Embed Size (px)
Citation preview

PRÓ-REITORIA DE PÓS-GRADUAÇÃO, PESQUISA E EXTENSÃO
ÁREA DE CIÊNCIAS TECNOLÓGICAS
Curso de Mestrado em Nanociências
MARCOS ANDRÉ PEREIRA DOS SANTOS
INTERAÇÃO DE MOLÉCULAS DE TiCl 3 E TiCl4 COM GRAFENO: UMA
SIMULAÇÃO DE PRIMEIROS PRINCÍPIOS
Santa Maria, RS
2009

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
MARCOS ANDRÉ PEREIRA DOS SANTOS
INTERAÇÃO DE MOLÉCULAS DE TiCl 3 E TiCl 4 COM
GRAFENO: UMA SIMULAÇÃO DE PRIMEIROS
PRINCÍPIOS
Dissertação apresentada ao Curso de
Mestrado em Nanociências do Centro
Universitário Franciscano de Santa Maria
como requisito parcial para obtenção do
título de Mestre em Nanociências.
Orientadora: Profa Dra SOLANGE BINOTTO FAGAN
Santa Maria, RS
2009

3
CENTRO UNIVERSITÁRIO FRANCISCANO
PRÓ-REITORIA DE PÓS-GRADUAÇÃO, PESQUISA E EXTENSÃO
CURSO DE MESTRADO EM NANOCIÊNCIAS
A COMISSÃO EXAMINADORA, ABAIXO ASSINADO, APROVA A
DISSERTAÇÃO:
INTERAÇÃO DE MOLÉCULAS DE TiCl 3 E TiCl4 COM GRAFENO VIA
SIMULAÇÃO DE PRIMEIROS PRINCÍPIOS
Elaborada por:
MARCOS ANDRÉ PEREIRA DOS SANTOS
COMISSÃO EXAMINADORA
______________________________________________
Profa. Dra. Solange Binotto Fagan - UNIFRA
Presidente
________________________________________________
Prof. Dr. Antônio Gomes de Souza Filho - UFC
________________________________________________
Prof. Dr. Sérgio Roberto Mortari - UNIFRA
Santa Maria-RS, 26 de março de 2009.

4
Dedico este trabalho aos meus pais, Amaury (in memorian) e Therezinha,
a quem tudo devo.

5
AGRADECIMENTOS
Dedico meus agradecimentos a todos que de alguma forma colaboraram com a realização
deste trabalho:
– a Deus, sou grato por todas as oportunidades que me foram dadas;
– aos colegas de graduação e pós-graduação da UNIFRA, pelo incentivo, troca de saberes e
amizade;
– aos demais professores do Mestrado em Nanociências, que colaboraram na minha
formação acadêmica;
– à UNIFRA pelo apoio financeiro durante minha formação;
– ao CENAPAD – SP, pelo espaço disponível para a realização dos cálculos e suporte
técnico;
– aos meus familiares, pelo apoio nas minhas decisões;
– à Profa. Dra. Ivana Zanella, pela co-orientação neste trabalho;
–à Profa. Dra. Solange B. Fagan, pela orientação, tolerância, paciência, amizade e pelos
ensinamentos.

6
RESUMO
Nesta Dissertação apresentamos um estudo da interação das moléculas TiCl4 e TiCl3
com grafeno de camada simples e bicamada fazendo uso da teoria do funcional da
densidade. Avaliamos as alterações nas propriedades estruturais, eletrônicas e magnéticas
do grafeno interagindo com as moléculas, as quais podem se comportar como sistemas
doadores ou aceitadores de carga eletrônica. Foram avaliadas as configurações mais
estáveis para as moléculas de TiCl3 e TiCl4 em três sítios utilizando uma molécula por
célula unitária e fazendo uso de condições periódicas de contorno. Em ambos os casos, as
moléculas possuem maior estabilidade química na configuração em que essas são colocadas
no centro de um hexágono do grafeno. Após a simulação foram observadas alterações
significativas nas propriedades eletrônicas do sistema. A molécula TiCl4 apresenta caráter
de ácido de Lewis (receptor de carga), enquanto, no caso da molécula estar confinada entre
dois planos de grafeno, comporta-se como uma base de Lewis (doador de carga). Também
se observa que o TiCl4 permanece não magnético em todas as configurações estudadas. A
adsorção da molécula causa distorções geométricas na folha de grafeno, originando uma
pequena mudança nos estados eletrônicos do grafeno, visto na abertura de um gap de
energia. Por outro lado, a molécula de TiCl3 apresenta caráter de base de Lewis, visto
diretamente no aumento do valor do nível de Fermi. O TiCl3 apresenta energia de ligação
bem superior quando comparado ao TiCl4 no grafeno, este fato mostra também que as
propriedades eletrônicas também se modificam. Observa-se que o momento magnético do
TiCl3 adsorvido no grafeno apresenta polarização de spin, dependente da configuração
estrutural. Desta forma, este trabalho busca, de forma inédita, demonstrar as propriedades
associadas com a interação de moléculas doadoras e aceitadoras de carga eletrônica
adsorvidas no grafeno fomentando o potencial de aplicação tecnológica deste material
inovador.
Palavras-chave: grafeno, teoria do funcional da densidade, doador/aceitador de carga
eletrônica

7
ABSTRACT
This work presents a study on the interaction of TiCl4 and TiCl3 molecules with a
single- and bi-layer graphene by using the density functional theory. It is evaluated the
changes in structural, electronic and magnetic properties of graphene after the adsorption of
these molecules, that can behave donors or acceptors of electronic charge. We evaluated the
arrangements for TiCl4 and TiCl3 molecules at three different sites using one molecule per
unit cell, making use of periodic boundary conditions. In both cases the molecules have
more stable chemical configuration when they are placed in the center of a hexagonal site.
Significant changes in the electronic properties of the systems are observed. The molecule
TiCl4 behaves like a Lewis acid (electronic charge acceptor), while in the case of the
confined molecule between two graphene planes the molecule behaves as a Lewis basis
(electronic charge donor). The TiCl4 on graphene remains non-magnetic in all
configurations studied here. The adsorption of the molecule causes distortions in the
graphene sheet, they originating a small change in the original electronic states, as a gap
opening. TiCl3 molecule presents a Lewis basics behavior, which is seen directly on the
lifting of the Fermi level. The TiCl3 presents a stronger binding energy when compared to
the TiCl4 in graphene; this fact also shows that the electronic properties also change. It is
observed that the magnetic moment of the adsorbed TiCl3 graphene associated with the spin
polarization, dependent on the adsorption configuration. Therefore, this innovative work
predicts the properties associated with the interaction of molecules with donors or accepted
electronic charge behavior adsorbed on graphene this contributing for optimizing and
developing potential application of this novel material.
Keywords: graphene, density functional theory, donor/acceptor

8
SUMÁRIO
1) INTRODUÇÃO............................................................................................................9
2) GRAFENO: PROPRIEDADES E APLICAÇÕES.................................................10
2.1) Carbono.....................................................................................................................10
2.2) Alótropos do carbono................................................................................................12
2.3) Obtenção do Grafeno................................................................................................15
2.4) Propriedades do Grafeno...........................................................................................17
2.5) Aplicações do grafeno...............................................................................................22
2.6) Moléculas: TiCl4 e TiCl3 ..........................................................................................23
3) METODOLOGIA .....................................................................................................25
3.1) Aproximação de Born-Oppenheimer........................................................................26
3.2) Teoria Funcional da Densidade.................................................................................27
3.2.1) Teoremas de Hohenberg e Kohn............................................................................28
3.2.2) Equações de Kohn-Sham.......................................................................................30
3.2.3) Termo de troca e correlação...................................................................................32
3.3) Pseudopotencial........................................................................................................33
3.3.1) Pseudopotencial Troullier-Martins........................................................................37
3.4) Orbitais Atômicos.....................................................................................................38
4) RESULTADOS..........................................................................................................41
4.1) Procedimentos de Cálculo.........................................................................................41
4.2) Grafeno.....................................................................................................................42
4.3) TiCl4 interagindo com uma monocamada de grafeno..............................................43
4.4) Interação de duas moléculas de TiCl4 com uma monocamada de grafeno..............47
4.5) Configuração TiCl4 confinada em uma bicamada de grafeno..................................49
4.6) Interação da molécula TiCl3 em uma monocamada de grafeno...............................50
5) CONCLUSÕES..........................................................................................................54
REFERÊNCIAS BIBLIOGRÁFICAS…………………………………… ………….56

9
1) INTRODUÇÃO
O carbono é um dos elementos mais intrigantes na tabela periódica. Forma muitos
alótropos, alguns conhecidos de tempos antigos (diamante e grafita) e outros descobertos
dez a vinte anos atrás (fulerenos, nanotubos). Uma das formas alotrópicas interessantes é a
estrutura bidimensional, grafeno, que só foi obtido muito recentemente e, imediatamente,
atraiu a atenção dos pesquisadores da área de Física da Matéria Condensada. Elétrons no
grafeno obedecem uma relação de dispersão linear, se comportando como se fossem
partículas relativísticas sem massa, que resultam em um número de propriedades
eletrônicas muito intrigantes, observadas neste material bidimensional tais como, o efeito
Hall quântico à temperatura ambiente. O grafeno também prevê uma ponte entre físicas da
matéria condensada e eletrodinâmica quântica abrindo, desta forma, novas perspectivas
para uma eletrônica baseada no carbono.
O grafeno é formado por uma única camada de átomos de carbono dispostos na
forma de uma rede de hexágonos e exibe alta qualidade cristalina e melhor o transporte
elétrico balístico em baixas temperaturas (NOVOSELOV et al, 2005). Além disto, possui
uma alta área superficial, sendo que cada átomo de carbono pode ser um centro de
interação.
Neste trabalho avaliou-se a interação das moléculas TiCl4 e TiCl3 com planos de
grafeno organizados em uma ou duas camadas. O objetivo é avaliar as propriedades de
transferência de carga das moléculas e do grafeno fazendo uma conexão com os resultados
da literatura, teóricos e experimentais, tendo em vista o potencial para aplicação como
sensores químicos. Da mesma forma, as propriedades eletrônicas também são sensíveis às
interações químicas resultantes da transferência de carga, podendo gerar aplicações
inovadoras para sistemas híbridos a base de grafeno. Para este estudo será empregado o
método ab inito, utilizando a Teoria da Densidade Funcional implementada no código
computacional SIESTA. Com os resultados, as propriedades estruturais, eletrônicas e
magnéticas dos sistemas resultantes serão analisadas.

10
2) GRAFENO: PROPRIEDADES E APLICAÇÕES
2.1) Carbono
O átomo de carbono pode estar presente tanto em materiais inorgânicos como
orgânicos devido à sua versatilidade. O elemento carbono, pertence ao grupo 4A na tabela
periódica, ao lado do qual se encontram o boro e o nitrogênio. O átomo de carbono possui 6
elétrons (Z = 6) e a estrutura eletrônica do estado fundamental é 1s2 2s2 2p2. No orbital 1s2
encontramos os elétrons fortemente ligados, chamados elétrons de caroço. Os 4 elétrons
restantes ocupam os orbitais atômicos 2s2 2p2 e são denominados elétrons de valência.
Devido à diferença de energia entre os níveis 2s e 2p ser pequena, se comparada à energia
das ligações químicas, esses podem facilmente se combinar. Esta combinação é chamada
hibridização spn dos orbitais do átomo de carbono. Existem três possíveis hibridizações no
caso do carbono: sp , sp2 e sp3.
Hibridização sp: Um exemplo de material formado por carbono que apresenta a
hibridização sp é o acetileno (C2H2). Esta é uma molécula linear, cuja tripla ligação
corresponde a uma ligação σ ao longo do plano da molécula e duas ligações π
perpendiculares ao plano da molécula e formadas pelos orbitais py e pz (Figura 2.1).
Figura 2.1: Diagrama esquemático dos orbitais híbridos sp.
Hibridização sp2: O etileno (H2C = CH2) apresenta a hibridização sp2. A dupla
ligação é formada por uma ligação σ ao longo do plano xy da molécula mais um orbital π
para cada átomo de carbono perpendicular ao plano xy (Figura 2.2).

11
Figura 2.2: Diagrama esquemático dos orbitais híbridos sp2.
Hibridização sp3: A molécula de metano (CH4) é um exemplo da hibridização sp3.
Nesse caso, temos quatro orbitais σ ao longo das ligações entre o carbono e os hidrogênios.
Essa molécula apresenta-se espacialmente numa estrutura tetragonal estando o átomo de
carbono ao centro (Figura 2.3).
Figura 2.3: Diagrama esquemático dos orbitais híbridos sp3.
Essa disponibilidade de ligações também possibilita a combinação dos átomos de
carbono com outros elementos multiplicando, ainda mais, o número de materiais com as
mais diversas propriedades físico-químicas. Além do acetileno, etileno e metano, o carbono
pode constituir estruturas ainda mais complexas como: grafite, diamante, fulerenos,
nanotubos de carbono, entre outros. Nesse Capítulo, nosso interesse se concentra no
grafeno, que apresenta ligações com hibridizações sp2. A seguir, descrevemos brevemente
as propriedades dos alótropos do carbono.

