Embed Size (px)
Citation preview

Desenvolvimento de processos de adsorção molecular
para remoção de impurezas genotóxicas de ingredientes
farmacêuticos ativos
Mariana Duarte de Pina
Dissertação para a obtenção do Grau de Mestre em
Bioengenharia e Nanossistemas
Orientador: Doutor Frederico Castelo Alves Ferreira
Coorientador: Professor Duarte Miguel de França Teixeira dos Prazeres
Júri
Presidente: Professor Luís Joaquim Pina da Fonseca
Orientador: Doutor Frederico Castelo Alves Ferreira
Vogal: Doutora Ana Isabel Ferreira Franco Vicente
Dezembro 2014

ii
Agradecimentos
Antes de mais, um grande agradecimento ao meu orientador, Doutor Frederico Ferreira, pelo
acompanhamento e conselhos dados ao longo do desenvolvimento desta tese.
À Hovione e aos membros da FFUL e da FCT-UNL que participaram neste projeto.
Aos meus colegas de laboratório, principalmente à Marisa Santos, Clara Lopes, Filipe
Carvalho, Rui Leitão, Nuno Lourenço e Aldo Arévalo, por me terem proporcionado um ambiente de
trabalho tão agradável e por toda a ajuda que me deram. Um agradecimento enorme à Sara Matias,
ao Ângelo Rocha e ao Rodrigo Raposo por tudo o que ensinaram e pela paciência que tiveram
comigo ao longo deste tempo.
À Doutora Teresa Esteves que, para além de ser responsável pela síntese e caracterização
dos MIPs, acompanhou o desenvolvimento desta tese estando sempre disponível para qualquer
esclarecimento.
Aos meus colegas, em especial Andreia Cuco, Ana Rita Valente, Gonçalo Adriano e
Guilherme Figueiredo pelos bons momentos proporcionados ao longo destes dois anos.
Ao Sérgio Paiva pelo apoio e ajuda que me deu, bem como pela sua paciência infinita.
Um agradecimento muito especial aos meus pais, António Pina e Isabel Pina, e à minha irmã,
Catarina Pina, por todo o apoio e motivação ao longo de toda a minha vida.
Por fim, à FCT – Fundação para a Ciência e Tecnologia pelo apoio financeiro através do
projeto PTDC/QEQ-PRS/2757/2012, “Removal of Genotoxic Impurities from Active Pharmaceutical
Ingredients”.

iii
Resumo
A maioria dos fármacos disponíveis no mercado é obtida através da utilização, como
reagentes ou catalisadores, de moléculas reativas que podem ser genotóxicas ou mesmo
carcinogénicas. Estas moléculas podem permanecer no API ou fármaco final como impurezas. O
risco que representam para a saúde dos pacientes tem-se tornado uma preocupação para a indústria
farmacêutica e agências reguladoras.
Vários compostos de diferentes famílias químicas, já foram catalogados como genotóxicos.
Estes compostos têm a capacidade de interagir com o DNA, impedindo a sua replicação, resultando
num risco carcinogénico. A produção de APIs através de uma síntese química que não recorresse a
compostos genotóxicos seria desejável. No entanto, esta abordagem nem sempre é exequível, uma
vez que estes compostos são muito úteis sinteticamente. Assim, é necessário controlar a quantidade
de GTIs presentes no API final, garantindo que o valor se encontra abaixo do TTC estabelecido pelas
agências reguladoras (1,5 µg/dia), motivando a procura de metodologias simples, robustas e
económicas para remover os GTIs dos APIs.
Durante o desenvolvimento desta tese foram estudados processos de purificação de APIs
convencionais (recristalização e resinas de troca iónica e adsorventes) e tecnologias emergentes
(nanofiltração, polímeros de impressão molecular e MIPs). Os resultados obtidos sugerem que a
recristalização não é um processo economicamente rentável para a indústria, pelo que devem ser
encontradas alternativas para aumentar o seu rendimento, sendo sugerido no âmbito desta tese, o
desenvolvimento de processos eficientes combinando em vários passos, recristalização ou
nanofiltração com resinas de troca iónica e/ou MIPs.
Palavras-chave: Genotóxico, purificação, recristalização, impressão molecular,

iv

v
Abstract
Most of the drugs available on the market are synthesized using highly reactive molecules.
These molecules may be present in the final API as impurities, that may be genotoxic or even
carcinogenic. The risk for patient’s health caused by these impurities has become an increasing
concern of pharmaceutical companies and regulatory authorities.
A broad range of unrelated chemicals from several chemical families, have been categorized
as genotoxic. These compounds have the ability to react with DNA, preventing its normal replication,
resulting in an associated carcinogenic risk. Although it is desirable to avoid the use of GTIs in the
manufacture of APIs, this is not always possible, since these compounds are synthetically useful. It is
fundamental to produce API with low GTI content, controlled below the Threshold of Toxicological
Concern (TTC) established by regulatory authorities (1,5 µg/day). So, it is necessary to find simple,
robust and economical routes do remove GTIs from APIs.
During the development of this thesis conventional purification techniques (recrystallization
and ionic exchange resins and adsorbents) as well as emergent techniques (nanofiltration, molecularly
imprinted polymers (MIPs)) were studied. The obtained results suggest that recrystallization is not a
cost-effective process. In that sense, it is necessary to find new ways to increase its yield through the
development of processes that combine recrystallization or nanofiltration with ionic exchange resins
and/or MIPs.
Keywords: genotoxic, purification, recrystallization, molecular imprinting, DNA

vi
Índice
Agradecimentos ...................................................................................................................................... 2
Resumo .................................................................................................................................................... 3
Abstract ................................................................................................................................................... 5
Lista de figuras ......................................................................................................................................... 8
Lista de tabelas ...................................................................................................................................... 11
Lista de abreviações .............................................................................................................................. 12
1. Introdução ....................................................................................................................................... 1
1.1. Genotoxicidade – Definição e Mecanismo de Ação ................................................................ 1
1.2. Regulamentação ...................................................................................................................... 3
1.2.1. Conferência Internacional para a Harmonização (ICH) ................................................... 3
1.2.2. Pharmaceutical Research and Manufacturers of America (PhRMA) .............................. 4
1.2.3. Agência Europeia do Medicamento (EMA) ..................................................................... 5
1.2.4. Food and Drug Administration (FDA) .............................................................................. 7
1.2.5. ICH M7 ............................................................................................................................. 8
1.2.6. ICH S2 ............................................................................................................................... 8
1.3. Classes de compostos genotóxicos ......................................................................................... 9
1.3.1. GTIs usados como reagentes ........................................................................................... 9
1.3.1.1. Haletos de alquilo e ésteres .................................................................................... 9
1.3.1.2. Sulfatos de alquilo ................................................................................................. 10
1.3.1.3. Epóxidos ................................................................................................................ 10
1.3.1.4. Hidrazinas .............................................................................................................. 10
1.3.1.5. TEMPO ................................................................................................................... 10
1.3.1.6. Aminas aromáticas ................................................................................................ 10
1.3.1.7. Ácidos borónicos ................................................................................................... 11
1.3.2. GTIs formados em reações secundárias ........................................................................ 11
1.3.2.1. Sulfonatos e os seus precursores .......................................................................... 11
1.3.3. Solventes orgânicos ....................................................................................................... 13
1.4. Remoção de impurezas genotóxicas ..................................................................................... 13
1.5. Purificação de APIs ................................................................................................................ 13
1.6. Objetivo ................................................................................................................................. 16
2. Materiais e Métodos ..................................................................................................................... 17
2.1. Materiais ................................................................................................................................ 17
2.2. Métodos experimentais ........................................................................................................ 17
2.2.1. Técnicas analíticas ......................................................................................................... 17

vii
2.2.1.1. High Performance Liquid Chromatography (HPLC) ............................................... 17
2.2.1.2. Espectroscopia UV/Vis ........................................................................................... 20
2.2.1.3. SEM ........................................................................................................................ 21
2.2.1.4. BET ......................................................................................................................... 22
2.2.1.5. Espectroscopia IV .................................................................................................. 24
2.2.2. Síntese de polímeros ..................................................................................................... 25
2.2.2.1. Polímeros Molecularmente Impressos (MIPs) ...................................................... 25
2.2.3. Processos ....................................................................................................................... 28
2.2.3.1. Recristalização ....................................................................................................... 28
2.2.3.2. Resinas ................................................................................................................... 29
2.2.3.3. Nanofiltração (OSN)............................................................................................... 34
3. Resultados e Discussão.................................................................................................................. 35
3.1. Estratégia ............................................................................................................................... 35
3.2. Recristalização ....................................................................................................................... 35
3.1. Polímeros de impressão molecular ....................................................................................... 38
3.1.1. Caracterização dos polímeros MIP4 e NIP4 .................................................................. 41
3.1.2. Isotérmica de adsorção do MIP4 ................................................................................... 43
3.1.3. Substituição do carvão ativado por MIPs ...................................................................... 43
3.2. Resinas de troca iónica e adsorventes .................................................................................. 43
3.2.1. DMAP em água .............................................................................................................. 44
3.2.2. DMAP em água:metanol (1:1) ....................................................................................... 48
3.2.3. DMAP em MeOH ........................................................................................................... 52
3.2.4. Purificação das soluções provenientes da recristalização ............................................ 55
3.3. Nanofiltração (OSN) .............................................................................................................. 56
3.4. Mitigação do MPTS ................................................................................................................ 57
3.4.1. Resinas de troca iónica e adsorventes .......................................................................... 57
4. Conclusões ..................................................................................................................................... 61
Referências ............................................................................................................................................ 62
Anexos ................................................................................................................................................... 70
Anexo 1 – Retas de calibração ........................................................................................................... 70
Anexo 2 – Isotérmicas de adsorção (variação da concentração inicial) ............................................ 73
Anexo 3 – Quantidade de GTI adsorvido em função da quantidade inicial de resina ...................... 80

viii
Lista de figuras
Figura 1 - Centros nucleofílicos das bases de DNA, indicados pelas setas vermelhas (adaptado de [8]).
................................................................................................................................................................. 1
Figura 2 - Numeração dos átomos das bases azotadas de DNA (adaptado de [3]). ............................... 2
Figura 3 - Sulfonilação catalisada pelo DMAP durante a síntese de Meta (à direita). .......................... 16
Figura 4 – Meta (esquerda), DMAP (meio) e MPTS (direita)................................................................. 16
Figura 5 - Sistema de HPLC (Adaptado de [88]). ................................................................................... 18
Figura 6 - Espectrofotómetro UV/Vis (Adaptado de [89]). ................................................................... 21
Figura 7 - Principais bandas no espectro IV. ......................................................................................... 24
Figura 8 - Representação esquemática do processo de impressão molecular. .................................... 27
Figura 9 - Processo de recristalização (adaptado de [106]). ................................................................. 29
Figura 10 - Isotérmicas de adsorção mais comuns................................................................................ 31
Figura 11 - Balanço de massa do processo de recristalização a 10ºC. .................................................. 35
Figura 12 - Balanço de massa do processo de recristalização a 4ºC. .................................................... 36
Figura 13 - Quantidade de Meta recuperado em função da quantidade de DMAP removido. ............ 36
Figura 14 - API removido ao longo do processo a 10ºC (painel superior) e a 4ºC (painel inferior). ..... 37
Figura 15 - GTI recuperado ao longo do processo de recristalização a 10ºC (painel superior) e a 4ºC
(painel inferior). ..................................................................................................................................... 38
Figura 16 - Remoção do DMAP com os diferentes MIPs. ...................................................................... 39
Figura 17 - Quantidade de DMAP removida pelo MIP4 e pelo NIP4. .................................................... 40
Figura 18 - Quantidade de Meta e DMAP removidos com o MIP4. ...................................................... 40
Figura 19 - Micrografias de SEM do MIP4. ............................................................................................ 41
Figura 20 - Micrografias de SEM do NIP4. ............................................................................................. 41
Figura 21 - Espectro de FT-IR para o MIP4 e NIP4. ................................................................................ 42
Figura 22 - Isotérmica do MIP4. ............................................................................................................ 43
Figura 23 - Substituição do carvão ativado por MIPs. ........................................................................... 43
Figura 24 - Processo de recuperação do Meta das soluções de lavagem. ............................................ 44
Figura 25 - Protonação do DMAP [118]................................................................................................. 44
Figura 26 - Variação da concentração de DMAP protonado em função do pH. ................................... 45
Figura 27 - Influência do pH na remoção do DMAP de uma solução aquosa. ...................................... 45
Figura 28 - Influência da temperatura na remoção do DMAP de uma solução aquosa. ...................... 46
Figura 29 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução
aquosa de DMAP. .................................................................................................................................. 46
Figura 30 - Isotérmica de adsorção com carvão ativado, variando a concentração inicial de uma
solução aquosa de DMAP. ..................................................................................................................... 47
Figura 31 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma
solução aquosa de DMAP. ..................................................................................................................... 47
Figura 32 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma
solução aquosa de DMAP. ..................................................................................................................... 47
Figura 33 - Capacidade de adsorção da resina AG 50W-X2 em função do tempo para uma solução
aquosa de DMAP. .................................................................................................................................. 48
Figura 34 - Influência do pH na remoção do DMAP em água:metanol. ................................................ 49
Figura 35 - Influência da temperatura na remoção do DMAP em água:metanol. ................................ 49
Figura 36 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução
de DMAP em água:metanol. ................................................................................................................. 50
Figura 37 - Isotérmica de adsorção com carvão ativado, variando a concentração inicial de uma
solução de DMAP em água:metanol. .................................................................................................... 50

ix
Figura 38 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma
solução de DMAP em água:metanol. .................................................................................................... 51
Figura 39 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma
solução de DMAP em água:metanol. .................................................................................................... 51
Figura 40 - Capacidade de adsorção da resina AG 50W-X2 em função do tempo. ............................... 52
Figura 41 - Influência do pH na remoção do DMAP em MeOH. ........................................................... 52
Figura 42 - Influência da temperatura na remoção do DMAP MeOH. .................................................. 53
Figura 43 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução
de DMAP em MeOH. ............................................................................................................................. 53
Figura 44 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma
solução de DMAP em MeOH. ................................................................................................................ 54
Figura 45 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma
solução de DMAP em MeOH. ................................................................................................................ 54
Figura 46 - Capacidade de adsorção em função do tempo. .................................................................. 55
Figura 47 - Quantidade de Meta e DMAP removidos com as resinas AG 50W-X2 e Amberlite IRC50. 55
Figura 48 - Recristalização com resinas e MIPs. .................................................................................... 56
Figura 49 - Remoção de DMAP e Meta por OSN em função do número de diavolumes. .................... 56
Figura 50 - Nanofiltração seguida da utilização de MIPs. ..................................................................... 57
Figura 51 – Estrutura química do MPTS. ............................................................................................... 57
Figura 52 – Quantidade de MPTS adsorvido pelas resinas. .................................................................. 58
Figura 53 - Influência da temperatura na remoção do MPTS em MeOH. ............................................. 58
Figura 54 - Isotérmica de adsorção com Amberlite IRA68, variando a concentração inicial de uma
solução de MPTS em MeOH. ................................................................................................................. 59
Figura 55 - Capacidade de adsorção da Amberlite IRA68 em função do tempo numa solução de MPTS
em MeOH. ............................................................................................................................................. 59
Figura 56 - Reta de calibração do DMAP em H2O. ................................................................................ 70
Figura 57 - Reta de calibração do DMAP em H2O:MeOH. ..................................................................... 70
Figura 58 - Reta de calibração do DMAP em MeOH. ............................................................................ 71
Figura 59 - Reta de calibração do DMAP em DCM. ............................................................................... 71
Figura 60 - Reta de calibração do MPTS em MeOH. ............................................................................. 71
Figura 61 - Reta de calibração do MPTS em DCM. ................................................................................ 72
Figura 62 - Reta de calibração do Meta em DCM. ................................................................................ 72
Figura 63 - Isotérmicas de adsorção para uma solução aquosa de DMAP com a resina AG 50W-X2. . 73
Figura 64 - Isotérmicas de adsorção para uma solução aquosa de DMAP com carvão ativado. .......... 73
Figura 65 - Isotérmicas de adsorção para uma solução aquosa de DMAP com Amberlite IRC50. ....... 74
Figura 66 - Isotérmicas de adsorção para uma solução aquosa de DMAP com Amberlite IRC86. ....... 74
Figura 67 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com AG 50W-X2. ... 75
Figura 68 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com carvão ativado.
............................................................................................................................................................... 75
Figura 69 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com Amberlite IRC50.
............................................................................................................................................................... 76
Figura 70 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com Amberlite IRC86.
............................................................................................................................................................... 76
Figura 71 - Isotérmicas de adsorção para uma solução DMAP em metanol com AG 50W-X2. ............ 77
Figura 72 - Isotérmicas de adsorção para uma solução DMAP em metanol com Amberlite IRC50. .... 77
Figura 73 - Isotérmicas de adsorção para uma solução DMAP em metanol com Amberlite IRC86. .... 78
Figura 74 - Isotérmicas de adsorção para uma solução de MPTS em metanol com Amberlite IRA68. 78
Figura 75 - Isotérmicas de adsorção para o MIP4. ................................................................................ 79

x
Figura 76 – Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução aquosa de DMAP com AG 50W-X2.
............................................................................................................................................................... 80
Figura 77 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução aquosa de DMAP com carvão
ativado. .................................................................................................................................................. 80
Figura 78 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução aquosa de DMAP com Amberlite
IRC50. .................................................................................................................................................... 80
Figura 79 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução aquosa de DMAP com Amberlite
IRC86. .................................................................................................................................................... 81
Figura 80 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com AG
50W-X2. ................................................................................................................................................. 81
Figura 81 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com
carvão ativado. ...................................................................................................................................... 81
Figura 82 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com
Amberlite IRC50. ................................................................................................................................... 82
Figura 83 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com
Amberlite IRC86. ................................................................................................................................... 82
Figura 84 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em metanol com AG
50W-X2. ................................................................................................................................................. 82
Figura 85 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em metanol com
Amberlite IRC50. ................................................................................................................................... 83
Figura 86 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de DMAP em metanol com
Amberlite IRC86. ................................................................................................................................... 83
Figura 87 - Quantidade de MPTS adsorvido determinado a partir da isotérmica de adsorção com
variação da quantidade inicial de resina obtida para uma solução de MPTS em metanol com
Amberlite IRA68. ................................................................................................................................... 83

xi
Lista de tabelas
Tabela 1 - Classificação das impurezas genotóxicas [19]. ....................................................................... 4
Tabela 2 - Implementação do modelo de impurezas em cinco classes. ................................................. 5
Tabela 3 - Ingestão diária permitida proposta (µg/dia) para GTIs durante o desenvolvimento clínico. 5
Tabela 4 - Limites propostos no documento Q&A da EMA [23]. ............................................................ 7
Tabela 5 – Guia da FDA para caracterizar e minimizar o risco. ............................................................... 7
Tabela 6 - Diferenças e semelhanças entre as diretrizes da FDA e da EMA. .......................................... 8
Tabela 7 - Limites de staged TTC propostos pela FDA. ........................................................................... 8
Tabela 8 - Precursores de alguns sulfonatos [1]. .................................................................................. 12
Tabela 9 - Radiação IV [92]. ................................................................................................................... 24
Tabela 10 – Condições das reações de polimerização. ......................................................................... 27
Tabela 11 - Propriedades das resinas estudadas. ................................................................................. 29
Tabela 12 - Grupos funcionais das resinas de troca iónica mais comuns. ............................................ 30
Tabela 13 - Análise BET do MIP4 e NIP4................................................................................................ 42
Tabela 14 - Parâmetros cinéticos obtidos para os modelos aplicados numa de DMAP em MeOH...... 59
Tabela 15 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de
DMAP com a resina AG 50W-X2. ........................................................................................................... 73
Tabela 16 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de
DMAP com carvão ativado. ................................................................................................................... 73
Tabela 17 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de
DMAP com Amberlite IRC50. ................................................................................................................ 74
Tabela 18 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de
DMAP com Amberlite IRC86. ................................................................................................................ 74
Tabela 19 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
água:metanol com AG 50W-X2. ............................................................................................................ 75
Tabela 20 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
água:metanol com carvão ativado. ....................................................................................................... 75
Tabela 21 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
água:metanol com Amberlite IRC50. .................................................................................................... 76
Tabela 22 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
água:metanol com Amberlite IRC86. .................................................................................................... 76
Tabela 23 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água
com AG 50W-X2. ................................................................................................................................... 77
Tabela 24 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
metanol com Amberlite IRC50. ............................................................................................................. 77
Tabela 25 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em
metanol com Amberlite IRC86. ............................................................................................................. 78
Tabela 26 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução de MPTS em
metanol com Amberlite IRA68. ............................................................................................................. 78
Tabela 27 - Parâmetros dos modelos de adsorção teóricos obtidos para o MIP4................................ 79

xii
Lista de abreviações
AA Ácido acrílico
ADI Ingestão diária aceitável (do inglês acceptable daily intake)
AIBN Azoisobutironitrilo
ALARP Tão baixo quanto razoavelmente possível (do inglês as low as reasonably practicable)
API Ingrediente farmacêutico ativo (do inglês active pharmaceutical ingredient)
ASAP Accelerated Surface Area and Porosimetry System
BET Brunauer, Emmett e Teller
BSE Eletrões retro difundidos (do inglês back scattered electrons)
C Citosina
CCD Dispositivo de carga acoplada (do inglês charge-coupled device)
CL Catodoluminescência
CMPH Comité de Medicamentos para Uso Humano
CPMP Comité de Especialidades Farmacêuticas
DCM Diclorometano
DEREK Deductive Estimation of Risk from Existing Knowledge
DMAP 4-dimetilaminopiridina
DMF Dimetilformamida
DMS Dimetilsulfato
DMSO Dimetilsulfóxido
DNA Ácido desoxirribonucleico
dsDNA DNA de cadeia dupla
DVB Divinilbenzeno
EDGMA Dimetacrilato de etilenoglicol
EDS Espectroscopia de raios X por dispersão em energia (do inglês energy-dispersive X-ray
spectroscopy)
EMA Agência Europeia do Medicamento (do inglês European Medicines Agency)
EWG Grupo de Trabalho de Peritos (Expert Working Group)
FDA Administração de Alimentos e Medicamentos (Food and Drug Administration)
FEG-SEM Microscópio eletrónico de varrimento de emissão de campo (Field Emission Gun
Scanning Electron Microscope)
FT-IR Espectroscopia de infravermelho por transformada de Fourier (Fourier transform
infrared spectroscopy)
G Guanina
GTI Impureza genotóxica (genotoxic impurity)
HEMA 2-hidroxietil metacrilato
HPLC Cromatografia líquida de elevada performance (High Performance Liquid
Chromatography)
ICH Conferência Internacional para a Harmonização (do inglês International Conference on

xiii
Harmonisation of Technical Requirements for Registration of Pharmaceuticals for
Human Use)
IV Infravermelho
LEDs Díodos emissores de luz
LOEL Nível com menor efeito observado (do inglês lowest observed effect level)
MAA Ácido metacrílico
MBAA N,N-metileno-bis-acrilamida
MeOH Metanol
Meta Furoato de mometasona
Min Minuto
MIP Polímero molecularmente impresso (do inglês molecularly imprinted polymer)
MMS Metanossulfonato de metil (do inglês methyl methanesulfonate)
MPTS P-toluenossulfonato de metilo (do inglês methyl p-toluenesulfonate)
NAT N-Acetiltransferase
NIP Polímero não impresso (do inglês non imprinted polymer)
NIR Infravermelho próximo (do inglês near infrared)
NMR Ressonância magnética nuclear (do inglês nuclear magnetic resonance)
NOEL Nível sem efeitos observáveis (do inglês no-observed-effect level)
OSN Nanofiltração com solventes orgânicos (do inglês organic solvent nanofiltration)
PDE Exposição diária permitida (do inglês permitted daily exposure)
PhRMA Investigadores e Fabricantes Farmacêuticos da América (do inglês Pharmaceutical
Research and Manufacturers of America)
PQRI Instituto de Investigação da Qualidade do Produto (do inglês Product Quality Research
Institute)
Q&A Perguntas e respostas (do inglês Questions & Answers)
QbD Quality by Design
RP-HPLC HPLC de fase reversa (do inglês reverse phase HPLC)
SAR Relação estrutura-atividade (do inglês structure-activity relationship)
SE Eletrões secundários (do inglês secondary electrons)
SEM Microscopia eletrónica de varrimento (do inglês scanning electron microscopy)
ssDNA DNA de cadeia simples
ST Sulfotransferase
SWP Grupo de Trabalho de Segurança (do inglês Safety Working Party)
T Timina
TEMPO N-oxil-2,2,6,6-tetrametilpiperidina
TRIM Trimetacrilato de trimetilolpropano
TTC Limiar de risco toxicológico (do inglês threshold of toxicological concern)
UV Ultravioleta
UV/Vis Ultravioleta/visível

1
1. Introdução
1.1. Genotoxicidade – Definição e Mecanismo de Ação
A carcinogénese inclui três estágios – iniciação, promoção e progressão. Os eventos
mutacionais ocorrem, por norma, na iniciação. Estes eventos são prontamente corrigidos pelos
mecanismos de reparação do DNA; no entanto, estes mecanismos nem sempre são eficazes e as
células mutadas começam a proliferar – estágio de promoção. Durante a replicação celular certas
anomalias podem resultar na criação de novos genes; as células mutadas são transportadas pela
corrente sanguínea, invadindo os tecidos saudáveis. A este processo dá-se o nome de metástase e
ocorre no estágio de progressão [1, 2]. O termo mutagenicidade indica a capacidade de induzir danos
genéticos transmissíveis, que incluem mutações génicas, aberrações cromossómicas e variações no
número de cromossomas. O termo genotoxicidade é um conceito mais abrangente que inclui todos os
danos genéticos, incluindo as alterações genéticas que podem resultar de mutações que não são
transmitidas à descendência [3, 4].
James e Elizabeth Miller perceberam, em 1981, que os agentes alquilantes carcinogénicos
eram eletrofílicos. Deste então descobriu-se que uma série de agentes acilantes eram carcinogénicos
e também estes eram eletrofílicos. James e Elizabeth Miller ficaram intrigados com a variedade de
carcinogénicos químicos com diferentes estruturas que eram eletrofílicos o que os levou a sugerir que
“a maioria, se não todos, dos carcinogénicos químicos são ou poderão ser convertidos in vivo em
derivativos reativos eletrofílicos que combinam com os grupos nucleofílicos de componentes com
ácidos nucleicos ou proteínas” [3, 5, 6]. De acordo com esta teoria, os locais que podem
potencialmente interagir com as espécies eletrofílicas são os centros nucleofílicos do DNA: átomos de
azoto e oxigénio da pirimidina e da purina e o esqueleto de fosfato-desoxirribose (Figura 1) [1, 3, 7].
Figura 1 - Centros nucleofílicos das bases de DNA, indicados pelas setas vermelhas (adaptado de [8]).

