Embed Size (px)
Citation preview

Escola Politécnica da Universidade de São Paulo
DANILO HENRIQUE STAVRO DUARTE
Síntese e caracterização físico-química de nanopartículas proteína-
DNA-metal visando à aplicação em estudos de entrega gênica
São Paulo
2012

2

3
DANILO HENRIQUE STAVRO DUARTE
Síntese e caracterização físico-química de nanopartículas proteína-
DNA-metal visando à aplicação em estudos de entrega gênica
Trabalho de conclusão do curso de Engenharia
Química da Escola Politécnica da Universidade de
São Paulo
Orientadores: Prof. Dr. Adriano Rodrigues Azzoni
Prof. Dr. Marcelo Seckler
São Paulo
2012

4
DEDICATÓRIA
Dedico este trabalho ao meu avô Constantino (em memória).

5
AGRADECIMENTOS
Aos professores orientadores, pela paciência, dedicação e prontidão no
auxílio à execução deste trabalho.
À amiga Renata Lippi pela grande contribuição na primeira etapa deste TCC.
Ao Roberto Ângelo, mestrando em síntese de nanopartículas de prata, pelo
auxílio com o tema de sua especialidade.
Ao Rafael Alves, aluno de mestrado em bioquímica, pelo acompanhamento e
instrução.
À Juliana Grijo, aluna de graduação, pelo auxílio nos experimentos realizados.
Ao Marcelo Szymanski, doutorando em bioquímica na Unicamp, pela ajuda
com a transfecção das células.
Aos amigos e família pelo apoio moral, por estarem sempre prontos a me
ajudar e pelo carinho. Especialmente minha mãe, Cláudia, minha namorada,
Gabriele e minha avó Denilde.

6
RESUMO
A terapia gênica tem sido apontada como uma alternativa promissora para o
tratamento de diversas doenças. Essa técnica baseia-se na inserção de genes em
células de indivíduos, carregados por meio de diferentes vetores (virais e não virais).
Os vetores não virais são complexos formados pela associação de material genético
(pDNA) com proteínas, lipídeos, polímeros, nanopartículas metálicas, etc. Apesar de
menos eficientes, tem a vantagem de serem de simples manipulação, podendo ser
sintetizados de acordo com a finalidade do uso. Como característica fundamental,
estes complexos procuram mimetizar as estratégias utilizadas pelos vírus para
superar as barreiras físicas, enzimáticas e difusionais que limitam a entrega gênica.
Contudo, a falta de estudos sistemáticos sobre a estabilidade e formação destes
complexos dificulta seu emprego e a reprodutibilidade de resultados. Este trabalho
tem como objetivo o estudo sistemático da formação de complexos pDNA-proteína-
nano partículas metálicas, utilizando-se um pDNA modelo (com gene repórter), uma
proteína modelo (Protamina) e nano partículas sintetizadas a partir de sais de prata
(Lee, et al., 1982). A cinética de formação, a estabilidade e parâmetros físico-
químicos, como tamanho e a carga dos complexos, foram analisados, através de
experimentos como o DLS, eletroforese e potencial Zeta, e relacionados com a
entrada das partículas nas células. Como resultados, a eletroforese em gel de
agarose confirma a formação do complexo. O potencial Zeta, ainda que preliminar,
indicou que as partículas são carregadas positivamente. Os ensaios de microscopia
eletrônica de varredura não foram conclusivos, por causa de possíveis
sobreposições de partículas. O DLS nos forneceu um tamanho de complexo maior
para as razões mássicas maiores de AgNPs, mas não está condizente com a
transfecção, que resultou numa eficiência igual para todas as amostras com prata.
Podemos afirmar com certeza que as nanopartículas de prata influenciam na
entrega gênica, pois os resultados de transfecção para essas amostras são maiores
que as restantes. É difícil, por enquanto, dizer o mecanismo ou o por quê isso
acontece, mas é efetivo.
Palavras-chave: entrega gênica, nanopartículas de prata, complexo
pDNA:Protamina:Metal.

7
ABSTRACT
The gene therapy has been pointed as a promising alternative with respect to
the treatment of diverse illnesses. This technique is based on the insertion of genes
in cells of individuals, carried by different vectors (viral and non viral). The non viral
vectors are complexes formed by the association of genetic material (pDNA) with
proteins, lipids, polymers, metallic nanoparticles, etc. Although less efficient, it has
the advantage to be of simple manipulation, being able to be synthesized in
accordance with the purpose of the use. As characteristic basic, these complexes try
to copy the strategies used for the viruses to surpass the physical, enzymatic and
diffusional barriers that limit the gene delivery. However, the lack of systematic
studies on the stability and formation of these complexes make it difficult its job and
the reproducibility of results. This work has as objective the systematic study of the
formation of complexes pDNA-protein-metallic nanoparticles, using one model pDNA
(with a reporter gene), a model protein (Protamina) and nanoparticles synthecized
from silver salts (Lee, et al., 1982). The formation kinetic, the stability and the
physicist-chemistries parameters, as the complexes’ size and load, had been
analyzed, through experiments as the DLS, electrophoresis and Zeta potential, and
related with the entrance of particles into the cells. As results, we have that
electrophoresis is only a data that confirms the formation of the complex. The Zeta
potential make us conclude that the complex is positive. The scanning electron
microscopy is not conclusive, because of the overlapping particles. The DLS has
provided us a bigger size of complex for the bigger mass reasons of AgNPs, but he is
not consistent with the transfection, that resulted in an equal efficiency for all the
samples with silver. We can affirm with certainty that silver nanoparticles influences
in the gene delivery, therefore the results of transfection for these samples are better
that the remains. It is difficult to say the mechanism or the reason it happens, but it is
effective.
Key words: gene delivery, silver nanoparticles, pDNA:Protemine:Metal complex.

8
LISTA DE FIGURAS
Figura 1 - Tipos de vetores de terapia gênica usados em testes clínicos (Park, et al.,
2011), maioria vetores virais. .................................................................................... 16
Figura 2 - Principais barreiras à expressão gênica durante o tráfego de vetores de
pDNA: (1) degradação por nucleases extracelulares; (2) entrada na célula; (3)
endocitose; (4) degradação no endossoma; (5) escape do endossoma; (6)
degradação por nucleases citosólicas; (7) entrada no núcleo; (8) transcrição; (9)
exportação do mRNA; (10) tradução. ........................................................................ 16
Figura 3 - Caminhos do vetor de entrega gênica. (a) Polinfecção e (b) Lipofecção. . 18
Figura 4 - Mecanismo de transfecção, suas dificuldades e caminhos (Cartier, et al.,
2002) ......................................................................................................................... 20
Figura 5 - Entrada de um vetor viral na célula alvo ................................................... 22
Figura 6 - a) Partículas carregadas se repelem umas das outras, enquanto b)
partículas sem carga ficam livres para colidirem e agregarem.................................. 31
Figura 7 - Ilustração das camadas de Stern e da camada Difusa. ............................ 32
Figura 8 - Potencial Zeta e potencial de superfície. .................................................. 33
Figura 9 - Amostra a ser analisada ........................................................................... 35
Figura 10 - Demonstração da dependência do espalhamento em relação à fase da
onda. ......................................................................................................................... 35
Figura 11 - Diferença da variação da intensidade com o tempo para partículas
grandes e pequenas. ................................................................................................. 36
Figura 12 - Intensidade de sinal no tempo. ............................................................... 36
Figura 13 - Coeficiente de correlação em função do tempo. ..................................... 37
Figura 14 - Dependência do volume e da intensidade com o tamanho das partículas
para duas populações com igual quantidade. ........................................................... 38
Figura 15 - Equipamento de eletroforese em gel. ..................................................... 38
Figura 16 - Gel de agarose após electroforese de fragmentos de DNA. ................... 40
Figura 17 - Amostra de nanopartículas de prata com 15 minutos de reação ............ 42
Figura 18 - Curva espectrofotométrica das nanopartículas de prata ......................... 43
Figura 19 - Amostra de nanopartículas de prata centrifugadas ................................. 44
Figura 20 - MEV das nanopartículas de prata a pH = 9 e 90°C ................................ 47
Figura 21 - Eletroforese em agarose de partículas pDNA e protamina ..................... 51

9
Figura 22 - Amostra de complexo pDNA-Protamina-Metal para ensaio de
eletroforese ............................................................................................................... 53
Figura 23 - Eletroforese em gel dos complexos ternários ......................................... 54
Figura 24 - MEV para razão mássica 1:5:0,5 ............................................................ 57
Figura 25 – Microscopia eletrônica de varredura de amostra diluída e razão mássica
1:5:0,5 ....................................................................................................................... 59
Figura 26 - Resultados de transfecção celular, nas diferentes razões mássicas de
pDNA:protamina:NpAg estudas, utilizando-se células HeLa cultivadas in vitro. ....... 62
Figura 27 - Laudo 1 do Zeta para nanopartículas em pH = 7,4 ................................. 69
Figura 28 - Laudo 2 do Zeta para nanopartículas em pH = 7,4 ................................. 69
Figura 29 - Laudo 3 do Zeta para nanopartículas em pH = 7,4 ................................. 70
Figura 30 - Laudo 1 do Zeta para nanopartículas em pH = 9 .................................... 70
Figura 31 - Laudo 2 do Zeta para nanopartículas em pH = 9 .................................... 71
Figura 32 - Laudo 3 do Zeta para nanopartículas em pH = 9 .................................... 71
Figura 33 - Laudo 1 do Zeta para complexos ternários razão 1:5:0,5 ....................... 72
Figura 34 - Laudo 2 do Zeta para complexos ternários razão 1:5:0,5 ....................... 72
Figura 35 - Laudo 3 do Zeta para complexos ternários razão 1:5:0,5 ....................... 73
Figura 36 - Laudo 1 do Zeta para complexos ternários razão 1:5:2 .......................... 73
Figura 37 - Laudo 2 do Zeta para complexos ternários razão 1:5:2 .......................... 74
Figura 38 - Laudo 3 do Zeta para complexos ternários razão 1:5:2 .......................... 74
Figura 39 - Laudo 1 do Zeta para complexos ternários razão 1:5:4 .......................... 75
Figura 40 - Laudo 2 do Zeta para complexos ternários razão 1:5:4 .......................... 75
Figura 41 - Laudo 3 do Zeta para complexos ternários razão 1:5:4 .......................... 76
Figura 42 - Laudo 1 do DLS para complexos ternários razão 1:5:0,5 ....................... 77
Figura 43 - Laudo 2 do DLS para complexos ternários razão 1:5:0,5 ....................... 77
Figura 44 - Laudo 3 do DLS para complexos ternários razão 1:5:0,5 ....................... 78
Figura 45 - Laudo 1 do DLS para complexos ternários razão 1:5:2 .......................... 78
Figura 46 - Laudo 2 do DLS para complexos ternários razão 1:5:2 .......................... 79
Figura 47 - Laudo 3 do DLS para complexos ternários razão 1:5:2 .......................... 79
Figura 48 - Laudo 1 do DLS para complexos ternários razão 1:5:4 .......................... 80
Figura 49 - Laudo 2 do DLS para complexos ternários razão 1:5:4 .......................... 80
Figura 50 - Laudo 3 do DLS para complexos ternários razão 1:5:4 .......................... 81
Figura 51 - MEV para razão mássica 1:5:0,5 aumentada ......................................... 82

10
Figura 52 - MEV para razão mássica 1:5:4 ............................................................... 83
Figura 53 - MEV para razão mássica 1:5:4 aumentada ............................................ 83
Figura 54 – Microscopia eletrônica de varredura de amostra diluída e razão mássica
1:5:0 .......................................................................................................................... 84
Figura 55 – Microscopia eletrônica de varredura de amostra diluída e razão mássica
1:5:0,5 com zoom ...................................................................................................... 84
Figura 56 – Microscopia eletrônica de varredura de amostra diluída e razão mássica
1:5:4 com zoom ......................................................................................................... 85
Figura 57 – Microscopia eletrônica de varredura de amostra diluída e razão mássica
1:5:4 .......................................................................................................................... 85
Figura 58 - EDS da amostra de razão mássica 1:5:0 ................................................ 86
Figura 59 - EDS do aglomerado da amostra de razão mássica 1:5:0 ....................... 86
Figura 60 - EDS 1 da amostra de razão mássica 1:5:0,5 .......................................... 87
Figura 61 - EDS 2 da amostra de razão mássica 1:5:0,5 .......................................... 87
Figura 62 - EDS 3 da amostra de razão mássica 1:5:0,5 .......................................... 88
Figura 63 - EDS 1 da amostra de razão mássica 1:5:4 ............................................. 88
Figura 64 - EDS 2 da amostra de razão mássica 1:5:4 ............................................. 89
Figura 65 - EDS 3 da amostra de razão mássica 1:5:4 ............................................. 89

11
LISTA DE TABELAS
Tabela 1 - Comparação dos métodos de transfecção. .............................................. 21
Tabela 2 - Comparação entre tipos de vetores de entrega gênica. ........................... 23
Tabela 3 - Resultado do ensaio de potencial Zeta para as nanopartículas de prata . 45
Tabela 4 - Amostras de ensaio de eletroforese ......................................................... 51
Tabela 5 - Volume das composições das amostras .................................................. 52
Tabela 6 - Valores do potencial Zeta para os complexos ternários ........................... 55
Tabela 7 - Resultados do ensaio DLS ....................................................................... 55
Tabela 8 - Ensaio de EDS das amostras de MEV com diluição ................................ 60

