Embed Size (px)
Citation preview

TERMODINÂMICA QUÍMICA
CAPÍTULO DA QUÍMICA FÍSICA QUE ESTUDA A VARIAÇÃO DE ENERGIA DOS SISTEMAS ENVOLVIDOS EM REACÇÕES QUÍMICAS
Dissolução do NH4Cl e do CaCl2 parece contrariar esta afirmação
“QUALQUER SISTEMA EVOLUI ESPONTANEAMENTE DE UM ESTADO DE MAIOR ENERGIA PARA UM ESTADO DE MENOR ENERGIA”

SISTEMA:
PORÇÃO DO UNIVERSO A QUE NOS REFERIMOS
ISOLADO:
NÃO TROCA ENERGIA NEM MASSA COM O EXTERIOR

SISTEMA:
PORÇÃO DO UNIVERSO A QUE NOS REFERIMOS
FECHADO:
NÃO TROCA MASSA COM O EXTERIOR

SISTEMA:
PORÇÃO DO UNIVERSO A QUE NOS REFERIMOS
ABERTO:
TROCA ENERGIAE MASSA COM O EXTERIOR

ENERGIA TOTAL E INTERNA DE UM SISTEMA
interna) U(energiaEEE potencialcinética total ++=
Num sistema em repouso (Ecinética = 0)e sujeito a um campo exterior constante (campo gravítico; Epotencial = cte)
(sistema mais comum em laboratórios)
UE ∆=∆
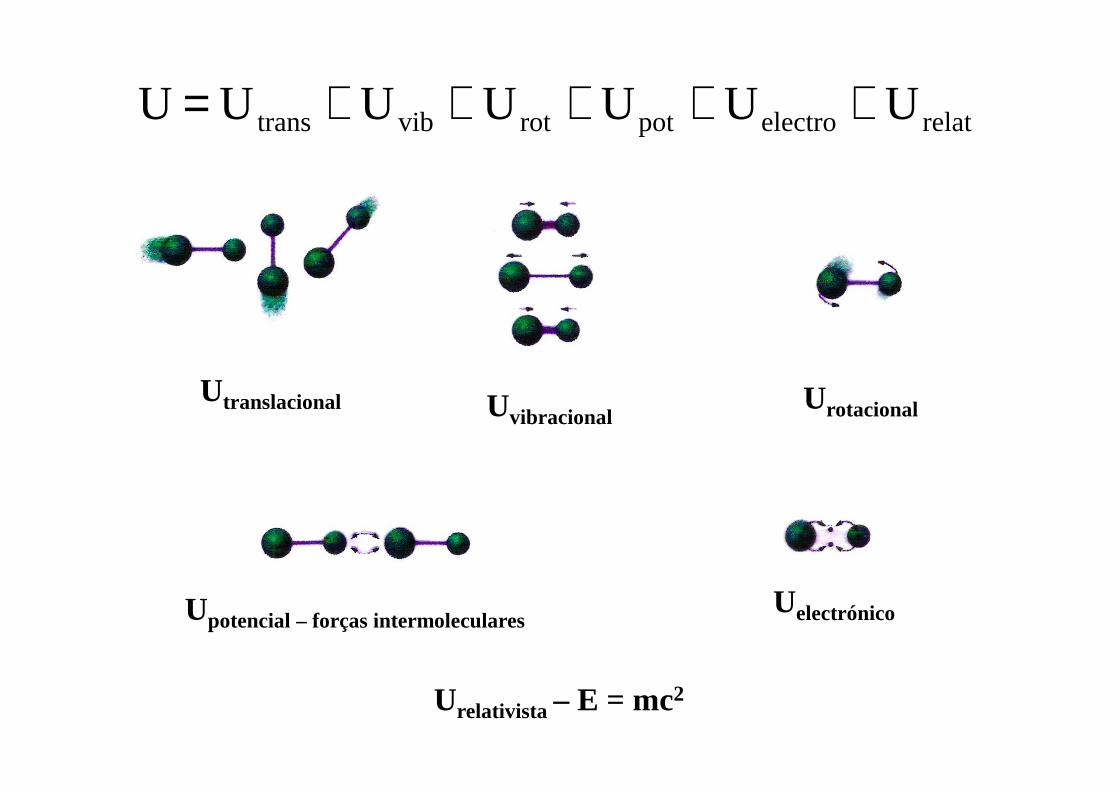
relatelectropotrotvibtrans UUUUU U U +++++=
Utranslacional UvibracionalUrotacional
Upotencial – forças intermolecularesUelectrónico
Urelativista – E = mc2

1ª LEI DA TERMODINÂMICA(LEI DA CONSERVAÇÃO DA ENERGIA)
A Energia interna do Universo permanece constante -embora haja transformações de tipos de Energia
Ex: Queda de um corpo: E potencial⇒ E cinética
Num sistema fechado ocorrem trocas de calor e realização de trabalho por interacção com o exterior
∆U = q + w
Convenções: ∆U = Ufinal - Uinicialq > 0, transformação endotérmicaw > 0, trabalho realizado sobre o sistema

