Embed Size (px)
Citation preview

INSTITUTO NACIONAL DE PESQUISAS DA AMAZÔNIA – INPA
Programa de Pós-Graduação em Genética, Conservação e Biologia Evolutiva (GCBEv)
UNIVERSIDADE FEDERAL DO AMAZONAS – UFAM
Laboratório de Evolução e Genética Animal (LEGAL)
SANDRA MARCELA HERNÁNDEZ RANGEL
MANAUS – AMAZONAS
JULHO, 2015
DINÂMICA E ESTRUTURA POPULACIONAL
DO JACARÉ-AÇU (Melanosuchus niger) NA
AMAZÔNIA

SANDRA MARCELA HERNÁNDEZ RANGEL
ORIENTADORA: DRA. IZENI PIRES FARIAS
COORIENTADOR: DR. TOMAS HRBEK
Apoio: Laboratório de Evolução e Genética Animal – LEGAL
Financiamento: CNPq-SISBIOTA/FAPEAM-SISBIOTA
MANAUS – AMAZONAS
JULHO, 2015
Dissertação apresentada ao Instituto Nacional
de Pesquisas da Amazônia, como parte dos
requisitos para obtenção do título de mestre
em Genética, Conservação e Biologia
Evolutiva.
DINÂMICA E ESTRUTURA POPULACIONAL
DO JACARÉ-AÇU (Melanosuchus niger) NA
AMAZÔNIA

ii
FICHA CATALOGRÁFICA
R196d Rangel, Sandra Marcela Hernández
Dinâmica e estrutura populacional do jacaré-açu (Melanosuchus
niger) na Amazônia / Sandra Marcela Hernández Rangel. ---
Manaus: [s.n.], 2015.
xiv, 59 f. : il. color.
Dissertação (Mestrado) --- INPA, Manaus, 2015.
Orientador : Izeni Pires Farias
Coorientador : Tomas Hrbek
Área de concentração : Genética, Conservação e Biologia
Evolutiva
1. Diversidade genética. 2. Melanosuchus niger. 3. Isolamento
por distância. 4. Barreiras ao fluxo gênico. I.Título
CDD 597.98
Sinopse:
Estudou-se a distribuição da diversidade genética e a estrutura de populações de
Melanosuchus niger na Amazônia utilizando o marcador mitocondrial Citocromo b e
marcadores SNPs. O isolamento por distância determina a diferenciação genética ao
longo da bacia amazônica e existe uma forte estruturação genética dentro das bacias
devido as barreiras geográficas (corredeiras). As corredeiras do rio Madeira restringem o
fluxo gênico das populações.
Palavras-chave: isolamento por distância, diversidade genética, fluxo gênico, barreiras
geográficas

iii
A minha grande amiga Úrsula,
sempre estarás no meu pensamento e no meu coração.

iv
AGRADECIMENTOS
Ao Programa de Estudante-Convênio de Pós-Graduação (PEC-PG) da CAPES/CNPq-
Brasil pela oportunidade de vir ao Brasil para continuar meus estudos e pelo financiamento da
bolsa.
Ao CNPq e à FAPEAM pelo financiamento do projeto "Rede de pesquisa para
ampliação do conhecimento sobre a biodiversidade de vertebrados da Amazônia brasileira"
(CNPq-FAPEAM 563348/2010-0) que financiou esse trabalho.
Ao Programa de Pós-Graduação em Genética, Conservação e Biologia Evolutiva
(GCBEv) do Instituto de Pesquisas da Amazônia (INPA). Especialmente, às professoras
Gislene e Eliana pela confiança e apoio que me deram.
À Universidade Federal do Amazonas (UFAM), pelo apoio logístico por meio do
Laboratório de Evolução e Genética Animal (LEGAL). E a todos os que contribuíram na
coleta das amostras depositadas na Coleção de Tecido de Genética Animal (CTGA) e que
foram usadas no presente estudo.
Ao Programa de Desenvolvimento Tecnológico em Insumos para Saúde - PDTIS -
FIOCRUZ pelo uso de suas instalações. E especialmente a Victor Souza pela disposição e
ajuda nos dias de desespero e por me mostrar que sempre existe uma solução.
Aos meus pais por sempre ter me apoiado, por confiar em mim e em minhas
capacidades e por sempre estarem do meu lado. Amo vocês!!!!
A minha irmã por me considerar como exemplo na vida. E apesar das circunstancias,
por sempre me amar sem condições.
Ao Nico por ser a força para acordar todos os dias e continuar o caminho. Pelo apoio
sempre, as visitas e o amor incondicional.

v
A minhas adoradas tias Cris, Mariu e Cos por sempre me mandarem a melhor energia,
por se lembrar de mim todos os dias e por cada detalhe de amor.
A minha família toda por me mandar sempre as melhores energias para terminar com
sucesso o mestrado. Especialmente, a minha prima Bibi por confiar em mim, por ser minha
amiga e confidente e sempre me fazer rir.
A Izeni por me aceitar no LEGAL sem me conhecer e por sempre confiar em mim.
Pelas palavras de animo quando me sentia perdida e por me orientar durante os momentos de
dúvidas. E ao Tomas por acreditar em mim e por ter a paciência de me explicar as questões
genéticas.
Aos amigos do LEGAL por serem um apoio constante. Elcio, Pedro, Fabrício,
Roberto, Ju, Mário, Joice, Jessica, Erick, Lu, Deyla, João, José, Wal, Val, Beta, Guta, Vini,
Fabinho, Priscila, Ana Paula, Israela, Aline.
A Dina pelas longas e difíceis horas de trabalho; pela ajuda incondicional e a
disposição constante. A Israela e a Valcita pela ajuda no laboratório nos últimos momentos de
bancada. Ao Vini por ser meu companheiro de captura. A Wal e a Beta por terem a paciência
de ler meu trabalho e corrigir meu português.
A José por ser uma pedacinho da Colômbia no Brasil. Por compartilhar todo o seu
conhecimento e sempre estar disposto a aclarar minhas dúvidas. Pela ajuda no NGS e pelos
abraços de felicidade quando as coisas funcionavam no laboratório.
A meu colega e amigo jacarólogo Fabinho pelas longas conversas em espanhol e por
sempre estar disposto a me ensinar alguma coisa sobre os bichinhos. E à professora Zilca pela
disposição para me ajudar e pelo apoio na distância.
Aos amigos de vida por sempre estarem presentes para me escutar ou dar um
conselho. Anita, Xime, Mónica, Dianis, Sergini, Charlie, Jucita, Galileu, Arthur, Lucita,
Francy, Vanessa, Wandis, Erika, Guta, Beta, Vini.

vi
A minhas amigas de dança por serem mulheres maravilhosas, por acreditar em mim e
sempre me mandar as melhores energias. Especialmente a Marce por me mostrar que a dança
é a existência em movimento.
Ao Rosse por ser meu anjo da guarda.
A todos os que de uma ou outra forma fizeram parte deste processo de crescimento
acadêmico, profissional, pessoal e espiritual. Agradeço!

vii
"Descobri que minha obsessão por cada coisa em seu lugar, cada assunto em seu tempo, cada palavra
em seu estilo, não era o prêmio merecido de uma mente em ordem, mas, pelo contrário, todo um
sistema de simulação inventado por mim para ocultar a desordem da minha natureza"
Gabriel García Márquez

viii
RESUMO
Melanosuchus niger é uma das 23 espécies da ordem Crocodylia. Recebe o nome
popular de jacaré-açu devido a seu grande tamanho, atingindo uma média de quatro metros
para os machos e 2.8 para as fêmeas. Está distribuído em toda a Amazônia e pode ser
encontrado nos rios da bacia do Amazonas e naqueles que drenam para a costa atlântica. M.
niger é encontrado nos remansos dos rios e principalmente em lagos e áreas alagáveis. A
espécie era muito abundante, mas devido a caça comercial entre os anos 1930-1980, chegou
quase a ser extinto. Porem, devido à proteção governamental e estratégias de conservação, as
suas populações tem se recuperado consideravelmente. Já foram desenvolvidos alguns
trabalhos sobre a estrutura e diversidade genética, entretanto com resultados parciais devido a
amostragem restrita. Nesse trabalho tentamos abranger a maior área possível da distribuição
da espécie, e mediante o uso do marcador mitocondrial Citocromo b (Cytb) e marcadores
SNPs (Single Nucleotide Polymorphisms), analisamos as populações avaliando os padrões de
variabilidade genética, a estrutura das populações e testamos o efeito de corredeiras como
barreira para a dispersão. Assim, foram analisadas 245 sequências do gene Cytb de 17
localidades, para calcular os índices de diversidades genética (Ĥ e π) e o equilíbrio genético
(D de Tajima e Fs de Fu). Para determinar a estrutura genética, foi realizada uma AMOVA,
comparações de Fst par a par e análises Bayesianas para estabelecer o número de grupos
biológicos. Usando sequenciamento de próxima geração (Next Generation Sequencing),
foram desenvolvidos os SNPs com a metodologia de ddRADseq para 81 indivíduos de 10
localidades. Os resultados mostraram que existe uma forte estruturação nas populações de
jacaré-açu determinada por dois fatores: isolamento por distância e presença de barreiras
geográficas. O isolamento por distância determina um padrão de distribuição da diversidade
Oeste-Leste, representado na sua maioria por dois haplótipos mais comuns. As corredeiras
dos rios Araguaia, Branco e Madeira/Guaporé são barreiras para a dispersão das populações,
por isso as populações que estão a montante delas devem ter colonizado essas áreas antes da
sua formação, gerando grupos biológicos estruturados. Testando as corredeiras do rio Madeira
como barreira, foi observado que elas restringem o fluxo gênico, que é dado de forma
unidirecional (de cima para baixo das corredeiras). Em nível geral, as populações de M. niger
estão em expansão, provavelmente após uma redução de tamanho drástica como a que
sofreram no século passado. Finalmente, foram propostas sete áreas para conservação: para as
áreas Oeste, Leste e Centro deveria existir pelo menos uma unidade operacional para o
manejo e conservação da espécie, e para as outras quatro áreas foi possível delimitar unidades
de manejo (UMs) para as populações dos rios Araguaia, Guaporé, Uraricoera e Napo.
Palavras-chave: variabilidade genética, padrões de dispersão, Cytb, SNPs, fluxo gênico,
barreiras geográficas.

ix
ABSTRACT
Melanosuchus niger is one of the 23 species of Crocodylia order. Receives the popular
name of black caiman due to its predominant color, averaging four meters for males and 2.8
for females. It is distributed in the entire Amazon and can be found in the rivers of the
Amazon basin and those that drain to the Atlantic coast. It is found in the backwaters of rivers
and especially in lakes and wetlands. The species was abundant, but due to commercial
hunting during 1930-1980, came close to being extinct. However, with government protection
and conservation strategies, their populations has recovered considerably. Some works on the
structure and genetic diversity have been developed, obtaining partial results due to restricted
sample. In this study, we try to cover the largest possible area of species distribution, and
through the use of a mitochondrial marker cytochrome b (Cytb) and SNPs markers (Single
Nucleotide Polymorphisms), we analyzed the populations evaluating the genetic variability
patterns, the structure of populations and tested the effect of rapids as a barrier to dispersion.
Thus, 245 Cytb sequences from 17 localities were analyzed to calculate the rates of genetic
diversity (H and π) and genetic equilibrium (Tajima´s D and Fu´s Fs). To determine the
genetic structure, AMOVA was performed; Fst pairwise comparisons and Bayesian analysis
were performed to determine the number of biological groups. Using next-generation
sequencing (NGS), SNPs were developed with the ddRADseq methodology using 81
individuals from 10 localities. The results showed that there is a strong structure in black
caiman populations determined by two factors: isolation by distance and presence of
geographical barriers. Isolation by distance determines a pattern of diversity distribution
West-East, represented mostly by the two most common haplotypes. The rapids of the rivers
Araguaia, Branco and Madeira/Guaporé are barrier to the dispersion of the population, so the
upstream populations must have colonized these areas prior to its formation, generating
biological groups isolated. Testing the rapids on the Madeira River as a barrier, it was
observed that they restrict gene flow, which is unidirectional (upstream to downstream the
rapids). At a general level, M. niger populations are expanding, probably after a drastic
reduction of size as that suffered in the past century. Finally, we proposed seven areas for
conservation: for West, East and Centre areas, should be defined at least one operating unit
for the management and conservation of the species; and for the other four areas, it was
possible to define management units (MUs) to the population of the Araguaia, Guaporé,
Uraricoera and Napo rivers.
Keywords: genetic variability, dispersal patterns, Cytb, SNPs, gene flow, geographical
barriers.

x
SUMÁRIO
RESUMO ................................................................................................................................ viii
ABSTRACT .............................................................................................................................. ix
LISTA DE TABELAS ............................................................................................................. xii
LISTA DE FIGURAS ............................................................................................................. xiii
INTRODUÇÃO .......................................................................................................................... 1
1.1 Crocodilianos .................................................................................................................................1
1.2 História natural de Melanosuchus niger .........................................................................................2
1.3 Conservação e estado do jacaré-açu ...............................................................................................4
1.4 Genética, genômica e conservação .................................................................................................6
1.5 Estudos genéticos em Melanosuchus niger ..................................................................................10
OBJETIVOS ............................................................................................................................. 12
2.1 Objetivo geral ...............................................................................................................................12
2.2 Objetivos específicos....................................................................................................................12
MATERIAL E MÉTODOS ...................................................................................................... 12
3.1 Área de estudo ..............................................................................................................................12
3.2 Amostragem .................................................................................................................................13
3.3.1 DNA mitocondrial .................................................................................................................14
3.3.2 DNA genômico ......................................................................................................................16
3.4 Análises de dados .........................................................................................................................21
3.4.1 DNA mitocondrial .................................................................................................................21
3.4.2 DNA genômico ......................................................................................................................23
RESULTADOS ........................................................................................................................ 26
4.1 DNA mitocondrial ........................................................................................................................26
4.1.1 Diversidade genética intra-populacional ..............................................................................26
4.1.2 Estrutura entre populações ...................................................................................................26
4.1.3 Grupos biológicos .................................................................................................................29
4.1.4.Genealogia de haplótipos ......................................................................................................29
4.2 DNA genômico ............................................................................................................................31

xi
4.2.1 Diversidade genética intra-populacional ..............................................................................31
4.2.4 Grupos biológicos .................................................................................................................33
4.3 Análises genéticas: Rio Madeira ..................................................................................................34
DISCUSSÃO ............................................................................................................................ 37
5.1 Estrutura genética na Amazônia ...................................................................................................37
5.2 Diversidade genética ....................................................................................................................42
5.3 Rio Madeira ..................................................................................................................................43
5.4 Conservação .................................................................................................................................45
CONCLUSÃO .......................................................................................................................... 49
REFERÊNCIAS BIBLIOGRÁFICAS ..................................................................................... 50

xii
LISTA DE TABELAS
Tabela 1. Avaliações populacionais realizadas em várias áreas do Brasil que evidenciam a recuperação local da
espécie M. niger segundo a "Avaliação do risco de extinção do jacaré-açu no Brasil" (Marioni et al., 2013).
........................................................................................................................................................................ 5
Tabela 2. Informação sobre o N amostral usado para a obtenção dos dados genéticos. Os códigos das localidades
serão usados ao longo do trabalho. ............................................................................................................... 14
Tabela 3. Primers usados para amplificação e sequenciamento do gene mitocondrial Cytb. Fonte: modificado de
(Hrbek et al., 2008). ...................................................................................................................................... 16
Tabela 4. Índices de diversidade genética e teste de neutralidade das populações de M. niger. N, número de
indivíduos por localidade; Hap, número de haplótipos; S, número de sítios segregantes (polimórficos); π,
diversidade nucleotídica de Nei; Ĥ, diversidade haplotípica de Nei; D (p), teste D de Tajima com valor de p
(α = 0.05); teste Fs de Fu com valor de p (α = 0.02). A localidade Tap (Tapajós) não foi incluída nas
análises pelo baixo número de amostras. N.A.: não se aplica.*Valores significativos. ................................ 27
Tabela 5. Matriz dos valores de Fst nas comparações par a par entre as populações de M. niger a partir dos dados
mitocondriais (Cytb). Os valores significativos (p < 0.04) estão em negrito. .............................................. 28
Tabela 6. Matriz dos valores de Fst nas comparações par a par entre as populações de M. niger a partir dos dados
genômicos (SNPs). Os valores significativos (p < 0.02) estão em negrito. ................................................. 33

xiii
LISTA DE FIGURAS
Figura 1. Etapas do ciclo de vida de Melanosuchus niger (ovo, filhote, adulto). Fotos ovo e adulto: Sandra
Hernández; Foto filhote: Peter Oxford. .......................................................................................................... 3
Figura 2. Distribuição da espécie Melanosuchus niger. Fonte: (Thorbjarnarson, 2010). ........................................ 3
Figura 3. Mapa genético do genoma mitocondrial de um crocodiliano. No círculo vermelho está ressaltado o
gene Citocromo b (Cytb). Fonte: (Janke e Arnason, 1997)............................................................................. 7
Figura 4. Um SNP (Single Nucleotide Polymorphism) é uma variação em um sítio da sequência de DNA. Essa
variação deve ocorrer no mínimo em 1% da população para ser considerada como SNP, pelo contrário é
considerada simplesmente como uma mutação. Fonte: (http://www.ibbl.lu/) ................................................ 8
Figura 5. Distribuição das localidades de jacaré-açu analisadas no presente trabalho, representadas pelas bolinhas
amarelas. As linhas tracejadas vermelhas indicam a presença de corredeiras: 1) Corredeiras do Bem-
Querer; 2) Cachoeira de Teotônio; 3) Cachoeira de Jirau; 4) Corredeiras de Itaboca (Tucurui); 5)
Corredeiras de Santa Isabel. As coordenadas geográficas de cada localidade são: RN - 0°38'S 75°48'O; PS -
5°16'S 74°26'O; Mam - 2° 3'S 65°17'O ; Pur - 4°43'S 62°21'O; Jan - 3°26'S 60°17'O; Bra - 3°21'N 61°25'O;
Ana - 2°32'S 60°55'O; BM - a) 3°53'S 59° 9'O b) 4°24'S 59°47'O c) 5°47'S 61°23'O d) 7°35'S 62°54'O e)
8°12'S 62°45'O; EM - a) 9°24'S 64°25'O b) 9°10'S 64°31'O; AM - a) 12°29'S 64°6'O b) 13°28'S 61° 2'O;
Tro - 1°25'S 56°47'O; Tap - 3°17'S 55°18'O; Xin - 2°52'S 51°59'O; Ara - a) 9°16'S 49°56'O b) 10°55'S
50°37'O c) 11°39'S 50°41'O; FG - 4°16'N 52°10'O; Uac - 3°45'N 51°36'O; LT - 3°39'N 51°18'O. ............ 15
Figura 6. As enzimas de restrição SdaI e Csp6 fragmentam o DNA e os adaptadores P1 e A são ligados aos
fragmentos. Nos adaptadores, está indicado o sítio de reconhecimento das enzimas (vermelho). O
adaptador A contém o sítio de anelamento para o primer de PCR (amarelo), a sequência barcode (laranja) e
o barcode adapter (verde); o adaptador P1 contém o sítio de anelamento para o primer de PCR e
sequenciamento (azul). Fonte: modificada de (Canton, 2014). .................................................................... 17
Figura 7. Gel da amplificação da digestão/ligação ............................................................................................... 18
Figura 8. PCR clonal ou de emulsão: os fragmentos são ligados às IonSpheres (pequenas esferas) através de
primers P1 (sequências complementares ao adaptador P1) na superfície das esferas. Um único fragmento se
liga a uma determinada esfera. Elas são capturadas individualmente em gotículas oleosas (microreatores)
onde a PCR em emulsão ocorre. Nesse processo, são geradas milhares de cópias de cada fragmento. Fonte:
(Mardis, 2008). ............................................................................................................................................. 19
Figura 9. a) As ISPs são incorporadas nos poços do chip Ion 318 V2. Uma ISP entrará por cada poço (esq.); b)
Durante o sequenciamento, a Taq polimerase incorpora uma base e é liberado um fosfato e um H+. Esse
último é detectado por um pHmêtro que transforma a mudança de pH em um sinal elétrico (dir.). Fonte: a)
(Mardis, 2008); b) (Varuzza, 2013). ............................................................................................................. 20
Figura 10. O Sistema Ion PGM está conformado pelos seguintes equipamentos: a) Ion OneTouch 2 (esq.), b) Ion
OneTouch ES (centro), c) Ion Personal Genome Machine (PGM) (dir.), e o servidor Ion Torrent.............. 21
Figura 11. Gráficos gerados no programa BAPS que mostram o número de grupos biológicos para M. niger
baseado nas sequências de mtDNA. Cada barra corresponde a um indivíduo e a sua cor representa o grupo
ao qual pertence. As localidades estão organizadas geograficamente Oeste-Leste (RN-LT) e Norte-Sul
(Bra-AM). a) Gráfico gerado incluindo todas as localidades. São determinados três grupos: grupo Oeste
(vermelho), que predomina ao Oeste; grupo Guiana (azul), que predomina no eixo Norte-Sul; grupo Leste
(verde), que predomina no Leste. b) Gráfico gerado retirando as localidades da Costa Atlântica (FG, Uac,
LT). São determinados quatro grupos, os três anteriores mais um novo grupo, grupo Bolívia (amarelo) que
corresponde as localidades EM e AM. c) Esquema que mostra a distribuição dos grupos biológicos na bacia
Amazônica e Costa Atlântica e os padrões encontrados. As linhas tracejadas vermelhas indicam a presença
de corredeiras. ............................................................................................................................................... 30

xiv
Figura 12. a) Rede de haplótipos baseada nas sequências de mtDNA, que mostra as relações genealógicas entre
os indivíduos, gerada no programa HaploViewer. Cada círculo representa um haplótipo e o número dentro
dele, indica quantos indivíduos possuem esse haplótipo. A letra H com um número representa os haplótipos
mais frequentes. Os nós azuis representam passos mutacionais; b) Padrão de distribuição Leste-Oeste dos
grupos biológicos gerado no programa BAPS e a correspondência com os haplótipos encontrados; c)
Padrão de distribuição Norte-Sul dos grupos biológicos gerado no programa BAPS e a correspondência
com os haplótipos encontrados. .................................................................................................................... 32
Figura 13. Estrutura populacional determinada no programa Structure mediante inferência Bayesiana. Foram
analisados 441 SNPs de 76 indivíduos de 8 localidades. A maior probabilidade posterior está associada
com três grupos biológicos. a) Gráfico individual; cada barra vertical representa um indivíduo; b) gráfico
populacional.................................................................................................................................................. 34
Figura 14. a) Gráfico gerado no programa BAPS que mostra o número de grupos biológicos para M. niger
baseado nas sequências de mtDNA das localidades do rio Madeira. Cada barra corresponde a um indivíduo
e a sua cor representa o grupo biológico ao qual pertence. São determinados quatro grupos: amarelo no rio
Guaporé (AM) e entre as corredeiras do Madeira/Guaporé (EM); rosa entre as corredeiras (EM); lilás e
vermelho no baixo Madeira (BM); b) Mapa que mostra a distribuição dos grupos biológicos no rio
Madeira. As linhas brancas representam as corredeiras mais importantes; no sentido do rio em direção Sul-
Norte: Guajará-Mirim, Jirau, Teotônio e Santo Antônio. ............................................................................. 35
Figura 15. Árvore filogenética não enraizada gerada no programa RAxML pelo método de máxima
verossimilhança. As sombras representam os grupos determinados mediante a análise Bayesiana; b)
Estrutura populacional determinada no programa Structure mediante inferência Bayesiana. Foram
analisados 910 SNPs de 30 indivíduos de 3 localidades do rio Madeira. A maior probabilidade posterior
está associada com dois grupos biológicos. .................................................................................................. 36
Figura 16. Modelo de fluxo gênico proposto para explicar o efeito das corredeiras do Rio Madeira como barreira
para M. niger. AM e EM formam um grupo diferente de BM, possivelmente devido a presença da
cachoeira de Teotônio. O fluxo gênico é unidirecional, dado que os indivíduos descem mas não conseguem
subir através das corredeiras. ........................................................................................................................ 44
Figura 17. Mapa das sete áreas definidas para conservação do jacaré-açu (M. niger) ao longo da sua de
distribuição. As interrogações indicam ausência de dados. A distribuição da espécie está delimitada pela
área branca. Camada da distribuição tomada de (IUCN, 2000). ................................................................... 48