12
2.2) Alótropos do Carbono
O carbono é a base da química orgânica. Devido à flexibilidade de suas ligações
químicas os sistemas à base de carbono mostram um ilimitado número de estruturas com
uma larga variedade de propriedades físicas e químicas. Essas propriedades são, em grande
parte, a definição da dimensionalidade dessas estruturas. São conhecidas quatro formas
alotrópicas do carbono, além da amorfa, que são: grafite, nanotubos, diamante e fulerenos,
como mostram a Figura 2.4 (a), (b), (c) e (d), respectivamente.
Figura 2.4: Representação das diferentes formas do carbono: (a) grafite; (b) nanotubo de carbono; (c) diamante e (d) fulereno.
Em 1985 observou-se um novo alótropo do carbono, o fulereno, que foi descoberto
pelo radioastrônomo Harold Kroto e seus colaboradores (KROTO et al, 1985). Por esta
descoberta, Kroto e seus colaboradores, foram agraciados com o prêmio Nobel de química
de 1996. Os fulerenos são uma das formas alotrópicas mais estáveis do carbono, tendo uma
estrutura formada de hexágonos e pentágonos curvada em planos permitindo formas
esférica, elipsoidal e cilíndrica. São estruturas tridimensionais similares a uma bola de
futebol, com 12 pentágonos e um número arbitrário de hexágonos. Os carbonos

13
apresentam, nestas estruturas, hibridizações do tipo sp2 formando ligações do tipo σ com os
três átomos de carbono mais próximos. O fulereno forma um sistema zero-dimensional
(0D), com relação ao confinamento, com alta simetria, assim como a esfera, podendo ser
considerado como a molécula de maior simetria. Seu estudo é intensificado para utilização
em dispositivos eletrônicos, sensores químicos e aplicações biológicas (DRESSELHAUS et
al, 1996).
Os nanotubos de carbono (Figura 2.4(b)) foram observados pela primeira vez em
1991 por Sumio Iijima no Japão (IIJIMA, 1991). Este pesquisador observou a existência de
materiais formados de múltiplas camadas de folhas de grafeno enroladas de forma
cilíndrica, através de microscopia eletrônica de transmissão. Estes novos alótropos do
carbono foram chamados de nanotubos de carbono devido à forma tubular apresentada. Em
1993 também foram obtidos nanotubos de carbono de uma única camada (BETHUNE et al,
1993; IIJIMA, 1993), abrindo a possibilidade de comparações entre propriedades previstas
teoricamente e os resultados experimentais. Os nanotubos de carbono abriram novas
perspectivas para inovações na indústria eletrônica, tornando possível uma eletrônica
baseada no carbono. Atualmente os nanotubos de carbono também têm sido alvos de
inúmeros estudos para aplicação biológica a partir da veiculação de fármacos ou terapias de
doenças como o câncer.
Já o diamante (Figura 2.4 (c)) apresenta hibridizações do tipo sp3, sendo que cada
carbono forma quatro ligações covalentes, originando uma das estruturas mais rígidas
encontradas na natureza. O diamante é também um material isolante e transparente.
A grafite (Figura 2.4 (a)) é uma das formas alotrópicas do carbono que apresenta
maior estabilidade. A estrutura cristalina é formada de planos de redes hexagonais
(grafeno) empilhados. Um único plano de carbono é chamado de grafeno de camada
simples, sendo que, cada átomo de carbono, neste plano, se liga a outros três átomos
vizinhos a ele através de ligações com hibridização sp2, formando ligações fortes do tipo σ
de comprimento aproximado de 1,42 Å. Entre os planos, há ligações fracas do tipo π, de
comprimento 3,35 Å. As camadas que compõem a grafite são ligadas por forças de van der
Walls, responsáveis pela fragilidade deste. No caso do grafeno, o limite para que seja um
sistema bi-dimensional é o número de 10 camadas (PARTOENS et al, 2006).
O grafeno foi manipulado e estudado no laboratório apenas em 2004

14
(NOVOSELOV et al, 2004). O grafeno faz parte da estrutura bidimensional do grafite, com
hibridização sp2. Devido a sua dimensão (2D), apresenta extraordinárias propriedades
físicas e químicas além de ser abundante na natureza e de baixo custo. Há cerca de 70 anos,
Landau e Peierls argumentaram que cristais 2D eram termodinamicamente instáveis e não
poderiam existir (PEIERLS et al, 1935; LANDAU et al, 1937). O argumento foi estendido
por (MERMIN et al, 1968) e era fortemente suportado por uma quantidade de observações
experimentais. Sabe-se que a temperatura de fusão dos filmes finos decresce rapidamente
com a espessura, tornando os filmes instáveis (VENABLES et al, 1984; EVANS et al,
2006). Desta forma, o grafeno era conhecido como parte integrante do grafite, sendo
impossível em estruturas bidimensionais. Mas, em 2004, um grupo de pesquisadores
liderados por Geim conseguiu isolar em laboratório uma única camada de grafeno
(NOVOSELOV et al, 2004), gerando novas perspectivas para uma nova tecnologia baseada
em estruturas bidimensionais de carbono.
De forma resumida, a Tabela 2.1 apresenta as principais propriedades dos alótropos
do carbono distribuídos em estruturas com dimensão zero (0-D), unidimensional (1-D),
bidimensional (2-D) e tridimensional (3-D).
Tabela 2.1: Exemplos de estruturas alotrópicas do carbono e suas propriedades com diferentes dimensionalidades (SAITO et al, 1998). Dimensão 0D 1D 2D 3D
Isômero C60
fulerenos
Nanotubo,
carbina
Grafite,
grafeno, fibra
Diamante,
C-amorfo
Hibridização sp2 sp2(sp) sp2 sp3
Densidade
(g/cm3)
1,72 1,20-2,00
2,68-3,13
2,26
~2,00
3,52
2-3
Comprimento
da ligação (Å)
1,40 (C=C)
1,46 (C-C)
1,44 (C=C) 1,42 (C=C)
1,44 (C=C)
1,52 (C-C)
Propriedades
eletrônicas
semicondutor
Egap=1,90 eV
metal ou
semicondutor
Egap~variável
semimetal
Efeito Hall
Quântico
isolante
Egap=5,47 eV
Na próxima seção apresentamos as principais formas de obtenção do grafeno a

15
partir do grafite.
2.3) Obtenção do Grafeno
Em 2004 a equipe de Geim preparou filmes de grafeno por meio de esfoliação
mecânica, obtendo de forma termodinamicamente estável, as primeiras estruturas isoladas
bidimensionais de carbono (NOVOZELOV et al, 2005). Inicialmente, eles utilizaram uma
plaqueta de 1mm de espessura de HOPG (Highly Ordered Pyrolytic Graphite). Usando a
técnica de gravura seca com plasma de oxigênio produziram blocos de grafite com 5µm de
profundidade no topo da plaqueta (mesas quadradas de comprimentos variando de 20µm a
2mm). A superfície estruturada foi, então, prensada contra um fotoresistor disposto sobre
um substrato de vidro. Após a secagem, as bases ficam presas a camada de fotoresistor,
possibilitando a sua clivagem do resto da amostra de HOPG. Com uma fita adesiva, os
pesquisadores iniciaram uma esfoliação repetida de fragmentos de grafite dessas amostras.
Os finos fragmentos, que restaram no fotoresistor foram colocados em acetona. Quando um
wafer (amostra) de Si foi mergulhado na solução e então lavado com água e propanol,
alguns fragmentos foram capturados na superfície do substrato. Após isto, fez-se uma
limpeza, eliminando a maioria dos fragmentos mais espessos. Os fragmentos mais delgados
(d < 10nm) foram encontrados adsorvidos no SiO2, devido a forças de van der Waals.
Para avaliar a espessura do grafeno, os pesquisadores usaram uma combinação de
microscópios óticos, de feixe eletrônico e de força atômica (Figura 2.5). Filmes grafíticos
com espessura menor que 50nm são transparentes à luz visível, entretanto, sobre a
superfície de SiO2 eles são, facilmente, visíveis devido à adição de caminho ótico que altera
a cor da interface. O SiO2, com uma espessura de 300nm, tem cor violeta, a espessura
extra-proveniente do filme delgado de grafite- provoca sua mudança para o azul. À medida
que a espessura da folha de grafeno tende para uma única camada, ou seja, quando se têm
filmes de grafeno com poucas camadas, ela vai ficando invisível para o microscópio ótico.

16
Figura 2.5: Filmes de grafeno. (A) Espectroscopia óptica de uma lâmina de
múltiplas camadas de grafeno com espessura 3 nm no topo do óxido de Si. (B) Imagem de um microscópio de força atômica [AFM] da amostra próxima à borda. A cor marrom
escura [laranja] é a superfície do SiO2 [3 nm acima do óxido]. (C) imagem de AFM de uma única camada de grafeno. A cor marrom escura é a superfície de SiO2.(D) Imagem de
microscópio eletrônico de varredura de um dispositivo experimental com filme de grafeno de poucas camadas (E) Figura esquemática do dispositivo (D) (NOVOSELOV et al, 2004).
Embora folhas de grafeno com espessuras menores que nm.d 51≈ não sejam
visíveis através de microscópio ótico podem ser vistas, claramente, por microscópios de
alta resolução de varredura SEM (Scanning Electron Microscopy). A Figura 2.6 mostra
com clareza a imagem da folha de grafeno em microscopia atômica e microscopia
eletrônica de varredura.
A forma mais fácil de isolar o grafeno é por meio de esfoliação química. Para isto, a
amostra de grafite é inicialmente intercalada, de forma que as folhas de grafeno ficam
separadas por uma camada de átomos ou moléculas. Esse procedimento, em geral, cria um
novo composto tridimensional (3D). Mas, em alguns casos, é possível introduzir moléculas
grandes de forma a forçar espaçamentos entre os planos de grafeno, resultando, assim, em
grafeno isolado envolvido numa matriz em três dimensões. O inconveniente desta técnica é
que é comum, por meio de uma reação química, a intercalação ser desfeita formando folhas
de grafeno enroladas e re-empilhadas (DRESSELHAUS et al, 2002).

17
Figura 2.6: a) Grafeno visualizado através de microscopia de força atômica. A região exibe uma altura relativa de 4 Å, claramente indica que é uma única camada. b) A
imagem de microscopia de transmissão eletrônica de uma folha de grafeno livremente suspenso em um metal (GEIM et al, 2007).
Para obtenção de melhor qualidade, os pesquisadores têm usado amostras obtidas
pela técnica de clivagem micromecânica, a mesma usada no primeiro isolamento do
grafeno (NOVOSELOV et al, 2004). Após um refinamento da técnica, consegue-se
produzir cristais de grafeno de alta qualidade com mais de 100 µm, sendo suficiente para a
maioria das pesquisas propostas.
Atualmente, diversas técnicas têm sido desenvolvidas e aprimoradas para obtenção
de grafeno de camadas isoladas, buscando uma qualidade cada vez melhor para aplicação
no desenvolvimento de novas tecnologias (GEIM et al, 2007).
2.4) Propriedades do Grafeno
Grafeno é o nome dado a uma monocamada planar de átomos de carbono dispostos
em uma rede bidimensional (2D) hexagonal e, é base para estruturas grafíticas de outras
dimensões, como mostra a Figura 2.7.

18
Figura 2.7: Plano de grafeno originando diferentes estruturas alotrópicas do carbono (GEIM et al, 2007).
O grafeno é formado por átomos de carbono organizados em uma estrutura
hexagonal (Figura 2.7). A estrutura atômica do grafeno não é uma rede de bravais, mas
pode ser visto como uma rede triangular com uma base de dois átomos por célula unitária.
Os vetores da rede direta do grafeno podem ser escritos como:
( )3,321
aa =r
, ( )3,322 −= a
ar
, (2.1)
onde a = 2,40 Å é o parâmetro de rede da estrutura hexagonal. A rede recíproca é dada
pelos vetores:
( )3,13
21 a
bπ=
r, ( )3,1
3
22 −=
ab
πr. (2.2)
A Figura 2.8 mostra a estrutura hexagonal na rede direta e recíproca com os vetores
correspondentes.