FUNÇÃO DE ESTADO: que não depende do modo como o estado final foi atingido – apenas dependem do estado inicial e do estado final, não dependem do caminho percorrido
∆U = Ufinal - Uinicial
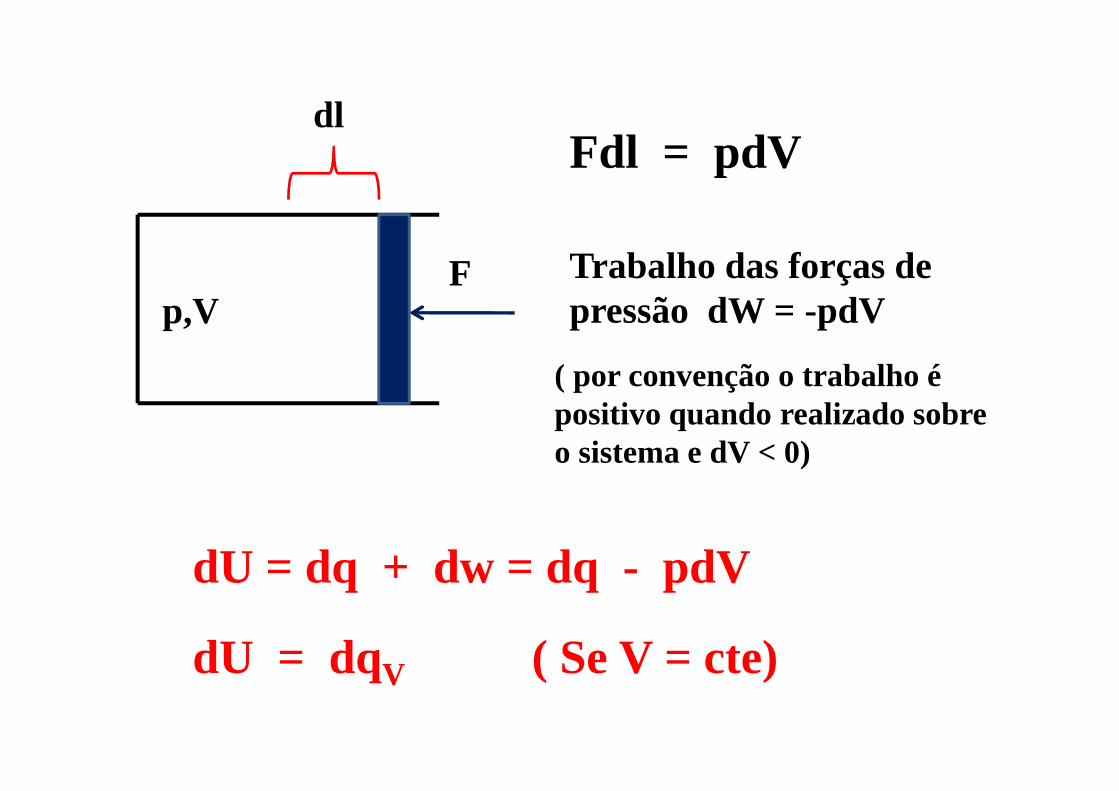
p,VF
dlFdl = pdV
Trabalho das forças de pressão dW = -pdV
( por convenção o trabalho é positivo quando realizado sobre o sistema e dV < 0)
dU = dq + dw = dq - pdV
dU = dqV ( Se V = cte)

