Embed Size (px)
Citation preview

Bruno Max de Souza Melo
Estrutura eletronica de folhas de grafeno
onduladas
Niteroi
2015

Bruno Max de Souza Melo
Estrutura eletronica de folhas de grafeno
onduladas
Dissertacao apresentada ao Curso de Pos-Graduacao em Fısica da Universidade Fede-ral Fluminense, como requisito parcial paraobtencao do Tıtulo de Mestre em Fısica.
Orientador:
Prof. Dr. CAIO HENRIQUE LEWENKOPF
Universidade Federal FluminenseInstituto de Fısica
Niteroi
2015

M528 Melo, Bruno Max de Souza. Estrutura eletrônica de folhas de grafeno onduladas / Bruno Max de Souza Melo ; orientador: Caio Henrique Lewenkopf–- Niterói, 2015.
60 p. : il.
Dissertação (Mestrado) – Universidade Federal Fluminense, Instituto de Física, 2015. Bibliografia: p.58-60.
1. GRAFENO. 2.ESTRUTURA ELETRÔNICA. 3.DEFORMAÇÃO. 4.MODELO DE LIGAÇÕES FORTES. I.Lewenkopf, Caio Henrique, Orientador. II.Universidade Federal Fluminense. Instituto de Física, Instituição responsável. III.Título.
CDD 530.41

Agradecimentos
Aos meus pais por terem me educado para a vida.
A minha esposa Iane Nogueira por me apoiar em tudo o que faco, por me dar forcas nos
momentos mais difıceis, por me amar incondicionalmente e tornar minha vida maravilhosa.
Ao meu orientador, Caio Lewenkopf, por todo apoio e incentivo durante essa longa jor-
nada, por sua inestimavel contribuicao a minha formacao, por sua generosidade e paciencia
com meus interminaveis questionamentos e por ser um grande exemplo para mim do que
e ser um cientista.
Ao professor Jorge Sa Martins, um grande mestre, por todo apoio e pelas importantes
palavras de incentivo para que eu realizasse o mestrado.
Ao professor Tome Schmidt pelas ricas discussoes e por sua enorme contribuicao a rea-
lizacao deste trabalho.
Aos meus amigos de grupo Daiara, Mariana, Leandro, Ronald, Jesus, Vladimir, Ricardi-
nho e Emılia pela valiosas discussoes e trocas de ideias e pelos momentos de descontracao
e risadas.

Abstract
There is a great interest in finding methods that allow for opening and tuning gapsin the band structure of graphene, since this is a fundamental feature for applicationsof graphene in electronics. Several attempts of achieving this goal have already beenproposed in the literature. However in most of the proposals problems appear due tothe presence of disorder, such as impurities, edge roughness (as is the case of graphenenanoribbons) that seriously affect the electronic mobility in graphene. Mechanisms thatnot introduce these kind of problems in the band structure of graphene such as in planedeformations and corrugations in the graphene sheet, have attracted great attention. Suchsuggestions are called band gap and/or strain engineering. Our study critically analyzeshow the band structure changes in the presence of a deformation field and in the presenceof ripples. For this purpose, we calculate the band structure of rippled graphene sheetsusing the tight-binding model, its low energy continuum limit (Dirac hamiltonian) andcompare with DFT results. We analyze two regimes: long and short wavelength ripples.For the former we study the cases with and without in-plane relaxation of the atoms in theunit cell. Besides, we study graphene sheets with only in-plane deformations. We concludethat for smooth deformations (with small curvatures) the tight-binding approximationreproduces with great accuracy the DFT results, while the low energy efective Hamiltoniangives only good qualitative results. For strong deformations (large curvatures) the tight-binding approach is no longer efficient.

Resumo
Existe um grande interesse por mecanismos que permitam a abertura e o controlede gaps na estrutura de bandas do grafeno, pois essa caracterıstica e fundamental paraaplicacoes deste material na eletronica. Diversas maneiras de se alcancar este objetivoja foram propostas, porem, em muitos casos, problemas relacionados com a presenca dedesordens como impurezas e defeitos de bordas (caso das nanofitas de grafeno) preju-dicam seriamente a mobilidade eletronica no grafeno. Mecanismos que nao introduzamestes tipos de problema na estrutura de bandas do grafeno, como deformacoes no planoe tambem corrugacoes na folha de grafeno tem atraıdo forte atencao dos pesquisadores.Tais propostas sao denominadas “band gap engineering” e/ou “strain engineering”. Nossoestudo analisa criticamente como se modifica a estrutura de bandas na presenca de umcampo de deformacoes e tambem na presenca de ondulacoes. Para isso, calculamos aestrutura de bandas de folhas de grafeno onduladas usando o modelo de ligacoes fortes, olimite contınuo de baixas energias (hamiltoniano de Dirac) e comparamos com resultadosde calculos de primeiros princıpios (density functional theory, DFT). Os calculos foramfeitos para ondulacoes de comprimentos de onda longo e curto, sendo que, para as primei-ras foram ainda estudadas as situacoes com e sem relaxacao in-plane dos atomos na celulaunitaria. Alem disso calculamos a estrutura de bandas de folhas de grafeno submetidas auma deformacao no plano da folha. Concluımos que para deformacoes suaves e no planoda folha o modelo de ligacoes fortes reproduz com grande precisao os resultados de DFT,enquanto que a teoria de perturbacao usando o limite contınuo (hamiltoniano de Dirac)funciona apenas qualitativamente. Para deformacoes com grandes curvaturas o modelode ligacoes fortes deixa de ser eficiente.

Lista de Figuras
1 Esquerda: estrutura cristalina do grafite, mostrando as ligacoes de van
der Waals entre os planos e as ligacoes covalentes entre os atomos de
carbono nos planos. Direita: o grafeno corresponde a uma unica camada
de atomos de carbono. . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 11
2 Um nanotubo de carbono pode ser visto como uma folha de grafeno
enrolada(extraıdo da Ref. [1]). . . . . . . . . . . . . . . . . . . . . . . . p. 12
3 Rede colmeia e sua primeira zona de Brillouin. Esquerda: estrutura
cristalina do grafeno construıda a partir de duas subredes triangulares
intercaladas. A subrede A e identificada pelos sıtios de cor branca e a
subrede B, pelos de cor azul. Os vetores de rede sao a1 and a2 e δi onde
i=1,2 e 3 sao os vetores de translacao para os sıtios primeiros vizinhos.
Direita: vetores da rede recıproca b1 e b2 e primeira zona de Brillouin
do grafeno. Os cones de Dirac estao centrados nos pontos K e K′ (figura
adaptada da Ref. [13]). . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 15
4 Hibridizacao sp2 do atomo de carbono. . . . . . . . . . . . . . . . . . . p. 17
5 Estrutura de bandas do grafeno. No detalhe, uma ampliacao da relacao
de dispersao na vizinhanca do ponto onde as bandas de valencia e conducao
se tocam (chamado ponto de Dirac). Ilustracao extraıda da Ref. [18]. . p. 18
6 Componentes do tensor de tensao em um volume infinitesimal do corpo. p. 23
7 (a)Angulo θ, (b) vetor dij e normais ni e nj usados na expressao que
fornece os hoppings considerando os efeitos de curvaturas nas folhas de
grafeno. Ilustracoes adaptadas (a) da Ref. [3] e (b) da Ref. [22]. . . . . p. 28
8 Deformacao uniaxial no grafeno monocamada e convencao de orientacoes
da rede cristalina e do vetor ~T . Ilustracao da Ref. [7]. . . . . . . . . . . p. 29
9 Relacao de dispersao para grafeno tensionado na direcao θ = 0 calculada
para alguns valores do parametro u. . . . . . . . . . . . . . . . . . . . . p. 30

10 Folha de grafeno com ondulacoes ao londo da direcao zigue-zague. . . . p. 40
11 Celula unitaria do grafeno com 12 atomos. . . . . . . . . . . . . . . . . p. 41
12 Primeira zona de Brillouin da supercelula unitaria (retangulo) dentro da
primeira zona de Brillouin convencional do grafeno. . . . . . . . . . . . p. 42
13 (a) Estrutura de bandas na direcao ky = 0 e (b) densidade de estados para
uma supercelula com 12 atomos mostrando que, de fato, e a densidade
de estados para o grafeno monocamada. . . . . . . . . . . . . . . . . . p. 42
14 Visualizacao do processo de band-folding olhando para o que acontece
com a primeira zona de Brillouin convencional do grafeno. A primeira
zona de Brillouin hexagonal e dobrada para dentro da primeira zona de
Brillouin retangular. A sequencia esta representada pelos esquemas (a)
a (d) e os rebatimentos correspondentes pelas setas. Nesse processo, os
pontos K e K′ sao alocados no centro comum das duas zonas (ponto Γ). p. 43
15 Celula corrugada mostrando o comprimento de onda λ da corrugacao e
quais atomos sao fixos no processo de relaxacao das posicoes atomicas. p. 44
16 Exemplos de corrugacao de comprimento de onda curto (12 atomos na
celula unitaria) e longo (60 atomos na celula unitaria). As ondulacoes
estao ao longo da direcao zigue-zague. . . . . . . . . . . . . . . . . . . . p. 45
17 (a)Relacao de dispersao para uma supercelula com 60 atomos, calculada
em ky = 0. O numero de vezes em que a estrutura de bandas e dobrada
e bem maior que na Fig. 13 pois a primeira zona de Brillouin e menor
na direcao kx para esta supercelula. (b) Os cones de Dirac, porem,
continuam situados no centro da primeira zona de Brillouin. . . . . . . p. 46
18 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 2.03 A. O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 47
19 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 3.48 A). O grafico da esquerda corresponde ao calculo tight-
binding e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . p. 47
20 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 3.85 A). O grafico da esquerda corresponde ao calculo tight-
binding e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . p. 48

21 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 4.43 A). O grafico da esquerda corresponde ao calculo tight-
binding e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . p. 48
22 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 4.80 A). O grafico da esquerda corresponde ao calculo tight-
binding e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . p. 48
23 Estrutura de bandas em torno do ponto Γ para corrugacao suave com
hmax = 5.71 A). O grafico da esquerda corresponde ao calculo tight-
binding e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . p. 49
24 Graficos de (a) ux versus x e (b) h versus x para corrugacao suave com
hmax = 3.85 A. Os pontos pretos correspondem aos valores obtidos a
partir das posicoes atomicas obtidos por DFT. A curva em vermelho
corresponde a funcao polinomial ajustada. . . . . . . . . . . . . . . . . p. 50
25 Estrutura de bandas em torno do ponto Γ para corrugacao suave nao
relaxada com (a) hmax = 2.03 A, (b) hmax = 3.48 A, (c) hmax = 3.85 A,
(d) hmax = 4.43 A, (e) hmax = 4.80 A, (f) hmax = 5.71 A. . . . . . . . . p. 52
26 Estrutura de bandas em torno do ponto Γ para corrugacao abrupta com
λ = 6.62 A). O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 54
27 Estrutura de bandas em torno do ponto Γ para corrugacao abrupta com
λ = 6.76 A). O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 54
28 Estrutura de bandas em torno do ponto Γ para corrugacao abrupta com
λ = 6.89 A). O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 54
29 Estrutura de bandas em torno do ponto Γ para corrugacao abrupta com
λ = 7.03 A). O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 55
30 Estrutura de bandas em torno do ponto Γ para corrugacao abrupta com
λ = 7.17 A). O grafico da esquerda corresponde ao calculo tight-binding
e o da direita ao calculo DFT. . . . . . . . . . . . . . . . . . . . . . . . p. 55

Conteudo
1 Introducao p. 11
2 Revisao Teorica - Aspectos Basicos p. 14
2.1 Estrutura Cristalina . . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 14
2.2 Estrutura Eletronica . . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 16
3 Grafeno Deformado p. 21
3.1 Teoria da Elasticidade . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 21
3.1.1 Tensor de deformacao . . . . . . . . . . . . . . . . . . . . . . . p. 21
3.1.2 Tensor de tensao . . . . . . . . . . . . . . . . . . . . . . . . . . p. 22
3.1.3 Energia livre e lei de Hooke . . . . . . . . . . . . . . . . . . . . p. 24
3.1.4 Teoria da elasticidade aplicada a uma membrana fina . . . . . . p. 25
3.2 Grafeno deformado: estrutura eletronica . . . . . . . . . . . . . . . . . p. 26
3.2.1 Grafeno deformado: deformacao uniaxial . . . . . . . . . . . . . p. 28
3.3 Grafeno deformado: limite de baixas energias. Hamiltoniano de Dirac . p. 31
3.4 Grafeno deformado: resultados analıticos . . . . . . . . . . . . . . . . . p. 35
4 Estrutura eletronica de grafeno ondulado p. 40
4.1 Metodologia de calculo . . . . . . . . . . . . . . . . . . . . . . . . . . . p. 43
4.2 Resultados para curvaturas suaves . . . . . . . . . . . . . . . . . . . . . p. 46
4.3 Resultados para curvaturas abruptas . . . . . . . . . . . . . . . . . . . p. 52
5 Conclusoes e Perspectivas p. 56

Referencias p. 58

11
1 Introducao
O grafeno e um cristal bidimensional composto por atomos de carbono. A semelhanca
no nome com o grafite indica uma ıntima relacao entre os dois materiais. Na Fig. 1
mostramos a estrutura cristalina em 3D do grafite que e composta por varios planos
ou camadas de atomos de carbono organizados em um padrao hexagonal (que lembra
uma colmeia). Cada plano e ligado aos vizinhos por ligacoes de van der Waals. O grafeno
monocamada corresponde justamente a uma dessas camadas de atomos de carbono. Pode-
se dizer, portanto, que o grafite e constituıdo por varias camadas de grafeno empilhadas.
Quando se utiliza um lapis para escrever, o que ocorre e um desprendimento de folhas
de grafeno multicamadas do grafite e sua transferencia para o papel, gracas ao fato de
as ligacoes entre elas serem muito fracas. Embora folhas de grafeno assim produzidas
apresentem muitas imperfeicoes e, raramente, sejam monocamadas, a tecnica usada para
produzir amostras de grafeno de excelente qualidade para pesquisa de ponta e a exfoliacao
mecanica que guarda semelhanca com o processo de escrita com o lapis e e tambem
conceitualmente muito simples [1, 2].
Figura 1: Esquerda: estrutura cristalina do grafite, mostrando as ligacoes de van derWaals entre os planos e as ligacoes covalentes entre os atomos de carbono nos planos.Direita: o grafeno corresponde a uma unica camada de atomos de carbono.

