lingüística, memória e
funções de planejamento mental do comportamento (Lent, 2001). Lesões no córtex
frontal podem levar o individuo a ter crises tônicas, clônicas e espasmos (Manrezza,
2003), assim como déficits cognitivos específicos, como na memória (Schlindwein-
Zanini et al., 2007). Pode-se sugerir que os resultados deste estudo são mediados por
danos em córtex frontal e cerebelo. Onde nessas áreas, a parte relacionada a motora

67
foi comprometida. Não se descarta também que outras áreas cerebrais tenham sido
afetadas, o fato é que o estudo foi focado na área motora com relação ao cerebelo e
córtex frontal. Não se pode justificar como fato a perda de memória, pois nem um teste
relacionado foi realizado. Mas pode-se pensar que a memória e funções relacionadas
ao planejamento motor e cognitivo tenham sido também afetadas após o trauma.
Mesmo porque o córtex frontal e o cerebelo também podem estar relacionados ao
aprendizado e a memória.
O trauma moderado através de um aparato de queda de peso em formato de um
projétil produziu neste estudo déficit motor nos camundongos. Estes dados estão de
acordo com as observações clínicas que mostram o prejuízo motor do TCE moderado
(Adelson et al., 1997; Piot-Grosjean et al., 2001).
Os resultados desta pesquisa comprovam com o footprint que após o trauma
moderado os prejuízos motores são significativos e que o uso do fármaco NMDA
causou o pré-condicionamento esperado em relação à coordenação motora. Observou-
se diminuição na largura do passo traseiro e no comprimento do passo no grupo TCE
quando comparado com o controle e com o grupo NMDA+TCE. Carter et al. (1999)
observaram em estudos que a largura do passo também estava diminuída em
camundongos transgênicos. Camundongos expostos ao metilmercúrio (MeHg) tiveram
prejuízo motor com relação o footprint devido ao dano cerebelar (Dietrich, et al., (2005).
O comportamento funcional motor, tanto na coordenação, velocidade ou tempo de um
determinado movimento, é afetado nas crianças após TBI moderado ou severo (Kuhtz-
Buschbeck, et al., 2003).
Observou-se que o pré-condicionamento com NMDA foi eficaz e preveniu os
prejuízos motores contra o TCE no footprint. Algumas destas estratégias de

68
neuroproteção se aproximam com o uso de antagonistas de receptores NMDA (Faden
et al., 1989; Temple et al., 2001). A administração de dose subconvulsivante de NMDA
via intraperitoneal tem sido utilizada como modelo de pré-condicionamento químico
frente a insultos no cerebro (Boeck et al., 2004). Resultados de pesquisas de Bernert &
Turski (1995) indicam que os antagonistas de NMDA possuem melhores propriedades
neuroprotetoras em processos excitotóxicos provocados por ferimentos devido a
traumas no cérebro em ratos jovens quando comparados a ratos adultos. Alguns
estudos em modelo animais de trauma mostraram efeitos benéficos dos antagonistas
do receptor NMDA como por exemplo MK-801, fenilciclidina, dextrometorfan, D-CPP-
ene, Pfizer PC 101-606 (Hamm et al., 1993) e magnésio (Heath & Vink, 1999). Outra
substância antagonista de receptores do glutamato, topiramate, estão sendo estudadas
como agente neuroprotetor do cérebro de eventos secundários resultantes do TCE. Foi
demonstrado em um modelo animal de TCE que após o tratamento com topiramate
houve uma recuperação neurológica mais rápida em relação ao desempenho motor e
da memória (O'Dell & Hamm, 1995).
Observamos também que com relação ainda ao footprint a largura do passo e o
comprimento do passo são os mais afetados após o trauma e estão diminuídos, isto
pode-se justificar de acordo com a área atingida. As sequelas neurofisiológicas do TCE
são consequência da combinação de lesões cerebrais focais e difusas. Os focos do
abalo envolvem geralmente os lóbulos frontais.
As lesões Fronto-basais produzem mudanças importantes como na
personalidade e no comportamento e o prejuízo dorsolateral das lesões das funções de
execução (falta do planejamento, da flexibilidade e do uso de estratégias).