A maioria das transformações químicas ocorre a P constante.
Surge assim uma nova função de estado –ENTALPIA –Calor transferido a pressão constante
H = U + pV
∆U = qP - p∆V
U2 - U1 = qP - P (V2 - V1)
(U2 + PV2) - (U1 + PV1) = qP
∆H = H2 - H1 = qP
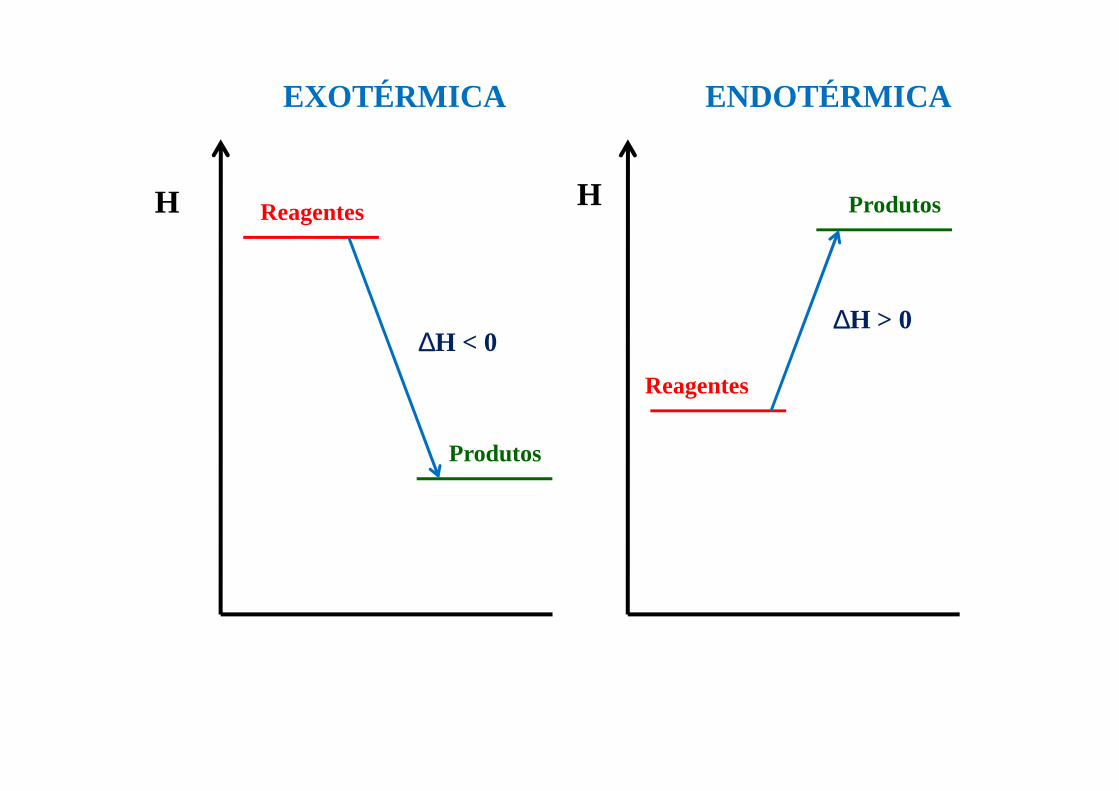
H HReagentes
Reagentes
Produtos
Produtos
EXOTÉRMICA ENDOTÉRMICA
∆H < 0∆H > 0

ENTALPIA DE TRANSFORMAÇÃO
Define se uma transformação é exotérmica (∆H<0) ou se é endotérmica (∆H>0)
∑∑ −=∆j
ji
i reagenteHprodutoHH )()(
H = f(composto, P, T, estado físico)
É necessário definir CONDIÇÕES PADRÃOT = 298 KP = 1 bar
H2(g) + ½ O2(g) → H2O(liq)
∆Hº (1 bar, 298K) = -285 kJ/mole
A Entalpia da reacção inversa tem o mesmo valor, mas com sinal contrário ⇒ 285 kJ/mole

ENTALPIA DE FORMAÇÃO PADRÃO
Este ∆Hº corresponde à Entalpia de Formação da água líquida em condições padrão:
∆Hºf (H2O, liq, 298 K, 1 bar) = - 285.83 kJ/mole
H2(g) + ½ O2(g) → H2O(liq)
∆Hº (1 bar, 298K) = -285 kJ/mole
Entalpia de formação padrão: Entalpia da reacção de formação de 1 mole do composto, no estado padrão, a partir dos seus elementos constituintes , também no estado padrão
A Entalpia de formação de um elemento no seu estado de referência é zero
As Entalpias de formação padrão estão tabeladas

ENTALPIA DE REACÇÃO
Como vimos ∆H é uma Função de Estado: não depende do caminho seguido
LEI DE HESS: ∆H só depende do estado inicial e do final, qualquer que seja o caminho seguido
∑ ∆∑ −∆=∆j
jfji
ifir reagenteHprodutoHH )(º)(ºº νν

