Embed Size (px)
Citation preview

i
UNIVERSIDADE FEDERAL DE PERNAMBUCO CENTRO DE CIÊNCIAS BIOLÓGICAS
DOUTORADO EM CIÊNCIAS BIOLÓGICAS
Síntese, Modelagem Molecular e Atividade
Hipoglicemiante de Novas Arilideno-Tiazolidinadionas
ROSA HELENA VERAS MOURÃO
RECIFE - BRASIL 2006

ii
ROSA HELENA VERAS MOURÃO
Síntese, Modelagem Molecular e Atividade
Hipoglicemiante de Novas Arilideno-Tiazolidinadionas
Tese de Doutoramento apresentada ao Programa de Doutorado em
Ciências Biológicas do Centro de Ciências Biológicas da
Universidade Federal de Pernambuco para obtenção do título de
Doutora em Ciências Biológicas, área de concentração Farmacologia,
Fisiologia e Química Medicinal.
ORIENTADORES:
Prof. Dr. Ivan da Rocha Pitta
Prof. Dr. Mário José Abdalla Saad
CO-ORIENTADOR:
Profª. Drª. Vera Lúcia Menezes de Lima
RECIFE
2006

iii

iv

v
DEDICO:
A minha mãe, Raimunda. Que sempre teve fé na vida, amor pelas pessoas e que sozinha se esforçou na minha educação. Cada vez mais, amo você.
A Fernanda, minha filha. Um dia você irá entender a ausência da mamãe, o computador ligado, os livros espalhados, as mudanças necessárias para concluir esse trabalho. Você acompanhou todo meu trabalho, e demonstrou a guerreira que é, enfrentando doenças, mudanças, ausências e colaborando com o trabalho da mamãe. Hoje com 6 anos é parte dessa história. Você é a criatura mais linda que um dia eu conheci. Eu te amo.
Ao Aloísio, a quem um dia prometi contribuir para o avanço do conhecimento na área de diabetes. Infelizmente você morreu das conseqüências dessa doença, mas acredite, eu me esforcei muito para dar essa pequena contribuição ao mundo da Ciência. A todas as pessoas que sofrem de diabetes e acreditam que na Ciência poderá ser encontrada uma solução.

vi
ANDO DEVAGAR PORQUE JÁ TIVE PRESSA, LEVO ESSE SORRISO PORQUE JÁ CHOREI DEMAIS. HOJE, ME SINTO MAIS FORTE, MAIS FELIZ QUEM SABE. SÓ LEVO A CERTEZA DE QUE MUITO POUCO EU SEI, EU NADA SEI (...) CADA UM DE NÓS COMPÕE A SUA HISTÓRIA, CADA SER EM SI CARREGA O DOM DE SER CAPAZ E SER FELIZ (...)
(Almir Sater – Tocando em frente)

vii
AGRADECIMENTOS
Ao Prof. Dr. Ivan da Rocha Pitta, pela oportunidade de desenvolver esta pesquisa,
amizade, provisão, além da valiosa orientação. Muito obrigada.
Agradeço ao Prof. Dr. Mário Saad, pela orientação segura e competente, amizade e
por ter propiciado condições necessárias para a realização deste trabalho.
A Profa Dra. Suely Galdino, amiga e professora, pelo crescimento pessoal e
profissional. Pela orientação e generosidade em todas as etapas desse trabalho.
A Profa Dra. Maria da Guia Lima, que nos meus primeiros anos de iniciação
cientifica me ensinou a fazer ciência de maneira séria e ao mesmo tempo divertida e sempre
me ofereceu a sua sincera amizade.
Aos profs Dr. Marcelo Zaldini (UFPE) e Dra. Lucia Fernanda Leite (Universidade
Católica de Pernambuco) pela complementação desse trabalho com a parte de Modelagem
Molecular.
As Profas Dra. Vera Menezes e Dra. Ana Dulce, (UFPE) por terem gentilmente
cedido seus laboratórios para realização de alguns experimentos.
Aos professores Dr. Lício Velloso e Dr. José Barreto Carvalheira (UNICAMP),
pelas valiosas orientações e sugestões.
A profa Dra. Maria do Carmo Lima (UFPE), pela sua ajuda competente e constante
na síntese das novas moléculas tiazolidínicas.
À Mirian Ueno, pela eficiente e agradável orientação de bancada, e principalmente
pela grande amizade nascida durante a realização dos experimentos.
A todos os amigos do Laboratório de Biologia Molecular (UNICAMP), Marciane,
Eliana, Talita, Fernanda, Maria Helena, Maristela, Graciela, Juliana, Patrícia Prada,
Patykaroll, Karen, Daniela, Cris, Vivian, Andressa, Adriana, Esméria, Kellen, Marcel,

viii
Rodrigo, Cafu, Marcelo, Dennys, Eli, Josénilson e Eduardo, pelas discussões e troca de
informações necessárias para uma convivência pacífica e formadora de opiniões.
Aos funcionários do biotério, em especial ao Márcio pelos zelosos cuidados
dispensados aos animais, e pelo auxilio oferecido durante a experimentação animal.
Aos funcionários Sr. Luiz e Jósimo, não só pelos serviços prestados e apoio, mas
também pela boa amizade.
A todos os colegas do laboratório de Síntese e Planejamento de Fármacos (UFPE),
especialmente à Teresinha, Lina, Flavia e Leila, pelo carinho e amizade cultivada ao longo
desses 4 anos.
Aos colegas do curso de pós-graduação da UFPE pelas conversas e horas de estudos.
Aos alunos de iniciação científica, especialmente Joíce Nunes, Clécio, Iane, Rodrigo e
Luciana pela amizade e colaboração nesse trabalho.
A Adenilda, pelo trabalho realizado junto a secretária do Programa de Pós-graduação.
Meu agradecimento e respeito aos ANIMAIS DE LABORATÓRIO, que doaram a
vida à pesquisa.
Ao CNPq pela concessão da bolsa de Doutorado sanduíche, e a UFPA pela concessão
de licença integral das minhas atividades de docência, o que propiciou a dedicação exclusiva
ao curso de Doutorado em Ciências Biológicas.
A todos que de uma forma ou de outra contribuíram para a realização desse trabalho,
meu muito OBRIGADA.

ix
SUMÁRIO
pag RESUMO xiv ABSTRACT xvi 1. INTRODUÇÃO 18
1.1. Obesidade como fator de risco para diabetes mellitus tipo 2 19
1.2. Mecanismos moleculares da ação da onsulina 21
1.3. Tiazolidinadionas e mecanismo de ação 24 2. OBJETIVOS 29
3. MATERIAL E MÉTODOS 32
3.1. PARTE QUÍMICA 32
3.1.1. Material 33
3.1.1.1. Reagentes e solventes 33
3.1.2. Equipamentos 34
3.1.2.1. Espectroscopia 34
3.1.2.2. Ponto de fusão 34
3.1.2.3. Cromatografia 34
3.1.3. Métodos 35
3.1.3.1. Obtenção da tiazolidina-2,4-diona 36
3.1.3.2. Obtenção da 3-(4-metil-benzil)-tiazolidina-2,4-diona 36
3.1.3.3. Obtenção dos etil-(2-ciano-3-fenil)-acrilatos de etila 36
3.1.3.4. Método geral para a obtenção dos derivados
5-benzilideno-3-(4-metil-benzil)-tiazolidina-2,4-dionas 37
3.2. PARTE MODELAGEM MOLECULAR 38
3.2.1. Material 39
3.2.2. Métodos 39

x
3.3. PARTE BIOLÓGICA 40
3.3.1. Modelo animal e tratamento 41
3.3.2. Western blot e imunoprecipitação 42
3.3.2.1. Anticorpos, reagentes químicos e material 42
3.3.2.2. Soluções utilizadas 42
3.3.2.3. Extração de tecidos, imunoblot e imunoprecipitação 44
3.3.3. Efeito de GQ2 no peso corporal, consumo de alimento e
quantidade de água em camundongos com obesidade 45
3.3.4. Teste de tolerância à insulina (ITT) 46
3.3.5. Parâmetros Bioquímicos 46
3.3.6. Determinação da massa de tecido epididimal 47
3.3.7. Toxicidade aguda e subcrônica 47
3.3.8. Atividade de NF-kB 47
3.3.9. Análise estatística 48
4. RESULTADOS 49
4.1. Síntese e modelagem molecular de novos derivados
tiazolidínicos (agonistas de PPARγ) 50
4.2. Efeito de GQ2 no peso corporal, ingestão alimentar, massa
adiposa epididimal e volume de água em camundongos com
obesidade induzida por dieta hiperlipídica 53
4.3. Parâmetros Bioquímicos 54
4.4. Teste de tolerância à insulina (ITT) 54
4.5. Efeito de GQ2 nas etapas iniciais e intermediárias da ação
insulínica em tecido adiposo de camundongos com resistência
à insulina induzida por dieta hiperlipidica 57
4.6. Efeito de GQ2 nas etapas iniciais e intermediárias da ação
insulínica no músculo de camundongos com resistência à
insulina induzida por dieta hiperlipidica 62
4.7. Efeito de GQ2 nas etapas iniciais e intermediárias da ação
insulínica no fígado de camundongos com resistência à insulina
induzida por dieta hiperlipidica 66

xi
4.8. Efeito de GQ2 na toxicidade aguda e subcrônica de
camundongos com obesidade induzida por dieta hiperlipídica 70
5. DISCUSSÃO 71 6. CONCLUSÃO 82 7. REFERÊNCIAS BIBLIOGRÁFICAS 84 8. ANEXOS 96
ANEXO 1 (Relacionado a tese)
Synthesis and Biological Activity of Novel Acridinylidene and Benzylidene
thiazolidinediones.
European Journal of Medicinal Chemistry, 40 (11): 1129-1133, 2005.
ANEXO 2 (Relacionado a tese)
Synthesis, Biological Evaluation and Molecular Modeling Studies of Arylidene-
Thiazolidinediones with Potential Hypoglycemic and Hypolipidemic Activities.
European Journal of Mecdicinal Chemistry (submetido).
ANEXO 3(NÃO Relacionado a tese)
Synthesis and Anti-Inflammatory Activity of Some New N and S-Alkylated Arylidene-
Thioxo-Imidazolidinones
Heterocyclic Communications, 11 (5): 433-440, 2005.

xii
LISTA DE ABREVIATURAS SIGLAS
Akt/PKB Proteína serina/treonina quinase B
α Alfa
β Beta
γ Gamma
ADA Associação América de Diabetes
AG Ácidos graxos
ALT Alanina aminotransferase
Anti-pY Anti-fosfotirosina
ATZD Arilideno-tiazolidinadionas
BSA Soro de albumina bovina
Cbl Protooncogene que exerce função de substrato do
receptor de insulina
DH Dieta Hiperlipídica
DNA Ácido desoxirribonucléico
DTT Ditiotreitol
EDTA Ácido etilenodiaminotetracético
ERKs Extracelular signal-regulated kinases
Quinases regulada por sinal extracelular
Gab-1 Substrato do receptor de insulina
GLUT4 Transportador de glicose 4
GQ2 5-(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-diona
GSK3 Glicogênio sintetase 3 quinase

xiii
IkB- α Quinase inibidora do fator nuclear Kb
IL-1β Interleucina-1 beta
IL-6 Interleucina-6
iNOS/NOS2 Óxido Nítrico Sintase induzível
IR Receptor de insulina
IRS-1 Substrato 1 do receptor de insulina
IRS-2 Substrato 2 do receptor de insulina
IRSs Substratos do receptor de insulina
ITT Teste de tolerância à insulina
JNK c-jun serina quinase
kDa Quilodalton
Kitt Constante de decaimento de glicose
MAPK Mitogen-activated protein Kinase
Proteína quinase ativada por sinal extracelular
Ncor Receptor nuclear co-repressor
NFκB Fator de transcrição kappa B
ob/ob Camundongo obeso com mutação no gene da leptina
p60dok Substrato do receptor de insulina
PDK-1 Phosphoinositide-dependent Kinase-1
Quinase dependente de fosfoinositideo-1
PEPCK Fosfoenolpiruvato carboxilase
PH Sítio de homologia a plecstrina
PI3K Fosfatildilinositol 3-quinase
PKC Proteína quinase C
PMSF Fluoreto de fenilmetil sulfonila
PPARγ Peroxisome proliferator-activated receptor gamma

xiv
Receptor ativado por proliferadores de peroxissoma gama
PPRE Elementos de resposta ao PPAR
PTP 1B Proteína tirosina fosfatase 1B
PTPases Proteínas tirosinas fosfatases
Raf-1 Proteína da cascata de ativação da MAP quinase
Ras Proteína originalmente identificada como oncogene, têm participação
na regulação do metabolismo e crescimento celular
RG Rosiglitazona
RXR Receptor retinóide X
SDS-PAGE Eletroforese em gel de poliacrilamida com dodecil sulfato de sódio
SH2 Domínios protéicos com homologia ao proto oncogene Src2
(Segunda homologia do src)
SH3 Terceira homologia do src
Shc Molécula adaptadora e substrato do receptor de insulina
SOS Son-of-sevenles - Fator de troca de nucleotídeo guanina
Src Oncogene originalmente definido como produto do sarcoma vírus Rous
SRC-1 Receptor de esteróide
TG Triclicerideos
TNF-α Fator de necrose de tumor –alfa
Tris Tri(hidroximetil)-aminometano
TZDs Tiazolidinadionas
WHO Organização Mundial de Saúde

xv
RESUMO

xvi
RESUMO
As glitazonas, também conhecidas como sensibilizadores de insulina, é a denominação genérica
de uma série de tiazolidinadionas (TZDs) possuidoras de atividade hipoglicemiante administrada
por via oral em pacientes com diabetes mellitus tipo 2. Dentre essas inclui a rosiglitazona e
pioglitazona. No presente estudo, sintetizamos 12 derivados arilideno-tiazolidinadionas (ATZDs)
da 5-benzilideno-3-(4-metil-benzil)-thiazolidina-2,4-diona com potencial atividade
hipoglicemiante. Em seguida, investigamos os efeitos metabólicos e moleculares do composto 5-
(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-diona (GQ2) em camundongos “swiss”
com obesidade induzida por dieta hiperlipidica (DH). As novas ATZDs, foram sintetizadas pela
reação de adição nucleofílica da 3-(4-metil-benzil)-tiazolidina-2,4-diona com etil-(2-ciano-3-
fenil)-acrilatos substituídos. De acordo com os estudos de modelagem molecular, os complexos
formados entre as novas ATZDs e o domínio LBD do receptor PPARγ apresentaram boa
estabilidade quando comparadas com a energia de ligação da rosiglitazona. O tratamento de
camundongos obesos com GQ2 durante 14 dias reduziu os níveis basais de glicose, insulina e
leptina associados a um aumento da sensibilidade à insulina, redução da gordura epididimal, com
menor retenção de fluídos e concomitante redução no ganho de peso corporal. Quando nesse
mesmo período foi avaliada a quantidade de ração ingerida, não foi detectada diferença
significativa no consumo de ração durante 24 horas entre os animais tratados com GQ2 e DH. A
via de sinalização da insulina, IR/IRSs/PI3K/Akt, que foi reduzida pela DH, apresentou
recuperação completa após tratamento com GQ2 em fígado, músculo e tecido adiposo,
acompanhados de redução da fosforilação do IRS-1ser307 em tecido adiposo e hepático. O
tratamento com GQ2 levou ainda a redução da expressão da enzima iNOS em músculo e tecido
adiposo, redução do TNF-α em tecido adiposo, paralelo a uma redução da ativação das serinas
quinases IKK e JNK nos 3 tecidos estudados. Sendo assim, o novo agonista de PPARγ, GQ2,
reduz a resistência à insulina e parece levar a um aumento da sensibilidade à insulina no fígado,
músculo esquelético e tecido adiposo através de alterações da expressão e fosforilação de
proteínas envolvidas na via de transmissão do sinal da insulina, via IR/IRSs/PI3K/Akt, em
animais com resistência insulínica e obesidade induzida por dieta hiperlipídica.
xvii
ABSTRACT

xviii
ABSTRACT
The glitazones, also referred to as insulin sensitizers, are generically a class of thiazolidinediones
(TZDs) that possess hypoglycemic activity and are orally administered in patients with type II
diabetes mellitus. Two of these thiazolidinediones are rosiglitazone and pioglityazone. We
synthesized 12 arylideno-thiazolidinediones (ATZDs) of 5-benzylideno-3-(4-methyl-benzyl)-
thiazolidine-2.4-dione with potential hypoglycemic activity. We later investigated the metabolic
and molecular effects of 3-(4-methyl-benzyl)-5-(4-chloro-benzylidene)-thiazolidine-2.4-dione
(GQ2) in Swiss mice with hyperlipidic diet (HD) induced obesity. The new ATZDs were
synthesized by the reaction that resulted from the nucleophilic addition of 3-(4- methyl-benzyl)-
5-(4-chloro-benzylidene)-thiazolidine-2.4-dione to substituted ethyl - (2-cyano-3-phenyl)-
acrylates. According to molecular modeling studies, the combinations between the new ATZDs
and the LBD domination of the PPARγ nuclear receptor present good stability (negative binding
energy) when compared with the binding energy of rosiglitazone, an insulin sensitizer, used as a
standard substance. GQ2 treatment of obese mice for a period of 14 days reduced the basal level
of glucose, insulin and leptin, increased insulin sensitivity, reduced peri-epididymal fat, lowered
fluid retention and concomitantly reduced weight gain. When the quantity of food intake during
this same period was evaluated, no significant difference was observed between animals treated
with GQ2 and those on DH during a period of 24 hours. The IR/IRSs/PI3K/Akt insulin-signaling
pathway in the liver, muscle and adipose tissue reduced by DH, recovered completely after
treatment with GQ2, together with a reduction in IR/IRSs/PI3K/Akt phosphorylation in the
adipose and hepatic tissue. Treatment with GQ2 led to reduction in the iNOS enzyme in the
muscle and adipose tissue, reduction of TNF-α in the adipose tissue and at the same time a
reduction of IKK and JNK serine kinases in the three tissues under study. Therefore, the new
PPARγ agonist, GQ2, reduces the insulin resistance and seems to increase the insulin sensibility
of the liver, muscle and apipose tissue through alterations on expression and fosforilation of
signal transmision related proteins (IR/IRSs/PI3K/Akt) on induced obesety and insulin resistance
animals through hyperlipidic diet.

19
INTRODUÇÃO

20
1. INTRODUÇÃO
1.1. Obesidade como fator de risco para diabetes mellitus tipo 2
A obesidade, antes considerada sinal de fartura, saúde e padrão de beleza, deixou de ser
vista como uma condição desejável, diante das evidências de morbimortalidade elevada em
indivíduos obesos (PI-SUNYER, 1993).
Hoje, a obesidade é considerada uma epidemia mundial independente de condições
econômicas e sociais (WHO, 1998; OGDEN, et al., 2003; POPKIN & DOAK, 1998;
KOPELMAN, 2000). Esta alteração patológica comumente associada a um conjunto de doenças
metabólicas, como dislipidemia, resistência à insulina, diabetes mellitus tipo 2 e hipertensão
arterial, condições intimamente relacionadas com doenças cardiovasculares, principal causa de
morte em paises industrializados (MANSON et al., 1990; RYAN et al., 1994; STERN &
COLDITZ, 2004; RAO et al., 2001; MYKKANEN et al., 1992; ECKEL et al., 2005).
A obesidade caracteriza-se pelo acúmulo de gordura corporal resultante do desequilíbrio
energético prolongado, que pode ser causado pelo excesso de consumo de calorias e/ou
inatividade física. Os fatores genéticos desempenham papel importante na determinação da
suscetibilidade do indivíduo para o ganho de peso, porém são os fatores ambientais e de estilo de
vida, tais como hábitos alimentares inadequados e sedentarismo, que geralmente levam a um
balanço energético positivo, favorecendo o surgimento da obesidade (BRAY & POPKIN, 1998).
Estudos têm demonstrado que a localização ou distribuição da quantidade de gordura corporal,
particularmente na região central ou intra-abdominal (gordura visceral) contribui mais para os
distúrbios metabólicos e risco cardiovascular (VAGUE, 1956; HAFFNER et al., 1987).
Embora nos últimos 10 anos, as pesquisas relacionadas à obesidade tenham tido grandes
avanços, incluindo a descoberta da leptina (ZHANG et al., 1994) e o gene do receptor da leptina
(TARTAGLIA et al., 1995) o mecanismo molecular da obesidade ainda não é completamente
conhecido.
A resistência à insulina, definida como a perda relativa da capacidade deste hormônio em
exercer seus efeitos biológicos (SALTIEL & KAHN, 2001; SHULMAN, 2004), é um possível
elo entre obesidade e outros fatores de risco cardiovascular, tais como hipertensão e diabetes
(COLDITZ & WILLETT, 1990; JONES et al., 1994) que são componentes da síndrome
metabólica. Embora a resistência à insulina possa ocorrer na ausência da obesidade
(FERRANNINI et al., 1987; GRUNFELD et al., 1994) a prevalência de hiperinsulinemia e

21
resistência à insulina aumenta de acordo com o aumento do índice de massa corporal.
(FERRANNINI et al., 1997).
A resistência à insulina acontece em vários tecidos envolvidos com seus efeitos
metabólicos (DEFRONZO, 1988). Em tecido muscular há uma menor captação de glicose
induzida por insulina. No fígado, há maior produção de glicose e gliconeogênese
(GASTALDELLI et al., 2001). No tecido adiposo, a captação de glicose está diminuída, mas os
efeitos de hipertrofia continuam, contribuindo para o desenvolvimento da obesidade (KAHN,
1994). O mecanismo pelo qual a distribuição central da adiposidade causa resistência à insulina é
em parte, porque os depósitos viscerais de triglicerídeos possuem turnover mais acelerado que o
de outras regiões, aumentando a oferta de ácidos graxos livres no sistema porta, que estimulam a
gliconeogênese e inibem a depuração hepática da insulina contribuindo para elevar a glicemia, a
insulinemia e a resistência insulínica (KISSEBAH et al., 1982; KROTKIEWSKI et al., 1983).
Tanto em pacientes obesos quanto em modelos animais de obesidade observamos
resistência à insulina (SWINBURN, NYOMBA et al.,. 1991; MONTAGUE, FAROOQI et al.,
1997; CLEMENT, VAISSE et al., 1998). Desta forma, o aumento da prevalência de obesidade
deverá acompanhar-se do aumento da prevalência de diabetes mellitus tipo 2. Com o ganho de
peso e desenvolvimento da resistência à insulina, há um mecanismo compensatório de aumento
da secreção pancreática de insulina, levando a hiperinsulinemia, para manter a glicemia em níveis
normais (POLONSKY & GIVEN ET al. 1988).
O tecido adiposo, principalmente o branco, não é simplesmente um reservatório de
gordura como fonte de energia acumulada, mas principalmente um órgão extremamente ativo do
ponto de vista tanto metabólico como secretório, liberando para a circulação grande número de
peptídeos ativos, fatores do complemento e citocinas (AHIMA & FLIER, 2000; KERSHAW &
FLIER, 2004). Algumas citocinas podem influenciar profundamente o metabolismo e o gasto
energético. A expressão do fator de necrose tumoral-alfa (TNF-α), está aumentada na gordura de
roedores e de humanos obesos e pode contribuir para a resistência à insulina (HOTAMISLIGIL et
al., 1996).
Sendo a resistência à insulina uma característica comum da obesidade muitos estudos com
o objetivo de esclarecer os mecanismos envolvidos nesta relação têm demonstrado que as
alterações na sensibilidade à insulina associada à obesidade são o resultado de anormalidades na
sinalização deste hormônio nos tecidos alvos.
Os primeiros estudos indicaram que a resistência à insulina na obesidade era atribuída à
regulação quantitativa dos transportadores de glicose sensíveis à insulina e dos seus receptores.
Entretanto, as pesquisas realizadas recentemente têm contribuído para o grande avanço no

22
entendimento da via molecular da ação da insulina, com a observação da atividade catalítica
intrínseca do receptor deste hormônio, bem como os eventos sinalizadores em cascata. Os
resultados destes estudos demonstraram que animais obesos e indivíduos com diabetes mellitus
tipo 2 apresentam alterações na via de sinalização da insulina resultando em um estado de
resistência à insulina (KAHN et al., 2001).
1.2. Mecanismos moleculares da ação insulínica
A insulina é um hormônio polipeptídico anabólico produzido pelas células beta do
pâncreas, cuja síntese é ativada pelo aumento dos níveis circulantes de glicose e aminoácidos
após as refeições. A insulina age em vários tecidos periféricos, incluindo músculo, fígado e tecido
adiposo. Seus efeitos metabólicos imediatos incluem: aumento da captação de glicose, aumento
da síntese de proteínas, ácidos graxos e glicogênio, bem como bloqueio da produção hepática de
glicose, da lipólise e proteólise. Além disso, a insulina tem efeitos tardios na expressão de genes e
síntese protéica, assim como na proliferação e na diferenciação celulares. Outras funções da
insulina incluem o aumento da produção de óxido nítrico no endotélio, a prevenção da apoptose
ou morte celular e a promoção da sobrevida celular.
A sinalização intracelular da insulina em tecidos insulino-sensíveis inicia-se com a ligação
do hormônio a um receptor específico de membrana, o receptor de insulina. O receptor de
insulina é uma glicoproteína constituída de duas subunidades α, extracelulares, onde localiza-se o
sítio de ligação da insulina, e duas subunidades β, intracelulares, estas últimas ligadas à
membrana que faz a transdução do sinal da insulina à célula (SCHWARTZ et al., 2000). Uma vez
ativado, o receptor de insulina fosforila vários substratos em tirosina. Atualmente, dez substratos
do receptor de insulina já foram identificados. Quatro desses pertencem à família dos substratos
do receptor de insulina, as proteínas IRS (WHITE, 1998). Os outros substratos incluem Shc, Gab-
1, p60dok, Cbl, JAk2 e APS (VELLOSO et al., 1998)
A ligação da insulina à subunidade α do receptor de insulina permite que a subunidade β
adquira atividade quinase, levando à alteração conformacional e à autofosforilação do receptor
nas subunidades β em múltiplos resíduos de tirosina (1158, 1162, 1163), o que aumenta ainda
mais sua atividade tirosina quinase (PATTI & KAHN, 1998). Uma vez fosforilado, o receptor de
insulina fosforila outras proteínas ou substratos sinalizadores citoplasmáticos intracelulares,
dentre eles o substrato 1 do receptor de insulina (IRS-1) e o substrato 2 do receptor de insulina
(IRS-2) (WHITE, 1998). A fosforilação em tirosina das proteínas IRSs cria sítios de
reconhecimentos para moléculas contendo domínios com homologia à Src 2 (SH2)

23
(WHITE,1998; PESSIN & SATIEL, 2000; CARVALHEIRA et al., 2001; VELLOSO et al,
1998).
As proteínas IRS-1 e 2 desempenham função essencial na transmissão do sinal insulínico
e a fosforilação desses substratos permite a interação com diversas proteínas adaptadoras com
atividade enzimática. Há uma estreita associação entre a enzima fosfatidilinositol 3-quinase
(PI3K) com IRS-1 e 2 após estimulo com insulina (FOLLI et al., 1992). A PI3K é uma enzima
que contém dois sítios SH2 e um SH3 (CARPENTER & CANTLEY, 1990). A PI3K tem um
papel importante em muitos processos celulares incluindo: proliferação celular, regulação da
mitogênese, transporte de glicose estimulado pela insulina e a inibição da fosfoenolpiruvato-
carboxilase (PEPCK), a enzima chave para a gliconeogênese (FOLLI et al., 1992; SHEPHERD et
al., 1995; CHEATHAM & KANH, 1995). A molécula de IRS-1, quando fosforilada em tirosina,
permite a sua associação ao domínio SH2 da subunidade reguladora da PI3K (p85), levando a
ativação desta (BACKER et al., 1992). Esta enzima catalisa a fosforilação dos fosfoinositídeos na
posição 3 do anel inositol, produzindo fosfatidilinositol-3-fosfato, fosfatidilinositol-3,4-bifosfato
e fosfatidilinositol-3,4,5-trifosfato (LIETZKE et al., 2000). Este último produto liga-se aos
domínios PH (“pleckstrin homology”) de diversas moléculas sinalizadoras, alterando sua
atividade e localização subcelulares (KESSLER et al., 2001).
Algumas proteínas, como p70 e Akt (proteína quinase B) são ativadas pela via ligada a
PI3K (KOHN et al., 1996; KROOK et al., 1998). A ativação da Akt é uma etapa importante na
ativação da enzima glicogênio sintetase quinase (GSK-3) e, portanto no armazenamento de
glicogênio. No tecido adiposo e muscular a Akt em resposta ao estímulo da insulina, encontra-se
ligada a vesículas contendo o transportador de glicose (GLUT4) (KUPRIYANOVA &
KANDOR, 1999). As Vesículas intracelulares que contêm o GLUT4 dependente de insulina é
translocado a superfície celular e atua permitindo a entrada de glicose e seu posterior
metabolismo (WHITE, 1997).
Assim como na sinalização de outros hormônios e fatores de crescimento, a insulina tem a
capacidade de estimular a via de sinalização da proteína ativada pela mitógeno (MAP) quinase e
da ERK. Esta via pode ser iniciada pela fosforilação em tirosina das proteínas IRS e/ou da Shc, as
quais por sua vez interagem com Grb2 recrutando a proteínas “son-of-sevenless” (SOS) para a
membrana plasmática e assim ativando Ras. Uma vez ativada, Ras se comporta como um
interruptor molecular, ativando uma cascata de serinas-quinase por meio da ativação de Raf,
MEK e ERK. ERK ativada pode se translocar para o núcleo, onde catalisa a fosforilação de
fatores de transcrição que levam à proliferação e à diferenciação celular (BOULTON et al, 1991).

24
A FIGURA 1 mostra um esquema simplificado das etapas de sinalização intracelular
desde a ligação da insulina a seu receport até a ativação do transporte de glicose. Os eventos que
ocorrem após a ligação da insulina a seu receptor são altamente regulados e específicos (PESSIN
& SATIEL, 2000).
FIGURA 1. As vias de sinalização da insulina. O receptor de insulina é uma tirosina quinase que se autofosforila e catalisa a fosforilação de proteínas intracelulares como as proteínas IRS, Shc e Cbl. Após a fosforilação essas proteínas se ligam a outras moléculas de sinalização através de seus domínios SH2, resultando na ativação de vias de sinalização intracelular como a via da PI(3)K, a cascata da MAPK e a ativação do TC10 via CAP/Cbl. Essas vias regulam o transporte de glicose, a síntese de glicogênio, lipídeos e proteínas, coordenando e integrando o metabolismo intermediário (CARVALHEIRA et al., 2002).
Em se tratando de obesidade, a resistência à insulina é manifestada pela redução do
transporte e metabolismo da glicose estimulado pela insulina no adipócito e músculo esquelético,
entre outros tecidos, bem como pelo aumento da liberação da glicose hepática. Estas alterações
funcionais podem resultar, em parte, de alterações nas vias de transmissão do sinal da insulina.
Tanto no músculo quanto no tecido adiposo a ligação da insulina ao seu receptor na membrana,
bem como a fosforilação e a atividade quinase deste receptor, estão reduzidas em indivíduos com
resistência à insulina (KAHN & FLIER, 2000).
Um importante mecanismo de modulação do sinal da insulina ocorre quando IRS-1 é
fosforilado em serina por serinas quinases intracelulares. Uma vez fosforilado em serina, torna-se
incapaz de ser fosforilado em tirosina, impedindo a ativação da PI3K, bloqueando a transmissão

25
do sinal insulínico (HOTAMISLIGIL et al., 1996). Essas fosforilações inibitórias causam
"feedback" negativo na sinalização insulínica e podem provocar resistência à insulina
(CARVALHEIRA et al., 2003). IRS-1 fosforilado em serina, poderá inibir retrogradamente a
ativação do receptor de insulina, pois uma vez ligado ao receptor, inibe sua capacidade tirosina-
quinase, impedindo a transmissão do sinal de insulina para outras moléculas de IRS-1, ou ainda
para outros substratos deste receptor (HOTAMISLIGIL & PERALDI et al. 1996).
A ação da insulina também pode ser atenuada por meio de proteínas tirosina-fosfatases
(PTPases), as quais catalisam a rápida desfosforilação do receptor de insulina e de alguns de seus
substratos. Algumas dessas proteínas são expressas em células sensíveis à insulina ou expressas
em maior quantidade em estado de resistência à ação desse hormônio. A mais estudada tem sido a
fosfatase PTP1B, cujo “knockout” gênico leva ao aumento da fosforilação em tirosina do IR e
IRSs em músculo, aumentando assim a sensibilidade à insulina (ELCHEBLY et al, 1999).
Várias serinas quinases incluindo c-jun N-terminal quinase (JNK) (LEE, et al., 2003) e
quinase inibidora do fator nuclear kB (IKKβ) foram identificadas como capazes de fosforilar IRS-
1 em resíduos de serina. É possível que a JNK e a IKK possam contribuir para a resistência à
insulina porque estas quinases são ativadas em algumas situações patológicas em que ocorre
aumento de produção de citocinas e espécies reativas de oxigênio, como na obesidade.
Analisando estes dados em conjunto entendemos que bloqueios na transmissão do sinal de
insulina intracelular podem estar relacionados à resistência às ações fisiológicas deste hormônio.
E a reversão destes bloqueios à sinalização podem resultar em melhora da sensibilidade à insulina
e assim evitar a progressão do diabetes mellitus tipo 2.
1.3. Tiazolidinadionas e mecanismo de ação
A introdução das tiazolidinadionas (TZDs), que atuam ligando-se e ativando o fator
nuclear PPARγ (Peroxisome proliferator-activated receptor gamma), tem demonstrado efeito
altamente benéfico, reduzindo a resistência à insulina em pacientes com diabetes mellitus tipo 2
bem como em vários modelos de animais com diabetes e obesidade (OLEFSKY & SALTIEL,
2000; BALFOUR & PLOSKER, 1999; PLOSKER & FAULDS, 1999). Estes fármacos foram
desenvolvidos a partir de análogos do ácido clofibrico como antioxidante, no inicio dos anos 80,
no Japão (KAWAMATSU et al., 1980; YOSHIOKA et al., 1989) sem haver conhecimentos
prévios de quais os seus alvos moleculares.
Inicialmente, Sohda e colaboradores (1982) sintetizaram várias TZDs e observaram
propriedades hipoglicemiantes, constatando que a ciglitazona apresentava-se como a mais ativa

26
da série, demonstrando uma redução na glicemia em modelos animais, particularmente em
animais geneticamente resistente à insulina tal como camundongos KK, db/db, e ob/ob e ratos
fa/fa (FUJITA et al., 1983; FUJIWARA et al., 1991), sendo posteriormente, abandonada, devido
a hepatoxicidade (GALE, 2001).
Em 1997, a Food and Drug Administration (FDA) aprovou a troglitazona (Rezulin®), a
primeira tiazolidinadiona avaliada na clínica nos Estados Unidos e Japão. Ela foi responsável por
alguns casos de necroses hepática maciça, seguida de óbito (WATKINS & WHITCOMB, 1998;
GITLIN et al., 1998) tendo sido retirada do mercado norte-americano, em março de 2000.
Os sérios efeitos colaterais ocorridos com troglitazona e sua interdição em alguns países
motivaram os pesquisadores a desenvolver novas glitazonas. Assim, em 1999, a FDA aprovou
dois novos fármacos da classe das TZDs: o maleato de rosiglitazona (Avandia®) e o hidrocloreto
de pioglitazona (Actos®). Atualmente, rosiglitazona e pioglitazona são os dois membros da
classe das TZDs usados na prática clínica em vários países incluindo os Estados Unidos, Japão,
Brasil e países da Europa.
Embora o mecanismo molecular da ação das TZDs não esteja precisamente conhecido,
hoje, sabe-se que os efeitos terapêuticos das TZDs parece resultar da alta afinidade de união das
TZDs ao PPARγ (Peroxisome Proliferator-Activated Receptor gamma) (LEHMANN et al., 1995;
SPIEGELMAM, 1998; SCHOONJANS et al., 2000), encontrado em vários tecidos-alvos para a
ação da insulina, incluindo o tecido adiposo, músculo esquelético, fígado e endotélio vascular
(LAW et al., 2000; IIJIMA et al., 1999). Ao contrário dos receptores de membrana plasmática,
que ativam cascatas de segundos mensageiros, os receptores intracelulares (citoplasmáticos ou
nucleares) são fatores de transcrição que exercem sua função regulatória diretamente no promotor
de genes alvos.
O PPARγ quando ativado, regula a transcrição de genes responsivos à insulina, que
controlam o metabolismo, tanto da glicose quanto de lipídios (KERSTEN et al., 1999; CHAWLA
et al., 1994; SPIEGELMAM, 1998; LOWELL, 1999; HSUEH, et al., 2001; GLASS &
ROSENFELD, 2000)
Três isoformas de PPARs foram identificadas PPPAR α, β/δ e γ. Estes receptores foram
originalmente identificados em 1990 por Issemann e Green e, assim, designados em virtude de
serem receptores ativados por drogas que induzem a proliferação de peroxissomas (ISSEMANN
& GREEN, 1990). Diversas moléculas como postaglandinas e vários compostos lipídicos
constituem ligantes naturais dos receptores PPARγ (YU et al.,1995; FORMAN, et al. 1995).

