Embed Size (px)
Citation preview

I
UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de fevereiro de 1808
Monografia
Med B51
Caracterização clínico-epidemiológica da fibrose cística
em um centro pediátrico de referência do Nordeste
brasileiro
Milena Silva Reis
Salvador (Bahia)
Abril, 2016

II
FICHA CATALOGRÁFICA (elaborada pela Bibl. Tatiana Bonfim Souza, da Bibliotheca Gonçalo Moniz : Memória da Saúde
Brasileira/SIBI-UFBA/FMB-UFBA)
R375
Reis, Milena Silva
Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico de referência do Nordeste brasileiro / Milena Silva Reis. (Salvador, Bahia): MSR, Reis , 2016
Viii, 47 p.: il.
Monografia, como exigência parcial e obrigatória para conclusão do Curso de Medicina da Faculdade de Medicina da Bahia (FMB), da Universidade Federal da Bahia (UFBA)
Professor orientador: Edna Lúcia Santos de Souza
Palavras chaves: 1.Fibrose Cística. 2.Crianças e adolescentes. 3. Perfil clínico-
epidemiológico. I. Souza, Edna Lúcia Santos. II. Universidade Federal da Bahia.
Faculdade de Medicina da Bahia. III. Título.
CDU: 616-036.22

III
UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de fevereiro de 1808
Monografia
MED B51
Caracterização clínico-epidemiológica da fibrose
cística em um centro pediátrico de referência do
Nordeste brasileiro
Milena Silva Reis
Professor orientador: Edna Lúcia Souza
Monografia de Conclusão do
Componente Curricular MED-
B60/2015.1, como pré-requisito
obrigatório e parcial para conclusão
do curso médico da Faculdade de
Medicina da Bahia da Universidade
Federal da Bahia, apresentada ao
Colegiado do Curso de Graduação
em Medicina.
Salvador (Bahia)
Abril, 2016

IV
Monografia: Caracterização clínico-epidemiológica da fibrose cística em
um centro pediátrico de referência do Nordeste brasileiro, de Milena Reis.
Professor orientador: Edna Lucia Souza
COMISSÃO REVISORA Edna Lucia Santos de Souza, Professora do Departamento de Pediatria da
Faculdade de Medicina da Bahia da Universidade Federal da Bahia
Margarida Célia Costa Neves, Professora do Departamento de Medicina Interna
e Apoio Diagnóstico da Faculdade de Medicina da Bahia da Universidade Federal
da Bahia
Lauro Antonio Porto, Professor do Departamento de Medicina Preventiva e
Social da Faculdade de Medicina da Bahia da Universidade Federal da Bahia
Membro suplente Regina Terse Trindade Ramos, Professor do Departamento Pediatria da Faculdade de
Medicina da Bahia da Universidade Federal da Bahia

V
“A utopia está lá no horizonte. Me aproximo dois passos,
ela se afasta dois passos. Caminho dez passos e o horizonte
corre dez passos. Por mais que eu caminhe, jamais
alcançarei. Para que serve a utopia? Serve para isso: para
que eu não deixe de caminhar.” (Eduardo Galeano).

VI
Aos Meus Pais, Edna e José Reis, e a
todos aqueles que estiveram ao meu
lado nesta longa caminhada.

VII
EQUIPE Milena Silva Reis, Faculdade de Medicina da Bahia/UFBA. Correio-e:
Professor orientador: Edna Lucia Santos de Souza. Correio-e:
Paloma Horejs, discente da Faculdade de Medicina da Bahia/UFBA;
Bianca Sampaio, discente da Faculdade de Medicina da Bahia/UFBA;
Carolina Godoy Almeida, nutróloga do Complexo Hospitalar Universitário
Professor Edgar Santos;
Ana Paula de Brito Aguiar, nutricionista do Complexo Hospitalar Universitário
Professor Edgar Santos;
Renata Lúcia Ferreia Lima, professora adjunto do Instituto de Biologia
Universidade Federal da Bahia;
Laís Ribeiro Mota, biomédica e mestre em Genética e Biodiversidade do
Laboratório de Gnética Humana e Mutagênese (LGHM) – IBIO/UFBA;
Tatiane da Anunciação Ferreira, pneumopediatra do Complexo Hospitalar
Universitário Professor Edgar Santos.
Marlene Amália da Silva, técnica de laboratório do Complexo Hospitalar
Universitário Professor Edgar Santos;
INSTITUIÇÕES PARTICIPANTES
UNIVERSIDADE FEDERAL DA BAHIA
Faculdade de Medicina da Bahia (FMB) Complexo Hospital Universitário Professor Edgard Santos (HUPES) Ambulatório Multidisciplinar de Fibrose Cística - AMFC
FONTES DE FINANCIAMENTO
1. Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB);
2. Recursos próprios.

VIII
AGRADECIMENTOS
À Professora orientadora, Doutora Edna Lucia Santos de Souza, pela
presença constante e todo apoio acadêmico e pessoal, além da disponibilidade
e boa vontade.
À minha Colega Paloma Horejs, pela colaboração no levantamento e análise
estatística dos dados;.
À minha Colega Bianca Sampaio, pela colaboração no levantamento dos dados
do estudo;
À minha mãe, Edna Reis, que me auxíliou na construção dos gráficos e tabelas,
além de todo apoio durante essa caminhada.
Ao meu pai, José Reis, por todo amor e carinho, além de apoio acadêmico na
minha vida.
Ao meu namorado, Petrônio Cangussu, pelo seu companheirismo, compreensão
e ajuda na formatação do trabalho.
Aos meus amigos, que me apoiaram incondicionalmente e sempre estiveram ao
meu lado neste processo.
Aos Doutores Margarida Célia Costa Neves e Lauro Antônio Porto, membros
da Comissão Revisora desta Monografia, os quais se dispuseram a fazer parte
da minha banca revisora, com toda boa vontade, e corrigiram prontamente a
minha Monografia.
À equipe multiplicinar do AMFC, pelo trabalho e pela dedicação de anos, sem
os quais eu não poderia desenvolver esse projeto.
Aos pacientes estudados, os quais foram e continuam sendo a motivação para
tantas pesquisas, a fim de melhorar a qualidade de vida deles.

1
SUMÁRIO
LISTA DE SIGLAS
2 ÍNDICE DE FIGURAS, GRÁFICOS, QUADROS E TABELAS 3 I. RESUMO 4 II. OBJETIVO 5 III. FUNDAMENTAÇÃO TEÓRICA 6 IV. METODOLOGIA 15 V. RESULTADOS 19
VI. DISCUSSÃO
26
VII. CONCLUSÕES
VIII. SUMMARY
IX. REFERÊNCIAS
X. APÊNDICES
31
32
33
APÊNDICE I: Formulário de Coleta de Dados 40
APÊNDICE II: Termo de Consentimento Livre e Esclarecido (TCLE)
XI. ANEXOS
44
ANEXO I: Parecer do Comitê de Ética em Pesquisa do HUPES 46

2
LISTA DE SIGLAS
AMFC Ambulatório Multidisciplinar de Fibrose Cística
AMN Ambulatório Magalhães Neto
CFF Cystic Fibrosis Foundation
CFTR Cystic Fibrosis Transmembrane Conductance Regulator
DP Desvio Padrão
EUA Estados Unidos da América
HUPES Hospital Universitário Professor Edgar Santos
FC Fibrose Cística
IMC/I Índice de massa corpórea por idade
MSSA Staphylococcus aureus meticilino sensível
MRSA Staphylococcus aureus meticilino resistente
PA Pseudomonas aeruginosa
RNAm Ácido ribonucleico mensageiro
OMS Organização Mundial de Saúde
PCR Reação em Cadeia da Polimerase
P/I Peso por idade
REBRAFC Registro Brasileiro de Fibrose Cística
VEF1 Volume Expiratório Forçado no primeiro segundo
TCLE Termo de Consetimento Livre e Esclarecido

3
ÍNDICE DE FIGURAS, GRÁFICOS, QUADROS E TABELAS
GRÁFICOS GRÁFICO 1. Procedência dos Pacientes com Fibrose Cística do Ambulatório
Multidisciplinar de Fibrose Cística. Salvador, 2016.
GRÁFICO 2. Faixas etárias dos pacientes admitidos no estudo. Salvador, 2016.
GRÁFICO 3. Idade de manifestação dos primeiros sintomas. Salvador, 2016.
GRÁFICO 4. Idade dos pacientes no momento do diagnóstico. Salvador, 2016.
GRÁFICO 5. Frequência das médias de valores de cloro do suor dos pacientes no
diagnóstico. Salvador, Bahia.
GRÁFICO 6. Presença da mutação p.Phe508del na população estudada. Salvador,
2016.
19
20
20
21
23
24
TABELAS TABELA 1. Frequências relativas e absolutas dos sinais e sintomas sugestivos para
o diagnóstico de Fibrose Cística de pacientes do Ambulatório Multidisciplinar de
Fibrose Cística.
TABELA 2. Classificações dos pacientes quanto ao estado nutricional, utilizando
o z escore -2 como ponto de corte (OMS, 2011), no diagnóstico. Salvador, 2016.
TABELA 3. Micro-organismos isolados em espécimes respiratórios de pacientes
com Fibrose Cística do Ambulatório Multidisciplinar de Fibrose Cística entre
2008 e 2015. Salvador, 2016.
22
22
25

4
I. RESUMO
CARACTERIZAÇÃO CLÍNICO-EPIDEMIOLÓGICA DA FIBROSE CÍSTICA
EM UM CENTRO DE REFERÊNCIA DO NORDESTE BRASILEIRO. A fibrose
cística (FC) é uma doença genética que apresenta diversas mutações no gene que codifica
uma proteína transmembrana, a Cystic Fibrosis Transmembrane Conductance Regulator
(CFTR), essencial no transporte de íons através da célula. As alterações na proteína levam
ao comprometimento multissistêmico, principalmente nos tratos respiratório e digestivo.
São escassos os trabalhos sobre a FC no Nordeste do Brasil. Objetivo: Descrever as
características clínico-epidemiológicas de crianças e adolescentes com fibrose cística
acompanhados em um centro de referência de Salvador-BA. Métodos: Foi realizado um
estudo seccional. Após a inclusão, foi preenchido um formulário para os dados clínico-
epidemiológicos, seguido de coleta de sangue para investigação da mutação p.Phe508del
e obtidas amostras de escarro para determinar colonização bacteriana do trato respiratório.
Resultados: Foram incluídos 59 pacientes, 52,5% do sexo masculino, todos não brancos.
As medianas de idade dos pacientes na admissão do estudo, início dos sintomas e no
diagnóstico foram: 123, 5 e 61 meses, respectivamente. O tempo mediano entre início
dos sintomas e o diagnóstico foi 36 meses. Os dados clínicos que sugeriram o diagnóstico
mais frequentes foram: manifestações respiratórias, os relacionados à insuficiência
pancreática e ao retardo do crescimento. Havia desnutrição em 36,4% dos pacientes no
momento do diagnóstico. p.Phe508del foi identificada em 37,2% dos pacientes,
frequência alélica 25,4% (27/106). De 2008 a 2015, os principais microorganismos
isolados foram: Staphylococcus aureus meticilino sensível (MSSA) – 25 a 80%;
Pseudomonas aeruginosa (PA) – 11,5 a 36,4%; Haemophilus sp. – 6,7 a 28,6%. A idade
mediana da primeira colonização por PA foi 60 meses. Conclusão: A população estudada
foi composta apenas por não brancos, o diagnóstico da doença foi tardio e houve baixa
frequência da mutação p.Phe508del.
Palavras-chave: 1. Fíbrose Cística; 2. Crianças e adolescentes; 3. Perfil clínico-
epidemiológico

5
II. OBJETIVOS
PRINCIPAL
Descrever as características clínico-epidemiológicas de crianças e adolescentes
com fibrose cística acompanhadas em um centro de referência de Salvador-BA.
SECUNDÁRIOS
Em pacientes com fibrose cística acompanhados no Ambulatório Multidisciplinar de
Fibrose Cística do Complexo Hospitalar Universitário Professor Edgard Santos:
1. Descrever as características demográficas;
2. Identificar a idade do diagnóstico da doença;
3. Determinar a idade do início dos primeiros sintomas, calculando o tempo entre os
primeiros sintomas e o diagnóstico;
4. Descrever as principais manifestações clínicas que motivaram o diagnóstico;
5. Classificar o estado nutricional no momento do diagnóstico;
6. Determinar a frequência da mutação p.Phe508del na população estudada;
7. Registrar o histórico de colonizações bacterianas do trato respiratório.
8. Identificar a idade dos pacientes no primeiro isolamento de Pseudomonas
aeruginosa.