12
LISTA DE ABREVIATURA E SIGLAS
AgNP ou NPAg Nanopartículas de prata
pDNA DNA plasmidial
MEV Microscopia eletrônica de varredura
Rcf Força centrífuga relativa
PBS Tampão fosfato salino
TAE Tris-Acetato-EDTA
EDTA Ácido etilenodiamino tetra-acético
DLS Dynamic Light Scattering
EDS Espectroscopia de raios X por dispersão em energia

13
SUMÁRIO
2.1. DIFICULDADES DE ENTREGA GÊNICA .................................................... 18
2.2. VETORES VIRAIS ....................................................................................... 21
2.3. VETORES NÃO VIRAIS ............................................................................... 23
2.3.1. Proteína e pDNA .................................................................................... 24
2.3.2. Nano partículas como vetores ............................................................... 25
3.1. MATERIAIS UTILIZADOS ............................................................................ 27
3.1.1. Método para síntese das AgNPs ........................................................... 27
3.1.2. Método de síntese de partículas de pDNA-Proteína .............................. 27
3.1.3. Preparo de complexos pDNA-Proteína- AgNP ...................................... 28
3.1.4. Transfecção em células animais ............................................................ 29
3.2. CARACTERIZAÇÃO DOS VETORES ......................................................... 30
3.2.1. Potencial Zeta ........................................................................................ 30
3.2.2. Tamanho das partículas por DLS (Dynamic Light Scattering) ............... 33
3.2.3. Eletroforese em gel de agarose ............................................................. 38
4.1. CARACTERIZAÇÃO DAS NANOPARTÍCULAS DE PRATA ....................... 42
4.1.1. Síntese das nanopartículas ................................................................... 42
4.1.2. Potencial Zeta ........................................................................................ 44
4.1.3. Tamanho das partículas por DLS (Dynamic Light Scattering) ............... 45
4.1.4. Microscopia eletrônica de varredura ...................................................... 46
4.2. CARACTERIZAÇÃO DOS COMPLEXOS TERNÁRIOS .............................. 49
4.2.1. Eletroforese ........................................................................................... 49
1. INTRODUÇÃO .................................................................................................... 15
2. REVISÃO BIBLIOGRÁFICA ............................................................................... 18
3. MATERIAIS E MÉTODOS .................................................................................. 27
4. RESULTADOS E DISCUSSÕES ........................................................................ 42

14
4.2.2. Potencial Zeta ........................................................................................ 54
4.2.3. Tamanho das partículas por DLS (Dynamic Light Scattering) ............... 55
4.3. MICROCOPIA ELETRÔNICA DE VARREDURA ......................................... 56
4.4. TRANSFECÇÃO CELULAR ......................................................................... 61
Em pH = 7,4 ........................................................................................................ 69
Em pH = 9 ........................................................................................................... 70
ANEXO B – LAUDOS DO ZETA PARA OS COMPLEXOS TERNÁRIOS .............. 72
Razão 1:5:0,5 ..................................................................................................... 72
Razão 1:5:2 ........................................................................................................ 73
Razão 1:5:4 ........................................................................................................ 75
ANEXO C – LAUDOS DO DLS PARA OS COMPLEXOS TERNÁRIOS ................ 77
Razão 1:5:0,5 ..................................................................................................... 77
Razão 1:5:2 ........................................................................................................ 78
Razão 1:5:4 ........................................................................................................ 80
ANEXO D – MICROSCOPIA ELETRÔNICA DE VARREDURA DOS COMPLEXOS
TERNÁRIOS .......................................................................................................... 82
Amostras sem diluição ........................................................................................ 82
Amostras com diluição ........................................................................................ 84
EDS das amostras com diluição ......................................................................... 86
5. CONCLUSÃO ..................................................................................................... 64
6. SUGESTÃO PARA FUTUROS TRABALHOS .................................................... 65
REFERÊNCIAS ......................................................................................................... 66
ANEXOS ................................................................................................................... 69
ANEXO A - LAUDOS DO ZETA DAS NANOPARTÍCULAS ...................................... 69

15
1. INTRODUÇÃO
O desenvolvimento da transferência genética de forma segura e eficiente é
indispensável para o sucesso da terapia gênica (Shim, et al., 2010). Moléculas de
DNA não entram na célula de modo eficiente devido ao tamanho e natureza
aniônica. Além disso, são também muito suscetíveis a degradação por enzimas
(nucleases). O grande desafio da terapia gênica é realizar essa entrega de forma
eficiente e segura (Al-Dosari, et al., 2009).
Embora os vetores virais sejam mais usados para este fim, as preocupações
com segurança (os vetores virais são de difícil produção e podem causar sérias
reações inflamatórias em organismos) mudaram o foco para as técnicas de vetores
não virais. Estes vetores além de serem mais seguros são também mais versáteis,
pois podem ser construídos de acordo com o seu destino (Shim, et al., 2010). A
Figura 1 apresenta os diferentes tipos de vetores utilizados até o momento em testes
clínicos.
Alguns vetores não virais tem se mostrado eficientes para a transposição das
barreiras físicas e químicas que o impedem de realizar a transfecção gênica (Al-
Dosari, et al., 2009). Dentre as barreiras, temos a matriz extracelular, a membrana
da célula, a matriz intracelular e a membrana nuclear. Segundo (Shim, et al., 2010) e
(Cartier, et al., 2002), dentre as várias barreiras enfrentadas pelo vetor, o
rompimento das vesículas endossomais (após a entrada na célula por endocitose) e
a dissociação dos ácidos nucleicos do vetor de entrega quando este chega ao
interior da célula são as principais. Para ultrapassar esses obstáculos os vetores não
virais são complexos formados por diferentes combinações de DNA (normalmente
na forma de pDNA plasmidial), elementos catiônicos para a condensação deste,
compostos lipídicos para aumentar a proteção e a afinidade com a membrana da
célula, e ligantes adicionais para ajudar na entrega.
Os parâmetros a serem considerados para avaliar a eficácia dos vetores na
entrega gênica são o tamanho, formato, carga superficial e estabilidade (Adler, et al.,
2010).
16
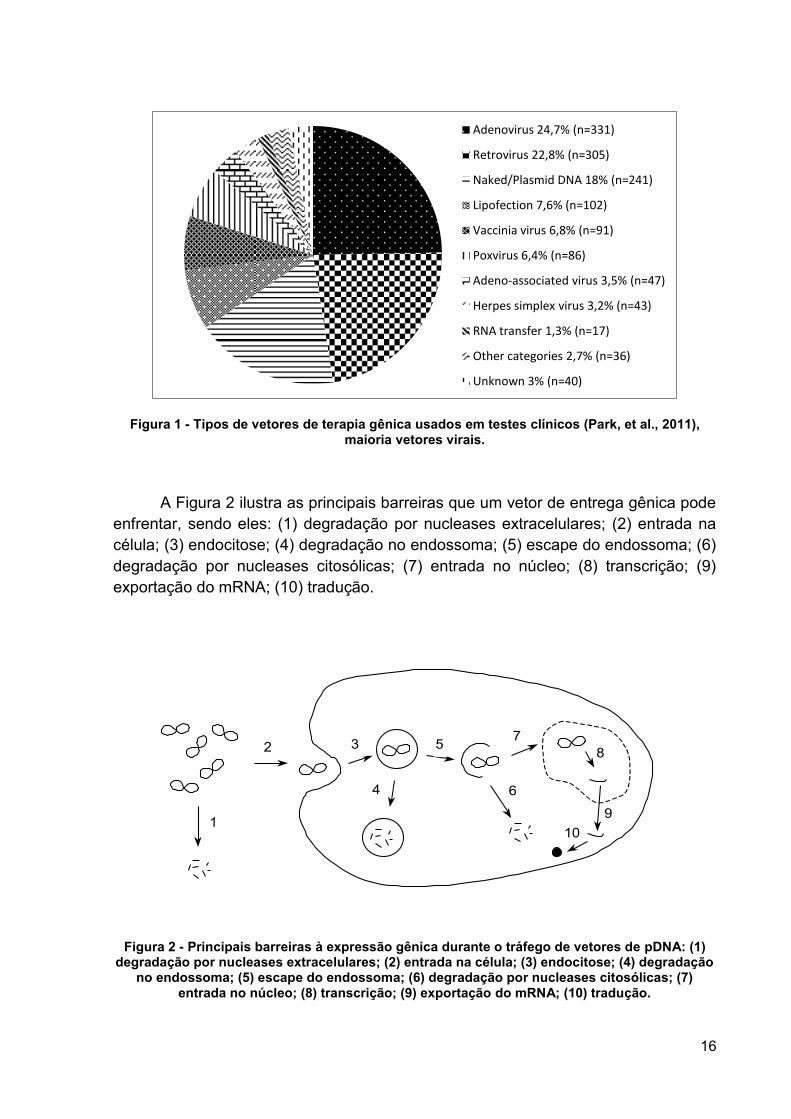
Figura 1 - Tipos de vetores de terapia gênica usados em testes clínicos (Park, et al., 2011), maioria vetores virais.
A Figura 2 ilustra as principais barreiras que um vetor de entrega gênica pode
enfrentar, sendo eles: (1) degradação por nucleases extracelulares; (2) entrada na
célula; (3) endocitose; (4) degradação no endossoma; (5) escape do endossoma; (6)
degradação por nucleases citosólicas; (7) entrada no núcleo; (8) transcrição; (9)
exportação do mRNA; (10) tradução.
1
2 3
4
5
6
7
8
9
10
Figura 2 - Principais barreiras à expressão gênica durante o tráfego de vetores de pDNA: (1) degradação por nucleases extracelulares; (2) entrada na célula; (3) endocitose; (4) degradação
no endossoma; (5) escape do endossoma; (6) degradação por nucleases citosólicas; (7) entrada no núcleo; (8) transcrição; (9) exportação do mRNA; (10) tradução.
Adenovirus 24,7% (n=331)
Retrovirus 22,8% (n=305)
Naked/Plasmid DNA 18% (n=241)
Lipofection 7,6% (n=102)
Vaccinia virus 6,8% (n=91)
Poxvirus 6,4% (n=86)
Adeno-associated virus 3,5% (n=47)
Herpes simplex virus 3,2% (n=43)
RNA transfer 1,3% (n=17)
Other categories 2,7% (n=36)
Unknown 3% (n=40)

17
Uma tecnologia que começou a ser usada na entrega de genes é a adição de
nano partículas metálicas ao complexo pDNA - proteína. Partículas de ouro, por
exemplo, conferem maior resistência à degradação da protease e à temperatura
(DeLong, et al., 2009).
Com isso, vamos focar nossos estudos nas caracterizações de parâmetros
que influenciam essa entrega gênica, tanto em complexos formados pela associação
de pDNA - proteína (complexos binários) como, principalmente, complexos ternários
formados por pDNA-proteína-metal.

18
2. REVISÃO BIBLIOGRÁFICA
2.1. DIFICULDADES DE ENTREGA GÊNICA
A terapia gênica se baseia em entrega de um gene em uma célula para que
esta possa realizar uma função nova ou para a substituição de um gene defeituoso.
Algumas entregas de RNA no citoplasma da célula são suficientes para realizar a
tarefa desejada, mas na maioria dos casos um DNA deve ser entregue no núcleo
para produzir um RNA que produzirá uma proteína de interesse, por exemplo
(Wagner, 1999).
Diversas barreiras são enfrentadas e afetam a eficiência da entrega do DNA.
Dentre essas barreiras temos: epitélio e endotélio (para inserção in vivo), matriz
extracelular, membrana celular, matriz citoplasmática, membrana nuclear e matriz
nuclear (Al-Dosari, et al., 2009).
A matriz extracelular pode ter nucleases que destruiriam facilmente o DNA,
logo este deve estar protegido. A etapa mais crítica, considerada limitante do
processo, é a passagem pela membrana da célula. Segundo a figura abaixo, o vetor
tem dois caminhos de interação com membranas. A: transferência direta do DNA na
membrana celular por fusão das membranas celulares e vetoriais ou passagem do
vetor através da membrana celular. B: entrada através de endossomo ou outra
vesícula interna, seguido de uma passagem para o citoplasma com rompimento da
membrana vesicular (Wagner, 1999) (Al-Dosari, et al., 2009) (Cartier, et al., 2002).
Figura 3 - Caminhos do vetor de entrega gênica. (a) Polinfecção e (b) Lipofecção.

19
Para os vetores capturados via endocitose, a vesícula é transformada em
vacúolo digestivo. Para escapar desta barreira, dois mecanismos são propostos. O
primeiro envolve o uso de membranas ativas ou moléculas que possam romper com
os vacúolos, tais como proteínas ou lipídios com porções hidrofóbicas. O segundo é
o uso de pressão osmótica, que é feita ligando-se o DNA com componentes de
amina ou polímeros catiônicos que absorvem prótons e diminuem a acidificação que
é essencial na transformação de endossomo para lisossomo. Consequentemente,
contra íons cloreto produzem uma pressão osmótica no endossomo.
A próxima barreira é o citoplasma celular. Nele o vetor encontra alguns
obstáculos, tais como, proteínas e o citoesqueleto celular, além de nucleases que
podem degradar o material genético. DNAs sem ligantes são ineficientes nesta
passagem e precisam de algum ligante para chegar ao núcleo com maior eficiência.
Em contra partida, o núcleo tem uma membrana dupla que possui proteínas, de
acesso ao seu interior, acopladas. Esta membrana não permite a passagem de
moléculas grandes. Estas são transportadas pelas proteínas acopladas que fazem,
por transporte ativo, a passagem de moléculas para o interior. Já quando a célula
está em fase de duplicação, ela deixa entrar no núcleo a maioria do DNA que está
dentro dela (Al-Dosari, et al., 2009) (Adler, et al., 2010).

