Embed Size (px)
Citation preview

Instituto Politécnico de Coimbra
Instituto Superior de Engenharia de Coimbra
Departamento de Engenharia Informática e de Sistemas
Mestrado em Informática e SistemasEstágio/Projecto Industrial
Relatório Final
Sistema Inteligente para Extracção deSistema Inteligente para Extracção de
Características e Classificação deCaracterísticas e Classificação de
ProteínasProteínas
Francisco Carlos Gaspar Branco Caridade
Orientador
Professor Doutor Carlos Pereira
ISEC – Departamento de Engenharia Informática e de Sistemas
Coimbra, Dezembro de 2010


The Computer is incredibly fast, accurate and
stupid. Man is unbelievably slow, inaccurate and
brilliant. The marriage of the two is a challenge
and an opportunity beyond imagination.
Walesh, 1989
3

4

Agradecimentos
Embora esta dissertação seja, pela sua �nalidade académica, um trabalho individual, foi
no entanto o culminar de um ciclo do Mestrado em Informática e Sistemas. Naturalmente
que este trabalho em muito se deve ao mérito do meu orientador, no entanto foi também
resultado de todo um conjunto de vivências e conhecimentos adquiridos ao longo dos anos
e principalmente no decorrer deste mestrado.
Quero por isso agradecer em primeiro lugar ao meu orientador Professor Doutor Carlos
Pereira, pedra angular neste trabalho, pelos seus conhecimentos, sugestões e disponibilidade
que sempre demonstrou ao longo deste período, que de outra forma não teria sido atingido.
Agradeço igualmente a todo o corpo docente do departamento de Informática e de Sistemas
do Instituto Superior de Engenharia de Coimbra.
Quero também agradecer ao Departamento de Engenharia Informática e de Sistemas, do Ins-
tituto Superior de Engenharia de Coimbra e ao bioink Project, pelos recursos disponibilizados
para a execução deste trabalho.
Agradeço também aos meus colegas, que nos momentos mais difíceis souberam sempre dar
uma palavra de apoio.
Por último, agradeço a toda a minha família pela motivação e carinho, com que sempre
me acompanharam, com especial destaque para os meus �lhos e para a minha esposa, pela
compreensão que sempre demonstraram nos momentos mais difíceis.
5

6

Resumo
O interesse na classi�cação de proteínas emergiu como resultado do aumento exponencial
de sequências conhecidas não acompanhando o conhecimento da estrutura da proteína. As
abordagens experimentais para a determinação da estrutura são morosas e dispendiosas.
Consequentemente, a utilização de classi�cadores inteligentes de proteínas, tornou-se num
dos problemas mais relevantes na área da biologia computacional.
Este trabalho resulta acima de tudo de um processo de investigação, tendo por base todo
um conhecimento produzido por inúmeras pessoas, de forma a dar mais um contributo na
área.
Desenvolvemos uma nova metodologia para a extracção de características, combinando
técnicas de extracção ao nível das propriedades físico-químicas das proteínas e técnicas de
Text-Mining com segmentação de palavras, que posteriormente foi integrado no classi�cador.
Os classi�cadores implementados tiveram por base máquinas de vectores de suporte e redes
neuronais.
Como resultado, esta metodologia foi implementada numa ferramenta computacional desig-
nada por Intelligent System for Feature Extraction and Classi�cation of Proteins - ISFECP.
Diferentes técnicas e combinações foram testadas durante a fase de estudo, sempre com
o objectivo de desenvolver o classi�cador com melhor desempenho. Os resultados obtidos
evidenciaram um claro aumento de desempenho, com um menor menor número de caracte-
rísticas, quando comparado com resultados de referência.
7

8

Abstract
The interest in proteins classi�cation has emerged as the result of the exponential increase
of known sequences that are not accompanied by knowledge of protein structure. The
experimental approaches for determining the structure are slow and expensive. In this sense,
the use of automatic classi�ers, has become one of the most important problems in the area
of computational biology.
This thesis results from a research work, based on all the knowledge produced by numerous
people, in order to give a further contribution in the area.
We developed a new methodology of features extraction, combining techniques of extraction
at the level of physical and chemical properties of proteins, Text-Mining techniques with word
segmentation, which was later integrated in each classi�er. The classi�ers were implemented
based on support vector machines and neural networks.
As result of the study, the methodology was implemented in a computational tool called
Intelligent System for Feature Extraction and Classi�cation of Proteins - ISFECP.
Di�erent techniques and combinations were tested during the experimental phase, always
with the aim of developing the classi�er with best performance. The results showed a clear
increase in performance, with less computational e�ort when compared to benchmark results.
9

10

Índice
Agradecimentos 5
Resumo 7
Abstract 9
1 Introdução 23
1.1 Motivação . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
1.2 Objectivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
1.3 Estrutura do relatório . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2 Conceitos Básicos 25
2.1 DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.2 Aminoácido . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.3 Proteínas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
2.4 Ponto isoeléctrico (pI) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
11

2.5 Cristalogra�a de raios X . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
2.6 Ressonância magnética nuclear . . . . . . . . . . . . . . . . . . . . . . . . 29
2.7 Formato Fasta . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.8 Blast . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.9 Exactidão global (Q3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.10 Matriz de confusão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.11 Taxa de verdadeiros positivos ou sensibilidade . . . . . . . . . . . . . . . . 33
2.12 Taxa de falsos positivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
2.13 Especi�cidade . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
2.14 Taxa de acerto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
2.15 Análise ROC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3 O longo caminho da sequência à sua estrutura 37
3.1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.2 A importância do conhecimento da estrutura . . . . . . . . . . . . . . . . . 38
4 Classi�cação de Proteinas 43
4.1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
4.2 A importância da previsão . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
5 Da informação ao conhecimento 47
5.1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
5.2 Classi�cadores em máquinas de aprendizagem . . . . . . . . . . . . . . . . 48
5.3 Alguns algoritmos em máquinas de aprendizagem . . . . . . . . . . . . . . 49
5.3.1 Redes neuronais . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
5.3.2 Máquinas de vectores de suporte (MVS) . . . . . . . . . . . . . . . 54
12

6 Dados usados na fase de aprendizagem do classi�cador 59
6.1 Extracção de características na previsão da estrutura das proteínas . . . . . 59
6.2 Selecção de características . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
7 Estado da arte 63
8 Metodologia 67
8.1 Leitura das sequências . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
8.2 Leitura conjunto de treino/teste . . . . . . . . . . . . . . . . . . . . . . . . 68
8.3 Extracção de características a partir da sequência . . . . . . . . . . . . . . 69
8.3.1 Contagem aminoácidos, Composição atómica, Peso molecular . . . . 70
8.3.2 Ponto isoeléctrico . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
8.3.3 Dicionário de Tuplos. . . . . . . . . . . . . . . . . . . . . . . . . . 71
8.3.3.1 Possíveis critérios de escolha dos tuplos: . . . . . . . . . . 71
8.3.3.2 Construção do dicionário. . . . . . . . . . . . . . . . . . . 73
8.3.3.3 Construção do vector de características. . . . . . . . . . . 73
8.4 Selecção de características . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
8.5 Treino do método de máquina de aprendizagem . . . . . . . . . . . . . . . 75
8.6 Execução do método da máquina de aprendizagem . . . . . . . . . . . . . . 77
8.7 Aplicação computacional . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
9 Bases de dados utilizadas 79
10 Resultados e sua análise 83
10.1 Validação dos resultados. . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
10.2 Desempenho individual em função do critério de extracção de características. 85
13

10.3 Desempenho dos tuplos de ordem K. . . . . . . . . . . . . . . . . . . . . . 87
10.4 Desempenho dos tuplos de ordem 4 em função do número de características
extraídas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
10.5 Desempenho dos tuplos de ordem 4 em função do critério de selecção na
construção do dicionário . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
10.6 Desempenho em função das características seleccionadas. . . . . . . . . . . 91
10.7 Desempenho adicionando as possíveis combinações das características selec-
cionadas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
10.8 Desempenho da máquina de vectores de suporte para as características se-
leccionadas como as melhores. . . . . . . . . . . . . . . . . . . . . . . . . 94
10.9 Desempenho do melhor classi�cador com bases de dados de maior dimensão,
usando redes neuronais. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
11 Análise critica e trabalhos futuros 97
A Manual de utilizador 99
A.1 Caracterização . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
A.2 Entidades envolvidas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
A.3 Procedimento para a execução . . . . . . . . . . . . . . . . . . . . . . . . 100
A.4 Con�guração da aplicação . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
A.5 Con�guração da extracção de características . . . . . . . . . . . . . . . . . 101
A.6 Con�guração da rede neuronal . . . . . . . . . . . . . . . . . . . . . . . . 102
A.7 Con�guração da máquina de vectores de suporte . . . . . . . . . . . . . . . 104
A.8 Con�guração do classi�cador . . . . . . . . . . . . . . . . . . . . . . . . . 106
A.9 Con�guração do algoritmo de classi�cação . . . . . . . . . . . . . . . . . . 106
A.10 Execução da aplicação . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
A.11 Manual de utilizador . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
14

15
Lista de referências 113

16

Índice de �guras
2.1 Estrutura geral de um aminoácido [34]. . . . . . . . . . . . . . . . . . . . . 26
2.2 Estrutura 3D da mioglobina, proteína globular de 153 aminoácidos, criada a
partir PDB SimpleView (pdb id: 2JHO) [31]. . . . . . . . . . . . . . . . . . 28
2.3 Determinação de uma estrutura por cristalogra�a de raios X [35]. . . . . . . 29
2.4 Carga de amostra no espectrómetro RMN de alto campo magnético (800
MHz) do Paci�c Northwest National Laboratory [36]. . . . . . . . . . . . . 30
2.5 Exemplo de uma sequência em formato Fasta. . . . . . . . . . . . . . . . . 31
2.6 Estrutura da matriz confusão. . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.7 Exemplo de um grá�co ROC. . . . . . . . . . . . . . . . . . . . . . . . . . 34
2.8 Área abaixo das curvas ROC A e B. Em média o classi�cador B é melhor
que A, porém em alguns pontos A tem melhor desempenho que B. . . . . . 36
3.1 Diferentes representações da estrutura da proteína . . . . . . . . . . . . . 38
3.2 A insulina (à esquerda) é uma hormona. O factor VIII (ao centro) é uma
proteína que promove a coagulação do sangue. O factor p21Ras (à direita)
é uma proteína de sinalização (signaling). . . . . . . . . . . . . . . . . . . 39
17

18 ÍNDICE DE FIGURAS
3.3 Fibras de amilóide resultantes da interacção entre proteínas cuja dobragem
é "defeituosa". . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.4 Forma normal do prião PrPc. . . . . . . . . . . . . . . . . . . . . . . . . 40
3.5 O gene PrP contém informação para expressar a proteína inócua PrPc.
Uma mutação pontual neste gene dá origem à forma infecciosa PrPSc da
proteína. Por intermédio de uma terceira proteína, não representada na
�gura, a PrPSc induz uma alteração conformacional na qual uma α-hélice
da PrPc é convertida numa folha β transformando-a numa molécula da sua
própria espécie. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
4.1 Instantâneos da simulação por dinâmica molecular da dobragem da villin
headpiece levada a cabo por Duan e Kollman em 1998. . . . . . . . . . . . 45
4.2 Blue gene. Um super-computador criado pela IBM para estudar a dobragem
de proteínas por dinâmica molecular. . . . . . . . . . . . . . . . . . . . . . 46
5.1 Classi�cação da informação em 3 grupos (clusters). . . . . . . . . . . . . . 48
5.2 Imagem real, apresentando interconexões entre os neurónios. . . . . . . . . 50
5.3 Neurónio arti�cial de um perceptrão. . . . . . . . . . . . . . . . . . . . . . 51
5.4 Exemplos linearmente separáveis (esquerda), exemplos não linearmente se-
paráveis (direita). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
5.5 Topologia de uma rede com uma camada de entrada, uma de saída e uma
intermédia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
5.6 Possíveis rectas de separação. . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.7 Nas MVS, as fronteiras encontram-se de�nidas pelos dados (recta a azul).
A recta verde constitui a linha que garante a maior distância entre as classe,
(representadas por bolas e quadrados). . . . . . . . . . . . . . . . . . . . . 56
5.8 Etapas do algoritmo MVS. . . . . . . . . . . . . . . . . . . . . . . . . . . 58
8.1 Excerto do �cheiro SCOP40 Minidatabase em formato Fasta. . . . . . . . 68

ÍNDICE DE FIGURAS 19
8.2 Excerto do �cheiro SCOP40mini−sequenceminidatabase19.cast que repre-
senta a matriz Cast Matrix. . . . . . . . . . . . . . . . . . . . . . . . . . . 69
8.3 Metodologia seguida para a extracção de características usando tuplos de
ordem k. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
8.4 Écran principal da aplicação. Imagem composta a partir do site PDB [2]. . . 78
9.1 Resultados para SCOP40 Minidatabase (PCB00019) [4]. . . . . . . . . . . 80
9.2 Resumo do SCOP40 Minidatabase (PCB00019), em termos de per�l [4]. . 80
9.3 Excerto das primeiras 5 proteínas, com per�l detalhado do SCOP40 Mini-
database (PCB00019) para cada classe (55) [4]. . . . . . . . . . . . . . . . 80
9.4 Resultados para SCOP95−Superfamily−Family (PCB00001) [4]. . . . . . . 81
9.5 Resumo do SCOP95−Superfamily−Family (PCB00001), em termos de per-
�l [4]. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
9.6 Excerto das primeiras 5 proteínas, com per�l detalhado do SCOP95 Super-
family Family (PCB00001) para cada classe (246) [4]. . . . . . . . . . . . . 81
10.1 Avaliação de desempenho individual em função do critério de extracção de
características. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
10.2 Avaliação de desempenho dos tuplos de ordem k. . . . . . . . . . . . . . . 88
10.3 Avaliação de desempenho do tuplos de ordem 4 em função do número de
características extraídas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
10.4 Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção na construção do dicionário. . . . . . . . . . . . . . . . . . . . . . 90
10.5 Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção e das características seleccionadas. . . . . . . . . . . . . . . . . . 91
10.6 Avaliação de desempenho, adicionando possíveis combinações de caracterís-
ticas seleccionadas em relação ao melhor resultado obtido a partir do SFS
em 10.6. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
A.1 Écran principal da aplicação. Imagem composta a partir do site PDB [2]. . . 100

20 ÍNDICE DE FIGURAS
A.2 Écran onde são de�nidos os parâmetros usados ao nível da extracção de
características. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
A.3 Écran onde são de�nidos os parâmetros usados pelo algoritmo da rede neuronal.104
A.4 Écran onde são de�nidos os parâmetros usados pela máquina de vectores de
suporte. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
A.5 Con�guração do algoritmo de classi�cação a usar durante a fase de treino
do classi�cador. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
A.6 Selecção do �cheiro de sequências a classi�car, com a extensão .fasta. . . . 108
A.7 Selecção do �cheiro de treino e teste, com a extensão .cast. . . . . . . . . . 109
A.8 Apresenta as várias opções de execução por parte da aplicação. . . . . . . . 110
A.9 Apresenta o último algoritmo de classi�cação e o nome dos �cheiros sobre o
qual foi feito a última extracção de características. . . . . . . . . . . . . . . 110
A.10 Indica que um processo está a decorrer. . . . . . . . . . . . . . . . . . . . 111
A.11 Indica que um processo foi concluído. . . . . . . . . . . . . . . . . . . . . . 111

Índice de tabelas
2.1 Simbologia e nomenclatura. . . . . . . . . . . . . . . . . . . . . . . . . . . 27
7.1 Tabela de redução de 8− 3 usado no método PHD. As duas últimas colunas
correspondem à classi�cação DSSP. . . . . . . . . . . . . . . . . . . . . . . 64
7.2 Tabela de agrupamento em nove grupos de acordo com as propriedades físicas
de químicas dos aminoácidos. . . . . . . . . . . . . . . . . . . . . . . . . . 65
10.1 Análise comparativa entre o desempenho obtido pela nossa rede neuronal
(RN) em relação aos valores obtidos pelo ICGEB, usando a extracção de
características a partir do Blast. . . . . . . . . . . . . . . . . . . . . . . . . 84
10.2 Avaliação de desempenho individual em função do critério de extracção de
características. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
10.3 Avaliação de desempenho dos tuplos de ordem k. . . . . . . . . . . . . . . 87
10.4 Avaliação de desempenho do tuplos de ordem 4 (D=4) em função do número
de características extraídas. . . . . . . . . . . . . . . . . . . . . . . . . . . 88
10.5 Avaliação de desempenho dos tuplos de ordem 4 (D=4) em função do critério
de selecção na construção do dicionário. . . . . . . . . . . . . . . . . . . . 90
21

22 ÍNDICE DE TABELAS
10.6 Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção e das características seleccionadas. . . . . . . . . . . . . . . . . . 92
10.7 Avaliação de desempenho, adicionando possíveis combinações de caracterís-
ticas seleccionadas em relação ao melhor resultado obtido a partir do SFS
em 10.6. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
10.8 Comparação entre os resultados obtidos por uma rede neuronal e máquina
de vectores de suporte, usando o melhor resultado obtido a partir do SFS em
10.6. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
10.9 Resultados obtidos usando o melhor resultado obtido a partir do SFS em
10.6 usando bases de dados de maior dimensão. . . . . . . . . . . . . . . . 95

CAPÍTULO 1
Introdução
1.1 Motivação
A classi�cação de proteínas em grupos funcionais é um dos problemas mais relevantes
na área da biologia computacional, consistindo na aplicação de técnicas de inteligência
computacional para análise de dados biológicos. Esta área tem sido alvo de enorme interesse
e tem registado importantes progressos, contudo ainda necessita de uma base mais e�ciente
para a gestão da informação e extracção de conhecimento.
1.2 Objectivos
Dado o aumento exponencial de dados (informação) sobre proteínas conhecidas ao nível da
sequência, torna-se cada vez mais importante a concepção de sistemas que permitam em
tempo útil classi�car correctamente a informação. Nos últimos anos muitos trabalhos têm
vindo a ser desenvolvidos e avanços signi�cativos tem ocorrido, contudo ainda se está longe
de um sistema que permita classi�car correctamente toda a informação existente. No sentido
de tentar contribuir um pouco mais nesse avanço, pretendemos desenvolver uma nova me-
todologia, para a extracção de características a partir da sequência da proteína, combinando
23

24 1. INTRODUÇÃO
técnicas de extracção ao nível físico-químico das proteínas e técnicas de Text-Mining com
segmentação de palavras. A metodologia será implementada num sistema computacional
usando classi�cadores inteligentes, nomeadamente redes neuronais e máquinas de vectores
de suporte.
1.3 Estrutura do relatório
Esta dissertação encontra-se organizada em 11 capítulos. Após o primeiro capítulo intro-
dutório, são apresentados os conceitos fundamentais necessários ao logo deste trabalho.
No capítulo seguinte é descrita a importância do conhecimento da estrutura a partir da
sequência. No quarto capítulo são explicadas as di�culdades na obtenção da estrutura
da proteína, evidenciando a importância que o papel da previsão de proteínas apresenta
neste contexto. No quinto capítulo, em virtude da quantidade de informação existente
sobre sequências de proteínas conhecidas, mas não traduzindo-se em conhecimento sobre a
estrutura das mesmas, é apresentada a importância dos classi�cadores inteligentes usando
máquinas de aprendizagem. No sexto capítulo é sublinhada a importância da selecção de
características no desempenho dos classi�cadores. No sétimo capítulo é descrito um pouco
o estado da arte sobre a temática de que nos propusemos no início, e que serviram de ponto
de partida para este trabalho. No oitavo capítulo é apresentada a metodologia por nós
seguida na elaboração do classi�cador e no capítulo nove são descritas as bases de dados
usadas para a obtenção de resultados que são descritos no capítulo dez. Por último é feita
uma análise critica do trabalho desenvolvido e trabalhos futuros.

