Embed Size (px)
Citation preview

Novembro de 2008
Processamento de Dados Biológicos Utilizando Tecnologias Grid
SÉRGIO MENDES COSTA
Dissertação para obtenção do Grau de Mestre em
ENGENHARIA INFORMÁTICA E DE COMPUTADORES
Júri
Presidente: Professor Doutor Nuno João Mamede
Orientador: Professor Doutor Arlindo Manuel Limede de Oliveira
Vogais: Professora Doutora Ana Teresa Correia de Freitas
Professor Doutor Pedro Fernandes
2 cm 2 cm

i

ii
Agradecimentos
Agradeço à Patrícia e aos meus pais todo o apoio, carinho e amor que sempre me deram,
bem como as regalias que me proporcionaram e toda a paciência que tiveram comigo ao longo
destes anos de vida universitária. Sem vocês nada disto seria possível.
Agradeço ao Engenheiro Pedro Fernandes todo o apoio, orientação e amizade que me deu
ao longo deste trabalho. O meu muito obrigado por todos os conselhos, críticas e sugestões e pela
disponibilidade permanente em partilhar comigo o seu conhecimento, algo que me permitiu
aprender e crescer imenso como investigador e como pessoa.
Agradeço aos meus orientadores, o Professor Arlindo Oliveira e a Professora Ana Teresa
Freitas, a supervisão da investigação subjacente a esta tese e o facto de me terem proporcionado
a realização de um trabalho estimulante e desafiante. Sempre me interessei pelas ciências
biológicas e esta dissertação constituiu uma oportunidade única de aplicar os conceitos de
engenharia informática aprendidos ao longo da licenciatura e do mestrado na resolução de
problemas biológicos.
Agradeço ao Professor António Rito Silva o apoio dado na componente de testes baseados
em modelos, bem como a sua constante disponibilidade para esclarecer dúvidas e responder às
minhas muitas questões.

iii

iv
Resumo
Actualmente, verifica-se um interesse crescente no desenvolvimento de sistemas nos quais
possa ser realizada análise científica que apresente requisitos elevados de computação e/ou de
armazenamento e processamento de dados. As tecnologias de computação em cluster e em Grid
têm emergido como o suporte de eleição para este tipo de sistemas.
As ciências biológicas estão entre aquelas que mais vêm beneficiando com os avanços
nestas tecnologias, nomeadamente no estudo dos mecanismos de expressão dos genes. Neste
sentido, a descoberta de locais de ligação para factores de transcrição e a análise de dados de
expressão de genes adquirem particular relevância. Na primeira situação, procuram-se usualmente
segmentos bem conservados de pequena dimensão, designados motivos, em DNA. Na segunda
situação, analisam-se, com técnicas de data mining como o biclustering, dados registados em
microarrays.
No âmbito desta dissertação disponibilizaram-se, no cluster hermes do Instituto Gulbenkian
de Ciência, algoritmos eficientes para inferência de motivos em regiões promotoras e para análise
de dados de expressão de genes. Os algoritmos foram criados no âmbito do projecto “BioGrid –
Algoritmos Paralelos Para Anotação de Genes”. Ao longo deste trabalho, foram realizadas as
tarefas necessárias de implementação, instalação e teste, bem como a criação das interfaces Web
e documentação para as várias aplicações. Foi ainda desenvolvido um estudo onde a técnica de
testes baseados em modelos foi utilizada para avaliação do software.
Os algoritmos desenvolvidos no contexto do projecto BioGrid estão agora disponíveis, num
sistema robusto, integrado e de fácil utilização, para uma vasta comunidade de utentes de
Bioinformática.
Palavras-chave: Análise de Dados Biológicos, Computação em Cluster, Descoberta de Motivos,
Técnicas de Biclustering, Testes Baseados em Modelos.

v

vi
Abstract
At present there is a growing interest in the development of systems in which scientific
analysis with high computing or data storage and processing requirements can be performed. The
cluster and Grid computing technologies have emerged has the best support infrastructures for this
type of systems.
Biological sciences are among those who have been benefiting more from the
advancement of these technologies, namely in the study of gene expression mechanisms. In that
sense, the discovery of transcription factor binding sites and the analysis of gene expression data
are particularly relevant. In the first case, we usually search for short segments of DNA, known as
motifs, that are well conserved. In the second case, we usually analyze microarray data using data
mining techniques like biclustering.
In the context of this thesis, efficient algorithms for motif inference in gene promoter regions
and for the analysis of gene expression data were made available in the hermes cluster of Instituto
Gulbenkian de Ciência. The algorithms were developed in the context of the “BioGrid – Parallel
Algorithms for Gene Annotation” project. During this work, the necessary tasks of implementing,
installing and testing were performed, as well as the development of Web interfaces and
documentation for every program. In addition to that, a study was conducted in which the model-
based testing technique was used to evaluate the software.
The algorithms created in the context of the BioGrid project are now available in a reliable,
integrated and user-friendly system for a large community of Bioinformatics users.
Keywords: Biclustering Techniques, Biological Data Analysis, Cluster Computing, Model-Based
Testing, Motif Finding.

vii

viii
Índice
Agradecimentos ................................................................................................. 2
Resumo .............................................................................................................. 4
Abstract ............................................................................................................. 6
Índice ................................................................................................................. 8
Lista de Figuras ................................................................................................ 12
Lista de Abreviaturas ........................................................................................ 14
Introdução .......................................................................................................... 1
1.1 Contexto ........................................................................................................ 1
1.2 Objectivos ..................................................................................................... 2
1.3 Contribuições ................................................................................................ 3
1.4 Organização da tese ....................................................................................... 3
1.5 Convenções ................................................................................................... 4
Capítulo 2 Fundamentos de Biologia Molecular .................................................. 5
2.1 A molécula de DNA ........................................................................................ 5
2.2 Expressão dos genes ...................................................................................... 9
2.3 Regulação da Expressão dos Genes ............................................................... 11
Capítulo 3 Computação em Cluster e Grid ......................................................... 15
3.1 Clusters computacionais e malhas de computadores .................................... 15
3.2 Computação Paralela ................................................................................... 20
Capítulo 4 Algoritmos ...................................................................................... 23
4.1 Bioinformática e Biologia Computacional ..................................................... 23

ix
4.2 Descoberta de Motivos ................................................................................ 24
4.2.1 Descrição do problema .................................................................................................. 26
4.2.2 Estado da arte dos algoritmos de descoberta de motivos ............................................. 27
4.3 Análise de dados de expressão de genes ....................................................... 32
4.3.1 Microarrays .................................................................................................................... 32
4.3.2 Biclustering ..................................................................................................................... 35
4.3.3 Estado da arte dos algoritmos de biclustering ............................................................... 36
4.4 Algoritmos trabalhados durante a tese ......................................................... 38
4.4.1 PSMILE ............................................................................................................................ 38
4.4.2 RISO ................................................................................................................................ 43
4.4.3 MUSA ............................................................................................................................. 44
4.4.4 Cheng e Church .............................................................................................................. 46
4.4.5 CCC-Biclustering e e-CCC-Biclustering ............................................................................ 47
Capítulo 5 Deployment dos Algoritmos no Cluster Hermes ................................ 51
5.1 Visão geral do sistema ................................................................................. 51
5.2 Sun Grid Engine ........................................................................................... 52
5.3 BioTeam iNquiry .......................................................................................... 53
5.4 Bioprogram ................................................................................................. 54
5.5 Adicionar aplicações no cluster hermes ......................................................... 56
5.6 Trabalho realizado com os algoritmos .......................................................... 57
5.6.1 MUSA ............................................................................................................................. 57
5.6.2 PSMILE ............................................................................................................................ 58
5.6.3 RISO ................................................................................................................................ 58
5.6.4 CCC-Biclustering e e-CCC-Biclustering ............................................................................ 59
5.6.5 Cheng e Church .............................................................................................................. 61
5.7 Teste das aplicações e algoritmos ................................................................. 63
5.7.1 Testes Baseado em Modelos ......................................................................................... 63
5.7.2 Vantagens e desvantagens da técnica MBT ................................................................... 66
5.7.3 Aplicação da técnica MBT às aplicações de descoberta de motivos .............................. 66
5.7.4 Realização dos testes com as aplicações de descoberta de motivos ............................. 74
5.7.5 Resultados dos testes para as aplicações de descoberta de motivos ............................ 77

x
5.7.6 Aplicação da técnica MBT às aplicações de biclustering ................................................ 78
Capítulo 6 Conclusões e trabalho futuro ........................................................... 80
6.1 Conclusões .................................................................................................. 80
6.2 Trabalho Futuro ........................................................................................... 81
6.3 Perspectivas de futuro para a Bioinformática ................................................ 84
Referências ....................................................................................................... 88
Anexo 1 – Interfaces Pessoa-Máquina Desenvolvidas ........................................ 95
A1.1 Interface CCC-Biclustering ............................................................................ 95
A1.1.1 Parâmetros de carregamento ..................................................................................... 96
A1.1.2 Parâmetros de pré-processamento ............................................................................ 98
A1.1.3 Parâmetros de pós-processamento .......................................................................... 102
A1.1.4 Parâmetros de saída ................................................................................................. 107
A1.2 Interface e-CCC-Biclustering ........................................................................... 108
A1.2.1 Parâmetros de Biclustering ....................................................................................... 108
A1.3 Interface Cheng and Church ........................................................................... 109
A1.3.1 Parâmetros de Biclustering ....................................................................................... 109
A1.4 Interface MUSA ............................................................................................. 110
A1.4.1 Parâmetros Simples .................................................................................................. 110
A1.4.2 Parâmetros Avançados ............................................................................................. 111
A1.5 Interface PSMILE ........................................................................................... 113
A1.5.1 Parâmetros genéricos ............................................................................................... 113
A1.5.2 Parâmetros de alfabeto ............................................................................................ 115
A1.5.3 Parâmetros do método de avaliação “shuffling” ...................................................... 116
A1.5.4 Parâmetros do método de avaliação “against” ........................................................ 117
A1.5.5 Parâmetros da caixa n ............................................................................................... 117
A1.6 Interface RISO ............................................................................................... 119
A1.6.1 Parâmetros genéricos ............................................................................................... 119
A1.6.2 Parâmetros da caixa n ............................................................................................... 120
Anexo 2 – Formato de saída das várias aplicações .......................................... 122

xi
A2.1 Aplicações de Biclustering .............................................................................. 122
A2.2 Aplicações de Motif Finding ...................................................................... 123
A2.2.1 MUSA ............................................................................................................................ 123
A2.2.2 PSMILE ........................................................................................................................... 125
A2.2.3 RISO ............................................................................................................................... 126
Anexo 3 – Condições e Acções para a extracção de um motivo estruturado ..... 127

xii
Lista de Figuras
Figura 2.1 – Estrutura do DNA [1] ....................................................................................................... 7
Figura 2.2 – Comparação entre o RNA e o DNA, adaptado de [2]. .................................................... 8
Figura 2.3 – Dogma Central da Biologia Molecular – adaptado de [3].. ............................................. 9
Figura 2.4 – Expressão dos genes em eucariotas e procariotas, adaptado de [4]. .......................... 10
Figura 2.5 – As três fases da transcrição, adaptado de [5]. ............................................................. 12
Figura 2.6 – Esquema de regulação nos procariotas, adaptado de [6]. ........................................... 13
Figura 2.7 – Esquema de regulação nos eucariotas, adaptado de [6]. ............................................ 13
Figura 3.1 – Cenário típico de e-Science, adaptado de [7]. .............................................................. 16
Figura 3.2 – Organização em camadas de um ambiente Grid. ........................................................ 18
Figura 3.3 – Arquitectura da versão 4 do Globus Toolkit. ................................................................. 20
Figura 4.1 – Instanciação do problema da Descoberta de Motivos com 𝒍𝒍 = 𝟓𝟓,𝒆𝒆 = 𝟏𝟏, 𝒒𝒒 = 𝒕𝒕 = 𝟑𝟑. .... 26
Figura 4.2 – Exemplo de PWM/PSSM para um padrão de comprimento 7. .................................... 29
Figura 4.3 – Exemplo de uma matriz de dados de um microarray.. ................................................. 33
Figura 4.4 – Exemplo de conjunto de dados de microarray de cor dupla, correspondente ao
desenvolvimento embrionário e adulto do organismo Drosophila melanogaster [96]. ..................... 34
Figura 4.5 – Diferentes tipos de biclusters.. ...................................................................................... 36
Figura 4.6 – Árvore de sufixos generalizada para as strings TACTA$ e CACTCA$ [119].. ............. 39
Figura 4.7 – Motivo complexo composto por 𝒑𝒑 caixas.. .................................................................... 40
Figura 4.8 – Exemplo de extracção de motivos simples utilizando uma árvore lexicográfica virtual (à
esquerda), que representa todos os motivos de comprimento 𝒌𝒌 = 𝟐𝟐 no alfabeto 𝚺𝚺 = {𝑨𝑨,𝑪𝑪,𝑮𝑮,𝑻𝑻}, e
uma árvore de sufixos (à direita) representando o conjunto de sequências 𝑻𝑻𝑨𝑨𝑪𝑪,𝑪𝑪𝑪𝑪𝑪𝑪.. .................. 41
Figura 4.9 – Extracção de motivos complexos com motivos simples de comprimento 𝒌𝒌. ................ 42
Figura 4.10 – Exemplo de factor tree para a string AGACAGGAGGC$ com 𝒍𝒍 = 𝟑𝟑 [119]. ................ 43
Figura 4.11 – Exemplo de box-links (à esquerda) e factor tree (à direita). ....................................... 44
Figura 4.12 – Construção da matriz de co-ocorrências no algoritmo MUSA [125]. .......................... 45
Figura 4.13 – Matriz de dados de expressão antes (à esquerda) e depois (à direita) da
transformação efectuada no alfabeto [126]. ...................................................................................... 48
Figura 4.14 – Árvore de sufixos generalizada com suffix-links para a matriz de dados da Figura
4.13. ................................................................................................................................................... 49
Figura 4.15 – e-ccc-biclusters máximos e não triviais com 1 erro [127]. .......................................... 50

xiii
Figura 5.1 - Vista geral sobre o cluster hermes. ............................................................................... 52
Figura 5.2 – Organização física da arquitectura de portal iNquiry [131]. .......................................... 53
Figura 5.3 – Vista genérica sobre uma árvore de programas [135]. ................................................. 55
Figura 5.4 – Texto de ajuda e parâmetros obrigatórios/condicionais [135]. ..................................... 56
Figura 5.5 – Diagrama do pacote smadeira em UML. ...................................................................... 59
Figura 5.6 – Diagrama UML das classes relevantes na implementação do algoritmo Cheng e
Church. .............................................................................................................................................. 62
Figura 5.7 – Workflow da técnica de teste baseado em modelos, adaptado de [147]. .................... 64
Figura 5.8 – Estrutura básica de uma tabela de decisão, adaptado de [150]. ................................. 68
Figura 5.9 – Tabela de decisão para aplicações de extracção de motivos simples. ........................ 70
Figura 5.10 – Tabela de decisão para aplicações de extracção de motivos compostos (parte 1). .. 72
Figura 5.11 - Tabela de decisão para aplicações de extracção de motivos compostos (parte 2). ... 73
Figura 5.12 – Resultados dos testes para as aplicações de descoberta de motivos.. ..................... 76
Figura 6.1 – Exemplo de representação utilizando motif logos de um conjunto de famílias
identificadas com o algoritmo MUSA [125]. ...................................................................................... 82
Figura 6.2 – Exemplo de workflow utilizando a aplicação MUSA instalada no cluster hermes e a
versão 1.7 da Taverna Workbench. .................................................................................................. 87

xiv
Lista de Abreviaturas (P)SMILE – (Parallel) Structured Motif Inference and Evaluation
ACV – Average Column Variance
ADI – Abstract Device Interface
ADN/DNA – Ácido Desoxirribonucleico / Deoxyribonucleic Acid
API – Application Programming Interface
ARN/RNA – Ácido Ribonucleico / Ribonucleic Acid
ARV – Average Row Variance
ccc-bicluster – contiguous column coherent bicluster
CGI – Common Gateway Interface
CNM – Clauset Newman Moore
FTP – File Transfer Protocol
GO – Gene Ontology
GSI – Grid Security Infrastructure
HMM – Hidden Markov Model
HPC – High Performance Computing
HTTP – HyperText Transfer Protocol
JAR – Java ARchive
MBT – Model-Based Testing
MPI – Message Passing Interface
MSR – Mean Squared Residue
MUSA – Motif finding using an UnSupervised Approach
NAT – Network Address Translation
OGSA – Open Grid Services Architecture
PSSM – Position Specific Scoring Matrix
PWM – Position Weight Matrix
RDF – Resource Description Format
REST – REpresentation State Transfer
rNTP – riboNucleotide Tri-Phosphate
RPC – Remote Procedure Call
SGE – Sun Grid Engine
SOAP – Simple Object Access Protocol
UML – Unified Modeling Language
WSDL – Web Services Description Language
WSRF – Web Services Resource Framework
XML – eXtensible Markup Language

xv

1
Introdução
1.1 Contexto
A investigação que constitui a base deste documento decorreu no âmbito do projecto
“BioGrid – Algoritmos Paralelos Para Anotação de Genes”, executado pelo grupo ALGOS/KDBIO
(ALGorithms for Simulation and Optimization / Knowledge Discovery and BIOinformatics) do
INESC-ID, Lisboa, e que contou com a participação do Instituto Gulbenkian de Ciência. O projecto
em questão tinha como objectivo capitalizar o conhecimento que a equipa de investigação possuía,
sobre técnicas de data mining e Bioinformática, para melhorar abordagens existentes para a
descoberta de motivos em regiões promotoras e análise de dados de expressão de genes, bem
como propor novos métodos. Em particular, pretendia-se estudar as possibilidades de
paralelização deste tipo de algoritmos.
O trabalho subjacente a esta tese consistiu, essencialmente, na disponibilização, no cluster
hermes do Instituto Gulbenkian de Ciência, dos algoritmos desenvolvidos no projecto BioGrid. Este
cluster corresponde ao centro de computação de alta performance que suporta o Centro Português
de Bioinformática. Realizaram-se as tarefas necessárias de implementação, instalação e teste,
bem como a criação das interfaces Web e documentação para as várias aplicações. Foi ainda
desenvolvido um estudo onde a técnica de testes baseados em modelos foi utilizada para
avaliação do software.
Este trabalho foi suportado pelo projecto BioGrid POSI/SRI/47778/2002, financiado pela
Fundação para a Ciência e Tecnologia e pelo programa POSI.

2
1.2 Objectivos
Após o término de projectos de sequenciação de genomas de vários organismos, bem
como de outros projectos semelhantes, verificou-se uma explosão no volume de dados biológicos
disponíveis para análise. Perante a impossibilidade de analisar estes dados de forma manual, dado
o esforço avassalador a que tal obrigaria, têm vindo a multiplicar-se as iniciativas que visam
desenvolver métodos computacionais para processamento de dados biológicos.
A Bioinformática tem emergido como uma disciplina que integra contribuições de áreas
como a matemática, a informática, a química e a estatística, com o intuito de analisar de forma
eficiente o grande volume de dados biológicos disponíveis actualmente. O objectivo desta análise é
o de extrair informação biológica relevante destes dados que possibilite a anotação de genomas e,
em última instância, a modelação e simulação de sistemas biológicos complexos. Duas tarefas
importantes neste contexto são a identificação de locais de ligação para factores de transcrição e a
análise de dados de expressão de genes.
Os locais de ligação para factores de transcrição são pesquisados através da análise de
regiões reguladoras de genes de um mesmo organismo ou de genes relacionados de organismos
diferentes. A descoberta de motivos é o problema de descobrir estes locais de ligação,
normalmente designados motivos, sem conhecimento prévio sobre as suas características. A
identificação correcta destes locais de ligação e de sequências promotoras contribui para melhorar
a nossa capacidade de prever genes, abrindo caminho para a previsão, quando possível, da sua
função.
Os chips de DNA, usualmente designados microarrays, são utilizados para medir o nível de
expressão de um grande número de genes segundo um conjunto de condições experimentais.
Actualmente, a técnica de data mining designada por biclustering tem-se assumido como aquela
que possibilita a obtenção de melhores resultados na extracção de padrões de expressão locais
dos dados de microarrays. Estes padrões de expressão podem ser a chave para determinar a
função de muitos genes e para a classificação de condições.
O sucesso das ferramentas de análise de dados biológicos pode ser incrementado com a
sua integração num ambiente de computação distribuída e paralela, algo que é conferido pelos
paradigmas de computação em cluster e Grid. Não só se verifica um aumento ao nível do número
de experiências ou ensaios passíveis de serem realizados em simultâneo, como a capacidade de
processamento e armazenamento de dados é superior. Além disso, promove-se a integração de
dados e informação proveniente de diferentes recursos.
O trabalho realizado nesta dissertação, integrado no projecto “BioGrid - Algoritmos
Paralelos Para Anotação de Genes”, é o primeiro esforço no sentido de disponibilizar algoritmos
eficientes, de inferência de motivos e de análise de dados de expressão de genes, num cluster
computacional. Deste modo, o objectivo principal desta tese consiste na implementação, instalação

3
e teste, num cluster de máquinas, dos algoritmos desenvolvidos no âmbito do projecto BioGrid.
Pretendia-se que as aplicações estivessem disponíveis e fossem facilmente utilizáveis por uma
grande comunidade de utentes de Bioinformática, pelo que foram também criadas as necessárias
interfaces Web e a documentação de suporte.
Além dos pontos referidos no parágrafo anterior, pretendeu-se desenvolver métodos
formais para teste deste tipo de aplicações. Por fim, esta dissertação tinha ainda como objectivo
apresentar algumas directrizes para trabalho futuro nestas áreas.
1.3 Contribuições
Como resultado desta tese, algumas das mais sofisticadas e eficientes aplicações de
descoberta de motivos e de análise de dados de expressão de genes estão agora disponíveis para
uma vasta comunidade de utentes de Bioinformática, num sistema robusto, integrativo e de fácil
utilização. Todas as aplicações estão acompanhadas de documentação detalhada sobre a sua
utilização.
Além disso, apresentam-se nesta tese métodos formais de teste que poderão ser bastante
úteis na avaliação deste tipo de aplicações. O estudo realizado com a técnica de testes baseados
em modelos permitiu retirar ilações concretas sobre a qualidade do software e algoritmos
trabalhados. Por último, apresentam-se algumas directrizes e perspectivas de trabalho futuro.
1.4 Organização da tese
No próximo capítulo são apresentados os conceitos fundamentais de biologia molecular
que serão necessários para a compreensão do restante desta tese. É dada especial relevância aos
conceitos relacionados com a expressão dos genes e os mecanismos de regulação da mesma,
nomeadamente quando essa regulação se dá ao nível da transcrição.
No terceiro capítulo, descrevem-se os paradigmas de computação em cluster e em malha
de computadores, mormente no que toca à sua ligação aos conceitos de e-Science e experiência
in silico. Descreve-se ainda o conceito de computação paralela e de que forma este se relaciona
com os paradigmas de computação apresentados.
No quarto capítulo, começa-se por se fazer uma breve introdução às disciplinas de
Bioinformática e Biologia Computacional. Em seguida, apresentam-se os problemas de descoberta
de motivos e de biclustering, acompanhados do estado da arte nas respectivas áreas de
investigação. Por fim, descrevem-se em detalhe PSMILE, RISO, MUSA, Cheng e Church, CCC-
Biclustering e e-CCC-Biclustering, os algoritmos desenvolvidos no projecto BioGrid e trabalhados
no âmbito desta dissertação.

4
No quinto capítulo, começa-se por se fazer uma pequena introdução ao cluster hermes
disponível no Instituto Gulbenkian de Ciência, com especial foque no software e hardware que
suportam a execução de aplicações de Bioinformática. Seguidamente, descrevem-se os
procedimentos de instalação, configuração e teste, bem como de criação de interfaces Web e
documentação, que estiveram envolvidos no deployment dos algoritmos/aplicações no cluster.
Finalmente, apresenta-se um estudo onde a técnica de testes baseados em modelos é aplicada às
aplicações instaladas no cluster hermes.
No sexto e último capítulo, analisam-se as conclusões e limitações do trabalho realizado.
São também fornecidas algumas directrizes para trabalho futuro. Por fim, apresentam-se algumas
perspectivas de futuro para a investigação em Bioinformática e Biologia Computacional, bem como
para o desenvolvimento de sistemas de e-Science.
1.5 Convenções
Na escrita desta tese adoptaram-se as convenções habituais em textos de ciência
computacional e de Bioinformática. Contudo, há alguns aspectos que poderão causar dúvidas ou
confusão e, como tal, devem ser referidos.
Tal como acontece na maioria dos textos científicos de Bioinformática, os termos
sequência e string são utilizados como sinónimos um do outro. Por string entenda-se uma cadeia
de caracteres ou, por outras, palavras, um conjunto ordenado de símbolos de um alfabeto pré-
definido. De notar que substring e subsequência não são sinónimos. Uma substring corresponde a
uma porção consecutiva de uma cadeia de caracteres, ao passo que uma subsequência é também
uma porção de uma cadeia de caracteres, não tendo no entanto que ser consecutiva. Neste
sentido e como veremos nos capítulos seguintes, um motivo simples é uma substring de uma
sequência, enquanto um motivo composto é uma subsequência de uma sequência.
De referir ainda que os termos “motivo composto”, “motivo complexo” e “motivo
estruturado” são utilizados para designar motivos com mais do que um componente. Para designar
os componentes, utiliza-se, por vezes, o termo “caixa”. Igualmente, de realçar que nesta tese não
se utiliza o termo “modelo” para designar motivos, algo que acontece em alguns textos.
É importante mencionar também o termo “deployment”, que é frequentemente utilizado
como referência ao conjunto de tarefas que devem ser levadas a cabo para tornar uma aplicação
de software (ou conjunto de aplicações) disponível para utilização. Entre essas tarefas incluem-se
instalação, configuração, adaptação, actualização, entre outras.
Por fim, de referir o termo “data mining”, utilizado em ciência computacional para designar
um conjunto de técnicas que envolvem a exploração de grandes quantidades de dados, com o
objectivo de encontrar padrões consistentes que permitam identificar relacionamentos sistemáticos
entre variáveis.

5
Capítulo 2 Fundamentos de Biologia Molecular
Neste capítulo apresentam-se os conceitos fundamentais de biologia molecular que serão
necessários para a compreensão do restante desta tese.
2.1 A molécula de DNA
A humanidade tem preocupações muito antigas sobre a fenomenologia associada à
hereditariedade. Contudo, só em 1865, graças ao trabalho do monge agostiniano Gregor Mendel,
foi possível constatar que as características de um indivíduo são determinadas por factores
discretos herdados dos progenitores, factores esses que, mais tarde, seriam denominados genes.
As experiências de Mendel envolviam essencialmente as ervilhas de cheiro do jardim do mosteiro
onde vivia. Mendel concentrou-se em características específicas como a cor da semente,
concluindo que as formas alternativas de cada característica eram especificadas por formas
alternativas do mesmo gene, sendo cada uma destas “versões” designada por alelo. O monge
conseguiu ainda provar que os genes permanecem intactos durante a transmissão de pai para
filhos, apesar de a sua combinação poder resultar numa característica diferente da dos
progenitores.
Sensivelmente na mesma altura, o biólogo suíço Friedrich Miescher descobriu, no núcleo
celular, uma substância desconhecida até então e que designou por ácido nucleico. Mais tarde,
esta substância viria a ser conhecida como a molécula de ADN (ácido desoxirribonucleico), ou
DNA, segundo a designação anglo-saxónica.

6
Os resultados de Mendel foram ignorados durante quase 40 anos, até que três cientistas
europeus (Hugo de Vries, Carl Correns e Erick von Tschermak) reavivaram o seu trabalho e
confirmaram os seus resultados. Nessa época, existiam já evidências fortes de que as células
constituíam as unidades básicas da vida no planeta e de que estruturas designadas cromossomas
actuavam como “transportadoras da hereditariedade”.
No início do século 20, pensava-se que polímeros de aminoácidos denominados proteínas
constituíam as moléculas responsáveis pelo transporte de informação hereditária de geração para
geração. Tal noção devia-se sobretudo ao facto de as proteínas possuírem um alfabeto mais rico,
composto por 20 aminoácidos, relativamente ao alfabeto do DNA composto por apenas 4
nucleótidos. No entanto, nos anos 40, foi possível demonstrar, através de experiências com
bactérias virulentas, que os genes eram compostos por DNA. Concluiu-se que a molécula de DNA
transporta toda a informação genética necessária para controlar as actividades celulares. Em 1953,
Watson e Crick descobriram a estrutura tridimensional do ácido desoxirribonucleico.
O DNA é composto por duas cadeias de nucleótidos que formam uma hélice dupla (Figura
2.1). As duas cadeias da hélice estão presas por pontes de hidrogénio entre as bases. Um
nucleótido é uma molécula composta por uma base nitrogenada, uma pentose (desoxirribose no
caso do DNA) e um grupo fosfato. Existem quatro tipos de nucleótidos de DNA, os quais diferem
apenas nas bases nitrogenadas: Adenina (A), Citosina (C), Guanina (G) e Timina (T). Como a
Adenina faz sempre par com a Timina e a Citosina faz sempre par com a Guanina, as duas
cadeias de nucleótidos denominam-se complementares. Tendo em conta essa propriedade,
Watson e Crick propuseram que, durante a replicação do DNA, uma das cadeias serve de molde
para a recreação da outra cadeia. O mecanismo concreto para a cópia do material genético foi
determinado cinco anos depois com os trabalhos de Arthur Kornberg, Mattew Meselson e Frank
Stahl. Kornberg isolou a DNA polimerase, a enzima que sintetiza o DNA, adicionando nucleótidos
complementares ao molde fornecido por uma das cadeias. Meselson e Stahl demonstraram,
através de experiências com isótopos de azoto, que uma das cadeias de DNA é mantida intacta
durante a replicação, isto é, que o “novo DNA” é gerado através da cópia do “antigo”.
A composição do DNA é idêntica em todas as células, mas existem algumas diferenças
consoante o organismo em causa seja eucariota ou procariota. No primeiro caso, em que se
englobam organismos como animais, plantas e fungos, a molécula de DNA encontra-se dentro de
um núcleo individualizado e delimitado pela membrana celular, estando associado a determinadas
proteínas para formar um complexo designado cromatina. No segundo caso, correspondente a
organismos dos domínios Bacteria e Archaea, não existe núcleo celular definido e a molécula de
DNA encontra-se espalhada pelo citoplasma (área intra-celular).
Em termos moleculares, um gene corresponde a um segmento de DNA com a capacidade
de codificar proteínas. Ao conjunto de todos os genes de um organismo, isto é, a toda a
informação hereditária codificada no DNA de um organismo, dá-se o nome de genoma. Um
genoma, tal como cada gene individual, pode ser representado por uma sequência de caracteres

7
definida sobre um alfabeto composto pelos quatro tipos de nucleótidos referidos no parágrafo
anterior: Σ = {𝐴𝐴,𝐶𝐶,𝐺𝐺,𝑇𝑇}.
Os genes são responsáveis por codificar a estrutura das proteínas e, por esse motivo, as
regiões do genoma que correspondem a genes dizem-se codificantes. Normalmente, cada gene é
responsável por codificar uma proteína. As proteínas estão envolvidas em praticamente todas as
actividades biológicas e a sua função celular pode ser estrutural, hormonal, de defesa (anticorpos),
energética, enzimática, de condução de gases ou de regulação de genes. A função de uma
proteína está associada, em última instância, à sua estrutura e às conformações que adopta. É
importante conhecer a estrutura para podermos inferir sobre a possível função da proteína. Por
outro lado, o conhecimento da sequência específica de aminoácidos que compõe a proteína não
basta para conhecer a sua estrutura. Sabe-se apenas que certas estruturas e subestruturas estão
associadas a padrões observáveis ao nível da sequência.
Figura 2.1 – Estrutura do DNA [1]

8
Existem também regiões do genoma que não transportam informação genética e, portanto,
não codificam proteínas. Estas regiões dizem-se reguladoras e determinam quando e em que
células serão sintetizadas determinadas proteínas. Nas células eucariotas, encontramos
frequentemente genes com um elevado número de regiões não codificantes. As regiões
codificantes, designadas exões, estão separadas entre si por regiões não codificantes, designadas
intrões.
Um ácido nucleico cuja composição é muito semelhante à do DNA é o ARN, ou RNA,
segundo a designação anglo-saxónica. Existem, contudo, três diferenças principais entre as duas
moléculas. Em primeiro lugar, enquanto o DNA está organizado em duas cadeias, o RNA
apresenta, geralmente, apenas uma cadeia de nucleótidos e de menor dimensão
comparativamente às do DNA. Em segundo lugar, no que toca à pentose, o RNA contém a ribose
e o DNA contém a desoxirribose. Em terceiro lugar, o nucleótido complementar à Adenina não é a
Timina, mas sim o Uracilo, que é basicamente uma versão não metilada da Timina. Estas
diferenças estão esquematizadas na Figura 2.2.
Figura 2.2 – Comparação entre o RNA e o DNA, adaptado de [2].

9
2.2 Expressão dos genes
Quando falamos em “expressão de genes”, referimo-nos ao processo através do qual a
informação genética existente num gene é transformada num produto funcional, seja este uma
proteína ou RNA. Este processo pode ser entendido através do dogma central da biologia
molecular (Figura 2.3), que estabelece um percurso para o fluxo de informação genética: DNA
RNA proteína.
Figura 2.3 – Dogma Central da Biologia Molecular – adaptado de [3]. As interacções mais comuns estão identificadas com a cor azul. As interacções que ocorrem apenas em casos específicos estão identificadas com a cor vermelha. 1 – Transcrição da sequência de DNA para uma sequência complementar de RNA; 2 – Tradução do RNA numa proteína; 3 – Replicação de DNA, que permite a transmissão de informação genética; 4 – Replicação de RNA 1; 4 – Transcrição inversa de RNA para DNA2 ; 6 – Tradução directa de DNA para proteína 3
1 Pensa-se que a replicação do RNA é o mecanismo que os vírus de RNA (vírus que possuem RNA como material genético) utilizam para se replicarem.
2 Ocorre nos chamados retrovírus, sendo o HIV o exemplo mais conhecido.
3 Possível num ambiente desprovido de células (tubo de ensaio), segundo experiências realizadas com estratos da E. Coli.
.