27
Existe, pelo menos, duas isoformas de PPARγ, (PPARγ1 e PPARγ2). Os adipócitos
apresentam uma alta expressão de PPARγ1, porém também tem se encontrado em outros tecidos
tais como cólon, retina, baço e células hematopoéticas (CHAWLA et al., 1994; SPIEGELMAM,
1998; LOWELL, 1999; KERSTEN et al., 1999). No entanto, a isoforma PPARγ2 é expressa
exclusivamente em adipócitos (REGINATO & LAZAR, 1999; KERSTEN et al., 1999; VIDAL-
PUIG et al. 1996; FAJAS et al. 1997). A expressão de PPARγ em adipócitos é essencial para a
diferenciação de pré-adipócitos em adipócitos (KERSTEN et al., 1999).
Todas as três isoformas de PPARs (PPARγ, PPARα e PPARδ) possuem estrutura e
características semelhantes. Quatro domínios funcionais principais foram identificados, A/B, C, D
e E/F. O dominio A/B localizado na região N-terminal , é pouco conservado entre as três
isoformas de PPARs e contêm uma função de transativação independente de ligante (AF-1)
(WERMAN et al., 1997) O estado de fosforilação deste domínio contribui para a modulação da
atividade do PPARα e γ (SHALEV et al., 1996; ZHANG et al., 1996; ADAMS et al., 1997). O
domínio central (domínio C) é altamente conservado e contém a região do receptor que liga se ao
DNA, sendo também conhecido como domínio de ligação ao DNA (DBD – “DNA binding
domain”). Este domínio apresenta dois complexos denominados “zinc finger”, ou seja dois
arranjos protéicos constituídos de uma α-hélice e uma folha β-pregueada, mantidas unidas por um
íon zinco na região central. Esta estrutura confere ao receptor uma maior estabilidade de
dobramento, para que este se ajuste na fenda maior da dupla hélice do DNA e forme associações
firmes com as regiões regulatórias dos genes-alvos (SCHOONJANS et al., 1996; KLIEWER et
al., 1992). O domínio D é um local de ancoragem para cofatores. O domínio E/F ou LBD é a
porção do receptor que se liga ao ligante/ativador, sendo responsável pela função de transativação
dependente de ligante (AF-2), além de facilitar a heterodimerização do PPAR com o receptor
Retinóide X (RXR) (SCHOONJANS et al., 1996). O heterodimero resultante subsequentemente
se liga aos PPREs (elemento responsivo ao PPAR) que são seqüências repetidas diretas de
hexanucleotídeos, separadas por uma base (DR1) e localizadas na região regulatória (promotor)
dos genes que estão sob seu controle transcricional (GEARING et al., 1993).
Em estudos recentes foram identificadas proteínas co-repressoras e co-ativadoras que
interagem com os PPARs, desempenhando papel importante na atividade transcricional destes
receptores. A interação dos PPARs com os ligantes gera alteração conformacional do receptor,
que permite o recrutamento de co-ativadores (SRC-1, CBP/P300) e a liberação co-repressores
(NCoR) (DOWELL et al., 1999; GLASS; ROSENFELD, 2000; DOWELL et al., 1999;
HORLEIN et al., 1995; CHEN & EVANS, 1995). Embora as isoformas possuam características

28
estruturais semelhantes, cada isoforma varia em especificidade, distribuição de tecido e
conseqüentemente sequem propósitos biológicos diferentes.
Na FIGURA 2 está apresentada a estrutura funcional e mecanismo molecular de
transativação da isoforma PPARγ.
FIGURA 2. Domínio funcional de isoformas de PPARγ e mecanismo molecular de transativação. Na parte superior do desenho duas isoformas de PPARγ1 e PPARγ2 com domínios estrutural e funcional, mostrando algumas mutações que podem ocorrer em humanos na isoforma PPARγ1 ou PPARγ2. Na parte inferior do desenho ativação do PPARγ pelo ativador/ligante. O receptor ativado liga-se a seqüências específicas de genes-alvo (PPER) como heterodimero com o receptor do áciod 9-cis retinóico (RXR). O heterodímero PPARγ:RXR associa-se com cofatores, incluindo proteínas co-ativadoras e co-rpressoras, para regular o processo trancricional (TSAI & MAEDA, 2005).
As TZDs, agonistas de PPARγ aumentam a sensibilidade do músculo à insulina.
Entretanto, este tecido apresenta baixa expressão do PPARγ. Muitas hipóteses têm sido
elaboradas para explicar esta aparente discrepância.
A primeira hipótese considera que a ação das TZDs é indireta, ou seja mediada pelo tecido
adiposo, órgão com alta expressão de PPARγ (MARTIN et al., 1996). Este efeito poderia ser
exercido por dois diferentes processos: a) primeiro, no tecido adiposo, ativadores do PPARγ

29
modulam a expressão de TNF-α e leptina (RICOTE et al., 1999; COHEN et al., 1996), que são
moléculas sinalizadoras que afetam a sensibilidade do músculo à insulina; b) segundo, a ativação
do PPARγ poderia induzir um “clearence” de ácidos graxos (AG) pelo tecido adiposo, gerando
uma diminuição na concentração plasmática de AG e no transporte para o músculo (MARTIN et
al., 1996; OAKES et al., 1997). Uma segunda hipótese para explicar a ação das TZDs é que estas
podem agir diretamente nos tecidos sensíveis à insulina, mesmo que esses apresentem baixa
expressão de PPARγ. A ativação do PPARγ pode afetar a sensibilidade à insulina no músculo por
regular genes envolvidos no metabolismo de glicose. (RIBON et al., 1998).
Apesar das TZDs representarem uma inovação dentro do tratamento do diabetes
mellitus tipo 2, a retenção de fluidos, apresentada como rápido ganho de peso corporal e a
formação de edemas tem sido considerado o efeito colateral mais comum da rosiglitazona
(ZHANG et al., 2005). Muitas TZDs têm sido desenvolvidas (FUJIWARA et al., 1988; CLARK
et al., 1991; CANTELLO et al., 1994; MOURÃO et al., 2005) e muitas outras se encontram em
vários estágios pré-clínicos e clínicos de desenvolvimento (YANAGISAWA et al., 2002). Todas
as TZDs compartilham uma estrutura tiazolidina-2,4-diona substituída, na qual se tem realizado
modificações químicas seletivas, na tentativa de melhorar a eficácia farmacológica e minimizar os
efeitos colaterais desses agentes.
Dentre os métodos de obtenção de novos fármacos, a modificação ou variação
molecular, utilizando principalmente o conceito de bioisosterismo, é o mais utilizado
(KOROLKOVAS, 1977; BARREIRO & FRAGA, 2001). Com a aplicação de bioisosterismo
pode-se analisar a influência da modificação de um átomo ou de um grupo de átomos por seu
bioisóstero sobre a atividade biológica que o fármaco original apresenta, podendo ser de ação
idêntica ou mesmo antagônica.
Assim, na primeira etapa deste estudo nós sintetizamos 12 derivados arilideno-
tiazolidinadionas (ATZDs) com potencial atividade hipoglicemiante. Posteriormente,
investigamos os efeitos do composto 5-(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-
diona (GQ2) na sensibilidade e sinalização da insulina em animais com obesidade induzida por
dieta hiperlipidica. Camundongos “swiss” submetidos a uma dieta hiperlipídica por um período
de seis a oito semanas apresentam: obesidade, hiperglicemia, hiperinsilinemia e resistência à
insulina inclusive a nível molecular, o que é um bom modelo de diabetes mellitus tipo 2.

30
OBJETIVOS

31
2. OBJETIVOS
Os objetivos do presente estudo foram:
Sintetizar novos derivados arilideno-tiazolidínicos com substituições no anel benzilideno;
Elucidar as estruturas químicas dos compostos sintetizados pelos métodos espectroscópicos
de infravermelho, ressonância magnética nuclear e espectrometria de massas;
Elucidação do padrão de interação entre o receptor PPARγ e as novas tiazolidinadionas
através de estudos de "Docking" realizados com o programa Autodock 3.
Avaliar o efeito de um novo agonista de PPARγ (GQ2) no tratamento de camundongos
“swiss” com obesidade induzida por dieta hiperlipídica sobre:
• A variação de massa visceral e da massa corporal total e a ingestão alimentar;
• Sensibilidade à insulina através do teste de tolerância à insulina (ITT);
• Os níveis de expressão e fosforilação do receptor de insulina, do IRS-1, do
IRS-2, bem como a interação dos IRSs (1 e 2) com a PI3K, e conseqüente
ativação da Akt e das ERKs (1 e 2) em tecido hepático, muscular e adiposo;
• A fosforilação do substrato IRS-1 em serina 307;
• Inibição ou ativação de proteínas envolvidas na resposta inflamatória tais
como: JNK, complexo IKK e TNF-α e INOS;

32
MATERIAL E MÉTODOS
PARTE QUÍMICA

33
3.1. PARTE QUÍMICA
3.1.1. Material
3.1.1.1. Reagentes e solventes
Todos os reagentes (Sigma ou Aldrich) e solvente (Vetec ou Quimis) utilizados possuíam
especificação P.A.
Ácido monocloroacético
Acetato de etila
Ácido sulfúrico
Benzeno
4-Clorobenzaldeído
4-Hidróxidobenzaldeído
2-Clorobenzaldeído
4-Metoxibenzaldeído
2,4-Dimetoxibenzaldeído
3-Clorobenzaldeído
2-Bromobenzaldeído
Tolueno
Cloreto de 4-metil-benzil
4-Nitrobenzaldeído
Etanol absoluto
Éter dietílico
Hidróxido de potássio
Indol-3-carboxaldeído
Metanol
n-Hexano
-4-Dimetilaminobenzaldeído
4-Benziloxibenzaldeído
Piperidina
Tiuréia
Cianoacetato de etila

34
3.1.2. Equipamentos
3.1.2.1. Espectroscopia
Os espectros das novas ATZDs foram realizados nos seguintes aparelhos:
Espectrofotometria de absorção no infravermelho (IV) – Perkin-Elmer 1310 ou
espectrofotômetro FTIR Bruker Modelo IFS 66, em pastilhas de KBr; e expressos em cm-1;
Espectroscopia de ressonância magnética nuclear de hidrogênio (RMN1H) –
espectrofotômetro Bruker AC 200 ou em aparelho Varian Modelo Plus 300 MHz;
Espectrometria de Massas (MS) - espectrômetro Delsi-Nermag, modelo R-1010C,
acoplado a CPG HP 5987.
3.1.2.2. Ponto de fusão
Os pontos de fusão foram determinados em aparelho Quimis Modelo 340.27.
3.1.2.3. Cromatografia
As cromatografias analíticas em camada delgada foram efetuadas em placas de sílica gel
60 Merck F254, de 0,25 mm de espessura. As revelações foram feitas por luz ultravioleta (254 ou
366nm) ou através de vapores de iodo.
As cromatografias “flash” (sob pressão) foram realizadas em sílica gel 60 Vetec (230-400
Mesh).
35
3.1.3. Métodos
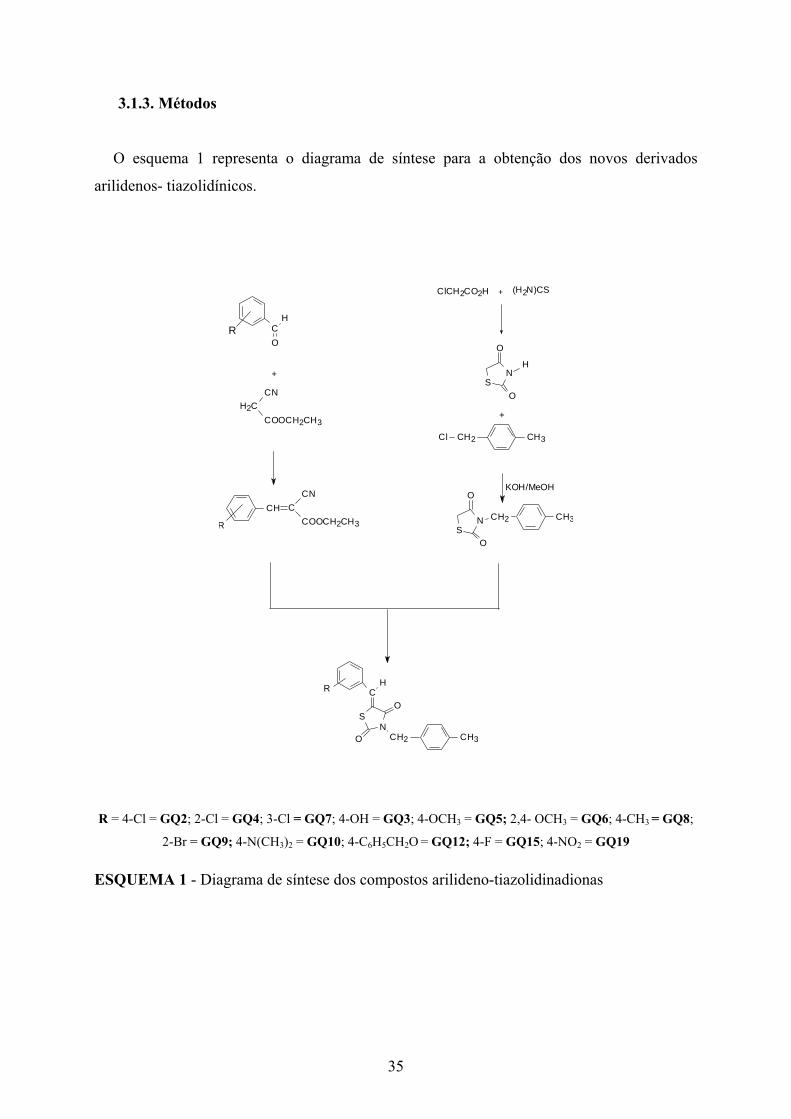
O esquema 1 representa o diagrama de síntese para a obtenção dos novos derivados
arilidenos- tiazolidínicos.
CCN
COOCH2CH3
CH
RCH3N
S
O
O
CH2
R
H2CCN
COOCH2CH3
+
CO
H
CH3
CH
(H2N)CS+ClCH2CO2H
CH3CH2Cl
NS
O
O
H
+
KOH/MeOH
SN
O
O CH2
R = 4-Cl = GQ2; 2-Cl = GQ4; 3-Cl = GQ7; 4-OH = GQ3; 4-OCH3 = GQ5; 2,4- OCH3 = GQ6; 4-CH3 = GQ8;
2-Br = GQ9; 4-N(CH3)2 = GQ10; 4-C6H5CH2O = GQ12; 4-F = GQ15; 4-NO2 = GQ19
ESQUEMA 1 - Diagrama de síntese dos compostos arilideno-tiazolidinadionas
R

36
3.1.3.1. Obtenção da tiazolidina-2,4-diona
Em um balão adicionou-se a tiouréia (5 g - 0,0658 mols), o ácido cloroacético (6,335 g -
0,0673 mols) previamente dissolvido em água. Aqueceu-se a mistura por 18 horas. Em seguida,
deixou-se o produto obtido em repouso por 24 horas na geladeira. Formaram-se cristais brancos,
cuja purificação foi feita por recristalizações sucessivas em água destilada.
3.1.3.2. Obtenção da 3-(4-metil-benzil)-tiazolidina-2,4-diona
A tiazolidina-2,4-diona (5,5712 g - 0,0476 mols) em 10 ml de etanol absoluto, adiciona-
se hidróxido de potássio (1,9 g - 0,0476 mols) dissolvido em 10 mL de etanol absoluto. Deixa-se
reagir à temperatura ambiente por 10 minutos. Ao sal de potássio da tiazolidina-2,4-diona
formado adiciona-se 7 ml do cloreto de 4-metil-benzil. Após 4 horas de reação uma temperatura
de 70 ºC. A mistura reacional adiciona-se gelo picado, o precipitado formado é filtrado e
purificado através de cromatografia sob pressão em sílica gel 60 através de um gradiente de
eluição CHCl3 e CHCl3/CH3OH 92:08 (SHIVAIKA et al., 1983).
3.1.3.3. Obtenção dos etil-(2-ciano-3-fenil)-acrilatos de etila
Em um balão de fundo redondo colocam-se o aldeído aromático substituído e o
cianoacetato de etila, em presença de piperidina como catalisador e benzeno como solvente. A
mistura reacional é aquecida a uma temperatura de 110 0C, durante 4 horas. O produto é mantido
na geladeira por 12 horas. Os ésteres são purificados através de recristalizações sucessivas em
solventes adequados ou por cromatografia sob pressão em sílica gel conforme descrito a seguir
(COPE et al., 1941).

37
3.1.3.4. Método geral para a obtenção dos derivados 5-benzilideno-3-(4-metil-benzil)-
tiazolidina-2,4-dionas
A mistura reacional da 3-(4-metil-benzil)-tiazolidina-2,4-diona e do éster cianocinâmico
substituído dissolvido em etanol seco, em presença de piperidina como catalisador, é aquecida a
refluxo durante 4 horas. Após resfriamento em banho de gelo ocorre a cristalização dos
derivados 5-benzilideno-3-(4-metil-benzil)-tiazolidina-2,4-diona.

38
MATERIAL E MÉTODOS
MODELAGEM MOLECULAR

39
3.2. MODELAGEM MOLECULAR
3.2.1. Material
Microcomputador Athlon XP 2600+
1G DDR
HD 80 GB (7200 rpm)
Programas Computacionais
BioMedCache;
Autodock 3.0;
ADT (AutoDockingTools);
Microcal TM Origin TM 7.0;
3.2.2. Métodos
O alvo molecular das TZDs é o receptor nuclear PPARγ (peroxisome proliferator-
activated receptor gamma), cuja estrutura foi determinada por cristalografia de raios-X, estando
disponível no banco de dados PDB sob o código 2PRG.
A busca conformacional inicial para a estrutura das novas ATZDs foi feita com o
programa BioMedCache. Depois de retirado o ligante co-cristalizado na estrutura 2PRG, foram
realizados os estudos de “docking” para as novas ATZDs, utilizando-se os programas Autodock
3.0 e ADT (AutoDockingTools).

40
MATERIAL E MÉTODOS
PARTE BIOLÓGICA

41
3.3. PARTE BIOLÓGICA
3.3.1. Modelo animal e tratamento
Foram utilizados camundongos “swiss” machos com cerca de quatro semanas de idade
fornecidos pelo Biotério Central da UNICAMP (CEMIB).
Os camundongos foram divididos randomicamente em dois grupos, a saber: aqueles que
receberam dieta comercial da Purina (Labina – ração padrão para animais de laboratório
(grupo controle (C)) e os que foram alimentados por um período de aproximadamente seis
semanas, com uma dieta hiperlipídica, (dieta contendo 35% de banha de porco).
Após seis semanas, os animais que receberam a dieta hiperlipídica (DH) e apresentavam
glicemia de jejum acima de 140mg/dl, foram subdivididos ao acaso em três novos grupos: grupo
controle da DH e os outros dois grupos tratados com agonistas de PPARγ diariamente durante
14 dias por via oral através de gavagem: rosiglitazona (RG = 10mg/kg/dia - grupo padrão) e
GQ2 (20mg/kg/dia – grupo teste). Os animais controles (C e DH) receberam somente o veículo
(solução salina contendo 0,25% de tween - 0,1ml/kg de peso corporal).
A escolha do composto GQ2, e a dose utilizada nesta etapa experimental foi baseada em
estudos preliminares com camundongos com diabetes induzida por aloxana (ANEXO 1).
Os camundongos durante o período experimental, foram mantidos em gaiolas individuais
sob condição padronizada de iluminação (ciclo claro/escuro de 12 horas), temperatura de 22 ±
2ºC e recebendo água in libidum, dieta padrão e/ou dieta hiperlipídica.
O procedimento experimental foi realizado sempre pela manhã. No sétimo dia de
tratamento, foi realizado teste de tolerância à insulina – ITT. Décimo dia de tratamento, amostras
de sangue foram coletadas para dosagens Bioquímicas. No décimo quinto dia de tratamento,
extração de tecidos (fígado, músculo e adiposo) para estudo das etapas iniciais e intermediarias
da sinalização da insulina. Os animais permaneceram em jejum 12 horas (uma noite) prévias aos
experimentos.
Todos os experimentos com animais foram aprovados pelo Comitê de Ética de Animal de
Experimentação da Unicamp (CEEA/IB-UNICAMP).

42
3.3.2. Western blot e imunoprecipitação
3.3.2.1. Anticorpos, reagentes químicos e materiais
Os reagentes e aparelhos para eletroforese em gel de poliacrilamida com dodecil sulfato
de sódio (SDS-PAGE) foram adquiridos da Bio-Rad (Richmond, CA). Metano
hidroximetilamina (TRIS), fenil-metilsulfonilfluoreto (PSMF), aprotinina e ditiotreitol (DTT),
Triton X-100, Tween 20, e glicerol foram fornecidos pela Sigma Chemical Co. (St. Louis, Mo.
USA). Kit de quimilunninecence foi adquirido da Amersham (Amersham, UK), e a Proteína A
Sepharose 6 MB da Pharmacia (Uppsala, Suécia). A membrana de nitrocelulose (Hybond ECL,
0.45 µm) foi obtida da Amersham (Aylesbury, UK). O agente anestésico KETAMIN (Cloridrato
de S(+) cetamina) foi adquirido da Cristália (Itapira/SP, Brasil) e a insulina regular humana
(IOLIN®) foi adquirida da Lilly. Os anticorpos anti-IR (sc-711, rabbit polyclonal), IRS-1 (sc-
559, rabbit polyclonal), IRS-2 (sc-8299, rabbit polyclonal), p-ERK (sc-7383, mouse polyclonal,
reconhecendo as subunidades 42 e 44 kDa da ERK fosforiladas em Tyr204), PPARγ (sc-7196,
rabbit polyclonal), iNOS (NOS2) (sc–7271, nonoclonal mouse), IkB-α (sc – 1643, nonoclonal
mouse), p-JNK (sc – 6254, nonoclonal mouse), e anti-fosfotirosina (sc-508, mouse monoclonal)
foram obtidos da Santa Cruz Biotechnology (CA, USA). Também foram utilizados os anticorpos
policlonais PI3-quinase (p85) e Anti-phospho-Akt (Ser473) adquiridas da Upstate Biochnology
(Lake Placid, NY, USA). Rosliglitazona (RG), droga sensibilisadora de insulina, usada como
padrão dos testes, foi obtida do laboratório Glaxo, Smith Kline (GSK).
3.3.2.2. Soluções utilizadas
• Solução tampão de extração (para extrato total e imunoprecipitação): utilizada para a
extração das proteínas celulares dos tecidos estudados, que foram posteriormente
imunoprecipitadas. Contém: Trisma base pH 7,5 (hidroximetil amino metano) 100 mM,
SDS (dodecil sulfato de sódio) 10%, EDTA (Ácido etileno-diamino tetracético) 10 mM,
fluoreto de sódio 100 mM, pirofosfato de sódio 10 mM, ortovanadato de sódio 10 mM,
fluoreto de fenilmetil sulfonila (PMSF) 2Mm (diluído em álcool etílico). Triton X- 100
1% e 0,1mg/mL de aprotinina. A solução foi mantida a 40C, sendo que o ortovanadato,
PMSF e a aprotinina foram acrescidos no momento de utilização do tampão.
• Solução tampão para lavagem do imunoprecipitado: contém: Trisma base 100 mM,
EDTA 10 mM, ortovanadato de sódio 2 mM e triton X-100 0,5%.

43
• Tampão de Laemmli (5X): usado para estocar o material extraído e sua posterior
aplicação no gel de poliacrilamida para eletroforese (SDS-PAGE), contém: azul de
bromofenol 0,1%, fosfato de sódio 1M pH 7,0, glicerol 50% e SDS 10%.
• Solução tampão utilizada na eletroforese em gel de poliacrilamida (SDS-PAGE):
contém: Trisma base 200 mM, glicina 1,52 M, EDTA 7,18 mM e SDS 0,4%. Para uso, a
solução é diluída 1:4.
• Solução tampão para transferência: empregada para a transferência das proteínas
separadas no SDS-PAGE para a membrana de nitrocelulose contém: Trisma base 25 mM,
glicina 192 mM, Metanol 20% e SDS 0,02% para facilitar a eluição de proteínas de alto
peso molecular. Mantida estocada a 4ºC.
• Solução tampão para SDS-PAGE - Gel de resolução (resolving): tampão composto de
EDTA 4 mM, SDS 2%, trisma base 750 mM, com pH ajustado para 8,9 com ácido
clorídrico.
• Solução tampão para SDS-PAGE - Gel da fase de empilhamento (stacking) das
proteínas: contém: EDTA 4 mM, SDS 2%, trisma base 50 mM, com pH ajustado para
6,7 com ácido fosfórico.
• Solução basal: solução básica utilizada para o manuseio da membrana de nitrocelulose
após transferência das proteínas, contém: Cloreto de sódio 150 mM, Trisma base 10 mM,
Tween 20 0,02%.
• Solução bloqueadora: utilizada para incubar a membrana de nitrocelulose, após a
transferência, contém: 5% de leite em pó desnatado e azida sódica 0,02%, dissolvidos em
solução basal.
• Solução para anticorpos: solução contendo anticorpos específicos que identificam as
proteínas transferidas para a membrana de nitrocelulose. Contém 0,3% de leite em pó
desnatado e azida sódica 0,02%, diluídos em solução basal.

44
• Solução com proteína A marcada com 125I: permite a visualização das bandas em auto-
radiografia, contém 0,1% de leite desnatado, dissolvido em solução basal com 2 μCi de
proteína A 125I.
• Tampão de extração nuclear - tampão A (extrato citoplasmático): Hepes 10mM, pH
7.9, KCl 10mM, MgCl2 1.5mM, dithiotheithol 1mM, ortovanadato de sódio 1mM,
phenymethylsulfony fluoride 1mM, aprotinina 10μg/mL. Tampão B (extrato nuclear):
Hepes 20mM, pH 7.9, NaCl 10.42M, KCl 10mM, glicerol 20%, dithiotheithol 1mM,
ortovanadato de sódio 1mM e aprotinina 10μg/mL.
3.3.2.3. Extração de tecidos, imunoblot e imunoprecipitação
Para o estudo das etapas iniciais da ação insulínica, os animais foram anestesiados
intraperitonealmente com ketamina (50mg/kg) e diazepan (10mg/kg) e submetidos à extração
dos tecidos, logo após a perda dos reflexos corneano e caudal.
Inicialmente foi aberta a cavidade abdominal e retirados fragmentos dos tecidos a serem
estudados (grupo negativo – sem estímulo de insulina). Para os animais do grupo positivo (com
estímulo agudo de insulina), foi injetada por via intraperitoneal insulina regular em concentração
de 1,5U/kg de peso. Após 5 minutos da injeção de insulina foram retirados fragmentos de fígado,
tecido adiposo branco (epididimal), músculo esquelético (gastrocnêmio) que foram
imediatamente homogeneizados e solubilizados em tampão de extração adequado a 40C com
Polytron durante 30 segundos. Após a homogeneização as amostras foram centrifugadas a
12.000 rpm por 40 minutos a 40C. O “pellet” foi desprezado e amostras do sobrenadante foram
usadas para medida da concentração protéica, utilizando-se o reagente para o ensaio Bio-Rad
Protein Assay-Dye Reagent Concentrate (Melville, NY). Foi utilizado como referencial, uma
curva-padrão de albumina. Amostras contendo 1mg de proteína total foram usadas para
imunoprecipitar durante período noturno com anticorpo anti IR, IRS-1, IRS-2. A seguir, as
proteínas foram separadas por SDS-PAGE, transferidas para a membrana de nitrocelulose,
utilizando-se o equipamento de eletro-transferência do minigel da Bio-Rad, sendo realizadas
durante 130 min a 120 V em gelo, banhadas com tampão de transferência. No mesmo gel foi
aplicada uma amostra padrão de proteínas, ou seja, marcador de peso molecular conhecidos:
miosina (205kDa), beta galactosidase (116 kDa), albumina sérica bovina (80kDa) e ovalbumina
(49 kDa). As proteínas apareciam sob coloração azul no gel de eletroforese e na membrana de
nitrocelulose, permitindo a orientação quanto ao peso molecular das bandas observadas. Após

45
transferência, as membranas foram bloqueadas com 5% de leite desnatado em solução Tris-
salina durante duas horas, a temperatura ambiente, para diminuir a ligação inespecífica de
proteínas. A seguir, as membranas foram lavadas com solução Tris-salina em três sessões de
cinco minutos, e incubada com anticorpos específicos sob agitação constante por uma noite a
40C. Foram então lavadas novamente com solução Tris-salina em três sessões de cinco minutos e
incubadas a seguir com anticorpo secundário anti rabbit ou anti-mouse (peroxidase conjugada
anti-rabbit ou anti-mouse – diluição 1/6000) por duas horas a temperatura ambiente. As
membranas foram então lavadas e os anticorpos foram detectados usando kit de
quimiluminecence. As membranas foram expostas ao filme de RX (Kodak XAR-Rochester, NY)
com intensificador (Cronex Lightining Plus intensifying screens-DuPont, Wilmington, DE) por
aproximadamente 20 minutos. Os filmes foram revelados na forma convencional.
A intensidade das bandas foi determinada através da leitura das autoradiografias
reveladas por densitometria ótica, utilizando um scanner (HP 3400) e o programa Scion Image
(Scion Corporation). Os dados numéricos obtidos, correspondentes às bandas protéicas foram
comparados estatisticamente como descrito abaixo.
Em experimentos de “imunoblot” direto alíquotas contendo 100μg de proteína por
amostra de extratos protéicos obtidos de cada tecido foram separados por SDS-PAGE,
transferidos para membrana de nitrocelulose e então foram adicionados anticorpos específicos e
analisada como descrito acima.
3.3.3. Efeito do composto GQ2 no peso corporal, consumo de alimento e quantidade de
água em camundongos com obesidade induzida por dieta hiperlipídica
O ganho de peso dos camundongos tratados com GQ2 e RG e/ou veiculo foi
acompanhado desde o primeiro dia de tratamento ao último dia do experimento. As pesagens
foram realizadas sempre pela manhã em balança digital de precisão.
Durante todo o período experimental (14 dias), a dieta hiperlipídica ou ração purina era
trocada diariamente entre 8 e 9 horas da manhã e as sobras pesadas. Controlando-se as
quantidades oferecidas e sobras, foi possível determinar a ingestão alimentar diária individual
dos animais tratados com GQ2 e RG e/ou controles.
No final do tratamento suas carcaças foram secas (620C) até obtenção de peso constante.
As diferenças entre o peso total e peso seco representava o ganho de água de cada animal.

46
3.3.4. Teste de tolerância à insulina (ITT)
Após os animais terem sido tratados como descrito anteriormente, foram mantidos em
jejum por 12 horas, e depois de coletada amostra de sangue da cauda (tempo zero), foi injetada
insulina (1,5 U/kg de peso corporal) na cavidade peritoneal e a seguir coletadas amostras de
sangue nos tempos 5, 10, 15, 20, 25 e 30 minutos para determinação da glicose plasmática. A
razão da constante de decaimento da glicose, (Kitt) foi calculada usando a fórmula 0.693/t1/2. A
glicose t1/2 foi calculada, usando a análise da queda do quadrado da concentração de glicose
plasmática durante o decaimento da fase linear (BONORA et al, 1987).
3.3.5. Parâmetros Bioquímicos
Amostras de sangue foram coletadas através de punção no plexo orbitário sob leve
anestesia com ketamina (50mg/kg) e diazepan (10mg/kg) centrifugadas, alíquotadas e mantidas
no freezer para analise das concentrações de insulina, leptina, citocinas inflamatórias (TNF –α,
Il-6 e IL-1β) e glicose.
• Determinação dos níveis de insulina: A insulina foi determinada por radioimunoensaio
e, a curva padrão determinada com insulina de rato (SCOTT et al, 1981). Os resultados foram
expressos em ng/ml.
• Determinação de leptina: A leptina das amostras de soro foi dosada utilizando-se o kit
comercial de ELISA segundo a indicação do fabricante (Linco Research Inc, St Charles, MO,
USA) Os resultados foram expressos em pg/ml.
• Determinação de TNF-α, IL-6 e IL-β1: As citocinas TNF-α, IL-6 e IL-β1 das amostras
de soro foram avaliadas através de kit comercial de ELISA segundo a indicação do fabricante
(Pierce). Os resultados foram expressos em pg/ml.
• Glicemia A glicemia foi determinada antes e após 14 dias de tratamento com os
agonistas de PPARγ e/ou veículo por meio de tiras reativas (Avantage, Roche Diagnostics
Corporation) analisadas em glicosímetro (marca Avantage® Eli Lily, Boehringer Mannheim). As

47
amostras de sangue foram coletadas da cauda dos animais sob leve anestesia com ketamina +
diazepan (20mg/kg). Os resultados foram expressos em mg/dl.
3.3.6. Determinação da massa de tecido peri-epididimal
Após 14 dias de tratamento com os agonistas de PPARγ (RG e GQ2 ) e/ou veículo, os
camundongos foram anestesiados e a cavidade abdominal foi aberta e toda a massa de tecido
adiposo peri-epididimal foi cuidadosamente dissecada e pesada. A variação do peso absoluto da
massa de tecido adiposo epididimal de cada grupo foi utilizada para cálculos e comparações. O
resultado foi expresso em gramas (g) por 100g de peso corpóreo.
3.3.7. Toxicidade aguda e subcrônica
Camundongos Swiss machos normoglicêmicos, pesando entre 28 a 30g foram usados
para determinar a toxicidade aguda da nova ATZD. A toxicidade aguda foi avaliada pela
administração oral de até 2000mg/kg de GQ2. Os animais foram observados por 48 horas e no
fim deste período à mortalidade foi anotada para cada grupo (DIETRICHLORKE, 1983). O
grupo controle recebeu apenas o veículo.
Para avaliar a toxicidade subcrônica de GQ2, os níveis de ALT foram determinados de
amostras de sangue coletadas após 14 dias de tratamento com GQ2 e/ou veículo. Os resultados
foram expressos em ng/ml.
3.3.8. Atividade do NF-kB
A atividade do fator de transcrição NF-kB foi determinada em extrato nuclear de
amostras de gordura epididimal, músculo gastrocnêmio e fígado por ELISA (kit Pierce número
89858 - p50) de acordo com as instruções do fabricante. Os extratos nucleares dos tecidos foram
obtidos por homogeneização em tampão contendo: Hepes 10mM, pH 7.9, KCl 10mM, MgCl2
1.5mM, dithiotheithol 1mM, ortovanadato de sódio 1mM, phenymethylsulfony fluoride 1mM,
aprotinina 10μg/mL. Após 10 minutos de incubação no gelo, as amostras foram centrifugadas
(2000xg por 10 minutos a 40C) e lavadas com o mesmo tampão e os pellets foram resuspendidos
com um novo tampão contendo: Hepes 20mM, pH 7.9, NaCl 10.42M, KCl 10mM, glicerol 20%,
dithiotheithol 1mM, ortovanadato de sódio 1mM e aprotinina 10μg/mL. As amostras foram
incubadas durante 30 minutos a 40C sob agitação constante e novamente centrifugadas a

48
16000xg por 30 minutos a 40C (SIEGRIST-KAISER et al., 1997). As concentrações de proteínas
dos sobrenadantes contendo os extratos nucleares foram determinadas pelo método de Bradford
(BRADFORD, 1976).
3.3.9. Análise estatística
Todos os resultados numéricos estão expressos como média ± erro padrão da média
seguido do número de experimentos. Os resultados dos “Western blot” estão apresentados como
comparações diretas das bandas protéicas nas auto-radiografias, as quais foram quantificadas
através de densitometria usando o programa “Scion Image” (Scion Corp). Os dados foram
analisados através de teste “t Student’s”, quando comparados dois grupos, e análise de variância
(Anova) seguida por análise de significância (Bonferroni) quando apropriado, comparando
grupos-controle e experimental. A significância estatística adotado foi p<0,05

49
RESULTADOS

50
4. RESULTADOS
4.1. Síntese e modelagem molecular de novos derivados tiazolidínicos
Na primeira etapa deste estudo, sintetizamos 12 derivados da série 5-arilideno-3-(4-
metil-benzil)-tiazolidina-2,4-dionas com substituições na posição dois, três e/ou quatro do anel
benzilideno, por cloro (GQ2 = 4-Cl, GQ4 = 2-Cl e GQ7 = 3-Cl), hidroxi (GQ3 = 4-OH), metóxi
(GQ5 = 4-OCH3), dimetóxi (GQ6 =2,4-CH3O), metil (GQ8 = 4-CH3), bromo (GQ9 = 2-Br),
dimetil-amina (GQ10 = 4-N(CH3)2), benziloxi (GQ12 = 4-C6H5CH2O), flúor (GQ15 = 4-F) e
nitro (GQ19 = 4-NO2), (ESQUEMA 1 e ANEXOS 1 e 2). Em seguida, avaliamos a interação
dessas novas moléculas arilideno-tiazolidinadionas (ATZDs) com o domínio LBD do receptor
PPARγ através dos estudos de “Docking”. Os complexos formados entre as novas ATZDs e o
receptor PPARγ apresentaram uma boa estabilidade (energia de ligação negativa) quando
comparadas com a energia de ligação da rosiglitazona (RG). Com o intuito de verificar a possível
influência do pré-metabolismo destas moléculas nos estudos de “Docking”, além das moléculas
originais, também foram usadas como possíveis ligantes para “Docking” as moléculas
hidrolisadas no nitrogênio do anel heterocíclico (TABELA 1 e ANEXO 2).
As interações da RG com o receptor PPARγ ocorrem através das formações de
ligações de hidrogênio entre o átomo de oxigênio do anel tiazolidínico com os resíduos de
aminoácidos histitina (his323) e serina (ser289) do domínio LBD do receptor. As novas ATZDs
também apresentaram este tipo de ligação, entre o átomo de oxigênio do anel heterocíclico e o
resíduo de ser289 do domínio LBD do receptor. A FIGURA 3 mostra que assim como a RG, o
novo agonista de PPARγ (GQ2), também interage com o domínio LBD do receptor PPARγ,
apresentando uma boa interação com o domínio LBD através da ligação de hidrogênio entre o
átomo de oxigênio do anel heterocíclico e o resíduo de ser289 do domínio LBD (ANEXO 2).
As novas ATZDs obtidas, após purificação apresentaram rendimentos satisfatórios,
variando de 36,5 a 88,9%. Apenas as TZDs GQ4 e GQ9 apresentaram baixo rendimento 18, 7 e
20,5% respectivamente. Os resultados da análise espectroscópica de ressonância magnética
nuclear de hidrogênio (RMN'H) e do infravermelho, além da espectrometria de massas dos
derivados tiazolidínicos obtidos se encontram nas publicações em anexo (ANEXO 1 e 2).