12
O grafeno monocamada e considerado a membrana mais fina que existe pois possui
apenas um atomo de espessura. alem de ser extremamente flexıvel [1, 3, 4]. Alem disso
e um material extremamente flexıvel, algo que pode ser verificado quando observamos
os nanotubos de carbono, por exemplo, que podem ser vistos como folhas de grafeno
enroladas (Fig. 2). A despeito de sua flexibilidade, o grafeno e muito resistente a ruptura
por tracao [4].
Figura 2: Um nanotubo de carbono pode ser visto como uma folha de grafeno enro-lada(extraıdo da Ref. [1]).
Alem de notaveis propriedades mecanicas, o grafeno tambem possui propriedades
eletronicas muito interessantes [1] que tem sido alvo de intensa pesquisa nos ultimos anos.
Existe um grande esforco para produzir dispositivos eletronicos com o grafeno. Este feito
proporcionaria uma revolucao na industria eletronica. Contudo, a aplicacao do grafeno
na eletronica ainda encontra algumas limitacoes. Uma destas limitacoes motiva o nosso
estudo: apesar de suas propriedades eletronicas notaveis, o grafeno nao possui gap em
sua estrutura de bandas. O gap e importante pois por meio dele e possıvel controlar as
propriedades eletronicas do material [5]. Este controle e fundamental e constitui elemento
basico no funcionamento de dispositivos eletronicos como os transistores, por exemplo.
A literatura tem varias propostas para a introducao de gaps na estrutura de bandas do
grafeno: nanofitas de grafeno e funcionalizacao (adicao de oxigenio e/ou hidrogenio, por
meio de tratamento quımico). Porem estas abordagens prejudicam seriamente a mobili-
dade dos portadores de corrente no grafeno, pois introduzem desordem, como imperfeicoes
nas bordas e impurezas [6]. Por outro lado, alguns autores mostraram que e possıvel obter
gap no grafeno deformando sua rede cristalina [6, 7, 8], o que pode ser feito por meio da

13
aplicacao de tensoes mecanicas ou introduzindo-se corrugacoes (ou ondulacoes) na folha
de grafeno. A proposta de controle das propriedades eletronicas por meio das deformacoes
vem sendo denominada (em ingles) de strain engineering [5, 9, 10].
Neste trabalho, estudamos como as tensoes mecanicas e as corrugacoes afetam a es-
trutura de bandas do grafeno. Comparamos resultados obtidos por calculos de primeiros
princıpios com calculos utilizando o modelo de ligacoes fortes e resultados analıticos.
Esta dissertacao esta organizada da seguinte forma. No Capıtulo 2, faremos uma re-
visao teorica de aspectos basicos relacionados a estrutura cristalina e eletronica do grafeno
monocamada. No Capıtulo 3, apresentamos os principais resultados que discutem o que
acontece com a estrutura de bandas do grafeno (incluindo o limite de baixas energias)
quando ele e deformado. O Capıtulo 4 contem os resultados ineditos deste trabalho. Nele
discutiremos como foi feito o nosso trabalho de calculo da estrtura de bandas de grafeno
com corrugacao. Comparamos os resultados obtidos por metodos diferentes para cor-
rugacoes suaves e abruptas. Finalmente, no Capıtulo 5, apresentamos nossas conclusoes.

14
2 Revisao Teorica - AspectosBasicos
Neste capıtulo, vamos revisar alguns aspectos basicos da teoria e modelamento de
monocamadas de grafeno necessarios para o nosso estudo. Inicialmente, vamos descrever
a estrutura cristalina do grafeno para, em seguida, apresentar os modelos teoricos mais
simples que reproduzem suas propriedades eletronicas.
2.1 Estrutura Cristalina
O grafeno monocamada possui uma estrutura cristalina composta por uma rede trian-
gular com uma base de dois atomos ou, de forma equivalente, por duas redes triangulares
entrelacadas, lembrando uma colmeia de abelhas [1, 3, 11, 12]. Consequentemente, a
celula primitiva unitaria possui dois atomos, um em cada subrede triangular, que sao
costumeiramente denotados por A e B (Fig. 3).
A estrutura cristalina do grafeno e descrita no espaco real por dois vetores de rede,
ja que se trata de um cristal bidimensional. Usando a geometria da Fig. 3 obtem-se os
vetores primitivos de rede:
a1 =a0
2(√
3, 3) a2 =a0
2(−√
3, 3) , (2.1)
onde a0 ≈ 1, 42 A e a distancia entre atomos de carbono indicada na Fig. 3. Cada atomo
da subrede A possui tres vizinhos (chamados comumente de primeiros vizinhos por razoes
obvias) pertencentes a subrede B e vice-versa. Os vetores de translacao para os primeiros
vizinhos sao dados por:
δ1 =a0
2(√
3, 1), δ2 =a0
2(−√
3, 1) e δ3 = −a0(0, 1). (2.2)
A celula unitaria primitiva usada convencionalmente para o grafeno e dada pelo lo-
sango sombreado na Fig. 3. Note que cada celula primitiva possui dois sıtios, um de cada

15
Figura 3: Rede colmeia e sua primeira zona de Brillouin. Esquerda: estrutura cristalinado grafeno construıda a partir de duas subredes triangulares intercaladas. A subrede Ae identificada pelos sıtios de cor branca e a subrede B, pelos de cor azul. Os vetores derede sao a1 and a2 e δi onde i=1,2 e 3 sao os vetores de translacao para os sıtios primeirosvizinhos. Direita: vetores da rede recıproca b1 e b2 e primeira zona de Brillouin dografeno. Os cones de Dirac estao centrados nos pontos K e K′ (figura adaptada da Ref.[13]).
subrede. A area desta celula e dada por Acelula = ||a1 × a2|| = a0
√3 · a0
√3 · sin 60◦ =
3√
3a20/2. Outra escolha comum para celula primitiva e o menor hexagono formado pelos
atomos de carbono. Esta celula tem o mesmo numero de atomos em seu interior e a
mesma area que a que escolhemos.
Dados os vetores da rede no espaco real, pode-se determinar os vetores primitivos de
rede no espaco recıproco ou, simplesmente, vetores da rede recıproca usando-se a condicao
bi · aj = 2πδij [14, 15]:
b1 =2π
3a0
(√
3, 1) e b2 =2π
3a0
(−√
3, 1) . (2.3)
Estes vetores estao ilustrados na Fig. 3. Alem disso, os pontos de alta simetria no espaco
recıproco estao girados de 90 graus em relacao aos pontos de alta simetria na rede direta.
A primeira zona de Brillouin do grafeno tambem esta ilustrada na Fig. 3. Observe que
existem tres pares de pontos identificados por K e K′. Esses sao pontos de alta simetria
da primeira zona de Brillouin e sao muito importantes para o estudo do grafeno, como
ficara evidente a partir das proximas secoes. As coordenadas destes pontos sao dadas por

16
[11]:
K(K′) = ξK(K′)b1 − b2
3+mb1 + nb2 (2.4)
onde m e n sao numeros inteiros, ξK = +1 e ξK′ = −1 fornecem as posicoes dos pontos
K e K′, respectivamente. Sao chamados usualmente de ındices de vale [11]. Os pontos
da rede recıproca com mesmo ξ sao equivalentes.
2.2 Estrutura Eletronica
Para entender a estrutura eletronica do grafeno e importante discutir a configuracao
dos atomos de carbono neste material [3, 12]. Quando em sua forma atomica e no estado
fundamental, os eletrons do carbono estao distribuıdos segundo a configuracao 1s22s22p2.
Porem quando os atomos se agrupam para formar o grafeno, ocorrem mudancas significa-
tivas em alguns orbitais. O orbital 1s nao e alterado, formando uma camada em volta do
nucleo do atomo de carbono. Porem o orbital 2s se agrupa com dois orbitais 2p em um
processo chamado de hibridizacao. Como resultado aparecem tres novos orbitais denota-
dos por sp2 (Fig. 4). Os orbitais sp2 sao os responsaveis pelas ligacoes entre os atomos de
carbono no plano e a ligacao covalente e chamada de ligacao σ. O outro orbital 2p que nao
participa da hibridizacao e perpendicular a estrutura planar e o eletron deste orbital pode
formar ligacoes covalentes com eletrons dos atomos de carbono vizinhos dando origem as
bandas π no grafeno. As bandas σ estao muito distantes do ponto de neutralidade de
carga e nao contribuem para a conducao de corrente eletrica. As ligacoes σ estao associ-
adas com as propriedades mecanicas da estrutura cristalina do grafeno. Por outro lado,
as ligacoes π dao origem as bandas de valencia e conducao do grafeno. Dessa forma, para
o calculo da estrutura eletronica, o foco sera dado aos orbitais π.
A estrutura eletronica do grafeno para energias proximas ao ponto de neutralidade de
carga e muito bem descrita pelo modelo de ligacoes fortes (em ingles, tight-binding). O
hamiltoniano eletronico do grafeno, considerando que um eletron π pode ser transferido
diretamente de um sıtio apenas para seus sıtios vizinhos, e escrito como [3, 11, 16]:
H = −t0∑〈i,j〉
(a†ibj + H.c.
), (2.5)
onde a†i (ai) e o operador que cria (aniquila) um eletron no sıtio Ri da subrede A e b†j(bj)
cria (aniquila) um eletron no sıtio Rj da subrede B, onde Rj = Ri+δ3 [11]. O elemento da
integral de transferencia entre primeiros vizinhos e t0 ≈ 2, 7 eV e 〈· · · 〉 indica que a soma
deve ser tomada entre todos os primeiros vizinhos. Nesta descricao estamos omitindo o

17
orbital pz não hibridizado
orbitais sp2 no plano da folha de grafenoátomo de carbono hibridizado
(vista lateral)
Figura 4: Hibridizacao sp2 do atomo de carbono.
spin do eletron, devido a degenerescencia associada a esta propriedade.
O hamiltoniano da Eq. (2.5) esta escrito na representacao da base de sıtios. A
invariancia translacional torna conveniente escreve-lo na base de momentos, que sao bons
numeros quanticos do problema. Para isso, basta usar as expressoes que relacionam
os operadores de criacao das duas bases (com relacoes analogas para os operadores de
aniquilacao):
a†i =1√N
∑k
e−ik·Ria†k , (2.6)
b†i =1√N
∑k
e−ik·Rib†k , (2.7)
onde N e o numero de celulas unitarias da rede cristalina. Substituindo estas expressoes
na Eq. (2.5), obtem-se:
H = −t0∑k
φ(k)a†kbk +H.c. , (2.8)
onde φ(k) ≡∑
n e−ik·δn e os vetores δn foram definidos na Eq. (2.2). Para obter a Eq.
(2.8) usamos que as integrais de transferencia conectam apenas primeiros vizinhos. O
hamiltoniano pode ser escrito na forma matricial:
H =
(0 φ(k)
φ∗(k) 0
). (2.9)
A relacao de dispersao, obtida a partir da diagonalizacao desta matriz, e dada pela

18
expressao [3, 11, 12, 17]:
E(k) = ±t0√|φ(k)|2 = ±t0
√3 + f(k) (2.10)
na qual define-se:
f(k) = 2 cos(√
3kya0) + 4 cos
(√3
2kya0
)cos
(3
2kxa0
). (2.11)
Na Fig. 5 e mostrado o grafico correspondente a esta relacao de dispersao. Nele
aparecem duas bandas: a banda de valencia correspondente a superfıcie com E(k) ≤ 0
e a banda de conducao com E(k) ≥ 0. O espectro e simetrico, E(k) = E(−k), ou seja,
possui simetria partıcula-buraco. No estado fundamental a banda de valencia encontra-se
completamente ocupada, pois em um cristal com N celulas primitivas, existem N vetores
k (e, logo, 2N estados, considerando a degenerescencia de spin) disponıveis dentro da
primeira zona de Brillouin. Como cada celula unitaria possui dois eletrons no orbital π,
logo 2N eletrons preenchem 2N estados dentro da primeira zona de Brillouin do grafeno.
Note tambem que nao existe gap na estrutura de bandas do grafeno. Dessa forma, o
grafeno e classificado como semicondutor sem gap [1].
Figura 5: Estrutura de bandas do grafeno. No detalhe, uma ampliacao da relacao de dis-persao na vizinhanca do ponto onde as bandas de valencia e conducao se tocam (chamadoponto de Dirac). Ilustracao extraıda da Ref. [18].
Os vetores k para os quais E(k) = 0 sao justamente os vetores K e K′ definidos na
Eq. (2.4). No grafico da Fig. 5 eles correspondem aos seis pontos dentro da primeira zona
de Brillouin onde a banda de valencia e conducao se tocam. Com base nas consideracoes
acima sobre a ocupacao de cada banda, conclui-se que, no estado fundamental, os eletrons
mais energeticos do grafeno encontram-se justamente nestes pontos definindo, portanto,

19
a energia e a “superfıcie”de Fermi para o grafeno neutro.
Quando o sistema e excitado, os eletrons deixam a banda de valencia e ocupam os
estados disponıveis na banda de conducao. Note, porem, que estes estados, na banda de
conducao, situam-se muito proximos dos estados definidos pelos pontos K e K′. Sendo
assim, pode-se dizer que a fısica de baixas energias do grafeno acontece em torno destes
pontos. Por isso justifica-se a expansao do hamiltoniano, Eq. (2.5), em torno de K (K′)
fazendo k = K + q (ou k = K′ + q).
Expandimos a funcao φ(k) que aparece na Eq.(2.8):
φ(K + q) =∑n
e−i(K+q)·δn ≈∑n
e−iK·δn(1− iq · δn)
=∑n
e−iK·δn︸ ︷︷ ︸=0
−i∑n
e−iK·δnq · δn (2.12)
que e uma boa aproximacao enquanto q · δn � 1, ou seja, q � 1/a0. Um par de pontos
usualmente empregados nessa expansao e dado por:
K =4π
3√
3a0
(1, 0) K′ =4π
3√
3a0
(−1, 0) .
Usando os valores de K(K′) acima e δn dados na Eq. (2.2) obtem-se que:
φ(q) = −i∑n
e−iK·δnq · δn =3a0
2(−qx + iqy) =
3a0
2~(−px + ipy) . (2.13)
onde px(y) = ~qx(y), e a componente x do momento efetivo do eletron. Portanto, o hamil-
toniano expandido em torno do ponto K, que descreve efetivamente a dinamica eletronica
de baixas energias, e dado por:
HK =3t0a0
2~
(0 px − ipy
px + ipy 0
), (2.14)
que com a substituicao p→ −i~∇ pode ser escrito:
HK = −i~vFσ · ∇ (2.15)
onde σ = (σx, σy) sao as matrizes de Pauli e:
vF =3t0a0
2~. (2.16)

20
Efetuando a expansao em torno do ponto K′ obtem-se:
HK′ =3t0a0
2~
(0 px + ipy
px − ipy 0
)= i~vFσT · ∇ , (2.17)
onde T denota transposicao: σT = (σTx , σTy ). A relacao de dispersao para ambos os vales
e dada por:
E = ±~vF |q| , (2.18)
com autovetores:
ψq(r) =1√A
(1
seiθq
)eiq·r , (2.19)
onde A e a area da folha de grafeno, s = + ou − e o sinal da energia representando
partıculas ou buracos, respectivamente, e θq = tan−1(qy/qx).
E comum reunir as duas expansoes em um unico hamiltoniano:
H =
(HK 0
0 HK′
)= vF
0 px − ipy 0 0
px + ipy 0 0 0
0 0 0 px + ipy
0 0 px − ipy 0
, (2.20)
Nesse caso, a funcao de onda e escrita como um vetor de 4 componentes:
Ψ =
ψKA
ψKB
ψK′A
ψK′B
, (2.21)
onde, por exemplo, ψKA e a componente da funcao de onda correspondente ao vale K e
a subrede A.
A Eq. (2.14) e analoga a equacao de Dirac em duas dimensoes para fermions sem
massa, onde vF faz o papel da velocidade da luz [1, 12]. Uma vez que o estado do eletron
em torno do ponto K e composto por uma combinacao de estados pertencentes as subredes
A e B, e necessaria uma funcao de onda de duas componentes (spinor), Eq. (2.19), para
descreve-lo. Este grau de liberdade associado a cada subrede e chamado de pseudospin,
em analogia com o spin da partıcula presente na equacao de Dirac “verdadeira”[1, 19].
No proximo capıtulo, veremos como estes resultados sao modificados por deformacoes
e um campo de tensao na folha de grafeno.