CAPÍTULO 2
Conceitos Básicos
Não seria possível elaborar esta dissertação sem que conhecimentos fundamentais fossem
assimilados, tendo sempre em mira os objectivos propostos. Assim sendo torna-se necessários
descrever e rever conceitos básicos de forma a estabelecer a ponte entre o nosso problema
na área da biologia e o nosso objectivo na área da informática.
2.1 DNA
Uma sequência de DNA ou sequência genética é uma série de letras representando a
estrutura primária de uma molécula ou cadeia de DNA. As letras possíveis são A, C, G
e T, representando os quatro nucleotídeos de uma cadeia de DNA - as bases Adenina,
Citosina, Guanina, Timina.
2.2 Aminoácido
Um aminoácido é uma molécula orgânica formada por átomos de Carbono, Hidrogénio,
Oxigénio, e Azoto unidos entre si (Fig. 2.1). Alguns aminoácidos podem igualmente conter
25

26 2. CONCEITOS BÁSICOS
enxofre. Os aminoácidos são divididos em quatro grupos: o grupo amina (NH2), carboxílico
(COOH), hidrogénio, carbono alfa, e um substituinte característico de cada aminoácido. Os
Figura 2.1: Estrutura geral de um aminoácido [34].
aminoácidos unem-se através de ligações peptídicas, formando as proteínas. Para que as
células possam produzir as proteínas, necessitam de aminoácidos, que podem ser obtidos
a partir da alimentação ou serem produzidos pelo próprio organismo. A nomenclatura dos
aminoácidos é apresentada na tabela 2.1.
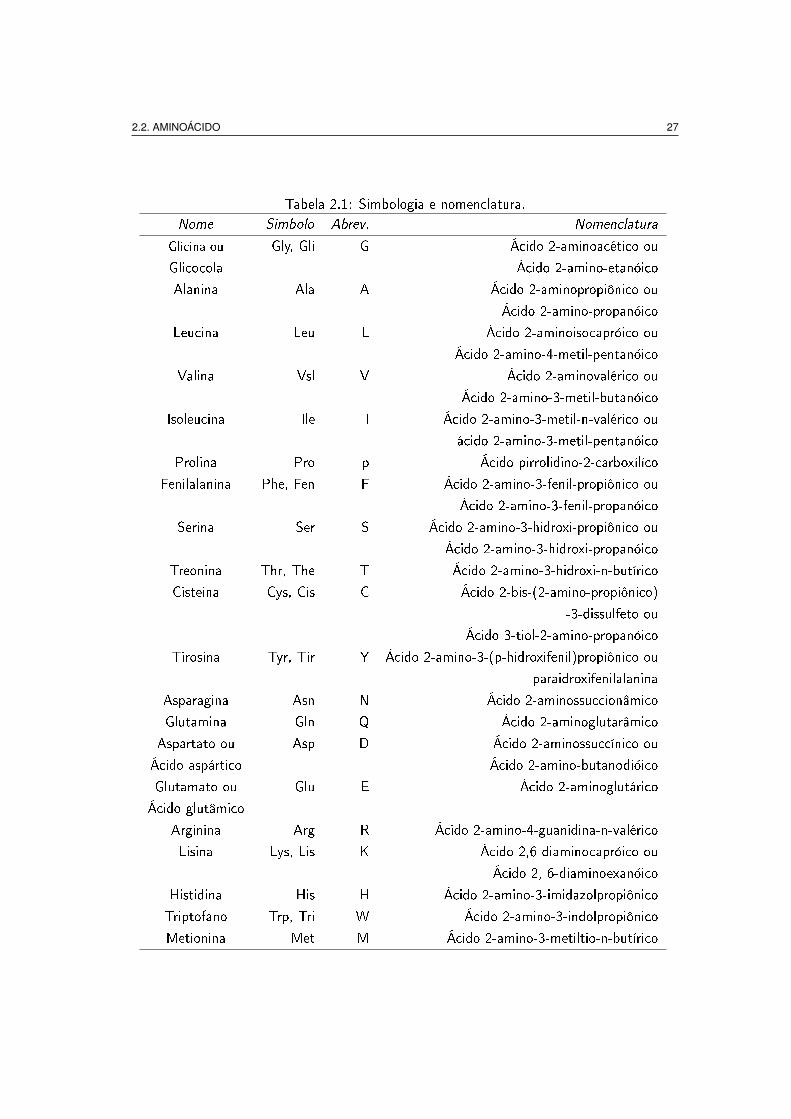
2.2. AMINOÁCIDO 27
Tabela 2.1: Simbologia e nomenclatura.Nome Simbolo Abrev. Nomenclatura
Glicina ou Gly, Gli G Ácido 2-aminoacético ou
Glicocola Ácido 2-amino-etanóico
Alanina Ala A Ácido 2-aminopropiônico ou
Ácido 2-amino-propanóico
Leucina Leu L Ácido 2-aminoisocapróico ou
Ácido 2-amino-4-metil-pentanóico
Valina Vsl V Ácido 2-aminovalérico ou
Ácido 2-amino-3-metil-butanóico
Isoleucina Ile I Ácido 2-amino-3-metil-n-valérico ou
ácido 2-amino-3-metil-pentanóico
Prolina Pro p Ácido pirrolidino-2-carboxílíco
Fenilalanina Phe, Fen F Ácido 2-amino-3-fenil-propiônico ou
Ácido 2-amino-3-fenil-propanóico
Serina Ser S Ácido 2-amino-3-hidroxi-propiônico ou
Ácido 2-amino-3-hidroxi-propanóico
Treonina Thr, The T Ácido 2-amino-3-hidroxi-n-butírico
Cisteina Cys, Cis C Ácido 2-bis-(2-amino-propiônico)
-3-dissulfeto ou
Ácido 3-tiol-2-amino-propanóico
Tirosina Tyr, Tir Y Ácido 2-amino-3-(p-hidroxifenil)propiônico ou
paraidroxifenilalanina
Asparagina Asn N Ácido 2-aminossuccionâmico
Glutamina Gln Q Ácido 2-aminoglutarâmico
Aspartato ou Asp D Ácido 2-aminossuccínico ou
Ácido aspártico Ácido 2-amino-butanodióico
Glutamato ou Glu E Ácido 2-aminoglutárico
Ácido glutâmico
Arginina Arg R Ácido 2-amino-4-guanidina-n-valérico
Lisina Lys, Lis K Ácido 2,6-diaminocapróico ou
Ácido 2, 6-diaminoexanóico
Histidina His H Ácido 2-amino-3-imidazolpropiônico
Triptofano Trp, Tri W Ácido 2-amino-3-indolpropiônico
Metionina Met M Ácido 2-amino-3-metiltio-n-butírico

28 2. CONCEITOS BÁSICOS
2.3 Proteínas
As Proteínas (Fig. 2.2) são compostos orgânicos de estrutura complexa e massa molecular
elevada (de 5.000 a 1.000.000 ou mais unidades de massa atómica), sintetizadas pelos
organismos vivos através da condensação de um grande número de moléculas de aminoácidos,
através de ligações denominadas ligações peptídicas. Uma proteína é um conjunto de pelo
menos 80 aminoácidos, mas sabemos que possui muito mais que essa quantidade, sendo os
conjuntos menores denominados polipeptídeos.
Figura 2.2: Estrutura 3D da mioglobina, proteína globular de 153 aminoácidos, criada a
partir PDB SimpleView (pdb id: 2JHO) [31].
2.4 Ponto isoeléctrico (pI)
Ponto isoeléctrico é o valor de pH onde uma molécula, por exemplo, um aminoácido ou uma
proteína, apresenta carga eléctrica líquida igual a zero. O pI é o pH no qual há equilíbrio
entre as cargas negativas e positivas dos grupos de um aminoácido ou de uma proteína.
2.5 Cristalogra�a de raios X
A cristalogra�a de raios X é uma técnica que consiste em fazer passar um feixe de raios
X através de um cristal da substância sujeita ao estudo. O feixe difunde-se em várias
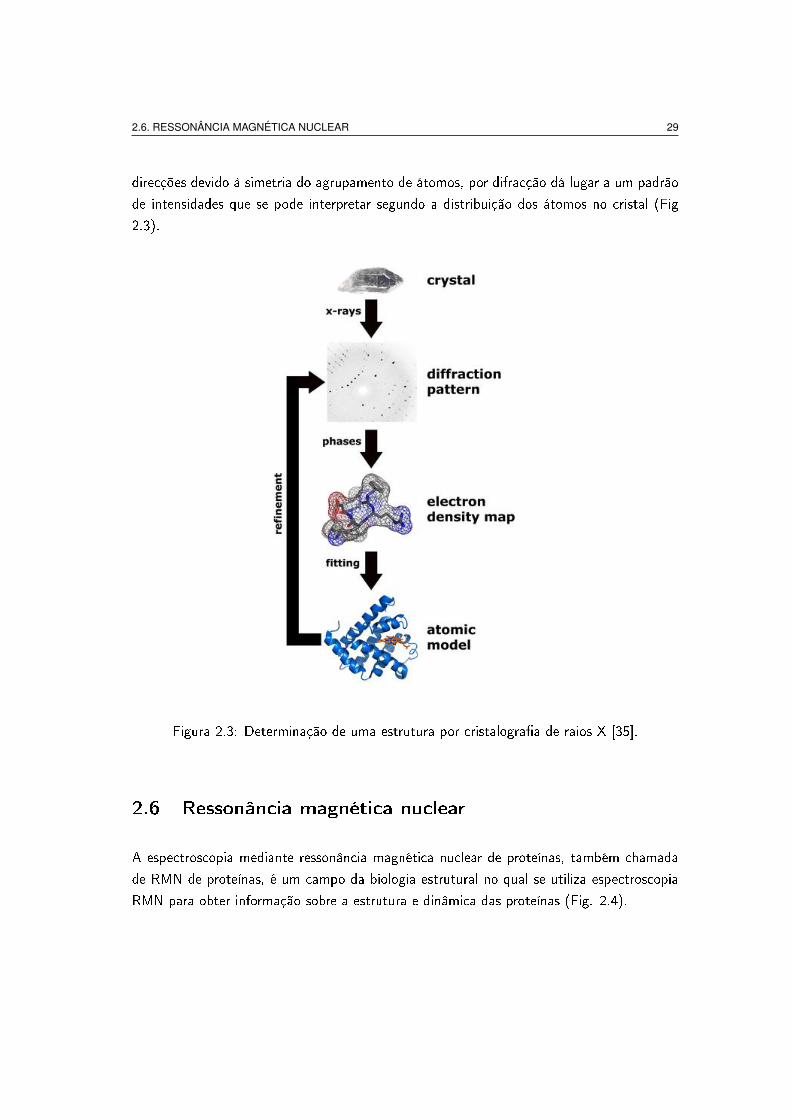
2.6. RESSONÂNCIA MAGNÉTICA NUCLEAR 29
direcções devido à simetria do agrupamento de átomos, por difracção dá lugar a um padrão
de intensidades que se pode interpretar segundo a distribuição dos átomos no cristal (Fig
2.3).
Figura 2.3: Determinação de uma estrutura por cristalogra�a de raios X [35].
2.6 Ressonância magnética nuclear
A espectroscopia mediante ressonância magnética nuclear de proteínas, também chamada
de RMN de proteínas, é um campo da biologia estrutural no qual se utiliza espectroscopia
RMN para obter informação sobre a estrutura e dinâmica das proteínas (Fig. 2.4).

30 2. CONCEITOS BÁSICOS
Figura 2.4: Carga de amostra no espectrómetro RMN de alto campo magnético (800 MHz)
do Paci�c Northwest National Laboratory [36].
2.7 Formato Fasta
Formato de �cheiro informático baseado em texto, podendo conter sequências quer de DNA,
quer de aminoácidos, podendo incluir o nome das sequências e comentários que precedem
a sequência em si.
Uma sequência em formato Fasta começa com uma descrição em uma única linha (linha de
cabeçalho), seguida por linhas com os dados da sequência. A linha de descrição distingue-se
dos dados de sequência por um símbolo '>' (maior que) na primeira coluna. A palavra
seguinte a este símbolo é o identi�cador da sequência, o resto da linha é a descrição (ambos
são opcionais). Não deverá existir espaço entre o '>' e a primeira letra do identi�cador.
Recomenda-se que todas as linhas de texto devam possuir menos de 80 caracteres. A
sequência termina quando surge outra linha começando com o símbolo '>' (isto indica o
começo de outra sequência). A Fig. 2.5 apresenta um exemplo simples de uma sequência
no formato Fasta.
Não há uma extensão de �cheiro standard para um �cheiro de texto contendo sequências
formatadas em Fasta. Os �cheiros neste formato têm com frequência extensões do tipo: .fa,
.mpfa, .fna, .fsa, .fas ou .fasta. Na aplicação desenvolvida, a extensão por defeito é .fasta,

2.7. FORMATO FASTA 31
Figura 2.5: Exemplo de uma sequência em formato Fasta.
permitindo contudo a selecção de outro tipo.

32 2. CONCEITOS BÁSICOS
2.8 Blast
BLAST (Basic Local Alignment Search Tool), é um algoritmo para comparar sequências de
nucleotídos ou proteínas, inferindo relações entre as sequências [1].
2.9 Exactidão global (Q3)
Exactidão global (muitas vezes representada por Q3 em problemas relacionados com previsão
de estrutura secundária em três classes), mede a proporção de exemplos correctamente
classi�cados em toda a amostra. O valor Q3, calcula-se dividindo o número total de
exemplos correctamente classi�cados (a soma dos elementos principais da matriz confusão)
pela dimensão da amostra.
2.10 Matriz de confusão
Os possíveis resultados obtidos pelo classi�cador são quatro e podem ser formulados numa
matriz 2 × 2, conhecida por matriz confusão, como mostra a Fig. 2.6. Os valores na
Figura 2.6: Estrutura da matriz confusão.
diagonal principal representam as decisões obtidas correctamente, e os valores na diagonal

2.11. TAXA DE VERDADEIROS POSITIVOS OU SENSIBILIDADE 33
secundária representam os erros (confusão) entre as classes, P classes positivas e N classes
negativas. Na Fig. 2.6, p e n representam os valores correctos das classe, em classes
positivas e negativas respectivamente, enquanto p' e n', representam os valores obtidos
após classi�cação na classe positiva e negativa respectivamente. Desta matriz, resultam
então:
• Verdadeiros Positivos (VP), em que o classi�cador obteve o valor positivo e a classe
é positiva.
• Verdadeiros Negativo (VN), em que o classi�cador obteve o valor negativo e a classe
é negativa.
• Falsos Positivos (FP), em que o classi�cador obteve o valor positivo e a classe é
negativa.
• Falsos Negativos (FN), em que o classi�cador obteve o valor negativo e a classe é
positiva.
2.11 Taxa de verdadeiros positivos ou sensibilidade
Sensibilidade ou taxa de verdadeiros positivos (TVP) é dada pela expressão:
TV P =V P
P=
V P
V P + FN(2.11.1)
2.12 Taxa de falsos positivos
A taxa de falsos positivos (TFP) é dada pela expressão:
TFP =FP
N=
FP
V N + FP(2.12.1)
2.13 Especi�cidade
Especi�cidade ou taxa de verdadeiros negativos (TVN) é dada pela expressão:
TV N =V N
N=
V N
V N + FP(2.13.1)
Por sua vez a especi�cidade pode ser obtida a partir de 1 - TFP

34 2. CONCEITOS BÁSICOS
2.14 Taxa de acerto
Taxa de acerto (TA) é dada pela expressão:
TA =V N + V P
P +N(2.14.1)
2.15 Análise ROC
A taxa de acerto de um classi�cador é a opção mais óbvia e mais utilizada. Uma taxa
de acerto de 100% é o objectivo ideal, contudo pode ser visto com uma grande incerteza.
Considere-se por exemplo um conjunto constituído essencialmente por casos negativos, em
que os exemplos positivos são na ordem de 1%. Obter uma taxa de acerto de 89%, poderá
signi�car que não estamos na presença de um classi�cador muito e�caz, basta o classi�cador
classi�car maioritariamente o exemplos como negativos para obter uma taxa de acerto
elevada. Uma alternativa consiste na comparação do desempenho de um classi�cador a partir
do grá�co ROC (Receiver Operating Characteristic). O grá�co ROC é uma técnica para
selecção de classi�cadores binários baseado na sua performance num espaço bidimensional,
em que a taxa de verdadeiros positivos (TVP) é representada no eixo do YY e a taxa de
falsos positivos (TFP) é representada no eixo do XX, como podemos ver na Fig. 2.7:
Figura 2.7: Exemplo de um grá�co ROC.