1
INTRODUÇÃO
1.1 Crocodilianos
Os crocodilianos modernos, da ordem Crocodilya, apareceram há 130 milhões de anos
aproximadamente. Eles têm seus ancestrais nos Protosuchios do Triássico superior, que eram
um grupo de pequenos crocodiliformes terrestres. Esse grupo desapareceu há 195 milhões de
anos e só no Jurássico inferior surgiram novos crocodilos do clado Mesosuchia. Esses animais
experimentaram uma grandiosa irradiação adaptativa, mas desapareceram dando lugar aos
Eusuchios durante o Cretáceo inferior, considerados as formas mais avançadas, os quais
incluem o grupo dos atuais crocodilianos (Rodríguez, 2000).
Existem 23 espécies reconhecidas atualmente pelo Grupo de Especialistas de
Crocodilianos (CSG - Crocodile Specialist Group), agrupadas em três linhagens evolutivas e
classificadas como famílias (King e Burke, 1997): Crocodylidae que inclui os crocodilos dos
gêneros Crocodylus, Mecistops e Osteolaemus; Alligatoridae com os jacarés dos gêneros
Alligator, Caiman, Paleosuchus e Melanosuchus; Gavialidae representada pelos gêneros
Gavialis e Tomistoma (Thorbjarnarson, 1996; Rodríguez, 2000; Brochu, 2003; Gatesy et al.,
2004; McAliley et al., 2006; Rueda-Almonacid et al., 2007).
Os crocodilianos são animais ectodérmicos e noturnos; são predadores generalistas
que dependem da água e distribuem-se nas regiões tropicais e subtropicais da América,
África, Ásia e Oceania. Representam um pequeno grupo, relativamente homogêneo, de répteis
caracterizados por seu corpo longo, cauda bastante musculosa, membros curtos, membrana
nictitante transparente o que permite a visão embaixo da água, maturidade demorada, vida
reprodutiva longa, oviparidade e uma ou poucas posturas por ano (Thorbjarnarson, 1996).
Caracterizam-se por serem os maiores predadores dos seus hábitats; eles exercem um
efeito importante nos seus ambientes porque mantêm a estrutura e função do ecossistema,
devido as suas atividades como seleção de peixes, reciclagem de nutrientes e manutenção de
refúgios durante os períodos secos (Ross, 1998). Todas as espécies têm sofrido uma
diminuição das suas populações por causa da caça para alimentação, comércio, tráfico,
incompatibilidade com o homem e seus animais domésticos, destruição, alteração e
contaminação do seu hábitat (Plotkin et al., 1983). Dado o valor do seu couro, elas suportam
um comércio internacional acima de 500 milhões de dólares anualmente. Por conseguinte, a

2
perda de qualquer espécie de crocodiliano representa uma perda significativa de
biodiversidade, potencial econômico e equilíbrio do ecossistema (Ross, 1998).
Suas características biológicas proporcionam grande potencial de resiliência às suas
populações, permitindo sua recuperação a partir de um estado crítico. Contudo, a caça não
regulada de adultos pode levar a um rápido declínio das populações, especialmente quando
combinado com a perda de hábitat. Assim, a conservação dos crocodilianos depende da
possibilidade de proporcionarmos incentivos para a manutenção de suas populações e seu
hábitat, e disposição para aceitar práticas de manejo que permitam a coexistência entre
humanos e crocodilianos (Ross, 1998).
Na Amazônia ocorrem cinco espécies de crocodilianos da família Alligatoridae: duas
espécies do gênero Caiman, que atingem até três metros de comprimento, C. crocodilus
conhecido como jacaré-tinga e C. yacare conhecido como jacaré-do-pantanal; duas espécies
do gênero Paleosuchus caracterizadas por serem as menores e as mais primitivas dentre os
jacarés, P. palpebrosus conhecida como jacaré-paguá e P. trigonatus como jacaré-coroa. E
por fim, Melanosuchus niger, a única espécie do gênero e a de maior tamanho da família. Das
cinco espécies, é a única cuja distribuição está restrita à Amazônia e a que mais sofreu pressão
devido a caça ilegal, durante o século passado. O jacaré-açu, como é conhecido popularmente,
é a espécie objeto de estudo do presente trabalho.
1.2 História natural de Melanosuchus niger
Etimologicamente, o nome Melanosuchus niger deriva do grego “melanos” (negro),
“souchus” (crocodilo) e “niger” (negro), dada a cor predominante desta espécie (Vasquez,
1991). No Brasil, é conhecida popularmente como jacaré-açu, uma palavra que vem do tupi
guarani e significa jacaré grande ou maior jacaré.
Melanosuchus niger é considerado o maior predador da bacia amazônica. Os machos
atingem uma média de quatro metros de comprimento total (Vasquez, 1991), ainda que
existam registros de espécimes de até seis metros (Medem, 1983; Plotkin et al., 1983). As
fêmeas chegam à maturação sexual aos dois metros de comprimento total e o seu tamanho
médio quando adultas é 2.8 m. O tempo de geração, ou seja, o tempo no qual os indivíduos
alcançam a maturação sexual é de 10 a 15 anos. Os ninhos são feitos de folhas, ramos e
galhos em morrinhos, onde são colocados entre 35-50 ovos. Os filhotes medem

3
aproximadamente 30 cm de comprimento quando nascem e recebem o cuidado da mãe por
um tempo (Thorbjarnarson, 1996) (Figura 1).
Figura 1. Etapas do ciclo de vida de Melanosuchus niger (ovo, filhote, adulto). Fotos ovo e adulto: Sandra
Hernández; Foto filhote: Peter Oxford.
O jacaré-açu possui um focinho largo, liso e uma proeminência óssea que se estende
desde acima dos olhos para baixo. Sua coloração dorsal é predominantemente preta quando
adulto, tem 3 a 5 manchas redondas e pretas sobre os lados das mandíbulas e o ventre
totalmente branco ou amarelado. A íris é verde-amarelada. Os filhotes e juvenis são pretos
com listras amareladas no corpo (Rueda-Almonacid et al., 2007).
Existe registro da espécie na Bolívia, Brasil, Colômbia, Equador, Guiana, Guiana
Francesa e Peru (Plotkin et al., 1983) (Figura 2). No Brasil, ela se distribui nos estados do
Acre, Amapá, Amazonas, Mato Grosso, Pará, Rondônia, Roraima e Tocantins (Medem, 1983;
Plotkin et al., 1983; Ross, 1998). Vasquez (1991) descreve a ocorrência de M. niger nos rios
Juruá, Purus, Madeira, Tapajós, Xingu, Araguaia, Tocantins, Negro, Trombetas e Amazonas,
incluindo as ilhas próximas à foz do rio Amazonas. Encontra-se com maior frequência em
águas com pouca correnteza, portanto pode ser observado nos remansos dos rios e, sobretudo
em lagos, bosques alagados e áreas pantanosas pouco profundas (Medem, 1963).
Figura 2. Distribuição da espécie Melanosuchus niger. Fonte: (Thorbjarnarson, 2010).

4
1.3 Conservação e estado do jacaré-açu
Esta espécie era muito abundante na Amazônia. Bates (1863; em (Plotkin et al., 1983))
ficou impressionado com as suas altas densidades no rio Solimões. Mas durante o último
século, suas populações foram reduzidas e algumas extintas na sua área de distribuição
natural. Hoje é pouco comum ver o jacaré-açu no canal principal do rio Solimões (Plotkin et
al., 1983). A principal causa do seu declínio foi o interesse comercial para a obtenção de
couro, sofrendo superexploração dada a qualidade superior do seu couro (Plotkin et al., 1983;
Brazaitis et al., 1996; Thorbjarnarson, 1998; Da Silveira e Thorbjarnarson, 1999; De Thoisy
et al., 2006).
A caça comercial de M. niger começou na década de 1930, porém chegou ao máximo
nos anos 50s, quando eram caçados aproximadamente 1.200.000 espécimes por ano (Ojasti,
1996). Entre 1950 e 1965, mais de 7.5 milhões de peles de jacaré-açu foram exportadas só do
estado do Amazonas, Brasil (Da Silveira e Thorbjarnarson, 1999). A grande demanda de peles
na Europa e nos Estados Unidos, junto com a perda de hábitat afetou as populações naturais
até o limite da extinção entre 1960 e 1969, alcançando um alto nível de fragmentação (CSG,
1995; Britton, 2012). Só depois da Lei N° 5.197 de 1967, a fauna silvestre no Brasil foi
protegida oficialmente, porém a caça ilegal de jacaré-açu continuou até 1980 (Plotkin et al.,
1983).
A espécie tem se recuperado substancialmente no Brasil devido a estratégias para a
conservação e uso dos jacarés que incluíram investigação em nível da biologia, genética,
densidade populacional e estrutura de tamanho, seleção de hábitat, padrões de movimento,
ecologia, alimentação e reprodução, que geraram recomendações apropriadas para o manejo
(Da Silveira e Thorbjarnarson, 1999).
Mas, as populações naturais dessa espécie continuam sendo afetadas por ações
antrópicas tais como construção de barragens, desmatamento e caça. O aumento da
construção de usinas hidroelétricas (UHE) pode ter efeito sobre o estado de conservação do
jacaré-açu devido aos impactos ambientais e ecológicos que geram nas regiões onde são
instaladas (Marioni et al., 2013). A fragmentação dos rios e portanto a perda de conectividade
é um dos maiores impactos; as usinas também podem causar o alagamento de áreas florestais
para a criação de reservatórios assim como desmatamento devido à construção de vias (Finer
e Jenkins, 2012).
Além disso, o uso de carne de jacaré-açu, jacaré-tinga e botos como isca para capturar
piracatinga (Calophysus macropterus) é uma nova forma de pescaria desenvolvida na

5
Amazônia. Embora essa atividade possivelmente começou na década de 90, foi reportada por
primeira vez no ano 2000, quando chamou a atenção para os problemas ecológicos devido ao
impacto sobre as populações de botos e jacarés (Estupiñán et al., 2003; Brum et al., 2015).
Estima-se que só em 2013, foram mortos uns 2.300 jacarés nos rios Solimões e Japurá.
Recentemente, o governo brasileiro criou uma normativa que proíbe a pescaria da piracatinga
durante cinco anos, a qual entrou em vigor em janeiro de 2015 (Botero-Arias et al., 2014).
O Estado do Amazonas é o maior produtor de carne salgada e seca de jacarés. Entre os
anos 1980 e 1999, estima-se que foram extraídas 65 toneladas de carne por ano da Reserva de
Desenvolvimento Sustentável Mamirauá. No ano 2005, foram extraídas aproximadamente 50
toneladas (5000 indivíduos) da Reserva de Desenvolvimento Sustentável Piagaçu-Purus
(Marioni et al., 2013).
Atualmente, os dados mostram que o jacaré-açu ocorre na sua área de distribuição
histórica no Brasil e que em algumas localidades é abundante (Jelden et al., 2007; Marioni et
al., 2013) (Tabela 1). Por isso, M. niger consta na categoria de baixo risco, dependente de
conservação da Lista Vermelha de Espécies Ameaçadas da IUCN (International Union for
Conservation of Nature) desde 2000 (IUCN, 2014). Encontra-se listado no Apêndice II da
CITES (Convention on International Trade in Endangered Species of Wild Fauna and Flora)
desde 2007, o que facilita o manejo do uso comercial (Thorbjarnarson, 2010).
Tabela 1. Avaliações populacionais realizadas em várias áreas do Brasil que evidenciam a recuperação local da espécie M.
niger segundo a "Avaliação do risco de extinção do jacaré-açu no Brasil" (Marioni et al., 2013).
ANO AVALIAÇÕES POPULACIONAIS FONTE
1994-1999 Reserva de Desenvolvimento Sustentável (RDS) Mamirauá
(AM): número de jacarés-açu aumentou de 556 a 3789 (580%);
número de ninhos em um lago aumentou de 1 a 22 ninhos
(Da Silveira, 2001)
2004-2005 85 localidades AM, AP, RO, TO e GO (768 km): 37.000 jacarés-
açu (2.1-467 ind./km)
(CITES, 2007)
2004-2008 Reserva Extrativista (RESEX) Lago Cuniã (RO) (467km): 16782
jacarés (5.1-100.4 ind./km)
(Mendoça e
Coutinho, 2009)
2005-2012 Reserva de Desenvolvimento Sustentável (RDS) Piagaçu-Purus
(AM): número de jacarés-açu adultos aumentou de 5.4% a 13.4%
(Marioni, resultados
não publicados)
2006-2010 Área de Proteção Ambiental (APA) Meandros do Araguaia (TO):
aumentou número de jacarés-açu
(Andrade e Coutinho,
2007; 2009)
2008 Região Lago Badajós (AM) (51km): 1.038 jacarés-açu (14-38
ind./km)
(Andrade e Coutinho,
2011)
2008 Parque Nacional (PARNA) Cabo Orange (AP) (43.5km): 370
jacarés-açu
(Andrade e Coutinho,
2011)

6
É importante ressaltar que entre os critérios biológicos para a inclusão de espécies na
Lista Vermelha da IUCN e nos Apêndice da CITES, vários parâmetros da diversidade
genética contribuem na determinação da categoria de uma espécie (Frankham et al., 2004).
Por isso, tem sido desenvolvidos vários estudos que utilizam técnicas moleculares para
resolver questões genéticas em nível populacional ou individual que contribuem com o
manejo, conservação e recuperação dos crocodilianos.
1.4 Genética, genômica e conservação
Na última década, os avanços dos métodos moleculares permitiram a utilização de
dados genéticos em estudos de processos populacionais e de questões ecológicas com mais
eficiência (Selkoe e Toonen, 2006). Assim, surge a genética da conservação como ferramenta
para entender os processos genéticos e evolutivos e delinear os padrões que são importantes
para o manejo das populações ameaçadas, preservando assim as espécies como entidades
dinâmicas capazes de lidar com mudanças ambientais (Frankham et al., 2004).
O estudo da estrutura populacional através de técnicas moleculares talvez seja a parte
mais importante da genética da conservação e têm sido útil tanto no estudo de populações
exploradas comercialmente (ou seja, abundantes, mas com riscos populacionais devido a
superexploração), como nas espécies já ameaçadas de extinção (Frankham et al., 2004).
Especificamente, a identificação de populações, a resolução da estrutura populacional,
a definição de unidades de conservação, a quantificação da depressão por endogamia, o
tamanho efetivo populacional, o tamanho mínimo viável da população, os níveis de variação
genética e o fluxo gênico em populações naturais entre outros, fornecem medidas específicas
e comparáveis dos processos que afetam as populações, e permitem entender os efeitos das
forças que geram mudanças evolutivas no tempo (DeSalle e Amato, 2004).
Assim, para o manejo e restauração de populações é importante preservar a
diversidade adaptativa e os processos evolutivos ao longo da distribuição geográfica da
espécie. Isso depende do grau e natureza das perturbações recentes e requer que as
recomendações feitas sejam baseadas em uma amostragem adequada e análises apropriadas
(Crandall et al., 2000).
Para isso, são utilizados diversos marcadores moleculares, ou seja, fragmentos de
DNA que permitem diferenciar indivíduos, que são herdados geneticamente e facilmente

7
detectáveis no genoma. Eles proveem informações sobre o polimorfismo de um determinado
loco e podem ser provenientes do DNA mitocondrial (mtDNA) ou do DNA nuclear (nDNA).
O mtDNA têm um papel importante em estudos filogenéticos e de genética de
populações devido a suas características. Possui herança uniparental (linhagem materna) e
ausência de recombinação e rearranjos genéticos, o que facilita o monitoramento de
transmissão ao longo das linhas evolutivas começando em uma evolução antiga (Avise et al.,
1987); tem um alto nível de variabilidade e uma alta taxa de evolução de 5-10 vezes maior
quando comparado com o nDNA (Brown et al., 1979).
O mtDNA dos aligatorídeos é uma molécula de DNA de aproximadamente 17.000
pares de bases (pb). Assim, como em outros metazoários, possui 22 tRNAs (RNAs de
transferência), 13 genes que codificam proteínas, 2 rRNAs (RNAs ribossomais) e uma região
conhecida como região controle ou D-loop que controla os processos de replicação e
transcrição. O arranjo dos tRNAs varia em relação a outros vertebrados, mas a organização
dos genes codificadores de proteínas é o mesmo que em mamíferos, anfíbios e peixes (Figura
3). Os estudos tem mostrado que a taxa evolutiva do mtDNA dos crocodilianos é
significativamente maior do que o resto de répteis (Janke e Arnason, 1997; Janke et al., 2001;
Roos et al., 2007), o que contradiz a hipótese de uma correlação entre a taxa de evolução
molecular e o tempo de geração (Martin e Palumbi, 1993; Janke et al., 2001).
Figura 3. Mapa genético do genoma mitocondrial de um crocodiliano. No círculo vermelho está ressaltado o
gene Citocromo b (Cytb). Fonte: (Janke e Arnason, 1997).

8
O gene Citocromo b (Cytb) é um dos marcadores mitocondriais mais utilizados para
análises moleculares em diferentes tipos de estudos em vertebrados (desde ecológicos até
evolutivos). Desde as primeiras publicações, foi sugerido que ele era variável o suficiente para
questões de populações e conservado o suficiente para questões filogenéticas. É um dos genes
mais sequenciados em vertebrados; a sua dinâmica, estrutura e função são bem conhecidas e
caracterizadas; e ainda que a sua taxa de substituição seja lenta, a taxa de evolução de
substituições silenciosas na terceira posição do códon é similar com outros genes
mitocondriais (Meyer, 1994; Johns e Avise, 1998).
Além das técnicas clássicas, o desenvolvimento da tecnologia genômica que envolve
sequenciamento de alto rendimento, tem permitido o uso de novos marcadores moleculares,
tais como SNPs (Single Nucleotide Polymorphisms), como as principais ferramentas para
detectar de forma rápida e eficaz a variabilidade genética (DeSalle e Amato, 2004). A
genômica da conservação aparece como uma nova ferramenta que, através de centenas de
marcadores moleculares, permite abordar questões muito importantes da conservação em um
nível populacional detalhado, que antes não era possível de serem tratadas por limitações dos
marcadores ou falta de informação genética (Allendorf et al., 2010; Catchen et al., 2013a).
Um SNP é a mudança de uma base em uma sequência de DNA. Embora qualquer das
quatro bases possa estar presente nessa posição, na prática os SNPs são bialélicos, ou seja,
existem duas alternativas ou possíveis nucleotídeos (Figura 4). Para tal posição ser
considerada como SNP, o alelo menos frequente deve estar presente pelo menos em 1% da
população. A sua evolução é por modelos de mutação simples, como o modelo de sítios
infinitos, e dado que a sua origem é através da substituição de um nucleotídeo, estima-se que
a sua taxa de mutação está entre 1x10-9
- 5x10-9
por nucleotídeo por ano para posições
neutrais no caso dos mamíferos (Vignal et al., 2002).
Figura 4. Um SNP (Single Nucleotide Polymorphism) é uma variação em um sítio da sequência de DNA. Essa
variação deve ocorrer no mínimo em 1% da população para ser considerada como SNP, pelo contrário é
considerada simplesmente como uma mutação. Fonte: (http://www.ibbl.lu/)

9
Os SNPs estão amplamente distribuídos no genoma e são as formas mais frequentes de
variações genéticas. Sua alta densidade permite estudar a herança de regiões genômicas. Uma
forma rápida e econômica de identificá-los é o screening de pequenos segmentos do genoma
de vários indivíduos, que são adjacentes ao sítio de reconhecimento de uma enzima de
restrição. Esses segmentos são conhecidos como RADs (Restriction-Site Associated DNA) e o
seu desenvolvimento permite ter uma subamostra do genoma de regiões homólogas para a
descoberta de milhares de SNPs e um alto rendimento na genotipagem de populações (Baird
et al., 2008). Um dos métodos mais eficientes é o ddRADseq (double digest RAD sequencing),
que consiste em gerar bibliotecas genômicas que possuem fragmentos obtidos mediante
digestão com duas enzimas de restrição e que estão dentro de uma faixa de tamanho
selecionada, e que serão posteriormente sequenciadas mediante sequenciamento de próxima
geração (NGS - Next Generation Sequencing) (Peterson et al., 2012).
Dessa forma, a genômica de populações que usa RADs e genotipagem por
sequenciamento, permite que questões clássicas em genética, ecologia e evolução, tais como
identificação de paternidade e parentesco, migração e fluxo gênico, estrutura populacional,
filogeografia, reconstrução filogenética, detecção de seleção e adaptação molecular possam
ser abordadas com uma potência e precisão sem precedentes (Morin et al., 2004; Catchen et
al., 2013b).
A combinação de distintos marcadores moleculares e métodos analíticos
complementares são relevantes para identificar sinais de diferentes tempos no passado a partir
dos dados genéticos, dando um maior suporte aos resultados e uma medida da força de
determinado sinal (Pearse et al., 2006). Assim, é possível uma adequada interpretação da
história evolutiva das populações e o acesso aos níveis de diversidade bem como à estrutura
genética delas.
Nesse estudo foram usados os dois marcadores moleculares mencionados
anteriormente: o gene Cytb e os SNPs para estabelecer as relações entre os indivíduos,
determinar a diversidade genética e conhecer a estrutura populacional do jacaré-açu na
Amazônia. O gene Cytb já foi usado em estudos anteriores em crocodilianos amazônicos
(Glenn et al., 2002; Farias et al., 2004; De Thoisy et al., 2006; Hrbek et al., 2008;
Vasconcelos et al., 2008; Muniz, 2012), e o uso de SNPs é uma nova abordagem para estudos
populacionais nesse grupo.

