Embed Size (px)
DESCRIPTION
Gotardo
Citation preview

UNESP - UNIVERSIDADE ESTADUAL PAULISTA INSTITUTO DE QUÍMICA
CAMPUS DE ARARAQUARA PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
Desenvolvimento de métodos para análise de medicamentos utilizando reflectância difusa e
espectrofotometria
MARA ANDRÉIA GOTARDO
Tese de Doutorado 2006

1
MARA ANDRÉIA GOTARDO
Desenvolvimento de métodos para análise de medicamentos utilizando reflectância difusa e
espectrofotometria
Tese apresentada ao Instituto de Química, Universidade Estadual Paulista, como parte dos requisitos para obtenção do título de Doutor em Química.
Orientadora: Profª Drª Helena Redigolo Pezza
ARARAQUARA 2006

2

3
DADOS CURRICULARES
1. Formação Acadêmica 1.1. Graduação
Farmácia - Bioquímica
Faculdade de Ciências Farmacêuticas de Ribeirão Preto - USP
Período: 1995 a 1999
1.2. Pós-Graduação Mestrado em Ciências Farmacêuticas
Faculdade de Ciências Farmacêuticas - UNESP - Araraquara
Período: 2000 a 2002
Doutorado em Química
Instituto de Química - UNESP - Araraquara
Período: 2003 a 2006
2. Artigos Publicados 2.1. GOTARDO, M. A.; MONTEIRO, M. Migration of diethylhexyl phthalate from PVC bags into intravenous cyclosporine solutions. J. Pharm. Biomed. Anal., v. 38, n. 4, p. 709-713, July 2005.
2.2. GOTARDO, M. A.; PEZZA, L.; PEZZA, H. R. Determination of hydrochlorothiazide in pharmaceutical formulations by diffuse reflectance spectroscopy, Eclet. Quím., v. 30, n. 2, p. 17-24, 2005.
2.3. CIAPINA, E. G.; SANTINI, A. O.; LOS WEINERT, P.; GOTARDO, M. A.; PEZZA, H. R.; PEZZA, L. Spectrophotometric determination of diclofenac in pharmaceutical preparations assisted by microwave oven, Eclet. Quím., v. 30, n. 1, p. 29-36, 2005.
2.4. GOTARDO, M. A.; GIGANTE, A. C.; PEZZA, L.; PEZZA, H. R. Determination of furosemide in pharmaceutical formulations by diffuse reflectance spectroscopy. Talanta, v. 64, n. 2, p. 361-365, Oct. 2004.

4
2.5. GIGANTE, A. C.; GOTARDO, M. A.; TOGNOLLI, J. O.; PEZZA, L.; PEZZA, H. R. Spectrophotometric determination of formaldehyde with chromotropic acid in phosphoric acid medium assisted by microwave oven. Microchem. J., v. 77, n. 1, p. 47-51, May 2004.
2.6. QUEIROZ, R. H. C.; PEREIRA, R. C.; GOTARDO, M. A.; CORDEIRO, D. S.; MELCHIOR. E. Determination of clofazimine in leprosy patients by high-performance liquid chromatography. J. Anal. Toxicol., v. 27, n. 6, p. 377-380, Sept. 2003.
2.7. GOTARDO, M. A.; TOGNOLLI, J. O.; PEZZA, H. R.; PEZZA, L. Determination of propranolol in pharmaceutical formulations by combined spot test-diffuse reflectance spectroscopy, 2006 (submetido).
3. Trabalhos apresentados em Congresso 3.1. GOTARDO, M. A.; DORETTO, K. M.; PEZZA, H. R.; PEZZA, L. Determinação de metoclopramida em medicamentos utilizando a combinação espectroscopia de reflectância difusa – spot test. In: REUNIÃO ANUAL DA SOCIEDADE BRASILEIRA DE QUÍMICA, 29., 2006, Águas de Lindóia. p. 101.
3.2. GOTARDO, M. A.; MONTEIRO, M. Migration of DEHP from PVC bags containing large parenteral solutions with cyclosporine. In: COLACRO: CONGRESSO LATINO-AMERICANO DE CROMATOGRAFIA E TÉCNICAS AFINS, 10., 2004, Campos do Jordão., v. 1, p. 140.
3.3. GOTARDO, M. A.; L. PEZZA; H.; R. PEZZA. Spot test quantitativo para a determinação de furosemida em formulações farmacêuticas. In: REUNIÃO ANUAL DA SOCIEDADE BRASILEIRA DE QUÍMICA, 27.; CONGRESSO LATINO AMERICANO DE QUÍMICA, 26., 2004, Salvador, p. QA239.
3.4. RIBEIRO, P. R. S.; GOTARDO, M. A.; L. PEZZA; H.R. PEZZA. Spot test quantitativo para a determinação de metildopa em formulações farmacêuticas por espectroscopia de reflectância difusa. In: REUNIÃO ANUAL DA SOCIEDADE BRASILEIRA DE QUÍMICA, 27.; CONGRESSO LATINO AMERICANO DE QUÍMICA, 26., 2004, Salvador, p. QA238.
3.5. GOTARDO, M. A.; MONTEIRO, M. PVC bags containing large parenteral solutions with cyclosporine: should DEHP migrate? In: CONGRESS OF PHARMACEUTICAL SCIENCES, 4., 2003, Ribeirão Preto. Revista Brasileira de Ciências Farmacêuticas, v. 39, p. 123, 2003.

5
3.6. GOTARDO, M. A.; MONTEIRO, M. Uso da cromatografia gasosa de alta resolução na determinação de DEHP. In: CONGRESSO BRASILEIRO DE QUÍMICA, 42., 2002, Rio de Janeiro. p. 171. 3.7 GOTARDO, M. A.; MONTEIRO, M. Plasticizers identification in plastic bag used to large-volume parenteral solution (LVPS) by high-resolution gas chromatography. In: CONGRESS OF PHARMACEUTICAL SCIENCES, 3rd, 2001, Águas de Lindóia. European Journal of Pharmaceutical Sciences, v. 13, p. S49, 2001.
3.8. GOTARDO, M. A.; SANTIAGO-SILVA, M.; MONTEIRO, M. PVC plastic bag for large-volume parenteral solution (LPVS): is DEHP present? In: PHARMATECH, 6th., ANNUAL MEETING OF THE SBTF, 3rd, 2001, Recife. Proceedings. Recife: [s.n.], 2001, p.221-222.
3.9. GOTARDO, M. A.; CORDEIRO, D. S.; DREOSSI, S. A. C.; QUEIROZ, R. H. C. A rapid, specific and sensitive HPLC analysis of clofazimine in human plasma samples. In: CONGRESSO LATINO-AMERICANO DE CROMATOGRAFIA E TÉCNICAS AFINS, 7., 1998, Águas de São Pedro. Livro de Resumos. São Carlos: Acta Eventos, 1998. p.175-175.
3.10. GOTARDO, M. A.; QUEIROZ, R. H. C. HPLC analysis of clofazimine in plasma. In: CONGRESS OF PHARMACEUTICAL SCIENCES, 1st, 1997, Ribeirão Preto. Bolletino Chimico Farmaceutico Rivista di Scienze Farmaceutiche e Biologiche. Milano: Società Editoriale Farmaceutica, v. 136, n. 2, p. 50, 1997.

6
DEDICATÓRIA Aos meus pais João e Dirce,
por todo amor e essencial apoio para a minha vida.
Aos meus irmãos Miriam e João Roberto,
por quem tenho muito carinho e admiração.
Ao Henrique,
que surgiu na minha vida para torná-la mais alegre
e iluminada, pelo apoio, carinho e amor.

7
AGRADECIMENTOS
A CAPES pela bolsa de Doutorado.
À Profª Helena Redigolo Pezza pela orientação, confiança e preocupação em ajudar,
sempre com muita energia e perseverança.
Ao Prof. Leonardo Pezza pela confiança, apoio e amizade.
Ao Prof. João Olímpio Tognolli pela colaboração na parte estatística deste trabalho.
Á Profª Drª Adriana Vitorino Rossi (Unicamp – Campinas) pela atenção e auxílio na
parte de montagem e uso do sistema portátil.
Aos membros da banca examinadora pela atenção e importante colaboração na
apreciação crítica deste trabalho.
Ao Prof. Massao Ionashiro pelos valiosos ensinamentos, incentivo e sincera
amizade.
Aos amigos do Grupo Fritz Feigl: Alberto (Fritz), Paulo, Elaine, Luciana, Keity,
Liliane, Patrícia, Zé Luiz, Fernanda, Sahra, Suelen, Joel, Marcão, Fabrícia, Tuane e
Thiago.
Aos amigos do Grupo Latig: Adriano, Cláudio, Elias, Emanuel, Gilbert e Marcelo.
Aos funcionários da biblioteca, seção de pós-graduação e do Departamento de
Química Analítica pela atenção e gentileza com que sempre me atenderam.

8
RESUMO
Este trabalho propõe métodos por espectroscopia de reflectância difusa utilizando spot
test para determinação de furosemida, hidroclorotiazida, propranolol e atenolol em
formulações farmacêuticas e também um método espectrofotométrico no visível para
determinação de metildopa em formulações farmacêuticas. Os métodos reflectométricos
foram baseados em reações em papel de filtro, utilizando p-dimetilaminocinamaldeído
(PDAC) como reagente cromogênico para furosemida e hidroclorotiazida; 2,6-
dicloroquinona-4-cloroimida (DCQ) para propranolol e p-cloranil para atenolol. Estas reações
produziram compostos coloridos com máximo de AR (log 1/R) em 585 nm para furosemida e
hidroclorotiazida; 500 nm para propranolol e 550 nm para atenolol. Planejamentos
experimentais foram empregados no desenvolvimento de todos os métodos e, as condições
otimizadas para as respectivas reações de spot test foram: 10 μL de solução de furosemida
em acetona, 20 μL de HCl 6,3% (m/v) em metanol e 20 μL de PDAC 0,4% (m/v)
em metanol, nesta ordem, com aquecimento a 80ºC por 5 minutos; 20 μL de solução de
hidroclorotiazida em acetona, 20 μL de HCl 10% (m/v) em metanol e; 20 μL de PDAC 0,4%
(m/v) em metanol, nesta ordem, com aquecimento a 80ºC por 8 minutos; 30 μL de solução de
propranolol em etanol 35% (v/v) e 30 μL de solução de DCQ 70 mg/mL em acetona e; 20 μL
de solução de atenolol em metanol e 20 μL de p-cloranil 8,00 × 10-2 mol L-1 em dioxano. As
curvas analíticas foram lineares nas faixas de concentração de 7,56 × 10-3 – 6,05 × 10-2 mol L-1
(r=0,9987) para furosemida, 3,36 × 10-2 – 1,01 × 10-1 mol L-1 (r=0,9979) para
hidroclorotiazida, 1,35 × 10-2 – 8,45 × 10-2 mol L-1 (r=0.9991) para propranolol e 1,13 × 10-2 –
7,88 × 10-2 mol L-1 (r=0,9992) para atenolol. O método espectrofotométrico para a
determinação de metildopa em formulações farmacêuticas utilizou p-cloranil como reagente
cromogênico e H2O2 como acelerador da reação. Planejamentos experimentais foram

9
realizados e a condição ótima para a reação foi obtida com a adição de 770 μL de p-cloranil e
85 μL de H2O2 4,55 mol L-1 para um volume final de 5,00 mL, em meio de metanol. A lei de
Beer foi obedecida na faixa de concentração de 4,20 × 10-4 a 2,48 × 10-3 mol L-1, com r =
0.9993. Tal método foi adaptado, com sucesso, a um dispositivo portátil baseado em medidas
fotométricas. Todos os métodos desenvolvidos neste trabalho foram aplicados na análise de
diversas marcas comerciais de formulações farmacêuticas e, comparados estatisticamente com
métodos descritos nas farmacopéias. Os resultados, para todos os métodos, foram
concordantes com relação à precisão e exatidão, demonstrando a possibilidade de se utilizar
métodos alternativos, vantajosos e confiáveis para a determinação de furosemida,
hidroclorotiazida, propranolol, atenolol e metildopa em formulações farmacêuticas.
Palavras-chave: espectroscopia de reflectância difusa; spot test; espectrofotometria;
furosemida; hidroclorotiazida; propranolol; atenolol; metildopa.

10
ABSTRACT
This work proposes methods by diffuse reflectance spectroscopy using spot tests for
the determination of furosemide, hydrochlorothiazide, propranolol and atenolol in
pharmaceutical formulations and also a visible spectrophotometric method for the
determination of methyldopa in pharmaceutical formulations. The reflectometric methods
were based on spot test reactions on filter paper, using p-dimetylaminocinnamaldehyde (p-
DAC) as chromogenic reagent for furosemide and hydrochlorothiazide, 2,6-dichloroquinone-
4-chloroimide (DCQ) for propranolol and, p-chloranil for atenolol. These reactions produced
colored compounds with maximum AR (log 1/R) at 585 nm for furosemida and
hydrochlorothiazide; 500 nm for propranolol and 550 nm for atenolol. Experimental designs
were employed in the development of all methods and, the conditions optimized for the
respective spot test reactions were: 10 μL of furosemide solution in acetone, 20 μL of HCl
6.3% (w/v) in methanol and 20 μL of PDAC 0.4% (w/v) in methanol, in this order,
with heating at 80ºC for 5 minutes; 20 μL of hydrochlorotiazide solution in acetone, 20 μL of
HCl 10% (w/v) in methanol and 20 μL of p-DAC 0.4% (w/v) in methanol, in this order, with
heating at 80ºC for 8 minutes; 30 μL of propranolol solution in ethanol 35% (v/v) and 30 μL
of DCQ solution at 70 mg/mL in acetone and, 20 μL of atenolol solution in methanol and 20
μL of p-cloranil at 8.00 × 10-2 mol L-1 in dioxane. The analytical curves were linear in the
concentration ranges of 7.56 × 10-3 – 6.05 × 10-2 mol L-1 (r=0.9987) for furosemide, 3.36 × 10-2 –
1.01 × 10-1 mol L-1 (r=0.9979) for hydrochlorothiazide, 1.35 × 10-2 – 8.45 × 10-2 mol L-1
(r=0.9991) for propranolol and (r=0.9992) for atenolol. The spectrophotometric method for
the determination of methyldopa in pharmaceutical formulations used p-chloranil as
chromogenic reagent and H2O2 as accelerator of the reaction. Experimental designs were
carried out and the optimal condition for the reaction was obtained with the addition of 770

11
μL of p-chloranil and 85 μL of H2O2 at 4.55 mol L-1 for a final volume of 5.00 mL, in
methanol medium. Beer´s law was obeyed in a concentration range from 4.20 × 10-4 to 2.48 ×
10-3 mol L-1, with r = 0.9993. Such method was successfully adapted to a portable device
based on photometric measurements. All methods developed in this work were applied for
analysis of some commercial brands of pharmaceuticals and statistically compared with
methods described in the pharmacopeias. The results for all methods were in agreement in
relation to precision and accuracy, demonstrating the possibility of using alternative,
advantageous and reliable methods for the determination of furosemida, hydrochlorothiazide,
propranolol, atenolol and methyldopa in pharmaceutical formulations.
Keywords: diffuse reflectance spectroscopy; spot test; spectrophotometry;
furosemida; hydrochlorothiazide; propranolol; atenolol; methyldopa.

12
LISTA DE ILUSTRAÇÕES
Figura 1. Estrutura química da furosemida 24
Figura 2. Estrutura química da hidroclorotiazida 25
Figura 3. Estrutura química do propranolol 27
Figura 4. Estrutura química do atenolol 28
Figura 5. Estrutura química da metildopa 29
Figura 6. Vista superior do acessório de reflectância Labsphere modelo RSA-HP-53
67
Figura 7. Espectros de reflectância obtidos a partir do produto da reação de spot test entre butenamida e p-dimetilaminocinamaldeído, em meio ácido. As análises foram realizadas utilizando um acessório de reflectância Labsphere, modelo DRA-CA-3300. Concentração de butemanida = 1,37 × 10-3 mol L-1. Figura 7a. Espectro de reflectância obtido a partir dos valores de %R. Figura 7b. Espectro de reflectância obtido a partir dos valores de )/1log( R . Figura 7c. Espectro de reflectância obtido a partir dos valores de )(Rf
69
Figura 8. Distribuição das publicações de métodos por espectroscopia de reflectância difusa na região do visível para análises quantitativas. Período: 1960-2006
71
Figura 9. Suporte utilizado na realização dos spot tests (Baseado em: TUBINO et al., 1997)
78
Figura 10. Espectro de reflectância do produto formado na reação entre furosemida e PDAC, em meio ácido, após aquecimento a 80ºC por 5 min. O comprimento de onda máximo foi 585 nm. Solução padrão de furosemida: 3,78 × 10-2 mol L-1
83
Figura 11. Gráficos tridimensionais obtidos para estabelecer condições ótimas de tempo e temperatura de aquecimento (A) e de volumes de PDAC e de ácido adicionados (B)
88
Figura 12. Espectro de reflectância do produto formado na reação entre furosemida e PDAC, em meio ácido, após aquecimento a 80ºC por 5 min. O comprimento de onda máximo foi 585 nm. Solução padrão de furosemida: 3,78 × 10-2 mol L-1
89
Figura 13. Curva analítica para a determinação de furosemida. Coeficiente de
correlação linear (r) = 0,9987. Valores de AR foram obtidos em 585 nm. Faixa de concentração das soluções padrão de furosemida: 7,56 × 10-3 a 6,05 × 10-2 mol L-1. As barras representam os desvios padrão para 3 repetições
90

13
Figura 14. Espectro de reflectância do produto formado na reação entre hidroclorotiazida e PDAC, em meio ácido, após aquecimento a 80ºC por 8 min. O comprimento de onda máximo foi 585 nm. Solução padrão de hidroclorotiazida: 4,20 × 10–2 mol L-1
94
Figura 15. Gráfico de Pareto do planejamento fatorial fracionário para a reação entre hidroclorotiazida e PDAC, sobre papel de filtro
97
Figura 16. Gráfico tridimensional obtido no estabelecimento das melhores condições de tempo e temperatura de aquecimento para a reação entre hidroclorotiazida e PDAC sobre papel de filtro
99
Figura 17. Estudo da estabilidade óptica do produto da reação entre hidroclorotiazida e PDAC, em meio ácido, sobre papel de filtro. As análises dos spot tests foram realizadas a cada 5 minutos até 40 minutos. Solução padrão de hidroclorotiazida: 1,01 × 10-1 mol L-1
100
Figura 18. Curva analítica para a determinação de hidroclorotiazida. Coeficiente de correlação linear (r) = 0,9979. Valores de AR foram obtidos em 585 nm. Concentrações das soluções padrão de hidroclorotiazida: 3,36 × 10-2 a 1,01 × 10-1 mol L-1. As barras representam os desvios padrão para 3 repetições
101
Figura 19. Espectro de reflectância do produto formado na reação entre propranolol e DCQ. Os spot tests foram analisados 10 min. após a adição das soluções sobre o papel de filtro. O comprimento de onda máximo foi em 500 nm. Solução padrão de propranolol: 8,45 × 10–2 mol L-1
106
Figura 20. Gráfico de Pareto do planejamento fatorial completo para a reação entre propranolol e DCQ, sobre papel de filtro
119
Figura 21. Superfície de resposta obtida no planejamento composto central para valores de AR em função da concentração de DCQ e mistura etanol-água (%, v/v) como solvente para propranolol
112
Figura 22. Estudo da estabilidade óptica do produto da reação entre propranolol e DCQ. As análises dos spot tests foram realizadas a cada 2 minutos até 60 minutos. Solução padrão de propranolol: 8,45 × 10-2 mol L-1
113
Figura 23. Curva analítica para determinação de propranolol. Coeficiente de correlação linear (r) = 0,9991. Valores de AR foram obtidos em 500 nm. Concentrações das soluções padrão de propranolol: 1,35 × 10-2 a 8,45 × 10-2 mol L-1. As barras representam o desvio padrão para 3 repetições
114
Figura 24. Espectro de reflectância do produto formado na reação entre atenolol e p-cloranil. O comprimento de onda máximo foi em 550 nm. Solução padrão de atenolol: 7,88 × 10–2 mol L-1
120

14
Figura 25. Efeito da concentração de p-cloranil na resposta (AR). As barras representam os desvios padrão para 3 repetições
121
Figura 26. Estudo da estabilidade óptica do produto da reação entre atenolol e p-cloranil. As análises dos spot tests foram realizadas no tempo zero, após 1 minuto e a cada 2 min. até 40 min. Solução padrão de atenolol: 7,88 × 10-2 mol L-1
122
Figura 27. Curva analítica para a determinação de atenolol. Coeficiente de correlação linear (r) = 0,9992. Valores de AR foram obtidos em 550 nm. Concentrações das soluções padrão de atenolol: 1,13 × 10-2 a 7,88 × 10-2 mol L-1. As barras representam os desvios padrão para 3 repetições
123
Figura 28. Espectro de absorção do produto da reação entre metildopa e p-cloranil. O comprimento de onda analítico foi 535 nm. Concentração final de metildopa = 1,07 × 10–3 mol L-1; caminho óptico: 1,0 cm
135
Figura 29. Estudo da estabilidade óptica do produto da reação entre metildopa e p-cloranil. As medidas de absorbância foram realizadas a cada 2 minutos até 60 minutos. Concentração final de metildopa: 7,14 × 10–4 mol L-1
135
Figura 30. Superfície de resposta obtida a partir dos valores de absorbância em função do volume adicionado de p-cloranil e volume de H2O2
138
Figura 31. Curva analítica para a determinação espectrofotométrica de metildopa. Valores de absorbância foram obtidos em 535 nm. Coeficiente de correlação linear (r) = 0,9993. As barras representam o desvio padrão para 3 repetições
139
Figura 32. Dispositivo portátil de medidas fotométricas: (a) Montagem do sistema para medidas fotométricas. (b) Esquema da cela de Teflon® (Fonte: CRIVELENTE, 2003)
143
Figura 33. Curva analítica para a determinação de metildopa por medidas fotométricas utilizando dispositivo portátil. Coeficiente de correlação linear (r) = 0,9986. As barras representam o desvio padrão para 3 repetições
144

15
LISTA DE TABELAS
Tabela 1. Métodos por espectrofotometria no visível descritos na literatura para a determinação de furosemida emedicamentos
33
Tabela 2. Métodos analíticos por CLAE descritos na literatura para a determinação de furosemida associada ou não a outros fármacos em comprimidos
36
Tabela 3. Métodos por espectrofotometria UV derivativa descritos na literatura para a determinação de hidroclorotiazida em formulações farmacêuticas combinadas
40
Tabela 4. Métodos analíticos por CLAE descritos na literatura para a determinação de hidroclorotiazida associada ou não a outros fármacos em comprimidos
42
Tabela 5. Comparação entre alguns métodos espectrofotométricos e seus reagentes cromogênicos descritos na literatura para a determinação de propranolol em formulações farmacêuticas
46
Tabela 6. Métodos espectrofotométricos no visível descritos na literatura para a determinação de metildopa em formulações farmacêuticas
54
Tabela 7. Número de marcas comerciais de formulações farmacêuticas analisadas em cada método desenvolvido e conteúdo declarado de fármaco por comprimido
77
Tabela 8. Quantidades adicionadas de fármaco padrão dadas em mg e em concentração (mol L-1)
81
Tabela 9. Combinações das variáveis estudadas e resultados dos planejamentos 22 com ponto central
87
Tabela 10. Resultados de recuperação de furosemida padrão adicionada em amostras de três formulações farmacêuticas comerciais diferentes
91
Tabela 11. Determinação de furosemida em formulações farmacêuticas comerciais 92
Tabela 12. Matriz do planejamento fatorial fracionário 96
Tabela 13. Matriz do planejamento fatorial completo com ponto central 98
Tabela 14. Resultados de recuperação de hidroclorotiazida padrão adicionada em três formulações farmacêuticas comerciais diferentes
102
Tabela 15. Determinação de hidroclorotiazida em formulações farmacêuticas comerciais
103
Tabela 16. Matriz e resultados do planejamento fatorial completo 108
Tabela 17. Matriz e resultados obtidos no planejamento composto central 111

16
Tabela 18. Resultados de recuperação de propranolol adicionado em formulações farmacêuticas
116
Tabela 19. Determinação de propranolol em formulações farmacêuticas comerciais 117
Tabela 20. Resultados de recuperação de atenolol padrão adicionado em três formulações farmacêuticas comerciais diferentes
125
Tabela 21. Determinação de atenolol em formulações farmacêuticas comerciais 126
Tabela 22. Matriz do planejamento composto central e valores de absorbância referentes a cada experimento
137
Tabela 23. Resultados de recuperação de metildopa adicionada em formulações farmacêuticas
141
Tabela 24. Determinação de metildopa em formulações farmacêuticas comerciais 142
Tabela 25. Determinação de metildopa em formulações farmacêuticas comerciais 146

17
LISTA DE ABREVIATURAS E SIGLAS AAS espectrometria de absorção atômica
CCD cromatografia em camada delgada
CCDAE cromatografia em camada delgada de alta eficiência
CLAE cromatografia líquida de alta eficiência
CLS mínimos quadrados clássicos
DCQ 2,6-dicloroquinona-4-cloroimida
DDQ 2,3-dicloro-5,6-diciano-1,4-benzoquinona
DNA ácido desoxirribonucléico
FIA análise por injeção em fluxo
ILS mínimos quadrados inverso
LDR light dependent resistor (resistor dependente de luz)
LED light emitting diode (diodo emissor de luz)
ND não descrito
PCR regressão de componentes principais
PDAC p-dimetilaminocinamaldeído
PLS mínimos quadrados parciais
PTFE politetrafluoretileno
PVC cloreto de polivinila
RDC resolução da diretoria colegiada
RMN ressonância magnética nuclear
SDS dodecil sulfato de sódio
TCNE tetracianoetileno
TCNQ 7,7,8,8-tetracianoquinodimetano
UV Ultravioleta

18
LISTA DE SÍMBOLOS A absorbância
RA densidade óptica para medida de reflectância
b caminho óptico
C concentração
I intensidade da radiação refletida
0I intensidade da radiação incidente
r coeficiente de correlação linear
R reflectância ou poder de reflexão
s coeficiente de dispersão
ε absortividade molar
k coeficiente de absorção molar
λ máx. comprimento de onda de máxima absorção

19
Sumário
CAPÍTULO I - INTRODUÇÃO .............................................................................22
I. 1. CONSIDERAÇÕES GERAIS ..............................................................................23 I. 1.1. Furosemida ..................................................................................................................................... 23 I. 1.2. Hidroclorotiazida........................................................................................................................... 24 I. 1.3. Propranolol...................................................................................................................................... 26 I. 1.4. Atenolol ............................................................................................................................................ 27 I. 1.5. Metildopa ......................................................................................................................................... 28
CAPÍTULO II. REVISÃO BIBLIOGRÁFICA - MÉTODOS ANALÍTICOS PARA QUANTIFICAÇÃO DOS FÁRMACOS EM ESTUDO................................29
II. 1. FUROSEMIDA .......................................................................................................30 II. 1.1. Espectrofotometria na Região do Visível ........................................................................... 30 II. 1.2. Espectrofotometria na região do UV ................................................................................... 32 II. 1.3. Cromatografia .............................................................................................................................. 33 II. 1.4. Titulometria .................................................................................................................................. 36 II. 1.5. Outras Técnicas Analíticas ...................................................................................................... 36
II. 2. HIDROCLOROTIAZIDA ...................................................................................37 II. 2.1. Espectrofotometria na região do UV ................................................................................... 37 II. 2.2. Cromatografia Líquida de Alta Eficiência (CLAE)............................................................ 40 II. 2.3. Outras Técnicas Analíticas ...................................................................................................... 43
II. 3. PROPRANOLOL ...................................................................................................44 II. 3.1. Espectrofotometria na Região do Visível ........................................................................... 44 II. 3.2. Espectrofluorimetria .................................................................................................................. 47 II. 3.3. Cromatografia Líquida de Alta Eficiência ........................................................................... 48 II. 3.4. Outras Técnicas........................................................................................................................... 48
II. 4. Atenolol...................................................................................................................49 II. 4.1. Espectrofotometria na Região do Visível ........................................................................... 49 II. 4.2. Outras Técnicas........................................................................................................................... 50
II. 5. Metildopa ...............................................................................................................52 II. 5.1. Espectrofotometria na Região do Visível ........................................................................... 52 II. 5.2. CLAE ................................................................................................................................................ 55 II. 5.3. Outras Técnicas........................................................................................................................... 55
CAPÍTULO III. JUSTIFICATIVA E OBJETIVOS............................................57
CAPÍTULO IV - ESPECTROSCOPIA DE REFLECTÂNCIA DIFUSA EM COMBINAÇÃO COM SPOT TEST PARA ANÁLISES QUANTITATIVAS ......61
IV. 1. SPOT TEST ............................................................................................................62

20
IV. 2. ASPECTOS TEÓRICOS DA ESPECTROSCOPIA DE REFLECTÂNCIA DIFUSA .............................................................................................64
IV. 2.1. Fundamentos............................................................................................................................... 65 IV. 2.2. Teoria de Kubelka-Munk ......................................................................................................... 66 III. 2.3. Instrumentos .............................................................................................................................. 67 IV. 2.4. Representação do espectro de reflectância..................................................................... 68 III. 2.5. Aplicações da espectroscopia de reflectância difusa para análises quantitativas.......................................................................................................................................................................... 70
CAPÍTULO V. DESENVOLVIMENTO DE MÉTODOS ANALÍTICOS POR ESPECTROSCOPIA DE REFLECTÂNCIA DIFUSA UTILIZANDO SPOT TEST PARA A DETERMINAÇÃO DE FUROSEMIDA, HIDROCLOROTIAZIDA, PROPRANOLOL E ATENOLOL EM FORMULAÇÕES FARMACÊUTICAS. ........74
V. 1. PARTE EXPERIMENTAL....................................................................................75 V. 1.1. Equipamentos ............................................................................................................................... 75 V. 1.2. Material e Solventes................................................................................................................... 75 V. 1.3. Reagentes e Soluções ............................................................................................................... 76 V. 1.4. Amostras ........................................................................................................................................ 77
V. 2. PROCEDIMENTO EXPERIMENTAL..............................................................78 V. 2.1. Spot test ......................................................................................................................................... 78 V. 2.2. Preparação das Amostras ........................................................................................................ 79 V. 2.3. Estudo de Interferências .......................................................................................................... 80 V. 2.4. Estudo de Adição e Recuperação .......................................................................................... 81 V. 2.5. Determinação de peróxido de hidrogênio em dioxano: método modificado ........ 82
V. 3. RESULTADOS E DISCUSSÃO.........................................................................83 V. 3.1. Determinação de furosemida em formulações farmacêuticas ................................... 83 V. 3.2. Determinação de hidroclorotiazida em formulações farmacêuticas ........................ 94
V. 3.2.2.1. Planejamento Fatorial Fracionário: triagem de fatores............................... 96 V. 3.2.2.2. Efeitos do tempo e temperatura de aquecimento......................................... 98
V. 3. 3. Determinação de propranolol em formulações farmacêuticas ............................... 105 Esquema 3 ............................................................................................................................................. 105
V. 3. 4. Determinação de atenolol em formulações farmacêuticas ...................................... 119
CAPÍTULO VI. DESENVOLVIMENTO DE MÉTODO ANALÍTICO POR ESPECTROFOTOMETRIA NA REGIÃO DO VISÍVEL PARA A DETERMINAÇÃO DE METILDOPA EM FORMULAÇÕES FARMACÊUTICAS128
VI. 1. PARTE EXPERIMENTAL ...............................................................................129 VI. 1.1. Equipamentos ........................................................................................................................... 129 VI. 1.2. Reagentes e Soluções............................................................................................................ 129 VI. 1.4. Amostras..................................................................................................................................... 129
VI. 2. PROCEDIMENTO ..............................................................................................130 VI. 2. 1. Curva analítica ........................................................................................................................ 130 VI. 2. 2. Estudo de interferências...................................................................................................... 130 VI. 2.3. Preparação da amostra ......................................................................................................... 131 V. 2.4. Estudo de Adição e Recuperação ........................................................................................ 131
VI. 3. RESULTADOS E DISCUSSÃO ....................................................................132

21
VI. 3.1. Planejamento Fatorial de Experimentos ......................................................................... 136 VI. 3.3. Curva Analítica ......................................................................................................................... 139 VI. 3.4. Estudo de interferências ....................................................................................................... 140 VI. 3.5. Estudo de Adição e Recuperação....................................................................................... 140 VI. 3.6. Aplicação do método proposto e comparação com o método de referência.... 141 VI. 3.7. Medidas em dispositivo portátil.......................................................................................... 142 VI. 3.7.1. Curva Analítica ..................................................................................................................... 144 VI. 3.7.2. Análise das amostras de comprimidos ........................................................................ 145 VI. 3.8. Conclusões ................................................................................................................................. 146
CAPÍTULO VII. CONCLUSÕES GERAIS ..........................................................148
CAPÍTULO VIII. PERSPECTIVAS FUTURAS...................................................151
CAPÍTULO IX. REFERÊNCIAS...........................................................................153

22
Capítulo I - Introdução

23
I. 1. CONSIDERAÇÕES GERAIS
Este trabalho apresenta o desenvolvimento de métodos analíticos para a determinação
de diferentes fármacos em formulações farmacêuticas. Os fármacos em estudo foram:
furosemida, hidroclorotiazida, propranolol, atenolol e metildopa. Considerações gerais sobre
cada um dos fármacos são dadas a seguir:
I. 1.1. Furosemida
A furosemida, ácido 5-(aminossulfonil)-4-cloro-2-[(2-furanilmetil) amino] benzóico,
(Figura 1) apresenta ponto de fusão de 206ºC, é solúvel em acetona, metanol,
dimetilformamida e soluções aquosas com pH maior que 8,0. É levemente solúvel em água,
clorofórmio e éter; e pouco solúvel em etanol (THE MERCK..., 2001). A furosemida é um
derivado do ácido antranílico ou o-aminobenzóico, pertencente à classe dos diuréticos de alça
(JACKSON, 2003; RANKIN, 2002). Os substituintes cloreto e sulfonamida presentes na
molécula de furosemida são característicos em algumas outras classes de diuréticos e
essenciais para a atividade diurética (RANKIN, 2002). Estes fármacos atuam primariamente
sobre o ramo ascendente da alça de Henle, inibindo o sistema de transporte Na+/K+/2Cl-, com
conseqüente diminuição do volume sangüíneo e do débito cardíaco (JACKSON, 2003).
Figura 1. Estrutura química da furosemida
Cl
COOH
NH
H2NO2S
CH2O

24
Os diuréticos de alça são os diuréticos mais potentes e, por isso, devem ser utilizados
com cautela. A furosemida é indicada no tratamento de edemas relacionados com os sistemas
cardíaco, renal e hepático e é usada na hipertensão arterial em situações especiais como
estados edematosos ou em emergências hipertensivas (JACKSON, 2003). A furosemida
apresenta um efeito salurético (excreção de íons sódio e cloreto) de 8 a 10 vezes maior que os
diuréticos tiazídicos e uma duração da ação mais curta, cerca de 6-8 horas. Ela é excretada,
em cerca de 80%, na sua forma inalterada, e uma pequena parte (cerca de 20%) é
metabolizada. A furosemida causa excreção de íons sódio, cloreto, potássio, cálcio, magnésio
e bicarbonato, podendo levar a desequilíbrios eletrolíticos. A furosemida também pode causar
como efeitos adversos ototoxicidade e distúrbios gastrintestinais. Este fármaco é efetivo por
via oral, mas pode ser usado parenteralmente em casos emergenciais. A dose de furosemida,
20 a 80 mg por dia, deve ser administrada em doses divididas devido a sua curta duração da
ação e cuidadosamente aumentadas até o máximo de 600 mg por dia (RANKIN, 2002).
I. 1.2. Hidroclorotiazida
A hidroclorotiazida (1,1-dióxido-6-cloro-3,4-diidro-2H-1,2,4-benzotiadiazino-7-
sulfonamida) (Figura 2) apresenta ponto de fusão de 273-275ºC, é solúvel em acetona, amônia
diluída, metanol e etanol e é praticamente insolúvel em água (THE MERCK..., 2001).
Figura 2. Estrutura química da hidroclorotiazida
N
NHS
Cl
H2NO2SO O
H

25
A hidroclorotiazida pertence à classe dos farmácos tiazídicos, os quais possuem um
núcleo benzotiadiazina 1,1-dióxido (RANKIN, 2002). Tais fármacos constituem uma
importante classe de agentes diuréticos que atuam no túbulo distal, diminuindo a reabsorção
ativa dos íons sódio e cloreto. Entretanto, estes fármacos apresentam ação diurética moderada
quando comparados aos diuréticos de alça. Os diuréticos tiazídicos levam a uma perda
significativa de potássio, causando hipocalemia (diminuição nos níveis plasmáticos de
potássio) que pode ser evitada através da administração concomitante de diuréticos
poupadores de potássio (triantereno, amilorida, por exemplo) ou suplementos de potássio.
Assim, algumas formulações farmacêuticas contendo hidroclorotiazida em associação com
diuréticos poupadores de potássio são comercialmente disponíveis. A hidroclorotiazida é
efetiva por via oral e é excretada na urina em sua forma inalterada, a duração da ação é de 8-
12 horas (RANKIN, 2002). Os principais efeitos adversos da hidroclorotiazida resultam de
algumas ações renais, sendo a depleção de potássio a mais importante. Outros efeitos incluem
alcalose metabólica e aumento dos níveis séricos de ácido úrico. A hidroclorotiazida é
indicada no tratamento de edemas associados a cardiopatias (insuficiência cardíaca
congestiva), hepatopatias (cirrose hepática) e doenças renais (síndrome nefrótica,
insuficiência renal crônica, glomerulonefrite aguda). Este fármaco também é muito usado no
controle da hipertensão (JACKSON, 2003). A diminuição da pressão arterial pode ser
atribuída à redução no volume sanguíneo e ao relaxamento direto do músculo liso vascular
(RANKIN, 2002). Os diuréticos tiazídicos podem ser administrados uma vez ao dia e são bem
tolerados (JACKSON, 2003).