2
Os agentes bifuncionais podem interagir com outro centro nucleofílico (i) na mesma base,
formando um sistema bicíclico ou tricíclico, (ii) numa base diferente na mesma cadeia de DNA ou na
cadeia oposta, originando ligações intra e intercadeias, ou (iii) numa proteína, induzindo a ligação
entre o DNA e a proteína [3].
A especificidade das reações nos diferentes locais depende muito da nucleofilicidade dos
centros de DNA e de factores estereoquímicos. Os locais mais nucleofílicos são os átomos de azoto
endocíclicos, como os átomos N3 E N7 da guanina e da adenina, enquanto os átomos de oxigénio de
bases exocíclicas são menos nucleofílicos [9]. O azoto N7 da guanina, que está exposto no major
groove da hélice de DNA, está mais acessível e, por isso, mais disponível para reagir com eletrófilos;
já a posição N3 da adenina está orientada para o minor groove, estando por isso menos acessível.
Pela mesma razão, alguns locais, como a posição N1 da adenina e a N3 da citosina não reagem
devido ao impedimento estereoquímico. [3].
Figura 2 - Numeração dos átomos das bases azotadas de DNA (adaptado de [3])1.
Os agentes alquilantes, após ativação metabólica, ficam suscetíveis ao ataque nucleofílicos
por parte das bases de DNA [10]. Os agentes alquilantes pequenos, com carga positiva e baixa
polarizabilidade (hard alkylating agents) apresentam maior reatividade com os nucleófilos de oxigénio
duros. Já os agentes alquilantes maiores, sem carga e polarizáveis (soft alkylating agents) preferem
reagir com os átomos de azoto. Os grupos amina exocíclicos não são alvo dos agentes alquilantes
[11].
Os agentes acilantes transferem um radical acilo para as bases de DNA. Já foi descrita a
formação de adutos na posição O6 da guanina e nos grupos amina da citosina, adenina e guanina
[12, 13].
As arilaminas são ativadas metabolicamente para poderem reagir com o DNA. A maioria
destes compostos forma adutos com o DNA nas posições N2 e C8 da guanina [14, 15].
Os compostos químicos podem também interagir com o DNA através de inserção intercalar
no espaço entre pares de bases adjacentes. A intercalação é uma interação não-covalente cuja
afinidade é regida pela força das interações eletrostáticas e de van der Waals. As estruturas
planarem aromáticas é agentes intercaladores capazes de se inserirem, parcial ou completamente,
1 No texto, os sobrescritos indicam átomos de oxigénio ou azoto exocíclicos, enquanto o formato regular indica átomos endocíclicos.

3
entre pares de bases adjacentes. Além destas, já foram identificadas outras moléculas capazes de
atuarem como agentes intercaladores [16]. A intercalação pode provocar alterações estruturais locais
no DNA, incluindo desenrolamento da dupla hélice e alongamento da cadeia de DNA danificada,
quebra da cadeia dupla e mutações frameshift em bactérias [3].
Os adutos gerados por compostos químicos que reagem com o DNA são sujeitos a uma série
de processos de reparação nas células. Quando estes sistemas falham, podem ocorrer mutações
durante a replicação do DNA. Dependendo de onde foram formados os adutos, a probabilidade de
ocorrerem lesões mutagénicas varia. Os adutos formados em posições codificantes, como a posição
N1 da guanina e da adenina ou a posição N3 da citosina, podem ser classificados como
promutagénicos, já que ocorrem mutações caso o processo de reparação falhe [17]. Os adutos
formados na posição N7 da guanina e da adenina e na posição N3 da adenina são menos estáveis,
podendo depurinar e produzir sítios apurínicos [18]. Além dos adutos formados nas posições
codificantes, também os adutos que alterem a estrutura do DNA ou previnam a sua replicação podem
ter efeitos genotóxicos.
1.2. Regulamentação
As impurezas genotóxicas (GTIs) têm recebido cada vez mais atenção por parte das agências
reguladoras bem como da indústria farmacêutica.
1.2.1. Conferência Internacional para a Harmonização (ICH)
A Conferência Internacional para a Harmonização (ICH) dos requisitos técnicos para registo
de produtos farmacêuticos para uso humano juntou as entidades reguladoras do Japão, Europa e
Estados Unidos da América, com o objectivo de minimizar as diferenças existentes nos requisitos
necessários para o desenvolvimento de fármacos. A ICH desenvolveu grande parte das diretrizes que
controlam as impurezas.
Em 1995, as diretrizes Q3 da ICH referiam apenas compostos com uma “toxicidade invulgar”,
que era uma clara referência a impurezas genotóxicas, e mencionavam a necessidade de estabelecer
limites mais apertados para essas impurezas. A diretriz ICH Q3A regula as impurezas em substâncias
ativas novas; apresenta limites para reportar, identificar e qualificar impurezas. A diretriz ICH Q3B é
igual à anterior, mas para novos medicamentos. Já a diretriz ICH Q3C controla solventes residuais;
sendo a primeira vez que a ICH aplica limites específicos a substâncias. De acordo com o risco que
representam para a saúde humana, os solventes residuais podem ser divididos em três classes. Os
solventes de Classe I devem ser evitados, os de Classe II têm limites de exposição diária permitida e
os solventes de Classe III não têm limite de exposição definido desde que a exposição diária seja
inferior a 50 mg/dia. A diretriz ICH Q3D estabelece limites de metais pesados nos fármacos [19].
As normas da ICH publicadas não são apropriadas para a maioria das impurezas
genotóxicas. Normalmente, a síntese de fármacos envolve a utilização de materiais reativos que têm
a capacidade de interagir com o DNA humano causando mutações e cancro, mesmo estando
presente em concentrações bastante baixas. Assim, as impureza genotóxicas devem ser evitadas ou,
se tal não for possível, reduzidas para um nível abaixo de um limite.

4
1.2.2. Pharmaceutical Research and Manufacturers of America (PhRMA)
Em 2004, Pharmaceutical Research and Manufacturers of America (PhRMA) formaram um
grupo de trabalho para discutir impurezas genotóxicas; desta discussão resultou a publicação de um
artigo na revista “Regulatory Toxicology and Pharmacology” intitulado “A Rationale for Determining,
Testing and Controlling Specific Impurities in Pharmaceuticals that Possess Potential for Genotoxicity”
[19].
Este artigo introduziu dois conceitos novos muito importantes:
1. Um sistema de classificação das impurezas genotóxicas em 5 classes (Tabela 1). Foi
elaborada uma estratégia para a classificação de impurezas baseada neste sistema
de classificação. Esta estratégia tornou-se uma norma para a avaliação de riscos
relacionados com impurezas genotóxicas. A estratégia proposta pela PhRMA pode
ser implementada em três passos, como descrito na Tabela 2;
2. Implementação do staged TTC em ensaios clínicos; para o cálculo deste parâmetro é
tido em consideração a dose e a duração dos ensaios clínicos, resultando num TTC
mais baixo para doses mais elevadas e num TTC mais elevado para tempos de
exposição menores (Tabela 3) [20]. Os valores estimados para o staged TTC devem
aplicar-se a todos os estágios de desenvolvimento e para cada composto individual
em casos em que estão presentes várias impurezas genotóxicas [19].
Estes conceitos permitiram à indústria ter uma estratégia de mitigação de risco durante o
desenvolvimento de novos fármacos. O staged TTC é também um elemento essencial nas diretrizes
da Agência Europeia do Medicamento (EMA) e da Food and Drug Administration (FDA).
Tabela 1 - Classificação das impurezas genotóxicas [19].
Classe Descrição
1 Impurezas genotóxicas (mutagénicas) e carcinogénicas
2 Impurezas genotóxicas (mutagénicas) com potencial carcinogénico desconhecido
3 Impurezas com alertas estruturais, não relacionadas com a estrutura do API, e com
potencial genotóxico (mutagénico) desconhecido
4 Impurezas com estruturas de alerta relacionadas com o API
5 Impurezas sem alertas estruturais ou cuja inexistência de genotoxicidade não esteja
comprovada

5
Tabela 2 - Implementação do modelo de impurezas em cinco classes.
Passo Ação Comentários
1
Identificar alertas estruturais no
composto original e impurezas
esperadas; as impurezas devem ser
classificadas de acordo com a Tabela
1.
Requer uma revisão da via sintética para
identificar compostos potencialmente
perigosos, incluindo impurezas do
processo, reagentes e intermediários
2
Estabelecer uma estratégia de
qualificação das impurezas de acordo
com a classificação. Este passo define
o potencial genotóxico de cada
impureza ou estabelece limites de
especificação permitidos para a
impureza no fármaco.
A informação sobre o potencial
carcinogénico baseia-se na literatura
suportada por dados experimentais obtidos
em testes de genotoxicidade, como o teste
de Ames.
3
Estabelecer limites para a impureza
baseados na ingestão diária aceitável
(ADI do inglês allowable daily
ingestion) e no conceito de TTC.
A determinação de quantidades aceitáveis
na substância ativa e no fármaco é baseada
na qualificação e avaliação do risco.
Tabela 3 - Ingestão diária permitida proposta (µg/dia) para GTIs durante o desenvolvimento clínico.
Duração da exposição
≤1 mês >1-3 meses >3-6 meses >6-12 meses >12 meses
Ingestão
diária
permitida
(µg/dia)
1202 ou 0,5%,
o que for mais
baixo
402 ou 0,5%,
o que for mais
baixo
202 ou 0,5%,
o que for mais
baixo
102 ou 0,5%,
o que for mais
baixo
1,53
1.2.3. Agência Europeia do Medicamento (EMA)
A Agência Europeia do Medicamento (EMA) é a autoridade responsável pela imposição de
normas detalhadas sobre como lidar com impurezas genotóxicas [21]. A preparação de propostas
destas normas esteve a cargo do Grupo de Trabalho de Segurança (SWP) do Comité de
Especialidades Farmacêuticas (CPMP), agora denominado Comité de Medicamentos para Uso
Humano (CMPH). O SWP considerou que as impurezas com potencial genotóxico eram um caso
particular que as diretrizes Q3A e Q3B não referiam. A primeira versão do artigo, “Position Paper on
the Limits of Genotoxic Impurities”, foi disponibilizada para consulta em 2002, e publicada em 2004.
2 A probabilidade de não exceder o risco de 10-6 é 93%. 3 A probabilidade de não exceder o risco de 10-5 é 93%. Outros limites (mais baixos ou mais pequenos) podem ser
apropriados e as abordagens usadas para identificar, qualificar e controlar impurezas devem ser aplicadas.

6
Em Junho de 2006 o CHMP publicou a versão final da “Guideline on the Limits of Genotoxic
Impurities” [22]. O SWP reforçou substancialmente a diretriz com a publicação de vários documentos
Q&A para clarificar todas as questões [23]. O documento aplica-se a impurezas genotóxicas em
novas substâncias ativas. Pode também ser aplicado a novas aplicações de substâncias ativas já
existentes, em que a avaliação da via de síntese, controlo de processos e perfil de impurezas não
garanta que os níveis de impurezas genotóxicas sejam inferiores aos autorizados. A classificação de
um composto como genotóxico significa, geralmente, que foram obtidos resultados positivos nos
ensaios de genotoxicidade in vitro ou in vivo, sendo o principal foco as substâncias que reagem com
o DNA e que têm potencial para o danificar diretamente. A diretriz recomenda que os compostos
genotóxicos sejam divididos em duas categorias:
1. Compostos genotóxicos para os quais existem evidências suficientes que suportem
um mecanismo baseado em limites;
2. Compostos genotóxicos para os quais não é possível estabelecer um mecanismo
baseado em limites.
A diretriz recomenda também que as experiências sejam limitadas a impurezas cuja
genotoxicidade é esperada, baseando-se na estrutura molecular, tipo e quantidade de impureza na
matéria-prima e dados de genotoxicidade existentes.
As impurezas que suportem um mecanismo baseado em limites são abordadas na diretriz
ICH Q3C (R3) para solventes de Classe 2. Esta abordagem permite calcular uma “exposição diária
permitida” (PDE), que é derivada do “nível sem efeitos observáveis” (NOEL) ou, em alternativa, do
“nível com o menor efeito” (LOEL); estes níveis são obtidos a partir de estudos animais e incorporam
uma série de fatores de incerteza.
Para o caso das impurezas genotóxicas para as quais não se podem definir mecanismos
baseados em limites é sugerida a utilização do princípio ALARP (As Low As Reasonably Praticable)
[19]; esta abordagem especifica que devem ser feitos todos os esforços para prevenir a formação
destas impurezas durante a síntese de fármacos e, se tal não for possível, devem ser feitos esforços
pós-síntese para reduzir os seus níveis.
Quando é proposta uma estratégia de formulação/produção devem ter-se em consideração
todas as substâncias químicas usadas como reagentes, ou que estejam presentes como
intermediários ou produtos secundários, que se saiba que são genotóxicos e/ou carcinogénicos. De
uma forma geral, as substâncias reativas e as que têm uma estrutura de alerta que não esteja
relacionada com a substância ativa podem ser consideradas. Se possível, devem ser utilizadas
alternativas que não levem à presença de resíduos genotóxicos no produto final. Se não for
encontrada uma alternativa viável, é necessária uma estratégia que inclua vias de síntese ou
formulação alternativas e o uso de reagentes alternativos. No entanto, as agências reguladoras
reconhecem que, em muitos casos, a eliminação completa de impurezas genotóxicas não é possível.
Se não for possível eliminar a impureza genotóxica, devem ser tomadas medidas para reduzir a
quantidade de impureza na substância ativa ou no fármaco em conformidade com as necessidades
de segurança do paciente ou para um nível tão baixo quanto possível.

7
Nos casos em que não é possível evitar as impurezas genotóxicas, a diretriz recomenda a
implementação de uma avaliação de risco. A norma propõe a utilização de um “limiar de risco
toxicológico” (TTC) para impurezas genotóxicas; esta abordagem já tinha sido referida no relatório do
grupo de trabalho da PhRMA [22]. Estimou-se que o valor numérico do TTC seria 1,5 µg/dia, o que
corresponde a um risco de cancro de 1 em 1.000.000. Em certos casos como exposições de curta
duração, tratamento de doenças que colocam a vida do paciente em risco, casos em que a
esperança de vida não ultrapassa os 5 anos ou quando a impureza é uma substância conhecida e se
sabe que o risco resultante da exposição a esta substância através de outras fontes é
significativamente maior que a associada ao consumo dos fármacos, seria possível estabelecer
limites mais elevados do que o TTC estipulado. O documento Q&A inclui uma tabela (Tabela 4) com
valores de TTC recomendados de acordo com a duração da exposição em ensaios clínicos.
Tabela 4 - Limites propostos no documento Q&A da EMA [23].
Duração da exposição
Dose única ≤1 mês ≤3 meses ≤6 meses ≤12 meses
Ingestão diária
permitida (µg) 120 60 20 10 5
1.2.4. Food and Drug Administration (FDA)
Em Dezembro de 2008, a US FDA publicou uma versão preliminar de um guia intitulado
“Genotoxic and Carcinogenic Impurities in Drug Substances and Products – Recommended
Approaches” [24]. A versão final do guia ainda não foi publicada e pode ser substituído pela diretriz
ICH M7. Este guia fornece recomendações específicas sobre a qualificação de segurança de
impurezas com potencial carcinogénico ou genotóxico suspeito ou conhecido. As abordagens são
semelhantes à diretriz da EMA e incluem três passos (Tabela 5).
Tabela 5 – Guia da FDA para caracterizar e minimizar o risco.
Passo Ação
1 Alterar as vias de síntese e/ou purificação para minimizar a formação e/ou
maximizar a remoção da impureza relevante.
2 Estabelecer como objectivo uma exposição máxima diária 1,5 µg/dia.
3 Caracterizar o risco genotóxico e carcinogénico para estabelecer
especificações apropriadas.
A orientação fornecida pela FDA é muito semelhante às abordagens descritas na diretriz da
EMA; no entanto, existem também várias diferenças. A Tabela 6 mostra algumas diferenças e
semelhanças entre as duas abordagens.

8
Tabela 6 - Diferenças e semelhanças entre as diretrizes da FDA e da EMA.
Semelhanças Diferenças
Recomendam a mesma abordagem para a
identificação e qualificação de impurezas
potencialmente genotóxicas. Recomendam
também a mesma abordagem para lidar com as
impurezas genotóxicas e carcinogénicas.
A diretriz da FDA inclui impurezas
carcinogénicas.
Estabelecem um TTC de 1,5 µg/dia
A FDA permite um TTC de 120 µg para uma
exposição inferior a 14 dias, enquanto a EMA
sugere esse limite para uma dose única.
Sugerem valores de TTC mais elevados para
exposições curtas durante os ensaios clínicos.
A norma da FDA não permite valores mais
elevados de TTC, baseado na exposição a curto
prazo, para produtos comerciais.
Os valores de TTC estabelecidos pela FDA para ensaios clínicos estão descritos na Tabela 7.
Tabela 7 - Limites de staged TTC propostos pela FDA.
Duração da exposição
14 dias >14 dias-1
mês >1-3 meses >3-6 meses >6-12 meses
Ingestão diária
permitida (µg) 120 60 20 10 5
1.2.5. ICH M7
Em 2013 foi publicada pela ICH a diretriz ICH M7 “Assessment and Control of DNA Reactive
(Mutagenic) Impurities” [25]. O objetivo desta norma é oferecer orientação sobre a análise das
relações estrutura-atividade (SAR) para a genotoxicidade. Além disso, pretende clarificar se as
impurezas com alertas semelhantes e que, potencialmente, tenham mecanismos de ação
semelhantes devem ou não ser combinadas para efeitos de cálculo do TTC, bem como se o TTC
deve variar consoante o tempo de utilização aprovado.
O Grupo de Trabalho de Peritos (EWG) reuniu-se pela primeira vez em Novembro de 2010
em Fukuoka, Japão. No “ICH Step 1 Working Draft”, resultante desta reunião, os princípios gerais
foram semelhantes aos existentes nas diretrizes da EMA, especialmente no que respeita aos alertas
estruturais, testes de Ames, aplicações do TTC e avaliação do risco.
1.2.6. ICH S2
A principal diretriz sobre os testes de genotoxicidade para produtos farmacêuticos é a ICH S2
(R1) intitulada “Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human
Use”. Combina duas diretrizes anteriores, a S2A (“Specific Aspects for Regulatory Genotoxicity Tests
for Pharmaceuticals”) e a S2B (“Genotoxicity: A Standard Battery for Genotoxicity Testing of

9
Pharmaceuticals”). A diretriz foca-se apenas em testes para moléculas de substância ativa, não
podendo ser aplicada a moléculas biológicos.
A diretriz está dividida em cinco partes:
A Parte I especifica o âmbito da diretriz e inclui a definição e princípios gerais de
testes de genotoxicidade. Os testes de genotoxicidade são definidos como testes in
vivo e in vitro designados para detetar compostos que induzem danos genéticos por
diversos mecanismos. Estes testes permitem a identificação de perigos no que toca a
danos do DNA e a sua fixação.
A Parte II descreve um conjunto de testes para as substâncias ativas e inclui testes
para mutações génicas em bactérias in vivo e in vitro.
As Partes III a V contêm recomendações específicas para testes in vitro, testes in
vivo e para a avaliação dos resultados obtidos.
1.3. Classes de compostos genotóxicos
Os compostos genotóxicos apresentam estruturas diversas e pertencem a famílias químicas
diferentes. Do ponto de vista químico, os GTIs não têm propriedades físico-químicas ou elementos
estruturais comuns que permitam uma identificação rápida e fácil. Existem algumas moléculas cujo
efeito genotóxico é conhecido, enquanto outras são perigosas devido à presença de grupos reativos
que podem levar à genotoxicidade; estes grupos reativos estão catalogados como “alertas
estruturais” [26]. Estima-se que cerca de 20 a 25% de todos os intermediários utilizados na síntese
farmacêutica padrão contêm “alertas estruturais”. É possível utilizar sistemas in silico, como o DEREK
(Deductive Estimation of Risk from Existing Knowledge), para determinar alertas estruturais para
atividade genotóxica [27-29]. Muitas vezes os alertas estruturais sobrestimam o potencial mutagénico,
uma vez que não têm em consideração factores como o elevado peso molecular, hidrofilicidade,
elevada reatividade, impedimento estereoquímico ou simetria molecular [30, 31].
Cada GTI tem um papel específico na via química utilizada na síntese de API, pelo que a sua
presença na produção de APIs não é estocástica; os GTIs estão presentes na reação química devido
à sua introdução na mesma, como solventes, reagentes ou catalisadores, ou devido à sua formação
como produtos secundários.
1.3.1. GTIs usados como reagentes
A reatividade elevada dos reagentes que os torna atrativos para a síntese química, pode
também ser responsável pela genotoxicidade dos mesmos. Muitas vezes os reagentes não são
consumidos completamente e continuam na mistura de reação, podendo seguir com o API ou
intermediário.
1.3.1.1. Haletos de alquilo e ésteres
Os haletos de alquilo são muito usados como agentes alquilantes [32]. O seu mecanismo de
toxicidade não é completamente conhecido, mas sabe-se que têm a capacidade de alquilar

10
diretamente macromoléculas biologicamente ativas, como as proteínas e o DNA [33]. Os haletos de
alquilo estão presentes no API devido à sua utilização direta, mas também devido a reações
secundárias entre álcoois, usando por exemplo como solventes, e haletos de hidrogénio. Os haletos
de alquilo, como o cloreto de metilo ou de etilo, que derivam de álcoois com baixo peso molecular são
voláteis e removidos do API durante o processo de secagem; mas podem por vezes também ficar
presos na matriz cristalina do API, sendo necessário o seu controlo [34, 35].
1.3.1.2. Sulfatos de alquilo
O dimetil sulfato (DMS) é um agente metilante forte utilizado para introduzir um grupo metilo
em átomos que contenham pares de eletrões não partilhados. Comparativamente com os haletos de
alquilo metilantes que funcionam como agentes metilantes, as reações com o DMS são mais
favoráveis devido à taxa de reação mais elevada e menor possibilidade de formação de produtos
secundários [1].
1.3.1.3. Epóxidos
Os epóxidos são éteres cíclicos com três átomos, dois átomos de carbono e um átomo de
oxigénio, que formam um anel; este anel é responsável pela elevada reatividade das moléculas.
Estes compostos participam em reações de adição com álcoois, aminas, haletos, compostos
organometálicos, cianetos, compostos aromáticos e grupos metileno ativos, em que o anel é aberto.
A reatividade elevada destes compostos torna-os genotóxicos, pois os seus dois átomos de carbono
eletrofílicos podem reagir com os centros nucleofílicos do DNA, originando produtos alquilados [3,
36].
1.3.1.4. Hidrazinas
A hidrazina e os seus derivativos são reagentes ou intermediários importantes em muitos
processos de síntese industriais. A hidrazina é uma base altamente reativa que atua como um agente
redutor; a sua toxicidade foi atribuída à geração de carbocatiões, radicais centrados em carbono e
radicais centrados em oxigénio, que são consideradas espécies altamente reativas. Estes
intermediários reativos têm a capacidade de alquilar o DNA e induzir outros tipos de danos [37, 38].
1.3.1.5. TEMPO
O composto N-oxil-2,2,6,6-tetrametilpiperidina (TEMPO) é um reagente geralmente utilizado
como um radical nitroxil estável na indústria química e bioquímica; é principalmente usado para a
oxidação de álcoois para gerar aldeídos e cetonas ou ácidos carboxílicos. O seu potencial genotóxico
já foi avaliado e, tendo por base os dados publicados, este composto é considerado genotóxico [39].
1.3.1.6. Aminas aromáticas
As aminas aromáticas não são genotóxicas de forma direta; isto é, não danificam o DNA por
si só, requerendo ativação metabólica. A ativação metabólica inicia-se com a oxidação da arilamina
na hidroxilamina correspondente, que é catalisada pela citrocromo P450 [40]. Segue-se a
esterificação em N-acetoxiarilamina pela N-acetiltransferase (NAT) ou num sulfato por uma
sulfotransferase (ST). Durante a ativação formam-se (ArN+H); estes iões são muito reativos e

11
ligam-se covalentemente a biomoléculas, como o DNA, gerando derivativos aminoaril [1, 3, 41-44]. As
reações com os iões nitrénio ocorrem, normalmente, na posição C8 da 2-deoxiguanosina (dG) e, em
menor escala, da 2-deoxiadenosina (dA); esta reação leva à formação de adutos C8 [1, 3, 14, 41, 42,
45-47].
As aminas aromáticas estão frequentemente presentes na síntese farmacêutica como
matéria-prima, intermediários ou reagentes [1, 3].
1.3.1.7. Ácidos borónicos
Os ácidos borónicos foram recentemente identificados como mutagénicos bacterianos. No
entanto, não é possível comprovar que haja uma ligação direta covalente entre estes compostos e
aberrações na replicação do DNA [48]. Os ácidos borónicos e os seus derivados tipo éster são
importantes blocos de construção na síntese orgânica, pois são fáceis de manusear e atuam como
ácidos de Lewis. Estas propriedades tornam-nos intermediários chave na produção de APIs [49].
1.3.2. GTIs formados em reações secundárias
Os GTIs podem-se formar como produtos secundários durante a síntese de API. Os
sulfonatos são a classe de GTIs mais estudada; esta classe de moléculas é formada em reações
secundárias com álcoois (utilizados como solventes, por exemplo) e, por isso, a sua presença na
síntese de APIs pode não ser óbvia. No entanto, graças ao trabalho desenvolvido pelo Instituto de
Investigação da Qualidade do Produto (PQRI), a indústria e academia estão agora cientes deste
desafio [50].
1.3.2.1. Sulfonatos e os seus precursores
Os sais sulfonatos dos APIs são muito úteis e os álcoois são muitas vezes utilizados como
solventes na cristalização nos processos de isolamento dos sais de sulfonatos. Quando um ácido
sulfónico e um álcool estão ambos presentes numa corrente de processo, há uma possibilidade de se
formar uma impureza: sulfonato de alquilo. [51, 52]. Os sulfonatos são agentes alquilantes capazes
de, por si só ou após ativação metabólica, adicionar resíduos alquilo aos locais nucleofílicos reativos
do DNA [1]. Os sulfonatos compreendem uma grande gama de estruturas químicas, desde simples
sulfonatos alquilo até estruturas mais complexas com sistemas aromáticos com diversos grupos
funcionais. Normalmente estes compostos não são introduzidos diretamente no processo de síntese
do API, formando-se em reações secundárias.

