Embed Size (px)
Citation preview

Centro Brasileiro de Pesquisas Físicas
Programa de Pós-Graduação em Física
Coordenação de Matéria Condensada, Física Aplicada e Nanociências
TESE DE DOUTORADO
Revestimentos nanométricos e cristalinos de hidroxiapatita em
substratos metálicos e poliméricos a temperatura ambiente:
Produção e Espectroscopia óptica de plasmas por
Laser pulsado de Nd:YAG
Gabriela Cerqueira Gomes
Orientador: Prof. Dr. Alexandre Mello de Paula Silva
Coorientador: Prof. Dr. Fábio de Oliveira Borges
Rio de Janeiro – RJ
Dezembro de 2017

2
Dedicado à minha família, por todo amor, apoio e confiança.
E em memória do professor Ademarlaudo França Barbosa.

3
Agradecimentos
A Deus, minha fonte de sabedoria, fé e principalmente resiliência para concluir essa
etapa da minha vida. Apesar da perda lastimável do prof. Laudo no início do doutorado,
eu tive muito apoio da minha família, dos meus amigos e dos meus atuais orientadores
para recomeçar. Eu agradeço a Deus por ter tudo isso.
Aos meus pais, Valter Gomes e Célia Lima, e meus irmãos, Rodrigo e Beatriz,
simplesmente por serem amor e me apoiarem em todos os momentos. À minha grande
família também, pelo apoio e por compreender minha ausência durante os períodos de
intensa dedicação à tese. Ao Max e sua família, por todo seu apoio e carinho.
Aos meus orientadores Alexandre Mello e Fábio Borges, primeiro por me permitirem
recomeçar, e depois por todo apoio para que eu pudesse terminar. Agradeço a confiança,
respeito e compreensão para me conduzir em um tema de pesquisa na qual eu não tinha
experiência prévia.
Aos pós-docs Fabrício Borghi e Rogélio Ospina, duas pessoas que também me deram
muito suporte e ensinamentos valiosos.
Aos professores que muito colaboraram: Cleo Martins, Elena Mavropoulos, Alexandre
Rossi.
Aos profs. coordenadores Sinnecker, Sarthour, Roditti, pelo apoio para participar da
Conferência de Ablação a Laser (COLA 2015), pois foi quando eu tive uma percepção
muito valiosa sobre o meu trabalho de tese.
A todos do grupo do Laboratório de Biomateriais, pelo fornecimento da matéria prima
desse trabalho: a hidroxiapatita.
Às meninas do Laboratório de Cultivo de Células por todo carinho, principalmente à
Suzana e Melissa pela dedicação durante os testes biológicos.
Aos meus amigos do CBPF pelo grande apoio e amizade durante esses últimos anos, e
àqueles que passaram pelo Laboratório de Plasma Aplicado e pelo Laboratório de
Superfícies e Nanoestruturas, pela troca de experiências.
A todos que me deram suporte com eficiência e prontidão nos laboratórios e
equipamentos do CBPF: Mariana Giffoni e Paula Marques, do Laboratório de Química
e Preparação de Amostras; Cilene Labre, do LabNano; Carlos Albuquerque e Anderson
Franco, do Laboratório de Criogenia; Francisco de Assis e Thiago Palhares, do
Laboratório de Espectrofotometria de FTIR/AA, à Luisa pelo treinamento no XPS, ao
Prof. Gomes pelo treinamento no AFM e à toda a equipe do Laboratório de
Instrumentação e Materiais Mecânicos (oficina mecânica).
À Bete Vicente, Gil, Viviane Vicente e Ronaldo, da secretaria da COMAN. Às amigas
da biblioteca: Edileuza, Rosa e Cida.
A todos os pós-graduandos do CBPF que estiveram à frente da APG-José Leite Lopes
durante esse período que, junto com Denise Coutinho, me ajudaram muito.
Aos professores Herman Lima, Paulo Renato e José Brant, e aos meninos Rafael Gama
e Victor Ferraz pelo grande apoio no primeiro ano de doutorado.
Ao LNLS, pelos dias de intenso trabalho e aprendizado.
Ao CNPq, pela bolsa de pesquisa.

4
Resumo
Os fosfatos de cálcio (CaP) formam uma classe de materiais biocompatíveis
frequentemente usados como recobrimentos de implantes ósseos. Dentre as fases de
CaP, a hidroxiapatita (HAP) se destaca devido a sua estrutura cristalina similar ao tecido
ósseo, baixa solubilidade e osteocondutividade. A técnica de deposição a laser pulsado
(PLD – Pulsed Laser Deposition) permite produzir recobrimentos de HAP com boa
adesão ao substrato e cristalinidade controlada, dependendo dos parâmetros escolhidos
para a ablação. Entretanto, a cristalinidade é frequentemente obtida com tratamento
térmico durante e/ou após a deposição por PLD, e isso pode provocar a formação de
outras fases indesejadas de CaP e óxido de cálcio nos recobrimentos. Além disso, os
tratamentos térmicos afetam a adesão e provocam rachaduras e descolamento devido às
tensões térmicas na interface entre o implante e o recobrimento.
Neste trabalho, recobrimentos cristalinos de HAP foram produzidos por PLD
com alta fluência (30 J/cm2) do laser Nd:YAG no comprimento de onda de 532 nm. A
cristalinidade foi alcançada em apenas 5 minutos de deposição. A morfologia dos
recobrimentos consistiu de partículas micrométricas (com até 10 µm de largura e 4 µm
de altura) depositadas sobre uma camada densa de 150 nm de espessura. Os filmes finos
cristalinos de HAP foram depositados sobre superfícies de Ti e de ácido polilático (PLA
- polylactic acid), um polímero bioabsorvível sensível à temperatura. Após a deposição
à temperatura ambiente, as superfícies de PLA/HAP se tornaram hidrofílicas e mais
apropriadas para aplicações biomédicas. Além disso, os testes de adesão com fita (tape
test) apresentaram máxima aderência dos filmes sobre os substratos de Si e PLA,
seguindo os critérios da norma ASTM D3359. A estrutura cristalina dos recobrimentos
foi investigada por Difração de raios X com Incidência Rasante (GIXRD – Grazing
Incidence X-ray Diffraction), apresentando uma fase única de HAP. Os difratogramas
das amostras com tratamento térmico in situ de 200 °C e 800 °C confirmaram a
ausência de outras fases ocultas sob a componente nanocristalina presente nos
recobrimentos não tratados. Outras caracterizações foram realizadas por Espectroscopia
de Fotoelétrons de raios X (XPS – X-ray Photoelectron Spectroscopy), Espectroscopia
de Infravermelho por Transformada de Fourier (FTIR – Fourier Transform Infrared
Spectroscopy), Microscopia Atômica (AFM – Atomic Force Microscopy) e Microscopia

5
Eletrônica de Varredura (SEM – Scanning Electron Microscopy). Testes biológicos
preliminares foram realizados para futuras aplicações biomédicas desses recobrimentos.
A transferência da estequiometria do alvo ao recobrimento é apontada como uma
das maiores vantagens do PLD. Nesse trabalho, a técnica de Espectroscopia de Plasma
Induzido por Laser com Calibração por um Ponto (OPC-LIBS – One-Point Calibration
Laser Induced Breakdown Spectroscopy) foi usada para analisar a estequiometria e Ca/P
dos alvos de HAP. Essa técnica não necessita de preparação da amostra, de forma que o
alvo pode ser usado para PLD imediatamente depois da análise por OPC-LIBS. Os
alvos de HAP também foram analisados por Espectroscopia de Absorção Atômica (AAS
- Atomic Absorption Spectroscopy), Espectroscopia UV-visível (UV-Vis – Ultraviolet
Visible Spectroscopy) e Fluorescência de raios X (XRF – X-ray Fluorescence), que são
técnicas de análises químicas que necessitam de digestão parcial das amostras. Os
resultados obtidos por OPC-LIBS foram comparados às técnicas analíticas, e
apresentaram bom ajuste dos dados.
Palavras–chave: filmes finos, hidroxiapatita, ablação a laser pulsado;
temperatura ambiente, ácido polilático, espectroscopia óptica, método OPC-LIBS (One-
Point Calibration - Laser Induced Breakdown Spectroscopy).

6
Abstract
Calcium phosphates (CaP) form a class of biocompatible materials often used as
bone implant coatings. Among the CaP phases, hydroxyapatite (HAP) stands out
because of its crystalline structure similar to bone tissue, low solubility and
osteoconductivity. The Pulsed Laser Deposition (PLD) technique allows the production
of crystalline HAP coatings with good adhesion to the substrate and controlled
morphology depending on the parameters chosen for ablation. However, crystallinity is
often obtained with heat treatment during and/or after PLD deposition, and this may
cause the formation of other undesired phases of CaP and calcium oxide in the coatings.
In addition, heat treatments affect adhesion and cause cracking and detachment due to
thermal stresses at the interface between the implant and the coating.
In this work, crystalline HAP coatings were produced by PLD with high fluence
(30 J/cm2) of Nd:YAG laser at 532 nm wavelength. The coatings crystallinity was
achieved in only 5 minutes of deposition time. The morphology of the coatings
consisted of micrometric particles (up to 10 μm wide and 4 μm high) deposited on a
dense layer of 150 nm thickness. The crystalline thin films of HAP were deposited on
Ti and polylactic acid (PLA) surfaces, which are temperature sensitive bioabsorbable
polymers. After deposition at room temperature, the PLA/HAP surfaces have become
hydrophilic and more suitable for biomedical applications. In addition, tape test showed
good adhesion of the films on the substrates. The crystalline structure of the coatings
was investigated by X-ray diffraction with grazing incidence (GIXRD), presenting a
single phase of HAP. GIXRD measurements in samples with 200 °C and 800 °C in situ
heat treatment confirmed the absence of other hidden phases under the nanocrystalline
component (without diffraction peaks) present in the untreated coatings. Further
characterizations were performed with X-ray Photoelectron Spectroscopy (XPS),
Fourier Transform Infrared Spectroscopy (FTIR), Atomic Microscopy (AFM) and
Scanning Electron Microscopy (SEM). Preliminary biological tests were performed for
future biomedical applications of these coatings.
The ability to transfer the target stoichiometry to the coating is pointed out as one of the
major advantages of the PLD technique. In this work, the laser-induced plasma
spectroscopy One-Point Calibration - Laser Induced Breakdown Spectroscopy (OPC-
LIBS) technique was used to analyze the stoichiometry and Ca/P of HAP targets. This

7
technique does not require preparation of the sample, so that the target can be used for
PLD immediately after analysis by OPC-LIBS. HAP targets produced in this work were
also analyzed by Atomic Absorption Spectroscopy (AAS), Ultraviolet Visible
Spectroscopy (UV-Vis) and X-ray Fluorescence (XRF), which are chemical analysis
techniques that require partial digestion of the samples. The results obtained by OPC-
LIBS were compared to the analytical techniques, and presented a good data fit.
Keywords: thin films, hydroxyapatite, pulsed laser deposition, room temperature,
polylactic acid, optical spectroscopy, One-Point Calibration - Laser Induced Breakdown
Spectroscopy method.

8
LISTA DE ABREVIATURAS E SÍMBOLOS
AAS Espectroscopia de Absorção Atômica
AFM Microscopia Atômica
ASTM Sociedade Americana de Testes e Materiais (American Society for
Testing and Materials)
CaP Fosfato de cálcio
CF-LIBS Espectroscopia de Plasma Induzido por Laser sem Calibração (Calibration
Free Laser-Induced Breakdown Spectroscopy)
FRX Fluorescência de raios X
FTIR Espectroscopia de Infravermelho por Transformada de Fourier
GIXRD Difração de raios X com Incidência Rasante
HAP Hidroxiapatita
LIBS Espectroscopia de Plasma Induzido por Laser (Laser-Induced Breakdown
Spectroscopy)
LIPS Espectroscopia de Plasma Induzido por Laser (Laser-Induced Plasma
Spectroscopy)
LTE Equilíbrio Termodinâmico Local (Local Termodynamic Equilibrium)
MAPLE Evaporação de Matriz Assistida por Laser Pulsado (Matrix-Assisted Pulsed
Laser Evaporation)
MS Magnetron Sputtering
Nd:YAG Neodímio-YAG (neodymium-doped yttrium aluminium garnet)
NIST Instituto Nacional de Padrões e Tecnologia (National Institute of Standards
and Technology)
OPC-
LIBS
Espectroscopia de Plasma Induzido por Laser com Calibração por um Ponto
(One-Point Calibration Laser Induced Breakdown Spectroscopy)
PLA Ácido polilático
PLD Deposição a laser pulsado (Pulsed Laser Deposition)
PS Plasma Spray
SEM Microscopia Eletrônica de Varredura
Si Silício
Ti Titânio
UV Ultravioleta

9
UV-Vis Espectroscopia UV/visível
XPS Espectroscopia de Fotoelétrons de raios X

10
Sumário
Resumo ....................................................................................................................... 4
Abstract ....................................................................................................................... 6
1. INTRODUÇÃO .............................................................................................. 12
2. OBJETIVOS ................................................................................................... 17
3. REVISÃO BIBLIOGRÁFICA ..................................................................... 19
3.1. Biomateriais e Hidroxiapatita ................................................................... 19
3.2. Deposição a laser pulsado (PLD – Pulsed Laser Deposition) .................. 21
3.2.1. Fundamentos da técnica de PLD ....................................................... 21
3.2.2. Vantagens e limitações ...................................................................... 23
3.2.3. Interação da radiação laser com o alvo: processos térmicos e
hipertérmicos .......................................................................................................... 24
3.2.4. Formação dos aglomerados na produção de filmes finos por PLD... 27
3.2.5. Nucleação e crescimento dos filmes sob influência de processos
hipertérmicos .......................................................................................................... 28
3.2.6. PLD aplicado a biomateriais ............................................................. 30
3.3. Análise da estequiometria dos alvos de HAP por espectroscopia de plasma
induzido por laser ....................................................................................................... 32
3.3.1. Princípios básicos da análise ............................................................. 33
3.3.2. Análise quantitativa por CF-LIBS..................................................... 37
3.3.3. O método de calibração por um ponto OPC-LIBS ........................... 41
4. MATERIAIS E MÉTODOS ......................................................................... 45
4.1. Síntese da HAP ......................................................................................... 45
4.2. Preparação dos alvos de HAP ................................................................... 45
4.3. Análise da estequiometria dos alvos de HAP ........................................... 45
4.3.1. Análise estequiométrica por XRF, UV-VIS e AAS .......................... 46
4.3.2. Análise estequiométrica por OPC-LIBS ........................................... 47
4.4. Preparação e tratamento dos substratos de Si, Ti e PLA .......................... 48
4.5. Produção dos recobrimentos de HAP por PLD ........................................ 49
4.6. Caracterização físico-química dos filmes produzidos .............................. 51
4.6.1. Difração de raios-X (XRD) ............................................................... 51
4.6.2. Espectroscopia de infravermelho por transformada de Fourier (FTIR)
...........................................................................................................52
4.6.3. Espectroscopia de Fotoelétrons por Raios-X (XPS) ......................... 53
4.6.4. Microscopia Eletrônica de Varredura (MEV) ................................... 53

11
4.6.5. Microscopia de Força Atômica (AFM) ............................................. 53
4.6.6. Teste de adesão dos recobrimentos ................................................... 54
4.7. Ensaios preliminares de caracterização biológica .................................... 54
5. RESULTADOS E DISCUSSÕES ................................................................. 56
5.1. Análise estequiométrica dos alvos de HAP por OPC-LIBS ..................... 56
5.1.1. Análise qualitativa e seleção das linhas ............................................ 56
5.1.2. Determinação dos parâmetros do plasma: temperatura e densidade . 59
5.1.3. Cálculo da composição da amostra padrão com a técnica CF-LIBS 63
5.1.4. Determinação dos parâmetros P(λ) com a amostra padrão ............... 65
5.1.5. Determinação da composição dos alvos de HAP .............................. 66
5.1.6. Comparação com outras técnicas ...................................................... 69
5.2. Influência dos parâmetros de deposição sobre a cristalinidade dos filmes
..................................................................................................................71
5.3. Caracterização físico-química dos filmes produzidos à temperatura
ambiente ..................................................................................................................77
5.3.1. Cristalinidade e composição de fase ................................................. 77
5.3.2. Composição e estequiometria de superfície ...................................... 79
5.3.3. Morfologia e composição estrutural .................................................. 85
5.3.4. Teste de adesão do filme ao substrato ............................................... 87
5.3.5. Teste de ângulo de contato ................................................................ 89
5.4. Ensaios preliminares de adesão e proliferação celular ............................. 90
6. CONCLUSÕES .............................................................................................. 95
7. SUGESTÕES DE TRABALHOS FUTUROS ............................................. 97
8. REFERÊNCIAS BIBLIOGRÁFICAS ......................................................... 98
9. APÊNDICES ................................................................................................ 104
9.1. APÊNDICE I: Publicações deste trabalho de tese.................................. 104
9.2. APÊNDICE II: Protocolo dos ensaios biológicos .................................. 105

12
1. INTRODUÇÃO
Atualmente, novos materiais e tecnologias são desenvolvidos para aumentar a
aceitação de implantes ortopédicos e dentais no corpo humano [1-2]. Muitos trabalhos
na literatura científica propõem diferentes modificações da superfície de implantes
comerciais, geralmente de Ti e suas ligas, com o objetivo de aumentar eficientemente
sua função biológica e integração óssea [3-4]. Os fosfatos de cálcio (CaP) formam uma
classe de materiais com composição química similar à fase inorgânica do osso humano,
por isso eles são adequadamente escolhidos para integrar e reparar danos nos ossos. Os
recobrimentos à base de CaP podem ser produzidos com determinado controle sobre
suas propriedades, tais como: espessura, composição, cristalinidade, razão Ca/P,
microestrutura, porosidade e rugosidade [4-6]. A adesão dos recobrimentos ao substrato
também é uma característica muito importante e que pode ser obtida dependendo dos
processos e parâmetros de produção.
A hidroxiapatita (HAP), que apresenta a fórmula química Ca10(PO4)6(OH)2, é muito
utilizada para o recobrimento de implantes ósseos devido à sua biocompatibilidade e
osteocondutividade [2,6-8]. Um implante metálico recoberto com um filme de HAP se
beneficia das propriedades de atividade biológica do recobrimento com as propriedades
mecânicas dos metais, promovendo a biointegração desse implante em tecidos ósseos
[3,8].
Diversas técnicas são usadas para a deposição de filmes finos e recobrimentos de
implantes com materiais de CaP, especialmente a HAP [8-9]. As metodologias de
recobrimento por plasma mais estudadas atualmente são as técnicas de Plasma Spray
(PS), Magnetron Sputtering (MS) e de Deposição por Laser Pulsado (PLD – Pulsed
Laser Deposition) [10]. A técnica de PS é usada comercialmente devido ao seu baixo
custo e alta taxa de deposição, produzindo recobrimentos relativamente espessos (5-300
µm), porosos e com espessura não uniforme [10]. Porém, novos processos de aspersão
térmica são desenvolvidos para permitir maior controle da espessura, cristalinidade e
homogeneidade dos recobrimentos [10]. A técnica de MS por alvos opostos é apontada
como uma alternativa para produzir recobrimentos cristalinos sem a necessidade de
tratamento térmico [11-12]. Entretanto, apesar de oferecer menor rugosidade de
superfície, essa técnica ainda apresenta baixa taxa de deposição quando comparada à PS

13
e PLD [10,13]. Por outro lado, a técnica de PLD é conhecida como uma opção bastante
eficiente para materiais de estrutura complexa, por preservar e transferir a
estequiometria do material do alvo ao recobrimento. A versatilidade do sistema PLD
também é comumente citada como uma grande vantagem, pois a possibilidade de
modificar os diversos parâmetros do processo permite obter maior controle das
características do recobrimento, como a estequiometria, cristalinidade e rugosidade da
superfície [14].
Recentemente, avanços significativos foram alcançados para obter implantes
biomédicos de alta qualidade com recobrimentos de CaP e HAP por PLD [15-16]. Em
geral, a deposição de recobrimentos de HAP por PLD é realizada com tratamentos
térmicos in situ, para aumentar o processo de difusão e nucleação durante o crescimento
do filme, e com tratamentos térmicos após a deposição, para recuperar a cristalinidade
do recobrimento [14-15]. Em muitos trabalhos publicados na literatura, esses
tratamentos térmicos durante e/ou após a deposição são considerados necessários e até
fundamentais para induzir a cristalinidade, com o objetivo de promover menor
solubilidade do recobrimento quando implantado [15-16]. No entanto, o uso de
tratamento térmico in situ pode provocar a formação de uma camada de óxido no
implante metálico e de tensões mecânicas induzidas termicamente entre o recobrimento
e o implante. Como resultado, fissuras e rachaduras podem ser formadas no
revestimento, danificando a adesão do filme e favorecendo sítios para o crescimento de
bactérias. Em ambos os casos, os tratamentos térmicos podem formar fases não
desejadas de CaP e causar tensões mecânicas nos implantes [17]. Outras desvantagens
associadas ao uso de tratamentos térmicos em PLD são o aumento no consumo de
tempo e energia durante o processo de produção dos recobrimentos e a limitação na
escolha dos substratos.
Na literatura, poucos trabalhos abordam a produção de recobrimentos de HAP por
PLD com laser Nd:YAG no comprimento de onda de 532 nm, pois a HAP absorve
pouco nesse comprimento de onda e a ablação não é coerente. O uso de baixa fluência
de irradiação laser (energia/área) é outra característica em comum encontrada nesses
trabalhos [16,18-23].
Neste trabalho, recobrimentos cristalinos de HAP foram produzidos em apenas 5
minutos de deposição por PLD e sem necessidade de tratamentos térmicos. Os filmes
foram depositados à temperatura ambiente sobre substratos de Si (100), Ti e substratos
poliméricos de PLA (ácido polilático), que são sensíveis a altas temperaturas.

14
Considerando a vantagem do PLD de transferir a estequiometria do alvo ao
recobrimento, a alta densidade dos alvos cerâmicos produzidos com HAP
estequiométrica e altamente cristalina foi combinada à alta fluência de irradiação de um
laser pulsado. Dessa forma, os recobrimentos cristalinos de HAP foram produzidos
usando o segundo harmônico do laser Nd:YAG laser (532 nm) e mais alta fluência de
irradiação, em comparação com trabalhos publicados na literatura. A estrutura
cristalográfica dos recobrimentos foi investigada por Difração de raios X de luz
síncrotron em Incidência Rasante (GIXRD – Grazing Incidence X-ray Diffraction). As
propriedades químicas foram analisadas por Espectroscopia de Infravermelho por
Transformada de Fourier (FTIR – Fourier Transform Infrared Spectroscopy) e
Espectroscopia de Fotoelétrons de raios X (XPS – X-ray Photoelectron Spectroscopy).
Também foram investigadas as propriedades morfológicas por Microscopia de Força
Atômica (AFM – Atomic Force Microscopy) e Microscopia Eletrônica de Varredura
(SEM – Scanning Electron Microscopy). A biocompatibilidade dos filmes foi
investigada por experimentos in vitro. Como resultado, os recobrimentos foram
formados por partículas micrométricas (com até 10 µm de largura e 4 µm de altura)
depositadas sobre uma camada densa de 150 nm de HAP, e apresentaram boa adesão
sobre os substratos.
Para garantir a transferência de estequiometria do alvo ao recobrimento durante o
processo de PLD, os parâmetros da técnica devem ser adequadamente ajustados. Além
disso, também é importante certificar que o alvo utilizado tenha a estequiometria
desejada. Os alvos produzidos nesse trabalho, e utilizados para a ablação durante a
produção dos filmes por PLD, foram caracterizados por uma técnica não destrutiva de
análise química elementar. A técnica de Espectroscopia de Plasma Induzido por Laser
LIPS (Laser-Induced Plasma Spectroscopy), ou a sigla mais usada LIBS (Laser-Induced
Breakdown Spectroscopy), é um tipo de espectroscopia de emissão atômica na qual a
composição química é investigada através da interação (atomização e excitação) de um
pulso laser sobre a superfície da amostra. Essa técnica é recente e ainda se encontra em
desenvolvimento. Nesse trabalho, as vantagens de usar a técnica LIBS para caracterizar
os alvos estão na possibilidade de usar o mesmo laser e configuração experimental do
sistema de PLD, na rapidez e praticidade da medida, na possibilidade de análise
qualitativa e quantitativa in situ, na característica de ser uma técnica praticamente não
destrutiva e na possibilidade de ser utilizada para uma ampla variedade de substâncias
[24].

15
Em uma espectroscopia LIBS, o espectro de energia da radiação luminosa emitida
pela pluma de um plasma (gerado pela interação do feixe laser sobre a superfície da
amostra) carrega informações sobre a composição elementar da amostra analisada, pois
cada elemento emite um único conjunto de linhas espectrais com comprimentos de onda
bem definidos. Em geral, uma análise elementar quantitativa com a técnica LIBS é feita
usando curvas de calibração, onde são necessárias várias amostras com a mesma matriz
(ou seja, amostras compostas pelos mesmos elementos) em diferentes concentrações do
elemento da amostra a ser analisada [25]. Em alguns casos, o sinal LIBS obtido de um
elemento apresenta uma resposta linear com a concentração do elemento na amostra, o
que permite fazer uma análise quantitativa usando curvas de calibração obtidas de
amostras de referência com composição conhecida [25].
De um modo geral, a técnica LIBS ainda apresenta outras vantagens: a flexibilidade
na montagem experimental, a rápida análise dos dados, a não necessidade de preparação
da amostra, a possibilidade de realizar análises em diversos ambientes: ar, vácuo, fluido,
com alta temperatura e pressão, além da possibilidade de ser realizada in situ e de forma
remota [26]. Além disso, possui sensibilidade da ordem de partes por milhão (ppm) e
praticamente não danifica a amostra pois a quantidade de material removido é da ordem
de alguns nanogramas. No entanto, essa técnica também possui algumas desvantagens,
tais como: a sua dependência com as condições de equilíbrio do plasma, a saturação do
sinal devido aos efeitos de auto-absorção (que ocorre para os elementos de alta
concentração), o efeito matriz (para amostras muito heterogêneas), o grande número de
fenômenos físico-químicos complexos envolvidos nos processos de ablação, formação e
evolução da pluma de plasma e a interação do plasma com o ambiente circundante [26].
Com o objetivo de superar as dificuldades em medidas de análise quantitativa por
LIBS, alguns pesquisadores italianos criaram em 1999 um método conhecido como
Espectroscopia de Plasma Induzido por Laser sem Calibração (CF-LIBS – Calibration
Free Laser-Induced Breakdown Spectroscopy) [27]. Esse método não exige curvas de
calibração ou amostras de referência, pois se baseia em um modelo matemático que
descreve a emissão do plasma. Desde então, várias outras abordagens e aproximações
foram testadas por grupos de diversas nacionalidades e os resultados foram comparados
com outras técnicas de análise existentes [28-30]. Recentemente, no ano de 2013, uma
variante para a técnica CF-LIBS conhecida como Espectroscopia de Plasma Induzido
por Laser usando um Ponto para Calibração (OPC-LIBS – One Point Calibration Laser-
Induced Breakdown Spectroscopy) foi proposta por G. H. Cavalcanti e co-autores [31].

