Embed Size (px)
Citation preview

I n t rodução à B io in fo rmát i ca
Marcelo Reis
Centro APTA Citros “Sylvio Moreira”
18 de julho de 2007
Duração estimada: ~ 2,5h (manhã)~ 2,5h (tarde)

Agenda
● Manhã:– Que trem é esse, bioinformática?!– Bioinfo do Citrus EST Project (CitEST);– Linux: o S.O. de bioinfo por excelência;– Instalando e utilizando a distro Ubuntu.
● Tarde:– Bioinfo em análise de sequências;– Identificação de sequências I : BLAST;– Identificação de sequências II : Pfam;– Alinhamento múltiplo (ClustalW).

B io in fo rmát i ca
Definição:
“Bioinformática é o uso de técnicas de matemática, computação, química e estatística, visando solucionar problemas biológicos, tipicamente de biologia molecular”.

Exemplos de Ap l i cações
● Extrair a sequência de DNA do arquivo gerado pela máquina sequenciadora (cromatograma);
● Eliminação de de contaminações de sequências (adaptadores, vetores, etc);
● Clusterização, montagem;● Armazenar e organizar as sequências;● Análise de Sequências;● Taxonomia, Predição de estruturas, etc.

B io in fo rmát i ca no Cent roAPTA C i t ros
● No Centro APTA Citros a bioinformática auxilia em todos os passos anteriormente ilustrados (eliminação de contaminações, montagem, análise de sequências).
● Existe um portal, chamado CitEST (Citrus EST Project), onde boa parte das ferramentas estão disponíveis via web. Além disso, várias outras análises são feitas via terminal.

Por ta l C i tEST
● Web site do Citrus EST Project (CitEST) e de todos os projetos relacionados (vírus, Phytophthora, bactérias, etc).
● Diversos serviços, destacando-se:– Genômica Comparativa (BLAST);– Sistema de submissão de sequências;– Editores (Gene Projects).
● “CitEST” pode ser uma referência tanto ao portal quanto ao conjunto das bibliotecas de cDNA do projeto.

Ed i to res : GP e Un igenes
● Editores servem como ferramenta ao pesquisador para:– Anotação e data mining de uma pequena
porção dos transcritos do CitEST (Gene Projects);
– Anotação e data mining de todos os transcritos do CitEST (Editor de Unigenes).
● O editor de Unigenes trabalha com uma montagem global (eliminação de transcritos repetidos).
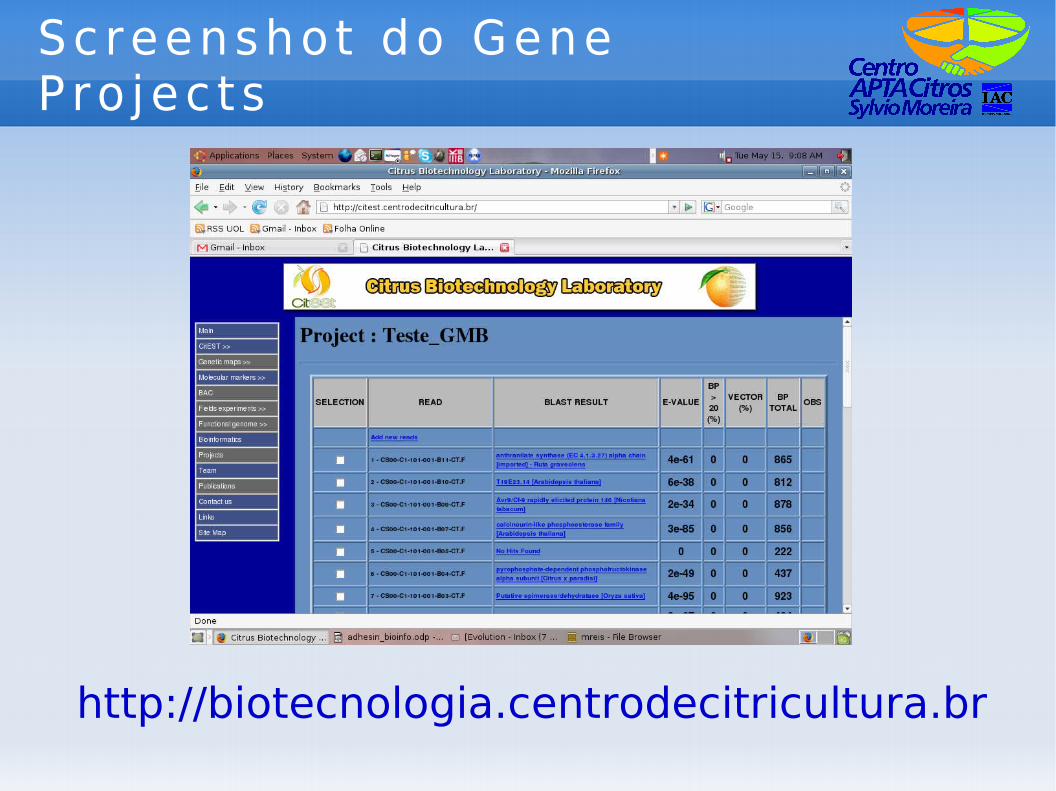
Screenshot do Gene P ro jec ts
http://biotecnologia.centrodecitricultura.br