12
Tabela 8 - Precursores de alguns sulfonatos [1].
Precursor GTI
Mesilato (Ms)
Triflato (Tf)
Nosilato (Ns)
Tosilato (Ts)
Os precursores dos sulfonatos na síntese de APIs são os ácidos sulfónicos, os haletos e os
anidridos correspondentes bem como os próprios sulfonatos. Em muitas sínteses de API não é
possível substituir álcoois enquanto solventes, devido à sua capacidade de solubilizar tanto o API
como os sais [53]. Além disso, o controlo do processo de cristalização de forma a obter o API
recristalizado com polimorfismo correto pode implicar a utilização de um álcool como solvente. Na
Tabela 8 estão representados alguns exemplos de precursores de sulfonatos.
Os sulfonatos podem estar presentes no API ou nos seus intermediários devido à sua
produção em reações secundárias entre ácidos sulfónicos e álcoois ou como reagentes que
permanecem em solução devido a reações incompletas. Quando é utilizado algum dos precursores
de sulfonatos na síntese do API, há possibilidade de formação de sulfonatos genotóxicos. Já foi
documentado que os sulfonatos se decompõem através de alcoólise para obter ácido sulfónico e um
éter; esta reação, juntamente com a reversibilidade da formação de ésteres, podem limitar a
quantidade de ésteres genotóxicos produzidos [51, 54]. A Farmacopeia Europeia obriga a que os
APIs comercializados como sais de ácidos sulfónicos comprovem que todo o éster sulfonato formado
é removido durante o processo de purificação [55]. Assim, é importante dedicar uma atenção especial
à monitorização de precursores de sulfonatos durante a síntese de APIs.

13
1.3.3. Solventes orgânicos
A indústria farmacêutica é um dos maiores consumidores de solventes orgânicos por
quantidade de produto produzido. Os solventes orgânicos estão sempre presentes nos processos de
produção de APIs [56]. Como mencionado na secção 1.2.1, a diretriz Q3C aconselha a divisão dos
solventes em quatro classes [57].
1.4. Remoção de impurezas genotóxicas
Durante a produção de APIs, os reagentes altamente reativos são muitas vezes utilizados
[58]. Por isso, é natural que o API final contenha vestígios de reagente ou produtos secundários como
impurezas; estas impurezas podem conter toxicidades indesejadas, incluindo genotoxicidade e
carcinogenicidade, tendo um impacto negativo na avaliação de risco do produto [24]. Apesar de ser
preferível evitar essas fontes de GTIs, nem sempre tal é possível. Assim, para ultrapassar este
desafio, é necessário identificar os GTIs num estágio inicial, desenvolver métodos analíticos para os
detetar e implementar processos sintéticos para os controlar.
A prevenção é a abordagem preferida e inclui uma otimização cuidada da síntese química. A
primeira estratégia para limitar a quantidade de GTIs na produção de APIs é evitar a sua utilização e
o seu aparecimento através da alteração da via sintética; tal pode ser conseguido através da
utilização de diferentes sínteses químicas para obter o mesmo API ou intermediário, ou através da
otimização da via sintética já existente. Muitas vezes, os reagentes e os intermediários são úteis
sinteticamente, não podendo ser evitados. Neste caso, deve ser utilizada uma abordagem baseada
na prevenção – Quality by Design (QbD) – que inclui o ajuste de parâmetros como o pH, temperatura,
tempo de reação ou matriz. Nesta abordagem, os químicos tentam evitar a utilização ou o
aparecimento de GTIs ao longo da síntese de APIs, procurando diferentes vias sintéticas para obter o
mesmo API ou otimizando a via já existente [59, 60]. Em alguns casos é possível aplicar esta
estratégia com sucesso, não havendo grandes repercussões no rendimento. No entanto, muitas
vezes não é prático alterar a via de síntese de APIs para controlar ou reduzir os GTIs, principalmente
quando o processo já está numa fase avançada. Neste caso é utilizada uma outra estratégia que se
baseia na eliminação ou redução da concentração de GTI durante o passo sintético crítico. Isto pode
ser conseguido através da alteração de certas condições da reação como: (i) proporção dos
componentes na reação, (ii) alteração da ordem de adição dos reagentes, ou (iii) alteração da
qualidade ou do método de preparação das matérias-primas. Com estas medidas preventivas, o
método QbD pode contribuir para a diminuição dos GTIs em APIs [61].
1.5. Purificação de APIs
A síntese de APIs inclui alguns passos de purificação que contribuem também para a
remoção de GTIs, apesar de não ser esse o seu principal propósito.
Para a remoção específica de GTIs, a seleção do método de purificação depende das
propriedades químico-físicas do composto, como a reatividade, solubilidade, volatilidade e

14
ionizabilidade do GTI [62]. É necessário garantir que, durante os passos de purificação, as perdas de
API não são significantes; outro cenário é inaceitável à escala industrial.
Para a remoção específica de GTIs, a seleção do método de purificação depende das
propriedades físico-químicas do GTI. Normalmente, quanto maior a seletividade de um processo de
separação para uma determinada impureza, menor é a perda de API e maior é a eficácia de remoção
da impureza em questão. Muitas vezes, para obter um API com níveis de segurança aceitáveis para o
paciente, é necessário aplicar uma estratégia de purificação capaz de reduzir os níveis de GTI até
valores aceitáveis. As Equações 1 a 4 permitem calcular a ingestão diária de GTI permitida [1, 63].
𝐸𝑓𝑖𝑐𝑖ê𝑛𝑐𝑖𝑎 𝑟𝑒𝑚𝑜çã𝑜 𝑑𝑜 𝐺𝑇𝐼 =𝐺𝑇𝐼 𝑛𝑜 𝑓𝑖𝑛𝑎𝑙 𝑑𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜
𝐺𝑇𝐼 𝑛𝑜 𝑖𝑛í𝑐𝑖𝑜 𝑑𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜 (1)
𝑃𝑒𝑟𝑑𝑎 𝑑𝑒 𝐴𝑃𝐼 =𝐴𝑃𝐼 𝑛𝑜 𝑓𝑖𝑛𝑎𝑙 𝑑𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜 𝑑𝑒 𝑝𝑢𝑟𝑖𝑓𝑖𝑐𝑎çã𝑜
𝐴𝑃𝐼 𝑎𝑙𝑖𝑚𝑒𝑛𝑡𝑎𝑑𝑜 𝑛𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜 𝑑𝑒 𝑝𝑢𝑟𝑖𝑓𝑖𝑐𝑎çã𝑜 (2)
𝑇𝑒𝑜𝑟 𝑑𝑒 𝐺𝑇𝐼 =𝐺𝑇𝐼 𝑛𝑜 𝑓𝑖𝑛𝑎𝑙 𝑑𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜 𝑑𝑒 𝑝𝑢𝑟𝑖𝑓𝑖𝑐𝑎çã𝑜
𝐴𝑃𝐼 𝑛𝑜 𝑓𝑖𝑛𝑎𝑙 𝑑𝑜 𝑝𝑟𝑜𝑐𝑒𝑠𝑠𝑜 𝑑𝑒 𝑝𝑢𝑟𝑖𝑓𝑖𝑐𝑎çã𝑜 (3)
𝐼𝑛𝑔𝑒𝑠𝑡ã𝑜 𝑎𝑐𝑒𝑖𝑡á𝑣𝑒𝑙 𝑑𝑖á𝑟𝑖𝑎 𝑑𝑒 𝐺𝑇𝐼 =𝐷𝑜𝑠𝑒 𝑑𝑖á𝑟𝑖𝑎 𝑑𝑒 𝐴𝑃𝐼
𝑘𝑔 (𝑝𝑎𝑐𝑖𝑒𝑛𝑡𝑒)× 𝑡𝑒𝑜𝑟 𝑑𝑒 𝐺𝑇𝐼 × 𝑃𝑒𝑠𝑜 𝑑𝑜 𝑝𝑎𝑐𝑖𝑒𝑛𝑡𝑒 (4)
Os baixos níveis descritos nas especificações representam um desafio para uma purificação
eficiente. A remoção de grandes quantidades de impurezas pode ser conseguida através do aumento
do número de ciclos do processo de purificação. No entanto, esta estratégia leva também a perdas
indesejáveis de API ao longo dos ciclos.
Alguns dos processos de purificação convencionais incluem a cristalização, precipitação,
extração, cromatografia em coluna, tratamento com carvão ativado, resinas e destilação. A eficiência
da separação é baseada nas diferenças das propriedades dos compostos a serem separados e/ou as
suas afinidades relativas para um agente seletivo. Durante a última década foram desenvolvidas
outras técnicas, como a separação por membranas ou polímeros molecularmente impressos [64].
A cristalização consiste na formação de cristais sólidos em solução. Neste processo, o API é
isolado como fase sólida e as impurezas mantêm-se dissolvidas na fase líquida. Esta técnica permite
a determinação da pureza e o conteúdo de impureza residual do API. A filtração pode ser usada para
isolar os sólidos cristalinos. Dependendo da otimização do processo, até 30% do API pode ser
perdido durante a lavagem dos sólidos ou durante a filtração, mantendo-se na solução mãe. As
impurezas podem também ser lavadas juntamente com o API ou manter-se como parte da rede
cristalina dependendo da eficiência do processo de lavagem [65-67].
A extração líquido-líquido é usada para separar compostos de acordo com a sua solubilidade
em dois solventes diferentes. Uma estratégia seguida com alguma frequência consiste em
transformar o API seletivamente num sal que é retido na fase aquosa enquanto as impurezas são
removidas na fase orgânica. O sal orgânico é então convertido nas suas espécies neutras por

15
acidificação ou basificação e extraído novamente num segundo solvente orgânico. Note-se que os
solventes orgânicos são imiscíveis em água. A eficiência do processo depende do coeficiente de
partição relativo do API e do GTI nos diferentes solventes [1, 65-67].
A precipitação pode ser promovida pela adição de um não-solvente a uma solução de API ou
vice-versa. As impurezas mantêm-se no líquido e o API na fase sólida. O sistema de solventes deve
ter uma solubilidade maior para as impurezas do que para o API. A filtração pode ser usada para
separar a fase líquida do sólido e a destilação é usada para evaporar solventes com baixos pontos de
ebulição.
A destilação é uma opção para purificar APIs voláteis através da evaporação de solventes
com baixo ponto de ebulição; pode também ser usada para remover solventes ou GTIs voláteis e
para efetuar trocas de solventes, quando se pretende mudar de um solvente com baixo ponto de
ebulição para outro com um ponto de ebulição maior [68].
Os adsorventes são utilizados para remover cor e impurezas, sendo baseados nas diferentes
afinidades dos diferentes compostos para o adsorvente. É desejável combinar uma grande afinidade
para o GTI com uma baixa afinidade para o API [69, 70]. As resinas de troca iónica são matrizes
poliméricas com grupos funcionais que têm um ião móvel que pode ser trocado com outros iões
presentes em solução para ser purificado [71]. A cromatografia em coluna é uma técnica pós-reação
que é usada na síntese de química orgânica para remover impurezas [72-74]. Os componentes são
retidos na fase estacionária distintamente e separam-se uns dos outros enquanto percorrem a coluna
a diferentes velocidades. O eluente pode ser um único solvente ou uma mistura. Parâmetros como o
tamanho da partícula, dimensões da coluna, fluxo e pressão do eluente são críticos.
Na extração supercrítica, os solventes de extração são fluídos supercríticos. Estes fluídos têm
um poder de solvatação elevado e a difusividade aumentada de um gás. Indo apenas de um estado
supercrítico para o estado gasoso é possível isolar o soluto. O dióxido de carbono é um solvente
supercrítico ideal devido ao seu ponto crítico relativamente baixo (304 K e 72,9 atm). A extração
supercrítica tem o potencial de proporcionar uma rota limpa e efetiva para remover GTIs de APIs.
A nanofiltração com solventes orgânicos (do inglês organic solvent nanofiltration – OSN)
baseia-se principalmente nas diferenças do tamanho da molécula, apesar de outras propriedades
também poderem contribuir. A eficiência desta técnica depende da membrana selecionada e da
respetiva curva de rejeição [75-78]. A OSN também pode ser útil para troca de solventes quando se
pretende passar de um solvente com elevado ponto de ebulição para outro com um ponto de ebulição
mais baixo [79, 80].
Os MIPs são preparados através da incorporação da molécula alvo numa matriz polimérica. O
alvo é removido deixando um local de reconhecimento molecular na matriz de modo a que a estrutura
final do polímero mostre uma afinidade aumentada para a remoção de moléculas usadas como molde
[81-83].

16
1.6. Objetivo
O objetivo desta tese é desenvolver processos de adsorção molecular que permitam remover
compostos genotóxicos das correntes de API, evitando perdas significativas de API.
Os compostos modelo selecionados para o desenvolvimento desta tese foram o furoato de
mometasona (Meta) como API e a 4-dimetilaminopiridina (DMAP) e o p-toluenossulfonato de metilo
(MPTS) enquanto genotóxicos. O Meta é um glucorticóide que pode ser utilizado no tratamento de
doenças inflamatórias [87]. Durante a sua síntese, o DMAP é utilizado como catalisador numa reação
de sulfonilação, que ocorre em DCM. Nesta reação participa também o cloreto de mesilo que, na
presença de MeOH, pode formar o composto genotóxico mesilato de metilo Figura 3.
Figura 3 - Sulfonilação catalisada pelo DMAP durante a síntese de Meta (à direita).
Neste trabalho, para ser possível quantificar o composto por HPLC-UV, foi utilizado o MPTS
em substituição do mesilato de metilo.
Figura 4 – Meta (esquerda), DMAP (meio) e MPTS (direita).
Para tal serão estudados processos de purificação convencionais – recristalização,
adsorventes e nanofiltração – e desenvolvidos polímeros específicos para os GTIs de interesse –
MIPs para o DMAP. Será também avaliada a possibilidade de utilizar resinas e MIPs para aumentar o
rendimento da recristalização.

17
2. Materiais e Métodos
2.1. Materiais
O DMAP foi comprado na Sigma Aldrich, o MPTS foi comprado na Acros Organic e o Meta foi
cedido pela Hovione. As resinas Amberlite são da Sigma-Aldrich, a resina AG 50W-X2 foi adquirida
na BioRad e o carvão ativado é da Merck.
As análises de HPLC (do inglês High Performance Liquid Chromatography) foram efetuadas
num cromatógrafo LaChrom Elite composto por uma bomba, um injetor L-2200 e um detetor de UV L-
2400. Foi utilizada uma coluna Nucleosil C18 da Macherey-Nagel.
Os espectros UV (ultravioleta) foram obtidos num espectrofotómetro U-2000 da Hitachi.
O pH foi medido num medidor 702 MS Titrino.
A análise SEM (do inglês Scanning Electron Microscoopy) foi realizada no MicroLab - Electron
Microscopy Laboratory of IST, tendo sido utilizado um microscópio eletrónico de varrimento de
emissão de campo (FEG-SEM) equipado com espetrómetro de raios X de energia dispersiva e com
detetor de eletrões retro difundidos (do inglês back scattered electrons – BSE), o JEOL JSM-
7001F/Oxford 250/HKL INCA Energy.
Para a análise BET (Brunauer, Emmett, Teller) foi utilizado um porosímetro gasoso, marca
Micromeritics, modelo ASAP 2000 (Accelerated Surface Area and Porosimetry System); esta análise
foi realizada na Faculdade de Ciências e Tecnologias da Universidade Nova de Lisboa.
A análise IV (infravermelho) foi feita na Faculdade de Farmácia da Universidade de Lisboa,
tendo sido usado um espectrofotómetro Nicolet 6700. FT-IR da Thermo Electron Corporation.
2.2. Métodos experimentais
2.2.1. Técnicas analíticas
2.2.1.1. High Performance Liquid Chromatography (HPLC)
O HPLC é uma técnica analítica utilizada para separar componentes de uma mistura,
identificar cada componente e quantificá-los. Um solvente líquido é usado como eluente, fase móvel,
e alimentado, a um caudal fixo, a uma coluna preenchida com material adsorvente. A amostra é
injetada no eluente em microlitros. Cada soluto na amostra interage com o material adsorvente de
forma diferente, fazendo com que os diferentes compostos tenham tempos de residência diferentes;
os componentes com mais afinidade com o enchimento demoram mais tempo a sair da coluna,
enquanto os componentes com menor afinidade com o enchimento viajam mais rapidamente através
desta. Estas diferenças no fluxo dos componentes permitem a separação dos mesmos à medida que
eles saem da coluna.
Existem diferentes tipos de coluna, com enchimentos com diferentes tamanhos de partícula e
diferentes químicas de superfície. A utilização de materiais de empacotamento com partículas mais
pequenas requer uma pressão operacional mais elevada e, tipicamente, melhora a resolução

18
cromatográfica. Em termos de química de superfície, as partículas de adsorvente podem ser
hidrofóbicas ou polares.
A cromatografia pode ser considerada um processo de transferência de massa que envolve
adsorção. O composto ativo da coluna, o adsorvente, é um material granular feito de partículas
sólidas de 2 a 50 µm. Os componentes da amostra são separados uns dos outros devido aos
diferentes graus de interação com as partículas de adsorvente. A fase móvel é, normalmente,
constituída por uma mistura de solventes, como acetonitrilo e água. A composição da fase móvel
pode ser mantida constante (modo de eluição isocrático) ou variar (gradiente) durante a análise
cromatográfica. A eluição isocrática é eficaz na separação de componentes cuja afinidade com a fase
estacionária não seja muito diferente. A composição e a temperatura da fase móvel têm um papel
importante no processo de separação, pois influenciam as interações entre os componentes da
amostra e o adsorvente. Estas interações podem ser hidrofóbicas, dipolo-dipolo ou iónicas.
O HPLC distingue-se da cromatografia líquida típica através da pressão significativamente
mais elevada que utiliza, podendo atingir os 350 bar; na cromatografia líquida típica, a fase móvel
atravessa a coluna através da ação da força da gravidade. Uma coluna típica de HPLC tem um
diâmetro de 2,1-4,6 mm e um comprimento de 30-250 mm. Como as partículas do adsorvente são
extremamente pequenas, o HPLC tem um poder de resolução maior na separação de misturas.
Figura 5 - Sistema de HPLC (Adaptado de [88]).
Na Figura 5 está representado um sistema de HPLC; o equipamento inclui um sampler,
bombas e um detetor. O sampler introduz a amostra na fase móvel, que a transporta até à coluna. A
maioria dos equipamentos de HPLC têm um forno para de coluna que permite ajustar a temperatura à
qual é feita a separação. A bomba permite regular o fluxo e a composição da fase móvel; algumas
bombas mecânicas permitem alterar o rácio dos solventes ao longo do tempo, gerando um gradiente
de composição na fase móvel. O detetor gera um sinal proporcional à quantidade de composto que
sai da coluna, permitindo a análise quantitativa dos componentes da amostra. Um microprocessador
digital e um software controlam o instrumento de HPLC e permitem a análise dos dados Os detetores
mais comuns são os de UV/Vis, de índice de refração ou espectrometria de massa.

19
HPLC de fase reversa (RP-HPLC)
O RP-HPLC tem uma fase estacionária não-polar e uma fase móvel aquosa moderadamente
polar. Uma fase estacionária comum é uma sílica com a superfície modificada com RMe2SiCl, sendo
R um grupo alquilo com cadeia linear. Com esta fase estacionária o tempo de retenção é maior para
compostos menos polares, enquanto os compostos polares eluem mais rapidamente. Os tempos de
retenção podem ser aumentados ou diminuídos através da adição de água ou solvente orgânico
miscível com água à fase móvel, respetivamente. A adição de água faz com que a afinidade do
composto hidrofóbico com a fase estacionária hidrofóbica seja mais forte em relação à fase móvel
agora mais hidrofílica.
O RP-HPLC baseia-se em interações hidrofóbicas, que são originadas a partir da elevada
simetria da estrutura dipolar da água; este tipo de cromatografia permite medir estas forças
interativas. A ligação do analito à fase estacionária é proporcional à área da superfície de contacto à
volta do segmento não-polar da molécula após a associação do ligando na fase estacionária. Este
efeito solvofóbico é dominado pela força da água para reduzir a cavidade à volta do analito. A energia
libertada neste processo é proporcional à tensão superficial do eluente e à superfície hidrofóbica do
analito e do ligando, respetivamente. Pode diminuir-se a retenção através da adição de um solvente
menos polar (miscível com água), como metanol ou acetonitrilo, à fase móvel para reduzir a tensão
superficial da água. O gradiente de eluição utiliza este efeito através da redução automática da
polaridade e da tensão superficial da fase móvel durante o decurso da análise.
As propriedades estruturais do analito têm um papel fundamental nas características da
retenção. Um analito que tenha uma área de superfície hidrofóbica maior é retida durante mais tempo
porque não interage com a estrutura da água. Já os analitos com áreas de superfície mais polares
ficam retidos durante menos tempo pois interagem melhor com a água.
Um fator muito importante é o pH da fase móvel, pois este pode modificar o carácter
hidrofóbico do analito. Por este motivo, muitas vezes são utilizadas soluções-tampão para controlar o
pH. As soluções-tampão têm várias finalidades desde controlar o pH até neutralizar a carga da
superfície de sílica da fase estacionário, passando por neutralizar a carga do analito.
Procedimento experimental
A quantificação dos componentes foi efetuada por RP-HPLC. Como fase móvel foram
utilizados água e acetonitrilo, ambos com 0,1% de ácido fórmico.
Para a quantificação do DMAP foi utilizado um fluxo de 1 mL/min e a deteção foi efetuada a
280 nm. O gradiente era linear dos 60% de H2O até aos 20% em 3 minutos, seguido por 60% de H2O
e 40% de CH3CN durante 1 minuto e, por fim, um período de reequilíbrio de 4 minutos.
Para a quantificação do MPTS o fluxo era de 2 mL/min e a deteção foi efetuada a 215 nm. Foi
utilizado um método isocrático com 50% de cada um dos solventes.
Todas as análises foram feitas em duplicado.

20
2.2.1.2. Espectroscopia UV/Vis
O UV/Vis refere-se à espectroscopia de absorção na região espectral do ultravioleta-visível.
Nesta região do espectro eletromagnético, as moléculas sofrem transições eletrónicas.
As moléculas que contêm eletrões π ou eletrões não-ligantes podem absorver energia na
forma de luz UV ou visível para excitar estes eletrões para orbitais moleculares antiligantes mais
elevadas. Quanto mais facilmente os eletrões forem excitados, maior o comprimento de onda de luz
que absorve.
Um espectrofotómetro UV/Vis pode ser utilizado como detetor no HPLC. A presença de um
analito produz uma resposta proporcional à sua concentração.
Lei de Beer-Lambert
A Lei de Beer-Lambert relaciona linearmente, dentro de um determinado intervalo, a
absorvância de uma solução com a concentração de espécies absorventes em solução e o
comprimento da célula (Equação 5). Assim, fixando o comprimento, a espectroscopia UV/Vis pode
ser utilizada para determinar a concentração de absorvente em solução.
𝐴 = log10 (𝐼0
𝐼) = 𝜀𝑐𝐿 (5)
Sendo A a absorvância medida em unidades de absorvância (AU), I0 a intensidade da luz
incidente a um dado comprimento de onda, I a intensidade transmitida, L o comprimento da célula e c
a concentração da espécie adsorvente. ε é uma constante conhecida como absortividade molar ou
coeficiente de extinção. Esta constante é uma propriedade molecular fundamental num dado solvente
a uma determinada temperatura e pressão e tem unidades de 1 𝑀 × 𝑐𝑚⁄ ou 𝐴𝑈 𝑀 × 𝑐𝑚⁄ .
Espectrofotómetro UV/Vis
O espectrofotómetro UV/Vis mede a intensidade de luz que passa através de uma amostra (I)
e compara-a com a intensidade de luz antes de passar através da amostra (I0). O rácio 𝐼 𝐼0⁄
denomina-se transmitância e é, normalmente, expresso em percentagem (%T). A absorvância é
baseada na transmitância (Equação 6).
𝐴 = − log%𝑇
100% (6)
O espectrofotómetro utilizado é constituído por uma fonte de luz, um suporte para a amostra,
uma rede de difração num monocromador para separar diferentes comprimentos de onda de luz e um
detetor (Figura 6). A fonte de radiação é uma lâmpada de deutério, que é contínua na região do UV
(190-400 nm). O detetor é um fotodíodo de silício que é utilizado com um monocromador, que filtra a
luz de modo a que apenas a luz de um único comprimento de onda atinja o detetor de cada vez. O
espectrofotómetro é de feixe duplo, pelo que a luz é separada em dois feixes antes de chegar à
amostra. Um feixe é usado como referência enquanto o outro feixe passa através da amostra.

21
Figura 6 - Espectrofotómetro UV/Vis (Adaptado de [89]).
Procedimento experimental
O espectrofotómetro UV foi utilizado para obter os espectros das espécies que iriam ser
analisadas por HPLC, de modo a determinar o seu comprimento de onda de máxima absorvância.
Para obter os espectros foram preparadas soluções de cada um dos compostos e estas
foram analisadas entre os 400 e os 190 nm.
2.2.1.3. SEM
O microscópio eletrónico de varrimento (SEM) é um microscópio eletrónico capaz de produzir
imagens de uma amostra através do varrimento da mesma com um feixe de eletrões. Os eletrões vão
interagir com os átomos da amostra, produzindo diversos sinais que podem ser detetados e que
contêm informação sobre a topografia da superfície e sobre a composição da amostra. Os sinais do
SEM incluem eletrões secundários (SE), eletrões retro difundidos (BSE), raios-X característicos,
catodoluminescência (CL), tensão do espécime e eletrões transmitidos.
As amostras são varridas por um feixe de eletrões acelerado por uma tensão que varia entre
0 e 40 kV, finamente focado através de um sistema de lentes eletromagnéticas. O feixe eletrónico
interage com a amostra, resultando na emissão de diversos tipos de radiação e eletrões, como os
eletrões secundários (SE) que são utilizados para a obtenção da imagem da amostra. Os SE são
eletrões da amostra que são excitados e são refletidos da superfície. Os SE provêm da interação
não-elástica entre os eletrões primários e os eletrões retro difundidos (BSE) com os eletrões de maior
energia de ligação. As imagens obtidas através da deteção de SE têm um forte contraste topográfico
devido à emissão dos BSE, cuja intensidade aumenta com o número atómico médio da amostra. Os
BSE identificam os eletrões da superfície da amostra com energia elevada. A emissão de BSE resulta
da interação elástica ou de perdas de energia. A utilização destes eletrões permite observar a
rugosidade das amostras em estudo devido ao efeito de sombra. Os BSE não são afetados por
efeitos locais de má condutividade dos materiais. A localização da amostra em relação ao detetor é
muito importante pois influencia a qualidade do contraste topográfico. Como os BSE provêm de
camadas profundas do material, a resolução das imagens depende fortemente do feixe incidente e do
número atómico médio local do material.

