Embed Size (px)
Citation preview

Publicado em:
Deficiência Mental: Abordagem Multidisciplinar
Fascículo no. 4, 2005
APAE de São Paulo
ENFOQUE GENÉTICO DA CRIANÇA COM DEFICIÊNCIA
MENTAL E SUA FAMÍLIA
Prof. Dr. Sérgio D.J. Pena
GENE – Núcleo de Genética Médica de Minas Gerais
Professor Titular do Departamento de Bioquímica e Imunologia da UFMG
Endereço para correspondência:
Dr. Sérgio D.J. Pena
GENE - Núcleo de Genética Médica de Minas Gerais
Av. Afonso Pena, 3111, 9º andar. Belo Horizonte, CEP 30130-909
Tel.: 31 – 3284 8000
Fax: 31 – 3227 3792
Email: [email protected]

2
I. INTRODUÇÃO
A Deficiência Mental (DM) é definida pela Organização Mundial de Saúde como um QI inferior a
70. Embora números precisos sejam difíceis de obter, estima-se que ela afete 2-3% das crianças,
sendo leve (QI 50-70) em aproximadamente três quartos dos casos e moderada-grave (QI < 50) nos
25% restantes. Alguns autores questionam o uso do QI como critério único para definir a
deficiência mental. Uma caracterização mais detalhada seria de um déficit importante em dois ou
mais domínios do comportamento neuropsicosocial, cognição, linguagem, sociabilidade, execução
de atividades cotidianas e controle motor. Entende-se aqui por déficit importante uma performance
abaixo de dois desvios padrão abaixo da média para a idade.
Deve ser enfatizado que a deficiência mental não constitui um diagnóstico per se, mas apenas o
resultado final de uma série de processos patológicos com etiologia genética, ambiental ou
multifatorial. A sua heterogeneidade etiológica pode ser ilustrada pelo banco de dados OMIM
(Online Mendelian Inheritance in Man; http://www.ncbi.nlm.nih.gov/entrez/
query.fcgi?db=OMIM) que lista nada menos de 1280 doenças gênicas humanas associadas com
deficiência mental. E dentro deste número não estão incluídas nem as cromossomopatias nem as
múltiplas causas ambientais de deficiência mental.
A identificação de urna criança com deficiência mental é sempre um evento dramático para a
família. Obviamente, a primeira preocupação do médico e dos pais é com o diagnóstico e
tratamento. Entretanto, a problemática é muito mais complexa. Há de se lidar com a família e seus
sentimentos de culpa, frustração e desespero, suas esperanças e ambições não realizadas, o estigma
da doença e o temor de recorrência em gestações futuras. Assim, a tarefa do médico não é fácil,
pois frequentemente extrapola seu treinamento convencional. Além disso, ele tem de controlar seus
próprios sentimentos e preconceitos para poder dar à família, de forma equilibrada e madura, o
suporte técnico e emocional necessários.
Neste capítulo, tentaremos dar um esquema geral para abordagem da criança com deficiência
mental e de sua família, começando pelo estabelecimento fundamental e indispensável do
diagnóstico preciso, passando pela comunicação com a família e terminando no aconselhamento
genético. Pela limitação de espaço, o tópico não poderá ser tratado com extensão e profundidade
ideais. Entretanto, esperamos que sirva de introdução e motivação para estudos mais aprofundados
em textos especializados.

3
II. ABORDAGEM DIAGNÓSTICA
A preocupação primordial do médico perante a criança com deficiência mental deve ser o
estabelecimento de um diagnóstico preciso, o qual é absolutamente essencial para que se possa:
estabelecer um tratamento, estabelecer um prognóstico clinico e de qualidade de vida, orientando
os pais quanto à evolução da doença e eventuais complicações, estabelecer um prognóstico
reprodutivo para a família, com base no risco de ocorrência da mesma ou de outra patologia em
gestações futuras do casal (assim, é feito o aconselhamento genético). Um diagnóstico etiológico
definitivo e convincente da deficiência mental leve só é estabelecido em menos de 30% o dos
casos, o restante permanecendo sob a rubrica de "deficiência mental idiopática". Já nos casos
moderados e graves é possível estabelecer um diagnóstico etiológico em aproximadamente 60%
dos casos.
De uma forma esquemática podemos dividir a abordagem diagnostica em três etapas: coleta de
dados (que inclui a anamnese, o exame físico e os exames complementares), avaliação dos
dados e estabelecimento do diagnóstico definitivo. A primeira preocupação do médico deve ser a
elucidação da época de inicio do problema: pré-natal, perinatal ou pós-natal, lembrando que estas
categorias não são necessariamente mutuamente exclusivas. Uma outra importante preocupação é
determinar se trata-se de uma DM sindrômica ou não-sindrômica, o que vai então orientar o
diagnóstico diferencial.
A. Coleta de dados
1. Anamnese
A anamnese, embora semelhante à realizada nas demais consultas pediátricas deve ser direcionada
para a história do desenvolvimento, história familial, história da gravidez e do parto.
a. A história do desenvolvimento deve esclarecer, além da época de inicio do problema, a
gravidade da deficiência mental e também se ela é de natureza estática ou progressiva. Deve ser
obtida uma história detalhada da idade em que foram atingidos os marcos principais de
desenvolvimento: quando a criança firmou a cabeça, quando sorriu, quando se assentou, quando
engatinhou, quando começou a passar itens de uma mão para outra, quando começou a balbuciar,
quando começou a "estranhar" pessoas de fora da família, quando começou a andar, quando falou
as primeiras palavras, quando começou a colocar palavras juntas, etc. A partir dai podemos
estabelecer a natureza da doença: estática, ou seja, um atraso consistente em alcançar os marcos de