2 NO (g) + O2 (g) → 2 NO2 (g)
∆Hrº = 2 ∆H fº (NO2, g) - 2 ∆H fº (NO, g)
2 NO (g) + O2 (g) → 2 NO2 (g) ∆Hrº = 2 ∆H fº (NO2, g) - 2 ∆H fº (NO, g)
2 NO (g) → N2 (g) + O2 (g) - 2 ∆H fº (NO, g)
2 O2 (g) + N2 (g) → 2 NO2 (g) 2 ∆H fº (NO2, g)
EXEMPLO 1
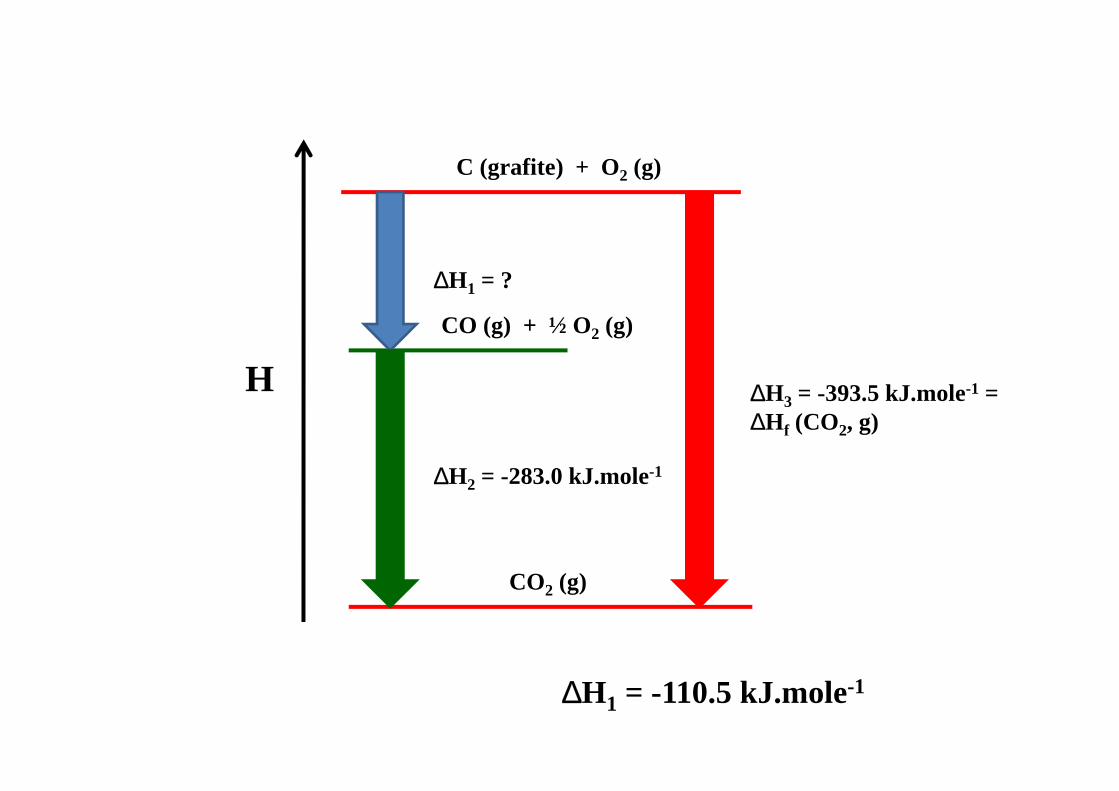
C (grafite) + O2 (g)
CO (g) + ½ O2 (g)
CO2 (g)
H
∆H1 = ?
∆H2 = -283.0 kJ.mole-1
∆H3 = -393.5 kJ.mole-1 = ∆H f (CO2, g)
∆H1 = -110.5 kJ.mole-1

EXEMPLO 2: Calcular ∆Hrº de
2 N2H4 (liq) + N2O4 (liq) → 3 N2(g) + 4 H2O (liq)
DADOS: ∆H fº (N2O4, liq) = -19.56 kJ/mole
∆Hcombustãoº (N2H4, liq) = - 622.91 kJ/mole
N2H4 (liq) + O2 (g) → N2(g) + 2 H2O (liq)
N2(g) + 2 O2 (g) → N2O4(g)

2 N2H4 (liq) + N2O4 (liq) → 3 N2(g) + 4 H2O (liq)
2 X [N2H4 (liq) + O2 (g) → N2(g) + 2 H2O (liq)]
N2(g) + 2 O2 (g) → N2O4(g)
INVERTER O SENTIDO DA REACÇÃO DE FORMAÇÃO
N2O4(g) → N2(g) + 2 O2 (g)
∆H1 = 2 x ∆Hcombustão(N2H4)
∆H2 = - ∆H fº (N2O4)

2 N2H4 (liq) + 2 O2 (g) → 2 N2(g) + 4 H2O (liq)]
N2O4(g) → N2(g) + 2 O2 (g)
2 N2H4 (liq) + N2O4 (liq) → 3 N2(g) + 4 H2O (liq)
∆Hr = ∆H1 + ∆H2 = 2 x ∆Hcombustão(N2H4) - ∆H fº (N2O4) =2 x (-622.91) – (-19.56) = - 1226.26 kJ/mol

CAPACIDADE CALORÍFICA MOLAR
A quantidade de calor transferida é proporcional à variação de temperatura do sistema
∆TnCq pp = ∆TnCq vv =
Cp,v = Capacidade calorífica molar a P e V constanteJ.K -1.mol-1
∆Tmcq pp = ∆Tmcq vv =
cp,v = Calor específico a P e V constante J.K -1.g-1

