Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de Fevereiro de 1808
Monografia
Correlação Clínico-Radiológico-Patológica das Vasculites ANCA-
Associadas com Envolvimento Pulmonar
Camila Tavares Joau e Silva
Salvador (Bahia)
Junho, 2012

Ficha catalográfica elaborada pela Biblioteca Universitária de Saúde, SIBI - UFBA.
S586 Silva, Camila Tavares Joau e
Correlação clínico-radiológico-patológica das vasculites ANCA-associadas com envolvimento pulmonar / Camila Tavares Joau e Silva. – Salvador, 2012.
90 f.
Orientador: Prof. Dr. Jorge Luiz Pereira e Silva Trabalho de Conclusão de Curso (Graduação) –
Universidade Federal da Bahia. Faculdade de Medicina da Bahia, 2012.
1. Medicina. 2. Vasculites. 3. Saúde. I. Silva, Jorge Luiz Pereira e. II. Universidade Federal da Bahia. III. Título.
CDU 616

UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de Fevereiro de 1808
Monografia
Correlação Clínico-Radiológico-Patológica das Vasculites ANCA-
Associadas com Envolvimento Pulmonar
Camila Tavares Joau e Silva
Professor-orientador: Jorge Luiz Pereira e Silva
Monografia de conclusão do componente curricular MED-B60, do currículo médico da Faculdade de Medicina da Bahia (FMB) da Universidade Federal da Bahia (UFBA), apresentada ao Colegiado do Curso de Graduação em Medicina da FMB-UFBA.
Salvador (Bahia), 2012
iii

Monografia: Correlação Clínico-Radiológico-Patológica das Vasculites ANCA-Associadas com Envolvimento Pulmonar. Camila Tavares Joau e Silva
Professor-orientador: Jorge Luiz Pereira e Silva
COMISSÃO EXAMINADORA Membros Titulares:
• Jorge Luiz Pereira e Silva (Presidente), Professor Associado do Departamento de Medicina Interna e Apoio Diagnóstico, FMB-UFBA.
• César Augusto de Araújo Neto, Professor Assistente do Departamento de Medicina Interna e Apoio Diagnóstico, FMB-UFBA.
• Antônio Raimundo Pinto de Almeida, Professor Associado do Departamento de Medicina Interna e Apoio Diagnóstico, FMB-UFBA
Salvador (Bahia), 2012
iv

Dedico este trabalho aos meus pais: à minha
querida mãe, Isabella Amaral Tavares Joau e
Silva, médica competente e inspiradora, mãe
amorosa e grande incentivadora de sua filha; e
ao meu querido pai, Sérgio Augusto Pereira
Joau e Silva, meu maior exemplo de ser
humano e verdadeiro orientador para a vida.
À minha família, avós, tios, primos e, em
especial, à minha irmã Giulia e a Marcelio, a
minha gratidão e o meu reconhecimento por
todo o apoio e carinho.

AGRADECIMENTOS
À Faculdade de Medicina da Bahia (FMB), professores e funcionários da
Universidade Federal da Bahia (UFBA), com especial destaque para a
Professora Dra. Lorene Louise Silva Pinto , Diretora da FMB, bem como a
todos aqueles que direta ou indiretamente contribuíram para a realização desta
obra, o meu muito obrigado pelos ensinamentos e orientações.
Ao estimado Professor Dr. José Tavares-Neto, Coordenador do Núcleo de
Formação Científica da FMB-UFBA, pela criteriosa orientação e irrestrito apoio.
Ao querido Professor Dr. Jorge Luiz Pereira e Silva da FMB-UFBA, estimado
amigo, orientador e grande incentivador, pelo estímulo, partilha de saber e
apoio dados na elaboração deste meu trabalho.
Aos estimados Professores Doutores César Augusto de Araújo Neto e
Antônio Raimundo Pinto de Almeida , Professores do Departamento de
Medicina Interna e Apoio Diagnóstico, FMB-UFBA, pela anuência e honrosa
participação na banca examinadora deste trabalho.
Aos funcionários da FMB-UFBA por seus préstimos, em especial ao senhor
Antônio Damasceno e ao dedicado pessoal da Biblioteca, em especial à
senhora Rita , pelo apoio constante.
Aos colegas e amigos de turma do curso de Medicina da UFBA pelas
demonstrações de apreço, amizade e consideração, bem como pelo rico
aprendizado, fruto da sadia convivência ao longo de todo o curso.
Aos meus pais, companheiros e fiéis revisores desta obra, pela paciência e
pelo apoio incondicional e incentivo.
Finalmente, ao Deus da minha fé que guarda os meus dias e ilumina os meus
caminhos. A todos, o meu muito obrigado!

RESUMO
Esta monografia aborda a correlação clínico-radiológico-patológica das
vasculites ANCA-associadas (VAA) com envolvimento pulmonar. O objetivo do
trabalho é, a partir do estudo das vasculites sistêmicas e das VAA com
envolvimento pulmonar, verificar a importância da abordagem integrada clínico-
radiológico-patológica no diagnóstico e tratamentos dessas doenças. A
metodologia adotada comportou uma pesquisa bibliográfica, documental e
exploratória, predominantemente fundamentada em livros e periódicos, visando
a obter o adequado embasamento teórico para o estudo em tela. Mediante a
apresentação de casos ilustrativos bem-documentados, este estudo discorre
sobre as vasculites sistêmicas e suas características clínicas; descreve os
fundamentos da classificação atual e dos critérios diagnósticos das vasculites
sistêmicas; e apresenta os aspectos clínicos, radiológicos e histopatológicos
das VAA, com especial atenção ao acometimento pulmonar. Os principais
tópicos são: definição e classificação das vasculites sistêmicas e a sua
perspectiva histórica; vasculites dos grandes, médios e pequenos vasos; e a
epidemiologia, etiologia e patogênese, manifestações clínicas, características
histopatológicas e radiológicas, diagnóstico, prognóstico e o tratamento das
VAA. A conclusão demonstra a importância da combinação dos aspectos
clínicos, radiológicos, laboratoriais e histopatológicos particulares de cada
doença para o correto diagnóstico das vasculites, em especial das ANCA-
associadas com envolvimento pulmonar, o que confirma a hipótese da autora
da imprescindibilidade dessa abordagem integrada, não apenas para o
adequado diagnóstico, mas também para o eficaz tratamento dessas doenças.
Como corolário dessa conclusão, afirma, ainda, que as VAA estudadas,
embora guardem entre si algumas características comuns, podem ser
diferenciadas com base na correlação multidisciplinar.
Palavras chave: Vasculites. Vasculites sistêmicas. Vasculites ANCA-
associadas. Síndrome de Churg-Strauss. Poliangiite granulomatosa (Wegener).
Poliangiite microscópica.

ABSTRACT
This monograph addresses the clinical-radiologic-pathologic ANCA-associated
vasculitis (AAV) with pulmonary involvement. This work is based on the study of
systemic vasculitis and AAV with pulmonary involvement. It verifies the
importance of an integrated clinical-radiologic-pathologic correlation in
diagnosis and treatment of these diseases. The methodology involved a
literature review, documentary and exploratory, predominantly based on books
and periodicals in order to achieve adequate theoretical basis for this study.
Upon the presentation of well-documented illustrative cases, this study
discusses the systemic vasculitis and their clinical characteristics, describes the
principles of the current classification and diagnostic criteria of systemic
vasculitis, and presents the clinical, radiological and pathological findings of the
AAV, particularly with pulmonary involvement. The main topics are: definition
and classification of systemic vasculitis and its historical perspective; vasculitis
of large, medium and small vessels, and the epidemiology, etiology and
pathogenesis, clinical manifestations, histopathological and radiological
features, diagnosis, prognosis and treatment of AAV. The conclusion
demonstrates the importance of the combination of clinical, radiological,
laboratory and histopathology individual features of each disease for the
diagnosis of vasculitis, particularly the ANCA-associated with pulmonary
involvement, which confirms the author’s hypothesis about the indispensability
of this integrated approach, not only suitable for diagnosis, but also for the
effective treatment of these diseases. As a corollary of this conclusion, this work
also states that the AAV, although keep common characteristics, can be
differentiated based on a multidisciplinary correlation.
Keywords: Vasculitis. Systemic vasculitis. ANCA-associated vasculitis. Churg-
Strauss syndrome. Granulomatosis with polyangiitis (Wegener). Microscopic
polyangiitis.

LISTA DE ILUSTRAÇÕES
QUADRO 1
FIGURA 1
Classificação das Vasculites ...................... .................................... 15
ANCA-C por imunofluorescência indireta ............ ......................... 27
FIGURA 2 ANCA-P por imunofluorescência indireta . .................................... 28
TABELA 1
TABELA 2
FIGURA 3
Tipos de ANCA associados com as Vasculites Primária s de
Pequenos Vasos .................................... .......................................... 29
Manifestações Clínicas da SCS ..................... ................................. 35
Imagem de biópsia (pneumonia eosinofílica e granulo ma
extravascular) .................................... ............................................... 38
FIGURA 4
FIGURA 5
FIGURA 6
FIGURA 7
FIGURA 8
QUADRO 2
QUADRO 3
TABELA 3
FIGURA 9
FIGURA 10
FIGURA 11
FIGURA 12
FIGURA 13
FIGURA 14
FIGURA 15
QUADRO 4
FIGURA 16
TABELA 4
FIGURA 17
Imagem de biópsia (capilarite necrosante) ......... .......................... 39
Radiografia torácica de paciente com SCS .......... ......................... 40
Tomografia computadorizada de tórax de paciente com SCS .... 41
Tomografia computadorizada de tórax de paciente com SCS .... 41
Tomografia computadorizada de tórax de paciente com SCS .... 42
Critérios do ACR de 1990 para a Classificação de SC S ............... 43
Five-Factor Score (FFS) – Escore de Cinco Fatores . ................... 47
Principais Manifestações da PAG ................... ............................... 52
Radiografia torácica de paciente com hemorragia alv eolar
secundária a PAG .................................. .......................................... 53
Capilarite e hemorragia pulmonar em PAG ........... ........................ 54
Nariz em sela em paciente com poliangiite granuloma tosa ........ 55
Fotomicrografia de biópsia renal (GNRP) de paciente com PAG 56
Imagem de biópsia (arterite granulomatosa e venulit e) em PAG 57
Imagem de biópsia (microabscesso e granuloma necros ante) em
PAG ................................................................................................... 58
Exemplos de manifestações radiológicas da PAG ..... .................. 59
Critérios do ACR de 1990 para a classificação da PA G ............... 60
Características histológicas da PAG ............... .............................. 62
Manifestações clínicas da PAM ..................... ................................. 66
Imagem de biópsia de tecido renal (glomerulonefrite necrosante
segmentar) em PAM ................................. ........................................ 67

FIGURA 18
FIGURA 19
FIGURA 20
QUADRO 5
TABELA 5
Imagem de biópsia pulmonar (hemorragia alveolar e c apilarite
neutrofílica) em PAM .............................. ......................................... 68
Radiografia torácica de paciente com PAM .......... ........................ 68
Tomografia computadorizada de tórax de paciente com PAM ... 69
Quadro comparativo entre as VAA com envolvimento pu lmonar 71
Avaliação da gravidade das vasculites e tratamento ................... 72

LISTA DE ABREVIATURAS E SIGLAS
ACG
ACR
ALC
ANCA
-
-
-
-
Arterite de Células Gigantes
American College of Rheumatology
Angiite Leucocitoclástica Cutânea
Anticorpo Anticitoplasma de Neutrófilos
Anti-MBG
ASN
AT
-
-
-
Anticorpo Anti-Membrana Basal Glomerular
Sociedade Americana de Nefrologia
Arterite de Takayasu
CCH - Consenso de Chapel Hill
CG - Catepsina G
DK - Doença de Kawasaki
ELISA - Enzyme-Linked Immunoabsorbent Assay
EULAR - Liga Européia contra o Reumatismo
EUVAS - European Vasculitis Study Group
FR - Fator Reumatóide
FFS - Five-Factor Score
FMB - Faculdade de Medicina da Bahia
GN
GNRP
HAD
HAS
H&E
HLA
HLE
IFI
-
-
-
-
-
-
-
-
Glomerulonefrite
Glomerulonefrite Rapidamente Progressiva
Hemorragia Alveolar Difusa
Hipertensão Arterial Sistêmica
Hematoxilina e Eosina
Human Leucocyte Antigen
Elastase
Imunofluorescência Indireta
IgA
IgE
IgG
IgM
-
-
-
-
Imunoglobulina A
Imunoglobulina E
Imunoglobulina G
Imunoglobulina M
LBA - Lavado Broncoalveolar
LF - Lactoferrina
MPO - Mieloperoxidase
PAG - Poliangiite Granulomatosa

PAM - Poliangiite Microscópica
PAN - Poliarterite Nodosa
PAS
PCR
-
-
Periodic acid-Schiff (do inglês) Proteína C Reativa
PHS
PR3
-
-
Púrpura de Henoch-Schönlein
Proteinase 3
SCS - Síndrome de Churg-Strauss
SHE - Síndrome Hipereosinofílica
TC
TCAR
-
-
Tomografia Computadorizada
Tomografia Computadorizada de Alta Resolução
UFBA - Universidade Federal da Bahia
VAA
VAS
VC
VHS
-
-
-
-
Vasculites ANCA-associadas
Vias Aéreas Superiores
Vasculite Crioglobulinêmica
Velocidade de Hemossedimentação

1
1
SUMÁRIO
1 INTRODUÇÃO .............................................................................................. 03
2 AS VASCULITES SISTÊMICAS ................................................................... 05
2.1 VASCULITES SISTÊMICAS: DEFINIÇÃO E CLASSIFICAÇÃO ................... 05
2.2 VASCULITES SISTÊMICAS: PERSPECTIVA HISTÓRICA DAS
CLASSIFICAÇÕES ................................................................................................... 07
2.2.1 Vasculites dos Grandes Vasos .................................................................. 09
2.2.1.1 Arterite de Células Gigantes (Arterite Temporal) ........................................... 09
2.2.1.2 Arterite de Takayasu ..................................................................................... 10
2.2.2 Vasculites de Vasos de Médio Porte ......................................................... 11
2.2.2.1 Poliarterite Nodosa ........................................................................................ 11
2.2.2.2 Doença de Kawasaki ..................................................................................... 12
2.2.3 Vasculites de Pequenos Vasos .................................................................. 13
2.2.3.1 Púrpura de Henock-Schönlein ....................................................................... 14
2.2.3.2 Vasculite Crioglobulinêmica .......................................................................... 15
2.2.3.3 Síndrome do Anticorpo anti-Membrana Basal Glomerular ........................... 16
2.2.3.4 Vasculites ANCA-Associadas ........................................................................ 17
3 VASCULITES ANCA-ASSOCIADAS ............................................................... 20
3.1 SÍNDROME DE CHURG-STRAUSS ................................................................. 20
3.1.1 Epidemiologia ................................................................................................. 21
3.1.2 Etiologia e Patogênese .. ................................................................................ 22
3.1.3 Manifestações Clínicas .................................................................................. 24
3.1.4 Achados Histopatológicos ............................................................................ 28
3.1.5 Apresentação Radiológica .. .......................................................................... 30
3.1.6 Diagnóstico . .................................................................................................... 33
3.1.7 Prognóstico .. .................................................................................................. 36
3.2 POLIANGIITE GRANULOMATOSA............... ................................................... 38
3.2.1 Substituição do Epônimo .............................................................................. 39
3.2.2 Epidemiologia .. ............................................................................................... 39
3.2.3 Etiologia e Patogênese .................................................................................. 41
3.2.4 Manifestações Clínicas .................................................................................. 41

2
2
3.2.5 Achados Histopatológicos .. .......................................................................... 47
3.2.6 Apresentação Radiológica . ........................................................................... 48
3.2.7 Diagnóstico .. ................................................................................................... 50
3.2.8 Prognóstico .. .................................................................................................. 53
3.3 POLIANGIITE MICROSCÓPICA ....................................................................... 54
3.3.1 Epidemiologia ................................................................................................. 54
3.3.2 Etiologia e Patogênese .. ................................................................................ 55
3.3.3 Manifestações Clínicas .................................................................................. 55
3.3.4 Achados Histopatológicos ............................................................................ 56
3.3.5 Apresentação Radiológica .. .......................................................................... 58
3.3.6 Diagnóstico . .................................................................................................... 59
3.3.7 Prognóstico .. .................................................................................................. 61
3.4 TRATAMENTO DAS VASCULITES ANCA-ASSOCIADAS ............................... 62
4 CONCLUSÃO ................................................................................................... 65
REFERÊNCIAS ................................................................................................. 67

3
1 INTRODUÇÃO
“As vasculites pulmonares primárias são doenças raras e seus diagnósticos estão entre os mais desafiadores da medicina, por serem os sinais e sintomas inespecíficos e comuns a diversas outras doenças, (...); o diagnóstico das vasculites depende, sobretudo, do reconhecimento de combinações de características clínicas, radiológicas, laboratoriais e histopatológicas particulares” (tradução livre).
Doutora Eva Castañer, Membro do Serviço de Radiologia do Hospital del Parc Taulí de Sabadell, Espanha (CASTAÑER, 2010).
O termo vasculite compreende uma série de condições clínicas
heterogêneas, caracterizadas por um processo inflamatório destrutivo
acometendo a parede dos vasos sanguíneos. Diferentes tipos de vasos em
diversos órgãos podem estar envolvidos, podendo afetar artérias, arteríolas,
capilares, vênulas e veias, isto é, vasos de grande, médio e pequeno calibres.
Dessa forma, costumam apresentar-se com manifestações clínicas variadas,
com sinais e sintomas determinados pelo tipo, tamanho e localização dos
vasos acometidos.
As vasculites podem ser secundárias a condições subjacentes, como
infecções, drogas, distúrbios de hipersensibilidade, doenças do tecido
conjuntivo e doenças neoplásicas, sendo a inflamação vascular, nestes casos,
uma manifestação da doença de base. Nas situações em que a vasculite é o
processo primário, são denominadas de vasculites primárias. Estas são
doenças sistêmicas raras e de etiologia desconhecida, sendo classificadas de
acordo com o calibre dos vasos predominantemente acometidos.
As vasculites paucimunes ANCA-associadas1 são classificadas como
vasculites de pequenos vasos e caracterizadas por inflamação necrosante
afetando principalmente capilares, vênulas, arteríolas e pequenas artérias. No
cenário das vasculites sistêmicas, o envolvimento pulmonar é mais freqüente
nesse subgrupo, foco principal desse estudo. Além da baixa prevalência, seu
diagnóstico representa um grande desafio devido à baixa especificidade dos
sinais e sintomas, por vezes sobrepostos e facilmente confundidos com os de
outras condições. Dessa forma, o diagnóstico das vasculites baseia-se na
1 Anticorpos anticitoplasma de neutrófilos, ou ANCA, são imunoglobulinas dirigidas contra
enzimas constituintes dos grânulos citoplasmáticos de neutrófilos. Sua presença está associada a determinado grupo de vasculites (mais informações a respeito serão detalhadas ao longo do trabalho).

4
correlação entre os achados clínicos, radiológicos, laboratoriais e
histopatológicos peculiares a cada uma dessas entidades, que requer um alto
índice de suspeição diagnóstica.
O presente trabalho foi elaborado por meio de uma pesquisa
bibliográfica e exploratória, predominantemente fundamentada em livros e
periódicos, com base na seguinte premissa: o diagnóstico específico das
vasculites ANCA-associadas (VAA) depende fundamentalmente de um alto
índice de suspeição diagnóstica fundamentado na correlação clínico-
radiológico-patológica, complementada por exames laboratoriais específicos.
Mediante a apresentação de casos ilustrativos bem-documentados, este
estudo aborda o tema vasculites ANCA-associadas com envolvimento
pulmonar, considerando os aspectos clínicos, radiológicos e histopatológicos
destas doenças; bem como discorre sobre as vasculites sistêmicas e suas
características clínicas; descreve os fundamentos da classificação atual e dos
critérios diagnósticos das vasculites sistêmicas; apresenta os aspectos clínicos,
radiológicos e histopatológicos das VAA, com especial atenção ao
acometimento pulmonar; e advoga a importância da correlação clínico-
radiológico-patológica para o diagnóstico específico das VAA com
envolvimento pulmonar.
Embora haja outros tipos de vasculites de pequenos vasos com
acometimento pulmonar, no presente trabalho serão abordadas apenas as
vasculites ANCA-associadas: síndrome de Churg-Strauss (SCS), poliangiite
granulomatosa (PAG) e poliangiite microscópica (PAM).
No desenvolvimento desta monografia, o primeiro capítulo após a
introdução é dedicado ao estudo das vasculites sistêmicas (definição e
classificação). A seguir, no Capítulo 3, são apresentadas as VAA, com
destaque para a epidemiologia, etiologia e patogênese, manifestações clínicas,
características histopatológicas e radiológicas, diagnóstico, prognóstico e
tratamento das mesmas. Finalmente, são apresentadas conclusões que
permitirão ao leitor perceber como a abordagem integrada clínico-radiológico-
patológica ao paciente com vasculite, em especial as ANCA-associadas com
envolvimento pulmonar, é de fundamental importância para o seu adequado
diagnóstico e, dessa forma, entender a sua imprescindibilidade no tratamento
dessas doenças.