20
Figura 4 - Mecanismo de transfecção, suas dificuldades e caminhos (Cartier, et al., 2002)
Depois que o gene está entregue, ele não é expresso de forma normal por
todo o tempo. Ele começa num nível alto de expressão e vai caindo com o tempo.
Diversas razões para isso podem ser citadas. O DNA injetado fica no núcleo na
forma de DNA episomal, sem chances de integrar o genoma celular. Como o
epissomo não é duplicado quando a célula se duplica, este não existirá em todas as
células filha e se diluirá entre as células. Outra razão para a diminuição da
expressão do transgene é que a célula pode se destruir ao ser infectada pelo vetor,
fazendo com que o número de células transfectadas caia. Para processo in vivo a
resposta imunológica pode fazer com que células sejam perdidas ou descartadas na
tentativa de controle de inflamação e com isso a expressão gênica também cairá.
Por outro lado, já foram encontrados plasmídeos em células de animais até dois
anos depois de sua transfecção (Al-Dosari, et al., 2009).
Na terapia gênica, os vetores virais, os métodos físicos de inserção de gene e
os métodos químicos (com as abordagens não virais inclusas) estão sendo

21
explorados, como mostra a Tabela 1 (Wagner, 1999). Vamos abordar a seguir os
vetores virais e não viras.
Tabela 1 - Comparação dos métodos de transfecção.
Método de transfecção
Vantagens Desvantagens
Métodos virais
Altamente eficiente Inflações, imunogênico,
cancerígeno
Métodos físicos
eletroporação Fácil desempenho
eficiente
Otimização para cada linhagem de célula requerida; grande
quantidade de DNA necessária
Micro injeção Exato direcionamento do ácido nucleico em
uma célula
Método sequencial e lento; abrange apenas
uma célula por vez
Biobalística Útil para vacina
gênica Penetração rasa do DNA
no tecido Métodos químicos
Compostos catiônicos
Fácil preparo Tóxico
Proteínas
recombinantes Alta
biocompatibilidade Alto custo
Nano
partículas poliméricas
Fácil preparo, tamanho controlável, fácil funcionalização
Eficiência limitada; algumas são tóxicas
Nano
partículas inorgânicas
Fácil preparo, tamanho controlável, fácil funcionalização
Eficiência limitada; algumas são tóxicas
2.2. VETORES VIRAIS
Os primeiros e mais usados vetores de entrega genica são os vetores virais.
Estes são mais eficientes que os vetores não virais por terem todo o maquinário
preciso para entrar nas celulas (Mastrobattista, et al., 2005). Entre suas
desvantagens podemos citar duas em especial: reação inflamatório para processos
in vivo e especificidade em relação às celulas (Smith, et al., 1998) Os adenovirus
são um exemplo de alta eficiencia com o pesar de provocar uma alta resposta imune
nas celulas.

22
Os vetores virais entram nas celulas através de endossomos. A proteina da
membrana da celula faz o reconhecimento do virus e este entra envolto por uma
vesicula, que tem a vantagem de conseguir passar pelas barreiras postas pelo
citoesqueleto do citoplasma celular. O esquema de entrada do virus na celula esta
ilustrado na figura abaixo.
Figura 5 - Entrada de um vetor viral na célula alvo
Um meio termo entre os vetores virais e não virais, é o vírus artificial. Este é
um vetor não viral que possui proteínas ou lipídios de alguns vírus. Outro método de
se fazê-lo é tirar uma proteína altamente infecciosa de um vírus para que este cause
menos respostas imunológicas. O intuito dessa técnica é aliar a segurança dos
vetores não virais com a eficiência dos virais e criar uma categoria com perspectivas
melhores para o futuro da entrega gênica (Mastrobattista, et al., 2005).

23
Tabela 2 - Comparação entre tipos de vetores de entrega gênica.
Vetores virais Vetores não virais Vírus artificiais
Prós
Transferência gênica muito eficiente;
expressão gênica persistente possível
Não infecciosos; manufatura de baixo custo; sem limite de
armazenagem de DNA
Não infecciosos; manufatura de baixo custo; sem limite de
armazenagem de DNA; transfecção de tipos
específicos de células
Contras
Imunogênico; potencialmente
infeccioso; alguns vírus são cancerígenos; manufatura de alto custo; capacidade
limitada de armazenagem de DNA
Transferência gênica ineficiente; expressão
gênica transiente
Potencialmente imunogênico; expressão
gênica transiente
2.3. VETORES NÃO VIRAIS
O estudo de vetores não virais para entrega gênica começou há quatro
décadas e esses estudos mostraram que a transfecção pode ser alcançada com a
junção de DNA com proteína, lipídio (o meio mais usado para em terapia gênica com
vetores não virais (Al-Dosari, et al., 2009)) ou polímero. Devido ao pequeno tamanho
(40 nm – 200 nm) e carga positiva, essas partículas são eficientes na entrega
genética.
Entrega gênica realizada por genes não virais são atrativas pelas seguintes
razões:
i. Eles podem ser gerados por alguns poucos componentes;
ii. Eles podem ser muito flexíveis em relação ao tamanho do DNA a ser
transportado.
iii. Plasmídeo e reagentes de transfecção podem ser produzidos em grande
escala com custos bastante baixos.
iv. Testes de segurança de um material sintético são menos trabalhosos do que
os testes de material recombinante.
Algumas características têm de ser olhadas com atenção na hora de construir
um vetor não viral para que ele tenha sucesso na entrada do núcleo. A química da
partícula é uma característica que influi nisso. Um complexo com lipídio é mais

24
eficiente na travessia do epitélio quanto menor sua porção hidrofóbica. O tamanho
de partícula influi na chegada do gene e seu método de medição deve ser
padronizado para possibilitar comparação. A carga da partícula possibilita que esta
seja positiva para entrar na membrana celular que é negativa. O formato do
complexo facilita a endocitose da célula, formatos cônicos não são tão absorvidos
por vacúolos como as esferas. A adição de ligantes para facilitar a ultrapassagem
das barreiras de entrada na célula (Adler, et al., 2010).
Mesmo com todo esse conhecimento sobre os vetores não virais, apenas 0,1
– 0,001% das partículas chegam intactas no núcleo de uma célula. Isso motiva os
estudos na área, não somente para a entrada na célula, mas também para o
rompimento do endossomo e entrada no núcleo (Mastrobattista, et al., 2005).
Neste trabalho iremos concentrar-nos nos complexos pDNA - Proteína e
pDNA - Proteína - Metal e vamos descrevê-los nos capítulos seguintes.
2.3.1. Proteína e pDNA
Na busca por algo que entregue genes às células com uma eficiência tão
grande quanto os vírus o fazem, surgiu o complexo pDNA-proteína. Os sistemas de
sítios ativos de enzimas, receptores e anticorpos envolvem de 5 a 20 aminoácidos,
ou seja, um complexo deste tipo pode ser construído com um peptídeo pequeno,
para poder interagir com essas estruturas.
As vantagens de se usar esse tipo de complexo é que a estrutura molecular e
a pureza dos reagentes já são conhecidas. Esse sistema sintético é muito versátil e
importantíssimo para provar que podem ocorrer mudanças significativas no cenário
de entrega gênica no sentido de estabilizar o complexo pDNA - proteína para que
este atinja uma maior eficiência.
A parte mais importante em se estudar esse tipo de partícula é poder
caracterizá-los, pois existem poucos estudos ao redor disso. Pouco se sabe sobre a
influência dos fatores na travessia de cada uma das barreiras do processo de
transfecção. As proteínas virais são conhecidas por ajudarem no rompimento do

25
endossomo e na captação nuclear, mas deve haver outras funções para elas que
ainda não são conhecidas. Ter o conhecimento sobre essas proteínas pode
estender os conhecimentos sobre como os vírus entram e se replicam numa célula
(Smith, et al., 1998).
2.3.2. Nano partículas como vetores
As possibilidades de se explorar as estruturas e processos das biomoléculas
para novas aplicações em materiais, biossensores e em atividades médicas, criou
um rápido crescimento no campo da nanobiotecnologia (Niemeyer, et al., 2004) e
consequentemente surgem os estudos referentes a nanopartículas. Como
característica principal, as nanopartículas (NPs) possuem o tamanho reduzido, o que
lhes permite penetrar em paredes e membranas celulares para fins terapêuticos
(Epple, et al., 2008).
As NPs são promissoras para a entrega de drogas como também podem ter
grande utilidade como ferramentas de diagnóstico. Diferentes sistemas de NPs que
possuem potencial para serem utilizadas em entrega de medicamentos são
estudados no intuito de diminuir a distância do descobrimento de uma droga até a
entrega eficiente desta droga no organismo (Parveen, et al., 2011).
As NPs metálicas podem ser sintetizadas em diversos tamanhos, podendo
chegar a tamanhos inferiores a 25 nm an e , et al., 2006). Sua grande superfície
pode proporcionar o transporte de doses relativamente altas de drogas (Parveen, et
al., 2011). A química de nanopartículas metálicas está sendo largamente estudada,
particularmente as NPs de metais nobres como ouro, prata, paládio e platina (Epple,
et al., 2008). As NPs de ouro, chamadas de AuNPs, são as mais comumente
utilizadas (Parveen, et al., 2011), no entanto existem muitos estudos sobre as NPs
de prata, também citadas como AgNPs an e , et al., 2006) (Yu, et al., 2007), as
quais serão utilizadas no trabalho de conclusão de curso em questão.
Existem estudos que citam NPs de prata (AgNPs) com atividade
antimicrobiana (Pan e , et al., 2006). No entanto, a atividade biocida depende de
várias características morfológicas e físico-químicas das partículas. A ação

26
bactericida das NPs de prata tem ocorrido para as menores partículas, ou seja, as
com menos de 10 nm (Martinez-Castanon, et al., 2008). O grupo de pesquisas do
Prof. Marcelo Seckler já obteve resultados sobre o controle da síntese das AgNPs
para diferentes condições de pH, além de ter desenvolvido um método novo para a
síntese das AgNPs, o qual permite a extinção da reação de síntese das NPs pelo
resfriamento da solução. O grupo também já conseguiu resultados de atividade
antibacteriana das AgNPs.
Este trabalho de conclusão de curso terá como objetivo estudar a viabilidade
bioquímica da utilização de AgNPs como vetores de entrega gênica, estudando
quais propriedades são necessárias para tanto.

27
3. MATERIAIS E MÉTODOS
3.1. MATERIAIS UTILIZADOS
3.1.1. Método para síntese das AgNPs
O método de Turkevich (Turkevich, et al., 1951), foi inicialmente desenvolvido
para partículas de ouro e depois foi modificado por Lee e Meisel (Lee, et al., 1982)
para nano partículas de prata (AgNPs), este último método será utilizado com
algumas modificações.
Nanopartículas de prata (AgNPs) serão sintetizadas pela redução química de
nitrato de prata com citrato de sódio em solução aquosa num reator encamisado sob
temperatura constante e agitação. Inicialmente, uma solução de 1 mM de citrato de
sódio deverá ter o pH ajustado ao valor desejado com hidróxido de sódio. Logo
após, 500 mL desta solução será adicionado ao reator e aquecido até a temperatura
desejada. A seguir 10 mL de solução de nitrato de prata 50 mM será adicionada,
como resultado a concentração de citrato e de sódio no reator devem ser de
aproximadamente 1 mM. O reator será mantido sob refluxo durante todo o curso da
reação, para reduzir a evaporação de água. A reação deverá ser conduzida por
períodos de tempo pré-determidados, sendo posteriormente resfriada pelo rápido
contato do meio de reação com um banho de gelo. Durante a reação, alíquotas de 5
mL serão retiradas a cada 5 minutos para caracterização da partícula. Os
experimentos devem ser feitos sob temperaturas entre 85°C e 95°C e sob um
intervalo de pH entre 7 e 9. O resfriamento será feito após 20 min e 40 min.
3.1.2. Método de síntese de partículas de pDNA-Proteína
3.1.2.1. DNA plasmidial (pDNA)
Será utilizado o plasmídeo modelo pVAX1GFP (3697 bp) purificado segundo
o kit Roche Miniprep (Roche, Alemanha). Este vetor, já disponível no laboratório, foi
construído a partir do vetor comercial pVAX1LacZ (Invitrogen), desenhado para o
desenvolvimento de vacinas de pDNA (descrito em detalhes por Azzoni et al. (2007)
(Azzoni, et al., 2007)). Em resumo, o vetor possui um promotor do citomegalovirus

28
humano CMV), gene repórter “green fluorescent protein” - GFP), gene de
resistência a Kanamicina para seleção em E. coli, e origem de replicação procariota
pMB1. O pDNA tem um tamanho de 500nm.
3.1.2.2. Preparação dos complexos binários pDNA-Proteína
Para a formação da do complexo binário pDNA-Proteína, o DNA plasmidial
modelo pVAX1GFP será primeiramente associado à proteína nuclear Protamina
(Sigma-Aldrich) em diferentes razões molares pDNA:Protamina (1:100, 1:500 e
1:1000, por exemplo).
Esta proteína já vem sendo estudada no grupo de pesquisa do Prof. Adriano
Azzoni. A interação entre estas proteínas de fusão e o pDNA (pVAX1GFP) é
conhecida por ser muito forte. Com isso, a preparação deste tipo de complexo será
feita pipetando as duas amostras em uma cubeta e esperando 10 minutos para que
a interação total entre elas aconteça.
A protamina utilizada tem tamanho de 1 nm e massa molar de 5100 g/gmol
(5,1 kDa). A Protamina é muito menor que o pDNA, por este motivo muitas proteínas
conseguem se acoplar ao pDNA e a razão molar tem essa alta diferença entre a
quantidade de ambos os compostos. Para ficar mais fácil, são usadas razões
mássicas, que são números mais próximos e mais fáceis de manipular.
O pDNA e a protamina usadas nos nossos experimentos têm concentrações
de 77,5 ng/µl ou 676,4 ng/µl para o pDNA e 1 µg/µl para a Protamina. Essas
concentrações servirão de base para o cálculo do volume de amostra que será
adicionado.
3.1.3. Preparo de complexos pDNA-Proteína-AgNP
Ao pDNA:Protamina será também adicionado a nano partícula metálica em
diferentes razões mássicas pDNA:Proteína:nano partícula metálica (1:5:1, 1:5:2 e
2:5:1, por exemplo). O procedimento abaixo foi retirado de um artigo cuja associação