CALORES LATENTES
Quantidade de calor não associadas a diferenças de temperatura
Entalpias de transformação, uma vez que se referem a transformações a P constante
qp = n Cp(sol)∆T1 + n ∆H fusão + n Cp(liq)∆T2 + n ∆Hvap + n Cp(gas)∆T3
qp = m cp(sol)∆T1 + n ∆H fusão + m cp(liq)∆T2 + n ∆Hvap+ m cp(gasl)∆T3
n = 2 moles
m = 2 x 18 g
∆T1 = 10 (-10 a 0ºC)
∆T2 = 100 (0 a 100ºC)
∆T3 = 10 (100 a 110ºC)
H2O (-10ºC) H2O (110ºC)2 moles

ESPONTANEIDADE E EQUILÍBRIO
Saber se um dado acontecimento ocorre naturalmente (sem ajuda exterior) - espontâneo
Exemplos de acontecimentos expontâneos:
- Fusão de gelo à temperatura ambiente
- Formação de ferrugem
- Reacções ácido-base
- Miscibilidade de líquidos

ESPONTANEIDADE E EQUILÍBRIO
Termodinâmica e Cinética
A termodinâmica apenas se preocupa com o sentido em que se dá o acontecimento até se atingir o equilíbrio
A cinética preocupa-se com a velocidade com que se atinge o equilíbrio
No equilíbrio a velocidade do processo directo é igual à velocidade do processo inverso
Processos rápidos: Fusão do gelo e Reacções ácido-baseProcessos lentos: Formação de ferrugem

FACTORES QUE AFECTAM A ESPONTANEIDADE
A -ENERGIA ∆E < 0
Ex: FOGO – libertação de calor (∆q < 0)
(∆U = ∆qV)
MAS
- Dissolução de NH4Cl é endotérmica e espontânea*
- Fusão ou evaporação dos lagos é endotérmica e espontânea*
* Nestes processos tem lugar uma maior desorganização do sistema no estado final

FACTORES QUE AFECTAM A ESPONTANEIDADE
B -ENTROPIA
ENTROPIA:
Nº de configurações possíveis de um sistema
lnωkSB
=kB – cte de Boltzman
ω = multiplicidade de estados microscópicos possíveis num dado sistema macroscópico
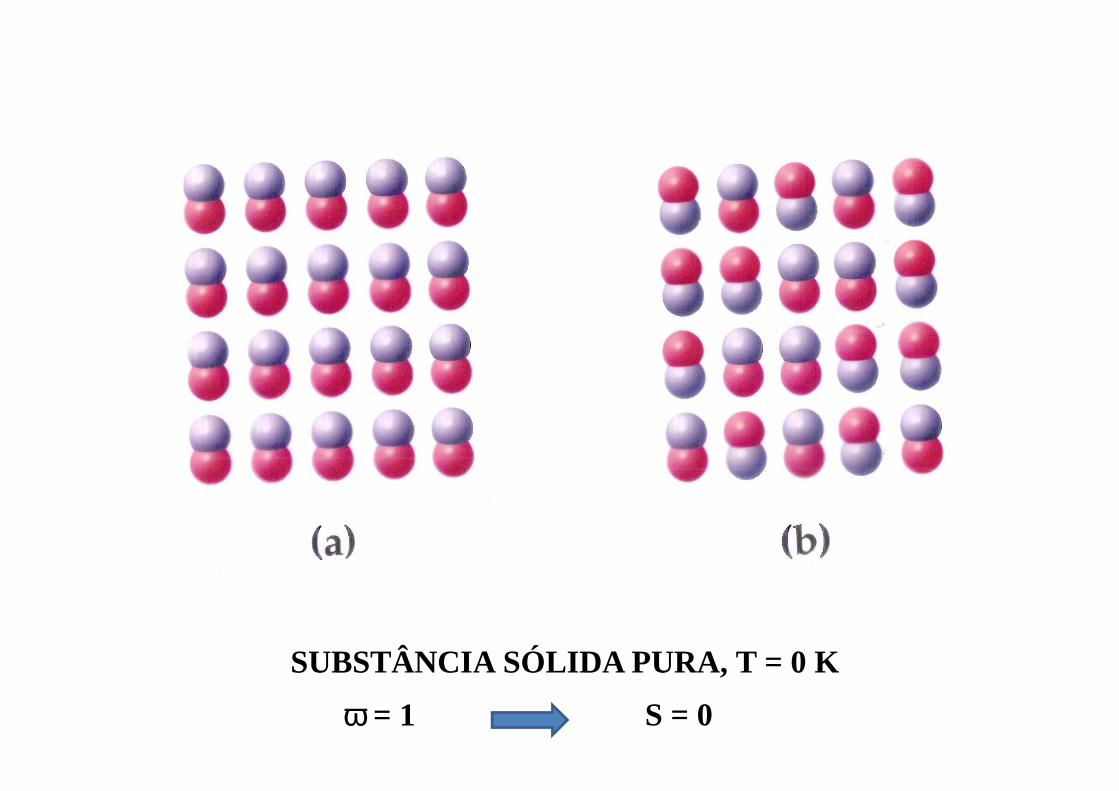
SUBSTÂNCIA SÓLIDA PURA, T = 0 K
ω = 1 S = 0
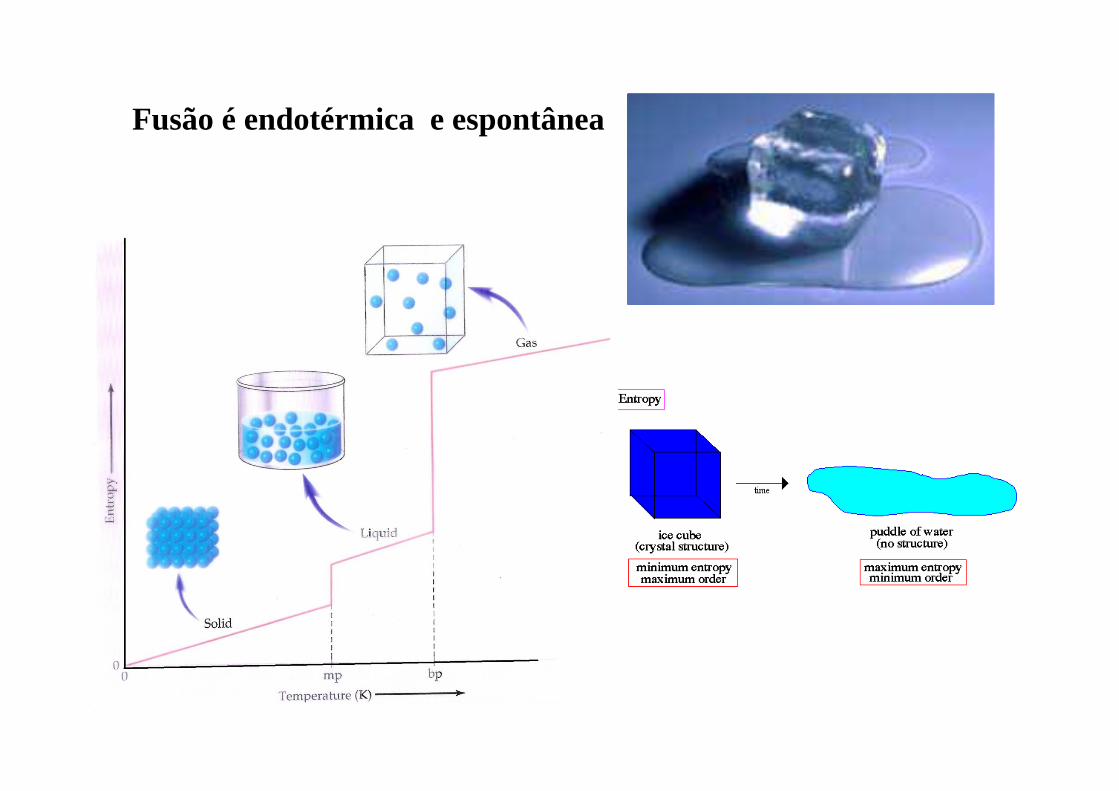
Fusão é endotérmica e espontânea