2.15. ANÁLISE ROC 35
Qualquer curva ROC é gerada por um número �nito de instâncias, dando origem a uma
função discreta no espaço ROC. Quanto maior o número de instâncias, mais a função se
aproxima de uma função contínua.
Para obter a curva ROC, são apenas necessários os TVP e os TFP. Uma vez que a TVP
é igual à sensibilidade e a TFP é igual a 1-especi�cidade , a curva de ROC é muitas vezes
entendida como sensibilidades vs (1- especi�cidade), conforme mostra a Fig.2.7.
A análise ROC é muito importante para comparar classi�cadores em problemas de classes
não balanceadas, exemplo dos casos por nós estudados, em que o número de exemplos
da classe negativa é normalmente muito superior ao número de casos da classe positiva.
Uma curva ROC é uma representação bidimensional do desempenho de um classi�cador.
Para comparar classi�cadores é preciso reduzir a curva ROC a um escalar. Um método
comum para realizar esta redução é calcular a área abaixo da curva ROC o chamado AUC
(Area Under Curve). Como o AUC é uma parte da área do quadrado unitário, (espaço
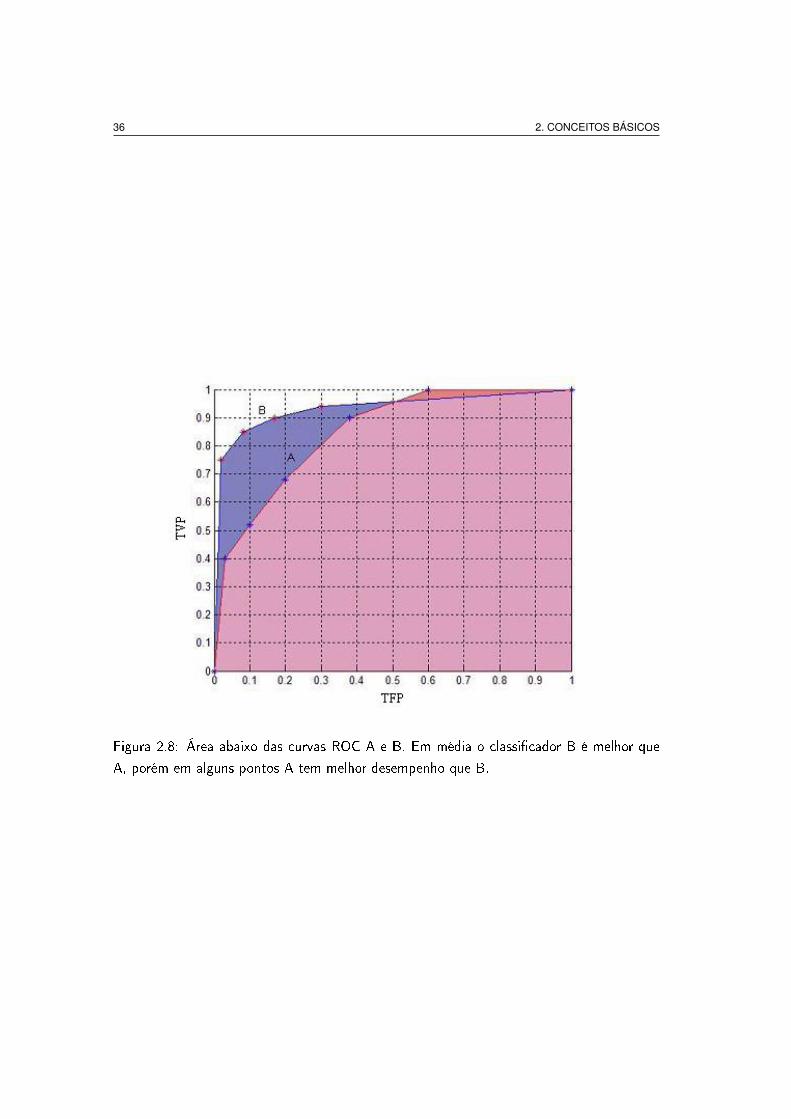
ROC) o resultado encontra-se entre 0.0 e 1.0. A Fig. 2.8 apresenta a área abaixo de
duas curvas ROC, A e B. O classi�cador B possui uma área maior e, portanto, tem um
melhor desempenho médio. Contudo, é possível que em algumas regiões do espaço ROC
um classi�cador seja melhor que outro. Na Fig. 2.8 o classi�cador B é geralmente melhor
que A excepto para TFP > 0.5 onde o classi�cador A apresenta uma pequena vantagem. O
cálculo do AUC apresentado nos nossos resultados é obtido a a partir da função do Matlab
perfcurv pertencente toolbox de estatística do Matlab.
36 2. CONCEITOS BÁSICOS
Figura 2.8: Área abaixo das curvas ROC A e B. Em média o classi�cador B é melhor que
A, porém em alguns pontos A tem melhor desempenho que B.

CAPÍTULO 3
O longo caminho da sequência à sua estrutura
3.1 Introdução
O Projecto Genoma Humano, levou à identi�cação de mais de 30 mil genes no genoma
humano, podendo codi�car 100.000 proteínas como resultado de splicing alternativo. Para
entender as funções biológicas como mecanismos funcionais dessas proteínas, o conheci-
mento das suas estruturas 3D é fundamental. O ambicioso projecto Structural Genomics
lançado pelo NIH (National Institute of General Medical Sciences) [22] em 1999, traçou
como objectivo decifrar a estruturas destas proteínas.
As proteínas são cadeias de polímeros de unidades repetitivas de polipeptídeos com cadeias
laterais ligadas a cada unidade polipeptídica. As cadeias laterais, também conhecidas como
resíduos, são aminoácidos com características diferentes. Existem 20 aminoácidos diferentes
(A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V). A sequência de aminoácidos
de uma cadeia de proteína é dada pela estrutura primária (Fig. 3.1). Uma proteína típica
contém 200/300 aminoácidos, mas pode aumentar até cerca de 30 mil numa única cadeia.
No seu ambiente nativo a cadeia de aminoácidos (ou resíduos) de uma proteína dobra-se
(fold) em estruturas secundárias locais, classi�cadas como α-hélices, folhas β, e estruturas
não regulares [24, 25]. A estrutura secundária é determinada pela sequência, classi�cando
37

38 3. O LONGO CAMINHO DA SEQUÊNCIA À SUA ESTRUTURA
cada aminoácido no elemento da estrutura, por exemplo α-hélice.
Figura 3.1: Diferentes representações da estrutura da proteína
A estrutura terciária resulta de interacções entre a cadeia principal/cadeia principal, cadeia
lateral/solventes, características dos aminoácidos, restrições de mobilidade dos componen-
tes, de forma a encontrar a estrutura mais estável [26]. A estrutura terciária é descrita
no espaço (x, y, z) pelas coordenadas de todos os átomos de uma proteína. Finalmente,
as cadeias de proteínas podem interagir ou unir-se para formar proteínas mais complexas.
Estas proteínas mais complexas correspondem à estrutura quaternária das proteínas (Fig.
3.1). A estrutura quaternária é representada através das coordenadas de todos os átomos
e de todas as cadeias principais. Numa célula, as proteínas interagem com outras proteínas
e com outras moléculas para realizar vários tipos de funções biológicas como a catálise
enzimática, regulação de genes, controle do crescimento e diferenciação, transmissão de
impulsos nervosos. Extensos estudos de bioquímica provaram que a função de uma proteína
é determinada pela sua estrutura. No entanto, o inverso não é verdadeiro, poderíamos ter
proteínas com a mesma função, mas com estruturas diferentes.
3.2 A importância do conhecimento da estrutura
Patologias comuns, como diabetes de tipo I e a hemo�lia, ocorrem porque o organismo é
incapaz de produzir a insulina (Fig. 3.2) e o factor VIII, respectivamente. Já certos tipos
de cancro resultam da produção "defeituosa" de uma proteína que participa na regulação
da divisão celular, o factor p21 Ras [3]. Outras patologias como a doença de Parkinson,
a doença de Alzheimer e as encefalopatias espongiformes, têm origem num processo de
dobragem do qual resultam proteínas defeituosas que tendo uma grande a�nidade entre si
se agregam para formar �bras (Fig. 3.3).

3.2. A IMPORTÂNCIA DO CONHECIMENTO DA ESTRUTURA 39
Figura 3.2: A insulina (à esquerda) é uma hormona. O factor VIII (ao centro) é uma
proteína que promove a coagulação do sangue. O factor p21Ras (à direita) é uma proteína
de sinalização (signaling).
Figura 3.3: Fibras de amilóide resultantes da interacção entre proteínas cuja dobragem é
"defeituosa".

40 3. O LONGO CAMINHO DA SEQUÊNCIA À SUA ESTRUTURA
Os priões são proteínas que estão na origem de algumas doenças neurodegenerativas como
é o caso da BSE, conhecida como a doença das vacas loucas. Estas proteínas podem
existir em duas formas. A forma normal PrPc, é inócua e existe na membrana celular
das células do sistema nervoso central, mas a sua função biológica não é bem conhecida.
Esta proteína pode sofrer uma alteração da sua conformação nativa na qual uma α-hélice é
convertida numa folha β formando-se a forma PrPSc esta sim, infecciosa que se multiplica
através de uma reacção em cadeia em que mais moléculas de PrPSc se formam à custa
das moléculas percursores PrPc. Quando existem moléculas de PrPSc em número su�ciente
Figura 3.4: Forma normal do prião PrPc.
formam-se longos agregados �lamentosos que se depositam no tecido neuronal conduzindo
gradualmente à sua degradação (Fig. 3.4).
Sabe-se hoje que a infecção por priões ocorre espontaneamente, devido a uma mutação
pontual da proteína PrPc, ou através da exposição a um agente patogénico externo (Fig.
3.5). Perante este factos, a estrutura da proteína é a chave para compreender a sua função,
que é essencial para todas as aplicações biológicas relacionadas, biotecnológicas, médicas ou
farmacêuticas. O número de sequências proteicas identi�cadas aumentou drasticamente nos
últimos anos devido à extensa pesquisa na área. No entanto, a maioria dessas sequências não
é acompanhada por qualquer informação sobre a sua função, consequência dos procedimen-
tos experimentais morosos e dispendiosos para a determinação da estrutura. Abordagens
experimentais, tais como cristalogra�a de raios X [6] e ressonância magnética nuclear [29],
são as técnicas mais relevantes para a determinação da estrutura das proteínas.

3.2. A IMPORTÂNCIA DO CONHECIMENTO DA ESTRUTURA 41
Figura 3.5: O gene PrP contém informação para expressar a proteína inócua PrPc. Uma
mutação pontual neste gene dá origem à forma infecciosa PrPSc da proteína. Por intermédio
de uma terceira proteína, não representada na �gura, a PrPSc induz uma alteração
conformacional na qual uma α-hélice da PrPc é convertida numa folha β transformando-a
numa molécula da sua própria espécie.

42 3. O LONGO CAMINHO DA SEQUÊNCIA À SUA ESTRUTURA

CAPÍTULO 4
Classi�cação de Proteinas
4.1 Introdução
Desde a determinação das duas primeiras estruturas de proteínas (mioglobina e hemoglobina)
usando cristalogra�a de raios X, o número de proteínas com estruturas conhecidas aumentou
rapidamente. Em 9 de Fevereiro de 2009, a base de dados do Protein Data Bank (PDB)
[2] continha 58.396 proteínas conhecidas e respectivas estruturas, em 21 de Agosto de
2010 o valor passou para 67.322. Este conjunto crescente de estruturas conhecidas fornece
informações valiosas para ajudar a entender melhor como uma cadeia de proteína se dobra
na sua estrutura 3D original, como as cadeias interagem na forma quaternária, e como prever
estruturas a partir da sequência (estrutura primária) [17]. Desde as experiências pioneiras
[28], mostrando que a estrutura de uma proteína é determinada pela sua sequência, prever a
estrutura da proteína a partir da sua sequência tornou-se num dos problemas fundamentais
da biologia estrutural. Isto não é somente um desa�o teórico fundamental, mas também
uma prática devido à discrepância entre o número de sequências de proteínas e o número de
estruturas conhecidas. Uma percentagem miníma das sequências actualmente conhecidas
tem a sua estruturas de�nida e cada dia que passa este fosso não pára de aumentar.
Apesar dos progressos na área da robótica e noutras áreas, a determinação experimental
de uma estrutura de proteínas ainda é muito dispendiosa, morosa, e nem sempre possível.
43

44 4. CLASSIFICAÇÃO DE PROTEINAS
Assim, uma maneira de entender a sua função é de uma forma comparativa com proteínas
conhecidas, com bases de dados anotadas, cuja estrutura é conhecida.
4.2 A importância da previsão
Perante estes elementos, a previsão da estrutura das proteínas constitui um enorme desa�o
em termos cientí�cos visto não ser claro como uma estrutura se encontra codi�cada ao nível
da sequência. Conforme referido existe uma relação directa entre a sequência e a estrutura
da proteína, pelo que a análise da sequência em termos de características adquire um papel
fundamental.
Muitas técnicas computacionais têm vindo a ser desenvolvidas para prever a estrutura das
proteínas, tirando partido de proteínas já conhecidas. Os métodos de previsão da estrutura
podem ser classi�cados como: ab initio ou baseados em conhecimento.
Métodos que explicam a estrutura das proteínas com base em cálculos de minimização de
energia, no sentido de obter uma "estrutura estável", são chamados métodos ab initio [12].
Estes métodos utilizam as teorias da mecânica quântica e termodinâmica estatística para a
previsão, de forma a minimizar a energia potencial. Para além disso, há que ter em conta que
o resultado do cálculo depende não só do tipo de átomo, mas também do ambiente químico
em que se encontram os átomos participantes na interacção, o que gera complicações
adicionais. Naturalmente este tipo de cálculo faz-se com recurso a computadores no contexto
das simulações por dinâmica molecular.
As simulações por dinâmica molecular são extremamente exigentes do ponto de vista compu-
tacional. Se pensarmos que precisamos de um dia de CPU para simular um nanosegundo (ns)
do processo de dobragem, o que é uma estimativa razoável tendo em conta as capacidades
de cálculo dos computadores de que dispomos actualmente. Se uma proteína consumir
10000 nanossegundos para encontrar o seu estado nativo, então precisaríamos de 10000 dias
de CPU, ou seja cerca de 30 anos, para simular o processo de dobragem na totalidade. Isto
é obviamente muito tempo para se esperar por apenas um resultado [3].
É claro que existem super-computadores capazes de simular mais do que um nanossegundo
por dia e foi com esse recursos que os dois cientistas americanos Yong Duan e Peter Kollman
recorreram em 1998 para fazerem a simulação por dinâmica molecular que é ainda hoje
considerada o estado da arte nesta área. Estes investigadores simularam, ainda que apenas
parcialmente, o processo de dobragem de uma pequena proteína com 36 resíduos (villin

4.2. A IMPORTÂNCIA DA PREVISÃO 45
headpiece) e doze mil átomos, na presença de água, durante 1000 ns o que correspondeu,
na prática à utilização de quatro meses de cálculo de CPU. O Blue gene é um super-
Figura 4.1: Instantâneos da simulação por dinâmica molecular da dobragem da villin
headpiece levada a cabo por Duan e Kollman em 1998.
computador construído pela IBM para estudar o processo de dobragem das proteínas. Esta
máquina ocupa metade de um campo de ténis. A 14 de Novembro de 2005 executava
280.6× 1012 operações matemáticas por segundo (Fig. 4.2) [3].
Perante estes argumentos de peso, este processo torna-se impraticável devido ao envolvi-
mento de vários milhares de átomos, para o qual os cálculos de energia teriam que ser
feitos, implicando a utilização de modelos ab initio bastante simpli�cados, naturalmente
com resultados bastante falíveis.
Métodos baseados no conhecimento tentam prever a estrutura, usando informações das
bases de dados de estruturas de proteínas conhecidas. Modelagem comparativa também
chamada de modelagem por homologia, é um dos muitos métodos de previsão e produz
resultados precisos dando os melhores alinhamentos da sequência target (sequência da
proteína desconhecida) em relação a um ou mais template(s) de sequências conhecidas.
Entre as abordagens por homologia para prever estruturas de proteínas, as mais bem su-
cedidas incluem modelos de redes neuronais, ferramentas de pesquisa em bases de dados,

46 4. CLASSIFICAÇÃO DE PROTEINAS
Figura 4.2: Blue gene. Um super-computador criado pela IBM para estudar a dobragem de
proteínas por dinâmica molecular.
alinhamento múltiplo da sequência, alinhamento local de sequências, threading, modelo de
cadeias de Markov, métodos do vizinho mais próximo, programação dinâmica e abordagens
combinando diferentes métodos de previsão.

CAPÍTULO 5
Da informação ao conhecimento
5.1 Introdução
O aparecimento dos computadores e das tecnologias de informação, permitiu a recolha e
armazenagem de enormes quantidades de informação, infelizmente quantidade de informação
não é sinónimo de conhecimento. A informação que actualmente se consegue recolher, é de
tal maneira vasta e complexa, que se corre o risco de não conseguir extrair todo o conhe-
cimento que a informação representa, quer pelo tempo envolvido, quer pela incapacidade
de encontrar signi�cado, dada a sua complexidade. Encontrar signi�cado em algo de novo,
através da inteligência, é uma característica associada ao ser humano. A capacidade de
aprendizagem tem servido de mote, no sentido de tentar aliar a "inteligência", à capacidade
de cálculo dos computadores. É neste contexto que surge uma área interdisciplinar das
Ciências da Computação, a Inteligência Arti�cial, que por sua vez contém um sub-campo
fundamental no nosso trabalho: Máquinas de aprendizagem (Machine Learning).
47

48 5. DA INFORMAÇÃO AO CONHECIMENTO
5.2 Classi�cadores em máquinas de aprendizagem
Máquinas de aprendizagem, é uma área cientí�ca em que a tónica se encontra no desenvolvi-
mento de algoritmos de forma que os computadores adquiram capacidade de aprendizagem,
a partir de um dado conjunto de informação, sobre o qual se conhece o signi�cado, conhe-
cimento.
Neste contexto, surgem os classi�cadores, em que a capacidade de aprendizagem advém
das características extraídas da informação fornecida, de forma que em nova informação, o
classi�cador seja capaz de correctamente classi�car a informação, tendo por base o processo
de aprendizagem anterior.
Normalmente, os algoritmos de classi�cação, referem-se a algoritmos supervisionados, que
são sobre os quais os nosso trabalho irá incidir. Contudo, existem igualmente os não
supervisionados, por exemplo os algoritmos de clustering, em que a classi�cação em grupos
é feita segundo uma dada métrica, por exemplo a distância entre os elementos (vectores)
num dado espaço vectorial (Fig. 5.1). Conforme referimos, o nosso interesse inside sobre
Figura 5.1: Classi�cação da informação em 3 grupos (clusters).
algoritmos supervisionados, em que a tarefa de aprendizagem reside na forma que a máquina
possa inferir uma função a partir dos dados de treino. No processo de treino supervisionado,
a máquina recebe como entrada um conjunto de dados, normalmente um vector, sobre o

5.3. ALGUNS ALGORITMOS EM MÁQUINAS DE APRENDIZAGEM 49
qual se conhece o valor correcto de saída, por vezes designado por sinal de supervisão.
O algoritmo de aprendizagem avalia os dados de treino e produz uma função, designada
por classi�cador. A função inferida, deverá então prever correctamente o valor de saída a
partir de novos dados de entradas, considerados válidos para o algoritmo. Os classi�cadores,
podem ser divididos em dois tipos:
• Classi�cadores multi-classe.
• Classi�cadores binários.
Enquanto no classi�cador multi-classe a tarefa consiste em classi�car as entradas em várias
classes, o classi�cador binário classi�ca os membros de um determinado conjunto de entradas
em dois grupos com base em se eles têm alguma propriedade ou não.
Como exemplos de classi�cadores binários podemos considerar:
• Testes médicos para determinar se um paciente tem uma determinada doença ou não
(a propriedade de classi�cação é a doença).
• Controle de qualidade em fábricas, ou seja, decidir se um novo produto tem qualidade
su�ciente para ser vendido ou não (a propriedade de classi�cação é ter qualidade
su�ciente).
Conforme referimos, a implementação da máquina de aprendizagem depende do algoritmo
de aprendizagem, de forma a construir correctamente o classi�cador.
5.3 Alguns algoritmos em máquinas de aprendizagem
• Árvores de Decisão (Decision tree)
• Redes Neuronais (Arti�cial neural networks)
• Programação Genética (Genetic programming)
• Máquinas de Vectores de Suporte (Support vector machines)
• Agrupamento (Clustering)
• Redes Baysianas (Bayesian networks)

50 5. DA INFORMAÇÃO AO CONHECIMENTO
• Redes Fuzzy (Fuzzy Networks)
• Modelos de Cadeias de Markov (Markov models)
No nosso trabalho, iremo-nos debruçar sobre as redes neuronais e sobre as máquinas de
vectores de suporte.
5.3.1 Redes neuronais
Redes neuronais são sistemas computacionais estruturados inspirados no paradigma de
realizar tarefas cognitivas à semelhança do sistema biológico que é o cérebro humano.
A chave principal deste paradigma é baseada no largo número de interconexões entre os
diversos elementos (neurónios Fig. 5.2), trabalhado num todo na resolução de um dado
problema.
Figura 5.2: Imagem real, apresentando interconexões entre os neurónios.
À semelhança dos humanos, a ideia das redes neuronais, é possuírem a capacidade de
aprendizagem. Em termos biológicos esta capacidade reside nos ajustamentos das regiões
de comunicação entre os neurónios, sinapses. O mesmo se pretende que aconteça ao nível
das redes neuronais, convém no entanto sublinhar que as redes neuronais, são modelos
extremamente simpli�cados em relação ao cérebro humano.

