Embed Size (px)
Citation preview

1
SILICATOS TÉCNICOS – CRISTAIS E VIDROS
de Armin F. Isenmann, CEFET-MG Campus Timóteo
Índice
1. Classes de silicatos técnicos ............................................................................................2
2. Silicatos de álcali ............................................................................................................2
3. Silicatos cristalinos na natureza.......................................................................................5
4. Vidros contra cristais ......................................................................................................6
4.1. Termodinâmica da cristalização ..............................................................................6
4.2. Cinética da cristalização. ....................................................................................... 14
4.3. Aspecto dos cristais formados ............................................................................... 20
5. Conclusões práticas para a produção de vidro ............................................................... 21
6. Conceito de formadores de rede, modificadores de rede e quebradores de rede ............. 23
6.1. "Formadores de rede" ............................................................................................ 23
6.2. "Quebradores de rede"........................................................................................... 24
6.3. "Modificadores de rede" ........................................................................................ 24
7. Tipos de vidros ............................................................................................................. 25
7.1. Vidro comum: história, matérias-primas e princípios de manufatura ...................... 25
7.2. Composição do vidro comum: quartzo, cal e soda ................................................. 27
7.3. Ataque químico por ácidos e bases ........................................................................ 29
8. Vidro de potassa e outros vidros técnicos ...................................................................... 30
8.1. Vidros especiais. ................................................................................................... 32
8.2. Coloração (artística) de vidros ............................................................................... 35
8.3. Opacidade, nebulosidade – atributos dos esmaltes. ................................................ 36
9. Literatura ...................................................................................................................... 37
10. Anexo ....................................................................................................................... 37

2
1. Classes de silicatos técnicos Os silicatos de relevância técnica podem ser classificados em:
Silicatos de álcali, também chamados de vidros d´água,
Vidros comuns,
Cerâmicas, louças.
Além destes, têm-se peneiras moleculares (estruturas de alumossilicatos, em forma de
pequenas gaiolas, que servem para absorver pequenas moléculas; uso no laboratório: secagem
de solventes orgânicos) e cimentos (silicatos amorfos que reagem irreversivelmente com
água, formando sólidos de elevados módulos).
Todos em comum têm o ânion silicato que podemos construir em sequência dos seus íons
homólogos da composição EO4n-
. Observamos em todos esses íons complexos um arranjo
tetraédrico que só no silicato é perfeitamente equilátero:
Cl
O
O
O
O S OO
O
O
P OO
O
O
Si O
O
O
O
2 3 4
Perclorato Sulfato Fosfato Silicato
2. Silicatos de álcali Geralmente são feitos pela co-fusão de areia (= quartzo impuro) e barrilha (carbonato de sódio
/ carbonato de potássio). Suas fórmulas idealizadas são M4SiO4, M2SiO3, M2Si2O5 e M2Si4O9,
dependendo da composição das matérias primas, sendo essa 1:2, 1:1, 2:1 e 4:1,
respectivamente. As seguintes equações representam o acontecimento, ao mesmo tempo
indicam a matéria-prima usada para tais reações: areia (ou quartzo) e soda (= carbonato de
sódio):
SiO2 + 2 M2CO3 M4SiO4 + 2 CO2
SiO2 + M2CO3 M2SiO3 + CO2
2 SiO2 + M2CO3 M2Si2O5 + CO2
4 SiO2 + M2CO3 M2Si4O9 + CO2
Observações:
1) Essas fórmulas sumárias não permitem nenhuma conclusão sobre a estrutura. Na verdade,
em todos esses vidros têm-se os tetraedros de [SiO4].
2) Uma consequência da última afirmação é que em todos esses produtos o silício apresenta-
se com NOX +4. Em nenhum caso ocorreu uma reação redox.

3
3) Esses materiais se obtêm na forma cristalina e pura, somente quando mantidos logo abaixo
da temperatura de amolecimento, por um tempo prolongado ("têmpera"). Daí, os tetraedros de
[SiO4] organizam-se em estruturas específicas, dependendo do tipo e da quantidade de cátions
presentes:
Isoladamente ("silicato de ilhas"), no caso do Li4SiO4 e Na4SiO4;
Unidimensional ("silicato de catena"), no caso do Li2SiO3, Na2SiO3 e K2SiO3;
Bidimensional ("silicato de camada"), no caso do Li2Si2O5, Na2Si2O5 e K2Si2O5;
Tridimensional (polissilicato), no caso do K2Si4O9.
Algumas destas estruturas cristalinas encontram-se no anexo (Fig. 16 a Fig. 22).
4) Todos esses cristais têm temperaturas de fusão, em torno de 1000 °C. Bem menos energia
térmica é preciso para fundir um produto da mesma composição, mas vitrificado, já que o
material amorfo tem muito mais energia interna (e também mais entropia = bagunça
estrutural), do que o cristal.
A formação desses vidros geralmente não é somente uma transformação física da fase (fusão),
mas é acompanhada pela condensação dos grupos [SiO4], eliminando-se água. Isso é
representado no seguinte esquema, a partir do ácido silícico, H4SiO4, mas vale igualmente
para seus sais, M+[H3SiO4], M
+2[H2SiO4] e M
+3[HSiO4]. A particularidade do ácido silícico é
que todos os grupos -OH podem ser condensados desta maneira, enquanto a presença de
cátions inibe a etapa da condensação (ver "quebradores de rede", na p. 24). Note também que
a presença de certos cátions favorece a cristalização em estruturas especiais (efeito de
gabarito).
Como a unidade [SiO4] tem estrutura tetraédrica, então chegamos a uma rede tridimensional.
Em caso de uma condensação perfeita do ácido H4SiO4, a fórmula se aproxima à composição
[SiO2] - um polímero tridimensional que identificamos como quartzo.
Fig. 1. Reações de condensação do ácido silícico e dos silicatos
Si
OH
OH
OH
HO Si
OH
OH
OH
HO- H2O
Si
OH
O
OH
HO Si
OH
OH
OH
Ácido monossilícico = ác. ortossilícico Ácido dissilícico
Si
OH
OH
OH
HO+
- H2O
Si
OH
O
OH
HO Si
OH
O
OH
Si
OH
OH
OH
Ácido trissilícico
Ácido tetrassilícico
Ácido polissilícico(um isopoli-ácido)
A tendência dos silicatos de condensar conforme este esquema é tão pronunciada que o
próprio ortossilicato somente é conhecido em solução bastante diluída e em ambiente alcalino.
Quando se acidifica cuidadosamente essa solução ela se torna aos poucos num gel. Sua
transparência diminui gradativamente e o material se torna turvo, nos últimos estágios da

4
condensação. O processo da geleificação gradativa, dos ortossilicatos para os isopoli-ânions, a
formação de uma rede frouxa destes íons complexos, até estruturas tridimensionais
reforçados, é esboçado nas Fig. 23 e Fig. 24, no anexo.
Ao contrário da condensação organizada que se conhece no ácido fosfórico, a condensação do
ácido silícico ocorre bastante randômica. Em sequência arbitrária se desenvolvem, além de
alongamentos de cadeias, o fecho de cadeias para anéis e a ramificação de cadeias curtas. A
consequência desta desordem é uma estrutura largamente amorfa que consiste de unidades
finais [H3SiO4], elos lineares [H2SiO4] e ramificações [HSiO4]; nos estágios prorrogados
dominam as duplas ramificações do tipo [SiO4].
Fig. 2. Unidades estruturais dentro de polissilicatos
SiO
OH
OH
OH
Unidade final
SiO
OH
O
OH
Unidade linear
SiO
OH
O
O
Ramificação
SiO
O
O
O
Dupla ramif icação
Na escala industrial se obtém o vidro pela co-fusão de areia e soda intimamente triturados e
misturados, um material mal cristalizado ou totalmente amorfo, isto é, sem ordem de longo
alcance. Resultam pedaços de ± 1 Kg, transparentes e podem ter coloração azulada, verde,
amarela ou marrom, devido às impurezas, principalmente por Fe (III). É conhecido como
"vidro de água" porque é solúvel na água - especialmente se for alcalina. Esses materiais são
comercializados em forma de solução aquosa que se obtém por dissolver os sólidos em água
superaquecida (150 °C; 5 atm) 1.
Composições ricas em sílica são usadas como:
cola mineral, usada na indústria de porcelanas (ou também para consertar porcelanas e
vidros quebrados),
para impregnar, reforçar e branquear papel,
para facilitar o processamento de fibras naturais na fiadeira: vidro d´água deixa a fibra
da seda mais pesada e aglutina as fibras da lã,
protege tecidos contra a deposição de sujeira,
como carga em sabão,
para conservação de ovos (em forma de gel, ver Fig. 23 e Fig. 24 no anexo),
retardar a queima de madeira e tecidos em prédios.
base em tintas minerais de paredes interiores e exteriores (onde acarretam o efeito
"fosco"; em mistura com ácido bórico é usado como pintura anti-mofo),
na construção civil o vidro d´água de lítio fecha os poros do concreto,
aglutinantes em revestimentos de eletrodos de solda,
material abrasivo em pasta de dente,
1 Mais comercializada é a solução do vidro d´água de sódio, da composição Na2O : SiO2 = 1 : (3,4 a 3,5), usado
principalmente em formulações de sabão em pó.

5
carga em plásticos,
entre outros.
Os vidros d´água também são usados como:
Matéria-prima para a produção de zeólitos (ver Fig. 25, no anexo)
Géis inorgânicos,
Suporte para catalisadores,
Material absorvente para gases (vapores de solventes orgânicos), água (em
exsicadores) e partículas (filtro de cigarros), etc.
A aplicação mais espetacular é provavelmente a absorção da nitroglicerina em silicagel que
leva à dinamite. Muitas destas aplicações se devem à grande superfície interna dos materiais
solidificados - uma área comparável à do carvão ativado. Os materiais com as maiores
superfícies são conhecidos como hidrogel (poros grandes) ou xerogel (poros pequenos).
Os vidros d´água ricos em soda são principalmente usados em produtos de limpeza, por
exemplo, como aditivo anti-corrosão em sabão de máquinas de lavar roupa e louça. O vidro
d´água exerce um efeito protetor frente todas as peças metálicas - menos alumínio que está
sendo atacado pelo elevado grau de alcalinidade. Lembre-se que sua hidrólise libera uma
quantidade notável de hidróxidos, tais como NaOH ou KOH. Seus complexos hidratados
podem ser obtidos em forma de cristais, de baixos pontos de fusão (Sesquissilicatos e
metassilicatos Na2H2SiO4 . 5 H2O de Tfus = 72 °C e Na2H2SiO4
. 8 H2O de Tfus = 47 °C).
3. Silicatos cristalinos na natureza Os silicatos cristalinos junto ao quartzo, [SiO2]x , representam cerca de 90% da crosta terrestre
sólida! Sendo assim, não é surpresa que é considerada como sendo a família mais
diversificada e mais bem estudada, de todos os compostos inorgânicos cristalinos. A
mineralogia pode ser considerada uma sub-disciplina da inorgânica que quase exclusivamente
estuda o fascinante mundo dos silicatos cristalinos. O imenso número de possíveis arranjos
dos ânions silicatos e a grande flexibilidade (em quantidade e tipo) dos cátions fornece um
grande número de diferentes cristais. Esse número aumenta mais uma vez, por que pequenos
cátions, tais como Al3+
, B3+
ou Be2+
, podem repor em parte os átomos de silício das unidades
[SiO4], levando assim aos alumossilicatos, borossilicatos e berilossilicatos, respectivamente
(ver também ""Modificadores de rede", p. 24).
Cerca de 70% dos silicatos cristalinos formam uma rede tridimensional, onde as unidades
[SiO4] podem compartilhar cantos, arestas ou faces. Já as estruturas bidimensionais (=
“filossilicatos”; feitos em camadas) são a segunda maior família, nos quais constam os
membros da família dos alumossilicatos. A fase de mica dentro do granito e muitas argilas são
representantes deste grupo (ver Fig. 20 e Fig. 21). Nestes, parte das unidades tetraédricas de
[SiO4] são repostas por octaedros de [AlO6] e um cátion alcalino. Relativamente raras são
estruturas pseudo-unidimensionais (fibras); as mais prominentes provêm também dos
alumossilicatos que por si têm a forte tendência de arranjo em camadas. Sob certos critérios
de organização, no entanto, essas camadas são fortemente curvadas, já que a unidade [SiO4] é
menor do que sua contraparte [AlO6]. Portanto, a camada de sílica fica recolhida em
comparação à camada de aluminato – o que provoca uma forte curvatura da dupla-camada
(Fig. 22). Macroscopicamente resulta uma fibra (muitas vezes oca) que conhecemos como
“amianto”, até hoje um importante material de construção em telhas. Note que o manuseio

6
deste material pode acarretar um elevado risco de silicose (doença nos pulmões) e, igualmente
grave, potencializa o risco de desenvolver câncer, a longo prazo. Explicação: as fibras podem
apresentar-se em forma de agulhas ultra-finas que podem ser inaladas junto à poeira do ar.
Dentro do corpo, no entanto, acham situações favoráveis a uma longa vida. As agulhas
penetram os alveolos pulmonares onde permanecem como corpos estranhos, por dezenas de
anos sem degradação notável. Antirreações do corpo humano levam finalmente à
desnaturação de partes dos pulmões, insuficiência respiratória e a desnaturação maligna das
células. O uso deste material no setor de construção civil está fortemente regressivo desde a
consolidação dessas evidências medicinais.
Algumas das estruturas mais prominentes, tri, bi e pseudo-unidimensionais, encontram-se no
anexo deste artigo.
4. Vidros contra cristais Sob "vidro" se entende, em geral, material fundido que solidificou sem cristalização. Resulta
então um corpo amorfo, fora do equilíbrio termodinâmico, ou seja, metaestável. Também
podemos afirmar que o vidro é nada outro do que o líquido congelado, com as mesmas
propriedades físicas, tais como densidade ou capacidade calorífica. Já o cristal é
fundamentalmente diferente do estado líquido ou vítreo.
No vidro os átomos têm uma ordem de curto alcance: número e tipo de átomos vizinhos são
definidos. Mas não têm ordem a longo alcance, isto é, incerteza sobre posição, número e tipo
do átomo além da primeira esfera de coordenação.
Fig. 3. Modelos dos silicatos cristalino, amorfo e do vidro d´água.
Para entender as condições favoráveis à vitrificação, consideremos primeiro os processos da
cristalização (= concorrente!), simplesmente por serem melhores estudados.
4.1. Termodinâmica da cristalização
A termodinâmica descreve a tendência para cristalização, em função da temperatura - mais
corretamente, o quanto a temperatura deve chegar abaixo do ponto de fusão.
Primeiramente, temos que realizar que a temperatura de fusão é uma função do raio, quando
falamos em pequenos cristalitos.