10
1.5 Estudos genéticos em Melanosuchus niger
Os primeiros estudos sobre diversidade genética e estrutura populacional em jacaré-
açu foram realizados por Farias et al. (2004) através do marcador mitocondrial Cytb. Os
autores estudaram indivíduos de quatro localidades: três da Amazônia Central e uma da
Guiana Francesa, encontrando uma alta diversidade genética do marcador, quanto a número
de haplótipos, homozigosidade observada e diversidade gênica, quando comparada com a
diversidade encontrada em Alligator mississippiensis (Glenn et al., 2002). Além disso, o
estudo mostrou estruturação populacional, determinada por isolamento por distância nas
populações da costa atlântica e aquelas da bacia amazônica, e possivelmente pelo tipo de água
nas populações da Amazônia central. Adicionalmente, a expansão demográfica também pode
ser parte importante da dinâmica populacional em algumas áreas. No entanto, M. niger
precisará de tempo para se recuperar completamente, mas com o manejo contínuo, suas
populações poderão apresentar uma recuperação real.
Alguns marcadores microssatélites das espécies Caiman latirostris (sete) e A.
mississipiensis (28) foram testados em M. niger, dos quais só oito amplificaram com sucesso
e foram polimórficos (De Thoisy et al., 2006). Os autores reportaram uma alta diversidade
genética em todas as sete populações estudadas (Guiana Francesa, Brasil e Equador),
considerando o número de alelos, heterozigosidade observada e esperada e diversidade
gênica. Eles observaram estruturação entre populações geograficamente próximas mas
ecologicamente distintas, assim como foi sugerido por Farias et al. (2004). O estudo indicou
que as populações atuais de M. niger se originaram da região Amazônica central devido a
distribuição da diversidade genética encontrada.
Posteriormente, o estudo realizado por Vasconcelos et al. (2008), mostrou também
uma alta diversidade genética do marcador Cytb, inclusive maior que a observada por Farias
et al. (2004), além de expansão demográfica em algumas áreas. Foi encontrada correlação
significativa entre a diversidade genética e a distância geográfica (entre populações da costa
atlântica e da bacia amazônica). Entretanto, ao contrário dos estudos anteriores (Farias et al.,
2004; De Thoisy et al., 2006), não foram observadas diferenças significativas relacionadas
com o tipo de água, embora a diferenciação genética dentro do tipo de água seja menor do que
entre os tipos de água.
Hrbek et al. (2008) realizaram uma análise filogenética dos jacarés sul americanos
usando o gene mitocondrial Cytb e os genes nucleares RAG1 (Recombination-Activating Gene
1) e MYC (gene regulador que codifica para um fator de transcrição). No que se refere a M.

11
niger, encontraram que é o grupo irmão do gênero Caiman. Mas, o mais relevante do estudo
foi com relação as espécies do gênero Caiman, C. yacare e C. crocodilus. Encontrou-se que a
área das corredeiras do Rio Madeira é uma área de transição entre as duas espécies, fato
explicado por duas possíveis hipóteses: houve contato secundário seguido de hibridização ou
diferenciação ao longo de um cline. Então, dado que M. niger ocorre em ambos lados das
corredeiras, tanto a montante quanto a jusante, pode significar que as corredeiras não atuam
como barreira à migração de indivíduos.
Finalmente, Muniz et al. (2011) realizaram um estudo sobre paternidade múltipla em
ninhadas de M. niger utilizando os marcadores microssatélites definidos por De Thoisy et al.
(2006). Os seus resultados mostraram que existe poliandria ou paternidade múltipla em M.
niger.
Os estudos prévios com M. niger mostraram um padrão de estruturação consistente
entre as populações da Amazônia central e da costa Atlântica. Dado que o jacaré-açu é uma
espécie de ampla distribuição, é necessário ter uma amostragem o mais abrangente possível
para evitar limitações geográficas e assim detectar padrões de distribuição da variabilidade
genética mais completos. Aparentemente, na calha principal do rio Amazonas não existem
barreiras geográficas que limitem a dispersão de espécies aquáticas de grande porte, o que
torna interessante o fato de incluir localidades dos rios que o drenam e que têm outras
dinâmicas hídricas e geológicas. Por exemplo, vários estudos tem indicado que as corredeiras
do Rio Madeira representam uma divisão biogeográfica total ou parcial para alguns grupos de
organismos aquáticos, tais como botos (Banguera-Hinestroza et al., 2002; Gravena et al.,
2014; Gravena et al., 2015), quelônios (Pearse et al., 2006), peixes (Farias et al., 2010; Willis
et al., 2012) e jacarés (Hrbek et al., 2008; Muniz, 2012). O que leva a perguntas como: qual é
a estrutura genética real da espécie ao longo da sua área de distribuição? Qual é o efeito das
barreiras geográficas nas populações de jacaré-açu? Existem outras barreiras na Amazônia
além das encontradas no rio Madeira?
Considerando os trabalhos realizados anteriormente, esse trabalho visa determinar
níveis de variabilidade genética a partir da associação de dois tipos de marcadores genéticos
com sinais evolutivos diferentes, para ter uma visão mais completa da dinâmica e distribuição
da diversidade genética da espécie M. niger, usando análises complementares.

12
OBJETIVOS
2.1 Objetivo geral
Detectar padrões de distribuição da variabilidade genética em Melanosuchus niger.
2.2 Objetivos específicos
Estimar os parâmetros de diversidade genética de M. niger.
Determinar a estrutura genética das populações de M. niger utilizando dois tipos de
marcadores moleculares (Cytb e SNPs).
Avaliar o efeito de barreiras naturais, tais como as corredeiras do alto rio Madeira, alto
rio Branco e médio rio Araguaia na dispersão de M. niger.
MATERIAL E MÉTODOS
3.1 Área de estudo
A Amazônia é uma região caracterizada pela grande quantidade de água que alberga,
seu clima quente e úmido e por estar coberta pela mais extensa e contínua floresta úmida
tropical. Ela possui a maior bacia de drenagem com uma área de 7 milhões de km2, formada
por rios, igarapés e córregos que drenam no rio mais volumoso do mundo: o rio Amazonas.
Esse sistema fluvial tropical abrange vários países da América do Sul, com 60% no Brasil,
13% no Peru e o resto dividido entre a Bolívia, Colômbia, Equador, Guiana, Guiana Francesa,
Suriname e Venezuela (Sioli, 1967; Lundberg et al., 1998).

13
A rio Amazonas nasce na parte ocidental da cordilheira dos Andes no Peru e deságua
no Oceano Atlântico, onde descarrega quase 175 mil m3/seg. que correspondem a 20% da
água doce que entra nos oceanos. Os maiores afluentes que vêm do Escudo Brasileiro são o
Araguaia-Tocantins, o Xingu e o Tapajós. O rio Madeira também recebe afluentes da parte
norte e oeste do Escudo Brasileiro, mas a maioria das águas do Madeira vem dos Andes
peruanos e bolivianos. Outros afluentes principais com origem nos Andes são o Purus, Juruá,
Ucayali, Huallaga, Marañón, Pastaza, Napo, Iça (Putumayo), Japurá (Caquetá). O rio Negro e
Branco são os maiores tributários do norte, e drenam grande parte da região sudoeste do
Escudo das Guianas. Outros rios que drenam pelo norte, são os que correm pelo sul do
Escudo das Guianas: o Uatumã, Trombetas, Paru e Jari (Lundberg et al., 1998).
O bioma amazônico, inclui rios que não drenam o rio Amazonas. Eles nascem no
Escudo das Guianas, mas deságuam no oceano Atlântico diretamente; geralmente são curtos e
bastante caudalosos. No Brasil, no estado do Amapá encontra-se o rio Uaça e o rio Oiapoque,
que marca a fronteira com a Guiana Francesa, onde os rios mais importantes são o Mana,
Sinnamary e Approuague. Na Guiana, os rios mais importantes são o Essequibo, Demerara,
Berbice, Corentyne e Rupununi.
3.2 Amostragem
As amostras foram coletadas através de procuras noturnas, retirando uma escama
caudal dos indivíduos para as posteriores análises no laboratório. Todas as amostras foram
armazenadas em álcool 95% e guardadas no freezer a -20 °C. Foram utilizadas amostras da
Coleção de Tecidos de Genética Animal (CTGA), da Universidade Federal do Amazonas –
UFAM, de várias localidades dos rios Amazonas, Araguaia, Branco, Japurá, Madeira, Negro,
Tapajós, Trombetas e Xingu.
Para gerar as sequências de mtDNA foram usadas 113 amostras. Foram incluídas as
132 sequências obtidas por Vasconcelos et al. (2006), dos rios Amazonas, Napo, Pacaya,
Japurá, Purus, Negro, Uaçá e Approuague, para um total de 245 sequências. Para o
desenvolvimento dos SNPs foram usadas 81 amostras dos rios: Amazonas, Araguaia, Branco,
Japurá, Madeira, Tapajós e Xingu (Tabela 2 e Figura 5).

14 Tabela 2. Informação sobre o N amostral usado para a obtenção dos dados genéticos. Os códigos das localidades
serão usados ao longo do trabalho.
Pais Localidade Código N para mtDNA N para SNPs
Equador Rio Napo RN 15 -
Peru Pacaya-Samiria, Rio Pacaya PS 6 -
Brasil
RDS Mamirauá, Rio Japurá Mam 14 10
RDS Piagaçu-Purus, Rio Purus Pur 9 -
Lago Janauacá, Rio Amazonas Jan 13 6
ESEC Maracá, Rio Uraricoera (afluente do
Rio Branco)
Bra 22 3
Anavilhanas, Rio Negro Ana 27 10
Baixo Madeira, Rio Madeira: a) Nova
Olinda; b) Borba; c) Manicoré; d) Humaitá;
e) Lago Cuniã
BM 17 10
Entre corredeiras, Rio Madeira: a) Jaci-
Paraná; b) Ilha do Búfalo
EM 17 10
Acima das corredeiras, Rio Guaporé: a)
Costa Marques b) Pimenteiras do Oeste
AM 13 10
Rio Trombetas Tro 8 -
Rio Tapajós Tap 3 2
Rio Xingu Xin 11 10
Rio Araguaia: a) Caseara; b) Ilha do
Bananal; c) São Felix
Ara 11 10
Rio Uaçá, Amapá Uac 18 -
Lago Txipok, Amapá LT 26 -
Guiana
Francesa
Rio Approuague FG 15 -
Total 17 245 81
3.3 Coleta de dados
3.3.1 DNA mitocondrial
Extração de DNA
O DNA genômico (gDNA) total foi extraído das escamas caudais dos indivíduos
amostrados usando o protocolo de CTAB 2% (Doyle e Doyle, 1987). A qualidade da extração
foi analisada através da eletroforese horizontal em gel de agarose 1% e tampão TBE. Os
DNAs genômicos foram quantificados no espectrofotômetro NanoDrop 2000 (Thermo
Scientific) para verificar a concentração e qualidade das amostras.

15
Figura 5. Distribuição das localidades de jacaré-açu analisadas no presente trabalho, representadas pelas
bolinhas amarelas. As linhas tracejadas vermelhas indicam a presença de corredeiras: 1) Corredeiras do Bem-
Querer; 2) Cachoeira de Teotônio; 3) Cachoeira de Jirau; 4) Corredeiras de Itaboca (Tucurui); 5) Corredeiras de
Santa Isabel. As coordenadas geográficas de cada localidade são: RN - 0°38'S 75°48'O; PS - 5°16'S 74°26'O;
Mam - 2° 3'S 65°17'O ; Pur - 4°43'S 62°21'O; Jan - 3°26'S 60°17'O; Bra - 3°21'N 61°25'O; Ana - 2°32'S
60°55'O; BM - a) 3°53'S 59° 9'O b) 4°24'S 59°47'O c) 5°47'S 61°23'O d) 7°35'S 62°54'O e) 8°12'S 62°45'O; EM
- a) 9°24'S 64°25'O b) 9°10'S 64°31'O; AM - a) 12°29'S 64°6'O b) 13°28'S 61° 2'O; Tro - 1°25'S 56°47'O; Tap -
3°17'S 55°18'O; Xin - 2°52'S 51°59'O; Ara - a) 9°16'S 49°56'O b) 10°55'S 50°37'O c) 11°39'S 50°41'O; FG -
4°16'N 52°10'O; Uac - 3°45'N 51°36'O; LT - 3°39'N 51°18'O.
Amplificação de DNA
O fragmento mitocondrial amplificado foi o gene Cytb, juntamente com o tRNA
treonina completos e os tRNAs glutamina e prolina parciais, através da reação em cadeia da
polimerase (PCR) usando os primers GluCRf.1 e ProCRr.1 publicados por Hrbek et al. (2008)
(Tabela 3). As reações de PCR foram feitas para um volume final de 15 μL, contendo 7.25 μL
de ddH2O, 1.25 μL de tampão 10X (100mM Tris-HCl, 500mM KCl), 1.25 de MgCl2 25mM,
1.25 μL de dNTP 2.5 mM; 1,25 μL de cada primer 2μM, 0.5 de Taq 1U/μL e 1.0 μL de DNA.
As condições da reação no termociclador foram: preaquecimento a 92°C por 60 segundos,
desnaturação a 92°C por 50 seg., anelamento a 53.5°C por 60 seg. e extensão a 72°C por 90
seg. Esses últimos três passos foram repetidos 35 vezes, seguidos de uma extensão final a
72°C por cinco minutos. O produto da PCR foi visualizado em gel de agarose 1%.

16
Sequenciamento
O produto de PCR foi purificado com ExoSAP-IT seguindo o protocolo do fabricante.
As reações de sequenciamento foram realizadas contendo: 1.0 μL do produto de PCR
purificado, 2.0 μL do primer de sequenciamento (Tabela 3), 0.3 μL do BigDye Terminator
v3.1, 1.45 μL do tampão do BigDye e 5.5 μL de ddH2O. As condições da reação foram as
sugeridas no protocolo do fabricante do Kit BigDye.
O produto da reação de sequenciamento foi precipitado com etanol e EDTA, e
posteriormente ressuspendido em 10 μL de formamida deionizada para ser injetados e
analisados no sequenciador automático ABI3500 (Applied Biosystems).
Tabela 3. Primers usados para amplificação e sequenciamento do gene mitocondrial Cytb. Fonte: modificado
de (Hrbek et al., 2008).
Primer Sequência 5'-3' Uso
GluCRf.1 CAACCAAAACCTGAGGYCTGA Amplificação
ProCRr.1 ATTAGAAYGTCGGCTTTGGGG Amplificação
CytbCRf.3 CCATACATYGGAGACACCAT Sequenciamento
CytbCRr.2 AAGATYAGGTGGGTKATGAG Sequenciamento
CytbCRf.1 ATGACCCACCAACTACGAAAA Sequenciamento
3.3.2 DNA genômico
Construção de biblioteca genômica
Para a construção das biblioteca genômicas é importante o uso de gDNA de alta
qualidade. O método descrito a seguir foi baseado no proposto por Peterson et al. (2012) com
uma adaptação simplificada e algumas modificações da metodologia proposta por (Martínez
et al., (in prep)).
Extração de DNA
O gDNA total foi extraído do tecido usando o protocolo adaptado de CTAB 2%
(Doyle e Doyle, 1987). As extrações foram visualizadas em gel de agarose 1% para verificar a
integridade do DNA obtido e a presença de contaminantes. Posteriormente, foram
quantificados no NanoDrop 2000 (Thermo Scientific) e foi feito o cálculo para obter 200
ng/μl de cada uma das amostras.

17
Digestão do DNA e ligação de adaptadores (Figura 6).
O DNA foi digerido usando duas enzimas de restrição, SdaI (corte raro) e Csp6I (corte
frequente), e simultaneamente foram ligados os adaptadores A1 e P1 (adaptador normal). Para
cada reação de digestão/ligação foi usado um volume final de 50 μL contendo: 5.0 μL do
tampão TANGO, 2.0 μL do adaptador P1, 2.0 μL de um adaptador A (um diferente para cada
amostra), 0.1 μL de SdaI, 0.1 μL de Csp6I, 0.5 μL de DNA T4 ligase, 0.5 μL de ATP e 38.8
μL de ddH2O. As condições da reação foram: 37 °C por 3 horas e 68 °C por 15 minutos.
Figura 6. As enzimas de restrição SdaI e Csp6 fragmentam o DNA e os adaptadores P1 e A são ligados aos
fragmentos. Nos adaptadores, está indicado o sítio de reconhecimento das enzimas (vermelho). O adaptador A
contém o sítio de anelamento para o primer de PCR (amarelo), a sequência barcode (laranja) e o barcode
adapter (verde); o adaptador P1 contém o sítio de anelamento para o primer de PCR e sequenciamento (azul).
Fonte: modificada de (Canton, 2014).
PCR da digestão/ligação
A partir do produto da digestão/ligação foi feita uma PCR com cinco réplicas por cada
amostra para diminuir o desvio padrão dos alelos representativos de cada indivíduos gerado
pelo viés da PCR. A reação teve um volume final de 25 μL contendo: 12.4 μL de ddH2O, 2.0
μL de MgCl2 25mM, 2.0 μL de dNTPs 2.5 mM, 2.5 μL de tampão 10X (750mM Tris-HCl,
200mM (Na4)2SO4), 2.5 μL do primer P1, 2.5 μL do primer A-amp, 0.1 μL de KlenTaq (uma
1 O adaptador A tem estrutura divergente em "Y" com um único barcode por indivíduo. O formato em "Y"
significa que na extremidade 3' possui uma sequência de nucleotídeos diferente que não é com a extremidade 5',
a qual possui o sítio de anelamento para o primer de amplificação. Além disso, possui uma sequência de dez
pares de bases conhecida como barcode que serve para identificar cada indivíduo utilizado na construção da
biblioteca genômica. Portanto, é usado um adaptador A diferente para cada amostra.

18
Taq de alta fidelidade que corrige erros de amplificação) e 1.0 μL do produto da
digestão/ligação. Os ciclos de amplificação foram: preaquecimento a 68°C por 60 seg.,
seguido de 18 ciclos de desnaturação a 93°C por 10 seg., anelamento a 52°C por 35 seg. e
extensão a 68°C por 90 seg.; e uma extensão final a 68°C por sete minutos. As cinco réplicas
por amostra foram unidas e visualizadas em gel de agarose 1% para verificar a reação, onde o
padrão esperado é um rastro como o observado na Figura 7.
Figura 7. Gel da amplificação da digestão/ligação
Quantificação e purificação das amostras
As amostras foram quantificadas no fluorômetro Qubit 2.0 (Invitrogen) e foi feito um
pool delas, colocando a mesma quantidade de DNA de cada uma. O pool de amostras foi
purificado com o kit de purificação AMPure (Agencourt) em uma proporção de 0.8V AMPure
beads : 1V DNA.
Seleção de tamanho e diluição da biblioteca
A seleção de tamanho dos fragmentos da biblioteca (pool de amostras) foi feita no
Pippin Prep (Sage Science) com cassetes de agarose 2% dye-free, internal standard mix,
seguindo as instruções do fabricante. A faixa de corte escolhida foi determinada mediante
uma simulação de corte enzimático do genoma mais próximo à espécie de estudo, para
conhecer a distribuição dos tamanhos dos fragmentos depois do corte e assim determinar a
faixa mais apropriadas para ser sequenciada. A média do tamanho foi 415pb, com início em
374pb e fim em 456pb. Depois da corrida, a biblioteca foi coletada, purificada com o kit
AMPure em uma proporção de 0.7V AMPure beads : 1V DNA e quantificada novamente no
Qubit 2.0. Com base nesse valor, foi feito o cálculo da diluição necessária para a preparação
do template (cada um dos fragmentos da biblioteca genômica).