19
Figura 2.8: À esquerda, estrutura de grafeno feito de duas redes triangulares (ar
1 e ar
2 são os vetores unitários da rede). À direita, corresponde a zona de Brillouin (CASTRO et al, 2007).
Devido a simetria da estrutura hexagonal do grafeno, alguns pontos de simetria da
zona de Brillouin são especiais na descrição das propriedades eletrônicas deste material,
como os pontos Γ, K, K´ e M. Os pontos K e K´ também são chamados de pontos de Dirac
e suas posições são dadas por:
=aa
K33
2,
3
2 ππ,
−=aa
K33
2,
3
2'
ππ. (2.3)
Fazendo uso de um modelo de tight-binding para o grafeno e considerando no
Hamiltoniano que esses elétrons possuem interação somente com seus primeiros e
segundos vizinhos, temos:
( ) ( )∑∑ ++−+−= ⊥⊥⊥
σσσσσ
σσσ
,,,,,,
,,,, .'.
jijiji
jiji chbbaatchbatH , (2.4)
onde ( )⊥σσ ,, ii aa destrói (cria) elétrons com spin ( )↓=↑,σσ no sítio
iR da sub-rede A. Uma
definição equivalente é usada na sub-rede B. t = 2,8 eV e t′=0,1 eV são as energias para os
elétrons saltarem para os primeiros e segundos vizinhos, respectivamente (REICH et al,
2002). As bandas de energia deste Hamiltoniano devem ter a forma (WALLACE, 1947):

20
( ) ( ) ( )kf'tkftkErrr
−+±=± 3 , (2.5)
( ) ( )
+= akcosakcosakcoskf
xyy 2
3
2
3432
r, (2.6)
onde o sinal positivo representa a banda de energia superior (π) ou banda de condução e o
sinal negativo, a banda inferior (π*) ou banda de valência.
A Figura 2.9 apresenta o espectro de energia para toda a zona de Brillouin calculada
usando a equação (2.5). A estrutura de bandas próxima aos pontos de Dirac tem uma
dispersão cônica:
( ) ( )2qOqvqEF
+±≈±
rr, (2.7)
onde qr
é o momento medido a partir dos pontos de Dirac e F
ν é a velocidade de Fermi
dada por 23 /taF
=ν , com um valor por s/mxF
6101≅ν (WALLACE, 1947).
Figura 2.9: À esquerda, bandas de energia de valores finitos de t e t’ , com eVt 7,2= e tt 2,0'= . À direita, ampliação em um dos pontos de Dirac (CASTRO et al,
2007).
A dispersão linear das bandas de energia próximo do ponto K a partir da equação
(2.7) tem semelhança com a energia de partículas sem massa chamadas de ultra-
relativísticas; a energia destas partículas é descrita quanticamente pela equação de Dirac.
Uma conseqüência imediata destas partículas de Dirac é a dispersão cíclotron de massa,
(Figura 2.10) que depende da densidade eletrônica como uma raiz quadrada
(NOVOSELOV et al, 2005).
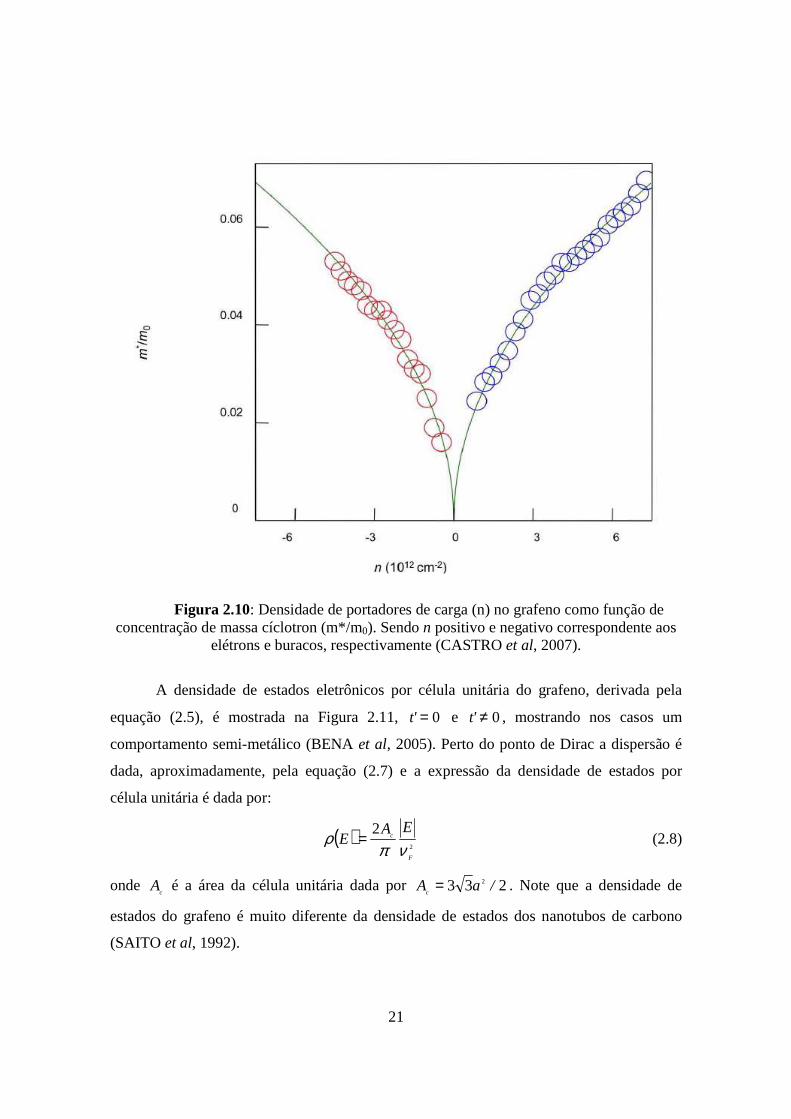
21
Figura 2.10: Densidade de portadores de carga (n) no grafeno como função de concentração de massa cíclotron (m*/m0). Sendo n positivo e negativo correspondente aos
elétrons e buracos, respectivamente (CASTRO et al, 2007).
A densidade de estados eletrônicos por célula unitária do grafeno, derivada pela
equação (2.5), é mostrada na Figura 2.11, 0='t e 0≠'t , mostrando nos casos um
comportamento semi-metálico (BENA et al, 2005). Perto do ponto de Dirac a dispersão é
dada, aproximadamente, pela equação (2.7) e a expressão da densidade de estados por
célula unitária é dada por:
( )2
2
F
cEA
Eνπ
ρ = (2.8)
onde c
A é a área da célula unitária dada por 233 2 /aAc
= . Note que a densidade de
estados do grafeno é muito diferente da densidade de estados dos nanotubos de carbono
(SAITO et al, 1992).
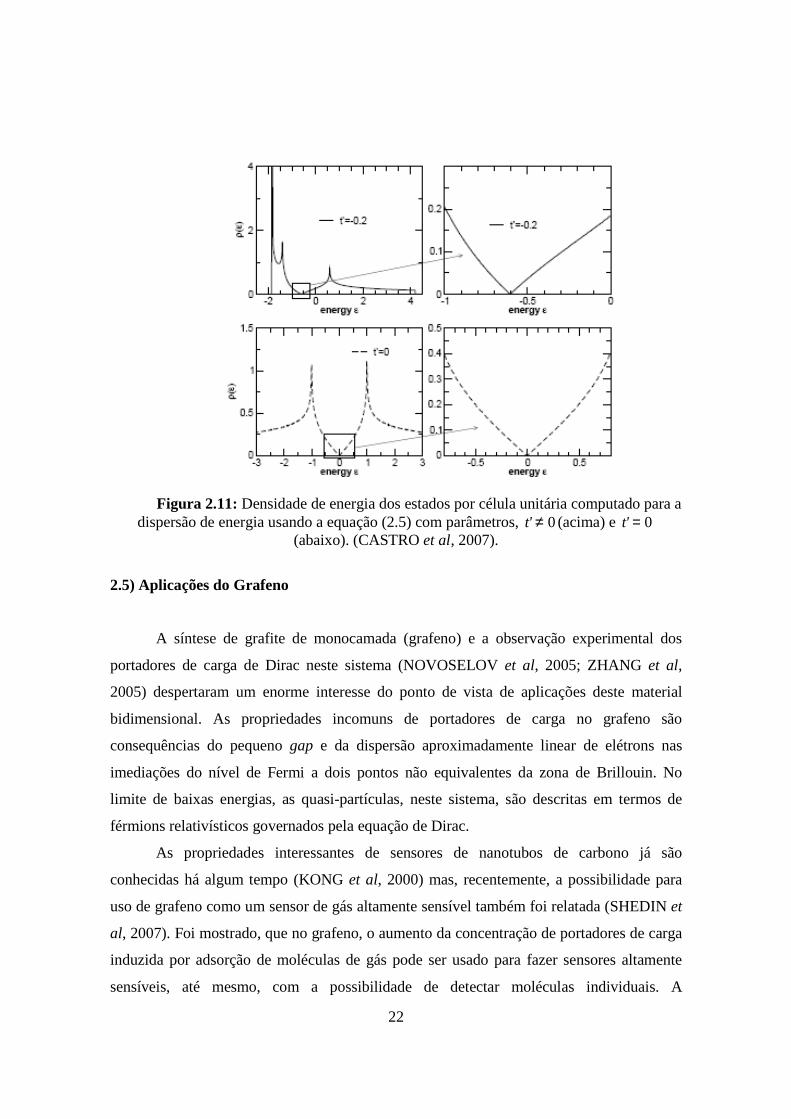
22
Figura 2.11: Densidade de energia dos estados por célula unitária computado para a dispersão de energia usando a equação (2.5) com parâmetros, 0≠'t (acima) e 0='t
(abaixo). (CASTRO et al, 2007).
2.5) Aplicações do Grafeno
A síntese de grafite de monocamada (grafeno) e a observação experimental dos
portadores de carga de Dirac neste sistema (NOVOSELOV et al, 2005; ZHANG et al,
2005) despertaram um enorme interesse do ponto de vista de aplicações deste material
bidimensional. As propriedades incomuns de portadores de carga no grafeno são
consequências do pequeno gap e da dispersão aproximadamente linear de elétrons nas
imediações do nível de Fermi a dois pontos não equivalentes da zona de Brillouin. No
limite de baixas energias, as quasi-partículas, neste sistema, são descritas em termos de
férmions relativísticos governados pela equação de Dirac.
As propriedades interessantes de sensores de nanotubos de carbono já são
conhecidas há algum tempo (KONG et al, 2000) mas, recentemente, a possibilidade para
uso de grafeno como um sensor de gás altamente sensível também foi relatada (SHEDIN et
al, 2007). Foi mostrado, que no grafeno, o aumento da concentração de portadores de carga
induzida por adsorção de moléculas de gás pode ser usado para fazer sensores altamente
sensíveis, até mesmo, com a possibilidade de detectar moléculas individuais. A

23
sensibilidade está baseada em mudanças na resistividade Hall, devido às moléculas
adsorvidas na folha de grafeno, que atuam como doadores ou aceitadores de carga.
Outra possibilidade, seria o uso do pó do grafeno em baterias elétricas, que já é um
dos principais mercados para o grafite. A razão de superfície por volume e a alta
condutividade do grafeno podem conduzir para significantes melhorias na eficiência de
baterias. Os nanotubos de carbono foram cogitados para aplicação em baterias, mas a
produção de pó de grafeno é muito mais barata (STANKOVICH et al, 2006).
Desta forma, torna-se essencial o conhecimento das propriedades resultantes do
grafeno com moléculas doadoras ou aceitadoras de carga, tornando o grafeno com
propriedades, ainda mais, interessantes do ponto de vista de aplicações. Neste estudo,
especificamente, avaliaremos as propriedades do grafeno quando interagindo com
moléculas como TiCl3 e TiCl4, as quais descreveremos suas propriedades quando isoladas,
na seção a seguir. Estas moléculas têm um interesse especial devido ao comportamento
como ácido ou base de Lewis na presença de compostos de carbono, como o grafite.
2.6) Moléculas: TiCl4 e TiCl3
O tetracloreto de titânio (TiCl4) é um líquido denso e tóxico, solúvel em água e com
um ponto de ebulição aos 136°C. É usado para obtenção de titanatos, sal de titânio para
tintura, catalisador de polimerização e, é um importante intermediário na produção de
titânio metálico e outras combinações. O TiCl4 também é conhecido como cloreto de titânio
(HOLLEMAM et al, 2001). Ele não é um usual exemplo de líquido-metal-haleto, pois é
muito volátil no ar, onde forma uma espetacular nuvem opaca de dióxido de titânio (TiO2) e
ácido clorídrico (HCl) (GREENWOOD et al, 1997). É um dos raros metais de transição
clorados que está em estado líquido a baixa temperatura. Esta propriedade, surge do fato
que TiCl4 é molecular; ou seja, cada molécula de TiCl4 interage fracamente com as
moléculas vizinhas (GREENWOOD et al, 1997). O TiCl4 adota estruturas semelhantes a
TiBr4 e TiI4; (WEBB et al, 1999).
Da mesma forma, o TiCl4 é considerado um versátil ácido de Lewis, com íons Cl-
doadores de carga sendo que o TiCl4 reage de forma consecutiva em espécies [Ti2Cl9]-,
[Ti2Cl10]2- e [TiCl6]
2- (CALDERAZZO et al, 1996).