6
III FUNDAMENTAÇÃO TEÓRICA
A fibrose cística (FC) é uma doença genética, autossômica recessiva, cuja
disfunção ocorre por mutações no gene localizado no braço longo do cromossomo 7, o
qual tem a propriedade de codificar um RNAm que transcreve uma proteína
transmembrana reguladora de transporte iônico, chamada de CFTR ( Cystic Fibrosis
Transmembrane Conductance Regulator). Esta proteína é encontrada na membrana
apical de células epiteliais do trato respiratório, de glândulas submucosas, do pâncreas
exócrino, do fígado, dos ductos sudoríparos e do trato reprodutivo, entre outros sítios,
(Reis et al., 1998) e é essencial para o transporte de íons através da membrana celular,
estando envolvida na regulação do fluxo de cloro, sódio e água.
Os estudos sobre a FC têm se ampliado, principalmente por essa ser a mais
importante doença hereditária, potencialmente fatal e incidente na raça branca (Ribeiro et
al., 2002). O conhecimento da sua fisiopatogenia, bem como a identificação e
sequenciamento do gene dessa doença permitiram novas formas de aconselhamento
genético e o tratamento de suas complicações. Contudo, existem poucos trabalhos
analisando a população de fibrocísticos no Brasil e em outros países em desenvolvimento.
Uma vez que o tratamento e as intervenções de saúde pública são, em sua maioria,
baseados em dados internacionais, as peculiaridades da população brasileira geralmente
não são levadas em conta, o que pode interferir, por exemplo, na escolha de medicamentos
fornecidos à população pela Rede Pública de Saúde (Alvarez et al., 2004).
A incidência observada entre os europeus e seus descendentes varia entre 1:2.000
e 1:5.000, nascidos vivos. Já entre os africanos é de 1:40.000 (Hamosh et al., 1998). No
Brasil, a incidência estimada é de 1:10.000 nascidos vivos (Bernadino et al., 2000).
Estudos já foram realizados em alguns estados brasileiros: estimativa da incidência de FC
no Estado do Rio Grande do Sul é de 1:2.000 nascidos vivos (Lemos et al., 2004), em São
Paulo 1:32.258, Santa Catarina 1:12.195, Paraná 1:6.803, Minas Gerais 1:21.277 nascidos
vivos (Raskin et al., 1999). No Rio de Janeiro, em estudo de Cabello et al. (1999) a
incidência foi estimada em 1: 6.902 nascidos vivos. A incidência na Bahia é
desconhecida. A inclusão da FC na triagem neonatal do Estado da Bahia, a partir de 2013,
permitirá conhecer esta frequência. Deste modo, estudos são necessários para avaliar as

7
características clínicas e epidemiológicas da população da região Nordeste do Brasil e
com isso determinar condutas terapêuticas mais adequadas à população nordestina.
Histórico e genética:
Em 1905, Landsteiner fez a primeira descrição anatomopatológica da FC em
recém-nascido falecido no quinto dia de vida devido a íleo meconial (Ribeiro et al., 2002).
Essa doença tornou-se conhecida cientificamente em 1938 através de um estudo
publicado por Dorothy H. Andersen, da Universidade de Columbia. A cientista realizou
a primeira descrição detalhada da FC, bem como das alterações produzidas nos órgãos.
Di’Santagnese e colaboradores documentaram, em 1953, o excesso de sódio e cloro no
suor, o que constituiu a chave para o desenvolvimento do teste do suor, ainda hoje o
principal exame utilizado para o diagnóstico da FC (Firmida et al., 2011). Até o momento,
o método de Gibson & Cooke (Gibson, 1959), em que se emprega o estimulo cutâneo da
face palmar do antebraço através da iontoforese com pilocarpina, continua sendo o
método mais usado em todo o mundo (Firmida et al., 2011).
Concomitante ao surgimento do teste diagnóstico, houve grande empenho de
cientistas em identificar o possível gene responsável pela FC. Os esforços culminaram
com a determinação do componente genético e de seu produto, a proteína CFTR (Cystic
Fibrosis Condutance Regulator). Nos últimos 15 anos, estudos com biologia molecular
permitiram melhor entendimento dessa entidade, já tendo sido descritas 2007 mutações
segundo o Cystic Fibrosis Mutation Database (2011), evidenciando a grande
heterogeneidade clínica da doença. As mutações podem ser divididas em 6 classes, de
acordo com suas alterações funcionais (O’Sullivan et al., 2009). A categorização está
representada no Quadro 1. As mutações classes I, II e III associam-se às formas mais
graves da FC, enquanto as classes IV, V, VI estão relacionadas a espectros mais brandos
da doença.

8
Quadro 1- Classes funcionais das mutações genéticas da fibrose cística. Adaptado de
Olveira G, Olveira C. Nutrición, fibrosis quística y aparato digestivo.Nutr Hosp.
2008;23(Supl. 2):71-86.
Classe
Funcional
Disfunção molecular em nível de CFTR Exemplos de mutações
I Ausência de produção G542X
II Defeito no processamento P.Phe508del
III Defeito na regulação do transporte iônico G551D
IV Defeito na condutância R334W
V Produção e transformação inadequada 3849+10kb C > T
VI Porção C-terminal ausente C1412X
A mutação mais frequentemente observada e estudada pertence à classe funcional
II e ocorre por uma deleção de três pares de bases, acarretando a perda de um aminoácido
(fenilalanina) na posição 508 (p.Phe508del) da proteína CFTR, o que impede seu
funcionamento adequado (Morral et al., 1994). Apesar do elevado número de mutações,
muitas são raras. Estima-se que a prevalência da mutação P.Phe508del em pacientes FC
em pacientes caucasianos seja de aproximadamente 70% (Bobadilla et al., 2002).
Contudo, sua frequência varia bastante de acordo com o grupo étnico (De Boeck et al.,
2014). No Brasil, esta mutação tem uma frequência que varia de 7,1% (Viana e Mesquita,
2003) a 52,6% (Firedrich et al., 2007) dos pacientes com FC, a depender do estado
estudado. As outras mutações para FC são menos frequentes e de prevalência variada
entre populações de diferentes origens étnicas e localizações geográficas. Menos de 20
mutações têm frequência mundial maior que 0,1%, cada.
Fisiopatogenia e Manifestações clínicas:
Como a CFTR está expressa em diversos órgãos, a FC pode ter manifestações
clínicas diversas. As diferentes classes das mutações causam alterações funcionais
distintas. Cada órgão que depende da proteína CFTR – pulmões, pâncreas, intestino,
glândulas sudoríparas e vasos deferentes – expressa esta disfunção de forma variada, de
acordo com a sensibilidade de cada um deles ao déficit funcional. Os tecidos dos canais
deferentes são os que mais exigem o funcionamento adequado da CFTR, seguidos das

9
glândulas sudoríparas e do pâncreas (Davis et al., 2006). As manifestações clínicas podem
ocorrer tanto precocemente (na infância), quanto na vida adulta.
O acometimento do trato respiratório associa-se com a maior morbidade e é causa
de morte em mais de 90% dos pacientes. Embora já tenham sido evidenciadas algumas
alterações pulmonares pré-natais, os pulmões são considerados praticamente normais ao
nascimento. Posteriormente, ocorre um ciclo de obstrução, infecção e inflamação que
acarreta danos progressivos a este órgão (Firmida et al., 2011). Na FC, devido à disfunção
da CFTR, a membrana apical do epitélio pulmonar torna-se impermeável ao cloro,
dificultando a sua saída. Como este eletrólito se acompanha normalmente de
movimentação de água, a hidratação da superfície celular fica comprometida. Para manter
a neutralidade elétrica, ocorre um influxo compensatório de sódio para as células, seguido
da água, o que acentua ainda mais a desidratação da superfície celular. O resultado deste
processo é o espessamento do muco, característica marcante desta doença, por este
motivo, denominada mucoviscidose (Farber et al., 1944). O espessamento da secreção
leva à obstrução das vias aéreas, comprometendo os mecanismos locais de defesa,
facilitando a ocorrência de infecções. Por isso, é comum que os pacientes fibrocísticos
apresentem infecção/colonização por diversas bactérias, as quais podem ser mais ou
menos predominantes a cada faixa etária (Gibson et al., 2003). De acordo com uma
publicação oficial da American College of Chest Physicians, de 2014, nos primeiros 5
anos de vida, os microorganismos predominantes são Staphylococcus aureus e
Haemophilus influenzae, embora algumas crianças possam ser infectadas por
Pseudomonas aeruginosa com semanas de nascimento. Na infância mais tardia, a
Pseudomonas aeruginosa torna-se a bactéria predominante (importante
morbimortalidade) (Ciofu et al., 2013). Em adultos, complexo de Burkholderia cepacia
Achromobacter xylosoxidans, Stenotrophomonas maltophilia também são encontradas
(Salsgiver et al., 2014). De todos esses microorganismos já citados, apenas a bactéria S.
aureus costuma ser patogênica em indivíduos imunocompetentes, sendo todos outros
considerados patógenos oportunistas (Gibson et al., 2003).
O comprometimento funcional pulmonar decorre da obstrução das vias aéreas e o
aprisionamento de ar, resultando em aumento na relação do volume residual com a
capacidade pulmonar total (Zemanick et al., 2011). Em estágios mais avançados da
doença, pode ocorrer fibrose intersticial, somando-se um componente restritivo à doença

10
pulmonar ocasionando a redução da capacidade pulmonar total (Firmida et al., 2011). O
envolvimento pulmonar e de seios da face são crônicos, com períodos de reagudizações
ou exacerbações: sinusites (muitas vezes associadas a polipose nasal), bronquites,
pneumonias e bronquectasias. A sintomatologia respiratória é geralmente constituída de
tosse crônica persistente, excesso de produção de escarro mucoso, muito espesso e, muitas
vezes, francamente purulento (Reis et al., 1998).
O comprometimento do pâncreas resulta na redução do conteúdo hídrico da
secreção pancreática e em maior acidez da mesma. Estas alterações contribuem para
obstrução dos ductos pancreáticos e autólise e fibrose do pâncreas. A intensidade deste
processo determina a progressão da doença pancreática. A lesão pancreática pode
começar intrauterinamente e continuar após o nascimento, na infância ou mais tarde, ate
que todo o tecido pancreático acinar seja destruído (Augarten et al., 2008). Os sinais de
insuficiência pancreática ocorrem, na maioria das vezes, bem cedo, sendo vista em,
aproximadamente, 90% dos pacientes com fibrose cística até o final do primeiro ano de
vida (Park et al., 1981). Alguns sintomas da insuficiência pancreática são: diarreia
crônica, má-absorção, esteatorreia, perda de apetite, baixo ganho de peso, com níveis
variáveis de má nutrição (Weintraub et al., 2009). A manifestação mais sugestiva de
disabsorção é a esteatorreia, a qual se caracteriza por fezes volumosas, oleosas e de odor
forte. Às vezes, os sinais clínicos de esteatorreia não são evidentes e a única manifestação
de disabsorção é a dificuldade de ganho ponderal.
O comprometimento nutricional é uma das manifestações iniciais mais comuns
da FC. Quando o paciente apresenta apenas sintomas inespecíficos, como a desnutrição,
poucas vezes inclui-se essa doença nas suspeitas diagnósticas. Supõe-se que o ganho
inadequado de peso seja causado por balanço energético negativo, resultante não só da
má absorção de nutrientes causada pela insuficiência pancreática exócrina, mas também
da doença respiratória crônica e do aumento da taxa de metabolismo basal, além da
ingesta calórica insuficiente. A insuficiência do pâncreas exócrina também pode se
manifestar por sinais e sintomas relacionados à perda proteica e à deficiência de vitaminas
lipossolúveis (Firmida et al., 2011). Uma vez iniciada a perda de função pancreática, não
há remissão.