19
Preparo do template
-PCR clonal do template
A biblioteca diluída foi utilizada seguindo o protocolo do fabricante “Prepare
Template-Positive Ion PGM Template OT2 400 Ion Sphere Particles (ISPs)”. Para esse
procedimento foram usados os reagentes e materiais dos kits “Ion PGM Template OT2
Reagents 400”, “Ion PGM Template OT2 Solutions 400”, “Ion PGM Template OT2 Reactions
400” e “Ion PGM Template OT2 Supplies 400”. A reação de PCR clonal foi realizada no
equipamento Ion OneTouch 2 (Figura 10a).
O objetivo da amplificação clonal ou PCR de emulsão é gerar muitas cópias de cada
fragmento da biblioteca. Nesse processo, cada fragmento é amplificado em microreatores
formados por emulsão em óleo, os quais contêm os reagentes de PCR e pequenas esferas
(IonSpheres Particles - ISPs) ou beads cobertas com o primer P1 (sequência complementar ao
adaptador P1), que servem para fixar os clones do template (Figura 8). O ideal é que cada
esfera incorpore um só fragmento para ter uma porcentagem de policlonalidade baixa
(Varuzza, 2013).
Figura 8. PCR clonal ou de emulsão: os fragmentos são ligados às IonSpheres (pequenas esferas) através de
primers P1 (sequências complementares ao adaptador P1) na superfície das esferas. Um único fragmento se liga
a uma determinada esfera. Elas são capturadas individualmente em gotículas oleosas (microreatores) onde a PCR
em emulsão ocorre. Nesse processo, são geradas milhares de cópias de cada fragmento. Fonte: (Mardis, 2008).
-Enriquecimento do template
O produto da PCR clonal foi recuperado e lavado, para ser usado no enriquecimento
do template seguindo o protocolo do fabricante “Enrich the Template-Positive Ion PGM
Template OT2 400 Ion Sphere Particles (ISPs)”. Para esse procedimento foram usados os
reagentes e materiais dos kits “Ion PGM Template OT2 Solutions 400”, “Ion PGM Template
OT2 Supplies 400” e “Ion PGM Enrichment Beads”. O enriquecimento foi feito no
equipamento Ion OneTouch ES (Figura 10b).

20
Durante essa fase, as esferas que incorporaram fragmentos hibridizam com beads
magnéticas de enriquecimento que são separadas do resto mediante um ímã. Aquelas que não
incorporaram fragmentos, são eliminadas mediante lavagens.
-Sequenciamento
Depois da corrida do passo anterior, foi obtida uma amostra com as ISPs enriquecidas,
a qual foi usada para o carregamento do chip Ion 318 V2 e posterior sequenciamento no
sistema Ion Personal Genome Machine (PGM) (Figura 10c). Para esse procedimento foram
usados os reagentes e materiais dos kits “Ion PGM Sequencing Reagents 400”, “Ion PGM
Sequencing Solutions 400” e “Ion PGM Sequencing Supplies 400”.
Uma vez feito o carregamento, as esferas enriquecidas são incorporadas
individualmente em um dos poços do chip onde será realizado o sequenciamento (Figura 9a).
No Ion PGM, a reação de polimerização gera naturalmente um H+ (próton) que altera o pH do
meio, o que é detectado diretamente por um transistor, que converte a alteração em um sinal
elétrico (Figura 9b). Cada poço tem seu pHmêtro e a detecção é dada por fluxo contínuos das
bases, onde a injeção de cada uma se da individualmente seguida de uma lavagem (Varuzza,
2013).
Figura 9. a) As ISPs são incorporadas nos poços do chip Ion 318 V2. Uma ISP entrará por cada poço (esq.); b)
Durante o sequenciamento, a Taq polimerase incorpora uma base e é liberado um fosfato e um H+. Esse último é
detectado por um pHmêtro que transforma a mudança de pH em um sinal elétrico (dir.). Fonte: a) (Mardis,
2008); b) (Varuzza, 2013).
Os procedimentos foram realizados no Laboratório de Evolução e Genética Animal -
LEGAL e no Laboratório de Genômica do Centro de Apoio Multidisciplinar - CAM, da
Universidade Federal do Amazonas - UFAM e na Plataforma de Genômica do Instituto
Leônidas e Maria Deane da Fundação Oswaldo Cruz - ILMD/FIOCRUZ.

21
Figura 10. O Sistema Ion PGM está conformado pelos seguintes equipamentos: a) Ion OneTouch 2 (esq.), b) Ion
OneTouch ES (centro), c) Ion Personal Genome Machine (PGM) (dir.), e o servidor Ion Torrent.
3.4 Análises de dados
3.4.1 DNA mitocondrial
Edição e alinhamento das sequências
As sequências foram editadas e alinhadas no programa CodonCode Aligner v.5.1.4.
(CodonCode Corporation). Elas foram traduzidas em aminoácidos usando o programa MEGA
ver.6 (Tamura et al., 2013) para verificar a presença de códons de parada putativos. Não
foram observados códons de parada, inserções, deleções nem outras anomalias.
Índices de diversidade a nível intra-populacional
Para estimar os níveis de diversidade genética foram usadas duas medidas: a
diversidade gênica ou haplotípica (Ĥ), que representa a probabilidade de que duas sequências
escolhidas aleatoriamente sejam diferentes (Li, 1987; Nei, 1987). Esta medida é equivalente à
heterozigosidade esperada em marcadores diplóides; e a diversidade nucleotídica (π), que faz
referência à probabilidade de que dois sítios homólogos (nucleotídeos) escolhidos ao acaso
sejam diferentes, o que é equivalente à diversidade gênica em nível nucleotídico (Tajima,
1983; Nei, 1987). Esses parâmetros foram estimados através do programa Arlequin ver.
3.5.1.3 (Excoffier e Lischer, 2010).
Neutralidade e equilíbrio genético
Para testar a seleção neutral e o equilíbrio genético das populações foram usados os
testes de D de Tajima (Tajima, 1989) e Fs de Fu (Fu, 1997). Os dois se baseiam no modelo de
sítios infinitos sem recombinação que é apropriado para sequências curtas de DNA. O modelo
assume que os sítios ao longo de uma sequência de DNA sofrem mutações independentes

22
dado que atuam sobre sítios que previamente não tinham sido afetados (Kimura, 1969). As
significância dos valores para ambos os testes pode indicar que as sequências não estão
evoluindo neutralmente e que outros fatores podem estar afetando as populações como
resultado de algum evento demográfico ou heterogeneidade nas taxas de mutações.
Especialmente, a estatística Fs de Fu é muito sensível à expansão demográfica populacional e
ao efeito carona (hitchhiking), gerando valores negativos (Fu, 1997). No caso do D de Tajima,
valores negativos estão relacionados com expansão demográfica (por exemplo, depois de um
gargalo populacional recente), eventos fundadores ou efeito carona; enquanto que valores
positivos podem estar relacionados com seleção balanceadora e subdivisão ou contração
populacional (Tajima, 1989). Os dois testes foram realizados no programa Arlequin ver.
3.5.1.3 (Excoffier e Lischer, 2010).
Estrutura populacional
A diferenciação entre e dentro das localidades estudadas, foi estimada mediante a
análise de variância molecular (AMOVA), que se baseia na variância das frequências gênicas
levando em consideração o número de mutações entre haplótipos. Os grupos são definidos a
priori e é feita uma análise hierárquica dos componentes da variância devido a diferenças
entre indivíduos, e entre ou dentro das populações, calculando o índice ΦST, análogo ao índice
de fixação (Fst) (Excoffier et al., 1992). O Fst de Wright (1969) foi usado para medir a
quantidade de diferenciação genética entre populações. Ambas as análises foram
implementadas no programa Arlequin ver. 3.5.1.3 (Excoffier e Lischer, 2010).
A hipótese de isolamento por distância foi testada mediante análise da relação entre
distâncias genéticas (Fst) e geográficas (calculadas seguindo o curso do rio), utilizando o teste
de Mantel (Mantel, 1967). Foram feitos dois testes: o primeiro com todas as localidades; o
segundo retirando as localidades Bra, EM, AM e Ara, dado que elas estão separadas por
barreira geográfica do restante das populações. Esse teste também foi realizado no programa
Arlequin ver. 3.5.1.3 (Excoffier e Lischer, 2010).
Análise Bayesiana de estrutura genética
Para inferir a estrutura genética das populações a partir das sequências de DNA
mitocondrial, foi usado um método que visa à formação de grupos biológicos baseado em um
modelo bayesiano e implementado no programa BAPS v.6.0 (Bayesian Analysis of Population
Structure) (Corander et al., 2003; Corander et al., 2008). Esse programa utiliza um algoritmo
de busca estocástica que calcula a distribuição posterior dos parâmetros do modelo

23
condicionados pelos dados observados, nesse caso as frequências nucleotídicas das sequências
de DNA. Para determinar o número de populações mais provável (K), foi realizada uma
análise de mistura de populações. O K é incluído como parâmetro a ser estimado e a melhor
partição (número de clusters) dos dados é identificada como a de maior probabilidade.
Distribuição e frequência dos haplótipos
Foram geradas redes de haplótipos para visualizar as relações genealógicas entre os
indivíduos usando o programa HaploViewer (Salzburger et al., 2011). O programa converte
as árvores geradas a partir de métodos de reconstrução filogenéticas tradicionais em
genealogias de haplótipos. As árvores foram geradas no programa TreeFinder v.2011 (Jobb et
al., 2004), usando o modelo de substituição mais apropriado, determinado pela ferramenta
"propose model" (HKY[{3,1,1,1,1,3},Empirical]) e 200.000 réplicas. Utilizando o método de
máxima verossimilhança para a inferência filogenética, o qual tenta achar a árvore que
possivelmente explica melhor a evolução dos dados observados.
3.4.2 DNA genômico
Edição dos dados genômicos
As leituras obtidas a partir do sequenciamento foram processadas pelo software
Torrent Suite (da plataforma Ion Torrent) e colocadas em formato BAM, um formato binário
para armazenar dados de sequências. Esse arquivo foi transformado em um formato FASTAQ
com todas as sequências brutas e os seus scores de qualidade. Posteriormente, o arquivo
FASTAQ foi processado, retirando as leituras menores de 200 pb mediante um script na
linguagem de programação Python.
O arquivo FASTAQ foi analisado no pipeline Stacks (Catchen et al., 2011; Catchen et
al., 2013b), que está composto por uma série de programas escritos na linguagem C++ e Perl.
A interfase dele é implementada em PHP que armazena a informação em um banco de dados
no MySQL. Os componentes do pipeline que foram usados são:
Process_radtags: limpa as sequências brutas. As leituras menores que 200pb, com
algum erro de sequenciamento e/ou com baixa qualidade (< 90% de confiança) foram
descartadas. As leituras processadas foram demultiplexadas (separadas por indivíduo
mediante a identificação do barcode).

24
Ustacks: constrói loci de novo2 e detecta haplótipos em cada indivíduo. Os arquivos
por indivíduo foram inseridos no programa que agrupa as leituras idênticas em stacks (pilhas)
levando em consideração a cobertura mínima (número mínimo de leituras exigido para formar
um stack). Os stacks são associados a um mesmo loco com uma distância máxima de dois
pares de bases e cada um deles poderá possuir no máximo dois stacks. Também é usado um
modelo de SNPs que procura polimorfismos em cada nucleotídeo de cada loco. Para
identificar se as diferenças encontradas entre dois stacks são um verdadeiro SNP, o programa
utiliza um modelo estatístico de máxima probabilidade que estima o valor mais provável da
taxa de erro de sequenciamento de cada posição para cada genótipo. Ele cria uma sequência
consenso e armazena os dados de SNPs e haplótipos.
Cstacks: une os locos de todos os indivíduos para criar um catálogo a partir de uma
sequência consenso para cada loco.
Sstacks: cruza os locos de cada indivíduo contra o catálogo para determinar o estado
alélico de cada loco em cada indivíduo.
Populations: analisa as populações e calcula vários parâmetros populacionais
(heterozigosidade esperada e observada, Π, Fis, Fst). Os SNPs de cada loco do catálogo
foram extraídos no formato Genepop. Para isso, foram considerados vários parâmetros como
a porcentagem mínima de indivíduos em que aquele loco deve estar presente em uma
população, o número mínimo de populações onde deve estar presente um loco, a cobertura
mínima de um loco para cada indivíduo e a mínima frequência alélica.
Diversidade genética e estrutura populacional
Com ajuda do programa PGDSpider (Lischer e Excoffier, 2012), o arquivo foi
transformado no formato específico para correr no programa Arlequin ver. 3.5.1.3 (Excoffier
e Lischer, 2010). De igual forma que com o mtDNA, foi feita uma AMOVA e o nível de
estruturação das populações foi identificado através da comparação par a par dos valores de
Fst. O isolamento por distância foi testado mediante o teste de Mantel.
Análise Bayesiana de estrutura genética
Com ajuda do programa PGDSpider (Lischer e Excoffier, 2012), o arquivo foi
transformado no formato específico para análise no programa STRUCTURE v. 2.3.4
(Pritchard et al., 2000) que identifica estrutura populacional a partir de genótipos multilocos.
2 Loci de novo: a identificação dos locos é dada a partir das leituras dado que não existe um genoma de
referência para a espécie.

25
Esse programa determina grupos biológicos atribuindo indivíduos às populações empregando
as frequências alélicas dos locos. Essa estimativa é feita mediante um algoritmo de
agrupamento baseado em um modelo Bayesiano que calcula a probabilidade posterior para
diferente número de grupos usando o modelo de mistura (admixture model) e frequências
alélicas correlacionadas (correlated allele frequencies). O primeiro modelo assume que os
indivíduos podem ter uma ancestralidade misturada, ou seja, que sua herança genética pode
ser derivada de mais de uma população; o segundo modelo considera que as frequências
alélicas de populações relacionadas, são similares (Falush et al., 2003).
O programa foi implementado com os seguintes pressupostos: 100.000 gerações de
"burn-in", 1.000.000 de gerações de cadeia de Markov e Monte Carlo (MCMC) para cada K.
As simulações foram repetidas dez vezes para cada K, calculando a proporção de atribuição
(Q) das localidades amostradas nos grupos detectados e o coeficiente individual de
agrupamento (q: proporção do genoma ancestral em cada indivíduo no grupo). Esse resultados
foram importados para o programa STRUCTURE HARVESTER (Earl e vonHoldt, 2012),
onde o número ótimo de grupos biológicos foi determinado pelo método de Evanno (Evanno
et al., 2005). Os resultados foram permutados no programa CLUMPP (Jakobsson e
Rosenberg, 2007). Por fim, as matrizes de cada valor de K foram visualizadas no programa
DISTRUCT (Rosenberg, 2004).
Análise filogenética
O arquivo FASTAQ também foi processado no pipeline PyRAD (Eaton, 2014). Ele
está escrito em Python e tenta maximizar a informação filogenética obtida a partir de
ddRADs. Para agrupar os dados, ele usa similaridade de sequência com base em um
alinhamento global de sequências através do programa USEARCH. Para concatenar as
sequências usa um método hierárquico de agrupamento para obter uma única sequência por
amostra, que é dada em arquivo Phylip.
Foi feita uma árvore filogenética a partir do arquivo Phylip no programa RAxML-HPC
v.8 (Randomized Axelerated Maximum Likelihood) (Stamatakis, 2014) na plataforma CIPRES
(Cyberinfrastructure for Phylogenetic Research) (Miller et al., 2010). Este é um programa
para inferência de grandes árvores filogenéticas baseada em máxima verossimilhança.