51
TABELA 1. Energia de ligação das novas ATZDs e rosiglitazona (RG) frente ao receptor PPARγ.
COMPOSTOS
CHS
N
O
OCH2 CH3
R
EBind
Kcal/mol
EBind(H)
Kcal/mol
EBind(OH)
Kcal/mol
GQ2 (4-Cl)
- 8,36
-7,01
-7,32
GQ3 (4-OH) - 8,39
-6,96 -7,24
GQ4 (2-Cl) - 8,61
-7,34 -7,71
GQ5 (4-CH3O) - 8,36
-6,68 -6,92
GQ6 (2,4- CH3O) - 8,22
-6,63 -6,97
GQ7 (3-Cl) - 8,61
-7,29
-7,64
GQ8 (4- CH3) - 8,39
-6,96
-7,24
GQ9 (2-Br) - 8,73
-7,3 -7,83
GQ10 (4- N(CH3) 2 ) - 8,85
-7,05 -7,44
GQ12 (4-C6H5CH2O) - 9,58 -8,45 -8,73
GQ15 (4-F) -8,22
-7,05
-7,45
GQ19 (4-NO2) -10,45
-8,16 -8,43
RG - 10,22
EBind, energia de ligação para a molécula não hidrolisada, EBind(H), energia de ligação para a molécula hidrolisada (H-N), EBind(OH), energia de ligação para a molécula hidrolisada (N-OH)

52
A
B
FIGURA 3. Interação entre tiazolidinadinas e o domínio LBD do receptor PPARγ A) PPARγ/rosiglitazona; B) PPARγ/GQ2
De acordo com resultados encontrados em estudos preliminares (dados dos ANEXOS 1 e
2), selecionamos o composto 5-(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-diona

53
(GQ2), para verificar o efeito deste, na sensibilidade e sinalização da insulina em camundongos
com obesidade induzida por dieta hiperlipídica. Nesse sentido estudamos quatro grupos de
animais: animais controles (tratados com dieta padrão – C), animais que receberam dieta
hiperlipídica (DH), animais que receberam DH e GQ2, e finalmente animais que receberam DH
mais RG, que é uma glitazona de uso rotineiro e que foi utilizada como parâmetro de
comparação à GQ2. As características químicas deste composto se encontram resumidas no
ANEXO 2.
4.2. Efeito de GQ2 no peso corporal, ingestão alimentar, massa adiposa peri-epididimal e
volume de água em camundongos com obesidade induzida por dieta hiperlipídica
A observação de que o tratamento de roedores e humanos com TZDs (rosiglitazona e
pioglitazona) leva a um ganho de peso corporal devido em parte a uma retenção de fluidos
(ZHANG et al., 2005), levou-nos a avaliar a implicação deste fenômeno em camundongos
“swiss” obesos submetidos ao tratamento com o novo agonista de PPARγ (GQ2), por um
período de 14 dias. Grupos controles: C e DH receberam o veículo (solução salina contendo
0,25% de tween – 0,1 ml/kg/dia) e RG (10mg/kg/dia) por gavagem durante o mesmo período.
Nas FIGURAS 4 A, B, C, D e E estão apresentados os dados referentes ao peso
corporal, ingestão alimentar, massa adiposa peri-epididimal e quantidade de água dos animais
dos grupos controles e tratados com os agonistas de PPARγ (GQ2 e RG).
A média do ganho de peso corporal dos animais tratados com GQ2 foi
significativamente menor (p< 0,05) (0,7 ± 0,35g - n = 21) em comparação aos animais dos
grupos controles: C (1,3 ± 0,45 – n = 19), DH (1,5 ± 0,31g – n = 22) e tratados com RG (2,9 ±
0,46g - n = 12). RG, além de maior ganho de peso corporal apresentou também uma progressão
no ganho de peso diferente dos demais grupos (FIGURA 4 A e B). No entanto, quando nesse
mesmo período foi avaliada a quantidade de ração ingerida, não foi detectada diferença
significativa (p<0,05) no consumo de ração de 24 horas entre os grupos estudados (C = 7,7 ±
0,38g – n = 5; DH = 8 ± 0,4g – n = 6; RG = 8,5 ± 0,3g – n = 6; GQ2 = 7,6 ± 0,35g – n = 7),
(FIGURA 4 C). Por outro lado, houve uma redução significativa (p<0,05) da massa adiposa peri-
edididimal, dos animais tratados com GQ2 (32%) e RG (37%) em comparação aos animais do
grupo DH (C = 1,77 ± 0,37; DH = 5,9 ± 0,28; RG = 3,7 ± 0,13; GQ2 = 4,0 ± 0,16g/100g de peso
corporal). Como esperado a massa adiposa peri-epididimal do grupo de animais submetidos a
dieta padrão, purina, (C) foi estatisticamente menor (p<0,05) do que os animais submetidos a

54
dieta hiperlipídica tratados ou não com agonistas de PPARγ, (FIGURA 4 D). Embora não tenha
havido diferença significativa (p<0,05) na massa adiposa entre GQ2 e RG, observamos que a
quantidade de água medida através do peso seco da carcaça foi significativamente menor
(p<0,05) no grupo que recebeu GQ2 em comparação ao grupo tratado com RG e similar aos
grupos controles (C = 14,1 ± 0,6g – n = 5; DH = 15 ± 0,45g – n= 6; RG = 16,5 ± 0,6g – n= 6;
GQ2 = 14,7 ± 0,3g - n - 6), (FIGURA 4 E).
4.3. Parâmetros Bioquímicos
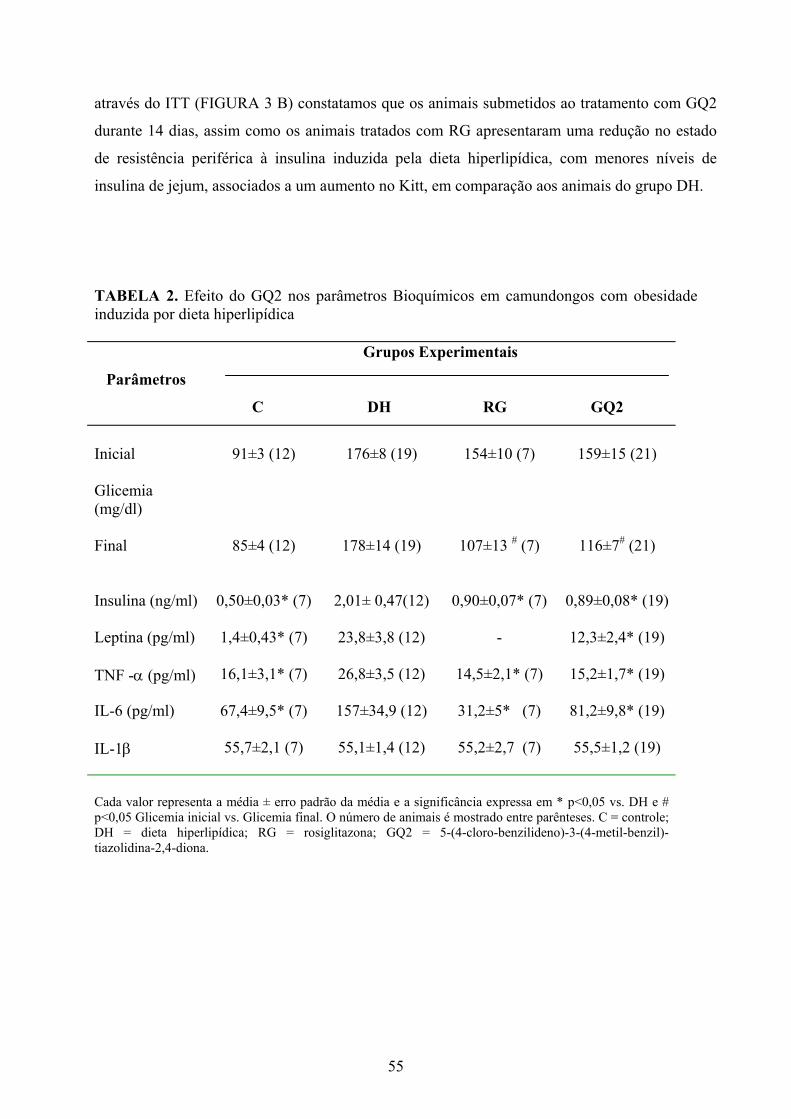
A TABELA 2 resume os dados basais dos níveis de glicose plasmática, insulinemia,
leptina, TNF-α, IL-6 e IL-1β obtidos após 10 dias de tratamento com agonistas de PPARγ e
grupos controles.
Os camundongos submetidos ao tratamento com RG e GQ2 apresentaram uma
redução significativa (p<0,05) da glicemia de jejum de 30 e 27 % respectivamente (TABELA 2,
FIGURA 5 A), e a insulina sérica, dosada no período de jejum, também se revelou diminuída nos
animais tratados com RG e GQ2 em comparação ao grupo DH (C = 0,5 ± 0,03; DH= 2,01 ±
0,47; RG = 0,90 ± 0,07 GQ2 = 0,89 ± 0,08 ng/ml - p< 0,05). Da mesma maneira, observamos
uma redução significativa nos níveis de leptina (C = 1,4 ± 0,43; DH = 23,8 ± 3,8; GQ2 = 12,3 ±
2,4 ng/ml - p<0,05), TNF-α (C = 16,1 ± 3,1; DH = 26,8 ± 3,5; RG = 14,5 ± 2,1; GQ2 = 15,2 ±
1,7 pg/ml - p<0,05 ) e IL-6 (C = 67,4 ± 9,5; RG = 31 ± 5 DH = 156,9 ± 34; GQ2 = 81,2 ± 9,8
pg/ml - p<0,05) nos animais tratados com RG e GQ2 em comparação ao grupo DH. Por outro
lado, os níveis de IL-1β não diferiram entre os grupos experimentais (C = 55,7 ± 2,1; DH = 55,1
± 1,4; RG = 55,5 ± 2,7; GQ2 = 55,5 ± 1,2 pg/ml - p<0,05), (TABELA 2).
4.4. Teste de tolerância à insulina (ITT)
Após a administração de 1,5U/kg de insulina, foi verificado um decréscimo da glicemia
em todos os animais do grupo controle (C) e tratados com RG e GQ2. A partir das glicemias
obtidas durante o ITT (30 minutos), foi calculado a velocidade constante do decaimento da
glicose (Kitt) para cada animal dos diferentes grupos experimentais (FIGURA 3 B). A dieta
hiperlipídica reduziu a velocidade de decaimento de glicose (C = 4,8 ± 0,28; DH = 3,14 ± 0,31) e
esta redução foi completamente revertida pelo tratamento com GQ2 (RG = 5,8 ± 0,47; GQ2 =
4,75 ± 0,2). Considerando uma redução na insulinemia (TABELA 2) e a ação da insulina medida
55
através do ITT (FIGURA 3 B) constatamos que os animais submetidos ao tratamento com GQ2
durante 14 dias, assim como os animais tratados com RG apresentaram uma redução no estado
de resistência periférica à insulina induzida pela dieta hiperlipídica, com menores níveis de
insulina de jejum, associados a um aumento no Kitt, em comparação aos animais do grupo DH.
TABELA 2. Efeito do GQ2 nos parâmetros Bioquímicos em camundongos com obesidade induzida por dieta hiperlipídica
Parâmetros
Grupos Experimentais
C DH RG GQ2
Inicial
Glicemia (mg/dl) Final
91±3 (12)
85±4 (12)
176±8 (19)
178±14 (19)
154±10 (7)
107±13 # (7)
159±15 (21)
116±7# (21)
Insulina (ng/ml) 0,50±0,03* (7) 2,01± 0,47(12)
0,90±0,07* (7) 0,89±0,08* (19)
Leptina (pg/ml) 1,4±0,43* (7)
23,8±3,8 (12)
- 12,3±2,4* (19)
TNF -α (pg/ml) 16,1±3,1* (7) 26,8±3,5 (12) 14,5±2,1* (7) 15,2±1,7* (19)
IL-6 (pg/ml) 67,4±9,5* (7) 157±34,9 (12) 31,2±5* (7) 81,2±9,8* (19) IL-1β
55,7±2,1 (7)
55,1±1,4 (12)
55,2±2,7 (7)
55,5±1,2 (19)
Cada valor representa a média ± erro padrão da média e a significância expressa em * p<0,05 vs. DH e # p<0,05 Glicemia inicial vs. Glicemia final. O número de animais é mostrado entre parênteses. C = controle; DH = dieta hiperlipídica; RG = rosiglitazona; GQ2 = 5-(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-diona.

56
16 16
16
16 16
16
3,00
10,0
8,0
6,0
4,0
2,0
18,0
6,05,04,03,02,0
1,0
*
2,00
15,0
1,00
12,09,06,03,0
0,0
0,0
0
0,0
DH
DH
DH
DH
RG
RG
RG
RG
GQ2
GQ2
GQ2
GQ2
C
C
C
C
A. Ganho de peso corporal
C. Ingestão alimentar
E. Quantidade de água
B. Variação do peso corporal
D. Variação da gordura epididimal
Gan
ho d
epe
so c
orpo
ral (
g)In
gest
ão a
limen
tar
24 h
oras
(g)
Qua
ntid
ade
deág
ua (g
)
Varia
ção
de g
ordu
raep
idid
imal
g/1
00g
peso
FIGURA 4. -gamaA e B C D
EDH
Efeito do tratamento de camundongos obesos com TZDs (RG = roglitazona e GQ2 = novo agonista de PPAR ) sobre variação de peso corporal ( ), ingestão de alimento em 24 hs ( ), variação da gordura peri-epididimal ( ) e quantidade de água medida através do peso seco da carcaça ( ). Os valores são expressos como média ± erro padrão da média e a significância expressa como *p<0,05
GQ2, RG e controle (C) ou GQ2 RG. vs. vs
16 1616 16
6,0
4,0
2,0
0,0DH RG GQ2CInício Final
A. Glicemia basal B. ITT (Kitt)
Nív
eis
de g
licos
e(m
g/dl
)
Kitt
(% m
in)
507090
110130150170190
~30%
FIGURA 5. -gamaA B
Efeito do tratamento de camundongos obesos com TZDs (RG = roglitazona e GQ2 = novo agonista de PPAR ) sobre a glicemia de jejum ( ) e ação da insulina medida através de ITT ( ). Camundongos controle (C) verde (n=12), DH vermelho (n=19), azu para os camundongos tratados com RG (n=7 para animais tratados com GQ2 (n=21). *p<0,05 DH .
l) e laranja Gq2, RG e controle (C)vs
Características Metabólicas
0
1
2
3
4
2 4 6 8 10 12 14
Tempo de tratamento (dias)
Var
iaçã
ode
peso
corp
óreo
(g)
CDHRGGQ2

57
4.5. Efeito de GQ2 nas etapas iniciais e intermediárias da ação insulínica em tecido adiposo
de camundongos com resistência à insulina induzida por dieta hiperlipidica
Para avaliar o efeito de GQ2 sobre as etapas iniciais e intermediárias da via de
sinalização da insulina, camundongos “swiss” foram submetidos a uma dieta hiperlipidica (DH)
por aproximadamente seis semanas, e em seguida foram tratados com 20mg/kg/dia de GQ2
através de gavagem durante 14 dias. Os resultados encontrados foram sempre comparados aos
camundongos do grupo DH, animais com obesidade induzida por dieta hiperlipídica, tratados
somente com veiculo (solução salina + 0,25% de tween – 0,1ml/kg), durante o mesmo período.
Usamos como substância padrão em nossos ensaios a rosiglitazona (RG), substância usada na
prática clínica para o tratamento de pacientes com diabetes mellitus tipo 2. As diferenças entre os
grupos foram consideradas significativas para p<0,05 (DH vs grupos tratados com GQ2, RG e
controle C).
Quando avaliamos a expressão do receptor de insulina em tecido adiposo peri-epididimal,
houve uma redução significativa do nível protéico deste receptor em animais que receberam DH.
O tratamento com agonistas de PPARγ reverteu os níveis teciduais deste receptor, como
determinado pelo immunobloting com anticorpo contra a porção COOH-terminal do receptor de
insulina. A FIGURA 6 A, mostra que o nível tecidual do receptor de insulina, avaliado por
densitometria óptica de 8 experimentos foi de 100 ± 5% para o grupo C, 50 ± 7% no grupo DH,
150 ± 10% RG e de 120 ± 6% para o grupo GQ2 .
Quando estas amostras foram submetidas a immunoblotting com anticorpo
antifosfotirosina (PY), observou-se que houve uma redução do grau de fosforilação, após
estimulo agudo com insulina, nos animais que receberam DH em comparação aos animais
tratados com RG e GQ2. O aumento no grau de fosforilação do receptor de insulina avaliado por
densitometria óptica de 8 experimentos, foi de aproximadamente de 30% para as TZDs (RG e
GQ2) comparado ao grupo que recebeu DH. (C= 100 ± 3; DH = 90 ± 5; RG = 130 ± 2; GQ2 =
120 ± 3,5%), FIGURA 6 B.
Utilizando anticorpos específicos anti-IRS-1, observamos que não houve alteração da
expressão desta proteína em tecido adiposo de camundongos que receberam DH em relação aos
grupos tratados com agonistas de PPARγ (C = 100 ± 4; DH = 96 ± 6; RG = 97 ± 5; GQ2 = 98 ±
3%), FIGURA 6 C. Avaliando o grau de fosforilação deste substrato com anticorpo anti-
fosfotirosina (PY), observamos que os animais que receberam DH apresentaram redução
significativa do grau de fosforilação do IRS-1, induzido por insulina, e esta redução foi
completamente revertida pelo tratamento com GQ2, mas não com RG (C = 100 ± 2; DH = 50 ±

58
5; RG = 50 ± 6; GQ2 = 100 ± 1,5%). No estado basal, os resultados foram similares para os
grupos controles e tratados com RG e GQ2 (FIGURA 6 D). Os dados foram obtidos de 10
experimentos, avaliados por densitometria ótica.
Tendo em vista a importância da associação das proteínas IRSs com a enzima
fosfatidilinositol 3-quinase (PI3K) no transporte de glicose e síntese de glicogênio, verificamos
esta associação, através da incubação das membranas cujas amostras foram previamente
imunoprecipitadas com anticorpo anti-IRS-1 ou anti-IRS-2, e em seguida submetidas à
immunobloting com anticorpo específico contra a subunidade reguladora de 85 kDa da PI3K.
Como mostrado na FIGURA 6 E, no estado basal o grau de associação IRS-1/PI3K foi reduzido
nos animais que receberam DH, e o tratamento com GQ e RG reverteu esta redução. Após
estímulo agudo com insulina, os animais que receberam DH apresentaram uma redução de
aproximadamente 60% da associação deste substrato à PI3K, e esta redução foi completamente
revertida pelo tratamento com GQ2 e RG (C = 100 ± 5; DH = 40 ± 3; RG = 97 ± 2; GQ2 = 95 ±
4%).
O nível protéico do IRS-2 em tecido adiposo, determinado por immunobloting com
anticorpo específico para este substrato foi similar entre os grupos estudados. A densitometria
ótica de 10 experimentos demonstrou que a quantidade de IRS-2 foi de (C = 100 ± 4,5; DH =
103 ± 5; RG = 105 ± 10; GQ2 = 107 ± 6%), FIGURA 6 F. Diferente dos resultados da
fosforilação do IRS-1, quando amostras de tecido adiposo peri-epididimal foram
imunoprecipitadas com anticorpo anti-IRS-2, e incubadas com anticorpo antifosfotirosina (PY),
observamos que após estímulo agudo com insulina, não houve diferença significativa do grau de
fosforilação em tirosina deste substrato entre os animais que receberam DH e os animais tratados
com GQ2, mas nos animais tratados RG houve uma redução na fosforilação deste substrato (C =
100 ± 2; DH = 108 ± 3; RG = 35 ± 6; GQ2 = 110 ± 10%), FIGURA 6 G. No entanto, a
associação do IRS-2 à PI3K após estimulo agudo com insulina, foi aproximadamente 40% menor
nos animais que receberam DH, e esta redução foi revertida pelo tratamento com GQ2 e RG (C =
100 ± 3; DH = 60 ± 2; RG = 93 ± 4; GQ2 = 91 ± 2%), FIGURA 4 H.
Investigamos também o efeito de GQ2 na expressão e fosforilação da proteína
serina/treonina quinase, Akt. Extrato total de tecido adiposo peri-epididimal foi submetido a
immunobloting com anticorpo anti-Akt e/ou pAkt (Akt Ser473). Como mostra a FIGURA 7 A,
não houve diferença significativa no nível tecidual da Akt avaliado por densitometria óptica de 6
experimentos entre os grupos experimentais (C = 100 ± 8; DH = 94 ± 12; RG = 98 ± 7; GQ2 =
105 ± 10%). Porém, após estímulo agudo com insulina, animais que receberam DH apresentaram
uma redução significativa do grau de fosforilação da Akt, e esta redução foi revertida e até

59
elevou-se o grau de fosforilação pelo tratamento com GQ2 e RG (C = 100 ± 7; DH = 47± 2; RG
= 130 ± 4; GQ2 = 132 ± 1%), FIRURA 7 B.
Uma vez que diversas serinas quinases sensíveis à insulina, principalmente a família das
MAP –quinase, exercem importante papel na via de controle mitogênico, avaliamos a
fosforilação de uma subfamília da MAP–quinase, as proteínas ERKs (1/2). Quando amostras de
tecido adiposo peri-epididimal foram submetidas ao immunobloting com anticorpo anti-pERK
(1/2), observamos por densitometria ótica de 6 experimentos, que animais que receberam DH
apresentaram redução significativa da fosforilação das ERKs após estimulo com insulina, e esta
redução foi completamente revertida pelo tratamento com GQ2 e RG (C = 100 ± 5; DH = 52±
1,5; RG = 110 ± 4; GQ2 = 115 ± 4,5%) FIGURA 7 C.
Como já citado, os efeitos terapêuticos das TZDs resultam da alta afinidade de união
das TZDs ao receptor nuclear PPARγ, presente em altas concentrações neste tecido. Para avaliar
o efeito de GQ2 sob a expressão deste fator nuclear, extrato total de tecido adiposo foi submetido
a immunobloting com anticorpo anti-PPARγ. Observamos por densitometria ótica de 6
experimentos, que os animais que receberam DH apresentaram um aumento de 50% da
expressão deste fator em comparação aos animais controle (C), o tratamento com GQ2 e RG
elevou os níveis protéicos do PPARγ em mais 50% (C = 100 ± 3; DH = 150± 7; RG = 210 ± 10;
GQ2 = 200 ± 5%), FIGURA 7 D.
Quando avaliamos o grau de fosforilação do IRS-1 em resíduos de serina 307 (IRS-
1SER307) em tecido adiposo, observamos que a DH induz aumento da fosforilação do IRS-1SER307, e
este aumento foi revertido pelo tratamento com GQ2, mas não com RG (FIGURA 8 A1).
A etapa seguinte constitui-se da avaliação da atividade das proteínas serinas quinases,
JNK e IKK, e dos níveis teciduais do TNF-α e iNOS em tecido adiposo de animais que
receberam DH. Como esperado, nos animais que receberam DH observou-se aumento da
atividade da JNK e IKK. Essa maior ativação da JNK e IKK, foi acompanhada pela alta
expressão de TNF-α e iNOS observado nesses animais, em relação ao grupo controle (C). O
tratamento com GQ2, além de reduzir a fosforilação do IRS-1SER307reduz também a ativação da
JNK e IKK, em paralelo a diminuição da expressão de TNF-α e iNOS em tecido adiposo
(FIGURA 8 A2 a A5). A atividade do fator de transcrição NF-kB foi avaliado em extrato nuclear
de amostras de gordura peri-epididimal através de teste de ELISA (subunidade p50 do NF-kB).
Os animais que receberam DH apresentaram um aumento na atividade deste fator nuclear e o
tratamento com GQ2 e RG reverteu esta alteração (FIGURA 8 B).

60
DH RG GQ2
IP:IR / IB:pY
CInsulina
Insulina
Insulina
+ + + +- - - -
150
90
60
120
30
0
B.
% U
nida
des
arbi
trária
s
100
80
20
40
60
0
DH RG GQ2
IP:IRS-1 / IB:pY
C+ + + +- - - -
1616D.
% U
nida
des
arbi
trária
s
DH RG GQ2
IP:IRS-2 / IB:PI3K
C+ + + ++ + + +- - - -- - - -
150
90
60
120
30
0
H.
% U
nida
des
arbi
trária
s
150
100
50
0DH RG GQ2
IP:IRS-2 / IB:IRS-2
C
F.
% U
nida
des
arbi
trária
s
1616
200
150
50
100
0DH RG GQ2C
IP:IR / IB:IR
Insulina
Insulina
A.%
Uni
dade
s ar
bitrá
rias
1616100
80
20
40
60
0DH RG GQ2C
IP:IRS-1 / IB:IRS-1
C.
% U
nida
des
arbi
trária
s
120100
4020
6080
0
DH RG GQ2
IP:IRS-2 / IB:pY
C
1616G.
% U
nida
des
arbi
trária
s
+ + + +- - - -
100
80
20
40
60
0
DH RG GQ2
IP:IRS-1 / IB:PI3K
C
1616E.
% U
nida
des
arbi
trária
s
+ + + +- - - -
FIGURA 6.A B
C D E
F - G H
Efeito do tratamento de camundongos obesos com GQ2 sobre os níveis protéicos e grau de fosforilação de IR, IRS-1 e IRS-2 e a associação dos IRSs com PI3K em tecido adiposo. - Avaliação do nível protéico do IR. Avaliação do grau de fosforilação do IR, antes (-) e após (+) estímulo agudo com insulina. - Avaliação do nível protéico do IRS-1. - Avaliação do grau de fosforilação do IRS-1, antes (-) e após (+) estímulo agudo com insulina. Avaliação da associação de IRS-1/PI3K, antes (-) e após (+) estímulo agudo com insulina. Avaliação do nível protéico do IRS-2. - Avaliação do grau de fosforilação do IRS-2, antes (-) e após (+) estímulo agudo com insulina. - Avaliação da associação de IRS-2/PI3K, antes (-) e após (+) estímulo agudo com insulina. Amostras de tecido adiposo de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2) foram previamente imunoprecipitadas (IP) com anticorpos anti-IR, IRS-1 e IRS-2, e submetidas a (IB)com anticorpos anti- IR, IRS-1, IRS-2, anti-fosfotirosina (PY) e anti-PI3K. Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 DH GQ2, RG e controle (C).
immunoblotting vs.
Adiposo

61
150
90
60
120
30
0DH
DH
DH
RGRG
RG
GQ2GQ2
GQ2
CC
C
120
120
100
100
40
40
20
20
60
60
80
80
0
0
1616
+ + + +- - - -
+ + + +- - - -
IB:Akt IB:pAkt (SER 473)
Insulina
Insulina
IB:pERK (½)
A. B.
C.
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s Adiposo
FIGURA 7.A B
C D
Efeito do tratamento de camundongos obesos com GQ2 sobre os níveis protéicos da Akt e grau de fosforilação Akt e ERK (1/2) em tecido adiposo. - Avaliação do nível protéico da Akt. Avaliação do grau de fosforilação em serina (ser 473) da Akt antes (-) e após (+) estímulo agudo com insulina. - Avaliação do grau de fosforilação das ERKs (1/2), antes (-) e após (+) estímulo agudo com insulina. - Avaliação da ativação de PPAR , após estimado com insulina. Extrato total de tecido adiposo de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a (IB) com anticorpos anti-Akt, anti-pAkt e anti-pERK (1/2). Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 DH s. GQ2, RG e controle (C).
immunoblotting v
A4 - IB:iNOS
A3 - IB:TNF-
A2 - IB:P-JNK(1/2)
A5 - IB:I B-
A1 - IB:IRS-1SER307
DH
DH
RG
RG
GQ2
GQ2
C
C
180
12090
150
60300
A.
DH RG GQ2C
250
100
50
150
200
0
1616D.
% U
nida
des
arbi
trária
s
B.
IB:PPAR
Uni
dade
s re
lativ
as
de lu
z (p
50)
FIGURA 8.A1 A2 A3
A4 A5
B .
Efeito do tratamento de camundongos obesos com GQ2 sobre a ativação de proteínas envolvidas na via inflamatória em tecido adiposo. - Avaliação da fosforilação de IRS-1 em serina 307. - Avaliação da ativação da quinase pJNK. - Avaliação da expressão de TNF- . - Avaliação da expressão da enzima iNOS. . - Avaliação da degradação da proteína quinase IkB- . Extrato total de tecido adiposo de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a (IB)com anticorpos anti-IRS-1 , anti-pJNK(1/2), anti-TNF- , anti- iNOS e anti- IkB- . = Atividade do NF-kB em extrato nuclear (p50)
immunoblotting ser307
específicas foram analisadas por meio de densitometria e a significância expressa como *p<0,05 DH . GG2, RG e controle (C).vsAs bandas

62
4.6. Efeito de GQ2 nas etapas iniciais e intermediárias da ação insulínica no músculo de
camundongos com resistência à insulina induzida por dieta hiperlipidica
Assim como no tecido adiposo, o efeito de GQ2 foi avaliado sobre as etapas iniciais da
ação insulínica, que levam a ativação da Akt, em músculo de camundongos com obesidade
induzida por dieta hiperlipídica. Amostras de músculo gastrocnêmio foram rapidamente
homogeneizadas e a seguir empregadas em métodos de imunoprecipitação e imunobloting.
Diferente do tecido adiposo, não houve variação significativa no nível protéico do receptor de
insulina nos diferentes grupos estudados (C = 100 ± 5; DH = 95 ± 7; RG = 105 ± 8; GQ2 = 108 ±
10%), FIGURA 9 A. Não observamos também, diferença significativa do grau de fosforilação do
receptor entre os grupos no estado basal (C = 100± 2,5; DH = 105±3,5; RG = 107 ± 5; GQ2 =
100 ± 1,5% - p<0,05) e após estímulo com insulina (C = 100 ± 5; DH = 97± 3,5; RG = 108 ± 2,5;
GQ2 = 110 ± 7%), FIGURA 9 B.
Os camundongos dos quatro grupos estudados não diferiram entre si quanto ao nível
tecidual do IRS-1 no músculo (C = 100 ± 1; DH = 103± 3; RG = 100 ± 2; GQ2 = 105 ± 3%),
FIGURA 9 C. Por outro lado, os animais que receberam DH apresentaram redução significativa
do grau de fosforilação em tirosina do substrato, induzida por insulina, e esta redução foi
completamente revertida pelo tratamento com GQ2 e RG (C = 100 ± 2,5; DH = 50 ± 5; RG =
103 ± 1,5; GQ2 = 100 ± 3%), sem alteração da .fosforilação deste substrato no estado basal,
entre os quatro grupos (FIGURA 9 D).
Quando avaliamos a associação IRS-1/PI3K no músculo, os animais que receberam DH
apresentaram redução significativa da associação desse substrato a PI3K, induzida por insulina, e
esta redução foi revertida completamente pelo tratamento com GQ2 e RG (C = 100 ± 2,5; DH =
50 ± 7; RG = 98 ± 1,5; GQ2 = 102 ± 2,5 %), FIGURA 9 E.
Quando amostras de tecido muscular foram submetidas a immunobloting com anticorpo
anti-IRS-2, não houve alteração no nível tecidual desta proteína entre os grupos estudados (C =
100 ± 5; DH = 108 ± 10; RG = 98 ± 7; GQ2 = 95 ± 3%), FIGURA 9 F. O grau de fosforilação do
IRS-2 nos grupos tratados com RG e GQ2 não diferem dos grupos controles no estado basal (C
= 30 ± 5; DH = 35 ± 3,5; RG = 45 ± 7; GQ2 = 46 ± 10%), mas após estímulo agudo com
insulina, os animais que receberam DH apresentaram redução significativa do grau de
fosforilação em tirosina desse substrato, e esta redução foi completamente revertida pelo
tratamento com GQ2 e RG (C = 100 ± 10; DH = 48 ± 7; RG = 100 ± 5; GQ2 = 105 ± 3,5%),
FIGURA 9 G.

