Embed Size (px)
Citation preview

UNIVERSIDADE DE SÃO PAULO
INSTITUTO DE FÍSICA DE SÃO CARLOS
ANA PAULA DA SILVA CITADINI ESTUDOS DA DINÂMICA ESTRUTURAL DA PROTEÍNA LIGANTE DE
CÁLCIO S100A12 HUMANA E DA LISOZIMA T4
São Carlos 2011


ANA PAULA DA SILVA CITADINI
ESTUDOS DA DINÂMICA ESTRUTURAL DA PROTEÍNA LIGANTE DE CÁLCIO S100A12 HUMANA E DA LISOZIMA T4
Versão Original
São Carlos 2011
Tese apresentada ao Programa de Pós-Graduação do Instituto de Física de São Carlos, da Universidade de São Paulo, para obtenção do título de Doutor em Ciências.
Área de concentração: Física Aplicada. Orientador: Prof. Dr. Antônio José da Costa Filho Co-orientadora: Profa. Dra. Maria Cristina Nonato

AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE.
Ficha catalográfica elaborada pelo Serviço de Biblioteca e Informação IFSC/USP
Citadini,Ana Paula da Silva Estudos da dinâmica estrutural da proteína ligante de cálcio S100A12 humana e da lisozima T4./Ana Paula da Silva Citadini; orientador Antonio José da Costa Filho; co-orientadora Maria Cristina Nonato.-- São Carlos, 2011.
182 p.
Tese (Doutorado–Programa de Pós-Graduação em Ciências. Área de concentração: Física Aplicada ) – Instituto de Física de São Carlos,Universidade de São Paulo. 1. Marcação de spin sitio dirigida. 2. Ressonância paramagnética eletronica. 3. Proteinas ligantes de calcio. 4. S100A12 humana. 5.Lisozima T4. I.Título.


Ao meu pai…


AGRADECIMENTOS
Primeiramente a Deus, por ter me dado sabedoria e paciência para a
realização deste trabalho;
Um agradecimento MUITO especial para o meu orientador e amigo Prof. Dr.
Antonio José da Costa Filho, Jábah, pelo apoio, incentivo constante e orientação
durante a realização deste trabalho e por sempre ter acreditado em mim... Agradeço,
não só pelo orientador que foi, mas pelo grande amigo que representas para mim,
presente sempre com palvras certas, nas horas certas... OBRIGADA, realmente por
tudo!!!!!
A minha mamys e meus irmãos, Juliana e Marcos Paulo, por me
acompanharem sempre e serem meu incentivo para continuar;
Ao Edgar, pelo carinho e paciência que teve comigo nestes últimos meses;
A toda minha família (avôs, avós, tios, tias, primos e primas), que sempre
torceram por mim e me acompanharam durante esses 5 anos;
Ao Prof. Dr. Wayne Hubbell (Universidade da Califórnia-UCLA), por ter me
acolhido em seu laboratório tão prontamente e ter me proporcionado uma
experiência maravilhosa;
Ao Carlos López, pela ajuda no trabalho com a enzima Lisozima T4;
Ao Dr. Christian Altenbach, por ter me cedido sua casa por alguns meses,
pelas discussões fundamentais para a realização deste trabalho, pelas caronas até
UCLA e também pelos barbecues;
À Profa. Dra. Maria Cristina Nonato (FCFRP-USP) por ter me ensinado biologia
molecular e purificação de proteínas;
À Dra. Thatyane Nobre pela imensa contribuição na realização do estudo com
monocamadas de Langmuir e principalmente por ser uma pessoa maravilhosa;
Ao Assuero Garcia, pela ajuda nos experimentos de biomembranas e pelas
conversas produtivas no laboratório;
Ao José Fernando, pela ajuda nas simulações das distâncias e pela paciência
que sempre teve comigo;
Ao Felipe, meu aluninho de IC, por ter me ajudado muito na produção dos
mutantes;
Ao Matheus Pinheiro e Patricia Feliciano da USP- Ribeirão Preto, por ter me
ensinado a fazer atividade com a proteína DHODH, a usar o pymol e pelas

discussões a respeito do trabalho. Matho, obrigada por me ajudar nas figuras das
estruturas desde 2005...
À Juliana Cheleski e Helton Wiggers, Química–USP, por me ensinarem o
pouco que sei a respeito de ITC e por me ajudarem na obtenção das distâncias
teóricas;
Ao Luis Guilherme Mansor Basso, Militaaaa, por sempre me socorrer nas horas
em que mais precisava, pela paciência, pelas discussões a respeito de tudo, de tudo
mesmo e por “aparecer” na minha vida;
Ao Fábio, pelas questões respondidas com tanta inteligência e também, pelas
risadas e fofocas no laboratório.
À Cherie Hubbell e Miyeon Kim, UCLA-EUA, por serem meus anjos da guarda
em Los Angeles;
Aos meus amigos do Grupo de Biofísica: Zé, meu querido, pelas agradáveis
conversas e pela companhia maravilhosa nas nossas noitadas; Débora, por ter me
aguentado 2 anos debaixo do mesmo teto; Célia, por ter me dado tanto apoio
durante esses 5 anos e simplesmente, por ser minha amiga; Lia, pela presença ao
meu lado nas horas difíceis; Joci, pelas conversas a respeito da vida e do futuro.
Enfim, a todos do Grupo de Biofísica e do laboratório da UCLA que de alguma forma
me ajudaram na realização deste trabalho;
Ao Prof. Dr. Otaciro pela atenção e carinho que sempre teve comigo e por
muito me ensinar;
À Bel, Andressa, João e Gi, por providenciar todas as coisas para o andamento
deste trabalho;
À Esther, que com agilidade sempre me ajudava nas horas de entregar os
relatórios da FAPESP e mantinha tudo organizado;
À FAPESP pelo apoio financeiro para a realização deste trabalho.

RESUMO
CITADINI, A. P. Estudos da Dinâmica Estrutural da Proteína Ligante de Cálcio S100A12 Humana e da Lisozima T4. 2011. 182p. Tese (Doutorado em Ciências)- Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, 2011.
O trabalho ora apresentado foi concebido como tendo dois objetivos. O primeiro,
mais geral, foi implementar uma nova metodologia para o estudo de mudanças
conformacionais em proteínas, ou seja, de sua dinâmica estrutural. A técnica de
marcação de spin sítio dirigida aliada à ressonância paramagnética eletrônica (SDSL-
RPE) são os pilares desse novo método que faz, agora, parte do conjunto de técnicas
disponíveis no Grupo de Biofísica Molecular Sérgio Mascarenhas do Instituto de
Física de São Carlos (USP). O segundo objetivo, mais específico, representou o
caminho efetivamente tomado para que se alcançasse o objetivo geral. Para isso, foi
proposto o estudo da correlação estrutura e função de dois sistemas biológicos muito
interessantes. O primeiro deles envolveu o estudo do movimento das hélices que
compõem a estrutura da proteína ligante de cálcio S100A12 humana (HS100A12)
induzido pelos íons cálcio e zinco. Sabendo que a proteína S100A12 humana além
de ligar íons Ca+2, apresenta afinidade por outros metais divalentes, como os íons
Zn+2 e Cu+2, e que a formação de diferentes oligômeros da proteína é governada
pela concentração dos íons Ca+2 e Zn+2, realizamos estudos espectroscópicos
utilizando a técnica de dicroísmo circular a fim de investigarmos a estabilidade
térmica da proteína HS100A12 na presença e ausência dos íons cálcio e zinco.
Mudanças conformacionais na estrutura da HS100A12 foram monitoradas através
da construção de uma série de mutantes (simples e duplos) em que resíduos nas
hélices B, C e D foram trocados por cisteínas, subsequentemente marcadas com a
sonda magnética MTSSL e submetidas às análises de SDSL-RPE. Estas consistiram
na medida do espectro de RPE dos vários mutantes em temperatura ambiente para
estudarmos os efeitos da presença dos íons sobre a dinâmica experimentada pela
sonda nas diversas posições. Além disso, efetuamos medidas de distância entre
duas sondas seletivamente inseridas na estrutura protéica, procurando assim
complementar o entendimento acerca do efeito da presença dos íons sobre a
proteína. Por fim, devido ao fato da proteína HS100A12 estar envolvida em alguns
eventos de sinalização celular e interação com o receptor para produtos de
glicosilação (RAGE), decidimos também, estudar a interação da proteína com

modelos de biomembranas, utilizando monocamadas de Langmuir. O outro
problema de interesse utilizou a lizosima do fago T4, uma proteína “padrão”, da qual
uma variedade de mutantes é produzida rotineiramente a fim de obtermos mais
detalhes a respeito da sua correlação estrutura e função e tornar mais sólido o
entendimento da técnica SDSL. Inicialmente, realizamos um estudo com a suposta
criação de uma cavidade no “core” hidrofóbico da porção C-terminal da enzima,
quando mutamos a Leu 133 por Ala e/ou Gly, ou seja, quando trocamos um resíduo
grande por um de menor volume, pois se acredita que a proteína sofra um reajuste
estrutural com o intuito de preencher o espaço vazio criado por essa substituição.
Para isso, propusemos estudar por SDSL o movimento da α-hélice H inserindo o
marcador de spin na posição vizinha ao resíduo mutado. Adicionalmente, realizamos
um experimento de “transmutação” com a enzima T4L, a fim de investigar a natureza
das contribuições para os diferentes modos dinâmicos experimentados pelo
marcador de spin quando introduzido em sítios topologicamente semelhantes.
Palavras-chave: Marcação de spin sítio dirigida. Ressonância paramagnética eletrônica. Proteínas ligantes de cálcio. S100A12 humana. Lisozima T4.

ABSTRACT
CITADINI, A. P. Structural Dynamics Studies of Human Calcium Binding Protein S100A12 and T4 Lysozyme. 2011. 182p. Thesis (Ph.D.)- Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, 2011. The work presented here was conceived with two main objectives. The first one,
more general, involved the implementation of a new methodology for the study of
conformational changes in proteins, i.e., its structural dynamics. The technique of
Site-directed Spin Labeling combined with Electronic Paramagnetic Resonance
(SDSL-EPR) are the pillars of this new method, which is now part of the set of
techniques available at the Grupo de Biofísica Molecular Sérgio Mascarenhas,
Instituto de Física de São Carlos (USP). The second objective, more specific,
represented the path actually taken to achieve the overall goal. Therefore, it was
proposed to study the structure-function correlation in two interesting biological
systems. The first involved the study of the movement of the helices that form the
structure of the human calcium binding protein S100A12 (HS100A12) induced by
calcium and zinc ions. Knowing that, besides Ca+2, human S100A12 has also affinity
for other divalent metals, such as Zn+2 and Cu+2 ions, and that the formation of
different protein oligomers is governed by the concentration of Ca+2 and Zn+2, we
performed spectroscopic studies using circular dichroism (CD) to investigate the
thermal stability of protein HS100A12 in the presence and absence of calcium and
zinc. Conformational changes in the structure of HS100A12 were monitored by
producing a series of mutants (singles and doubles) in which residues in helices B, C
and D were replaced by cysteine and subsequently labeled with a magnetic probe
MTSSL and then analyzed via SDSL-EPR. The latter consisted of the EPR spectra
measurement of many mutants at room temperature to study the effects of the
presence of ions on the dynamics experienced by the probe in different positions. In
addition, we performed measurements of the distance between two probes inserted
in the protein structure, thereby, seeking to improve the understanding of the effect of
the ions presence on the protein. Finally, due to the fact that HS100A12 is involved in
some events of cell signaling and interaction with the Receptor for Advanced
Glycation End Products (RAGE), we also decided to study the interaction of protein
with models of biomembranes using Langmuir monolayers. In the other problem of
interest, we used a variety of mutants of the enzyme T4 lysozyme, a protein

“standard”, in order to obtain more details about its structure-function correlation and
make more solid the understanding of SDSL technique. Initially, we conducted a
study about the alleged creation of a cavity in the hydrophobic C-terminal portion of
the enzyme, when we replaced the Leu 133 by Ala and/or Gly, or when we changed
a large residue for a smaller one, because it is believed that the protein undergoes a
structural adjustment in order to fill the gap created by this substitution. For this, we
studied by SDSL the α-helix H motion, inserting the spin label in a neighbor position
of the mutated residue. Additionally, we performed an experiment of “transmutation”
with the enzyme T4L in order to investigate the nature of contributions for different
dynamic modes experienced by the spin label when it is introduced in topologically
similar sites.
Keywords: Site-directed spin labeling. Electronic Paramagnetic Resonance. Calcium binding proteins. Human S100A12. T4 Lysozyme.

Lista de Figuras
Figura 1- Reação através da qual é obtida a proteína marcada com
MTSSL. ............................................................................................................. 30
Figura 2- Estrutura química do radical nitróxido. (a): Estrutura da cadeia
lateral R1 indicando os ângulos diédricos χχχχ1 a χχχχ5. (b): Estrutura
da cadeia lateral R1 localizada em resíduo na superfície de
hélice αααα .............................................................................................................. 31
Figura 3- Mudanças na forma de linha de RPE em função da estrutura
local onde se encontra o marcador de spin................................................ 32
Figura 4- Estrutura do motivo “EF-hand” de proteínas ligantes de Ca+2 com
o íon cálcio em vermelho ............................................................................... 45
Figura 5- Motivo “EF-hand” ............................................................................................. 46
Figura 6- Estrutura do domínio “EF-hand” de proteínas ligantes de Ca+2
com o íon cálcio em vermelho ...................................................................... 47
Figura 7- Representação esquemática da estrutura secundária de uma
proteína S100 .................................................................................................. 49
Figura 8- Dímero da HS100A12 nativa, indicando as hélices A e D de cada
monômero ........................................................................................................ 50
Figura 9- Alinhamento sequencial entre CAGC humana (HS100A12) e
CAGC porcina.................................................................................................. 51
Figura 10- Comparação dos sítios de ligação dos íons cobre e zinco na
proteína HS100A12 ........................................................................................ 53
Figura 11- Esquema ilustrando uma possível rota para a transição dímero-
tetrâmero-hexâmero ....................................................................................... 54
Figura 12- Estrutura do monômero da proteína HS100A12 (PDB 1E8A),
indicando os resíduos mutados por cisteínas para posterior
marcação de spin ............................................................................................ 55
Figura 13- Estruturas das proteína HS100A12 apo (PDB 2WCE) em cinza,
com cálcio (PDB 1E8A) em azul e com zinco (PDB 2WCB) em
rosa, evidenciando a mudança conformacional na hélice C. .................. 56
Figura 14- Sequência codificadora da proteína HS100A12.......................................... 57
Figura 15- Amplificação do oligonucleotídeo que codifica o mutante HS100
A80C ................................................................................................................. 61

Figura 16- Origem do efeito do dicroísmo circular ..........................................................68
Figura 17- (A) Níveis de energia do elétron desemparelhado do grupo
nitróxido NO em um campo magnético externo. (B) Espectro de
RPE de um marcador de spin livre ...............................................................72
Figura 18- Ilustração de uma cuba de Langmuir equipada com um sensor
de tensão superficial e duas barreiras móveis ...........................................73
Figura 19- Isoterma de pressão de superfície para uma monocamada de
DPPC puro........................................................................................................74
Figura 20- Estrutura dos fosfolipídios utilizados para o estudo de
monocamadas de Langmuir ..........................................................................74
Figura 21- Expressão da proteína HS100A12. Proteínas visualizadas em
gel de poliacrilamida (SDS-PAGE 16%) em tampão tricina .....................75
Figura 22- Expressão dos mutantes A80C e F57C/A80C a 370C. Proteínas
visualizadas em gel SDS- PAGE 16% em tampão tricina ........................76
Figura 23- Purificação da proteína HS100A12 por cromatografia de
interação hidrofóbica e de exclusão molecular. Proteínas
visualizadas em gel SDS- PAGE 16% em tampão tricina ........................78
Figura 24- Análise por eletroforese em SDS-PAGE 16% em tampão tricina
da purificação dos mutantes da proteína HS100A12 ................................79
Figura 25- Cromatografia por exclusão molecular da proteína HS100A12
nativa e dos padrões de peso molecular .....................................................81
Figura 26- Espectros de CD da proteína HS100A12 na presença e
ausência dos íons cálcio e zinco em tampão 50mM Tris, 25mM
NaCl pH 7,5 a 200C .........................................................................................83
Figura 27- Análise da desnaturação térmica da proteína HS100A12
utilizando dicroísmo circular. (A): HS100A12 Apo. (B):
HS100A12 na presença de cálcio. (C): HS100A12 na presença
de zinco. (D): HS100A12 na presença de cálcio e zinco ..........................85
Figura 28- Transição da desnaturação térmica com a elipticidade
monitorada a 208 nm. (A): HS100A12 Apo. (B): HS100A12 na
presença de cálcio. (C): HS100A12 na presença de zinco. (D):
HS100A12 na presença de cálcio e zinco...................................................86
Figura 29- Desnaturação térmica da proteína HS100A12 na presença e
ausência dos íons cálcio e/ ou zinco ............................................................88

Figura 30- Gráficos ln(ln(1/fN) versus 1/T. (A): HS100A12 Apo. (B):
HS100A12 na presença de cálcio. (C): HS100A12 na presença
de zinco. (D): HS100A12 na presença de cálcio e zinco ......................... 89
Figura 31- Estrutura da TnC cardíaca humana (PDB 1SPY) com a
indicação das hélices A, B, C e D e N-terminal. ........................................ 91
Figura 32- Sobreposição das estruturas das proteínas TnC e HS100A12,
indicando os resíduos da proteína HS100A12 que
correspondem aos resíduos mutados na TnC ........................................... 92
Figura 33- Estrutura do dímero da proteína HS100A12 (PDB 2WCE),
indicando os resíduos mutados por cisteínas para posterior
marcação de spin ............................................................................................ 93
Figura 34- Espectros de RPE em banda X dos mutantes da proteína
HS100A12 com o marcador de spin MTSSL na ausência e na
presença de cálcio e zinco a 250C ............................................................... 96
Figura 35- Espectros normalizados de RPE em banda X do mutante
simples da proteína HS100A12 com o marcador de spin
MTSSL na posição 80 (preto) e dos duplos mutantes nas
posições (A) K33C/A80C, (B) I53C/A80C, (C) D54C/A80C, (D)
F57C/A80C, (E) L60C/A80C, (F) D61C/A80C (vermelho) a -
1000C, sem cálcio, na presença de cálcio e com zinco.......................... 100
Figura 36- Estrutura da proteína HS100A12 apo (PDB 2WCE)- (A) e com
cálcio (PDB 1E8A)- (B), indicando os resíduos marcados com
MTSSL e os valores das distâncias entre eles ........................................ 102
Figura 37- Cinética de adsorção da proteína HS100A12 sobre
monocamada de DPPC ............................................................................... 105
Figura 38- Cinética de adsorção da proteína HS100A12 sobre
monocamada de DPPG ............................................................................... 106
Figura 39- Isoterma de pressão de superfície para o DPPC com tampão,
HS100A12 apo, HS100A12 + Ca+2, HS100A12 + Zn+2 e
HS100A12 + Ca+2 + Zn+2 . ........................................................................... 110
Figura 40- Isoterma de pressão de superfície para o DPPG com tampão,
HS100A12 apo, HS100A12 + Ca+2, HS100A12 + Zn+2 e
HS100A12 + Ca+2 + Zn+2 . ........................................................................... 111

Figura 41- Alinhamento seqüencial entre Lisozima do bacteriófago T4
(T4Lysozyme) e Lisozima de clara de ovo de galinha
(eggLysozyme)...............................................................................................116
Figura 42- Estrutura da Lisozima do bacteriófago T4, indicando a região N
e C-terminal e as hélices C e H...................................................................117
Figura 43- Estrutura da Hélice C e da Hélice H indicando seus respectivos
resíduos e destacando os ambientes estruturais em torno da
posição 72 e 131 que serão explorados nos estudos por SDSL...........119
Figura 44- Seqüência codificadora da Lisozima T4......................................................120
Figura 45- Expressão do mutante V131C/ L133A. Proteínas visualizadas
em gel SDS-PAGE, gradiente 8-25 ............................................................125
Figura 46- Expressão do mutante D127N/E128Q/V131C. Proteínas
visualizadas em gel SDS-PAGE, gradiente 8-25 .....................................125
Figura 47- Expressão do mutante D127N/E128Q/V131C/K135R. Proteínas
visualizadas em gel SDS-PAGE, gradiente 8-25 ....................................126
Figura 48- Purificação do mutante da proteína T4L V131C por
cromatografia de interação iônica. Proteínas visualizadas em
SDS-PAGE, gradiente 8-25 .........................................................................127
Figura 49- Espectros de RPE em banda X da proteína T4L com o marcador
de spin MTSSL na posição 131 e dos mutantes da enzima T4L
L133A e L133G a 298K. ...............................................................................129
Figura 50- Espectros de RPE em banda X da proteína T4L com o marcador
de spin R1 nas posições 72 e 131..............................................................131
Figura 51- Espectros de RPE em banda X da proteína T4L com o marcador
de spin MTSSL na posição 131 e do mutante da enzima T4L
D127N a 273K, 298K e 313K, respectivamente.......................................132
Figura 52- Espectros de RPE em banda X da proteína T4L com o marcador
de spin MTSSL na posição 131 e do mutante da enzima T4L
D127N/ E128Q a 273K, 298K e 313K, respectivamente. .......................133
Figura 53- Espectros de RPE em banda X da proteína T4L com o marcador
de spin MTSSL na posição 131 e do mutante da enzima T4L
D127N/ E128Q/K135R a 273K, 298K e 313K, respectivamente...........134
Figura 54- Espectros de RPE em banda X da proteína T4L com o marcador
de spin MTSSL na posição 131 e do mutante da enzima T4L