11
Outras manifestações clínicas são: íleo meconial, manifestação mais precoce da
FC que ocorre em recém-nascidos; doença hepatobiliar; distúrbios hidroeletrolícos;
prolapso retal; infertilidade; hipertensão pulmonar com cor pulmonale; hipovitaminoses
A, D ou E; diabetes dellitus; osteoartropatia hipertrófica; dentre outras, de acordo com a
faixa etária do paciente (Firmida et al., 2011). Com o início da triagem neonatal, espera-
se que os sintomas mais graves sejam cada vez mais retardados e mais facilmente
controlados.
Diagnóstico:
Em mais de 90% dos casos, o diagnóstico da FC é baseado na suspeita clínica,
sendo confirmada por dois testes do suor positivos e/ou a identificação de duas mutações
no gene do cromossoma 7 (Wallis et al., 2006). A Cystic Fibrosis Foundation (CFF),
organização americana sem fins lucrativos com o objetivo de assegurar meios para cura
e controle da doença, além de melhorar a qualidade de vida destes pacientes, sintetizou
os critérios para o diagnóstico da doença, os quais estão sumarizados no Quadro II. A
confirmação do diagnóstico de FC deve ser baseada na presença de uma ou mais
características clínicas fenotípicas, história familiar de FC ou teste de triagem neonatal
(tripsina imunorreativa [IRT]) positivo (critérios A no quadro II) associados à evidência
laboratorial de disfunção da CFTR, documentada por níveis elevados de cloro no suor ou
identificação de mutações causadoras de FC em cada gene da CFTR, ou, ainda,
demonstração in vivo de anormalidades no transporte iônico no epitélio nasal, no teste da
diferença de potencial nasal (critérios B do quadro II) (Rosenstein et al., 1998).
Quadro II: Critérios para o diagnóstico de FC segundo a CFF. A presença de um dos
critérios A associado a um dos critérios B determina a doença. Adaptado de Rosenstein
et al., 1998.
Critérios A Critérios B
Uma ou mais manifestações fenotípicas
consistentes com FC
Duas dosagens de cloro no suor positivas
pelo método de Gibson e Cooke
História famíliar de FC Identificação de 2 mutações do gene FC
Teste de triagem neonatal positivo Demonstração de alteração no transporte
iônico no epitélio nasal
12
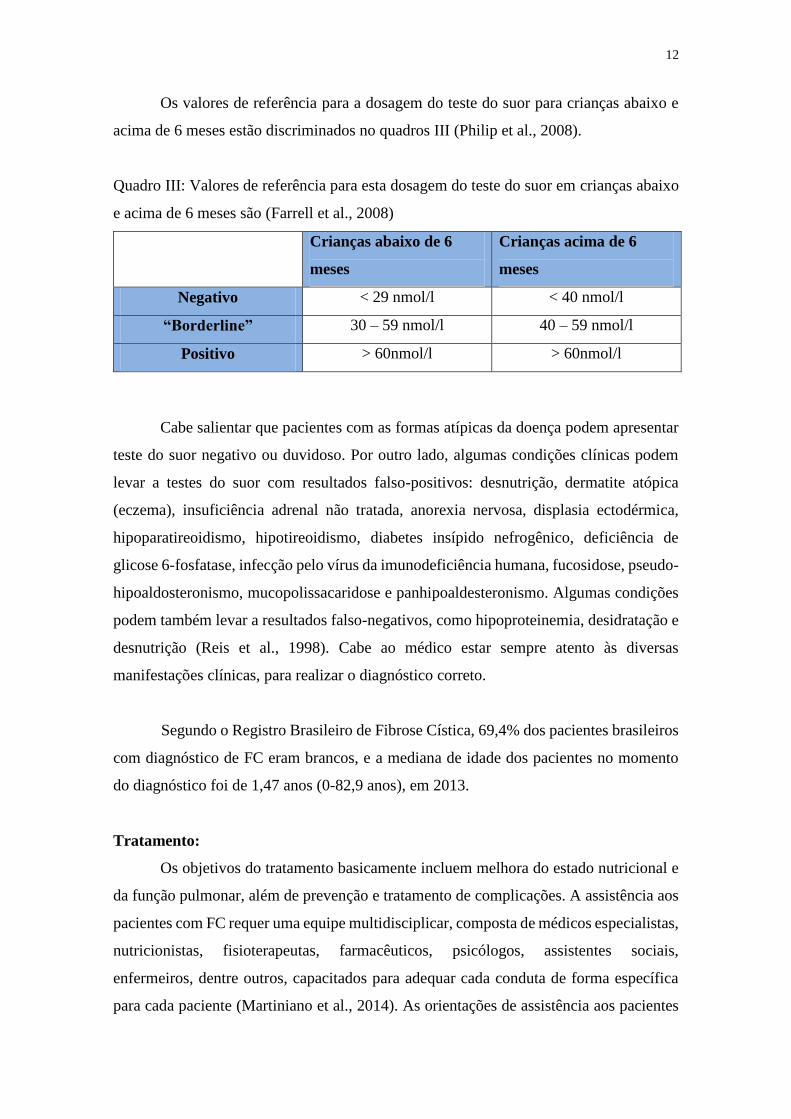
Os valores de referência para a dosagem do teste do suor para crianças abaixo e
acima de 6 meses estão discriminados no quadros III (Philip et al., 2008).
Quadro III: Valores de referência para esta dosagem do teste do suor em crianças abaixo
e acima de 6 meses são (Farrell et al., 2008)
Crianças abaixo de 6
meses
Crianças acima de 6
meses
Negativo < 29 nmol/l < 40 nmol/l
“Borderline” 30 – 59 nmol/l 40 – 59 nmol/l
Positivo > 60nmol/l > 60nmol/l
Cabe salientar que pacientes com as formas atípicas da doença podem apresentar
teste do suor negativo ou duvidoso. Por outro lado, algumas condições clínicas podem
levar a testes do suor com resultados falso-positivos: desnutrição, dermatite atópica
(eczema), insuficiência adrenal não tratada, anorexia nervosa, displasia ectodérmica,
hipoparatireoidismo, hipotireoidismo, diabetes insípido nefrogênico, deficiência de
glicose 6-fosfatase, infecção pelo vírus da imunodeficiência humana, fucosidose, pseudo-
hipoaldosteronismo, mucopolissacaridose e panhipoaldesteronismo. Algumas condições
podem também levar a resultados falso-negativos, como hipoproteinemia, desidratação e
desnutrição (Reis et al., 1998). Cabe ao médico estar sempre atento às diversas
manifestações clínicas, para realizar o diagnóstico correto.
Segundo o Registro Brasileiro de Fibrose Cística, 69,4% dos pacientes brasileiros
com diagnóstico de FC eram brancos, e a mediana de idade dos pacientes no momento
do diagnóstico foi de 1,47 anos (0-82,9 anos), em 2013.
Tratamento:
Os objetivos do tratamento basicamente incluem melhora do estado nutricional e
da função pulmonar, além de prevenção e tratamento de complicações. A assistência aos
pacientes com FC requer uma equipe multidisciplicar, composta de médicos especialistas,
nutricionistas, fisioterapeutas, farmacêuticos, psicólogos, assistentes sociais,
enfermeiros, dentre outros, capacitados para adequar cada conduta de forma específica
para cada paciente (Martiniano et al., 2014). As orientações de assistência aos pacientes

13
são baseados em evidências científicas de ensaios clínicos randomizados, consensos de
especialistas e "benchmarking", uma ferramenta usada pela Cystic Fibrosis Foundation
para identificar práticas de cuidado na saúde associadas aos melhores resultados, para que
essas técnicas sejam disseminadas com o objetivo de melhorar cuidado dos fibrocísticos
em todos os lugares (Schechter et al., 2012).
O tratamento para a doença pulmonar na FC tem como objetivo principal oferecer
a melhor conduta para os seguintes aspectos: o ciclo progressivo de obstrução por muco,
infecções pulmonares crônicas, resposta inflamatória excessiva (para manter a função
pulmonar), reduzir exacerbações respiratórias e diminuir a progressão da bronquiectasia
e do dano da estrutura pulmonar. O uso de drogas que diminuem a obstrução das vias
aéreas, os antibióticos e as medicações inaladas na nebulização para aumentar a liberação
de muco têm levado a significantes melhoras na sobrevida dos pacientes (Martiniano et
al., 2014). Os antibióticos podem ser administrados por via oral, intravenosa ou inalatória.
Para a infecção/colonização por pseudomonas, muito prevalente nos pacientes, o uso é
direcionado sob a forma de demanda (quando houver sinais de exacerbação), profilaxia
(antes que a bactéria seja detectada, para se evitar infecção/colonização) ou de
manutenção (por tempo prolongado, contínuo ou intermitente), além da erradicação da
colonização. Baseia-se nas definições de colonização (sem sinais e sintomas diretos de
infecção e dano pulmonar, podendo ser aguda ou crônica) e infecção (com as
manifestações, também classificada de acordo com o tempo) ou quando existirem
exacerbações das manifestações pulmonares (sinais e sintomas: aumento da tosse e da
produção de escarro, febre, anorexia e perda de peso, absenteísmo na escola ou trabalho,
diminuição da tolerância aos exercícios, diminuição na saturação de oxigênio, novos
achados à ausculta pulmonar, ao raio X de tórax e redução de mais de 10% no Volume
Expiratório Forçado no primeiro segundo – VEF1). As quinolonas orais e
aminoglicosídeos inalatórios têm sido utilizados. Para S. aureus e pseudomonas
resistentes, utiliza-se vancomicina e imipenen, respectivamente. Para Burkholderias
cepacia, podem ser administrados tobracina e amiloride, ambos por via inalatória. Outras
classes de drogas são os moduladores inflamatórios (corticoesteroides e os anti-
inflamatórios não hormonais), os broncodilatadores e os mucolíticos (dornase-alfa, salina
hipertônica e manitol). Além disso, a fisioterapia respiratória regular é recomendada
(Ribeiro et al., 2002).

14
A CFF, em colaboração com companhias biofarmacêuticas, vem desenvolvendo
e testando uma nova classe de drogas chamadas moduladores de CFTR, as quais têm
como alvo mutações de CFTR específicas. Essas drogas têm o potencial de modificação
do curso da doença, estendendo a expectativa de vida dos fibrocísticos em anos e,
possivelmente, décadas (Martiniano et al., 2014). Embora pouco frequente, o transplante
pulmonar bilateral é uma opção a ser considerada em pacientes com FC que desenvolvem
bronquiectasias e doença pulmonar terminal (Braun et al., 2011). Na terapia nutricional
da FC, o objetivo é manter a velocidade de crescimento normal durante a infância e o
peso adequado na vida adulta. Baseando-se em estudos que mostram que o crescimento
proporcional à idade está associado à melhora da função pulmonar, o objetivo
estabelecido para crianças menores de 2 anos é manter o índice Peso/Idade (P/I) maior
que o percentil 50 e para os maiores de 2 anos, o índice IMC/idade (IMC/I) maior que o
percentil 50. Para alcançar os objetivos do crescimento, as crianças com FC devem ter
uma dieta hipercalórica e rica em gordura. Suplementos orais ou por tubos gastrostomia
são frequentemente usados para atingir essas metas calóricas. Para pacientes insuficientes
pancréaticos, a terapia de reposição enzimática é utilizada, com o objetivo de manter a
absorção intestinal adequada de carboidratos, gorduras e proteínas. As enzimas são
tomadas antes de todas as refeições e calculadas de acordo com o peso do paciente ou
pela quantidade de gordura da dieta (Martiniano et al., 2014). É importante também
adicionar polivitamínicos, ricos em vitamina A, D e E, na dieta de fibrocísticos que não
estão ganhando peso. A FC, ainda que não tenha cura, apresenta melhora significativa
do prognóstico com o tratamento sintomático adequado (Tauber et al., 2002). Wang et al.
(2002) demonstraram que o diagnótico precoce pode diminuir a morbidade e Wagener et
al. (2003) destacaram a importância do tratamento precoce e adequado da insuficiência
pancreática e da desnutrição, assim como a fisioterapia e respiratória, com o objetivo de
manter a via aérea desobstruída.
Assim, o presente estudo tem o objetivo de descrever clínica e
epidemiologicamente os pacientes pediátricos de um centro de referência do Nordeste
brasileiro, a fim de conhecer melhor as características peculiares desta população. Uma
vez traçado o perfil populacional, melhores estratégias governamentais acerca do
tratamento e da qualidade de vida dos fibrocísticos podem ser criadas ou modificadas,
atendendo às demandas específicas dessa população tão miscigenada do nordeste
brasileiro.