29
envolve NPs de ouro (DeLong, et al., 2009), no entanto, o método será testado para
NPs de prata. Para o caso de complexos binários pDNA-Metal, a adição da proteína
deverá ser ignorada.
As AgNPs poderão ser associadas a proteínas e pDNA sendo adicionadas (à
vazão de 1 mL/s) em bateladas de 3-3,5 mg misturadas manualmente ou agitadas
num vórtex, adiciona-se então a protamina na razão mássica pDNA:Protamina (ou
RP3) 1:1, 1:2 e 1:5, por exemplo, a seguir adiciona-se pDNA na razão mássica
pDNA:nano partícula metálica 1:1, 1:2 e 2:1. As partículas serão precipitadas com
solução de etanol 70%, ou estocadas em solução de etanol 70% até o uso. Em
alguns casos, as NPs poderão ser separadas por centrifugação (DeLong, et al.,
2009).
No nosso caso, adicionamos as AgNps aos complexos pDNA:Protamina
(após eles interagirem por 10 minutos) e aguardamos mais 10 minutos para que o
complexo ternário seja formado, para que, após isso, possamos fazer qualquer
análise ou experimento. As AgNps estão armazenadas em seu próprio meio de
reação, ou seja, meio contendo citrato (agente estabilizante), sódio, nitrato e íons
prata. Assim o complexo ternário fica armazenado em tampão PBS (pH = 7,4), mas
com algumas substâncias do meio reativo das AgNps. Outra maneira é centrifugar
as nano partículas e resuspendê-las em meio PBS.
Os complexos ternários assim formados serão então analisados também por
métodos, como o potencial zeta, espalhamento de luz e eletroforese em gel de
agarose.
3.1.4. Transfecção em células animais
Células HeLa “Human epitheloid carcinoma”), serão crescidas em meio F-12
(Ham - nutrient mixture, Gibco, Inglaterra) contendo 10% (v/v) de soro fetal bovino
(Gibco, Inglaterra). As células serão crescidas em frascos de cultura para células
aderentes de 75 cm2 e incubadas em ambiente umidificado contendo 5% de CO2 e
a temperatura de 37ºC. Após atingirem a confluência, as células serão tripsinizadas
e semeadas em placas de cultura de 24 poços (5 x 104 células por poço). As células

30
serão incubadas por 24 horas e então transfectadas com as diferentes soluções
contendo os complexos carreadores em diferentes razões pDNA/proteína ou
pDNA/proteína/Metal. Transfecções controle serão feitas utilizando-se o reagente de
transfecção (lipídeo catiônico) Lipofectamine 2000TM Invitrogen, EUA), 1 μg
pDNA/proteína mais 1.5 μL lipídeo em 100 μL de meio por poço). A avaliação e
quantificação do nível de transfecção das células em cultura serão baseadas na
quantificação dos níveis de transfecção por citometria de fluxo: Vinte e quatro horas
após a transfecção, células em cultura serão lavadas, tripsinizadas, centrifugadas
(160g por 8 min) e suspensas em tampão PBS (tampão a pH = 7,4 de cloreto de
sódio, cloreto de potássio, fosfato de sódio e fosfato de potássio, isotônica em
relação ao corpo humano) (Azzoni, et al., 2007). As células transfectadas e não
transfectadas (controle negativo) serão então analisadas em um citômetro de fluxo
FACscan Scalibur (Becton-Dickinson, EUA), que analisará o espalhamento de luz
frontal (forward scatter - FSC), lateral (side scatter - SSC), e fluorescência em verde
FL1). A partir disso, os dados relativos às células serão isolados dos “debris”
através das características FSC versus SSC, e o nível de transfecção será
determinado através da subtração dos dados de background FL1 (células não
transfectadas) da população celular total. Os dados serão analisados utilizando-se o
software CellQuest Pro (Becton-Dickinson, EUA).
3.2. CARACTERIZAÇÃO DOS VETORES
3.2.1. Potencial Zeta
As forças superficiais na interface da partícula e do líquido possuem um efeito
eletrocinético e são responsáveis tanto por possíveis aglomerações entre as
partículas quanto pela repulsão entre elas. Quanto menores as cargas das
partículas, estas ficam livres para se colidirem e aglomerarem, enquanto partículas
carregadas tendem a se repelir, tendo probabilidade maior de ficarem dispersas
(como mostra a Figura 6).

31
Figura 6 - a) Partículas carregadas se repelem umas das outras, enquanto b) partículas sem carga ficam livres para colidirem e agregarem.
A carga de uma partícula em suspensão pode ser controlada modificando-se
o pH ou a natureza de espécies iônicas na solução. Uma técnica comum é o uso de
tensoativos que adsorvem diretamente na superfície da partícula, mudando suas
características.
O modelo de dupla camada é utilizado para a descrever a região iônica na
vizinhança da partícula carregada e explica como as forças de repulsão ocorrem. Se
usarmos uma partícula carregada negativamente, podemos identificar o efeito sobre
os íons positivos (contra-íons) na solução. Inicialmente, a atração da partícula
negativa sobre os contra-íons forma uma camada firmemente anexada à volta da
superfície da partícula, essa camada de contra-íons é conhecida como Camada de
Stern. Devido à quantidade de íons positivos na camada de Stern outros íons
positivos são repelidos ao tentar se aproximar da partícula negativa, esse equilíbrio
dinâmico resulta na formação de uma camada difusa de contra-íons, pois íons
negativos podem ser atraídos pela quantidade de íons positivos nas proximidades
da partícula (A Figura 7 ilustra a distribuição de íons em torno de uma partícula).
Portanto o modelo de dupla camada diz que existem duas camadas, uma formada
apenas por contra-íons, a camada de Stern, e outra mais distante da partícula, a
camada difusa, formada por contra-íons e co-íons onde a concentração de contra-
íons cai com o distanciamento da partícula e a concentração de co-íons aumenta
com o distanciamento da partícula até o equilíbrio.

32
Figura 7 - Ilustração das camadas de Stern e da camada Difusa.
A dupla camada é formada para neutralizar a carga da partícula, no entanto,
causa um potencial eletrocinético entre a superfície da partícula e qualquer ponto na
massa do liquido de suspenção. A diferença de voltagem é da ordem de milivolts e
se refere ao potencial da superfície. A magnitude do potencial de superfície é
relacionado à carga superficial e à espessura da dupla camada. Conforme nos
distanciamos da superfície o potencial cai linearmente na camada de Stern e
exponencialmente pela camada difusa chegando a zero no limite imaginário da
dupla camada. A camada de Stern é considerada sendo rigidamente presa à
partícula, enquanto a camada difusa não é. Como resultado o potencial elétrico
nessa união de camadas está relacionado com a mobilidade da partícula e é
chamado de potencial zeta (Zeta-Meter).
O potencial zeta, apesar de ser uma medida de potencial elétrico, pode ser
ainda relacionado com a carga da partícula, visto que é mais facilmente medido (ver
Figura 8).

33
Figura 8 - Potencial Zeta e potencial de superfície.
O potencial zeta é de grande importância, pois os vetores devem ter carga
positiva para interagirem e serem endocitados pela célula, caso contrário haverá
repulsão entre os vetores e as membranas celulares, as quais possuem carga
negativa.
3.2.2. Tamanho das partículas por DLS (Dynamic Light Scattering)
A técnica de DLS é usada para medir o tamanho de partículas pequenas.
Esta técnica mede o movimento browniano (o movimento browniano é o movimento
aleatório de partículas macroscópicas num fluido como consequência dos choques
das moléculas do fluido nas partículas) e o relaciona com o tamanho das partículas.
Quanto maior a partícula menor o movimento browniano, partículas grandes são
difíceis de serem movidas pelo meio. A temperatura deve ser precisa, pois ela pode
causar correntes de convecção que causariam movimentos ordenados e a
viscosidade (que depende da temperatura) também deve ser fixa, pois influi no meio.
(Instruments)
O diâmetro hidrodinâmico reflete como a partícula se difunde no meio e é
calculado pela equação abaixo. O coeficiente de difusão translacional depende do
tamanho da partícula e da concentração e tipos de íons do meio.

34
Onde:
d(H) = diâmetro hidrodinâmico;
D = coeficiente de difusão translacional;
k = constante de Boltzman;
T = temperatura absoluta do meio;
η = viscosidade do meio.
O tipo de íon e sua força e concentração podem interferir na velocidade de
difusão da partícula mudando a espessura da dupla camada elétrica. Num meio de
baixa condutividade a difusão é menor e o diâmetro H é maior aparentemente, o
inverso é verdadeiro. Para minimizar estas interferências usa-se um meio padrão
que diminui a dupla camada elétrica e mede-se o diâmetro hidrodinâmico esperado.
A intensidade de espalhamento de luz se relaciona com o diâmetro de
partícula e com o comprimento de onda da seguinte maneira:
Onde:
I = intensidade de espalhamento de luz;
d = diâmetro de partícula;
λ = comprimento de onda da luz.
Com isso podemos perceber que uma partícula de 50nm de diâmetro espalha
1.000.000 de vezes mais luz que uma de 5 nm.
A medição é feita incidindo-se um feixe de luz na amostra e capturando de
novo esta luz através de um arranjo ótico como na figura abaixo.

35
Figura 9 - Amostra a ser analisada
As partes pretas representam as ondas destrutivas e as brancas representam
as ondas construtivas, que se somam (Figura 10). Esta configuração está em
constante modificação, visto que o movimento browniano move as partículas.
Figura 10 - Demonstração da dependência do espalhamento em relação à fase da onda.
Quanto menor elas são, mais rápido elas se movem (Figura 11).

36
Figura 11 - Diferença da variação da intensidade com o tempo para partículas grandes e pequenas.
O sistema ótico capta o sinal de luz, mas é necessário um comparador de
sinais para relacionar a intensidade de sinais em um pequeno δt. Com isso,
podemos relacionar dois sinais bem parecidos, pois o tempo entre eles é curto
(Figura 12).
Figura 12 - Intensidade de sinal no tempo.

37
Se o sinal é comparado com ele mesmo o coeficiente de correlação é perfeito
e vale 1, se é totalmente imperfeito vale 0. Se um sinal é comparado com um sinal
padrão em um tempo “t” ele começa perfeito e no infinito tende a ser 0. Se as
partículas são grandes o sinal tende a mudar devagar e a curva demora a cair
(Figura lado esquerdo), e vice versa (figura lado direito). Quanto mais rápido o
decaimento, mais monodispersa é a amostra, pelo contrário, quanto mais devagar o
decaimento, mais polidispersa a amostra.
Figura 13 - Coeficiente de correlação em função do tempo.
Para duas amostras de mesma população com diâmetros de 5nm e 50nm o
volume delas fica na proporção de 1:1.000 por causa da dependência com d3 do
volume e a intensidade do sinal fica na proporção de 1:1.000.000, pois a
dependência é com d6 (Figura 14). Assim o equipamento consegue medir o tamanho
das partículas.

38
Figura 14 - Dependência do volume e da intensidade com o tamanho das partículas para duas populações com igual quantidade.
3.2.3. Eletroforese em gel de agarose
A eletroforese, método de separação de moléculas carregadas sob a
influência de um campo elétrico, é um processo que tem sido extensivamente
utilizado, principalmente no campo da bioquímica, desde a década de 30 (Santos, et
al., 2000).
Figura 15 - Equipamento de eletroforese em gel.