D127N/E128Q/K135R/N132A a 273K, 298K e 313K,
respectivamente. ........................................................................................... 136


Lista de Tabelas
Tabela 1- Resíduos de aminoácidos de HS100A12 que foram mutados por
resíduos de cisteína, gerando mutantes simples e duplos da
proteína............................................................................................................. 58
Tabela 2- Oligonucleotídeos da enzima HS100A12 que foram utilizados na
amplificação por PCR..................................................................................... 59
Tabela 3- Distância spin-spin e variação da distância dos duplos mutantes
da proteína HS100A12 marcados com MTSSL....................................... 101
Tabela 4- Valores da pressão de superfície de dois modelos de membrana
na presença da proteína HS100A12 à concentração de 0,5 e
2,5 µg/ml, na presença e ausência dos íons cálcio e/ ou zinco. ........... 107
Tabela 5- Oligonucleotídeos da enzima T4L que foram utilizados na
amplificação por PCR................................................................................... 121
Tabela 6- Condições de expressão para os mutantes da proteína T4L ................. 126


Lista de Abreviaturas e Siglas
ACB Anidrase carbônica
BSA Albumina de soro bovino
CaBP Proteínas ligantes de cálcio (Calcium Binding Protein)
CD Dicroísmo Circular (Circular Dichroism)
Cit C Citocromo C
DHO Diidroorotato
DHODH Diidroorotato desidrogenase
DNA Ácido desoxirribonucleico
dNTP Desoxirribonucleotídeos trifosfatos
DO Densidade ótica
DPPC 1,2-dipalmitoil-sn-glicero-3-fosfatidil colina
DPPG 1,2-dipalmitoil-sn-glicero-3-fosfatidil glicerol
DTT DL-ditiotreitol
Ea Energia de ativação
EcDHODH Diidroorotato desidrogenase de Escherichia coli
EDTA Ácido etileno diamino tetracético
ExPASy Expert Protein Analysis System
FMN Flavina mononucleotídeo
FN Fração de proteína nativa
G gasoso
HsDHODH Diidroorotato desidrogenase humana
HS100A12 S100A12 humana
IPTG Isopropil-β-D-tiogalactopiranosídeo
ITS Inibidor de Tripsina de soja
LB Luria- Bertani
LC Líquida- condensada
LE Líquida- expandida
LlDHODH Diidroorotato desidrogenase de Lactococcus lactis
LmDHODH Diidroorotato desidrogenase de Leishmania major
MTSSL (1-oxi-2,2,5,5-tetrametilpirrolina-3-metil)metanetiosulfonato
MSC Múltiplos sítios de clonagem

OVA Ovalbumina
PCR Reação em Cadeia da Polimerase (Polymerase Chain Reaction)
PDB Protein Data Bank
pET28a Vetor de clonagem
pHSeS Vetor de clonagem
pI Ponto isoelétrico
PM-IRRAS Espectroscopia de absorção e reflexão no infravermelho com
modulação por polarização (Polarization-Modulation Infrared
Reflection Absorption Spectroscopy)
PMSF fluoreto de fenilmetano sulfonila
pQE60 Vetor de clonagem
R1 Cadeia lateral do aminoácido marcado, contendo o radical nitróxido
RAGE Receptor para produtos de glicosilação (Receptor for Advanced
Glycation End Products)
RMN Ressonância Magnética Nuclear
RNA Ácido ribonucleico
RPE Ressonância Paramagnética Eletrônica
S fase sólida
SDS Dodecil sulfato de sódio
SDSL Marcação de Spin Sítio- dirigida (Site-Directed Spin Labeling)
SDS- PAGE Eletroforese em gel de poliacrilamida - SDS (Sodium Dodecyl
Sulphate - Polyacrilamide Gel Electrophpresis)
T4L Lisozima do fago T4
TcDHODH Diidroorotato desidrogenase de Trypanosoma cruzi
TnC Troponina cardíaca humana
Tm Temperatura de transição (melting temperature)
UV Ultra violeta

SUMÁRIO
1 Introdução............................................................................................. 27
1.1 Ressonância Paramagnética Eletrônica (RPE) de Sistemas
Biológicos............................................................................................. 28
1.1.1 Dinâmica do Marcador de Spin............................................................ 30
2 Objetivos............................................................................................... 35
3 Materiais............................................................................................... 39
3.1 Materiais e Equipamentos.................................................................... 39
3.2 Reagentes e Resinas........................................................................... 40
4 As Proteínas Ligantes de Cálcio (CaBP- “Calcium Binding Protein”)... 45
4.1 Proteínas da Família S100................................................................... 47
4.2 S100A12 Humana (HS100A12)........................................................... 51
4.3 Metodologia.......................................................................................... 57
4.3.1 Construção dos Oligonucleotídeos e Amplificação dos Genes............ 57
4.3.2 Reação de Dupla Digestão................................................................... 60
4.3.3 Subclonagem dos Genes no Plasmídeo.............................................. 61
4.3.4 Transformação da Bactéria Escherichia coli DH5α com o Plasmídeo
Recombinante pQE60.......................................................................... 62
4.3.5 Extração e Análise Sequencial do Plasmídeo Recombinante pQE60
de Escherichia coli DH5 α.................................................................... 63
4.3.6 Transformação da Bactéria Escherichia coli Rosetta com o
Plasmídeo Recombinante pQE60........................................................ 64
4.3.7 Expressão da Proteína HS100A12....................................................... 64
4.3.8 Purificação da Proteína HS100A12...................................................... 65
4.3.9 Separação dos Oligômeros da Proteína HS100A12............................ 65
4.3.10 Determinação da Concentração da Proteína HS100A12..................... 66
4.3.11 Dicroísmo Circular................................................................................ 66
4.3.12 Marcação de Spin Sítio Dirigida dos Mutantes da Proteína
HS100A12............................................................................................ 70
4.3.13 Ressonância Paramagnética Eletrônica............................................... 71
4.3.14 Monocamadas de Langmuir................................................................. 72
4.4 Resultados e Discussões..................................................................... 75
4.4.1 Expressão Heteróloga da Proteína HS100A12.................................... 75

4.4.1.1 Expressão dos Mutantes da Proteína HS100A12................................ 75
4.4.2 Purificação da Proteína HS100A12...................................................... 77
4.4.2.1 Purificação dos Mutantes da Proteína HS100A12............................... 78
4.4.3 Influência dos Íons Cálcio e Zinco no Estado de Oligomerização da
HS100A12............................................................................................ 79
4.4.4 Dicroísmo Circular................................................................................ 81
4.4.4.1 Fatores Estruturais da Proteína HS100A12 e dos seus Mutantes....... 82
4.4.4.2 Estudo da Estabilidade Térmica da Proteína HS100A12..................... 84
4.4.5 Estudo de Ressonância Paramagnética Eletrônica por marcação de
spin sítio dirigida da transição estrutural induzida pelos íons cálcio e
zinco na S100A12 humana.................................................................. 90
4.4.6 Interação da proteína HS100A12 com fosfolipídios utilizando
modelos de biomembranas.................................................................. 103
5 As Proteínas Lisozimas........................................................................ 115
5.1 Lisozima do Bacteriófago T4 (T4L)...................................................... 116
5.2 Metodologia.......................................................................................... 119
5.2.1 Produção dos mutantes da proteína T4L............................................. 119
5.2.2 Expressão da Enzima Lisozima do Bacteriófago T4............................ 122
5.2.3 Purificação da Enzima Lisozima do Bacteriófago T4........................... 123
5.2.4 Determinação da Concentração da Proteína T4L................................ 123
5.2.5 Medidas de RPE................................................................................... 123
5.3 Resultados e Discussões..................................................................... 124
5.3.1 Expressão dos Mutantes da Enzima T4L............................................. 124
5.3.2 Purificação dos Mutantes da Enzima T4L............................................ 126
5.3.3 Resposta Estrutural da Lisozima T4 a Substituições de Aminoácidos
Grandes por Pequenos no Interior da Proteína.................................... 127
5.3.4 SDSL na determinação de diferenças estruturais entre sítios
topologicamente equivalentes da Enzima T4L..................................... 129
6 Conclusões........................................................................................... 139
Referências.......................................................................................................... 145
Apêndice A.......................................................................................................... 155
Apêndice B.......................................................................................................... 167

INTRODUÇÃO


Introdução 27
1 Introdução
As proteínas são moléculas com estruturas dinâmicas que desempenham uma
variedade de funções importantes nas células de organismos vivos, incluindo seres
humanos. O entendimento sobre sua relação estrutura-função nos fornece
informações essenciais para aplicações práticas nas áreas de medicina, agricultura,
nutrição e em indústrias (1). Com isso, há um intenso esforço em se investigar os
modos dinâmicos de proteínas em seu estado nativo. Para tal, a união entre
estruturas estáticas, obtidas a partir de cristalografia de proteínas, e técnicas que
devolvam o aspecto dinâmico ao problema (como é o caso de espectroscopias
diversas) oferece uma oportunidade única para que se faça uma abordagem ampla
do problema de interesse.
Mudanças estruturais em proteínas entre duas diferentes conformações é um
processo dinâmico que muitas vezes regula a atividade enzimática ou a função
biológica da macromolécula e que tem sido observado para proteínas pequenas
através de várias técnicas experimentais, em particular, as de ressonância
magnética nuclear (RMN) e eletrônica (RPE).
Acredita-se que há uma hierarquia desses modos dinâmicos (2, 3), onde a
proteína que se encontra em um determinado estado, consiste de vários subestados
conformacionais em equilíbrio, com tempo de meia vida entre micro e milisegundos
cada um, antes de migrarem para outro subestado (mudança conformacional) (4).
Esses subestados conformacionais são também chamados de subestados
taxonômicos porque, geralmente, ocorrem em pequena quantidade e possuem
tempo de vida suficientemente longo para serem descritos com detalhes
moleculares.
Por outro lado, cada subestado conformacional pode ser formado por um
conjunto de outros subestados, chamado de subestados estatísticos, que são
numerosos e de vida curta, variando entre pico e nanosegundos. As transições entre
esses subestados estatísticos correspondem a movimentos de alta frequência da
cadeia principal e lateral das proteínas (4), por exemplo, a abertura e fechamento de
uma “tampa” importante para a ligação do ligante na proteína (5).

28
Dentre os métodos experimentais para se investigar alterações
conformacionais em estruturas de macromoléculas biológicas que ocorrem nessa
escala de tempo, observamos um crescimento e consolidação da técnica conhecida
como marcação de spin sítio dirigida. O emprego da técnica de marcação de spin
sítio dirigida aliada à ressonância paramagnética eletrônica (SDSL-RPE) a fim de
investigar a estrutura e dinâmica de proteínas se deu apenas no final da década de
80 (6),com sua utilização de forma mais corriqueira em diversos laboratórios
conseguida no final dos anos 90. Exemplos recentes incluem interações de diversas
proteínas com membrana biológica (7, 8); bases estruturais da transdução de
energia (9); mudanças conformacionais induzidas por Ca+2 (10); mudanças
relacionadas com o movimento de domínios que controlam o acesso de substrato e
produto ao sítio ativo de enzimas (11) e mudanças ativadas por pH (12).
Contudo, no Brasil ainda está sendo conduzido um esforço para que a técnica
SDSL-RPE se estabeleça como uma ferramenta poderosa na análise
conformacional de proteínas, podendo ajudar a desvendar a relação entre estrutura,
dinâmica e função em situações diversas. Portanto, como um dos objetivos
primordiais do presente trabalho de tese tivemos a implementação da metodologia de
"site-directed spin labeling" no Grupo de Biofísica Molecular do Instituto de Física de
São Carlos (IFSC/USP).
Para isso, é proposto o estudo da correlação estrutura e função através da
técnica SDSL-RPE de dois sistemas biológicos muito interessantes. O primeiro deles
é o estudo do movimento das hélices que compõem a estrutura da proteína
S100A12 humana (HS100A12) induzido pelos íons cálcio e zinco. O outro utiliza a
lizosima do fago T4, uma proteína “padrão”, da qual uma variedade de mutantes é
produzida rotineiramente, a fim de se obter mais detalhes a respeito da sua
correlação estrutura e função e tornar mais sólido o entendimento da técnica SDSL.
1.1 Ressonância Paramagnética Eletrônica (RPE) de Sistemas Biológicos
A técnica de RPE é uma técnica poderosa, que faz uso de elétrons
desemparelhados presentes em diversos compostos. Como a maioria dos
compostos de interesse biológico é diamagnética, ou seja, não possui elétrons

Introdução 29
desemparelhados, foi desenvolvido por Ohnishi e McConnell (13) o método do
marcador de spin, que se baseia na introdução de um marcador ou molécula
repórter no sistema a ser estudado por RPE. Em geral, esse grupamento é o radical
nitróxido (N-O) ligado covalentemente a moléculas como fosfolipídios ou resíduos de
cisteína em proteínas, para monitorar mudanças na vizinhança do grupo repórter
(14, 15, 16).
A técnica de “Site-Directed Spin Labeling” ou marcação de spin sítio dirigida,
aliada à ressonância paramagnética eletrônica (SDSL-RPE), surgiu como uma
ferramenta bastante promissora na determinação de estruturas de proteínas em
nível da cadeia polipeptídica. Tanto proteínas solúveis quanto aquelas presentes em
membranas e de peso molecular arbitrário são passíveis de estudo por SDSL com
concentrações de amostra na ordem de 100 pmol (17,16).
A estratégia da SDSL envolve a introdução de um radical nitróxido em uma
posição específica da proteína e uma mutação sítio dirigida é normalmente usada
para esse fim, sendo um resíduo nativo trocado por um de cisteína, para a posterior
reação do seu grupo sulfidrílico com um marcador de spin adequado (Figura 1). Até
hoje, o reagente metanotiosulfonato (MTSSL), que gera a chamada cadeia lateral
R1, tem sido o mais utilizado para fins de SDSL, embora outros tipos também já
tenham sido empregados (16). Perturbações no enovelamento, estabilidade térmica
ou funcional devido à introdução do marcador de spin parecem não ser um problema
limitante para a maioria dos casos investigados (16, 17, 18).
A partir de uma cadeia lateral contendo um radical nitróxido podemos obter
informações como: parâmetros de acessibilidade, Π, que é determinado a partir da
frequência de colisão entre o nitróxido e um reagente paramagnético em solução e
que, por sua vez, é proporcional à acessibilidade do solvente à determinada cadeia
lateral; mobilidade da cadeia lateral; topografia da cadeia polipeptídica (18, 19, 20);
podemos identificar estruturas secundárias regulares e sua orientação dentro da
proteína (18); medir a distância entre dois sítios marcados (21); identificar sítios
onde existam interações entre estruturas terciárias e investigar mudanças estruturais
em função do tempo dos parâmetros acima (22, 23, 24).

30
Figura 1- Reação através da qual é obtida a proteína marcada com MTSSL. A cadeia lateral marcada é
denominada R1. Figura adaptada de Frazier et al, 2002 (25).
1.1.1 Dinâmica do Marcador de Spin
O movimento da cadeia lateral contendo o radical nitróxido (R1) em uma escala
de tempo entre picosegundos e nanosegundos resulta em relaxação magnética que
determina o espectro de RPE. Com isso, informações da dinâmica da proteína que
ocorre nessa escala de tempo são obtidas por SDSL através da forma da linha
espectral, que resulta da superposição de três movimentos executados pela
estrutura protéica: rotação da proteína inteira (“tumbling”), rotação em torno das
ligações que conectam o nitróxido à cadeia principal da proteína e movimento local
da cadeia principal onde está o nitróxido.
Entretanto, as informações buscadas por SDSL são obtidas apenas pelos
movimentos em torno das ligações que conectam o nitróxido à cadeia principal e o
movimento da cadeia principal da proteína. O movimento rotacional da proteína
como um todo é uma constante para as cadeias laterais de todos os resíduos na
estrutura da proteína nativa. A contribuição desse movimento para o espectro de
RPE é pequena no caso de proteínas de massa molecular elevada ou pode ser
reduzida, no caso de estruturas pequenas, através do artifício de se fazer as
medidas de RPE em solvente contendo, por exemplo, 30% de sacarose (aumento
da viscosidade do meio). Esta é a condição ideal de trabalho já que não nos
interessa o movimento de “tumbling” da proteína inteira, mas sim flutuações locais
de sua cadeia principal.
O tempo de correlação, τi, que está associado ao movimento em torno das
ligações que conectam o nitróxido à cadeia principal, depende tanto da estrutura do
marcador de spin (tamanho da cadeia lateral), quanto das estruturas primária,
secundária e terciária da proteína, ao passo que o tempo de correlação associado

Introdução 31
ao movimento da cadeia principal, τB, é determinado pela flexibilidade do segmento
de cadeia principal em estudo.
Rotações da cadeia lateral R1 acontecem em torno das ligações entre o grupo
N-O e a cadeia principal (Figura 2A). Essas ligações formam ângulos arbitrários
entre si, o que torna muito difícil o ajuste dos espectros experimentais aos modelos
usualmente empregados em simulação espectral, já que estes envolvem, em geral,
movimentos em torno de eixos mutuamente ortogonais. Sendo assim, uma
descrição semiquantitativa acabou por tornar-se comum para análise dos espectros
obtidos e que permite interpretar os dados experimentais em termos da relação
mobilidade da cadeia lateral e estrutura protéica. O termo mobilidade é usado com
um significado mais geral para incluir efeitos de ordenamento molecular e taxa de
movimento. Assim, um estado de baixa mobilidade pode se dever tanto a uma
situação de movimento de grande amplitude e baixa taxa de difusão quanto a um
movimento restrito executado com alta freqüência (26).
Figura 2- Estrutura química do radical nitróxido. (a): Estrutura da cadeia lateral R1 indicando os ângulos
diédricos χ1 a χ5. (b): Estrutura da cadeia lateral R1 localizada em resíduo na superfície de hélice α. Está conformação indica que rotações moleculares são feitas em torno dos ângulos diédricos χ4 e χ5. As linhas pontilhadas indicam os eixos em torno dos quais essas rotações ocorrem. Figura adaptada de Pistolesi et al, 2006 (27).
A referida descrição semiquantitativa envolve uma análise da dinâmica
experimentada pela cadeia lateral R1 nos sítios de interesse da proteína. Como já
dito, esta dinâmica se reflete na forma de linha do espectro de RPE de uma maneira
dependente da estrutura local experimentada pela sonda paramagnética. A relação
entre estrutura local e dinâmica pode ser resumida pelos espectros típicos
mostrados na figura 3. Neste caso, os espectros se referem ao marcador MTSSL em
regiões de loop, de superfície de hélice, de contatos terciários e sítios enterrados na
estrutura da lisozima T4 (18). A classe englobando sítios em contatos terciários é a

32
que possui maior grau de heterogeneidade no que diz respeito à forma de linha dos
espectros e mobilidade da cadeia lateral porque tanto o grau de contatos estéricos
quanto a mobilidade da cadeia principal mudam com a posição do sítio. Neste ponto,
cabe definir o que entendemos por sítios em contatos terciários (ou sítios em
interações terciárias) como sendo sítios nos quais a substituição com a cadeia R1
cria conflitos estereoquímicos com átomos das cadeias laterais ou da cadeia
principal de resíduos adjacentes na estrutura tridimensional (18, 28). Portanto, sítios
envolvidos em contatos terciários podem apresentar espectros que variam desde
aqueles obtidos para sítios enterrados até espectros vistos para sítios em superfícies
de hélices. A figura 3 mostra, portanto, como medidas de RPE podem distinguir a
dinâmica de diferentes elementos de estrutura secundária.
Figura 3- Mudanças na forma de linha de RPE em função da estrutura local onde se encontra o marcador de
spin. Figura adaptada de Mchaourab et al, 1999 (28).

OBJETIVOS

34

Objetivos 35 2 Objetivos
O objetivo deste trabalho foi implementar, utilizando de forma intensiva e em
diferentes vertentes, a metodologia de marcação de spin sítio dirigida no Grupo de
Biofísica Molecular Sérgio Mascarenhas do Instituto de Física de São Carlos (USP).
Para tal, realizamos estudos estruturais e funcionais das proteínas S100A12
humana (HS100A12) e lisozima do bacteriófago T4 (T4L). Os objetivos específicos
foram:
• Estudos por SDSL para medidas de distância entre duas hélices da proteína
HS100A12 que se movimentam quando da ligação com os íons cálcio e
zinco.
• Estudos da mudança conformacional experimentada por uma hélice
específica da proteína HS100A12 na presença dos íons cálcio e/ou zinco
utilizando a técnica de marcação de spin sítio dirigida (SDSL).
• Estudos por SDSL para investigação da suposta criação de uma cavidade
no “core” hidrofóbico da porção C-terminal da enzima T4L quando trocamos
um resíduo grande por um de menor volume.
• Estudo da interação entre as cadeias laterais do marcador de spin e dos
resíduos vizinhos de uma dada hélice da lisozima do fago T4 com o intuito
de consolidar a técnica SDSL e acrescentar informações no banco de dados
de SDSL de proteínas, isto é, uma biblioteca que possibilita a correlação da
forma da linha do espectro com a estrutura de proteínas.
Adicionalmente, como objetivos secundários, mas não menos importantes,
realizamos:
• Estudos da estabilidade estrutural da HS100A12 recombinante e seus dos
mutantes com o uso da espectroscopia de dicroísmo circular, avaliando o
papel exercido pelo íons Ca+2 e Zn+2 sobre a estabilidade estrutural da
proteína.
• Investigação da interação da proteína HS100A12 com fosfolipídios
utilizando monocamada de Langmuir como modelo de membrana.