21
3 Grafeno Deformado
Neste capıtulo iremos apresentar os efeitos de um campo de deformacoes na estrutura
eletronica do grafeno. Vamos iniciar a discussao analisando a situacao onde o grafeno e
submetido a uma tensao uniaxial, para depois generalizar para deformacoes arbitrarias.
A estrutura eletronica e obtida atraves do modelo de ligacoes fortes com um termo de
hopping modificado pelas deformacoes na rede cristalina. Vamos mostrar como a partir
do tensor de deformacoes e da teoria elastica e possıvel obter um hamiltoniano efetivo
de baixas energias para o modelo de tight-binding. Deformacoes suaves, numa escala
de comprimento muito maior que o parametro de rede, dao origem a campos de calibre
(pseudo-magneticos) na equacao de Dirac.
Na descricao do grafeno deformado sao usadas as ferramentas da teoria da elasticidade.
A seguir revisaremos alguns elementos basicos desta teoria que serao usados na analise
da relacao entre propriedades mecanicas e eletronicas do grafeno.
3.1 Teoria da Elasticidade
A teoria da elasticidade apresentada a seguir e um resumo escrito com base nos pri-
meiros capıtulos da Ref. [20]. Apenas os topicos mais relevantes para a aplicacao no
grafeno deformado foram destacados.
3.1.1 Tensor de deformacao
Consideremos um corpo elastico dividido em elementos infinitesimais cujos pontos
possuem coordenadas dadas pelo vetor posicao:
r = xx + yy + zz (3.1)

22
Quando o corpo sofre uma deformacao, as posicoes dos dos seus elementos de volume sao
alteradas,
x′i = xi + ui i = 1, 2, 3
ou seja, o deslocamento de um dado elemento e dado pela expressao r′ − r = u, onde o
vetor u e chamado de vetor deslocamento.
Para um corpo nao deformado, a distancia entre um elemento na posicao r e outro
na posicao r + dr e dada por (estamos adotando a convencao da soma de Einstein):
||dr||2 = dl2 = dxidxi.
Para um corpo deformado, as projecoes cartesianas das distancias sao:
dx′i = dxi + dui
O que nos leva a expressao:
dl′2 = dx′idx′i = (dxi + dui)(dxi + dui) = dxidxi + 2duidxi + duidui
Vamos considerar que u = u(r) e uma funcao suave da posicao, assim ui = ui(xi) e
dui =∂ui∂xj
dxj , (3.2)
logo:
dl′2 = dl2 + 2∂ui∂xj
dxidxj +∂ui∂xj
∂ui∂xk
dxjdxk
= dl2 + 2uijdxidxj
onde
uij ≡1
2
(∂ui∂xj
+∂uj∂xi
+∂uk∂xi
∂uk∂xj
)(3.3)
e chamado de tensor de deformacoes, que descreve as modificacoes no elemento de com-
primento quando o corpo e deformado. O tensor de deformacoes e simetrico, ou seja
uij = uji.
3.1.2 Tensor de tensao
Consideremos um corpo deformado e analisemos as forcas que atuam em um dado
volume de interesse deste corpo. A soma de tais forcas (forca total) e dada pela integral∫fdV , onde f e a forca por unidade de volume e fdV e a forca sobre o elemento dV .

23
Sabemos que as diversas partes que compoem este volume exercem forcas umas sobre as
outras de maneira que, pela 3a Lei de Newton, a soma destas forcas e zero. Logo apenas as
forcas exercidas pelas partes que circundam o volume de interesse contribuem para a forca
total. Tais forcas atuam apenas na superfıcie deste volume. Desta forma, e conveniente
transformar a integral∫
fdV em uma integral de superfıcie.
Consideramos a i-esima componente da forca total∫fidV e vamos usar o teorema da
divergencia. Como f e um vetor, e possıvel escrever cada componente fi como o divergente
de um tensor de segunda ordem:
fi =∂σij∂xj
. (3.4)
Com isto podemos transformar a integral:∫fidV =
∫∂σik∂xk
dV =
∮σikdsk , (3.5)
onde σik e o tensor de tensao. Observe que a quantidade σikdsk e a i-esima componente
da forca sobre o elemento de superfıcie ds. Nesse caso, podemos dizer que σik e a i-esima
componente da forca sobre uma unidade de area cuja normal tem a direcao xk. Por
exemplo, σxx e a componente x da forca aplicada sobre um elemento de area com normal
na direcao x, σyx e a componente y nesse mesmo elemento, etc., como mostrado na Fig.
6. Em um elemento infinitesimal de volume, a aceleracao angular e nula, logo o torque
Figura 6: Componentes do tensor de tensao em um volume infinitesimal do corpo.
tambem e nulo. Isso implica que o tensor de tensao tambem e simetrico, ou seja σik = σki.
No caso de o corpo estar submetido a forca externas (gravitacional ou eletrica, por

24
exemplo), a condicao de equilıbrio local e expressa por:
∂σik∂xk
+ f(e)i = 0 (3.6)
onde f(e)i e a i-esima componente da forca externa por unidade de volume.
Suponha agora que uma forca por unidade de area P seja exercida na superfıcie desse
corpo. De fato, esta e a situacao que, em geral, causa a deformacao do corpo. Em uma
situacao de equilıbrio, as forcas na superfıcie sao equilibradas pelas forcas de tensao do
corpo. Em um elemento de superfıcie do corpo tem-se que a i-esima componente da forca
externa e dada por Pids enquanto que a mesma componente das forcas de tensao e dada
por σikdsk = σiknkds. Logo, no equilıbrio:
σiknk = Pi . (3.7)
3.1.3 Energia livre e lei de Hooke
E possıvel escrever a energia livre por unidade de volume como funcao das compo-
nentes do tensor de deformacao por meio de uma expansao em serie dada por:
F = F0 +1
2λu2
ii + µu2ij (3.8)
onde os coeficientes λ e µ sao chamados coeficientes de Lame.
Vamos escrever o tensor de tensao como funcao do tensor de deformacao com o auxılio
da diferencial dF :
dF =Kukkdukk + 2µ(uij −
1
3δijukk
)d(uij −
1
3δijukk
)=Kukkdukk + 2µ
(uij −
1
3δijukk
)duij + 2µ
(− 1
3uijδij +
1
9δijδijukk
)dukk
onde K = λ + 2µ/3 e chamado modulo de elasticidade volumetrica. Como δijδij =∑ij δijδij = 3, entao:
dF =[Kukkδij + 2µ
(uij −
1
3δijukk
)]duij (3.9)
Da termodinamica, sabemos que:
dF = −SdT + σikduik . (3.10)

25
Logo:
σij = Kukkδij + 2µ(uij −
1
3δijukk
). (3.11)
O tensor de tensao e linear no tensor de deformacao. Esta e a lei de Hooke, valida para
deformacoes pequenas. A partir da Eq. (3.11), e possıvel obter o tensor de deformacao.
Basta calcular a soma dos termos diagonais de σij:
σkk = 3Kukk ou ukk =σkk3K
, (3.12)
e isolar uij na Eq. (3.11) usando a Eq. (3.12):
uij =σkk9K
δij +1
2µ
(σij −
1
3δijσkk
). (3.13)
3.1.4 Teoria da elasticidade aplicada a uma membrana fina
Aqui vamos considerar uma membrana muito fina e supor que as deformacoes sejam
pequenas. Dessa forma, aproximamos uij por:
uij =1
2
(∂ui∂xj
+∂uj∂xi
)(3.14)
Vamos supor ainda que nao ha forcas externas atuando sobre a membrana. Matematica-
mente isso corresponde a condicao de contorno para a superfıcie da membrana:
σijnj = 0 . (3.15)
Vamos parametrizar a deformacao na membrana pela funcao z = h(x, y). O vetor normal
a superfıcie descrita por esta funcao e dado por:
n =1√
1 + |∇h|2
(∂h
∂x,∂h
∂y,−1
)(3.16)
Para deformacoes pequenas, |∇h| � 1 , o vetor normal e praticamente paralelo ao eixo
z. Consequentemente, usando a Eq. (3.15):
σxz = σyz = σzz = 0 . (3.17)
A Eq. (3.17) vale para ambas as superfıcies da membrana. Para uma membrana muito
fina, a Eq. (3.17) deve valer dentro da membrana. Usando as Eq. (3.11), (3.14) e (3.17)
obtem-se:
0 = σxz = 2µuxz ⇒ ∂ux∂z
= −∂uz∂x
(3.18)

26
0 = σyz = 2µuyz ⇒ ∂uy∂z
= −∂uz∂y
(3.19)
0 = σzz = K(uxx + uyy + uzz) + 2µ(2
3uzz −
1
3uxx −
1
3uyy
)(3.20)
⇒ (−3K − 4µ)uzz = (3K − 2µ)(uxx + uyy) (3.21)
⇒ uzz = −3K − 2µ
3K + 4µ(uxx + uyy) = − σ
1− σ
(∂ux∂x
+∂uy∂y
)(3.22)
Vamos supor que uz = h(x, y) nao depende de z dentro da membrana (aproximacao
de membrana ultra-fina, que e apropriada). Assim, integrando as Eq. (3.18) e (3.19)
obtem-se:
ux = −z∂h∂x
e uy = −z∂h∂y
(3.23)
Consequentemente, as componentes do tensor de deformacao sao:
uxx =∂ux∂x
= −z∂2h
∂x2; uyy =
∂uy∂y
= −z∂2h
∂y2(3.24)
uxy =1
2
(∂ux∂y
+∂uy∂x
)= −z ∂
2h
∂x∂y(3.25)
uxz =1
2
(∂uz∂z
+∂uz∂x
)= 0 = uyz (3.26)
e, finalmente:
uzz = zσ
σ + 1
(∂2h
∂x2+∂2h
∂y2
). (3.27)
3.2 Grafeno deformado: estrutura eletronica
Nas secoes anteriores, apresentamos conceitos basicos da teoria da elasticidade. Estes
conceitos sao usados para construir modelos que descrevem as modificacoes na estrutura de
bandas do grafeno devido a presenca de um campo de deformacoes em sua rede cristalina.
O modelo mais simples e uma generalizacao do modelo de ligacoes fortes discutido
no Capıtulo 2, o qual e extensamente discutido nas Refs. [7, 4, 16]. A ideia e que os
deslocamentos da rede cristalina influenciam a estrutura eletronica alterando as integrais
de transferencia do modelo de tight-binding.

27
O hamiltoniano de ligacoes fortes de primeiros vizinhos e reescrito:
H = −∑〈i,j〉
tija†ibj +H.c. , (3.28)
onde tij = t(ri, rj), com ri = Ri + u(Ri). No caso geral, u depende da posicao do sıtio.
Uma parametrizacao eficiente para tij, extraıda de calculos de primeiros princıpios [7]
e:
tij = t0e−β(
dija0−1
)(3.29)
onde β = ∂ log t/∂ log a0 ≈ 3 e dij = |ri − rj|, que e o modulo da distancia entre sıtios
vizinhos. Alternativamente, podemos escrever:
dij ≡ dn(ri) ∼= (I + u)δn , (3.30)
onde u = u(Ri) e o tensor local de deformacoes,
u =
(uxx uxy
uyx uyy
). (3.31)
A Eq. (3.29) leva em conta apenas o efeito de modificacao nas distancias entre os atomos
de carbono nas integrais de transferencia.
O proximo passo no estudo de folhas de grafeno com ondulacoes e discutir o efeito das
curvaturas sobre os hoppings [3, 21, 22]. Em uma folha de grafeno curvada, os orbitais
pz nao sao mais paralelos. Isso implica uma modificacao na integral de transferencia
dependente do angulo θ entre os orbitais, mostrado na Fig. 7a. Nesse caso pode-se
decompor a integral de transferencia em uma componente referente a uma ligacao π e
outra referente a uma ligacao σ. Definimos:
cos θ = ni · nj (3.32)
onde ni e o vetor normal a superfıcie no sıtio i, dado pela Eq. (3.16). Em uma folha de
grafeno curvada os hoppings sao dados por [22]:
tij =a2
0
d4ij
[(Vppσ − Vppπ)(ni · dij)(nj · dij) + Vppπd2ijni · nj] (3.33)
onde dij e o vetor que conecta os sıtios i e j, Vppπ = t0 e Vppσ = 7.13 eV.
Note que a expressao acima nao e obtida de um ajuste com DFT. Ela nao e inteira-
mente consistente com a Eq. (3.29), como pode ser facilmente constatado no caso planar,

28
orbitais pz
(a)
(b)
Figura 7: (a)Angulo θ, (b) vetor dij e normais ni e nj usados na expressao que for-nece os hoppings considerando os efeitos de curvaturas nas folhas de grafeno. Ilustracoesadaptadas (a) da Ref. [3] e (b) da Ref. [22].
ni ·nj = 1. Neste trabalho nao consideramos explicitamente correcoes devido a curvaturas
em nossos calculos de estruturas de banda (Cap. 4).
3.2.1 Grafeno deformado: deformacao uniaxial
O caso mais simples que captura a fısica discutida acima e discutido no importante
trabalho de Pereira e colaboradores [7]. Os autores se concentraram em deformacoes
uniformes no plano da folha de grafeno apenas. A forma como o grafeno e deformado esta
ilustrada na Fig. 8. No sistema de coordenadas x e y, a direcao x esta alinhada com a
direcao zig-zag da folha de grafeno. No sistema de coordenadas x′ e y′, a direcao da forca
T esta alinhada com a direcao x′.
A tensao e aplicada em uma direcao arbitraria que faz um angulo θ com a direcao
x. Esta tensao deforma a folha e modifica as posicoes atomicas. As deformacoes sao
descritas, segundo a teoria da elasticidade pelos tensores de tensao σij e deformacao uij,
que se relacionam pela lei de Hooke. No sistema de coordenadas Ox′y′, temos que:
u′ij = Sijklσ′kl = TSijklδkxδlx = TSijxx. (3.34)
De acordo com a Ref. [23], apenas as constantes elasticas Sxxxx, Sxxyy, Sxxzz, Szzzz, Syzyz
e Sxyxy sao nao nulas no grafite. Consequentemente, as unicas componentes nao-nulas do
tensor de deformacao sao:
u′xx = TSxxxx e u′yy = TSxxyy, (3.35)