16
Essa aproximação descreve o uso de uma única amostra padrão para corrigir os erros
encontrados nos parâmetros atômicos e na curva de calibração. Essa correção é feita
antes de se iniciar a análise da amostra desconhecida, o que melhora consideravelmente
a exatidão da técnica. Depois das correções realizadas com a amostra padrão, os
procedimentos de análise com a aproximação OPC-LIBS passam a ser iguais aos da
técnica CF-LIBS e comparáveis a medidas realizadas em equipamentos comerciais de
fluorescência de raios-X (XRF – X-Ray Fluorescence) [30]. Neste trabalho, a
abordagem OPC-LIBS é desenvolvida para análise elementar dos alvos de HAP,
regularmente usados para a produção dos recobrimentos de HAP.

17
2. OBJETIVOS
O presente trabalho aborda a produção de recobrimentos de fosfatos de cálcio pela
técnica de deposição a laser pulsado (PLD) à temperatura ambiente, para serem
aplicados em implantes metálicos e poliméricos comumente utilizados em substituições
ósseas e reparos ortopédicos. Em geral, para a produção de filmes por PLD, o
comprimento de onda do laser deve ser escolhido de acordo com a faixa de absorção do
material que compõe o alvo, e assim garantir uma ablação congruente. No caso deste
trabalho, os parâmetros da técnica de PLD foram explorados na condição em que o
material do alvo, a HAP, absorve pouco no comprimento de onda do laser em 532 nm.
Portanto, um dos objetivos específicos foi observar a influência dos parâmetros da
técnica sobre as propriedades dos filmes produzidos, e assim, encontrar uma condição
ideal de produção que satisfaça condições de uso e aplicação em implantes. Além disso,
nos trabalhos comumente encontrados na literatura, tratamentos térmicos durante e/ou
após a produção dos filmes de HAP são considerados essenciais para garantir a
cristalinidade dos filmes obtidos. Por outro lado, a temperatura das espécies do plasma
também fornece uma quantidade de energia que pode contribuir no crescimento,
nucleação e formação dos filmes.
Outros objetivos específicos desse trabalho podem ser citados como: analisar as
propriedades dos filmes produzidos através de técnicas de caracterização estrutural,
morfológica e físico-química; investigar a possibilidade de produção sobre diferentes
tipos de superfícies de interesse biológico, como por exemplo, substratos metálicos (Ti
e ligas de Ti) e poliméricos sensíveis à temperatura (ácido polilático); além de verificar
a propriedade de adesão dos recobrimentos produzidos à temperatura ambiente às
diferentes superfícies, sendo esta uma característica muito importante para a aplicação
proposta.
De forma complementar a produção e caracterização dos recobrimentos, os alvos de
HAP produzidos e utilizados neste trabalho devem ter sua composição validada a fim de
garantir a formação de filmes estequiométricos, pois a transferência de estequiometria é
uma das principais vantagens na produção de filmes pela técnica de PLD. Entretanto, a
análise da composição química dos alvos não pode ser feita por técnicas convencionais
que envolvem procedimentos destrutivos de preparação e digestão da amostra. Nesse

18
contexto, o presente trabalho também tem como objetivo investigar a relação Ca/P dos
alvos de HAP com uma variante recente da técnica LIBS, a técnica de espectroscopia
óptica OPC-LIBS, através de equações que descrevem as propriedades de plasmas
produzidos por laser. Nesse caso, o controle sobre a estequiometria e a relação Ca/P dos
alvos de HAP deve ser estabelecido de modo não destrutivo, de forma que as amostras
possam ser usadas posteriormente para ablação por PLD. A partir desse propósito, a
técnica OPC-LIBS foi aplicada e seus resultados foram validados por comparação com
técnicas convencionais de análise elementar (e que exigem digestão parcial da amostra)
como as técnicas de AAS e XRF.

19
3. REVISÃO BIBLIOGRÁFICA
3.1. Biomateriais e Hidroxiapatita
A característica de biocompatibilidade pode ser descrita como a habilidade de um
material desempenhar satisfatoriamente uma desejada função em uma aplicação
biológica, sem provocar efeito sistêmico ou local indesejado [32]. Em geral, os
biomateriais podem ser divididos em três classes primárias: (i) materiais bioativos, que
são biocompatíveis e se aderem facilmente aos tecidos do corpo formando ligações
químicas e biológicas logo nos estágios iniciais após a implantação. Essa classe
compreende a HAP sintética e os vidros cerâmicos. (ii) Os materiais bioinertes, que
embora sejam biocompatíveis, não induzem ligações químicas na interface entre o
tecido e o implante. Eles também podem exibir adesão e proliferação celular, porém em
um grau muito menor do que os materiais bioativos. Exemplos dessa classe de materiais
são os implantes compostos de aço inoxidável, zircônia e ligas de titânio. (iii) Os
materiais biodegradáveis, que se degradam gradativamente após a implantação e são
substituídos por novos tecidos. Exemplos comuns incluem o fosfato tricálcico e
polímeros sintéticos biodegradáveis como o PLA (ácido polilático) e o PGA (ácido
poliglicólico) [32].
A Hidroxiapatita (HAP) é um fosfato de cálcio hidratado cristalino, sua composição
química e estrutura cristalográfica são semelhantes à fase mineral dos ossos e dentes
(aproximadamente 70% em volume). Dentre todos os fosfatos de cálcio, a HAP é a mais
estável e menos solúvel em meio aquoso [33]. A solubilidade e estabilidade térmica da
HAP dependem fortemente da sua composição e da incorporação de impurezas por
substituições iônicas e catiônicas na sua estrutura [34, 35]. A decomposição da estrutura
em β e α tricálcio fosfato (TCP) ocorre em temperaturas muito acima de 1100 ºC para
uma HAP estequiométrica (Ca/P=1,67) e em temperaturas inferiores para HAPs com
deficiência em cálcio. O processo de sinterização da HAP e sua transformação em
material cerâmico ocorre a partir de 900 ºC, com a redução da área específica e
porosidade e formação de estrutura granular [34].
A hidroxiapatita HAP, de fórmula unitária Ca10(PO4)6(OH)2, cristaliza no sistema
hexagonal, grupo espacial P63/m com parâmetros de rede: a = b = 0,9432 nm e c =
0,6881 nm. A célula unitária da HAP é formada por dez íons de cálcio Ca2+, seis íons de

20
fosfato PO43- e dois íons de hidroxila OH-. Os íons Ca2+ estão dispostos em dois sítios:
quatro íons no sítio do Ca I, e seis íons no sítio do Ca II. Os 24 íons de oxigênio ocupam
os sítios OI, OII e OIII na estrutura da HAP. Os 6 íons de fosfato e o grupo OH- ocupam
um sítio cada [34]. Considerando os diferentes sítios onde estão o Ca e o O, a fórmula
pode ser reescrita como [35]:
Ca(I)4Ca(II)6[PO(I)O(II)O(III)2](OHH)2 (3.1)
O arranjo desses átomos nos planos perpendiculares ao eixo c é mostrado na Figura
3.1(a). Ao longo das colunas paralelas ao eixo c, estão os íons hidroxila (eixo OH) e os
4 íons de Ca nos sítios CaI (eixo CaI). Cada CaI ao longo do eixo é conectado com o
outro CaI vizinho acima e abaixo, através de 3 átomos de O compartilhados, com sítios
de OI de um lado e sítios OII de outro (Figura 3.1b). Os outros 6 átomos de CaII são
associados com os grupos OH, e formam coordenações com 1 átomo de OII, 4 átomos
de OIII e 1 de OH. O P tem coordenação com OI, OII e 2OIII formando a estrutura do
fosfato PO4, que por sua vez está coordenado com 2 ou 3 átomos de Ca [35, 36].
Figura 3.1: (a) Estrutura hexagonal da célula unitária da HAP no plano ab e (b) colunas de CaI
ligadas ao OI e OII ao longo do eixo c [36].
A característica específica da HAP é que os íons OH- formam canais internos ao
longo do eixo c. Os grupos OH também podem ser substituídos por cloreto ou carbonato

21
(Ca5(PO4)3(Cl,CO3)), levando à formação de outras formas de apatita com mais alta
solubilidade [37]. A hidroxiapatita biológica, presente em tecidos ósseos naturalmente
mineralizados, contém íons carbonato (CO3)2-, sendo metade deles adsorvidos na
superfície do cristal e a outra metade presente no volume, nas posições do (PO4)3- e do
OH-. A substituição é do tipo A quando os íons carbonato substituem parcialmente os
íons hidroxila, e do tipo B quando a substituição ocorre no sítio do fosfato [38]. A
hidroxiapatita biológica é principalmente do tipo B com uma pequena quantidade do
tipo A [32].
3.2. Deposição a laser pulsado (PLD – Pulsed Laser
Deposition)
3.2.1. Fundamentos da técnica de PLD
A técnica de deposição a laser pulsado (PLD) é considerada simples do ponto de
vista experimental. O sistema de PLD consiste de dois suportes, um para o alvo e outro
para o substrato, e ambos localizados dentro de uma câmara de vácuo. Um laser de alta
potência é usado como uma fonte de energia externa à câmara de deposição. Um
conjunto de componentes ópticos é usado para focar o feixe laser sobre a superfície do
alvo. Para a entrada do feixe na câmara, é usada uma janela com alta transmitância para
todos os comprimentos de onda. O suporte do alvo deve ser acoplado a um sistema de
rotação a fim de evitar formação de cratera com a incidência de pulsos sucessivos, e o
suporte do substrato pode ser aquecido resistivamente para permitir melhor difusão do
material depositado no substrato. Dependendo dos parâmetros do sistema, o
recobrimento pode ter a mesma composição do alvo. A Figura 3.2 apresenta um
diagrama esquemático de montagem experimental para um sistema de PLD.
A deposição a laser pulsado é um processo de deposição provocado a partir da
interação de um pulso laser com um alvo e envolve fenômenos físicos complexos. O
pulso laser com alta densidade de energia é absorvido por um pequeno volume da
superfície do alvo. Devido à forte interação, ocorre ejeção de material em direção
perpendicular à superfície do alvo. Cada pulso emitido vaporiza e sublima uma pequena
quantidade de material criando uma pluma de plasma (Figura 3.3). O plasma geralmente

22
é formado por átomos, íons, moléculas, além de fragmentos do alvo. Em seguida, o
plasma se expande e se condensa no substrato produzindo uma camada fina de
recobrimento.
Figura 3.2: Esquema de montagem experimental para um sistema de PLD.
O primeiro experimento de PLD foi realizado por Smith e Turner em 1965 com um
laser de rubi, e o desenvolvimento da técnica foi impulsionado pela descoberta e
evolução dos lasers [14]. Atualmente, o PLD é usado na deposição de óxidos,
semicondutores, metais, polímeros e materiais biológicos, e inclusive na produção de
nanopartículas em vácuo ou em meio líquido [32]. A simplicidade e flexibilidade do
PLD oferecem um grande número de possibilidades de modificar as condições
experimentais. Por esse motivo, são diversos os fatores que influenciam a formação e as
propriedades dos filmes produzidos por PLD, como por exemplo: as características do
material do alvo, os parâmetros do laser (que incluem comprimento de onda, frequência,
energia e largura de pulso), a área de interação, a geometria do sistema, a distância alvo-
substrato, a atmosfera no interior da câmara de ablação, o tempo de deposição e o tipo e
a temperatura do substrato. Dependendo desses parâmetros, o PLD pode fornecer tanto
uma deposição a nível atômico quanto altas taxas de deposição.

23
(a) (b) (c) (d) (e) (f)
Figura 3.3: Diagrama representativo do processo de interação de um pulso laser com a
superfície de um alvo sólido, como ocorre no método de deposição a laser pulsado em vácuo:
(a) interação do feixe, (b) absorção e aquecimento: formação de um volume de material
derretido, (c) vaporização e ejeção do material: atomização, fragmentação e sublimação, (d)
expansão do plasma, (e) ejeção e deposição do material no substrato, (f) formação da cratera no
alvo.
3.2.2. Vantagens e limitações
A versatilidade da técnica de PLD é comumente apontada como uma grande
vantagem, pois o fato de ter a fonte de energia (o laser) independente da câmara de
ablação permite escolher diferentes configurações para o sistema. Ele também pode ser
operado em conjunto com outras fontes de evaporação em uma abordagem híbrida.
Outra vantagem normalmente destacada como uma das mais importantes é a capacidade
de transferir a estequiometria do alvo ao recobrimento, inclusive para materiais com
estequiometria complexa. Além disso, o PLD pode ser utilizado com atmosfera de gás
reativo no interior da câmara de ablação. O gás reativo pode ser usado para participar
ativamente da composição química do filme depositado ou para reduzir a energia
cinética das espécies do plasma. Geralmente são usados gases inertes ou gases que tem
grande influência na formação do filme, como no caso de oxigênio para produção de
filmes finos de óxidos. O tipo e a quantidade de gás constituem alternativas que podem
ser modificadas para produzir uma fase desejada do recobrimento [14, 32].
Algumas características apontadas como limitações da técnica de PLD são a não
uniformidade na espessura do recobrimento produzido e a formação de aglomerados e
gotículas no filme. No PLD, a distribuição de espessura dos filmes é não uniforme

24
devido à natureza de transferência de material da pluma de plasma, mas é simétrica com
relação à normal da superfície do alvo e pode ser descrita em termos de uma
distribuição cosn(θ). Entretanto, várias alternativas são propostas para produzir um PLD
de grande área e permitir o seu uso em larga escala. Para citar alguns exemplos, o
sistema de ablação pode ser adaptado para permitir uma varredura do feixe no alvo.
Outra alternativa é manter o feixe fixo e configurar um movimento de translação em
eixos ortogonais x e y para o alvo ou para o substrato [14]. A questão dos aglomerados
é abordada com mais detalhes nas Seções 3.2.4 e 3.2.5.
3.2.3. Interação da radiação laser com o alvo: processos
térmicos e hipertérmicos
No processo de ablação a laser, ocorre uma rápida interação do pulso laser com
o alvo. Isso envolve mecanismos de absorção do laser, de dissipação da energia e a
consequencia no material do alvo depois da irradiação. Todo esse processo depende da
duração do pulso laser, e a abordagem é baseada em escalas de tempo de interação. A
resposta do material à interação com o feixe laser pode ocorrer em dois regimes [39]:
1- Regime térmico, que envolve a abordagem clássica de equilíbrio termodinâmico:
aquecimento, derretimento, e consequentemente a evaporação térmica.
2- Regime hipertérmico, que envolve processos fora do equilíbrio termodinâmico:
superaquecimento, explosão de fase e instabilidades entre a superfície e o volume do
alvo.
Em processos térmicos o material pode ser analisado como um meio contínuo
que, sob absorção da energia do pulso laser ∆E, sofre um aumento da sua energia
interna ∆U dependente da capacidade térmica c e da massa m do alvo: ∆E=∆U=mc∆T.
O aumento da temperatura ∆T é provocado pelas vibrações e quebra das ligações entre
átomos e moléculas e favorece as transições de fase do material.
Do ponto de vista atômico, a temperatura é uma medida da energia cinética
média dos constituintes do alvo. Depois da primeira excitação por absorção de um
fóton, ocorrem muitas colisões de elétrons na rede até que o equilíbrio térmico por
colisões seja estabelecido. Isso ocorre em escalas de tempos típicos de 10 a 100 ps. A
incidência do laser aumenta a energia dos elétrons do alvo, que por sua vez equilibram
as colisões elétron-elétron. As colisões elétron-fônon transferem a energia para a rede,

25
que equilibra as colisões fônon-fônon. Geralmente, essa transferência de energia dos
elétrons para a rede ocorre em escala de tempo muito maior do que a transferência de
energia entre elétrons, ou seja, é um processo muito mais lento. Essa abordagem é
descrita por equações acopladas no modelo de duas temperaturas que consideram a
constante de acoplamento elétron-fônon, e as constantes de capacidade e condutividade
térmica para os elétrons e para a rede do cristal. Nessas equações, as condutividades
térmicas não são consideradas constantes, mas dependem das temperaturas do elétron e
da rede [39].
Do ponto de vista clássico, a transferência de calor e consequentemente a fusão e
evaporação, começam na superfície e é dissipado por difusão térmica no volume do
alvo. A excitação acontece no volume de absorção, que é dado pela área de seção
transversal do feixe laser e a profundidade de penetração. No entanto, o volume de
absorção não corresponde ao volume de ablação, porque nem toda a energia absorvida é
usada para evaporar o material. Por isso, a transferência de energia dos elétrons para a
rede deve ser considerada em mais detalhes. A energia absorvida pelos elétrons é
localmente transferida para os fônons no volume de absorção. A excitação por fônon é
dissipada para o volume do alvo por colisões fônon-fônon, aquecendo um volume cada
vez maior por difusão térmica, que é dada como um gradiente da temperatura [39].
No PLD, a ablação vai vaporizar a superfície e depositar o filme fino a partir de
um valor limiar para a densidade de potência do laser. Dizemos que ocorre evaporação
congruente se o volume aquecido, com comprimento de difusão térmica L, é menor ou
igual à espessura da camada ablacionada por cada pulso. O comprimento de difusão
térmica L, na direção perpendicular à área de interação do feixe, é dado por [14]:
𝐿[𝑚] = 2√𝐷[𝑚2 𝑠⁄ ] × 𝜏[𝑠] (3.2)
onde D é a difusividade térmica e τ é o tempo de interação da radiação no alvo,
determinado pela duração do pulso. Como a evaporação congruente ocorre para um
menor volume aquecido, essa condição pode ser obtida com uso de pulsos laser mais
curtos e favorece a conservação de estequiometria durante a transferência de massa. Um
menor comprimento de onda também fornece menor profundidade de absorção e
redução dos aglomerados [14].

26
Esse modelo assume que existe um tempo entre o início da absorção laser e a
vaporização e a duração desse tempo é determinado pela taxa de difusão térmica até que
uma temperatura critica seja atingida. Esse é o modelo mais simples para descrever
interações laser-sólido baseado no regime térmico, no entanto não é o que acontece em
PLD com alta densidade de potência da radiação laser (tipicamente, acima de 108
W/cm2) [14]. No PLD, essas escalas de tempo dependem muito da duração e
intensidade do pulso laser. Sob alta potência do laser, a forte excitação pode fornecer
uma alta energia cinética aos elétrons, que penetram no volume do alvo antes que sua
energia seja transferida ao fônon e depois convertida em calor. Eles são chamados de
elétrons balísticos, pois não perdem sua energia cinética na superfície, mas podem
penetrar no volume do alvo. Essa energia fica confinada numa região mais profunda do
alvo e rapidamente surge um volume de alta energia envolvido por uma rede fria, que
significa um gradiente brusco em termos de ordem atômica (Figura 3.4). O aumento de
pressão, por sua vez, aumenta a temperatura e ocorre um superaquecimento e liberação
de energia de forma explosiva. Embora esse processo possa ser explicado por princípios
termodinâmicos, ele não está em equilíbrio termodinâmico. Esse é um típico processo
hipertérmico, de situações que ocorrem em um tempo mais curto do que o tempo
necessário para o equilíbrio. Os processos hipertérmicos são caracterizados por rápida
acumulação de energia em um volume cercado por uma região mais fria, e a relaxação é
rápida e provocada de modo explosivo. Eles são observados para ablação com lasers
ultracurtos (<ps) ou com muito alta energia [39].
Figura 3.4: Diagrama representativo de um volume de alta energia confinado em uma rede
mais fria, típico de um processo em regime hipertérmico [39].
Transferência de energia
dentro do volume do alvo

27
3.2.4. Formação dos aglomerados na produção de filmes
finos por PLD
No PLD, a pluma de plasma consiste de uma mistura de constituintes altamente
energéticos, incluindo átomos, moléculas, elétrons, íons, aglomerados sólidos de
tamanho micrométrico e aglomerados fundidos também chamados de gotículas. A
ocorrência dos aglomerados e gotículas sobre os filmes produzidos por PLD é uma
característica intrínseca do processo e foi observada desde os primeiros experimentos.
Por muito tempo, ela foi considerada como um grande problema e apontada como a
causa que impedia o desenvolvimento da técnica. Os aglomerados sólidos ou fundidos
podem induzir a formação de defeitos durante o crescimento dos filmes e provocar uma
morfologia rugosa indesejada. Existem pelo menos três mecanismos que, combinados
ou não, causam a formação e deposição dos aglomerados nos substratos [14]:
1 – Aquecimento em subcamada do alvo: ocorre se o tempo necessário para
transferir a energia do laser em calor é mais curto do que o necessário para evaporar
uma camada da superfície. Nessa condição, uma camada abaixo da superfície é
superaquecida antes da superfície atingir a fase vapor. Quando a camada superficial
vaporiza, micro gotículas da subcamada do alvo são expelidas no substrato.
2 – Pressão de recuo de uma onda de choque: Uma onda de choque provocada
pela pluma de plasma exerce uma pressão de recuo e expulsa uma camada aquecida da
superfície do substrato. Esse mecanismo também forma gotículas micrométricas que se
condensam no substrato.
3 – Esfoliação: Os aglomerados formados por esfoliação são expelidos do alvo em
formato sólido. A taxa de ejeção e o tamanho deles dependem tanto da densidade de
potência do laser quanto da morfologia de superfície do alvo. Para a maioria dos
materiais, e também para alvos cerâmicos sinterizados, como é o caso do alvo de HAP,
a superfície é desgastada pelo acúmulo dos pulsos e pode formar microestruturas de
formato alongado nos alvos. Esses detritos podem ser novamente quebrados, ejetados e
embutidos na superfície do filme.
Dada a flexibilidade do sistema de PLD, muitas abordagens são propostas para
melhorar a morfologia dos filmes reduzindo ou eliminando completamente a ocorrência
dos aglomerados, como por exemplo: por uso de filtro mecânico, por manipulação da

28
geometria de interação entre o feixe e o alvo, aumentando a densidade e superfície do
alvo, e outros [14].
3.2.5. Nucleação e crescimento dos filmes sob influência de
processos hipertérmicos
Em regime de alto vácuo, devido à expansão da pluma de plasma, a cinética de
nucleação e crescimento dos filmes, a evolução da sua microestrutura e as suas
propriedades são altamente dependentes de características relacionadas às espécies que
chegam ao substrato. Essas características são a taxa de deposição e o tipo e energia das
partículas condensadas no plasma durante a ablação. Imediatamente após a ablação, as
partículas do plasma tem alta energia cinética e podem influenciar muito na formação e
crescimento do filme.
As interações entre essas partículas e os sólidos (substratos) que controlam as
propriedades dos filmes estão resumidas na Figura 3.5. Essas partículas representam
qualquer tipo de espécie atômica que atinge o substrato, como átomos neutros, íons e
pequenos aglomerados. Geralmente, as partículas térmicas são aquelas que possuem
energia abaixo de 1 eV, enquanto que aquelas que tem energia maior do que 1 keV são
os íons acelerados. No intervalo entre elas, na faixa de energia de 1 eV a 1 keV, estão as
partículas hipertérmicas e podem ser consideradas as mais típicas em processos de PLD.
Alguns dos efeitos provocados por partículas hipertérmicas sobre os filmes produzidos
por PLD são [32]:
1- Pré-limpeza de superfície
Energias típicas de desorção física são < 1 eV e de desorção química são de 1-10
eV. O bombardeamento de partículas com essas energias pode remover contaminantes
da superfície e aumentar a adesão do filme no substrato.
2- Crescimento do filme em camadas
O colapso de aglomerados instáveis com energia 0,1 eV e o deslocamento dos
átomos da superfície por colisões balísticas a 10 eV aumentam a mobilidade de átomos
adsorvidos, podendo interromper o crescimento por ilhas e favorecer o crescimento do

29
filme em camadas. Como conseqüência, pode evitar a absorção de moléculas de gases e
água entre as colunas.
3- Pulverização
O limiar de energia das partículas para ejeção de átomos do alvo é inferior a 30
eV para a maioria dos materiais. A pulverização de espécies fracamente ligadas ao filme
contribui para formação de uma microestrutura mais densa. Além disso, o material
retroespalhado não tem direção de deposição e podem preencher alguns vales que se
formam pela ausência de bombardeamento de íons. Esse efeito pode conduzir a
mudanças na composição do filme.
4- Defeitos estruturais
O bombardeamento de íons pode formar defeitos estruturais na superfície e no
volume, afetando a microestrutura dos filmes em crescimento.
5- Implantação
As energias de deslocamentos de átomos em metais são da ordem de 20 eV. Para
um íon com energia mais alta do que a de energia do deslocamento, eles penetram cada
vez mais profundamente no substrato. Isso pode provocar uma nucleação com alta
densidade e implantação de mistura de partículas incidentes. Como conseqüência, pode
aumentar a adesão do filme no substrato.
6- Modo de crescimento do filme por implantação superficial
Em vez de serem depositadas na superfície do substrato, as partículas
hipertérmicas exibem implantação superficial, ou seja, em poucas camadas inferiores à
superfície do material. Como o coeficiente de difusão do volume é bem menor do que o
da camada superficial, não pode ocorrer redistribuição significativa dos átomos
implantados, em comparação com os átomos na superfície.