36

MATERIAIS

38

Materiais 39 3 Materiais
Nesta seção estão listados os materiais e equipamentos utilizados na
realização de todos os experimentos apresentados neste trabalho.
3.1 Materiais e Equipamentos
• Termocicladores RobocyclerR 40 (Stratagene), Mastercycler (Eppendorf) e
Veriti 96 Well- Applied Biosystems
• Transiluminador BioAgency
• Sequenciadores automático ABI Prism 3100 Genetic Analyser (Hitachi) e
ABI Prism 377-96 Collection (Applied Biosystem)
• Sistema para eletroforese Bio-rad e PhastGel
• Centrífuga refrigerada Sorval modelo 5B e RC5C, centrífuga refrigerada
Eppendorf modelo 5804R e centrífuga de bancada Eppendorf modelo
5414D
• Agitador refrigerado Forma Scientific
• Sonicador Fisher Scientific modelo 500
• Sistema para cromatografia de baixa pressão ÄKTA Purifier (GE Healthcare)
• Espectrofotômetros Hitachi modelos U-2001 e U-2801
• Concentradores Amicon Ultra-15 Centrifugal Filter Devices (Milipore)
• Cubeta retangular de quartzo – caminho ótico de 0,1 cm
• Espectropolarímetro modelo J-720 (Jasco) equipado com banho de água
(TC100) e controlador de temperatura Jasco PFD 425S
• Espectrômetros de RPE modelo E109 (Varian) operando em modo de onda
contínua e equipado com ponte de microondas em banda X (9,5 GHz) e
Bruker ESP300, equipado com uma cavidade de alta sensibilidade
• Controlador de temperatura VT Controler E257-X (Varian)
• Sistema de fluxo de nitrogênio (Bruker, Rheinstetten, Germany)

40
• Cela chata de quartzo Avanti
• Cuba de Langmuir Mini- KSV (KSV Instruments, Finlândia) equipada com
um sensor que determina a tensão superficial (método Wilhelmy)
• Capilares de vidro (VitroCom Inc., Mountain Lakes, NJ).
3.2 Reagentes e Resinas
Os reagentes utilizados para reação em cadeia da polimerase (PCR),
oligonucleotídeos, enzimas de restrição BamHI, HindIII, NcoI e NdeI e seus
respectivos tampões foram obtidos da Invitrogen Life Technologies (EUA). A enzima
Pfu Turbo DNA polimerase, tampão Pfu, foram obtidos da Fermentas (Canadá). A
enzima T4 DNA Ligase foi adquirida da Promega (EUA). Os quites para extração de
DNA “Kit QIAquick Gel Extraction”, “Kit QIAquick PCR Purification” e “kit QIAprep
Spin MiniPrep” foram adquiridos da Qiagen. Os oligonucleotídeos específicos
(primers) foram obtidos da Integrated DNA Technologies (Coralville, IA, EUA).
As linhagens de Escherichia coli (E. coli) utilizadas nos experimentos de
clonagem e expressão foram DH5α, XL1, BL21(DE3) e Rosetta obtidas da Promega
e Novagen (San Diego, CA, EUA). O antibiótico ampicilina e o indutor isopropil-β-D-
tiogalactopiranosideo (IPTG) foram comprados da Calbiochem (La Jolla, CA, EUA).
As colunas cromatográficas HiTrap SP HP, HiTrapTM Desalting, Hiprep Fenil-
Sefarose FF e Superdex 75 10/300 GL, géis PhastGel- Gradiente 8-25 e tampão
PhastGel SDS Buffer Strips foram comprados da GE Healthcare (Uppsala, Suécia).
Os reagentes Tris, MOPS, NaCl, MgCl2, CaCl2, ZnCl2, acrilamida, NaOH, HCl,
glicerol, β-mercaptoetanol, ácido acético, agarose, ácido etileno diamino tetracético
(EDTA), fluoreto de fenilmetano sulfonila (PMSF), brometo de etídeo, clorofórmio,
acetonitrila, DL-ditiotreitol (DTT), meio de cultura Luria-Bertani (LB), albumina do
soro bovino (BSA – 66 kDa), ovalbumina (OVA – 43 kDa), anidrase carbônica (ACB
– 30 kDa), inibidor de tripsina de soja (ITS - 20,1 kDa), e citocromo C (cit C - 12,4
kDa) foram obtidos da empresa Sigma-Aldrich Chemical (EUA). O marcador de spin
MTSSL ((1-oxi-2,2,5,5-tetrametilpirrolina-3-metil)metanetiosulfonato) foi comprado da
empresa Toronto Research Chemicals (Canadá).

Materiais 41
Os fosfolipídios DPPC (1,2-dipalmitoil-sn-glicero-3-fosfatidil colina) e DPPG
(1,2-dipalmitoil-sn-glicero-3-fosfatidil glicerol) foram adquiridos comercialmente da
empresa Avanti Polar Lipids (Alabaster, EUA).

42

S100A12 HUMANA


HS100A12 45 4 As Proteínas Ligantes de Cálcio (CaBP- “Calcium Binding
Protein”)
O cálcio é um dos mais importantes, se não o mais importante metal para a
vida de todos os organismos, pois controla processos vitais que englobam desde
mineralização dos ossos à sinalização celular. Ele interage com um grande número
de proteínas, mas a variedade de configurações com sítios de ligação de cálcio é
quase limitada. O mais comum entre estes é o motivo “EF- hand” (Figura 4).
Figura 4- Estrutura do motivo “EF-hand” de proteínas ligantes de Ca+2 com o íon cálcio em vermelho.
Figura adaptada de Bhattacharya et al, 2004 (29).
O termo “EF-hand” foi criado por Kretsinger e Nockolds (30) há 25 anos para a
descrição gráfica do motivo ligante de cálcio observado na parvalbumina, uma
pequena proteína ligante de cálcio isolada do músculo de carpa. Foi uma ótima
escolha do nome, pois ele descreve não somente o enovelamento do polipeptídio,
mas também o potencial movimento que é induzido pela ligação do cálcio (Figura
5A). Desde então, esse motivo estrutural foi encontrado em um grande número de
família de proteínas, com 66 subfamílias conhecidas até hoje (31). Na grande
maioria de proteínas que foram estudadas, o motivo certamente liga o cálcio ou, em
alguns casos, o magnésio (32, 33, 34).
As funções das proteínas que possuem o motivo “EF-hand” são tão diversas
quanto: tamponamento do cálcio no citosol, homeostase (29), transdução de sinal
entre compartimentos celulares (35), exocitose, metabolismo, transcrição, fertilização
e proliferação celular e contração muscular (36). Esse motivo estrutural, que pode
acomodar tanto o íon cálcio, como o magnésio com pequenas diferenças na

46
afinidade e na variação induzida pela mudança conformacional (37), é adotado de
uma maneira muito pragmática pelas bactérias (38), sempre que uma ou outra
característica seja vantajosa.
O clássico “EF-hand” é um motivo do tipo hélice-loop-hélice, caracterizado pela
sequência de 12 resíduos com o padrão X•Y•Z•–Y•–X••–Z, onde X, Y, Z, –X, –Y e –Z
são os ligantes que participam na coordenação do metal e os pontos representam os
resíduos entre eles (Figura 5B) (39). Essa sequência forma um loop que pode
acomodar cálcio ou magnésio, diferindo apenas na formação geométrica: magnésio
é usualmente ligado por seis ligantes em um octaedro, enquanto sete ligantes do
vértice de uma pirâmide pentagonal coordenam o cálcio.
(A) (B)
Figura 5- Motivo “EF-hand”. (A): Representação esquemática do motivo. A hélice E é representada pelo dedo
indicador, enquanto a hélice F é representada pelo polegar da mão direita. Quando o Ca+2 se liga, a hélice F se move de uma conformação “fechada” (cinza claro) para uma conformação “aberta” (cinza escuro). (B): geometria da ligação do Ca+2 com a proteína. Nas posições X e Y, encontram-se normalmente cadeias laterais de ácido aspártico ou asparagina; na posição Z encontram-se cadeias laterais de ácido aspártico, asparagina e serina e o oxigênio da carbonila do peptídeo encontra-se em –Y. –X normalmente é uma molécula de água e –Z é um ligante bidentado, ácido glutâmico ou ácido aspártico. Figura adaptada de Lewit- Bentley et al, 2000 (39).
A unidade estrutural/ funcional básica das proteínas ligantes de cálcio é um par
de motivos “EF-hand” (30) que juntos formam um domínio de quatro hélices estável
(Figura 6) (40, 41). O pareamento dos motivos “EF-hand” permite cooperatividade na
ligação dos íons Ca+2, que é essencial para o fornecimento de uma resposta clara,
mesmo sob uma variação relativamente modesta na concentração do Ca+2 durante a
sinalização (42). Estudos mostram que todo domínio “EF-hand” deve ser tratado
como uma unidade única, globalmente cooperativa (40, 41). Com isso, para se
entender a transdução do sinal de cálcio por proteínas “EF-hand” é também

HS100A12 47 importante reconhecer se a afinidade por Ca+2, respostas conformacionais devido à
ligação com íons e interação com moléculas alvos são energeticamente favoráveis
(43). Muitas proteínas do tipo “EF-hand” possuem mais de um único domínio “EF-
hand” e as variações estruturais quanto à organização desses domínios contribuem
significativamente para a seletividade de moléculas alvos e para a diversidade de
funções dessas proteínas.
A
B
C
D
A
B
C
D
Figura 6- Estrutura do domínio “EF-hand” de proteínas ligantes de Ca+2 com o íon cálcio em vermelho.
Diferentes cores são usadas para distinguir os parres “EF-hand”. Figura adaptada de Bhattacharya, et al, 2004 (29).
4.1 Proteínas da Família S100
As proteínas pertencentes à família S100 são pequenas proteínas “EF-hand”
de ligação com cálcio com peso molecular entre 10 e 14 kDa. O nome S100 foi
introduzido porque várias proteínas desse grupo eram solúveis em 100% de sulfato
de amônia.
O primeiro membro dessa família foi identificado como uma mistura não
fracionada das proteínas S100B e S100A1, que foram isoladas de cérebro bovino e
definida como específica do cérebro (44). Vários anos depois, descobriu-se que a
proteína S100 de cérebro bovino consistia de duas proteínas estritamente
relacionadas (S100B e S100A1) (45, 46) e não eram, de fato, específicas do cérebro
(47).A análise da sequência indicou que a S100B e S100A1 eram proteínas que
ligavam Ca+2 em um motivo “EF-hand” (45, 46), estruturalmente relacionadas com as
proteínas de ligação de cálcio “EF-hand” já conhecidas, como as calmodulina,
troponina C e parvalbumina (48).

48
Até o momento, aproximadamente 25 membros da família de proteínas S100
foram identificados em humanos, formando a maior família de proteínas ligantes de
cálcio pertencentes à superfamília “EF-hand” (49). Quase todas as proteínas S100
tendem a formar dímeros, considerados como essenciais para suas funções e
ligações com alvo (50, 51, 52). As exceções encontradas para isso são: a
monomérica calbindina D9K, que possui um papel de tamponamento de cálcio (53) e
p26olf, que é uma molécula de cadeia única consistindo de dois domínios quase
idênticos aos encontrados nas proteínas S100 (54, 55). Adicionalmente, temos
também como exceções duas proteínas com alto peso molecular, que são proteínas
associadas a filamentos intermediários precursoras de profilagrina (56) e tricoialina
(57) e que possuem domínios como os encontrados nas proteínas S100. Em alguns
casos, as proteínas S100 podem formar também, heterodímeros. Por exemplo,
S100B é encontrada no cérebro na forma dimérica (80%) e heterodimérica (20%
S100B + S100A1) (58).
O número de estruturas cristalográficas de proteínas da família S100 está
crescendo rapidamente. Atualmente, encontram-se aproximadamente coordenadas
atômicas de oitenta estruturas depositadas no Banco de Dados de Proteínas
(Protein Data Bank –PDB, http://www.pdb.org/pdb/home/home.do), sendo elas
correspondetes às formas protéicas apo ou na presença de diferentes ligantes:
Calbidina D9K bovina (59), Calcilclina de coelho (60), Calprotectina (S100A8/
S100A9) (estrutura submetida), S100P humana (61), S100B bovina (62), S100B de
humana (63), S100A1 de rato (64), S100A2 (65), S100A3 humana (66), S100A4
(67), S100A5 (68), S100A6 humana (69), S100A7 humana (70), S100A8 humana
(71), S100A9 (72), S100A10 humana (73), S100A11 humana (52), S100A12 humana
(74, 75), S100A16 humana (76) e S100A13 humana (77).
Nas proteínas da família S100, os dois motivos ligantes de Ca+2 do tipo “EF-
hand” possuem características diferentes. O motivo N-terminal (EF-1) é composto
por duas α-hélices (A e B) ligadas por um “loop” ligante de cálcio (LI); o segundo
motivo, C-terminal (EF-2), possui as hélices C e D ligadas por um “loop” ligante de
cálcio (LIII); os dois motivos “EF” são conectados por um “loop linker” LII, chamado
de hinge (Figura 7).

HS100A12 49
A B C DA B C D
Figura 7- Representação esquemática da estrutura secundária de uma proteína S100. Cada “loop” ligante de Ca+2 (LI e LIII, no meio do N- e C- terminal, respectivamente) é conectado por α-hélices (A e B, C e D). A região “linker” conecta as hélices B e C, dos motivos EF-1 e EF-2, respectivamente. Figura adaptada de Donato, et al, 2001 (78).
O motivo N-terminal ligante de Ca+2 (EF-1), característico de proteínas da
família S100, é uma região rica em aminoácidos básicos, com uma sequência de 31
resíduos (14 resíduos no “loop” que liga Ca+2) com o Ca+2 ligado principalmente
através dos átomos de oxigênio carbonil da cadeia principal (79). EF-1 tem baixa
afinidade pelo cálcio quando comparada com a calmodulina e troponina C e mais
ainda quando comparada com EF-2 das proteínas S100. Sua conformação quase
não é alterada quando ocorre a ligação do cálcio (80).
Por outro lado, o motivo C-terminal ligante de Ca+2 (EF-2), que contém um
“loop canônico”, é muito similar ao motivo “EF-hand” da calmodulina (81), troponina
C (82) e parvalbumina (83). Assim como elas, ele apresenta alta afinidade pelo Ca+2
e sofre mudanças conformacionais significativas após ligação com o íon. O motivo
EF-2 contém 29 resíduos, sendo doze deles do “loop canônico” que liga Ca+2 (84).
No EF-2, o Ca+2 é ligado por cinco átomos de oxigênio de cadeias laterais, um
átomo de oxigênio carbonil da cadeia principal e uma molécula de água.
Provavelmente, a diferença na estrutura e na afinidade pelo íon Ca+2 dos dois
motivos “EF-hand” contribui para a função de regulação do cálcio na célula (85, 69).
Uma grande mudança conformacional na proteína é induzida pela ligação do
cálcio, passando da conformação “fechada” para “aberta”, expondo assim sua
superfície hidrofóbica e interagindo com moléculas alvos, receptores extracelulares e
outras proteínas (86).
A identidade sequencial entre as proteínas S100 está entre 35 e 45%, com a
região de menor identidade sequencial entre as hélices B e C, chamada de “hinge”
(Figura 6) (75). Baseado nessa baixa conservação da sequência, se propõe que o
“hinge” seja o responsável pela especificidade no reconhecimento da molécula alvo,
como por exemplo, nas estruturas da S100 complexada com peptídeos alvos, onde
esses peptídeos estão localizados entre o “hinge” e o C-terminal da hélice D
(52,73,75).

50
Com a ligação do cálcio há um movimento significativo na hélice C da proteína,
expondo assim uma superfície hidrofóbica constituída de resíduos presentes na
região “hinge”, hélice C e C-terminal. A interface de dimerização é formada pelas
hélices A e D de cada uma das subunidades monoméricas e o conjunto das quatro
hélices envolvidas na dimerização é altamente similar nas proteínas S100 apo (sem
íon) e com cálcio (Figura 8). Contudo, há uma diferença significativa entre as
estruturas desses dois estados e isso provavelmente explica porque as estruturas da
proteína na presença de cálcio não foram resolvidas com sucesso por modelos de
substituição molecular a partir dos dados da estrutura apo (65,66,69).
A
D
A
D
A
D
A
D
Figura 8- Dímero da HS100A12 nativa, indicando as hélices A e D de cada monômero.
Uma característica adicional das proteínas S100 é a afinidade que podem
apresentar por outros metais divalentes, como os íons Zn+2 e Cu+2, sendo os sítios
de ligação para estes, distintos dos sítios de ligação para o Ca+2. A ligação destes
íons divalentes pode desempenhar um papel importante na quimiotaxia e
homeostase de íons tóxicos para células (29).
Algumas proteínas da família S100 influenciadas pelo íon Zn+2 tem sua
afinidade pelo íon Ca+2 afetada e essa afinidade por diferentes íons divalentes
contribui certamente para a diversidade funcional observada para proteínas S100.
Há um grande interesse sobre o papel do íon Zn+2 na funcionalidade das proteínas
S100, com vários estudos sugerindo mecanismos para a regulação de zinco,
incluindo interações dependentes de zinco da proteína S100B com proteínas τ,
formação de heterotetrâmeros das proteínas S100A8/ S100A9 induzida pelo zinco e
inibição da sinalização do cálcio pelo zinco em proteínas S100 (87).
Embora muitas funções tenham sido propostas para proteínas S100, o seu
papel biológico permanece obscuro. Há muitas evidências de que as proteínas da

HS100A12 51 família S100 participam do crescimento e diferenciação celular, estando associadas
ao desenvolvimento de tumores e a indução de metástase, atuando em processos
inflamatórios e, ainda, possuindo atividades antiparasitária, antifúngica e
antimicrobiana.
4.2 S100A12 Humana (HS100A12)
A proteína S100A12 humana, também chamada de Calgranulina C, é um dos
25 membros da família S100 de proteínas “EF-hand” ligantes de cálcio. A proteína
HS100A12 foi caracterizada por Guignard et al. (1995) (88) e após 6 anos sua
estrutura cristalográfica na presença de Ca+2 foi determinada por Moroz et al. (2001)
(74). Ela é uma proteína pequena, de baixo peso molecular (10,5 kDa), sem
resíduos nativos de cisteína, o que a torna alvo ideal para estudos pela técnica de
marcação de spin sítio dirigida (ver seção 1.1.) e possui proteínas homólogas em
granulócitos de porco (89, 90) com identidade sequencial de 70% (Figura 9).
Figura 9- Alinhamento sequencial entre CAGC humana (HS100A12) e CAGC porcina. Alinhamento e figura
foram feitos utilizando o programa MULTALIN (91).
A HS100A12 juntamente com S100A8 e S100A9 pertence à subfamília das
Calgranulinas, que são expressas exclusivamente em granulócitos e são
relativamente abundantes neste tipo de leucócito (92). Foi observado que a
S100A12 é necessária para o funcionamento apropriado dos granulócitos e forte
evidência para isso é sua abundância nas células e sua resposta muito específica
para a sinalização do cálcio quando a proteína se move do citosol para o
citoesqueleto e membranas (93).
Novas informações sobre o possível papel biológico da S100A12 foram obtidas
estudando-se a ativação do receptor para produtos de glicosilação (“Receptor for
Advanced Glycation End Products”- RAGE), que possui envolvimento com
crescimento e propagação de tumores, sendo que a interação com esse receptor

52
parece desempenhar uma função crucial em inflamações relacionadas à S100A12
(94, 95). Sabe-se também que a HS100A12 liga-se a drogas antialérgicas (96),
indicando que está também envolvida em eventos e caminhos de sinalização
diversos.
O enovelamento do monômero da S100A12 humana (Figuras 8 e 10) é
semelhante a outras estruturas conhecidas para proteínas S100, ou seja, dois
motivos “EF-hand” unidos (v. seção 4). Como a maioria das proteínas da família
S100, a HS100A12 é um dímero em solução, com a interface entre as duas
subunidades unidas por interações hidrofóbicas. A análise de dados estruturais do
complexo S100-moléculas alvo indica que a dimerização é essencial para o
reconhecimento da molécula alvo pelas proteínas S100. A comparação estrutural e
das sequências primárias das proteínas S100A10-anexinaII e S100A11-anexinaI
com HS100A12 sugere que a região de ligação da HS100A12 com moléculas alvo é
formada pelo “loop” LII e a região C-terminal de uma subunidade e a região N-
terminal da outra subunidade do dímero. A comparação mostra que os resíduos Glu
4 e Glu 8, encontrados na região N-terminal, estão na mesma conformação que no
complexo S100A10 e S100A11 e são altamente conservados na família S100,
particularmente nas calgranulinas, sugerindo que esses resíduos participam da
ligação do alvo com as proteínas S100 (74).
Como já mencionado na seção 4.1., muitas proteínas S100 apresentam sítio de
ligação para os íons Zn+2 e Cu+2, essenciais para muitos processos biológicos. Um
único sítio de ligação para o Zn+2 por monômero foi identificado na S100A12 porcina
por titulação fluorimétrica (89). Com isso, a análise da sequência das proteínas
S100A12 humana e porcina mostrou a presença do motivo de ligação do zinco
HisxxxHis localizado no C-terminal (97). Esse sítio de ligação, com estequiometria
de um íon Zn+2 por subunidade de proteína, foi muito bem caracterizado
observando-se a estrutura 3D do complexo S100A12 e zinco (75), indicando que os
resíduos de His e Asp no N-terminal, também participam da ligação com o íon
(Figura 10A). Além disso, há evidências que a proteína HS100A12 também liga
Cu+2, contendo um íon cobre por monômero, os quais são encontrados nos sítios de
ligação do zinco, sugerindo que os sítios de ligação para os dois íons são os
mesmos (86) (Figura 10B).