29
Figura 8: Deformacao uniaxial no grafeno monocamada e convencao de orientacoes darede cristalina e do vetor ~T . Ilustracao da Ref. [7].
que correspondem a uma dilatacao e a uma contracao transversa (chamada de contracao
de Poisson). Logo podemos escrever o tensor:
u′ = u
(1 0
0 −σ
)(3.36)
onde u = TSxxxx e σ = −u′yy/u′xx e a razao de Poisson. Por fim, devemos expressar o tensor
de deformacao no sistema de coordenadas Oxy, o que corresponde a efetuar uma rotacao
no tensor escrito nas coordenadas Ox′y′
u = u
(cos2 θ − σ sin2 θ (1 + σ) cos θ sin θ
(1 + σ) cos θ sin θ sin2 θ − σ cos2 θ
). (3.37)
Com este tensor podemos calcular as novas posicoes atomicas e, por sua vez, como se
modificam os vetores que conectam os atomos na rede cristalina. Na rede nao deformada
eles foram denotados por δn, Eq. (2.2). Na rede deformada, sao denotados por dn e
calculados de acordo com a Eq. (3.30). Com isto, as distancias entre os atomos primeiros
vizinhos na rede deformada, sao dadas por:
|d1| = 1 +3
4u11 −
√3
2u12 +
1
4u22 (3.38)
|d2| = 1 + u22 (3.39)
|d3| = 1 +3
4u11 +
√3
2u12 +
1
4u22 (3.40)
Para os casos especiais θ = 0◦ (tensao ao longo da direcao zig-zag) e θ = 90◦ (tensao ao
30
longo da direcao arm-chair), temos
|d1| = |d3| = 1 +3
4u− 1
4uσ, |d2| = 1− uσ (3.41)
Ao contrario do caso geral , discutido acima, e representado pelo hamiltoniano da Eq.
(3.28), como estamos tratando de uma situacao especıfica de deformacao uniaxial, e ne-
cessario considerar apenas tres hoppings diferentes, que serao denotados por t1, t2 e t3,
relacionados as mudancas nos vetores δ1, δ2 e δ3. As integrais de transferencia sao dadas
por:
tn = t0e−β(
|dn|a0−1), (3.42)
A relacao de dispersao e obtida analiticamente e dada por:
E(k) = ±|t2 + t3e−ik·a1 + t1e
−ik·a2| . (3.43)
Para a tensao ao longo da direcao zigue-zague, calculamos E(k) cujo grafico esta repre-
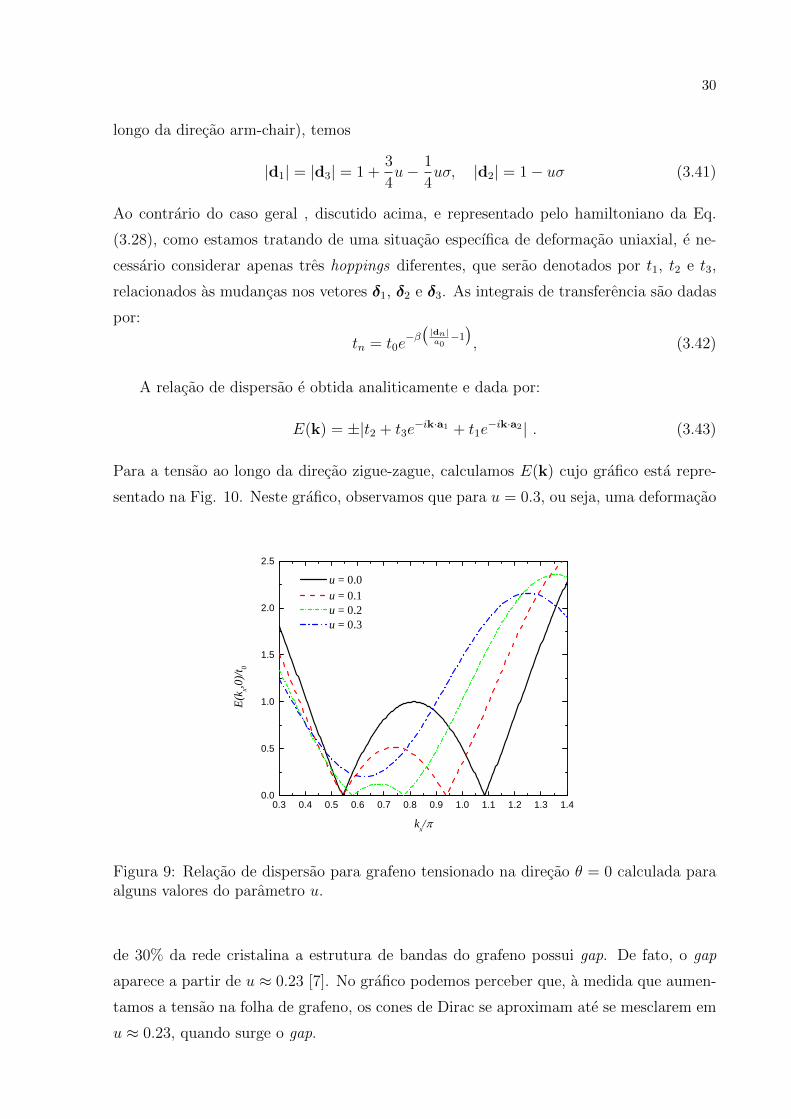
sentado na Fig. 10. Neste grafico, observamos que para u = 0.3, ou seja, uma deformacao
u = 0 . 0 u = 0 . 1 u = 0 . 2 u = 0 . 3
0 . 3 0 . 4 0 . 5 0 . 6 0 . 7 0 . 8 0 . 9 1 . 0 1 . 1 1 . 2 1 . 3 1 . 40 . 0
0 . 5
1 . 0
1 . 5
2 . 0
2 . 5
E(kx,0)
/t 0
k x / �
Figura 9: Relacao de dispersao para grafeno tensionado na direcao θ = 0 calculada paraalguns valores do parametro u.
de 30% da rede cristalina a estrutura de bandas do grafeno possui gap. De fato, o gap
aparece a partir de u ≈ 0.23 [7]. No grafico podemos perceber que, a medida que aumen-
tamos a tensao na folha de grafeno, os cones de Dirac se aproximam ate se mesclarem em
u ≈ 0.23, quando surge o gap.

31
E importante destacar que u ≈ 0.23 representa uma deformacao muito grande e muito
trabalhosa de estabelecer. Por isso, deformar o grafeno da maneira sugerida nesta secao
nao e considerado um modo eficiente de induzir gaps na sua estrutura de bandas. Contudo
neste trabalho de Pereira e colaboradores, e estabelecida a importante conexao entre o
controle das propriedades eletronicas atraves de deformacoes mecanicas e a possibilidade
de que outros tipos de deformacoes mais faceis de reproduzir possam ser utilizadas visando
este controle.
3.3 Grafeno deformado: limite de baixas energias.
Hamiltoniano de Dirac
Para o grafeno monocamada neutro no estado fundamental os eletrons preenchem as
bandas ate a energia de Fermi, que corresponde aos estados nos pontos K e K′. Para
baixas dopagens e excitacoes de baixas energias, apenas estados com vetores de onda
proximos a estes pontos sao relevantes para a conducao eletronica. Nesse sentido, e
conveniente estudar o que acontece na vizinhanca de K e K′, quando deformamos o
grafeno. Varios autores [4, 10, 16, 24, 25] se concentraram nesta tarefa usando o modelo
de ligacoes fortes para deformacoes homogeneas. Para obter um hamiltoniano efetivo
modificamos a Eq. (2.8):
H = −∑k
(∑n
tne−ik·dn
)a†kbk +H.c. (3.44)
para levar em conta as alteracoes nos hoppings e tambem nos vetores de rede. O efeito
das deformacoes nas integrais de transferencia e calculado usando-se a Eq. (3.42). Os
vetores dn sao calculados de acordo com a Eq. (3.30). Logo, em primeira ordem:
dn =√
dn · dn =√δ2n + 2δn · uδn + (uδn)2 ≈ a0
(1 +
δn · uδna2
0
)(3.45)
onde δ2n = a2
0 e o termo de segunda ordem no tensor de deformacao foi desprezado.
Portanto, podemos reescrever a Eq. (3.42) como:
tn ≈ t0 + δtn = t0
(1− β
a20
δn · uδn), δtn = −t0β
a20
δn · uδn . (3.46)
Visando obter um hamiltoniano efetivo de baixas energias, efetuamos a expansao do

32
hamiltoniano da Eq. (3.44) fazendo k = K + q, com q � 1/a0:
H = −∑n
tn
(0 e−i(K+q)·dn
ei(K+q)·dn 0
)
= −∑n
tn
(0 e−iK·δn(1− iK · uδn)(1− iq · dn)
eiK·δn(1 + iK · uδn)(1 + iq · dn) 0
)
= −∑n
tn
(0 e−iK·δn
eiK·δn 0
)(1 + iK · uδn 0
0 1− iK · uδn
)(1 + iq · dn 0
0 1− iq · dn
)
= −∑n
tn
(0 e−iK·δn
eiK·δn 0
)(1 + iσzK · uδn)(1 + iσzq · δn + iσzq · uδn). (3.47)
Usando o ponto K da Eq.(2.2) pode-se escrever a matriz abaixo em uma forma mais
compacta [25]: (0 e−iK·δn
eiK·δn 0
)=
i
a0
(σ · δn)σz . (3.48)
Desse modo:
H = −∑n
t0
(1− β
a20
δn · uδn
)[i
a0
(σ · δn)σz
](1 + iσzK · uδn)(1 + iσzq · δn + iσzq · uδn) .
(3.49)
Vamos agora analisar o hamiltoniano da Eq. (3.49) relendo todos os termos ate ordem
linear em u. Assim como no caso sem deformacao, H e simplificado pela identidade:∑n
(σ · δn)σz = 0
o que nos permite escrever:
H = H0 +HA +HB , (3.50)
onde
H0 = −∑n
t0
( ia0
(σ · δn)σz
)(iσzq · δn) = vFσ · p,
corresponde ao hamiltoniano efetivo usual para o grafeno sem deformacao, Eq. (2.14),
HA representa os termos de H que nao dependem de q e HB representa os termos que
dependem de q.
O termo HA da origem ao largamente discutido pseudo-potencial vetor induzido por
deformacao [3, 4, 9, 10, 16, 26]. O primeiro termo
HA1 =∑n
t0β
a20
δn · uδn( ia0
(σ · δn)σz
)(3.51)

33
pode ser escrito, apos simplificacoes:
HA1 =3t0a0
2
[− β
2a0
(uxx − uyy)σx +β
a0
uxyσy
]. (3.52)
Usando HA1 = vFσ · eA definimos as quantidades:
A(1)x =
~e
β
2a0
(uxx − uyy)
A(1)y = −~
e
β
2a0
(−2uxy) (3.53)
de forma que o hamiltoniano H0, Eq. (3.3), e reescrito substituindo q por q − e/~A, de
maneira analoga ao que acontece na mecanica classica quando uma partıcula esta sujeita
a um campo magnetico. E com base nessa analogia que se conclui que as deformacoes
no grafeno induzem um pseudo-potencial vetor com componentes dadas pela Eq. (3.53)
[3, 4, 9, 10, 16, 26]. Este pseudo-potencial vetor surge devido as mudancas nos hoppings
decorrentes das deformacoes no grafeno, o que se conclui da Eq. (3.51), onde apenas
o termo correspondente a modificacao nos hoppings esta presente. Por outro lado, ao
considerarmos o segundo termo:
HA2 = −∑n
t0
( ia0
σ · δn)σz
)(−iK · uδn) , (3.54)
a utilizacao do mesmo procedimento realizado para HA1 permite definir:
A(2)x = −~
e
4π√
3
9a0
uxx ,
A(2)y = −~
e
4π√
3
9a0
uxy (3.55)
que constituem uma correcao ao pseudo-potencial vetor, somando-se as componentes da
Eq. (3.53). E facil perceber pela Eq. (3.54) que esta correcao e decorrente das mudancas
nos vetores da rede cristalina do grafeno deformado [4]. Esta contibuicao tem sido fre-
quentemente ignorada na literatura, embora seja da mesma ordem da primeira. Logo, ao
levarmos em conta as mudancas nos hoppings e nos vetores da rede no grafeno deformado
temos o pseudo-potencial vetor em primeira ordem no tensor de deformacao:
A = A(1) + A(2) (3.56)
E importante destacar que apenas o termo A(1) contribui para o pseudo-campo magnetico

34
pois e possıvel mostrar que [4, 9, 16]:
A2 ∝ ∇(K · u) , (3.57)
o que implica que B2 = ∇×A2 = 0.
Ainda na Eq. (3.49), existem termos lineares no tensor de deformacao e que dependem
de q, HB = HB1 +HB2 +HB3 . O primeiro deles e:
HB1 =3∑
n=1
t0
[1
a0
(σ · δn)σz
](σzq · uδn) = vFσ · up (3.58)
que e escrito na representacao de posicoes substituindo o produto uijpl pela combinacao
simetrica [27]:
uijpl → −i~
[uij
∂
∂xl+
1
2
∂uij∂xl
](3.59)
Logo:
HB1 = −i~vF
(σ · u∇+
1
2σ · ∇T u
). (3.60)
Outros termos apresentados em [16] sao:
HB2 =3∑
n=1
t0
(β
a20
δn · uδn)(
1
a0
(σ · δn)σz
)(σzq · δn) =
βvF4σ · (2u + Tr(u)I) · p
HB3 = −3∑
n=1
t0
(i
a0
(σ · δn)σz
)(iσzK · uδn) (iσzq · δn) = i
vFa0
2σ · (K · u · ω) · p (3.61)
Usando a Eq. (3.59), escrevemos:
HB2 = −i~βvF4σ ·[2u · ∇+ Tr(u)I · ∇+∇T · u +
1
2∇T · Tr(u)I
]HB3 = ~
vFa0
2σ ·[(K · u · ω) · ∇+
1
2∇ · (K · u · ω)
](3.62)
onde σ = (σx, σy) sao matrizes de Pauli e ω = (−σz, σx). Os termos lineares em p
contribuem para uma renormalizacao de vF .
Deformacoes no grafeno que envolvem dilatacao da rede cristalina, ou seja uxx+uyy 6=0, modificam o hamiltoniano de baixas energias introduzindo uma quantidade usualmente
chamada de potencial escalar dado por [10]:
V = g(uxx + uyy) . (3.63)
Este termo aparece na diagonal do hamiltoniano de Dirac e, em um primeiro momento,

35
foi obtido no estudo das vibracoes termicas da rede cristalina, no contexto da interacao
eletron-fonon [26, 28]. Porem pode-se mostrar que modificacoes dos hoppings entre se-
gundos vizinhos tambem levam a uma expressao semelhante para o potencial escalar [3].
3.4 Grafeno deformado: resultados analıticos
Nesta secao vamos apresentar as expressoes analıticas para as correcoes aos autova-
lores de energia devido a deformacao no grafeno. Vamos usar teoria de perturbacao de
primeira e segunda ordem considerando o hamiltoniano de baixas energias para o grafeno
deformado, dado por:
H = H0 +HA +HB (3.64)
Nesta abordagem, vamos desprezar HB. De acordo com a teoria de perturbacao, o auto-
valor Eq do problema com perturbacao pode ser escrito como a soma:
Eq = E(0)q + E(1)
q + E(2)q + . . . (3.65)
onde E(0)q e o autovalor do problema sem perturbacao e E
(1)q e E
(2)q sao as correcoes de
primeira e segunda ordem, respectivamente, ao autovalor Eq.
E importante notar que em cada um dos vales K e K′ ha dois estados que possuem
a mesma energia para q = 0, um oriundo de estados de partıcula e outro de buracos.
Temos assim quatro estados degenerados, dois no vale K:
ψ1(r) =1√A
1
0
0
0
, ψ2(r) =1√A
0
1
0
0
, (3.66)
e dois no vale K′:
ψ3(r) =1√A
0
0
1
0
e ψ2(r) =1√A
0
0
0
1
. (3.67)
Portanto devemos usar teoria de perturbacao para o caso degenerado, para o calculo da
relacao de dispersao em q → 0. Nesse caso, as correcoes em energia para cada vale sao

36
dadas por [29]:
E(1)q =
1
2
[W11 +W22 ±
√(W11 −W22)2 + 4|W12|2
](3.68)
onde Wij = 〈ψi|HA|ψj〉. Usando um estado arbitrario para um dos vales,
ψi(r) =1√A
(ψAi
ψBi
)eiq·r , (3.69)
podemos escrever:
〈ψi|HA|ψj〉 =−∫
dr1√Ae−iq·r
(ψ∗Ai
, ψ∗Bi
)vFσ · eA
(ψAj
ψBj
)1√Aeiq·r
=− evFA
∫dr(ψ∗Ai
, ψ∗Bi
)( 0 Ax − iAyAx + iAy 0
)(ψAj
ψBj
)
=− evFA
[ψ∗Ai
ψBj
∫dr(Ax − iAy) + ψAj
ψ∗Bi
∫dr(Ax + iAy)
]. (3.70)
Usando os estados da Eq. (3.66), concluımos que:
W11 = W22 = 0
W12 = −evFA
∫dr(Ax − iAy)
W21 = −evFA
∫dr(Ax + iAy) . (3.71)
Logo, para cada vale:
E(1)q = ±evF
A
{[∫dr(Ax − iAy)
][∫dr(Ax + iAy)
]}1/2
(3.72)
A integral espacial e tomada sobre a celula unitaria extendida de area A. O pseudo-
potencial vetor A e calculado das Eqs. (3.53) e (3.55), onde o tensor de deformacao uij(r)
e uma representacao contınua obtida a partir dos vetores de deslocamento dos sıtios da
rede Ri −R0i .
Vamos agora usar teoria de perturbacao de segunda ordem para obter uma expressao
analıtica para o gap na estrutura de bandas de uma folha de grafeno corrugado, seguindo
o trabalho das Refs. [12, 30].