26
I. 1.3. Propranolol
O propranolol, 1-(isopropilamino)-3-(1-naftiloxi)-2-propanol, (Figura 3) apresenta
ponto de fusão de 163-164ºC, é solúvel em água e praticamente insolúvel em éter, benzeno e
acetato de etila (THE MERCK..., 2001).
Figura 3. Estrutura química do propranolol
O propranolol pertence à classe dos β-bloqueadores muito utilizados no tratamento da
hipertensão associada à doença arterial coronária ou a arritmias cardíacas. Os β-bloqueadores
exercem seus efeitos farmacológicos bloqueando os receptores β-adrenérgicos, levando à
redução na contratilidade miocárdica e na freqüência cardíaca. O propranolol é um β-
bloqueador não seletivo, uma vez que interage com os receptores β1 e β2 com igual afinidade.
Este fármaco também apresenta eficácia para reduzir a gravidade e a freqüência dos ataques
de angina de esforço e para melhorar a sobrevida de pacientes que sofreram infarto do
miocárdio (HOFFMAN, 2003).
O N
OHH

27
I. 1.4. Atenolol
O atenolol, 4-(2-hidroxi-3-[(1-metiletil)-amino] propoxi) benzenoacetamida, (Figura 4)
apresenta ponto de fusão de 146-148ºC, é solúvel em metanol, acetona e dimetilsulfóxido;
levemente solúvel em etanol 96%; pouco solúvel em água, isopropanol; muito pouco solúvel
em acetona, dioxano e praticamente insolúvel em acetonitrila, acetato de etila e clorofórmio
(THE MERCK..., 2001).
Figura 4. Estrutura química do atenolol
O atenolol é um agente β-bloqueador seletivo, isto é, exibe afinidade ligeiramente
maior pelos receptores β1 do que pelos receptores β2. Os β-bloqueadores, entre eles, o
atenolol, exercem seu efeito farmacológico bloqueando os receptores β-adrenérgicos, levando
à redução na contratilidade miocárdica e na freqüência cardíaca. Da mesma forma que o
propranolol, o atenolol é muito utilizado em todos os graus de hipertensão e também no
tratamento da angina e arritmias cardíacas (HOFFMAN, 2003).
H3C
CH3
NOH
O
NH2
O
H

28
I. 1.5. Metildopa
A metildopa, α-metil-3,4-diidroxifenilalanina, (Figura 5) é solúvel em água a 25ºC
(~10 mg/ml), em ácidos minerais diluídos e é praticamente insolúvel nos solventes orgânicos
mais comuns (THE MERCK..., 2001). A metildopa é quimicamente relacionada às
catecolaminas e possui atividade anti-hipertensiva. Tal atividade é atribuída ao seu
metabolismo no sistema nervoso central à α-metilnorepinefrina, a qual ativa os receptores α2
adrenérgicos centrais, reduzindo o tônus simpático e conseqüentemente a pressão arterial.
Efeitos adversos da metildopa incluem sonolência, hipotensão postural, retenção de fluidos
com edemas e distúrbios gastrintestinais. A dose inicial habitual de metildopa é de 250 mg, 2
vezes ao dia (HOFFMAN, 2003).
Figura 5. Estrutura química da metildopa
HO
HO
COOH
CH3H2N

29
Capítulo II. Revisão Bibliográfica - Métodos analíticos
para quantificação dos fármacos em estudo

30
Levantamento bibliográfico foi realizado nas principais bases de dados sobre os
métodos analíticos publicados nas revistas científicas de maior circulação para a determinação
de furosemida, hidroclorotiazida, propranolol, atenolol e metildopa em formulações
farmacêuticas. Para melhor compreensão, este levantamento foi descrito em tópicos
subdivididos, como segue:
II. 1. FUROSEMIDA
II. 1.1. Espectrofotometria na Região do Visível
A espectrofotometria na região do visível tem sido amplamente descrita na literatura
como técnica analítica utilizada na determinação de furosemida em preparações farmacêuticas
(AGATONOVIC et al., 1990; CASASSAS; FABREGAS, 1979; GARCÍA et al., 1997;
MOHAMED, 1989; SASTRY et al., 1988; SASTRY; SURYANARAYANA; TIPIRNENI,
1989; SEVILLANO-CABEZA; CAMPÍNS-FALCÓ; SERRADOR-GARCÍA, 1997;
ZIVANOVIC; AGATONOVIC; RADULOVIC, 1990). O método descrito por Agatonovic et
al. (1990) foi baseado na reação entre furosemida e cloreto de paládio (II). A mesma reação
foi utilizada por García et al. (1997) para a determinação espectrofotométrica de furosemida
em preparações farmacêuticas utilizando análise por injeção em fluxo (FIA) (GARCIA, et al.,
1997). O método desenvolvido por Zivanovic, Agatonovic e Radulovic (1990) envolveu a
formação de um complexo colorido a partir da reação entre furosemida e cloreto férrico, em
pH 5,2-6,2. Sastry et al. (1988) descreveram o uso de 3-metil-2-benzotiazolinona hidrazona
(MBTH) na determinação espectrofotométrica de diversos fármacos diuréticos, entre eles a
furosemida, em amostras de comprimidos. A reação entre furosemida e MBTH ocorreu na
presença de um agente oxidante - cloreto férrico - com aquecimento a 85ºC por 5 minutos. A
furosemida também foi determinada espectrofotometricamente a partir da reação com
cloramina T, sob aquecimento em banho-maria a 70ºC por 10 minutos, seguida da adição de

31
N,N-dimetilfenilenodiamina (SASTRY; SURYANARAYANA; TIPIRNENI, 1989). O
método descrito por Sevillano-Cabeza, Campíns-Falcó e Serrador-García (1997) foi baseado
na reação entre furosemida e 1,2-naftoquinona-4-sulfonato com aquecimento a 70ºC por 30
minutos e envolveu um procedimento de extração do produto colorido utilizando álcool
isoamílico como solvente extrator. Outros reagentes cromogênicos citados na literatura para a
determinação espectrofotométrica no visível de furosemida em medicamentos foram 7,7,8,8-
tetracianoquinodimetano (MOHAMED, 1989) e 1,3,5-benzenotriol (floroglucinol)
(CASASSAS; FABREGAS, 1979). A Tabela 1 sumariza as condições analíticas dos métodos
espectrofotométricos acima mencionados. A maioria dos métodos espectrofotométricos
relatados apresenta algumas desvantagens como, por exemplo, necessidade de tempos de
espera para o aparecimento da cor, etapas de aquecimento ou de extração por solvente.

32
Tabela 1. Métodos por espectrofotometria no visível descritos na literatura para a determinação de
furosemida em medicamentos.
Reagente Cromogênico λ máx.
(nm)
Condições para a reação Referência
Pd (II) 527 pH 10,0 AGATONOVIC et al., 1990
Pd (II) 527 pH 5,0 - aquecimento a 55ºC
Análise por injeção em fluxo
GARCÍA et al., 1997
FeCl3 513 pH 5,2-6,2 ZIVANOVIC; AGATONOVIC; RADULOVIC, 1990
FeCl3 e 3-metil-2-benzotiazolinona hidrazona
630 Aquecimento a 85ºC por 5 min.
SASTRY et al., 1988
Cloramina T e Dicloridrato de N-N-dimetilfenilenodiamina
540 Aquecimento a 70ºC por 10 min.
SASTRY; SURYANARAYANA; TIPIRNENI, 1989
1,2 - naftoquinona-4-sulfonato de sódio
498 Aquecimento a 70ºC por 30 min.
Extração com álcool isoamílico
SEVILLANO-CABEZA; CAMPÍNS-FALCÓ; SERRADOR-GARCÍA, 1997
7,7,8,8-tetracianoquinodimetano
578 pH 9,0-9,5 MOHAMED, 1989
Floroglucinol 558 Tempo de espera de 20 min.
Produto colorido é estável por 20 min.
CASASSAS; FABREGAS, 1979
II. 1.2. Espectrofotometria na região do UV
O método descrito na Farmacopéia Britânica para a determinação de furosemida em
comprimidos emprega a espectrofotometria na região do UV (271 nm). O procedimento
recomendado requer diversas etapas de diluição e filtração lenta em papel de filtro
(BRITISH…, 2001). A espectrofotometria no UV também foi empregada no método
desenvolvido por Moustafa e Abdel-Moety (1987) para a determinação de formulações
farmacêuticas contendo somente furosemida. Os métodos espectrofotométricos na região do

33
UV, entretanto, apresentam pouca seletividade uma vez que compostos insaturados
apresentam uma ou mais bandas nesta região do espectro.
A espectrofotometria derivativa tem sido amplamente descrita para a análise de
formulações farmacêuticas contendo furosemida em associação com outros fármacos. Alguns
métodos utilizando esta técnica são descritos na literatura para a determinação simultânea de
furosemida e amilorida (TORAL et al., 2002; FERRARO; CASTELLANO; KAUFMAN,
2001) e também de furosemida e espironolactona em formulações farmacêuticas combinadas
(MILLERSHIP; PARKER; DONNELLY, 2005). Dias, Martins e de Oliveira Neto (2005)
propuseram um método por espectrofotometria derivativa na região do UV para a
determinação de furosemida em medicamentos na presença de seus produtos de
fotodegradação.
II. 1.3. Cromatografia
Devido a sua seletividade e sensibilidade, a cromatografia líquida de alta eficiência
(CLAE) tem sido empregada para a quantificação de furosemida em formulações
farmacêuticas contendo somente furosemida ou em associação com outros fármacos e
também em estudos de estabilidade (ANAPURE; KHANNA; DIGHE, 1989; BARROSO et
al., 1996; BARROSO; ALONSO; JIMENEZ, 1996; BROCH; ROMERO; COQUE, 2000;
RAPAKA; ROTH; PRASAD, 1982; SEMAAN et al., 2005).
A CLAE em fase reversa utilizando detector UV (254 nm) foi empregada por
Anapure, Khanna e Dighe (1989) para quantificar furosemida e espironolactona em
medicamentos que combinam estes dois fármacos. Rapaka, Roth e Prasad (1982) descreveram
um método por CLAE com detector UV (280 nm) utilizando extração com hidróxido de sódio
para quantificar ácido 4-cloro-5-sulfamoilantranílico - produto de degradação da furosemida -
e furosemida em matérias-primas e em formulações farmacêuticas. Outro método por CLAE
com detector UV (237 nm) para a determinação de furosemida em comprimidos empregou

34
uma coluna C18 homemade (SEMAAN et al., 2005). A CLAE empregando fases móveis
micelares também tem sido descrita para a determinação de furosemida. Broch, Romero e
Coque (2000) utilizaram a CLAE com detector UV (274 nm), coluna C18 e fase móvel
constituída por dodecil sulfato de sódio (SDS) 0,06 mol L-1 e propanol 8% para a
determinação de furosemida em medicamentos. Este método também foi empregado para o
estudo da estabilidade da furosemida. Neste caso, a separação de furosemida e dos seus
produtos de fotodegradação foi conseguida utilizando-se uma fase móvel com força de
eluição menor (SDS 0,04 mol L-1 e propanol 2%) (BROCH; ROMERO; COQUE, 2000).
A CLAE com detector amperométrico tem sido empregada para a determinação de
furosemida, principalmente, quando se requer grande sensibilidade. Barroso et al. (1996)
utilizaram CLAE com detector amperométrico para quantificar furosemida e piretanida em
formulações farmacêuticas e em amostras de urina. Outro método utilizando CLAE com
detector amperométrico foi aplicado à determinação simultânea de furosemida e triantereno
em amostras de comprimido contendo os dois fármacos e também em amostras de urina
(BARROSO; ALONSO; JIMENEZ, 1996). Os métodos por CLAE, entretanto, apresentam
algumas desvantagens, tais como o alto custo dos cromatógrafos e colunas cromatográficas e
a necessidade de grandes volumes de solventes orgânicos, levando à geração de resíduos
tóxicos.
A cromatografia em camada delgada (CCD) foi descrita para a determinação
simultânea de amilorida, bumetanida, politiazida e furosemida em formulações farmacêuticas
(ZIVANOVIC; AGATONOVIC; RADULOVIC, 1989). Tal método empregou placas de
sílica gel, NH3 25% : propanol (3:7) ou clorofórmio : isopropanol : metanol : ácido acético
(5:3:1:1) como fases móveis e a detecção foi em 254 nm.
Algumas informações sobre os métodos por CLAE descritos acima estão apresentadas
na Tabela 2.

35
Tab
ela
2. M
étod
os a
nalít
icos
por
CLA
E de
scrit
os n
a lit
erat
ura
para
a d
eter
min
ação
de
furo
sem
ida
asso
ciad
a ou
não
a o
utro
s fár
mac
os e
m c
ompr
imid
os.
Col
una
Fase
Móv
el
Det
ecçã
o Fá
rmac
o A
ssoc
iado
Ref
erên
cia
C18
A
ceto
nitri
la:h
idro
geno
fosf
ato
diam
ônio
0,02
mol
L-1
(55:
45, v
/v),
pH 4
,0
UV
(254
nm
) Es
piro
nola
cton
a A
NA
PUR
E; K
HA
NN
A;
DIG
HE,
198
9
C18
A
ceto
nitri
la:Á
cido
Fos
fóric
o 0,
08 m
ol L
-1
(10:
90, v
/v)
UV
(280
nm
) -
RA
PAK
A; R
OTH
;
PRA
SAD
, 198
2
C18
M
etan
ol:T
ampã
o Fo
sfat
o (1
0 m
mol
L-1
,
pH 5
,5) (
30:7
0, v
/v)
UV
(237
nm
) -
SEM
AA
N e
t al.,
200
5
C18
SD
S 0,
04 m
ol L
-1 :
prop
anol
2%
SDS
0,06
mol
L-1
: pr
opan
ol 8
%
UV
(230
, 268
e 3
30 n
m)
- B
RO
CH
; RO
MER
O;
CO
QU
E, 2
000
C18
A
ceto
nitri
la:á
gua
(40:
60, v
/v) T
ampã
o
Fosf
ato
5,0
mm
ol L
-1 p
H 5
,5
Elet
roqu
ímic
a Pi
reta
nida
B
AR
RO
SO e
t al.,
199
6
C18
A
ceto
nitri
la:á
gua
(30:
70, v
/v) T
ampã
o
Fosf
ato
5,0
mm
ol L
-1 p
H 5
,5
Elet
roqu
ímic
a Tr
iant
eren
o B
AR
RO
SO; A
LON
SO;
JIM
ENEZ
, 199
6
SDS:
dod
ecil
sulfa
to d
e só
dio

36
II. 1.4. Titulometria
A titulometria tem sido empregada em alguns estudos envolvendo a determinação de
furosemida em sua forma pura ou em medicamentos (KULICHENKO; FESENKO, 2002;
OSSMAN et al., 1988).
Ossman et al. (1988) propuseram métodos titulométricos indiretos para a determinação
de furosemida em sua forma pura. O método foi baseado na reação entre furosemida e N-
bromosuccinimida ou N-clorosuccinimida. À solução contendo excesso de reagente foi
adicionado iodeto de potássio. Esta solução foi, então, titulada com tiossulfato de sódio,
utilizando amido como indicador. O tempo ótimo para a reação se completar foi de 20
minutos para N-bromosuccinimida e de 30 minutos para N-clorosuccinimida. Kulichenko e
Fesenko (2002) estudaram a solubilidade e as propriedades ácido-base da furosemida e
propuseram um método titulométrico utilizando tensoativos catiônicos com determinação
potenciométrica ou utilizando azul de bromotimol como indicador.
II. 1.5. Outras Técnicas Analíticas
A cromatografia eletrocinética capilar micelar - técnica eletroforética na qual há a
introdução de micelas na solução tampão - foi utilizada para separar e quantificar furosemida,
hidroclorotiazida, clortalidona e triantereno em comprimidos (LUIS et al., 2002). Um método
por espectroscopia de fluorescência associada à calibração multivariada (método dos mínimos
quadrados parciais) foi descrito para a determinação simultânea de furosemida e triantereno
em amostras de urina e em formulações farmacêuticas (LUIS et al., 2004). Medicamentos
contendo furosemida foram analisados por espectroscopia de ressonância magnética nuclear
(RMN) (ABOUL-ENEIN; AL-BADR; RASHED, 1979). Barroso, Alonso e Jimenez (1995)
desenvolveram métodos por voltametria de pulso diferencial e voltametria de onda quadrada
para determinar furosemida em amostras de urina e em formulações farmacêuticas. Um

37
eletrodo de membrana de PVC incorporado com o par iônico furosemida-cloreto de
tricaprilmetilamônio foi desenvolvido por Dias et al., 2004 para quantificar furosemida em
medicamentos.
II. 2. HIDROCLOROTIAZIDA
II. 2.1. Espectrofotometria na região do UV
A maioria dos métodos analíticos para a determinação de hidroclorotiazida em
medicamentos emprega a espectrofotometria no UV (ALBERO et al., 2002; CARLUCCI; DI
GIUSEPPE; MAZZEO, 1993; DINC; USTUNDAG, 2003; DINC, 2002; EL-WALILY et al.,
1995; ERK; ONUR, 1996; ERK, 1999, 2002; FERRARO; CASTELLANO; KAUFMAN,
2002, 2004; JOSEPH-CHARLES et al., 2003; KARGOSHA; SARRAFI, 2001; MAHADIK,
2000; MARTIN et al. 1995, 1998; PANDERI, 1999; PANZADE; SALAMA et al., 2000;
SASTRY et al., 1988; URBÀNYI; O´CONNELL, 1972). A Farmacopéia Britânica descreve
um método por espectrofotometria no UV em 273 nm para a determinação de
hidroclorotiazida em comprimidos (BRITISH…, 2001). Os métodos por espectrofotometria
no UV descritos na literatura são, principalmente, para a determinação de hidroclorotiazida
em associação com um ou mais fármacos. Entretanto, medidas diretas na região do UV estão
sujeitas às interferências provenientes de fármacos e/ou de excipientes presentes na
formulação farmacêutica, resultando em baixa seletividade, uma vez que todos os compostos
insaturados apresentam uma ou mais bandas na região UV do espectro eletromagnético. Desta
forma, os métodos por espectrofotometria no UV para a determinação de hidroclorotiazida
associada a outros fármacos presentes em formulações farmacêuticas são baseados em
medidas derivativas a fim de se obter métodos com maior seletividade do que os métodos
espectrofotométricos convencionais. Além disso, muitos destes métodos aplicam ferramentas
quimiométricas, tais como: mínimos quadrados clássicos (CLS), mínimos quadrados inverso

38
(ILS), regressão de componentes principais (PCR) e mínimos quadrados parciais (PLS) sobre
os dados dos espectros UV e sobre suas derivadas de primeira ou segunda ordem. Algumas
informações sobre as determinações de hidroclorotiazida associada a outros fármacos por
espectrofotometria derivativa com ou sem aplicação de métodos quimiométricos estão
apresentadas na Tabela 3.

39
Tabela 3. Métodos por espectrofotometria UV derivativa descritos na literatura para a determinação
de hidroclorotiazida em formulações farmacêuticas combinadas.
Fármaco(s) associado(s) Método(s) quimiométrico(s)
empregado(s)
Referência
Amilorida, atenolol e timolol CLS, PCR, PLS FERRARO; CASTELLANO; KAUFMAN, 2004
Amilorida PLS FERRARO; CASTELLANO; KAUFMAN, 2002
Amilorida Regressão linear múltipla MARTIN et al., 1995
Benazepril, triantereno, cilazapril Método de Vierordt ERK, 1999
Benazepril Derivada 2ª ordem PANDERI, 1999
Benazepril CLS, ILS, PCR DINC, 2002
Captopril - PANZADE; MAHADIK, 2000
Captopril, enalapril, fosinopril Derivada 2ª ordem SALAMA et al., 2000
Cilazapril Derivada 1ª ordem ERK; ONUR, 1996
Enalapril Derivada 1ª ordem EL-WALILY et al., 1995
Enalapril Derivada 2ª ordem CARLUCCI; DI GIUSEPPE; MAZZEO, 1993
Espironolactona CLS, ILS, PCR, PLS DINC; USTUNDAG, 2003
Espironolactona, canrenona PLS MARTIN et al., 1998
Fosinopril Derivada 1ª ordem ERK, 2002
Irbesartan Derivada 1ª ordem JOSEPH-CHARLES et al., 2003
Irbesartan Derivada 1ª ordem ALBERO et al., 2002
Losartan Derivada 1ª ordem ERK, 2001
Triantereno PLS e PCR KARGOSHA; SARRAFI, 2001
Valsartan Derivada 1ª ordem SATANA et al., 2001 CLS: mínimos quadrados clássicos; ILS: mínimos quadrados inverso; PCR: regressão de componentes principais; PLS: mínimos quadrados parciais.

40
II. 2.2. Cromatografia Líquida de Alta Eficiência (CLAE)
Dentre os métodos cromatográficos, a CLAE, em fase reversa, com detector UV tem
sido a mais descrita para a determinação de hidroclorotiazida em formulações farmacêuticas
(ATAY; TAMER; ARIKAN, 2001; CETIN; SUNGUR, 2005; ERK; KARTAL, 1999; ERK,
1999, 2001; KARTAL; ERK, 1999; OZKAN, 2001; PANDERI; PARISSI-POULOU, 1999).
Com o objetivo de reduzir o consumo de solventes e tornar os métodos por CLAE mais
seletivos, colunas micro-bore (com diâmetro interno entre 1,0 a 4,6 mm) têm sido utilizadas
(PANDERI; PARISSI-POULOU, 1999). CLAE com detecção por quimiluminescência
também tem sido empregada para a determinação de hidroclorotiazida em formulações de
comprimidos (OUYANG et al., 1999). Neste caso, a detecção foi baseada na reação entre
hidroclorotiazida e cério (IV) em meio de ácido sulfúrico, sensibilizada pelo corante
fluorescente rodamina 6G (OUYANG et al., 1999). Os dados sobre as determinações por
CLAE estão apresentados na Tabela 4.

41
Tab
ela
4. M
étod
os a
nalít
icos
por
CLA
E de
scrit
os n
a lit
erat
ura
para
a d
eter
min
ação
de
hidr
oclo
rotia
zida
ass
ocia
da o
u nã
o a
outro
s fár
mac
os e
m c
ompr
imid
os.
Col
una
Fase
Móv
el
Det
ecçã
o (n
m)
Padr
ão In
tern
oFá
rmac
o(s)
Ass
ocia
do(s
)R
efer
ênci
a
C18
M
etan
ol:T
ampã
o Fo
sfat
o pH
4,0
(70:
30, v
/v)
209
feno
barb
ital
Cila
zapr
il C
ETIN
; SU
NG
UR
, 20
05
C18
Ta
mpã
o fo
sfat
o 0,
01 m
ol L
-1 :
Met
anol
(53:
47, v
/v) p
or 5
m
in. e
(36:
64, v
/v) p
or 1
5 m
in.
254
trim
etro
pina
C
ilaza
pril
ATA
Y; T
AM
ER;
AR
IKA
N, 2
001
C18
K
H2P
O4
0,01
mol
L-1
: ac
eton
itrila
(65
:35,
v/v
) pH
3,1
aj
usta
do c
om H
3PO
4 23
2 fu
rose
mid
a Lo
sarta
n O
ZKA
N, 2
001
C18
–
mic
robo
re
Tam
pão
Fosf
ato
0,02
5 m
ol L
-1 p
H 4
,8 :
ace
toni
trila
(5
5:45
, v/v
) 25
0 na
prox
eno
Ben
azep
ril
PAN
DER
I; PA
RIS
SI-
POU
LOU
, 199
9
C18
ac
eton
itrila
: H
2O (2
0:80
, v/v
) - p
H 3
,8
213
- Li
sino
pril
ERK
; KA
RTA
L,
1999
C18
Á
cido
o-f
osfó
rico
0,02
5 m
ol L
-1 p
H 3
,0 a
just
ado
com
tri
etila
min
a : a
ceto
nitri
la (8
4:16
, v/v
) 27
8 -
Am
ilorid
a K
AR
TAL;
ER
K,
1999
C18
N
aH2P
O4
0,01
mol
L-1
: M
etan
ol :
Ace
toni
trila
(8:
2:1,
v/
v/v)
pH
5,5
aju
stad
o co
m H
3PO
4 26
5 -
Losa
rtan
ERK
, 200
1
C18
M
etan
ol 1
0% -T
ampã
o ac
etat
o de
sód
io 5
0 m
mol
L-1
pH
3,
5 co
nten
do o
ctan
osul
fona
to d
e só
dio
25m
mol
L-1
C
L tio
pron
ina
Cap
topr
il O
UY
AN
G e
t al.,
19
99
CL:
Qui
milu
min
escê
ncia

43
II. 2.3. Outras Técnicas Analíticas
A Farmacopéia Brasileira descreve um método para quantificar hidroclorotiazida em
comprimidos utilizando titulação em meio não-aquoso (FARMACOPÉIA..., 1988).
Um método por FIA baseado na reação de quimiluminescência entre hidroclorotiazida
e cério (IV) foi desenvolvido por Ouyang et al. (1998). A mesma reação foi utilizada em outro
método utilizando quimiluminescência para a determinação de hidroclorotiazida em
formulações farmacêuticas (PULGARIN; MOLINA; OLIVARES NIETO, 2004).
A cromatografia eletrocinética capilar micelar foi utilizada por Luis et al. (2002) para
a separação e quantificação de clortalidona, furosemida, triantereno e hidroclorotiazida e
também no método desenvolvido por Quaglia et al. (2002) para a determinação de losartan e
hidroclorotiazida em formulações farmacêuticas.
Poucos métodos por espectrofotometria na região do visível são citados na literatura
para a determinação de hidroclorotiazida em formulações farmacêuticas (SASTRY et al.,
1989; SASTRY; SURYANARAYANA; TIPIRNENI, 1988). Os métodos mencionados
envolveram reações químicas com dicloridrato de N,N-dimetilfenilenodiamina e cloramina T
(SASTRY et al., 1989) e 3-metil-2-benzotiazolinona hidrazona após hidrólise alcalina
(SASTRY; SURYANARAYANA; TIPIRNENI, 1988).

44
II. 3. PROPRANOLOL
II. 3.1. Espectrofotometria na Região do Visível
A maioria dos métodos analíticos descritos na literatura para a determinação de
propranolol em formulações farmacêuticas emprega a espectrofotometria na região do visível.
Estes métodos são baseados em reações entre propranolol e diferentes reagentes
cromogênicos. Salem (2002) desenvolveu métodos baseados na reação do propranolol
(doador de elétrons) e reagentes aceptores de elétrons, tais como: iodo, p-cloranil, bromanil,
2,3-dicloro-5,6-diciano-1,4-benzoquinona (DDQ), tetracianoetileno (TCNE) e 7,7,8,8-
tetracianoquinodimetano (TCNQ), produzindo complexos de transferência de carga coloridos
(SALEM, 2002). Gölcü, Yücesoy e Serin (2004) descreveram dois métodos por
espectrofotometria no visível para determinar propranolol em formulações farmacêuticas.
Tais métodos foram baseados em reações de complexação do fármaco com Cu (II) e Co (II).
Um método indireto utilizando a espectrofotometria no visível foi descrito para a
determinação de propranolol e piroxicam em formulações farmacêuticas e em matérias-
primas (GOWDA; SEETHARAMAPPA; MELWANKI, 2002). Este método foi baseado na
oxidação de propranolol por um excesso de N-bromosuccinimida, em meio ácido, seguida
pela reação do excesso de oxidante com cloridrato de prometazina e cloridrato de
metidilazina, resultando em um produto colorido. Outro método indireto foi proposto por
Sastry, Srinivas e Prasad (1996). Tal método foi baseado na reação do propranolol com N-
bromosuccinimida, a qual foi adicionada em concentração conhecida e em excesso. A
concentração de N-bromosuccinimida que não reagiu foi determinada pela diminuição na
absorbância do corante celestine blue. O método proposto por El-Eman et al. (2003) foi
baseado na reação de acoplamento oxidativo entre propranolol e 3-metil-2-benzotiazolinona
hidrazona (MBTH), na presença de Ce (IV). Outros métodos espectrofotométricos para a
determinação de propranolol em formulações farmacêuticas envolveram reações com azul de

45
bromotimol (RADULOVIC; JOVANOVIC; ZIVANOVIC, 1986) e ácido 4-amino-3,5-
dinitrobenzóico diazotizado (IDOWU; ADEGOKE; OLANIYI, 2004). O método descrito por
Zivanovic, Radulovic e Jovanovic (1988) envolveu a reação entre propranolol e cloreto
férrico, em meio ácido, na presença de tiocianato de amônio. O complexo colorido formado
foi extraído com clorofórmio.
A Tabela 5 apresenta as condições analíticas dos métodos espectrofotométricos acima
mencionados, bem como os reagentes cromogênicos envolvidos nas reações químicas para
determinação de propranolol em formulações farmacêuticas. A maioria dos métodos
espectrofotométricos descritos apresenta algumas desvantagens, tais como tempo de espera
para o aparecimento da cor, etapas de derivatização ou extração com clorofórmio e alguns são
indiretos.

46
Tab
ela
5. C
ompa
raçã
o en
tre a
lgun
s m
étod
os e
spec
trofo
tom
étric
os e
seu
s re
agen
tes
crom
ogên
icos
des
crito
s na
lite
ratu
ra p
ara
a de
term
inaç
ão d
e pr
opra
nolo
l
em fo
rmul
açõe
s far
mac
êutic
as.
Rea
gent
e D
etec
ção
(nm
)C
omen
tári
os
Ref
erên
cia
Iodo
p-
Clo
rani
l B
rom
anil
DD
Q
TCN
E TC
NQ
365
440
450
470
420
840
D
esen
volv
imen
to d
a co
r apó
s 5, 5
, 15,
20,
20
e 40
m
in.,
resp
ectiv
amen
te.
SA
LEM
, 200
2
Cu
(II)
C
o (I
I)
548
614
Des
envo
lvim
ento
da
cor a
pós 3
0 m
in.
GÖ
LCÜ
; YÜ
CES
OY
; SER
IN,
2004
N-b
rom
osuc
cini
mid
a
515
Mét
odo
indi
reto
G
OW
DA
; SEE
THA
RA
MA
PPA
; M
ELW
AN
KI,
2002
N-b
rom
osuc
cini
mid
a e
cele
stin
e bl
ue
540
Mét
odo
indi
reto
SA
STR
Y; S
RIN
IVA
S;
PRA
SAD
, 199
6
3-m
etilb
enzo
tiazo
lina-
2-on
a hi
draz
ona
496
Req
uer t
rata
men
to c
om su
lfato
cér
ico
amon
iaca
l EL
-EM
AM
et a
l., 2
003
Azu
l de
brom
otim
ol
414
Extra
ção
com
clo
rofó
rmio
R
AD
ULO
VIC
; JO
VA
NO
VIC
; ZI
VA
NO
VIC
, 198
6
Áci
do 4
-am
ino-
3,5-
dini
trobe
nzói
co
470
Req
uer e
tapa
de
deriv
atiz
ação
ID
OW
U; A
DEG
OK
E;
OLA
NIY
I, 20
04
FeC
l 3 e
NH
4CN
S 47
7 Ex
traçã
o co
m c
loro
fórm
io
ZIV
AN
OV
IC; R
AD
ULO
VIC
; JO
VA
NO
VIC
, 198
8 D
DQ
: 2,3
-dic
loro
-5,6
-dic
iano
-1,4
-ben
zoqu
inon
a; T
CN
E: te
traci
anoe
tilen
o; T
CN
Q: 7
,7,8
,8-te
traci
anoq
uino
dim
etan
o.

47
II. 3.2. Espectrofluorimetria
Alguns métodos fluorimétricos têm sido desenvolvidos para a determinação de
propranolol em formulações farmacêuticas. Tais métodos foram baseados na fluorescência
natural da molécula de propranolol (FERNÀNDEZ-SÀNCHEZ et al., 2003; LA PEÑA;
SALINAS; DURÁN, 1991; RUIZ et al., 1998) ou em reações de derivatização (RAMESH et
al., 2003). Um método por espectrofluorimetria foi descrito para a determinação simultânea
de propranolol e hidralazina em formulações farmacêuticas. Os espectros foram registrados
após aquecimento da amostra em meio aquoso a 70ºC por 15 minutos e resfriamento a 20ºC e
suas derivadas (1ª ordem) foram utilizadas (LA PEÑA; SALINAS; DURÁN, 1991). Outro
método empregando espectrofluorimetria derivativa foi descrito por Ruiz et al. (1998) para a
determinação simultânea de propranolol e pindolol em formulações farmacêuticas. Neste
método, os espectros foram registrados a 25ºC e suas derivadas (1ª ordem) foram utilizadas.
Um sensor óptico sensível à fluorescência foi desenvolvido por Fernàndez-Sànchez et al.
(2003) para determinar propranolol e pindolol em formulações farmacêuticas e em amostras
de urina. Ramesh et al. (2003) propuseram um método por espectrofluorimetria para a
determinação simultânea de piroxican e propranolol em formulações farmacêuticas. O método
foi baseado na reação de oxidação entre o fármaco e excesso de N-bromosuccinimida, seguida
pela reação do excesso de N-bromosuccinimida com cloridrato de metdilazina, resultando em
espécies fluorescentes (RAMESH et al., 2003).