15
IV METODOLOGIA
IV.1. CAMPO DE ESTUDO
O estudo foi realizado no Ambulatório Multidisciplinar de Fibrose Cística do
Serviço de Pneumologia Pediátrica do Ambulatório Magalhães Neto (AMN), o qual faz
parte do Complexo-HUPES da Universidade Federal da Bahia (UFBA), em Salvador,
Bahia. Trata-se de um serviço exclusivamente pediátrico, que presta assistência a
pacientes na faixa etária de 0 a 20 anos de idade, os quais são, primordialmente, de baixa
renda. O Complexo-HUPES funciona na condição de hospital escola, contribuindo para
a formação de estudantes de diversas áreas e estágios de especialização, incluindo alunos
de graduação advindos dos cursos de Enfermagem, Nutrição, Fisioterapia e Medicina,
bem como residentes de diversas áreas de saúde. A população fibrocística é atendida
semanalmente por equipe composta por médicos e profissionais das áreas de fisioterapia,
nutrição, farmácia, assistência social e psicologia, que atendem os pacientes desde os
primeiros meses de vida, buscando oferecer-lhes atenção integral e multidisciplinar à
saúde.
IV.2. POPULAÇÃO ESTUDADA
A população em estudo foi composta por todos os pacientes portadores de Fibrose
cística, de 0 a 20 anos, acompanhadas no Ambulatório Multidisciplinar de Fibrose Cística
do Serviço de Pneumologia Pediátrica do Ambulatório Magalhães Neto (Complexo –
HUPES) da Universidade Federal da Bahia, Salvador-BA, entre dezembro de 2008 e
setembro de 2015, perfazendo um total de 59 pacientes.
IV.3. DESENHO DO ESTUDO
O presente estudo trata-se de um estudo seccional, sem grupo de comparação.
IV.4. CRITÉRIOS DE INCLUSÃO
Foram incluídos no estudo: todos os pacientes acompanhandos no AMFC que
apresentaram pelo menos 2 testes do suor positivos ou que tiveram o diagnóstico
confirmado pelo estudo genético ou pacientes com teste do suor normal e com doença
pulmonar supurativa crônica e colonização respiratória crônica por Pseudomonas
aeruginosa cepa mucoide e cujo responsável tenha aceitado assinar o termo de

16
consentimento livre e esclarecido (TCLE) para participação do estudo (em caso de
pacientes com idade inferior a 18 anos). Para maiores de 18 anos, foi considerada a
assinatura do próprio paciente.
IV.5. CRITÉRIOS DE EXCLUSÃO
Foram excluídos do estudo os pacientes enquadrados em ao menos uma das
situações abaixo:
a) Desistiram da participação no estudo;
b) Apresentaram teste do suor normal ou duvidoso e sem colonização respiratória crônica
por Pseudomonas aeruginosa cepa mucoide;
c) Tiveram acompanhamento inferior a 3 meses.
IV.6. DEFINIÇÃO DE TAMANHO AMOSTRAL
Como toda a população-alvo foi estudada, não houve técnica de amostragem nem
cálculo de tamanho amostral, trata-se de uma amostra por conveniência. Todas as crianças
de 0-20 anos de idade acompanhadas no ambulatório de FC no período de Janeiro de 2008
a Setembro de 2015 foram convidadas a participar da pesquisa.
IV.7 VARIÁVEIS:
As variáveis estudadas foram as de caráter demográfico (como sexo, etnia,
procedência, idades na admissão do estudo, consaguinidade e grau de parentesco) e
clínico: idade de aparecimento dos primeiros sintomas, e no momento do diagnóstico;
sintomas que contribuíram com o diagnóstico: desconforto respiratório, história familiar,
ritmo instetinal alterado, retardo de ganho de peso, desidratação, doença hepato-biliar,
distensão abdominal, distúrbio metabólico, doença sinusal, edema ou anemia e
esteatorreia; estado nutricional no diagnóstico (medido através de z escores de
IMC/idade); frequência alélica para a mutação P.Phe508del; e registro de culturas
positivas para Pseudomonas aeruginosa, P. cepa mucoide, S. aureus, MSSA, MRSA, H.
influenza. A etnia foi definida pelo pesquisador principal, de acordo com as carcterísticas
fenotípicas dos pacientes e dos pais.

17
IV.8 COLETA DOS DADOS:
Após a inclusão no estudo, a coleta dos dados clínicos foi realizada pela avaliação
do prontuário médico, no período de Junho de 2012 a Setembro de 2015, de forma
restrospectiva. As informações foram colhidas por mais de um investigador, após
treinamento pelo orientador do projeto, e, se necessário, os responsáveis pelos pacientes
foram entrevistados. Os dados clínicos foram colhidos de forma concomitante aos dados
laboratoriais. Um profissional capacitado realizou as coletas das amostras, através da
técnica do Swab ou da coleta do escarro para cultura das bactérias estudadas, realizadas
a cada consulta. A cada consulta, que é realizada em média a cada 3 meses, são coletadas
espécimes respiratórias, preferencialmente As amostras foram semeadas em ágar
chocolate, ágar sangue, ágar suplementado, meio de cultura MacConkey (específico para
Pseudomonas) e tioglicolato. As cepas mucosas de Pseudomonas aeruginosa foram
identificadas visualmente pela morfologia característica (presença de muco líquido). Foi
coletado 1-2 ml de sangue pela técnica de laboratório, sendo a amostra armazenada sob
refrigeração e encaminhada para o laboratório de gnética do Instituto de Biologia da
Universidade Federal da Bahia, para pesquisa da mutação p.Phe508del. A análise foi feita
através de reação em cadeia da polimerase (PCR) e análise em gel de poliacrilamida 8%,
segundo a técnica de Sanguinetti et al.. (1994). A avaliação do estado nutricional seguiu
os critérios da OMS, que determina pontos de corte segundo z escores, para cada faixa
etária. O índice utilizado foi o de IMC/idade, de acordo com o sexo, classificando os
pacientes estudados em desnutridos e nutridos. Para a avaliação desse indicador, foi
utilizado o programa WHO Anthro 2005 (OMS, Genebra, Suíça). Os dados obtidos foram
registrados em um formulário padrão (ANEXO I).
IV.9. ANÁLISE ESTATÍSTICA
Os dados obtidos foram armazenados em um banco de dados no programa Epidata
e analisados através do programa Epitada Analysis. A análise descritiva foi realizada
através do cálculo da média, mediana, frequência simples e relativa e amplitude de
variação/amplitude interquartílica das variáveis estudadas. A frequência alélica para a
mutação p.Phe508del foi estimatida dividindo-se o número de alelos mutantes pelo
número total de alelos da amostra. Os pacientes com parentesco foram excluídos deste
cálculo.

18
IV.10. ASPECTOS ÉTICOS
O protocolo da pesquisa foi aprovado pelo Comitê de Ética Médica do Hospital
Universitário Professor Edgar Santos (CEP-HUPES), cadastro nº 121, com aprovação em
05 de Junho de 2012. (ANEXO III) O Termo de Consentimento Livre e Esclarecido
(TCLE) foi assinado pelos pais do paciente ou responsáveis legais. (ANEXO III).
19
V RESULTADOS
Foram incluído 59 pacientes no estudo, 31 (52,5%) do sexo masculino. Sete
crianças foram excluídas por apresentarem teste do suor normal ou duvidoso e sem
colonização respiratória crônica por P. aeruginosa cepa mucoide. Todos os pacientes
estudados foram considerados não-brancos.
Oito (13,6%) pacientes tinham pais consanguíneos (os quais eram primos de até
terceiro grau). Havia história familiar para FC em 21,1% (12/57) dos pacientes, sendo
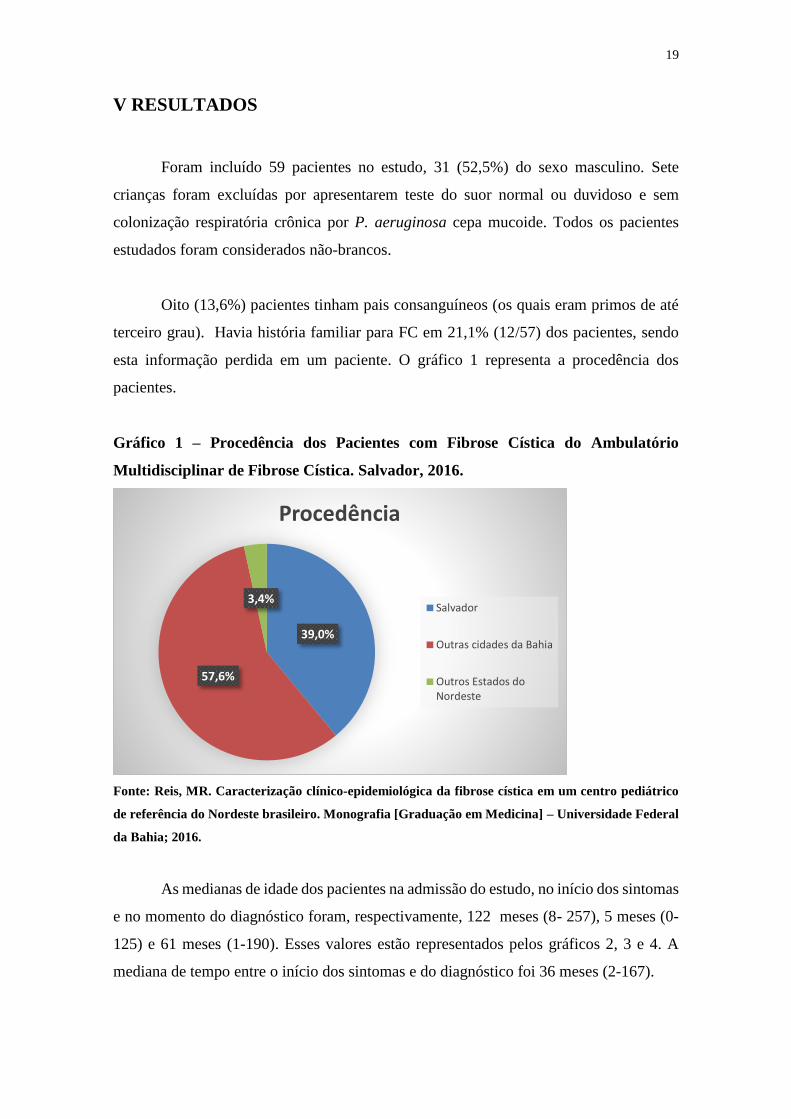
esta informação perdida em um paciente. O gráfico 1 representa a procedência dos
pacientes.
Gráfico 1 – Procedência dos Pacientes com Fibrose Cística do Ambulatório
Multidisciplinar de Fibrose Cística. Salvador, 2016.
Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico
de referência do Nordeste brasileiro. Monografia [Graduação em Medicina] – Universidade Federal
da Bahia; 2016.
As medianas de idade dos pacientes na admissão do estudo, no início dos sintomas
e no momento do diagnóstico foram, respectivamente, 122 meses (8- 257), 5 meses (0-
125) e 61 meses (1-190). Esses valores estão representados pelos gráficos 2, 3 e 4. A
mediana de tempo entre o início dos sintomas e do diagnóstico foi 36 meses (2-167).
39,0%
57,6%
3,4%
Procedência
Salvador
Outras cidades da Bahia
Outros Estados doNordeste

20
Gráfico 2: Faixas etárias dos pacientes admitidos no estudo. Salvador, 2016.
Count significa contagem. Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística
em um centro pediátrico de referência do Nordeste brasileiro. Monografia [Graduação em Medicina]
– Universidade Federal da Bahia; 2016.
Gráfico 3: Idade de manifestação dos primeiros sintomas. Salvador, 2016.
Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico
de referência do Nordeste brasileiro. Monografia [Graduação em Medicina] – Universidade Federal
da Bahia; 2016.

21
Gráfico 4: Idade dos pacientes no momento do diagnóstico. Salvador, 2016.
Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico
de referência do Nordeste brasileiro. Monografia [Graduação em Medicina] – Universidade Federal
da Bahia; 2016.
As principais manifestações clínicas que motivaram o diagnóstico foram: queixas
respiratórias (81,4%); dificuldade ganho de peso (72,9%); esteatorreia (54,2%); ritmo
intestinal alterado (49,2%) e retardo do crescimento (23,7%). Nenhum paciente
apresentou íleo meconial (Tabela 1).