5
2 AS VASCULITES SISTÊMICAS
2.1 VASCULITES SISTÊMICAS: DEFINIÇÃO E CLASSIFICAÇÃO
As vasculites sistêmicas compreendem um grupo de doenças
caracterizadas por um processo inflamatório que acomete vasos sanguíneos,
acompanhado de destruição das paredes vasculares e necrose tecidual
(ARAS, 2011). O termo vasculite compreende diversas entidades nosológicas
(QUADRO 1), que podem ser secundárias a condições subjacentes - como
infecções, uso de drogas, distúrbios de hipersensibilidade, doenças do tecido
conjuntivo e enfermidades neoplásicas (BARBAS, 2005) - sendo a inflamação
vascular, a manifestação preponderante da doença de base. Nas situações em
que a vasculite é o processo fundamental, elas são denominadas vasculites
primárias.
CLASSIFICAÇÃO DAS VASCULITES
VASCULITES IDIOPÁTICAS PRIMÁRIAS
Grandes vasos: Arterite de células gigantes Arterite de Takayasu
Médios vasos: Poliarterite nodosa Doença de Kawasaki
Pequenos vasos: Síndrome de Churg-Strauss Poliangiite granulomatosa Poliangiite microscópica
VASCULITES PRIMÁRIAS MEDIADAS POR IMUNOCOMPLEXOS Púrpura de Henoch-Schönlein Vasculite Crioglobulinêmica Síndrome de Goodpasture
VASCULITES SECUNDÁRIAS Doenças autoimunes:
Lúpus eritematoso sistêmico Artrite reumatóide Polimiosite, dermatomiosite Esclerose sistêmica progressiva

6
Síndrome de anticorpos antifosfolípidas Doença inflamatória intestinal Reação medicamentosa Síndromes paraneoplásicas Infecções
Quadro 1 – Classificação das Vasculites
Fonte: Castañer, 2010.
As vasculites primárias, por sua vez, são doenças sistêmicas raras e de
etiologia desconhecida. Diferentes tipos de vasos em diversos órgãos são
envolvidos, podendo afetar artérias, arteríolas, capilares, vênulas e veias, cuja
distribuição preponderante constitui a base da classificação das vasculites de
grande, médio e pequeno calibre (WIIK, 2003). As manifestações clínicas são
variadas, com sinais e sintomas determinados pelo tipo, tamanho e localização
dos vasos acometidos. As apresentações clínicas normalmente incluem sinais
e sintomas constitucionais - como febre, mialgia, artralgia, mal-estar geral -
acompanhadas de manifestações locais de isquemia ou sangramento, bem
como de sintomas sistêmicos que refletem o comprometimento vascular.
A classificação das vasculites sistêmicas mais amplamente utilizada diz
respeito ao calibre dos vasos afetados. Os grandes vasos geralmente denotam
a artéria aorta e seus principais ramos, sendo as principais vasculites deste
grupo a arterite de células gigantes (ACG) e arterite de Takayasu (AT). Os
vasos de médio calibre dizem respeito às artérias viscerais (por exemplo:
artérias renais, hepáticas, mesentéricas e coronárias), estando compreendidas
neste grupo, dentre outras, a poliarterite nodosa (PAN) e a doença de
Kawasaki (DK). E, por fim, os pequenos vasos, incluindo pequenas artérias,
arteríolas, vênulas e capilares. Integram este último grupo as vasculites
mediadas por imunocomplexos – púrpura de Henoch-Schönlein (PHS) e a
vasculite crioglobulinêmica (VC) –, a angiite leucocitoclástica cutânea (ALC), as
associadas ao anticorpo anti-membrana basal glomerular (anti-MBG) e as
chamadas vasculites paucimunes ou ANCA-associadas – síndrome de Churg-
Strauss (SCS), poliangiite granulomatosa (PAG) e poliangiite microscópica
(PAM).

7
2.2 VASCULITES SISTÊMICAS: PERSPECTIVA HISTÓRICA DAS
CLASSIFICAÇÕES
Desde as primeiras descrições da poliarterite nodosa, por Kussmaul e
Maier em 1866 (KUSSMAUL e MAIER, 1866), muitos buscaram desenvolver
uma nomenclatura unificada e um sistema de classificação para as vasculites
sistêmicas. No entanto, as definições e classificações propostas são ainda
objeto de controvérsia. Estudos epidemiológicos e um maior entendimento
acerca das características clínicas, do prognóstico e da resposta terapêutica
destas doenças ficavam, por conseguinte, comprometidos, em face da
dificuldade em se realizar estudos científicos frente à ausência de uma forma
padronizada para classificá-las (KALLENBERG, 2008).
A classificação empregada inicialmente considerava o tamanho dos
vasos acometidos e achados histopatológicos na biópsia da lesão vascular
(BRUCE e BELL, 1997). Era, todavia, bastante abrangente, por vezes
admitindo dúvidas quanto ao diagnóstico específico, não sendo incomum que
diferentes nomes fossem usados para se referir a uma mesma doença
(JENNETTE e FALK, 1994). Fazia-se necessária, portanto, uma classificação
mais uniforme e eficaz, que definisse critérios apropriados a serem preenchidos
para o estabelecimento do diagnóstico adequado da doença.
Dois principais sistemas de classificação foram então propostos – os
critérios do Colégio Americano de Reumatologia (American College of
Rheumatology, ACR) de 1990 e o Consenso de Chapel Hill (CCH) de 1994 –,
permanecendo amplamente utilizados por muito tempo (LIU, 2008).
Os critérios do ACR de 1990 surgiram a partir da análise de um vasto
número de casos de vasculites, baseada em características clínicas e exames
complementares (BRUCE e BELL, 1997). Foram concebidos notadamente com
o intuito de apresentar uma abordagem diagnóstica padronizada e eficaz que
permitisse a realização de estudos epidemiológicos (HUNDER, 1990). Apesar
de oferecer grande valor discriminatório entre uma síndrome vasculítica e
outra, não se adequava, porém, para distinguir vasculite de outras doenças.
Além disso, deixava de incluir algumas doenças, como a poliangiite
microscópica, tampouco considerava os anticorpos anticitoplasma de
neutrófilos (ANCA) nos critérios de classificação (LANE, 2002).

8
O Consenso de Chapel Hill (CCH) para a Nomenclatura das Vasculites
Sistêmicas, realizada na cidade americana de Chapel Hill, no estado da
Carolina do Norte (EUA), em 1992 e publicada em 1994, contou com a
participação de um grupo seleto de clínicos e patologistas de seis países e de
diversas especialidades, tais como reumatologistas, nefrologistas e
imunologistas, com vasta experiência no diagnóstico de vasculites (JENNETTE
e FALK, 1994). O documento que resultou desse encontro multidisciplinar tinha
por objetivo determinar um sistema unificado de nomenclatura aliado a
conceitos relacionados aos principais tipos de vasculites (JENNETTE e FALK,
1994). Como principal limitação, não estabelecia critérios diagnósticos, fato que
restringia o seu uso na prática clínica (KALLENBERG, 2008). Outro avanço
apresentado pelo Consenso de Chapel Hill foi o reconhecimento da importância
do ANCA para o diagnóstico das chamadas vasculites paucimunes (WATTS,
2007), ou vasculites ANCA-associadas (VAA), o principal escopo deste
trabalho, que serão pormenorizadas no próximo capítulo.
Posteriormente, a Nova Classificação Européia, atualmente utilizada,
surgiu sob a forma de algoritmo para o diagnóstico específico das vasculites
sistêmicas. Esta combinava os critérios do ACR, os conceitos do Consenso de
Chapel Hill e os critérios de Lanham2 para a síndrome de Churg-Strauss
(WATTS, 2007). Este algoritmo trouxe vantagens, destacando sua classificação
clínica simplificada para as VAA e para a poliarterite nodosa (PAN)3
(KALLENBERG, 2008), facilitando assim a realização de estudos
epidemiológicos e comparativos, e sua aplicação clínica (LIU, 2008). De acordo
com Liu et al, com o algoritmo de Watts, reduzir-se-ia o número de casos que
permaneceriam sem classificação definida, bem como a ocorrência de
sobreposição de diagnósticos.
2 Critérios de Lanham: critérios clínicos publicados em 1984 por Lanham et at para o diagnóstico clínico da síndrome de Churg-Strauss. Os pacientes analisados em seu estudo possuíam o diagnóstico de SCS, porém não era possível classificá-los segundo os critérios da ACR nem pelos critérios do CCH (LANHAM, 1984; REID, 1998). Mais informações sobre os critérios retro mencionados constam do subitem 3.1 - Síndrome de Churg-Strauss, deste trabalho. 3 Poliarterite nodosa (PAN): esta foi incluída no algoritmo de Watts devido a ocorrência de sobreposição de diagnósticos. Pacientes classificados como PAN, de acordo com os critérios do ACR, eram considerados como portadores de poliangiite microscópica, segundo os conceitos do CCH.

9
A seguir, são apresentadas considerações sobre cada grupo das
vasculites de grandes, médios e de pequenos vasos.
2.2.1 Vasculites dos Grandes Vasos As grandes artérias elásticas, a saber, a aorta e seus grandes ramos,
contribuem para estabilizar o fluxo sanguíneo, tornando-o mais uniforme e,
assim, contribuindo para manter a pressão arterial e a perfusão tecidual. Estas
artérias são as principais acometidas nas vasculites que compõem este grupo.
As principais vasculites dos vasos de grande calibre - a arterite de células
gigantes e a arterite de Takayasu - são caracterizadas pela presença de
inflamação granulomatosa com células gigantes acometendo a aorta e seus
ramos principais.
2.2.1.1 Arterite de Células Gigantes (Arterite Temp oral)
A arterite de células gigantes (ACG), também conhecida como arterite
temporal, é uma vasculite sistêmica que acomete artérias de médio a grande
calibres, na presença de inflamação granulomatosa nas paredes vasculares
(WANG, 2008). Com incidência de 200casos/1.000.000/ano, é considerada a
vasculite primária mais comum (SAVAGE, 2000). Sua ocorrência está
relacionada à idade, sendo rara em indivíduos com menos de 50 anos, e
predomina no gênero feminino e na raça branca, especialmente na
Escandinávia e populações norte-americanas (SMITH, FIDLER e PINALS,
1983). Além da artéria temporal, outras são frequentemente acometidas, como
as ciliares, a oftálmica e a artéria central da retina.
O aparecimento de cefaléia é o sintoma mais frequente desta condição e
possui caráter variável, constante ou intermitente, lancinante ou pulsátil.
Também são comuns sintomas constitucionais - febre, perda ponderal, fadiga,
sudorese noturna e anorexia - além de claudicação da mandíbula, disfunção da
língua e otalgia (CALAMIA, 1980). Perda visual permanente, acidentes
vasculares cerebrais isquêmicos e aneurismas de aorta torácica e abdominal
são complicações temidas da ACG.

10
Possui importante associação com a polimialgia reumática, síndrome
clínica caracterizada por dor e rigidez no pescoço e nas cinturas escapular e
pélvica, estando presente em 50% dos pacientes com ACG e, portanto,
contribuindo para o diagnóstico (EVANS, 2000).
O diagnóstico é confirmado por meio de biópsia da artéria temporal,
demonstrando vasculite com infiltrado predominantemente mononuclear ou
granulomas, geralmente com células gigantes multinucleadas. O tratamento é
realizado com corticosteróides em altas doses, devendo ser iniciado no
momento do diagnóstico (CALVO-ROMERO, 2003).
2.2.1.2 Arterite de Takayasu
Doença sem pulso, tromboaortopatia oclusiva ou síndrome de Martorell,
como também é conhecida a arterite de Takayasu (AT), é uma arterite
inflamatória crônica que afeta os grandes vasos, com particular predominância
a aorta e seus principais ramos. A inflamação dos vasos acometidos leva ao
espessamento da parede, possibilitando o aparecimento de estenoses,
obstruções e a formação de trombos (JOHNSTON, LOCK e GOMPELS, 2002).
É uma doença rara, afetando mais frequentemente mulheres jovens.
Possui distribuição mundial, porém predomina no Japão, sudeste asiático, Índia
e México (LANDE, 1976).
A apresentação clínica é variável, com sintomas que refletem o
comprometimento da perfusão orgânica secundária às estenoses e formações
trombóticas. Alguns achados, observados com maior freqüência, são: pulsos
com amplitude diminuída ou impalpáveis, claudicação de membros inferiores e
sopros arteriais. Manifestações não específicas também são comuns e incluem
febre, sudorese noturna, perda ponderal, artralgia, mialgia e anemia moderada
(HALL, 1985).
Hipertensão arterial sistêmica (HAS), muitas vezes presente, geralmente
reflete a estenose da artéria renal, vista em cerca de 38% dos pacientes. A
HAS é, com frequência, grave e pode levar à insuficiência cardíaca congestiva
e até à encefalopatia hipertensiva (KERR, 1994). Demais complicações
importantes são a formação de aneurismas de aorta, resultante da destruição
da camada média arterial, retinopatia e envolvimento neurológico.

11
A angiografia permanece como método de referência para o diagnóstico,
enquanto o tratamento é feito com corticosteróides e/ou agentes
imunossupressores.
2.2.2 Vasculites de Vasos de Porte Médio
Acometem predominantemente vasos de médio calibre, as artérias
viscerais principais, como as coronárias, hepática e mesentérica, e os seus
ramos. As duas principais categorias são a poliarterite nodosa e a doença de
Kawasaki (EDGELL e JENNETTE, 2002). Ambas as entidades causam arterite
necrosante aguda que pode ser complicada por trombose e hemorragia
(JENNETTE E FALK, 2005).
2.2.2.1 Poliarterite Nodosa
A poliarterite nodosa (PAN) é, por definição adotada no Consenso de
Chapel Hill de 1994, uma vasculite caracterizada por inflamação necrosante
das artérias de pequeno ou médio calibres, sem glomerulonefrite ou vasculite
nas arteríolas, capilares ou vênulas (JENNETTE e FALK, 1994).
Doença relativamente rara, de etiologia e patogenia desconhecidas, é
mais comum em homens, entre 40 e 60 anos de idade, atingindo uma
proporção de cerca de 2:1 em relação às mulheres (NEVES, 2001).
São descritos numerosos casos de associação de PAN com hepatite
pelo vírus B (GUILLEVIN, 2005). Segundo McMahon et al., esta associação é
uma complicação séria e com risco de morte que ocorre no início do curso de
uma infecção pelo vírus B, descrevendo melhora com terapia
imunossupressora e possibilidade de prevenção pela vacina contra o vírus
(MCMAHON, 1989).
O quadro clínico é variável, geralmente contemplado por sintomas
gerais, como febre, astenia e emagrecimento, além de demais sintomas, dentre
eles: articulares, neurológicos, cutâneos, digestivos, renais e cardíacos,
refletindo o acometimento sistêmico da doença.
Comprometimento articular, com artralgia ou artrite, está presente em
cerca de 50% dos casos, sendo a mais frequente a poliartrite assimétrica das

12
grandes articulações de membros inferiores (VALENTE e CONN, 2000).
Neuropatia periférica pode ser observada em 50 a 75% dos casos e, por vezes,
como uma manifestação inicial da doença. A apresentação mais comum é sob
a forma de mononeurite multiplex (polineuropatia assimétrica) (NEVES, 2001).
Em 60 a 80% dos casos, observa-se comprometimento renal, havendo
isquemia, hemorragia ou infarto renal, podendo levar à insuficiência do rim
afetado e até à hipertensão maligna. Raramente é descrita a ocorrência de
glomerulonefrite ou de sintomas pulmonares como hemoptise ou hemorragia
alveolar difusa. Tais achados, por sua vez, sugerem o acometimento de vasos
de pequeno calibre, denotando, portanto, uma vasculite de pequenos vasos,
como a poliangiite microscópica (PAM) ou a poliangiite granulomatosa (PAG)
(GUILLEVIN e LHOTE, 1995), síndromes vasculíticas estas que serão
discutidas posteriormente.
O diagnóstico é baseado na presença de aneurismas arteriais na
angiografia das circulações hepática, renal ou esplênica. A biópsia do músculo
ou do nervo afetado pode confirmar a presença de vasculite (SAVAGE, 2000).
Se não tratada, a PAN é fatal na maioria dos casos, geralmente pela
insuficiência renal crônica ou por complicações da hipertensão. O tratamento é
realizado com corticosteróides e agentes imunossupressores, como a
ciclofosfamida, resultando em sobrevida maior que 5 anos em 80% dos casos
(SILVA JUNIOR, 2010).
2.2.2.2 Doença de Kawasaki
A doença de Kawasaki (DK) apresenta-se como uma condição febril
aguda, com inflamação difusa das mucosas, rash cutâneo e linfadenopatia
cervical, sendo observada mais frequentemente em crianças menores de cinco
anos de idade (KAWASAKI, 1967; BURNS, 1996). Considerada uma das
principais causas de doença cardíaca adquirida em crianças nos Estados
Unidos e no Japão, é mais comum em meninos, exibindo relação de 1,5:1
(DAJANI, 1993; TAUBERT, 1994).
É uma síndrome vasculítica de etiologia desconhecida, que acomete
principalmente artérias de pequeno a médio calibres, em especial as
coronárias. Dentre suas principais complicações, destacam-se a formação de

13
aneurismas, oclusão trombótica, progressão para doença isquêmica miocárdica
e aterosclerose precoce, sendo o infarto do miocárdio a principal causa de
morte (KATO, 1986).
2.2.3 Vasculites de Pequenos Vasos
As vasculites de pequenos vasos distinguem-se das demais por
apresentarem manifestações clínicas que refletem o envolvimento capilar,
como a glomerulonefrite, a púrpura ou a capilarite alveolar. No entanto, diferem
entre si quanto à predominância da localização dos vasos acometidos. Como
consequência, diferentes padrões de manifestações clínicas são reconhecidos,
que se traduzem nas diversas síndromes que compõem este grupo
(JENNETTE e FALK, 2005).
Constituem este grupo as vasculites mediadas por imunocomplexos – a
púrpura de Henoch-Schönlein e a vasculite crioglobulinêmica –, a angiite
leucocitoclástica cutânea, as associadas ao anticorpo anti-membrana basal
glomerular (anti-MBG) e as chamadas vasculites paucimunes, ou ANCA-
associadas.
As vasculites que são denominadas paucimunes não apresentam
evidências de acúmulo de imunoglobulinas na parede vascular. São também
conhecidas como vasculites ANCA-associadas (VAA), por estarem
habitualmente associadas aos anticorpos anticitoplasma de neutrófilos (ANCA),
fazendo deste um útil marcador diagnóstico. Incluem a síndrome de Churg-
Strauss (SCS), a poliangiite granulomatosa (PAG)4 e a poliangiite microscópica
(PAM) (EDGELL e JENNETTE, 2002).
As VAA são caracterizadas por vasculite necrosante, afetando
principalmente os capilares (especialmente os capilares alveolares e
glomerulares), vênulas (vênulas dérmicas pós-capilares) e arteríolas e
pequenas artérias (pequenas artérias epineurais e viscerais). O envolvimento
renal e pulmonar é mais freqüente nas mesmas, enquanto o envolvimento renal
4 Poliangiite granulomatosa é a denominação atualmente recomendada para a Granulomatose de Wegener. Em novembro de 2010, um consenso entre o Colégio Americano de Reumatologia (ACR), a Sociedade Americana de Nefrologia (ASN) e a Liga Européia contra o Reumatismo (EULAR) recomendou que o termo “granulomatose de Wegener” fosse gradualmente substituído por “poliangiite granulomatosa (Wegener)”, abreviado como PAG.

14
e cutâneo (síndrome cutâneo-renal) é mais comumente observado nas
vasculites por imunocomplexos.
À microscopia óptica, as vasculites ANCA-associadas não são
claramente distinguidas das vasculites por imunocomplexos; no entanto, a
imunohistoquímica distingue as duas, demonstrando a presença de
imunocomplexos nas paredes vasculares nas últimas.
2.2.3.1 Púrpura de Henoch-Schönlein
A púrpura de Henoch-Schönlein (PHS) é uma vasculite sistêmica de
pequenos vasos, de etiologia até então desconhecida, caracterizada por
depósito predominantemente de IgA perivascular, acometendo tipicamente
pequenos vasos cutâneos, renais e intestinais. Afeta tipicamente crianças,
podendo, no entanto, manifestar-se em qualquer idade.
O quadro clínico inclui o aparecimento de púrpura palpável associada a
uma combinação variável de sintomas, como artralgia, nefrite e dor abdominal;
sintomas estes pouco específicos por serem também encontrados nos demais
tipos de vasculites de pequenos a médios vasos. O envolvimento cutâneo está
quase sempre presente, sendo encontrado em cerca de 90% dos pacientes,
enquanto o acometimento renal, principal determinante prognóstico, ocorre na
metade dos casos (JENNETTE e FALK, 1997; ALMEIDA, 2007).
Dessa forma, baseando-se apenas em sinais clínicos, o diagnóstico de
PHS pode não ser facilmente percebido, havendo risco de insuficiência renal
crônica de instalação insidiosa principalmente para aqueles pacientes que
permaneceram sem o diagnóstico e que, por conseguinte, não receberam
tratamento adequado (DAVIN, 2003).
Para o diagnóstico de PHS, é condição sinequa non o achado de
depósitos de IgA na parede dos vasos associado a sinais característicos de
vasculite de pequenos vasos (DAVIN, 2003). Assim, biópsia de pele deve ser
realizada para estudos histológicos e de imunofluorescência para que o
diagnóstico seja definido no momento do aparecimento dos sintomas e da
suspeita clínica. Na histopatologia, encontra-se angiite leucocitoclástica dos
vasos sanguíneos superficiais, lesão não específica que pode estar presente
em qualquer tipo de vasculite de pequenos vasos (JENNETTE e FALK, 2005).

15
Finalmente, pela imunofluorescência encontra-se depósito de IgA, padrão
granular, na parede dos vasos da pele.
2.2.3.2 Vasculite Crioglobulinêmica
Vasculite crioglobulinêmica é uma vasculite sistêmica mediada por
imunocomplexos que acomete predominantemente pequenos vasos,
comprometendo, também, em alguns casos, vasos de médio calibre
(JENNETTE e FALK, 1994; CARSON, 1998). Consoante o próprio nome indica,
é acompanhada de crioglobulinemia5, condição na qual estão presentes no
sangue proteínas anormais (crioglobulinas) que se precipitam em baixas
temperaturas. Como resultado, ao serem expostos ao frio, alguns pacientes
experimentam sintomas como artralgia, púrpura, fenômeno de Raynaud,
cianose e até ulcerações e necrose cutâneas, embora existam aqueles que se
mantenham assintomáticos (BROUET, 1974).
Quanto a sua etiologia, a crioglobulinemia pode ocorrer sem uma
condição subjacente identificável (crioglobulinemia “essencial”) ou em
associação com um distúrbio definido (crioglobulinemia secundária), como
doenças autoimunes, doenças linfoproliferativas ou em resposta a infecções.
São classificadas em tipos I, II e III. A crioglobulinemia do tipo I contém
anticorpos monoclonais (IgG ou IgM), sem presença de fator reumatóide (FR).
Estão associadas a algumas doenças hematológicas malignas, como o
mieloma múltiplo. Já a crioglobulinemia dos tipos II e III são consideradas
formas mistas, por conterem anticorpos IgG e IgM. O componente IgG é
sempre policlonal, enquanto o IgM pode ser tanto monoclonal (tipo II) quanto
policlonal (tipo III). Em ambas, o FR é positivo. Ocorrem em associação a
doenças como hepatite C, síndrome de Sjögren e lúpus eritematoso sistêmico
e podem causar vasculite de pequenos a médios vasos, resultante da
deposição de imunocomplexos nas paredes vasculares e ativação do
complemento (LAMPRECHT, 1999; D’AMICO, 1998).
5 Crioglobulinemia: Crioglobulinas são proteínas séricas anormais que se precipitam em
condições de baixa temperatura, dissolvendo-se após serem aquecidas. Sua presença no sangue (crioglobulinemia) é usualmente associada com diversas condições sistêmicas, podendo levar a complicações que incluem as vasculites.