30
Figura 3.5: Intervalos de energia para interações entre partículas e sólidos que influenciam mais
significativamente as propriedades de filmes finos [32].
Muitos fenômenos induzidos por partículas energéticas podem ocorrer no
intervalo de energia hipertérmica (1 a 100 eV). O plasma produzido por PLD não
reativo sob alto vácuo, como o produzido neste trabalho para obter os filmes de HAP,
representa uma fonte de partículas energéticas na região de energia hipertérmica.
Portanto, sob condições que permitem a formação dos aglomerados, o PLD pode ser
considerado uma técnica de deposição por feixe de partículas hipertérmicas, que
favorecem o crescimento e nucleação dos filmes.
3.2.6. PLD aplicado a biomateriais
O uso da técnica de PLD para produção de recobrimentos biocompatíveis teve
início com o trabalho de Cottel em 1992. Desde então muitos trabalhos de PLD com
HAP produziram resultados importantes e recobrimentos sobre diversos tipos de
substratos. Atualmente, na área de biomateriais, o PLD é utilizado com gases inertes,
oxigênio, água ou misturas deles, e também com óxido nitroso N2O que é mais reativo
do que o oxigênio. A espessura típica de um recobrimento é menor do que 1 μm, e a

31
morfologia da superfície pode ser rugosa ou não, uma vez que é fortemente dependente
dos parâmetros de deposição e das propriedades do alvo [32].
As principais vantagens associadas à produção de recobrimentos por PLD são: a
capacidade de crescer filmes de HAP puros, com alta densidade e cristalinidade, com
estequiometria apropriada, razão Ca/P bem controlada, e com excelente aderência e por
ser um processo limpo que possibilita controle de temperatura durante o processamento.
Além disso, também possui flexibilidade para controlar a morfologia, fase,
cristalinidade e composição química de outros componentes de CaP. Como essas
características influenciam principalmente as propriedades biológicas, tais como a
bioabsorção (ou dissolução envolvida na osseointegração dos recobrimentos), o PLD
pode ser usado para fazer biodispositivos em multicamadas, consistindo de diferentes
materiais nanoestruturados. Além disso, cerâmicas baseado em HAP nanocristalina
(com pequenos tamanhos de grãos) podem apresentar maior resistência e tenacidade, e
coeficientes de expansão térmica que praticamente correspondem aos do substrato (Si
ou liga de Ti), devido à grande fração de volume de átomos situados nos limites de grão
[40]. As principais limitações ainda estão na deposição em grande área e na
uniformidade de espessura.
Os parâmetros críticos da técnica para a produção de filmes de CaP são o
comprimento de onda do laser, a fluência e a duração do pulso. Os mais diversos tipos
de laser com diferentes comprimentos de onda já foram usados para produção desses
filmes. No entanto, poucos trabalhos abordaram deposições com fluência acima de 10
J/cm2 [23,41-43]. Além disso, a maioria utilizou tratamentos térmicos durante e/ou após
a deposição. Para os trabalhos produzidos à temperatura ambiente e sem tratamento
térmico após a deposição, os recobrimentos não apresentaram a cristalinidade desejada.
Abordagens híbridas usando dois feixes laser também já foram testadas. O método de
PLD ainda é considerado eficiente e muitos trabalhos nessa área estão concentrados no
uso do laser de fs.
O comportamento biológico de um implante é fortemente influenciado por sua
química de superfície, que pode ser ajustada através das propriedades dos filmes. Com
recentes resultados na área de osseointegração, muitos esforços estão sendo conduzidos
em direção de uma nova abordagem do PLD que oferece deposição simultânea de
material orgânico e inorgânico. Uma nova geração de recobrimentos bioativos são
produzidos por evaporação de matriz assistida por laser pulsado (MAPLE – Matrix-
Assisted Pulsed Laser Evaporation). Nessa abordagem, o feixe laser interage com um

32
alvo congelado. O alvo é uma matriz que contém moléculas de proteínas em solução
(por exemplo, um solvente volátil) e é mantido congelado através de um sistema de
refrigeração com nitrogênio líquido. O alvo absorve a energia do feixe UV e o vapor da
solução transporta as proteínas em direção ao substrato. As moléculas da solução são
eliminadas pelo sistema de vácuo, enquanto os aglomerados de proteínas e sais são
transferidos e se condensam no substrato, formando um filme fino. Geralmente a
temperatura do substrato é mantida a 30°C para melhor adesão das moléculas e para
evaporar as moléculas de água que podem atingir o substrato. Um esquema desse
processo pode ser observado na Figura 3.6(a) MAPLE e na Figura 3.6(b) C-MAPLE
(Combinatorial MAPLE), uma configuração que usa dois lasers para ablação de dois
alvos concêntricos. Essa técnica pode ser usada para deposição de drogas, enzimas,
citocinas e substâncias farmacêuticas com atividade específica em dispositivos
biomédicos, para estimulação direcionada e/ou cicatrização de micro sítios lesionados
[44].
Figura 3.6: Novas abordagens para uso do PLD na área de biomateriais: (a) MAPLE e (b) C-
MAPLE (combinatorial laser evaporation) para deposição simultânea de biopolímeros e
proteínas [45].
3.3. Análise da estequiometria dos alvos de HAP por
espectroscopia de plasma induzido por laser
Nesta seção serão apresentados os princípios básicos da análise quantitativa
elementar com a técnica LIBS (Laser Induced Breakdown Spectroscopy) e suas

33
variantes CF-LIBS (Calibration Free – LIBS) e OPC-LIBS (One Point Calibration –
LIBS). A metodologia de análise elementar por OPC-LIBS foi utilizada para cálculo da
razão estequiométrica de Ca e P presente nos alvos de HAP.
3.3.1. Princípios básicos da análise
A técnica LIBS é um método de diagnóstico elementar a partir da análise da radiação
emitida pelo plasma que se origina da interação de um feixe laser intenso com a
superfície da amostra. Essa técnica é essencialmente rápida e não destrutiva. Ela não
exige a preparação da amostra, que pode ter qualquer formato e estar em estado sólido,
líquido ou gasoso.
Uma análise padrão com a técnica LIBS é realizada através de curvas de calibração
obtidas de amostras de referência com composição conhecida. Isso só é possível quando
a intensidade da radiação emitida por um elemento atômico apresenta uma resposta
linear à concentração do elemento na amostra. Essa relação de linearidade nem sempre é
satisfeita, devido à propriedade de transitoriedade do plasma. Em alguns casos, é difícil
quantificar os componentes de mais alta concentração por causa do chamado efeito-
matriz, que são fenômenos não lineares induzidos na intensidade da linha espectral
devido a natureza da amostra. Outro fenômeno que representa um problema na análise
LIBS é a auto-absorção, que ocorre quando os fótons emitidos pelas espécies atômicas
são absorvidos por outros átomos da mesma espécie no plasma. Isso altera a intensidade
e o perfil da linha espectral, o que influencia diretamente a determinação da temperatura
do plasma e dificulta a análise quantitativa dos elementos principais. Vários métodos
foram criados para avaliar e corrigir o efeito da auto-absorção [46-49]. Dessa forma, em
uma análise com a técnica LIBS, deve-se selecionar com precisão as linhas de emissão a
serem analisadas. Alguns critérios de seleção foram propostos na literatura e são
brevemente comentados abaixo [50]:
Todas as linhas que envolvem transição para o estado fundamental (linhas
ressonantes) devem ser excluídas, pois são as mais afetadas pelos fenômenos de
auto-absorção.
Todas as linhas do espectro que apresentem mais alta intensidade quando
comparada às demais devem ser consideradas com certo cuidado.

34
Todas as linhas com probabilidade de transição inferior a 2×106 s-1 não devem
ser consideradas para análise quantitativa, uma vez que os correspondentes
tempos de emissão podem ser comparáveis ao tempo em que ocorre variações no
plasma [51].
A escolha e análise das linhas espectrais devem ser realizadas com muito cuidado,
evitando alguns fenômenos não lineares pertinentes ao plasma produzido por laser. Em
geral, três aspectos fundamentais devem ser verificados sempre que for realizada uma
análise quantitativa ou semi-quantitativa com a técnica LIBS:
1- Ablação estequiométrica e homogeneidade do plasma
Durante a ablação, podem ocorrer efeitos de fracionamento de alguns elementos em
função da diferença do ponto de fusão e vaporização dos elementos que compõem o
material analisado. Isso depende muito da matriz em estudo e da escolha dos parâmetros
de ablação. Para minimizar esses efeitos e realizar uma análise confiável, o processo de
remoção de material do alvo para formar o plasma deve ser de forma explosiva
(ablação), na qual a temperatura de vaporização da superfície é ultrapassada dentro de
uma fração do tempo de duração do pulso laser. Antes que a superfície irradiada possa
vaporizar, o material subjacente se aquece e alcança a temperatura de sublimação. O
aumento da pressão no pequeno volume irradiado provoca uma explosão sobre a
superfície do alvo, a expulsão de material e a formação da pluma de plasma com a
mesma composição química do alvo. Sendo assim, a estequiometria do plasma
representa a composição da amostra. A definição apropriada dos parâmetros do laser
como fluência, comprimento de onda e duração do pulso, é essencial para garantir uma
ablação estequiométrica [52][53].
Por outro lado, a homogeneidade do plasma é uma questão mais complexa. De fato,
os gradientes espaciais e temporais de temperatura e densidade surgem no plasma
devido à sua dinâmica rápida e sua interação com o meio em que foi formado. As zonas
periféricas do plasma, mais frias, apresentam um aumento dos processos de
recombinação entre íons e elétrons. Como consequência, podem surgir diferenças entre
os parâmetros físicos das zonas externa e interna do plasma, mas esses efeitos podem
ser contornados através da escolha dos tempos de aquisição do sinal óptico [52].

35
2- Equilíbrio Termodinâmico Local
Para um sistema em equilíbrio termodinâmico, uma mesma temperatura T é válida
para todas as leis de distribuição: de Planck, de população de Boltzmann, de
velocidades de Maxwell, e a equação de Saha, que descreve o equilíbrio químico do
plasma. Um plasma produzido por laser é dito ser transiente no tempo, ou seja, após
receber a energia de um pulso, ele é criado em poucos nanossegundos e evolui em
alguns microssegundos até se apagar. Devido a essa natureza transitória, o plasma
manifesta um desvio do equilíbrio termodinâmico decorrente da perda de radiação
através da emissão espectral e de sua expansão. Com isso, a temperatura e a densidade
eletrônicas do plasma gerado por laser também evoluem com o tempo. Se, no plasma,
os processos colisionais são mais importantes que os radiativos, ou seja, se pode
desprezar a perda por radiação, o plasma é dito estar em equilíbrio termodinâmico local
(LTE) [52,54,55]. Nessa condição, se pode considerar que as partículas estão
localmente e instantaneamente em equilíbrio que é estabelecido num tempo muito curto
quando comparado ao tempo de expansão do plasma. Um plasma transiente pode ser
modelado como sendo estacionário ao ser observado depois de decorrido um tempo
suficiente em que sua evolução é mais lenta (≈ 1 μs), com uma janela temporal pequena
o suficiente (≈ 200 ns) para que a perda de energia seja pequena (Figura 3.7). Nesse
estado, digamos quase “estacionário”, a temperatura eletrônica Te e a densidade
eletrônica ne não devem variar apreciavelmente dentro do intervalo de tempo ∆t, ou
seja:
∆𝑇𝑒
∆𝑡≈ 0 𝑒
∆𝑛𝑒
∆𝑡≈ 0. (3.3)
Em um plasma no estado de equilíbrio termodinâmico local (LTE), os processos de
colisão dominam sobre os processos radiativos, ou seja, um estado excitado tem maior
probabilidade de desexcitação por colisão do que por radiação espontânea. As
populações e velocidades ainda estão descritas pelas relações de equilíbrio, embora a
radiação não seja mais descrita como de corpo negro. Nesse caso, é possível encontrar
uma temperatura T que satisfaça as relações de equilíbrio descritas pelas equações de
Maxwell, Boltzmann e Saha.

36
Figura 3.7: Diagrama representativo da evolução do plasma no tempo.
No estado de LTE, deve existir uma densidade mínima de elétrons com maior
mobilidade, e as colisões desses elétrons com os átomos e íons do plasma são as
responsáveis pela distribuição da energia no interior do plasma. O valor para essa
densidade de elétrons, ne, pode ser calculado pelo critério de McWhirter dado pela
equação [55, 56]:
𝑛𝑒(𝑐𝑚−3) ≥ 1,6 × 1012 √𝑇(𝐾)[∆𝐸𝑖𝑗(𝑒𝑉)]3
, (3.4)
onde T é a temperatura do plasma e ΔEij é o intervalo de energia entre os níveis superior
i e inferior j envolvidos na transição. O critério de McWhirter é apenas uma condição
necessária e não suficiente para o LTE, mas seu uso é amplamente documentado [55,
57].
3- Plasma opticamente fino
Durante a ablação, o material removido do alvo se expande em direção ortogonal ao
alvo e o plasma evolui com o passar do tempo. No início da expansão, o plasma é
altamente denso, emite uma radiação contínua e se encontra fora do equilíbrio. Logo
depois, o plasma que está evoluindo passa para um regime de equilíbrio colisional com
baixa absorção de radiação, onde a maior parte de sua energia é distribuída entre seus
átomos e íons pelas colisões com os elétrons livres. Nesse momento o plasma se torna
opticamente fino e pode ser descrito por equações de equilíbrio termodinâmico. Um
plasma é opticamente fino quando ele é transparente à sua própria radiação, ou seja, a

37
radiação emitida pelas espécies no plasma o atravessa e escapa sem experimentar uma
absorção ou dispersão significativa [52, 57, 58].
Quando um plasma possui alta densidade, ele absorve suas próprias linhas de
emissão, e essa auto-absorção tem um efeito negativo no cálculo dos parâmetros do
plasma. Esse efeito é conduzido por uma distorção no perfil e na intensidade da linha
espectral, aumentando a largura da linha e diminuindo sua altura. Quando isso ocorre
com uma linha espectral, dizemos que o plasma é opticamente espesso a esse
determinado comprimento de onda. O fenômeno da auto-absorção geralmente afeta as
linhas que possuem valores altos de probabilidade de transição e as linhas ressonantes,
linhas que estão ligadas ao estado fundamental e possuem baixa energia de excitação.
Isso ocorre particularmente para elementos que apresentam uma alta concentração no
plasma. Nesse trabalho, a presença de auto-absorção nas linhas é muito pequena e é
corrigida pelo emprego do modelo OPC-LIBS, como será visto na Seção 3.3.3.
3.3.2. Análise quantitativa por CF-LIBS
A intensidade integrada Iij de uma linha de emissão óptica de comprimento de onda
ij está relacionado ao número de portadores no nível superior ou estado excitado i, ni,
por:
𝐼𝑖𝑗 = 𝐹𝑛𝑖𝐴𝑖𝑗 , (3.5)
onde Aij é o coeficiente de emissão espontânea da transição considerada (probabilidade
de transição entre os níveis i e j) e F é um fator experimental que leva em conta a
eficiência óptica do sistema de coleta. Para um espectro radiométrico, esse fator é
independente do comprimento de onda [59]. Esta relação é válida apenas quando a
transição considerada, ij, é opticamente fina. Em resumo, a intensidade de uma
determinada linha de emissão é proporcional à densidade do número de emissores, que
por sua vez, é proporcional à concentração do elemento emissor presente na amostra
irradiada. Portanto, a concentração de uma dada espécie está linearmente relacionada à
intensidade da radiação emitida. Porém, dependendo da matriz da amostra, a linearidade
da reta de calibração na análise LIBS pode ser afetada pelos efeitos discutidos
anteriormente. Além disso, a reta de calibração exige a preparação de padrões

38
compatíveis com a matriz da amostra, e essa preparação pode ser inviável ou de difícil
fabricação.
Uma alternativa à abordagem LIBS é o método de calibração livre (CF-LIBS), que
ainda se baseia na suposição de que o plasma está em LTE, mas a informação
quantitativa é obtida através das equações que descrevem o plasma [52, 60]. Na
condição de LTE, a densidade do número de emissores é dada pela distribuição de
Boltzmann [56]:
𝑛𝑖 =𝑔𝑖𝑛𝑠
𝑄𝑠(𝑇)𝑒𝑥𝑝 (
−𝐸𝑖
𝑘𝑇), (3.6)
onde gi é o peso estatístico do nível excitado com energia Ei do qual ocorre a
transição, ns é a densidade numérica total da espécie s considerada, 𝑄𝑠(𝑇) =
∑ 𝑔𝑖𝑖 𝑒𝑥𝑝(−𝐸𝑖 𝑘𝑇⁄ ) é a função de partição da espécie s à temperatura T, e k é a
constante de Boltzmann. Substituindo a Equação (3.6) na Equação (3.5), temos a
relação de dependência da intensidade da linha Iij com a densidade da espécie no
plasma, nS:
𝐼𝑖𝑗 = 𝐹𝑛𝑠
𝐴𝑖𝑗𝑔𝑖
𝑄𝑠(𝑇)𝑒𝑥𝑝 (
−𝐸𝑖
𝑘𝑇), (3.7)
que pode ser reescrita na forma logarítmica:
𝑙𝑛 (𝐼𝑖𝑗
𝐴𝑖𝑗𝑔𝑖) =
−𝐸𝑖
𝑘𝑇+ 𝑙𝑛 (
𝐹𝑛𝑠
𝑄𝑠(𝑇)) (3.8)
É possível relacionar a Equação (3.8) com a equação da reta 𝑦 = −𝑎𝑥 + 𝑏𝑠,
identificando as seguintes variáveis:
𝑦 = 𝑙𝑛 (𝐼𝑖𝑗
𝐴𝑖𝑗𝑔𝑖) ; 𝑎 =
1
𝑘𝑇 ; 𝑥 = 𝐸𝑖 ; 𝑏𝑆 = 𝑙𝑛 (
𝐹𝑛𝑠
𝑄𝑠(𝑇)) (3.9)
Essa linearização é comumente conhecida como ajuste de Boltzmann, onde a
temperatura eletrônica do plasma pode ser obtida através da inclinação da reta, e a

39
concentração da espécie pode ser determinada através do coeficiente linear bS. Nesse
ajuste, todas as espécies do plasma devem ter a mesma temperatura e o diagrama de
Boltzmann para diferentes espécies de um mesmo plasma deve fornecer linhas paralelas
com a mesma inclinação e diferentes pontos de interceptação com o eixo y. Esse método
permite a determinação do fator Fns, que é obtido a partir dos valores de bS. Sendo
assim, ns, que é a concentração percentual relativa de cada espécie, pode ser obtida pela
relação [52, 60]:
𝑛𝑠 =𝑄𝑠(𝑇)𝑒𝑥𝑝(𝑏𝑠)
𝐹 (3.10)
O fator experimental F é uma constante que leva em consideração a concentração de
todas as espécies constituintes da amostra. Ele pode ser determinado pelo procedimento
de normalização para as concentrações percentuais de todas as espécies atômicas do
plasma [52].
∑ 𝑛𝑆
𝑆
=1
𝐹∑ 𝑄𝑆
𝑆
(𝑇) 𝑒𝑥𝑝(𝑏𝑆) = 100. (3.11)
Geralmente, a energia do laser aplicada nos experimentos não é suficientemente alta
para remover mais de um elétron dos átomos. Então a concentração total de um
elemento do plasma nT é dada pela soma das densidades da sua espécie neutra nI com a
espécie uma vez ionizada nII,
𝑛𝑇 = 𝑛𝐼 + 𝑛𝐼𝐼 . (3.12)
Quando são identificadas apenas as linhas espectrais atômicas, ou seja, as linhas das
espécies ionizadas não aparecem na faixa espectral observada, é possível ainda
encontrar a densidade das espécies iônicas pela equação de Saha [52]:
𝑛𝐼𝐼
𝑛𝐼=
(2𝜋𝑚𝑒𝑘𝑇)3
2⁄
𝑛𝑒ℎ3
2𝑄𝐼𝐼(𝑇)
𝑄𝐼(𝑇)𝑒𝑥𝑝 (−
𝐸𝑖𝑜𝑛
𝑘𝑇), (3.13)

40
onde me é a massa do elétron, ne é a densidade de elétrons, h é a constante de Planck,
QI(T) e QII(T) são as funções de partição das espécies neutra e iônica, respectivamente, e
Eion é a energia de ionização do elemento neutro.
A precisão na determinação da temperatura do plasma afeta fortemente os resultados
da composição da amostra obtida pelo método de CF-LIBS. Geralmente, a temperatura
do plasma é determinada através de um ajuste de Boltzmann, que fornece um valor com
grande imprecisão por ter em conta apenas as linhas de uma espécie atômica durante os
cálculos. Um método mais confiável e que apresenta uma maior precisão é o diagrama
de Saha-Boltzmann [52, 61]. Ele é obtido ao se combinar as Equações (3.8) e (3.13),
onde são consideradas, no mesmo ajuste, as linhas espectrais atômicas e iônicas. Nesse
ajuste, as coordenadas são dadas por:
𝑦 = 𝑙𝑛 (𝐼𝑖𝑗
𝐴𝑖𝑗𝑔𝑖) − 𝑧𝑙𝑛 [
2
𝑛𝑒(
2𝜋𝑚𝑒𝑘𝑇
ℎ2)
32⁄
] (3.14)
e
𝑥 = 𝐸𝑖 + 𝑧𝐸𝑖𝑜𝑛 (3.15)
onde z = 0 para átomos neutros e z = 1 para íons.
No diagrama de Saha-Boltzmann, a energia de ionização do átomo é adicionada aos
valores de energia para os níveis superiores dos íons (Equação 3.15). Assim, a distância
no eixo da energia, entre o grupo de pontos ligados aos átomos neutros e aos íons,
aumenta, afastando os pontos, e permitindo um ajuste linear mais preciso. Como o valor
da temperatura está ligado ao coeficiente angular da reta, isso acarretará na
determinação de uma temperatura com uma maior precisão. No entanto, para obter o
valor da temperatura a partir da inclinação da reta no diagrama de Saha-Boltzmann, se
faz necessário conhecer o valor da densidade do plasma ne. Como a segunda parte da
Equação (3.14) depende da temperatura, a única forma de resolvê-la é de uma forma
iterativa em que o processo é iniciado com uma temperatura inicial Te1, mais imprecisa.
Este valor inicial é obtido por um ajuste de Boltzmann. O diagrama de Saha-Boltzmann
é bem condicionado e geralmente converge em aproximadamente cinco iterações [62].
Para se realizar um diagrama de Saha-Boltzmann se faz necessário que um dentre os
elementos presentes no plasma apresente linhas de emissão atômica e iônica na faixa

41
espectral observada, e que estas linhas possuam probabilidades de transição
experimentalmente determinadas, além do conhecimento prévio da densidade eletrônica
do plasma. A densidade eletrônica do plasma é calculada a partir do alargamento Stark
associado à linha Hα [63]. Isso é possível porque o alargamento das linhas emitidas pelo
átomo de hidrogênio depende fortemente da densidade das partículas carregadas em seu
entorno [56]. A partir de uma aproximação semi-empírica, a seguinte equação foi
determinada [64]:
𝑛𝑒 ≈ 9,77 × 1022(∆𝜆𝑆𝑡𝑎𝑟𝑘𝐻𝛼 )
1,39 (3.16)
onde ∆𝜆𝑆𝑡𝑎𝑟𝑘𝐻𝛼 é a largura à meia altura (FWHM) da componente lorentziana da
transição.
Para que a abordagem CF-LIBS seja utilizada em medidas quantitativas, as
premissas mencionadas na Seção 3.3.1 (de ablação estequiométrica, de condição LTE,
de homogeneidade do plasma e de o plasma ser opticamente fino) devem ser
rigorosamente obedecidas. A precisão das medidas quantitativas com o método CF-
LIBS depende muito do conjunto de suposições apresentadas e pode fornecer apenas
resultados semi-quantitativos quando elas não são rigorosamente satisfeitas [65, 66].
Em uma recente publicação de 2016 [67], um grupo italiano fez uma comparação entre
três métodos baseados na técnica CF-LIBS. Nesse artigo, foi demonstrado que o método
de calibração por um ponto OPC-LIBS apresenta os melhores resultados analíticos.
Esse método, descrito na Seção 3.3.3, foi utilizado para obter a estequiometria dos alvos
de HAP através de uma análise quantitativa dos elementos Ca e P e posterior verificação
da relação Ca/P.
3.3.3. O método de calibração por um ponto OPC-LIBS
O ponto de partida do método OPC-LIBS é a equação de Boltzmann (Equação 3.7),
que sob a condição de LTE descreve a dependência da intensidade das linhas de
emissão com a concentração elementar da espécie analisada [31]. No modelo CF-LIBS,
o fator experimental F é identificado como uma constante de proporcionalidade entre a
intensidade medida da linha e a intensidade "real" emitida pelo plasma. Ele é
determinado através de uma calibração radiométrica e carrega a eficiência espectral do

42
sistema de detecção e sua dependência com o comprimento de onda. A abordagem de
calibração por um ponto OPC-LIBS parte da consideração de que o parâmetro F poderia
ser redefinido para incluir correções de incerteza dos parâmetros Aij, correções devido a
erros de calibração e pequenas correções de auto-absorção. Sendo assim, a definição de
um novo parâmetro P(λ) dependente da transição considerada e que carrega todas essas
correções, é determinado a partir de uma única amostra padrão de composição
conhecida e permite realizar uma análise mais precisa das amostras com composição
desconhecida.
Para determinar o parâmetro P() de uma linha de emissão, partimos da hipótese de
que a densidade de uma espécie do plasma determinada por CF-LIBS, nS, é diferente do
valor real, n´S, e a relação entre elas pode ser expressa por [31]:
𝑛𝑠′ =
𝑛𝑇′
𝑛𝑇𝑛𝑠 , (3.17)
onde nT é a densidade do elemento obtida por CF-LIBS e n´T é a densidade real do
elemento na amostra analisada. Então o termo n’T/nT é apresentado como a razão que
corrige o valor da densidade obtida por CF-LIBS do real valor da densidade da espécie
na amostra.
Se multiplicarmos os dois lados da Equação (3.7) por n’T/nT e linearizarmos em um
gráfico de Boltzmann, temos:
𝑙𝑛 (𝐼𝑖𝑗
𝐴𝑖𝑗𝑔𝑖
𝑛𝑇′
𝑛𝑇) =
−𝐸𝑖
𝑘𝑇+ 𝑙𝑛 (
𝐹𝑛𝑆
𝑄𝑠(𝑇)) + 𝑙𝑛 (
𝑛𝑇′
𝑛𝑇), (3.18)
que pode ser identificada como a equação de uma reta, y’=-ax+bs+ln(n’T/nT). Essa nova
reta é um deslocamento da primeira reta apresentada na Equação (3.8), com variação no
coeficiente linear. A diferença entre as duas retas é dada por:
𝑦′ − 𝑦 = 𝛥𝑦 = 𝑙𝑛 (𝑛𝑇
′
𝑛𝑇), (3.19)
onde y é identificado como o deslocamento do ponto 𝑦 = 𝑙𝑛(𝐼𝑖𝑗 𝐴𝑖𝑗𝑔𝑖⁄ ) para o novo
valor corrigido 𝑦′ = 𝑙𝑛(𝐼𝑖𝑗𝑛𝑇′ 𝐴𝑖𝑗𝑔𝑖𝑛𝑇⁄ ), como pode ser visto na Figura 3.8. Esse

43
deslocamento dá origem a uma nova reta, e terá um valor diferente corrigindo os erros
experimentais para cada ponto no diagrama de Boltzmann. Esse valor é usado para
determinar o fator P(λ) correspondente a cada linha espectral estudada:
𝑃(𝜆) = 𝐴𝑖𝑗𝑔𝑖𝑒𝑥𝑝(−∆𝑦). (3.20)
Portanto, encontrar os valores y com uma amostra padrão é o mesmo que
determinar o fator de correção 𝑛𝑇′ 𝑛𝑇⁄ = 𝑒𝑥𝑝(∆𝑦) para cada linha espectral das espécies
do plasma. Uma vez obtidos os valores de P(λ), as amostras desconhecidas são
analisadas por CF-LIBS convencional através de um novo gráfico de Boltzmann
produzido com os P() encontrados no lugar do giAij:
𝑙𝑛 (𝐼𝑖𝑗
𝑃(𝜆)) =
−𝐸𝑖
𝑘𝑇+ 𝑙𝑛 (
𝑐𝑡𝑒𝑛𝑠
𝑄𝑠(𝑇)). (3.21)
Na análise por OPC-LIBS, as possíveis variações de correção dos parâmetros do
plasma (temperatura, densidade, composição) são levadas em consideração e são
totalmente compensados dentro dos valores de P(λ). Como essas variações estavam
também presentes na amostra padrão, e a amostra padrão fornece um novo valor de y’,
então os resultados obtidos para as amostras desconhecidas não terão a contribuição
dessas variações.