4
desenvolvimento; progressiva, isto é, uma gravidade crescente. De especial interesse é a evidência
de perda de habilidades, ou seja, regressão, que é uma das características da síndrome de Rett.
Uma perda crescente de habilidades motoras sugere pode sugerir também a possibilidade de uma
doença neurodegenerativa, principalmente se associada a convulsões (doença de matéria cinzenta)
ou de espasticidade e hiperreflexia (doença de matéria branca). Outras características de
comportamento que devem ser especificamente pesquisadas na anamnese são: comportamento
inapropriadamente alegre, sorriso imotivado, hiperatividade, agressividade, automutilação,
comportamento obstinado (teimosia), ausência ou outros distúrbios da fala (coprolalia, ecolalia, fala
perseverativa), sonolência, apetite exagerado, movimentos inapropriados com as mãos e distúrbios
do sono. Além disso, alguns elementos que devem ser explorados são: história de hipotonia em
qualquer época, dificuldades de alimentação e reações idiossincráticas a determinados alimentos,
episódios de ataxia, episódios de coma, dificuldade auditiva e odores estranhos ou peculiares da
urina ou da própria criança.
b. Na história familiar quatro perguntas são fundamentais:
• Há história de casos similares na família?
• Há história de outras doenças na família?
• Há consangüinidade entre os pais
• A idade do pai ou da mãe é elevada?
A história de casos similares na família alerta para a possibilidade de uma etiologia genética e o
padrão de relacionamento familial dos afetados, estudados no heredograma freqüentemente permite
o estabelecimento do modo de herança. A pesquisa de outras doenças na família é relevante
porque várias doenças genéticas apresentam expressividade variável. Assim, crianças que
aparentemente têm problemas diferentes podem ter a mesma doença genética. Em especial deve ser
investigada a ocorrência de casos adicionais de doença neuropsiquiátrica, de malformações
congênitas e de doenças dermatológicas. Por exemplo, a neurofibromatose tipo 1 pode manifestar-
se como deficiência mental, como macrocefalia, como uma doença dermatológica ou como um
tumor neuroendócrino. Também, alguns casais portadores de translocações cromossômicas
equilibradas podem ter filhos com tipos diferentes de doenças cromossômicas e,
conseqüentemente, com quadros clínicos diferentes que podem envolver deficiência mental e,'ou
malformações congênitas. A consangüinidade entre os pais deve alertar para a possibilidade
especifica de doenças autossômicas recessivas. Enquanto a elevação da idade paterna é vista nos

5
casos de mutações autossômicas dominantes, a elevação da idade materna é vista em doenças
cromossômicas, particularmente nas trissomias.
c. A história da gravidez deve ser a mais completa possível. Dois aspectos devem ser explorados
com cuidado:
• Exposição a agentes teratogênicos
• Evolução da gravidez e intercorrências clinicas
A história de exposição a agentes teratogênicos deve ser completa, mas colhida com sensibilidade
para evitar o agravamento de possíveis sentimentos de culpa dos pais. Há uma tendência grande da
família em fixar-se, por vezes neuroticamente, em episódios de ingestão de medicamentos ou
exposição á radiação para tentar explicar o retardo do desenvolvimento, principalmente quando de
início pré-natal e acompanhado de malformações congênitas. Nessa perspectiva, é importante que o
médico exerça o seu julgamento para não valorizar indevidamente elementos de menor
importância. Por exemplo, há boas evidências na literatura de que a ingestão de medicamentos,
como a doxilamina, o ácido acetilsalisílico, a dipirona, os corticosteróides, os antidepressivos
tricíclicos, as anfetaminas, não está associada a um aumento significativo da taxa de problemas
fetais. Igualmente, raios-X diagnósticos, ou seja, com doses fetais inferiores a l0 Rads (p. ex., uma
urografia excretora tem uma dose fetal máxima de 1 Rad) não apresentam risco significativos. Já a
ingestão de talidomida, álcool, anticonvulsivantes dicumarinicos, litio, ácido retinóico e
metotrexato e também o uso de radiação terapêutica têm sido definitivamente associados a defeitos
de desenvolvimento fetal. Quando a criança apresenta malformações múltiplas é importante
explorar a possibilidade da ingestão de teratógenos no período critico de organogênese (18-60 dias
pós-concepção). Já nos casos de deficiência mental não-sindrômica podem estar associados com
exposição no período fetal, no qual ocorrem a proliferação e migração de neurônios para
histogênese do sistema nervoso central.
Na evolução da gravidez é importante saber se houve aceleração ou atraso do crescimento fetal, se
houve ameaças de abortamento e se houve poliidrâninio ou oligoidrâmnio. Determinar o nível de
movimentação fetal é importante, pois a sua diminuição pode sugerir uma patologia neuromuscular.
Todas as intercorrências clinicas devem ser cuidadosamente anotadas, incluindo episódios de
febre alta, doenças infecciosas (especialmente toxoplasmose, rubéola e citomegalovirus), diabetes
gestacional, etc.
d. Na história do parto e período neonatal deve ser especificamente investigado se ocorreram
prematuridade, aspiração de mecônio ou de liquido amniótico, asfixia ou hipoglicemia. As

6
apresentações pélvica e transversa estão significativamente associadas com um aumento de
complicações e partos muito rápidos podem estar associados a hemorragias cerebrais. O índice de
Apgar baixo, na ausência de complicações perinatais, sugere uma doença em evolução desde a vida
intra-uterina. Peso, comprimento e perímetro cefálico trazem informações sobre o crescimento pré-
natal, que costuma estar alterado nas síndromes dismórficas, especialmente as cromossômicas. O
status neurológico no período neonatal deve ser escrutinizado, incluindo tônus muscular. postura,
estado de alerta, posicionamento, vigor, movimentação, esforço respiratório, caráter do choro e
sucção.
2. Exame físico
O exame físico da criança com deficiência mental não difere do exame de rotina, exceto na ênfase
posta em obter alguns dados especiais. Nós fazemos rotineiramente as seguintes medidas: peso,
comprimento, envergadura, perímetro cefálico, distâncias intercantais interna e externa,
comprimento das orelhas, perímetro torácico e comprimento da mão e comprimento do dedo
médio. Em adolescentes deve ser estimado o volume testicular (de preferência com o uso do
orquidômetro de Prader). A determinação dos percentis de cada medida é útil para averiguação de
assimetrias e distúrbios de proporção corporal e para quantificação de impressões clinicas. Estas
quantificação é fundamental para estabelecer a presença de microcefalia, hipertelorismo, telecanto,
rizomelia, braquidactilía, etc. No exame dos diversos segmentos e sistemas corporais, o sistema
nervoso central obviamente merece atenção especial.
Terminado o exame físico o médico deve ser capaz de distinguir se há presença de uma deficiência
mental sindrômica ou não-sindrômica. Na primeira, é a constelação de achados no exame físico
que vai permitir o diagnóstico preciso. Assim, devem ser pesquisados os seguintes elementos:
Comportamento: interação com o examinador, incluindo contato ocular, interação com os pais,
interação com o ambiente.
Sistema nervoso: exame completo, com especial atenção para tônus (hipotonia, espasticidade),
reflexos (hiporreflexia, hiporreflexia), respostas a estímulos visuais e auditivos (ausência, resposta
exageradas), paralisias, paresias.
Pele: padrões de pigmentação localizados ou generalizados, especialmente manchas hipocrômicas
(melhor visualizadas com luz ultravioleta de comprimento de onda longo, i.e., lâmpada de Woods)
e hipercrômicas. Todas a manchas devem ser enumeradas e cada uma, especialmente as de
coloração café com leite, devem ter suas dimensões e estabelecida.
Crânio: tamanho, forma, simetria, fontanelas.