16
A detecção de crioglobulinas no sangue confirma a crioglobulinemia,
porém não necessariamente indica a existência da doença clínica. Havendo a
doença vasculítica, na análise imunohistoquímica, serão demonstrados
depósitos de IgG e IgM na parede vascular, diferentemente da púrpura de
Henoch-Schönlein (na qual são observados imunocomplexos de IgA), e da
poliangiite microscópica (na qual não são observados imunocomplexos).
Hipocomplementenemia é também um achado importante na vasculite
crioglobulinêmica, encontrado em 90% dos pacientes, e que ajuda a distinguí-la
das vasculites ANCA-associadas, nas quais o complemento está normal ou há
hipercomplementenemia (MONTI, 1995).
2.2.3.3 Síndrome do Anticorpo anti-Membrana Basal G lomerular
A síndrome do anticorpo anti-membrana basal glomerular (anti-MBG) foi
inicialmente descrita pelo patologista norte-americano, Ernest Goodpasture, em
1919 (GOODPASTURE, 1919), tornando-se posteriormente também conhecida
pelo epônimo Síndrome de Goodpasture (SG) (STANTON & TANGE, 1958).
Esta síndrome pode apresentar-se como doença limitada aos rins
(glomerulonefrite anti-MBG) ou como uma síndrome pulmão-rim (KELLY,
1994), com glomerulonefrite rapidamente progressiva e hemorragia pulmonar,
caracterizando a SG.
Sua incidência ocorre geralmente em dois picos de idade (FISCHER,
2006), com o primeiro entre as segunda e terceira décadas de vida, havendo
predominância no sexo masculino e maior frequência de hemorragia pulmonar.
O segundo pico é nas sexta e sétima décadas, com predominância em
mulheres e da doença limitada aos rins.
O diagnóstico é feito por meio da detecção de anticorpos anti-MBG
circulantes e da demonstração de deposição linear de anticorpos IgG ao longo
da membrana basal glomerular pela imunofluorescência, na amostra de biópsia
renal (LIM, 2011).
17
2.2.3.4 Vasculite ANCA-associadas
Anticorpos anticitoplasma de neutrófilos ou ANCA são imunoglobulinas
dirigidas contra enzimas constituintes de grânulos citoplasmáticos de
neutrófilos. São principalmente direcionadas contra as enzimas proteinase 3
(PR3) e mieloperoxidase (MPO), sendo, porém, outras enzimas – como
elastase (HLE), lactoferrina (LF) e catepsina G (CG) – também descritas
(FREIRE, 1997).
Os ANCA foram primeiramente descritos em 1982 por Davies et al. ao
demonstrarem pela imunofluorescência indireta (IFI) coloração granular com
padrão citoplasmático em neutrófilos (ANCA-C) fixados com etanol, em
pacientes com glomerulonefrite e sinais de hemorragia alveolar difusa
(DAVIES, 1982). Dois anos mais tarde, Hall et al. confirmaram este achado em
quatro pacientes com vasculite de pequenos vasos (HALL, 1984). Hoje, são
reconhecidos dois padrões clássicos de ANCA, pela IFI: o padrão
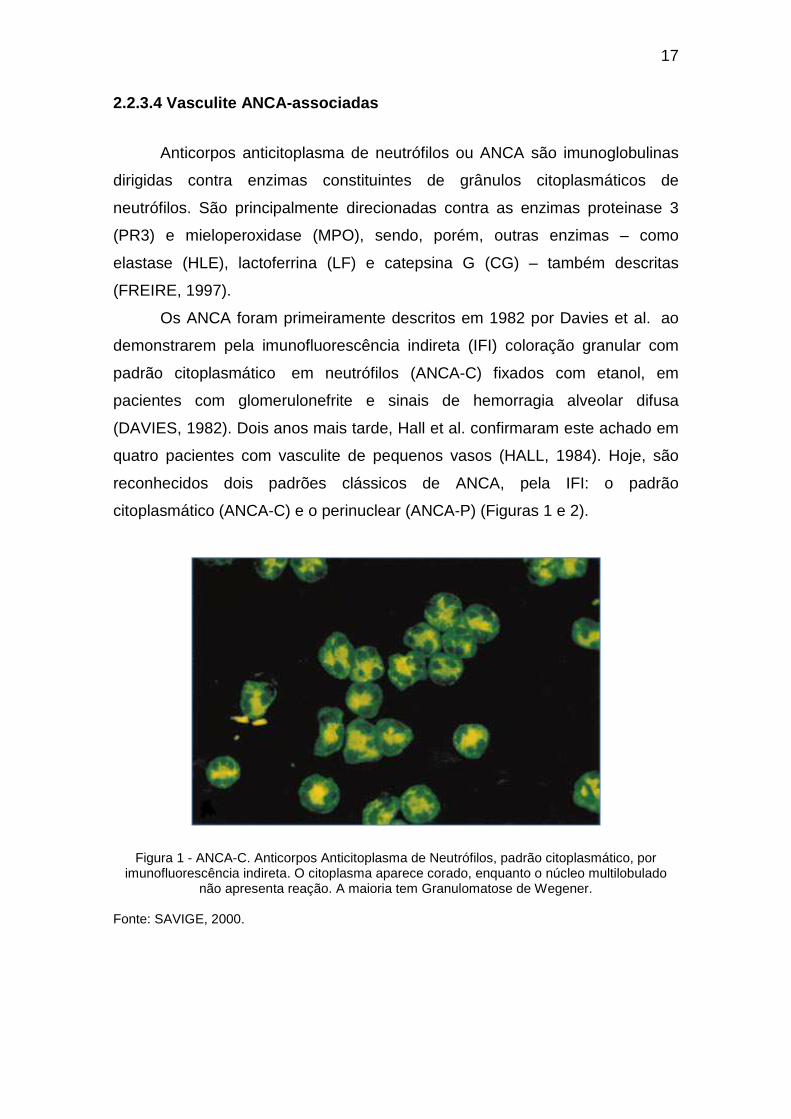
citoplasmático (ANCA-C) e o perinuclear (ANCA-P) (Figuras 1 e 2).
Figura 1 - ANCA-C. Anticorpos Anticitoplasma de Neutrófilos, padrão citoplasmático, por imunofluorescência indireta. O citoplasma aparece corado, enquanto o núcleo multilobulado
não apresenta reação. A maioria tem Granulomatose de Wegener. Fonte: SAVIGE, 2000.

18
Figura 2 - ANCA-P. Anticorpos Anticitoplasma de Neutrófilos, padrão perinuclear, por imunofluorescência indireta. O corante se restringe à porção perinuclear, enquanto o
citoplasma é não-reativo. Outras enfermidades sem vasculites podem apresentar ANCA-P (+) por outros anticorpos.
Fonte: SAVIGE, 2000.
Estudos posteriores de Van der Woude et al. e Falk et al. demonstraram
que o ANCA estaria intimamente associado a três principais síndromes
vasculíticas de pequenos vasos – a síndrome de Churg-Strauss, a poliangiite
granulomatosa e a poliangiite microscópica (VAN DER WOUDE, 1985; FALK,
1988). Estas formas de vasculites foram posteriormente agrupadas devido a
sua positividade para este anticorpo, sendo atualmente referidas como
vasculites ANCA-associadas (VAA) (KALLENBERG, 1994).
A pesquisa de ANCA é, atualmente, um exame indispensável na
investigação de casos suspeitos de vasculites. Positividade para ANCA é
encontrada em 75 a 95% dos casos de poliangiite granulomatosa e poliangiite
microscópica em atividade (JENNETTE, 1997; SAVAGE, 1997), sendo a
sensibilidade aumentada conforme a atividade da doença Tabela 1
(FINKIELMAN, 2007).

19
Tabela 1 – Tipos de ANCA associados com as Vasculites Primárias de Pequenos
Vasos
Fonte: Castañer, 2010.
TIPOS DE ANCA ASSOCIADOS COM AS VASCULITES PRIMÁRIA S DE PEQUENOS VASOS
VASCULITES DE PEQUENOS VASOS
TIPO DE ANCA COMENTÁRIOS
Poliangiite granulomatosa (PAG)
ANCA-C (anti-PR3) Sensibilidade de 85%-90% para PAG ativa generalizada Sensibilidade de 60% para doença pulmonar limitada Sensibilidade de 40% para doença em remissão
Síndrome de Churg-Strauss
ANCA-P (anti-MPO) Sensibilidade de 35%-50%
Poliangiite microscópica ANCA-P (anti-MPO) Sensibilidade de 35%-70%
É importante lembrar que positividade para ANCA é também encontrada
em algumas outras enfermidades, como doenças inflamatórias intestinais,
doenças hepáticas e reumatológicas. Sendo, portanto, pouco específico,
resultados positivos do exame devem ser interpretados em conjunto com
achados clínicos, radiológicos e histológicos.
A IFI é considerada o melhor método de triagem para a pesquisa de
ANCA, utilizando, como substrato, neutrófilos humanos normais fixados em
etanol. Pela IFI, são identificados dois padrões clássicos: ANCA-C, ou padrão
citoplasmático, e ANCA-P, ou perinuclear. Em casos dúbios, um teste positivo
deve ser confirmado pela pesquisa dos anticorpos específicos anti-PR3
(ANCA-C) e anti-MPO (ANCA-P), por meio de imunoensaio enzimático
(ELISA). O ANCA, por conseguinte, atua como importante marcador
diagnóstico para as vasculites sistêmicas. No entanto, seu papel como
marcador prognóstico, para avaliação de recidivas e para definir a manutenção
do tratamento, ainda é objeto de controvérsia.
Os capítulos que seguem serão destinados às supracitadas vasculites
associadas aos anticorpos anticitoplasma de neutrófilo (ANCA).

20
3 VASCULITES ANCA-ASSOCIADAS
3.1 SÍNDROME DE CHURG-STRAUSS
Em 1951, os médicos Jacob Churg e Lotte Strauss autopsiaram treze
pacientes que apresentavam em comum quadro semelhante de asma grave,
febre e eosinofilia, juntamente com manifestações sistêmicas de uma vasculite
necrosante. Apresentaram, então, o que seria o primeiro relato de uma
interessante síndrome vasculítica, a qual denominaram de angiite
granulomatosa alérgica (CHURG e STRAUSS, 1951). Mais tarde, a síndrome
tornou-se mais conhecida pelos nomes de seus descobridores, e o termo
“síndrome de Churg-Strauss” (SCS) passou a ser o título aceito desta distinta
forma de vasculite sistêmica.
A SCS é uma vasculite necrosante sistêmica incomum, que envolve
preferencialmente pequenos vasos sanguíneos dos pulmões, pele, nervos
periféricos e, menos frequentemente, do coração e do trato gastrointestinal
(ABRIL, 2003), podendo acometer também vasos de médio calibre.
Caracteriza-se histologicamente pela presença de infiltrado inflamatório
extravascular rico em eosinófilos com formação de granulomas e vasculite
necrosante. Clinicamente, é caracterizada pela presença de rinossinusite de
longa data e asma de início tardio, eosinofilia importante em sangue periférico
e sintomas sistêmicos da vasculite (ROSE, 1957). Os pulmões geralmente são
acometidos, exibindo lesões no parênquima pulmonar à imagem radiográfica.
Mais recentemente, foram descritas formas frustas da síndrome de Churg-
Strauss, em que a reação eosinofílica encontra-se atenuada pelo uso de
corticóide sistêmico dirigido à asma e à rinossinusite alérgica.
Cerca de 40% dos pacientes apresentam ANCA positivo, geralmente
com o padrão perinuclear (ANCA-P) e especificidade ao antimieloperoxidase
(anti-MPO). A apresentação clínica dos pacientes ANCA-positivos pode diferir
significativamente daqueles pacientes ANCA-negativos, podendo apresentar
com maior freqüência mononeurite multiplex e glomerulonefrite no primeiro
(SABLÉ-FOURTASSOU, 2005; SINICO, 2005).
O manejo terapêutico depende principalmente da terapia com
corticosteróides e, quando necessário, normalmente para pacientes com

21
quadros mais graves, lança-se mão do tratamento combinado com drogas
imunossupressoras, especialmente a ciclofosfamida. O tratamento da SCS é
semelhante ao das outras VAA, razão pela qual as considerações sobre este
assunto serão feitas no subitem 3.4 - Tratamento das Vasculites ANCA-
associadas.
3.1.1 Epidemiologia
A SCS tem sido cada vez mais reconhecida durante as últimas décadas,
no entanto, são poucos os estudos epidemiológicos exclusivos a esta
síndrome, a maioria deles a respeito das vasculites sistêmicas primárias em
geral. Por outro lado, até recentemente pouco se sabia acerca da incidência e
prevalência das vasculites, no entanto os estudos epidemiológicos têm
aumentado nos últimos anos, principalmente na Europa (WATTS, 2005).
Sendo uma das mais raras vasculites, a SCS ocorre em média na quinta
década de vida, podendo ocorrer em todas as idades (GUILLEVIN, 2004). Há
relatos em crianças desde os quatro anos de idade (LOUTHRENOO, 1999); no
entanto, permanece incomum na faixa pediátrica e, quando ocorre, parece
seguir curso mais agressivo com graves manifestações pulmonares e
cardiovasculares (ZWERINA, 2009). Não há diferença significativa entre os
gêneros, embora alguns estudos indiquem uma ligeira predominância em
homens (HARROLD, 2005; CHUMBLEY, 1977).
Em relação as três vasculites ANCA-associadas, a SCS é a menos
comum (KEOGH, 2006), com uma prevalência na população geral variando de
10,7 a 13 casos por milhão de habitantes (MAHR, 2004; HAUGEBERG, 1998)
e uma incidência anual de 0,5 a 6,8 casos novos por milhão de habitantes
(MARTIN, 1999; WATTS, 2000; WATTS, 2005), variando com a localização
geográfica e com os critérios de classificação adotados em cada estudo
(DUNOGUÉ, 2011).

22
3.1.2 Etiologia e Patogênese
Nos últimos anos, diversos estudos foram realizados a respeito da
patogenia da SCS. No entanto, os mecanismos patológicos e a etiologia da
doença ainda permanecem desconhecidos, com várias teorias propostas,
sugerindo, dessa forma, uma natureza multifatorial.
ASSOCIAÇÃO COM FÁRMACOS
Na década de 1990, foram relatados casos de SCS após o tratamento
com antagonistas de leucotrienos6 em pacientes com asma, sendo a
associação entre a terapia da asma e a SCS amplamente discutida desde
então (WELLER, 2001). Observou-se, no entanto, que em 88% dos pacientes,
os sintomas apareceram quando corticosteróides orais estavam sendo
reduzidos ou interrompidos. Uma das teorias para o desenvolvimento da
síndrome nessa situação considera que a redução ou a interrupção da terapia
com corticóides a partir do controle efetivo da asma poderia propiciar o
surgimento dos sinais e sintomas da vasculite sistêmica previamente
mascarados (WELLER, 2001; KNOELL, 1998; WECHSLER, 1998; D’CRUZ,
1999).
ASMA E ATOPIA
A associação com asma e atopia é típica, sugerindo uma possível
etiologia alérgica. No entanto, nem todos os pacientes apresentam o curso
clássico da doença, havendo casos descritos em que não há história de asma
prévia (LANHAM, 1990; CHEN, 1992). Há também sugestões de que um
agente etiológico infeccioso ou um antígeno exógeno, possivelmente inalado,
poderia desencadear resposta alérgica inflamatória em indivíduo
geneticamente suscetível, levando ao desenvolvimento de rinossinusite e
asma, seguido de infiltração tecidual de eosinófilos (GUILLEVIN, 1991).
6 Antagonistas de leucotrienos são antagonistas específicos que competem com os receptores
para cisteinil-leucotrienos, e têm efeito agonista ao efeito dos corticosteróides, assim como a teofilina e o salmeterol, sendo usados em associação com o corticóide inalatório no tratamento da asma.

23
EOSINÓFILOS e EOSINOFILIA
Alguns estudos sugerem que a presença de grande número de
eosinófilos depositados nos vasos e nos tecidos de pacientes com SCS teria
um papel direto na patogênese da vasculite (HALL, 1993; COTTIN, 1995).
Devido aos seus efeitos pró-inflamatórios e citotóxicos (MARTIN, 1999),
estariam relacionados aos mecanismos responsáveis pelos danos teciduais
(NOTH, 2003). No entanto, os mecanismos patogênicos dos eosinófilos não
foram completamente elucidados.
ANCA
Devido à associação com o ANCA, também tem sido aventada a
participação deste na gênese das síndromes vasculíticas a ele associadas.
Alguns estudos sugerem um mecanismo patogênico pelo qual os ANCA
causariam, ou ao menos facilitariam, a ativação neutrofílica, a adesão às
células endoteliais e a consequente lesão e destruição do endotélio vascular
(SAVIGE, 1999; SUNDY, 1995). Especula-se que esses autoanticorpos
poderiam ativar neutrófilos que, então, promoveriam a apoptose e necrose das
células endoteliais. Além disso, a adesão endotelial com consequente ativação
de leucócitos conduz à inflamação vascular e ao processo inflamatório nos
vasos, levando ao processo vasculítico necrosante nos órgãos e sistemas
afetados (NOTH, 2003). No entanto, uma vez que cerca de um terço dos
pacientes não apresentam esses autoanticorpos, acredita-se que estes não
constituam uma causa primária da síndrome (NOTH, 2003).
GENÉTICA
Vaglio et al propuseram a existência de uma predisposição genética
para o desenvolvimento da SCS, ao identificarem o gene HLA-DRB4 como um
fator de risco genético para a doença (VAGLIO, 2007).
Embora avanços tenham sido feitos no entendimento da patogenia da
SCS, nenhum mecanismo patogênico único foi estabelecido. Ressalta-se,
dessa forma, a possível existência de diferentes padrões patogênicos,
representando a heterogeneidade histológica, clínica e biológica da SCS.

24
3.1.3 Manifestações Clínicas
A história natural da doença consiste tipicamente em três fases
parcialmente sobrepostas (LHOTE, 1995; LANHAM, 1984). Um quadro inicial
de rinossinusite e/ou asma, com sintomas inicialmente leves e que se agravam
progressivamente, por vezes, refratários ao tratamento. Essa seria a primeira
fase da doença, ou fase prodrômica. Pólipos nasais e sinusite recorrentes
também podem estar presentes nesta fase inicial. Manifestações mais graves,
como perfuração nasal e rinorréia purulenta ou sanguinolenta, são raras na
SCS, estando, no entanto, mais associadas a poliangiite granulomatosa
(LANHAM, 1984). Esta fase prodrômica é comum e pode durar vários anos. A
asma, quando presente, precede o aparecimento da vasculite em uma média
de 3 a 8 anos, com relatos de intervalo de tempo de até 30 anos (CHUMBLEY,
1977; FRUSTACI, 1998; ERZURUM; 1989). A rinite alérgica é também uma
característica comum desta primeira fase, estando presente em 70% dos casos
(CHUMBLEY, 1977).
Segue-se, então, uma segunda fase – eosinofílica –, caracterizada por
eosinofilia periférica e infiltração eosinofílica de tecidos, especialmente nos
pulmões e no trato gastrointestinal (LANHAM, 1984). A taxa de eosinófilos no
sangue periférico é geralmente superior a 10% do total de leucócitos, ou
superior a 1500 células/mm3 (MASI, 1990; LANHAM, 1990).
Por fim, geralmente anunciada por sintomas constitucionais
inespecíficos – febre, mal-estar, cansaço e perda de peso –, surge a fase de
vasculite (LANHAM, 1990; TRAVIS, 1996). A fase vasculítica é caracterizada
por manifestações diversas de vasculite sistêmica acometendo vasos de
pequenos e médios calibres.
A Tabela 2 ilustra algumas manifestações sistêmicas da SCS com sua
freqüência de acometimento.

