Embed Size (px)
Citation preview

Universidade Estadual de Campinas Instituto de Química
Departamento de Química Orgânica
Estudos Visando a Elucidação Estrutural
de uma diidro-2H-piranona Natural
Dissertação de Mestrado
Mayra Beloti Salvador
Orientador: Prof. Dr. Ronaldo Aloise Pilli
Campinas, 2007

ii
FICHA CATALOGRÁFICA ELABORADA PELA BIBLIOTECA DO INSTITUTO DE
QUÍMICA DA UNICAMP
Salvador, Mayra Beloti.
Sa38e Estudos visando a elucidação estrutural de uma diidro-2H-piranona natural / Mayra Beloti Salvador. -- Campinas, SP: [s.n], 2007.
Orientador: Ronaldo Aloise Pilli.
Dissertação – Universidade Estadual de Campinas, Instituto de Química.
Alilação. 2. RCM. 3. Lactona. I. Pilli, Ronaldo Aloise. II. Universidade Estadual de Campinas. Instituto de Química. III. Título.
Título em inglês: Studies toward the structural elucidation of a natural diidro-2H-
piranone.
Palavras-chave em inglês: Allylation, RCM (Ring closing metathesis), Lactone.
Área de concentração: Química Orgânica.
Titulação: Mestre em Química na área de Química Orgânica.
Banca Examinadora: Ronaldo Aloise Pilli (orientador), Carlos Roque Duarte Correia
(IQ-UNICAMP), Alcindo Aparecido dos Santos (DQ-UFSCAR).
Data da defesa: 28/09/2007

v
“Para ser grande, sê inteiro: nadaTeu exagera ou exclui.
Sê todo em cada coisa. Põe quanto ésNo mínimo que fazes.
Assim como em cada lago a lua todaBrilha, porque alta vive.”
Fernando Pessoa
DEDICO ESTA TESE
À MINHA QUERIDA FAMÍLIA:
PAI, MÃE, DI E LARA
EUVALDO, ÂNGELA E NINA
VALDINHO
E À DEUS

vii
Agradecimentos
Gostaria de agradecer ao Prof. Pilli por contribuir tanto para minha formação
desde os meus primeiros anos na universidade até a conclusão de meu mestrado. Com
suas aulas cheias de entusiasmo a química passou a me fascinar. Nunca conheci uma
pessoa tão dedicada e competente. Um modelo para qualquer tipo de profissional que
deseje uma carreira de sucesso. Meus agradecimentos aos professores Roque, Coelho
e Anita e seus respectivos grupos de pesquisa. Gostaria de agradecer ao Prof. Eberlin e
ao Leonardo Santos pela colaboração nos estudos mecanísticos de alilação realizados
através da espectrometria de massas. Agradeço também ao Lindolfo, D. Gilda, Elaine e
Rinaldo e também a Sônia, Soninha e Paula.
Gostaria de fazer um agradecimento geral ao grupo do professor Pilli,
extremamente atualizado e dedicado, tornando o ambiente de laboratório propício ao
aprendizado contínuo.
Ao Fernandinho, Adão, Ramon, Lápis, Patrícia, Giovanni, Clécio, Diogo, Manoel,
César, Roberta, Cilene, Leila, Luiz, Beto, Lili, Ivan, Léo Steil, Ilton, Nilton, Gustavito,
Cilene, Dedéia e Ângelo.
Gostaria muito de agradecer a minha família pelo grande apoio. Obrigado Ângela
e Euvaldo, por torcerem e acreditarem no meu potencial. Meus queridos irmãos Lara,
Diego e Nina, pelo carinho, admiração e pela nossa união. Obrigada Marina e André por
todo apoio. Obrigado pai e mãe, por estarem sempre olhando por mim, pelo meu futuro,
pela minha formação, educação, saúde e felicidade. Eu sei que provavelmente jamais
terei como retribuir tudo aquilo que vocês me deram e vêm me dando durante todos
esses anos, mas saibam que vocês tem com todo o meu amor a minha mais sincera e
profunda gratidão.
Euvaldinho: Obrigada pela confiança, por acreditar em mim, por me admirar, e
por estar sempre ao meu lado. Obrigada por me guiar durante os momentos difíceis do
meu mestrado, por dialogar comigo e me apontar o caminho do que é correto. Obrigada
por me ensinar o caminho do sucesso, me ajudando a traçar metas pessoais,
estimulando de maneira saudável a minha capacidade de competitir, ganhar e perder. A
vida sem você não seria a mesma. Te amo.
Agradeço acima de tudo a Deus!

ix
Súmula Curricular
Mayra Beloti Salvador
Experiência Profissional
Abril/2007-presente- Supervisora de Ciência e Tecnologia da Glasshield Security
Products, empresa do grupo espanhol Rioglass que atua no ramo de vidros laminados.
Julho/2006-Dezembro/2007: Trainee em pesquisa e desenvolvimento na indústria
farmacêutica Altana Pharma AG (atual Nycomed).
Julho/2005-Setembro/2005: Trainee em pesquisa e desenvolvimento na indústria
farmacêutica Altana Pharma AG (atual Nycomed).
Formação Acadêmica
Março/2004 – presente: Desenvolvimento do projeto de mestrado “Estudos Visando a
Elucidação Estrutural de uma dihidro-2H-piranona Natural” no Instituto de Química da
Universidade Estadual de Campinas (Unicamp), Departamento de Química Orgânica
sob a orientação do Prof. Dr. Ronaldo Aloise Pilli e com apoio financeiro da Fundação
de Amparo à Pesquisa do estado de São Paulo (FAPESP).
Março/2002 – Dezembro/2003: Desenvolvimento do projeto de Iniciação Científica
“Alilação Catalítica e Enantiosseletiva de Aldeídos. Aplicação na Síntese de Lactonas
Insaturadas” no Instituto de Química da Universidade Estadual de Campinas (Unicamp),
Departamento de Química Orgânica sob a orientação do Prof. Dr. Ronaldo Aloise Pilli e
com apoio financeiro da Fundação de Amparo à Pesquisa do estado de São Paulo
(FAPESP).
Janeiro/2000 – Dezembro/2003: Bacharelado em Química na Universidade Estadual
de Campinas (Unicamp).

x
Cursos
17 a 18 de fevereiro de 2005: “QSAR e Modelagem Molecular” ministrado pelo Prof.
Dr. Marcelo Zaldini da Universidade Federal de Pernambuco (UFPe) na Universidade
Federal do Rio de Janeiro (UFRJ) Rio de Janeiro-RJ. Carga horária: 8h.
14 a 16 de fevereiro de 2005: “Antitumor drugs” ministrado pelo Prof. Dr. Hugo
Cerecetto (Universidad de La Republica – Uruguai) na Universidade Federal do Rio de
Janeiro (UFRJ) Rio de Janeiro-RJ. Carga horária: 12h.
14 a 18 de fevereiro de 2005 – “Highlights in Medicinal Chemistry”ministrado por Dr.
Jörg Senn-Bilfinger (Altana Pharma AG, Alemanha), Dr. John R. Proudfoot (Boehinger
Ingelheim Pharmaceuticals, Inc., EUA), Dr. Mukund S. Chorghade (Chorghade
Enterprises, EUA), Prof. Dr. Robin Ganellin (University of London, Inglaterra), Prof. Dr.
Paul W. Erhardt (University of Toledo, EUA), Dr. Janos Fisher (Gideon-Ritcher, Hungria)
e Prof. Dr. Eli Breuer (Hebrew University of Jerusalem, Israel) na Universidade Federal
do Rio de Janeiro (UFRJ) Rio de Janeiro-RJ. Carga horária: 20h.
19 a 23 de julho de 2004: “Nuclear Magnetic Ressonance (NMR) Concepts” ministrado
pelo Professor Dr. Daniel Traficante (University of Rhode Island –EUA) no Instituto de
Química (IQ) da UNICAMP, Campinas-SP. Carga horária: 40h.

xi
Resumo
Esta dissertação de mestrado trata da síntese da Criptomoscatona D2, uma
lactona isolada pelo grupo de pesquisa dos Profs. Cavalheiro e Yoshida a partir da
Cryptocarya moschata, planta encontrada em território brasileiro, cujas configurações
relativa e absoluta ainda não foram determinadas. Além de auxiliar em sua elucidação
estrutural a síntese desta molécula nos permitiria realizar estudos sobre sua atividade
citotóxica, dando prosseguimento a estudos anteriores desenvolvidos em nosso
laboratório com essa classe de compostos. Partindo-se do benziloxiacetaldeído obteve-
se o álcool homoalílico quiral correspondente através de uma reação de alilação
assimétrica com alilestanana e (S)-binaftol. Clivagem oxidativa da dupla ligação e
reação de alilação mediada por InCl3 e estanho metálico na presença de brometo de
alila forneceu uma mistura de álcoois homoalílicos sin/anti 1:1 cuja separação
cromatográfica permitiu o prosseguimento da síntese racêmica com cada um dos
diasteroisômeros. A reação de proteção das hidroxilas com o grupo TBS, seguida de
clivagem oxidativa da dupla ligação e reação de alilação com BF3.Et2O e alilestanana
forneceu o terceiro álcool homoalílico com mistura diastereoisomérica de cerca de 2:1
em ambas as rotas. Por fim, uma reação de esterificação do álcool remanescente na
forma de acrilato seguida de reação de metátese de olefinas para formação do anel
lactônico nos possibilitou o mapeamento de grande parte da rota sintética da
Criptomoscatona D2 em sua forma racêmica.
BnO
O
H(S)-Binaftol/TiCl4
SnBu3
BnO
OH
BnO
OTBS OTBS
1) OsO4/NaIO4
2) InCl3, Sn(0),Br
3) TBSOTf
BnO
OTBS OTBS OH2) BF3.Et2O,
SnBu3
1) OsO4/NaIO4
O
Cl
1)
2) Cat. Grubbs
BnO
OTBS OTBS O
O

xiii
Abstract
This work describes the preliminary studies on the racemic and the asymmetric
synthesis of Cryptomoscatone D2 based on sequential allylation reactions for the
construction of its three stereogenic centers and ring-closing methatesis reaction to
construct the lactone scaffold. Besides allowing the structure elucidation of the molecule
isolated from a typical brazilian plant by the research groups of Profs. Cavalheiro and
Yoshida, the synthesis of such a lactone would allow us to carry new cytotoxic studies
which are being lately developed with this class of compound.
The synthesis started with the allylation reaction of benzyloxyacetaldehyde under
the conditions described by Keck and coworkers to furnish corresponding homoallylic
alcohol. After an oxidative cleavage of the double bond, an InCl3 promoted allylation
reaction allowed the preparation a 1:1 mixture of syn/anti homoallylic alcohols which
were as the TBS ethers and submitted separately to double bond oxidative cleavages.
These aldehydes were used as substrates for another allylation reaction with BF3.Et2O
and allyltri-n-butyltin and the homolallylic alcohols (2:1 diatereoisomeric mixtures) were
converted to the corresponding acrylates in order to carry out the planned RCM reaction.
Several allylation reactions were tested and the homoallylic alcohols were
prepared in 1:1 diasteroisomeric excesses. Efforts will be carried out in order to enhance
the distereoselectivity of the allylation reactions for an efficient approach to
Cryptomoscatone D2 (12) backbone.
BnO
O
H(S)-Binaftol/TiCl4
SnBu3
BnO
OH
BnO
OTBS OTBS
1) OsO4/NaIO4
2) InCl3, Sn(0),Br
3) TBSOTf
BnO
OTBS OTBS OH2) BF3.Et2O,
SnBu3
1) OsO4/NaIO4
O
Cl
1)
2) Cat. Grubbs
BnO
OTBS OTBS O
O

xv
Índice
Lista de Tabelas....................................................................................................
Lista de Figuras.....................................................................................................
Lista de Esquemas................................................................................................
Lista de Símbolos e Abreviaturas..........................................................................
1. Introdução..................................................................................................
2. Objetivos....................................................................................................
3. Resultados e Discussões ...........................................................................
3.1. Preparação do benzilóxiacetaldeído (19) ............................................
3.2. Preparação do (+)-1-benziloxipent-4-en-2-ol (17).................................
3.2.1. Versão Racêmica ......................................................................
3.2.2. Versão Assimétrica ...................................................................
3.3. Preparação do 2-fenil-2-trifluorometil-2-metóxi etanoato de (1-
benzilóxi)-pentenila (25).......................................................................................
3.4. Preparação do 1-O-benzil-2-O-metóxietóximetil-1,2-dihidroxipente-4-
eno (26) .................................................................................................................
3.5. Preparação do 4-O-benzil-3-O-metóxietóximet- 3,4-dihidroxibutan-4-
al (27).....................................................................................................................
3.6. Preparação do 1-O-benzil-2-O-metóxietóximetil-1,2-dihidroxihep-6-en-
4-ol (28)...................................................................................... ………................
3.6.1. Metodologia de Keck .................................................................
3.6.2. Metodologia de Maruoka............................................................
3.6.3. Metodologia de Kurosu ..............................................................
3.7. Preparação do 1-benzilóxi-hept-6-eno-2,4-diol (+)-16 ……………........
3.7.1. Uso de BF3.Et2O …………………………………………………….
3.7.2. Índio metálico ……………………………………………………….
3.7.3. Estanho metálico e InCl3…………………………………………...
3.8. Preparação do 1-O-benzil-2,4-O,O-isopropilideno-1,2,4-tributóxihept-
6-eno (31)………………………………………………………………….......................
3.9. Preparação do 4-allil-6-[(benzilóxi)metil]-1,3-dioxan-2-ona (+)-(42)….
1
7
8
8
11
11
14
16
20
21
22
22
23
24
26
28
28
33
35
38
x
xiii
xiiv
xix

xvi
3.10. Preparação do {6-[(benzilóxi)metil]-2-oxo-1,3-dioxan-4-il}
acetaldeído (+)-(35)………………………………………………………………..........
3.11. Preparação do anti-1-benzilóxi-2,4-bis-(terc-butildimetilsililóxi)
hept-6-eno anti-(37)...............................................................................................
3.12. Preparação do isômero anti-benzilóxi-3,5-bis-
(tercbutildimetilsililoxi) hexanal anti-(38)…………………………………………….
3.13. Preparação do anti-1-benzilóxi-2,4-bis-(terc-butildimetilsililóxi)
non-8-en-6-ol 2,4-anti-(15) ……………………………………………………….......
3.14. Preparação do [1-benzilóxi-2,4-bis(terc-butyldimetilsililoxi)-non-8-
en-6-il] acrilato 2,4-anti-(+)-(39) ……………………………………………...............
3.15. Estudos sintéticos com a série sin…………………………………....
3.16. Preparação do 2’,4’-sin-6- [5-(benzilóxi)-2,4-bis(terc-
butildimetilsililóxi) pentil] -5,6-dihidropira-2-ona 2’,4’-sin-(+)-14……....................
4. Conclusões……………………………………………………………………....
5. Parte Experimental ………………………………………………………….....
5.1. Instrumental…………………………………………………….................
5.2. Procedimentos Experimentais …………………………………………..
5.2.1.Preparação de 1-benzilóxiprop-2-eno (18)……………………...
5.2.2. Preparação de 2-benzilóxiacetaldeído (19) ……………………
5.2.3. Preparação de 1-benzilóxi-pent-4-en-2-ol (17)………………...
5.2.3.1. Versão Racêmica: Preparação de (+)-(17)…………...
5.2.3.2. Versão Assimétrica: Preparação de (S)-17................
5.2.4. Preparação de (2S*,2’R)- 2- fenil-2-trifluorometil-2-
metoxietanoato de (1-benzilóxi)-pent-enila-2 (2S*,2’R)-(25).................
5.2.5. Preparação 1-O-benzil-2-O-metóxietóximetil-1,2-dihidroxi-
pent-4-eno (26)......................................................................................
5.2.6. Preparação d o 4-O-benzil-3-O-metóxietóximetil-3,4-
dihidroxi-butanal (27) ............................................................................
5.2.7. Preparação de (+)-1-benzilóxi-hep-6-en-2,4-diol (+)-(16)....
5.2.7.1. BF3.Et2O e alil-tri-n-butilestanho ...........................
5.2.7.2. Índio Metálico.........................................................
41
43
44
45
46
47
48
50
51
51
52
52
53
53
54
52
55
56
57
58
58
58

xvii
5.2.7.3. Estanho Metálico e InCl3 .........................................
5.2.8. Preparação do sin e do anti-1-O-benzil-2,4-O,O-
isopropilideno-1,2,4-trihidroxi-hept-6-eno sin-(31) e anti-(31)..............
5.2.9. Preparação do 4-alil-6-[(benzilóxi)metil]-1,3-dioxan-2-ona
(+)-32..................................................................................................
5.2.10. Preparação de anti-1-benzilóxi-2,4-bis-(terc-
butildimetilsililóxi) hept-6-eno anti-(37)...................................................
5.2.11. Preparação sin-1-benzilóxi-2,4-bis-(terc-butildimetilsililóxi)-
hept-6-eno sin-(37) ................................................................................
5.2.12. Preparação anti-1-benzilóxi-2,4-bis-(terc-butildimetilsililóxi)-
hexanal anti-(38) ...................................................................................
5.2.13. Preparação anti-1-benzilóxi-2,4-bis-(terc-butildimetilsililóxi)-
hexanal sin-(38) ....................................................................................
5.2.14. Preparação do 2,4 -anti-(+)-1-benzilóxi- 2,4-bis- (terc-
butildimetilsililóxi)-oct-8-en-6-ol 2,4-anti-(+)-(15) …………………………
5.2.14.1. Aliltri-n-butilestanho (22) e BF3.Et2O .......................
5.2.14.2. Brometo de alilmagnésio em Et2O ..........................
5.2.14.3. Brometo de Índio metálico e brometo de alila .........
5.2.15. Preparação do 2,4-sin-(+)-8-benzilóxi-6,8-bis-(terc-
butildimetilsililóxi)-oct-1-em-4-ol, 2,4-sin-(+)-15.......................................
5.2.16. Preparação [9-benzilóxi-6,8-bis(terc-butyldimetilsililóxi)-non-
1-em-4-il] acrilato 2,4-anti-(39) ...............................................................
5.2.17. Preparação [9-benzilóxi-6,8-bis(terc-butyldimetilsililóxi)-non-
1-em-4-il] acrilato 2,4-sin-(39) ...............................................................
5.2.18. Preparação da 2’,4’-sin-5-[6-(benzilóxi)-2,4-bis(terc-
butildimetilsililóxi) pentil]-5,6-dihidropira-2-ona 2’,4’-sin-(+)-14 ..............
6. Espectros.....................................................................................................
59
61
63
64
65
66
67
68
68
69
69
70
71
72
73
74

xix
Lista de Tabelas
Tabela 1: Relação de sinais no espectro de 1H-RMN do alceno 18 .........................
Tabela 2: Relação de sinais no espectro de 1H-RMN do álcool homoalílico (+)-17...
Tabela 3: Relação de sinais no espectro de 1H-RMN do álcool homoalílico (+)-26...
Tabela 4: Relação de sinais no espectro de 1H-RMN do diol 16a.............................
10
13
21
32

xxi
Lista de Figuras
Figura 1: Reagentes utilizados por Brown, Cossy e por Leighton............................
Figura 2: Passiflorcina A (5), Estrictiofoliona (6), Obolactona (7) e
(+)-Goniotalamina (8)...................................................................................
Figura 3: (+)-Pinatoxina A (9), o (-)-Microcarpalídeo (10), (+)-Cyantiwigina U (11)
e a Antascomicina B (12)..........................................................................
Figura 4: Criptomoscatona D2 (13)...........................................................................
Figura 5: 2-benziloxiprop-2-eno (18).........................................................................
Figura 6: 2-benziloxiacetaldeído (19)........................................................................
Figura 7: Efeito de Hiperconjugação.........................................................................
Figura 8: Diagrama de energia dos orbitais moleculares da ligação C-Si. ∆E1 é a
diferença de energia entre σ C-Si e p C+ e, ∆E2 é a diferença de energia
entre σ C-C e p C+.........................................................................................
Figura 9: (+)-2-benziloxipent-4-em-2-ol (+)-17...........................................................
Figura 10: Proposta de estrutura do complexo de Ti (IV)/(S)-BINOL 24..................
Figura 11: Coordenação do aldeído 19 ao complexo 24..........................................
Figura 12: Ésteres de Mosher. A configuração do centro estereogênico do
resíduo do álcool secundário foi atribuída assumindo-se que os
grupos –OOC, R1 e R2 possuem respectivamente prioridades 1,2 e 3
segundo as regras de Cahn-Ingold-Prelog ........................................... .
Figura 13: Espectros de 19F-RMN (470 MHz, C6D6) dos compostos a (2S,2’R)- 25
e (2S*,2’R)-25, respectivamente ............................................................
Figura 14: Composto (+)-26 ......................................................................................
Figura 15: Catalisador de Maruoka 29 .....................................................................
Figura 16: 1-benziloxi-hep-6-en-2,4-diol (16)…………………………………………..
Figura 17: Determinação da estereoquímica de 1,3 – acetonídeos pelo método de
Rychnovsky ............................................................................................
Figura 18: Acetais 39a ou 39b..................................................................................
Figura 19: Estruturas de ressonância, momentos de dipolo (µ) e densidades
eletrônicas (δ) do composto (+)-32..........................................................
1
3
5
6
9
10
12
12
13
15
15
18
19
20
23
31
35
36
39

xxii
Figura 20: Interações orbitalares que fortalecem as ligações C=O de ésteres e
carbonatos..............................................................................................
Figura 21: 4-allil-6-[(benziloxi)metil]-1,3-dioxan-2-ona (+)-(32).................................
Figura 22: Espectro de 1H-RMN (300 MHz, CDCl3) do composto 18.......................
Figura 23: Espectro de 13C-RMN (75 MHz, CDCl3) do composto 18........................
Figura 24: Espectro de IV do composto 18...............................................................
Figura 25: Espectro de 1H-RMN (300Mz, CDCl3) do composto 19...........................
Figura 26: Espetro de 13C-RMN (75 Mz, CDCl3) do composto 19............................
Figura 27: Espetro de IV do composto 19................................................................
Figura 28: Espectro de 1H-RMN (75Mz, CDCl3)do composto (S)-17........................
Figura 29: Espectro de 13C-RMN (75Mz, CDCl3) do composto (S)-17.....................
Figura 30: Espectro de IV do composto (S)-17.........................................................
Figura 31: Espectro de 1H-RMN (300Mz, CDCl3) do composto (2S,2’R)-25............
Figura 32: Espectro de 13C-RMN (75Mz, CDCl3) do composto (2S, 2R)-25.............
Figura 33: Espectro de IV do composto (2S, 2R)-25................................................
Figura 34: Espectro de 1H-RMN (300Mz, CDCl3) do composto 26...........................
Figura 35: Espectro de 13C-RMN (75Mz, CDCl3) do composto 26...........................
Figura 36: Espectro de IV do composto 26...............................................................
Figura 37: Espectro de 1H-RMN (300Mz, CDCl3) do composto 27...........................
Figura 38: Espectro de 13C-RMN (75Mz, CDCl3) do composto 27...........................
Figura 39: Espectro de IV do composto 27..............................................................
Figura 40: Espectro de 1H-RMN (300Mz, CDCl3) do composto anti-16...................
Figura 41: Espectro de 13C-RMN (75Mz, CDCl3) do composto anti-16....................
Figura 42: Espectro de IV do composto anti-16.......................................................
Figura 43: Espectro de 1H-RMN (300Mz, CDCl3) do composto sin-16....................
Figura 44: Espectro de 13C-RMN (75Mz, CDCl3) do composto sin-16.....................
Figura 45: Espectro de IV do composto sin-16........................................................
Figura 46: Espectro de 1H-RMN (300Mz, CDCl3) do composto (+)-32.....................
Figura 47: Espectro de 13C-RMN (75Mz, CDCl3) do composto (+)-32......................
Figura 48: Espectro de IV do composto (+)-32..........................................................
Figura 49: Espectro de 13C-RMN (75 Mz, CDCl3) do composto anti-31...................
Figura 50: Espectro de 13C-RMN (75Mz, CDCl3) do composto anti-31....................
40
40
74
74
75
75
76
76
77
77
78
78
79
79
80
80
81
81
82
82
83
83
84
84
85
85
86
86
87
87
88

xxiii
Figura 51: Espectro de IV do composto anti-31.......................................................
Figura 52: Espectro de 1H-RMN (300Mz, CDCl3) do composto sin-31....................
Figura 53: Espectro de 13C-RMN (75 Mz, CDCl3) do composto sin-31....................
Figura 54: Espectro de IV do composto sin-31........................................................
Figura 55: Espectro de 1H-RMN (300Mz, CDCl3) do composto anti-37...................
Figura 56: Espectro de 13C-RMN (75Mz, CDCl3) do composto anti-37....................
Figura 57: Espectro de IV do composto anti-37.......................................................
Figura 58: Espectro de 1H-RMN (300Mz, CDCl3) do composto sin-37....................
Figura 59: Espectro de 13C-RMN (75Mz, CDCl3) do composto sin-37.....................
Figura 60: Espectro de IV do composto sin-37........................................................
Figura 61: Espectro de 1H-RMN (300Mz, CDCl3) do composto anti-38...................
Figura 62: Espectro de 13C-RMN (75Mz, CdCl3) do composto anti-38....................
Figura 63: Espectro de IV do composto anti-38.......................................................
Figura 64: Espectro de 1H-RMN (300Mz, CDCl3) do composto sin-38....................
Figura 65: Espectro de 13C-RMN (75Mz, CDCl3) do composto sin-38.....................
Figura 66: Espectro de IV do composto sin-38........................................................
Figura 67: Espectro de 1H-RMN (300Mz, CDCl3) do composto 2,4-anti-(+)-15........
Figura 68: Espectro de 13C-RMN (300Mz, CDCl3) do composto 2,4-anti-(+)-15.......
Figura 69: Espectro de IV do composto 2,4-anti-(+)-15............................................
Figura 70: Espectro de IV do composto 2,4-sin-(+)-15.............................................
Figura 71: Espectro de 13C-RMN (75Mz, CDCl3) do composto 2,4-sin-(+)-15..........
Figura 72: Espectro de IV do composto 2,4-sin-(+)-15.............................................
Figura 73: Espectro de 1H-RMN (300Mz, CDCl3) do composto 2,4-anti-(+)-39........
Figura 74: Espectro de 13C-RMN (75Mz, CDCl3) do composto 2,4-anti-(+)-39.........
Figura 75: Espectro de IV do composto 2,4-anti-(+)-39............................................
Figura 76: Espectro de 1H-RMN (300Mz, CDCl3) do composto 2,4-sin-(+)-39.........
Figura 77: Espectro de 13C-RMN (75Mz, CDCl3) do composto 2,4-sin-(+)-39..........
Figura 78: Espectro de IV do composto 2,4-sin-(+)-39.............................................
Figura 79: Espectro de 1H-RMN (300Mz, CDCl3) do composto 2’,4’-sin-(+)-14.......
Figura 80: Espectro de 13C-RMN (75Mz, CDCl3) do composto 2’,4’-sin-(+)-14........
Figura 81: Espectro de IV do composto 2’,4’-sin-(14)..............................................
88
89
89
90
90
91
91
92
92
93
93
94
94
95
95
96
97
97
98
98
99
99
100
100
101
101
102
102
103
103

xxv
Lista de Esquemas
Esquema 1: Diasterosseletividade de reações de alilação e crotilação de β-
hidroxialdeídos.....................................................................................
Esquema 2: Mecanismo de metátese de olefinas proposto por Chauvin.................
Esquema 3: Métateses de olefinas do tipo RCM (Ring-Closing Metathesis),
ADMET (Acyclic Diene Metathesis Polymerization), ROMP (Ring-
Opening Metathesis Polymerization), ROM (Ring - Opening
Metathesis) e CM (Cross-Metathesis)…………………………………...
Esquema 4: Retrossíntese proprosta para Criptomoscatona D2 (13)......................
Esquema 5: Reação para obtenção de 19................................................................
Esquema 6: Mecanismo de reação para obtenção de 21........................................
Esquema 7: Obtenção de (+)-17...........................................................
Esquema 8: Mecanismo de reação para obtenção de (+)-17....................................
Esquema 9: Hidrólise da ligação Ti-O.......................................................................
Esquema 10: Reação para obtenção de (S)-17 através da metodologia de Keck...
Esquema 11: Reação para obtenção do éster de Mosher 25...................................
Esquema 12: Mecanismo de Reação para obtenção de (S)-25...............................
Esquema 13: Preparação de (+)-26..........................................................................
Esquema 14: Reação para preparação de (+)-27 utilizando-se o método de
Lemieux-Johnson ..............................................................................
Esquema 15: Reação para obtenção de (+)-28 através da metodologia de Keck....
Esquema 16: Reação para obtenção de (+)-33 através da metodologia de
Maruoka.............................................................................................
Esquema 17: Preparação de (+)-28 a partir do método de
Kurosu................................................................................................
Esquema 18: Reação de retroaldol para β-hidroxi aldeídos.....................................
Esquema 19: Preparação de (+)-30 utilizando-se de OsO4 (2 mol %) e NaIO4 (4
equiv.)........................................................................................................................
Esquema 20: Preparação dos álcoois (+)-16 utilizando-se OsO4/ NaIO4 na etapa a
e BF3.Et2O como ácido de Lewis na etapa b.............................................................
2
4
4
8
8
9
11
11
13
14
16
17
20
21
22
24
24
25
25
27

xxvi
Esquema 21: Obtenção dos dióis (+)-16 utilizando-se OsO4/ NaIO4 na
etapa a e brometo de alila (21) na etapa b...........................................................
Esquema 22: Mecanismo de reação do tipo Barbier que leva à oxidacao do In
Metálico .............................................................................................
Esquema 23: Mecanismo de adicao do grupamento alil à carbonila de (+)-36.........
Esquema 24: Reação para obtenção dos dióis (+)-16 utilizando-se os método de
Jin (etapa 1) e de Loh (etapa 2).................................................................................
Esquema 25: Mecanismo da reação para obtenção do diol (+)-16 utilizando-se o
método de Loh...................................................................................
Esquema 26: Obtenção dos cetais 31a e 31b..........................................................
Esquema 27: Obtenção do carbonato (+)-32 ...........................................................
Esquema 28: Mecanismo de reação para obtenção do carbonato (+)-32 a partir
do trisfosgênio (33)....................................................................................................
Esquema 29: Obtenção do aldeído (+)-35 sob as condições de Jin.........................
Esquema 30: Mecanismo para obtenção do aldeído α,β-insaturado 36...................
Esquema 31: Obtenção do composto anti-37 a partir da reação do diol anti-16
com TBSOTf..............................................................................................................
Esquema 32: Preparação do aldeído anti-38...........................................................
Esquema 33: Preparação do álcool 2,4-anti-(+)-15..................................................
Esquema 34: Preparação do éster 2,4-anti-(+)-39....................................................
Esquema 35: Etapas para síntese do acrilato 2,4-sin-54 a partir do diol sin-16.....
Esquema 36 Reação para preparação da lactona 2’,4’-sin-(+)-14...........................
Esquema 37: Ciclo catalítico de RCM (Ring Closing Metathesis)............................
29
29
29
34
34
36
38
38
41
42
44
45
45
47
48
48
49

xxvii
Lista de abreviações e símbolos
ADMET: Acyclic Diene Metathesis Polymerization
Ac: acetato
BINOL: binaftol
Bn: benzila
CCD: Cromatografia em camada delgada
CG: Cromatografia gasosa
CM: Cross-Metathesis
CSA: ácido canforsufônico
Cy: cicloexila
d: dubleto
dd: duplo dubleto
ddd: duplo duplo dubleto
ddt: duplo duplo tripleto
dt: duplo tripleto
dq: duplo quarteto
δ: deslocamento químico
DCC: dicicloexilcarbodiimida
DCM: diclorometano
DIPEA:diisopropiletilamina
DMAP: dimetilaminopiridina
DMF: dimetilformamida
E: energia
e.e.: excesso enantiomérico
EMAR: Espectrometria de massas de alta resolução
Et: etila
Ipc: isopinocanfeil iPr: isopropila
IV: infravermelho
J: constante de acoplamento
HPLC: cromatografia líquida de alta performance

xxviii
L: ligante
m: multipleto
M: metal
Me: metila
MEM: metoxietoximetil
MEMCl: cloreto de metoxietoximetila
MOMCl: cloreto de metoximetila
MOM: metoximetil
MTPA: ácido methoxi(trifluorometil)phenil acético
Ph: fenila
ppm: partes por milhão
Pr: propila
q: quarteto
qt: quinteto
RCM: metátese para fechamento de anel
r.d.: razão diastereoisomérica
RMN: ressonância magnética nuclear
ROM: Ring-Opening Metathesis
ROMP: Ring-Opening Metathesis Polymerization
s: singleto
sl: singleto largo
t: tripleto
TBS: terc-butildimetilsilil
TBSCl: cloreto de terc-butildimetilsilil
THF: tetraidrofurano
Tf: triflato