HS100A12 53
A BA B
Figura 10 - Comparação dos sítios de ligação dos íons cobre e zinco na proteína HS100A12. (A):
Sobreposição do sítio de ligação do zinco das estruturas Zn-HS100A12 e Ca/Cu- HS100A12 (amarelo). Resíduos das duas subunidades do dímero contribuem para a ligação com o Zn+2, uma subunidade está em azul claro e a outra em verde. O íon zinco está em azul e o cobre está em marrom. (B): Estrutura do dímero da proteína HS100A12 complexada com zinco ou cobre (marrom) e cálcio (amarelo). Figura adaptada de Moroz, et al, 2009 (75).
Adicionalmente, estudos recentes evidenciaram que as proteínas S100 formam
oligômeros que são importantes funcionalmente, por exemplo, a formação de
tetrâmeros e oligômeros maiores da proteína S100A4 estimula o crescimento neural
(98) e tem importância em artrite reumatóide (99). A oligomerização dependente de
metais das proteínas S100B (63), S100A8/ A9 (100) e S100A12 (101, 102) também
já demonstrou sua relevância funcional.
No caso da HS100A12, a transição dímero-tetrâmero-hexâmero é governada
pela concentração de zinco e/ou cálcio, ou seja, a formação do tetrâmero é
observada na presença de Ca+2, mas a proporção da população de tetrâmeros
aumenta lentamente com o aumento da concentração do íon Ca+2, ao passo que a
adição de uma pequena quantidade de Zn+2 (10 µM) aumenta drasticamente a
fração de tetrâmeros. Quando é adicionada uma quantidade maior de Ca+2 na
proteína tetramérica contendo Zn+2, os hexâmeros são a única espécie observada
(75, 102).
De posse desses resultados e analisando as estruturas cristalográficas da
proteína HS100A12 Apo (PDB 2WCE), na presença de cálcio (PDB 1E8A) e na
presença de zinco (PDB 2WCB), Moroz et al. (75) propuseram que a ligação do Zn+2
na proteína livre de outro metal causa um alongamento na hélice D e uma mudança
conformacional na hélice A, estabilizando a interface de tetramerização e
possibilitando a formação de tetrâmeros (Figura 11). Outra consequência da ligação

54
do Zn+2 na proteína HS100A12 é a reestruturação da hélice C, conduzindo a uma
mudança no EF-2, explicando, com isso, porque a ligação do Zn+2 aumenta a
afinidade da proteína pelo íon Ca+2. Na ausência de Zn+2, o motivo EF-2 é estável,
com a densidade eletrônica bem definida nas duas subunidades do dímero, mas
essa situação é alterada pela ligação com o Zn+2. A densidade eletrônica para a
região do EF-2 é pobremente definida para todas as subunidades da proteína na
presença do metal, concluindo que essa região é muito flexível quando o Zn+2 está
ligado e adota facilmente a estereoquímica necessária para a ligação do Ca+2 (75).
Como resultado, HS100A12 na presença de Zn+2 necessita de menos íon Ca+2
que a proteína Apo para chegar ao estado saturado de Ca+2. Uma vez que o “EF-
hand” está totalmente ocupado com Ca+2, íons cálcio adicionais podem se ligar entre
os dímeros, levando à formação de hexâmeros (Figura 11).
A
A B C
DE
Ca+2 adicionais
+ Ca+2
Ca+2
Zn+2
F
tetramerização
A
A B C
DE
Ca+2 adicionais
+ Ca+2
Ca+2
Zn+2
F
tetramerização
Figura 11 - Esquema ilustrando uma possível rota para a transição dímero-tetrâmero-hexâmero. (A)
Dímero da proteína HS100A12 Apo. (B) Ligação do íon zinco (vermelho) permite um rearranjo da interface de dimerização e um alongamento da hélice D. (C) As mudanças conformacionais observadas em B estabilizam a interface de tetramerização, levando a formação de tetrâmeros. (D) Ligação do íon cálcio (amarelo) nos motivos EF-hands da proteína já ligada com zinco. O complexo Zn-HS100A12 precisa de uma concentração muito menor de cálcio para ocorrer a saturação. (E) Íons cálcio adicionais se ligam nos sítios entre os dímeros, levando à formação de hexâmeros. (F) Quando o hexâmero é formado, a interface de tetramerização é rompida, restando somente HS100A12 na forma hexamérica. Figura adaptada de Moroz, et al., 2009 (75).

HS100A12 55
Diante disso, decidimos investigar o papel dos íons Ca+2 e Zn+2 na relação
estrutura-função da proteína HS100A12, utilizando as técnicas de marcação de spin
sítio dirigida (SDSL) e espectroscopia de RPE. A utilização com sucesso da técnica
de SDSL passa pelo conhecimento detalhado da estrutura da proteína em estudo.
Etapas como a escolha dos resíduos a serem mutados para posterior marcação e
das regiões de interesse na estrutura protéica dependem fortemente desse
conhecimento detalhado.
Com isso, iniciamos escolhendo resíduos adequadamente posicionados de
forma a monitorarmos a abertura/fechamento da região de ligação do íon Ca+2. Os
pares de resíduos escolhidos foram: lisina 33 (hélice B) e alanina 80 (hélice D)
originando o duplo mutante K33C/A80C; fenilalanina 57 (hélice C) e alanina 80
(hélice D) construindo o duplo mutante F57C/A80C (Figura 12). Devemos salientar,
ainda, que produzimos também o “single” mutante A80C, que é a base para as
medidas de distância. Com os duplos mutantes esperamos conseguir detectar
mudanças conformacionais que envolvam a aproximação/afastamento das cisteínas
introduzidas quando do funcionamento da proteína.
Figura 12 - Estrutura do monômero da proteína HS100A12 (PDB 1E8A), indicando os resíduos mutados
por cisteínas para posterior marcação de spin. As quatro hélices são indicadas pelas letras de A-D.
Além disso, examinando as estruturas cristalográficas da proteína HS100A12
na forma apo (ausência de ligantes) (PDB 2WCE), na presença de cálcio (PDB
1E8A) e na presença de zinco (PDB 2WCB) observamos uma grande mudança
conformacional na hélice C (Figura 13). Diante dessa diferença estrutural, decidimos
estudar a dinâmica de afastamento e aproximação das hélices C e D como um todo,
A
B
C
D
K33 A80
F57

56
quando Ca+2 e Zn+2 estão ligados à proteína, inserindo o marcador de spin em cinco
posições diferentes da hélice C. Os duplos mutantes construidos foram: isoleucina
53 (hélice C) e alanina 80 (hélice D) originando o mutante I53C/ A80C, ácido
aspártico 54 (hélice C) e alanina 80 (hélice D) construindo o mutante D54C/ A80C,
fenilalanina 57 (hélice C) e alanina 80 (hélice D)- F57C/ A80C, leucina 60 (hélice C)
e alanina 80 (hélice D)- L60C/ A80C, ácido aspártico 61 (hélice C) e alanina 80
(hélice D)- D61C/ A80C.
Figura 13 - Estruturas das proteína HS100A12 apo (PDB 2WCE) em cinza, com cálcio (PDB 1E8A) em azul
e com zinco (PDB 2WCB) em rosa, evidenciando a mudança conformacional na hélice C.
Finalmente, devido ao fato da proteína HS100A12 estar envolvida em alguns
eventos de sinalização celular e interação com o receptor para produtos de
glicosilação (RAGE) (94, 101), decidimos também, estudar a interação da proteína
com modelos de biomembranas.
Para tal, escolhemos utilizar as monocamadas de Langmuir, que são modelos
“alternativos” aos modelos de bicamadas, devido ao fato de não mimetizarem a
membrana como um todo e sim modelarem apenas metade dela. Entretando, as
monocamadas possuem algumas vantagens, como: permitir controle rigoroso da
composição das membranas; permitir controle do estado de compactação e, com
isso, controlar a estruturação da monocamada e são modelos planares, que se
aproximam ao formato de uma superfície celular, ao contrário de lipossomos, por
exemplo, que possuem uma curvatura na superfície (103, 104).

HS100A12 57 4.3 Metodologia
As etapas nesta seção descrevem as metodologias em biologia molecular
utilizadas para a obtenção, caracterização, expressão e purificação dos mutantes da
proteína Calgranulina C (S100A12) humana e da proteína nativa, além dos
experimentos de dicroísmo circular (CD), ressonância paramagnética eletrônica
(RPE) e monocamada de Langmuir.
4.3.1 Construção dos Oligonucleotídeos e Amplificação dos Genes
O plasmídeo pQE60 contendo o gene que codifica a proteína HS100A12 nos
foi gentilmente cedido pela Dra. Olga Moroz da Universidade de York, Inglaterra. A
sequência que codifica a proteína HS100A12 utilizada na estratégia de amplificação
para obtenção dos genes de interesse e a sequência dos sítios de restrição para as
enzimas NcoI e HindIII são apresentados na figura 14.
1
AGGAGAAATTAACCATGGGAGGATCCATGGGAGGATCCACAAAACTTGAAGAGCATCTGGAGGGAATT
GTCAATATCTTCCACCAATACTCAGTTCGGAAGGGGCATTTTGACACCCTCTCTAAGGGTGAGCTGAA
GCAGCTGCTTACAAAGGAGCTTGCAAACACCATCAAGAATATCAAAGATAAAGCTGTCATTGATGAAA
TATTCCAAGGCCTGGATGCTAATCAAGATGAACAGGTCGACTTTCAAGAATTCATATCCCTGGTAGCC
ATTGCGCTGAAGGCTGCCCATTACCACACCCACAAAGAGAAGCTTAATTAGCTGAGCTTGGACTC
285
Figura 14 - Sequência codificadora da proteína HS100A12. A sequência mostra 337 nucleotídeos sendo que 285 nucleotídeos codificam para a proteína Calgranulina C humana. Os nucleotídeos em negrito vermelho representam o início e o fim da sequência que codifica para a proteína. Os oligonucleotídeos sublinhados representam os sítios de clivagem para as enzimas de restrição NcoI e HindIII.
A estratégia da mutação sítio dirigida e, consequentemente, da amplificação
resumiu-se à aplicação do protocolo descrito na metodologia de extensão
sobreposta (“Overlap Extension”) por PCR (v. apêndice A). A primeira reação em
cadeia da polimerase é realizada utilizando-se dois tubos, A e B, que serão
amplificados ao mesmo tempo, utilizando o mesmo programa de PCR. Tubo A:
primer forward 5’ e o primer reverse com a mutação de interesse. Tubo B: primer

58
forward, contendo a mesma mutação e o primer reverse 3’. Em seguida, os
produtos amplificados são analisados, excisados do gel e purificados. Em uma
segunda reação de PCR, quantidades equivalentes de cada produto são usados
como molde em uma segunda reação de PCR. Nesta segunda reação, inicialmente
os dois fragmentos serão anelados entre si nas porções complementares e estes
servirão tanto como molde, como primers, onde as regiões de fita simples serão
preenchidas. A seguir, os primers forward 5’ e reverse 3’ irão amplificar os
fragmentos de DNA completos, contendo a mutação.
As mutações sítio dirigidas foram introduzidas na sequência de nucleotídeos
que codifica a HS100A12 através da utilização de oligonucleotídeos planejados
especificamente para alterar a sequência de códons que codificam os resíduos de
interesse (Tabela 1 e 2).
Tabela 1 - Resíduos de aminoácidos de HS100A12 que foram mutados por resíduos de cisteína, gerando mutantes simples e duplos da proteína.
MUTAÇÃO POSIÇÃO NOME DO MUTANTE
1 Lys33 – Cys33 33 K33C
2 Ile53 – Cys53 53 I53C
3 Asp54 – Cys54 54 D54C
4 Phe57 – Cys57 57 F57C
5 Leu60 – Cys60 60 L60C
6 Asp61 – Cys61 61 D61C
7 Ala80 – Cys80 80 A80C
8 Lys33 – Cys33 e Ala80 – Cys80 33 e 80 K33C/A80C
9 Ile53 – Cys53 e Ala80 – Cys80 53 e 80 I53C/A80C
10 Asp54 – Cys54 e Ala80 – Cys80 54 e 80 D54C/A80C
11 Phe57 – Cys57 e Ala80 – Cys80 57 e 80 F57C/A80C
12 Leu60 – Cys60 e Ala80 – Cys80 60 e 80 L60C/A80C
13 Asp61 – Cys61 e Ala80 – Cys80 61 e 80 D61C/A80C

HS100A12 59
Os oligonucleotídeos contendo as mutações de interesse para a proteína
HS100A12 e também os primers forward e reverse que foram desenhados e
sintetizados são listados na Tabela 2.
Tabela 2 - Oligonucleotídeos da enzima HS100A12 que foram utilizados na amplificação por PCR. Os nucleotídeos em azul representam as trincas dos códons que codificam para a mutação de interesse (aminoácido cisteína).
CLONES MUTAÇÃO OLIGONUCLEOTÍDEO SEQÜÊNCIA
Forward- 5’ 5’-CATAAAAAATTTATTTGCTTTGTGAGCGG-3’
Reverse- 3’ 5’-CTTTACGGTCTTTAAAAAGGCCG-3’
HS100 K33C Sense 5’-GGTGAGCTGTGCCAGCTGCTTAC -3’ Lys33 – Cys33 K33C
HS100 K33C AS 5’-GTAAGCAGCTGGCACAGCTCACC -3’
HS100 I53C Sense 5’-GCTGTCTGCGATGAAATATTCC -3´ Ile53 – Cys53
I53C
HS100 I53C AS 5’-GGAATATTTCATCGCAGACAGC -3´
HS100 D54C Sense 5’-GCTGTCATTTGCGAAATATTCCAAG -3´ Asp54 – Cys54
D54C
HS100 D54C AS 5’-CTTGGAATATTTCGCAAATGACAGC -3´
HS100 F57C Sense 5'-GATGAAATATGCCAAGGCCTG -3’ Phe57 – Cys57
F57C
HS100 F57C AS 5'-CAGGCCTTGGCATATTTCATC -3’
HS100 L60C Sense 5'-CCAAGGCTGCGATGCTAATC -3’ Leu60 – Cys60
L60C
HS100 L60C AS 5'-GATTAGCATCGCAGCCTTGG -3’
HS100 D61C Sense 5'-CAAGGCCTGTGCGCTAATCAAG -3’ Asp61 – Cys61
D61C
HS100 D61C AS 5'-CTTGATTAGCGCACAGGCCTTG -3’
HS100 A80C Sense 5'-GTAGCCATTTGCCTGAAGGC -3’ Ala80 – Cys80
A80C
HS100 A80C AS 5'-GCCTTCAGGCAAATGGCTAC -3’
A amplificação via PCR foi realizada em um termociclador Mastercycler
(Eppendorf) empregando-se um volume final de 50 µL constituído de 5 µL de cada
oligonucleotídeo em uma concentração de 5 µM, 5 ng de DNA molde, 5 mM de

60
dNTP’s, 5 µL de Pfu buffer (10X), 1 µL de Pfu Turbo DNA polimerase (2.5U) e 25
mM de MgCl2.
As condições de amplificação compreenderam uma desnaturação inicial a 95oC
por 5 minutos, seguida de 30 ciclos constituídos de desnaturação a 95oC por 60 s,
hibridização a 49oC por 60 s e extensão dos “primers” a 72oC por 60 s; com
extensão final a 72oC por 10 min. Ao final da reação, resfriou-se a amostra a 6oC.
Os produtos das primeiras reações de PCR foram separados aplicando-se 50
µL das misturas de reação em gel de agarose 1%, contendo 0,5 µg/mL de brometo
de etídeo, em tampão 1X TAE (80 mM Tris; 40 mM ácido acético glacial; 2,5 mM
EDTA, pH 8,3) e, em seguida, foram submetidos a uma tensão de 100 V em um
intervalo de 30 a 45 min. Após a migração, foram visualizados sob luz UV (Figura
15). Uma vez confirmado o tamanho correto dos fragmentos de DNA obtidos, os
produtos de PCR foram excisados do gel utilizando-se o Kit QIAquick Gel Extraction
(Qiagen) de acordo com as instruções do fabricante. O resultado da extração foi
misturado entre si e com os “primers forward” (5’) e “reverse” (3’), para a realização
da segunda reação em cadeia da polimerase (PCR), nas mesmas condições citadas
para a primeira PCR.
Por fim, os produtos das amplificações dos mutantes S100A12 humana foram
purificados utilizando o Kit QIAquick PCR Purification (Qiagen) para, em seguida,
realizar-se a reação de dupla digestão.
4.3.2 Reação de Dupla Digestão
Partindo do ponto que o novo DNA produzido está puro, o próximo passo é
realizar a reação de dupla digestão, que ocorre na presença de duas enzimas de
restrição que “cortam” o DNA em dois pontos específicos, chamados de sítios de
restrição. A reação de dupla digestão é feita tanto no inserto de DNA purificado
quanto no plasmídeo pQE 60, para posterior reação de ligação de ambos. A
digestão de DNA por enzimas de restrição é um processo bastante simples;
colocamos o DNA ou o plasmídeo em contato com as enzimas de restrição a 37oC,
por duas horas e a enzima inicia imediatamente o processo de digestão.

HS100A12 61
Para a proteína HS100A12, misturamos 5 µL de Buffer 2, específico para as
enzimas utilizadas, 1 µL de cada enzima de restrição, NcoI e HindIII e 1,2 ng de
inserto do DNA (contém a mutação) ou 9,2 ng de plasmídeo pQE 60. O conjunto de
fragmentos gerados por digestão com enzimas de restrição é, então, detectado por
eletroforese em gel de agarose (Figura 15) e excisados utilizando-se o Kit QIAquick
Gel Extraction (Qiagen), como descrito na seção 4.3.1 e quantificado por
espectrofotometria no UV-visível.
Na Figura 15, como ilustração, é apresentado o gel de agarose das etapas de
amplificação do gene que codifica o mutante HS100 A80C e do resultado da dupla
digestão do inserto de interesse e do plasmídeo pQE 60. O produto de amplificação
de 285 pares de bases, como esperado, não apresentou nenhuma banda
inespecífica e resultou em quantidades suficientes para posterior clonagem.
1 2 3 4 5 6 7
bp
1000
500
300
100
bp
10000
3000
1000
500
250
1 2 3 4 5 6 7
bp
1000
500
300
100
bp
10000
3000
1000
500
250
Figura 15 - Amplificação do oligonucleotídeo que codifica o mutante HS100 A80C. Os fragmentos de DNA
foram separados por gel de agarose 1% e visualizados em um transiluminador UV a 365nm. Poços: 1- padrão de massa molecular 100bp; 2- produto de amplificação (5´) da primeira PCR; 3- produto de amplificação (3´) da primeira PCR; 4- produto da dupla digestão do inserto; 5- produto da dupla digestão do plasmídeo e 7- padrão de massa molecular 1 kb.
4.3.3 Subclonagem dos Genes no Plasmídeo
O plasmídeo do sistema pQE 60 utilizado possui um gene que confere
resistência a ampicilina. As sequências codificantes de interesse dos mutantes da
enzima HS100A12 foram inseridas no vetor pQE 60 através de uma reação de

62
ligação. A mistura de ligação foi preparada empregando-se um volume final de 10 µL
constituídos de 1,2 ng do inserto, 1,0 µL do vetor pQE 60 a 9,2 ng, 1 µL do tampão
de ligação para a enzima T4 DNA ligase (10X), 1 µL da enzima T4 DNA ligase (6U)
e água Milli-Q que foi utilizada para completar o volume total. Esta reação foi
mantida a 4oC por aproximadamente 12 horas. Essa mistura de ligação da enzima
HS100A12 foi utilizada na transformação por choque térmico de células
competentes de E. coli DH5α para propagação plasmidial.
4.3.4 Transformação da Bactéria Escherichia coli DH5αααα com o Plasmídeo
Recombinante pQE 60
As suspensões de células competentes DH5α estocadas a -80oC foram
mantidas durante pelo menos 10 min em banho de gelo. A cada 100 µL de células
DH5α foram adicionados 5 µL da amostra da reação de ligação. A mistura resultante
foi homogeneizada gentilmente e mantida em gelo por 30 min. Em seguida, esta
mistura foi submetida à incubação em banho de água a 42oC por 45 segundos e
subsequentemente colocada no gelo por 2 min (choque térmico). Após este período,
250 µL de meio LB foram adicionados no tubo contendo as células transformadas
com a reação de ligação. A nova mistura foi mantida a 37oC por 1 hora e então
plaqueada em placas de Petri contendo 20 mL de meio seletivo Luria-Bertani sólido
suplementado com 20 µL do antibiótico de seleção ampicilina a 100 mg/mL e
incubada “overnight” a 37oC em estufa.