25
Tabela 2 – Manifestações Clínicas da SCS
Fonte: BROWN, 2006.
MANIFESTAÇÕES CLÍNICAS FREQUENCIA
(%)
ASMA 98-100
SINTOMAS CONSTITUCIONAIS (febre, perda de peso,
mialgia)
70-80
NEUROLÓGICA (mononeurite multiplex, hemorragia
cerebral)
50-80
CUTÂNEA (púrpura, urticária, nódulos subcutâneos,
exantema)
50-80
RINOSSINUSITE 20-70
CARDÍACA (pericardite, insuficiência cardíaca, vasculite
coronariana)
35-50
GASTROINTESTINAL (diarréia, sangramento digestivo,
colite, dor abdominal)
30-60
RENAL (proteinúria, hematúria) 10-50
MANIFESTAÇÕES PULMONARES
O envolvimento pulmonar é quase universal na SCS. A asma, presente
em 98 a 100% dos pacientes, geralmente precede a vasculite sistêmica em
aproximadamente oito a dez anos, podendo ocorrer após ou até
simultaneamente (GUILLEVIN, 1999). A idade média de início da asma é
tardia, normalmente em adultos jovens (SABLÉ-FOURTASSOU, 2005). Muitas
vezes é grave, corticosteróide-dependente e associada com manifestações de
vias aéreas superiores, especialmente a rinossinusite (70%) com obstrução e
polipose nasal (62,5%) (GUILLEVIN, 1999). Com o desencadeamento da fase
vasculítica, a gravidade da asma e o número de exacerbações por vezes
sofrem um aumento.
A hemorragia alveolar secundária à vasculite de pequenos vasos
pulmonares é uma manifestação grave, porém menos comum, ocorrendo em
4% dos casos (GUILLEVIN, 1999). Ocorre predominantemente entre os anti-

26
MPO e ANCA-positivos (SINICO, 2005). É caracterizada por hemoptise,
anemia e infiltrados alveolares aos exames de imagem.
Finalmente, as manifestações pleurais existem, mas são raras e de
pouca expressão clínica. Derrames pleurais são assintomáticos e
caracterizados principalmente por exsudato rico em eosinófilos (LANHAM,
1990).
MANIFESTAÇÕES NEUROLÓGICAS
O sistema nervoso é, com freqüência, envolvido, sendo especialmente
acometidos os nervos periféricos em 50 a 70% dos pacientes (MOORE, 1994;
SEHGAL, 1995). A vasculite da SCS tipicamente envolve o vasa vasorum dos
nervos periféricos, resultando em mononeurite multiplex, apresentação mais
comum da neurite periférica nesta doença (OLIVEIRA, 2005). Pacientes podem
manifestar fraqueza súbita, juntamente com déficits sensoriais na distribuição
de um ou mais nervos distais. Menos comumente, uma polineuropatia também
pode estar presente, sendo reconhecida nos critérios para SCS, os quais serão
apresentados a seguir no tópico destinado ao diagnóstico (CHUMBLEY, 1977).
O envolvimento do sistema nervoso central é, por sua vez, pouco
freqüente, ocorrendo em 25% dos casos com sintomas neurológicos (ABRIL,
2003). Acidentes vasculares cerebrais isquêmicos e hemorrágicos são os mais
comuns, constituindo importante causa de morbidade e mortalidade.
(CHUMBLEY, 1977).
MANIFESTAÇÕES CUTÂNEAS
O aparecimento de lesões cutâneas é comum na fase vasculítica e está
presente em 40% a 70% dos casos, apresentando-se de formas diversas, com
lesões eritematosas, máculo-papulares, pústulas ou nódulos (RODRIGUES e
ARAÚJO, 2011). Púrpura é descrita em cerca de metade dos casos e afeta
predominantemente os membros inferiores. Nódulos subcutâneos contendo
granulomas extravasculares estão presentes em cerca de um terço dos
pacientes com SCS. Clinicamente, são nódulos vermelhos ou violáceos, às
vezes dolorosos, preferencialmente nas superfícies extensoras do cotovelo,
dedos e couro cabeludo (GUILLEVIN, 1999).

27
MANIFESTAÇÕES GASTROINTESTINAIS
A infiltração eosinofílica da parede do intestino pode produzir sintomas
semelhantes aos de uma gastroenterite eosinofílica, com dor abdominal (30 a
60%), náuseas, vômitos, diarréia e até sangramento digestivo. (LANHAM,
1990). Qualquer porção do trato gastrointestinal pode ser afetada, inclusive a
vesícula biliar (LIE, 1993). Além disso, a ocorrência de vasculite acometendo
vasos mesentéricos acarreta lesões ulcerativas isquêmicas, podendo ser
grave, com complicações potencialmente fatais de hemorragia e perfuração
intestinal (GUILLEVIN, 1999).
MANIFESTAÇÕES CARDÍACAS
As complicações cardíacas respondem por aproximadamente 50% das
mortes causadas pela doença (ERZURUM, 1989; TRAVIS, 1996; DEREMEE,
2002), devendo-se principalmente a infarto agudo do miocárdio, pericardite
aguda ou constritiva (BARROS, 2005). Não é incomum o desenvolvimento de
insuficiência cardíaca congestiva, presente em 25% dos casos, secundária à
miocardite eosinofílica ou à vasculite coronariana (CHUMBLEY, 1977).
Derrame pericárdico também é comum, podendo causar sintomas constritivos.
(CONRON e BEYNON, 2000). Além disso, outras manifestações cardíacas,
como hipertensão arterial sistêmica e alterações eletrocardiográficas, são
também relatadas (LANHAM, 1984).
MANIFESTAÇÕES RENAIS
O envolvimento renal é menos frequente do que nas demais vasculites
ANCA-associadas e normalmente não é grave (DEREMEE, 2002). Apesar de
evidência laboratorial de doença renal ser comum, com elevação de creatinina
e uréia séricas, proteinúria e hematúria microscópica, geralmente há boa
resposta aos corticosteróides, sendo incomum a evolução para insuficiência
renal (LANHAM, 1990; CHUMBLEY, 1977).
MISCELÂNEA
O comprometimento ocular é raro na SCS. Manifestações oculares
incluem episclerite, nódulos conjuntivais (composto de granulomas

28
extravasculares), ceratite, uveíte posterior, paralisias oculares, e neuropatia
óptica isquêmica (SOLANS, 2001). Manifestações otológicas foram também
relatadas, com otorréia recorrente, mastoidite e até perda auditiva, as quais
geralmente respondem bem aos corticosteróides (ISHIYAMA, 2001).
3.1.4 Achados Histopatológicos
As alterações patológicas típicas incluem vasculite necrosante de
pequenos vasos, infiltrado celular rico em eosinófilos e granuloma necrosante,
podendo ocorrer em qualquer órgão (LIE, 1990; LANHAM, 1990). Os
granulomas caracterizam-se por um centro necrótico envolto por histiócitos em
paliçada e células gigantes multinucleadas, como observados na Figura 3
abaixo (CHURG e STRAUSS, 1951; RODRIGUES, 2011). Este achado, no
entanto, não é considerado patognomônico da doença, nem sempre sendo
encontrado (LIE, 1990; JENNETTE, 1997).
Figura 3 - Imagem ampliada de amostra obtida por meio de biópsia por videotoracoscopia mostrando espaços alveolares preenchidos por eosinófilos (setas, pneumonia eosinofílica).
Pequeno granuloma extravascular (cabeças de seta) constituído de histiócitos e células gigantes multinucleadas. Hematoxilina e eosina; ampliação original em 100x.
Fonte: CHUNG, 2010.
A coexistência dos três tipos de lesões características da doença –
vasculite ou capilarite necrosante (Figura 4), infiltração eosinofílica e

29
granulomas extravasculares – é rara, não sendo mandatória para o diagnostico
histológico (LANHAM, 1984; LHOTHE, 2008).
Figura 4 - Imagem ampliada de amostra obtida mediante biópsia por videotoracoscopia
mostrando espessamento bronquiolar devido ao infiltrado inflamatório celular (eosinofílico e linfocítico) (setas) e hiperplasia folicular linfóide (cabeça de seta). Capilarite necrosante (setas,
paredes espessadas com infiltração eosinofílica e necrose) nas paredes alveolares. Hematoxilina e eosina; ampliação original em 100x.
Fonte: CHUNG, 2010.
Alterações histopatológicas características pulmonares incluem
bronquite asmática, pneumonia eosinofílica e granulomas extravasculares.
Demais alterações pulmonares comuns à doença incluem a bronquite
asmática (hialinização da membrana basal, aumento de secreção mucosa e
infiltrado eosinofílico nas paredes brônquicas), pneumonia eosinofílica
(alvéolos preenchidos por eosinófilos, macrófagos e células gigantes
multinucleadas, podendo encontrar cristais de Charcot-Leyden,
microabscessos eosinofílicos e infiltrado inflamatório intersticial, com
arquitetura pulmonar geralmente preservada) e granulomas extravasculares
(CHURG e STRAUSS, 1951; ABRIL, 2003).
Churg e Strauss, em 1951, observaram ainda inflamação eosinofílica
das paredes vasculares com progressivo espessamento e evolução para

30
fibrose, resultando em diminuição e até obliteração do lúmen (CHURG e
STRAUSS, 1951).
A presença de eosinófilos no interior das paredes vasculares pode
ocorrer em outras formas de vasculites. No entanto, a presença de eosinófilos
nos tecidos extravasculares é mais específica para a SCS (MASI, 1990). Deve
ser realizada biópsia preferencialmente nos supostos órgãos afetados, estando
as manifestações histológicas da SCS mais comumente localizadas na pele
(67,4%), nos nervos (65,7%) e nos músculos (47,9%) (LHOTHE, 2008).
3.1.5 Apresentação Radiológica
Alterações à radiografia torácica estão presentes em cerca de 60% dos
pacientes e incluem opacidades segmentares e infiltrados pulmonares
transitórios bilaterais multifocais, sem distribuição lobar ou segmentar
específica (Figuras 5 e 6) (LANHAM, 1984). Os infiltrados pulmonares ocorrem
com maior freqüência na periferia dos campos pulmonares, geralmente são
assimétricos, e, ocasionalmente, nodulares (LHOTE, 1995).
Figura 5 - Síndrome de Churg-Strauss em paciente do gênero feminino, 63 anos. Radiografia
torácica em projeção frontal mostra opacidades pulmonares extensas bilaterais. Ambos os ápices estão relativamente poupados.
Fonte: CHUNG, 2010.

31
Figura 6 - Síndrome de Churg-Strauss em uma mulher com 63 anos de idade. Imagem de TC com janela para pulmão mostrando opacidades em vidro fosco extensas e irregulares,
grosseiramente simétricas, ocupando a região medular de ambos os pulmões. Fonte: CHUNG, 2010.
A presença de nódulos pulmonares múltiplos é eventual (Figuras 7 e 8)
(LANHAM, 1984), sendo mais comum, no entanto, na poliangiite
granulomatosa. Estes nódulos, ao contrário do que acontece na PAG,
raramente apresentam escavações.
Figura 7 - Síndrome de Churg-Strauss em paciente masculino, 51 anos. TC com janela para pulmão mostrando espessamento de paredes brônquicas, árvore em brotamento (setas) e
opacidades em vidro fosco (cabeças de seta) em ambos os pulmões. Fonte: (CHUNG, 2010).

32
Figura 8 - Síndrome de Churg-Strauss em paciente do sexo masculino, 51 anos. TC repetida após nove meses de acompanhamento, evidenciando doença recorrente em ambos os pulmões, com áreas de consolidação, opacidades em vidro fosco e lesões nodulares.
Fonte: CHUNG, 2010.
As anormalidades radiográficas de tórax em pacientes com SCS são
diversas e incluem (CHOI, 2000; ERZURUM, 1989; BUSCHMAN, 1990;
WORTHY, 1998):
• Opacidades transitórias e irregulares (75% dos pacientes), sem
distribuição lobar ou segmentar;
• Opacidades simétricas em distribuição periférica e axilar;
• Opacidades irradiando-se do hilo com linfonodomegalia hilar;
• Opacidades intersticiais difusas ou com padrão miliar;
• Hemorragia pulmonar causando vidro-fosco difuso;
• Nódulos bilaterais não escavados; e
• Derrames pleurais, encontrados em 30% dos casos, geralmente
exsudatos eosinofílicos.
Achados típicos da tomografia computadorizada de alta resolução
(TCAR) na SCS incluem: infiltração peribroncovascular, espessamento septal,
e opacidades difusas e irregulares. Estes achados, apesar de não serem
específicos para a SCS, podem orientar a escolha do local para a biópsia de
pulmão, além de caracterizar a extensão da doença. (BUSCHMAN, 1990).

33
3.1.6 Diagnóstico
Critérios diversos têm sido propostos para o diagnóstico da SCS. Os
critérios diagnósticos descritos por Churg e Strauss, em 1951, baseados
principalmente em séries post-mortem, não mais serviam à prática clínica.
Dentre as novas classificações propostas, as mais comumente utilizadas são:
os critérios de Lanham e os critérios do Colégio Americano de Reumatologia
(ACR), sendo o último utilizado preferencialmente (LANHAM, 1984; MASI,
1990).
Lanham et al estabeleceram três critérios que baseavam-se na presença
de manifestações clínicas: (1) asma, (2) eosinofilia em sangue periférico
(eosinófilos acima de 1500 células/mm3 ou acima de 10% da contagem total de
leucócitos) e (3) evidência de uma vasculite sistêmica envolvendo dois ou mais
órgãos extrapulmonares (LANHAM, 1984). No entanto, esta definição sofreu
algumas críticas, como o fato do quadro clínico nem sempre apresentar-se de
forma clássica. Além disso, o quadro de vasculite, apesar de apresentar uma
clínica característica, pode ser difícil confirmá-lo sem a realização de biópsia.
Alguns anos mais tarde, um grupo de especialistas em vasculites do
ACR estabeleceu seis critérios que indicariam o diagnóstico de SCS em um
paciente com quadro de vasculite já estabelecido (MASI, 1990). A presença de
quatro ou mais destes seis critérios, observados no Quadro 2 abaixo, tem
sensibilidade de 85% e especificidade de 99,7% para a SCS.
CRITÉRIOS DEFINIÇÃO
ASMA História de sibilância ou constatação de sibilos
expiratórios agudos e difusos.
EOSINOFILIA Eosinófilos >10% da contagem total de
leucócitos.
MONONEUROPATIA OU
POLINEUROPATIA
Desenvolvimento de mononeuropatia, incluindo
mononeurite multiplex, ou polineuropatia
atribuível a vasculite.
INFILTRADOS
PULMONARES
TRANSITÓRIOS
Infiltrados pulmonares migratórios ou transitórios
à radiografia de tórax.

34
ANORMALIDADE EM
SEIOS PARANASAIS
História de rinossinusite aguda ou crônica ou
opacificação de seios paranasais à radiografia.
EOSINOFILIA
EXTRAVASCULAR
Biópsia de artéria, arteríola ou vênula mostrando
acumulação de eosinófilos em áreas
extravasculares.
Quadro 2 – Critérios do Colégio Americano de Reumatologia de 1990 para a Classificação de
Síndrome de Churg-Strauss.
Fonte: ACR, 1990.
Novos critérios diagnósticos atualmente em discussão incluem o ANCA
ou anti-MPO, além de redefinições clínicas e biológicas da SCS (DUNOGUÉ,
2011).
A suspeita diagnóstica da SCS é, geralmente, estabelecida clinicamente.
Asma e/ou rinossinusite associada à eosinofilia periférica e sintomas
sugestivos de vasculite é um quadro bastante sugestivo. No entanto, a análise
histopatológica por meio de biópsia dos locais afetados (geralmente, pulmão,
pele ou nervos periféricos), bem como uma avaliação complementar mais
detalhada, fazem-se fundamentais para a confirmação diagnóstica e avaliação
do quadro clínico geral do paciente.
A confirmação do diagnóstico, no entanto, é por vezes dificultada. Além
das manifestações clássicas da doença, descritas anteriormente, nem sempre
estarem presentes, algumas delas podem se apresentar de forma isolada, anos
antes do surgimento de características típicas adicionais. Ademais, os sintomas
facilmente se confundem com os de outras doenças, e até mesmo de outras
vasculites sistêmicas, principalmente as associadas ao ANCA.
Os achados histológicos clássicos – vasculite necrosante, infiltrado
eosinofílico e granulomas extravasculares –, por sua vez, embora bastante
sugestivos de SCS, nem sempre são encontrados (ROUFOSSE, 2003). Além
disso, não são patognomônicos da doença (REID; 1998), vez que também
podem ser encontrados tanto em outras vasculites, como a poliangiite
granulomatosa, quanto em algumas doenças autoimunes.

35
LABORATÓRIO
A avaliação laboratorial revela tipicamente uma anemia normocítica e
normocrômica, leucocitose, trombocitose e uma resposta de fase aguda com
aumento da proteína C reativa (PCR) e da velocidade de hemossedimentação
(VHS) (SOLANS, 2001). A eosinofilia é observada em praticamente todos os
pacientes, assim como os níveis de IgE que encontram-se elevados em 75%
dos casos. Pode ainda haver fator reumatóide positivo, porém em baixos
títulos.
Cerca de 40 a 60% dos pacientes com SCS possuem ANCA positivo,
geralmente com padrão de coloração perinuclear (ANCA-P) (HASLEY, 1990;
SINICO, 2005). Embora o padrão citoplasmático também possa ser visto, é
pouco comum. A maioria dos pacientes ANCA-positivos, entre 70 a 75%,
apresenta anticorpos dirigidos contra a mieloperoxidase (KEOGH, 2003;
SINICO, 2005).
DIAGNÓSTICO DIFERENCIAL
O diagnóstico diferencial da SCS é amplo e variável conforme os
sintomas manifestados pelo paciente. As principais doenças a serem
consideradas são a pneumonia eosinofílica crônica, a aspergilose
broncopulmonar alérgica e a síndrome hipereosinofílica, bem como as outras
vasculites ANCA-associadas (PAG e PAM) (DUNOGUÉ, 2011; COTTIN, 2005).
A síndrome hipereosinofílica (SHE) também é uma condição clínica que
pode ser de difícil diferenciação com a SCS. Na primeira os pacientes podem
apresentar neuropatia, embora a mononeurite multiplex seja rara, cardiopatia e
sintomas respiratórios, sendo a presença de infiltrados pulmonares e/ou asma
pouco comum (CHURG e STRAUSS, 1951). Alguns achados, que falariam
mais a favor da SCS são: positividade para o ANCA, surgimento de
manifestações de vasculite ou a identificação de granulomas. Além disso, a
eosinofilia da SHE é geralmente refratária à terapia com corticosteróides.
A pneumonia eosinofílica crônica, por sua vez, é mais comum em
mulheres, está limitada aos pulmões, não se associando a vasculite sistêmica
ou a presença de granulomas. Entretanto, cerca de 50% dos casos de
pneumonia eosinofílica crônica têm asma associada, a distribuição periférica e

36
o caráter recidivante das opacidades pulmonares são variáveis que se
superpõem ao quadro de SCS.
A aspergilose broncopulmonar alérgica também tem características de
asma, manifesta-se por opacidades pulmonares fugazes e eosinofilia no
sangue periférico e em secreções respiratórias; no entanto, não causa
acometimento sistêmico.
A asma é em si uma doença associada com eosinofilia, porém menos
pronunciada que na SCS. Valores acima de 800 células/mm3 não são
frequentes na asma, e por isso mais sugestivos de SCS. Convêm, contudo,
lembrar que os eosinófilos circulantes podem ser suprimidos pela corticoterapia
(NOTH, 2003).
Quando os sintomas de vasculite se tornam evidentes, os principais
diagnósticos diferenciais passam a ser as outras vasculites sistêmicas,
especialmente as ANCA-associadas. PAG e PAM costumam acometer os
pulmões, todavia, a presença de asma e de eosinofilia de grande monta são
achados típicos da SCS, sendo incomuns nas demais.
Compartilhando numerosas particularidades clínicas com a PAM, como
a mononeurite multiplex e sintomas gastrointestinais, a SCS é, mais uma vez,
sugerida na vigência de asma, história de atopia, eosinofilia e infiltrados
pulmonares.
Pacientes portadores de PAG também podem apresentar sintomas
comuns à SCS, como a rinossinusite crônica e sintomas respiratórios; no
entanto, a presença de asma, eosinofilia, envolvimento não-destrutivo dos
seios paranasais, mononeurite multiplex, e, quando presentes, ANCA-P ou
anti-MPO (ANCA-C ou anti-PR3 na PAG), favoreceria o diagnóstico de SCS.
Em cada caso, detalhes do quadro clínico devem ajudar a distinguir
todas estas entidades da SCS.
3.1.7 Prognóstico
Antes do emprego de terapia imunossupressora, a taxa de mortalidade
da SCS era cerca de 50% em três meses após o diagnóstico da vasculite
(LANHAM, 1984; MASI, 1990). Após o estabelecimento da terapêutica com
corticosteróides, a sobrevida aumentou consideravelmente, passando para

37
90% em um ano, 62 a 75% em cinco anos, e 50% em sete anos (CHUMBLEY,
1977). Hoje, a sobrevida geral em 5 anos pode chegar a 97% (BOURGARIT,
2005).
No entanto, nem todos os pacientes com SCS partilham o mesmo
prognóstico, o qual, por sua vez, depende do grau inicial da extensão da
doença e do envolvimento de órgãos vitais.
AVALIAÇÃO DA GRAVIDADE
Um sistema foi desenvolvido para avaliar a gravidade da doença em
pacientes com SCS: o Five Factor Score (FFS)7, ou “Escore de Cinco Fatores”,
visto no Quadro 3 (GUILLEVIN, 1996; GAYRAUD, 2001). Este sistema de
pontuação tem apresentado forte associação com prognóstico, incluindo
mortalidade, sendo, portanto, útil para orientar a terapêutica inicial
(GUILLEVIN, 1996).
Permitindo uma contagem de 0 a dois, a pontuação zero é atribuída
quando nenhum fator está presente; um ponto para apenas um fator presente,
e dois para a ocorrência de dois ou mais fatores.
FIVE FACTOR SCORE (FFS) – ESCORE DE CINCO FATORES
Envolvimento cardíaco
Doença gastrointestinal (sangramento, perfuração, infarto ou
pancreatite)
Lesão renal (creatinina sérica >1,58mg/dL)
Proteinúria (>1g/dia)
Envolvimento do sistema nervoso central
Quadro 3 – Five-Factor Score (FFS) – Escore de Cinco Fatores
Fonte: MASI, 1990.
7 O Five-Factor Score (FFS) – Escore de Cinco Fatores - (GUILLEVIN, 1996) é um escore prognóstico obtido por meio de análises uni e multivariada de 342 pacientes com vasculites, incluindo 82 deles com SCS (GUILLEVIN, 1996). Constitui-se, este, um forte indicador prognóstico de mortalidade, como ficou demonstrado em pacientes com pontuações de 0, 1 e 2, os quais apresentaram diferentes taxas de mortalidade em 5 anos, sendo respectivamente de 12%, 26%, ou 46%. O FFS ajuda a identificar quais os pacientes possuem maiores riscos de recaída e mortalidade, exigindo, dessa forma, tratamento imunossupressor mais agressivo.

