Embed Size (px)
Citation preview

CASO CLÍNICO Joana Ramos Lopes, M. Carmo Macário
Serviço de Neurologia, Centro Hospitalar e Universitário de Coimbra
XIV Curso Básico Doenças Hereditárias Metabolismo Coimbra 11,12 e 13 de Dezembro de 2017

Género feminino
• Antecedentes pessoais: sem relevância
• Sem medicação habitual
• Sem história familiar de doenças neurodegenerativas ou outras
• Sem consanguinidade na família
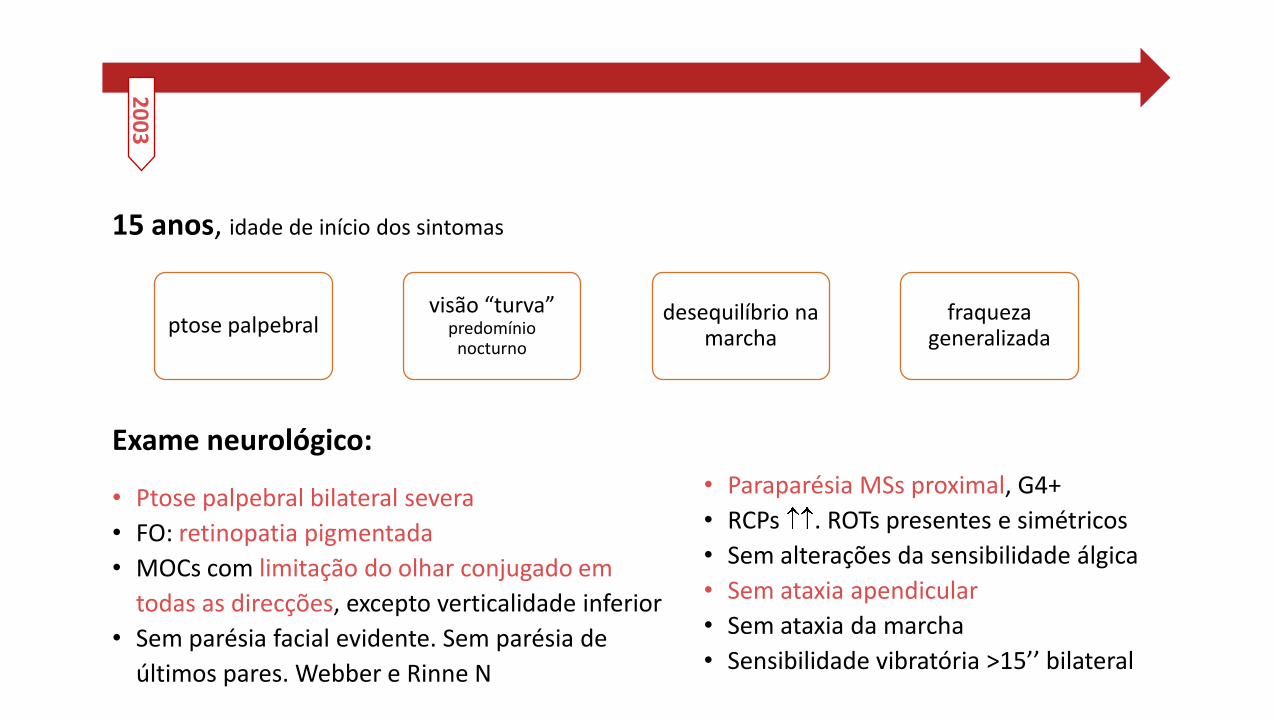
Exame neurológico:
• Ptose palpebral bilateral severa
• FO: retinopatia pigmentada
• MOCs com limitação do olhar conjugado em
todas as direcções, excepto verticalidade inferior
• Sem parésia facial evidente. Sem parésia de
últimos pares. Webber e Rinne N
20
03
ptose palpebral visão “turva”
predomínio nocturno
desequilíbrio na marcha
fraqueza generalizada
15 anos, idade de início dos sintomas
• Paraparésia MSs proximal, G4+
• RCPs . ROTs presentes e simétricos
• Sem alterações da sensibilidade álgica
• Sem ataxia apendicular
• Sem ataxia da marcha
• Sensibilidade vibratória >15’’ bilateral