69
O dano axonal difuso é relacionado ao prejuízo da atenção, à velocidade do
processamento mental e às funções do lóbulo frontal (Junque, 1999). E as lesões no
cérebro e focais, devido ao TCE, conduzem não somente aos déficits funcionais na
área da lesão, mas igualmente perturbam a rede neuronal estrutural intacta que está
conectada ao local da lesão (Wiese et al., 2004). Em muitos casos como sequela
motora do TCE tem-se espasticidade em membros inferiores (MMII), isso pode justificar
o fato de o comprimento da passada estar alterado assim como a diminuição da
passada traseira. Pois se sabe que além do córtex frontal o cerebelo também está
envolvido na coordenação dos movimentos voluntários, manter o equilíbrio, na
regulação do tônus muscular e no aprender das habilidades motoras (Veiga Neto &
Segura, 2002).
Os resultados obtidos neste trabalho com o rotarod demonstram que após o
trauma moderado os prejuízos motores são significativos. Mas que o uso do fármaco
NMDA não causou o pré-condicionamento esperado em relação ao equilíbrio motor no
rotarod. Com relação aos achados desta pesquisa os estudos de Clark al de et., (2000)
que utilizaram o fármaco z-DEVD-fmk como neuroprotetor cerebral, observando que
este fármaco protegeu as células do cérebro, mas que em relação ao prejuízo motor
não se observou melhora. Outros estudos relatam que após o TCE pode-se haver
amnésia pós-traumática (Dixon et al., 1996), ou déficits de memória por ate 30 dias
(Hamm et al., 1993). Considerando que os déficits motores são evidentes nos tempo
01h30minh, 6h e que não houve a neuroproteção esperada pelo NMDA no rotarod e
que 24 horas após o trauma não houve diferença entre o controle e os grupos TCE e
NMDA+TCE sugere-se que este déficit motor possa estar associado à memória,
necessitando de uma análise clínica e comportamental mais detalhada. A

70
aprendizagem, a memória e o desenvolvimento do cérebro são associados com as
modificações duradouras das sinapses e essas modificações incluem a plasticidade. A
plasticidade é uma propriedade intrínseca do cérebro humano e representa uma
estratégia da evolução para capacitar o sistema nervoso para resistir as restrições de
seu próprio genoma e assim adaptar-se às pressões ambientais, mudanças fisiológicas
e experiências. Flutuações dinâmicas na potência de conexões pré-existentes através
de redes neurais distribuídas, modificações na coerência córtico-cortical e córtico-
subcortical relacionada a tarefas e modificações na correspondência referente ao
comportamento e à atividade neural ocorrem em resposta a modificações do input
aferente ou da demanda eferente. Tais modificações rápidas e contínuas podem ser
seguidas pelo estabelecimento de novas conexões através do crescimento e da
arborização dendríticos. Entretanto, elas trazem o perigo de que o padrão evolutivo da
ativação neural possa por si mesmo levar a um comportamento anormal (Pascual-
Leone et al., 2005).
Diversas outras mudanças celulares e moleculares estão implicadas na
homeostase sináptica, mas uma característica comum de muitos estudos é na
plasticidade homeostática onde existe uma alteração no número ou no complemento de
receptores NMDA (Watt et al., 2000; Pérez-Otaño & Ehlers, 2005). Embora esse
mecanismo ainda seja mal compreendido, esses resultados podem estar também
relacionados com a ativação ou inibição dessa plasticidade pelos receptores NMDA
após o TCE. E ainda segundo Pascual-Leone et al., (2005) as modificações plásticas
procuram minimizar o dano cerebral e que circuitos motores paralelos podem ser
ativados para estabelecer algum input alternativo para os neurônios motores. Sendo
que a recuperação do equilíbrio e a destreza motora fina dos camundongos após 24h

71
do trauma estejam relacionadas não necessariamente no local da lesão, mas
paralelamente a ela e mais se a regulação dessa homeostase, que é desestabilizada
após o TCE, pode ser mantida ou reorganizada devido ao pré-condicionamento com
NMDA.
Pode-se concluir que o pré-condicionamento com NMDA é eficaz quando
relacionado ao footprint e que o pré-condicionamento não foi eficaz quando relacionado
ao rotarod.
Ainda não se sabe o porquê e como os mecanismos que estão envolvidos no
trauma dão tais respostas e são poucos os estudos com fármacos para pré-condicionar
o cérebro contra o TCE. As variáveis genéticas podem explicar em diferenças
individuais da parte no grau de prejuízo motor ou memória e no relacionamento do TCE
com a doença de Alzheimer (Junque, 1999). Mas podemos perceber que o NMDA
causou um pré-condicionamento no cérebro contra o TCE, mas como os mecanismos
envolvidos respondem a essa atividade ainda não se sabe. O modelo de TCE
moderado executado em camundongos permite o estudo da interdependência entre
dano estrutural, mudanças da memória e motora, fatores genéticos, e os efeitos do
tratamento farmacológico utilizado.
Este estudo teve como objetivo verificar as possíveis alterações de dano celular
no córtex frontal e cerebelo e avaliar o conteúdo das subunidades NMDA (NR1, NR2a,
NR2b) em cerebelo de camundongos submetidos ao TCE e pré-condicionados com
NMDA. Pode-se concluir que a única alteração observada e pesquisada foi em NR1a,
NR2a e NR2b no cerebelo, não ocorrendo alterações em DNA e MTT no córtex e
cerebelo após o trauma. Outro objetivo verificado foi quanto à coordenação motora em

72
camundongos pré-condicionados ou não com NMDA e expostos ao TCE e observou-se
que o trauma causa alteração na coordenação motora.
Este estudo proporciona assim mais referências com informações relevantes
sobre o TCE e o pré-condicionamento com NMDA e dispõe a estudos diversos
questionamentos frente a neuroproteção cerebral e os mecanismos envolvidos no TCE
tendo como perspectivas realizar a detecção de imunoproteinas para córtex frontal em
6h; cerebelo e córtex frontal em 24h como também a verificação da memória após o
TCE em diversos tempos.