38
REATIVAÇÃO
Outra grande preocupação da SCS é a ocorrência de reativação da
doença. Embora 90% dos pacientes entrem em remissão, a taxa de recidiva
geral é cerca de 26%, atingindo até 73,8% em relação àqueles com
prognóstico pior (GUILLEVIN, 1999). A maioria das recidivas ocorre durante o
primeiro ano de acompanhamento (BOURGARIT, 2005; GUILLEVIN, 2010),
sendo geralmente precedidas por elevação da contagem de eosinófilos
(GUILLEVIN, 1999).
MORTALIDADE
A mortalidade é relacionada a complicações cardíacas, responsáveis por
46,7% dos casos de óbito, como insuficiência cardíaca, anormalidades de
condução e doença coronária secundária à vasculite. Complicações
gastrointestinais, especialmente infarto mesentérico (8,7%), constituem a
segunda causa mais comum (GUILLEVIN, 1999). Demais importantes causas
de óbito são atribuídas ao sangramento gastrointestinal, hemorragia cerebral,
estado de mal asmático e insuficiência respiratória.
3.2 POLIANGIITE GRANULOMATOSA
A poliangiite granulomatosa (PAG), também conhecida como
granulomatose de Wegener, é uma vasculite sistêmica de pequenos e médios
vasos, caracterizada por necrose e formação de granulomas. É a mais comum
dentre as vasculites ANCA-associadas, sendo tipicamente relacionada à tríade
clássica: envolvimento renal e dos tratos respiratórios superior e inferior
(CHUNG, 2010). Pode, no entanto, afetar praticamente qualquer órgão, com
variáveis graus de acometimento (HOFFMAN, 1992).
Cerca de 80 a 90% dos pacientes com PAG possuem ANCA positivo,
com especificidade para a proteinase 3 (PR3) em 80%, e em até 20%, para a
mieloperoxidase (MPO). Com a doença em atividade, sensibilidade e
especificidade podem chegar a 90% (HOMER, 1998). No entanto, o ANCA só é
detectado em metade dos pacientes com a forma limitada da doença (HAGEN,
1998).

39
O diagnóstico é desafiador devido ao quadro clínico inespecífico,
devendo ser realizado diagnóstico diferencial com diversas outras
enfermidades. Além disso, o diagnóstico precoce é de fundamental importância
e com implicação direta no desfecho clínico, exigindo, portanto, um alto grau de
suspeição do médico, bem como uma abordagem diagnóstica adequada.
O tratamento da PAG é semelhante ao das outras VAA, razão pela qual
as considerações sobre este assunto serão consolidadas no subitem 3.4 -
Tratamento das Vasculites ANCA-associadas.
3.2.1 Substituição do Epônimo
O termo poliangiite granulomatosa foi recentemente proposto em
substituição à denominação tradicional, granulomatose de Wegener, com a
ideia de que o epônimo seja gradualmente omitido com o passar do tempo.
Em novembro de 2010, um consenso entre o Colégio Americano de
Reumatologia (ACR), a Sociedade Americana de Nefrologia (ASN) e a Liga
Européia contra o Reumatismo (EULAR) recomendou que o termo
“granulomatose de Wegener” fosse substituído por “poliangiite granulomatosa
(Wegener)”, abreviado como PAG (FALK, 2011). A mudança foi motivada pela
evidência de que o Dr. Friedrich Wegener, o qual descreveu e estudou a
doença, fora membro do partido Nazista antes e depois da Segunda Guerra
Mundial (WOYWODT, 2006). Essa mudança reflete ainda a tendência de
substituir termos epônimos honoríficos por termos que descrevam a doença ou
que sejam etiologicamente relacionados.
3.2.2 Epidemiologia
INCIDÊNCIA NA POPULAÇÃO GERAL
Como visto anteriormente, a PAG é a mais comum dentre as vasculites
ANCA-associadas (KEOGH, 2006). Sua incidência na população geral vem
aumentando ao longo dos anos, com estimativas atuais de 4 a 12 casos por
milhão de habitantes (WATTS, 2004; KOLDINGSNES, 2000). Sua prevalência
na população geral é variável conforme a localização geográfica, sendo maior
nos países de mais altas latitudes, tanto ao norte quanto ao sul da linha do
Equador (GATENBY, 2009).

40
INCIDÊNCIA SEGUNDO IDADE E GÊNERO
A idade média no momento do diagnóstico é de cerca de 40 a 55 anos,
podendo ocorrer, no entanto, em qualquer faixa etária (GONZALEZ-GAY, 2001;
REINHOLD-KELLER, 2000). Além disso, a PAG é mais comum em
caucasianos (80-97%), sem haver predileção por gênero (LANGFORD, 1992).
FATORES AMBIENTAIS: INFECÇÃO
Alguns estudos sugerem um evento infeccioso relacionado à PAG,
devido a relatos circunstanciais do aparecimento da doença precedido por
infecção. Além disso, soma-se também ao fato da maior incidência de PAG
durante os meses de outono e inverno, estações com maiores taxas de
infecções respiratórias. No entanto, há controvérsia quanto a esta presumível
sazonalidade no início da doença (MAHR, 2006).
OCUPACIONAL e ESTILO DE VIDA
Exposição à sílica cristalina destaca-se como um convincente fator de
risco ambiental, conferindo um risco aumentado em duas a quatorze vezes
para as vasculites ANCA-associadas, SCS, PAG e PAN (NUYTS 1995;
HOGAN, 2007).
São ainda sugeridas outras associações para a PAG, incluindo a
pecuária, o contato com solventes orgânicos, a inalação de partículas e gases,
e a exposição ao chumbo, cádmio ou mercúrio (ALBERT, 2004). O papel das
exposições ocupacionais como fatores de risco, no entanto, é ainda
questionado, como em um estudo recente que não encontrou relação entre a
ocupação profissional e PAG (KNIGHT, 2010).
Haubitz et al. estudaram a associação do tabagismo com as vasculites
ANCA-associadas. Descreveram uma proporção de fumantes ativos
significativamente menor no grupo dos portadores de vasculites do que na
população geral, sugerindo que o fumo estaria relacionado a um risco
diminuído para as VAA (HAUBITZ, 2005).

41
3.2.3 Etiologia e Patogênese
A patogênese das vasculites ANCA-associadas não é completamente
compreendida. No entanto, há evidências de que os anticorpos anticitoplasma
de neutrófilos (ANCA) desempenhem um papel direto na indução ao dano
tecidual (JENNETTE, 1993).
A presença de granulomas e processo vasculítico sugere uma resposta
celular imune exagerada ou resposta de hipersensibilidade; todavia, nenhum
agente etiológico foi identificado (LANGFORD, 1999). Além disso, a
predisposição da doença em acometer tratos respiratórios inferior e superior
apóia a hipótese de que antígenos, provavelmente inalados, devem iniciar a
resposta celular imune.
Agentes infecciosos também têm sido implicados, mas em nenhum
deles ficou definitivamente comprovada a associação. Ainda apoiando uma
causa infecciosa, Stegeman et al. demonstraram que a presença de
colonização nasal crônica por Staphylococcus aureus em pacientes com PAG
estaria associada a uma maior taxa de recaídas. Este mesmo grupo também
relatou uma diminuição na taxa de recidivas de manifestações em via aérea
superior (orelha, nariz, orofaringe), mas não renal ou pulmonar, em pacientes
que recebem terapia antibacteriana crônica com sulfametoxazol-trimetoprima
(STEGEMAN, 1996).
3.2.4 Manifestações Clínicas
Sintomas constitucionais, os quais incluem febre (40%), emagrecimento
(70%) e astenia, podem estar presentes no início dos sintomas.
Até 40% dos pacientes manifestam, à apresentação inicial, envolvimento
renal. Todavia, cerca de 80 a 90% dos casos desenvolvem doença renal em
algum momento.
O ANCA padrão citoplasmático (ANCA-C) está positivo em mais de 90%
dos pacientes com a forma generalizada da PAG, mas só é detectado em
metade dos pacientes com a doença limitada.
Na Tabela 3 abaixo, estão as principais manifestações da poliangiite
granulomatosa, refletindo o amplo espectro de apresentação da doença.

42
Tabela 3 – Principais Manifestações da PAG
Fonte: LEHMANN, 1990.
SISTEMA ENVOLVIDO PRINCIPAIS SINTOMAS INCIDÊNCIA
Pulmões Dispnéia, hemoptise 85%
Cabeça e pescoço Epistaxe, rinossinusite, crostas
nasais, perda auditiva, estenose
subglótica
95%
Glomerulonefrite Edema, hematúria microscópica 80%
Musculoesquelético Artralgia, artrite, mialgia 70%
Olhos Episclerite, conjuntivite, granuloma
retrobulbar
- 60%
Sintomas gerais Febre, perda de peso, sudorese
noturna
> 50%
Pele Púrpura, necrose, nódulos - 50%
Polineuropatia Paralisia do nervo fibular, parestesia 20%
Sistema nervoso
central
Epilepsia, psicose < 10%
Gastrointestinal Hemorragia digestiva, tenesmo < 10%
MANIFESTAÇÕES PULMONARES
O envolvimento das vias aéreas inferiores e do parênquima pulmonar é
muito frequente, manifestando-se por tosse (habitualmente produtiva),
hemoptise, dispnéia, sibilância e desconforto torácico (GÓMEZ-PUERTA, 2009;
ANTUNES, 2005). Estes sintomas muitas vezes estão acompanhados de
estenose traqueal ou subglótica e derrame pleural, além de alterações
radiológicas, as quais são encontradas em mais de 70% dos casos durante
todo o curso da doença.
HEMORRAGIA ALVEOLAR DIFUSA
A hemorragia alveolar difusa (HAD), devido à capilarite da
microvasculatura pulmonar, é uma complicação da PAG ameaçadora à vida,
exigindo tratamento imunossupressor imediato e agressivo (LYNCH, 1997). Em
mais de 90% dos casos, está associada à glomerulonefrite rapidamente

43
progressiva (GNRP) (LYNCH, 1997; TRAVIS, 1990). São relatadas taxas de
mortalidade de 60%, seis vezes maior que a mortalidade da PAG em que não
ocorreu HAD (GREEN, 1996).
O diagnóstico é clínico, e pode ser estabelecido ao se observar infiltrado
alveolar bilateral difuso aos exames de imagem do tórax (Figura 9) e uma
redução do hematócrito (anemia), acompanhados ou não de hemoptise
(CHUNG, 2010). Na ausência de hemoptise, o lavado broncoalveolar (LBA)
persistentemente hemorrágico ou sero-sanguinolento (fase aguda), ou com
macrófagos repletos de hemossiderina (fase subaguda) consubstancia o
diagnóstico de hemorragia alveolar difusa.
Figura 9: Hemorragia alveolar secundária a poliangiite granulomatosa. Radiografia torácica em
incidência póstero-anterior de um indivíduo do gênero masculino, 67 anos, demonstrando infiltrado alveolar bilateral e difuso. Recuperação completa alcançada com corticosteróide e
ciclofosfamida.
Fonte: LYNCH, 2011.
A biópsia pulmonar cirúrgica é prescindível ao diagnóstico de
hemorragia alveolar difusa, mostrando achados inespecíficos de hemorragia e
inflamação alveolar e de necrose de capilares (Figura 10) (TRAVIS, 1990).
Além disso, a biópsia pode representar um grande risco, especialmente em
casos graves de HAD. Todavia, mostra-se útil na distinção entre PAG e PAM,

44
uma vez que somente a PAG costuma apresentar granulomas, ao lado da
necrose e vasculite.
Figura 10: Fotomicrografia. Capilarite e hemorragia pulmonar em poliangiite granulomatosa. (A) Numerosos neutrófilos sobre os capilares alveolares associados a recente hemorragia alveolar.
(B) Necrose fibrinóide precoce da parede alveolar associada a capilarite. (C) Hemorragia pulmonar com capilarite.
Fonte: TAZELAAR & LYNCH, 2011.
MANIFESTAÇÕES DAS VIAS AÉREAS SUPERIORES
Sintomas de vias aéreas superiores (VAS) são frequentes, ocorrendo em
mais de 90% dos pacientes com a doença (GUBBELS, 2003). Comumente são
encontrados rinorréia, epistaxe, rinossinusite crônica, otite, úlceras mucosas e
até lesões ósseas destrutivas (JENNETTE e FALK, 1997), com anormalidades
na TC de seios da face em mais de 85% dos casos (HOFFMAN, 1992).
A ocorrência de nariz em sela (Figura 11), deformidade secundária à
destruição da cartilagem nasal e do vômer, é rara; no entanto, apesar de não
ser patognomônico, é bastante característico da PAG. Estas lesões são
frequentemente infectadas secundariamente por Staphylococcus aureus
(ANTUNES, 2005; JONES, 1999).

Figura 11: Nariz em sela em paciente com poliangiite granulomatosa.
Fonte: GOTTSCHLICH, 2006
Otalgia e otorréia também são bastante f
auditiva. Também podem estar presentes úlceras orais dolorosas e aumento
doloroso das glândulas submandibulares e parótidas
2005).
MANIFESTAÇÕES RENAIS
O acometimento renal ocorre em 70 a 85% dos pacientes durante o
curso da doença (LYNCH,
manifestações mais graves da doença, vez que pode evoluir rapidamente para
insuficiência renal, mesmo na ausência de sintomas (
clínico deve-se atentar em achados laboratoriais anormais, como proteinúria,
hematúria microscópica e cilindros hemáticos, bem como aumento da
creatinina sérica ou redução da taxa de filtração glomerular, que são, pois,
sugestivos de uma complicação renal
Figura 11: Nariz em sela em paciente com poliangiite granulomatosa.
2006.
Otalgia e otorréia também são bastante frequentes, assim como perda
auditiva. Também podem estar presentes úlceras orais dolorosas e aumento
doloroso das glândulas submandibulares e parótidas (SEO, 2004;
MANIFESTAÇÕES RENAIS
O acometimento renal ocorre em 70 a 85% dos pacientes durante o
(LYNCH, 2011). A glomerulonefrite (GN) é uma das
manifestações mais graves da doença, vez que pode evoluir rapidamente para
insuficiência renal, mesmo na ausência de sintomas (FAUCI, 1983). Assim, o
se atentar em achados laboratoriais anormais, como proteinúria,
hematúria microscópica e cilindros hemáticos, bem como aumento da
creatinina sérica ou redução da taxa de filtração glomerular, que são, pois,
uma complicação renal manifesta.
45
Figura 11: Nariz em sela em paciente com poliangiite granulomatosa.
requentes, assim como perda
auditiva. Também podem estar presentes úlceras orais dolorosas e aumento
2004; ANTUNES,
O acometimento renal ocorre em 70 a 85% dos pacientes durante o
2011). A glomerulonefrite (GN) é uma das
manifestações mais graves da doença, vez que pode evoluir rapidamente para
, 1983). Assim, o
se atentar em achados laboratoriais anormais, como proteinúria,
hematúria microscópica e cilindros hemáticos, bem como aumento da
creatinina sérica ou redução da taxa de filtração glomerular, que são, pois,

46
Os achados histopatológicas vão desde formas leves, como a
glomerulonefrite segmentar e focal, lesão renal característica da PAG (LYNCH,
1992; HOFFMAN, 1992), até a glomerulonefrite rapidamente progressiva com a
formação de crescentes (Figura 12), com frequência associada à insuficiência
renal dialítica.
Figura 12: Fotomicrografia. Biópsia renal percutânea de paciente do gênero masculino, 65 anos
de idade, com poliangiite granulomatosa, apresentando-se com hemorragia alveolar, glomerulonefrite rapidamente progressiva (GNRP) e otite média serosa. Notar a típica formação
de crescentes indicativa da GNRP.
Fonte: LYNCH, 1989.
MANIFESTAÇÕES CUTÂNEAS
Manifestações cutâneas aparecem em até 50% dos pacientes durante o
curso da doença (LYNCH, 2011), e incluem úlceras, púrpuras palpáveis,
nódulos subcutâneos, pápulas e rash cutâneo. Pioderma gangrenoso e
fenômeno de Raynaud são raramente relatados. Nódulos granulomatosos
refletem a inflamação granulomatosa, enquanto púrpura, petéquias e lesões
hemorrágicas são indicativas do processo vasculítico (LYNCH, 2004).
A biópsia de pele pode mostrar vasculite granulomatosa com necrose;
no entanto, vasculite leucocitoclástica é um achado mais comum
(BARKSDALE, 1995). Lesões cutâneas na PAG estão associadas a uma maior
incidência de envolvimento renal e articular, comparativamente àqueles
pacientes sem alterações cutâneas (LYNCH, 2004).

47
3.2.5 Achados Histopatológicos
O estudo histopatológico na PAG é de fundamental importância para seu
manejo clínico adequado, vez que esta doença requer confirmação histológica
(LEAVITT, 1990). Os principais achados histopatológicos da PAG incluem uma
vasculite necrosante acometendo pequenos vasos, necrose “geográfica”
extensa e infiltrado inflamatório com formação de granulomas (Figura 13)
(TRAVIS, 2004).
Figura 13: Variedade de lesões vasculares na poliangiite granulomatos. (A) Arterite
granulomatosa com células gigantes multinucleadas no lúmen arterial (H&E, 100x). (B) Venulite com necrose fibrinóide da parede venosa (seta) (H&E, 200x). (C) Arterite com infiltrado linfocítico e hiperplasia proeminente da íntima (H&E, 100x). (D) Arterite com eosinófilos
presentes. (H&E, 200x). Fonte: FISHBEIN, 2011.
A necrose do parênquima pode apresentar-se sob a forma de
microabscessos neutrofílicos ou necrose “geográfica”. Os granulomas, por sua
vez, podem aparecer como dispersos aglomerados de células gigantes,
histiócitos em paliçada, células gigantes alinhadas envolvendo a área de
necrose geográfica ou microabscessos, como mostra a Figura 14 (TAZELAAR
& LYNCH, 2011).
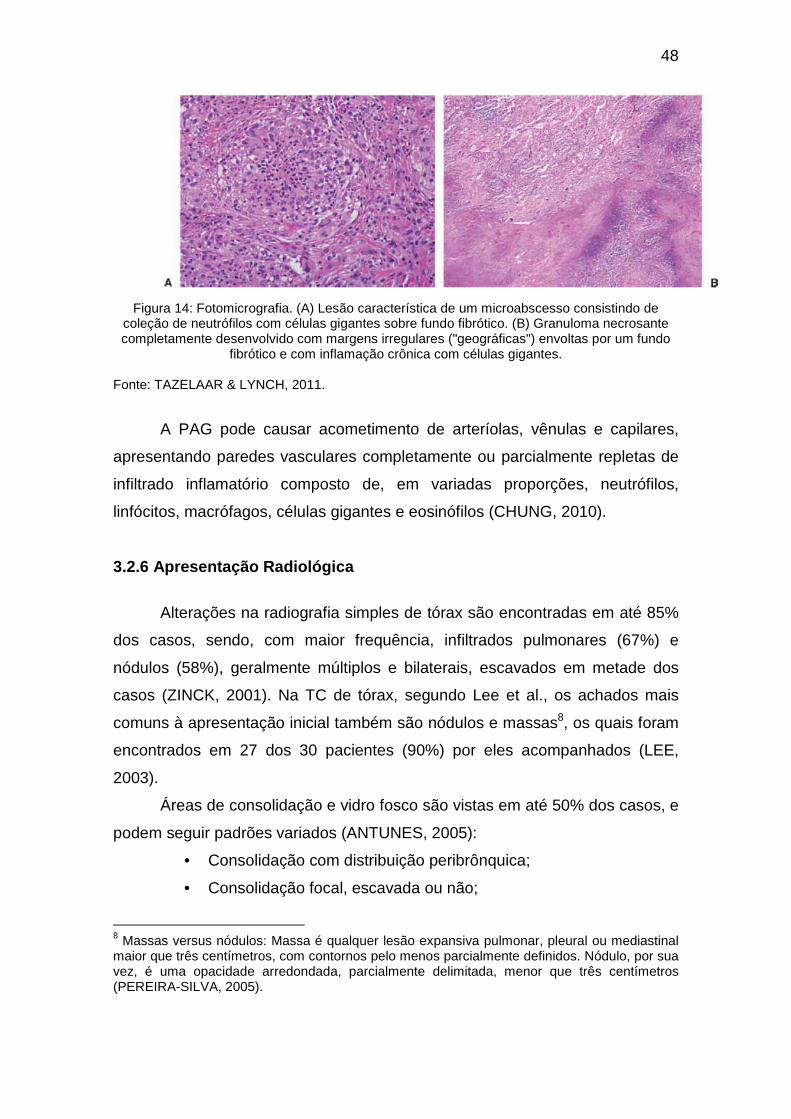
48
Figura 14: Fotomicrografia. (A) Lesão característica de um microabscesso consistindo de
coleção de neutrófilos com células gigantes sobre fundo fibrótico. (B) Granuloma necrosante completamente desenvolvido com margens irregulares ("geográficas") envoltas por um fundo
fibrótico e com inflamação crônica com células gigantes. Fonte: TAZELAAR & LYNCH, 2011.
A PAG pode causar acometimento de arteríolas, vênulas e capilares,
apresentando paredes vasculares completamente ou parcialmente repletas de
infiltrado inflamatório composto de, em variadas proporções, neutrófilos,
linfócitos, macrófagos, células gigantes e eosinófilos (CHUNG, 2010).
3.2.6 Apresentação Radiológica
Alterações na radiografia simples de tórax são encontradas em até 85%
dos casos, sendo, com maior frequência, infiltrados pulmonares (67%) e
nódulos (58%), geralmente múltiplos e bilaterais, escavados em metade dos
casos (ZINCK, 2001). Na TC de tórax, segundo Lee et al., os achados mais
comuns à apresentação inicial também são nódulos e massas8, os quais foram
encontrados em 27 dos 30 pacientes (90%) por eles acompanhados (LEE,
2003).
Áreas de consolidação e vidro fosco são vistas em até 50% dos casos, e
podem seguir padrões variados (ANTUNES, 2005):
• Consolidação com distribuição peribrônquica;
• Consolidação focal, escavada ou não;
8 Massas versus nódulos: Massa é qualquer lesão expansiva pulmonar, pleural ou mediastinal maior que três centímetros, com contornos pelo menos parcialmente definidos. Nódulo, por sua vez, é uma opacidade arredondada, parcialmente delimitada, menor que três centímetros (PEREIRA-SILVA, 2005).

49
• Bandas parenquimatosas;
• Áreas de consolidação periférica (mimetizando infartos
pulmonares); e
• Áreas de vidro fosco difusas e bilaterais, em geral representando
hemorragia alveolar.
Alguns achados radiológicos comuns estão ilustrados na Figura 15.
Figura 15: Exemplos de manifestações radiológicas da PAG: (A e B) Radiografia simples de
tórax com massas escavadas, com velamento adjacente em B; (C a E): Tomografia computadorizada; (C) Áreas de vidro despolido; (D) Massa escavada; (E) Grande caverna com
nível líquido e duas massas escavadas coalescentes na região inferior; (F) Reconstrução bidimensional por tomografia computadorizada de vias aéreas, com evidência de estenose de
cerca de 50% da luz da traquéia
Fonte: ANTUNES, 2005.