63
A associação do IRS-2 à PI3K no tecido muscular após estimulo agudo com insulina,
encontra-se reduzida nos animais que receberam DH, e esta redução foi completamente revertida
pelo tratamento com GQ2 e RG (C = 100 ± 5,5; DH = 40 ± 7; RG = 98 ± 1,5; GQ2 = 100 ±
2,5%), sem alteração no estado basal, FIGURA 9 H.
Semelhante ao tecido adiposo, quando realizamos immunobloting para o estudo da
proteína Akt em tecido muscular, observamos que foi similar o nível tecidual dessa proteína
entre os grupos controles e tratados com RG e GQ2, embora tenha sido observado uma leve
redução nos animais que receberam DH em relação aos animais tratados com RG e GQ2 (C =
100 ± 7; DH = 95 ± 5; RG = 115 ± 8; GQ2 = 110 ± 2,5%), sem alteração também entre os grupos
no estado basal (C = 70 ± 2; DH = 60 ± 10; RG = 72 ± 10; GQ2 = 70 ± 8% ), FIGURAS 10 A e
B. Assim, como no tecido adiposo, após estímulo agudo com insulina, os animais que receberam
DH apresentaram redução significativa do grau de fosforilação em serina 473 da Akt, e esta
redução foi completamente revertida pelo tratamento com GQ2 e RG (C = 100 ± 2,5; DH = 80 ±
6; RG = 105 ± 3; GQ2 = 108 ± 2%), FIGURA 10 B.
Os resultados de immunobloting com anticorpo anti-pERK(1/2) em tecido muscular,
diferente do tecido adiposo, foram similares entre os grupos estudados após estímulo agudo com
insulina (C = 100 ± 4; DH = 90 ± 10; RG = 102 ± 4,5; GQ2 = 100 ± 5% - p<0,05), FIGURA 10
C.
Diferente do tecido adiposo, no músculo houve um discreto aumento do grau de
fosforilação do IRS-1SER307 nos animais que receberam DH. Nesses animais observou-se também
aumento da ativação da JNK, do complexo IKK e da enzima iNOS, e este aumento foi revertido
pelo tratamento com GQ2 e RG, (FIGURA 11 A). Assim como no tecido adiposo, a atividade do
fator de transcrição NF-kB foi avaliado em extrato nuclear de amostras de tecido muscular
através de teste de ELISA (subunidade p50 de NF-kB). Os animais que receberam DH
apresentaram um aumento na atividade deste fator nuclear em comparação aos animais tratados
com GQ2 e RG (FIGURA 11 B). A expressão de TNF-α não foi detectada em amostras de
tecido muscular com anticorpo anti-TNF-α por ensaios de immunobloting.
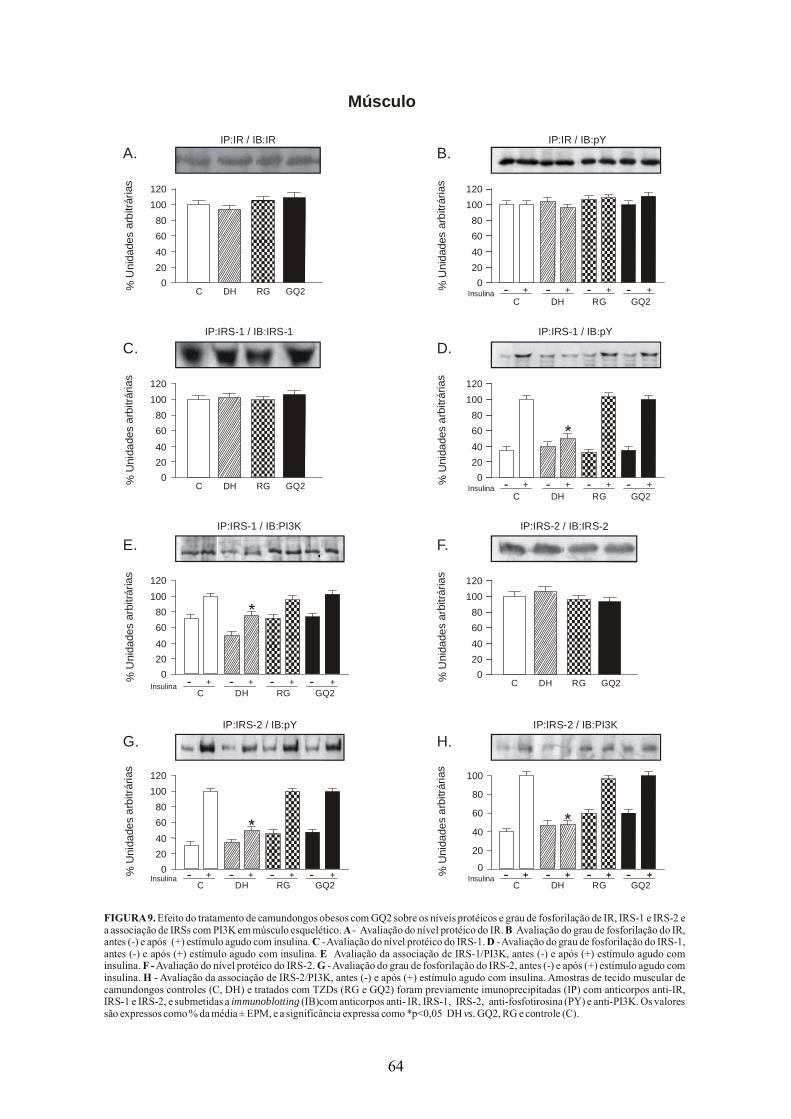
64
100
60
40
80
20
0
DH
DH
DH
DH
DH
DH
DH
DH
RG
RG
RG
RG
RG
RG
RG
RG
GQ2
GQ2
GQ2
GQ2
GQ2
GQ2
GQ2
GQ2
C
C
C
C
C
C
C
C
120
120
120
120
100
100
100
100
40
40
40
40
20
20
20
20
60
60
60
60
80
80
80
80
0
120100
4020
6080
0
120100
4020
6080
0
120100
4020
6080
0
0
0
0
16
16
16
16
+ + + ++ + + +
+ + + +- - - -
- - - -- - - -+ + + +
+ + + +
- - - -
- - - -
+ + + +- - - -
A. B.
C. D.
G. H.
E. F.
IP:IR / IB:IR IP:IR / IB:pY
IP:IRS-1 / IB:IRS-1 IP:IRS-1 / IB:pY
IP:IRS-2 / IB:pY
IP:IRS-1 / IB:PI3K IP:IRS-2 / IB:IRS-2
Insulina
Insulina
InsulinaInsulina
Insulina
IP:IRS-2 / IB:PI3K
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s %
Uni
dade
s ar
bitrá
rias
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s %
Uni
dade
s ar
bitrá
rias
% U
nida
des
arbi
trária
s
Músculo
FIGURA 9.A B
C D E
F - G H
Efeito do tratamento de camundongos obesos com GQ2 sobre os níveis protéicos e grau de fosforilação de IR, IRS-1 e IRS-2 e a associação de IRSs com PI3K em músculo esquelético. - Avaliação do nível protéico do IR. Avaliação do grau de fosforilação do IR, antes (-) e após (+) estímulo agudo com insulina. - Avaliação do nível protéico do IRS-1. - Avaliação do grau de fosforilação do IRS-1, antes (-) e após (+) estímulo agudo com insulina. Avaliação da associação de IRS-1/PI3K, antes (-) e após (+) estímulo agudo com insulina. Avaliação do nível protéico do IRS-2. - Avaliação do grau de fosforilação do IRS-2, antes (-) e após (+) estímulo agudo com insulina. - Avaliação da associação de IRS-2/PI3K, antes (-) e após (+) estímulo agudo com insulina. Amostras de tecido muscular de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2) foram previamente imunoprecipitadas (IP) com anticorpos anti-IR, IRS-1 e IRS-2, e submetidas a (IB)com anticorpos anti- IR, IRS-1, IRS-2, anti-fosfotirosina (PY) e anti-PI3K. Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 DH GQ2, RG e controle (C).
immunoblotting vs.

65
DHDH
DH
RGRG
RG
GQ2GQ2
GQ2
CC
C
120
120
120100
100
100
40
40
4020
20
20
60
60
6080
80
80
0
0
0
1616
+ + + +- - - -
+ + + +- - - -
IB:Akt IB:pAkt (SER 473)
Insulina
Insulina
IB:pERK
A. B.
C.
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
FIGURA 10.
A B
C
Efeito do tratamento de camundongos obesos com GQ2 sobre os níveis protéicos da Akt e grau de fosforilação das proteínas Akt e ERK (1/2) em músculo esquelético. - Avaliação do nível protéico da Akt. Avaliação do grau de fosforilação em serina (ser 473) da Akt antes (-) e após (+) estímulo agudo com insulina. - Avaliação do grau de fosforilação das ERKs (1/2), antes (-) e após (+) estímulo agudo com insulina. Extrato total de músculo grastrocnêmico de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a
(IB)com anticorpos anti-Akt, anti-pAkt e anti-pERK (1/2). Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 DH .GQ2, RG e controle (C).
immunoblotting
vs
A3 - IB:iNOS
A2 - IB:p-JNK
A4 - IB:I B-
A1 - IB:IRS-1SER307
DHDH
RGRG
GQ2GQ2
CC
150
90
60
120
30
0
A. B.
Uni
dade
s re
lativ
as
de lu
z (p
50)
Músculo
FIGURA 11.A B C
D D
Efeito do tratamento de camundongos obesos com GQ2 sobre a ativação de proteínas envolvidas na via inflamatória em tecido muscular. - Avaliação da fosforilação de IRS-1 em serina 307. Avaliação da ativação da quinase pJNK. - Avaliação da expressão de TNF-. . - Avaliação da expressão da enzima iNOS. . - Avaliação da degradação da proteína quinase IkB-. Extrato total de músculo de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a (IB)com anticorpos anti-IRS-1 , anti-pJNK(1/2), anti-TNF-, anti- iNOS e anti- IkB-. As bandas específicas foram analisadas por meio de densitometria e a significância expressa como *p<0,05 DH .
immunoblotting ser307
vs GQ2, RG e contole (C).

66
4.7. Efeito de GQ2 nas etapas iniciais e intermediárias da ação insulínica no fígado de
camundongos com resistência à insulina induzida por dieta hiperlipidica
Além do tecido adiposo e muscular, avaliamos também o efeito de GQ2 sobre as
etapas iniciais da ação insulínica, que levam a ativação da Akt, em tecido hepático de
camundongos com obesidade induzida por dieta hiperlipídica. A FIGURA 12 A mostra que não
houve alteração do nível protéico do receptor de insulina nos diferentes grupos experimentais (C
= 100 ± 7; DH = 103 ± 8; RG = 97 ± 5; GQ2 = 98 ± 3%). Os animais que receberam DH
apresentaram redução significativa do grau de fosforilação em tirosina do receptor de insulina,
após estímulo agudo com insulina no tecido hepático, e esta redução foi completamente revertida
pelo tratamento com GQ2 e RG (C = 100 ± 4; DH = 18 ± 2,5; RG = 102 ± 3,5; GQ2 = 93 ±
4,5%), FIGURA 12 B.
Quando amostras de tecido hepático foram submetidas a immunobloting com anticorpo
anti-IRS-1, não houve diferença significativa no nível protéico desta proteína entre os animais
dos grupos controles e tratados com as TZDs (C = 100 ± 7; DH = 110 ± 10; RG = 108 ± 9; GQ2
= 109 ± 8,5%), FIGURA 12 C. Por outro lado,os animais que receberam DH apresentaram
redução significativa do grau de fosforilação em tirosina do IRS-1, e esta redução foi
completamente revertida pelo tratamento com GQ2 e RG (C = 100 ± 2; DH = 50 ± 1,5; RG =
110 ± 7; GQ2 = 118 ± 6%), FIGURA 12 D, assim como, a associação deste substrato com a
enzima PI3K (C = 100 ± 4,5; DH = 20 ± 1,5; RG = 110± 7; GQ2 = 95 ± 3%), FIGURA 12 E..
Em relação ao IRS-2, houve redução do nível protéico desta proteína em tecido hepático
de camundongos que foram tratados DH, e esta redução foi completamente revertida pelo
tratamento com GQ2 e RG (C = 100 ± 7; DH = 80 ± 10; RG = 110 ± 7; GQ2 = 100 ± 5%),
FIGURA 12 F. Após estímulo agudo com insulina, os animais que receberam DH apresentaram
redução significativa do grau de fosforilação em tirosina deste substrato, induzido por insulina, e
esta redução foi completamente revertida pelo tratamento com GQ2 e RG (C = 100 ± 3,5; DH =
30 ± 5; RG = 180 ± 10; GQ2 = 120 ± 7,5%), FIGURA 12 G. Porém, não observamos diferença
significa entre os grupos com relação a associação deste substrato com PI3K, após estimulo
agudo com insulina (C = 100 ± 7; DH = 95 ± 4; RG = 93± 5; GQ2 = 108 ± 6%), FIGURA 12 H.
Quando realizamos immunobloting para estudo da fosforilação da Akt em serina 473, os
animais que receberam DH apresentaram redução de aproximadamente de 30% no estado basal.
Após estímulo agudo com insulina houve também redução da fosforilação da Akt nos animais

67
que receberam DH, e esta redução foi revertida pelo tratamento com GQ2 e RG (C = 100 ± 5;
DH = 103 ± 6,5; RG = 135± 4,5; GQ2 = 130 ± 6%), FIGURA 13 A.
A avaliação do grau de fosforilação das ERKs (1/2) no tecido hepático, foi semelhante ao
tecido muscular, os resultados dos grupos tratados com TZDs e DH foram similares antes (C =
95 ± 8; DH = 90 ± 6; RG = 97 ± 7,5; GQ2 = 98 ±6% - p<0,05), e após estímulo agudo com
insulina (C = 100 ± 10; DH = 97 ± 6; RG = 104 ± 7; GQ2 = 106 ± 8%), FIGURA 13 B.
No tecido hepático observamos maiores níveis de fosforilação do IRS-1SER307, em paralelo
com aumento da atividade do complexo IKK nos animais que receberam DH, e o tratamento com
GQ2 e RG reverte parcialmente este quadro de resistência à insulina. Quando avaliamos nesse
tecido a ativação da JNK, observamos um discreto mais significativo aumento, nos animais que
receberam DH, e o tratamento com GQ2 reverte a ativação da JNK (FIGURA 14 A). A atividade
do fator de transcrição NF-kB foi avaliado em extrato nuclear de amostras de tecido hepático
através de teste de ELISA (subunidade p50 de NF-kB). Os animais que receberam DH
apresentaram um aumento na atividade deste fator nuclear em comparação aos animais tratados
com GQ2 e RG (FIGURA 14 B). TNF-α e iNOS não foram detectadas em amostras de tecido
hepático com anticorpo anti-TNF-α e anti-inos respectivamente por ensaios de immunobloting.

68
100120
6040
80
200
DH
DH
DH
RG
RG
RG
GQ2
GQ2
GQ2
C
C
C
120
120
120
180
100
100
100
150
40
40
40
60
20
20
20
30
60
60
60
90
80
80
80
120
0
120100
4020
6080
0
100
40
20
60
80
0
0
0
0
16
16
16
16
150
90
60
120
30
0
DH
DH
DH
DH
DH
RG
RG
RG
RG
RG
GQ2
GQ2
GQ2
GQ2
GQ2
C
C
C
C
C+ + + ++ + + +
+ + + +- - - -
- - - -- - - -+ + + +
+ + + +
- - - -
- - - -
+ + + +- - - -
A. B.
C. D.
G. H.
E. F.
IP:IR / IB:IR IP:IR / IB:pY
IP:IRS-1 / IB:IRS-1 IP:IRS-1 / IB:pY
IP:IRS-2 / IB:pY
IP:IRS-2 / IB:IRS-2
Insulina
Insulina
InsulinaInsulina
Insulina
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s %
Uni
dade
s ar
bitrá
rias
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s %
Uni
dade
s ar
bitrá
rias
% U
nida
des
arbi
trária
s
IP:IRS-1 / IB:PI3K
IP:IRS-2 / IB:PI3K
FIGURA 12.A B
C D E
F - G H
Efeito do tratamento de camundongos obesos com GQ2 sobre os níveis protéicos e grau de fosforilação de IR, IRS-1 e IRS-2 e a associação de IRSs com PI3K em tecido hepático. - Avaliação do nível protéico do IR. Avaliação do grau de fosforilação do IR, antes (-) e após (+) estímulo agudo com insulina. - Avaliação do nível protéico do IRS-1. - Avaliação do grau de fosforilação do IRS-1, antes (-) e após (+) estímulo agudo com insulina. Avaliação da associação de IRS-1/PI3K, antes (-) e após (+) estímulo agudo com insulina. Avaliação do nível protéico do IRS-2. - Avaliação do grau de fosforilação do IRS-2, antes (-) e após (+) estímulo agudo com insulina. - Avaliação da associação de IRS-2/PI3K, antes (-) e após (+) estímulo agudo com insulina. Amostras de fígado de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2) foram previamente imunoprecipitadas (IP) com anticorpos anti-IR, IRS-1 e IRS-2, e submetidas a (IB)com anticorpos anti- IR, IRS-1, IRS-2, anti-fosfotirosina (PY) e anti-PI3K. Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 DH
immunoblotting . vs GQ2, RG e contole (C).
Fígado

69
100120
6040
80
200
150
60
30
90
120
0
DHDH RGRG GQ2GQ2 CC+ + + +- - - -+ + + +- - - -
A. B.IB:pAKT SER473 IB:pRK
InsulinaInsulina
% U
nida
des
arbi
trária
s
% U
nida
des
arbi
trária
s
A2 - p-JNK
A3 - I B-
A1 - IRS-1SER307
DH
DH
RG
RG
GQ2
GQ2
C
C
180150
6030
90120
0
B.A.
Uni
dade
s re
lativ
as
de lu
z (p
50)
Fígado
FIGURA 11.A B
C
Efeito do tratamento de camundongos obesos com GQ2 sobre o grau de fosforilação das proteínas Akt e ERK (1/2) em tecido hepático. - Avaliação do nível protéico da Akt. Avaliação do grau de fosforilação em serina (ser 473) da Akt antes (-) e após (+) estímulo agudo com insulina. - Avaliação do grau de fosforilação das ERKs (1/2), antes (-) e após (+) estímulo agudo com insulina. Extrato total de fígado de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a (IB)com anticorpos anti-pAkt e anti-pERK (1/2). Os valores são expressos como % da média ± EPM, e a significância expressa como *p<0,05 vs DH.
immunoblotting
FIGURA 12.A B C
D D
Efeito do tratamento de camundongos obesos com GQ2 sobre a ativação de proteínas envolvidas na via inflamatória em tecido hepático. - Avaliação da fosforilação de IRS-1 em serina 307. Avaliação da ativação da quinase pJNK. - Avaliação da expressão de TNF-. . - Avaliação da expressão da enzima iNOS. . - Avaliação da degradação da proteína quinase IkB-. Extrato total de fígado de camundongos controles (C, DH) e tratados com TZDs (RG e GQ2), foram submetidos a (IB)com anticorpos anti-IRS-1 , anti-pJNK(1/2), anti-TNF-, anti- iNOS e anti- IkB-. As bandas específicas foram analisadas por meio de densitometria e a significância expressa como *p<0,05 vs DH.
immunoblotting ser307

70
4.8. Efeito de GQ2 na toxicidade aguda e subcrônica de camundongos com obesidade
induzida por dieta hiperlipídica
Para avaliar a toxicidade aguda de GQ2, camundongos “swiss” normoglicemicos
receberam por gavagem doses crescentes de GQ2. Após 48 horas a mortalidade foi anotada.
Observamos que não houve letalidade dos camundongos tratados com GQ2, até a dose de
2000mg/kg, não sendo, portanto, necessário determinar a DL50 (dose letal que mata 50% dos
animais). Os dados apontam que essa substância apresenta uma baixa toxicidade, visto que doses
maiores de 1000mg/kg são consideradas de baixa toxicidade (DIETRICH, 1983). Quando
Avaliamos os níveis de ALT no soro de animais obesos e submetidos ao tratados com GQ2 por
14 dias esses, não foram diferentes dos animais do grupo DH (C = 22,3 ± 4; DH = 77,3 ± 7; RG
= 56 ± 8; GQ2 = 77 ± 9 - p<0,05).

71
DISCUSSÃO

72
5. DISCUSSÃO
A necessidade de novas substâncias terapêuticas mais eficazes, com baixa toxicidade e
maior especificidade, têm levado pesquisadores a intensificar os estudos para a descoberta de
novos fármacos ou melhoramento dos já existentes. O desenvolvimento da síntese orgânica
moderna tem proporcionado um aumento de substâncias sintéticas para uso medicinal, e têm sido
empregadas no controle de vários tipos de doenças.
Focalizando as tiazolidina-2,4-dionas, vários trabalhos utilizaram o conceito clássico de
bioisosterismo para obtenção de novos derivados das tiazolidinadionas (KOROLKOVAS, 1997).
Substâncias contendo este heterociclo apresentam diversas atividades biológicas, tais como
antifúngica (INAMORI et al., 1998), antibacteriana (MENEZES et al.,1992; SIM et al., 2002),
hipoglicemiante (MOMOSE et al., 2002; NEOGI et al., 2003) e antiinflamatória (BOSCHELLI
et al., 1992; DE LIMA et al., 1994), dentre outras.
A síntese de novos compostos pertencentes à classe das tiazolidinadionas (TZDs),
substâncias que melhoram a ação da insulina e controlam o diabetes mellitus tipo 2 constituem
um passo importante nessa linha. As TZDs são conhecidas como sensibilizadores de insulina, já
que sua ação farmacológica está dirigida para melhorar a ação da insulina mediante mecanismos
extrapancreáticos.
Na primeira etapa deste estudo sintetizamos uma série novas moléculas tiazolidinicas
com potencial atividade hipoglicemiante. Posteriormente, investigamos os efeitos metabólicos e
moleculares do composto 5-(4-cloro-benzilideno)-3-(4-metil-benzil)-tiazolidina-2,4-diona
(GQ2) em animais com obesidade induzida por dieta hiperlipidica.
É importante salientar, que camundongos “swiss” submetidos à dieta hiperlipidica (DH)
por seis semanas apresentam: 1) hiperglicemia de jejum com altos níveis de insulina e leptina, 2)
aumento da expressão de TNF-α no tecido adiposo com conseqüente aumento da massa adiposa
peri-epididimal, além de resistência à insulina inclusive a nível molecular em fígado, músculo
esquelético e tecido adiposo. Os nossos achados, estão em concordância com outros laboratórios
referindo que dieta hiperlipídica causa obesidade e resistência à insulina em roedores
(CHALKLEY et al., 2002; KIM et al., 2000; SINGH et al., 2003; STORLIEN et al., 1993).
De acordo com os estudos de modelagem molecular, o complexo formado entre o
composto GQ2 e o receptor nuclear PPARγ apresenta uma boa estabilidade (energia de ligação
negativa) quando comparada com a energia de ligação da rosiglitazona (RG).

73
O estudo de modelagem molecular tem sido aplicado ao planejamento racional de
fármacos de várias formas. Quando se conhece, por exemplo, a estrutura tridimensional do alvo
biológico, é possível compreender as interações importantes entre o ligante e o receptor através
de procedimentos de “Docking”. Por outro lado, quando se dispõe apenas da estrutura do ligante,
ou seja, quando não se conhece a estrutura tridimensional do alvo biológico, também pode se
utilizar à modelagem molecular na tentativa de obter parâmetros eletrônicos e estéricos que
funcionem como descritores para a elucidação das relações quantitativas (QSAR – Quantitative
Structure-Activity Relationship) ou qualitativas (SAR – Structure-Activity Relationship) entre
estrutura e atividade biológica (ANDREI e al., 2003).
No caso das TZDs, a estrutura tridimensional do alvo biológico já é conhecida, cuja
estrutura foi determinada por cristalografia de raios-X se encontra disponível no banco de dados
de proteínas sob o código 2PRG (“Protein Data Bank”- http://www.rcsb.org/pdb). Sendo assim,
ela foi usada como ponto de partida nos estudos de “Docking” para as nossas moléculas. Os
estudos de “Docking”, de uma maneira geral, são procedimentos de modelagem molecular que
tentam encontrar soluções ou encaixes estáveis para moléculas de ligantes nos sítios ativos destes
alvos biológicos (HUNTZ, 1999).
RG interage com o receptor PPARγ através da formação de ligações de hidrogênio entre
o átomo de oxigênio do anel tiazolidínico com os resíduos de aminoácidos histitina (His323) e
serina (ser289) do domínio LBD do receptor. Este tipo de ligação é importante na estabilidade e
ativação do receptor (IWATA et al., 2001).
Assim como RG, o composto GQ2 apresentou a formação de ligações de hidrogênio
entre o átomo de oxigênio do anel heterociclico e o resíduo de serina 289 do domínio LBD do
receptor, ligação importante na ativação e estabilidade deste receptor.
O efeito antidiabético das TZDs, está bem estabelecido. Embora, os tecidos alvos e o
mecanismo da atividade farmacológica dessas drogas não sejam completamente compreendidos.
As TZDs agem com alta afinidade de união e ativação dos receptores PPARγ (SPIEGELMAN,
1998), exercendo sua ação principal sobre a glicemia com ação secundária sobre o pérfil lipídico.
As pesquisas têm focado o efeito das TZDs principalmente no músculo esquelético e
tecido adiposo. O efeito das TZDs na gordura parece resultar da diferenciação de adipócitos
(TAKAMURA et al., 2001), redistribuição da gordura, em particular, gordura visceral para
tecido adiposo subcutâneo (MIYAZAKI et al., 2002; BAJAJ et al., 2004), redução da lipólise
(MIYAZAKI et al., 2001) e adipogênese (GURNELL et al., 2000).
No músculo esquelético, há aumento da sensibilidade à insulina (MIYAZAKI et al.,
2001) podendo ser um efeito indireto, ou seja, mediada pelo tecido adiposo, órgão com alta

74
expressão de PPARγ (MARTIN et al., 1996) ou por agir diretamente nos tecidos sensíveis à
insulina, mesmo que esses apresentem baixa expressão de PPARγ. A ativação do PPARγ pode
afetar a sensibilidade à insulina no músculo por regular genes envolvidos no metabolismo de
glicose. De fato, o mRNA do transportador de glicose, GLUT4, bem como o da proteína
associada c-Cbl, foram relatadas por serem induzidos por PPARγ (RIBON et al., 1998). Todavia,
outros estudos precisam ser realizados para identificar os PPREs nestes genes e demonstrar se
sua regulação é realmente mediada por PPARγ.
No fígado, embora não tenha sido investigado extensivamente como músculo e tecido
adiposo, o efeito das TZDs na sensibilidade à insulina nesse órgão é conseqüência da redução na
produção de glicose hepática, resultado da inibição das enzimas chave da gliconeogênese,
fosfoenolpiruvato carboxilase (PEPCK), glicose-6-fosfatase e piruvato carboxilase (WAY et al.,
2001). Além disso, o tratamento com TZDs reduz significativamente a liberação de ácidos
graxos livres (AGL) a partir do tecido adiposo (MIYAZAKI et al., 2001) e este é um sinal
inibitório para a via da gliconeogênese.
A ativação de PPARγ por agonistas parece também ter efeitos antiinflamatórios. Esses
efeitos ocorrem porque a atividade transcricional do PPARγ antagoniza a de outros fatores de
transcrição, como por exemplo, a do AP-1, STAT e NF-kB, responsáveis pelo controle da
expressão de genes pró-inflamatórios, tais como: 1) da gelatinase B, uma metaloproteínase, que
em macráfagos ativados, causa danos no tecido durante processos inflamatórios agudos e
crônicos, 2) a inibição da expressão do gene SR-A, que codifica uma proteína de superfície
celular associada a eventos de adesão celular e a entrada de macromoléculas incluindo LDL
oxidada e 3) da óxido nítrico sintase induzida (iNOS) (RICOTE et al., 1998).
Em concordância, o trabalho de Jiang e colaboradores mostraram que agonistas de
PPARγ (PGJ2, 15-deoxi-Δ12,14 e troglitazona) também inibem em monócito a síntese de outras
moléculas pro-inflamatórias, por diminuírem a expressão de citocinas como IL 1β, IL-6 e o TNF-
α (JIANG et al., 1998).
Na segunda etapa deste estudo, nós examinamos os efeitos metabólicos e moleculares do
tratamento de camundongos “swiss” obesos com um novo agonista de PPARγ (GQ2). Assim
como RG, GQ2, produz um modesto mais significativa redução da glicemia de jejum cerca de
27%, diminuindo o estado de resistência periférica à insulina induzida pela dieta hiperlipídica
com menores níveis de insulina e leptina de jejum associados a um aumento no Kitt em
comparação aos animais que receberam DH.

75
O teste de tolerância à insulina (ITT), é uma das maneiras de aferir a sensibilidade à
insulina experimentalmente, através da avaliação da curva de decaimento da glicemia após
infusão de insulina. Observamos que quanto maior é a sensibilidade à insulina, tanto maior é a
inclinação da curva de queda da glicemia em função do tempo, após infusão de sobrecarga de
insulina. A razão obtida a partir da inclinação desta curva define uma constante chamada Kitt
(BONORA, MOGHETTI et al. 1989). Animais que receberam DH apresentaram redução
significativa nos níveis de Kitt e esta redução foi completamente revertida pelo tratamento com
GQ2 e RG, refletindo um aumento na sensibilidade da insulina na periferia.
Nossos resultados mostraram que o composto GQ2 reduz os níveis de leptina em animais
obesos, em conformidade com outros estudos (DE VOS et al.,1996). A leptina produto de gene
ob é um fator de sinalização regulador do peso corporal e balanço energético. A expressão do
gene ob em roedores está aumentada na obesidade e é regulada pela alimentação e hormônios
tais como insulina e glicocorticoides. O tratamento de ratos com agonistas de PPARγ
(tiazolidinadionas) aumenta o consumo alimentar e massa adiposa enquanto reduz os níveis de
mRNA para o gene ob de maneira dose dependente. A ação inibitória das TZDs na expressão de
gene ob também foi observado in vitro (DE VOS et al.,1996).
Por outro lado, o ganho de peso corporal, dos animais tratados com GQ2 foi de
aproximadamente 4 vezes menor em relação aos animais tratados com RG e cerca de 2 vezes
menor em relação aos animais do grupo DH. No entanto, não observamos diferenças
significativa em relação ao consumo de alimento de 24 horas entre os animais controles e
tratados com RG e GQ2. Para investigar a razão do menor ganho de peso dos animais tratados
com essa nova tiazolidinadiona, analisamos a massa adiposa peri-epididimal dos animais tratados
com RG, GQ2 e animais controles (C e DH). Para nossa surpresa, não houve diferença
significativa entre os camundongos tratados com RG e GQ2, embora as TZDs tenham
apresentado uma redução de aproximadamente 33% em relação aos animais que receberam DH,
isso não explicaria totalmente o menor ganho de peso observado nos animais tratados com GQ2.
Nossos resultados demonstraram que o tratamento de camundongos ”swiss” obesos com
10mg/kg/dia de rosiglitazona durante 14 dias, induziu um ganho de peso corporal progressivo.
Esses dados estão em concordância com outros estudos. O rápido ganho de peso observado em
animais e humanos tratados com rosiglitazona e pioglitazona parece ser em parte conseqüência
da retenção de fluidos, e a formação de edemas (ZHANG et al., 2005). O mecanismo de retenção
de fluidos em pacientes tratados com TZDs não é completamente conhecido e pode envolver
vários fatores, incluindo redução na excreção urinária de sódio (BANDO et al., 1999), alteração
da permeabilidade endotelial (WALKER et al., 1998), aumento da atividade do sistema nervoso

76
simpático (YOSHIMOTO et al., 1997) ou alteração intesticial do transporte de íon
(HOSOKAWA et al., 1999). O mecanismo pelo qual as TZDs eleva a absorção de sódio e causa
edema permanece obscuro. Porém, alguns autores têm referido como sendo devido à presença de
alta quantidade de PPARγ predominantemente no ducto coletor renal (GUAN et al., 1997;
GUAN et al., 2005).
Com o intuito de verificar a associação de GQ2 com a retenção de fluidos, avaliamos a
quantidade de água medida através do peso seco da carcaça. Interessantemente, os animais
tratados com GQ2, apresentaram uma redução na quantidade de água em comparação ao grupo
tratado com RG, mas foi similar aos grupos controles (C e DH). Estes dados explicam em parte o
menor ganho de peso encontrado nos animais tratados com GQ2, todavia, outros experimentos
precisam ser realizados.
Estudos realizados em modelos de animais de obesidade e resistência à insulina, têm
demonstrado defeitos moleculares progressivos na maquinaria de sinalização da insulina em
músculo esquelético, tecido adiposo e fígado (SAAD et al, 1992; CARVALHO et al, 1996;
BJORNHOLM et al, 2002).
A sinalização intracelular da insulina começa com a sua ligação a um receptor específico
de membrana, uma proteína heterotetramérica com atividade tirosina quinase. A insulina ao se
ligar a seu receptor (IR) promove a ativação da função tirosina quinase da subunidade catalítica
β do receptor, que se autofosforila e ativa substratos intracelulares, dos quais o IRS-1 é o mais
estudado. Há seis anos, outro constituinte da banda pp185, chamado IRS-2 foi também
purificado e a seqüência cDNA foi determinada, tendo aproximadamente 45% de homologia
com o IRS-1. Parece que o IRS-2 atua sinergicamente com o IRS-1, tendo um papel importante
nos eventos que controlam o metabolismo da glicose (SUN et al., 1995).
Após a fosforilação do IRS-1 e/ou IRS-2, ocorre sua associação com a PI3K, ativando-a.
A ativação da PI3K aumenta os níveis teciduais de fosfatidilinositol 3-fosfato, que é um
intermediário essencial na ativação da proteína Akt/PKB. (SALTIEL & KAHN, 2001). A Akt é
uma serina/treonina quinase que modula diversas ações de crescimento e metabólicas da
insulina, incluindo transporte de glicose, síntese protéica e de glicogênio (UEKI, YAMAMOTO-
HONDA et al. 1998; VAN WEEREN, DE BRUYN et al. 1998; JIANG, ZHOU et al. 2003).
Vários estudos implicam a Akt como proteína importante para a translocação de GLUT4 para
superfície celular (BROZINICK, ROBERTS et al. 2003). Camundongos modificados que não
expressam a Akt2 estão associados a estados de resistência à insulina e diabetes. Este achado
reflete a importância da via IRS-1/PI3K/Akt2 para efetuação dos efeitos metabólicos da insulina
(CHO, MU et al. 2001).

77
Estudos recentes têm sugerido que vários fatores derivados da gordura corporal podem
interferir na sinalização da insulina. Uma grande variedade de citocinas pró-inflamatórias e
outros mediadores, como leptina, fator de necrose tumoral alfa (TNF-α), IL-6 e PAI-1 têm sido
identificados como marcadores inflamatórios que podem predizer o desenvolvimento do diabetes
mellitus tipo 2 e o ganho de peso em seres humanos (SCHMIDT & DUNCAN, 2001).
Dentre estes fatores, tem sido demonstrado que o TNF-α pode induzir resistência à
insulina através da fosforilação em resíduos de serina do IRS-1, provocando uma inibição da
sinalização da insulina em alguns modelos de obesidade e resistência à insulina
(HOTAMISLIGIL, 1999). Várias quinases serina/treonina são ativadas por estímulos
inflamatórios e contribuem para inibição do sinal da insulina, incluindo c-jun N-terminal quinase
1 (JNK1) (AGUIRRE, UCHIDA et al. 2000) e o complexo do IKK (GAO, HWANG et al.
2002). Estas quinases foram identificadas como capazes de fosforilar o IRS-1 em serina,
notadamente na posição 307 (IRS-1ser307). Esta fosforilação reduz de maneira importante a
capacidade de fosforilação de resíduos de tirosina após estímulo com insulina no receptor
(HOTAMISLIGIL et al., 1996).
JNK é um membro da família da MAPK (WESTON et al., 2002; DAVIS, 1999) e pode
ser ativada por TNF-α (HIROSUMI et al., 2002) e IL-1β (NIKULINA et al., 2003). Três
isorformas da JNK foram identificadas, JNK1, 2 e 3 (IP & DAVIS, 1998), das quais a JNK1 é a
mais envolvida na fisiopatologia da obesidade e resistência à insulina (HIROSUMI et al., 2002).
A ativação da JNK induz fosforilação do IRS-1ser307 e por sua vez, indiretamente aumenta o
número de resíduos de serina e treonina fosforilados no IRS-1, transformando-o em uma proteína
com ação inibitória sobre o sinal da insulina (BIRNBAUM, 2001).
O complexo IKK é composto de 2 quinases, IKK-α e IKK-β, as quais fosforilam IκB,
sendo a IKK-β mais potente do que a IKK-α. A IκB é uma proteína inibitória citoplasmática que
tem a função de seqüestrar o NF-κB, impedindo que ele migre para o núcleo da célula. O NF-κB
faz parte de uma família de fatores de transcrição celular e está envolvido na expressão de genes
que regulam a resposta inflamatória (BAUERLE & BALTIMORE, 1996). Quando as IKK são
estimuladas, elas fosforilam a IκB, promovendo sua degradação e permitindo a translocação do
NF-κB para o núcleo.
Alguns trabalhos sugerem que agentes anti-inflamatórios como a aspirina e o salicilato
sódico podem inibir especificamente a IKK-β gerando tanto uma menor resposta inflamatória
quanto uma melhora do sinal da insulina (pela menor fosforilação do IRS-1 em serina e
treonina).