7
Cabelos: pigmentação, linhas de implantação anterior e posterior, localização do redemoinho.
Face: impressão geral, simetria, paralisias.
Olhos: sobrancelhas, cílios, forma do olho, fendas palpebrais (inclinação, tamanho, distância entre
elas), ptose, coloração da esclera e íris, transparência do cristalino, pupilas, fundo de olho
(indispensável a visualização da papila ocular e da mácula).
Nariz: forma, comprimento, aspecto do dorso, das narinas e do septo.
Boca: forma, aparência dos lábios, fendas, macrostomia, exame dos alvéolos, do palato e língua.
Mandíbula: forma, tamanho e simetria.
Orelhas: localização, rotação, tamanho, anomalias da hélice e anti-hélice, tragus, lobo e conduto
auditivo.
Pescoço: presença de excesso de pele, seios, fistulas, torcicolo.
Tórax: forma, simetria, localização dos mamilos, mamilos acessórios.
Aparelho cardiovascular: pulsos, pressão arterial, ausculta.
Abdome: aparência do umbigo, número de vasos, tônus muscular, integridade da parede,
tumorações, visceromegalias.
Genitália: tamanho, aparência, ambigüidade, aspecto da bolsa escrotal, localização dos testículos.
Ânus: localização e patência.
Dorso: simetria, coluna, fosseta pilonidal
Extremidades: proporções, aparência, limitação de movimentos, presença de redução, desvio,
ausência de segmentos.
Mãos e pés: pregas palmares, dedos, articulações, unhas.
A documentação fotográfica de qualquer criança com anomalias congênitas deve ser rotineira. A
foto servirá como registro das anomalias ("uma figura vale mil palavras"), permitirá a apresentação
do caso a outros médicos e, no caso de morte da criança, será de grande valia como memória, para
o clínico e para os pais.
3. Exames complementares.
Há controvérsia na literatura sobre a melhor estratégia para investigação laboratorial de uma
criança com deficiência mental. Alguns autores favorecem a utilização de uma lista de exames que
devem ser solicitados em todos os casos (exames indispensáveis). Outros autores enfatizam a

8
necessidade de avaliar cada paciente individualmente, pedindo exclusivamente os exames que se
julguem necessários naquele caso específico. O problema com esta última conduta é que algumas
das principais síndromes genéticas associadas com deficiência mental, como a síndrome do X-
frágil, apresentam poucas características fenotípicas específicas e não serão diagnosticadas a não
ser que uma rotina de exames seja seguida. Recentemente van Karnebeek e colaboradores (2005)
fizeram uma rigorosa meta-análise de 219 estudos diagnósticos em pacientes com deficiência
mental. Com base em considerações de sensibilidade e custo-beneficio, estes autores identificaram
alguns exames que eles consideraram parte integral da avaliação de toda criança com deficiência
mental e outros exames cuja indicação deve ser especificamente avaliada em cada paciente.
Sugerimos que as recomendações destes autores sejam seguidas e apresentamos abaixo a lista de
exames indispensáveis (que devem ser feitos em todos os casos) e exames importantes (que
devem ser avaliados em cada caso).
a. Estudo cromossômico (cariótipo): Anomalias cromossômicas perfazem um importante grupo
etiológico dentro das deficiências mentais. Uma parte destes resultados é devida à síndrome de
Down, que é a causa genética mais comum de deficiência mental. Entretanto, outras anomalias
numéricas dos cromossomos têm sido encontradas em todos os estudos realizados até agora e
mesmo em crianças com manifestações fenotípicas muito discretas. Assim, um estudo
cromossômico é mandatório em todos os casos de deficiência mental, mesmo na ausência de
dismorfismo. Em geral, as anomalias de numero dos cromossomos sexuais são vistas
primariamente em casos de DM leve, enquanto aberrações de autossomos são mais frequentemente
vistas em pacientes com DM moderada-grave. Preferencialmente os estudos cromossômicos devem
ser feitos em alta resolução (500-600 bandas) para permitir também o diagnóstico de anomalias
estruturais, que são vistas em freqüências menores do que as anormalidades numéricas, mas ainda
em proporções significativas, especialmente na DM moderada-grave.
b. Exames de Citogenética Molecular (pela PCR ou FISH) devem ser solicitados quando se
suspeita de síndromes de genes contíguos ou microdeleções cromossômicas (por exemplo:
síndrome de Prader-Willi, síndrome de Angelman, síndrome Velo-Cárdio-Facial, síndrome de
Willíams, síndrome de Smith-Magenis, etc.). Um caso especial de anomalias estruturais são as
pequenas deleções nas extremidades dos cromossomos (regiões sub-telomérícas) que não são
visíveis a microscopia ótica. O diagnóstico destes rearranjos sub-teloméricos depende então de
técnicas de citogenética molecular (hibridização in situ fluorescente - FISH - ou estudos com
microssatélites) que são ainda bastantes dispendiosas. Por isto, embora alguns estudos mostrem
uma prevalência de 3-7% destas anomalias em crianças com DM moderado-grave, não se pode

9
recomendar o uso rotineiro da investigação de deleções subteloméricas, que devem ser
especificamente pesquisadas quando existe suspeita baseada na presença de malformações
associadas.
c. Estudos em DNA: Recomenda-se que a pesquisa molecular da síndrome do X-Frágil deva
ser feita em todos os meninos com DM, independente da gravidade. Esta síndrome, que tem
herança ligada ao X e compreende 2-5% dos meninos com DM, tem características fenotípicas
discretas, principalmente nos primeiros anos de vida. Por outro lado, o diagnóstico é essencial para
poder prevenir outros casos na família. Já existem testes moleculares simples e relativamente
baratos para o diagnóstico da síndrome de X-frágil em meninos baseados na reação em cadeira da
polimerase (PCR) dependente de metilação (ver abaixo).
Algumas portadoras da síndrome de X-frágil apresentam DM, geralmente leve. Os estudos
moleculares de X-frágil em meninas são mais complexos e dispendiosos do que em meninos e
consequentemente devem ser reservados para os casos em que há uma história familial sugestiva da
síndrome. A síndrome de Rett é, após a síndrome de Down, a segunda causa mais freqüente de DM
em meninas. Geralmente a síndrome tem urna história característica de regressão do
desenvolvimento e deve ser pesquisada especificamente nestes casos.
d. Tomografia axial computadorizada ou ressonância nuclear magnética: podem ser solicitadas
para avaliar a presença de anomalias estruturais do sistema nervoso central como causa dos
problemas apresentados pelo paciente ou como um achado que contribuirá para firmar o
diagnóstico definitivo. Estes estudos revelam anormalidades em aproximadamente 30% dos casos,
mas ocasionalmente a detecção da presença destas anormalidades não é determinante no
diagnóstico etiológico. Como esperado, a ressonância nuclear magnética é mais sensível do que a
tomografia axial computadorizada na detecção de alterações anatômicas. Uma desvantagem dos
estudos neuroradiológicos, especialmente a ressonância nuclear magnética, é a necessidade de
imobilização da criança, o que pode ser bastante traumático em alguns casos. Assim, estes exames
devem ser reservados para casos em que haja uma suspeita especifica.
e. Sorologia para infecções congênitas: somente quando houver suspeita clínica de infecções
congênitas.
f. Testes metabólicos ou bioquímicos: não devem ser usados rotineiramente, mas devem ser
solicitados nos casos com suspeita específica de doenças metabólicas. Deve-se pensar em doença
metabólica, quando houver: DM progressiva ou evidência de doença neurodegenerativa,
convulsões, hipotonia grave, catarata, alterações da pigmentação da retina, face grosseira, hepato-