44
Figura 3.8: Diagrama representativo para a determinação de Δy, que é a distância entre o valor
da medida realizada por CF-LIBS (ponto verde) e a reta calibrada pela amostra padrão no
método OPC-LIBS (linha azul).

45
4. MATERIAIS E MÉTODOS
4.1. Síntese da HAP
A síntese de HAP estequiométrica (Ca/P=1.67) foi produzida no Laboratório de
Materiais Biocerâmicos do CBPF, conforme procedimentos pré-estabelecidos [34]. O
material foi preparado através do método de precipitação por via úmida (T = 90 ºC e pH
= 11) a partir de soluções aquosas de nitrato de cálcio (Ca(NO3)2.4H2O) e fosfato
diamônico (NH4)2HPO4. Após 2 horas, o material precipitado foi separado por
filtragem, lavado repetidamente com água deionizada fervida (marca Milli-Q) e
submetido à secagem a 100°C durante 24 horas. Após a secagem, o pó foi manualmente
macerado e peneirado dentro de uma câmara de fluxo laminar. O material resultante
possui partículas com granulometria entre 77 µm e 210 µm.
4.2. Preparação dos alvos de HAP
Os alvos foram preparados com 5 g de HAP em pó prensada a 5,6 toneladas, em
matriz de diâmetro 3,5 cm, formando pastilhas cilíndricas de 0,4 cm de espessura. Em
seguida, essas pastilhas foram densificadas por sinterização em forno tipo mufla
Carbolite, modelo CWF1300, da Sala de Fornos do CBPF. O procedimento consistiu de
aquecimento com rampa de 3°/min, permanecendo em 700 °C por 30 minutos, seguido
de rampa de 1°/min até 1150°C por 2 horas. Em seguida, as pastilhas permaneceram por
16 horas sob a inércia de resfriamento do forno. Como resultado deste processo, foram
obtidas pastilhas cilíndricas de HAP (CaP=1,67) com diâmetro de 2,6 cm, espessura de
0,3 cm e densidade de 2,8 g/cm3.
4.3. Análise da estequiometria dos alvos de HAP
As pastilhas de HAP produzidas neste trabalho foram usadas para a produção de
filmes finos pela técnica de PLD. A característica de transferência da estequiometria do
alvo ao recobrimento é apontada como uma das principais vantagens dessa técnica. Por
esse motivo, investigar a estequiometria dos alvos é importante para garantir a

46
composição dos recobrimentos produzidos. A estrutura cristalina, composição química e
estequiometria da HAP são parâmetros controlados durante a síntese do pó, porém eles
podem mudar durante a prensagem e sinterização da pastilha. Assim, a estequiometria
das pastilhas já preparadas foi analisada de forma rápida e não destrutiva através de uma
variante da técnica CF-LIBS (Calibration Free - Laser Induced Breakdown
Spectroscopy), conhecida como OPC-LIBS (One Point Calibration - Laser Induced
Breakdown Spectroscopy). Nesse trabalho, a estequiometria de seis alvos de HAP foi
analisada e os resultados foram comparados com os obtidos por fluorescência de raios-
X (XRF), UV-Vis e absorção atômica (AAS). Devido à característica destrutiva dessas
últimas, ou seja, a necessidade de se utilizar uma parte da amostra para digestão em
solução, a técnica OPC-LIBS se faz mais vantajosa. A comparação dos resultados
obtidos com as diferentes técnicas permitiu a validação da metodologia de análise
elementar apresentada neste trabalho e que se aproveita da técnica não destrutiva OPC-
LIBS.
4.3.1. Análise estequiométrica por XRF, UV-VIS e AAS
As análises de Fluorescência de Raios-X (XRF), Espectrofotometria no ultravioleta
visível (UV-VIS) e Absorção Atômica (AAS) foram realizadas no Laboratório de
Espectrofotometria do CBPF. A técnica analítica de Espectrometria de Absorção
Atômica com chama (AAS) foi utilizada para quantificar o elemento cálcio, e UV-VIS
para quantificar o fósforo. Ambos os elementos também foram analisados por (XRF).
Nas análises por AAS foi utilizado o espectrômetro modelo AA-6800 da Shimadzu.
As amostras foram preparadas em triplicata, por solubilização total (digestão da
amostra) em ácido nítrico (HNO3). A análise do cálcio foi realizada em chama de gás
acetileno (Ar-C2H2) com fluxo de 2 litros/min no comprimento de onda de 422,7 nm. O
fósforo foi analisado por espectrômetro de UV-visível modelo UV-1800 da Shimadzu.
A amostra foi tratada com solução de metavanadato de amônio, medindo a absorbância
do P em 420 nm. Nas duas análises a leitura foi realizada com curvas de calibração
obtidas a partir de padrões de alta pureza da MERCK.
A análise por espectrometria de Fluorescência de Raios-X (XRF) foi realizada com
equipamento da Panalytical modelo AxiosmAX, com 3,0 kW de potência, detector
sequencial por dispersão de comprimento de onda. As amostras foram quantificadas em

47
um programa de análise semi-quantitativo utilizando o software Omnian. Os padrões de
calibração são preparados exclusivamente para esse programa, que abrange uma
varredura completa entre o F e U. A concentração dos elementos é obtida com base nas
intensidades de raios-X fluorescentes emitidos pelos elementos presentes dentro de uma
área pré-estabelecida da amostra. O detector de fluxo é um detector a gás metano usado
para analisar elementos leves. O detector e o cristal são escolhidos de acordo com os
elementos que se deseja quantificar e as condições usadas para dosagem do Ca e P são
apresentadas na Tabela 4.1:
Tabela 4.1: Condições para quantificação do cálcio e do fósforo por XRF.
Elemento Linha Cristal Colimador Detector kV mA ângulo(°)
Ca kα LiF 200 150 um Flow 30 70 113,048
P kα Ge 111 550 um Flow 24 90 140,985
4.3.2. Análise estequiométrica por OPC-LIBS
Para análise da estequiometria por OPC-LIBS, o primeiro passo é obter os espectros
LIBS de cada amostra. Esses espectros foram obtidos no Laboratório de Plasma e
Espectroscopia da Universidade Federal Fluminense, RJ. A montagem experimental
para uma análise por LIBS é apresentada na Figura 4.1. A configuração constitui de: (1)
uma fonte de radiação laser Nd:YAG Q-switched, com pulso de 6 ns e comprimento de
onda de 1064 nm; (2) duas lentes ópticas; (3) um monocromador tipo Czerny-Turner
que está acoplado a uma câmera intensificada (ICCD) e (4) um computador.
Durante os experimentos, um pulso laser de 1064 nm, 6 ns de largura e 100 mJ de
energia foi focalizado sobre o alvo de HAP com uma lente biconvexa de 200 mm,
formando um plasma sobre a sua superfície. A luz emitida pelo plasma é recolhida e
focada através de uma lente óptica (também de 200 mm) diretamente sobre a fenda de
entrada do monocromador. Os parâmetros de aquisição do sinal (tempo de aquisição,
tempo de retardo entre o início do plasma e a abertura da ICCD, e o número de
acumulações dos espectros) são ajustados via software por meio do computador que se
encontra conectado ao espectrógrafo. Nesse caso, os espectros ópticos foram obtidos
com uma janela temporal de 200 ns, retardo de 2 µs em relação à incidência do pulso
laser sobre a amostra e acumulação de 100 espectros.

48
Figura 4.1: Configuração experimental para análise da estequiometria dos alvos de HAP pela
técnica de OPC-LIBS.
Uma vez obtidos os espectros para cada amostra, uma análise CF-LIBS foi realizada
com os seguintes passos: 1) escolha das linhas espectrais dos elementos em análise; 2)
determinação das suas intensidades; 3) cálculo da densidade eletrônica do plasma; 4)
cálculo da temperatura eletrônica do plasma através do plot de Saha-Boltzmann; 5)
determinação dos valores de densidade percentual para cada espécie atômica do Ca e do
P; 6) quantificação elementar das amostras.
Na análise por OPC-LIBS, a amostra Ap foi escolhida como padrão, pois apresentou
Ca/P = 1,67 de acordo com a quantificação por XRF, AAS e UV. A partir dos valores
obtidos por CF-LIBS com a amostra padrão, o procedimento descrito na Seção 3.3.3
(OPC-LIBS) foi usado para calcular os parâmetros P() das demais amostras: A1, A2,
A3, A4, A5, A6 e A7.
4.4. Preparação e tratamento dos substratos de Si, Ti e PLA
O protocolo de limpeza dos substratos de Si (001) de 1,2 x 1,2 cm foi realizado com
água deionizada e detergente extran na proporção 1:1 em ultrassom por 10 min, 3
lavagens com água deionizada no ultrassom por 8 min cada, em acetona no ultrassom
por 10 minutos e secagem com gás nitrogênio comprimido. Em seguida, foram tratados
na seguinte sequência: imersão em solução de 5% de HF (ácido hidrofluorídrico) + água

49
deionizada por 30 segundos, três lavagens com água deionizada no ultrassom por 8 min
cada e secagem com gás nitrogênio comprimido [34, 68].
Para os substratos de Ti, pastilhas de 2 mm de espessura foram cortadas a partir de
uma peça cilíndrica de titânio puro comercial de 12 mm de diâmetro. O seguinte
procedimento de limpeza foi adotado [34, 68]: água deionizada e extran na proporção
1:1 em ultrassom por 10 min, secagem com ar nitrogênio comprimido, jateamento com
partículas de sílica, água deionizada no ultrassom por 15 min, em acetona no ultrassom
15 min, imersão em solução de 2% de HF (ácido fluorídrico) + 8% de HNO3 (ácido
nítrico) + água deionizada no ultrassom por 6 min, três lavagens em água deionizada no
ultrassom por 8 min cada e por fim, secagem com gás nitrogênio comprimido. Os
substratos de Ti serão usados para os testes biológicos de solubilidade e cultura celular.
Substratos de poliácido lático (PLA) com diâmetro de 1,2 cm foram obtidos por
tecnologia aditiva em impressora 3D do CBPF, usando filamento de biopolímero
comercial modelo 4043D da Ingeo TM [69]. Para a limpeza, utilizou-se apenas extran e
água deionizada por 15 min em ultrassom.
4.5. Produção dos recobrimentos de HAP por PLD
Todos os recobrimentos foram produzidos no Laboratório de Plasma Aplicado do
CBPF. A montagem experimental e os equipamentos utilizados são mostrados na Figura
3.10. As deposições foram feitas em câmara de alto vácuo de aço inox montada no
CBPF [68]. A câmara está equipada com um suporte de substrato acoplado a um
aquecedor resistivo, que permite aquecer o substrato até 800°C. Possui também um
suporte para seis alvos com movimentos de translação e rotação e uma janela de vidro
com alta transmitância em banda larga que permite realizar medidas de espectroscopia
de emissão do plasma. A pressão da câmara pode chegar a 10-6 Pa (10-8 mbar). A fonte
utilizada é um laser Nd:YAG Brilliant b da Quantel com duração de pulso de 5 ns e
frequência de até 10 Hz. Um sistema óptico com lentes e espelhos para o comprimento
de onda de 532 nm (laser verde) foi utilizado para orientar e direcionar o feixe com
ângulo de incidência de 45° e ponto focal de 1 mm2 sobre a superfície do alvo.

50
Figura 4.2: Montagem experimental para a produção dos recobrimentos de HAP: laser
Nd:YAG, espelhos e lentes para λ=532 nm e a câmara de deposição por PLD.
Considerando a aplicação proposta neste trabalho de tese, os parâmetros do sistema
de deposição foram explorados a fim de encontrar a melhor condição para a produção
de recobrimentos cristalinos de HAP por PLD com laser de Nd:YAG 532 nm.
Diferentes séries de filmes foram produzidas, enquanto variava-se um parâmetro
mantiveram-se fixos os demais (Tabela 4.2). Neste trabalho, os seguintes parâmetros
foram considerados: energias de 100 a 380 mJ, distâncias de 2 cm, 3 cm e 4 cm entre
alvo e substrato, tempos de deposição de 1 min a 20 min, pressão ambiente de 10-4 mbar
e 10-6 mbar e tratamento térmico in situ de 200 °C, 400 °C, 600 °C e 800 °C. Para
algumas amostras também foram realizados tratamentos térmicos após a deposição. A
melhor condição de deposição à temperatura ambiente foi usada para os ensaios
biológicos sobre substratos de Ti e PLA.

51
Tabela 4.2: Séries de amostras de Si/HAP produzidas sob diferentes condições de deposição.
Séries Energia (mJ) Tempo (min) Distância
(cm)
Pressão
(mbar)
Temperatura
(oC)
Primeira 100;150;200;
250;300;350;380 5 3 3x10-6 25
Segunda 250 0,25;0,5;1;2;3;4 3 3x10-6 25
Terceira 300 0,25;0,5;1;2;3;4 3 3x10-6 25
Quarta 250 5 2; 4 3x10-6 25
Quinta 300 5 2; 4 3x10-6 25
Sexta 250 5 2; 3; 4 10-4 25
Sétima 300 5 2; 3; 4 10-4 25
Oitava 250 5 3 3x10-6 200;400;600;800
Nona 300 5 3 3x10-6 200;400;600;800
4.6. Caracterização físico-química dos filmes produzidos
Os filmes foram caracterizados quanto à composição química, as fases minerais
presentes e o grau de cristalinidade através das técnicas de Difração de raios-X (DRX),
Espectroscopia de Infravermelho por Transformada de Fourier (FTIR) e Espectroscopia
de Fotoelétrons de Raios-X (XPS). A espessura, morfologia de superfície e rugosidade
foram analisadas por Microscopia Eletrônica de Varredura (MEV) e Microscopia de
Força Atômica (AFM). A adesão dos recobrimentos sobre os substratos de Si e PLA foi
analisada por método com fita (tape test).
4.6.1. Difração de raios-X (XRD)
A análise estrutural por Difração de Raios-X por Incidência Rasante foi
realizada no CBPF em equipamento X-Pert Pro da Panalytical com fonte de radiação Cu
(Kα) e λ = 0,154 nm e no Laboratório Nacional de Luz Síncrotron (LNLS) do Centro de
Pesquisas em Materiais (CNPEM) em Campinas-SP. No LNLS, a fonte de radiação
síncrotron monocromática da linha XRD2, com energia de 9 keV (λ = 0,13775 nm)
fornece maior incidência de fótons sobre a amostra. Dessa forma, o difratograma

52
apresenta maior relação sinal ruído e menor alargamento dos picos de difração, em
comparação com o sinal obtido por difratômetro comerciais com fonte de Cu (Kα). O
modo GIXRD (Grazing Incidence X-Ray Diffraction) foi utilizado com ângulo de
incidência fixo em θ = 0,5° e área da fenda incidente de 1,0 mm2 (2 mm × 0,5 mm).
Esse modo é geralmente usado para filmes de pouca espessura a fim de aumentar o
volume de interação dos fótons incidentes com o material do filme e evitar o sinal dos
substratos. Para cada amostra, o detector (2θ) com fenda de entrada de 0,5 mm varreu o
intervalo de 8° a 55° com passo de 0,025° no tempo de aproximadamente 40 minutos
(120.000 contagens/passo). A posição dos picos e a identificação dos índices de Miller
foram determinadas utilizando a ficha ICDD número 84-1998.
A cristalinidade relativa entre as amostras foi calculada através da razão entre a
contribuição de área dos picos de difração e a área total dentro de um intervalo 2θ entre
15° e 35°. Esse intervalo é suficiente porque compreende tanto a contribuição
nanocristalina quanto os principais picos de difração da HAP [70].
4.6.2. Espectroscopia de infravermelho por transformada de
Fourier (FTIR)A técnica de infravermelho por transformada de Fourier
(FTIR) é uma técnica de espectroscopia molecular por energia vibracional que permite
identificar características como a composição, algumas impurezas e grau de
cristalinidade dos filmes através dos modos de vibração dos grupos iônicos ou
moleculares. Foi utilizado o espectrômetro Shimadzu IR-Prestige 21 do CBPF. Para
operação no modo de transmissão, os filmes de HAP com aproximadamente 150 nm de
espessura foram depositados sobre uma fina pastilha de KBr, que é transparente ao
infravermelho. Os espectros foram obtidos em uma área 300 × 300 µm, com
comprimento de onda variando de 400 a 4000 cm-1 e com 300 varreduras por amostra.
Foram feitas as correções de smoothing e atmosférica (CO2) de acordo com cada
amostra.
As amostras de PLA/HAP foram analisadas com o espectrômetro conectado a
um microscópio AIM 8800 operando em modo de reflectância total atenuada (ATR –
attenuated total reflection). Os espectros foram obtidos em uma área 432 × 332 µm, 300
varreduras por amostra e com comprimento de onda variando de 450 a 4000 cm-1.

53
4.6.3. Espectroscopia de Fotoelétrons por raios-X (XPS)
A composição elementar e de ligações químicas entre os elementos na superfície
do recobrimento foi analisada por Espectroscopia de Fotoelétrons de Raios-X (XPS)
através do espectrômetro SPECS PHOIBOS 100/150 com um analisador hemisférico de
150 mm operando com fonte de raios-X de radiação policromática Al (Kα) de energia
1486,6 eV e ângulo de tomada de captura de 30°, Epass de 30,0 eV, com até 0,5 eV de
energia/passo para o espectro total (survey) e 0,02 eV de energia/passo para os espectros
de alta resolução sobre o pico de cada elemento analisado. A deconvolução dos picos
envelopes nos espectros de alta resolução foi realizada através do software CASA-XPS
e banco de dados NIST XPS para as energias de ligação dos picos C 1s, Ca 2p, P 2p, e
O 1s. A energia de calibração utilizada durante os experimentos foi do carbono orgânico
C 1s (284.6 eV). Previamente a resolução do instrumento foi aferida através da banda de
energia Ag 3d5/2, de uma amostra altamente pura (99,999%) de prata, após limpeza por
sputtering de íons de argônio.
4.6.4. Microscopia Eletrônica de Varredura (MEV)
A morfologia dos recobrimentos foi investigada em um Microscópio Eletrônico
de Varredura (SEM) JSM-6490LV JEOL com sinal de elétrons secundários. Devido ao
comportamento isolante da HAP, uma amostra de Si/HAP foi recoberta com uma
camada de 10 nm de ouro. Para análise da espessura, amostras de Si/HAP foram
clivadas imediatamente depois de serem congeladas sob imersão em nitrogênio líquido
e imagens da seção transversal foram obtidas com um Microscópio Eletrônico de
Varredura de Alta Resolução (FE-SEM) JSM-7100F.
4.6.5. Microscopia de Força Atômica (AFM)
A morfologia e topografia e rugosidade dos recobrimentos também foram
analisados por microscopia de força atômica (AFM) com microscópio BRUKER
MultiMode 8-HR. As medidas foram feitas em tapping mode com ponta de 10 nm. Os
parâmetros de calibração como a freqüência de ressonância da ponta, fase, amplitude e

54
ganho foram variados para cada amostra. A rugosidade foi obtida através de análise das
imagens com software Gwyddyon.
4.6.6. Teste de adesão dos recobrimentos
A qualidade da adesão dos recobrimentos foi avaliada através do método de adesão
por fita (tape test) seguindo o método B da norma ASTM D3359. A fita utilizada foi
uma fita filamentosa 3M modelo 8934, com força adesiva de 6,5 N/cm conforme
exigida pela norma [71]. Uma caneta com ponta de diamante foi usada para cortar
quadrados de área 1×1 mm2 sobre a superfície dos filmes, porém com pressão suficiente
para que o corte chegasse até os substratos. Após os cortes, as amostras passaram por
fluxo de ar para retirada de possíveis fragmentos e a fita foi manualmente pressionada
sobre a área dos cortes na superfície das amostras. Após um minuto, a fita foi retirada a
um ângulo de 180°. Imagens com microscópio óptico foram obtidas antes e depois do
teste de adesão.
4.7. Ensaios preliminares de caracterização biológica
Para a caracterização biológica, filmes de HAP foram produzidos em substratos de
Ti e PLA sob as mesmas condições de deposição: 30 J/cm-2 de fluência, 5 minutos de
deposição, 10-6 mbar de pressão e 3 cm de distância alvo-substrato. Ensaios de adesão e
proliferação celular foram realizados em amostras de PLA controle, PLA/HAP, Ti
controle e Ti/HAP em tempos de 4 h, 24 h e 48 h com o objetivo de observar a cinética
de tempo (veja Tabela 4.3). Antes dos testes, as amostras de Ti controle e Ti/HAP
foram colocadas em placas de cultura celular e esterilizadas em raios gama (fonte de
cobalto 60, 15 KGy, 19,72 Gy/min) por 12 horas, no laboratório da COPPE no Fundão.
As amostras de PLA controle e PLA/HAP foram esterilizadas através de óxido de
etileno pela empresa ACECIL, em São Paulo. A resposta biológica dos recobrimentos
foi analisada por ensaios de adesão e proliferação celular.

55
Tabela 4.3: Procedimento experimental para os ensaios biológicos.
1ª Placa 2ª Placa
Amostras em sextuplicata Amostras em sextuplicata
PLA controle PLA/HAP Ti controle Ti/HAP 4h
24h
48h
O tipo de célula usada foi SAOS-2 (célula humana de osso maduro) com densidade
inicial de 0,4×104 células. Para microscopia de fluorescência, as células foram semeadas
em placa de 24 poços com clone tubes sobre as amostras. Após os tempos de 4h e 24h
todas as amostras foram fixadas em 4% de paraformaldeído, permeabilizadas com 0,1%
de Triton X-100, bloqueadas com 3% de BSA e incubadas com o anticorpo DAPI
Fluorshield (marca o núcleo). Nas amostras do tempo de 48h foi realizada a marcação
tripla, que corresponde à fixação em 4% de paraformaldeído, permeabilizadas com
0,1% de Triton X-100, bloqueadas com 3% de BSA e incubadas com o anticorpo anti-
vinculina específico (1:200) durante a noite a 4°C. Alexa-Fluor 488 (1:500) foi usado
como anticorpo secundário durante 1 hora. Após as etapas de lavagens, as amostras
foram incubadas com o anticorpo de Alexa-Fluor 546 conjugado com faloidina (1:80)
durante 30 minutos e montadas em laminas de vidro com DAPI Fluorshield. A área de
células aderentes foi adquirida digitalmente para quantificação celular (5 campos
representativos por amostra), utilizando os filtros fluorescentes para marcações em
verdes, vermelhos e azuis. As visualizações foram feitas pelo microscópio Axio Vision
4.8.1 (Zeiss) e o software Imagem Pro Plus 6.0 foi usado para o processamento das
imagens e sobreposição. Todos os testes foram realizados no Laboratório de Cultura de
Células do CBPF e os procedimentos do cultivo celular e de marcação com os
anticorpos seguiram protocolos previamente estabelecidos (APÊNDICE II).

56
5. RESULTADOS E DISCUSSÕES
5.1. Análise estequiométrica dos alvos de HAP por OPC-
LIBS
Nesta seção, são apresentados os resultados de quantificação elementar para os alvos
de HAP produzidos e utilizados durante esse trabalho de tese. As seguintes subseções
determinam os passos para obtenção dos resultados por OPC-LIBS e a última subseção
é deixada para apresentar o resultado de comparação entre as técnicas.
5.1.1. Análise qualitativa e seleção das linhas
A análise começa com a realização de experimentos com uma amostra
certificada (que chamaremos de amostra padrão, Ap) que tem a mesma matriz e uma
composição semelhante a dos alvos a serem analisados. Inicialmente, irradiamos a
amostra com um pulso laser de 100 mJ, escolhendo os melhores parâmetros para o
experimento. Então, um espectro LIBS é obtido para posterior análise das linhas
espectrais de emissão dos elementos do alvo. A Figura 5.1 mostra o espectro de emissão
da amostra padrão, na faixa espectral de 205-600 nm com um retardo de 2 s em
relação a chegada do feixe laser e uma janela de observação de 200 ns. A partir da
análise qualitativa destes espectros podemos identificar linhas de emissão do oxigênio
(O I), nitrogênio (N I) e do hidrogênio (H I), que são elementos que provém do ar
circundante e da própria amostra que apresenta hidrogênio e oxigênio na sua
composição. O espectro apresenta também linhas de emissão características do cálcio
(Ca I e Ca II) e do fósforo (P I), elementos que serão quantificados.
A molécula de hidroxiapatita, Ca10(PO4)6(OH)2, apresenta uma razão de cálcio
por fósforo Ca/P = 1,67. Assim, um alvo será dito estequiométrico se a sua razão Ca/P
se aproximar do valor de 1,67. Neste trabalho, obtivemos um total de oito espectros
LIBS para serem analisados: um espectro da amostra padrão Ap e sete das amostras
com razão Ca/P desconhecida, que denominamos de A1 a A7. Em uma análise OPC-
LIBS se faz necessário que todos os experimentos sejam realizados com os mesmos

57
parâmetros e arranjo experimental para que todos os parâmetros do espectro óptico
apresentem valores semelhantes.
Figura 5.1: Espectros LIBS da amostra padrão Ap mostrando as linhas espectrais dos
elementos identificados na análise qualitativa.
Para se obter uma análise quantitativa, é necessário selecionar apropriadamente
as linhas espectrais correspondente a cada espécie atômica dos elementos presentes na
amostra. Uma boa linha espectral a ser usada na análise deve estar claramente separada
de outras linhas atômicas adjacentes e possuir probabilidade de transição e nível de
energia superior catalogados na literatura. Algumas linhas pertencentes às espécies
atômicas dos elementos Ca e P foram escolhidas durante a análise do espectro LIBS da
amostra padrão (Figura 5.1) e estão especificadas na Tabela 5.1. Nem todas as linhas
espectrais encontradas dentro da faixa espectral observada estavam bem isoladas, um
exemplo são as linhas 255,326 nm e 255,491 nm, pertencentes ao dupleto do fósforo
(ver Figura 5.2), que estão muito próximas devido a resolução do nosso espectrógrafo.
Quase todas as linhas do fósforo, usadas neste trabalho, apresentam uma pequena
sobreposição com outra adjacente. As linhas de fósforo foram deconvolucionadas para
determinar suas intensidades.