HS100A12 63 4.3.5 Extração e Análise Sequencial do Plasmídeo Recombinante pQE 60 de
Escherichia coli DH5αααα
Após o período de incubação, algumas colônias resistentes (possíveis
recombinantes ou clones positivos) foram selecionadas a fim de se realizar a
extração dos seus DNAs plasmidiais e, posteriormente, estes foram analisados por
sequenciamento.
As colônias selecionadas foram repicadas individualmente em tubos estéreis
para cultivo de bactérias contendo 20 mL de meio LB líquido contendo 2 µL de
ampicilina a 100 mg/mL (pré-inóculo). Os tubos foram incubados a 37oC por 16
horas, sob agitação de 250 rpm. A seguir, as células tiveram seus DNAs plasmidiais
extraídos através do kit QIAprep Spin MiniPrep (Qiagen) para análise destes DNAs
por sequenciamento e obtenção dos genes a serem posteriormente subclonados
nos vetores de expressão.
Os DNAs plasmidiais livres de contaminantes como DNA genômico, RNA,
proteínas e macromoléculas foram quantificados por espectrofotometria no UV-
visível e submetidos ao sequenciamento automático. O sequenciamento de DNA
(ver apêndice A) foi realizado utilizando-se sequenciador automático ABI Prism 377-
96 Collection (Applied Biosystem), seguindo o protocolo do fabricante. Após o
sequenciamento, para verificarmos a integridade da sequência obtida, esta foi
submetida ao programa Chromas 1.45
(http://www.technelysium.com.au/chromas.html) e comparada à sequência original.
Uma vez confirmada a presença dos DNAs plasmidiais que contêm as
sequências codificantes das enzimas mutantes nestas células, os mesmos foram
utilizados na transformação por choque térmico de células competentes de E. coli
BL21 para expressão heteróloga da enzima HS100A12.

64
4.3.6 Transformação da Bactéria Escherichia coli Rosetta com o Plasmídeo
Recombinante pQE 60
Após confirmada a presença dos DNAs mutados nas células é necessário
“inseri-los” em células que possiblitem a expressão. O procedimento empregado na
transformação de células competentes de E.coli Rosetta foi o mesmo descrito na
transformação de células DH5α (seção 4.3.4.).
4.3.7 Expressão da Proteína HS100A12
Para a expressão da proteína HS100A12 e dos seus mutantes em bactéria, o
pré-inóculo foi preparado utilizando a cepa Rosetta de Escherichia coli,
transformadas com o plasmídeo que contém o gene que codifica os mutantes da
proteína HS100A12 (pQE60), em meio de cultura LB (Luria-Bertani) sob agitação
(250 rpm), durante aproximadamente 16 horas, a 37°C e na presença de 5 µL de
Ampicilina (200 mg/mL), pois o gene codificador da enzima HS100A12 confere
resistência a esse antibiótico.
Após 16 h, o pré-inóculo foi utilizado para inocular 1 L de meio de cultura LB
fresco contendo 500 µL de Ampicilina (200 mg/mL) em uma diluição 1(pré-
inóculo):100(inóculo). As células foram mantidas sob 250 rpm de agitação, a 37oC,
até que a absorbância a 600 nm (DO600nm) atingisse valores entre 0,5-0,6. A
expressão da proteína foi, então, induzida pela adição de 1 mM de IPTG (isopropil-β-
D-tiogalactopiranosídeo). A cultura foi mantida a 37oC e sob a mesma agitação por 5
h. Para o isolamento das células, o material foi centrifugado a 5.000 rpm, 4oC, por 10
minutos e o “pellet” congelado a -20oC. A expressão da proteína nativa e dos
mutantes foi monitorada por eletroforese em gel de poliacrilamida (SDS-PAGE).

HS100A12 65 4.3.8 Purificação da Proteína HS100A12
A purificação da proteína HS100A12 nativa e dos mutantes se iniciou com o
descongelamento das células que foram ressuspensas em 50 mM Tris, 150 mM
NaCl pH 7,5 e lisadas por sonicação em 7 ciclos de 30 segundos com intervalos de
1 minuto. Os resíduos celulares e o material insolúvel foram removidos por
centrifugação a 12.000 rpm por 30 minutos. O sobrenadante foi incubado com 10
mM CaCl2 por 15 minutos e centrifugado por 10 minutos a 15.000 rpm. O
sobrenadante clarificado é aplicado na coluna Hiprep Fenil-Sefarose FF (GE
Healthcare) (interação hidrofóbica) previamente equilibrada com o tampão 50 mM
Tris, 150 mM NaCl, 5 mM DTT, 5 mM CaCl2 pH 7,5. A coluna foi lavada com o
mesmo tampão e subsequentemente a proteína foi eluída com 50 mM Tris, 150 mM
NaCl, 5 mM DTT, 5 mM EDTA (ácido etilenodiamino tetra-acético) pH 7,5.
As frações contendo HS100A12 foram aplicadas na coluna Superdex 75
10/300 GL (GE Healthcare) (exclusão molecular), previamente equilibrada com o
tampão 50 mM Tris, 25 mM NaCl, 5 mM DTT pH 7,5 e eluída com o mesmo tampão.
4.3.9 Separação dos Oligômeros da Proteína HS100A12
As frações de proteína eluída da coluna Superdex 75 foram incubadas com
quantidade de íon cálcio suficiente para saturar os sítios da proteína (4 moléculas de
Ca+2 por molécula de proteína,ou seja, 2 moléculas de Ca+2 por monômero) ou
dialisadas, pelo menos, quatro vezes contra o tampão 50 mM Tris, 25 mM NaCl, 40
µM ZnCl2 pH 7,5, utilizando o concentrador Amicon Ultra 5MWCO (Milipore).
A fim de separar os oligômeros da proteína e dos seus mutantes formados em
função da presença e/ou ausência e da concentração de cálcio e zinco, a proteína
foi aplicada novamente na coluna Superdex 75 10/300 GL (GE Healthcare) e eluída
com o mesmo tampão utilizado na primeira aplicação da proteína na coluna. A
purificação e a separação dos oligômeros foram realizadas utilizando-se o sistema

66
ÄKTA Purifier (GE Healthcare). O resultado do protocolo descrito acima foi avaliado
através de eletroforese em gel de poliacrilamida SDS-PAGE.
A técnica de cromatografia de exclusão molecular foi utilizada para quantificar
analiticamente o estado de oligomerização da proteína HS100A12 e dos seus
mutantes. Os padrões de peso molecular foram aplicados na mesma coluna
previamente equilibrada com o tampão (50 mM Tris, 25 mM NaCl, 5 mM DTT pH
7,5) utilizados para a purificação e separação dos oligômeros da proteína
HS100A12. O inibidor de tripsina de soja- ITS (20 kDa), ovalbumina (43 kDa) e
albumina (66 kDa) foram utilizados como padrões de peso molecular.
4.3.10 Determinação da Concentração da Proteína HS100A12
A estimativa da concentração da proteína foi feita pela medida de sua
densidade ótica (DO) em 280 nm, com uso do coeficiente de extinção molar teórico
ε= 2980 M-1 cm-1, calculado a partir da lei de Lambert-Beer (105) através do pacote
de programas Expert Protein Analysis System – ExPASy (http://bo.expasy.org).
4.3.11 Dicroísmo Circular
Desde a década de 1980, há uma necessidade crescente de se realizar
estudos estruturais sob as mesmas condições em que a proteína realmente se
encontra, isto é, geralmente em solução com parâmetros físico-químicos, como pH e
força iônica, controlados. Além disso, há bastante interesse em se poder variar tais
parâmetros com o subsequente acompanhamento de mudanças estruturais
induzidas na estrutura protéica, mudanças estas, em geral, essenciais a sua função
biológica. A técnica de dicroísmo circular (CD, do inglês circular dichroism) afirmou-
se reconhecidamente como uma técnica estrutural valiosa para direcionar esses
ensaios (106, 107), pois está intimamente relacionada com a conformação de uma

HS100A12 67 molécula, em particular, com o conteúdo e orientação de elementos de estrutura
secundária, como hélices α e folhas β, existentes em proteínas.
O dicroísmo circular é observado em moléculas oticamente ativas que
apresentam diferenças na absorção da luz circularmente polarizada à esquerda e à
direita. A técnica de dicroísmo circular é particularmente útil para estudos de
proteínas, pois estas possuem centros assimétricos naturais como o carbono
α, carbono β dos aminoácidos treonina e isoleucina, ligações peptídicas que
absorvem entre 195 e 240 nm, aminoácidos aromáticos que absorvem na região de
260 a 320 nm e pontes dissulfeto que possui uma banda centrada em 260 nm,
distribuídos ao longo de sua cadeia principal (107, 108, 109).
Nesse contexto, com a espectroscopia de CD é possível realizar estudos de
mudanças conformacionais, vizinhanças locais, interações de ligantes com
macromoléculas, “folding-refolding-unfolding” de proteínas, bem como se obter uma
estimativa do conteúdo de estrutura secundária através do espectro de CD medido
na região do ultravioleta distante (195 a 250 nm). Neste intervalo de comprimento de
onda, o cromóforo em investigação é o dipolo elétrico associado à ligação peptídica
e que se caracteriza por uma fraca, mas larga transição n → π∗ centrada em 220 nm
e uma transição π → π∗ mais intensa em torno de 190 nm (107).
O espectro de CD da proteína na região de UV próximo (“near UV”) fornece a
“impressão digital” da estrutura terciária da mesma, que pode ser usada para
comparar, por exemplo, a proteína selvagem com suas formas mutantes. O espectro
na região de 260-320 nm se refere aos aminoácidos aromáticos, cada qual com seu
perfil característico, como por exemplo, o triptofano que possui um pico em 290 nm;
a tirosina, cujo pico está entre 275 e 282 nm e a fenilalanina que apresenta uma
estrutura fina entre 255 e 270 nm. Contudo, a forma e a magnitude do espectro de
CD da proteína na região do ultravioleta próximo dependem do número de cada tipo
de aminoácido presente, da mobilidade dos mesmos, da natureza do meio (ligações
de hidrogênio, grupos polares e polarizabilidade) e de suas disposições espaciais na
proteína (107).
A técnica de CD consiste na aplicação de um feixe de luz circularmente
polarizado à direita, D, e um circularmente polarizado à esquerda, E, à amostra de
interesse. Ao passar através da amostra oticamente ativa, as componentes do
campo elétrico da luz polarizada não são absorvidas igualmente. Desse modo, o raio

68
do círculo traçado pelo vetor campo elétrico de cada componente é diferente.
Quando essas duas componentes, em sentidos opostos, atravessam a amostra e
são combinadas, o resultado é uma luz elipticamente polarizada (Figura 16). Assim,
o dicroísmo circular mede a diferença entre a absorção da luz circularmente
polarizada à direita e à esquerda (∆A) (105, 109).
Figura 16- Origem do efeito do dicroísmo circular. Decomposição do feixe plano polarizado em duas
componentes circulares à esquerda (E) e à direita (D) e depois de passar pela amostra. E = Ed – Ee.
A definição da absorbância A é dada pela lei de Lambert-Beer:
=
I
IA
0log , (1)
Sendo I0 a intensidade da luz incidente na amostra e I a intensidade da luz após
passar pela amostra. Aplicando a lei para ambos os feixes, temos:
=
e
e
I
IA
0log
=
d
d
I
IA
0log
Sendo Ae a absorbância medida da luz circularmente polarizada à esquerda e
Ad a absorbância da luz circularmente polarizada à direita. Sendo assim, ∆A é dado
por:
=−=∆
d
e
de
I
IAAA log (2)

HS100A12 69
Como já foi dito anteriormente, a luz resultante ao passar pela amostra é
elipticamente polarizada. Quando os vetores campo elétrico das duas componentes
circulares estão na mesma direção, sua soma resulta no semi-eixo maior da elipse,
e, quando estão em direções opostas, sua soma resulta no semi-eixo menor. Logo,
o CD é caracterizado pela razão entre os dois semi-eixos, ou seja, pela tangente do
ângulo θ, denominado elipticidade. Sendo θ um ângulo muito pequeno, a tangente
de θ pode ser aproximada para θ. A medida da elipticidade em função do
comprimento de onda λ da luz incidente resulta no espectro de CD.
Existe uma relação numérica entre ∆A e θ que é dada por (105):
( )
πθ
4
180303.2de
AA −= (3)
Os dados de CD em função do comprimento de onda foram convertidos em
elipticidade molar média por aminoácido (θ)λ (grau.cm2.dmol-1), utilizando a seguinte
relação:
(θ)λ=(θλ M0)/(10 d c) (4)
onde θλ é a elipticidade observada em 103 grau, M0 é o peso médio de um
resíduo (M0=108 Da), c é a concentração da proteína (mg/mL) e d é o caminho ótico
(cm).
As medidas de dicroísmo circular foram realizadas no espectropolarímetro
Jasco J-720, em um intervalo de 195 a 250 nm, utilizando cubetas de quartzo de 1
mm de caminho óptico e 8 varreduras. A fim de confirmarmos o conteúdo de
estrutura secundária e, consequentemente, o enovelamento correto dos mutantes da
proteína HS100A12 apo e na presença dos íons Ca+2 e/ou Zn+2, utilizamos 0,2
mg/mL da proteína em tampão 50 mM Tris, 25 mM NaCl, 5 mM DTT pH 7,5 a 20oC.
O íon cálcio foi adicionado à proteína em quantidade suficiente para saturar os seus
sítios e, no caso das proteínas contendo íon zinco, a saturação dos sítios levou à
precipitação da proteína, por isso, estas foram dialisadas contra o tampão 50 mM
Tris, 25 mM NaCl, 40 µM ZnCl2 pH 7,5.
A estabilidade da proteína HS100A12 nativa na ausência e presença dos íons
cálcio e/ou zinco foi estudada em função da temperatura, sendo esta variada de 10 a

70
100oC, em intervalos de 2oC. Após o processo de desnaturação por aquecimento, a
amostra foi imediatamente resfriada para 20oC e seu espectro novamente medido
para verificarmos a reversibilidade da reação.
4.3.12 Marcação de Spin Sítio Dirigida dos Mutantes da Proteína HS100A12
A proteína HS100A12 foi eluída da coluna cromatográfica com tampão
contendo 1,4-Ditiotreitol (DTT), uma molécula redutora que não permite que os
resíduos de cisteína presentes na proteína se liguem através de pontes dissulfetos,
assim, impossibilitando também a formação de dímeros ou moléculas oligoméricas
unidas por tais ligações. A fim de realizarmos a marcação da proteína com o
marcador de spin MTSSL, que se liga a resíduos de cisteína por uma ligação S-S, foi
necessário remover o DTT da solução. Para isso, a proteína eluída foi aplicada em
uma coluna HiTrapTM Desalting (GE Healthcare), previamente equilibrada com 50
mM Tris, 25 mM NaCl pH 7,5 e eluída com o mesmo tampão. Passo fundamental
para o sucesso do procedimento de marcação está na adição, no frasco de coleta da
proteína eluída, de um excesso do marcador MTSSL (10 vezes a concentração
molar da proteína). Em seguida, cobrimos o frasco com papel alumínio (pois o
marcador de spin é sensível a luz) e deixamos sob agitação constante a 4 oC, por 16
horas.
Com a finalidade de retirar o excesso de marcador e ao mesmo tempo separar
os diferentes oligômeros formados com a incubação dos íons, a amostra foi aplicada
em uma coluna Superdex 75 (GE Healthcare), previamente equilibrada com tampão
50 mM Tris, 25 mM NaCl, pH 7,5 e eluída com o mesmo tampão. Em seguida, foram
concentradas através de concentradores Amicon Ultra-15 (Millipore Corporation,
Bedford, MA, EUA) com membrana de 5 kDa, tamanho adequado à massa
molecular da proteína HS100A12. A concentração da proteína recombinante
purificada foi determinada medindo-se sua absorbância em espectrofotômetro UV-
Vis a 280 nm (seção 4.3.10).

HS100A12 71 4.3.13 Ressonância Paramagnética Eletrônica
Quando os elétrons desemparelhados do radical nitróxido presentes na
proteína são submetidos à ação de um campo magnético externo passam a se
comportar como se fossem pequenos ímãs e se orientam a favor (orientação
preferida pela maioria por ser a de menor energia) ou contra o campo, dando origem
a dois níveis com energias diferentes. Estes elétrons podem, então, receber energia
igual à diferença (∆E) entre as energias desses dois estados, levando assim a uma
mudança de sua orientação. Esta transição é acompanhada de uma absorção de
radiação eletromagnética com energia correspondente àquela diferença que pode,
então, ser detectada, gerando o sinal de RPE (110).
O sinal de RPE de cada centro paramagnético é bastante característico, uma
vez que é sensível a fatores como tipo de centro, seu estado de oxidação e suas
interações com vizinhos próximos ao elétron desemparelhado. Desta forma, provê
informações úteis não só sobre este elétron, mas também sobre os núcleos
atômicos próximos e que têm spin nuclear diferente de zero (como H, N e 13C),
assim como sobre a estrutura dinâmica de tal vizinhança.
Quando o momento magnético associado ao spin do elétron interage com o
momento magnético nuclear (I) de um núcleo próximo aparece a chamada estrutura
hiperfina (Figura 17A). No caso do nitrogênio encontrado no grupo nitróxido NO (I =
1) a multiplicidade do spin nuclear será 2I + 1 = 3. Deste modo, cada um daqueles
níveis de energia do elétron encontrados na presença de um campo magnético
externo será desdobrado em três, devido à interação hiperfina com o spin nuclear do
nitrogênio. A estrutura hiperfina do grupo nitróxido é, assim, observada pela
existência de três linhas no espectro de RPE (Figura 17B).

72
(A) (B) Figura 17- (A) Níveis de energia do elétron desemparelhado do grupo nitróxido NO em um campo
magnético externo. (a): somente interação Zeeman; (b): incluindo interação com spin nuclear I=1 do nitrogênio (hiperfina). (B) Espectro de RPE de um marcador de spin livre. Figura adaptada de Schreier (110).
Os mutantes marcados com MTSSL foram medidos à temperatura ambiente e
a -100oC no espectrômetro de RPE Varian E109, operando em banda X (9,5 GHz) e
utilizando uma potência de microondas de 5 mW para as medidas a temperatura
ambiente e 0,1 mW para as medidas a -100oC. Para os experimentos de RPE foram
utilizadas alíquotas de 100 µL de soluções das proteínas marcadas, que eram
colocadas em cela chata de quartzo para realizar as medidas a temperatura
ambiente e em tubos de quartzo para as medidas em baixa temperatura.
4.3.14 Monocamadas de Langmuir
As monocamadas de Langmuir são formadas espalhando uma solução
lipídica, preparada em solvente orgânico, sobre uma fase aquosa. Esses filmes
foram preparados utlizando uma cuba de Langmuir Mini- KSV (KSV Instruments,
Finlândia) equipada com um sensor que determina a tensão superficial pelo método
de Wilhelmy, e duas barreiras móveis, capazes de “varrer” as moléculas presentes
na interface (Figura 18) (111).

HS100A12 73
Figura 18 - Ilustração de uma cuba de Langmuir equipada com um sensor de tensão superficial e duas
barreiras móveis. Figura adaptada de <http://www.4engr.com/product/catalog/16626/index1.html>.
A variação da área altera o grau de compactação da monocamada e com isso,
as propriedades do filme podem ser estudadas. Nesse estudo, isotermas de pressão
superficial (π) em função da área por molécula (π-A) foram obtidas, bem como as
cinéticas de adsorção das proteínas nas monocamadas lipídicas.
Na figura 19, para efeito ilustrativo, é apresentada uma curva característica de
isoterma π-A para o fosfolipídio dipalmitoilfosfatidilcolina (DPPC), utilizando água
como subfase à temperatura de 210C, onde os diferentes estados físicos
bidimensionais do filme lipídico podem ser observados. Inicialmente, à pressão
superficial baixa indica que as moléculas encontram-se no estado gasoso
bidimensional (G), onde as moléculas apresentam elevada área por molécula
disponível, resultando em uma baixa interação entre as moléculas. Com o aumento
da compressão, a área disponível por molécula diminui, de modo que as moléculas
passam a realizar interações laterais, resultando nos estados líquido-expandido (LE)
e líquido-condensado (LC) bidimensionais. Em alguns casos, como na figura 19,
apresentada abaixo, um patamar na curva é observado, e corresponde a coxistência
das fases líquido-expandida e líquido- condensada, para o fosfolipídio DPPC.
Entretanto, muitos compostos não apresentam essa região bem definida em suas
isotermas de pressão superficial.
Finalmente, com a subseqüente compressão do filme lipídico, temos a máxima
compactação e orientação das moléculas com relação à normal da interface,
resultando no estado sólido bidimensional. Nesta condição determina-se o valor de
área mínima, através da tangente à fase sólida que intercepta o eixo da coordenada
x (área por molécula). Após este estágio de interação lateral entre as moléculas,
uma posterior compressão ocasiona o colapso da monocamada, onde as moléculas

74
não mais se encontram arranjadas como um filme monomolecular. Ocorrendo então,
a formação de agregados, bicamadas, multicamadas e dissolução na subfase (112).
Figura 19 - Isoterma de pressão de superfície para uma monocamada de DPPC puro. Representação
esquematizada das fases de uma monocamada de Langmuir com as diferentes regiões de compressão do filme. Região I: fase Gasosa (G), onde a pressão superficial é zero. Região II: fase Líquida- Expandida (LE). Região III: fase Líquida- Condensada (LC). Região IV: fase Sólida (S). Região V: colapso. Adaptada de Silva-Lucca, et al. 1999 (112).
As isotermas de pressão de superfície em função da área (π-A) foram obtidas
com a compressão da monocamada usando barreiras móveis a uma velocidade de
10 Å2/ molécula/ min. Inicialmente, a cuba de Langmuir, com capacidade total de 35
mL, foi preenchida com tampão 50 mM Tris, 25mM NaCl pH 7,5 e então 25 µL de
lipídio dissolvido em clorofórmio para DPPC ou clorofórmio:metanol (2:1) para
DPPG, na concentração de 1mM foram espalhados na interface ar- água (Figura
20). Depois da evaporação do clorofórmio, a proteína HS100A12 apo (sem íons
ligados) e na presença dos íons cálcio e/ ou zinco foi injetada na subfase. De modo
a acompanhar a adsorção da proteína na interface, medidas de cinética de adsorção
foram realizadas, e apenas após o equilíbrio ter sido atingido, a monocamada mista
foi comprimida. Os experimentos foram realizados com duas concentrações
diferentes da proteína, 0,5 µg/mL e 2,5 µg/mL.
DPPC DPPG DPPC DPPG Figura 20 - Estrutura dos fosfolipídios utilizados para o estudo de monocamadas de Langmuir. DPPC:
1,2-Dipalmitoil- sn- glicero- 3- fosfatidil colina. DPPG: 1,2- Dipalmitoil- sn- glicero- 3- fosfatidil glicerol.