7
Concentração limite, supersaturação e o crescimento dos cristais
A elevada energia em pequenos cristalitos tem sua explicação na termodinâmica, como já
vimos acima. Quantitativamente, isso foi estudado por B.W. Thomson; na verdade, ele estudou
a pressão de vapor sobre gotículas pequenas e tirou as conclusões para a solubilidade de
cristalitos, porque a única diferença entre esses dois fenômenos é o estado físico atingido
pelas transições: é líquido gasoso e líquido sólido, respectivamente.
Ele achou:
RT
M
rp
p 2ln Equação de Thomson, (1)
com p = pressão de vapor sobre a gotícula; p = pressão de vapor sobre uma lagoa
macroscópica; M = massa molar; = densidade; r = raio da gotícula; = tensão superficial;
R = constante dos gases; T = temperatura termodinâmica (o produto RT é também conhecido
como energia térmica).
Em toda analogia obtemos para a solubilidade de cristalitos:
RT
M
rcc
c
cr
r 2lnlnln , (2)
com cr = solubilidade de cristalitos do raio r; c = solubilidade de cristais macroscópicos, em
geral com r > 1 µm.
Para apenas pequenas elevações da solubilidade podemos simplificar:
como b
ba
b
aln :
r
VK
RT
M
rc
ccr 2. (3)
Nesta equação identificamos c
ccr como aumento relativo em solubilidade. Como se trata
do valor limite, isto é idêntico com a supersaturação relativa (em %). Expressa, ao mesmo
tempo, o grau de instabilidade termodinâmica. A supersaturação relativa não deve ser
confundida com a supersaturação absoluta (em °C) – que identificamos com o termo ccr .
V = volume molar M
V e os valores constantes são resumidos em RT
K2
, para o caso
de experimento isotérmico. Justamente essa restrição, de manter a temperatura constante,
deixamos cair a seguir.

8
Superresfriamento - a força propulsora da cristalização
Conforme o acima discutido, a concentração limite (medida na presença de cristais
macroscópicos) tem que ser ultrapassada por certo grau, para que ocorra a cristalização a
partir de uma fundição completa. Assumimos proporcionalidade entre concentração e a
temperatura, então podemos também afirmar que a cristalização requer temperaturas
notavelmente distantes da temperatura de fusão do cristal macroscópico 2. Como a
cristalização é um processo exotérmico (= libera energia), a reação está sendo promovida por
temperaturas baixas. Então podemos afirmar que a cristalização requer de temperaturas
bastante abaixo da temperatura TL (= temperatura líquidus = ponto de fusão dos cristais
macroscópicos), ou seja, um super-resfriamento.
A tendência da cristalização e da dissolução de cristais é descrita na termodinâmica, pelo
potencial da cristalização, µ.
L
Lfus
T
TTH )(; (5)
com Hfus = entalpia de fusão; TL = temperatura líquidus; )( TTL = super-resfriamento
absoluto; L
L
T
TT )( = super-resfriamento relativo. O super-resfriamento relativo pode então
ser visto como força propulsora da cristalização.
Nucleação - o início da cristalização
O nascimento de uma nova fase se conhece como nucleação (também é usada a expressão
germinação). Exemplos:
Cristalização: um líquido junta e organiza suas partículas de forma que possa crescer
um cristal.
Ebulição: dentro do líquido formam-se bolinhas de gás minúsculas
Condensação: gotículas começam crescer a partir de um vapor de alta concentração,
de preferência em superfícies sólidas. A gente conhece este fenômeno como orvalho -
de suma importância na meteorologia, na formação das nuvens de chuva.
Outra forma de condensação pode ser a transformação do gás diretamente em
cristalitos (= re-sublimação). Isso acontece, por exemplo, na formação de neve e
geada.
Diferenciamos entre:
Nucleação heterogênea: existem locais dentro da fase amorfa onde a organização
cristalina é induzida (= catalisada). Geralmente os núcleos heterogêneos são impurezas
2 O que está falado sobre a temperatura, certamente vale também para a pressão, aplicada a uma fase que deve
ser transformada em um cristal: a pressão deve ser bastante acima da pressão limite na qual os cristais
macroscópicos se dissolvem. O efeito da pressão é especialmente grande em transformações gasoso → sólido (=
re-sublimação), enquanto um líquido é naturalmente menos sensível a variações da pressão, devido sua baixa
compressibilidade. Mas a tendência é a mesma, já que o cristal geralmente tem densidade maior do que o líquido
(ou vidro), devido ao melhor empacotamento atômico. Exceção famosa: água a +4 ºC cuja densidade é maior do
que do gelo.

9
cristalinas, isto é, material diferente da fundição. Este material, no entanto, deve ter
semelhança com o sistema cristalino (cúbico, hexagonal, tetragonal, ortorrômbico,
romboédrico, monoclínico ou triclínico) e com as dimensões da célula unitária
(diferenças < 6%) do material a ser cristalizado.
Nucleação homogênea: na ausência de corpos estranhos e então sítios de nucleação
preferencial, a cristalização se inicia por nucleação homogênea. Na prática isso requer
de uma mistura completamente derretida, isenta de impurezas cristalinas, que se
encontra em um recipiente com superfície perfeitamente lisa. A nucleação homogênea
é espontânea e completamente randômica; a força propulsora são flutuações térmicas
randômicas dentro do material fundido (não-homogeneidades temporárias).
Note que a nucleação homogênea sempre é mais difícil do que a heterogênea, como será
mostrado a seguir.
Nucleação homogênea
Essa forma de nucleação ocorre com mais dificuldade, no interior de uma fase líquida e
límpida. No entanto, esta fase está submetida a flutuações estatísticas, no que diz respeito à
densidade, concentração e à temperatura (a temperatura pode ser interpretada sendo a soma
dos movimentos das partículas), dentro de um pequeno volume. Essas flutuações temporárias,
caso ocorrem em um volume suficientemente grande, podem finalmente desencadear a
formação do novo cristal.
A temperatura é de suma importância para a nucleação: um líquido deve ser resfriado abaixo a
temperatura máxima da nucleação heterogênea (identificamos esse limite como temperatura
de fusão), para que ocorra nucleação homogênea. Isso vale para o congelamento do líquido
puro que não tenha contato com uma superfície áspera. Um líquido que se encontra abaixo da
temperatura de fusão está no estado de super-resfriamento, isto é, fora do equilíbrio
termodinâmico. O super-resfriamento acarreta uma supersaturação que por sua vez induz a
nucleação. Em termos da energia livre por volume (que tem o significado físico de uma
pressão, pVGGV ), podemos afirmar que no estado de supersaturação a pressão dentro
da fase líquida se tornou superior à pressão interna dos cristalitos. A natureza prediz para este
caso uma vantagem da forma cristalina e a transformação de líquido em novos cristais deve
ser um processo espontâneo. Isto é, G é negativo.
O que contribui à energia livre da nova fase cristalina?
1. Ganho de um novo volume cuja energia interna fica mais baixa do que a da fase
original. Conhecemos o produto p.V como trabalho de volume, então esta parte da
energia conta negativamente. Sendo assim, a criação de novo volume em forma do
cristal é um processo favorável ( G < 0).
2. Criação de uma nova interface, líquido-sólido. Uma interface, A, sempre é uma região
de maior energia do que o interior de uma fase. Portanto, esta parte conta
positivamente à energia livre do processo ( G > 0); a tendência natural é minimizar a
interface – o que se opõe à formação do cristal.
Em um modelo muito simples (cálculos exatos mostraram sua validade) podemos assumir o
crescimento de um cristal em forma de esfera. O processo espontâneo da nucleação
homogênea se dá da soma das contribuições energéticas, que podemos formular como:
23 43
4rprAVpG . (6)
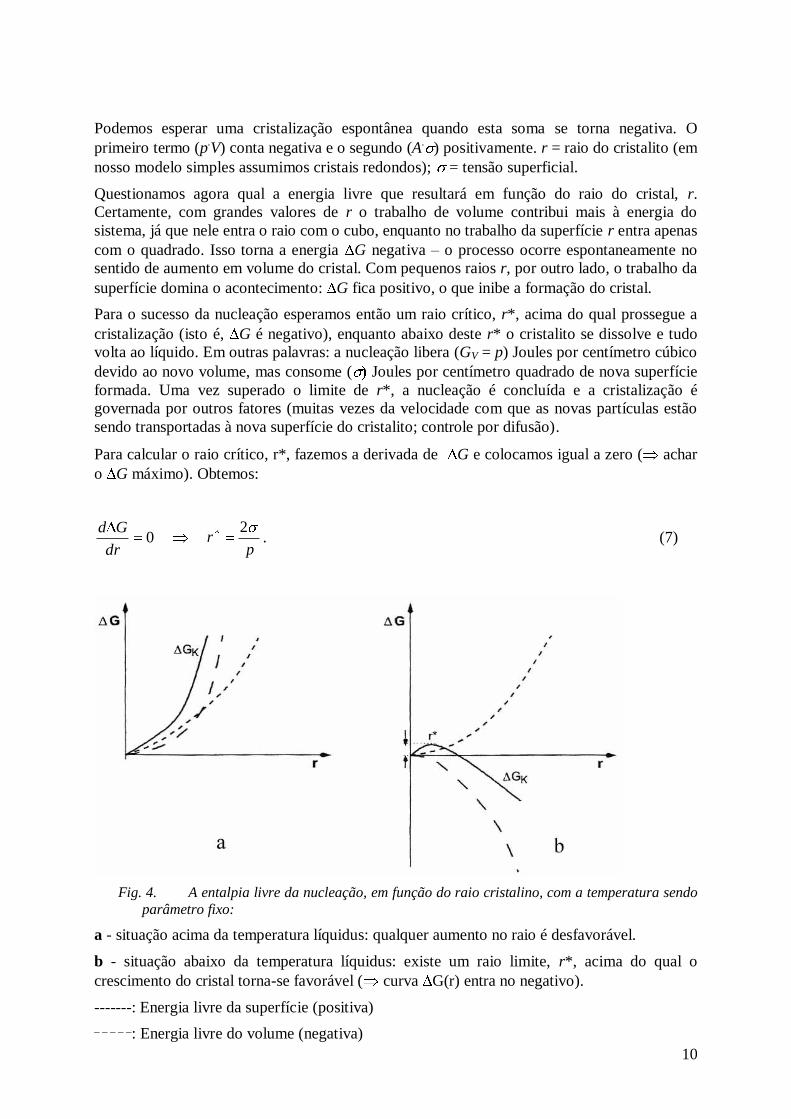
10
Podemos esperar uma cristalização espontânea quando esta soma se torna negativa. O
primeiro termo (p.V) conta negativa e o segundo (A
.) positivamente. r = raio do cristalito (em
nosso modelo simples assumimos cristais redondos); = tensão superficial.
Questionamos agora qual a energia livre que resultará em função do raio do cristal, r.
Certamente, com grandes valores de r o trabalho de volume contribui mais à energia do
sistema, já que nele entra o raio com o cubo, enquanto no trabalho da superfície r entra apenas
com o quadrado. Isso torna a energia G negativa – o processo ocorre espontaneamente no
sentido de aumento em volume do cristal. Com pequenos raios r, por outro lado, o trabalho da
superfície domina o acontecimento: G fica positivo, o que inibe a formação do cristal.
Para o sucesso da nucleação esperamos então um raio crítico, r*, acima do qual prossegue a
cristalização (isto é, G é negativo), enquanto abaixo deste r* o cristalito se dissolve e tudo
volta ao líquido. Em outras palavras: a nucleação libera (GV = p) Joules por centímetro cúbico
devido ao novo volume, mas consome ( Joules por centímetro quadrado de nova superfície
formada. Uma vez superado o limite de r*, a nucleação é concluída e a cristalização é
governada por outros fatores (muitas vezes da velocidade com que as novas partículas estão
sendo transportadas à nova superfície do cristalito; controle por difusão).
Para calcular o raio crítico, r*, fazemos a derivada de G e colocamos igual a zero ( achar
o G máximo). Obtemos:
0dr
Gd
pr
2. (7)
Fig. 4. A entalpia livre da nucleação, em função do raio cristalino, com a temperatura sendo
parâmetro fixo:
a - situação acima da temperatura líquidus: qualquer aumento no raio é desfavorável.
b - situação abaixo da temperatura líquidus: existe um raio limite, r*, acima do qual o
crescimento do cristal torna-se favorável ( curva G(r) entra no negativo).
-------: Energia livre da superfície (positiva)
_ _ _ _ _: Energia livre do volume (negativa)