78
A Óxido Nítrico Sintase induzível (iNOS) produz NO a partir da conversão de L-arginina
em L-citrulina, e é importante componente do sistema imune inato, tendo sua expressão tecidual
aumentada após estímulo com certas interleucinas. Inicialmente descrita em macrófagos, pode
ter sua expressão tecidual induzida em vários tecidos diferentes, inclusive em tecidos insulino-
sensíveis, como muscular, adiposo e hepático (KURREK, ZAPOL et al. 1995; RIBIERE,
JAUBERT et al. 1996; KAPUR, BEDARD et al. 1997). Em tecido muscular a indução da iNOS
parece estar associada ao desenvolvimento de resistência à insulina. O aumento da expressão da
iNOS em modelos experimentais de obesidade sugere que esta é outra via da resposta imune
inata que está ativada na obesidade, podendo ter participação fisiopatológica no desenvolvimento
da resistência à insulina, de forma semelhante às vias inflamatórias anteriormente descritas.
Analisando a via IRS-1/PI3K/Akt, entendemos que bloqueios na transmissão do sinal da
insulina nesta via como ocorre com IRS-1ser307 por proteínas quinases tais como JNK e IKK
podem estar relacionados à resistência às ações fisiológicas deste hormônio. E a reversão destes
bloqueios na sinalização pode resultar em melhora da sensibilidade à insulina nos tecidos
periféricos tais como músculo, fígado e tecido adiposo.
Estudos têm demonstrado que as TZDs exercem seus efeitos como sensibilizadores de
insulina parcialmente por potencializar a ativação da PI3K e Akt estimulada pela insulina
(HEVENER et al., 2000; ZHANG et al., 1994; SMITH et al., 2001). Contudo, não está claro se
os efeitos mediados pelas TZDs na ativação da PI3K/Akt são eventos precoces que contribuem
para a sensibilização da insulina ou são eventos tardios resultante da melhora da sensibilidade à
insulina in vivo.
Jiang e colaboradores observaram que o tratamento agudo (24 horas) de ratos Zucker
obesos (fa/fa) com rosigliazona reduz os níveis plasmáticos de ácidos graxos livres, níveis de
insulina, concomitante aumento da fosforilação da proteína Akt em treonina 308 (Akt-pT308)
estimulada pela insulina em músculo e tecido adiposo. Efeito similar na fosforilação da Akt foi
observado no fígado somente após sete dias de tratamento com rosiglitazona. Esses achados
sugerem que o efeito no músculo e tecido adiposo pode ser primário. Neste mesmo estudo, foi
observado também aumento da fosforilação da Akt em serina 473 (Akt-pS473), fosforilação em
resíduos de tirosina da subunidade β do receptor de insulina e do IRS-1 em ambos, músculo e
tecido adiposo (JIANG, DALLAS-YANG et al., 2002).
Como descrito anteriormente, a modulação do sinal da insulina, pode ocorrer quando
IRS-1 é fosforilado em resíduos de serina e treonina por serinas quinases intracelulares. Estudos
têm demonstrado que rosiglitazona reduz o grau de fosforilação do IRS-1 em resíduos de serina
307 e 612 em células HEK-293.IRS1 (HEK-293 células expressando proteína recombinante

79
IRS1), bem como em adipócitos 3T3-L1. Além disso, tem sido observado que em ratos obesos
Zucker, o grau de fosforilação do IRS-1ser307 está aumentado em tecido adiposo branco, e tal
aumento tem sido revertido com o tratamento com rosiglitazona (JIANG, DALLAS-YANG et al.,
2004; JIANG, DALLAS-YANG et al., 2002). O efeito inibitório da fosforilação do IRS-1ser307, é
um componente chave na resistência à insulina, e esta reversão contribui para o efeito
sensibilizador da insulina observado com agonistas de PPARγ. Estudos têm demonstrado que a
reversão sob o efeito inibitório do IRS-1ser307 por rosiglitazona é em parte devido a redução da
fosforilação da proteína quinase JNK1 (JIANG, DALLAS-YANG et al., 2004; KHANDOUDI,
DELERIVE et al., 2002).
Para avaliar o efeito de GQ2 sobre as etapas iniciais da ação insulínica, que levam a
ativação da Akt, realizamos estudo direto da expressão, grau de fosforilação em resíduos de
tirosina do IR, IRS-1 e IRS-2 bem como a associação e ativação dos IRSs com a enzima PI3K e
a fosforilação da proteína Akt através de métodos de imunuprecipitação e immunoblonting em
tecido adiposo peri-epididimal, músculo esquelético gastrocnêmio e fígado de camundongos
“swiss” submetidos a dieta hiperlipídica, e em seguida tratados por 14 dias com 20mg/kg/dia de
GQ2. Nos nossos ensaios usamos grupos controles (camundongos “swiss” alimentados com dieta
padrão (C) e dieta hiperlipídica (DH)) e grupo padrão, rosiglitazona (RG).
O aumento da sensibilidade à insulina, demonstrado pelos estudos das características
metabólicas dos animais tratados com GQ2 em relação aos animais que receberam DH, foi
acompanhado por recuperação da via IRS-1/PI3K/Akt de sinalização da insulina em todos os
tecidos estudados.
Camundongos “swiss” submetidos à dieta hiperlipidica apresentam redução substancial
da ativação da via IRS-1/PI3K/Akt estimulada pela insulina, em fígado, músculo e tecido
adiposo, contribuindo para a resistência à insulina desses animais, porque essa via tem sido
implicada no transporte de glicose e síntese de glicogênio (CROSS et al., 1995).
Conseqüentemente, esses animais, apresentaram também aumento do grau de fosforilação do
IRS-1ser307, em paralelo, ao aumento da atividade das serinas quinases, JNK e IKK, e dos níveis
teciduais do TNF-α e iNOS. Interessantemente, a redução da fosforilação em resíduos de tirosina
do IRS-1, sem mudança do IRS-2, em tecido adiposo, foi suficiente para reduzir a ativação da
Akt neste tecido. O tratamento com GQ2 aumentou a fosforilação do IRS-1 em resíduos de
tirosina, atividade da PI3K, e Akt. Embora não tenha sido observado, variação do grau de
fosforilação do IRS-2 após o tratamento com GQ2, houve um aumento da atividade desse
substrato a PI3K em comparação ao grupo que recebeu DH. A melhora na sensibilidade à

80
insulina observada após tratamento com GQ2 em tecido adiposo, parece resultar em parte da
redução da fosforilação do IRS-1ser307 associado aos menores níveis teciduais de TNF-α.
No fígado de animais que receberam DH, além da redução da via IR/IRS-1/PI3K/Akt,
observou-se também redução do nível protéico e grau de fosforilação em tirosina, estimulado
pela insulina, do IRS-2. O tratamento com GQ2, assim como no tecido adiposo, reverteu o
quadro de resistência à insulina, induzido pela DH no fígado. Isso provavelmente ocorreu devido
a uma redução do grau de fosforilação do IRS-1ser307 e da atividade das proteínas quinases JNK e
IKK nesse tecido.
E, finalmente, de maneira semelhante ao tecido adiposo e ao tecido hepático a DH,
alterou de maneira significativa a via IRS-1/PI3K/Akt após estímulo com insulina em tecido
muscular, demonstrando mais uma vez, a importância dessa via na sensibilidade da insulina.
Assim como no tecido hepático, no músculo houve também redução do grau de fosforilação em
tirosina do IRS-2. O tratamento com GQ2 levou a uma reversão dessa condição de resistência à
insulina induzida pela DH.
Ainda dentro dessa abordagem, é oportuno destacar que a ativação da JNK induz
fosforilação do IRS-1ser307 (AGUIRRE, et al., 2002), levando uma redução da ativação da PI3K,
estimulada pela insulina. Nossos dados demonstram aumento da ativação da JNK em
concordância com alteração da sinalização da insulina em tecido adiposo e hepático de
camundongos submetidos a DH, sugerindo, que essa serina quinase tem importante papel no
desenvolvimento da resistência à insulina nesses camundongos, e que o tratamento com GQ2 foi
importante na redução da atividade dessa proteína. Finalmente, é possível que a etiologia
molecular da resistência à insulina seja multifatorial, onde vários mecanismos como ativação da
JNK, IKK e aumento de iNOS contribuam para o fenômeno final. É interessante destacar que
tanto a RG quanto o GQ2 reverteram estes múltiplos mecanismos.
Nossos resultados estão em concordância com outros estudos quanto ao efeito de TZDs
na recuperação das etapas iniciais da ação insulínica, embora em nossos estudos tenhamos
encontrados alguns dados contraditórios em relação a rosiglitazona, provavelmente pelo tempo
de tratamento, dose utilizada e modelo experimental nesse estudo.
A sensibilidade à insulina, demonstrada pelo maior Kitt do grupo de camundongos
tratados com GQ2 quando comparado ao grupo que recebeu DH foi acompanhada a nível
molecular pela reversão total ou parcial da via IR/IRSs(1/2)/PI3K/Akt em tecido adiposo,
músculo esquelético e tecido hepático.
A respeito da ativação da via de sinalização da insulina relacionada ao crescimento
celular, a cascata da MAP-quinase, a qual foi avaliada através da determinação da fosforilação

81
em tirosina induzida por insulina da ERK1 e ERK2, surpreendeu-nos o fato de que o tratamento
de camundongos obesos com GQ2 ou RG levou a um aumento da atividade de ERK1 e ERK2
após estímulo com insulina somente no tecido adiposo. No músculo esquelético e fígado não
houve diferença entre os grupos experimentais. Há dados controversos na literatura, relatando
que a ERK desempenha função de crescimento e diferenciação em tecido adiposo. Alguns
estudos mostram que a ativação das proteínas ERK pela insulina promove mitogênese
acompanhado de inibição da diferenciação (FONT DE MORA et al, 1997; KIM et al, 2001),
enquanto outros mostram que a ERK promove diferenciação do pré-adipócito em adipócito
(ZHANG et al, 1996; MACHINAL-QUELIN et al, 2002). Em um estudo recente, Prusty e
colaboradores (PRUSTY et al 2002) observaram que, em pré-adipócitos 3T3-L1, a insulina induz
ativação precoce e transitória da ERK, a qual induz a expressão de C/EBPα and PPAR-γ, que são
fatores transcripcionais envolvidos, respectivamente, com a diferenciação de pré-adipócitos e a
adipogênese (ROSEN et al, 2000). Esses autores concluíram que, durante a diferenciação de
adipócitos a ERK exerce um efeito positivo participando de fases iniciais de sua indução. Porém,
a completa diferenciação de adipócitos pode ocorrer através da ativação do PPARγ que participa
do controle de etapas subseqüentes da adipogênese. No estudo realizado por ZHANG et al.
(1996), observou-se que a insulina pode promover a ativação do PPARγ através da ERK, em
adipócitos em cultura. É interessante ressaltar que em um estudo clínico recente avaliaram-se
1.152 genes em tecido adiposo omental de pacientes magros e obesos, revendo-se que, entre
outros, os genes que codificam as proteínas da família da MAP-quinase estavam
significativamente aumentados em pacientes obesos (GOMEZ-AMBROSI et al, 2004). De
acordo com esse estudo, a atividade das proteínas da família MAP-quinase em tecido adiposo de
pacientes obesos pode participar tanto de eventos relacionados com ganho de peso como no
controle da adipogênese.
Observamos que em tecido adiposo de camundongos obesos tratados com GQ2 e RG,
houve um aumento da expressão de PPARγ após estímulo com insulina. Sendo assim, a ativação
de PPARγ por GQ2 poderá está envolvida no controle da adipogênese.
Assim, neste estudo demonstramos que o novo agonista de PPARγ, GQ2, induz redução
importante da glicemia de jejum, do peso corporal, da gordura epididimal e da quantidade de
água. Em paralelo, houve recuperação da via IRS-1/PI3K/Akt induzida por insulina em todos os
tecidos estudados e concomitante redução da atividade das proteínas envolvidas na via
inflamatória. Sugerindo que o composto GQ2, é uma nova TZD com grande potencial
terapêutico para o tratamento do diabetes mellitus tipo 2.

82
CONCLUSÃO

83
6. CONCLUSÃO
A partir dos resultados encontrados neste estudo, podemos concluir que:
Os estudos de modelagem molecular sugerem que essas novas moléculas arilideno-
tiazolidinadionas (ATZDs) possuem uma boa interação com o receptor PPARγ através das
ligações de hidrogênio entre o átomo de oxigênio do anel heterocíclico das novas ATZDs e o
resíduo de aminoácido ser289 do domínio LBD do receptor PPARγ.
O tratamento de camundongos com obesidade induzida por dieta hipelipidica com
GQ2 por um período de 14 dias, induz redução importante da glicemia de jejum, do peso
corporal, da gordura epididimal e da retenção de fluídos. Em paralelo, melhora a via IRS-
1/PI3K/Akt induzida por insulina em fígado, músculo esquelético e tecido adiposo, concomitante
redução da atividade das proteínas envolvidas na via inflamatória.
Sugerindo que o composto GQ2, é uma nova ATZD com grande potencial terapêutico
para o tratamento do diabetes mellitus tipo tipo 2.
Os resultados obtidos até aqui são estimuladores e necessitam de investigações
adicionais mais aprofundadas no âmbito da atividade Biológica. Neste sentido pretende-se dar
continuidade aos estudos pré clínicos com o composto GQ2.

84
REFERÊNCIAS BIBLIOGRÁFICAS

85
7. REFERÊNCIAS BIBLIOGRÁFICAS
ADAMS M, REGINATO MJ, SHAO D, LAZAR MA, CHATTERJEE VK: Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272:5128–5132, 1997. AGUIRRE, V., T. UCHIDA, et al. "The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307)." J Biol Chem 275(12): 9047-54. 2000. AGUIRRE, V., E. D. WERNER, et al. "Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action." J Biol Chem 277(2): 1531-7, 2002. ARAKI, E.; LIPES, M.A.; PATTI, M.E. et al. Alternative pathway of insulin signaling in mice with targeted disruption of the IRS-1 gene. Nature, 372: 186-90, 1994. BACKER, J. M.; MYERS, M. G., JR.; SHOELSON, S. E.; CHIN, D. J.; SUN, X. J.; MIRALPEIX, M. et al. Phosphatidylinositol 3'-kinase is activated by association with IRS-1 during insulin stimulation. Embo J, 9: 3469-79, 1992. BALFOUR, J.A.; PLOSKER, G.L. Rosiglitazone. Drugs, 57: 921–30, 1999. BAJAJ M, SURAAMORNKUL S, PIPER P, HARDIES LJ, GLASS L, CERSOSIMO E, PRATIPANAWATR T, MIYAZAKI Y, DEFRONZO RA. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab 89:200-6, 2004. BARREIRO, E.J.; FRAGA, C.A.M. Química Medicinal: as bases moleculares da ação de fármacos, ARTMED: Porto Alegre, 2001. BAUERLE PA, BALTIMORE D. NF-κB: Ten years after. Cell 87:13-20, 1996. BIRNBAUM MJ. Turning down insulin signaling. J Clin Invest 108:655-659, 2001.
BJORNHOLM, M.; AL-KHALILI, L.; DICKER, A.; NASLUND, E.; ROSSNER, S.; ZIERATH, J. R. et al. Insulin signal transduction and glucose transport in human adipocytes: effects of obesity and low calorie diet. Diabetologia, 8: 1128-35, 2002. BONORA, E.; MANICARDI, V.; ZAVARONI, I.; COSCELLI, C.; BUTTURINI, U. Relationships between insulin secretion, insulin metabolism and insulin resistance in mild glucose intolerance. Diabete Metab, 2: 116-21, 1987. BOSCHELLI, D. H.; CONNOR, D. T.; KUIPERS, P. J.; WRIGHT, C. D.; Bioorg. Med. Chem. Lett. 2, 705, 1992.

86
BOULTON, T. G.; NYE, S. H.; ROBBINS, D. J.; IP, N. Y.; RADZIEJEWSKA, E.; MORGENBESSER, S. D.; DEPINHO, R. A.; PANAYOTATOS, N.; COBB, M. H.; YANCOPOULOS, G. D. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell, 4: 663-75, 1991. BRAY, G.A.; POPKIN, B.M. Dietary fat intake does affect obesity! Am. J. Clin. Nutr, 68: 1157-1173, 1998. BROZINICK, J. T., JR., B. R. ROBERTS, et al. "Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance." Diabetes 52(4): 935-41, 2003. CANTELLO, B.C.; CAWTHORNE, M.A.; COTTAM, G.P. et al. [omega-(heterocyclylamino)-alkoxy]benzyl]-2,4-thiazolidinediones as potent antihyperglycemic agents. J. Med Chem, 37: 3977-3985, 1994. CARVALHEIRA, J.B.; SILOTO, R.M.; IGNACCHITTI, I. et al. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus [letter]. FEBS, 500: 119-24, 2001. CARVALHEIRA, J.B.;.RIBEIRO, E.B. et al. "Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia, 46(12): 1629-40, 2003. CARVALHO, C. R.; BRENELLI, S. L.; SILVA, A. C.; NUNES, A. L.; VELLOSO, L. A.; SAAD, M. J. Effect of aging on insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of rats. Endocrinology, 1: 151-9, 1996. CARPENTER, C.L. & CANTLEY, I.C. - Phosphoinositide kinases. Biochem., 29:11147-11156, 1990. CHALKLEY, S.M., HETTIARACHCHI, M., CHISHOLM, D.J., KRAEGEN, E.W.,. Long-term high-fat feeding leads to severe insulin resistance but not diabetes in Wistar rats. American Journal of Physiology, 282 (6), E1231–E1238, 2002. CHAWLA, A.; SCHWARZ, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor g (PPARg ): adipose predominant expression and induction early in adipocyte differentiation. Endocrinology, 135: 798–800, 1994. CHEATHAM B, KAHN CR: Insulin action and insulin signaling network. Endocrine Reviews; 16:117-141, 1995. CHO, H., J. MU, et al. "Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta)”. Science, 292(5522): 1728-31, 2001. CLARK, D.A.;GOLDSTEIN, S.W.; VOLKMANN, R.A. et al., substituted dihydrobenzopyran and dihydrobenzofuran thiazolidine-2,4-diones as hypoglycemic agents, MED J Chem, 34: 319–325, 1991. COLDITZ, G.A; WILLETT, W.C.; STAMPFER, M.J, et al. Weight as a risk factor for clinical diabetes in women. Am J Epidemiol, 132: 501-13,1990.

87
CROSS DA, ALESSI DR, COHEN P, ANDJELKOVICH M, HEMMINGS BA Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789, 1995. DAVIS RJ., Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp 64:1–12, 1999. DE LIMA, J. G.; PERRISSIN, M.; CHANTEGREL, J.; LUU-DUC, C.; ROUSSEAU, A.; NARCISSE, G.; Arzneim.-Forsch./Drug Res., 44, 831, 1994. DEFRONZO, R.A. "Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM." Diabetes 37(6): 667-87, 1988. DE VOS, P., LEFEBVRE, A.M, STEPHEN G. MILLER, S.G. et al., Thiazolidinediones Repress ob Gene Expression in Rodents Via Activation of Peroxisome Proliferator–activated Receptor-γ. J. Clin. Invest. 98:(4) 1004-1009, 1996. DIETRICH, L. A new Approach to Pratical Acute Tooxicity Testing. Archives of toxicology, 54: 275-287, 1983. DOWELL, P.; ISHMAEL, J.E.; AVRAM, D. et al.,. Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor alfa interacting protein. J Biol Chem, 274: 15901-15907, 1999. DUBOIS M, PATTOU F, KERR-CONTE J, GMYR V, VANDEWALLE B, DESREUMAUX P, AUWERX J, SCHOONJANS K, LEFEBVRE J Expression of peroxisome proliferatoractivated receptor gamma (PPARgamma) in normal human pancreatic islet cells. Diabetologia 43:1165-9, 2000 Order EBINA, Y.; ELLIS, L. et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signaling. Cell,;40: 747-58, 1985. ECKEL, R.H.; GRUNDY, S.M.; ZIMMET, P.Z. The metabolic syndrome. Lancet 365: 1415–1428, 2005. EBDRUP, S.; PETTERSSON, I.; RASMUSSEN, H. B.; DEUSSEN, H-J.; JENSEN, A. F.; MORTENSEN, S. B.; FLECKNER, J.; PRIDAL, L.; NYGAARD, L.; SAUERBERG, P. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor α/γ agonist ragaglitazar, J. Med. Chem., 46, 1306-1317, 2003. ELCHEBLY, M.; PAYETTE, P.; MICHALISZYN, E.; CROMLISH, W.; COLLINS, S.; LOY, A. L. et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science, 5407: 1544-8, 1999. FERRANNINI, E., NATALI, A.; BELL, P.; CAVALLO-PERIN, P.; LALIC, N.; MINGRONE, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J Clin Invest, 100: 1166-73, 1997. FERRANNINI, E.; BUZZIGOLI, G.; BONADONNA, R. et al. Insulin resistance in essential hypertension. N Engl J Med, 317: 350-7, 1987.

88
FOLLI, F.; SAAD, M.J.; BACKER, J.M.; KAHN, C.R. Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J Biol Chem, 267: 22171-7, 1992. FONT DE MORA, J.; PORRAS, A.; AHN, N.; SANTOS, E. Mitogen-activated protein kinase activation is not necessary for, but antagonizes, 3T3-L1 adipocytic differentiation. Mol Cell Biol, 10: 6068-75, 1997. FORMAN, B.M. et al. 15-deoxy-Delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 83, 803–812, 1995. FUJITA, T.; SUGIYAMA, Y.; TAKETOMI, S. et al., Redution of insulin resistance in obese and/or diabetic animals by 5-[4-(1-methylcyclohexylmethoxy)benzyl]-thiazolidine-2,4-dione (ADD-3878, u-63, 287, ciglitazone), a new antidiabetic agent. Diabetes, 32: 804-810. 1983. FUJIWARA T, WADA M, FUKUDA K et al., Characterization of CS-045, a new oral antidiabetic agent. II effects on glycemic control and pancreatic islet structure at a late stage of the diabetic syndrome in C57BL/Ksj-db/db mice. Metabolism, 40: 1213-1218, 1991. FUJIWARA, T., YOSHIOKA, S., YOSHIOKA, T., et al. Characterization of a New oral antidiabetic agent CS-045: Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes, 37:1549-1558, 1988. GALE, E.A.M. Lessons from he glitazones, A story of drug development. Lancet, 1870-1875, 2001. GAO, Z., D. HWANG, et al. "Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex." J Biol Chem 277(50): 48115-21, 2002 GASTALDELLI A, TOSCHI E, PETTITI M, FRASCERRA S, QUINONES-GALVAN A, SIRONI A.M, NATALI A, FERRANNINI E. Effect of physiological hyperinsulinemia on gluconeogenesis in nondiabetic subjects and in type 2 diabetic patients. Diabetes 50:1807-12, 2001. GITLIN, N.; JULIE, N.L.; SPURR, C.L.; LIM, K.N.; JUARBE, H.M. Two cases of severe clinical and histological hepatotoxicity associated with troglitazone. Ann Intern Med, 129: 36–38, 1998. GLASS, C.K.; ROSENFELD, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev, 14: 121–41, 2000. GOMEZ-AMBROSI, J.; CATALAN, V.; DIEZ-CABALLERO, A.; MARTINEZ-CRUZ, L. A.; GIL, M. J.; GARCIA-FONCILLAS, J. et al. Gene expression profile of omental adipose tissue in human obesity. Faseb J, 1: 215-7, 2004. GRUNFELD, B.; BALZARETI, M.; ROMO, M.; GIMENEZ, M.; GUTMAN, R. Hyperinsulinemia in normotensive offspring of hypertensive parents. Hypertension, 23: I12-5, 1994.

89
GURNELL M, WENTWORTH JM, AGOSTINI M, ADAMS M, COLLINGWOOD TN, PROVENZANO C, BROWNE PO, RAJANAYAGAM O. et al A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J Biol Chem 275:5754-9, 2000. HAFFNER, S.M.; STERN, M.P; HAZUDA, H.P.; PUGH, J.; PATTERSON, J.K. Do upper-body and centralized adiposity measure different aspects of regional body-fat distribution? Relationship to non-insulin-dependent diabetes mellitus, lipids, and lipoproteins. Diabetes, 36: 43-51, 1987. HIROSUMI J, TUNCMAN G, CHANG L, GORGUN CZ, UYSAL KT, MAEDA K, KARIN M, HOTAMISLIGIL GS A central role for JNK in obesity and insulin resistance. Nature 420:333–336, 2002. HOTAMISLIGIL GS, SPIEGELMAN BM. Tumor necrosis factor-α: a key component of the obesity-diabetes link. Diabetes, 43: 1271-1278, 1994. HOTAMISLIGIL GS. Mechanism of TNF-α induced insulin resistance. Exp Clin Endocrinol Diabetes; 107: 119-125, 1999. HOTAMISLIGIL, G.S.; PERALDI, P.; BUDAVARI, A.; ELLIS, R.;WHITE, M.F.;SPIEGELMAN, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha and obesity-induced insulin resistance. Science, 271: 665-8, 1996. HSUEH, W.A.; JACKSON, S.; LAW, R.E. Control of cardiovascular cell proliferation and migration by PPARγ, a new approach to the macrovascular complications of diabetes. Diabetes Care, 24: 392-397, 2001. HUNTER, S.J.; GARVEY, W.T. Insulin action and insulin resistance: disease involving defects in insulin receptors, signal transduction, and the glucose transport effector system. Am J Med, 105: 331-345, 1998. INAMORI, Y.; OKAMOTO, Y.; TAKEGAWA, Y.; TSUJIBO, H.; SAKAGAMI, Y.; KUMEDA, Y.; SHIBATA, M.; NUMATA, A.; Biosci., Biotechnol., Biochem., 62, 1025, 1998. IP YT, DAVIS RJ Signal transduction by the c-Jun N-terminal kinase (JNK): from inflammation to development. Curr Opin Cell Biol 10:205–219 signaling in insulin sensitive tissues of MSG-rats. Life Sci 73:1369–1381, 1998. ISSEMANN, I.; GREEN, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature, 347: 645-50, 1990. IWATA, Y.; MIYAMOTO, S.; TAKAMURA, M.; YANAGISAWA, H.; KASUYA, A. Interaction between peroxisome proliferators-activated receptor γ and its agonist: docking study of oximes having 5-benzyl-2,4-thiazolidnedione. Journal of Molecular Graphics and Modelling, 19, 536-542, 2001.
JIANG, Z. Y., Q. L. ZHOU, et al"Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing." Proc Natl Acad Sci U S A 100(13): 7569-74, 2003.

90
JIANG, C., TING, A.T.; SEED, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82–86, 1998. JONES, D.W.; KIM, J.S; ANDREW, M.E. KIM, S.J; HONG, Y.P. Body mass index and blood pressure in Korean men and women: the Korean National Blood Pressure Survey. J Hypertens. 12: 1433-7, 1994. KAHN BB, FLIER JS. Obesity and insulin resistance. J Clin Invest; 106: 473-81, 2000. KAHN, C.R. "Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes." Diabetes 43(8): 1066-84, 1994. KAHN, S.E.; PRIGEON, R.L.; SCHWARTZ, R.S.; FUJIMOTO, W.Y.; KNOPP, R.H.; BRUNZELL, J.D. et al. Obesity, body fat distribution, insulin sensitivity and islet b -cell function as explanations for metabolic diversity. J Nutr, 131: 354S-60S, 2001. KAPUR, S., S. BEDARD, et al. "Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action." Diabetes 46(11): 1691-700, 1997. KAWAMATSU, Y., SARAIE, T., IMAMIYA, E., NISHIKAWA, K. AND HAMURO, Y. Studies on antihyperlipidemic agents. I. Synthesis and hypolipidemic activities of phenoxyphenyl alkanoic acid derivatives. Arzneim Forsch./Drug, 30: 454–459, 1980. KERSHAW, E.E; FLIER, J.S. Adipose tissue as an endocrine organ, J. Clin. Endocrinol. Metab, 89: 2548–2556, 2004. KERSTEN, S.; SEYDOUX. J.; PETERS, J.M.; GONZALEZ, F.J.; DESVERGNE, B.; WHALI, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest, 103: 1489-98, 1999. KIM, J.Y., NOLTE, L.A., HANSEN, P.A., HAN, D.H., FERGUSON, K., THOMPSON, P.A., HOLLOSZY, J.O. High-fat diet induced muscle insulin resistance: relationship to visceral fat mass. American Journal of Physiology, 279 (6), R2057–R2065, 2000. KIM, S. W.; MUISE, A. M.; LYONS, P. J.; RO, H. S. Regulation of adipogenesis by a transcriptional repressor that modulates MAPK activation. J Biol Chem, 13: 10199-206, 2001. KISSEBAH, A.; VYDELINGUM, N.; MURRAY, R.; EVANS, D.; HARTZ, A.; KALKHOFF, R.K. et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab, 54: 254-60, 1982. KLIEWER, S.A.; UMESONO, K.; NOONAN, D.J.; HEYMAN, R.A.; EVANS, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature, 358: 771–774, 1992. KOHN, A. D., S. A. SUMMERS, et al. "Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation." J Biol Chem 271(49): 31372-8, 1996. KOROLKOVAS, A; Fundamentos de farmacologia molecular: base para o planejamento de fármacos, 2º ed., EDART: São Paulo, 1977.

91
KROTKIEWSKI, M.; BJORNTORP, P.; SJOSTROM, L.; SMITH, U. Impact of obesity on the metabolism in men and women: importance of regional adipose tissue distribution. J Clin Invest, 72: 1150-62, 1983. KURREK, M. M., W. M. ZAPOL, et al. "In vivo lipopolysaccharide pretreatment inhibits cGMP release from the isolated-perfused rat lung." Am J Physiol 269(5 Pt 1): L618-24, 1995. LAW, R.E.; GOETZE, S.; XI, X.; JACKSON, S.; KAWANO, Y. et al. Expression and function of PPARg in rat and human vascular smooth muscle cells. Circ Res, 101: 1311–1318, 2000. LEE YH, GIRAUD J, DAVIS RJ, WHITE MFc-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278:2896–2902, 2003. LEFEBVRE PJ, SCHEEN AJ. Improving the action of insulin. Clin Inv Med 18(4):340-7, 1995. LEHMANN, J.M.; MOORE, L.B.; SMITH-OLIVER, T.A.; WILKISON, W.O. et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 270: 12953–12956, 1995. LIJIMA, K.; YOSHIZUMI, M.; AKO, J.; ETO, M. et al. Expression of peroxisome proliferator-activated receptor g PPARg in rat aortic smooth muscle cells. Biochem Biophys Res Commun, 247(2): 353–356, 1999. LOWELL, B.B. PPARgamma: an essential regulator of adipogenesis and modulator of fat cell function. Cell, 99: 239-242, 1999. MACHINAL-QUELIN, F.; DIEUDONNE, M. N.; LENEVEU, M. C.; PECQUERY, R.; GIUDICELLI, Y. Proadipogenic effect of leptin on rat preadipocytes in vitro: activation of MAPK and STAT3 signaling pathways. Am J Physiol Cell Physiol, 4: C853-63, 2002. MANSON, J.E.; COLDITZ, G.A.; STAMPFER, M.J. et al. A prospective study of obesity and risk of coronary heart disease in women. N Eng J Med. 322: 882-9, 1990. MENEZES, E. H. C.; GÓES, A. J. S.; DIU, M. B. S.; GALDINO, S. L.; PITTA, I. R.; LUU-DUC, C. ; Pharmazie, 46, 457, 1992. MIYAZAKI Y, MAHANKALI A, MATSUDA M, MAHANKALI S, HARDIES J, CUSI K, MANDARINO LJ, DEFRONZO RA Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 87:2784-91, 2002. MIYAZAKI Y, GLASS L, TRIPLITT C, MATSUDA M, CUSI K, MAHANKALI A, MAHANKALI S, MANDARINO LJ, DEFRONZO RA Effect of rosiglitazone on glucose and nonesterified fatty acid metabolism in Type II diabetic patients. Diabetologia 44:2210-2219, 2001. MOURÃO, R.H.; SILVA, T.G.; SOARES, A.L.M.; VIEIRA, E.S.; SANTOS, J.N.; LIMA, M.C.A.; LIMA, V.L.M.; GALDINO, S.L.; BARBE, J.; PITTA, I.R. Synthesis and biological activity of novel acridinylidene and benzylidene thiazolidinediones. Eur. J. Med. Chem. 40: 1129-33, 2005.

92
MYKKANEN, L; LAAKSO, M.; PYORALA, K. Association of obesity and distribution of obesity with glucose tolerance and cardiovascular risk factors in the elderly. Int J Obes Relat Metab Disord, 16: 695-704, 1992. NIKULINA M.A, SANDHU N, SHAMIM Z, ANDERSEN N.A, OBERSON A, DUPRAZ P, THORENS B, KARLSEN A.E, BONNY C, MANDRUP-POULSEN T. The JNK binding domain of islet-brain 1 inhibits IL-1 induced JNK activity and apoptosis but not the transcription of key proapoptotic or protective genes in insulinsecreting cell lines. Cytokine 24:13–24, 2003. OGDEN C.L, CARROLL MD, FLEGAL KM Epidemiologic trends in overweight and obesity. Endocrinol Metab Clin North Am 32:741–760, vii, 2003. OLEFSKY, J.M. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 106: 467–472, 2000. OLEFSKY, J.M.; SALTIEL, A. RPPAR gamma and the treatment of insulin resistance. Trends Endocrinol Metab, 11: 362–368, 2000. PATTI, M.E, KAHN, C.R. The insulin receptor — a critical link in glucose homeostasis and insulin action. J Basic Clin Physiol Pharmacol. 9: 89-109. 1998. PESSIN, J.E.; SALTIEL, A.R. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest, 106: 165-9, 2000. PI-SUNYER F. Medical hazards of obesity. Ann Intern Med, 119: 655-660, 1993. PLOSKER, G.L.; FAULDS, D. Troglitazone: a review of its use in the management of type 2 diabetes mellitus. Drugs, 57: 409–38, 1999. POPKIN, B.M.; DOAK, C.M. The obesity epidemic is a worldwide phenomenon. Nutr. Rev, 56: 106-114, 1998. PRUSTY, D.; PARK, B. H.; DAVIS, K. E.; FARMER, S. R. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem, 48: 46226-32, 2002. RAO, S.V.; DONAHUE, M.; PI-SUNYER, F.X.; FUSTER, V. Results of expert meetings: obesity and cardiovascular disease. Obesity as a risk factor in coronary artery disease, Am. Heart J. 142: 1102–1107, 2001. REGINATO, M.J.; LAZAR, M.A. Mechanisms by which thiazoldidinediones enhance insuline action. TEM, 10(1): 9-13, 1999. RIBIERE, C., A.M. JAUBERT, et al. "White adipose tissue nitric oxide synthase: a potential source for NO production." Biochem Biophys Res Commun 222(3): 706-12, 1996. RICOTE, M.; LI, A.C., WILLSON, T.M., KELLY, C.J.; GLASS, C.K. The peroxisome proliferator- activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82, 1998.