22
Uma amostra para poder ser analisada por SEM tem que satisfazer uma série de condições:
Ter boa condutividade elétrica superficial – caso contrário é necessário utilizar um
revestimento ultrafino de Au ou C;
Suportar vácuo;
Estabilidade física e química nas condições de observação e interação com o feixe
eletromagnético.
Consoante o tipo de deteção utilizado é possível caracterizar a amostra em termos de
topografia (SE e BSE), número atómico (BSE), propriedades cristalinas (BSE), composição química
elementar (BSE), campo magnético (BSE) e orientação cristalina local da amostra (BSE).
A esta técnica está muitas vezes associada a técnica dispersiva de raios-X (EDS), permitindo
a obtenção de imagens tridimensionais da amostra e mapas de composição dos elementos à
superfície da amostra [90, 91].
Procedimento experimental
As amostras de polímero foram fixas com uma fita de carbono num suporte metálico e
revestidas com ouro-paládio. As partículas que foram analisadas por SEM tinham entre 38 e 63 µm.
Foi utilizado um FEG-SEM equipado com espetrómetro de raios X de energia dispersiva, permitindo
obter imagens 3D da amostra. O equipamento estava também equipado com um detetor de BSE.
2.2.1.4. BET
A Teoria de BET permite descrever a adsorção física de moléculas de gás sobre uma
superfície sólida. Este método foi desenvolvido por Stephen Brunauer, Paul Hugh Emmett e Edward
Teller, sendo considerado uma extensão da Teoria de Langmuir.
Enquanto a Teoria de Langmuir relaciona a adsorção de uma única camada de moléculas de
adsorvato com a pressão do gás do meio a uma temperatura fixa, a teoria de BET introduz o conceito
de adsorção em multicamadas de acordo com três hipóteses:
1. As moléculas de gás são adsorvidas em camadas infinitamente;
2. As diferentes camadas não interagem entre si;
3. A teoria aplica-se a todas as camadas.
Tal como na Teoria de Langmuir, o método de BET requer a suposição de mecanismos
consequentes às três hipóteses consideradas.
1. A adsorção de cada uma das moléculas de gás na superfície do sólido está
relacionada com uma única energia de adsorção bem definida;
2. Uma molécula pode servir como sítio único de adsorção para outra molécula de uma
nova camada;
3. A camada mais externa está em equilíbrio com a fase gasosa, isto é, as taxas de
adsorção e desabsorção são semelhantes;
4. O processo de desabsorção é limitado cineticamente, sendo necessário ceder calor
de adsorção:
4.1. Este fenómeno é homogéneo em todas as moléculas da mesma camada;

23
4.2. O calor de adsorção referente à primeira camada é igual ao calor de adsorção
à superfície sólida (E1);
4.3. As camadas superiores são semelhantes entre si, podendo ser representadas
como espécies condensadas (fase líquida), pelo que o calor de adsorção
destas camadas é o calor de liquefação (El).
5. À pressão de saturação, o número de camadas adsorvidas tende para infinito; este
fenómeno é equivalente à superfície sólida estar imersa em fase líquida.
1
𝑣 [(𝑝𝑜
𝑝) − 1]
=𝑐 − 1
𝑣𝑚𝑐(
𝑝
𝑝𝑜
) +1
𝑣𝑚𝑐 (7)
Sendo p e p0 a pressão no equilíbrio e a pressão de saturação, respetivamente, dos
adsorvatos à temperatura de adsorção, v é a quantidade de gás adsorvido, vm é o volume de gás
adsorvido quando a superfície do sólido está completamente coberta por uma monocamada e c é a
constante de BET.
𝑐 = exp (𝐸1 − 𝐸𝐿
𝑅𝑇) (8)
Sendo E1 o calor de adsorção para a primeira camada e EL o calor de adsorção para as
camadas seguintes que é igual ao calor de liquefação.
A Equação de BET é uma isotérmica de adsorção e pode ser representada como uma linha
reta com 1
𝑣[(𝑝𝑜𝑝
)−1] em função de (
𝑝
𝑝0). Esta relação é linear para 0,05 < 𝑝 𝑝0⁄ < 0,35. Os valores do
declive (m) e da ordenada na origem (b) permitem determinar vm e a constante de BET, c.
𝑣𝑚 =1
𝑚 + 𝑏 (9)
𝑐 = 1 +𝑚
𝑏 (10)
A área de superfície total (Stotal) e a área de superfície específica (SBET) são determinadas
pelas Equações 11 e 12, respetivamente.
𝑆𝑡𝑜𝑡𝑎𝑙 =(𝑣𝑚𝑁𝑠)
𝑉 (11)
𝑆𝐵𝐸𝑇 =𝑆𝑡𝑜𝑡𝑎𝑙
𝑎 (12)
Sendo N o número de Avogadro, s a área da secção transversal de uma molécula de
adsorvato, V o volume molar do gás adsorvato e a a massa da amostra sólida ou adsorvente.

24
Procedimento experimental
A área de superfície e o diâmetro de poro das partículas poliméricas foram determinadas
através da adsorção de N2 a -196ºC, de acordo com o método BET.
2.2.1.5. Espectroscopia IV
As ligações químicas vibram a frequências específicas a níveis de energia bem definidos.
Estas frequências de vibração, ou de ressonância, são determinadas pela forma da molécula, pelos
seus níveis de energia e pela massa dos átomos que a constituem. Como as frequências de
ressonância de uma ligação química estão relacionadas com a força da ligação e com a massa dos
átomos em cada extremidade, cada frequência de vibração pode ser associada a um tipo específico
de ligação químico. Para que um modo vibracional seja ativo no infravermelho (IV) tem que estar
associado a variações do momento dipolar da molécula.
A radiação eletromagnética é constituída por um campo elétrico oscilante e um campo
magnético oscilante, perpendiculares um ao outro. O primeiro interfere com o momento dipolar da
molécula e esta interferência é detetada. Na Tabela 9 estão representadas as regiões
correspondentes a radiação no IV.
Tabela 9 - Radiação IV [92].
Região Comprimento de onda (µm) Número de onda (cm-1)
Próximo 0,78 – 2,5 12800 – 4000
Médio 2,5 – 50 4000 – 200
Longínquo 50 – 1000 200 - 10
As moléculas diatómicas simples têm apenas uma ligação que se pode estirar, enquanto as
moléculas com mais átomos têm maior número de ligações e as vibrações podem estar coordenadas.
Assim, podem-se observar absorções de IV a frequências características, relacionadas com os
grupos químicos que estão na constituição da molécula.
Na literatura é possível encontrar diversos quadros com as frequências típicas de absorção
de IV para cada ligação. Na Figura 7 estão representadas as principais bandas no espectro IV.
Figura 7 - Principais bandas no espectro IV.

25
Princípio de funcionamento
A amostra passa através de um feixe de luz infravermelha e é medida a quantidade de
energia absorvida pela amostra a cada comprimento de onda. A partir desta informação obtém-se o
espectro de transmissão ou de absorção, que mostra os comprimentos de onda do IV a que a
amostra absorve radiação. A partir do espectro obtido é possível interpretar que tipos de ligações
químicas estão presentes.
Só é possível utilizar esta técnica com ligações covalentes. Quanto mais ligações ativas no IV
existirem na molécula, mais complexo será o espectro.
Procedimento experimental
Durante a análise de FT-IR, as amostras de polímero foram analisadas sob a forma de
pastilhas de KBr. Os espetros foram obtidos na região de 4000 a 400 cm -1 com uma resolução de 2
cm-1.
2.2.2. Síntese de polímeros
2.2.2.1. Polímeros Molecularmente Impressos (MIPs)
A impressão molecular é uma técnica para obter locais de ligação à medida, conhecendo a
forma, tamanho e grupos funcionais da molécula molde [93]. Os MIPs podem ser sintetizados por
copolimerização dos monómeros funcionais e agente de reticulação na presença do molde. Após a
remoção das moléculas molde, são obtidos locais de ligação complementares ao molde em tamanho,
forma e funcionalidade química, onde se vão poder ligar as moléculas-alvo. Os MIPs apresentam
algumas vantagens como a síntese simples e elevada estabilidade em condições físicas e químicas
adversas, apresentando estabilidade em solventes orgânicos.
A impressão molecular pode ser conseguida através de ligações covalentes reversíveis [94]
ou através de interações não-covalentes entre os moldes e os monómeros funcionais [95]. Na
impressão covalente, os moldes estão ligados a monómeros apropriados, como o 4-vinilbenzilamina,
por ligações covalentes. Após a polimerização, essa ligação é quebrada e o molde é removido do
polímero. Quando o MIP se liga novamente à molécula utilizada como molde, forma-se a mesma
ligação covalente. Como as ligações covalentes são muito estáveis, é possível obter uma distribuição
mais homogénea de locais de ligação. No entanto, a impressão covalente é considerada menos
estável porque, para que a molécula-alvo se ligue ao polímero novamente é necessário que ocorram
rapidamente ligações covalentes reversíveis entre o molde e os monómeros funcionais. Como estas
ligações são muito fortes é complicado atingir o equilíbrio termodinâmico. No caso da impressão não-
covalente, os complexos molde-monómeros são formados por várias interações como pontes de
hidrogénio, interações iónicas, forças de van der Waals, interações π-π, entre outras. Após a
polimerização e remoção do molde, a matriz polimérica funcionalizada pode ligar-se novamente à
molécula-alvo (molde) através das mesmas ligações não-covalentes. Este processo é mais simples,
pois só requer que o molde e os monómeros sejam misturados num solvente apropriado [96]. Quando

26
o molde ligado covalentemente é removido, é possível que haja ligação não-covalente entre a
molécula-alvo e o polímero; esta abordagem é descrita como semi-covalente [97, 98].
Apesar das vantagens apresentadas, os MIPs estão associados a alguns problemas como
fuga do molde, incompatibilidade com meio aquoso, baixa capacidade de binding e transferência de
massa lenta.
Reagentes de polimerização
As reações de polimerização são processos complexos que podem ser afetados por diversos
factores, como o tipo e concentração do monómero, agente de reticulação e iniciador, temperatura e
tempo de polimerização, presença ou ausência de campo magnético e volume da mistura de
polimerização [93]. Para obter um MIP com boas propriedades é necessário estudar os efeitos das
propriedades de reconhecimento dos materiais poliméricos [99].
As moléculas molde devem satisfazer três requisitos [93]:
1. Não devem conter grupos que estejam envolvidos ou que possam impedir a
polimerização;
2. Devem ser quimicamente estáveis durante a reação de polimerização;
3. Devem ter grupos funcionais adequados para se ligarem aos monómeros funcionais.
O monómero serve para fornecer grupos funcionais capazes de formar complexos com a
molécula molde por interações covalentes ou não-covalentes. A força das interações entre o molde e
o monómero vão afetar a afinidade dos MIPs e determinar a precisão e seletivade dos locais de
reconhecimento [100-102]. Quanto mais forte for a interação, mais estável é o complexo, resultando
numa maior capacidade de ligação do MIP; a seleção dos monómeros funcionais é de extrema
importância [103]. Alguns dos monómeros mais utilizados são o ácido metacrílico (MAA), ácido
acrílico (AA), 2- ou 4-vinilpiridina (2- ou 4-VP), acrilamida, ácido trifluorometacrílico e 2-hidroxietil
metacrilato (HEMA). O MAA pode atuar como dador ou aceitador de pontes de hidrogénio e é
adequado para interações iónicas, pelo que tem sido usado como monómero funcional “universal”
[93, 104]. Normalmente, o rácio molar entre o molde e o monómero no processo de síntese afeta a
afinidade e a eficiência da impressão dos MIPs. Usando um rácio mais baixo haverão menos locais
de ligação no polímero devido ao menor número de complexos molde-monómero; pelo contrário, se o
rácio for demasiado elevado a ligação será menos específica, diminuindo a seletividade do binding. É
necessário otimizar o rácio molar entre o molde e o monómero para aumentar a eficiência da
impressão molecular [93].
O agente de reticulação serve para fixar os grupos funcionais dos monómeros funcionais em
torno das moléculas impressas e, assim, formar um polímero rígido altamente reticulado. Após a
remoção do molde, os locais de ligação devem ser complementares. O tipo e quantidade de agente
de reticulação influenciam a seletividade e a capacidade de ligação dos MIPs. Utilizando uma baixa
quantidade de agente de reticulação, os MIPs podem não conseguir manter as configurações da
cavidade estáveis devido ao baixo grau de reticulação; se for utilizada uma grande quantidade de
agente de reticulação, o número de locais de reconhecimento irá diminuir. Os agentes de reticulação
mais utilizados são o dimetacrilato de etilenoglicol (EGDMA), trimetacrilato de trimetilolpropano
(TRIM), N,N-metileno-bis-acrilamida (MBAA) e divinilbenzeno (DVB) [93].

27
O solvente porogénico, como o DCM, atua como porogénio e como solvente no processo de
separação. Este composto influencia a força de ligação entre os monómeros funcionais e os moldes.
Nos processos de polimerização não covalente, são normalmente utilizados solventes orgânicos
apróticos e com baixa polaridade, como acetonitrilo ou clorofórmio, para se obter uma boa eficiência
de impressão.
Processo de síntese
Os polímeros molecularmente impressos foram sintetizados pela Doutora Teresa Esteves no
Instituto Superior Técnico.
Figura 8 - Representação esquemática do processo de impressão molecular.
O DMAP foi utilizado como molde, o MAA como monómero, o EGDMA como agente de
reticulação e o azoisobutironitrilo (AIBN) como iniciador. Foram efetuadas várias sínteses com
diferentes quantidades de molde, monómero, agente de reticulação e iniciador.
Tabela 10 – Condições das reações de polimerização.
Reação Molde (mmol) Monómero
(mmol)
Agente de
reticulação
(mmol)
Condições de polimerização
MIP1 4 4 40 65ºC durante 24 horas
MIP2 4 4 40
12h a 40ºC, aumento de
5ºC de 30 em 30 minutos até aos
65ºC seguidos de 3h a 65ºC
MIP3 1 4 10
MIP4 1 4 20
NIP4 0 4 20
O DMAP foi utilizado como molde para a preparação dos MIPs, mas não foi utilizado na
preparação do polímero de controlo, o NIP. O MAA foi dissolvido em DCM, que atua como porogénio.
Para a preparação do MIP adicionou-se o molde à solução de MAA e deixou-se durante 5 minutos. O
agente de reticulação EGDMA e o iniciador AIBN foram adicionados à solução de polimerização, que
foi purgada com azoto seco. Os tubos de polimerização foram selados e deu-se início à
polimerização. A polimerização foi feita a diferentes temperaturas (Tabela 10) Após o término da
polimerização, os polímeros foram removidos dos tubos de polimerização e ligeiramente esmagados

28
com o auxílio de um almofariz e pilão. O molde foi então removido num Soxhlet com uma solução de
HCl 0,1 M em MeOH por 48h. O ácido que permaneceu nos polímeros foi lavado com MeOH num
aparelho de Soxhlet durante 24h. Os polímeros foram esmagados num moinho de bolas e
peneirados, sendo que se escolheu a gama de 38-63 µm para estudar as propriedades de binding
dos polímeros. Os polímeros foram então secos a 40ºC durante a noite. A mesma fração foi utilizada
para caracterizar química e morfologicamente os polímeros.
Os NIPs foram preparados da mesma maneira, mas sem a presença do molde.
Para estudar a capacidade destes polímeros se ligarem ao DMAP, estes foram colocados a
agitar em soluções de DMAP de 100 e 1000 ppm em DCM. Adicionaram-se 25 e 50 mg de polímero
em 1 mL de solução. As soluções foram agitadas a 60 rpm à temperatura ambiente. Após 24 horas
de agitação, foi determinada a concentração de DMAP em solução por HPLC-UV.
Para obter a isotérmica foram preparadas soluções de DMAP com concentrações de 10, 15,
20, 50, 150 e 250 ppm de DMAP e adicionaram-se 7,5 mg de MIP a 1,5 mL de solução de DMAP. As
soluções foram agitadas durante 24 horas a 60 rpm à temperatura ambiente, tendo sido depois
determinada a concentração de DMAP em solução por HPLC-UV.
Foram também realizados ensaios com o Meta para determinar a percentagem de ligação
não específica de API ao MIP. Para tal preparou-se uma solução com uma concentração de 100 ppm
de DMAP e 10000 ppm de Meta. Adicionaram-se 75 mg de MIP a 1,5 mL da solução preparada e
esta foi agitada durante 24 horas a 60 rpm à temperatura ambiente.
2.2.3. Processos
2.2.3.1. Recristalização
A recristalização é um processo de purificação de APIs. O API é isolado como fase sólida,
enquanto as impurezas se mantêm dissolvidas na fase líquida. Neste processo são criadas condições
termodinâmicas que levam as moléculas a aproximarem-se e a agruparem-se em estruturas
altamente organizadas, os cristais. As condições de operação não permitem obter cristais com 100%
de pureza, verificando-se inclusões de moléculas que também têm grande afinidade para o soluto.
O processo de cristalização inicia-se pela nucleação. A força motriz para a cristalização é a
sobressaturação da mistura líquida. Este estado é naturalmente muito instável, daí ser possível
ocorrer a nucleação. Para ocorrer a cristalização é necessário haver agitação ou convecção da
mistura líquida, provocando a aproximação e o choque entre as moléculas e, consequentemente
ocorre transferência de quantidade de movimento. Pequenos fragmentos dos primeiros cristais
(nucleação primária) podem transformar-se em novos núcleos (nucleação secundária). Após a
formação do núcleo, o cristal começa a crescer. A velocidade de agitação, grau de saturação e a
temperatura são alguns dos factores que condicionam a velocidade de crescimento dos cristais. Para
o sucesso deste processo é necessário conhecer as relações de equilíbrio entre a fase líquida e a
fase sólida.

29
Procedimento experimental
Na Figura 9 está representado o processo de recristalização.
Figura 9 - Processo de recristalização (adaptado de [106]).
Foi preparada uma solução de 50 mL com uma concentração de cerca de 1000 ppm de
DMAP (38,21 mg) e 10.000 ppm de Meta (560,89 mg) em DCM. A solução foi concentrada sob
pressão reduzida, no evaporador rotativo, até 10% do volume (5 mL). Foram adicionados 10 mL de
MeOH e a solução foi aquecida até aos 50ºC, sendo novamente concentrada até aos 5 mL. Este
procedimento foi repetido duas vezes, tendo-se observado formação de sólidos. A solução foi então
arrefecida até aos 20ºC durante 1 hora e depois até aos 10ºC. Após a solução estar a 10ºC, foi
agitada durante 2 horas a cerca de 220 rpm. O Meta foi filtrado e lavado com MeOH arrefecido a
10ºC. Adicionaram-se 0,3 g de carvão ativado, 10 mL de MeOH e 10 mL de DCM ao bolo húmido. O
API foi dissolvido a 50ºC, tendo-se agitado a mistura durante 30 minutos. O carvão ativado foi filtrado
e o equipamento de filtração foi lavado 2 vezes com 2 mL de DCM, que foi adicionado à solução de
API. A solução foi novamente concentrada sob pressão reduzida até 5 mL, tendo-se obtido uma
suspensão. Foram adicionados 10 mL de MeOH e a mistura foi novamente concentrada, sob pressão
reduzida, até aos 5 mL. A mistura foi arrefecida até aos 23ºC durante uma hora e depois arrefecida
até aos 10ºC. Seguiu-se agitação da amostra a 10ºC durante 2 horas. O Meta foi filtrado e lavado
com MeOH a 10ºC. O produto obtido foi seco na estufa a 70ºC durante 24 horas. Foi medida a
concentração de Meta e DMAP em todas as etapas por HPLC-UV.
Para verificar o efeito da temperatura no processo de cristalização repetiu-se o processo
descrito, mas as recristalizações foram efetuadas a 4ºC (em vez de 10ºC). Neste caso, a solução
inicial foi preparada com 500,80 mg de Meta e 56,02 mg de DMAP.
2.2.3.2. Resinas
Com o intuito de melhorar o rendimento do processo de recristalização foram estudadas
diversas resinas com diferentes características (Tabela 11).
Tabela 11 - Propriedades das resinas estudadas.

30
Resina Grupo funcional Tipo
AG 50W-X2 Ácido sulfónico Ácido forte
Carvão ativado
Adsorção
CG400
Base forte
IRA458 Sal de amónio quaternário Base forte
IRA68 Amina terciária Base fraca
IRC50 Ácido carboxílico Ácido fraco
IRC86 Ácido carboxílico Ácido fraco
XAD16 Não tem grupos funcionais ionizáveis Adsorção
XAD7 Não tem grupos funcionais ionizáveis Adsorção
Grande parte das resinas estudadas é de permuta iónica, que consiste na troca
estequiométrica de um ião por outro. O principal componente estrutural da matriz das resinas de troca
iónica é o poliestireno que forma uma estrutura reticulada com o divinilbenzeno, impedindo a
dissolução das resinas. Além disso, consoante a quantidade de DVB presente na estrutura, determina
a porosidade da matriz, o grau de inchamento e a mobilidade dos iões na resina [107, 108].
Dependendo da componente funcional da resina, i.e. do grupo funcional iónico que está fixo na
matriz, as resinas podem ser do tipo ácido (catiónicas) ou do tipo base (aniónicas), forte ou fraca
[107].
A adsorção é um fenómeno em que as moléculas da fase líquida ficam retidas na superfície
de um sólido, o adsorvente, por forças de van der Waals, formando uma camada (monocamada) ou
multicamadas de moléculas. A capacidade de adsorção depende do tipo e tamanho dos poros, da
sua distribuição e da natureza da superfície do adsorvente [107].
Tabela 12 - Grupos funcionais das resinas de troca iónica mais comuns.
Tipo de resina Grupo funcional
Catiónica do tipo ácido forte Sulfónico: R-SO3 H
Catiónica do tipo ácido fraco Carboxílico: R – COOH
Aniónica do tipo I base forte
Aniónica do tipo II base forte
Aniónica do tipo base fraca
Amino: R-N+(CH3)3Cl–
R-N+(CH3)2(CH2)2OHCl
R-N+(CH3)
As isotérmicas de adsorção permitem descrever o equilíbrio de fases subjacente à partição do
soluto entre a fase fluída e a fase adsorvida. Para traçar uma isotérmica de adsorção representa-se a
quantidade de soluto adsorvido por quantidade de adsorvente em função da concentração do soluto
no equilíbrio.
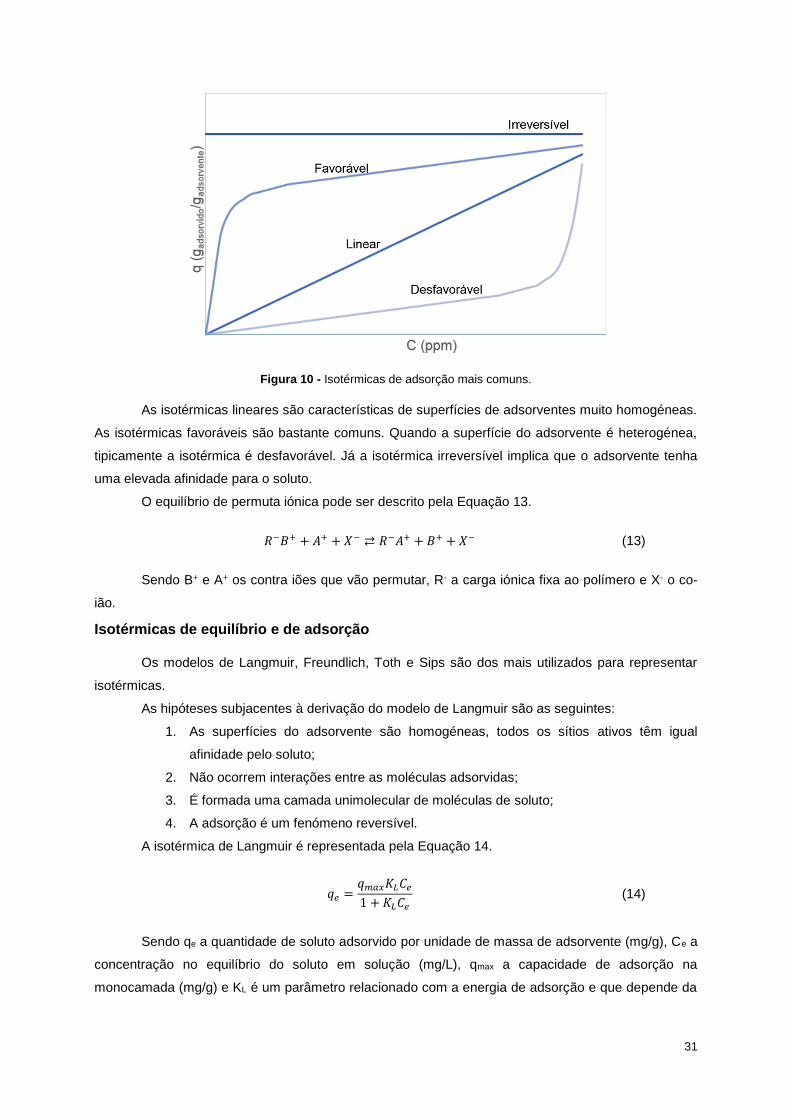
31
Figura 10 - Isotérmicas de adsorção mais comuns.
As isotérmicas lineares são características de superfícies de adsorventes muito homogéneas.
As isotérmicas favoráveis são bastante comuns. Quando a superfície do adsorvente é heterogénea,
tipicamente a isotérmica é desfavorável. Já a isotérmica irreversível implica que o adsorvente tenha
uma elevada afinidade para o soluto.
O equilíbrio de permuta iónica pode ser descrito pela Equação 13.
𝑅−𝐵+ + 𝐴+ + 𝑋− ⇄ 𝑅−𝐴+ + 𝐵+ + 𝑋− (13)
Sendo B+ e A+ os contra iões que vão permutar, R- a carga iónica fixa ao polímero e X- o co-
ião.
Isotérmicas de equilíbrio e de adsorção
Os modelos de Langmuir, Freundlich, Toth e Sips são dos mais utilizados para representar
isotérmicas.
As hipóteses subjacentes à derivação do modelo de Langmuir são as seguintes:
1. As superfícies do adsorvente são homogéneas, todos os sítios ativos têm igual
afinidade pelo soluto;
2. Não ocorrem interações entre as moléculas adsorvidas;
3. É formada uma camada unimolecular de moléculas de soluto;
4. A adsorção é um fenómeno reversível.
A isotérmica de Langmuir é representada pela Equação 14.
𝑞𝑒 =𝑞𝑚𝑎𝑥𝐾𝐿𝐶𝑒
1 + 𝐾𝐿𝐶𝑒
(14)
Sendo qe a quantidade de soluto adsorvido por unidade de massa de adsorvente (mg/g), Ce a
concentração no equilíbrio do soluto em solução (mg/L), qmax a capacidade de adsorção na
monocamada (mg/g) e KL é um parâmetro relacionado com a energia de adsorção e que depende da

32
temperatura (L/mg). Os valores de KL e qmax podem ser obtidos por linearização da Equação 26,
obtendo-se a Equação 15.
1
𝑞𝑒
=1
𝑞𝑚𝑎𝑥𝐾𝐿𝐶𝑒
+1
𝑞𝑚𝑎𝑥
(15)
Representado 1 𝑞𝑒⁄ em função de 1 𝐶𝑒⁄ obtém-se uma reta com declive 1 𝑞𝑚𝑎𝑥𝐾𝐿𝐶𝑒⁄ e
ordenada na origem 1 𝑞𝑚𝑎𝑥⁄ .
A isotérmica de Freundlich permite descrever a adsorção em superfícies heterogéneas, sendo
representada pela Equação 16.
𝑞𝑒 = 𝐾𝐹𝐶𝑒1 𝑛⁄
(16)
Onde KF e n são constantes. O valor de n representa o grau de heterogeneidade da
superfície, sendo superior a 1 quando a isotérmica é favorável e inferior a 1 quando a isotérmica é
desfavorável. Este modelo assume que a quantidade adsorvida tende para infinito, correspondendo à
formação de multicamadas de moléculas adsorvidas. Tal como no caso anterior, é possível linearizar
a equação de Freundlich para determinar os valores de KF e n.
log 𝑞𝑒 = log 𝐾𝐹 +1
𝑛log 𝐶𝑒 (17)
A representação de log 𝑞𝑒 em função de log 𝐶𝑒 permite obter uma reta com declive 1/n e
ordenada na origem log 𝐾𝐹. A isotérmica de Freundlich só deve ser utilizada numa faixa de
concentrações moderadas de soluto.
A isotérmica de Sips é descrita pela Equação 18.
𝑞𝑒 =𝑞𝑚𝑎𝑥𝐾𝐿𝐶𝑒
1 𝑛⁄
1 + 𝐾𝐿𝐶𝑒1 𝑛⁄
(18)
Quando a concentração de adsorvato é baixa, esta equação reduz-se à isotérmica de
Freundlich; pelo contrário, quando a concentração de adsorvato é elevada, descreve o processo de
adsorção em monocamada, que é uma característica da isotérmica de Langmuir. Se 𝑛 = 1 obtém-se
a equação de Langmuir.
A isotérmica de Toth é expressa pela Equação 19.
𝑞𝑒 =𝑞𝑚𝑎𝑥𝐶𝑒
(𝑏 + 𝐶𝑛)1 𝑛⁄ (19)
Sendo b uma constante relacionada com a afinidade para o adsorvente.