48
II. 3.3. Cromatografia Líquida de Alta Eficiência
A CLAE com detector UV (270 nm) foi utilizada por DasGupta (1985) para a
quantificação de propranolol em formulações farmacêuticas. O método empregou uma coluna
C18 e verapamil como padrão interno. Outro método por CLAE no UV (290 nm) foi proposto
por Hitscherich et al. (1987). Este método empregou coluna de cianopropilsilano para separar
e quantificar propranolol e hidroclorotiazida em formulações farmacêuticas. Martinez, Coque
e Camanas (1997) desenvolveram um método por CLAE com detector UV (225 nm) com o
propósito de separar e quantificar diversos β-bloqueadores, entre eles o propranolol, em
formulações farmacêuticas. O método empregou fases móveis micelares em diferentes valores
de pH.
II. 3.4. Outras Técnicas
A Farmacopéia Britânica descreve um método por espectrofotometria na região do UV
(290 nm) para a determinação de propranolol em comprimidos (BRITISH…, 2001). Um
método por quimiluminescência e análise por injeção em fluxo foi desenvolvido por Tsogas et
al. (2005) para a determinação de propranolol em formulações farmacêuticas. Tal método foi
baseado na reação com pirogalol oxidado por periodato. Trata-se de um método indireto, uma
vez que o propranolol é oxidado por periodato que é adicionado em excesso e a detecção é
feita pela medida de quimiluminescência gerada pela oxidação de pirogalol com o excesso de
periodato. Outro método por quimiluminescência e FIA foi baseado na quimiluminescência
produzida pela reação entre propranolol e permanganato de potássio em meio de ácido
sulfúrico (TOWNSHEND; PULGARIN; PARDO, 2005).
Outras técnicas citadas para a determinação de propranolol em formulações
farmacêuticas têm sido a polarografia de pulso diferencial (EL-RIES; ABOU-SEKKINA;
WASSEL, 2002), a fosforescência à temperatura ambiente (DIAZ et al., 2002; MARTINEZ;
CAMANAS; COQUE, 1994) e a espectrometria de absorção atômica (AAS) (EL-RIES;

49
ATTIA; IBRAHIM, 2000; KHALIL; BORHAM, 2000; KHALIL; EL-RABIEHI, 2000). Um
método titulométrico direto utilizando N-bromosuccinimida como titulante também foi
descrito para determinar propranolol em formulações farmacêuticas (PATHAKA; SHUKLA;
SHUKLA, 1982). Briguenti e Bonato (2005) empregaram a eletroforese capilar para a análise
dos β-bloqueadores: atenolol, metoprolol, pindolol e propranolol em preparações
farmacêuticas. Métodos potenciométricos utilizando eletrodos íon-seletivos também têm sido
descritos na literatura para a determinação de propranolol em formulações farmacêuticas
(ABOUL-ENEIN; SUN, 2000; HASSAN et al., 2003; KARTAMYSHEV; RYASENSKII;
GORELOV, 2002).
II. 4. Atenolol
II. 4.1. Espectrofotometria na Região do Visível
Diversos métodos espectrofotométricos na região do visível têm sido descritos para
quantificar atenolol em formulações farmacêuticas (AGRAWAL et al., 1992; AL-
GHANNAM; BELAL, 2002; AL-GHANNAM, 2006; AMIN; RAGAB; SALEH, 2002;
GÖLCÜ; YÜCESOY; SERIN, 2004; SALEM, 2002). Um método espectrofotométrico
indireto foi desenvolvido por Agrawal et al. (1992). O método foi baseado na reação de
atenolol com cloridrato de hidroxilamina, produzindo um derivado do ácido hidroxâmico, o
qual formou um complexo colorido com Fe (III) que foi determinado em 510 nm. Métodos
para a determinação de diversos agentes β-bloqueadores, entre eles o atenolol, baseados em
reações que formam complexos de transferência de carga foram descritos por Salem (2002) e
empregaram iodo, p-cloranil, bromanil, TCNE e TCNQ como reagentes cromogênicos.
Golcü, Yücesoy e Serin (2004) descreveram dois métodos por espectrofotometria no visível
para determinar acebutolol, propranolol e atenolol em formulações farmacêuticas. Tais
métodos foram baseados em reações de complexação dos fármacos com Cu (II) e Co (II). Um
método espectrofotométrico cinético para a determinação de atenolol em formulações

50
farmacêuticas foi proposto por Al-Ghannam e Belal (2002). O método foi baseado na reação
entre atenolol e 4-cloro-7-nitrobenzeno-2-oxa-1,3-diazol em pH 8,0 e com aquecimento por
30 minutos, produzindo um composto colorido (460 nm). O método descrito por Al-Ghannam
(2006) para a determinação de atenolol envolveu o uso de corantes ácidos, tais como: azul de
bromofenol, azul de bromotimol e vermelho de bromocresol. O complexo colorido formado
foi analisado por espectrofotometria em 415 nm.
II. 4.2. Outras Técnicas
Martinez, Coque e Camanas (1997) desenvolveram um método por CLAE com
detector UV (225 nm) com o propósito de separar e quantificar diversos β-bloqueadores, entre
eles o atenolol, em formulações farmacêuticas. O método empregou fases móveis micelares
em diferentes valores de pH.
A cromatografia em camada delgada de alta eficiência (CCDAE) foi empregada para a
determinação de atenolol e nitrendipina em formulações farmacêuticas (ARGEKAR;
SAWANT, 1999). Tal método empregou placas de sílica gel sobre folhas de alumínio como
fase estacionária e clorofórmio: metanol : tolueno : amônia 25% (2:2:5,5:0,1, v/v) como fase
móvel. A detecção foi realizada em 233 nm (ARGEKAR; SAWANT, 1999). Um método
semelhante utilizando CCDAE foi aplicado à determinação de atenolol e amlodipina em
amostras de comprimidos (ARGEKAR; POWAR, 2000). Tal método empregou placas de
sílica gel como fase estacionária e cloreto de metileno : metanol : amônia 25% (8,8:1,3:0,1,
v/v) como fase móvel. A detecção foi realizada em 230 nm.
A Farmacopéia dos Estados Unidos descreve um método por CLAE com detector UV
(226 nm) para a quantificação de atenolol em comprimidos (UNITED..., 2003). O método
recomendado pela Farmacopéia Britânica envolve a espectrofotometria no UV em 275 nm
(BRITISH…, 2001).

51
A espetrofotometria derivativa no UV utilizando métodos quimiométricos foi descrita
por Ferraro, Castellano e Kaufman (2004) para a determinação simultânea de amilorida,
atenolol, hidroclorotiazida e timolol em formulações farmacêuticas e em misturas sintéticas.
A espectrofotometria UV derivativa também foi empregada nos métodos desenvolvidos por
Bonazzi et al. (1996) para a análise de formulações farmacêuticas contendo atenolol,
metoprolol e suas associações, tais como: atenolol-clortalidona, metoprolol-clortalidona e
metoprolol-hidroclorotiazida.
Entre os métodos potenciométricos para a determinação de atenolol está o uso de um
biossensor contendo membrana de bicamada lipídica com superfície estabilizada com DNA
desenvolvido para triagem rápida de atenolol em preparações farmacêuticas (NIKOLELIS;
PETROPOULOU; MITROKOTSA, 2002). Outro método potenciométrico descrito para a
determinação de atenolol em formulações farmacêuticas empregou sensores de membrana de
matriz polimérica de PVC incorporada com o par iônico atenolol-tungstofosfato (HASSAN et
al., 2003). Um método potenciométrico utilizando eletrodo íon-seletivo de membrana de PVC
foi descrito na literatura para a determinação de atenolol em formulações farmacêuticas. O
eletrodo foi construído pela incorporação na matriz de PVC com o par iônico atenolol-tetrakis
(p-clorofenil) borato (SHAMSIPUR; JALALI, 2005).
Briguenti e Bonato (2005) empregaram a eletroforese capilar para a análise dos β-
bloqueadores atenolol, metoprolol, pindolol e propranolol em preparações farmacêuticas.
Outros métodos por eletroforese capilar foram descritos por Shafaati e Clark (1996) para a
determinação de atenolol, na presença de substâncias relacionadas, em formulações
farmacêuticas e por Maguregui, Jimenez e Alonso (1998) para a determinação simultânea de
atenolol e de outros agentes anti-hipertensivos em formulações farmacêuticas e em amostras
de urina.

52
II. 5. Metildopa
II. 5.1. Espectrofotometria na Região do Visível
O método recomendado pela Farmacopéia Brasileira para a análise de comprimidos
contendo metildopa emprega a espectrofotometria na região do visível. O método é baseado
na reação entre metildopa e tartarato ferroso em pH 8,5 (FARMACOPÉIA..., 1988). Outros
métodos espectrofotométricos, na região do visível, descritos para a determinação de
metildopa são baseados em diferentes tipos de reações químicas. O método desenvolvido por
Nagaraja et al. (1998) envolveu uma reação de oxidação da metildopa por N-
bromosuccinimida, seguida pelo acoplamento oxidativo com isoniazida. A metildopa também
foi determinada pela reação com sulfanilamida diazotizada, na presença de íons molibdato,
em meio ácido (NAGARAJA; VASANTHA; SUNITHA, 2001). O método desenvolvido por
Aman et al. (1998) empregou a reação entre metildopa e ácido barbitúrico, em meio alcalino.
Outros reagentes citados têm sido: periodato (AFKHAMI; KHATAMI, 2003), p-
dimetilaminocinamaldeído (WALASH; ABOU-OUF; SALEM, 1985), vanilina (SALEM,
1985), nitrato de cério (IV) (HELALEH; RAHMAN; ABU-NAMEH, 1997) e cloreto férrico
(ZIVANOVIC; VASILJEVIC; RADULOVIC, 1991). Alguns métodos espectrofotométricos
utilizando FIA também têm sido descritos na literatura (NEVADO; GALLEGO; LAGUNA,
1995; TUBINO; RODRIGUES-JR; VILA, 2004; ABDULRAHMAN; AL-ABACHI; AL-
QAISSY, 2005; RIBEIRO et al., 2005). Tais métodos envolveram reações entre metildopa e
metaperiodato (NEVADO; GALLEGO; LAGUNA, 1995), p-aminofenol em meio alcalino
(TUBINO; RODRIGUES-JR; VILA, 2004), p-toluidina e periodato de sódio
(ABDULRAHMAN; AL-ABACHI; AL-QAISSY, 2005) e molibdato de amônio (RIBEIRO
et al., 2005). Ainda, um método espectrofotométrico para a determinação de metildopa em
formulações farmacêuticas empregou extrato bruto de raiz de batata doce (Ipomoea batatas
(L.) Lam.) como fonte da enzima polifenol oxidase, a qual catalisa a reação de oxidação de

53
catecolaminas levando a formação de compostos coloridos (VIEIRA; FATIBELLO-FILHO,
1998). Dados adicionais sobre os métodos espectrofotométricos citados acima estão
apresentados na Tabela 6. A maioria dos métodos espectrofotométricos citados apresenta
algumas desvantagens, tais como necessidade de tempo de espera para o aparecimento da cor,
etapa de aquecimento e envolvem procedimentos complexos.

54
Tab
ela
6. M
étod
os e
spec
trofo
tom
étric
os n
o vi
síve
l des
crito
s na
liter
atur
a pa
ra a
det
erm
inaç
ão d
e m
etild
opa
em fo
rmul
açõe
s far
mac
êutic
as.
Rea
gent
e D
etec
ção
(nm
) L
ei d
e B
eer
(μg
mL
-1)
R
Com
entá
rios
R
efer
ênci
a
N-b
rom
osuc
cini
mid
a –
Ison
iazi
da
480
5,0
– 16
,0
0,99
94
Tem
po d
e es
pera
de
5 m
in.
NA
GA
RA
JA e
t al.,
199
8
Áci
do b
arbi
túric
o 54
0 1,
0 –
250
0,99
98
Aqu
ecim
ento
por
20
min
. A
MA
N e
t al.,
199
8 p-
Tolu
idin
a –
Perio
dato
de
sódi
o 48
0 1,
0 –
50,0
0,
9898
FI
A
AB
DU
LRA
HM
AN
; AL-
AB
AC
HI;
AL-
QA
ISSY
, 200
5 Pe
rioda
to
665
0,10
– 2
,5
0,99
45
Mét
odo
indi
reto
A
FKH
AM
I; K
HA
TAM
I, 20
03
Sulfa
nila
mid
a di
azot
izad
a -
íons
mol
ibda
to
500
0,5
– 7,
0 0,
9895
Te
mpo
s de
espe
ra d
e 10
m
in.
NA
GA
RA
JA; V
ASA
NTH
A;
SUN
ITH
A, 2
001
p-D
imet
ilam
inoc
inam
alde
ído
440
4,0
– 60
,0
1,0
Tem
po d
e es
pera
de
5 m
in.
WA
LASH
; AB
OU
-OU
F; S
ALE
M,
1985
V
anili
na
420
12 –
144
N
D
Tem
po d
e es
pera
de
20
min
. SA
LEM
, 198
5
Nitr
ato
de C
e (I
V)
550
125
– 25
0 N
D
Aqu
ecim
ento
a 8
0ºC
por
20
-25
min
. H
ELA
LEH
; RA
HM
AN
; AB
U-
NA
MEH
, 199
7 Fe
Cl 3
423
19,0
– 7
6,2
0,99
8 Te
mpo
de
espe
ra d
e 10
m
in.
ZIV
AN
OV
IC; V
ASI
LJEV
IC;
RA
DU
LOV
IC, 1
991
Met
aper
ioda
to
473
4,76
– 4
7,6
0,99
99
FIA
- A
quec
imen
to a
45
ºC
NEV
AD
O; G
ALL
EGO
; LA
GU
NA
, 19
95
p-A
min
ofen
ol
608
47,6
4 –
714,
6 0,
9999
FI
A
TUB
INO
; RO
DR
IGU
ES-J
R; V
ILA
, 20
04
Mol
ibda
to d
e A
môn
io
410
50 –
100
0,
9999
FI
A
RIB
EIR
O e
t al.,
200
5 Ex
trato
de
bata
ta d
oce
(pol
ifeno
l oxi
dase
) 48
0 47
,64
– 14
29,2
0,
9990
R
eaçã
o en
zim
átic
a V
IEIR
A; F
ATI
BEL
LO-F
ILH
O, 1
998
ND
: Não
des
crito

55
II. 5.2. CLAE
A CLAE também tem sido descrita para a análise de formulações farmacêuticas
contendo metildopa. Um método por CLAE com detector UV (280 nm) foi descrito por Ting
(1983). O método empregou uma coluna C18, uma mistura de ácido acético 3% : metanol
(96:4, v/v) como fase móvel e teobromina como padrão interno (TING, 1983). A CLAE
utilizando fase móvel micelar foi empregada no método proposto por Camanas et al. (1995)
para a determinação das catecolaminas: L-dopa, metildopa, epinefrina, dopamina e
isoproterenol, bem como de outros compostos freqüentemente associados com essas
catecolaminas, em formulações farmacêuticas (CAMANAS et al., 1995). A CLAE com
detector UV (286 nm) foi empregada para a determinação simultânea de metildopa, amilorida
e hidroclorotiazida em comprimidos (ZECEVIC et al., 2001). As condições ótimas de
separação foram estabelecidas utilizando metodologia de superfície de resposta. A fase móvel
consistiu de água : metanol (75:25, v/v) pH 3,60, a coluna foi uma C18 e o padrão interno
cafeína (ZECEVIC et al., 2001).
II. 5.3. Outras Técnicas
Badawy et al. (1996) descreveram um método potenciométrico para a determinação de
L-dopa, carbidopa, metildopa and aspartame utilizando um eletrodo seletivo
trinitrobenzenosulfonato. A polarografia de pulso diferencial também foi descrita para a
determinação de metildopa em formulações farmacêuticas (BALLANTINE; WOOLFSON,
1979). Um método por titulometria foi descrito para a determinação de metildopa em
comprimidos. Neste método, N-bromosuccinimida foi utilizada para titular a solução
contendo amostra, iodo e vermelho de metila (PATHAK; SHUKLA; SHUKLA, 1982). Um
método utilizando espectrofotometria no UV foi descrito por Wahbi et al. (1978). Tal método
empregou a reação com cobaltinitrito de sódio e foi aplicado na determinação de alguns

56
fármacos fenólicos, entre eles a metildopa, em amostras de comprimidos. Outro método
espectrofotométrico no UV (358 nm) para a determinação de metildopa em formulações
farmacêuticas utilizou reação com p-cloranil (KORANY; WAHBI, 1979). Entretanto, este
método apresenta problemas relacionados à necessidade de aquecimento por tempo
prolongado (30 minutos) e à possibilidade de interferências provenientes da matriz, os quais
também absorvem na região do UV (KORANY; WAHBI, 1979).

57
Capítulo III. Justificativa e Objetivos

58
A hipertensão arterial é a mais comum das doenças cardiovasculares. Em 1998, as
doenças cardiovasculares foram responsáveis por 27% dos óbitos registrados no país
(MINISTÉRIO DA SAÚDE, 2006). O tratamento farmacológico de pacientes hipertensos
visa reduzir a morbidade e a mortalidade decorrentes de doenças cardiovasculares. A terapia
com anti-hipertensivos previne, consideravelmente, acidentes vasculares cerebrais
hemorrágicos, insuficiência cardíaca e insuficiência renal resultantes da hipertensão
(KANNEL, 1996). Dentre os medicamentos indicados para o tratamento da hipertensão
arterial destacam-se a furosemida, a hidroclorotiazida, o propranolol, o atenolol e a metildopa,
os quais são utilizados por grande parte da população brasileira de hipertensos.
A RDC nº 33 da Agência Nacional de Vigilância Sanitária/Ministério da Saúde, que
institui o Regulamento Técnico sobre Boas Práticas de Manipulação de Medicamentos em
Farmácias, em seu Anexo I (Boas Práticas de Manipulação em Farmácias) (AGÊNCIA
NACIONAL DE VIGILÂNCIA SANITÁRIA, 2000) estabelece que todos os itens que
comprovem a especificação das matérias primas e que garantam seu teor, pureza e integridade
devem ser re-analisados pelo setor de Controle de Qualidade da Farmácia para certificação do
laudo de análise do fornecedor (AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA,
2000). Um grande problema mundial é a falsificação e adulteração de medicamentos
consumidos pela população. O uso destes medicamentos representa um risco para a saúde das
pessoas. No Brasil, este problema foi detectado em 1998, quando uma variedade de
medicamentos, tais como contraceptivos orais, antibióticos, antineoplásicos, e antipiréticos
foram falsificados com sérias consequências. Assim, a análise de formulações farmacêuticas
tem como objetivo não somente o controle de qualidade, mas também demonstrar a
idoneidade do produto.
Diversos métodos têm sido desenvolvidos para a análise de furosemida,
hidroclorotiazida, propranolol, atenolol e metildopa em formulações farmacêuticas.

59
Entretanto, a maioria dos métodos descritos na literatura, de modo geral, envolve métodos por
CLAE convencional, a qual requer instrumentação complexa e de alto custo, além de um
excessivo tempo de análise e necessidade de grandes volumes de solventes orgânicos tóxicos.
Outra técnica muito utilizada para a determinação dos fármacos envolvidos neste
estudo é a espectrofotometria no UV e no visível. Todavia, os métodos espectrofotométricos
baseados em medidas diretas no UV estão sujeitos a interferências provenientes da matriz.
Assim, a maioria dos métodos por espectrofotometria no UV é derivativa e envolve,
geralmente, a determinação de diversos fármacos simultaneamente, os quais empregam
análise multivariada, apresentando o inconveniente de necessitar conhecer todos os
componentes presentes na amostra que poderiam influenciar na resposta de absorbância. Os
métodos por espectrofotometria no visível descritos para a determinação dos fármacos em
estudo apresentam problemas relacionados à necessidade de etapas de aquecimento e de
derivatização, tempo de análise relativamente longo, instabilidade do composto colorido
formado e alguns são métodos indiretos.
A partir das considerações descritas acima, a necessidade de métodos simples, rápidos,
de baixo custo e com baixo consumo de reagentes e solventes torna-se evidente,
especialmente para análises de controle de qualidade de furosemida, hidroclorotiazida,
propranolol, atenolol e metildopa.
A combinação da espectroscopia de reflectância difusa com spot test torna-se muito
atraente no desenvolvimento de métodos analíticos, uma vez que esta combinação fornece
medidas rápidas, simples, com baixo consumo de reagentes e solventes e baixa geração de
resíduos. Além disso, pouca atenção havia sido dada à espectroscopia de reflectância difusa
como técnica para análises quantitativas. Atualmente esta técnica tem sido empregada,
principalmente, em métodos para a análise de metais. No entanto, relatos do uso desta técnica
para análises de medicamentos ainda são incipientes.

60
Quando envolve reações rápidas e medidas diretas, a espectrofotometria de absorção
molecular na região do visível consiste de uma técnica muito útil para análises de fármacos.
Além disso, existe a possibilidade de se adaptar métodos espectrofotométricos para
dispositivo portátil de medidas fotométricas, tornando-os práticos para serem usados, por
exemplo, em farmácias de manipulação.
Desta forma, os objetivos deste trabalho foram:
• Desenvolver métodos analíticos para a determinação de furosemida,
hidroclorotiazida, propranolol e atenolol em formulações farmacêuticas por espectroscopia de
reflectância difusa utilizando spot test.
• Desenvolver método analítico para a determinação de metildopa em formulações
farmacêuticas por espectrofotometria na região do visível e adaptação do método para uso em
dispositivo portátil de medidas fotométricas.
• Aplicar os métodos desenvolvidos na análise de formulações farmacêuticas
disponíveis comercialmente e comparar os resultados obtidos pelos métodos propostos com
àqueles obtidos por métodos oficiais.

61
Capítulo IV - Espectroscopia de reflectância difusa em
combinação com spot test para análises quantitativas

62
IV. 1. SPOT TEST
Há muitas décadas os spot tests têm sido discutidos e explorados, principalmente, em
análises clínicas, testes de controle da qualidade do ar, em análises de água e alimentos, em
prospecções geoquímicas e em análises forenses (JUNGREIS, 1997). Os spot tests envolvem,
basicamente, a mistura de algumas gotas de uma solução contendo a substância de interesse
com algumas gotas de uma solução contendo o reagente formando um produto colorido. O
meio para a reação pode ser um suporte de superfície porosa, por exemplo, papel de filtro ou
sílica gel (JUNGREIS, 1997). Historicamente, Friedlieb Ferdinand Runge, químico alemão, é
considerado um dos importantes pioneiros no uso de papel de filtro como suporte para reações
químicas (ANFT, 1955). Schönbein e Goppelsroeder (1861) e Schiff (1859) também são
citados na literatura como pioneiros, mas a reação mais antiga descrita é àquela realizada por
Runge em 1822 (ANFT, 1955). Tal reação foi desenvolvida a partir de um estudo para avaliar
o poder de branqueamento de soluções alvejantes através de um teste para detectar a presença
de cloro em tais soluções. O teste ocorreu sobre uma superfície de papel de filtro impregnado
com amido e iodeto de potássio. Quando gotas da solução alvejante foram adicionadas sobre o
papel, uma mancha azul apareceu na superfície devido à liberação de iodo e sua reação com o
amido (ANFT, 1955). Esta é, provavelmente, a reação de spot test mais antiga utilizando
papel de filtro impregnado e antecede o teste desenvolvido por Schiff, o qual detectou ácido
úrico utilizando papel impregnado com carbonato de amônio (ANFT, 1955; JUNGREIS,
1997). Posteriormente, Fritz Feigl se destacou como um grande estudioso das análises por
spot test de substâncias orgânicas e inorgânicas, tendo estudado reações mais sensíveis e
seletivas e publicado diversos livros científicos (FEIGL, 1966, 1958).
Uma vez que os spot tests levam à formação de um produto colorido, eles podem ser
usados para análises qualitativas - quando a detecção da substância de interesse se dá
visualmente pela presença de cor; para análises semi-quantitativas – na quais escalas de cores,

63
feitas com padrões analíticos de diferentes concentrações, são utilizadas para comparação
com a intensidade de cor do teste e para análises quantitativas, utilizando, por exemplo,
instrumentos para medir a luz refletida, neste caso, a espectroscopia de reflectância difusa.
Para análises semi-quantitativas ou quantitativas, é importante que o procedimento para a
realização do spot test seja bem elaborado, visando à obtenção de uniformidade na cor da
mancha formada e, conseqüentemente, uma boa precisão das medidas. Detalhes importantes
da execução do spot test envolvem o controle do volume da solução a ser adicionada
utilizando micropipetas ou microsseringas e da velocidade da adição das soluções, bem como
a escolha da ordem de adição que resulte em maior uniformidade da mancha colorida formada
(GHAUCH et al., 2000; TUBINO; ROSSI; MAGALHÃES, 1997; JUNGREIS, 1997).
O fato dos spot tests serem procedimentos especialmente rápidos, simples e que
apresentam baixo consumo de reagentes e solventes o tornam muito interessantes para o
desenvolvimento de métodos analíticos, principalmente do ponto de vista da Química Verde,
a qual tem interesse por métodos e técnicas que reduzem ou eliminam o uso e a geração de
substâncias prejudiciais à saúde humana ou ao meio ambiente (ANASTAS, 1999).
Atualmente, a espectroscopia de reflectância difusa utilizando spot tests tem sido utilizada
com sucesso em análises quantitativas (ARENA; PORTER; FRITZ, 2003; DMITRIENKO et
al., 2002; FRITZ et al., 2003; GAZDA; FRITZ; PORTER, 2004a, 2004b; GHAUCH et al.,
1999, 2000; MATIAS; VILA; TUBINO, 2003, 2004; MOLINER-MARTÍNEZ; CAMPÍNS-
FALCÓ, 2005; MOLINER-MARTÍNEZ; HERRÀEZ-HERNANDEZ; CAMPÍNS-FALCÓ,
2005; NADZHAFOVA et al., 2005; NAKANO et al., 1993; NARAYANASWAMY, 1993;
OLIVEIRA; SALIBA, 1994; TUBINO; ROSSI; MAGALHÃES, 1997; TUBINO; SOUZA,
2006; ZAPOROZHETS; GAWER; SUKHAN, 1998; ZAPOROZHETS et al., 1999;
ZAPOROZHETS; TSYUKALO, 2002).

64
IV. 2. ASPECTOS TEÓRICOS DA ESPECTROSCOPIA DE REFLECTÂNCIA DIFUSA
A percepção óptica dada por uma substância química é baseada em sua interação com
a luz. Absorção, emissão e reflexão ou espalhamento são os três fenômenos mais comumente
empregados para medidas analíticas (GHAUCH et al., 2000).
Ao contrário da espectroscopia de absorção que há muito tempo vêm apresentando
amplo uso como técnica de análise quantitativa, a espectroscopia de reflectância difusa tem
sido pouco estudada. Durante muitos anos, o uso da espectroscopia de reflectância difusa foi
limitado às áreas têxteis, de papel, cerâmica, tintas e pigmentos como técnica analítica de
controle de qualidade para avaliar cor, brancura e brilho das superfícies brancas ou coloridas.
Por exemplo, na indústria têxtil, durante a produção de tecidos tingidos, amostras de tecidos
são retiradas para avaliar se a tonalidade está sendo mantida, sempre tendo como referência o
espectro de reflectância. Na indústria de papel, além da cor, a brancura e brilho também são
avaliados (WENDLANDT; HECHT, 1966). A espectroscopia de reflectância difusa foi
considerada uma técnica inviável para análises quantitativas, uma vez que não era possível
alcançar boa precisão das medidas obtidas a partir de spot tests convencionais (KEALEY,
1972). De acordo com Kealey (1972) as medidas de reflectância obtidas a partir de spot tests
convencionais apresentavam precisão relativa entre 10 e 20% e, portanto, resultados
quantitativos foram considerados inviáveis. Entretanto, com os avanços tecnológicos na área
de dispositivos ópticos, incluindo fibras ópticas e esferas de integração, medidas de
reflectância puderam ser obtidas com bons resultados de precisão (GHAUCH et al., 2000;
TUBINO; ROSSI; MAGALHÃES, 1997).

65
IV. 2.1. Fundamentos
Segundo as teorias discutidas por Wendlant e Hecht (1966) a reflexão da radiação
sobre uma superfície pode ser de dois tipos: especular ou tipo espelho e difusa. Para que se
possa utilizar a reflectância como técnica analítica é necessário entender os tipos de reflexão.
A reflexão especular ou tipo espelho ocorre em superfícies lisas ou polidas e pode ser
definida pela lei de reflexão de Fresnel. Neste tipo de processo os ângulos de incidência e de
reflexão são idênticos e não há transmissão da radiação através da superfície (WENDLANT;
HECHT, 1966).
Ao contrário da reflexão especular, a reflexão difusa acontece em superfícies foscas ou
opacas e ocorre por reflexões múltiplas a partir das superfícies das partículas que constituem o
meio. A reflexão difusa compreende um processo complexo que ocorre quando a radiação
penetra em um substrato sólido. Parte desta radiação retorna à superfície do substrato,
ocorrendo também dispersão múltipla e absorção parcial da radiação pelas partículas ou fibras
que constituem o substrato sólido (WENDLANT; HECHT, 1966).
A reflectância ou poder de reflexão (R) é dada por:
0IIR = , onde: (Equação 1)
R é o sinal que representa a radiação refletida e que pode ser registrado por detectores, tais
como uma fotomultiplicadora ou arranjo de fotodiodos.
=0I intensidade da radiação incidente
=I intensidade da radiação refletida
Os valores de R variam entre 0 e 1 e são normalmente expressos em termos de
reflectância percentual (%)R , isto é:
1000
×=IIR (Equação 2)

66
Considera-se R igual a 1 quando o sinal de reflectância é igual ao da radiação
incidente, condição que pode ser conseguida utilizando um padrão de reflectância máxima,
geralmente pastilhas de BaSO4 e MgO (WENDLANT; HECHT, 1966).
IV. 2.2. Teoria de Kubelka-Munk
Diversos modelos foram desenvolvidos para descrever, em termos quantitativos, a
intensidade da radiação refletida difusamente (SKOOG, 2002). A teoria de Kubelka-Munk é o
modelo mais aceito que relaciona concentração da amostra e reflectância. Esta teoria descreve
a relação entre coeficiente de absorção molar, coeficiente de dispersão e poder de reflectância
em um meio semi-infinito (infinitamente fino) de acordo com a seguinte equação:
sk
RRRf =
−=
.2)1()(
2
, onde: (Equação 3)
=k coeficiente de absorção molar
=s coeficiente de dispersão
Para amostras diluídas, k está relacionado à absortividade molar (ε) e à concentração
(C) pela relação:
Ck .303,2 ε= (Equação 4)
Para a aplicação adequada da equação de Kubelka-Munk é necessária a determinação
da reflectância absoluta do material usado como referência (REINECKE et al., 1988).
Entretanto, a relação linear entre )(Rf e concentração somente é observada na prática
para substâncias que absorvem fracamente e quando o tamanho das partículas que constituem
o meio é relativamente pequeno (1 μm de diâmetro interno) (FREI; MACNEIL, 1973).
Atualmente, diversos gráficos têm sido descritos para análises quantitativas envolvendo
transformações matemáticas da variável dependente (sinal) e/ou independente (concentração),

67
tais como: Rlog versus RC −2 ; R versus ])1/([ 2CC − ; Rlog versus C/1 ; e R versus
Clog (TUBINO; ROSSI; MAGALHÃES, 1997).
Vale mencionar que em análises por espectroscopia de reflectância difusa, o sinal de
reflectância pode ser convertido em densidade óptica para medida de reflectância ( RA ), que é
dada por:
)/1log( RAR = (Equação 5)
III. 2.3. Instrumentos
Os instrumentos para medida de reflectância podem ser acessórios acoplados a um
espectrofotômetro ou podem ser equipamentos portáteis. Em geral, os instrumentos para
medidas de reflectância contêm os seguintes componentes: fonte de luz, suporte para amostra
e para padrão branco usado como referência, esfera de integração, espelhos, detector e
registrador. A Figura 6 apresenta um esquema de um acessório de reflectância Labsphere,
modelo RSA-HP-53.
Figura 6. Vista superior do acessório de reflectância Labsphere modelo RSA-HP-53
Fonte: LABSPHERE. RSA-HP-53 instruction manual. North Sutton, [1999?]. p. 4.

68
A esfera de integração consiste de uma esfera revestida internamente por uma camada
de material altamente refletor. Antigamente, as esferas de integração eram fabricadas com
material metálico revestido com tinta branca ou pó de MgO ou de BaSO4 prensado. Os
revestimentos utilizados atualmente são materiais brancos de politetrafluoroetileno (PTFE) ou
outros termoplásticos (Spectralon®) (STORM; SPRINSTEEN, 2006). As propriedades de
reflectância de uma amostra envolvem a determinação da distribuição espacial da radiação
refletida, com relação à intensidade e ao estado de polarização. A esfera de integração tem a
função de coletar e integrar espacialmente o fluxo de radiação refletida por uma superfície.
Assim, a intensidade da luz refletida em qualquer ponto da esfera corresponde à medida do
fluxo total de radiação refletida pela superfície. Desta forma é possível obter o fluxo de luz
refletida integrado total, o qual é utilizado para as medidas de reflectância (LABSPHERE,
2006).
Os equipamentos para medidas de reflectância podem apresentar esferas de integração
de diferentes tamanhos. Esferas com diâmetro interno maior são mais eficazes na integração
da luz e as medidas são mais exatas do que esferas de menor diâmetro interno. Os
equipamentos para medidas de reflectância também podem apresentar diferentes geometrias,
podendo levar a valores de reflectância diferentes para uma mesma amostra em dois
equipamentos com geometrias distintas. Isto ocorre devido a diferenças nos ângulos de
incidência e de reflexão, os quais são muito importantes para as medidas da reflectância
difusa (WENDLANDT; HECHT, 1966).
IV. 2.4. Representação do espectro de reflectância
Os espectros de reflectância podem ser apresentados de várias maneiras. Os gráficos
são comumente obtidos a partir de (%)R , )(Rf ou )/1(log( RAR em função do comprimento
de onda (nm). A Figura 7 apresenta três espectros obtidos a partir de um mesmo equipamento
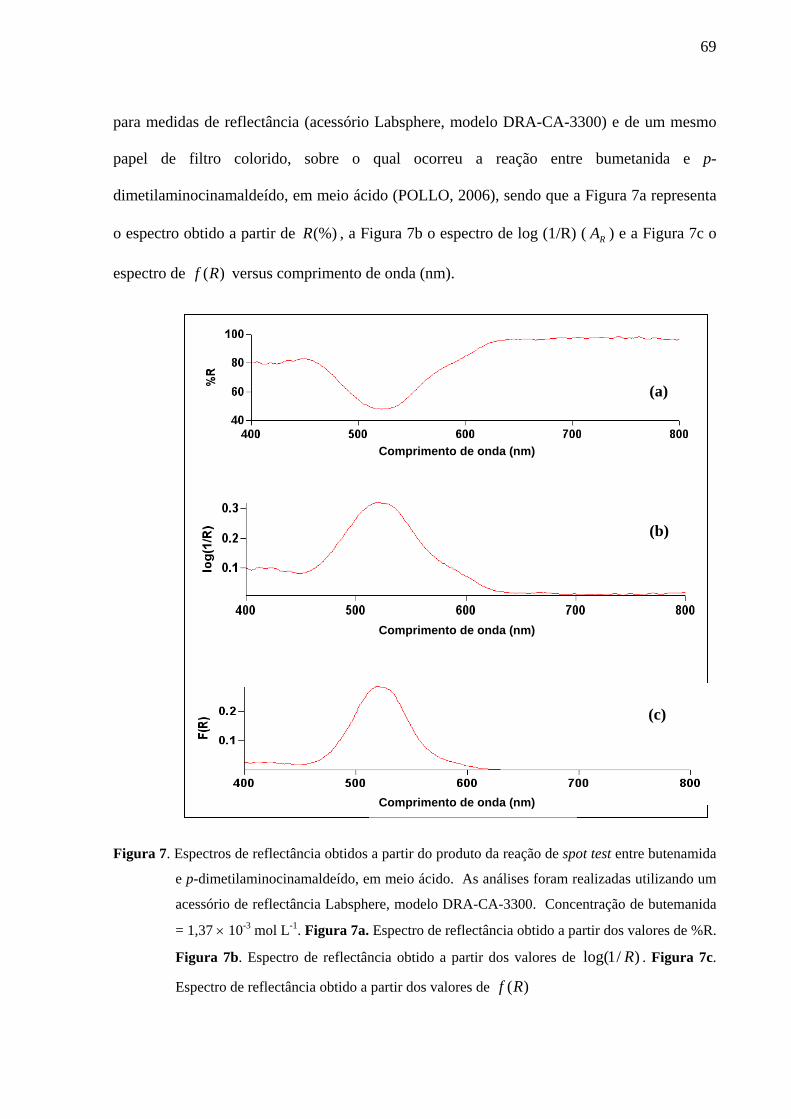
69
para medidas de reflectância (acessório Labsphere, modelo DRA-CA-3300) e de um mesmo
papel de filtro colorido, sobre o qual ocorreu a reação entre bumetanida e p-
dimetilaminocinamaldeído, em meio ácido (POLLO, 2006), sendo que a Figura 7a representa
o espectro obtido a partir de (%)R , a Figura 7b o espectro de log (1/R) ( RA ) e a Figura 7c o
espectro de )(Rf versus comprimento de onda (nm).
Figura 7. Espectros de reflectância obtidos a partir do produto da reação de spot test entre butenamida
e p-dimetilaminocinamaldeído, em meio ácido. As análises foram realizadas utilizando um
acessório de reflectância Labsphere, modelo DRA-CA-3300. Concentração de butemanida
= 1,37 × 10-3 mol L-1. Figura 7a. Espectro de reflectância obtido a partir dos valores de %R.
Figura 7b. Espectro de reflectância obtido a partir dos valores de )/1log( R . Figura 7c.
Espectro de reflectância obtido a partir dos valores de )(Rf
Comprimento de onda (nm)
Comprimento de onda (nm)
Comprimento de onda (nm)
(a)
(b)
(c)

70
Para compreender as diferenças entre os espectros apresentados na Figura 7, basta
converter (%)R em )(Rf e em )/1log( R , de acordo com as Equações 3 e 5. Desta forma,
pode-se entender porque o espectro de R (Fig. 7a) deve apresentar-se invertido em relação aos
outros espectros (Fig. 7b e 7c).
III. 2.5. Aplicações da espectroscopia de reflectância difusa para análises
quantitativas
Diversos métodos por espectroscopia de reflectância difusa, na região do visível, para
análises quantitativas têm sido descritos na literatura (ARENA; PORTER; FRITZ, 2003;
BEROZA; HILL; NORRIS, 1968; BRADDOCK; MAREC, 1966; DMITRIENKO et al.,
2002; FRITZ et al., 2003; FRODYMA; FREI, 1964, 1965, 1969; GAZDA; FRITZ; PORTER,
2004a, 2004b; GHAUCH et al., 1999, 2000; KEALEY, 1972, 1974, 1976, 1977; KIRK;
MOSS; VALENTIN, 1968; LIEU et al., 1967; LIEU; ZAYE; FRODYMA, 1969; LIEU;
FRODYMA, 1966; MATIAS; VILA; TUBINO, 2003, 2004; MOLINER-MARTÍNEZ;
CAMPÍNS-FALCÓ, 2005; MOLINER-MARTÍNEZ; HERRÀEZ-HERNANDEZ;
CAMPÍNS-FALCÓ, 2005; NADZHAFOVA et al., 2005; NAKANO et al., 1993;
NARAYANASWAMY; SEVILLA III; 1986; NARAYANASWAMY, 1993; OLIVEIRA;
SALIBA, 1994; TUBINO; ROSSI; MAGALHÃES, 1997; TUBINO; SOUZA, 2006;
ZAPOROZHETS; GAWER; SUKHAN, 1998; ZAPOROZHETS et al., 1999;
ZAPOROZHETS; TSYUKALO, 2002; ZAYE; FREI; FRODYMA, 1967). O gráfico
apresentado na Figura 8 apresenta o resultado de um levantamento realizado, no período de
1960 até 2006, nas principais bases de dados, sobre métodos que empregaram a
espectroscopia de reflectância difusa, na região visível do espectro, para análises
quantitativas.