73
5. CONCLUSÃO
Observou-se que o trauma causa alterações na coordenação motora que foi
prevenida pelo pré-condicionamento com NMDA nas avaliações realizadas no teste
footprint.
As alterações observadas no conteúdo das subunidades NR1, NR2A e NR2B no
cerebelo após o TCE foram mantidas nos animais pré-condicionados.
O pré-condicionamento com NMDA levou a mesma modificação observada pelo
TCE, porém as vias celulares envolvidas provavelmente são diferentes;
O estudo de vias celulares que possam modular a transmissão glutamatérgica é
de fundamental importância para a compreensão do desenvolvimento de mecanismos
fisiológicos e fisiopatológicos no SNC.
Tendo em vista o papel protetor do pré-condicionamento com NMDA, entende-se
que este mecanismo possua funções preventivas ao SNC e/ou de manutenção contra
danos causados pelo TCE, que envolvam o sistema glutamatérgico.

74
6. REFERÊNCIAS
ADAMS JH; DOYLE D; FORD I; GENNARELLI TA; GRAHAM DI; MCLELLAN DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15: 49–59. 1989.
ADELSON PD; DIXON CE; ROBICHAUD P; KOCHANEK, P. M. Motor and cognitive
functional deficits following diffuse traumatic brain injury in the immature rat. Journal of Neurotrauma 14: 99–108. 1997.
ADELSON PD; ROBICHAUD P; HAMILTON RL; KOCHANEK PM. A model of diffuse
traumatic brain injury in the immature rat. Journal of Neurosurgery 85: 877–884. 1996.
ALVES CM; MARZOCCHI-MACHADO CM; CARVALHO IF; LUCISANO VALIM YM.
Application of the chemiluminescence systems to evaluate the role of Fcgamma and complement receptors in stimulating the oxidative burst in neutrophils. Talanta 60(2-3): 601-608. 2003.
ARUNDINE M; TYMIANSKI M. Molecular mechanisms of calcium-dependent
neurodegeneration in excitotoxicity. Cell Calcium 34: 325-337. 2003. BERNERT H; TURSKI L. Traumatic brain damage prevented by the non-N-methyl-D-
aspartate antagonist 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f] quinoxaline. Proceedings of the National Academy of Sciences of the United States of America 93(11): 5235-5240. 1996.
BESSHO Y; NAWA H; NAKANISHI S. Selective up-regulation of an NMDA receptor
subunit mRNA in culture cerebellar granule cells by K(+)-induced depolarization an NMDA treatment. Neuron 12(1): 87-95. 1994.
BLISS TVP; COLLINGRIDGE GL. A synaptic model of memory; long-term potentiation
in the hippocampus. Nature 361: 31-39. 1993. BOECK C; GANZELLA M; LOTTERMANN A; VENDITE D. NMDA preconditioning
protects against seizures and hippocampal neurotoxicity induced by quinolinic acid in mice. Epilepsia 45: 745-750. 2004.
.BOYDEN ES; KATOH A; RAYMOND JL. Cerebellum-Dependent Learning: The Role Of
Multiple Plasticity Mechanisms. Annual Review of Neuroscience 27: 581–609. 2004.
BRASHERS-KRUG T; SHADMEHR R; BIZZI E. Consolidation in human motor memory.
Nature 382: 252–255. 1996.