1
1. Introdução
A alilação assimétrica de aldeídos é uma das reações mais importantes de
adição à carbonilas para a síntese de álcoois homoalílicos secundários opticamente
ativos. O mais clássico dos métodos foi desenvolvido por Brown1 na década de 80 e
emprega a adição de quantidade equimolar de H2CCHCH2B(-)Ipc (1) a aldeídos. Esta
metodologia ainda encontra extensa aplicação em sínteses totais2. Outros métodos
estequiométricos de alilação podem ser citados dentre eles aquele desenvolvido por
Duthaler e Hafner 3 , amplamente utilizado por Cossy 4 na França e que utiliza o
composto de aliltitânio (2) ao invés de boro ou aqueles desenvolvidos por Leighton5 (3)
e (4) que estende ainda mais a aplicação para compostos de silício.
B
2
TiCp
OO
Ph
PhO
O
Ph
Ph
**
SiO
N Cl
Me
Ph
Me NSi
N
Cl
Bn
Bn
(R,R)-2
3 4
1
Figura 1: Reagentes para alilação enantiosseletiva de aldeídos.
Reações do tipo Barbier mediadas por metais como estanho e índio também
estão no escopo de metodologias desenvolvidas por Loh6, Paquette7 e recentemente
por Singaram8.
1 a) Brown, H.C.; Bhat, K.S.; Randad, R.S. J.Org.Chem. 1987, 2, 319. b) Brown, H.C.; Bhat, K.S J. Am. Chem. Soc. 1986, 108, 5919. 2 Brittain, D.E.A.; Jones, C.M.G-.; Linder, M.R.; Smith, M.D.; McCusker, C.; Barlow, J.S.; Akiyama, R.; Yasuda, K.; Ley, S.V. Angew. Chem. Int. Ed. 2005, 44, 2732. 3 a) Riediker, M.; Duthaler, R.O. Angew. Chem. Int. Ed. Engl. 1989, 28, 494. b) Duthaler, R.O.; Hafner, A.; Riediker, M. Pure Appl. Chem. 1990, 62, 631. 4a) Cossy, J.; BouzBouz, S. Org. Lett. 2000, 2, 501. b) Cossy, J.; BouzBouz, S.; Pradaux, F.; Willis, C.; Bellosta,V. Synlett 2002, 10, 1595. 5 Kinnaird, J.W.A.; Ng, P.Y.; Kubota, K.; Wang, X.; Leighton, J.L. J.Am.Chem.Soc. 2002, 124, 7920. 6 Li, X.-R.; Loh, T.-P. Tetrahedron Assym. 1996, 7, 1535. 7 Paquette, L.A.; Mitzel, T.M. J.Am.Chem.Soc.1996, 118, 1931. 8 Hirayama, L.C.; Gamsey, S.; Knueppel, D.; Steiner, D.; DeLaTorre, K.; Singaram, B.Tetrahedron Lett., 2005, 46, 2315.

2
Dentre os métodos de alilação catalítica e assimétrica, pode-se destacar
aqueles desenvolvidos independentemente por Keck 9 , Tagliavini-Umani-Ronchi 10 e
Maruoka11 que empregam adição de reagentes de alilestanho a aldeídos na presença
de complexos de Ti(IV)-binaftol ou o método desenvolvido por Yamamoto12 que utiliza
complexos de prata (I) em suas alilações. Mais recentemente, Loh 13 também
desenvolveu métodos de alilação catalítica via complexo de índio (III) quiral,
abrangendo ainda mais seus estudos com esse tipo de metal.
Reações de alilação e crotilacão de α- e β-hidroxialdeídos fornecem produtos
intermediários úteis na síntese de açucares e outros compostos altamente oxigenados
com potenciais atividades farmacológicas14. O controle quiral dessas reações possibita
a construção de sistemas 1,3-dióis com alta diasterosseletividade4a,15,16.
O
H
OH
R
R
OHOH
OHOH
*
* *
**
Esquema 1: Diastereosseletividade de reações de alilação e crotilação de ββββ-
hidróxi-aldeídos.
Outros laboratórios de pesquisa vêm empregando com sucesso a reação de
alilação assimétrica de aldeídos na síntese de produtos naturais com atividades
biológicas variadas. Exemplos de literatura recente são as sínteses da Passifloricina A17
9 Keck, G.E.; Tarbet, K.H.; Geraci, L.S. J.Am.Chem.Soc. 1993,115, 8467.; b) Keck, G.E.; Geraci, L.S. Tetrahedron Lett. 1993, 34, 7827. 10 Costa, A.L.; Piazza, M.G.; Tagliavini, E.; Trombini, C.; Umani-Ronchi,A. J.Am.Chem.Soc. 1993,115, 7001. 11 Hanawa, H.; Hashimoto, T.; Maruoka, K. J.Am.Chem.Soc. 2003, 125, 1708. 12 Yanagisawa, A.; Nakashima, H.; Ishiba, A.; Yamamoto, H. J.Am.Chem.Soc. 1996, 118, 4723. 13 a) Teo, C.Y-, Tan, T.K-, Loh, P.T- Chem. Comm. 2005, 1318; b) Lu, J.; Ji, S.-J.; Teo, Y.-C.; Loh, T.-P. Org.Lett. 2005, 7, 159. 14 Prasad, K.R.; Shivajirao, G.L J. Org. Chem. 2007, asap. 15 Panek, J.S.; Jain, N.F.; J.Org.Chem. 2001, 66, 2747. 16 Bode, S.E.; Wolberg, M.; Müller, M. Synthesis 2006, 4, 557. 17 a) Murga, J.; Fortanet.-, J.G.; Carda, M.; Marco, J.A. J.Org.Chem.2004, 69, 7277;b) Hunter, T.J.; O’Doherty, G.A. Org. Lett. 2001, 3, 2777.

3
(5), a (+)-Estrictiofoliona18 (6) e da Obolactona (7)19 , polióis contendo um resíduo
lactônico α,β-insaturado cujos centros estereogênicos no anel são em sua maioria de
configuração absoluta R.
Em virtude da extensa aplicação de metodologias de alilação na síntese de
compostos com algum potencial biológico nosso grupo de pesquisa tem desenvolvido
alguns projetos voltados para este tipo de química 20 . Recentemente em nosso
laboratório realizou-se a síntese total da (R) e da (S)- Goniotalamina21 (8) e de outros
análogos a fim de avaliar suas atividades biológicas frente a diversas linhagens de
células cancerígenas.
O
O
8
O
O
OH OH
OH12
O
O
OH OH
5 6
O
O
O
O
H
7
Figura 2: Passifloricina A17 (5), Estrictiofoliona18 (6), Obolactona19 (7) e (+)-
Goniotalamina21 (8)
Uma vez estabelecidos os centros estereogênicos dos sistemas 1,3-dióis é
possível construir os anéis lactônicos4,21 através de reações de metátese de olefinas
utilizando-se o catalisador de Grubbs.
As reações de metátese de olefinas já vêm tendo extensa aplicação em
processos industriais petroquímicos desde a década de 50 tendo atraído o interesse
dos químicos orgânicos a partir do fim dos anos 70. Seu mecanismo foi desvendado por
18 Bouz-Bouz, S.; Cossy, J. Org. Lett. 2003, 5, 1995. 19 Zhang, J.; Li, Y.; Wang, W.; She, X.; Pan, X. J. Org. Lett. 2006, 7, 2918. 20 Marco, J.A.; Carda, M.; Murga, J.; Falomir, E Tetrahedron, 2007, 63, 2929. 21 a) De Fátima, A.; Pilli, R.A.Arkivoc 2003, 10, 118. b) De Fátima, A.; Pilli, R.A. Tetrahedron Lett. 2003, 44, 8721. c) De Fátima, A; Kohn, L.K.; Antônio, M.A.; Carvalho, J.E.; Pilli, R.A. Bioorg.Med.Chem. 2004,12, 5437.

4
Chauvin22, que a partir de então possibilitou o desenvolvimento de novos catalisadores
metálicos alquilidênicos em fase homogênia por Schrock 23 e Grubbs 24 , dois
pesquisadores de enorme expressão nessa área. Segundo Chauvin22, uma cicloadição
do tipo [2+2] possibilita cicloconversões e polimerizações de variados tipos.
[M]R
R1 R2
+[M]
R
R2R1
[M]
R1
R
R2
+
Esquema 2: Mecanismo de metátese de olefinas proposto por Chauvin22.
O controle regioquímico de reações de metátese de olefinas em sistemas
altamente insaturados ainda é um desafio para os pesquisadores da área que procuram
tornar o método uma ferramenta ainda mais versátil e eficiente em sínteses totais.
X
RCM
XX
n
ADMET
ROMP
X+ R
ROMX
R
R1 R2CM R1 R2
-C2H4 -C2H4
-C2H4
Esquema 3: Métateses de olefinas do tipo RCM (Ring-Closing Metathesis), ADMET
(Acyclic Diene Metathesis Polymerization), ROMP (Ring-Opening Metathesis
Polymerization), ROM (Ring-Opening Metathesis) e CM (Cross-Metathesis).
22 Hérisson, J.-L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161. 23 Wallace, K.C.; Liu, A.H.; Dewan, J.C.; Scrock, R.R. J. Am. Chem. Soc. 1988, 110, 4964. 24 Grubbs, R.H.; Tumas, W. Science 1989, 243, 907.
5
A metátase de olefinas é uma reação tão importante nos dias atuais que
contemplou seus inventores Grubbs e Schrock, bem como Chauvin, com o renomado
Prêmio Nobel em Química de 2005.
Em função de sua extrema eficiência, tornou-se muito comum a utilização dessa
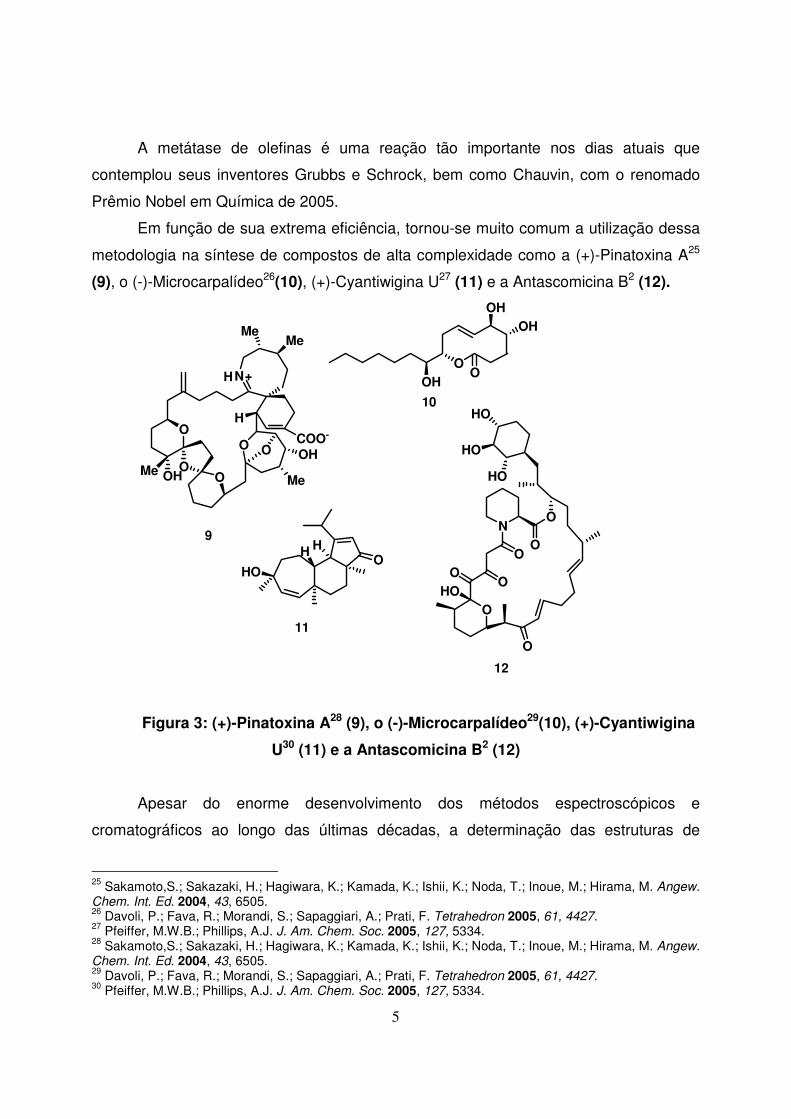
metodologia na síntese de compostos de alta complexidade como a (+)-Pinatoxina A25
(9), o (-)-Microcarpalídeo26(10), (+)-Cyantiwigina U27 (11) e a Antascomicina B2 (12).
N
MeMe
O
OO
O OCOO-
H
Me OH
+
OH
Me
H
9
OHO
O
OHOH
10
OHH
HO
11
NO
O
O
OO
O
O
HO
HO
HO
HO
12
Figura 3: (+)-Pinatoxina A28 (9), o (-)-Microcarpalídeo29(10), (+)-Cyantiwigina
U30 (11) e a Antascomicina B2 (12)
Apesar do enorme desenvolvimento dos métodos espectroscópicos e
cromatográficos ao longo das últimas décadas, a determinação das estruturas de
25 Sakamoto,S.; Sakazaki, H.; Hagiwara, K.; Kamada, K.; Ishii, K.; Noda, T.; Inoue, M.; Hirama, M. Angew. Chem. Int. Ed. 2004, 43, 6505. 26 Davoli, P.; Fava, R.; Morandi, S.; Sapaggiari, A.; Prati, F. Tetrahedron 2005, 61, 4427. 27 Pfeiffer, M.W.B.; Phillips, A.J. J. Am. Chem. Soc. 2005, 127, 5334. 28 Sakamoto,S.; Sakazaki, H.; Hagiwara, K.; Kamada, K.; Ishii, K.; Noda, T.; Inoue, M.; Hirama, M. Angew. Chem. Int. Ed. 2004, 43, 6505. 29 Davoli, P.; Fava, R.; Morandi, S.; Sapaggiari, A.; Prati, F. Tetrahedron 2005, 61, 4427. 30 Pfeiffer, M.W.B.; Phillips, A.J. J. Am. Chem. Soc. 2005, 127, 5334.

6
produtos naturais através da síntese total ainda encontra aplicação. Vários são os
exemplos onde uma estrutura incorreta foi atribuída com base na análise
espectroscópica sendo posteriormente revisada através da síntese total. A
Passifloricina A17 (5), por exemplo, teve sua estrutura inicial completamente revista
quando Cossy4, ao final de sua síntese, detectou incoerências nos espectros de RMN
do composto sintetizado e do composto isolado por Echeverri e colaboradores 31 .
Exatamente por isso tem se estudado atualmente maneiras de alcançar-se a síntese de
diversos diastereoisomeros por meio de uma técnica conhecida como tagging.
Curran32 e colaboradores reproduziram recentemente a síntese da Passifloricina
A (5) proposta por Marco17 preparando seus 8 possíveis diasteroisômeros através de
uma “etiquetagem” dos álcoois homoalílicos intermediários com grupos protetores de
silício contendo variadas quantidades de átomos de flúor.
Através deste protocolo é possível trabalhar com uma mistura complexa de
diastereoisômeros “etiquetados” que, ao afinal, podem ser separados em uma coluna
de HPLC preparativa do tipo PF-C8 e utilizados na determinação da configuração
absoluta do produto natural. Entretanto muitos estudos ainda devem ser conduzidos até
que este se torne um método aplicável, afinal colunas do tipo PF-C8 e grupos
protetores contendo flúor ainda são de difícil acesso aos químicos orgânicos sintéticos.
Em razão do interesse de nosso laboratório na síntese de diidropiranonas para
avaliação biológica e como ferramenta para a determinação das configurações relativa
e absoluta de produtos naturais, decidimos estudar a síntese da Criptomoscatona D2
(13) mediante aplicação de metodologias de alilação de aldeídos para a construção de
seus 3 centros estereogênicos e da metátese de olefinas para construção do anel da
lactona.
OH OH O
O
6R
13
Figura 4: Criptomoscatona D2 (13).
31 Echeverri, F.; Arango, V.; Quiñones, W.; Torres, F.; Escobar, G.; Rosero, Y.; Archbold, R.; Phytochemistry 2001, 56, 881. 32 Curran, D.P.; Moura-Letts, G.; Pohlman, M. Ang.Chem. Int. Ed. 2006, 45, 2423.

7
A Criptomoscatona D2 (13) é uma molécula isolada pelos Prof. Yoshida e
Cavalheiro33 da UNESP de Araraquara a partir de uma planta encontrada em território
brasileiro conhecida como Cryptocarya moschata e que não teve sua estereoquímica
atribuída pelos autores do isolamento. Com base na presença de um efeito Cotton
positivo nas curvas de dicroísmo circular na região de 254-272 nm, Cavalheiro e
Yoshida33 propõem a configuração absoluta R para o centro estereogênico do anel
lactônico das piranonas isoladas da C. moschata.
2. Objetivos
Este trabalho tem como objetivo desenvolver uma rota de síntese eficiente para
um dos possíveis isômeros da Criptomoscatona D2 (13).
Admitindo-se a configuração R do centro esterogênico no carbono 6 da
Criptomoscatona D2 poderíamos focar o trabalho na síntese de apenas quatro
diastereoisômeros diminuindo o número de alvos sintéticos possíveis4,21,17. Conhece-se
atualmente a configuração relativa anti das hidroxilas 13 através de estudos por
cristalografia de raios-x 34 mas ao longo do desenvolvimento desta síntese não
possuíamos tal informação.
Optamos propor inicialmente a síntese do isômero anti,sin-13 cujos centros
estereogênicos seriam construídos a partir de três reações de alilação assimétrica de
aldeídos e a dupla ligação do anel lactônico poderia ser construída através de uma
reação de metátese de olefinas utilizando-se o catalisador de Grubbs. A dupla ligação
do resíduo estirênico poderia ser construída através de uma reação de Julia-Kocienski35
segundo procedimento já otimizado em nosso laboratório de pesquisa.
33 Cavalheiro, A.J.; Yoshida, M. Phytochemistry 2000, 53, 811 34 Murphy, B.T.; Brodie, P.; Miller, J.S.; Razafitsalama, R.A.; Rasamison, V.E.; Kingston, D.G.I. Abstracts of the 48th Annual Meeting of the American Society of Pharmacognosy, Portland, Maine, EUA, 2007 35 Blakemore, P.R.; Cole, W.J.; Kocienski, P.J.; Morley, A. Synlett 1997, 26.

8
OH OH O
O
OP OP O
O
OBn
PO OP OH
OBn
OH OH
OBn
OHBnO BnO
13 14
15 16
17 18
P =TBS
P =TBS
Esquema 4: Retrossíntese proprosta para Criptomoscatona D2 (13).
3. Resultados e Discussões
3.1. Preparação do benziloxiacetaldeído, (19)
O alceno 18, preparado através da reação entre o álcool benzílico (20) e o
brometo de alila (21) na presença de NaH e em DMF (92% de rendimento) foi
submetido a uma reação de clivagem oxidativa na presença de OsO4 (2 mol %) e NaIO4
(4 eq.) em mistura de éter etílico e água segundo a metodologia de Lemieux-Johnson36
em 67 % de rendiemento.
OH
20
O
18
OH
19
Oa b
a) NaH, DMF, 2h então CH2CHCH2Br (21) , 92%; b)OsO4 (2 mol%)NaIO4 (4 equiv.), Et2O / H2O 1:1,67%
Esquema 5: Reação para obtenção de 19.
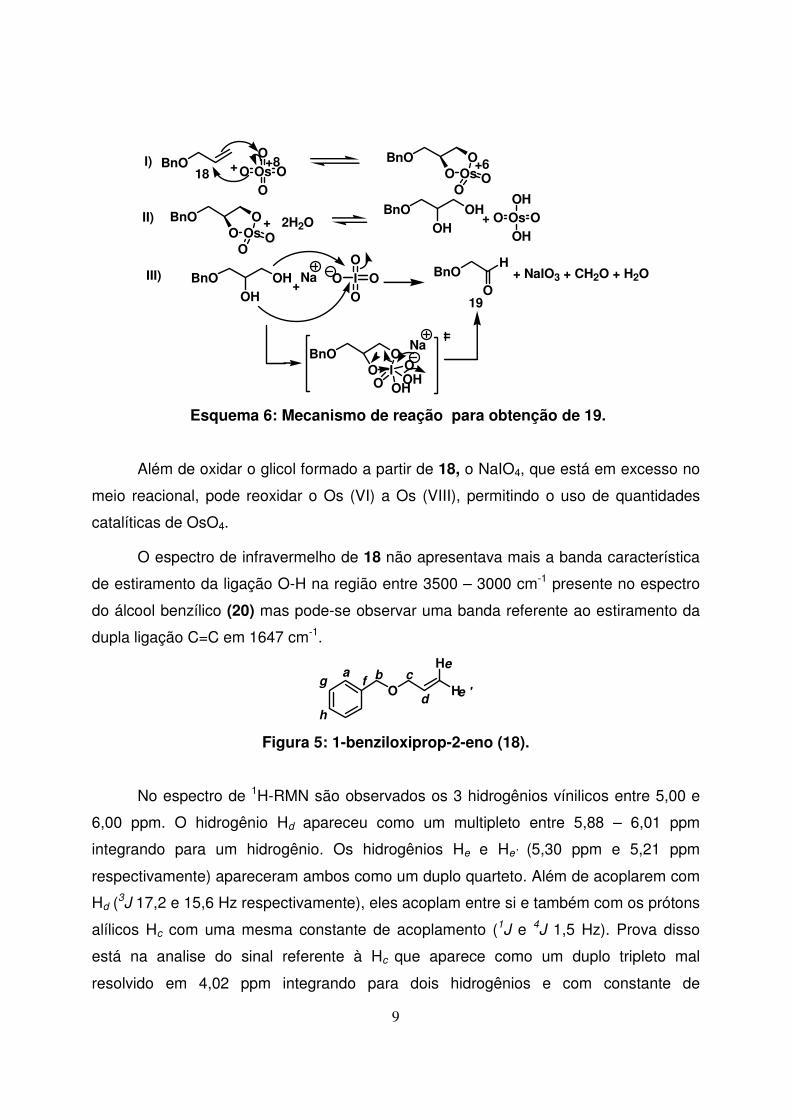
Na reação de clivagem oxidativa, o tetróxido de ósmio reage com alcenos via
uma adição 1,3-dipolar de modo a formar um éster monomérico de ósmio (VI) que, nas
condições empregadas, sofre hidrólise e clivagem oxidativa pelo periodato de sódio.
36 Pappo, R.; Jr. Allen, D.S. Lemieux, R.U.; Johnson, W.S. J. Org. Chem. 1956, 21, 478
9
BnO
BnOH
O
OOs OOO
+BnO
OO
Os OO
BnOO
OOs O
O
+ 2H2OBnO
OHOH
+
OHOsO OOH
I)
II)
III) BnOOH
OHOI OO
ONa+
BnO OO I OO OH
OH
=
+ NaIO3 + CH2O + H2O
+8 +618
19
Na
Esquema 6: Mecanismo de reação para obtenção de 19.
Além de oxidar o glicol formado a partir de 18, o NaIO4, que está em excesso no
meio reacional, pode reoxidar o Os (VI) a Os (VIII), permitindo o uso de quantidades
catalíticas de OsO4.
O espectro de infravermelho de 18 não apresentava mais a banda característica
de estiramento da ligação O-H na região entre 3500 – 3000 cm-1 presente no espectro
do álcool benzílico (20) mas pode-se observar uma banda referente ao estiramento da
dupla ligação C=C em 1647 cm-1.
O
a b c
d
e
fe '
H
Hg
h
Figura 5: 1-benziloxiprop-2-eno (18).
No espectro de 1H-RMN são observados os 3 hidrogênios vínilicos entre 5,00 e
6,00 ppm. O hidrogênio Hd apareceu como um multipleto entre 5,88 – 6,01 ppm
integrando para um hidrogênio. Os hidrogênios He e He’ (5,30 ppm e 5,21 ppm
respectivamente) apareceram ambos como um duplo quarteto. Além de acoplarem com
Hd (3J 17,2 e 15,6 Hz respectivamente), eles acoplam entre si e também com os prótons
alílicos Hc com uma mesma constante de acoplamento (1J e 4J 1,5 Hz). Prova disso
está na analise do sinal referente à Hc que aparece como um duplo tripleto mal
resolvido em 4,02 ppm integrando para dois hidrogênios e com constante de

10
acoplamento de 3J 5,5 Hz e 4J 1,5 Hz. O acoplamento alílico, muito embora ocorra a 4
ligações, é bastante comum e é observado nos espectros de outros compostos por nós
sintetizados. Pudemos encontrar também um singleto integrando para dois hidrogênios
referentes aos hidrogênios benzílicos Hb em 4,52 ppm e um multileto entre 7,34 e 7,22
ppm integrando para cindo hidrogênios referente aos aromáticos.
Tabela 1: Relação de sinais no espectro de 1H-RMN do alceno 18.
Hidrogênios δδδδ (ppm) Multiplicidade IntegraçãoHa, Hg, Hh 7,34-7,22 m 5H
Hb 4.52 s 2H Hc 4.02 dt 2H Hd 5,88 – 6,01 m 1H He 5,30 dq 1H He’ 5,21 dq 1H
No espectro de 13C-RMN do éter 18 pudemos observar 8 sinais, 6 deles acima
de 100 ppm referentes aos carbono de hibridização sp2. Os carbinólicos Cb e Cc foram
encontrados em uma região um pouco mais protegida (72,1 e 71,1 ppm).
No espectro de IV do aldeído 19 foram observadas duas bandas muito
características de aldeídos: em 1736 cm-1, referente ao estiramento da ligação C=O, e
em 2711 cm-1 referente ao estiramento da ligação C-H do grupo funcional CHO. A
banda de estiramento C=C em 1647 cm-1 não foi observada, indício de que a clivagem
oxidativa foi bem sucedida.
OO
Ha b cde
g
f h
Figura 6: 2-benziloxiacetaldeído (19).
No espectro de 1H-RMN do aldeído foi possível observar um sinal característico
em 9,72 ppm integrando para um hidrogênio referente ao hidrogênio Hd. No espectro de 13C-RMN foram observados 5 sinais acima de 100 ppm referentes aos Csp
2 aromáticos
e carbonílico e dois sinais na região típica dos carbonos Cc e Cb.