MCDTs REALIZADOS:
20
06
Estudo analítico completo
sem alterações
TC-ce
moderada atrofia cerebelosa média
e superior
ECG
sem alterações
Ecocardiograma TT
sem alterações
ECG-Holter 24h
normal RM-ce
marcada atrofia cortical cerebral e
cerebelosa de predomínio
vermiano inferior

20
07
Biópsia de músculo:
• fibras rotas e vermelhas e fibras COX negativas dispersas, com aspecto compatível com Doença Mitocondrial.
Estudo na cadeia respiratória nos leucócitos e músculos:
• diminuição da actividade do complexo IV (37% da actividade em relação ao normal).
DNA mitocondrial:
• delecção de 4977pb nos leucócitos e no músculo.

20
10
• Emagrecimento
P= 37kg »» IMC <18,5
• Anorexia, enfartamento pós-prandial
• Alteração do padrão intestinal
Excluídos distúrbios alimentares
• doseamento de timidina soro: N
• mutação do gene TYM em heterozigotia
• gene POLG: sem alterações
Investigação para Síndrome MNGIE (encefalomiopatia
neuro-gastro-intestinal mitocondrial)

AGRAVAMENTO PROGRESSIVO DAS QUEIXAS
P = 35kg 2
01
7
Exame neurológico:
• Sem alterações cognitivas • Sem disartria • Ptose palpebral severa (já operada). • OEP severa, com incapacidade de MOCs em
qualquer direcção, com olhar fixo em frente Diplopia ocasional
• Acuidade visual 4/10 OD, 5/10 OE, com retinopatia pigmentar
• Diparésia facial • Disfonia. Disfagia ocasional. • Sem surdez • Teraparésia G4, predomínio proximal • Sem alterações da sensibilidade • Sem ataxia
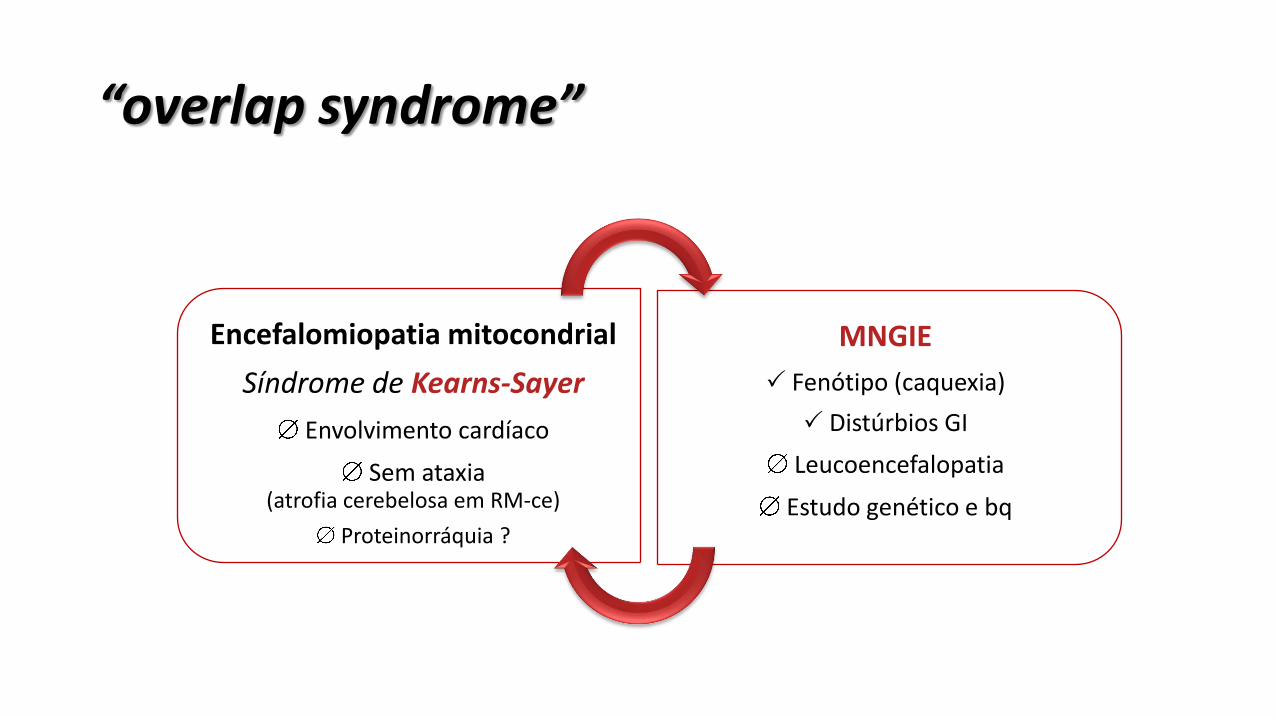
“overlap syndrome”
Encefalomiopatia mitocondrial
Síndrome de Kearns-Sayer
Envolvimento cardíaco
Sem ataxia (atrofia cerebelosa em RM-ce)
Proteinorráquia ?
MNGIE
Fenótipo (caquexia)
Distúrbios GI
Leucoencefalopatia
Estudo genético e bq