33
Procedimento experimental
Inicialmente foi estudada a influência do pH na quantidade de DMAP adsorvido por cada
resina. O pKa do DMAP é 9,7; foram selecionados valores de pH acima e abaixo deste valor de modo
a verificar se o estado protonado ou desprotonado do DMAP influenciava a adsorção. Os diferentes
valores de pH podem também interferir com os grupos funcionais das resinas utilizadas.
Preparou-se uma solução de DMAP de 1000 ppm em água. Foram adicionados 20 mg de
cada uma das resinas a 4 mL de solução. Foram testados 4 valores de pH: 11.55, 10.88, 8.18 e 6.53.
O pH foi ajustado com soluções de HCl 1M e NaOH 1M. As amostras foram agitadas a cerca de 220
rpm, à temperatura ambiente, durante 24 horas, tendo-se depois determinado a concentração de
DMAP em solução. De seguida foi estudada a influência da temperatura no processo de adsorção.
Para tal, foi novamente preparada uma solução de DMAP de 1000 ppm e adicionaram-se 20 mg de
resina a 4 mL de solução. Neste caso, as amostras foram agitadas durante 24h a 220 rpm à
temperatura de 25ºC, 35ºC e 45ºC. Após 24 horas foi determinada a quantidade de DMAP que
permanecia em solução. Após a seleção da temperatura ideal para a adsorção, foram determinadas
as isotérmicas. As isotérmicas foram determinadas de duas formas: variando a quantidade de resina
que é colocada um contacto com uma solução de 1000 ppm e colocando 20 mg de resina em
contacto com soluções com diferentes concentrações. No primeiro caso, foi preparada uma solução
de DMAP de 1000 ppm. A 4 mL de solução foram adicionados 10, 20, 40, 60, 80, 100, 200 e 400 mg
de resina e estas soluções foram agitadas durante 24 horas a 220 rpm. Na segunda abordagem
foram adicionados 20 mg de resina a 4 mL de soluções de DMAP com concentrações de 100, 250,
500, 750 e 1000 ppm e estas foram agitadas durante 24 horas a 220 rpm. Ao fim de 24 horas, foi
determinada a concentração de DMAP em solução. Finalmente, foi estudada a cinética do processo
de adsorção com a resina que apresentava melhor capacidade de remover o DMAP da solução. Para
tal, foi preparada uma solução de 1000 ppm e adicionada uma quantidade de resina que foi
determinada através das isotérmicas de adsorção. A solução foi continuamente agitada a 220 rpm
durante 24 horas, tendo sido retiradas amostras de cerca de 1,5 mL em intervalos de tempo
determinados. Foram tiradas amostras aos 2, 4, 6, 10, 15, 30, 60, 120, 240, 480 e 1440 minutos e foi
determinada a quantidade de DMAP adsorvida em cada um dos casos. A concentração de DMAP foi
determinada por HPLC-UV depois das amostras terem sido filtradas com filtro de seringa.
Este estudo foi repetido com soluções de DMAP numa mistura de H2O e MeOH (1:1), DMAP
em MeOH e MPTS em MeOH.

34
2.2.3.3. Nanofiltração (OSN)
A nanofiltração é um processo de separação por membranas que se rege pelos princípios de
exclusão molecular. O solvente passa através de uma membrana semipermeável através da
aplicação de pressão [110].
As propriedades da membrana e o fluxo utilizado são parâmetros fundamentais para um
correto funcionamento do processo. A equação geral do fluxo de permeado, JP (L/m2/h), é dado pela
Equação 32.
𝐽𝑃 = 𝐿𝑃(Δ𝑃 − Δ𝜋) (32)
Sendo LP o coeficiente de permeabilidade hidráulica (L/h/m2/bar), Δ𝑃 é a pressão aplicada e
Δ𝜋 o gradiente de pressão osmótica transmembranar. O caudal de permeado, QP (L/h), pode também
ser utilizado para determinar o fluxo de permeado (Equação 33).
𝐽𝑃 =𝑄𝑃
𝐴 (33)
Sendo A a área total da membrana (m2).
Este processo caracteriza-se pela rejeição de moléculas de acordo com o tamanho, carga e
forma da molécula em questão [111]. A rejeição da membrana pode ser determinada pela Equação
34.
𝑅 = (1 −𝐶𝑃
𝐶𝐴
) × 100 (34)
Sendo CP a concentração do permeado e CA a concentração na alimentação.
Uma grande desvantagem das membranas é a colmatação, que se caracteriza pela criação
gradual de uma camada de solutos retidos sobre a sua superfície. Esta camada representa uma nova
barreira e vai diminuir progressivamente o fluxo de permeado [112, 113].
O estudo de nanofiltração, operada em modo de diafiltração, foi sustentado por um modelo
teórico tendo por base balanços de massa e rejeição dos solutos a separar.

35
3. Resultados e Discussão
3.1. Estratégia
O trabalho desenvolvido nesta tese procura estudar um dos processos de purificação de APIs
mais utilizados na indústria farmacêutica, a recristalização. Durante este processo, uma parte
significativa do API é perdido nas soluções de lavagem descartadas no isolamento de cristais, pelo
que é interessante procurar melhorar a eficiência dos processos através de estratégias que
recuperem seletivamente API destas soluções, sem sacrificar a pureza final do API. Nesse sentido,
foram testadas soluções mais convencionais – resinas de troca iónica e de adsorção.
Alternativamente, o processo de recristalização, ou passos deste processo, podem ser substituídos
por métodos de separação mais eficientes. Seguindo esta estratégia foram desenvolvidas novas
soluções baseadas em MIPs.
A quantidade de GTI permitida nos APIs é determinada através do valor de TTC (1,5 µg/dia) e
da dose máxima diária (g/dia) [22]. Considerando que um paciente consome 5 mg de Meta por dia, a
quantidade máxima de DMAP é dada pela Equação 35.
𝑄𝑢𝑎𝑛𝑡𝑖𝑑𝑎𝑑𝑒 𝑑𝑒 𝐷𝑀𝐴𝑃 𝑝𝑒𝑟𝑚𝑖𝑡𝑖𝑑𝑎 (𝑝𝑝𝑚) =1,5 𝜇𝑔 𝑑𝑒 𝐷𝑀𝐴𝑃/𝑑𝑖𝑎
5 𝑚𝑔 𝑑𝑒 𝑀𝑒𝑡𝑎/𝑑𝑖𝑎×
1000 𝑚𝑔
1 𝑔= 300 𝑝𝑝𝑚
= 0,3 𝑚𝑔𝐷𝑀𝐴𝑃
𝑔𝑀𝑒𝑡𝑎
(35)
Assim, começando o processo com uma solução com 10 g/L de Meta e 1 g/L de DMAP, é
necessário remover 99,7% do GTI. No entanto, sabe-se que quanto maior a pureza desejada, maior é
a perda de API. Assim, é necessário haver um balanço entre o grau de pureza e a perda de API
[106].
3.2. Recristalização
O processo de recristalização foi efetuado a 10ºC e a 4ºC de forma a quantificar o efeito da
temperatura na eficiência de separação e rendimento do processo (secção 2.2.3.1, patente
WO9800437). Este processo inclui duas recristalizações e uma adsorção em carvão em
metanol/diclorometano, sendo necessário proceder a trocas de solventes por evaporação entre
operações. A Figura 11 representa o balanço de massa do processo de recristalização a 10ºC.
Figura 11 - Balanço de massa do processo de recristalização a 10ºC.

36
𝑄𝑢𝑎𝑛𝑡𝑖𝑑𝑎𝑑𝑒 𝑑𝑒 𝐷𝑀𝐴𝑃 𝑛𝑜 𝑀𝑒𝑡𝑎 =0,64 𝑚𝑔 𝐷𝑀𝐴𝑃
160,46 𝑚𝑔 𝑀𝑒𝑡𝑎= 3,99
𝑚𝑔𝐷𝑀𝐴𝑃
𝑔𝑀𝑒𝑡𝑎
O balanço de massa do processo de recristalização a 4ºC está representado na Figura 12. A
utilização de uma temperatura mais baixa durante a recristalização permite diminuir as perdas de API,
sendo possível recuperar 58,28% do Meta inicial, mas sacrificando a quantidade de remoção de GTI
para um valor de 78,92%, correspondendo a um quociente de 40,46 mgDMAP/gMeta, valor bastante
superior ao valor alvo.
Figura 12 - Balanço de massa do processo de recristalização a 4ºC.
𝑄𝑢𝑎𝑛𝑡𝑖𝑑𝑎𝑑𝑒 𝑑𝑒 𝐷𝑀𝐴𝑃 𝑛𝑜 𝑀𝑒𝑡𝑎 =11,81 𝑚𝑔 𝐷𝑀𝐴𝑃
291,86 𝑚𝑔 𝑀𝑒𝑡𝑎= 40,46
𝑚𝑔𝐷𝑀𝐴𝑃
𝑔𝑀𝑒𝑡𝑎
Na Figura 13 está representada a quantidade de API recuperado em função da remoção de
GTI.
Figura 13 - Quantidade de Meta recuperado em função da quantidade de DMAP removido.
A temperatura influencia a nucleação e o crescimento dos cristais através da manipulação da
solubilidade e supersaturação da amostra [114-117]. A solubilidade do API diminui com a temperatura
sendo a fração de API nos cristais maior e perdendo-se apenas 0,35% do Meta nas soluções de
lavagem na 1ª recristalização a 4ºC, mas 15,75% quando esta recristalização decorre a 10ºC.
No entanto, a uma temperatura mais alta, a formação da rede cristalina acontece de forma
mais lenta, permitindo uma remoção de DMAP mais eficiente a 10ºC, enquanto este GTI parece ficar
preso na rede cristalina quando a primeira recristalização acontece a 4ºC. Caso a recristalização a
10ºC não tenha ainda atingido o equilíbrio, aumentar o tempo de recristalização poderia permitir um
aumento do rendimento em termos de cristais obtidos, sem comprometer a pureza destes. Também a
1ª Recristalização
Adsorção com carvão
2ª Recristalização
1ª Recristalização
Adsorção com carvão 2ª
Recristalização
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100
% A
PI
recu
pe
rad
o
% GTI removido
10ºC 4ºc

37
adição de cristais de Meta poderia conduzir a um processo de recristalização mais rápido e
organizado, eventualmente expulsando o DMAP da rede cristalina.
Na 2ª recristalização, as perdas de Meta são mais uma vez maiores para temperaturas mais
altas, tendo sido perdido 12,51% do Meta na recristalização a 10ºC e 8,48% na recristalização a 4ºC.
No caso da recristalização a 10ºC, 97,46% do DMAP foi removido na 1ª recristalização, pelo
que os passos seguintes de uso do carvão e a segunda recristalização conduzem a maiores perdas
de API sem que haja um aumento da sua pureza. A 4ºC só se conseguiu remover 33,85% do DMAP;
este resultado advém do facto de os cristais de Meta se formarem rapidamente promovendo
inclusões de DMAP nos cristais e não permitindo a sua difusão para a solução. Assim, a segunda
recristalização tem um papel importante na remoção do GTI.
Observando o gráfico da Figura 13 verifica-se que se a recristalização for efetuada a uma
temperatura mais elevada é possível remover uma maior quantidade de DMAP, enquanto se a
recristalização acontecer a uma temperatura mais baixa consegue-se recuperar uma maior
quantidade de GTI, mas os cristais serão mais impuros. A uma temperatura mais baixa o processo de
nucleação e formação dos cristais é mais rápido, tornando os cristais mais impuros.
Na Figura 14 e na Figura 15 é possível observar a remoção de DMAP e de Meta ao longo do
processo, respetivamente.
Figura 14 - API removido ao longo do processo a 10ºC (painel superior) e a 4ºC (painel inferior).
0
100
200
300
400
500
600
Início Recristalização1
Carvão Recristalização2
Ma
ss
a d
e A
PI
(mg
)
Estágio do processo
API recuperado API perdido nas lavagens Outras perdas de API
0
100
200
300
400
500
600
Início Recristalização1
Carvão Recristalização2
Ma
ss
a d
e A
PI
(mg
)
Estágio do processo
API recuperado API perdido nas lavagens Outras perdas de API

38
No processo de adsorção com carvão a quantidade de Meta perdido é considerável em
ambas as situações. Neste caso, a adsorção com carvão foi feita a 50ºC em ambos os processos.
Neste estágio é perdido 51,98% de Meta, correspondendo a 64,78% do Meta total perdido, e apenas
0,60% de DMAP no processo cuja cristalização foi feita a 10ºC. No segundo caso, perdeu-se 32,75%
de Meta, correspondendo a 78,76% do Meta total perdido e 19,40% de DMAP. Destes resultados é
possível concluir que o carvão consegue adsorver tanto o Meta como o DMAP, não sendo o mais
indicado para este estágio. Seria de todo o interesse procurar alternativas, capazes de remover o
DMAP da solução sem remover uma grande quantidade de Meta, para substituir este passo.
Figura 15 - GTI recuperado ao longo do processo de recristalização a 10ºC (painel superior) e a 4ºC (painel
inferior).
3.1. Polímeros de impressão molecular
Em ambos os processos de recristalização há uma grande perda de Meta no passo de
adsorção com carvão em diclorometano. Para a indústria farmacêutica, perdas de Meta desta
magnitude são inaceitáveis, já que isso representa um custo. Além de se perder uma grande
quantidade de Meta, o API após este estágio está mais enriquecido em DMAP. Uma alternativa para
diminuir as perdas de Meta e aumentar a remoção de DMAP é utilizar polímeros seletivos para o GTI.
Nesse sentido, foram sintetizados MIPs específicos para DMAP.
0
5
10
15
20
25
30
35
40
45
Início Recristalização 1 Carvão Recristalização 2
Ma
ss
a d
e G
TI
(mg
)
Estágio do processo
GTI recuperado GTI removido nas lavagens Outras perdas de GTI
0
10
20
30
40
50
60
Início Recristalização 1 Carvão Recristalização 2
Ma
ss
a d
e G
TI
(mg
)
Estágio do processo
GTI recuperado GTI removido nas lavagens Outras perdas de GTI

39
Tal como foi mencionado na secção 2.2.2, inicialmente foram sintetizados vários polímeros
para verificar qual o rácio agente de reticulação/molde mais indicado. Foram realizados ensaios para
quantificar a remoção de DMAP, em soluções de DCM, pelos diferentes MIPs.
Figura 16 - Remoção do DMAP com os diferentes MIPs.
A título de exemplo, para a análise da influência dos diferentes reagentes de polimerização
será estudo o caso em que temos uma solução de DMAP a 100 ppm em contacto com 50 mg de
polímero.
Analisando a Tabela 10 e o gráfico da Figura 16 é possível concluir que a quantidade de
molde e agente de reticulação presentes na síntese dos MIPs influencia a capacidade de remoção do
DMAP.
Aumentando a quantidade de agente de reticulação, de 10 mmol (MIP3) para 40 mmol (MIP1
e MIP2), verifica-se um aumento na quantidade de DMAP adsorvido de 71%, no caso do MIP2 e 66%
no caso do MIP1, para 84%, no caso do MIP3. Quando a quantidade de agente de reticulação é
demasiado baixa, como é o caso do MIP3, o grau de reticulação é baixo e impede que o polímero
mantenha a configuração da cavidade para o reconhecimento estável [93]. Também o rácio
molde/monómero influencia a quantidade de DMAP que é removido da solução. Quando se utiliza
mais monómero em relação à quantidade de molde (1:4), como no MIP4, a quantidade de DMAP
removido é maior, 93%, do que quando é usada a mesma quantidade de monómero e molde (4:4),
como é o caso do MIP1 (66%). Esta diferença pode dever-se ao facto de o rácio molde:monómero
utilizado influenciar a quantidade de locais de ligação disponíveis bem como a seletividade da ligação
[93].
Além dos reagentes de polimerização, também as condições da reação de polimerização
influenciam a capacidade do polímero de remover o DMAP. Observando a Figura 16 é possível
concluir que o MIP1 é que o que apresenta pior capacidade de remoção do DMAP. Este resultado
pode dever-se ao facto de a reação de polimerização ter ocorrido inteiramente a 65ºC. Quando foi
utilizado um gradiente de temperatura, as cadeias poliméricas puderam formar-se convenientemente
em torno das cavidades onde terá lugar o reconhecimento molecular. No caso do MIP1, é possível
que não tenham sido criadas as condições necessárias para que o mesmo acontecesse.
De um modo geral, para as soluções de 100 ppm e 1000 ppm, o MIP4 é o que apresenta
melhores resultados, pelo que foi o selecionado para os restantes estudos. Foi preparado o NIP4,
0
20
40
60
80
100
MIP4 MIP3 MIP2 MIP1
% d
e D
MA
P a
ds
orv
ido
Polímero
100 ppm 50 mg 100 ppm 25 mg 1000 ppm 50 mg 1000 ppm 25 mg

40
correspondente ao MIP4, que tem exatamente a mesma proporção de agentes de polimerização
utilizada na síntese do MIP4 sem o molde. Na Figura 17 está representada a diferença na quantidade
de DMAP adsorvido pelo MIP4 e NIP4.
Figura 17 - Quantidade de DMAP removida pelo MIP4 e pelo NIP4.
O NIP4 consegue adsorver o DMAP, uma vez que tem o grupo carboxílico dos monómeros
disponíveis para formar ligações não covalentes no DMAP. No entanto, é possível observar que a
quantidade de DMAP removido pelo MIP4 é maior do que com o NIP4. Este resultado demonstra que
a introdução do DMAP durante a preparação do MIP influencia a seletividade da ligação. Foi
determinado o valor-p, tendo-se obtido um valor de 0,0007; pode assim afirmar-se que estes
resultados têm significância estatística.
Foi também estudada a especificidade do MIP4 para o DMAP. Foi preparada uma solução
com 100 ppm de DMAP e 10.000 ppm de Meta e 75 mg de MIP em 1,5 mL de solução.
Figura 18 - Quantidade de Meta e DMAP removidos com o MIP4.
O MIP4 foi eficaz na remoção do DMAP (98,7%), perdendo-se 9,63% de Meta. Esta diferença
permite afirmar que o MIP4 é específico para o DMAP, não tendo capacidade de adsorver Meta.
0
20
40
60
80
100
MIP4 NIP4
% d
e D
MA
P r
em
ovid
o
Polímero
0
20
40
60
80
100
% d
e r
em
oçã
o d
e M
eta
e
DM
AP
MIP4
DMAP Meta

41
3.1.1. Caracterização dos polímeros MIP4 e NIP4
O desempenho dos MIPs não resulta apenas da interação do molde com o monómero
funcional, mas também das suas características morfológicas que incluem a forma e tamanho da
partícula e a textura porosa, que podem afetar as propriedades de reconhecimento molecular. Para a
caracterização morfológica do MIP e do NIP foi utilizada a técnica de SEM. Nas Figuras 19 e 20 estão
representadas as micrografias obtidas para o MIP4 e NIP4, respetivamente.
Figura 19 - Micrografias de SEM do MIP4.
Figura 20 - Micrografias de SEM do NIP4.
Observando as micrografias do MIP4 e do NIP4 é possível observar diferenças consideráveis
na sua morfologia, o que indica que o molde afeta a morfologia do polímero. O MIP4 apresenta
clusters de polímero maiores.
Os polímeros foram também caracterizados por FT-IR; os espectros obtidos para o MIP4 e
NIP4 estão representados na Figura .

42
Figura 21 - Espectro de FT-IR para o MIP4 e NIP4.
O MIP4 e o NIP4 têm espetros IV semelhantes indicando que a sua constituição estrutural é
semelhante.
No espetro IV é possível observar absorção devido ao estiramento do grupo OH do MAA
(2960 cm-1), estiramento do grupo carbonilo (1730 cm-1), estiramento do grupo C-O (1150 cm-1) e
diversas vibrações C-H. Apesar de possuírem a mesma estrutura, o processo de impressão
molecular confere especificidade, o que é comprovado pela maior quantidade de DMAP adsorvido
pelo MIP4 comparativamente com o NIP4.
A área BET dos polímeros foi obtida a partir dos dados de adsorção usando 0,162 nm2 como
área de secção reta para as moléculas de azoto adsorvidas. Os resultados obtidos estão
representados na Tabela 13.
Tabela 13 - Análise BET do MIP4 e NIP4.
Área de superfície BET
(m2·g-1)
Volume do poro
(cm3·g-1)
Diâmetro médio do
poro (nm)
MIP4 207 0.024 5.6
NIP4 242 0.025 7.3
Os resultados representados na Tabela 13 indicam que quando o molde é introduzido na
polimerização, a área BET e o diâmetro médio do poro diminuem. Não se verifica uma diferença
considerável no volume do poro. A diferença nos valores pode dever-se às cavidades do MIP4 após a
remoção do molde. Estes valores sugerem também que as diferenças encontradas na capacidade de
remoção de DMAP pelo MIP4 e NIP4 devem-se ao facto de o MIP4 ser específico para o DMAP e
não por ter maior área de superfície.

43
3.1.2. Isotérmica de adsorção do MIP4
Foi obtida a isotérmica do DMAP com o MIP4, tendo sido colocados 75 mg de MIP4 em
contacto com 1,5 mL de soluções de DMAP com concentrações de 10, 15, 20, 50, 150 e 250 ppm.
Figura 22 - Isotérmica do MIP4.
Verificou-se que o modelo de Freundlich é o mais indicado para representar a isotérmica do
MIP4. Pode assim concluir-se que a quantidade de DMAP adsorvido pelo MIP tende para infinito,
havendo formação de multicamadas. Além disso, este modelo reflete também a heterogeneidade das
partículas de MIP4, resultante do processo de esmagamento e peneiração do polímero.
Os restantes modelos e os parâmetros obtidos para cada modelo teórico estão representados
no Anexo 2.
3.1.3. Substituição do carvão ativado por MIPs
Após a 1ª recristalização a 4ºC a razão entre DMAP e Meta é de 74,23 mgDMAP/gMeta,
aumentando para 78,12 mgDMAP/gMeta após o passo de adsorção com carvão
Figura 23 - Substituição do carvão ativado por MIPs.
Substituindo o carvão ativado por MIPs é possível, em apenas um passo, baixar a razão
mgDMAP/gMeta para 18,86 mgDMAP/gMeta. Este valor ainda está bastante longe do objectivo final (0,3
mgDMAP/gMeta), pelo que este passo pode ser repetido tantas vezes quantas forem necessárias.
3.2. Resinas de troca iónica e adsorventes
A recristalização a 10ºC é muito eficiente na remoção de DMAP, mas como foi discutido a
perda de Meta durante este passo não é aceitável. Assim, uma possibilidade de melhoramento do
processo seria a recuperação do Meta descartado nas soluções de lavagem e realimentá-lo ao
processo de purificação, após troca de solventes de metanol por diclorometano por destilação. No
entanto, estas soluções de lavagem têm uma razão de 421,55 mgDMAP/gMeta, consideravelmente
0
1
2
3
4
5
6
3,50 4,00 4,50 5,00 5,50 6,00 6,50
qe
(mg
DM
AP/g
MIP
)
Ceq (ppm)Isotérmica de adsorção Aproximação de Freundlich
44
superior à razão 100 mgDMAP/gMeta da solução alimentada ao processo, sendo necessário que esta
solução de metanol seja purificada para remover DMAP até atingir uma razão mgDMAP/gMeta inferior a
100.
Figura 24 - Processo de recuperação do Meta das soluções de lavagem.
Processos de purificação usando resinas de troca iónica e adsorventes encontram-se
desenvolvidos à escala industrial e podem ser rapidamente implementados pela indústria para
purificar o Meta perdido no licor mãe, usando resinas e adsorventes disponíveis comercialmente.
O solvente destas soluções de lavagem é o MeOH, mas a maioria das resinas estão
concebidas para serem utilizadas em solução aquosa. Assim, um dos desafios para implementar a
estratégia proposta decorre do desempenho destes processos de adsorção em metanol. De forma a
ganhar sensibilidade para a eficiência deste sistema, numa primeira fase foram utilizadas soluções
aquosas de DMAP. Depois usou-se uma mistura metanol:água (1:1) e, finalmente, metanol como
solvente nos ensaios de quantificação de eficácia das resinas na remoção deste GTI.
3.2.1. DMAP em água
Começou por se estudar a influência do pH no processo de adsorção. O DMAP é uma base
forte e é protonado no azoto (sp2) que é perpendicular ao sistema π (Figura 25) [118]., podendo o
seu grau de protonação ter um efeito na disponibilidade de cargas nesta molécula para interação com
resinas de troca iónica.
Figura 25 - Protonação do DMAP [118].
O DMAP tem um pKa de 9,7. O valor de pKa indica o valor de pH ao qual existe um equilíbrio
entre a fase protonada e a fase desprotonada. O DMAP é uma base forte pelo que, mesmo num meio
com uma baixa concentração de H+, vai continuar a exercer a sua função de base e mantém-se
ligado aos protões. Quando o pH é inferior ao pKa, o DMAP está maioritariamente protonado, e se o
pH for inferior ao pKa, o DMAP está maioritariamente protonado (Figura 25).