39
A eletroforese em gel de agarose consiste no método padrão usado para
separar, identificar, analisar, caracterizar e purificar fragmentos de DNA. A técnica é
simples, rápida e capaz de separar misturas de fragmentos de DNA que não podem
ser separados por outros métodos, tais como centrifugação com densidade de
gradiente ou por velocidade de sedimentação. A localização do DNA no gel pode ser
determinada diretamente. As bandas no gel são coloridas principalmente por
Brometo de Etídeo em baixa concentração, que colore por intercalar-se na dupla fita
de DNA. Quantidades de até 1 ng de DNA podem ser visualizadas por exame direto
de gel na luz ultravioleta. Na Figura 16, o DNA é visível como bandas de cor
alaranjada, a cor se deve à fluorescência do corante usado, o brometo de etídio.
Uma molécula de DNA, quando exposta a um campo elétrico, migra para o
eletrodo na velocidade ou mobilidade eletroforética, proporcional a força do campo e
a carga líquida da molécula. A mobilidade eletroforética é também inversamente
proporcional ao coeficiente friccional da molécula, que, por sua vez, é função do
tamanho e forma da molécula, e da viscosidade do meio (ZE=vf). Portanto, uma
mistura de moléculas diferentes pode ser separada eletroforeticamente com base
em:
tamanho da molécula;
forma ou conformação da molécula;
magnitude das cargas elétricas líquida na molécula;
Quando aplicadas na mesma posição num campo elétrico, as moléculas
serão separadas em bandas e migrarão a velocidades diferentes para posições
diferentes no meio.
Sob condições fisiológicas, os grupos fosfatos dos ácidos nucléicos
encontram-se ionizados. Cadeias de polinucleotídeos de DNA e RNA são chamados
de poli-ânions e migram para o eletrodo positivo (anodo) quando colocados em
campo elétrico. Devido à natureza repetitiva dos fosfatos, o DNA dupla fita possuem
aproximadamente a mesma relação de massa e carga líquida. Ajustando a
viscosidade do meio, entretanto, os efeitos da fricção e o formato das moléculas
podem ser usados, permitindo separar os ácidos nucléicos eletroforeticamente por

40
tamanho. A viscosidade do meio pode ser determinada pelo tamanho dos poros do
meio através da concentração de agarose.
Figura 16 - Gel de agarose após electroforese de fragmentos de DNA.
A agarose consiste num polissacarídeo linear de galactose e um derivativo de
galactose associados através de ponte de hidrogênio. Agarose é composta de
unidades de b-D-galactopiranose com ligação 1,3 e 3,6 anidro-a-L-galactopiranose
com ligação 1,4. Essa unidade básica de agarobiose é repetida cerca de 4000
vezes, formando longas cadeias com peso molecular médio de 120.000 Daltons.
Também podem ocorrer no polissacarídeo, grupos com carga, tipicamente piruvato e
sulfato. Esses grupos são responsáveis por várias propriedades da agarose, e a
seleção cuidadosa de matéria prima para o preparo da agarose controla a qualidade
para fins específicos (Compri-Nardy, et al., 2009).
Para o ensaio de eletroforese foram misturados 0,4g de agarose com 50ml de
tampão TAE (solução tampão usada em eletroforese em agarose, tipicamente para
a separação de ácidos nucleicos tais como o DNA e RNA. Ele é feito de tampão
acetato da base Tris, normalmente a pH 8.0, e EDTA, os quais sequestram cátions
divalentes) em um erlenmeyer para fazer o gel. A agarose é misturada e aquecida
até dissolver totalmente para depois ser colocada no equipamento.
Após solidificado o gel, basta colocar as amostras nos espaços
correspondentes e ligar o equipamento a 60V. As amostras a serem analisadas são

41
misturadas com 5 µl de glicerol. Depois de corrido o ensaio (2 horas depois),
colocamos o gel em um corante chamado SYBR Safe, que é um corante
fluorescente marcador de DNA para a leitura dos resultados, por 20 minutos. Para
finalizar colocamos o gel em água destilada por 20 minutos para tirar o excesso de
corante. Agora basta ler os resultados em camara de ultravioleta.

42
4. RESULTADOS E DISCUSSÕES
4.1. CARACTERIZAÇÃO DAS NANOPARTÍCULAS DE PRATA
4.1.1. Síntese das nanopartículas
As partículas foram feitas nas condições de pH = 9 e temperatura de reação =
90⁰C em reator encamisado com agitação moderada.
A solução começa incolor e vai se tornando amarelada ou caramelo escuro,
de acordo com a conversão (Figura 17). A coloração se dá porque as nanopartículas
de prata têm um tamanho tal que elas entram em ressonância com as ondas do
comprimento da luz amarela, emitindo luz neste comprimento de onda.
Figura 17 - Amostra de nanopartículas de prata com 15 minutos de reação

43
O teste de espectrofotometria das partículas nos mostra o comprimento de
onda que a solução mais absorve. Este máximo de comprimento de onda é
característico de cada substância e o da prata é perto dos 390 nm.
Figura 18 - Curva espectrofotométrica das nanopartículas de prata
De acordo com a curva acima, o máximo se dá em 430 nm. A curva foi feita
subtraindo-se os valores de absorbância da amostra de água dos valores da
amostra das partículas, para eliminar os ruídos. O pico foi deslocado para a direita
em relação ao valor de referência, possivelmente por causa da oxidação da prata
diante da luz, devido á forma não esférica das partículas, ou ainda pela presença de
agregados (Lok, et al., 2007) (Yan, et al., 2006) (Wan, et al., 2005) (Santana, et al.).
As nanopartículas de prata estão imersas em meio contendo citrato de sódio
e nitrato de prata. Como o complexo ternário pDNA-Protamina-Metal será formado
em solução do tampão PBS, decidimos centrifugar a amostra e tentar suspendê-la
novamente no tampão desejado sem a interferência dos componentes da dispersão
original. A centrifugação foi feita a 9000 rcf (unidades de força g) por 10 minutos.
Depois da centrifugação a solução ficou límpida e com um aglomerado de partículas
no fundo (Figura 19).
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
200 300 400 500 600 700 800
Ab
sorb
ânci
a
Comprimento de onda (nm)
Espectrofotometria de AgNps

44
Figura 19 - Amostra de nanopartículas de prata centrifugadas
O líquido sobrenadante foi retirado com pipeta e o PBS foi acrescentado.
Aparentemente as nanopartículas tinham resuspendido, pois a solução voltou a ficar
amarela como antes. Mas, no dia seguinte, a solução tinha ficado transparente como
água, o que indica que as nanopartículas não estavam em solução. Era possível ver
pequenos pontos metálicos na parede do tubo.
Uma possível explicação é que a alta salinidade do PBS comparada ao outro
meio, que também continha citrato que é um bom agente estabilizante, provocou a
aglomeração das partículas de prata. Com isso, para os ensaios posteriores,
procuramos resuspender as partículas logo antes de fazer os experimentos, ou
simplesmente não resuspender.
4.1.2. Potencial Zeta
A carga resultante do complexo pDNA:Protamina:AgNPs deve ser positiva
para entrar na membrana da célula que é negativa. Sabemos que o pDNA é
negativo e a protamina é positiva, fazendo com que um complexo binário de razão
mássica 1:5 resulte numa partícula positiva a pH = 7,4 (+24 mV segundo ensaios já
realizados pelo grupo).
Para as AgNPs, o ensaio de potencial Zeta foi realizado a pH = 7,4 (em
tampão PBS) e a pH = 9 (em solução de citrato). As medições foram realizadas em
triplicata para cada pH. De acordo com a tabela abaixo temos os resultados.

45
Tabela 3 - Resultado do ensaio de potencial Zeta para as nanopartículas de prata
Amostra (mV)
pH 1 2 3 Média Desvio Padrão
9,0 -18,6 -14,7 -23,6 -19,2 6,3
7,4 -49,7 -51,1 -50,0 -50,3 0,7
De acordo com os resultados podemos ver que com a diminuição do pH a
partícula fica mais negativa. A carga da partícula a pH = 7,4 é, em média, 2,5 vezes
mais negativo que a pH = 9. O desvio padrão é muito alto para as amostras a pH
mais alto, requerendo mais amostras.
Como a partícula é negativa, principalmente para pH = 7,4 que é a condição
do tampão PBS e na qual se realizarão os experimentos, agora podemos determinar
razões mássicas para os ensaios seguintes.
Esta carga sendo muito negativa a pH = 7,4 nos remete aos resultados de
excesso de protamina em relação às AgNPs. Esta carga é, em módulo, o dobro da
carga da protamina e faz com que uma partícula tenha mais “força” que uma de
protamina, devido às altas forças eletrostáticas.
Os laudos do experimento são encontrados no ANEXO A - LAUDOS DO ZETA
DAS NANOPARTÍCULAS.
4.1.3. Tamanho das partículas por DLS (Dynamic Light Scattering)
No ensaio de DLS apareceram duas populações de partículas, com tamanhos
médios de 3,4 e 75 nm. Não foi possível estimar a quantidade de cada uma das
populações.
A população menor (3,4 nm) é praticamente desprezível no ponto de vista de
aumentar o tamanho do complexo ternário. Por outro lado, se apenas uma
nanopartícula de 75 nm interagir com o complexo pDNA-protamina, ela já aumentará

46
significativamente o seu tamanho (o tamanho do complexo binário é da ordem de
500 nm).
Em contrapartida, a quantidade de partículas menores pode ser maior,
aumentando a probabilidade de interação.
Este tamanho realmente é da escala nanométrica como esperado. De acordo
com a cor da solução (amarelada) já tínhamos uma noção de que o experimento
tinha produzido as nanopartículas, mas o DLS veio para quantificar e qualificar isso.
O tamanho de partícula segundo o ensaio de DLS será comparada com a
medição a seguir para o ensaio de microscopia eletrônica de varredura.
4.1.4. Microscopia eletrônica de varredura
Foi realizada a microscopia eletrônica de varredura das nanopartículas de
prata. O ensaio é feito pingando-se umas gotas da solução de nanopartículas numa
superfície metálica apoiadora de amostra, com posterior evaporação do líquido e
sobra da amostra sólida. Com isso, podemos fazer medições de tamanho de
partícula de acordo com a escala da foto. Além disso, este teste serve para
confirmar o ensaio de DLS, pois para que este seja válido as partículas devem ser
esféricas, de acordo com as aproximações feitas para se chegar ao resultado.

47
Figura 20 - MEV das nanopartículas de prata a pH = 9 e 90°C
De acordo com a figura acima, podemos dizer que as partículas são
aproximadamente esféricas e o resultado do DLS não pode ser rejeitado por este
quesito. Algumas partículas, às vezes, podem ficar em formato de agulha, o que
afetaria o resultado do DLS.
Como mostra a figura, temos algumas partículas com medições de tamanho
em vermelho. Pegando uma grande amostra destas, ou seja, não podemos
considerar apenas as medições desta foto, conseguimos um valor da média do
tamanho das partículas de prata. O tamanho médio das partículas por este método é
64 nm.
Além desta população, temos uma outra população de partículas menores,
cujo tamanho médio é de 18 nm segundo a foto.
Comparando com o DLS, o erro, em relação à população maior, é pequeno
de aproximadamente 15%. Já o erro da população menor é maior da ordem de
500%. Levando em conta que as medições são feitas à mão, a principal fonte de
erro neste método é a imprecisão do olho humano ao fazer o traço que liga os
extremos das partículas, principalmente no que diz respeito às partículas menores.
Existem erros, também, ligados ao fato de que a amostra é pequena em relação ao
conteúdo do reator, podendo não representar o conjunto das partículas. Finalmente,

48
como as partículas não são totalmente esféricas, não existe uma dimensão ideal
para se medir este “tamanho” de partícula.
O ensaio de microscopia nos revela que as partículas de prata não estão
altamente aglomeradas, o que seria um efeito indesejado, pois não seria viável a
transfecção de complexos muito grandes.
Um cálculo simples foi feito para avaliar se há excesso de AgNPs ou de
Protamina nas nossas amostras. Como o complexo binário é formado e não corre no
gel a partir da razão mássica 1:1 (Figura 21), podemos pensar que ao se
acrescentar a protamina até a nossa razão mássica de estudo (1:5) temos um
excesso de partículas positivas.
Hipóteses e dados iniciais do cálculo:
massa de pDNA = 0,6 µg;
massa de protamina = 3 µg (segundo razão mássica 1:5) sendo que
2,4 µg estão em excesso, pois a razão 1:1 já é positiva;
massas de AgNPs 0,3 µg (1:5:0,5), 1,2 µg (1:5:2) e 2,4 µg (1:5:4);
partículas de prata são esféricas de tamanho médio 3,4 nm (este
tamanho de partícula é o tamanho médio de uma das distribuições de
população dadas pelo DLS);
50% da prata usada para sintetizar as nanopartículas de prata são
convertidas nelas, valor assumido desde o início, inclusive para definir
os volumes de amostra para adição no complexo binário.
O cálculo de excesso de protamina segue como abaixo:
Onde:
QP é a quantidade de protamina em excesso;
NA é a constante de Avogadro;
ME é a massa de protamina em excesso;

49
MMP é a massa molar da protamina;
O cálculo da quantidade de AgNPs é dado abaixo:
Onde:
dp é o diâmetro das nanopoartículas (3,4 nm);
Vp é o volume de uma partícula de prata;
QAgNPs é a quantidade de nanopartículas de prata;
VA é o volume de amostra utilizado na razão mássica 1:5:4, que é a
que mais possui prata (5,74 µl);
µ é a massa específica da prata (10,5 g/ml);
MMAg é a massa molar da prata que é 108 g/mol;
CAg é a concentração de prata na solução = 1 mM;
Conv é a taxa de conversão de prata em nanopartículas = 50%.
Avaliando as quantidades de protamina e nanopartículas vemos que a
primeira esta em excesso de 200 vezes em relação a esta última.
4.2. CARACTERIZAÇÃO DOS COMPLEXOS TERNÁRIOS
4.2.1. Eletroforese
O objetivo do experimento é caracterizar se o complexo
pDNA:Protamina:AgNPs foi ou não formado e se a sua carga é positiva, para que a
sua entrada na célula seja possivel, já que a carga da membrana celular é negativa.
Para isso, as bandas formadas na eletroforese da solução de complexo foram
comparadas com as bandas formadas na eletroforese da solução de pDNA puro e
pDNA:Protamina de mesma concentração e de marcador ou ladder (fragmentos de

50
DNA com tamanho e concentração conhecidos, que permite determinar o tamanho
dos fragmentos de DNA presentes na amostra).
A migração do DNA na direção do polo positivo ocorre porque, em meio
neutro ou básico, o DNA assume carga negativa. Assim, ele penetra no gel em
direção ao polo positivo, devido ao efeito eletroforético que atua sobre suas
moléculas. O fator de maior influência na migração do DNA através do gel de
agarose é a massa molecular ou o tamanho dos fragmentos de DNA. Isso porque o
gel funciona como uma peneira molecular, o que significa que moléculas com
massas moleculares mais elevadas são retidas pelas malhas do gel, tendo sua
migração retardada. A mobilidade de um fragmento é inversamente proporcional ao
log10 de sua massa molecular. Assim, grupos de mesmo tamanho migram juntos e
assumem da forma do poço, formando as chamadas bandas de DNA. Apesar disso,
a linearidade e a condensação do DNA também influenciam na velocidade de
migração deste.
O DNA utilizado no experimento é plasmidial (circular) e pode assumir três
formas diferentes: relaxada ou Nick (com corte), superenovelada ou supercoiled
(condensado) e circular (DNA não condensado em forma de anel). Como foi utilizado
apenas um tipo de pDNA, e não fragmentos de massas moleculares diferentes, a
velocidade de migração do pDNA puro só foi afetada pelo formato do DNA. Sendo
assim, na solução de pDNA puro é esperada a formação de 3 bandas. A primeira
corresponde à forma relaxada (menor velocidade de migração), a segunda à forma
circular e a terceira à forma superenovelada (maior velocidade de migração). A
formação de complexos faz com que o DNA plasmidial em solução não assuma as
três formas citadas acima. Ao invés disso, ele interage com as proteínas, formando
as partículas do complexo. Logo, é esperado que, após a formação completa das
partículas de complexo, deixe de haver três bandas, e ocorra a formação de uma
única banda extensa (devido à distribuição de tamanhos das partículas do
complexo).
Resultados de eletroforese para diferentes razões mássicas pDNA:Protamina
são mostrados na Figura 21. Observa-se, as bandas DNA1 e DNA2 representam o
DNA puro e a primeira amostra é o controle mássico com DNA de vários tamanhos.
Nas diferentes proporcoes pDNA-proteina, observa-se que não há migração.