10
De acordo com este princípio, o RNA é sintetizado a partir de um molde de DNA com um
processo que se designa por transcrição. Durante este processo, a sequência de DNA é copiada
por uma enzima designada por RNA polimerase para uma sequência complementar de RNA. Ao
RNA resultante dá-se o nome de RNA mensageiro (mRNA). No citoplasma, o mRNA é
transformado em aminoácidos através de um processo designado por tradução. Deste modo e
como referimos anteriormente, as moléculas de RNA são intermediárias na expressão da
informação genética.
A ocorrência da transcrição e da tradução dá-se de forma distinta consoante o organismo
seja eucariota ou procariota. Nos procariotas, os dois processos ocorrem praticamente em
simultâneo e ambos no citoplasma. Nos eucariotas, os dois processos ocorrem em locais distintos
da célula. A transcrição dá-se no núcleo, ao passo que a tradução ocorre no citoplasma, após o
transporte do mRNA para esse local (Figura 2.4).
Figura 2.4 – Expressão dos genes em eucariotas e procariotas, adaptado de [4].
Sendo as proteínas os produtos finais da expressão dos genes, tem de existir,
naturalmente, uma correspondência entre o alfabeto dos nucleótidos e o alfabeto dos aminoácidos.
A essa correspondência dá-se o nome de código genético. Deste modo, cada grupo de três
nucleótidos, designado codão, codifica um aminoácido, excepção feita ao codão de terminação,
que indica o fim da síntese da proteína. Existe ainda o codão de iniciação, que indica o início da
síntese da proteína, mas que também codifica um aminoácido. Um aspecto interessante é o facto
de vários codões poderem codificar o mesmo aminoácido, mas nenhum codão poder ser utilizado
para codificar mais do que um aminoácido. É por este motivo que o código genético se classifica
como degenerado. O facto de o código genético ser degenerado permite a existência de mutações

11
“silenciosas”, ou seja, um codão é transformado noutro codão e este último codifica a mesma
proteína que o original.
Sobre as mutações, de referir que estas se tratam de erros ocorridos durante a replicação
do DNA. As mutações podem afectar desde um simples nucleótido a vários milhões.
Independentemente do número de nucleótidos que afectam, as mutações poderão ser de três tipos
distintos: inserções, substituições ou remoções. No primeiro caso, um novo nucleótido é
introduzido na sequência. No segundo caso, um nucleótido existente na sequência é substituído
por outro. Por fim, no terceiro caso, um nucleótido é removido da sequência.
2.3 Regulação da Expressão dos Genes
Em organismos multicelulares existem, geralmente, tipos de células diferentes, sendo que
as diferenças ocorrem, tanto em termos de estrutura, como em termos de função. Estas células
contêm o mesmo DNA, mas sintetizam diferentes tipos de RNA e proteínas. A explicação para
estas diferenças está nas variações do conjunto de genes que cada célula expressa. Cada célula
tem necessidades metabólicas específicas e apenas expressa um subconjunto dos genes que
constituem o seu genoma. Este subconjunto apresenta carácter variável e pode ser alterado, por
exemplo, como resposta a um estímulo exterior.
Se atentarmos na Figura 2.4, verificamos que existem vários passos do processo de
expressão dos genes em que podemos exercer controlo. Contudo, a regulação ao nível da
transcrição, o primeiro passo na expressão de um gene, adquire particular relevância e tem sido
alvo de intensa análise e investigação. Na secção anterior, indicámos que, durante a transcrição, o
mRNA é sintetizado a partir de um molde de DNA. Esta síntese dá-se em três fases, iniciação,
elongação e terminação, como indicado na Figura 2.5.
Como também já foi referido, todos os genes apresentam regiões que regulam a
transcrição, estando estas situadas, geralmente, a montante dos genes na sequência de DNA.
Uma região reguladora partilhada por todos os genes é conhecida como promotor, providenciando
uma posição que é reconhecida pelo mecanismo de transcrição quando um gene está prestes a
ser expresso. As regiões reguladoras podem apresentar carácter simples ou complexo, mas todas
funcionam como interruptores. Estas regiões contêm locais de ligação (binding sites) aos quais se
ligam proteínas específicas, designadas por factores de transcrição. Algumas destas proteínas
podem funcionar como repressoras ou activadoras, inibindo ou activando a expressão do gene,
respectivamente. À sequência de nucleótidos existente num local de ligação dá-se o nome de
motivo. Esta ligação é possível devido ao facto de a superfície da proteína ser, na maioria da sua
extensão, complementar às características superficiais do DNA na região onde se encontra o
motivo. Tal como referimos na secção anterior, o processo de transcrição é catalisado pela enzima
RNA polimerase, que separa as duas cadeias.
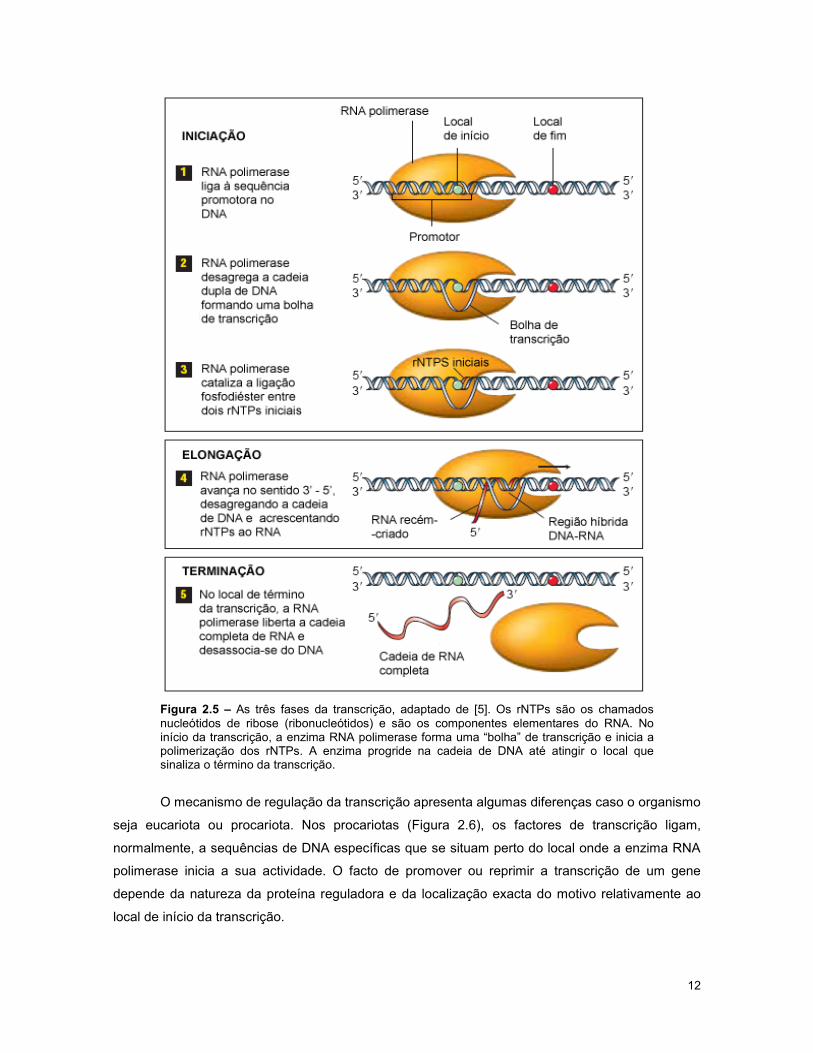
12
Figura 2.5 – As três fases da transcrição, adaptado de [5]. Os rNTPs são os chamados nucleótidos de ribose (ribonucleótidos) e são os componentes elementares do RNA. No início da transcrição, a enzima RNA polimerase forma uma “bolha” de transcrição e inicia a polimerização dos rNTPs. A enzima progride na cadeia de DNA até atingir o local que sinaliza o término da transcrição.
O mecanismo de regulação da transcrição apresenta algumas diferenças caso o organismo
seja eucariota ou procariota. Nos procariotas (Figura 2.6), os factores de transcrição ligam,
normalmente, a sequências de DNA específicas que se situam perto do local onde a enzima RNA
polimerase inicia a sua actividade. O facto de promover ou reprimir a transcrição de um gene
depende da natureza da proteína reguladora e da localização exacta do motivo relativamente ao
local de início da transcrição.
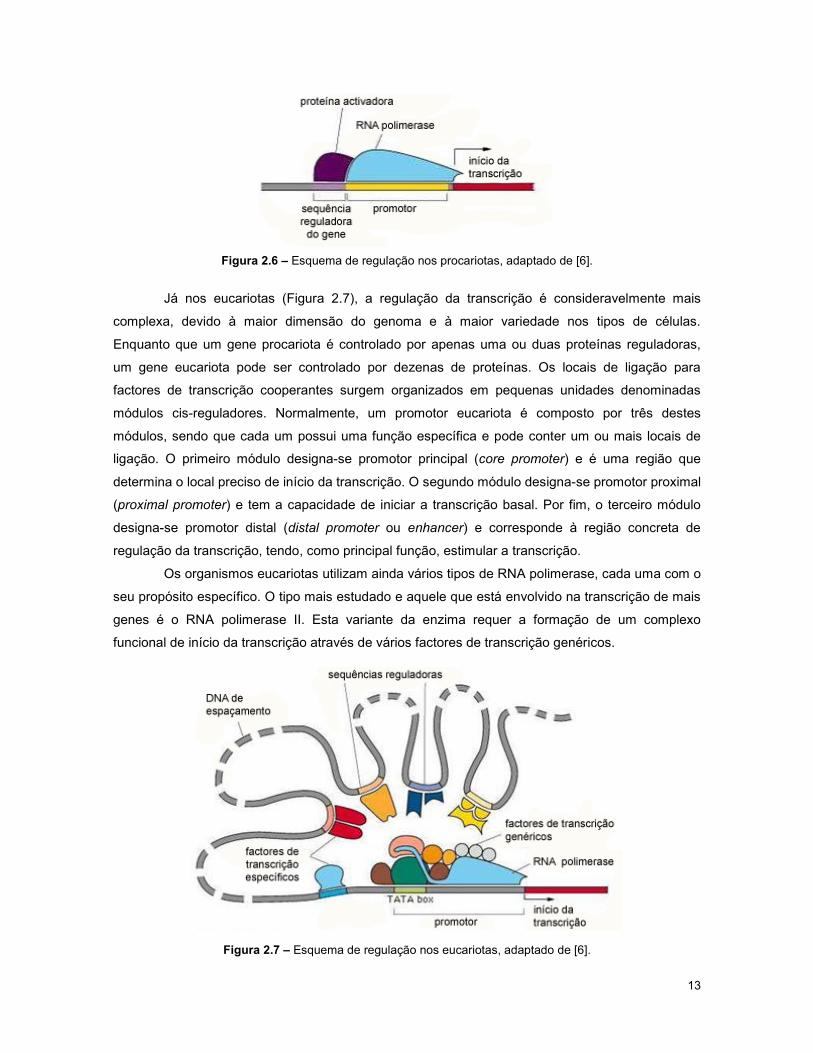
13
Figura 2.6 – Esquema de regulação nos procariotas, adaptado de [6].
Já nos eucariotas (Figura 2.7), a regulação da transcrição é consideravelmente mais
complexa, devido à maior dimensão do genoma e à maior variedade nos tipos de células.
Enquanto que um gene procariota é controlado por apenas uma ou duas proteínas reguladoras,
um gene eucariota pode ser controlado por dezenas de proteínas. Os locais de ligação para
factores de transcrição cooperantes surgem organizados em pequenas unidades denominadas
módulos cis-reguladores. Normalmente, um promotor eucariota é composto por três destes
módulos, sendo que cada um possui uma função específica e pode conter um ou mais locais de
ligação. O primeiro módulo designa-se promotor principal (core promoter) e é uma região que
determina o local preciso de início da transcrição. O segundo módulo designa-se promotor proximal
(proximal promoter) e tem a capacidade de iniciar a transcrição basal. Por fim, o terceiro módulo
designa-se promotor distal (distal promoter ou enhancer) e corresponde à região concreta de
regulação da transcrição, tendo, como principal função, estimular a transcrição.
Os organismos eucariotas utilizam ainda vários tipos de RNA polimerase, cada uma com o
seu propósito específico. O tipo mais estudado e aquele que está envolvido na transcrição de mais
genes é o RNA polimerase II. Esta variante da enzima requer a formação de um complexo
funcional de início da transcrição através de vários factores de transcrição genéricos.
Figura 2.7 – Esquema de regulação nos eucariotas, adaptado de [6].

14
A extracção de motivos em regiões reguladoras oferece importantes dados para a
investigação em biologia molecular. Genes que partilham motivos e são regulados pelo mesmo
factor de transcrição ou pelo mesmo conjunto de factores de transcrição (sendo este o caso mais
usual) dizem-se co-regulados. A existência de co-regulação num dado conjunto de genes permite-
nos inferir importantes informações sobre a função de outros genes e identificar genes em
sequências de DNA. No primeiro caso, se um gene, para o qual se desconhece a função biológica,
partilha motivos com um conjunto de genes de função biológica conhecida, então podemos propor
que a função do primeiro será semelhante à dos restantes. No segundo caso, referimo-nos ao
problema de gene finding ou gene prediction, onde se pretende encontrar pedaços de DNA com
função biológica relevante, nomeadamente com capacidade de codificar proteínas. Ao encontrar
um motivo conhecido numa dada região de uma sequência de DNA, podemos propor que essa
região é codificante e, portanto, corresponde a um gene.
Pelas razões apontadas no parágrafo anterior, a identificação de motivos tem sido alvo de
intensa investigação, nomeadamente utilizando ferramentas computacionais, como veremos no
capítulo 4.

15
Capítulo 3 Computação em Cluster e Grid
Neste capítulo descrevem-se os paradigmas de computação em cluster e Grid, bem como
o conceito de computação paralela.
3.1 Clusters computacionais e malhas de computadores
Nos últimos tempos, temos assistido a um aumento na computação realizada em
computadores convencionais (commodity computers), bem como na performance da rede. Tal
deve-se essencialmente à criação de software mais sofisticado e hardware mais rápido. Com base
na agregação destes computadores convencionais, são construídos sistemas computacionais de
baixo custo e alta performance, os quais se designam por clusters. A computação em cluster
consiste assim na utilização conjunta de dois ou mais computadores com vista a resolver um
problema ou executar uma dada tarefa.
Este tipo de sistemas tem vindo a ganhar importância em aplicações científicas, pois são
vários os problemas que requerem computação de alta performance (High Performance Computing
– HPC). Recentemente, esta classe de problemas tem vindo a tornar-se cada vez mais dependente
do uso colaborativo de recursos distribuídos em larga-escala, tais como bases de dados,
instrumentos científicos, clusters computacionais, entre outros. Neste sentido, surge o termo e-
Science, que corresponde à realização de experiências científicas em ambientes que podem ser
altamente distribuídos e onde existem requisitos elevados de computação ou de armazenamento e
processamento de dados. A Figura 3.1 ilustra um cenário típico de e-Science. Um conceito

16
relacionado é o de experiência in silico, que consiste num procedimento que utiliza informação e/ou
recursos computacionais para testar uma hipótese ou demonstrar um facto. Estas experiências
são, normalmente, realizadas através de workflows4 nos quais se passam dados de um recurso
para outro até que seja atingido o objectivo analítico pretendido.
Figura 3.1 – Cenário típico de e-Science, adaptado de [7].
Do ponto de vista do investigador, o ambiente de e-Science deve permitir [8]: a criação e
descoberta de workflows que satisfaçam os objectivos das experiências científicas a realizar; a
execução, monitorização e controlo de workflows, nomeadamente a inspecção de saídas
intermédias; interacção com ferramentas de terceira parte; integração de informação e recursos;
assistência na visualização e manipulação dos resultados; notificação da chegada de novos dados,
recursos e ferramentas; registo de hipóteses, materiais, métodos, decisões e conclusões; e a
capacidade de personalização do ambiente de trabalho, das anotações e dos métodos de acordo
com o contexto de investigação e/ou organizacional em que o cientista está inserido.
Para além da computação em cluster, outro paradigma tem emergido como resposta aos
requisitos de e-Science: a computação em malha de computadores (Grid computing). Existem
várias definições para uma malha de computadores. O projecto Globus [9] define Grid como “uma
infra-estrutura que permite o uso colaborativo e integrado de computadores de elevada gama,
redes, bases de dados e instrumentos científicos possuídos e geridos por múltiplas organizações”.
Já o projecto Gridbus [10] apresenta a seguinte definição: “Grid é um tipo de sistema distribuído
4 Um workflow consiste numa sequência de operações ou fluxo de eventos em que as saídas de um recurso podem servir de entradas para outros recursos.

17
paralelo que permite a partilha, selecção e agregação dinâmica, em tempo de execução, de
recursos autónomos distribuídos, dependendo da sua disponibilidade, capacidade, performance,
custo e requisitos de qualidade de serviço dos utilizadores”. Em suma, trata-se de uma infra-
estrutura onde recursos computacionais autónomos e distribuídos estão virtualizados.
Clusters e grids apresentam algumas diferenças importantes. Num cluster, os nós5 estão
tightly-coupled, isto é, coexistem todos na mesma sub-rede e estão conectados por ligações de
alto débito. Já uma grid é um sistema inerentemente distribuído e os nós estão loosely-coupled:
podem existir em redes, domínios ou regiões geográficas distintas. Não são necessárias ligações
de alto débito e, normalmente, são utilizadas as ligações de rede disponíveis. As interfaces de
integração realizam o mínimo de considerações sobre os nós cooperantes e, por esse motivo,
estes sistemas apresentam uma maior facilidade em lidar com alterações nos recursos
computacionais disponíveis. É frequente numa grid existirem, não só alterações ao nível do
hardware e software dos nós, como também adições e remoções de nós. São sistemas em que o
dinamismo é uma característica dominante, o que contribui para a sua escalabilidade6
Outra diferença importante reside nas capacidades de computação paralela (o conceito
será explicado com maior detalhe na próxima secção). Numa grid, o poder computacional ou de
armazenamento, existente nos vários nós, é partilhado durante a execução de tarefas. Para tal,
não é necessário que as aplicações estejam paralelizadas: como já foi referido, os recursos
computacionais estão virtualizados e são alocados consoante as necessidades. Contudo, a
realização de uma tarefa numa grid não pode, normalmente, beneficiar da execução de múltiplas
instruções em simultâneo (na secção seguinte veremos que a MPICH-G2 surge como resposta a
este problema) e neste aspecto encontramos uma vantagem dos clusters. De facto, num cluster
pode existir um ambiente paralelo que permita que diferentes porções da mesma tarefa sejam
executadas em paralelo, comunicando entre si e partilhando informação, com vista a uma maior
rapidez de execução. Deste modo, uma grid é mais aconselhável para problemas paralelos
compostos essencialmente por tarefas independentes (usualmente designados problemas de
“granularidade grossa”), ao passo que um cluster, com o seu hardware dedicado, ligações de alto
.
Os nós de um cluster são, geralmente, homogéneos: possuem o mesmo software, o
mesmo hardware e a mesma configuração. Já os recursos computacionais que compõem uma grid
podem ser heterogéneos. Além disso, cada nó de um cluster é um recurso dedicado e o sistema é
gerido por um gestor de recursos centralizado. Numa grid, todos os nós são autónomos e
comportam-se como entidades independentes.
5 Cada recurso computacional existente numa grid ou num cluster toma, usualmente, a designação de “nó”.
6 A escalabilidade é a capacidade de manter a qualidade de serviço quando ocorre aumento de carga, através da adição de recursos com um acréscimo de custo aproximadamente constante.

18
débito e ambiente paralelo, constitui uma solução eficiente para problemas paralelos com tarefas
interdependentes (usualmente designados problemas de “granularidade fina”).
Um aspecto interessante é o facto de uma grid poder incorporar vários clusters. Este tipo
de organização tem vindo a ganhar popularidade e é, hoje em dia, utilizado em muitos sistemas.
As tarefas podem ser alocadas aos nós que melhor se adequam às mesmas: tarefas que sejam
interdependentes, ou onde a comunicação e a cooperação são necessárias, são atribuídas a um
dos clusters; tarefas independentes são atribuídas a todos os nós da grid, inclusivamente aos que
estiverem disponíveis nos clusters que fazem parte do sistema. Esta organização é particularmente
útil se atendermos ao facto de que muitos problemas científicos actuais requerem um misto de
computação de granularidade grossa e de granularidade fina, isto é, tratam-se de problemas
compostos por tarefas independentes que por sua vez são compostas por subtarefas
interdependentes.
As implementações de Grid têm por base a introdução de uma nova camada de software,
designada middleware (Figura 3.2), entre a camada aplicacional e a infra-estrutura “física”
(computadores, redes, entre outros). Do ponto de vista dos programadores, aplicações e
utilizadores, esta camada permite esconder a complexidade e diversidade da Grid, oferecendo
acesso transparente aos recursos de computação nela disponíveis. Entre as funcionalidades
disponíveis no middleware encontram-se: autenticação e autorização, balanceamento da carga e
localização de recursos.
Figura 3.2 – Organização em camadas de um ambiente Grid.

19
A principal solução de middleware Grid existente actualmente é o Globus Toolkit [9].
O Globus Toolkit consiste num conjunto de ferramentas open source que permitem a
construção de grids computacionais e aplicações baseadas no paradigma de malha de
computadores. Com estas ferramentas, é possível a partilha segura de poder computacional e de
armazenamento entre recursos heterogéneos. Estas ferramentas satisfazem a maior parte dos
requisitos definidos na OGSA (Open Grid Services Architecture). A OGSA tem vindo a ser
desenvolvida pelo Global Grid Forum [11] (agora designado Open Grid Forum) e é um esforço no
sentido de criar interfaces standards para praticamente todos os serviços que encontramos numa
aplicação Grid (gestão de trabalhos e de recursos, autenticação e autorização, entre outros). A
OGSA requer serviços com estado7
A WSRF constitui a infra-estrutura que sustenta a maior parte dos serviços do Globus
Toolkit, mas existem também serviços que não utilizam a tecnologia de Web Services, como
podemos observar na
e a equipa de desenvolvimento do Globus Toolkit utilizou a
WSRF (Web Services Resource Framework) para providenciar esse tipo de serviços. A
especificação WSRF foi desenvolvida pelo consórcio OASIS [12] (Organization for Advancement of
Structured Information Standards) e define a forma de implementar os serviços definidos na OGSA
utilizando a tecnologia de Web Services [13], um standard definido pela W3C. Em particular,
especifica como transformar os Web Services em serviços com estado.
Figura 3.3.
Além desta divisão horizontal, existe uma divisão vertical em cinco categorias de
componentes. Os componentes de “Common Runtime” providenciam as ferramentas e bibliotecas
necessárias para a criação de serviços. A categoria de segurança engloba um conjunto de
componentes baseados na GSI (Grid Security Infrastructure), a qual possui métodos para
autenticação e autorização que tornam as comunicações em Grid seguras. Na categoria “Data
Management” encontramos componentes que providenciam as funcionalidades de transmissão e
armazenamento de dados (como podemos observar na imagem, nestes componentes inclui-se o
GridFTP, uma extensão do protocolo FTP, File Transfer Protocol, que possibilita o movimento de
dados de forma robusta e segura num ambiente Grid). A categoria “Information Services”
corresponde aos componentes de descoberta e monitorização de recursos (Monitoring and
Discovery Services). Por fim, na categoria de “Execution Management” encontramos componentes
que tratam da alocação de recursos, da recolha de resultados e da submissão, monitorização e
escalonamento de trabalhos num ambiente Grid.
7 Serviços com estado mantêm informação entre chamadas e são úteis em situações onde é necessário realizar um diálogo em vários passos.

20
Figura 3.3 – Arquitectura da versão 4 do Globus Toolkit.
3.2 Computação Paralela
A computação paralela refere-se a uma forma de computação onde várias instruções são
executadas em simultâneo. Para tal é necessário começar por dividir problemas complexos em
problemas mais simples, encontrando um conjunto de problemas de menor complexidade que
podem ser resolvidos concorrentemente.
A computação em paralelo introduz vários desafios ao nível da programação, visto que
passam a ser necessárias preocupações com concorrência, comunicação e sincronização. Além
destes aspectos, é muitas vezes difícil dividir um problema em unidades que possam ser
executadas em paralelo e existem mesmo problemas que não são paralelizáveis. A contrapartida
é, naturalmente, um acréscimo na velocidade de execução das aplicações, daí a importância do
paralelismo na computação de alta performance.

21
A computação paralela apresenta várias variantes. O chamado paralelismo ao nível do bit
consiste no aumento do tamanho da palavra do processador. O paralelismo ao nível das instruções
consiste na reordenação das instruções de um programa, de modo a criar grupos de instruções
que possam ser executados de forma individual e em paralelo. O paralelismo ao nível dos dados
tem por base a existência de ciclos programáticos onde sequências semelhantes de operações
são executadas sobre elementos de uma estrutura de dados de grande dimensão. Neste caso, a
estrutura de dados pode ser dividida por vários nós computacionais (é este o método seguido na
paralelização do algoritmo SMILE, como veremos no capítulo seguinte). Finalmente, o paralelismo
ao nível das tarefas contrasta com o paralelismo ao nível dos dados, no sentido em que explora a
distribuição de diferentes tarefas (usualmente threads) por vários nós computacionais. Deste modo,
cada nó executa uma tarefa diferente sobre os mesmos dados.
Em termos de hardware, o paralelismo pode ser concretizado de diversas formas. Uma das
hipóteses consiste em utilizar processadores multi-core, isto é, com múltiplos núcleos de execução.
Uma outra hipótese consiste em utilizar multiprocessamento simétrico, ou seja, trabalhar com
sistemas computacionais com múltiplos processadores idênticos que partilham memória. Outra
hipótese consiste em recorrer a computação distribuída, onde existem vários nós computacionais
ligados por uma rede. Dentro da computação distribuída podemos considerar os já referidos
clusters, processamento paralelo em massa 8
A implementação mais popular do standard MPI designa-se MPICH [15]. Trata-se de uma
implementação completa do standard MPI-1 que possui extensões para suportar as
e a também já referida computação em Grid. Por fim,
existem também sistemas paralelos mais específicos, normalmente aplicáveis apenas a um
pequeno número de classes de problemas paralelos.
Para terminar e no que toca ao software, têm vindo a ser desenvolvidas variadíssimas
linguagens de programação paralelas, bibliotecas, API’s e modelos de programação em paralelo.
Estas ferramentas podem ser divididas em três tipos: memória partilhada (comunicam através da
manipulação de variáveis em memória partilhadas), memória distribuída (comunicam através da
troca de mensagens) e memória distribuída partilhada.
A especificação mais utilizada actualmente designa-se MPI (Message Passing Interface)
[14]. Esta especificação para uma interface de comunicação de dados em computação paralela
baseia-se na arquitectura paralela de passagem de mensagens. Na especificação MPI, uma
aplicação é constituída por um ou mais processos que podem comunicar entre si (tanto com
comunicação ponto-a-ponto como com comunicação colectiva), utilizando para o efeito funções de
envio e recepção de mensagens. A maioria das implementações da especificação assume que a
mesma aplicação é executada em todos os processos (correspondendo, portanto, a um modelo de
execução SPMD – Single Program Multiple Data), mas, recentemente, têm vindo a ser propostas
várias implementações que permitem a execução de diferentes programas num único job MPI.
8 Um único computador com um grande número de processadores ligados em rede.

22
funcionalidades de entradas/saídas paralelas definidas no standard MPI-2. Os seus autores
tiveram como preocupação principal combinar portabilidade com alta performance. Esse objectivo
é alcançado através de uma organização em camadas: na camada de topo encontramos uma
interface MPI de acordo com o standard. Na camada imediatamente inferior encontramos uma
implementação da interface MPI cujas funções são independentes dos dispositivos de rede e
gestores de processos. A restante funcionalidade necessária é atribuída a camadas inferiores por
meio da ADI (Abstract Device Interface). A ADI tem como função realizar a comunicação entre o
subsistema de rede e as camadas de topo.
Recentemente, foi desenvolvida a MPICH-G2 [16], uma implementação do standard MPI
desenhada especificamente para grids que utilizem o middleware Globus. Esta implementação
utiliza como base a MPICH na qual foi modificada a interface ADI. O objectivo é providenciar num
ambiente Grid as capacidades de comunicação e cooperação oferecidas pela especificação MPI.
Cabe ao Globus Toolkit lidar com a heterogeneidade do sistema, através das suas funções de
autenticação, autorização, alocação de recursos, comunicação e de criação, controlo e
monitorização de processos.

23
Capítulo 4 Algoritmos
Tal como foi referido no primeiro capítulo, a investigação subjacente a esta tese tinha como
objectivo principal a migração dos algoritmos e aplicações de Bioinformática desenvolvidos no
âmbito do projecto BioGrid para um cluster computacional. Neste capítulo apresentam-se os
algoritmos sobre os quais incidiu o trabalho. Começa-se, no entanto, por se fazer uma pequena
introdução às disciplinas de Bioinformática e Biologia Computacional e apresenta-se o estado da
arte dos algoritmos de descoberta de motivos e biclustering.
4.1 Bioinformática e Biologia Computacional
A Bioinformática e a Biologia Computacional envolvem a aplicação de técnicas de
matemática, informática, inteligência artificial, química e estatística com vista à resolução de
problemas biológicos, usualmente ao nível molecular. As duas disciplinas são habitualmente
confundidas, mas existe uma diferença substancial entre elas: a Bioinformática debruça-se sobre a
criação, desenvolvimento e aplicação de algoritmos e técnicas estatísticas e computacionais para a
resolução de problemas formais ou práticos resultantes da análise de informação biológica,
enquanto a Biologia Computacional se refere, genericamente, à investigação de problemas
biológicos baseada em hipóteses e realizada por meios computacionais. O principal objectivo da
Biologia Computacional é o de aumentar e melhorar o conhecimento biológico através do trabalho
com hipóteses, enquanto a Bioinformática se baseia essencialmente na análise de informação
biológica.

24
As principais áreas de investigação em Bioinformática e Biologia Computacional são a
análise de sequências, anotação de genomas, biologia evolucionária computacional, medição de
biodiversidade, análise de expressão de genes, análise do esquema de regulação dos genes,
análise da expressão de proteínas, análise de mutações em patologias cancerígenas, predição da
estrutura de proteínas, modelação de sistemas biológicos, análise de imagens biomédicas e
predição de interacções entre proteínas. Neste capítulo, serão abordadas as áreas de análise de
sequências, análise de expressão de genes e análise dos mecanismos de regulação dos genes.
4.2 Descoberta de Motivos
Como vimos no capítulo 2, a descoberta de locais de ligação para factores de transcrição é
uma das tarefas mais importantes no estudo dos mecanismos que regulam a expressão dos
genes. Esta descoberta consiste, usualmente, na procura de segmentos bem conservados de
pequena dimensão, os chamados motivos, em DNA.
O problema da descoberta/extracção/pesquisa de motivos consiste na descoberta de
motivos sem conhecimento prévio sobre as características dos mesmos. Um outro problema
relacionado, que não será abordado nesta tese, é o da localização de motivos, que consiste em
descobrir estes segmentos com conhecimento prévio sobre as suas características.
A pesquisa de motivos pode ser feita recorrendo a dois métodos: analisando as regiões
reguladoras de genes de organismos diferentes ou analisando as regiões reguladoras de genes do
mesmo organismo.
O primeiro método é conhecido por “footprinting filogenético” [17] e considera que as
mutações em regiões codificantes dos genes se acumulam mais lentamente do que em regiões
não codificantes, devido à pressão evolutiva para manter a função biológica. Deste modo,
pressupõe-se que os elementos reguladores evoluem a um ritmo mais lento do que as sequências
não codificantes em seu redor, estando, por conseguinte, mais conservados no DNA de
organismos considerados próximos na árvore de evolução filogenética. O “footprinting filogenético”
efectua assim comparações de sequências genómicas no sentido de encontrar possíveis
elementos reguladores. Naturalmente, deverá existir uma cuidadosa selecção das sequências a
incluir na análise, nomeadamente no sentido de garantir que as referidas sequências
correspondem a regiões reguladoras de genes que estão relacionados de forma evolutiva (genes
ortólogos).
O segundo método baseia-se na noção de que motivos comuns a um determinado número
de regiões reguladoras têm fortes possibilidades de possuir um papel relevante na regulação da
expressão dos genes considerados. Duas importantes ferramentas auxiliares para este método
são a análise proteómica quantitativa (estudo em larga escala da presença de proteínas) e as
experiências com microarrays. Ambos os métodos permitem agrupar genes que são expressos de

25
forma idêntica segundo determinadas condições experimentais. Alguns destes genes serão co-
regulados e, por essa razão, é possível que apresentem elementos reguladores em comum.
Seleccionado um conjunto de genes através de um dos dois métodos descritos
anteriormente, o próximo passo consiste em identificar as regiões reguladoras. Existem, no
entanto, várias considerações a ter em conta no que toca à localização dos motivos. Nos
eucariotas, os elementos reguladores podem estar tanto a montante como a jusante do local de
início da transcrição. Já nos procariotas, os motivos situam-se usualmente perto do local de início
da transcrição, existindo, contudo, situações em que tal não ocorre e a análise se complica.
Outro ponto a ter em conta é o facto de o mecanismo de transcrição reconhecer motivos
que não são exactos, isto é, locais de ligação que sofreram mutações [18] (podendo estas ser
inserções, remoções ou substituições de nucleótidos, como vimos no capítulo 2). A possibilidade
de algumas sequências terem sido corrompidas leva a que uma abordagem computacional para a
descoberta de motivos tenha de considerar a hipótese de nem todas as sequências em análise
possuírem a mesma sequência de nucleótidos para um dado motivo.
De referir ainda que os motivos não têm de corresponder a sequências contíguas. De
facto, existem dois tipos de sequências de nucleótidos reconhecidas pelos factores de transcrição:
sequências contíguas, designadas por motivos simples; e sequências interrompidas por
espaçamentos, designadas por motivos complexos, compostos ou estruturados.
A generalidade dos primeiros algoritmos para identificação de locais de ligação dos
factores de transcrição considera apenas a extracção de motivos simples. Deste modo, estes
algoritmos pesquisam sequências contíguas de nucleótidos comuns a várias regiões promotoras,
tendo em conta a possibilidade de existirem mutações.
Recentemente, têm sido propostos algoritmos que identificam motivos complexos. Existem
algumas vantagens em considerar motivos complexos. Por um lado, alguns factores de transcrição
apresentam uma estrutura complexa, no sentido em que reconhecem sequências não-contíguas
de nucleótidos. Os motivos complexos constituem assim um modelo mais adequado para estes
locais de ligação. Além disso, os factores de transcrição podem ligar-se de forma cooperativa à
região promotora, o que envolve o reconhecimento de várias sequências contíguas de nucleótidos
separadas por espaçamentos. Por outro lado, a imposição de espaçamentos entre as sequências
facilita a tarefa de distinguir entre motivos biologicamente relevantes para o mecanismo de
transcrição e motivos que estão presentes nas várias regiões promotoras, mas não são
importantes neste contexto.
Qualquer algoritmo para descoberta de motivos deverá ser capaz de lidar com os desafios
apresentados nos parágrafos anteriores, tendo, como já foi referido, informação muito escassa ou
nula sobre os motivos a encontrar.

26
4.2.1 Descrição do problema
O problema da Descoberta de Motivos sem informação prévia, também designado por
Extracção de Motivos ab initio, pode ser enunciado do seguinte modo:
Dado um conjunto de sequências de DNA, 𝑆𝑆 = {𝑆𝑆1,𝑆𝑆2, … , 𝑆𝑆𝑡𝑡}, pretende-se encontrar um
conjunto de motivos, simples e/ou compostos, com comprimento contido num conjunto
{𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 } que ocorre num número 𝑞𝑞 ≤ 𝑡𝑡 de sequências com o máximo de 𝑒𝑒 erros. O parâmetro
𝑞𝑞 é usualmente designado por quorum e o número de erros 𝑒𝑒 corresponde ao número de mutações
ocorridas nos motivos (por outras palavras, pretende-se que a distância de Hamming9
Figura 4.1
entre a
subsequência encontrada e o motivo de consenso seja no máximo 𝑒𝑒). No caso dos motivos
compostos, é comum considerar também que cada componente está separado por uma distância
contida num conjunto {𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 }. A ilustra uma possível instância deste problema
com três sequências:
Figura 4.1 – Instanciação do problema da Descoberta de Motivos com 𝑙𝑙 = 5, 𝑒𝑒 = 1, 𝑞𝑞 = 𝑡𝑡 =3. Um algoritmo de descoberta de motivos deveria encontrar AATAT, TATAT, GATAT e CATAT, estando o nucleótido onde ocorreu a mutação representado com a cor verde e os nucleótidos conservados identificados com a cor vermelha.
Existem muitas outras formulações para o problema, nomeadamente tendo em conta a
perspectiva probabilística, que será mencionada na secção seguinte. Praticamente todas as
variantes do problema da Descoberta de Motivos são NP-Difíceis10
9 A distância de Hamming entre duas cadeiras de caracteres corresponde ao número mínimo de erros (substituições de caracteres) que transformam uma cadeia na outra.
10 Um problema é NP-Difícil se todos os problemas da classe NP forem redutíveis a ele. Os problemas da classe NP são todos aqueles que podem ser resolvidos em tempo polinomial por uma máquina não determinística.
. Tal deve-se essencialmente ao
facto de as características dos motivos “óptimos” (comprimento dos motivos simples/compostos,
número de sequências em que os motivos ocorrem, número de mutações) não serem conhecidas a
priori. Outro ponto que contribui para essa classificação é o requerimento da optimalidade da
solução. De facto, a grande maioria dos algoritmos existentes actualmente sacrifica a certeza de

27
encontrar a melhor solução em favorecimento da rapidez de execução. Além disso, tem sido feito
um esforço bastante considerável no desenvolvimento de métodos para estruturar a procura,
aspecto onde reside parte da ineficiência dos algoritmos mais básicos.
Na secção seguinte apresenta-se o estado da arte relativo aos algoritmos de descoberta
de motivos.
4.2.2 Estado da arte dos algoritmos de descoberta de motivos
Nos últimos anos, foram propostos mais de uma centena de algoritmos para identificação
de motivos, bem como alguns métodos para classificação dos primeiros. As várias abordagens têm
como pontos de variação a representação escolhida para os motivos, a função objectiva utilizada
para determinar a “qualidade” de um motivo, o procedimento de procura adoptado e a optimalidade
da solução.
A divisão mais comum encontrada na literatura distribui os algoritmos por dois grandes
grupos: algoritmos combinatórios e algoritmos probabilísticos [19].
Os algoritmos combinatórios baseiam-se na enumeração implícita ou explícita de todos os
padrões possíveis e podem seguir duas abordagens distintas: enumerar todos os padrões
possíveis com um comprimento fixo 𝑙𝑙 (𝑙𝑙-ímeros) e verificar a ocorrência de cada padrão nas
sequências de entrada com um máximo de 𝑒𝑒 erros; ou gerar a vizinhança-𝑒𝑒11 de todas os 𝑙𝑙-ímeros
nas sequências de entrada.
Os algoritmos probabilísticos baseiam-se na utilização de modelos probabilísticos para os
motivos, sendo que os parâmetros livres dos modelos são estimados utilizando o princípio da
máxima verosimilhança12 ou inferência Bayesiana13
De um modo geral, podemos dizer que os métodos probabilísticos são muito bons na
descoberta de motivos “fortes”, isto é, motivos que ocorrem numa porção elevada de sequências,
. O objectivo destes algoritmos é assim o de
encontrar o motivo ou conjunto de motivos que “melhor explica” as sequências de entrada.
Usualmente consistem num procedimento iterativo dividido em dois passos: no primeiro, são
determinadas as ocorrências mais prováveis do motivo com base no modelo calculado na iteração
anterior; no segundo, actualiza-se o modelo do motivo com base nas ocorrências determinadas no
passo anterior.
11 A vizinhança- 𝑒𝑒 de uma cadeia de caracteres 𝑠𝑠 corresponde ao conjunto de strings que estão a uma distância (de Hamming ou Levenshtein) de 𝑠𝑠 igual ou inferior a 𝑒𝑒.
12 O princípio da máxima verosimilhança estabelece que o melhor o conjunto de parâmetros para um dado modelo desconhecido é aquele que tornar os dados observáveis mais verosímeis segundo aquele modelo.
13 A inferência Bayesiana permite actualizar ou inferir a probabilidade de uma hipótese ser verdadeira com base num conjunto de observações.