5.3. ALGUNS ALGORITMOS EM MÁQUINAS DE APRENDIZAGEM 51
Um pouco de história
Em 1943 McCulloch e Pitts desenvolveram modelos de redes neuronais, baseadas no seu
conhecimento em neurologia. Estes modelos assentavam acima de tudo no seu entendimento
de como os neurónios funcionavam, onde o neurónio possuia apenas uma saída, que era uma
soma do valor das suas entradas. Em 1958, Rosenblatt, devido ao seu grande interesse pela
área, desenvolve o "Perceptrão", no Cornell Aeronautical Laboratory, conjunto de neurónios
arti�ciais e sinapses que os interligam, que pode ser visto como o tipo mais simples de rede
neuronal feedforward. O perceptão é um classi�cador binário que mapeia a entrada x (um
vector de valor real) para um valor de saída f(x) (um valor binário simples) através de:
f(x) =
{1 se w.x+ b > 0
0 caso contrário(5.3.1)
onde w um vector de peso real w.x o produto escalar (que calcula uma soma com pesos) e b
representa a inclinação, um termo constante que não depende de qualquer valor de entrada.
Cada neurónio calcula a média ponderada dos sinais que recebe como entrada (Fig. 5.3), em
que as ponderações ou pesos estão armazenados nas respectivas sinapses. O resultado desta
ponderação é usado como entrada de uma função de activação (inicialmente linear) e por
sua vez, a saída desta é o resultado do neurónio. Esta saída poderá ou não ser processada
por outro neurónio.
Figura 5.3: Neurónio arti�cial de um perceptrão.
Contudo, um perceptrão só consegue aprender com exemplos que sejam linearmente sepa-
ráveis, ou seja, separáveis por um hiperplano (Fig. 5.4). A limitação do modelo, exposto
em 1969 por Minsky e Papert, provocou um enorme desinteresse na área [30]. É na década
de 80, que surge novamente o interesse nas redes neuronais, nomedamente a descrição
do algoritmo de treino com backpropagation (Rumelhart, Hinton, & Williams 1986). O

52 5. DA INFORMAÇÃO AO CONHECIMENTO
Figura 5.4: Exemplos linearmente separáveis (esquerda), exemplos não linearmente separá-
veis (direita).
termo backpropagation surge do facto de o algoritmo se basear na retropropagação dos
erros para realizar os ajustes de pesos das camadas intermédias. A forma de calcular as
derivadas parciais do erro de saída em relação a cada um dos pesos da rede caracteriza o
backpropagation
Topologia
A topologia de uma rede neuronal é de�nida pela forma como os neurónios se encontram
interligados entre si. Tipicamente uma rede supervisionada, é constituída por uma camada
de neurónios de entrada, uma ou mais camadas intermédias e uma camada de saída Fig.
5.5.
Figura 5.5: Topologia de uma rede com uma camada de entrada, uma de saída e uma
intermédia.

5.3. ALGUNS ALGORITMOS EM MÁQUINAS DE APRENDIZAGEM 53
Aprendizagem
A propriedade mais importante das redes neuronais, é sua capacidade de aprender no seu
ambiente e com isso melhorar seu desempenho. Isso é conseguido através de um processo
iterativo de ajustes aplicado aos seus pesos, ou seja o chamado treino. O processo de
aprendizagem pode por sua vez ser dividido em duas formas:
• Aprendizagem supervisionada - neste caso o processo decorre pela existência de
dados de entrada e saída, em que à partida se conhecem as saídas.
• Aprendizagem não supervisionada - neste caso o processo decorre pela existência de
dados de entrada e saída, mas não se conhecem as saídas. O processo de aprendizagem
ocorre tentando descobrir padrões nos dados.
Por outro lado a aprendizagem é in�uenciada pelos algoritmos de aprendizagem, em que o
mais conhecido é o backpropagation.
Backpropagation - Propagação de erros
No algoritmo backpropagation, em cada ciclo de aprendizagem, os erros obtidos pelas
diferenças entre as saídas da rede e os valores de treino são propagados para trás, desde os
neurónios de saída até aos de entrada, ocorrendo depois um reajuste dos pesos das conexões.
O treino termina usualmente quando se obtém o erro mínimo à saída.
Em termos gerais o algoritmo backpropagation, decorre da seguinte forma:
1. Rede = Iniciar a rede com pesos entre neurónios aleatórios
2. Enquanto o critério de paragem não for satisfeito (por cada exemplo apresentado à
rede)
i Output = Resposta da rede ao exemplo
ii Erro = Resposta esperada ao exemplo - saída
iii Backpropagate (erro, rede) - propagar erro para as camadas intermédias
iv Actualizar pesos (rede)
3. Retorna a rede

54 5. DA INFORMAÇÃO AO CONHECIMENTO
Estando a falar de redes supervisonadas, em que conhecemos os valores de saída para uma
dada entrada, podemos calcular o erro obtido a partir dos valores conhecidos e o valor obtido
pela nossa rede.
O passo seguinte consiste em ajustar os pesos das camadas intermédias, após a determinação
do erro da camada de saída.
Backpropagation - Criterio de paragem
Sendo um processo iterativo, evidentemente que carece de critérios de paragem que poderão
ser:
• Um número prede�nido de iterações.
• Efectuar um Feed-Forward e veri�car a diferença entre a resposta obtida e a esperada.
O treino da rede termina quando esta diferença for menor que uma determinada
margem de erro.
• Outros critérios.
5.3.2 Máquinas de vectores de suporte (MVS)
Apoiando-se na teoria estatística da aprendizagem, as máquinas de vectores de suporte
(Support Vector Machines normalmente designado por SVM), surgiram pela primeira vez
na década de setenta com o trabalho de Vapnik em 1979 [32]. A fundamentação das
máquinas de vectores de suporte, baseia-se no algoritmo de "se suportar" em alguns dados
de forma a de�nir a distância entre as classes. As �guras seguintes pretendem ilustrar as
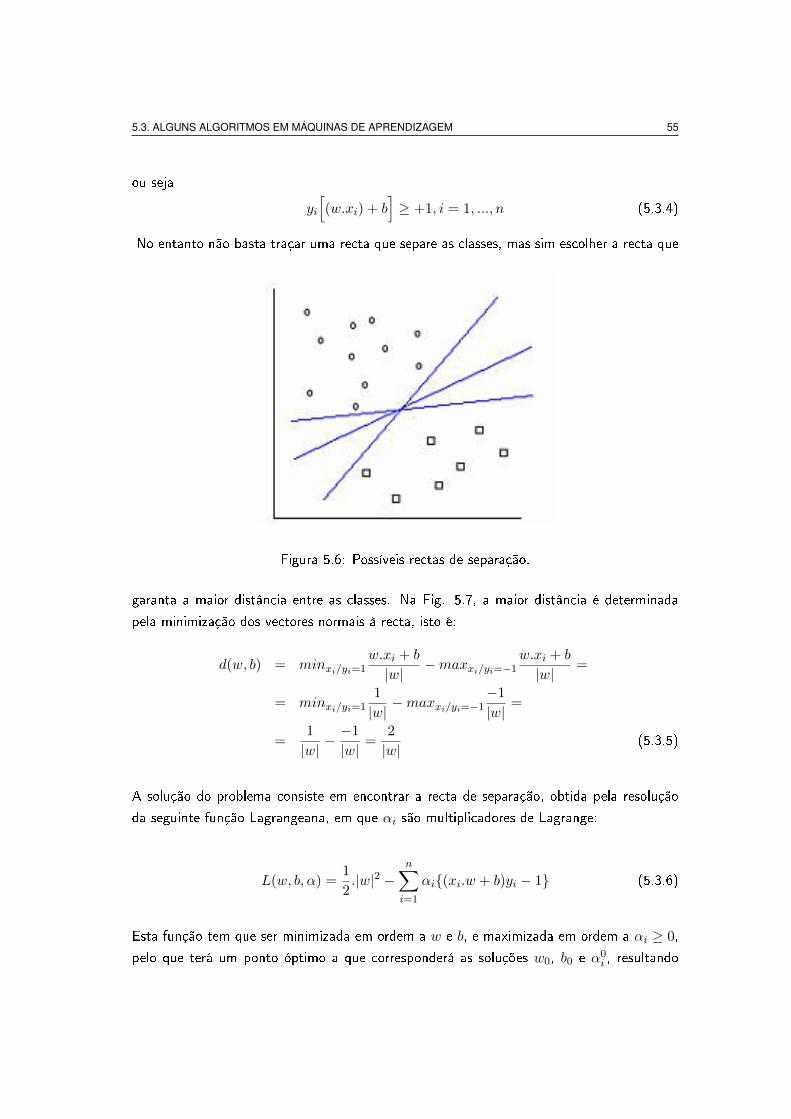
bases matemáticas das máquinas de vectores de suporte. Na Fig 5.6 está representado um
conjunto de dados do tipo (xi; yi), com xi ∈ <n , e yi ∈ {−1;+1}, em que os pontos
representados por quadrados correspondem a yi = −1 e os outros a yi = +1. No exemplo
aqui apresentado, a classi�cação é óbvia pois visualmente podemos estabelecer várias linhas
de fronteira para separar as classes. Uma recta de separação é do tipo w.xi+b = 0 (equação
da recta neste exemplo), com w a inclinação da recta, e b a ordenada na origem. Qualquer
uma das rectas representadas na (Fig. 5.6), poderia ser uma recta de separação desde sejam
satisfeitas as seguintes condições [32]
(w.xi) + b ≥ +1⇒ yi = +1 (5.3.2)
(w.xi) + b ≤ +1⇒ yi = −1 (5.3.3)
5.3. ALGUNS ALGORITMOS EM MÁQUINAS DE APRENDIZAGEM 55
ou seja
yi
[(w.xi) + b
]≥ +1, i = 1, ..., n (5.3.4)
No entanto não basta traçar uma recta que separe as classes, mas sim escolher a recta que
Figura 5.6: Possíveis rectas de separação.
garanta a maior distância entre as classes. Na Fig. 5.7, a maior distância é determinada
pela minimização dos vectores normais à recta, isto é:
d(w, b) = minxi/yi=1w.xi + b
|w|−maxxi/yi=−1
w.xi + b
|w|=
= minxi/yi=11
|w|−maxxi/yi=−1
−1|w|
=
=1
|w|− −1|w|
=2
|w|(5.3.5)
A solução do problema consiste em encontrar a recta de separação, obtida pela resolução
da seguinte função Lagrangeana, em que αi são multiplicadores de Lagrange:
L(w, b, α) =1
2.|w|2 −
n∑i=1
αi{(xi.w + b)yi − 1} (5.3.6)
Esta função tem que ser minimizada em ordem a w e b, e maximizada em ordem a αi ≥ 0,
pelo que terá um ponto óptimo a que corresponderá as soluções w0, b0 e α0i , resultando

56 5. DA INFORMAÇÃO AO CONHECIMENTO
Figura 5.7: Nas MVS, as fronteiras encontram-se de�nidas pelos dados (recta a azul). A
recta verde constitui a linha que garante a maior distância entre as classe, (representadas
por bolas e quadrados).
numa linha de separação com as seguintes propriedades:
n∑i=1
α0i .yi = 0, α0
i ≥ 0 (5.3.7)
w0 =n∑i=1
α0i .yi.xi, α
0i ≥ 0 (5.3.8)
Segundo o teorema de Kühn-Tucker, é necessário ainda veri�car a seguinte condição:
α0i .([
(xi.w0) + b0].yi − 1
)= 0, i = 1, ..., n (5.3.9)
substituindo na função Lagrangeana resulta em:
W (α) =
n∑i=1
αi −1
2
n∑i=1,j=1
αi.αj .yi.yj .(xi.xj), αi ≥ 0 (5.3.10)
Em que função está restringida a∑n
i=1 αi.yi = 0. A solução é α0 = (α01, ..., α
0n), podendo
escrever-se a função de decisão:
f(x) = sign(∑
V ect.sup
α0i .yi.(xi.x)− b0) (5.3.11)

5.3. ALGUNS ALGORITMOS EM MÁQUINAS DE APRENDIZAGEM 57
sendo xi os vectores de suporte b0 = 12
[(w0.x
∗(1))+(w0.x
∗(−1))], com x∗(1) um vector de
suporte pertencente à primeira classe, e x∗(−1) um qualquer vector de suporte pertencente
à segunda classe. Contudo, os problemas nem sempre são lineares. Assim, é necessário
fazer uma transformação nos dados para que estes possam ser linearmente separáveis. As
máquinas de vectores de suporte fazem uma transformação não linear nos dados para um
espaço multi-dimensional onde �cará uma imagem dos dados que permita uma separação
linear.
Algoritmo da MVS
O algoritmo, apresentado na Fig. 5.8, que implementa uma MVS é constituído pelas
seguintes etapas:
• Mapping.
• De�nição do hiperplano.
• Análise.
Mapping
O objectivo do mapping é permitir a separação linear dos dados que terá lugar no hiper-
espaço, de forma que a dimensionalidade permita uma separação linear, ou seja um hiper-
plano. O mapping é conseguido com recurso aos métodos de Kernel.
De�nição do hiperplano
A de�nição do hiperplano é sustentada com recurso a vectores. Os vectores são construí-
dos a partir de algumas características dos dados de entrada, após a etapa de mapping,
denominadas de características. A selecção destas características tem como objectivo que o
hiperplano consiga a separação com a maior distância possível entre as classes.
Métodos de Kernel
Os métodos de Kernel tem como objectivo a separação linear de um conjunto de dados, num
espaço de dimensionalidade su�ciente, de forma a garantir a separação linear. Do ponto de

58 5. DA INFORMAÇÃO AO CONHECIMENTO
Figura 5.8: Etapas do algoritmo MVS.
vista teórico, a transformação levada a cabo pelos métodos de Kernel é de enorme potencial,
permitindo que um algoritmo destes seja capaz de separar qualquer conjunto de dados.
Os métodos mais utilizados são:
• Linear: K(x, y) = x.y
• Polinomial: K(x, y) = (a.x.y + b)d, com a > 0, d=1,...
• Radial Basis Function (RBF): K(x, y) = exp(− (x−y)2σ2 )
• Multi-Layer Perceptron (MLP): K(x, y) = tanh(a.x.y + b)
Análise
Esta última etapa, consiste como o próprio nome indica, na avaliação e interpretação dos
resultados obtidos.