51
Figura 21 - Eletroforese em agarose de partículas pDNA e protamina
Dentre as amostras colocadas todas são positivas, inclusive 1:1. Com isso,
podemos dizer que a amostra 1:5, que contém uma maior proporção de moléculas
positivas de proteína, é muito positiva. Esta condição no complexo binário é
desejável, pois assim, mesmo após a adição das AgNPs negativas, os complexos
ternários continuem positivos, sendo assim capazes de adentrar nas células.
Após a etapa de escolha da quantidade de protamina a ser adicionada,
devemos avaliar a quantidade de AgNPs. Esta etapa foi mais complicada, pois não
tínhamos nenhuma informação de eletroforese em amostras de complexos ternários
com prata, por isso não sabíamos em que quantidades as nossas amostras
continuariam positivas. Decidimos montar o experimento com valores pequenos de
prata devido à sua carga ser muito negativa (Tabela 3). O ensaio comporta oito
amostras, com isso, montamos a seguinte disposição, segundo a Tabela 4.
Tabela 4 - Amostras de ensaio de eletroforese
Amostra
1 2 3 4 5 6 7 8
Controle de massa Controle pDNA 1:5:0 1:5:0,5 1:5:1 1:5:2 1:5:4 1:5:6
DNA1 1:20 DNA2 1:10 1:5 1:2 1:1

52
Onde:
Controle de massa são pedaços pequenos de pDNA corados em azul,
representando a menor massa entre as amostras, ou seja, enquanto ela
continuar percorrendo o gel as outras também estão lá. Serve para garantir a
validade do ensaio.
Controle de pDNA representa o resultado para o pDNA puro, serve de
comparação.
As demais proporções são representadas por pDNA:protamina:NpAg. Com
isso, a amostra 3 é a comparação de amostra sem NpAg e as demais estão
em relação crescente de partícula de prata.
Com isso, temos 3 controles e 5 amostras de complexo ternário. Aumentamos
a razão de NpAg gradativamente, para obtermos complexos positivos e talvez poder
quantificar o ponto de viragem para carga negativa.
De acordo com a concentração de pDNA e protamina que temos, a razão de
proteína escolhida e a estimativa de conversão das nanopartículas de prata (50%),
já que não temos esta informação, podemos determinar os volumes a ser misturado
para a formação dos complexos.
Os volumes de pDNA e protamina são fixos de acordo com a razão mássica,
os volumes de NpAg variam com a sua razão e o volume de PBS usado serve para
completar o volume da amostra para 20µl (Tabela 5) (Figura 22).
Tabela 5 - Volume das composições das amostras
Volumes de amostra (µl)
Amostras pDNA Protamina NpAg PBS Total
2 7,75 0,00 0,00 12,25 20,00
3 7,75 3,00 0,00 9,25 20,00
4 7,75 3,00 0,72 8,53 20,00
5 7,75 3,00 1,43 7,82 20,00
6 7,75 3,00 2,90 6,35 20,00
7 7,75 3,00 5,74 3,51 20,00
8 7,75 3,00 8,61 0,64 20,00

53
Figura 22 - Amostra de complexo pDNA-Protamina-Metal para ensaio de eletroforese
De acordo com a Figura 23 vemos que os controles correram no gel,
validando o ensaio. Tendo e vista que apenas as partículas negativas correm no gel
e as positivas ficam retidas no local onde foram inseridas, devido à diferença de
potencial colocada no gel, podemos afirmar que o complexo permaneceu formado,
pois nenhum pDNA foi encontrado no gel, em comparação com a amostra 1
(controle).
Esse resultado indica que, nas razões mássicas estudadas, o complexo
pDNA-protamina se manteve estável, formando uma partícula que não percorre a
malha do gel, independentemente da quantidade de NpAgs adicionada.
No entanto, conforme os nossos cálculos de quantidade de protamina em
excesso e quantidade de partículas de prata, pode ter ocorrido aglomeração dessas
duas substâncias, fazendo eventualmente com que a prata não interagisse com o
complexo.

54
Figura 23 - Eletroforese em gel dos complexos ternários
Para completar a analise precisamos do ensaio de potencial Zeta e DLS para
avaliar se a carga e o tamanho dos complexos, respectivamente, mudaram. Com
isso poderemos ter mais ferramentas para tentar observar a formação do complexo
ternário.
4.2.2. Potencial Zeta
O ensaio de potencial Zeta foi realizado para complexos ternários em
diferentes razões de nanopartículas de prata. O intuito deste ensaio era avaliar a
mudança de carga dos complexos com o aumento da concentração de NpAg, visto
que, o potencial para uma partícula binária com razão pDNA:Protamina de 1:5 o
potencial era de +24mV. Os laudos deste experimento foram anexados no final do
relatório no ANEXO B – LAUDOS DO ZETA PARA OS COMPLEXOS TERNÁRIOS.

55
Tabela 6 - Valores do potencial Zeta para os complexos ternários
Teste (mV)
Razões mássicas
1 2 3 Média Desvio Padrão
1:5:0,5 21,0 22,5 21,9 21,8 0,8
1:5:2 19,1 17,5 20,2 18,9 1,4
1:5:4 20,7 23,1 21,8 21,9 1,2
De acordo com a Tabela 6, não obtemos o resultado esperado, que seria o
abaixamento do potencial do complexo com a adição de nanopartículas de prata.
Podemos dizer que, de acordo com os valores de desvio padrão, os valores
de carga são iguais para as três condições do ensaio. Mais do que isso,
dependendo do valor do desvio padrão da carga do complexo pDNA:Protamina
(+24mV) poderíamos dizer que as nanopartículas não afetaram a carga deste
complexo binário.
As amostras como estão dadas confirmam os nossos cálculos de excesso de
protamina. Isso é notável, pois mesmo após a adição das nanopartículas de prata
(que são negativas) as cargas do complexo ternário continuam positivas, e
praticamente invariáveis para todas as razões mássicas utilizadas.
4.2.3. Tamanho das partículas por DLS (Dynamic Light Scattering)
O ensaio de DLS foi feito para comprovar o tamanho das partículas do
complexo ternário.
Tabela 7 - Resultados do ensaio DLS
1° população 2° população
Razão mássica
Número de pontos
Média de tamanho (nm)
Desvio padrão (nm)
Média de quantidade (%)
Número de pontos
Média de tamanho (nm)
Desvio padrão (nm)
Média de quantidade (%)
1:5:0,5 2,00 435,50 23,76 100,00 3,00 4581,00 411,25 0,00
1:5:2 3,00 845,07 182,27 100,00 3,00 4781,67 482,81 0,00
1:5:4 3,00 623,30 9,73 100,00 1,00 5560,00 0,00 0,00

56
A intensidade do sinal do DLS é inversamente proporcional ao diâmetro da
partícula elevado à 6ª potência. Como os sinais obtidos para as 2 populações
mostradas na Tabela 7 são similares (ver distribuição de tamanhos completa no
ANEXO C – LAUDOS DO DLS PARA OS COMPLEXOS TERNÁRIOS), concluímos
que a 1ª população, a das partículas menores, corresponde a quase a totalidade das
partículas.
A expectativa é que quanto maior a razão de NpAg maior o tamanho final do
complexo, pois mais partículas se agregarão nele. Excluindo a amostra de razão
1:5:2, que teve alto desvio padrão, podemos dizer que o tamanho do complexo
aumenta 50% da razão 1:5:0,5 para a 1:5:4, ou seja, com o aumento de 8 vezes das
partículas de prata.
A nossa razão de pDNA:Protamina tem um excesso de proteína, que é muito
positiva. Com isso, podemos dizer que com a adição de poucas nanopartículas de
prata, estas interagem fortemente com o excesso de proteínas e formam agregados
de partículas, que podemos evidenciar, pois aparecem dois picos no resultado.
Estes aglomerados podem ser, não apenas, resultados de interações destes dois
tipos de compostos, mas na verdade uma aglomeração de complexos binários com
AgNPs e protaminas.
Para a primeira razão mássica temos que a média de tamanho é a menor de
todas, já as outras duas, se levarmos em conta o alto desvio padrão da amostra 2,
são praticamente iguais. Essa análise pode ser completada dizendo que para a
primeira razão temos poucas AgNPs, ou seja, temos poucas chances de
aglomeração e por isso o tamanho menor. Já para as outras temos maior chance de
aglomeração pela maior quantidade de partículas de prata, mas sem distinção entre
elas.
4.3. MICROCOPIA ELETRÔNICA DE VARREDURA
O ensaio de MEV foi feito pensando em visualizar o complexo ternário, apesar
de não terem sido encontrados artigos que fazem uso deste tipo de teste para este

57
tipo de amostra. Realizamos os ensaios (ANEXO D – MICROSCOPIA ELETRÔNICA
DE VARREDURA DOS COMPLEXOS TERNÁRIOS) em duas condições, uma sem
diluição da amostra e outra com diluição da mesma.
No primeiro ensaio, as amostras foram feitas sem diluição do sal contido no
PBS, 50 mM. Com isso, as imagens não ficaram boas e o sal predomina. Podemos
constatar isso pelo aparecimento de partículas facetadas com dimensões de
aproximadamente 1000 nm, características de sal (Figura 24). As microscopias
foram realizadas para as razões 1:5:0,5 e 1:5:4, mas não houve diferença entre elas,
ou seja, elas apresentaram partículas de mesma forma e aparência.
Figura 24 - MEV para razão mássica 1:5:0,5
De acordo com o tamanho e formato das formas que aparecem na
microscopia eletrônica de varredura, observamos aglomerações, mas não sabemos