28
mas não são tão bons na identificação de motivos “fracos”, isto é, que ocorrem numa porção
reduzida de sequências. Têm também a vantagem de apenas necessitarem da especificação de
um número reduzido de parâmetros. Em termos de desvantagens, há ainda que referir o facto de
serem bastante sensíveis a pequenas variações nos dados de entrada, o facto de não oferecerem
a garantia de convergir para um máximo global e o facto de, geralmente, apenas considerarem a
ocorrência de um único motivo nas sequências de entrada.
Os métodos combinatórios, quando executados com parâmetros adequados, são bons
para identificar motivos “fortes” e “fracos”. Contudo, apresentam as desvantagens de requererem a
especificação de um elevado número de parâmetros e reportarem muitas vezes um conjunto
enorme de motivos. Sobre este último ponto, tem sido realizada investigação intensa no sentido de
providenciar métodos, nomeadamente estatísticos, que permitam distinguir as sequências
biológicas relevantes de entre todos os motivos reportados pelos algoritmos combinatórios.
Existem, contudo, várias propostas de classificação alternativas para os métodos de
pesquisa de motivos. O estudo de Pavesi et al. [20] divide os algoritmos em duas categorias:
baseados em consenso e baseados em alinhamento. Já Wasserman e Krivan [21] dão maior
ênfase aos conceitos de biologia molecular em que se baseia a descoberta de motivos em regiões
reguladoras, mencionando aspectos como as ilhas CpG e filogenia. Brazma et al. [22] distinguem
os métodos para descoberta de motivos tendo em conta o facto de estes usarem explicitamente
sequências negativas, expressividade dos modelos de padrões, se os padrões são determinísticos
ou estatísticos e se os algoritmos são dirigidos por padrões ou por sequências. Maclsaac e
Fraenkel [23] focam no workflow da descoberta de motivos. Num estudo recente, Das e Dai [24]
dividem os métodos em três grandes classes: (1) algoritmos que utilizam sequências promotoras
de genes co-regulados de um único genoma, (2) algoritmos que utilizam sequências promotoras
ortólogas de um único gene em múltiplas espécies (footprinting filogenético) e (3) algoritmos que
utilizam sequências promotoras de genes co-regulados em conjunto com o footprinting filogenético.
Existem ainda estudos mais específicos, que focam, por exemplo, em filogenia ou num genoma em
particular.
O esquema mais completo para a classificação de algoritmos de descoberta de motivos, ou
seja, aquele que oferece uma maior cobertura para a multiplicidade de algoritmos existentes, é a
framework proposta por Sandve e Drabløs [25]. Esta framework divide-se em quatro níveis.
O nível mais básico (Nível 1) representa a ligação de factores de transcrição a pequenos
segmentos contíguos. Para modelar estes segmentos, utilizam-se modelos de motivos simples,
sendo atribuída uma pontuação distinta a cada um dos segmentos identificados na região
reguladora. A este nível existem dois elementos a considerar: o modelo de match, que especifica o

29
grau de semelhança entre a substring14
Já os modelos de match probabilísticos recorrem a pontuações ponderadas. O modelo
mais utilizado é o Position Weight Matrix (PWM), também conhecido como Position Specific
Scoring Matrix (PSSM) [40]. Uma PWM contém uma linha para cada símbolo do alfabeto e uma
coluna para cada posição no padrão. Por outras palavras, assumindo que o alfabeto é Σ e que as
sequências têm comprimento 𝑙𝑙, a matriz PWM será |Σ| × 𝑙𝑙 e conterá, para cada posição no motivo,
a pontuação de cada carácter no alfabeto. A
que está a ser tratada pelo algoritmo e o modelo de
consenso de base; e o occurrence prior, que fornece o contexto genético para um elemento
regulador.
Em termos de modelos de match, estes podem ser determinísticos ou probabilísticos. Os
modelos determinísticos atribuem pontuações binárias às substrings consideradas. O modelo
determinístico mais simples é o oligo, que devolve o valor 1 para uma substring específica e o valor
0 para todas as outras substrings. O modelo oligo foi utilizado nos primeiros algoritmos de
descoberta de motivos e, recentemente, tem sido utilizado em métodos de contagem de palavras
[26-28] e métodos baseados em dicionário [29]. Além dos oligos, podem ser utilizados modelos de
expressão regular (que devolvem o valor 1 se a substring está contida na linguagem denotada por
uma expressão regular) [27, 30-33] e modelos de mismatch (que devolvem o valor 1 se a distância
de Hamming entre a substring considerada e a substring de consenso estiver abaixo de um dado
limite) [34-39].
Figura 4.2 corresponde a um exemplo de uma PWM.
Figura 4.2 – Exemplo de PWM/PSSM para um padrão de comprimento 7.
A pontuação de um motivo pode ser obtida da PWM através do cálculo da soma das
pontuações para cada símbolo e posição. Note-se que este tipo de modelo assume independência
entre posições no padrão. Exemplos de algoritmos que utilizam PWM’s são AlignACE [41],
BioProspector [42], GibbsDNA [43] e MEME [44-46]. Em [47] encontramos uma abordagem que
utiliza as PWM’s para agrupar motivos individuais em comunidades/famílias de motivos. De referir
que têm sido propostas variadíssimas extensões às PWM’s básicas, nomeadamente considerando
dependências posicionais no interior de um motivo [48-50].
Além das PWM’s, existem outros modelos probabilísticos, como as redes Bayesianas.
Qualquer destes modelos é mais expressivo do que qualquer um dos modelos determinísticos,
visto que todos os oligos, expressões regulares e modelos mismatch podem ser representados
14 Uma substring de uma cadeia de caracteres 𝑇𝑇 = 𝑡𝑡1 … 𝑡𝑡𝑚𝑚 é uma string 𝑇𝑇′ = 𝑡𝑡1+𝑚𝑚 … 𝑡𝑡𝑚𝑚+𝑚𝑚 em que 𝑚𝑚 ≥ 0 e 𝑚𝑚 + 𝑚𝑚 ≤ 𝑚𝑚.

30
como PWM’s. Contudo, o grande benefício da utilização de modelos determinísticos consiste no
facto de estes permitirem, geralmente, a descoberta exaustiva de motivos óptimos.
Em termos de occurrence priors, existem várias formas de modelar o contexto genético de
um elemento regulador. A distância ao local de início da transcrição, a conservação de sequência
em genes ortólogos, a estrutura do DNA e a presença de ilhas CpG podem ser factores relevantes
para a actividade do elemento regulador em estudo. A forma mais simples da função occurrence
prior é o rácio de abundância de motivos [51], que influencia apenas o número de substrings que
contam como ocorrências. Em [39, 52-56] utiliza-se um occurrence prior binário que explora a
noção de que as regiões altamente conservadas têm uma probabilidade muito superior (320 vezes,
segundo o estudo de Krivan e Wasserman [57]) de conter elementos reguladores. Já em [58] é tida
em conta a estrutura do DNA durante a descoberta de motivos. Por fim, em [59] é tido em conta o
facto de que uma elevada concentração de nucleótidos C e G ou a existência de ilhas CpG indica a
presença de elementos reguladores.
O segundo nível da framework representa módulos, isto é, aglomerados de factores de
transcrição cujos locais de ligação ao DNA são próximos, existindo, contudo, uma certa
flexibilidade no que toca à distância entre os locais de ligação. Para modelar estes aglomerados
utilizam-se modelos de motivo compostos, ou seja, modelos que consistem num conjunto de
motivos simples. A pontuação do motivo composto é calculada a partir das pontuações dos motivos
simples e das distâncias entre esses motivos. Geralmente, essa pontuação corresponde à soma ou
ao produto das pontuações e distâncias dos motivos simples.
Por conseguinte, existem dois aspectos a considerar a este nível: a função de distância a
utilizar e a forma de combinar as pontuações e distâncias. No que toca à função de distância
escolhida, podem, por exemplo, ser estabelecidas restrições para as distâncias entre os motivos
simples. Em [27, 42] utilizam-se distâncias de valor fixo, em [60-62] consideram-se distâncias
abaixo de um dado limite e em [27, 31, 38, 42, 63] recorre-se a distâncias contidas em intervalos.
Em vez de se considerarem restrições entre distâncias, pode-se requerer que todos os
motivos estejam contidos numa janela de um dado comprimento [53, 64-66], o que equivale a
considerar um valor limite na distância máxima entre os motivos. Uma abordagem mais genérica
consiste em definir pontuações não binárias entre motivos simples. Esta abordagem pode ser
concretizada através de funções que crescem linearmente com a distância [67].
Foram também propostas várias formas de combinar as pontuações de distâncias e
motivos simples numa única medida. Nos métodos que utilizam modelos de match determinísticos
e restrições nas distâncias, todas as pontuações componentes são binárias. A pontuação do
motivo composto é assim a intersecção das pontuações componentes [54, 68-70]. Para métodos
que utilizam pontuações não binárias, a abordagem mais comum consiste em calcular a soma das
pontuações das distâncias e dos motivos simples [67, 71]. Alguns métodos requerem que todas as

31
funções de distância tomem o valor 1, pelo que a pontuação do motivo composto é a soma das
pontuações dos motivos simples [64, 65, 72, 73].
O terceiro nível da framework representa a interacção entre módulos de modo a regular um
determinado gene. Estes modelos consistem em vários modelos de motivos compostos e utilizam
uma função de pontuação para o gene que combina a pontuação dos módulos ao longo da região
reguladora.
A pontuação ao nível do gene é normalmente definida como a pontuação máxima de entre
todos os motivos na região reguladora de um gene. Tal corresponde a assumir que existe uma
única ocorrência relevante do motivo na região reguladora [64, 74-77]. A alternativa é, obviamente,
considerar todas as pontuações de motivos compostos no cálculo da pontuação do gene. Como as
pontuações de motivos são normalmente logarítmicas, a maior parte dos métodos soma as
exponenciais das pontuações [63, 65, 78-80]. Além destas abordagens, muitas variações têm sido
utilizadas no cálculo da pontuação do gene. Caselle et al. [81] e Cora et al. [56, 82] calculam a
pontuação do gene como o p-value15
Vários métodos utilizam directamente a sobre-representação dos motivos, comparando
pontuações observadas com pontuações de um modelo de base [27, 32, 34, 66]. Em particular,
recorrem-se a medidas p-value e z-value
do conjunto observado de pontuações de motivos. Curran et
al. [83] calculam as pontuações dos genes com base numa regressão logística e Hong et al. [84]
utilizam uma tangente hiperbólica na soma das pontuações dos motivos. Por fim, Beiko et al. [58]
utilizam uma rede neuronal para combinar pontuações de motivos.
Podemos também considerar diferentes módulos ao nível do gene, isto é, assumir que o
gene tem várias regiões reguladoras, definindo restrições apertadas para a distância entre locais
de ligação no interior de uma região reguladora e distâncias mais flexíveis para diferentes regiões
reguladoras. Xing et al. [85] utilizam um Modelo de Markov Não-observável (Hidden Markov Model
– HMM) que pode representar diferentes módulos de locais de ligação com diferentes distribuições
geométricas no interior e entre módulos.
O quarto nível da framework representa a interacção de vários conjuntos de módulos que
regulam vários conjuntos de genes, sendo, por esse motivo, uma análise ao nível de genoma. As
pontuações são utilizadas para avaliar novos motivos, tendo por base a sua sobre-representação
ou a correspondência entre pontuações de motivos e dados experimentais.
16
15 No teste estatístico de hipóteses, o p-value corresponde à probabilidade de obter, segundo a hipótese nula, um resultado pelo menos tão elevado como um dado ponto nos dados. No caso da descoberta de motivos, corresponde à probabilidade de um motivo observado aparecer por puro acaso, assumindo uma hipótese nula para a distribuição das bases no DNA.
. O modelo de base é normalmente um modelo de
16 Em estatística, o z-value, também designado por pontuação standard, corresponde ao valor obtido pela subtracção de uma dada pontuação bruta à média da população, efectuando depois a divisão deste valor pelo desvio padrão da população.

32
Markov. Uma terceira hipótese consiste em utilizar uma medida de significância que está
relacionada com o conteúdo de informação das PWM’s descobertas. É este o método utilizado
pelo algoritmo MEME [44-46] que consiste basicamente num algoritmo de expectativa-
maximização em que, no passo de expectativa, se procuram boas correspondências (matches) do
motivo actual e, no passo de maximização, se actualiza a PWM com os motivos candidatos,
construindo o novo motivo actual.
Relativamente à correspondência com dados experimentais, podem ser utilizados dados
de microarrays. Os microarrays são utilizados para medir os níveis de expressão relativos de
genes num conjunto de experiências. Pensa-se que os genes, que apresentam mudanças
sincronizadas nos níveis de expressão, partilham elementos de regulação da transcrição, por
exemplo, factores de transcrição. Deste modo, podem ser utilizadas técnicas de data mining em
conjuntos de genes que evidenciem co-regulação para extrair motivos [86].
Para a identificação fiável de regiões com locais de ligação, têm sido utilizadas as
experiências ChIP/chip [87, 88]. Numa destas experiências, é possível obter a localização de locais
de ligação para reguladores específicos, apesar de a relevância desta informação estar limitada
pelo conjunto específico de condições experimentais utilizadas e pela resolução da experiência.
Além de experiências ChIP/chip e de microarrays, são normalmente formados grupos de
genes através de genes ortólogos conservados [74, 80, 89, 90] ou através de genes com
semelhanças na função anotada [28, 56]. O agrupamento de genes pode ser feito com base na
semelhança verificada durante as experiências, atribuindo cada gene a um único grupo de genes
co-regulados. Durante a descoberta de motivos, todos os genes são tratados de forma idêntica e
não se tem em conta o grau de semelhança entre um gene e o resto do grupo [60, 83, 84, 91, 92].
Em Park et al. [93] é feito o inverso, isto é, começa-se por se descobrir motivos nas regiões
reguladoras de todos os genes e depois formam-se grupos de genes que partilham motivos
comuns. A significância dos motivos é medida com base na similaridade da expressão dos genes
dentro de um grupo. Por fim, Holmes e Bruno [94] calculam a verosimilhança conjunta, tanto dos
motivos partilhados, como da similaridade das expressões dos hipotéticos grupos de genes.
4.3 Análise de dados de expressão de genes
4.3.1 Microarrays
Tal como foi referido na secção 4.2.2, os microarrays (também conhecidos como chips de
DNA, chips de genes ou arrays de genes) são utilizados para medir os níveis de expressão
relativos de genes num conjunto de experiências (também designadas por amostras) [95]. As
diferentes amostras podem corresponder a instantes temporais distintos, a tecidos cancerígenos e
saudáveis, a órgãos distintos, a diferentes fases do ciclo celular, entre outros. Cada segmento de

33
DNA toma a designação de sonda e um único microarray pode conter milhares destes segmentos.
Deste modo, cada experiência microarray corresponde a realizar milhares de testes genéticos em
paralelo, o que permite acelerar substancialmente este tipo de investigação. Naturalmente, coloca-
se o problema de como extrair informação biológica relevante através da análise de microarrays.
A organização habitual dos dados em microarrays corresponde a uma matriz semelhante à
da Figura 4.3.
Figura 4.3 – Exemplo de uma matriz de dados de um microarray. Cada linha 𝑚𝑚 da matriz corresponde ao nível de expressão do gene 𝑚𝑚 nas diferentes amostras. Cada coluna 𝑗𝑗 da matriz corresponde ao nível de expressão de todos os genes segundo as condições da amostra 𝑗𝑗. Cada posição (𝑚𝑚, 𝑗𝑗) da matriz de dados contém um valor em vírgula flutuante que corresponde, usualmente, ao logaritmo da abundância relativa de mRNA do gene 𝒊𝒊 segundo a condição 𝑗𝑗.

34
Figura 4.4 – Exemplo de conjunto de dados de microarray de cor dupla, correspondente ao desenvolvimento embrionário e adulto do organismo Drosophila melanogaster [96]. Genes com expressão acima da média (up-regulated) estão identificados a vermelho. Genes com expressão abaixo da média (down-regulated) estão identificados a verde.
Os microarrays podem ser de cor dupla ou cor única. Os de cor dupla são usualmente
hibridados com DNA complementar, cDNA17, retirado de duas amostras que se pretende comparar.
Um possível exemplo seria uma amostra de tecido saudável e outra de tecido cancerígeno. Cada
amostra é marcada com um fluoróforo18
Figura 4.4
diferente. Normalmente utilizam-se os fluoróforos cianina 3
(Cy3) e cianina 5 (Cy5) que correspondem, respectivamente, às componentes verde e vermelha do
espectro visível ( ). Deste modo, as intensidades relativas de cada fluoróforo podem ser
utilizadas para identificar genes up-regulated ou down-regulated [97].
17 O DNA complementar, ou cDNA, corresponde a DNA sintetizado a partir de um molde maduro de RNA mensageiro, através de uma reacção catalisada pela enzima DNA polimerase (também designada por transcriptase inversa).
18 Componente de uma molécula que a torna fluorescente.

35
Já os microarrays de cor única são desenhados de modo a providenciar estimativas dos
níveis absolutos da expressão dos genes. A comparação de duas condições requer duas
hibridações separadas.
4.3.2 Biclustering
Tal como referido em [98], a análise de dados de expressão de genes pode ter vários
objectivos: agrupar genes de acordo com a sua expressão em múltiplas condições; classificar um
novo gene de acordo com a expressão de outros genes cuja classificação é conhecida; agrupar
condições com base na expressão de um grupo de genes; e classificar uma nova amostra, dado o
nível de expressão dos genes nessa condição experimental.
Para alcançar estes objectivos, podemos recorrer a uma técnica de data mining designada
por clustering. O clustering consiste no agrupamento automático de dados segundo o seu grau de
semelhança. Neste caso, esta técnica pode ser utilizada tanto para agrupar genes como para
agrupar condições. Existem, contudo, algumas limitações do clustering tais como identificadas em
[98]. De facto, alguns padrões de activação de um grupo de genes só se verificam em
determinadas condições experimentais. A análise unidimensional realizada através da técnica de
clustering é assim insuficiente para a procura de informação biológica relevante em dados de
expressão de genes.
Para ultrapassar esta dificuldade, surge o conceito de biclustering, onde se efectua o
agrupamento automático de dados a duas dimensões. No caso em análise, podemos agrupar
simultaneamente genes e condições. Por outras palavras e se pensarmos em termos de uma
matriz de dados de microarray, estaremos a realizar agrupamento simultâneo de linhas e colunas.
O conceito de biclustering surge pela primeira vez no trabalho de Cheng e Church [99],
inserido no âmbito da análise de dados de expressão, mas tem vindo a ser utilizado em outras
áreas. O problema de biclustering pode ser formulado do seguinte modo: considera-se uma matriz
de dados 𝑚𝑚 por 𝑚𝑚, em que cada elemento 𝑚𝑚𝑚𝑚𝑗𝑗 é um valor real que representa a expressão do gene 𝑚𝑚
segundo a condição 𝑗𝑗. A matriz é designada por 𝐴𝐴 ou (𝑋𝑋,𝑌𝑌), sendo o conjunto de linhas designado
por 𝑋𝑋 e o conjunto de colunas designado por 𝑌𝑌. Um bicluster 𝐴𝐴𝐼𝐼𝐼𝐼 = (𝐼𝐼, 𝐼𝐼) corresponde à sub-matriz
formada pelo subconjunto de linhas 𝐼𝐼 e o subconjunto de colunas 𝐼𝐼. Por outras palavras,
corresponde a um subconjunto de linhas que exibe comportamento semelhante num determinado
subconjunto de colunas e vice-versa. Dada a matriz 𝐴𝐴, o problema de biclustering consiste na
identificação de um conjunto de biclusters 𝐵𝐵𝑘𝑘 = (𝐼𝐼𝑘𝑘 , 𝐼𝐼𝑘𝑘) tal que cada bicluster 𝐵𝐵𝑘𝑘 satisfaz
características específicas de homogeneidade. Este problema, na maioria das suas variantes, é
NP-completo19
.
19 Um problema que pertence simultaneamente a NP e a NP-difícil diz-se NP-completo.

36
4.3.3 Estado da arte dos algoritmos de biclustering
O estudo realizado em [98] classifica os algoritmos de biclustering de acordo com quatro
características: tipo de biclusters considerados, estrutura, forma de descoberta e abordagem.
No que toca ao tipo de biclusters, os algoritmos podem ter como objectivo procurar
biclusters com valores constantes, isto é, submatrizes onde todas as entradas apresentam o
mesmo valor. Este é o caso dos algoritmos de Hartigan [100], Tibshirani [101] e Cho [102]. Uma
outra hipótese consiste em considerar biclusters com valores constantes nas linhas ou colunas, isto
é, biclusters em que o valor de cada linha ou coluna é sempre o mesmo. Este é o caso dos
trabalhos realizados em Getz [103], Califano [104], Sheng et al. [105] e Segal et al. [106]. Os
algoritmos de biclustering podem também procurar biclusters com valores coerentes. Neste caso,
considera-se que existe um modelo aditivo ou multiplicativo de base que determina os valores de
todas as entradas. Nos trabalhos realizados por Cheng e Church [99], Cho [102], Hartigan [100],
Getz [103], Kluger et al. [107], Segal et al. [108] e Tang et al. [109] encontramos exemplos de
utilizações de biclusters com valores coerentes. Uma última hipótese consiste em procurar
biclusters com evolução coerente, sendo que essa evolução pode ser ao nível das colunas, ao
nível das linhas ou ao na totalidade da matriz. Este é o caso nos trabalhos de Ben-Dor [110], Liu e
Wang [111], Murali e Kasif [112] e Tanay et al.[113]. A Figura 4.5 sumariza os diferentes tipos de
biclusters:
Figura 4.5 – Diferentes tipos de biclusters. (a) bicluster constante; (b) bicluster com linhas constantes; (c) bicluster com colunas constantes; (d) bicluster com valores coerentes segundo um modelo aditivo; (e) bicluster com valores coerentes segundo um modelo multiplicativo; (f) bicluster com evolução coerente na totalidade da matriz; (g) bicluster com evolução coerente nas linhas; (h) bicluster com evolução coerente nas colunas.
Relativamente à estrutura dos biclusters, existem duas hipóteses: considerar que a matriz
de dados contém um único bicluster; ou considerar que a matriz de dados contém múltiplos

37
biclusters, sendo que o número de biclusters, 𝑘𝑘, é definido a priori. [110, 112] são exemplos do
primeiro caso, enquanto que em [99, 100, 102, 106-108, 113] encontramos exemplos do segundo
caso. De notar que os algoritmos onde se pretende procurar um único bicluster também podem ser
aplicados na procura de múltiplos biclusters, visto que a única diferença nestes algoritmos é o facto
de incluírem um último passo que escolhe o melhor bicluster de um conjunto com base em
determinados critérios de avaliação. Quando aos algoritmos que procuram mais do que um
bicluster, há ainda que distinguir entre aqueles que permitem biclusters coincidentes e os que não
permitem.
Uma outra dimensão da análise dos algoritmos de biclustering é a estratégia algorítmica
utilizada. Algumas abordagens procuram identificar um bicluster de cada vez, enquanto que outras
procuram múltiplos biclusters em simultâneo. Um exemplo do primeiro caso é o trabalho de Cheng
e Church [99] que, como veremos no final deste capítulo, identifica um bicluster, mascara-o com
valores aleatórios e depois repete o procedimento na matriz alterada. Em Hartigan [100] temos um
exemplo de descoberta de um conjunto de biclusters em simultâneo, mais concretamente, um
conjunto composto por dois biclusters. Já em [114, 115] temos um exemplo de um algoritmo que
procura todos os biclusters em simultâneo.
Outro ponto de variação nos algoritmos é a abordagem heurística utilizada, que pode ser
uma combinação de clustering iterativo de colunas e linhas, uma abordagem “dividir-para-
conquistar”, procura “gananciosa”, procura exaustiva ou identificação da distribuição de
parâmetros.
A primeira abordagem, utilizada em [103, 109], consiste em aplicar os métodos usuais de
clustering nas linhas e nas colunas da matriz, combinando posteriormente os resultados, de modo
a obter os biclusters finais.
A abordagem “dividir-para-conquistar” procura dividir a matriz de dados em várias
submatrizes onde depois se aplica uma técnica de extracção de biclusters. A grande desvantagem
desta abordagem está na dificuldade em definir critérios de divisão da matriz, o que pode fazer
com que o algoritmo falhe bons biclusters. O primeiro algoritmo deste tipo foi o “Block Clustering”,
introduzido em [100] e desenvolvido posteriormente em [101, 116].
Já com a técnica de procura greedy criam-se biclusters através da adição/remoção de
colunas/linhas dos mesmos, com base num critério de maximização do ganho local. O primeiro
trabalho onde se usou esta abordagem foi o de Cheng e Church [99], seguindo-se os trabalhos
[102, 110, 114, 115].
Em contraste com as soluções heurísticas apresentadas, a procura exaustiva de biclusters
é completa, no sentido em que encontra sempre o melhor bicluster. Esta técnica passa pela
enumeração exaustiva de todos os biclusters existentes na matriz de dados e requer a definição
prévia de restrições na dimensão dos biclusters, pois, caso contrário, a enumeração exaustiva não

38
seria exequível. O algoritmo SAMBA de Tanay et al. [113] constitui um exemplo de um algoritmo de
procura exaustiva. Outro exemplo pode ser encontrado em [111].
Por fim, temos as abordagens de identificação de parâmetros de distribuição. Estas
abordagens têm por base um modelo estatístico e tentam encontrar os parâmetros de distribuição
usados para gerar os dados, através da minimização iterativa de um determinado critério de
avaliação. Exemplos de algoritmos que utilizam estas abordagens podem ser encontrados em
[105-108].
4.4 Algoritmos trabalhados durante a tese
4.4.1 PSMILE
O algoritmo SMILE (Structured Motif Inference and Evaluation) [37, 117] é um método
combinatório que utiliza árvores de sufixos generalizadas para a extracção de motivos complexos.
SMILE utiliza um modelo de match baseado em mismatches, combina motivos simples através da
intersecção de componentes e impõe restrições na distância entre os componentes de um motivo
complexo.
Uma árvore de sufixos é uma estrutura de dados que expõe a estrutura interna de uma
cadeia de caracteres, identificando claramente o conjunto de sufixos da mesma. Esta
representação é aquela que permite soluções mais eficientes para os problemas de descoberta de
padrões em sequências de caracteres. Uma árvore de sufixos 𝑇𝑇 para uma sequência de
𝑚𝑚 caracteres, 𝑆𝑆 = 𝑠𝑠1, 𝑠𝑠2, … , 𝑠𝑠𝑚𝑚 ,, é uma árvore dirigida, com raiz e que possui 𝑚𝑚 folhas [118]. Cada nó
interno tem pelo menos dois filhos. Cada ramo é etiquetado com uma substring não vazia de 𝑆𝑆.
Dois ramos com origem no mesmo nó não podem ter etiquetas que comecem pelo mesmo
carácter. Para cada folha 𝑓𝑓, a concatenação das etiquetas dos ramos que se encontram no
caminho da raiz até 𝑓𝑓 corresponde ao sufixo de 𝑆𝑆 que se inicia em 𝑓𝑓, isto é, corresponde à
substring 𝑠𝑠𝑓𝑓 … 𝑠𝑠𝑚𝑚 .
Uma árvore de sufixos generalizada é uma estrutura semelhante, mas que pode ser
aplicada a um conjunto de sequências. Para cada sequência, define-se um carácter diferente que
funciona como um marcador e distingue os sufixos dessa sequência na árvore. Outra possibilidade
consiste em anotar cada nó da árvore com uma string de 𝑘𝑘 bits (sendo 𝑘𝑘 o número de sequências
de caracteres consideradas), estrutura usualmente designada por Colors, em que o 𝑓𝑓-ésimo bit
está a 1 se a substring faz parte da 𝑓𝑓-ésima sequência. A construção de uma árvore de sufixos
generalizada tem uma complexidade temporal e espacial de 𝑂𝑂(𝑁𝑁) [118], em que 𝑁𝑁 representa o

39
comprimento das sequências. A anotação da árvore requer um tempo adicional de 𝑂𝑂(𝑘𝑘𝑁𝑁). A Figura
4.6 corresponde a um exemplo de árvore de sufixos generalizada com a estrutura Colors.
Figura 4.6 – Árvore de sufixos generalizada para as strings TACTA$ e CACTCA$ [119]. O array 𝑪𝑪𝑪𝑪𝒍𝒍𝑪𝑪𝑪𝑪𝑪𝑪 contém apenas duas entradas correspondentes às duas sequências consideradas.
Qualquer palavra que surja em, pelo menos, uma sequência do conjunto considerado,
pode ser obtida através de um caminho único com início na raiz da árvore. Deste modo, para
investigar se um dado padrão 𝑝𝑝 está no conjunto de sequências, basta seguir o caminho indicado
pela árvore até que não existam mais caracteres a considerar em 𝑝𝑝 ou até que não seja possível
seguir mais nenhum ramo da árvore. Caso o padrão seja encontrado na totalidade, a string de bits
do próximo nó da árvore indica quais as sequências onde podemos encontrar o padrão.
Para que se considere a possibilidade de existirem 𝑒𝑒 erros, executa-se a procura do
padrão 𝑝𝑝 em diferentes caminhos. Durante este procedimento, registam-se o número de erros em
cada caminho e descartam-se os caminhos que excederem o limite 𝑒𝑒. Se for possível encontrar a
totalidade do padrão, os caminhos obtidos representam as ocorrências de 𝑝𝑝 nas sequências, com
o máximo de 𝑒𝑒 erros. As sequências onde o padrão se encontra podem ser obtidas através da
operação lógica OR entre as strings de bits dos caminhos obtidos. Para considerar a existência de
um quorum 𝑞𝑞, basta verificar se o resultado da operação OR tem um número de bits a 1 igual ou
superior a 𝑞𝑞.
Em [37] são descritas quatro variantes do problema que a família de algoritmos SMILE
pretende resolver. O ponto de variação está na generalidade dos modelos de motivos complexos
considerados. No primeiro problema, o motivo complexo consiste em dois motivos simples
separados por um espaçamento contido num intervalo. No segundo problema, o motivo complexo
consiste em 𝑝𝑝 motivos simples, separados por 𝑝𝑝 − 1 espaçamentos contidos em intervalos. No
terceiro problema, o motivo complexo consiste em dois motivos simples separados por um
espaçamento contido num intervalo e com um sub-intervalo permitido. Por fim, o quarto problema
considera motivos simples separados por 𝑝𝑝 − 1 espaçamentos contidos em intervalos, sendo que

40
cada um destes intervalos tem um sub-intervalo permitido. A Figura 4.7 ilustra um exemplo de um
modelo correspondente ao segundo problema.
Figura 4.7 – Motivo complexo composto por 𝑝𝑝 caixas. Na caixa 𝑚𝑚 é permitido um máximo de 𝑒𝑒𝑚𝑚 erros. A distância mínima (máxima) entre a caixa 𝑚𝑚 e a caixa 𝑚𝑚 + 1 é 𝑑𝑑𝑚𝑚 ,𝑚𝑚𝑚𝑚𝑚𝑚 (𝑑𝑑𝑚𝑚 ,𝑚𝑚𝑚𝑚𝑚𝑚 ).
São propostos dois algoritmos que visam resolver o primeiro e segundo problemas. Ambas
as abordagens passam por percorrer uma árvore de sufixos generalizada 𝑇𝑇 com base numa
procura em profundidade-primeiro feita numa árvore lexicográfica virtual 𝑀𝑀. Uma árvore
lexicográfica é uma árvore em que as strings representadas nas folhas aparecem em ordem
lexicográfica [120]. No caso do SMILE, esta estrutura representa todos os motivos simples de um
dado comprimento 𝑘𝑘. A árvore diz-se virtual, pois, ao contrário da árvore de sufixos, a estrutura de
dados correspondente nunca chega a ser construída durante a execução do algoritmo.
A fase de extracção de motivos simples é idêntica para ambos os algoritmos e está
ilustrada na Figura 4.8 com 𝑘𝑘 = 𝑞𝑞 = 2 e 𝑒𝑒 = 1. Quando já não existe nenhum caminho em 𝑇𝑇 que
satisfaça o limite de erros ou o quorum considerados, a árvore inferior ao nó actual é descartada.
No que toca à extracção de motivos compostos e para o primeiro algoritmo proposto,
efectuam-se saltos na árvore de sufixos com tantos níveis de profundidade adicionais quanto a
distância especificada entre cada parte (o intervalo [𝑑𝑑𝑚𝑚 ,𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚 ,𝑚𝑚𝑚𝑚𝑚𝑚 ]). Por outras palavras, após a
extracção de um motivo simples 𝑚𝑚1, o algoritmo inicia a extracção de todos os motivos simples 𝑚𝑚2
que poderiam formar um motivo estruturado,{(𝑚𝑚1,𝑚𝑚2), (𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 )}, com 𝑚𝑚1. É efectuado um salto
na árvore de sufixos para os descendentes do nó actual que estão a uma distância contida em
[𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]. A Figura 4.9 ilustra este processo.
Já o segundo algoritmo utiliza um processo um pouco diferente para a extracção de
motivos compostos. Depois de extraído um motivo simples 𝑚𝑚1, é efectuado um salto na árvore de
sufixos para os descendentes do nó actual que estão a uma distância contida no intervalo [𝑘𝑘 +
𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , 𝑘𝑘 + 𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]. A este nível, o algoritmo analisa as estruturas 𝐶𝐶𝐶𝐶𝑙𝑙𝐶𝐶𝐶𝐶𝑠𝑠 dos nós alcançados e, em
seguida, regressa ao nível 𝑘𝑘 seguindo suffix links. Um suffix link é uma ligação existente entre um
nó interno que não é a raiz e outro nó interno. Se existe um caminho da raiz para o nó onde se
completa a string 𝜒𝜒𝜒𝜒, sendo 𝜒𝜒 um carácter simples e 𝜒𝜒 uma string possivelmente vazia, então esse
nó tem um suffix link para o nó que representa 𝜒𝜒. De regresso ao nível 𝑘𝑘, a informação recolhida
no nível inferior é utilizada para actualizar temporariamente as estruturas Colors. Esta alteração
leva à criação de uma árvore 𝑇𝑇’ que contém todas as sub-árvores 𝑡𝑡 com profundidade máxima

41
igual a 𝑘𝑘 e que têm início em nós que representam possíveis segundos componentes para o
motivo complexo iniciado em 𝑚𝑚1. A extracção de 𝑚𝑚2 prossegue depois desde a raiz de 𝑇𝑇’. Após a
extracção de todos os motivos {(𝑚𝑚1,𝑚𝑚2), (𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 )} válidos, a árvore de sufixos volta ao estado
inicial, considerando, naturalmente, que estamos a extrair motivos compostos formados por apenas
dois motivos simples.
O segundo algoritmo apresenta um ganho exponencial em termos de complexidade
temporal relativamente ao primeiro. Contudo, a extracção de motivos simples encontra-se limitada
aos primeiros 𝑘𝑘 níveis da árvore.
Figura 4.8 – Exemplo de extracção de motivos simples utilizando uma árvore lexicográfica virtual (à esquerda), que representa todos os motivos de comprimento 𝑘𝑘 = 2 no alfabeto Σ = {𝐴𝐴,𝐶𝐶,𝐺𝐺,𝑇𝑇}, e uma árvore de sufixos (à direita) representando o conjunto de sequências {𝑇𝑇𝐴𝐴𝐶𝐶,𝐶𝐶𝐶𝐶𝐶𝐶}. Na travessia da árvore de sufixos admite-se um máximo de 𝑒𝑒 = 1 erros. (1) Situação inicial; (2) O nucleótido A é identificado na árvore de sufixos com 0 erros no primeiro ramo (assinalado a vermelho com “(A,0)”) e 1 erro no segundo e terceiro ramos que emanam da raiz. (3) O motivo AA é identificado com 1 erro, mas apenas na sequência TAC, pelo que o quorum não se verifica. (4) Os motivos AC e CC são identificados em ambas as sequências de entrada com 0 e 1 erro, respectivamente.

42
Figura 4.9 – Extracção de motivos complexos com motivos simples de comprimento 𝑘𝑘. O nó identificado com a cor azul é o nó actual. Os nós identificados com a cor verde correspondem aos nós para os quais podemos saltar a partir do nó actual. A distância entre estes nós e o nó de cor azul está contida no intervalo [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ].
Ao contrário de muitos algoritmos de descoberta de motivos, SMILE garante tanto a
convergência como a completude. Contudo, a sua complexidade espacial e temporal variam
exponencialmente com o número de motivos simples.
De forma a minorar este problema, é proposto em [121] um algoritmo paralelo, PSMILE,
que particiona o espaço de procura por vários nós. Deste modo, um dos pontos fulcrais do
algoritmo é justamente a construção de uma partição balanceada que divida o esforço
computacional pelos vários nós disponíveis. Para alcançar estes objectivos, faz-se uma
generalização do problema 3-PARTITION, designada por PARTITION UP TO ε. Como este novo
problema é NP-completo, utiliza-se um algoritmo de optimização para obter a partição da árvore de
procura. A partição é feita na árvore lexicográfica virtual, ficando cada nó responsável por fazer a
travessia de uma parte desta árvore. A árvore de sufixos é construída na totalidade em todos os
nós (um possível melhoramento do algoritmo consiste em cada nó construir apenas a parte da
árvore de sufixos que vai efectivamente analisar). A extracção prossegue sem necessidade de
comunicação entre os vários nós, sendo, contudo, necessária uma fase de junção dos resultados
obtidos por cada unidade computacional.
Para terminar, de referir que, tanto no SMILE como no PSMILE, é necessário um passo
adicional de avaliação estatística, de modo a discriminar os verdadeiros locais de ligação de entre
todos os motivos reportados pelo algoritmo. Este passo é executado depois de finalizada a fase de
extracção.