CAPÍTULO 6
Dados usados na fase de aprendizagem do classi�cador
Conforme referido anteriormente, máquinas de aprendizagem, é uma área cientí�ca em que
a tónica se encontra no desenvolvimento de algoritmos de forma que os computadores
adquiram capacidade de aprendizagem, a partir dum dado conjunto de dados, informação,
sobre o qual se conhece o signi�cado, conhecimento. Neste sentido torna-se imperativo, a
extracção de características e posteriormente a selecção das que vamos usar no processo de
aprendizagem.
6.1 Extracção de características na previsão da estrutura
das proteínas
Tipicamente os desenvolvimentos nesta área têm seguido duas vertentes: baseado na sequên-
cia e baseado em bases de dados anotadas [37]. Os métodos baseados em sequências
podem-se subdividir em três categorias:
• Previsão baseada na composição dos aminoácidos.
• Previsão baseada em sequências conhecidas.
59

60 6. DADOS USADOS NA FASE DE APRENDIZAGEM DO CLASSIFICADOR
• Previsão em outros métodos de extracção de conhecimento.
Os trabalhos pioneiros sobre previsão baseada na composição dos aminoácidos foram feitos
por Nishikawa [23], Reinhardt e Hubbard [27] tendo sido posteriormente realizados vários
estudos. Outra forma de previsão é baseada em comparação com sequências conhecidas
[10]. Estes métodos orientam-se no sentido de comparar com sequências conhecidas e daí
deduzir o seu grau de similaridade. Contudo tem um grande handicap, pelo simples facto
de muitas vezes não termos sequências conhecidas para puder comparar, com resultados
satisfatórios. Gabriela e seus colegas [14] desenvolveram um método (Complex Object
Feature Extraction - COFE), baseada na distância de similaridade ao nível da estrutura.
Uma outra vertente, tenta tirar partido de bases de dados e anotações sobre proteínas
(protein annotation databases). Estes métodos baseiam-se no facto de que estas bases
de dados tem vindo a crescer em informação, sendo capazes de fornecer "pistas", sobre
a identidade das proteínas ou sua homologia, tais como motivos (motifs), antologia de
genes (gene antology) [33] e domínio da função da proteína. Chou e os seus colegas [7�9],
propuseram um método híbrido o Go-FunD-PseAA. O principal domínio de conhecimento
deriva de um �ltro à base de dados contendo informação relacionada com o per�l da proteína
e sua localização celular, sendo posteriormente re�nado pelas correspondentes anotações
(annotation matching). Esta abordagem, possui evidentemente uma lacuna em virtude da
ausência de anotações sobre proteínas novas. No entanto à medida que as bases de dados
de anotações vão crescendo, é espectável um ganho de desempenho do método. Hoglund e
seus colegas [13] propõem igualmente um método híbrido combinando targeting sequence,
composição de aminoácidos e motivos.
6.2 Selecção de características
A selecção de características (feature selection), é uma das principais abordagens na re-
dução de dimensionalidade, seleccionando um subconjunto de características originais para
representação de padrões observados. A selecção de características ganha especial im-
portância já que o desempenho dos algoritmos de classi�cação (classi�cador) se degrada
rapidamente não só com a inserção de características irrelevantes mas também com a
inclusão de características relevantes que estejam relacionadas. Por outro lado, à medida que
aumenta a dimensionalidade do espaço de características, o esforço computacional envolvido
é incrementado, colocando muitas vezes em risco a obtenção de soluções em tempo "útil".
O processo de classi�cação ocorre num conjunto de dados, num dado espaço de�nido pelas
características. É nesse espaço de características que a decisão sobre a classe de dados

6.2. SELECÇÃO DE CARACTERÍSTICAS 61
desconhecida é tomada, por exemplo a de�nição de um hiperplano separador do espaço
mapeado, feito pela MVS. Desta forma a solução de características pode igualmente ser
considerada um processo computacional que ocorre numa etapa anterior ao processo de
treino do classi�cador, tendo como objectivo a selecção de características relevantes no
espaço e de retirar características irrelevantes ou danosas ao processo de classi�cação. São
conhecidos vários métodos para selecção de características. O objectivo �nal é obter o
melhor subconjunto "óptimo" de características, que melhor se adapte ao problema em
questão. Podemos dividir a selecção de características em três segmentos: Wrappers, Filters
e Embedded. Os Wrappers usam algoritmos de pesquisa sobre o espaço de procura, de
forma a tentar obter a solução óptima do sub-conjunto de características segundo uma dada
função objectivo. Como é sabido, os problemas de optimização padecem, de uma forma
geral, da seguinte problemática: esforço computacional versus qualidade da solução obtida.
Além de que muitas vezes não é obtido um óptimo global mas sim de um óptimo local. O
algoritmo mais simples de selecção de características é conhecido como Best Feature (BF).
Cada característica é usada individualmente para treinar o classi�cador e o resultado da sua
avaliação é tomado em função do seu desempenho. São então consideradas como melhores
características as que obtiverem melhor desempenho em relação a uma dada métrica. O
BF é útil no sentido de analisar a importância de uma característica em termos individuais
em detrimento da avaliação das interdependências entre características. A complexidade
da classi�cação reside nas interdependências que surgem entre características. Visto que
o esforço computacional aumenta de uma forma exponencial à medida que o número de
características aumenta, torna-se imperativo o uso de heurísticas a �m de explorar o espaço
de procura. Ao longo destes anos vários algoritmos tem vindo a ser propostos, tendo
sempre como meta o melhor conjunto de características. Os algoritmos mais conhecidos
são o SFS (sequencial forward selection) e o SBS (sequencial backward selection), ambos
baseados no algoritmo de Greedy. O SFS é um algoritmo botton-up, iniciado com uma
única característica, obtida a partir do BF. Iterativamente vai acrescentando características
do espaço de procura (características não seleccionadas), através da medição de desempenho
do classi�cador, de forma que a qualidade da solução obtida não pior. O SBS é um algoritmo
top-down em que a diferença para o SFS é que é iniciado com o conjunto de características
completo e vai eliminando as menos importantes, ou seja as que menos alteram a função
objectivo. Naturalmente surgem variantes do SFS e do SBS, nomeadamente o SFFS
(sequencial forward �oating selection) e o SBFS (sequencial backward �oating selection).
Apesar de existir um acréscimo de esforço computacional, segundo Jain et al. [15, 16], estes
são os métodos que melhor combinam tempo de execução com qualidade dos resultados.
Dando o exemplo do SFFS, após a inclusão de uma nova característica é analisado o novo

62 6. DADOS USADOS NA FASE DE APRENDIZAGEM DO CLASSIFICADOR
conjunto no sentido de veri�car a possibilidade de excluir uma característica do conjunto
(sem ser naturalmente a característica incluída nessa iteração) de forma a termos um ganho
por parte da função objectivo. Os Filters são semelhantes aos Wrappers na abordagem
de procura, mas em vez de usarem a avaliação segundo um modelo, é feito segundo um
�ltro. Um dos �ltros mais usados é o coe�ciente de correlação que indica a relação entre
as características. Os Embedded como o próprio nome indica, estão inseridos no próprio
modelo, como exemplo temos: Random forest (RF), Random multinomial logit (RMNL),
Ridge Regression, Decision Tree, Memetic Algorithm.

CAPÍTULO 7
Estado da arte
No capitulo 5, enumerámos alguns algoritmos de máquinas de aprendizagem, tendo sido
referido que no nosso trabalho nos iríamos debruçar sobre redes neuronais e máquinas de
vectores de suporte. Não faz parte dos objectivos traçados, estabelecer qualquer tipo de
comparação entre algoritmos, tendo sido escolhidos os atrás numerados, pelo simples facto de
constituírem os métodos mais usados ao longo da vasta literatura consultada. Vamos a seguir
descrever algumas das abordagens [18], apresentado sempre que possível o desempenho e
o método usado. Sublinhe-se que alguns resultados não são comparáveis, quer porque as
bases de dados utilizadas não são as mesmas, quer porque os critérios de comparação dos
resultados também não o são. Veja-se o exemplo de resultados apresentados em termos de
performance Q3 e de AUC.
O método, PHD é o primeiro método a ultrapassar o limite de 70% de Q3, dos métodos de
previsão de estrutura secundária (classi�cando em 3 classe folhas β, α-hélice e estruturas não
regulares), usando a tabela de redução de 8 para 3, a partir do código DSSP (Dictionary of
Protein Secondary Structure), apresentado na tabela 7.1, é usada uma rede neuronal de dois
níveis que tem sido adoptada por vários outros métodos (tal como JNet e PSIPRED), em que
fundamentalmente varia a tabela de redução. Bastante mais bem sucedido é o método GOR
(Garnier-Osguthorpe-Robson), baseado na ideia de que a previsão da estrutura secundária
não é mais do que um processo de tradução de uma mensagem (estrutura primária) para
63

64 7. ESTADO DA ARTE
Tabela 7.1: Tabela de redução de 8 − 3 usado no método PHD. As duas últimas colunas
correspondem à classi�cação DSSP.Redução Código DSSP Descrição
H H Alpha-helix
H G 3-helix (3/10 helix)
H I 5-helix (pi helix)
C B residue in isolated beta-bridge
E E Extended strand, participates in beta ladder
C T Hydrogen bonded turn
C S Bend
C (no code) Loop/coil
outra (estrutura secundária). Estatísticas bem fundamentadas, incluem não somente a
composição de aminoácidos mas igualmente tuplos de ordem 2 e 3 resultando num método
robusto e teoricamente correcto, cuja terceira versão (GOR III) atinge níveis de exactidão
um pouco acima dos 60%. A versão GOR V atinge valores na ordem de 73.5% de Q3
[11]. O Pretator, é um método baseado numa rede neuronal que considera que os padrões
de aminoácidos podem formar ligações de hidrogénio entre as estruturas folhas β e α-
hélice, considera substituições encontrados em alinhamentos da sequência e os gaps são
considerados como hélices. Os resultados de Q3 foram 71, 9% para o PHD e 68, 6% para
o PREDATOR [21]. Lukasz Kurgan e Ke Chen [20], propõe um método LLSC-PRED, que
inclui a extracção de 58 características descrevendo as propriedades da composição e físico-
químicas das sequências. Podemos evidenciar, as características isolectricas, hidrofóbicas,
composição de aminoácidos, tuplos de ordem 2 e grupos químicos. A taxa de acerto foi da
ordem de 62%, utilizando uma máquina de vectores de suporte. Mary Qu Yang, Jack Y.
Yang e YiZhi Zhang [38], propõem um método de extracção de 81 características a partir
da sequência, nomeadamente da composição dos amionoácidos, do enquadramento de cada
um dos aminoácidos na tabela 9-gram (tabela 7.2), hidrofobicidade e complexidade (obtida
a partir da medida de Shannon´s [19]) Os autores não apresentam no artigo resultados
quantitativos.
Yang Yang, Bao-Liang Lu, e Wen-Yun Yang [39], propõem um algoritmo inspirado em
classi�cação de textos e técnicas baseadas em segmentação de palavras chinesas. As
sequências são analisadas e extraídas as palavras (sub-palavras da sequência) que são tuplos
de ordem k, com k a variar entre 2 e 4, que representam algum signi�cado, como por exemplo
a frequência, dando origem ao chamado dicionário. Além destas palavras é acrescentado

65
Tabela 7.2: Tabela de agrupamento em nove grupos de acordo com as propriedades físicas
de químicas dos aminoácidos.Grupos Residuos Descrição
1 C Cysteine, remains strongly during evolution
2 M Hydrophobic, sulfur containing
3 N,Q Amides, polar
4 D,E Acids, polar, charged
5 S,T Alcohols
6 P,A,G Small
7 I,V,L Aliphatic
8 F,Y,W Aromatic
9 H,K,R Bases, charged, positive
ao dicionário os 20 aminoácidos conhecidos. É igualmente adicionado ao dicionário alguns
motivos que contenham signi�cado biológico e que constem em bases de dados públicas.
No caso foram acrescentado os motivos que constam na base de dados PROSITE [5]. Após
a construção do dicionário, as sequências são segmentadas de forma a de�nir o vector de
características.
O algoritmo da máquina de aprendizagem usando é uma máquina de vectores de suporte,
tendo sido testado em dois grupos, da familia SCOP e outro da familia GPCR (G- Protein
Coupled Receptors), tendo obtido taxas de acerto de 92.6% e de 88.8% em função dos
grupos testados.
É neste trabalho que a metodologia por nós desenvolvida é baseada.

66 7. ESTADO DA ARTE

CAPÍTULO 8
Metodologia
A ferramenta computacional para classi�cação de proteínas foi implementada em Matlab e
está estruturada em 6 partes distintas:
1. Leitura das sequências
2. Leitura do conjunto de treino e teste
3. Extracção de características
4. Selecção de características
5. Treino do método
6. Execução do método
8.1 Leitura das sequências
As sequências usadas em testes encontram-se em formato Fasta. Além da sequência de
aminoácidos, este formato poderá conter informação adicional sobre a mesma. Na aplicação
desenvolvida, além da sequência, é guardado o seu identi�cador. Dando o exemplo da
67

68 8. METODOLOGIA
SCOP40 Minidatabase, temos o �cheiro SCOP40mini.fasta com informação do tipo (Fig.
8.1)
Figura 8.1: Excerto do �cheiro SCOP40 Minidatabase em formato Fasta.
8.2 Leitura conjunto de treino/teste
De forma a treinar e testar as máquinas implementadas, torna-se necessário ler o �cheiro
que contêm todo o conjunto de sequências (identi�cado pelo seu identi�cador) e a sua clas-
si�cação em relação a cada classe. É um �cheiro de texto, designado por Cast Matrix, com
a extensão .cast , em que na primeira coluna se encontra o identi�cador da sequência e na
primeira linha a identi�cação da classe. A restante informação encontra-se estruturada da se-
guinte forma: 1 - Treino Positivo, 2 - Treino Negativo, 3 - Teste Positivo e 4 - Teste Negativo,
para uma dada sequência de uma dada classe. Para os exemplo da SCOP40 Minidatabase,
temos a matriz Cast Matrix, designada por SCOP40mini−sequenceminidatabase19.cast, com
uma estrutura do tipo representado na Fig. 8.2. Neste caso é indicado que a sequência com
o identi�cador "d1c75a" é um teste positivo (4), d1ctj é um treino negativo (2) ambos
para a classe "a.118.1.".
Sublinhe-se que a aplicação estabelece a relação entre a informação (exemplo sequências
e casos de treino ou teste) tirando partido da posição ao nível das linhas das matrizes.
Exempli�cando, na linha 25 de um �cheiro que contém as sequências está uma sequência
com um dado identi�cador, por outro lado no �cheiro da Cast Matrix a linha 25 corresponde
ao mesmo identi�cador contendo a informação respectiva de treino ou teste para cada uma
das classes numeradas nessa linha (representadas em colunas). Foi aceite esta premissa no
sentido de tirar partido da relação da informação usando matrizes. Esta premissa em si não

8.3. EXTRACÇÃO DE CARACTERÍSTICAS A PARTIR DA SEQUÊNCIA 69
Figura 8.2: Excerto do �cheiro SCOP40mini−sequenceminidatabase19.cast que representa
a matriz Cast Matrix.
constitui uma restrição, já que caso a informação não se encontre relacionada por posição,
facilmente seria relacionada usando o identi�cador da sequência.
8.3 Extracção de características a partir da sequência
Conforme indicamos no início desta dissertação, o objectivo deste trabalho, consiste no
desenvolvimento de uma nova metodologia para a extracção de características a partir da
sequência da proteína, no sentido de dar um contributo na classi�cação mais e�ciente das
proteínas a partir da sequência.
A nova metodologia combinando técnicas de extracção ao nível físico-químico das proteínas
e técnicas de Text-Mining com segmentação de palavras.
Neste estudo, e tendo como objectivo conseguir o melhor classi�cador, iremos avaliar
individualmente o desempenho de cada uma das seguintes características:
1. Contagem aminoácidos
2. Composição atómica
3. Peso molecular
4. Ponto isoeléctrico

70 8. METODOLOGIA
5. Dicionário de tuplos
8.3.1 Contagem aminoácidos, Composição atómica, Peso molecular
Descrição e sua relevância
Conforme já referido, uma proteína é composta por aminoácidos, representados na sequência
que pretendemos classi�car.
Por outro lado, cada aminoácido é uma molécula orgânica em termos atómicos constituída
por átomos de Carbono, Hidrogénio, Oxigénio e Azoto unidos entre si, podendo em algumas
situações conter Enxofre.
Cada átomo, constituinte de cada aminoácido da proteína, possui um peso molecular.
Número de características extraídas:
Contagem aminoácidos: iremos obter 20 características, que correspondem à contagem
de cada um dos aminoácidos na sequência da proteína.
Composição atómica: iremos obter 5 características , que correspondem ao número de
cada um dos átomos, Carbono, Hidrogénio, Oxigénio, Azoto e Enxofre, presentes em cada
aminoácido constituinte da proteína.
Peso molecular: iremos obter 1 característica, o valor do peso de todas as moléculas
da proteína, correspondente ao somatório de peso molecular de cada um dos átomos dos
aminoácido constituintes da proteína.
8.3.2 Ponto isoeléctrico
Descrição e sua relevância
O pI de uma proteína corresponde ao valor de pH em que a molécula se encontre electrica-
mente neutra, ou seja, quando o número de cargas positivas for igual ao número de cargas
negativas. O pI das proteínas é determinado experimentalmente, sendo considerado o pH
no qual a molécula não migra quando submetida a campo eléctrico (processo conhecido
de eletroforese). Cada proteína possui um pI característico, que re�ecte a proporção entre
aminoácidos ácidos e básicos.

8.3. EXTRACÇÃO DE CARACTERÍSTICAS A PARTIR DA SEQUÊNCIA 71
Número de características extraídas:
Iremos obter 1 característica, que correspondem ao valor do pH onde a proteína tem carga
nula.
8.3.3 Dicionário de Tuplos.
Descrição e sua relevância.
Muitos métodos têm sido desenvolvidos ao longo destes anos, muitos deles baseados na
contagem de aminoácidos, ou extensões da contagem não de 1 aminoácido mas sim de k
aminoácidos, sendo k um número inteiro maior que 1.
Yang Yang [39], propõe um método de extracção de características inspirado na classi�cação
de textos e técnicas de segmentação de palavras chinesas.
A nossa metodologia de construção de tuplos, baseia-se neste trabalho, acima de tudo nas
ideias mestres associadas à classi�cação de textos, aplicando técnicas de Text-Mining. São
essa técnicas, descritas no trabalho de Yang Yang [39], que adoptamos para a selecção dos
tuplos que ocorrem ao longo da sequência da proteína.
A selecção dos tuplos que posteriormente irão servir de base para a construção do dicionário,
pode ocorrer segundo um dos critérios apresentados a seguir. Na fase de estudo, e com
o objectivo de obter o classi�cador com o melhor desempenho, foram testados os vários
critérios, contudo sublinhe-se que na construção do dicionário somente um deles é tido em
linha de conta, função da escolha efectuada.
8.3.3.1 Possíveis critérios de escolha dos tuplos:
• Frequência (TF)
São escolhidos os tuplos que mais frequentemente ocorrem ao longo de todas as
sequências de treino, partindo da assunção que pelo facto de aparecem mais vezes, do
que outros tuplos que aparecem raramente, tenham mais importância em termos da
estrutura das proteínas.
Seja f a frequência com que o tuplo t aparece na sequência s, wt o peso do tuplo t,
e N a dimensão do conjunto de treino.
O valor (TF), é o peso que o tuplo t apresentada ao longo de todas as sequências de

72 8. METODOLOGIA
treino, é obtido pela fórmula 8.3.1.
wt =N∑s=1
ft,s (8.3.1)
O valor ft,s, que também será usado nos critérios seguintes, é normalizado dividindo
o valor pelo possível número de tuplos para a sequência s.
• tf-idf
Este critério de selecção, tem em linha de conta a distribuição de cada tuplo ao longo
de todas as sequências do conjunto de treino. O valor é obtido, a partir da proporção
entre o número de ocorrências de um tuplo t, ou seja a frequência do tuplo numa
sequência s, e o inverso da proporção do número de sequências do conjunto de treino
em que o tuplo aparece pelo menos uma vez. O valor do tf-idf é obtido pelas fórmulas
8.3.2 e 8.3.3.
wt,s = ft,s × logN
nt(8.3.2)
O peso de t, designado por wt, é de�nido pelo valor máximo de wt,s, isto é:
wt = maxs∈Twt,s (8.3.3)
onde T representa todo o conjunto de dados.
• Entropia.
A física usa o termo entropia para descrever a quantidade de desordem associada a
um sistema. No presente contexto, este termo tem uma signi�cado semelhante, ele
mede o grau de desordem de um conjunto de dados. O valor da entropia é obtido
pelas fórmulas 8.3.4 e 8.3.5
.wt,s = log(ft,s + 1.0)× (1 +1
logN
∑s∈T
[ft,sntlog
fs,tnt
]) (8.3.4)
O peso de t, wt, é de�nido como o valor máximo de wt,s:
wt = maxs∈Twt,s (8.3.5)
onde T representa todo o conjunto de dados.
Os valores obtidos wt, a partir de cada um dos critérios, servirão de base para construção
do dicionário.