Dig i ta l Nor thern
● Existem diversos métodos para determinar expressão diferencial de um gene entre diferentes bibliotecas de cDNA:– Microarray -> $$$+;– qPCR -> $$+;– bibliotecas subtrativas -> $+;– digital northern (hibridização in silico) -> $--!!

Exemplo de Ap l i cação D ig i ta l Nor thern
● Digital Northern compara, aplicando uma estatística, a abundância de transcritos de um dado gene entre 2 ou mais bibliotecas.

S i s temas de B io in fo
● Todos os sistemas de Bioinformática do CitEST (e do Centro APTA Citros como um todo) utiliza soluções gratuitas (software livre):– Sistema Operacional: Linux;– Banco de Dados Relacional: MySQL;– Linguagem de Programação: Perl, CGI;– Servidora Web: Apache.
● Dessas soluções acima, vamos conhecer um pouco mais do S.O. Linux.

L inux x M ic ro$o f t

GNU-L inux
● GNU-Linux é um Sistema Operacional, tal como o Window$ ou o MacOS.
● Ele é muito usado para bioinformática por várias razões, dentre elas:– Por ser software livre, ele é gratuito e pode
ser livremente modificado;– Não tem problemas de vírus, apresenta maior
confiabilidade e robustez;– É mais adequado para disponibilizar
ferramentas web ou executar ferramentas de linha de comando.

Dis t ros de L inux
● Distro é a abreviação de “distribuidoras”. ● Como o Linux é software livre, ele
funciona análogo à um livro que esteja no domínio público: assim como qualquer editora pode publicar, digamos o Les Misérables, de Victor Hugo, qualquer empresa pode “empacotar” e distribuir o “seu Linux”.
● Exemplos de distros: Red Hat, Debian, Mandriva, Fedora, Ubuntu.

L inux - Exerc í c io
1. Utilize o CD do Ubuntu fornecido para instalar o sistema em sua máquina; acompanhe o instrutor;
2. Ao terminar a instalação, examine o Sistema Operacional, procurando se familiarizar com o ambiente gráfico;
3. Abra o terminal e acompanhe os comandos explicados pelo instrutor (ls, cd, mkdir, clear, nano, etc).

Par te I I – Aná l i se de Sequênc ias

Formato Fas ta
● Após o sequenciamento, os cromatogramas são convertidos em um arquivo texto, num formato chamado “fasta”.
● Pode ser arquivo de nucleotídeos ou de aminoácidos.
● O arquivo pode ter 1 ou mais sequências, sempre iniciada na primeira linha por um cabeçalho começando com o caracter “>”.

Exemplos de A rqu ivos no Formato Fas ta
● Aminoácidos:>gi|532319|pir|TVFV2E|TVFV2E envelope protein
ELRLRYCAPAGFALLKCNDADYDGFKTNCSNVSVVHCTNLMNTTVTTGLLLNGSYSENRT
QIWQKHRTSNDSALILLNKHYNLTVTCKRPGNKTVLPVTIMAGLVFHSQKYNLRLRQAWC
HFPSNWKGAWKEVKEEIVNLPKERYRGTNDPKRIFFQRQWGDPETANLWFNCHGEFFYCK
● Nucleotídeos:>BTBSCRYR
tgcaccaaacatgtctaaagctggaaccaaaattactttctttgaagacaaaaactttca
aggccgccactatgacagcgattgcgactgtgcagatttccacatgtacctgagccgctg
caactccatcagagtggaaggaggcacctgggctgtgtatgaaaggcccaattttgctgg
gtacatgtacatcctaccccggggcgagtatcctgagtaccagcactggatgggcctcaa

Genômica Compara t i va
● Os métodos que vamos estudar (BLAST, Clustalw, Pfam) também são chamados de Genômica Comparativa, pois as identificações são feitas através da comparação, da sequência que você tem em mãos, com uma ou mais sequências.
● Exceto pelo alinhamento múltiplo, normalmente a comparação é feita através da comparação com sequências de um banco de dados biológicos.