10
esplenomegalia, letargia ou coma, vômitos persistentes, odor peculiar, acidose metabólica, testes de
função hepática alterados, hiperbilirrubinemía persistente, hiperamonemia, hipercolesterolemia,
hipoglicemia.
g. Eletroencefalograma: o eletroencefalograma não contribui para o diagnóstico etiológico de uma
criança com DM.
B. As três causas genéticas mais comuns de deficiência mental
l. Síndrome de Down.
A síndrome de Down, causada por trissomia total ou parcial do cromossomo 21, tem uma
incidência aproximada de 1 em cada 600 recém-nascidos. O quadro clínico, bem conhecido, é
caracterizado por dismorfismo craniofacial discreto, que inclui braquicefalia, inclinação
mongolóide das fissuras palpebrais com pregas epicantais, nariz pequeno, orelhas pequenas e
pescoço curto. Há instabilidade atlantoaxial, com luxação (geralmente sub-clínica) em 12-20% dos
casos. As íris apresentam um padrão estrelado (manchas de Brushfield) e há pequenas opacidades
lenticulares em 59% dos casos (cataratas em 30-60% dos adultos). As mãos são pequenas, com
hipoplasia da falange média do 5° dedo (60%), clinodactilia (50%) e prega palmar única (50%).
Nos pés há uma separação aumentada entre o primeiro e o segundo artelhos e um padrão
dermatoglífico aberto na região halucal das solas (50%). Há uma anomalia congênita cardíaca em
40% dos casos, sendo mais específicos os defeitos do coxim endocárdico. Atresia do esôfago ou
duodeno é vista em 12% dos casos. Pacientes masculinos são estéreis, mas as mulheres são férteis.
Os bebês com síndrome de Down são tipicamente hipotônicos, com tendência a manter a boca
aberta e haver protusão da língua.
A deficiência mental é vista em todos os pacientes com síndrome de Down e é geralmente de grau
moderado. Entretanto, deve ser destacado que no passado os pacientes com síndrome de Down
eram socialmente marginalizados e frequentemente institucionalizados, o que certamente contribuía
para um pior ritmo de desenvolvimento e inteligência final. Programas de estimulação precoce e
enriquecimento ambiental têm resultado em um aumento significativo da capacidade intelectual e
da qualidade de vida.
O diagnóstico da síndrome de Down, geralmente feito no berçário, é um evento de enorme impacto
para a família. Os esforços iniciais da equipe médica devem ser sempre no sentido de minimizar o
trauma para os pais e de facilitar a aceitação do bebê (ver abaixo). O envolvimento dos pais com
programas de estimulação precoce deve ser promovido assim que o diagnóstico for feito. Nós
geralmente recomendamos que a estimulação precoce seja feita somente pelos pais nos primeiros 3-

11
6 meses de vida, estimulando a formação dos laços afetivos. Após seis meses de idade recomenda-
se acompanhamento por fisioterapeutas e fonoaudiólogos.
A síndrome de Down é causada por trissomia livre do cromossomo 21 em 94% dos casos. Esta
trissomia tem origem preponderantemente na meiose feminina e tende a aumentar com a idade
materna, provavelmente pela supressão de recombinação meiótica em ovócitos mais velhos. A
frequência de recém-nascidos com síndrome de Down é de 1/250 aos 35 anos, 1/100 aos 40 anos e
1/25 aos 45 anos. Aproximadamente 70% das concepções com trissomia 21 terminam em perdas
fetais - apenas 30% chegam ao nascimento.
Em 2% dos casos é observada a presença de uma linhagem celular trissômica conjuntamente com
uma linhagem normal (mosaicismo). Alguns destes pacientes apresentam um quadro clínico mais
leve, o que pode em alguns casos dificultar o diagnóstico. Por isto, é necessário que um estudo
cromossômico seja feito sempre que houver qualquer suspeita diagnóstica da síndrome de Down.
Em aproximadamente 4% dos casos a trissomia é devida a uma translocação cromossômica, que
pode ser familial. Na presença de translocação deve ser feito o estudo cromossômico dos pais e o
aconselhamento genético deve ser adequado á presença ou não de uma translocação balanceada em
um dos pais. Quando um dos pais tem uma translocação balanceada o risco de recorrência é de
2030%. Quando a translocação é de novo, o risco é inferior a 1%.
O risco de recorrência de uma trissomia em gravidezes futuras é de aproximadamente 1%. O
diagnóstico pré-natal (ver abaixo) deve ser sempre oferecido e é instrumental em ajudar os pais a
lidarem com este risco.
2. Síndrome do X-Frágil.
A síndrome do retardo mental associado ao X-frágil é a causa mais comum de deficiência
mental hereditária e, depois da síndrome de Down, a segunda causa genética mais comum de DM
na infância, com uma incidência aproximada de 1 em 1500 em meninos e 1 em 2000 em meninas.
Clinicamente, além do retardo mental nos meninos, a síndrome está associada a um dismorfismo
facial discreto, aumento do tamanho das orelhas e desenvolvimento pós-puberal de
macroorquidismo. Não há dismorfismo nas mulheres afetadas. A grande importância do
reconhecimento clínico e diagnóstico específico da síndrome do X-frágil vem do fato de que
virtualmente todos os casos são hereditários e familiais. O diagnóstico de uma criança com a
síndrome do X-frágil estabelece uma oportunidade valiosa de fazer estudos na família, para
identificar outros afetados e portadoras, e de fazer um aconselhamento genético eficiente levando à