11
_______: Energia livre, G, resultante da soma das duas anteriores.
Inserimos esse valor do raio crítico na equação (6) e obtemos a energia livre necessária para
chegar ao raio crítico do cristalito:
2
3
2
3
3
16
3
16
VGpG . (8)
G da cristalização pode ser expressa em termos da entalpia de fusão, Hfus, e o grau de
super-resfriamento, T, como será mostrado a seguir. Iniciamos com a dependência da
entalpia livre de Gibbs, de um termo entálpico e entrópico:
fusfus STHG . (9)
Aplicamos essa equação ao ponto crítico, na temperatura líquidus, TL (= temperatura de
fusão), onde G é zero. Obtemos uma expressão para a desconhecida entropia da fusão:
L
fus
fusT
HS (10)
Substituímos a entropia na equação acima:
L
fus
fusT
HTHG . (11)
Um denominador comum e a abreviação de TTT L leva à seguinte expressão para a
entalpia livre da cristalização:
L
fusT
THG . (12)
Essa relação já conhecemos (ver equação (5), expressão para o potencial químico, µ). Isso não
é surpresa, já que o potencial químico é nada outro do que a energia livre, relacionada à parte
n do cristalito dentro do sistema todo,
pTdn
dG
,
. (13)
12
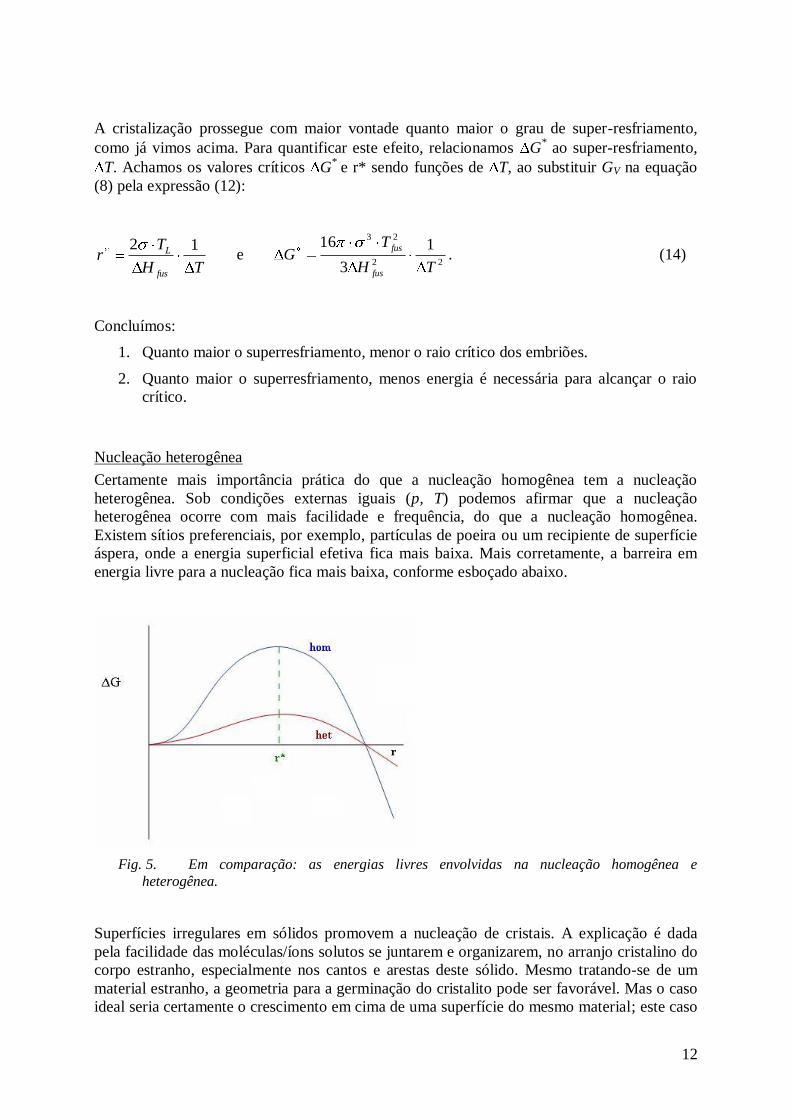
A cristalização prossegue com maior vontade quanto maior o grau de super-resfriamento,
como já vimos acima. Para quantificar este efeito, relacionamos G* ao super-resfriamento,
T. Achamos os valores críticos G*
e r* sendo funções de T, ao substituir GV na equação
(8) pela expressão (12):
TH
Tr
fus
L 12 e
22
231
3
16
TH
TG
fus
fus. (14)
Concluímos:
1. Quanto maior o superresfriamento, menor o raio crítico dos embriões.
2. Quanto maior o superresfriamento, menos energia é necessária para alcançar o raio
crítico.
Nucleação heterogênea
Certamente mais importância prática do que a nucleação homogênea tem a nucleação
heterogênea. Sob condições externas iguais (p, T) podemos afirmar que a nucleação
heterogênea ocorre com mais facilidade e frequência, do que a nucleação homogênea.
Existem sítios preferenciais, por exemplo, partículas de poeira ou um recipiente de superfície
áspera, onde a energia superficial efetiva fica mais baixa. Mais corretamente, a barreira em
energia livre para a nucleação fica mais baixa, conforme esboçado abaixo.
Fig. 5. Em comparação: as energias livres envolvidas na nucleação homogênea e
heterogênea.
Superfícies irregulares em sólidos promovem a nucleação de cristais. A explicação é dada
pela facilidade das moléculas/íons solutos se juntarem e organizarem, no arranjo cristalino do
corpo estranho, especialmente nos cantos e arestas deste sólido. Mesmo tratando-se de um
material estranho, a geometria para a germinação do cristalito pode ser favorável. Mas o caso
ideal seria certamente o crescimento em cima de uma superfície do mesmo material; este caso

13
identificamos como “crescimento do cristal macroscópico” – que será discutido na próxima
seção.
A facilidade de se estabelecer o contato entre um líquido e uma superfície sólida pode ser
descrita quantitativamente pelo fenômeno da molhabilidade. O experimento é bastante
simples: em cima de uma superfície horizontal, plana e lisa, coloca-se uma gota do líquido e
observa-se a forma da bolha. Ângulos de contato menores que 90° promovem a nucleação.
A energia livre necessária para induzir a nucleação heterogênea pode ser formulada como:
ohetero GfG hom)( . (15)
Nesta, o fator de correção f( ) 1. A expressão exata do fator é:
32cos
4
1cos
4
3
2
1cos1cos2
4
1)(f , (16)
onde é o ângulo de contato, conforme mostrado na Fig. 6.
Identificamos três casos típicos:
Ângulo de contato é grande. A superfície repele o líquido. Neste caso temos
f( ) 1; há dificuldade de nucleação heterogênea; a situação aproxima-se à da
nucleação homogênea.
Ângulo de contato é pequeno. A gota se espalha e fica achatada em cima do suporte.
A superfície atrai o líquido e resulta uma grande área onde a nucleação pode ocorrer.
Neste caso temos f( ) 0; há pouco impedimento para o contato heterogêneo;
nucleação fácil.
Num ângulo = 90° temos f( ) = ½. O líquido assume a forma de meia-esfera; em
comparação à nucleação homogênea precisa ser superada apenas a metade da energia
de superfície, para se conseguir nucleação.
Fig. 6. Definição do ângulo de contato, (obs.: neste esboço a tensão superficial é
representada por ). No esboço à esquerda tem-se uma gota numa superfície aderente, no
esboço à direita numa superfície repelente.

14
Quanto mais baixa a barreira energética da nucleação heterogênea, menos super-resfriamento
é necessário. O ângulo de contato indica a facilidade desta forma de nucleação. Pela nossa
surpresa o raio crítico dos embriões não varia muito, como já implicam as duas curvas de G
na Fig. 5. Porém, o volume crítico pode ser drasticamente reduzido, porque o ângulo de
contato afeta a forma do novo conjunto cristalino.
Ao contrário da nucleação homogênea onde se forma uma nova interface a partir de zero, a
nucleação heterogênea acontece a custo de outra interface já existente. Ou seja, com um grão
de poeira já introduzimos uma área de elevada energia. O sumiço da área, poeira/líquido, é
uma vantagem energética que compensar parte da energia necessária para formar a nova
interface líquido/cristal. A um estágio avançado da cristalização a formação de novos
cristalitos ocorre, portanto, sempre na zona intergranular, isto é, no encostamento de dois
grãos já formados. O crescimento em forma radial pode-se observar, por exemplo, numa
janela onde crescem cristais de gelo no inverno (ver Fig. 10a); em sala de aula isso pode ser
mostrado pelo experimento da cristalização de uma solução supersaturada de acetato de sódio 3.
4.2. Cinética da cristalização.
Cinética da cristalização segundo Tammann
Na temperatura de fusão 4 a velocidade da cristalização é nula. Ao abaixar a temperatura ela
aumenta, percorre um máximo e cai novamente a zero, ao chegar a temperaturas muito abaixo
do TL. Deste fenômeno podemos tirar uma conclusão importante para a prática da produção de
vidro: quem pretende produzir um vidro homogêneo, amorfo e transparente, deve percorrer o
intervalo térmico da cristalização, o mais rápido possível.
A cinética da cristalização foi descrita por Tammann 5. Segundo ele podemos separar
nitidamente o estágio da nucleação, da fase do crescimento. As velocidades destes
acontecimentos dependem tipicamente da temperatura – mais corretamente do
superresfriamento. A Fig. 7 fornece uma noção qualitativa destas velocidades; observamos
duas curvas bem semelhantes, enquanto a curva da nucleação (S) fica deslocada para
temperaturas menores, o que corresponde a um maior superresfriamento. Seu deslocamento
para a esquerda é especialmente grande quando a fusão/solução de partida é isenta de corpos
estranhos, pois neste caso esperamos nucleação homogênea. Certamente, o “nascer” de um
cristal é um processo mais difícil do que seu crescimento, então a curva S(T) fica sempre
deslocada para temperaturas mais baixas, em relação à curva R(T).
3 Um ensaio de sala de aula é mais impressionante quanto mais simples: dissolva a quente cerca de 100 g de
acetato de sódio, em apenas 20 mL de água. Tudo derrete e dá uma solução transparente. Em caso de impurezas
visíveis deve-se filtrar a solução à quente, através de um filtro preguado. Durante o resfriamento à temperatura
ambiente a solução não deve ser sacudida. Cristalização espontânea pode ser desencadeada, por meio de
ranhuras na parede interna do copo, melhor por introdução de um grãozinho de acetato de sódio cristalino.
4 A temperatura de fusão também é chamada de temperatura líquidus; TL, ela é idêntica com o ponto de
solidificação, a ser determinado no cristal macroscópico.
5 Fonte histórica: Tammann, Z. Phys. Chem. B 25 (1898) 441.
Uma descrição da teoria da cristalização atual, ver http://en.wikipedia.org/wiki/Nucleation

15
Fig. 7. Velocidades de nucleação e crescimento de cristais, em dependência da temperatura.
Tanto a nucleação (S) como o crescimento (R) percorrem um máximo e ambas as velocidades
são nulas na temperatura líquidus, TL. A curva de crescimento do cristal macroscópico, R(T),
tem valores positivos abaixo da TL, isto é, os cristais crescem. Acima da TL o crescimento é
negativo, ou seja, o cristal se dissolve. Um valor negativo da curva S(T), por outro lado, não
faz sentido, pois o embrião começa crescer a partir de zero.
A queda das duas curvas em ambos os lados do seu máximo tem explicações diferentes: ao
lado de altas temperaturas tem motivos energéticos (= entálpicos e entrópicos); ao lado de
baixas temperaturas se explica com a viscosidade do ambiente amorfo, fusão/solução, que
sobe rapidamente quanto mais baixa a temperatura. Daí os movimentos moleculares são muito
lentos e o transporte de uma molécula soluta até a posição certa da superfície do cristalito, é
cada vez mais demorado.
Logo abaixo da TL observamos uma faixa (estreita), o "Intervalo de Ostwald-Miers", onde a
nucleação é infinitamente lenta, isto é, não se formam embriões de cristais, mas já temos
condições favoráveis para o crescimento de um cristal maior. A largura deste intervalo prediz
o quanto um líquido puríssimo pode ser resfriado abaixo do ponto TL sem cristalizar. O estado
físico de um líquido nesta situação é meta-estável, quer dizer, fora do equilíbrio
termodinâmico.
A dependência térmica da velocidade de cristalização, R(T), pode ser descrita por uma
equação empírica e simples, do tipo
)( TTK
R La, (17)
com R = velocidade de crescimento dos cristais; T = temperatura atual; TL = Temperatura
líquidus; = viscosidade; a, K = constantes, enquanto a geralmente tem um valor perto de 1.
Olhando somente na equação (17) na sua forma anotada acima, não deduzimos uma curva
R(T) em forma de sino, conforme esboçada na Fig. 7. Em vez disso esperamos uma

16
velocidade cada vez maior quando mais longe do ponto TL. Mas junto ao fato que a própria
viscosidade da fase líquida também depende caracteristicamente da temperatura, finalmente
fornece a imagem encontrada no experimento. Essa dependência, igualmente empírica, pode
ser formulada como
T
A
eBT )( , (18)
onde B e A são constantes 6. Note que a viscosidade aumenta de maneira exponencial, e esta
dependência certamente dominará, quanto mais frio o líquido. Portanto, resulta uma queda da
curva de cristalização ao lado esquerdo do seu máximo, conforme mostrado na Fig. 7.
TL , a e K dependem da composição da mistura fundida. Desta fórmula concluímos que o
superresfriamento absoluto, )( TTL , é o fator principal para a velocidade da cristalização.
Perguntamos agora quanto ao tamanho do cristal, sob a condição de um resfriamento de
velocidade (vresfr) constante. O tamanho é mais facilmente determinado através do
comprimento (médio) de aresta, l.
Temos então as duas definições:
dt
dlR , para a velocidade da cristalização, e
dt
dTvresfr , para a velocidade do resfriamento.
Após o resfriamento completo temos então cristais do tamanho
LT
resfr
dTRv
dtRl00
1. (19)
A última integral é simplesmente a área abaixo da curva R(T), do crescimento dos cristais.
Vamos chamá-la de F, então podemos indicar o tamanho dos cristais após o resfriamento total
como
resfrv
Fl . (20)
Esperamos então cristais grandes caso a fundição/solução for resfriada lentamente. Também
podemos afirmar: quanto maior a área abaixo da curva R(T), mais fácil se obter cristais
grandes. Vamos inverter essas afirmações para chegar às condições favoráveis de se formar
um vidro: a velocidade do resfriamento deve ser escolhida grande o suficiente, para que o
tamanho dos cristalitos fique abaixo do limite de detecção. Em termos práticos podemos
afirmar que a cristalização fica ausente quando a fundição/solução é submetida a um
resfriamento choque. Daí a mistura solidifica sem cristalização, isto é, vitrifica-se.
Vamos definir um limite superior para o tamanho dos cristais que podem ocorrer em vidros
ópticos de qualidade: com l < 200 nm a transparência do corpo é garantida, pois o
6 Os valores de B e A, para os solventes mais comuns, são referidos no Cap. 2.3.2 de: A. Isenmann, “Operações
Unitárias na Indústria Química”; disponível em
http://www.timoteo.cefetmg.br/site/sobre/cursos/quimica/repositorio/livros/ou/index.html