50
Outras formas de apresentação compreendem nódulos pulmonares
isolados ou múltiplos, escavados ou não. Estas são relativamente frequentes,
enquanto linfonodomegalias e derrame pleural são menos comuns (5% a 20%
dos pacientes) (ABERLE, 1990).
3.2.7 Diagnóstico
O diagnóstico é desafiador devido ao quadro clínico inespecífico,
devendo ser realizado diagnóstico diferencial com diversas outras entidades.
Além disso, o diagnóstico precoce é de fundamental importância pelo impacto
que produz nos diversos desfechos clínicos, inclusive na mortalidade, para o
qual é necessário um alto grau de suspeição clínica, seguida de uma
abordagem diagnóstica adequada.
Os achados laboratoriais são os de uma doença inflamatória sistêmica,
com anemia normocrômica e normocítica, trombocitose e velocidade de
hemossedimentação elevada. Alterações nos níveis de uréia e creatinina e no
sedimento urinário (com proteinúria e hematúria) podem ocorrer na vigência de
envolvimento renal. Testes de função pulmonar podem mostrar padrões
obstrutivos, restritivos e até mistos (LYNCH, 2004).
O diagnóstico da PAG é determinado ao serem combinados
manifestações clínicas, radiológicas e achados anatomopatológicos, apoiando-
se ainda na análise do ANCA e de exames laboratoriais complementares.
Segundo os critérios do ACR de 1990, a presença de dois ou mais dos
fatores expostos no Quadro 4 estaria associada a uma sensibilidade de 88% e
especificidade de 92% para o estabelecimento do diagnóstico de PAG
(LEAVITT, 1990).
CRITÉRIOS DO
COLÉGIO AMERICANO DE REUMATOLOGIA (ACR)
ÚLCERAS ORAIS OU RINORRÉIA (purulenta ou sanguinolenta)
ACHADOS ANORMAIS NA RADIOGRAFIA DE TÓRAX (nódulos, cavernas,
infiltrados fixos).
SEDIMENTO URINÁRIO ANORMAL (cilindros hemáticos ou >5 hemácias por
campo)

51
INFLAMAÇÃO GRANULOMATOSA NA PAREDE ARTERIAL OU NA REGIÃO
PERIVASCULAR, OU EXTRAVASCULAR NA ANÁLISE HISTOLÓGICA DA
BIÓPSIA (presença de hemoptise, se biópsia não estiver disponível).
Quadro 4 – Critérios do Colégio Americano de Reumatologia de 1990 para a classificação da
poliangiite granulomatosa (ACR 1990)
Fonte: LEAVITT, 1990.
Segundo Leavitt et al., o diagnóstico da poliangiite granulomatosa seria
confirmado ao encontrar-se vasculite na amostra de biópsia ou à angiografia e
associando este dado com a presença dos critérios supracitados. (LEAVITT,
1990).
Posteriormente, surgiu a classificação européia, definindo um novo
algoritmo para o diagnóstico de PAG, o qual associa os critérios e conceitos já
estabelecidos pelo ACR e pelo CCH (WATTS, 2007). Foi sugerido que a
presença das seguintes manifestações poderia permitir o diagnóstico da PAG
na ausência de biópsia:
• Vias aéreas inferiores: evidência radiográfica de infiltrados pulmonares
fixos, nódulos ou cavernas por mais de um mês, ou estenose brônquica;
• Vias aéreas superiores: rinorréia sanguinolenta e crostas por mais de
um mês ou ulceração nasal, rinossinusite crônica, otite média ou
mastoidite por mais de três meses; massa retrorbitária, estenose
subglótica; nariz sela ou doença destrutiva nasal;
• Glomerulonefrite: hematúria associada a cilindros hemáticos ou >10
hemácias dismórficas por campo, ou hematúria ou proteinúria (duas
cruzes) utilizando dipsticks; e
• ANCA também foi incluído como um critério diagnóstico. Quando
positivo em paciente apresentando as supracitadas manifestações, pode
ser estabelecido o diagnóstico de PAG sem a realização de biópsia.
O uso deste algoritmo permitiu a classificação dos pacientes com uma
concordância interobservador de 91,5% (WATTS, 2007) e resultou em redução
do número de casos que ainda permaneciam sem classificação de acordo com
os critérios do ACR ou definições do CCH (LIU, 2008).

52
No entanto, não sendo preenchidos os supracitados critérios, porém
havendo cenário clínico compatível e após excluídas outras enfermidades (ver
tópico Diagnóstico Diferencial, a seguir), o diagnóstico pode ser confirmado
pela observação de vasculite, inflamação granulomatosa e necrose na amostra
de biópsia (WALTON, 1958), exemplificada na Figura 16.
Figura 16: Parênquima pulmonar demonstrando características histológicas da poliangiite
granulomatosa: necrose, inflamação granulomatosa e vasculite. Fonte: LANGFORD, 1999.
Estas características histológicas, por sua vez, geralmente apresentam
uma distribuição irregular, sendo a possibilidade de se obter uma biópsia
conclusiva influenciada pelo local onde foi realizada e, particularmente, pela
quantidade de tecido obtido na biópsia (TRAVIS, 1991).
A biópsia pulmonar cirúrgica é método ideal para se firmar um
diagnóstico de PAG com envolvimento pulmonar. Quando infiltrados
pulmonares focais ou nódulos estão presentes, a observação de vasculite e
granulomas necrosantes na amostra de biópsia é possível em mais de 90%
dos pacientes (TRAVIS, 2004; TRAVIS, 1991).

53
DIAGNÓSTICO DIFERENCIAL
O diagnóstico diferencial da PAG baseia-se no conjunto das
manifestações clínicas apresentadas por cada paciente. Geralmente inclui
infecções, neoplasia, doenças do tecido conjuntivo e outras vasculites
sistêmicas (LANGFORD, 1999).
Havendo a presença de imagens nodulares escavadas à radiografia
torácica deve ser aventada a possibilidade de uma causa infecciosa, em
especial infecções micobacterianas e fúngicas, comuns em nosso meio. Ainda,
este achado de imagem também pode sugerir a possibilidade de metástases
escavadas ou uma forma multinodular do anteriormente denominado
carcinoma bronquioloalveolar (CHUNG, 2010).
Também é importante considerar outras doenças que cursam com
acometimento concomitante de rins e pulmões, como a leptospirose, a
síndrome do anticorpo anti-MBG, lúpus eritematoso sistêmico e as outras
vasculites ANCA associadas (PAM e SCS).
Devemos incluir ainda condições clínicas que cursem com lesões
destrutivas em face, como neoplasias (linfoproliferativas, em geral), infecções
micobacterianas e fúngicas e leishmaniose mucocutânea.
Em resumo, PAG, PAM e SCS, embora guardem entre si algumas
características comuns, é possível firmar o diagnóstico específico com base na
correlação clínico-radiológico-patológica com auxílio de exames laboratoriais
comprobatórios. A classificação da PAG baseada nas formas de apresentação,
restritas e difusas, permite estabelecer o prognóstico e definir o nível da
intervenção terapêutica.
3.2.8 Prognóstico
Uma vez que as medicações para o tratamento da PAG são
potencialmente tóxicas, por vezes, com efeitos adversos importantes, faz-se
necessário estabelecer o diagnóstico definitivo de forma precoce.
Após a introdução da ciclofosfamida na terapia imunossupressora,
remissão completa tem sido obtida em 70 a 90% dos casos, apesar de
recidivas ainda serem relativamente frequentes (LAMPRECHT, 2004). Um pior
prognóstico está associado à presença de hemorragia alveolar, dano renal

54
grave, idade avançada e proteinase 3 (PR3) positiva. A forma limitada da
doença apresenta melhor prognóstico que a forma difusa.
3.3 POLIANGIITE MICROSCÓPICA
A poliangiite microscópica (PAM) é uma vasculite necrosante sistêmica
paucimune caracterizada clinicamente por uma fase prodrômica de sintomas
constitucionais, seguida do desenvolvimento de glomerulonefrite necrosante
focal rapidamente progressiva, associada a envolvimento pulmonar em 30%
dos casos (FRANKEL, 2006). Em pacientes que desenvolvem doença
pulmonar, HAD com capilarite é a manifestação mais comum. Acometimento
de pele, articulações, sistema nervoso periférico e gastrointestinal também são
relativamente comuns.
Esta vasculite ANCA-associada acomete vasos de pequeno calibre,
predominantemente, e também vasos de médio porte, podendo causar arterite
necrosante histologicamente semelhante à causada pela poliarterite nodosa
(PAN). Inicialmente, inclusive, a PAM era reconhecida como uma forma
particular de PAN (DAVSON, 1948). Após o CCH em 1994, no entanto, ambas
foram reconhecidas como entidades distintas, diferindo, pela ausência de
vasculite acometendo veias em pacientes com PAN, bem como pela presença
de vasculite em vasos de pequeno calibre (arteríolas, vênulas e capilares),
presente na PAM (JENNETTE, 1994; WATTS, 1996).
O ANCA-P é positivo em 50 a 75% dos casos, com anti-MPO positivo
em 35 a 65%; porém, ANCA-C também pode ser encontrado em 10 a 15%
(JENNETTE, 2001). É histologicamente caracterizada por vasculite focal,
necrosante e segmentar, com infiltrado inflamatório misto, e ausência de
formação de granulomas, presentes na PAG (SANTOS, 2004).
3.3.1 Epidemiologia
É uma vasculite necrosante paucimune, relativamente rara na população
geral, com uma incidência anual variando entre 2,1 a 17,5 por milhão de
habitantes (GIBELIN, 2011).
Comumente acomete pacientes com média de idade de 50 anos, ao
início dos sintomas, embora possa ocorrer em indivíduos de qualquer idade

55
(SAVAGE, 1985; JENNETTE, 1997). Além disso, apesar de leve predomínio
em homens ter sido aventado, uma incidência relacionada ao gênero
permanece incerta (SAVAGE, 1985; D’AGATI, 1986). Algumas evidências
sugerem maior incidência da PAM em asiáticos, comparativamente a europeus
(FUJIMOTO, 2006; WATTS, 2008).
3.3.2 Etiologia e Patogênese
Alguns autores estudam uma etiologia infecciosa para as vasculites
sistêmicas, especialmente em relação às VAA, as quais possuem
acometimento pulmonar frequente. O desenvolvimento da doença justificar-se-
ia por infecções causadas por patógenos inalados (GIBELIN, 2011).
Devido a relação com o ANCA, também tem sido aventada a
participação deste na patogênese da PAM. No entanto, permanece ainda
incerto se estes anticorpos estão diretamente envolvidos na patogênese das
vasculites ou se seriam apenas um fenômeno associado (JENNETTE, 1994).
Fatores genéticos também tem sido implicados, especialmente durante
as últimas décadas. Em estudo realizado no Japão, foi relatada associação da
PAM com o alelo HLA-DRB1*0901; no entanto, este achado ainda carece de
novos estudos (TSUCHIYA, 2006).
3.3.3 Manifestações Clínicas
O rim é o órgão alvo mais frequentemente acometido e a
glomerulonefrite é um evento quase universal, sendo observada em 90% dos
pacientes (CHUNG, 2010). Está associada a envolvimento pulmonar em até
30% dos casos (AKIKUSA, 1997), sendo ambas as mais importantes causas
de morbidade e mortalidade da doença. Podem ocorrer ainda alterações
cutâneas (púrpura e nódulos cutâneos), neurológicas e gastrointestinais. Vias
aéreas superiores também são acometidas pelo processo vasculítico, sendo,
todavia, muito menos comum do que na PAG (GUILLEVIN, 1999). Ainda, a
hemorragia alveolar difusa é uma séria complicação da PAM, que ocorre
devido à capilarite pulmonar.

56
Algumas das principais manifestações clínicas da PAM estão listadas na
Tabela 4 abaixo, com suas respectivas frequências de acometimento.
Tabela 4 – Manifestações clínicas da poliangiite microscópica
Fonte: BROWN, 2006.
MANIFESTAÇÕES CLÍNICAS INCIDÊNCIA
Glomerulonefrite rapidamente progressiva 100%
Pulmões (hemorragia, hemoptise) 10-30%
Sintomas constitucionais (febre, perda de peso, calafrios,
artralgia, mialgias)
70-80%
Pele (púrpura, urticária, exantema, nódulos subcutâneos) 50-65%
Sistema nervoso central (mononeurite multiplex) 15-50%
Gastrointestinal (dor, hemorragia digestiva, perfuração) 30-45%
Ocular (Conjuntivite, uveíte) 0-30%
Cardíaca 10-20%
Vias aéreas 0-15%
3.3.4 Achados Histopatológicos
Constituem o padrão anatomopatológico da PAM a vasculite necrosante
segmentar ou focal, associada a infiltrado inflamatório misto (neutrófilos e
monócitos), sem formação de granuloma, (SAVAGE, 2000).
Glomerulonefrite é observada em 90% dos pacientes (CHUNG, 2010),
sendo caracterizada por necrose focal, formação de crescentes e ausência ou
escassez de depósito de imunocomplexos (Figura 17).
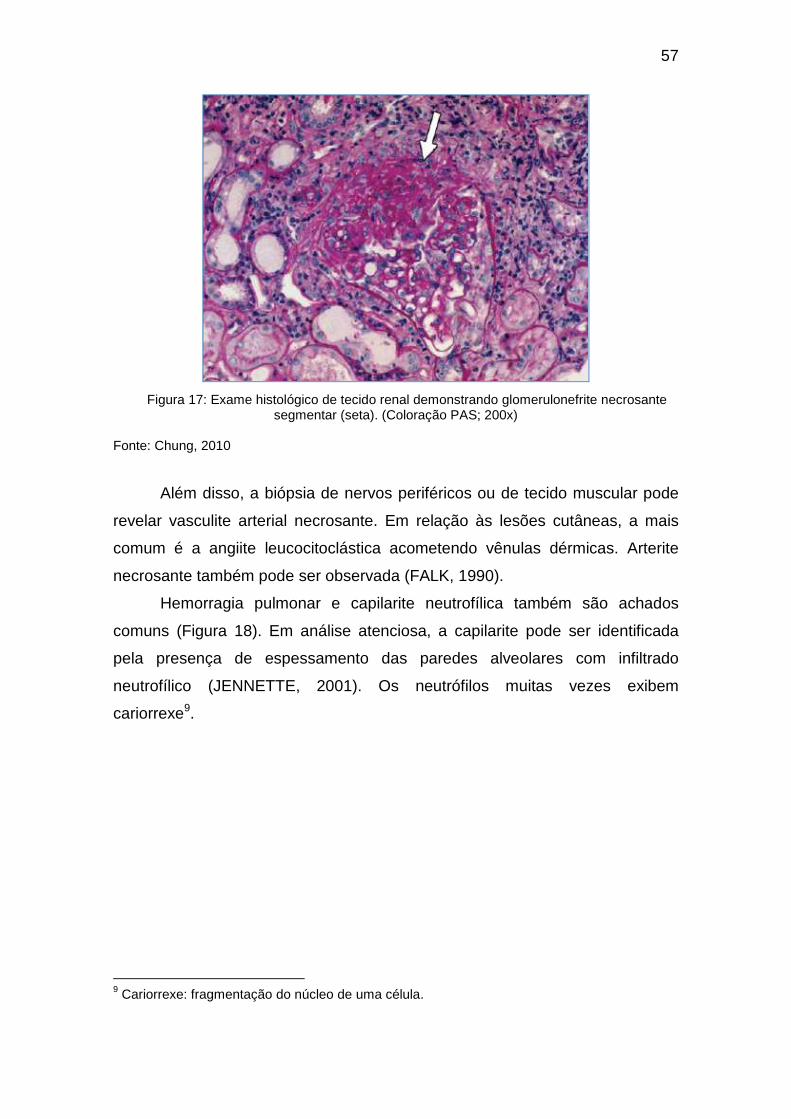
57
Figura 17: Exame histológico de tecido renal demonstrando glomerulonefrite necrosante
segmentar (seta). (Coloração PAS; 200x) Fonte: Chung, 2010
Além disso, a biópsia de nervos periféricos ou de tecido muscular pode
revelar vasculite arterial necrosante. Em relação às lesões cutâneas, a mais
comum é a angiite leucocitoclástica acometendo vênulas dérmicas. Arterite
necrosante também pode ser observada (FALK, 1990).
Hemorragia pulmonar e capilarite neutrofílica também são achados
comuns (Figura 18). Em análise atenciosa, a capilarite pode ser identificada
pela presença de espessamento das paredes alveolares com infiltrado
neutrofílico (JENNETTE, 2001). Os neutrófilos muitas vezes exibem
cariorrexe9.
9 Cariorrexe: fragmentação do núcleo de uma célula.

58
Figura 18: Biópsia pulmonar obtida por videotoracoscopia mostrando hemorragia alveolar e
espessamento das paredes dos alvéolos com infiltrado inflamatório neutrofílico (setas). Detalhe no canto inferior esquerdo demonstrando capilarite neutrofílica e cariorrexe (H&E 100x).
Fonte: Chung, 2010.
3.3.5 Apresentação Radiológica
Inicialmente, o radiograma de tórax pode mostrar opacidades alveolares
bilaterais sugestivas de hemorragia alveolar (Figura 19) (CHUNG, 2010).
Figura 19: Poliangiite microscópica em paciente do sexo feminino, 33 anos. Radiografia
de tórax mostrando áreas extensas de consolidação bilaterais em zonas pulmonares médias e inferiores.
Fonte: CHUNG, 2010.

59
O uso da tomografia computadorizada é recomendado para aqueles
pacientes com suspeita de HAD ou para aqueles com queda aguda da função
renal. A TC tem sensibilidade elevada, comparativamente à radiografia;
pacientes sem achados anormais na radiografia torácica podem apresentar
imagens de vidro fosco difuso na TC (Figura 20) (LAUQUE, 2000).
Figura 20: Poliangiite microscópica em paciente do sexo feminino, 33 anos. TC com
janela para pulmão mostrando opacidades pulmonares extensas bilaterais, por vezes com o padrão de perfusão em mosaico. Nódulo mal definido (seta) é observado no lobo superior
esquerdo.
Fonte: CHUNG, 2010. 3.3.6 Diagnóstico
O diagnóstico de poliangiite microscópica pode ser estabelecido ao se
observar alterações histológicas características da doença em paciente com
quadro clínico compatível. Biópsia a céu aberto é o procedimento com maior
rendimento diagnóstico, sendo, todavia, associado com maior mortalidade
(CORDIER, 1996). Um teste positivo para o ANCA, com imunofluorescência indireta
indicando padrão perinuclear (ANCA-P) e confirmação da presença de
anticorpos antimieloperoxidase (anti-MPO) por ELISA, somando-se a um
quadro clínico compatível, também é altamente sugestivo de PAM
(O'SULLIVAN, 2002).

60
DIAGNÓSTICO DIFERENCIAL
O diagnóstico diferencial da PAM é amplo, sendo importante realizar
uma abordagem clínico-radiológico-patológica, bem como análise laboratorial,
para que se estabeleça o diagnóstico específico.
A PAM deve ser diferenciada não somente das demais vasculites de
pequenos vasos, como também das vasculites de médios vasos, como a
poliarterite nodosa e a doença de Kawasaki.
A PAM pode causar arterite necrosante histologicamente semelhante a
causada pela PAN. O quadro clínico, bem como fatores epidemiológicos e
algumas particularidades histológicas, permitem a diferenciação entre ambas.
Assim, a ocorrência de glomerulonefrite e/ou hemorragia alveolar difusa,
sugestivas de capilarite renal e pulmonar, respectivamente, indicaria PAM, vez
que a PAN, por definição, não acomete capilares arteríolas e vênulas
(JENNETTE, 1994).
A ausência ou escassez de depósitos de imunocomplexos nas paredes
vasculares pode ajudar a diferenciar a PAM das vasculites de pequenos vasos
mediadas por imunocomplexos, como a púrpura de Henoch-Schönlein e a
vasculite crioglobulinêmica. Assim como a ausência de anticorpo anti-MBG,
que falaria contra a síndrome do anticorpo anti-MBG (ou síndrome de
Goodpasture).
A vasculite presente em pacientes com PAM é histopatologicamente
indistinguível da PAG ou da SCS. No entanto, a observação de inflamação
granulomatosa (ausente na PAM) e uma positividade para o ANCA-C
assinalaria PAG. Da mesma forma, se há evidência de inflamação
granulomatosa associada a quadro clínico de asma e eosinofilia, o diagnóstico
mais apropriado é de SCS.
A glomerulonefrite necrosante, ocasionada pela PAM, pode ser
observada em diversas doenças inflamatórias incluindo, além das vasculites
ANCA-associadas, a nefrite lúpica. Nesta última, no entanto, é observada
importante deposição de imunocomplexos, geralmente ausente na PAM.
O Quadro 5, a seguir, traz uma comparação entre importantes aspectos
das VAA.