24
O tricloreto de titânio (TiCl3) é um sal hidratado, solúvel em água, ponto de fusão
425°C, corrosivo e comporta-se como base de Lewis. É um dos mais comuns haletos de
titânio e é um importante catalisador na fabricação de poleofinas. O TiCl3 sólido pode ser
encontrado quatro formas ou polimorfes (STAR et al, 1968).
O TiCl4 pode ser reduzido a TiCl3 usando rota eletroquimicamente. O TiCl3 é um
catalisador, embora a atividade catalítica varie com o método de preparação (UENO et al,
2000). As Figuras 2.12 (a) e (b) apresentam a estrutura química com as distâncias de
ligação e ângulos entre os átomos das moléculas TiCl4 e TiCl3, respectivamente. O ângulo
entre os átomos de cloro de TiCl4 é igual a 109,4° e 119,83° para os de TiCl3.
Figura 2.12: Estrutura molecular das moléculas (a) TiCl4 – estrutura tetragonal (b)
TiCl3 – estrutura planar.

25
3) METODOLOGIA
Simulações não relativísticas das propriedades eletrônicas e estruturais de sistemas
com milhares de átomos tornaram-se rotina nos últimos anos em vários laboratórios. Tais
sistemas requerem uma solução precisa da equação de Schrödinger, que em si é facilmente
construída para um sistema de muitos corpos (FAGAN, 2003). Porém, é impossível
resolvê-la diretamente além dos sistemas mais simples sem fazer algumas aproximações,
devido às limitações metodológicas. Algumas destas aproximações, que são utilizadas no
programa SIESTA (Spanish Initiative for Electronic Simulations with Thousand of Atoms)
(SOLER et al, 2002) para a realização dos cálculos desta Dissertação e serão brevemente
discutidas ao longo deste Capítulo.
A teoria estabelecida a partir da mecânica quântica ressalta que a equação de
Schrödinger de um sistema de partículas (ℓ elétrons e m núcleos) a ser resolvida tem a
forma:
( ) ( )RrERrRrHrrrrrr
,,),(ˆ Φ=Φ (3.1)
onde H)
é o operador Hamiltoniano total não relativístico, ( )rrΦ é a função de estado do
sistema, e rr
e Rr
são, respectivamente, as coordenadas dos elétrons e dos núcleos. O
Hamiltoniano, no sistema de unidades atômicas (au) (sistema utilizado em toda a
dissertação), é dado por:
( ) ( ) ( ) ( )rVR,rVRVRT)r(THeeNeNNNe
rrrrrr ++++= , (3.2)
onde e
T)
é o operador energia cinética eletrônica, N
T)
é o operador energia cinética nuclear,
NNV)
o operador energia potencial nuclear, ee
V)
é o operador energia potencial eletrônico e
NeV)
é o operador referente à interação elétron-núcleo. A resolução da equação (3.1)
depende diretamente do número de partículas envolvidas, pois matematicamente sistemas a
partir do átomo de hidrogênio não possuem solução analítica exata. As aproximações
tornam-se essenciais para a descrição das propriedades físicas dos sistemas. A seguir

26
apresentamos a aproximação de Born-Oppenheimer que reduz, consideravelmente, o
número de partículas envolvidas no cálculo das propriedades do sistema.
3.1) Aproximação de Born-Oppenheimer
Sabe-se que a massa nuclear é bem maior que a massa dos elétrons, sendo assim, a
velocidade dos elétrons é muito maior que a dos núcleos. Então, para qualquer movimento
dos núcleos, há uma reação quase que instantânea dos elétrons.
A aproximação de Born-Oppenheimer (BO) desacopla o movimento dos elétrons e
núcleos, ou seja, o movimento dos núcleos é tratado de maneira clássica, supondo os
núcleos estejam em posições fixas, reduzindo o problema de muitos corpos resolvendo de
forma quântica a dinâmica dos elétrons acoplada ao movimento nuclear.
Essa aproximação é introduzida escrevendo a função de onda total do sistema como
( ) ( ) ( )RR,rR,rrrrrr
ΘΨ=Φ (3.3)
onde ( )Rr
Θ é a função de onda nuclear e ( )R,rrr
Ψ é a função de onda eletrônica com R
apenas como parâmetro.
Com a aproximação BO, a equação de Schrödinger (3.1) pode ser reescrita como:
( ) ( ) ( ) ( ) ( )( ) ( ) ( )RrERrRrVrVrTRVRrH ieNeeeeNNie
rrrrrrrrrrr.,,ˆˆˆˆ,ˆ Ψ=Ψ+++=Ψ ++ (3.4)
e
( )[ ] ( ) ( )RERrET ieN
rrr Θ=Θ+ +ˆˆ (3.5)
A energia ieE + é a energia de BO do sistema. A segunda equação é resolvida
utilizando algoritmos clássicos de integração e equações de Newton. Portanto, a energia
total do sistema pode ser escrita como:

27
( ) ( )RTREEiie
rr+= + (3.6)
(3.7)
onde ( )RTe
r é a energia cinética dos elétrons, ( )REee
ré a energia de interação elétron-elétron,
( )REie
r é a energia de interação elétron-íon, ( )REii
r é a energia de interação íon-íon (fixas
para as posições atômicas dadas) e ( )RTi
r é a energia cinética dos íons.
A resolução exata da equação (3.1) depende da solução de várias equações
matemáticas que são inexistentes para sistemas de muitos corpos e que geram a necessidade
da inclusão de aproximações, como veremos a seguir.
3.2) Teoria do Funcional da Densidade (DFT)
O método DFT tem sido considerado, na física do estado sólido, como uma
ferramenta bem sucedida entre os métodos ab initio (primeiros princípios) devido à alta
eficiência computacional e aos bons resultados fornecidos (KOHN, 1999).
A partir da mecânica quântica sabemos que a função de onda total, que depende das
coordenadas dos N elétrons, é o objeto fundamental de estudo e pode gerar todos os
observáveis do sistema. Existe outra maneira de resolver o problema, em que o objeto
fundamental é a densidade eletrônica total ( )rnr
(THOMAS, 1927; FERMI, 1928), ou seja, a
grandeza fundamental não é a função de onda e sim a densidade. A equação de n elétrons
com a função de onda de 3N variáveis (se não considerarmos o spin) pode ser escrita como
uma equação da densidade eletrônica com somente três variáveis.
Thomas (1927) e Fermi (1928) (TF), independentemente, desenvolveram um
procedimento simplificado para determinar densidade eletrônica do sistema quando o
potencial varia suavemente com rr
. Baseando-se nas seguintes suposições:
(a) Desprezavam-se correções relativísticas;
(b) No átomo havia um campo efetivo dado por um potencial ν , dependendo
somente da distância r dos núcleos, tal que:
0→ν quando ∞→rr
( ) ( ) ( ) ( ) ( ),RTRERERERTiiiieeee
rrrrr++++=

28
Zer →ν (carga nuclear) quando 0→rr
;
(c) Os elétrons estariam distribuídos uniformemente num espaço de fase de
dimensão 6, ou seja, faz-se uso da densidade uniforme de elétrons;
(d) O potencial ν é por si mesmo determinado pela carga nuclear e sua
distribuição eletrônica.
A aproximação de TF apresenta um sistema onde a densidade eletrônica é uma
grandeza a ser calculada. Esta densidade é calculada para um gás de elétrons livres e não
interagentes. TF não leva em conta cálculos variacionais, bem como não trata o termo de
correlação eletrônica, sendo que a parte de exchange (troca) foi adicionada à teoria de TF
por Dirac em 1931. Mesmo a adição do exchange, a teoria não é adequada para cálculos de
energia total, pois desconsidera o uso da autoconsistência no cálculo da energia total.
3.2.1) Teoremas de Hohenberg e Kohn
A DFT esta baseada em dois teoremas propostos em 1964 por Hohenberg e Kohn
(HK) (HOHENBERG et al, 1964). Neste caso, podemos considerar um sistema com N
partículas e escrevemos o Hamiltoniano na forma geral:
( )ij
N
jiiext
N
i
iN
i rrVH
1
2 1
2
1∑∑∑
<==
++
∇−= r
. (3.8)
Deste Hamiltoniano concluímos que N e )(rVext
r determinam todas as propriedades
do sistema para o estado fundamental, onde N (número de partículas) e a densidade ( )rnr
estão relacionadas através da condição de normalização:
∫= rdrnN 3)(r
. (3.9)
Ao invés de usar N (elétrons) e )(rVext
r, o primeiro teorema de HK impõe o uso da
densidade ( )rnr
como uma variável básica. Temos então:
Teorema 1:

29
A densidade de carga do estado fundamental ( )rnr
com o potencial )(rVext
r é
determinada de modo unívoco, a menos de uma constante aditiva, a partir do potencial
externo )(rVext
r.
Corolário 1: Uma vez que o Hamiltoniano é inteiramente determinado, exceto pela
variação constante na energia, a densidade eletrônica de muitos corpos para todos os
estados são determinadas. Consequentemente, todas as propriedades do sistema são
completamente determinadas, dada somente a densidade do estado fundamental.
Portanto, o funcional de energia para um dado ( )rν é definido por:
[ ] ( ) ( ) [ ]∫ += nFrdrnrnErrr 3ν , (3.10)
onde [ ]nF é um funcional universal, é dado por:
[ ] [ ] [ ] [ ]nEnVnTnFxcHe
++= , (3.11)
onde [ ]nTe
representa a energia cinética de um sistema de partículas não interagentes,
[ ]nVH
é o potencial de interação elétron-elétron (potencial de Hartree) e [ ]nExc
inclui todas
as correlações, ou seja, efeitos não clássicos de troca e correlação (termo ausente na
formulação de TF).
O segundo teorema de HK baseia-se no princípio variacional da energia, sendo
escrito como:
Teorema 2:
O valor mínimo do funcional da energia ][nE é a energia do estado fundamental e a
densidade com a qual se obtém esse mínimo é a densidade exata de uma partícula no
estado fundamental.
Corolário 2: O funcional ][nE sozinho é suficiente para determinar a exata energia e
densidade do estado fundamental. Geralmente, estados excitados de elétrons devem ser
determinados por outros meios (MARTIN, 2004).
Ressalta-se que esses dois teoremas nos garantem encontrar a densidade eletrônica
do sistema no estado fundamental resolvendo-se as equações de Kohn-Sham, as quais serão

30
descritas a seguir.
3.2.2 Equações de Kohn-Sham
Pelo fato das interações Coulombianas serem de longo alcance, é conveniente
separar do funcional universal [ ]nF a parte clássica, escrevendo-o da seguinte forma:
( ) ( )∫∫ +
−= ]['
'
'
2
1][ 33 nGrrdd
rr
rnrnnF rr
rr
, (3.12)
tal que
[ ] ( ) ( ) ( ) ( )∫∫∫ +
−+= ]['
'
'
2
1 333 nGrrddrr
rnrnrdrnrnE rr
rrrrν ; (3.13)
onde ][nG também é um funcional universal. Em 1965, Kohn e Sham (KS) (KOHN et al,
1965) foram os primeiros a apresentar uma estratégia para o cálculo de estrutura eletrônica
de sistemas envolvendo muitas partículas com o uso de [ ]nE . O funcional ][nG pode ser
escrito na forma,
[ ] ][][0
nEnTnGxc
+≡ , (3.14)
onde ][0
nT é a energia cinética de um sistema de elétrons não interagentes com densidade
( )rnr
e ][nExc
contém a energia de troca (exchange) e energia de correlação do sistema
interagente com densidade ( )rnr
.
De acordo com o teorema variacional, tomando a variação de ][nExc
, com o vínculo
que a carga eletrônica total seja fixa temos,
( ) Nrdrn =∫3r
. (3.15)
Da condição de extremo, temos,
[ ] ( )[ ]( ) 03 =−µ−δ ∫ NrdrnnEr
, (3.16)
obtemos

31
( ) ( ) ( ) [ ] 0330 =
−+−
++∫ ∫ rdn'rd'rr
'rnr
n
Trn
xcµνν
δδδ r
rr
rrr
(3.17)
aqui [ ]nxc
ν é o potencial de troca-correlação, dado por
[ ]n
En xc
xc δδν = . (3.18)
Kohn e Sham (KOHN, 1999) propuseram uma prática para complementar as idéias
expostas nos teoremas de Hohenberg e Kohn. A idéia fundamental consiste em representar
a densidade de carga através da soma dos quadrados dos módulos das funções de onda de
uma partícula:
( ) ( ) 2
1rrn
m
ocup
m
rr ψ∑=
= (3.19)
onde ( )rrψ é o spin-orbital. Então, para o estado fundamental temos:
( ) ( ) ( ) ( )rrn,rV2
1rH
iiieff
2
iKS
rrrr ψεψψ =
+∇−= , (3.20)
onde
( ) ( ) ( )∫ +−
+= n'rd'rr
'rnr
xceffννν r
rr
rr 3 , (3.21)
deve-se lembrar que as funções ( )ri
rψ sejam ortonormais, ou seja, ijji
| δψψ >=< .
Utilizando esta condição de ortonormalidade das funções de onda, a densidade de
partículas obtidas no estado fundamental é exatamente a mesma que teríamos resolvendo
um problema de muitos corpos.
O potencial, ( )nxc
ν , de troca e correlação origina-se da seguinte derivação:
( ) [ ]( )rn
nEn,rV xc
xc rr
δδ
= . (3.22)
Através do esquema proposto por Kohn e Sham (KOHN et al, 1965), foi possível
demonstrar a possibilidade de representar o problema multieletrônico por um conjunto
equivalente de equações autoconsistentes de um elétron que se move sob ação de um
potencial efetivo produzido pelos outros elétrons. Conhecendo a energia de troca e
correlação de maneira exata, sua derivada funcional em relação à densidade, nos dará um
potencial que permite a inclusão de maneira exata os efeitos de troca e correlação.