45
Figura 26 - Variação da concentração de DMAP protonado em função do pH.
A reta de calibração utilizada para determinar as concentrações de DMAP está representada
no Anexo 1.
Foram estudados quatro valores de pH – 11.55, 10.88, 8.18 e 6.53 (Figura ), estando o DMAP
maioritariamente protonado nos dois primeiras casos e maioritariamente desprotonado nos dois
últimos.
Figura 27 - Influência do pH na remoção do DMAP de uma solução aquosa.
As resinas AG 50-X2, Amberlite IRC50, Amberlite IRC86 bem como o carvão ativado foram
eficientes na remoção do DMAP, tal como seria expectável, dada a potencial interação iónica entre os
grupos eletronegativos das resinas (como por exemplo, os grupos carboxílicos) e os grupos
eletropositivos (aminas). As resinas mencionadas são catiónicas do tipo ácido forte (AG 50W-X2),
sendo o seu grupo funcional ácido sulfónico, e catiónicas do tipo ácido fraco (Amberlite IRC50 e
Amberlite IRC86), com ácido carboxílico como grupo funcional. O processo de adsorção é mais eficaz
quando o DMAP está desprotonado. O processo de adsorção é mais eficaz quanto o DMAP está
desprotonado, tendo o grupo amina disponível para estabelecer pontes de hidrogénio ou ligações ião-
dipolo com a resina. O tipo de ligação depende do estado de protonação da resina.
No caso do carvão ativado, também se verifica que a adsorção é mais eficaz quando o pH é
mas elevado. O pH e a força iónica da solução determinam a capacidade de adsorção e a natureza
do processo de adsorção [119].
0
20
40
60
80
100
0 2 4 6 8 10 12 14 16
% D
MA
P p
roto
na
do
e n
ão
p
roto
na
do
pH
DMAP protonado DMAP não protonado
0
20
40
60
80
100
AG 50W-X2 IRC50 IRC86 Carvão XAD16 XAD7 CG400 IRA458 IRA68
% D
MA
P a
ds
orv
ido
Resina
pH 6,53 pH 8,18 pH 10,88 pH 11,55

46
Assim, é possível concluir que o DMAP deve estar desprotonado para conseguir removê-lo
mais eficazmente da solução.
Após se ter estudado o efeito do pH na capacidade de adsorção das resinas, foi estudado o
efeito da temperatura (Figura ).
Figura 28 - Influência da temperatura na remoção do DMAP de uma solução aquosa.
Pela observação da Figura conclui-se que a temperatura utilizada não afeta particularmente
o processo de adsorção.
Uma vez que a temperatura não tem influência sobre o processo de adsorção, foram
determinadas as isotérmicas de adsorção a 25ºC. Foram estudados os modelos de Langmuir,
Freundlich, Toth e Sips. Foram colocados 20 mg de resina em 4 mL de solução aquosa de DMAP
com concentrações de 100, 250, 500, 750 e 1000 ppm. As isotérmicas obtidas estão representadas
nas Figuras 29 a 30. Nas Figuras estão representados apenas os modelos que se aplicam às
isotérmicas obtidas, estando as restantes representadas no Anexo.2.
Figura 29 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução aquosa de
DMAP.
0
10
20
30
40
50
60
70
80
90
100
AG 50W-X2
Carvão IRC50 IRC86
% D
MA
P a
ds
orv
ido
Resina
25ºC 35ºC 45ºC
0
50
100
150
200
250
0 5 10 15 20 25
Cap
acid
ad
e d
e a
ds
orç
ão
(m
gD
MA
P/g
resin
a)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Freundlich

47
Figura 30 - Isotérmica de adsorção com carvão ativado, variando a concentração inicial de uma solução aquosa
de DMAP.
Figura 31 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma solução
aquosa de DMAP.
Figura 32 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma solução
aquosa de DMAP.
Os parâmetros dos modelos teóricos obtidos estão descritos no Anexo 2.
0
20
40
60
80
100
120
140
160
0 20 40 60 80 100
Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
0
50
100
150
200
0 10 20 30 40 50 60
Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
0
50
100
150
200
0 10 20 30 40 50
Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
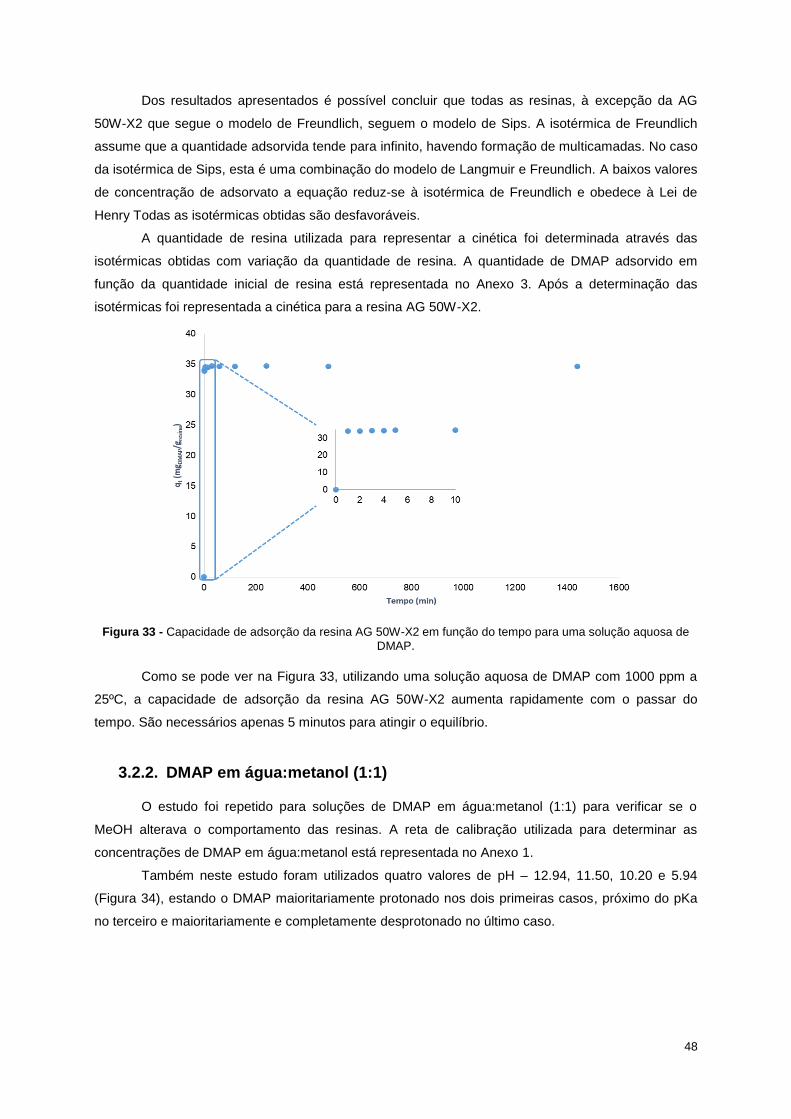
48
Dos resultados apresentados é possível concluir que todas as resinas, à excepção da AG
50W-X2 que segue o modelo de Freundlich, seguem o modelo de Sips. A isotérmica de Freundlich
assume que a quantidade adsorvida tende para infinito, havendo formação de multicamadas. No caso
da isotérmica de Sips, esta é uma combinação do modelo de Langmuir e Freundlich. A baixos valores
de concentração de adsorvato a equação reduz-se à isotérmica de Freundlich e obedece à Lei de
Henry Todas as isotérmicas obtidas são desfavoráveis.
A quantidade de resina utilizada para representar a cinética foi determinada através das
isotérmicas obtidas com variação da quantidade de resina. A quantidade de DMAP adsorvido em
função da quantidade inicial de resina está representada no Anexo 3. Após a determinação das
isotérmicas foi representada a cinética para a resina AG 50W-X2.
Figura 33 - Capacidade de adsorção da resina AG 50W-X2 em função do tempo para uma solução aquosa de
DMAP.
Como se pode ver na Figura 33, utilizando uma solução aquosa de DMAP com 1000 ppm a
25ºC, a capacidade de adsorção da resina AG 50W-X2 aumenta rapidamente com o passar do
tempo. São necessários apenas 5 minutos para atingir o equilíbrio.
3.2.2. DMAP em água:metanol (1:1)
O estudo foi repetido para soluções de DMAP em água:metanol (1:1) para verificar se o
MeOH alterava o comportamento das resinas. A reta de calibração utilizada para determinar as
concentrações de DMAP em água:metanol está representada no Anexo 1.
Também neste estudo foram utilizados quatro valores de pH – 12.94, 11.50, 10.20 e 5.94
(Figura 34), estando o DMAP maioritariamente protonado nos dois primeiras casos, próximo do pKa
no terceiro e maioritariamente e completamente desprotonado no último caso.

49
Figura 34 - Influência do pH na remoção do DMAP em água:metanol.
Tal como anteriormente, as resinas AG 50-X2, Amberlite IRC50, Amberlite IRC86 bem como
o carvão ativado foram as mais eficientes na remoção do DMAP. Verifica-se uma diminuição na
remoção de DMAP no caso do carvão ativado, o que pode ser justificado pelo facto de o carvão
ativado ser capaz de adsorver MeOH, diminuindo a capacidade de adsorver DMAP. É também de
notar que as resinas continuam a adsorver o DMAP mais facilmente quando este está desprotonado.
O facto de não haver adsorção ao valor de pH mais elevado não era esperado. A valores de pH
elevados podem estar a formar-se espécies iónicas que competem com o DMAP pela ligação à
resina. Outra possibilidade é que a valores de pH muito alcalinos, tanto o DMAP como a resina estão
completamente desprotonados pelo que não há protão para fazer a interação entre as resinas e o
DMAP. É também possível concluir que o MeOH compete com o DMAP para se ligar às resinas.
Após se ter estudado o efeito do pH na capacidade de adsorção das resinas, foi estudado o
efeito da temperatura (Figura 35).
Figura 35 - Influência da temperatura na remoção do DMAP em água:metanol.
Ao contrário do que se observou nas soluções aquosas, verifica-se aqui que a temperatura
pode influenciar o processo de remoção do DMAP. No caso da resina AG 50W-X2, a sua capacidade
de remoção do DMAP diminui significativamente a 35ºC, enquanto com o carvão diminui aos 45ºC e
com a Amberlite IRC86 diminui a 25ºC. Esta diferença pode dever-se ao facto de a temperatura
alterar as constantes de equilíbrio.
0
20
40
60
80
100
AG 50W-X2 IRC50 IRC86 Carvão XAD16 XAD7 CG400 IRA458 IRA68
% D
MA
P a
dso
rvid
o
Resina
pH 5,94 pH 10,20 pH 11,50 pH12,94
0
10
20
30
40
50
60
70
80
90
100
AG 50W-X2
Carvão IRC50 IRC86
% D
MA
P a
ds
orv
ido
Resina
25ºC 35ºC 45ºC

50
Neste caso, as isotérmicas de adsorção foram determinadas à temperatura em que
removeram uma maior quantidade de DMAP; 25ºC para a resina AG 50W-X2, 35ºC para o carvão
ativado e 45ºC para as resinas Amberlite IRC50 e Amberlite IRC86.
Foram colocados 20 mg de resina em 4 mL de solução aquosa de DMAP com concentrações
de 100, 250, 500, 750 e 1000 ppm. As isotérmicas obtidas estão representadas nas Figuras 36 a 37.
Figura 36 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução de DMAP
em água:metanol.
Figura 37 - Isotérmica de adsorção com carvão ativado, variando a concentração inicial de uma solução de
DMAP em água:metanol.
0
20
40
60
80
100
120
140
160
0 2 4 6 8 10 12
Ca
pa
cid
ad
e d
e a
ds
orç
ão
(m
gD
MA
P/g
res
ina)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
0
10
20
30
40
50
60
70
80
90
0 50 100 150 200 250 300 350
Cap
ac
ida
de
de
ad
so
rçã
o
(mg
DM
AP/g
res
ina)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
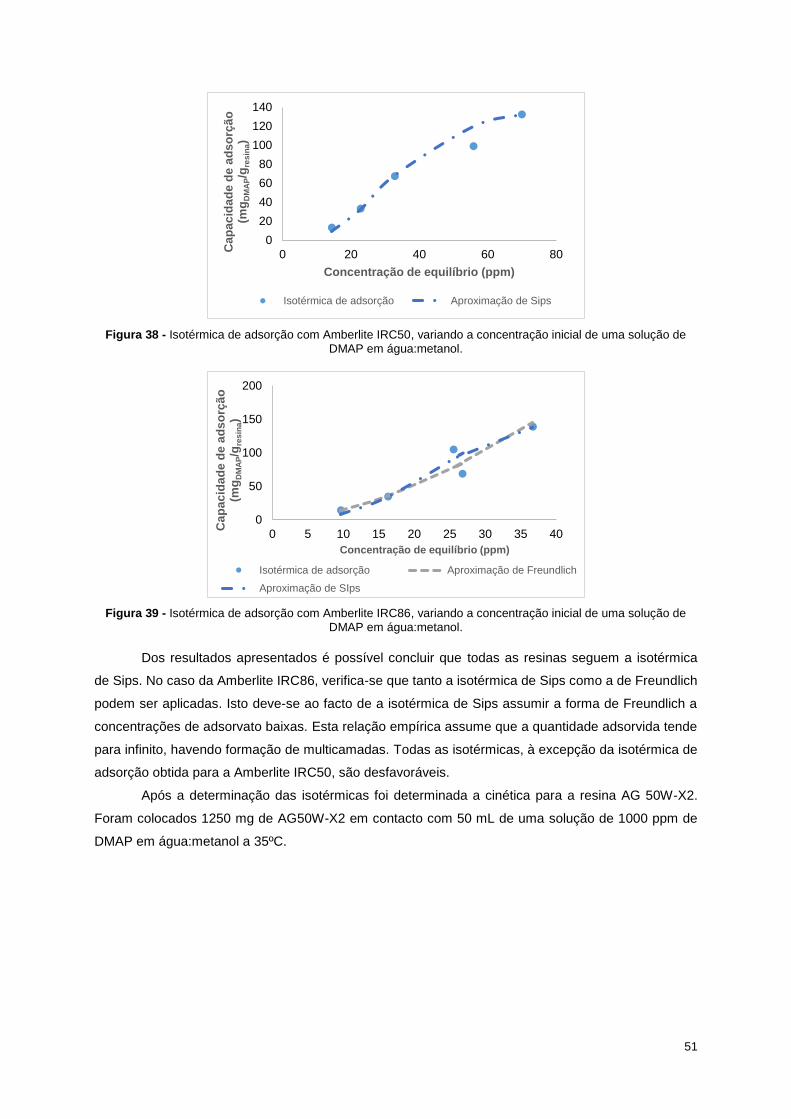
51
Figura 38 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma solução de
DMAP em água:metanol.
Figura 39 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma solução de
DMAP em água:metanol.
Dos resultados apresentados é possível concluir que todas as resinas seguem a isotérmica
de Sips. No caso da Amberlite IRC86, verifica-se que tanto a isotérmica de Sips como a de Freundlich
podem ser aplicadas. Isto deve-se ao facto de a isotérmica de Sips assumir a forma de Freundlich a
concentrações de adsorvato baixas. Esta relação empírica assume que a quantidade adsorvida tende
para infinito, havendo formação de multicamadas. Todas as isotérmicas, à excepção da isotérmica de
adsorção obtida para a Amberlite IRC50, são desfavoráveis.
Após a determinação das isotérmicas foi determinada a cinética para a resina AG 50W-X2.
Foram colocados 1250 mg de AG50W-X2 em contacto com 50 mL de uma solução de 1000 ppm de
DMAP em água:metanol a 35ºC.
0
20
40
60
80
100
120
140
0 20 40 60 80
Ca
pa
cid
ad
e d
e a
ds
orç
ão
(m
gD
MA
P/g
res
ina)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Sips
0
50
100
150
200
0 5 10 15 20 25 30 35 40
Ca
pa
cid
ad
e d
e a
ds
orç
ão
(m
gD
MA
P/g
res
ina)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Freundlich
Aproximação de SIps

52
Figura 40 - Capacidade de adsorção da resina AG 50W-X2 em função do tempo.
Tal como se verificou anteriormente, a capacidade de adsorção da resina AG 50W-X2
aumenta muito rapidamente com o tempo. Ao fim de 1 minuto já é atingida a sua capacidade de
adsorção máxima.
3.2.3. DMAP em MeOH
Finalmente foi efetuado o estudo em MeOH para analisar a possibilidade de esta solução ser
utilizada para purificar o Meta nas soluções de lavagem. A reta de calibração utilizada para
determinar as concentrações de DMAP em MeOH está representada no Anexo 1.
Neste estudo foram apenas utilizadas as resinas que foram eficazes na remoção do DMAP
nos ensaios anteriores – AG 50W-X2, Amberlite IRC50 e Amberlite IRC86 e o carvão ativado. Os
valores de pH estudados foram 12.05, 10.08, 8.50 e 5.52 (Figura 41).
Figura 41 - Influência do pH na remoção do DMAP em MeOH.
No ensaio realizado com MeOH verifica-se uma diminuição geral da quantidade de DMAP
adsorvido, o que confirma que o uso de MeOH com estas resinas é desafiante. A quantidade de
DMAP removido pelo carvão ativado diminuiu drasticamente, pelo que não foi utilizado nos restantes
-20
0
20
40
60
80
100
AG 50W-X2 IRC50 IRC86 Carvão
% D
MA
P a
dso
rvid
o
ResinapH 5,52 pH 8,50 pH 10,08 pH 12,05

53
ensaios. As resinas conseguem remover mais DMAP quando o pH da solução está próximo do pKa
do DMAP; quando o DMAP está completamente desprotonado (pH 12,05); quando está
completamente protonado (pH 5,52), a quantidade de remoção do DMAP é consideravelmente mais
baixa. Novamente se verifica que a valores de pH na ordem de 12, a quantidade de DMAP adsorvido
é praticamente nula. Este resultado sugere, de facto, a existência de espécies iónicas a valores de pH
mais elevados que competem com o DMAP para se ligarem à resina. Também aqui se confirma que
esta tendência não é aplicável ao carvão ativado.
Após se ter estudado o efeito do pH na capacidade de adsorção das resinas, foi estudado o
efeito da temperatura (Figura 42).
Figura 42 - Influência da temperatura na remoção do DMAP MeOH.
Tal como no ensaio em que foi utilizada a água como solvente, não se verifica qualquer
influência da temperatura no processo de remoção do DMAP.
As isotérmicas de adsorção foram obtidas a 25ºC. Foram colocados 20 mg de resina em 4 mL
de solução aquosa de DMAP com concentrações de 100, 250, 500, 750 e 1000 ppm. As isotérmicas
obtidas estão representadas nas Figuras 43 a 44.
Figura 43 - Isotérmica de adsorção com AG 50W-X2, variando a concentração inicial de uma solução de DMAP
em MeOH.
0
10
20
30
40
50
60
70
80
90
100
AG 50W-X2 IRC50 IRC86
% D
MA
P a
ds
orv
ido
Resina
25ºC 35ºC 45ºC
0
50
100
150
200
250
300
75 80 85 90Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilibrio (ppm)
Isotérmica de adsorção Aproximação de Freundlich
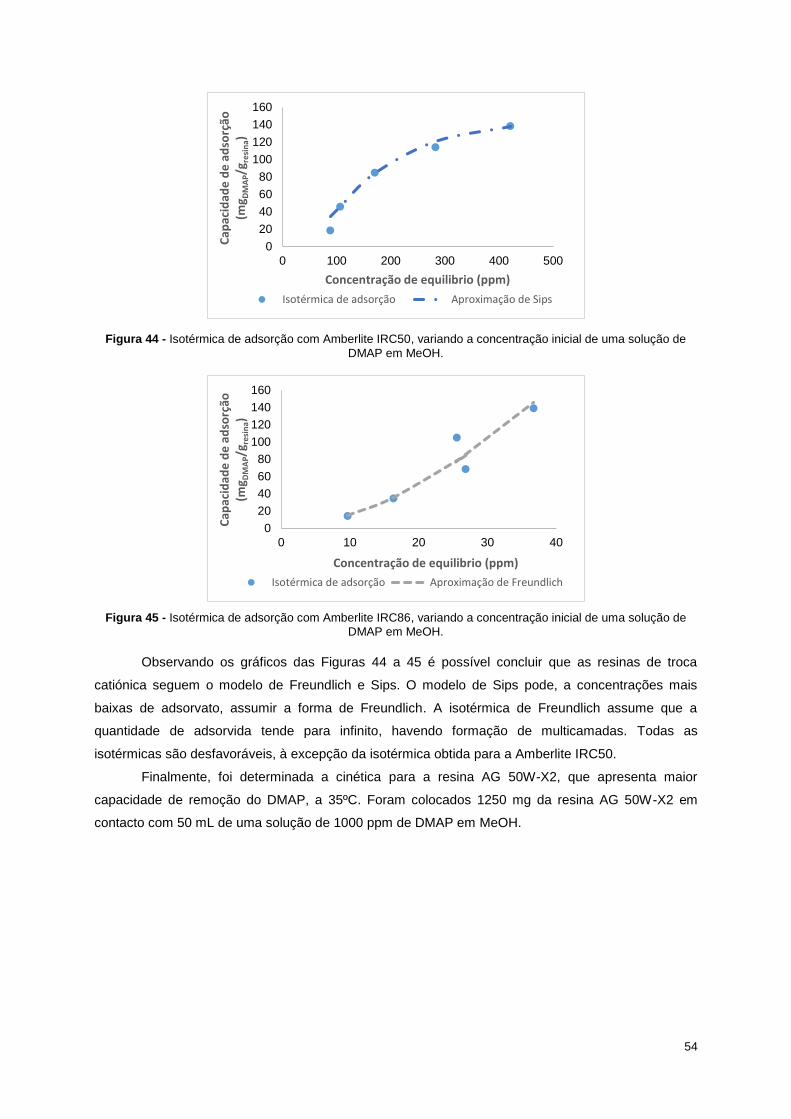
54
Figura 44 - Isotérmica de adsorção com Amberlite IRC50, variando a concentração inicial de uma solução de
DMAP em MeOH.
Figura 45 - Isotérmica de adsorção com Amberlite IRC86, variando a concentração inicial de uma solução de
DMAP em MeOH.
Observando os gráficos das Figuras 44 a 45 é possível concluir que as resinas de troca
catiónica seguem o modelo de Freundlich e Sips. O modelo de Sips pode, a concentrações mais
baixas de adsorvato, assumir a forma de Freundlich. A isotérmica de Freundlich assume que a
quantidade de adsorvida tende para infinito, havendo formação de multicamadas. Todas as
isotérmicas são desfavoráveis, à excepção da isotérmica obtida para a Amberlite IRC50.
Finalmente, foi determinada a cinética para a resina AG 50W-X2, que apresenta maior
capacidade de remoção do DMAP, a 35ºC. Foram colocados 1250 mg da resina AG 50W-X2 em
contacto com 50 mL de uma solução de 1000 ppm de DMAP em MeOH.
0
20
40
60
80
100
120
140
160
0 100 200 300 400 500
Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilibrio (ppm)
Isotérmica de adsorção Aproximação de Sips
0
20
40
60
80
100
120
140
160
0 10 20 30 40
Cap
acid
ade
de
ad
sorç
ão
(mg D
MA
P/g
resi
na)
Concentração de equilibrio (ppm)
Isotérmica de adsorção Aproximação de Freundlich

55
Figura 46 - Capacidade de adsorção em função do tempo.
Em MeOH, a capacidade de adsorção da resina AG 50W-X2 vai aumentando ao longo do
tempo, sendo necessárias 2 horas para que o equilíbrio seja atingido.
3.2.4. Purificação das soluções provenientes da recristalização
Das resinas mencionadas anteriormente capazes de remover o DMAP em MeOH, foram
selecionadas duas com diferentes grupos funcionais e tentou perceber-se se estas também removiam
Meta (Figura ). A solução utilizada neste ensaio foi a proveniente da 1ª filtração da recristalização a
10ºC, que tinha uma concentração de 2955,56 ppm de DMAP e 7011,11 ppm de Meta (421,55
mgDMAP/gMeta).
Figura 47 - Quantidade de Meta e DMAP removidos com as resinas AG 50W-X2 e Amberlite IRC50.
Pela observação do gráfico da Figura facilmente se concluiu que as resinas são ideais para
remoção do DMAP sem haver perda significativa de Meta. A resina AG50W-X2 removeu 92,94% do
DMAP e apenas 1,86% Meta, permitindo obter uma solução com 29,28 mgDMAP/gMeta. No caso da
Amberlite IRC50 esta não foi tão eficaz na remoção de DMAP (63,36%), mas também não removeu
qualquer Meta da solução; foi possível obter uma solução com 154,55 mgDMAP/gMeta.
Uma vez que se pretendia conseguir uma razão mgDMAP/gMeta inferior a 100 para permitir a
sua reintrodução no processo de recristalização, a resina AG 50W-X2 é a mais indicada.
O processo de recristalização sugerido está representado na Figura 53.
0
5
10
15
20
25
30
35
40
45
0 500 1000 1500 2000
qt(m
gD
MA
P/g
res
ina)
Tempo (min)
-20
0
20
40
60
80
100
AG50 IRC50%D
MA
P e
Me
ta r
em
ovid
os
Resina
Meta DMAP

56
Figura 48 - Recristalização com resinas e MIPs.
3.3. Nanofiltração (OSN)
O estudo de nanofiltração foi feito com base num modelo teórico. Os dados utilizados foram
retirados da literatura para a membrana GMT-oFN-2 [106]. A rejeição de DMAP é de 16,5% e de
Meta é de 99,1%; a membrana é eficaz a reter o API, que tem um peso molecular muito elevado,
enquanto o GTI consegue atravessar a membrana. Na Figura 49 está representada a quantidade de
Meta e DMAP removidos com a membrana em função do número de diavolumes.
Figura 49 - Remoção de DMAP e Meta por OSN em função do número de diavolumes.
Pela análise do gráfico da Figura 49 é possível concluir que com 6 diavolumes se consegue
remover 99,85% do DMAP com uma perda de 5,29% de Meta, permitindo obter uma solução com
0,16 mgDMAP/gMeta (inferior a 0,3 mgDMAP/gMeta) No entanto, um elevado número de diavolumes implica
um elevado consumo de solventes e uma maior perda de API, contribuindo ambos para um aumento
do custo do processo. Pode então combinar-se nanofiltração com MIPs, permitindo diminuir o número
de diavolumes (Figura 50). Com apenas 3 diavolumes é possível remover 96,09% do DMAP
perdendo apenas 2,68% de Meta, conseguindo obter uma solução com 4,02 mgDMAP/gMeta. Seguindo-
se um passo de adsorção com MIPs, é possível baixar a razão mgDMAP/gMeta para 0,33, que é o
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
% d
e G
TI
rem
ovid
o e
AP
I p
erd
ido
Diavolumes
Remoção de GTI Perdas de API
20% perdas de API
95% de remoção de GTI

57
objetivo inicial de modo a cumprir o TTC. No entanto, o passo de adsorção com MIPs pode ainda ser
repetido.
Figura 50 - Nanofiltração seguida da utilização de MIPs.
3.4. Mitigação do MPTS
Além do DMAP, havia um outro genotóxico na reação modelo utilizada – o MPTS. Os
sulfonatos não são genotóxicos muito óbvios, uma vez que se formam em reações secundárias na
presença de MeOH. Tal como para o DMAP, é necessário diminuir a quantidade deste genotóxico em
solução de modo a ser cumprido o TTC.
3.4.1. Resinas de troca iónica e adsorventes
Tal como no DMAP, o primeiro estudo foi a influência do pH no processo de adsorção e na
resina. O MPTS é um ácido forte, estando esta espécie essencialmente na sua forma neutra.
Figura 51 – Estrutura química do MPTS.
A reta de calibração utilizada para determinar as concentrações de MPTS está representada
no Anexo 1.
Foram estudados dois valores de pH – 9.67e 1.18 (Figura 52).