11
3.2. Preparação do (+)-1-benziloxipent-4-en-2-ol (17)
3.2.1. Versão Racêmica
Inicialmente, decidimos trabalhar em uma versão racêmica da síntese com o
intuito de mapear a rota sintética bem como adquirir padrões para determinação de
excessos enantioméricos no momento da implementação da rota assimétrica.
A reação de alilação do aldeído 19 foi conduzida em CH2Cl2, a -78°C com
aliltrimetilsilano (22) na presença de TiCl4 (1 equiv.) como ácido de Lewis. Após
purificação do material bruto de reação por cromatografia em coluna de sílica gel, (+)-17
foi isolado em 77 % de rendimento.
Si(CH3)3 BnOH
O+
a BnOOH
22 19 (+)-17
a) TiCl4, CH2Cl2, -78ºC, 40 min., 77%
Esquema 7: Obtenção de (+)-17.
A formação do produto pode ser explicada pela ativação do aldeído 19 através
de sua complexação ao titânio seguida de ataque nucleofílico do aliltrimetilsilano 22.
Si(CH3)3
BnOO
H+ TiCl4
BnOO
H
TiClCl ClCl
δδδδ++++
BnOO
H
TiClCl ClCl
δδδδ++++
+ BnOOTiCl3
Si(CH3)3Cl
BnOOTiCl3
Si(CH3)3
Cl
BnOOTiCl3
I)
II)
III) ClSi(CH3)3+
Esquema 8: Mecanismo de reação para obtenção de (+)-17.
A reação é facilitada pela formação de um carbocátion estabilizado pelo átomo
de sílicio através de hiperconjugação. Segundo a Teoria do Orbital Molecular (TOM),
quando o orbital σC-Si está perfeitamente alinhado com o orbital p do carbocátion, há
uma propagação da nuvem eletrônica (σC-Si→pC+) capaz de promover uma estabilização
de carga. Trata-se de uma espécie de delocalização eletrônica característica do

12
fenômeno de ressonânica previsto pela Teoria de Ligação de Valência (TLV) com a
particularidade de não promover quebras e formações efetivas de ligações químicas.
H
MeMeMe σσσσC-Si
SiMe
MeMe
p
Figura 7: Efeito de Hiperconjugação.
Esta interação orbitalar também é favorecida em termos energéticos. Através da
combinação linear dos orbitais atômicos do carbono e do silício (que têm maior energia
devido a menor eletronegatividade do silício) gera-se dois orbitais moleculares σC-Si e
σ*C-Si. Como os orbitais p C+ (deficiente de elétrons) e σ C-Si (rico em elétrons) estão
mais próximos em energia (∆E1< ∆E2), a sobreposição será mais efetiva do que no caso
das interações orbitalares σ C-C → p C+
. Esse efeito é conhecido como hiperconjugação
e é através dele que o carbono β propaga sua carga positiva para o silício.
Figura 8: Diagrama de energia dos orbitais moleculares da ligação C-Si. ∆∆∆∆E1 é a
diferença de energia entre σσσσ C-Si e p C+ e, ∆∆∆∆E2 é a diferença de energia entre σσσσ C-C e
p C+.
Após tratamento aquoso da reação ocorre a hidrólise da ligação Ti-O, a liberação
de Ti(OH)4 e HCl e a formação do produto (+)-17.
ECCCC SSSSiiii CCCC CCCC
σ *C-Si
σ C-Si
σ*C-C
σ C-C
ppppCCCC
∆E1 ∆E2

13
BnOOTiCl3
+ 4 H2O BnOOH
(+)-17
+ Ti(OH)4 + 3HCl
Esquema 9: Hidrólise da ligação Ti-O.
Em uma primeira análise, feita através do espectro de infravermelho, pudemos
verificar a formação de (+)-17 pela observação de bandas características de álcool e
alceno, em 3444 e 1643 cm-1 respectivamente, e pelo desaparecimento da banda típica
do aldeído 19 em 1736 cm-1. Não obstante, os espectros de 1H-RMN e 13C-RMN é que
nos forneceram resultados mais concretos a respeito da formação de (+)-17.
a b cd
e g
fO
OHh
ij
k
Figura 9: (+)-2-benziloxipent-4-em-2-ol (+)-17.
Além dos hidrogênios aromáticos e benzílicos, pudemos observar o
aparecimento de sinais característicos de hidrogênios vinílicos Hg e Hf entre 5,00 e 6,00
ppm. Os hidrogênios alílicos He apareceram com um deslocamento químico δ 2,26
como um tripleto integrando para dois hidrogênios e com um 3J 6,6 Hz. Os hidrogênios
carbinólicos Hc e Hd apareceram em uma região de campo mais baixo. Ambos
forneceram multipletos em repectivamente 3,54 – 3,35 ppm e 3,91 – 3,85 ppm. O
hidrogênio hidroxílico Hh também foi encontrado como um dubleto em 2,34 ppm em
função do acoplamento com Hd (3J 3,3 Hz).
Tabela 2: Relação de sinais no espectro de 1H-RMN do álcool homoalílico (+)-17
Hidrogênios δδδδ (ppm) Multiplicidade IntegraçãoHa, Hj, Hk 7,39 – 7,24 m 5H
Hb 4,54 s 2H Hc 3,54 – 3,35 m 2H Hd 3,91 – 3,85 m 1H He 2,26 t 2H Hf 5,80 – 5,74 m 1H Hg 5,14 – 5,05 m 2H Hh 2,34 d 1H

14
No espectro de 13C-RMN, como era esperado, foram observados 10 sinais.
Acima de 100 ppm apareceram 6 sinais referentes aos carbonos a, f, g, i, j e k. Os
carbinólicos b, c e d foram observados na região esperada (entre 60 e 80 ppm) e o
carbono alílico Ce em um região mais protegida (37,9 ppm).
Tendo em mãos o padrão racêmico (+)-17, decidimos iniciar os estudos para
obtenção do composto enantiomericamente enriquecido (S)-17.
3.2.2. Versão Assimétrica
O álcool (S)-17 foi obtido através da adição de alil-tri-n-butilestanho 23 ao
aldeído 19 em CH2Cl2 a –20 °C e na presença do catalisador de (S)-BINOL/Ti(OiPr)4 24
(10 mol %), segundo metodologia descrita por Keck9 e colaboradores. Após purificação
do bruto de reação por cromatografia em coluna de sílica gel, (S)-17 foi isolado em 75
% de rendimento e com um excesso enantiomérico >95 %. A determinação do excesso
enantiomérico desta reação bem como a determinação do isômero majoritário formado,
serão explicitadas mais adiante. O valor de rotação óptica [α]D medido para este
composto foi de –2.18 (c 2.59, CHCl3)37.
BnOOH
BnOO
H S
19 (S)-17
a) CH2Cl2, (S)-BINOL,Ti(OiPr)4,peneira molecular 4A, refluxo, 1h, ta, então 19, -78 ºC então CH2CHCH2SnBu3 (23), -20 ºC , 7 dias, 75 %, >95 % e.e.
a
Esquema 10: Reação para obtenção de (S)-17 através da metodologia de Keck9.
O mecanismo desta reação é análogo aqueles descritos nos esquemas 8 e 9.
Neste caso também há a formação de um carbocátion estabilizado por
hiperconjugação. O estanho também é mais eletropositivo que o carbono e favorece a
delocalização eletrônica do tipo σ C-Sn→ pC+. O complexo de titânio 24 também ativa o
aldeído através de coordenação, tornando-o mais eletrofílico e portanto mais suscetível
ao ataque do nucleófilo alil-tri-n-butilestanho 23.
37 [α]D Reportado para (S)-17: -2.20 (c 2.64, CHCl3): Ghosh, A.K.; Lei, H. J.Org.Chem. 2000, 65, 4779.

15
Os dados espectroscópios do composto racêmico (+)-17 foram idênticos aos do
composto enentiomericamente enriquecido (S)-17.
O tetraisopropóxido de titânio (IV), na presença de (S)-binaftol tende a formar um
complexo tetraédrico pois devido ao caráter oxofílico do metal é possível ocorrer a troca
de ligantes oxigenados facilitada pela natureza bidentada do BINOL.
OO
TiOiPrOiPr
24
Figura 10: Proposta de estrutura do complexo de Ti (IV)/(S)-BINOL 24.
O catalisador 24, de simetria C2, não possui centro estereogênico mas sua
quiralidade resulta da ausência de um plano de simetria. O eixo C2 torna as valências
ocupadas pelos dois grupos OiPr equivalentes. Acreditamos que na presença do
aldeído 19 ocorra a troca de ligantes com eliminação de um grupo isopropóxido e
coordenação do aldeído 19 pelo par de elétrons anti ao seu grupamento R de modo que
o hidrogênio da carbonila se localize mais próximo dos anéis aromáticos deixando a
face Si mais desimpedida, independentemente das duas possibilidades de
coordenação. Desta forma, o aliltributilestanho 23 ataca o carbono da carbonila de 19
pela face Si, fornecendo neste caso o isômero de configuração S do álcool 17.
Figura 11: Coordenação do aldeído 19 ao complexo 24.
H
OOBn
19
Par de elétrons anti ao grupo -OBn

16
3.3. Preparação do 2-fenil-2-trifluorometil-2-metoxi etanoato de (1-benziloxi)-pent-
enila-2 (25).
A determinação do excesso enantiomérico do produto da reação de alilação
assimétrica descrita no esquema 10 foi possível através da análise dos espectros de 19F-RMN dos ésteres de Mosher derivados dos álcoois homoalílicos (+)-17 e (S)-17.
Os ésteres de Mosher (2S*,2’R)-25 e (2S,2’R)-25 foram preparados a partir da
reação entre os respectivos álcoois (+)-17 e (S)-17 e (R)-MTPA na presença de DCC e
DMAP, em CH2Cl2, em 55 % de rendimento.
F3CO
OHOMePh
R
BnOOH
O
OBnO
F3C
MeO Ph
R(+)-17
a
a) DCC, DMAP, CH2Cl2, 18h, 55%; b)DCC, DMAP, CH2Cl2, 18h, 55%
(2S,2'R)-25
BnOOH
(S)-17b
O
OBnO
F3C
MeO Ph
R
(2R,2'R)-25
+
O
OBnO
F3C
MeO Ph
R
(2S,2'R)-25
(1:1)
Esquema 11: Reação para obtenção dos ésteres de Mosher 25.
Dentre os compostos carbonilados, os ácidos carboxílicos são os menos
eletrofílicos e têm portanto dificuldade em sofrer reações de adição de nucleófilos. Um
ácido é mais facilmente esterificado com um álcool na presença de um agente
desidratante como o DCC sendo este convertido a um derivado da uréia ao final do
processo38. O DMAP além de agir como base promove a formação do sal de piridínio
intermediário que sofre ataque nucleofílico pelo álcool secundário 17.
38 March, J. Advanced Organic Chemistry, JohnWiley & Sons, New York, 1985, p. 349.

17
F3CO
OHOMePh
+ CyN=C=NCyDMAP (cat.)
F3CO
OOMePh
R R + CyN=C=NCyH
F3CO
OOMePh
RH O
O
CF3
Ph
MeO
C NCyCyNH
R
OO
CF3
Ph
MeO
C NCyCyNH
R N
N
+F3C
O
N
NMeO Ph
+
O
NCyCyNH
BnOOH BnO
OH
+
S
S
OS
OBnO
F3C
MeO Ph
R
R
I)
II)
III)
IV)
V) - DMAP
Derivado da Uréia
+
31
+
CyN
O
NCyH H
(2S,2'R)-25
(S)-17
CyN=C=NCy+
Esquema 12: Mecanismo de Reação para obtenção de (2S, 2’R)-25.
Os ésteres (2S*,2’R)-25 e (2S,2’R)-25 foram caracterizados por espectroscopia
de RMN e infravermelho.
Em ambos os espectros de infravermelho foram encontradas bandas
características de estiramento da ligação C=O de ésteres (1748 cm-1). Os espectros de 1H-RMN e 13C-RMN dos compostos (2S*,2’R)-25 e (2S,2’R)-25 apresentaram grande
semelhança. Pudemos observar que, na região de prótons aromáticos do espectro de 1H-RMN, a integração era de 10H em função da presença do anel benzílico e da fenila
proveniente do ácido de Mosher. O sinal referente a metoxila apareceu sobreposto ao
dos hidrogênios carbinólicos (BnOCH2R) entre 3,55 –3,53 ppm. Para estes compostos
(25) continuamos observando os sinais referentes aos prótons vinílicos entre 5,00 e
6,00 ppm.

18
A determinação da configuração absoluta de álcoois secundários
enantiomericamente puros foi estudada em profundidade por Mosher39 que, em 1969,
publicou pela primeira vez um método de preparação de ésteres derivados desses
álcoois a partir do ácido (R) ou (S)-α-(feniltrifluorometilmetoxi) acético (MTPA). Esses
ésteres, em sua maioria, apresentam diferentes deslocamentos químicos (δ) nos
espectros de RMN e, através dos resultados obtidos, Mosher propôs um modelo para
correlacionar as configurações absolutas dos respectivos álcoois. Segundo Mosher a
conformação mais estável para os ésteres é aquela em que Ha, a carbonila e o grupo
CF3 (figura 12) encontram-se coplanares, assim a determinação da configuração
absoluta poderia ser realizada através da derivatização de um único enantiômero do
álcool com ambas as formas do MTPA ou pela derivatização de uma mistura racêmica
de álcoois com MTPA enantiomericamente puro. A fenila, quando justaposta com um
dos grupos R, é capaz de exercer um efeito anisotrópico de blindagem deslocando os
prótons deste grupo para um campo mais alto.40
F3CO
OOMePh
R
F3CO
OPhMeO
a
c
F3CO
OOMePh
R
F3CO
OPhMeO
R
S S
b
d
Ha
R2
R1S
Ha
R1R2
S
Ha
Ha
R1
R1R2
R2R
Figura 12: Ésteres de Mosher. A configuração do centro estereogênico do resíduo
do álcool secundário foi atribuída assumindo-se que os grupos –OCO-, R1 e R2
possuem respectivamente prioridades 1,2 e 3 segundo as regras de Cahn-Ingold-
Prelog.
No caso em questão, estávamos interessados apenas em determinar o excesso
enantiomérico do produto obtido na reação de alilação assimétrica descrita no esquema
39 Dale, J.A.; Dull, D.L.; Mosher, H.S. J. Org. Chem. 1969, 34, 2543. 40 D’Oca, M.G.M. O Uso de Auxiliares Quirais Cicloexílicos na Adição de Nucleófilos a Íons N-Acilimínios Cíclicos. A Reação de Aliltrimetilsilano com Íons N-Acilimínios em Fase Gasosa, Tese de Doutorado, Universidade Estadual de Campinas, Brasil,2000.

19
10 pois já encontrava-se relatado na literatura os dados espectroscópicos de (S)-17
bem como seu valor de rotação óptica específica [α]D37. Leighton5 e colaboradores
prepararam os ésteres de Mosher39 derivados de (+)-17 e (S)-17 concluindo que,
através dos espectros de 19F-RMN, era possível adquirir uma boa separação dos sinais
relativos a cada um dos enantiômeros. Logo, já tínhamos em mãos um método válido
para determinação do excesso enantiomérico do nosso produto de reação de alilação
descrita no esquema 10 além de uma confirmação da estereoquímica do composto (S)-
17 formado.
Na figura 14 encontram-se os espectros de 19F-RMN dos ésteres de Mosher
(2S,2’R)-25 e (2S*,2’R)-25 derivados, respectivamente, dos álcoois homoalílicos (S)-17
e (+)-17. Pode-se verificar através da integração dos sinais, um excesso enantiomérico
> 95 % e.e.
Figura 13: Espectros de 19F-RMN (470 MHz, C6D6)41 dos compostos a (2S,2’R)-25 e
(2S*,2’R)-25, respectivamente.
41 O padrão interno utilizado foi TFA = Ácido trifluoracético

20
3.4. Preparação do 1-O-benzil-2-O-metoxietoximetil-1,2-dihidroxipent-4-eno (26)
Com base nos estudos de alilação desenvolvidos por Kurosu 42 , que utiliza
compostos de zircônio como ácidos de Lewis, decidimos proteger a hidroxila do
composto (+)-17 com o grupo MEM.
O composto 26 foi preparado a partir da reação entre o álcool (+)-17 e o MEMCl
em CH2Cl2 e DIPEA a 0 °C. A reação foi relativamente longa (18 h) e forneceu o
produto (+)-(26) em 58% rendimento.
(+)-17 (+)-26
BnOOH
BnOOMEM
a
a) MEMCl, DIPEA, CH2Cl2, 18h, 58%
Esquema 13: Preparação de (+)-26.
O composto (+)-26 foi caracterizado por análises de IV, 1H-RMN e 13C-RMN. A
informação mais interessante que nos foi fornecida pelo espectro de infravermelho foi
com relação ao desaparecimento da banda de OH em torno de 3550 cm-1. Pouca
evidência de formação de (+)-26 é atestada a partir das demais bandas. Os espectros
de 1H-RMN e 13C-RMN forneceram as informações conclusivas.
OO O
O
a b cd
e
f
g
h
i
jk
H Hh'
HHb'
Figura 14: Composto (+)-26.
Entre 3,00 e 5,00 ppm pudemos encontrar os sinais referentes aos prótons
carbinólicos provenientes do MEM. Os prótons Hh e Hh’ forneceram dubletos distintos e
superpostos em 4,82 e 4,79 ppm (não respectivamente nesta ordem), isto em razão de
serem diastereotópicos e acoplarem entre si, com uma constante de acoplamento bem
alta (1J 15,7 Hz). Com os prótons benzílicos Hb aconteceu algo semelhante. Foi
possível observar um conjunto de quatro sinais com um padrão típico de quarteto entre
42 Kurosu, M.; Lorca, M. Tetrahedron Lett. 2002, 43, 1765.

21
4,57 e 4,49 ppm. Na verdade cada próton benzílico Hb fornece um dubleto típico de
sistemas AB onde ∆ν/J ≈ 1,5. Os demais sinais referentes aos prótons vinílicos, alílicos,
aromáticos etc., também foram observados com integrações, multiplicidades e
deslocamentos químicos (δ) que pouco diferiam do espectro do álcool (+)-17. O
espectro de 13C-RMN só veio a confirmar a formação de (+)-26. Observamos exatos
quatorze sinais: seis acima de 100 ppm referentes aos carbonos de hibridização sp2 (os
2 carbonos vinílicos e os quatro aromáticos orto, meta, para e C0), sete sinais entre 50 e
100 ppm referentes aos carbonos carbinólicos e um sinal em campo mais alto (35,51
ppm) referente ao carbono alílico, o único da cadeia alifática que não encontra-se
diretamente ligado a um heteroátomo.
Tabela 3: Relação de sinais no espectro de 1H-RMN do álcool homoalílico (+)-26
Hidrogênios δδδδ (ppm) Multiplicidade Integração Ha 7,36 – 7,23 m 5H Hb 4,57 ou 4,49 m 1H Hb’ 4,49 ou 4,57 m 1H Hc 3,60 m 2H Hd 3,90 – 3,83 m 1H He 2,41 – 2,30 m 2H Hf 5,80 ddt 1H Hg 5,11 – 5,02 m 2H Hh 4,82 ou 4,79 d 1H Hh’ 4,82 ou 4,79 d 1H
Hi e Hj 3,52 – 3,40 m 4H Hk 3,37 s 3H
3.5. Preparação do 4-O-benzil-3-O-metoxietoximetil-3,4-dihidroxibutan-4-al (27)
De posse de (+)-26 pudemos prosseguir nossos estudos e realizar a segunda
clivagem oxidativa da rota sintética que visa a preparação do aldeído 27 necessário à
segunda alilação utilizada na construção de mais um centro estereogênico de um do
isômero 13 da Criptomoscatona D2.
O aldeído (+)-27 foi preparado através da reação entre o alceno (+)-26, NaIO4 (4
equiv.) e quantidade catalítica de OsO4 (2 mol %), em éter etílico e água numa
proporção de 1:1 em 74 % de rendimento.

22
(+)-26
BnOOMEM
BnOH
MEMO O
(+)-27
a
a) OsO4, NaIO4, Et2O / H2O 1:1, 5h, 74%
Esquema 14: Reação para preparação de (+)-27 utilizando-se o método de
Lemieux-Johnson36.
O composto (+)-27 foi caracterizado por Espectrometria de Massas de Alta
Resolução (EMAR), IV, 1H-RMN e 13C-RMN.
No espectro de infravermelho do composto (+)-27 encontramos o aparecimento
de uma banda em 1723 cm-1 bastante característica de estiramento da ligação C=O de
aldeídos. O espectro de massas de alta resolução de (+)-27, de fórmula molecular
C15H22O5, mostrou o sinal do íon molecular em m/z 282.14673 sendo que a massa
calculada para este composto também foi m/z 282.14673.
No espectro de 1H-RMN pudemos observar o aparecimento de um tripleto típico
de hidrogênio CHO em 9,78 ppm integrando para 1H (3J 1,8 Hz), em função de seu
acoplamento com os dois hidrogênios α à carbonila. Os demais sinais apareceram com
deslocamentos químicos, multiplicidade e integrações semelhantes àqueles
encontrados no espectro do alceno (+)-26. No espectro de 13C-RMN encontramos o
sinal referente ao carbono da carbonila acima de 200 ppm.
Tendo em mãos o aldeído (+)-27 iniciamos os testes da segunda reação de
alilação.
3.6. Preparação do 1-O-benzil-2-O-metoxietoximetil-1,2-dihidroxihep-6-en-4-ol (28)
3.6.1. Metodologia de Keck9
Em uma primeira tentativa, a reação entre o aldeído (+)-27 e o alil-tri-n-
butilestanho 23 nas condições de Keck9 não forneceu o produto de alilação (+)-28 sendo
o aldeído (+)-27 completamente recuperado após purificação do material bruto de
reação por cromatografia em coluna de sílica gel utilizando-se hexano/EtOAc 1:1 como
eluente.

23
BnOH
MEMO O
(+)-27
BnOMEMO OH
(+)-28
a
a) (S)-BINOL, Ti(OiPr)4 CH2Cl2, peneira molecular 4A, refluxo, 1h ii) ta, então (+)-27, iii) - 78ºC, então 23, iv) -20 °C, 60 h
Esquema 15: Reação para obtenção de (+)-28 através da metodologia de Keck9.
Em uma segunda tentativa, modificamos o procedimento experimental elevando
o tempo reacional para sete dias ao invés de 60 h. Mais uma vez o aldeído (+)-27 não
havia sido consumido.
3.6.2. Metodologia de Maruoka11
O primeiro método alternativo de alilação catalítica e assimétrica a ser testado foi
o de Maruoka.11
Em primeiro lugar preparamos o catalisador de Maruoka11 29 através da reação
entre quantidades catalíticas de TiCl4, Ti(OiPr)4, Ag2O e (S)-BINOL em CH2Cl2. O
catalisador 29 deveria ser preparado imediatamente antes da reação de alilação não
podendo ser armazenado por muito tempo. Segundo experiência do grupo, após 5 h
sob intensa agitação em um balão protegido da luz, o catalisador deve adquirir uma
coloração rosa para que possa ser utilizado, caso contrário, ele deve ser descartado. A
estrutura proposta para o catalisador formado segundo Maruoka11 encontra-se ilustrada
na figura 15.
OO
TiOiPrO
TiO
PrOi O
29
Figura 15: Catalisador de Maruoka (29).
O catalisador de Maruoka11 tem um princípio de funcionamento semelhante ao
de Keck9 24. A única diferença está na estrutura do complexo metálico que no caso do
catalisador de Maruoka11 29 trata-se de um dímero.

24
Após resfriamento do balão contendo o catalisador a – 10 °C adicionou-se o
aldeído (+)-27 e o alil-tri-n-butilestanho 23.
BnOH
MEMO O
(+)-27
BnOMEMO OH
(+)-28
a
a) i) TiCl4, Ti(OiPr)4, CH2Cl2, 1h, ii) Ag2O, 5h, iii) (S)-BINOL iv) - 10 °C , então (+)-27 e 23, -20 ºC, 7 dias
Esquema 16: Reação para obtenção de (+)-33 através da metodologia de
Maruoka.11
Após sete dias de reação a –20 °C, não houve consumo do material de partida
(+)-27 e a reação foi interrompida.
3.6.3. Metodologia de Kurosu42
A metodologia de Kurosu42 foi testada assim que tivemos acesso ao Zr(OtBu)4.
Alguns parâmetros reacionais tiveram que ser adaptados em função da
indisponibilidade de parte dos reagentes necessários à reação de alilação do aldeído
(+)-27 através deste método. Em primeiro lugar, utilizamos acetonitrila ao invés de
pivalonitrila como co-solvente da reação e, em segundo lugar, o grupo protetor da
hidroxila do álcool (+)-17 era o MEM e não MOM pois não possuímos o reagente
MOMCl em nosso laboratório de pesquisa.
O (S)-BINOL e o Zr(OtBu)4 foram submetidos à reação por 1 h em presença de
peneira molecular 4Ǻ em tolueno seco e acetonitrila de forma que o catalisador de
zircônio fosse preparado. A mistura foi então levada a –78 °C para que o alil-tri-n-
butilestanho 23 e o aldeído (+)-27 fossem adicionados. A reação foi mantida por cinco
dias a -20°C e então submetida a tratamento aquoso. Desta vez houve decomposição
do material de partida (+)-27.
BnOMEMO
H
O
BnOMEMO OH
(+)-27 (+)-28
a) i) Zr(OtBu)4 , (S)-BINOL, Peneira molecular 4A, acetonitrila, tolueno ii) - 78 ºC, então 23 e (+)-27, -20 ºC, 5 dias
a
Esquema 17: Preparação de (+)-28 a partir do método de Kurosu42.

25
Por ora, não se pode afirmar que a metodologia de Kurosu42 seja ineficiente para
nosso sistema uma vez que o procedimento original foi modificado. Em primeiro lugar, a
hidroxila em C3 encontra-se protegida com MEM e não com o MOM que, como relatado
anteriormente, tratava-se do grupo protetor mais adequado para reações de alilação de
β-alcoxialdeídos segundo estudos metodológicos realizados pelo próprio Kurosu42 Em
segundo lugar, utilizamos acetonitrila ao invés de pivalonitrila como co-solvente da
reação. Embora ambos tenham a função de aumentar a polaridade do meio reacional,
cada qual tem sua particularidade.
Frente a tantas dificuldades optamos por modificar a nossa rota de síntese. Em
primeiro lugar decidimos eliminar a etapa de proteção com MEM ou qualquer outro
grupo protetor porque acreditamos que a coordenação do aldeído (+)-27 aos
catalisadores metálicos estava sendo prejudicada em função do fator estéreo. Os
catalisadores de Keck9 24, de Maruoka11 29 e de Kurosu42 são espécies volumosas por
natureza e talvez, diminuindo-se o tamanho do aldeído (+)-27 pudéssemos contornar o
problema do impedimento estéreo.
Um das alternativas seria executar a reação de clivagem oxidativa diretamente
no substrato (+)-17 sem que houvesse proteção de sua hidroxila. A princípio não
queríamos seguir por esse caminho pois, segundo experiência do grupo, a síntese de β-
hidroxialdeídos é dificultada pela espontaneidade da reação inversa de retro-aldol.
H
OOH
Retro-aldol
O
H+
O
H
Esquema 18: Reação de retroaldol para ββββ-hidroxi aldeídos.
Com base em alguns trabalhos de Cossy 4que tratam justamente de reações de
alilação a β-hidroxialdeídos, decidimos realizar alguns testes de clivagem oxidativa do
alceno (+)-17.

26
Na tentativa de prepararmos o aldeído (+)-30, utilizamos uma quantidade
catalítica de OsO4 (2 mol %) e NaIO4 (4 equiv.) na reação de clivagem oxidativa do
alceno (+)-17 em éter etílico e água 1:1.
H
OOHOHBnO BnO
(+)-17 (+)-30
a) OsO4 (2mol %), NaIO4(4 equiv.), 5h.
a
Esquema 19: Preparação de (+)–30 utilizando-se de OsO4 (2 mol %) e NaIO4 (4
equiv.).
O material bruto de reação foi isolado e prontamente utilizado na reação de
alilação seguinte devido a sua extrema instabilidade.
3.7. Preparação do 1-benziloxi-hept-6-eno-2,4-diol (+)-16
Diversos métodos de alilação catalítica e assimétrica foram testados na tentativa
de obter o diol (+)-16 dentre eles os de Keck9, de Maruoka11 e de Kurosu42 que
encontram-se descritos anteriormente. A metodologia de Augè, que utiliza um sistema
catalítico de In(0) na presença de brometo de alila (21) e Mn(0)/TMSCl como sistema
redutor, também não forneceu o produto desejado. Daí a decisão de abandonarmos
neste ponto do trabalho os métodos catalíticos de alilação para averiguarmos os
métodos equimolares disponíveis.
A construção do segundo centro estereogênico do isômero 13 da
Criptomoscatona D2 poderia ser diasterosseletivamente controlado pelo centro
remanescente em (+)-17, por isso que a princípio não nos preocupamos muito em
utilizar um método que envolvesse um ácido de Lewis quiral ou um alilmetal quiral nesta
etapa. Os ácidos de Lewis aquirais mais comumente usados em reações de alilação
são o TiCl4 e o BF3.Et2O sendo este último mais brando e portanto mais adequado no
momento devido à instabilidade do aldeído (+)-30.

27
3.7.1. Uso de BF3.Et2O
Uma vez preparado o aldeído (+)-30 bruto a partir da clivagem oxidativa do
alceno (+)-17, este foi utilizado na reação de alilação com o alil-tri-n-butilestanho (23)
em CH2Cl2 na presença de BF3. Et2O a –42 °C.
OHBnO
(+)-17
OHOHBnO
(+)-16a
a) OsO4(15mol %), NaIO4 (4 equiv.), b) i) CH2Cl2, - 42 ºC ii) alil-tri-n-butilestanho (23)iii)BF3.Et2O, 50 min, 31% para duas etapas.
a, b
OHOHBnO
(+)-16b+
Esquema 20: Preparação dos álcoois (+)-16 utilizando-se as OsO4/NaIO4 na etapa a
e BF3.Et2O como ácido de Lewis na etapa b.
Pela primeira vez obtivemos resultados interessantes. Após purificação do
material bruto de reação conseguimos isolar um composto em 56 % rendimento. Seu
espectro de IV forneceu sinais extremamente intensos na região de estiramentos C-H
que de certa forma camuflaram as bandas de estiramento C=C e O-H do composto (+)-
16 fazendo-as aparecer juntamente aos ruídos. Já no espectro de 1H-RMN pudemos
observar nitidamente os sinais característicos dos hidrogênios vinílicos na região entre
5,00 e 6,00 ppm e dois singletos de diferentes integrações quase sobrepostos em 4,56
e 4,55 ppm referentes aos hidrogênios benzílicos (pertencentes provavelmente a cada
um dos dois possíveis diasteroisômeros de 16 formados). Não obstante, entre 0 e 2,00
ppm foram encontrados sinais extremamente intensos que nos fizeram suspeitar da
presença de impurezas de estanho provenientes do resíduo de SnBu3, que justificariam
inclusive as intensidades das bandas de estiramento C-H no espectro de IV. A análise
de um espectro de 119Sn deste composto veio a confirmar nossa suspeita. Neste
espectro foi possível identificar um intenso sinal de estanho em 156,02 ppm. O resíduo
de estanho encontrava-se aparentemente ligado a uma hidroxila e coordenado a outra
no composto (+)-16 porque análises de CCD indicavam a presença de um único
composto na amostra analisada.
Mesmo sabendo que com a incorporação de resíduos de estanho o rendimento
da reação era inferior aos 56 % anteriormente relatados decidimos repetir a reação e

28
modificar o seu tratamento de modo que os resíduos –SnBu3 fossem removidos neste
processo.
Em um primeiro momento, havíamos tratado a reação descrita no esquema 20
com uma solução saturada de NaHCO3. Com base em um artigo de Renaud43 a
respeito de tratamentos que visam a remoção de resíduos de estanho passamos a
utilizar NaOH 1molL-1no tratamento reacional. Após uma hora sob agitação em meio
aquoso básico (NaOH 1molL-1) o resíduo de estanho foi hidrolisado e convertido a
Bu3Sn-OH o qual pôde ser removido através de uma purificação do composto bruto (+)-
16 por cromatografia em coluna de sílica gel. O diol (+)-16 foi isolado em 31 % de
rendimento para duas etapas.
No espectro de 1H-RMN foi possível observar os sinais dos hidrogênios
aromáticos, vinílicos e benzílicos. Todos com integrações e multiplicidades coerentes.
Contudo, abaixo de 5,00 ppm começamos a observar sobreposição de sinais que nos
impossibilitaram de fazer uma atribuição precisa dos mesmos. Isso já era esperado,
afinal tínhamos em mãos uma mistura diasteroisomérica de (+)-16.No espectro de 1H-
RMN ainda era possível observar pequenos sinais provenientes de resíduos de estanho
entre 0 e 2,00 ppm, além de um pequeno singleto em 9,8 ppm, típico de aldeído. O
espectro de 13C-RMN não nos trouxe muitas informações adicionais neste momento.
Até então estávamos muito satisfeitos com os resultados alcançados. Embora o
rendimento da reação não tenha sido significativo (31 % para duas etapas) tivemos,
pela primeira vez, a garantia da formação do aldeído (+)-30, o qual havia sido utilizado
em sua forma bruta na reação de alilação seguinte e, em nenhum momento, pôde ser
caracterizado em função de sua grande instabilidade.
3.7.2. Índio metálico
A segunda metodologia equimolar de alilação a ser testada foi a de Paquette7
que trata da adição de alilíndio a aldeídos substituídos com um heteroátomo nas
posições α ou β.
O aldeído (+)-30, preparado a partir da clivagem oxidativa da dupla ligação de (+)-
17, foi utilizado em sua forma bruta na reação de alilação com BrCH2CHCH2 (21) e 43
Renaud, P.; Lacôte, E.; Quaranta, L. Tetrahedron Lett.1998, 39, 2123.