58
se são realmente partículas aglomeradas ou até que ponto houve sobreposição de
moléculas ou partículas quando as amostras foram evaporadas.
Foram observadas partículas com as seguintes características:
- partículas facetadas com dimensões de aproximadamente 1000 nm, que são
sais oriundos da solução tampão, formados pela evaporação da solução sobre o
porta-amostras.
- partículas irregulares com dimensões entre 100 a 500 nm, que
provavelmente corresponde ao tamanho do complexo DNA- proteína (Figura 51 do
ANEXO D).
- partículas irregulares com dimensões variáveis, possivelmente aglomerados
de complexos DNA-proteína.
Possivelmente, parte das AgNPs encontram-se aderidas à superfíce dos
complexos DNA-proteína, mas este fenômeno não pode ser observado devido ao
pequeno aumento.
Por outro lado, algumas partículas parecem ter o tamanho e a aparência de
complexos, até mesmos com as nanopartículas grudadas. Mas mais uma vez não
podemos dizer se eles já estavam formados em solução ou se foram sobrepostos na
evaporação da água.
No segundo ensaio, utilizamos as razões 1:5:0, 1:5:0,5 (por exemplo na
Figura 25) e 1:5:4. Além disso, fizemos a diluição dessas amostras para 50 vezes,
em volume, com água destilada para que o sal não atrapalhe.
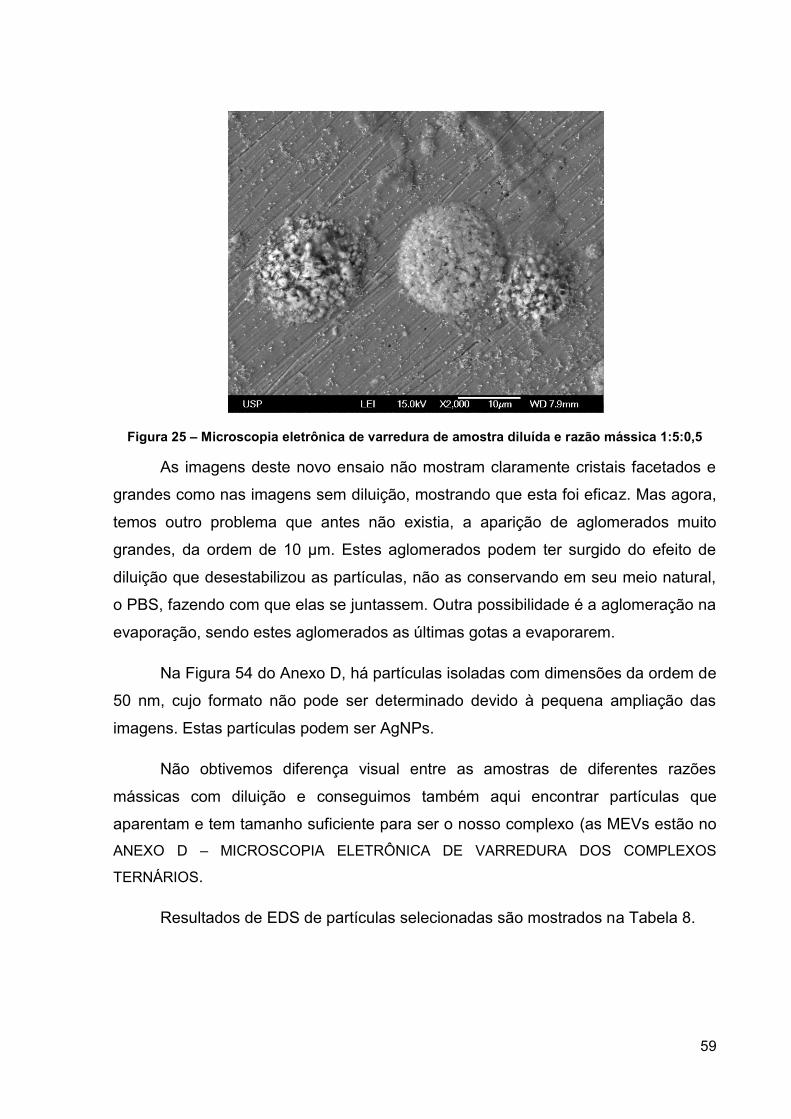
59
Figura 25 – Microscopia eletrônica de varredura de amostra diluída e razão mássica 1:5:0,5
As imagens deste novo ensaio não mostram claramente cristais facetados e
grandes como nas imagens sem diluição, mostrando que esta foi eficaz. Mas agora,
temos outro problema que antes não existia, a aparição de aglomerados muito
grandes, da ordem de 10 µm. Estes aglomerados podem ter surgido do efeito de
diluição que desestabilizou as partículas, não as conservando em seu meio natural,
o PBS, fazendo com que elas se juntassem. Outra possibilidade é a aglomeração na
evaporação, sendo estes aglomerados as últimas gotas a evaporarem.
Na Figura 54 do Anexo D, há partículas isoladas com dimensões da ordem de
50 nm, cujo formato não pode ser determinado devido à pequena ampliação das
imagens. Estas partículas podem ser AgNPs.
Não obtivemos diferença visual entre as amostras de diferentes razões
mássicas com diluição e conseguimos também aqui encontrar partículas que
aparentam e tem tamanho suficiente para ser o nosso complexo (as MEVs estão no
ANEXO D – MICROSCOPIA ELETRÔNICA DE VARREDURA DOS COMPLEXOS
TERNÁRIOS.
Resultados de EDS de partículas selecionadas são mostrados na Tabela 8.

60
Tabela 8 - Ensaio de EDS das amostras de MEV com diluição
% Atômica
Razão mássica
Ponto Figura Carbono Oxigênio Fósforo Cloro Chumbo Berílio
1:5:0 1 Figura 58 45.64 51.21 2.97 0.17
1:5:0 2 Figura 58 47.90 49.31 1.81 0.98
1:5:0 1 Figura 59 39.49 50.56 0.94 1.97 7.03
1:5:0,5 1 Figura 60 43.74 52.96 3.25 0.04
1:5:0,5 1 Figura 61 39.02 43.19 2.31 5.29 10.20
1:5:0,5 1 Figura 62 46.98 48.47 3.38 1.17
1:5:0,5 2 Figura 62 46.30 49.85 3.85
1:5:4 1 Figura 63 47.31 49.76 2.92
1:5:4 2 Figura 63 51.84 45.99 2.17
1:5:4 3 Figura 63 46.28 49.55 4.17
1:5:4 1 Figura 64 54.44 37.17 1.44 6.95
1:5:4 1 Figura 65 47.49 9.31 1.01 42.19
1:5:4 2 Figura 65 46.98 50.62 2.28 0.11
1:5:4 3 Figura 65 49.89 47.86 2.26
1:5:4 4 Figura 65 54.29 45.71
1:5:4 5 Figura 65 44.42 55.14 0.43
A nossa amostra contém os componentes que foram adicionados na solução
de AgNPs (que contém citrato, sódio, nitrato e prata), o tampão PBS (sódio, fosfato,
cloreto), o pDNA, a Protamina, menos a água destilada, que foi evaporada.
Estes componentes fazem a nossa amostra ser rica em carbono, oxigênio,
nitrogênio e fósforo. Alguns componentes de menor quantidade são a prata, cloreto
e sódio.
A tabela acima vem quantificar as nossas amostras e confirmar algumas de
nossas suposições. Realmente a quantidade de carbono e oxigênio são maioria,
com isso, podemos dizer que nessas partículas contém Protamina ou pDNA. Uma
surpresa foi a não detecção de nitrogênio, muito presente nas duas substâncias,
apesar da menor quantidade.
O fósforo encontrado pode ter vindo tanto do pDNA como do tampão PBS,
com isso não podemos afirmar a procedência exata delas. O cloro encontrado em
pequena quantidade também veio do tampão, cristalizado pela evaporação da água
na preparação da amostra.

61
Podemos notar o aparecimento de picos de chumbo, que teoricamente não
teria de onde sair, mas uma solução pensada para este pico é a contaminação do
porta amostra de latão com chumbo. O único pico de berílio encontrado também é
estranho e não tem procedência e nem explicação para a sua aparição.
A maior surpresa deste teste foi o não aparecimento de prata em nenhuma
das imagens. A explicação para isso é a pequena quantidade de prata colocada
frente aos outros componentes.
4.4. TRANSFECÇÃO CELULAR
O ensaio final e mais importante deste trabalho é a transfecção celular. Ele
nos dirá se as nanopartículas de prata tem algum efeito sobre a entrada de genes
nas células, comparativamente com amostras sem prata. O resultado da transfecção
é indicado pela quantificação da atividade da enzima repórter luciferase no extrato
celular 24 horas após a transfecção. O gene que codifica a luciferase só expressa
quando o pDNA chega ao núcleo das células, sendo um indicativo da eficiência da
transfecção.
As amostras estão dispostas como na Figura 26. São seis amostras, células
somente, células com pDNA, células com pDNA:Protamina, células com complexo
ternário de razões mássicas 1:5:0,5; 1:5:2 e 1:5:4.
Estes resultados são preliminares, não tendo sido ainda normalizados pela
concentração de proteína total nos extratos celulares. Precisamos de um valor que
nos quantifique o número de células que continuaram vivas em cada ensaio para
dividir os resultados por este valor. Apesar de visualmente não serem constatadas
células mortas, a prata pode ter algum efeito tóxico para as células, o que não é do
escopo do trabalho, e aumentaria os nossos valores para as amostras com prata.
Podemos constatar claramente que as amostras que contem AgNPs são mais
eficientes na transfecção de genes para o interior dos núcleos das células. Os
valores chegam a ser 100 vezes mais eficientes que a amostra que possui células

62
com o complexo binário pDNA:Protamina e 1000 vezes melhor que a amostra de
células com pDNA.
Figura 26 - Resultados de transfecção celular, nas diferentes razões mássicas de pDNA:protamina:NpAg estudas, utilizando-se células HeLa cultivadas in vitro.
As amostras que possuem AgNPs não podem ser diferenciadas entre si em
relação à eficiência da entrega gênica. A barra de erros mostrada no gráfico nos
mostra isso, e os resultados se equivalem. Se tivéssemos mais amostras
poderíamos melhorar os resultados e tirar alguma conclusão.
Essa equivalência pode ser explicada pela pequena quantidade de prata nas
três amostras, fazendo com que elas causem o mesmo efeito. Outra possível
explicação pode vir do fato de a prata produzir algum mecanismo, desconhecido por
nós, de entrada do complexo nas células não importando a quantidade adicionada.
A variável de tamanho pode ser relacionada aqui de maneira fácil, mas
contraditória. Como os valores de entrega gênica são iguais entre as 3 amostras
com AgNPs e o tamanho médio das partículas é menor para a amostra de razão
1:5:0,5, temos uma contradição, pois teoricamente as menores partículas
1,55E+02 3,21E+02 3,09E+03
1,53E+05 1,76E+05
2,84E+05
-5,00E+04
0,00E+00
5,00E+04
1,00E+05
1,50E+05
2,00E+05
2,50E+05
3,00E+05
3,50E+05
4,00E+05
Un
idad
es
de
em
issã
o d
e lu
z
Transfecção
Transfecção Celular

63
transfectam melhor. Isso pode ser uma indicação de que quanto mais prata melhor a
entrega dos genes, pois as razões mássicas maiores de prata tem maior tamanho
(dificultador de transfecção).

64
5. CONCLUSÃO
Foram caracterizados, preliminarmente, alguns complexos ternários de
acordo com sua carga, tamanho e eficiência de transfecção, alem da tentativa de
visualização através de microscopia eletrônica de varredura.
A formação dos complexos binários foi confirmada pelo ensaio de
eletroforese. Estes ensaios também são consistentes com complexos
pDNA:proteína:AgNPs, muito embora eles não permitam diferenciar a adesão das
AgNPs nos complexos.
As MEVs permitiram visualizar partículas dos complexos, mas sem distinguir
entre partículas isoladas e sobrepostas.
O EDS é um ensaio considerado interessante, pois pode quantificar as
composições de alguns pontos de interesse na imagem de MEV.
O ensaio de potencial Zeta mostrou que as AgNPs são carregadas
negativamente e que os complexos ternários têm carga positiva. Já o DLS nos
fornece tamanhos diferentes destas partículas, sendo as razões mássicas de 1:5:2
(830 nm ± 180) e 1:5:4 (630 nm ± 10) sendo praticamente iguais, devido ao grande
desvio padrão e pequena quantidade de amostras e, pelo menos, 1,5 vezes maior
que a 1:5:0,5 (430 nm ± 20). Este ensaio pode indicar a formação de agregados,
explicando o aumento de tamanho.
A transfecção dos complexos foi realizada e foi constatado um aumento na
sua eficiência para as amostras que continham as AgNPs. Esta eficácia não teve
variação entre as diferentes razões mássicas de nanopartículas de prata.
A partir dos resultados acima, pode-se resumir os achados deste trabalho.
Complexos pDNA:proteína, com dimensões de cerca de 500 nm e carga positiva,
quando associados em proporções adequadas a AgNPs com dimensões de cerca
de 50 nm e carga negativa, formam partículas de 500 nm e carga positiva. Os
complexos triplos apresentam eficiência de transfecção muito superior aos
complexos sem AgNPs, cerca de cem vezes maior. Estes resultados preliminares
revelam uma rota promissora para entrega gênica baseada em AgNps.

65
6. SUGESTÃO PARA FUTUROS TRABALHOS
Como já foi dito, este trabalho encontrou muitas dificuldades e a maior delas
foi a falta de conhecimento na área. Com isso, nos preocupamos neste item em dar
algumas diretrizes para os trabalhos que futuramente poderão surgir.
1. Avaliar dados de conversão para as nanopartículas de prata, a fim de ter as
razões mássicas calculadas de forma correta;
2. Avaliar a eficiência da transfecção para diferentes tamanhos de AgNPs
acoplada;
3. Utilizar menor razão mássica pDNA:Protamina, preferencialmente menor que
1:1;
4. Usar maior quantidade de AgNPs na construção dos complexos para tentar
ver o ponto de viragem de carga, de positivo para negativo;
5. Avaliar a estabilidade tanto das AgNPs quanto dos complexos, para ver se
eles são estáveis e até quando são.
6. Tentar usar algum outro método de visualização das partículas, que não a
MEV;
7. Normalizar os dados de transfecção para obter os valores corretos e
utilizados no mundo acadêmico.

66
REFERÊNCIAS
Adler, Andrew F. and Leong, Kam W. 2010. Emerging links between surface
nanotechnology and endocytosis: Impact on nonviral gene delivery. Nano Today.
2010, Vol. 5, pp. 553-569.
Al-Dosari, Mohammed S. and Gao, Xiang. 2009. Nonviral Gene Delivery: Principle,
Limitations, and Recent Progress. The AAPS Journal. 2009, Vol. 11, 4, pp. 671-681.
Azzoni, A. R., et al. 2007. The impact of polyadenylation signals on plasmid
nuclease-resistance and transgene expression. The Journal of Gene Medicine. 2007,
Vol. 9, 5, pp. 392-402.
Cartier, Régis and Reszka, Regina. 2002. Biological and Cellular Barriers Limiting
the Clinical Application of Nonviral Gene Delivery Systems. Gene Therapy. 2002,
Vol. 9, 3, pp. 157–167.
Compri-Nardy, M., Stella, M. B. and Oliveira, C. 2009. Práticas de Laboratório de
Bioquímica e Biofísica. s.l. : Guanabara Koogan, 2009.
DeLong, Robert K., et al. 2009. Characterization and performance of nucleic acid
nanoparticles combined with protamine and gold. Biomaterials. 2009, Vol. 30, pp.
6451-6459.
Epple, Matthias and Sokolova, Viktoriya. 2008. Inorganic Nanoparticles as
Carriers of Nucleic Acids into Cells. Angewandte Chemie International Edition. 2008,
Vol. 47, pp. 1382-1395.
Instruments, Malvern. Dynamic Light Scattering: An Introduction in 30 Minutes
(Technical note).