61
Quadro 5 – Quadro comparativo entre as vasculites ANCA-dependentes com envolvimento
pulmonar
Fonte: PEREIRA-SILVA, 2011.
3.3.7 Prognóstico
A morbidade e mortalidade dependem diretamente da rapidez com a
qual foi instituída a terapia com corticosteróides (JENNETTE, 2001).
Mais de 25% dos pacientes vão a óbito durante o primeiro episódio de
hemorragia alveolar difusa, a despeito de tratamento adequado (SANTOS,
2004). Em relação aos demais que sobrevivem, exibem boa resposta
terapêutica, mas episódios recorrentes são esperados. Episódios recorrentes
de hemorragia alveolar difusa podem levar a importantes complicações
crônicas, tendo sido descritas a ocorrência de fibrose pulmonar e doença
pulmonar obstrutiva (MARVIN, 2000).
VARIÁVEIS PAG SCS PAM
VASCULITE NECROSANTE ++++ ++++ ++++
CAPILARITE +++ +++ ++++
GRANULOMAS ++++ +++ ----
VIAS AÉREAS SUPERIORES ++++ ++++ ----
RINS +++ ++ ++++
INFILTRADO EOSINOFÍLICO + ++++ +
EOSINOFILIA ---- ++++ ----
ANCA C (PR3) P (MPO) P (MPO)

62
3.4 TRATAMENTO DAS VASCULITES ANCA-ASSOCIADAS
O tratamento das vasculites ANCA-associadas é basicamente o mesmo,
sendo realizado com imunossupressores. Antes do emprego da terapia
imunossupressora, as taxas de mortalidade das vasculites sistêmicas
chegavam a 75% e o tempo médio de sobrevida era de cinco meses (BROWN,
2006).
Os principais objetivos da terapia são eliminar os sintomas da doença e
evitar ou minimizar danos orgânicos irreversíveis. A intensidade do tratamento
é determinada pela atividade da doença e gravidade das manifestações
presentes. Para se estabelecer a gravidade da doença ativa, são considerados
o número de órgãos acometidos, o grau de disfunção renal e a presença de
hemorragia alveolar difusa. Assim, um útil sistema de classificação clínica dos
pacientes com seus respectivos esquemas terapêuticos foi desenvolvido pelo
Grupo Europeu de Estudos sobre as Vasculites (EUVAS), exposto na Tabela 5
abaixo (JAYNE, 2000).
Tabela 5: Avaliação da gravidade das vasculites e tratamento
Fonte: BROWN, 2006.
CLASSIFICAÇÃO CLÍNICA
SINTOMAS GERAIS
CREATININA SÉRICA
ÓRGÃO VITAL
AFETADO
OPÇÃO TERAPÊUTICA
Limitada Não < 1,4 mg/dL Não Corticosteróides ou metotrexato ou azatioprina
Inicial generalizada
Sim < 1,4 mg/dL Não
Ciclofosfamida ou metotrexato
+ Corticosteróides
Ativa generalizada Sim < 5,7 mg/dL Sim Ciclofosfamida
+ Corticosteróides
Grave Sim > 5,7 mg/dL Sim
Ciclofosfamida +
Corticosteróides +
plasmaferese
Refratária Sim Qualquer Sim Novas opções terapêuticas

63
O tratamento é geralmente dividido em duas etapas:
• Remissão-indução: Etapa inicial que objetiva o controle da atividade da
doença; e
• Manutenção: Etapa menos agressiva; objetivo de manter a doença em
remissão.
Durante a fase de remissão-indução de tratamento, em pacientes com
doença limitada, o tratamento pode ser feito com apenas uma droga. Já
naqueles que apresentam doença generalizada, com ou sem comprometimento
de órgão vital, a recomendação é utilizar a combinação de corticosteróides e
ciclofosfamida (BROWN, 2006).
Para as vasculites que apresentam evolução mais grave, preconiza-se o
tratamento combinado com corticosteróides em altas doses e ciclofosfamida,
com relatos de resposta superior a 90% para casos de PAG e PAM. O
esquema utiliza metilprednisolona intravenosa em doses de 1g/dia durante 3
dias consecutivos seguida de prednisona oral (0,75 a 1,0mg/Kg/dia),
concomitante à ciclofosfamida oral (2 mg/kg/dia) ou intravenosa (0,5 a 1 g/m2),
iniciada ao mesmo tempo que a metilprednisolona e repetida após 1 e 4
semanas (SCHWARZ, 2000). Ainda que a resposta terapêutica à
ciclofosfamida oral usada diariamente seja superponível à da aplicação mensal
intravenosa, a dose cumulativa é menor com a aplicação venosa do que oral,
com o potencial de causar menos efeitos adversos e complicações.
Em formas graves de vasculites, onde há dano renal acentuado ou
qualquer outra condição potencialmente fatal, a associação de plasmaferese à
combinação corticosteróides + ciclofosfamida tem se mostrado superior ao uso
de pulsoterapia com corticosteróides em altas doses no controle do dano renal
(GASKIN, 2001; FRASCA, 2003).
A plasmaferese terapêutica, por sua vez, deve ser reservada a casos
refratários ao tratamento com drogas imunossupressoras e a pacientes com
formas graves da doença, com ou sem disfunção renal rapidamente
progressiva.
A transição da fase de indução para a fase de manutenção costuma
ocorrer, em média, entre três e seis meses, devendo considerar sempre as
evidências clínicas de atividade de doença. A terapia da fase de manutenção,
cuja meta é manter o controle da doença, exige doses menores de

64
imunossupressores e está associada a menor incidência de efeitos adversos,
porém, deve se considerar o risco de recidivas (SCHWARZ, 2000).
Como regra geral, as dose de corticosteróides orais não devem ser
reduzidas até pelo menos um mês após a remissão da doença. Os esquemas
inadequados de tratamento costumam ser a principal causa de falência
terapêutica. Se não houver recidivas, o tratamento de manutenção deve ser
continuado por, pelo menos, doze a dezoito meses.
Em relação àqueles casos que não respondem ao esquema combinado
de corticosteróides + ciclofosfamida, são formas raras e comumente
relacionadas a quadros fulminantes. Para este grupo de pacientes, devem ser
consideradas novas modalidades de tratamento. Estas ainda estão sendo
melhor estudadas; incluem anticorpos antifator de necrose tumoral, anticorpos
anticélulas T ou células B, imunoglobulina intravenosa e outros agentes
quimioterápicos (FRANKEL, 2006).
COMPLICAÇÕES E RECIDIVA DA DOENÇA
É necessário manter o acompanhamento dos pacientes, com atenção
especial a possíveis complicações do tratamento, bem como a ocorrência de
reativação da doença. Pacientes que apresentem deterioração clínica devem
ser suspeitados de infecção, toxicidade por drogas, reativação da doença ou
um novo problema não relacionado (BROWN, 2006). Ainda que o ANCA não
se deva prestar para monitoração de atividade de doença, a tendência de
ascensão depois de um período de estabilidade clínica e laboratorial, alerta
para a possibilidade de recidiva de doença. Por outro lado, pacientes em
remissão clínica podem ter o ANCA persistentemente reagente.

65
4 CONCLUSÃO
O termo vasculite compreende uma série de condições clínicas
heterogêneas, caracterizadas por um processo inflamatório destrutivo
acometendo a parede dos vasos sanguíneos. Diferentes tipos de vasos em
diversos órgãos podem estar envolvidos, podendo afetar artérias, arteríolas,
capilares, vênulas e veias.
As vasculites podem ser secundárias a condições subjacentes de
infecções e outros distúrbios, sendo a inflamação vascular, nestes casos, uma
manifestação da doença de base. Nas situações em que a vasculite é o
processo primário, elas são denominadas de vasculites primárias.
Vimos, ao longo do presente trabalho, que a classificação mais
amplamente utilizada diz respeito ao calibre dos vasos afetados, os quais
podem ser grandes, médios ou pequenos. Dessa forma, costumam apresentar-
se com manifestações clínicas variadas, com sinais e sintomas determinados
pelo tipo, tamanho e localização dos vasos acometidos.
Nesse contexto, encontram-se as vasculites paucimunes ANCA-
associadas, classificadas como vasculites de pequenos vasos e caracterizadas
por inflamação necrosante afetando principalmente capilares, vênulas,
arteríolas e pequenas artérias. No cenário das vasculites sistêmicas, o
envolvimento pulmonar é mais freqüente nesse subgrupo, foco principal do
estudo elaborado. Além da baixa prevalência, seu diagnóstico representa um
grande desafio devido à baixa especificidade dos sinais e sintomas, por vezes
sobrepostos e facilmente confundidos com os de outras condições.
Embora haja outros tipos de vasculites de pequenos vasos com
acometimento pulmonar, o presente trabalho abordou apenas as vasculites
ANCA-associadas, onde é mais comumente visto, sendo elas a síndrome de
Churg Strauss (SCS), a poliangiite granulomatosa (PAG, antiga granulomatose
de Wegener) e a poliangiite microscópica (PAM), considerando os seus
aspectos clínicos, radiológicos e histopatológicos.
A epidemiologia, etiologia e patogênese, manifestações clínicas,
características histopatológicas e radiológicas, diagnóstico, prognóstico e o
tratamento das VAA apresentados no decorrer deste trabalho monográfico,
permitem asseverar que se tratam de doenças raras e cujo diagnóstico é

66
desafiador, devido ao quadro clínico inespecífico, que impõe a realização de
um diagnóstico diferencial com diversas outras entidades. Além disso, o
diagnóstico precoce é de fundamental importância pelo impacto que produz
nos diversos desfechos clínicos, inclusive na mortalidade, para o qual é
necessário um alto grau de suspeição clínica, seguida de uma abordagem
diagnóstica adequada.
Nesse contexto, desponta a importância da combinação dos aspectos
clínicos, radiológicos, laboratoriais e histopatológicos particulares de cada
doença para o correto diagnóstico das vasculites, em especial das ANCA-
associadas com envolvimento pulmonar, confirmando a hipótese da autora da
imprescindibilidade dessa abordagem integrada, não apenas para o adequado
diagnóstico, mas também para o eficaz tratamento dessas doenças.
Como corolário dessa conclusão, podemos afirmar que SCS, PAG e
PAM, embora guardem entre si algumas características comuns, podem ser
diferenciadas com base na correlação clínico-radiológico-patológica e auxílio
de exames laboratoriais comprobatórios.
Finalmente, as ideias que consubstanciam as conclusões apresentadas
e corroboram a hipótese desta monografia podem ser assim sintetizadas: a
abordagem do paciente com presumível vasculite é multidisciplinar; os padrões
radiológicos descritos, ainda que possam fornecer pistas diagnósticas, são
muitas vezes inespecíficos; e o esforço conjunto de clínicos, radiologistas e
patologistas é imprescindível para o adequado diagnóstico e tratamento das
VAA.

67
REFERÊNCIAS
ABERLE, D.R.; GAMSU, G.; LYNCH, D. Thoracic manifestations of Wegener granulomatosis: diagnosis and course. Radiology 1990; 174(3 pt 1):703–709. ABRIL, Andy; CALAMIA, Kenneth T.; COHEN, Marc D. The Churg Strauss Syndrome (allergic granulomatous angiitis): review and update. Seminars in Arthritis and Rheumatism 2003; Volume 33, Issue 2, pp 106–114. AKIKUSA, B.; SATO, T.; OGAWA, M.; et al. Necrotizing alveolar capillaritis in autopsy cases of microscopic polyangiitis: incid ence, histopathogenesis and relationship with systemic va sculitis. Arch Pathol Lab Med, 1997; 121:144–149. ALMEIDA J.L.J.; CAMPOS, L.M.A.; PAIM, L.B.; LEONE, C.; KOCH, V.H.K.; SILVA, C.A.A. Renal involvement in Henoch-Schönlein purpura: a multivariate analysis of initial prognostic factors . J Pediatr. 2007; 83(3): 259-266. ANTUNES, T.; BARBAS, C.S.V. Granulomatose de Wegener. J Bras Pneumol. 2005; 31(Supl 1):S21-S6. ARAS, Gülfidan. Recent Aspects of Vasculitis and Future Direction . Internal Medicine 2011. Vol. 50, No. 18 pp.1869-1877. BARBAS, Carmen Sílvia Valente; BORGES, Eduardo da Rosa; ANTUNES, Telma. Vasculites pulmonares: quando suspeitar e como faze r o diagnóstico. J. bras. pneumol. 2005, vol.31, suppl.1. BARKSDALE, S.K.; HALLAHAN, C.W.; KERR, G.S.; FAUCI, A.S.; STERN, J.B.; TRAVIS, W.D. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 75 biopsies in 46 patien ts. Am J Surg Pathol 1995; 19(2):161–172. BOURGARIT, A.; LE TOUMELIN, P.; PAGNOUX, C.; et al; French Vasculitis Study Group. Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and fa ctors predictive of mortality based on 595 patients. Medicine (Baltimore) 2005; 84(5):323–330. BROUET, J.C.; CLAUVEL, J.P.; DANON, F.; KLEIN, M.; SELIGMANN, M. Biologic and clinical significance of cryoglobulins : a report of 86 cases. Am J Med 1974; 57:775–88. BROWN, Kevin K. Pulmonary Vasculitis. Proc Am Thorac Soc, 2006; 3:48–57.

68
BRUCE, I.N., BELL, A.L. A comparison of two nomenclature systems for primary sistemic vasculitis . British Journal of Rheumatology 1997; 36:453-458. BURNS, J.C. et al. Sequelae of Kawasaki Disease in Adolescents and Young Adults . J Am Coll Cardiol 1996; 28:253-7. BUSCHMAN, D.L.; WALDRON, J.A. Jr.; KING, T.E. Jr. Churg-Strauss pulmonary vasculitis. High-resolution computed tomo graphy scanning and pathologic findings. Am Rev Respir Dis 1990; 142:458. CALAMIA, K.T.; HUNDER, G.G. Clinical manifestations of giant-cell (temporal) arteritis. Clin Rheum Dis 1980; 6: 389-403. CALVO-ROMERO, J,M. Giant cell arteritis . Postgrad Med J 2003; 79:511–515. CHOI, Y.H.; IM, J.G.; HAN, B.K.; et al. Thoracic manifestation of Churg-Strauss syndrome: radiologic and clinical findings. Chest 2000; 117:117. CHUMBLEY, L.C.; HARRISON, E.G.; DEREMEE, R.A. Allergic granulomatosis and angiitis (Churg-Strauss Syndrome ): Report and analysis of 30 cases. Mayo Clin Proc 1977;52:477-84. CHUNG, Man Pyo; YI, Chin A; LEE, Ho Yun; HAN, Joungho; LEE, Kyung Soo. Imaging of Pulmonary Vasculitis. Radiology, May 2010; 255:2. CHURG, J.; STRAUSS, L. Allergic granulomatosis, allergic angiitis and periarteritis nodosa. Am J Pathol 1951;27:277-301. CHURG, A. Recent advances in the diagnosis of Churg-Strauss s yndrome. Mod Pathol 2001; 14:1284. CONRON, M.; BEYNON, H.L.C. Churg-Strauss syndrome. Thorax 2000; 55:870–877. COTTIN, V.; CORDIER, J.F. Eosinophilic pneumonias. Allergy 2005; 60(7):841–857. D’AGATI, V.; CHANDER, P.; NASH, M.; MANCILLA-JIMENEZ, R. Idiopathic microscopic polyarteritis nodosa: ultrastructural o bservations on the renal vascular and glomerular lesions . Am J Kidney Dis 1986, 7:95–110. DAJANI, A.S.; TAUBERT, K.A.; GERBER, M.A. et al. Diagnosis and Therapy of Kawasaki Disease in Children . Circulation 1993; Vol 87, No 5. D’AMICO, G. Renal involvement in hepatitis C infection: cryoglo bulinemic glomerulonephritis . Kidney Int 1998;54:650–71.

69
DAVIES, D.J.; MORAN, J.E.; NIALL, J.F.; RYAN, G.B. Segmental necrotosing glomerulonephritis with antineutrophil antibody: po sible arbovirus aetiology . BMJ 1982; 285: 606.4. DAVIN, J.C.; WEENING, J.J. Diagnosis of Henoch-Schönlein purpura: renal or skin biopsy? Pediatr Nephrol 2003; 18(12):1201-3. DAVIS, M.D.; DAOUD, M.S.; MCEVOY, M.T.; SU, W.P. Cutaneous manifestations of Churg-Strauss syndrome: a clinico pathologic correlation. J Am Acad Dermatol.1997; 37(2 Pt 1):199-203. DAVSON, J.; BALL, J.; PLATT, R. The kidney in periarteritis nodosa. QJM, 1948; 17:175–202. D’CRUZ, D.P.; BARNES, N.C.; LOCKWOOD, C.M. Difficult asthma or Churg-Strauss Syndrome? Br Med J 1999; 318:475-6. DUNOGUÉ, Bertrand; PAGNOUX, Christian; GUILLEVIN, Loïc. Churg-Strauss Syndrome: Clinical Symptoms, Complementary Investig ations, Prognosis and Outcome, and Treatment. Semin Respir Crit Care Med 2011; 32:298–309. ERZURUM, S.C.; UNDERWOOD, G.A.; HAMILOS, D.L.; WALDRON, J.A. Pleural effusion in Churg-Strauss Syndrome. Chest 1989; 95: 1357-9. EVANS, J.M.; HUNDER, G.G. Polymyalgia rheumatica and giant cell arteritis . Rheum Dis Clin North Am 2000;26:493–515. FALK, R.J.; HOGAN, S.; CAREY, T.S. Clinical course of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephri tis and systemic vasculitis . The Glomerular Disease Collaborative Network . Ann Intern Med 1990; 113: 656-63. FALK, R.J.; JENNETTE, J.C. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients wi th systemic vasculitis and idiopathic necrotizing and crescentic glomerulo nephritis . N Engl J Med 1988; 318: 1651-1657. FALK, R.J.; GROSS, W.L.; GUILLEVIN, L.; et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for W egener’s granulomatosis. Ann Rheum Dis 2011; 70(4):704. FINKIELMAN, Javier D. et al. ANCA in Active Wegener’s Granulomatosis . The American Journal of Medicine 2007; Vol 120, No 7. FISCHER, E.G.; LAGER, D.J. Anti-glomerular basement membrane glomerulonephritis: a morphologic study of 80 cases . Am J Clin Pathol 2006; 125(3):445–450.

70
FRANKEL, Stephen K.; COSGROVE, Gregory P.; FISCHER, Aryeh; MEEHAN, Richard T.; BROWN, Kevin K. Update in the Diagnosis and Management of Pulmonary Vasculitis. Chest, Fev 2006; 129(2). FRASCA, G.M.; SOVERINI, M.L.; FALASCHINI, A.; et al. Plasma exchange treatment improves prognosis of antineutrophil cyto plasmic antibody-associated crescentic glomerulonephritis: a case-co ntrol study in 26 patients from a single center . Ther Apher Dial 2003; 7:540–546. FREIRE, Beatriz Funayama Alvarenga. Anticorpos anticitoplasma de neutrófilos (ANCA) . Botucatu 1997; s.n, 116 p. FRUSTACI, A.; GENTILONI, N.; CHIMENTI, C.; NATALE, L.; GASBARRINI, G.; MASERI, A. Necrotizing myocardial vasculitis in Churg-Strauss syndrome: Clinicohistogic evaluation of steroids and immunosu ppressive therapy. Chest 1998; 114:1484-9. GASKIN, G.; CLUTTERBUCK, E.J.; PUSEY, C.D. Renal disease in Churg-Strauss Syndrome. Contrib Nephrol 1991; 94:58-65. GASKIN, G.; PUSEY, C. Plasmapheresis in antineutrophil cytoplasmic antibodyassociated systemic vasculitis . Ther Apher 2001;5:176–181. GATENBY, P.A.; LUCAS, R.M.; ENGELSEN, O.; PONSONBY, A.L.; CLEMENTS, M. Antineutrophil cytoplasmic antibody-associated vasculitides: could geographic patterns be explaine d by ambient ultraviolet radiation? Arthritis Rheum 2009; 61(10):1417–1424. GAYRAUD, M.; GUILLEVIN, L.; LE TOUMELIN, P.; et al. Long-term followup of polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: analysis of four prospective trials inclu ding 278 patients. Arthritis Rheum 2001; 44:666. GIBELIN, Aude; MALDINI, Carla; MAHR, Alfred. Epidemiology and Etiology of Wegener Granulomatosis, Microscopic Polyangiitis , Churg-Strauss Syndrome and Goodpasture Syndrome: Vasculitides wit h Frequent Lung Involvement . Semin Respir Crit Care Med, 2011; 32:264–273. GÓMEZ-PUERTA, J.A.; HERNÁNDEZ-RODRÍGUEZ, J.; LÓPEZ-SOTO, A.; BOSCH, X. Antineutrophil cytoplasmic antibody-associated vasc ulitides and respiratory disease. Chest 2009; 136:1101. GONZALEZ-GAY, M.A.; GARCIA-PORRUA, C. Epidemiology of the vasculitides. Rheum Dis Clin North Am. 2001;27(4):729-49. GOODPASTURE, Ernest W. The Significance of Certain Pulmonary Lesions in Relation to the Etiology of Influenza. American Journal of the Medical Sciences, Nov 1919; 158(6):863-870.

71
GOTTSCHLICH, S.; AMBROSCH, P.; KRAMKOWSKI, D.; LAUDIEN, M.; BUCHELT, T.; GROSS, W.L.; B. HELLMICH. Head and neck manifestations of Wegener’s granulomatosis. Rhinology, 2006; 44, 227-233. GREEN, R.J.; RUOSS, S.J.; KRAFT, S.A.; BEMY, G.J.; RAFFIN, T.A. Pulmonary capillaritis and alveolar hemorrhage – Up date on diagnosis and management. Chest 1996; 110: 1305-16. GUBBELS, S.P.; BARKHUIZEN, A.; HWANG, P.H. Head and neck manifestations of Wegener’s granulomatosis. Otolaryngol Clin North Am 2003; 36(4):685–705. GUILLEVIN, Loïc. Clinical trials on systemic necrotizing vasculitides. Presse Med 2010; 39(6):653–659. GUILLEVIN, Loïc.; COHEN, P.; GAYRAUD, M.; LHOTE, F.; JARROUSE, B.; CASSAUS, P. Churg-Straus Syndrome: Clinical study and long term follow-up of 96 patients. Medicine 1999; 78:26-37. GUILLEVIN, Loïc; DURAND-GASSELIN, B.; CEVALLOS, R.; et al. Microscopic polyangiitis. Arthritis Rheum 1999; 42:421–430. GUILLEVIN, Loïc.; LHOTE, F.; GAYRAUD, M.; et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome: a prospective study in 342 patients. Medicine (Baltimore) 1996; 75(1):17–28. GUILLEVIN, Loïc.; LHOTE, F. Polyarteritis nodosa and microscopic polyangiitis . Clin Exp Immunol, 1995; 101(Suppl 1): 22–23. GUILLEVIN, Loïc; MAHR, Alfred; CALLARD, Patrice; GODMER, Pascal; PAGNOUX, Christian; LERAY, Emmanuelle; COHEN, Pascal. Hepatitis B Virus-Associated Polyarteritis Nodosa: Clinical Cha racteristics, Outcome, and Impact of Treatment in 115 Patients . Medicine, 2005; Volume 84, Issue 5, 313-322. GUILLEVIN, Loïc; PAGNOUX, Christian; MOUTHON, Luc. Churg-Strauss Syndrome. Semin Respir Crit Care Med 2004; 25(5):535-545. GUILLEVIN, L.; PAGNOUX, C.; SEROR, R.; MAHR, A.; MOUTHON, L.; LE TOUMELIN, P. French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necr otizing vasculitides based on the French Vasculitis Study Group (FVSG) c ohort. Medicine (Baltimore) 2011; 90(1):19–27. HAGEN, E.C.; DAHA, M.R.; HERMANS, J.; et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int 1998; 53 (3):743–753.