29
índio metálico em THF/H2O 1:1. O diol (+)-16 foi obtido com um rendimento de 93 %
para duas etapas.
OHBnO
(+)-17
OHOHBnO
(+)-16a
a, b
a) OsO4(15mol %), NaIO4 (4 equiv.), Et2O / H2O 1:1, 5h, b) In, CH2CHCH2Br (21), THF:H2O, 18h, 93 %, para duas etapas
OHOHBnO
(+)-16b+
Esquema 21: Obtenção dos dióis (+)-16 utilizando-se OsO4/NaIO4 na etapa a e In
metálico e bromteo de alila (21) na etapa b.
O mecanismo desta reação, segundo Paquette7, ainda não está elucidado.
Acredita-se, com base em estudos mecanísticos de reações análogas, haver
primeiramente a formação de um ânion radical alílico na superfície do metal que leva a
oxidação do In(0) a In3+.
Br + In (0)In3+
3 3 + 3 Br -
Esquema 22: Mecanismo de reação do tipo Barbier que leva à oxidacao do In
metálico.
A espécie InL3 formada (onde L = -CH2CHCH2), após concomitante coordenação
do índio aos oxigênios hidroxílico e carbonílico de (+)-30, promove a adição do ânion
alílico a carbonila de aldeídos.
H
HO OIn
L L
(+)-30
BnO
L =
HO OIn
L L
BnO
Esquema 23: Mecanismo de adicão do grupamento alil à carbonila de (+)-36.
Após purificação do material bruto de reação por cromatografia em coluna de
sílica gel utilizando-se CH2Cl2 / MeOH 9:1 como eluente, isolou-se a mistura
diastereoisomérica de (+)-16 a qual foi caracterizada por técnicas de Ressonânica
Magnética Nuclear (RMN), Infravermelho (IV) e Espectrometria de Massas de Alta
Resolução (EMAR).

30
No espectro de IV pudemos observar uma banda intensa e larga em 3399 cm-1
características das duas hidroxilas de (+)-16.
No espectro de 1H-RMN pudemos observar com clareza sinais característicos de
hidrogênios vinílicos entre 5,00 e 6,00 ppm, benzílicos em 4,55 ppm (dubleto) e
aromáticos em 7,38 – 7,25 ppm (multipleto). Mais uma vez, observamos uma enorme
sobreposição de sinais na região entre 1,00 e 4,50 ppm o que não nos possibilitou uma
atribuição precisa dos mesmos. A maior evidência de formação de (+)-16 extraída do
espectro de 1H-RMN foi com relação as integrações que se encaixavam perfeitamente
para número de hidrogênios de nossa estrutura (+)-16.
No espectro de 13C-RMN observamos a duplicidade da maioria dos sinais em
uma proporção de 1:1, indício de que a reação não havia sido seletiva. Mais adiante,
relataremos com clareza como foi estimado o excesso diastereoisomérico desta reação.
Entre 140 e 100 ppm, região típica de carbonos com hibridização sp2, encontramos
exatamente dez sinais sendo que, na mistura dos compostos sin e anti-16 poderíamos
ter observado até doze carbonos quimicamente distintos, indicando que havia dois
pares com o mesmo deslocamento químicos de outros 2 carbonos. Na região dos
carbonos carbinólicos encontramos exatamente oito sinais de acordo nossa expectativa
e, outros três sinais, referentes aos carbonos que não encontram-se diretamente
ligados a um heteroátomo, foram encontrados em campo mais alto.
No espectro do DEPT 135 ° foi muito interessante observar que, na região dos
carbonos carbinólicos foram encontrados quatro sinais de metileno CH2 em um campo
mais baixo referentes aos carbonos benzílicos e -BnOCH2 de sin-16 e anti-16, além de
quatro sinais acima da linha base (provenientes de metinos CH) numa região um pouco
mais protegida (referentes aos carbonos diretamente ligados as hidroxilas).
A análise do espectro de EMAR nos deu uma forte evidência da formação do diol
(+)-16. A massa calculada para este composto C14H20O3 é de m/z 236,14125 sendo que
sua massa medida foi de m/z 236,13783. A medida está dentro do erro tolerado que
pode ser calculado através da seguinte expressão:
|Valor calculado – Valor Medido| x 10 6 ≤ 50 ppm
Valor calculado

31
Com todos esses resultados em mãos, nosso próximo passo foi o de encontrar
um sistema de solventes adequado para promover a separação dos isômeros sin e anti
em uma coluna cromatográfica. Após diversas análises por CCD, concluímos que o
melhor eluente seria Hexano /EtOAC / Et3N 7,5 :7,5: 1 quando a diferença de Rf entre
as duas manchas foi de apenas ∆Rf 0,05.
Após purificação de (+)-16 por cromatografia em coluna de sílica flash, separou-
se grande parte dos isômeros sin-16 e anti-16 os quais foram caracterizados por
técnicas de IV, RMN e EMAR. Inevitavelmente, obteve-se frações contendo mistura de
isômeros o que não nos possibilitou determinar o excesso diasteroisomérico da reação
em função das massas de sin-16 e anti-16 isoladas.
OHOHO
a
b
c
d
e
f
g
h
i
j kl
nm
**
H Hc'
Figura 16: 1-benzilóxi-hep-6-en-2,4-diol, (16a).
No espectro de 1H-RMN do composto 16a com Rf 0,29 foram observados os
sinais característicos dos hidrogênios aromáticos como um multipleto entre 7,38 – 7,25
ppm integrando para cinco hidrogênios. Os hidrogênios vinílicos Hh e Hi forneceram
dois multipletos em respectivamente 5,88 – 5,74 ppm e 5,14 – 5,12 ppm e com
integrações de um e dois hidrogênios respectivamente. Observamos também um
singleto em 4,55 ppm integrando para dois hidrogênios referentes aos benzílicos Hb.
Entre 3,00 e 4,00 ppm encontramos dois multipletos integrando para um hidrogênio
cada um. Não sabemos distinguir qual deles é Hd e qual é Hf. Cada um dos hidrogênios
Hc forneceu um sinal distinto porque são diastereotópicos. Em 3,45 ppm apareceu um
duplo dubleto com constantes de acoplamento 2J 9,5 Hz e 3J 4,0 Hz. Em 3,38 vimos
outro duplo dubleto com 2J 9,5 Hz e 3J 7,0 Hz. Através destes resultados podemos
concluir que o hidrogênios Hc acoplam entre si com 2J 9,5 Hz e também acoplam com
os hidrogênios Hd (3J 7,0 Hz e 3J 4,0 Hz) que encontram-se a 3 ligações de distância.
Em 3,29 e 3,21 ppm pudemos observar a presença de dois singletos largos muito
próximos (∆δ 0,08 ppm) integrando para 1H, possivelmente relativos aos prótons
hidroxílicos Hj e Hk (não necessariamente nesta ordem). O hidrogênio He forneceu um

32
tripleto em 2,24 ppm (3J 6,9 Hz) em função do acoplamento com Hd e Hf. Os
hidrogênios alílicos Hg forneceram um multipleto integrando para dois hidrogênios em
uma região mais protegida do espectro (entre 1,62 – 1,50 ppm).
Tabela 4: Relação de sinais no espectro de 1H-RMN do diol 16a.
Hidrogênios δ δ δ δ (ppm) Multiplicidade IntegraçãoHa, Hm, Hn 7,38 – 7,25 m 5H
Hb 4,55 s 2H Hc 3,45 ou 3,38 dd 1H Hc’ 3,38 ou 3,45 dd 1H He 2,24 t 2H Hg 1,62 – 1,50 m 2H Hh 5,88 – 5,74 m 1H
Hj ou Hk 3,29 sl 1H Hj ou Hk 3,21 sl 1H
No espectro de 1H-RMN do composto 16b com Rf 0,24 foram observados
basicamente os mesmos sinais encontrados no espectro de 16a, com alguma variação
nos valores de deslocamento químico. Os sinais mais característicos foram aqueles
referentes às hidroxilas Hj e Hk que deslocaram-se para um campo mais alto (2,87 e
2,57 ppm) e afastaram-se um do outro (∆δ 0,30 ppm).
Os íons moleculares dos espectros de EMAR dos compostos 16a e 16b de
fórmula molecular C14H20O3 forneceram picos em respectivamente m/z 236,14073 e m/z
236,14801 sendo a massas calculadas para esses compostos de m/z 236,14125.
Os espectros de IV não nos forneceram informações tão enriquecedoras. Em
ambos os espectros, pudemos observar basicamente o aparecimento de uma intensa
banda de estiramento O-H.
Nos espectros de 13C-RMN das espécies 16a e 16b foram observados doze
sinais: seis acima de 100 ppm referentes aos carbonos aromáticos e vinílicos, quatro
entre 65 e 75 ppm referentes aos carbonos b,c,d e f e dois abaixo de 45 ppm referentes
ao carbonos e e g. Os dois espectros eram muito parecidos e havia apenas dois sinais
muito particulares para cada um dos isômeros sin-16 e anti-16. Os sinais em 71,05 e
71,16 ppm observados no espectro de 16a apareciam em uma região mais protegida no
espectro de 16b (67,81 e 67,88 ppm). Como já havíamos determinado através da
análise do DEPT 135° da mistura diastereoisomerica de (+)-16, esses sinais pertenciam

33
aos carbonos carbinólicos Cd e Cf. Naquele momento não tínhamos como estimar com
precisão o excesso diastereoisomérico da reação através do espectro de 13C-RMN de
(+)-16 já que desconhecimos a proveniência dos sinais, mas agora, conhecendo-se a
atribuição dos mesmos podemos garantir, através da comparação de suas intensidades
(71,05 versus 67,81 e 71,16 versus 67,88) que na não houve favoreciemento na
formação dos diasteroisômeros sin e anti. Esta estimativa pode ser feita com base na
análise do espectro de 13C-RMN por estarmos comparando carbonos análogos de dois
diastereoisômeros, para os quais não é esperado diferença apreciável nos tempos de
relaxação e no incremento de sinal devido à tranferência de polarização pelo hidrogênio
diretamente ligado a este carbono.
A reação não foi tão seletiva quanto desejávamos mas ainda assim julgamos
adequado prosseguir com a síntese a partir de um dos diastereoisômero muito embora
ainda não conhecêssemos sua configuração relativa. Desta maneira poderíamos
mapear a síntese e determinar a princípio a configuração relativa de um dos possíveis
isômeros da Criptomoscatona D2 (13) ficando a determinação da configuração absoluta
a posteriori.
3.7.3. Estanho metálico e InCl313
Outra metodologia de alilação que forneceu bons resultados foi aquela
desenvolvida por Loh13 para a alilação de aldeídos derivados de açúcares.
O aldeído (+)-30, preparado a partir da reação de clivagem oxidativa da dupla
ligação do alceno (+)-17 utilizando-se OsO4 (1 mol %), NaIO4 (4 equiv.) e 2,6-lutidina em
dioxana / H2O 3:1, foi utilizado em sua forma bruta na reação de alilação com brometo
de alila (20) na presença de estanho metálico (2 equiv.) e InCl3 (1 equiv.) em H2O. O
produto (+)-16 foi isolado em 86 % de rendimento porém mais uma vez como uma
mistura equimolar de isômeros.

34
OHBnO
(+)-17
OHOHBnO
(+)-16a
a, b
a) OsO4(1mol %), NaIO4(4 equiv.), 1,4-dioxana / H2O, 2,6-lutidina, b)i) Sn (2 equiv.), InCl3 (1 equiv.), H2O e 21, 4h, ii) então (+)-30, 1h 86 %, 2 etapas
OHOHBnO
(+)-16b
+
Esquema 24: Obtenção dos dióis (+)-16 utilizando-se os métodos de Jin44 e de
Loh13.
Segundo Loh13 a reação de alilação de aldeídos α-oxigenados é promovida pela
espécie InCl3 e mediada por estanho metálico. É possível que esta proposta também se
aplique ao nosso sistema β-oxigenado.
InCl2+
OH O
HBnO
HO O
HBnO
InCl Cl
Br
InCl
ClCl InCl
ClSnBrCl +
HO O
HBnO
InCl Cl
H2O OH OHBnO
(+)-16
I)
II)
III)
IV)+ InCl2OH
Sn (0) +BrSn
BrSn
(+)-30
(+)-30
Esquema 25: Mecanismo da reação para obtenção do diol (+)-16 utilizando-se o
método de Loh13.
44 Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett .2004, 6, 3217

35
3.8. Preparação do 1-O-benzil-2,4-O,O-isopropilideno-1,2,4-tributoxihept-6-eno (31)
A determinação da configuração relativa dos compostos 16a e 16b foi feita com
base no método de Rychnovsky45,46que, através de análise de RMN de 13C, possibilita
uma determinação segura da configuração sin ou anti em 1,3-dióis.
H
OOMe
Me HR2
H (Me)
Me(H)R1δδδδ 30,0
δδδδ 98,1
δδδδ 19,3
sin (cadeira)
OO
MeMe
Me(H)H(Me)
H
H
R1
R2
δδδδ 100,6
δδδδ 24,6
anti (barco torcido)
Figura 17: Determinação da estereoquímica de 1,3-acetonídeos pelo método de
Rychnovsky45,46.
O método de Rychnovsky é baseado na variação dos deslocamentos químicos
(δ) no espectro de 13C-RMN, para o carbono do cetal e seus substituintes metila,
associada as diferenças conformacionais existentes para os 1,3 – acetonídeos sin
(cadeira) e anti (barco torcido). Conforme a generalização observada, os isômeros sin
apresentam deslocamentos de carbono em 19 e 30 ppm para as metilas do acetonídeo,
enquanto que para os isômeros anti a faixa singular de ressonância é de 24 – 25 ppm.
À primeira vista, parece desfavorável a presença de um confôrmero de tão alta
energia em solução como o barco torcido. Em nossa opinião é mais provável que o
isômero anti esteja em um equilíbrio dinâmico tão rápido entre suas diversas
conformações que o que enxergamos no espectro é a média de sinais metílicos
coincidindo em um único valor de δ.
Os acetais 31a e 31b foram preparados a partir da reação de cada uma das
espécies 16a e 16b com 2,2-dimetoxipropano (2,2-DMP) em acetona anidra e na
presença (+)-CSA (ácido canforsulfônico). Os rendimentos das reações foram ambos de
90 %.
45 a) Rychnovsky, S.D.; Skalitzky, D.J. Tetrathedron Lett. 1990, 31, 945. b) Evan, D.A.; Rieger, D.L.; Gage, J.R. Tetrahedron Lett. 1990, 31, 7099. c) Rychnovsky, S.D.; Rogers, B.; Yang, G. J.Org.Chem.1993, 58, 3511. 46 d) Corrêa Júnior, I.R. Síntese Total e Elucidação Estrutural da Delactomicina,Tese de Doutorado, 2003, Instituto de Química Unicamp.

36
BnOOH OH
BnOO O
16a 31a
a
a) 2,2-DMP, acetona, (+)-CSA, 2h , 90 %
BnOOH OH
BnOO O
a
16b 31b
ou
Esquema 26: Obtenção dos cetais 31a e 31b.
Os compostos 31a e 31b foram caracterizados por análises espectroscópicas e
espectrométricas (RMN, IV e EMAR).
OO O
a b cd
e fg
h
i
j k
l
mn
o
H Hb'
* *
Figura 18: Acetais 39a ou 39b.
No espectro de IV dos compostos 31a e 31b observamos o desaparecimento das
bandas largas em torno de 3400 cm-1 referentes aos estiramentos das ligações O-H.
Os sinais observados no espectro de 1H-RMN de 31a e 31b foram basicamente
os mesmos com alguma ou outra variação de deslocamentos químicos (δ). Entre 5,00 e
6,00 ppm foram observados os hidrogênios vinílicos (Hh e Hi) e entre 1,30 e 1,60 ppm
dois singletos integrando para três hidrogênios cada um referentes aos hidrogênios
metílicos Hj e Hk. Em ambos os espectros, os hidrogênios benzílicos Hb e Hb’
forneceram dubletos distintos que, de tão próximos um do outro, aparentavam um
quarteto com sinais de intensidades relativas iguais a 1 :3: 3: 1 (típico do triângulo de
Pascal). Na verdade os sinais tinham tal aparência pois tratavam-se de um sistema AB
onde ∆ν/J ≈ 1,5.
As regiões típicas de carbonos sp2 e carbonos carbinólicos dos espectros de 13C-
RMN de 31a e 31b foram muito semelhantes. Acima de 100 ppm encontramos os seis

37
sinais referentes aos carbonos aromáticos e vinílicos e na região de carbinólicos (entre
60 e 100 ppm) pudemos encontrar os cinco sinais esperados. Na região entre 10 e 50
ppm é que os espectros apresentaram alguma diferença. No espectro de 31b foram
observados apenas três sinais nesta região ao passo que, no espectro de 31a, foram
observados quatro. Decidimos então realizar análises de DEPT 90° e de DEPT 135°
para que pudéssemos fazer uma atribuição precisa desses sinais.
Nos espectros de DEPT 90° de 31a e 31b foram observados seis sinais: quatro
na região de carbonos Csp2 referentes aos metinos a, n ,o e h e dois na região de
carbinólicos referentes ao carbonos Cd e Cf. Nos espectros de DEPT 135° pudemos
observar cinco sinais abaixo da linha base: um sinal na região de Csp2 (referente ao
metileno Ch), dois sinais na região de carbinólicos referentes aos Cb e Cc e outros dois
sinais em uma região mais protegida no espectro referentes aos Ce e Cg. Por fim
tivemos a garantia de que, os dois últimos sinais do espectro de 31a de DEPT 135°
localizados acima da linha base em uma região de campo mais alto, pertenciam aos
seus dois únicos carbonos do tipo CH3 (Cj eCk). Já no espectro de 31b apenas um sinal
de CH3 foi observado em 25,00 ppm.
Com base no método de Richnowsky45 e nos espectros de 13C-RMN dos
compostos 31a e 31b podemos afirmar com garantia que tais compostos
correspondem, respectivamente, aos isômeros sin e anti os quais foram sintetizados a
partir dos dióis 16a de Rf 0,29 e 16b de Rf 0,24 (Hexano /EtOAC / Et3N 7,5 :7,5: 1).
Com a finalidade de contarmos com um grupo de proteção do sistema 1,3-diol
em 16a e 16b compatível com condições ácidas previstas para serem empregadas em
etapas porsteriores do trabalho, decidimos preparar os carbonatos correspondentes a
16a e 16b cuja desproteção requer condições básicas47.
Demos continuidade à síntese do isômero 13 da Criptomoscatona D2 em sua
versão racêmica para que pudéssemos mapear a rota sintética.
47 Greene, T.W.; Wutz, P.G.M. Protective Group in Organic Synthesis, John Wiley & Sons Inc., New York, 1991.

38
3.9. Preparação do 4-alil-6-[(benziloxi)metil]-1,3-dioxan-2-ona (+)-(32)
O carbonato (+)-32 foi preparado a partir do tratamento da mistura
diastereoisomérica do diol (+)-16 com trisfosgênio (33) em CH2Cl2 e piridina seca a -70
°C, segundo metodologia de Burk48, em 69 % de rendimento.
BnOOH OH
BnOO O
(+)-16a + (+)-16ab (+)-32a + (+)-32bO
Cl3CO
O
OCCl3 +
33
a) piridina, CH2Cl2, - 70 °C, 20 min, 69 %
a
Esquema 27: Obtenção do carbonato (+)-32.
O mecanismo desta reação encontra-se descrito no esquema 28.
Cl3CO
O
OCCl3+ ROH Cl3CO OCCl3
OHRO
Cl3CO
O
ORH
OCCl3+
Cl3CO
O
ORH
Cl O
Cl ClCOCl2 (g) + Cl
+ Cl HCl + Cl3CO
O
OR
Cl3CO
O
OR+ ROH COCl2 + HCl +
RO
O
OR
I)
II)
III)
IV)
RO
O
ORCl3CO
O
OCCl3+ 2 ROH 2 HCl + 2 COCl2 (g)
33
33
+
H
34
34
34
Esquema 28: Mecanismo de reação para obtenção do carbonato (+)-32 a partir do
trisfosgênio (33).
O álcool se adiciona à carbonila do trisfosgênio (33), promovendo a liberação do
gás fosgênio (34) em solução (etapa II) deslocando a reação de equílibrio da etapa I
para a direita. Mesmo o gás fosgênio (34) formado é reativo em solução. Na verdade
um equivalente de trisfosgênio (33) é suficiente para reagir com três hidroxilas.
48 Burk, R.M.; Roof, M.B. Tetrahedon Lett, 1993, 34, 395.

39
Os carbonatos (+)-32a e (+)-32b foram caracterizados por técnicas de
Infravermelho (IV) e Ressonância Magnética Nuclear (RMN).
OO O
O
µµµµ µµµµ
µµµµ
BnOO O
O
BnOO O
O
Estruturas Canônicas
Momentos de Dipolo
BnOO O
O
δδδδ−−−− Densidade de Carga (δδδδ -) sob o Carbono (Blindagem)
Figura 19: Estruturas de ressonância, momentos de dipolo (µµµµ) e densidades
eletrônicas (δδδδ) do composto (+)-32.
No espectro de IV de (+)-32 pudemos observar uma intensa banda de
estiramento da ligação C=O em 1743 cm-1 e o desaparecimento da banda característica
O-H por volta de 3400 cm-1. É interessante observar que a freqüência de estiramento da
ligação C=O da carbonila do carbonato (+)-32 aparece em um número de onda maior do
que os de aldeído (1725 cm-1) e cetona (1712 cm-1). Segundo a Teoria da Ligação de
Valência (TLV), isso não era esperado, pois a ressonância de carga presente no grupo
carbonato (+)-32 (figura 19) deveria enfraquecer a ligação C=O que necessitaria de uma
menor energia de vibração. Como energia é diretamente proporcional ao número de
onda (E = h.c /λ = hcū), esperaríamos para o carbonato (+)-32 um valor de número de
onda menor do que aqueles observados para aldeídos e cetonas, que não apresentam
uma ressonância de carga tão pronunciada. A Teoria do Orbital Molecular (TOM) pode
explicar essa discrepância com base em fatores orbitalares.

40
O
O
Orbital não liganten do oxigênio
Orbital σσσσ*C-O
O
O
Orbital ππππ*C=O
Orbital não ligante n do oxigênio
Figura 20: Interações orbitalares das ligações C=O de ésteres e carbonatos.
De forma análoga ao que se observa em ésteres (número de onda da ligação
C=O em torno de 1735 cm-1), as interações orbitalares n→σC-O e n→πC=O presentes em
carbonatos dão à ligação C=O um caráter de tripla. Essas interações orbitalares devem
ser inclusive mais pronunciadas em carbonatos do que em ésteres, em função da
presença de uma ligação simples C-O a mais nestes compostos. Isso poderia explicar a
presença de banda de estiramento C=O do carbonato (+)-32 em 1743 cm-1.
OO O
O
a b cd
ef
g
h
i
j
kl
m
OO O
O
a b cd
ef
g
h
i
j
kl
m
Figura 21: 4-allil-6-[(benziloxi)metil]-1,3-dioxan-2-ona (+)-32a e (+)-32b.
No espectro de 1H-RMN de (+)-32a e (+)-32b pudemos observar o
desaparecimento dos hidrogênios hidroxílicos dos dióis (+)-16a e (+)-16b entre 2.00 e
3.50 ppm e uma sobreposição dos hidrogênios carbinólicos d e f que, neste espectro
apareceram em uma região mais desprotegida quando comparados aos mesmos sinais
do espectro dos dióis (+)-16a e (+)-16b. Isso porque o grupo carbonato exerce um efeito
retirador de elétrons que aparentemente se propaga ao longo das ligações σ e π
afetando os hidrogênios Hd e Hf próximos ao grupo protetor -OCOO. Os demais sinais
(alílico, vinílicos, aromáticos etc.) apareceram com valore de δ multiplicidades e
integrações coerentes. Contudo a sobreposição de sinais foi muito grande devido a
presença de dois diasteroisômeros na amostra analisada.
No espectro de 13C-RMN observamos duplicação de sinais (contabilizados em
26). Na região típica de Csp2 (entre 100 e 150 ppm) encontramos quatorze sinais, cada

41
sete provenientes de um isômero, incluindo os carbonos dos carbonatos . Carbonos de
hibridização sp2 ligados a um heteroátomo como o oxigênio (como é o caso de
carbonilas de aldeídos e cetonas) tendem a sofrer mais os efeitos de anisotropia
magnética e os efeitos indutivos retiradores de elétrons perdendo sua blindagem natural
por parte desses e aparecendo em regiões de campo mais baixo (>150 ppm).
Entretanto, o efeito de ressonância do grupo carbonato é tão acentuado que diminui
densidade de carga negativa (δ +) sobre o carbono carbonílico favorecendo uma maior
blindagem deste carbono que acaba por sentir um pouco menos o efeito do campo
magnético externo. Essa é uma possível explicação para o aparecimento dos sinais
referentes aos carbonos dos carbonatos em uma região atípica para maioria das
carbonilas. Entre 70 e 80 ppm pudemos observar oito sinais, típicos de carbonos de
hibridização sp3 ligados a um heteroátomo (Cb, Cc, Cd e Cf). Os sinais dos carbonos Ce
e Cg apareceram abaixo de 40 ppm.
3.10. Preparação do {6-[(benziloxi)metil]-2-oxo-1,3-dioxan-4-il} acetaldeído (+)-(35)
Em uma primeira tentativa de obter-se o aldeído (+)-35, realizamos a reação
entre o alceno (+)-32 com OsO4 (1 mol %) e NaIO4 (4 equiv.) na presença de 2,6-lutidina
em 1,4-dioxana / H2O 3:1.
BnOO O
O
BnOO O
O
O
H
(+)-32a (+)-35a
a
a) OsO4 (1 mol %), NaIO4 (4 equiv.), 2,6-lutidina, 1,4-dioxana / H2O 3:1
BnOO O
O(+)-32b
BnO
O O
O
O
H
+ +
(+)-35b
Esquema 29: Obtenção do aldeído (+)-35 sob as condições de Jin44.
Segundo Jin44, o uso de 2,6-lutidina promove uma melhoria do método de
Lemieux-Johnson36 devido a neutralização do meio reacional que contém pequenas
quantidades de ácido iódico
Após purificação do material bruto da reação por cromatografia em coluna de
sílica gel, isolou-se um composto cuja caracterização por RMN forneceu resultados

42
inesperados. No espectro de 1H-RMN deste composto (+)-35 encontraram-se sinais
característicos de hidrogênios benzílicos e aromáticos além de muitos sinais de
impurezas. Pudemos observar também um dubleto na região típica de hidrogênios –
CHO. Esta multiplicidade nos intrigou muito pois esperávamos observar um tripleto (em
função do acoplamento entre os hidrogênios CHO e CH2CHO) ou até mesmo um
singleto largo caso a constante de acoplamento com grupo metilênico fosse de
pequena magnitude. Além disso observamos dois multipletos entre 6,00 e 7,00 ppm
com integrações imprecisas. Os sinais típicos de hidrogênios vinílicos na região entre
5,00 e 6,00 ppm haviam desaparecido. O espectro de IV do composto isolado
apresentava uma intensa banda em 1335 cm –1 (típica de estiramento O-H) e outra em
1687 cm-1, ou seja, com uma freqüência de estiramento de C=O menor do que
esperávamos para aldeídos (~ 1725 cm-1). A banda característica do grupo carbonato
(1743 cm-1) também havia desaparecido. Após uma análise detalhada dos espectros
concluímos ter obtido o aldeído α,β-insaturado 6-benziloxi-5-hidroxi-hex-2-enal 36.
A medida que são formados, os aldeído (+)-35a e (+)-35b vão sendo consumidos
em uma reação paralela segundo mecanismo descrito no esquema abaixo.
BnOO O
O
BnOO O
O
OO
H H
BnOO O
O
O
H
OHHαααα Hαααα
BnOO O
H
+ H2O
(+)-35
(+)-36
+ CO2 (g)
I)
II)
BnOO O
H+ H2OIII)
BnOOH O
H+ HO
Esquema 30: Mecanismo para obtenção do aldeído α,βα,βα,βα,β-insaturado (+)-36.
Os hidrogênios α a carbonila, que são de natureza ácida, podem ser abstraídos
por uma espécie básica presente no meio reacional para promover a eliminação de CO2
numa reação favorecida termodinamicamente.

43
Isso explica a ausência da banda de estiramento da ligação C=O do carbonato
em 1743 cm-1 e o aparecimento da banda em 1687 cm-1 no espectro de IV do composto
(+)-36 pois esta banda é típica de estiramento C=O de aldeídos α,β-insaturados. A
ligação C=O nesses aldeídos é mais fraca que o normal em função do fenômeno de
ressonância que de certa forma enfraquece a ligação C=O (figura 22). A banda
referente ao estiramento O-H encontrada no espectro também pode ser justificada com
base na estrutura molecular do composto (+)-36.
3.11. Preparação do anti-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi) hept-6-eno,
anti-(37).
Decidimos prosseguir a síntese utilizando-se o isômero anti-16 pois possuíamos
uma maior quantidade pura deste composto em detrimento de sin-16, lembrando que
os dois centros estereogênicos em questão (C7 e C9 figura 4) são desconhecidos na
molécula alvo Criptomoscatona D2, tornando a priori indiferente a natureza do
diastereoisômero com o qual trabalhamos.
O novo grupo protetor escolhido para o diol anti-16 foi o TBS. Apesar de ser
extremamente volumoso e poder dificultar a etapa de alilação o TBS é aparentemente o
grupo mais resistente às nossas futuras condições de reações. Não podemos deixar de
enfatizar que as hidroxilas do diol (+)-16 devem estar protegidas até o último momento,
sendo sua desproteção o desfecho da síntese. O problema de impedimento estéreo da
próxima reação de alilação talvez não fosse um empecílho tão grande neste momento
uma vez que utilizaríamos métodos equimolares de alilação. Anteriormente utilizávamos
condições catalíticas de alilação, onde o fator estéreo era muito significativo devido à
presenca de catalisadores metálicos complexados ao binaftol, volumoso por natureza.
Embora inicialmente avessos à utilização de TBS como grupo de proteção na versão
catalítica, não poderíamos deixar de realizar uma reação teste para averiguar o seu
potencial como grupo protetor neste caso.
O composto anti-37 foi preparado a partir da reação entre diol anti-16 e TBSOTf
(2,5 equiv.) na presença de 2,6–lutidina em CH2Cl2 a O°C. Após purificação do material

44
bruto de reação por cromatografia em coluna de sílica flash o produto anti-37 foi isolado
em 88 % de rendimento.
BnOOH OH
BnOTBSO OTBS
anti-16 anti-37
a
a) TBSOTf (2,5 equiv.), 2,6-lutidina, CH2Cl2, 0 ºC, 12 h, 88%
* ** *
Esquema 31: Obtenção do composto anti-37 a partir da reação do diol anti-16 com
TBSOTf.
O composto anti-37 foi caracterizado por técnicas espectroscópicas de
Ressonância Magnética Nuclear e Infravermelho.
No espectro de IV de anti-37 observou-se um aumento na intensidade das
bandas referentes aos estiramentos de ligações C-H, em função da presença das
tercbutilas e das metilas provenientes dos grupos TBS. Houve também o
desaparecimento da banda típica de hidroxilas em torno de 1500 cm -1.
No espectro de 1H-RMN pudemos observar os sinais típicos de hidrogênios
aromáticos, vinílicos e benzílicos com integrações, multiplicidades e valores de
δ coerentes. Os sinais dos hidrogênios metílicos característicos do grupo TBS
apareceram em uma região de campo alto como esperado (entre 0 e 1,00 ppm).
No espectro de 13C-RMN observou-se os sinais acima de 100 ppm típicos de
carbonos de hibridização sp2 e entre 60 e 80 ppm foram observados os sinais
referentes aos carbonos carbinólicos. Em -4, 18 e 26 ppm foram observados os
carbonos típicos de TBS.
3.12. Preparação do isômero anti--benzilóxi-3,5-bis-(terc-butildimetilsililoxi)
hexanal, anti-(38).
Após a reação de proteção das hidroxilas do diol anti-16, promovemos a terceira
reação de clivagem oxidativa prevista na rora sintética.