12
prevenção de novos casos. Infelizmente, o diagnóstico clínico da síndrome do X-frágil na infância é
difícil por causa da inespecificidade dos sinais clínicos.
A mutação que causa a síndrome do X-frágil é a expansão de um microssatélite com repetições
CGG na região promotora do gene FMR1. Em indivíduos normais, esta região varia de 5 a 42
repetições. Em pacientes com a síndrome do X-frágil, há considerável expansão para mais de 200
repetições. Estas grandes expansões causam metilação da região promotora e conseqüente
repressão do gene FMR1. Expansões na faixa de 42 a 200 repetições são chamadas de pré-
mutações. Uma pré-mutação pode evoluir para uma mutação completa, mas isto só ocorre na
meiose feminina. Uma pequena proporção de pacientes com a síndrome do X-frágil apresenta
mutações na sequência do gene FMR1.
Há basicamente dois tipos de testes laboratoriais que permitem o diagnóstico da síndrome do X-
frágil: testes cromossômicos e testes moleculares. O estudo cromossômico completo com pesquisa
específica adicional de X-frágil tem a vantagem teórica de também permitir o diagnóstico de outras
alterações cromossômicas que podem ser a causa do retardo mental. Entretanto, este teste tem
níveis significativos de resultados falso-positivos e falso-negativos para a síndrome do X-frágil e
hoje foi quase totalmente substituído pelos testes moleculares.
Pesquisas recentes realizadas no GENE - Núcleo de Genética Médica permitiram o
desenvolvimento de novos testes moleculares, baseados na PCR (Reação em Cadeia da
Polimerase), que apresentam sensibilidade e especificidade de virtualmente 100% para diagnóstico
da síndrome do X-frágil em afetados do sexo masculino As grandes vantagens destes testes são
rapidez e baixo custo. Também, como os testes são baseados na PCR, quantidades muito pequenas
de DNA são necessárias, permitindo usar a coleta de esfregaço bucal, fácil e indolor, ao invés da
coleta de sangue. Como dito acima, é nossa opinião estes testes de triagem deveriam ser realizados
em toda criança masculina com retardo mental.
A síndrome do X-frágil tem herança ligada ao X. A mutação completa tem uma penetrância quase
completa em indivíduos masculinos e uma penetrância ao redor de 50% em mulheres. As pré-
mutações não estão associadas com doença clínica. Por causa da natureza progressiva das
expansões do microssatélite (CGG)n praticamente todas as mães de meninos afetados são
portadoras. Assim, em casais que têm um filho com DM, o risco para gravizedes futuras vai variar
com o tamanho da pré-mutação na mãe, de acordo com a tabela abaixo, baseada em Saul e Tarleton
(2005):
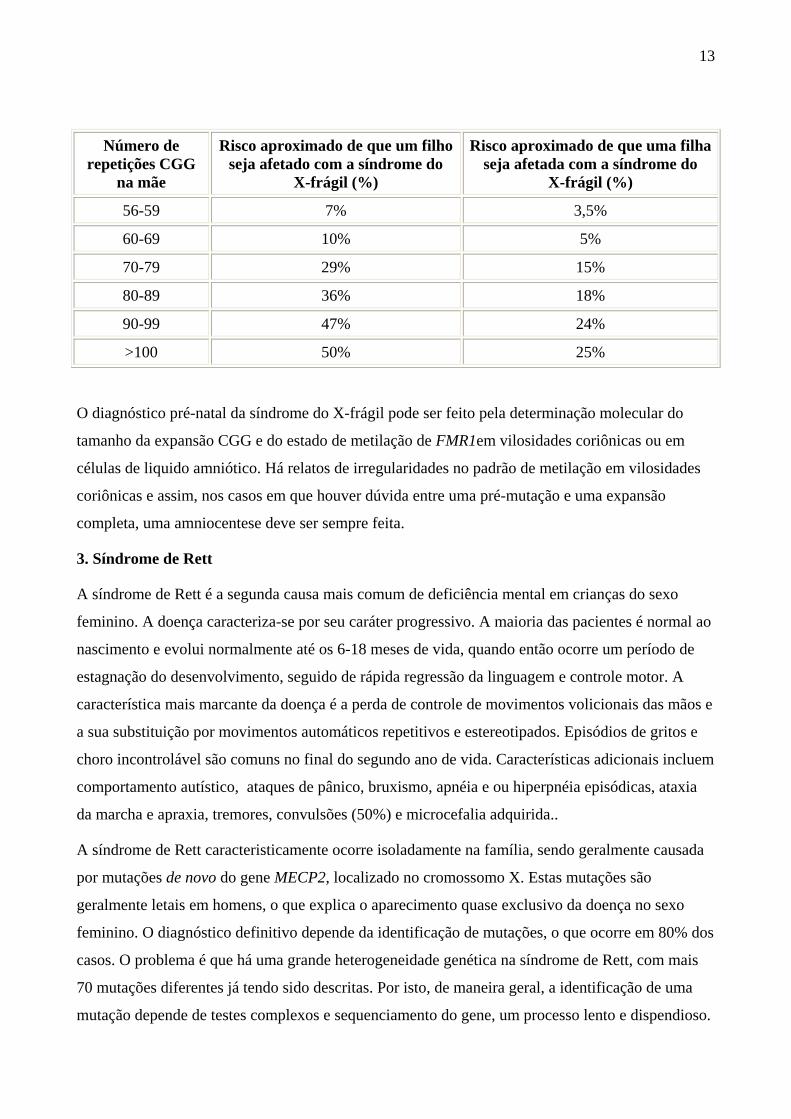
13
Número de repetições CGG
na mãe
Risco aproximado de que um filho seja afetado com a síndrome do
X-frágil (%)
Risco aproximado de que uma filha seja afetada com a síndrome do
X-frágil (%)
56-59 7% 3,5%
60-69 10% 5%
70-79 29% 15%
80-89 36% 18%
90-99 47% 24%
>100 50% 25%
O diagnóstico pré-natal da síndrome do X-frágil pode ser feito pela determinação molecular do
tamanho da expansão CGG e do estado de metilação de FMR1em vilosidades coriônicas ou em
células de liquido amniótico. Há relatos de irregularidades no padrão de metilação em vilosidades
coriônicas e assim, nos casos em que houver dúvida entre uma pré-mutação e uma expansão
completa, uma amniocentese deve ser sempre feita.
3. Síndrome de Rett
A síndrome de Rett é a segunda causa mais comum de deficiência mental em crianças do sexo
feminino. A doença caracteriza-se por seu caráter progressivo. A maioria das pacientes é normal ao
nascimento e evolui normalmente até os 6-18 meses de vida, quando então ocorre um período de
estagnação do desenvolvimento, seguido de rápida regressão da linguagem e controle motor. A
característica mais marcante da doença é a perda de controle de movimentos volicionais das mãos e
a sua substituição por movimentos automáticos repetitivos e estereotipados. Episódios de gritos e
choro incontrolável são comuns no final do segundo ano de vida. Características adicionais incluem
comportamento autístico, ataques de pânico, bruxismo, apnéia e ou hiperpnéia episódicas, ataxia
da marcha e apraxia, tremores, convulsões (50%) e microcefalia adquirida..
A síndrome de Rett caracteristicamente ocorre isoladamente na família, sendo geralmente causada
por mutações de novo do gene MECP2, localizado no cromossomo X. Estas mutações são
geralmente letais em homens, o que explica o aparecimento quase exclusivo da doença no sexo
feminino. O diagnóstico definitivo depende da identificação de mutações, o que ocorre em 80% dos
casos. O problema é que há uma grande heterogeneidade genética na síndrome de Rett, com mais
70 mutações diferentes já tendo sido descritas. Por isto, de maneira geral, a identificação de uma
mutação depende de testes complexos e sequenciamento do gene, um processo lento e dispendioso.