71
Figura 8. Distribuição das publicações de métodos por espectroscopia de reflectância difusa na região
do visível para análises quantitativas. Período: 1960-2006.
Podemos observar (Figura 8) que muitos artigos foram publicados na década de 60 e,
talvez pela dificuldade em obter resultados com boa precisão naquela época, o número de
publicações diminuiu e só voltou a aumentar na década de 90, provavelmente, devido aos
avanços tecnológicos e à facilidade de se obter medidas mais precisas, além da preocupação
em se desenvolver métodos simples, rápidos e com baixo consumo de reagentes e solventes.
Atualmente, o número de publicações envolvendo a espectroscopia de reflectância difusa na
região do visível para análises quantitativas vem aumentando consideravelmente (Figura 8).
Algumas destas análises envolvem a determinação de metais sobre superfície de papel de
filtro (GAZDA; FRITZ; PORTER, 2004b; GHAUCH et al., 1999, 2000; TUBINO; ROSSI;
MAGALHÃES, 1997).
0
2
4
6
8
10
12
14
16
Núm
ero
de p
ublic
açõe
s
60 70 80 90 2000-2006
Décadas

72
O método descrito por Tubino, Rossi e Magalhães (1997) foi proposto para a
determinação de Fe(III), Cr(VI) e Ni(II). Ghauch et al. (1999) descreveram um método
semelhante para a determinação de Co(II), Cu(II), Fe(III) e Ni(II). Fosfato, amônio e Cu(II)
também foram determinados por espectroscopia de reflectância difusa utilizando spot test em
papel de filtro (GHAUCH et al., 2000).
A espectroscopia de reflectância difusa associada à extração em fase sólida (EFS)
também tem sido descrita na literatura. Gazda, Fritz e Porter (2004b) desenvolveram um
método para a determinação de Ni (II) em amostras de água a bordo da Estação Espacial
Internacional. Outros métodos utilizando EFS acoplada a espectroscopia de reflectância difusa
foram propostos para a quantificação de aminas alifáticas (MOLINER-MARTÍNEZ;
CAMPÍNS-FALCÓ, 2005), iodo (GAZDA; FRITZ; PORTER, 2004a) e iodeto, Ag (I), Cu
(II), Ni (II), Fe (III), Cr (VI) e outros cátions (ARENA; PORTER; FRITZ, 2003; FRITZ et al.,
2003; MOLINER-MARTÍNEZ; HERRÀEZ-HERNANDEZ; CAMPÍNS-FALCÓ, 2005) em
amostras de água. Sílica gel impregnada com reagentes analíticos também tem sido associada
à espectroscopia de reflectância difusa para a determinação de diversos metais
(NADZHAFOVA et al., 2005; ZAPOROZHETS et al., 1999; ZAPOROZHETS; GAWER;
SUKHAN, 1998; ZAPOROZHETS; TSYUKALO, 2002).
Além disso, a espectroscopia de reflectância difusa tem oferecido a possibilidade da
construção em laboratório de dispositivos portáteis para análises quantitativas (MATIAS;
OLIVEIRA; MOSCHIM, 1997; MATIAS; VILA; TUBINO, 2003). Um sensor de fibra óptica
portátil foi desenvolvido por Matias, Oliveira e Moschim (1997) para analisar a fumaça
proveniente de motores de veículos a diesel. O sensor funciona por amostragem da fumaça
sobre a superfície de uma fita adesiva branca, seguida da medida da luz refletida pela fita
marcada. Um outro dispositivo portátil empregando o fenômeno da reflectância difusa foi
desenvolvido para a determinação de Ni (II) em catalisadores (MATIAS; VILA; TUBINO,

73
2003). O método foi baseado na clássica reação entre Ni (II) e dimetilglioxima, em meio
alcalino (NH4OH), a detecção foi realizada por um LDR (light dependent resistor) e a medida
da resistência por um multímetro, a qual foi correlacionada com a reflectância.
Recentemente, a espectroscopia de reflectância difusa utilizando spot test sobre papel
de filtro foi empregada para a determinação de alguns fármacos em formulações
farmacêuticas (GOTARDO et al., 2004; GOTARDO; PEZZA; PEZZA, 2005; MATIAS;
VILA; TUBINO, 2004; TUBINO; SOUZA, 2006).
O método descrito por Matias, Vila e Tubino (2004) para a determinação de ácido
acetilsalicílico envolveu uma reação de complexação do ácido salicílico, obtido a partir da
hidrólise alcalina do fármaco, com íons Fe (III). O complexo colorido foi submetido à análise
em um reflectômetro homemade, composto por um LED (light emitting diode) e um LDR e
medidas de resistência foram obtidas. Um método utilizando um reflectômetro semelhante foi
descrito por Tubino e Souza (2006) para a determinação de diclofenaco em formulações
farmacêuticas.
Os estudos citados acima revelaram que o uso apropriado da espectroscopia de
reflectância difusa forneceu resultados quantitativos confiáveis, demonstrando o potencial
desta técnica para análises quantitativas.

74
Capítulo V. Desenvolvimento de métodos analíticos por
espectroscopia de reflectância difusa utilizando spot
test para a determinação de furosemida,
hidroclorotiazida, propranolol e atenolol em
formulações farmacêuticas.

75
V. 1. PARTE EXPERIMENTAL
V. 1.1. Equipamentos
Acessório para reflectância Labsphere RSA-HP-53, contendo esfera de integração (76
mm de diâmetro interno) e lâmpada de tungstênio-halogênio (5W), acoplado a um
espetrofotômetro de arranjo de diodos HP-8453A.
Os métodos de referência foram realizados utilizando espectrofotômetro UV-Visível
Cary 1E, com cubetas de quartzo de 1,0 cm de caminho óptico.
Estufa com controle de temperatura - Tecnal, Modelo TE-392.
Agitador magnético Corning.
Micropipetas Eppendorf (10 a 100 μL) e Brand (100 a 1000 μL).
V. 1.2. Material e Solventes
Papel de filtro quantitativo Whatman 41 e 42 foram usados como suporte sólido no
método para a determinação de furosemida e hidroclorotiazida, respectivamente. Papel de
filtro qualitativo Whatman 42 foi usado nos métodos para a determinação de propranolol e
atenolol.
Os excipientes utilizados nos estudos de interferências foram de grau farmacêutico.
Os solventes utilizados nos métodos para a determinação de furosemida e
hidroclorotiazida foram acetona (grau p.a.) e metanol (grau HPLC) (Mallinckrodt, Xalostoc,
México). No método para a determinação de atenolol foi usado metanol grau HPLC (J.T.
Baker, Phillipsburg, EUA) e dioxano (grau p.a.) (Tedia, Fairfield, EUA). Os solventes usados
no método para a determinação de propranolol foram acetona (grau p.a.), etanol (grau HPLC)
(Mallinckrodt, Xalostoc, México) e água desionizada.

76
V. 1.3. Reagentes e Soluções
Nos métodos para a determinação de furosemida e hidroclorotiazida, p-
dimetilaminocinamaldeído (PDAC) (p.a., Riedel-de haën, Alemanha) foi usado para preparar
a solução de PDAC 2,23 × 10-1 mol L-1 em metanol. Esta solução foi mantida refrigerada por,
no máximo, uma semana. Soluções de ácido clorídrico 6,3% (m/v) (1,73 mol L-1) e 10% (m/v)
(2,75 mol L-1) foram preparadas por diluição adequada do ácido clorídrico concentrado (37%)
(p.a., Mallinckrodt, Xalostoc, México) em metanol e empregadas nos métodos para a
determinação de furosemida e hidroclorotiazida, respectivamente.
No método para a determinação de atenolol foi utilizado p-cloranil (Sigma, St Louis,
EUA) no preparo de uma solução 8,00 × 10-2 mol L-1 em dioxano. Esta solução foi preparada
diariamente.
No método para a determinação de propranolol, 2,6-dicloroquinona-4-cloroimida
(DCQ) ou reagente de Gibbs (p.a., Sigma, St. Louis, E.U.A.) foi usado para preparar uma
solução 3,33 × 10-1 mol L-1 (70 mg/ml) em acetona. Esta solução foi preparada diariamente.
Solução estoque de furosemida (Purifarma, São Paulo, Brasil), grau de pureza de
100,1%, foi preparada na concentração de 1,52 × 10-1 mol L-1 em acetona. Soluções padrão
foram obtidas a partir de diluições da solução estoque de furosemida em acetona e usadas na
construção da curva analítica (7,56 × 10-3 a 6,05 × 10-2 mol L-1).
Solução estoque de hidroclorotiazida (Purifarma, São Paulo, Brasil), grau de pureza de
99,85%, foi preparada na concentração de 1,68 × 10-1 mol L-1 em acetona. Soluções padrão
foram obtidas a partir de diluições da solução estoque de hidroclorotiazida em acetona e
usadas na construção da curva analítica (3,36 × 10-2 a 1,01 × 10-1 mol L-1).
Solução estoque de propranolol (Purifarma, São Paulo, Brasil), grau de pureza de
99,80%, foi preparada na concentração de 1,68 × 10-1 mol L-1 em solução de etanol 35% (v/v)
em água. Soluções padrão foram obtidas a partir de diluições da solução estoque de

77
propranolol em etanol 35% (v/v) e usadas na construção da curva analítica (1,35 × 10-2 a 8,45
× 10-2 mol L-1).
Solução estoque de atenolol (Purifarma, São Paulo, Brasil), grau de pureza de 99,90%,
foi preparada na concentração de 2,25 × 10-1 mol L-1 em metanol. Soluções padrão foram
obtidas a partir de diluições da solução estoque de atenolol em metanol e usadas na
construção da curva analítica (1,13 × 10-2 a 7,88 × 10-2 mol L-2).
Todas as soluções estoque foram preparadas no momento do uso.
V. 1.4. Amostras
As amostras comerciais das formulações farmacêuticas (comprimidos) analisadas
foram obtidas em farmácias locais e analisadas dentro dos seus prazos de validade.
O número de marcas comerciais de formulações farmacêuticas analisadas para cada
um dos fármacos estudados e o conteúdo de fármaco declarado pelo fabricante em cada
comprimido estão apresentados na Tabela 7.
Tabela 7. Número de marcas comerciais de formulações farmacêuticas analisadas em cada método
desenvolvido e conteúdo declarado de fármaco por comprimido.
Furosemida Hidroclorotiazida Propranolol Atenolol
Nº Marcas Analisadas 6 6 6 5
Conteúdo Declarado
(mg/comprimido)
40 50 40 25, 50 e 100

78
V. 2. PROCEDIMENTO EXPERIMENTAL
V. 2.1. Spot test
Para a realização dos spot tests, as soluções foram adicionadas sobre a superfície de
papel de filtro, cortado em quadrados com área de, aproximadamente, 2 cm2. O papel foi
colocado na parte inferior de um suporte (Figura 9), de acordo com o procedimento descrito
por Tubino, Rossi e Magalhães (1997) com o auxílio de uma pinça. As soluções foram
adicionadas no centro do papel de filtro utilizando uma micropipeta adaptada ao suporte.
Figura 9. Suporte utilizado na realização dos spot tests (Baseado em: TUBINO; ROSSI;
MAGALHÃES, 1997).
A seguir são descritos os procedimentos para a realização dos spot tests em cada
método desenvolvido:
Furosemida:
Inicialmente, 10 μL da solução contendo o fármaco foram adicionados sobre a
superfície do papel de filtro, seguidos da adição de 20 μL da solução de HCl (6,3%, m/v) e de
20 μL da solução de PDAC (0,4%, m/v), nesta ordem. Em seguida, o papel de filtro foi
aquecido a 80ºC por 5 minutos em uma estufa com controle de temperatura. O aquecimento
foi necessário para a formação do produto colorido, que foi analisado por espectroscopia de
reflectância difusa em 585 nm.

79
Hidroclorotiazida:
Inicialmente, 20 μL da solução contendo o fármaco foram adicionados sobre a
superfície do papel de filtro, seguidos da adição de 20 μL da solução de HCl (10%, m/v) e de
20 μL da solução de PDAC (0,4%, m/v), nesta ordem. O papel de filtro foi então aquecido a
80ºC por 8 minutos em uma estufa com controle de temperatura. O aquecimento foi
necessário para a formação do produto colorido, que foi analisado por espectroscopia de
reflectância difusa em 585 nm.
Propranolol:
Inicialmente, 30 μL da solução contendo o fármaco foram adicionados sobre a
superfície do papel de filtro, seguidos da adição de 30 μL da solução de DCQ (3,33 × 10-1 mol
L-1). O produto colorido foi analisado por espectroscopia de reflectância difusa em 500 nm.
Atenolol:
Inicialmente, 20 μL da solução contendo o fármaco foram adicionados sobre a
superfície do papel de filtro, seguidos da adição de 20 μL da solução de p-cloranil (8,00 × 10-2
mol L-1). O produto colorido foi analisado por espectroscopia de reflectância difusa em 550
nm.
V. 2.2. Preparação das Amostras
Em todos os métodos desenvolvidos, vinte comprimidos (do mesmo lote), de cada
uma das amostras comerciais estudadas, foram pesados em balança analítica e finamente
triturados em gral de ágata. O valor da massa de um comprimido foi expresso como a média
de 20 determinações, com variação menor que 2% (FARMACOPÉIA..., 1988).

80
Quantidades de amostra equivalentes a 100, 150, 80 e 120 mg de furosemida,
hidroclorotiazida, propranolol e atenolol, respectivamente, foram pesadas com precisão de
0,0001 g e submetidas à agitação com o solvente adequado (acetona para furosemida e
hidroclorotiazida; etanol 35% (v/v) para propranolol e metanol para atenolol) em um agitador
magnético por 10 minutos. Após a agitação, as soluções foram adicionadas em balões
volumétricos de 10,00 mL e o volume foi completado com o solvente adequado. Alíquotas
destas soluções foram retiradas para a realização dos spot tests, conforme descrito no item
V.2.1. O branco contendo apenas o solvente e os reagentes foi utilizado como referência e
analisado nas mesmas condições.
V. 2.3. Estudo de Interferências
Uma vez que o objetivo deste trabalho foi determinar o fármaco (furosemida,
hidroclorotiazida, propranolol ou atenolol) em formulações farmacêuticas (comprimidos), o
efeito dos excipientes mais comumente utilizados foi cuidadosamente avaliado.
Em todos os métodos desenvolvidos, os excipientes estudados foram amido, talco,
lactose, estearato de magnésio, etilcelulose, dióxido de silício e croscarmelose sódica. No
método para a determinação de atenolol foram ainda estudados os excipientes lauril sulfato de
sódio e celulose microcristalina.
O procedimento para o estudo de interferências, em todos os métodos desenvolvidos, é
descrito a seguir:
Uma determinada quantidade de fármaco (50, 120, 80 ou 120 mg de furosemida,
hidroclorotiazida, propranolol ou atenolol, respectivamente) e cada um dos excipientes,
separadamente, em quantidade igual e dez vezes maior à de fármaco foram submetidos ao
procedimento descrito no item V. 2.2.

81
V. 2.4. Estudo de Adição e Recuperação
Em todos os métodos desenvolvimentos foi realizado um estudo de adição e
recuperação de fármaco padrão, o qual envolveu o seguinte procedimento:
Às amostras contendo massa equivalente a 50, 100, 80 e 60 mg de furosemida,
hidroclorotiazida, propranolol e atenolol, respectivamente, foram adicionadas quantidades de
fármaco padrão, de forma a se obter soluções com concentrações finais que abrangessem a
faixa de linearidade da curva analítica (THOMPSON; ELLISON; WOOD, 2002). Para todos
os métodos, as quantidades adicionadas de fármaco padrão corresponderam a 50, 100, 150 e
200% da massa de fármaco presente na amostra. Ao béquer contendo amostra e fármaco
padrão foi adicionado o solvente adequado, seguindo o procedimento descrito no item V. 2.2.
A Tabela 8 apresenta as quantidades - em massa (mg) e em concentração final (mol L-1) - de
fármaco padrão adicionado nas amostras.
Tabela 8. Quantidades adicionadas de fármaco padrão dadas em mg e em concentração (mol L-1).
Quantidades adicionadas de fármaco padrão Fármaco (mg) (mol L-1)
Furosemida 25 50 75 100
7,56 × 10-3 1,51 × 10-2 2,27 × 10-2 3,02 × 10-2
Hidroclorotiazida
50 100 150 200
1,68 × 10-2 3,36 × 10-2 5,04 × 10-2 6,72 × 10-2
Propranolol
40 80 120 160
1,35 × 10-2 2,70 × 10-2 4,05 × 10-2 5,40 × 10-2
Atenolol
30 60 90 120
1,13 × 10-2 2,25 × 10-2 3,38 × 10-2 4,50 × 10-2

82
V. 2.5. Determinação de peróxido de hidrogênio em dioxano: método modificado
No desenvolvimento do método reflectométrico para a determinação de atenolol foi
realizada a quantificação de peróxido de hidrogênio no dioxano utilizado como solvente para
o p-cloranil. Para tal, foi inicialmente empregado o método espectrofotométrico descrito por
Benatsky e Tomik (1988) para determinação de peróxidos em solventes orgânicos, o qual é
baseado na reação de peróxido com metavanadato de amônio e ácido nítrico concentrado,
formando um composto colorido (λ máx. = 450 nm) (BENATSKY; TOMIK, 1988). Entretanto,
durante o emprego do método observou-se que a etapa de extração descrita não era necessária,
uma vez que o dioxano e a solução contendo metavanadato de amônio misturaram-se de
forma homogênea. Além disso, a quantidade de peróxido na amostra de dioxano não era
conhecida. Desta forma, a quantificação foi realizada pelo método das adições de padrão e
com algumas modificações no método descrito na literatura (BENATSKY; TOMIK, 1988),
como segue:
A solução reagente foi preparada a partir da dissolução de 0,6250 g de metavanadato
de amônio em 3,75 mL de ácido nítrico concentrado. O volume foi completado com água
desionizada em balão volumétrico de 25,00 mL. A 5,00 mL de amostra de dioxano foram
adicionados individualmente 0,50, 1,00, 2,00, 3,00 e 4,00 mL de uma solução padronizada de
peróxido de hidrogênio (2,37 × 10-2%, m/m). Em seguida, adicionou-se 10,00 mL da solução
de reagente e o volume foi completado para 25,00 mL em balão volumétrico. As soluções
foram analisadas por espectrofotometria em 450 nm.
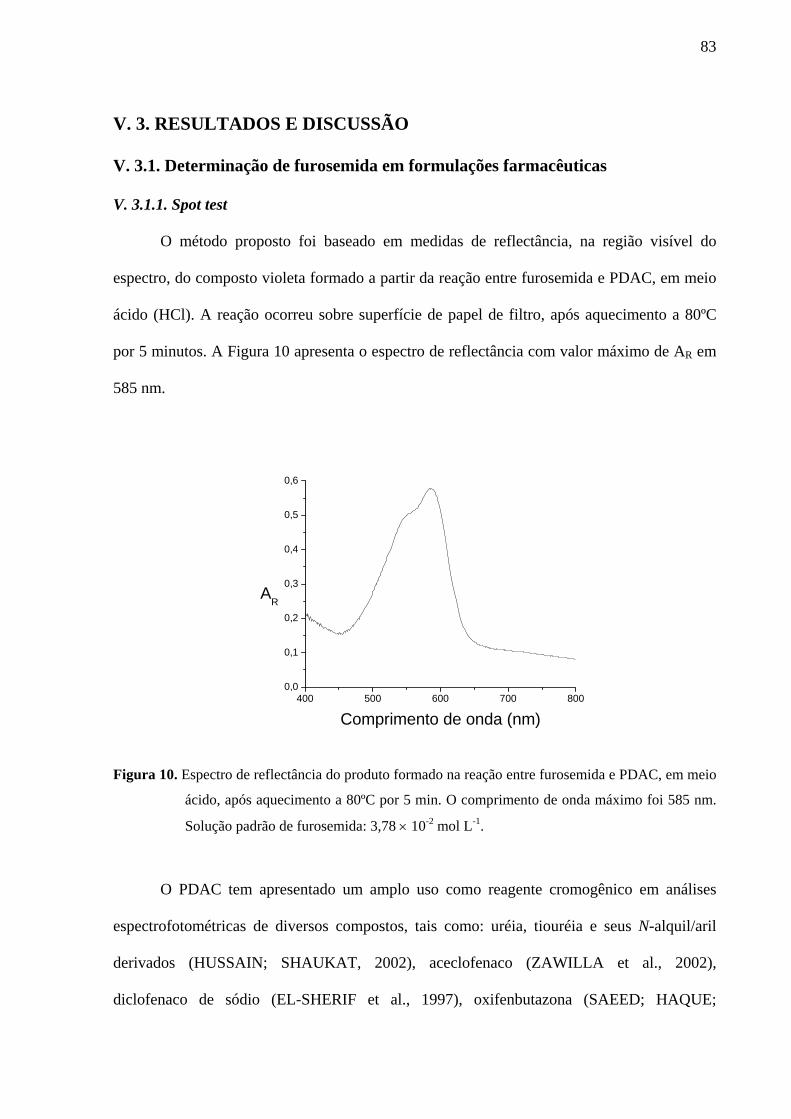
83
V. 3. RESULTADOS E DISCUSSÃO
V. 3.1. Determinação de furosemida em formulações farmacêuticas
V. 3.1.1. Spot test
O método proposto foi baseado em medidas de reflectância, na região visível do
espectro, do composto violeta formado a partir da reação entre furosemida e PDAC, em meio
ácido (HCl). A reação ocorreu sobre superfície de papel de filtro, após aquecimento a 80ºC
por 5 minutos. A Figura 10 apresenta o espectro de reflectância com valor máximo de AR em
585 nm.
Figura 10. Espectro de reflectância do produto formado na reação entre furosemida e PDAC, em meio
ácido, após aquecimento a 80ºC por 5 min. O comprimento de onda máximo foi 585 nm.
Solução padrão de furosemida: 3,78 × 10-2 mol L-1.
O PDAC tem apresentado um amplo uso como reagente cromogênico em análises
espectrofotométricas de diversos compostos, tais como: uréia, tiouréia e seus N-alquil/aril
derivados (HUSSAIN; SHAUKAT, 2002), aceclofenaco (ZAWILLA et al., 2002),
diclofenaco de sódio (EL-SHERIF et al., 1997), oxifenbutazona (SAEED; HAQUE;
400 500 600 700 8000,0
0,1
0,2
0,3
0,4
0,5
0,6
AR
Comprimento de onda (nm)

84
QURESHI, 1993), glafenina e metoclopramida (MOUSSA, 2000). O PDAC também tem sido
usado para análises, em micro-placas, de ácido p-aminohipúrico em amostras de plasma e
urina (AGARWAL, 2002) e para testes rápidos de benzodiazepinas em medicamentos
(LAUDSZUN; KOVAR, 1991). A maioria dos métodos mencionados acima envolve reação
em meio ácido com aquecimento, produzindo compostos coloridos (variando de laranja a
vermelho ou rosa).
O PDAC também foi descrito na literatura para a determinação espectrofotométrica de
furosemida (SHIMDDA; NAGASE, 1977). Uma provável reação entre aminas secundárias
aromáticas e PDAC envolve a condensação do grupo amino secundário protonado com o
grupo carbonila do reagente, formando um sal imínio ou base de Schiff (EL-SHERIF et al.,
1997; ZAWILLA et al., 2002; MOUSSA, 2000). A reação apresentada no Esquema 1 foi
baseada em reações entre outras aminas secundárias aromáticas e PDAC descritas na
literatura (EL-SHERIF et al., 1997; ZAWILLA et al., 2002; MOUSSA, 2000).

85
Esquema 1
Um fato interessante no método para a determinação de furosemida foi que a cor do
spot test obtido a partir da adição da solução de furosemida, PDAC e HCl, nesta ordem, foi
mais intensa nas bordas do que no centro da mancha formada. Observou-se também que
quando a solução de reagente foi adicionada por último, a cor do spot test foi mais uniforme e
muito mais intensa.
Sal Imínio
Cl
H2NO2S
NHCH2
COOH
OH+
OCH2
Cl
H2NO2S COOH
NH2+
+CH3
Ch3CHOCH NCH
Rápido
Cl
H2NO2S COOH
OCH2
NH NCHCHCCH3
CH3
++HO
H
Rápido+
NH
OCH2
Cl
H2NO2S COOH
+ CH3
CH3CHC
H
OHNCH
NCHCHCCH3
CH3
Cl
H2NO2S COOH
OCH2
NH
Rápido
OCH2
Cl
H2NO2S
N
COOH
CH3
CH3NCHC CH+
+
Furosemida PDAC

86
De acordo com Wendlandt e Hecht (1966) a cor do spot test deve ser uniforme sobre
toda a superfície sólida para se obter melhor precisão das medidas de reflectância. Neste
sentido, cabe ressaltar que para a elaboração dos spot tests envolvidos em todos os métodos
desenvolvidos neste trabalho levou-se em consideração alguns detalhes, tais como: ordem e
velocidade de adição das soluções, qualidade do papel de filtro utilizado e volume das
soluções adicionadas. Todos estes detalhes mostraram ser importantes para a uniformidade da
cor do spot test e, conseqüentemente, para a obtenção de boa precisão das medidas de
reflectância.
V. 3.1.2. Planejamento Fatorial de Experimentos
Estudos foram realizados para estabelecer as condições mais favoráveis para a reação
entre furosemida e PDAC, em meio ácido (HCl), visando alcançar a intensidade máxima da
cor do spot test, sobre papel de filtro, em 585 nm. Uma vez que a necessidade de aquecimento
para o desenvolvimento da cor foi observada, os fatores inicialmente estudados foram tempo e
temperatura de aquecimento. Utilizando um planejamento fatorial de 2 níveis em torno das
condições utilizadas para a reação em testes preliminares (10 μL de solução de furosemida
3,02 × 10-2 mol L-1, 10 μL de solução de HCl 6,3%, m/v, 10 μL de solução de PDAC 0,4%
m/v e aquecimento à aproximadamente 70ºC por 3 minutos) foi possível variar os dois fatores
(tempo e temperatura de aquecimento) simultaneamente. A partir dos resultados obtidos neste
planejamento, foi realizado um planejamento de modo similar, no qual os fatores avaliados
foram volumes adicionados de PDAC e de HCl. Desta forma, os valores de AR (respostas)
obtidos com volumes fixos das soluções de furosemida, PDAC e de ácido adicionados foram
otimizados em função do tempo e da temperatura de aquecimento. Do mesmo modo, para
tempo e temperatura de aquecimento e volume de furosemida fixos, as respostas (AR) foram
otimizadas em função dos volumes adicionados de PDAC e de ácido. As condições avaliadas
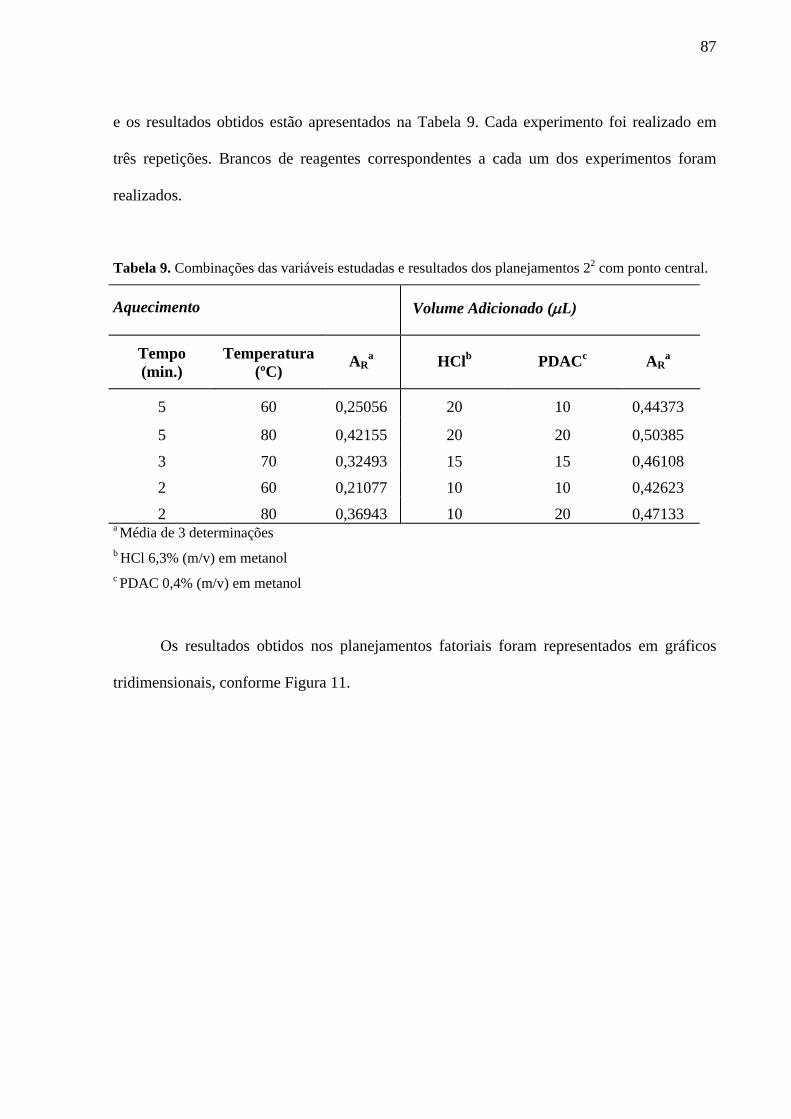
87
e os resultados obtidos estão apresentados na Tabela 9. Cada experimento foi realizado em
três repetições. Brancos de reagentes correspondentes a cada um dos experimentos foram
realizados.
Tabela 9. Combinações das variáveis estudadas e resultados dos planejamentos 22 com ponto central.
Aquecimento Volume Adicionado (μL)
Tempo (min.)
Temperatura (ºC) AR
a HClb PDACc ARa
5 60 0,25056 20 10 0,44373
5 80 0,42155 20 20 0,50385
3 70 0,32493 15 15 0,46108
2 60 0,21077 10 10 0,42623
2 80 0,36943 10 20 0,47133 a Média de 3 determinações b HCl 6,3% (m/v) em metanol c PDAC 0,4% (m/v) em metanol
Os resultados obtidos nos planejamentos fatoriais foram representados em gráficos
tridimensionais, conforme Figura 11.

88
Figura 11. Gráficos tridimensionais obtidos para estabelecer condições ótimas de tempo e temperatura
de aquecimento (A) e de volumes de PDAC e de ácido adicionados (B).
Comparações entre médias foram realizadas utilizando ANOVA e a significância
estatística das diferenças entre as médias foi estimada utilizando teste de Tukey (p≤0,05). Os
resultados demonstraram que as maiores medidas de reflectância sem perda de uniformidade
da cor foram obtidas com aquecimento a 80ºC por 5 minutos, diferindo significativamente
daquelas obtidas com outros tempos de aquecimento e temperaturas. Cabe mencionar que spot
tests obtidos com aquecimento a 90ºC apresentaram reversão de cor do papel e não puderam
ser analisados. Por esta razão, o planejamento foi descontinuado. A partir dos resultados do
planejamento para avaliar volumes de PDAC e de HCl adicionados foi constatado que as
medidas de reflectância obtidas com 20 μL de PDAC e 20 μL de HCl foram
significativamente maiores do que as outras condições estudadas. Analisando os gráficos
tridimensionais é possível observar pontos referentes às condições escolhidas, as quais
mostraram os maiores valores de reflectância (Figura 11).
(A) (B)

89
V. 3.1.3. Estabilidade Óptica
Com o objetivo de avaliar a estabilidade óptica do produto da reação entre furosemida
e PDAC, em meio ácido, foi realizado um acompanhamento cinético do valor de AR em 585
nm a cada 5 minutos até 40 minutos após a adição das soluções no papel de filtro. Os
resultados obtidos demonstraram que não houve diferença significativa entre os valores de AR
obtidos nos diferentes períodos de tempo estudados. Portanto, a estabilidade óptica do produto
formado foi de, pelo menos 40 minutos, nas condições estudadas (Figura 12).
Figura 12. Espectro de reflectância do produto formado na reação entre furosemida e PDAC, em meio
ácido, após aquecimento a 80ºC por 5 min. O comprimento de onda máximo foi 585 nm.
Solução padrão de furosemida: 3,78 × 10-2 mol L-1.
V. 3.1.4. Curva Analítica
A Figura 13 apresenta a curva analítica construída a partir de soluções padrão de
furosemida na faixa de concentração de 7,56 × 10-3 a 6,05 × 10-2 mol L-1. Uma relação linear
(r = 0,9987) foi obtida pela representação gráfica de AR versus log da concentração de
furosemida (mo L-1). O fator 103 foi usado para ajustar a curva analítica com valores de log
maiores do que zero. Valores de AR para esta faixa de concentração foram ajustados pela
5 10 15 20 25 30 35 40 45
0,3
0,4
0,5
0,6
0,7
AR
Tempo (minutos)

90
equação: AR = -0,17768 + 0,41338 × C, onde C = log [furosemida] × 103 (mol L-1). O limite
de detecção foi 1,05 × 10–3 mol L-1 ⎟⎠⎞
⎜⎝⎛ ×
aDPB3 , sendo a = coeficiente angular da curva
analítica e BDP = desvio padrão do branco para 10 repetições (LONG; WINEFORDNER,
1983).
Figura 13. Curva analítica para a determinação de furosemida. Coeficiente de correlação linear (r) =
0,9987. Valores de AR foram obtidos em 585 nm. Faixa de concentração das soluções
padrão de furosemida: 7,56 × 10-3 a 6,05 × 10-2 mol L-1. As barras representam os desvios
padrão para 3 repetições.
V. 3.1.5. Estudo de Interferências
O estudo de interferências foi realizado visando avaliar o efeito dos excipientes mais
comumente presentes na amostra. A porcentagem de furosemida encontrada nas soluções
contendo o fármaco padrão e cada um dos excipientes, separadamente, foi de 96 a 104%, com
coeficientes de variação menores que 5% para três repetições. Esses resultados foram
considerados aceitáveis e, portanto, nenhuma interferência foi constatada a partir destes
excipientes, nas condições estudadas.
0.8 1.0 1.2 1.4 1.6 1.8
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
AR
log ([furosemida]/mol L-1 x 103)

91
V. 3.1.6. Estudo de Adição e Recuperação
Com o objetivo de avaliar efeitos de matriz (formulação farmacêutica, a qual contém
todos os excipientes juntos) foram realizados testes de adição e recuperação de fármaco
padrão utilizando 3 amostras de diferentes marcas comerciais de comprimidos. Os resultados
obtidos estão apresentados na Tabela 10. Para todas as amostras, os valores médios de
recuperação (n=3) variaram de 96,7 a 104,6%, com desvios padrão relativos entre 2,1 a 5,1%.
Essas variações foram consideradas aceitáveis, indicando que os componentes das
formulações farmacêuticas analisadas não influenciaram nas análises.
Tabela 10. Resultados de recuperação de furosemida padrão adicionada em amostras de três
formulações farmacêuticas comerciais diferentes.
Amostras Valor Adicionado (mol L-1)
Valor Encontradoa
(mol L-1) Recuperaçãoa
(%)
A -
7,56 × 10-3
1,51 × 10-2
2,27 × 10-2
3,02 × 10-2
(1,51±0,07) × 10-2
(7,39±0,35) × 10-3
(1,46±0,05) × 10-2
(2,21±0,08) × 10-2
(3,02±0,14) × 10-2
-
97,8 ±4,6
96,7±3,3
97,4±3,5
100,0±4,6
B -
7,56 × 10-3
1,51 × 10-2
2,27 × 10-2
3,02 × 10-2
(1,50±0,04) × 10-2
(7,42±0,24) × 10-3
(1,58±0,05) × 10-2
(2,21±0,10) × 10-2
(2,97±0,06) × 10-2
-
98,1±3,1
104,6±3,3
97,4±4,4
98,3±2,0
C -
7,56 × 10-3
1,51 × 10-2
2,27 × 10-2
3,02 × 10-2
(1,51±0,05) × 10-2
(7,51±0,38) × 10-3
(1,48±0,06) × 10-2
(2,25±0,11) × 10-2
(3,05±0,13) × 10-2
-
99,3±5,1
98,0±4,0
99,1±4,9
101,0±4,3 a Média ± Desvio padrão (DP), n = 3.