45
BnOOP OP
anti-37
BnOOP OP
Hanti-38
Oa
a) OsO4, NaIO4, Dioxana / H2O 3:1, 2,6-Lutidina, 5h, 97 %
P=TBS P=TBS
* * * *
Esquema 32: Preparação do aldeído anti-38.
O alceno anti-37 sofreu a reação de clivagem oxidativa pelo tratamento com
tetróxido de ósmio e periodato de sódio em dioxana/H2O 3:1 na presença de 2,6-lutidina
como base, segundo metodologia de Jin44. O aldeído anti-38, após purificação por
cromatografia em coluna, foi isolado em 97 % de rendimento.
A caracterização do aldeído anti-38 foi feita mediante análise de espectros de
infravermelho, 1H-RMN e 13C-RMN. No espectro de IV pudemos observar uma intensa
banda em 1732 cm-1 referente a freqüência de estiramento de ligação C=O de aldeídos.
No espectro de 1H-RMN encontrou-se sinais típicos do hidrogênio do grupo CHO em
9,79 ppm integrando para 1H. Embora a resolução deste sinal não esteja muito boa,
observa-se uma espécie de duplo dubleto para este hidrogênio em função de seu
acoplamento com os dois hidrogênios vizinhos α a carbonila. Não foram observados
sinais de hidrogênios vinílicos entre 5,00 e 6,00 ppm. No espectro de 13C-RMN deste
composto observou-se um sinal de baixa intensidade em 202,02 ppm referente ao
carbono no aldeído de hibrização sp2.
3.13. Preparação do anti-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi) non-8-en-6-ol,
2,4-anti-(15).
BnOPO OP
2,4-anti-(+)-15
BnOPO OP
H
anti-38
O OHa
a) BF3.Et2O, -42ºC, CH2Cl2, 23, 30 min., 94% 2.1:1 d.e.
** * *
P=TBS P=TBS
Esquema 33: Preparação do álcool 2,4-anti-(+)-15.
O aldeído anti-38, após ativação por parte do ácido de Lewis BF3.Et2O em
diclorometano a -42 ºC, sofre uma reação de alilação na presença de aliltri-n-

46
butilestanho (23) fornecendo o álcool 2,4-anti-(+)-15 em 94% de rendimento,
apresentando uma mistura diastereoisomérica de 2,1:1.
No espectro de IV de 2,4-anti-(+)-15 observou-se a banda larga da hidroxila em
3490 cm-1. No espectro de 1H-RMN observamos dois multipletos integrando
respectivamente para 1 e 2 hidrogênios entre 5,00 e 6,00 ppm, típicos de hidrogênios
vinílicos. No espectro de 13C-RMN encontramos sinais de carbonos sp2 acima de 100
ppm e sinais de carbonos carbinólicos entre 60 e 70 ppm que em sua maioria
apresentaram-se duplicados em razão da presença de dois diastereoisômeros na
amostra analisada. Através das intensidades dos sinais duplicados no espectro de 13C-
RMN pode-se estimar a razão diastereoisomérica da reação (2,1:1). Neste momento
não foi possível separar os isômeros formados por métodos cromatográficos pois
trabalhávamos com quantidade muito pequenas de material.
Outras metodologias de alilação foram testadas nesta etapa, dentre elas, a de
Grignard que forneceu o produto desejado em 25% de rendimento e a de Paquette7 que
utiliza índio metálico e brometo de alila em THF/H2O 1:1 e que havia fornecido bons
rendimentos na ocasião em que testávamos a segunda alilação prevista na síntese de
um dos isômeros da Criptomoscatona D2. Através do método de Paquette7 obteve-se o
álcool 2,4-anti-(+)-15 em 63 % de rendimento mas a razão distereoisómerica alcançada
foi de 1:1.
Dado aos inúmeros testes realizados com a série de compostos anti, não
possuíamos material suficiente para condução do estudo de metátese seguinte neste
ponto do trabalho, ficando a otimização desta etapa para o momento em que
trabalhássemos com a série sin.
3.14. Preparação do [1-benziloxi-2,4-bis(terc-butyldimetilsililoxi)-non-8-en-6-il]
acrilato, 2,4-anti-(+)-(39)
A mistura de álcoois diastereoisoméricos 2,4-anti-(+)-15 sofreu uma reação de
esterificação na presença de cloreto de acriloíla em CH2Cl2 e Et3N a 0ºC fornecendo a
mistura de acrilatos 2,4-anti-(+)-39 em rendimento quantitavo após 1h de reação.

47
BnOPO OP
2,4-anti-(+)-15
OH
BnOPO OP
2',4'-anti-(+)-39
O
O
a
a) Et3N, 0ºC, CH2Cl2, cloreto de acriloíla, 1h, 75%.P=TBS P=TBS
* * * *
Esquema 34: Preparação do éster 2,4-anti-(+)-39.
No espectro de IV do composto 2,4-anti-(+)-39 observamos uma absorção em
1726 cm-1 típica de estiramento C=O de ésteres α,β-insaturados bem como o
desaparecimento da banda larga típica de hidroxila acima de 3500 cm-1. No espectro de 1H-RMN foram observados cinco multipletos entre 5,00 e 7,00 ppm referentes aos seis
hidrogênios olefínicos da molécula 2,4-anti-(+)-39. No espectro de 13C-RMN pudemos
verificar novamente a duplicação de sinais devido a presença de dois diasteroisômeros
na amostra analisada, contudo os sinais encontram-se em regiões características para
os grupos funcionais –COOR. Muitos sinais encontravam-se sobrepostos devido à
mistura de diasteroisômeros presente na amostra analisada.
Até este momento prosseguimos a síntese com materiais contendo misturas
diasteroisoméricas para que pudéssemos apenas mapear a rota sintética.
3.15. Estudos sintéticos com a série sin
Todos os estudos sintéticos foram executados com os compostos da série sin a
partir do diol sin-16. As condições reacionais utilizadas para a série sin foram as
mesmas da série anti.

48
BnOOH OH
BnOOP OP
P=TBS
BnOOP OP
H
O
P=TBS
BnOOP OP OH
* * * * * *
* *
P=TBS
BnOOP OP O
* *
OP=TBS
a b
c d
a) TBSOTf (2,5 equiv.), 2,6-lutidina, CH2Cl2, 0 ºC, 18 h, 86%, b) OsO4, NaIO4, dioxana/H2O 3:1, 2,6-lutidina, 5h, 95 %,c) BF3.Et2O, -42ºC, CH2Cl2, 23, 1,5 h, 81% 2:1 r.d. d) Et3N, 0ºC, CH2Cl2, cloreto de acriloíla, 1h, 73%.
sin-16 sin-37 sin-38
2,4-sin-(+)-15 2',4'-sin-(+)-39
Esquema 35: Etapas para síntese do acrilato 2,4-sin-54 a partir do diol sin-16.
Um vez preparado o acrilato 2,4-sin-(+)-39 prosseguiu-se os estudos de
metátese de olefinas para o fechamento do anel da lactona.
3.16. Preparação do 2’,4’-sin-6- [5-(benziloxi)-2,4-bis(terc-butildimetilsililoxi) pentil]
-5,6-dihidropira-2-ona, 2’,4’-sin-(+)-14.
BnOPO OP
2,4-sin-(+)-39
O
O
BnOPO OP
2',4'-sin-(+)-14
O
O
a
a) Catalisador de Grubbs, CH2Cl2, 2,5h, 83%
P=TBS P=TBS
* * * *
Esquema 36 Reação para preparação da lactona 2’,4’-sin-(+)-14.
O composto 2,4-sin-(+)-39 foi submetido a uma reação de metátese de olefinas
intramolecular na presença do catalisador de Grubbs de 1ª geração em CH2Cl2 sob
refluxo. O produto 2’,4’-sin-(+)-14 foi obtido em 83% de rendimento após purificação do
material bruto de reação por cromatografia em coluna.
O ciclo catalítico da reação encontra-se descrito no esquema 37:

49
Ru
( )n
( )n
( )n
RuCl
ClP
PH
Ph
RuCl
ClP
CH2
+
Ru
P
P= PCy3
ClCl
( )n
H
H
( )n
Cl
P
Cl
Ru
PCl
Cl
H
Ph
Ru
( )n
Cl
P
Cl
H
( )n
-P
CH2=CH2
Esquema 37: Ciclo catalítico de RCM (Ring Closing Metathesis)49.
No primeiro ciclo, o catalisador de Grubbs de primeira geração libera o resíduo
de estireno do catalisador ao invés de etileno. Somente a partir do segundo ciclo é que
o H2C=CH2 começa a ser liberado. É por isso que o catalisador não pode ser
recuperado ao final da reação. Uma vez formado o ciclo de quatro membros, pode
ocorrer um rearranjo eletrônico que promove “migração de duplas ligações”. A fim de se
evitar metátese cruzada (CM_Cross Metathesis) utiliza-se condições reacionais
diluídas.
No espectro de 1H-RMN do composto obtido observamos dois multipletos
integrando para um hidrogênio entre 6,00 e 7,00 ppm referentes aos hidrogênios
olefícos da lactona α,β−insaturada 2’,4’-sin-(+)-14. Vale observar que os sinais de
hidrogênios de duplas ligações pobres em elétrons aparecem em uma região mais
desprotegida do espectro do que as duplas ligações dos álcoois homoalílicos com os
quais trabalhamos até então, que aparecem em uma região de campo mais alto, entre
49 Frederico, D.; Brocksom, U. e Brocksom, T.J. Química Nova, 2005, 28, 692.

50
5,00 e 6,00 ppm. Essa deficiência eletrônica da dupla ligação se dá devido à sua
conjugação com a carbonila da lactona, que reduz a sua densidade eletrônica pelo
efeito de ressonância. No espectro de 13C-RMN, observamos novamente duplicação de
sinais dada à mistura diasteroisomérica de nossa amostra. No espectro de IV
observamos a banda de estiramento C=O da lactona α,β-insaturada em 1727 cm-1.
Ao avaliarmos o eluente utilizado para purificação da lactona 2’,4’-sin-(+)-14,
verificamos que a mistura hexano/EtOAc 9:1 também proporciona uma boa separação
dos seus diasteroisômeros, segundo análises por CCD. A separação dos mesmos
poderia ser alcaçada caso possuíssemos uma maior quantidade de material.
4. Conclusão
Realizamos 10 das 14 etapas da síntese racêmica da Criptomoscatona D2 (13).
A primeira reação de alilação foi feita através da metodologia catalítica e assimétrica
desenvolvida por Keck9 e colaboradores em 75 % de rendimento e com um excesso
enantiomérico maior que 95%. O excesso enantiomérico foi determinado através da
análise dos espectros de 19F-RMN dos ésteres de Mosher derivados dos álcoois
homoalílicos (S)-17 e (+)-17. A segunda reação de alilação foi alcançada através da
utilização das metodologias de Paquette7 e Loh6 em respectivamente 97 e 92 % de
rendimento para duas etapas. Em ambos os casos observou-se a formação dos dois
possíveis isômeros sin-16 e anti-16 em uma proporção de 1:1 segundo análises de 13C-RMN dos dois compostos. A terceira reação de alilação foi feita em 94% de
rendimento e 2,1:1 r.d. para série anti e 81 % de rendimento e 2:1 r.d. para série sin,
utilizando-se BF3.Et2O e aliltri-n-butilestanho (23) em CH2Cl2 a -42°C.
A reação de metátese de olefinas realizada com o acrilato 2,4-sin-(+)-39 foi
efetuada em excelente rendimento. Embora não tenha sido possível separar os
isômeros 2,4-sin-(+)-39 formados após a terceira reação de alilação devido a pequena
quantidade de material disponível neste ponto da síntese sabe-se, através de análises
cromatográficas por CCD, de que é possível separar os isômeros utilizando-se uma
mistura de Hexano/EtOAc 9:1.
Através deste trabalho pôde-se mapear grande parte da síntese racêmica
proposta para a Criptomoscatona D2 (13).

51
5. Parte Experimental
5.1. Instrumentação
Todas as reações envolvendo reagentes sensíveis a umidade foram realizadas
sob atmosfera de argônio em um sistema previamente flambado. Todos os solventes
utilizados foram previamente secos.
As sílicas utilizadas nas colunas cromatográficas eram da ACROS (0.035–0.070
mm e 0.060–0200 mm). A alumina básica utilizada para o tratamento de alguns
solventes eram da ACROS (50 – 200 microns).
Os espectros de 1H-RMN e 13C-RMN foram obtidos em aparelhos Varian Gemini
300 e Inova 500. O padrão interno utilizado foi tetrametilsilano. Os solventes utilizados
foram clorofórmio ou benzeno deuterado. Os deslocamentos químicos (δ) foram
expressos em ppm e as constantes de acoplamento (J) em Hertz (Hz). As abreviações
utilizadas têm os seguintes significados: s (singleto), d (dubleto), t(tripleto), q (quarteto),
qt (quinteto) dd(duplo dubleto), dt (duplo tripleto), dq (duplo quarteto), ddd (duplo duplo
dubleto), ddt (duplo duplo tripleto), m(multipleto), sl (singleto largo). Os sobrescritos nas
constantes de acoplamento (1J, 2J, 3J, 4J) tratam-se das constantes a respectivamente
uma, duas, três e quatro ligações.
Os espectros de Infravermelho foram obtidos em aparelhos Nicolet Impact 410
(FTIR), utilizando-se cela de NaCl para filme.
As análises cromatográficas gasosas foram realizadas em um cromatógrafo
Hewlett Packard 5890 usando uma coluna semi-capilar HP-5 (5 % PhMe silicone, 30 m
x 0,53 mm x 1,3 µL). As condições de forno para análises cromatográficas em fase
gasosa foram de 100°C (1 min), 10°C min-1, 250 °C (20 min) e os volumes de amostra
injetados eram de aproximadamente 0,5 µL.
Os valores de rotação óptica foram obtidos em um polarímetro Polamat A (Carl
Zeiss/Jena).

52
5.2. Procedimentos Experimentais
5.2.1 Preparação de 1-benziloxiprop-2-eno, (18).
O
a b c
d
e
e'
H
H
À uma solução de NaH (510 mg; 21,3 mmol) em 17 ml de DMF a 0°C, adicionou-
se, gota a gota, o álcool benzílico (20) (1,00 mL; 9,70 mmoL). Após verificação do total
desprendimento de H2 através do borbulhardor adicionou-se à solução KI (423 mg; 2.50
mmol) e brometo de alila (21) (1.00mL; 11,5 mmol) previamente destilado. A reação foi
mantida sob agitação por cerca de 18 h e então diluída em CH2Cl2 (25 mL) e lavada
com água destilada (25mL). A fase aquosa foi extraída com CH2Cl2 (3 x 25 mL) e a fase
orgânica com água até remoção de boa parte do DMF. Os extratos orgânicos foram
secos sob MgSO4 e concentrados à vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica gel utilizando-se hexano/CH2Cl2 1:1 como eluente.
Produto final 18: 1,32 g; 8,90 mmol
Rendimento: 92 %
IV (filme): 3066, 3032, 2854, 1647, 1454, 1358, 1092, 922, 743 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,34 – 7,22 (m, 5H, Ha); 5,88 – 6,01 (m, 1H, Hd); 5,30 (dq, 2J 1,5Hz, 3J 17,2Hz, 1H, He); 5,21 (dq, 2J 1,5Hz, 3J 15,6Hz, 1H, He’); 4,52 (s, 2H, Hb);
4,02 ( dt, 4J1,5Hz, 3J 5,5Hz, 2H, Hc). 13C-RMN (75 MHz, CDCl3): δ 138,1; 134,6; 128,2; 127,6; 127,4; 117,0; 72,1; 71,1.
Rf: 0,51 (hexano/CH2Cl2 1:1)
CG/Tr (min): 2,750 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)

53
5.2.2. Preparação de 2-benziloxiacetaldeído, (19).
OH
O
a b cd
A uma solução de 1-benziloxiporp-2-eno (18) (1,02 g; 6,87 mmol) em 12 mL de
água e 12 mL de éter etílico foi adicionado 0.5 mL de uma solução de tetróxido 67
mg.mL-1 de ósmio em água destilada (330 mg; 0,13 mmol). Adicionou-se em seguida e
lentamente NaIO4 (3,18 g; 14,9 mmol) previamente macerado com o auxílio de um
almofariz e de um pistilo. A mistura reacional foi mantida sob agitação por cerca de 4 h
e então diluída em CH2Cl2 (30mL) e água destilada (20 mL). A fase aquosa foi extraída
com CH2Cl2 (3 x 30 mL). Os extratos orgânicos foram reunidos, secos sob MgSO4 e
concentrados à vácuo. O produto bruto de reação foi purificado por cromatografia em
coluna de sílica gel utilizando-se CH2Cl2 como eluente.
Produto final 19: 678 mg; 4,51 mmol
Rendimento: 67 %
IV (filme): 3032; 2920; 2862; 2711; 1736; 1462; 1377; 1120; 759 cm-1
1H-RMN (300 MHz, CDCl3): δ 9,69 (s,1H, Hd); 7,32 (m, 5H, Ha); 4,61 (s, 2H, Hb); 4,08 (s,
2H, Hc). 13C-RMN (75 MHz, CDCl3): δ 200,0; 136,6; 128,4; 128,0; 127,8; 75,2; 73,5.
Rf: 0,31 (CH2Cl2)
CG/Tr (min): 3,516 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
5.2.3. Preparação de 1-benziloxi-pent-4-en-2-ol, (17).
5.2.3.1. Versão Racêmica: Preparação de (+)-(17)
OOH
a b cd
e
f
g
h
h'
H
H
Preparou-se uma solução de 2-benziloxietanal (19) (256 mg; 1,70 mmol) em 20
mL de CH2Cl2 anidro a –78 °C. adicionou-se a essa solução TiCl4 (0,19 mL; 1,73 mmol)
e, após 10 min, aliltrimetilsilano (22) (0,32 mL; 2,05 mmol). A reação foi mantida sob

54
agitação por 40 min e após este período adicionou-se à mistura 17 mL de H2O
destilada,17 mL de CH2Cl2 e 10 mL de uma solução saturada de NaHCO3. A fase
aquosa foi extraída com CH2Cl2 (3 x 20 mL). Os extratos orgânicos foram reunidos,
secos sob MgSO4 e concentrados à vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica gel utilizando-se a hexano/EtOAc 8:2 como eluente.
Produto Final (+)-(17): 254 mg; 1,32 mmol
Rendimento: 77 %
5.2.3.2. Versão assimétrica: Preparação de (S)-(17)
OOH
a b cd
e
f
g
h
h'
H
H
A um balão previamente flambado e sob atmosfera de argônio adicionou-se (S)-
BINOL (54 mg ; 0,2 mmol) e 1,9 mL de CH2Cl2 seco. Após a dissolução do catalisador
adicionou-se à solução 760 mg de peneira molecular 4 Å ativada em forno a 250 °C por
24 h e armazenada em estufa. Adicionou-se à mistura reacional Ti (OiPr)4 (56 µL; 0,2
mmol) e mais 0,2 mL de CH2Cl2. após 1 h sob refluxo a reação foi levada a ta e neste
ponto adicionou-se o 2-benziloxiacetaldeído (19) (294 mg; 1,96 mmol) com o auxílio de
uma seringa, a qual foi posteriormente lavada com CH2Cl2 ( 2 x 0.3 mL). Cinco minutos
após a adição do aldeído, a reação foi resfriada a cerca –78 °C. Adicionou-se então o
aliltri-n-butilestanho (23) (0,74 mL; 2,35 mmol) e reação foi transferida para um freezer
aonde ficou, a -20°C por 1 semana. Após este período a reação foi finalizada
adicionando-se a ela uma solução saturada de NaHCO3 e mais 5 mL de CH2Cl2. Duas
horas depois a mistura foi filtrada à vácuo sob celite em um funil de placa sinterizada. A
fase aquosa foi extraída com CH2Cl2 (3 x 10 mL). Os extratos orgânicos foram reunidos,
secos sob MgSO4 e concentrados à vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica gel utilizando-se a princípio hexano como solvente a
fim de que fosse removido todo o resíduo de estanho da reação e em seguida
hexano/EtOAc 4:1.
Produto Final (S)-17: 283 mg; 1,47 mmol
Rendimento: 75 %, > 95 % e.e.

55
IV (filme): 3444; 3072; 3038; 2908; 2862; 1643; 1450; 1365; 1207; 1107; 914; 741; 698
cm-1
[αααα]D : - 2,18 (c 2.59, CHCl3) 1H-RMN (300 MHz, CDCl3): δ 7,39 – 7,24 (m, 5H, Ha); 5,80 – 5,74 (m, 1H, Hg); 5,14 –
5,05 (m, 2H, Hh); 4,54 (s, 2H, Hb); 3,90 – 3,83 (m, 1H, Hd); 3,52 – 3,33 (m, 2H, Hc); 2,55
(s, 1H, He); 2,26 (tt, 4J 1.1Hz, 3J 1.1Hz, Hf). 13C-RMN (75 MHz, CDCl3): δ 137.7; 134.0; 128.2; 127.5; 117.4; 73.8; 73.3; 69.6; 37.9.
Rf: 0,33 (hexano/EtOAc 4:1)
CG/Tr (min): 6,70 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
5.2.4. Preparação de (2S*,2’R)- 2- fenil-2-trifluorometil-2-metoxi etanoato de (1-
benziloxi)-pent-enila-2, (2S*,2’R)-(25).
OO
OCF3
OMePh
a b cd f
g
h
eH
a
Hg
A um balão previamente flambado e sob atmosfera de argônio adicionou-se o
álcool (S)-(17) (20 mg; 0.1 mmol), 0.3 mL de CH2Cl2 seco, DCC (20 mg; 0.092 mmol),
DMAP (ponta de espátula) e o (R)-MTPA (30 mg; 0.1 mmol). A reção foi mantida sob
agitação por cerca de 18h, diluída em CH2Cl2 (2 mL) e tratada com 1mL de uma
solução saturada de NaHCO3. A fase aquosa foi extraída com CH2Cl2 (3 x 3 mL). Os
extratos orgânicos foram reunidos, secos sob MgSO4 e concentrados à vácuo. O
produto bruto de reação foi purificado por cromatografia em coluna de sílica gel
utilizando-se a princípio hexano como solvente a fim de que fosse removido todo o
resíduo de estanho da reação e em seguida hexano/EtOAc 19:1.
Produto Final (2S,2’R)-(25): 20 mg; 0.049 mmol
Rendimento: 55 %
IV (filme): 2984; 2934; 2861; 1742; 1450; 1374; 1243; 1048; 919; 837; 735 cm-1

56
1H-RMN (500 MHz, CDCl3): δ 7,56 – 7,22 (m, 10H, Ha); 5,81 – 5,73 (m, 1H, Hf); 5,43 –
5,38 (m, 1H, Hg); 5,16 – 5,11 (m,1H,Hg’); 4,46 (d, 2J 11,9 Hz,1H, Hb); 4,41 (d, 2J 11,9
Hz, 1H, Hb’); 3,55 – 3,53 (m, 5H, Hc e Hh); 2,51 – 2,42 (m,2H,He). 19F (470 Hz, C6D6):::: δ -72,1; -72,3.
Rf: 0,21 (hexano/EtOAc 9.5:0.5)
CG/Tr (min): 15.1 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
5.2.5. Preparação 1-O-benzil-2-O-metoxietoximetil-1,2-dihidroxi-pent-4-eno, (26).
OO O
O
a b cd
e
f
g
h
i
jk
H Hh'
A um balão contendo o álcool (+)-(17) (49 mg; 0.3 mmol) em 0.4 mL de CH2Cl2
seco e DIPEA (50 µL; 0.3 mmol) a 0°C adicionou-se MEMCl (30 �L; 0.3 mmol). Após
cerca de 18 h a mistura foi diluída em CH2Cl2 (2 mL) e tratada com uma solução de
NaHCO3 10% (2 mL). A fase aquosa foi extraída com CH2Cl2 (3 x 5 mL). Os extratos
orgânicos foram reunidos, secos sob MgSO4 e concentrados à vácuo. O produto bruto
de reação foi purificado por cromatografia em coluna de sílica gel utilizando-se a
hexano/EtOAc 8:2 como eluente.
Produto Final (+)-26: 42 mg; 0.15 mmol
Rendimento: 58 %
IV (filme): 2922; 2879; 1453; 1365; 1201; 1177; 1111; 1039; 916; 850; 737; 698 cm_1
1H-RMN (300 MHz, CDCl3): δ 7,36 – 7,23 (m, 5H, Ha); 5,80 (ddt,3J 7,0 Hz, 10,0 Hz, 24,2
Hz, 1H, Hf); 5,11 – 5,02 (m, 2H, Hg); 4,82 (d, 2J 15,7 Hz, 1H, Hh); 4,79 (d, 2J 15,7 Hz,
1H, Hh); 4,57 – 4,49 (m, 2H, Hb); 3,90 – 3,83 (m, 1H, Hd); 3,73 – 3,60 (m, 2H, Hc); 3,52
– 3,40 (m, 4H, Hi e Hj); 3,37 (s, 3H, Hk); 2,41 – 2,30 (m,2H,He). 13C-RMN (75 MHz, CDCl3): δ 138.1; 134.3; 128.2; 127.4; 127.4; 117.2; 94.7; 75.5; 73.2;
72.1; 71.7; 66.9; 59.0; 36.5.
Rf: 0,40 (hexano/EtOAc 8:2)
CG/Tr (min): 11.8 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)

57
5.2.6. Preparação d o 4-O-benzil-3-O-metoxietoximetil-3,4-dihidroxi-butanal, (27).
O CHOO O
O
ab
cd f
gh
i j
eb'
g'
H H
H H
A um balão contendo o alceno 26 (70 mg; 0.3 mmol), 0.5 mL de Et2O e 0.5 mL de
água destilada adicionou-se 20 mL de uma solução de OsO4 67 mg mL-1 (1.7 mg; 0.01
mmol) em água. Em cerca de 3 min a reação passou de incolor a preta. Adicionou-se
NaIO4 (118 mg; 0.55 mmol) macerado com o auxílio de um almofariz e de um pistilo.
Lentamente a reação foi clareando até adquirir uma tonalidade branca. Após 5h sob
intensa agitação a mistura foi diluída em CH2Cl2 (3 mL) e água destilada (1 mL). A fase
aquosa foi extraída com CH2Cl2 (3 x 3 mL). Os extratos orgânicos foram reunidos,
secos sob MgSO4 e concentrados à vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica gel utilizando-se a hexano/EtOAc 1:1 como eluente.
Produto Final (+)-27: 52,0 mg; 0.18 mmol
Rendimento: 74 %
IV (filme): 3089; 3060; 3027; 2929; 2885; 2815; 2733; 1723; 1454; 1366; 1244; 1200;
1098; 1036; 849; 740; 700 cm_1
1H-RMN (300 MHz, CDCl3): δ 9,78 (t, 3J 1,8 Hz, 1H, Hf); 7,37 – 7,28 (m, 5H, Ha); 4,82
(d, 2J 14,6 Hz, 1H, Hg ou Hg’); 4,80 (d, 2J 14,6 Hz, 1H, Hg ou Hg’); 4,55 (d, 2J 14,3 Hz,
1H, Hb ou Hb’); 4,51 (d, 2J 14,3 Hz, 1H, Hb ou Hb’); 4,32 (qt, 3J 5,8 Hz,1H, Hd); 3,73 –
3,66 (m, 2H, Hc); 3,62 – 3,37 (m, 4H, Hh e Hi); 3,37 (s, 3H, Hj); 2,73 – 2,70 (m, 2H, He). 13C-RMN (75 MHz, CDCl3): δ 200,4; 137,7; 128,3; 127,6; 127,5; 95,1; 73,4; 71,8; 71,7;
67,2; 59,0; 46,6.
Rf: 0,48 (hexano/EtOAc 1:1)

58
5.2.7. Preparação de (+)-1-benziloxi-hep-6-en-2,4-diol, (+)-(16).
5.2.7.1. BF3.Et2O e alil-tri-n-butilestanho
OOH OH
a bc
dc'
ef
g
h
iHH
j k
A um balão contendo a olefina (+)-17 (99 mg; 0.5 mmol), 5.5 mL de Et2O e 5.5
mL H2O, 3 mL de uma solução de OsO4 67mg mL-1 em água destilada (17.3 mg; 0.07
mmol). Após 5 min a mistura reacional adquiriu uma coloração marron e, neste ponto
adicionou-se pouco a pouco NaIO4 (385 mg; 1.80 mmol) macerado com o auxílio de
uma almofariz e de um pistilo. Após 5 h a solução foi diluída em 10 mL de CHCl3 e 5 mL
de água destilada. A fase aquosa foi extraída com CHCl3 (5 x 10 mL). Os extratos
orgânicos foram reunidos, secos sob MaSO4 e concentrados rapidamente a vácuo. O
aldeído bruto 30 da reação foi imediatamente utilizado na reação de alilação seguinte.
A um balão contendo uma solução de 30 (~ 0.45 mmol) em CH2Cl2 (5 mL) a –42
°C adiconou-se BF3.Et2O (60 µL; 0.50 mmol) e, após 5 min, alil-tri-n-butilestanho (23)
(0.3 mL, 0.90 mmol). Após 3h, adicionou-se uma solução de NaOH 1M (10 mL) à
reação a qual foi mantida sob agitação rigorosa por 1h. A fase aquosa foi extraída com
EtOAc (4 x 8 mL) e os extratos orgânicos foram reunidos, secos sob MaSO4 e
concentrados a vácuo. O produto bruto de reação foi purificado por cromatografia em
coluna de sílica flash utilizando-se CH2Cl2/MeOH 9:1 como eluente.
Produto Final (+)-16: 33 mg; 0.14 mmol
Rendimento: 31 %
5.2.7.2. Índio Metálico
A um balão contedo o alceno (+)-1-benziloxi-pent-4-en-2-ol (+)-(17) (266 mG; 1,38
mmol) adicionou-se 16 mL de Et2O e 16 mL de água. A essa mistura adicionou-se 0,81
mL de uma solução de OsO4 62 mg mL-1 em água (50 mg; 0,2 mmol). Após 15 min, a
solução inicialmente incolor adquiriu uma cor escura. Neste ponto adicionou-se pouco a
pouco NaIO4 (1,112 g; 5,200 mmol) previamente macerado com o auxílio de um
almofariz e um pistilo. Cerca de 5h depois a mistura reacional havia adquirido uma