43
4.4.2 RISO
RISO [122] é um algoritmo combinatório que melhora o desempenho do SMILE em dois
aspectos.
Em primeiro lugar, ao invés de construir toda a árvore de sufixos a partir das sequências de
entrada, no RISO constrói-se a árvore apenas até um determinado nível de profundidade 𝑙𝑙. Esta
estrutura é designada por factor tree [123] e representa um ganho em termos de complexidade
espacial relativamente ao SMILE. A Figura 4.10 corresponde a um exemplo de uma factor tree com
𝑙𝑙 = 3:
Figura 4.10 – Exemplo de factor tree para a string AGACAGGAGGC$ com 𝑙𝑙 = 3 [119].
Em segundo lugar, o RISO utiliza uma nova estrutura de dados designada por box-link que
armazena informação sobre regiões conservadas que ocorrem de uma forma bem ordenada e
espaçada nas sequências consideradas. Por outras palavras, as box-links ligam todos os 𝑝𝑝 motivos
simples que compõem um possível motivo complexo. Esta informação é utilizada para guiar o
“salto” de um motivo simples (caixa - box) para o componente seguinte num motivo estruturado.
Podemos dizer que existe uma box-link da folha 𝑓𝑓1 para a folha 𝑓𝑓2, se, a partir da folha 𝑓𝑓1 à
profundidade 𝑘𝑘, for possível atingir a folha 𝑓𝑓2 seguindo um único caminho de sufixos de todos os
filhos de 𝑓𝑓1 à profundidade 2𝑘𝑘 + 𝑑𝑑.
Depois de construída a factor tree generalizada, são construídas as box-links através da
enumeração exaustiva de todos os motivos estruturados possíveis com as sequências
consideradas. Em seguida, essas box-links são adicionadas à árvore como folhas. Cada box-link
indica os saltos a efectuar e é um tuplo com número de nós igual ao número de componentes dos
motivos que se pretendem extrair.
Consideremos a sequência AAACCCCCGGGGGT para um possível exemplo de utilização
de factor trees e box-links. Assumindo que estamos a extrair motivos com 𝑝𝑝 = 3 componentes de
comprimento 𝑘𝑘 = 3 e separados por uma distância 𝑑𝑑 = 2, existiriam apenas duas box-links para a
sequência considerada, atendendo a que existem, no máximo, dois motivos estruturados. As box-
links necessárias e a factor tree com 𝑙𝑙 = 3 estão ilustradas na Figura 4.11

44
Figura 4.11 – Exemplo de box-links (à esquerda) e factor tree (à direita).
O processo de extracção recursivo inicia-se com a extracção de motivos simples,
realizando uma procura em profundidade-primeiro na factor tree. A extracção da segunda caixa
procede de forma semelhante ao segundo algoritmo SMILE. Desta vez, em vez de se seguir as
suffix-links, seguem-se as box-links e continua-se a procura sobre uma secção temporariamente
modifica da árvore. Após a extracção de todos os motivos {(𝑚𝑚1,𝑚𝑚2), (𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 )} válidos, a árvore
de sufixos volta ao estado inicial, considerando, mais uma vez, que estamos a extrair motivos
compostos formados por apenas dois motivos simples.
Após a extracção de todos os motivos estruturados, estes são classificados utilizando uma
medida de significância estatística. Os resultados experimentais e a análise da complexidade
mostram ganhos exponenciais, tanto em termos espaciais como temporais, relativamente ao
SMILE.
4.4.3 MUSA
O algoritmo MUSA (Motif finding using an UnSupervised Approach) [124] surge como
resposta a um dos principais problemas dos métodos de descoberta de motivos actuais, isto é, o
facto de existir uma grande dependência entre o output e os parâmetros fornecidos como input.
Por um lado, permitindo um elevado grau de degeneração, especificando um intervalo
largo de comprimentos, ou mesmo especificando um quorum reduzido, podemos obter um número
muito elevado de motivos, complicando a identificação daqueles que serão mais relevantes do
ponto de vista biológico. Por outro lado, a utilização de parâmetros muito rígidos pode levar a que
não seja descoberto nenhum motivo. Além disso, quando não existe conhecimento prévio sobre as
sequências a analisar, poder-se-á dizer que a especificação de valores para os parâmetros é
apenas especulação. Este problema é ainda mais relevante na descoberta de motivos complexos,
onde, normalmente, é necessário especificar o número de motivos simples que compõe o motivo
complexo, a distância entre estes motivos e o comprimento dos mesmos.

45
Neste sentido, MUSA não faz qualquer tipo de suposições sobre o número de
componentes de um motivo composto ou a distância entre os mesmos. De forma semelhante aos
algoritmos já descritos, um motivo complexo é definido como um conjunto de 𝑝𝑝 motivos simples,
separados por distâncias (𝑑𝑑𝑚𝑚 , 𝜀𝜀)1≤𝑚𝑚≤𝑝𝑝−1. Uma ocorrência de um motivo complexo é o conjunto de
ocorrências exactas dos motivos que o compõem e estas ocorrências encontram-se separadas por
um espaçamento de comprimento contido no intervalo [𝑑𝑑𝑚𝑚 − 𝜀𝜀,𝑑𝑑𝑚𝑚 + 𝜀𝜀]. O algoritmo requer apenas a
especificação de 𝜀𝜀, pois tanto 𝑝𝑝 como as distâncias 𝑑𝑑𝑚𝑚 são calculadas em tempo de execução.
O primeiro passo do algoritmo (ilustrado na Figura 4.12) consiste na identificação de todas
as ocorrências de um dado comprimento 𝜆𝜆 e na construção de uma estrutura designada por matriz
de co-ocorrências. Esta matriz permite-nos tirar conclusões sobre a abundância das configurações
mais comuns para cada par de 𝜆𝜆-ímeros.
Figura 4.12 – Construção da matriz de co-ocorrências no algoritmo MUSA [125].
O segundo passo consiste em utilizar a técnica de biclustering sobre a matriz de co-
ocorrências. Este passo inicia-se com os elementos que apresentam a pontuação mais elevada,
que se situarão na diagonal principal ou serão um par de elementos do triângulo superior e inferior.
Cada um destes elementos é já um bicluster, ao qual são adicionados linhas ou colunas de forma
greedy enquanto a pontuação do bicluster resultante for superior à do inicial. Dado que o segundo
passo progride heuristicamente, é possível que algumas boas soluções não sejam identificadas.
O último passo do MUSA corresponde à junção de motivos individuais em famílias de
motivos. Esta junção é feita observando os motivos que podem ser justapostos a motivos com

46
significância mais elevada, através da troca de uma única base. Por fim, é avaliada a significância
estatística dos motivos encontrados.
4.4.4 Cheng e Church
Como referido anteriormente, o conceito de biclustering surge pela primeira vez no trabalho
de Cheng e Church [99], onde o objectivo é o de encontrar conjuntos coerentes de genes e
condições em dados de expressão.
Cheng e Church utilizam como medida o resíduo médio quadrático, mean squared residue,
ou MSR. Pretende-se encontrar biclusters cujo valor do resíduo médio quadrático seja inferior a um
dado limite. Quanto mais reduzido for o valor de MSR, mais coerentes serão os dados no interior
de um bicluster. Considera-se uma matriz 𝐴𝐴 em que a entrada da linha 𝑚𝑚 e coluna 𝑗𝑗 é representada
por 𝑚𝑚𝑚𝑚𝑗𝑗 . O resíduo do elemento 𝑚𝑚𝑚𝑚𝑗𝑗 num bicluster cujo conjunto de linhas é representado por 𝐼𝐼 e o
conjunto de colunas é representado por 𝐼𝐼 é dado por:
𝑚𝑚𝑚𝑚𝑗𝑗 − 𝑚𝑚𝑚𝑚𝐼𝐼 − 𝑚𝑚𝐼𝐼𝑗𝑗 + 𝑚𝑚𝐼𝐼𝐼𝐼 (1)
Onde 𝑚𝑚𝑚𝑚𝐼𝐼 é a média dos valores na linha 𝑚𝑚 do bicluster, 𝑚𝑚𝐼𝐼𝑗𝑗 é a média dos valores na coluna
𝑗𝑗 do bicluster e 𝑚𝑚𝐼𝐼𝐼𝐼 é a média de todos os valores do bicluster. O resíduo médio quadrático do
bicluster (𝐼𝐼, 𝐼𝐼) é dado por:
𝐻𝐻(𝐼𝐼, 𝐼𝐼) =1
|𝐼𝐼||𝐼𝐼|� �𝑚𝑚𝑚𝑚𝑗𝑗 − 𝑚𝑚𝑚𝑚𝐼𝐼 − 𝑚𝑚𝐼𝐼𝑗𝑗 + 𝑚𝑚𝐼𝐼𝐼𝐼 �2
𝑚𝑚∈𝐼𝐼,𝑗𝑗∈𝐼𝐼
(2)
Quando 𝐻𝐻(𝐼𝐼, 𝐼𝐼) = 0, estamos na presença de um bicluster em que todas as entradas têm o
mesmo valor, isto é, um bicluster constante. Quando 𝐻𝐻(𝐼𝐼, 𝐼𝐼) ≠ 0, é sempre possível retirar uma
linha ou coluna para diminuir o resíduo médio quadrático.
O algoritmo de Cheng e Church assume uma estratégia “gananciosa” e identifica um
bicluster por iteração. Os parâmetros relevantes são os seguintes:
• 𝐴𝐴: matriz com os dados de expressão. As entradas são números reais e podem existir
valores em falta.
• 𝛿𝛿: o resíduo médio quadrático máximo permitido.
• 𝜒𝜒: parâmetro a ser utilizado no passo de multiple node deletion.
• 𝜏𝜏: número mínimo de colunas e linhas para a execução do passo de multiple node deletion.
• 𝜂𝜂: número de biclusters que se pretende extrair.

47
O algoritmo começa por considerar que o primeiro bicluster candidato corresponde à
totalidade da matriz original. Em seguida, inicia-se o processo de remoção de múltiplos nós
(multiple node deletion).
A remoção de múltiplos nós vai retirar linhas e colunas ao bicluster candidato até que este
satisfaça a seguinte propriedade:
𝐻𝐻(𝐼𝐼, 𝐼𝐼) ≤ 𝛿𝛿 (3)
Caso o número de linhas (colunas) seja inferior a 𝜏𝜏, não é aplicado o passo de multiple
node deletion nas linhas (colunas), optando-se pelo passo de remoção de um único nó (single
node deletion), que remove uma única linha ou coluna do bicluster. A escolha do elemento a
remover tem por base a linha ou coluna que mais contribuir para a diminuição do valor do resíduo
médio quadrático.
Após a remoção de colunas e/ou linhas, o bicluster identificado pode não ser máximo, no
sentido em podemos adicionar linhas e/ou colunas sem aumentar o valor do resíduo médio
quadrático. Para atender a este facto, é realizado um passo de adição de nós (node addition). Este
passo é repetidamente executado até que não possam ser adicionadas mais linhas ou colunas
sem aumentar o valor de 𝐻𝐻(𝐼𝐼, 𝐼𝐼) do bicluster. Terminado este passo, obtemos um bicluster máximo.
O bicluster resultante é reportado pelo algoritmo e é mascarado na matriz de dados com
valores aleatórios (caso este último passo não fosse executado, o algoritmo iria identificar o mesmo
bicluster em todas as iterações). Em seguida, repete-se o processo, utilizando a matriz alterada,
tantas vezes quando o número de biclusters, 𝜂𝜂, que se pretende extrair.
4.4.5 CCC-Biclustering e e-CCC-Biclustering
Existe uma restrição importante no problema de biclustering que permite a criação de
algoritmos lineares eficientes para a descoberta de biclusters máximos. Esta restrição pode ser
aplicada quando os dados de expressão de genes correspondem aos níveis de expressão numa
sequência de instantes temporais. A ideia de base consiste no facto de a activação de um conjunto
de genes corresponder à activação de um dado processo biológico. Neste caso, usualmente
designado por biclustering em séries temporais, pretende-se encontrar biclusters cujas colunas são
contíguas na matriz de dados original. Estas colunas correspondem, deste modo, a amostras
obtidas em instantes temporais consecutivos onde os genes exibem níveis de expressão
coerentes.
Os algoritmos CCC-Biclustering [126] e e-CCC-Biclustering [127] são métodos de
biclustering em séries temporais que se baseiam na utilização de árvores de sufixos generalizadas
(ver secção 4.4.1).

48
A base de funcionamento de ambos os algoritmos é idêntica. Em primeiro lugar, os níveis
de expressão que constam na matriz de dados são discretizados para um conjunto de símbolos.
Na situação mais simples, esse conjunto de símbolos é igual a
Σ = {𝐷𝐷,𝑁𝑁,𝑈𝑈} = {𝐷𝐷𝐶𝐶𝐷𝐷𝑚𝑚𝐷𝐷𝑒𝑒𝐷𝐷𝐷𝐷𝑙𝑙𝑚𝑚𝑡𝑡𝑒𝑒𝑑𝑑,𝑁𝑁𝐶𝐶𝐶𝐶ℎ𝑚𝑚𝑚𝑚𝐷𝐷𝑒𝑒,𝑈𝑈𝑝𝑝𝐷𝐷𝑒𝑒𝐷𝐷𝐷𝐷𝑙𝑙𝑚𝑚𝑡𝑡𝑒𝑒𝑑𝑑}, isto é, corresponde aos vários níveis
de activação possíveis (regulação negativa, “down regulated”, sem alteração “no change” e
regulação positiva, “up regulated”). É ainda realizada uma transformação no alfabeto que adiciona
a cada símbolo a coluna a que este pertence (Figura 4.13).
Os autores introduzem o conceito de contiguous column coherent bicluster (ccc-bicluster),
como um conjunto de linhas 𝐼𝐼 = {𝑚𝑚1, … , 𝑚𝑚𝑘𝑘} e um conjunto contíguo de colunas 𝐼𝐼 = {𝑗𝑗𝐶𝐶 , 𝑗𝑗𝐶𝐶+1, … , 𝑗𝑗𝑠𝑠−1, 𝑗𝑗𝑠𝑠}
da matriz de dados tal que 𝐴𝐴𝑚𝑚𝑗𝑗 = 𝐴𝐴𝐼𝐼𝑗𝑗 para todo o 𝑚𝑚 ∈ 𝐼𝐼 e 𝑗𝑗 ∈ 𝐼𝐼.Tal significa que estamos a considerar
colunas contíguas e constantes.
Cada ccc-bicluster define uma cadeia de caracteres que corresponde ao padrão de
expressão comum a todas as linhas do bicluster, como se pode ver na Figura 4.13.
Figura 4.13 – Matriz de dados de expressão antes (à esquerda) e depois (à direita) da transformação efectuada no alfabeto [126]. Estão também representados dois biclusters, 𝐵𝐵1 e 𝐵𝐵2. A string definida por 𝐵𝐵1 é UDU, enquanto que a string definida por 𝐵𝐵2 é UN. Depois da transformação de alfabeto, as strings passam a U2D3U4 e U4N5, respectivamente.
O resultado principal do artigo [126] é o de que, sendo 𝑇𝑇 uma árvore de sufixos
generalizada para as strings definidas por cada linha da matriz de dados, então cada nó interno da
árvore que satisfaça uma condição particular define um ccc-bicluster. Tendo como ponto de partida
o exemplo da Figura 4.13, podemos construir a árvore de sufixos generalizada com suffix-links da
Figura 4.14.

49
Figura 4.14 – Árvore de sufixos generalizada com suffix-links para a matriz de dados da Figura 4.13. Os biclusters 𝐵𝐵1 e 𝐵𝐵2, os únicos ccc-biclusters máximos e não triviais existentes na matriz, estão também representados.
Na Figura 4.14 verificamos que existem seis nós internos na árvore, sendo que cada um
deles, como já foi referido, corresponde a um ccc-bicluster. De igual modo, cada nó folha cujo ramo
tenha um comprimento superior a 1 é também um ccc-bicluster. No entanto, alguns destes ccc-
biclusters são triviais, no sentido em que representam biclusters com apenas uma coluna ou
correspondem a folhas, e outros não são máximos, pois têm um suffix-link com origem num nó com
o mesmo número de folhas. No exemplo proposto, apenas os ccc-biclusters 𝐵𝐵1 e 𝐵𝐵2 são máximos
e não triviais.
Deste modo, o algoritmo CCC-Biclustering recebe como entrada uma matriz de dados,
discretiza-a, efectua a transformação de alfabeto, constrói a árvore de sufixos generalizada e
efectua uma procura em profundidade-primeiro na árvore que identificará todos os biclusters
máximos e não triviais. Tanto a construção da árvore de sufixos como a procura em profundidade-
primeiro são feitas em tempo linear.
Há um detalhe importante que poderá afectar a qualidade dos resultados obtidos com o
algoritmo CCC-Biclustering. De facto, é comum a existência de ruído nas experiências com
microarrays e podem também ocorrer erros durante o processo de discretização. Deste modo,
torna-se interessante adicionar ao algoritmo a capacidade de lidar com erros nas entradas da
matriz de dados de expressão. Para resolver este problema, surge o algoritmo e-CCC-Biclustering
[127].
Este novo algoritmo admite a existência de um máximo de 𝑒𝑒 erros por gene (isto é, por
linha) em cada ccc-bicluster (agora designados e-ccc-biclusters). A Figura 4.15 ilustra este
aspecto. Estes erros podem, de um modo geral, corresponder a substituições de um símbolo no
padrão de expressão por outros símbolos do alfabeto (erros de medição) ou restritos aos símbolos
lexicograficamente mais próximos (erros de discretização).

50
Figura 4.15 – e-ccc-biclusters máximos e não triviais com 1 erro [127].
O algoritmo e-CCC-Biclustering é baseado no algoritmo SPELLER [128], que também está
na base do SMILE. SPELLER extrai motivos de um conjunto de 𝑁𝑁 sequências utilizando uma
árvore de sufixos generalizada, tendo em conta a possibilidade de existirem erros e de os motivos
não estarem presentes em todas as sequências. O algoritmo pode ser adaptado ao problema de
biclustering do seguinte modo: dado o conjunto de strings definido pelas linhas da matriz de dados
de expressão, pretende-se identificar todos os modelos 𝑚𝑚 (padrões de expressão) que estão
presentes em pelo menos 𝑞𝑞 linhas distintas (quorum) que começam e terminam na mesma coluna.
O algoritmo e-CCC-Biclustering começa assim por armazenar todos os modelos com um
máximo de 𝑒𝑒 erros que correspondem a right-maximal e-ccc-biclusters [127]. Para verificar se um
modelo corresponde a um right-maximal e-ccc-bicluster, analisam-se os modelos que resultam da
extensão do primeiro e verifica-se se estes possuem o mesmo número de genes que o
ascendente. Em caso afirmativo, não estamos na presença de um right-maximal e-ccc-bicluster.
O segundo passo consiste em remover, dos modelos armazenados, aqueles que não
correspondem a left-maximal e-ccc-biclusters [127]. Um e-ccc-bicluster é left-maximal se o padrão
de expressão não puder ser estendido para a esquerda, adicionando um símbolo no início do
padrão.
O terceiro passo consiste em remover, dos modelos armazenados, aqueles que
correspondem a biclusters repetidos. Por fim, reportam-se todos os e-ccc-biclusters máximos
encontrados.
A análise de complexidade revela que o algoritmo e-CCC-Biclustering encontra e reporta
todos os e-ccc-biclusters máximos em tempo polinomial relativamente à dimensão da matriz.
Através da selecção dos e-ccc-biclusters mais significativos do ponto de vista estatístico, torna-se
possível identificar processos biológicos potencialmente relevantes. De facto, os resultados obtidos
pelos autores revelam que os biclusters encontrados apresentam maior relevância biológica do que
os identificados por qualquer outro algoritmo de biclustering.

51
Capítulo 5 Deployment dos Algoritmos no Cluster Hermes
Neste capítulo descreve-se o trabalho realizado com os algoritmos descritos no capítulo
anterior. Começa-se, no entanto, por fazer uma pequena introdução ao cluster hermes disponível
no Instituto Gulbenkian de Ciência.
5.1 Visão geral do sistema
O campus do Instituto Gulbenkian de Ciência (IGC) possui um Centro de Computação de
Alta Performance (High Performance Computing Centre – HPCC), denominado hermes, que
suporta o Centro Português de Bioinformática.
O HPCC foi construído com base num cluster de máquinas de média dimensão. O cluster é
dedicado à investigação em Bioinformática e permite aos utilizadores obterem resposta rápida aos
seus pedidos, apesar da simplicidade da interface pessoa-máquina. O sistema é um conjunto
heterogéneo de computadores interligados que possui os seguintes componentes:
• hermes, uma máquina baseada num duplo processador Intel Xeon, acessível através
da rede local do IGC e que serve como nó centralizado de gestão;
• Um servidor de disco, com um array de discos de alta performance, partilhados dentro
do cluster, totalizando 4200 GB de espaço;

52
• 60 nós computacionais baseados na arquitectura JS20 PowerPc, com 2 GB de
memória RAM e discos locais de 80 GB.
A Figura 5.1 providencia uma vista geral sobre estes componentes:
Figura 5.1 - Vista geral sobre o cluster hermes.
O conjunto de máquinas está interligada utilizando a sua rede Gigabit local e todos os nós
correm o sistema operativo Suse Linux Enterprise Server. Os trabalhos no cluster são geridos pelo
scheduler Sun Grid Engine que distribui permanentemente a carga.
5.2 Sun Grid Engine
O Sun Grid Engine (SGE) é o sistema de gestão de recursos utilizado no cluster hermes e
tem como responsabilidades a aceitação, ordenamento, processamento e gestão de trabalhos
remotos, podendo estes ser paralelos e interactivos. O SGE é ainda responsável pela gestão e
alocação de recursos distribuídos tais como processadores, memória, espaço em disco e licenças
de software.
Existem dois conceitos fundamentais no universo SGE: trabalhos (jobs) e filas (queues).
Os utilizadores submetem trabalhos que são colocados em filas e posteriormente executados,
estando essa execução dependente da carga do sistema e da prioridade dos trabalhos.
Extensa documentação sobre o software SGE pode ser encontrada em [129].

53
5.3 BioTeam iNquiry
O sistema hermes tem instalado o software Bioteam iNquiry, que consiste simultaneamente
num instalador de clusters e num portal Web para aplicações de Bioinformática, sendo este portal
gerido pelo SGE. As aplicações disponíveis são um misto de programas de domínio público e
programas contribuídos pelo IGC, sendo que estes últimos vão sendo criados como resultado das
colaborações de investigação feitas com o Instituto.
O software iNquiry utiliza uma arquitectura de “portal”. Existe uma máquina com o portal
que serve de interface para todas as outras máquinas que formam o sistema. O portal pode ser
acedido em [130] e é composto por duas interfaces de rede: a ligação pública à rede local e a
ligação privada à rede interna do cluster. As restantes máquinas são nós computacionais que
estão “sequestrados” pela rede privada. Os nós computacionais não estão ligados à rede pública e
só podem ser acedidos através do portal. Os nós computacionais acedem ao exterior através de
um serviço de tradução de endereço de redes (Network Address Translation – NAT) existente no
portal. A Figura 5.2 ilustra a organização física da arquitectura de portal iNquiry:
Figura 5.2 – Organização física da arquitectura de portal iNquiry [131].
Os utilizadores e administradores interagem apenas com o portal. Os administradores
efectuam alterações nos nós utilizando batches20
20 Os ficheiros batch são utilizados para automatizar tarefas. O processamento batch é usualmente mais eficiente do que o processamento online para situações onde existem operações rotineiras e de elevado volume.
, enquanto que os utilizadores submetem
trabalhos para a fila. Deste modo, os utilizadores são alheios a actualizações do sistema, nós
indisponíveis e outros problemas comuns. Os utilizadores vêem os nós computacionais como um

54
conjunto de recursos aos quais podem aceder e não como um conjunto de máquinas distintas. Os
administradores beneficiam com este modelo de interacção na medida em que o tempo necessário
para a gestão do cluster não cresce linearmente com o número de nós existentes.
5.4 Bioprogram
O módulo Bioprogram providencia a interface do portal iNquiry e consiste numa re-
implementação completa do software PISE [132] desenvolvido por Catherine Letondal.
PISE e Bioprogram permitem a especificação da interface de aplicações de Bioinformática
através de ficheiros XML (eXtensible Markup Language). Para cada aplicação é criado um ficheiro
XML contendo a descrição dos parâmetros e da forma como estes estarão organizados na linha de
comandos. Bioprogram lê estes ficheiros e apresenta a interface ao utilizador. Além da interface
HTML (HyperText Markup Language) e CGI (Common Gateway Interface) apresentada no portal, é
também gerada automaticamente uma interface programática para ser utilizada com Web Services,
baseada na WSDL (Web Services Description Language) [133], e no protocolo de comunicação
SOAP (Simple Object Access Protocol) [134].
Em Bioprogram, todas as ferramentas/programas são organizadas em pastas de acordo
com as suas funções. Além disso, todas as aplicações têm uma forma “simples” e uma forma
“avançada”, isto é, estão definidos dois níveis de utilização: no nível simples, a interface exibe
apenas os parâmetros obrigatórios; no nível avançado, a interface exibe todos os parâmetros
disponíveis na aplicação.
Bioprogram e Pise admitem tipos de parâmetros variados. Alguns exemplos são: ficheiros
de entrada/saída, descrições textuais de sequências de aminoácidos/nucleótidos, parâmetros de
valor inteiro ou em vírgula flutuante, cadeias de caracteres, listas de valores e parâmetros binários.
Todos os diferentes tipos de parâmetros têm associado um conjunto de atributos. Esses atributos
permitem, por exemplo, definir valores limite para os parâmetros, definir a obrigatoriedade de
especificação dos parâmetros, definir textos de ajuda associados aos parâmetros, definir a posição
dos parâmetros nas invocações em linha de comandos, definir condições para a utilização dos
parâmetros, entre outros.
Os parâmetros de cada aplicação encontram-se organizados em grupos, de acordo com a
documentação específica. Para parâmetros textuais, é possível introduzir os dados directamente
ou efectuar o carregamento de um ficheiro. Um aspecto interessante relativo aos ficheiros com
sequências de nucleótidos/aminoácidos é a possibilidade de Bioprogram converter
automaticamente entre vários formatos. Esta funcionalidade torna-se bastante útil no caso de
estarmos a trabalhar com aplicações que aceitam apenas um formato de sequências (ex: Fasta),
dando ao utilizador a possibilidade de introduzir dados no formato que lhe é mais familiar.

55
Os resultados da execução dos vários programas são exibidos no navegador Web e
podem também ser enviados por correio electrónico para o utilizador. A Figura 5.3 sumariza todos
estes pontos.
Figura 5.3 – Vista genérica sobre uma árvore de programas e a interface de uma aplicação [135].
Parâmetros identificados com um círculo vermelho são obrigatórios, no sentido em que o
seu valor deve ser sempre especificado pelo utilizador. Parâmetros identificados com um círculo
azul são condicionais, no sentido em que o seu valor apenas deve ser especificado pelo utilizador
no caso de se verificar uma ou mais condições específicas. Na verdade, o que acontece na prática
é que o valor de um parâmetro condicional é ignorado caso a condição (ou condições) associada
ao parâmetro não se verifique. Parâmetros opcionais são todos aqueles que não estão
identificados por um círculo vermelho ou azul, sendo a especificação do seu valor, obviamente,
opcional.
A maioria dos parâmetros tem ainda um texto de ajuda acoplado, como ilustrado na Figura
5.4.

56
Figura 5.4 – Texto de ajuda e parâmetros obrigatórios/condicionais [135].
Bioprogram suporta ainda computação paralela, disponível através de um ambiente
paralelo especializado no software SGE. Do ponto de vista do programador e assumindo que a
aplicação alvo já está paralelizada, é apenas necessária a inclusão de uma tag adicional no
ficheiro XML, onde é definida a implementação paralela a ser utilizada (ex: MPICH) e o número de
nós a serem envolvidos na computação.
5.5 Adicionar aplicações no cluster hermes
Existem duas formas de adicionar novas aplicações ao cluster hermes ou, de uma forma
genérica, em sistemas baseados no software iNquiry. A primeira recorre directamente ao Sun Grid
Engine, enquanto que a segunda disponibiliza a aplicação no portal, envolvendo por isso a criação
de uma interface pessoa-máquina.
Ambos os métodos requerem, naturalmente, a compilação prévia da aplicação ou
aplicações que se pretendem utilizar. No caso do primeiro método, o passo seguinte consiste em
criar um script executável que fornece ao SGE as instruções necessárias para a execução do
programa. Em seguida, utilizam-se comandos específicos para lançar o trabalho na fila de
execução e para monitorizar o mesmo. De notar que cabe ao software SGE decidir qual o nó ou
nós para os quais será enviado o trabalho.
O segundo método, aquele que foi adoptado na investigação subjacente a esta tese,
também começa por compilar e testar as aplicações, mas prossegue com a criação de uma
interface utilizando o módulo Bioprogram. Depois de criada a interface, resta apenas activá-la no
portal. Esta activação é bastante simples, sendo apenas necessária a colocação dos ficheiros XML
em directórios específicos.
Uma descrição pormenorizada do processo de deployment de aplicações no cluster
hermes pode ser encontrada em [136].

57
5.6 Trabalho realizado com os algoritmos
Nesta secção, apresentam-se os procedimentos adoptados no deployment no cluster
hermes dos algoritmos descritos no quarto capítulo.
5.6.1 MUSA
O trabalho realizado com o algoritmo MUSA teve como base a implementação codificada
por Nuno D. Mendes na linguagem C. A saída da aplicação original consistia num ficheiro com a
lista dos motivos identificados nas sequências de entrada ordenados pela medida de significância
estatística p-value.
Esta implementação foi posteriormente modificada por Alexandre P. Francisco para que
fossem agrupados os motivos simples reportados pelo algoritmo em famílias. Este trabalho baseia-
se no algoritmo CNM (Clauset-Newman-Moore) descrito em [47]. O algoritmo CNM tem como
objectivo processar a saída dos métodos combinatórios para a descoberta de motivos, agrupando
motivos em famílias. Considera-se que todos os motivos que pertencem à mesma família são
variações de um único motivo e podem assim ser vistos como um todo. Esta organização em
famílias facilita consideravelmente a análise da saída dos algoritmos combinatórios, que, como
referimos no capítulo anterior, tem, geralmente, grande dimensão.
O algoritmo CNM funciona em três passos: no primeiro passo, é construído um grafo que
representa as relações entre os motivos encontrados. Cada nó/vértice do grafo corresponde a um
motivo. Existe um arco entre dois nós se os dois motivos em causa têm uma sobreposição
significativa, isto é, um mínimo de caracteres comuns nas sequências de entrada. Não é tida em
conta a similaridade entre os motivos, mas sim o grau de co-ocorrência dos mesmos.
No segundo passo, encontram-se comunidades no grafo de motivos, com base no conceito
de modularidade. A modularidade mede a qualidade da divisão de um grafo em comunidades
através da avaliação do número de arcos existentes no interior das comunidades e o número de
arcos que ligam vértices de diferentes comunidades. Este passo do algoritmo consiste num ciclo
onde, em cada iteração, se escolhe, de forma greedy, a divisão que provoca uma maior mudança
no valor da modularidade.
O terceiro passo do método consiste em calcular a PWM de cada família identificada. Tal é
feito através do alinhamento múltiplo dos vários motivos que compõem uma família.
O primeiro passo no deployment do MUSA consistiu em realizar pequenas adaptações no
código de modo a prepará-lo para a arquitectura PowerPc. O passo seguinte consistiu na criação
de uma interface em XML utilizando o módulo Bioprogram. Foram definidos os diversos
parâmetros, textos de apoio ao utilizador e criadas as instruções necessárias à verificação dos
valores introduzidos para os parâmetros.

58
5.6.2 PSMILE
O trabalho realizado com o algoritmo PSMILE teve por base a implementação codificada
por Alexandra M. Carvalho na linguagem C. Esta implementação concretiza a paralelização do
SMILE através da MPICH-G2. Dado que esta investigação incidiu sobre um cluster computacional
e não sobre uma grid, foi necessário substituir a implementação MPICH-G2 pela MPICH.
No caso do PSMILE, e como vimos no capítulo anterior, a componente de comunicação
entre os vários nós oferecida pela especificação não é necessária. A paralelização é realizada do
seguinte modo: através de funções específicas do protocolo MPI, cada processo obtém as
informações necessárias para realizar a partição da árvore lexicográfica. A função
MPI_Comm_size permite conhecer o número de nós/processos envolvidos numa execução
paralela e a função MPI_Get_processor_name permite obter o identificador único do nó/processo
actual numa execução paralela. Com estes dados, cada nó fica responsável por fazer a travessia
de uma porção da árvore lexicográfica. Como referido no capítulo anterior, a árvore de sufixos é
construída na totalidade em todos os nós.
O primeiro passo no deployment desta aplicação consistiu em realizar pequenas
adaptações no código de modo a prepará-lo para a arquitectura PowerPc. A implementação tinha,
contudo, um problema, o facto de todos os nós envolvidos na computação registarem os motivos
encontrados concorrentemente no mesmo ficheiro de saída. Foram então codificadas as alterações
necessárias de modo a que cada nó escrevesse num ficheiro independente, sendo os resultados
posteriormente combinados recorrendo a uma pequena e simples aplicação em C desenvolvida
para o efeito.
O passo seguinte consistiu na criação da interface da aplicação recorrendo ao módulo
Bioprogram. Foram definidos os diversos parâmetros, textos de apoio ao utilizador e criadas as
instruções necessárias à verificação dos valores introduzidos para os parâmetros.
5.6.3 RISO
O trabalho realizado com o algoritmo RISO teve por base a implementação, codificada por
Alexandra M. Carvalho na linguagem C, da variante descrita em [137]. Esta variante designa-se
RISOTTO e baseia-se na noção de extensibilidade máxima (max extensibility). O objectivo desta
variante é o de melhorar o desempenho do algoritmo original na pesquisa de motivos longos. A
modificação introduzida consiste no armazenamento de informação relativa à extensibilidade
máxima dos motivos, de modo a evitar a exploração repetida de motivos que não poderão ser
estendidos de acordo com os parâmetros de entrada. Por exemplo, se durante a extracção for
identificado um motivo 𝑚𝑚1 que, de acordo com o valor considerado para o quorum, pode ser
estendido até um dado comprimento 𝑀𝑀𝑚𝑚𝑚𝑚𝑀𝑀𝑚𝑚𝑡𝑡(𝑚𝑚1), e, mais tarde, for identificado um motivo 𝑚𝑚2 que

59
tem 𝑚𝑚1 como sufixo, então podemos usar a informação 𝑀𝑀𝑚𝑚𝑚𝑚𝑀𝑀𝑚𝑚𝑡𝑡(𝑚𝑚1) para determinar que 𝑚𝑚2 só
poderá ser estendido com no máximo 𝑀𝑀𝑚𝑚𝑚𝑚𝑀𝑀𝑚𝑚𝑡𝑡(𝑚𝑚1) símbolos. Para motivos complexos, regista-se a
informação 𝑀𝑀𝑚𝑚𝑚𝑚𝑀𝑀𝑚𝑚𝑡𝑡 apenas para o primeiro componente.
O primeiro passo no deployment desta aplicação consistiu em realizar pequenas
adaptações no código de modo a prepará-lo para a arquitectura PowerPc. Em seguida, foi criada a
interface da aplicação recorrendo ao módulo Bioprogram.
5.6.4 CCC-Biclustering e e-CCC-Biclustering
O trabalho realizado com os algoritmos CCC-Biclustering e e-CCC-Biclustering teve por
base a implementação codificada por Sara Madeira na linguagem Java. A principal contribuição de
Sara Madeira consiste no pacote smadeira, cujo diagrama em UML (Unified Modelling Language)
se apresenta em seguida:
Figura 5.5 – Diagrama do pacote smadeira em UML.
O módulo ontologizer [138] foi desenvolvido pelo Charité University Hospital e oferece a
capacidade de gerar listagens de anotações Gene Ontology (GO) [139-141] 21
21 O projecto Gene Ontology tem como objectivo a criação de um vocabulário controlado para descrever genes e produtos de genes em qualquer organismo, de modo a promover a interoperabilidade entre bases de dados. A esse vocabulário dá-se o nome de ontologia. A principal oportunidade do projecto consiste na transferência automática de anotações de organismos tratáveis experimentalmente para organismos não tratáveis experimentalmente, sendo essa transferência efectuada com base na similaridade genética e proteica.
. As anotações GO

60
são ordenadas de acordo com a frequência em cada cluster. A ferramenta pode ainda ser utilizada
para gerar representações gráficas da sobre-representação dos termos GO, apesar de esta
funcionalidade não ter sido utilizada no decorrer do trabalho subjacente a esta tese.
O módulo utils contém todos os métodos necessários para ordenar os biclusters extraídos
com os dois algoritmos, para discretizar a matriz de dados de expressão e para cálculo das
medidas relevantes (ex: p-value). Contém ainda as definições dos modelos (padrões de
expressão).
Os restantes módulos possuem os métodos necessários para a execução dos algoritmos,
nomeadamente para a construção e manipulação das árvores de sufixos generalizadas (conforme
descrito no capítulo anterior).
Para disponibilizar os algoritmos no cluster hermes foi desenvolvido um ficheiro jar (Java
ARchive) que contém o pacote smadeira, juntamente com os pacotes jargs, flanagan e smcosta
(este último criado de raiz com o trabalho realizado durante esta tese).
O pacote jargs [142] foi criado por Steve Prucell e é actualmente mantido por Ewan Mellor.
Este pacote oferece funcionalidades de parsing de opções em linhas de comandos. No caso em
análise, permite a invocação dos algoritmos do pacote smadeira através de uma linha de
comandos, bem como a especificação, com recurso a flags, dos parâmetros de entrada para os
algoritmos.
O pacote flanagan [143] tem vindo a ser desenvolvido por Michael Thomas Flanagan. Este
pacote contém classes e métodos que permitem lidar com distribuições e funções estatísticas,
funções matemáticas, regressão, transformadas de Fourier, interpolação, geração de números
aleatórios, optimização e criação de gráficos.
O pacote smcosta implementa duas classes: a primeira, contendo a definição de todos os
parâmetros de entrada dos algoritmos CCC-Biclustering e e-CCC-Biclustering. A segunda,
contendo as instruções necessárias para, com base no pacote jargs, fazer o parsing das opções
fornecidas na linha de comandos e invocar os métodos apropriados no pacote smadeira.
Depois de criado e integrado o pacote smcosta, procedeu-se à criação de duas interfaces
em XML (uma para cada algoritmo) recorrendo ao módulo Bioprogram.