17
espalhamento excessivo da luz (= espalhamento de Rayleigh) somente começa quando chegar
à região do comprimento da onda. No caso da luz visível, isto é entre 400 e 800 nm. Sendo
assim, a velocidade de resfriamento fica limitada a nm
F
l
Fvresfr
200.
Em analogia à discussão da curva R(T), podemos também analisar a curva da nucleação, S(T).
Essa curva reflete o número de cristalitos que se formam a certa temperatura. O número dos
cristais após o resfriamento total é o quociente de área abaixo da curva S, dividida pela
velocidade de resfriamento, vresfr, em analogia à equação (19).
Com essas evidências afirmamos para o resfriamento de uma solução supersaturada ou uma
fundição superresfriada:
1) A condição (19) para o tamanho dos cristais somente vale para os cristais maiores.
2) Por outro lado, os embriões que não foram formados logo no início da elevação da
curva S(T), mas numa temperatura mais baixa ainda, levam a cristalitos menores e não
têm chance de crescer.
3) Um fenômeno interessante pode-se observar nas imediações de um cristal maior, onde
todos os cristalitos miúdos estão sendo "comidos". Em volta de cada cristal maior tem-
se um pátio isento de cristalitos, conforme mostrado na micrografia da Fig. 8.
Fig. 8. Micrografia (1 : 120) de dipicrilaminato de tálio:
a) cristalização a partir da solução mais diluída e aos poucos;
b) cristalização a partir de alta supersaturação e rápida. Os cristais maiores formaram-se a custo de uma multidão de menores.
Os cristais menores têm uma superfície maior em relação ao seu volume, do que os cristais
grandes. Como a superfície possui elevada energia em comparação ao interior de uma fase,
então a tendência natural é a criação de uma fase maciça e reduzir a superfície, conforme a
equação (6). Isso é realizado num cristal maior (= pequena superfície e grande volume). A
explicação quantitativa fornece a termodinâmica, mais especificamente a relação de Thomson
(equações 1 e 2).

18
A equação de Avrami
A equação de Avrami 7 descreve a velocidade de transformações de fases em geral, à
temperatura constante (= isotérmica). Ela tem importância, tanto na físico-química (descrição
da cristalização em sólidos amorfos, especialmente em polímeros), quanto na metalurgia onde
se calculam os tamanhos dos grãos em ligas metálicas. Ela pode ser aplicada ao nosso
problema: a transformação de uma mistura fundida para a fase cristalina. Através da equação
obtemos uma noção da velocidade de cristalização. Os resultados não são muito exatos
porque a equação tenta descrever a transformação completa, desde 100% líquido até perto de
100% cristalino, usando apenas duas variáveis: a velocidade de nucleação S e a velocidade do
crescimento dos cristais, R.
O começo da mudança de uma fase para outra é um processo imprevisível, no que diz respeito
ao local. Em poucos pontos dentro da fase A começa, devido a flutuações estatísticas de
temperatura e densidade, a nucleação, também chamada de nucleação homogênea. Alternativa
seria a existência de impurezas ou superfícies ásperas que representam pontos fixos onde a
cristalização pode prosseguir ("nucleação heterogênea"; não é descrita pela equação de
Avrami).
A partir destes poucos pontos a nova fase (em nosso caso: os cristais) começa crescer. Ao
decorrer do tempo também formam-se em outros locais novos pontos de nucleação. Assim
prossegue até que todo o volume é ocupado pela nova fase, B. A equação de Avrami descreve
então quanto à porcentagem da nova fase, em função do tempo. Um desenho simples do
acontecimento presume um crescimento isotrópico, ou seja, cristais redondos.
Fig. 9. Estados da nucleação de cristais
Pressupondo uma velocidade de nucleação S constante, daí a distribuição arbitrária de núcleos
esféricos dentro do volume, a parte da nova fase B, se calcula por:
7 M. Avrami, Kinetics of phase change. III: Granulation, Phase Change and Microstructure, J. Chem. Phys., 9
(1941), 177–184.

19
43
31tRS
edt
dBB , (20)
com a velocidade do crescimento R, dentro do tempo t. Conforme essa equação, não têm-se
restrições nos tempos (vale para processos lentos e rápidos), nem nas partes de B (pouca fase
B até quase completo).
Essa equação parece ser bastante complexa. Para o estágio inicial da cristalização (Fig. 9,
imagem de cima), no entanto, ela se simplifica. Para partes de 1B podemos escrever
ze z1 . Então:
43
3tvNBini
. (22)
Interpretação: o número de núcleos cresce com S.t, também o tamanho de cada cristal,
tRl , cresce linearmente, então seu volume cresce com 3
tR . Levando em conta esses
dois fatores juntos, podemos afirmar que o volume total de todos os cristais cresce com t4.
Somente quando os cristais tornam-se maiores (concrescência) e o volume onde podem
formar-se novos núcleos se torna cada vez mais raro, o volume de B aumenta mais devagar do
que t4.
A formulação mais em geral da equação de Avrami é, portanto:
ntkeB 1 , (23)
onde n é o expoente de Avrami e pode ter valores entre 1 e 4. Com esta equação todas as
dependências do tempo podem ser descritas. Por exemplo, o crescimento em apenas duas
dimensões, isto é, cristalização em camadas muito finas: o expoente n = 3. Tanto a velocidade
da nucleação S como a velocidade do crescimento R, estão contidas na constante k. As duas
dependem fortemente da temperatura, portanto k = k(T).
Geralmente a equação de Avrami descreve bem o início da cristalização, enquanto no final
deste processo se evidencia uma discrepância cada vez maior entre cálculo e realidade. Isto se
deve principalmente a dois fatos:
1. Os cristais grandes, de diferentes orientações, encostam e formam interfaces
energeticamente desfavoráveis (beiradas intergranulares; ver Fig. 9 imagem de baixo).
2. Todas as novas peças a serem implementadas no novo cristal chegam à superfície via
difusão, atravessando uma fina camada ao redor do cristal em crescimento, também
chamada de "pátio do cristal" (Fig. 8). Sendo assim, a velocidade da cristalização
depende inteiramente da velocidade deste transporte (“controle por difusão”); isso
afeta o lado esquerdo da curva R(T), na Fig. 7. Quando os pátios de dois cristais
vizinhos se tocam, não há mais gradiente em concentração e a difusão pára.

20
4.3. Aspecto dos cristais formados
O ensaio da cristalização do acetato de sódio, feito em sala de aula, mostrou que a
cristalização libera uma quantidade notável de energia. A direção através da qual essa energia
pode ser dissipada 8, influencia no aspecto dos cristais, ou seja: seu hábito.
O que determina a forma do cristal macroscópico é a velocidade de crescimento, por sua vez
governada pela facilidade com que o calor pode ser levado para fora da zona de cristalização.
As condutividades térmicas dependem, além do estado físico, também da direção no espaço.
É claro que num líquido não temos preferências a longa distância, mas num cristal podemos
identificar direções preferenciais, em quais as distâncias interatômicas são significativamente
menores do que em qualquer outra direção. O fenômeno é conhecido como “anisotropia”.
Identificamos dois casos fundamentalmente diferentes que influenciam notavelmente na
aparência do cristal:
a) A dissipação ocorre mais facilmente pela fundição. Isto significa que o gradiente térmico é
maior dentro da fundição do que no cristal. Podemos esperar uma temperatura inferior na fase
amorfa do que no cristal, promovendo neste local o crescimento do cristal, R(T). Este é o
caso, em geral, ao se cristalizar sob super-resfriamento rápido ou quando a fase líquida se
apresenta como filme fino. Uma ponta que cresceu acha condições cada vez mais favoráveis,
portanto continua crescendo. Em consequência forma-se um cristal com grande superfície,
com aspecto de agulhas que podem ter muitas ramificações. Conhece-se este cristal como
dentrito 9 (ver Fig. 10 a). Exemplos são, além do gelo nas janelas no inverno, a etringita
formada no gesso e o composto C3A no concreto, que ambos provocam a pega devido à
interpenetração das suas agulhas.
b) A dissipação ocorre principalmente através do próprio cristal. Isto significa que o gradiente
térmico é maior dentro do cristal do que na fundição. Daí a frente do crescimento chega a uma
região de temperatura mais alta - ela se recua. Desta forma, nenhum canto avança muito e
forma-se uma face lisa e reta. O cristal macroscópico tem uma área superficial mínima, dentro
dos planos cristalinos principais - ele aparece compacto. Essa é uma característica de cristais
que cresceram lentamente, não muito longe das condições de equilíbrio com a fundição ou
solução saturada. Como exemplos podemos referir a maioria dos metais e suas ligas, onde há
formação de grãos de aspecto bem redondo.
8 Dissipar = conduzir o calor gerado num local, ao seu redor.
9 Ao extremo, a forma anisotrópica da cristalização leva aos esferulitos, ou seja, “cristais redondos”, ver Fig. 14.

21
Fig. 10. "Habitus" é chamada a forma que o cristal macroscópico toma por fora:
(a) Dentrito: gelo formado em cima de uma janela fria. (b) Cristal maciço: fluorita de uma
caverna.
5. Conclusões práticas para a produção de vidro Após este excurso ao mundo dos cristais, vamos voltar para o objetivo principal deste artigo:
a produção de vidros. Um fato importante para a prática, não só na produção de vidro, mas
também em todos os processos onde se forma material cristalino, é a solubilidade elevada de
cristalitos muito pequenos (diâmetros tipicamente < 100 nm). Por isso, conseguimos um
precipitado, somente a partir de uma solução supersaturada. O valor da saturação/fundição
saturada, conforme indicado na literatura, no entanto, sempre vale para sistemas onde há
cristais macroscópicos. O sistema entra em uma região meta-estável que, na exclusão de
corpos estranhos e cantos de elevada energia, pode ser mantida por muito tempo. Por outro
lado, o primeiro sinal de um embrião de cristal leva ao crescimento espontâneo do mesmo.
Número e tamanho dos cristais formados dependem dos seguintes fatores:
A probabilidade S da formação de um novo embrião é proporcional à supersaturação
relativa, c
ccr , enquanto
A velocidade R do crescimento dos cristalitos é apenas proporcional à supersaturação
absoluta, ccr .
Para a produção de vidro é de suma importância evitar as condições favoráveis à formação de
embriões e, ao mesmo tempo, evitar condições favoráveis de crescimento dos mesmos. Na
Fig. 7 anotamos duas curvas, R(T) e S(T), que têm formas bem semelhantes, mas as duas são
deslocadas uma da outra, por um valor característico que chamamos de Intervalo de Ostwald-
Miers. Sempre observamos que a germinação requer temperaturas mais baixas, por ser um
processo mais difícil do que o crescimento. Em outras palavras: a largura do intervalo de
Ostwald-Miers determina a facilidade de produzirmos um vidro. Quanto mais largo o
intervalo, mais fácil se obter um vidro.
A partir da cinética de cristalização de Tammann resulta uma afirmação remarcável: um vidro
frio, quando aquecido aos poucos, vai chegando à nucleação, logo abaixo da temperatura TL -
22
mesmo se um crescimento de cristais de tamanho notável não for observado. Isso fornece a
explicação do princípio de produzirmos uma cerâmica vítrea (ver p. 32), ao manter um vidro
numa faixa térmica logo abaixo da TL, isto é, onde a velocidade de crescimento dos cristais é
notável enquanto a taxa de crescimento ainda está muito pequena. Durante o reaquecimento
de um vidro passamos então pelo intervalo de nucleação, S(T), sem necessariamente correr o
perigo de formar cristais grandes. Cristalização que é provocada por este caminho pode
também ser chamada de "desvitrificação". Isso nem sempre é sinônimo de defeito na
produção de vidro; em alguns produtos, nas cerâmicas vítreas, é justamente esse o processo
que, quando feito de maneira bem controlada, leva ao sucesso (mais sobre isso na p. 32).
O caminho direto, quer dizer, o resfriamento lento da fundição, não leverá ao mesmo
resultado, já que passamos primeiro pela região do crescimento rápido e só depois, numa
temperatura mais baixa, na região da nucleação.
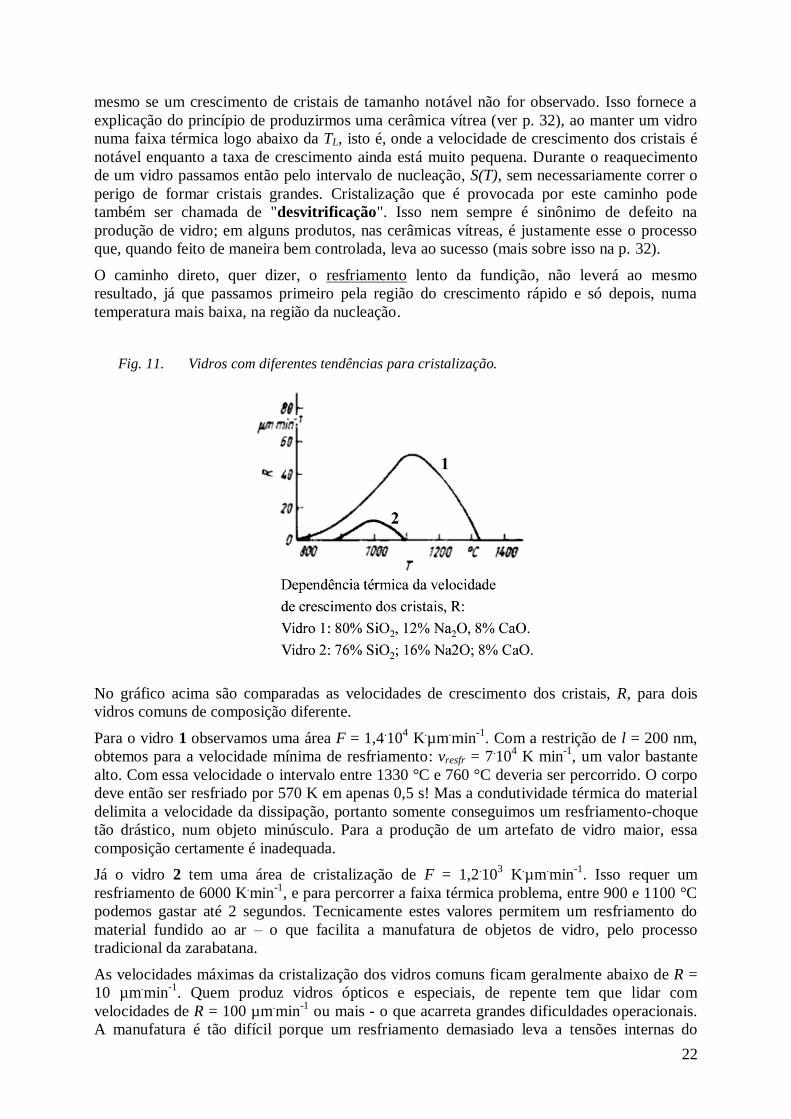
Fig. 11. Vidros com diferentes tendências para cristalização.
No gráfico acima são comparadas as velocidades de crescimento dos cristais, R, para dois
vidros comuns de composição diferente.
Para o vidro 1 observamos uma área F = 1,4.10
4 K
.µm
.min
-1. Com a restrição de l = 200 nm,
obtemos para a velocidade mínima de resfriamento: vresfr = 7.10
4 K min
-1, um valor bastante
alto. Com essa velocidade o intervalo entre 1330 °C e 760 °C deveria ser percorrido. O corpo
deve então ser resfriado por 570 K em apenas 0,5 s! Mas a condutividade térmica do material
delimita a velocidade da dissipação, portanto somente conseguimos um resfriamento-choque
tão drástico, num objeto minúsculo. Para a produção de um artefato de vidro maior, essa
composição certamente é inadequada.
Já o vidro 2 tem uma área de cristalização de F = 1,2.10
3 K
.µm
.min
-1. Isso requer um
resfriamento de 6000 K.min
-1, e para percorrer a faixa térmica problema, entre 900 e 1100 °C
podemos gastar até 2 segundos. Tecnicamente estes valores permitem um resfriamento do
material fundido ao ar – o que facilita a manufatura de objetos de vidro, pelo processo
tradicional da zarabatana.
As velocidades máximas da cristalização dos vidros comuns ficam geralmente abaixo de R =
10 µm.min
-1. Quem produz vidros ópticos e especiais, de repente tem que lidar com
velocidades de R = 100 µm.min
-1 ou mais - o que acarreta grandes dificuldades operacionais.
A manufatura é tão difícil porque um resfriamento demasiado leva a tensões internas do