26
RESULTADOS
4.1 DNA mitocondrial
4.1.1 Diversidade genética intra-populacional
Foram analisadas 245 sequências do gene mtDNA Cytb, com um tamanho de 1038 pb
(o gene Cytb possui 1140 pb). As sequências mostraram 58 sítios polimórficos e um total de
51 haplótipos (Tabela 4). Níveis relativamente altos de diversidade foram identificados em
sete populações (PS, Pur, Jan, Ana, BM, Uac e LT: 0.7051 - 0.8154). As populações RN,
Mam, EM, Tro e FG mostraram valores intermediários (0.4286 - 0.6264), enquanto que nas
populações com um único haplótipo (Bra, AM, Xin e Ara) não foi observada diversidade
nenhuma. A diversidade gênica total foi relativamente alta (0.8718 ± 0.0122). Porém, as
diversidades nucleotídicas foram baixas em todas as populações (0.000000 - 0.002709) assim
como a diversidade nucleotídica total (0.002424).
O teste D de Tajima foi significativo para RN, Uac e LT, enquanto que o Fs de Fu foi
significativo para RN, PS, Jan, Ana, Uac e LT. Quando analisadas todas as localidades,
ambos os teste mostraram valores negativos e diferenças significativas (D = -2.17 (p < 0.05);
Fs = -26.51 (p < 0.02)).
4.1.2 Estrutura entre populações
A análise de variância molecular (AMOVA) mostrou que existe um alto nível de
diferenciação genética entre as localidades amostradas (Fst = 0.50, p < 0.0001), onde 50.53%
da variação está distribuída entre as populações. Esse resultado foi confirmado com as
comparações par a par dos valores de Fst, as quais foram significativas para 82% dos casos
(Tabela 5). Para as localidades RN, Bra, EM e AM e Ara todos os valores de Fst foram altos
e significativos; especialmente Bra que apresentou a maioria (13/15) de Fst acima de 0.80. A
localidade Xin apresentou valores altos e significativos, com exceção daqueles que
envolveram as localidades da Costa Atlântica (FG, Uac e LT), os quais não foram
significativos (p > 0.1). Essas últimas localidades não mostraram diferenças entre si. As
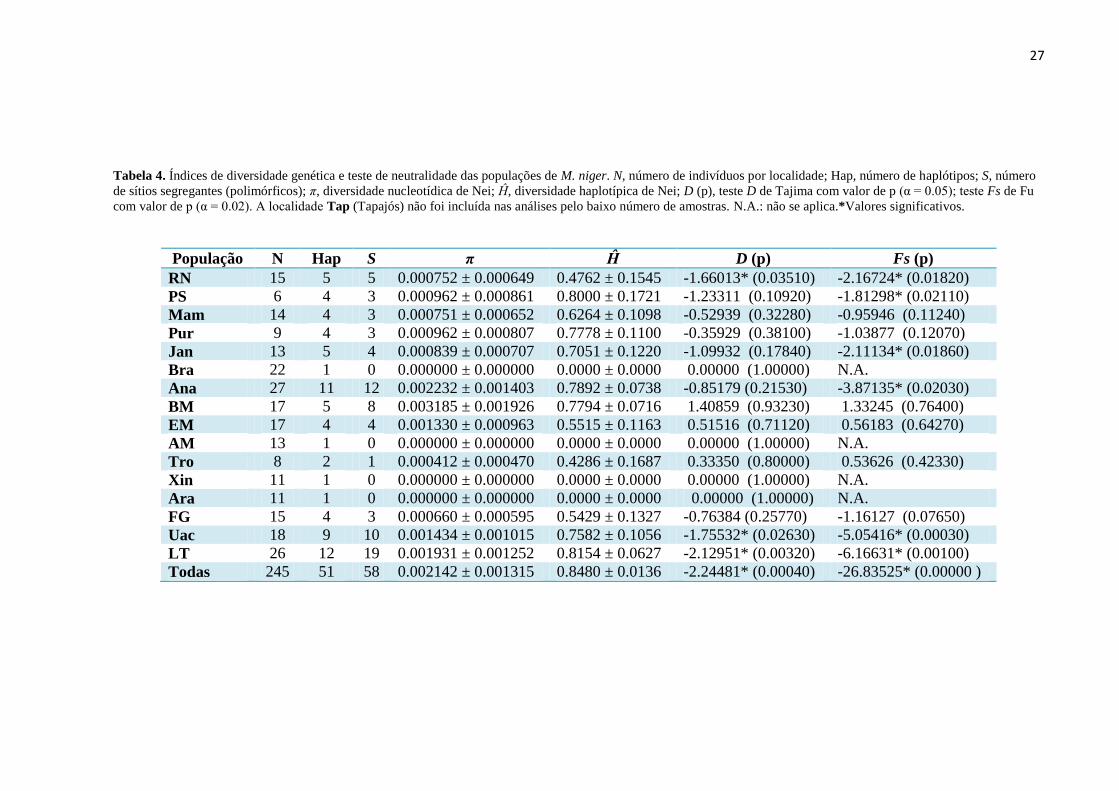
27
Tabela 4. Índices de diversidade genética e teste de neutralidade das populações de M. niger. N, número de indivíduos por localidade; Hap, número de haplótipos; S, número
de sítios segregantes (polimórficos); π, diversidade nucleotídica de Nei; Ĥ, diversidade haplotípica de Nei; D (p), teste D de Tajima com valor de p (α = 0.05); teste Fs de Fu
com valor de p (α = 0.02). A localidade Tap (Tapajós) não foi incluída nas análises pelo baixo número de amostras. N.A.: não se aplica.*Valores significativos.
População N Hap S π Ĥ D (p) Fs (p)
RN 15 5 5 0.000752 ± 0.000649 0.4762 ± 0.1545 -1.66013* (0.03510) -2.16724* (0.01820)
PS 6 4 3 0.000962 ± 0.000861 0.8000 ± 0.1721 -1.23311 (0.10920) -1.81298* (0.02110)
Mam 14 4 3 0.000751 ± 0.000652 0.6264 ± 0.1098 -0.52939 (0.32280) -0.95946 (0.11240)
Pur 9 4 3 0.000962 ± 0.000807 0.7778 ± 0.1100 -0.35929 (0.38100) -1.03877 (0.12070)
Jan 13 5 4 0.000839 ± 0.000707 0.7051 ± 0.1220 -1.09932 (0.17840) -2.11134* (0.01860)
Bra 22 1 0 0.000000 ± 0.000000 0.0000 ± 0.0000 0.00000 (1.00000) N.A.
Ana 27 11 12 0.002232 ± 0.001403 0.7892 ± 0.0738 -0.85179 (0.21530) -3.87135* (0.02030)
BM 17 5 8 0.003185 ± 0.001926 0.7794 ± 0.0716 1.40859 (0.93230) 1.33245 (0.76400)
EM 17 4 4 0.001330 ± 0.000963 0.5515 ± 0.1163 0.51516 (0.71120) 0.56183 (0.64270)
AM 13 1 0 0.000000 ± 0.000000 0.0000 ± 0.0000 0.00000 (1.00000) N.A.
Tro 8 2 1 0.000412 ± 0.000470 0.4286 ± 0.1687 0.33350 (0.80000) 0.53626 (0.42330)
Xin 11 1 0 0.000000 ± 0.000000 0.0000 ± 0.0000 0.00000 (1.00000) N.A.
Ara 11 1 0 0.000000 ± 0.000000 0.0000 ± 0.0000 0.00000 (1.00000) N.A.
FG 15 4 3 0.000660 ± 0.000595 0.5429 ± 0.1327 -0.76384 (0.25770) -1.16127 (0.07650)
Uac 18 9 10 0.001434 ± 0.001015 0.7582 ± 0.1056 -1.75532* (0.02630) -5.05416* (0.00030)
LT 26 12 19 0.001931 ± 0.001252 0.8154 ± 0.0627 -2.12951* (0.00320) -6.16631* (0.00100)
Todas 245 51 58 0.002142 ± 0.001315 0.8480 ± 0.0136 -2.24481* (0.00040) -26.83525* (0.00000 )
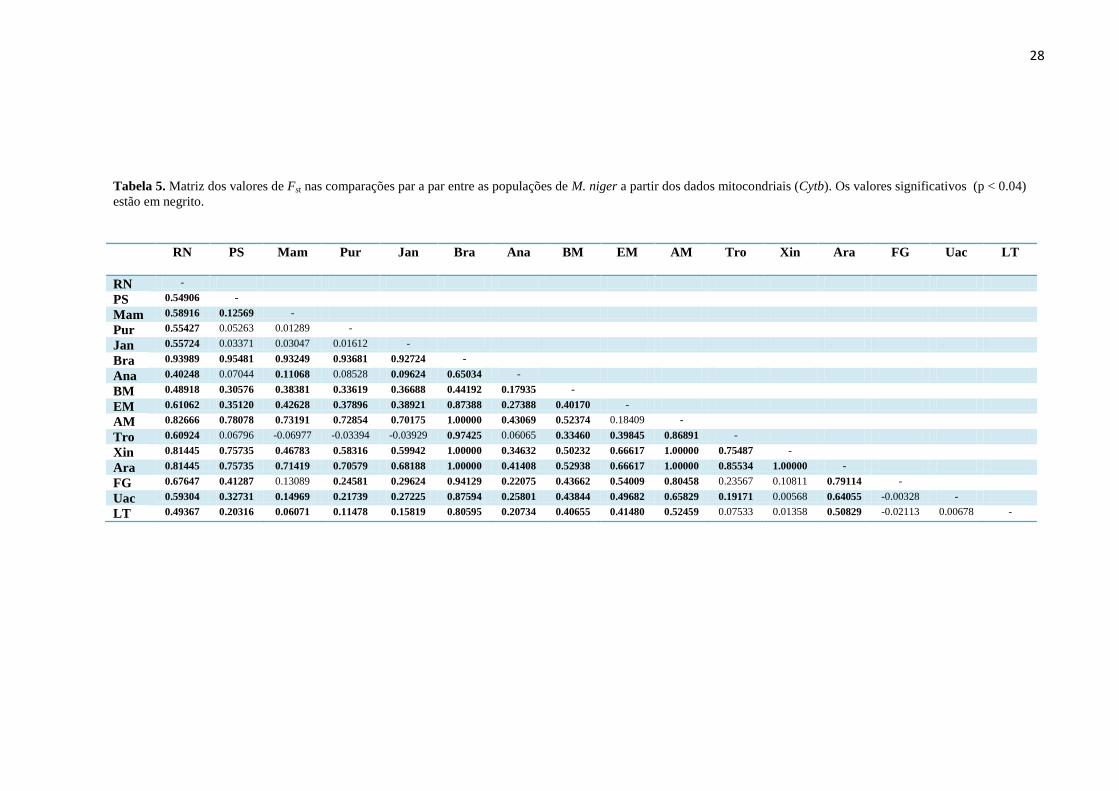
28
Tabela 5. Matriz dos valores de Fst nas comparações par a par entre as populações de M. niger a partir dos dados mitocondriais (Cytb). Os valores significativos (p < 0.04)
estão em negrito.
RN PS Mam Pur Jan Bra Ana BM EM AM Tro Xin Ara FG Uac LT
RN -
PS 0.54906 -
Mam 0.58916 0.12569 -
Pur 0.55427 0.05263 0.01289 -
Jan 0.55724 0.03371 0.03047 0.01612 -
Bra 0.93989 0.95481 0.93249 0.93681 0.92724 -
Ana 0.40248 0.07044 0.11068 0.08528 0.09624 0.65034 -
BM 0.48918 0.30576 0.38381 0.33619 0.36688 0.44192 0.17935 -
EM 0.61062 0.35120 0.42628 0.37896 0.38921 0.87388 0.27388 0.40170 -
AM 0.82666 0.78078 0.73191 0.72854 0.70175 1.00000 0.43069 0.52374 0.18409 -
Tro 0.60924 0.06796 -0.06977 -0.03394 -0.03929 0.97425 0.06065 0.33460 0.39845 0.86891 -
Xin 0.81445 0.75735 0.46783 0.58316 0.59942 1.00000 0.34632 0.50232 0.66617 1.00000 0.75487 -
Ara 0.81445 0.75735 0.71419 0.70579 0.68188 1.00000 0.41408 0.52938 0.66617 1.00000 0.85534 1.00000 -
FG 0.67647 0.41287 0.13089 0.24581 0.29624 0.94129 0.22075 0.43662 0.54009 0.80458 0.23567 0.10811 0.79114 -
Uac 0.59304 0.32731 0.14969 0.21739 0.27225 0.87594 0.25801 0.43844 0.49682 0.65829 0.19171 0.00568 0.64055 -0.00328 -
LT 0.49367 0.20316 0.06071 0.11478 0.15819 0.80595 0.20734 0.40655 0.41480 0.52459 0.07533 0.01358 0.50829 -0.02113 0.00678 -

29
comparações AM-Bra, Xin-Bra, Xin-AM, Ara-Bra, Ara-AM, Ara-Xin mostraram valores
de Fst = 1.00.
As distâncias entre as localidades estiveram entre 77.8 e 5445 quilômetros. O teste de
correlação entre distâncias geográficas e genéticas não foi significativo (0.128935, p > 0.1)
quando todas as localidades foram analisadas juntas. Retirando as localidades que estão
separadas por uma barreira física (nesse caso presença de corredeiras nos rios Branco,
Madeira e Araguaia), o teste foi significativo (0.379968, p < 0.05), mostrando assim um
padrão de isolamento por distância. Quando foram consideradas só as localidades da bacia
Amazônica sem as anteriores, o teste de Mantel também foi significativo (0.446988, p <
0.05).
4.1.3 Grupos biológicos
Em nível geral, o número de grupos biológicos estimado pelo BAPS foi de k = 3. Eles
correspondem a: grupo Oeste (vermelho), com predominância de indivíduos do Oeste da
bacia Amazônica até o rio Trombetas, incluindo a localidade Ara no Leste; grupo Leste
(verde), com predominância de indivíduos do Leste da bacia, desde o rio Tapajós até as
localidades da Costa Atlântica; grupo Guiana (azul) que incorpora todos os indivíduos da
localidade Bra, predominância de indivíduos do BM e parcialmente indivíduos de Ana
(Figura 11a). Quando foram analisadas só as localidades da bacia Amazônica, retirando
aquelas da Costa Atlântica, o k foi 4. Apareceram os mesmos grupos anteriormente descritos,
e um novo grupo no rio Madeira/Guaporé, grupo Bolívia (amarelo) nas localidades AM e
EM, com uns poucos indivíduos em Ana e BM (Figura 11b). Com isso, observamos um
padrão de distribuição dos grupos biológicos Oeste-Leste, determinado pelos grupos Oeste e
Leste, e um padrão Norte-Sul determinado pelos grupos Guiana e Bolívia, com alguns grupos
menores (Figura 11c).
4.1.4.Genealogia de haplótipos
Dos 51 haplótipos observados, nove foram os mais representativos e o restante foi
encontrado só em um indivíduo ou no máximo dois, mostrando uma grande quantidade de
haplótipos raros. A rede de haplótipos revela que provavelmente o haplótipo H2 é o mais

30
Figura 11. Gráficos gerados no programa BAPS que mostram o número de grupos biológicos para M. niger
baseado nas sequências de mtDNA. Cada barra corresponde a um indivíduo e a sua cor representa o grupo ao
qual pertence. As localidades estão organizadas geograficamente Oeste-Leste (RN-LT) e Norte-Sul (Bra-AM).
a) Gráfico gerado incluindo todas as localidades. São determinados três grupos: grupo Oeste (vermelho), que
predomina ao Oeste; grupo Guiana (azul), que predomina no eixo Norte-Sul; grupo Leste (verde), que predomina
no Leste. b) Gráfico gerado retirando as localidades da Costa Atlântica (FG, Uac, LT). São determinados quatro
grupos, os três anteriores mais um novo grupo, grupo Bolívia (amarelo) que corresponde as localidades EM e
AM. c) Esquema que mostra a distribuição dos grupos biológicos na bacia Amazônica e Costa Atlântica e os
padrões encontrados. As linhas tracejadas vermelhas indicam a presença de corredeiras.
ancestral. Os haplótipos H1 e H2 foram os mais frequentes nas populações de jacaré-açu e os
que correspondem ao padrão de distribuição Oeste-Leste. H1 inclui a maior parte dos
indivíduos das localidades do Leste e alguns poucos indivíduos de Ana e BM, com exceção
de Ara que aparece como um haplótipo diferente (H8) e mais próximo de H2. O haplótipo H2

31
inclui a maioria dos indivíduos do Oeste, com vários de Ana e BM, com exceção de RN que
corresponde a H3 e quatro haplótipos únicos derivados dele (Figura 12a-b).
Os outros cinco haplótipos representativos são exclusivos do eixo Norte-Sul. Os mais
frequentes foram H4 e H9. O haplótipo H4 representa totalmente a localidade Bra e inclui
alguns indivíduos de Ana. A partir dele, deriva-se o H5 que corresponde a indivíduos só de
BM. Os haplótipos H6 e H7 estão separados por dois passos mutacionais do haplótipo H2. H6
está formado por indivíduos de BM e Ana, e H7 só por indivíduos de Ana. Por fim, o
haplótipo H9 inclui todos os indivíduos de AM, vários de EM e alguns de BM (Figura 12a-c).
4.2 DNA genômico
Foram sequenciados quatro chips, cada um deles com 20 amostras (um deles com 21).
A média do número de leituras geradas por corrida foi de aprox. 4 milhões, por tanto foram
produzidas mais de 16 milhões de leituras, das quais 10.705.968 passaram o controle de
qualidade e tiveram um tamanho maior que 200 pb. Depois de processar os dados de 76
indivíduos3 no programa Stacks, foram obtidos um total de 68.089 locos, dos quais 6.319
foram polimórficos (contendo um ou mais SNPs).
Para gerar o arquivo usado nas análises posteriores, foi usado o componente
Populations do Stacks, no qual foi estabelecido que para a escolha dos SNPs, eles deviam
estar presentes em pelo menos 90% dos indivíduos (-r 0.90), a frequência dos seus alelos
devia ser maior que 1% (-a 0.01) e só devia ser anotado um único SNPs por locos (--
write_single_snp). Assim, o arquivo final continha 441 SNPs.
4.2.1 Diversidade genética intra-populacional
A análise de variância molecular (AMOVA) mostrou que existe um alto nível de
diferenciação genética entre as localidades amostradas (Fst = 0.30, p < 0.0001), onde 30.19%
da variação está distribuída entre as populações. Todos os valores de Fst das comparações par
a par foram significativos menos um (Mam-Jan) (Tabela 6), porém alguns desses valores são
próximos de zero, portanto não são significativos biologicamente. A população Ara é
3 Foram descartados cinco indivíduos de duas localidades por alto missing data e baixo número de amostras.
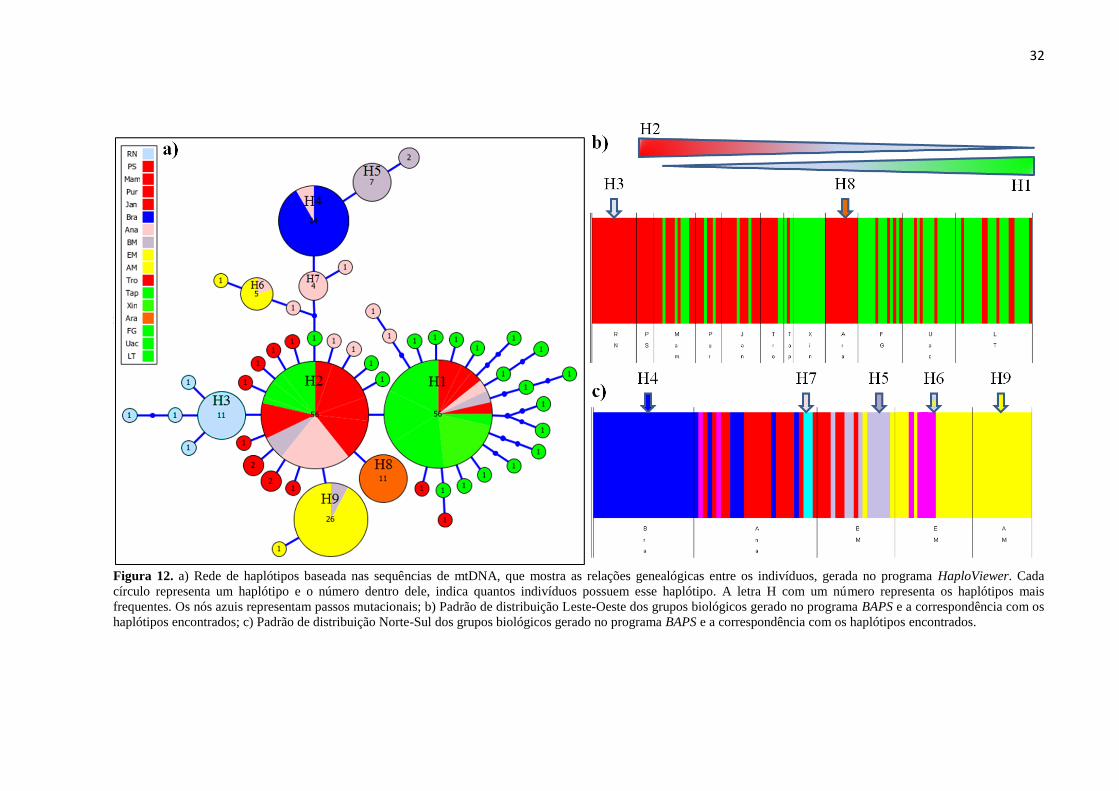
32
ibui
Figura 12. a) Rede de haplótipos baseada nas sequências de mtDNA, que mostra as relações genealógicas entre os indivíduos, gerada no programa HaploViewer. Cada
círculo representa um haplótipo e o número dentro dele, indica quantos indivíduos possuem esse haplótipo. A letra H com um número representa os haplótipos mais
frequentes. Os nós azuis representam passos mutacionais; b) Padrão de distribuição Leste-Oeste dos grupos biológicos gerado no programa BAPS e a correspondência com os
haplótipos encontrados; c) Padrão de distribuição Norte-Sul dos grupos biológicos gerado no programa BAPS e a correspondência com os haplótipos encontrados.

33
claramente divergente do resto das populações analisadas com valores de Fst entre 0.42 e
0.63. Xin mostrou uma alta diferenciação com EM e AM (Fst de 0.34 e 0.45,
respectivamente), mas quando comparada com o resto das populações foi baixa (Fst ~ 0.1).
As populações BM, Ana, Jan e Mam ficam no mesmo grupo, com uma baixa diferenciação
entre elas (valores de Fst entre 0.04 e 0.09). AM mostrou uma alta estruturação em relação as
outras populações menos EM, cujo valor de Fst é 0.09.
Tabela 6. Matriz dos valores de Fst nas comparações par a par entre as populações de M. niger a partir dos dados
genômicos (SNPs). Os valores significativos (p < 0.02) estão em negrito.
Mam Jan Ana BM EM AM Xin Ara
Mam -
Jan -0.00626 -
Ana 0.05394 0.04782 -
BM 0.09025 0.06550 0.05011 -
EM 0.23554 0.25175 0.22685 0.16786 -
AM 0.33593 0.37935 0.33623 0.25200 0.09135 -
Xin 0.13831 0.15524 0.12133 0.14007 0.34135 0.45515 -
Ara 0.47205 0.50731 0.45495 0.42322 0.54722 0.62912 0.42969 -
As distâncias entre as localidades estiveram entre 210 e 4527 quilômetros. O teste de
correlação entre distâncias geográficas e genéticas foi significativo (0.884752, p < 0.0001)
quando todas as localidades foram analisadas juntas.
4.2.4 Grupos biológicos
O programa Structure foi usado para determinar o número de grupos biológicos mais
provável, usando o modelo de mistura, que permite que os indivíduos tenham ancestrais em
mais de um grupo biológico. O programa identificou três grupos biológicos. As populações
Mam e Jan, AM e Ara pertencem a um grupo biológico puro (Mam: q > 0.98 de ser do
grupo verde; Jan: q > 0.90 de ser do grupo verde; AM: q > 0.99 de ser do grupo roxo; Ara: q
> 0.99 de ser do grupo amarelo). As outras mostraram diferentes níveis de mistura (Ana: q >
0.87 verde; BM: q > 0.55 verde; EM: q > 0.84 roxo; Xin: q > 0.78 verde) (Figura 13).

34
Figura 13. Estrutura populacional determinada no programa Structure mediante inferência Bayesiana. Foram
analisados 441 SNPs de 76 indivíduos de 8 localidades. A maior probabilidade posterior está associada com três
grupos biológicos. a) Gráfico individual; cada barra vertical representa um indivíduo; b) gráfico populacional.
4.3 Análises genéticas: Rio Madeira
Usando as 47 sequências mitocondriais do rio Madeira (BM: Nova Olinda, Borba,
Manicoré, Humaitá e Lago Cuniã; EM: Ilha do Búfalo e Jaci-Paraná; AM: Costa Marques e
Pimenteiras do Oeste), foi feita a análise Bayesiana para estabelecer o número de grupos
biológicos no programa BAPS, e foram observados quatro grupos. O grupo Oeste (vermelho)
que corresponde ao haplótipo mais comum e ancestral H2, que aparece só no BM até a
localidade de Humaitá. O grupo Bolivia (amarelo) que está formado por indivíduos do
haplótipo H9, e é o único que representa AM, das localidades Pimenteiras do Oeste e Costa
Marques, e domina em EM. Os dois grupos menores, o grupo Baixo Madeira (lilás) e o grupo
Alto Madeira (rosa) que correspondem a H5 e H6. O grupo lilás está restrito ao BM, desde o
Lago Cuniã até Borba; e por fim o grupo rosa que está nas localidades Jaci-Paraná e Ilha do
Búfalo (EM), com dois indivíduos em Ana (Figura 12 e Figura 14).
Depois de processar os dados dos 30 indivíduos do rio Madeira no programa Stacks,
foram obtidos um total de 42.786 locos, dos quais 3.948 foram polimórficos (contendo um ou
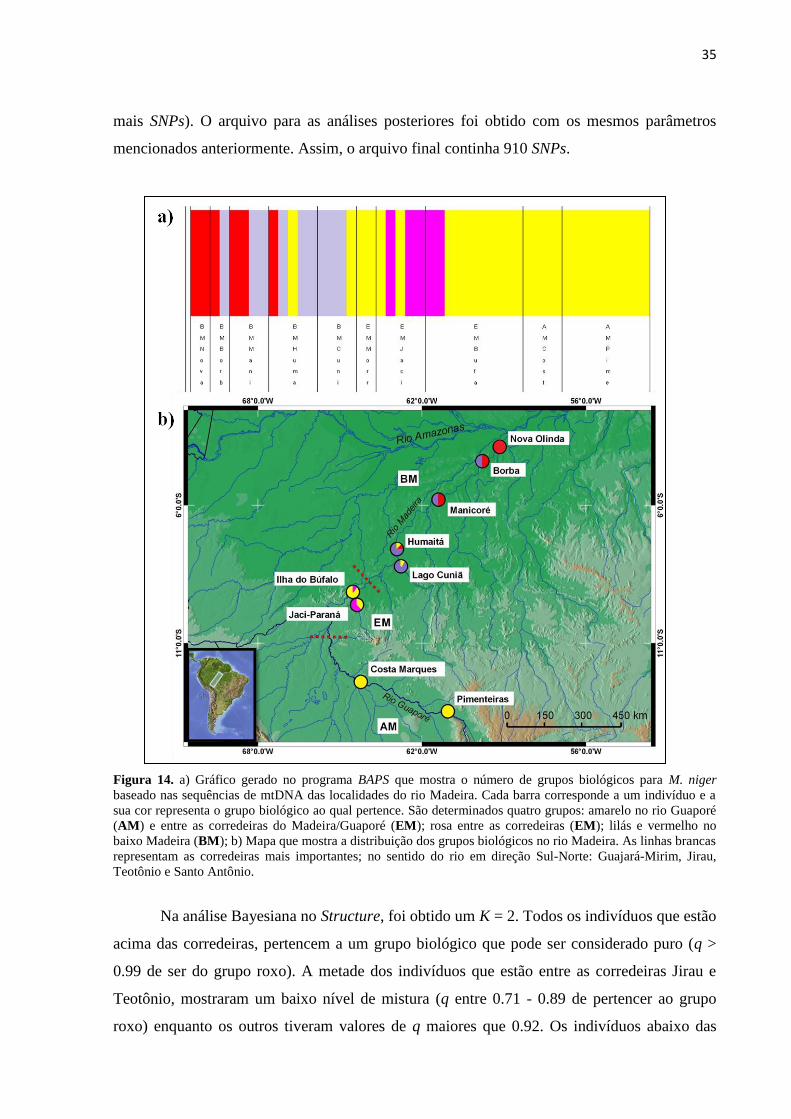
35
mais SNPs). O arquivo para as análises posteriores foi obtido com os mesmos parâmetros
mencionados anteriormente. Assim, o arquivo final continha 910 SNPs.
Figura 14. a) Gráfico gerado no programa BAPS que mostra o número de grupos biológicos para M. niger
baseado nas sequências de mtDNA das localidades do rio Madeira. Cada barra corresponde a um indivíduo e a
sua cor representa o grupo biológico ao qual pertence. São determinados quatro grupos: amarelo no rio Guaporé
(AM) e entre as corredeiras do Madeira/Guaporé (EM); rosa entre as corredeiras (EM); lilás e vermelho no
baixo Madeira (BM); b) Mapa que mostra a distribuição dos grupos biológicos no rio Madeira. As linhas brancas
representam as corredeiras mais importantes; no sentido do rio em direção Sul-Norte: Guajará-Mirim, Jirau,
Teotônio e Santo Antônio.
Na análise Bayesiana no Structure, foi obtido um K = 2. Todos os indivíduos que estão
acima das corredeiras, pertencem a um grupo biológico que pode ser considerado puro (q >
0.99 de ser do grupo roxo). A metade dos indivíduos que estão entre as corredeiras Jirau e
Teotônio, mostraram um baixo nível de mistura (q entre 0.71 - 0.89 de pertencer ao grupo
roxo) enquanto os outros tiveram valores de q maiores que 0.92. Os indivíduos abaixo das