59
coloração amarelo-esbranquiçada e o consumo total do alceno (+)-(17) havia sido
averiguado através de análise por CCD. A mistura reacional foi então diluída em CH2Cl2
(5 mL) e a fase aquosa rapidamente extraída com CH2Cl2 (5 x 25 mL). Os extratos
orgânicos foram reunidos, secos sob MaSO4 e concentrados rapidamente a vácuo.
A um balão contendo raspas de índio metálico (233 mg; 2,03 mmol) adicionou-se
7,5 mL de água destilada, 7,5 mL de THF (previamente passado por uma coluna de
Al2O3 básica de modo a remover óxidos) e brometo de alila (21) (0,2 mL; 2,3 mmol).
Após 10 min adicionou-se 312 mg do aldeído bruto (+)-30 (aproximadamente 266 mg;
1,38 mmol do aldeído 30 assumindo-se um rendimento máximo de 100 % na reação
anterior). A reação foi mantida por cerca de 18h. Observou-se o total consumo de In (0)
e através de análise por CCD verificou-se o total consumo do material de partida. A
reação foi diluída em EtOAc (60 mL) e matida sob intensa agitação por cerca de 1h. A
fase aquosa foi então extraída com EtOAc (3 x 30 mL). Os extratos orgânicos foram
reunidos, secos sob MaSO4 e concentrados à vácuo. O produto bruto de reação foi
purificado por cromatografia em coluna de sílica flash utilizando-se a
hexano/EtOAc/Et3N 1,5:1,5:02 como eluente. Ao final foi possível isolar separadamente
os isômeros sin e anti-16 em uma proporção sin/anti 1:1 a qual havia sido inicialmente
determinada por análise do espectro de 13C-RMN de uma amostra contendo a mistura
dos dois diasteroisômeros.
Produto Final (+)-16: 302 mg ; 1,28 mmol (massa total)
Rendimento: 93 % para duas etapas, sin-16/anti-16 1:1
5.2.7.3. Estanho Metálico e InCl3
A um balão contendo a olefina (+)-17 (55 mg; 0,3 mmol), 0.7 mL de 1,4–dioxana,
2,0 mL H2O e 60 µL de 2.,6 lutidina adicionou-se
21 µL de uma solução de OsO4 62mg mL-1 em água destilada (1.4 mg; 0.005 mmol).
Após 5 min a mistura reacional adquiriu uma coloração rosada e, neste ponto
adicionou-se pouco a pouco NaIO4 (222 mg; 1.04 mmol) macerado com o auxílio de
uma almofariz e de um pistilo. Após 45 min a solução foi diluída em 5 mL de CHCl3 e 5
mL de água destilada. A fase aquosa foi extraída com CHCl3 (5 x 7 mL). Os extratos

60
orgânicos foram reunidos, secos sob MaSO4 e concentrados rapidamente a vácuo. O
aldeído bruto 30 da reação foi imediatamente utilizado na reação de alilação seguinte.
A um balão contendo uma solução de Sn (62 mg; 0,5 mmol), InCl3 (58 mg; 0,26
mmol) em H2O (2,6 mL), adicionou-se o brometo de alila (21) (45 µL; 0,52 mmol). Após
4h sob intensa agitação adicionou-se, com o auxílio de uma pipeta, essa mistura
reacional turva e viscosa ao balão contendo o aldeído bruto 30 a reação foi mantida por
2h e tratada com uma solução saturada de NaHCO3 (5mL). Após neutralização do meio
ácido a mistura foi diluída em EtOAc (8 mL) e mantida sob agitação por 30 min. A fase
aquosa foi extraída com EtOAc (4 x 8 mL) e os extratos orgânicos foram reunidos,
secos sob MaSO4 e concentrados a vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc/Et3N 7,5:7,5:1
como eluente. Ao final foi possível isolar separadamente os isômeros syn e anti em uma
proporção sin:anti 1:1 a qual foi determinada por análise do espectro de 13C-RMN de
uma amostra contendo a mistura dos dois diasteroisômeros.
Produto Final (+)-16: 58 mg; 0.25 mmol
Rendimento: 86 %
Sin-16:
IV (filme): 3406; 3068; 3030; 2910; 2862; 1641; 1496; 1452; 1365; 1315; 1217; 1099;
1028; 997; 916; 771; 739; 698 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,25 (m, 5H, Ha); 5,88 – 5,74 (m, 1H, Hh); 5,14 –
5,08 (m, 2H, Hi); 4,55 (s, 2H, Hb); 4,05 (sl, 2H, Hd ou Hf); 3,95 – 3,88 (m, 1H, Hd ou Hf);
3,45 (dd,2J 9,5Hz, 3J 4,0 Hz, 1H, Hc ou Hc’); 3,38 (dd, 2J 9,5Hz, 3J 7,0 Hz, 1H, Hc ou
Hc’); 3,29 (sl, 1H, Hj ou Hk); 3,21 (sl, 1H, Hj ou Hk); 2,24 (t, 3J 6,9 Hz, 2H, He); 1,62 –
1,50 (m, 2H, Hg). 13C-RMN (75 MHz, CDCl3): δ 137,7; 134,4; 128,3; 127,7; 127,6; 117,8; 74,3; 73,4; 71,2;
71,1; 42,3; 38,7.
Rf: 0,29 (hexano/EtOAc/Et3N 7,5:7,5:1)
CG/Tr (min): 11,05 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
EMAR (IE, 70 eV): m/z 236,14081 (valor calculado para C14H20O3 m/z 236, 14125)

61
Anti-16 :
IV (filme): 3406; 3066; 3030; 2976; 2912; 2860; 1641; 1496; 1454; 1435; 1363; 1311;
12.05; 1093; 1076; 1028; 997; 914; 833; 739; 698 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,27 (m, 5H, Ha); 5,88 – 5,74 (m, 1H, Hh); 5,15 –
5,09 (m, 2H, Hi); 4,55 (s, 2H Hb); 4,51 (sl, 2H, Hd ou Hf); 4,14 (sl, 1H, Hd ou Hf); 3,50
(dd,2J 9,5Hz, 3J 3,7 Hz, 1H, Hc ou Hc’); 3,40 (dd, 2J 9,5Hz, 3J 7,7 Hz, 1H, Hc ou Hc’);
2,87 (sl, 1H, Hj ou Hk); 2,57 (sl, 1H, Hj ou Hk); 2,28 – 2,18 (m, 2H, He); 1,70 – 1,50 (m,
2H, Hg). 13C-RMN (75 MHz, CDCl3): δ 137,7; 134,5; 128,3; 127,7; 127,6; 117,9; 74,4; 73,3; 67,9;
67,8; 42,2; 38,7.
Rf: 0,24 (hexano/EtOAc/Et3N 1,5:1,5:02)
CG/Tr (min): 11,05 (100°C por 1 min, 10°C min-1, 250 °C por 20 min).
EMAR (IE, 70 eV): m/z 236,14073 (valor calculado para C14H20O3 m/z 236, 14125)
5.2.8. Preparação do sin e do anti-1-O-benzil-2,4-O,O-isopropilideno-1,2,4-
trihidroxi-hept-6-eno, sin-(31) e anti-(31).
OO O
a cd f
e g
h
i
j k
HHb b'
A um balão contendo o diol sin-17 ou anti-17 (21 mg; 0.09 mmol) em 0.6 mL de
acetona seca adicionou-se 0.6 mL de 2,2-dimetoxipropano e uma ponta de espátula
de CSA. Após 2h de reação a mistura foi diluída em CHCl3 (2 mL) e tratada com uma
solução saturada de NaHCO3 (1mL). . A fase aquosa foi extraída com CHCl3 (3 x 3 mL)
e os extratos orgânicos foram reunidos, secos sob MaSO4 e concentrados a vácuo. O
produto bruto de reação foi purificado por cromatografia em coluna de sílica flash
dopada com Et3N utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final sin-31 ou anti-31: 22 mg; 0.080 mmol
Rendimento: 90 %

62
Sin-31
IV (filme): 3072; 2981; 2920; 2866; 1722; 1639; 1603; 1456; 1379; 1263; 1200; 1111;
1024; 912; 742 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,37 – 7,24 (m, 5H, Ha); 5,79 (ddt, 3J 6,9 Hz, 9,9 Hz e
17,2 Hz; 1H, Hh); 5,11 – 5,03 (m, 2H, Hi); 4,60 (d, 2J 12.,1Hz, 1H, Hb ou Hb’); 4.53 (d, 2J
12,1Hz, 1H, Hb ou Hb’); 4,12 – 4,03 (m, 1H, Hd ou Hf); 3,94 – 3,85 (m, 1H, Hd ou Hf);
3,50 (dd, 2J 9.9 Hz, 3J 5,9 Hz, 1H, Hc ou Hc’); 3,37 (dd, 2J 9,9 Hz, 3J 4,8 Hz, 1H, Hc ou
Hc’); 2,36 – 2,27 (m, 1H, Hg ou Hg’); 2,20 – 2,10 (m, 1H, Hg ou Hg’); 1,55 (dt, 3J 2,6 Hz, 2J
12,8 Hz 2H, He); 1,46 (s, 3H, Hi ou Hk); 1,42 (s, 3H, Hl ou Hk). 13C-RMN (125 MHz, CDCl3): δ 138,1; 134,0; 128,2; 127,6; 127,5 ; 117,1; 98,6; 73,7;
73,4; 68,5; 68,3 ;40,9; 33,4; 30,2; 19,9.
Rf: 0,43 (hexano/EtOAc 9:1)
CG/Tr (min): 10,64 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
EMAR (IE, 70 eV): m/z 276,17022 (valor calculado para C17H24O3 m/z 276,17255)
Anti-31
IV (filme): 3070; 3030; 2989; 2937; 2910; 2864; 1714; 1666; 1641; 1452; 1379; 1263;
1200; 1111; 1026; 995; 912; 751; 698 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,37 – 7,24 (m, 5H, Ha); 5,79 (ddt, 3J 7,0 Hz, 10,3 Hz e
17,3 Hz; 1H, Hh); 5,12 – 5,03 (m, 2H, Hi); 4,62 (d, 2J 12,1Hz, 1H, Hb ou Hb’); 4,54 (d, 2J
12,1Hz, 1H, Hb ou Hb’); 4,10 – 4,01 (m, 1H, Hd ou Hf); 3,91 – 3,82 (m, 1H, Hd ou Hf);
3,53 – 3,38 (m, 1H, Hc); 2,36 – 2,14 (m, 2H, Hg); 1,70 – 1,46 (m, 2H, He); 1,39 (s, 3H, Hi
ou Hk); 1,37 (s, 3H, Hl ou Hk). 13C-RMN (125 MHz, CDCl3): δ 138,2; 134,2; 128,2; 127,6; 127,5 ; 116,8; 100,3; 73,3;
72,7; 66,3; 66,2 ;40,2; 34,2; 25,0.
Rf: 0,43 (hexano/EtOAc 9:1)
CG/Tr (min): 10,64 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)
EMAR (IE, 70 eV): m/z 276,17072 (valor calculado para C17H24O3 m/z 276,17255)

63
5.2.9. Preparação do 4-alil-6-[(benziloxi)metil]-1,3-dioxan-2-ona, (+)-32.
OO O
O
a b c d fe g
h
i
A uma solução do diol (+)-17 (49 mg; 0.21 mmol) em 0.7 mL CH2Cl2 seco e
piridina seca (0.12 mL;1.47 mmol), resfriada a –70 °C canulou-se uma solução de
trisfogênio (33) (37 mg; 0.12 mmol) em 0.5 mL de CH2Cl2 seco. Rapidamente, trocou-se
o septo do balão por uma tampa de vidro e levou-se a temperatura da reação a ta. A
reação incolor tornou-se amarelada em 20 min e após este período observou-se o
consumo do material de partida através de análise por CCD. A reação diluída em
CH2Cl2 (2 mL) e tratada com 2 mL de uma solução saturada de NH4Cl. A fase aquosa
foi extraída com CH2Cl2 (3 x 4 mL) e os extratos orgânicos foram reunidos, secos sob
MgSO4 e concentrados a vácuo. O produto bruto de reação foi purificado por
cromatografia em coluna de sílica flash uilizando-se CH2Cl2/MeOH 19:1 como eluente.
Produto Final (+)-32: 38 mg; 0.15 mmol
Rendimento: 69 %
IV (filme): 3080; 3030; 2925; 2864; 1743; 1643; 1496; 1450; 1385; 1412; 1255; 1219;
1192; 1117; 1026; 920; 802; 741; 700 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,28 (m, 5H, Ha); 5,85 – 5,70 (m, 1H, Hh); 5,21 –
5,14 (m, 2H, Hi); 4,68 – 4,57 (m, 1H, Hd ou Hf); 4,56 (s,2H, Hb); 4,55 – 4,32 (m, 1H, Hd
ou Hf ); 3,67 (dd, 2J 4,4 Hz, 3J 2,0 Hz, 1H, Hc ou Hc’); 3,62 (dd, 2J 4,4 Hz, 3J 3,3 Hz, 1H,
Hc ou Hc’); 2,59 – 2,32 (m, 1H, He); 2,18 – 1,70 (m, 2H, Hg). 13C-RMN (125 MHz, CDCl3): δ 148,9; 148,8; 137,4; 137,2; 131,5; 131,2; 128,5; 128,4;
128,0; 127,9; 127,7; 127,7; 119,5; 77,7; 77,4; 75,6; 74,9; 73,7; 73,6; 70,8; 70,6; 39,3;
38,9; 29,1; 27,4.
Rf: 0,65 (CH2Cl2/MeOH 9.5:0.5)
CG/Tr (min): 10,5 (100°C por 1 min, 10°C min-1, 250 °C por 20 min)

64
5.2.10. Preparação de anti-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi) hept-6-eno,
anti-(37).
OPO OP
a b cd
gf
h
ie
* *
P=TBS
O diol anti-17 (104 mg, 0,44 mmol) foi dissolvido em 4 mL de CH2Cl2 seco. Foi
adicionado à solução 0,34 mL de 2,6-lutidina (311 mg, 2,90 mmol) e a mesma foi
resfriada a 0°C com o auxílio de um banho de gelo. O reagente TBSOTf (0,2 mL, 0,88
mmol) foi então lentamente adicionado à solução a qual foi mantida sob agitação a ta
por 12 h. A mistura reacional foi então diluída em CH2Cl2 e H2O. A fase aquosa foi
extraída com diclorometano (3 x 8 mL). Os extratos orgânicos foram reunidos, secos
sob Na2SO4, filtrados e concentrados a vácuo. O material bruto de reação foi purificado
por cromatografia em coluna de sílica gel utilizando-se hexano/EtOAc 18:1 como
eluente.
Produto Final anti-37: 190 mg, 0,41 mmol
Rendimento: 88 %.
IV (filme): 2954, 2929, 1893, 2856, 1472, 1361, 1253, 1099, 1004, 913, 834, 774, 734,
697 cm-1
1H-RMN (300 MHz, CdCl3): δ 7,35 – 7,29 (m, 5H, Ha); 5,86 – 5,74 (m, 1H, Hh); 5,07 –
5,02 (m, 2H, Hi); 4,52 (s, 2H, Hb); 4,00 – 3,86 (m, 2H, Hd e Hf); 3,44 – 3,35 (m, 2H, Hc);
2,27 – 2,23 (m, 2H, Hg); 1,65 – 1,61 (m, 2H, Hc); 0,89 – 0,88 (m, Ht-Bu); 0,08- 0,06 (m,
HSiCH). 13C-RMN (75 MHz, CDCl3): δ 138,45; 134,96; 128,24; 127,53; 127,40; 116,92; 75,41;
73,12; 69,49; 44,40; 42,72; 42,65; 25,94; 25,91; 25,70; 18,09; -2,96; -3,95; -4,00; -4,30;
-4,58.
Rf: 0,38 (Hexano/EtOAc 18:1)
CG / Tr (min): 15,8 (100°C por 1 min, 10°C/min; 250°C por 20 min)

65
5.2.11. Preparação sin-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi)-hept-6-eno, sin-
(37).
OPO OP
a b cd
gf
h
ie
* *
P=TBS
A uma balão sob atmosfera inerte adicionou-se o sin-17 (56 mg, 0.24 mmol), 2
mL de CH2Cl2 seco e 0.2 mL de 2,6-lutidina (180 mg, 1.68 mmol). Essa mistura foi
resfriada a 0°C com o auxílio de um banho de gelo e água e então adcionou-se 0.12 mL
de TBSOTf (180 mg, 1.68 mmol). A mistura foi mantida sob agitação a ta por 18h e
então diluída em CH2Cl2 (4 mL) e H2O (2 mL). A fase aquosa foi extraída com
diclorometano (3 x 4 mL). Os extratos orgânicos foram reunidos, secos sob Na2SO4,
filtrados e concentrados a vácuo. O material bruto de reação foi purificado por
cromatografia em coluna de sílica gel utilizando-se hexano/EtOAc 18:1 como eluente.
Produto Final sin-37: 102 mg, 0.22 mmol
Rendimento: 86 %
IV (filme): 2954, 2929, 1893, 2856, 1472, 1361, 1253, 1099, 1004, 913, 834, 774, 734,
697 cm-1
1H-RMN (300 MHz, CdCl3): δ 7,35 – 7,29 (m, 5H, Ha); 5,86 – 5,74 (m, 1H, Hh); 5,07 –
5,02 (m, 2H, Hi); 4,52 (s, 2H, Hb);4,00 – 3,86 (m, 2H, Hd e Hf);3,44 – 3,35 (m, 2H,
Hc);2,27 – 2,23 (m, 2H, Hg);1,65 – 1,61 (m, 2H, Hc);0,89 – 0,88 (m, Ht-Bu);0,08- 0,06 (m,
HSiCH). 13C-RMN (75 MHz, CDCl3): δ 138,45; 134,96; 128,24; 127,53; 127,40; 116,92; 75,41;
73,12; 69,49; 44,40; 42,72; 42,65; 25,94; 25,91; 25,70; 18,09; -2,96; -3,95; -4,00; -4,30;
-4,58.
Rf: 0,38 (Hexano/EtOAc 18:1)
CG / Tr (min): 15,8 (100°C por 1 min, 10°C/min; 250°C por 20 min)

66
5.2.12. Preparação anti-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi)-hexanal, anti-
(38).
OPO OP
Ha b cd
gf h
e
O* *
P=TBS
O alceno anti-37 (189 mg, 0.41 mmol) foi dissolvido em 3 mL de 1,4-dioxana e 1
mL de H2O destilada. Adicionou-se à solução 2,6-lutidina (0.1 mL, 0.8 mmol) e 30 µL de
uma solução de OsO4 67 mg/mL em H2O ( 2 mg, 0.01 mmol). Após 5 min, adicionou-se
rapidamente NaIO4 (348 mg, 1.63 mmol) previamente macerado. Após 5h de reação a
mistura foi diluída em CH2Cl2 (7 mL) e H2O (4 mL). A fase aquosa foi extraída com
CH2Cl2 (3 x 8 mL), os extratos orgânicos foram reunidos, secos sob MaSO4 e
concentrados a vácuo. O material bruto de reação foi purificado por cromatografia em
coluna de sílica flash utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final anti-(38): 150 mg, 0.32 mmol
Rendimento: 97%
IV (filme): 3023, 2954, 2925, 2891, 2852, 2713, 1732, 1468, 1363, 1254, 1103, 1007,
835, 808, 775, 737, 679 cm-1
1H-RMN (300 MHz, CDCl3): δ 9,83 – 9,79 (m, 1H, Hh); 7,58 – 7,29 (m, 5H, Ha); 4,52 (s,
2H, Hb); 4,40 – 4,32 (m, 1H, Hd ou Hf); 3,98 – 3,91 (m, 1H, Hd ou Hf); 3,46 – 3,36 (m, 2H,
Hc); 2,62 (ddd, 2J 15,7, 3J 5,1 e 2,2, 1H, He ou He’); 2,53 (ddd, 2J 15,7, 3J 6,3 e 2,9, 1H,
He e He’); 1,93 – 1,65 (m, 2H, Hg); 0,89 (s, HtBu), 0,88 (s, HtBu); 0,10 – 0,07 (m, Hi). 13C-RMN (75 MHz, CDCl3): δ 202,02; 138,19; 128,31; 127,58; 75,01; 73,26; 69,25;
66,06; 51,70; 43,70; 29,69; 25,89; 25,76; 18,14; 17,95; -4,09; -4,02; -4,42; -4,59.
Rf: 0,55 (Hexano/EtOAc 9:1)
CG/ Tr (min): 13,6 (100ºC por 1 min, 10°C/min, 250°C por 20 min

67
5.2.13. Preparação anti-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi)-hexanal sin-
(38).
OPO OP
Ha b cd
gf h
e
O* *
P=TBS
O alceno sin-37 (910 mg, 1.96 mmol) foi dissolvido em 15 mL de 1,4-dioxana e 5
mL de H2O destilada. Adicionou-se à solução 2,6-lutidina (0.5 mL, 3.9 mmol) e 0.2 mL
de uma solução de OsO4 62.5 mg/mL em H2O ( 10 mg, 0.04 mmol). Após 5 min,
adicionou-se rapidamente NaIO4 (1.675 g, 7.832 mmol) previamente macerado. Após
5h de reação a mistura foi diluída em CH2Cl2 (25 mL) e H2O (8 mL). A fase aquosa foi
extraída com CH2Cl2 (3 x 20 mL), os extratos orgânicos foram reunidos, secos sob
MgSO4 e concentrados a vácuo. O material bruto de reação foi purificado por
cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final sin-38: 870 mg, 1.86 mmol
Rendimento: 95 %
IV (filme): 2954, 2929, 2887, 2856, 2713, 1726, 1471, 1454, 1361, 1254, 1101, 1057,
1030, 1005, 835, 808, 775, 735, 696 cm-1
1H-RMN (300 MHz, CDCl3): δ 9,80 (dd, 3J 2,9 e 1,8 Hz, 1H, Hh); 7,38 – 7,29 (m, 5H, Ha);
4,53 (s, 2H, Hb); 3,94 – 3,86 (m, 1H, Hd ou Hf); 3,90 (qt, 1H, Hd ou Hf); 3,45 – 3,36 (m,
2H, Hc); 2,60 (ddd, 2J 15,6, 3J 4,4 e 2,2, 1H, Hg ou Hg’); 2,52 (ddd, 2J 15,6, 3J 7,0 e 2,9,
1H, Hg e Hg’); 1,77 (m, 2H, He); 0,90 – 0,88 (m, 18H, HtBu); 0,07 – 0,06 (m, 12H, SiMe).
13C-RMN (75 MHz, CDCl3): δ 201,94; 138,11; 128,24; 127,55; 127,50; 74,67; 73,36;
68,75; 65,55; 50,58; 42,88; 25,95; 25,84; 18,20; -4,06; -4,22; -4,59; -4,64.
Rf: 0,55 (Hexano/EtOAc 9:1)
CG/ Tr (min): 4,5 (200ºC por 1 min, 10°C/min, 250°C por 20 min)

68
5.2.14. Preparação 2,4-anti-(+)-1-benziloxi-2,4-bis-(terc-butildimetilsililoxi)-oct-8-en-
6-ol, 2,4-anti-(+)-(15).
5.2.14.1. Aliltri-n-butilestanho (22) e BF3.Et2O
OPO OP
a b cd
gf h
e
OH
i j k
l
* *
P=TBS
O aldeído anti-38 (137 mg, 0.29 mmol) foi dissolvido em CH2Cl2 seco (3 mL) e a
solução foi resfriada a -42°C com o auxílio de um banho de acetonitrila e gelo seco.
Adicionou-se BF3.Et2O (40 µL, 0.29 mmol) à solução e então aliltri-n-butilestanho (23).
Após 30 min a reação foi diluída em CH2Cl2 (3mL) e lavada com H2O (3mL). A fase
aquosa foi extraída com CH2Cl2 (3 x 6 ml) e os extratos orgânicos foram reunidos,
secos sob MgSO4 e concentrados a vácuo. O material bruto de reação foi purificado por
cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final 2,4-anti-(+)-(15): 140 mg, 0.28 mmol
Rendimento: 94 % e 2.1:1 r.d.
IV (filme): 3480, 2953, 2928, 2856, 1640, 1466, 1363, 1254, 1102, 1004, 913, 835, 776,
737, 698 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,29 (m, 5H, Ha); 5,91 – 5,76 (m, 1H, Hj); 5,14 –
5,09 (m, 2H, Hk); 4,53 (s, 1H, Hb); 4,49 – 3,77 (m, 4H, Hd, Hf, Hh, Hl); 3,41 – 3,38 (m, 2H,
Hc); 1,94 – 1,51 (m, 2H, Hc e Hg); 1,28 (t, 3J 7 Hz, 2H, Hi); 0,99 – 0,89 (m, HtBu); 0,69 –
0,03 (H, HSiCH).
13131313CCCC----RMN (75 MHz, CDClRMN (75 MHz, CDClRMN (75 MHz, CDClRMN (75 MHz, CDCl3333):):):): δ 138,2; 135,0; 134,9; 128,3; 127,6; 127,5; 117,5; 117,3;
75,0; 74,9; 73,3; 70,6; 69,8; 69,5; 69,6; 67,8; 44,3; 43,9; 42,3; 42,2; 42,1; 41,9; 29,7;
25,9; 25,8; 18,1; 17,9; -3,9; -4,1; -4,2; -4,5; -4,6; -4.6.
Rf: 0,38 (Hexano/EtOAc 9:1)
CG / Tr (min): 8,7 (200ºC por 1 min, 10°C/min, 250°C por 20 min)

69
5.2.14.2. Brometo de alilmagnésio em Et2O
A uma balão previamente flambando e sob atmosfera inerte adicionou-se o
aldeído anti-38 (15 mg, 0.03 mg), 2 mL de Et2O previamente seco e 0.04 mL solução
de brometo de alilmagnésio 1 M em Et2O (0.38 mmol). A mistura reacional foi mantida
sob intensa agitação por 1h. Após este período adicionou-se à solução 4 mL de CH2Cl2
e 2 mL de salmoura. A fase aquosa foi extraída com CH2Cl2 (3 x 4mL) e os extratos
orgânicos foram reuinidos, secos sob MgSO4, filtrados e concentrados vácuo. O
material bruto de reação foi purificado por cromatografia em coluna de sílica gel
utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final 2,4-anti-(+)-(15): 4 mg, 0.008 mmol
Rendimento: 25 %
5.2.14.3. Brometo de Índio metálico e brometo de alila
A um balão contendo In0 (6 mg, 0.048 mmol) e THF/H2O 50% (0.4 mL)
adicionou-se brometo de alila (4 µL, 0.048 mmol). Após 5 minutos transferiu-se essa
mistura reacional a um balão contendo o aldeído anti-38 (16 mg, 0.03 mmol). Após 30
minutos de reação, a mistura foi diluída em EtOAc (2 mL) e H2O (1 mL) mantida sob
agitação por 1h. A fase aquosa foi extraída com EtOAc (3 x 2 mL) e os extratos
orgânicos foram reunidos, secos sob MgSO4 e concentrados a vácuo. O material bruto
de reação foi purificado por cromatografia em coluna de sílica flash utilizando-se
hexano/EtOAc 9:1 como eluente.
Produto Final 2,4-anti-(+)-(15): 10 mg, 0.02 mmol
Rendimento: 63 %, 1:1 r.d.