75
BRESSAN RA; PILOWSKY LS. Hipótese glutamatérgica da esquizofrenia. Revista Brasileira de Psiquiatria 25 (3 ): 177-183. 2003.
BRINK JD; GARRETT AL; HALE WR; NICKEL VL; WOO–SAM J. Recovery of motor and intellectual function in children sustaining severe head injuries. Developmental Medicine & Child Neurology 12: 565–571. 1970.
BUONOMANO D; MAUK M. The neural basis of temporal processing. Annual Review
of Neuroscience 27: 307–340. 2004. BUTEFISCH CM. Neurobiological bases of rehabilitation. Neurological Sciences 27:
18-23. 2006. CARLOS TM. Expression of endothelial adhesion molecules and recruitment of
neutrophils after traumatic brain injury in rats. In:. Administration of adenosine receptor agonists or antagonists after controlled cortical impact in mice: effects on function and histopathology. (Varma, MR. Dixon CE, Jackson EK, Peters GW, Melick JA, Griffith RP, Vagni VA, Clark RSB, Jenkins LW, Kochanek PM, eds.). Brain Research 951: 191-201. 2002.
CARMICHAEL ST. Plasticity of cortical projections after stroke. Neuroscientist 9: 64–
75. 2003. CARTER RJ; LIONE LA; HUMBY T; MANGIARINI L; MAHAL A; BATES GP; CLARK
RS; KOCHANEK PM; WATKINS SC; CHEN M; DIXON CE; SEIDBERG NA; MELICK J; LOEFFERT JE; NATHANIEL PD; JIN KL; GRAHAM SH. Caspase-3 mediated neuronal death after traumatic brain injury in rats. Journal of Neurochemistry 74: 740-753. 2000.
CENTRO DE PESQUISAS EM EDUCAÇÃO E PREVENÇÃO DA REDE SARAH.
Traumatismo crânio encefálico . Disponível em: <http://www.sarah.br/>. Acesso em: 10 nov. 2008.
CLAUSEN T; BULLOCK R. Medical Treatment and Neuroprotection in Traumatic Brain
Injury. Current Pharmaceutical Design 7: 1517-1532. 2001. COTMAN, C.W., KAHLE, J.S., MILLER, S.E., ULAS, J., BRIDGES, R.J. Excitatory
amino acid neurotransmission. In: Psychopharmacology: The Fourth Generation of Progress. ( Floyd, E., Bloom, F.E., Kupfer, D.J. eds.). Raven Press, New York, pp. 75-85. 1995.
DALE N; ROBERTS A. Dual- component amino-acid-mediated synaptic potentiais:
excitatory drive for awimming in Xenopus embryos. Journal of Physiology 363: 35-59. 1985.

76
DEBERNARDI R; MAGISTRETTI PJ; PELLERIN L. Trans-innibition of glutamate transport prevents excitatory amino acid-induced glycolysis in astrocytes. Brain Research 850: 39-46. 1999.
DIETRICH MO; MANTESE CE; ANJOS G; SOUZA DO; FARINA M. Motor impairment
induced by oral exposure to methylmercury in adult mice. Environmental Toxicology and Pharmacology 19: 169–175. 2005.
DIXON CE; BAO J; LONG DA; HAYES RL. Reduced evoked release of acetylcholine in
the rodent hippocampus following traumatic brain injury. Pharmacology, Biochemistry, Behavior 53(3): 679-86. 1996.
DUNNETT SB; MORTON AJ. Characterization of Progressive Motor Deficits in Mice
Transgenic for the Human Huntington’s Disease Mutation. The Journal of Neuroscience 19(8): 3248–3257. 1999.
FADEN AI; DEMEDIUK P; PANTER SS; VINK R. The role of excitatory amino acids and
NMDA receptors in traumatic brain injury. Science 244: 798–800. 1989. FAIRMAN WA; VANDENBERG RJ; ARRIZA JL. An excitatory amino-acid transporter
with properties of a ligand-gated chloride channel. Nature 375: 599-603. 1995. FEI Z; ZHANG X; BAI H; JIANG X; LI X; ZHANG W; HU W. Posttraumatic secondary
brain insults exacerbates neuronal injury by altering Metabotropic Glutamate Receptors. Neuroscience 8: 1471-2202. 2007.
FLECK MW; BARRIONUEVO G; PALMER AM. Synaptosomal and vesicular
accumulation of L- glutamate, L-aspartate and D- aspartate. Neurochemistry International 39: 217-225. 2001.
FRANCESC X; PAPADIA SS; HOFMANN F; NEIL R; BADING HH; HARDINGHAM GE.
Preconditioning Doses of NMDA Promote Neuroprotection by Enhancing Neuronal Excitability. The Journal of Neuroscience 26(17): 4509–4518. 2006.
GAGLIARD RJ. Neuroprotection, excitotoxicity and NMDA antagonists. Arquivos de
Neuropsiquiartria . 58(2B): 583-588. 2000. GENNARELLI GA; GRAHAM DI. Neuropathology. In Textbook Of Traumatic Brain
Injury ( Silver JM; Mcallister TW; Yudofsky SC, eds.). American Psychiatric Association, Washington, pp. 27-34. 2005.
GENNARELLI TA; THIBAULT LE; ADAMS JH; GRAHAM DI; THOMPSON CJ;
MARCINCIN RP. Diffuse axonal injury and traumatic coma in the primate. Annals of Neurology 12(6): 564–572. 1982.
GENTRY LR; GODERSKY JC; THOMPSON BH. Traumatic. brain stem injury: MR
imaging. Radiology 171: 177-87. 1989.