58
Figura 52 – Quantidade de MPTS adsorvido pelas resinas.
Pela observação da Figura 52 é possível concluir que apenas a resina Amberlite IRA68 é
eficaz na remoção do MPTS da solução. A resina Amberlite IRA68 é uma resina aniónica do tipo base
fraca, sendo o seu grupo funcional uma amina terciária: 𝑅 − 𝑁+(𝐶𝐻3)2. Verifica-se que a variação do
pH não influencia de forma significativa a permuta iónica entre o MPTS e a resina uma vez que, como
foi mencionado anteriormente, o MPTS está desprotonado nesta gama de pH. Uma vez que as
aminas terciárias têm um pKa entre 9 e 12, a resina estará protonada, podendo ligar-se ao MPTS. No
entanto, verifica-se um ligeiro aumento da quantidade de DMAP adsorvido com a diminuição do pH e
consequente aproximação ao pKa do MPTS.
Após se ter estudado o efeito do pH na capacidade de adsorção das resinas, foi estudado o
efeito da temperatura (Figura 53).
Figura 53 - Influência da temperatura na remoção do MPTS em MeOH.
Verifica-se que, apesar de a quantidade de MPTS removido aumentar com a temperatura,
este aumento não é muito significativo.
A isotérmica de adsorção foi obtida a 45ºC, temperatura à qual a quantidade de MPTS
removido é maior. Foram colocados 20 mg de resina em 4 mL de solução aquosa de MPTS com
concentrações de 100, 500, 750 e 1000 ppm. A isotérmica obtida está representada na Figura 54.
0
20
40
60
80
100
AG 50W-X2 IRC50 IRC86 Carvão XAD16 XAD7 CG400 IRA458 IRA68
% D
MA
P a
dso
rvid
o
Resina
pH 1,18 pH 9,67
0
20
40
60
80
100
IRA68
% d
e D
MA
P a
ds
orv
ido
Resina
25ºC 35ºC 45ºC
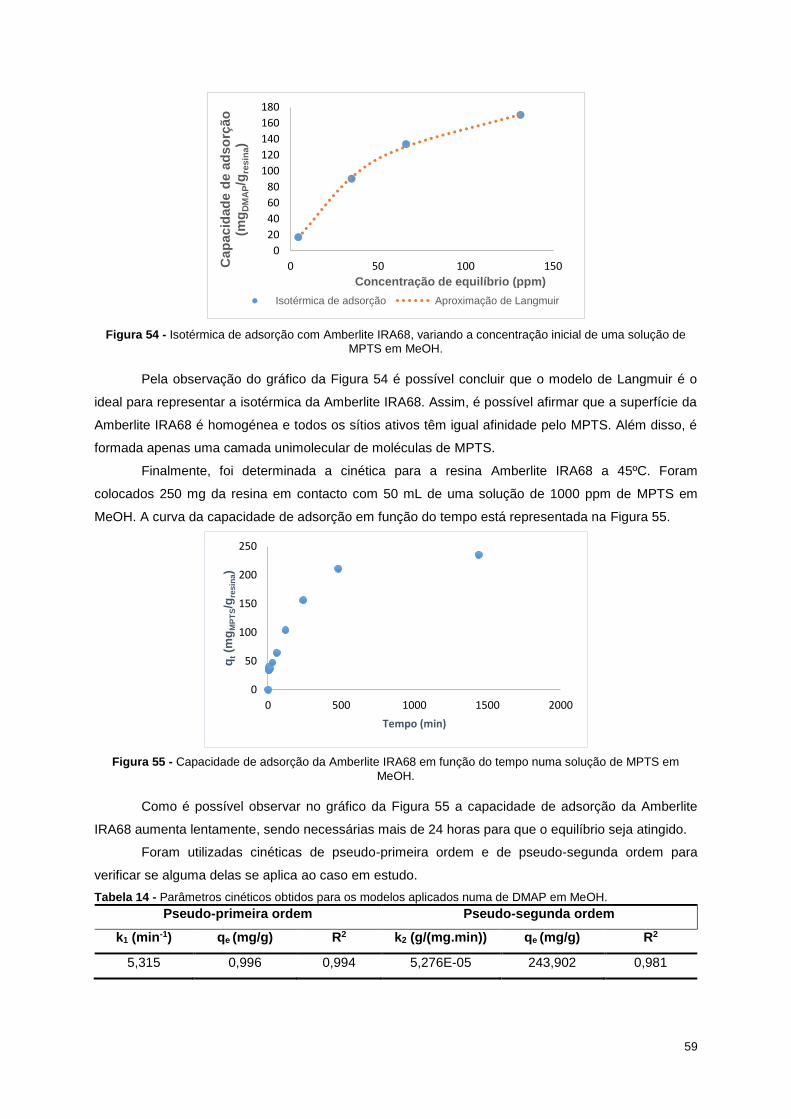
59
Figura 54 - Isotérmica de adsorção com Amberlite IRA68, variando a concentração inicial de uma solução de
MPTS em MeOH.
Pela observação do gráfico da Figura 54 é possível concluir que o modelo de Langmuir é o
ideal para representar a isotérmica da Amberlite IRA68. Assim, é possível afirmar que a superfície da
Amberlite IRA68 é homogénea e todos os sítios ativos têm igual afinidade pelo MPTS. Além disso, é
formada apenas uma camada unimolecular de moléculas de MPTS.
Finalmente, foi determinada a cinética para a resina Amberlite IRA68 a 45ºC. Foram
colocados 250 mg da resina em contacto com 50 mL de uma solução de 1000 ppm de MPTS em
MeOH. A curva da capacidade de adsorção em função do tempo está representada na Figura 55.
Figura 55 - Capacidade de adsorção da Amberlite IRA68 em função do tempo numa solução de MPTS em
MeOH.
Como é possível observar no gráfico da Figura 55 a capacidade de adsorção da Amberlite
IRA68 aumenta lentamente, sendo necessárias mais de 24 horas para que o equilíbrio seja atingido.
Foram utilizadas cinéticas de pseudo-primeira ordem e de pseudo-segunda ordem para
verificar se alguma delas se aplica ao caso em estudo.
Tabela 14 - Parâmetros cinéticos obtidos para os modelos aplicados numa de DMAP em MeOH.
Pseudo-primeira ordem Pseudo-segunda ordem
k1 (min-1) qe (mg/g) R2 k2 (g/(mg.min)) qe (mg/g) R2
5,315 0,996 0,994 5,276E-05 243,902 0,981
0
20
40
60
80
100
120
140
160
180
0 50 100 150Cap
acid
ad
e d
e a
dso
rção
(m
gD
MA
P/g
resin
a)
Concentração de equilíbrio (ppm)
Isotérmica de adsorção Aproximação de Langmuir
0
50
100
150
200
250
0 500 1000 1500 2000
qt(m
gM
PT
S/g
res
ina)
Tempo (min)

60
A analisar pelos valores dos coeficientes de correlação poderia afirmar-se que ambas as
cinéticas são adequadas para descrever a cinética da Amberlite IRA68. No entanto apenas o valor de
qe determinado a partir da linearização da equação de pseudo-segunda ordem se aproxima do valor
experimental (243,91 mgDMAP/gresina).
Todos os modelos teóricos aplicados, bem como os parâmetros obtidos para cada isotérmica
e as isotérmicas determinadas variando a quantidade inicial de resina estão representados nos
Anexos 2 e 3.

61
4. Conclusões
Durante o desenvolvimento desta tese foram estudados processos convencionais de
purificações de API – recristalização e resinas de troca iónica e adsorventes – e novos processos –
MIPs e OSN. É muito importante que estes processos sejam viáveis para a indústria farmacêutica;
isto é, os processos devem ser capazes de remover os GTIs sem que haja uma perda considerável
de API, já que as perdas de API não são economicamente exequíveis para a indústria.
No processo de recristalização verificou-se que se perde uma grande quantidade de Meta ao
longo do processo, e a pureza dos cristais obtidos não satisfaz os requisitos da indústria
farmacêutica. Neste sentido, é necessário encontrar alternativas para aumentar o rendimento do
processo de recristalização. Um dos estágios críticos do processo de recristalização é o estágio de
adsorção com carvão. É neste estágio que se perde mais de metade do Meta total perdido ao longo
do processo. Os polímeros de impressão molecular podem ser utilizados em DCM, pelo que são uma
boa alternativa ao carvão neste estágio, sendo capazes de remover o DMAP em solução sem que a
perda de Meta seja muito significativa. As resinas de troca iónica e adsorventes foram estudadas
como alternativas para purificar as soluções provenientes das lavagens após as filtrações. Foi
possível identificar três resinas de troca iónica capazes de remover o DMAP eficazmente de uma
solução em MeOH – a AG 50W-X2, Amberlite IRC50 e Amberlite IRC86. Estas resinas, além de
serem eficazes na remoção do DMAP, não removem uma quantidade significativa de Meta.
Além da recristalização foi também estudada OSN como meio de purificação de APIs. Este
processo permite remover uma grande quantidade de DMAP e recuperar quase todo o Meta
alimentado ao processo. Mesmo assim, nem todo o DMAP passa através da membrana
contaminando o Meta que também ficou retido. Através da utilização de MIPs, é possível recuperar o
Meta que tenha ficado retido na membrana. O Meta perdido no filtrado pode ser purificado através da
realimentação desta solução ao processo de OSN.

62
Referências
1. Székely, G.S., Miriam; Ferreira, Frederico C.; Gil, Marco; Heggie, William, Sources of
genotoxic impurities in drug synthesis: a systematic review. To be submitted.
2. Woo, Y.-t.L., David Y., (Q)SAR ANALYSIS OF GENOTOXIC AND NONGENOTOXIC
CARCINOGENS: A STATE-OF-THE-ART OVERVIEW, in Cancer Risk Assessment: Chemical
Carcinogenesis, Hazard Evaluation, and Risk Quantification, Wiley, Editor. 2010. p. 517.
3. Benigni, R. and C. Bossa, Mechanisms of chemical carcinogenicity and mutagenicity: a review
with implications for predictive toxicology. Chem Rev, 2011. 111(4): p. 2507-36.
4. Zeiger, E., History and rationale of genetic toxicity testing: An impersonal, and sometimes
personal, view. Environmental and Molecular Mutagenesis, 2004. 44(5): p. 363-371.
5. Miller, E.C. and J.A. Miller, Searches for ultimate chemical carcinogens and their reactions
with cellular macromolecules. Cancer, 1981. 47(10): p. 2327-2345.
6. Miller, E.C.M., J. A. , Mechanisms of chemical carcinogenesis. Cancer, 1981. 47: p. 10.
7. Pullman, B.P., Alberte, Nucleophilicity of DNA. Relation to Chemical Carcinogenesis., in
Carcinogenesis: Fundamental Mechanisms and Environmental Effects. 1980, Springer
Netherlands. p. 55-66.
8. Ball, M., Chemical structure of DNA. 2013: http://madprime.org/articles/2013/01/23/cc0-all-the-
media/.
9. Carcinogenesis: Fundamental Mechanisms and Environmental Effects. in Thirteenth
Jerusalem Symposium on Quantum Chemistry and Biochemistry. 1980. Jerusalem, Israel:
Springer Netherlands.
10. Garte, S.J., et al., Carcinogen specificity in the activation of transforming genes by direct-
acting alkylating agents. Carcinogenesis, 1985. 6(12): p. 1709-1712.
11. Drablos, F., et al., Alkylation damage in DNA and RNA--repair mechanisms and medical
significance. DNA Repair (Amst), 2004. 3(11): p. 1389-407.
12. Beyerbach, A.B.F., Peter; Sabbioni, Gabriele, Biomarkers for Isocyanate Exposure: Synthesis
of Isocyanate DNA Adducts. Chem. Res. Toxicol., 2006. 19(12): p. 8.
13. Segal, A.S., J. J.; Maté, U; Van Dureen; B. L., Formation of 6-dimethylcarbamyloxy-dGuo, 6-
dimethylamino-dGuo and 4-dimethylamino-dThd following in vitro reaction of
dimethylcarbamyl chloride with calf thymus DNA and 6-diethylcarbamyloxy-dGuo following in
vitro reaction of diethylcarbamyl chloride with calf thymus DNA. Chemico-Biological
Interactions, 1982. 40(2): p. 23.
14. Beland, F.A., et al., Arylamine-DNA adducts in vitro and in vivo: their role in bacterial
mutagenesis and urinary bladder carcinogenesis. Environ Health Perspect, 1983. 49: p. 125-
34.
15. Takamura-Enya, T., et al., Development for an efficient synthesis method of 2'-
deoxyguanosine-C8 adducts with several amino/nitro-arenes. Nucleic Acids Res Suppl,
2003(3): p. 23-4.

63
16. Snyder, R.D.M., J.; Zairov, G.; Ewing, D.E.; Hendry, L.B., The influence of N-dialkyl and other
cationic substituents on DNA intercalation and genotoxicity. Mutation Research/Fundamental
and Molecular Mechanisms of Mutagenesis, 2005. 578(1-2): p. 12.
17. La, D.K.S., James A., DNA adducts: biological markers of exposure and potential applications
to risk assessment. Mutation Research/Reviews in Genetic Toxicology, 1996. 365(1-3): p. 18.
18. Boysen, G.P., Brian F.; Nakamura, Jun; Swenberg, James A., The formation and biological
significance of N7-guanine adducts. Mutation Research/Genetic Toxicology and
Environmental Mutagenesis, 2009. 678(2): p. 19.
19. Müller, L., et al., A rationale for determining, testing, and controlling specific impurities in
pharmaceuticals that possess potential for genotoxicity. Regulatory Toxicology and
Pharmacology, 2006. 44(3): p. 198-211.
20. Humfrey, C.D.N., Recent Developments in the Risk Assessment of Potentially Genotoxic
Impurities in Pharmaceutical Drug Substances. Toxicological Sciences, 2007. 100(1): p. 24-
28.
21. (CHMP), C.f.H.M.P., The potential risks of carcinogens, mutagens and substances toxic to
reproduction when these substances are used as excipients of medicinal products for human
use, in EMEA/CHMP/SWP/146166/2007, E.M. Agency, Editor. 2008.
22. Guidelines On The Limits Of Genotoxic Impurities, in EMEA/CHMP/QWP/251344/2006,
C.F.M.P.F.H.U. (CHMP), Editor. 2006.
23. Questions and answers on the 'Guideline on the limits of genotoxic impurities'. 2010.
24. Jacobson-Kram, D., Genotoxic and Carcinogenic Impurities in Drug Substances and Products:
Recommended Approaches, U.S.D.o.H.a.H. Services, Editor. 2008.
25. ICH guideline M7 on assessment and control of DNA reactive (mutagenic) impurities in
pharmaceuticals to limit potential carcinogenic risk, in EMA/CHMP/ICH/83812/2013, E.M.
Agency, Editor. 2013.
26. Sawatari, K., Y. Nakanishi, and T. Matsushima, Relationships between chemical structures
and mutagenicity: a preliminary survey for a database of mutagenicity test results of new work
place chemicals. Ind Health, 2001. 39(4): p. 341-5.
27. Snyder, R.D., et al., Assessment of the sensitivity of the computational programs DEREK,
TOPKAT, and MCASE in the prediction of the genotoxicity of pharmaceutical molecules.
Environmental and Molecular Mutagenesis, 2004. 43(3): p. 143-158.
28. Snyder, R.D. and M.D. Smith, Computational prediction of genotoxicity: room for
improvement. Drug Discovery Today, 2005. 10(16): p. 1119-1124.
29. Benigni, R.B., Cecilia, Structural Alerts of Mutagens and Carcinogens. Current Computer-
Aided Drug Design, 2006. 2(2): p. 169-176.
30. Raillard, S.P., et al., Prediction of Drug Degradation Pathways leading to Structural Alerts for
Potential Genotoxic Impurities. Organic Process Research & Development, 2010. 14(4): p.
1015-1020.

64
31. Snodin, D.J., Genotoxic Impurities: A Regulatory Toxicology Commentary on Recent Articles
in Organic Process Research & Development. Organic Process Research & Development,
2011. 15(6): p. 1243-1246.
32. Rajski, S.R. and R.M. Williams, DNA Cross-Linking Agents as Antitumor Drugs. Chemical
Reviews, 1998. 98(8): p. 2723-2796.
33. Bolt, H.M. and B. Gansewendt, Mechanisms of Carcinogenicity of Methyl Halides. Critical
Reviews in Toxicology, 1993. 23(3): p. 237-253.
34. Guengerich, F.P., Activation of dihaloalkanes by thiol-dependent mechanisms. J Biochem Mol
Biol, 2003. 36(1): p. 20-7.
35. Yang, Q., et al., Controlling the Genotoxins Ethyl Chloride and Methyl Chloride Formed During
the Preparation of Amine Hydrochloride Salts from Solutions of Ethanol and Methanol.
Organic Process Research & Development, 2009. 13(4): p. 786-791.
36. Koskinen, M. and K. Plná, Specific DNA adducts induced by some mono-substitued epoxides
in vitro and in vivo. Chemico-Biological Interactions, 2000. 129(3): p. 209-229.
37. Kovacic, P. and J.D. Jacintho, Mechanisms of carcinogenesis: focus on oxidative stress and
electron transfer. Curr Med Chem, 2001. 8(7): p. 773-96.
38. Poso, A., A. von Wright, and J. Gynther, An empirical and theoretical study on mechanisms of
mutagenic activity of hydrazine compounds. Mutat Res, 1995. 332(1-2): p. 63-71.
39. Smith, B.J.G.G., R.; Wagner, V. O.; Madraymootoo, W.; Van Velsen, F.; Van Gompel, J.;
Bigger, A. , Genetic toxicity assessment of the nitroxide radical 2,2,6,6-tetramethylpiperidin-l-
oxyl (TEMPO), in Genetic Toxicology Association Meeting. 2009: University of Delaware,
Newark, USA.
40. Kim, D. and F.P. Guengerich, Cytochrome P450 activation of arylamines and heterocyclic
amines. Annu Rev Pharmacol Toxicol, 2005. 45: p. 27-49.
41. Neumann, H.G., The role of DNA damage in chemical carcinogenesis of aromatic amines. J
Cancer Res Clin Oncol, 1986. 112(2): p. 100-6.
42. Meier, C. DNA Damage by Aromatic Amines – Are Arylamine Adducts responsible for the
Induction of the Chemical Carcinogenesis? 2014.
43. Suji, G. and S. Sivakami, DNA damage by free radical production by aminoguanidine. Ann N
Y Acad Sci, 2006. 1067: p. 191-9.
44. Snodin, D.J., Genotoxic Impurities: From Structural Alerts to Qualification. Organic Process
Research & Development, 2010. 14(4): p. 960-976.
45. Balasubramanian, M.R., R. Neethu; Gayathri, V.; John, K.M. Maria, In vitro analysis of
Calotropis peroxidase transformed diamine compounds. Journal of Environmental Treatment
Techniques, 2013. 1(1): p. 38-45.
46. Beland, F.A. and F.F. Kadlubar, Formation and persistence of arylamine DNA adducts in vivo.
Environ Health Perspect, 1985. 62: p. 19-30.
47. Sengupta, J. and R.H. Duncan Lyngdoh, Relative preferences of carcinogenic aromatic
nitrenium ions for adduct formation with different DNA base sites: Theoretical elucidations.
Journal of Biological Physics, 1995. 21(4): p. 285-294.

65
48. O’Donovan, M.R., et al., Boronic acids—A novel class of bacterial mutagen. Mutation
Research/Genetic Toxicology and Environmental Mutagenesis, 2011. 724(1–2): p. 1-6.
49. Hall, D.G., Structure, Properties, and Preparation of Boronic Acid Derivatives, in Boronic
Acids. 2011, Wiley-VCH Verlag GmbH & Co. KGaA. p. 1-133.
50. Elder, D., et al., An Approach to Control Strategies for Sulfonate Ester Formation in
Pharmaceutical Manufacturing Based on Recent Scientific Understanding. Organic Process
Research & Development, 2012. 16(11): p. 1707-1710.
51. Teasdale, A., et al., A Detailed Study of Sulfonate Ester Formation and Solvolysis Reaction
Rates and Application toward Establishing Sulfonate Ester Control in Pharmaceutical
Manufacturing Processes. Organic Process Research & Development, 2010. 14(4): p. 999-
1007.
52. Gerber, C. and H.-G. Toelle, What happened: The chemistry side of the incident with EMS
contamination in Viracept tablets. Toxicology Letters, 2009. 190(3): p. 248-253.
53. Elder, D.P., et al., The utility of sulfonate salts in drug development. Journal of Pharmaceutical
Sciences, 2010. 99(7): p. 2948-2961.
54. Teasdale, A., et al., Mechanism and Processing Parameters Affecting the Formation of Methyl
Methanesulfonate from Methanol and Methanesulfonic Acid: An Illustrative Example for
Sulfonate Ester Impurity Formation. Organic Process Research & Development, 2009. 13(3):
p. 429-433.
55. Pierson, D.A., et al., Approaches to Assessment, Testing Decisions, and Analytical
Determination of Genotoxic Impurities in Drug Substances. Organic Process Research &
Development, 2008. 13(2): p. 285-291.
56. Slater, C.S.S., M. J.; Hesketh, R. P. Measurement and Reduction of Organic Solvents in
Pharmaceutical Manufacture. in Green Chemistry and Engineering for Sustainability. 2006.
San Francisco: American Institute of Chemical Engineers Anual Meeting.
57. ICH, Impurities: Guideline For Residual Solvents Q3C(R5), I.C.O.H.O.T.R.F.R.O.P.F.H. Use,
Editor. 2011.
58. Meng, K.W., Hartmund; Horstmann, Haral, Sulphonamides. 1966, Bayer: United Kingdom.
59. Raman, N.V., A.V. Prasad, and K. Ratnakar Reddy, Strategies for the identification, control
and determination of genotoxic impurities in drug substances: a pharmaceutical industry
perspective. J Pharm Biomed Anal, 2011. 55(4): p. 662-7.
60. Vogt, F.G. and A.S. Kord, Development of quality-by-design analytical methods. Journal of
Pharmaceutical Sciences, 2011. 100(3): p. 797-812.
61. Cimarosti, Z., et al., Application of Quality by Design Principles to Support Development of a
Control Strategy for the Control of Genotoxic Impurities in the Manufacturing Process of a
Drug Substance. Organic Process Research & Development, 2009. 14(4): p. 993-998.
62. Teasdale, A., et al., Risk Assessment of Genotoxic Impurities in New Chemical Entities:
Strategies To Demonstrate Control. Organic Process Research & Development, 2013. 17(2):
p. 221-230.

66
63. Looker, A.R., et al., Risk Assessment of Potentially Genotoxic Impurities within the Framework
of Quality by Design. Organic Process Research & Development, 2010. 14(4): p. 1032-1036.
64. Arai, T., Chiral separation of pharmaceuticals possessing a carboxy moiety. Journal of
Chromatography B: Biomedical Sciences and Applications, 1998. 717(1–2): p. 295-311.
65. Rohani, S., S. Horne, and K. Murthy, Control of Product Quality in Batch Crystallization of
Pharmaceuticals and Fine Chemicals. Part 1: Design of the Crystallization Process and the
Effect of Solvent. Organic Process Research & Development, 2005. 9(6): p. 858-872.
66. Shekunov, B.Y. and P. York, Crystallization processes in pharmaceutical technology and drug
delivery design. Journal of Crystal Growth, 2000. 211(1–4): p. 122-136.
67. Kim, S., C. Wei, and S. Kiang, Crystallization Process Development of an Active
Pharmaceutical Ingredient and Particle Engineering via the Use of Ultrasonics and
Temperature Cycling. Organic Process Research & Development, 2003. 7(6): p. 997-1001.
68. Yang, Y. and R. Tjia, Process modeling and optimization of batch fractional distillation to
increase throughput and yield in manufacture of active pharmaceutical ingredient (API).
Computers & Chemical Engineering, 2010. 34(7): p. 1030-1035.
69. Lee, C., et al., Removal of Electrophilic Potential Genotoxic Impurities Using Nucleophilic
Reactive Resins. Organic Process Research & Development, 2010. 14(4): p. 1021-1026.
70. Iverlund, N., A. Parsons, and F. Muller, Carbon cartridges and their use as a purification step
in pharmaceutical API processes. Chemical Engineering Research and Design, 2009. 87(6):
p. 852-858.
71. Welch, C.J., et al., Adsorbent Screening for Metal Impurity Removal in Pharmaceutical
Process Research. Organic Process Research & Development, 2005. 9(2): p. 198-205.
72. Guest, D.W., Evaluation of simulated moving bed chromatography for pharmaceutical process
development. Journal of Chromatography A, 1997. 760(1): p. 159-162.
73. Kecili, R., et al., Fast identification of selective resins for removal of genotoxic aminopyridine
impurities via screening of molecularly imprinted polymer libraries. J Chromatogr A, 2014.
1339: p. 65-72.
74. Kecili, R., et al., Selective scavenging of the genotoxic impurity methyl p-toluenesulfonate from
pharmaceutical formulations. Separation and Purification Technology, 2013. 103: p. 173-179.
75. Valadez-Blanco, R., et al., A membrane bioreactor for biotransformations of hydrophobic
molecules using organic solvent nanofiltration (OSN) membranes. Journal of Membrane
Science, 2008. 317(1–2): p. 50-64.
76. Meier, J., T. Melin, and L.H. Eilers, Nanofiltration and adsorption on powdered adsorbent as
process combination for the treatment of severely contaminated waste water. Desalination,
2002. 146(1–3): p. 361-366.
77. Székely, G., et al., Organic solvent nanofiltration: A platform for removal of genotoxins from
active pharmaceutical ingredients. Journal of Membrane Science, 2011. 381(1-2): p. 21-33.
78. Geens, J., B. De Witte, and B. Van der Bruggen, Removal of API's (Active Pharmaceutical
Ingredients) from Organic Solvents by Nanofiltration. Separation Science and Technology,
2007. 42(11): p. 2435-2449.