8.3. EXTRACÇÃO DE CARACTERÍSTICAS A PARTIR DA SEQUÊNCIA 73
8.3.3.2 Construção do dicionário.
Conforme referimos anteriormente,o dicionário é construído em função de um dos critérios
anteriormente apresentados (TF, tf-idf, Entropia). Na construção do dicionário, só iremos
considerar tuplos que apresentem um valor, dado pelos critérios referidos, superior a um
dado número.
Tome-se por exemplo o critério TF, em que para tuplos de ordem 3, apresentam os seguintes
valores em termos de TF:
{GLM=0.4, PFL=0.36, GTK=0.2, HTS=0.34, LLM=0.12, MLM=0.13, KHR=0.62,
SMM=0.39, LRP=0.52, LSQ=0.73, FLA=0.44, WTT=0.31}.
De�nindo como valor mínimo 0.3, para o critério TF, os tuplos que seriam integrados no
dicionário seria então:
{GLM, PFL, HTS, KHR, SMM, LRP, LSQ, FLA, WTT}.
Na aplicação computacional desenvolvida, esta abordagem é implementada de forma a
controlar o número máximo de tuplos que se pretende que o dicionário venha a conter,
ou seja farão parte do dicionário os n tuplos, que apresentem os valores mais elevados
segundo o critério escolhido.
Seguindo o exemplo anterior teríamos então para o critério TF o seguinte dicionário (D):
D = {GLM, PFL, HTS, KHR, SMM, LRP, LSQ, FLA, WTT}.
8.3.3.3 Construção do vector de características.
Considerando a seguinte sequência de treino:
s=GLMHGLMMTKHRSSMLRPLSQLRQTTWTTTLRQLSQHTSMM
iríamos construir o vector de características para os tuplos de ordem 3, determinando o
número de ocorrências de cada elemento do dicionário na sequência s.
s=GLMHGLMMTKHRSSMLRPLSQLRQTTWTTTLRQLSQHTSMM
A bold encontra-se as palavras que constam no dicionário D.
Veri�camos por exemplo que o tuplo GLM do dicionário, ocorre na sequência 2 vezes, o
tuplo PFL corre 0 vezes, o tuplo WTT ocorre 1 vez. Seguindo este critério teríamos então

74 8. METODOLOGIA
o vector de características de�nido por:
vc = (2, 0, 1, 1, 2, 1, 1, 0, 1).
Na metodologia por nós implementada, consideramos sobreposições entre tuplos, já que não
existe qualquer fundamento em termos biológicos para que tal não venha a acontecer. Assim
sendo, os tuplos HTS e SMM, encontram-se sobrepostos na sequência.
De forma a evitar encontrar palavras na sequências que não constem no dicionário, são
adicionadas ao dicionário os 20 aminoácidos conhecidos. Do exposto resulta que o dicionário
�nal D, será então constituído por:
D = {G,A,L,V,I,P,F,S,T,C,Y,N,Q,D,E,R,K,H,W,M,GLM,PFL,HTS,KHR,SMM LRP,LSQ,
FLA,WTT}.
Consequentemente o vector de características �nal será dado por:
vc = (0, 0, 6, 0, 0, 0, 0, 5, 7, 0, 0, 0, 1, 0, 0, 0, 4, 3, 1, 6, 2, 0, 1, 1, 2, 1, 1, 0, 1).
Por último, sublinhe-se que o valor de k altera signi�cativamente o valor do espaço de
procura, para k = 10, o espaço de procura é de 2010 (= 10240000000000), traduzindo-se
evidentemente num esforço computacional bastante elevado. Num estudo apresentado por
Wen-Yun Yang [37] (k variando de 1 a 5), a taxa de acerto mais elevada é obtida para k = 5
em algumas situações, contudo em termos médios o melhor valor é para k = 4. Re�ra-se
que passar de k = 4, para k = 5, é passar de um espaço de soluções de 160.000, para
3.200.000, traduzindo logicamente num grande acréscimo computacional.
Número de características extraídas
O número de características, vai depender do número de tuplos adicionados ao dicionário.
No caso usado como exemplo, teríamos 29 características.
Apresentamos de seguida o �uxograma e a Fig. 8.3, representativos da metodologia seguida
para a extracção de características usando tuplos de ordem k.
Fluxograma.
1. Extracção a partir da sequência dos tuplos de ordem k
2. Selecção do critério de escolha dos tuplos
a. Frequência
b. tf-idf

8.4. SELECÇÃO DE CARACTERÍSTICAS 75
c. Entropia
3. Construção do dicionário a partir do critério de escolha em 2.
4. Segmentação da sequência a partir do dicionário
5. Construção do vector de características a partir do dicionário e da segmentação
6. Aplicação do classi�cador
Figura 8.3: Metodologia seguida para a extracção de características usando tuplos de ordem
k.
8.4 Selecção de características
Conforme referimos anteriormente, existem vários métodos de selecção de características,
tendo como objectivo aumentar a performance do classi�cador, reduzindo a dimensionalidade
do espaço de características. O algoritmo por nós usado foi o SFS por nós referido em 6.2,
em que cada característica é usada individualmente para treinar o classi�cador e o resultado
da sua avaliação é tomado em função do seu desempenho. O passo seguinte, consiste
em acrescentar de uma forma iterativa ao classi�cador as características que apresentavam,
melhor desempenho e não diminuíam a performance do classi�cador. Tendo perfeita noção
que este processo leva a obtenção de óptimos locais, foram testadas outras combinações de
forma aleatória no sentido de tentar obter melhores resultados.
8.5 Treino do método de máquina de aprendizagem
O treino do classi�cador, foi feito de uma forma interactiva, tendo como objectivo a obtenção
do melhor valor para a métrica usada. Conforme referido na secção 2.15, a análise da taxa de

76 8. METODOLOGIA
acerto, em conjunto de dados não balanceados, constitui por si só uma métrica que poderá
não re�ectir o correcto desempenho do classi�cador. Assim sendo, tomou-se como métrica
de avaliação a análise da curva ROC e respectivo cálculo de AUC. De�niu-se um valor
(representado na aplicação por Goal) em que enquanto este não fosse atingido, o processo
de treino era repetido, até um dado número de iterações (representado na aplicação por
MaxIteracoes).
Na fase de pesquisa, foram testadas várias con�gurações ao nível dos métodos, quer de
número de camadas, parâmetros, funções de treino, etc. Não fazia parte dos objectivos deste
trabalho determinar as variações de desempenho, pelo que em determinado momento do
trabalho determinaram-se as con�gurações que serviriam para comparar os desempenhos dos
diversos classi�cadores. Com isto, estamos a dizer que variando os parâmetros ( por exemplo
a topologia no caso da rede neuronal), ou outros elementos, o resultado �nal certamente
será diferente, podendo mesmo ser superior. No entanto neste caso não permitiria comparar
classi�cador ao nível da selecção de características.
Para os testes por nós efectuados foram de�nidos os seguintes parâmetros:
Rede Neuronal
• Número de camadas: 3
• Número de neurónios: 20
• Método de treino : traincgb
� A função de treino actualiza os valores de "weights" e "biases", conjugando o
backpropagation gradiente com o Powell-Beale restarts
• Epoch = 500 (número máximo de épocas por treino)
• Goal = 0 (objectivo a atingir)
• Lr = 0.01 (taxa de aprendizagem em cada época)
• Maxfail = 100 (número máximo de falhas em validação)
• Mc = 0.9 (quantidade de momentum, permitindo à rede ajustar o peso em função
do estado anterior)
• MinGrad = 1e−10 (performance mínima do gradiente)

8.6. EXECUÇÃO DO MÉTODO DA MÁQUINA DE APRENDIZAGEM 77
Máquinas de Vectores de Suporte
• Função de Kernel: rbf
• RBFSigmaValue: 1
• PolyorderValue: 3
• MlpParamsValue: [1,−1]
• MethodValue: SMO
8.6 Execução do método da máquina de aprendizagem
Após a fase de treino, é executado o classi�cador que melhor performance obteve na fase
de treino, segundo a métrica usada.
8.7 Aplicação computacional
A metodologia desenvolvida foi implementada numa ferramenta computacional designada
por ISFECP - Intelligent System for Feature Extraction and Classi�cation of Proteins, do
qual apresentamos na Fig. 8.4 o écran principal.
A aplicação permite o uso de diferentes algoritmos de máquinas de aprendizagem, no-
meadamente máquinas de vectores de suporte e redes neuronais. Por outro lado permite
diferentes con�gurações quer ao nível das máquinas de aprendizagem referidas, quer ao nível
da extracção de características. A forma de utilização encontra-se detalhada no anexo A,
respeitante ao manual de utilização.
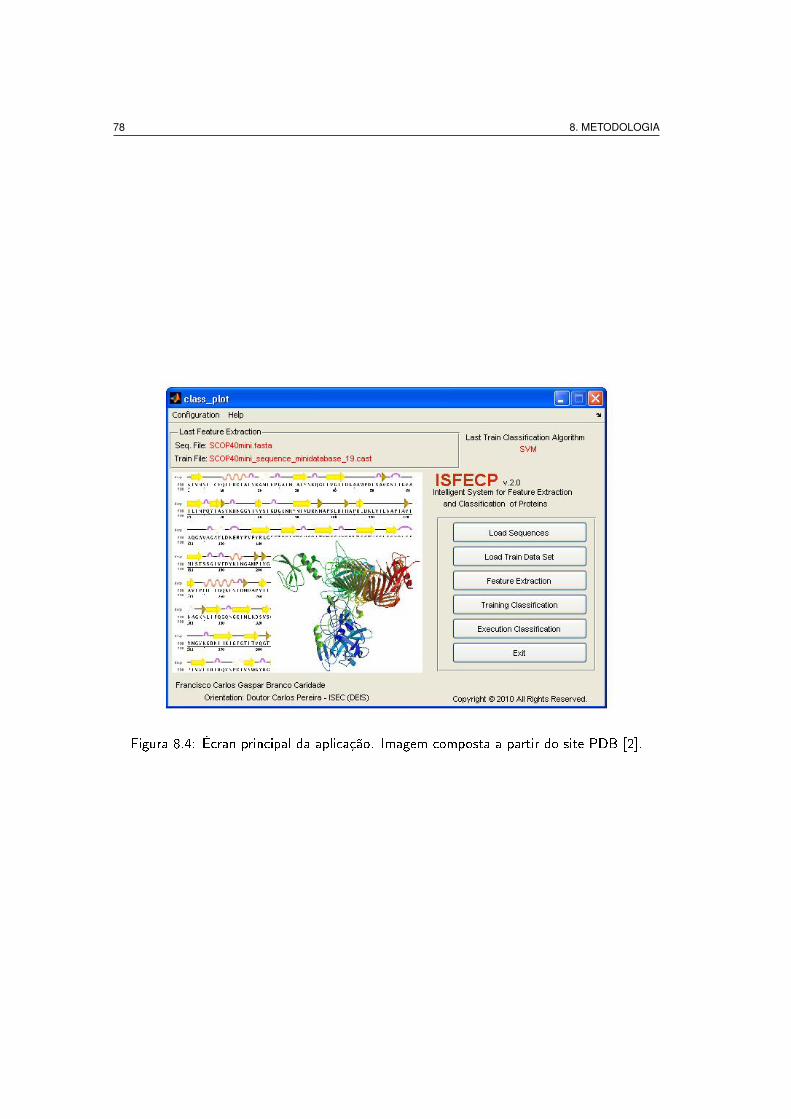
78 8. METODOLOGIA
Figura 8.4: Écran principal da aplicação. Imagem composta a partir do site PDB [2].

CAPÍTULO 9
Bases de dados utilizadas
De forma a avaliar o desempenho dos métodos das máquinas de aprendizagem por nós
implementadas, tornou-se imperativo o uso de dados padrão, de forma a comparar os
resultados obtidos. As bases de dados escolhidas foram do PCBC (Protein Classi�cation
Benchmark Collection) [4], que contém um conjunto de sequências subdivididas em treino
positivo/negativo e respectivo conjunto de teste positivo/negativo, baseadas em famílias de
acordo com a classi�cação obtida pelo SCOP e CATH. O conjunto de classes são codi�cadas
num formato simples de matriz (cast matrix) conforme explicado na secção 8.2. Por outro
lado, encontra-se um conjunto de matrizes, que contêm a distância de cada sequência em
relação a todas as outras usando métodos como Blast e Smith-Waterman. A avaliação de
um método na base de dados consiste em calcular uma medida de desempenho, a partir da
curva de ROC, o AUC. Os resultados obtidos são diferenciados em relação a cada método de
classi�cação, tais como: 1NN (Nearest neighbour), SVM (Support Vector Machines), ANN
(Arti�cial Neural Network), RF (Random Forests), conforme podemos ver na Fig. 9.1. Um
dos problemas com que nos deparamos, foi o facto de não estarmos perante bases de dados
balanceadas, ou seja o número de casos positivos e negativos serem relativamente similares,
quer para treino quer para teste.
No nosso trabalho usamos por base 2 bases de dados, a SCOP40−Minidabase (código
interno no PCBC: PCB00019), e a SCOP95−Superfamily−Family (código interno no PCBC:
79

80 9. BASES DE DADOS UTILIZADAS
PCB00001). A primeira de dimensão reduzida, 1357 sequências e 55 classes, em que os re-
sultados, resumo e per�l do PCBC são apresentados nas Fig. 9.1, 9.2 e 9.3 respectivamente.
A segunda de dimensão mais elevada, 11944 sequências e 246 classes, em que os resultados,
resumo e per�l do PCBC são apresentados nas Fig. 9.4, 9.5 e 9.6 respectivamente.
Figura 9.1: Resultados para SCOP40 Minidatabase (PCB00019) [4].
Figura 9.2: Resumo do SCOP40 Minidatabase (PCB00019), em termos de per�l [4].
Figura 9.3: Excerto das primeiras 5 proteínas, com per�l detalhado do SCOP40
Minidatabase (PCB00019) para cada classe (55) [4].

81
Figura 9.4: Resultados para SCOP95−Superfamily−Family (PCB00001) [4].
Figura 9.5: Resumo do SCOP95−Superfamily−Family (PCB00001), em termos de per�l
[4].
Figura 9.6: Excerto das primeiras 5 proteínas, com per�l detalhado do SCOP95 Superfamily
Family (PCB00001) para cada classe (246) [4].

82 9. BASES DE DADOS UTILIZADAS

CAPÍTULO 10
Resultados e sua análise
Para a obtenção de resultados foram usados os parâmetros referidos na secção 8.5, em
que a rede neuronal, é treinada de forma iterativa tendo como objectivo a obtenção de um
dado valor de AUC (de�nido na aplicação como Goal), até um número máximo de iterações
(de�nido na aplicação como MaxIteracoes). Os valores usados no treino foram de Goal=0.9
e MaxIteracoes=100. Quando atingido o valor do Goal ou o de MaxIteracoes, é guardada a
rede que obteve melhor AUC. Após a fase de treino é executada a rede anterior (de melhor
AUC), guardando no �cheiro resultados.xls os valores obtidos, nomeadamente o número de
características envolvidas na classi�cação, a taxa de verdadeiros positivos (TVP), a taxa de
falsos positivos (TFP), a taxa de acerto (TA) e o valor de AUC.
Os valores apresentados nas tabelas seguintes, são valores médios em função do número de
classe envolvidas no processo de classi�cação, neste caso 55 classes ou 246 conforme a base
de dados escolhida.
Conforme referimos no inicio deste trabalho, o objectivo consiste em desenvolver um sistema
pela combinação de métodos de extracção de características a partir da sequência de uma
proteína, usando classi�cadores inteligentes, nomeadamente redes neuronais e máquinas
de vectores de suporte. Visto que o objectivo se baseia no método de extracção de
características e não no método do classi�cador em si, optou-se por comparar os diferentes
83

84 10. RESULTADOS E SUA ANÁLISE
métodos de extracção de características usando uma rede neuronal.
10.1 Validação dos resultados.
De forma a validarmos os resultados por nós obtidos, houve necessidade de executar em
primeira instância a rede neuronal, usando as características extraídas a partir do vector das
distâncias/similaridade entre cada uma das proteínas e as proteínas do conjunto, obtidas
a partir do Blast [1]. O objectivo consiste em obter valores de referência em relação aos
obtidos pelo ICGEB (International Center for Genetic Engineering and Biotechnology) no
site de avaliação de desempenho em relação ao conjuntos de dados de testes [4].
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.
• Critério de extracção de características
� Distâncias entre as proteínas, obtidas a partir do Blast.
A variação entre a rede por nós implementada e a rede do site é de −2.75%, conforme
pudemos constar na tabela 10.1, ou seja os resultados por nós obtidos situam-se 2.75%
abaixo do valor obtido no site.
Além do valor do AUC, foram igualmente determinados para a rede por nós implementada,
os seguintes valores médios: TVP=0.184, TFP=0.003, Taxa de acerto=0.979, obtidos a
partir das 55 classes, para o conjunto de dados testados. Os valores obtidos pela nossa
Tabela 10.1: Análise comparativa entre o desempenho obtido pela nossa rede neuronal (RN)
em relação aos valores obtidos pelo ICGEB, usando a extracção de características a partir
do Blast.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Site[4] 1357 - - - - - - 0.798 -
RN 1357 0.184 - 0.003 - 0.980 - 0.777 -2.75%

10.2. DESEMPENHO INDIVIDUAL EM FUNÇÃO DO CRITÉRIO DE EXTRACÇÃO DE CARACTERÍSTICAS. 85
rede, usando o vector de características das distâncias entre as proteínas, obtido a partir
do Blast, irão ser usados como referência de forma a avaliar o desempenho dos diferentes
critérios de extracção de características por nós implementados. As tabelas seguintes serão
apresentadas recorrendo ao uso de abreviaturas, como objectivo de visualizar em cada linha
todos os valores obtidos. Assim sendo, as abreviaturas são as seguintes:
• NC - Número de características usadas no processo de classi�cação.
• D(k=x) - Dicionário construído pelo processo anteriormente descrito, usando tuplos
de ordem x.
• NA - Características extraídas a partir do número de aminoácidos.
• CA - Características extraídas a partir da composição atómica.
• PM - Características extraídas a partir do peso molecular.
• PI - Características extraídas a partir do ponto isoeléctrico.
• RN - Rede neuronal.
• MVS - Máquina de vectores de suporte.
• Todas - Constituído por todas as características D+CA+PM+PI.
Os desvios apresentados nas diversas tabelas, são resultantes da diferenças entre os valores
apresentados a bold (primeira linha), e o respectivo valor apresentado, para cada coluna.
10.2 Desempenho individual em função do critério de ex-
tracção de características.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.