58
Os oito espectros experimentais foram analisados e cada linha que está incluída
na Tabela 5.1 foi ajustada com uma função analítica Voigt [64,72,73], obtendo sua área
(intensidade) e seu perfil espectral com o programa de ajuste gráfico Origin.
Tabela 5.1: Parâmetros das linhas espectrais consideradas na análise quantitativa das amostras.
Espécie (nm) gi Aij (108 s-1) Ei (cm-1) P() (108 s-1) Ref.
Ca I 428,301 5 0,434 38551,558 3,591 [74]
Ca I 428,937 3 0,6 38464,808 3,103 [74]
Ca I 431,855 3 0,74 38464,808 3,778 [74]
Ca I 612,222 3 0,287 31539,495 0,726 [74]
Ca I 649,378 5 0,44 35730,454 0,658 [74]
Ca I 649,965 5 0,081 35730,454 0,224 [74]
Ca II 210,324 4 0,82 72722,23 8,135 [74]
Ca II 211,276 6 0,97 72730,93 15,043 [74]
Ca II 315,887 4 3,1 56858,25 5,511 [74]
Ca II 370,603 2 0,88 52166,93 1,116 [74]
Ca II 373,690 2 1,7 52166,93 1,370 [74]
P I 214,914 2 3,18 57876,574 5,308 [74]
P I 215,294 4 0,485 65156,242 2,754 [74]
P I 215,408 6 0,58 65157,126 4,774 [74]
P I 253,397 4 0,2 58174,366 0,8202 [74]
P I 253,560 4 0,95 58174,366 3,043 [74]
P I 255,326 2 0,71 57876,574 1,397 [74]
P I 255,491 2 0,3 57876,574 0,915 [74]
Os principais efeitos que contribuem para o perfil espectral de uma linha são: o
alargamento Stark, o alargamento instrumental e o alargamento Doppler. Devido ao
peso atômico apresentado pelos elementos em análise, o alargamento Doppler se faz
desprezível e as linhas são efetivamente alargadas pela contribuição do efeito Stark e
pela função instrumental. O alargamento Stark tem um perfil lorentziano e, de acordo
com nossas medidas, o alargamento instrumental segue um perfil gaussiano. Assim, as
linhas estudadas possuem um perfil Voigt que é dado pela convolução de uma gaussiana
e uma lorentziana.

59
255,00 255,15 255,30 255,45 255,60
0
2
4
6
8
10
12
Inte
nsid
ad
e (
x1
05 u
.a.)
Comprimento de Onda (nm)
Dubleto do
Fósforo
Figura 5.2: Deconvolução das linhas de emissão do dubleto do fósforo: 255,326 e 255,491 nm.
5.1.2. Determinação dos parâmetros do plasma: temperatura
e densidade
A temperatura do plasma é o parâmetro mais importante na análise pela técnica
LIBS. Uma análise quantitativa precisa passa pela determinação exata da temperatura
que, através do método de Saha-Boltzmann, depende do valor da densidade de elétrons.
A densidade foi calculada pela Equação 3.16 da Seção 3.3.2, aonde usamos o
alargamento Stark da linha 656,279 nm do hidrogênio (Hα). Devido à degenerescência
do átomo que a emite, essa linha apresenta um alargamento muito mais pronunciado
para a resolução do nosso espectrógrafo, fornecendo um valor preciso para a largura a
meia altura. Nos experimentos LIBS, o efeito Doppler é desprezível para a maioria dos
átomos, o que não acontece para o hidrogênio, o mais leve e de maior mobilidade dentre
os átomos. Devido a esse fato, para calcular um valor para a densidade eletrônica,
devemos separar os alargamentos instrumental e Doppler do alargamento Stark. A linha
espectral Hα apresenta um perfil Voigt gerado pela convolução de um perfil gaussiano,
provenientes dos alargamentos Doppler e instrumental, com um perfil lorentziano que
provém do alargamento Stark. Usando o software de ajuste gráfico Origin,

60
deconvolucionamos as oito linhas Hα, pertencente aos espectros de cada amostra. A
deconvolução da linha Hα do espectro da amostra padrão é mostrada na Figura 5.3. Os
valores para o alargamento Stark e a densidade eletrônica são apresentados na Tabela
5.2.
Figura 5.3: Função Voigt ajustada aos dados experimentais da linha Hα presente no espectro da
amostra padrão Ap.
Tabela 5.2: Alargamento Stark e densidade de elétrons do plasma para os diferentes espectros
investigados. Estes valores foram obtidos através da linha Hα presente nos espectros das
amostras.
Amostra Alargamento (nm) Densidade (1023 m-3)
Ap 1,35 0,07 1,44 0,03
A1 1,42 0,08 1,56 0,04
A2 1,40 0,02 1,52 0,02
A3 1,27 0,02 1,34 0,04
A4 1,23 0,02 1,28 0,03
A5 1,42 0,01 1,56 0,03
A6 1,46 0,02 1,62 0,03
A7 1,41 0,02 1,55 0,02

61
Figura 5.4: Fluxograma para o algoritmo de iteração utilizado para resolver o diagrama de
Saha-Boltzmann.
O passo seguinte foi obter a intensidade das linhas de emissão das espécies
neutra e ionizada do Ca para calcular a temperatura de excitação do plasma. Para isso, o
plasma é suposto estar em equilíbrio termodinâmico local durante a janela de
observação e o valor para a temperatura será único e igual para todos os demais
elementos presentes na pluma. As linhas espectrais do Ca I e Ca II, que foram usadas no
cálculo, estão apresentados na Tabela 5.1. Para o cálculo do valor da temperatura,
realizamos um diagrama de Saha-Boltzmann assumindo que existe uma relação linear
entre as variáveis x e y (ver Seção 3.3.2). Uma temperatura inicial é então obtida através
de um ajuste de Boltzmann do Ca I com a Equação 3.8. Este valor é utilizado para
iniciar os cálculos com o diagrama de Saha-Boltzmann. Como o diagrama de Saha-

62
Boltzmann (Equações 3.14 e 3.15) depende da temperatura, a única forma de resolvê-lo
é através de um código iterativo. Este código é iniciado com a primeira temperatura,
dada pelo ajuste de Boltzmann, e depois realizamos um ajuste por Saha-Boltzmann. E
seguida, o código entrega um valor mais preciso para a temperatura, que é novamente
usado para reiniciar o código e calcular um novo valor de temperatura. Este processo
iterativo é realizado até que o critério de parada seja satisfeito ( = 0,001). O valor
entregue pelo código após a convergência é a temperatura do plasma Te. Na Figura 5.4
apresentamos o fluxograma usado para resolver o diagrama de Saha-Boltzmann. Na
Figura 5.5 apresentamos o diagrama de Saha-Boltzmann para as linhas de Ca I e Ca II
da amostra padrão. A temperatura encontrada foi utilizada para determinar os valores
dos parâmetros P() de calibração OPC-LIBS.
O passo seguinte foi obter a intensidade das linhas de emissão das espécies neutra e
ionizada do Ca para calcular a temperatura de excitação do plasma. Para isso, o plasma
é suposto estar em equilíbrio termodinâmico local durante a janela de observação e o
valor para a temperatura será único e igual para todos os demais elementos presentes na
pluma. As linhas espectrais do Ca I e Ca II, que foram usadas no cálculo, estão
apresentados na Tabela 5.1. Para o cálculo do valor da temperatura, realizamos um
diagrama de Saha-Boltzmann assumindo que existe uma relação linear entre as
variáveis x e y (ver Seção 3.3.2). Uma temperatura inicial é então obtida através de um
ajuste de Boltzmann do Ca I com a Equação 3.8. Este valor é utilizado para iniciar os
cálculos com o diagrama de Saha-Boltzmann. Como o diagrama de Saha-Boltzmann
(Equações 3.14 e 3.15) depende da temperatura, a única forma de resolvê-lo é através de
um código iterativo. Este código é iniciado com a primeira temperatura, dada pelo
ajuste de Boltzmann, e depois realizamos um ajuste por Saha-Boltzmann. E seguida, o
código entrega um valor mais preciso para a temperatura, que é novamente usado para
reiniciar o código e calcular um novo valor de temperatura. Este processo iterativo é
realizado até que o critério de parada seja satisfeito ( = 0,001). O valor entregue pelo
código após a convergência é a temperatura do plasma Te. Na Figura 5.4 apresentamos
o fluxograma usado para resolver o diagrama de Saha-Boltzmann. Na Figura 5.5
apresentamos o diagrama de Saha-Boltzmann para as linhas de Ca I e Ca II da amostra
padrão. A temperatura encontrada foi utilizada para determinar os valores dos
parâmetros P() de calibração OPC-LIBS.

63
Figura 5.5: Diagrama de Saha-Boltzmann para as linhas de cálcio presentes no espectro da
amostra padrão.
5.1.3. Cálculo da composição da amostra padrão com a
técnica CF-LIBS
Com base na temperatura encontrada para a amostra padrão na Seção 3.3.3, a
densidade das espécies presentes na pluma de plasma foi avaliada através do coeficiente
linear do ajuste de Boltzmann. A densidade relativa do número de átomos pode ser
obtida através dos coeficientes lineares bs da Equação 3.9. Para encontrarmos os valores
destes coeficientes, realizamos ajustes de Boltzmann com as intensidades de cada
espécie atômica de P e Ca presentes no espectro experimental. Nesse ajuste, a
inclinação da reta é dada por -1/kT. Então, uma vez obtidos os valor da temperatura
para o espectro observado, fica estabelecido o coeficiente angular das retas (-1/kT) a
serem ajustadas. Se o plasma está em LTE a sua temperatura é a mesma para todas as
espécies atômicas presentes na pluma e a inclinação da reta no ajuste de Boltzmann de
cada espécie deve ser a mesma. Então, fixamos o valor conhecido para a inclinação da
reta no ajuste de cada espécie separadamente e determinamos os coeficientes lineares da
reta bs. No espectro da amostra padrão foram realizados três ajustes para as espécies de
Ca I, Ca II e P I e estão apresentados na Figura 5.6.

64
Figura 5.6: Ajuste de Boltzmann dos dados experimentais obtidos com a amostra
padrão.
Uma vez realizados os ajustes de cada espécie, devemos determinar a densidade
relativa do número de átomos através das Equações 3.10 a 3.12. Para isso, é necessário
conhecer os coeficientes bs de cada uma das espécies atômicas dos elementos presentes
na análise. Entretanto, muitas vezes isso não é possível por algumas razões, tais como:
as linhas espectrais de uma das espécies podem estar fora da faixa espectral observada;
as linhas podem estar sobrepostas às demais linhas do espectro experimental ou as
linhas podem apresentar muito baixa intensidade e ser confundida com o ruído de
fundo. Quando isso acontece, recorremos à equação de Saha (Equação 3.13) para
calcular uma relação entre as densidades das espécies neutras e ionizadas de um mesmo
elemento. Neste trabalho, o elemento P apresentou somente linhas de emissão da
espécie neutra (P I) devido à limitação da faixa espectral observada, e usamos a equação
de Saha para calcular a densidade da espécie ionizada P II.
Os resultados quantitativos para a densidade relativa do número de átomos dos
elementos Ca e P foram determinados somando os valores encontrados para as
densidades das espécies neutras e ionizadas. Esses resultados estão presentes na Tabela
5.3. Podemos observar que a razão Ca/P não corresponde ao valor real da amostra
padrão que tem estequiometria com Ca/P = 1,67. Este resultado já era esperado porque
CF-LIBS é considerada uma técnica semi-quantitativa.

65
Tabela 5.3. Densidade do número de átomos obtida pela técnica CF-LIBS para os elementos Ca
e P da amostra padrão.
Elemento Densidade de átomos (%)
Ca 58,64 12,31
P 41,36 6,87
Ca/P 1,42 0,38
5.1.4. Determinação dos parâmetros P(λ) com a amostra
padrão
A abordagem da técnica OPC-LIBS parte dos desvios de valores entre a
quantificação real da amostra padrão e o valor de quantificação obtido por CF-LIBS. A
idéia básica da aproximação é a possibilidade de determinar valores para o parâmetro
P() (Equação 3.20) a partir do valor real da composição da amostra padrão. Estes
valores são então usados para uma análise usual CF-LIBS de amostras desconhecidas.
Para determinar os parâmetros P() é necessário reconstruir uma equação para o ajuste
de Boltzmann cuja reta está deslocada verticalmente devido ao acréscimo de uma
constante. Essa constante é obtida com os valores certificados e os valores calculados
por CF-LIBS da amostra padrão (ver equação 3.18). Em seguida encontramos a
diferença entre o valor de y, para cada ponto do ajuste de Boltzmann, e a coordenada y’
para o mesmo ponto com a equação corrigida (Equação 3.19). Este procedimento pode
ser visualizado na Figura 3.8. A partir dessas diferenças ∆y calculamos o parâmetro
P() (Equação 3.20) para cada linha espectral presente neste trabalho. Esses valores de
P(), mostrados na Tabela 5.1, foram usados nas análises das amostras A1 a A7.
Para confirmar os valores de P(), produzimos uma análise por OPC-LIBS na
amostra padrão, o que deve fornecer um resultado igual ao certificado. Com a
temperatura encontrada anteriormente, ajustamos a Equação (3.21). Esse novo ajuste foi
realizado com os parâmetros P() no lugar do giAij, e carrega possíveis correções sobre
os parâmetros do plasma. Procedendo como na técnica CF-LIBS, calculamos as
concentrações atômicas dos elementos a partir do coeficiente linear dessa reta e
obtivemos as densidades para Ca = 62,51% e P = 37,49%. Essa análise forneceu uma
razão Ca/P = 1,67 ± 0,03, que concorda com o valor real da amostra padrão. Na Figura
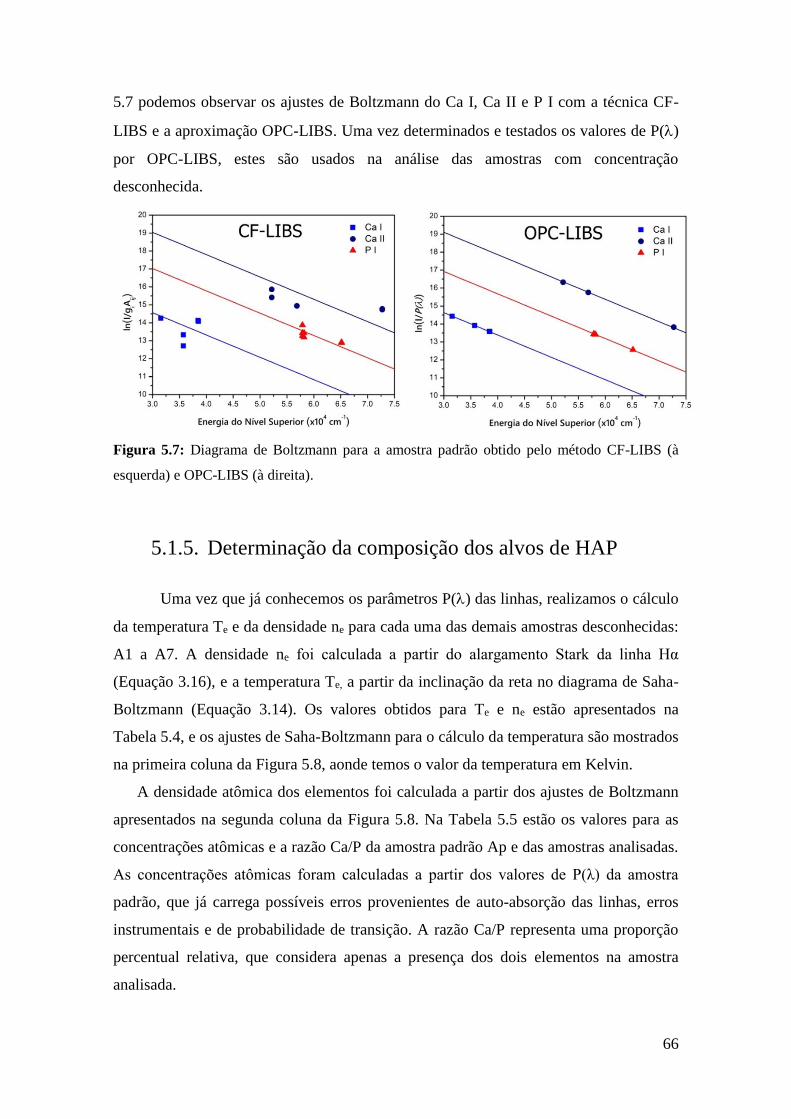
66
5.7 podemos observar os ajustes de Boltzmann do Ca I, Ca II e P I com a técnica CF-
LIBS e a aproximação OPC-LIBS. Uma vez determinados e testados os valores de P()
por OPC-LIBS, estes são usados na análise das amostras com concentração
desconhecida.
Figura 5.7: Diagrama de Boltzmann para a amostra padrão obtido pelo método CF-LIBS (à
esquerda) e OPC-LIBS (à direita).
5.1.5. Determinação da composição dos alvos de HAP
Uma vez que já conhecemos os parâmetros P() das linhas, realizamos o cálculo
da temperatura Te e da densidade ne para cada uma das demais amostras desconhecidas:
A1 a A7. A densidade ne foi calculada a partir do alargamento Stark da linha Hα
(Equação 3.16), e a temperatura Te, a partir da inclinação da reta no diagrama de Saha-
Boltzmann (Equação 3.14). Os valores obtidos para Te e ne estão apresentados na
Tabela 5.4, e os ajustes de Saha-Boltzmann para o cálculo da temperatura são mostrados
na primeira coluna da Figura 5.8, aonde temos o valor da temperatura em Kelvin.
A densidade atômica dos elementos foi calculada a partir dos ajustes de Boltzmann
apresentados na segunda coluna da Figura 5.8. Na Tabela 5.5 estão os valores para as
concentrações atômicas e a razão Ca/P da amostra padrão Ap e das amostras analisadas.
As concentrações atômicas foram calculadas a partir dos valores de P(λ) da amostra
padrão, que já carrega possíveis erros provenientes de auto-absorção das linhas, erros
instrumentais e de probabilidade de transição. A razão Ca/P representa uma proporção
percentual relativa, que considera apenas a presença dos dois elementos na amostra
analisada.

67
Tabela 5.4: Valores da temperatura eletrônica Te e densidade eletrônica ne para todas as
amostras.
Amostra Te (eV) ne (1023 m-3)
Ap 1,02 ± 0,01 1,44 ± 0,03
A1 0,89 ± 0,03 1,56 ± 0,04
A2 0,97 ± 0,02 1,52 ± 0,02
A3 1,06 ± 0,03 1,34 ± 0,04
A4 1,01 ± 0,02 1,28 ± 0,03
A5 0,93 ± 0,02 1,56 ± 0,04
A6 0,98 ± 0,02 1,62 ± 0,03
A7 0,98 ± 0,03 1,55 ± 0,02
Tabela 5.5: Valores das densidades de Ca e P e razão Ca/P medidos por OPC-LIBS.
Amostra Ca (%) P (%) Ca/P
Ap 62,51 ± 0,87 37,49 ± 0,67 1,67 ± 0,03
A1 61,53 ± 2,90 38,47 ± 0,91 1,60 ± 0,08
A2 63,45 ± 2,60 36,55 ± 0,75 1,74 ± 0,08
A3 62,97 ± 0,99 37,03 ± 1,26 1,70 ± 0,06
A4 61,99 ± 3,11 38,01 ± 1,06 1,63 ± 0,09
A5 62,42 ± 1,15 37,58 ± 0,58 1,66 ± 0,04
A6 63,10 ± 2,65 36,90 ± 0,74 1,71 ± 0,08
A7 62,82 ± 1,21 37,18 ± 0,75 1,69 ± 0,05