67
Lee, P. C. and Meisel, D. 1982. Adsorption and surface-enhanced Raman of dyes
on silver and gold sols. The Journal of Physical Chemistry. 1982, Vol. 86, 17, pp.
3391–3395.
Lok, Chun-Nam, et al. 2007. Silver nanoparticles: partial oxidation and antibacterial
activities. 2007.
Martinez-Castanon, G. A., et al. 2008. Synthesis and antibacterial activity of silver
nanoparticles with different sizes. J. Nanopart. Res. 2008, Vol. 10, pp. 1343-1348.
Mastrobattista, Enrico, et al. 2005. Nonviral gene delivery systems: From simple
transfection agents to artificial viruses. Drug Discovery Today: Technologies. 2005,
Vol. 2, 1, pp. 103-109.
Niemeyer, Christof M. and Mirkin, Chad A. 2004. Nanobiotechnology: concepts,
applications and perspectives. s.l. : Wiley-VCH, 2004.
, Al š, t l. 2006. Silver Colloid Nanoparticles: Synthesis,
Characterization, and Their Antibacterial Activity. The Journal of Physical Chemistry
B. 2006, Vol. 110, 33, pp. 16248-16253.
Park, Hyun-Ji, Yang, Fan and Cho, Seung-Woo. 2011. Nonviral delivery of genetic
medicine for therapeutic angiogenesis. Advanced Drug Delivery Reviews. 2011, Vol.
Disponível online.
Parveen, Suphiya, Misra, Ranjita and Sahoo, Sanjeeb K. 2011. Nanoparticles: a
boon to drug delivery, therapeutics, diagnostic and imaging. Nanomedicine:
Nanotechnology, Biology, and Medicine. 2011, Vol. Disponível online.
Santana, Henrique de, et al. Preparação e caracterização de substratos SERS
ativos: um estudo da adsorção do cristal violeta sobre nanopartículas de prata.

68
Santos, Marcia R., Tavares, Marina F. M. and Rubim, Joel C. 2000.
Implementação de um Sistema de Eletroforese Capilar com Detecção de
Fluorescência Induzida por Lase. Química Nova. 2000, Vol. 23, 5, pp. 585-589.
Shim, Min Suk and Kwon, Young Jik. 2010. Acid-transforming polypeptide micelles
for targeted nonviral gene delivery. Biomaterials. 2010, Vol. 31, pp. 3404–3413.
Smith, Louis C., et al. 1998. Synthetic peptide-based DNA complexes for nonviral
gene delivery. Advanced Drug Delivery Reviews. 1998, Vol. 30, pp. 115-131.
Turkevich, John, Stevenson, Peter Cooper and Hillier, James. 1951. A study of
the nucleation and growth processes in the synthesis of colloidal gold. Discussions of
the Faraday Society. 1951, Vol. 11, pp. 55-75.
Wagner, Ernst. 1999. Application of membrane-active peptides for nonviral gene
delivery. Advanced Drug Delivery Reviews. 1999, Vol. 38, 3.
Wan, Hongshui, et al. 2005. Preparation of silver nanoparticles by chemical
reduction method. 2005.
Yan, Ya, Kang, Shi-Zhao and Mu, Jin. 2006. Preparation of high quality Ag film
from Ag nanoparticles. 2006.
Yu, Da-Guang, Lin, Wen-Ching and Yang, Ming-Chien. 2007. Surface Modification
of Poly(L-lactic acid) Membrane via Layer-by-Layer Assembly of Silver Nanoparticle-
Embedded Polyelectrolyte Multilayer. Bioconjugate Chemistry. 2007, Vol. 18, 5, pp.
1521-1529.
Zeta-Meter, Inc. Zeta Potential: A Complete Course in Five Minutes.

69
ANEXOS
ANEXO A - LAUDOS DO ZETA DAS NANOPARTÍCULAS
Em pH = 7,4
Figura 27 - Laudo 1 do Zeta para nanopartículas em pH = 7,4
Figura 28 - Laudo 2 do Zeta para nanopartículas em pH = 7,4

70
Figura 29 - Laudo 3 do Zeta para nanopartículas em pH = 7,4
Em pH = 9
Figura 30 - Laudo 1 do Zeta para nanopartículas em pH = 9
71
Figura 31 - Laudo 2 do Zeta para nanopartículas em pH = 9
Figura 32 - Laudo 3 do Zeta para nanopartículas em pH = 9

72
ANEXO B – LAUDOS DO ZETA PARA OS COMPLEXOS
TERNÁRIOS
Razão 1:5:0,5
Figura 33 - Laudo 1 do Zeta para complexos ternários razão 1:5:0,5
Figura 34 - Laudo 2 do Zeta para complexos ternários razão 1:5:0,5

73
Figura 35 - Laudo 3 do Zeta para complexos ternários razão 1:5:0,5
Razão 1:5:2
Figura 36 - Laudo 1 do Zeta para complexos ternários razão 1:5:2
74
Figura 37 - Laudo 2 do Zeta para complexos ternários razão 1:5:2
Figura 38 - Laudo 3 do Zeta para complexos ternários razão 1:5:2

75
Razão 1:5:4
Figura 39 - Laudo 1 do Zeta para complexos ternários razão 1:5:4
Figura 40 - Laudo 2 do Zeta para complexos ternários razão 1:5:4

76
Figura 41 - Laudo 3 do Zeta para complexos ternários razão 1:5:4

77
ANEXO C – LAUDOS DO DLS PARA OS COMPLEXOS
TERNÁRIOS
Razão 1:5:0,5
Figura 42 - Laudo 1 do DLS para complexos ternários razão 1:5:0,5
Figura 43 - Laudo 2 do DLS para complexos ternários razão 1:5:0,5

78
Figura 44 - Laudo 3 do DLS para complexos ternários razão 1:5:0,5
Razão 1:5:2
Figura 45 - Laudo 1 do DLS para complexos ternários razão 1:5:2

79
Figura 46 - Laudo 2 do DLS para complexos ternários razão 1:5:2
Figura 47 - Laudo 3 do DLS para complexos ternários razão 1:5:2

80
Razão 1:5:4
Figura 48 - Laudo 1 do DLS para complexos ternários razão 1:5:4
Figura 49 - Laudo 2 do DLS para complexos ternários razão 1:5:4

81
Figura 50 - Laudo 3 do DLS para complexos ternários razão 1:5:4

82
ANEXO D – MICROSCOPIA ELETRÔNICA DE VARREDURA DOS
COMPLEXOS TERNÁRIOS
Amostras sem diluição
Figura 51 - MEV para razão mássica 1:5:0,5 aumentada

83
Figura 52 - MEV para razão mássica 1:5:4
Figura 53 - MEV para razão mássica 1:5:4 aumentada

84
Amostras com diluição
Figura 54 – Microscopia eletrônica de varredura de amostra diluída e razão mássica 1:5:0
Figura 55 – Microscopia eletrônica de varredura de amostra diluída e razão mássica 1:5:0,5 com zoom

85
Figura 56 – Microscopia eletrônica de varredura de amostra diluída e razão mássica 1:5:4 com zoom
Figura 57 – Microscopia eletrônica de varredura de amostra diluída e razão mássica 1:5:4
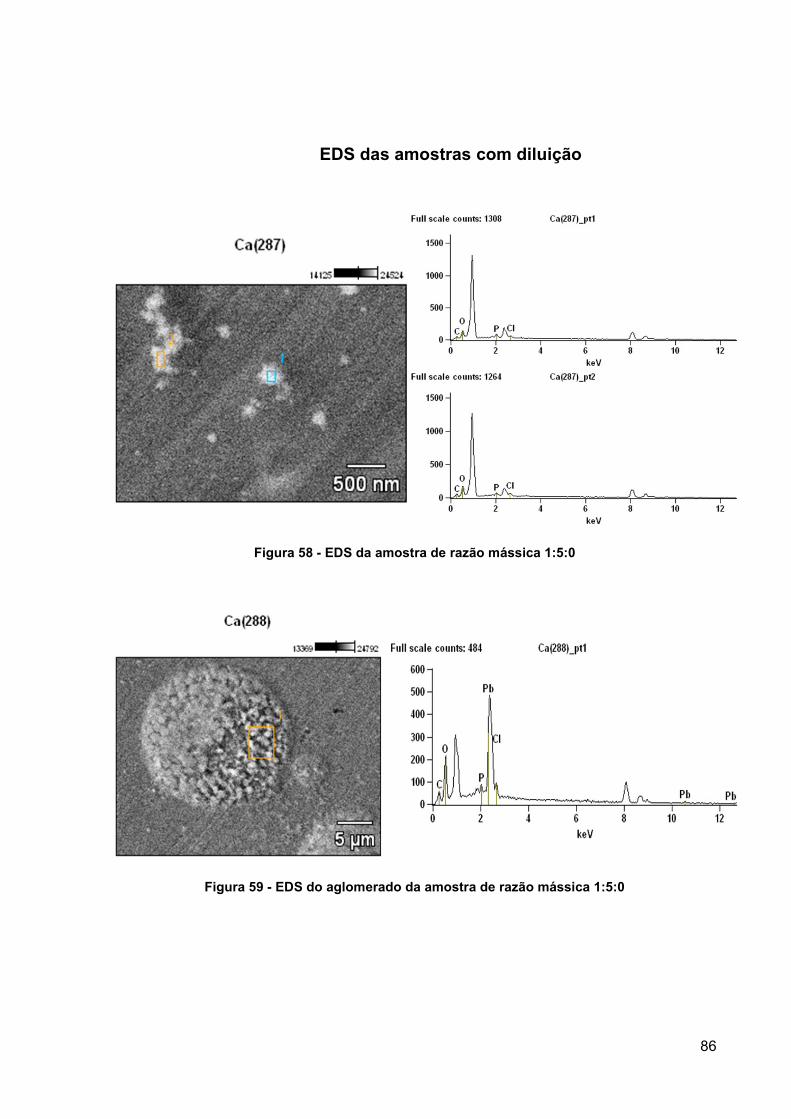
86
EDS das amostras com diluição
Figura 58 - EDS da amostra de razão mássica 1:5:0
Figura 59 - EDS do aglomerado da amostra de razão mássica 1:5:0

87
Figura 60 - EDS 1 da amostra de razão mássica 1:5:0,5
Figura 61 - EDS 2 da amostra de razão mássica 1:5:0,5
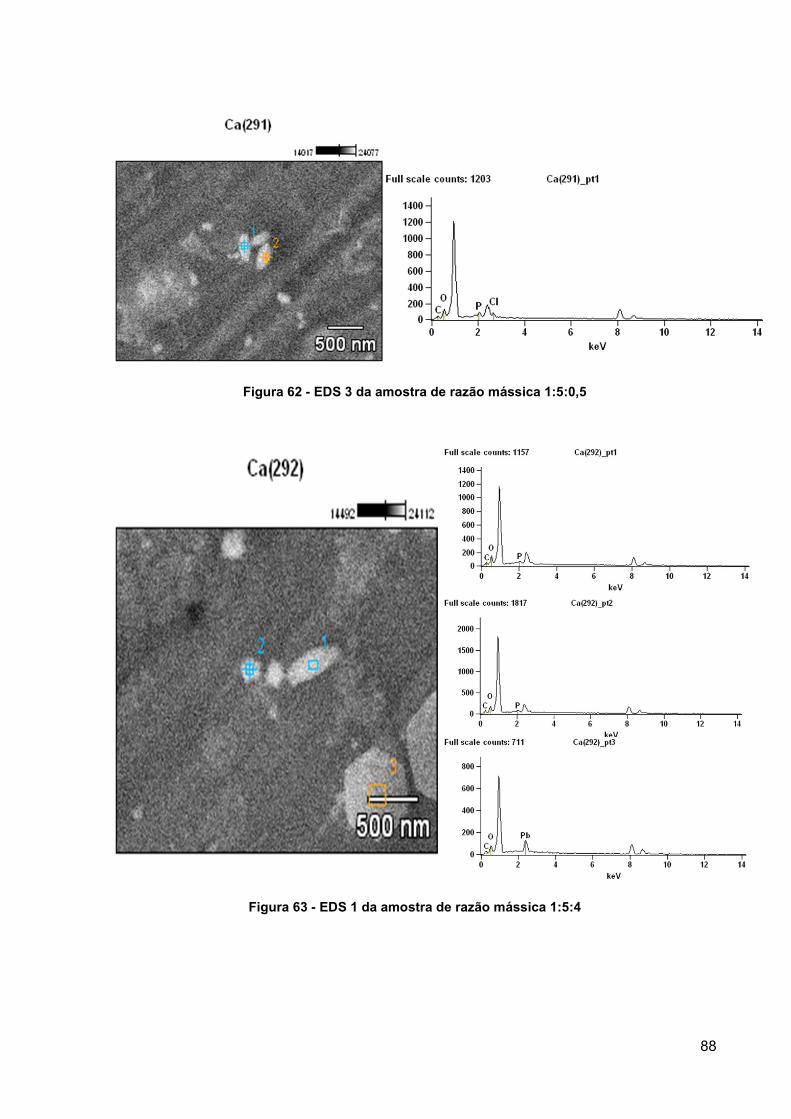
88
Figura 62 - EDS 3 da amostra de razão mássica 1:5:0,5
Figura 63 - EDS 1 da amostra de razão mássica 1:5:4

89
Figura 64 - EDS 2 da amostra de razão mássica 1:5:4
Figura 65 - EDS 3 da amostra de razão mássica 1:5:4





