86 10. RESULTADOS E SUA ANÁLISE
Tabela 10.2: Avaliação de desempenho individual em função do critério de extracção de
características.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Blast 1357 0.184 - 0.003 - 0.980 - 0.777 -
CA 5 0.051 -72.53% 0.002 -33.09% 0.977 -0.25% 0.767 -1.27%
NA 20 0.079 -57.33% 0.002 -28.01% 0.978 -0.19% 0.759 -2.30%
PM 1 0.032 -82.53% 0.005 58.31% 0.974 -0.62% 0.716 -7.89%
PI 1 0.014 -92.36% 0.003 -11.66% 0.976 -0.42% 0.590 -24.10%
Figura 10.1: Avaliação de desempenho individual em função do critério de extracção de
características.

10.3. DESEMPENHO DOS TUPLOS DE ORDEM K. 87
Da análise dos resultados obtidos na tabela 10.2 e na Fig. 10.1, constatamos que os
resultados obtidos a partir do Blast são superiores aos obtidos pelos outros métodos de
extracção. No entanto a diferença entre o Blast, a contagem de aminoácidos e a composição
atómica não é signi�cativa, com um número de características bastante inferior. Sublinhe-se
que estamos a falar de uma variação de 1.27%, em que o número de características é de
1357 para o Blast e de 5 para a contagem de aminoácidos.
10.3 Desempenho dos tuplos de ordem K.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.
• Critério de selecção na construção do dicionário
� TF.
Da análise dos resultados evidenciados na tabela 10.3 e da Fig. 10.2, constatamos que os
tuplos de ordem 4 (k=4) obtém o melhor desempenho quando comparado com a extracção
de tuplos para k=2 e k=3. Sublinhe-se que passar de k=2 para k=3, não apresenta
um incremento de desempenho signi�cativo, no entanto a passagem de k=3 para k=4, o
incremento é bastante notório. Conforme já referido, à medida que o k aumenta, o esforço
computacional igualmente aumenta, no entanto é perfeitamente justi�cado face ao aumento
de desempenho apresentado.
Tabela 10.3: Avaliação de desempenho dos tuplos de ordem k.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Blast 1357 0.184 - 0.003 - 0.980 - 0.777 -
D(k=2) 34 0.062 -66.54% 0.002 -26.22% 0.978 -0.20% 0.719 -7.99%
D(k=3) 33 0.060 -67.31% 0.002 -34.87% 0.978 -0.20% 0.722 -7.03%
D(k=4) 32 0.113 -38.81% 0.003 -21.57% 0.979 -0.13% 0.777 0.04%

88 10. RESULTADOS E SUA ANÁLISE
Figura 10.2: Avaliação de desempenho dos tuplos de ordem k.
10.4 Desempenho dos tuplos de ordem 4 em função do
número de características extraídas.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.
Tabela 10.4: Avaliação de desempenho do tuplos de ordem 4 (D=4) em função do número
de características extraídas.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Blast 1357 0.184 - 0.003 - 0.980 - 0.777 -
D TF 32 0.132 -38.81% 0.003 -21.57% 0.979 -0.13% 0.777 0.04%
D TF 75 0.133 -27.85% 0.003 0.52% 0.979 -0.14% 0.765 -1.47%
D t�df 28 0.101 -45.08% 0.002 -35.79% 0.979 -0.12% 0.887 14.20%
D t�df 44 0.101 -45.08% 0.002 -22.06% 0.978 -0.12% 0.892 14.44%
D Entropia 41 0.108 -41.27% 0.003 -22.06% 0.979 -0.12% 0.889 14.44%
D Entropia 91 0.104 -41.27% 0.003 -35.79% 0.979 -0.12% 0.887 14.20%

10.5. DESEMPENHO DOS TUPLOS DE ORDEM 4 EM FUNÇÃO DO CRITÉRIO DE SELECÇÃO NACONSTRUÇÃO DO DICIONÁRIO 89
Figura 10.3: Avaliação de desempenho do tuplos de ordem 4 em função do número de
características extraídas.
Tendo sempre presente a importância do esforço computacional versus desempenho, os
resultados apresentados na tabela 10.4 e na Fig. 10.3, foram obtidos no sentido de retirar
ilações sobre o aumento do número de características ao nível dos tuplos de ordem 4. Nesse
sentido para cada critério usado na construção do dicionário, o número de características foi
aproximadamente duplicado e analisado o seu desempenho. Quer para o critério TF, quer
no critério tf-idf, a duplicação do número de características não evidenciou o aumento de
desempenho, alias no caso do TF até piorou o desempenho. No entanto, re�ra-se que uma
variação de 1.47% não é de todo signi�cativa. No caso da entropia, existe algum ganho de
desempenho, mas que não se revela expressivo.
Constatamos então que a duplicação do número de características nos tuplos de ordem 4,
não constitui uma boa opção.
10.5 Desempenho dos tuplos de ordem 4 em função do
critério de selecção na construção do dicionário
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).

90 10. RESULTADOS E SUA ANÁLISE
• Método
� Rede Neuronal.
Figura 10.4: Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção na construção do dicionário.
Conforme referido anteriormente, a construção poderá ser feita em função de 3 critérios, TF,
tf-idf e entropia. Da análise da tabela 10.5 e da Fig. 10.4, veri�ca-se que o dicionário obtido
a partir dos critérios tf-idf e da entropia apresentam um desempenho signi�cativo quando
comparado com o critério TF, cerca de 14%. Na comparação entre o tf-idf e a entropia,
o ganho de desempenho é cerca de 0.24%, não constituindo um acréscimo expressivo. No
entanto, é através do critério da entropia que é obtido o melhor desempenho.
Tabela 10.5: Avaliação de desempenho dos tuplos de ordem 4 (D=4) em função do critério
de selecção na construção do dicionário.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Blast 1357 0.184 - 0.003 - 0.980 - 0.777 -
D TF 32 0.113 -38.81% 0.003 -21.57% 0.979 -0.13% 0.777 0.04%
D t�df 28 0.101 -45.08% 0.002 -35.79% 0.979 -0.12% 0.887 14.20%
D Entropia 41 0.108 -41.27% 0.003 -22.06% 0.979 -0.12% 0.889 14.44%
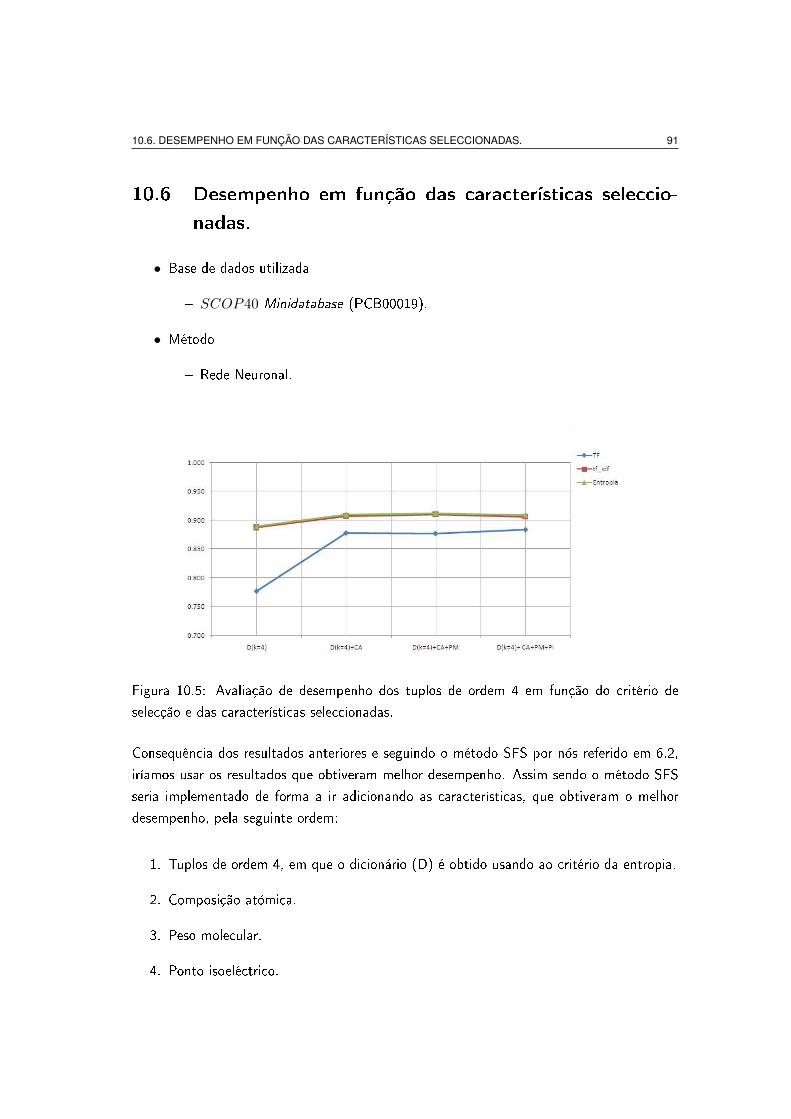
10.6. DESEMPENHO EM FUNÇÃO DAS CARACTERÍSTICAS SELECCIONADAS. 91
10.6 Desempenho em função das características seleccio-
nadas.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.
Figura 10.5: Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção e das características seleccionadas.
Consequência dos resultados anteriores e seguindo o método SFS por nós referido em 6.2,
iríamos usar os resultados que obtiveram melhor desempenho. Assim sendo o método SFS
seria implementado de forma a ir adicionando as características, que obtiveram o melhor
desempenho, pela seguinte ordem:
1. Tuplos de ordem 4, em que o dicionário (D) é obtido usando ao critério da entropia.
2. Composição atómica.
3. Peso molecular.
4. Ponto isoeléctrico.

92 10. RESULTADOS E SUA ANÁLISE
Tabela 10.6: Avaliação de desempenho dos tuplos de ordem 4 em função do critério de
selecção e das características seleccionadas.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
Blast 1357 0.184 - 0.003 - 0.980 - 0.777 -
TF
D 32 0.113 -38.81% 0.003 -21.57% 0.979 -0.13% 0.777 0.04%
D+CA 37 0.166 -9.60% 0.003 -8.03% 0.980 -0.14% 0.878 13.02%
D+CA+PM 38 0.172 -6.77% 0.003 -9.65% 0.980 -0.02% 0.877 12.85%
Todas 39 0.156 -15.22% 0.003 -23.33% 0.9809 0.00% 0.884 13.75%
tf-idf
D 28 0.101 -45.08% 0.002 -35.79% 0.979 -0.12% 0.887 14.20%
D+CA 33 0.147 -19.98% 0.003 -20.96% 0.980 -0.02% 0.907 16.76%
D+CA+PM 34 0.161 -12.40% 0.004 8.73% 0.979 -0.07% 0.910 17.17%
Todas 35 0.155 -15.86% 0.003 -14.64% 0.980 -0.02% 0.906 16.64%
Entropia
D 41 0.108 -41.27% 0.003 -22.06% 0.979 -0.12% 0.889 14.44%
D+CA 46 0.185 0.50% 0.004 4.72% 0.980 -0.03% 0.909 17.06%
D+CA+PM 47 0.186 1.28% 0.004 20.22% 0.980 -0.01% 0.912 17.35%
Todas 48 0.159 -13.76% 0.003 2.73% 0.979 -0.06% 0.909 16.99%
No entanto, razão da qual a frase anterior se encontrar no condicional, iremos da mesma
forma aplicar o SFS partindo da construção do dicionário usando o critério TF e tf-idf, na
mira de encontrar variações de desempenho. Da análise da tabela 10.6 e da Fig. 10.5,
constatamos que quer no caso da construção do dicionário usando o critério tf-idf, quer
usando a entropia, o ganho de desempenho ocorre até ao momento em que se adiciona o
ponto isoeléctrico (PI). Em ambos piorou o desempenho após a inserção da característica
PI, pelo que o algoritmo SFS deve ser interrompido. No caso dos resultados obtidos a partir
do TF, houve variações quer positivas, quer negativas, no que concerne ao desempenho, à
medida que as características iam sendo adicionadas. Usando o algoritmo SFS, o mesmo seria
interrompido após adicionarmos o peso molecular (PM), visto ter provocado um decréscimo
no desempenho.
Fazendo agora uma análise do melhor resultado, concluímos que o melhor desempenho ocorre
com a seguinte combinação:
1. Tuplos de ordem 4, em que o dicionário é obtido atendendo ao critério da entropia.
2. Composição atómica.

10.7. DESEMPENHO ADICIONANDO AS POSSÍVEIS COMBINAÇÕES DAS CARACTERÍSTICASSELECCIONADAS. 93
3. Peso molecular.
traduzindo-se num ganho de desempenho de 17.35%, quando comparado com os resultados
obtidos a partir do Blast. Por outro lado, e tendo em conta o número de características
envolvidas, no caso do Blast é de 1357, enquanto que no nosso método é de 47, ou seja
signi�cativamente inferior e com um ganho de desempenho notável.
10.7 Desempenho adicionando as possíveis combinações
das características seleccionadas.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Rede Neuronal.
• Critério de selecção na construção do dicionário
� Entropia.
De forma a veri�car se o SFS, não tinha originado um óptimo local, foram testadas as
possíveis combinações entre características.
Da análise dos resultados apresentados na tabela 10.7 e na Fig. 10.6 constatamos que
a melhor combinação de características continua a ser a obtida anteriormente a partir do
método SFS.
Tabela 10.7: Avaliação de desempenho, adicionando possíveis combinações de características
seleccionadas em relação ao melhor resultado obtido a partir do SFS em 10.6.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
D+CA+PM 47 0.186 1.28% 0.004 20.22% 0.980 -0.01% 0.912 -
D+CA 46 0.185 -0.77% 0.004 -12.9% 0.980 -0.03% 0.909 -0.25%
D+PM 42 0.133 -28.43% 0.002 -47.44% 0.980 -0.00% 0.987 -1.55%
D+PI 47 0.112 -39.72% 0.003 -28.91% 0.979 -0.13% 0.882 -3.30%
D+CA+PI 48 0.160 -14.04% 0.003 -23.65% 0.979 -0.06% 0.903 -0.90%
D+PM+PI 47 0.160 -14.04% 0.004 -23.65% 0.979 -0.06% 0.903 -0.90%

94 10. RESULTADOS E SUA ANÁLISE
Figura 10.6: Avaliação de desempenho, adicionando possíveis combinações de características
seleccionadas em relação ao melhor resultado obtido a partir do SFS em 10.6.
10.8 Desempenho da máquina de vectores de suporte para
as características seleccionadas como as melhores.
• Base de dados utilizada
� SCOP40 Minidatabase (PCB00019).
• Método
� Máquina de Vectores de Suporte.
• Características extraídas
� Dicionário (D=4) construído a partir do critério da entropia.
� Composição atómica.
� Peso molecular.
Da análise dos resultados apresentados na tabela 10.8 veri�camos que a MVS apresenta um
incremento de desempenho em relação à RN, no que se refere ao AUC. O incremento, na
ordem de 5.47% é bastante signi�cativo visto que estamos a passar de um AUC de 0.912
obtido pela RN, para um valor de AUC de 0.961 na MVS. Contudo, no que se refere à taxa
de acerto, é inferior em 0.17%, o que não é signi�cativo.

10.9. DESEMPENHO DO MELHOR CLASSIFICADOR COM BASES DE DADOS DE MAIOR DIMENSÃO, USANDOREDES NEURONAIS. 95
Tabela 10.8: Comparação entre os resultados obtidos por uma rede neuronal e máquina de
vectores de suporte, usando o melhor resultado obtido a partir do SFS em 10.6.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
RN 47 0.186 - .004 - 0.980 - 0.912 -
MVS 47 0.006 -96.97% 0.001 -86.06% 0.978 -0.22% 0.961 5.47%
10.9 Desempenho do melhor classi�cador com bases de
dados de maior dimensão, usando redes neuronais.
• Base de dados utilizada
� SCOP95−Superfamily−Family (PCB00001).
• Método
� Rede Neuronal.
• Características extraídas
� Dicionário (D=4) construído a partir do critério da entropia.
� Composição atómica.
� Peso molecular.
O resultado por nós obtido, tabela 10.9, não é passível de comparar em virtude da informação
não se encontrar disponibilizada ao nível do algoritmo de classi�cação usado no presente
teste. A razão talvez se prenda com a dimensão dos dados envolvidos, 11 944 sequências e
246 classes.
Contudo, e em virtude dos valores apresentados, taxa de acerto de 0.998 e AUC 0.880,
pensamos que pudemos inferir que os resultados se a�guram como bons resultados.
Tabela 10.9: Resultados obtidos usando o melhor resultado obtido a partir do SFS em 10.6
usando bases de dados de maior dimensão.NC TVP Desvio TFP Desvio TA Desvio AUC Desvio
RN 43 0.002 - 0.002 - 0.998 - 0.880 -

96 10. RESULTADOS E SUA ANÁLISE

CAPÍTULO 11
Análise critica e trabalhos futuros
Conscientes da importância da classi�cação de proteínas em grupos funcionais na área da
biologia computacional, das di�culdades envolvidas, comuns a qualquer projecto de inves-
tigação, de�nimos como objectivo dar mais um contributo na área supra citada, que ainda
se encontra longe de um sistema que permita classi�car correctamente toda a informação
existente ao nível das proteínas.
Neste sentido, desenvolveu-se uma nova metodologia para a extracção de características,
combinando técnicas de extracção ao nível físico-químico das proteínas e técnicas de Text
Mining com segmentação de palavras. Os resultados foram comparados com bases de
dados de referência, tendo sido conseguido um aumento de desempenho de 17.35%, com
um número de características signi�cativamente inferior, um vector de 47 características em
vez de 1357.
Do exposto, pensamos que os principais objectivos foram alcançados, tendo resultado a im-
plementação da nova metodologia numa ferramenta computacional, designada por Intelligent
System for Feature Extraction and Classi�cation of Proteins - ISFECP.
Neste trabalho foi usada a base de dados de referência [4], ao longo dos vários testes
efectuados, de forma a validar os resultados obtidos. No �nal foi usada uma outra base de
dados, esta de maiores dimensões, mas que não foi possível comparar o resultado obtido, em
97

98 11. ANÁLISE CRITICA E TRABALHOS FUTUROS
virtude da informação não se encontrar disponibilizada ao nível do algoritmo de classi�cação
por nós usado nesse teste (rede neuronal). A razão talvez se prenda com a dimensão
apresentada pela base de dados.
No entanto, torna-se fundamental testar a metodologia desenvolvida em outras bases de
dados de referência de forma a mais correctamente validar os resultados até então obtidos.
Por outro lado, seria de todo o interesse complementar este trabalho, ao nível da meto-
dologia, em testar algumas características que nos pareceram relevantes durante a fase de
estudo. Nomeadamente a hidrofobicidade e a classi�cação segundo a tabela 9-gram (tabela
7.2), dos aminoácidos.
Por último, a integração da "LIBSVM" na aplicação desenvolvida, visto constituir uma
referência ao nível das máquinas de vectores de suporte.