70
5.2.15. Preparação do 2,4-sin-(+)-8-benziloxi-6,8-bis-(terc-butildimetilsililoxi)-oct-1-
em-4-ol, 2,4-sin-(+)-15.
OPO OP OH
a b cd
gf h
e i j k
l
* *
P=TBS
O aldeído sin-38 (870 mg, 1.86 mmol) foi dissolvido em CH2Cl2 seco (20 mL) e a
solução foi resfriada a -42°C com o auxílio de um banho de acetonitrila e gelo seco.
Adicionou-se BF3.Et2O (0.25 mL, 1.86 mmol) à solução e então aliltri-n-butilestanho.
Após 1,5 h a reação foi diluída em CH2Cl2 (5mL) e lavada com H2O (10mL). A fase
aquosa foi extraída com CH2Cl2 (3 x 20 ml) e os extratos orgânicos foram reunidos,
secos sob MgSO4 e concentrados a vácuo. O material bruto de reação foi purificado por
cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc 9:1 como eluente.
Produto Final 2,4-sin-(+)-15: 769 mg, 1.51 mmol
Rendimento: 81 %, 2:1 r.d.
IV (filme): 3491, 3066, 3030, 2956, 2929, 2897, 2856, 1728, 1641, 1471, 1464, 1410,
1387, 1361, 1101, 1028, 1005, 937, 914, 837, 808, 775, 735, 698, 667 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,29 (m, 5H, Ha); 5,91 – 5,76 (m, 1H, Hj); 5,14 –
5,08 (m, 2H, Hk); 5,52 (s, 2H, Hb); 4,27 – 4,21 (m, 1H, Hh); 4,19 – 4,02 (m, 1H, Hd); 3,88
– 3,76 (m, 1H, Hf); 3,54 – 3,31 (m, 2H, Hc); 2,33 – 2,13 (m, 1H, Hi); 1,96 – 1,64 (m, 1H,
Hi’); 1,49 – 1,32 (m, 2H, Hg); 1,27 – 1,02 (m, 2H, He); 0,94 – 0,89 (m, 18H, HSitBu); 0,13
– 0,05 (m, 12H, SiMe). 13C-RMN (75 MHz, CDCl3): δ138,2; 138,1; 138,0; 134,9; 128,3; 128,2; 127,6; 127,6;
127,5; 117,4; 117,3; 75,1; 75,0; 73,3; 73,3; 70,7; 70,5; 69,0; 68,9; 68,8; 67,6; 73,7; 72,
3; 42,2; 41,9; 70,8; 40,0; 25,8; 25,8; -4,0; -4,1; -4,2; -4,6; -4,7; -4,8; -4,9.
Rf: 0,38 (Hexano/EtOAc 9:1)
CG / Tr (min): 7,3 (200ºC por 1 min, 10°C/min, 250°C por 20 min)

71
5.2.16. Preparação [9-benziloxi-6,8-bis(terc-butyldimetilsililoxi)-non-1-em-4-il]
acrilato, 2,4-anti-(39).
OPO OP
a b cd
gf h
e
O
i j k
l
O
m
P=TBS
* *
Uma solução do álcool 2,4-anti-(+)-15 (110 mg, 0.22 mmol) em CH2Cl2 seco (0.5
mL) foi resfriada a 0°C com o auxílio de um banho de gelo e água. Adicionou-se à
solução o cloreto de acriloíla (32 µL, 0.389 mmol) e em seguida a base Et3N (0.1 mL,
0.778 mmol). A reação foi mantida sob agitação por 1h e então diluída em CH2Cl2 (2mL)
e H2O (1 mL). A fase aquosa foi extraída com CH2Cl2 (3 x 2 mL) e os extratos orgânicos
foram reunidos, secos sob MgSO4 e concentrados a vácuo. O material bruto de reação
foi purificado por cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc
4:1 como eluente.
Produto Final 2,4-sin-(+)-15: 92 mg, 0.16 mmol
Rendimento: 75 %
IV (filme): 3076, 3030, 2952, 2929, 2856, 2893, 1726, 1462, 1406, 1387, 1362, 1255,
1192, 1101, 1047, 1005, 968, 918, 835, 808, 775, 734, 698 cm-1
1H-RMN (300 MHz, CDCl3): δ�7.34 – 7.29 (m, 5H), 6.42 – 6.32 (m, 1H), 6.13 – 6.01 (m,
1H), 5.83 – 5.69 (m, 2H), 5.10 – 5.05 (m, 2H), 4.57 – 4.48 (m, 2H), 4.15 – 3.84 (m, 2H),
3.43 – 3.32 (m, 2H), 2.41 – 2.32 (m, 2H), 2.06 – 1.54 (m, 7H), 0.90 – 0.86 (m, 18H), 0.09
- -0.01 (m, 12H).
Rf: (Hexano/EtOAc 9:1)
CG / Tr (min): 10.67 (200ºC por 1 min, 10°C/min, 250°C por 20 min)

72
5.2.17. Preparação [9-benziloxi-6,8-bis(terc-butyldimetilsililoxi)-non-1-em-4-il]
acrilato, 2,4-sin-(39).
OOP OP O
O
a b cd
gf h
e i j k
lm
* *
P=TBS
Uma solução do álcool 2,4-sin-(+)-15 (720 mg, 1.415 mmol) em CH2Cl2 seco (2
mL) foi resfriada a 0°C com o auxílio de um banho de gelo e água. Adicionou-se à
solução o cloreto de acriloila (0,2 mL, 2,547 mmol) e em seguida a base Et3N (0,7 mL,
5,094 mmol). A reação foi mantida sob agitação por 1h e então diluída em CH2Cl2 (2mL)
e H2O (1 mL). A fase aquosa foi extraída com CH2Cl2 (3 x 5 mL) e os extratos orgânicos
foram reunidos, secos sob MgSO4 e concentrados a vácuo. O material bruto de reação
foi purificado por cromatografia em coluna de sílica flash utilizando-se hexano/EtOAc
4:1 como eluente.
Produto Final 2,4-sin-(39): 583 mg, 1,04 mmol
Rendimento: 73 %
IV (filme): 3078, 3030, 2956, 2954, 2927, 2893, 2856, 1726, 1637, 1620, 1471, 1462,
1406, 1387, 1361, 1296, 1255, 1192, 1097, 1065, 1047, 1005, 984, 970, 939, 918, 837,
808, 775, 735, 698, 678 cm-1
cm-1
1H-RMN (300 MHz, CDCl3): δ 7.38 – 7.29 (m, 5H, Ha); 6.42 – 6.32 (m, 1H, Hm); 6.13 –
6.01 (m, 1H, Hm’); 5.83 – 5.12 (m, 2H, Hj e Hl); 5.10 – 5.05 (m, 3H, Hk e Hh); 4.52 – 4.50
(m, 2H, Hb); 3.96 – 3.83 (m, 2H, Hd e Hf); 3.43 – 3.32 (m, 2H, Hc); 2.40 – 2.34 (m, 2H,
Hi); 2.32 – 1.62 (m, 4H, H2 e Hg); 0.89 – 0.88 (m, 18H, HtBu); 0.08 – -0.12 (m, 12H,
SiMe). 13C-RMN (75 MHz, CDCl3): δ 165.76, 165.45, 138.50, 138.30, 133.52, 133.37, 130.32,
128.87, 128.78, 128.31, 128.23, 127.61, 127.56, 127.53, 127.38, 117.90, 75.14, 74.92,
73.32, 73.19, 70.96, 70.64, 68.95, 68.74, 66.53, 65.83, 43.66, 42.20, 40.91, 40.79,
39.03, 25.91, 25.85, 18.04, 17.93, -4.11, -4.14, -4.25, -4.44, -4.53, -4.67, -4.78, -4.88.
Rf: 0.55 (Hexano/EtOAc 9:1)
CG / Tr (min): 11.53 (200ºC por 1 min, 10°C/min, 250°C por 20 min)

73
EMAR (70 eV): m/z 562.32829
5.2.18. Preparação da 2’,4’-sin-5-[6-(benziloxi)-2,4-bis(terc-butildimetilsililoxi)
pentil]-5,6-dihidropira-2-ona, 2’,4’-sin-(+)-14.
OPO OP O
O
a b cd
gf he i j
k
P=TBS
* *
A uma solução do éster 2,4-sin-39 (583 mg, 0.66 mmol) em 130 mL de CH2Cl2
seco adicionou-se o catalisador de Grubbs de 1ª geração (583 mg, 1.04 mmol). A
mistura reacional de cor roxa foi mantida sob refluxo e após 2.5h tornou-se marron. O
solvente foi evaporado e o material bruto de reação foi purificado por cromatografia em
coluna de sílica flash utilizando-se hexano/EtOAc 4:1 como eluente.
Produto final 2’,4’-sin-(+)-14: 458 mg, 0.86 mmol
Rendimento: 83 %
IV (filme): 3062, 3049, 2952, 2927, 2856, 2887, 1734, 1471, 1462, 1387, 1361, 1252,
1107, 1061, 1032, 1005, 837, 775, 737, 698, 663 cm-1
1H-RMN (300 MHz, CDCl3): δ 7,38 – 7,28 (m, 5H, Ha); 6,87 – 6,83 (m, 1H, Hj); 6,03 –
6,00 (m, 1H, Hk); 4,64 – 4,49 (m, 3H, Hb e Hh); 4,22 – 4,05 (m, 1H, Hd); 3,97 – 3,88 (m,
1H, Hf); 3,45 – 3,35 (m, 2H, Hc); 2,41 – 2,26 (m, 2H, Hi); 2,08 – 1,99 (m, 2H, Hg); 1,76 –
1,72 (m, 2H, Hl); 0,90 – 0,88 (m, 18H, HtBu); 0,10 – 0,05 (m, 12H, SiMe). 13C-RMN (75 MHz, CDCl3): δ 164,14; 163,90; 144,86; 144,70; 138,24; 128,21; 127,55;
127,45; 127,40; 121,51; 76,58; 74,86; 74,48; 73,34; 73,26; 68,55; 65,04; 43,46; 42,68;
42,00; 41,62; 30,16; 29,89; 25,97; 25,91; 25,76; 18,22; 18,14; 18,07; -4,01; -4,09; -4,14;
-4,21; -4,45; -4,50; -4,59.
Rf: 0,48 (Hexano/EtOAc 4:1)
CG / Tr (min): 7,4 (200ºC por 1 min, 10°C/min, 250°C por 20 min)
EMAR (70 eV): m/z 534,31340

74
6. Espectros
5.251.00
2.142.29
2.48
ppm-0123456789
Figura 22: Espectro de 1H-RMN (300 MHz, CDCl3) do composto 18.
ppm708090100110120130140
Figura 23: Espectro de 13C-RMN (75 MHz, CDCl3) do composto 18.
BnO
BnO

75
Figura 24: Espectro de IV do composto 18.
0.000.00
0.000.00
ppm-8-7-6-5-4-3-2-1-012
Figura 25: Espectro de 1H-RMN (300 MHz, CDCl3) do composto 19.
BnO
BnOH
O

76
ppm20406080100120140160180200
Figura 26: Espetro de 13C-RMN (75 MHz, CDCl3) do composto 19.
BnOH
O
Figura 27: Espetro de IV do composto 19.
BnOH
O
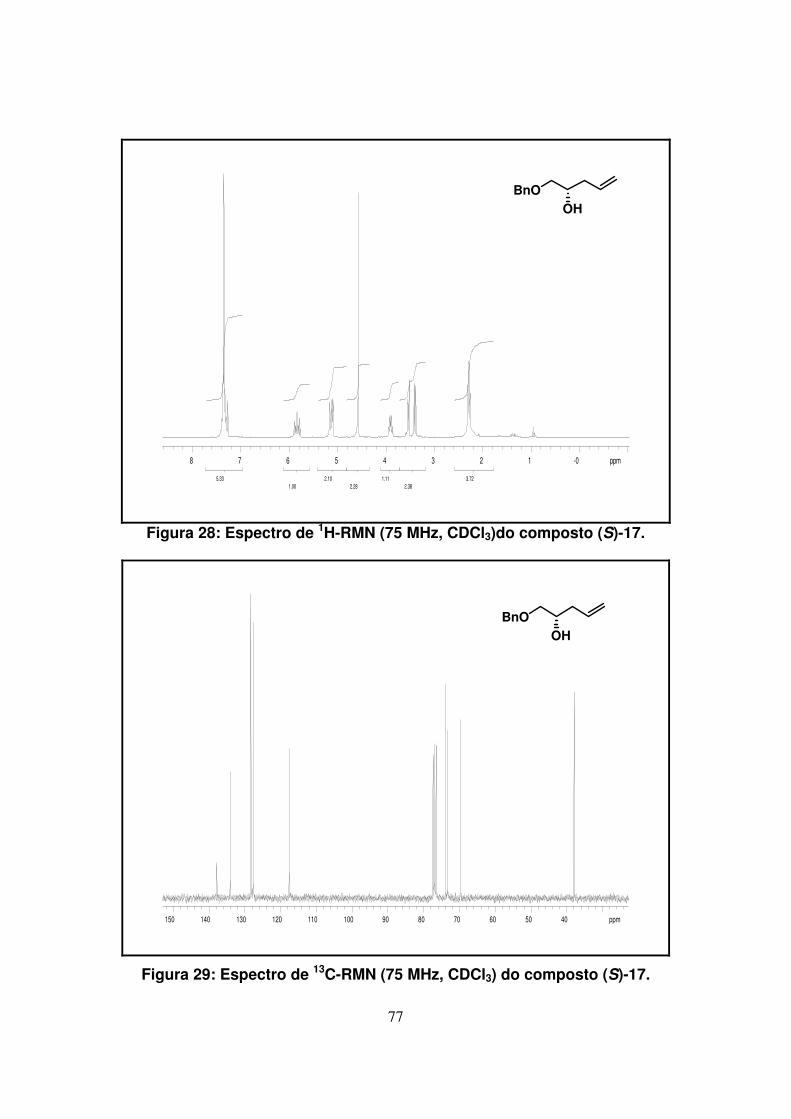
77
5.331.00
2.102.28
1.112.38
3.72
ppm-012345678
Figura 28: Espectro de 1H-RMN (75 MHz, CDCl3)do composto (S)-17.
ppm405060708090100110120130140150
Figura 29: Espectro de 13C-RMN (75 MHz, CDCl3) do composto (S)-17.
BnOOH
BnOOH

78
1.968.95
1.671.87
1.992.90
2.142.00
0.00
ppm-1-012345678910
Figura 31: Espectro de 1H-RMN (300 MHz, CDCl3) do composto (2S,2’R)-25.
BnOO
CF3
O
OMePh
Figura 30: Espectro de IV do composto (S)-17.
BnOOH
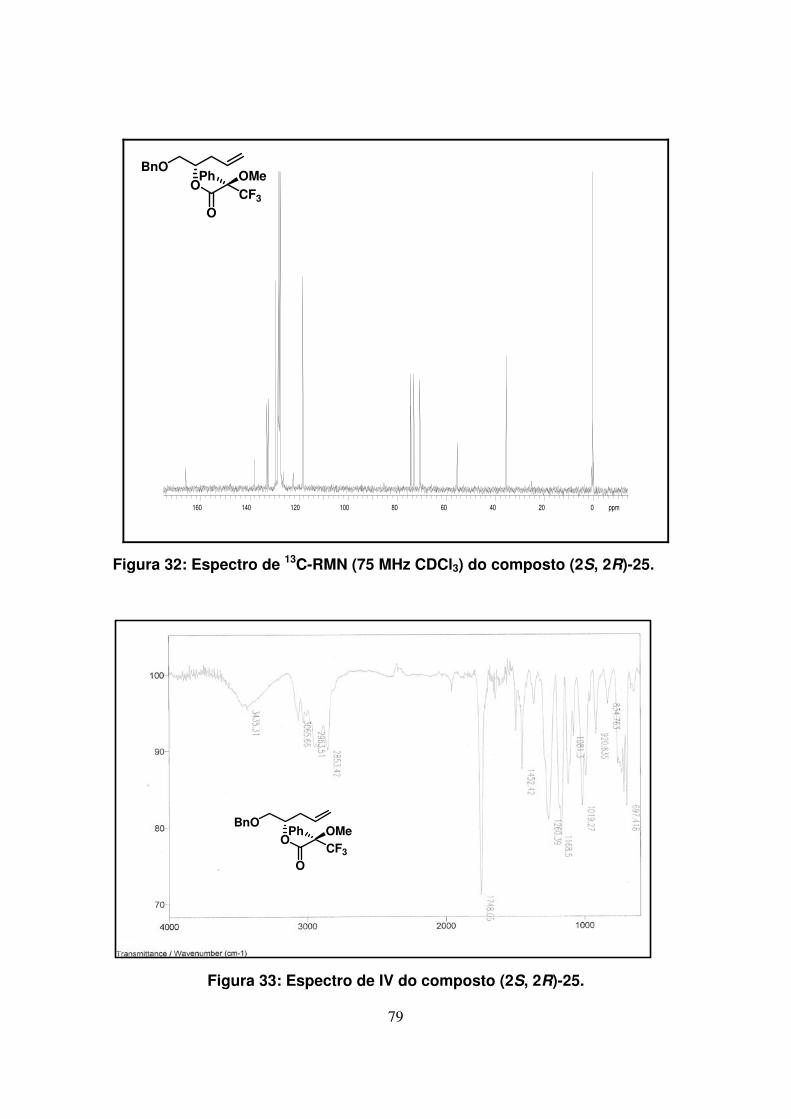
79
ppm020406080100120140160
Figura 33: Espectro de IV do composto (2S, 2R)-25.
BnOO
CF3
O
OMePh
Figura 32: Espectro de 13C-RMN (75 MHz CDCl3) do composto (2S, 2R)-25.
BnOO
CF3
O
OMePh
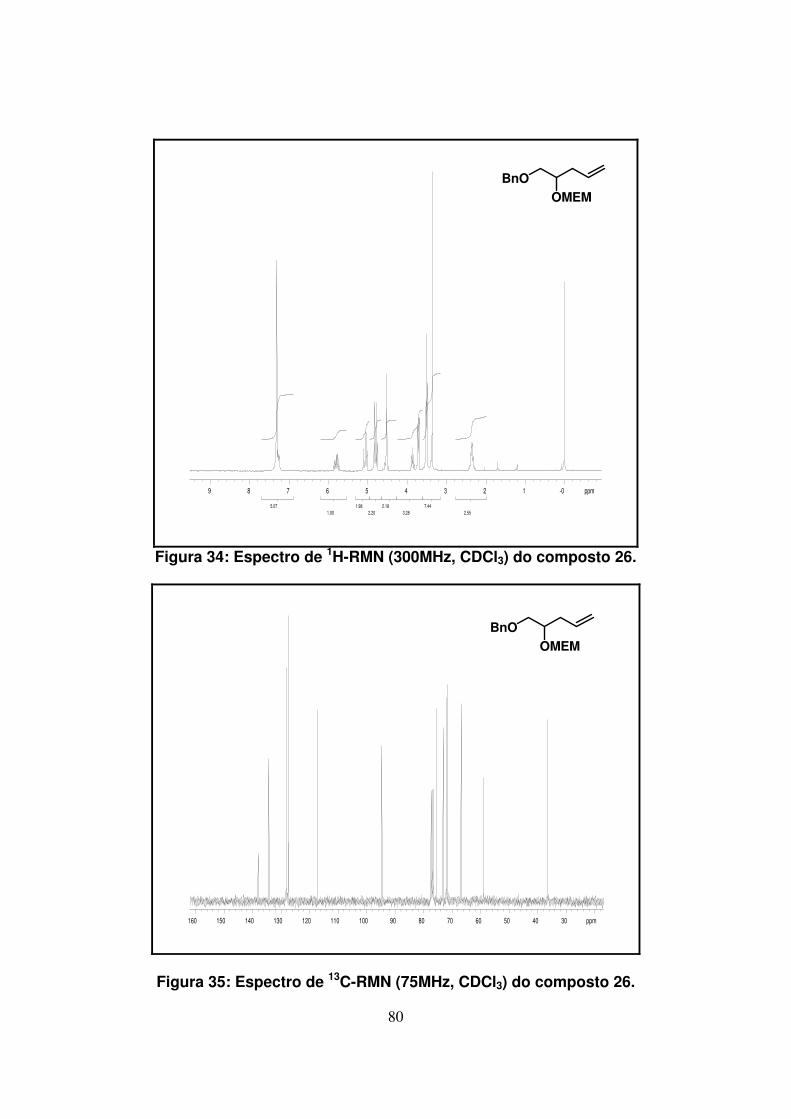
80
5.071.00
1.982.20
2.183.28
7.442.55
ppm-0123456789
Figura 34: Espectro de 1H-RMN (300MHz, CDCl3) do composto 26.
ppm30405060708090100110120130140150160
Figura 35: Espectro de 13C-RMN (75MHz, CDCl3) do composto 26.
BnOOMEM
BnOOMEM
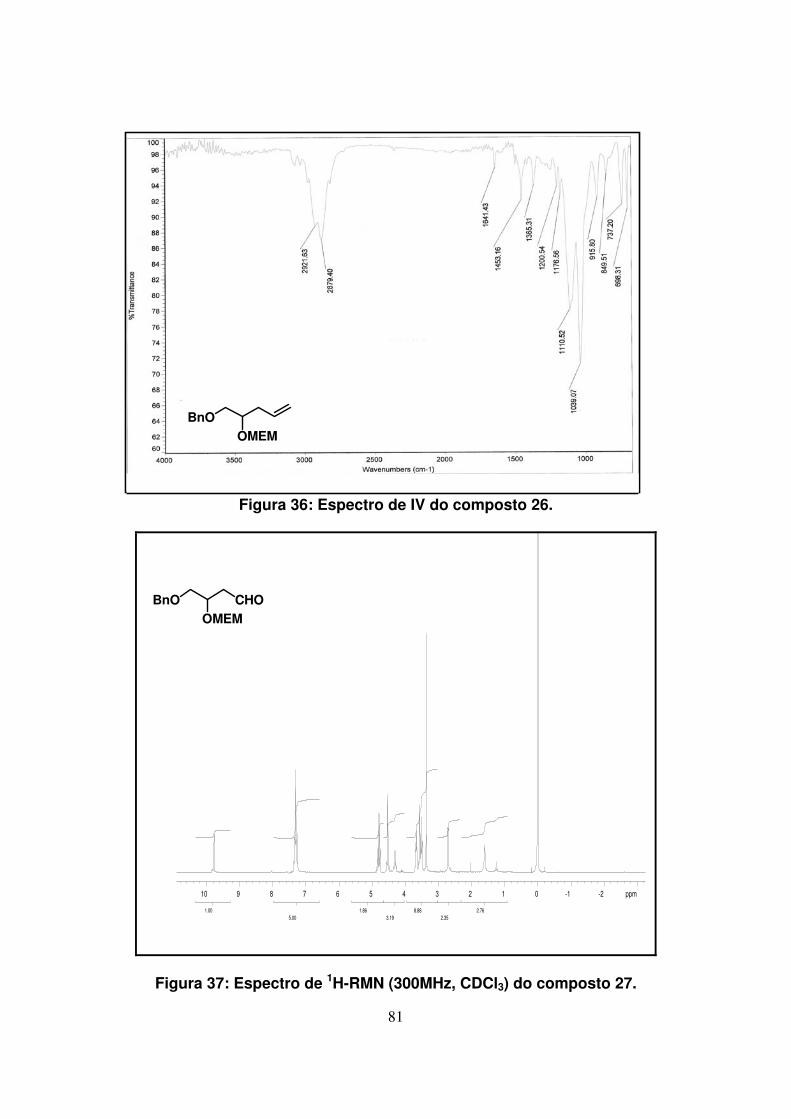
81
Figura 36: Espectro de IV do composto 26.
1.005.00
1.863.19
8.882.35
2.76
ppm-2-1012345678910
Figura 37: Espectro de 1H-RMN (300MHz, CDCl3) do composto 27.
BnOOMEM
CHO
BnOOMEM
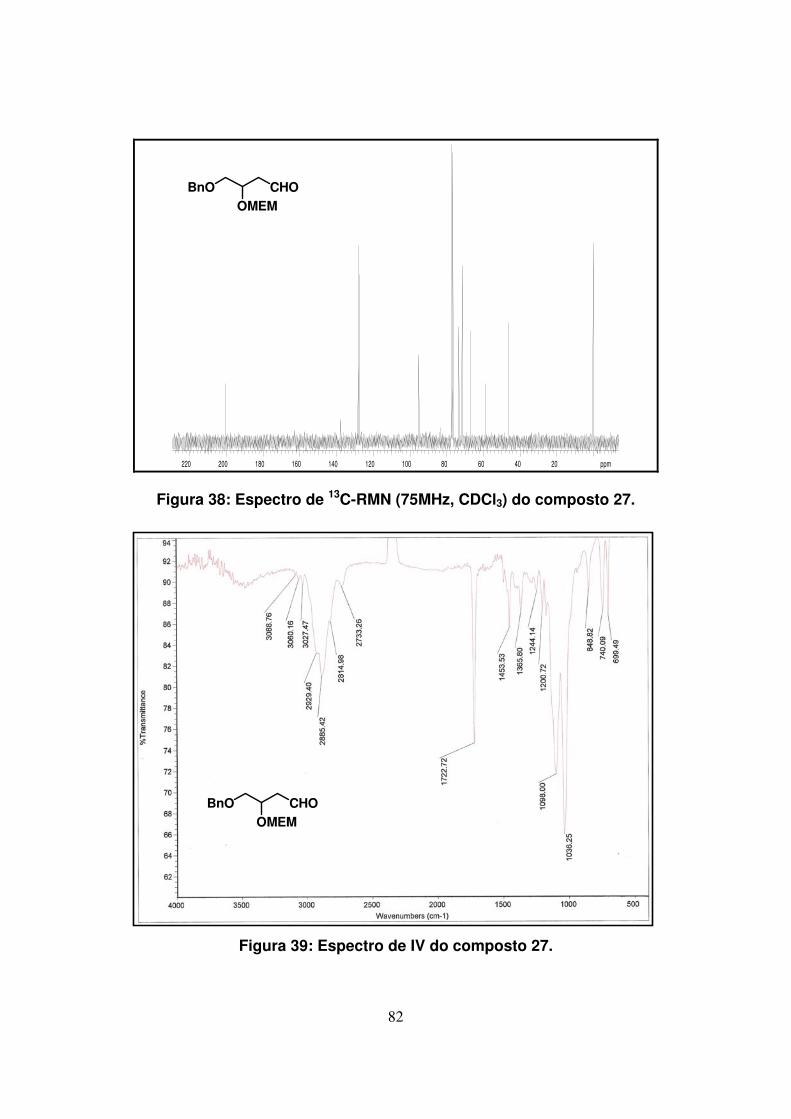
82
ppm20406080100120140160180200220
Figura 38: Espectro de 13C-RMN (75MHz, CDCl3) do composto 27.
Figura 39: Espectro de IV do composto 27.
BnOOMEM
CHO
BnOOMEM
CHO

83
5.281.00
2.152.30
2.332.58
2.132.51
3.48
ppm-1-012345678
Figura 40: Espectro de 1H-RMN (300MHz, CDCl3) do composto anti-16.
ppm405060708090100110120130140150160
Figura 41: Espectro de 13C-RMN (75MHz, CDCl3) do composto anti-16.
BnOOH OH
* *
BnOOH OH
* *
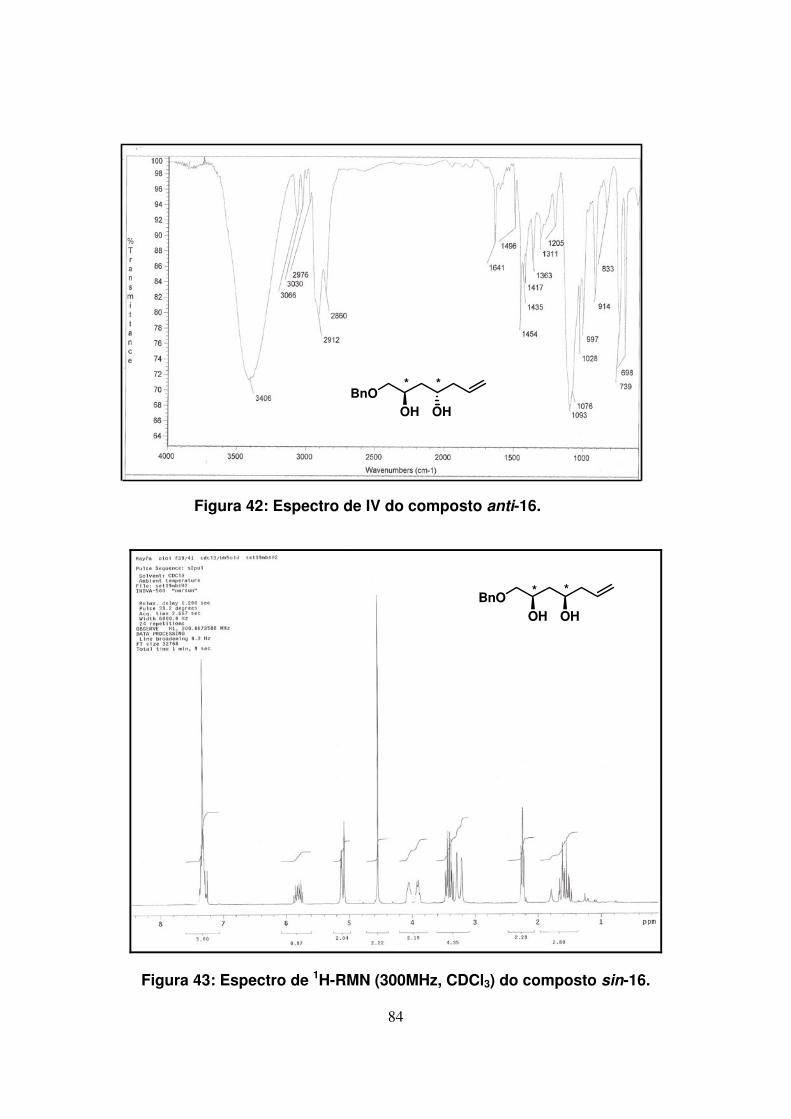
84
Figura 43: Espectro de 1H-RMN (300MHz, CDCl3) do composto sin-16.
Figura 42: Espectro de IV do composto anti-16.
BnOOH OH
* *
BnOOH OH
* *

85
ppm30405060708090100110120130140150160
Figura 44: Espectro de 13C-RMN (75MHz, CDCl3) do composto sin-16.
Figura 45: Espec
Figura 45: Espectro de IV do composto sin-16.
BnOOH OH
* *
BnOOH OH
* *
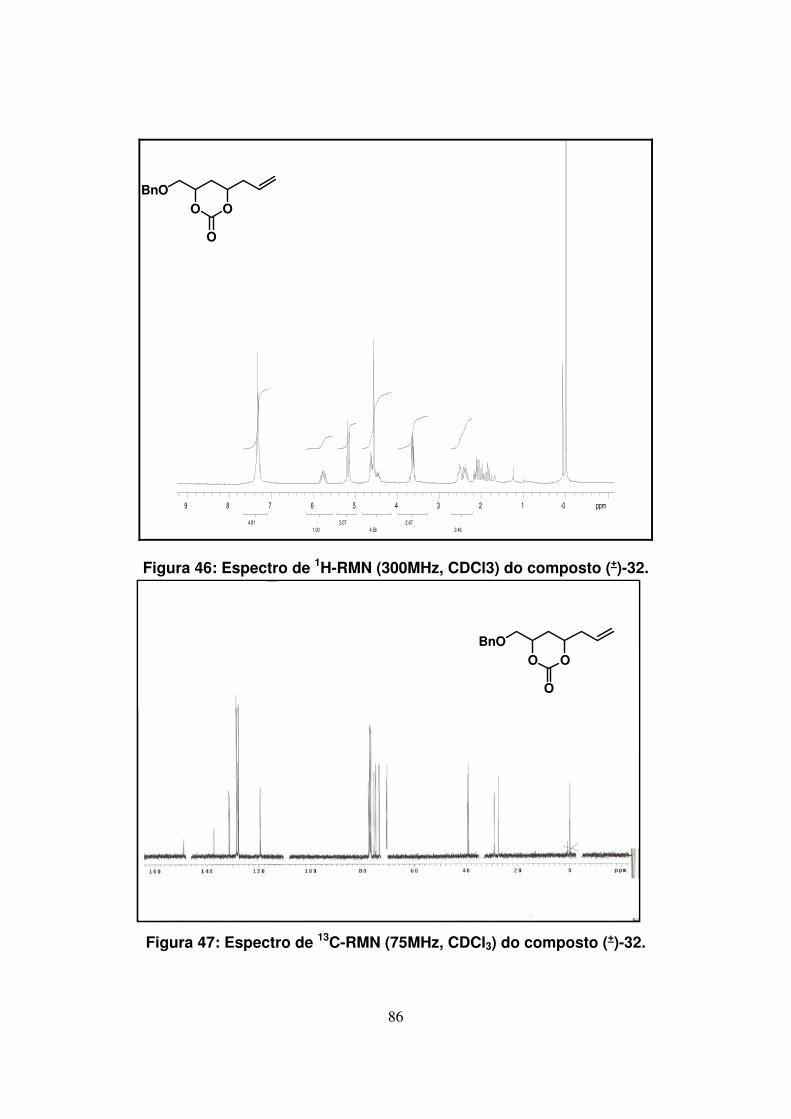
86
4.811.00
2.074.56
2.672.46
ppm-0123456789
Figura 46: Espectro de 1H-RMN (300MHz, CDCl3) do composto (+)-32.
Figura 47: Espectro de 13C-RMN (75MHz, CDCl3) do composto (+)-32.
BnOO O
O
BnOO O
O

87
5.341.00
2.182.33
2.482.53
2.7113.46
ppm-012345678910
Figura 49: Espectro de 13C-RMN (75 MHz, CDCl3) do composto anti-31.
Figura 48: Espectro de IV do composto (+)-32.
BnOO O
O
BnOO O
* *