23
objeto. Essas tensões abaixam drasticamente sua resistência mecânica (vibrações harmônicos,
batidas, armação torta, etc.) e facilitam sua rachadura catastrófica. Além disso, essas tensões
acarretam um efeito óptico conhecido como birrefringência 10
, um fenômeno bastante
prejudicial em lentes, prismas e espelhos, nas diversas aplicações de óculos, microscópios,
telescópios, aparelhos de espectroscopia, etc. Somente ao tiver objetos de pequenas
dimensões, um resfriamento-choque pode ser levado em consideração, sem correr o perigo de
perdê-los por trinco. Sob essa promessa, até metais podem ser levados ao estado vítreo. Os
mais famosos exemplos são os aços austenítico e martensítico, onde o carbono está soluto
randômico dentro das fases Fe-γ e Fe-α, respectivamente.
A vitrificação é então mais fácil em materiais onde nucleação e/ou crescimento dos cristalitos
é devagar. Em misturas de vidro que foram aquecidas apenas pouco acima da zona de fusão,
têm-se suficientes núcleos ("embriões") de cristais nos vidros técnicos, devido às impurezas
sólidas contidas na fundição ou à superfície áspera do recipiente da mistura. Neste caso pode-
se esperar uma nucleação S heterogênea, fácil e rápida, e o crescimento dos cristais R se torna
o fator delimitante para os vidros técnicos.
6. Conceito de formadores de rede, modificadores de rede e
quebradores de rede
6.1. "Formadores de rede"
A produção de um vidro requer um sistema de cristalização lenta, como o exemplo acima
deixou claro. Geralmente, quanto mais pura e homogênea a mistura sólida dos ingredientes
minerais (equipamento mais utilizado: moinho de bolas), mais fácil e rápida sua cristalização.
Portanto, é mais fácil produzir um vidro a partir de uma mistura de diferentes ingredientes que
dificultam mutuamente sua cristalização.
O ingrediente principal de cada mistura tecnicamente interessante é um óxido de semi-metal
(“óxidos ácidos” 11
). Dentre estes são apenas a metade das fórmulas que se destacam por uma
taxa de cristalização suficientemente baixa. São essas principalmente os óxidos das fórmulas:
A2O3 (p. ex. B2O3, Al2O3, As2O3, Sb2O3),
AO2 (p. ex. SiO2, GeO2, também o isostérico BeF2)
A2O5 (p. ex. P2O5, As2O5).
Todos esses óxidos são feitos por poliedros de coordenação [AOp], com p < 6, e esses
poliedros são interconectados somente através dos seus cantos. Sendo assim, cada poliedro
[AOp] tem três ou mais cantos em comum com os poliedros vizinhos, mas nenhuma vértice ou
até face. Para o oxigênio isso significa que tem, no máximo, dois vizinhos de semi-metal.
Significado técnico hoje somente têm os vidros a base do quartzo, SiO2.
10 O fenômeno é a dupla aparência de um objeto quando observado através do corpo de vidro. Explica-se a
separação do feixe de luz com diferentes velocidades da luz (polarizada), dependendo da direção com que
atravessa o corpo. Isso não é só um fenômeno de cristais de certa geometria (a calcita da Islândia é o exemplo
mais famoso), mas também em vidros e até em líquidos em movimento. A física explica a birrefrigência com a
forte anisotropia dentro do material iluminado, provocada por tensão e/ou pressão localizada.
11 Essa expressão se entende quando interpretar os óxidos como anidrido. Com B2O3, As2O3, SiO2 e P2O5, após a
adição de água chegamos no ácido bórico, ácido arsênico, ácido silícico, ácido fosfórico, respectivamente.

24
Por outro lado, os óxidos das fórmulas AO, AO3, AO4 e AO5, não são adequados como base
de vidros de qualidade, pela tendência pronunciada de cristalização rápida e espontânea.
Especialmente fácil é a produção de um vidro, a partir da co-fusão de uma mistura de óxidos
ácidos, sendo essas SiO2, B2O3, Al2O3, P2O5, em primeira linha. Seus poliedros formam uma
rede tridimensional e compacta, portanto o nome "formadores de rede".
6.2. "Quebradores de rede"
Quando os óxidos especificados acima são misturados com óxidos básicos (= óxidos de
metais), tais como Na2O, K2O, MgO, CaO, PbO ou ZnO, ocorre um afrouxamento da rede
tridimensional.
A estrutura principal dos vidros técnicos é providenciada por tetraedros de [SiO4] não
orientados que são interligados através dos seus cantos. Algumas destas pontes simples de
siloxano, Si-O-Si, podem então ser quebradas, por meio de íons O2-
adicionais, introduzidos
pelos óxidos básicos.
Si O Si + O2-Si O-
Si-O+
Por este motivo, os óxidos básicos são chamados de "quebradores de rede". Em geral,
podemos observar quanto mais pontes de siloxano quebradas, mais baixa a temperatura de
amolecimento do vidro, Tg. Comparando os óxidos dos álcalis, M2O, com os álcalis terrosos
MO, podemos afirmar que os primeiros acarretam uma maior depressão da temperatura Tg do
que quantidades equivalentes de óxidos MO, já que a ligação iônica no primeiro,
Si O-Si
-O2 M+
, fica mais fraca do que no segundo caso, Si O-
Si-OM2+
,
devido às distâncias inter-iônicas maiores (tem 2 cátions) e então forças de Coulomb menores.
6.3. "Modificadores de rede"
Os átomos tetravalentes de Si, estrutura predominante nos vidros comuns, podem ser
substituídos parcialmente por outros íons, igualmente capazes de sustentar uma rede
tridimensional. O arranjo dos íons O2-
fica inalterado, ou seja, continuam formando tetraedros
em volta dos novos cátions. São estes: o boro trivalente, o alumínio trivalente e o fósforo
pentavalente. Portanto, estes íons são chamados de "modificadores de rede". Note que a
substituição de cada Si4+
por B3+
ou Al3+
aumenta a carga negativa da rede, enquanto a
implementação de P5+
abaixa sua carga por unidade. Sendo assim, cada modificador da
primeira categoria (B ou Al) consome um quebrador de rede. Já um modificador da segunda
categoria (P, As ou Sb) induz mais uma ponte quebrada de siloxano. Tais vidros modificados
de silicato destacam-se por intervalos térmicos de amolecimento, especialmente largos. Além
disso, têm coeficientes de expansão térmica, bastante pequenos. Para a produção de vidros
isso tem efeitos benéficos:
Durante o largo intervalo térmico onde o material está ainda fundido, a viscosidade do
material é bastante alta. Isso impede a reorganização da rede cristalina, ou seja, essas
misturas se destacam por cristalização lenta.
As velocidades de resfriamento podem ser escolhidas mais altas, sem correr o risco de
trincar o objeto devido a tensões internas. Então é possível resfriar o objeto novo de
vidro simplesmente ao ar, em vez de serem necessários "fornos de resfriamento", que
garantem um perfil térmico mais suave, lento e controlado.
No produto acabado: menos tensões internas, então melhor qualidade óptica; vidros de
laboratório ou da cozinha, que não trincam mesmo se aquecidos bruscamente ou
resfriados por choque.

25
7. Tipos de vidros Os vidros inorgânicos podem ser discriminados, conforme seu uso:
vidro comum: vidro de chapa e vidro de recipientes,
vidro especial de elevada resistência química,
vidro óptico (para lentes, óculos, fibras de condução de luz, etc.),
vidros do setor eletro-eletrônico,
vidro de solda,
vidro de cerâmica (entre outros, que aguentam altas temperaturas, mudanças drásticas
de temperatura, ferroelétricos, fotossensível).
7.1. Vidro comum: história, matérias-primas e princípios de manufatura
Já os povos primitivos tinham contato com figurinhas de vidro, sem saber de onde vêm.
Foram usados para cultos religiosos, principalmente. Hoje se sabe que foram formados
quando descargas de tempestade, com milhares de graus por curto tempo, caíram em bancos
de areia. Os grãos da areia, isto é, quartzo com até 10% de impurezas, fundiram e, devido ao
resfriamento na ordem de poucos segundos, vitrificaram. A forma interessante dessas
figurinhas depende da facilidade de fundir, então principalmente da composição local das
areias, além de desvios do caminho que o raio sofreu em minerais magnéticos ou em pedras
compactas.
Os primeiros vidros que foram fabricados pelo homem datam da época dos egípcios (3400
a.C.).
As matérias-primas usadas hoje são pouco diferentes da antiguidade:
Nome trivial Fórmula do mineral Fórmula dentro do vidro
Areia de quartzo SiO2 SiO2
Soda ou sulfato de sódio
mais carvão 12
Na2CO3 ou Na2SO4/C Na2O
Potassa (originada de cinzas
de madeira)
K2CO3 K2O
Calcário CaCO3 CaO
Zarcão 13
Pb3O4 PbO
Bórax Na2B4O7 B2O3
Caolinita 14
ou feldspato Al2(OH)4[Si2O5] ou
M1+
[AlSi3O8]
Al2O3
12 Na2SO4 + C Na2O + SO2 + CO.
13 Pb3O4 3 PbO + 0,5 O2.
14 Barro ou argila, são misturas complexas onde a Caolinita é o componente principal. Também contêm Ilita,
(K+/H3O+)y{Al2(OH)2[Si4-yAlyO10]}, com y = 0,7 a 0,9.