BLAST
● BLAST significa “Basic Local Alignment Search Tool”.
● Funciona comparando sequências através de alinhamentos locais; vantagens:– identificação de domínios;– rapidez;
● Diversos “sabores” (blastx, blastn, etc);● Serviço de BLAST local disponível tanto
via web (local ou no NCBI) quanto em versão de linha de comando.

BLAST loca l x BLAST NCB I
● Rodar o BLAST no NCBI, (ou qualquer outra ferramenta) com sequências recém sequenciadas e novas (cujo paper ainda não foi publicado), é algo NÃO recomendável (confidencialidade).
● Nesse caso, pode-se configurar um BLAST web em alguma máquina do laboratório, ou rodar o BLAST em linha de comando.
● Rodar em linha de comando traz a vantagem de permitir maior controle sobre o processo.

Bancos do BLAST
● Diversos bancos de dados existentes. Alguns dos principais deles:
– GenBank (o banco do NCBI é maior e mais abrangente, mas menos preciso);
– Swiss-Prot (banco pequeno, mas contendo apenas sequências verificadas experimentalmente – comprovação proteômica).
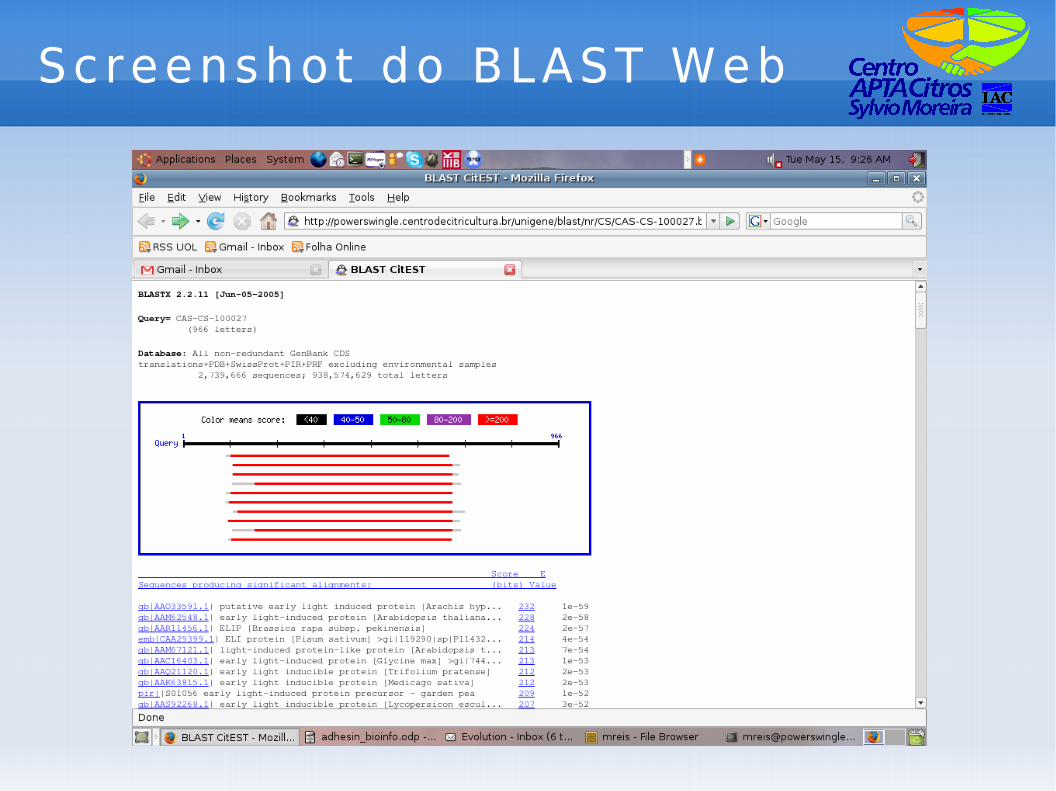
Screenshot do BLAST Web

BLAST - Exerc í c io
1. Instale o BLAST em seu computador, via apt. Para isso, digite:sudo aptget install blast2
2. Copie do CD (ou do servidor fornecido) o banco de dados Swiss-Prot;
3. Dispare o comando BLAST, da mesma forma demonstrada pelo instrutor;
4. Abra com algum editor de texto o arquivo de saída; vamos analisar o “report”.