32
3.2.3 Termo de troca e correlação
Mesmo que os teoremas de Hohenberg e Kohn mostrem que a energia total pode ser
escrita como um funcional único da densidade eletrônica do estado fundamental, há um
fator desconhecido, que é o funcional de energia de troca e correlação. Para contornar essa
dificuldade serão discutidos, a seguir, os dois tipos de aproximações mais utilizados na
literatura e utilizados neste trabalho.
- Aproximação da densidade local (LDA)
Nesta aproximação, a energia de troca e correlação para um sistema de gás de
elétrons homogêneo de densidade ( )rnr
no ponto rr
é assumida como sendo igual à energia
de troca-correlação de um gás de elétrons homogêneo com mesma densidade. Nesta
aproximação supõe-se que a densidade varia suavemente nas proximidades do ponto rr
. O
termo de troca e correlação é descrito como:
[ ] ( )( ) ( ) rdrnrnnExcxc
rrr∫= ε (3.23)
onde xc
ε é a energia de troca-correlação por partícula para um gás de elétrons homogêneo
de densidade ( )rnr
(KOHN et al, 1965).
A parte de troca (exchange) é dada em unidades atômicas por:
[ ]s
xx r
.n
4580−=ε , (3.24)
onde sr é o raio de uma esfera contendo um elétron,
Já a parte de correlação foi primeiramente estimada por E.P.Wigner em 1938:
[ ]87
440
.r
.n
s
c +−=ε . (3.25)
As estimativas mais recentes e confiáveis para a parte de correlação foram dadas por

33
D.M. Ceperley e Alder em1980 (CEPERLEY et al, 1980).
A aproximação da densidade local assume que o funcional da energia de troca e
correlação é puramente local, e para sistemas com densidade eletrônica uniforme, ela é
exata. Portanto, espera-se que ela descreva bem sistemas onde a densidade eletrônica varie
lentamente com a posição. Se a densidade eletrônica for fortemente não uniforme, a energia
de troca e correlação calculada através da densidade local não é uma boa aproximação.
- Aproximação de gradientes generalizados (GGA)
É um refinamento da aproximação LDA, muito utilizado no formalismo DFT para
expressar o funcional [ ]nExc
em termos do gradiente da densidade de carga, a qual é
conhecida como aproximação de gradientes generalizados (GGA). Esta aproximação pode
ser de dois tipos: semi-empíricos e não empíricos (ou ab initio), que satisfazem a vínculos
teóricos. A aproximação de Perdew, Burke e Ernzerhof (PERDEW et al, 1996) é uma das
mais utilizadas, sendo da classe dos não empíricos. Assim, descrevemos a energia de troca-
correlação na forma denominada GGA:
[ ] ( )( ) ( ) ( )( ) ( ) ...|. 22 +∇ε+ε= ∫∫ rdrnrnrdrnrnnExcxcxc
rrrrrr (3.26)
Empiricamente, são conhecidas determinadas tendências para LDA: subestima o
valor da constante de rede entre 1-3%, os bulk modulus são superestimados entre 8 e 18% e
não é capaz de reproduzir com precisão as energias de coesão devido ao erro cometido ao
calcular a energia por átomo como um sistema homogêneo. Tanto LDA e GGA subestimam
o tamanho do gap nos semicondutores e isolantes na ordem de 50%. Como citado
anteriormente, o GGA corrige alguns problemas em relação à LDA, mas isso não é válido
necessariamente para todos os sistemas (FUCHS et al, 1998).
3.3 Pseudopotencial
Os elétrons em um sólido dividem-se basicamente em dois tipos:

34
i) Os elétrons próximos aos núcleos atômicos que sentem um forte potencial
atrativo, tendo pouca participação nas ligações químicas. A região que é formada pelo
núcleo atômico e os elétrons mais internos denominados de caroço. A principal
contribuição das funções de onda dos elétrons do caroço para a ligação química é forçar as
funções de onda dos elétrons de valência a serem ortogonais aos auto-estados de caroço;
ii) Os elétrons de valência têm uma grande participação nas ligações químicas,
determinando, portanto a maior parte das propriedades físicas de um sólido ou de uma
molécula. Estes elétrons de valência sentem um potencial bem menos atrativo e seus
orbitais apresentam formas mais suaves.
Em estrutura eletrônica, a maioria dos pseudopotenciais são gerados a partir do
cálculo da função de onda atômica de todos os elétrons. Dentro do DFT isto é feito
assumindo a aproximação da esfera blindada e resolvendo, autoconsistentemente, a equação
radial de KS (KERKER et al, 1978).
( ) [ ] ( ) ( )rrRrrRr;nVrdr
dnnn
rrrlllll
ε=
−++−22
2
2
1
2
1 (3.27)
onde ln
ε é o autovalor da energia do elétron de valência com o número quântico orbital l ,
[ ]r;nVr
é o potencial auto-consistente de um elétron dado por:
[ ] [ ] [ ]nVnVr
Zr;nV
xcH++−=r
(3.28)
onde ( )rnr
é a densidade eletrônica para as funções de ondas ocupadas ( )rRnl
r, [ ]nV
xc é o
potencial de troca e correlação e [ ]nVH
o potencial de Hartree.
A maioria dos pseudopotenciais construídos satisfaz quatro condições gerais:
(i) as pseudofunções de onda de valência (PS) geradas usando-se o pseudopotencial
não devem conter nodos. Isto vem do fato de que desejamos construir pseudofunções de
onda mais suaves, sendo, portanto, a oscilação associada aos nodos, indesejável;
(ii) a pseudofunção de onda radial normalizada (PS) com momento angular l deve
ser igual para a função de onda radial normalizada para um cálculo envolvendo todos os
elétrons (AE) a partir do raio de corte:

35
( ) ( )rRrR AEps rrll
= para c
rr > (3.29)
(iii) a carga dentro do raio de corte deve ser igual para as duas funções de onda PS e
AE
( ) ( )∫∫ =cr
0
22AE22cr
0
ps rdrrRrdrrRrrrr
ll. (3.30)
(iv) Os autovalores de energia do pseudopotencial e de todos os elétrons de valência
devem ser iguais
AEps
llεε = . (3.31)
Se o pseudopotencial obedecer às quatro condições acima ele é chamado de
pseudopotencial de norma conservada e sua transferibilidade será assegurada (BACHELET
et al, 1982).
Assumindo a aproximação da esfera blindada e resolvendo a equação radial de
Schrödinger autoconsistentemente, o pseudopotencial é construído dentro da DFT.
( ) [ ] ( ) ( )rrRrrRr;nVrdr
dnnnl
rrrllll
ε=
−++−22
2
2
1
2
1, (3.32)
onde [ ]r;nVr
é o potencial autoconsistente de um elétron dado por:
[ ] [ ] [ ]nVnVr
Zr;nV
xcH++−=r
(3.33)
onde ( )rnr
é a densidade eletrônica para as funções de ondas ocupadas ( )rRn
rl
.
Invertendo-se a pseudofunção de onda equação radial de Schrödinger (3.32),
obtemos o pseudopotencial blindado:

36
( ) ( )( ) ( )[ ]rrR
dr
d
rrRrrV ps
ps
ps
,scr
rr
llrl
l
ll 2
2
2 2
1
2
1 ++−= ε . (3.34)
Para evitar um pseudopotencial com uma singularidade na origem, a pseudofunção
de onda deve comportar-se com lr próximo à origem. A condição para que o potencial seja
contínuo é que a pseudofunção de onda tenha derivadas primeiras e segundas contínuas.
Removendo-se o efeito dos elétrons de valência e gerando um potencial iônico, será
possível usá-lo num procedimento autoconsistente para determinar a blindagem eletrônica
em outros ambientes. Assim, retirando-se os potenciais de Hartree e de troca e correlação,
cada componente do momento angular da PS sentirá um potencial diferente:
( ) ( ) [ ] [ ]nVnVrVrV ps
H
ps
,scr
ps
,ion−−= rr
ll. (3.35)
Há uma explícita dependência do pseudopotencial iônico com o momento angular
da pseudofunção de onda, sendo que cada momento angular l sentirá um potencial
diferente, o qual pode ser escrito como:
( ) ( ) ( )∑+=l
nlocal
ps
local,ion
ps
,ionPrVrVrV
ll
)rrr (3.36)
onde l
)P é um operador de projeção para o momento angular l .
Separando-se o termo local do termo não local nlocalV aumentamos a eficiência dos
cálculos computacionais. O termo local ( )rV pslocalion
r, que é de longo alcance pode ser
escolhido de forma arbitrária, porém, é vinculado a reproduzir as propriedades e
espalhamento atômico.
O termo semilocal é de curto alcance e é escrito da seguinte forma:
( ) ( ) ( )rVrVrV ps
local,ion
ps
,ion,nlocal
rrvll
−= (3.37)

37
Usando o procedimento de Kleinmann e Bylander (KLEINMANN et al, 1982) o
potencial semilocal dado na equação (3.37) pode ser transformado em um termo não local:
( ) ( ) ( ) ( ) ( )( ) ( ) ( )⟩⟨
⟩⟨=
rrVr
rVrrrVrV
O,ps
,nlocal
O,ps
,nlocal
O,psO,ps
,nlocalKB
l,nlocal rrr
rrrrr
lll
llll
φφφφ
, (3.38)
onde ( )rO,ps rl
φ é a pseudofunção de onda atômica.
3.3.1 Pseudopotencial Troullier-Martins
Um exemplo de pseudopotencial suave é o de Troullier-Martins (TROULLIER et
al, 1991), mostrando uma rápida convergência na energia total calculada do sistema, com
uma rápida convergência das propriedades do sistema, respeitando o aumento das funções
base. Esta convergência deve ser obtida sem considerar a escolha particular da estrutura
representativa do cristal, de sua constante de rede ou de alguma posição atômica interna.
No desenvolvimento de um pseudopotencial mais suave, começamos pela
generalização do procedimento de Kerker (KERKER, 1978), no qual podemos gerar e
parametrizar pseudopotenciais de norma conservada (BACHELET et al, 1982). O primeiro
passo é fazer com que a pseudofunção de onda dentro do raio de corte cr seja uma função
analítica, que se comporta como lr para r pequeno e que não tenha nodos. A
pseudofunção de onda de Kerker é definida como:
( ) ( )[ ]
=rpr
RrR
AE
ps
rr
l
l
l exp
c
c
r r se
rr se
≤≥
(3.39)
onde p(r) é um polinômio de ordem n = 4
∑=
+=n
2i
i
i0rcc)r(p . (3.40)
O coeficiente 1
c é omitido para evitar a singularidade do pseudopotencial blindado

38
em r =0. Os quatro outros coeficientes são determinados pelas condições de
pseudopotencial de norma conservada (BACHELET et al, 1982).
O pseudopotencial blindado é obtido pela inversão da equação radial de Schrödinger
( )( )( ) ( ) ( ) ( )[ ]
++++=
22
12
r'pr"p
r
r'p
rV
rV
AE
ps
scr l
r
r
lε
c
c
r r se
rr se
≤≥
(3.41)
A vantagem desse procedimento é que a pseudofunção de onda ( )rRps
l e o
pseudopotencial blindado ( )rV ps
,scr l são funções analíticas dentro do
cr .
A construção de Troullier-Martins (TM) (TROULLIER et al, 1991), que é uma
generalização do método de Kerker, foi de aumentar a ordem n do polinômio p(r), pois os
coeficientes adicionais darão o grau de liberdade variacional necessário para aumentar a
suavidade dos pseudopotenciais, sem aumentar o raio de corte.
O polinômio ( )rp usado por TM é ordem n=6 em potências2r , ou seja,
( ) 1212
1010
88
66
44
220 rcrcrcrcrcrccrp ++++++=r
. (3.42)
O motivo pelo qual usamos este pseudopotencial, é porque os resultados convergem
mais rapidamente.
3.4) Orbitais Atômicos
Para resolvermos a equação de Kohn-Sham (KOHN et al, 1965) é necessário
utilizar uma base para descrever os orbitais ( )ri
rφ . O programa SIESTA (Spanish Initiative
for Electronic Simulations with Thousand of Atoms) faz uso, como base, funções de onda
atômicas localizadas, as quais possuem dois parâmetros importantes, número de orbitais
por átomo e alcance desses orbitais (ARTACHO et al, 1999). As bases são uma boa