HS100A12 75 4.4 Resultados e Discussões
4.4.1 Expressão Heteróloga da Proteína HS100A12
A produção da proteína recombinante em quantidade e na ausência de
contaminantes se faz necessária para a realização dos estudos espectroscópicos
propostos neste trabalho. Para tal, a síntese da proteína HS100A12 foi conduzida
em E. coli, cepa Rosetta e induzida por IPTG, conforme protocolo descrito na seção
4.3.7.
A expressão da proteína foi monitorada por eletroforese em gel de
poliacrilamida SDS-PAGE 16% em tampão tricina (Figura 21), onde são observadas
bandas de expressão adicionais frente ao conteúdo de proteínas totais de E.coli. A
presença da proteína HS100A12 é confirmada pela banda em aproximadamente
10,57 kDa.
1 2 3kDa
16,914,410,7
6,21
8,15
1 2 31 2 3kDa
16,914,410,7
6,21
8,15
1 2 3kDa
16,914,410,7
6,21
8,15
1 2 3
Figura 21 - Expressão da proteína HS100A12. Proteínas visualizadas em gel de poliacrilamida (SDS-PAGE
16%) em tampão tricina. Colunas: 1- marcadores de massa molecular; 2- cultura antes da indução e 3- cultura induzida com 1 mM de IPTG após 5 horas a 370C. A posição da HS100A12 está indicada pela seta vermelha.
4.4.1.1 Expressão dos Mutantes da Proteína HS100A12
Primeiramente, foi realizado teste de expressão de cada mutante da proteína
HS100A12 e confirmadas as melhores condições para a produção de grande

76
quantidade e estabilidade da proteína para, então, procedermos com a expressão
em maior escala (2 litros). Os procedimentos utilizados para realizar o teste de
expressão e a expressão foram os mesmos para todos os diferentes mutantes da
proteína.
Os testes de expressão foram feitos em células da cepa Rosetta de E. coli
inoculadas em 10 mL de meio LB e induzidas pela adição de solução 1 mM de IPTG.
O nível de expressão das proteínas recombinantes foi avaliado retirando-se
alíquotas de hora em hora das culturas de células mantidas nas temperaturas de
30oC e 37oC (dados não mostrados).
Todos os precipitados (“pellets”) provenientes das alíquotas retiradas durante a
etapa de expressão protéica foram adequadamente ressuspendidos e preparados
para análises por eletroforese em gel de poliacrilamida SDS-PAGE. As frações dos
mutantes A80C e F57C/A80C foram escolhidas para efeito de ilustração, pois todos
os mutantes da proteína apresentam resultados de expressão similares.
Na figura 22, é apresentado o gel eleforético de poliacrilamida do teste de
expressão a 37oC dos mutantes A80C e F57C/A80C da proteína HS100A12,
evidenciando a existência de frações induzidas com o tamanho esperado para a
proteína (aproximadamente 10,5 kDa). As frações induzidas nessa temperatura por
5 horas revelam a existência de maior quantidade de proteína tanto para o mutante
A80C quanto para F57C/A80C. Com isso, optamos por expressar estes e todos os
outros mutantes em 2 L de meio LB, a 37oC por 5 horas, como descrito para a
proteína nativa (seção 4.3.7.), obtendo-os em quantidade e estáveis para posterior
purificação.
kDa
16,914,410,7
6,21
8,15
1 2 3 4 5 6 7 8
kDa
16,914,410,7
6,21
8,15
1 2 3 4 5 6 7 8
Figura 22 - Expressão dos mutantes A80C e F57C/A80C a 370C. Proteínas visualizadas em gel SDS- PAGE
16% em tampão tricina. Colunas: 1- marcadores de massa molecular; 2- amostra não induzida; 3- Mutante A80C induzido a 370C por 2 horas; 4- Mutante A80C induzido a 370C por 5 horas; 5- Mutante A80C induzido a 370C “overnight”; 6- Mutante F57C/A80C induzido a 370C por 2 horas; 7- Mutante F57C/A80C induzido a 370C por 5 horas e 8- Mutante F57C/A80C induzido a 370C “overnight”. As posições dos mutantes da proteína HS100A12 estão indicadas pelas setas vermelhas.

HS100A12 77 4.4.2 Purificação da Proteína HS100A12
O protocolo inicial de purificação da proteína HS100A12 nativa foi adaptado do
procedimento utilizado por Moroz et al (75), onde fazia-se uso de duas etapas
cromatográficas, sendo a primeira, uma cromatografia por interação hidrofóbica,
seguida por uma cromatografia de troca iônica. Contudo, o intenso manuseio da
amostra para a retirada do excesso do sal NaCl após a etapa de eluição da coluna
de interação hidrofóbica levou à precipitação da proteína. Com isso, o sucesso
esperado não foi alcançado com o uso do protocolo original, sendo necessário que
estabelecéssemos um novo procedimento, descrito ao longo desta seção, para
obtermos a proteína pura.
A purificação da proteína HS100A12 inicia-se incubando o sobrenadante que
resultou do rompimento das células (extrato bruto) com CaCl2 que, em seguida, é
aplicado na coluna de interação hidrofóbica Hiprep Fenil-Sefarose FF e eluída com
tampão contendo EDTA. A incubação com CaCl2 é necessária pois, como
mencionado na introdução deste capítulo, o íon Ca+2 provoca mudanças
conformacionais na proteína HS100A12, expondo seu interior hidrofóbico e com
isso, aumentando a interação da proteína com a resina.
A análise dos resultados (Figura 23) indica que esta etapa de purificação foi
importante para retirar uma grande parte de contaminantes do material bruto, mas
não foi suficientemente eficiente para purificar a proteína, exigindo a utilização de
uma segunda etapa cromatográfica. A escolha desta segunda etapa se baseou na
observação de que a maioria dos contaminantes presentes na amostra possuía
pesos moleculares maiores que a proteína de interesse, assim levando-nos a decidir
por uma cromatografia de exclusão molecular, em que se separa as moléculas por
diferença de tamanho. Com isso, a proteína HS100A12 eluída da primeira etapa de
purificação foi aplicada à coluna Superdex 75 e o resultado foi a obtenção da
proteína em quantidade e grau de pureza suficientes para as medidas
espectroscópicas (Figura 23).

78
1 2 3 4 5 6 7 8 9
kDa
16,9
8,1510,7
6,21
1 2 3 4 5 6 7 8 9
kDa
16,9
8,1510,7
6,21
Figura 23 - Purificação da proteína HS100A12 por cromatografia de interação hidrofóbica e de exclusão
molecular. Proteínas visualizadas em gel SDS- PAGE 16% em tampão tricina. Colunas: 1- marcadores de massa molecular; 2- amostra induzida; 3- amostra não induzida; 4- pellet; 5- fração solúvel das células lisadas; 6- material que não se ligou à coluna Fenil Sefarose; 7- amostra da proteína de interesse eluída da coluna Fenil Sefarose; 8- fração que compõem um dos picos do cromatograma de exclusão molecular e 9- amostra da proteína eluída da coluna Superdex 75. A posição da proteína HS100A12 está indicada pela seta vermelha.
4.4.2.1 Purificação dos Mutantes da Proteína HS100A12
As etapas cromatográficas empregadas na purificação dos mutantes da
proteína HS100A12 foram similares àquelas padronizadas para obtenção da
proteína nativa, exceção feita à adição do reagente 1,4-Ditiotritol (DTT) nos tampões
de todas as etapas da purificação. Esta modificação foi introduzida no protocolo de
purificação dos mutantes para evitar a formação de pontes dissulfetos entre os
resíduos de cisteína inseridos pelas mutações.
Assim como para a proteína nativa, o nível de pureza obtido para todos os
mutantes foi bastante adequado com variações apenas em termos de rendimento,
que foi muito pequeno para alguns mutantes em estudo.
Na figura 24 são apresentados os géis eletroforéticos (SDS-PAGE 16% em
tampão tricina) dos mutantes A80C, F57C/A80C e K33C/A80C para efeito de
ilustração, pois todos os mutantes da proteína apresentam o mesmo perfil de
purificação. A presença dos mutantes da proteína HS100A12 é confirmada pela
banda em aproximadamente 10,575 kDa.

HS100A12 79
1 2 3 4 5 6 7 8kDa
16,9
14,4
10,7
6,21
1 2 3 4 5 6 7 8kDa
16,9
14,4
10,7
6,21
1 2 3 4 5 6 7
1 6, 914 ,4
10 ,78,1 5
6,2 1
KDa1 2 3 4 5 6 7
1 6, 914 ,4
10 ,78,1 5
6,2 1
KDa
A B1 2 3 4 5 6 7 8
kDa
16,9
14,4
10,7
6,21
1 2 3 4 5 6 7 8kDa
16,9
14,4
10,7
6,21
1 2 3 4 5 6 7
1 6, 914 ,4
10 ,78,1 5
6,2 1
KDa1 2 3 4 5 6 7
1 6, 914 ,4
10 ,78,1 5
6,2 1
KDa
A B
Figura 24 - Análise por eletroforese em SDS-PAGE 16% em tampão tricina da purificação dos mutantes
da proteína HS100A12. (A): Mutante A80C. Colunas: 1- marcadores de massa molecular; 2- amostra induzida; 3- amostra não induzida; 4- pellet; 5- fração solúvel das células lisadas; 6- material que não se ligou à coluna Fenil Sefarose; 7- amostra do mutante eluída da coluna Fenil Sefarose e 8- amostra da proteína eluída da coluna Superdex 75. (B): Colunas: 1- marcadores de massa molecular; 2- material não ligado à coluna Fenil Sefarose; 3- amostra do mutante F57C/A80C eluída da coluna Fenil Sefarose; 4- amostra do mutante F57C/A80C eluída da coluna Superdex 75; 5- material não ligado à coluna Fenil Sefarose; 6- amostra do mutante K33C/A80C eluída da coluna Fenil Sefarose; 4- amostra do mutante K33C/A80C eluída da coluna Superdex 75. As posições da proteína HS100A12 estão indicadas pelas setas vermelhas.
4.4.3 Influência dos Íons Cálcio e Zinco no Estado de Oligomerização da
HS100A12
Estudos cristalográficos mostraram que o cálcio possui um papel importante na
ativação de mudanças conformacionais e na oligomerização da proteína HS100A12
(101), onde o excesso de Ca+2 faz com que os dímeros da proteína HS100A12 se
arranjem de forma hexamérica (v. seção 4.2.). Adicionalmente, em 2009, Moroz et al
(75) determinaram as estruturas cristalográficas da HS100A12 apo (forma dimérica)
e complexada com Zn+2 (forma tetramérica), que mostrou-se também importante no
enovelamento da proteína e nas interações com seus alvos específicos.
Ainda com o objetivo de explorar os fatores que influenciam a oligomerização
da HS100A12 e da sua interação com moléculas alvo, como por exemplo, receptor
extracelular RAGE, Moroz et al (102) realizaram uma série de experimentos que
indicaram mudanças no estado oligomérico da proteína HS100A12 quando se
variava a concentração de cálcio e de zinco na solução de proteína. Com a técnica
de ressonância plasmônica de superfície (SPR), os autores observaram que a
presença de cálcio e zinco é essencial para a interação da proteína com o receptor.
Diante disso, decidimos verificar o estado de oligomerização da proteína
HS100A12 nativa e dos seus mutantes utilizando a técnica de cromatografia de

80
exclusão molecular. Inicialmente, reproduzimos e confirmamos o experimento feito
por Moroz et al (102), aplicando a proteína apo (ausência de íons) na coluna
Superdex 75 10/300 GL (GE Healthcare) (exclusão molecular) previamente
equilibrada com o tampão 50 mM Tris, 200 mM NaCl, pH 7,5 e observamos, após
eluição com o mesmo tampão, a presença da HS100A12 na forma dimérica. O
mesmo procedimento foi realizado com a proteína na presença de 1 mM Ca+2, 10
µM Zn+2 e na presença de ambos (1 mM Ca+2 e 10 µM Zn+2), sendo eluídas da
coluna na forma dimérica, tetramérica e hexamérica, respectivamente (dados não
mostrados).
Em seguida, realizamos um estudo utilizando a técnica de cromatografia de
exclusão molecular variando-se a concentração de cloreto de sódio (NaCl) presente
no tampão e observamos que os perfis de eluição da proteína HS100A12 com e sem
íons não eram reprodutíveis quando comparados com os resultados de Moroz et al
(102). A dependência dos estados de oligomerização da HS100A12 em função da
concentração de NaCl, tipos de tampões e valores de pHs, apesar de interessante,
nos levaria a desviar do objetivo principal do presente trabalho, qual seja a
preparação de vários mutantes para posteriores experimentos de RPE e
implementação da técnica de marcação de spin sítio dirigida. Portanto, devido à
impossibilidade de conseguirmos estudar e caracterizar todas essas diferentes
condições por cromatografia de exclusão molecular e dicroísmo circular, optamos
por escolher uma única condição e, assim, podermos caracterizar a proteína para
realizarmos as medidas por ressonância paramagnética eletrônica.
As condições de trabalho escolhidas para todas as medidas a serem realizadas
a partir de então foram 50 mM Tris, 25 mM NaCl, pH 7,5 e se deu pela possibilidade
da amostra ser pouco manuseada, já que é nesta condição que a proteína é eluída
no ultimo passo de purificação e também pela pouca quantidade de NaCl, ideal para
a realização de medidas de CD.
Na figura 25 é apresentado o perfil da cromatografia de exclusão molecular da
proteína HS100A12 nativa, para efeito de ilustração, pois todos os mutantes da
proteína apresentam o mesmo perfil cromatográfico. Analisando o cromatograma,
observamos que o perfil de eluição da proteína HS100A12 apo mostrou um pico
correspondente à massa molecular de 18 ± 3 kDa, enquanto a HS100A12 com Ca+2
apresentou um pico correspondente à massa molecular de 20 ± 3 kDa, a proteína
com Zn+2 e na presença de Ca+2 e Zn+2 eluiram em picos correspondente a 43 ± 3

HS100A12 81 kDa (Figura 25). Os valores obtidos para a proteína apo e na presença de cálcio são
aproximadamente o dobro do esperado para o monômero (~10,5 kDa), indicando
assim, que a proteína é homodimérica nas condições analisadas (50 mM Tris, 25mM
NaCl pH 7,5). Já a proteína na presença de Zn+2 e também na presença dos dois
íons possuiram o quádruplo do peso esperado para o monômero, sendo, portanto,
tetramérica nas mesmas condições.
4 5 6 7 8 9 10 11 12 13 14 15 16 17 180
50
100
150
200
250
300
350
400
HS100A12 Apo HS100A12 com Ca+2
HS100A12 com Zn+2
HS100A12 com Ca+2 e Zn+2
Padrao- 20 kDa Padrao- 43 kDa Padrao- 66 kDa
Abs
orbâ
ncia
280n
m(m
AU
)
Volume de eluiçao (mL)
Figura 25 - Cromatografia por exclusão molecular da proteína HS100A12 nativa e dos padrões de peso
molecular. Apo: preto; com cálcio: vermelho; com zinco: azul e com cálcio e zinco: marrom. Padrões: ITS (20 kDa): roxo tracejado; Ovalbumina (43 kDa): verde tracejado e albumina (66 kDa): rosa tracejado.
4.4.4 Dicroísmo Circular
O conteúdo de estrutura secundária dos mutantes da proteína HS100A12 e a
estabilidade térmica da proteína nativa foram investigados como descrito na seção
4.3.11. A influência dos íons Ca+2 e Zn+2 na estabilidade da proteína também foi
estudada por dicroísmo circular, adicionando-se 38 µM de CaCl2 e dialisando a
amostra por 4 vezes com tampão contendo 40 µM ZnCl2.

82
4.4.4.1 Fatores Estruturais da Proteína HS100A12 e dos seus Mutantes
A influência dos íons cálcio e zinco na estrutura da proteína HS100A12 e dos
seus mutantes foi investigada por espectroscopia de dicroísmo circular. O espectro
de CD da HS100A12 nativa em sua forma apo e em tampão 50 mM Tris, 25 mM
NaCl pH 7,5 a 20oC é mostrado na figura 26 e é caracterizado por dois mínimos
negativos em 220 e 208 nm e um máximo positivo em torno de 197 nm. Os mínimos
em 208 e 220 nm são atribuídos a transições encontradas em estruturas helicoidais
(106), o que indica conteúdo de α-hélice na estrutura da S100A12 humana. No
entanto, em estruturas com alto conteúdo de α-hélice, a banda em 197 nm é
usualmente muito mais intensa do que as bandas negativas do espectro. Este fato
sugere que há uma mistura de espectros associados a α-hélice com espectros
correspondentes a estruturas do tipo “loop” ou “turns”. Estes últimos apresentam
uma banda negativa em torno de 190 nm, cuja soma com o máximo em 197 nm
relativo a α-hélice, levaria a um decréscimo desta última. Assim, uma análise
qualitativa do espectro de CD da HS100A12 indica que esta possui conteúdo de
estrutura secundária com α-hélice e regiões flexíveis (desordenadas), o que está de
acordo com a estrutura cristalográfica previamente reportada (74).
Para estudarmos a influência dos íons cálcio e zinco quando ligados na
estrutura da HS100A12 também realizamos experimentos de dicroísmo circular da
proteína nativa na presença de Ca+2 e/ou Zn+2. Esses íons são conhecidos por se
ligarem em sítios distintos da proteína e possuirem um papel biológico importante.
Por isso, também mostrado na figura 26, estão os espectros de CD da proteína
HS100A12 na presença dos dois íons, separadamente e juntos, sendo a
concentração de cálcio suficiente para saturar os sítios de ligação da proteína (38
µM) e a concentração de zinco resulta de quatro diálises em amicon com tampão
contendo 40 µM de ZnCl2. Características espectrais similares são observadas
nesses casos quando comparadas ao espectro de CD da HS100A12 apo (sem íons)
indicando, portanto, que a composição de estrutura secundária da proteína não é
significativamente alterada pela ligação dos íons.
Entretanto, algumas diferenças espectrais podem ser detectadas como, por
exemplo, a variação entre a intensidade das curvas, que provavelmente se deve a

HS100A12 83 diferentes estados de oligomerização da proteína. O espectro da proteína na
presença de Zn+2 possui tanto as bandas negativas (208 e 220 nm) quanto a banda
em 197 nm mais intensas que nos demais espectros, devido ao fato de apresentar-
se no estado tetramérico, onde pode ocorrer um aumento do conteúdo de α- hélice
ou um rearranjo estrutural das hélices quando da ligação com o zinco. O contrário
ocorre com a proteína na presença dos dois íons ao mesmo tempo, em que a
diminuição da intensidade do espectro pode ser atribuída a um rearranjo
desfavorável de hélices quando comparado com o espectro da proteína apo (sem
íon ligado). Em relação à variação entre as intensidades das curvas da proteína apo
e da proteína na presença de Ca+2, estas podem ser devidas ao rearranjo estrutural,
principalmente da hélice C, que sofre uma mudança conformacional de
aproximadamente 90 graus na presença de cálcio, comparada a estrutura apo
(Figura 13).
200 210 220 230 240 250-30
-20
-10
0
10
20
30
HS100A12 Apo HS100A12 com Ca+2
HS100A12 com Zn+2
HS100A12 com Ca+2 e Zn+2
[θ] (
103 de
g.cm
2 . dm
ol-1)
Comprimento de onda (nm)
Figura 26 - Espectros de CD da proteína HS100A12 na presença e ausência dos íons cálcio e zinco em
tampão 50mM Tris, 25mM NaCl pH 7,5 a 200C. Preto: espectro da proteína sem íons; vermelho: espectro da proteína na presença de cálcio; azul: espectro da proteína na presença de zinco e verde: espectro da proteína na presença de cálcio e zinco.
A fim de caracterizar os mutantes da proteína HS100A12 analisando o
conteúdo de estrutura secundária (dados não mostrados), observamos
características espectrais similares quando comparados ao espectro de CD da
HS100A12 nativa indicando, portanto, que a composição estrutural secundária da
proteína não é significativamente alterada pelas mutações, confirmando a
integridade estrutural dos mutantes da proteína que foram expressos, um pré-
requisito essencial para estudos futuros mais detalhados.