ENTROPIA :
- Calor posto em jogo numa transformação reversível
(que se realiza tão lentamente que a variação das Funções de Estado é diminuta)
(que passa por sucessivos estados de equilíbrio)
Ex: Expansão infinitamente lenta de um gás
T
qS rev=∆

2ª LEI DA TERMODINÂMICA
Um processo é espontâneo quando provoca um aumento de entropia do Universo
0>∆+∆=∆ extsistUniv SSS
∑ ∆∑ −∆=∆j
jji
ii reagenteSprodutoSS )()( νν
∑ ∆∑ −∆=∆j
jji
ii reagenteSprodutoSS )(º)(ºº νν
Nas condições padrão
A Entropia é uma Função de Estado

ENERGIA LIVRE DE GIBS
Considerando um processo reversível a P = cte
HqP ∆=T
qS rev=∆ 0>∆+∆=∆ extsistUniv SSS
0>∆+∆=∆T
HSS ext
sistUniv
0>
∆−+∆T
HS sist
sist
Temos
Como este calor é trocado com o exterior

0>
∆−+∆T
HS sist
sist 0<∆−∆ STHSurge assim uma nova Função de Estado
ENERGIA LIVRE DE GIBBS
STHG ∆−∆=∆
∑ ∆∑ −∆=∆j
jji
ii reagenteGprodutoGG )()( νν
∑ ∆∑ −∆=∆j
jji
ii reagenteGprodutoGG )(º)(ºº νν