93
RYAN, A.S.; ROCHE, A.F.; WELLENS, R.; GUO, S. Relationship of blood pressure to fatness and fat patterning in Mexican American adults from the Hispanic Health and Nutrition Examination Survey (NHANES, 1982-1984). Coll Antropol, 18: 89-99, 1994. ROSEN, E. D.; WALKEY, C. J.; PUIGSERVER, P.; SPIEGELMAN, B. M. Transcriptional regulation of adipogenesis. Genes Dev, 11: 1293-307, 2000. SAAD MJA, FOLLI F, KAHN JA, KAHN CR: Modulation of insulin receptor, insulin receptor substrate 1 and PI 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest, 92: 2065-2072, 1993. SAAD, M.J., E. ARAKI, et al. "Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance”. J Clin Invest 90(5): 1839-49, 1992. SALTIEL, A.R.; KAHN, C. R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 6865: 799-806, 2001. SCHOONJANS, K.; STAELS, B.; AUWERX, J. Role of the peroxisome proliferators-activated receptor (PPPAR) in mediating the effectcs of fibrates and fatty acids on gene expression. J Lipid Res, 37: 907-25, 1996. SCHWARTZ, M.W.; WOODS, S.C.; PORTE, D.; SEELEY, R.J.; BASKIN, D.G. Central nervous system control of food intake. Nature, 404: 661-71, 2000. SCHMIDT MI, DUNCAN BB. Chronic activation of the innate immune system may underlie the metabolic syndrome. São Paulo Med J 119(3):122-127, 2001. SHEPHERD, P.R.; NAVE, B.T.; SIDDLE, K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3-L1 adipocytes: evidence for the involvement of phosphoinositide 3-kinase and p70 ribosomal protein-S6 kinase. Biochem J, 305: 25-8, 1995. SHULMAN, G. I. Unraveling the cellular mechanism of insulin resistance in humans: new insights from magnetic resonance spectroscopy. Physiology (Bethesda), 183-90, 2004. SINGH, M.K., KRISAN, A.D., CRAIN, A.M., COLLINS, D.E., YASPELKIS, B.B. High-fat diet and leptin treatment alter skeletal muscle insulin-stimulated phosphatidylinositol 3-kinase activity and glucose transport. Metabolism 52 (9), 1196–1205, 2003. SOHDA, T.; MIZUNO, K. IMAMIYA, E. SUGIYAMA, Y.; FUJITA, T.; KAWAMATSU, Y. Studies on antidiabetic agents. II. Synthesis of 5-[4-(1-methylcyclohexylmethoxy)-benzyl]thiazolidine-2,4-dione (ADD-3878) and its derivatives. Chem Pharm Bull (Tokyo), 30(10): 3580-600, 1982.
SUN XJ, WANG LM, ZHANG Y, YENUSH L, MYERS Jr MG, GLASHEEN EM, LANE WS, PIERCE JH AND WHITE MF. Role of the IRS-2 in insulin and cytokine signaling. Nature; 377: 173-177, 1995.

94
SPIEGELMAN, B.M. PPARg: Adipogenic regulator and thiazolidinedione receptor. Diabetes, 47(4): 507-14, 1998. STEIN, C. J.; COLDITZ, G. A. The epidemic of obesity. The Journal of Clinical Endocrinology & Metabolism, 89: 2522–2525, 2004. STERN, M. Epidemiology of obesity and its link to heart disease. Metab Clin Exper, 44/9: 1-3, 1995. STERN, M.P.; HAFFNER, S.M. Body fat distribution and hyperinsulinemia as risk factors for diabetes and cardiovascular disease. Arteriosclerosis, 6: 123-30, 1996. STORLIEN LH, PAN DA, KRIKETOS AD, BAUR LA. High fat diet-induced insulin resistance. Lesson and implications from animal studies. In: Klimes I, Howard BV, Storlien LH, Sebokova E, editors. Dietary lipids and insulin action. Second International Smolenice Insulin Symposium, vol 683. Ann N Y Acad Sci. p. 82– 90, 1993. TARTAGLIA, L.A.; DEMBSKI, M.; WENG, X.; DENG, N.; CULPEPPER, J.; DEVOS, R. et al. Identification and expression cloning ofa leptin receptor OB-R. Cell, 83: 1263-1271, 1995. TAKAMURA T, NOHARA E, NAGAI Y, KOBAYASHI K Stage-specific effects of a thiazolidinedione on proliferation, differentiation and PPARgamma mRNA expression in 3T3-L1 adipocytes. Eur J Pharmacol 422:23-9, 2001. UEKI, K., R. YAMAMOTO-HONDA, et al. "Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis." J Biol Chem, 273(9): 5315-22, 1998. VAGUE, J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout and uric calculus disease. Am J Clin Nutr, ;4: 20-34, 1956. VAN WEEREN, P. C., K. M. DE BRUYN, et al. "Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB." J Biol Chem 273(21): 13150-6, 1998. VELLOSO, L.A.; CARVALHO, C.R.; ROJAS, F.A.; FOLLI, F.; SAAD, M.J. Insulin signaling in heart involves insulin receptor substrates-1 and -2, activation of phosphatidylinositol 3-kinase and the JAK 2-growth related pathway. Cardiovasc Res, 40: 96-102, 1998. VESSB, Y.B. Dietery fat and insulin action in humans. Brit J. Nutrr, 83: S91-6, 2000. VIDAL-PUIG A, JIMENEZ-LINAN M, LOWELL BB, HAMANN A, HU E, SPIEGELMAN B, FLIER JS, MOLLER DE Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 97:2553-61, 1996. WATKINS, P.B.; WHITCOMB, R.W. Hepatic dysfunction associated with troglitazone. New Engl. J. Med, 338: 916–917, 1998.

95
WAY JM, HARRINGTON WW, BROWN KK, GOTTSCHALK WK, SUNDSETH SS, MANSFIELD TA, RAMACHANDRAN RK, WILLSON TM, KLIEWER SA Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology 142:1269-77, 2001. WESTON CR, LAMBRIGHT DG, DAVIS RJ Signal transduction: MAP kinase signaling specificity. Science, 296:2345–2347, 2002. WHITE, M.F. The IRS-signaling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem, 182: 03-11, 1998. WHITE MF. The insulin signaling system and the IRS proteins. Diabetologia; 40:S2-S17, 1997. WORLD HEALTH ORGANIZATION (WHO) Obesity – preventing and managing the global epidemic. Geneva: Report of a WHO Consultation on Obesity, 1998. YAMAUCHI, T.; TOBE, K.; TAMEMOTO, H.; UEKI, K.; KABURAGI, Y.; YAMAMOTO-HONDA, R. et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol, 6: 3074-84, 1996. YOSHIOKA T, FUJITA T, KANAI T, et al., Studies on hindered phenols and analogues. 1. Hipolipidemic and hypolycemic agents wuth ability to inhibit lipid peroxidation. J Med Chem, 32: 421-428, 1989. YU, K. et al. Differential activation of peroxisome proliferatoractivated receptors by eicosanoids. J Biol Chem, 270: 23975–23983, 1995. YUAN, M.; KONSTANTOPOULOS, N.;LEE, J. et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science, 293: 1673-7, 2001. ZHANG, H.; ZHANG, A.; KOHAN, E.D. et al. Collecting duct-specific deletion of peroxisome proliferator-activated recepto γ blocks thiazolidinedione-induced fluid retention. PNAS, 102: 9406-11, 2005. ZHANG,Y.; PROENCA, R.; MAFFEI, M.; BARONE, M.; LEOPOLD, L.; FRIEDMAN, J. M. Positional cloning of the mouse obese gene and its human homologue. Nature, 372: 425–43, 1994. ZHANG, B.; BERGER, J.; ZHOU, G. et al., Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor c, J. Biol. Chem. 271: 31771–31774, 1996. ZECCHIN, C.H.; CARVALHEIRA, J.B.C.; SAAD, M.J.A. Mecanismos moleculares de resistência à insulina na síndrome metabólica. Rev Soc Cardiol Estado de São Paulo 4: 574-89, 2004.

96
ANEXOS

97
Synthesis and Biological Activity of Novel Acridinylidene and Benzylidene thiazolidinediones
R.H. Mourãoa, T.G. Silvaa, A.L.M. Soaresa, E.S.Vieiraa, J.N. Santosa, M.C.A. Limaa, V.L.M. Limaa, S.L. Galdinoa, J. Barbeb and I.R. Pittaa,*
a Universidade Federal de Pernambuco, Centro de Ciências Biológicas
50.670-901 Recife, Brasil b GERCTOP – UMR CNRS 6178, Université de la Méditerranée
Faculté de Pharmacie, 13385 Marseille cedex 5, France.
Corresponding author : I.R. Pitta, Dpto de antibioticos – Universidade federal de Pernambuco Centro de ciencias biologicas 50670-901 Recife Brasil e-mail: [email protected]
Abstract- A novel set of acridinylidene thiazolidinediones and benzylidene
thiazolidinediones was synthesized by nucleophilic addition of cyanoacrylates. Some of these
compounds were evaluated for their glucose lowering capability and their effects on the
triglyceride level in alloxan diabetic mice.
Key-words- Thiazolidinediones – hypoglycaemic agents
1.Introduction
The thiazolidinediones (TZDs) or glitazones are oral hypoglycaemic agents, which act mainly
by increasing tissue sensitivity especially adipose tissue, to insulin [1-4]. Various studies
report the effects of TZDs on the metabolism of lipids, on cell differentiation and on some
cardiovascular risk factors [5-6]. The first compound described was ciglitazone, which
produced a dramatic decrease in the glycaemia level in animal models but showed poor
clinical effect. In 1997 troglitazone was launched onto the market, but had to be withdrawn in

98
the USA in march 2000 because of idiosyncratic liver failure. Since 1999, rosiglitazone and
pioglitazone have been available in Europe as second line drugs restricted to combination
therapy while in the United States, their use has been allowed as first line agents in
monotherapy or in combination with other drugs [4]. It was demonstrated that the molecular
target of the TZDs is part of the nuclear receptor called Peroxisome Proliferator-Activated
Receptor-gamma (PPAR-γ) which controls the differentiation of adipocytes [3], the
metabolism of fatty acids and alters the expression of genes that are also regulated by insulin
[7]. This paper describes the synthesis and gives the structural characteristics of several
derivatives of the 5-benzylidene-3-(4-methyl-benzyl)-thiazolidine-2,4-dione substituted on the
benzylidene moiety and those of the 3-benzyl-5-acridinylidene-thiazolidine-2,4-dione
substituted on the benzyl ring branched on the thiazolidine moiety. Some of these compounds
were evaluated in after oral administration at doses of 10 mg/kg or 30 mg/kg in alloxan
diabetic mice. Plasma glucose (PG) and triglyceride (TG) levels were measured and compared
with rosiglitazone (10 mg/kg p.o.) as the reference drug.
2.Chemistry
The 5-benzylidene-3-(4-methyl-benzyl-)-thiazolidine-2,4-diones, 3, were prepared by a
nucleophilic addition of the 3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 2, on selected aryl-
substituted ethyl-(2-cyano-3-phenyl)-acrylates [8] according to Daboun et al. [9]. Synthetic
pathways are portrayed in fig. 1. Thiazolidine-2,4-dione, 1, was N-(3)-alkylated in the
presence of potassium hydroxide which leads to the thiazolidine potassium salt which reacts
with benzyl or phenacyl halides in hot alcoholic medium [10]. The 3-benzyl (or phenacyl)-
thiazolidine-2,4-diones, 2 or 7, were obtained in this way. The acridinylidene
thiazolidinedione derivatives, 8a-8d, were synthesized by a nucleophilic addition of
substituted thiazolidinediones, 7, on 9-[ethyl-(2'-cyano)-acrylate]-acridine, 6. Direct

99
condensation of 9-acridinaldehyde, 5, with the substituted thiazolidinediones, 7, did not led to
the expected 3-benzyl-5-acridinylidene-thiazolidine-2,4-diones, 8a-8d. 9-Methyl-acridine, 4,
was prepared from diphenylamine treated with zinc dichloride in acetic acid medium
according to Tsuge et al. [11]. Subsequent oxidation of 4 with pyridinium chlorochromate
according to Mosher et al. [12], gave 9-acridinaldehyde, 5. Condensation of 5 with ethyl
cyanoacetate in the presence of piperidine in hot anhydrous benzene led to the 9-[ethyl-(2'-
cyano)-acrylate]-acridine, 6. Synthetic pathways are portrayed in fig. 2. Benzylidene-
thiazolidinediones were isolated in a single isomeric form. X-ray crystallographic studies and
13C NMR have demonstrated the preferred Z configuration for 5-arylidene-thiazolidinones
[13]. In contrast, acridinylidene-thiazolidinedione derivatives, 8a-8d, were isolated as
isomeric mixtures. The isomers were readily identified by 1H NMR, the ethylene proton being
more deshielded in the isomer Z than it is in the isomer E, owing to the cis-position of the
exocyclic carbonyl function. Chemical shifts listed in the experimental part are those of the Z
isomer, which is the prevalent isomer. MS data fully agree with the structure proposed.
3.Results and Discussion
The glucose and triglyceride lowering effects after oral administration of new thiazolidine
analogues, 3a-3f, in alloxan diabetic mice are summarized in Table 1. All the compounds
tested show a dose-dependent hypoglycaemic activity during the 15 days treatment. The
decrease in the level of plasma glucose observed with these compounds was progressive
throughout the treatment, especially at a 30mg/kg/day dose. After 15 days of treatment with
this dose, compound 3c showed a better biological activity than other compounds do in both
parameters analysed, i.e. around 50% decrease in PG and 59% decrease in TG. However, this
compound was recognized to be very toxic, with a 50% mortality rate during the treatment at
the two doses tested. In contrast, the other compounds, although they produced less reduction

100
in PG and TG rates, had a very low if not zero mortality rate (Tables 1 and 2). The percentage
for mortality observed in the case of a 10mg/kg/day dose of 3a,3b,3d-3f was also very low.
The mortality observed with the 10mg/kg/day dose could not be due to the compounds under
evaluation but to the hyperglycaemia caused by alloxan, as the mortality rate for diabetic
animals treated with this vehicle (CMC 0.5%) is in the same range. The presence of the two
methoxy groups in position 2 and 4 of the benzylidene ring in 3d led to a reduction in
hypoglycaemia (35%) and hypolipidemia (46%) activity, although this compound did not
show toxicity and there were no deaths at the 30mg/kg/day dose as observed with 3c which is
only substituted by one methoxy group. The change in the position of the chlorine atom on
the phenyl ring led to a significant hypoglycaemic activity (P< 0.05). This is clearly
demonstrated by comparing the 4-chloro derivative 3a (PG =23%; TG = 48%) with the 2-
chloro substituted one 3b (PG = 43%; TG = 26%). However, the opposite is seen for the
hypolipidemic activity which is greater with 3a than it is with 3b.
4.Conclusion
The new 5-arylidene-3-(4-methyl-benzyl)-thiazolidine-2,4-diones investigated have shown
promising glucose lowering activity. Their activity on the triglyceride level is close to that of
the rosiglitazone when used at the same concentration but activity is much better at higher
concentration (30 mg/kg) which can be used safely because of the low toxicity of compounds
evaluated. These results indicate the need for additional investigations, chiefly about the
agonistic effect of these drugs on PPAR-γ and PPAR-α.

101
5.Experimental protocols
5.1Chemistry
Melting points were measured in a capillary tube on a Buchi (or Quimis) apparatus. Thin
Layer Chromatography was performed on silicagel plates Merck 60F254. Infrared spectra of
1% KBr pellets were recorded on a Bruker IFS66 spectrometer (or Perkin Elmer 1310
spectrometer for compounds 3a-3d). 1H NMR spectra were recorded on a Bruker AC 300 P
spectrophotometer in DMSO-d6 as solvent, with tetramethylsilane as internal standard. Mass
spectra were recorded on Delsi-Nermag R 1010 C spectrometer (or HP 5897 for compounds
8a-8d). The published chemical data on 2,4,5,6 [14], are not reported here.
Analyses of all the compounds prepared were within ± 0.4% of the theoretical values.
5-Benzylidene-3-(4-methyl-benzyl)-thiazolidine-2,4-diones, 3a-3h: general procedure.
A mixture of 3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 2, (0.59 g, 2.5 mmol) and ethyl-(2-
cyano-3-phenyl)-acrylate (2.7 mmol) is refluxed for 2-6 h in absolute ethanol (20 mL) with
piperidine (0.25 mL) added. After cooling, precipitates are purified by column
chromatography or crystallized in suitable solvents.
5-(4-Chloro-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3a.
C18H14ClNO2S, yield: 0.427 g (89%). Mp: 184°C. TLC benzene : ethyl acetate (95:5) Rf: 0.8.
IR cm-1 (KBr): υ 1730, 1670, 1600, 1374, 1335, 1138, 820. 1H NMR (δ ppm, DMSO-d6):
2.27 (s, CH3), 4.79 (s, NCH2), 7.97 (s, CH), 7.17 (m, 4H benzyl), 7.63 (m, 4H benzylidene).
Ms, m/z (%) : 343(M+ 100), 344(22.2), 345(24.6),105(96.9), 103(6).
5-(2-Chloro-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3b.

102
C18H14ClNO2S, yield: 0.087 g (30%). Mp: 116°C. TLC benzene : ethyl acetate (95:5) Rf:
0.74. IR cm-1 (KBr): υ 1738, 1682, 1598, 1370, 1310, 755. 1H NMR (δ ppm, DMSO-d6): 2.28
(s, CH3), 4.8 (s, NCH2), 8.06 (s, CH), 7.16 (d, 2H benzyl, J = 8.1Hz), 7.23 (d, 2H benzyl, J =
8.1Hz), 7.51-7.54 (m, 2H benzylidene), 7.59-7.62 (m, 1H benzylidene), 7.64-7.67 (m, 1H
benzylidene). Ms, m/z (%): 343(M+ 45.1), 344(5.7), 345(8), 308(52.8), 280(5.2), 105(100),
94(7.8).
5-(4-Methoxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3c.
C19H17NO3S, yield : 0.354 g (77%). Mp: 135°C. TLC benzene : ethyl acetate (95:5) Rf: 0.83.
IR cm-1 (KBr): υ 1738, 1672, 1588, 1503, 1365, 1260, 820. 1H NMR (δ ppm, DMSO-d6):
2.28 (s, CH3), 3.84 (s, OCH3), 4.79 (s, NCH2), 7.93 (s, CH), 7.16 (d, 2H benzyl, J = 8.4Hz),
7.21 (d, 2H benzyl, J = 8.1Hz), 7.12 (d, 2H benzylidene, J = 9Hz), 7.61 (d, 2H benzylidene, J
= 9Hz). Ms, m/z (%): 339(M+ 58.3), 340(15), 341(4),105(100).
5-(2,4-Dimethoxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3d.
C20H19NO4S, yield: 0.197 g (77%). Mp: 161°C. TLC benzene : ethyl acetate (95:5) Rf: 0.76.
IR cm-1 (KBr): υ 1720, 1675, 1575, 1367, 1267, 1147, 830. 1H NMR (δ ppm, DMSO-d6):
2.27 (s, CH3), 3.85 (s, OCH3), 3.9 (s, OCH3), 4.77 (s, NCH2), 8.06 (s, CH), 6.7 (s, 1H
benzylidene), 6.71 (d, 1H benzylidene, J = 8.4Hz), 7.39 (d, 1H benzylidene, J = 8.4Hz), 7.15
(d, 2H benzyl, J = 8.1Hz), 7.19 (d, 2H benzyl, J = 8.1Hz). Ms, m/z (%): 369(M+ 100),
370(12.7), 371(14.1), 264(29.2), 105(14.5).
5-(4-Dimethylamino-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3e.
C20H20N2O2S, yield: 0.215 g (47%). Mp: 180°C. TLC benzene : ethyl acetate (95:5) Rf: 0.75.
IR cm-1 (KBr): υ 1726, 1673, 1593, 1380, 811. 1H NMR (δ ppm, DMSO-d6): 2.27 (s, CH3),
3.02 (s, N(CH3)2 ), 4.77 (s, NCH2), 8.06 (s, CH), 6.82 (d, 2H benzylidene, J = 9Hz), 7.46 (d,

103
2H benzylidene, J = 9Hz), 7.14 (d, 2H benzyl, J = 8.1Hz), 7.19 (d, 2H benzyl, J = 8.4Hz). Ms,
m/z (%): 352(M+ 41.7), 353(2.1), 177(46.6), 176(34.6), 161(12), 105(100), 89(23.3),
77(37.2).
5-(4-Benzyloxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3f.
C25H21NO3S, yield: 0.323 g (58%). Mp: 157°C. TLC benzene : ethyl acetate (95:5) Rf: 0.86.
IR cm-1 (KBr): υ 1736, 1677, 1593, 1263, 826. 1H NMR (δ ppm, DMSO-d6): 2.28 (s, CH3),
4.79 (s, NCH2), 5.21 (s, OCH2), 7.93 (s, CH), 7.15-7.21 (m, 4H benzyl, 2H benzylidene),
7.35-7.46 (m, 5H benzyloxy), 7.61 (d, 2H benzylidene, J = 9Hz). Ms, m/z (%): 415(M+ 17.3),
416(2.5), 91(100), 77(6.9).
5-(4-Fluoro-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3g.
C18H14FNO2S, yield: 0.387 g (87%). Mp: 124°C. TLC benzene : ethyl acetate (95:5) Rf: 0.81.
IR cm-1 (KBr): υ 1736, 1685, 1613, 1376, 1291, 680. 1H NMR (δ ppm, DMSO-d6): 2.27 (s,
CH3), 4.8 (s, NCH2), 7.96 (s, CH), 7.15 (d, 2H benzyl, J = 8.1Hz), 7.21 (d, 2H benzyl, J =
8.4Hz), 7.33-7.38 (m, 1H benzylidene), 7.45-7.5 (m, 2H benzylidene), 7.56-7.64 (m, 1H
benzylidene). Ms, m/z (%): 327(M+ 100), 328(16.4), 329(6.6), 105(91.8), 91(4.2).
5-(5-Bromo-2-methoxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione, 3h.
C19H16BrNO3S, yield: 0.352 g (62%). Mp: 147°C. TLC n-hexane : ethyl acetate (70:30) Rf:
0.78. IR cm-1 (KBr): υ 1733, 1684, 1604, 1339, 1283, 808. 1H NMR (δ ppm, DMSO-d6): 2.27
(s, CH3), 3.89 (s, OCH3), 4.78 (s, NCH2), 7.96 (s, CH), 7.15 (d, 2H benzyl, J = 8.1Hz), 7.2 (d,
2H benzyl, J = 8.4Hz), 7.14 (d, 1H benzylidene, J = 9.3Hz), 7.53 (d, 1H benzylidene, J =
2.4Hz), 7.65 (dd 1H benzylidene, J = 9,3Hz). Ms, m/z (%): 417(M+ 60), 386(7.1), 227(13),
199(11), 105(100), 91(31), 77(23.5).
3-Benzyl (or phenacyl)-5-acridinylidene-thiazolidine-2,4-diones, 8a-8d : general procedure.

104
Benzyl (or phenacyl) thiazolidine, 7 (0.9 mmol) and 9-[ethyl-(2'-cyano)-acrylate]-acridine, 6,
(0.9 mmol) are dissolved in absolute ethanol (8 mL). The solution is refluxed for 4 h in the
presence of a small amount of piperidine as catalyst. The precipitate obtained is filtered and
washed with water. Compounds isolated are of acceptable purity and were analysed without
further recrystallization.
5-Acridin-9-yl-methylene-3-benzyl-thiazolidine-2,4-dione, 8a.
C24H16N2O2S, yield: 0.124 g (88%). Mp: 150-152oC. TLC n-hexane : ethyl acetate (7:3) Rf:
0.41. IR cm-1 (KBr): υ 1750, 1694, 1623, 1381, 1339, 1149, 761. 1H NMR (δ ppm, DMSO-
d6): 4.89 (s, NCH2), 7.41-7.4 (m, 4H benzyl), 7.39-7.34 (m, 1H benzyl), 7.72-7.66 (m, 2H
acridine), 7.94-7.89 (2H acridine), 8.14 (d, 2H acridine, J = 8.4Hz), 8.24 (d, 2H acridine, J =
8.4Hz), 8.79 (s, 1H, CH). Ms, m/z (%): 396(M+ 4.2), 397(1.9), 305(8), 235(100), 232(26.6),
231(20.4), 91(10.1).
5-Acridin-9-yl-methylene-3-(4-fluoro-benzyl)-thiazolidine-2,4-dione, 8b.
C24H15FN2O2S, yield: 0.095 (56%). Mp: 200-202oC. TLC n-hexane : ethyl acetate (7:3) Rf:
0.58. IR cm-1 (KBr): υ 1749, 1693, 1604, 1382, 1337, 1152, 763. 1H NMR (δ ppm, DMSO-
d6): 4.88 (s, NCH2), 7.23 (t, 2H benzyl, J = 9Hz), 7.45-7.5 (m, 2H benzyl), 7.71-7.66 (m, 2H
acridine), 7.94-7.89 (m, 2H acridine), 8.14 (d, 2H acridine, J = 8.7Hz), 8.24 (d, 2H acridine, J
= 8.7Hz), 8.78 (s, 1H, CH). Ms, m/z (%): 414(M+ 12.9), 415(4), 305(34.3), 261(19.9),
235(100), 234(44.1), 231(68.8), 191(16.9), 109(32.6).
5-Acridin-9-yl-methylene-3[2-(4-nitro-phenyl)-2-oxo-ethyl]-thiazolidine-2,4-dione, 8c.
C25H15N3O5S yield: 0.136 g (70%). Mp: 229-230oC. TLC n-hexane : ethyl acetate (7:3) Rf:
0.3. IR cm-1 (KBr): υ 1747, 1699, 1629, 1603, 1530, 1346, 1222, 855, 761. 1H NMR (δ ppm,
DMSO-d6): 5.49 (s, NCH2), 7.77-7.71 (m, 2H acridine), 7.97-7.92 (m, 2H acridine), 8.13 (d,

105
2H acridine, J = 8.7Hz), 8.26 (d, 2H acridine, J = 9Hz), 8.88 (s, 1H, CH). Ms, m/z (%):
469(M+ 50), 470(16.2), 235(93.1), 234(30.8), 230(79.2), 150(100), 120(35.5), 104(40).
5-Acridin-9-yl-methylene-3[2-(4-fluoro-phenyl)-2-oxo-ethyl]-thiazolidine-2,4-dione, 8d.
C25H15FN2O3S, yield: 0.084 g (48%). Mp: 213-214oC. TLC n-hexane : ethyl acetate (7:3) Rf:
0.41. IR cm-1 (KBr): υ 1754, 1701, 1596, 1411, 1381, 1229, 1154, 759. 1H NMR (δ ppm,
DMSO-d6): 5.39 (s, NCH2), 7.46 (t, 2H benzyl, J = 8.7Hz), 7.76-7.71 (m, 2H acridine), 7.97-
7.91 (m, 2H acridine), 8.12 (d, 2H acridine, J = 8.7Hz), 8.26 (d, 2H acridine, J = 8.4Hz), 8.26-
8.21 (m, 2H benzyl), 8.87 (s, 1H, CH). Ms, m/z (%): 442(M+ 20.2), 443(5.8), 235(31.5),
234(14.1), 230(40.5), 191(9.5), 123(100), 113(12.2).
5.2Animals and treatment
The experiment used 8 to 9 week-old Swiss albino mice of both sexes, with weight between
22 and 30g. Animals were provided by the Bioterium of the Antibiotics Department of the
Federal University of Pernambuco (UFPE) ; they were kept in a small colony in the animal
house at a temperature of 25 ± 30C, with 12 hour cycles of light and darkness, receiving
standard feed (Purina®) and water as required. The experimental procedure was approved by
the Local Committee for Animal Ethics. (CCB – UFPE). Drugs 3a-3f, were suspended at a
concentration of 10 and 30mg/kg/day, in a solution of 0.5% carboxymethylcelulose (CMC)
and they were administered orally for 15 days to groups of hyperglycaemic Swiss mice (N =
6, three males and three females). Hyperglycaemia was induced by alloxan monohydrate at a
80mg/kg p.o. dose. After a 10 days treatment, mice with a 200-350 mg/dL PG level and a
100-250 mg/dL TG level were selected for further experiments. By comparison, the standard
level was then around 100-130 mg/dL. Blood samples were regularly collected from the
animals after they had been fed between 8h and 10h in the morning, the blood being taken
from the retro-orbital plexus under a light anaesthetic (ethyl ether) one hour after the
administration of the compounds on different days throughout the treatment (1, 3, 10 and 15

106
days). Levels of plasma glucose and triglycerides were analysed using commercially available
kits (LABTEST- Brazil), based on enzymatic methods [15]. Control groups of diabetic and
normoglycaemic animals were treated with the vehicle solution (CMC 0.5% 10mL/kg).
Rosiglitazone 10mg/kg/day was used as standard drug. Effects on the glucose and triglyceride
levels of the compounds, vehicle and rosiglitazone were calculated as a percentage of
reduction, using the formula 1-[(TT/OT)/(TC/OC)] x 100, where TT = treated test on the day;
OT= treated test at day zero; TC = control test on the day; OC = control at day zero, according
to Gurram et al. [16]. The diabetic animals treated with 0.5% CMC were used as controls in
the above formula.
Acknowledgments
The CAPES/COFECUB (Coordenação de Aperfeiçoamento de Pessoal do Ensino
Superior/Comité Français d’Evaluation et de Coopération Universitaire avec le Brésil) and
CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) are gratefully
acknowledged for their support to the join research programme.
References and Notes
[1] A.R. Saltiel, J.M. Olefsky, Diabetes, 45 (1996) 1661-1669.
[2] R.J. Jha, Clin. Exp. Hypertens. 21 (1999) 157-166.
[3] J..M. Lehmann, L.B. Moore, T.A. Smith-Oliver, W.O. Wilkison, T.M. Wilson, S.A.
Kliewer, J. Biol. Chem. 270 (1995) 12953-12956.
[4] A L. Peters, Am. J. Manag. Care 7 (2001) 587-595.
[5] R.E. Law, W.P. Meehan, K. Graf, D.A. Wuthric, W. Coats, J. Clin. Invest. 98 (1996)
1897-1905 .

107
[6] AR. Collions, W.P. Meehan, U. Kintscher, S. Jackson, S. Wakino, G. Noh, et al.,
Arterioscl. Throm. Vasc. Biol. 21 (2001) 365-371.
[7] AC. Li, K.K. Brown, M.J. Silvestre, T.M. Wilson, W. Palinski, C.K. Glass, J. Clin. Invest.
106 (2000) 523-531.
[8] W.M. Phillips, D.J. Currie, Can. J. Chem. 47 (1969) 3137-3141.
[9] H.A.F. Daboun, S.E. Abdou, M.M. Hussein, M.H. Enalgdi, Synthesis (1982) 502-504.
[10] O.P. Shvaika, N.I. Korotkikh, A.Y Chervinskii, N. Artemov, Zh. Org. Khim. 39 (1983)
1533-1542.
[11] O. Tsuge, M. Nishinohara, M. Tashiro, Bull. Chem. Soc. Japan, 36 (1963) 1477-1485.
[12] M. D. Mosher, N.R. Natale, J. Heterocyclic Chem., 32 (1995) 779-781.
13. V.L.M. Guarda, M.A. Pereira, C.A. De Simone, J.C. Albuquerque, S.L. Galdino, J.
Chantegrel, M. Perrissin, C. Beney, F. Thomasson, I.R. Pitta, C. Luu-Duc, Sulfur Letters 26
(2003) 17-27.
14. T.G Silva, F.S.V. Barbosa, S.S.F. Brandão, M.C.A. Lima, S.L. Galdino, I.R. Pitta, J.
Barbe, Heterocycl. Comm. 7 (2001) 523-528
15. D. Braham, P. Trinder, Analyst, 97 (1972) 142-145.
16. R. M. Gurram, R. Chakrabarti, R.K. Vikramadithyan, R.N.V.S. Mamidi, V. Balraju, B.M.
Rajesh, P. Misra, S.K.B. Kumar, B.B. Lohray, V.B. Lohray, R. Rajagopalan, Bioorg. Med.
Chem. 10 (2002) 2671-2680

108
Table 1 - Effect of oral administration of benzylidene-thiazolidinedione derivatives on the
glucose (PG) and triglyceride (TG) levels in mice with alloxan-induced hyperglycaemia with
reference to rosiglitazone.
Compound Dose
(mg/kg/day
p.o.)
% of decrease (*) in
PG (**) at day
_______________________________________
1 3 10 15
TG (**) at day
15
3a
10
30
4
1.5
12
7
17
18.5
19
23
22
48
3b
10
30
0.5
9
10
12.5
10
24.5
19
43
15
26
3c
10
30
4
16.5
1
21
24
29
21
51
38
59
3d
10
30
NE
9
NE
17
17
30
22
35
40
46.5
3e
10
30
3
10
NE
12
23
23
15
41
8
44
3f
10
30
NE
8
7
NE
11
14
9
20
22
33
Rosiglitazone 10 23 24.5 22.5 37 43
(*) standard deviation is less than 0.05 in all cases (**) in mg/dL

109
Table 2 - Effect of oral administration of benzylidene-thiazolidinedione derivatives and rosiglitazone (10mg/kg/day), on the change in body weight and mortality of mice with alloxan-induced hyperglycaemia.
Tested Group
Dose
(mg/kg p.o.)
Body weight (g) ± SD
% %
Initial Final of corporal of mortality (day 15) loss
Normoglycaemic - 27.0±1.0 28.5±1.7 - 0
Diabetic + CMC 10(*) 25.2±0.8 22.4±2.6 11 20
“ + 3a
10
30
25.3±0.6
28.0±0.8
23.3±1.4
24.9±1.1
8
11
0
17
“ + 3b 10
30
26.4±0.4
25.0±0.9
26.2±0.8
21.1±0.6
0.75
16
20
14
“ + 3c 10
30
21.3±0.7
26.4±1.2
21.7±1.6
22.7±2.0
-
14
50
50
“ + 3d 10
30
27.9±2.8
24.0±0.8
23.3±2.7
21.3±1.5
16
11
17
0
“ + 3e 10
30
22.7±1.5
24.4±0.7
20.2±0.9
21.9±0.7
11
10
17
0
“ + 3f 10
30
29.1±3.4
25.3±0.4
27.2±3.0
24.0±0.9
6.5
5
0
0
“ + Rosiglitazone 10 26.5±0.4 27.4±1.3 - 17
* this value is given in mL/kg p.o.

110
NH
S O
ON
S O
O CH2
CH3
N
S O
O CH2
HCH3
R
4-CH3C6H4CH2Cl R-ArCH=(CN)COOC2H5
1 23a - h ( Z )
Fig. 1 Synthetic pathways of benzylidene-thiazolidinediones
1
7
7
8c, d ( Z + E )
8a, b ( Z + E )
654
N
N
S
O
O R
N
N
S O
O
O
R
R
N
S
O
OO
N
S
O
O R
RC6H4COCH2Cl
RC6H4CH2ClNH
S
O
O
NCCH2COOC2H5
N
CN
COOC2H5
N
CHO
PCC
N
CH3
Fig. 2 Synthetic pathways of acridinylidene-thiazolidinediones

111
Synthesis, Biological Evaluation and Molecular Modeling Studies of Arylidene-
Thiazolidinediones with Potential Hypoglycemic and Hypolipidemic Activities
Leite1 , L.F.C.C.; Mourão2 , R.H.V.; Vieira2, E. S.; Lima2, M.C.A. de, Suely Lins Galdino2,
Marcelo Zaldini Hernandes3, Francisco de Assis Rocha Neves4, Stephanie Vidal5, Jacques
Barbe5, Ivan da Rocha Pitta*2
1Departamento de Química - Universidade Católica de Pernambuco, Brasil,.2Departamento de
Antibióticos and 3Ciências Farmacêuticas - Universidade Federal de Pernambuco, CEP
50670-901 Recife, Pernambuco, Brasil, 4Universidade de Brasília, Brasil, 5GERCTOP - UMR
CNRS 6178, Université de la Méditerranée, Faculté de Pharmacie, 13385 Marseille cedex 5,
France.
RECEIVED DATE (to be automatically inserted after your manuscript is accepted if
required according to the journal that you are submitting your paper to)
TITLE RUNNING HEAD: ATZDs with Hypoglycemic and Hypolipidemic Activities
The synthesized arylidene-thiazolidinediones compounds - ATZDs - were evaluated in the
alloxan-induced hyperglycemia in mice. The molecular target was a nuclear receptor PPARγ
(peroxisome proliferator-activated receptor) whose crystallographic structure is available on
the PDB database under code 2PRG. After removing the co-crystallized ligand present in
structure 2PRG, docking studies of ATZDs compounds were carried out using the Autodock
3.0.5 and ADT 1.1 (Auto Docking Tools) programs. We also achieved a correlation between
* [email protected], Departamento de Antibióticos, Universidade Federal de Pernambuco, CEP 50670-901, Recife-PE, Brasil.