61
5.6.5 Cheng e Church
O pacote smadeira, referido na secção anterior, continha uma implementação de uma
variante do algoritmo de Cheng e Church. Essa variante designa-se por CC_TSB [144] e pretende
aplicar a mesma ideia do algoritmo original mas considerando apenas biclusters cujas colunas são
contíguas, isto é, pretende tratar o problema de biclustering em séries temporais. Foi com base
nesta implementação e no pacote jargs que se desenvolveu, nos pacotes smadeira e smcosta, a
implementação do algoritmo Cheng e Church. O diagrama UML das classes relevantes é ilustrado
na Figura 5.6.
A classe abstracta Abstract_CC_Biclustering define as variáveis e métodos comuns aos
dois algoritmos, nomeadamente os relativos ao cálculo de resíduos. Os métodos para
remover/inserir colunas e para inserir linhas são definidos como métodos abstractos e a sua
implementação é concretizada nas classes CC_TSB_Biclustering e ChengAndChurch_Biclustering
conforme os algoritmos descritos em [144] e [99], respectivamente.
A classe ChengAndChurchOptions contém todas as opções de linha de comandos
relevantes para o algoritmo de Cheng e Church. O pacote jargs, referido na secção anterior, é
novamente o responsável por efectuar o parsing das opções na linha de comandos oriunda do
módulo Bioprogram.
A classe ChengAndChurchConsole faz a ligação entre as opções especificadas na linha de
comando e a chamada dos métodos da classe ChengAndChurchBiclustering que implementam os
diferentes passos do algoritmo.
Concluído o desenvolvimento dos pacotes Java relevantes, procedeu-se à criação de uma
interface em XML recorrendo ao módulo Bioprogram.

62
Figura 5.6 – Diagrama UML das classes relevantes na implementação do algoritmo Cheng e Church.

63
5.7 Teste das aplicações e algoritmos
As aplicações e sistemas informáticos são muitas vezes testados e avaliados apenas com
base na intuição dos programadores sobre o conjunto de casos de teste relevantes. No caso das
aplicações de Bioinformática, normalmente recorre-se a informação biológica real onde os
elementos em análise são conhecidos. Para teste e avaliação de algoritmos/aplicações de
descoberta de motivos, por exemplo, recorre-se a conjuntos de sequências biológicas com locais
de ligação conhecidos e comparam-se os motivos extraídos pelos algoritmos com os motivos reais.
Esta abordagem, seja qual for a natureza do software em teste, não oferece a garantia de
uma boa cobertura, isto é, não garante que todos os caminhos relevantes da aplicação são
exercitados. Como referido em [145], o teste de software deve ser (1) sistemático, no sentido em
que todos os caminhos possíveis são exercitados, (2) objectivo, isto é, utilizar informação
disponível sobre a localização mais provável para as falhas, e (3) automatizado, pois a
automatização permite a criação e execução de um maior número de testes repetíveis e
consistentes. A técnica de teste baseado em modelos (Model Based Testing – MBT) [146] permite
alcançar os três objectivos descritos no parágrafo anterior. Nesta secção, apresenta-se a técnica
MBT e descreve-se uma possível aplicação da mesma ao teste das aplicações e algoritmos
instalados no cluster hermes.
5.7.1 Testes Baseado em Modelos
A técnica de teste baseado em modelos consiste em derivar casos de teste, em todo ou
em parte, de um modelo que representa o comportamento esperado do sistema a testar. Esta é
uma técnica de teste de “caixa negra”, no sentido em que não são tidos em conta ou não se
conhecem os detalhes internos do item sobre teste. Os testes são derivados do modelo e não do
código fonte. É possível e recomendável, contudo, combinar o teste baseado em modelos com o
teste ao nível do código fonte.
A técnica divide-se num conjunto de actividades ilustradas na Figura 5.7:

64
Figura 5.7 – Workflow da técnica de teste baseado em modelos, adaptado de [147]. Podemos identificar seis actividades principais: construção do modelo, geração de entradas esperadas, geração de saídas esperadas, execução dos testes, comparação das saídas obtidas com as saídas esperadas e decisão de acções futuras.
O primeiro passo desta técnica consiste na construção do modelo e esta construção
começa com a análise dos requisitos do sistema. A documentação de requisitos deve especificar
tanto as características funcionais como as características não funcionais do sistema. Para
sistemas complexos, é recomendada a utilização de uma linguagem de modelação como o UML
para a especificação dos requisitos.
Concluída a análise de requisitos, deverá então ser desenvolvida uma representação
mental da funcionalidade do sistema, que pode ser vista como um pré-modelo. Com base no pré-
modelo, procede-se então à construção do modelo formal, utilizando uma técnica de modelação de
comportamento. As técnicas mais usuais são as tabelas de decisão (conjuntos de condições e
acções resultantes), as máquinas de estados finitas (conjunto de estados finito, transições entre
estados e acções), as gramáticas (conjunto de regras para linguagens) e as cadeias de Markov
(processo estocástico discreto com a propriedade de Markov, isto é, processo em que a
probabilidade de estar num determinado estado depende exclusivamente do estado imediatamente
anterior). As máquinas de estados finitas constituem a solução mais usual. Podem utilizar-se,

65
contudo, muitas outras técnicas. O importante é que o modelo criado permita a geração de casos
de teste de forma simples e, geralmente, automatizada. A experiência no domínio da aplicação,
não sendo essencial, constitui uma preciosa aliada na criação do modelo. Em [148] podemos
encontrar um guia para auxílio na escolha da melhor técnica de modelação consoante a aplicação
que se pretende testar.
Os dois passos seguintes consistem na geração das entradas e saídas esperadas, tendo
por base o modelo criado no passo anterior. Esta geração pode ser efectuada automaticamente,
desde que o modelo se encontre bem definido segundo uma das técnicas de modelação mais
usuais. Tal deve-se ao facto de, normalmente, apenas os tipos de modelos mais comuns serem
reconhecidos pelas aplicações existentes para geração de casos de teste. Em [147] pode ser
encontrada uma lista das ferramentas existentes para geração de testes com base em modelos.
Quando o modelo criado consiste numa máquina de estados finita ou, pelo menos, pode
ser interpretado como tal (o que acontece na maioria das situações), os casos de teste são
identificados através da análise dos caminhos executáveis da máquina de estados. Cada caminho
executável corresponde, geralmente, a um caso de teste. No entanto, dependendo da
complexidade do sistema e do modelo, o número de configurações possíveis do sistema pode ser
enorme e, consequentemente, o número de caminhos executáveis é também elevadíssimo. É
importante, por isso, possuir métodos que guiem a identificação dos melhores casos de teste. Para
alcançar este objectivo, podem ser empregues vários métodos distintos para derivação algorítmica
de casos de teste, tais como: prova de teoremas, programação em lógica com restrições, métodos
de verificação de modelos, manipulação simbólica e geração de casos de testes através de
modelos de fluxo de eventos [149].
Um ponto importante nesta fase do processo MBT é o conceito de “oráculo de teste” que
consiste no mecanismo que determina se os resultados da execução de um teste estão correctos
ou não. Por outras palavras, e como referido em [147], é o critério utilizado para verificar a
correcção da saída da aplicação em teste. Nem sempre é fácil obter um oráculo de teste adequado
e, no caso de isso ser impossível, a solução consiste em verificar apenas a plausibilidade das
saídas das aplicações (por exemplo, se as saídas estão dentro de certos limites considerados
razoáveis ou se verificam determinadas condições de consistências).
Com a definição das entradas e das saídas esperadas, obtemos um conjunto de casos de
teste derivado do modelo. Este conjunto contém testes funcionais ao mesmo nível de abstracção
do modelo. Por este motivo, o conjunto de testes gerado a partir do modelo não é directamente
executável, sendo necessária a criação de um conjunto de testes executáveis. Este novo conjunto
de testes pode comunicar directamente com o sistema que se pretende testar.

66
O próximo passo é, naturalmente, executar os casos de teste criados. Muitas aplicações de
model based testing geram automaticamente scripts executáveis que facilitam bastante este passo.
Após a execução dos casos de teste, procede-se à comparação das saídas esperadas com as
saídas observadas. Deverá existir nesta fase um processo automático (baseado no oráculo de
teste) que efectua esta comparação e alerta a equipa de testes para eventuais falhas.
O último passo da técnica é decidir sobre as acções futuras a realizar tendo em conta os
resultados obtidos com os casos de teste criados. A equipa de testes pode decidir realizar
alterações no modelo proposto para o sistema, gerar mais casos de teste, criar uma estimativa da
robustez do sistema ou dar a fase de testes por terminada.
5.7.2 Vantagens e desvantagens da técnica MBT
A grande vantagem da técnica MBT é sem dúvida o facto de possibilitar um elevado grau
de automatização na fase de testes do desenvolvimento de software, permitindo assim a execução
de muitos testes não-repetitivos e úteis. Outra grande vantagem é o facto de permitir uma
cobertura mais alargada da funcionalidade do sistema, oferecendo, à equipa de desenvolvimento,
mais garantias sobre a robustez do sistema. Além destas vantagens, podemos referir que esta
técnica é muito boa para testes de correcção/funcionais, facilita a comunicação entre a equipa de
teste e a equipa de desenvolvimento, facilita a exposição de ambiguidades na especificação do
sistema e os modelos são facilmente adaptáveis a mudanças. Esta adaptabilidade do modelo leva
a uma maior adaptabilidade dos casos de teste às eventuais mudanças nos requisitos.
A técnica MBT tem também algumas desvantagens. A mais óbvia será o facto de ser
necessária a criação de um modelo formal que representa o comportamento do sistema, algo que
pode ser bastante complicado de se efectuar. Além disso e como vimos na secção anterior, pode
ocorrer uma explosão em termos do número de casos de teste gerados a partir do modelo,
tornando-se necessário recorrer a técnicas adicionais que permitam seleccionar os melhores casos
de teste. Pode ser, contudo, complicado encontrar critérios objectivos para realizar esta filtragem
de casos de teste. Outro problema é o facto de o modelo ser, por vezes, muito sensível a
pequenas mudanças, ou seja, pequenas alterações no modelo podem originar um conjunto de
casos de teste completamente diferente.
5.7.3 Aplicação da técnica MBT às aplicações de descoberta de motivos
Seguindo o workflow da técnica de teste baseado em modelos, apresentado na Figura 5.7,
o primeiro passo consiste em determinar os requisitos da aplicação. Esses requisitos podem ser

67
facilmente e formalmente obtidos através da descrição do problema de descoberta de motivos,
apresentada na secção 4.2.1 e que reproduzimos aqui:
Dado um conjunto de sequências de DNA, 𝑆𝑆 = {𝑆𝑆1,𝑆𝑆2, … , 𝑆𝑆𝑡𝑡}, pretende-se encontrar um
conjunto de motivos, simples e/ou compostos, com comprimento contido num conjunto
{𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 } que ocorre num número 𝑞𝑞 ≤ 𝑡𝑡 de sequências com o máximo de 𝑒𝑒 erros. No caso dos
motivos compostos, é comum considerar também que cada componente está separado por uma
distância contida num conjunto {𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 }.
Por questões de simplicidade, vamos assumir que existe apenas um motivo 𝑚𝑚 no conjunto
de sequências 𝑆𝑆. O motivo 𝑚𝑚 pode ser simples ou composto, mas inicialmente vamos considerar
apenas motivos simples, isto é, com um único componente. Partindo então da descrição do
problema, podemos sumarizar o comportamento das aplicações do seguinte modo:
Dado um conjunto de sequências de DNA, 𝑆𝑆 = {𝑆𝑆1,𝑆𝑆2, … , 𝑆𝑆𝑡𝑡}, que contém um motivo simples
𝑚𝑚 com quorum 0 < 𝑞𝑞 ≤ 100 e comprimento 𝑙𝑙 > 0, fornecemos à aplicação um conjunto de
parâmetros que representam um conjunto de motivos simples 𝑚𝑚′. Os motivos em 𝑚𝑚′ têm quorum
de valor igual ou superior a 𝑞𝑞′, comprimento contido no intervalo [𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ] e admite-se um
número de mutações igual ou inferior a 𝑒𝑒. As várias situações que podem ocorrer são as seguintes:
• Se 𝑞𝑞 ≥ 𝑞𝑞′, 𝑙𝑙 ∈ [𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ] e 𝑒𝑒 = 0, pretende-se que a aplicação devolva uma lista que
contém o motivo 𝑚𝑚. Um ponto importante: assume-se que a aplicação tem a capacidade
de combinar motivos sobrepostos num único motivo. Na situação em que 𝑙𝑙 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , a
aplicação irá identificar apenas o motivo 𝑚𝑚, mas, quando 𝑙𝑙 ∈ ]𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ], serão
identificados o conjunto de motivos parciais de 𝑚𝑚. Dado 𝑙𝑙, o conjunto dos motivos parciais
de 𝑚𝑚 = 𝑚𝑚1 …𝑚𝑚𝑚𝑚 contém todos os motivos 𝑚𝑚′′ = 𝑚𝑚1+𝑚𝑚 …𝑚𝑚𝑗𝑗+𝑚𝑚 em que 0 ≤ 𝑚𝑚 e 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤
𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 . Queremos que a aplicação seja capaz de combinar esses motivos parciais de modo a
reportar apenas o motivo 𝑚𝑚, pelo que não se faz distinção entre a situação em que 𝑙𝑙 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚
e a situação em que 𝑙𝑙 ∈ ]𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ].
• Se 𝑞𝑞 ≥ 𝑞𝑞′, 𝑙𝑙 ∈ [𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ] e 𝑒𝑒 > 0, pretende-se que a aplicação devolva uma lista que
contém o motivo 𝑚𝑚 e todos os motivos que estão a uma distância de Hamming do motivo
𝑚𝑚 igual ou inferior ao número de erros 𝑒𝑒. Por outras palavras, a aplicação deverá devolver
todos os motivos 𝑚𝑚′′ em que 𝑑𝑑𝑚𝑚𝑠𝑠𝑡𝑡𝑚𝑚𝑚𝑚𝑑𝑑𝑚𝑚𝑚𝑚𝐻𝐻𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝐷𝐷(𝑚𝑚,𝑚𝑚′′ ) ≤ 𝑒𝑒 . Mais uma vez, considera-se
que a aplicação tem a capacidade de combinar motivos parciais num único motivo.
• Se 𝑞𝑞 < 𝑞𝑞′, pretende-se que a aplicação devolva uma lista que não contém o motivo 𝑚𝑚.
Trata-se da situação em que o parâmetro quorum fornecido à aplicação tem um valor
superior ao quorum do motivo 𝑚𝑚, isto é, 𝑚𝑚 está presente em menos sequências do que
aquelas que são pretendidas.

68
• Se 𝑙𝑙 < 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , pretende-se que a aplicação devolva uma lista que não contém o motivo 𝑚𝑚. O
motivo 𝑚𝑚 tem um comprimento inferior ao mínimo pretendido.
• Se 𝑞𝑞 ≥ 𝑞𝑞′, 𝑙𝑙 > 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 e 𝑒𝑒 = 0, pretende-se que a aplicação devolva uma lista que contém o
conjunto de motivos parciais de 𝑚𝑚 tais que 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 (ver primeiro ponto desta lista).
Queremos 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 para considerarmos apenas os motivos parciais máximos.
• Se 𝑞𝑞 ≥ 𝑞𝑞′, 𝑙𝑙 > 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 e 𝑒𝑒 > 0, pretende-se que a aplicação devolva uma lista que contém o
conjunto de motivos parciais de 𝑚𝑚, tais que 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , e todos os motivos que estão a
uma distância de Hamming desses motivos parciais igual ou inferior ao número de erros 𝑒𝑒.
Por outras palavras, a aplicação deverá devolver também todos os motivos 𝑚𝑚′′′ em que
𝑑𝑑𝑚𝑚𝑠𝑠𝑡𝑡𝑚𝑚𝑚𝑚𝑑𝑑𝑚𝑚𝑚𝑚𝐻𝐻𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝐷𝐷(𝑚𝑚′′,𝑚𝑚′′′ ) ≤ 𝑒𝑒 .
• Se 𝑞𝑞′ < 0 ou 𝑞𝑞′ > 100, a aplicação deverá retornar com erro.
• Se 𝑒𝑒 < 0, a aplicação deverá retornar com erro.
• Se 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 > 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ou 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 < 0, aplicação deverá retornar com erro.
Com base nesta descrição de comportamento, podemos agora criar o modelo. O tipo de
modelo escolhido foi a tabela de decisão [150]. Este tipo de modelo permite representar relações
complexas entre os parâmetros de entrada, sumarizando a lógica de um programa de forma
semelhante aos mapas de Karnaugh22
Figura 5.8
. Uma vantagem importante das tabelas de decisão
relativamente a outros métodos de representação é o facto de não requererem que as condições
sejam independentes entre si (algo que acontece, por exemplo, com as classes de equivalência). A
ilustra a estrutura básica de uma tabela de decisão.
Figura 5.8 – Estrutura básica de uma tabela de decisão, adaptado de [150]. Uma tabela de decisão está dividida em quatro secções: stubs de condições, onde se incluem todas as condições que podem ocorrer no problema em estudo. São geralmente as entradas da aplicação; entradas de condições – onde estão presentes todas as combinações de condições que podem ocorrer; stubs de acções, onde se incluem todas as acções possíveis no problema em análise. São geralmente as saídas da aplicação; e entradas de acções – onde estão presentes as acções a tomar para cada situação específica. Cada coluna na secção de entradas denomina-se regra.
22 Um mapa de Karnaugh é um diagrama que auxilia na minimização de funções booleanas.

69
As condições escolhidas para a situação em que pretendemos extrair apenas motivos
simples das sequências de entrada foram as seguintes:
c1: O motivo simples 𝑚𝑚 com quorum 𝑞𝑞 e comprimento 𝑙𝑙 está presente em 𝑆𝑆;
c2: 𝑞𝑞 ≥ 𝑞𝑞′
c3: 𝑙𝑙 ∈ [𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ]
c4: 𝑒𝑒 = 0
c5: 𝑙𝑙 > 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚
c6: 𝑞𝑞′ ≤ 0
c7: 𝑞𝑞′ > 100
c8: 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 > 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚
c9: 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 0
c10: 𝑒𝑒 < 0
As condições c6 a c10 poderiam ser combinadas numa única condição, mas pretendemos
que a aplicação seja capaz de identificar e reportar as diferentes situações de erro que podem
ocorrer.
As possíveis acções para a situação em análise serão as seguintes:
a1: Devolver uma lista que contém o motivo 𝑚𝑚
a2: Devolver uma lista que não contém o motivo 𝑚𝑚
a3: Devolver uma lista que contém os motivos a uma distância de Hamming de 𝑚𝑚
igual ou inferior a 𝑒𝑒
a4: Devolver uma lista que contém os motivos parciais de 𝑚𝑚 tais que, sendo 𝑚𝑚′′ =
𝑚𝑚1+𝑚𝑚 …𝑚𝑚𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑚𝑚, então 0 ≤ 𝑚𝑚 e 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 .
a5: Devolver uma lista que contém os motivos a uma distância de Hamming igual ou
inferior a 𝑒𝑒 dos motivos parciais de 𝑚𝑚 tais que 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚
a6: Devolver erro para quorum com valor igual ou inferior a 0
a7: Devolver erro para quorum com valor superior a 100
a8: Devolver erro para comprimento mínimo superior ao comprimento máximo
a9: Devolver erro para comprimento mínimo igual ou inferior a zero
a10: Devolver erro para número de erros com valor negativo
Uma tabela de decisão para esta situação está representada na Figura 5.9. As entradas
referentes às condições tomam os valores binários true – T (verdadeiro) e false – F (falso). As
entradas preenchidas com o símbolo “-“ denominam-se don’t care entries. Tratam-se de entradas
cujo valor não influencia a saída da aplicação. Um ponto importante sobre as tabelas de decisão é

70
o facto de não serem imperativas, isto é, não é imposta qualquer ordem sob as condições ou
acções. De referir ainda que esta é uma tabela de decisão limitada, no sentido em que utiliza
condições binárias (as tabelas de decisão estendidas utilizam múltiplas condições).
c1 - - - - - T T T T T T F
c2 - - - - - T T F - T T -
c3 - - - - - T T - F F F -
c4 - - - - - T F - - T F -
c5 - - - - - F F - F T T -
c6 T F F F F F F F F F F F
c7 - T F F F F F F F F F F
c8 - - T F F F F F F F F F
c9 - - - T F F F F F F F F
c10 - - - - T F F F F F F F
a1 X X
a2 X X X
a3 X
a4 X X
a5 X
a6 X
a7 X
a8 X
a9 X
a10 X
Figura 5.9 – Tabela de decisão para aplicações de extracção de motivos simples.
Para verificar a completude da tabela de decisão, utiliza-se o conceito de contagem de
regras [150]. Cada coluna equivale a uma regra, mas cada entrada “don’t care” duplica o número
de regras para a coluna respectiva. Neste caso temos 29 + 28 + 27 + 26 + 25 + 1 + 1 + 23 + 22 +
1 + 1 + 24 = 1024. O número de regras para uma tabela de decisão limitada é dado pela fórmula:
𝑚𝑚𝐷𝐷𝑚𝑚𝑒𝑒𝐶𝐶𝐶𝐶 𝑑𝑑𝑒𝑒 𝐶𝐶𝑒𝑒𝐷𝐷𝐶𝐶𝑚𝑚𝑠𝑠 = 2𝑚𝑚𝐷𝐷𝑚𝑚𝑒𝑒𝐶𝐶𝐶𝐶 𝑑𝑑𝑒𝑒 𝑑𝑑𝐶𝐶𝑚𝑚𝑑𝑑𝑚𝑚 çõ𝑒𝑒𝑠𝑠 (4)
Vem 210 = 1024 regras, o que coincide com o valor calculado com o outro método. Este
facto permite-nos concluir que temos uma tabela de decisão completa. Quando estes dois valores
não coincidem (algo que acontece com muita frequência nos estágios iniciais de desenvolvimento
do modelo) a tabela de decisão não é completa e, consequentemente, necessita de revisão.

71
O próximo passo consiste em extrair casos de teste do nosso modelo. Geralmente, o
número de regras corresponde ao número de casos de teste a extrair da tabela de decisão, pelo
que nesta situação teríamos 1024 casos de teste. Podemos contudo assumir que as entradas
“don’t care” representam condições para as quais basta testar um único valor de verdade e, deste
modo, teremos apenas um caso de teste por coluna da tabela23
Vamos agora considerar a situação em que 𝑚𝑚 é um motivo composto. Mais uma vez por
questões de simplicidade, vamos considerar que 𝑚𝑚 tem no máximo dois componentes:
. Como é possível observar, os 12
casos de teste obtidos são semelhantes à descrição feita nas páginas 66 e 67.
Dado um conjunto de sequências de DNA, 𝑆𝑆 = {𝑆𝑆1,𝑆𝑆2, … , 𝑆𝑆𝑡𝑡}, que contém um motivo 𝑚𝑚 com
quorum 0 < 𝑞𝑞 ≤ 10, composto por 𝑑𝑑 ∈ {1,2} componentes (o componente 𝑚𝑚 é designado por 𝑑𝑑𝑚𝑚) de
comprimentos 𝑙𝑙 = {𝑙𝑙1, … , 𝑙𝑙𝑑𝑑}, tal que ∀𝑧𝑧∈{1,2}∶ 𝑙𝑙𝑧𝑧 > 0, e, quando 𝑑𝑑 = 2, separados por uma distância
𝑑𝑑 > 0, fornecemos à aplicação um conjunto de parâmetros que representam um conjunto de
motivos 𝑚𝑚′. Os motivos em 𝑚𝑚′ têm quorum de valor igual ou superior a 𝑞𝑞′, são compostos por 𝑑𝑑′
componentes (o componente 𝑚𝑚 é designado por 𝑑𝑑′𝑚𝑚 ) de comprimentos mínimos
𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 = {𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑑𝑑′ ,𝑚𝑚𝑚𝑚𝑚𝑚 }, comprimentos máximos 𝑙𝑙𝑚𝑚𝑚𝑚𝑚𝑚 = {𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙𝑑𝑑′ ,𝑚𝑚𝑚𝑚𝑚𝑚 }, número de erros
𝑒𝑒 = {𝑒𝑒1, … , 𝑒𝑒𝑑𝑑′} e, quando 𝑑𝑑′ = 2, separados por uma distância contida no intervalo [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ].
As condições e acções identificadas para esta situação apresentam-se no Anexo 3. Com
base nestas condições e acções, construiu-se a tabela de decisão representada na Figura 5.10 e
na Figura 5.11. Vamos novamente verificar a completude da tabela de decisão utilizando o método
descrito na página anterior. Temos um total de 24 condições, pelo que 2𝑚𝑚𝐷𝐷𝑚𝑚𝑒𝑒𝐶𝐶𝐶𝐶 𝑑𝑑𝑒𝑒 𝑑𝑑𝐶𝐶𝑚𝑚𝑑𝑑𝑚𝑚 çõ𝑒𝑒𝑠𝑠 = 224 =
16777216. A contagem de regras para as diferentes colunas leva-nos a esse mesmo valor:
223 + 222 + 221 + 220 + 219 + 218 + 217 + 216 + 215 + 214 + 27 + 25 + 3 × 27 + 25 + 26 +
213 + 211 + 27 + 14 × 25 + 4 × 24 + 25 + 29 + 4 × 23 + 12 × 24 + 212 = 16777216.
Resta agora extrair os casos de teste do modelo criado. Tal como na situação em que
apenas considerávamos motivos simples, vamos assumir que as entradas “don’t care” representam
condições para as quais basta testar um único valor de verdade. Teremos assim um caso de teste
por cada coluna da tabela o que resulta em 58 casos de teste.
23 De notar que, normalmente, uma entrada “don’t care” representa uma condição cujo valor não influencia a saída da aplicação. Tal não significa que a aplicação vai ignorar essa condição e que esse valor não deve ser testado. De facto, se não assumíssemos que basta testar um dos valores para cada entrada “don’t care”, teríamos de testar todas as combinações possíveis para essas entradas. Para a primeira coluna da tabela, por exemplo, teríamos 512 casos de teste diferentes.

72
c1 - - - - - - - - - - T - T T T - - - T T - - - - - - - -
c2 - - - - - - - - - - - T - - - T T - - - T T T T T T T T
c3 - - - - - - - - - - T T T T T T T - F T T T T T T T T T
c4 - - - - - - - - - - F F F F F F F - T F F F F F F F F F
c5 - - - - - - - - - - T T T T T T T F - T T T T T T T T T
c6 - - - - - - - - - - T T T F F T F - - F F T F T T F F F
c7 - - - - - - - - - - - T - - - T F - - - T F F F F F T T
c8 - - - - - - - - - - - - - - - - - - - - - - - - - - - -
c9 - - - - - - - - - - T T F T F F - - - T T T T F F F T F
c10 - - - - - - - - - - - - - - - - - - - - - - - - - - - -
c11 - - - - - - - - - - F F F T T F F - - F T F T T F T F F
c12 - - - - - - - - - - - F - - - F F - - - F T T F T T F F
c13 - - - - - - - - - - - - - - - - - - - - - - - - - - - -
c14 - - - - - - - - - - - - - - - - - - - - - - - - - - - -
c15 T F F F F F F F F F F F F F F F F F F F F F F F F F F F
c16 - T F F F F F F F F F F F F F F F F F F F F F F F F F F
c17 - - T F F F F F F F F F F F F F F F F F F F F F F F F F
c18 - - - T F F F F F F F F F F F F F F F F F F F F F F F F
c19 - - - - T F F F F F F F F F F F F F F F F F F F F F F F
c20 - - - - - T F F F F F F F F F F F F F F F F F F F F F F
c21 - - - - - - T F F F F F F F F F F F F F F F F F F F F F
c22 - - - - - - - T F F F F F F F F F F F F F F F F F F F F
c23 - - - - - - - - T F F F F F F F F F F F F F F F F F F F
c24 - - - - - - - - - T F F F F F F F F F F F F F F F F F F
a1
a2 X X X X X X X X X X X X X X X
a3 X X X X X X
a4 X X X X X X X X X X X X
a5 X X X X X
a6 X X X X X X X X
a7 X X
a8 X
a9 X
a10 X X X X X X X
a11 X X X X
a12 X X X
a13 X X
a14
a15
a16
a17
a18
a19
a20
a21
a22
a23
a24
a25
a26
a27
a28
a29 X
a30 X
a31 X
a32 X
a33 X
a34 X
a35 X
a36 X
a37 X
a38 X
Figura 5.10 – Tabela de decisão para aplicações de extracção de motivos compostos (parte 1).

73
c1 - - - - - - F F F F - - - - - - - - - - - - - - - - - - F
c2 T T T T T T T T T T T T T T T T T T T T T T T T T T T T F
c3 T T T T T T F F F F F F F F F F F F F F F F F F F F F F -
c4 F F F F F F T T T T T T T T T T T T T T T T T T T T T T -
c5 T T T T T T T T T T T T T T T T T T T T T T T T T T T T -
c6 F F T T F F F F T F F - T T T T F F F F T T T T F F F F -
c7 F F F F F F - - - - - - - - - - - - - - - - - - - - - - -
c8 - - - - - - T F F F F - T T T T T T T T F F F F F F F F -
c9 T F T F T F - - - - - - T F T F T T F F T F T F T F T F -
c10 - - - - - - - - - - - - T T F F T F T F T T F F T T F F -
c11 F F F F T T F F F T F - F F F F T T T T F F F F T T T T -
c12 T T F F F F - - - - - - - - - - - - - - - - - - - - - - -
c13 - - - - - - F T F F F - F F F F F F F F T T T T T T T T -
c14 - - - - - - T T T T T F T T T T - - - - - - - - - - - - -
c15 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c16 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c17 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c18 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c19 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c20 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c21 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c22 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c23 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
c24 F F F F F F F F F F F F F F F F F F F F F F F F F F F F F
a1 X X X X
a2 X X X X X X X X X X X X X X X X X X X X X X X X X
a3 X X
a4 X X X X X X X X X X X X X X X X X X X X X X X X X X X
a5
a6 X X X X X X X X X X X X X X X X X X X X X X X X X X X X X
a7 X
a8
a9
a10 X X
a11 X X
a12 X
a13 X
a14 X
a15 X
a16 X
a17 X
a18 X
a19 X
a20 X
a21 X
a22 X
a23 X
a24 X
a25 X
a26 X
a27 X
a28 X
a29
a30
a31
a32
a33
a34
a35
a36
a37
a38
Figura 5.11 - Tabela de decisão para aplicações de extracção de motivos compostos (parte 2).

74
5.7.4 Realização dos testes com as aplicações de descoberta de motivos
De modo a testar as aplicações de descoberta de motivos trabalhadas durante esta tese,
criou-se uma aplicação em Java para a geração de sequências de DNA aleatórias com motivos
implantados. A aplicação aceita um ficheiro com a especificação dos motivos, simples e/ou
compostos, a serem implantados, ficheiro esse que tem o seguinte formato:
# RATE POS1 MOTIF1 ERRORS1 POS2 MOTIF2 ERRORS3 ... POSN MOTIFN ERRORSN
A coluna “RATE” corresponde ao quorum do motivo e toma valores entre 0 e 1. As colunas
seguintes correspondem às características dos diferentes componentes do motivo: “POS” indica a
posição de início do componente, “MOTIF” corresponde ao conjunto de nucleótidos do componente
e “ERRORS” indica o número de mutações ocorridas no componente. Naturalmente, as mutações
ocorridas são aleatórias no sentido em que diferentes sequências podem ter o mesmo componente
com diferentes mutações. Um possível exemplo seria:
.4 20 CTGC 0 40 ATTCTTT 0
.2 30 CCTGC 1 50 TTTCA 0 60 AATTTAC 2
Neste caso, teríamos dois motivos compostos. O primeiro motivo tem quorum 0.4 (o que
significa que, se tivermos 10 sequências, o motivo estará presente em 4 dessas sequências) e é
composto por dois componentes. O primeiro componente contém os caracteres “CTGC”, tem início
na posição 20 da sequência (o primeiro carácter da sequência está na posição 0) e tem 0
mutações. O segundo componente contém os caracteres “ATTCTTT”, tem início na posição 40 da
sequência e tem 0 mutações.
O segundo motivo tem quorum 0.2 e é composto por três componentes. O primeiro
componente contém os caracteres “CCTGC”, tem início na posição 30 da sequência e tem 1
mutação. O segundo componente contém os caracteres “TTTCA”, tem início na posição 50 da
sequência e tem 0 mutações. Por fim, o terceiro componente contém os caracteres “AATTTAC”,
tem início na posição 60 da sequência e tem 2 mutações.
A aplicação é invocada com três parâmetros: nseq, lseq e specfile. O parâmetro nseq
corresponde ao número de sequências que se pretende gerar. O parâmetro lseq corresponde ao
comprimento das sequências. Finalmente, o parâmetro specfile corresponde ao ficheiro com a
especificação dos motivos a serem implantados. Um exemplo de invocação seria:
> 10 100 exemplo.spec

75
Neste caso iriam ser geradas 10 sequências com 100 caracteres cada e nas quais estão
implantados os motivos descritos no ficheiro exemplo.spec.
Dado que apenas os parâmetros da aplicação RISO obedecem exactamente ao modelo
desenvolvido, foi necessário efectuar umas pequenas alterações/simplificações nos testes das
restantes aplicações (no Anexo 1 pode ser encontrada uma listagem e descrição pormenorizada
de todos os parâmetros das diferentes aplicações trabalhadas no âmbito desta tese). No caso da
aplicação PSMILE, existem três parâmetros adicionais: número total de substituições, comprimento
mínimo total e comprimento máximo total. Na fase de execução dos testes, estes parâmetros foram
definidos como as características correspondentes do motivo a encontrar (caso este exista,
naturalmente).
A aplicação MUSA apresenta uma série de diferenças relativamente ao modelo criado. Em
particular, não é possível especificar o número de componentes de um motivo, o comprimento
mínimo e máximo de cada componente, o número de erros admitido em cada componente e a
distância entre cada par de componentes. É possível, contudo, especificar a maioria destes
parâmetros tendo em conta a totalidade do motivo e procurou-se realizar os testes tendo em conta
esse aspecto. Relativamente ao parâmetro “pesquisar em ambas as cadeias”, optou-se por
efectuar a pesquisa em apenas uma das cadeias de DNA.
Para todos os casos de teste foram geradas 10 sequências de 100 nucleótidos cada. Os
motivos implantados variaram consoante o caso de teste em estudo. O método de avaliação
estatística utilizado com o PSMILE e com o RISO foi sempre o método de shuffling com os
parâmetros definidos por defeito (consultar Anexo 2). Os testes foram realizados num dos nós
computacionais do cluster hermes.
Uma tabela sumarizando os resultados obtidos está ilustrada na Figura 5.12, onde os
casos de teste foram numerados segundo a ordem pela qual se apresentam as colunas da tabela
de decisão das páginas 72 e 73.
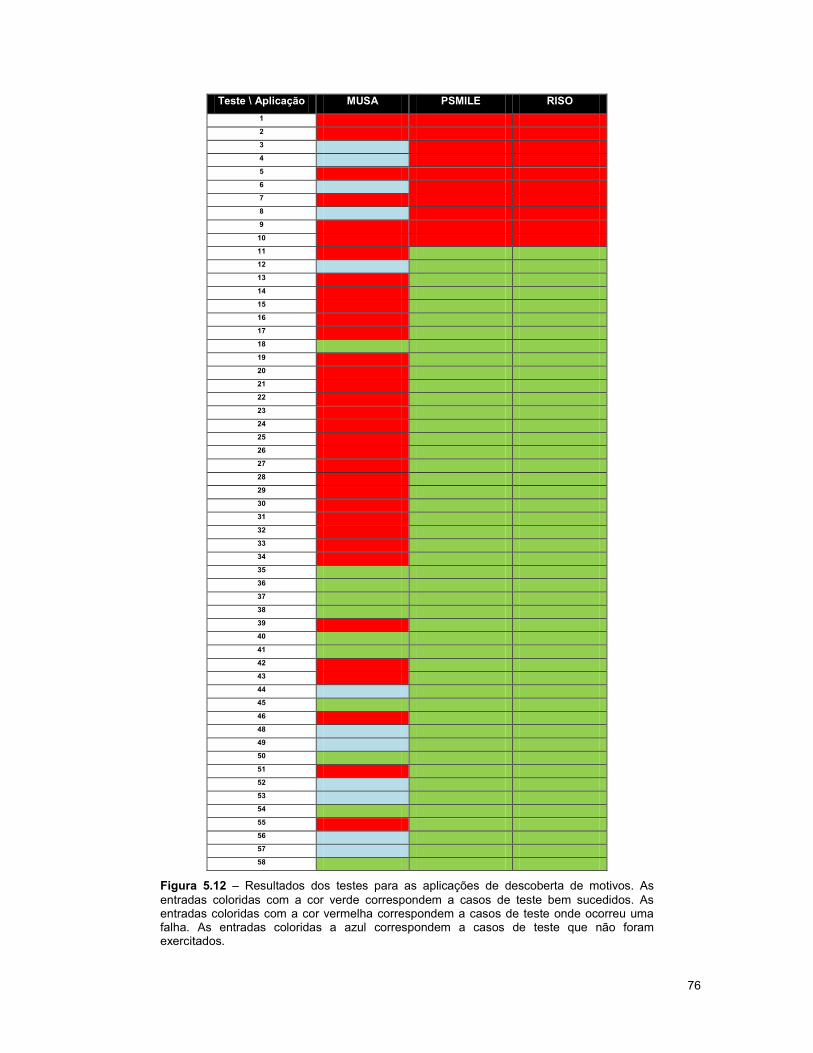
76
Teste \ Aplicação MUSA PSMILE RISO 1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
48
49
50
51
52
53
54
55
56
57
58
Figura 5.12 – Resultados dos testes para as aplicações de descoberta de motivos. As entradas coloridas com a cor verde correspondem a casos de teste bem sucedidos. As entradas coloridas com a cor vermelha correspondem a casos de teste onde ocorreu uma falha. As entradas coloridas a azul correspondem a casos de teste que não foram exercitados.