84
4.4.4.2 Estudo da Estabilidade Térmica da Proteína HS100A12
Com o intuito de investigarmos a estabilidade térmica da proteína HS100A12
na presença e na ausência dos íons cálcio e zinco, utilizamos um tratamento
termodinâmico para determinarmos as temperaturas e energias envolvidas na
transição.
Os espectros de CD da HS100A12 apo e na presença dos íons Ca+2, Zn+2 e
Ca+2/Zn+2 em função da variação de temperatura são mostrados na figura 27. À
medida que a temperatura aumenta gradativamente de 10 para 100oC, as mudanças
no espectro de CD da proteína, com e sem íons, revelam uma diminuição na
elipticidade. O ponto isodicróico em 203 nm (ponto de intersecção de todos os
espectros) que está presente nos espectros da proteína apo é perdido quando a
proteína está ligada aos íons, dando indícios de que a perda de estrutura secundária
(desnaturação) é um processo diferente para a proteína com e sem íons.
A desnaturação térmica da proteína HS100A12 apo e na presença dos íons é
um processo de desenovelamento irreversível, já que as amostras resfriadas para
20oC imediatamente após o processo de aquecimento originaram espectros de CD
bastante diferentes daqueles inicialmente obtidos para a mesma temperatura,
concluindo que a proteína não retornou ao seu estado nativo inicial após o
aquecimento-resfriamento.

HS100A12 85 A B
200 210 220 230 240 250
-20
-10
0
10
20
30
40 100C 140C 180C 220C 260C 300C 340C 380C 420C 460C 500C 540C 580C 620C 660C 700C 740C 780C 820C 860C 900C 940C 980C
HS100A12 Apo
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
200 210 220 230 240 250-30
-20
-10
0
10
20
30
100C 140C 180C 220C 260C 300C 340C 380C 420C 460C 500C 540C 580C 620C 660C 700C 740C 780C 820C 860C 900C 940C 980C
HS100A12 com Calcio
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
C D
200 210 220 230 240 250
-30
-20
-10
0
10
20
30
100C 140C 180C 220C 260C 300C 340C 380C 420C 460C 500C 540C 580C 620C 660C 700C 740C 780C 820C 860C 900C 940C 980C
HS100A12 com Zinco
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
200 210 220 230 240 250
-15
-10
-5
0
5
10
15
20
25
30
100C 140C 180C 220C 260C 300C 340C 380C 420C 460C 500C 540C 580C 620C 660C 700C 740C 780C 820C 860C 900C 940C 980C
HS100A12 com Calcio e Zinco[θ
] (10
3 deg.
cm
2 . dm
ol-1)
Comprimento de onda (nm)
Figura 27- Análise da desnaturação térmica da proteína HS100A12 utilizando dicroísmo circular. (A):
HS100A12 Apo. (B): HS100A12 na presença de cálcio. (C): HS100A12 na presença de zinco. (D): HS100A12 na presença de cálcio e zinco.
Resultados similiares são observados quando a elipticidade das amostras é
monitorada em 208 nm (Figura 28), comprimento de onda considerado um indicativo
prático de estrutura secundária da proteína (113). A desnaturação térmica da
HS100A12 exibe um comportamento do tipo sigmoidal, típico de processos
cooperativos, que se inicia em torno de 72oC e é completado próximo a 98oC, para a
HS100A12 apo (sem íons ligados) e começa em torno de 72oC, 86oC e 92oC e que
está quase completamente finalizado em torno de 88oC, 96oC e 100oC para a
proteína na presença de Ca+2, Zn+2 e Ca+2/Zn+2, respectivamente.

86
A B
0 20 40 60 80 100-22
-20
-18
-16
-14
-12
-10
-8
-6
-4
θ20
8(103 d
eg.c
m2 .d
mol
-1)
Temperatura (0C)
0,00 0,02 0,04 0,06 0,08 0,10-25
-20
-15
-10
-5
0
θ20
8 (1
03 deg.
cm
2 .dm
ol-1)
Temperatura (0C)
C D
0 20 40 60 80 100-30
-25
-20
-15
-10
-5
θ20
8(10
3 deg
. cm
2 . dm
ol-1)
Temperatura (0C)
0 20 40 60 80 100
-14
-12
-10
-8
-6
-4
θ20
8 (1
03 deg.
cm
2 . dm
ol-1)
Temperatura (0C)
Figura 28 - Transição da desnaturação térmica com a elipticidade monitorada a 208 nm. (A): HS100A12
Apo. (B): HS100A12 na presença de cálcio. (C): HS100A12 na presença de zinco. (D): HS100A12 na presença de cálcio e zinco. A diminuição do módulo da elipticidade está associada com a perda de estrutura secundária. As linhas sólidas são ajustes obtidos com funções do tipo sigmóide.
Como colocado acima, a desnaturação térmica da HS100A12 na presença e
ausência dos íons cálcio e zinco pode ser descrita como um processo irreversível de
dois estados, descrito por:
N →k D
onde N e D são as formas nativa e desnaturada da enzima, respectivamente e k é a
constante de primeira ordem da reação, cuja dependência com a temperatura é
representada pela equação de Arrhenius,
k=A exp (−Ea/RT) (5)
onde T é a temperatura em Kelvin e Ea é a energia de ativação da reação.
As frações de proteína desnaturada, fD, e nativa, fN, são dadas por

HS100A12 87
FD = (θT−θN)/(θN−θD) e fD + fN =1 (6)
onde θT é a elipticidade da amostra a uma temperatura particular; θD e θN são
valores característicos de elipticidade dos estados desnaturado e nativo tomados em
alguma temperatura bem abaixo ou bem acima da transição térmica. A fração da
proteína nativa, fN, está relacionada com a temperatura de acordo com a seguinte
equação (114):
ln(ln(1/fN))=(Ea/R)(1/Tm − 1/T) (7)
onde Tm é a temperatura de transição. Então, a equação representada acima pode
ser usada para estimarmos a energia de ativação da reação, Ea, e a temperatura de
transição, Tm, construindo-se um gráfico de ln(ln(1/fN)) versus 1/T.
Os valores da elipticidade foram transformados em frações de proteína nativa e
desnaturada, de acordo com a Eq. 6 e a variação com a temperatura da fD é
apresentada na figura 29. Esses valores foram então usados para construir os
gráficos de ln(ln(1/fN)) versus 1/T, que foram ajustados para uma função linear (Eq.
7). Os ajustes resultaram em uma boa relação linear (Figura 30) e, a partir do
coeficiente angular desses gráficos, a energia de ativação aparente foi calculada
para HS100A12 apo (198±9 kJ/mol), para HS100A12 na presença do Ca+2 (303± 20
kJ/mol), para a proteína na presença do Zn+2 (650±80 kJ/mol) e para HS100A12
ligada com Ca+2 e Zn+2 (720± 90 kJ/mol).
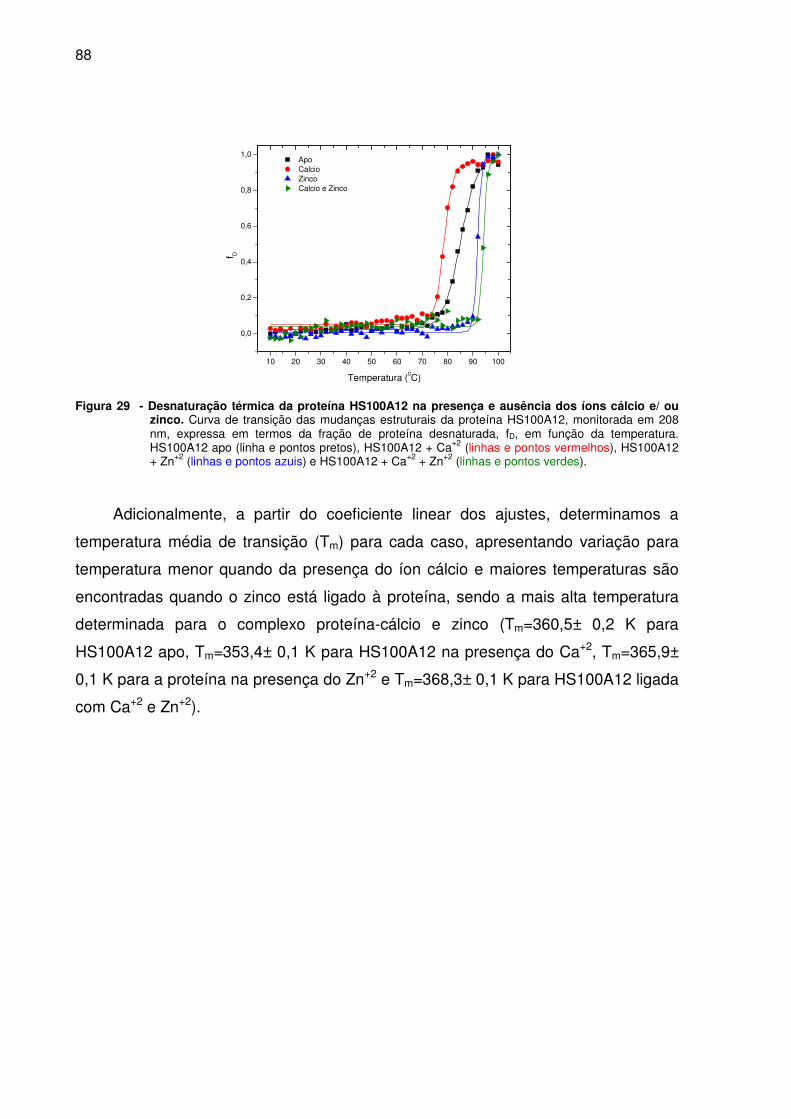
88
10 20 30 40 50 60 70 80 90 100
0,0
0,2
0,4
0,6
0,8
1,0
Apo Calcio Zinco Calcio e Zinco
f D
Temperatura (0C)
Figura 29 - Desnaturação térmica da proteína HS100A12 na presença e ausência dos íons cálcio e/ ou
zinco. Curva de transição das mudanças estruturais da proteína HS100A12, monitorada em 208 nm, expressa em termos da fração de proteína desnaturada, fD, em função da temperatura. HS100A12 apo (linha e pontos pretos), HS100A12 + Ca+2 (linhas e pontos vermelhos), HS100A12 + Zn+2 (linhas e pontos azuis) e HS100A12 + Ca+2 + Zn+2 (linhas e pontos verdes).
Adicionalmente, a partir do coeficiente linear dos ajustes, determinamos a
temperatura média de transição (Tm) para cada caso, apresentando variação para
temperatura menor quando da presença do íon cálcio e maiores temperaturas são
encontradas quando o zinco está ligado à proteína, sendo a mais alta temperatura
determinada para o complexo proteína-cálcio e zinco (Tm=360,5± 0,2 K para
HS100A12 apo, Tm=353,4± 0,1 K para HS100A12 na presença do Ca+2, Tm=365,9±
0,1 K para a proteína na presença do Zn+2 e Tm=368,3± 0,1 K para HS100A12 ligada
com Ca+2 e Zn+2).

HS100A12 89
A B
0,00270 0,00275 0,00280 0,00285 0,00290-3
-2
-1
0
1
HS100A12 Apoln
(ln(1
/f N))
1/T (K)
Valor
Coef. linear 66,1564
Coef. angular -23849,75356
0,00280 0,00285 0,00290-3
-2
-1
0
1
ln(ln
(1/f N
))
1/T (K)
HS100A12 com Calcio
ValorCoef. linear 103,0209Coef. angular -36402,45
C D
0,00270 0,00273 0,00276-4
-3
-2
-1
0
1
2
ln(ln
(1/f N
))
1/T (K)
Valor
Coef. linear 214,83028
Coef. angular -78611,24711
HS100A12 com Zinco
0,00267 0,00270 0,00273 0,00276-3
-2
-1
0
1
2
3
4HS100A12 com Calcio e Zinco
ln(ln
(1/f N
))
1/T (K)
Valor
Coef. linear 235,77051
Coef. angular -86839,83044
Figura 30 - Gráficos ln(ln(1/fN) versus 1/T. (A): HS100A12 Apo. (B): HS100A12 na presença de cálcio. (C):
HS100A12 na presença de zinco. (D): HS100A12 na presença de cálcio e zinco. As tabelas indicam valores dos coeficientes linear e angular. As linhas sólidas são ajustes obtidos com funções lineares.
Os dois parâmetros, energia de ativação e temperatura de transição, indicam
que a ocupação dos sítios ligantes na estrutura da HS100A12 pelo zinco confere ao
complexo proteína-zinco uma estabilidade maior do que aquela observada apenas
para a HS100A12. Isto é compatível com os dados de Moroz et al (75), onde os
autores propõem que a ligação do zinco na proteína livre de metal causa um
rearranjo na hélice D e uma mudança conformacional na hélice A, resultando na
estabilização da interface de tetramerização e formação de tetrâmeros.
Adicionalmente, podemos aventar a hipótese de que a HS100A12 com os dois íons
ligados (cálcio e zinco) se torne ainda mais estável devido à facilidade de formação
de oligômeros maiores (hexâmeros), pois com a ligação do zinco a afinidade da
proteína pelo íon cálcio aumenta em torno de 1500 vezes, fazendo com que todos

90
os sítios de ligação sejam rapidamente ocupados pelo cálcio, com o cálcio adicional
podendo se ligar entre os dímeros, levando à formação de hexâmeros.
Já a desestabilização do complexo proteína-cálcio se deve ao conhecido
mecanismo de proteínas S100 em que a ligação com o metal é a responsável por
expor o interior hifrofóbico da proteína, aumentando o conteúdo eletrostático
carregado negativamente e com isso, deixando a proteína mais sensível à variações
de temperatura e mudanças conformacionais.
4.4.5 Estudo de Ressonância Paramagnética Eletrônica por marcação de spin
sítio dirigida da transição estrutural induzida pelos íons cálcio e zinco na
S100A12 humana
Como dito anteriormente, o sucesso da técnica de SDSL passa pelo
conhecimento detalhado da estrutura da proteína em estudo. Um dos primeiros
objetivos que tentamos alcançar com o presente trabalho, foi monitorar mudanças
conformacionais na HS100A12 quando da ligação dos íons cálcio e zinco. Um ponto
crucial da estratégia de SDSL-RPE está exatamente no planejamento de onde serão
introduzidos os resíduos de cisteína de forma a sermos capazes de detectar as
mudanças de interesse. Para satisfazer a esta condição, apresentamos nesta seção
uma comparação entre as estruturas da Troponina C cardíaca humana (TnC) e
S100A12 humana (HS100A12). Assim como a enzima HS100A12, a TnC cardíaca é
uma proteína ligante de cálcio, que pertence à superfamília das calmodulinas e,
consequentemente, liga-se a íons Ca+2 através de motivos “EF-hand”. Além dessas
características em comum, a TnC foi alvo de estudos por SDSL-RPE e, assim, os
resíduos da HS100A12 em posições análogas àquelas que foram sensíveis a
mudanças na estrutura da TnC foram escolhidos como ponto de partida para nossos
estudos.
O monômero da TnC possui dois sítios de ligação de cálcio, o sítio I que é um
motivo “EF-hand” e consiste da hélice A-“loop”-hélice B e o sítio II que consiste da
hélice C-“loop”-hélice D (Figura 31), sendo a afinidade do sítio I pelo íon Ca+2 muito
mais baixa que do sítio II (10). A ligação do cálcio induz uma grande mudança

HS100A12 91 conformacional na proteína, passando de uma conformação “fechada” para uma
“aberta”, expondo assim sua superfície hidrofóbica e interagindo com moléculas
alvos, receptores extracelulares e outras proteínas.
A
B
CD
N
A
B
CD
N
Figura 31 - Estrutura da TnC cardíaca humana (PDB 1SPY) com a indicação das hélices A, B, C e D e N-terminal.
Como dito anteriormente, escolhemos utilizar para efeito de comparação a TnC
cardíaca porque, além de ser uma proteína que liga íons Ca+2, muitos estudos estão
sendo feitos, como por exemplo, aquele realizado por Ueki et al (10) em que se
examina o movimento induzido pelo íon Ca+2 nas hélices B e C relativo à hélice D do
estado monomérico da TnC utilizando ressonância paramagnética eletrônica (10).
Nesse trabalho, os autores examinam a mudança da distância interspin (distância de
um marcador de spin para outro marcador de spin) quando ocorre a ligação do íon
cálcio nos duplos mutantes da TnC. Dois mutantes da enzima TnC foram utilizados,
cada um contendo dois resíduos de cisteína. Em um caso, os resíduos de cisteína
estavam presentes nas hélices B e D e no outro, eles estavam presentes na hélice C
e D; o resíduo escolhido na hélice D foi a cisteína nativa 84. A fim de examinar o
movimento da hélice B, os autores mutaram a Glicina 42 (Gly-42) por uma cisteína,
porque este é um resíduo que está exposto ao solvente e sua polaridade é parecida
com a polaridade do resíduo de cisteína. A glutamina 58 (Gln-58) foi selecionada
para a mutação na hélice C, usando o mesmo critério utilizado para a escolha da
Gly-42 (10).
A partir dessas informações, fizemos uma comparação da estrutura da enzima
TnC cardíaca humana com a enzima HS100A12 (Figura 32) e identificamos alguns
resíduos para a realização das primeiras mutações sítio dirigidas utilizadas para o

92
estudo da mudança conformacional da proteína HS100A12 induzida pelos íons
cálcio e zinco.
Cys- 84
Ala- 80
Gly- 42Lys- 33
Phe- 57
Gln- 58
Cys- 84
Ala- 80
Gly- 42Lys- 33
Phe- 57
Gln- 58
Figura 32 - Sobreposição das estruturas das proteínas TnC (verde) e HS100A12 (rosa), indicando os
resíduos da proteína HS100A12 que correspondem aos resíduos mutados na TnC por Ueki et al (10).
Inicialmente, assim como Ueki et al (10), três mutantes da proteína HS100A12
foram construídos (Figura 12 e Tabela 1): no primeiro deles substituímos a alanina
80 (A80) por uma cisteína, onde foi inserido o marcador de spin; no segundo, além
da alanina 80 presente na hélice D, substituímos a lisina 33 (K33) presente na hélice
B por uma cisteína e, por fim, substituímos a fenilalanina 57 (F57), presente na
hélice C, também por uma cisteína, construindo, assim, dois duplos mutantes
(K33C/A80C e F57C/A80C) que nos possibilitaram examinar a mudança da distância
interspin quando ocorre a ligação dos íons cálcio e zinco.
A fim de mapear e até mesmo propor um modelo para a dinâmica de
afastamento e aproximação das hélices C e D como um todo, quando Ca+2 e Zn+2
estão ligados à proteína, construímos também os seguintes duplos mutantes:
isoleucina 53 (hélice C) e alanina 80 (hélice D) originando o mutante I53C/A80C,
ácido aspártico 54 (hélice C) e alanina 80 (hélice D) construindo o mutante
D54C/A80C, fenilalanina 57 (hélice C) e alanina 80 (hélice D) (F57C/ A80C), leucina
60 (hélice C) e alanina 80 (hélice D) (L60C/A80C), ácido aspártico 61 (hélice C) e
alanina 80 (hélice D) (D61C/A80C) (Figura 33).