68

69
Figura 5.8: Resultados dos diagramas produzidos para as amostras A1 a A7. À esquerda, ajuste
de Saha-Boltzmann com informação da temperatura eletrônica do plasma e, à direita, ajuste de
Boltzmann produzido com o método OPC-LIBS, cujo coeficiente linear fornece informação da
concentração atômica dos elementos.
5.1.6. Comparação com outras técnicas
A amostra Ap é uma amostra de composição conhecida e que concorda com o valor
teórico de razão Ca/P = 1,67. As amostras A1 a A5 são pastilhas de HAP prensadas e
sinterizadas a 1150°C, enquanto que as amostras A6 e A7 são alvos prensados, mas não
sinterizados. Essas sete amostras tinham sua composição desconhecida, pois foram
tratadas para formar os alvos usados para PLD. As amostras A6 e A7 representam os
lotes originais do material, dos quais todos os alvos usados nesse trabalho de tese foram
produzidos: A6 (lote 267) e A7 (lote 274). A Tabela 5.6 apresenta os valores da
densidade de Ca e P e a razão Ca/P obtidos por OPC-LIBS e esses resultados foram
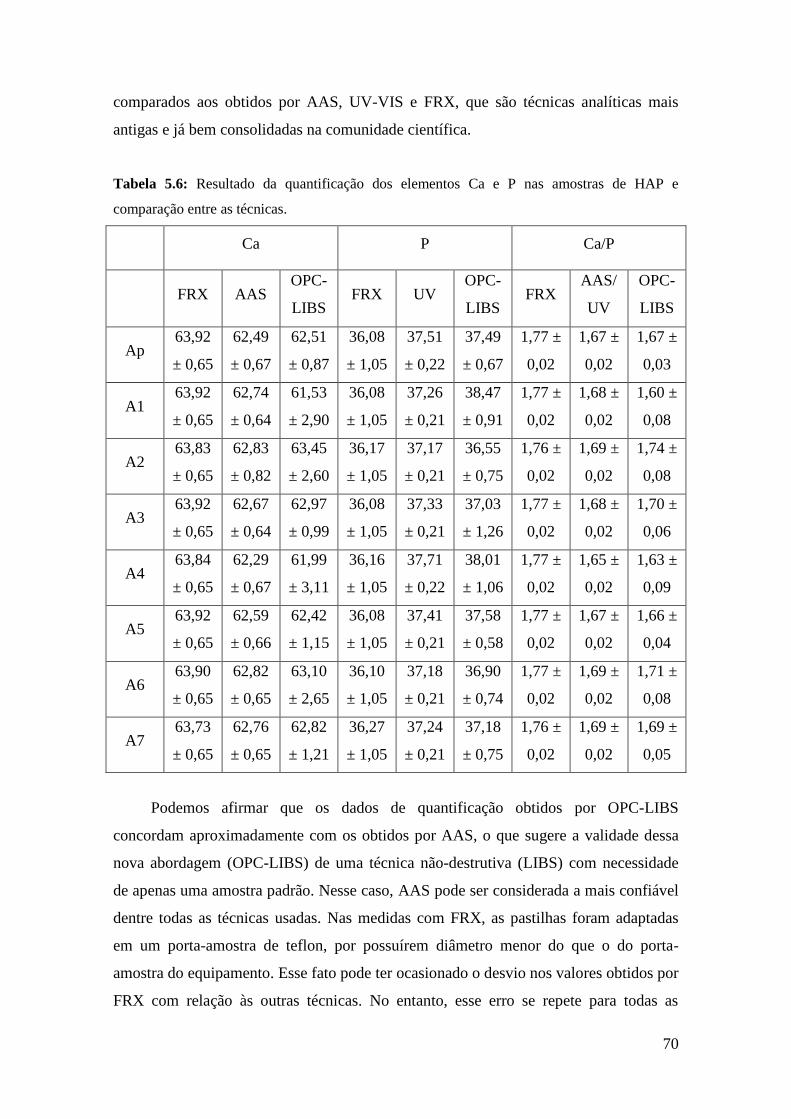
70
comparados aos obtidos por AAS, UV-VIS e FRX, que são técnicas analíticas mais
antigas e já bem consolidadas na comunidade científica.
Tabela 5.6: Resultado da quantificação dos elementos Ca e P nas amostras de HAP e
comparação entre as técnicas.
Ca P Ca/P
FRX AAS OPC-
LIBS FRX UV
OPC-
LIBS FRX
AAS/
UV
OPC-
LIBS
Ap 63,92
± 0,65
62,49
± 0,67
62,51
± 0,87
36,08
± 1,05
37,51
± 0,22
37,49
± 0,67
1,77 ±
0,02
1,67 ±
0,02
1,67 ±
0,03
A1 63,92
± 0,65
62,74
± 0,64
61,53
± 2,90
36,08
± 1,05
37,26
± 0,21
38,47
± 0,91
1,77 ±
0,02
1,68 ±
0,02
1,60 ±
0,08
A2 63,83
± 0,65
62,83
± 0,82
63,45
± 2,60
36,17
± 1,05
37,17
± 0,21
36,55
± 0,75
1,76 ±
0,02
1,69 ±
0,02
1,74 ±
0,08
A3 63,92
± 0,65
62,67
± 0,64
62,97
± 0,99
36,08
± 1,05
37,33
± 0,21
37,03
± 1,26
1,77 ±
0,02
1,68 ±
0,02
1,70 ±
0,06
A4 63,84
± 0,65
62,29
± 0,67
61,99
± 3,11
36,16
± 1,05
37,71
± 0,22
38,01
± 1,06
1,77 ±
0,02
1,65 ±
0,02
1,63 ±
0,09
A5 63,92
± 0,65
62,59
± 0,66
62,42
± 1,15
36,08
± 1,05
37,41
± 0,21
37,58
± 0,58
1,77 ±
0,02
1,67 ±
0,02
1,66 ±
0,04
A6 63,90
± 0,65
62,82
± 0,65
63,10
± 2,65
36,10
± 1,05
37,18
± 0,21
36,90
± 0,74
1,77 ±
0,02
1,69 ±
0,02
1,71 ±
0,08
A7 63,73
± 0,65
62,76
± 0,65
62,82
± 1,21
36,27
± 1,05
37,24
± 0,21
37,18
± 0,75
1,76 ±
0,02
1,69 ±
0,02
1,69 ±
0,05
Podemos afirmar que os dados de quantificação obtidos por OPC-LIBS
concordam aproximadamente com os obtidos por AAS, o que sugere a validade dessa
nova abordagem (OPC-LIBS) de uma técnica não-destrutiva (LIBS) com necessidade
de apenas uma amostra padrão. Nesse caso, AAS pode ser considerada a mais confiável
dentre todas as técnicas usadas. Nas medidas com FRX, as pastilhas foram adaptadas
em um porta-amostra de teflon, por possuírem diâmetro menor do que o do porta-
amostra do equipamento. Esse fato pode ter ocasionado o desvio nos valores obtidos por
FRX com relação às outras técnicas. No entanto, esse erro se repete para todas as
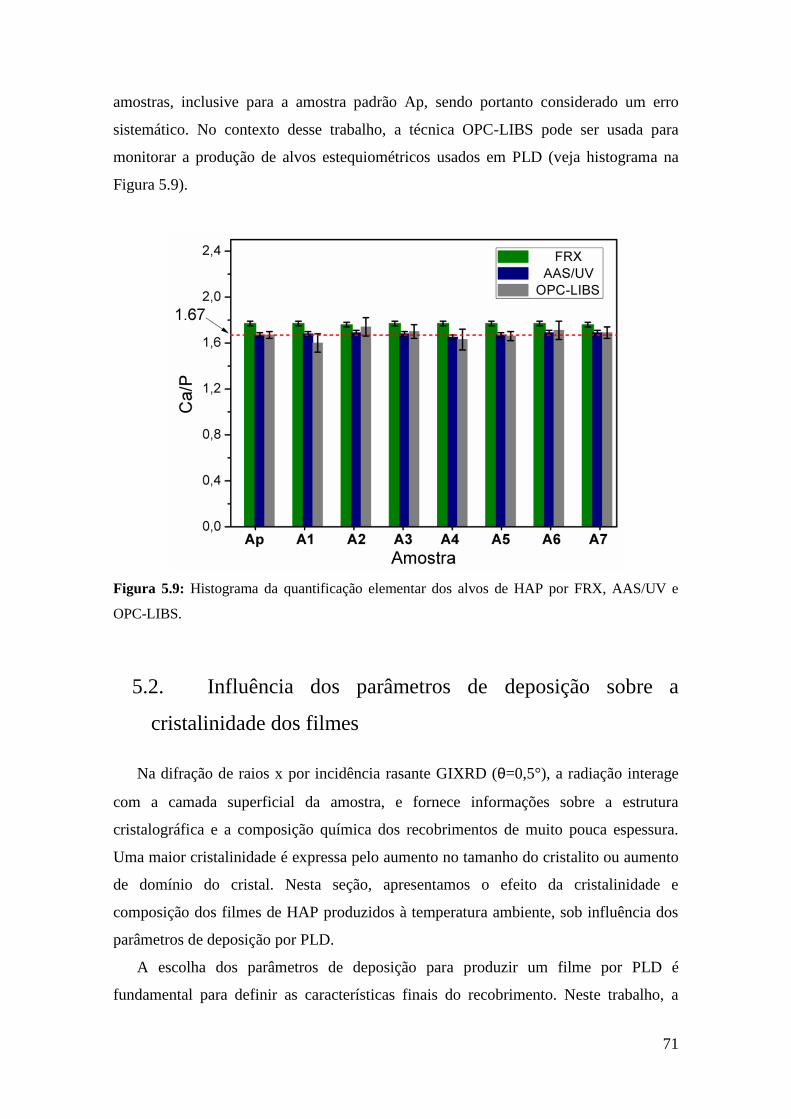
71
amostras, inclusive para a amostra padrão Ap, sendo portanto considerado um erro
sistemático. No contexto desse trabalho, a técnica OPC-LIBS pode ser usada para
monitorar a produção de alvos estequiométricos usados em PLD (veja histograma na
Figura 5.9).
Figura 5.9: Histograma da quantificação elementar dos alvos de HAP por FRX, AAS/UV e
OPC-LIBS.
5.2. Influência dos parâmetros de deposição sobre a
cristalinidade dos filmes
Na difração de raios x por incidência rasante GIXRD (θ=0,5°), a radiação interage
com a camada superficial da amostra, e fornece informações sobre a estrutura
cristalográfica e a composição química dos recobrimentos de muito pouca espessura.
Uma maior cristalinidade é expressa pelo aumento no tamanho do cristalito ou aumento
de domínio do cristal. Nesta seção, apresentamos o efeito da cristalinidade e
composição dos filmes de HAP produzidos à temperatura ambiente, sob influência dos
parâmetros de deposição por PLD.
A escolha dos parâmetros de deposição para produzir um filme por PLD é
fundamental para definir as características finais do recobrimento. Neste trabalho, a

72
energia do laser foi o primeiro parâmetro a variar, pois a energia fornecida ao alvo
durante a ablação tem implicação direta na dinâmica de vaporização do material antes
mesmo que o material ejetado entre em contato com o substrato. Para todas as
deposições, a área de ablação foi mantida fixa em 1 mm2 (área de interação do feixe na
superfície do alvo). Dessa forma, uma série de amostras foi produzida variando a
fluência de deposição (energia/área) e mantendo fixos os demais parâmetros: t = 5 min,
d = 3 cm, P = 3x10-6 mbar e temperatura ambiente (veja Tabela 4.2). A Figura 5.10
apresenta os difratogramas para os filmes produzidos de 10 a 38 J/cm2. O resultado
confirma o aumento na cristalinidade dos recobrimentos devido ao aumento da fluência,
pois mais energia está sendo fornecida para formação e cristalização dos filmes. Os
filmes dessa série consistiram da fase de HAP com diferentes graus de cristalinidade e
nenhuma outra fase foi formada nessas condições de deposição.
10 20 30 40
Inte
nsid
ade
2( °)
38 J/cm²
35 J/cm²
30 J/cm²
25 J/cm²
20 J/cm²
15 J/cm²
10 J/cm²
Figura 5.10: Variação na cristalinidade dos filmes em função do aumento da fluência do laser,
para os filmes produzidos à temperatura ambiente. Esses difratogramas exibem apenas os picos
de fase cristalina referente à HAP.
Para conhecer a influência do tempo de deposição na formação da fase cristalina,
os filmes produzidos com 25 J/cm2 apresentaram cristalinidade a partir de 2 minutos de
deposição nos 3 planos mais intensos para a HAP, como mostram os difratogramas da

73
Figura 5.11.O recobrimento produzido em 2 minutos tem aproximadamente 60 nm de
espessura.
Figura 5.11: Difratogramas com variação no tempo de deposição para 25 J/cm2. Os picos mais
intensos da HAP aparecem a partir de 2 min de deposição. Todos os picos correspondem à fase
cristalina da HAP.
Tanto a pressão de deposição quanto a distância entre alvo e substrato
influenciam diretamente na formação do filme através do livre caminho médio das
partículas da pluma de plasma. Nesse trabalho, considerando que a deposição foi
realizada sob alto vácuo e com atmosfera não-reativa, a influência da pressão de
deposição pode ser verificada na Figura 5.12 (a) e (b) para os filmes produzidos com 25
e 30 J/cm2, respectivamente. Sob baixo vácuo de 10-2 mbar, a quantidade de material
depositado e a cristalinidade decrescem significativamente quando a distância alvo-
substrato passa de 2 para 3 cm, pois aumenta a interação do plasma com as partículas no
interior da câmara, diminuindo o livre caminho médio e aumentando a distância que as
partículas energéticas tem que percorrer para alcançar o substrato. De forma análoga,
para mais alto vácuo e distância de 3 cm, o plasma tem maior expansão e as partículas
chegam ao substrato sem grandes interferências com partículas na atmosfera de
deposição.

74
Figura 5.12: Influência da pressão de deposição e da distância entre alvo-substrato para filmes
depositados com (a) 25 J/cm2 e (b) 30 J/cm2. Os resultados apresentam maior quantidade de
material depositado sob mais baixa pressão P = 10-6 mbar e distância fixa de 3 cm. Todos os
filmes apresentaram fase cristalina da HAP.
Para menor distancia alvo-substrato as partículas ejetadas da ablação chegam ao
substrato com mais alta energia. Essa energia pode ser suficiente para provocar
deposição por implantação de íons e partículas e/ou aumentar a mobilidade dos átomos
na superfície dos filmes, produzindo assim filmes mais densos, cristalinos e altamente
aderentes ao substrato (veja Seção 3.2.5). Por outro lado, a incidência de partículas com
alta energia nos substratos também pode provocar defeitos estruturais durante o
crescimento do filme, pulverização por íons retroespalhados e retirar átomos fracamente
adsorvidos na camada superficial do recobrimento. Essas características podem saturar a
taxa de ablação. Além disso, a energia pode ser suficiente para induzir a formação de
outras fases. Isso foi observado quando depositamos o filme sob os mesmo parâmetros,
porém com 1 cm de distância entre alvo e substrato. Como consequencia desses efeitos,
o recobrimento apresentou decomposição da HAP em fases de CaO (ICDD 01-082-
1690) e β-Tcp (ICDD 04-008-8714). O resultado é mostrado na Figura 5.13, e o mesmo
efeito foi observado para fluências de 10 J/cm2 e de 30 J/cm2. Portanto, nesse caso, a
decomposição da HAP em outras fases foi provocada por partículas hipertérmicas (1 -
103 eV) produzidas como resultado direto dos parâmetros de deposição [43].

75
Figura 5.13: Difratogramas para dois filmes produzidos com distância alvo-substrato de 1 cm e
fluências de (a) 10 J/cm2 e (b) 30 J/cm2. Sob essas condições de deposição, os filmes
apresentaram decomposição de fase em β-Tcp e CaO.
Figura 5.14: e ausência de fases ocultas no recobrimento produzido à temperatura ambiente.
Para os recobrimentos produzidos à temperatura ambiente e que apresentaram alto
grau de cristalinidade, verificamos a estabilidade com tratamentos térmicos durante e
após a deposição.
Os filmes cristalinos produzidos à temperatura ambiente (Figura 5.10) foram
submetidos a tratamento térmico após deposição. Os resultados para as amostras de
mais baixa (10 J/cm2) e mais alta fluência (38 J/cm2) são mostrados na Figura 5.14(a) e
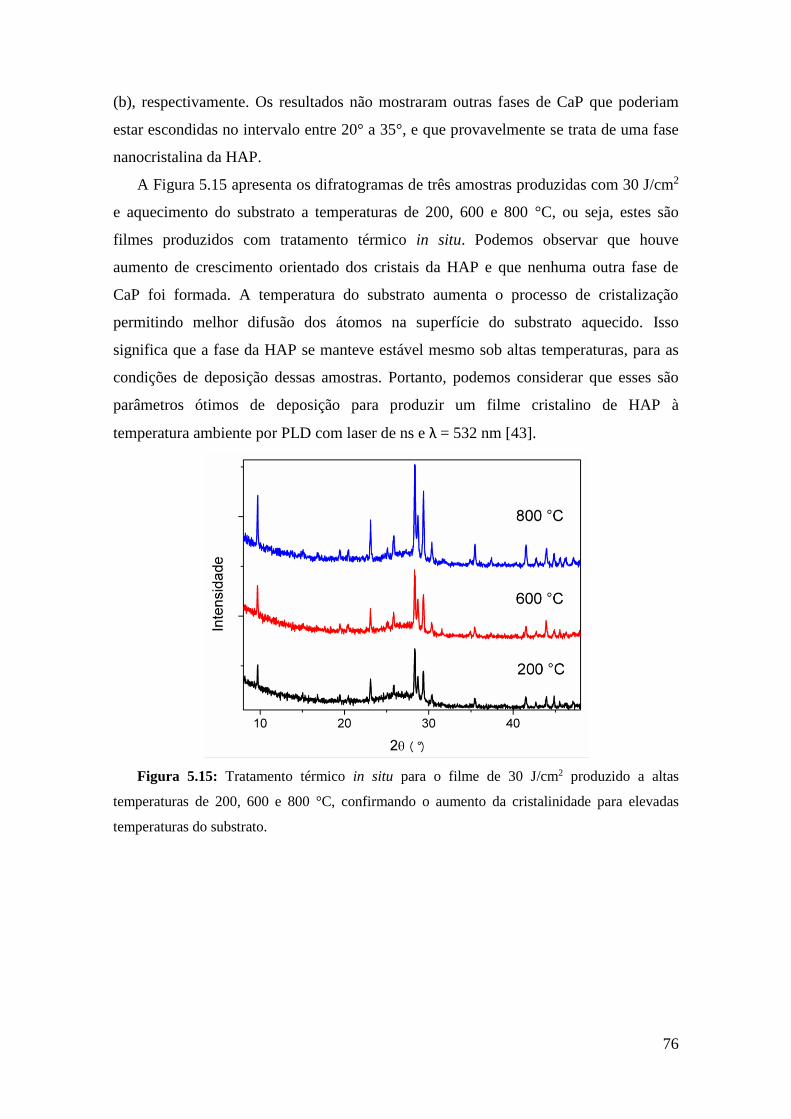
76
(b), respectivamente. Os resultados não mostraram outras fases de CaP que poderiam
estar escondidas no intervalo entre 20° a 35°, e que provavelmente se trata de uma fase
nanocristalina da HAP.
A Figura 5.15 apresenta os difratogramas de três amostras produzidas com 30 J/cm2
e aquecimento do substrato a temperaturas de 200, 600 e 800 °C, ou seja, estes são
filmes produzidos com tratamento térmico in situ. Podemos observar que houve
aumento de crescimento orientado dos cristais da HAP e que nenhuma outra fase de
CaP foi formada. A temperatura do substrato aumenta o processo de cristalização
permitindo melhor difusão dos átomos na superfície do substrato aquecido. Isso
significa que a fase da HAP se manteve estável mesmo sob altas temperaturas, para as
condições de deposição dessas amostras. Portanto, podemos considerar que esses são
parâmetros ótimos de deposição para produzir um filme cristalino de HAP à
temperatura ambiente por PLD com laser de ns e λ = 532 nm [43].
Figura 5.15: Tratamento térmico in situ para o filme de 30 J/cm2 produzido a altas
temperaturas de 200, 600 e 800 °C, confirmando o aumento da cristalinidade para elevadas
temperaturas do substrato.

77
5.3. Caracterização físico-química dos filmes produzidos à
temperatura ambiente
Após deposição e caracterização dos filmes de HAP em substratos de Si, os
recobrimentos foram produzidos sobre substratos de PLA e Ti. Nesta seção, são
apresentados os principais resultados sobre os recobrimentos produzidos nas condições
ideais de deposição proposta nesta tese.
5.3.1. Cristalinidade e composição de fase
Os padrões de difração do alvo e dos filmes de Si/HAP são apresentados na Figura
5.16 (b-e). Esses difratogramas exibem apenas os picos de fase cristalina referente à
HAP, de acordo com a ficha ICDD nº 84-1998, mostrado na Figura 5.16-a. Os filmes
apresentam crescimento preferencial ao longo do plano do Si (100) quando comparado
ao padrão de difração do alvo (Figura 5.16-b) e uma estrutura larga entre 20° e 35°, que
geralmente está relacionada à existência de fosfato de cálcio amorfo. Entretanto, isso
também pode representar uma cristalinidade de curto alcance formado por cristais da
ordem de 3 a 4 nm [75]. Os difratogramas dos filmes produzidos com substrato
aquecido a 200 °C (Figura 5.16-d) e 800 °C (Figura 5.16-e) também mostraram que não
há qualquer outra fase de CaP além da HAP. Isso significa que a parte nanocristalina
dos recobrimentos é também HAP e não oculta fases de decomposição, como tricálcio
fosfato (β-TCP), óxido de cálcio (CaO) e outras.
Para cada amostra, é possível estimar uma quantidade de cristalinidade relativa pela
razão entre a área dos picos e a contribuição nanocristalina [70]. Essa quantidade
percentual de cristalinidade relativa, apresentada à direita dos difratogramas na Figura
5.16, mostrou que uma temperatura in situ mais alta é responsável pelo aumento da
cristalinidade a longo alcance (de 44% para 68%), pois a energia disponível permite o
aumento do domínio de cristal e reduz a fase nanocristalina.

78
Figura 5.16: (a) difratograma padrão de acordo com a ficha ICDD nº 84-1998. Padrões de
difração e respectivas cristalinidades relativas para (b) alvo de HAP, (c) recobrimento de HAP
depositado a temperatura ambiente (RT – room temperature) com fluência de 30 J/cm2 e quando
depositado com tratamento térmico in situ a (d) 200°C e (e) 800°C.
Os filmes produzidos na condição ideal foram depositados sobre substratos de PLA
durante 20 minutos. O padrão de difração da amostra PLA/HAP foi obtido com
difratômetro comercial com fonte Cu (Kα) e confirma a existência de uma fase referente
a HAP (ICDD nº 09-0432), conforme mostra a Figura 5.17. Os picos de HAP são
mostrados no intervalo 2θ entre 20° e 40° junto com fortes picos cristalinos do substrato
de PLA em 16,4° e 18,7° [71]. Estruturas largas são observadas entre 15° e 25° e entre
28° e 40° e podem ser atribuídos a uma estrutura amorfa de PLA ou uma fase com
cristalinidade de curto alcance de HAP. Esses resultados, combinados com o espectro
EDS da Figura 5.27f confirmam a deposição de HAP cristalina sobre substrato de PLA.

79
Figura 5.17: Recobrimento de PLA/HAP produzido em 20 min de deposição nas condições
ideais de deposição.
5.3.2. Composição e estequiometria de superfície
A estequiometria e composição de superfície dos recobrimentos foram analisadas
por XPS. O espectro total e os espectros de alta resolução de energia, para os
recobrimentos produzidos a temperatura ambiente, são apresentados nas Figuras 5.18 e
5.19, respectivamente. A Figura 5.18 mostra a presença dos elementos típicos que
compõem a estequiometria da HAP, ou seja, Ca 2p, P 2p e O 1s [77-81]. A partir dos
espectros de alta resolução, as energias de ligação correspondentes ao Ca 2p3/2, P 2p, e
O 1s foram 346,9 eV, 132,9 eV, and 530,9 eV, respectivamente. O pico correspondente
ao C 1s em 284,6 eV também foi observado, uma vez que não foi feita uma limpeza
iônica da superfície (sputtering) antes da medida. O pico C1s é atribuído à
contaminação orgânica na superfície, devido à exposição da superfície da amostra ao
carbono de origem orgânica presente no ar. Além disso, o espectro de XPS ainda
apresenta as linhas espectrais dos elétrons Auger do oxigênio e do cálcio presentes na
amostra.

80
Figura 5.18: Espectro total de XPS do recobrimento de HAP produzido por 30 J.cm-2 com a
identificação dos elementos presentes na superfície.
Figura 5.19: Espectros de XPS de alta resolução e respectivos ajustes para o recobrimento de
HAP produzido com 30 J.cm-2: (a) Ca 2p, (b) P 2p, (c) O 1s, e (d) C 1s.
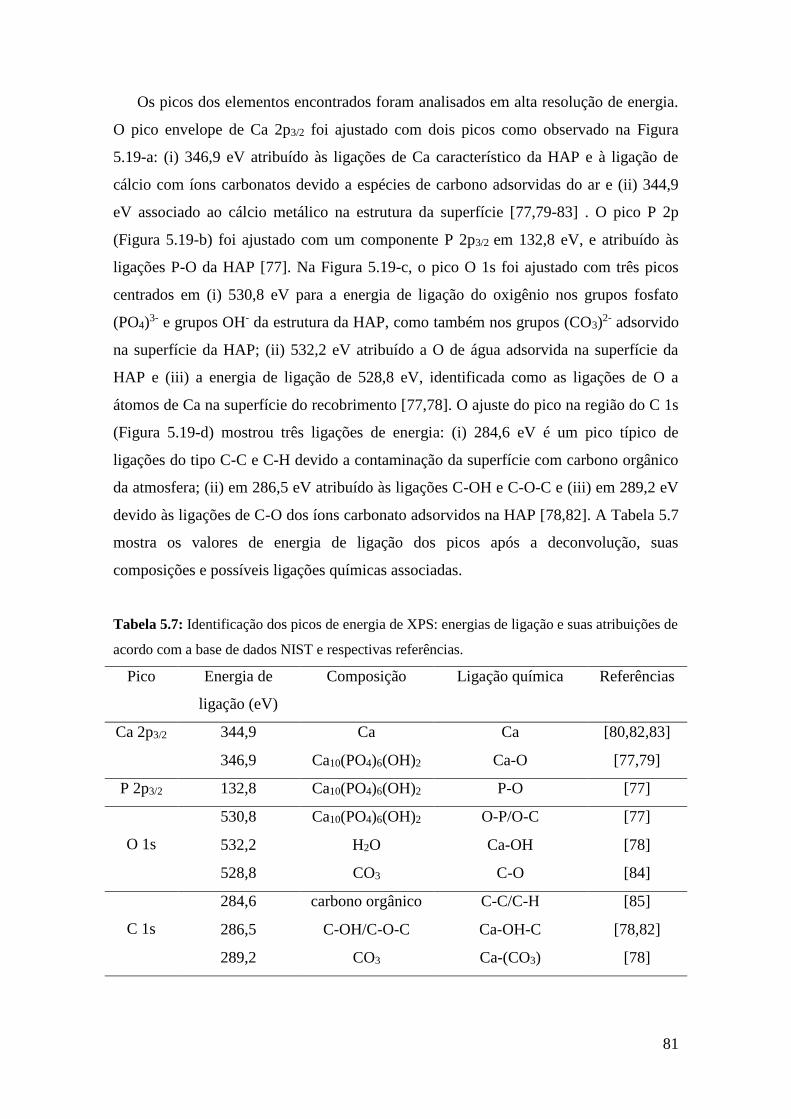
81
Os picos dos elementos encontrados foram analisados em alta resolução de energia.
O pico envelope de Ca 2p3/2 foi ajustado com dois picos como observado na Figura
5.19-a: (i) 346,9 eV atribuído às ligações de Ca característico da HAP e à ligação de
cálcio com íons carbonatos devido a espécies de carbono adsorvidas do ar e (ii) 344,9
eV associado ao cálcio metálico na estrutura da superfície [77,79-83] . O pico P 2p
(Figura 5.19-b) foi ajustado com um componente P 2p3/2 em 132,8 eV, e atribuído às
ligações P-O da HAP [77]. Na Figura 5.19-c, o pico O 1s foi ajustado com três picos
centrados em (i) 530,8 eV para a energia de ligação do oxigênio nos grupos fosfato
(PO4)3- e grupos OH- da estrutura da HAP, como também nos grupos (CO3)
2- adsorvido
na superfície da HAP; (ii) 532,2 eV atribuído a O de água adsorvida na superfície da
HAP e (iii) a energia de ligação de 528,8 eV, identificada como as ligações de O a
átomos de Ca na superfície do recobrimento [77,78]. O ajuste do pico na região do C 1s
(Figura 5.19-d) mostrou três ligações de energia: (i) 284,6 eV é um pico típico de
ligações do tipo C-C e C-H devido a contaminação da superfície com carbono orgânico
da atmosfera; (ii) em 286,5 eV atribuído às ligações C-OH e C-O-C e (iii) em 289,2 eV
devido às ligações de C-O dos íons carbonato adsorvidos na HAP [78,82]. A Tabela 5.7
mostra os valores de energia de ligação dos picos após a deconvolução, suas
composições e possíveis ligações químicas associadas.
Tabela 5.7: Identificação dos picos de energia de XPS: energias de ligação e suas atribuições de
acordo com a base de dados NIST e respectivas referências.
Pico Energia de
ligação (eV)
Composição Ligação química Referências
Ca 2p3/2 344,9 Ca Ca [80,82,83]
346,9 Ca10(PO4)6(OH)2 Ca-O [77,79]
P 2p3/2 132,8 Ca10(PO4)6(OH)2 P-O [77]
O 1s
530,8 Ca10(PO4)6(OH)2 O-P/O-C [77]
532,2 H2O Ca-OH [78]
528,8 CO3 C-O [84]
C 1s
284,6 carbono orgânico C-C/C-H [85]
286,5 C-OH/C-O-C Ca-OH-C [78,82]
289,2 CO3 Ca-(CO3) [78]