77
5.7.5 Resultados dos testes para as aplicações de descoberta de motivos
Na situação em estudo, o teste das aplicações tem uma dupla utilidade: se assumirmos
que a implementação de um dado algoritmo está totalmente correcta, podemos tirar ilações sobre
a “qualidade” do algoritmo relativamente ao problema que este pretende resolver; por outro lado,
podemos também, sendo este o objectivo principal da técnica de testes baseados em modelos,
tirar ilações sobre a qualidade da implementação dos algoritmos, isto é, sobre a qualidade do
software.
Em todas as aplicações é evidente que falta realizar algum trabalho em termos de
verificação dos parâmetros de entrada. No caso do MUSA, a aplicação nunca reporta os erros nos
parâmetros de entrada que especificam as características dos motivos a procurar. O algoritmo é
executado com os parâmetros incorrectos e a saída do programa é, naturalmente, uma lista vazia.
É no mínimo questionável se esta é a melhor opção. Se, por um lado, o utilizador percebe que algo
correu mal ao deparar-se com uma lista vazia, por outro lado não lhe é dada qualquer informação
sobre o parâmetro ou parâmetros com valor incorrecto.
No caso do PSMILE e do RISO, verifica-se, através da análise do código fonte, que a
verificação dos parâmetros de entrada está implementada, não estando, contudo, funcional.
De referir ainda que, não obstante o facto de as interfaces desenvolvidas no âmbito desta
tese possuírem os mecanismos necessários para a verificação dos parâmetros de entrada, é
sempre importante que essa verificação esteja também nas aplicações, nomeadamente se estas
forem portadas para ambientes onde não existe pré-processamento de parâmetros.
É facilmente perceptível pela configuração da tabela de resultados que a grande maioria
dos testes realizados com o MUSA resultaram em falha. O facto de o MUSA utilizar uma
abordagem heurística é sem dúvida uma das principais razões para o elevado número de casos de
teste falhados, dado que as soluções pretendidas podem não ser identificadas. Além disso, é
complicado forçar a procura por motivos compostos ou motivos simples. Outra das razões mais
óbvias é o facto de o modelo base do MUSA ser menos específico que o modelo utilizado para os
testes. De referir ainda que, devido ao facto de o algoritmo não suportar mutações no interior dos
motivos, todos os testes onde foram inseridos erros resultaram em falha. Podem ser levantadas
duas questões relativamente a este aspecto: por um lado, se faz sentido considerar esses casos
de teste com a aplicação MUSA, visto que já sabíamos à partida que esta não iria extrair os
motivos implantados; por outro lado, se faz sentido considerar que a aplicação falhou, atendendo a
que se comportou de acordo com aquilo que esperamos para o algoritmo. Relativamente à
primeira questão, optou-se, no caso do MUSA, por não se considerar apenas os casos de teste
que envolviam a especificação de parâmetros referentes a mais do que um componente (algo que
não é suportado pelo modelo subjacente ao algoritmo). Relativamente à segunda questão e por

78
uma questão de rigor, considerámos que a aplicação falhou nos casos de teste em análise, pois,
de acordo com o modelo utilizado para os testes, não exibiu o comportamento esperado.
O programa apresentou problemas principalmente na situação em que as sequências
continham apenas um motivo simples. Nessa situação, observaram-se erros de acesso a memória,
o retorno de uma lista vazia ou (situação mais frequente) o ignorar do comprimento especificado
para os motivos. Este último problema fez com que muitas vezes a aplicação reportasse os
motivos na sua totalidade e não os motivos parciais pretendidos.
Nas situações em que um motivo com dois componentes e sem mutações tinha sido
implantado nas sequências, a aplicação comportou-se de forma satisfatória, não apresentando
problemas inclusivamente na identificação de motivos parciais.
Relativamente ao modelo utilizado, poderia ter sido criado um modelo específico para cada
aplicação, que tivesse em conta as particularidades de cada algoritmo. Por uma questão de
simplicidade, optou-se por se recorrer a um modelo mais genérico o que acaba por funcionar bem
com as aplicações de descoberta de motivos, dado que a maioria destas tem como problema de
base o apresentado nesta tese (ou variações muito semelhantes).
Para terminar, é importante mencionar que o oráculo de teste utilizado pode ser refinado.
Considerou-se que para um dado teste ser bem sucedido apenas era necessário que os motivos
pretendidos estivessem contidos na lista devolvida pela aplicação. Este critério pode ser refinado
se tivermos em conta que a lista retornada pelas aplicações vem ordenada segundo medidas de
significância estatística. Podemos, por exemplo, considerar que um caso de teste apenas é bem
sucedido se os motivos pretendidos estiverem entre os 10 primeiros da lista. Podemos também
considerar valores máximos para o p-value dos motivos pretendidos. Outro aspecto que poderá ser
incluído no oráculo de teste é o tempo de execução. Podemos considerar que um teste é bem
sucedido apenas quando este é executado com sucesso num tempo inferior a um valor pré-
estabelecido.
5.7.6 Aplicação da técnica MBT às aplicações de biclustering
Essencialmente por questões de espaço e de complexidade, optou-se por não se realizar
nesta tese um estudo sobre utilização da técnica MBT nas aplicações de biclustering instaladas no
cluster hermes. Deixam-se, no entanto, algumas directrizes para a realização desse estudo.
A ideia será, naturalmente, partir da definição do problema de biclustering, apresentada no
capítulo anterior. Será útil desenvolver uma aplicação que crie matrizes de dados aleatórios onde
estão implantados biclusters de características bem definidas.

79
Dado que todas as aplicações de biclustering trabalhadas nesta tese têm o seu
processamento dividido em várias fases distintas, cada uma com entradas e saídas bem definidas,
será útil dividir o estudo a realizar com a técnica MBT segundo essas diferentes fases. Poderá ser
criado um modelo para cada uma dessas fases e posteriormente os vários modelos poderão ser
combinados de modo a obter uma representação completa do comportamento de cada aplicação.
No caso das aplicações CCC-Biclustering e e-CCC-Biclustering, os modelos para carregamento de
parâmetros, pré-processamento, pós-processamento e output serão idênticos para as duas
aplicações, sendo apenas necessário criar um modelo distinto para a fase de extracção de
biclusters, na medida em que a segunda aplicação admite erros nos dados de expressão.
A análise poderá também ser realizada de uma forma mais genérica, à semelhança do que
foi feito para as aplicações de descoberta de motivos. Deste modo, será criado apenas um modelo
para as três aplicações dos quais serão derivados casos de teste onde se pretende extrair
biclusters com determinadas características que foram implantados numa matriz de dados
aleatórios. Esta análise é naturalmente mais simples, tendo a desvantagem de não trabalhar sobre
as especificidades de cada algoritmo/aplicação.

80
Capítulo 6 Conclusões e trabalho futuro
6.1 Conclusões
No âmbito desta dissertação disponibilizaram-se, num cluster computacional, métodos
eficientes para inferência de motivos em regiões promotoras de genes e para análise de dados de
expressão de genes. Ao longo do trabalho foram realizadas as tarefas necessárias de
implementação, instalação e teste, bem como a criação das interfaces Web para as várias
aplicações. Os algoritmos desenvolvidos no âmbito do projecto “BioGrid – Algoritmos Paralelos
Para Anotação de Genes” estão agora disponíveis, num sistema robusto e de fácil utilização, para
uma vasta comunidade de utentes de Bioinformática.
Como vimos nos capítulos 2 e 4, os problemas que estes algoritmos se propõem a resolver
têm associado um conjunto de desafios e dificuldades que a ciência actual está ainda longe de
dominar. No caso da descoberta de motivos, as principais dificuldades residem na inexistência de
informação prévia sobre os motivos a extrair, na identificação da região reguladora a pesquisar, na
possibilidade de existirem mutações nos locais de ligação e na necessidade de considerar motivos
complexos. No caso da análise de dados de expressão de genes, podemos identificar como
principais dificuldades os erros sistemáticos na análise dos dados de microarrays (quantidades
distintas de mRNA de experiência para experiência, problemas experimentais ou protocolares,
fluoróforos com eficiência de incorporação diferente, entre outros) e a escolha dos melhores
métodos para detectar e minimizar esses erros. Além destes aspectos, há ainda que considerar,
tanto no caso da pesquisa de motivos como no caso da análise de dados de expressão, a

81
possibilidade de as aplicações identificarem motivos ou biclusters que resultam de ocorrências
aleatórias e não possuem qualquer relevância biológica. Apesar da vasta maioria das aplicações
modernas de Bioinformática possuir um último passo de avaliação estatística, que ordena os
elementos extraídos de acordo com uma medida de significância estatística, tal é ainda insuficiente
para isolar verdadeiramente os elementos importantes do ponto de vista biológico. De facto, um
dos grandes problemas que hoje se coloca na investigação em Bioinformática e Biologia
Computacional é a compreensão da relação entre o significado biológico e o significado estatístico.
Apesar destas dificuldades, verifica-se, nomeadamente através de estudos como o realizado no
capítulo 5 com a técnica de testes baseados em modelos, que as várias abordagens trabalhadas
durante esta tese são capazes de, na maioria dos casos, identificar os elementos pretendidos,
tanto em dados sintéticos como em dados reais.
Sobre o estudo realizado no capítulo 5 com a técnica de teste baseado em modelos, há
alguns aspectos que importa mencionar. Para a aplicação desta técnica, recorreu-se naturalmente
a dados sintéticos onde foram implantados elementos de características conhecidas. Esta é uma
condição necessária para se avaliar a qualidade do software desenvolvido. No entanto, este estudo
nada nos diz sobre a “qualidade biológica” dos algoritmos, isto é, sobre a capacidade destes
métodos em identificar elementos biologicamente relevantes. De facto, o controlo total sobre os
elementos implantados, proporcionado pela utilização de dados sintéticos, tem como contrapartida
o facto de sequências reais não poderem ser modeladas apenas com motivos importantes para a
regulação da transcrição que se encontram rodeados de nucleótidos insignificantes. Na realidade,
e como vimos em parte no capítulo 2, as regiões reguladoras são o resultado da interferência de
vários processos e a posição e distribuição dos nucleótidos nestas regiões não é aleatória, mas
sim o resultado de milhões de anos de evolução.
Este estudo não nos permite também tirar quaisquer ilações sobre a qualidade das
interfaces pessoa-máquina desenvolvidas. Neste sentido, apresentam-se na próxima secção
algumas directrizes para trabalho futuro.
6.2 Trabalho Futuro
O trabalho desenvolvido no âmbito desta dissertação pode ser complementado ou
melhorado em diversos pontos.
Neste momento, o último passo da execução das aplicações RISO e PSMILE consiste na
avaliação estatística dos motivos identificados, utilizando o método shuffling ou against (ver Anexo
1). Este último passo pode ser melhorado se substituirmos os métodos presentemente utilizados
por um procedimento que aglomera os motivos reportados em famílias, à semelhança do que já

82
acontece com a aplicação MUSA. Esta alteração está prevista e em execução, mas não foi
possível conclui-la em tempo útil para esta tese.
Este último passo pode ainda ser melhorado adoptando uma representação mais gráfica
das famílias identificadas, como é realizado em [125], onde se encontram disponíveis os algoritmos
RISO e MUSA. Essa representação consiste num sequence logo [151], também designado por
motif logo, para cada família de motivos extraída das sequências de entrada. Este motif logo é
desenhado com base nas PWM’s que representam a família e que, como vimos no capítulo
anterior, são geradas pelo procedimento que aglomera motivos. As famílias reportadas estão
ordenadas segundo a medida de significância estatística p-value. A Figura 6.1 ilustra o tipo de
representação pretendido.
Figura 6.1 – Exemplo de representação utilizando motif logos de um conjunto de famílias identificadas com o algoritmo MUSA [125].
Também esta alteração está prevista e em execução, não tendo, contudo, sido possível
terminá-la em tempo útil para esta tese. Será utilizado um script escrito na linguagem PHP para
desenhar os motif logos após a fase de extracção. O script pode ser executado localmente e
recebe os parâmetros através de HTTP GET.
Em [125] existe ainda uma funcionalidade que permite a comparação das famílias de
motivos identificadas com os locais de ligação para factores de transcrição descritos na base de
dados YEASTRACT. Esta funcionalidade poderá eventualmente ser integrada nas aplicações de

83
descoberta de motivos instaladas no cluster hermes, possivelmente utilizando diferentes bases de
dados de locais de ligação.
Em termos algorítmicos, será importante explorar as possibilidades de paralelização de
MUSA, RISO, CCC-Biclustering e e-CCC-Biclustering. A distribuição do esforço computacional
pelos vários nós poderá incidir sobre a travessia da árvore de sufixos ou factor tree, tal como
acontece com o algoritmo PSMILE.
Relativamente à avaliação do sistema, esta pode ser aperfeiçoada, complementando o
estudo, realizado com a técnica de testes baseados em modelos, com a avaliação das interfaces.
A avaliação das interfaces pode ser também realizada com a técnica de testes baseados
em modelos, recorrendo para o efeito a um tipo de modelo específico denominado fluxo de eventos
[152]. No entanto, a usabilidade de uma interface é normalmente avaliada recorrendo a métodos
analíticos ou empíricos [153]. Com os métodos analíticos, os utilizadores finais do sistema não são
integrados na avaliação. Já os métodos empíricos baseiam-se na experiência de utilização dos
utilizadores do sistema. Nos métodos analíticos surgem dois tipos de avaliação: a avaliação
heurística, que é realizada por peritos, e a avaliação preditiva, que é realizada com base em
modelos de utilização. No primeiro caso, um pequeno conjunto de avaliadores (entre 3 a 5)
examinam a interface com o utilizador, tendo por base a aderência dessa mesma interface a
heurísticas de usabilidade (as chamadas heurísticas de Nielsen [154]). A avaliação heurística é
mais aconselhável para a avaliação de designs iniciais e protótipos, mas, como é óbvio, também
pode ser utilizada na avaliação de produtos finais.
No caso específico das aplicações instaladas no cluster hermes, o método mais útil será a
avaliação empírica, por permitir obter dados mais concretos sobre a usabilidade do sistema. Para
alcançar esse fim, poder-se-á recorrer a um conjunto de técnicas que vão desde entrevistas e
questionários até á simples observação da realização das tarefas. Durante a realização dos testes,
deverão ser recolhidas medidas de desempenho, como, por exemplo, o tempo necessário para
completar uma tarefa, o número de erros cometidos, a frequência de utilização de manuais ou
ajudas, entre outros.
De referir que para garantir o sucesso desta avaliação de usabilidade é necessária a
colaboração próxima com a BioTeam, visto que a base das interfaces foi desenvolvida por esta
empresa.
Por fim, de referir que, apesar desta tese se intitular “processamento de dados biológicos
utilizando tecnologias Grid”, na realidade exercitou-se maioritariamente o conceito de computação
em cluster. A migração destas aplicações para um ambiente Grid beneficiará das vantagens
inerentes à utilização do paradigma da malha de computadores relativamente à computação em
cluster: suporte para heterogeneidade entre os nós da malha; possibilidade de utilizar recursos

84
computacionais disponíveis em diferentes nós, mormente na situação em que as aplicações não
estão paralelizadas; maior dinamismo do sistema e maior autonomia dos nós.
6.3 Perspectivas de futuro para a Bioinformática
A investigação continuada, nas áreas da Bioinformática e Biologia Computacional referidas
no capítulo 4, promete novos desafios, bem como um conjunto de oportunidades inesperadas e
sem precedentes.
O estudo dos mecanismos de expressão dos genes, recorrendo a técnicas como a análise
de dados de microarrays e a utilização de RNA de interferência (RNAi)24
A análise de haplótipos
, permitirá a classificação
de tumores, bem como uma melhor compreensão dos processos de evolução das doenças e a
criação de modelos formais para esses processos. Em teoria, todas as patologias relacionadas
com genes “defeituosos” deixarão de ser uma ameaça.
Os avanços na investigação dos mecanismos de regulação dos genes permitirão também
um melhor entendimento do funcionamento do cérebro humano, a rede neuronal mais complexa de
entre as conhecidas. Num futuro próximo ocorrerá uma sinergia entre neurologia, informática e
biologia, dando origem a uma nova disciplina denominada Neurobioinformática [155], a qual
investigará possíveis melhoramentos das capacidades intelectuais humanas.
No que toca à imunologia, o melhoramento dos estudos e bases de dados actuais terá
várias aplicações ao nível da saúde, tais como o combate ao bioterrorismo e a doenças (como a
asma e as doenças auto-imunes), através do desenho de vacinas e do controlo de alergias. 25
24 O RNAi é uma técnica recente que utiliza RNA de cadeia dupla (dsRNA) para inibir a expressão de um ou mais genes alvo. Apesar de só em 1998 ter sido descoberto por Andrew Fire e Craig Mello (galardoados com o prémio Nobel de Medicina de 2006), o RNAi é uma mecanismo antigo que foi conservado durante a evolução em organismos como o do ser humano e das leveduras.
25 Os cromossomas não passam de geração para geração como cópias idênticas, sofrendo um processo denominado recombinação. No entanto, à medida que vão surgindo novas gerações, existem segmentos dos cromossomas ancestrais que não são quebrados e são misturados em repetidos eventos de recombinação. Alguns destes segmentos, os haplótipos, correspondem a sequências de DNA partilhadas por múltiplos indivíduos e que contêm a maior parte da informação sobre as variações genéticas.
é outra área bastante promissora. O estudo destes elementos
permitirá a descoberta das variantes genéticas de cada doença e a análise de respostas
individuais a agentes terapêuticos. Com a descoberta dessas variantes, os cientistas poderão obter
uma melhor compreensão sobre as origens das doenças e criar formas de preveni-las, diagnosticá-
las e tratá-las. Além disso, os tratamentos médicos poderão ser personalizados de acordo com o
contexto genético do paciente, de modo a maximizar a eficiência destes procedimentos e minimizar
os seus efeitos secundários. De referir o projecto HapMap [156], que resulta de uma colaboração

85
conjunta de vários países e onde se pretende desenvolver um mapa de haplótipos para o genoma
humano que descreva os padrões de variação genéticos mais comuns. Também no grupo KDBIO
do INESC-ID se realiza neste momento investigação relacionada com a inferência de haplótipos,
nomeadamente utilizando técnicas de satisfação booleana (SAT).
Outra área com boas perspectivas de futuro é a modelação de proteínas por homologia,
que consiste em utilizar como referência para o modelo de uma dada proteína uma ou mais
proteínas homólogas de estrutura terciária conhecida. Pensa-se que através da combinação de
técnicas de modelação, de alinhamento múltiplo de sequências e de previsão de estrutura
secundária, será possível determinar a sequência, estrutura e função das proteínas e relacionar
estes três aspectos de modo a construir modelos de processos biológicos complexos.
Naturalmente, o objectivo maior da investigação em Bioinformática e Biologia
Computacional é a criação de modelos computacionais integrados que sejam capazes de simular,
de uma forma realista, o comportamento dinâmico de sistemas biológicos complexos. Estes
modelos permitirão compreender e, em última instância, modificar o comportamento de redes de
regulação complexas, bem como testar hipóteses sobre o funcionamento de processos biológicos.
Neste sentido, é importante evoluir de uma investigação centrada em aspectos específicos (genes,
haplótipos, proteínas) para uma investigação mais geral e integrante que tenha como alvo redes de
regulação, interacções entre proteínas, subsistemas celulares, entre outros. Pensa-se que os
desenvolvimentos no estudo das redes complexas [157] em muito contribuirão para esta transição.
O futuro próximo da Bioinformática é a integração. Não apenas a integração das diferentes
áreas de investigação, como referido no parágrafo anterior, mas também dos recursos
computacionais disponíveis.
Até há algum tempo atrás, a integração de recursos era conseguida em parte graças a
métodos de screen-scraping26
Actualmente, um dos meios que oferece melhores condições para a comunicação e
combinação de vários recursos é o conjunto de tecnologias de Web services. O termo “Web
service” é comummente associado ao standard da W3C [13], mas, hoje em dia, qualquer método
de acesso programático na Web toma essa designação. Existem três tecnologias principais de
Web services [158]: os Web Services da W3C, os serviços REST (Representational State Transfer)
e os serviços XML-RPC (Remote Procedure Call). A tecnologia mais promissora corresponde ao
de páginas Web ou métodos manuais (copy-paste). Estes métodos
são claramente problemáticos: o screen-scraping está destinado a falhar quando ocorrem
modificações, ainda que pequenas, nas páginas a analisar; os métodos manuais são demasiado
trabalhosos e a sua correcção é de difícil verificação.
26 O screen-scraping consiste em utilizar um programa ou script que extrai informação da saída de outro programa. É mais complicado do que o parsing usual, no sentido em que os elementos a analisar foram criados com vista a serem compreendidos por humanos, não estando estruturados de forma a facilitar a extracção automática de informação dos mesmos.

86
standard da W3C. Esta tecnologia permite aos fornecedores de recursos computacionais, sejam
estes ferramentas, bases de dados ou outros, a descrição dos métodos de acesso e manipulação
desses recursos através da WSDL (Web Services Description Language). É esta linguagem
“universal” que possibilita a interoperabilidade e integração fácil de recursos heterogéneos. A
comunicação entre recursos processa-se utilizando o protocolo SOAP (Simple Object Access
Protocol) e as mensagens são escritas em XML.
Existem ainda, contudo, muitas dúvidas sobre qual será a melhor forma de integrar os
dados. Neste sentido, encontramos duas abordagens distintas [158]: a integração sintáctica que se
baseia no esforço de estandardização; e a integração semântica, em que a interoperabilidade é
alcançada através da partilha de um modelo semântico para os dados. No primeiro caso, o formato
de eleição tem sido o FASTA, apesar de esforços recentes incidirem sobre o XML. No segundo
caso, os modelos semânticos são actualmente formalizados com formatos semi-estruturados como
o RDF [159] (Resource Descritption Format), a OWL [160] (Web Ontology Language) ou, para
descrever serviços, a OWL-S [161]. A integração semântica pode ser realizada de forma
centralizada num servidor, realizada segundo uma metodologia par-a-par ou realizada do lado do
cliente. Este último método tem vindo a ganhar relevância dados os benefícios para os
investigadores que advêm da sua utilização. Os recursos que o investigador tem à sua disposição
são-lhe apresentados num ambiente intuitivo e integrativo, com metáforas de interacção
conhecidas e, mais importante do que tudo, não são necessárias preocupações com as
idiossincrasias tecnológicas. Um exemplo de ferramenta para integração do lado do cliente é a
Taverna Workbench [162].
A Taverna Workbench foi criada no âmbito do projecto myGrid [163] e providencia um
ponto de acesso único para os recursos disponíveis, sejam estes locais ou remotos, permitindo a
criação e execução de workflows com base nesses recursos. A Taverna Workbench é distribuída
com informação sobre um vasto conjunto de Web services, mas qualquer serviço que exiba uma
interface programática pode ser adicionado ao painel da aplicação. Na Figura 6.2, mostra-se um
exemplo de workflow utilizando a aplicação MUSA instalada no cluster hermes. Para a versão 2.0,
a ser lançada brevemente, estão previstos alguns melhoramentos, tais como o suporte a workflows
de longa duração recorrendo a tecnologias Grid e um registo semântico onde os serviços estão
anotados com termos de uma ontologia standard.
A Taverna é apenas uma das vertentes do projecto myGrid. Na realidade, este projecto
constitui o primeiro esforço no sentido de oferecer um conjunto de serviços e ferramentas que
satisfaçam todos os requisitos de e-Science descritos no capítulo 3. Um dos elementos mais
importantes do projecto é a rede social myExperiment [164] que oferece a funcionalidade de
pesquisa de workflows, tutoriais ou mensagens/tópicos interessantes. É importante mencionar
também a ferramenta Feta [165] que permite aos investigadores a descoberta semântica (ou seja,
não apenas por designação, mas também por função) de serviços que satisfaçam os seus
requisitos. De referir ainda a UTOPIA [166], uma colecção de aplicações para análise da sequência
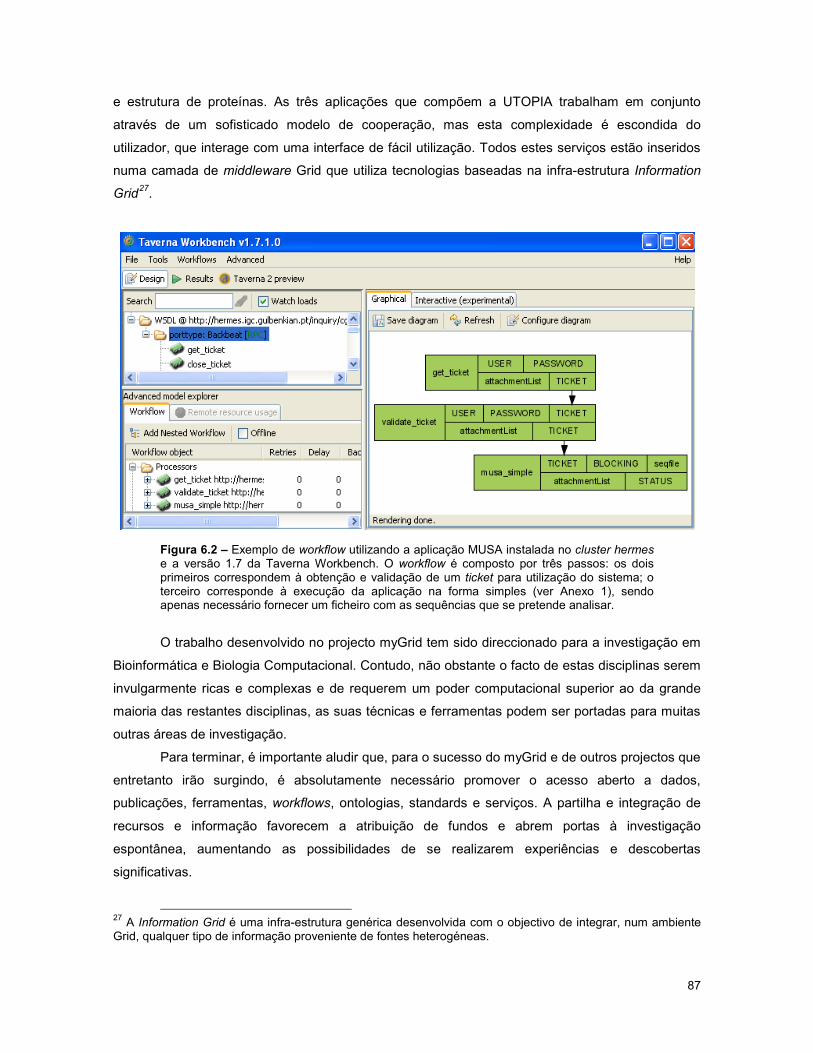
87
e estrutura de proteínas. As três aplicações que compõem a UTOPIA trabalham em conjunto
através de um sofisticado modelo de cooperação, mas esta complexidade é escondida do
utilizador, que interage com uma interface de fácil utilização. Todos estes serviços estão inseridos
numa camada de middleware Grid que utiliza tecnologias baseadas na infra-estrutura Information
Grid27
.
Figura 6.2 – Exemplo de workflow utilizando a aplicação MUSA instalada no cluster hermes e a versão 1.7 da Taverna Workbench. O workflow é composto por três passos: os dois primeiros correspondem à obtenção e validação de um ticket para utilização do sistema; o terceiro corresponde à execução da aplicação na forma simples (ver Anexo 1), sendo apenas necessário fornecer um ficheiro com as sequências que se pretende analisar.
O trabalho desenvolvido no projecto myGrid tem sido direccionado para a investigação em
Bioinformática e Biologia Computacional. Contudo, não obstante o facto de estas disciplinas serem
invulgarmente ricas e complexas e de requerem um poder computacional superior ao da grande
maioria das restantes disciplinas, as suas técnicas e ferramentas podem ser portadas para muitas
outras áreas de investigação.
Para terminar, é importante aludir que, para o sucesso do myGrid e de outros projectos que
entretanto irão surgindo, é absolutamente necessário promover o acesso aberto a dados,
publicações, ferramentas, workflows, ontologias, standards e serviços. A partilha e integração de
recursos e informação favorecem a atribuição de fundos e abrem portas à investigação
espontânea, aumentando as possibilidades de se realizarem experiências e descobertas
significativas.
27 A Information Grid é uma infra-estrutura genérica desenvolvida com o objectivo de integrar, num ambiente Grid, qualquer tipo de informação proveniente de fontes heterogéneas.

88
Referências
1. MundoVestibular.com. DNA. 2007 [citado; Disponível em: http://www.mundovestibular.com.br/materias/imagens/DNA2.gif.
2. Kessler, C.C. DNA vs RNA. 2007 [citado; Disponível em: http://www.algosobre.com.br/images/stories/biologia/dna_rna.jpg.
3. Jones, M. Flow of information in biological systems. 2006 [citado; Disponível em: http://en.wikipedia.org/wiki/Image:CDMB2.png.
4. Alberts, B., et al., Figure 3-15: The transfer of information from DNA to protein, in Molecular Biology of the Cell. 2002, Garland.
5. Lodish, H., et al., Figure 4-10: Three stages in transcription, in Molecular Cell Biology. 2003, W. H. Freeman. p. 110.
6. Mendes, N.D., Inference of Complex Motifs using Biclustering Techniques. 2005, IST, Universidade Técnica de Lisboa.
7. Buyya, R. and S. Venugopal, A Gentle Introduction to Grid Computing and Technologies, in CSI Communications. 2005: Mumbai, India. p. 9-19.
8. Stevens, R., et al., Performing in silico experiments on the Grid: a users perspective, in Proceedings of the UK e-Science All Hands Conference. 2003: Nottingham, UK Cox. p. 43-50.
9. The Globus Alliance. 2007 [citado; Disponível em: www.globus.org. 10. The Grid Computing and Distributed Systems (GRIDS) Laboratory, University of
Melbourne. 2007 [citado; Disponível em: www.gridbus.org. 11. Open Grid Forum. 2007 [citado; Disponível em: www.ggf.org. 12. OASIS Web Services Resource Framework (WSRF) TC. 2008 [citado; Disponível em:
http://www.oasis-open.org/committees/wsrf. 13. W3C. Web Services @ W3C. 2008 [citado; Disponível em: http://www.w3.org/2002/ws/. 14. Message-Passing-Forum, MPI: A Message-Passing Interface Standard. 1994, University of
Tennessee. 15. William, G., et al., A high-performance, portable implementation of the MPI message
passing interface standard. Parallel Comput., 1996. 22(6): p. 789-828. 16. Karonis, N.T., B. Toonen, and I. Foster, MPICH-G2: a Grid-enabled implementation of the
Message Passing Interface. J. Parallel Distrib. Comput., 2003. 63(5): p. 551-563. 17. Blanchette, M., B. Schwikowski, and M. Tompa, Algorithms for phylogenetic footprinting. J
Comput Biol, 2002. 9(2): p. 211-23. 18. Vanet, A., L. Marsan, and M.-F. Sagot, Promoter sequences and algorithmical methods for
identifying them. Res Microbiol, 1999. 150(9-10): p. 779-99. 19. Stormo, G.D., DNA binding sites: representation and discovery. Bioinformatics, 2000. 16(1):
p. 16-23. 20. Pavesi, G., G. Mauri, and G. Pesole, In silico representation and discovery of transcription
factor binding sites. Brief Bioinform, 2004. 5(3): p. 217-36. 21. Wasserman, W.W. and W. Krivan, In silico identification of metazoan transcriptional
regulatory regions. Naturwissenschaften, 2003. 90(4): p. 156-66.

89
22. Brazma, A., et al., Approaches to the automatic discovery of patterns in biosequences. J Comput Biol, 1998. 5(2): p. 279-305.
23. MacIsaac, K.D. and E. Fraenkel, Practical strategies for discovering regulatory DNA sequence motifs. PLoS Comput Biol, 2006. 2(4): p. e36.
24. Das, M. and H. Dai, A survey of DNA motif finding algorithms. BMC Bioinformatics, 2007. 8(Suppl 7).
25. Sandve, G.K. and F. Drablos, A survey of motif discovery methods in an integrated framework. Biol Direct, 2006. 1: p. 11.
26. van Helden, J., B. Andre, and J. Collado-Vides, Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol, 1998. 281(5): p. 827-42.
27. Sinha, S. and M. Tompa, A statistical method for finding transcription factor binding sites. Proc Int Conf Intell Syst Mol Biol, 2000. 8: p. 344-54.
28. Jensen, L.J. and S. Knudsen, Automatic discovery of regulatory patterns in promoter regions based on whole cell expression data and functional annotation. Bioinformatics, 2000. 16(4): p. 326-33.
29. Bussemaker, H.J., H. Li, and E.D. Siggia, Building a dictionary for genomes: identification of presumptive regulatory sites by statistical analysis. Proc Natl Acad Sci U S A, 2000. 97(18): p. 10096-100.
30. Xie, X., et al., Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature, 2005. 434(7031): p. 338-45.
31. van Helden, J., A.F. Rios, and J. Collado-Vides, Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res, 2000. 28(8): p. 1808-18.
32. Takusagawa, K.T. and D.K. Gifford, Negative information for motif discovery. Pac Symp Biocomput, 2004: p. 360-71.
33. Shinozaki, D. and O. Maruyama, A Method for the Best Model Selection for Single and Paired Motifs. Genome Informatics, 2002. 13: p. 432-433.
34. Tompa, M., An exact method for finding short motifs in sequences, with application to the ribosome binding site problem. Proc Int Conf Intell Syst Mol Biol, 1999: p. 262-71.
35. Pevzner, P.A. and S.H. Sze, Combinatorial approaches to finding subtle signals in DNA sequences. Proc Int Conf Intell Syst Mol Biol, 2000. 8: p. 269-78.
36. Pavesi, G., G. Mauri, and G. Pesole, An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics, 2001. 17 Suppl 1: p. S207-14.
37. Marsan, L. and M.-F. Sagot, Algorithms for extracting structured motifs using a suffix tree with an application to promoter and regulatory site consensus identification. J Comput Biol, 2000. 7(3-4): p. 345-62.
38. Eskin, E. and P.A. Pevzner, Finding composite regulatory patterns in DNA sequences. Bioinformatics, 2002. 18 Suppl 1: p. S354-63.
39. Baldwin, N.E., et al., High Performance Computational Tools for Motif Discovery, in 18th International Parallel and Distributed Processing Symposium (IPDPS'04) – Workshop 9. 2004, IEEE Computer Society: Los Alamitos, CA, USA. p. 192a.
40. Stormo, G.D., et al., Use of the 'Perceptron' algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res, 1982. 10(9): p. 2997-3011.
41. Roth, F.P., et al., Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol, 1998. 16(10): p. 939-45.
42. Liu, X., D.L. Brutlag, and J.S. Liu, BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput, 2001: p. 127-38.
43. Lawrence, C., et al., Detecting Subtle Sequence Signals: A Gibbs Sampling Strategy for Multiple Alignment. 1993.
44. Bailey, T.L. and C. Elkan, Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol, 1994. 2: p. 28-36.
45. Timothy, L.B. and E. Charles, Unsupervised Learning of Multiple Motifs in Biopolymers Using Expectation Maximization. Mach. Learn., 1995. 21(1-2): p. 51-80.