STHG ∆−∆=∆

CRITÉRIO DE ESPONTANEIDADE
Para P e T ctes o sistema evolui espontaneamente quando
Uma transformação espontânea é acompanhada de
uma diminuição da Energia Livre de Gibbs do sistema
0<∆−∆=∆ STHGQuando ∆G = 0 o sistema está em equilíbrio
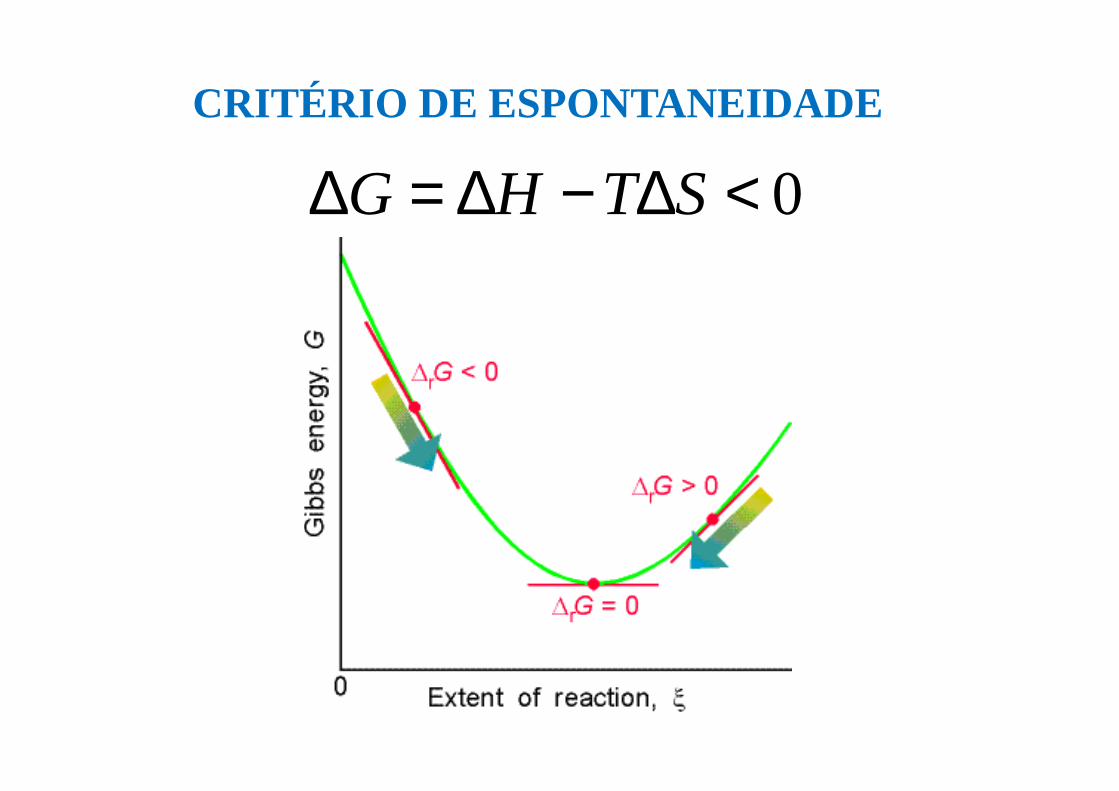
0<∆−∆=∆ STHG
CRITÉRIO DE ESPONTANEIDADE

PROBLEMA: Calcule a T mínima a que a seguinte reacção é espontânea
2 Fe2O3 (s) + 3 C (grafite) → 4 Fe (s) + 3 CO2 (g)
DADOS (25˚C) ∆G ˚ = 300.0 kJ/mole
∆H ˚ = 467.9 kJ/mole
∆S ˚ = 560.3 J/mole.K
0ººº =∆−∆=∆ STHG
ºº STH ∆=∆
KS
HT 835
ºº =
∆∆=
Acima de T = 835 K, ∆G ˚ < 0 e a reacção é espontânea

DEPENDÊNCIA DE G COM A PRESSÃO
=−
1
212 ln
P
PnRTGG
Se P1 = P0 = 1 atm (padrão)
PnRTGP
PnRTGG lnºlnº
0+=
+=

DEPENDÊNCIA DE G COM A CONCENTRAÇÃO
nRTPV = RTcV
nRTP =
=
1
2
11
22
1
2
)/()/(
c
c
VnRT
VnRT
P
P ==

DEPENDÊNCIA DE G COM A CONCENTRAÇÃO
=
=−
1
2
1
212 lnln
c
cnRT
P
PnRTGG
Se c1 = c0 = 1 mole dm-3 (padrão)
cnRTGc
cnRTGG lnºlnº
0+=
+=

VARIAÇÃO DE G NUMA TRANSFORMAÇÃO QUÍMICA
POTENCIAL QUÍMICOVariação elementar de G por variação elementar do nº de moles a P e T ctes
dn
dG=µ dndG µ=
Como P e T são ctes
VARIAÇÃO DE G DE UMA MISTURA REACCIONAL
PRTdn
dG
dn
dGln
º += PRT lnº+= µµ
∑∑ −=j
jji
ii reagentednprodutodndG )()( µµ
PnRTGG lnº+=