26
Quantidades controladas destes ingredientes são co-moídos. Uma granulação mais fina abaixa
a temperatura da primeira fusão da mistura, a partir das matérias-primas. Mas também o
material reciclado é geralmente finamente moído, para estabelecer homogeneidade perfeita no
produto acabado.
Note que a produção de vidro envolve uma alta taxa de reciclados: em média 60% do novo
material de vidro já era vidro! Essa taxa de reciclagem pode ser aumentada até ~90%.
A fusão é feita em grandes panelas de barro de 400 a 800 kg de conteúdo, em um forno onde
cabem até 16 destas panelas ao mesmo tempo. Alternativa é num leito de capacidade de até
300 t, onde é fundido a 1000 °C e clarificado (= purificado; remoção das impurezas sólidas e
das bolhas de gases). Note que a clarificação requer temperaturas muito acima da faixa de
fusão, de 1450 a 1550 °C, onde a fundição é um líquido ralo.
A propriedade de aumentar em viscosidade aos poucos até a solidificação total, permite um
manuseio confortável do vidro fundido. As principais técnicas de transformação mecânica
são:
Dobrar tubos e barras,
Fechar ampolas a partir de tubos infinitos,
Soprar objetos ocos através de um tubo comprido ("zarabatana") e ar comprimido, seja
dos pulmões do vidreiro ou de um compressor mecânico (vidro oco, tais como
garrafas, vasos - em geral, recipientes baratos);
Grandes partes de vidro fundido são calandradas (vidro de janela e espelho) ou
Prensadas em moldes (telha de vidro; no caso do tijolo de vidro as duas metades
moldadas são fechadas e seladas a quente, daí o resfriamento acarreta uma atmosfera
rarefeita no interior do tijolo que, afinal, promove as propriedades isolantes do
material em construção civil);
Despejar o vidro fundido em cima de uma lagoa de estanho fundido (vidro Float, do
inglês = boiar)
Muito usadas são técnicas combinadas, de sopro usando vácuo e moldagem (informe-
se sobre o processo de Ingle e Smith). Por essa técnica se produzem vidros baratos
usados no dia a dia, tais como pratos, copos de cerveja, garrafas e copos de conserva.
Hoje somente por fins artísticos: sopro com a zarabatana. Na época da fabricação de
vidro para as janelas das antigas igrejas esse era o único método de se fazer chapas de
vidro. Cortava-se o fundo da bolha soprada e se achatava o restante do vidro por
rotações rápidas da zarabatana. Por isso, as peças foram pequenas e redondas, sempre
mais grossas no centro.
Um tratamento posterior da superfície do artefato pode ser necessário: lixamento grosso com
areia de quartzo, lixar fino com lixa ou esmeril, polimento com pastas minerais. Na maioria
dos objetos visa-se uma superfície extremamente lisa e lustrosa e um interior claro e
transparente. Mas também o oposto pode ser desejado: uma aparência fosca pode ser
propiciada, ou na hora da produção do vidro ou em uma etapa posterior. No primeiro caso se
consegue um efeito em massa, no segundo um efeito na superfície. Os processos mais
aplicados são:
Suspensão de um material fino, de alto ponto de fusão (ver p. 36);
Clareamento insuficiente (isto é, a falta da etapa de altíssima temperatura na
produção); daí a inclusão de bolinhas de gás abaixam a transparência;

27
Desvitrificação demasiada (ver p. 32; cerâmicas vítreas);
Jateamento abrasivo com areia;
Por meios químicos, especialmente a caustificação da superfície por ácido fluorídrico,
HF (ver p. 29), mas também por NaOH em alta concentração.
7.2. Composição do vidro comum: quartzo, cal e soda
É de composição simples, pois contém além de quartzo apenas soda e cal. Sendo assim, sua
composição média é:
Na2O . CaO
. 6 SiO2.
Em massa, isto são: 12,9% de Na2O, 11,6% CaO e 75,5% SiO2. Esse vidro atende a maioria
das aplicações, sejam essas: janelas, copos, garrafas e espelhos baratos. Os vidros com
suficiente resistência química deveriam ter a seguinte fórmula:
s = 3 (n² + 1), (23)
onde s = número de mols de SiO2; n = relação molar de Na2O / CaO.
Interpretando isso num exemplo:
Aumentar a razão Na2O / CaO, deve ter a consequência de aumentar também a porcentagem
em quartzo. A composição do vidro referido acima, Na2O . CaO
. 6 SiO2, é então um caso
especial desta fórmula, onde n = 1. Nos mais diversos vidros técnicos a relação Na2O / CaO
pode variar entre n = 0,6 e 1,8. Como já vimos acima (p. 2), um vidro isento de cal é solúvel
na água. Também aqueles vidros comuns que são ricos em álcali e pobres em cal, são
vulneráveis frente à hidrólise. Parte do seu álcali é dissolvida da superfície do objeto de vidro,
especialmente quando estiver em contato com água a altas temperaturas. Daí forma-se uma
camada mais rica em SiO2 e CaO, por sua vez bem fechada e protetora do material interior do
objeto. Portanto, é aconselhado expor vidraria nova do laboratório, à água quente, melhor
ainda a vapores d´água em um autoclave, antes do seu primeiro uso. Ainda mais eficazes
neste sentido do que água pura são ácidos minerais diluídos (lembre-se: Na2O tem caráter
básico).
Vidro comum é feito a partir das mais baratas matérias-primas. Qualquer outro ingrediente do
que quartzo, soda e cal significará custo a mais. Dentro destes três (ver diagrama das fases
abaixo 15
), a soda cáustica é a mais cara.
15 Para a interpretação de um diagrama das fases ternário, estude o seguinte gráfico:

28
Fig. 12. Diagrama das fases ternário, de quartzo, soda e cal.
Então, tenta-se minimizar a porcentagem do Na2O no vidro, dando preferência a vidros ricos
em SiO2 e CaO.
A resistência química do vidro depende, em primeira linha, da razão Ca/Na; ela é maior em
vidros ricos em cálcio. Podemos afirmar que os vidros mais baratos também são os
quimicamente mais resistentes. Da Fig. 13, porém, vemos que essas duas vantagens se
compram com um processamento mais difícil, pois a tendência à cristalização aumenta numa
composição pobre em álcali. Tanto a temperatura líquidus, TL, como a velocidade da
cristalização, ambas aumentam, acarretando maiores custos no aquecimento e maiores perdas
devido às tensões internas formadas durante o resfriamento do objeto de vidro. Portanto, os
teores em SiO2 mais realizados são entre 70 e 75%, enquanto o teor em álcali raramente cai
abaixo de 15%.
Fig. 13. Diagrama das fases ternário, quartzo, soda e cal, destacando as fases minerais
identificadas após a têmpera prolongada.

29
Contudo, um emprego da quantidade mínima de álcali leva a composições, perto do vinco nas
linhas pontilhadas da Fig. 12, que assinalam as composições da mesma velocidade máxima de
cristalização. Conforme a Fig. 13, isto fica aproximadamente na beirada do campo de
existência da -volastonita. Em cima desta curva, procura-se então o teor máximo em CaO.
7.3. Ataque químico por ácidos e bases
Sais alcalinos e suas soluções geralmente são os atacantes número Um para a maioria dos
vidros. Eles dissolvem o SiO2 da rede e o transformam em silicatos alcalinos que, como já
elucidado no capítulo "Silicatos de álcali", são solúveis na água.
Uma solução de NaOH, nem mesmo diluída, jamais deve ser guardada em frascos de vidro
(balões, garrafas, tubos de ensaio etc.). Mais indicado seria um recipiente de polietileno.
Especialmente quando a finalidade da solução é o Laboratório de Analítica, pode-se contar
com uma falsificação da concentração de álcali e uma crescente presença de silicatos solutos.
Ambas as concentrações prejudicam exatidão e confiabilidade das medições. Ao deixar
pastilhas de NaOH somente um dia num vidro de relógio, caustificam a vidraria e a tornam
inútil.
Mas também certos ácidos têm potencial agressivo frente ao vidro. Ácido fosfórico,
concentrado e quente, ataca aos poucos a vidraria comum de laboratório. Mais corrosivo ainda
do que álcali concentrado é o ácido fluorídrico (que está sendo produzido in situ, ao acidificar
uma solução contendo um sal fluoreto, com ácidos fortes!). A ligação Si-F é especialmente
estável: com 595 kJ.mol
-1 é a segunda mais forte ligação covalente (somente perde para a
ligação B-F). Em comparação, a ligação simples de Si-O “somente” conta com 444 kJ. Por
isso o HF dissolve o silício em forma de SiF4 – que é um gás fumegante. O artefato de vidro é
rapida e irreversivelmente caustificado. Aproveita-se deste efeito ao aplicar gravuras e
produzir vidros foscos ("vidro leitoso").
30
Note que os demais ácidos, quer orgânicos ou minerais, não atacam notavelmente o vidro
comum.
8. Vidro de potassa e outros vidros técnicos A próxima tabela mostra as composições médias (valores dados em % mássicos), de
diferentes fabricantes e de vidros de diferentes destinos. Observa-se que todas as receitas
contêm mais ou menos Al2O3 na intenção de elevar a resistência química do produto.
Além disso, podemos ver que o óxido de cálcio pode ser reposto por MgO ou BaO. Tanto
MgO quanto BaO levam ao aumento em viscosidade do vidro a altas temperaturas. Daí se
abre uma janela térmica mais ampla para a sua usinabilidade, em comparação ao vidro de cal.
Além disso, o bário e o magnésio abaixam a tendência da cristalização.
Além do óxido de sódio aplica-se como “quebrador de rede” também K2O (potassa) - que é
somente pouco mais cara do que Na2O. A potassa usa-se para abaixar a dureza do vidro a ser
lapidado; também melhora o aspecto do artefato, devido ao maior brilho e menor dispersão 16
,
portanto é chamado de "vidro de coroa" ou "vidro de lustre" (crown glass). Também as partes
ópticas em aparelhos, óculos, telescópios etc., são feitos de vidro de coroa. Uma composição
típica de vidro de coroa é:
73% SiO2; 5% Na2O; 17% K2O; 3% CaO; 2% Al2O3.
A grande semelhança entre as composições apresentadas abaixo revela-se ao comparar os
teores em álcalis-terrosos total e álcalis total.
Nome comercial SiO2 Na2O K2O CaO MgO BaO Al2O3
Vidro de cabo 72 13 6,5 6,5 0,2 0,2 1,0
Vidro chato 72 14 1 8 4 - 1,3
Vidro de recipiente 72 15,5 0,4 8,5 2 - 1,4
Vidro de lâmpadas 72,5 16,3 - 6,5 3 - 1,3
Vidro óptico de lustre 72 10 7 11 - - -
Vidro de aparelhos 68 13 4 9 - - 5,5
Vidro de tela de TV 68 9 7 0,3 - 11 4
16 Dispersão se chama a dependência da refração, do comprimento da onda da luz. Sendo assim, a luz azul é mais
fortemente quebrada do que a luz vermelha. Uma alta dispersão geralmente é bastante desvantajosa, na maioria
dos aparelhos ópticos, pois dificulta o focamento preciso por lentes. O vidro de coroa, neste aspecto, é mais
vantajoso do que o vidro de flint (vidro comum de soda), onde o ponto de focamento fica mais borrado:

31
Vidro romano do 1° século 70 16,5 1 7 0,6 - 5
Já mais difícil de fundir do que os vidros de cal e soda, são os de barrilha, onde parte ou todo
sódio é reposto por potássio. Ao manter a razão de M2O / CaO, os vidros de barrilha são mais
vulneráveis contra o ataque da água. Portanto, recomenda-se modificar a composição para
maiores teores em quartzo, sendo satisfeita a fórmula:
s = 4 (n² + 1), (25)
onde s = mols de SiO2; n = relação molar de K2O / CaO.
Desta fórmula resulta uma composição média de K2O . CaO
. 8 SiO2. O tradicional vidro de
louça da Boémia é um exemplo clássico da sua aplicação. Neste, as lapidações aparecem
especialmente bonitas. Também no laboratório químico se dá preferência a tubos deste vidro
quando se exige alta resistência térmica e química, por exemplo, tubos de combustão usados
na análise elementar (C, N, H) de compostos orgânicos. Note que hoje se conhecem vidros
melhores para esta finalidade (vidro Supremax, ver abaixo). Finalidade óptica e decorativa
tem o vidro de lustre - um vidro de barrilha e cal. Outro vidro que contém tanto Na como K, é
o vidro de Turingua: com temperaturas de amolecimento entre 550 e 600 °C o artefato pode
ser usado numa temperatura de uso permanente até 400 °C.
A resistência do vidro contra água, ácidos, bases e variações térmicas, aumenta bastante ao
repor parte do quartzo por B2O3 e/ou Al2O3, daí o nome "vidros de bórax” e “vidro de barro".
O óxido de boro reduz drasticamente o coeficiente de expansão térmica do vidro (apenas 3 a 5 .10
-6 K
-1; compare com outros vidros de qualidade referidos na nota de rodapé 19), então o
torna adequado a ser aquecido por uma chama direta de um lado e resfriado por água fria no
outro lado. Além disso, aumenta a resistência à hidrólise e contra o ataque por ácidos. Durante
a produção do vidro o bórax facilita a fusão das matérias-primas, especialmente as misturas
ricas em Al2O3. O óxido de alumínio reduz o quebradiço do vidro (aumento em elasticidade) e
abaixa o perigo da desvitrificação (expressão ver p. 22).
Um famoso vidro desta classe é o Duran®
da Schott, cuja composição é:
74,5% SiO2; 8,5% Al2O3; 4,6% B2O3; 7,7% Na2O; 3,9% BaO; 0,8% CaO e 0,1% MgO.
Seu amolecimento ocorre entre 600 e 700 °C. Foi originalmente feito para o uso no
laboratório químico, mas hoje é muito disseminado nas cozinhas, também. Composições e
qualidades comparáveis têm os vidros de Jena, as marcas Pyrex®
, Silex®
, Resista®
e Durax®
.
Importante para o químico ainda é o vidro Supremax, com temperatura de amolecimento
acima de 1000 °C:
56,4% SiO2; 20,1% Al2O3; 8,9% B2O3; 8,7% MgO; 4,8% CaO; 0,6% K2O; 0,6% Na2O.
Ao repor num vidro de barrilha e cal, parte ou toda cal por óxido de chumbo, obtém-se um
produto de fácil fusão, chamado de "vidro de cristal de chumbo” 17
. Este ganhou o nome
devido ao alto brilho nas suas superfícies planas. Isto se deve ao alto índice de refração e alta
densidade (3,5 a 4,8 g.cm
-3; compare: quartzo tem uma densidade de 2,65 g
.cm
-3). É usado
para objetos de uso e luxo. Um vidro rico em K e Pb é usado em lentes e prismas.
Especialmente rico em PbO 18
e com algum B2O3 se apresenta o "strass". Seu índice de
refração é semelhante ao do diamante, portanto é usado na bijuteria, na imitação de pedras
nobres.
17
Atenção: esta denominação popular é uma aberração - já que sabemos que um vidro é o oposto de um cristal!
18 Curiosidade: misturas com até 80% de PbO podem ser solidificados em forma de vidro! Os vidros de chumbo mais produzidos, porém, contêm entre 24 e 30% m/m de PbO.