77
GIZA CC; MARIA NSS; HOVDA DA. N-Methyl-D-Aspartate Receptor Subunit Changes
after Traumatic Injury to the Developing Brain. Journal of Neurotrauma 23(6): 950–961. 2006.
GOLDMAN-RAKIC PS; PORRINO, LJ. The primate mediodorsal (MD) nucleus and its
projection to the frontal lobe. Journal of Comparative Neurology 242: 535–560. 1985.
GROG L; HEINE M; COUSINS SL; STEPHENSON FA; LOUNIS B; COGNET L;
CHOQUET D. NMDA receptor surface mobility depends on NR2A-2B subunits. Neuroscience 103(49): 18769–18774. 2006.
GUYER B; ELLERS B. Childhood injuries in the United States: mortality, morbidity, and
cost. American Journal of Diseases of Children 144: 649–652. 1990. HAMM RJ; O'DELL DM; PIKE BR; LYETH B. Cognitive impairment following traumatic
brain injury: the effect of pre- and post-injury administration of scopolamine and MK-801. Brain Research 1(4): 223-226. 1993.
HARDINGHAM GE; BADING H. Coupling of extrasynapticNMDAreceptors to a CREB
shut-off pathway is developmentally regulated. Biochimica et biophysica acta 1600: 148 –153. 2002.
HARDINGHAM GE, BADING H. The yin and yang of NMDA receptor signaling. Trends
in Neurosciences 26: 81– 89. 2003. HARDINGHAM GE; FUKUNAGA Y; BADING H. Extrasynaptic NMDARs oppose
synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature Neuroscience 5: 405– 414. 2002.
HEATH DL; VINK R. Concentration of brain free magnesium following severe brain
injury correlates with neurologic motor outcome. Journal of Neurosurgery 6(6): 504-509. 1999.
HOLMIN S. Intracerebral inflammation after human brain contusion. Journal
Neurosurgery. Brain Research 951: 191-201. 2002. HORAK M; CHANG K; WENTHOLD RJ. Masking of the endoplasmic reticulum retention
signals during assembly of the NMDA receptor. The Journal of Neuroscience 28(13): 3500-3509. 2008.
IKONOMIDOU C; TURSKI L. Why did NMDA receptor antagonists fail clinical trials for
stroke and traumatic brain injury? Lancet Neurology 1: 383–386. 2002.

78
JENSEN JB; PICKERING DS; SCHOUSBOE A. Depolarization-induced realese of 3 H D- aspartate from GABAergic neurons caused by reversal of glutamate transporters. International Journal of Developmental Neuroscience 18: 309-315. 2000.
JUNQUE C. Neuropsychological sequelae of head injury. Revista de Neurologia
28(4): 423-9. 1999. KANAI Y; HEDIGER MA. Primary structure and functional characterization of a high-
affinity glutamate transporter. Nature 360: 467-471. 1992. KATZ DI; WHITE DK; ALEXANDER MP; KLEIN RB. Recovery of ambulation after
traumatic brain injury. Archives of Physical Medicine and Rehabilitation 85: 865–869. 2004.
KATZ DI; ALEXANDER MP. Traumatic brain injury: Predicting course of recovery and
outcome for patients admitted to rehabilitation. Archives of Neurology 51: 661–670. 1994.
KIM J; AVANTS B; PATEL S; WHYTE J; COSLETT BH; PLUTA J; DETRE, JA; GEE
JC. Structural consequences of diffuse traumatic brain injury: A large deformation tensor-based morphometry study. Neuroimage 39: 1014-1026. 2008.
KREUTZ MR; BÖCKERS TM; BOCKMANN J; SEIDENBECHER CI; KRACHT B; VORWERK CK; WEISE J; SABEL BA. Axonal injury alters alternative splicing of the retinal NR1 receptor: the preferential expression of the NR1b isoforms is crucial for retinal ganglion cell survival. The Journal of Neuroscience 18(20): 8278-8291. 1998.
KUHTZ-BUSCHBECK JP; HOPPE B; GOLGE M; DREESMANN M; DAMM-STUNITZ U; RITZ A. Sensorimotor recovery in children after traumatic brain injury: analyses of gait, gross motor, and fine motor skills. Developmental Medicine and Child Neurology 45: 821–828. 2003.
KUHTZ-BUSCHBECK JP; STOLZE H; GÖLGE M; RITZ A. Analyses of Gait, Reaching,
and Grasping in Children After Traumatic Brain Injury. Archives of Physical Medicine and Rehabilitation 84: 424-430. 2003.
LENT R. Cem bilhões de neurônios . São Paulo: Atheneu, 226-246 pp., 2001. LAEMMLI UK. Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature Neuroscience 227(5259): 680-685. 1970 LAPIN LP. Stimulant and convulsant effects of Kynurenines injected into brain ventricles
in mice. Journal of Neural Transmission 42: 37-43. 1978.