37
Ja vimos que o hamiltoniano:
H0 = ~vF (σxqx + σyqy)
possui relacao de dispersao sem gap E(q) = ~vF |q|. A inclusao de um termo diagonal do
tipo ∆σz leva a:
H = ~vF (σxqx + σyqy) + ∆σz ,
modificando a relacao de dispersao:
Ep ∼ ±∆
(1 +
p2
2∆2/v2F
).
Claramente, nesta situacao, surge um gap dado por Eg = 2∆.
Logo, para obter informacoes sobre gaps na relacao de dispersao devemos focar em
termos que estejam multiplicados por σz (os chamados “termos de massa”), pois o gap e
consequencia da presenca de tais termos. Uma interpretacao fısica que e dada para esta
modificacao que acabamos de descrever corresponde a dizer que houve uma quebra de
simetria de subrede no grafeno. Esta quebra leva ao desaparecimento da degenerescencia
existente no ponto K(K′) no problema original sem perturbacao e a abertura do gap.
Vamos estimar ∆ em teoria de perturbacao para uma folha de grafeno corrugada [12, 30].
Para o problema sem perturbacao, a funcao de Green retardada e:
Gr0 =
1
E −H0 + iδ=
E +H0
(E + iδ)2 −H20
onde δ e um infinitesimal positivo e para o vale K:
H0 = ~vFσ · q (3.73)
onde H20 = (~vF q)2. Para o vale K′, H0 e dado pela Eq. (2.17). Para o hamiltoniano com
perturbacao, a funcao de Green total e dada por:
Gr =1
E −H + iδ=
1
E −H0 − Σ(E)
onde Σ(E) e a auto-energia e
H = H0 +HA +HV
HA = −vFσ · eA
HV = V (r) (3.74)

38
Com base na Eq. (3.4), podemos entender a auto-energia como uma correcao a energia do
problema sem perturbacao. No que segue vamos estimar Σ em segunda ordem e examinar
seus termos diagonais para discutir mecanismos de geracao de massa.
A correcao de segunda ordem, no espaco de momentos, e dada por:
Σ(2)(k,k′, E) =∑k′
Wk−k′Gr0(k′, E)Wk′−k (3.75)
onde:
Wq = Vq + σ ·Aq .
E costumeiro escrever E = ~vFk, de modo que:
Gr0 =
1
~vFk + σ · q
(k + iδ)2 − q2. (3.76)
Vale a pena ressaltar que a dimensao de Gr0 (e Gr) e 2 × 2, pois nos restringimos a um
unico vale. Isto e justificado pois perturbacoes suaves nao misturam vales.
O produto de W e Gr0 resulta em termos multiplicados por σz. Note que tanto W
quanto Gr0 possuem termos em σx e σy e ainda:
σxσy = −σyσx = iσz
Como estamos interessados em obter o gap no ponto de neutralidade de carga, calculamos
Σr tomando E = 0:
Σ(2) =∑q
(Vk−q + σ ·Ak−q)(− σ · q
~vq2
)(Vq−k + σ ·Aq−k)
=∑q
(Vk−q + σxAxk−q + σyA
yk−q)
(− σxqx + σyqy
~vq2
)(Vq−k + σxA
xq−k + σyA
yq−k)
Em seguida, agrupamos os termos com σz que sao os termos de gap:
Σ(2) = − 1
~v∑q
1
q2
[. . .+ iσz
(Axk−qqyVq−k − A
yk−qqxVq−k − Vk−qqyA
xq−k + Vk−qqxA
yq−k)
+ . . .]
= − 1
~v∑q
1
q2
[. . .+ 2iσzVk−q
(qxA
yq−k − qyA
xq−k)
+ . . .]
onde usamos que Ak−q = A∗q−k.
Dessa forma obtem-se para o gap a expressao [12, 30]:
∆k =2
~v∑q
Im[Vk−q(qxAyq−k − qyAxq−k)]
q2(3.77)

39
Para k = 0 (correspondente ao ponto de Dirac):
∆k =2
~v∑q
Im[V−q(qxAyq − qyAxq)]
q2, (3.78)
usando
qxAyq − qyAxq = Bz
q = Bq ,
chega-se a [12, 30]:
∆k=0 =2
~v∑q
Im[V−qBq]
q2.
Vamos aplicar esta expressao ao problema de uma folha de grafeno com uma cor-
rugacao descrita por uma funcao h(x, y). Admitiremos que foi permitida a relaxacao nas
posicoes dos atomos de modo a se alcancar o mınimo da energia elastica que surge em de-
correncia da corrugacao. Nessas condicoes o tensor de deformacoes e dado pela expressao
[12, 30]:
uij(k) =λ+ µ
λ+ 2µ
kikj[k2xfyy(k) + k2
yfxx(k)− 2kxkyfxy(k)]
|k|4(3.79)
onde
fαβ(k) = −∑k′
k′α(kβ − k′β)hk′hk−k′ (3.80)
sao as componentes de Fourier do tensor [31]:
fαβ(r) ≡ ∂h
∂xα
∂h
∂xβ
Substituindo as Eq. (3.79), (3.63) e (3.53) na Eq. (3.4) obtem-se [12, 30]:
∆k =gβ
a
(λ+ µ)2
(λ+ 2µ)2
∑k
∣∣k2xfyy(k) + k2
yfxx(k)− 2kxkyfxy(k)∣∣2 cos(3θk)
|k|4(3.81)
onde θk e o angulo polar do vetor k. O valor desta expressao e zero. Ao fazermos a
substituicao k → −k o cosseno muda de sinal pois θ−k = π + θk e como fαβ(r) e real,
temos fαβ(−k) = f ∗αβ(k) . Isso nos leva a expressao ∆k = −∆−k . Como o gap e uma
quantidade escalar (energia), o unico resultado possıvel para essa condicao e zero.
Ou seja, potencial escalar e vetor decorrentes da mesma deformacao nao induzem gap
na estrutura de bandas. De acordo com as Refs. [12, 30], para abrir o gap e necessario
aplicar um potencial eletrostatico externo nao homogeneo a uma folha de grafeno com
deformacoes .

40
4 Estrutura eletronica de grafenoondulado
Neste capıtulo, vamos estudar quantitativamente como ondulacoes e tensoes modi-
ficam a estrutura eletronica de uma folha de grafeno, mais especificamente em torno
do ponto de Dirac. Com este proposito, vamos comparar resultados obtidos a partir
de calculos de primeiros princıpios com o modelo de tight-binding e o limite de baixas
(equacao de Dirac). Estes sao os resultados originais desta dissertacao.
Vamos considerar ondulacoes periodicas ao longo de uma direcao apenas, a direcao
zigue-zague, que vamos convencionar paralela a direcao x. Isto significa que o sistema
possui invariancia translacional na direcao y. A Fig. 10 ilustra a situacao estudada. Note
Figura 10: Folha de grafeno com ondulacoes ao londo da direcao zigue-zague.
que, agora, a estrutura cristalina nao e mais a do grafeno perfeito, sem ondulacoes. A
celula unitaria passa a ser extendida, compreendendo toda a ondulacao. Isto requer uma
modificacao no calculo da estrutura de bandas. E preciso definir uma nova celula unitaria
e novos vetores de rede. Obviamente, a celula unitaria sera maior que a celula unitaria

41
primitiva do grafeno perfeito (que possui 2 atomos)e, consequentemente, a primeira zona
de Brillouin correspondente sera menor que a do grafeno nao corrugado.
Vamos discutir agora o que acontece quando usamos uma celula extendida para ana-
lisar a estrutura de bandas do grafeno sem ondulacoes. Nesse caso, a estrutura de bandas
sera dobrada ou rebatida dentro desta primeira zona de Brillouin menor, o assim chamado
band folding. Observe que podemos definir uma supercelula unitaria de tal forma que a
mesma possua Nb > 2 atomos. Tomemos como exemplo a supercelula ilustrada na Fig.
11. Nesta situacao definimos seguintes vetores de rede:
Figura 11: Celula unitaria do grafeno com 12 atomos.
a1 = (3√
3a0, 0) e a2 = (0, 3a0). (4.1)
A rede de Bravais subjacente e retangular e, de fato, transformamos o problema inicial
do grafeno monocamada em um problema de uma rede de Bravais retangular, com uma
base de 12 atomos. Consequentemente, a rede recıproca tambem e um retangulo com
vetores de rede:
b1 =
(2π
3√
3a0
, 0
)e b2 =
(0,
2π
3a0
). (4.2)
Dessa forma, a primeira zona de Brillouin passa a ser o retangulo de vertices:
A =π
3a0
(− 1√
3,−1
)B =
π
3a0
(1√3,−1
)C =
π
3a0
(1√3, 1
)D =
π
3a0
(− 1√
3, 1
)(4.3)
que possui uma area menor que a primeira zona de Brillouin do grafeno perfeito monoca-
mada, conforme ilustrado pela Fig. 12. A estrutura de bandas usualmente representada
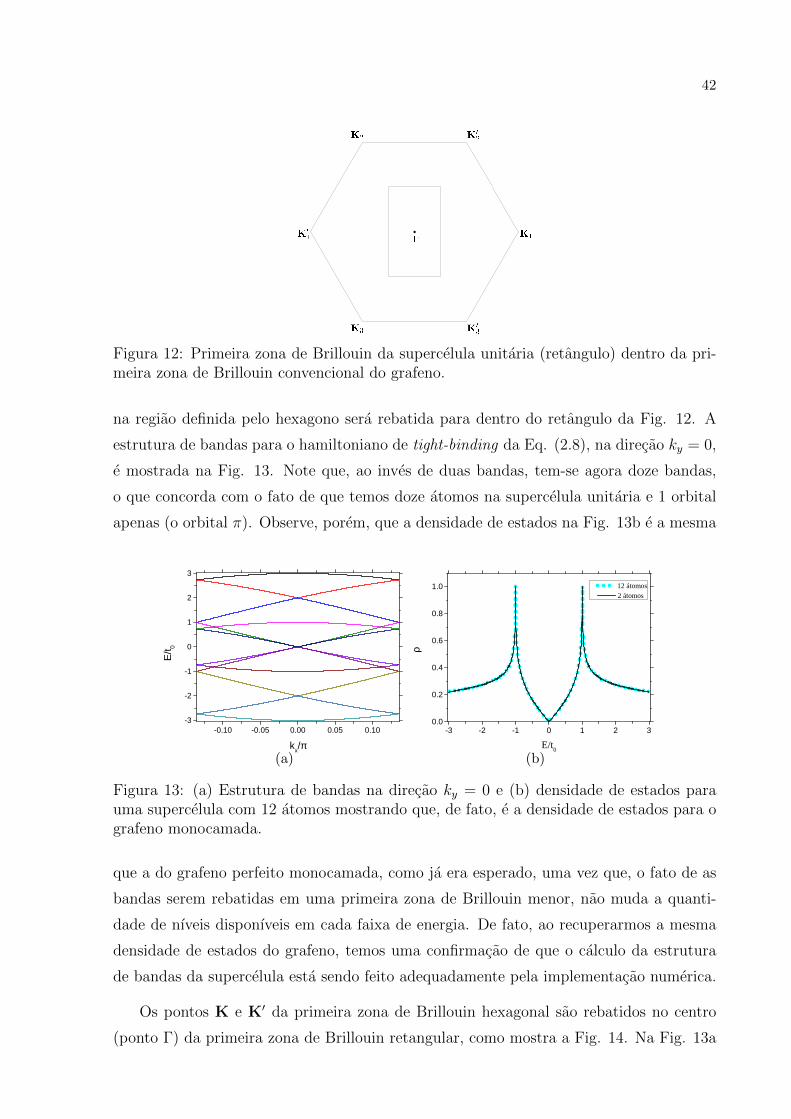
42
Figura 12: Primeira zona de Brillouin da supercelula unitaria (retangulo) dentro da pri-meira zona de Brillouin convencional do grafeno.
na regiao definida pelo hexagono sera rebatida para dentro do retangulo da Fig. 12. A
estrutura de bandas para o hamiltoniano de tight-binding da Eq. (2.8), na direcao ky = 0,
e mostrada na Fig. 13. Note que, ao inves de duas bandas, tem-se agora doze bandas,
o que concorda com o fato de que temos doze atomos na supercelula unitaria e 1 orbital
apenas (o orbital π). Observe, porem, que a densidade de estados na Fig. 13b e a mesma
- 0 . 1 0 - 0 . 0 5 0 . 0 0 0 . 0 5 0 . 1 0- 3
- 2
- 1
0
1
2
3
E/t0
k x / π(a)
- 3 - 2 - 1 0 1 2 30 . 0
0 . 2
0 . 4
0 . 6
0 . 8
1 . 0
ρ
E / t 0
1 2 á t o m o s 2 á t o m o s
(b)
Figura 13: (a) Estrutura de bandas na direcao ky = 0 e (b) densidade de estados parauma supercelula com 12 atomos mostrando que, de fato, e a densidade de estados para ografeno monocamada.
que a do grafeno perfeito monocamada, como ja era esperado, uma vez que, o fato de as
bandas serem rebatidas em uma primeira zona de Brillouin menor, nao muda a quanti-
dade de nıveis disponıveis em cada faixa de energia. De fato, ao recuperarmos a mesma
densidade de estados do grafeno, temos uma confirmacao de que o calculo da estrutura
de bandas da supercelula esta sendo feito adequadamente pela implementacao numerica.
Os pontos K e K′ da primeira zona de Brillouin hexagonal sao rebatidos no centro
(ponto Γ) da primeira zona de Brillouin retangular, como mostra a Fig. 14. Na Fig. 13a
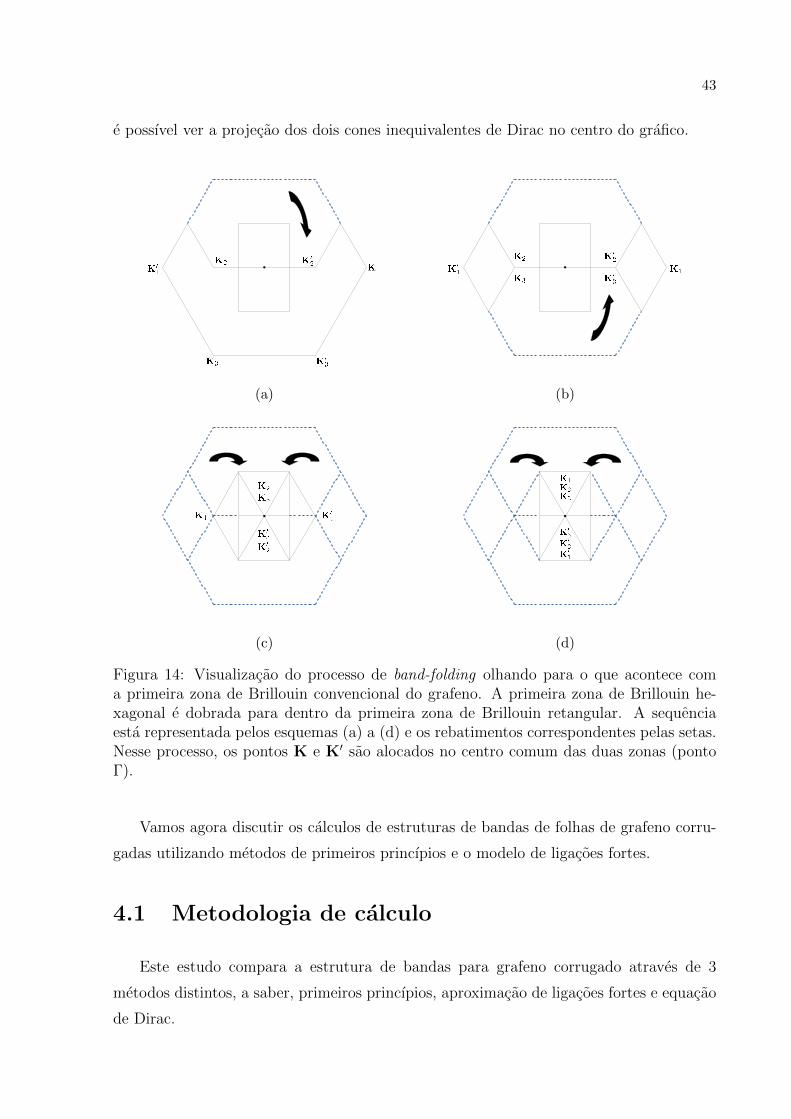
43
e possıvel ver a projecao dos dois cones inequivalentes de Dirac no centro do grafico.
(a)
(b)
(c)
(d)
Figura 14: Visualizacao do processo de band-folding olhando para o que acontece coma primeira zona de Brillouin convencional do grafeno. A primeira zona de Brillouin he-xagonal e dobrada para dentro da primeira zona de Brillouin retangular. A sequenciaesta representada pelos esquemas (a) a (d) e os rebatimentos correspondentes pelas setas.Nesse processo, os pontos K e K′ sao alocados no centro comum das duas zonas (pontoΓ).
Vamos agora discutir os calculos de estruturas de bandas de folhas de grafeno corru-
gadas utilizando metodos de primeiros princıpios e o modelo de ligacoes fortes.
4.1 Metodologia de calculo
Este estudo compara a estrutura de bandas para grafeno corrugado atraves de 3
metodos distintos, a saber, primeiros princıpios, aproximacao de ligacoes fortes e equacao
de Dirac.