breve revisão teórica

síndromes de delecção mitocondrial Kearns-Sayre
Syndrome
PEO Pearson
Syndrome
Doença multissistémica
Tríade característica
Diagnóstico: clínica + biópsia + diminuição da actividade de complexos respiratórios +
delecção mtDNA
++ SNC, músculo esquelético, coração
Idade início <20 anos
Retinopatia pigmentada
OEP
“delecção comum” = delecção de 4977 pb
Ke
arn
s-Sa
yre
Sy
nd
rom
e

síndromes de deplecção mitocondrial
Encefalomiopatia neuro-gastro-intestinal mitocondrial
-MNGIE-
• Mutações AR no gene TYMP (timidina fosforilase)
• Clínica: ptose + oftamolparésias + caquexia + dismotilidade GI + neuropatia periférica + leucoencefalopatia difusa (RM-ce)
• Patologia mitocondrial: aumento de lactatos, biópsia muscular + , diminuição da actividade da c. respiratória, deplecções ou delecções múltiplas do mtDNA
• MNGIE-like syndromes ( leucoencefalopatia na RM-ce) têm sido associados a mutações patogénicas no
mtDNA, assim como mutações noutros genes nucleares responsáveis pela manutenção do mt DNA (ex. POLG).
Défice de TP nucleosídeos plasmáticos (timidina e deoxiuridina) desequilíbrio no pool de
nucleótidos instabilidade do mtDNA

• Saneto RP, Parikh S, Cohen BH. Mitochondrial Case Studies: Underlying Mechanisms and Diagnosis. Elsevier Inc., 2015. 338 p. • Suh, B. C., Jeong, H., Yoon, B. S., et al. Compound heterozygous mutations of TYMP as underlying causes of mitochondrial neurogastrointestinal
encephalomyopathy (MNGIE). Molecular Medicine Reports 8.1 (2013): 17-22. • Khambatta S, Nguyen DL, Beckman TJ, Wittich CM. Kearns-Sayre syndrome: a case series of 35 adults and children. Int J Gen Med. 2014 Jul 3;7:325-32. • DiMauro S, Hirano M. Mitochondrial DNA Deletion Syndromes. GeneReviews. 2011, May 3.
Ref
erê
nci
as