22
Tabela 1: Frequências relativas e absolutas dos sinais e sintomas sugestivos para o
diagnóstico de Fibrose Cística de pacientes do Ambulatório Multidisciplinar de
Fibrose Cística. Salvador, 2016.
SINTOMAS PRESENTE
QTD %
Sintomas respiratorios 47 88,7%
Dificuldade de ganho de Peso 43 81,1%
Ritmo intestinal alterado 27 50,9%
Esteatorreia 26 49,1%
Retardo do crescimento 13 24,5%
Disturbio metabolico 8 15,1%
Edema ou anemia 8 15,1%
Historia familiar 7 13,2%
Desidratacao 7 13,2%
Distensao abdominal 7 13,2%
Doenca hepato-biliar 5 9,4%
Doenca sinusal 3 5,7%
Outros(*) 5 9,4% Outros (*): DRGE, regurgitação, vômitos, dores abdominais, obstipação, inapetência. Nenhum
paciente apresentou íleo meconial. Fonte: Reis, MR. Caracterização clínico-epidemiológica da
fibrose cística em um centro pediátrico de referência do Nordeste brasileiro. Monografia [Graduação
em Medicina] – Universidade Federal da Bahia; 2016.
No momento do diagnóstico, 55 pacientes tiveram avaliação do estado nutricional,
destes, 20 (36,4%) foram classificados como desnutridos, dos quais 55¨% tinham idade
inferior a 24 meses. A informação nutricional de 4 pacientes foi perdida. A tabela 2
representa esses dados.
Tabela 2: Classificações dos pacientes quanto ao estado nutricional, utilizando o z
escore -2 como ponto de corte (OMS, 2011). Salvador, 2016.
Qtd %
NUTRIDO (IMC/I > -2) 35 63,6%
DESNUTRIDO (IMC/I < -2) 20 36,4%
A informação quanto ao estado nutricional de 4 pacientes no momento do diagnóstico foi perdida.
Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico
de referência do Nordeste brasileiro. Monografia [Graduação em Medicina] – Universidade Federal
da Bahia; 2016.
23
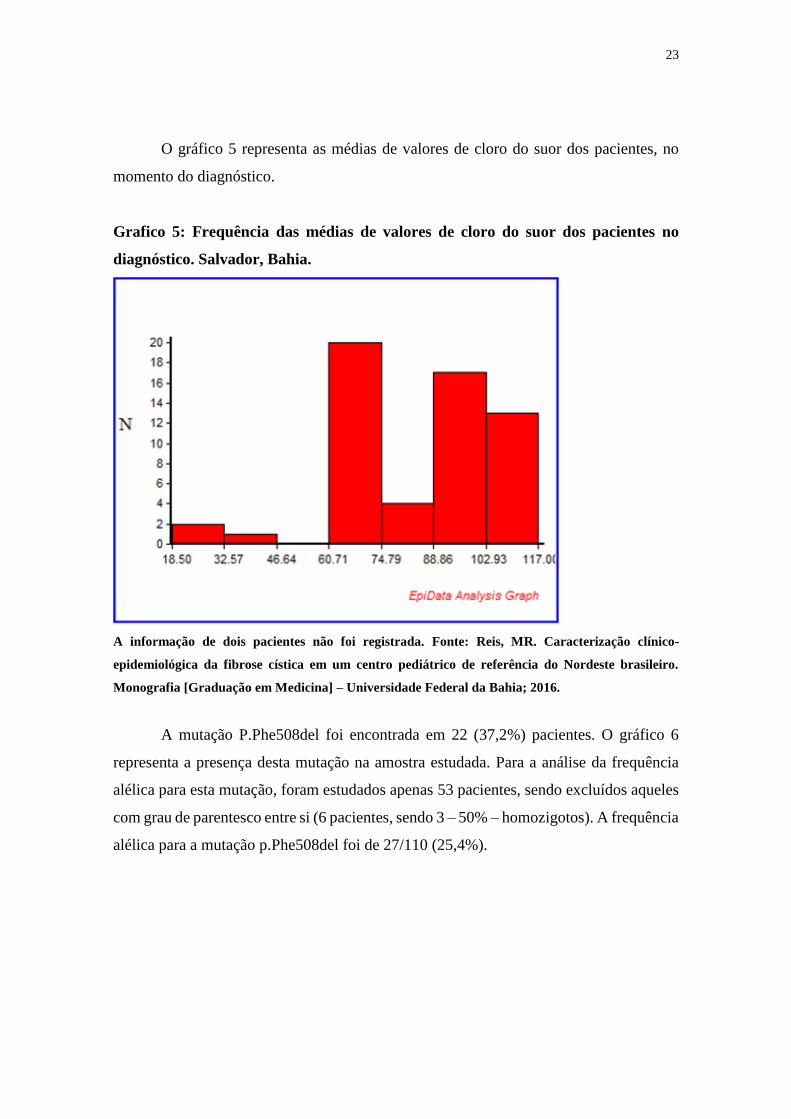
O gráfico 5 representa as médias de valores de cloro do suor dos pacientes, no
momento do diagnóstico.
Grafico 5: Frequência das médias de valores de cloro do suor dos pacientes no
diagnóstico. Salvador, Bahia.
A informação de dois pacientes não foi registrada. Fonte: Reis, MR. Caracterização clínico-
epidemiológica da fibrose cística em um centro pediátrico de referência do Nordeste brasileiro.
Monografia [Graduação em Medicina] – Universidade Federal da Bahia; 2016.
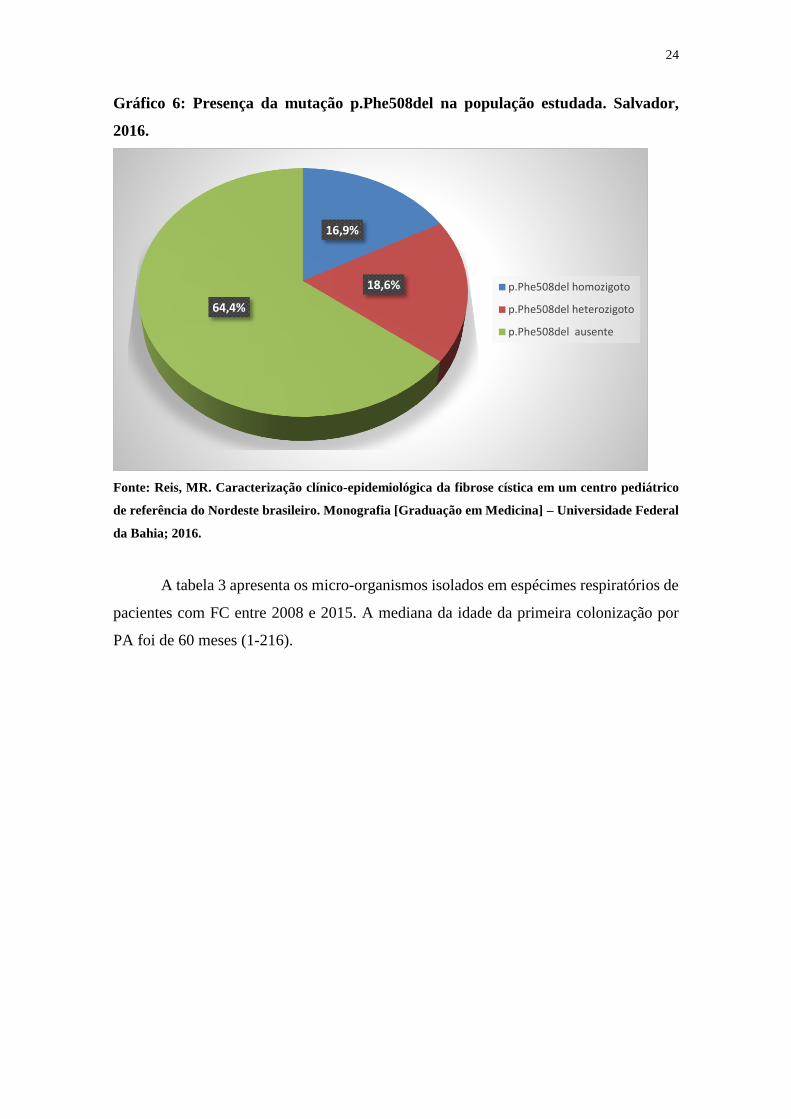
A mutação P.Phe508del foi encontrada em 22 (37,2%) pacientes. O gráfico 6
representa a presença desta mutação na amostra estudada. Para a análise da frequência
alélica para esta mutação, foram estudados apenas 53 pacientes, sendo excluídos aqueles
com grau de parentesco entre si (6 pacientes, sendo 3 – 50% – homozigotos). A frequência
alélica para a mutação p.Phe508del foi de 27/110 (25,4%).
24
Gráfico 6: Presença da mutação p.Phe508del na população estudada. Salvador,
2016.
Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose cística em um centro pediátrico
de referência do Nordeste brasileiro. Monografia [Graduação em Medicina] – Universidade Federal
da Bahia; 2016.
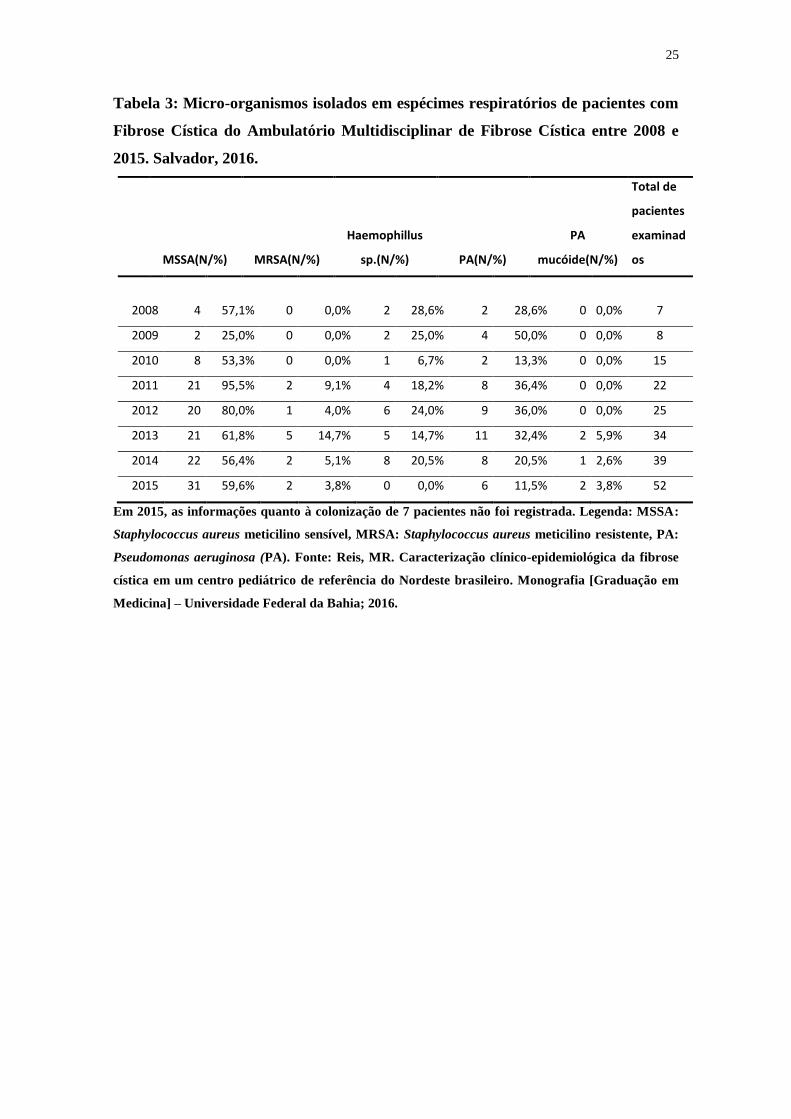
A tabela 3 apresenta os micro-organismos isolados em espécimes respiratórios de
pacientes com FC entre 2008 e 2015. A mediana da idade da primeira colonização por
PA foi de 60 meses (1-216).
16,9%
18,6%
64,4%
p.Phe508del homozigoto
p.Phe508del heterozigoto
p.Phe508del ausente
25
Tabela 3: Micro-organismos isolados em espécimes respiratórios de pacientes com
Fibrose Cística do Ambulatório Multidisciplinar de Fibrose Cística entre 2008 e
2015. Salvador, 2016.
MSSA(N/%) MRSA(N/%)
Haemophillus
sp.(N/%) PA(N/%)
PA
mucóide(N/%)
Total de
pacientes
examinad
os
2008 4 57,1% 0 0,0% 2 28,6% 2 28,6% 0 0,0% 7
2009 2 25,0% 0 0,0% 2 25,0% 4 50,0% 0 0,0% 8
2010 8 53,3% 0 0,0% 1 6,7% 2 13,3% 0 0,0% 15
2011 21 95,5% 2 9,1% 4 18,2% 8 36,4% 0 0,0% 22
2012 20 80,0% 1 4,0% 6 24,0% 9 36,0% 0 0,0% 25
2013 21 61,8% 5 14,7% 5 14,7% 11 32,4% 2 5,9% 34
2014 22 56,4% 2 5,1% 8 20,5% 8 20,5% 1 2,6% 39
2015 31 59,6% 2 3,8% 0 0,0% 6 11,5% 2 3,8% 52
Em 2015, as informações quanto à colonização de 7 pacientes não foi registrada. Legenda: MSSA:
Staphylococcus aureus meticilino sensível, MRSA: Staphylococcus aureus meticilino resistente, PA:
Pseudomonas aeruginosa (PA). Fonte: Reis, MR. Caracterização clínico-epidemiológica da fibrose
cística em um centro pediátrico de referência do Nordeste brasileiro. Monografia [Graduação em
Medicina] – Universidade Federal da Bahia; 2016.