112
the binding energy results calculated for the pair ATZD-PPARγ and experimental results of
plasmatic glucose reduction (ED40).
KEYWORDS: arylidene-thiazolidinediones, hypoglycemic, hypolipidemic, molecular
modeling, docking
BRIEFS: The hypoglycemic and hypolipidemic activities of new arylidene-thiazolidinediones
(ATZDs) compounds were evaluated and compared with the docking results obtained from
the molecular modeling studies using the PPARγ and PPARα receptors.
INTRODUCTION
Diabetes mellitus (DM) is a chronic multifactorial metabolic disease characterized by
insulin resistance, hyperglycemia, and often hyperlipidemia. TZDs are known to be insulin
sensitizers and have been developed and clinically used as antidiabetic agents. The maleate of
rosiglitazone (Avandia®), a medicine of the TZDs class, showed significant clinical efficacy
against DM type 2. The disease is associated with obesity, dislipidemia and hypertension
leading to increased cardiovascular risks1. Untreated, type 2 diabetes leads to several chronic
diseases such as retinopathy, nephropathy, and cardiovascular diseases, the latter leading to
increased mortality2. Owing to the forecasted epidemic in DM type 2, the increasing financial
and social costs, and the complicated pathology of the disease, new therapies are needed that
address both insulin resistance and the dislipidemic components of the disease3-5. It was
discovered empirically decades ago that two classes of compounds known as
thiazolidinediones (TZDs) and fibrates possess the ability to lower blood glucose and lipids in
rodent models of insulin resistance and hyperlipidemia, respectively. In humans, the fibrates
are effective at lowering serum triglycerides and raising HDL cholesterol levels, primarily

113
through increased clearance and decreased synthesis of triglyceride-rich VLDL. Fibrates have
been shown to slow the progression of atherosclerosis and reduce the number of coronary
events in secondary prevention studies and in patients with normal levels of LDL cholesterol
and, recently, in diabetic patients2. Thiazolidinediones (TZDs) have been shown to reduce
plasma glucose, lipid, and insulin levels, and can be used for treatment of DM type 2 6-7. The
target of the TZDs has been identified as peroxisome proliferator-activated receptor γ
(PPARγ) and the glucose-lowering action of the TZDs has been shown to be closely related to
their PPARγ agonist activity. TZDs enhance adipocyte differentiation and increase the
insulin-sensitivity of tissues8. Insulin sensitizers PPARγ agonists, such as pioglitazone and
rosiglitazone (Chart 1) have a range of clinical effects including improvement of insulin
sensitivity and glucose tolerance and lowering of blood glucose levels in type 2 diabetic
patients9,10. Several structures of the PPARγ ligand-binding domain (LBD) have been
obtained using X-ray crystallography and have been reported in the literature. Some examples
include the structures of the apo-PPARγ LDB, a ternary complex of LDB with a TZD
(rosiglitazone) and an 88 amino acid fragment of the coactivator SRC1 11,12 and LDB
complexes with a partial agonist GW0072 13. The identification of the nuclear peroxisome
proliferator activated receptor γ (PPARγ) and α (PPARα) as the primary targets for the
normoglycemic TZDs and the lipid lowering fibrates, respectively, has provided new
opportunities for the identification of novel compounds for the treatment of type 2 diabetes14.
This present work describes the synthesis and gives the structural characteristics of 12
arylidene-thiazolidinediones (ATZDs) derivatives of 5-benzylidene-3-(4-methyl-benzyl)-
thiazolidine-2,4-dione substituted on the benzylidene moiety15. These compounds were
evaluated after oral administration at different doses in hyperglycemic mice with alloxan-
induced diabetes. The plasmatic glucose (PG) and the triglyceride levels (TG) were measured
and compared with rosiglitazone as a reference drug. In spite of the pharmacological

114
mechanism was not determined for these ATZDs, based upon the similarity between these
molecule and ATZDs, our results strongly support the hypothesis that these compounds could
be acting through PPARγ receptor. The structure of the complexes formed between these
compounds and PPARγ and PPARα LDB were modelled and their interactions analysed,
using Autodock 3.0.5 and ADT 1.1 (Auto Docking Tools) programs.
N O NH
S
OO N N
ONH
S
O
O
(a) (b)
Chart 1: Pioglitazone (a) and rosiglitazone (b)
EXPERIMENTAL SECTION
The 5-arylidene-3-(4-methyl-benzyl)-thiazolidine-2,4-diones - ATZDs, 1-12, were prepared
by nucleophilic addition of the 3-(4-methyl-benzyl)-thiazolidine-2,4-dione to selected aryl-
substituted ethyl-(2-cyano-3-phenyl)-acrylates. The synthetic pathways are illustrated in
Scheme 1. Thiazolidine-2,4-dione, was N-(3)-alkylated in the presence of potassium
hydroxide, which enables the thiazolidine potassium salt to react with benzyl halide in a hot
alcohol medium.
HNS
O
OKOH
4-CH3C6H4CH2Cl NS
O
O
NS
O
O
H
piperidine
RArCH=(CN)COOC2H5
R
(Z)1 - 12

115
Scheme 1. Synthetic pathways of arylidene-thiazolidinediones
Arylidene-thiazolidinediones were isolated in a single isomer form. X-ray crystallographic
studies and 13C NMR have demonstrated the preferred Z configuration for 5-arylidene-
thiazolidinones16-18. Melting points were measured in a capillary tube on Buchi (or Quimis)
apparatus. Thin layer chromatography was performed on silica gel plates Merck 60F254.
Infrared spectra of 1% KBr pellets were recorded on a Perkin Elmer 1310 spectrometer for
2,6,7 compounds or Bruker IFS66 spectrometer for 8,12. 1H NMR spectra were recorded on a
Bruker AC 300 P spectrophotometer in DMSO-d6 as solvent, with tetramethylsilane as
internal standard. Mass spectra were recorded on Delsi-Nermag R 1010 C spectrometer. The
published chemical data on 1,3-5,9-11 19, are not reported here.
(Z) 5-Benzylidene-3-(4-methyl-benzyl)-thiazolidine-2,4-diones: general procedure.
A mixture of 3-(4-methyl-benzyl)-thiazolidine-2,4-dione, (0.59 g, 2.5 mmol) and ethyl-(2-
cyano-3-phenyl)-acrylate (2.7 mmol) is refluxed for 2-6 h in absolute ethanol (20 mL) with
added piperidine (0.25 mL). After cooling, precipitates are purified by column
chromatography or crystallized in suitable solvents.
(Z) 5-(4-Hydroxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 2.
C18H15NO3S, yield: 38%. Mp: 194-196°C. TLC chloroform Rf: 0.68. IR cm-1 (KBr): ט
3325, 1720, 1655, 1575, 1510,1365,1330, 1130, 825. 1H NMR (δ ppm, DMSO-d6): 2.25 (s,
CH3), 4.76 (s, NCH2), 7.85 (s, CH), 7.16 (m, 4H benzyl), 6.92 (d, 2H benzylidene, J=8.1 Hz),
7.48 (d, 2H benzylidene, J=8.4 Hz), 10.4 (sl, OH). Ms, m/z (%): 325(M+. 100), 326(29.1),
312(6.3), 105(61.7).
(Z) 5-(3-Chloro-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 6.
C18H14ClNO2S, yield: 50%. Mp: 143-145°C. TLC n-hexane : ethyl acetate (70:30) Rf: 0.8.
IR cm-1 (KBr): 920 ,1135 ,1320 ,1370 ,1420 ,1605 ,1675 ,1730 ,2935 ,3040 ט. H1NMR (δ

116
ppm, DMSO-d6): 2.27 (s, CH3), 4.79 (s, CH2), 7.16 (d, 2H, benzyl, J=8.4 Hz), 7.21 (d, 2H,
benzyl, J=8.4 Hz), 7.58 (m, 3H, benzylidene), 7.73 (s, 1H, benzylidene), 7.96 (s, CH). Ms,
m/z (%): 343(M+. 55.4), 344(11.6), 345(16.6),105(100).
(Z) 3-(4-Methyl-benzyl)-5-(4-methyl-benzylidene)- thiazolidine-2,4-dione 7.
C19H17NO2S, yield: 45%. Mp: 139-140°C. TLC n-hexane : ethyl acetate (70:30) Rf: 0.78. IR
cm-1 (KBr): 805 ,1135 ,1320 ,1370 ,1425 ,1590 ,1675 ,1730 ,2930 ,3010 ט. H1NMR (δ ppm,
DMSO-d6): 2.27 (s, CH3), 2.36 (s, CH3), 4.79 (s, CH2), 7.15 (d, 2H, benzyl, J=8.1 Hz), 7.2 (d,
2H, benzyl, J=8.4 Hz), 7.36 (d, 2H, benzylidene, J=8.1 Hz), 7.53 (d, 2H, benzylidene, J=8.1
Hz), 7.93 (s, CH). Ms, m/z (%): 323(M+. 100), 324(18.7), 325(7.3), 105(45.3), 91(6.3).
(Z) 5-(3-Bromo-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 8.
C18H14BrNO2S, yield: 48%. Mp: 109-111°C. TLC benzene : ethyl acetate (95:5) Rf: 0.9. IR
cm-1 (KBr): 758 ,1325 ,1376 ,1427 ,1600 ,1692 ,1743 ,2911 ט. H1NMR (δ ppm, DMSO-d6):
2.28 (s, CH3), 4.79 (s, CH2), 7.16 (d, 2H, benzyl, J=8.1 Hz), 7.22 (d, 2H, benzyl, J=8.1 Hz),
7.4-7.45 (m, 1H, benzylidene), 7.54-7.6 (m, 2H, benzylidene), 7.82 (d, 1H, benzylidene, J=8.4
Hz), 8.02 (s, CH). Ms, m/z (%): 387(M+. 34.6), 389(21.6), 308(25.7), 212(6.1), 132(19.8),
105(100), 89(80.4), 77(14.4).
(Z) 3-(4-methyl-benzyl)-5-(4-nitro-benzylidene)- thiazolidine-2,4-dione 12.
C18H14N2O4S, yield: 39%. Mp: 189-191°C. TLC n-hexane : ethyl acetate (70:30) Rf: 0.56.
IR cm-1 (KBr): 840 ,1509,1380,1148 ,1614 ,1692 ,1743 ,2360 ט. H1NMR (δ ppm, DMSO-d6):
2.28 (s, CH3), 4.81 (s, CH2), 7.16 (d, 2H, benzyl, J=8.1 Hz), 7.22 (d, 2H, benzyl, J=7.8 Hz),
7.9 (d, 2H, benzylidene, J=9 Hz), 8.35 (d, 2H, benzylidene, J=9 Hz), 8.08 (s, CH). Ms, m/z
(%): 354(M+. 100), 355(13.5), 326(5.6),105(35.7), 89 (6.6).

117
BIOLOGICAL ACTIVITY
The effects of the new ATZDs summarized and characterized were evaluated using an
experimental model of diabetes induced by alloxan. The biological activity in vivo of the
ATZDs was evaluated with daily doses of 5 to 40mg/kg administered orally for 15 days of
treatment, and specific reduction in levels of PG (plasmatic glucose) and TG (plasmatic
triglyceride) was measured for the diabetic groups treated with the ATZDs compared with the
diabetic group treated with vehicle (CMC 0,25%). Rosiglitazone was used as a reference drug
at doses of 5 to 30mg/kg/day. Hyperglycemia was induced by alloxan monohydrate at a
80mg/kg p.o. dose. After a 10 days treatment, mice with a 200-350 mg/dL PG level and a
100-250 mg/dL TG level were selected for further experiments. Blood samples were regularly
collected from the animals after they had been fed between 8h and 10h in the morning, the
blood being taken from the retro-orbital plexus under a light anaesthetic - ethyl ether, one
hour after the administration of the compounds on different days throughout the treatment (1,
3, 10 and 15 days). Levels of plasmatic glucose and triglycerides were analysed using
commercially available kits (LABTEST- Brazil), based on enzymatic methods.
The hypoglycemic and hypolipidemic effects after oral administration of the ATZDs in
mice with alloxan-induced diabetes are summarized in Table 1 and represented in graphs 1
and 2, which shows the results of the biochemical parameters in terms of percentage reduction
in GP and TG levels, obtained from samples of blood collected before and after 15 days of
treatment, the animals treated with ATZDs, rosiglitazone and vehicle. The values represent on
average ± standard error (N=6) for each experimental group. With the exception of 5-(3-
chloro-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 6, the dose of 10mg/kg/dia
reduced the levels of GP significantly (p<0.05) after 15 days of treatment.

118
Table 1. Effect of the dose-response of ATZDs: 1-12 (Scheme 1) in mice with alloxan-
induced diabetes
# R Dose
(mg/kg/day)
% Reduction
PG (mg/dL)
% Reduction
TG (mg/dL)
10 18.7 p<0.002 NS
30 22.8 p<0.002 48 p<0.001
1
4-Cl
40 31.5 p<0.02 27.1 p<0.01
10 19.9 p<0.008 NS
30 37.3 p<0.04 NS
2
4-OH
40 37.2 p<0.001 NS
10 19.2 p<0.05 14.8 p<0.05
20 47 p<0.001 20 p<0.01
3
2-Cl
30 43.3 p<0.05 26 p<0.01
5 27.2 p<0.001 32.2 p<0.01
10 20.6 p<0.07 37.6 p<0.01
4
4-OCH3
30 50.8 p<0.00003 58.8 p<0.04
10 21.8 p<0.01 40 p<0.01
30 34.6 p<0.001 46.5 p<0.01
5
2.4-OCH3
40 20.1 p<0.025 NS
6 3-Cl 10 NS NS
7 4-CH3 10 16.3 p<0.025 41.7 p<0.020
8 3-Br 10 21.3 p<0.001 17.6 p<0.05
10 14.9 p<0.01 NS
20 35.8 p<0.025 17.4 p<0.01
9
4-N(CH3)2
30 40.8 p<0.001 44.1 p<0.001
10 9.37 p<0.05 43.6 p<0.05

119
20 27.7 p<0.001 45.2 p<0.025 10 4-C6H5CH2O
30 20.2 p<0.0001 42.1 p<0.01
11 4-F 30 13.5 p<0.05 NS
12 4-NO2 30 11.42 p<0.05 7.1 p<0.05
5 26.3 p<0.001 15.9 p<0.05
10 36.7 p<0.001 43.3 p<0.001
Rosiglitazone
30 11.6 p<0.05 6.4 p<0.05
Diabetic + CMC
Average of 11 groups with n = 6
0.1mL/10g
T0 462.4±25
T15 474.9±22.3
NS
T0 209.6±31.3
T15 199.8±21.5
NS Normoglycemic +
CMC
Average of 11 groups with n = 6
0.1mL/10g
T0 126.5±7.6
T15 124.2±6.8
NS
T0 118.2±10.2
T15 111.4±9
NS
Normoglycemic
n = 6
-
T0 129.5±5
T15 131±7
NS
T0 128.7±7.3
T15 125±14
NS
Diabetic mice treated with the ATZDs orally for 15 days with the doses mentioned. Reduction percentage calculated using the equation: 1-[(TT/0T)/(TC/0C)] x 100; TT: treated test on the day; 0T: treated test at the day zero; TC: control of the day, 0C: control of the day zero; NS: not significant, T 0: time zero, T 15: after 15 days of treatment. Each value represents on average a ± standard error of n = 6 animals. Differences between time zero and 15 days of treatment were considered significant for p≤0.05.
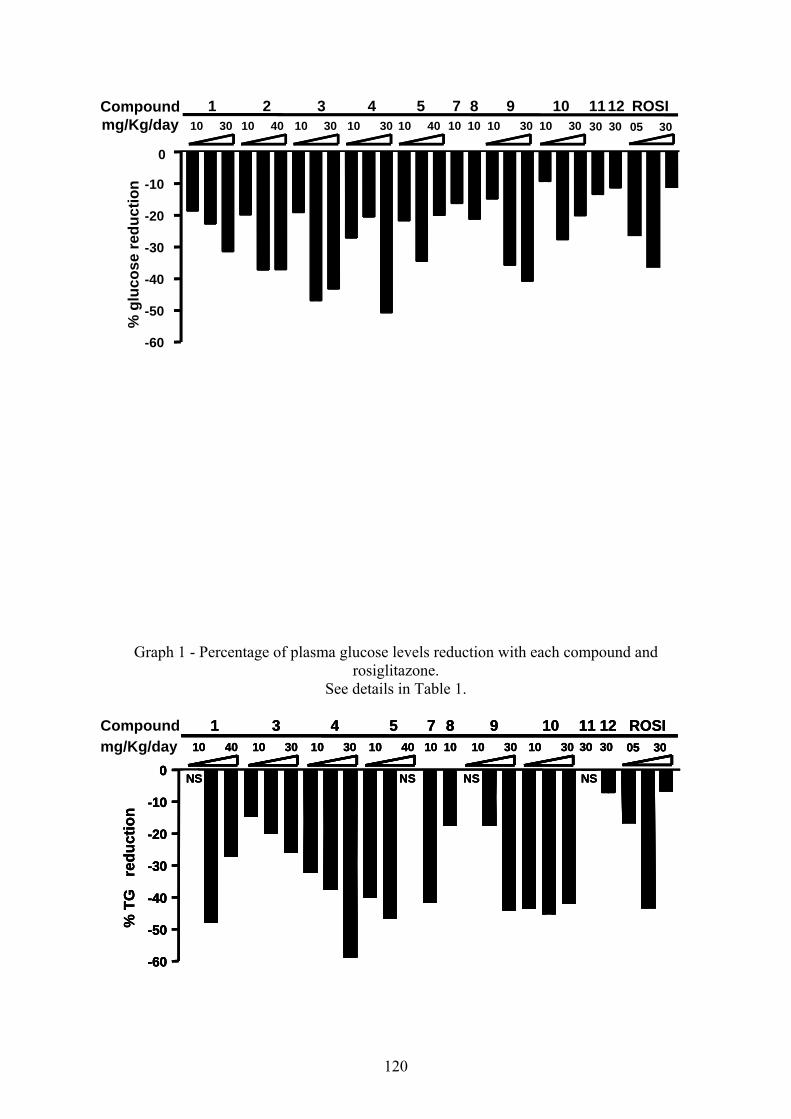
120
Graph 1 - Percentage of plasma glucose levels reduction with each compound and
rosiglitazone. See details in Table 1.
-60
-50
-40
-30
-20
-10
0%
glu
cose
redu
ctio
nCompoundmg/Kg/day 10 30 10 40 10 30 10 30 10 30 10 3010 40
1 2 3 4 5 7 8 9 10 10 10
111230 30 05 30
ROSI
Compoundmg/Kg/day
-60
-50
-40
-30
-20
-10
0
% T
G
redu
ctio
n
10 40 10 30 10 30 10 40 10 30 10 3010 10
1 3 4 5 7 8 9 10 11 12 ROSI30 30
NS NS NS NS
05 30
-60
-50
-40
-30
-20
-10
0
% T
G
redu
ctio
n
-60
-50
-40
-30
-20
-10
0
-60
-50
-40
-30
-20
-10
0
% T
G
redu
ctio
n
10 40 10 30 10 30 10 40 10 30 10 3010 10
1 3 4 5 7 8 9 10 11 12 ROSI30 30
NS NS NS NS
05 3010 40 10 30 10 30 10 40 10 30 10 3010 10
1 3 4 5 7 8 9 10 11 12 ROSI30 30
NS NS NS NS
05 30

121
Graph 2 - Percentage of plasma triglycerides (TG) reduction with each compound and
rosiglitazone (ROSI). See details in Table 1. Observation: results with compound 2 was not
shown because it was not significant.
THEORETICAL CALCULATIONS
The structures and conformational analysis of arylidene-thiazolidinediones 1-12 compounds
were obtained by the application of AM120 method available in BioMedCache software
[BioMedCAChe version 6.1, Copyright ©2000-2003 Fujitsu Limited, Copyright©1989-2000,
Oxford Molecular Ltd., http:///www.CACheSoftware.com], using internal default settings for
convergence criteria. The results obtained in these calculations confirm the greater stability of
the Z isomer in all cases.
The docking analysis was firstly carried out on the PPARγ binding site (structure 2PRG11,
from RCSB Protein Data Bank, http://www.rcsb.org/pdb), where the residues were bonded
more closely to the rosiglitazone agonist, co-crystallized with PPARγ. In this crystal structure,
the Ligand Binding Domain (LBD) forms a homodimer in which both monomers have nearly
identical Cα conformations. The structure of the “A” monomer of the LBD homodimer was
chosen as the target for docking studies. The rosiglitazone agonist was extracted from the
complex, and, during the calculations, the active-site was determined within a cubic box
centered on the co-crystallized ligand (coordinates: X= 50.140; Y=-38.202; Z=19.559) with
sides of 22.5 Å in length, thereby ensuring that the active site region was covered. Within this
pre-defined region, the grids of probe atom interaction energies were computed with a default
resolution of 0.375 Å and the ligand probes were then docked by Genetic Algorithm followed
by a Local Search procedure (GA_LS), also know as a Lamarckian Genetic Algorithm
(LGA)21, and the 50 lowest energy structures were stored for further analysis.

122
The docking analysis of arylidene-thiazolidinediones 1-12 compounds and rosiglitazone
was carried out by means of the AutoDockTools22 (ADT) v1.1 and Autodock v3.0.5 program
[AutoDock, AutoGrid, AutoTors, Copyright©1991-2000, The Scripps Research Institute,
http://www.scripps.edu/mb/olson/doc/autodock], and using the internal default parameters for
all the variables, except for the number of docking runs (50), the maximum number of energy
evaluations on Genetic Algorithm-GA (25000000) and the maximum number of generations
in GA (5000). Preliminary calculations indicate that these tree specific parameter
modifications give a significant improvement in the results obtained by the docking
procedure, contributing to more stables conformations of the ligands. The active site was
treated as a rigid molecule, whereas the ligands were treated as being flexible, i.e., all non-
ring torsions are allowed (active). This approach seems to be coherent with the structural
similarity observed between rosiglitazone and the arylidene-thiazolidinediones 1-12
compounds.
The docking procedure for the PPARα binding site basically follows the same setup shown
for PPARγ. The “A” chain of the structure 1K7L23 (RCSB Protein Data Bank) was used as the
target for docking in this case. The GW409544 agonist co-crystallized with PPARα in
structure 1K7L was used to define the active site, centered at coordinates (X=-17.866; Y=-
13.599 ; Z=-3.726).
RESULTS AND DISCUSSION
The most stables docking solutions for the arylidene-thiazolidinediones 1-12 compounds
complexed with the PPARγ LBD and PPARα LBD are presented in Table 2, along with the
Free Energy of Binding (ΔG) values.

123
Table 2. Docking results for arylidene-thiazolidinediones (ATZDs) ligands in PPARγ and
PPARα
PPARγ PPARα
ATZD #
R Free Energy of Binding (Kcal/mol)
1 4-Cl -8.36 -8.56
2 4-OH -8.90 -8.13
3 2-Cl -8.61 -7.95
4 4-OCH3 -8.36 -7.63
5 2.4-OCH3 -8.22 -7.70
6 3-Cl -8.61 -8.49
7 4-CH3 -8.39 -8.44
8 3-Br -8.73 -8.01
9 4-N(CH3)2 -8.85 -8.69
10 4-C6H5CH2O -9.58 -9.26
11 4-F -8.22 -8.26
12 4-NO2 -10.45 -8.62
Rosiglitazone -10.22 -13.49
In the crystal structures of the rosiglitazone at the LBD of PPARγ, the thiazolidinic ring
establishes several specific interactions with neighboring amino acids of the LBD. These
interactions include hydrogen bonding with amino acids H323, H449 and Y473, as reported
in the literature1,8.
The docking studies started with rosiglitazone, for which the binding mode has been
determined experimentally8. Autodock was successful in reproducing the binding position for
rosiglitazone, showing an RMS deviation of 0.8 Å in comparison with the experimental
geometry co-cristalized in PPARγ and the same patterns for hydrogen bonding.

124
The binding profile of the 5-arylidene-3-benzyl-thiazolidine-2,4-dione was compared with
the profile of the rosiglitazone molecule. The ligand 5-(4-hydroxy-benzylidene)-3-(4-methyl-
benzyl)-thiazolidine-2,4-dione 2 interacts mainly with PPARγ trough the formation of two
hydrogen bonds between the para hydroxyl group of the benzylidene ring of the ligand
molecule and the histidine H449(A) and the glutamine Q286(A) residues, respectively (Figure
1). The histidine (H449) residue also interacts with rosiglitazone in crystal structure (2PRG)
trough hydrogen bonding8.
Figure 1. Important hydrogen bonds between (Z) 5-(4-hydroxy-benzylidene)-3-(4-methyl-
benzyl)-thiazolidine-2,4-dione 2 and PPARγ.

125
In Figure 2 the ligand (Z) 5-(4-methoxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-
dione 4 shows a hydrogen bond between the oxygen atom of the thiazolidine ring in 4-
position and the histidine His440(A) residue of the PPARα receptor. The LIGPLOT
program24 was used to produce Figures 1 and 2.
Figura 2. Hydrogen bond between (Z) 5-(4-methoxy-benzylidene)-3-(4-methyl-benzyl)-
thiazolidine-2,4-dione 4 and the His440(A) residue of PPARα.
Figure 3 shows the ligand (Z) 5-(4-hydroxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-
2,4-dione 2 (ball&stick model) interacting with the His449(A) and Gln286(A) PPARγ
residues (strong line model) through hydrogen bonding, at measured distance of 1.897Ǻ and
2.162Ǻ, respectively. The crystallographic structure of the rosiglitazone ligand (weak line

126
model) shows interactions with the Ser289(A) and His323(A) residues through hydrogen
bonding at distances of 1.932Ǻ and 1.859Ǻ respectively.
(a) (b)
Figure 3. Front (a) and back (b) view of hydrogen bonds between the ligands (Z) 5-(4-
hydroxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 2 (ball&stick model) and
co-crystallized rosiglitazone (weak line model), and PPARγ (strong line model).
A correlation was observed between the ATZDs (1-4, 9) free energy of binding (PPARγ)
and the values of –log (ED40) for PG after 15 days of treatment (Table 3 and Figure 4). This
correlation corroborates the expectation that as more stable is the ligand-receptor complex, or
smaller is the free energy of binding, greater is the value of -log (ED40) or greater is the
potency of the respective molecule.

127
Table 3. Effective dose for a 40% reduction (ED40) of GP and TG levels for ATZD’s 1-4 and
9 in mice with alloxan-induced diabetes and the Free Energy of Binding (ΔG) for PPARγ and
PPARα receptors.
ATZD
#
PM ED40 PG
10-5 mol/L
ΔG (Kcal/mol)
PPARγ
ED40 TG
10-5 mol/L
ΔG (Kcal/mol)
PPARα
1 343 6.9 -8.36 5.8 -8.56
2 325 4.2 -8.90 - -8.13
3 343 3.3 -8.61 8.8 -7.95
4 339 5.6 -8.36 0.97 -7.63
9 352 4.3 -8.85 5.8 -8.69

128
Figure 4. Correlation between –log (ED40) for the PG, with ED40 in mol/l, of 1-4 and 9
compounds and the PPARγ free energy of binding. The point labels represent the “R” group
for each molecule (see Table 1).
The effective dose (mg/kg/day) for a reduction of 40% (ED40) in the levels of GP and TG
for 1-4 and 9, compounds was calculated from the dose-response curve for the three doses.
The compound 3 (2-Cl) showed the highest potency to reduce the glucose levels. It was 2.09
times more potent than compound 1, 1.69, 1.27 and 1,30 more potent than compounds 4 (4-
OCH3), 2 (4-OH) and 9 (4-N(CH3)2) respectively. However, the compound 4 showed the best
efficacy since it reduced the blood glucose levels to 50.8% from baseline, a higher value than
it was observed with rosiglitazone treatment (36.7 %). We did not analyze whether the
hypoglycemic effect of our compounds were derived from a PPAR agonist effect, since we
did not evaluated their direct effect or binding affinity to PPARγ receptor. However, since
these ligands show a very similar chemical structure to rosiglitazone, our results strongly
suggested that these compounds should have some effect through PPAR receptor. Preliminary
transcription assay, performed U937 cells where we co-transfected with PPARγ expression
vector together with a PPARγ response element driving a luciferase reporter gene showed that
these compounds have an agonist PPARγ effect (not shown). Further experiments are going
on to address the PPARγ and PPARα binding affinity together with a dose-response curve in
transcription reporter gene assay and PPAR� crystal structure with these ligands.
CONCLUSIONS
After the analysis of the docking and biological data, it was possible to conclude that the
presence of the chlorine in 4 or in 2-position in the phenyl ring as observed in (Z) 5-(4-chloro-

129
benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione 1 and (Z) 5-(2-chloro-benzylidene)-
3-(4-methyl-benzyl)-thiazolidine-2,4-dione 3 compounds seems to play an important role in
explaining the hypoglycaemic and hypolipidemic activity. The chlorine in the 3-position plays
the opposite role, as observed in (Z) 5-(3-chloro-benzylidene)-3-(4-methyl-benzyl)-
thiazolidine-2,4-dione 6. At a dose of 10mg/kg/dia, the chlorine in the 2-position reduces the
levels of GP by 19.2%, in the 4-position by 18.7% and in 3-position it does not show
significant results. It was observed that, generally speaking, that the radicals present in the
arylidene ring contribute significantly to its biological activity, while the ATZDs with
electrons donor groups in the 4-position produce a significant reduction in glycose levels.
Further efforts of our group will be done in such a way to continue the improvement of the
hypoglycemic and hypolipidemic activities of these ATZD class of compounds that are being
synthesized and modelled in our laboratory.
ACKNOWLEDGMENT
We thank the CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico,
Brasil) and the CAPES/COFECUB (Fundação Coordenação de Aperfeiçoamento de Pessoal
de Ensino Superior/Comité Français d’Évaluation de la Coopération Universitaire avec le
Brésil) for their support.
REFERENCES
(1) Ebdrup, S.; Pettersson, I.; Rasmussen, H. B.; Deussen, H-J.; Jensen, A. F.; Mortensen, S.
B.; Fleckner, J.; Pridal, L.; Nygaard, L.; Sauerberg, P. Synthesis and biological and structural

130
characterization of the dual-acting peroxisome proliferator-activated receptor α/γ agonist
ragaglitazar, J. Med. Chem. 2003, 46, 1306-1317.
(2) Sauerberg, P.; Pettersson, I.; Jeppesen, L.; Bury, P.S.; Mogensen, J.P.; Wassermann, K.;
Brand, C.L.; Sturis, J.; Woldike, H.F.; Fleckner, J.; Andersen, A.S.; Mortensen, S.B.;
Svensson, L.A.; Rasmussen, H.B.; Lehmann, S.V.; Polivka, Z.; Sindelar, K.; Panajotova, V.;
Ynddal, L.; Wulff, E.M. Novel tricyclic-alpha-alkyloxyphenylpropionic acids: dual
PPARalpha/gamma agonists with hypolipidemic and antidiabetic activity. J Med Chem. 2002,
45, 789-804.
(3) Cobb, J.; Dukes, I. Recent advances in the development of agents for treatment of type 2
diabetes. Annu. Rep. Med. Chem. 1998, 33, 213-221.
(4) Grover, S.A.; Coupal, L.; Zowall, H.; Dorais, M. Cost–effectiveness of treating
hyperlipidemia in presence of diabetes. Circulation 2000, 102, 722-727.
(5) Balley, C.J. Potencial new treatments for type 2 diabetes. Trends in Pharmacological
Sciences 2000, 21, 259-265.
(6) Day, C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet. Med. 1999, 16,
179-792.
(7) Spiegelman, B.M. PPARγ: adipogenic regulator and thiazolidinedione receptor.
Diabetes 1998, 47, 507-514.
(8) Iwata, Y.; Miyamoto, S.; Takamura, M.; Yanagisawa, H.; Kasuya, A. Interaction
between peroxisome proliferators-activated receptor γ and its agonist: docking study of
oximes having 5-benzyl-2,4-thiazolidnedione. Journal of Molecular Graphics and Modelling
2001, 19, 536-542.

131
(9) Boyle, P.J.; King, A. B.; Olansky, L.; Marchetti, A.; Lau, H.; Magar, R.; Martin, J.
Effects of pioglitazone and rosiglitazone on blood lipid levels and glycemic control in patients
with type 2 diabetes mellitus: a retrospective review of randomly selected medical records.
Clin. Ther. 2002, 24, 378-396.
(10) Chilcott, J.; Tappenden, P.; Jones, M.L.; Wight, J.P. A systematic review of the clinical
effectiveness of pioglitazone in the treatment of type 2 diabetes mellitus. Clin. Ther. 2001, 23,
1792-1823.
(11) Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.;
Rosenfeld, M.G.; Willson, T.M.; Glass, C. Ligand binding and co-activator assembly of the
peroxisome proliferator-activated receptor-γ. Nature, 1998, 395, 137-143.
(12) Uppenberg, J.; Svensson, C.; Jaki, M.; Bertilsson, G.; Jendeberg, L.; Berkenstam, A.
Crystal structure of the ligand binding domain of the human nuclear receptor PPARγ. J. Biol.
Chem. 1998, 273, 31108-31112.
(13) Oberfield, J. L.; Collins, J. L.; Holmes, C. P.; Goreham, D. M.; Cooper, J. P.; Cobb, J.
E.; Lenhard, J. M.; Hull-Ryde, E. A.; Mohr, C. P.; Blanchard, S. G.; Parks, D. J.; Moore, L.
B.; Lehmann, J. M.; Plunket, K.; Miller, A. B.; Milburn, M. V.; Kliewer, S. A.; Willson, T.
M. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte
differentiation. P. Natl. Acad. Sci. USA 1999, 96, 6102-6106.
(14) Willson, T.M.; Cobb, J.E.; Cowan, D.J.; Wiethe, R.W.; Correa, I.D.; Prakash, S.R.;
Beck, K.D.; Moore, L.B.; Kliewer, S.A.; Lehmann, J.M. The structure-activity relationship
between peroxisome proliferator-activated receptor gamma agonism and the
antihyperglycemic activity of thiazolidinediones. J. Med. Chem. 1996, 39, 665-668.

132
(15) Pitta, I.R.; Galdino, S.L.; Lima, M.C.A.; Barbe, J. Compostos
arilidenotiazolidinadiônicos com atividade hipoglicêmica. Brazilian patent PI-0300997-1,
Revista da Propriedade Intelectual 2003, nº 1695.
(16) Guarda, V.L.M.; Pereira, M.A.; De Simone, C.A.; Albuquerque, J.C.; Galdino, S.L.;
Chantegrel, J.; Perrissin, M.; Beney, C.; Thomasson, F.; Pitta, I.R.; Luu-Duc, C. Synthesis
and structural study of arylidene thiazolidine and benzothiazine compounds. Sulfur Letters
2003, 26, 17-27.
(17) Tan, S.F.; Ang, K.P.; Fong, Y.F. (Z)- and (E)-5-Arylmethylenehydantoins:
spectroscopic properties and configurayion assignment. J. Chem. Soc. Perkin Trans. II 1986,
12, 1941-1944.
(18) Albuquerque, J.F.C.; Albuquerque, A.; Azevedo, C.C.; Thomasson, F.; Galdino, S.L.;
Chantegrel, J.; Catanho, M.T.J.; Pitta, I.R.; Luu-Duc, C. Substituted thiazolidinediones and
thio-imidazolidinones: Synthesis, structural study and pharmacological activity. Pharmazie
1995, 50, 387-389.
(19) Mourão, R.H.; Silva, T.G.; Soares, A.L.M.; Vieira, E.S.; Santos, J.N.; Lima, M.C.A.;
Lima, V.L.M.; Galdino, S.L.; Barbe, J.; Pitta, I.R. Synthesis and biological activity of novel
acridinylidene and benzylidene thiazolidinediones. Eur. J. Med. Chem. 2005, June (in press).
(20) Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A new general
purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902-3909.
(21) Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.;
Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical
binding free energy function. Journal of Computational Chemistry 1998, 19, 1639-1662.