92
V. 3.1.7. Aplicação do método proposto e comparação com o método de referência.
O método proposto foi aplicado na determinação de furosemida em algumas
formulações farmacêuticas disponíveis comercialmente e comparado com o método para a
determinação de furosemida em comprimidos descrito na Farmacopéia Britânica
(BRITISH…, 2001), o qual é baseado em medidas por espectrofotometria no UV (271 nm) e
envolve sucessivas diluições da amostra com NaOH 0,1 mol L-1. As análises foram realizadas
em três repetições e os resultados estão apresentados na Tabela 11.
Tabela 11. Determinação de furosemida em formulações farmacêuticas comerciais.
Método Proposto Método de Referência
Amostrasa Valor Encontradob
(mg/comprimido)
Valor de t
(2,78)c
Valor de F
(19,00)c
Valor Encontradob
(mg/comprimido)
A 39,4±0,5 1,72 1,96 39,6±0,7
B 39,6±1,1 0,45 3,31 40,2±2,0
C 40,8±1,5 0,09 1,86 40,9±1,1
D 39,3±1,5 1,17 1,78 41,0±2,0
E 39,2±1,0 0,97 4,00 39,4±0,5
F 39,3±0,8 0,72 2,56 39,2±0,5 a Valor Declarado: 40mg furosemida/comprimido. b Média ± Desvio padrão (DP), n=3. c Valores teóricos de t e F em nível de confiança de 95%.
Os valores médios de furosemida obtidos pelo método proposto variaram de 39,2 a
40,8 mg/comprimido e os desvios padrão de 0,5 a 1,5 mg/comprimido (Tabela 11). Para todas
as formulações analisadas, os resultados obtidos pelo método de referência e pelo método
proposto foram comparados aplicando-se testes t e F, com nível de confiança de 95%. Em
todos os casos, os valores de t e de F calculados não excederam os valores teóricos, indicando
que não houve diferença significativa entre os métodos com relação à precisão e exatidão
(Tabela 11).

93
V. 3.1.8. Conclusões
Este estudo revelou, pela primeira vez, a possibilidade de se empregar a
espectroscopia de reflectância difusa, na região do visível, utilizando spot test na
determinação de furosemida em formulações farmacêuticas. A ordem de adição e os volumes
das soluções de PDAC e de HCl influenciaram significativamente a reação. As melhores
condições para a reação foram obtidas a partir de 10 μL de solução de furosemida, 20 μL da
solução de HCl e 20 μL da solução de PDAC, nesta ordem de adição, e aquecimento a 80ºC
por 5 minutos. Este método foi aplicado com sucesso na análise de furosemida em amostras
de comprimidos de diferentes marcas comerciais. A espectroscopia de reflectância difusa
utilizando spot test oferece vantagens sobre outras técnicas usadas atualmente como
simplicidade, rapidez e baixo consumo de reagentes. Assim, o desenvolvimento de métodos
por espectroscopia de reflectância difusa para a análise de medicamentos é encorajado neste
estudo.

94
V. 3.2. Determinação de hidroclorotiazida em formulações farmacêuticas
V. 3.2.1. Spot test
O método proposto foi baseado em medidas de reflectância, na região do visível, do
composto rosa formado a partir da reação entre hidroclorotiazida e PDAC, em meio ácido
(HCl). A reação ocorreu sobre superfície de papel de filtro, com aquecimento a 80ºC por 8
minutos. A Figura 14 apresenta o espectro de reflectância com valor máximo de AR em 585
nm.
Figura 14. Espectro de reflectância do produto formado na reação entre hidroclorotiazida e PDAC, em
meio ácido, após aquecimento a 80ºC por 8 min. O comprimento de onda máximo foi 585
nm. Solução padrão de hidroclorotiazida: 4,20 × 10–2 mol L-1.
Este trabalho descreve, pela primeira vez, o uso de PDAC como reagente cromogênico
para a determinação de hidroclorotiazida. Uma provável reação entre hidroclorotiazida e
PDAC é apresentada no Esquema 2, tendo como base aquelas descritas na literatura para a
determinação de aminas secundárias aromáticas (EL-SHERIF et al., 1997; ZAWILLA et al.,
2002; MOUSSA, 2000), as quais envolvem a condensação do grupo amino secundário
400 500 600 700 8000,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
0,18
AR
Comprimento de onda (nm)

95
protonado com o grupo carbonila do PDAC, formando um sal imínio ou base de Schiff
(Esquema 2).
H2NO2S
Cl
SNH
NH
O2
H+O2
H2NO2S
Cl
SNH2
NH
+
+ NCHCHOCHCh3
CH3
Rápido
H2NO2S
Cl
SNH
NH
O2 CH3
Ch3CHC
HNCH+
+OH
NCHCh3
CH3O2H2NO2S
Cl
SNH
NHH
OHC CH
Rápido
+
Lento
+O2 CH3
Ch3NCH
H2NO2S
Cl
SNH
NH
CH CHH2NO2S
Cl
SN
NHCh3
CH3O2
NCHCH CH+
Hidroclorotiazida PDAC
Esquema 2
Sabendo que a ordem de adição das soluções pode influenciar a uniformidade e a
intensidade da cor do spot test, este detalhe foi considerado, estudando-se todas as possíveis
combinações de ordens de adição das soluções. Da mesma forma que no método para a
determinação de furosemida, foi observado que o spot test apresentou uma cor mais uniforme
e intensa quando a solução do analito foi inicialmente adicionada, seguida pela adição da
solução do ácido e, por último, da solução de reagente.
Sal Imínio ou Base de Schiff

96
V. 3.2.2. Planejamento Fatorial de Experimentos
V. 3.2.2.1. Planejamento Fatorial Fracionário: triagem de fatores
Presumindo que a reação entre hidroclorotiazida e PDAC, em meio ácido (HCl)
poderia ser influenciada por vários fatores, as condições mais favoráveis para a reação sobre
papel de filtro foram investigadas visando alcançar a intensidade máxima da cor em 585 nm.
Planejamento fatorial fracionário permitiu o estudo simultâneo dos efeitos que diversos
fatores poderiam ter sobre a reação de spot test. Os fatores avaliados foram: tempo de
aquecimento, temperatura de aquecimento, volume de ácido e volume de reagente. Todos os
experimentos foram realizados com solução padrão de hidroclorotiazida com concentração de
6,72 × 10-2 mol L-1. Brancos de reagentes correspondentes a cada um dos experimentos foram
realizados. A Tabela 12 apresenta a matriz de planejamento e os valores de AR obtidos no
planejamento fatorial fracionário para cada um dos experimentos.
Tabela 12. Matriz do planejamento fatorial fracionário.
Fatores
Experimentos Temperatura (ºC)
Tempo (min.)
Volume de Ácido (μL)a
Volume de PDAC (μL)b
AR
1 60 10 20 10 0,18859
2 60 5 20 20 0,17124
3 60 5 10 10 0,17355
4 90 5 10 20 0,19092
5 60 10 10 20 0,18475
6 90 10 20 20 -
7 90 10 10 10 -
8 90 5 20 10 0,19232 a Solução de HCl 10% (m/v) b Solução de PDAC 0,4% (m/v)

97
Deve-se mencionar que houve dificuldades para se realizar todos os experimentos,
uma vez que o papel não resistiu a temperaturas relativamente elevadas (próximas a 90ºC),
ocorrendo reversão de cor do papel e o espectro de reflectância do produto colorido foi
diferente daquele apresentado pelo composto rosa formado com aquecimento em
temperaturas mais baixas. Por esta razão, os experimentos 6 e 7 (Tabelas 12) não foram
executados.
A partir dos resultados do planejamento fatorial fracionário, o gráfico de Pareto
(Figura 15) foi obtido para visualização dos efeitos estimados dos fatores principais. Embora
nenhum dos fatores tivesse influência significativa – nenhuma barra (fator) ultrapassou a linha
de significância – pode ser notado que os efeitos estimados dos fatores tempo e temperatura
de aquecimento foram maiores do que os fatores volume de ácido e volume de PDAC.
Portanto, tempo e temperatura de aquecimento foram considerados fatores mais importantes
do que volume de ácido e de reagente e foram estudados em seguida.
Figura 15. Gráfico de Pareto do planejamento fatorial fracionário para a reação entre hidroclorotiazida
e PDAC, sobre papel de filtro.
0,0123232
-0,031754
-1,4085
-1,49178
p=.05
Efeito Estimado (Valor Absoluto)
(3)Volume de Ácido (uL)
(4)Volume de PDAC (uL)
(1)Temperatura (ºC)
(2)Tempo (min.)

98
V. 3.2.2.2. Efeitos do tempo e temperatura de aquecimento
Para investigar os fatores tempo e temperatura de aquecimento foi realizado um
planejamento fatorial completo. A Tabela 13 mostra a matriz de planejamento que abrangeu
as mesmas condições utilizadas no planejamento anterior, porém, com um maior número de
níveis entre os valores mínimos e máximos e também contendo um ponto central (Tabela 13).
Tabela 13. Matriz do planejamento fatorial completo com ponto central.
Experimentos 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Temperatura (ºC) 80 90 60 60 70 80 70 80 80 70 90 60 90 90 60 70
Tempo (min) 5 8 7 8 5 7 7 8 10 8 5 5 10 8 10 10
Vale mencionar que o aquecimento do papel de filtro contendo as soluções não pôde
ser mantido a 90ºC por mais do que 5 minutos. Por isso, as combinações referentes à
temperatura de 90ºC por 7, 8 e 10 minutos não foram realizadas.
A Figura 16 apresenta o gráfico tridimensional obtido a partir dos resultados do
planejamento com ponto central. Analisando a superfície obtida, é possível observar os pontos
referentes às condições escolhidas, os quais mostraram valores de AR mais altos e que
forneceram spot tests com maior uniformidade na cor.

99
Figura 16. Gráfico tridimensional obtido no estabelecimento das melhores condições de tempo e
temperatura de aquecimento para a reação entre hidroclorotiazida e PDAC sobre papel de
filtro.
Como pode ser visto na Figura 16, as respostas de reflectância mais altas foram
alcançadas com o aquecimento a 80ºC por 8 minutos. O formato da superfície analisada
indicou a direção, na qual novos experimentos deveriam ser realizados. Entretanto, devido a
limitações experimentais (tempos e temperaturas de aquecimento relativamente altas) o
planejamento não pôde ser estendido para outras condições experimentais.
0,2 0,18 0,16 0,14 0,12

100
V. 3.2.3. Estabilidade Óptica
Com o objetivo de avaliar a estabilidade óptica do produto da reação entre
hidroclorotiazida e PDAC, em meio ácido, foi realizado um acompanhamento cinético do
valor de AR em 585 nm a cada 5 minutos até 40 minutos após a adição das soluções no papel
de filtro. Os resultados obtidos demonstraram que não houve diferença significativa entre os
valores de AR obtidos nos diferentes períodos de tempo. Portanto, a estabilidade óptica do
produto formado foi de pelo menos 40 minutos, nas condições estudadas (Figura 17).
Figura 17. Estudo da estabilidade óptica do produto da reação entre hidroclorotiazida e PDAC, em
meio ácido, sobre papel de filtro. As análises dos spot tests foram realizadas a cada 5
minutos até 40 minutos. Solução padrão de hidroclorotiazida: 1,01 × 10-1 mol L-1.
V. 3.2.4. Curva Analítica
A Figura 18 apresenta a curva analítica construída a partir das soluções padrão de
hidroclorotiazida na faixa de concentração de 3,36 × 10-2 a 1,01 × 10-1 mol L-1. Uma relação
linear (r = 0,9979) foi obtida representando-se graficamente AR versus log da concentração de
hidroclorotiazida (mol L-1). O fator 102 foi usado para ajustar a curva analítica com valores
de log maiores do que zero. Valores de AR para esta faixa de concentração foram
0 5 10 15 20 25 30 35 40 450,12
0,14
0,16
0,18
0,20
0,22
AR
Tempo (minutos)
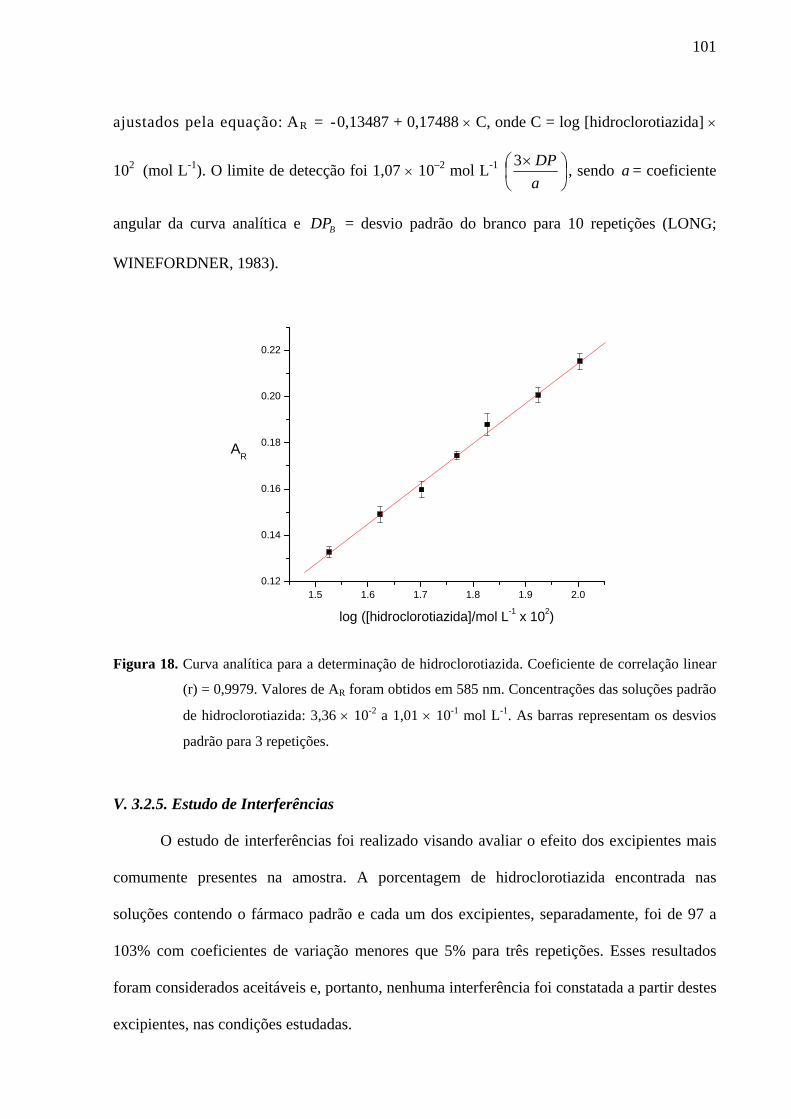
101
ajustados pela equação: AR = -0,13487 + 0,17488 × C, onde C = log [hidroclorotiazida] ×
102 (mol L-1). O limite de detecção foi 1,07 × 10–2 mol L-1 ⎟⎠⎞
⎜⎝⎛ ×
aDP3 , sendo a = coeficiente
angular da curva analítica e BDP = desvio padrão do branco para 10 repetições (LONG;
WINEFORDNER, 1983).
Figura 18. Curva analítica para a determinação de hidroclorotiazida. Coeficiente de correlação linear
(r) = 0,9979. Valores de AR foram obtidos em 585 nm. Concentrações das soluções padrão
de hidroclorotiazida: 3,36 × 10-2 a 1,01 × 10-1 mol L-1. As barras representam os desvios
padrão para 3 repetições.
V. 3.2.5. Estudo de Interferências
O estudo de interferências foi realizado visando avaliar o efeito dos excipientes mais
comumente presentes na amostra. A porcentagem de hidroclorotiazida encontrada nas
soluções contendo o fármaco padrão e cada um dos excipientes, separadamente, foi de 97 a
103% com coeficientes de variação menores que 5% para três repetições. Esses resultados
foram considerados aceitáveis e, portanto, nenhuma interferência foi constatada a partir destes
excipientes, nas condições estudadas.
1.5 1.6 1.7 1.8 1.9 2.00.12
0.14
0.16
0.18
0.20
0.22
AR
log ([hidroclorotiazida]/mol L-1 x 102)
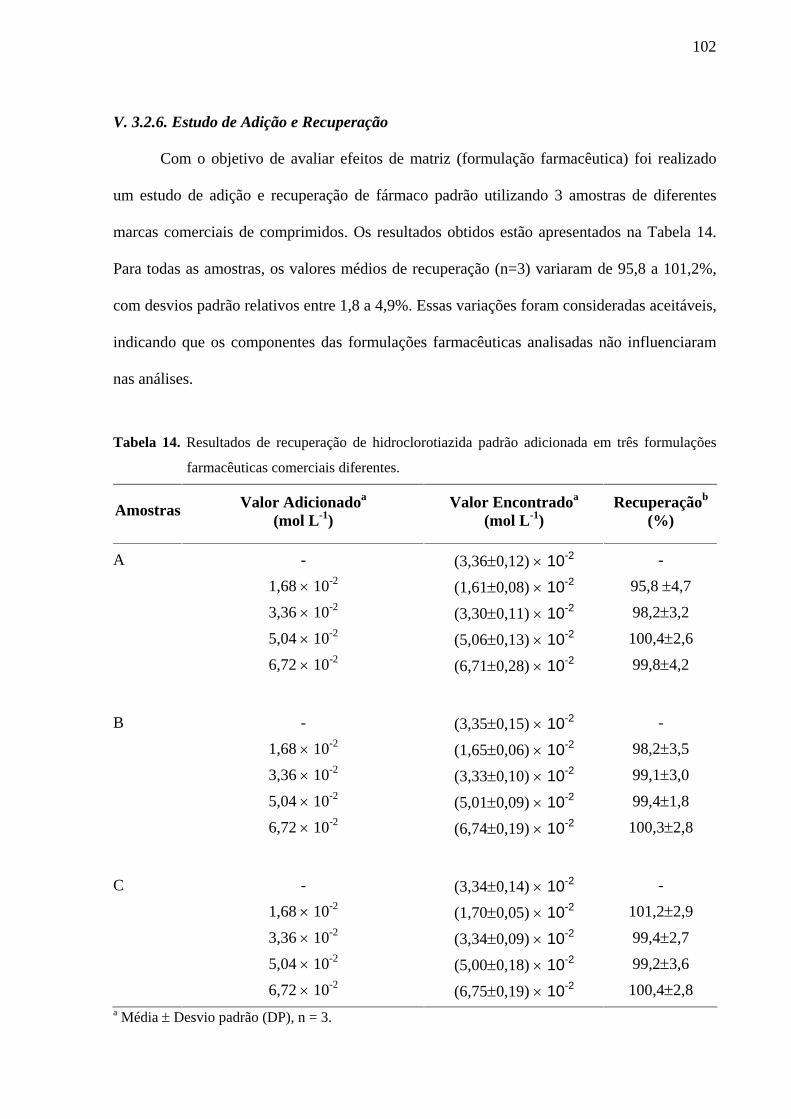
102
V. 3.2.6. Estudo de Adição e Recuperação
Com o objetivo de avaliar efeitos de matriz (formulação farmacêutica) foi realizado
um estudo de adição e recuperação de fármaco padrão utilizando 3 amostras de diferentes
marcas comerciais de comprimidos. Os resultados obtidos estão apresentados na Tabela 14.
Para todas as amostras, os valores médios de recuperação (n=3) variaram de 95,8 a 101,2%,
com desvios padrão relativos entre 1,8 a 4,9%. Essas variações foram consideradas aceitáveis,
indicando que os componentes das formulações farmacêuticas analisadas não influenciaram
nas análises.
Tabela 14. Resultados de recuperação de hidroclorotiazida padrão adicionada em três formulações
farmacêuticas comerciais diferentes.
Amostras Valor Adicionadoa (mol L-1)
Valor Encontradoa
(mol L-1) Recuperaçãob
(%)
A - 1,68 × 10-2
3,36 × 10-2 5,04 × 10-2 6,72 × 10-2
(3,36±0,12) × 10-2 (1,61±0,08) × 10-2
(3,30±0,11) × 10-2 (5,06±0,13) × 10-2
(6,71±0,28) × 10-2
- 95,8 ±4,7
98,2±3,2 100,4±2,6 99,8±4,2
B
-
1,68 × 10-2 3,36 × 10-2
5,04 × 10-2 6,72 × 10-2
(3,35±0,15) × 10-2 (1,65±0,06) × 10-2 (3,33±0,10) × 10-2
(5,01±0,09) × 10-2 (6,74±0,19) × 10-2
-
98,2±3,5 99,1±3,0
99,4±1,8 100,3±2,8
C
-
1,68 × 10-2
3,36 × 10-2 5,04 × 10-2 6,72 × 10-2
(3,34±0,14) × 10-2 (1,70±0,05) × 10-2
(3,34±0,09) × 10-2 (5,00±0,18) × 10-2
(6,75±0,19) × 10-2
-
101,2±2,9
99,4±2,7 99,2±3,6 100,4±2,8
a Média ± Desvio padrão (DP), n = 3.

103
V. 3.2.7. Aplicação do método proposto e comparação com o método de referência.
O método proposto foi aplicado na determinação de hidroclorotiazida em seis
formulações farmacêuticas disponíveis comercialmente e comparado com o método para a
determinação de hidroclorotiazida em comprimidos descrito na Farmacopéia Britânica
(BRITISH…, 2001), o qual é baseado em medidas por espectrofotometria no UV (273 nm) e
envolve sucessivas diluições da amostra com NaOH 0,1 mol L-1. As análises foram realizadas
em três repetições e os resultados estão apresentados na Tabela 15.
Tabela 15. Determinação de hidroclorotiazida em formulações farmacêuticas comerciais.
Método Proposto Método de ReferênciaAmostrasa Valor Encontradob
(mg/comprimido) Valor de t
(2,78)c Valor de F
(19,00)c Valor Encontradob (mg/comprimido)
A 49,2±2,2 0,23 18,66 49,5± 0,5
B 48,5±1,8 0,11 1,92 48,3±1,3
C 49,5±2,6 0,62 13,80 48,6± 0,7
D 51,5±2,6 0,53 1,69 50,5± 2,0
E 50,0±1,0 1,62 6,25 49,1±0,4
F 48,8±1,7 0,38 2,52 48,1±2,7 a Valor Declarado: 50 mg hidroclorotiazida/comprimido. b Média ± Desvio padrão (DP), n = 3. c Valores teóricos de t e F em nível de confiança de 95%.
Os valores médios de hidroclorotiazida obtidos pelo método proposto variaram de 48,5
a 51,5 mg/comprimido e os desvios padrão de 1,0 a 2,6 mg/comprimido (Tabela 15). Para
todas as formulações analisadas, os resultados obtidos pelo método de referência e pelo
método proposto foram comparados aplicando-se testes t e F, com nível de confiança de 95%.
Em todos os casos, os valores de t e de F calculados não excederam os valores teóricos,

104
indicando que não houve diferença significativa entre os métodos com relação à precisão e
exatidão (Tabela 15).
V. 3.2.8. Conclusões
O método proposto demonstrou ser uma interessante alternativa aos outros
procedimentos existentes para a análise de comprimidos contendo hidroclorotiazida. Os
resultados do planejamento fatorial de experimentos indicaram que tempo e temperatura de
aquecimento foram os fatores mais importantes para a reação de spot test, influenciando
significativamente na resposta de reflectância. As melhores condições para a reação foram
obtidas a partir de 20 μL de solução de hidroclorotiazida, 20 μL de solução de HCl e 20 μL de
solução de PDAC, nesta ordem de adição, e aquecimento a 80ºC por 8 minutos. Este método
foi aplicado com sucesso na análise de hidroclorotiazida em amostras de comprimidos de
diferentes marcas comerciais. Comparando o método proposto com os outros métodos
descritos para a determinação de hidroclorotiazida em comprimidos, este método oferece
vantagens relacionadas à simplicidade, rapidez e baixo consumo de reagentes, preenchendo os
requisitos para o controle de qualidade de medicamentos.

105
V. 3. 3. Determinação de propranolol em formulações farmacêuticas
V. 3.3.1. Spot Test
Desde 1927, a 2,6-dicloroquinona-4-cloroimida (DCQ) ou reagente de Gibbs tem sido
comumente utilizada para a determinação de fenóis (GIBBS, 1927). O reagente de Gibbs
também tem tido aplicação para a detecção de aminas e hidrocarbonetos aromáticos por
cromatografia em camada delgada (ROSS, 1968). Recentemente, o uso de DCQ tem sido
descrito para a detecção de produtos oxigenados resultantes da biotransformação de substratos
aromáticos (QUINTANA; DIDION; DALTON, 1997) e para a determinação de fármacos, tais
como: ranitidina (EMMANUEL; HALDANKAR, 1989), cefadroxil (SASTRY; RAO;
PRASAD, 1997), mequitazina (EL-RAGEHY; BADAWEY; EL-KHATEEB, 2002),
tolnaftato (SASTRY et al., 1993) e D-penicilamina (AL-MAJED, 1999), que é um produto
caracterítisco da degradação ácida dos antibióticos β-lactâmicos. Vale ressaltar que este
trabalho descreve, pela primeira vez, o uso de DCQ como reagente cromogênico para a
determinação de propranolol.
Considerando que o propranolol possui um grupo naftol substituído, uma possível
reação entre propranolol e DCQ, com base na literatura (SASTRY et al., 1993), é apresentada
no Esquema 3.
Esquema 3

106
A reação apresentada no Esquema 3 foi baseada na reação entre tolnaftato e DCQ
(SASTRY et al., 1993), a qual envolve um acoplamento oxidativo que ocorre com o ataque
eletrofílico do reagente de Gibbs na posição orto do radical naftol substituído presente no
propranolol, formando um produto colorido.
Neste trabalho, a reação entre propranolol e DCQ produziu um composto colorido na
superfície do papel de filtro, cujo espectro de reflectância, com valor máximo de AR em 500
nm, é apresentado na Figura 19.
Figura 19. Espectro de reflectância do produto formado na reação entre propranolol e DCQ. Os spot
tests foram analisados 10 minutos após a adição das soluções sobre o papel de filtro. O
comprimento de onda máximo foi 500 nm. Solução padrão de propranolol: 8,45 × 10–2
mol L-1.
V. 3.3.2. Planejamento Fatorial de Experimentos
Estudos foram realizados para estabelecer as condições mais favoráveis para a reação
entre propranolol e DCQ, sobre papel de filtro, visando alcançar a intensidade máxima da cor
do spot test em 500 nm. Todos os experimentos foram realizados com solução de propranolol
na concentração de 8,45 × 10-2 mol L-1.
400 500 600 700 8000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
AR
Comprimento de onda (nm)

107
Inicialmente, um planejamento fatorial completo foi realizado, o qual permitiu estudar
simultaneamente três fatores que poderiam ter um efeito importante na reação de spot test. Os
fatores de interesse foram: solvente para DCQ, solvente para propranolol e concentração de
DCQ. O solvente para DCQ foi fixado em 3 níveis: acetona, dioxano e acetonitrila; o solvente
para propranolol também foi fixado em 3 níveis: água, etanol e metanol; e a concentração de
DCQ em 2 níveis: 20 e 60 mg mL-1. Os experimentos não foram realizados na ordem em que
aparecem na Tabela 16 e sim de forma aleatória dentro do conjunto de experimentos, em 2
repetições. Vale mencionar que, em todo o planejamento, brancos correspondentes a cada um
dos experimentos foram realizados. A Tabela 16 apresenta a matriz de planejamento, bem
como os valores de resposta obtidos no planejamento fatorial completo gerado a partir das
combinações dos três fatores estudados, em todos os níveis. Os valores reais correspondentes
aos níveis de cada fator foram codificados com algarismos arábicos (1 a 3) visando remover
as unidades e facilitar a execução dos experimentos.
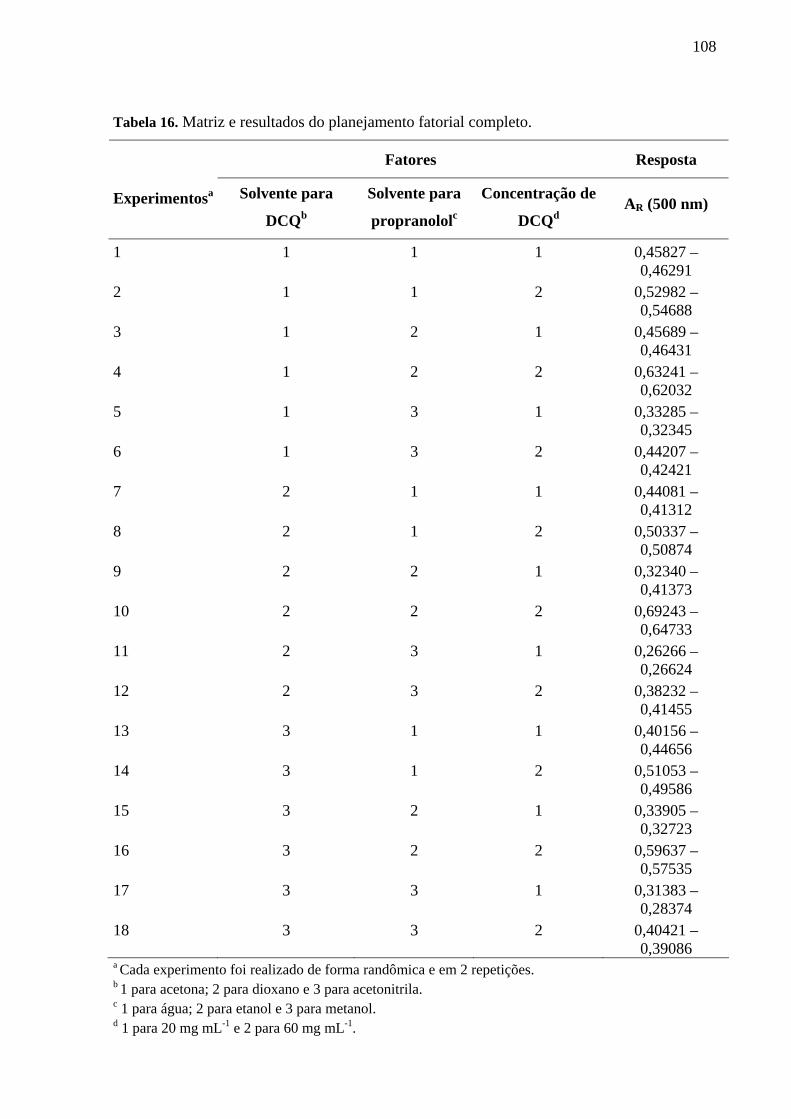
108
Tabela 16. Matriz e resultados do planejamento fatorial completo.
Fatores Resposta
Experimentosa Solvente para
DCQb
Solvente para
propranololc
Concentração de
DCQd AR (500 nm)
1 1 1 1 0,45827 – 0,46291
2 1 1 2 0,52982 – 0,54688
3 1 2 1 0,45689 – 0,46431
4 1 2 2 0,63241 – 0,62032
5 1 3 1 0,33285 – 0,32345
6 1 3 2 0,44207 – 0,42421
7 2 1 1 0,44081 – 0,41312
8 2 1 2 0,50337 – 0,50874
9 2 2 1 0,32340 – 0,41373
10 2 2 2 0,69243 – 0,64733
11 2 3 1 0,26266 – 0,26624
12 2 3 2 0,38232 – 0,41455
13 3 1 1 0,40156 – 0,44656
14 3 1 2 0,51053 – 0,49586
15 3 2 1 0,33905 – 0,32723
16 3 2 2 0,59637 – 0,57535
17 3 3 1 0,31383 – 0,28374
18 3 3 2 0,40421 – 0,39086
a Cada experimento foi realizado de forma randômica e em 2 repetições. b 1 para acetona; 2 para dioxano e 3 para acetonitrila. c 1 para água; 2 para etanol e 3 para metanol. d 1 para 20 mg mL-1 e 2 para 60 mg mL-1.

109
A partir dos resultados do planejamento fatorial completo, o gráfico de Pareto (Figura
20) foi obtido para visualização dos efeitos estimados dos fatores principais. O gráfico de
Pareto fornece uma representação gráfica para estes fatores e permite olhar a magnitude e a
importância de um efeito. Neste gráfico, as barras (fatores) que graficamente ultrapassam a
linha de significância exercem uma influência estatisticamente significativa sobre o resultado.
Assim, pode ser claramente observado na Figura 20, que todos os fatores exerceram efeito
significativo sobre a resposta. Concentração de DCQ foi o fator que teve maior influência,
seguida pelo solvente para propranolol e, em menor extensão, solvente para DCQ.
-2,56093
-8,78132
11,09
p=.05
Efeito Estimado (Valor Absoluto)
(1)Solvente para DCQ
(2)Solvente para Propranolol
(3)Concentração de DCQ
Figura 20. Gráfico de Pareto do planejamento fatorial completo para a reação entre propranolol e
DCQ, sobre papel de filtro.
Concentração de DCQ foi o fator mais importante entre todos os fatores investigados e
teve um elevado valor positivo. Este fato indicou que as maiores respostas foram obtidas com
solução de DCQ na concentração de 60 mg mL-1. Assim, valores de concentração mais altos
foram investigados no planejamento experimental seguinte, o qual foi do tipo composto
central.

110
Observou-se também no gráfico de Pareto (Figura 20) que o solvente para propranolol
teve um efeito significativo e negativo, indicando que as maiores respostas foram obtidas com
o uso de água. Uma interessante observação foi que misturas etanol-água dissolviam melhor o
propranolol do que estes solventes separadamente. Por esta razão, no planejamento composto
central misturas etanol-água, em proporções variando de 0 a 70% (v/v) foram avaliadas e a
possibilidade de se utilizar metanol como solvente para propranolol foi eliminada.
O gráfico de Pareto (Figura 20) também demonstrou que o solvente para DCQ teve um
significativo, mas baixo efeito negativo sobre a resposta, indicando que as maiores respostas
foram obtidas com acetona. Considerando estes resultados e lembrando que se tratam de
fatores qualitativos, a acetona foi então selecionada para ser o solvente para DCQ.
Com base nos resultados obtidos no planejamento fatorial completo, um planejamento
composto central foi então delineado visando obter as melhores condições para a reação. Os
fatores estudados foram concentração de DCQ e solvente para propranolol. Os pontos (níveis)
de um planejamento composto central estão a uma distância de 2 unidade codificada do
ponto central (normalmente codificado como ponto zero); desta forma todos os pontos estão
sobre uma circunferência com raio 2 . A Tabela 17 apresenta a matriz usada para o
planejamento composto central e os valores de resposta para cada um dos experimentos. O
experimento correspondente ao ponto central foi realizado em quatro repetições.

111
Tabela 17. Matriz e resultados obtidos no planejamento composto central.
Fatores Resposta Experimentos
Concentração de DCQ
(mg mL-1)a
Solvente para Propranolol
% (v/v) Etanol em água a AR (500 nm)
1 20 (-1) 10% (-1) 0,46570
2 80 (+1) 10% (-1) 0,53543
3 20 (-1) 60% (+1) 0,46486
4 80 (+1) 60% (+1) 0,54286
5 50 (0) 35% (0) 0,55065
6 50 (0) 35% (0) 0,56264
7 50 (0) 35% (0) 0,55828
8 50 (0) 35% (0) 0,56023
9 10 (- 2 )b 35% (0) 0,38359
10 90 (+ 2 )b 35% (0) 0,53933
11 50 (0) 0% (água) (- 2 )b 0,43494
12 50 (0) 70% (+ 2 )b 0,43496 a Os valores codificados estão entre parênteses. b Os valores experimentais foram aproximados a 2 para facilitar a preparação das soluções.
A metodologia de superfície de resposta é uma poderosa ferramenta experimental para
otimizar parâmetros múltiplos e inter-relacionados. No desenvolvimento deste método,
experimentos foram conduzidos de forma a alcançar valores de cada um dos fatores estudados
(variáveis independentes) que fornecessem maiores valores de AR (variável dependente). Os
valores da variável dependente foram medidos para cada uma das condições da variável
independente (concentração de DCQ e solvente para propranolol). Em seguida, uma
superfície de resposta foi obtida.
A Figura 21 apresenta o gráfico tridimensional obtido a partir dos dados experimentais
e ajustados a uma superfície de resposta.