26
VI. DISCUSSÃO
O presente estudo descreveu características clínico-epidemiológicas de 59
pacientes com fibrose cística acompanhados em um centro de referência de Salvador-BA.
Embora a FC seja mais comum entre os brancos, chamou a atenção que a
população estudada foi composta, exclusivamente, por pacientes não brancos, pois, ainda
que um número pequeno de pacientes apresentassem traços caucasoides, estes ou seus
pais também apresentavam características tipicamente de outros grupos étnicos, como os
negros e os ameríndios. Estes resultados contrastam com os dados do Registro Brasileiro
de Fibrose Cística (REBRAFC), de 2013, segundo o qual cerca de 70% dos pacientes
eram brancos. Reis et al. (2000) e Alvarez et al. (2004) encontraram dados semelhantes
aos nacionais, em estudos realizados no Sudeste e Sul respectivamente. A miscigenação
que ocorre na população brasileira é resultado principalmente da mistura de três grupos
étnicos: o europeu, o africano e o ameríndio. Por sua distribuição heterogênea ao longo
do território brasileiro, encontramos contribuições quantitativamente diferentes dos
colonizadores nos diferentes estados. (Viana e Mesquita, 2003). A contribuição africana
foi importante na Bahia (Moura et al., 2008), uma vez que no estado encontram-se sete
das dez cidades com maior concentração de negros do país, em termos relativos (IBGE,
2013). Desta forma, é possível sugerir que a forte influência afrodescente na composição
da população baiana possa explicar os achados do presente estudo. É importante destacar,
ainda, que a classificação étnica apenas pelos traços fenotípicos é falha, pois não
considera origens, apenas características visuais. (IBGE, 2013).
Ainda que os pacientes tenham sido considerados não brancos, houve frequência
alélica de 25,4% para a mutação p.Phe508del, a mais frequente em caucasianos, estando
presente em torno de 70% dos cromossomos dos pacientes dos países europeus e dos
Estados Unidos da América (EUA) (European Working Group on CF Genetics, 1990;
Okay et al., 2005). A presença dessa mutação, essencialmente de caucasoides, corrobora
com a ideia de miscigenação da população baiana, que, embora fenotipicamente
classifique-se afrodescente, revela ocorrência de mutação de origem caucasiana. No
Brasil, a frequência alélica média da mutação p.Phe508del é de 47%, variando de menos
de 9% a 55,2%, em diferentes estados e regiões geográficas, assim como dos diferentes
ancestrais dessas populações (Raskin et al., 1999; Raskin and Fauez, 2001; Cabello et al.,
1999, 2001; Streit et al., 2003; Viana e Mesquita, 2003; Araújo et al. 2005). Costa et al.

27
(2007) encontraram uma frequência alélica para esta mutação na população do estado da
Bahia de cerca de 9%, e supôs que esse baixo percentual pode ser devido à grande
miscigenação populacional, a qual é composta por muitos afrodescendentes. Nesta
casuística, a frequência encontrada foi de 25,4%, bem superior ao encontrado por Costa
et al. (2007), o que pode refletir formas mais graves em pacientes bem jovens, uma vez
que a mediana de idade deste estudo foi de 122 meses, ou seja, cerca de metade dos
pacientes tinham menos de 10 anos. Como há uma baixa frequência alélica, cabe salientar
a importância de pesquisas mais amplas das mutações genéticas, uma vez que a população
estudada deve apresentar outras mutações menos frequentes em brancos. Assim, deve-se
considerar todas as mutações para o diagnóstico genético da FC (Costa et al., 2007).
A maioria dos indivíduos estudados (57,6%) eram procedentes de outras cidades
do estado da Bahia. Os percentuais de pacientes não residentes na capital são ligeiramente
superiores ao observado por Pinto et al. (2009) que demonstraram que cerca de 40%
dos pacientes atendidos num centro de referência de Recife-PE eram de outros
municipios. A FC é uma doença complexa, que exige acompanhamento multidisciplinar,
o qual muitas vezes não é oferecido em cidades menores e com poucos recursos, de forma
que muitos brasileiros com FC deslocam-se para as regiões metropolitanas em busca de
assistência adequada. Os centros de referência para a FC concentram-se nas metrópoles
regionais e na capital. Estes locais oferecem melhores condições de assistência e
tratamento, fornecendo medicamentos e insumos aos fibrocísticos, entretanto, é
importante considerar o ônus socioeconômico deste deslocamento.
Mais de 75% da população estudada tinha menos de 14 anos, ou seja, os pacientes
eram muito jovens. Os dados do REBRAFC 2013 revelaram que 75,2% dos pacientes
com FC tinham menos de 18 anos de idade, destacando a importância do
acompanhamento pediátrico no curso da doença e, possivelmente, a baixa sobrevida
destes pacientes no Brasil .
É importante ressaltar que os sintomas relacionados à doença surgiram
precocemente, com mediana de 5 meses, dado que enfatiza as conclusões de estudos
prévios (Vidigal et al., 2008, Alvarez et al., 2004 e Reis et al., 2000). Pacientes que
manifestam a FC muito cedo tendem a ter piores prognósticos (Fernández et al., 2014).
O diagnóstico e a intervenção precoce alteram significativamente a expectativa de vida

28
dos pacientes (Robinson P, 2001). Nos EUA, Canadá e países da Europa, o diagnóstico
da FC é feito precocemente, antes do primeiro ano de vida, o que proporciona às crianças
fibrocísticas serem tratadas e monitoradas quanto às variáveis que influenciam
diretamente no prognóstico da doença, como, por exemplo, o acompanhamento da curva
de peso/altura e a presença de colonização de vias aéreas superiores por patógenos, que
têm relação íntima com pior prognóstico da doença. (Robinson P, 2001) Em nosso país,
a média de idade ao diagnóstico varia de 1,6 a 9,6 anos, o que leva a prejuízos no
acompanhamento desses pacientes ( Farias et al., 1997; Rabbi-Bortolini et al.; 1998). Essa
variação pode ser explicada pelas diferenças na implementação da Triagem Neonatal no
país, a qual se iniciou nos estados do Sul e Sudeste, sendo muito recente em outro estados,
como os do Nordeste (Mota LR., 2015). Nesta casuística, o tempo mediano dos sintomas
ao diagnóstico foi de 36 meses, semelhante a estudo brasileiro prévio (Alvarez et al.,
2004). Algumas hipóteses podem ser levantadas para a explicar o retardo do diagnóstico:
a não realização de triagem neonatal, que só foi iniciada na Bahia em 2013 e que, portanto,
ainda não pode ser refletida neste trabalho e o baixo grau de suspeição clínica por
pediatras (Kabra et al., 2006).
O diagnóstico precoce da FC pode melhorar a sobrevida dos pacientes. Para
avaliar o impacto do diagnóstico precoce, realizado por rastreamento neonatal antes do
aparecimento de sintomas clínicos, estudo de coorte retrospectiva realizado em New
South Wales, estado mais populoso da Austrália, comparou as evoluções clínicas de
pacientes com diagnóstico realizado antes e depois da introdução do screening neonatal
de rotina. Não foram encontradas diferenças significantes no estado nutricional entre os
dois grupos. Entretanto, o escore de Shwachman-Kulczycki, que avalia a gravidade da
doença como um todo, foi melhor no grupo com diagnóstico precoce. Também foram
significantemente melhores a avaliação radiológica e a função pulmonar (Mckay et al.,
2005). Collins et al. (2008) acompanharam pacientes nascidos em Connecticut, EUA,
entre 1983 e 1997 até a adolescência e mostraram vantagens do diagnóstico precoce por
meio de screening neonatal. Os autores compararam a evolução clínica e da função
pulmonar até os 15 anos de idade, entre crianças cujo diagnóstico de FC foi realizado por
screening neonatal ou por teste do suor. Após o início dos sintomas, os índices de peso e
estatura foram mais adequados tanto na idade do diagnóstico quanto aos 15 anos nas
crianças diagnosticadas por triagem neonatal, enfatizando a importância do diagnóstico
precoce no estado nutricional e no crescimento. Este mesmo grupo também apresentou
melhores parâmetros de função pulmonar aos 15 anos e evolutivamente. Dos 6 aos 15

29
anos, as crianças do grupo screening neonatal apresentaram aumento do VEF1 e da CVF
de 4% e 13%, respectivamente, enquanto o grupo com diagnóstico tardio apresentou
redução destes parâmetros de 14% e 5%, respectivamente. A colonização por
Pseudomonas aeruginosa foi semelhante nos dois grupos (Collins et al., 2008). A triagem
neonatal configura uma ferramenta importante a na detecção precoce da FC (Santos et
al., 2005), bem como capacitação adequada dos pediatras e médicos em geral, afim de
reduzir esse intervalo de tempo entre o início dos sintomas e do diagnóstico. Espera-se
que após o completo estabelecimento da triagem neonatal da FC na Bahia, o curso clínico
da FC nesse estado melhore.
As manifestações clínicas da FC podem ser muito variáveis e ocorrerem
precocemente ou se manifestarem mais tardiamente. Segundo a literatura, as principais
queixas descritas no diagnóstico na FC são: tosse persistente, diarreia crônica e déficit
nutricional (Alvarez et al., 2004, Reis et al., 2000, e Farias et al., 1997). Até 90% dos
pacientes apresentam insuficiência pancreática no primeiro ano de vida (Bronstein et al.,
1992). Dentre os dados da história clínica, os mais frequentes encontrados neste estudo
foram as manifestações respiratórias, os relacionadas à insuficiência pancreática e aos
índices antropométricos (dificuldade de ganho de peso e retardo de crescimento), o que
corrobora os achados prévios da literatura (Pinto et al., 2009; REBRAFC, 2013), embora
não tenha ocorrido íleo meconial nos indivíduos estudados.
A desnutrição é um sinal clínico muito importante na FC, o qual pode piorar a
sobrevida dos pacientes. Ela está diretamente relacionada à presença de insuficiência
pancreática (Burowitz et al., 2009). Nesta pesquisa, a maioria dos pacientes desnutridos
tinham idade inferior aos 24 meses no momento do diagnóstico, o que sugere maior
gravidade da FC neste subgrupo. Estudos prévios determinaram frequência maior de
desnutrição no diagnóstico da doença (Reis et al., 2000; Pinto et al., 2009; Fernández et
al., 2014). Dados do REBRAFC (2013) mostram que os valores de z escore, segundo os
critérios da OMS (2011), caem à medida que o paciente envelhece, mostrando uma
tendência ao aumento de desnutrição nas populações mais velhas. Uma vez que a
população do estudo foi composta por pacientes muito jovens, a desnutrição poderia não
ter se estabelecido ainda no momento do diagnóstico, pois apenas 36,4% dos pacientes
estudados estavam desnutridos neste período.