133
(22) Sanner, M. F. Python: a programming language for software integration and
development. Journal of Molecular Graphics and Modelling 1999, 17, 57-61.
(23) Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.;
Oplinger, J.A.; Kliewer, S.A.; Gampe, Jr. R.T.; Mckee, D.D.; Moore, J.T.; Willson, T.M.
Structural determinants of ligand binding selectivity between the peroxisome proliferator-
activated receptors, P. Natl. Acad. Sci. USA 2001, 98, 13919-13924.
(24) Wallace, A. C.; Laskowski, R. A.; Thornton, J. M. LIGPLOT: a program to generate
schematic diagrams of protein-ligand interactions. Protein Engineering 1995, 8, 127-134.

134
Synthesis and anti-inflammatory activity of some new N and S-alkylated arylidene-thioxo-imidazolidinones
L.C. SANTOS*, R.H.V. MOURÃO*, F.T. UCHOA*, T.G. SILVA*, D.J.N. MALTA*, R.O.MOURA*, M.C.A. LIMA*, S.L. GALDINO*, I.R. PITTA*, and J. BARBE** * Universidade Federal de Pernambuco, Departamento de Antibióticos 50.670-901 Recife, Brasil
** GERCTOP – UMR CNRS 6178, Université de la Méditerranée, Faculté de Pharmacie, 13385 Marseille cedex 5, France. Abstract: New arylidene-thioxo-imidazolidinones and S-alkylated arylidene-imidazolidinone
derivatives were prepared from substituted 2-thioxo-imidazolidin-4-one by nucleophilic
addition of cyanoacrylates. N and S-alkylation was achieved treating 5-arylidene-2-thioxo-
imidazolidin-4-ones with benzyl or phenyloxoethyl chlorides under alkaline conditions. The
anti-inflammatory activity of the synthesized imidazolidines was evaluated by the
carrageenin-induced paw edema test.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used in the treatment of
inflammatory diseases. The main limitations in using NSAIDs consist in their side effects,
including gastrointestinal ulceration and renal toxicity. Thiazolidines and bioisosteric
imidazolidines are known to carry anti-inflammatory activity as it was demonstrated with
some 5-arylidene-4-thioxothiazolidinones and 5-arylidene-thiazolidine-2,4-diones substituted
at position 3 by 4-chlorobenzyl group [1]. Synthesis of some 5-arylidene-
thioxoimidazolidinones and thioxothiazolidinones substituted at position 3 by a benzyl group
or a phenyloxoethyl one, was previously reported [2-4]. Synthesis and physicochemical data
of new 3-benzyl-5-benzylidene-2-thioxo-imidazolidin-4-ones, 10-15, or 5-benzylidene-3-(2-
phenyl-2-oxo-ethyl)-2-thioxo-imidazolidin-4-ones, 16-21, and 5-benzylidene-3-(4-

135
fluorobenzyl)-2-(4-bromobenzylsulfanyl)-3,5-dihydro-imidazol-4-ones, 22-23, 2-[(4-
bromophenyl)-2-oxo-ethylsulfanyl]-3-(4-fluorobenzyl)-5-(4-methoxybenzylidene)-3,5-
dihydro-imidazol-4-one, 24, or 5-benzylidene-3-[2-(4-bromophenyl)-2-oxo-ethyl]-2-[2-(4-
bromophenyl)-2-oxo-ethylsulfanyl]-3,5-dihydro-imidazol-4-ones, 25, are now reported
(Figure 1). These compounds were obtained from various (2-cyano-3-phenyl)-ethyl acrylates,
by a nucleophilic Michael addition on the 2-thioxo-imidazolidin-4-one 1. The 5-benzylidene-
2-thioxo-imidazolidin-4-ones 2-9, formed were S- and N-alkylated at position 2 or 3 with
benzyl or phenyloxoethyl chlorides under alkaline conditions.
R1CH=C(CN)COOCH2CH3
4BrC6H4COCH2Br
N
N
S
O H
H
N
N
S
O H
H
H
N
N
S
O H
H
RR
R1
R1C6H4CH2BrN
N
S
OH
H
O
Br
R1
R
R
N
N
O
H
O
S
O
Br
BrN
N
O
H
S
R2C6H4CH2Br
R2
4BrC6H4COCH2Br
1
2 R = H
6 R = 4OCH3
8 R = 3,4,5OCH3
3 R = 2Br4 R = 2Cl
7 R = 2,4OCH3
12 R=2Br, R1=4NO2
10 R=H, R1=4F 11 R=4OCH3, R1=4F
13 R=2Cl, R1=4NO214 R=3Cl, R1=4NO215 R=2,4OCH3, R1=4NO2
17 R=4OCH318 R=2,4OCH319 R=4Br20 R=3,4,5OCH3
16 R=3Cl
21 R=H
10-15
16-212-9
22-24 25
22 R=H, R1=4F, R2=4Br, n = CH2
23 R=4OCH3, R1=4F, R2=4Br, n = CH2
5 R = 3Cl
9 R = 4Br
(n)
24 R=4OCH3, R1=4F, R2=4Br, n = CH2CO
R2C6H4COCH2Br

136
Figure 1. Imidazolidinones: synthetic pathway
Chemistry and Molecular structure Compounds 22-25, were synthesized in three steps: (i), At first, 5-arylidene-2-thioxo-imidazolidin-4-ones 2-9, were prepared in the presence of piperidine by a 1,4-nucleophilic addition between 2-thioxo-imidazolidin-4-one 1, and (2-cyano-3-phenyl)-ethyl acrylates. Acrylates were prepared by refluxing equimolar mixture of aldehyde and ethyl cyanoacetate with piperidine in benzene. (ii) Then compounds 2-9, were N alkylated at position 3 in the presence of potassium carbonate that leads to the imidazolidine potassium salt. Then, benzyl or phenyloxoethyl chlorides reacted in alcoholic medium [5] to yield the 3-benzyl-5-benzylidene-2-thioxo-imidazolidin-4-ones 10-15, and the 5-benzylidene-3-(2-phenyl-2-oxo-ethyl)-2-thioxo-imidazolidin-4-ones 16-21. (iii) The 5-benzylidene-3-(4-fluorobenzyl)-2-(4-bromo-benzylsulfanyl)-3,5-dihydro-imidazol-4-ones 22-23, 2-[(4-bromophenyl)-2-oxo-ethylsulfanyl]-3-(4-fluorobenzyl)-5-(4-methoxybenzylidene)-3,5-dihydro-imidazol-4-one 24, and 5-benzylidene-3-[2-(4-bromophenyl)-2-oxo-ethyl]-2-[2-(4-bromophenyl)-2-oxo-ethylsulfanyl]-3,5-dihydro-imidazol-4-ones 25, were prepared in the presence of sodium methoxyde. Finally, one must emphasize that compounds 2-9, were isolated in a single isomer form which is the Z derivative as shown comparing spectroscopic data with those of imidazolidinones previously investigated [6,7]. EXPERIMENTAL SECTION Biological activity The synthesized imidazolidines 22 and 24 were assayed for general effects according to the method described by De Luca [8]. These compounds (1000 mg/kg) were dissolved in Tween 80/saline (0.2:10) mixture and were administered intraperitonealy in female Swiss mice. On the whole, animals were depressed and not stimulated. No lethal effects were observed during the assay. The animals had been observed firstly at 1 h time then each 12 h till 48 h were completed. Neither significant physiological alterations nor behavioral changes were evidenced. Thus, it can be considered that these compounds are only slightly toxic. During the pharmacological assay the maximum dose used was 0.5% of that tested for evaluating the acute toxicity [9]. The anti-inflammatory activity was prophylactically evaluated according to two different methodologies. The first one was the air-pouch test [10]. Pouches were formed by subcutaneous injections in male and female Swiss mice on the basis of a 2,5 mL air volume on day 0 and day 3. Carrageenin powder was dissolved in saline to a 10 mg/mL concentration and the solution was sterilized and homogenized by storing in oven where temperature is brought at 90°C for about 1 h before to be maintained at 37°C. The carrageenin solution (1 mL) was injected into the pouch six days after the initial injection of air with the aim to induce inflammation. The control group of mice only received the saline. Compounds 22 and 24, and etoricoxib as reference drug were administered orally 1 h before injection of carrageenin. After 4h, mice were killed by ether exposure and pouches were washed thoroughly with 3 mL of phosphate buffer solution containing 50mUI/mL heparine. The total number of polymorphonuclear leukocytes infiltrated was measured using an improved Newbauer heamocytometer.

137
Proceeding thus the injection of carrageenin in the air pouch causes a fast influx of leukocytes (4 h after injection), whilst that of the saline solution produces only a non significant leukocyte infiltration (Table 1). The anti-inflammatory effect of the drugs is expressed in terms of percentage of inhibition. Pre-treatment with compounds 22 and 24 at doses from 0.63 to 5 mg/kg, as well as that with etoricoxib at a 20mg/kg dose, produced a strong decrease in the migration of leukocytes when compared to the control group. Moreover, the lesser dose tested (0.63 mg/kg) the better the result. The second method used was the classical WINTER’s one [11]. Each male and female Wistar rat received orally either compounds 22, 24, or etoricoxib at a 2.5 mg/kg dose. The control group received only the vehicle. One hour after the drug administration, 0.1 mL of a 1% carrageenin aqueous solution as inflammatory agent was injected subcutaneously in the foot arch of the right hindleg. The volume of the leg was measured using a manual pletsmometer. Results are collected in Table 2. Compounds assayed show a significant prophylactic anti-inflammatory activity when compared to the control group. Table 1: Number of cells found in the pouch 4 hours after the inflammation be produced and inhibitory effects on the leukocyte cellular influx Compounds Doses (mg/kg) Number of cells/μL Cellular influx Control
inhibition (%) group 24 0.63 0.41 ± 0.05 x 103 71 Z
24 1.25 1.49 ± 0.37 x 103 65 Y
24 2.5 1.99 ± 0.39 x 103 53 Y
24 5 2.22 ± 0.25 x 103 25 X
22 0.63 0.46 ± 0.15 x 103 67 Z 22 1.25 1.36 ± 0.37 x 103 54 X 22 2.5 1.92 ± 0.47 x 103 35 X 22 5 1.97 ± 0.23 x 103 34 X Etoricoxib 20 2.31 ± 0.41 x 103 46 Y Control X 2.97 ± 0.45 x 103 - Control Y 4.26 ± 0.84 x 103 - Control Z 1.40 ± 0.08 x 103 - Control (without lesion) 0.07 ± 0.01 x 103 -
Table 2: Inhibition of the leg edema (1) (%)
time after induction of the edema Compounds 1 h 2 h 4 h 22 0 59 47 24 32 41 47 Etoricoxib 24 28 42

138
(1) The control group is considered to show 100% of edema. Compound 24 presents a 32% inhibition at the early stage of the development of edema. This is probably due to the local production of bradykinin. Around 45% inhibition is observed in the second stage (2 and 4 h) where the synthesis of prostaglandins is involved. Compound 22 is only active in the second step, suggesting that this compound is more selective than 24 and perhaps only disrupts the prostaglandin way.
Chemistry Melting points were measured on a Buchi apparatus. Thin layer chromatography was performed on Merck 60 F254 silica gel plates with a 0.2 mm thickness. Compounds were powdered, mixed with KBr at 1% concentration and pressed into pellets before infrared spectra be recorded on a IFS 66 Bruker spectrometer, apart from compounds 2,10,11,14,18,21-25 which were studied on a MB 100 M Bomem. 1H NMR spectroscopy was carried out on a Bruker AC 300 P spectrophotometer apart from compounds 2,10,11,21,24-25 which were studied on a Bruker ARX 200MHz spectrometer. DMSO-d6 was used as solvent and TMS as reference. Chemical shifts (δ) are given in parts per million (ppm), and coupling constants (J) are given in hertz (Hz), 70eV Electronic impact mass spectra were recorded on a Delsi-Nermag R-1010c spectrometer, apart from compound 13 which was carried out on a Finnigan GCQ Mat Quadrupole Ion-Trap. Intensity of molecular peaks is given with reference to the most intense peak M+(%). The already published data of compounds 3,5 [3], 4,9 [12] and 6,7 [1] are not reported here. Analyses of all the compounds prepared were within ± 0.4% of the theoretical values. (5Z)-5-Arylidene-2-thioxo-imidazolidin-4-ones, 2,8: general procedure. An equimolar (4.3mmol) mixture of 2-thioxoimidazolidin-4-one, 1 (0.5g) and (2-cyano-3-phenyl)-ethyl acrylate dissolved in ethanol (10mL) with piperidine (250µL) added was heated at 80-90°C for 4-8h. After cooling, the precipitate was collected and washed with water or recrystallized from ethanol or methanol. (5Z)-5-Benzylidene-2-thioxo-imidazolidin-4-one, 2 C10H8N2OS. Yield 72%. M.p. 268-270°C. TLC, (n-hex:AcOEt, 70:30) Rf 0,53. IR (KBr; υ, cm-1) 3237, 1724, 1643, 1499, 1478, 1187, 768. H1 NMR (δ ppm, DMSO-d6): 6.47 (s, CH ethylenic), 7.38-7.41 (m, 3H benzylidene), 7.71-7.75 (dd, 2H benzylidene, J=7.8-1.6 Hz), 12.17 (s, NH), 12.37 (s, NH). MS EI m/z(%): 204 (M+ 100%), 205 (M+1 21%), 117(70), 90(72), 89(58), 59(65). (5Z)-2-Thioxo-5-(3,4,5-trimethoxybenzylidene)-imidazolidin-4-one, 8 C13H14N2O4S. Yield 48%. M.p. 218-220°C. TLC, (n-hex.:AcOEt, 50:50) Rf 0,58. IR (KBr; υ, cm-1) 3117, 1725, 1653, 1577, 1502, 1333, 1127, 961, 638. H1 NMR (δ ppm, DMSO-d6): 3.69 (s, 3H OCH3), 3.85 (s, 6H OCH3), 6.45 (s, CH ethylenic), 6.97 (s, 2H benzylidene), 12.26 (s, NH), 12.39 (s, NH). MS EI m/z(%): 294 (M+ 100%), 295 (M+1 16%), 279(35), 192(17), 91(9), 78(18), 77(18). (5Z)-3-Benzyl-5-benzylidene-2-thioxo-imidazolidin-4-ones, 10-15, and (5Z)-5-benzylidene-3-(2-phenyl-2-oxo-ethyl)-2-thioxo-imidazolidin-4-ones, 16-21: general procedure. A solution of 5-substituted 2-thioxo-imidazolidin-4-one 2-9, (3.8mmol), potassium carbonate (5.5mmol) in methanol (10mL) was stirred at room temperature for 1h. Then 1-bromomethyl-4-fluoro-benzene or 1-bromomethyl-4-nitro-benzene and 2-bromo-1-(4-fluoro-phenyl)-

139
ethanone or 1-(4-bromo-phenyl)-ethanone (4.2mmol) was added and the mixture was stirred for 12-18h. Upon cooling in an ice bath, the product precipitated was collected and washed with water or recrystallized from ethanol or methanol. (5Z)- 5-Benzylidene-3-(4-fluorobenzyl)- 2-thioxo-imidazolidin-4-one, 10 C17H13FN2OS. Yield 97%. M.p. 185-187°C. TLC, (n-hex:AcOEt, 70:30) Rf 0,53. IR (KBr; υ, cm-1) 3067, 1708, 1633, 1509, 1410, 1159. H1 NMR (δ ppm, DMSO-d6): 4.55 (s, CH2), 6.74 (s, CH ethylenic), 7.16 (t, 2H benzyl, J=8.6 Hz), 7.55 (dd, 2H benzyl J=8.6 5.6 Hz), 7.43 (t, 3H benzylidene J=8.2 Hz), 8.18 (d, 2H benzylidene J=8.2 Hz), 11.84 (s, NH). MS EI m/z(%): 312 (M+ 26%), 313 (M+1 4%), 116(8), 109(100), 89(11), 83(17). (5Z)-3-(4-Fluorobenzyl)-5-(4-methoxybenzylidene)-2-thioxo-imidazolidin-4-one, 11 C18H15FN2O2S. Yield 92%. M.p. 213-215°C. TLC, (n-hex:AcOEt, 60:40) Rf 0,59. IR (KBr; υ, cm-1) 3073, 1704, 1630, 1597, 1511, 1264, 1170. H1 NMR (δ ppm, DMSO-d6): 3.8 (s, OCH3), 4.53 (s, CH2), 6.72 (s, CH ethylenic), 7.01 (d, 2H benzylidene, J=9.1 Hz), 7.16 (t, 2H benzyl, J=8.8 Hz), 7.54 (dd, 2H benzyl J=8.8 5.8 Hz), 8.16 (d, 2H benzylidene, J=9.1 Hz), 11.71 (s, 1H NH). MS EI m/z(%): 342 (M+ 43%), 343 (M+1 14%), 309(15), 174(34), 146(13), 109(100), 83(40). (5Z)- 5-(2-Bromobenzylidene)-3-(4-nitrobenzyl)-2-thioxo-imidazolidin-4-one, 12 C17H12BrN3O3S. Yield 47%. M.p. 194-196°C. TLC, (n-hex.:AcOEt, 60:40) Rf 0,5. IR (KBr; υ, cm-1) 3120, 3058, 1706, 1630, 1514, 1492, 1407, 1342, 1186, 763. H1 NMR (δ ppm, DMSO-d6): 4.68 (s, CH2), 6.96 (s, CH ethylenic), 7.33 (dt, 1H benzylidene, J=7.5 1,5 Hz), 7.54 (t, 1H benzylidene, J=7.5 Hz), 7.73 (d, 1H benzylidene, J=8.1 Hz), 7.8 (d, 2H benzyl, J=8.7 Hz), 8.22 (d, 2H benzyl, J=8.7 Hz), 8.73 (d, 1H benzylidene, J=8.1 Hz), 12.05 (s, NH). MS EI m/z(%): 417 (M+ 4), 419 (M+2 6%), 338(6), 203(84), 116(38), 115(52), 90(51.1), 89(100). (5Z)- 5-(2-Chlorobenzylidene)-3-(4-nitrobenzyl)-2-thioxo-imidazolidin-4-one, 13 C17H12ClN3O3S. Yield 67%. M.p. 224-226°C. TLC, (benzene:AcOEt, 80:20) Rf 0,5. IR (KBr; υ, cm-1) 3122, 3057, 1706, 1631, 1599, 1512, 1410, 1340, 1186, 759. H1 NMR (δ ppm, DMSO-d6): 4.68 (s, CH2), 7 (s, CH ethylenic), 7.41 (dt, 1H benzylidene, J=7.5 1,8 Hz), 7.48 (dt, 1H benzylidene, J=7.5 1.5 Hz), 7.54 (dd, 1H benzylidene, J=7.8 1.5 Hz), 7.78 (d, 2H benzyl, J=8.4 Hz), 8.21 (d, 2H benzyl, J=8.7 Hz), 8.73 (d, 1H benzylidene, J=8.1 Hz), 11.98 (s, NH). MS m/z(%): 373 (M+ 2), 372(6), 337(100), 304(13), 292(11), 202(13), 150(10), 89(8). (5Z)- 5-(3-Chlorobenzylidene)-3-(4-nitrobenzyl)-2-thioxo-imidazolidin-4-one, 14 C17H12ClN3O3S. Yield 72%. M.p. 200-201°C. TLC, (benzene:AcOEt, 70:30) Rf 0,6. IR (KBr; υ, cm-1) 3142, 3074, 1714, 1609, 1520, 1348, 1184. H1 NMR (δ ppm, DMSO-d6): 4.69 (s, CH2), 6.75 (s, CH ethylenic), 7.44-7.51 (m, 2H benzylidene), 7.83 (d, 2H benzyl, J=8.4 Hz), 8.02 (d, 1H benzylidene, J=6.6 Hz), 8.21 (d 2H, benzyl, J=8.4 Hz), 8.34 (s, 1H benzylidene), 12.01 (s, NH). MS EI m/z(%): 373 (M+ 7%), 375 (M+2 4%), 150(26), 106(17), 90(58), 89(100), 78(86), 63(41). (5Z)- 5-(2,4-Dimethoxyibenzylidene)-3-(4-nitrobenzyl)-2-thioxo-imidazolidin-4-one, 15

140
C19H17N3O5S. Yield 79%. M.p. 212-214°C. TLC, (benzene:AcOEt, 70:30) Rf 0,52. IR (KBr; υ, cm-1) 3058, 2820, 1721, 1604, 1509, 1348, 1289, 1258, 1186, 951. H1 NMR (δ ppm, DMSO-d6): 3.85 (s, OCH3), 3.87 (s, OCH3), 4.65 (s, CH2), 6.6 (d, 1H benzylidene, J=2.4 Hz), 6.68 (dd, 1H benzylidene, J=8.7; 2.4 Hz), 7.05 (s, CH ethylenic), 7.78 (d, 2H benzyl, J=8.7 Hz), 8.21 (d, 2H benzyl, J=8.7 Hz), 8.61 (d, 1H benzylidene, J=9 Hz), 11.68 (s, NH). MS EI m/z(%): 399 (M+ 24%), 400 (M+1 4%), 264(91), 204(100), 169(32), 136(47), 106(25), 89(57), 78(51). (5Z)-3-[2-(4-Bromophenyl)-2-oxo-ethyl]-5-(3-chlorobenzylidene)-2-thioxo-imidazolidin-4-one, 16 C18H12BrClN2O2S. Yield 78%. M.p. 139-140°C. TLC, (n-benzene:AcOEt, 80:20) Rf 0,5. IR (KBr; υ, cm-1) 3132, 3074, 1704, 1633, 1503, 1415, 1183, 779. H1 NMR (δ ppm, DMSO-d6): 4.98 (s, CH2), 6.63 (s, CH ethylenic), 7.1 (t, 1H benzylidene, J=7.8 Hz), 7.29-7.32 (m, 1H benzylidene), 7.77-7.82 (m, 1H benzylidene), 7.81 (d, 2H phenacyl, J=8.4 Hz), 8.03-8.06 (m, 1H benzylidene), 8.04 (d, 2H phenacyl, J=8.7 Hz), 11.97 (s, NH). MS m/z(%): 434 (M+ 2%), 436(M+2 2%), 402(4), 238(59), 240(30), 183(100), 185(84), 151(51), 116(27), 89(56). (5Z)-3-[2-(4-Bromophenyl)-2-oxo-ethyl]-5-(4-methoxyibenzylidene)-2-thioxo-imidazolidin-4-one, 17 C19H15BrN2O3S. Yield 94%. M.p. 176-177°C. TLC, (n-hex.:AcOEt, 60:40) Rf 0,46. IR (KBr; υ, cm-1) 3140, 3069, 1711, 1633, 1597, 1509, 1255, 1181, 1168, 830. H1 NMR (δ ppm, DMSO-d6): 3.76 (s, OCH3), 4.90 (s, CH2), 6.54 (d, 2H benzylidene, J=9 Hz), 6.63 (s, CH ethylenic), 7.75 (d, 2H benzylidene, J=9 Hz), 7.88 (d, 2H phenacyl, J=8.7 Hz), 8.09 (d, 2H phenacyl, J=8.7 Hz), 11.78 (s, NH). MS EI m/z(%): 430 (M+ 23%), 432 (M+2 29%), 247(74), 234(27), 183(100), 185(89), 155(63), 155(63), 157(43), 146(82), 132(39), 78(48). (5Z)-3-[2-(4-Bromophenyl)-2-oxo-ethyl]-5-(2,4-dimethoxybenzylidene)-2-thioxo-imidazolidin-4-one, 18 C20H17BrN2O4S. Yield 88%. M.p. 187°C. TLC, (n-hex.:AcOEt, 55:45) Rf 0,44. IR (KBr; υ, cm-1) 3063, 3129, 1708, 1604, 1493, 1284, 1183, 948, 830. H1 NMR (δ ppm, DMSO-d6): 3.78 (s, OCH3), 3.82 (s, OCH3), 4.88 (s, CH2), 5.8 (dd, 1H benzylidene, J=9 2.4 Hz), 6.51 (d, 1H benzylidene, J=2.4 Hz), 6.95 (s, CH ethylenic), 7.87 (d, 2H phenacyl, J=8.4 Hz), 8.04 (d, 1H benzylidene, J=9 Hz), 8.08 (d, 2H phenacyl, J=8.4 Hz), 11.73 (s, NH). MS EI m/z(%): 460 (M+ 1%), 462 (M+2 1%), 428(1), 430(1), 264(18), 183(54), 185(75), 155(20), 157(23), 76(62), 64(100). (5Z)-5-(4-Bromobenzylidene)-3-[2-(4-bromophenyl)-2-oxo-ethyl]-2-thioxo-imidazolidin-4-one, 19 C18H12Br2N2O2S. Yield 64 M.p. >270°C. TLC, (n-hex.:AcOEt, 60:40) Rf 0,5. IR (KBr; υ, cm-
1) 3063, 1713, 1689, 1633, 1583, 1505, 1179 1072, 819. H1 NMR (δ ppm, DMSO-d6): 4.93 (s, CH2), 6.65 (s, CH ethylenic), 7.16 (d, 2H benzylidene, J=8.7 Hz), 7.73 (d, 2H benzylidene, J=8.7 Hz), 7.87 (d, 2H phenacyl, J=8.4 Hz), 8.08 (d, 2H phenacyl, J=8.4 Hz), 11.95 (s, NH). MS EI m/z(%): 478 (M+ 12%), 480 (M+2 20%), 183(52), 185(69), 157(32), 155(35), 137(100), 116(22), 115(21), 89(20), 76(27). (5Z)-3-[2-(4-Bromophenyl)-2-oxo-ethyl]-5-(3,4,5-trimethoxybenzylidene)-2-thioxo-imidazolidin-4-one, 20 C21H19BrN2O5S. Yield 52%. M.p. 174°C. TLC, (n-hex.:AcOEt, 50:50) Rf 0,57. IR (KBr; υ, cm-1) 3063, 1708, 1633, 1576, 1503, 1461, 1245, 1130, 993, 809. H1 NMR (δ ppm, DMSO-d6): 3.6 (s, 6H, OCH3), 3.68 (s, 3H, OCH3), 5.11 (s, CH2), 6.69 (s, CH ethylenic), 7.48 (s, 2H

141
benzylidene),7.8 (d, 2H phenacyl J=8.7 Hz), 7.97 (d, 2H phenacyl, J=8.7 Hz), 11.89 (s, NH). MS EI m/z(%): 490 (M+ 1%), 492 (M+2 1%), 294(17), 183(89), 185(100), 157(30), 155(42), 128(11), 76(63). (5Z)-3-[2-(4-Bromophenyl)-2-oxo-ethyl]-5-benzylidene-2-thioxo-imidazolidin-4-one, 21 C18H13BrN2O2S. Yield 46%. M.p. 199-200°C. TLC, (n-hex.:AcOEt, 50:50) Rf 0,79. IR (KBr; υ, cm-1) 3064, 1717, 1633, 1584, 1509, 1176, 989. H1 NMR (δ ppm, DMSO-d6): 4.92 (s, CH2), 6.65 (s, CH ethylenic), 7.02 (t, 2H benzylidene J=7.4 Hz), 7.24 (t, 1H benzylidene J=7.4 Hz), 7.80 (d, 2H benzylidene J=7.4 Hz), 7.83 (d, 2H phenacyl, J=8.6Hz), 8.06 (d, 2H phenacyl, J=8.6Hz), 11.90 (s, 1H NH). MS EI m/z(%): 400 (M+ 17%), 402 (M+2 10%), 217(16), 183(100), 185(98), 155(22), 76(35), 64(14). (5Z)-5-Benzylidene-2-(4-bromobenzylsulfanyl)-3-(4-fluorobenzyl)-3,5-dihydro-imidazol-4-ones, 22-23, and 2-[(4-bromophenyl)-2-oxo-ethylsulfanyl]-3-(4-fluorobenzyl)-5-(4-methoxibenzylidene)-3,5-dihydro-imidazol-4-one, 24: general procedure. A mixture of 10 or 11 (0.45 mmol), 1-bromomethyl-4-bromo-benzene or 2-bromo-1-(4-bromophenyl)-ethanone (0.67 mmol) and sodium methoxide (0.54 mmol) in acetonitrile (10 ml) was stirred at room temperature for 3 at 12 h. After cooling, the fluffy product was collected by filtration and washed with water. (5Z)-5-Benzylidene-2-(4-bromobenzylsulfanyl)-3-(4-fluorobenzyl)-3,5-dihydro-imidazol-4-one, 22 C24H18BrFN2OS. Yield 72%. M.p. 132-133°C. TLC, (n-hex.:AcOEt, 80:20) Rf 0,64. IR (KBr; υ, cm-1) 3411, 3067, 1716, 1635, 1509, 1493, 1361, 1220, 1188, 1071, 972. H1 NMR (δ ppm, DMSO-d6): 4.6 (s, CH2), 4.74 (s, CH2), 6.97 (s, CH ethylenic), 7.17 (t, 2H benzyl, J=9 Hz), 7.21(d, 2H benzylsulfanyl, J=8,4 Hz), 7.42-7.52 (m, 3H benzylidene), 7.52-7.56 (m, 2H benzyl), 7.55 (d, 2H benzylsulfanyl, J=8.4 Hz), 8.23-8.25 (m, 2H benzylidene). MS EI m/z(%): 480 (M+ 1%), 482 (M+2 1%), 447(1), 449(1), 371(2), 373(2.5), 311(5), 280(2), 169(8.4), 171(8), 109(100), 89(20.7). (5Z)-2-(4-Bromobenzylsulfanyl)-3-(4-fluorobenzyl)-5-(4-methoxybenzylidene)-3,5-dihydro-imidazol-4-one, 23 C25H20BrFN2O2S. Yield 74%. M.p. 141-142°C. TLC, (n-hex.:AcOEt, 80:20) Rf 0,53. IR (KBr; υ, cm-1) 3440, 2837, 1702, 1635, 1599, 1509, 1499, 1257, 1171, 1029. H1 NMR (δ ppm, DMSO-d6): 3.82 (s, OCH3), 4.58 (s, CH2), 4.72 (s, CH2), 6.95 (s, CH ethylenic), 7.05 (d, 2H benzylidene, J=9 Hz), 7.17 (t, 2H benzyl, J=9 Hz), 7.19 (d, 2H benzylsulfanyl, J=8.4 Hz), 7.51-7.55 (m, 2H benzyl), 7.54 (d, 2H benzylsulfanyl, J=8.1 Hz), 8.23 (d, 2H benzylidene, J=9 Hz). MS m/z(%): 510 (M+ 10%), 512 (M+2 9%), 402(6), 404(7), 341(6), 169(20), 171(18), 146(22), 109(100), 89(24). 2-[(4-bromophenyl)-2-oxo-ethylsulfanyl]-3-(4-fluorobenzyl)-5-(4-methoxybenzylidene)-3,5-dihydro-imidazol-4-one, 24 C26H20BrFN2O3S. Yield 79%. M.p. 201-202°C. TLC, (n-hex.:AcOEt, 70:30) Rf 0,6. IR (KBr; υ, cm-1) 3433, 1702, 1634, 1597, 1511, 1260, 1173. H1 NMR (δ ppm, DMSO-d6): 3.82 (s, OCH3), 4.60 (s, CH2), 5.22 (s, CH2), 6.93 (s, CH ethylenic), 7.05 (d, 2H benzylidene, J=8.8 Hz), 7.17 (t, 2H benzyl, J=8.7 Hz), 7.79 (d, 2H benzylsulfanyl, J=8.5 Hz), 7.54 (dd, 2H benzyl, J=8.7 5.6 Hz), 7.98 (d, 2H benzylsulfanyl, J=8.5 Hz), 8.24 (d, 2H benzylidene, J=8.8 Hz). MS m/z(%): 538 (M+ 7%), 540 (M+2 8%), 341(38), 183(100), 174(48), 146(61), 109(66), 83(18).

142
(5Z)-5-benzylidene-3-[2-(4-bromophenyl)-2-oxo-ethyl]-2-[2-(4-bromophenyl)-2-oxo-ethylsulfanyl]-3,5-dihydro-imidazol-4-ones, 25 A mixture of 21 (0.05 g, 0.13 mmol), 1-bromomethyl-4-bromo-benzene (0.05 g, 0.19 mmol) and sodium hydrure (0.013 g, 0.58 mmol) in acetonitrile (5 mL) was stirred at room temperature for 24 h. After cooling, the fluffy product was collected by filtration and washed with water. C26H18Br2N2O3S. Yield 30%. M.p. 136-138°C. TLC, (n-hex:AcOEt, 70:30) Rf 0,6. IR (KBr; υ, cm-1) 3426, 2920, 1710, 1697, 1635, 1584, 1498, 1362, 1229, 1191, 1070, 994. H1 NMR (δ ppm, DMSO-d6): 5 (s, CH2), 5.34 (s, CH2), 6.85 (s, CH ethylenic), 6.98-7.07 (m, 2H benzylidene), 7.28 (t, 1H benzylidene, J=7.3 Hz), 7.42-7.61 (m, 2H benzylidene), 7.83 (d, 2H phenacyl, J=8.4 Hz), 7.85 (d, 2H phenacyl, J=8.6 Hz), 8.04 (d, 2H phenacyl, J=8.8 Hz), 8.08 (d, 2H phenacyl, J=8.8 Hz), 8.08 (d, 2H phenacyl, J=8.8 Hz). MS EI m/z(%): 596 (M+ 1%), 598(M+2 1%), 22 4(9), 183(100), 185(94), 160(41), 155(29), 157(25), 128(26), 76(13), 64(37). Acknowledgements The authors thank the CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brasil) and the CAPES/COFECUB (Fundação Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior / Comité Français d’Évaluation de la Coopération Universitaire avec le Brésil) for their support. References [1] L.C. Santos, F.T. Uchôa, A.R.P.A. Cañas, I.A. Sousa, R.O. Moura, M.C.A. Lima, S.L.
Galdino, I.R. Pitta and J. Barbe, Heterocycl. Comm., in press.
[2] A.M.C. Andrade, W.T. Lima, M.P.A. Rocha, M.C.A. Lima, S.L. Galdino, J.M. Barbosa
Filho, A.J.S. Góes and I.R. Pitta, Boll. Chim. Farm. 141, 428-433 (2002).
[3] S.S.F. Brandao, A.M.C.Andrade, D.T.M.Pereira, J.M.Barbosa Filho, M.C.A.Lima, S.L.Galdino, I.R.Pitta, J. Barbe, Heterocycl. Comm. 10, 9-14 (2004). [4] V.L.M.Guarda, M.A. Pereira, C.A. De Simone, J.C. Albuquerque, S.L. Galdino, J.
Chantegrel, M. Perrissin, C. Beney, F. Thomasson, I.R.Pitta, C.Luu-Duc, Sulfur Lett. 26, 17-
27 (2003).
[5] C.K. Bradsher, F.C. Brown and E.F. Sinclair, J. Amer. Chem. Soc. 78, 6189-6192 (1956). [6] J.F.C. Albuquerque, A. Albuquerque, C.C. Azevedo, F. Thomasson, S.L. Galdino, J. Chantegrel, M.T.J. Catanho, I.R. Pitta and C. Luu-Duc, Pharmazie 50, 387-389 (1995). [7] S.S.F. Brandão, V.L. Guarda, I.R. Pitta, J. Chantegrel, M. Perrissin, V.M. Souza, S.L. Galdino, F. Thomasson, M.C.A. Lima, F.F.C.C Leite and C. Luu-Duc, Boll. Chim. Farm. 139, 54-58 (2000). [8] R.R. De Luca,.; S.R. Alexandre,. T. Marques, N.L. Souza, J.L.B. Merusse, S.P. Neves, Manual Para Técnicas em Bioterismo, 2ª ed. São Paulo: Winnes Graph., 1996.

143
[9] G.H. Gosselin, H.C. Hodge, R.P. Smith, M.N. Gleason, Clinical toxicological of commercial products. 4th Ed., Williams and Wilkins Co., Baltimore, 1976. [10] P. Klemm, H.J. Harris, M. Perreti, Eur. J. Phamacol. 281, 69-74 (1995). [11] C.A. Winter, E.A. Risley, G.W. Nuss, Proc. Soc. Exp. Biol. Med. 111, 544-547 (1962). [12] S.M. Oliveira, M.C.P.A. Albuquerque, M.G.R. Pitta, E. Malagueño, J.V. Santana, M.C.A. Lima, I.R. Pitta, Acta Farm. Bonaer. 23, 343-8 (2004).




