14
Assim, é importante ter uma suspeita clínica forte antes de pedir os testes moleculares e para tal foi
criado um critério de pontuação diagnóstica que facilita a identificação dos casos (Huppke et al.,
2003).
No GENE - Núcleo de Genética Médica decidimos abordar a questão do diagnóstico da Síndrome
de Rett por um ângulo diferente, desenhando um teste molecular mais simples, mais rápido e, bem
menos dispendioso, mas que detecta apenas as dez mutações mais comuns da síndrome de Rett
(cerca de 60% do total). A disponibilidade deste teste permitirá testar pacientes que não tenham
quadros clínicos absolutamente específicos a um baixo custo (C.M.B. Carvalho, W. Camargo e
S.D.J. Pena, manuscrito em preparação). Por exemplo, recentemente nós identificamos uma
mutação de MECP2 em uma paciente que tinha um quadro muito sugestivo de Síndrome de
Angelman. Mutações de MECP2 já foram descritas em pacientes com autismo e também em outros
com deficiência mental isolada.
O risco de recorrência na síndrome de Rett na família é muito baixo. Mesmo assim, nos casos em
que uma mutação de MECP2 foi identificada, deve ser feito o diagnóstico pré-natal em gravidezes
futuras, porque algumas mães fenotipicamente normais apresentam mosaicismo germinativo.
C. Estabelecimento do diagnóstico final.
Recapitulando, deve ser feito um grande esforço para estabelecer um diagnóstico etiológico preciso
em cada criança com DM, já que o mesmo é essencial para orientar o tratamento e permitir o
aconselhamento genético. Recomendamos a seguinte estratégia diagnóstica em todos os casos:
1. Em todas as crianças com DM deve ser obtida uma história clínica detalhada e um exame
físico minucioso deve ser feito, incluindo um exame neuropediátrico completo e uma avaliação
dismorfológica. A anamnese e o exame físico vão estabelecer a época de início do problema e
classificar a deficiência mental como sindrômica ou não-sindrômica, constituindo a melhor
ferramenta clínica para um diagnóstico etiológico preciso e para guiar as investigações
laboratoriais.
2. Em todas a crianças com DM deve ser feito um estudo citogenético com 500-600 bandas,
independente de gravidade, história familial e a presença ou não de dismorfismo.
3. Em todas as crianças com DM do sexo masculino deve ser feito um estudo molecular para
detecção da síndrome do X-frágil. Os estudos citogenéticos para diagnóstico da síndrome dão
resultados falso-positivos e falso-negativos e devem ser sempre que possível substituídos pelos
testes em nível de DNA.

15
4. Estudos metabólicos, sorologia para infecções perinatais e estudos neuroradiológicos não devem
ser feitos em todos os casos e devem ser pedidos quando houver indicação com base no exame
clínico.
III. COMUNICAÇÃO COM OS PAIS.
Lamentavelmente, nenhum aspecto da conduta perante a criança com deficiência mental é mais
negligenciado no nosso meio do que a comunicação com pais. Essa comunicação tem como
objetivo promover a aceitação da criança, dar suporte emocional, estimular o entendimento do
problema e prover as coordenadas para decisões racionais e para o planejamento do futuro. A
comunicação com os pais começa com uma fase aguda, logo após a identificação do problema, na
qual o diagnóstico é informado aos pais e as decisões imediatas são tomadas. Segue-se um período
prolongado, no qual o médico vai assistir a família no ajuste psicológico à sua nova realidade.
A. Como dar a noticia aos pais
Não há maneira boa de se contar uma noticia ruim. Mas há muitas maneiras ruins, as quais devem
ser evitadas.
1. Quando contar? Este é o primeiro dilema. Certamente, quando a deficiência mental é
diagnosticada, a notícia deve ser dada o mais cedo possível.
2. A quem contar? De forma ideal, a noticia deve ser dada aos dois pais juntos. O médico nunca
deve pedir ao pai que ele sozinho dê a noticia à mãe, ou vice-versa. E comum que a pessoa reaja
com sentimento de raiva ou rejeição a quem lhe transmitiu uma má noticia.
3. Quem vai contar? Varia de caso para caso. Funciona bem uma estratégia gradual, na qual mais
de uma pessoa dá a noticia. Por exemplo, o pediatra, que geralmente é chegado à família, pode
informar em linhas gerais uma suspeita, indicar que será necessário um acompanhamento mais
cuidadoso e fazer um encaminhamento para um neurologista (nos casos de deficiência mental
isolada) ou um geneticista (nos casos de deficiência mental sindrômica). De qualquer maneira,
devem ser dadas instruções claras a todos auxiliares de saúde no sentido de que toda a informação à
família deverá ser canalizada através de um ou dois médicos.
4. O que contar? Durante a fase inicial, o foco da comunicação com os pais deve ser a criança e a
ênfase deve ser dada em relação a aspectos imediatos da conduta. Por exemplo, nessa altura a
informação poderá ser propositadamente vaga quanto ao prognóstico do desenvolvimento
neuropsicomotor no futuro distante (por exemplo, com relação a escolaridade), com detalhes sendo