39
escolha para um reduzido custo computacional e cálculos precisos. O ajuste das bases para
cada átomo deve ser cuidadoso, pois há, neste método, desvantagem da convergência.
O programa SIESTA faz uso de orbitais atômicos numéricos (NAO) e estes são
obtidos através da resolução da equação de Schrödinger para os pseudo-átomos isolados,
com as mesmas aproximações para sólidos ou moléculas (mesmo xc
V e PS) (SOLER et al,
2002; ARTACHO et al, 1999). A localização restrita nas funções de base está atribuída nas
condições de contorno, adicionando ao PS um potencial confinante divergente ou
multiplicando a função de um átomo livre por uma função de corte.
Usando os NAO’s podemos ter bases mais simples, como uma single-ζ (SZ), até
bases mais completas, como double-ζ (DZ), multiple-ζ (MZ), ou adicionando também uma
flexibilização angular, o que chamamos de função de polarização (P).
Uma base mínima, ou SZ, tem somente uma função radial para cada canal de
momento angular e somente para os estados ocupados na valência do átomo isolado.
Permitindo cálculos rápidos com um grande número de átomos e as tendências qualitativas
das ligações químicas e uma boa descrição da banda de valência são obtidas. Essa base é
muito rígida para ser usada em cálculos que requerem uma grande flexibilidade, tanto radial
como angular, como por exemplo, sistemas onde há transferência de carga. Com uma base
mínima podemos otimizar a curvatura para uma dada configuração, mas se pretendemos
descrever o sistema como um conjunto de funções para várias geometrias diferentes é
preciso flexibilizá-la.
É necessário adicionar uma segunda função por canal de momento angular à base
SZ gerando então uma base double-ζ (DZ) com a qual se obtém uma melhor descrição da
parte radial. A ideia básica é adicionar um segundo orbital numérico, que reproduza a
função de onda a partir de um determinado raio DZ
R e seja suave na origem com
( )2brar −l , onde os parâmetros a e b se ajustam de modo que esta segunda função e sua
derivada sejam contínuas em DZ
R . A vantagem dessa segunda função é que ela está
estritamente localizada em um raio DZ
R , menor que o raio de corte original, o que reduz o
custo computacional. Desta mesma forma, este esquema é utilizado para calcular o
multiple-ζ, bastando escolher outros valores para o raio DZ
R .
A flexibilidade angular se obtém adicionando camadas de momentos angulares mais

40
altos do que o estritamente necessário para descrever os elétrons dos átomos isolados. O
procedimento para gerar esses orbitais polarizados utilizados no SIESTA é o uso de
polarização perturbativa, ou seja, na presença de um campo elétrico uniforme, as soluções
do problema atômico original de momento angular l se desacoplam em componente
( )1−l e ( )1+l pelo efeito da perturbação. Dessa forma, podemos resolver a equação de
Schrödinger para o átomo na presença de um campo elétrico e isolar a componente do
momento angular que nos interessa.
Para as bases estritamente localizadas o problema é encontrar uma maneira
sistemática de definir todos os raios das funções de base. O esquema proposto, no qual
todos os raios são definidos em função de um só parâmetro, é a correção na energia (energy
shift), isto é um incremento na energia que sofre o orbital quando está confinado (SOLER
et al, 2002). Este processo cresce a curvatura do orbital e, portanto, sua energia cinética. Se
cortarmos todos os raios de maneira que este incremento seja o mesmo para todos eles,
geramos uma base compensada que evita transferências de cargas espúrias.
No próximo Capítulo apresentaremos os resultados obtidos para moléculas de TiCl3
e TiCl4 interagindo com grafeno de uma ou duas camadas, fazendo uso da metodologia
explicitada neste Capítulo.

41
4) RESULTADOS
Neste Capítulo será descrito o estudo da interação das moléculas TiCl4 e TiCl3 em
camadas de grafeno. Inicialmente, apresentaremos os procedimentos de cálculo, a seguir as
propriedades do grafeno isolado e, finalmente, os resultados com a adsorção das moléculas
no grafeno, avaliando as propriedades estruturais, eletrônicas e magnéticas.
4.1) Procedimentos de Cálculo
O estudo das propriedades estruturais, eletrônicas e magnéticas foi realizado por
cálculos de primeiros princípios e foram executados, fazendo uso da teoria do funcional da
densidade (HOHENBERG et al, 1964; KOHN et al, 1965), utilizando o código
computacional SIESTA (SOLER et al, 2002). As equações de Kohn-Sham (KS) foram
resolvidas de forma autoconsistente usando pseudo-orbitais atômicos numéricos como base.
Em todas as simulações foram utilizadas bases numéricas do tipo double-ζ mais a função de
polarização (DZP) sendo que o alcance da base, definido pelo energy shift, foi de 0,05 eV
para os orbitais atômicos de C, Ti e Cl.
Os termos de troca-correlação foram descritos pela aproximação GGA, que satisfaz
aos vínculos teóricos de Perdew, Burke e Ernzerhof (PERDEW et al,1996) e para efeitos de
comparação dos resultados também avaliamos os resultados com a aproximação LDA
(modelo proposto por Perdew e Zunger (PERDEW et al, 1981)) para a molécula de TiCl4
adsorvida em uma monocamada de grafeno.
O pseudopotencial suave não-local de norma conservada utilizado para descrever a
interação entre eletron-íon foi o de Troullier e Martins (TROULLIER et al, 1991).
Os cálculos foram realizados com um parâmetro de convergência de que as forças
sobre cada coordenada de todos os átomos fossem menores que 0,05 eV/Å.
Para os cálculos da energia de ligação utilizamos a equação:
[ ]moleculagrafenomoléculagrafenob
EEEE −−−= + ][ (4.1)

42
onde b
E é a energia de ligação, grafeno
E é a energia total do grafeno puro, molécula
E é a energia
total da molécula e ]moléculagrafeno[
E + é a energia total do grafeno e da molécula.
Figura 4.1: Supercélula do grafeno usada na simulação para aproximação nas diferentes configurações moleculares (H, B e T).
Foram analisadas três configurações para cada molécula de TiCl3 ou TiCl4, sobre a
ligação carbono-carbono denominada (B), sobre um átomo de carbono, denominada (T) e
sobre o centro do hexágono, denominada (H), como pode ser visto na Figura 4.1.
A seguir apresentaremos os principais resultados para a simulação ab initio para o
grafeno puro, para efeitos de comparação, e a seguir a molécula de TiCl4 e TiCl3 adsorvida
no grafeno.
4.2) Grafeno
A supercélula usada para a simulação de uma folha de grafeno infinita contém 50
átomos de carbono, constituída de 5x5 células primitivas de uma rede hexagonal
bidimensional (Figura 4.2 (a)). O plano descrito encontra-se a uma distância de,
aproximadamente, 12 Å em relação aos planos de grafeno vizinhos. A distância de ligação

43
entre carbonos é de 1,42 Å após a otimização de geometria, valor similar ao encontrado na
literatura (CASTRO et al, 2007).
A estrutura eletrônica de bandas do grafeno puro (Figura 4.2 (b)) mostra um gap
tendendo a zero no ponto K, o que dá ao grafeno o caráter semimetálico (CASTRO et al,
2007).
Figura 4.2: (a) Supercélula de grafeno com 50 átomos de carbono. (b) Estrutura de bandas do grafeno puro. A linha horizontal tracejada em (b) corresponde a energia de
Fermi.
A seguir serão apresentados os resultados das simulações de grafeno puro a partir de
uma monocamada e bicamada interagindo com a molécula de TiCl4 e somente para uma
monocamada para o caso do TiCl3.
4.3) TiCl4 interagindo com uma monocamada de grafeno
Inicialmente consideramos a molécula de TiCl4 interagindo com uma monocamada
de grafeno em três configurações (B, T e H), fazendo uso das aproximações GGA e LDA,
com cálculos via polarização de spin. As três configurações mais estáveis e as
correspondentes distâncias de ligação para o TiCl4 nos sítios H, B e T são apresentadas nas
Figuras 4.3 (a), (b) e (c), respectivamente fazendo uso da aproximação GGA.

44
Figura 4.3: Configuração da estrutura otimizada da molécula de TiCl4 adsorvida em uma monocamada de grafeno nos casos: a) no sítio H; b) no sítio B e c) b no sítio T.
A Tabela 4.1 apresenta os resultados para as energias de ligação, distâncias mais
próximas de ligação e transferência de carga para o TiCl4 adsorvido em uma monocamada
de grafeno.
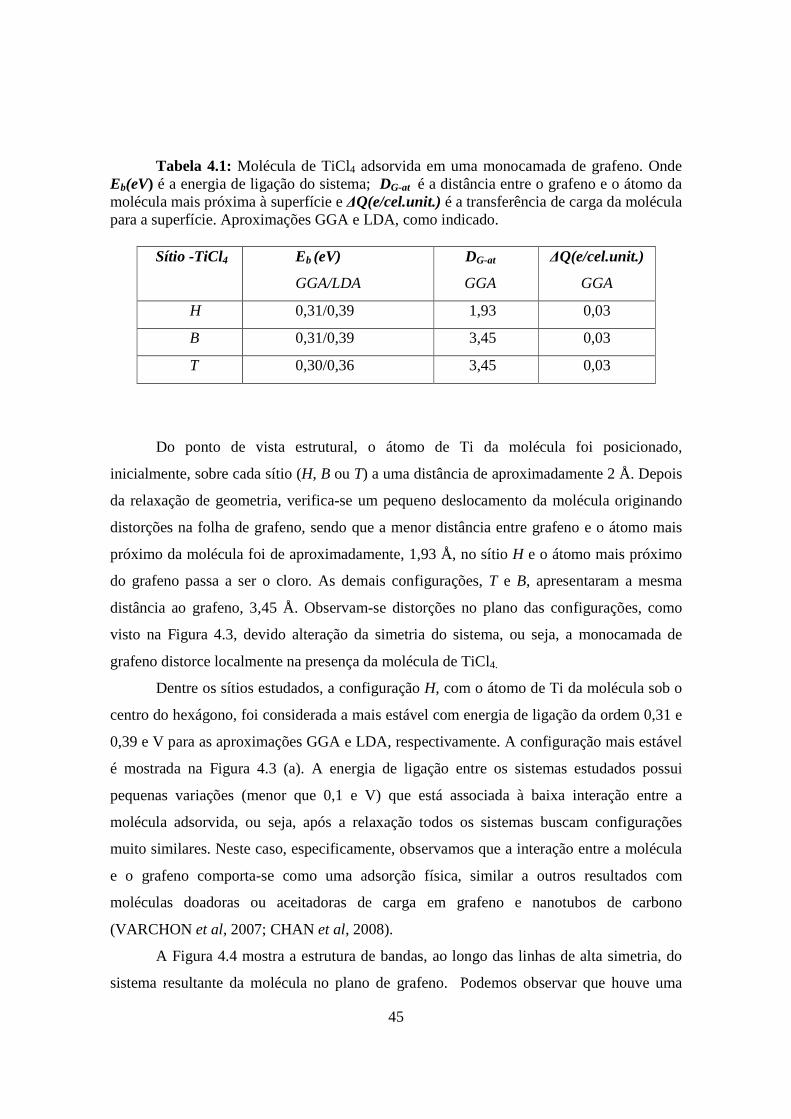
45
Tabela 4.1: Molécula de TiCl4 adsorvida em uma monocamada de grafeno. Onde Eb(eV) é a energia de ligação do sistema; DG-at é a distância entre o grafeno e o átomo da molécula mais próxima à superfície e ∆Q(e/cel.unit.) é a transferência de carga da molécula para a superfície. Aproximações GGA e LDA, como indicado.
Sítio -TiCl4 Eb (eV)
GGA/LDA
DG-at
GGA
∆Q(e/cel.unit.)
GGA
H 0,31/0,39 1,93 0,03
B 0,31/0,39 3,45 0,03
T 0,30/0,36 3,45 0,03
Do ponto de vista estrutural, o átomo de Ti da molécula foi posicionado,
inicialmente, sobre cada sítio (H, B ou T) a uma distância de aproximadamente 2 Å. Depois
da relaxação de geometria, verifica-se um pequeno deslocamento da molécula originando
distorções na folha de grafeno, sendo que a menor distância entre grafeno e o átomo mais
próximo da molécula foi de aproximadamente, 1,93 Å, no sítio H e o átomo mais próximo
do grafeno passa a ser o cloro. As demais configurações, T e B, apresentaram a mesma
distância ao grafeno, 3,45 Å. Observam-se distorções no plano das configurações, como
visto na Figura 4.3, devido alteração da simetria do sistema, ou seja, a monocamada de
grafeno distorce localmente na presença da molécula de TiCl4.
Dentre os sítios estudados, a configuração H, com o átomo de Ti da molécula sob o
centro do hexágono, foi considerada a mais estável com energia de ligação da ordem 0,31 e
0,39 e V para as aproximações GGA e LDA, respectivamente. A configuração mais estável
é mostrada na Figura 4.3 (a). A energia de ligação entre os sistemas estudados possui
pequenas variações (menor que 0,1 e V) que está associada à baixa interação entre a
molécula adsorvida, ou seja, após a relaxação todos os sistemas buscam configurações
muito similares. Neste caso, especificamente, observamos que a interação entre a molécula
e o grafeno comporta-se como uma adsorção física, similar a outros resultados com
moléculas doadoras ou aceitadoras de carga em grafeno e nanotubos de carbono
(VARCHON et al, 2007; CHAN et al, 2008).
A Figura 4.4 mostra a estrutura de bandas, ao longo das linhas de alta simetria, do
sistema resultante da molécula no plano de grafeno. Podemos observar que houve uma