30
Habitualmente, crianças com fibrose cística são precocemente colonizadas por
Haemophilus influenzae e Staphilococus aureus, ou pelos dois microorganismos. Em
poucos anos, PA torna-se o organismo mais identificado nas vias aéreas. (Rosenfeld et
al., 2003) A infecção por PA contribui consideravelmente para morbidade e mortalidade
dos fibrocísticos, devido à intensa resposta inflamatória, que leva a uma perda de função
pulmonar irreversível (Ratjen FA, 2009). Bush (2002) demonstrou a importância de tratar
agressivamente a primeira colonização por PA, pois os pacientes que não tratados
precocemente, em seu estudo, tinham pior sobrevida. Burns et al., (2001) demonstraram
em uma coorte realizada com 40 pacientes com FC que 98% apresentavam evidência
sorológica ou de cultura de infecção por PA aos 3 anos de idade. O presente estudo
determinou uma mediana de idade da primeira colonização por Pseudomonas aeruginosa
de 60 meses (1-216). Quanto às colonizações, as mais frequentes nos período de 2008 a
2015 foram as por MSSA, Haemophillus sp. e PA, em ordem decrescente. Porém, as
frequências encontradas ficaram abaixo das encontradas em um estudo no estado de São
Paulo, no qual mais de 80% dos pacientes estavam colonizados por MSSA, 76% por PA
mucoide e não mucoide e 5,2% por Bulkhoderia cepacia (Alvarez et al., 2004). O
REBRAFC, 2013, demonstrou que as colonizações mais frequentes na população
brasileira com FC, naquele ano, foram: MSSA, PA, PA mucoide, Haemophillus sp e
MRSA, estando de acordo com os resultados encontrados no presente estudo. O Registro
Brasileiro de FC apresentou, ainda, porcentagens mais altas de colonização por MSSA
até os 20 anos, idade a partir da qual se aumentou a frequência de colonização por PA
(tornando-se a colonização mais frequente). Algumas limitações do estudo das
colonizações nesta pesquisa foram a falta de meios de cultura adequados para a
Bulkhoderia cepacia; os recursos muitas vezes escassos, o que poderia prejudicar a
realização constante de culturas do escarro e da orofaringe; além da dificuldade de acesso
a alguns prontuários, no hospital onde foi realizada a pesquisa. Além disso, a população
estudada correspondeu, principalmente, a crianças pequenas e não supurativas, limitando
a utilização do exame do escarro para isolamento de micro-organismos, que apresenta
maior sensibilidade quando comparado ao swab da orofaringe que foi realizado para a
amioria dos pacientes.

31
VII. CONCLUSÕES
1. A população estudada foi composta apenas por pacientes não brancos.
2. O diagnóstico da FC foi tardio.
3. Houve um retardo no tempo entre a idade de início dos sintomas e o diagnóstico.
4. Os sintomas respiratórios e os gastrointestinais foram os mais frequentes no
momento do diagnóstico.
5. Cerca de 40% dos pacientes desta casuística tinham desnutrição.
6. A frequência da mutação p.Phe508del encontrada foi inferior ao descrito no Sul
e Sudeste brasileiros.
7. As colonizações por Staphylococcus aureus, Haemophillus sp. e Pseudomonas
aeruginosa foram as mais frequentes na população estudada.
8. A idade para a primeira colonização por Pseudomonas aeruginosa variou de 1 a
216 meses.

32
VIII. SUMMARY
Clinical and epedimiological caracterization of cystic fibrosis at a reference center
in northeastern Brazil. Cystic fibrosis (CF) is a genetic disease caused by several
mutations at the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)’s gene
that encodes this transmembrane protein, essential for the ionic transportation through
cell. Defects in the protein lead to multissistemic manifestations, mainly at respiratory
and intestinal tracts. Studies about CF in northeastern Brazil are scarce.
Objective: To describe clinical and epidemiological profile of children with CF under the
care of a reference center from Salvador, Bahia.
Methods: A cross-sectional study was performed. After inclusion, a form has been
completed with the clinical and epidemiological data, followed by blood samples
collection for p.Phe508del mutation research and obtained sputum samples to determine
bacterial colonization of the respiratory tract. Results: 59 patients were included, 52,5%
male, all non-white. The median age of the patients at the admission, onset of symptoms
and diagnosis was: 12,5 e 61 months, respectively. The median time between the onset of
symptoms and diagnosis was 36 months. The most common clinical data that suggested
the diagnosis were: respiratory manifestations, the ones related to pancreatic insufficiency
and growth retardation. There was malnutrition in 36,4% of patients by the time of
diagnosis. p.Phe508del was identified in 37,2%, allelic frequency of 27/106 (25,4%).
From 2008 to 2015, the main microorganisms isolated were methicillin-sensitive
Staphylococcus aureus (MSSA) - 25-80%; Pseudomonas aeruginosa (PA) - 11.5 to
36.4%; Haemophilus sp. - 6.7 to 28.6%. The median age of first colonization by PA was
60 months. Conclusion: The study population was composed by only non-white patients,
with late diagnosis and low frequency of p.Phe508del.
Key-words: 1. Cystic fibrosis; 2. Children and adolescents; 3. Clinical and
epidemiological profile

33
IX REFERÊNCIAS
1. Alvarez AE, Ribeiro AF, Hessel G, Bertuzzo CS, Ribeiro JD. Fibrose cística em
um centro de referência no Brasil: características clínicas e laboratoriais de 104
pacientes e sua associação com o genótipo e a gravidade da doença. J Pediatr
2004;80:371-9.
2. Augarten A, Tov AB, Madgar I, et al.. The changing face of the exocrine pancreas
in cystic fibrosis: the correlation between pancreatic status, pancreatitis and cystic
fibrosis genotype. Eur J Gastroenterol Hepatol 2008; 20:164-8.
3. Bernardino AL, Ferri A, Passos-Bueno MR et al.. Molecular analysis in Brazilian
cystic fibrosis patients reveals five novel mutations. Genet Test 2000; 4:69-74.
4. Bobadilla JF, Macek Júnior M, Fine JP, Farrel PM. Cystic Fibrosis: A Worldwide
Analysis of CFTR Mutations—Correlation With Incidence Data and Application to
Screening. Hum Mutat 2002; 19:575–606.
5. Braun AT, Merlo CA. Cystic fibrosis lung transplantation. Curr Opin Pulm Med
2011; 17:467–72.
6. Bronstein MN, Sokol RJ, Abman SH, Chatfield BA, Hammond KB, Hambridge
KM, et al. Pancreatic insufficiency, growth, and nutrition in infants identified by
newborn screening as having cystic fibrosis. J Pediatr 1992; 120:533-40.
7. Burns J, Gibson R, McNamara S, et al. Longitudinal assessment of Pseudomonas
aeruginosa in young children with cystic fi brosis. J Infect Dis 2001; 183: 444–
52.
8. Burowitz et al. Cystic Fibrosis Foundation Evidence-Based Guidelines for
Management of Infants with Cystic Fibrosis. J Pediatr 2009; 155 (6): 73-93.
9. Bush A. Decisions facing the cystic fibrosis clinician at the first isolation of
Pseudomonas aeruginosa. Paediatr Respir Rev 2002; 3:82-8.

34
10. Cabello GMK, Moreira AF, Horovitz D. Cystic fibrosis: low frequency of D508
mutation in 2 population samples from Rio de Janeiro, Brazil. Human Biolology.
Detroit 1999; 71:189-196.
11. Ciofu et al.. Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm
Med 2013, 19:251–258.
12. Collins MS, Abbott MA, Wakefield DB, Lapin CD, Drapeau G, Hopfer SM, et al.
Improved Pulmonary and Growth Outcomes in Cystic Fibrosis by Newborn
Screening. Pediatr Pulmonol 2008;43:648-55.
13. Cystic Fibrosis Mutation Database. Homepage: www.genet.sickkids.on.ca.
Access in: 07/10/2015.
14. Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med 2006; 173:475-
82.
15. De Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L. The relative frequency of
CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros 2014;
13 (4):403–409.
16. European Working Group on CF Genetics. Gradient of distribution in Europe of
the major CF mutation and of its associated haplotype. Hum Genet 1990; 85: 436-
45.
17. Farber, S. Pancreatic function and disease in early life: pathologic changes
associated with pancreatic insuficiency in early life. Arch Path 1944; 37:231-50.
18. Farias L, Rosário Filho NA, Kovalhuk L, Miasaki N, Chaves SM, Recco RA, et
al. Aspectos clínicos da fibrose cística: experiência no Hospital de Clínicas da
UFPR, 1980-1996. Pediatria (São Paulo). 1997;19(4):241-8.

35
19. Farrell PM, Rosenstein BJ, White TB, et al.. Guidelines for diagnosis of cystic
fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus
report. J Pediatr 2008; 153: S4–14.
20. Fernández GF, Suárez GA, Brunet APP, Valdés JAG, Leyva RP. Caracterización
de la fibrosis quística en el primer año de vida. Revista Cubana de Pediatría. 2014;
86(4):423-432.
21. Firmida MC, Marques BL, Costa CH. Artigo 4: Fisiopatologia e Manifestações
Clínicas da Fibrose Cística. Revista do Hospital Universitário Pedro Ernesto,
UERJ 2011.
22. Friedrich, DC. Análise molecular de mutações frequentes em pacientes com suspeita
clínica de Fibrose Cística. 2007. 69 f. Dissertação (Mestrado em Biociências).
Instituto de Biociências. Universidade Federal do Rio Grande do Sul. Porto Alegre.
Rio Grande do Sul.
23. Gibson, L.E.; Cooke, R.E. test for concentration of eletctrolytes in sweat in cystic
fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics,1959;
23:23-62.
24. Gibson LR, Burns JL, Ramsey BW. Pathophysiology and Management of
Pulmonary Infections in Cystic Fibrosis. Am J Respir Crit Care Med 2003; 168:
918–951.
25. Hamosh A, Fitzsimmons SC, Macek MJr, Knowles MR, Rosenstein BJ, Cutting GR.
Comparison of the clinical manifestations of cystic fibrosis in black and white
patients. J Pediatr 1998; 132(2):255–9.
26. IBGE (Instituto Brasileiro de Geografia e Estatísticas). Características da população
e domicílios do Censo 2010. URL: www.ibge.gov.br. Acesso: 28 de mar de 2016.
27. Kabra SK, Kabra M. Diagnosing and managing cystic fibrosis in the developing
world. Paed Res Ver 2006; 7S: 147–150.

36
28. Lemos AC, Matos E, FRANCO R. Fibrose Cística em adultos: aspectos clínicos
e espirométricos. Jornal Brasileiro de Pneumologia 2004; 30(1): 9-13.
29. Martiniano SL, Hoppe JE, Sagel SD. Advances in the Diagnosis and Treatment
of Cystic Fibrosis. Advances in Pediatrics 2014; 61:225–243.
30. Mckay KO, Waters DL, Gaskin KJ. The influence of newborn screening for cystic
fibrosis on pulmonary outcomes in New South Wales. J Pediatr 2005; 147:47-50.
31. Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Giménez J et al.. The
origin of the major cystic fibrosis mutation (D508) in european populations.
Nature Genet 1994; 7:169-75.
32. Mota LR. Estudo de Mutações no Gene CFTR em Pacientes com Fibrose Cística
de um Centro Universitário de Referência em Salvador-BA. Bahia. Dissertação
[Mestrado em Genética e Biodiversidade] – Universidade Federal da Bahia; 2015.
33. Moura Costa FM, Santana MA, Lemos ACM, Acosta AX. Low frequency of the
Δ508 mutation of the CFTR gene in a highly admixed population in Bahia, Brazil.
Hum Biol. 2007; 79(3):293-8.
34. OKAY TS et al. Frequency of the D508 mutation in 108 cystic fibrosis patients
in São Paulo: comparison with reported Brazilian data. CLINICS 60(2):131-134,
2005.
35. Olveira G, Olveira C. Nutrición, fibrosis quística y aparato digestivo. Nutr Hosp.
2008; 23(2): 71-86.
36. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373: 1891–904.
37. Park RW, Grand RJ. Gastrointestinal manifestations of cystic fibrosis: a review.
Gastroenterology 1981;81:1143-61.