P fam
● Pfam -> Protein families.● Serve para identificação de domínio de
proteínas.● Ao contrário do BLAST, serve para
identificação a partir de uma lista de domínios de diversas famílias de proteínas.
● BLAST serve para identificar através da “anotação” da sequência mais parecida.
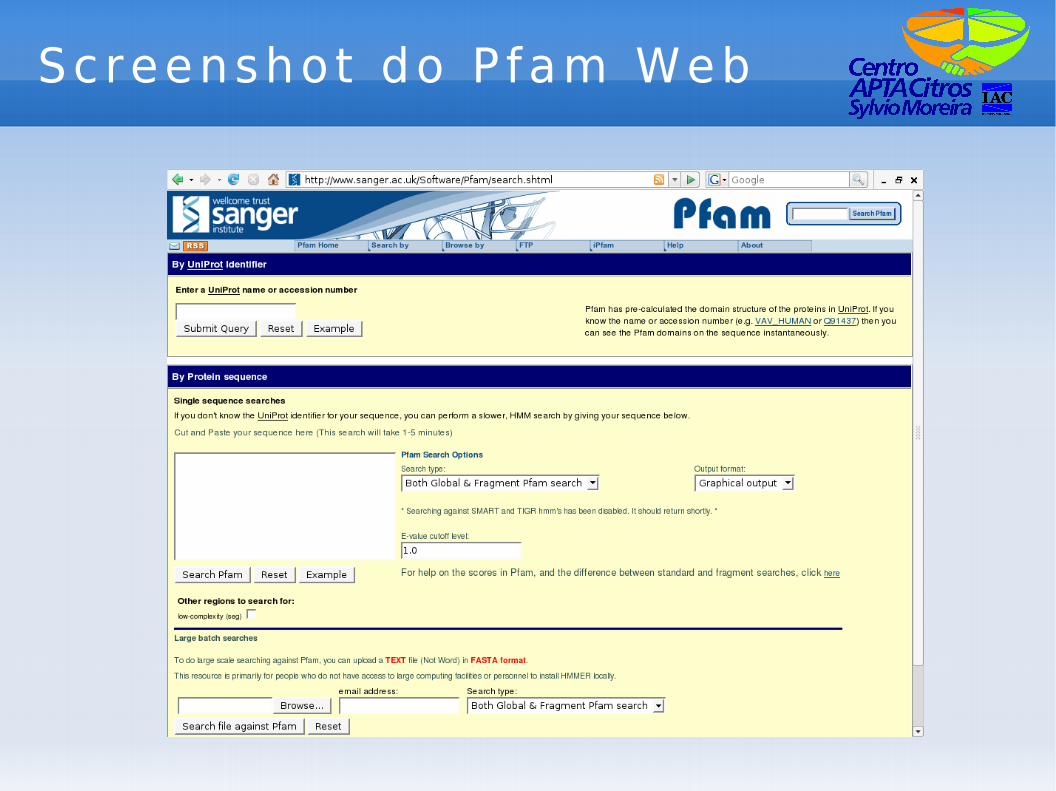
Screenshot do P fam Web

P fam - Exerc í c io
1. Instale o software HMMER em seu computador, via apt. Para isso, digite:sudo aptget install hmmer
2. Copie do CD (ou do servidor fornecido) o banco de dados do Pfam (Pfam_fs);
3. Dispare o comando hmmpfam, da mesma forma demonstrada pelo instrutor;
4. Abra com algum editor de texto o report produzido.

A l inhamento Mú l t ip lo
● Também chamado de alinhamento global, serve para alinhar mais de 2 sequências ao mesmo tempo (BLAST compara apenas 2 de cada vez).
● Serve para diversos tipos de inferências, tais como:– detectar regiões conservadas (paper);– filogenia;– busca de SNPs, etc.

C lus ta lW
● Ferramentas:– ClustalW e variantes (Clustalx, etc).

Clus ta lw - Exerc í c io
1. Instale o software clustalw em seu computador, via apt. Para isso, digite:sudo aptget install clustalw
2. Copie o arquivo multifasta (i.e., arquivo fasta com várias sequências), a ser indicado, do GenBank (NCBI):
http://www.ncbi.nlm.nih.gov/Genbank
3. Dispare o comando clustalw;
4. Tal qual em BLAST e Pfam, abrir o report gerado para entender o resultado obtido.

Resumo da ópera
● Bioinformática é a exatas (computação, matemática, etc) auxiliando a biologia (sendo mais exato, a biologia molecular).
● Linux é um sistema operacional, software livre, e indicado para realizar análises de bioinformática.
● Os métodos de análise de sequências normalmente utilizam uma ferramenta (web ou de terminal) e um banco de dados biológicos.

Dúv idas?
?

Obr igado!
!





