88
ppm020406080100120140160
Figura 50: Espectro de 13C-RMN (75MHz, CDCl3) do composto anti-31.
BnOO O
* *
Figura 51: Espectro de IV do composto anti-31.
BnOO O
* *
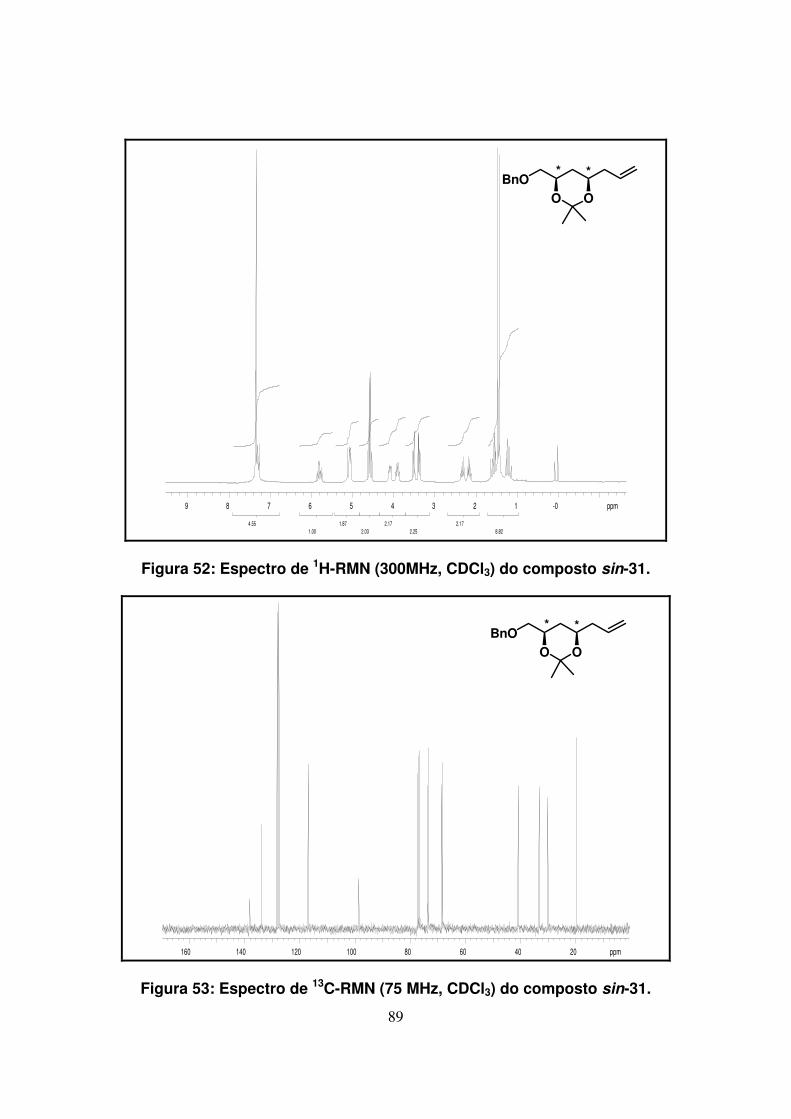
89
4.551.00
1.872.03
2.172.25
2.178.82
ppm-0123456789
Figura 52: Espectro de 1H-RMN (300MHz, CDCl3) do composto sin-31.
ppm20406080100120140160
Figura 53: Espectro de 13C-RMN (75 MHz, CDCl3) do composto sin-31.
BnOO O
* *
BnOO O
* *
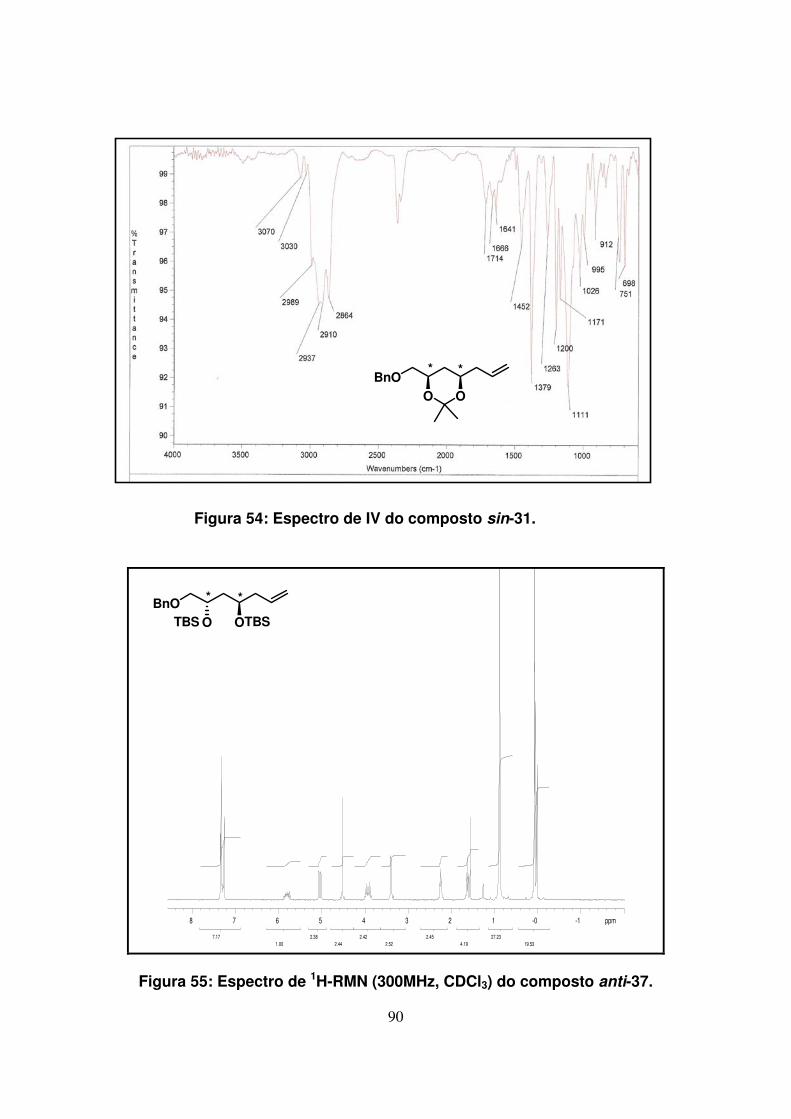
90
7.171.00
2.382.44
2.422.52
2.454.19
27.2319.53
ppm-1-012345678
Figura 55: Espectro de 1H-RMN (300MHz, CDCl3) do composto anti-37.
BnOO OTBS TBS
* *
Figura 54: Espectro de IV do composto sin-31.
BnOO O
* *
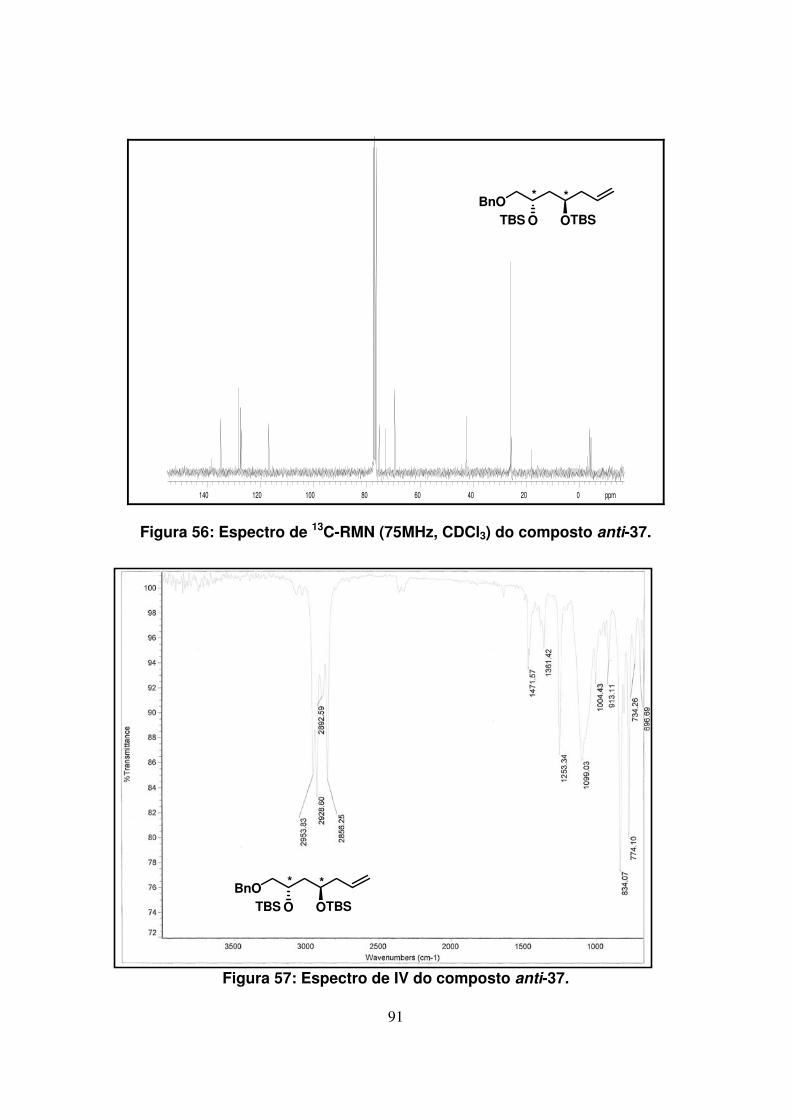
91
ppm020406080100120140
Figura 56: Espectro de 13C-RMN (75MHz, CDCl3) do composto anti-37.
Figura 57: Espectro de IV do composto anti-37.
BnOO OTBS TBS
* *
BnOO OTBS TBS
* *
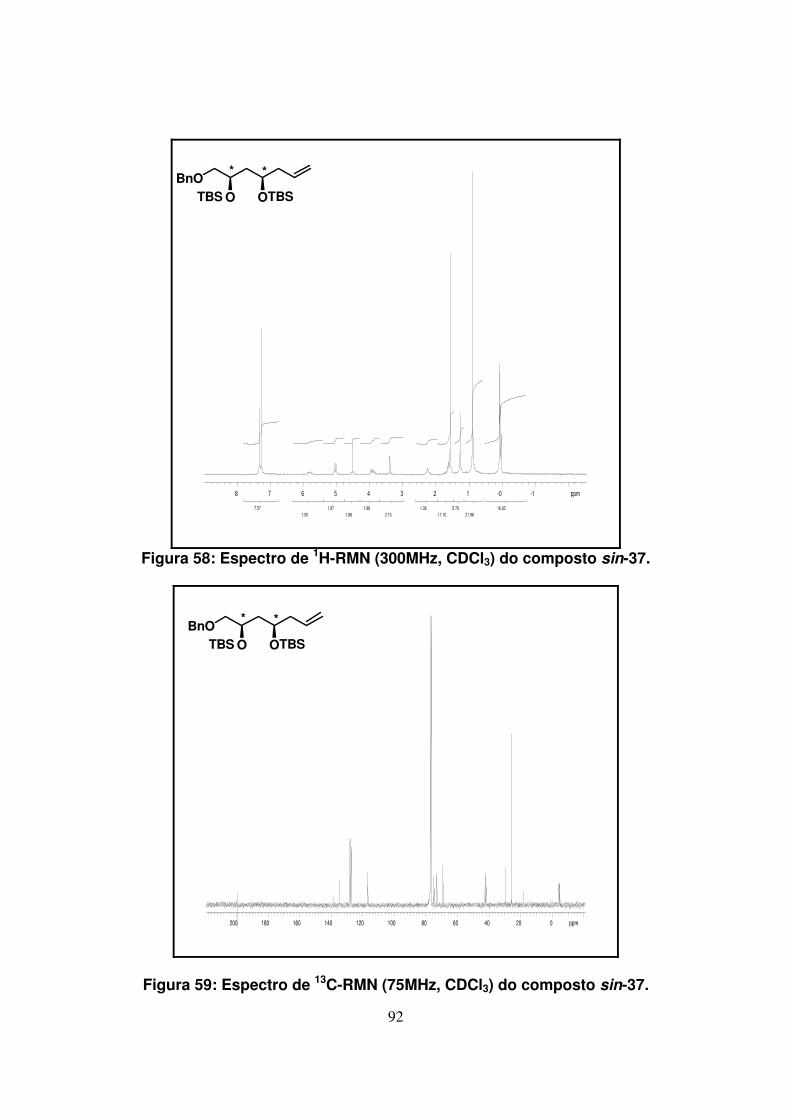
92
7.571.00
1.971.88
1.982.15
1.3611.10
5.7921.98
16.83
ppm-1-012345678
Figura 58: Espectro de 1H-RMN (300MHz, CDCl3) do composto sin-37.
ppm020406080100120140160180200
Figura 59: Espectro de 13C-RMN (75MHz, CDCl3) do composto sin-37.
BnOO OTBS TBS
* *
BnOO OTBS TBS
* *
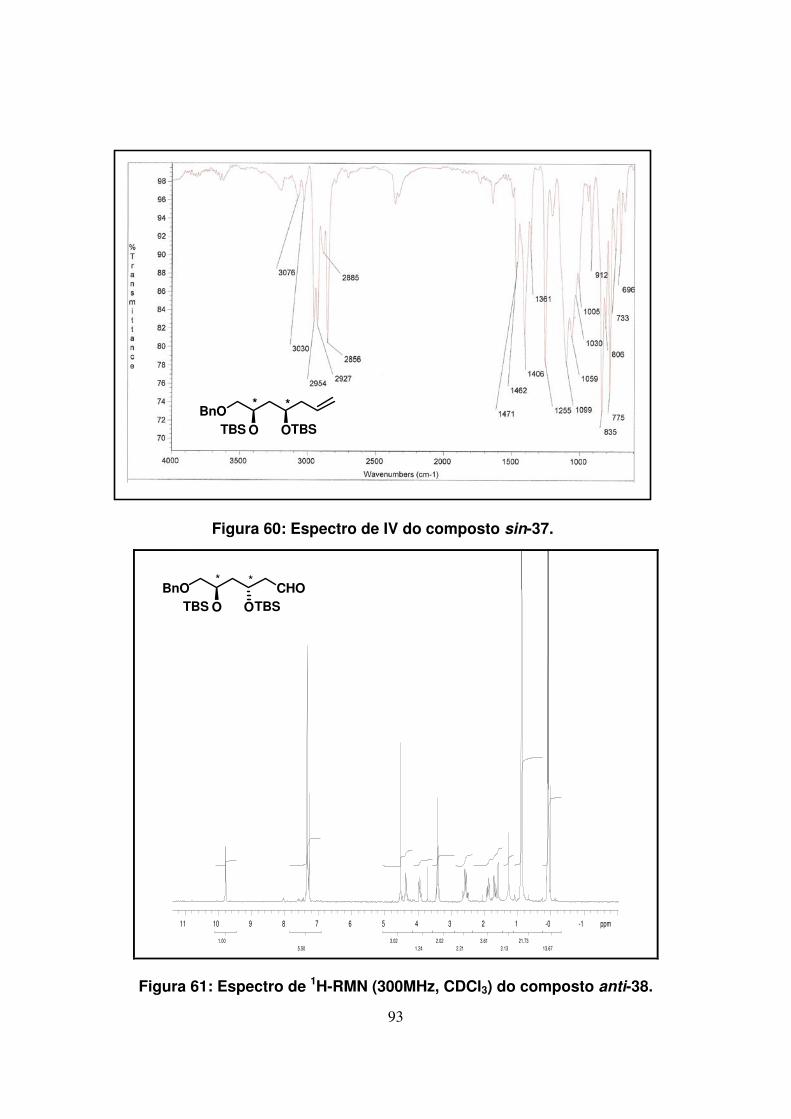
93
1.005.50
3.021.24
2.022.21
3.612.13
21.7313.67
ppm-1-01234567891011
Figura 61: Espectro de 1H-RMN (300MHz, CDCl3) do composto anti-38.
BnOO OTBS TBS
CHO* *
Figura 60: Espectro de IV do composto sin-37.
BnOO OTBS TBS
* *

94
ppm20406080100120140160180200
Figura 62: Espectro de 13C-RMN (75MHz, CdCl3) do composto anti-38.
Figura 63: Espectro de IV do composto anti-38
BnOO OTBS TBS
CHO* *
BnOO OTBS TBS
CHO* *

95
1.005.94
2.941.06
2.171.87
5.0923.19
20.97
ppm-1-01234567891011
Figura 64: Espectro de 1H-RMN (300MHz, CDCl3) do composto sin-38.
ppm020406080100120140160180200
Figura 65: Espectro de 13C-RMN (75MHz, CDCl3) do composto sin-38.
BnO CHOO OTBS TBS
* *
BnO CHOO OTBS TBS
* *
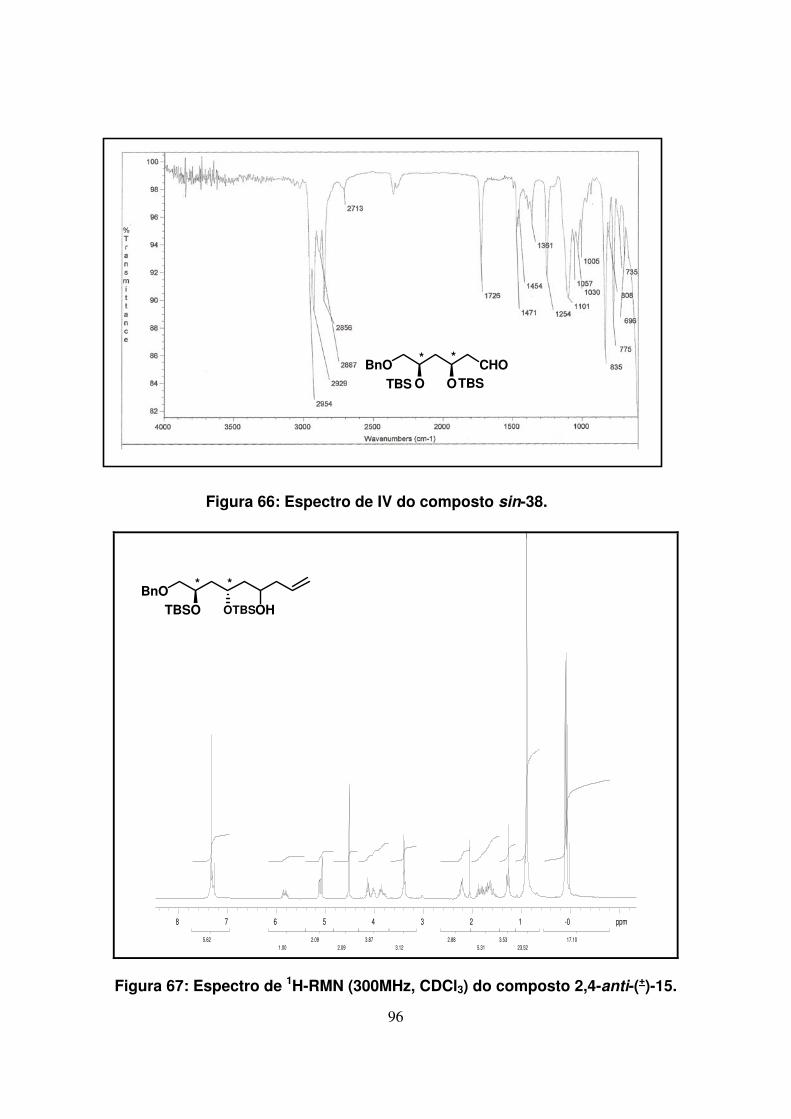
96
5.621.00
2.092.09
3.873.12
2.885.31
3.5323.52
17.10
ppm-012345678
Figura 67: Espectro de 1H-RMN (300MHz, CDCl3) do composto 2,4-anti-(+)-15.
BnOTBSO OTBSOH
* *
Figura 66: Espectro de IV do composto sin-38.
BnO CHOO OTBS TBS
* *

97
ppm020406080100120140
Figura 68: Espectro de 13C-RMN (300MHz, CDCl3) do composto 2,4-anti-(+)-15.
Figura 69: Espectro de IV do composto 2,4-anti-(+)-15.
BnOTBSO OTBSOH
* *
BnOTBSO OTBSOH
* *
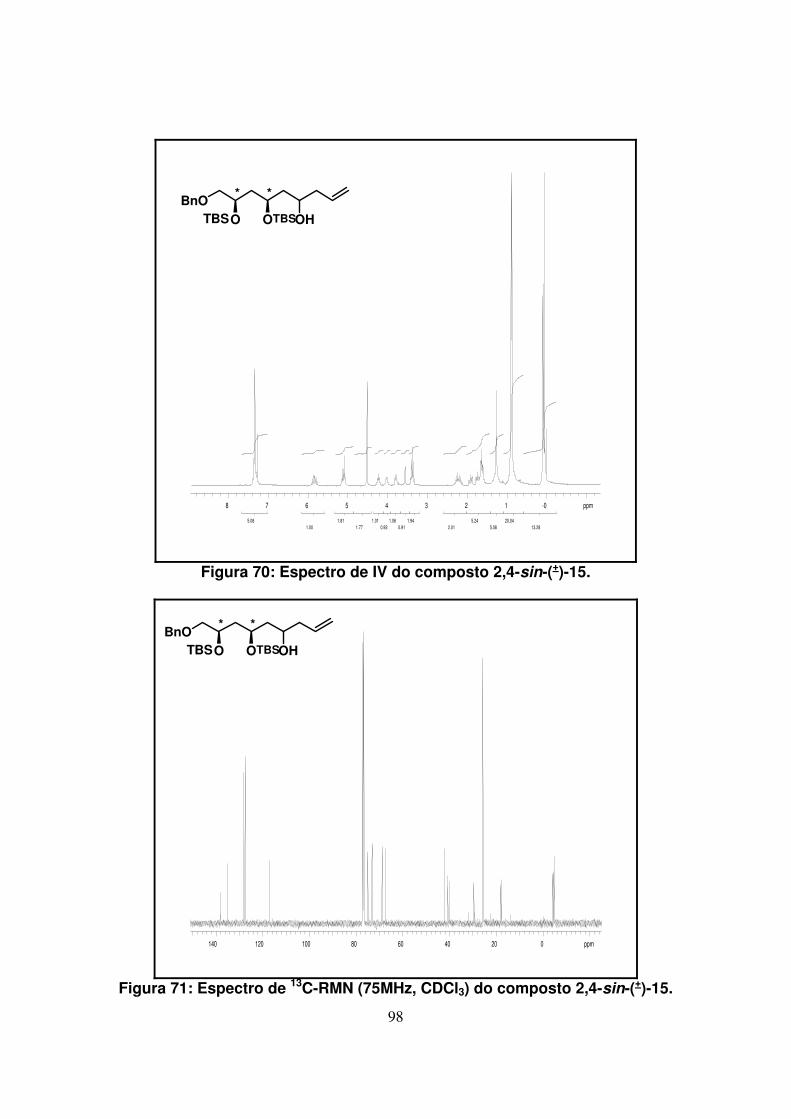
98
5.081.00
1.811.77
1.010.93
1.060.91
1.942.01
5.245.08
20.0413.28
ppm-012345678
Figura 70: Espectro de IV do composto 2,4-sin-(+)-15.
ppm020406080100120140
Figura 71: Espectro de 13C-RMN (75MHz, CDCl3) do composto 2,4-sin-(+)-15.
BnOO OTBS TBSOH
* *
BnOO OTBS TBSOH
* *
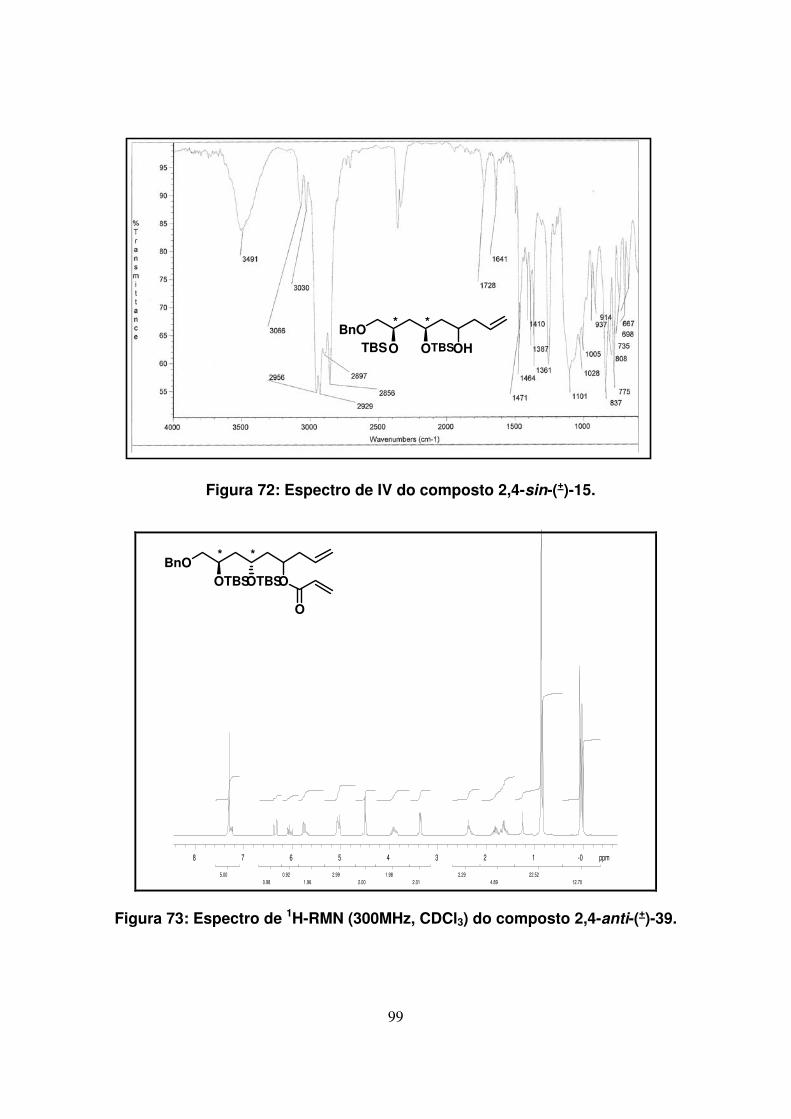
99
5.000.98
0.921.96
2.992.00
1.982.01
2.294.89
22.5212.70
ppm-012345678
Figura 73: Espectro de 1H-RMN (300MHz, CDCl3) do composto 2,4-anti-(+)-39.
Figura 72: Espectro de IV do composto 2,4-sin-(+)-15.
BnOO OTBS TBSOH
* *
BnOOTBSOTBSO
O
* *

100
ppm020406080100120140160
Figura 74: Espectro de 13C-RMN (75MHz, CDCl3) do composto 2,4-anti-(+)-39.
Figura 75: Espectro de IV do composto 2,4-anti-(+)-39.
BnOOTBSOTBSO
O
* *
BnOOTBSOTBSO
O
* *

101
5.120.99
0.861.77
2.702.00
2.102.11
1.935.97
16.7911.34
ppm-012345678
Figura 76: Espectro de 1H-RMN (300MHz, CDCl3) do composto 2,4-sin-(+)-39.
ppm020406080100120140160180
Figura 77: Espectro de 13C-RMN (75MHz, CDCl3) do composto 2,4-sin-(+)-39.
BnOO OTBS TBSO
O
* *
BnOO OTBS TBSO
O
* *

102
3.151.00
0.861.77
1.611.48
4.5512.72
10.53
ppm-0123456789
Figura 79: Espectro de 1H-RMN (300MHz, CDCl3) do composto 2’,4’-sin-(+)-14.
BnOO OTBS TBSO
O
* *
Figura 78: Espectro de IV do composto 2,4-sin-(+)-39.
BnOO OTBS TBSO
O
* *
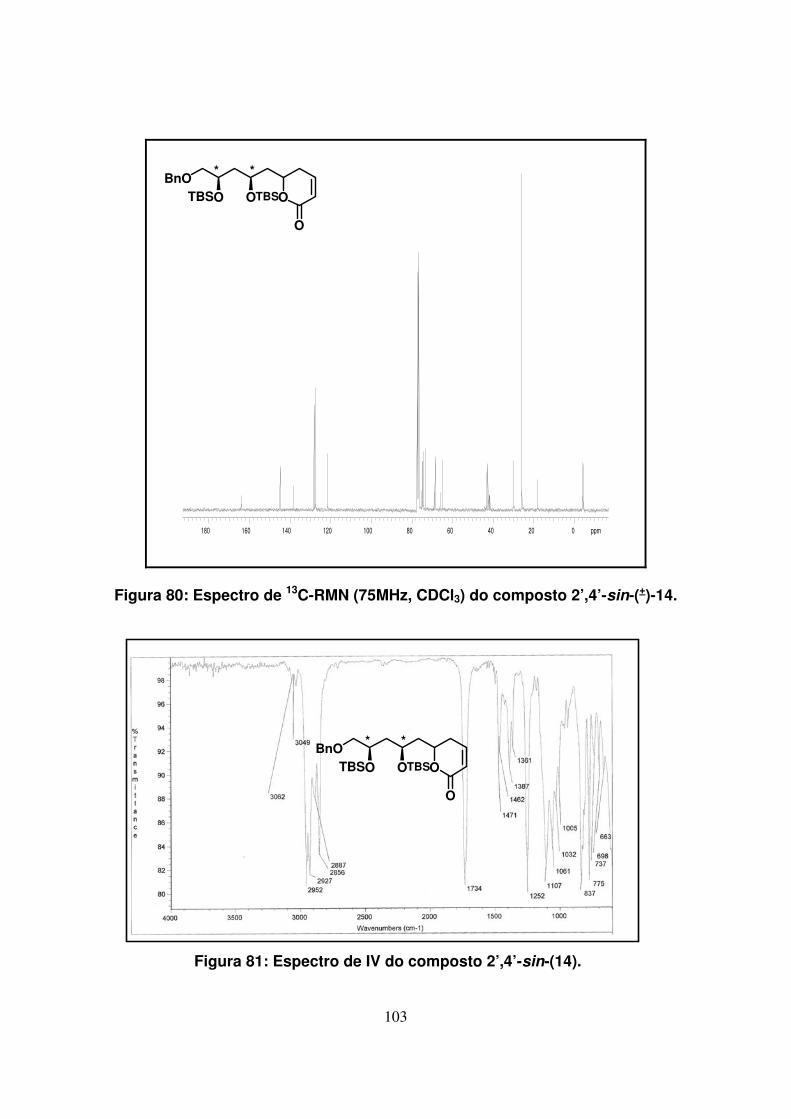
103
ppm020406080100120140160180
Figura 80: Espectro de 13C-RMN (75MHz, CDCl3) do composto 2’,4’-sin-(+)-14.
BnOO OTBS TBSO
O
* *
Figura 81: Espectro de IV do composto 2’,4’-sin-(14).
BnOO OTBS TBSO
O
* *






![DISSERTAÇÃO DE MESTRADO - UNICAMPbiq.iqm.unicamp.br/arquivos/teses/vtls000415865.pdf · DABCO 1,4-diazobiciclo[2.2.2]octano DIBAL hidreto de diisobutil alumínio ... PMB p-metoxi](https://img.document.onl/doc/110x75/5bf9f11609d3f2e7208cd4dd/dissertacao-de-mestrado-dabco-14-diazobiciclo222octano-dibal-hidreto.jpg)






