44
Figura 15: Celula corrugada mostrando o comprimento de onda λ da corrugacao e quaisatomos sao fixos no processo de relaxacao das posicoes atomicas.
Os resultados de primeiros princıpios sao baseados na teoria do funcional da densi-
dade (DFT, do ingles density functional theory). Estes calculos foram realizados pelo
nosso colaborador Prof. Tome Schmidt da Universidade Federal de Uberlandia. Os resul-
tados foram obtidos usando um programa computacional para modelagem de materiais
em escala atomica chamado VASP e a aproximacao conhecida como GGA (Generalized
Gradient Approach) para os funcionais de troca e correlacao [32]. A interacao entre os
ıons e os eletrons de valencia e tratada usando o metodo PAW (Projected Augmented
Wave) [33]. As funcoes de onda de uma partıcula de Kohn-Sham sao representadas por
expansoes em ondas planas com cutoff de 350 eV.
As corrugacoes estudadas neste trabalho estao agrupadas em dois tipos: ondulacoes
de comprimentos de onda longo (mais suaves) e curto (mais abruptas). O tipo de ge-
ometria estudada esta mostrado Fig.15. Para folhas de grafeno com ondulacoes suaves
a supercelula unitaria possui 60 atomos, enquanto que para ondulacoes abruptas a su-
percelula unitaria possui 12 atomos, como mostra a Fig. 16. A corrugacao e gerada
fazendo-se os atomos, dentro da celula, seguirem uma forma gaussiana com altura hmax
e largura λ, mantendo-se o numero de atomos constante. A celula corrugada ocupa um
volume menor, entao a variacao do tamanho da celula forca a corrugacao. Os atomos das
bordas da celula sao fixados no plano da mesma (Fig. 15). Em seguida, as posicoes dos
atomos livres sao relaxadas repetidamente ate atingirem um mınimo de energia, usando
tecnicas padrao de calculos de primeiros princıpios. Nesse processo de relaxacao, a coor-
denada y tambem e mantida fixa, ou seja, a relaxacao ocorre apenas para as coordenadas
x e z dos atomos livres da Fig. 15. Apos o reposicionamento dos atomos para uma confi-
guacao de equilıbrio, a estrutura de bandas e calculada. As posicoes dos atomos na celula
unitaria, sao usadas como parametros de entrada para o calculo da estrutura de bandas

45
Figura 16: Exemplos de corrugacao de comprimento de onda curto (12 atomos na celulaunitaria) e longo (60 atomos na celula unitaria). As ondulacoes estao ao longo da direcaozigue-zague.
usando o modelo de tight-binding.
No calculo da estrutura de bandas usando o modelo de tight-binding resolvemos,
numericamente, a equacao:
Hψk = Eψk , (4.4)
onde o auto-estado ψk(r) e dado por:
ψk(r) =∑i
ck,iχk,i(r) (4.5)
onde i = 1, ..., Nb denota os atomos dentro da celula unitaria. No nosso estudo Nb = 60
para as ondulacoes suaves e Nb = 12 para as ondulacoes abruptas. As funcoes χk,i(r) sao
expressas por:
χk,i(r) =1√Nat
∑R
eik·Rφ(r− ti −R) (4.6)
onde ti e a posicao do atomo i dentro da celula unitaria, R sao vetores da rede e φ e a
funcao de onda do eletron no orbital π do grafeno.
Note que temos um problema de autovalor com uma matriz de dimensao N ×N . Os
elementos de matriz do hamiltoniano sao dados por:
〈χk,i|H|χk,i〉 = ε→ 0 (elementos da diagonal) (4.7)
〈χk,i|H|χk,j〉 =∑Rv
ti,jeik·Rv (elementos fora da diagonal) (4.8)
onde ti,j sao os hoppings entre os atomos i e j e Rv 6= 0 sao os vetores de rede no caso de
i e j estarem em celulas vizinhas (v). Os hoppings sao calculados usando-se a Eq. (3.29).
As distancias dij sao calculadas usando a expressao:
|dij| = |ti − tj| =[(xi − xj)2 + (yi − yj)2 + (zi − zj)2
]1/2. (4.9)
Nas proximas secoes vamos comparar os resultados de primeiros princıpios com calculos

46
de tight-binding. E importante enfatizar que o modelo tight-binding usado nao descreve
hibridizacao de ligacoes devido a curvaturas , restringindo-se a modelar o efeito de modi-
ficacoes do comprimento de ligacoes. Assim, a comparacao dos modelos da uma medida
da importancia das curvaturas na estrutura eletronica.
Finalmente , conhecendo as posicoes ti dos atomos e possıvel construir o vetor u(r)
ajustando uma funcao suave aos ti. No nosso estudo, usamos polinomios de grau 9.
A partir de u(r) calculamos numericamente os tensores de deformacao uij(r), que nos
permitem estimar as quantidades discutidas no capıtulo anterior usando a equacao de
Dirac e teoria de perturbacao (secao 3.4).
4.2 Resultados para curvaturas suaves
Vamos analisar primeiramente o caso onde as curvaturas sao suaves λ/hmax > 5.
Na analise dos resultados nos concentramos em estudar o que acontece na estrutura de
bandas para pontos proximos ao ponto Γ, ou seja, em torno do centro da primeira zona
de Brillouin. Nossa analise e feita considerando a direcao ky = 0. Assim como no caso
discutido em detalhe de Nb = 12, para ondulacoes com Nb = 60 e possıvel mostrar atraves
de band-folding que os cones de Dirac K e K′ caem no ponto Γ da supercelula. A Fig.
17 mostra a evidencia numerica de que isto e verdadeiro. As Fig. 18 a 23 mostram a
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2
- 2
0
2
E/t0
k x / π(a)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2
- 0 . 4
- 0 . 2
0 . 0
0 . 2
0 . 4
E/t0
k x / π(b)
Figura 17: (a)Relacao de dispersao para uma supercelula com 60 atomos, calculada emky = 0. O numero de vezes em que a estrutura de bandas e dobrada e bem maior que naFig. 13 pois a primeira zona de Brillouin e menor na direcao kx para esta supercelula. (b)Os cones de Dirac, porem, continuam situados no centro da primeira zona de Brillouin.
evolucao da relacao de dispersao para baixas energias para valores crescentes de hmax. As
figuras mostram que os resultados obtidos pelos dois metodos apresentados possuem um
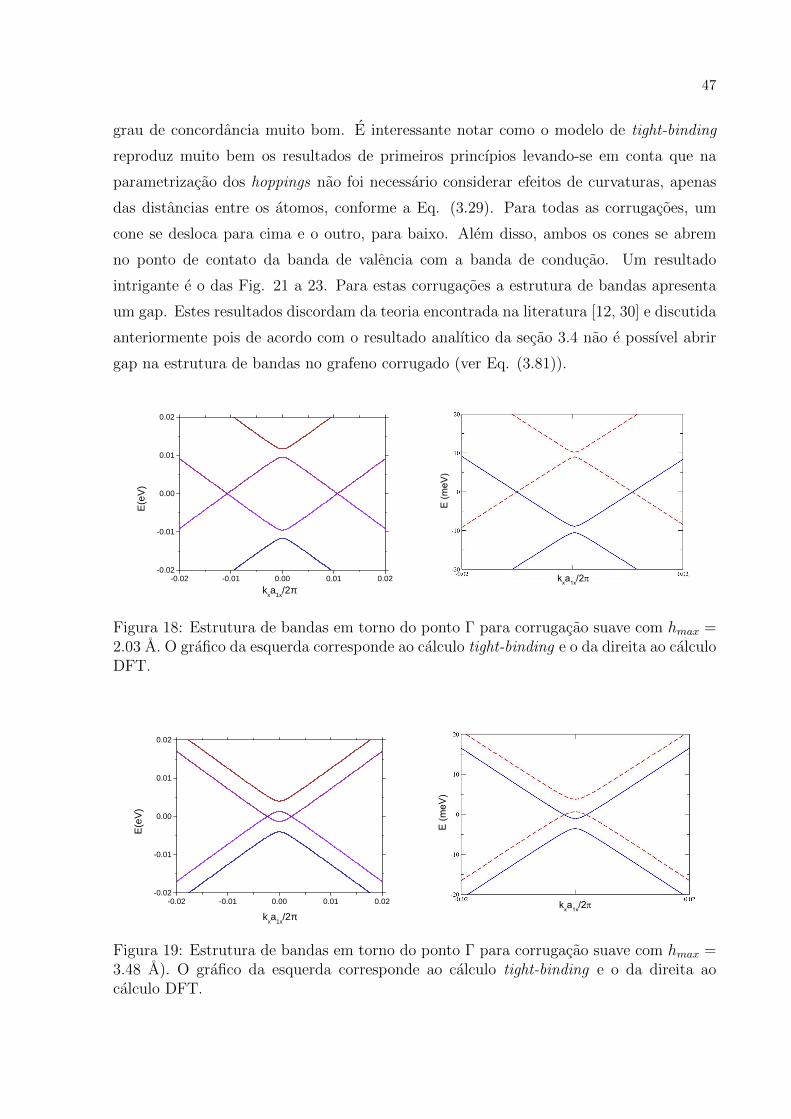
47
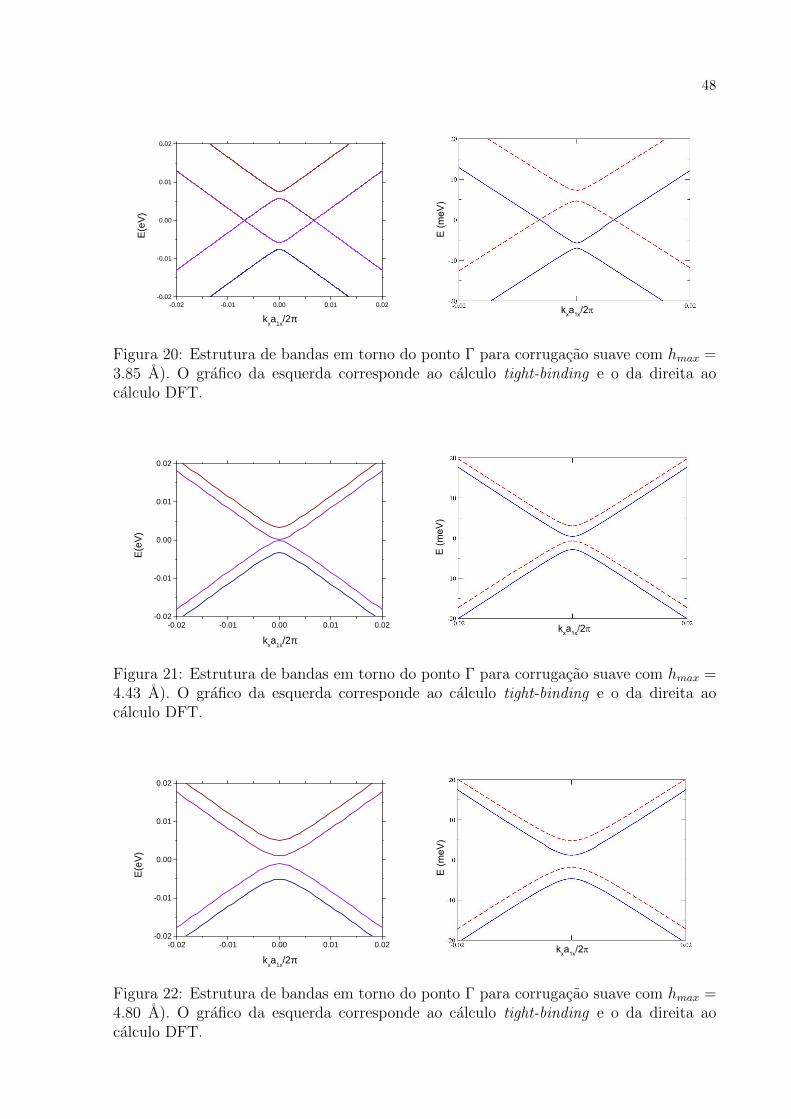
grau de concordancia muito bom. E interessante notar como o modelo de tight-binding
reproduz muito bem os resultados de primeiros princıpios levando-se em conta que na
parametrizacao dos hoppings nao foi necessario considerar efeitos de curvaturas, apenas
das distancias entre os atomos, conforme a Eq. (3.29). Para todas as corrugacoes, um
cone se desloca para cima e o outro, para baixo. Alem disso, ambos os cones se abrem
no ponto de contato da banda de valencia com a banda de conducao. Um resultado
intrigante e o das Fig. 21 a 23. Para estas corrugacoes a estrutura de bandas apresenta
um gap. Estes resultados discordam da teoria encontrada na literatura [12, 30] e discutida
anteriormente pois de acordo com o resultado analıtico da secao 3.4 nao e possıvel abrir
gap na estrutura de bandas no grafeno corrugado (ver Eq. (3.81)).
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
kxa
1x/2p
E (
meV
)
Figura 18: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =2.03 A. O grafico da esquerda corresponde ao calculo tight-binding e o da direita ao calculoDFT.
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
E (
meV
)
kxa
1x/2p
Figura 19: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =3.48 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
48
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(e
V)
k x a 1 x / 2 π
E (
meV
)
kxa
1x/2p
Figura 20: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =3.85 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
E (
meV
)
kxa
1x/2p
Figura 21: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =4.43 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
E (
meV
)
kxa
1x/2p
Figura 22: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =4.80 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
49
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(e
V)
k x a 1 x / 2 π
E (
meV
)
kxa
1x/2p
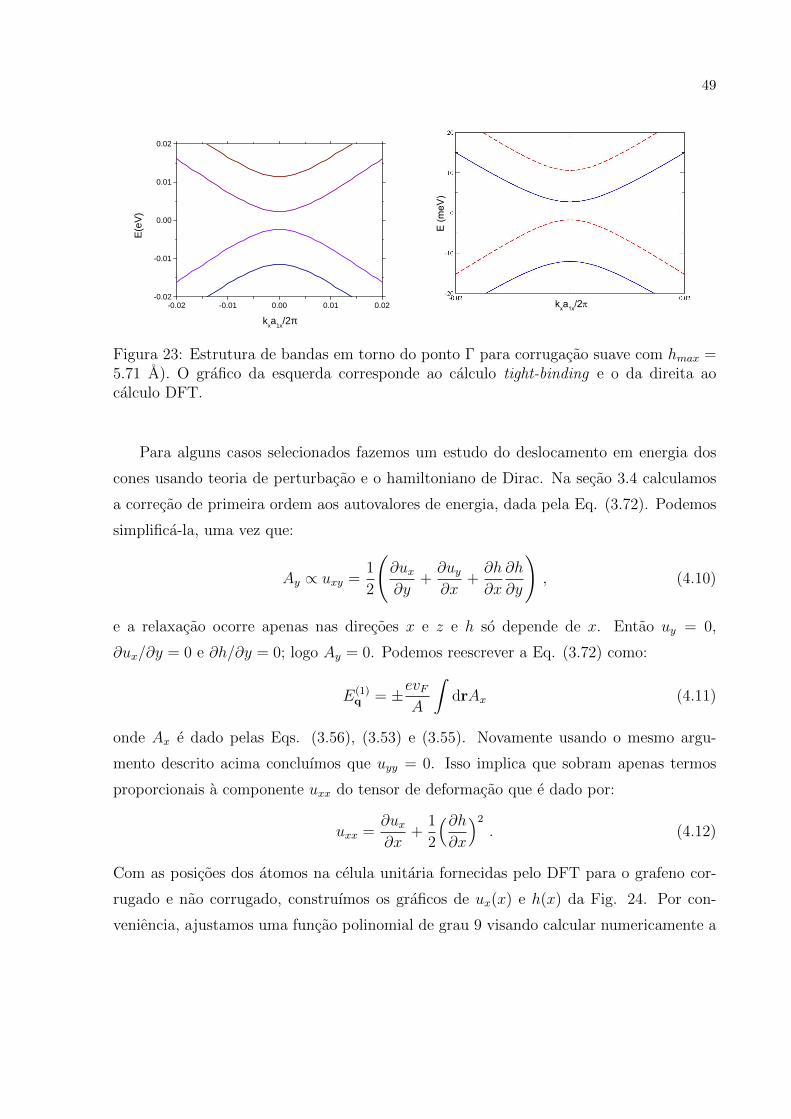
Figura 23: Estrutura de bandas em torno do ponto Γ para corrugacao suave com hmax =5.71 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
Para alguns casos selecionados fazemos um estudo do deslocamento em energia dos
cones usando teoria de perturbacao e o hamiltoniano de Dirac. Na secao 3.4 calculamos
a correcao de primeira ordem aos autovalores de energia, dada pela Eq. (3.72). Podemos
simplifica-la, uma vez que:
Ay ∝ uxy =1
2
(∂ux∂y
+∂uy∂x
+∂h
∂x
∂h
∂y
), (4.10)
e a relaxacao ocorre apenas nas direcoes x e z e h so depende de x. Entao uy = 0,
∂ux/∂y = 0 e ∂h/∂y = 0; logo Ay = 0. Podemos reescrever a Eq. (3.72) como:
E(1)q = ±evF
A
∫drAx (4.11)
onde Ax e dado pelas Eqs. (3.56), (3.53) e (3.55). Novamente usando o mesmo argu-
mento descrito acima concluımos que uyy = 0. Isso implica que sobram apenas termos
proporcionais a componente uxx do tensor de deformacao que e dado por:
uxx =∂ux∂x
+1
2
(∂h∂x
)2
. (4.12)
Com as posicoes dos atomos na celula unitaria fornecidas pelo DFT para o grafeno cor-
rugado e nao corrugado, construımos os graficos de ux(x) e h(x) da Fig. 24. Por con-
veniencia, ajustamos uma funcao polinomial de grau 9 visando calcular numericamente a
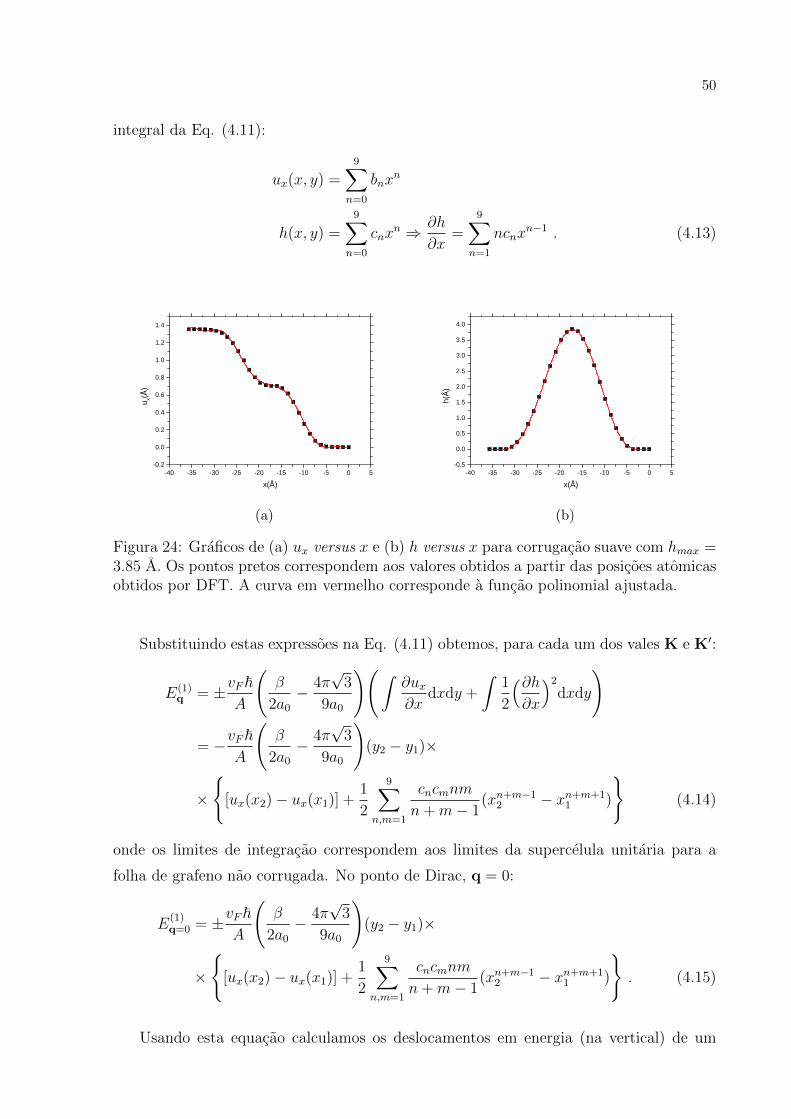
50
integral da Eq. (4.11):
ux(x, y) =9∑
n=0
bnxn
h(x, y) =9∑
n=0
cnxn ⇒ ∂h
∂x=
9∑n=1
ncnxn−1 . (4.13)
- 4 0 - 3 5 - 3 0 - 2 5 - 2 0 - 1 5 - 1 0 - 5 0 5- 0 . 2
0 . 0
0 . 2
0 . 4
0 . 6
0 . 8
1 . 0
1 . 2
1 . 4
u x(Å)
x ( Å )
(a)
- 4 0 - 3 5 - 3 0 - 2 5 - 2 0 - 1 5 - 1 0 - 5 0 5- 0 . 50 . 00 . 51 . 01 . 52 . 02 . 53 . 03 . 54 . 0
h(Å)
x ( Å )
(b)
Figura 24: Graficos de (a) ux versus x e (b) h versus x para corrugacao suave com hmax =3.85 A. Os pontos pretos correspondem aos valores obtidos a partir das posicoes atomicasobtidos por DFT. A curva em vermelho corresponde a funcao polinomial ajustada.
Substituindo estas expressoes na Eq. (4.11) obtemos, para cada um dos vales K e K′:
E(1)q = ±vF~
A
(β
2a0
− 4π√
3
9a0
)(∫∂ux∂x
dxdy +
∫1
2
(∂h∂x
)2
dxdy
)
= −vF~A
(β
2a0
− 4π√
3
9a0
)(y2 − y1)×
×
{[ux(x2)− ux(x1)] +
1
2
9∑n,m=1
cncmnm
n+m− 1(xn+m−1
2 − xn+m+11 )
}(4.14)
onde os limites de integracao correspondem aos limites da supercelula unitaria para a
folha de grafeno nao corrugada. No ponto de Dirac, q = 0:
E(1)q=0 = ±vF~
A
(β
2a0
− 4π√
3
9a0
)(y2 − y1)×
×
{[ux(x2)− ux(x1)] +
1
2
9∑n,m=1
cncmnm
n+m− 1(xn+m−1
2 − xn+m+11 )
}. (4.15)
Usando esta equacao calculamos os deslocamentos em energia (na vertical) de um