36
corredeiras tiveram valores variáveis de mistura, com maior aporte do grupo verde (q entre
0.69 - 0.93 de pertencer ao grupo verde) (Figura 15).
Figura 15. Árvore filogenética não enraizada gerada no programa RAxML pelo método de máxima
verossimilhança. As sombras representam os grupos determinados mediante a análise Bayesiana; b) Estrutura
populacional determinada no programa Structure mediante inferência Bayesiana. Foram analisados 910 SNPs de
30 indivíduos de 3 localidades do rio Madeira. A maior probabilidade posterior está associada com dois grupos
biológicos.
Dado que temos localidades acima, abaixo e entre as corredeiras, foi testado o efeito
das mesmas na estruturação das populações de jacaré-açu. Para o mtDNA, os valores de Fst
observados foram significativos e mostraram uma forte estruturação entre o BM em relação a
EM e AM (Fst = 0.40170 e Fst = 0.52374, p < 0.00001, respectivamente). Quando
comparamos EM e AM, não houve diferenças significativas entre as duas áreas.
A AMOVA mostrou um valor de Fst significativo (0.51621, p < 0.00001), com uma
porcentagem de variação entre grupos de 48%. As comparações par a par dos Fst baseadas
nos SNPs, foram significativas, corroborando que AM e BM são divergentes, e que entre AM
e EM não existe uma estruturação forte. Esses resultados são apoiados pela árvore
filogenética que evidencia que as populações que estão acima e entre as corredeiras formam

37
uma grupo monofilético em relação as do Baixo Madeira (Figura 15). O teste de Mantel não
foi significativo para nenhum dos dois marcadores moleculares utilizados.
DISCUSSÃO
Mediante o uso de marcadores mitocondriais e genômicos, foram analisadas
populações de grande parte da área de distribuição da espécie M. niger. Foi visto que a
aplicação de métodos complementares permite uma visão mais completa e clara dos padrões
da variabilidade genética.
5.1 Estrutura genética na Amazônia
Com base nos resultados obtidos a partir do mtDNA e nDNA, podemos observar a
existência clara de um padrão de distribuição da diversidade genética nas populações de
jacaré-açu na Amazônia. As populações da calha principal do rio Amazonas e as que se
encontram na Costa Atlântica constituem um padrão de diferenciação genética em sentido
Oeste-Leste determinado por isolamento por distância (teste de Mantel significativo) (Figura
11). Essas populações formam duas unidades não discretas, com uma distribuição contínua e
uma diferenciação gradual. O grupo Oeste parece ser o mais ancestral devido a que
corresponde a um dos haplótipos mais comuns, geograficamente é o grupo mais amplamente
distribuído e na configuração da rede de haplótipos, aparece como o centro da rede a partir da
qual se derivam os outros oito haplótipos mais frequentes (Avise et al., 1987). Assim,
possivelmente a espécie se originou nessa região e posteriormente colonizou o resto da
Amazônia no sentido Oeste-Leste. Porém, devido à ampla distribuição da espécie e a uma
restrição ao fluxo gênico sem barreiras evidentes, as populações mais distantes foram se
diferenciando devido à fixação de um novo haplótipo que permitiu o surgimento de um novo
grupo biológico, o grupo Leste (verde na Figura 11). Esse grupo se separou do mais ancestral,
e recolonizou a Amazônia no sentido Leste-Oeste.
Os valores de Fst par a par, corroboram esse padrão. A maioria dos Fst das localidades
do Oeste (RN-Tro) não são significativos, assim como também não são para as localidades do
Leste (Tro-LT); e os que são significativos, mostram pouca diferenciação genética. Já,

38
quando é feita a comparação entre esses dois grupos, os valores de Fst são maiores e mostram
um nível de estruturação mais forte (Tabela 5 e Tabela 6).
Esse padrão já foi encontrado em outras espécie de fauna aquática na Amazônia, em
estudos que possuíam uma amostragem similar. Para o pirarucu (Arapaima gigas) foi
suportada a hipótese de fluxo gênico restrito com isolamento por distancia (Hrbek et al.,
2005). Para a tartaruga da Amazônia (Podocnemis expansa) e o peixe-boi (Trichechus
inunguis), os estudos (Cantanhede et al., 2005; Pearse et al., 2006) mostraram que há uma
relação fraca entre distancias geográficas e diferenciação genética, mas que existe uma
restrição ao fluxo gênico e/ou dispersão em longa distância. Por tanto, parece que vertebrados
aquáticos de grande porte apresentam esse padrão de livre dispersão na calha principal do rio
Amazonas, que atua como um corredor com algumas restrições ao fluxo gênico.
Para o jacaré-açu, já tido sido observado que as populações da Amazônia e da Costa
Atlântica estão estruturadas devido a isolamento por distância (Farias et al., 2004; De Thoisy
et al., 2006; Vasconcelos et al., 2008); no entanto no presente estudo, além de confirmar esse
padrão, foi visto que o isolamento por distância também existe entre as populações
amazônicas de M. niger.
O outro padrão visível está relacionado com a possível fragmentação e posterior
isolamento de algumas populações devido a barreiras geográficas, nesse caso a presença de
corredeiras nos rios que drenam o Amazonas, que determinam uma forte estruturação das
populações. Essa situação gera um padrão Norte-Sul devido as drenagens dos escudos
Brasileiro e Guianense, no caso dos rios Branco, Negro e Madeira, onde é evidente que as
corredeiras no rio Branco e a série de corredeiras entre Porto Velho e Guajará-Mirim no rios
Madeira/Mamoré restringem o fluxo gênico e deixam as populações que estão acima delas
praticamente isoladas.
A população do rio Uraricoera (grupo Guyana), afluente do rio Branco, constitui o
grupo mais diferenciado geneticamente do resto, como evidenciam os dados de mtDNA
(Figura 12a). Essa população possui um haplótipo único que a deixa como um grupo
biológico separado. Possivelmente, essa separação é mais antiga que a própria separação dos
grupos da calha principal do Amazonas, dado que possui uma maior quantidade de diferenças
genéticas e pode estar relacionada com o fato de que o rio Branco fazia parte de outra bacia
hidrográfica, o paleo-drenagem Proto-Berbice. Até o início do Pleistoceno, o rio fluiu rumo
norte drenando no rio Berbice e finalmente no Mar Caribe. A partir do Oligoceno, o paleo-
divisor Proto-Berbice/Amazonas, uma antiga cadeia de montanhas onde o sistema fluvial
tinha suas cabeceiras, começou ser erodido na sua porção mais ao sul, na altura do que hoje

39
constitui as corredeiras de Bem-Querer. Isso gerou a captura da drenagem dessa área em uma
única calha hidrográfica formando assim o rio Branco, que desemboca no rio Negro, o maior
tributário da margem esquerda do rio Amazonas (Schaefer e Dalrymple, 1996; Schaefer e
Vale Júnior, 1997; Barbosa et al., 2007).
Possivelmente, essa população diferenciada está distribuída até o rio Rupununi e
talvez a separação esteja determinada pelas corredeiras de Bem-Querer, as quais são o
afloramento do Escudo das Guianas no leito do rio Branco. Alguns indivíduos desse grupo
estão presentes no Arquipélago de Anavilhanas; e um ponto interessante é que indivíduos com
um haplótipo único derivado do anterior são encontrados exclusivamente no Baixo Madeira.
O que pode significar que existe fluxo gênico unidirecional dado que os indivíduos
conseguem atravessar a barreira no sentido montante-jusante.
No rio Guaporé também existe um haplótipo único compartilhado por todos os
indivíduos dessa população e configura um grupo biológico diferente (grupo Bolivia). Esse
haplótipo também está presente na maior parte dos indivíduos que estão entre as corredeiras
do Madeira/Mamoré e pode ser observado até o Baixo Madeira, onde está representado por
pouquíssimos indivíduos. De igual forma, outras espécies já evidenciaram resultados
similares. Os estudos com ariranha (Pteronura brasiliensis), usando marcadores
microssatélites e mitocondriais também revelaram o isolamento das populações do rio
Guaporé (Pickles et al., 2011; Pickles et al., 2012). Duas populações estudadas do rio
Guaporé da tartaruga da Amazônia formam um grupo biológico diferente, determinado
mediante marcadores microssatélites e compartilham um haplótipo de mtDNA que não é
encontrado fora da bacia desse rio. As duas encontram-se acima das corredeiras do
Madeira/Mamoré, que podem ser uma barreira para o fluxo gênico nessa espécie (Pearse et
al., 2006). A diferença com o presente estudo, é que as tartarugas ainda compartilham o
haplótipo mais comum, enquanto que no nosso caso, as populações dessa mesma região
possuem um haplótipo próprio o que pode estar indicando que a divergência entre as
populações de jacaré-açu é mais antiga que a das tartarugas.
Assim, parece que as corredeiras restringem o fluxo gênico entre as populações de
jacaré-açu, permitindo a passagem de indivíduos em apenas um sentido: descendo as
corredeiras, ou seja, os indivíduos conseguem atravessá-las para baixo mas não subir através
das mesmas. O que pode indicar que essas populações se estabeleceram nesses locais antes do
surgimento daquelas formações geológicas. No caso do rio Branco seria interessante uma
amostragem a montante e a jusante das corredeiras do Bem-Querer para confirmar realmente
que elas são as barreiras que estão impedindo a passagem dos jacarés.

40
Os resultados do padrão anteriormente descrito, podem estar mostrando que houve
uma conexão antiga entre o rio Negro e o rio Madeira que permitia a migração direta de
indivíduos entre essas duas bacias, e portanto o compartilhamento dos diferentes haplótipos.
Esse compartilhamento genético já foi evidenciado em outros grupos de fauna aquática como
boto (Inia geoffrensis) (Farias, 2015), jacaré-paguá (Paleosuchus palpebrosus) (Muniz et al.,
(in prep)), tucunaré (Willis et al., 2010) e acará-disco (Symphysodon) (Farias e Hrbek, 2008).
Analisando a população do rio Araguaia com os dois tipos de marcadores que foram
usados nesse estudo (mtDNA e SNPs) podemos ver que os resultados são concordantes. Ela
está representada por um único haplótipo e a sua diferenciação genética é alta com relação às
outras populações (Tabela 5 e Tabela 6). De igual forma, ela constitui um grupo biológico
diferente baseado nas suas frequências alélicas e não mostra evidencia de mistura recente
(Figura 13). O interessante é que ela foi originaria do grupo Oeste, ainda que ela esteja no
Leste da Amazônia, possivelmente devido a que o seu haplótipo só possui uma mutação, o
que a deixa ainda próxima do haplótipo ancestral, devido a uma separação relativamente
recente. Os dados mostram que essa população é um grupo biológico distinto que compartilha
um sistema reprodutivo entre seus indivíduos, mas não com outras populações. Mas talvez,
como nos casos anteriores, alguns indivíduos consigam migrar para outras populações.
Possivelmente, esse fato pode estar relacionado com a separação da bacia Araguaia-
Tocantins da bacia Amazônica ao redor de 1.8 milhões de anos. Durante o Pleistoceno as duas
bacias ficaram separadas devido a depósitos de sedimentos que preencheram o paleovale.
Mais tarde durante o Holoceno com a formação da Ilha do Marajó, foi criada uma pequena
conexão entre elas: o canal Amapá, um fino canal que liga o rio Amazonas com o rio Pará, no
qual drena o rio Araguaia-Tocantins (Rossetti e Valeriano, 2007). Porém, atualmente a
conexão entre as duas bacias é limitada pela presença de uma série de corredeiras na parte
baixa de ambos rios, reflexo da transição entre o Escudo Brasileiro e a planície amazônica.
Entretanto, dado que a diferencia entre haplótipos está determinada por uma mutação só, pode
ser que a colonização dessa bacia seja um evento muito mais recente que ocorreu depois da
configuração atual da bacia Araguaia-Tocantins e esteja relacionado com os períodos de
incursões marinhas. Durante o último milhão de anos, tem ocorrido oscilações do nível do
mar, a maioria levando à diminuição do nível e algumas ao incremento deste. Os maiores
aumentos do nível do mar foram aproximadamente 400.000 e 50.000 anos atrás. Altos níveis
marinhos podem ter gerado incursões marinhas que destruíram habitas de água doce e criaram
outros adequados para serem colonizados, ou podem ter feito um backup do rio Amazonas,
criando um enorme lago de água doce na parte baixa e central da Amazônia e portanto a

41
expansão da planície de inundação (Billups, 2004; Farias e Hrbek, 2008; Farias et al., 2010).
Dessa forma, o aumento do nível do mar pode ter permitido a conectividade entre regiões,
neste caso entre as bacia do Amazonas e do Araguaia-Tocantins e facilitado o passo do
jacarés através das corredeiras e cachoeiras dessa área, indicando uma colonização recente do
tipo efeito fundador.
Esse padrão de separação dos indivíduos do rio Araguaia em relação ao resto da
Amazônia foi evidenciado também para outras espécies. A tartaruga da Amazônia, P.
expansa, apresentou as populações do rio Araguaia, como o grupo mais claramente
diferenciado do resto de populações estudadas (Pearse et al., 2006). As populações de
pirarucu das localidades do alto rio Araguaia mostraram sinais de colonização recente (Leão,
2009). No boto vermelho (Inia sp.) as diferenças foram ainda mais fortes, chegando à
descrição de uma nova espécie, I. araguaiaensis sp. nov., para essa bacia (Hrbek et al., 2014).
O rio Napo apresentou o mesmo padrão que o rio Araguaia. Ficou incluso no grupo
vermelho, mas possui um haplótipo diferente (com alguns derivados) do resto das populações.
Um trabalho anterior mostrou que essa população corresponde a um grupo biológico
diferente, mediante inferência Bayesiana usando marcadores microssatélites (De Thoisy et al.,
2006); os autores sugeriram que o padrão de distribuição dessa população poderia estar
relacionado com a hipótese do "Lago Amazônica" (Marroig e Cerqueira, 1997) que propõe a
expansão do ecossistema de água doce do rio Amazonas devido ao aumento do nível da água
em 50-150 metros, formando um lago entre 35.000 e 5.000 BC, que pôde ter influenciado na
distribuição de várias espécies animais. Uma explicação alternativa para a sua diferenciação, é
que a população pode ter ficado isolada durante os períodos secos do Pleistoceno, que
geraram a fragmentação da floresta amazônica em vários blocos de bosque úmido (refúgios),
separados por savanas, bosques secos e outros tipos de vegetação de clima seco que podem ter
sido barreiras efetivas para a dispersão de plantas e animais da floresta úmida (Haffer e
Prance, 2002). No casso do jacaré-açu, cujo hábitat preferido parecem ser os lagos de várzea e
áreas alagáveis (Thorbjarnarson, 2010), esses teriam desaparecido durante os períodos secos
impedindo a dispersão dos indivíduos. Existem evidencias palineológicas que indicam que a
região do Napo foi um refúgio florestal das terras baixas amazônicas do Equador (Haffer e
Prance, 2002).
Os resultados obtidos são de grande importância para entender a distribuição
geográfica da espécie, que junto com sua história natural permitem ter uma visão mais clara
dos padrões e dinâmica das suas populações. Um fato interessante é que as corredeiras além
de serem barreira para o jacaré-açu restringindo o fluxo gênico, constituem o limite para a sua

42
distribuição em vários casos. Na Colômbia, por exemplo, a sua distribuição para o norte, está
limitada pela corredeira "La Libertad" no rio Apaporis, "El Depósito" no rio Miriti-Paraná,
"La Gamitana" no rio Yari e "Araracuara" no rio Caquetá (Japurá). No rio Guainía (alto rio
Negro) também não é encontrado, dado que a montante de Cucuí, o rio tem corredeiras e
cachoeiras (Medem, 1963), o que possivelmente explica sua ausência na Venezuela, onde não
tem registros da espécie. Igualmente, a outra alternativa para ele chegar na Venezuela, poderia
ser o rio Uraricoera, mas o rio é extremamente encachoeirado na sua parte alta. Além da
existência dessas barreiras, é possível que a presença de Crocodilus intermedius na região do
Orinoco também evite a dispersão de M. niger por exclusão competitiva.
De igual forma, para o sul, não é encontrado na bacia do Paraguai, talvez pela falta de
conexão entre as bacias. Caso contrário do que ocorre com Caiman yacare, que consegue
andar rapidamente por terra firme e já foi encontrado dentro da floresta, longe da água, entre
as cabeceiras dos rios Jaurú (tributário do rio Paraguai) e Aguapeí (tributário do rio Guaporé)
que ficam relativamente próximas (Medem, 1963). Nesse sentido, os resultados são
consistentes com os hábitos do jacaré-açu, que prefere lagos e áreas pantanosas, e devido a
seu tamanho e peso, sempre fica próximo dos ambientes aquáticos.
5.2 Diversidade genética
Como já tinha sido avaliada nos trabalhos anteriores (Farias et al., 2004; De Thoisy et
al., 2006; Vasconcelos et al., 2008), a diversidade genética da espécie M. niger é alta em
relação a outros crocodilianos. A diversidade gênica total foi Ĥ = 0.848, o que pode ser
explicado devido a que espécies com grandes populações geralmente possuem alta
diversidade genética (Frankham et al., 2004). Porém, o fato da espécie ter sofrido uma
diminuição drástica do número de indivíduos não se evidencia à nível genético, dado que a
diversidade se manteve, possivelmente devido aos grandes tamanhos populacionais que
existiam antes da época de caça do jacaré-açu na década dos 30, a uma diminuição
demográfica recente e ao longo tempo de geração da espécie. Essa mesma situação já foi
encontrada para outras espécies, como no caso do rinoceronte-indiano (Rhinocerus unicornis),
que constitui um exemplo clássico de genética da conservação. Os resultados do estudo para
essa espécie, mostraram a importância de considerar a distribuição histórica, a demografia e
os parâmetros da história natural para avaliar os efeitos de gargalo nas populações silvestres
(Dinerstein e McCracken, 1990).