90
46. Bailey, T.L. and C. Elkan, The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol, 1995. 3: p. 21-9.
47. Franscisco, A.P., A.L. Oliveira, and A.T. Freitas, Identification of Transcription Factor Binding Sites in Promoter Regions by Modularity Analysis of the Motif Co-Occurrence Graph. 2008, INESC-ID/ IST, Technical University of Lisbon, Portugal.
48. Zhou, Q. and J.S. Liu, Modeling within-motif dependence for transcription factor binding site predictions. Bioinformatics, 2004. 20(6): p. 909-16.
49. O'Flanagan, R.A., et al., Non-additivity in protein-DNA binding. Bioinformatics, 2005. 21(10): p. 2254-63.
50. Benos, P.V., M.L. Bulyk, and G.D. Stormo, Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res, 2002. 30(20): p. 4442-51.
51. Jensen, S.T., et al., Computational Discovery of Gene Regulatory Binding Motifs: A Bayesian Perspective. Statistical Science, 2004. 19(1): p. 188-204.
52. Thompson, W., E.C. Rouchka, and C.E. Lawrence, Gibbs Recursive Sampler: finding transcription factor binding sites. Nucleic Acids Res, 2003. 31(13): p. 3580-5.
53. Sinha, S., E. van Nimwegen, and E.D. Siggia, A probabilistic method to detect regulatory modules. Bioinformatics, 2003. 19 Suppl 1: p. i292-301.
54. Sharan, R., et al., CREME: a framework for identifying cis-regulatory modules in human-mouse conserved segments. Bioinformatics, 2003. 19 Suppl 1: p. i283-91.
55. McCue, L., et al., Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res, 2001. 29(3): p. 774-82.
56. Cora, D., et al., Ab initio identification of putative human transcription factor binding sites by comparative genomics. BMC Bioinformatics, 2005. 6: p. 110.
57. Krivan, W. and W.W. Wasserman, A predictive model for regulatory sequences directing liver-specific transcription. Genome Res, 2001. 11(9): p. 1559-66.
58. Beiko, R.G. and R.L. Charlebois, GANN: genetic algorithm neural networks for the detection of conserved combinations of features in DNA. BMC Bioinformatics, 2005. 6: p. 36.
59. Pudimat, R., E.G. Schukat-Talamazzini, and R. Backofen. Feature Based Representation and Detection of Transcription Factor Binding Sites. in German Conference on Bioinformatics. 2004: GI.
60. Thompson, W., et al., Decoding human regulatory circuits. Genome Res, 2004. 14(10A): p. 1967-74.
61. Kel, A.E., et al. ClusterScan: A Tool for Automatic Annotation of Genomic Regulatory Sequences by Searching for Composite Clusters. in German Conference on Bioinformatics. 2001: German Research Center for Biotechnology.
62. Hu, Y.J., Finding subtle motifs with variable gaps in unaligned DNA sequences. Comput Methods Programs Biomed, 2003. 70(1): p. 11-20.
63. Bussemaker, H.J., H. Li, and E.D. Siggia, Regulatory element detection using correlation with expression. Nat Genet, 2001. 27(2): p. 167-71.
64. Aerts, S., et al., Computational detection of cis -regulatory modules. Bioinformatics, 2003. 19 Suppl 2: p. ii5-14.
65. GuhaThakurta, D. and G.D. Stormo, Identifying target sites for cooperatively binding factors. Bioinformatics, 2001. 17(7): p. 608-21.
66. Rebeiz, M., N.L. Reeves, and J.W. Posakony, SCORE: a computational approach to the identification of cis-regulatory modules and target genes in whole-genome sequence data. Site clustering over random expectation. Proc Natl Acad Sci U S A, 2002. 99(15): p. 9888-93.
67. Bailey, T.L. and W.S. Noble, Searching for statistically significant regulatory modules. Bioinformatics, 2003. 19 Suppl 2: p. ii16-25.
68. Brazma, A., et al., Data mining for regulatory elements in yeast genome. Proc Int Conf Intell Syst Mol Biol, 1997. 5: p. 65-74.
69. Scherf, M., A. Klingenhoff, and T. Werner, Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: a novel context analysis approach. J Mol Biol, 2000. 297(3): p. 599-606.

91
70. Wagner, A., Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics, 1999. 15(10): p. 776-84.
71. Frech, K. and T. Werner, Specific modelling of regulatory units in DNA sequences. Pac Symp Biocomput, 1997: p. 151-62.
72. Aerts, S., et al., A genetic algorithm for the detection of new cis-regulatory modules in sets of coregulated genes. Bioinformatics, 2004. 20(12): p. 1974-6.
73. Klingenhoff, A., et al., Functional promoter modules can be detected by formal models independent of overall nucleotide sequence similarity. Bioinformatics, 1999. 15(3): p. 180-186.
74. Blanchette, M. and M. Tompa, Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res, 2002. 12(5): p. 739-48.
75. Liu, X.S., D.L. Brutlag, and J.S. Liu, An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol, 2002. 20(8): p. 835-9.
76. Segal, E., et al., From promoter sequence to expression: a probabilistic framework in Proceedings of the sixth annual international conference on Computational biology. 2002: Washington, DC, USA. p. 263-272.
77. Workman, C.T. and G.D. Stormo, ANN-Spec: a method for discovering transcription factor binding sites with improved specificity. Pac Symp Biocomput, 2000: p. 467-78.
78. Conlon, E.M., et al., Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci U S A, 2003. 100(6): p. 3339-44.
79. Frith, M.C., et al., Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res, 2004. 32(4): p. 1372-81.
80. Wang, T. and G.D. Stormo, Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics, 2003. 19(18): p. 2369-80.
81. Caselle, M., F. Di Cunto, and P. Provero, Correlating overrepresented upstream motifs to gene expression: a computational approach to regulatory element discovery in eukaryotes. BMC Bioinformatics, 2002. 3: p. 7.
82. Cora, D., et al., Computational identification of transcription factor binding sites by functional analysis of sets of genes sharing overrepresented upstream motifs. BMC Bioinformatics, 2004. 5: p. 57.
83. Curran, M.D., et al., Statistical methods for joint data mining of gene expression and DNA sequence database. SIGKDD Explor. Newsl., 2003. 5(2): p. 122-129.
84. Hong, P., et al., A boosting approach for motif modeling using ChIP-chip data. Bioinformatics, 2005. 21(11): p. 2636-43.
85. Xing, E.P., et al., Logos: a modular bayesian model for de novo motif detection. J Bioinform Comput Biol, 2004. 2(1): p. 127-54.
86. Rustici, G., et al., Periodic gene expression program of the fission yeast cell cycle. Nat Genet, 2004. 36(8): p. 809-17.
87. Buck, M.J. and J.D. Lieb, ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics, 2004. 83(3): p. 349-60.
88. Ren, B., et al., Genome-wide location and function of DNA binding proteins. Science, 2000. 290(5500): p. 2306-9.
89. Mironov, A.A., et al., Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res, 1999. 27(14): p. 2981-9.
90. Qin, Z.S., et al., Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat Biotechnol, 2003. 21(4): p. 435-439.
91. McGuire, A.M., J.D. Hughes, and G.M. Church, Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res, 2000. 10(6): p. 744-57.
92. Thijs, G., et al., A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J Comput Biol, 2002. 9(2): p. 447-64.
93. Park, P.J., A.J. Butte, and I.S. Kohane, Comparing expression profiles of genes with similar promoter regions. Bioinformatics, 2002. 18(12): p. 1576-84.

92
94. Holmes, I. and W.J. Bruno, Finding Regulatory Elements Using Joint Likelihoods for Sequence and Expression Profile Data, in Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. 2000. p. 202-210.
95. Baldi, P. and G.W. Hatfield, DNA Microarrays and Gene Expression: From Experiments to Data Analysis and Modeling. 2002: Cambridge University Press.
96. Ramos, E., et al., Genomic organization of gypsy chromatin insulators in Drosophila melanogaster. Genetics, 2006. 172(4): p. 2337-49.
97. Tang, T., et al., Expression ratio evaluation in two-colour microarray experiments is significantly improved by correcting image misalignment. Bioinformatics, 2007. 23(20): p. 2686-91.
98. Madeira, S.C. and A.L. Oliveira, Biclustering algorithms for biological data analysis: a survey. IEEE/ACM Trans Comput Biol Bioinform, 2004. 1(1): p. 24-45.
99. Cheng, Y. and G.M. Church, Biclustering of Expression Data, in Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. 2000, AAAI Press.
100. Hartigan, J.A., Direct Clustering of a Data Matrix. Journal of the American Statistical Association, 1972. 67(337): p. 123-129.
101. Tibshirani, R., et al., Clustering methods for the analysis of DNA microarray data. 1999, Department of Health Research and Policy, Stanford University.
102. Cho, H., et al., Minimum sum squared residue co-clustering of gene expression data. 2004. 103. Getz, G., E. Levine, and E. Domany, Coupled two-way clustering analysis of gene
microarray data. Proc Natural Academy of Sciences USA, 2000. 97(22): p. 12079-12084. 104. Califano, A., G. Stolovitzky, and Y. Tu, Analysis of gene expression microarrays for
phenotype classification. Proceedings of the International Conference on Intelligent Systems for Molecular Biology, 2000. 8: p. 75-85.
105. Sheng, Q., Y. Moreau, and B. De Moor, Biclustering microarray data by Gibbs sampling. Bioinformatics, 2003. 19 Suppl 2: p. ii196-205.
106. Segal, E., et al., Rich probabilistic models for gene expression. Bioinformatics, 2001. 17 Suppl 1: p. S243-52.
107. Kluger, Y., et al., Spectral biclustering of microarray data: coclustering genes and conditions. Genome Res, 2003. 13(4): p. 703-716.
108. Segal, E., A. Battle, and D. Koller, Decomposing gene expression into cellular processes. Pac Symp Biocomput, 2003: p. 89-100.
109. Tang, C., et al., Interrelated Two-way Clustering: An Unsupervised Approach for Gene Expression Data Analysis, in Proceedings of the 2nd IEEE International Symposium on Bioinformatics and Bioengineering 2001, IEEE Computer Society. p. 41.
110. Ben-Dor, A., et al. Discovering local structure in gene expression data: the order-preserving submatrix problem. in RECOMB '02: Proceedings of the sixth annual international conference on Computational biology. 2002: ACM.
111. Liu, J. and W. Wang, OP-Cluster: Clustering by Tendency in High Dimensional Space, in Proceedings of the Third IEEE International Conference on Data Mining. 2003, IEEE Computer Society. p. 187.
112. Murali, T.M. and S. Kasif, Extracting conserved gene expression motifs from gene expression data. Pacific Symposium on Biocomputing, 2003: p. 77-88.
113. Tanay, A., R. Sharan, and R. Shamir, Discovering statistically significant biclusters in gene expression data. Bioinformatics, 2002. 18 Suppl 1: p. S136-44.
114. Yang, J., et al., Enhanced Biclustering on Expression Data, in Proceedings of the 3rd IEEE Symposium on BioInformatics and BioEngineering. 2003, IEEE Computer Society.
115. Yang, J., et al. delta-cluster: Capturing Subspace Correlation in a Large Data Set. in ICDE. 2002.
116. Duffy, D. and A. Quiroz, A permutation-based algorithm for block clustering. Journal of Classification, 1991. 8(1).
117. Marsan, L. and M.-F. Sagot, Extracting structured motifs using a suffix tree\—algorithms and application to promoter consensus identification, in Proceedings of the fourth annual international conference on Computational molecular biology. 2000, ACM: Tokyo, Japan.
118. Weiner, P., Linear Pattern Matching Algorithms, in Proceedings of the 14th IEEE Annual Symposium on Switching and Automata Theory. 1973, IEEE. p. 1-11.

93
119. Carvalho, A.M., Efficient algorithms for structured motif extraction in DNA sequences. 2004, IST, Universidade Técnica de Lisboa.
120. Andersson, A., N.J. Larsson, and K. Swanson, Suffix Trees on Words, in Proceedings of the 7th Annual Symposium on Combinatorial Pattern Matching. 1996, Springer-Verlag. p. 102-115.
121. Carvalho, A.M., et al., A parallel algorithm for the extraction of structured motifs, in Proceedings of the 2004 ACM symposium on Applied computing. 2004, ACM: Nicosia, Cyprus.
122. Carvalho, A.M., et al., An Efficient Algorithm for the Identification of Structured Motifs in DNA Promoter Sequences. IEEE/ACM Trans. Comput. Biol. Bioinformatics, 2006. 3(2): p. 126-140.
123. Allali, J. and M.-F. Sagot, The at-most k-deep factor tree. 2004, Institut Gaspard Monge. 124. Mendes, N.D., et al., MUSA: a parameter free algorithm for the identification of biologically
significant motifs. Bioinformatics, 2006. 22(24): p. 2996-3002. 125. Monteiro, P.T., et al., YEASTRACT-DISCOVERER: new tools to improve the analysis of
transcriptional regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res, 2007.
126. Madeira, S.C. and A.L. Oliveira, A Linear Time Biclustering Algorithm for Time Series Gene Expression Data, in 5th Workshop on Algorithms in Bioinformatics (WABI). 2005, Springer: Palma de Mallorca. p. 39-52.
127. Madeira, S.C. and A.L. Oliveira, An Efficient Biclustering Algorithm for finding Genes with Similar Patterns in Time-Series Expression Data, in Asia Pacific Bioinformatics Conference. 2007, Imperial College Press: Hong Kong. p. 67-80.
128. Sagot, M.-F., Spelling Approximate Repeated or Common Motifs Using a Suffix Tree, in Proceedings of the Third Latin American Symposium on Theoretical Informatics. 1998, Springer-Verlag.
129. Sun Grid Engine. 2007 [citado; Disponível em: http://gridengine.sunsource.net. 130. hermes - the High Performance Computing Centre that supports the Centro Português de
Bioinformática. 2008 [citado; Disponível em: http://hermes.igc.gulbenkian.pt. 131. BioTeam, iNquiry Administrator's Guide. 2006. 132. Letondal, C., A Web interface generator for molecular biology programs in Unix.
Bioinformatics, 2001. 17(1): p. 73-82. 133. W3C. Web Services Description Language. 2007 [citado; Disponível em:
http://www.w3.org/2002/ws/desc/. 134. W3C. Simple Object Access Protocol. 2007 [citado; Disponível em:
http://www.w3.org/2000/xp/Group/. 135. BioTeam. hermes - iNquiry Bioinformatics Portal - Instituto Gulbenkian de Ciência. 2007
[citado; Disponível em: http://hermes.igc.gulbenkian.pt/inquiry. 136. Costa, S., Hermes Cluster: deploying and launching computationally intensive jobs. 2008,
Instituto Gulbenkian de Ciência. 137. Pisanti, N., et al. RISOTTO: Fast extraction of motifs with mismatches. in Proceedings of
the 7th Latin American Theoretical Informatics Symposium. 2006. Chile, Valdivia: Springer. 138. Robinson, P.N., et al., Ontologizing gene-expression microarray data: characterizing
clusters with Gene Ontology. Bioinformatics, 2004. 20(6): p. 979-81. 139. Camon, E., et al., The Gene Ontology Annotation (GOA) Database--an integrated resource
of GO annotations to the UniProt Knowledgebase. In Silico Biol, 2004. 4(1): p. 5-6. 140. Camon, E., et al., The Gene Ontology Annotation (GOA) Database: sharing knowledge in
Uniprot with Gene Ontology. Nucleic Acids Res, 2004. 32(Database issue): p. D262-6. 141. Harris, M.A., et al., The Gene Ontology (GO) database and informatics resource. Nucleic
Acids Res, 2004. 32(Database issue): p. D258-61. 142. Purcell, S. and E. Mellor. JArgs command line option parsing suite for Java. 2005 [citado;
Disponível em: http://jargs.sourceforge.net. 143. Flanagan, M.T. Michael Thomas Flanagan's Java Scientific Library. 2008 [citado;
Disponível em: http://www.ee.ucl.ac.uk/~mflanaga/java. 144. Ya, Z., Z. Hongyuan, and C. Chao-Hisen, A Time-Series Biclustering Algorithm for
Revealing Co-Regulated Genes, in Proceedings of the International Conference on

94
Information Technology: Coding and Computing (ITCC'05) - Volume I - Volume 01. 2005, IEEE Computer Society.
145. Binder, R.V., Testing Object-Oriented Systems: Models, Patterns and Tools. The Addison-Wesley Object Technology Series. 2000: Addison-Wesley Professional.
146. Utting, M. and B. Legeard, Practical Model-Based Testing: A Tools Approach. 2006: Morgan Kaufmann Publishers Inc.
147. DACS. Software Acquisition Gold Practice: Model-Based Testing. Software Acquisition Gold Practice 2008 [citado; Disponível em: https://www.goldpractices.com/practices/mbt/.
148. El-Far, I.K. Enjoying the Perks of Model-Based testing. in Software Testing, Analysis and Review Conference (STARWEST 2001). 2001. San Jose, CA.
149. Wikipedia. Model-based testing --- Wikipedia{,} The Free Encyclopedia. 2008 [citado; Disponível em: http://en.wikipedia.org/wiki/Model-based_testing.
150. Ferriday, C., A Review Paper on Decision Table-Based Testing. 2007, Swansea University. 151. Schneider, T.D. and R.M. Stephens, Sequence logos: a new way to display consensus
sequences. Nucleic Acids Res, 1990. 18(20): p. 6097-6100. 152. Atif, M.M., An event-flow model of GUI-based applications for testing: Research Articles.
Softw. Test. Verif. Reliab., 2007. 17(3): p. 137-157. 153. Dix, A., et al., Evaluation techniques, in Human-Computer Interaction. 2004, Prentice Hall. 154. Nielsen, J., Heuristic evaluation, in Usability inspection methods. 1994, John Wiley \&
Sons, Inc. p. 25-62. 155. Costa, L.d.F., Neurobioinformática: quando mentes e genes se encontram, in ComCiência.
2003, SPBC/Labjor. 156. The International HapMap Project. Nature, 2003. 426(6968): p. 789-796. 157. Newman, M.E.J., The Structure and Function of Complex Networks. SIAM Review, 2003.
45(2): p. 167-256. 158. Stockinger, H., et al., Experience using web services for biological sequence analysis.
Briefings in bioinformatics, 2008. 159. W3C. Resource Description Framework (RDF) / W3C Semantic Web Activity. 2008
[citado; Disponível em: http://www.w3.org/RDF/. 160. W3C. OWL Web Ontology Language Overview. 2008 [citado; Disponível em:
http://www.w3.org/TR/owl-features/. 161. W3C, OWL-S: Semantic Markup for Web Services. 2008. 162. Hull, D., et al., Taverna: a tool for building and running workflows of services. Nucleic Acids
Res, 2006. 34(Web Server issue). 163. Stevens, R.D., A.J. Robinson, and C.A. Goble, myGrid: personalised bioinformatics on the
information grid. Bioinformatics, 2003. 19 Suppl 1: p. i302-i304. 164. Goble, C. and D. De Roure. myExperiment: social networking for workflow-using e-
scientists. in WORKS '07: Proceedings of the 2nd workshop on Workflows in support of large-scale science. 2007: ACM Press.
165. Lord, P., et al. Feta: A light-weight architecture for user oriented semantic service discovery. in Proceedings of the European Semantic Web Conference 2005. 2005: Springer-Verlag.
166. Pettifer, S., et al., myGrid and UTOPIA: An Integrated Approach to Enacting and Visualising in Silico Experiments in the Life Sciences, in Data Integration in the Life Sciences. 2007. p. 59-70.
167. Costa, S., BioGrid Motif Finding: Efficient Algorithms for finding common motifs in DNA sequences, User Manual. 2007, Instituto Gulbenkian de Ciência.
168. Costa, S., BioGrid Biclustering: Biclustering algorithms for gene expression data, User Manual. 2007, Instituto Gulbenkian de Ciência.
169. Zvelebil, M.J., et al., Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J Mol Biol, 1987. 195(4): p. 957-61.
170. Smith, R.F. and T.F. Smith, Automatic generation of primary sequence patterns from sets of related protein sequences. Proc Natl Acad Sci U S A, 1990. 87(1): p. 118-22.

95
Anexo 1 – Interfaces Pessoa-Máquina Desenvolvidas
Neste anexo apresenta-se uma descrição detalhada das interfaces pessoa-máquina
desenvolvidas para as diferentes aplicações instaladas no cluster hermes. Esta informação faz
parte dos manuais de utilizador [167] e [168], desenvolvidos no âmbito desta tese.
Para cada parâmetro, fornece-se a seguinte informação:
• Descrição: descrição do parâmetro.
• Tipo: indica se o parâmetro é obrigatório, condicional ou opcional.
• Domínio: indica o conjunto de valores possíveis para o parâmetro.
• Valor por defeito: indica o valor atribuído ao parâmetro por defeito.
A1.1 Interface CCC-Biclustering
A interface da aplicação CCC-Biclustering é composta por quatro blocos ou parágrafos:
• Parâmetros de carregamento;
• Parâmetros de pré-processamento;
• Parâmetros de pós-processamento;
• Parâmetros de saída;

96
A1.1.1 Parâmetros de carregamento
A1.1.1.1 Dataset File
• Descrição: ficheiro que contém a matriz de dados de expressão de genes.
• Tipo: obrigatório.
• Domínio: O utilizador poderá carregar um ficheiro ou introduzir os dados directamente na
caixa de texto fornecida para o efeito. Qualquer que seja o método escolhido, os dados
deverão ser estruturados do seguinte modo:
Onde cada Gx é o nome de um gene, cada Cy é o nome de uma condição (instante
temporal) e cada Exy representa o valor do gene Gx sob a condição Cy. A matriz de dados
de expressão pode ter alguns valores em falta.
• Valor por defeito: não especificado
A1.1.1.2 Data Separator
• Descrição: os valores no ficheiro contendo o conjunto de dados a analisar deverão estar
separados por um carácter específico, definido por este parâmetro.
• Tipo: obrigatório.
• Domínio: tabulação (‘\t’), espaço (‘ ‘) ou ponto e vírgula (‘;’).
• Valor por defeito: tab.
A1.1.1.3 Organism
• Descrição: este parâmetro especifica o organismo que constitui o alvo da análise.
• Tipo: obrigatório.
• Domínio: Os organismos disponíveis são aqueles que foram anotados pelo projecto Gene
Ontology (GO):
o Anaplasma phagocytophilum HZ
o Arabidopsis thaliana

97
o Bacillus anthracis Ames
o Bos taurus
o Caenorhabditis elegans
o Campylobacter jejuni RM1221
o Candida albicans
o Coxiella burnetii RSA 493
o Danio rerio
o Dehalococcoides ethenogenes 195
o Dictyostelium discoideum
o Drosophila melanogaster
o Ehrlichia chaffeensis Arkansas
o Gallus gallus
o Geobacter sulfurreducens PCA
o Homo sapiens
o Leishmania major
o Listeria monocytogenes 4b F2365
o Methylococcus capsulatus Bath
o Mus musculus
o Neorickettsia sennetsu Miyayama
o Oryza sativa
o Plasmodium falciparum
o Pseudomonas aeruginosa PAO1
o Pseudomonas syringae DC3000
o Rattus norvegicus
o Saccharomyces cerevisiae
o Schizosaccharomyces pombe
o Shewanella oneidensis MR-1
o Silicibacter pomeroyi DSS-3
o Trypanosoma brucei
o Trypanosoma brucei chr 2
o Vibrio cholerae
• Valor por defeito: Saccharomyces cerevisiae
A1.1.1.4 Log
• Descrição: realizar uma transformação logarítmica nos dados.
• Tipo: opcional.

98
• Domínio: activo ou inactivo.
• Valor por defeito: inactivo.
A1.1.2 Parâmetros de pré-processamento
A1.1.2.1 Filter
• Descrição: filtrar genes;
• Tipo: opcional (mas deverá ser especificado sempre que a matriz de dados apresentar
valores em falta).
• Domínio:
o No filtering (não realizar filtragem);
o Filter all genes with missing values (remover todos os genes com valores em falta);
o Filter only genes with more than a percentage of missing values (remover genes
com uma percentagem de valores em falta superior a um dado valor)
• Valor por defeito: Filter all genes with missing values.
A1.1.2.2 Percentage of missing values
• Descrição: genes com uma percentagem de valores em falta superior a este valor serão
filtrados.
• Tipo: condicional – só deverá ser especificado se se pretende remover genes com uma
percentagem de valores em falta superior a um dado valor.
• Domínio: Valor em vírgula flutuante, maior ou igual a 0.0 e menor ou igual a 100.0.
• Valor por defeito: não especificado.
A1.1.2.3 Fill remaining
• Descrição: preencher os valores em falta que não foram filtrados.
• Tipo: condicional – só deverá ser especificado se se pretende remover genes com uma
percentagem de valores em falta superior a um dado valor.
• Domínio:
o Missing values with a fixed value: preencher os valores em falta com um valor fixo.
o Missing with average over k neighbors: preencher os valores em falta com a média
da expressão génica de k vizinhos.

99
o Missing with average over the same row: preencher os valores em falta com a
média da expressão génica.
Valor por defeito: não especificado.
A1.1.2.4 Value to fill missing ones
• Descrição: o valor que irá substituir os valores em falta.
• Tipo: condicional – só deverá ser especificado se se pretende preencher os valores em
falta com um valor fixo.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: -10000.
A1.1.2.5 Number of neighbors
• Descrição: número de vizinhos a considerar para o cálculo da média.
• Tipo: condicional – só deverá ser especificado se se pretende preencher os valores em
falta com a média da expressão génica de k vizinhos.
• Domínio: Inteiro.
• Valor por defeito: 5.
A1.1.2.6 Normalization
• Descrição: normalizar a matriz de dados de expressão.
• Tipo: opcional.
• Domínio:
o by overall values – normalizar a matriz de dados tendo em conta a sua distribuição
global de valores.
o by gene – normalizar a matriz de dados tendo em conta a distribuição de valores
para cada gene.
• Valor por defeito: não especificado.

100
A1.1.2.7 Smooth
• Descrição: realizar um alisamento da matriz de dados de expressão.
• Tipo: opcional.
• Domínio: Inteiro que especifica a dimensão da janela de alisamento.
• Valor por defeito: 5.
A1.1.2.8 Discretization
• Descrição: realizar uma discretização da matriz de dados de expressão.
• Tipo: opcional.
• Domínio:
o Não realizar discretização;
o Expression average – discretizar a matriz de dados utilizando o valor médio da
expressão dos genes em toda a matriz. Este método de discretização calcula o
nível de expressão médio da totalidade da matriz, bem como o seu desvio padrão;
o Gene expression average – discretizar a matriz de dados utilizando o valor médio
da expressão de cada gene. Este método de discretização calcula o nível de
expressão médio e o desvio padrão para cada linha da matriz;
o Mid ranged – discretizar a matriz de dados de expressão utilizando a mediana da
totalidade da matriz:
o Gene mid range – discretizar a matriz de dados utilizando a mediana de cada
gene. Este método de discretização calcula, para cada gene, o valor mais elevado
e mais reduzido do nível de expressão, bem como o valor mediano;
o Max-Minus Percent Max – discretizar a matriz de dados aplicando o método max –
X% max à totalidade da matriz. Esta técnica considera como valor de corte o nível
de expressão máximo encontrado na matriz;
o Gene Max-Minus Percent Max – discretizar a matriz de dados aplicando o método
max – X% max para cada gene. Esta técnica considera como valor de corte o nível
de expressão máximo encontrado para cada gene;
o Equal frequency – discretizar a matriz de dados utilizando o princípio “equal
frequency” na totalidade da matriz. Este método de discretização considera um
dado conjunto de símbolos no qual os valores de expressão serão transformados e
selecciona símbolos de modo a que exista o mesmo número de pontos de dados
para todos os símbolos. Quando são considerados apenas dois símbolos, os
valores de expressão são ordenados por ordem ascendente ou descendente e são
depois atribuídos ao intervalo que corresponde a um dos símbolos considerados;

101
o Equal frequency by gene - discretizar a matriz de dados utilizando o princípio
“equal frequency” para cada gene;
o Equal width – discretizar a matriz de dados utilizando discretização de largura
idêntica com dois símbolos e tendo em conta a totalidade da matriz;
o Equal width by gene – discretizar a matriz de dados utilizando discretização de
largura idêntica com dois símbolos e tendo em conta cada gene individualmente;
o Expression mean and standard deviation – discretizar a matriz de dados utilizando
o valor médio (mean) e o desvio padrão (std) da totalidade da matriz. Este método
de discretização utiliza um parâmetro α e calcula 𝑚𝑚𝑒𝑒𝑚𝑚𝑚𝑚 ± 𝜒𝜒 × 𝑠𝑠𝑡𝑡𝑑𝑑;
o Gene expression mean and standard deviation – – discretizar a matriz de dados
utilizando o valor médio (𝑚𝑚𝑒𝑒𝑚𝑚𝑚𝑚) e o desvio padrão (𝑠𝑠𝑡𝑡𝑑𝑑) da expressão de cada
gene. Este método de discretização utiliza um parâmetro 𝜒𝜒 e calcula 𝑚𝑚𝑒𝑒𝑚𝑚𝑚𝑚 ± 𝜒𝜒 ×
𝑠𝑠𝑡𝑡𝑑𝑑;
o Transitional State Discrimination (2 symbols) – discretizar a matriz de dados
utilizando o método “transitional state discrimination”. Este método estandardiza a
matriz para z-scores e discretiza a expressão de cada gene com base em duas
transições de estado;
o Variation between time points (2 symbols) – discretizar a matriz de dados com
base nas variações entre instantes temporais e utilizando dois símbolos. Este
processo de discretização começa por calcular o desvio padrão do instante
temporal 0, 𝑠𝑠𝑡𝑡𝑑𝑑(0), e, com um parâmetro 𝜒𝜒, calcula um limite de discretização
𝑡𝑡 = 𝑠𝑠𝑡𝑡𝑑𝑑(0) × 𝜒𝜒. Em seguida, a matriz discretizada é calculada com base nesse
limite;
o Variation between time points (3 symbols) – discretizar a matriz de dados com
base nas variações entre instantes temporais e utilizando três símbolos. Este
método de discretização calcula variações entre instantes temporais sucessivos e
considera essas variações significantes se excederem um dado limite, estando
este relacionado com um parâmetro 𝜒𝜒.
• Valor por defeito: Variation between time points (3 symbols).
A1.1.2.9 Max Percentage
• Descrição: a percentagem máxima a utilizar durante a discretização.
• Tipo: condicional – só tem de ser especificado se se pretende usar os métodos de
discretização “max-minus percent max” e “gene max-minus percent max”.
• Domínio: Valor em vírgula flutuante, maior ou igual a 0.0, menor ou igual a 100.0.
• Valor por defeito: 10.0.

102
A1.1.2.10 Number of symbols
• Descrição: o número de símbolos a ser usado por alguns métodos de discretização.
• Tipo: condicional - só tem de ser especificado se se pretende usar os métodos de
discretização “equal frequency”, “equal frequency by gene”, “equal width” e “equal width by
gene”.
• Domínio: Inteiro.
• Valor por defeito: 3.
A1.1.2.11 Alpha
• Descrição: um limite a ser usado por alguns métodos de discretização.
• Tipo: condicional - só tem de ser especificado se se pretende usar os métodos de
discretização “expression mean and standard deviation”, “gene expression mean and
standard deviation”, “variation between time points (2 symbols)” e “variation between time
points (3 symbols)”.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: 0.08.
A1.1.3 Parâmetros de pós-processamento
A1.1.3.1 Filter
• Descrição: filtrar os biclusters extraídos.
• Tipo: opcional.
• Domínio:
o Não aplicar filtragem;
o (Filters all the) biclusters with less than x genes – filtrar todos os biclusters com
menos de x genes;
o (Filters all the) biclusters with less than y conditions – filtrar todos os biclusters com
menos de y condições;
o (Filters all the) biclusters with Average Column Variance (ACV) greater than c –
filtrar todos os biclusters com variância media por coluna maior que c;
o (Filters all the) biclusters with Average Row Variance (ARV) greater than r – filtrar
todos os biclusters com variância media por linha maior que r;

103
o (Filters all the) biclusters with ARV greater than r and ACV greater than c – filtrar
todos os biclusters com ARV maior que r e ACV maior que c;
o (Filters all the) biclusters with Mean Squared Residue (MSR) greater than m -
filtrar todos os biclusters com resíduo médio quadrático maior que m;
o (Filters all the) biclusters with size lesser than s -– filtrar todos os biclusters com
dimensão inferior a s;
o (Filters all the) biclusters with size lesser than s, ACV greater than c and ARV
greater than r – filtrar todos os biclusters com dimensão inferior a s, ACV maior que
c e ARV maior que r;
o (Filters all the) biclusters with number of conditions lesser than y, number of genes
lesser than x, ACV greater than c and ARV greater than r – filtrar todos os
biclusters com número de condições inferior a y, número de genes inferior a x,
ACV maior que c e ARV maior que r;
o (Filters all the) biclusters with number of conditions lesser than y, number of genes
lesser than x, ACV greater than c, MSR greater than m and ARV greater than r –
filtrar todos os biclusters com número de condições inferior a y, número de genes
menor que x, AVC maior que c, MSR maior que m e ARV maior que r.
o (Filters all the) biclusters with a row similarity greater than z – filtrar todos os
biclusters com uma similaridade entre linhas superior a z;
o (Filters all the) biclusters with a row and column similarity greater than w – filtrar
todos os biclusters com uma similaridade entre linhas e entre colunas maior que w;
o (Filters all the) biclusters with number of highly significant (Bonferroni corrected)
GO p-values equal to 0 - filtrar todos os biclusters sem GO p-values altamente
significantes (com a correcção de Bonferroni). Os GO p-values são calculados
utilizando a distribuição hipergeométrica ou a distribuição binomial, como a
probabilidade de x em n genes terem uma dada anotação, dado que G em N
apresentam essa anotação no genoma. A correcção de Bonferroni indica que, se
um experimentador está a testar n hipóteses independentes num determinado
conjunto de dados, então o nível de significância estatística que deverá ser usado
para cada hipótese é o nível utilizado para testar uma única hipótese multiplicado
por 1/n. Por exemplo, ao testar duas hipóteses, em vez de um p-value de 0.05,
utilizaríamos um valor mais apertado de 0.025 (0.05/2). Um p-value altamente
significante tem de apresentar valor inferior a 0.01.
o (Filters all the) biclusters with number of highly significant GO p-values equal to 0 –
filtrar todos os biclusters sem GO p-values altamente significantes.
o (Filters all the) biclusters with number of significant (Bonferroni corrected) GO p-
values equal to 0 – filtrar todos os biclusters sem GO p-values significantes (com a
correcção de Bonferroni). Um p-value significante apresenta valor inferior a 0.05.

104
o (Filters all the) biclusters with number of significant GO p-values equal to 0 – filtrar
todos os biclusters sem GO p-values significantes.
• Valor por defeito: (Filters all the) biclusters with a row and column similarity greater than
w.
A1.1.3.2 Number of genes
• Descrição: o número de genes para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação do número de genes.
• Domínio: Inteiro.
• Valor por defeito: 5.
A1.1.3.3 Number of conditions
• Descrição: o número de condições para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação do número de condições.
• Domínio: Inteiro.
• Valor por defeito: 2.
A1.1.3.4 Average Column Variance (ACV)
• Descrição: a variância média por coluna para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação da ACV.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: 0.0.
A1.1.3.5 Average Row Variance (ARV)
• Descrição: a variância média por linha para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação da ARV.
• Domínio: Valor em vírgula flutuante.

105
• Valor por defeito: 1.0.
A1.1.3.6 Mean Squared Residue (MSR)
• Descrição: o resíduo médio quadrático para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação de MSR.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: 0.0.
A1.1.3.7 Size
• Descrição: a dimensão dos biclusters para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação da dimensão dos biclusters.
• Domínio: Inteiro.
• Valor por defeito: 10.
A1.1.3.8 Threshold
• Descrição: um limite para a filtragem em pós-processamento.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar um método de
filtragem que requer a especificação da similaridade entre linhas e/ou colunas.
• Domínio: Valor em vírgula flutuante, maior ou igual a 0.0, menor ou igual a 1.0.
• Valor por defeito: 0.25.
A1.1.3.9 Sort
• Descrição: ordenar os biclusters extraídos da matriz de dados.
• Tipo: opcional.
• Domínio:
o Não ordenar os biclusters;
o (Sort by) ACV in descending order – ordenar por ordem decrescente de ACV;
o (Sort by) ARV in ascending order – ordenar por ordem crescente de ARV;
o (Sort by) MSR in ascending order – ordenar por ordem crescente de MSR;

106
o (Sort by) ARV and MSR in ascending order – ordenar por ordem crescente de ARV
e MSR;
o (Sort by) number of genes in ascending order – ordenar por ordem crescente do
número de genes;
o (Sort by) number of genes in descending order – ordenar por ordem decrescente
do número de genes;
o (Sort by) number of conditions in ascending order – ordenar por ordem crescente
do número de condições;
o (Sort by) number of conditions in descending order – ordenar por ordem
decrescente do número de condições;
o (Sort by) size in ascending order – ordenar por ordem crescente da dimensão dos
biclusters;
o (Sort by) size in descending order – ordenar por ordem decrescente da dimensão
dos biclusters;
o (Sort by) variance in descending order – ordenar por ordem decrescente da
variância;
o (Sort by) best GO p-values in ascending order – ordenar por ordem crescente dos
melhores GO p-values;
o (Sort by) best GO Bonferroni corrected p-values in ascending order – ordenar por
ordem crescente dos melhores GO p-values com a correcção de Bonferroni;
o (Sort by) highly significant GO Bonferroni Corrected p-values in descending order -
ordenar por ordem decrescente dos melhores GO p-values altamente significantes
e com a correcção de Bonferroni;
o (Sort by) highly significant GO p-values in descending order - ordenar por ordem
decrescente dos melhores GO p-values altamente significantes;
o (Sort by) significant GO Bonferroni Corrected p-values in descending order -
ordenar por ordem crescente dos melhores GO p-values significantes e com a
correcção de Bonferroni;
o (Sort by) significant GO p-values in descending order - ordenar por ordem
decrescente dos melhores GO p-values significantes;
o (Sort by) significant GO p-values with user threshold in descending order - ordenar
por ordem decrescente dos melhores GO p-values, tendo por base um limite
definido pelo utilizador. Neste caso, os p-values são considerados significantes se
o seu valor for menor do que o limite especificado;
o (Sort by) bicluster pattern p-values in ascending order – ordenar por ordem
crescente dos p-values padrão de cada bicluster;
o (Sort by) bicluster pattern with columns p-values in ascending order – ordenar por
ordem crescente dos p-values padrão de cada bicluster e com colunas;

107
• Valor por defeito: (Sort by) bicluster pattern with columns p-values in ascending order.
A1.1.3.10 Markov Chain Order
• Descrição: especifica a ordem da cadeia de Markov.
• Tipo: condicional – só tem de ser especificado se se pretende utilizar os métodos de
ordenação “bicluster pattern”.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 1.
A1.1.3.11 GO User Threshold
• Descrição: um limite para os GO p-values.
• Tipo: condicional – só deverá ser especificado se se pretende utillizar os métodos de
ordenação “Significant GO Bonferroni Corrected P-values” e “Significant Go P-values with
user threshold”.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: não especificado.
A1.1.4 Parâmetros de saída
A1.1.4.1 Number of Biclusters to print
• Descrição: o número máximo de biclusters a serem escritos no ficheiro de saída.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 20.
A1.1.4.2 Output file
• Descrição: nome do ficheiro de saída.
• Tipo: obrigatório.
• Domínio: cadeia de caracteres.
• Valor por defeito: output.txt.