51
hmax (A) λ (A) E(1)q(TP )
(meV) E(1)q(DFT )
(meV) E(1)q(TB)
(meV)
2.03 27.87 15.4 9.6 10.63.48 27.19 8.1 2.3 2.73.85 26.94 19.8 6.1 6.74.43 26.52 21.0 1.2 1.74.80 26.22 29.4 1.6 3.05.71 25.39 47.7 4.5 6.9
Tabela 1: Comparacao entre os valores dos deslocamentos em energia dos cones de Di-rac calculados usando teoria de perturbacao (E
(1)q(TP )), calculos de primeiros princıpios
(E(1)q(DFT )
) e modelo de tight-binding (E(1)q(TB)
).
dos cones para as folhas de grafeno corrugadas. Os resultados sao mostrados na tabela
1, que apresenta tambem os deslocamentos obtidos numericamente a partir dos graficos
produzidos pelos metodos tight-binding e DFT (Figs. 18 a 23). A proximidade entre
os valores dos deslocamentos E(1)q(DFT )
e E(1)q(TB)
e mais uma evidencia da concordancia
entre os resultados produzidos pelos dois metodos. Contudo, os valores calculados usando
teoria de perturbacao nao fornecem resultados quantitativamente consistentes com o que
e observado nos graficos das Figs. 18 a 23. A tendencia e superestimar as correcoes para
deformacoes onde λ/hmax & 10. Ja para os casos onde λ/hmax . 10, a unica semelhanca
entre os resultados da teoria de perturbacao e dos graficos e que ambos variam de forma
bastante irregular, em decorrencia da variacao das corrugacoes.
Por fim, calculamos a estrutura de bandas para as folhas de grafeno corrugadas com
as posicoes atomicas nao relaxadas. Nesse caso o programa que faz o calculo de primeiros
princıpios calcula E(k) a partir das posicoes atomicas dadas, sem o uso do processo de
relaxacao descrito na secao 4.1. Os graficos estao mostrados na Fig. 25 e mostram que
para algumas corrugacoes, a relaxacao modifica consideravelmente a estrutura de bandas
como e o caso, por exemplo, da ondulacao com hmax = 5.71 A. Nesse caso, para a celula
relaxada a estrutura de bandas possui um gap enquanto que, na celula nao relaxada, ele
nao existe.
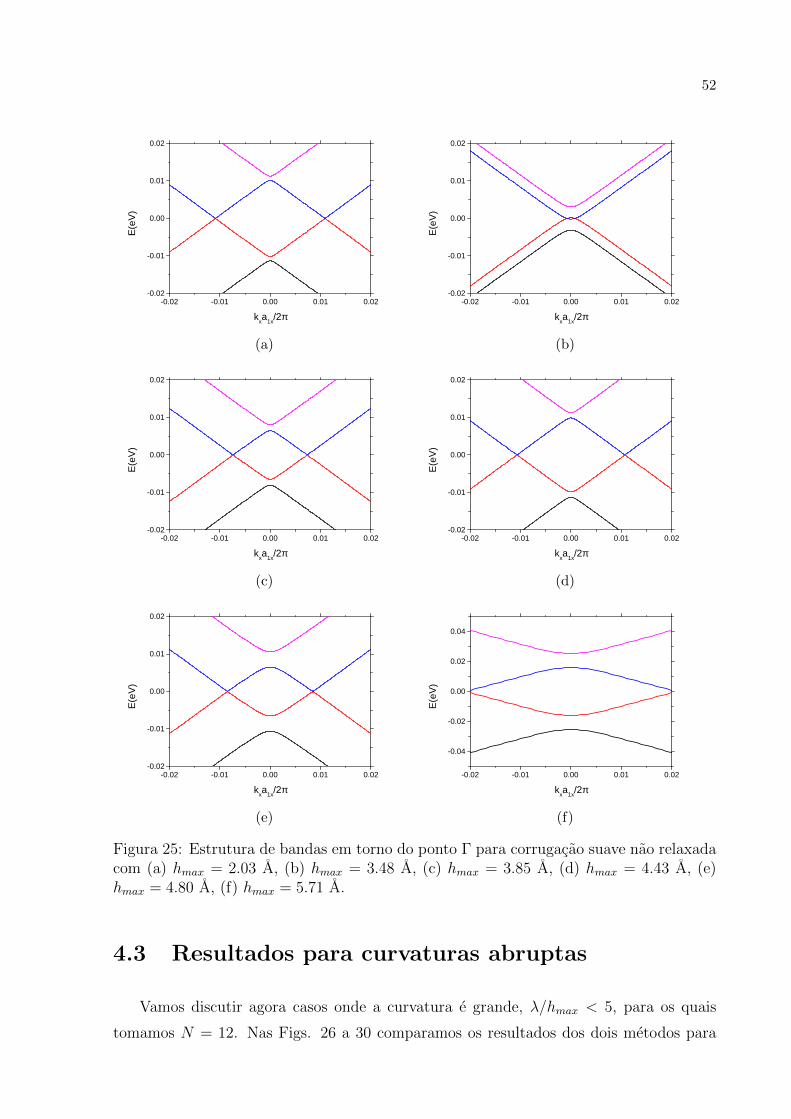
52
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(e
V)
k x a 1 x / 2 π
(a)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
(b)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
(c)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
(d)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2- 0 . 0 2
- 0 . 0 1
0 . 0 0
0 . 0 1
0 . 0 2
E(eV)
k x a 1 x / 2 π
(e)
- 0 . 0 2 - 0 . 0 1 0 . 0 0 0 . 0 1 0 . 0 2
- 0 . 0 4
- 0 . 0 2
0 . 0 0
0 . 0 2
0 . 0 4
E(eV)
k x a 1 x / 2 π
(f)
Figura 25: Estrutura de bandas em torno do ponto Γ para corrugacao suave nao relaxadacom (a) hmax = 2.03 A, (b) hmax = 3.48 A, (c) hmax = 3.85 A, (d) hmax = 4.43 A, (e)hmax = 4.80 A, (f) hmax = 5.71 A.
4.3 Resultados para curvaturas abruptas
Vamos discutir agora casos onde a curvatura e grande, λ/hmax < 5, para os quais
tomamos N = 12. Nas Figs. 26 a 30 comparamos os resultados dos dois metodos para