72
HALL, S.; BARR, W.; LIE, J.T.; et al. Takayasu arteritis . A study of 32 North American patients. Medicine 1985;64:89–99. HALL, J.B.; WADHAM, B.M.; EOOD, C.J.; ASHTON, V.; ADAM, W.R. Vasculitis and glomerulonephritis: a subgroup with na antineutrophil cytoplasmic antibody. Aust N Z J Med 1984; 14: 277-278. HOFFMAN, G.S.; KERR, G.S.; LEAVITT, R.Y.; HALLAHAN, C.W.; LEBOVICS, R.S.; TRAVIS, W.D.; et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992; 116(6):488-98. HOMER, R.J. Antineutrophil cytoplasmic antibodies as markers fo r systemic autoimmune disease. Clin Chest Med. 1998; 19(4):627-39. WOYWODT, A.; HAUBITZ, M.; HALLER, H.; MATTESON E.L. Wegener’s granulomatosis. Lancet 2006; 367(9519):1362–1366. HUNDER, G.G.; AREND, W.A; BLOCH, D.A.; et al. The American College of Rheumatology 1990 criteria for the classification o f vasculitis. Introduction. Arthritis Rheum 1990; 33:1065–7. HARROLD, L.R.; ANDRADE, S.E.; GO, A.S.; BUIST, A.S.; EISNER, M.; VOLLMER, W.M.; CHAN, K.A.; FRAZIER, E.A.; WELLER, P.F.; WECHSLER, M.E.; YOOD, R.A.; DAVIS, K.J.; PLATT, R. Incidence of Churg-Strauss syndrome in asthma drug users: a population-based p erspective. J Rheumatol 2005; 32(6):1076-1080. HASLEY, P.B.; FOLLANSBEE, W.P.; COULEHAN, J.L. Cardiac manifestations of Churg-Strauss Syndrome: Report of a case and review of the literature. Am Heart J 1990; 120:996-9. HATTORI N, ICHIMURA M, NAGAMATSU M, LI M, YAMAMOTO K, KUMAZAWA K, et al. Clinicopathological features of Churg-Strauss syndrome-associated neuropathy. Brain 1999;122:427-39. HAUGEBERG, G.; BIE, R.; BENDVOLD, A.; LARSEN, A.S.; JOHNSEN, V. Primary vasculitis in a Norwegian community hospita l: a retrospective study. Clin Rheumatol 1998;17(5):364–368. JAYNE, D. Evidence-based treatment of systemic vasculitis. Rheumatology (Oxford) 2000; 39:585–595. JENNETTE, J.C.; FALK R.J. Pathogenic potential of antineutrophil cytoplasmic autoantibodies. Adv Exp Med Biol 1993; 336:7–15. JENNETTE, J.C.; FALK, R.J.; et al. Nomenclature of systemic vasculitis. Arthritis Rheum. 1994, 37: 187-192. JENNETTE, J.C.; FALK, R.J. Small-vessel vasculitis. N Engl J Med 1997; 337:1512-23.

73
JENNETTE, J.C.; FALK, R.J.; ANDRASSY, K.; BACON, P.A.; CHURG, J.; GROSS, W.L.; et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum, 1994; 37:187–92. JENNETTE, J.C.; FALK, R.J.; GREENBERG, A. Primer on Kidney Diseases, "Vasculitis (Polyarteritis Nodosa, Microscopic Poly angiitis, Wegener's Granulomatosis, Henoch-Schönlein Purpura)". National Kidney Foudation 2005; 4ª Edição, p. 2.0-2.13. JENNETTE, J.C.; THOMAS, D.B.; FALK, R.J. Microscopic polyangiitis (microscopic polyarteritis). Semin Diag Pathol 2001; 18:3-13. JOHNSTON, S.L.; LOCK, R.J.; GOMPELS, M.M. Takayasu arteritis: a review. J Clin Pathol 2002;55:481–486. JONES, N.S. Nasal manifestations of rheumatic diseases. Ann Rheum Dis 1999; 58:589-90. JUNQUEIRA, L.C.; CARNEIRO, J. Sistema circulatório. In. Junqueira LC. Histologia Básica. 8ª edição. Rio de Janeiro: Guanabara, 1985. ISHIYAMA, A.; CANALIS, R. Otological manifestations of Churg-Strauss Syndrome. Laryngoscope 2001; 111:1619-24 KALLENBERG, Cees G. M. The Last Classification of Vasculitis. Clinic Rev Allerg Immunol 2008; 35:5–10. KALLENBERG, Cees G.M.; BROUWER, E.; WEENING, J.J.; COHEN TERVAERT, J.W. Antineutrophil cytolasmic antibodies: current diagn ostic and pathophysiologic potential. Kidney Int 1994; 46: 1-15. KATO, H.; ICHINOSE, E.; KAWASAKI, T. Myocardial infarction in Kawasaki disease: Clinical analyses in 195 cases. J Pediatr 1986; 108:923-927. KAWASAKI, T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the finge rs and toes in children. Jpn J Allergy.1967;16:178-222. KELLY, P.T.; HAPONIK, E.F. Goodpasture syndrome: molecular and clinical advances. Medicine (Baltimore) 1994; 73(4):171–185. KEOGH, K.A.; SPECKS, U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148. KERR, G.S.; HALLAHAN, C.W.; GIORDANO, J.; et al. Takayasu arteritis. Ann Intern Med 1994;120:919–29.

74
KOLDINGSNES, W.; NOSSENT, H. Epidemiology of Wegener’s granulomatosis in northern Norway. Arthritis Rheum 2000; 43(11):2481–2487. KUSSMAUL, A.; MAIER, R. Ueber eine bisher micht beschriebene eigenthümliche Arterienerkrankung (periarteritis no dosa) die mit Morbus Brightu und rapid fortschreitender allgemeiner Musk ellahmung einhergeht. Dtsch Arch Klin Med 1866; 1:484-518. LAMPRECHT, P.; GAUSE, A.; GROS, W.L. Cryoglobulinemic vasculitis. Arthritis Rheum 1999; Vol. 42, No. 12, pp 2507–251. LAMPRECHT, P.; GROSS, W.L. Wegener’s granulomatosis. Herz 2004; 29(1):47-56. LANDE, A.; BARD, R.; ROSSI, P.; PASSARIELLO, R.; CASTRUCCI, A. Takayasu’s arteritis. A world-wide entity. NY State J Med 1976; 76: 1477-82. LANE, S.E.; WATTS, R.A.; BARKER, T.H.W.; SCOTT, D.G.I. Evaluation of the Sorensen diagnostic criteria in the classification systemic vasculitis. Rheumatology 2002;41:1138–41. LANGFORD, Carol A.; HOFFMAN, Gary S. Rare diseases.3: Wegener's granulomatosis. Thorax 1999; 54(7):629-637. LANGFORD, Caro A.; SNELLER, M.C. Biologic therapies in the vasculitides. Curr Opin Rheumatol. 2003; 15(1):3-10. Review. LANHAM, J.G.; CHURG, J. Churg-Strauss syndrome. In: CHURG A, CHURG J, eds. Systemic Vasculitides. Tokyo: Igaku-Shoin, 1990. LANHAM, J.G.; ELKON, K.B.; PUSEY, C.D.; HUGHES, G.R. Systemic vasculitis in asthma and eosinophilia: a clinical a pproach to the Churg Strauss Syndrome. Medicine (Baltimore) 1984; 63:65–81. LAUQUE, D.; CADRANEL, J.; LAZOR, R.; et al. Microscopic polyangiitis with alveolar hemorrhage: a study of 29 cases and review of the literature. Groupe d’Etudes et de Recherche sur les Maladies “O rphelines” Pulmonaires (GERM”O”P) . Medicine (Baltimore) 2000 ; 79 ( 4 ): 222 – 233 . LEAVITT, R.; FAUCI, A.; BLOCH, D.; et al. The American College of Rheumatology 1990 criteria for the classification o f Wegener’s granulomatosis. Arthritis Rheum 1990; 33(8):1101–1107. LEHMANN, H.; KIEFER, B. Clinical manifestations of Wegener's. granulomatosis. APMIS 1990; 98: 19-20. LHOTHE, F.; COHEN, P.; GUILLEVIN, L. Syndrome de Churg et Strauss. Maladies et syndromes systemiques. 5e edition. Medecine-Sciences, Flammarion. Paris, 2008; 652-673.

75
LHOTE, F.; GUILLEVIN, L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome: Clinical aspects and tr eatment. Rheum Dis Clin North Am 1995; 21:911-47. LHOTE, F.; GUILLEVIN, L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Semin Respir Crit Care Med, 1998; 19:27–45. LIE, J.T. Illustrated histopathological classification criter ia for selected vasculitis syndromes. Arthritis Rheum 1990; 33:1074-87. LIM, L.C.L.; TAYLOR, J.G.; SCHMITZ, J.L.; FOLDS, J.D.; WILKMAN, A.S.; FALK, R.J.; et al. Diagnostic usefulness of antineutrophil cytoplasmic autoantibody serology: Comparative evaluation of co mmercial indirect fluorescent antibody kits and enzyme immunoassays k its . Am J Clin Pathol 1999; 111:363-9. LIM, Ming Y.; GREENE, Eddie L. 65-Year-Old Woman With Shortness of Breath and Dark Urine. Mayo Clin Proc, Set 2011; 86(9):912-915. LIU, L-J.; CHEN, M.; YU, F.; ZHAO, M-H.; WANG, H-Y. Evaluation of a new algorithm in classification of systemic vasculitis. Rheumatology 2008; 47:708–712. LOUTHRENOO, W.; NORASETTHADA, A.; KHUNAMORNPONG, S.; SRESHTHAPUTRA, A.; SUKITAWUT, W. Childhood Churg-Straus Syndrome. J Rheum 1999; 26:1387-93. LYNCH, J.P.III; MATTESON, E.; MCCUNE, J.W. Wegener’s granulomatosis: evolving concepts. Medical Rounds 1989; 2:67–80. LYNCH, J.P.III; WHITE, E.; TAZELAAR, Henry.; LANGFORD, C.A. Wegener’s granulomatosis: evolving concepts in treatment. Semin Respir Crit Care Med 2004; 25(5):491–521. LYNCH, Joseph P.; TAZELAAR, Henry. Wegener Granulomatosis (Granulomatosis with Polyangiitis): Evolving Concep ts in Treatment. Semin Respir Crit Care Med 2011;32:274–297. MAHR, A.; GUILLEVIN, L.; POISSONNET, M.; AYMÉ, S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Weg ener’s granulomatosis, and Churg-Strauss syndrome in a Fre nch urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004; 51(1):92–99. MAHR, A.; ARTIGUES, N.; COSTE, J.; AOUBA, A.; PAGNOUX, C.; GUILLEVIN, L.; French Vasculitis Study Group. Seasonal variations in onset of Wegener’s granulomatosis: increased in summer? J Rheumatol 2006; 33(8):1615–1622.

76
MAHR, A.D.; NEOGI, T.; MERKEL, P.A. Epidemiology of Wegener’s granulomatosis: Lessons from descriptive studies an d analyses of genetic and environmental risk determinants. Clin Exp Rheumatol 2006; 24(2, suppl 41):S82–S91. MARTIN, R.M.; WILTON, L.V.; MANN, R.D. Prevalence of Churg-Strauss syndrome, vasculitis, eosinophilia and associated c onditions: retrospective analysis of 58 prescription-event mon itoring cohort studies. Pharmacoepidemiol Drug Saf 1999; 8(3):179–189. MARVIN, I.S.; KEVIN, K.B. Small vessel vasculitis of the lung. Thorax 2000; 55:502-10. MASI, A.T.; HUNDER, G.G.; LIE, J.T.; MICHEL, B.A.; BLOCH, D.A.; ARENDT, W.P.; et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss Syndrome (Allergic Granulomatosis and Angiitis). Arthritis Rheum 1990; 33:1094-100 MCMAHON, B.J.; HEYWARD, W.L.; TEMPLIN, D.W.; CLEMENT, D.; LANIER, A.P. Hepatitis B-associated polyarteritis nodosa in Alas kan eskimos: clinical and epidemiologic features and long-term follow up. Hepatology 1989; 9: 97-101. MONTI, G.; GALLI, M.; INVERNIZZI, F.; PIOTELLI; SACCARDO, F.; MONTEVERDE, A.; et al. Cryoglobulinaemias: a multi-centre study on the early clinical and laboratory manifestations of pri mary and secondary disease. QJM 1995; 88:115–26. MOORE, P.M.; CALABRESE, L.H. Neurologic manifestations of systemic vasculitides. Semin Neurol 1994;14:300-6. NEVES, Sofia; MOTA, Margarida; DIAS, Manuela; RELVAS, Moura; VALENTE, João. Poliarterite nodosa – a propósito de dois casos clí nicos. Medicina Interna 2001; Vol. 8, N. 3. NOTH, I.; STREK, M.E.; LEFF, A.R. Churg-Strauss syndrome. Lancet. 2003; 361(9357):587-594. OLIVEIRA, S.; MENDONCA, C.; AMBAR, J.; MENDES, B. Síndrome de Churg-Strauss – a propósito de um caso clinico. Rev Port Pneumol. 2005; XI (1): 73-83. O'SULLIVAN, Brian P.; ERICKSON, Lori A.; NILES, John L. Case 30-2002 —An Eight-Year-Old Girl with Fever, Hemoptysis, and Pulmonary Consolidations . N Engl J Med, 2002; 347:1009-1017. PEREIRA-SILVA, Jorge Luiz. Vasculites sistêmicas com envolvimento pulmonar . Aula ministrada no Encontro do Clube Manoel de Abreu de São José do Rio Preto, São Paulo, 2011.

77
PEREIRA-SILVA, Jorge Luiz; KAVAKAMA, Jorge; PORTO, Nelson da S.; SOUZA JUNIOR, Arthur Soares; MARCHIORI, Edson; TERRA FILHO, Mario; ARAUJO NETO, Cesar de; CHAVES, Marcelo; IRION, Klaus L.; JASINOVODOLINSK, Dany; DALTRO, Pedro; NOBRE, Luiz Felipe; FUNARI, Marcelo; ESCUISSATO, Dante L. Diretrizes da SBPT. Consenso Brasileiro sobre a Terminologia dos Descritores de Tomografia Computadorizada do Tórax. Jornal de Pneumologia, São Paulo, 2005; v. 31, n. 2, p. 149-156. REID, A.J.C.; HARRISON, B.D.W.; WATTS, R.A.; WATKIN, S.W.; MCCANN, B.G.; SCOTT, D.G.I. Churg Strauss Syndrome in a district hospital. Q J Med 1998; 91:219–29. REINHOLD-KELLER, E.; BEUGE, N.; LATZA, U.; DE GROOT, K.; RUDERT, H.; NOLLE, B.; et al. An interdisciplinary approach to the care of patien ts with Wegener's granulomatosis: long-term outcome in 155 patients. Arthritis Rheum. 2000;43(5):1021-32. REZENDE, Carlos E. B. et al. Granulomatose de Wegener: relato de caso. Rev. Bras. Otorrinolaringol, 2003; 69(2). RODRIGUES, B.F.; ARAÚJO, J.M. Síndrome de Churg-Strauss. 2011; 18(3). ROSE, G.A.; SPENCER, H. Polyarteritis nodosa. Q J Med 1957;26:43-81. ROUFOSSE, F.; COGAN, E.; GOLDMAN, M. The hypereosinophilic syndrome revisited. Annu Rev Med 2003; 54:169–184. SABLÉ-FOURTASSOU, R.; COHEN, P.; MAHR, A.; et al. French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143(9):632–638. SANTOS, J.W.A.; MICHEL, G.T.; PEREIRA, C.E.L.; CAPELOZZI, V.L.; MILETO, J.N.; FIORINI, C.A.; Poliangeíte microscópica com hemorragia alveolar difusa. Jornal Brasileiro de Pneumologia, Mar/Abr 2004; 30(2). SAVAGE, C.O.S.; HARPER, L.; ADU, D. Primary systemic vasculitis. Lancet 1997; 349:553–8. SAVAGE, C.O.; HARPER, L.; COCKWELL, P.; ADU, D.; HOWIE, A.J. ABC of arterial and vascular disease: P Vasculitis. BMJ VOLUME 320 13 MAY 2000. SAVAGE C.O.; WINEARLS, C.G.; EVANS, D.J.; REIS, A.J.; LOCKWOOD, C.M. Microscopic polyarteritis: presentation, pathology and prognosis . Q J Med 1985; 56:467-83. SAVIGE, JUDY.; DAVIES, DAVID.; FALK, RONALD J.; JENNETTE, J. CHARLES.; AND WIIK, ALLAN. Antineutrophil cytoplasmic antibodies and associated diseases: A review of the clinical and l aboratory features. Kidney International, 2000; 57:846–862.

78
SCHWARZ, Marvin I.; BROWN, Kevin K. Small vessel vasculitis of the lung. Thorax 2000; 55:502–510. SEHGAL, M.; SWANSON, J.W.; DEREEME, R.A.; COLBY, T.V. Neurologic manifestations of Churg-Strauss Syndrome. Mayo Clin Proc 1995; 70:337-41. SILVA JUNIOR, O.F.; FREIRE, E.A.M.; BANDEIRA, F.C.; LEITE, R.R.C.; AMORIM, F.D.B.; LEITE, D.R.C.; COURY, U.L.M.S.M. Poliarterite nodosa: revisão de literatura a propósito de um caso clínic o. J Vasc Bras. 2010;9(1):86-89. SINICO, R.A.; DI TOMA, L.; MAGGIORE, U.; et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodi es in Churg-Strauss syndrome. Arthritis Rheum 2005; 52(9):2926–2935. SMITH, C.A.; FINDLER, W.J.; PINALS, R.S. The epidemiology of giant cell arteritis. Report of a Ten-Year Study in Shelby Cou nty, Tennessee. Arthritis and Rheumatism 1983;26(10):1214-1219. SOLANS, R.; BOSCH, J.A.; PEREZ-BOCANEGRA, C.; SELVA, A.; HUGUET, P.; ALIJOTAS, J.; et al. Churg-Strauss syndrome: Outcome and long term follow-up of 32 patients. Rheumatology 2001; 40:763-71. SULLIVAN, E.J.; HOFFMAN, G.S. Pulmonary vasculitis. Clin Chest Med. 1998;19(4):759-76, ix. Review. TAUBERT, K.A.; ROWLEY, A.H.; SHULMAN, S.T. Seven-year national survey of Kawasaki disease and acute rheumatic feve r. Pediatr Infect Dis J. 1994;13:704-708. TRAVIS, W.D. Pathology of pulmonary granulomatous vasculitis. Sarcoidosis Vasculitis Diffuse Lung Dis 1996; 13:14-27. TRAVIS, W.D. Pathology of pulmonary vasculitis. Semin Respir Crit Care Med 2004; 25(5):475–482. TRAVIS, W.D.; HOFFMAN, G.S.; LEAVITT, R.Y.; et al. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–33. TSUCHIYA, N.; KOBAYASHI, S.; HASHIMOTO, H.; OZAKI, S.; TOKUNAGA, K. Association of HLA-DRB1*0901-DQB1*0303 haplotype wi th microscopic polyangiitis in Japanese . Genes Immun 2006; 7(1):81–84. VAN DER WOUDE, F.J.; RASMUSSEN, N.; LOBATTO, S.; WIIK, A.; PERMIN, H.; VAN ES LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity i n Wegener´s granulomatosis. Lancet 1985; 1: 425-429.

79
WALTON, E. Giant cell granuloma of the respiratory tract (Wege ner’s granulomatosis). BMJ 1958; 2:265–70. WANG, X.; HU, Z.P.; LU, W.; TANG, X.Q.; YANG, H.P.; ZENG, L.W.; ZHANG, J.; LI, T. Giant cell arteritis . RHEUMATOLOGY INTERNATIONAL 2006; Volume 29, Number 1, 1-7. WATTS, R.A.; JOLLIFFE, V.A.; CARRUTHERS, D.M.; LOCKWOOD, M.; SCOTT, D.G.I. Effect of classification on the incidence of polyar teritis nodosa and microscopic polyangiitis. Arthritis Rheum, 1996; 39:1208–12. WATTS, R.A.; LANE, S.E.; BENTHAM, G.; SCOTT, D.G. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum 2000; 43(2):414–419. WATTS, R.A.; LANE, S.E.; HANSLIK, T.; HAUSER, T.; HELLMICH, B.; KOLDINGSNES, W.; MAHR, A.; SEGELMARK, M.; COHEN-TERVAERT, J.W.; SCOTT, D. Development and validation of a consensus methodolo gy for the classification of the ANCA- associated vasculit ides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007; 66:222–227. WATTS, R.A.; LANE, S.; SCOTT, D.G. What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol 2005; 19(2):191–207. WECHSLER, M.E.; GARPESTAD, E.; FLIER, S.R.; KOCHER, O.; WEILAND, D.A.; POLITO, A.J.; et al. Pulmonary infiltrates, eosinophilia, and cardiomyopathy following corticosteroid withdrawal in patients with asthma receiving zafirlukast. JAMA 1998; 279:455-7. WELLER, P.F.; PLAUT, M.; TAGGART, V.; TRONTELL, A. The relationship of asthma therapy and Churg-Strauss Syndrome: NIH work shop summary report. J Allergy Clin Immunol 2001;108: 175-83. WORTHY, S.A.; MÜLLER, N.L.; HANSELL, D.M.; FLOWER, C.D. Churg-Strauss syndrome: the spectrum of pulmonary CT find ings in 17 patients. AJR Am J Roentgenol 1998; 170:297. ZINCK S.E.; SCHWARTZ E.; BERRY, G.J.; LEUNG, A.N. CT of noninfectious granulomatous lung disease. Radiol Clin North Am. 2001; 39(6):1189-209. ZWERINA, J.; EGER, G.; ENGLBRECHT, M.; et al. Churg-Strauss syndrome in childhood: a systematic literature review and cl inical comparison with adult patients. Semin Arthritis Rheum 2009; 39:108.





