67
79. Abbott, E., T.D. Veenstra, and H.J. Issaq, Clinical and pharmaceutical applications of packed-
column supercritical fluid chromatography. Journal of Separation Science, 2008. 31(8): p.
1223-1230.
80. Sheth, J.P., et al., Nanofiltration-based diafiltration process for solvent exchange in
pharmaceutical manufacturing. Journal of Membrane Science, 2003. 211(2): p. 251-261.
81. Piletsky, S.A., S. Alcock, and A.P.F. Turner, Molecular imprinting: at the edge of the third
millennium. Trends in Biotechnology, 2001. 19(1): p. 9-12.
82. Owens, P.K., et al., Molecular imprinting for bio- and pharmaceutical analysis. TrAC Trends in
Analytical Chemistry, 1999. 18(3): p. 146-154.
83. Székely, G., et al., A hybrid approach to reach stringent low genotoxic impurity contents in
active pharmaceutical ingredients: Combining molecularly imprinted polymers and organic
solvent nanofiltration for removal of 1,3-diisopropylurea. Separation and Purification
Technology, 2012. 86: p. 79-87.
84. Biomimética, in Dicionário Priberam da Língua Portugues. 2013:
http://priberam.pt/dlpo/biomim%C3%A9tica.
85. LaBean, T.H. and H. Li, Constructing novel materials with DNA. Nano Today, 2007. 2(2): p.
26-35.
86. Yang, D., et al., Novel DNA materials and their applications. Wiley Interdiscip Rev Nanomed
Nanobiotechnol, 2010. 2(6): p. 648-69.
87. Bousquet, J., Mometasone furoate: an effective anti-inflammatory with a well-defined safety
and tolerability profile in the treatment of asthma. International Journal of Clinical Practice,
2009. 63(5): p. 806-819.
88. How Does High Performance Liquid Chromatography Work? . [cited 2014; Available from:
http://www.waters.com/waters/pt_PT/How-Does-High-Performance-Liquid-Chromatography-
Work%3F/nav.htm?cid=10049055.
89. Schematic of UV- visible spectrophotometer. 2013:
http://commons.wikimedia.org/wiki/File:Schematic_of_UV-_visible_spectrophotometer.png.
90. Sá, C., Digital Analysis of SEM Images for Materials Characterization and Interface/Surface
Studies of Biological Materials in Imaging techniques in biomaterials: digital image processing
applied to orthopaedic and dental implants 1994. p. 24.
91. Monteiro, A.J.S.C., Técnicas de Caracterização de Revestimentos Finos. 2005:
https://repositorium.sdum.uminho.pt/bitstream/1822/3280/5/Cap%C3%ADtulo%204.pdf.
92. Paiva, M., Espectroscopia de Infravermelhos. 2006:
www.dep.uminho.pt/mcpaiva/pdfs/IP_UCI_I/0607F01.pdf.
93. Chen, L., S. Xu, and J. Li, Recent advances in molecular imprinting technology: current status,
challenges and highlighted applications. Chemical Society Reviews, 2011. 40(5): p. 2922-
2942.
94. Wulff, G., Molecular Imprinting in Cross-Linked Materials with the Aid of Molecular
Templates— A Way towards Artificial Antibodies. Angew. Chem. Int. Ed. Engl., 1995. 34(17):
p. 20.

68
95. Mosbach, K., Molecular imprinting. Trends in Biochemical Sciences, 1994. 19(1): p. 9-14.
96. Okutucu, B.Ö., Seçil; Telefoncu, Azmi, Noncovalently galactose imprinted polymer for the
recognition of different saccharides. Talanta, 2009. 78(3): p. 4.
97. Cacho, C.T., E.; Martín-Esteban, A.; Ayala, D.; Pérez-Conde, C., Semi-covalent imprinted
polymer using propazine methacrylate as template molecule for the clean-up of triazines in
soil and vegetable samples. Journal of Chromatography A, 2006. 1114(2): p. 8.
98. Whitcombe, M.J.R., M. E.; Villar, Pablo; Vulfson, Evgeny N., A New Method for the
Introduction of Recognition Site Functionality into Polymers Prepared by Molecular Imprinting:
Synthesis and Characterization of Polymeric Receptors for Cholesterol. Journal of the
American Chemical Society, 1995. 117(27): p. 7.
99. Piletska, E.V., et al., Influence of the Polymerization Conditions on the Performance of
Molecularly Imprinted Polymers. Macromolecules, 2009. 42(14): p. 4921-4928.
100. Shamsipur, M.F., Javad; Khanchi, Alireza; Hassani, Rahim; Alizadeh, Kamal; Shamsipur,
Hosna, A stoichiometric imprinted chelating resin for selective recognition of copper(II) ions in
aqueous media. Analytica Chimica Acta, 2007. 599(2): p. 7.
101. Koohpaei, A.R.S., S. J.; Ganjali, M. R.; Forushani, A. R.; Golbabaei F., Application of
multivariate analysis to the screening of molecularly imprinted polymers (MIPs) for ametryn.
Talanta, 2008. 75(4): p. 9.
102. Zhang, H.S., Tao; Zong, Fulin; Chen, Tiechun; Pan, Canping, Synthesis and Characterization
of Molecularly Imprinted Polymers for Phenoxyacetic Acids. Int. J. Mol. Sci., 2008. 9(1): p. 9.
103. Karim, K.B., Florent; Rouillon, Regis; Piletska, Elena V.; Guerreiro, Antonio; Chianella, Iva;
Piletsky, Sergey A., How to find effective functional monomers for effective molecularly
imprinted polymers? Advanced Drug Delivery Reviews, 2005. 57(12): p. 14.
104. Feás, X.S., J.A.; Vázquez-Tato, M.P.; Regal, P.; Cepeda, A.; Fente, C., Syntheses of
molecularly imprinted polymers: Molecular recognition of cyproheptadine using original print
molecules and azatadine as dummy templates. Analytica Chimica Acta, 2009. 631(2): p. 8.
105. Vandezande, P., L.E.M. Gevers, and I.F.J. Vankelecom, Solvent resistant nanofiltration:
separating on a molecular level. Chemical Society Reviews, 2008. 37(2): p. 365-405.
106. Székely, G., et al., Environmental and economic analysis for selection and engineering
sustainable API degenotoxification processes. Green Chemistry, 2013. 15(1): p. 210.
107. Ferreira, A.B., Fernando P.; Granjo, José; Ferreira, Licínio; Carvalho, Maria; Rasteiro, Marian.
Adsorção e Permuta iónica.
108. Permuta Iónica. Available from:
http://paginas.fe.up.pt/~mgi00015/Ensdist/versao_web/teoria/teoria_base.htm.
109. Lito, P.F.C., Simão P.; Loureiro, José M.; Silva, Carlos M., Ion Exchange Equilibria and
Kinetics, in Ion Exchange Technology I, Inamuddin, Editor. 2012, Springer Netherlands. p. 51-
120.
110. Allgeier, S., Membrane filtration guidance manual. 2003: DIANE Publishing.
111. Wagner, J., Membrane Filtration Handbook - Practical Tips and Hints. 2001: Osmonics. 129.
112. Baker, R.W., Membrane Technology and Applications. 2012: Wiley. 588

69
113. Santos, J.L.C., et al., Analysis of solvent flux through nanofiltration membranes by
mechanistic, chemometric and hybrid modelling. Journal of Membrane Science, 2007. 300(1–
2): p. 191-204.
114. Giegé, R. and V. Mikol, Crystallogenesis of proteins. Trends in Biotechnology, 1989. 7(10): p.
277-282.
115. McPherson, A., Current approaches to macromolecular crystallization. European Journal of
Biochemistry, 1990. 189(1): p. 1-23.
116. Lorber, B. and R. Giege´, A versatile reactor for temperature controlled crystallization of
biological macromolecules. Journal of Crystal Growth, 1992. 122(1–4): p. 168-175.
117. Drenth, J., Concluding remarks. Journal of Crystal Growth, 1988. 90(1–3): p. 368-370.
118. Scudder, P.H., Electron Flow in Organic Chemistry: A Decision-Based Guide to Organic
Mechanisms. 2013: Wiley.
119. Bansal, R.C. and M. Goyal, Activated Carbon Adsorption. 2005: Taylor & Francis.

70
Anexos
Anexo 1 – Retas de calibração
Figura 56 - Reta de calibração do DMAP em H2O.
Figura 57 - Reta de calibração do DMAP em H2O:MeOH.
y = 305767x - 2E+06R² = 0,9996
0
50000000
100000000
150000000
200000000
250000000
0 100 200 300 400 500 600 700 800
Áre
a d
o p
ico
Concentração (ppm)
y = 323783x - 885517R² = 0,9998
0
20000000
40000000
60000000
80000000
100000000
120000000
140000000
160000000
180000000
0 100 200 300 400 500 600
Áre
a d
o p
ico
Concentração (ppm)

71
Figura 58 - Reta de calibração do DMAP em MeOH.
Figura 59 - Reta de calibração do DMAP em DCM.
Figura 60 - Reta de calibração do MPTS em MeOH.
y = 313582x - 8E+06R² = 0,9986
0
50000000
100000000
150000000
200000000
250000000
300000000
350000000
0 200 400 600 800 1000 1200
Áre
a d
o p
ico
Concentração (ppm)
y = 290118x - 1E+06R² = 0,9986
0
20000000
40000000
60000000
80000000
100000000
120000000
140000000
160000000
180000000
200000000
0 100 200 300 400 500 600 700
Áre
a d
o p
ico
Concentração (ppm)
y = 65720x + 583464R² = 0,9993
0
10000000
20000000
30000000
40000000
50000000
60000000
70000000
0 200 400 600 800 1000 1200
Áre
a d
o p
ico
Concentração (ppm)

72
Figura 61 - Reta de calibração do MPTS em DCM.
Figura 62 - Reta de calibração do Meta em DCM.
y = 127073x - 3E+06R² = 0,9734
0
20000000
40000000
60000000
80000000
100000000
120000000
140000000
0 200 400 600 800 1000 1200
Áre
a d
o p
ico
Concentração (ppm)
y = 8968,7x + 748586R² = 0,9965
0
10000000
20000000
30000000
40000000
50000000
60000000
70000000
80000000
90000000
100000000
0 2000 4000 6000 8000 10000 12000
Áre
a d
o p
ico
Concentração (ppm)
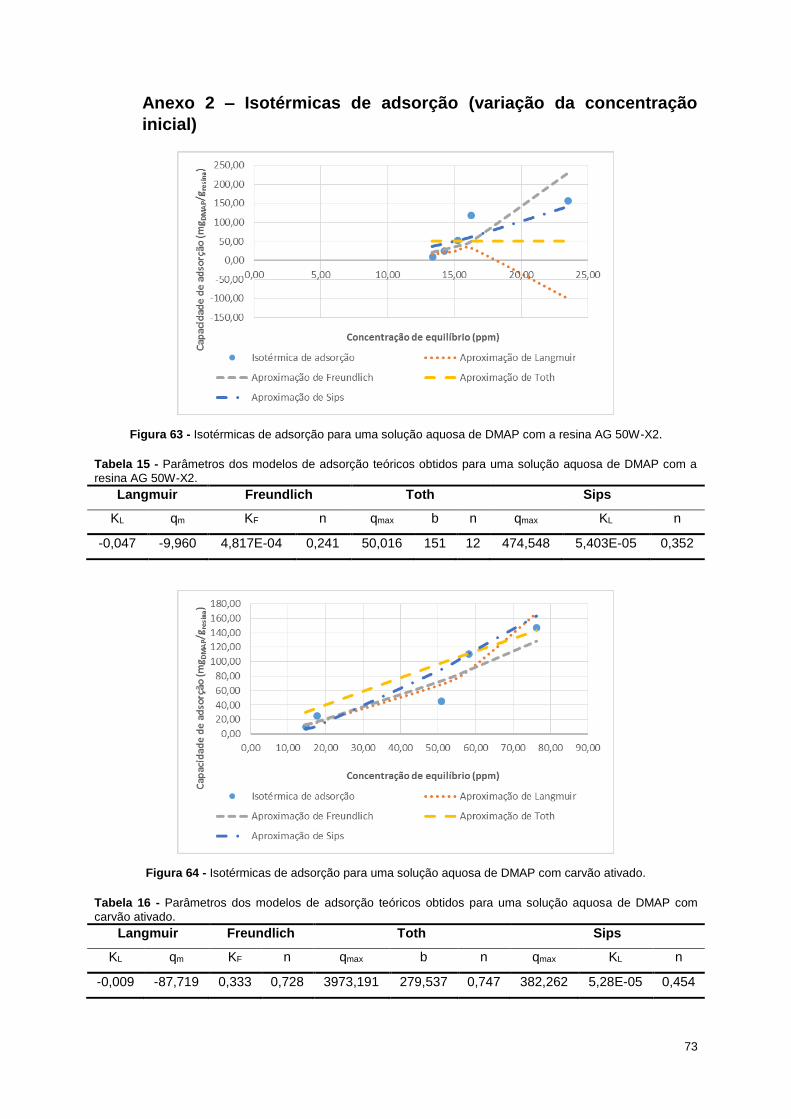
73
Anexo 2 – Isotérmicas de adsorção (variação da concentração
inicial)
Figura 63 - Isotérmicas de adsorção para uma solução aquosa de DMAP com a resina AG 50W-X2.
Tabela 15 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de DMAP com a
resina AG 50W-X2.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,047 -9,960 4,817E-04 0,241 50,016 151 12 474,548 5,403E-05 0,352
Figura 64 - Isotérmicas de adsorção para uma solução aquosa de DMAP com carvão ativado.
Tabela 16 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de DMAP com
carvão ativado.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,009 -87,719 0,333 0,728 3973,191 279,537 0,747 382,262 5,28E-05 0,454
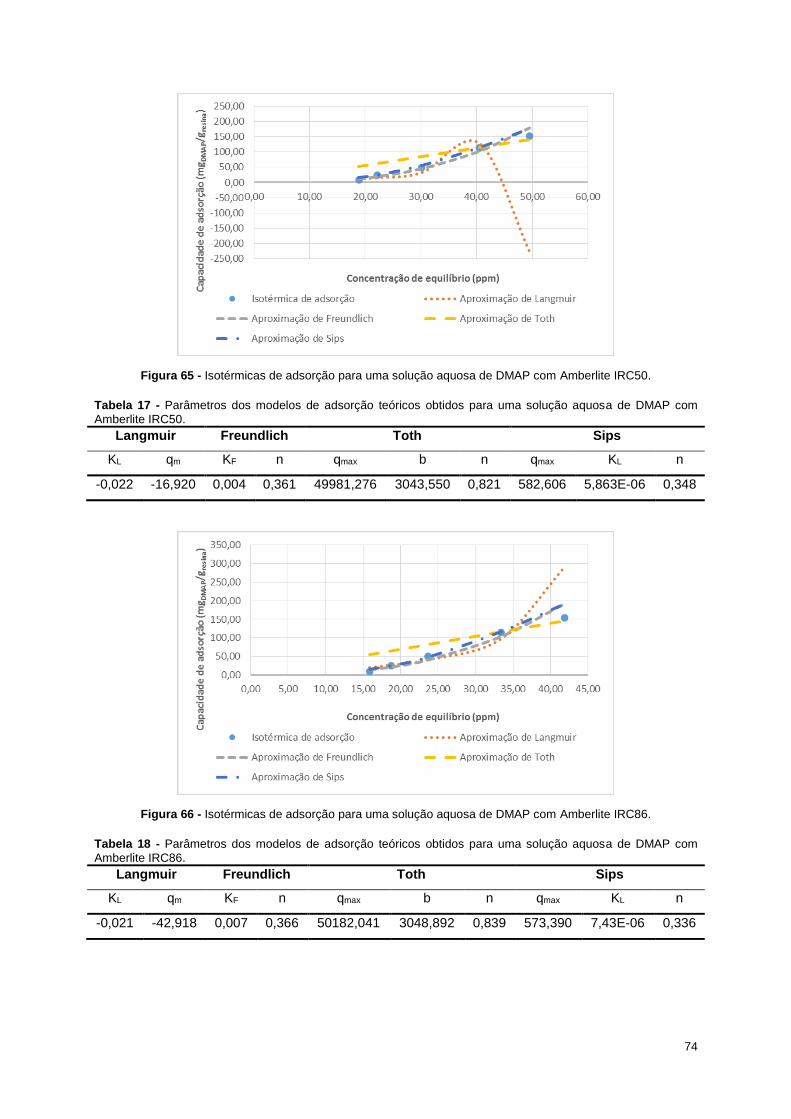
74
Figura 65 - Isotérmicas de adsorção para uma solução aquosa de DMAP com Amberlite IRC50.
Tabela 17 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de DMAP com
Amberlite IRC50.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,022 -16,920 0,004 0,361 49981,276 3043,550 0,821 582,606 5,863E-06 0,348
Figura 66 - Isotérmicas de adsorção para uma solução aquosa de DMAP com Amberlite IRC86.
Tabela 18 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução aquosa de DMAP com
Amberlite IRC86.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,021 -42,918 0,007 0,366 50182,041 3048,892 0,839 573,390 7,43E-06 0,336

75
Figura 67 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com AG 50W-X2.
Tabela 19 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água:metanol
com AG 50W-X2.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,073 -33,557 0,212 0,364 51001,487 3001,161 0,952 568,051 4,10E-04 0,355
Figura 68 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com carvão ativado.
Tabela 20 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água:metanol
com carvão ativado.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,004 -40,323 0,094 0,817 410120,929 3421,612 0,695 47,605 3,25E-04 0,526

76
Figura 69 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com Amberlite IRC50.
Tabela 21 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água:metanol
com Amberlite IRC50.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,013 -65,359 0,375 0,711 41903,710 3348,495 0,812 129,471 1,57E-04 0,405
Figura 70 - Isotérmicas de adsorção para uma solução DMAP em água:metanol com Amberlite IRC86.
Tabela 22 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água:metanol
com Amberlite IRC86.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,023 -51,020 0,279 0,576 44281,935 3207,281 0,863 102,615 2,06E-04 0,358
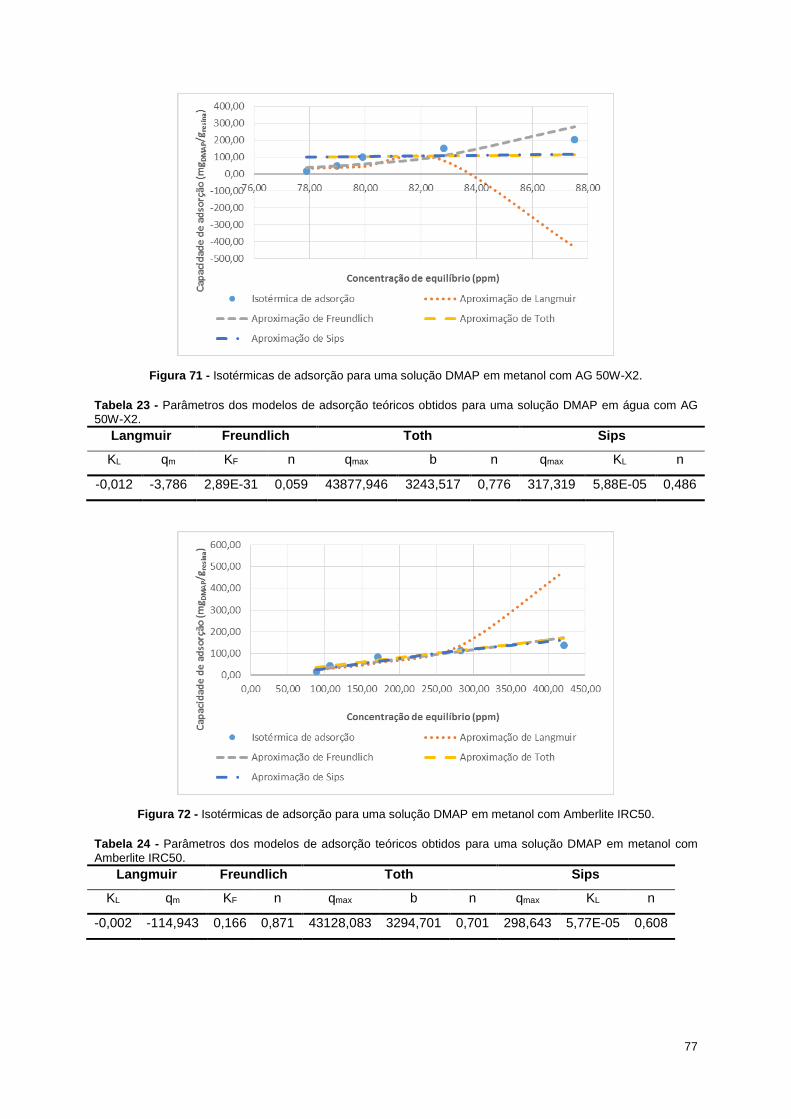
77
Figura 71 - Isotérmicas de adsorção para uma solução DMAP em metanol com AG 50W-X2.
Tabela 23 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em água com AG
50W-X2.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,012 -3,786 2,89E-31 0,059 43877,946 3243,517 0,776 317,319 5,88E-05 0,486
Figura 72 - Isotérmicas de adsorção para uma solução DMAP em metanol com Amberlite IRC50.
Tabela 24 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em metanol com
Amberlite IRC50.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,002 -114,943 0,166 0,871 43128,083 3294,701 0,701 298,643 5,77E-05 0,608
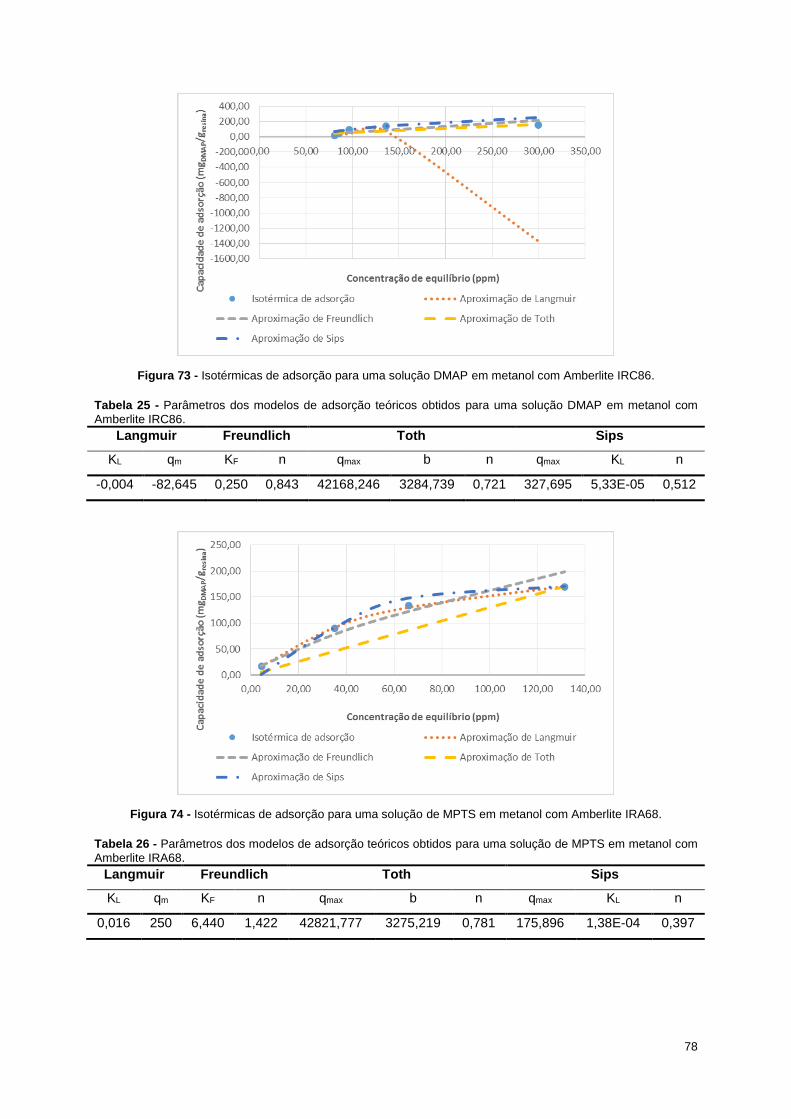
78
Figura 73 - Isotérmicas de adsorção para uma solução DMAP em metanol com Amberlite IRC86.
Tabela 25 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução DMAP em metanol com
Amberlite IRC86.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-0,004 -82,645 0,250 0,843 42168,246 3284,739 0,721 327,695 5,33E-05 0,512
Figura 74 - Isotérmicas de adsorção para uma solução de MPTS em metanol com Amberlite IRA68.
Tabela 26 - Parâmetros dos modelos de adsorção teóricos obtidos para uma solução de MPTS em metanol com
Amberlite IRA68.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
0,016 250 6,440 1,422 42821,777 3275,219 0,781 175,896 1,38E-04 0,397

79
Figura 75 - Isotérmicas de adsorção para o MIP4.
Tabela 27 - Parâmetros dos modelos de adsorção teóricos obtidos para o MIP4.
Langmuir Freundlich Toth Sips
KL qm KF n qmax b n qmax KL n
-1272,92 -0,067 6,75E-
06 0,133 39927,275 3413,499 0,678 130,345 1E-04 0,338
80
Anexo 3 – Quantidade de GTI adsorvido em função da quantidade
inicial de resina
Figura 76 – Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução aquosa de DMAP com AG 50W-X2.
Figura 77 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução aquosa de DMAP com carvão ativado.
Figura 78 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução aquosa de DMAP com Amberlite IRC50.
92
93
94
95
96
97
98
99
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
94
95
96
97
98
99
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
91
91
92
92
93
93
94
94
95
95
96
96
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)

81
Figura 79 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução aquosa de DMAP com Amberlite IRC86.
Figura 80 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com AG 50W-X2.
Figura 81 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com carvão ativado.
92
92
93
93
94
94
95
95
96
96
97
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
0
20
40
60
80
100
120
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
96
96
97
97
98
98
99
99
100
100
0,000 0,020 0,040 0,060 0,080 0,100
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)

82
Figura 82 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com Amberlite IRC50.
Figura 83 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em água:metanol com Amberlite IRC86.
Figura 84 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em metanol com AG 50W-X2.
78
80
82
84
86
88
90
92
94
96
98
0 0,02 0,04 0,06 0,08 0,1
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
84
86
88
90
92
94
96
98
0,000 0,020 0,040 0,060 0,080 0,100
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
92
93
94
95
96
97
98
99
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
83
Figura 85 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em metanol com Amberlite IRC50.
Figura 86 - Quantidade de DMAP adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de DMAP em metanol com Amberlite IRC86.
Figura 87 - Quantidade de MPTS adsorvido determinado a partir da isotérmica de adsorção com variação da
quantidade inicial de resina obtida para uma solução de MPTS em metanol com Amberlite IRA68.
91
91
92
92
93
93
94
94
95
95
96
96
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
92
92
93
93
94
94
95
95
96
96
97
0,000 0,050 0,100 0,150 0,200 0,250
% d
e D
MA
P a
dso
rvid
o
Massa de resina (g)
0
20
40
60
80
100
120
0,000 0,050 0,100 0,150 0,200 0,250
% d
e M
PTS
ad
sorv
ido
Massa de resina (g)





