46
pequena abertura de gap na estrutura eletrônica de bandas causada pela funcionalização da
molécula, a qual alterou a simetria (BARROS et al, 2006), além do abaixamento do nível
de Fermi e aparecimento de níveis desocupados. Este abaixamento do nível de Fermi deve-
se a transferência de carga do grafeno para a molécula de TiCl4. Para o caso da
configuração H a carga transferida do grafeno à molécula foi de 0,034-GGA(e/cél.unit.) e
de 0,028-GGA(e/cél.unit.) para as configurações T e B, assim, a molécula comporta-se
como um ácido de Lewis. A molécula não apresentou polarização de spin, ou seja, o
sistema permaneceu não magnético.
Figura 4.4: Estrutura de bandas ao longo das linhas de alta simetria ligando os pontos Γ M K Γ: (a) grafeno puro, e com a molécula de TiCl4 adsorvida no grafeno nos
sítios H, B e T, em (b), (c) e (d), respectivamente. As linhas horizontais pontilhadas representam o nível de Fermi.
Como já estabelecido na literatura, sabe-se que a aproximação LDA superestima a

47
energia de ligação e subestima a distância de ligação entre dois sistemas químicos. Por
outro lado, a aproximação GGA subestima a energia de ligação e superestima a distância de
ligação, aproximando-se, na maioria dos casos com mais precisão dos resultados
experimentais. Estes resultados foram observados na análise das propriedades da molécula
de TiCl4 em uma monocamada de grafeno. Portanto, a partir deste momento utilizaremos
somente a aproximação GGA para a descrição dos resultados deste trabalho.
4.4) Interação de duas moléculas de TiCl4 com uma monocamada de grafeno
Nesta configuração foi usado um plano de grafeno entre duas moléculas de TiCl4
(chamada de (TiCl4)2@grafeno), como mostra a Figura 4.5. Para os cálculos foi usado o
termo de troca e correlação GGA, com polarização de spin e relaxação total das
coordenadas atômicas do sistema. Cada molécula estava inicialmente a uma distância de
2,00 Å do plano de grafeno.
Figura 4.5: Configuração estrutural do (TiCl4)2@grafeno.

48
Depois de ocorrida a simulação, verifica-se um pequeno deslocamento das
moléculas, nenhuma mudança na estrutura da folha de grafeno e a menor distância entre
cada molécula e grafeno, de 3,75 e 3,91 Å, sendo o átomo mais próximo o Cl, como mostra
a Figura 4.5. Não se observa distorções no plano de grafeno, como visto na configuração
com somente uma molécula, pois é restabelecida a simetria do sistema devido a presença da
outra molécula, como consequência o fechamento do gap.
A configuração (TiCl4)2@grafeno apresenta energia de ligação do sistema de 0,86 e
V calculada através da equação 4.1. Esta energia é maior que duas vezes a interação de
uma molécula de TiCl4 com uma monocamada de grafeno. Este fato indica que duas
moléculas adsorvidas em faces opostas do grafeno geram uma estabilidade maior na ligação
devido à relaxação estrutural.
A Figura 4.6 (b) mostra a estrutura eletrônica de bandas, para os pontos de alta
simetria, do sistema resultante (TiCl4)2@grafeno em comparação ao grafeno puro (Figura
4.6 (a)). Podemos observar que o gap de energia permanece nulo na estrutura eletrônica de
bandas, além do abaixamento do nível de Fermi e aparecimento de níveis desocupados.
Este abaixamento do nível de Fermi deve-se a transferência de carga do grafeno à molécula
de TiCl4. A carga transferida foi de 0,064 (e/cél.unit.) do grafeno para a molécula,
demonstrando o comportamento de ácido de Lewis do TiCl4. O sistema não apresentou
polarização de spin, ou seja, o sistema permaneceu não magnético na configuração
(TiCl4)2@grafeno.
Figura 4.6: Estrutura eletrônica de bandas para a) grafeno puro e b) configuração (TiCl4)2@grafeno. As linhas horizontais tracejadas correspondem à energia de Fermi.

49
4.5) Configuração TiCl4 confinada em uma bicamada de grafeno
Nesta configuração foi usada uma molécula de TiCl4 confinada entre dois planos de
grafeno (denominada de TiCl4@bigrafeno). Para as simulações foi usado o termo de troca e
correlação GGA com otimização total de geometria. A molécula estava inicialmente a uma
distância de 2,00 Å de cada plano de grafeno da célula unitária. Depois da otimização
verifica-se um pequeno deslocamento dos planos afastando-se da molécula, mas com
ausência de alterações na configuração estrutural das camadas de grafeno.
A Figura 4.7 apresenta a configuração estrutural do sistema TiCl4@bigrafeno, sendo
que a menor distância entre cada folha de grafeno e a molécula é de 3,11 e 3,07 Å. Estes
valores para distância de ligação são inferiores quando comparados com a molécula de
TiCl4 em uma monocamada de grafeno ou na estrutura (TiCl4)2@grafeno.
Figura 4.7: Interação da molécula de TiCl4 confinada entre dois planos de grafeno (TiCl4@bigrafeno).
A energia de ligação da molécula de TiCl4 na configuração TiCl4@bigrafeno

50
fazendo uso da equação 4.1 resultou em 0,41 eV. Comparado com os resultados da Tabela
4.1 (resultados com GGA) para uma molécula em uma camada de grafeno, observamos que
o confinamento aumenta a estabilidade do sistema.
A Figura 4.8 mostra a estrutura de bandas ao longo das linhas de alta simetria, do
sistema resultante da molécula com os planos de grafeno em comparação ao grafeno puro.
Figura 4.8: Estrutura eletrônica de bandas para (a) grafeno puro e (b) configuração TiCl4@bigrafeno. As linhas horizontais tracejadas indicam a energia de Fermi.
Avaliando os dados de estrutura eletrônica, podemos observar que de gap de energia
permanece nulo após o confinamento do TiCl4 entre duas camadas de grafeno. Por outro
lado, observa-se uma elevação na energia de Fermi e aparecimento de níveis desocupados e
ocupados, que se deve a presença da molécula. Este levantamento do nível de Fermi deve-
se a transferência de carga da molécula de TiCl4 em relação aos planos de grafeno. A carga
transferida foi de 0,108 (e/cél.unit.), assim, a molécula demonstra seu comportamento de
uma base de Lewis, como aceitadora de carga, resultado contrário em relação a molécula de
TiCl4 não confinada entre planos de grafeno.
4.6) Interação da molécula TiCl3 em uma monocamada de grafeno
Avaliamos a interação da molécula de TiCl3 com o grafeno de forma a comparar as
variações nas propriedades estruturais e eletrônicas com o sistema com TiCl4. Utilizou-se o

51
termo de troca e correlação GGA, com polarização de spin e todas as coordenadas atômicas
foram permitidas relaxar. A molécula de TiCl3 foi inicialmente colocada no sistema a uma
distância de 2,00 Å da superfície do plano de grafeno.
Depois de ocorrida a simulação, observamos que a menor distância da molécula em
relação à superfície do plano ocorre na configuração H, ou seja, configuração com o Ti no
centro do hexágono, com valor de 2,52 Å. Para os sítios T e B as distâncias mínimas
obtidas foram de 3,59 e 3,56 Å, respectivamente. As Figuras 4.9 (a), (b) e (c) apresentam as
configurações otimizadas para o TiCl3 no grafeno nas posições H, B e T, respectivamente.
Figura 4.9: Estrutura final da interação da molécula TiCl3 com grafeno nos sítios
(a) H, (b) B e (c) T.
A Tabela 4.2 apresenta os resultados obtidos para energia de ligação, distância
mínima entre a molécula e o grafeno, variação de carga eletrônica e polarização de spin,
relacionados com as configurações da Figura 4.9.

52
Tabela 4.2: Molécula de TiCl3 adsorvida na superfície do plano de grafeno, onde Eb(eV) é a energia de ligação do sistema, DG-at é a distância entre o grafeno e o átomo da molécula mais próxima à superfície, ∆Q(e/cel.unit.) é a transferência de carga da molécula para o grafeno e Pol.Spin é a polarização de spin resultante.
SítioTiCl3 Eb(eV) DG-at(Å) ∆Q(e/cél.unit.) Pol. Spin (µB)
H 1,02 2,52 0,25 0,48
B 0,91 3,56 0,04 1,00
T 0,89 3,59 0,03 1,00
Como no caso do TiCl4, a menor energia de ligação para adsorção da molécula de
TiCl3 ocorreu no sítio H, no valor de 1,02 eV, enquanto que as outras, B e T, obtiveram os
valores 0,91 e 0,89 e V, respectivamente, como presente na Tabela 4.1. Os menores valores
(mais estáveis) de energia e a distância em relação à superfície do plano de grafeno são
referentes ao sítio H, sendo, assim, este sítio é o mais favorável para adsorção da molécula
TiCl3 no plano de grafeno. Os outros sítios mostram uma energia de ligação e distância
entre moléculas muito próximas, ou seja, são praticamente equivalentes. Ao contrário do
caso do TiCl4, na adsorção de TiCl3 a estrutura do plano de grafeno não sofreu alterações,
como pode ser visto na Figura 4.9. Nas configurações dos sítios B e T observamos uma
geometria planar da molécula, como mostra a Figura 4.9(b) e (c), respectivamente. As
estruturas de bandas de energia das três configurações em estudo são mostradas na Figura
4.10 e comparadas com o grafeno puro (Figura 4.2(b)).
Observamos que não existem alterações significativas na estrutura de bandas do
grafeno, em comparação ao puro, a não ser a elevação do nível de Fermi, que se deve a
transferência de carga, (0,25 (e/cel.unit.) da molécula ao plano de grafeno), comportando-se
como uma base de Lewis. Podemos também observar o surgimento de níveis desocupados
de energia acima do nível de Fermi, aos quais se devem a presença da molécula. Os níveis
presentes na banda de energia hibridizados originando uma interação química, como
observado através da energia de ligação.

53
Figura 4.10: Estrutura de bandas ao longo das linhas de alta simetria Γ M K Γ: (a)
grafeno puro, e com a molécula de TiCl3 adsorvida nos sítios H, B e T, em (b), (c) e (d), respectivamente. As linhas horizontais pontilhadas e tracejadas representam o nível de Fermi.
A molécula de TiCl3 no seu estado fundamental é magnética e o plano de grafeno
puro não magnético, mas os sistemas resultantes apresentaram polarização de spin. A partir
da Tabela 4.2, percebemos que o sítio mais favorável apresenta o valor de polarização
muito menor que os outros casos. Este fato está diretamente relacionado com a hibridização
resultante do sistema.