32
8.1. Vidros especiais.
Como visto acima, os vidros podem ser adaptados à sua finalidade, escolhendo ingredientes
diferentes (tais como ZnO, Sb2O3, P2O5, etc.) e estes numa faixa relativamente larga, que
substituem em parte os três ingredientes clássicos, sílica, cal e soda. Junto ao fato de que um
vidro não dispõe de uma estequiometria definida, faz com que existam inúmeras receitas de
vidros especiais. Aqui somente alguns poucos exemplos mais marcantes.
Vidro "Uviol": deixa passar, não só a luz visível, mas também raios ultravioletas até 253 nm.
Contém quantidades notáveis de fosfato de bário e óxido de cromo.
Vidros de proteção em usinas nucleares: para bloquear (na verdade: absorver) nêutrons lentos,
esses vidros contém além de borossilicatos, também óxido de cádmio e diversos fluoretos.
Efetivo na absorção de raios γ se mostrou um vidro contendo fosfato de tungstênio, WPO4.
Vidro temperado
O vidro temperado aumenta a segurança do manuseio do objeto: sua superfície é mais
resistente a ranhuras, tem elevada resistência mecânica contra batidas pontiagudas, e quando
realmente sofre estrago, o vidro temperado quebra em inúmeros pequenos cacos – em vez de
trincar irregularmente e deixar grandes pedaços com cantos afiados e perigosos, como é a
característica do vidro comum.
Hoje é aplicado em objetos de vidro comum - geralmente vidros de chapa - onde a quebra
pode afetar seriamente o usuário: portas de boxe de chuveiro, pára-brisas, portas de vidro em
edifícios públicos, etc. Um processamento em duas etapas:
1) Têmpera em aproximadamente 625 °C, num forno que tem tipicamente 80 m de
comprimento. Nesta temperatura não há cristalização notável; ela deve ser aplicada de
maneira mais uniforme possível.
2) Saindo do forno da têmpera o objeto é resfriado rapidamente por meio de ar soprado nos
dois lados da chapa. Esse último detalhe é de suma importância, se não o efeito oposto é
garantido (isto é, elevado quebradiço no lado que não foi resfriado).
Para entender isso, devemos levar em consideração a dilatação térmica do vidro comum: no
forno ele se dilata 19
uniformemente. Ao ser resfriado pelo jato de ar a camada externa do
objeto sofre um aumento drástico em viscosidade, o que impede a volta dos átomos na sua
posição conforme a temperatura. O resultado é uma camada externa que fica
permanentemente estendida. No interior do objeto, no entanto, a velocidade do resfriamento é
naturalmente muito mais baixa, devido à baixa condutividade térmica do vidro. Lá, o
resfriamento é mais suave, o material tem tempo suficiente para se recolher, de acordo com a
temperatura. No final do processo permanece uma tensão no objeto, no sentido que a camada
externa sempre fica comprimida pelo miolo do objeto. Isso faz com que a durabilidade do
objeto aumente, já que somente a superfície é sujeita ao desgaste mecânico.
19 O coeficiente de dilatação linear do vidro comum fica em torno de 9.10-6 K-1. Em comparação: metais comuns
entre 15 e 25.10-6 K-1; concreto 6,8.10-6 K-1; vidro Pyrex® 3,2.10-6 K-1; recordista entre os materiais de uso técnico
é o quartzo vitrificado, com apenas 0,6.10-6 K-1.
Notamos apenas como curiosidade física: existem pouquíssimos materiais cujo coeficiente de dilatação térmica
fica negativo (NTE = negative thermal expansion; por exemplo, observado no ZrW2O8).

33
O que acontece se o resfriamento rápido somente for aplicado de um lado, se vê no
experimento clássico da “Garrafa da Bolonha”: sua camada externa (= resfriado) se torna tão
resistente que até um prego pode ser martelado na madeira. Mas quando deixar cair o prego
dentro da garrafa ela quebra em inúmeros pequenos pedaços.
Desvantagem do vidro temperado: devido às tensões permanentes é impossível qualquer
tratamento mecânico após a têmpera. Recorte, lixamento, gravura, furos, etc. têm que ser
aplicados antes deste procedimento.
Distinguimos o vidro temperado, das cerâmicas vítreas. Embora os dois processos têm como
base a têmpera controlada, somente o último leva à cristalização notável do material.
Cerâmicas de vidro (também vitrocerâmica ou cerâmica vítrea):
Desvitrificação (expressão ver p. 22), por sua vez considerada sendo defeito na produção de
vidro comum, aqui está feita com propósito. Em geral, essas cerâmicas são feitas a partir dos
vidros da respectiva composição, submetendo-os à têmpera controlada. Nestas condições
ocorre a formação de muitos, pequenos cristais, a custo da fase amorfa.
Lembramos que o vidro representa um estado metaestável, então sua transformação em cristal
é um processo espontâneo ( G < 0). No capítulo "Vidros contra cristais" vimos que logo
abaixo da temperatura sólidus, TL, temos a condição termodinâmica para a formação
permanente de cristais (curva R na Fig. 7). Mas também vimos que nessa temperatura não
necessariamente é adequada para induzir a cristalização a partir de um material
completamente amorfo; lembramos que o intervalo de Ostwald-Miers dificulta o processo da
cristalização - até mesmo é responsável pela existência do estado vítreo. Para provocarmos
uma recristalização controlada, temos que procurar as condições certas para superar os
impedimentos cinéticos da nucleação (curva S na Fig. 7), sem entrar na região do crescimento
rápido dos embriões.
A nucleação somente ocorre em uma estreita faixa térmica (curva S na Fig. 7), isto é, existe
uma temperatura limite inferior e superior, fora das quais não ocorre uma notável formação de
cristalitos.
O limite inferior se deve à alta viscosidade do vidro, quando estiver a baixas
temperaturas; isso dificulta a difusão, um processo essencial para o crescimento de um
cristal. Portanto, existe um limite térmico abaixo do qual não prossegue a cristalização
em tempos hábeis.
O limite superior se deve à barreira energética frente à criação de uma nova interface – o
que é um processo altamente endotérmico. Essa barreira deve ser superada. No início da
cristalização, mais especificamente no processo da nucleação homogênea, o gasto em
energia devido à nova interface, supera de longe o ganho em energia devido à criação de
novo volume cristalino (Fig. 5). Em outras palavras: o início da vida de um cristal é
mais difícil do que o seu crescimento. Isto implica que a nucleação homogênea dos
cristais a partir de um líquido ou vidro sempre requer um ambiente de baixa energia, isto
é, uma situação de super-resfriamento. Portanto, existe uma temperatura limite superior
acima da qual não se formam embriões de cristais.
Essas duas considerações deixam claro que existe uma faixa térmica estreita onde se
prossegue a desvitrificação controlada, reaquecendo um material que foi 100% vitrificado por
resfriamento rápido. A composição química dos cristais se dá no diagrama das fases (Fig. 13).
Pelo menos na teoria, porque em nenhum caso real temos um sistema composto por
rigorosamente três componentes, nem a ausência completa de impurezas sólidas. Mesmo se
for a partir das três matérias-primas supracitadas, sempre acontece que o material fundido se

34
junte ao material do recipiente de cozimento; lembre-se que o revestimento interno do
recipiente da fábrica é feita de refratário, de composição semelhante à do próprio vidro.
Portanto, podemos levar em consideração também cristais de composição estranha, que não
constam dos diagramas ternários. No caso das cerâmicas vítreas devem-se evitar tais
impurezas, se não se corre perigo de perder controle sobre a cristalização. É evidente que a
elevada exigência à pureza também eleva o custo deste material, ao comparar com o vidro
comum.
Sob essa promessa e através de um controle rigoroso da temperatura, conseguimos controlar o
processo da desvitrificação tão bem que podem ser produzidas cerâmicas com diferentes
propriedades e para aplicações especificadas. Em dependência do programa térmico em que o
objeto de vidro for submetido:
Ao manter um vidro por longo tempo, no meio da região do superresfriamento de
Ostwald-Miers, formam-se poucos, mas grandes cristais. Estes geralmente são
perfeitamente esféricos, portanto chamados de "esferulitos", ver Fig. 14.
Ao manter a temperatura de um vidro na proximidade da velocidade máxima de
germinação (note que naquela região o crescimento dos cristais geralmente não é
particularmente rápido), formam-se muitos e pequenos cristais, a dizer micro-cristais
em alta concentração, enluvados no vidro contínuo. O material tratado assim não
perde muito da sua transparência óptica 20
; mas sua resistência mecânica aumenta
bastante.
Caso o vidro é mantido numa primeira etapa perto da velocidade máxima de
germinação e em seguida na temperatura (mais alta) da velocidade máxima de
crescimento dos cristais, então formam-se muitos cristais grandes e de tamanhos mais
ou menos iguais. É ainda possível variar os tempos de demora nestas duas
temperaturas características - uma largamente independente da outra. Desta maneira se
consegue o maior grau em cristalinidade. Esse procedimento fornece uma cerâmica
extremamente resistente.
(1) (2) (3)
20 Atenção: vidro opaco pode ser produzido por este processo, também. Basta submeter o vidro por mais tempo
nesta temperatura especificada. Há mais duas maneiras de se obter um material opaco, das quais uma já foi
mencionada na lista da p. 25: a participação de pequenas partículas sólidas de índice de refração diferente ao da
matriz. Mas também existem condições termodinâmicas onde pode ocorrer uma separação líquido-líquido. Daí
se fala em "lacuna de miscibilidade". A fase de maior volume forma a matriz, a de menor volume a fase discreta
(= gotículas finamente dispersas). Mesmo se cada uma destas fases permaneça amorfa, o produto sairá opaco,
devido aos diferentes índices de refração.

35
Fig. 14. (1) Estágios da formação de um esferulito; (2) Modelo de esferulito em “vidro
orgânico” (resinas acrílicas, PMMA, PVC, PS, POM, etc.); (3) Situação real da cristalização
em esferuitos (micrografia de TEM).
Após qualquer um destes métodos o resfriamento da peça ocorre de maneira mais suave
possível, em fornos de resfriamento constante e lento. Sendo assim, a peça leva as mínimas
tensões internas residuais – um fato que certamente contribui positivamente à sua estabilidade
mecânica.
As cerâmicas vítreas têm seu lugar intocável na cozinha, onde os fogões elétricos mais
avançados são recobertos por chapas deste material. Misturas típicas são:
Codierita, composição aproximada: 2 MgO . 2 Al2O3
. 5 SiO2,
-espodumena, composição aproximada: Li2O . Al2O3
. 4 SiO2 ,
-eucriptita, composição aproximada: Li2O . Al2O3
. 2 SiO2.
Destacam-se por altíssima resistência à tração e torção (batidas mecânicas) e, lógico, por
imensa resistência contra variações em temperatura.
As cerâmicas vítreas diferenciam-se das cerâmicas de barro, principalmente por serem mais
densas, impedindo a penetração de gases. Outras aplicações destas cerâmicas são: espelhos
astronômicos, revestimento de pontas de projéteis bélicos, bolas de moinhos, esmalte em
panelas de cozinha, etc.
8.2. Coloração (artística) de vidros
Existem duas estratégias de produzir vidro colorido:
por óxidos de certos metais,
por metais pesados finamente dispersos.
No primeiro caso trata-se de soluções verdadeiras, no segundo de dispersões coloidais.
Fig. 15. Vidros coloridos, expostos no comércio.

36
Colorações por óxidos:
Violeta por NiO
Violeta-azulado por Mn2O3
Azul por CoO
Verde-azulado por FeO
Verde por Cr2O3 e CuO
Marrom por Fe2O3 e MnO2; bastante usado também a coloração com pirita, FeS2 ou
uma mistura de FeSO4 e carvão.
Amarelo por Ag2O
Cor laranja por UO3
Vermelho por Cu2O.
Desta lista pode-se ver que, além do número de coordenação, o NOX do metal influencia
drasticamente a coloração do metal complexado. Neste contexto é importante lembrar-se que
o comportamento das cores nem sempre é aditivo! Isto se deve às mudanças no número de
coordenação e da formação de complexos de transferência de carga entre os metais ("charge-
transfer"). Também podem ocorrer, nestas misturas de sais, trocas de ligantes entre os
complexos que acarretam uma falsificação das cores puras.
Desde a idade média existe um vasto conhecimento empírico na área da coloração de vidro.
Além dos citados acima, os íons (+3) dos metais das terras raras provocam colorações que são
apreciadas pelos artistas. A quantidade do aditivo pode variar, na ordem de poucos gramas até
uns Kg, a cada 100 Kg de vidro.
Muitas vezes pequenas quantidades de certos óxidos podem ser adicionadas, para compensar
colorações indesejadas. Com este objetivo deve-se escolher o elemento que provoca a cor
complementar no vidro. Por exemplo, consegue-se compensar a coloração verde do vidro
comum, quase sempre presente devido a impurezas originadas por Fe (II), por adição de
dióxido de manganês (pirolusita, também chamado de "sabão do vidreiro").
Enquanto a coloração por óxidos aparece desde o material fundido, o efeito por metais
coloidais requer um tratamento térmico diferente. O artefato tem que ser reaquecido por meia
hora, a uma temperatura de 450 a 500 °C, somente daí o objeto até então quase incolor, torna-
se fortemente colorido. Mais famoso é o "vidro de rubi de ouro", um vermelho intenso
provocado por ouro coloidal. Parecido a este é o vidro de rubi de cobre; a prata coloidal
provoca tonalidades amarelas, o selênio uma cor de rosa.
Essas técnicas foram aperfeiçoadas, já na época da gótica, como pode-se ver nas janelas
magníficas das igrejas ocidentais.
8.3. Opacidade, nebulosidade – atributos dos esmaltes.
Para algumas finalidades precisa-se de vidro opaco (“vidro de alabastro”). Em geral, são
partículas finas, com índice de refração diferente em relação à matriz de vidro, que podem ser
embutidos na matriz vitrificada. Servem para este fim o fosfato de cálcio, Ca3(PO4)2, óxido de
estanho, SnO2 e criolita, Na3AlF6. Um vidro opaco famoso é o esmalte, com fim de proteção
ou decoração de metais. Um objeto de ferro é mergulhado em uma suspensão grossa de vidro
pulverizado (composição básica: álcali, ácido bórico e argila). Depois da secagem a camada
de pó é vitrificada em uma mufla de esmalte. Resulta uma camada lisa, dura e brilhante.
Agentes de nebulosidade aqui: TiO2 ou ZrO2.