16
fornecidos apenas para pautar a conduta imediata. Por outro lado, é errado tentar minimizar a
situação, o que só reforça a negação dos pais.
B. Adaptação psicológica dos pais ao diagnóstico.
A adaptação psicológica dos pais á sua nova realidade é um processo lento e doloroso. Durante
toda a gravidez e início de vida foi feito o planejamento de uma criança idealizada como perfeita e
subitamente há uma ruptura entre essa idealização e a realidade. Embora a reação de cada casal seja
diferente, psicólogos têm identificado fases consistentes que antecedem a obtenção do equilíbrio
emocional. Estas fases são: negação ("eu não"), culpa ('`por que eu?"), depressão ("pobre de mim")
e raiva. A passagem bem-sucedida por essas fases é um pré-requisito para a aceitação completa da
criança e para o planejamento racional do futuro. O médico deve aprender a reconhecer, sem
compactuar ou antagonizar, as manifestações das fases e ajudar o casal a vencê-las.
IV. ACONSELHAMENTO GENÉTICO.
A. O processo do aconselhamento genético
Todos os médicos estão envolvidos no processo de fazer o diagnóstico, estabelecer um
prognóstico clínico e, finalmente, chegar a um tratamento para seus pacientes. Quando lidamos
com doenças genéticas, tudo isso é necessário, mas não suficiente, pois o prognóstico genético
passa a ser de importância fundamental. Assim, o foco do médico não pode mais ser somente a
saúde do paciente especifico, sua responsabilidade passa a abranger a família como um todo,
incluindo indivíduos não ainda afetados e mesmo não ainda nascidos. Um casal que tem uma
criança com doença genética e que tem risco alto de recorrência da mesma em uma gestação futura
espera (e tem o direito de) ser informado sobre esses riscos. O processo de identificar e transmitir â
família os riscos genéticos presentes e futuros é o que chamamos de aconselhamento genético. O
aconselhamento genético não constitui um procedimento limitado a geneticistas; grande parte dos
casos de aconselhamento é relativamente simples e pode ser feito pelo próprio pediatra ou
neurologista. Por outro lado, existem situações complexas em que a família deve ser encaminhada a
um especialista e o médico tem de saber identificar essas situações. Na dúvida, o paciente deve ser
encaminhado a um geneticista, pois é melhor o pediatra não fazer o aconselhamento do que
aconselhar erradamente.
B. Cálculo do risco genético
Feito o diagnóstico etiológico preciso pelo exame clínico, propedêutica laboratorial e
heredograma, pode-se calcular o risco genético de recorrência da doença. Nos casos de doenças

17
gênicas, o cálculo do risco é simples, na maioria das vezes, já que se baseia na aplicação das regras
mendelianas. Assim, o aconselhamento é relativamente simples nas doenças autossômicas
dominantes, porque existe uma relação 1:1 entre a doença e o gene responsável. Se um dos pais é
afetado, o risco é de 50% para gestações subseqüentes. Se nenhum dos pais é afetado, a doença da
criança provavelmente representa uma mutação nova, e os pais podem ser tranqüilizados de que o
risco para crianças futuras é negligível. Entretanto, diante de uma doença que apresenta
expressividade variável ou não-penetrância, o aconselhamento deve ser mais cauteloso. Nas
doenças autossômicas recessivas presume-se que ambos os pais sejam portadores e o risco para
gestações subseqüentes é de 25%. O aconselhamento nas doenças recessivas ligadas ao X é
simples quando se sabe que há outros casos na família e que a mãe é portadora. Por outro lado, em
casos isolados há a possibilidade de mutações novas e o calculo pode torna-se bastante complexo.
Nesses casos, é sempre prudente pedir a ajuda de um geneticista.
O calculo do risco de recorrência não é difícil na maioria dos casos de doenças cromossômicas. As
cromossomopatias mais comuns são as trissomias, que quase sempre representam fenômenos de
não-disjunção, verdadeiros acidentes genéticos, e assim têm riscos baixos de recorrência, ao redor
de 1-2%. Não há indicação para estudo cromossômico dos pais em casos de trissomias, a não ser
para tranqüilizá-los quando há grande ansiedade. Por outro lado, os pais devem ser sempre
estudados quando a criança tem uma anomalia estrutural dos cromossomos (translocações,
deleções e duplicações). Se um dos pais apresenta o defeito estrutural em forma balanceada, o
risco para gestações futuras será alto, podendo alcançar 30 a 50% e, em casos raros, 100%. Já, se os
cariótipos dos pais são normais, evidencia-se uni rearranjo cromossômico de novo, de ocorrência
acidental e com risco de repetição inferior a 1%. De qualquer maneira, acreditamos que há
indicação para diagnóstico pré-natal (ver adiante) em todas as gestações futuras de casais que
tiveram uma criança com cromossomopatia. Como há uma correlação grande entre
cromossomopatias e abortos, natimortos e mortes neonatais Para fins de aconselhamento genético,
deve ser sempre feito um estudo cromossômico dos pais se há história de mais de uma perda fetal,
ou se houve um natimorto malformado ou morte neonatal sem as investigações apropriadas.
C. Risco empírico
Como mencionamos acima, em mais de 70% dos casos de deficiência mental leve e em 40% dos
casos de deficiência mental moderada-grave não é possível estabelecer um diagnóstico etiológico
preciso. Nestes casos o aconselhamento genético deve ser baseado em riscos empíricos. Os estudos
empíricos mostram que a severidade da deficiência mental afeta significantemente o risco de
recorrência para casais não consangüíneos que têm apenas uma criança afetada sem história

18
familiar. O risco de recorrência para DM leve em vários estudos fica ao redor de 20%, sugerindo
que herança mendeliana seja responsável por uma proporção significativa destes casos. Já nos casos
de DM moderada-grave o risco de recorrência parece ser menor, da ordem de 10%. Estes riscos
estão muito diminuídos em casais que já tiveram crianças normais. É interessante notar que o sexo
da criança afetada não influencia muito o risco de recorrência. Também é pouco significativa a
influência da epilepsia, paralisia cerebral, microcefalia e baixa estatura.
Deve ser estressado que o aconselhamento genético não é importante apenas nos casos em que há
um risco palpável de recorrência da doença em gravidezes futuras. Também nos casos em que ó. .
risco de recorrência é negligível o aconselhamento genético é fundamental, pois esta informação
vai ser valiosa para os pais. Assim, é tão importante aconselhar o casal que tem um filho com
síndrome do X-frágil com risco de 25% em gravidezes futuras (ou seja, risco de 50% para filhos
homens) quanto aconselhar um casal que tem uma filha com síndrome de Rett ou com rubéola
congênita, nas quais o risco de recorrência é essencialmente nulo.
Um outro aspecto importante do aconselhamento genético é o "exorcismo" de crenças errôneas.
Muitas vezes o casal acredita erroneamente que a doença da criança é devida a algo que foi feito
(ou deixado de ser feito) na gravidez ou no período perinatal, ou mesmo um "castigo divino" por
algum pecado ou erro do passado. Estas crenças às vezes não são explicitadas voluntariamente e o
aconselhador deve estar alerta para poder identificar sua presença e "exorcizá-la".
D. Comunicação do risco genético
Calculado o risco, este é comunicado à família. Esta é a etapa culminante do processo de
aconselhamento e, paradoxalmente, a menos cientifica; pois estão envolvidas inúmeras variáveis
psicológicas. Por isso, a entrevista deve ser conduzida em um ambiente calmo e sem pressa, já que
todas as dúvidas do casal terão de ser pacientemente explicadas. O médico tem de se conscientizar
de que os conceitos de risco e probabilidades não são claros e riscos numericamente iguais podem
ter significados diferentes. Os riscos devem ser dados sempre no contexto específico da doença
e da família e contrastados com o desejo de outras gestações. Correr um risco de 25% de ter um
filho com uma doença letal no período neonatal, é bem diferente de correr o mesmo risco de 25%
de ter uma criança com doença grave crônica, que pode se estender por décadas. Também, a
maneira de informar os riscos é de importância. Por exemplo, famílias reagem diferentemente se
são apresentados a eles riscos de recorrência numericamente idênticos de 25%, ou 0,25, ou uma
chance em quatro, ou em proporções de um para três. Os pais também podem interpretar
diferentemente o risco de 25% de repetição da doença verso a chance de 75% da próxima criança