79
LEE B; BUTCHER GQ; HOYT KR; IMPEY S; OBRIETAN K; Activitydependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. Journal of Neuroscience 25: 1137–1148. 2005.
LEINHASE I; HOLERS M; THURMAN JM; HARHAUSEN D; SCHMIDT OI; PIETZCKER
M; TAHA ME; RITTIRSCH D; HUBER-LANG M; SMITH WR; WARD PA; STAHEL PF. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. Neuroscience 7(55): 1471-2207. 2006.
LEVINE B; FUJIWARA E; O’CONNOR C. In Vivo Characterization of Traumatic Brain
Injury Neuropathology with Structural and Functional Neuroimaging. Journal of Neurotrauma 23: 1396–1411. 2006.
LEWINE JD; DAVIS JT; BIGLER ED; THOMA R; HILL D; FUNKE M; SLOAN JH; HALL
S; ORRISON WW. Objective documentation of traumatic brain injury subsequent to mild head with MEG. SPECT and MRI. The Journal of Head Trauma Rehabilitation 22 (3): 141-155. 2007.
LEWIN L; MATTSON MO; SELLSTROM A. Differences in the release of L- glutamate
and D- aspartate from primary neuronal chick cultures. Neurochemical Research 21: 79-85. 1996.
LINDNER MD; PLONE MA; CAIN CK; FRYDEL B; FRANCIS JM; EMERICH DF;
SUTTON RL. Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. Journal of Neurotrauma 15: 199–216. 1998.
LIPTON SA; ROSENBERG MD. Excitatory amino acids as a final common pathway for neurologic disorders. The New England Journal of Medicine 330: 613-622. 1994.
LIU Y; WONG TP; AARTS M. ROOYAKKERS A, LIU L; LAI T WU DC; LU J; TYMIANSKI M; CRAIG AM; WANG YT. NMDAReceptor Subunits Have Differential Roles in Mediating Excitotoxic Neuronal Death Both In Vitro and In Viv. The Journal of Neuroscience 27(11): 2846 –2857. 2007.
LIVINGSTON DH; LAVERY RF; MOSENTHAL AC. Recovery at one year following
isolated traumatic brain injury: a Western Trauma Association prospective multicenter trial. Journal of Trauma 59: 1298–1304. 2005.
LONGUEMARE MC; SWANSON RA. Excitatory amino acid release from astrocytes
during energy failure by reversal of sodium-dependent uptake. Journal of Neuroscience Research 40: 379-386. 1995.
MANREZA LA. Traumatismo cranioencefálico. In: Tratado das doenças neurológicas
(Melo-souza SE, ed.). Guanabara Koogan, Rio de Janeiro, pp. 509-511. 2000.

80
MANREZA, MLG. Epilepsia na infância e na adolescência . São Paulo: Lemos, 211
p., 2003. MAXWELL WL; POVLISHOCK JT; GRAHAM DL. A mechanistic analysis of
nondisruptive axonal injury: a review. Journal of Neurotrauma 14: 419–440. 1997. MARR D. Simple memory: a theory for archicortex. Philos. Philosophical transactions
of the Royal Society of London. Series B, Biologica l sciences 262: 23–81. 1971. MCALLISTER TW; SPARLING MB; FLASHMAN LA; SAYKIN AJ. Neuroimaging findings
in mild traumatic brain injury. Journal of Neuroscience Research 23: 775–791. 2001.
MCINTOSH TK. Neurochemical sequelae of traumatic brain injury: therapeutic
implications. Cerebrovascular and Brain Metabolism Reviews 6: 109-162. 1994. MORECRAFT RJ; HERRICK JL; STILWELL-MORECRAFT KS. Localization of arm
representation in the corona radiata and internal capsule in the non-human primate. Brain Research 125: 176–198. 2002.
MUIZELAAR JP; MARMAROU AAF. Cerebral blood flow and metabolism in severely
head injured children. Journal of Neurosurgery 71: 63-71. 1989. MUÑOZ-CÉSPEDES JM; PÁUL-LAPEDRIZA N; PELEGRÍN-VALERO C;TIRAPU-
USTARROZ J. Factores de prognóstico em los traumatismos craneoencefalicos. Revista de Neurologia 32(4): 351-364. 2001.
MUNSON S; SCHROTH E; ERNST M. The Role of Functional Neuroimaging in
Pediatric Brain Injury. Pediatrics 117: 1372-1381. 2006. MUSCARA F; CATROPPA C; EREN S; ANDERSON V. The impact of injury severity on
long-term social outcome following paediatric traumatic brain injury. Neuropsychological Rehabilitation 1: 1. 2008.
MITCHELL HL; FRISELLA WA; BROOKER RW; YOON KW. Attenuation of traumatic
cell death by an adenosine. A¹ agonist in rat hippocampal cells. Neurosurgery 36(5): 1003-1007. 1995.
NAKANISHI S. Molecular Diversity of Glutamate Receptors and Implications for Brain
Function. Science 258: 597-603. 1992. NIEMEIER JP; MARWITZ JH; LESHER K; WALKER WC; BUSHNIK T. Gender
differences in executive functions following traumatic brain injury. Neuropsychological Rehabilitation 17: 293–313. 2007.