108
A1.2 Interface e-CCC-Biclustering
Além dos quatro blocos de parâmetros apresentados na secção A1.1, a utilização do
algoritmo e-CCC-Biclustering implica a especificação de um bloco adicional: Parâmetros de
Biclustering.
A1.2.1 Parâmetros de Biclustering
A1.2.1.1 Number of errors
• Descrição: número máximo de erros permitidos durante a extracção dos biclusters.
• Tipo: obrigatório.
• Domínio: Inteiro.
• Valor por defeito: 1.
A1.2.1.2 Restricted errors
• Descrição: erros genéricos são úteis para identificar erros de medição que ocorreram
durante as experiências com microarrays. No entanto, se pretendemos identificar erros de
discretização, podemos considerar “restricted errors”, isto é, substituições de símbolos na
matriz de dados pelos símbolos lexicograficamente mais próximos no alfabeto. Por
exemplo, com o alfabeto ∑ = {𝐷𝐷,𝑁𝑁,𝑈𝑈}, o símbolo 𝑈𝑈 só pode ser substituído por 𝑁𝑁 se
estivermos a usar “restricted erros”, mas pode ser substituído tanto por 𝑁𝑁 como por 𝐷𝐷 se
estivermos a utilizar erros genéricos.
• Tipo: opcional.
• Domínio: activo ou inactivo.
• Valor por defeito: inactivo.
A1.2.1.3 Neighbors for restricted errors
• Descrição: número de vizinhos a considerar para a opção “restricted errors”. Por exemplo,
com o alfabeto ∑ = {𝐷𝐷,𝑁𝑁,𝑈𝑈} e neighbors = 1, 𝑈𝑈 só pode ser substituído por 𝑁𝑁. Contudo,
com o mesmo alfabeto e neighbors = 2, 𝑈𝑈 pode ser substituído tanto por 𝑁𝑁 como por 𝐷𝐷.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar a opção “restricted
errors”.
• Domínio: Inteiro.

109
• Valor por defeito: não especificado.
A1.3 Interface Cheng and Church
A interface Cheng and Church é composta por quatro blocos ou parágrafos:
• Parâmetros de carregamento – idênticos aos parâmetros de carregamento da
interface CCC-Biclustering e, como tal, não serão descritos novamente;
• Parâmetros de pré-processamento - idênticos a um subconjunto dos parâmetros
de pré-processamento da interface CCC-Biclustering e, como tal, não serão
descritos novamente;
• Parâmetros de Biclustering;
• Parâmetros de saída - idênticos aos parâmetros de saída da interface CCC-
Biclustering e, como tal, não serão descritos novamente.
A1.3.1 Parâmetros de Biclustering
A1.3.1.1 Threshold
• Descrição: número mínimo de linhas e colunas para o passo de eliminação de múltiplos
nós (multiple node deletion). Se o número de linhas ou colunas na matriz de dados for
inferior a este limite, o passo de eliminação de múltiplos nós não será executado. De notar
que o passo de eliminação de um único nó (single node deletion) é sempre executado.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.
A1.3.1.2 Alpha
• Descrição: o valor de alpha é um limite para o passo de eliminação de um único nó.
• Tipo: obrigatório.
• Domínio: Valor em vírgula flutuante, maior ou igual a 1.0 (ver [99]).
• Valor por defeito: não especificado.
A1.3.1.3 Delta
• Descrição: o resíduo médio quadrático mínimo, δ.
• Tipo: obrigatório.

110
• Domínio: Valor em vírgula flutuante, maior ou igual a 0.0.
• Valor por defeito: não especificado.
A1.4 Interface MUSA
A interface MUSA é composta por dois blocos ou parágrafos:
• Parâmetros Simples;
• Parâmetros Avançados;
A1.4.1 Parâmetros Simples
A1.4.1.1 Sequences file
• Descrição: ficheiro que contém as sequências de nucleótidos.
• Tipo: obrigatório.
• Domínio: o utilizador poderá carregar o ficheiro ou escrever os dados directamente na
caixa de texto fornecida para o efeito. Os dados deverão obedecer a um dos seguintes
formatos:
o IG/Stanford;
o GenBank/GB
o NBRF
o EMBL
o GCG
o DNAStrider
o Fitch
o Pearson/Fasta
o Zuker
o Olsen
o Phylip3.2
o Phylip
o Plain/Raw
o PIR/CODATA
o MSF
o ASN.1
o PAUP/NEXUS
• Valor por defeito: não especificado.

111
A1.4.2 Parâmetros Avançados
A1.4.2.1 Search both strands
• Descrição: esta opção permite efectuar a pesquisa de motivos em ambas as cadeias de
DNA;
• Tipo: opcional.
• Domínio: activo ou inactivo.
• Valor por defeito: activo.
A1.4.2.2 Lambda
• Descrição: o parâmetro lambda (λ) define o comprimento dos λ-ímeros que são utilizados
para construir os motivos durante a execução do algoritmo. Não será encontrado nenhum
motivo cujo comprimento seja inferior a λ. Deste modo, ao especificar um valor mais alto
(baixo) para λ, estamos a focar a procura em motivos de dimensão elevada (reduzida).
• Tipo: opcional.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 4.
A1.4.2.3 Epsilon
• Descrição: o parâmetro ε define a tolerância para a distância de Hamming entre os λ-
ímeros. No caso dos motivos complexos, este parâmetro permite que exista alguma
variação na distância de Hamming entre componentes nas várias ocorrências do mesmo
motivo.
• Tipo: opcional.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: 1.
A1.4.2.4 Minimum distance between λ-mers
• Descrição: este parâmetro define a distância mínima entre dois λ-ímeros em motivos
complexos. Podemos utilizar esta opção para forçar a procura de motivos complexos.
• Tipo: opcional.
• Domínio: Inteiro, maior ou igual a 1.

112
• Valor por defeito: 1.
A1.4.2.5 Maximum distance between λ-mers
• Descrição: este parâmetro define a distância máxima entre dois λ-ímeros em motivos
complexos. Podemos utilizar esta opção para focar a procura em porções limitadas da
sequência de nucleótidos.
• Tipo: opcional.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 50.
A1.4.2.6 Sieve
• Descrição: define a proporção de sequências na qual o motivo deverá ocorrer de modo a
ser identificado. Usualmente, este parâmetro toma a designação de quorum.
• Tipo: opcional.
• Domínio: Inteiro, maior ou igual a 0, menor ou igual a 100.
• Valor por defeito: 30.
A1.4.2.7 Maximum p-value for assessment of relevant motifs for family generation
• Descrição: especifica o p-value máximo do subconjunto de motivos que será considerado
para a geração das famílias.
• Tipo: opcional.
• Domínio: Valor em vírgula flutuante.
• Valor por defeito: 0.001.

113
A1.5 Interface PSMILE
A interface PSMILE é composta pelos seguintes blocos:
• Parâmetros genéricos;
• Parâmetros do alfabeto;
• Parâmetros do método de avaliação “shuffling”;
• Parâmetros do método de avaliação “against”;
• Parâmetros da caixa n (n є {1,4}).
A1.5.1 Parâmetros genéricos
A1.5.1.1 Extraction Program
• Descrição: este parâmetro especifica o programa que será utilizado para extrair motivos.
De notar que “smile without deltas” é a implementação paralela do algoritmo SMILE, ao
passo que “smile with deltas” não utiliza paralelismo. Ao utilizar “deltas”, o algoritmo SMILE
permite um espaçamento de d ± δ entre cada duas componentes de um motivo complexo.
• Tipo: obrigatório.
• Domínio: {“x-smile: smile without deltas”, “x-smile: smile with deltas”}.
• Valor por defeito: “x-smile: smile without deltas”.
A1.5.1.2 Evaluation Method
• Descrição: especifica o método que será utilizado para calcular a significância estatística
dos motivos encontrados. O método de avaliação “shuffling” compara as frequências dos
motivos encontrados com sequências aleatórias que apresentam uma composição de k-
ímeros idêntica às sequências originais. Os valores de significância estatística são depois
calculados com base nas diferenças das frequências observadas. Este método de
avaliação é útil quando temos apenas um conjunto de sequências. O método de avaliação
“against” calcula a significância estatística utilizando dois conjuntos de sequências: o
“correcto”, isto é, aquele que contém os motivos encontrados durante a fase de extracção
do algoritmo; e o “incorrecto”, que contém sequências que não possuem motivos em
comum com as sequências originais.
• Tipo: obrigatório.
• Domínio: {“e-smile_shuffling: shuffle method”, “e-smile_against: compare with other set of
sequences”}.

114
• Valor por defeito: “e-smile_shuffling: shuffle method”.
A1.5.1.3 Sequences File
Ver parâmetro A1.4.1.1.
A1.5.1.4 Output File
• Descrição: nome do ficheiro de saída.
• Tipo: obrigatório.
• Domínio: cadeia de caracteres.
• Valor por defeito: outfile.out.
A1.5.1.5 Quorum
Ver parâmetro A1.4.2.6.
A1.5.1.6 Total min length
• Descrição: o comprimento total mínimo para cada motivo complexo. Por exemplo, se um
motivo complexo apresenta três componentes com comprimento 2, 3 e 4, o seu
comprimento total será 2+3+4 = 9.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.
A1.5.1.7 Total max length
• Descrição: o comprimento total máximo para cada motivo complexo.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 1. Deverá ter um valor superior ao do parâmetro
A1.5.1.6.
• Valor por defeito: não especificado.

115
A1.5.1.8 Total substitutions
• Descrição: o número total máximo de substituições (erros) em cada motivo complexo. Por
exemplo, se um motivo complexo apresenta três caixas com 2, 3 e 4 substituições, o
número total de substituições será 2+3+4=9.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: não especificado.
A1.5.1.9 Number of boxes
• Descrição: o número de caixas/componentes permitidas para cada motivo complexo.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: não especificado.
A1.5.2 Parâmetros de alfabeto
A1.5.2.1 Alphabet Type
• Descrição: especifica o tipo de alfabeto a utilizar, podendo este ser um dos formatos pré-
definidos ou um formato definido pelo utilizado.
• Tipo: obrigatório.
• Domínio: {DNA, Degenerated DNA, Protein (Barton and Sternberg) [169], Protein (Smith
and Smith) [170], User Defined}.
• Valor por defeito: DNA.
A1.5.2.2 Alphabet File
• Descrição: o ficheiro contendo a especificação para o alfabeto definido pelo utilizador. O
ficheiro poderá ser carregado ou os dados poderão ser introduzidos directamente na caixa
de texto fornecida para o efeito.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar um tipo de alfabeto
diferente dos pré-definidos.
• Domínio: este ficheiro deverá conter:

116
o Um tipo de dados (“Nucleotides”, “Proteins”, “Other”) que permita ao algoritmo
SMILE reconhecer grupos de símbolos conhecidos.
o Um conjunto de símbolos, por exemplo:
AB
C
AD
… indica que SMILE irá gerar modelos/motivos num alfabeto de três
símbolos: [AB], C e D. De notar que é necessário especificar todos os
símbolos que se pretendem agrupar.
Por exemplo, se estivermos na presença de um conjunto de sequências de
DNA contendo A, C, G, T e R e pretendermos gerar modelos num alfabeto
contendo R e Y, o ficheiro deveria ser escrito do seguinte modo:
• Type: Nucleotides
• AGR // que será reconhecido como R
• CT // (ou CTY) que será reconhecido como Y.
• Valor por defeito: não especificado.
A1.5.3 Parâmetros do método de avaliação “shuffling”
A1.5.3.1 Shuffle output file
• Descrição: nome do ficheiro de saída para o método de avaliação “shuffling”.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar o método de
avaliação “shuffling”.
• Domínio: cadeia de caracteres.
• Valor por defeito: outfile.shuffle.
A1.5.3.2 Shufflings
• Descrição: o número de “shufflings” que será executado.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar o método de
avaliação “shuffling”.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 20.

117
A1.5.3.2 Size k-mer
• Descrição: dimensão dos k-ímeros a serem conservados durante a aplicação do método
“shuffling”.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar o método de
avaliação “shuffling”.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: 3.
A1.5.4 Parâmetros do método de avaliação “against”
A1.5.4.1 Input File
• Descrição: o nome do ficheiro de entrada para o método de avaliação “against”. As
frequências dos motivos nas sequências originais serão comparadas com as frequências
dos motivos neste ficheiro, onde se sabe que os motivos estão ausentes.
• Tipo: condicional – só deverá ser especificado se se pretende utilizar o método de
avaliação “against”.
• Domínio: ficheiro em formato Fasta.
• Valor por defeito: outfile.shuffle.
A1.5.5 Parâmetros da caixa n
SMILE permite um número máximo de quatro caixas. Para cada caixa, deverão ser
especificados o comprimento máximo e mínimo, o número de substituições permitido na caixa e o
comprimento do espaçamento entre a caixa actual e a próxima caixa. De notar que estes dois
últimos parâmetros não terão de ser especificados para a última caixa. Ao utilizar “deltas”, é
também necessário especificar o valor de δ.
A1.5.5.1 Min Length
• Descrição: comprimento mínimo da caixa.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa).
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.

118
A1.5.5.2 Max Length
• Descrição: comprimento máximo da caixa.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa).
• Domínio: Inteiro, maior ou igual a 1. O valor deverá ser igual ou superior ao do parâmetro
A1.5.5.1.
• Valor por defeito: não especificado.
A1.5.5.3 Substitutions
• Descrição: número de substituições permitidas na caixa.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa).
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: não especificado.
A1.5.5.4 Min spacer length
• Descrição: comprimento mínimo do espaçamento entre a caixa actual e a próxima.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa).
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: não especificado.
A1.5.5.5 Max spacer length
• Descrição: comprimento máximo do espaçamento entre a caixa actual e a próxima.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa).
• Domínio: Inteiro, maior ou igual a 0. O valor deverá ser igual ou superior ao do parâmetro
A1.5.5.4.
• Valor por defeito: não especificado.

119
A1.5.5.6 Delta
• Descrição: ao utilizar “deltas”, SMILE permite um “salto” de d ± δ entre cada duas caixas.
• Tipo: opcional (mas deverá ser especificado se se pretende utilizar a caixa respectiva e se
o programa de extracção seleccionado é “x-smile_delta”).
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.
A1.6 Interface RISO
A interface RISO é composta pelos seguintes blocos:
• Parâmetros genéricos;
• Parâmetros de alfabeto – ver secção A1.5.2;
• Parâmetros do método de avaliação “shuffling” – ver secção A1.5.3;
• Parâmetros do método de avaliação “against” – ver secção A1.5.4;
• Parâmetros da caixa n (n є {1,4}).
A1.6.1 Parâmetros genéricos
A1.6.1.1 Sequences File
Ver parâmetro A1.5.1.3.
A1.6.1.2 Output File
Ver parâmetro A1.5.1.4
A1.6.1.3 Quorum
Ver parâmetro A1.5.1.5.

120
A1.6.1.4 Number of boxes
• Descrição: o número de caixas/componentes permitidas para cada motivo complexo.
• Tipo: obrigatório.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.
A1.6.1.5 Evaluation Method
Ver parâmetro A1.5.1.2.
A1.6.2 Parâmetros da caixa n
A aplicação RISO permite a especificação de até quatro caixas. Para cada caixa, é
necessário especificar o comprimento mínimo e máximo, o número de substituições permitidas e o
comprimento do espaçamento entre a caixa actual e a próxima. De notar que estes dois últimos
parâmetros não têm de ser especificados para a última caixa.
A1.6.2.1 Min Length
• Descrição: comprimento mínimo da caixa.
• Tipo: obrigatório para a primeira caixa. Deverá ser especificado para todas as caixas
utilizadas.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.
A1.6.2.2 Max Length
• Descrição: comprimento máximo da caixa.
• Tipo: obrigatório para a primeira caixa. Deverá ser especificado para todas as caixas
utilizadas.
• Domínio: Inteiro, maior ou igual a 1.
• Valor por defeito: não especificado.

121
A1.6.2.3 Substitutions
• Descrição: o número de substituições permitidas na caixa.
• Tipo: obrigatório para a primeira caixa. Deverá ser especificado para todas as caixas
utilizadas.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: 0.
A1.6.2.4 Min spacer length
• Descrição: comprimento mínimo do espaçamento entre a caixa actual e a próxima.
• Tipo: obrigatório para a primeira caixa. Deverá ser especificado para todas as caixas
utilizadas.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: 0.
A1.6.2.5 Max spacer length
• Descrição: comprimento máximo do espaçamento entre a caixa actual e a próxima.
• Tipo: obrigatório para a primeira caixa. Deverá ser especificado para todas as caixas
utilizadas.
• Domínio: Inteiro, maior ou igual a 0.
• Valor por defeito: 0.

122
Anexo 2 – Formato de saída das várias aplicações
Neste anexo descrevem-se o formato de saída de cada uma das aplicações instaladas no
cluster hermes.
A2.1 Aplicações de Biclustering
O output das três aplicações de biclustering é idêntico e corresponde a um ficheiro
contendo os n melhores biclusters (sendo que n é especificado pelo utilizador). Para cada bicluster,
a seguinte informação é fornecida:
• Identificador do bicluster;
• Número de condições;
• Primeira e última condição;
• Número de genes;
• Dimensão do bicluster (número de genes × número de condições);
• MSR;
• Variância;
• ARV;
• ACV;
• ARV – MSR;
• Padrão p-value;
• Padrão p-value com colunas;
• Padrão p-value com a correcção de Bonferroni;

123
• Padrão p-value com colunas e a correcção de Bonferroni;
• Melhor p-value GO;
• Melhor p-value GO com a correcção de Bonferroni;
• Número de p-values GO altamente significantes (p-value < 0.01);
• Número de p-values GO significantes ( 0.01 ≤ p-value < 0.05)
• Lista de genes e o correspondente nível de expressão. Exemplo:
YBL023C U
YBR052C U
YBR053C U
YBR054W U
Podem ser encontrados alguns exemplos de execução destes algoritmos em [168].
A2.2 Aplicações de Motif Finding
A2.2.1 MUSA
O output da aplicação MUSA consiste num ficheiro denominado “file.musa” (para aceder ao
ficheiro, é necessário clicar na opção “all files” da página de resultados). Este ficheiro contém uma
lista dos motivos complexos identificados e do seu agrupamento em famílias:
# Input arguments: quiet=YES bothstrand=YES lambda=4 epsilon=1 sieverate=30% (2 in 4 seqs) mindist=1 maxdist=50 siglevel=0.001000 minoverlap=70% # Motifs: 506 # ID Motif Quorum P-value (832) CCAATG <6,8> CAGCA 3 of 4 1.83483e-10 (1230) TGCTG <6,8> CATTGG 3 of 4 1.83483e-10 (639) ACTGT <38,40> GATAA 3 of 4 5.29019e-10 (1261) TTATC <38,40> ACAGT 3 of 4 5.29019e-10 (831) CCAAT <7,9> CAGC 4 of 4 7.07568e-10 (1011) GCTG <7,9> ATTGG 4 of 4 7.07568e-10 (867) CGATA <37,39> TGTA 3 of 4 1.83915e-09 (1100) TACA <37,39> TATCG 3 of 4 1.83915e-09 (581) AAGA <27,29> GCTAAG 3 of 4 4.94989e-09 (919) CTTAGC <27,29> TCTT 3 of 4 4.94989e-09 (1151) TCAT <39,41> CCTTTT 3 of 4 7.13684e-09 (695) AGTAG <25,27> GATA 3 of 4 2.17954e-08 (1129) TATC <25,27> CTACT 3 of 4 2.17954e-08 (583) AAGA <32,34> GATA 4 of 4 3.87263e-08 (...) Number of families found: 5 (maxQ: 0.000000)

124
Family 1 (1 motifs and 0 pairs) (quorum stat: min 1.00 avg 1.00 max 1.00) CTGATGCT 0 3 1 3.5683e-05 A: 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 0.000000 C: 1.000000 0.000000 0.000000 0.000000 0.000000 0.000000 1.000000 0.000000 G: 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 0.000000 0.000000 T: 0.000000 1.000000 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 Family 2 (2 motifs and 1 pairs) (quorum stat: min 1.00 avg 1.00 max 1.00) AGCATCAG- 0 3 2 3.5683e-05 AGCATCAGC 1 2 1 0.000294312 A: 1.000000 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 0.000000 0.000000 C: 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 G: 0.000000 1.000000 0.000000 0.000000 0.000000 0.000000 0.000000 1.000000 0.000000 T: 0.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 0.000000 (...)
A primeira linha identifica os parâmetros de entrada fornecidos à aplicação. A segunda
linha indica o número de motivos complexos encontrados.
As linhas subsequentes apresentam informação detalhada sobre cada motivo complexo
extraído das sequências de entrada. A primeira coluna corresponde ao identificador do motivo. A
segunda linha corresponde ao motivo identificado. Se se trata de um motivo complexo, a distância
entre cada componente é dado entre os símbolos ‘<’ e ‘>’. A terceira coluna indica o quorum do
motivo. A terceira coluna corresponde ao valor do p-value, a medida de significância estatística
utilizada.
Se o utilizador especifica um valor de 𝜀𝜀 maior do que zero, a distância entre componentes
é apresentada como um par (distância mínima, distância máxima). Se o utilizador efectua uma
procura em ambas as cadeias de DNA, na lista de resultados constarão, normalmente, o motivo e
o seu complemento reverso.
Considerando como exemplo o primeiro motivo da lista, verificamos que este é composto
por dois componentes, CCAATG e CAGCA, estando estes separados por uma distância mínima de
6 caracteres e uma distância máxima de 8 caracteres. O motivo ocorre em 3 das 4 sequências de
entrada.
Cada família é representada por uma PWM. A primeira linha da lista de famílias identifica o
número de famílias encontradas. As linhas seguintes fornecem informação detalhada sobre as

125
famílias identificadas, nomeadamente os motivos que foram incluídos numa dada família e a PWM
que a representa.
A2.2.2 PSMILE
O output de PSMILE consiste em dois ficheiros. O primeiro contém uma lista dos motivos
complexos encontrados e o segundo especifica a significância estatística desses motivos.
Um possível exemplo do ficheiro que contém os motivos identificados seria o seguinte:
AAAAA_AAAAT 00000-00003 23 382
AAAAA_AAAGA 00000-00020 23 355
AAAAA_AAATC 00000-00031 23 196
AAAAA_AACAA 00000-00100 23 325
AAAAA_ACAAT 00000-01003 23 183
AAAAA_ACATG 00000-01032 23 88
AAAAA_ACTTT 00000-01333 23 146
(…)
A primeira coluna corresponde à sequência de nucleótidos que compõem o motivo. Os
diferentes componentes de um motivo complexo são separados pelo carácter (‘_’). A segunda
coluna corresponde à posição do motivo complexo. A terceira coluna indica o número de
sequências nas quais o motivo pode ser encontrado (neste caso, 23). A última coluna corresponde
ao comprimento do espaçamento entre os componentes do motivo complexo.
Um possível exemplo do ficheiro que contém os resultados da avaliação estatística através
do método “shuffling” seria o seguinte:
STATISTICS ON THE NUMBER OF SEQUENCES HAVING AT LEAST ONE OCCURRENCE
Model %right #right %shfl. #shfl. Sigma Chi2 Z-score
===========================================================================
ACATG_CCAAC 100.00% 23 67.83% 15.60 1.35 8.82 5.47
TCACT_TTACC 100.00% 23 69.13% 15.90 1.48 8.40 4.79
TTTTA_GTCCT 100.00% 23 74.78% 17.20 1.24 6.64 4.68
TCTTT_CTCAT 100.00% 23 85.43% 19.65 0.75 3.61 4.50
STATISTICS ON THE TOTAL NUMBER OF OCCURRENCES
Model #right #shfl. Sigma Chi2 Z-score

126
===========================================================================
TTCTT_TCTTT 384 126.05 11.32 131.92 22.80
TCTTT_TCTTT 397 126.85 13.18 140.92 20.49
TGTTT_TCTTT 344 110.00 11.63 121.81 20.12
Os motivos são ordenados de acordo com o valor do z-score. Quanto mais alto for o z-
score, mais está o motivo representado nas sequências originais. Se o valor for negativo, o motivo
não se encontra representado. Um valor igual a MAX_INT indica que a avaliação da representação
do motivo não pôde ser executada (o que significa que o motivo não pôde ser encontrado nas
sequências de entrada ou que foi encontrado em todas elas).
O valor Chi2 (𝜒𝜒2) é também uma medida da significância estatística. As colunas “right”
indicam a percentagem e número de ocorrências de cada motivo identificado nas sequências de
entrada. As colunas “shfl” fornecem a mesma informação, mas para as sequências “shuffled”.
Finalmente, a coluna “sigma” indica o desvio padrão.
O ficheiro que resulta da aplicação do método de avaliação “against” é muito semelhante
aquele que é criado pelo método “shuffling”. Contudo, as estatísticas para as sequências e para o
número total de ocorrências surgem na mesma linha (‘T’ corresponde a dados estatísticos sob o
número total de ocorrências). Além disso, a única medida estatística que pode ser aplicada com o
método de avaliação “against” é 𝜒𝜒2. De notar que, com esta medida, não podemos tirar conclusões
sobre o grau de representação do motivo, uma vez que o valor de 𝜒𝜒2 não apresenta sinal. Para
ultrapassar este problema, é escrito um sinal depois do valor de 𝜒𝜒2 que indica o grau de
representação do motivo:
Model #right #wrong #rightT #wrongT Chi^2 Chi^2 T
===========================================================================
TTAT_AAAT 842 0 2871 0 36.95 + 14.81 +
TTAT_AAAA 832 0 4089 0 34.99 + 21.22 +
TTTA_AAAA 827 0 4191 0 34.07 + 21.76 +
AAAT_TTTT 812 0 4123 0 31.52 + 21.40 +
TATT_AAAT 811 0 2836 0 31.37 + 14.63 +
TTTA_AAAT 808 0 2954 0 30.89 + 15.24 +
A2.2.3 RISO
Consultar a secção A2.2.2. O formato de saída da aplicação RISO é idêntica à da
aplicação PSMILE.
Exemplos de execução dos algoritmos de motif finding podem ser encontrados em [167].

127
Anexo 3 – Condições e Acções para a extracção de um motivo estruturado
As condições e acções identificadas para a extracção de um motivo estruturado com dois
componentes foram as seguintes:
c1: O motivo simples 𝑚𝑚 com quorum 𝑞𝑞 e um componente de comprimento 𝑙𝑙1 está
presente em 𝑆𝑆
c2: O motivo composto 𝑚𝑚 com quorum 𝑞𝑞, dois componentes de comprimentos 𝑙𝑙1 e 𝑙𝑙2
separados por uma distância 𝑑𝑑 > 0, está presente em 𝑆𝑆
c3: 𝑑𝑑′ = 1
c4: 𝑑𝑑′ = 2
c5: 𝑞𝑞 ≥ 𝑞𝑞′
c6: 𝑙𝑙1 ∈ [𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ]
c7: 𝑙𝑙2 ∈ [𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ]
c8: 𝑙𝑙2 ∈ [𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 , … , 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ]
c9: 𝑒𝑒1 = 0
c10: 𝑒𝑒2 = 0
c11: 𝑙𝑙1 > 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
c12: 𝑙𝑙2 > 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
c13: 𝑙𝑙2 > 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚
c14: 𝑑𝑑 ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]
c15: 𝑞𝑞′ ≤ 0
c16: 𝑞𝑞′ > 100

128
c17: 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 > 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
c18: 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 > 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚
c19: 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 0
c20: 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 0
c21: 𝑒𝑒1 < 0
c22: 𝑒𝑒2 < 0
c23: 𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 0
c24: 𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 > 𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚
As acções para a situação em análise serão as seguintes:
a1: Devolver uma lista que contém o motivo composto 𝑚𝑚
a2: Devolver uma lista que não contém o motivo composto 𝑚𝑚
a3: Devolver uma lista que contém o motivo simples 𝑑𝑑1
a4: Devolver uma lista que não contém o motivo simples 𝑑𝑑1
a5: Devolver uma lista que contém o motivo simples 𝑑𝑑2
a6: Devolver uma lista que não contém o motivo simples 𝑑𝑑2
a7: Devolver uma lista que contém os motivos a uma distância de Hamming de
𝑑𝑑1 igual ou inferior a 𝑒𝑒1
a8: Devolver uma lista que contém os motivos a uma distância de Hamming de
𝑑𝑑2 igual ou inferior a 𝑒𝑒1
a9: Devolver uma lista que contém os motivos a uma distância de Hamming de
𝑑𝑑2 igual ou inferior a 𝑒𝑒2
a10: Devolver uma lista que contém os motivos parciais de 𝑑𝑑1 tais que, sendo 𝑑𝑑1′ =
𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑑𝑑1, então 0 ≤ 𝑚𝑚 e 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
a11: Devolver uma lista que contém os motivos parciais de 𝑑𝑑2 tais que, sendo 𝑑𝑑2′ =
𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚 e 𝑚𝑚 + 𝑦𝑦 = 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
a12: Devolver uma lista que contém os motivos a uma distância de Hamming igual ou
inferior a 𝑒𝑒1 dos motivos parciais de 𝑑𝑑1 tais que, sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um
motivo parcial de 𝑑𝑑1, então 0 ≤ 𝑚𝑚 e 𝑗𝑗 + 𝑚𝑚 = 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
a13: Devolver uma lista que contém os motivos a uma distância de Hamming igual ou
inferior a 𝑒𝑒1 dos motivos parciais de 𝑑𝑑2 tais que, sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um
motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚 e 𝑚𝑚 + 𝑦𝑦 = 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚
a14: Devolver uma lista que contém os motivos de dois componentes cujo primeiro
componente está a uma distância de Hamming de 𝑑𝑑1 igual ou inferior a 𝑒𝑒1 e o
segundo componente é 𝑑𝑑2

129
a15: Devolver uma lista que contém os motivos de dois componentes cujo segundo
componente está a uma distância de Hamming de 𝑑𝑑2 igual ou inferior a 𝑒𝑒2 e o
primeiro componente é 𝑑𝑑1
a16: Devolver uma lista que contém os motivos de dois componentes cujo primeiro
componente está a uma distância de Hamming de 𝑑𝑑1 igual ou inferior a 𝑒𝑒1 e o
segundo componente está a uma distância de Hamming de 𝑑𝑑2 igual ou inferior a
𝑒𝑒2
a17: Devolver uma lista que contém os motivos de dois componentes cujo segundo
componente é 𝑑𝑑2 e o primeiro corresponde aos motivos parciais de 𝑑𝑑1 que
obedecem às seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de
𝑑𝑑1, então 0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ] �
a18: Devolver uma lista que contém todos os motivos de dois componentes cujo
segundo componente é 𝑑𝑑2, ou qualquer motivo a uma distância de Hamming igual
ou inferior a 𝑒𝑒2 de 𝑑𝑑2, e o primeiro componente corresponde aos motivos parciais
de 𝑑𝑑1 que obedecem às seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo
parcial de 𝑑𝑑1, então 0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e
(𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�
a19: Devolver uma lista que contém todos os motivos de dois componentes cujo
segundo componente é 𝑑𝑑2 e o primeiro componente corresponde aos motivos
parciais de 𝑑𝑑1 que obedecem às seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚
um motivo parcial de 𝑑𝑑1, então 0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈
[𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos que estão a uma distância de
Hamming desses motivos parciais igual ou inferior ao número de erro𝑠𝑠 𝑒𝑒1
a20: Devolver devolva uma lista que contém todos os motivos de dois componentes
cujo segundo componente é 𝑑𝑑2, ou qualquer motivo a uma distância de Hamming
igual ou inferior a 𝑒𝑒2 de 𝑑𝑑2, e o primeiro componente corresponde aos motivos
parciais de 𝑑𝑑1 que obedecem às seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚
um motivo parcial de 𝑑𝑑1, então 0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈
[𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos que estão a uma distância de
Hamming desses motivos parciais igual ou inferior ao número de erros 𝑒𝑒1
a21: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente é 𝑑𝑑1 e o segundo componente corresponde aos motivos
parciais de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦
um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑚𝑚) ∈
[𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�

130
a22: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente é 𝑑𝑑1, ou qualquer motivo a uma distância de Hamming igual
ou inferior a 𝑒𝑒1 de 𝑑𝑑1, e o segundo componente corresponde aos motivos parciais
de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um
motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e
(𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�
a23: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente é 𝑑𝑑1 e o segundo componente corresponde aos motivos
parciais de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦
um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑚𝑚) ∈
[𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos que estão a uma distância de
Hamming desses motivos parciais igual ou inferior ao número de erros 𝑒𝑒2
a24: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente é 𝑑𝑑1, ou qualquer motivo a uma distância de Hamming igual
ou inferior a 𝑒𝑒1 de 𝑑𝑑1, e o segundo componente corresponde aos motivos parciais
de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um
motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e
(𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos que estão a uma distância
de Hamming desses motivos parciais igual ou inferior ao número de erros 𝑒𝑒2
a25: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente corresponde aos motivos parciais de 𝑑𝑑1 que obedecem às
seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑑𝑑1, então
0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�; e o segundo
componente corresponde aos motivos parciais de 𝑑𝑑2 que obedecem às seguintes
condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚,
𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�
a26: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente corresponde aos motivos parciais de 𝑑𝑑1 que obedecem às
seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑑𝑑1, então
0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos
os motivos que estão a uma distância de Hamming desses motivos parciais igual
ou inferior ao número de erros 𝑒𝑒1; e o segundo componente corresponde aos
motivos parciais de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ =
𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e
(𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�

131
a27: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente corresponde aos motivos parciais de 𝑑𝑑1 que obedecem às
seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑑𝑑1, então
0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�; e o segundo
componente corresponde aos motivos parciais de 𝑑𝑑2 que obedecem às seguintes
condições: sendo 𝑑𝑑2′ = 𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚,
𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos
que estão a uma distância de Hamming desses motivos parciais igual ou inferior
ao número de erros 𝑒𝑒2
a28: Devolver uma lista que contém todos os motivos de dois componentes cujo
primeiro componente corresponde aos motivos parciais de 𝑑𝑑1 que obedecem às
seguintes condições: sendo 𝑑𝑑1′ = 𝑑𝑑1,1+𝑚𝑚 … 𝑑𝑑1,𝑗𝑗+𝑚𝑚 um motivo parcial de 𝑑𝑑1, então
0 ≤ 𝑚𝑚, 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑗𝑗 + 𝑚𝑚 ≤ 𝑙𝑙1,𝑚𝑚𝑚𝑚𝑚𝑚 e (𝑑𝑑 + 𝑙𝑙1 − (𝑗𝑗 + 𝑚𝑚)) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos
os motivos que estão a uma distância de Hamming desses motivos parciais igual
ou inferior ao número de erros 𝑒𝑒1; e o segundo componente corresponde aos
motivos parciais de 𝑑𝑑2 que obedecem às seguintes condições: sendo 𝑑𝑑2′ =
𝑑𝑑2,1+𝑚𝑚 … 𝑑𝑑2,𝑚𝑚+𝑦𝑦 um motivo parcial de 𝑑𝑑2, então 0 ≤ 𝑚𝑚, 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 ≤ 𝑚𝑚 + 𝑦𝑦 ≤ 𝑙𝑙2,𝑚𝑚𝑚𝑚𝑚𝑚 e
(𝑑𝑑 + 𝑚𝑚) ∈ [𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 , … ,𝑑𝑑𝑚𝑚𝑚𝑚𝑚𝑚 ]�, bem como todos os motivos que estão a uma distância
de Hamming desses motivos parciais igual ou inferior ao número de erros 𝑒𝑒2.
a29: Devolver erro para quorum com valor igual ou inferior a 0
a30: Devolver erro para quorum com valor superior a 100
a31: Devolver erro para comprimento mínimo do primeiro componente superior ao
comprimento máximo
a32: Devolver erro para comprimento mínimo do segundo componente superior ao
comprimento máximo
a33: Devolver erro para comprimento mínimo do primeiro componente com valor igual
ou inferior a zero
a34: Devolver erro para comprimento mínimo do segundo componente com valor igual
ou inferior a zero
a35: Devolver erro para número de mutações do primeiro componente com valor igual
ou inferior a zero
a36: Devolver erro para número de mutações do segundo componente com valor igual
ou inferior a zero
a37: Devolver erro para distância entre motivos com valor mínimo igual ou inferior a
zero
a38: Devolver erro para distância entre motivos com valor mínimo superior ao valor
máximo



![[ Services ] Grid Computing](https://img.document.onl/doc/110x75/5571f1bf49795947648b9f9b/-services-grid-computing.jpg)