19
ser normal. Recomendamos que as várias versões sejam explicadas á família para que eles possam
avaliar bem os riscos. Temos de estar conscientes que o próprio conceito de probabilidades e riscos
às vezes não é claro para o leigo.
É, também fundamental que o médico tente ser não-diretivo em seu aconselhamento, deixando que
o casal tome as suas próprias decisões. Afinal de contas, é a família, e não o médico, quem terá de
viver com o resultado dessas decisões.
E. Diagnóstico pré-natal.
O aconselhamento genético informa aos pais os seus riscos reprodutivos, e deixa a cargo deles
a decisão. Essa é uma situação de conflitos e o casal passa a viver uma atmosfera de "roleta
genética". É claro, então, que qualquer procedimento que possa a vir reduzir a incerteza pela
antecipação do conhecimento do genótipo (ou genótipo) se tomará extremamente relevante para o
casal.
Essa possibilidade de antecipação do conhecimento do estado de saúde do feto tornou-se possível
com a introdução das técnicas de diagnóstico pré-natal por amniocentese na 16ª semana de
gravidez e posteriormente da biópsia de vilosidades coriônicas na 12ª semana de gravidez. .
Através do exame laboratorial do liquido amniótico ou das células fetais presentes nele, passou a
ser possível o diagnóstico preciso de um número grande de anomalias cromossômicas, metabólicas
e estruturais do concepto ainda no segundo trimestre de gravidez. Dessa maneira, no caso de um
feto acometido de doença grave, o casal passou a ter opção de interromper a gravidez e evitar o
nascimento de um recém-nascido anormal. A atmosfera de "roleta genética" pode assim ser
neutralizada ou eliminada.
Hoje, outros métodos se incorporaram ao diagnóstico pré-natal, já sendo possível realizá-lo para
centenas de doenças e esse número cresce continuamente. No entanto, ao solicitar o diagnóstico
pré-natal, o médico precisa saber para qual doença o feto apresenta risco elevado. Quando se
suspeita de doenças gênicas (autossômicas dominantes, autossômicas recessivas e ligadas ao X) e
doenças multifatoriais, o diagnóstico é especifico e informa se o feto é acometido ou não por
determinada patologia. Assim, ao pedir o exame, é necessário avaliar conjuntamente o risco da
doença e a disponibilidade do diagnóstico.
V. REFERÊNCIAS E LEITURAS SUGERIDAS
Bundey S, Thake A, Todd J. (1989) The recurrence risks for mild idiopathic mental retardation. J
Med Genet. 26:260-6.

20
Costeff H, Weller L. (1987) The risk of having a second retarded child. Am J Med Genet. 27:753-
66.
Crawford DC, Acuna JM, Sherman SL. (2001) FMR1 and the fragile X syndrome: human genome
epidemiology review. Genet Med. 3:359-71.
Hunter AG. (2002) Medical genetics: 2. The diagnostic approach to the child with dysmorphic
signs. Can Med Ass J. 167:367-72.
Huppke P, Kohler K, Laccone F, Hanefeld F. (2003) Indication for genetic testing: a checklist for
Rett syndrome. J Pediatr. 142:332-5.
Jones, K.L. (1997) Smith's Recognizable Patterns of Human Malformations. 5th Edition.
Philadelphia.W.B. Saunders.
Pena SDJ. (1998) Molecular cytogenetics II: PCR-based Dagnosis ofchromosomal deletions and
microdeletion syndromes. Genet Mol Biol. 21: 453-460.
Pena, S.D.J. (2001) Diagnóstico molecular em pediatria. Em: Doenças Genéticas em Pediatria. (G.
Carakushansky, ed.) Rio de Janeiro, Ed. Guanabara Koogan, pp. 87-93.
Pena SDJ, Carakushansky G. (1999) A hora e vez da pediatria molecular. Temas de Pediatria
Nestlê, 71: 1-15.
Pena SDJ, Sturzeneker R. (1999) Diagnosis of the fragile X syndrome in males using methylation-
specific PCR of the FMR1 locus. Genet Mol Biol. 22: 169-172.
Polifka JE, Friedman JM. (2002) Medical genetics: 1. Clinical teratology in the age of genomics.
Can Med Ass J. 167:265-73.
Saul RA, Tarleton JC. (2005) GENE Reviews: FMRl-related disorders. www.genetests.org
Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD.
(2003) Quality Standards Subcommittee of the American Academy of Neurology; Practice
Committee of the Child Neurology Society. Practice parameter: evaluation of the child with global
developmental delay. Report of the Quality Standards Subcommittee of the American Academy of
Neurology and The Practice Committee of the Child Neurology Society. Neurology. 60:367-80.
van Karnebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC. (2005) Diagnostic
investigations in individuals with mental retardation: a systematic literature review of their
usefulness. Eur J Hum Genet. 13:6-25.

21
Vercesi AML, Pena SDJ (2001) Síndromes de microdeleções cromossômicas. Em: Doenças
Genéticas em Pediatria. (G. Carakushansky, ed.) Rio de Janeiro, Ed. Guanabara Koogan, pp. 145-
151.



![Implementac¸ao da t˜ ecnica de´ Displacement Mapping em ...dmt/DM.pdf · A tecnica de´ Displacement Mapping [Cook 1984], e uma t´ ecnica´ alternativa em contraste comtecnicas](https://img.document.onl/doc/110x75/600d71ef4ab88a271a2278a8/implementacao-da-toe-ecnica-de-displacement-mapping-em-dmtdmpdf-a-tecnica.jpg)

