43
Apesar dessa alta diversidade da espécie, os novos resultados obtidos nesse trabalho,
evidenciam que quatro das populações estudadas, apresentam uma diversidade genética
reduzida para o marcador Cytb. As populações dos rios Branco, Guaporé, Araguaia e Xingu
possuem um único haplótipo, portanto nenhum sítio variável. No caso do Xingu, só e
observado o haplótipo H1 que é um dos dois haplótipos mais comuns. Possivelmente, essa
população é produto de uma colonização bem recente, o qual pode ser suportado pelo fato de
que a espécie não ocorre a montante da Volta Grande do Xingu (Campos, comunicação
pessoal).
No caso das outras três populações, elas possuem um haplótipo próprio, talvez produto
do efeito fundador que levou à fixação daquele haplótipo em cada uma delas, perdendo a
variação genética da população original (Mayr, 1942). De igual forma, é evidente o aumento
da endogamia devido a que apresentam frequências alélicas diferentes das outras populações
(Figura 13).
As áreas geográficas com maior diversidade genética e com os alelos mais antigos, são
o centro de distribuição de uma espécie (Castelloe e Templeton, 1994). Como já tinha sido
sugerido por De Thoisy (2006), a região central da Amazônia é a mais diversa para M. niger
(grupo vermelho e haplótipo H2 na Figura 12); portanto, espera-se que as populações atuais
tenham sido originadas nesta região. De igual forma, Medem (1963) já tinha sugerido que os
centros de distribuição do jacaré-açu são o baixo e meio Amazonas, incluindo a ilha do
Marajó e Mexiana e a região denominada Solimões (Tabatinga-Manaus). Provavelmente,
estes são os sítios atuais de distribuição e por isso correspondem a os dois haplótipos mais
comuns (H1 e H2).
5.3 Rio Madeira
O rio Madeira representa um caso raro na Amazônia dado que é um rio grande de água
branca com trechos encachoeirados que marcam a transição abrupta entre o escudo brasileiro
e a bacia amazônica (De Queiroz et al., 2013). As corredeiras do Madeira/Guaporé são uma
série de 18 corredeiras em um trecho de 290 quilômetros (Cella-Ribeiro et al., 2013).
As corredeiras representam uma barreira para a dispersão do jacaré-açu diferenciando
as populações do rio Guaporé (AM) e Alto Madeira (EM) daquelas que estão no Baixo
Madeira (BM). Dos quatro haplótipos encontrados, dois são restritos ao BM, um a EM e

44
outro é compartilhado entre as três populações, mas com baixa frequência no BM, o que pode
significar que existe um fluxo gênico unidirecional na direção montante-jusante (Figura 16).
Figura 16. Modelo de fluxo gênico proposto para explicar o efeito das corredeiras do Rio Madeira como barreira
para M. niger. AM e EM formam um grupo diferente de BM, possivelmente devido a presença da cachoeira de
Teotônio. O fluxo gênico é unidirecional, dado que os indivíduos descem mas não conseguem subir através das
corredeiras.
No Baixo Madeira, existe um grupo biológico que é único para essa região e que se
encontra entre o lago Cuniã e Borba. Esse grupo corresponde ao haplótipo H5, que é derivado
do H4 do rio Uraricoera (Bra). Possivelmente, alguma conexão antiga existiu entre o rio
Madeira e o rio Negro, que permitiu que esses indivíduos colonizaram a parte baixa do rio
Madeira e se diferenciaram. Depois da conformação do curso atual do rio, esse grupo pode ter
ficado restrito a essa área devido algum evento histórico ainda não esclarecido, mas que
concorda com o padrão visto em outras espécies. Para boto, esse ponto é o limite entre a
distribuição das espécies I. geoffrensis e I. boliviana (Gravena et al., 2014), e para peixes é
uma quebra abrupta da ictiofauna presente (De Queiroz et al., 2013).
A chegada de indivíduos dos outros dois grupos no Baixo Madeira devem ser
migrações posteriores de indivíduos desde o rio Amazonas e alguns do Alto Madeira que
conseguiram atravessar as corredeiras, talvez durante alguma das grandes cheias do rio ou
arrastados pela correnteza, o que não é muito comum e explicaria a presença ocasional desses
indivíduos. O ambiente que eles preferem são as águas tranquilas, e são encontrados com
maior frequência nos rios com menos correnteza, nos remansos e especialmente nos lagos e
áreas pantanosas (Medem, 1963).
A população do rio Guaporé, que está acima das corredeiras provavelmente se separou
depois do surgimento delas, como consequência do levantamento do arco de Fitzcarrald
durante o Plioceno (~4 milhões de ano) (Pearse et al., 2006; Hrbek et al., 2008; Espurt et al.,
2010; Muniz, 2012; De Queiroz et al., 2013; Gravena et al., 2014; Gravena et al., 2015). A

45
população original ficou fragmentada e esse grupo ficou isolado, o que permitiu a sua
diferenciação. Os dados moleculares evidenciam que no rio Guaporé, existe um grupo
biológico puro (Figura 15) formado por indivíduos com um haplótipo só (Figura 14) com um
sistema endogâmico marcante sem fluxo gênico entrante.
Porém, parece que essa população pode ser fonte de indivíduos que migram para a
região entre as corredeiras e que se estabeleceram recentemente, dado que os dados
mitocondriais mostram que a maioria possuem o mesmo haplótipo (H9) mas existe um
pequeno grau de mistura no seu genoma nuclear. Possivelmente, os dois grupos determinados
pelo mtDNA (amarelo e rosa) estão se reproduzindo entre si. Esse pequeno grupo rosa, está
restrito ao trecho que está entre as cachoeiras de Jirau e Teotônio.
Basicamente, M. niger está divida em dois grupos determinados por uma forte
estruturação (ver valores de Fst). A população do Baixo Madeira se diferencia das outras duas
devido a uma grande restrição ao fluxo gênico que pode estar determinada principalmente
pela cachoeira de Teotônio, como já foi visto para outras espécies. A cachoeira de Teotônio
era de tipo composto, com 900 metros de largura e 600 de longitude, com uma queda de água
de 30 metros ao longo de um quilômetro. Devido a suas dimensões era a mais importante e
junto com a cachoeira de Jirau, a segunda mais importante (730 metros de largura e 1000 de
extensão) constituíam um limite natural para várias espécies ou populações de animais
aquáticos (Pearse et al., 2006; Hrbek et al., 2008; Muniz, 2012; De Queiroz et al., 2013;
Gravena et al., 2014; Gravena et al., 2015).
Portanto, é claro que para animais aquáticos o deslocamento está restrito aos caminhos
fluviais que conectam as diferentes áreas de uma região. Por isso, qualquer limitação na
movimentação pode ser chamada barreiras geográfica. Elas são importantes porque são um
fator que determina a composição da fauna regional e promove o endemismo (Rahel, 2007),
tendo efeito nos processos evolutivos das espécies e populações.
5.4 Conservação
O jacaré-açu é uma espécie catalogada como em Baixo Risco, dependente da
conservação (LR/CD) na Lista Vermelha da IUCN desde 2009 (IUCN, 2014), devido à
recuperação demográfica em várias áreas da sua distribuição. Além das avaliações em campo,
os dados genéticos indicam que a nível geral, o jacaré-açu está experimentando uma rápida
expansão populacional (D de Tajima e Fs de Fu significativos), possivelmente depois de uma

46
redução demográfica drástica. Como é bem sabido, a espécie experimentou uma forte pressão
por caça ilegal, entre 1930 e 1980 (Da Silveira e Thorbjarnarson, 1999), que a colocou quase
em risco de extinção.
A espécie tem um potencial de recuperação populacional alto, que pode estar
relacionado com a influência da paternidade múltipla na manutenção da variabilidade genética
e o incremento do tamanho efetivo populacional (Sugg e Chesser, 1994; Muniz et al., 2011),
que foi evidenciado nos alto valores de diversidade genética (Ĥ) encontrados nesse trabalho e
nos anteriores. Depois de 25 anos, as populações, principalmente de áreas protegidas, tem
conseguido aumentar o número de indivíduos consideravelmente. Contudo, elas ainda são
vulneráveis devido a caça ilegal para obtenção da carne e destruição de hábitat, precisando de
planos de manejo para a sua conservação.
A dinâmica demográfica e os padrões de distribuição da variabilidade genética são
importantes para a manutenção das espécies, e contribuem com as ações de conservação
fazendo com que sejam mais efetivas. Nesse sentido, os aportes da genética são vários, mas
dentre os mais importantes está o delineamento de unidades de conservação como entidades
biologicamente significativas para a conservação (Frankham et al., 2004; Funk et al., 2012).
No caso das populações que apresentam um padrão contínuo de distribuição da
variabilidade genética Oste-Leste, a definição de unidades de conservação pode se tornar
complicada devido a que não é possível delimitar unidades discretas. Então, é importante
estabelecer a melhor estratégia para definir as unidades em nível intraespecífico e poder
conservar a variabilidade genética da espécie. Para isso, podem ser definidas as posições
geográficas de unidades operacionais, como reservas, onde é possível focar os esforços de
conservação mediante a implementação de planos de manejo e monitoramentos constantes
(Diniz-Filho e Telles, 2002).
O jacaré-açu é um componente econômico importante e notável do ecossistema de
várzea da Amazônia. Como foi dito anteriormente, apresenta uma estrutura genética similar
com outras espécie de fauna aquática (tartaruga da Amazônia, peixe-boi e pirarucu), e nesse
sentido a proposta de unidades operacionais beneficia todas as espécies como membros
importantes da várzea (Hrbek et al., 2007). Para pirarucu já foram propostas estratégias de
conservação que podem ser aplicadas para o jacaré-açu, levando em consideração as
particularidades da espécie. Assim, em nível geral, pelo menos duas reservas deveriam existir
no ecossistema de várzea que abrange uns quatro mil quilômetros, quase a totalidade da
distribuição de M. niger, para conservar a maior parte da diversidade genética. Isso significa
que pelo menos deve haver uma reserva no grupo Oeste e outra no grupo Leste. O centro de

47
dispersão da espécie é o mais antigo e diverso, então é importante estabelecer uma reserva
nessa área correspondente ao grupo Centro.
Já para as populações estruturadas dos rios Araguaia (Ara), Guaporé (AM),
Uraricoera (Bra) e Napo (RN), podemos considerar que constituem unidades de manejo
(MUs) levando em consideração os critérios definidos para tal atribuição: não existe
monofilia recíproca no mtDNA, mas existe divergência nas frequências haplotípicas e
alélicas; representam populações biologicamente independentes, mas com fluxo gênico
restrito (Moritz, 1994; Funk et al., 2012). Além disso, as primeiras três populações
apresentam um único haplótipo e a sua diversidade genética é reduzida, o que as faz
potencialmente vulneráveis, merecendo uma consideração especial.
Devido a que barreiras são importantes para a estrutura e dinâmica da espécie, a
construção de barragens é uma problemática atual devido ao ritmo acelerado de construção no
Brasil e os países vizinhos. A maioria de populações estruturadas se encontram em regiões
que estão acima de corredeiras e que portanto são alvo para a construção de hidrelétricas. No
rio Tocantins, a hidrelétrica de Tucuruí está em funcionamento há quase 30 anos atrás; no rio
Madeira, as hidroelétricas de Jirau e Santo Antônio fecharam as barragens recentemente
(2011-2012) deixando isolado o trecho entre elas. No rio Branco, a hidrelétrica Bem Querer-
Paredão está em fase de projeto.
Não existe informação sobre a adaptabilidade de M. niger às represas (Marioni et al.,
2013), mas daqui a uns anos conheceremos os seus efeitos e consequências. Podemos intuir
que a modificação das características naturais do sistema terá efeitos sobre a conectividade ou
isolamento, que são reconhecidos como fatores fundamentais que determinam a distribuição
das espécies (Pringle, 2003); mas esses efeitos estarão dirigidos à perda de variabilidade
genética dado que as populações a montante da corredeiras já estão isoladas mas contribuem
com indivíduos que possuem características genéticas diferentes. Porém, com base nos
resultados deste trabalho, podemos dizer que as maiores consequências da construção de
hidrelétricas para as populações de jacaré-açu, serão de carater ambiental e ecológico. O
regime hidrológico mudará afetando os ciclos de enchente e vazante dos rios que estão
relacionados com a reprodução, alimentação e distribuição da espécie; e as áreas de
nidificaçaõ diminuirão ou desaparecerão devido ao alagamento de áreas florestais para
construção dos reservatórios. Desta maneira, recomenda-se que sejam feitos monitoramentos
genéticos ao longo dos anos, para que no futuro possamos mitigar quaisquer efeitos dessas
barreiras à saúde genética das populações de M. niger.

48
Portanto, nesse trabalho definimos sete áreas importantes para conservação que
abrangem quase a totalidade da distribuição da espécie. Os grupos Oeste, Centro, Leste,
Guyana, Bolivia, Napo e Araguaia são as áreas onde deveriam existir unidades operacionais
para implementação dos planos de manejo (
Figura 17). Ficam algumas localidades em branco, onde seria importante fazer
esforços de amostragem para completar a definição das áreas de conservação. Assim mesmo,
é importante que todas as áreas escolhidas sejam o suficientemente grandes (megareservas:
maiores a uma megahectar), levando em consideração a capacidade de deslocamento dos
indivíduos, calculada em pelo menos 38 km (Da Silveira et al., 2011); bem conservadas,
porque a manutenção das populações por migração em áreas degradadas não é viável; e com
áreas de nidificação, dado que áreas com densidades altas de ninhos também apresentam altas
densidades de adultos e subadultos (Da Silveira et al., 1997). Todos esses fatores permitirão o
desenvolvimento de grandes populações que sejam fonte de indivíduos para áreas menos
adequadas e sujeitas a forte pressão antrópica.
Figura 17. Mapa das sete áreas definidas para conservação do jacaré-açu (M. niger) ao longo da sua de
distribuição. As interrogações indicam ausência de dados. A distribuição da espécie está delimitada pela área
branca. Camada da distribuição tomada de (IUCN, 2000).

49
CONCLUSÃO
As populações de jacaré-açu (M. niger) apresentam padrões de distribuição da
variabilidade genética devido a diferenciação por isolamento por distância e estruturação por
presença de barreira geográficas, que constituem as dinâmicas populacionais mais importantes
para a espécie.
As corredeiras do rio Madeira/Guaporé atuam como barreira para a espécie
restringindo o fluxo gênico entre as populações que estão acima, entre e abaixo delas, e só é
dado de forma unidirecional, em sentido montante-jusante.
O isolamento por distância determina um padrão Oeste-Leste o que significa que é
necessário definir unidades operacionais, tais como reservas, para conservar a variabilidade
genética da espécie.
A diferenciação Norte-Sul evidencia uma antiga conexão entre o baixo rio Negro e o
baixo rio Madeira. Essas duas áreas constituem um centro de variabilidade genética da
espécie. Portanto, a definição de unidades operacionais permitirá a conservação da
diversidade genética assim como dos processos evolutivos e históricos que levaram ao
desenvolvimento desse padrão.
As populações dos rio Araguaia, Guaporé e Uraricoera constituem unidades de manejo
diferenciadas devido a que são grupos biológicos isolados, que compartilham um sistema de
reprodução dentro de cada um deles e não recebem indivíduos de outras populações.
A diversidade genética da espécie é alta quando comparada com outras espécies de
crocodilianos, embora existam populações isoladas com diversidade reduzida.
Para espécies de ampla distribuição é importante abranger grande parte dela para ter
informação mais completa sobre sua dinâmica e variabilidade genética.

50
As ameaças atuais tais como destruição de habitat por construção de hidrelétricas,
podem afetar os padrões de distribuição da espécie. Outras ameaças como caça ilegal podem
afetá-la demograficamente diminuindo a sua variabilidade genética.
REFERÊNCIAS BIBLIOGRÁFICAS
Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. 2010. Genomics and the future of
conservation genetics. Nat Rev Genet, 11: 697-709.
Andrade, T.; Coutinho, M.E. (Anais do III Congresso Brasileiro de Herpetologia).
2007. Ecologia populacional dos jacarés (Melanosuchus niger e Caiman crocodilus) na área
de proteção ambiental Meandros do Araguaia/GO-MT. Anais do III Congresso Brasileiro de
Herpetologia, Belém.
Andrade, T.; Coutinho, M.E. (Anais do IV Congresso Brasileiro de Herpetologia).
2009. Ecologia da nidificação de jacaré-açu (Melanosuchus niger) na Área de Proteção
Ambiental Meandros do Araguaia/GO-MT. Anais do IV Congresso Brasileiro de
Herpetologia, Pirenópolis.
Andrade, T.; Coutinho, M.E. (Anais do IX Congresso Latinoamericano de
Herpetologia e V Congresso Brasileiro de Herpetologia). 2011. Distribuição e abundância de
crocodilianos no Parque Nacional Cabo Orange, Amapá, Brasil. Anais do IX Congresso
Latinoamericano de Herpetologia e V Congresso Brasileiro de Herpetologia, Curitiba.
Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb,
C.A.; Saunders, N.C. 1987. INTRASPECIFIC PHYLOGEOGRAPHY: The Mitochondrial
DNA Bridge Between Population Genetics and Systematics. Annu Rev Ecol Syst, 18: 489-
522.
Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.;
Selker, E.U.; Cresko, W.A.; Johnson, E.A. 2008. Rapid SNP discovery and genetic mapping
using sequenced RAD markers. PLOS ONE, 3(10): e3376.
Banguera-Hinestroza, E.; Cárdenas, H.; Ruiz-García, M.; Marmontel, M.; Gaitán, E.;
Vázquez, R.; García-Vallejo, F. 2002. Molecular identification of evolutionarily significant
units in the Amazon river dolphin Inia sp.(Cetacea: Iniidae). J Hered, 93(5): 312-322.
Barbosa, R.I.; Campos, C.; Pinto, F.; Fearnside, P.M. 2007. The" Lavrados" of
Roraima: Biodiversity and Conservation of Brazil's Amazonian Savannas. Functional
Ecosystems and Communities, 1(1): 30-42.
Billups, K. 2004. Palaeoclimate: Low-down on a rhythmic high. Nature, 427(6976):
686-687.

51
Botero-Arias, R.; Franco, D.d.L.; Marmontel, M. 2014. A mortalidade de jacarés e
botos associada à pesca da piracatinga na região do Médio Solimões - Amazonas, Brasil.
Instituto de Desenvolvimento Sustantável Mamirauá, Tefé. 60pp.
Brazaitis, P.; Rebêlo, H.; Yamashita, C.; Odierna, E.A.; Walanabe, M.E. 1996. Threats
to Brazilian crocodilian populations. Oryx, 30(4): 275-284.
Britton, A. 2012. Crocodilian Species List: Melanosuchus niger (Spix, 1825).
http://crocodilian.com/cnhc/csp_mnig.htm. Acesso: 01/08 2013.
Brochu, C.A. 2003. Phylogenetic approaches toward crocodylian history. Annu Rev
Earth Planet Sci, 31(1): 357-397.
Brown, W.M.; George Jr, M.; Wilson, A.C. 1979. Rapid evolution of animal
mitochondrial DNA. Proc Natl Acad Sci USA, 76(4): 1967-1971.
Brum, S.M.; Silva, V.M.F.; Rossoni, F.; Castello, L. 2015. Use of dolphins and
caimans as bait for Calophysus macropterus (Lichtenstein, 1819) (Siluriforme: Pimelodidae)
in the Amazon. J Appl Ichthyol: 1-6.
Cantanhede, A.M.; Da Silva, V.M.F.; Farias, I.P.; Hrbek, T.; Lazzarini, S.M.; Alves-
Gomes, J. 2005. Phylogeography and population genetics of the endangered Amazonian
manatee, Trichechus inunguis Natterer, 1883 (Mammalia, Sirenia). Mol Ecol, 14: 401-413.
Canton, R.d.C. 2014. Análise da variação fenotípica e genotípica do complexo
Brotogeris sanctithomae (Aves: Psittaciformes). Dissertação de mestrado, Instituto Nacional
de Pesquisa da Amazônia INPA, Manaus. 52pp.
Castelloe, J.; Templeton, A.R. 1994. Root probabilities for intraspecific gene trees
under neutral coalescent theory. Mol Phylogen Evol, 3(2): 102-113.
Catchen, J.; Bassham, S.; Wilson, T.; Currey, M.; O´Brien, C.; Yeates, Q.; Cresko,
W.A. 2013a. The population structure and recent colonization history of Oregon threespine
stickleback determined using restriction-site associated DNA-sequencing. Mol Ecol, 22:
2864-2883.
Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. 2013b. Stacks:
an analysis tool set for population genomics. Mol Ecol, 22(11): 3124-3140.
Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. 2011.
Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes, Genom,
Genet, 1(3): 171-182.
Cella-Ribeiro, A.; Torrente-Vilara, G.; Hungria, D.B.; De Oliveira, M. 2013. As
Corredeiras do Rio Madeira. In: De Queiroz, L.J.; Torrente-Vilara, G.; Massaharu, W.; da
Silva, T.H.; Zuanon, J.; da Costa, C.R. (Eds). Peixes do Rio Madeira. Dialeto Latin American
Documentary, São Paulo. p. 57-63.
CITES. 2007. Considerations of Proposal for Amendment of Appendices I and II.
Fourteenth meeting of the Conference of the Parties. Convention on International Trade in
Endangered Species of Wild Fauna and Flora, The Hague (Netherlands), CoP14 Prop. 13.

52
Corander, J.; Marttinen, P.; Sirén, J.; Tang, J. 2008. Enhanced Bayesian modelling in
BAPS software for learning genetic structures of populations. BMC Bioinformatics, 9(1): 539.
Corander, J.; Waldmann, P.; Sillanpaa, M.J. 2003. Bayesian Analysis of Genetic
Differentiation Between Populations. Genetics, 163( ): 367-374.
Crandall, K.A.; Bininda-Emonds, O.R.P.; Mace, G.M.; Wayne, R.K. 2000.
Considering evolutionary processes in conservation biology. Trends Ecol Evol, 15(7): 290-
295.
CSG. 1995. Resurgence of illegal trade in skin of black caiman Melanosuchus niger.
CSG Crocodile Specialist Group Newsletter, 14(2): 14-15.
Da Silveira, R. 2001. Monitoramento, crescimento e caça de jacaré-açu
(Melanosuchus niger) e jacaré-tinga (Caiman crocodilus crocodilus). Tese de Doutorado,
Instituto Nacional de Pesquisas da Amazônia - INPA / Universidade Federal do Amazonas -
UFAM, Manaus. 145pp.
Da Silveira, R.; Amaral, J.V.; Magnusson, W.E.; Thorbjarnarson, J.B. 2011.
Melanosuchus niger (Black Caiman). Long distance movement. Herpetological Review,
42(3): 424-425.
Da Silveira, R.; Magnusson, W.E.; Campos, Z. 1997. Monitoring the distribution,
abundance and breeding areas of Caiman crocodilus crocodilus and Melanosuchus niger in
the Anavilhanas Archipelago, Central Amazonia, Brazil. J Herpetol, 31(4): 514-520.
Da Silveira, R.; Thorbjarnarson, J.B. 1999. Conservation implications of commercial
hunting of black and spectacled caiman in the Mamirauá Sustainable Development Reserve,
Brazil. Biol Conserv, 88(1): 103-109.
De Queiroz, L.J.; Torrente-Vilara, G.; Massaharu, W.; da Silva, T.H.; Zuanon, J.; da
Costa, C.R. 2013. Peixes do Rio Madeira. Dialeto Latin American Documentary, São Paulo.
400pp.
De Thoisy, B.; Hrbek, T.; Farias, I.P.; Vasconcelos, W.R.; Lavergne, A. 2006. Genetic
structure, population dynamics, and conservation of Black caiman (Melanosuchus niger). Biol
Conserv, 133(4): 474-482.
DeSalle, R.; Amato, G. 2004. The expansion of conservation genetics. Nat Rev Genet,
5: 702-712.
Dinerstein, E.; McCracken, G.F. 1990. Endangered greater one-horned rhinoceros
carry high levels of genetic variation. Conserv Biol, 4(4): 417-422.
Diniz-Filho, J.A.F.; Telles, M.P.D.C. 2002. Spatial autocorrelation analysis and the
identification of operational units for conservation in continuous populations. Conserv Biol,
16(4): 924-935.
Doyle, J.J.; Doyle, J.L. 1987. A rapid DNA isolation procedure for small quantities of
fresh leaf tissue. Phytochem Bull, 19: 11-15.