37
38. Pinto ICS, Silva CP, Britto MCA. Perfil nutricional, clínico e socioeconômico de
pacientes com fibrose cística atendidos em um centro de referência no nordeste
do Brasil J Bras Pneumol. 2009;35(2):137-143.
39. Rabbi-Bortolini E, Bernadino ALF, Lopes AL et al. Sweat electrolyte and cystic
fibrosis mutation analysis allows early diagnosis in brazilian children with clinical
signs compatible with cystic fibrosis. Am J Med Genet 1998; 76:288-90.
40. Raskin S, Phillips J A, Kaplan G. Geographic heterogeneity of 4 common
worldwide cystic fibrosis non-D508 mutations in Brazil. Human Biology. Detroit
1999; 71:111-121.
41. Registro brasileiro de Fibrose Cística: Relatório Anual de 2013. Grupo Brasileiro
de Estudos de Fibrose Cística. Disponível em:
http://www.gbefc.org.br/gbefc/Registro2013Portugues.pdf.
42. Reis FC, Damasceno N. Fibrose cística, Cystic fibrosis, J Pediatr (Rio J)
1998;74(1):76-94.
43. Reis FJC, Oliveira MCL, Penna FJ, Oliveira MGR, Oliveira EA, Monteiro APAF.
Quadro clínico e nutricional de pacientes com fibrose cística: 20 anos de
seguimento no HC-UFMG. Rev Ass Med Brasil 2000; 46(4): 325-30.
44. Ribeiro JD, Ludwig Neto N, Ribeiro AF, Camargos PAM. Fibrose Cística. In:
Lopez. FA, Campos Júnior D, eds. Tratado de Pediatria. São Paulo: Manole, 2007:
1846- 57.
45. Ribeiro JD, Ribeiro MAGO, Ribeiro AF. Controvérsias na fibrose cística – do
pediatra ao especialista.J Ped 2002 Jul; 78:171-185.
46. Rosenstein BJ, Cutting GR: The diagnosis of cystic fibrosis . A consensus
statement. Cystic fibrosis Foundation Consensus Panel. J pediatr 1998; 132:589-
595.

38
47. Rosenfeld M, Ramsey BW, Gibson RL. Pseudomonas acquisition in young
patients with cystic fibrosis: pathophysiology, diagnosis, and management. Curr
Opin Pulmonol Med 2003; 9: 492–97.
48. Robinson P. Cystic fibrosis. Review series: Paediatric origins of adult lung
disease. Thorax. 2001;56:237-41.
49. Salsgiver EL, Fink AK, Knapp EA, LiPuma JJ, Olivier KN, Marshall BC,
Saiman L. Changing Epidemiology of the Respiratory Bacteriology of Patients
with Cystic Fibrosis. North American Cystic Fibrosis Conference, Atlanta, GA,
October 2014.
50. Sanguinetti CJ, Dias Neto E, Simpson AJG. Rapid silver staining and recovery of
PCR products separated on polyacrylamide gels. Biotechniques. 1994; 17: 915-
919.
51. Santos GP, Domingos MT, Wittig EO, Riedi CA, Rosário NA. Programa de
triagem neonatal para fibrose cística no estado do Paraná: avaliação após 30 meses
de sua implantação. J Pediatr 2005; 81:240-4.
52. Schechter MS. Benchmarking to improve the quality of cystic fibrosis care. Curr
Opin Pulm Med 2012; 18:596–601.
53. Tauber E, Eichler I, Gartner C, Halmerbauer G, Gotz M, Rath R, et al.
Improvements of lung function in cystic fibrosis. Pediatr Pulmonol. 2002; 33:263-
8.
54. Viana AMH, Mesquita ERL. Análise de mutações do gene CFTR em indivíduos com
sinais clínicos sugestivos de Fibrose Cística. Revista do Hospital Universitário.
Universidade Federal do Maranhão. 2003;1-2(1):13-20.

39
55. Vidigal PVT, Reis FJC, Boson WLM et al. Δ508del in a heterogeneous cystic
fibrosis population from Minas Gerais, Brazil. Braz J Med Biol Res. 2008;
41:643-647.
56. Wagener JS, Headley AA. Cystic fibrosis: current trends in respiratory care.
Respir Care. 2003; 48:234-45.
57. Walllis C. Diagnosis and presentation of Cystic Fibrosis. In: Chernick V, Boat
TF, Wilmott RW, Bush A. Kendig`s disorders of the Respiratory Tract in
Children. Sauders Elsevier, 2006; 866- 87.
58. Wang SS, O.Leary LA, Fitzsimmons SC, Khoury MJ. The impact of early cystic
fibrosis on pulmonary function in children. J Pediatr. 2002; 141:804-10
59. Weintraub A, Blau H, Mussaffi H, et al.. Exocrine pancreatic function testing in
patients with cystic fibrosis and pancreatic sufficiency: a correlation study. J
Pediatr Gastroenterol Nutr 2009; 48:306–10.
60. World Health Organization [homepage on the Internet]. Geneva: World Health
Organization. [cited 2007 May 25]. WHO Anthro 2005 software and macros;
[about 2 screens]. Available from:
http://www.who.int/childgrowth/software/en/index.html.
61. Zemanick ET, Harris JK, Conway S, et al.. Measuring and improving respiratory
outcomes in cystic fibrosis lung disease: Opportunities and challenges to therapy.
J Cyst Fibros. 2010; 9:1-16.

40
X APÊNCIDES
APÊNDICE I: Formulário para Coleta de Dados
COMPLEXO HOSPITALAR PROFESSOR EDGARD SANTOS
PESQUISA FIBROSE CÍSTICA - ADMISSÃO DO ESTUDO (ADAPTADO)
Q.1 Número do protocolo: ____________ Q.2 Número do prontuário:
_____________
Q.3 Nome do responsável:_________________________________________________
Q.4 Nome do paciente:___________________________________________________
Q.5 Idade (ano e meses):_________________ Q.6 Data de Nascimento: ____________
Q.7 Procedência:_____________________ Q.8 Naturalidade: ____________________
Q.9 Nome da genitora:____________________________________________________
Q.10 Data de aplicação do questionário:___/___/___
Q.11 Telefone para contato: __________________
Q.13 Peso ao nascimento:___________________
Q.14 Etnia do paciente: 1- [ ] Branca 2-[ ] Negra 3- [ ] Amarela 4- [ ] Parda 5- [ ]
Indígena
Q.17 Consanguinidade: 1- [ ] Sim 2- [ ] Não 9- [ ]Sem informação
Q.18 Parentes com fibrose cística:
1- [ ] Ausente 2- [ ]pai 3- [ ]mãe 4 - [ ]irmão
5- [ ]primos 6- [ ]tios 7- [ ] Outros_____________
9-[ ] Sem Informação
Dados relativos à Fibrose Cística
Q.19 Idade dos primeiros sintomas: ______anos e _______ meses.
Q.20 Idade do diagnóstico: ____ anos e _____ meses.
Q.21 Diagnóstico sugerido por:
1-[ ] Sintomas respiratórios 2-[ ] História Familiar
3-[ ] Ritmo intestinal alterado (esteatorréia) 4-[ ] Retardo do crescimento

41
5-[ ] Dificuldade de ganho de peso 6-[ ] Desidratação
7-[ ] IRT elevado____________________ 8-[ ] Doença Hepato-biliar
9-[ ] Distensão abdominal 10-[ ] Distúrbio metabólico
11-[ ] Doença sinusal 12-[ ] Edema ou anemia
13-[ ] Outros_____________________ 99-[ ] Sem Informação
Q.26 Apresentou íleo meconial: 1- [ ] Sim 2- [ ] Não 9- [ ]Sem informação
Q.27 Presença de comorbidades:
(0) Ausente; (1) Doença celíaca; (2) Neuropatia; (3) Asma ; (4) Tuberculose; (5) Cílio
imóvel ; (6) Deficiência de IGA (7) Outros____________________
(9) Sem Informação
Questões referentes a sinais, sintomas e outras patologias do paciente no período
anterior ao diagnóstico de fibrose cística:
Q.28 Apresentava desconforto respiratório: (1)Sim (0)Não (99) Sem informação
Q.29 Apresentava dispnéia: (1)Sim (0)Não (99) Sem informação
Q.30 Apresentava chiado: (1)Sim (0)Não (99) Sem informação
Q.31 Apresentava tosse: (1)Sim (0)Não (99) Sem informação
Q.32 Se sim, tosse: (1)Seca (0)Produtiva (99) Sem informação
Q.33. Apresentava infecções pulmonares recorrentes: (1)Sim (0)Não (99) Sem
informação
Q.34 Apresentava sinusite: (1)Sim (0)Não (99) Sem informação
Q.35 Apresentava obstrução intestinal crônica: (1)Sim (0)Não (99) Sem informação
Q.36 Apresentava edema: (1)Sim (0) Não (99) Sem informação
Q.37 Apresentava pólipos nasais: (1) Sim (0)Não (99) Sem informação
Q.38 Apresentava esteatorreia: (1)Sim (0)Não (99) Sem informação
Q.39 Apresentava diarréia crônica: (1)Sim (0)Não (99) Sem informação
Q.40 Apresentava prolapso retal: (1)Sim (0) (99) Sem informação
Q.41 Apresentava pancreatite: (1)Sim (0)Não (99) Sem informação
Q.42 Apresentava litíase biliar: (1)Sim (0)Não (99) Sem informação
Q.43 Apresentava cianose: (1)Sim (0)Não (99) Sem informação
Q.44 Apresentava hipocratismo digital: (1)Sim (0)Não (99) Sem informação
Q.45 Apresentava dermatites: (1)Sim (0)Não (99) Sem informação

42
Q.46 Apresentava outros sinais (listar):_______________________________________
Q.47 Apresentava intolerância a glicose: (1)Sim (0)Não (99) Sem informação
Q.48 Apresentava alterações no exame da glicemia jejum: (1)Sim (0)Não (99) Sem
informação
Q.49 Escore de Shwachman (a época do diagnóstico):____________
Q.50 Escore de Shwachman atual: _______________
Estudo Genético
Q.51 Foi realizado o estudo genético?
1. [ ] Sim 2. [ ] Não 3. [ ] Não realizado 9. [ ] Sem informação
Q.52 Caso sim, mutação(ões) encontrada (s): _______________________________
Teste do Suor
Cloretos no suor, teste 1:
Q.53 1ª amostra: _______ MEq/l
Q.54 2ª amostra: _______ MEq/l
Avaliação Nutricional
Período referente ao diagnóstico:
Q.77 Peso:________________ Q.78 Altura:__________________
Q.79 IMC/I:_________________ Q.80 Peso/Altura: ___________
Q.81 Peso/Idade _____________ Q.82 Altura/Idade: ___________
Colonização Bacteriana
Q.124 Escarro 1
(Momento do
diagnóstico)
Data:__/__/__
Isolado:
0.[ ] Ausente 1.[ ] P. aeruginosa 2. [ ] Pseudomonas cepa
mucoide
3. [ ] Pseudomonas multiresistente 4.[ ]S. aureus 5. [ ] MSSA
6.[ ] MRSA 7.[ ]Klebsiella 8.[ ]H. influenza 9.[ ]Flora
saprófita
10.[ ]S. maltophilia 11.[ ] B. cepacea 12.[ ]Aspergillus

43
13.[ ]Micobactéria 14.[ ]Sem informação
15.[ ]Outros_________________________________________
Q.125 Escarro 2
(Momento da
introdução no
estudo)
Data:__/__/__
Isolado:
0.[ ] Ausente 1.[ ] P. aeruginosa 2. [ ] Pseudomonas cepa
mucoide
3. [ ] Pseudomonas multiresistente 4.[ ]S. aureus 5. [ ] MSSA
6.[ ] MRSA 7.[ ]Klebsiella 8.[ ]H. influenza 9.[ ]Flora
saprófita
10.[ ]S. maltophilia 11.[ ] B. cepacea 12.[ ]Aspergillus
13.[ ]Micobactéria 14.[ ]Sem informação
15.[ ]Outros_________________________________________
Q.126 Já foi colonizado por Pseudomonas: (1)Sim (0)Não (99) Sem informação
Q.127 Qual a idade do primeiro isolamento: ___ anos e _____ meses (99) não se aplica
Q. 128 Qual a idade do segundo isolamento: ___ anos e _____ meses (99) não se aplica
Q. 129 É colonizado crônico por pseudomonas: (1) Sim (0)Não (99) Sem informação
Data Resultado da cultura – microrganismo isolado (usar código)

44
APÊNDICE II: Termo de Consentimento Livre e Esclarecido (TCLE)

45
46
XI. ANEXOS
ANEXO I: Parecer do Comitê de Ética em Pesquisa do HUPES

47





