82
Os valores das razões atômicas Ca/P = 1,85 e C/Ca = 0,17 foram obtidos a partir
do valor da área do pico dividido pelo fator de sensibilidade (F.S.) do respectivo
elemento (Tabela 5.8). Os valores de F.S. são tabelados para cada elemento, e considera
também a função trabalho do equipamento de XPS utilizado [82]. As razões atômicas
representam a proporção atômica na superfície de aproximadamente 2,5 nm de
profundidade. Esse valor foi obtido a partir de simulações da trajetória livre média
inelástica dos fotoelétrons excitados pela fonte de raios X de Al(Kα) na estrutura do
recobrimento de HAP [81,86].
O alto valor da razão Ca/P (>1,67) pode ser explicado pela adsorção de CO3, que
entra na superfície do recobrimento e muda a sua estequiometria. Já se sabe que a HAP
tem grande afinidade para adsorver os gases CO2 formando uma HAP carbonatada na
sua superfície [87]. Além disso, a técnica de PLD produz recobrimentos rugosos e a
grande área de superfície também favorece a adsorção de contaminantes presentes no ar.
A partir desses resultados, a seguinte equação de equilíbrio pode ser sugerida para esta
superfície: Ca+210(PO4)
-35.4(CO3)
-21.7(OH)-1
0.4. Essa estequiometria é rica em substituição
do tipo A, onde os íons carbonato (CO3)-2
substituem principalmente os íons hidroxila
(OH)-1 na superfície do filme [38]. Isso é confirmado pelas medidas de FTIR, onde se
observa um decréscimo na intensidade da banda OH em 3570 cm-1 (Figura 5.20a) [88].
Tabela 5.8: Área obtida após os ajustes de deconvolução e área corrigida pelo fator de
sensibilidade de cada elemento.
Picos Área obtida F.S. Área corrigida
Ca2p 3/2 110030,1 3,35 32844,8
P2p 3/2 13979,4 0,789 17717,9
O 1s 182021,3 2,93 182021,3
C 1s 5485,6 1 5485,6
A composição química dos recobrimentos também foi analisada por FTIR. Os
espectros da HAP em pó e do recobrimento produzido com 30 J.cm-2 de fluência são
apresentados nas Figuras 5.20a e b, respectivamente. O recobrimento de HAP apresenta
os modos de vibração do fosfato de cálcio (PO4)3- em 1020 cm-1, 947 cm-1, 596 cm-1,
566 cm-1 e 470 cm-1 e do grupo iônico (OH) em 3570 cm-1. Esses modos são
tipicamente encontrados no espectro da HAP. Geralmente bandas mais largas como as
observadas na Figura 5.20b sugerem a formação de uma estrutura local desordenada.
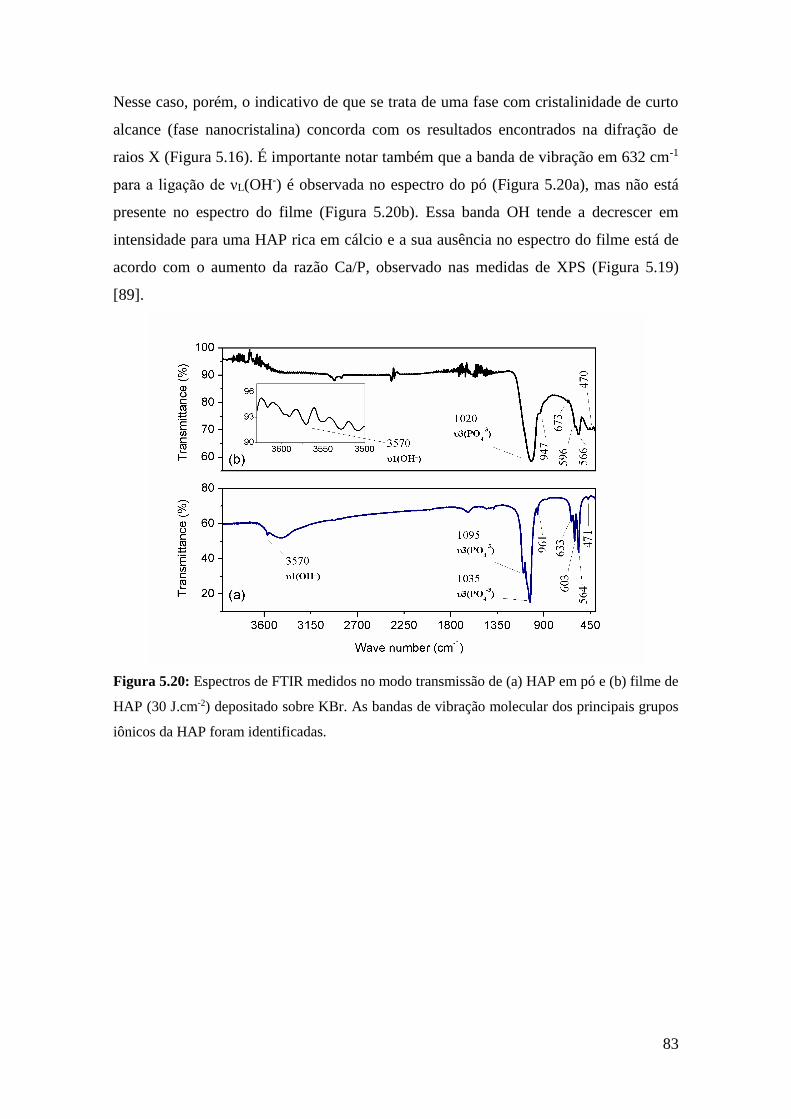
83
Nesse caso, porém, o indicativo de que se trata de uma fase com cristalinidade de curto
alcance (fase nanocristalina) concorda com os resultados encontrados na difração de
raios X (Figura 5.16). É importante notar também que a banda de vibração em 632 cm-1
para a ligação de νL(OH-) é observada no espectro do pó (Figura 5.20a), mas não está
presente no espectro do filme (Figura 5.20b). Essa banda OH tende a decrescer em
intensidade para uma HAP rica em cálcio e a sua ausência no espectro do filme está de
acordo com o aumento da razão Ca/P, observado nas medidas de XPS (Figura 5.19)
[89].
Figura 5.20: Espectros de FTIR medidos no modo transmissão de (a) HAP em pó e (b) filme de
HAP (30 J.cm-2) depositado sobre KBr. As bandas de vibração molecular dos principais grupos
iônicos da HAP foram identificadas.

84
Figura 5.21: Espectros de FTIR de (a) substrato de PLA e (b) PLA recoberto com HAP. As
atribuições das bandas de vibração molecular identificadas estão na Tabela 5.9.
Medidas de FTIR também foram realizadas em amostras de PLA/HAP. O
espectro de FTIR do substrato de PLA (Figura 5.21a) apresenta seus modos de vibração
típicos [90,91]. Quando recoberto usando o processo de PLD, os modos de vibração do
fosfato de cálcio (PO4)3- para uma HAP desordenada com carbonatos estão presentes em
uma banda larga centrada em 1018 cm-1 (Figura 5.21b) [89]. A Tabela 5.9 apresenta
todas as bandas vibracionais para o recobrimento PLA/HAP e suas atribuições de
ligação química.
Table 5.9: Identificação das bandas vibracionais para o espectro de infravermelho da amostra
PLA/HAP na Figura 5.21b.
Posição do pico
(cm-1) Atribuição Referência
1735 -C=O do PLA [90]
1450 -CH3 do PLA [90]
1381, 1357 -CH- do PLA [90]
1263 -C=O do PLA [90]
1182 -C-O- do PLA [90]
1018 P-O da HAP [89]
869 PLA [91]
754 PLA [91]

85
5.3.3. Morfologia e composição estrutural
O recobrimento de HAP produzido por PLD, nas condições de deposição
estabelecidas nesse trabalho, é indicado como um filme denso, não poroso, homogêneo
e rugoso, de acordo com imagens de microscopia eletrônica e de microscopia atômica.
A presença de partículas aderidas sobre a superfície do recobrimento é uma
característica inerente da técnica. Do ponto de vista morfológico, os filmes podem ser
descritos por três componentes: (1) uma camada densa que cobre completamente a
superfície do substrato, (2) partículas com tamanhos que variam de nanômetros até 10
μm distribuídas homogênea e uniformemente sobre o recobrimento denso. Essas
partículas foram condensadas em formato de roscas ou de discos finos. E (3) partículas
maiores agregadas ao filme por energia cinética durante a ejeção. Essas últimas
distribuem-se de forma mais espaçada do que as outras (Figura 5.22).
Figura 5.22: Imagens de MEV dos filmes de HAP depositados sobre substrato de Si com (a)
magnificação de 150x e (b) magnificação de 1000x. As barras de escala são de 100 μm e 10
μm, respectivamente.
Uma melhor representação das estruturas que formam o recobrimento é apresentada
na Figura 5.23, por três imagens observadas em um mesmo ponto em corte transversal,
apenas com diferença de distância focal entre elas. Na Figura 5.23a, o foco sobre a
superfície clivada permite observar a espessura do filme (~250 nm para 15 min de
deposição) que cobre completamente o substrato de Si. Ao variar a distância focal em
mais dois estágios sobre a superfície do filme, é possível observar a presença dos dois
tipos de partículas agregadas à superfície, que são as partículas ejetadas diretamente do
alvo (Figuras 5.23b) e as partículas que foram coalescidas e condensadas sobre a
superfície do filme (Figura 5.23c).

86
Figure 5.23: Imagens de seção transversal do recobrimento de HAP com: (a) uma camada
densa com espessura de ~250 nm, (b) uma partícula micrométrica supostamente não fundida e
(c) partícula que passou por processo de fusão antes de se aglomerar ao recobrimento.
A interação do laser pulsado com o alvo de HAP causa um aquecimento local e uma
rápida emissão de vapor, aglomerados e micro-grãos ou partículas. Por essa razão, a
existência das partículas sobre a superfície é um artefato inerente à técnica PLD,
especialmente nesse trabalho, já que o material do alvo tem baixa absorção de fótons no
comprimento de onda de 532 nm [20]. A distribuição de tamanho de partículas é uma
característica que depende do comprimento de onda do laser, além da temperatura e da
superfície do substrato. A morfologia apresentada nas Figuras 5.22 e 5.23 é muito
similar aos resultados encontrados na literatura [20].
A rugosidade e topografia de superfície também foram investigadas por AFM. A
Figura 5.24 mostra que a camada densa é formada pela aglomeração de partículas
fundidas, depositadas umas sobre as outras e que cobrem completamente o substrato.
Além disso, algumas partículas micrométricas apresentam coalescência, sendo o centro
uma região mais plana em relação às bordas. A rugosidade média de 100,4 nm para uma
área de 10x10 µm foi calculada com o software Gwyddion.

87
Figura 5.24: Medidas de AFM do recobrimento produzido à temperatura ambiente com
fluência de 30 J.cm-2: (a) representação 2D, (b) representação 3D e (c) perfil linear de
rugosidade.
5.3.4. Teste de adesão do filme ao substrato
A adesão do recobrimento ao substrato é considerada um fator importante para
fabricação de implantes de alta qualidade porque tem influencia direta na estabilidade a
longo prazo dos implantes em meio biológico. Testes de adesão por fita (tape test)
foram realizados sobre os filmes depositados em substratos de Si e PLA. O
procedimento foi realizado segundo o método B da norma ASTM D3359 [72] e
resultados apresentaram boa adesão dos filmes ao substratos, sendo classificados como
5B (nenhum descolamento) As imagens obtidas antes e depois do teste (Figura 5.25)
mostraram que alguns aglomerados foram removidos pela fita, mas o filme permaneceu
no substrato, como pode ser visto na fotografia da Figura 5.26.

88
Figure 5.25: Imagens dos fimes de HAP depositados sobre Si (a) antes e (b) depois, e sobre
PLA (c) antes e (d) depois do teste de adesão
Figura 5.26: Fotografia tirada após o teste de adesão dos recobrimentos de HAP produzidos à
temperatura ambiente e depositados sobre substratos de Si (à esquerda) e PLA (à direita). O
procedimento utilizado seguiu o método B da norma ASTM D3359 [72].

89
5.3.5. Teste de ângulo de contato
Depois de caracterizados em substratos de Si, os recobrimentos foram depositados
sobre substrato de PLA para teste de ângulo de contato. O resultado, apresentado na
Figura 5.27 a e b apresenta a mudança na hidrofobicidade da superfície devido a
existência do recobrimento de HAP no substrato de PLA. Considerando a topografia de
superfície, as imagens de MEV (Figuras 5.27 c e d) da amostra de PLA/HAP mostra
que o filme de HAP cobre completamente e uniformemente a superfície do substrato de
PLA. Na Figura 5.27d as partículas micrométricas previamente descritas são visíveis.
Na Figura 5.27 e-f, o espectro EDS mostra os principais elementos presentes nas
amostras de PLA e de PLA recoberto com HAP, respectivamente.
Figure 5.27: Caracterização do substrato de PLA (à esquerda) e após a deposição do
recobrimento de HAP (à direita): (a-b) teste de ângulo de contato (c-d) imagens de MEV e (e-f)
espectros EDS.
O rápido crescimento da HAP nos filmes produzidos por PLD pode ser associado a
alta molhabilidade da superfície. Uma superfície hidrofílica favorece a adesão, migração

90
e proliferação celular, e os osteoblastos se prendem mais facilmente a superfícies
hidrofílicas [44]
5.4. Ensaios preliminares de adesão e proliferação celular
O procedimento experimental para os primeiros ensaios biológicos (Seção 4.7 do
Capítulo 4) foi proposto para observar adesão e cinética de tempo de crescimento e
proliferação celular sobre a superfície dos filmes de HAP. Os testes foram realizados em
amostras de Ti/HAP (Ti filme), Ti controle (Ti puro), PLA/HAP (PLA filme) e PLA
controle (PLA puro) durante tempos de 4h, 24h e 48h.
A Figura 5.28 apresenta duas imagens representativas de cada amostra após 4h em
cultura celular. Para ambos os substratos, observamos uma diminuição de população de
células nas amostras com filme, quando comparadas às amostras sem o recobrimento de
HAP. Entretanto, essa característica é mais evidente entre as amostras de Ti filme e Ti
puro. Para as amostras de 24h, foi realizada quantificação por triplicata com cinco
imagens para cada amostra, total de 15 imagens. As imagens representativas para as
amostras de 24h com marcação DAPI estão apresentadas na Figura 5.29. Entre os
tempos de 4h e 24h houve aumento no número de células para as amostras de Ti puro e
Ti filme. Porém, o mesmo não se pode afirmar sobre as amostras de PLA puro e PLA
filme. A Figura 5.30 apresenta o histograma de contagem celular para as quatro
amostras no tempo de 24h. A contagem foi realizada com software Image-Pro Plus para
as amostras de Ti puro e Ti filme, e contagem manual/visual foi usada para as amostras
de PLA puro e PLA filme.
Figura 5.28: Imagens representativas das amostras de 4hs. Ampliação de 10x da lente objetiva.

91
Figura 5.29: Imagens representativas para as amostras de 24h com marcação DAPI. Barra de
escala de 100 μm.
Nas amostras de 48h, foi realizada marcação tripla para observar os filamentos de
actina formados na adesão celular. As imagens representativas para as quatro amostras
são apresentadas na Figura 5.31. Os resultados mostram que, com exceção da amostra
de Ti, não houve espraiamento celular. Na amostra de PLA filme, o marcador de
faloidina (vermelho) está sendo absorvido pelo filme, que pode estar absorvendo
também as propriedades ou nutrientes das células, impedindo sua proliferação.
Provavelmente, esse mesmo efeito ocorre entre as amostras de Ti puro e Ti filme. Nesse
sentido, um processo de cultura dinâmica pode fornecer resultados mais conclusivos.
(a)Ti puro
(c)PLA puro
(b) Ti filme
(d) PLA filme

92
Figura 5.30: Histograma de quantificação celular para as amostras de 24h.
Ao analisarmos os resultados, constatamos a presença de muitos fatores que
possivelmente influenciaram a baixa adesão celular nas amostras com o filme de HAP.
É importante considerar, por exemplo, a diferença na morfologia e rugosidade entre as
quatro amostras, que por sua vez modifica a energia de superfície. De fato, as amostras
de Ti foram tratadas com lixa de até 1500 e depois com ataque ácido, o que forneceu
rugosidade micrométrica ao substrato. Nas amostras de Ti filme, essa rugosidade foi
substituída pela presença do filme, cuja morfologia foi apresentada na Figura 5.22
(Seção 5.3.3) e apresenta mais baixa rugosidade, pois o filme depositado preenche as
ranhuras profundas do substrato de Ti lixado. Isso sugere que a maior adesão celular nas
amostras de Ti puro pode ser ter sido favorecida pela sua rugosidade.
Nas amostras de PLA, observamos a preferência das células em se alojarem nas
fendas da superfície, especialmente nas amostras de PLA puro. Os substratos de PLA
apresentam fendas ou sulcos micrométricos provenientes da deposição por filamentos
no processo de manufatura aditiva. A partir das imagens nas Figuras 5.28 e 5.29, fica
claro que a dimensão desses sulcos é conveniente à adesão celular, quando comparado à
“superfície dos filamentos”, apresentada por uma morfologia mais suave. Essa
superfície mais suave está mostrada na Figura 5.27d da Seção 5.3.5. Nessa figura, a área
foi cautelosamente escolhida para a quantificação por EDS, evitando os sulcos
micrométricos da superfície. De certa forma, a presença desses sulcos nos substratos de

93
PLA não teve influência na caracterização físico-química dos recobrimentos, mas
comprometeu a interpretação dos resultados nos testes biológicos. Com relação à
morfologia das amostras de PLA filme (PLA/HAP) observa-se maior quantidade de
células aderidas sobre a “superfície dos filamentos”, que nesse caso apresentam a
rugosidade do filme de HAP, como pode ser observado nas imagens da Figura 5.27(c-
d). Dessa forma, podemos afirmar que o PLA puro provocou adesão de maior
quantidade de células, porém o PLA filme permitiu maior dispersão da adesão celular.
A presença do recobrimento na amostra de PLA filme favoreceu uma distribuição
celular de forma mais homogênea, sugerindo que o filme depositado preenche os sulcos
existentes no PLA puro. As diferentes rugosidades entre as quatro amostras, e inclusive
entre as amostras controle, é uma questão importante que deve ser levada em conta na
interpretação dos resultados. De fato, resultados da literatura relatam o efeito relativo da
rugosidade na adesão e proliferação celular [92].
Além da cristalinidade e morfologia, a química e a energia de superfície também são
propriedades de governam as funcionalidades biológicas de um implante. Uma vez
introduzido no corpo humano, a superfície do implante é rapidamente coberta com
proteínas pelo sangue ou fluidos intersticiais que constroem uma camada adsorvida
uniforme através da qual as células interagem com o implante [44,92]. Em ensaios
biológicos, as células induzem a formação e deposição de uma camada de soro protéico
sobre a superfície da amostra. Essa camada protéica pode estar sendo absorvida pelo
substrato de PLA e/ou pelo filme de HAP. Tanto as características em escala micro
quanto em escala nano são importantes porque as células humanas primárias são
capazes de reagir com micro e nano topografias, enquanto que a adsorção de proteínas é
influenciada pela morfologia em escala nanométrica [92].
Finalmente, a metodologia de contagem de células por área para a quantificação
realizada na amostra de 24h não forneceu uma boa estatística. Foram consideradas cinco
imagens com área de 1,5 mm² cada, de forma que a área total observada para cada
amostra é de 7,5 mm². Sendo a amostra uma pastilha de raio 5 mm, então a área total é
de 78,54 mm². Portanto, a área observada para quantificação foi cerca de 10% da área
total da amostra. A quantificação de células por amostra que considere a área total,
obtida com microscópio automático, por exemplo, poderia fornecer uma estatística mais
confiável. Portanto, o fato de termos muitas variáveis envolvidas dificulta a
interpretação dos dados. Outros ensaios complementares são necessários para correta e
efetiva conclusão dos resultados.
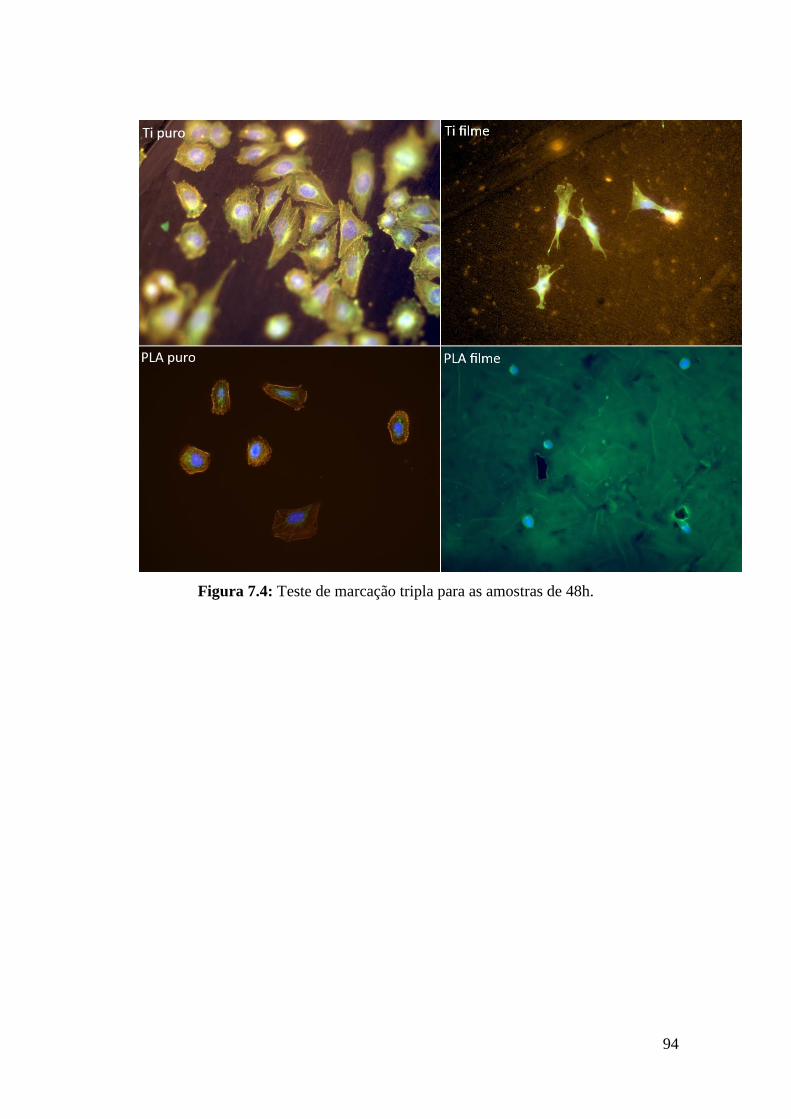
94
Figura 7.4: Teste de marcação tripla para as amostras de 48h.

95
6. CONCLUSÕES
1) Os objetivos propostos neste trabalho de tese foram alcançados e alguns desses
resultados foram publicados na literatura científica [43].
2) Esse trabalho descreve a produção de recobrimentos de HAP cristalina à
temperatura ambiente usando PLD com alta fluência do laser Nd:YAG 532 nm.
Os recobrimentos depositados são caracterizados por microestrutura compacta e
bem aderida ao substrato, formada por uma camada nanométrica homogênea
com uma superfície irregular que é característico do PLD.
3) Tratamentos térmicos realizados durante a deposição mostraram que a
componente não cristalina dos recobrimentos consiste de HAP nanocristalina e
não degrada em fases indesejadas de CaP e CaO. Tratamentos térmicos
realizados após a deposição mostraram que os filmes permaneceram estáveis sob
altas temperaturas de 800°C.
4) Os tratamentos térmicos foram realizados apenas para verificar a estabilidade
do material e a possível presença de outras fases ocultas na parte nanocristalina
ou amorfa presentes nos difratogramas, e que são comumente observados nos
filmes de HAP produzidos por PLD. Nesse trabalho, os recobrimentos
cristalinos de HAP foram produzidos à temperatura ambiente com taxa de
deposição de 30 nm/min. Esse resultado é essencial para depositar esses
recobrimetos em substratos sensíveis a temperatura como os biopolímeros.
5) Apesar de uma ablação conduzida sob baixa absorbância da HAP no
comprimento de onda do laser 532 nm, o processo realizado em regime
hipertérmico garantiu o mecanismo de expulsão de partículas de alta energia a
partir de uma ablação não coerente. Essa condição permite a produção de HAP
cristalina devido a transferencia de energia cinética das partículas e íons durante
a formação e crescimento do filme.

96
6) Um alvo denso e estequiométrico é desejado a fim de suportar a alta fluência do
laser e promover o crescimento de recobrimentos puros de HAP. A
estequiomeria dos alvos de HAP foi analisada por uma técnica não destrutiva de
espectroscopia óptica OPC-LIBS, que se mostrou válida quando comparada com
técnicas analíticas convencionais, mas que necessitam de digestão da amostra.
7) Os recobrimentos foram aplicados a substratos de Ti e de PLA sensível a
temperatura e apresentaram boa adesão ao substrato.
8) O uso de maior comprimento de onda e fluência gera mais alta taxa de ablação,
permitindo a deposição de 150 nm de HAP cristalina em apenas 5 minutos. Esse
resultado apresenta taxa de ablação de 30 nm/min que é no mínimo 6 vezes mais
alta do que comparado com a técnica de magnetron sputtering. Isso é um fator
importante quando consideramos a perspectiva de produção em larga escala,
como por exemplo a aplicação comercial desses recobrimentos em implantes
biomédicos.
9) Os ensaios biológicos realizados nesse trabalho de tese forneceram resultados
inconclusivos e mais testes devem ser realizados para efetiva caracterização
biológica dos filmes produzidos.

97
7. SUGESTÕES DE TRABALHOS FUTUROS
1) PLD com espectrômetro de massa.
2) PLD com RHEED.
3) Simulação de dinâmica molecular. Os mecanismos básicos da ablação a laser podem
ser investigados através de simulações baseadas em processos não lineares de
dinâmica molecular. A expansão rápida dos fragmentos em um processo de ablação
geralmente é estudada a fim de investigar os processos iniciais de formação de
filmes e de nanopartículas produzidas por PLD. As simulações são um meio
eficiente de prever e analisar os efeitos obtidos experimentalmente.
4) Investigar a dinâmica de crescimento do filme por microscopia eletrônica de
transmissão (TEM). Os filmes produzidos nesta tese podem ser preparados em FIB
(Focused Ion Beam) e observados em TEM, a fim de observar os fenômenos de
interação na interface entre o filme e os substratos de Si, Ti e PLA. Além disso, os
estágios iniciais de crescimento do filme podem ser observados ao depositar em
grades de carbono para TEM. Testes preliminares foram realizados e estão em
processo de análise.
5) Realizar ensaios mecânicos de adesão e dureza (scrash tests).
6) Espectroscopia do plasma produzido durante a ablação e crescimento dos filmes.
Relacionar as propriedades de temperatura e densidade do plasma às características
dos filmes produzidos.
7) Testes com células pré-osteoblásticas MC3T3. As células MC3T3 são células de
rato pré-osteoblásticas, e menos sensíveis do que as células SAOS.
8) Testes de adsorção de proteínas. Já se sabe que a HAP adsorve proteínas, tais como
albumina e osteocalcina, enquanto que o Ti e o PLA não adsorvem. Sugere-se que o
filme apresente maior aceitação de adesão e proliferação quando em contato prévio
com biofilme ou proteínas.
9) Testes in-vitro de cultura dinâmica. Um processo de cultura dinâmica tem melhor
representatividade de um meio biológico real, pois simula processos inflamatórios e
regenerativos com fluxo de células e proteínas, como ocorre dentro do corpo.
10) Testes biológicos in-vivo. Sugere-se que sejam realizados testes in-vivo em calvário
de ratos.