53
folhas de grafeno com ondulacoes abruptas. Neste caso, a discrepancia entre os calculos
de DFT e tight-binding e bem grande. A estrutura de bandas calculada pelo modelo de
tight-binding e simetrica em relacao ao eixo x em todos os graficos, o que nao acontece
com os resultados obtidos por primeiros princıpios das Figs. 26 a 29, onde um dos cones
de Dirac se abre de forma bastante assimetrica em relacao ao outro, que abre muito pouco.
Nos tres primeiros graficos DFT, um dos cones se desloca e o outro permanece centrado na
origem, enquanto que nos dois ultimos graficos, os dois cones se deslocam. Interpretamos
que estas propriedades sao oriundas da curvatura da folha, aspecto nao considerado pelo
modelo de tight-binding que usamos para calcular as bandas.
Curvaturas altas modificam e podem aumentar o overlap dos orbitais pz entre atomos
vizinhos, modificando os termos de hopping. Alguns autores sugerem que as curvaturas
provocam uma rehibridizacao dos orbitais pz com os orbitais sp2 [21, 22]. Investigamos
uma parametrizacao de tight-binding que leva em conta curvaturas, Eq. (3.33) [22]. Tal
modelo tambem falha em descrever os espectros de DFT e nao captura a quebra de
simetria partıcula-buraco. Isto sugere que seja necessario incluir hoppings entre vizinhos
mais distantes. Ainda nao investigamos essa possibilidade.
A medida que o comprimento de onda da corrugacao aumenta e a curvatura dimi-
nui, os resultados DFT e tight-binding parecem tender a concordar, como fica claro ao
acompanharmos os graficos em sequencia e ao notarmos a concordancia entre os dois re-
sultados evidenciada na Fig. 30. Para a configuracao escolhida neste caso, a relaxacao
dos atomos na celula unitaria produz uma folha de grafeno praticamente planar, porem
comprimida de aproximadamente 3%. Obviamente aqui, os efeitos de curvatura deixam
de ser importantes. Nesse caso, o modelo de tight-binding que usamos volta a ser eficiente
para modelar o problema e por isso temos a concordancia com os resultados DFT.
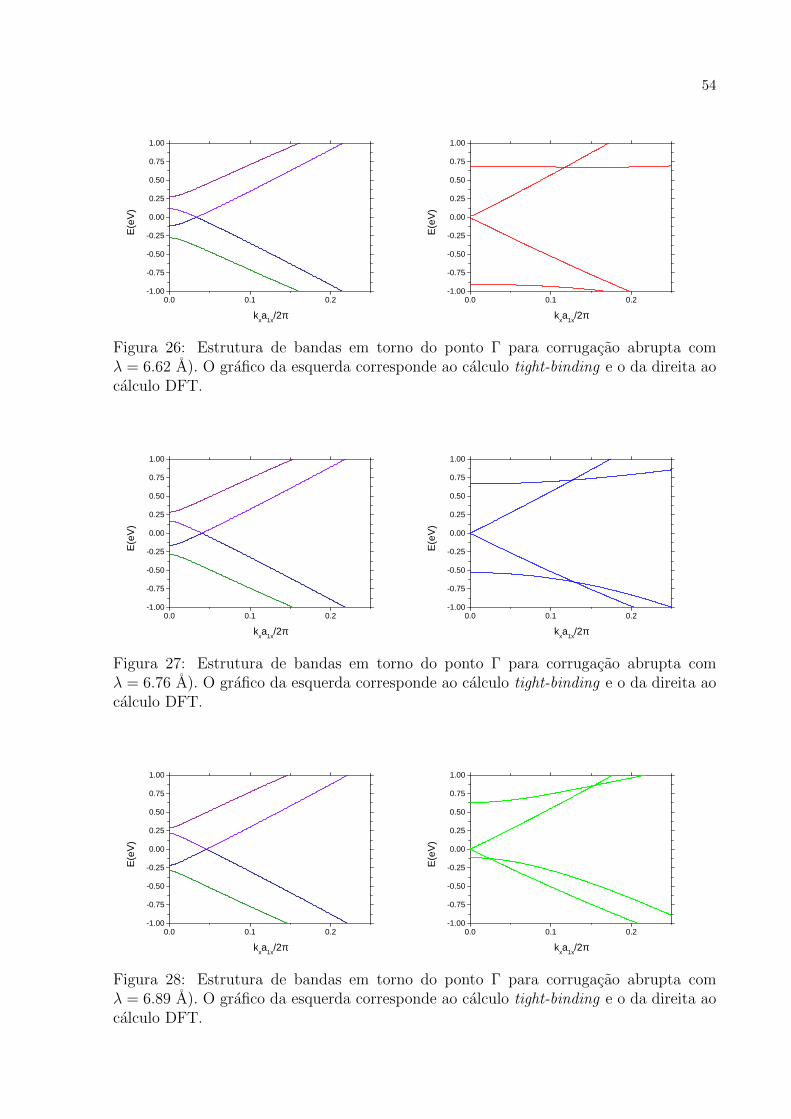
54
0 . 0 0 . 1 0 . 2- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(e
V)
k x a 1 x / 2 π0 . 0 0 . 1 0 . 2
- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π
Figura 26: Estrutura de bandas em torno do ponto Γ para corrugacao abrupta comλ = 6.62 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
0 . 0 0 . 1 0 . 2- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π0 . 0 0 . 1 0 . 2
- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(e
V)
k x a 1 x / 2 π
Figura 27: Estrutura de bandas em torno do ponto Γ para corrugacao abrupta comλ = 6.76 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
0 . 0 0 . 1 0 . 2- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π0 . 0 0 . 1 0 . 2
- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π
Figura 28: Estrutura de bandas em torno do ponto Γ para corrugacao abrupta comλ = 6.89 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
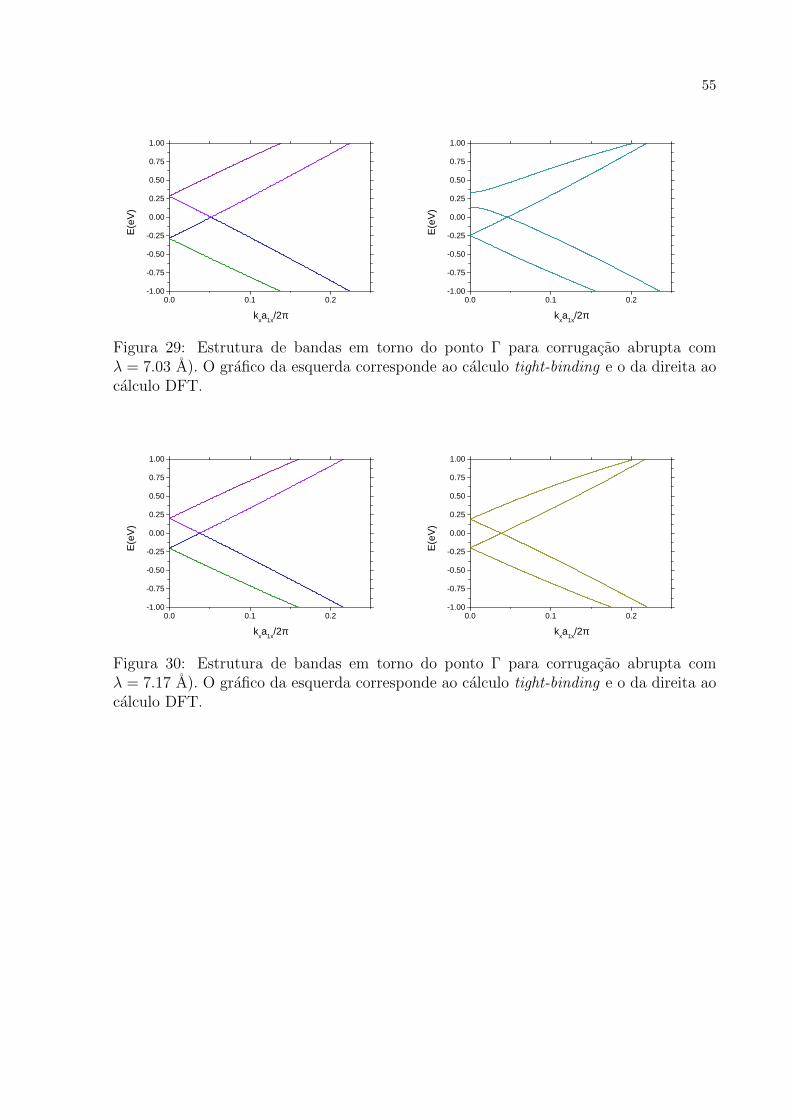
55
0 . 0 0 . 1 0 . 2- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(e
V)
k x a 1 x / 2 π0 . 0 0 . 1 0 . 2
- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π
Figura 29: Estrutura de bandas em torno do ponto Γ para corrugacao abrupta comλ = 7.03 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.
0 . 0 0 . 1 0 . 2- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(eV)
k x a 1 x / 2 π0 . 0 0 . 1 0 . 2
- 1 . 0 0- 0 . 7 5- 0 . 5 0- 0 . 2 50 . 0 00 . 2 50 . 5 00 . 7 51 . 0 0
E(e
V)
k x a 1 x / 2 π
Figura 30: Estrutura de bandas em torno do ponto Γ para corrugacao abrupta comλ = 7.17 A). O grafico da esquerda corresponde ao calculo tight-binding e o da direita aocalculo DFT.

56
5 Conclusoes e Perspectivas
Nesta dissertacao estudamos a estrutura eletronica de folhas de grafeno onduladas,
no contexto do que vem sendo chamado de strain engineering, ou seja, o controle das
propriedades eletronicas por meio de deformacoes mecanicas. Apresentamos resultados
numericos e analıticos da literatura e tambem discutimos as duas abordagens que utiliza-
mos para calcular a estrutura de bandas: modelo de tight-binding e calculo de primeiros
princıpios (DFT).
Vimos que para curvaturas suaves, o calculo da estrutura de bandas com o modelo de
tight-binding reproduz com bastante precisao os calculos efetuados com o uso de primeiros
princıpios. E interessante reiterar que para modelar as mudancas nos hoppings usados no
modelo de tight-binding, levou-se em conta apenas as distancias entre os atomos de car-
bono vizinhos, deprezando-se qualquer efeito associado as curvaturas da folha de grafeno.
Mesmo assim, os resultados concordam muito bem.
Ainda para curvaturas suaves, mostramos que a estrutura de bandas possui gap para
determinadas corrugacoes. Este resultado entra em conflito com o resultado analıtico da
literatura, reobtido na secao 3.4, que mostra que o gap e nulo para folhas de grafeno
corrugadas. Tambem mostramos o efeito da relaxacao das posicoes atomicas na estrutura
de bandas. Para a corrugacao com maior hmax a estrutura de bandas para as posicoes
relaxadas possui gap enquanto que na estrutura de bandas para as posicoes nao relaxadas
o gap desaparece. Por fim, vimos que os pequenos deslocamentos da estrutura de bandas
em torno do ponto Γ da primeira zona de Brillouin incentivaram-nos a usar a teoria de
perturbacao de primeira ordem para a construcao de modelos analıticos no limite de baixas
energias visando estimar estes deslocamentos. Contudo vimos que nossas estimativas nao
sao quantitativamente consistentes com os graficos obtidos.
Para curvaturas abruptas a descricao por meio do modelo de tight-binding e clara-
mente ineficiente para modelar o problema, fornecendo resultados bastante discrepantes
em relacao aos calculos de primeiros princıpios. Sugerimos que nesse regime talvez seja ne-

57
cessario considerar no modelo de tight-binding os efeitos das curvaturas sobre as integrais
de transferencia.
Nossa investigacao, portanto, continua e se concentra nos aspectos ainda nao es-
clarecidos. O principal e a discordancia entre os resultados para curvaturas suaves e o
resultado analıtico, o que inclui os deslocamentos em energia nos pontos K e K′ e tambem
a abertura do gap. Estudaremos tambem um modo eficiente de modelar as curvaturas
no modelo de tight-binding de modo a buscar um melhor ajuste entre resultados tight-
binding e DFT para curvaturas abruptas. Os resultados ja obtidos certamente estimulam
o aprofundamento da investigacao sobre o papel das corrugacoes na estrutura de bandas
do grafeno.

58
Referencias
[1] A. K. Geim and K. S. Novoselov. The rise of graphene. Nature materials, 6(3):183–91,March 2007.
[2] K S Novoselov, D Jiang, F Schedin, T J Booth, V V Khotkevich, S V Morozov, anda K Geim. Two-dimensional atomic crystals. Proceedings of the National Academyof Sciences of the United States of America, 102(30):10451–3, July 2005.
[3] A. H. Castro Neto, N. M. R. Peres, K. S. Novoselov, and A. K. Geim. The electronicproperties of graphene. Reviews of Modern Physics, 81(1):109–162, January 2009.
[4] Alexander L. Kitt, Vitor M. Pereira, Anna K. Swan, and Bennett B. Goldberg.Lattice-corrected strain-induced vector potentials in graphene. Physical Review B,85(11):115432, March 2012.
[5] Vitor Pereira and A. Castro Neto. Strain Engineering of Graphene’s ElectronicStructure. Physical Review Letters, 103(4):046801, July 2009.
[6] Gui Gui, Jianxin Zhong, and Zhenqiang Ma. Electronic properties of rippledgraphene. Journal of Physics: Conference Series, 402:012004, December 2012.
[7] Vitor Pereira, A. Castro Neto, and N. Peres. Tight-binding approach to uniaxialstrain in graphene. Physical Review B, 80(4):045401, July 2009.
[8] F. Guinea, M. I. Katsnelson, and A. K. Geim. Energy gaps and a zero-field quantumHall effect in graphene by strain engineering. Nature Physics, 6(1):30–33, September2009.
[9] Fernando de Juan, Juan L. Manes, and Marıa a. H. Vozmediano. Gauge fields fromstrain in graphene. Physical Review B, 87(16):165131, April 2013.
[10] M.A.H. Vozmediano, M.I. Katsnelson, and F. Guinea. Gauge fields in graphene.Physics Reports, 496(4-5):109–148, November 2010.
[11] Cristina Bena and Gilles Montambaux. Remarks on the tight-binding model ofgraphene. New Journal of Physics, 11(9):095003, September 2009.
[12] M.I. Katsnelson. Graphene: Carbon in Two Dimensions. Cambridge UniversityPress, 2012.
[13] Vitor M. Pereira, R. M. Ribeiro, N. M. R. Peres, and A. H. Castro Neto. Opticalproperties of strained graphene. EPL (Europhysics Letters), 92(6):67001, December2010.
[14] Neil W. Ashcroft and N. David Mermin. Solid State Physics. Cengage Learning,1976.

59
[15] Charles Kittel. Introduction to Solid State Physics. John Wiley & Sons, Inc., 8a ed.edition, 2004.
[16] M. Ramezani Masir, D. Moldovan, and F.M. Peeters. Pseudo magnetic field instrained graphene: Revisited. Solid State Communications, 175-176:76–82, December2013.
[17] P. Wallace. The Band Theory of Graphite. Physical Review, 71(9):622–634, May1947.
[18] Eduardo Vieira Castro. http://faraday.fc.up.pt/cfp/Members/evcastro/figs/imag.jpg/view.
[19] M.I. Katsnelson and K.S. Novoselov. Graphene: New bridge between condensedmatter physics and quantum electrodynamics. Solid State Communications, 143(1-2):3–13, July 2007.
[20] L. D. Landau and E. M. Lifshitz. Theory of Elasticity. Pergamon, third edition,1986.
[21] Eun-Ah Kim and A. H. Castro Neto. Graphene as an electronic membrane. EPL(Europhysics Letters), 84(5):57007, December 2008.
[22] A. Isacsson, L. Jonsson, J. Kinaret, and M. Jonson. Electronic superlattices incorrugated graphene. Physical Review B, 77(3):035423, January 2008.
[23] O. L. Blakslee et al. Elastic Constants of Compression-Annealed Pyrolytic Graphite.Journal of Applied Physics, 41(8):3373, 1970.
[24] Alexander L. Kitt, Vitor M. Pereira, Anna K. Swan, and Bennett B. Goldberg.Erratum: Lattice-corrected strain-induced vector potentials in graphene [Phys. Rev.B 85, 115432 (2012)]. Physical Review B, 87(15):159909, April 2013.
[25] Fernando de Juan, Mauricio Sturla, and Marıa a. H. Vozmediano. Space DependentFermi Velocity in Strained Graphene. Physical Review Letters, 108(22):227205, May2012.
[26] Hidekatsu Suzuura and Tsuneya Ando. Phonons and electron-phonon scattering incarbon nanotubes. Physical Review B, 65(23):235412, May 2002.
[27] T. L. Linnik. Effective Hamiltonian of strained graphene. Journal of physics. Con-densed matter : an Institute of Physics journal, 24(20):205302, May 2012.
[28] J. Manes. Symmetry-based approach to electron-phonon interactions in graphene.Physical Review B, 76(4):045430, July 2007.
[29] David J. Griffiths. Introduction to Quantum Mechanics. Pearson Education, secondedi edition, 2005.
[30] T. Low, F. Guinea, and M. I. Katsnelson. Gaps tunable by electrostatic gates instrained graphene. Physical Review B, 83(19):195436, May 2011.
[31] F. Guinea, Baruch Horovitz, and P. Le Doussal. Gauge field induced by ripples ingraphene. Physical Review B, 77(20):205421, May 2008.

60
[32] John P Perdew, Koblar A Jackson, Mark R Pederson, D J Singh, and Carlos Fio-lhais. Atoms, molecules, solids, and surfaces: Applications of the generalized gradientapproximation for exchange and correlation. Physical Review B, 46(11):6671–6687,September 1992.
[33] P. E. Blochl. Projector augmented-wave method. Physical Review B, 50(24):17953–17979, December 1994.





