112
Figura 21. Superfície de resposta obtida no planejamento composto central para valores de AR em
função da concentração de DCQ e mistura etanol-água (%, v/v) como solvente para
propranolol.
O modelo de regressão quadrático (valor de p < 0,0005), em termos de fatores
escalonados é dado por:
22 000075,0005151,0000039,0005377,0291554,0 yyxxZ −+−+= (Equação 7)
onde, Z é o fator de resposta correspondente ao valor de AR. Os fatores x e y são:
concentração de DCQ e solvente para propranolol, respectivamente.
O coeficiente de regressão calculado (R2) foi 81%, indicando que a equação obtida
explica a relação entre os resultados experimentais e os efeitos dos fatores estudados.
Nota-se pelo formato da superfície que a região ótima foi encontrada e também que as
respostas máximas foram alcançadas com concentração de DCQ 70 mg mL-1 e etanol 35%
(v/v) como o solvente para propranolol.
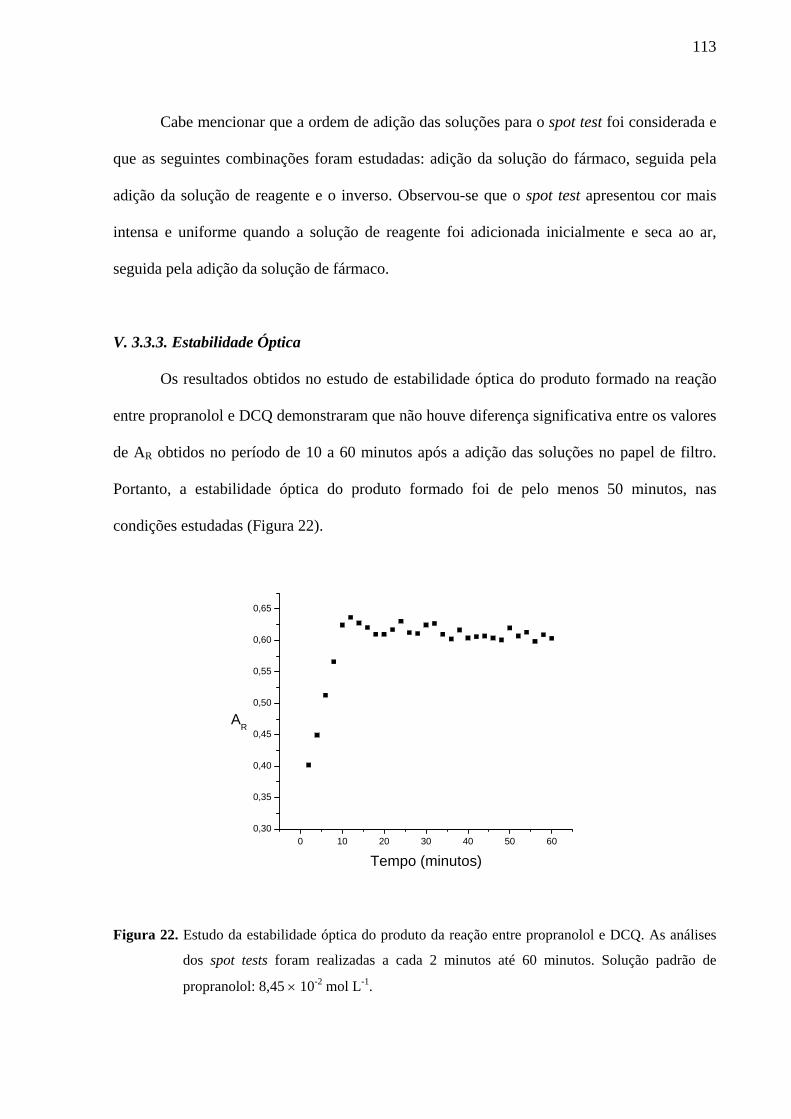
113
Cabe mencionar que a ordem de adição das soluções para o spot test foi considerada e
que as seguintes combinações foram estudadas: adição da solução do fármaco, seguida pela
adição da solução de reagente e o inverso. Observou-se que o spot test apresentou cor mais
intensa e uniforme quando a solução de reagente foi adicionada inicialmente e seca ao ar,
seguida pela adição da solução de fármaco.
V. 3.3.3. Estabilidade Óptica
Os resultados obtidos no estudo de estabilidade óptica do produto formado na reação
entre propranolol e DCQ demonstraram que não houve diferença significativa entre os valores
de AR obtidos no período de 10 a 60 minutos após a adição das soluções no papel de filtro.
Portanto, a estabilidade óptica do produto formado foi de pelo menos 50 minutos, nas
condições estudadas (Figura 22).
Figura 22. Estudo da estabilidade óptica do produto da reação entre propranolol e DCQ. As análises
dos spot tests foram realizadas a cada 2 minutos até 60 minutos. Solução padrão de
propranolol: 8,45 × 10-2 mol L-1.
0 10 20 30 40 50 600,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
AR
Tempo (minutos)

114
V. 3.3.4. Curva Analítica
A Figura 23 apresenta a curva analítica construída a partir das soluções padrão de
propranolol na faixa de concentração de 1,35 × 10-2 a 8,45 × 10-2 mol L-1. Uma relação linear
(r = 0,9991) foi obtida representando-se graficamente AR versus log da concentração de
propranolol (mol L-1). O fator 102 foi usado para ajustar a curva analítica para valores de log
maiores que zero. Valores de AR para a faixa de concentração estudada foram
ajustados pela equação: AR = 0,114360 + 0,421087 × C, onde C = log [propranolol] × 102
(mol L-1). O limite de detecção foi 8,16 × 10–3 mol L-1 ⎟⎟⎠
⎞⎜⎜⎝
⎛ ×
aPD B3
, sendo a = coeficiente
angular da curva analítica e BDP = desvio padrão do branco para 10 repetições (LONG;
WINEFORDNER, 1983).
Figura 23. Curva analítica para determinação de propranolol. Coeficiente de correlação linear (r) =
0,9991. Valores de AR foram obtidos em 500 nm. Concentrações das soluções padrão de
propranolol: 1,35 × 10-2 a 8,45 × 10-2 mol L-1. As barras representam o desvio padrão para
3 repetições.
0.0 0.2 0.4 0.6 0.8 1.0
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
AR
log ([propranolol]/mol L-1 x 102)

115
V. 3.3.5. Estudo de Interferências
O estudo de interferências foi realizado visando avaliar o efeito dos excipientes mais
comumente presentes na amostra. A porcentagem de propranolol encontrado nas soluções
contendo o fármaco padrão e cada um dos excipientes, separadamente, foi de 94 a 103% com
coeficientes de variação menores que 5% para três repetições. Esses resultados foram
considerados aceitáveis e, portanto, nenhuma interferência foi constatada a partir destes
excipientes, nas condições estudadas.
V. 3.3.6. Estudo de Adição e Recuperação
Com o objetivo de avaliar efeitos de matriz (formulação farmacêutica) foi realizado
um estudo de adição e recuperação de fármaco padrão utilizando 3 amostras de diferentes
marcas comerciais de comprimidos. Os resultados obtidos estão apresentados na Tabela 18.
Para todas as amostras os valores médios de recuperação (n=3) variaram de 95,0 a 104,7%,
com desvios padrão relativos entre 0,2 a 4,8%. Essas variações foram consideradas aceitáveis,
indicando que os componentes das formulações farmacêuticas analisadas não influenciaram
nas análises.

116
Tabela 18. Resultados de recuperação de propranolol adicionado em formulações farmacêuticas.
Amostras Valor Adicionadoa
(mol L-1)
Valor Encontradoa
(mol L-1)
Recuperaçãoa
(%)
A
-
1,35 × 10-2
2,70 × 10-2
4,05 × 10-2
5,40 × 10-2
(2,65±0,10) × 10-2
(1,29±0,02) × 10-2
(2,68±0,06) × 10-2
(4,24±0,08) × 10-2
(5,53±0,14) × 10-2
-
95,4±1,5
99,3±2,4
104,7±2,1
102,5±2,5
B
-
1,35 × 10-2
2,70 × 10-2
4,05 × 10-2
5,40 × 10-2
(2,68±0,10) × 10-2
(1,32±0,03) × 10-2
(2,56±0,120× 10-2
(4,14±0,14) × 10-2
(5,46±0,01) × 10-2
-
97,5±2,4
95,0±4,6
102,3±3,4
101,2±0,2
C
-
1,35 × 10-2
2,70 × 10-2
4,05 × 10-2
5,40 × 10-2
(2,70±0,10) × 10-2
(1,36±0,06) × 10-2
(2,66±0,12) × 10-2
(3,95±0,08) × 10-2
(5,32±0,15) × 10-2
-
100,7±4,6
98,4±4,4
97,5±1,9
98,6±2,8 a Média ± Desvio padrão (DP), n = 3.
V. 3.3.7. Aplicação do método proposto e comparação com o método de referência
O método proposto foi aplicado na determinação de propranolol em algumas
formulações farmacêuticas disponíveis comercialmente e comparado com o método para a
determinação de propranolol em comprimidos descrito na Farmacopéia Britânica
(BRITISH…, 2001), o qual é baseado na espectrofotometria no UV (290 nm) e envolve
sucessivas diluições da amostra com metanol. As análises foram realizadas em três repetições
e os resultados estão apresentados na Tabela 19.

117
Tabela 19. Determinação de propranolol em formulações farmacêuticas comerciais.
Método Proposto Método de Referência
Amostrasa Valor Encontradob
(mg/comprimido) Valor de t
(2,78)c Valor de F
(19,00)c Valor Encontradob (mg/comprimido)
A 38,9±0,3 1,06 2,67 38,5±0,5
B 41,0±1,5 0,88 1,86 40,1±1,1
C 39,7±0,4 1,60 6,02 38,6±1,1
D 38,0±1,1 1,10 9,16 38,1± 0,4
E 41,0±2,4 0,72 1,44 39,7±2,0
F 37,4±0,5 0,47 1,04 38,6±0,5
a Valor Nominal: 40 mg propranolol/comprimido b Média ± Desvio padrão (DP), n = 3. c Valores teóricos de t e F em nível de confiança de 95%.
Os valores médios de atenolol obtidos pelo método proposto variaram de 37,4 a 41,0
mg/comprimido, com desvios padrão de 0,30 a 2,38 mg/comprimido (Tabela 19). Para todas
as formulações analisadas, os resultados obtidos pelo método de referência e pelo proposto
foram comparados aplicando-se testes t e F, com nível de confiança de 95%. Em todos os
casos, os valores de F e de t calculados não excederam os valores teóricos, indicando que não
houve diferença significativa entre os métodos com relação à precisão e exatidão (Tabela 19).
V. 3.3.8. Conclusões
Este trabalho demonstrou, pela primeira vez, o uso de DCQ como reagente
cromogênico para a determinação de propranolol e também da espectroscopia de reflectância
difusa utilizando spot test como uma interessante alternativa aos outros procedimentos
existentes para a análise de comprimidos contendo propranolol. Planejamento fatorial foi
empregado a fim de se otimizar as condições para a reação de spot test e as melhores
condições foram obtidas a partir de 30 μL de solução de propranolol em etanol 35% (v/v) e 30

118
μL de solução de DCQ 70 mg/mL em acetona. O método proposto foi aplicado com sucesso
na análise de propranolol em amostras de comprimidos de diferentes marcas comerciais. Os
resultados obtidos pelo método proposto mostraram boa concordância quando comparados
com àqueles obtidos pelo método da Farmacopéia Britânica, indicando que não há diferença
significativa entre ambos métodos com relação à precisão e exatidão. O método proposto
oferece vantagens relacionadas à simplicidade, rapidez e baixo consumo de reagentes sobre os
outros métodos descritos para determinação de propranolol em comprimidos, podendo ser
recomendado para análises de rotina no controle de qualidade de medicamentos.

119
V. 3. 4. Determinação de atenolol em formulações farmacêuticas
V. 3.4.1. Spot Test
O p-cloranil (2,3,5,6-tetracloro-p-benzoquinona) tem sido usado como reagente
cromogênico para a determinação de diversos fármacos, tais como: sulfato de isoprenalina e
metildopa (KORANY; WAHBI, 1979), fluoxetina e sertralina (BEBAWY et al., 1999),
salbutamol (BAKRY et al., 1996), nortriptilina (ATTIA, 2000), antidepressivos (IBRAHIM et
al., 1983) e beta-bloqueadores, entre eles, o atenolol (SALEM, 2002). Estes métodos são
baseados na interação entre doadores de elétrons (fármacos) e p-cloranil, o qual age como
aceptor de elétrons formando complexos de transferência de carga intensamente coloridos
(FOSTER, 1968; SALEM, 2002).
Baseando-se na reação entre atenolol e p-cloranil em solução, previamente descrita na
literatura (SALEM, 2002), realizou-se esta reação em papel de filtro, a qual resultou no
aparecimento imediato de cor, a qual foi utilizada no desenvolvimento de spot tests
quantitativos. A Figura 24 apresenta o espectro de reflectância com valor máximo de AR em
550 nm.

120
Figura 24. Espectro de reflectância do produto formado na reação entre atenolol e p-cloranil. O
comprimento de onda máximo foi em 550 nm. Solução padrão de atenolol: 7,88 × 10–2
mol L-1.
Sabendo que reações envolvendo p-cloranil podem ser aceleradas com o uso de H2O2
(WEINERT et al., 2004) e que o dioxano é um solvente peroxidável (JACKSON et al., 1970),
a concentração de peróxido presente no dioxano utilizado neste estudo foi determinada. A
quantificação foi realizada com base no método espectrofotométrico descrito por Benatsky e
Tomik (1988) para a determinação de peróxido em solventes orgânicos, o qual envolve reação
com metavanadato de amônio e ácido nítrico concentrado, resultando em um composto
colorido (450 nm). Os resultados obtidos revelaram que o dioxano analisado continha 0,0016
g% (m/v) de H2O2, que foi uma quantidade menor que aquela descrita nas especificações no
rótulo que é de 0,005 g% (m/v) (dioxano p.a - Tedia, Fairfield, EUA).
400 500 600 700 8000,0
0,1
0,2
0,3
0,4
0,5
0,6
AR
Comprimento de onda (nm)

121
V. 3.4.2. Efeito da concentração de p-cloranil no spot test
O efeito da concentração de p-cloranil na resposta (AR) foi estudado utilizando
planejamento univariado, tendo sido fixada a concentração de atenolol em 7,88 × 10-2 mol L-1.
Os resultados obtidos no planejamento univariado estão apresentados na Figura 25.
Figura 25. Efeito da concentração de p-cloranil na resposta (AR). As barras representam os desvios
padrão para 3 repetições.
Observando o gráfico podemos notar que a adição da solução padrão de atenolol sem
p-cloranil (primeiro ponto do gráfico) não apresentou resposta de reflectância, indicando que
não houve reação entre o analito e componentes do papel. Também podemos observar que os
valores de AR obtidos aumentaram continuamente conforme o aumento da concentração de p-
cloranil até 8,00 × 10-2 mol L-1. Com a adição de soluções de p-cloranil em concentrações
maiores do que 8,00 × 10-2 mol L-1 a resposta de reflectância (AR) estabilizou, não tendo
0,00 0,02 0,04 0,06 0,08 0,10 0,12 0,14 0,16
0,0
0,1
0,2
0,3
0,4
0,5
AR
Concentração de p-cloranil (mol L-1)

122
ocorrido diferença significativa entre as concentrações de 8,00 × 10-2, 1,00 × 10-1 e 1,20 × 10-1
mol L-1. Porém, com a adição da solução de p-cloranil a 1,4 × 10-1 mol L-1 os valores de AR
foram menores e significativamente diferentes de todas as demais (p<0,05). Assim, os
resultados obtidos indicaram que a solução de p-cloranil na concentração de 8,00 × 10-2 mol
L-1 foi suficiente para que a reação de spot test se completasse, sendo, portanto, a
concentração escolhida.
V. 3.4.3. Estabilidade Óptica
Os resultados obtidos no estudo de estabilidade óptica do produto formado na reação
de spot test entre atenolol e p-cloranil demonstraram que não houve diferença significativa
entre os valores de AR obtidos nos diferentes períodos de tempo estudados. Portanto, a
estabilidade óptica do produto formado foi de pelo menos 40 minutos, nas condições
estudadas (Figura 26).
Figura 26. Estudo da estabilidade óptica do produto da reação entre atenolol e p-cloranil. As análises
dos spot tests foram realizadas no tempo zero, após 1 minuto e a cada 2 min. até 40 min.
Solução padrão de atenolol: 7,88 × 10-2 mol L-1.
0 5 10 15 20 25 30 35 40 450,15
0,20
0,25
0,30
0,35
0,40
0,45
AR
Tempo (minutos)

123
V. 3.4.4. Curva Analítica
A Figura 27 apresenta a curva analítica construída a partir das soluções padrão de
atenolol na faixa de concentração de 1,01 × 10-2 a 7,88 × 10-2 mol L-1. Uma relação linear
(r = 0,9992) foi obtida pela representação gráfica de AR versus log da concentração de
atenolol (mol L-1). O fator 102 foi usado para ajustar a curva analítica com valores de log
maiores do que zero. Valores de AR para esta faixa de concentração foram ajustados pela
equação: AR = 0,13469 + 0,32613 × C, onde C = log [atenolol] × 102 (mol L-1). O limite de
detecção foi 2,80 × 10–3 mol L-1 ⎟⎠⎞
⎜⎝⎛ ×
aDPB3 , sendo a = coeficiente angular da curva analítica e
BDP = desvio padrão do branco para 10 repetições (LONG; WINEFORDNER, 1983).
Figura 27. Curva analítica para a determinação de atenolol. Coeficiente de correlação linear (r) =
0,9992. Valores de AR foram obtidos em 550 nm. Concentrações das soluções padrão de
atenolol: 1,13 × 10-2 a 7,88 × 10-2 mol L-1. As barras representam os desvios padrão para 3
repetições.
0,0 0,2 0,4 0,6 0,8 1,0
0,15
0,20
0,25
0,30
0,35
0,40
0,45
AR
log [atenolol] x 102 (mol L-1)log ([atenolol]/mol L-1 × 102)

124
V. 3.4.5. Estudo de Interferências
O estudo de interferências foi realizado visando avaliar o efeito dos excipientes mais
comumente presentes na amostra. A porcentagem de atenolol encontrado nas soluções
contendo o fármaco padrão e cada um dos excipientes, separadamente, foi de 99 a 104%, com
coeficientes de variação menores que 5% para três repetições. Esses resultados foram
considerados aceitáveis e, portanto, nenhuma interferência foi constatada a partir destes
excipientes, nas condições estudadas.
V. 3.4.6. Estudo de Adição e Recuperação
Com o objetivo de avaliar efeitos de matriz (formulação farmacêutica) foi realizado
um estudo de adição e recuperação de fármaco padrão utilizando 3 amostras de diferentes
marcas comerciais de comprimidos. Os resultados obtidos estão apresentados na Tabela 20.
Para todas as amostras os valores médios de recuperação (n=3) variaram de 95,3 a 104,0%,
com desvios padrão relativos entre 0,6 a 4,0%. Essas variações foram consideradas aceitáveis,
indicando que os componentes das formulações farmacêuticas analisadas não influenciaram
nas análises.

125
Tabela 20. Resultados de recuperação de atenolol padrão adicionado em três formulações
farmacêuticas comerciais diferentes.
Amostras Valor Adicionadoa (mol L-1)
Valor Encontrado (mol L-1)a
Recuperação (%)a
A -
1,13 × 10-2
2,25 × 10-2
3,38 × 10-2
4,50 × 10-2
(2,23±0,06) × 10-2
(1,10±0,19) × 10-2
(2,17±0,08) × 10-2
(3,25±0,03) × 10-2
(4,58±0,13) × 10-2
-
97,0±0,2
96,7±3,8
96,1±0,9
101,9±2,9
B
-
1,13 × 10-2
2,25 × 10-2
3,38 × 10-2
4,50 × 10-2
(2,22±0,04) × 10-2
(1,08±0,01) × 10-2
(2,34±0,09) × 10-2
(3,27±0,03) × 10-2
(4,36±0,08) × 10-2
-
95,8±0,6
104,0±4,2
96,8±0,9
97,0±1,8
C
-
1,13 × 10-2
2,25 × 10-2
3,38 × 10-2
4,50 × 10-2
(2,16±0,08) × 10-2
(1,08±0,03) × 10-2
(2,22±0,04) × 10-2
(3,22±0,07) × 10-2
(4,41±0,12) × 10-2
-
95,3±3,0
98,7±1,8
95,4±2,0
98,1±2,7
a Média ± Desvio padrão (DP), n = 3.
V. 3.4.7. Aplicação do método proposto e comparação com o método de referência
O método proposto foi aplicado na determinação de atenolol em algumas formulações
farmacêuticas disponíveis comercialmente e comparado com o método para a determinação
de atenolol em comprimidos descrito na Farmacopéia Britânica (BRITISH…, 2001), o qual é
baseado na espectrofotometria no UV em 275 nm e envolve aquecimento a 60ºC por 15
minutos e diluições com metanol. As análises foram realizadas em três repetições e os
resultados estão apresentados na Tabela 21.

126
Tabela 21. Determinação de atenolol em formulações farmacêuticas comerciais.
Método Proposto Método de ReferênciaAmostrasa
Valor Encontradob
(mg/comprimido) Valor de t
(2,78)c Valor de F
(19,00)c Valor Encontradob
(mg/comprimido)
A 98,6±1,3 1,23 6,76 99,5± 0,5
B 98,4±1,5 0,21 9,00 98,6±0,5
C 47,8±1,3 0,25 10,56 48,0±0,4
D 49,1±1,6 0,01 3,16 49,1± 0,9
E 24,1±0,4 0,87 5,06 24,6±0,9 a Valor Declarado: Amostras A e B: 100 mg atenolol/comprimido; Amostras C e D: 50 mg de
atenolol/comprimido; Amostra E: 25 mg de atenolol/comprimido. b Média ± Desvio padrão (DP), n = 3. c Valores teóricos de t e F em nível de confiança de 95%.
Para todas as formulações analisadas, os resultados obtidos pelo método de referência
e pelo proposto foram comparados aplicando-se testes t e F, com nível de confiança de 95%.
Em todos os casos, os valores de F e de t calculados não excederam os valores teóricos,
indicando que não houve diferença significativa entre os métodos com relação à precisão e
exatidão (Tabela 21).
V. 3.4.8. Conclusões
Este estudo demonstrou o uso da espectroscopia de reflectância difusa utilizando spot
tests para o controle de qualidade de atenolol em comprimidos. O método envolveu um
procedimento de spot test muito simples, no qual 20 μL da solução contendo o fármaco e 20
μL da solução de reagente (p-cloranil) foram adicionados em papel de filtro. A reflectância do
produto colorido foi medida a 550 nm. Um planejamento univariado foi realizado, resultando
na escolha da melhor concentração de p-cloranil que foi de 8,0 × 10–2 mol L-1. O método foi
aplicado com sucesso na determinação de atenolol em amostras de comprimidos de diferentes

127
marcas comerciais, apresentando bons resultados de precisão e exatidão quando comparados
ao método da Farmacopéia Britânica. Desta forma, este trabalho apresenta uma proposta
viável de método para a análise de atenolol em comprimidos, oferecendo vantagens
relacionadas à simplicidade, rapidez e baixo consumo de reagentes e de solventes.

128
Capítulo VI. Desenvolvimento de método analítico por
espectrofotometria na região do visível para a
determinação de metildopa em formulações farmacêuticas

129
VI. 1. PARTE EXPERIMENTAL
VI. 1.1. Equipamentos
As medidas de absorbância, tanto para o método proposto como para o método da
Farmacopéia Brasileira, foram realizadas utilizando um espectrofotômetro UV-Visível Cary
1E, com cubetas de vidro de 1,0 cm de caminho óptico.
Agitador magnético Corning.
Micropipetas Eppendorf (10 a 100 μL) e Brand (100 a 1000 μL).
VI. 1.2. Reagentes e Soluções
Os excipientes usados no estudo de interferentes foram de grau farmacêutico. Os
solventes utilizados foram acetona (grau p.a.) e metanol (grau HPLC) (Mallinckrodt,
Xalostoc, México). p-Cloranil (p.a., Sigma, St. Louis, USA) foi usado para preparar
diariamente uma solução 4,07x10-2 mol L-1 (1,0%, m/v) em acetona. Solução de peróxido de
hidrogênio 4,55 mol L-1 foi preparada por diluição adequada de peridrol 30% (Merck,
Darmstadt, Alemanha) e padronizada conforme descrito na literatura (OHLWEILER, 1982).
Solução estoque de metildopa, grau de pureza de 99,50%, (Henrifarma, São Paulo, Brasil) foi
preparada na concentração de 1,68 × 10-2 mol L-1 (4000 ppm) em metanol. Soluções padrão
foram obtidas a partir de diluições da solução estoque de metildopa em metanol e usadas na
construção da curva analítica numa faixa de concentração de 4,20 × 10-4 a 2,48 × 10-3 mol L-1
(100 a 590 ppm).
VI. 1.4. Amostras
As amostras comerciais dos medicamentos contendo metildopa foram obtidas em
farmácias locais e foram analisadas dentro dos seus prazos de validade. Quatro formulações

130
farmacêuticas (comprimidos) contendo 250 mg de metildopa por comprimido, conforme
declarado no rótulo, foram analisadas.
VI. 2. PROCEDIMENTO
VI. 2. 1. Curva analítica
Alíquotas (125 a 740 μL) da solução estoque de metildopa (1,68 × 10-2 mol L-1) foram
transferidas para balões volumétricos de 5,00 mL visando a obtenção de uma curva analítica
na faixa de concentração de 4,20 × 10-4 a 2,48 × 10-3 mol L-1. Em seguida, 770 μL da solução
de p-cloranil (4,07 × 10-2 mol L-1) e 85 μL de H2O2 (4,55 mol L-1) foram adicionados. O
volume final (5,00 mL) foi completado com metanol. As medidas de absorbância foram
realizadas em 535 nm (b=1,0 cm). Branco de reagentes preparado de forma similar, porém
omitindo-se o fármaco foi utilizado como referência em todas as medidas. A curva analítica
foi obtida representando-se graficamente valores de absorbância versus concentração de
metildopa (mol L-1).
VI. 2. 2. Estudo de interferências
O efeito dos excipientes mais comumente empregados nas formulações farmacêuticas
de metildopa foi cuidadosamente estudado visando avaliar a presença de possíveis
interferentes. Os excipientes estudados foram: amido, talco, estearato de magnésio, ácido
tartárico, polietilenoglicol, polipropilenoglicol, celulose, hidroxipropilmetilcelulose, lactose,
etilcelulose, dióxido de silício e croscarmelose sódica. Para este estudo, soluções contendo
metildopa (4,0 mg/mL) e cada um dos excipientes, separadamente, em concentração igual e
dez vezes maior à concentração de metildopa foram submetidas à agitação com metanol em
um agitador magnético por 10 minutos. O volume foi completado para 10,00 mL e, após

131
filtração em papel de filtro Whatman 41, as soluções foram analisadas sob as mesmas
condições descritas no item VI. 2.1.
VI. 2.3. Preparação da amostra
Vinte comprimidos (do mesmo lote) de cada amostra das diferentes marcas comerciais
estudadas foram pesados em balança analítica e pulverizados em gral de ágata. O valor da
massa de um comprimido foi expresso como a média de 20 determinações, com variação
menor que 2% (FARMACOPÉIA..., 1988). Uma porção de amostra pulverizada equivalente a
100 mg de metildopa foi pesada com precisão de 0,0001 g e submetida à agitação com
metanol em agitador magnético por 10 minutos. Em seguida, foi realizada uma diluição em
balão volumétrico de 25,00 mL, completando-se o volume com metanol. Esta solução foi
filtrada em papel de filtro Whatman 41 e uma alíquota do filtrado foi submetida ao
procedimento descrito no item VI. 2.1.
V. 2.4. Estudo de Adição e Recuperação
Estudo de adição e recuperação de fármaco padrão foi realizado com o objetivo de
avaliar efeitos de matriz (formulação farmacêutica) nas análises. Para tanto, quantidades
conhecidas (2,52 × 10-4; 5,04 × 10-4; 7,55 × 10-4 e 1,00 × 10-3 mol L-1) de metildopa foram
adicionadas nas amostras previamente analisadas contendo o equivalente a 1,00 × 10-3 mol L-1
de metildopa. As quantidades de metildopa adicionadas corresponderam a 25, 50, 75 e 100%
da quantidade presente na amostra. O procedimento foi, então, seguido de acordo com o item
VI. 2.3.

132
VI. 3. RESULTADOS E DISCUSSÃO
Conforme discutido no item V. 3.4.1., o p-cloranil tem sido usado como reagente
cromogênico para a determinação de diversos fármacos, tais como: sulfato de isoprenalina e
metildopa (KORANY; WAHBI, 1979), fluoxetina e sertralina (BEBAWY et al., 1999),
salbutamol (BAKRY et al., 1996), nortriptilina (ATTIA, 2000), antidepressivos (IBRAHIM et
al., 1983), beta-bloqueadores (SALEM, 2002), sulfato de isoprenalina e metildopa
(KORANY; WAHBI, 1979). De acordo com a literatura (FOSTER, 1968; SALEM, 2002) o
p-cloranil interage com alguns fármacos por dois possíveis mecanismos de reação, um em que
ocorre uma reação de substituição e outro em que o p-cloranil (π-aceptor) interage com
doadores de elétrons formando complexos de transferência de carga como mostra o Esquema
4. No entanto, esses mecanismos não são bem elucidados existindo muita discussão na
literatura a respeito dos produtos formados, considerando-se inclusive que estão envolvidos
equilíbrios químicos complexos e com diversos produtos existindo concomitantemente.

133
Esquema 4
O uso de p-cloranil também foi descrito na literatura na detecção de metildopa em
formulações farmacêuticas por cromatografia em camada delgada (ADIKWU et al., 2002).
Como mencionado anteriormente, um método espectrofotométrico para a determinação de
sulfato de isoprenalina e metildopa utilizando p-cloranil como reagente cromogênico foi
publicado por Korany e Wahbi (1979) (KORANY; WAHBI, 1979). Entretanto, este método
apresenta alguns problemas, por exemplo, aquecimento por tempo prolongado (30 minutos) e
a medida da absorbância para metildopa é realizada na região do UV (358 nm). Além disso, a
p-cloranil Complexo de transferência de carga
em acetona
Par iônico
Amino-quinona (produto de substituição)
Proposta B
Proposta A

134
preparação da amostra requer o uso de HCl concentrado e solução tampão, exigindo várias
etapas de manipulação (KORANY; WAHBI, 1979).
Com o objetivo de desenvolver um procedimento mais simples, o método proposto
utilizou a reação entre metildopa e p-cloranil, a qual foi significativamente acelerada na
presença de H2O2. Esta reação produziu um composto colorido e a medida de absorbância foi
realizada em 535 nm, sem a necessidade de aquecimento. A implicação mais importante
destes resultados sobre àqueles já descritos para a análise de metildopa com p-cloranil
(KORANY; WAHBI, 1979) é que o aquecimento por 30 minutos pôde ser substituído pelo
uso de H2O2.
A Figura 28 apresenta o espectro do produto da reação entre metildopa e p-cloranil, na
presença de H2O2. Como pode ser observado, o espectro exibe bandas largas, as quais são
características de complexos de transferência de carga. Nota-se também na Figura 28 que o
espectro apresenta bandas de absorção máxima em 435 e em 535 nm. Assim, um estudo da
estabilidade óptica do complexo colorido foi realizado por acompanhamento cinético dos
valores de absorbância em 435 e em 535 nm, no tempo zero e a cada 2 minutos até 60
minutos (Figura 29).

135
Figura 28. Espectro de absorção do produto da reação entre metildopa e p-cloranil. O comprimento de
onda analítico foi 535 nm. Concentração final de metildopa = 1,07 × 10–3 mol L-1; caminho
óptico: 1,0 cm.
Figura 29. Estudo da estabilidade óptica do produto da reação entre metildopa e p-cloranil. As
medidas de absorbância foram realizadas a cada 2 minutos até 60 minutos. Concentração
final de metildopa: 7,14 × 10–4 mol L-1.
g g g 435 nm
g g g 535 nm
0 10 20 30 40 50 600,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0 10 20 30 40 50 600,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
Abs
orbâ
ncia
Tempo (min)
400 500 600 700 8000,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
Abs
orbâ
ncia
Comprimento de onda (nm)

136
Os resultados obtidos demonstraram que em 435 nm houve um aumento progressivo
da absorbância em função do tempo e que em 535 nm houve uma faixa de estabilidade óptica
de 15 a 35 minutos (Figura 29). Portanto, nas condições estudadas (535 nm), a estabilidade
óptica do complexo colorido foi de 20 minutos, o qual foi considerado tempo suficiente para a
execução das análises. Deve-se ressaltar que para a obtenção do complexo colorido (Figura
28), a presença de H2O2 foi essencial. Na ausência de H2O2, a reação foi extremamente lenta e
o produto não foi estável.
Em estudos anteriores realizados em nosso laboratório, foi desenvolvido um método
espectrofotométrico para a determinação de sacarina utilizando p-cloranil, o qual também
utilizou H2O2 como agente acelerador da reação (WEINERT et al., 2004). Nossos resultados
corroboram com aqueles já descritos para a análise de sacarina, sugerindo que reações
envolvendo p-cloranil e doadores de elétrons podem ser aceleradas com o uso de H2O2.
VI. 3.1. Planejamento Fatorial de Experimentos
Visando investigar as melhores condições para a reação entre p-cloranil e metildopa na
presença de H2O2 um planejamento fatorial utilizando planejamento composto central foi
empregado. Neste caso, as melhores condições para a reação significaram as concentrações
mínimas de reagentes que resultaram numa resposta máxima, isto é, a maior absorbância
possível.
Planejamento composto central permitiu estudar simultaneamente os dois fatores que
poderiam ter um efeito importante na reação. Os fatores de interesse foram concentração de p-
cloranil e concentração de H2O2. Estes fatores foram estudados na forma de volumes
adicionados das soluções de p-cloranil e de H2O2 em concentrações fixadas em 4,07 × 10-2
mol L-1 e 4,55 × 10-1 mol L-1, respectivamente. A Tabela 22 apresenta a matriz do
planejamento composto central e os resultados de absorbância. O experimento correspondente
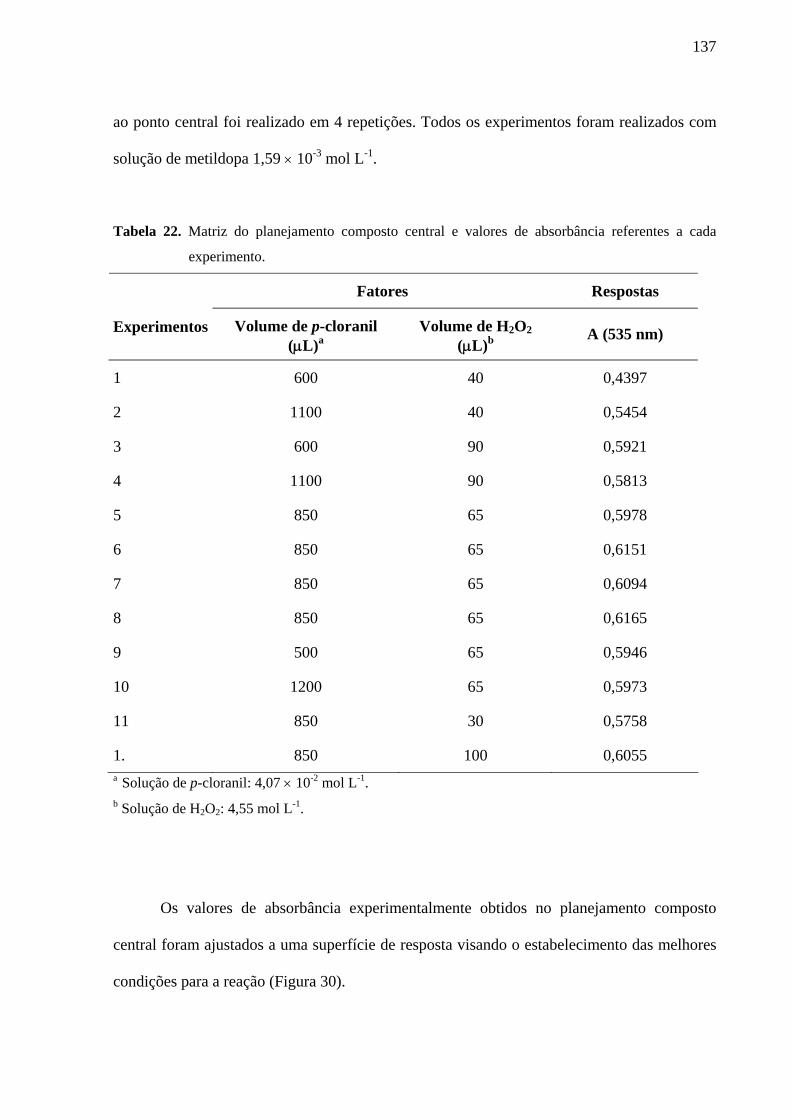
137
ao ponto central foi realizado em 4 repetições. Todos os experimentos foram realizados com
solução de metildopa 1,59 × 10-3 mol L-1.
Tabela 22. Matriz do planejamento composto central e valores de absorbância referentes a cada
experimento.
Fatores Respostas
Experimentos Volume de p-cloranil (μL)a
Volume de H2O2 (μL)b
A (535 nm)
1 600 40 0,4397
2 1100 40 0,5454
3 600 90 0,5921
4 1100 90 0,5813
5 850 65 0,5978
6 850 65 0,6151
7 850 65 0,6094
8 850 65 0,6165
9 500 65 0,5946
10 1200 65 0,5973
11 850 30 0,5758
1. 850 100 0,6055 a Solução de p-cloranil: 4,07 × 10-2 mol L-1. b Solução de H2O2: 4,55 mol L-1.
Os valores de absorbância experimentalmente obtidos no planejamento composto
central foram ajustados a uma superfície de resposta visando o estabelecimento das melhores
condições para a reação (Figura 30).