37
9. Literatura Monografias:
Nölle, Günther. Technik der Glasherstellung. Wiley-VCH, Stuttgart 1997
Schmelzer, J (Ed.), Fokin, Yuritsyn, Zanotto. Nucleation Theory and Applications. Nucleation
and Crystallization Kinetics in Silicate Glasses: Theory and Experiment. Wiley-VCH,
Stuttgart 2005
Holleman-Wiberg, Lehrbuch der anorganischen Chemie. 91a - 100
a edição, Walter de Gruyter,
Berlin 1985.
Publicações relacionadas ao assunto:
A. Isenmann "Construção de um reator de microondas e produção de vidro colorido", roteiro
de aula prática (6 páginas).
A. Isenmann "Alvenaria e Vidro - os Pilares da Civilização Moderna", painel didático (largura
2,05 m, altura 1,05 m).
Na internet:
http://www.usp.br/fau/deptecnologia/docs/bancovidros/vidro.htm (descrição detalhada dos
processos de vidros industriais; acesso em 12/2014)
http://www.joinville.udesc.br/portal/professores/carmeane/materiais/Aula_10___Vidro_carme
ane.pdf (apresentação powerpoint dos vidros usados na construção civil)
10. Anexo Fig. 16. Princípios de construção dos ácidos polissilícicos, formando cadeias, faixas ou
camadas.
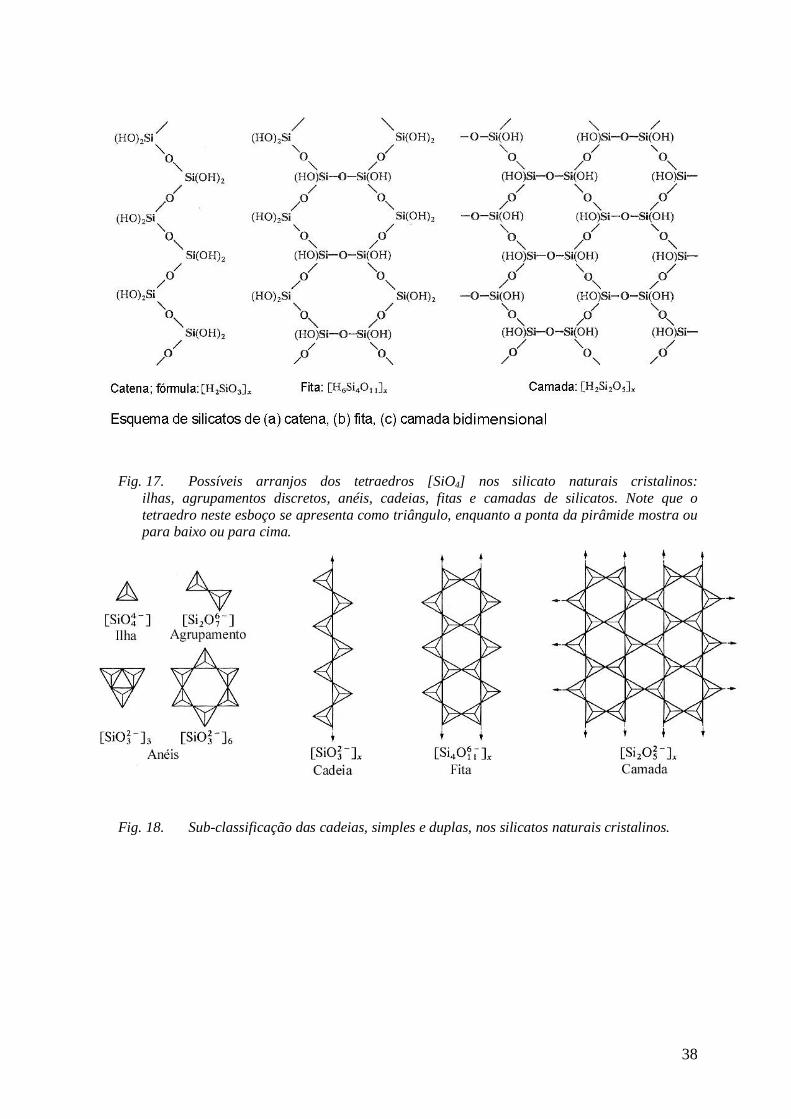
38
Fig. 17. Possíveis arranjos dos tetraedros [SiO4] nos silicato naturais cristalinos:
ilhas, agrupamentos discretos, anéis, cadeias, fitas e camadas de silicatos. Note que o
tetraedro neste esboço se apresenta como triângulo, enquanto a ponta da pirâmide mostra ou para baixo ou para cima.
Fig. 18. Sub-classificação das cadeias, simples e duplas, nos silicatos naturais cristalinos.
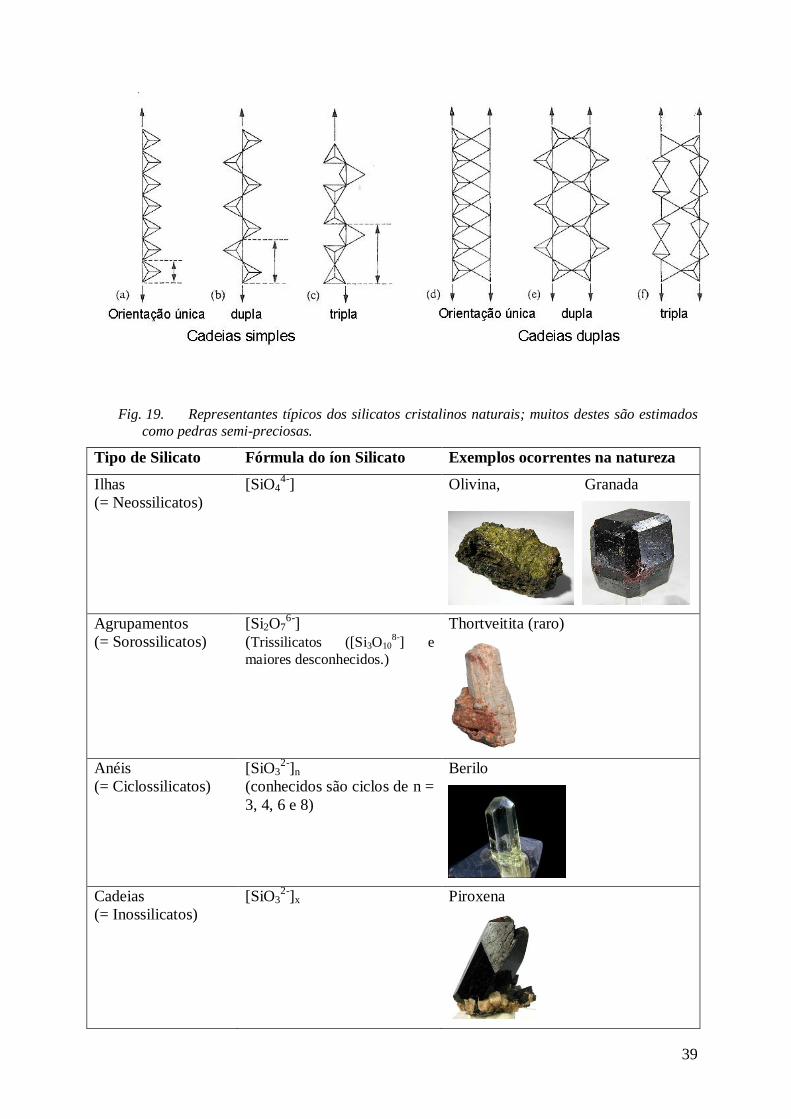
39
Fig. 19. Representantes típicos dos silicatos cristalinos naturais; muitos destes são estimados
como pedras semi-preciosas.
Tipo de Silicato Fórmula do íon Silicato Exemplos ocorrentes na natureza
Ilhas
(= Neossilicatos)
[SiO44-
]
Olivina, Granada
Agrupamentos
(= Sorossilicatos)
[Si2O76-
]
(Trissilicatos ([Si3O108-
] e
maiores desconhecidos.)
Thortveitita (raro)
Anéis
(= Ciclossilicatos)
[SiO32-
]n
(conhecidos são ciclos de n =
3, 4, 6 e 8)
Berilo
Cadeias
(= Inossilicatos)
[SiO32-
]x
Piroxena

40
Fita (= Inossilicatos) [Si4O116-
]x
(entre outros)
Anfíbola
Camada
(= Filossilicatos)
[Si2O52-
]x
(muitos representantes)
Asbesto (= Amianto), Caolinita,
Talco, Pirofilita, Mica.
Esqueleto
tridimensional
(= Tectossilicatos)
[AlySi1-yO2y-
]x Feldspato, Zeólitos
Fig. 20. Estrutura da serpentina, Mg3(OH)4[Si2O5], e da caolinita, Al2(OH)4[Si2O5]:
(a) Projeção do plano a-b (por fins de maior clareza a camada de OH que fica abaixo da
camada dos cátions Mg (Al) não é representada); (b) Projeção do plano b-c (se refere aos grupos destacados no desenho (a) em negrito).
Típicas nestas estruturas bidimensionais são as camadas de silicato (ver última estrutura na
Fig. 17, enquanto a ponta de cada tetraedro mostra na mesma direção).
Fig. 21. Silicatos em camadas (filossilicatos): Reações químicas idealizadas que levam às estruturas bidimensionais da pirofilita e da mica.
Projeções do plano b-c de:
(a) talco, Mg3(OH)2[Si2O5]2 e da pirofilita, Al2(OH)4[Si2O5]2 ; (b) esquema da mica (parte brilhante dentro do granito).
Os cátions alcalinos têm o papel de espaçadores entre as camadas destes alumossilicatos. A
forte anisotropia nestas estruturas se percebe em todos os aspectos físicos: aspecto visual, resistência mecânica, condutividade térmica, etc. A “moleza” do talco e sua aplicação como
lubrificante se deve ao fácil deslocamento entre as camadas, enquanto cada camada por si é
bastante rígida e estável.
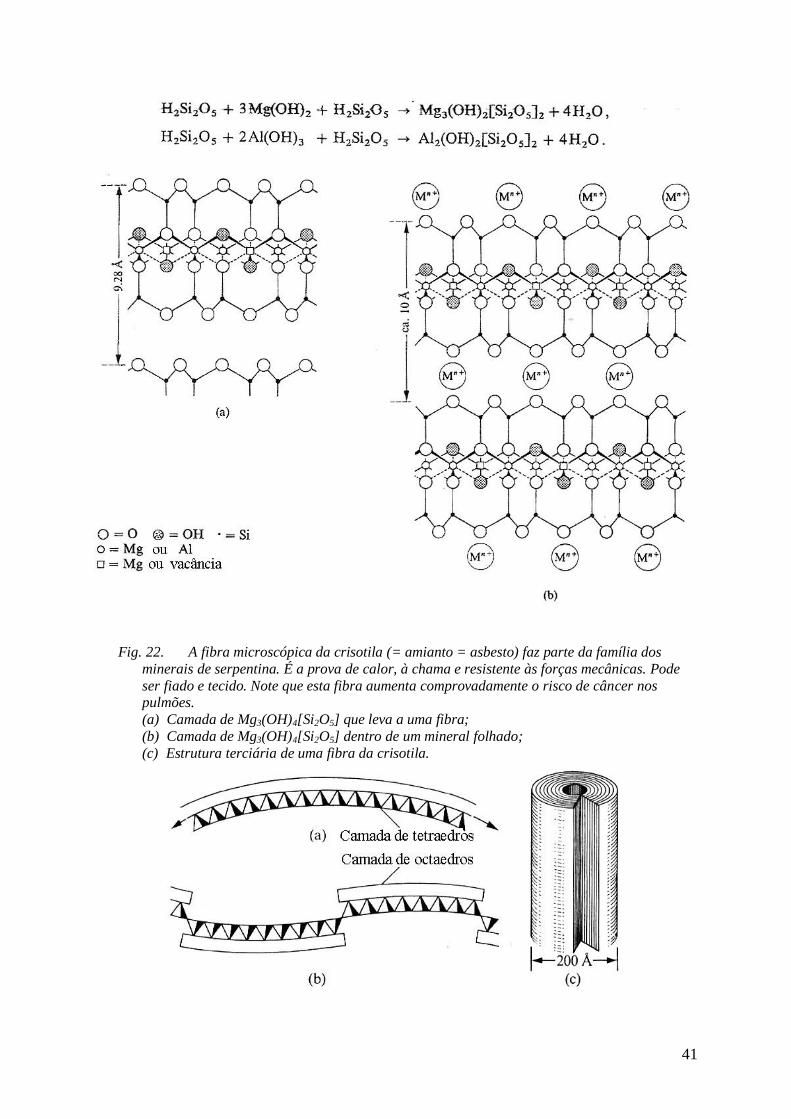
41
Fig. 22. A fibra microscópica da crisotila (= amianto = asbesto) faz parte da família dos
minerais de serpentina. É a prova de calor, à chama e resistente às forças mecânicas. Pode
ser fiado e tecido. Note que esta fibra aumenta comprovadamente o risco de câncer nos pulmões.
(a) Camada de Mg3(OH)4[Si2O5] que leva a uma fibra;
(b) Camada de Mg3(OH)4[Si2O5] dentro de um mineral folhado;
(c) Estrutura terciária de uma fibra da crisotila.

42
Fig. 23. Esquema da condensação (estágio inicial):
ácido monossilícico dissilícico tetrassilícico polissilícico
Fig. 24. Esquema da condensação (estágio intermediário e final): ocorre a transformação do
sol em gel. Cada bolinha representa um isopoli-ânion (última estrutura na Fig. 23).
Fig. 25. Estruturas de alguns zeólito, usado no laboratório como peneiras moleculares. Fonte: A.J.S. Mascarenhas, E.C. Oliveira, H.O. Pastore, Peneiras Moleculares: Selecionando as
Moléculas por seu Tamanho. Disponível em:
http://qnesc.sbq.org.br/online/cadernos/02/peneiras.pdf (acesso em 11/2014)









![Difusão aula MCM A.ppt [Modo de Compatibilidade] · – Resfriamento rápido dos aços produzindo estruturas de não equilíbrio – Formação de metais amorfos (“vidro metálico”)](https://img.document.onl/doc/110x75/5c44131c93f3c34c416c93a0/difusao-aula-mcm-appt-modo-de-compatibilidade-resfriamento-rapido-dos.jpg)



