53
Earl, D.A.; vonHoldt, B.M. 2012. STRUCTURE HARVESTER: a website and
program for visualizing STRUCTURE output and implementing the Evanno method. Conserv
Genet Resour, 4(2): 359-361.
Eaton, D.A.R. 2014. PyRAD: assembly of de novo RADseq loci for phylogenetic
analyses. Bioinformatics: 1-6.
Espurt, N.; Baby, P.; Brusset, S.; Roddaz, M.; Hermoza, W.; Barbarand, J. 2010. The
Nazca Ridge and uplift of the Fitzcarrald Arch: implications for regional geology in Northern
South America. In: Hoorn, C.; Wesselingh, F.P. (Eds). Amazonia: Landscape and Species
Evolution: a look into the past. Blackwell Publishing. p. 89-100.
Estupiñán, G.M.B.; Marmontel, M.; De Queiroz, H.L.; Roberto e Souza, P.; Valsecchi,
J.; Batista, G.d.S.; Pereira, S.B. 2003. A pesca da piracatinga (Calophysus macropterus) na
Reserva de Desenvolvimento Sustentável Mamirauá. Relatorio técnico, Ministério da Ciência
e Tecnologia e Instituto de Desenvolvimento Sustentável Mamirauá, Tefé. 14pp.
Evanno, G.; Regnaut, S.; Goudet, J. 2005. Detecting the number of clusters of
individuals using the software STRUCTURE: a simulation study. Mol Ecol, 14: 2611-2620.
Excoffier, L.; Lischer, H.E.L. 2010. Arlequin suite ver 3.5: a new series of programs
to perform population genetics analyses under Linux and Windows. Mol Ecol Resour, 10(3):
564-567.
Excoffier, L.; Smouse, P.E.; Quattro, J.M. 1992. Analysis of molecular variance
inferred from metric distances among DNA haplotypes: application to human mitochondrial
DNA restriction data. Genetics, 131(2): 479-491.
Falush, D.; Stephens, M.; Pritchard, J.K. 2003. Inference of population structure using
multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164(4):
1567-1587.
Farias, I.P.; Hrbek, T. 2008. Patterns of diversification in the discus fishes
(Symphysodon spp. Cichlidae) of the Amazon basin. Mol Phylogen Evol, 49(1): 32-43.
Farias, I.P.; Silveira, R.D.; Thoisy, B.d.; Monjela, L.A.; Thorbjarnarson, J.; Hrbek, T.
2004. Genetic diversity and population structure of Amazonian crocodilians. Anim Conserv,
7(3): 265-272.
Farias, I.P.; Torrico, J.P.; García-Dávila, C.; Santos, M.d.C.F.; Hrbek, T.; Renno, J.-F.
2010. Are rapids a barrier for floodplain fishes of the Amazon basin? A demographic study of
the keystone floodplain species Colossoma macropomum (Teleostei: Characiformes). Mol
Phylogen Evol, 56: 1129-1135.
Farias, J.G. 2015. Filogeografia e genética populacional de Inia geoffrensis
geoffrensis e Inia geoffrensis humboldtiana (Cetartiodactyla: Iniidae) nos rios Branco e
Negro e Sistema Orinoco. Dissertação de Mestrado, Universidade Federal do Amazonas -
UFAM, Manaus. 116pp.
Finer, M.; Jenkins, C.N. 2012. Proliferation of Hydroelectric Dams in the Andean
Amazon and Implications for Andes-Amazon Connectivity. PLOS ONE, 7(4): e35126.

54
Frankham, R.; Ballou, J.D.; Briscoe, D.A. 2004. A primer of conservation genetics.
Cambridge University Press, New York. 220pp.
Fu, Y.-X. 1997. Statistical tests of neutrality of mutations against population growth,
hitchhiking and background selection. Genetics, 147(2): 915-925.
Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. 2012. Harnessing
genomics for delineating conservation units. Trends Ecol Evol, 27(9): 489-496.
Gatesy, J.; Baker, R.H.; Hayashi, C. 2004. Inconsistencies in arguments for the
supertree approach: supermatrices versus supertrees of Crocodylia. Syst Biol, 53(2): 342-355.
Glenn, T.C.; Staton, J.L.; Vu, A.T.; Davis, L.M.; Bremer, J.R.A.; Rhodes, W.E.;
Brisbin, I.L.; Sawyer, R.H. 2002. Low mitochondrial DNA variation among American
Alligators and a novel non-coding region in Crocodilians. J Exp Zool B Mol Dev Evol, 294(4):
312-324.
Gravena, W.; Farias, I.P.; da Silva, M.N.F.; da Silva, V.M.F.; Hrbek, T. 2014.
Looking to the past and the future: were the Madeira River rapids a geographical barrier to the
boto (Cetacea: Iniidae)? Conserv Genet, 15(3): 619-629.
Gravena, W.; Silva, V.M.F.; Silva, M.N.F.; Farias, I.P.; Hrbek, T. 2015. Living
between rapids: genetic structure and hybridization in botos (Cetacea: Iniidae: Inia spp.) of
the Madeira River, Brazil. Biol J Linn Soc, 114: 764-777.
Haffer, J.; Prance, G.T. 2002. Climatic forcing of evolution in Amazonia during the
Cenozoic: on the refuge theory of biotic differentiation. Amazoniana, 16(3): 579-607.
Hrbek, T.; Crossa, M.; Farias, I.P. 2007. Conservation strategies for Arapaima gigas
(Schinz, 1822) and the Amazonian várzea ecosystem. Braz J Biol, 67(4): 909-917.
Hrbek, T.; da Silva, V.M.F.; Dutra, N.; Gravena, W.; Martin, A.R.; Farias, I.P. 2014.
A new species of river dolphin from Brazil or: how little do we know our biodiversity. PLOS
ONE, 9(1): e83623.
Hrbek, T.; Farias, I.P.; Crossa, M.; Sampaio, I.; Porto, J.I.R.; Meyer, A. 2005.
Population genetic analysis of Arapaima gigas, one of the largest freshwater fishes of the
Amazon basin: implications for its conservation. Anim Conserv, 8(3): 297-308.
Hrbek, T.; Vasconcelos, W.R.; Rebelo, G.; Farias, I.P. 2008. Phylogenetic
relationships of South American alligatorids and the Caiman of Madeira River. J Exp Zool A
Ecol Genet Physiol, 309(10): 588-599.
IUCN (Internacional Union for Conservation of Nature). 2000. Melanosuchus niger.
The IUCN Red List of Threatened Species. Version 2015.2. http://www.iucnredlist.org.
Acesso: 12 de junho de 2015.
IUCN (Internacional Union for Conservation of Nature). 2014. Melanosuchus niger.
The IUCN Red List of Threatened Species. Version 2014.3. http://www.iucnredlist.org.
Acesso: 4 de abril de 2015.

55
Jakobsson, M.; Rosenberg, N.A. 2007. CLUMPP: a cluster matching and permutation
program for dealing with label switching and multimodality in analysis of population
structure. Bioinformatics, 23(14): 1801-1806.
Janke, A.; Arnason, U. 1997. The Complete Mitochondrial Genome of Alligator
mississippiensis and the Separation Between Recent Archosauria (Birds and Crocodiles). Mol
Biol Evol, 14(12): 1266-1272.
Janke, A.; Erpenbeck, D.; Nilsson, M.; Arnason, U. 2001. The mitochondrial genomes
of the iguana (Iguana iguana) and the caiman (Caiman crocodylus): implications for amniote
phylogeny. Proc R Soc Lond, Ser B: Biol Sci, 268: 623-631.
Jelden, D.; Ross, J.P.; Thorbjarnarson, J.; TRAFFIC_SouthAmerica; Webb, G.
(CITES, CoP 14 Prop. 13). 2007. Transfer of the Black Caiman Melanosuchus niger
population of Brazil from Appendix I to Appendix II. CITES, CoP 14 Prop. 13. p. 1-7
Jobb, G.; von Haeseler, A.; Strimmer, K. 2004. TREEFINDER: a powerful graphical
analysis environment for molecular phylogenetics. BMC Evol Biol, 4: 18.
Johns, G.C.; Avise, J.C. 1998. A comparative summary of genetic distances in the
vertebrates from the mitochondrial cytochrome b gene. Mol Biol Evol, 15(11): 1481-1490.
Kimura, M. 1969. The number of heterozygous nucleotide sites maintained in a finite
population due to steady flux of mutations. Genetics, 61(4): 893-903.
King, F.W.; Burke, R.L. 1997. Crocodilian, tuatara, and turtle species of the world:
An online taxonomic and geographic reference. Association of Systematics Collections,
Washington, D.C. 217pp.
Leão, A.S.d.A. 2009. Análises da variabilidade genética das populações de pirarucu
(Arapaima gigas, Schinz 1822) dos principais tributários do rio Amazonas através do uso de
marcadores microssatélites. Dissertação de Mestrado, Instituto Nacional de Pesquisas da
Amazônia - INPA / Universidade Federal do Amazonas - UFAM, Manaus. 88pp.
Li, W.-H. 1987. Molecular Evolution. Sinauer Associates, Sunderland. 487pp.
Lischer, H.E.L.; Excoffier, L. 2012. PGDSpider: an automated data conversion tool
for connecting population genetics and genomics programs. Bioinformatics, 28(2): 298-299.
Lundberg, J.G.; Marshall, L.G.; Guerrero, J.; Horton, B.; Malabarba, M.C.S.L.;
Wesselingh, F. 1998. The stage for Neotropical fish diversification: a history of tropical South
American rivers. In: Malabarba, L.R.; Reis, R.E.; Vari, R.P.; Lucena, Z.M.; Lucena, C.A.S.
(Eds). Phylogeny and Classification of Neotropical Fishes. Edipucrs, Porto Alegre. p. 13-48.
Mantel, N. 1967. The detection of disease clustering and a generalized regression
approach. Cancer Res, 27(2): 209-220.
Mardis, E.R. 2008. Next-Generation DNA Sequencing Methods. Annual Review of
Genomics and Human Genetics, 9: 387-402.
Marioni, B.; Farias, I.; Verdade, L.M.; Bassetti, L.; Coutinho, M.E.; de Mendonça,
S.H.S.T.; Vieira, T.Q.; Magnusson, W.E.; Campos, Z. 2013. Avaliação do risco de extinção

56
do jacaré-açú Melanosuchus niger (Spix, 1825) no Brasil. Biodiversidade Brasileira, 3(1): 31-
39.
Marroig, G.; Cerqueira, R. 1997. Plio-Pleistocene South American history and the
Amazon Lagoon hypothesis: a piece in the puzzle of Amazonian diversification. Journal of
Comparative Biology, 2(2): 103-118.
Martin, A.P.; Palumbi, S.R. 1993. Body size, metabolic rate, generation time, and the
molecular clock. Proceedings of the National Academy of Sciences, 90(9): 4087-4091.
Martínez, J.G.; Hernández-Rangel, S.M.; Bertoul, F.; Farias, I.P.; Hrbek, T. (in prep).
Simplifying the Construction of Subgenomics Libraries for Next Generation Sequencing: an
Adaptation of ddRADs-based genotyping for Ion-Torrent in Non-model Amazonian Animal
Taxas.
Mayr, E. 1942. Systematics and the Origin of Species. Columbia University Press,
New York.
McAliley, L.R.; Willis, R.E.; Ray, D.A.; White, P.S.; Brochu, C.A.; Densmore, L.D.
2006. Are crocodiles really monophyletic?-Evidence for subdivisions from sequence and
morphological data. Mol Phylogen Evol, 39(1): 16-32.
Medem, F. 1963. Osteología craneal, distribución geográfica y ecología de
Melanosuchus niger (Spix), (Crocodylia: Alligatoridae). Rev Acad Colomb Cienc, 12(45): 5-
19.
Medem, F. 1983. Los Crocodylia de Sur America. Universidad Nacional de Colombia,
Fondo Colombiano de Investigaciones Científicas y Proyectos Especiales "Francisco José de
Caldas", Colciencias, Bogotá. 261pp.
Mendoça, S.; Coutinho, M.E. (Anais do IV Congresso Brasileiro de Herpetologia).
2009. Bases biológicas para o manejo do jacaré-açu (Melanosuchus niger) na Reserva
Extrativista do Lago do Cuniã, Rondônia. Anais do IV Congresso Brasileiro de Herpetologia,
Pirenópolis.
Meyer, A. 1994. Shortcomings of the cytochrome b gene as a molecular marker.
Trends Ecol Evol, 9(8): 278-280.
Miller, M.A.; Pfeiffer, W.; Schwartz, T. 2010. Creating the CIPRES Science Gateway
for inference of large phylogenetic trees. Gateway Computing Environments Workshop
(GCE), : 1-8.
Morin, P.A.; Luikart, G.; Wayne, R.K. 2004. SNPs in ecology, evolution and
conservation. Trends Ecol Evol, 19(4): 208-216.
Moritz, C. 1994. Defining "Evolutionarily Significant Unit" for conservation. Trends
Ecol Evol, 9(10): 373-375.
Muniz, F.L. 2012. Filogeografia e genética de populações de jacaré-paguá
(Paleosuchus palpebrosus) ao longo do rio Madeira e bacia do rio Paraguai (Pantanal).
Dissertação de Mestrado, Instituto Nacional de Pesquisas da Amazônia - INPA / Universidade
Federal do Amazonas - UFAM, Manaus. 95pp.

57
Muniz, F.L.; Campos, Z.; Hrbek, T.; Farias, I.P. (in prep). Unraveling the recent
history of the cryptic Paleosuchus palpebrosus from the Amazon to Pantanal basin.
Muniz, F.L.; Da Silveira, R.; Campos, Z.; Magnusson, W.E.; Hrbek, T.; Farias, I.P.
2011. Multiple paternity in the Black Caiman (Melanosuchus niger) population in the
Anavilhanas National Park, Brazilian Amazonia. Amphibia-Reptilia, 32(3): 428-434.
Nei, M. 1987. Molecular Evolutionary Genetics. Columbia University Press, New
York.
Ojasti, J. 1996. Wildlife utilization in Latin America: current situation and prospects
for sustainable management. FAO, Roma. 237pp.
Pearse, D.E.; Arndt, A.D.; Valenzuela, N.; Miller, B.A.; Cantarelli, V.; Sites Jr, J.W.
2006. Estimating population structure under nonequilibrium conditions in a conservation
context: continent-wide population genetics of the giant Amazon river turtle, Podocnemis
expansa (Chelonia; Podocnemididae). Mol Ecol, 15: 985-1006.
Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. 2012. Double
digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model
and non-model species. PLOS ONE, 7(5): e37135.
Pickles, R.S.A.; Groombridge, J.J.; Rojas, V.D.Z.; Van Damme, P.; Gottelli, D.;
Ariani, C.V.; Jordan, W.C. 2012. Genetic diversity and population structure in the endangered
giant otter, Pteronura brasiliensis. Conserv Genet, 13(1): 235-245.
Pickles, R.S.A.; Groombridge, J.J.; Rojas, V.D.Z.; Van Damme, P.; Gottelli, D.;
Kundu, S.; et al. 2011. Evolutionary history and identification of conservation units in the
giant otter, Pteronura brasiliensis. Mol Phylogen Evol, 61(3): 616-627.
Plotkin, M.J.; Medem, F.; Mittermeier, R.A.; Constable, I.D. 1983. Distribution and
conservation of the black caiman (Melanosuchus niger). In: Rhodin, A.G.J.; Miyata, K.
(Eds). Advances in Herpetology and Evolutionary Biology. Museum of Comparative Zoology,
Cambridge. p. 695-705.
Pringle, C. 2003. What is hydrologic connectivity and why is it ecologically
important? Hydrol Process, 17: 2685-2689.
Pritchard, J.K.; Stephens, M.; Donnelly, P. 2000. Inference of population structure
using multilocus genotype data. Genetics, 155(2): 945-959.
Rahel, F.J. 2007. Biogeographic barriers, connectivity and homogenization of
freshwater faunas: it's a small world after all. Freshwat Biol, 52(4): 696-710.
Rodríguez, M.A. 2000. Cocodrilos (Archosauria: Crocodylia) de la región neotropical.
Biota Colombiana, 1(2): 135-140.
Roos, J.; Aggarwal, R.K.; Janke, A. 2007. Extended mitogenomic phylogenetic
analyses yield new insight into crocodylian evolution and their survival of the Cretaceous-
Tertiary boundary. Mol Phylogen Evol, 45(2): 663-673.

58
Rosenberg, N.A. 2004. DISTRUCT: a program for the graphical display of population
structure. Mol Ecol Notes, 4(1): 137-138.
Ross, J.P. 1998. Crocodiles. Status survey and conservation action plan. IUCN/SSC
Crocodile Specialist Group. IUCN Gland, Switzerland and Cambridge. viii + 96pp.
Rossetti, D.F.; Valeriano, M.M. 2007. Evolution of the lowest amazon basin modeled
from the integration of geological and SRTM topographic data. Catena, 70: 253-265.
Rueda-Almonacid, J.V.; Carr, J.L.; Mittermeier, R.A.; Rodríguez-Mahecha, J.V.;
Mast, R.B.; Vogt, R.C.; et al. 2007. Las Tortugas y Cocodrilianos de los Paises Andinos del
Trópico. Conservación Internacional, Bogotá DC. 538pp.
Salzburger, W.; Ewing, G.B.; Von Haeseler, A. 2011. The performance of
phylogenetic algorithms in estimating haplotype genealogies with migration. Mol Ecol, 20(9):
1952-1963.
Schaefer, C.E.R.; Dalrymple, J. 1996. Pedogenesis and relict properties of soils with
columnar structure from Roraima, north Amazonia. Geoderma, 71(1): 1-17.
Schaefer, C.E.R.; Vale Júnior, J.F. 1997. Mudanças climáticas e evolução da paisagem
em Roraima: uma resenha do Cretáceo ao recente. In: Barbosa, R.I.; Ferreira, E.J.G.;
Castellón, E.G. (Eds). Homem, Ambiente e Ecologia no Estado de Roraima. INPA, Manaus.
p. 231-266.
Selkoe, K.A.; Toonen, R.J. 2006. Microsatellites for ecologists: a practical guide to
using and evaluating microsatellite markers. Ecol Lett, 9: 615-629.
Sioli, H. 1967. Studies in Amazonian waters. Atas do Simpósio sobre a Biota
Amazônica, 3: 9-50.
Stamatakis, A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-
analysis of large phylogenies. Bioinformatics, 30(9): 1312-1313.
Sugg, D.W.; Chesser, R.K. 1994. Effective population sizes with multiple paternity.
Genetics, 137(4): 1147-1155.
Tajima, F. 1983. Evolutionary relationship of DNA sequences in finite populations.
Genetics, 105(2): 437-460.
Tajima, F. 1989. Statistical Method for Testing the Neutral Mutation Hypothesis by
DNA Polymorphism. Genetics, 123: 585-595.
Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. 2013. MEGA6:
Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol, 30(12): 2725-2729.
Thorbjarnarson, J. 1998. Melanosuchus niger. In: Ross, J.P. (Ed.). Crocodiles. Status
survey and conservation action plan. IUCN/SSC Crocodile Specialist Group. IUCN Gland,
Switzerland and Cambridge. p. 23-28.
Thorbjarnarson, J.B. 1996. Reproductive characteristics of the order Crocodylia.
Herpetologica, 52(1): 8-24.

59
Thorbjarnarson, J.B. 2010. Black Caiman Melanosuchus niger. In: Manolis, S.C.;
Stevenson, C. (Eds). Crocodiles. Status survey and conservation action plan. Crocodile
Specialist Group: Darwin. p. 29-39.
Varuzza, L. 2013. Introdução à análise de dados de sequenciadores de nova geração:
Versão 2.0.1. http://lvaruzza.com/. Acesso: Maio 2015.
Vasconcelos, W.R.; Hrbek, T.; Da Silveira, R.; De Thoisy, B.; Ruffeil, L.A.S.; Farias,
I.P. 2008. Phylogeographic and conservation genetic analysis of the black caiman
(Melanosuchus niger). J Exp Zool A Ecol Genet Physiol, 309(10): 600-613.
Vasquez, P.G. 1991. Melanosuchus, M. niger. Reptilia: Crocodylia: Alligatoridae.
Catalogue of American Amphibians and Reptiles, 530: 1-4.
Vignal, A.; Milan, D.; SanCristobal, M.; Eggen, A. 2002. A review on SNP and other
types of molecular markers and their use in animal genetics. Genet Sel Evol, 34(3): 275-305.
Willis, S.C.; Macrander, J.; Farias, I.P.; Ortí, G. 2012. Simultaneous delimitation of
species and quantification of interspecific hybridization in Amazonian peacock cichlids
(genus Cichla) using multi-locus data. BMC Evol Biol, 12(1): 96.
Willis, S.C.; Nunes, M.; Montana, C.G.; Farias, I.P.; Orti, G.; Lovejoy, N.R. 2010.
The Casiquiare river acts as a corridor between the Amazonas and Orinoco river basins:
biogeographic analysis of the genus Cichla. Mol Ecol, 19(5): 1014-1030.





