138
Figura 30. Superfície de resposta obtida a partir dos valores de absorbância em função do volume
adicionado de p-cloranil e volume de H2O2.
O modelo de regressão quadrática (valor de p< 0,0005), em termos de fatores
escalonados é dado por:
2000037,0009891,0000965,0177376,0 yyxZ −++= (Equação 7)
onde, Z é o fator resposta correspondente ao valor de absorbância. Os fatores x e y são o
volume de p-cloranil e volume de H2O2, respectivamente. O valor do coeficiente do fator 2x ,
bem como dos coeficientes das interações dos fatores foram muito pequenos e por esta razão
não foram mostrados.
Pode-se observar pela forma da superfície (Figura 30) que a região ótima foi
encontrada e que as respostas máximas foram alcançadas com cerca de 770 μL da solução de
p-cloranil e 85 μL da solução de H2O2.
0,61 0,56 0,51 0,46 0,41 0,36 0,31

139
VI. 3.3. Curva Analítica
A Figura 31 apresenta a curva analítica obtida a partir dos valores de absorbância
versus concentração de metildopa (mol L-1). A lei de Beer é obedecida na faixa de
concentração de 4,20 × 10-4 a 2,48 × 10-3 mol L-1, com um coeficiente de correlação linear de
0,9993. Os valores de absorbância para esta faixa de concentração foram ajustados pela
equação: A = - 0,04006 + 364,315 × C, onde C = concentração de metildopa (mol L-1) e o
valor do coeficiente angular corresponde numericamente a absortividade molar (ε), que é 3,64
× 102 L. mol-1. cm-1. O limite de detecção foi 1,51 × 10-5 mol L-1 ⎟⎠⎞
⎜⎝⎛ ×
aDPB3 , sendo a =
coeficiente angular da curva analítica e BDP = desvio padrão do branco para 10 repetições e o
limite de quantificação 5,03 × 10-5 mol L-1 ⎟⎠⎞
⎜⎝⎛ ×
aDPB10 (LONG; WINEFORDNER, 1983).
Figura 31. Curva analítica para a determinação espectrofotométrica de metildopa. Valores de
absorbância foram obtidos em 535 nm. Coeficiente de correlação linear (r) =
0,9993. As barras representam o desvio padrão para 3 repetições.
0,0005 0,0010 0,0015 0,0020 0,0025
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
Abs
orbâ
ncia
(535
nm
)
Concentração de metildopa (mol L-1)

140
VI. 3.4. Estudo de interferências
O estudo de interferências foi realizado visando avaliar o efeito dos excipientes mais
comumente presentes na amostra (THOMPSON; ELLISON; WOOD, 2002). A porcentagem
de metildopa encontrada nas soluções contendo o fármaco padrão e cada um dos excipientes,
separadamente, foi de 94,0 a 103,0% com coeficientes de variação menores que 5% para três
repetições. Esses resultados foram considerados aceitáveis e, portanto, nenhuma interferência
foi constatada a partir destes excipientes, nas condições estudadas.
VI. 3.5. Estudo de Adição e Recuperação
Com o objetivo de avaliar efeitos de matriz (formulação farmacêutica, a qual contém
todos os excipientes juntos) foi realizado um estudo de adição e recuperação de fármaco
padrão utilizando 3 amostras de diferentes marcas comerciais de comprimidos. Os resultados
obtidos estão apresentados na Tabela 23. Para todas as amostras, os valores médios de
recuperação (n=3) variaram de 96,3 a 105,9%, com desvios padrão relativos entre 1,9 e 6,1%.
Essas variações foram consideradas aceitáveis, indicando que os componentes das
formulações farmacêuticas analisadas não influenciaram nas análises.

141
Tabela 23. Resultados de recuperação de metildopa adicionada em formulações farmacêuticas.
Amostras Valor Adicionado
(mol L-1)a
Valor Encontrado
(mol L-1)a
Recuperaçãoa
(%)
A
-
2,52 × 10-4
5,04 × 10-4
7,56 × 10-4
1,01 × 10-3
(9,83±0,45) × 10-4
(2,54±0,14) × 10-4
(5,26±0,10) × 10-4
(7,42±0,29) × 10-4
(1,05±0,05) × 10-3
-
100,8±5,6
104,2±0,2
98,1±3,8
104,0±5,0
B
-
2,52 × 10-4
5,04 × 10-4
7,56 × 10-4
1,01 × 10-3
(1,02±0,04) × 10-3
(2,48±0,10) × 10-4
(5,34±0,30) × 10-4
(7,46±0,30) × 10-4
(1,01±0,06) × 10-3
-
98,4±3,9
105,9±5,9
98,7±4,0
100,8±6,0
C
-
2,52 × 10-4
5,04 × 10-4
7,56 × 10-4
1,01 × 10-3
(1,04±0,02) × 10-3
(2,56±0,13) × 10-4
(5,30±0,17) × 10-4
(7,67±0,47) × 10-4
(9,73±0,30) × 10-4
-
101,6±5,2
105,2±3,3
101,5±6,2
96,3±3,0 a Média ± Desvio padrão (DP), n = 3.
VI. 3.6. Aplicação do método proposto e comparação com o método de referência.
O método proposto foi aplicado à determinação de metildopa em algumas formulações
farmacêuticas disponíveis comercialmente e comparado com o método para a determinação
de metildopa em comprimidos (FARMACOPÉIA..., 1988). Tal método é baseado em
medidas por espectrofotometria na região do visível (520 nm) e envolve a reação entre
metildopa e solução de tartarato ferroso. As análises foram realizadas em três repetições e os
resultados estão apresentados na Tabela 24.

142
Tabela 24. Determinação de metildopa em formulações farmacêuticas comerciais.
Método Proposto Método de Referência
Amostrasa Valor Encontradob
(mg/comprimido)
Valor de t
(2,78)c
Valor de F
(19,00)c
Valor Encontradob
(mg/comprimido)
A 250,3±3,1 0,67 2,97 249,0±1,8
B 248,1±0,6 2,32 4,00 249,9±1,2
C 248,6±2,3 0,26 1,83 248,9±1,7
D 258,5±3,1 0,02 4,90 258,5±1,4 a Valor Nominal: 250 mg metildopa/comprimido b Média ± Desvio padrão (DP), n = 3. c Valores teóricos de t e F em nível de confiança de 95%.
Os valores médios encontrados variaram de 248,1 a 258,5 mg/comprimido e os
desvios padrão de 0,6 a 3,1 mg/comprimido (Tabela 24). Para todas as formulações
analisadas, os resultados obtidos pelo método da Farmacopéia Brasileira (FARMACOPÉIA...,
1988) e pelo proposto foram comparados aplicando-se testes t e F, com nível de confiança de
95%. Em todos os casos, os valores de F e de t calculados não excederam os valores teóricos,
indicando que não houve diferença significativa entre os métodos com relação à precisão e
exatidão.
VI. 3.7. Medidas em dispositivo portátil
O método desenvolvido para a determinação de metildopa foi adaptado para o uso em
um sistema de medidas portátil descrito por Rossi, He e Tubino (2000). Tal sistema consiste
em um dispositivo para medidas fotométricas, o qual é baseado em uma relação entre a
resistência de um sensor (LDR) e a intensidade de luz que incide sobre sua superfície. Assim,
a resistência varia em função da intensidade de cor de uma solução. Este sistema é constituído
por um multímetro digital (ET-1502 – Minipa®), ao qual é acoplada uma cela de PTFE

143
(politetrafluoretileno) (Teflon®) preto contendo um compartimento para a inserção de um
tubo de vidro de fundo chato (5,0 cm de altura e 5,0 mm de diâmetro interno), o qual é
preenchido com a solução a ser analisada. O volume desta solução deve ser padronizado,
neste trabalho, o volume da solução a ser analisada foi de 1,2 mL. Utilizou-se uma luminária
(lâmpada compacta fluorescente 9 W – Startec) como fonte de luz incidindo continuadamente
sobre o sistema, a fim de se evitar variações da luz ambiente sobre o LDR. A cela de teflon foi
cedida pelos autores do dispositivo para a realização desta etapa do trabalho.
A Figura 32a apresenta uma foto do sistema montado para realizar as medidas
fotométricas no dispositivo portátil e a Figura 32b um esquema da cela de Teflon® para
medidas fotométricas, incluindo LDR, tubo de vidro, janela de vidro e contato elétrico.
Figura 32. Dispositivo portátil de medidas fotométricas: (a) Montagem do sistema para medidas
fotométricas. (b) Esquema da cela de Teflon® (Fonte: CRIVELENTE, 2003).
(a)
(b)

144
As condições experimentais adotadas para as análises de metildopa no dispositivo
portátil foram àquelas estabelecidas no desenvolvimento do método espectrofotométrico
descrito anteriormente.
VI. 3.7.1. Curva Analítica
A curva analítica foi construída na faixa de concentração de 7,14 × 10–4 a 2,48 × 10–3
mol L-1, utilizando o mesmo procedimento descrito no item VI. 2.1. A solução contendo o
produto colorido foi, então, transferida para o tubo de vidro (1,2 mL), o qual foi introduzido
na cela de PTFE para a medida da resistência. As medidas foram realizadas em triplicata. A
Figura 33 apresenta a curva analítica obtida a partir dos valores de resistência (ohm) versus
concentração de metildopa (mol L-1). Uma relação linear (r=0,9986) foi obtida a partir da
equação: y = 2,35 × 104 × C + 9,33543.
Figura 33. Curva analítica para a determinação de metildopa por medidas fotométricas utilizando
dispositivo portátil. Coeficiente de correlação linear (r) = 0,9986. As barras representam o
desvio padrão para 3 repetições.
0,0008 0,0012 0,0016 0,0020 0,00240
10
20
30
40
50
Res
istê
ncia
(103 oh
ms)
Concentração metildopa (mol L-1)

145
Comparando-se a curva analítica obtida por espectrofotometria (Figura 31) e a curva
obtida a partir das medidas de resistência no dispositivo portátil (Figura 33) nota-se uma
diferença na dispersão das medidas (barras verticais nos gráficos). Embora a mesma reação
tenha sido utilizada no método espectrofotométrico e no método adaptado ao dispositivo
portátil, as medidas espectrofotométricas apresentaram valores de desvio padrão relativos
(dispersão) menores (variando de 1,4 a 3,8%) do que as medidas de resistência, as quais
apresentaram valores de desvio padrão relativo de 2,5 a 7,9%. Entretanto, mesmo com menor
precisão de medidas, o coeficiente de correlação linear obtido (0,9986) foi considerado
adequado, indicando uma boa correlação entre concentração de metildopa e medidas de
resistência.
VI. 3.7.2. Análise das amostras de comprimidos
As amostras analisadas foram preparadas conforme o procedimento descrito no item
VI. 2.3. Os resultados obtidos a partir das medidas de resistência das soluções contendo o
produto colorido da reação entre metildopa e p-cloranil foram estatisticamente comparados
com aqueles obtidos pelo método da Farmacopéia Brasileira (FARMACOPÉIA..., 1988). Para
todas as formulações analisadas, os resultados obtidos pelo método da Farmacopéia e pelo
método adaptado para o dispositivo portátil foram comparados aplicando-se os testes t e F,
com nível de confiança de 95%. Em todos os casos, o valor de F e de t calculados não
excederam os valores teóricos, indicando que não há diferença significativa entre ambos
métodos com relação à precisão e exatidão (Tabela 25). Estes resultados evidenciaram a
aplicabilidade do sistema de medidas portátil para a determinação de metildopa em
formulações farmacêuticas.
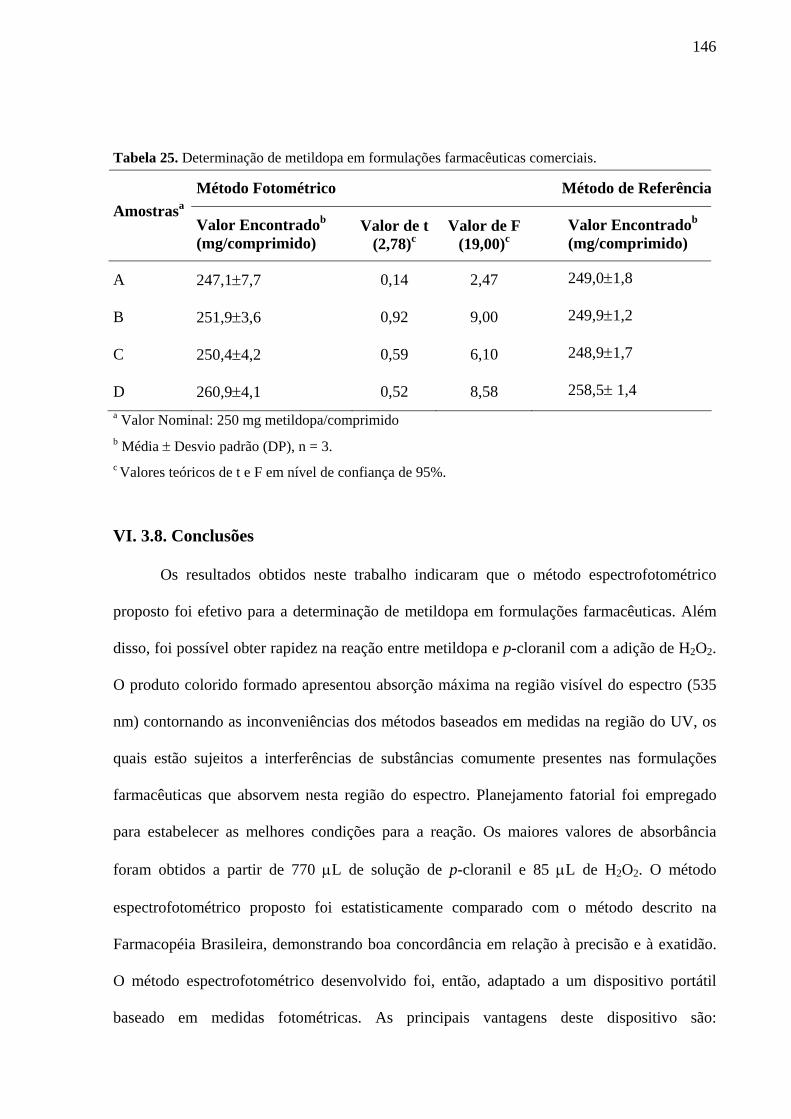
146
Tabela 25. Determinação de metildopa em formulações farmacêuticas comerciais.
Método Fotométrico Método de ReferênciaAmostrasa
Valor Encontradob (mg/comprimido)
Valor de t (2,78)c
Valor de F (19,00)c
Valor Encontradob (mg/comprimido)
A 247,1±7,7 0,14 2,47 249,0±1,8
B 251,9±3,6 0,92 9,00 249,9±1,2
C 250,4±4,2 0,59 6,10 248,9±1,7
D 260,9±4,1 0,52 8,58 258,5± 1,4 a Valor Nominal: 250 mg metildopa/comprimido b Média ± Desvio padrão (DP), n = 3. c Valores teóricos de t e F em nível de confiança de 95%.
VI. 3.8. Conclusões
Os resultados obtidos neste trabalho indicaram que o método espectrofotométrico
proposto foi efetivo para a determinação de metildopa em formulações farmacêuticas. Além
disso, foi possível obter rapidez na reação entre metildopa e p-cloranil com a adição de H2O2.
O produto colorido formado apresentou absorção máxima na região visível do espectro (535
nm) contornando as inconveniências dos métodos baseados em medidas na região do UV, os
quais estão sujeitos a interferências de substâncias comumente presentes nas formulações
farmacêuticas que absorvem nesta região do espectro. Planejamento fatorial foi empregado
para estabelecer as melhores condições para a reação. Os maiores valores de absorbância
foram obtidos a partir de 770 μL de solução de p-cloranil e 85 μL de H2O2. O método
espectrofotométrico proposto foi estatisticamente comparado com o método descrito na
Farmacopéia Brasileira, demonstrando boa concordância em relação à precisão e à exatidão.
O método espectrofotométrico desenvolvido foi, então, adaptado a um dispositivo portátil
baseado em medidas fotométricas. As principais vantagens deste dispositivo são:

147
simplicidade, facilidade de se obter e operar o equipamento e baixo custo. O método adaptado
ao dispositivo portátil foi estatisticamente comparado com o método da Farmacopéia
Brasileira apresentando bons resultados de precisão e exatidão. Portanto, este sistema
representa uma opção para que farmácias de manipulação possam realizar o controle de
qualidade de medicamentos com confiabilidade e baixo custo.

148
Capítulo VII. Conclusões Gerais

149
Os métodos analíticos desenvolvidos neste trabalho para a determinação de
furosemida, hidroclorotiazida, propranolol e atenolol utilizando a combinação espectroscopia
de reflectância difusa - spot test foram aplicados com sucesso na análise de formulações
farmacêuticas comerciais e foram considerados confiáveis, podendo ser úteis no controle de
qualidade de medicamentos. Além disso, os métodos por espectroscopia de reflectância difusa
demonstraram, de maneira inédita, o uso desta técnica combinada com spot test para a
quantificação de fármacos em formulações farmacêuticas. A combinação espectroscopia de
reflectância difusa - spot test oferece vantagens relacionadas principalmente à simplicidade,
rapidez e baixo consumo de reagentes e solventes, sendo muito interessante do ponto de vista
da Química Verde, a qual tem interesse por métodos e técnicas que reduzem e eliminam o uso
e a geração de substâncias prejudiciais à saúde humana e ao meio ambiente.
A falta de interferências dos excipientes avaliados demonstrou uma boa seletividade
dos métodos desenvolvidos no presente trabalho. Os métodos descritos na Farmacopéia
Britânica (2001) utilizados para comparação com os métodos reflectométricos propostos são
baseados em medidas diretas na região do UV e estão sujeitos a interferências provenientes de
excipientes presentes nas formulações ou possíveis falsificações e/ou adulterações, resultando
em baixa seletividade, uma vez que todos os compostos insaturados apresentam uma ou mais
bandas na região UV do espectro eletromagnético.
O método espectrofotométrico para a determinação de metildopa em formulações
farmacêuticas foi aplicado com sucesso na análise de formulações farmacêuticas comerciais.
O produto da reação entre metildopa e p-cloranil, na presença de peróxido de hidrogênio
apresentou absorção máxima na região do visível (535 nm), evitando possíveis interferências
de excipientes que absorvem na região do UV. Os resultados demonstraram que o uso de
peróxido de hidrogênio foi extremamente útil, uma vez que agiu como um acelerador da
reação, evitando a necessidade de aquecimento. Considerando estes resultados, sugere-se que

150
outros métodos envolvendo reações entre doadores de elétrons e p-cloranil podem ser
melhorados com o uso de peróxido de hidrogênio.
O método espectrofotométrico foi adaptado ao dispositivo portátil de medidas
fotométricas e os resultados da comparação com o método oficial indicaram que este sistema
representa uma boa opção para que farmácias de manipulação possam realizar controle de
qualidade destes medicamentos com confiabilidade e baixo custo.

151
Capítulo VIII. Perspectivas Futuras

152
Diante dos resultados obtidos no presente trabalho, surgem algumas perspectivas de
trabalhos futuros que dão continuidade à linha de pesquisa envolvendo o desenvolvimento de
métodos analíticos para a determinação de medicamentos. Entre elas, destacam-se:
a) Aplicação dos métodos analíticos desenvolvidos para a quantificação de
furosemida, hidroclorotiazida, propranolol e atenolol em fluidos biológicos, visando a
monitorização dos níveis terapêuticos destes fármacos e a análise de doping, uma vez que tais
fármacos estão incluídos na lista de substâncias proibidas pelo Comitê Olímpico
Internacional.
b) Aplicação do método espectrofotométrico para a determinação de metildopa em
amostras de fluidos biológicos visando a monitorização terapêutica.
c) Automação dos métodos propostos, tanto por espectroscopia de reflectância difusa
como por espectrofotometria, utilizando análise por injeção em fluxo.
d) Estudar mais detalhadamente a reação entre metildopa e p-cloranil, na presença de
peróxido de hidrogênio, com intuito de elucidar o mecanismo da reação e compreender a
formação do produto colorido.

153
Capítulo IX. Referências

154
ABDULRAHMAN, L. K.; AL-ABACHI, A. M.; AL-QAISSY, M. H. Flow injection-spectrophotometric determination of some catecholamine drugs in pharmaceutical preparations via oxidative coupling reaction with p-toluidine and sodium periodate. Anal. Chim. Acta, v. 538, n. 1/2, p. 331-335, May 2005.
ABOUL-ENEIN, H. Y.; AL-BADR, A. A.; RASHED, M. S. E. Application of NMR spectroscopy in pharmaceutical analysis. Assay of furosemide. Spectrosc. Lett., v. 12, n. 4, p. 323-331, 1979.
ABOUL-ENEIN, H. Y.; SUN, X. X. A novel ion selective PVC membrane electrode for determination of propranolol in pharmaceutical formulations. Analusis, v. 28, n. 9, p. 855-858, Nov. 2000.
ADIKWU, M. U.; AJALI, U.; OKIDE, G. B.; ODOH, U. E. Detection of α-methyldopa on thin-layer plates using ϖ- acceptors in 1,4-dioxane. Boll. Chim. Farm., v. 141, n. 4, p. 315-316, 2002.
AFKHAMI, A.; KHATAMI, H. A. Determination of some catecholamines based on their reaction with periodate. J. Anal. Chem., v. 58, n. 2, p. 135-138, Feb. 2003.
AGARWAL, R. Rapid microplate method for PAH estimation. Am. J. Physiol. Renal Physiol., v. 283, n. 2, p. F236-F241, Aug. 2002.
AGATONOVIC, S. K.; ZIVANOVIC, L.; RADULOVIC, D.; PECANAC, D. Spectrophotomeric determination of furosemide and its palladium (II) complex. J. Pharm. Biomed. Anal., v. 8, n. 8/12, p. 983-986, 1990.
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Aprova o Regulamento Técnico sobre Boas Práticas de Manipulação de Medicamenos em Farmácias. Resolução RDC n. 33 de 19 de abril de 2000. Diário Oficial da União, Brasília, DF, 24 abril 2000.
AGRAWAL, Y. K.; RAMAN, K.; RAJPUT, S.; MENON, S. K. Spectrophotometric determination of atenolol via hydroxamic acid formation. Anal. Lett., v. 25, n. 8, p. 1503-1510, 1992.
ALBERO, I.; RODENAS, V.; GARCIA, S.; SANCHEZ-PEDRENO, C. Determination of irbesartan in the presence of hydrochlorothiazide by derivative spectrophotometry. J. Pharm. Biom. Anal., v. 29, n. 1/2, p. 299-305, June 2002.

155
AL-GHANNAM, S.M.; BELAL, F. Kinetic spectrophotometric determination of atenolol in dosage forms. J. AOAC Int., v. 85, n. 4, p. 817-823, Jul./Aug. 2002.
AL-GHANNAM, S. M. A simple spectrophotometric method for the determination of β-blockers in dosage- forms. J. Pharm. Biom. Anal., v. 40, p. 151-156, n. 1, Jan. 2006.
AL-MAJED, A. A. Spectrophotometric estimation of D-penicillamine in bula and dosage forms using 2,6-dichloroquinone-4-chlorimide (DCQ). J. Pharm. Biom. Anal., v. 21, n. 4, p. 827-833, Dec. 1999.
AMAN, T.; KHAN, I. U.; ASLAM, N.; AHMAD, I. Spectrophotometric determination of methyldopa in pure and pharmaceutical preparations. Anal. Lett., v. 31, n. 6, p. 1007-1020, 1998.
AMIN, A. S.; RAGAB, G. H.; SALEH, H. Colorimetric determination of β-blockers in pharmaceutical formulations. J. Pharm. Biomed. Anal., v. 30, n. 4, p. 1347-1353, Nov. 2002.
ANAPURE, S. A.; KHANNA, S.; DIGHE, V. S. High performance liquid chromatographic method for frusemide and spirolactone combination. East. Pharm., p. 193-194, Aug. 1989.
ANASTAS, P. T. Green chemistry and the role of analytical methodology development. Crit. Rev. Anal. Chem., v. 29, p. 167-175, 1999.
ANFT, B. Friedlieb Ferdinand Runge: a forgotten chemist of the nineteenth century. J. Chem. Educ., v. 32, n. 11, p. 566-574, Nov. 1955.
ARGEKAR, A. P.; SAWANT, J. G. Simultaneous determination of atenolol and nitrendipine in pharmaceutical dosage forms by HPTLC. J. Liq. Chromatogr. Rel. Technol., v. 22, n. 10, p. 1571-1578, 1999.
ARGEKAR, A. P.; POWAR, S. G. Simultaneous determination of atenolol and amlodipine in tablets by high-performance thin-layer chromatography. J. Pharm. Biomed. Anal., v. 21, n. 6, p. 1137-1142, Jan. 2000.
ARENA, M. P.; PORTER, M. D.; FRITZ, J. S. Rapid, low level determination of silver (I) in drinking water by colorimetric-solid-phase extraction. Anal. Chim. Acta, v. 482, n. 2, p. 197-207, 2003.

156
ATAY, O.; TAMER, U.; ARIKAN, D. Determination of cilazapril and hydrochlorothiazide in pharmaceuticals by high performance liquid chromatography. Anal. Lett., v. 34, n. 7, p. 1153-1161, Apr. 2001.
ATTIA, F. M. A. Use of charge-transfer complex formation for the spectrophotometric determination of nortriptyline. Farmaco, v. 55. n. 3, p. 659-664, Nov./Dec. 2000.
BADAWY, S. S.; ISSA, Y. M.; TAG-ELDIN, A. S. Potentiometric determination of L-dopa, carbidopa, methyldopa and aspartame using a new trinitrobenzenesulfonate selective electrode. Electroanalysis, v. 8, n. 11, p. 1060-1064, Nov. 1996.
BAKRY, R. S.; RAZAK, O. A.; EL-WALILY, A. F. M.; BELAL, S. F. Spectrophotometric determination of salbutamol sulphate using chlorinate quinines in the presence or absence of acetaldehyde. J. Pharm. Biomed. Anal., v. 14, n. 3, p. 357-362, Jan. 1996.
BALLANTINE, J.; WOOLFSON, A. D. Determination of catecholamines in pharmaceutical preparations by differential pulse polarography at the glassy carbon electrode. Int. J. Pharm., v. 3, n. 4/5, p. 239-246, 1979.
BARROSO, B. M.; ALONSO, R. M.; JIMENEZ, R. M. Electrochemical determination of the loop diuretics piretanide and furosemide in pharmaceutical formulations and urine. Anal. Chim. Acta, v. 305, n. 1/3, p. 332-339, Apr. 1995.
BARROSO, M. B.; JIMENEZ, R. M.; ALONSO, R. M.; ORTIZ, E. Determination of piretanide and furosemide in pharmaceuticals and human urine by high-performance liquid chromatography with amperometric detection. J. Chromatogr., B: Biomed. Appl., v. 675, n. 2, p. 303-312, Jan. 1996.
BARROSO, M. B.; ALONSO, R. M.; JIMENEZ, R. M. Simultaneous determination of the diuretics triamterene and furosemide in pharmaceutical formulations and urine by HPLC-EC. J. Liq. Chromatogr. Relat. Technol., v. 19, n. 2, p. 231-246, 1996.
BEBAWY, L. I.; El-KOUSY, N.M.; SUDDIK, J. K.; SHOKRY, M. Spectrophotometric determination of fluoxetine and sertraline using chloranil, 2,3-dchloro-5,6-dicyano benzoquinone and iodine. J. Pharm. Biomed. Anal., v. 21, n. 1, p. 133-142, Oct. 1999.
BENATSKY, M.; TOMIK, B. Peroxides in organic solvents. Chem. Listy, v. 82, n. 7, p. 759-762, 1988.

157
BEROZA, M.; HILL, K. R.; NORRIS, K. H. Determination of reflectance of pesticide spots on thin-layer chromatograms using fiber optics. Anal. Chem., v. 40, n. 11, p. 1608-1613, Sept. 1968.
BONAZZI, D.; GOTTI, R.; ANDRISANO, V.; CAVRINI, V. Derivative UV spectrophotometric determination of atenolol and metroprolol in single-and multi-component pharmaceutical, dosage forms. Farmaco, v. 51, n. 11, p. 733-738, Nov. 1996.
BRITISH Pharmacopoeia. London: The Stationery Office, 2001. v. 2.
BRIGUENTI, A. C. C.; BONATO, P. S. Quantitative analysis of β-blockers in pharmaceutical preparations by capillary electrophoresis. Drug Dev. Ind. Pharm., v. 31, n. 2, p. 209-214, 2005.
BRADDOCK, L. I.; MAREC, N. Use of reflectance spectra for estimation of barbiturates by spot test techniques. Microchem. J., v. 11, n. 1, p. 121-138, Mar. 1966.
CAMANAS, R. M. V.; MALLOLS, J. M. S.; LAPASIO, J. R. T.; RAMIS-RAMOS, G. Analysis of pharmaceutical preparations containing catecholamines by micellar liquid chromatography with spectrophotometric detection. Analyst, v. 120, n. 6, p. 1767-1772, June 1995.
BROCH, S. C.; ROMERO, J. E.; COQUE, M. C. G. A. Furosemide assay in pharmaceuticals by micellar liquid chromatography: study of the stability of the drug. J. Pharm. Biom. Anal., v. 23, n. 5, p. 803-817, Oct. 2000.
CARLUCCI, G.; DI GIUSEPPE, E.; MAZZEO, P. Simultaneous determination of enalapril maleate and hydrochlorothiazide in tablets by derivative UV spectrophotometry and high-performance liquid chromatography. Int. J. Pharm., v. 93, n. 1/3, p. 245-248, May 1993.
CASASSAS, E.; FABREGAS, J. L. Spectrophotometric determination of furosemide with phloroglucinol. Anal. Chim. Acta, v. 106, n. 1, p. 151-154, 1979.
CETIN, G.; SUNGUR, S. The simultaneous determination of hydrochlorothiazide and cilazapril in tablet formulation by high performance liquid chromatography. Fresenius Environ. Bull., v. 14, n. 5, p. 353-356, 2005.
CRIVELENTE, W. C. T. Reações luminescentes e colorimétricas para determinação de hidrocortisona em fármacos. Dissertação (Mestrado em Química) - Instituto de Química, Universidade Estadual de Campinas, Campinas, 2003.

158
DASGUPTA, V. Quantitation of propranolol hydrochloride in pharmaceutical dosage forms by high-performance liquid chromatography. Drug Dev. Ind. Pharm., v. 11, n. 11, p. 1931-1937, 1985.
DIAS, I. L. T.; DE OLIVEIRA NETO, G.; VENDRAMINI, D. C.; SOMMER, C.; MARTINS, J. L. S.; KUBOTA, L. T. A poly(vinyl chloride) membrane electrode for the determination of the diuretic furosemide. Anal. Lett., v. 37, n. 1, p. 35-46, 2004.
DIAS, I. L. T.; MARTINS, J. L. S.; DE OLIVEIRA NETO, G. Furosemide determination by first-derivative spectrophotometric method. Anal. Lett., v. 38, n. 7, p. 1159-1166, 2005.
DIAZ, B. C.; BLANCO, C. C.; CARRETERO, A. S.; GUTIERREZ, A. F. Simple determination of propranolol in pharmaceutical preparations by heavy atom induced room temperature phosphorescence. J. Pharm. Biom. Anal., v. 30, n. 4, p. 987-992, Nov. 2002.
DMITRIENKO, S. G.; SVIRIDOVA, O. A.; PYATKOVA, L. N.; SENYAVIN, V. M. Polyurethane foams as solid chromogenic reagents for diffuse reflectance spectroscopy. Anal. Bioanal. Chem., v. 374, n. 3, p. 361-368, Oct. 2002.
DINC, E.; USTUNDAG, O. Spectrophotometric quantitative resolution of hydrochlorothiazide and spironolactone in tablets by chemometric analysis methods. Farmaco, v. 58, n. 11, p. 1151-1161, 2003.
DINC, E. Spectral analysis of benazepril hydrochloride and hydrochlorothiazide in pharmaceutical formulations by three chemometric techniques. Anal. Lett., v. 35, n. 6, p. 1021-1039, 2002.
EL-EMAM, A. A.; BELAL, F. F.; MOUSTAFA, M. A.; EL-ASHRY, S. M.; EL-SHERBINY, D. T.; HANSEN, S. H. Spectrophotometric determination of propranolol in formulations via oxidative coupling with 3-methylbenzothiazoline-2-one hydrazone. Farmaco, v. 58, n. 11, p. 1179-1186, 2003.
EL-RAGEHY, N. A.; BADAWEY, A. M.; EL-KHATEEB, S. Z. Stability indicating methods for assay of mequitazine in presence of its degraded. J. Pharm. Biomed. Anal., v. 29, n. 1/2, p. 121-137, June 2002.
EL-RIES, M. A.; ATTIA, F. M. A.; IBRAHIM, S. A. AAS and spectrophotometric determination of propranolol HCl and metoprolol tartrate. J. Pharm. Biomed. Anal., v. 24, n. 2, p. 179-187, Dec. 2000.

159
EL-RIES, M. A.; ABOU-SEKKINA, M. M.; WASSEL, A. A. Polarographic determination of propranolol in pharmaceutical formulation. J. Pharm. Biomed. Anal., v. 30, p. 837-842, Oct. 2002.
EL SHERIF, Z. A.; WALASH, M. I.; EL-TARRAS, M. F.; OSMAN, A. O. Colorimetric determination of two nonsteroidal anti-inflammatory drugs using p-dimethylaminocinnamaldehyde. Anal. Lett., v. 30, n. 10, p. 1881-1896, 1997.
EL WALILY, A. F. M.; BELAL, S. F.; HEABA, E. A.; EL KERSH, A. Simultaneous determination of enalapril maleate and hydrochlorothiazide by first-derivative ultraviolet spectrophotometry and high-performance liquid chromatography. J. Pharm. Biom. Anal., v. 13, n. 7, p. 851-856, June 1995.
EMMANUEL, J.; HALDANKAR, S. D. A simple and sensitive spectrocolorimetric method for the estimation of ranitidine hydrochloride and its formulations. Indian Drugs, v. 26, n. 5, p. 259-250, 1989.
ERK, N. Determination of active ingredients in the pharmaceutical formulations containing hydrochlorothiazide and its binary mixtures with benazepril hydrochloride, triamterene and cilazapril by ratio spectra derivative spectrophotometry and vierordt's method. J. Pharm. Biom. Anal., v. 20, n. 1/2, p. 155-167, June 1999.
ERK, N. Analysis of binary mixtures of losartan potassium and hydrochlorothiazide by using high performance liquid chromatography, ratio derivative spectrophotometric and compensation technique. J. Pharm. Biom. Analysis, v. 24, n. 4, p. 603-611, Feb. 2001.
ERK, N. Simultaneous determination of fosinopril and hydrochlorothiazide in pharmaceutical formulations by spectrophotometric methods. J. Pharm. Biom. Anal., v. 27, n. 6, p. 901-912, Mar. 2002.
ERK, N.; KARTAL, M. Comparison of high - performance liquid chromatography and absorbance ratio methods for the determination of hydrochlorothiazide and lisinopril in pharmaceutical formulations. Anal. Lett., v. 32, n. 6, p. 1131-1141, 1999.
ERK, N.; ONUR, F. Simultaneous determination of cilazapril and hydrochlorothiazide in tablets by spectrophotometric methods. Anal. Lett., v. 29, n. 11, p. 1963-1974, 1996.
FARMACOPÉIA Brasileira. 4. ed. São Paulo: Atheneu, 1988. Parte I.
FEIGL, F. Spot test in organic analysis. 7th ed. New York: Elsevier, 1966. 772 p.