APÊNDICE A
Manual de utilizador
ISFECP - Intelligent System for FeatureExtraction and Classi�cation of Proteins
A.1 Caracterização
A aplicação ISFECP v2.0, é um classi�cador inteligente, tendo por base uma nova meto-
dologia para extracção de características, combinando técnicas de extracção ao nível das
propriedades físico-químicas das proteínas, técnicas de Text-Mining com segmentação de
palavras.
É uma aplicação desenvolvida em Matlab R2010a no âmbito do mestrado em Informática e
de Sistemas.
A.2 Entidades envolvidas
• Instituto Superior de Engenharia de Coimbra - Departamento de Engenharia de Infor-
mática e de Sistemas.
99

100 A. MANUAL DE UTILIZADOR
• Projecto de investigação bioink Project.
A.3 Procedimento para a execução
Copiar a pasta "class_plot" para uma unidade do disco duro (por exemplo unidade D). Após
abrir o Matlab, seleccionar a current directory, o caminho para a unidade escolhida (usando
o exemplo anterior seria d:\class_plot) e na janela de comando digitar "class_plot". Deverá
surgir então o écran Fig. A.1.
Nota: A aplicação necessita que o Matlab tenha instaladas as toolbox's: "Bioinformatics
Toolbox", "Neural Network Toolbox" e "Statistics Toolbox".
Figura A.1: Écran principal da aplicação. Imagem composta a partir do site PDB [2].

A.4. CONFIGURAÇÃO DA APLICAÇÃO 101
A.4 Con�guração da aplicação
A aplicação já tem de�nido por defeito valores a usar, contudo o utilizador pode alterar as
con�guração ao aceder no menu ao item Con�guration, e alterar de acordo com os valores
pretendidos. O processo de con�guração decorre de acordo com o que apresentamos a
seguir.
A.5 Con�guração da extracção de características
No écran principal, o utilizador ao seleccionar a opção, "Con�guration⇒Feature Extraction",
é apresentado um écran conforme mostra a Fig. A.2. Sempre que se fazem alterações na
con�guração, passam os valores alterados a ser considerados valores por defeito.
Neste écran os possíveis parâmetros são:
• Dictionary Building Method.
� Esta opção de�ne o critério de escolha dos tuplos, mediante o critério seleccio-
nado, que irá constituir a base da construção do dicionário a usar na extracção
de características.
� Os valores possíveis nesta opção são:
∗ TF
∗ tf-idf
∗ Entropia
• N◦ Max Tuplos.
� Este parâmetro de�ne o número máximo de tuplos que pretendemos que o
classi�cador use ao nível da fase de treino.
� Os valores possíveis são números inteiros.
• Goal AUC.
� Este parâmetro de�ne o valor que pretendemos que o classi�cador atinja em
termos de AUC.

102 A. MANUAL DE UTILIZADOR
� Os valores possíveis são números compreendidos entre 0 e 1.
• Maximum Iteration.
� Este parâmetro de�ne o número máximo de iterações que o classi�cador executa
na fase de treino no sentido de atingir o valor de�nido no ponto anterior, Goal
AUC.
� Os valores possíveis são números inteiros positivos.
Figura A.2: Écran onde são de�nidos os parâmetros usados ao nível da extracção de
características.
A.6 Con�guração da rede neuronal
No écran principal, o utilizador ao seleccionar a opção, Con�guration⇒Neural Network, é
apresentado um écran conforme mostra a Fig. A.3. Sempre que se fazem alterações na
con�guração, passam os valores alterados a ser considerados valores por defeito.
Neste écran os possíveis parâmetros são:
• N◦ Layers.
� Este parâmetro permite de�nir o número de camadas da topologia da rede
neuronal.
� Os valores possíveis são números inteiros positivos.

A.6. CONFIGURAÇÃO DA REDE NEURONAL 103
• N◦ Neurons.
� Este parâmetro permite de�nir o número de neurónios da topologia da rede
neuronal.
� Os valores possíveis são números inteiros positivos.
• Training Function.
� Esta opção de�ne a função de treino usada pela rede neuronal.
� Os valores possíveis nesta opção são:
∗ traingdm
∗ traingda
∗ traingdx
∗ traincgp
∗ traincgf
∗ trainscg
∗ trainoss
∗ trainbfg
• N◦ Epoch.
� Este parâmetro permite de�nir o número máximo de épocas por treino.
� Os valores possíveis são números inteiros positivos.
• Goal.
� Este parâmetro permite de�nir o objectivo a atingir pela rede.
� Os valores possíveis são números positivos.
• Lr.
� Este parâmetro permite de�nir a taxa de aprendizagem.
� Os valores possíveis são números positivos entre 0 e 1.
• Max fail.
� Este parâmetro permite número máximo de falhas.
104 A. MANUAL DE UTILIZADOR
� Os valores possíveis são números inteiros positivos.
• Mc.
� Este parâmetro permite de�nir o valor de "momentum", permitindo ajustar a
rede em função do estado anterior.
� Os valores possíveis são números positivos entre 0 e 1.
• Min Grad.
� Este parâmetro permite de�nir a performance mínima do gradiente.
� Os valores possíveis são números positivos.
Figura A.3: Écran onde são de�nidos os parâmetros usados pelo algoritmo da rede neuronal.
A.7 Con�guração da máquina de vectores de suporte
No écran principal, o utilizador ao seleccionar a opção, Con�guration⇒Support Vector
Machine, é apresentado um écran conforme mostra a Fig. A.4. Sempre que se fazem

A.7. CONFIGURAÇÃO DA MÁQUINA DE VECTORES DE SUPORTE 105
alterações na con�guração, passam os valores alterados a ser considerados valores por
defeito.
• Kernel Funcion.
� Esta opção de�ne o kernel usado pela máquina de vectores de suporte.
� Os valores possíveis nesta opção são:
∗ rbf
∗ linear
∗ quadratic
∗ polynomial
∗ mlp
• RBF Sigma Value.
� Este parâmetro permite de�nir o valor do sigma quando é usado o kernel rbf.
� Os valores possíveis são números positivos.
• Method Value.
� Este parâmetro permite o método usado na de�nição do hiperplano.
� Os valores possíveis nesta opção são:
∗ SMO
∗ QP
∗ LS

106 A. MANUAL DE UTILIZADOR
Figura A.4: Écran onde são de�nidos os parâmetros usados pela máquina de vectores de
suporte.
A.8 Con�guração do classi�cador
O classi�cador tem por defeito os parâmetros que são apresentados nas Fig. A.2, A.3 e A.4,
no que respeita à con�guração da rede neuronal, da máquina de vectores de suporte e da
extracção de características, respectivamente. O algoritmo de classi�cação por defeito é a
rede neuronal.
Quando o utilizador alterar qualquer um dos parâmetros, no que respeita à rede neuronal,
máquina de vectores ou extracção de características, esses valores passarão a ser os valores
por defeito sempre que iniciar a aplicação.
A.9 Con�guração do algoritmo de classi�cação
Para alterar o algoritmo de classi�cação, o utilizador no écran principal, deve seleccionar
"Classi�cation Algorithm", de acordo com o pretendido, conforme é apresentado na Fig.
A.5. Esta selecção terá re�exo ao nível da fase de treino do classi�cador. Isto signi�ca
dizer que se o utilizador escolher a opção "Neural Network" na fase de treino, ao executar
o classi�cador, a aplicação irá usar a rede neuronal, mesmo que o utilizador tenha alterado
no écran principal o algoritmo de classi�cação para "Support Vector Machine".

A.9. CONFIGURAÇÃO DO ALGORITMO DE CLASSIFICAÇÃO 107
Figura A.5: Con�guração do algoritmo de classi�cação a usar durante a fase de treino do
classi�cador.

108 A. MANUAL DE UTILIZADOR
A.10 Execução da aplicação
Na Fig. A.8 estão representadas as diferentes operações que o utilizador poderá fazer. A
sequência normal é a seguinte:
Load Sequence⇒Load Train Data Set⇒Feature Extraction⇒⇒Training Classi�cation⇒Execution Classi�cation.
• Load Sequence.
� É apresentado ao utilizador uma janela para seleccionar o �cheiro que contêm as
sequências que pretende classi�car (Fig. A.6). Em caso de não ser seleccionado
nenhum �cheiro, é mostrada uma informação de erro.
� Por defeito a aplicação apresenta a selecção dos �cheiros de sequências a ler, na
pasta "Ficheiros". A extensão por defeito é .fasta.
Figura A.6: Selecção do �cheiro de sequências a classi�car, com a extensão .fasta.
• Load Train Data Set.
� É apresentado ao utilizador uma janela para seleccionar o �cheiro de treino e
teste que pretende usar para a execução do classi�cador (Fig. A.7). Em caso de
não ser seleccionado nenhum �cheiro, é mostrada uma informação de erro.
� Por defeito a aplicação apresenta a selecção dos �cheiro a ler, na pasta "Fichei-
ros". A extensão por defeito é a .cast.
A.10. EXECUÇÃO DA APLICAÇÃO 109
Figura A.7: Selecção do �cheiro de treino e teste, com a extensão .cast.
• Feature Extraction.
� Ao seleccionar esta operação, a aplicação irá fazer a extracção das características
de acordo com os parâmetros de�nidos na con�guração.
• Training Classi�cation.
� Ao seleccionar esta operação, a aplicação irá treinar o classi�cador de acordo
com os parâmetros de�nidos na con�guração.
• Execution Classi�cation.
� Ao seleccionar esta operação, a aplicação irá executar o melhor classi�cador
obtido na fase de treino.
A aplicação guarda o resultado das operações que o utilizador vai executando. Após a
leitura das sequências, da leitura do conjunto de treino e da execução da extracção de
características, a aplicação guarda o resultado das características extraída em �cheiro,
permitindo ao utilizador retomar o processo e classi�cação num outro momento. Assim
sendo quando a aplicação é iniciada, é apresentado no canto superior esquerdo da aplicação,
o nome dos �cheiros das sequências e conjunto de treino usados na última extracção de
características, conforme apresentado na Fig. A.9. Ao executar a operação de treino do

110 A. MANUAL DE UTILIZADOR
Figura A.8: Apresenta as várias opções de execução por parte da aplicação.
classi�cador, a aplicação guarda no �nal a informação necessária para a fase da execução
do classi�cador. Ou seja, mais uma vez o utilizador poderá retomar o processo de execução
do classi�cador numa fase posterior.
Sublinhe-se que a aplicação ao iniciar apresenta no canto superior direito o último algoritmo
usado ao nível da classi�cação, na fase de treino, conforme apresentado na Fig. A.9.
Figura A.9: Apresenta o último algoritmo de classi�cação e o nome dos �cheiros sobre o
qual foi feito a última extracção de características.
Durante a fase de treino e de execução, visto que são fases com alguma morosidade, a
aplicação apresenta ao utilizador uma janela indicativa que o processo está a decorrer (Fig.
A.10). Após a sua conclusão é apresentado a informação de conclusão do processo (Fig.
A.11).
Após a conclusão do processo da execução do classi�cador, os resultados são guardados no
�cheiro resultados.xls, que se encontra na directoria "Resultados".

A.10. EXECUÇÃO DA APLICAÇÃO 111
Figura A.10: Indica que um processo está a decorrer.
Figura A.11: Indica que um processo foi concluído.

112 A. MANUAL DE UTILIZADOR
A.11 Manual de utilizador
O utilizador poderá aceder ao manual seleccionando a opção "Help" no écran principal da
aplicação.
Copyright c© 2010 All Rights Reserved.

Lista de referências
[1] Blast, Dezembro 2010. URL http://blast.ncbi.nlm.nih.gov/Blast.cgi.
[2] PDB Protein Data Bank, Agosto 2010. URL http://www.pdb.org/pdb/home/
home.do.
[3] Novembro 2010. URL http://www.cftc.cii.ul.pt/prisma.
[4] Icgeb, 2010. URL http://hydra.icgeb.trieste.it/benchmark/index.php?
experiment=19#.
[5] Novembro 2010. URL http://www.expasy.org/prosite/.
[6] Lawrence Bragg. The development of x-ray analysis. The Rutherford Memorial Lecture,
1960.
[7] Y.D. Cai and K.C. Chou. Predicting 22 protein localizations in budding yeast.
Biochemical and Biophysical Research Communications, 323:425�428, 2004.
[8] K. C. Chou and Y. D. Cai. Predicting protein localization in budding yeast.
Bioinformatics, 21:944�950, 2005.
[9] K.C. Chou and Y.D. Cai. Prediction of protein subcellular locations by GO-FunD-
PseAA predictor. Biochemical and Biophysical Research Communications, 320:1236�
1239, 2004.
113

114 LISTA DE REFERÊNCIAS
[10] O. Emanuelsson, H. Nielsen, S. Brunak, and G. Heijn. Predicting subcellular localization
of proteins based on their n-terminal amino acid sequence. J. Mol. Biol., 300:1005�
1016, 2000.
[11] GOR, Novembro 2010. URL http://gor.bb.iastate.edu/.
[12] Corey Hardin, Taras V Pogorelov, and Zaida Luthey-Schulten. Ab initio protein
structure prediction.
[13] A. Hoglund, P. Donnes, T. Blum, H. Adoplh, and O. Kohlbacher. Using n-terminal
targeting sequences, amino acid composition, and sequence motifs for predicting protein
subcellular localization. Proceedings of the German Conference on Bioinformatics (GCB
'05), pages 45�59, 2005.
[14] G. Hristescu and M. Farach-Colton. Structure-based feature extraction from protein
databases.
[15] A.K. Jain and D. Zongker. Feature-selection: Evaluation, application, and small sample
performance. IEEE Transactions on Pattern Analysis and Machine Intelligence, 19(2):
152�157, 1997.
[16] A.K. Jain, R.P.W. Duin, and J. Mao. Statistical pattern recognition: A review. IEEE
Transactions on Pattern Analysis and Machine Intelligence, 22(1):4�37, 2000.
[17] John-Marc and Steven E. Brenner. The impact of structural genomics: Expectation
and outcomes. Review, SCIENCE, (Vol 311), 2006.
[18] Reer Kamal Kaur, Manpreet Singh, and Parvinder Singh Sandhu. Protein secondary
structure prediction. World Academy of Science, Engineering and Technology, 2008.
[19] Klaus Krippendor�. Mathematical Theory of Communication.
[20] Lukasz Kurgan and Ke Chen. Prediction of protein structural class for the twilight zone
sequences. 2007.
[21] Leong Lee, Cyriac Kandoth, Jennifer L. Leopold, and Ronald L. Frank. Protein
secondary structure prediction using parallelized rule induction from covering. World
Academy of Science, Engineering and Technology, 2009.
[22] NIH. Structural genomics, Agosto 2010. URL http://www.nigms.nih.gov/
initiatives/PSI.

LISTA DE REFERÊNCIAS 115
[23] K. Nishikawa, Y. Kubota, and T. Ooi. Classi�cation of proteins intogroups based on
amino acid compostion and other characters. J.Biochem., 94:997�1007, 1983.
[24] Linus Pauling and Robert B. Corey. Atomic coordinates and structure factors for two
helical con�guration of polypeptide chains. Proceedings of the National Academy of
Sciences, 37:235, 1951.
[25] Linus Pauling and Robert B. Corey. The pleated sheet, a new layer con�guration of
polypeptide chains. Chemistry, Pauling and Corey, (Vol. 37):251, 1952.
[26] Francisco Pereira. Apontamentos da Cadeira de Bioinformática. ISEC, 2009.
[27] A. Reinhardt and T. Hubbard. Using neural networks for prediction of subcellular
location of proteins. Nucleic Acids Research, 26:2230�2236, 1998.
[28] F. Sanger and E. O. P. Thompson. The investigation of peptides from enzymic
hydrolysates.
[29] S.J.Matthews. Nmr of proteins and nucleic acids. The Royal Society of Chemistry,
(Vol 32), 2003.
[30] Ray J. Solomono. Machine Learning - Past and Future. 2009.
[31] spdbv, Setembro 2010. URL http://spdbv.vital-it.ch/.
[32] M. O. stitson, J. A. E. Weston, A. Gammerman, V. Vovk, and V. Vapnic. Theory of
support vector machines.
[33] The Gene Ontology Website, 2010. URL http://www.geneontology.org/.
[34] Wikipédia, 2010. URL http://pt.wikipedia.org/wiki/Amino%C3%A1cido.
[35] Wikipédia. Cristalogra�a (raio x), Setembro 2010. URL http://en.wikipedia.org/
wiki/X-ray_crystallography.
[36] Wikipédia. Espectómetro RMN do Paci�c Northwest Laboratory, Setembro 2010.
URL http://en.wikipedia.org/wiki/Protein_nuclear_magnetic_resonance_
spectroscopy.
[37] W.Y.Yang, B.L. Lu, and Y. Yang. A comparative study on feature extraction from
protein sequences for subcellular localization prediction.

116 LISTA DE REFERÊNCIAS
[38] Mary Qu Yang, Jack Y. Yang, and YiZhi Zhang. Extracting features from protein
primary structure with feature selection to enhance protein structural and functional
prediction. .
[39] W.Y. Yang, B.L. Lu, and Wen.Y. Yang. Classi�cation of proteins sequences based on
words segmentation methods. .





