98
8. REFERÊNCIAS BIBLIOGRÁFICAS
[1] N. Eliaz, N. Metoki, Calcium Phosphate Bioceramics: A Review of Their History, Structure,
Properties, Coating Technologies and Biomedical Applications, Materials 10 (2017) 334.
[2] W. Habraken, P. Habibovic, M. Epple, M. Bohner, Calcium phosphates in biomedical applications:
materials for the future?, Mater. Today 19 (2016) 69–87.
[3] R. Bosco, J.V.D. Beucken, S. Leeuwenburgh and J. Jansen, Surface Engineering for Bone Implants-
A Trend from Passive to Active Surfaces, Coatings 2 (2012) 95-119.
[4] D. Duraccio, F. Mussano, M.G. Faga, Biomaterials for dental implants: current and future trends, J.
Mater. Sci. 50 (2015) 4779–4812.
[5] R.A. Surmenev, M.A. Surmeneva, A.A. Ivanova, Significance of calcium phosphate coatings for the
enhancement of new bone osteogenesis - a review, Acta Biomater. 10 (2014) 557–579.
[6] S.V. Dorozhkin, Calcium orthophosphate deposits: Preparation, properties and biomedical
applications, Mater. Sci. Eng. C 55 (2015) 272–326.
[7] S. Zhang, Hydroxyapatite Coatings for Biomedical Applications, CRC Press, 2013.
[8] A.H. Choi, B. Ben-Nissan, Applications of Hydroxyapatite Nanocoatings and Nanocomposite
Coatings in Dentistry, JSM Dent. Surg. 1, (2016) 1002.
[9] V.M. Ievlev, Coatings based on calcium phosphates for metallic medical implants, Russ. Chem. Rev.
82 (2013) 131–149.
[10] R.A. Surmenev, A review of plasma-assisted methods for calcium phosphate-based coatings
fabrication, Surf. Coatings Technol. 206 (2012) 2035–2056.
[11] A. Mello, Z. Hong, A.M. Rossi, L. Luan, M. Farina, W. Querido, J. Eon, J. Terra, G. Balasundaram,
T. Webster, A. Feinerman, D. E. Ellis, J. B. Ketterson, C.L. Ferreira, Osteoblast proliferation on
hydroxyapatite thin coatings produced by right angle magnetron sputtering, Biomed. Mater. 2 (2007)
67-77.
[12] E.O. López, A. Mello, H. Sendão, L.T. Costa, A.L. Rossi, R.O. Ospina, F.F. Borghi, J.G. Silva Filho,
A.M. Rossi, Growth of crystalline hydroxyapatite thin films at room temperature by tuning the
energy of the RF-magnetron sputtering plasma, ACS Appl. Mater. Interfaces 5 (2013) 9435–9445.
[13] V. Nelea, C. Morosanu, M. Iliescu, I. N. Mihailescu, Hydroxyapatite thin films grown by pulsed laser
deposition and radio-frequency magnetron sputtering: comparative study, Appl. Surf. Sci. 228 (2004)
346–356.
[14] D. B. Chrisey, G. K. Hubler, Pulsed Laser Deposition of Thin Films, first ed., John Wiley & Sons,
New York, 1994.
[15] D.G. Wang, W.L. Zhang, H.J. Li, J.H. Zhang, C.Z. Chen, HA/BG composite films deposited by pulse
laser under O2 atmosphere, Ceram. Int. 43 (2017) 672–676.
[16] A. Das, M. Shukla, Surface morphology, bioactivity, and antibacterial studies of pulsed laser
deposited hydroxyapatite coatings on Stainless Steel 254 for orthopedic implant applications, Proc.
Inst. Mech. Eng. Part L J. Mater. Des. Appl. (2016) 1–8.

99
[17] G. P. Dinda, J. Shin, J. Mazumder, Pulsed laser deposition of hydroxyapatite thin films on Ti-6Al-
4V: Effect of heat treatment on structure and properties, Acta Biomater. 5 (2009) 1821–1830.
[18] Y. Suda, H. Kawasaki, T. Ohshima, S. Nakashima, S. Kawazoe, and T. Toma, Hydroxyapatite
coatings on titanium dioxide thin films prepared by pulsed laser deposition method, Thin Solid Films,
506–507 (2006) 115–119.
[19] L.C. Nistor, C. Ghica, V.S. Teodorescu, S.V. Nistor, M. Dinescu, D. Matei, N. Frangis, N.
Vouroutzis, C. Liutas, Deposition of hydroxyapatite thin films by Nd:YAG laser ablation: A
microstructural study, Mater. Res. Bull. 39 (2004) 2089–2101.
[20] J. Beltrano, L. Torrisi, and D. Margarone, Biocompatible film deposition by using Nd:YAG pulsed
laser, Radiat. Eff. Defects Solids. 160 (2005) 545–552.
[21] J. J. Beltrano, L. Torrisi, A. M. Visco, N. Campo, and E. Rapisarda, Pulsed Laser Deposition (PLD)
Technique to Prepare Biocompatible Thin Films, Adv. Sci. Technol. 49 (2006) 56–61.
[22] J.V. Rau, I. Cacciotti, A. De Bonis, M. Fosca, V.S. Komlev, A. Latini, A. Santagata, R. Teghil, “Fe-
doped hydroxyapatite coatings for orthopedic and dental implant applications,” Appl. Surf. Sci. 307
(2014) 301–305.
[23] D. Ferro, S. M. Barinov, J. V. Rau, R. Teghil, A. Latini, Calcium phosphate and fluorinated calcium
phosphate coatings on titanium deposited by Nd:YAG laser at a high fluence, Biomaterials 26 (2005)
805-812.
[24] E. Tognoni, V. Palleschi, M. Corsi, G. Cristoforetti, Quantitative micro-Analysis by Laser-induced
Breakdown Spectroscopy: A Review of the Experimental Approaches. Spectrochim. Acta B 57
(2002) 1115-1130.
[25] R. Fantoni, L. Caneve, F. Colao, L. Fornarini, V. Lazic, V. Spizzichino, Methodologies for
laboratory Laser Induced Breakdown Spectroscopy semi-quantitative and quantitative analysis—A
review, Spectrochimica Acta Part B 63 (2008) 1097-1108.
[26] F. Anabitarte, A. Cobo, J. M. Lopez-Higuera, Laser-Induced Breakdown Spectroscopy:
Fundamentals, Applications, and Challenges 2012 (2012) 1-12.
[27] A. Ciucci, M. Corsi, V. Palleschi, S. Rastelli, A. Salvetti, E. Tognoni, New Procedure for
Quantitative Elemental Analysis by Laser-Induced Plasma Spectroscopy, Appl. Spectrosc. 53 (1999)
960-964.
[28] R. Gaudiuso, M. Dell'Aglio, O. De Pascale, S. Loperfido, A. Mangone, A. De Giacomo, Laser
induced breakdown spectroscopy of archaeological findings with calibration-free inverse method:
comparison with classical laser-induced breakdown spectroscopy and conventional techniques, Anal
Chim Acta. 813 (2014) 15-24.
[29] C. Aragón, J.A. Aguilera, C Sigma graphs: A new approach for plasma characterization in laser-
induced breakdown spectroscopy, JQRST 149 (2014) 90-102.
[30] E. Tognoni, G. Cristoforetti, S. Legnaioli, V. Palleschi, Calibration-Free Laser-Induced Breakdown
Spectroscopy: State of the art 65 (2010) 1-14.
[31] G.H. Cavalcanti, D.V. Teixeira, S. Legnaioli, G. Lorenzetti, L. Pardini, V. Palleschi, One-point
calibration for calibration-free laser-induced breakdown spectroscopy quantitative analysis,
Spectrochim. Acta B 87 (2013) 51-56.

100
[32] R. Eason, Pulsed Laser Deposition of Thin Films: Applications-Led Growth of Functional Materials,
John Wiley & Sons, New Jersey, 2007.
[33] J.C. Elliott, R.M. Wilson, S.E.P. Dowker, Apatite Structures, JCPDS-International Centre for
Diffraction Data, Advances in X-ray Analysis, 45 (2002) 172-181.
[34] A. Mello, Filmes finos cristalinos de hidroxiapatita: uma abordagem original com magnetron
sputtering de alvos opostos, tese de doutorado, Instituto Militar de Engenharia, 2007.
[35] J. Terra, M. Jiang, D.E. Elllis, Philos. Mag. A, 82 (2002) 2357-2377.
[36] E.O.L. Meza, Influência dos parâmetros do plasma de um magnetron sputtering de alvos opostos
(RAMS) no controle do crescimento de filmes finos de hidroxiapatita, com substituições iônicas para
aplicações biomédicas, tese de doutorado, Centro Brasileiro de Pesquisas Físicas, 2014.
[37] V.S. Bystrov et al., J. Phys. D: Appl. Phys. 48 (2015) 195-302.
[38] B. Ren, Y. Leng, Carbonated apatite, Type-A or Type-B?, Key Eng. Mater. 493–494 (2012) 293–
297.
[39] M. Castillejo, P.M.Ossi, L.Zhigilei, Lasers in Materials Science, Springer Series in Materials
Science, v.191, London, 2014.
[40] R.J. Narayan, Hidroxyapatite-diamond like carbon nanocomposite films, Mater. Sci. Eng. C 25
(2005) 398-404.
[41] E.N. Antonov, V.N. Bagratashvili, V.K. Popov, M.D. Ball, D.M. Grant, S.M. Howdle, C.A.
Scotchford, Properties of calcium phosphate coatings deposited and modified with lasers, J. Mater.
Sci. Mater. Med. 13 (2003) 151-155.
[42] M. Jelinek et al., Effect of processing parameters on the properties of hydroxylapatite films grown by
pulsed laser deposition, Thin Solid Films, 257 (1995) 125-129.
[43] G.C. Gomes, F.F. Borghi, R.O. Ospina, E.O. López, F.O. Borges, A. Mello, Nd:YAG (532 nm)
pulsed laser deposition produces crystalline hydroxyapatite thin coatings at room temperature.
Surface & Coatings Technology, 329 (2017) 174-183.
[44] F. Sima, C. Ristoscu, L. Duta, O. Gallet, K. Ancelme, I.N. Mihailescu, Laser Surface Modification of
Biomaterials (2016) 77-125.
[45] F. Sima et al. Combinatorial matrix-assisted pulsed laser evaporation: Single-step synthesis of
biopolymer compositional gradient thin film assemblies, Appl. Phys. Lett. 101(2012).
[46] H.-Y. Moon, K.K Herrera, N. Omenetto, B.W. Smith, J.D. Winefordner, On the Usefulness of a
Duplicating Mirror to Evaluate self-Absorption Effects in Laser Induced Breakdown Spectroscopy.
Spectrochim. Acta B 64 (2009) 702-713.
[47] H. Amamou, A. Bois, B. Ferhat, R. Redon, B. Rossetto, P. Matheron, Correction of self-Absorption
Spectral Line and Ratios of Transition Probabilities for Homogeneous and LTE Plasma. J. Quant.
Spectrosc. Radiat. 75 (2002) 747-763.
[48] D. Bulajic, M. Corsi, G. Cristoforetti, S. Legnaioli, V. Palleschi, A. Salvetti, E. Tognoni, A
Procedure for Correcting self-Absorption in Calibration Free-Laser Induced Breakdown
Spectroscopy. Spectrochim. Acta B 57 (2002) 339-353.
[49] F. Bredice, F. O. Borges, H. Sobral, M. Villagran-Muniz, H. O. Di Rocco, G. Cristoforetti, S.
Legnaioli, V. Palleschi, L. Pardini, A. Salvetti and E. Tognoni, Evaluation of self-absorption of

101
manganese emission lines in Laser-Induced Breakdown Spectroscopy measurements,
Spectrochimica Acta Part B 61(2006) 1294.
[50] A. De Giacomo, M. Dell’Aglio, O. De Pascale, S. Longo, M. Capitelli, Laser Induced Breakdown
Spectroscopy on Meteorites. Spectrochim. Acta B, 62 (2007) 1606-1611.
[51] A. De Giacomo, Experimental Characterization of Metallic Titanium-Laser Induced Plasma by Time
and Space Resolved Optical Emission Spectroscopy. Spectrochim. Acta B, 58 (2003) 71-83.
[52] F. O. Borges, Contribuição para o Estudo Teórico-Experimental de Sistemas Atômicos Complexos,
tese de doutorado, Universidade Federal Fluminense, 2007.
[53] R. Noll, Laser-Induced Breakdown Spectroscopy: Fundamentals and Applications, Springer,
Londres. 2012.
[54] L.J. Radziemski, D.A. Cremers, Laser-induced Plasma and Applications, Eds. Marcel Dekker: New
York, NY, USA, 1989.
[55] G. Cristoforetti, A. De Giacomo, M. Dell'Aglio, S. Legnaioli, E. Tognoni, V. Palleschi, N. Omenetto,
Local Thermodynamic Equilibrium in Laser-Induced Breakdown Spectroscopy: Beyond the
McWhirter Criterion, Spectrochimica Acta Part B 65 (2010) 86–95.
[56] H.R. Griem, Plasma Spectroscopy, Mc Graw Hill, New York, 1964.
[57] A.P. Thorne, Spectrophysics, Chapman and Hall Ltd, 2ª Edição, Londres, 1988.
[58] H. Amamou, A. Bois, B. Ferhat, R. Redon, B. Rossetto, M. Ripert, Correction of the self-absorption
for reversed spectral lines: application to two resonance lines of neutral aluminium. J. Quant.
Spectrosc. Radiat. Transfer 77 (2003) 365-372.
[59] P. Fauchais, J. F. Coudert, M. Sardelle, Diagnostic in Thermal Plasma em Plasma Diagnostics,
Academic Press, New York, NY, USA, 1989.
[60] A. Ciucci, M. Corsi, V. Palleschi, S. Rastelli, A. Salvetti, E. Tognoni, New Procedure for
Quantitative Elemental Analysis by Laser Induced Plasma Spectroscopy, Appl. Spectrosc., 53 (2009)
960-964.
[61] J. Aguilera e C. Aragón, Multi-element Saha-Boltzman and Boltzmann plots in laser-induced
plasmas, Spectrochimica Acta Part B: Atomic Spectroscopy, 62 (2007) 378—385
[62] B. Praher. V. Palleschi, R. Viskup, J. Heitz, J.D. Pedarnig, Calibration free laser-induced breakdown
spectroscopy of oxide materials, Sprectrochim. Acta B 65 (2010) 671-679.
[63] A.M. El Sherbini, H. Hegazy e T.M. El Sherbini, Measurement of electron density utilizing the Hα-
line from laser produced plasma in air. Spectrochim. Acta B 61(2006) 532-539.
[64] H.J. Kunze, Introduction to Plasma Spectroscopy, Springer Series on Atomic, Optical and Plasma
Physics, New York, 2009.
[65] E. Tognoni, G. Cristoforetti, S. Legnaioli, V. Palleschi, Calibration-Free Laser-induced Breakdown
Spectroscopy: State of the Art, Spectrochim. Acta B, 65 (2010) 1-14.
[66] E. Tognoni, G. Cristoforetti, S. Legnaioli, V. Palleschi, A. Salvetti, M. Mueller, U. Panne, I.
Gornushkin, A Numerical Study of Expected Accuracy and Precision in Calibration-Free
Laser-induced Breakdown Spectroscopy in the Assumption of Ideal Analytical Plasma,
Spectrochim. Acta B, 62 (2007) 1287-1302.

102
[67] E. Grifoni, S. Legnaioli, G. Lorenzetti, S. Pagnotta, F. Poggialini, V. Palleschi, From Calibration-
Free to Fundamental Parameters Analysis: A comparison of three recently proposed approaches,
Spectrochimica Acta Part B 124 (2016) 40–46.
[68] F. Borghi, Recobrimentos cristalinos de hidroxiapatita produzidos a temperatura ambiente por
ablação a laser pulsado de Nd:YAG – 532 nm, dissertação de mestrado, Centro Brasileiro de
Pesquisas Físicas, 2012.
[69] M. Applications and P. Information, Ingeo TM Biopolymer 4043D Technical Data Sheet 3D Printing
Monofilament – General Purpose Grade, 4 (2016) 1–4.
[70] P. Ahvenainen, I. Kontro, K. Svedström, Comparison of sample crystallinity determination methods
by X-ray diffraction for challenging cellulose I materials, Cellulose. 23 (2016) 1073–1086.
[71] R. Neppalli, V. Causin, A. Marigo, M. Meincken, P. Hartmann, A. J. van Reenen, Effect of the
electrospun ethylene vinyl alcohol copolymer (EVOH) fibres on the structure, morphology and
properties of poly (lactic acid) PLA, Polymer 54 (2013) 5909-5919.
[72] Standard Test Methods for Rating Adhesion by tape Test ASTM D3359 (2017).
[73] H.O. Di Rocco, D. I. Iriarte, and J. Pomarico, General Expression for the Voigt Function that is of
Special Interest for Applied Spectroscopy, Appl Spectrosc 55 (2001) 822-826.
[74] E.E. Whiting, An Empirical Approximation to the Voigt Profile, J Quant Spectros Radiat Tran 8
(1968) 1379-1384.
[75] J.E. Sansonetti, and W.C. Martin, Handbook of Basic Atomic Spectroscopic Data, J Phys Chem Ref
Data 34 (2005) 1559-2259.
[76] E.O. López, A. Mello, M. Farina, A.M. Rossi, A.L. Rossi, Nanoscale analysis of calcium phosphate
films obtained by RF magnetron sputtering during the initial stages of deposition, Surf. Coatings
Technol. 279 (2015) 16–24.
[77] T. Hanawa, M. Ota, Calcium phosphate naturally formed on titanium in electrolyte solution,
Biomaterials., 12 (1991) 767–774.
[78] A. Demri, D. Muster, XPS study of some calcium compounds, J. Mater. Process. Technol. 55 (1995)
311–314.
[79] M.A. Stranick, M.J. Root, Influence of strontium on monofluorophosphate uptake by hydroxyapatite
XPS characterization of the hydroxyapatite surface, Colloids and Surfaces, 55 (1991) 137–147.
[80] Y.A. Teterin, M.I. Sosulnikov, X-ray photoelectron study of Ca, Sr and Ba ion chemical states in
high-Tc superconductors, Phys. C Supercond. its Appl. 212 (1993) 306–316.
[81] P.J. Cumpson, M.P. Seah, Elastic Scattering Corrections in AES and XPS. II. Estimating Attenuation
Lengths and Conditions Required for their Valid Use in Overlayer/Substrate Experiments, Surf.
Interface Anal. 25 (1997) 430–446.
[82] B. Wagner, W.M. Riggs, L.E. Davis, J.F. Moulder, Handbook of X-Ray Photoelectron Spectroscopy,
first ed., Perkin-Elmer Corporation, Minnesota, 1979.
[83] S. Kaciulis, G. Mattogno, L. Pandolfi, M. Cavalli, G. Gnappi, A. Montenero, XPS study of apatite-
based coatings prepared by sol-gel technique, Appl. Surf. Sci. 151 (1999) 13–16.
[84] B. Ren, Y. Leng, Carbonated apatite, Type-A or Type-B?, Key Eng. Mater. 493–494 (2012) 293–
297.

103
[85] C. Hinnen, D. Imbert, J. M. Siffre, P. Marcus, An in situ XPS study of sputter-deposited on graphite,
Appl. Surf. Sci. 4332 (1994) 219–231.
[86] http://lasurface.com/accueil/index.php
[87] J. Hsu, P.W. Wang, C. Lee, X-ray photoelectron spectroscopy study of thin TiO 2 films cosputtered
with Al, App. Optics. 45 (2006) 4303–4309.
[88] J.C.V. Gilmore, I.S., Surface Analysis – The Principal Techniques, second ed. John Wiley & Sons,
Chichester, 2009.
[89] S. Koutsopoulos, Synthesis and characterization of hydroxyapatite crystals: A review study on the
analytical methods, Wiley Periodicals. February, (2002) 600–612.
[90] D. Garlotta, A literature review of poly(lactic acid), Journal of Polymers and the Environment 9
(2002) 63-84.
[91] B. Sibeko, Y. E. Shoonara, L. C. Toit, G. Modi, D. Naidoo, R. A. Khan, P. Kumar, V. M. K.
Ndesendo, S. E. Iyuke, V. Pillay, Composite Polylactic-Methacrylic Acid Copolymer Nanoparticles
for the Delivery of Methotrexate, Journal of Drug Delivery. 2012 (2012) 1-18.
[92] K. Anselme, L. Ploux, A. Ponche, Cell/material interfaces: Influence of surface chemistry and
surface topography on cell adhesion, J. Adhes. Sci. Technol. 24 (2010) 831–852.
[93] Prasad D K, Swaminathan AA, Prasad D A. Current trends in surface textures of implants. J Dent
Implant 2016;6:85-91

104
9. APÊNDICES
9.1. APÊNDICE I: Publicações deste trabalho de tese
1) Gomes, G.C.; Borghi, F.F.; Ospina, R.O.; López, E.O.; Borges, F.O.; Mello, A.
Nd:YAG (532 nm) pulsed laser deposition produces crystalline hydroxyapatite
thin coatings at room temperature. Surface & Coatings Technology, v. 329 p.174-
183, 2017.
2) Borges, Fábio O.; Cavalcanti, Gildo H.; Gomes, Gabriela C.; Palleschi,
Vincenzo; Mello, Alexandre. A fast method for the calculation of electron number
density and temperature in laser-induced breakdown spectroscopy plasmas using
artificial neural networks. Applied Physics. B, Lasers and Optics, v. 117 (1), p.
437-444, 2014.
3) Borges, Fábio; Ospina, Johnatann; Cavalcanti, Gildo; Farias, Eliel; Rocha,
Anderson; Ferreira, Paula; Gomes, Gabriela; Mello, Alexandre. Calibration-Free
Laser-Induced Breakdown Spectroscopy Analysis for Frozen Aqueous Solution
Samples by using a Standard Internal Reference and Correcting Self-Absorption
Effect. Journal of Analytical Atomic Spectrometry (Em revisão)

105
9.2. APÊNDICE II: Protocolo dos ensaios biológicos
Procedimento do cultivo celular
As células foram cultivadas rotineiramente em garrafas específicas para cultivo
celular e mantidas na incubadora a 37°C em atmosfera contendo 5% de CO2. Duas
vezes por semana as células SAOS-2 foram cultivadas em meio McCoy’s suplementado
com 10% de soro fetal bovino (SFB). Quando confluentes, foram expandidas em
garrafas de cultivo de 75 cm2. Para cada passagem, as células foram tripsinizadas em
fluxo laminar, com tripsina-EDTA a 0,2%. A tripsinização é um processo enzimático
que quebra os pontos de adesão das células e facilita a sua suspensão no meio de cultura
celular. O crescimento das células em cultura foi acompanhado diariamente pela
observação em microscópio invertido de contraste de fase. Para os experimentos, as
células foram quantificadas em câmara de Neubauer e as densidades específicas foram
semeadas em placas de 24 poços.
Protocolo da fixação dos anticorpos
Após os tempos de adesão, o meio de cultura foi retirado com cuidado para não
agredir as células e os seguintes procedimentos foram realizados:
1) Para as amostras de 30 min, 4hs e 24hs:
- Lavamos 3x com PBS pH 7.4 (o PBS é uma solução tampão fosfato-salino
comumente usado para manter um valor de pH constante);
- Fixamos as células com 0,5 ml de PFA 4% por 10 min em temperatura ambiente (o
paraformaldeído PFA serve para bloquear a membrana celular e fixar as células);
- Lavamos 3x com PBS pH 8;
- Adicionamos 500 µl de NH4Cl (cloreto de amônio) 20 mM por 10 min em
temperatura ambiente.
- Lavamos 3x com PBS pH 8;
- Adicionamos em 500 µl de 0,1% de Triton X-100 + BSA 1% por 10 min (para
permeabilizar a estrutura celular);

106
- Lavamos 3x com PBS pH 8;
- Adicionamos 500 µl BSA 3% por 30 min em temperatura ambiente.
- Lavamos 1x com PBS pH 8.
- Preparamos as amostras para visualização com selante fluorshield com DAPI
(solução que já contém o marcador para núcleo).
2) Para as amostras de 48hs: Procedimento de marcação tripla
- Realizamos os procedimentos anteriores antes da preparação com DAPI;
- Adicionamos 20 μl do 1° anti-corpo (Anti- Vinculina) 1:200, diluída em solução
de BSA 1% (PBS) + 0,1% Triton X-100 e incubada por 1h15 em temperatura ambiente.
Cálculo da quantidade necessária para a diluição da anti-vinculina:
15 poços × 20 μl de anticorpo = 300 μl de solução final.
𝐶𝑖 × 𝑉𝑖 = 𝐶𝑓 × 𝑉𝑓 → 𝑉𝑖 =1
200× 300 = 1,5 μl de anticorpo.
300 − 1,5 = 298,5 μl de BSA 1% (PBS) + 0,1% Triton X-100
- Lavamos 3x com PBS pH 8.
- Adicionamos 20 μl do 2° anti-corpo Alexa Fluor 488 (vinculina) 1:500, diluído em
solução BSA 1% (PBS) + 0,1% Triton X-100 por 1h em temperatura ambiente.
Cálculo da quantidade necessária para a diluição da vinculina:
𝐶𝑖 × 𝑉𝑖 = 𝐶𝑓 × 𝑉𝑓 → 𝑉𝑖 =1
500× 300 = 0,6 μl de anticorpo.
300 − 0,6 = 299,4 μl de BSA 1% (PBS) + 0,1% Triton X-100
- Lavamos 3x com PBS pH 8.

107
- Adicionamos 20 μl do 3° anti-corpo Alexa Fluor 546 (faloidina) 1:80, diluído em
solução BSA 1% (PBS) + 0,1% Triton X-100 por 30 min em temperatura ambiente.
Cálculo da quantidade necessária para a diluição da faloidina:
𝐶𝑖 × 𝑉𝑖 = 𝐶𝑓 × 𝑉𝑓 → 𝑉𝑖 =1
80× 300 = 3,75 μl de anticorpo.
300 − 3,75 = 296,25 μl de BSA 1% (PBS) + 0,1% Triton X-100
- Lavamos 3x com PBS pH 8.
- Preparamos as amostras para visualização com selante fluorshield com DAPI.
Os anticorpos foram adicionados em placa forrada com parafilme. Os
fluorocromos foram descongelados imersos em um suporte com gelo. Devido a sua
fotossensibilidade, os fluorocromos foram diluídos e encubados no escuro.





