81
NOVELLI A; REILLY JA; LYSKO PG. Glutamate becomes neurotoxic via the N-methyl- D- aspartate receptor when intracellular energy levels are reduced. Brain Research 451: 205-212. 1988.
NUDO RJ. Recovery after damage to motor cortical areas. Current Opinion in
Neurobiology 9: 740–747. 1999. NUDO RJ. Plasticity. NeuroRx: the journal of the American Society for
Experimental NeuroTherapeutics 3: 420–427. 2006. OBRENOVITCH TP; URENJAK J; ZILKHA E. Excitotoxicity in neurological disordens-
the glutamate paradox. International journal of developmental neuroscience : the official journal of the International Society f or Developmental Neuroscience 18: 281-287. 2000.
OBRIST WD; LANGFITT TW; JAGGI JL; CRUZ J; GENNARELLI TA. Cerebral blood
flow and metabolism in comatose patients with acute head injury. Journal of Neurosurgery 61(22): 241-253. 1984.
ODDY M; COUGHLAN T; TYERMAN A; JENKINS D. Social adjustement after closed
head injury: A further follow-up seven years after injury. Journal of neurology, Neurosurgery and Psychiatry 48: 564–568. 1985.
O'DELL DM; HAMM RJ. Chronic postinjury administration of MDL 26,479 (Suritozole), a
negative modulator at the GABAA receptor, and cognitive impairment in rats following traumatic brain injury. Journal of Neurosurgery 83(5): 878-83. 1995.
OLNEY JW; HO OL. Brain damage in infant mice following oral intake of glutamate,
aspatate or cysteine. Nature 227: 609-611. 1969. OLVERMAN HJ; JONES AW; MEWETT KN. Structure/activity relations of N-methyl-D-
aspartate receptor ligands as studied by their innibition of 3H D-amino-5- phosphopentanoic acid binding in rat brain membranes. Neuroscience 26: 17-31. 1998.
PANTER S; FADEN A. Pretreatment with NMDA antagonists limits release of excitatory
amino acids following traumatic brain injury. Neuroscience Letters 136(2): 165-8. 1992.
PAPADIA S; STEVENSON P; HARDINGHAM NR; BADING H; HARDINGHAM GE;
Nuclear Ca2_ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. Journal of Neuroscience 25: 4279–4287. 2005
PASCUAL-LEONE A; AMEDI A; FREGNI F; MERABET LB. The plastic human brain
cortex. Neuroscience 28: 377-401. 2005.

82
PÉREZ-OTAÑO I; EHLERS MD. Homeostatic plasticity and NMDA receptor trafficking. Neurosciences 28(5): 229-238. 2005.
PERRETT SP; RUIZ BP; MAUK MD. Cerebellar cortex lesions disrupt learning-
dependent timing of conditioned eyelid responses. The Journal of Neuroscience 13: 1708–1718. 1993.
PETERSON GL. A simplification of the protein assay method of Lowry et al. which is
more generally applicable. Analytical Biochemistry 83(2): 346-356. 1977. PIOT-GROSJEAN O; WAHL F; GOBBO O; STUTZMANN JM. Assessment of
Sensorimotor and Cognitive Deficits Induced by a Moderate Traumatic Injury in the Right Parietal Cortex of the Rat. Neurobiology of Disease 8: 1082-1093. 2001.
POVLISHOCK JT; CHRISTMAN CW. The pathobiology of traumatically induced axonal
injury in animals and humans: a review of current thoughts. Journal of Neurotrauma 12: 555–564. 1995.
RABACCHI S; BAILLY Y; DELHAYE- BOUCHAUD N. Involvement of the N- methyl- D
aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science 256: 1823-1825. 1992.
RAO VLR; DOGAN A; TODD KG; BOWER KK; DEMPSEY RJ. Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Research 911: 96-100. 2001.
RAO VLR; RAO AM; DOGAN A; BOWEN KK; HATCHER J; ROTHSTEIN JD; DEMPSEY RJ. Glial glutamate transporter GLT-1 down-regulation precedes delayed neuronal death in gerbil hippocampus following transient global cerebral ischemia. Neurochemistry 36(6): 531-537. 2000.
REES PM. Contemporary issues in mild traumatic brain injury. Review article. Archives
of physical medicine and rehabilitation 84: 1885-1894. 2003. RICHARDSON MR; SUN D; BULLOCK RM. Neurogenesis After Traumatic Brain Injury.
Neurosurgery Clinics 18: 169-181. 2007. ROMANSKI LM; GIGUERE M; BATES JF; GOLDMAN-RAKIC PS. Topographic
organization of medial pulvinar connections with the prefrontal cortex in the rhesus monkey. The Journal of Comparative Neurology 379: 313–332. 1997.
SAKAI K; HOMMA H; LEE JA. Emergence of D- aspartic acid en the differentiating
neurons of the rat central nervous system. Brain Research 808: 65-71. 1998. SCHELL MJ; COOPER OB; SNYDER SH. D- aspaartate localizations imply neuronal
and neuroendocrine roles. Proceedings of the National Academy of Sciences of the United States of America 94: 2013-2018. 1997.