160
FEIGL, F. Spot test in inorganic analysis. Amsterdam: Elsevier, 1958. 600 p.
FERNANDEZ-SANCHEZ, J. F.; CARRETERO, A. S.; CRUCES-BLANCO, C.; FERNANDEZ-GUTIERREZ, A. A sensitive fluorescence optosensor for analyzing propranolol in pharmaceutical preparations and a test for its control in urine in sport. J. Pharm. Biom. Anal., v. 31, n. 5, p. 859-865, Apr. 2003.
FERRARO, M. C.; CASTELLANO, P. M.; KAUFMAN, T. S. A spectrophotometric-partial least squares (PLS-1) method for the simultaneous determination of furosemide and amiloride hydrochloride in pharmaceutical formulations. J. Pharm. Biom. Anal., v. 26, n. 3, p. 443-451, Oct. 2001.
FERRARO, M. C. F.; CASTELLANO, P. M.; KAUFMAN, T. S. Simultaneous determination of amiloride hydrochloride and hydrochlorothiazide in synthetic samples and pharmaceutical formulations by multivariate analysis of spectrophotometric data. J. Pharm. Biom. Anal., v. 30, n. 4, p. 1121-1131, Nov. 2002.
FERRARO, M. C. F.; CASTELLANO, P. M.; KAUFMAN, T. S. Chemometric determination of amiloride hydrochloride, atenolol, hydrochlorothiazide and timolol maleate in synthetic mixtures and pharmaceutical formulations. J. Pharm. Biom. Anal., v. 34, n. 2, p. 305-314, Feb. 2004.
FOSTER, R. Organic charge-transfer complexes. 2nd ed. London: Academic Press, 1968. 470 p.
FREI, R. W.; MACNEIL, J. D. Diffuse reflectance spectroscopy in environmental problem-solving. Cleveland: CRC, 1973. 220 p.
FRITZ, J. S.; MATTEO, P.; ARENA, S. S. A.; PORTER, M. D. Rapid determination of ions by combined solid-phase extraction–diffuse reflectance spectroscopy. J. Chromatogr., A, v. 997, p. 41-50, May 2003.
FRODYMA, M. M.; FREI, R. W. Determination by reflectance spectrophotometry of amino acids resolved on thin-layer plates. J. Chromatogr., v. 15, n. 4, p. 501-509, 1964.
FRODYMA, M. M.; FREI, R. W. An improved method for determination of amino acids by spectral reflectance. J. Chromatogr., v. 17, n. 1, p. 131-139, 1965.
FRODYMA, M. M.; FREI, R. W. Reflectance spectroscopic analysis of dyes separated by thin layer chromatography. J. Chem. Educ., v. 46, n. 8, p. 522-524, Aug. 1969.

161
GARCIA, M. S.; SÁNCHEZ-PEDREÑO, C.; ALBERO, M. I. RÓDENAS, V. Flow injection spectrophotometric determination of frusemide or sulphathiazole in pharmaceuticals. J. Pharm. Biomed. Anal., v. 15, n. 4, p. 453-459, Jan. 1997.
GAZDA, D. B.; FRITZ, J. S.; PORTER, M. D. Investigation of the iodine–poly(vinylpyrrolidone) interaction employed in the determination of biocidal iodine by colorimetric solid-phase extraction. Anal. Chim. Acta, v. 510, n. 2, p. 241-247, May 2004a.
GAZDA, D. B.; FRITZ, J. S.; PORTER, M. D. Determination of nickel (II) as the nickel dimethylglyoxime complex using colorimetric solid phase extraction. Anal. Chim. Acta, v. 508, n. 1, p. 53-59, Apr. 2004b.
GHAUCH, A.; RIMA, J.; CHAREF, A.; SUPTIL, J.; FACHINGER, C.; MARTIN-BOUYER, M. Quantitative measurements of ammonium, hydrogenophosphate and Cu (II) by diffuse reflectance spectrometry. Talanta, v. 48, n. 2, p. 385-392, Feb. 1999.
GHAUCH, A.; TURNAR, C.; FACHINGER, C.; RIMA, J.; CHAREF, A.; SUPTIL, J.; MARTIN-BOUYER, M. Use of diffuse reflectance spectrometry in spot test reactions for quantitative determination of cations in water. Chemosphere, v. 40, n. 12, p. 1327-1333, June 2000.
GIBBS, H. D. Phenol tests. III. The indophenol test. J. Biol. Chem., v. 72, p. 649-664, 1927.
GÖLCÜ, A.; YÜCESOY, C.; SERIN, S. Spectrophotometric determination of some beta-blockers in dosage forms based on complex formation with Cu(II) and Co(II). Farmaco, v. 59, n. 6, p. 487-492, June 2004.
GOTARDO, M. A.; GIGANTE, A. C.; PEZZA, L.; PEZZA, H. R. Determination of furosemide in pharmaceutical formulations by diffuse reflectance spectroscopy. Talanta, v. 64, n. 2, p. 361-365, Oct. 2004.
GOTARDO, M. A.; PEZZA, L.; PEZZA, H. R. Determination of hydrochlorothiazide in pharmaceutical formulations by diffuse reflectance spectroscopy. Eclet. Quim., v. 30, n. 2, p. 17-24, 2005.
GOWDA, B. B.; SEETHARAMAPPA, J.; MELWANKI, M. B. Indirect spectrophotometric determination of propranolol hydrochloride and piroxicam in pure and pharmaceutical formulations. Anal. Sci., v. 18, n. 6, p. 671-674, June 2002.

162
HASSAN, S. S. M.; ABOU-SEKKINA, M. M.; EL-RIES, M. A.; WASSEL, A. A. Polymeric matrix membrane sensors for sensitive potentiometric determination of some β-blockers in pharmaceutical preparations. J. Pharm. Biomed. Anal., v. 32, n. 1, p. 175-180, Apr. 2003.
HELALEH, M. I. H.; RAHMAN, N.; ABU-NAMEH, E. S. M. Use of cerium(IV) nitrate in the spectrophotometric determination of levodopa and methyldopa in the pure form and pharmaceutical preparations. Anal. Sci., v. 13, n. 6, p. 1007-1010, Dec. 1997.
HITSCHERICH, M. E.; RYDBERG, E. M.; TSILIFONIS, D. C.; DALY, R. E. Simultaneous determination of hydrochlorothiazide and propranolol hydrochloride in tablets by high-performance liquid chromatography. J. Liquid Chromatogr., v. 10, n. 5, p. 1011-1021, 1987.
HOFFMAN, B. B. Catecolaminas, fármacos simpatomiméticos e antagonistas dos receptores adrenérgicos. In: HARDMAN, J. G.; LIMBIRD, L. E.; GOODMAN-GILMAN, A. (Ed.). As bases farmacológicas da terapêutica. 10. ed. Rio de Janeiro: McGraw-Hill, 2003. cap. 10, p. 163-203.
HUSSAIN, I.; SHAUKAT, U. Reactions of p-dimethylaminocinnamaldehyde with urea, thiourea and their N-alkyl/aryl derivatives. J. Chem. Society Pakistan, v. 24, n. 4, p. 282-290, Dec. 2002.
IBRAHIM, E. A.; ISSA, A. S.; SALAM, M. A. A.; MAHROUS, M. S. The use of chloranil for spectrophotometric determination of some tranquillizers and antidepressants. Talanta, v. 30, n. 7, p. 531-533, 1983.
IDOWU, O. S.; ADEGOKE, O. A.; OLANIYI, A. A. Colorimetric assay of propranolol tablets by derivatization: novel application of diazotized 4-amino-3,5-dinitrobenzoic acid (ADBA). J. AOAC Int., v. 87, n. 3, p. 573-578, May/June 2004.
JACKSON, E. K. Diuréticos. In: HARDMAN, J. G.; LIMBIRD, L. E.; GOODMAN-GILMAN, A. (Ed.). As bases farmacológicas da terapêutica. 10. ed. Rio de Janeiro: McGraw-Hill, 2003. cap. 29, p. 569-592.
JACKSON, H. L.; McCORMACK, W. B.; RONDESTVEDT, C. S.; SMELTZ, K. C.; VIELE, I. E. Control of peroxidizable compounds. J. Chem. Educ., v. 47, n. 3, Mar. 1970.
JOSEPH-CHARLES, J.; BRAULT, S.; BOYER, C.; LANGLOIS, M. H.; CABRERO, L.; DUBOST, J. P. Simultaneous determination of irbesartan and hydrochlorothiazide in tablets by derivative spectrophotometry. Anal. Lett., v. 36, n. 11, p. 2485-2495, 2003.

163
JUNGREIS, E. Spot test analysis: clinical, environmental, forensic, and geochemical applications. 2nd ed. New York: Wiley-Interscience, 1997. 377 p.
KANNEL, W. B. Blood pressure as a cardiovascular risk factor: prevention and treatment. JAMA, J. Am. Med. Assoc., v. 275, n. 20, p. 1571-1576, May 1996.
KARGOSHA, K.; SARRAFI, A. H. M. Spectrophotometric simultaneous determination of triamterene and hydrochlorothiazide in triamterene-H tablets by multivariate calibration methods. J. Pharm. Biom. Anal., v. 26, n. 2, p. 273-279, Sept. 2001.
KARTAL, M.; ERK, N. Simultaneous determination of hydrochlorothiazide and amiloride hydrochloride by ratio spectra derivative spectrophotometry and high-performance liquid chromatography. J. Pharm. Biom. Anal., v. 19, n. 3/4, p. 477-485, Mar. 1999.
KARTAMYSHEV, S. V.; RYASENSKII, S. S.; GORELOV, I. P. Pharmaceutical analysis using electrodes selective to a cation form of propranolol. Pharm. Chem. J., v. 36, n. 5, p. 274-276, 2002.
KEALEY, D. Quantitative reflectometry. 1. Principles and scope. Talanta, v. 19, n. 12, p. 1563-1569, 1972.
KEALEY, D. Quantitative reflectometry .2. Precision and Interferences. Talanta, v. 21, n. 6, p. 475-480, 1974.
KEALEY, D. Quantitative reflectometry – III. Determination of protein in aqueous media. Talanta, v. 23, p. 715-717, 1976.
KEALEY, D. Quantitative reflectometry – IV. Determination of copper in nickel-plating solutions. Talanta, v. 24, p. 255-258, 1977.
KHALIL, S.; BORHAM, N. Indirect atomic absorption spectrometric determination of pindolol, propranolol and levamisole hydrochlorides based on formation of ion-associates with ammonium reineckate and sodium cobaltinitrite. J. Pharm. Biom. Anal., v. 22, n. 2, p. 235-240, Mar. 2000.
KHALIL, S.; EL-RABIEHI, M. M. Indirect atomic absorption spectrometric determination of pindolol, propranolol and levamisole hydrochlorides based on formation of ion associates with manganese thiocyanate and potassium ferricyanide. J. Pharm. Biom. Anal., v. 22, n. 1, p. 7-12, Feb. 2000.

164
KIRK, A. D.; MOSS, K. C.; VALENTIN, J. G. Separation, identification, and estimation of chromium(III) complex ions. Thin-layer chromatography and diffuse reflectance spectroscopy. J. Chromatogr., v. 36, n. 3, p. 332-337, 1968.
KORANY, M. A.; WAHBI, A. M. Spectrophotometric determination of isoprenaline sulphate and methyldopa using chloranil. Analyst, v. 104, n. 1235, p. 146-148, Feb. 1979.
KULICHENKO, S. A.; FESENKO, S. A. Titrimetric determination of furosemide using aqueous - micellar solutions of surfactants. J. Anal. Chem., v. 57, n. 3, p. 231-234, Mar. 2002.
LABSPHERE. A guide to integrating sphere: theory and applications. Disponível em: <http:www.labsphere.com/tecdocs.aspx>. Acesso em: 30 jan. 2006.
LABSPHERE. RSA-HP-53 instruction manual. North Sutton, [1999?]. p. 4.
LA PEÑA, A. M.; SALINAS, F.; DURÁN, M. S. Simultaneous determination of propranolol and hydralazine by derivative synchronous spectrofluorimetry. Anal. Chim. Acta, v. 255, n. 2, p. 317-323, Dec. 1991.
LAUDSZUN, M.; KOVAR, K. A. Rapid testing method and mechanism of the reaction with 4-dimethylaminocinnamaldehyde (DMAC) - colored salts of benzodiazepines. Pharmaceut. Acta Helv., v. 66, n. 9/10, p. 268-273, 1991.
LIEU, V. T.; FRODYMA, M. M. Selection of optimum concentration range for reflectance spectrophotometric analysis. Talanta, v. 13, n. 9, p. 1319-1327, Sept. 1966.
LIEU, V. T.; FRODYMA, M. M.; HIGASHI, L. S.; KUNIMOTO, L. H. Analysis by reflectance spectroscopy of nucleotides resolved on chromatoplates. Anal. Biochem., v. 19, n. 3, p. 454-460, June 1967.
LIEU, V. T.; ZAYE, D. F.; FRODYMA, M. M. Differential reflectance spectrophotometry .I. high-reflectance method for determination of micro amounts of substances resolved on thin plates. Talanta, v. 16, n. 9, p. 1289-1296, Sept. 1969.
LONG, G. L.; WINEFORDNER, J. D. Limit of detection: a closer look at the IUPAC definition. Anal. Chem., v. 55, n. 7, p. 712A-724A, 1983.
LUIS, M. L.; CORUJEDO, S.; BLANCO, D.; FRAGA, J. M. G.; JIMÉNEZ, A. I.; JIMÉNEZ, F.; ARIAS, J. J. Micellar electrokinetic capillary chromatography analysis of diuretics in pharmaceutical formulations. Talanta, v. 57, n. 2, p. 223-231, May 2002.

165
LUIS, M. L.; FRAGA, J. M. G.; JIMENEZ, A. I.; JIMENEZ, F.; HERNANDEZ, O.; ARIAS, J. J. Application of PLS regression to fluorimetric data for the determination of furosemide and triamterene in pharmaceutical preparations and triamterene in urine. Talanta, v. 62, n. 2, p. 307-316, Feb. 2004.
MAGUREGUI, M. I.; JIMENEZ, R. M.; ALONSO, R. M. Simultaneous determination of the beta-blocker atenolol and several complementary antihypertensive agents in pharmaceutical formulations and urine by capillary zone electrophoresis. J. Chromatogr. Sci., v. 36, n. 10, p. 516-522, Oct. 1998.
MARTIN, E.; HERNANDEZ, O.; JIMENEZ, F.; ARIAS, J. J. Simultaneous spectrophotometric determination of hydrochlorothiazide and amiloride in pharmaceutical preparations. Anal. Lett., v. 28, n. 8, p. 1449-64, 1995.
MARTIN, E.; HERNANDEZ, O.; JIMENEZ, A. I.; JIMENEZ, F.; ARIAS, J. J. A partial least-squares multivariate calibration method for the simultaneous spectrophotometric determination of spironolactone, canrenone and hydrochlorothiazide. Anal. Lett., v. 31, n. 11, p. 1857-1877, 1998.
MARTINEZ, I. R.; CAMANAS, R. M. V.; COQUE, M. C. G. A. Micelle-stabilized room-temperature phosphorimetric procedure for the determination of naproxen and propranolol in pharmaceutical preparations. Analyst, v. 119, n. 5, p. 1093-1097, May 1994.
MARTINEZ, I. R.; COQUE, M. C. G. A.; CAMANAS, R. M. V. Liquid chromatographic procedure for the evaluation of β-blockers in pharmaceuticals using hybrid micellar mobile phases. J. Chromatogr., A, v. 765, n. 2, p. 221-231, Mar. 1997.
MATIAS, F. A. A.; OLIVEIRA, W. A.; MOSCHIM, E. Fibre optic sensor for diesel engine smoke measurement. Sens. Actuators, B, v. 41, n. 1/3, p. 159-162, June 1997.
MATIAS, F. A. A.; VILA, M. M. D. C.; TUBINO, M. A simple device for quantitative colorimetric diffuse reflectance mesurements. Sens. Actuators, B, v. 88, n. 1, p. 60-66, Jan. 2003.
MATIAS, F. A. A.; VILA, M. M. D. C.; TUBINO, M. Quantitative reflectance spot test for the determination of acetylsalicylic acid in pharmaceutical preparations. J. Braz. Chem. Soc., v. 15, n. 2, p. 327-330, Mar./Apr. 2004.
MILLERSHIP, J. S.; PARKER, C.; DONNELLY, D. Ratio spectra derivative spectrophotometry for the determination of furosemide and spironolactone in a capsule formulation. Farmaco, v. 60, n. 4, p. 333-338, 2005.

166
MINISTÉRIO DA SAÚDE. Sistema de informações sobre mortalidade. Disponível em: <http://tabnet.datasus.gov.br/cgi/idb2004/matriz.htm>. Acesso em: 7 jul. 2006.
MOHAMED, A. M. I. Spectrophotometric determination of some benzenesulfonamides with 7,7,8,8-tetracyanoquinodimethane. J. Assoc. Off. Anal. Chem., v. 72, n. 6, p. 885-889, Nov./Dec. 1989.
MOLINER-MARTINEZ, Y.; CAMPÍNS-FALCÓ, P. Detector supports: application to aliphatic amines in wastewater. Talanta, v. 65, n. 1, p. 217-222, Jan. 2005.
MOLINER-MARTÍNEZ, Y.; HERRÁEZ-HERNANDEZ, R.; CAMPÍNS-FALCÓ, P. Improved detection limit for ammonium/ammonia achieved by Berthelot’s reaction by use of solid-phase extraction coupled to diffuse reflectance spectroscopy. Anal. Chim. Acta, v. 534, n. 2, p. 327-334, Apr. 2005.
MOUSSA, B. A. Determination of some aminobenzoic acid derivatives: glafenine and metoclopramide. J. Pharm. Biomed. Anal., v. 23, n. 6, p. 1045-1055, Nov. 2000.
MOUSTAFA, A. A.; ABDEL-MOETY, E. M. Identification and quantification of frusemide (furosemide) in raw materials and in tablets. Farmaco, v. 42, n. 2, p. 51-57, 1987.
NADZHAFOVA, O. Y.; ZAPOROZHETS, O. A.; RACHINSKA, I. V.; FEDORENKO, L. L.; YUSUPOV, N. Silica gel modified with lumogallion for aluminum determination by spectroscopic methods. Talanta, v. 67, n. 4, p. 767-772, Oct. 2005.
NAGARAJA, P.; MURTHY, K. C. S.; RANGAPPA, K. S.; GOWDA, N. M. M. Spectrophotometric methods for the determination of certain catecholamine derivatives in pharmaceutical preparations. Talanta , v. 46, n. 1, p. 39-44, May 1998.
NAGARAJA, P.; VASANTHA, R. A.; SUNITHA, K. R. A sensitive and selective spectrophotometric estimation of catechol derivatives in pharmaceutical preparations. Talanta, v. 55, n. 6, p. 1039-1046, Dec. 2001.
NAKANO, N.; YAMAMOTO, A.; KOBAYASHI, Y.; NAGASHIMA, K. Development of a monitoring tape for hydrogen chloride gas using metanil yellow. Analyst, v. 118, p. 1539-1542, 1993.
NARAYANASWAMY, R. Optical chemical sensors: transduction and signal processing. Analyst, v. 118, p. 317-322, Apr. 1993.

167
NARAYANASWAMY, R.; SEVILLA III, F. Reflectometric study of the acid-base equilibria of indicators immobilised on a styrene/divinylbenzene copolymer. Anal. Chem. Acta, v. 189, p. 365-369, 1986.
NEVADO, J. J. B.; GALLEGO, J. M. L.; LAGUNA, P. B. Spectrophotometric determination of dopamine and methyldopa with metaperiodate by flow injection analysis. Fresenius J. Anal. Chem., v. 353, n. 2, p. 221-223, Sept. 1995.
NIKOLELIS, D. P.; PETROPOULOU, S. E.; MITROKOTSA, M. V. A minisensor for the rapid screening of atenolol in pharmaceutical preparations based on surface-stabilized bilayer lipid membranes with incorporated DNA. Bioelectrochemistry, v. 58, n. 1, p. 107-112, Nov. 2002.
OHLWEILER, O. A. Química analítica quantitativa. 3. ed. Rio de Janeiro: LTC, 1982. v. 3, p. 190.
OLIVEIRA, W. A.; SALIBA, P. R. Optosensing of carbon monoxide through paper impregnated with palladium (II) chloride. J. Braz. Chem. Soc., v. 5, n. 1, p. 39-42, 1994.
OSSMAN, A. E.; AHMAD, A. S.; EL-ZAHABY, M.; SALAMA, F. Investigation and application of the reaction between N-halosuccinimide and furosemide. Pharm. Ind., v. 50, n. 8, p. 986-989, Aug. 1988.
OUYANG, J.; BAEYENS, W. R. G.; DELANGHE, J.; VAN DER WEKEN, G.; CALOKERINOS, A. C. Cerium(IV)-based chemiluminescence analysis of hydrochlorothiazide. Talanta, v. 46, n. 5, p. 961-968, Aug. 1998.
OUYANG, J.; BAEYENS, W. R. G.; DELANGHE, J.; VAN DER WEKEN, G.; VAN DAELE, W.; DE KEUKELEIRE, D.; GARCIA CAMPANA, A. M. Chemiluminescence-based liquid chromatographic determination of hydrochlorothiazide and captopril. Anal. Chim. Acta, v. 386, n. 3, p. 257-264, Apr. 1999.
OZKAN, S. A. Simultaneous determination of losartan potassium and hydrochlorothiazide from tablets and human serum by RP-HPLC. J. Liq. Chromatogr. Relat. Technol., v. 24, n. 15, p. 2337-2346, 2001.
PANDERI, I. E.; PARISSI-POULOU, M. Simultaneous determination of benazepril hydrochloride and hydrochlorothiazide by micro-bore liquid chromatography. J. Pharm. Biom. Anal., v. 21, n. 5, p. 1017-1024, Dec. 1999.

168
PANDERI, I. E. Simultaneous determination of benazepril hydrochloride and hydrochlorothiazide in tablets by second-order derivative spectrophotometry. J. Pharm. Biom. Anal., v. 21, n. 2, p. 257-265, Nov. 1999.
PANZADE, P. D.; MAHADIK, K. R. Simultaneous spectrophotometric determination of captopril and hydrochlorthiazide in combined dosage forms. East. Pharm., v. 43, n. 512, p. 153-154, 2000.
PATHAK, V. N.; SHUKLA, M. S. R; SHUKLA, I. C. Direct titrimetry determination of the antyhypertensive drugs methyldopa and propranolol in pharmaceutical preparations. Analyst, v. 107, n. 1278, p. 1086-1087, Sept. 1982.
POLLO, F. Desenvolvimento de métodos analíticos para a determinação de bumetanida em medicamentos. Dissertação (Mestrado em Química) - Instituto de Química, Universidade Estadual Paulista, Araraquara, em desenvolvimento.
PULGARIN, J. A. M.; MOLINA, A. A.; OLIVARES NIETO, G. P. Determination of hydrochlorothiazide in pharmaceutical preparations by time resolved chemiluminescence. Anal. Chim. Acta, v. 518, n. 1/2, p. 37-43, Aug. 2004.
QUAGLIA, M. G.; DONATI, E.; CARLUCCI, G.; MAZZEO, P.; FANALI, S. Determination of losartan and hydrochlorothiazide in tablets by CE and CEC. J. Pharm. Biom. Anal., v. 29, n. 6, p. 981-987, Aug. 2002.
QUINTANA, M. G.; DIDION, C.; DALTON, H. Colorimetric method for a rapad detection of oxygenated aromatic biotransformation products. Biotechnol. Tech., v. 11, n. 8, p. 585-587, Aug. 1997.
RADULOVIC, D.; JOVANOVIC, M. S.; ZIVANOVIC, L. Spectrophotometric determination of propranolol hydrochloride as bromthymolbluelon pair in tablets. Pharmazie, v. 41, n. 6, p. 434-434, June 1986.
RAMESH, K. C.; GOWDA, B. G.; SEETHARAMAPPA, J.; KESHAVAYYA, J. Indirect spectrofluorimetric determination of piroxicam and propranolol hydrochloride in bulk and pharmaceutical preparations. J. Anal. Chem., v. 58, n. 10, p. 933-936, Oct. 2003.
RANKIN, G. O. Diuretics. In: WILLIAMS, D. A.; LEMKE, T. L. Foye’s principles of medicinal chemistry. 5th ed. Philadelphia: Lippincott Williams & Wilkins, 2002. cap. 22, p. 518-531.

169
RAPAKA, R. S.; ROTH, J.; PRASAD, V. K. Analysis of 4-chloro-5-sulfamoylanthranilic acid in the bulk material and pharmaceutical preparations of furosemide by a high performed liquid chromatographic method. Int. J. Pharm., v. 11, n. 3, p. 237-247, 1982.
REINECKE, D.; JENSEN, A.; FISTER, F.; SCHERNAU, U. Quantitative determination of organic compounds by diffuse reflectance Fourier transform infrared spectrometry. Anal. Chem., v. 60, n. 11, p. 1221-1224, June 1988.
RIBEIRO, P. R. S.; GOMES-NETO, J. A.; PEZZA, L.; PEZZA, H. R. Flow-injection spectrophotometric determination of methyldopa in pharmaceutical formulations. Talanta, v. 67, n. 1, p. 240-244, July 2005.
ROSS, J. H. 2,6-Dichloroquinone 4-chloroimide as a reagente for amines and aromatic hydrocarbons on thin-layer chromatograms. Anal. Chem., v. 40, n. 14, p. 2138-2143, Dec. 1968.
ROSSI, A. V.; HE, X.; TUBINO, M. A simple, portable and low cost device for a colorimetric spot test quantitative analysis. Anal. Lett., v. 33, n. 9, p. 1885-1898, 2000.
RUIZ, T. P.; MARTINEZ-LOZANO, C.; TOMAS, V.; CARPENA, J. Simultaneous determination of propranolol and pindolol by synchronous spectrofluorimetry. Talanta, v. 45, n. 5, p. 969-976, Mar. 1998.
SAEED, A.; HAQUE, S.; QURESHI, S. Z. Resin bead detection and spectrophotometric determination of oxyphenbutazone with p-dimethylaminocinnamaldehyde: application to bulk drug and dosage forms. Talanta, v. 40, n. 12, p. 1867-1871, Dec. 1993.
SALAMA, F. M.; ABD EL-SATTAR, O. I.; EL-ABASAWY, N. M.; FOAD, M. M. Second derivative spectrophotometric determination of captopril, enalapril maleate, and fosinopril sodium in presence of hydrochlorothiazide. J. Pharm. Sci., v. 26, p. 96-105, 2000.
SALEM, F. B. Colorimetric determination of certain sympathomimetic amines. Anal. Lett., v. 18, n. 89, p. 1063-1075, 1985.
SALEM, H. Spectrophotometric determination of β-adrenergic blocking agents in pharmaceutical formulations. J. Pharm. Biomed. Anal., v. 29, n. 3, p. 527-538, July 2002.
SASTRY, C. S. P.; PRASAD, T. N. V.; SASTRY, B. S.; RAO, E. V. Spectrophotometric methods for the determination of some diuretics using 3-methyl-2-benzothiazolinone hydrazone. Analyst, v. 113, n. 2, p. 255-258, Feb. 1988.

170
SASTRY, C. S. P.; SURYANARAYANA, M. V.; TIPIRNENI, A. S. R. P. Application of p-N,N-dimethylphenylenediamine dihydrochloride for the determination of some diuretics. Talanta, v. 36, n. 4, p. 491-494, Apr. 1989.
SASTRY, C. S. P.; SASTRY, B. S.; RAO, J. V.; KRISHNA, R. R. Spectrophotometric methods for the determination of tolnaftate. Talanta, v. 40, n. 4, p. 571-576, 1993.
SASTRY, C. S. P.; SRINIVAS, K. R.; PRASAD, K. M. M. Spectrophotometric determination of drugs in pharmaceutical formulations with N-bromosuccinimide and celestine blue. Mikrochim. Acta, v. 122, n. 1/2, p. 77-86, 1996.
SASTRY, C. S. P.; RAO, K. R.; PRASAD, D. S. Determination of cefadroxil by three simple spectrophotometric methods using oxidative coupling reactions. Mikrochim. Acta, v. 126, n. 1/2, p. 167-172, 1997.
SATANA, E.; ALTINAY, S.; GOGER, N. G.; OZKAN, S. A.; SENTURK, Z. Simultaneous determination of valsartan and hydrochlorothiazide in tablets by first-derivative ultraviolet spectrophotometry and LC. J. Pharm. Biom. Anal., v. 25, n. 5/6, p. 1009-1013, July 2001.
SEMAAN, F. S.; DOS SANTOS NETO, A. J.; LANÇAS, F. M.; CAVALHEIRO, E. T. G. Rapid HPLC-DAD determination of furosemide in tablets using a short home-made column. Anal. Lett., v. 38, n. 10, p. 1651-1658, 2005.
SEVILLANO-CABEZA, A.; CAMPÍNS-FALCÓ, P.; SERRADOR-GARCÍA, M. C. Extractive-spectrophotometric determination of furosemide with sodium 1,2-naphthoquinone-4-sulphonate in pharmaceutical formulations. Anal. Lett., v. 30, n. 1, p. 91-107, 1997.
SHAFAATI, A.; CLARK, B. J. Development and validation of a capillary zone electrophoretic method for the detn. of atenolol in presence of its related substances in bulk and tablet dosage form. J. Pharm. Biom. Anal., v. 14, n. 11, p. 1547-1554, Aug. 1996.
SHAMSIPUR, M.; JALALI, F. Preparation of an atenolol ion-selective electrode and its application to pharmaceutical analysis. Anal. Lett., v. 38, n. 3, p. 401-410, 2005.
SHIMDDA, K.; NAGASE, Y. Spectrophotometric determination of 4-aminoantipyrine with para-dimethylaminocinnamaldehyde. Bunseki Kagaku, v. 26, p. 712-715, 1977.
SKOOG, D. A. Princípios de análise instrumental. 5th ed. Porto Alegre: Bookman, 2002. p. 124-125.

171
STORM, S. L.; SPRINGSTEEN, A. The integrating sphere reflectance accessory. Disponível em: <http:www.labsphere.com/knownledge.asp>. Acesso em: 30 jan. 2006. Application note nº 03.
THE MERCK Index: an encyclopedia of chemical, drugs, and biology. 13th ed. Whitehouse Station: Merck & Co, 2001.
THOMPSON, M.; ELLISON, S. L. R.; WOOD, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl. Chem., v. 75, n. 5, p. 835-855, 2002.
TING, S. Liquid chromatographic determination of methyldopa and methyldopa-thiazide combinations in dosage forms. J. Assoc. Off. Anal. Chem., v. 66, n. 6, p. 1436-1442, 1983.
TORAL, M.; POPE, S.; QUINTANILLA, S.; RICHTER, P. Simultaneous determination of amiloride and furosemide in pharmaceutical formulations by first digital derivative spectrophotometry. Int. J. Pharm., v. 249, n. 1/2, p. 117-126, Dec. 2002.
TOWNSHEND, A.; PULGARIN, J. A. M.; PARDO, M. T. A. Flow injection-chemiluminescence determination of propranolol in pharmaceutical preparations. Anal. Chim. Acta, v. 488, n. 1, p. 81-88, July 2003.
TSOGAS, G. Z.; STERGIOU, D. V.; VLESSIDIS, A. G.; EVMIRIDIS, N. P. Development of a sensitive flow injection-chemiluminescence detection method for the indirect determination of propranolol. Anal. Chim. Acta, v. 541, n. 1/2, p. 151-157, June 2005.
TUBINO, M.; ROSSI, A. V.; MAGALHÃES, M. E. A. Quantitative spot tests of Fe (III), Cr (VI) and Ni (II) by reflectance measurements. Anal. Lett., v. 30, n. 2, p. 271-282, 1997.
TUBINO, M.; RODRIGUES-JR., A.; VILA, M. M. D. C. A simple flow injection spectrophotometric method for the determination of α-methyldopa in pharmaceutical preparations. J. Flow Injection Anal., v. 21, n. 2, p. 132-136, 2004.
TUBINO, M.; SOUZA, R. L. Determination of diclofenac in pharmaceutical preparations by diffuse reflectance photometry. Talanta, v. 68, n. 3, p. 776-780, Jan. 2006.
UNITED States Pharmacopeia: USP 26 the national formulary USP 26-NF21. 26th ed. Rockville, MD, 2003.

172
URBANYI, T.; O’CONNELL, A. Simultaneous automated determination of hydralazine hydrochloride, hydrochlorothiazide, and reserpine in single tablet formulations. Anal. Chem., v. 44, n. 3, p. 565-570, Mar. 1972.
VIEIRA, I. C.; FATIBELLO-FILHO, O. Spectrophotometric determination of methyldopa and dopamine in pharmaceutical formulations using a crude extract of sweet potato root (Ipomoea batatas (L.) Lam.) as enzymic source. Talanta, v. 46, n. 4, p. 559-564, Aug. 1998.
WAHBI, A.A. M.; ABDINE, H.; KORANY, M.; ABDEL-HAY, M. H. Sodium cobaltinitrite as a reagent for determining some phenolic drugs. J. Assoc. Off. Anal. Chem., v. 61, n. 5, p. 1113-1117, 1978.
WALASH, M. I.; ABOU-OUF, A.; SALEM, F. B. Colorimetric determination of some sympathomimetic amines. J. Assoc. Off. Anal. Chem., v. 68, n. 1, p. 91-95, 1985.
WEINERT, P. L.; PEZZA, H. R.; OLIVEIRA, J. E.; PEZZA, L. A simplified spectrophotometric method for routine analysis of saccharin in commercial noncaloric sweeteners. J. Agric. Food Chem., v. 52, p. 7788-7792, 2004.
WENDLANT, W. W.; HECHT, H. G. Reflectance spectroscopy. New York: Interscience, 1966. 297 p.
ZAPOROZHETS, O.; GAWER, O.; SUKHAN, V. Determination of Fe (II), Cu (II) and Ag (I) by using silica gel loaded with 1,10-phenanthroline. Talanta, v. 46, n. 6, p. 1387-1394, Aug. 1998.
ZAPOROZHETS, O.; PETRUNIOCK, N.; BESSARABOVA, O.; SUKHAN, V. Determination of Cu(II) and Zn(II) using silica gel loaded with 1-(2-thiasolylazo)-2-naphthol. Talanta, v. 49, n. 4, p. 899-906, July 1999.
ZAPOROZHETS, O. A.; TSYUKALO, L. Ye. Xylenol orange adorbed on silica surface as a solid phase reagent for lead determination using diffuse reflectance spectroscopy. Talanta, v. 58, n. 5, p. 861-868, 2002.
ZAYE, D. F.; FREI, R. W; FRODYMA, M. M. Cation analysis by thin-layer chromatography and reflectance spectroscopy .I. rapid identification of cations resolved on thin-layer plates. Anal. Chim. Acta, v. 39, n. 1, p. 13-18, 1967.

173
ZAWILLA, N. H.; MOHAMMAD, M. A. A.; EL KOUSY, N. M.; EL-MOGHAZY, A. S. M. Determination of aceclofenac in bulk and pharmaceutical formulations. J. Pharm. Biomed. Anal., v. 27, n. 1/2, p. 243-251, Jan. 2002.
ZECEVIC, M.; ZIVANOVIC, L.; AGATONOVIC, S. K.; MINIC, D. The use of a response surface methodology on HPLC analysis of methyldopa, amiloride and hydrochlorothiazide in tablets. J. Pharm. Biom. Anal., v. 24, n. 5/6, p. 1019-1025, Mar. 2001.
ZIVANOVIC, L.; RADULOVIC, D.; JOVANOVIC, M. Colorimetric determination of propranolol chloride in pharmaceutical preparations. Pharmaceut. Acta Helv., v. 63, n. 12, p. 350-352, 1988.
ZIVANOVIC, L.; AGATONOVIC, S.; RADULOVIC, D. Determination of diuretics in pharmaceutical preparations by thin-layer-chromatographic densitometry. Pharmazie, v. 44, n. 12, p. 864, Dec. 1989.
ZIVANOVIC, L.; AGATONOVIC, S. K.; RADULOVIC, D. Extraction spectrophotometric determination of furosemide as its iron(III) complex. Pharmazie, v. 45, n. 12, p. 935-935, Dec. 1990.
ZIVANOVIC, L.; VASILJEVIC, S.; RADULOVIC, D. Colorimetric assay of methyldopa bulk drug and tablets as its Fe(III) complex. Boll. Chim. Farm., v. 130, n. 5, p. 162-165, May 1991.