VARIAÇÃO DE G NUMA TRANSFORMAÇÃO QUÍMICA
αA (g) + βB (g) ↔ γC (g) + δD (g)
início nA nB nC nD
final nA-αdφ nB-βdφ nC+γdφ nD+δdφ
φ - grau de avanço da reacção = nº de unidades de reacção entre o instante inicial e um dado instante (final)
Variação do nº de moles por composto
dnA=-αdφ dnB=-βdφ dnC=+γdφ dnD=+δdφ

VARIAÇÃO DE G NUMA TRANSFORMAÇÃO QUÍMICA
αA (g) + βB (g) ↔ γC (g) + δD (g)
início nA nB nC nD
final nA-αφ nB-β φ nc+γ φ nD+δ φ
dnA=-αdφ dnB=-βdφ dnC=+γdφ dnD=+δdφvariação
∑∑ −=j
jji
ii reagentednprodutodndG )()( µµ
( )φβµφαµφδµφγµ dddddG BADC +−+=
( )BADCd
dG βµαµδµγµφ
+−+=

Como PRT lnº+= µµ
( )
( )[ ]BADC
BADC
PPPPRT
d
dG
lnlnlnln
ºººº
βαδγ
βµαµδµγµφ
+−+
++−+=
( )BADCd
dG βµαµδµγµφ
+−+=
( ) ( )( ) ( )βα
δγ
φφ BA
DC
PP
PPRT
d
dG
d
dGln
º +=
PQRTGG lnº+∆=∆( ) ( )( ) ( )βα
δγ
BA
DCP PP
PPQ =
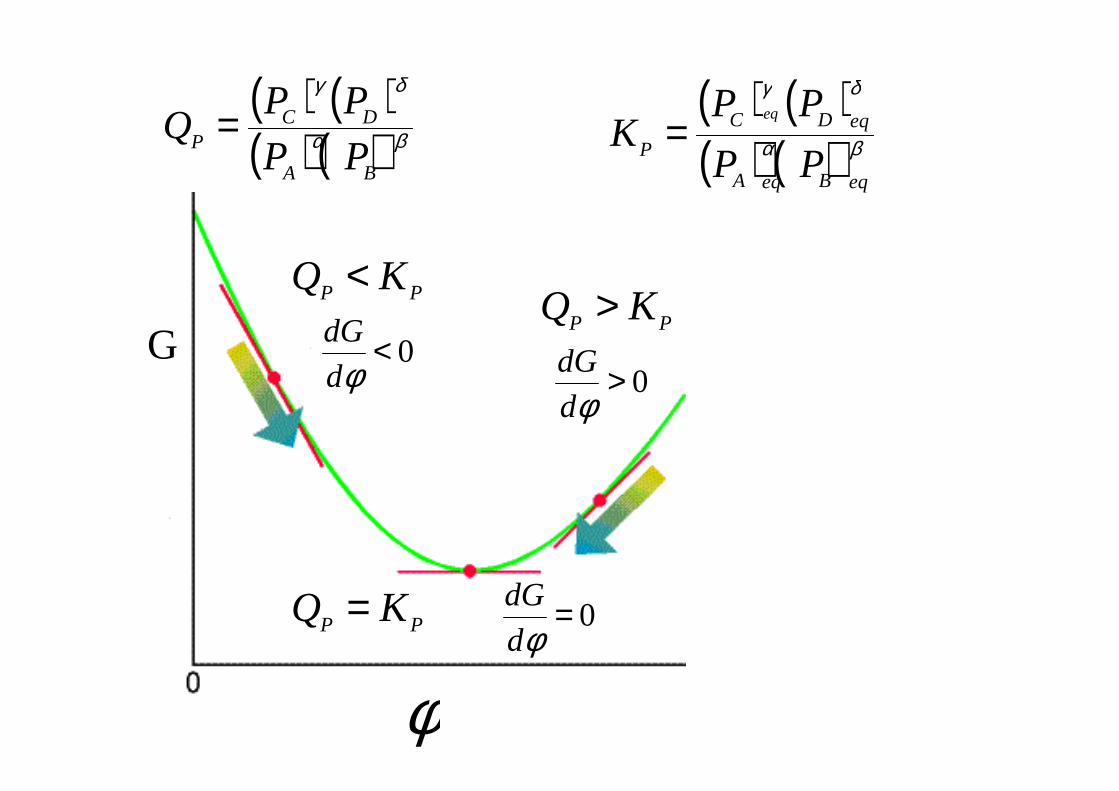
( ) ( )( ) ( )βα
δγ
BA
DCP PP
PPQ = ( ) ( )
( ) ( )βα
δγ
eqBeqA
eqDC
P PP
PPK
eq
=
G 0<φd
dG
φ
0=φd
dG
0>φd
dG
PP KQ <PP KQ >
PP KQ =





