83
SCHUMANN ; ALEXANDROVICH GA; BIEGON A; YAKA R. Inhibition of NR2B
Phosphorylation Restores Alterations in NMDA Receptor Expression and Improves Functional Recovery following Traumatic Brain Injury in Mice. Journal of Neurotrauma 25: 945–957. 2008.
SCHURR A; PAYNE RS; TSENG MT; GOZAL E; GOZAL D. Excitotoxic preconditioning
elicited by both glutamate and hypoxia and abolished by lactate transport inhibition in rat hippocampal slices. Neuroscience 307: 151–154. 2001.
SHADMEHR R; BRASHERS-KRUG T. Functional stages in the formation of human
long-term motor memory. The Journal of Neuroscience 17: 409–419. 1997. SHADMEHR R; HOLCOMB HH. Neural correlates of motor memory consolidation.
Science 277: 821–825. 1997. SHELTON FN; REDING MJ. Effect of lesion location on upper limb motor recovery after
stroke. Stroke 32: 107–112. 2001. STORK T; SCHLTE S; HOFMANN K. Structure, expression, and functional analysis of a
Na dependent glutamate/aspartate transporter from rat brain. Proceedings of the National Academy of Sciences of the United States o f América 89: 10955-10959. 1992.
TEMPLE M; O’LEARY D; FADEN A. The role of glutamate receptors in the
pathophysiology of traumatic brain injury. John Wiley & Sons 1: 87-113. 2001.
VEIGA NETO ER; SEGURA DCA. O cerebelo e as aferências da propiocepção inconsciente/ The cerebellum and the unconscious proprioception inputs. Arquivos Ciências Saúde UNIPAR 6(3): 145-149. 2002.
XIONG Y; MAHMOOD A; LU D; QU C; GOUSSEV A; SCHALLERT T; CHOPP M. Role of gender in outcome after traumatic brain injury and therapeutic effect of erythropoietin in mice. Brain Research 1185: 301-312. 2007.
XU GP; DAVE KR; VIVERO R; SCHMIDT-KASTNER R; SICK TJ; PEREZ-PINZON MA.
Improvement in neuronal survival after ischemic preconditioning in hippocampal slice cultures. Brain Research 952: 153–158. 2002
YAKOVLEV AG; KNOBLACH SM; FAN L; FOX GB; GOODNIGHT R; FADEN AI.
Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. Journal Neuroscience 17: 7415-7424. 1997.
WANG GJ; CHUNG HJ; SCNUER J. Dihydrokainate-sensitive neuronal glutamate
transport is required for protection of rat cortical neurons in culture against

84
synaptically release glutamate. The European Journal of Neuroscience 10: 2523-2531. 1998.
WATT AJ; VAN ROSSUM MC; MACLEOD KM; NELSON SB; TURRIGIANO GG.
Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26: 659–670. 2000.
WIESE H; STUDE P; NEBEL K; OSENBERG D; VOLZKE V; ISCHEBECK W; STOLKE D; DIENER HC; KEIDEL M. Impaired movement-related potentials in acute frontal traumatic brain injury. Clinical Neurophysiology 115(2): 289-98. 2004.
WOLOSKER H; D’ANIELLO A; SNYDER SH. D-aspartate disposition in neuronal and endocrine tissues: ontogeny, biosynthesis and release. Neurocience 100: 183-189. 2000.
ZOHAR O; SCHREIBER C; GETSLEV V; SCHWARTZ JP; MULLINS PG; PICK CG. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 118: 949-955. 2003.

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo








![Anafilaxia induzida em camundongos pelo veneno do peixe ... · Anafilaxia induzida em camundongos pelo veneno do peixe Thalassophryne nattereri. [Tese (Doutorado em Imunologia)] São](https://img.document.onl/doc/110x75/5e50954efd44283c861df033/anafilaxia-induzida-em-camundongos-pelo-veneno-do-peixe-anafilaxia-induzida.jpg)




