54
5) CONCLUSÕES
Neste trabalho, as interações das moléculas TiCl4 e TiCl3 em grafeno são estudadas
usando simulações de primeiros-princípios baseada na teoria funcional da densidade.
Cálculos de energia de ligação, geometria, densidade de estados e transferência de carga
foram realizadas.
Observa-se que a molécula TiCl4 apresenta caráter de ácido de Lewis quando
adsorvida em uma monocamada de grafeno, sendo um receptor de elétrons. Este resultado
pode ser observado pela transferência eletrônica de carga e pela variação da energia de
Fermi em relação ao grafeno puro (diminuição do seu valor). A adsorção de uma única
molécula de TiCl4 causa distorções localizadas na folha de grafeno, originando uma
pequena mudança nos estados eletrônicos do grafeno, visto na abertura de um gap de
energia.
Para a molécula de TiCl4 confinada entre dois planos de grafeno o comportamento
da mesma torna-se como uma base de Lewis, ou seja, ocorre a transferência de elétrons da
molécula para o grafeno. Este fato é claramente demonstrado pela estrutura eletrônica de
bandas, a partir da variação da energia de Fermi para valores superiores ao grafeno puro.
No caso da adsorção de duas moléculas de TiCl4 simétricas em relação a uma
monocamada de grafeno observamos o comportamento das mesmas como ácido de Lewis
(aceitadores de elétrons), recuperando a simetria do sistema, ou seja, não distorcendo
localmente a estrutura planar do grafeno. Da mesma forma, ressaltamos que o sistema
resultante possui o mesmo caráter semimetálico do grafeno puro, a não ser pela variação do
nível de Fermi devido a transferência de carga. Salienta-se ainda que para todos os casos
estudados para a adsorção do TiCl4 em grafeno não se observa polarização de spin nos
sistemas resultantes.
No caso da molécula de TiCl3 adsorvida em uma monocamada de grafeno,
observamos o comportamento da mesma como base de Lewis. Este fato é evidenciado pela
transferência de carga da molécula para o grafeno e pelo aumento da energia de Fermi em
relação ao grafeno puro. Também se observa que no sítio mais estável (Ti situado no centro
do hexágono de carbono do grafeno) a energia de ligação e a transferência de carga são

55
superiores quando comparadas com as outras configurações da mesma molécula. Também
nota-se que na configuração mais estável a molécula de TiCl3 não se apresenta planar,
demonstrando alterações estruturais devido à interação química. Ao mesmo tempo, ressalta-
se que a molécula de TiCl3 apresenta valores para energia de ligações bem superiores
quando comparado ao TiCl4 no grafeno, demonstrando a maior reatividade da molécula de
TiCl3. Notamos, da mesma forma, que os níveis eletrônicos do TiCl3 que aparecem na
banda de valência e banda de condução possuem uma maior hibridização com os níveis do
grafeno, quando comparados com os casos da molécula de TiCl4. Também se observa que
os sistemas resultantes com a molécula de TiCl3 possuem polarização de spin sendo que na
configuração mais estável ocorrem mudanças significativas devido a transferência de carga
e hibridizações entre os níveis da molécula e do grafeno.
Desta forma, podemos concluir que as moléculas de TiCl4 e TiCl3 possuem
comportamentos associados à transferência de carga quando adsorvido no grafeno. A
molécula de TiCl4 quando adsorvida em uma monocamada de grafeno comporta-se em uma
monocamada de grafeno como um ácido de Lewis (receptor de carga). Entretanto, quando a
molécula de TiCl4 esta confinada entre camadas de grafeno ou o TiCl3 em monocamadas de
grafeno, observamos o comportamento de base de Lewis (doador de carga). O
entendimento destes sistemas torna-se essencial para o uso do grafeno em sensores híbridos
associados com armazenamento de cargas para a nanoeletrônica.
A viabilidade experimental da interação destas moléculas, TiCl4 e TiCl3, são muito
complicados para manuseio. Qualquer experimento envolvendo esses compostos, precisa
ser realizado em atmosfera inerte, vidraria flambada e livre de água.

56
REFERÊNCIAS BIBLIOGRÁFICAS
ARTACHO, E.; SÁNCHEZ-PORTAL, D.; ORDEJÓN, P.; GARCIA A.; SOLER, J. M.; Linear-scaling ab initio calculations for large and complex systems Phys. Stat. Sol, 215, 809 (1999). ASCROFT, N. W.; MERMIN, N. D., Solid State Physics , Saunders College, Philadelphia, PA, (1976). BACHELET, G.B.; HAMANN, D.R.; SCHLUTER, M.; Pseudopotentials that work: From H to Pu ; Phys.Rev.B 26, 4199 (1982). BARROS E.B.; JORIO A., G. G.; SANSONIDZE, C. R. B.; SOUZA FILHO, A.G.; MENDES FILHO, J., DRESSELHAUS, G.; DRESSELHAUS, M.; Review on the symmetry-related properties of Carbon Nanotubes, Phys. Reports 431, 261 (2006). BENA, C.; Kivelson, S. A.; Quasiparticle scattering and local density of states in graphite, Phys. Rev, B 72, 125432, (2005). BETHUNE, D. S. ; KIANG, C. H.; VRIES, M. S.; GORMAN,G.; SAVOY,R.; VAZQUEZ, J.; BEYERS, R.; Cobalt-catalysed growth of carbon nanotubes with single-atomic-layer walls. Nature 363, 605 (1993). CALDERAZZO F.; FERRI, I.; PAMPALONI, G.; TROYANOV, S.; " η6-Arene Derivatives of Titanium(IV), Zirconium(IV) and Hafn ium(IV)" . J. of Organometallic Chemistry. 518, 189, (1996). CASTRO, H. N.; GUINEA, F.; PERES, N. M. R.; NOVOSELOV, K. S.; GEIM, A. K.; Electronic properties of bilayer and multilayer graphene. Phys. Rev. B, 78, 4 (2007). CEPERLEY, D.M.; ALDER, B. J.; Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566 (1980). CHAN K. T.; NEATON, J. B.; COHEN, M. L.; First-principles study of metal adatom adsorption on graphene, Phys. Rev. B 77, 23 (2008). DAVIE M. E.; FOERSTER, T.; PARSONS, S.; PULHAM, C.; RANKIN, D. W. H.; SMART, B. A. The Crystal Structure of Tetrakis (dimethylamino) titanium(IV) . Polyhedron 25, 923 (2005). DRESSELHAUS, M. S.; DRESSELHAUS, G.; EKLUND, P. C.; Science of Fullerenes and Carbon Nanotubes. New York, NY, San Diego, CA: Academic Press, 1996. DRESSELHAUS, M. S.; DRESSELHAUS, G.; Intercalation compounds of graphite.

57
Adv. Phys., 51, 1 (2002). E. FERMI; Eine statistische Methode zur Bestimmung einiger Eigenschaften des Atoms und ihre Anwendung auf die Theorie des periodischen Systems der Elemente. Z. Physik. 48, 73 (1928). EVANS, J. W.; THIEL, P. A. & BARTELT, M. C. Morphological evolution during epitaxial thin film growth: Formation of 2D islands and 3D mounds. Sur. Sci. Rep. 61, 1 (2006). FAGAN, S. B.; Funcionalização de Nanotubos de Carbono: uma Abordagem de Primeiros Princípios, Tese de Doutorado – Universidade Federal de Santa Maria (2003). FUCHS, M.; BOCKSTEDTE, M.; PEHLKE, E.; SCHEFFLER, M.; Pseudopotential study of binding properties of solids within generalized gradient approximations: The role of core-valence exchange correlation. Phys. Rev. B, 57, 2134 (1998). GEIM, A. K.; NOVOSELOV, K. S.; The rise of graphene. Nat. Mater., 6, 183 (2007). GREENWOOD, N. N.; EARNSHAW, A.; Chemistry of the Elements, 2nd Edition, Oxford:Butterworth-Heinemann, (1997). GUNDERSEN, L.-L.; RISE, F.; UNDHEIM, K.; Titanium(IV) chloride, Encyclopedia of Reagents for Organic Synthesis, L. Paquette, J. Wiley & Sons (2004). HOHENBERG. P.; KOHN, W.; Inhomogeneous electron gas. Phys. Rev. B, 13, 3B, B864 (1964). HOLLENMAN, A. F.; WIBERG, E. Inorganic Chemistry, Academic Press: San Diego, (2001). HOUAISS, A., VILLAR, M.S. – Dicionário Houaiss da língua portuguesa. Rio de Janeiro, Objetiva, 2001. IIJIMA, S.; Helical microtubules of graphitic carbon. Nature, 354, 56 (1991). IIJIMA, S.; ICHIHASHI, T.; Single-shell carbon nanotubes of 1-nm diameter. Nature 363, 603 (1993). JONGEN, L.; MEYER, G.; Caesium heptaiododititanate(III), CsTi2I 7. Zeitschrift für Anorganische und Allgemeine Chemie 630, 211 (2004). KERKER, G. P.; Non-singular atomic pseudopotentials for solid state applications. Journal of Physics C: Solid State Physics, 13, 189 (1980). KHANTHA, M. et al. Interaction of lithium with graphene: An ab initio study. Phys. Rev. B, 70, 125422 (2004).

58
KLEINMANN, L.; BYLANDER, D. M.; Efficacious form for model pseudopotentials. Phys. Rev. Lett. 48, 1425 (1982). KOHN, W.; Nobel lecture: Electronic structure of matter - Wave functions and density functional; Rev. Mod. Physics, 71, 1253 (1999). KOHN, W.; SHAM, L. J.; Self-consistent equations including exchange and correlation effects. Phys. Rev., 140, A1133 (1965). KONG, J.; FRANKLIN, N. R.; ZHOU, C.; CHAPLINE, M. G.; PENG, S.; CHO, K.; DAI, H.; Nanotube molecular wires as chemical sensors; Science, 287, 622 (2000). KROTO, H. W.; et al. C60 - buckminsterfullerene. Nature, 318, 162 (1985). L.H.THOMAS ; The calculation of atomic fields Proc.Camb. Phil. Soc. 23, 542 (1927). LANDAU; ZUR, L. D.; Theorie der phasenumwandlungen II. Phys. Z. Sowjetunion 11, 26 (1937). MARTIN, R. M.; Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press, Urbana-Champaign (2004). MERMIN, N. D.; Crystalline order in two dimensions. Phys. Rev. 176, 250 (1968). NOVOSELOV, K. S. et al. Electric Field Effect in Atomically Thin Carbon Fil ms. Science, 306,666 (2004). NOVOSELOV, K. S. et al. Two-dimensional atomic crystals. American Proc. Natl. Acad. Sci. U. S. A., 102, 10451 (2005). NOVOZELOV, K. S.; GEIM, A. K.; MOROZOV, S. V.; JIANG, D.; KATSNELSON, M. I.; GRIGORIEVA, I. V.; DUBONOS, S. V.; FIRSOV, A. A.; Two-dimensional gas of massless Dirac fermions in graphene. Nature (London) 438, 197 (2005). PARTOENS, B.; PEETERS, F. M.; From graphene to graphite: Electronic structure around the K point. Phys. Rev. B 74, 075404 (2006). PEIERLS, R. E.; Quelques proprietes typiques des corpses sollides. Anm. I. H. Poincare 5, 177 (1935). PERDEW, J. P.; BURKE, K.; ERNZERHOF, M.; Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996). PERDEW, J. P.; ZUNGER, A.; Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B, 23, 5048 (1981).

59
REICH, S.; MAULTZSCH, J.; THOMSEN, C.; ORDEJÓN, P.; Tight-binding description of graphene. Phys. Rev. B 66, 035412 (2002). SAITO, R.; FUJITA, M.; DRESSELHAUS, G.; DRESSELHAUS, M. S.; Electronic-structure of chiral graphene tubules. Appl. Phys. Lett. 60, 2204 (1992a). SAITO, R.; FUJITA, M.; DRESSELHAUS, G.; DRESSELHAUS, M. S.; Electronic structure of graphene tubules based on C60; Appl. Phys. Rev. B 46, 1804 (1992b). SCHEDIN, F.; GEIM, A. K.; MOROZOV, S. V.; HILL, E. W.; BLAKE, P.; KATSNELSON, M. I. NOVOSELOV, K. S.; Detection of individual gas molecules adsorbed on graphene ; Nat. Mater. 6, 652 (2007). SOLER, J. M., ARTACHO, E.; GALE, J. D.; GARCIA, A. ; JUNQUERA, J. ; ORDEJÓN P.; SANCHEZ-PORTAL, D., The SIESTA method for ab initio order-N materials simulation; J. Phys: Condens. Matter. 14, 2745 (2002). STANKOVICH, S. et al. Graphene-based composite materials, Nature 442, 282 (2006). STARR, C.; BITTER, F.; KAUFMAN, A.R.; LIPPARD, S. Halides & Halide Complexes in Progress in Inorganic Chemistry, Cotton (Ed.) 9, (John Wiley & Sons, Inc.) 6 (1968). TROULLIER, N. ; MARTINS, J.L. ; Efficient pseudopotentials for plane-wave calculations; Phys. Rev. B 43, 1993 (1991). UENO, H.; IMANISHI, K.; UEKI, S.; KOHARA, T. Kinetics Study of Propene Polymerization with Porous Titanium Trichloride . Chemical Society of Japan 7, 495 (2000). VARCHON, F.; FENG, R.; HASS, J.; LI, X.; NGUYEN, B. N.; NAUD, C.; MALLET, P.; et al Electronic Structure of Epitaxial Graphene Layers on SiC: Effect of the Substrate. Phys. Rev. Lett., 92, 12 (2007). VENABLES, J. A.; SPILLER, G. D. T.; HANBUCHEN, M.; Nucleation and growth of thin in films. Rep. Prog. Phys. 47, 399 (1984). WALLACE, P. R.; The band theory of graphite, Phys. Rev. 71, 622 (1947). WEBB, S. P.; GORDON, M. S. Intermolecular Self-Interactions of the Titanium Tetrahalides TiX4 (X = F, Cl, Br). J. Am. Chem. Soc. 121, 2552 (1999). ZHANG,Y.; TAN, Y.-W.; STORMER, H. L.; KIM, P.; Experimental observation of the quantum Hall effect and Berry's phase in graphene; Nature (London) 438, 201 (2005).

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo





