HS100A12 93
A80
I53
D54
F57
L60
D61K33
A80
I53
D54
F57
L60
D61K33
Figura 33 - Estrutura do dímero da proteína HS100A12 (PDB 2WCE), indicando os resíduos mutados por
cisteínas para posterior marcação de spin. Figura construída utilizando-se o programa Pymol (115).
Na Figura 34, apresentamos os espectros de RPE dos mutantes HS100 A80C,
K33C/A80C, I53C/A80C, D54C/A80C, F57C/A80C, L60C/A80C, D61C/A80C com o
marcador de spin (MTSSL) na ausência e na presença dos íons cálcio e zinco a
250C. Em vários dos casos estudados, os espectros dos mutantes em estudo,
principalmente na presença do íon Zn2+, apresentam uma componente que se refere
a uma população de nitróxidos (marcador de spin) em regime de movimento
praticamente isotrópico e devida principalmente à presença de moléculas de
marcadores de spin livres em solução mesmo após o procedimento de separação
descrito na seção 4.3.12. Essa componente livre é caracterizada por três linhas de
ressonância estreitas e de intensidade similares (por exemplo, Figura 34B no
espectro na presença de Zn2+).
Façamos uma análise dos espectros de cada mutante nas três situações
investigadas (proteína na forma apo, na presença de Ca2+ ou de Zn2+). O espectro
do mutante HS100A12 A80C (Figura 34A, linha preta), sonda no resíduo base para
as medidas de distância, contém duas componentes e não apresenta contribuição
significativa devida a marcadores de spin livres em solução. As duas componentes
observadas (duas ressonâncias na linha de campo baixo – v. setas na Figura 34A)
podem ser associadas a duas conformações experimentadas pela cadeia principal

94
da proteína que se alternam numa escala de tempo de micro a nanosegundos. A
adição do íon Ca2+ não altera o espectro do mutante A80C (Figura 34A, linha
vermelha). Por outro lado, na presença dos íons Zn2+, o espectro se altera
fortemente sendo agora constituído por apenas uma componente espectral que não
pode ser relacionada a nenhuma das componentes previamente observadas (Figura
34A, linha azul). Ou seja, o espectro medido nesse caso difere dos anteriores e
indica, portanto, que os íons zinco estabilizam uma conformação distinta da cadeia
protéica em que o marcador de spin sofre grande restrição de movimento, já que seu
espectro é típico de sondas em regime de movimento bastante lento (quase rígido).
Os espectros medidos para o duplo mutante HS100A12 K33/A80C (Figura
34B) seguem o mesmo padrão observado para o mutante A80C: espectros contendo
duas componentes associadas a marcadores com restrição média ou grande de
movimento quando a proteína está em sua forma apo e na presença de cálcio, com
estabilização de uma conformação estrutural diferente após adição de zinco. Neste
caso, observamos o aparecimento de uma componente espectral relacionada a
marcadores livres em solução na presença de zinco (linhas bastante finas). Vale
ressaltar que, apesar das grandes mudanças observadas na presença de zinco, em
nenhuma das três condições se vê alargamento das linhas de ressonância devido à
presença de duas sondas na estrutura protéica.
Neste ponto, cabe lembrar que quando dois momentos de dipolo estão
separados por distâncias inferiores a cerca de 20 Å, o efeito de sua interação dipolar
magnética aparece nos espectros de RPE-CW na forma de um alargamento de
linhas. A não observação de aumento nas larguras de linha é indicativo de que os
dois centros paramagnéticos estão localizados a distâncias superiores a 20 Å e, no
caso do duplo mutante K33C/A80C, não parece sofrer maiores alterações em
termos de separação quando da presença dos íons.
Esta situação é completamente distinta ao passarmos para os espectros do
duplo mutante HS100A12 I53C/A80C (Figura 34C). Neste caso e nas três situações
medidas, observamos facilmente grande alargamento das ressonâncias,
evidenciando o sucesso de nossa estratégia de duplas mutações e marcações,
sugerindo distâncias muito curtas entre as sondas e que não mudam de forma
significativa ao adicionarmos os íons divalentes.

HS100A12 95
3300 3350 3400 3450
Zn+2
Ca+2
Campo Magnético (Gauss)
(A) HS100A12 A80C
Apo
3300 3350 3400
Zn+2
Ca+2
Apo
Campo Magnético (Gauss)
(B) HS100A12 K33C/A80C
3200 3250 3300 3350
Zn+2
Ca+2
Campo Magnético (Gauss)
(C) HS100A12 I53C/ A80C
Apo
3200 3250 3300
Zn+2
Ca+2
Campo Magnético (Gauss)
(D) HS100A12 D54C/A80C
Apo
continua

96
3300 3350 3400 3450
(E) HS100A12 F57C/ A80C
Zn+2
Ca+2
Apo
Campo Magnético (Gauss)
3200 3250 3300 3350
Zn+2
Ca+2
Campo Magnético (Gauss)
(F) HS100A12 L60C/A80C
Apo
3200 3250 3300
Zn+2
Ca+2
Campo Magnético (Gauss)
(G) HS100A12 D61C/A80C
Apo
Figura 34 - Espectros de RPE em banda X dos mutantes da proteína HS100A12 com o marcador de spin
MTSSL na ausência e na presença de cálcio e zinco a 250C. (A) A80C. (B) K33C/A80C, (C) I53C/A80C, (D) D54C/A80C, (E) F57C/A80C, (F) L60C/A80C, (G) D61C/A80C .

HS100A12 97
Os espectros do duplo mutante HS100A12 D54C/A80C (Figura 34D) são
interessantes pois, apesar de não apresentarem alargamentos devidos à
proximidade das sondas contidas na estrutura da proteína, mostram mudanças
espectrais tanto após adição de cálcio quanto de zinco. O espectro da proteína na
forma apo é alterado para espectros associados a marcadores com restrição de
movimento na presença dos íons, sendo o zinco aquele que leva à maior
imobilização da sonda, o que indica que essa é uma posição sensível aos processos
de mudança conformacional ou oligomerização experimentados pela S100 na
presença de cálcio ou zinco.
No caso do mutante HS100A12 F57C/A80C (Figura 34E), vemos pela primeira
vez um caso em que a dupla marcação é sensível à alterações tanto na forma de
linha do espectro (mudança na mobilidade dos marcadores) quanto na distância
interspin (proximidade dos marcadores). O espectro da proteína na forma apo é, de
forma geral, mais largo que aquele observado para o mutante simples A80C (Figura
34A), indicando que os centros magnéticos se encontram a menos de 20 Å. A
adição de cálcio não leva a grandes diferenças espectrais, como antes observado
para outras posições, mas a presença de zinco induz variações na forma e no
padrão de alargamento, mais uma vez sugerindo conformações distintas adotadas
após adição dos íons.
Na figura 34F, observamos os espectros do mutante HS100A12 L60C/A80C.
Neste caso, vemos de maneira bastante clara o diferente efeito causado pelos íons
cálcio e zinco. Na forma apo e na presença de cálcio, não se vê claramente no
espectro de RPE-CW medido à 250C maiores alargamentos devido à interação
dipolar. No entanto, a adição de zinco causa profunda mudança evidenciada pelo
forte alargamento das linhas, mostrando que esse íon leva a diminuição da distância
entre as duas sondas presentes na estrutura da proteína, ou seja, a posição 60 da
hélice C se aproxima da posição 80 da hélice D.
Curiosamente, indo-se uma posição à frente na hélice C e marcando-se a
cisteína introduzida na posição 61, não detectamos alterações significativas nos
espectros de RPE (Figura 34G) quando comparamos as três condições em estudo.
O duplo mutante HS100A12 D61C/A80C tem espectro típico de marcadores em
movimento de dinâmica intermediária, sem alargamentos dipolares claramente
evidenciados. A presença dos íons não afeta esse padrão.

98
As interações spin-spin são melhor analisadas a partir dos espectros dos
mutantes da proteína HS100A12 medidos a -100oC, pois assim conseguimos
remover a influência da mobilidade da cadeia lateral presente no espectro a 250C e
nos concentrar nas alterações de distância. Com esse tipo de experimento,
selecionamos a característica que desejamos investigar, no caso a distância,
removendo as contribuições da dinâmica do marcador à largura e forma de linha dos
espectros. Assim, uma alteração na intensidade dos espectros de RPE-CW
normalizados e medidos a -100oC está associada a uma mudança na distância spin-
spin, de forma a termos ressonâncias mais ou menos intensas (linhas menos ou
mais largas) quando os centros magnéticos se afastam ou se aproximam,
respectivamente.
Na figura 35, apresentamos os espectros de RPE-CW a -100oC obtidos de
todos os duplos mutantes construídos e medidos nas três condições anteriormente
discutidas (forma apo, presença de cálcio ou de zinco). Em cada um dos painéis da
figura 35 é mostrado também o espectro do mutante simples A80C com o intuito de
evidenciarmos o sucesso da estratégia de dupla mutação e marcação, já que em
todos os casos a menor intensidade dos espectros é sinal claro da existência de
interação dipolar magnética. Outra vantagem da apresentação conjunta com o
espectro do mutante simples está na comparação imediata que pode ser feita entre
os diversos espectro, assim obtendo-se uma descrição qualitativa das mudanças em
investigação.
Analisando primeiramente os espectros de cada duplo mutante obtido com a
proteína na ausência de ligantes (primeira coluna na figura 35), podemos ordenar os
diversos pares de resíduos por distâncias, indo do mais próximo para o mais
distante, apenas comparando as intensidades (ou larguras) dos espectros. Assim,
temos que a distância entre a posição A80 aumenta na seguinte ordem: I57 ≈ F61 <
L64 < K37 < D58 < D65. Se comparamos esta sequência com a estrutura
cristalográfica da HS100A12 (Figura 33 acima), podemos perceber que a técnica de
SDSL é capaz de distinguir as distâncias entre resíduos consecutivos na hélice C,
caso dos I53 e D54, e que estejam posicionados no lado interno ou externo da
referida hélice. O espectro do duplo mutante I53C/A80C (Figura 35B) é mais largo
(menos intenso) do que aquele obtido para o mutante D54C/A80C (Figura 35C),
indicando maior proximidade no primeiro caso. Isto também pôde ser visto nos
espectros medidos à 25oC (Figura 34C e D).
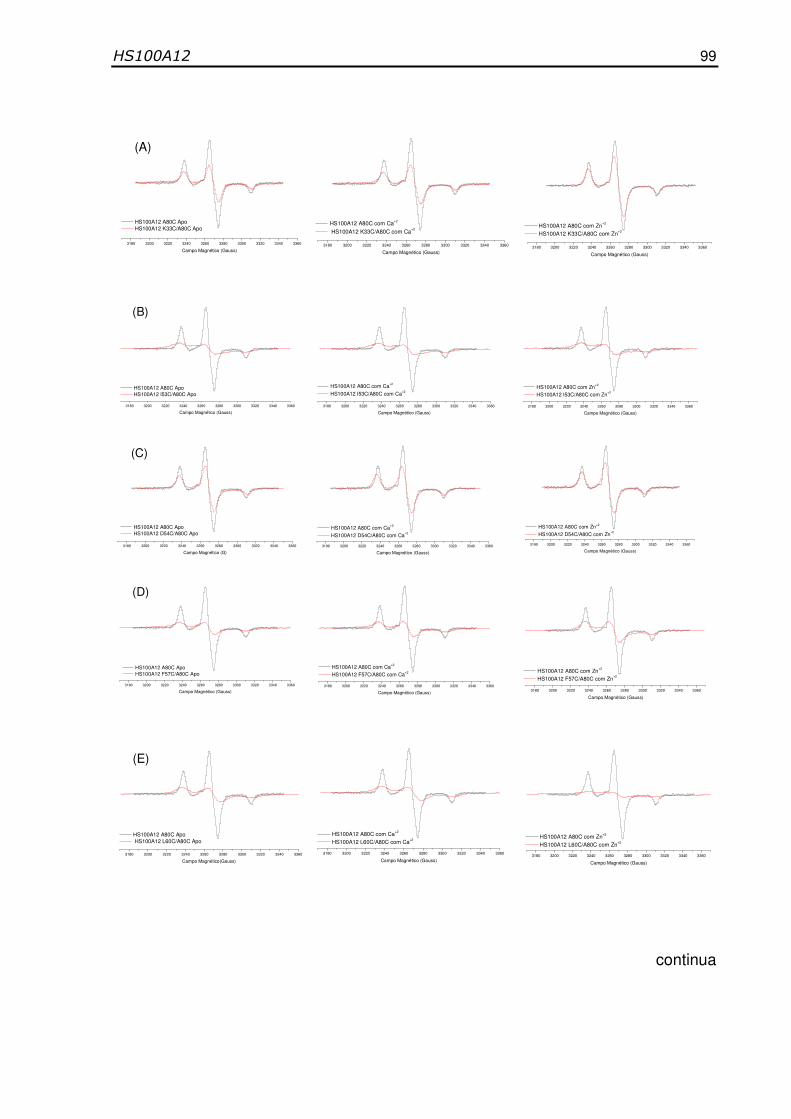
HS100A12 99
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 K33C/A80C Apo
Campo Magnético (Gauss)
(A)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 K33C/A80C com Ca+2
Campo Magnético (Gauss)3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 K33C/A80C com Zn+2
Campo Magnético (Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 I53C/A80C Apo
Campo Magnético (Gauss)
(B)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 I53C/A80C com Ca+2
Campo Magnético (Gauss) 3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 I53C/A80C com Zn+2
Campo Magnético (Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 D54C/A80C Apo
Campo Magnético (G)
(C)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 D54C/A80C com Ca+2
Campo Magnético (Gauss) 3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 D54C/A80C com Zn+2
Campo Magnético (Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 F57C/A80C Apo
Campo Magnético (Gauss)
(D)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 F57C/A80C com Ca+2
Campo Magnético (Gauss) 3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 F57C/A80C com Zn+2
Campo Magnético (Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 L60C/A80C Apo
Campo Magnético(Gauss)
(E)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 L60C/A80C com Ca+2
Campo Magnético (Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 L60C/A80C com Zn+2
Campo Magnético (Gauss)
continua

100
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C Apo HS100A12 D61C/A80C Apo
Campo Magnético (Gauss)
(F)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Ca+2
HS100A12 D61C/A80C com Ca+2
Campo Magnético(Gauss)
3180 3200 3220 3240 3260 3280 3300 3320 3340 3360
HS100A12 A80C com Zn+2
HS100A12 D61C/A80C com Zn+2
Campo Magnético (Gauss)
Figura 35 - Espectros normalizados de RPE em banda X do mutante simples da proteína HS100A12 com o
marcador de spin MTSSL na posição 80 (preto) e dos duplos mutantes nas posições (A) K33C/A80C, (B) I53C/A80C, (C) D54C/A80C, (D) F57C/A80C, (E) L60C/A80C, (F) D61C/A80C (vermelho) a -1000C, sem cálcio, na presença de cálcio e com zinco.
Comparando agora os espectros da figura 35 após a adição dos íons
divalentes, podemos observar que a presença de cálcio ou zinco leva sempre a
alguma mudança nas intensidades dos espectros dos duplos mutantes, o que havia
sido também percebido através dos espectros medidos em mais alta temperatura.
Apesar de, em muitos casos, a análise qualitativa acima descrita ser suficiente,
procuramos também determinar valores de distância para alguns dos duplos
mutantes (K33C/A80C e F57C/A80C) e, assim, fecharmos o que pode ser
considerado como o ciclo completo da estratégia de marcação de spin sítio dirigida
para estudo de mudanças conformacionais.
Partindo deste ponto, as distâncias entre os marcadores de spin para os dois
duplos mutantes foram estimadas através do método de desconvolução de Fourier
(116) utilizando-se o programa cedido gentilmente pela Profa. Dra. Christiane
Timmel (University of Oxford). Em linhas gerais, o referido método consiste em
assumir que o espectro do duplo mutante é o resultado da convolução entre um
espectro em que não há interação dipolar (espectro padrão de não-interação) e uma
função que toma conta do alargamento oriundo da interação dipolar (forma de um
dubleto de Pake). No nosso caso, o espectro de RPE do mutante A80C (em preto,
Figura 35) foi utilizado como padrão de espectro de não-interação. Na verdade, esse
não é um espectro de não-interação de um mutante duplo porque o espectro de não-
interação ideal deve conter a informação dos marcadores nos dois sítios, ou seja, a
soma dos espectros dos dois mutantes contendo marcador de spin em cada uma
das cisteínas separadamente ou um espectro medido utilizando uma amostra
contendo um marcador de spin e um análogo diamagnético. Contudo, em baixa
temperatura (-1000C), os movimentos do marcador de spin são praticamente

HS100A12 101 congelados (regime rígido) e passam a se comportar de maneira idêntica. Portanto,
o uso do espectro do mutante simples (A80C) é uma boa aproximação sob
condições rígidas e não altera os valores das distâncias calculados (10).
Na Tabela 3, são apresentados os valores das distâncias spin-spin dos
mutantes da proteína HS100A12 K33C/A80C e F57C/A80C calculados pelo método
de desconvolução de Fourier. Analisando o mutante K33C/A80C, podemos observar
que a distância da hélice B em relação à hélice D diminui 1,3 Å com a ligação do íon
cálcio, enquanto a distância da hélice C em relação à hélice D, observada no
mutante F57C/A80C aumenta 1,7 Å.
Tabela 3 - Distância spin-spin e variação da distância dos duplos mutantes da proteína HS100A12 marcados com MTSSL.
MUTANTES HS100A12 CONDIÇÕES DISTÂNCIA (Å) VARIAÇÃO DA
DISTÂNCIA (Å)
Sem Ca+2 13,86 ------- K33C/A80C
Com Ca+2 12,61 -1,25
Sem Ca+2 7,04 -------
F57C/A80C Com Ca+2 8,71 +1,67
Examinando as estruturas cristalográficas da proteína HS100A12 na forma apo
(75) e na presença de cálcio (74) (Figura 36), verificamos que os valores das
distâncias entre as hélices em estudo obtidos, utilizando a técnica de SDSL aliada à
RPE estão consistentes com os valores observados nas referidas estruturas.
Cabe aqui lembrar que discrepâncias entre os números oriundos das análises
de SDSL e dados estruturais de cristalografia de raios-X se devem ao fato de que a
medida das distâncias na estrutura cristalográfica é feita a partir da cadeia principal
da proteína, enquanto que nos mutantes ela se refere à separação entre os dois
grupos nitróxidos inseridos nas cadeias laterais dos resíduos de cisteína. Ou seja,
quando calculamos distância na estrutura cristalográfica estamos medindo a
separação entre átomos da cadeia principal, ao passo que por SDSL-RPE
monitoramos a separação entre as cadeias laterais marcadas que podem adotar
diferentes conformações e, com isso, levar a distâncias maiores ou menores do que
aquelas obtidas da estrutura tridimensional.

102
As distâncias entre K33R1 e A80R1 (resíduos ligados com o marcador de spin
R1) na ausência e na presença de cálcio, obtidas a partir dos dados cristalográficos,
são 12,05 Å e 14,87 Å, respectivamente, ao passo que, a partir de nossos
resultados, temos 13,9 Å e 12,6 Å, indicando, neste caso, uma incongruência entre
os valores oriundos das duas diferentes fontes. Com a ligação do cálcio seria
esperado que as hélices se distanciassem já que a hélice B, onde se encontra o
resíduo K33, constitui o domínio de ligação do Ca+2 EF-hand I e a hélice C, onde
está o resíduo A80, é uma das hélices do domínio EF-hand II. Com isso, esse
resultado carece de uma análise mais cuidadosa, assim como a produção de
mutantes adicionais da hélice B se faz necessário para uma análise completa da
dinâmica de afastamento/aproximação.
Já as distâncias entre F57R1 e A80R1 observadas na estrutura cristalográfica
são de 11,00 Å sem cálcio e 13,79 Å com cálcio e por SDSL-EPR são de
aproximadamente 7,0 Å na ausência de cálcio e 8,7 Å na presença do íon, assim
confirmando o mecanismo de ligação de proteínas EF-hand com o íon Ca+2, onde o
afastamento das duas hélices que compõem o domínio de ligação com o íon, resulta
na exposição dos resíduos hidrofóbicos da proteína.
(A) (B)
K37K37
F61
F61
A84 A84
(A) (B)(A) (B)
K33K33
F57
F57
A80 A80
(A) (B)
K37K37
F61
F61
A84 A84
(A) (B)(A) (B)
K33K33
F57
F57
A80 A80
(A) (B)
K37K37
F61
F61
A84 A84
(A) (B)(A) (B)
K33K33
F57
F57
A80 A80
Figura 36 - Estrutura da proteína HS100A12 apo (PDB 2WCE)- (A) e com cálcio (PDB 1E8A)- (B), indicando
os resíduos marcados com MTSSL e os valores das distâncias entre eles, (A): K33C/A80C- 12,05 A e F57C/A80C- 11,00 A. (B): K33C/A80C- 14,87 A e F57C/A80C- 13,79 A.

HS100A12 103 4.4.6 Interação da proteína HS100A12 com fosfolipídios utilizando modelos de
biomembranas
Os membros da família S100 possuem tanto função intracelular como
extracelular, desta forma, participando de diferentes processos, como, crescimento e
diferenciação celular, regulação do ciclo celular, transcrição e atuando também
como receptores de transdução de sinal. Como a proteína HS100A12 é expressa
principalmente em locais de inflamação crônica, acredita-se que ela possua um
papel muito importante nesta etapa do ciclo da célula. O mecanismo pelos quais
essa proteína modula o curso de processos inflamatórios está relacionado à sua
interação com o receptor para produtos de glicosilação (“Receptor for Advanced
Glycation End Products”- RAGE), que é um membro multiligante de moléculas da
superfície celular da superfamília da imunoglobulina (117).
Baseado nessas informações decidiu-se investigar os aspectos fundamentais
da interação proteína-célula em nível molecular, empregando monocamadas de
Langmuir como sistemas biomiméticos de membrana. Esses modelos de membrana
são bastante eficientes porque mimetizam metade de uma membrana biológica ao
mesmo tempo possibilitando um alto controle da densidade, arquitetura e
composição do filme.
Inicialmente, foi determinada a cinética de adsorção da proteína HS100A12 na
ausência e presença dos íons Ca+2 e/ou Zn+2 em monocamadas constituídas pelos
fosfolipídios DPPC e DPPG (Figuras 37 e 38). Como pode ser observado na figura
20, os dois fosfolipídios empregados neste estudo possuem características
diferentes, sendo o DPPC, zwitteriônico (carga total neutra) e o DPPG contendo um
grupo carregado negativamente. Com isso, também podemos investigar a influência
dos grupos carregados na interação da proteína com a monocamada lipídica.
O experimento de cinética foi realizado conforme descrito na seção 4.3.14,
onde a proteína foi injetada na subfase aquosa da monocamada lipídica em baixo
empacotamento, com pressão superficial zero.
Nas figuras 37 e 38 são apresentados os gráficos de variação da pressão de
superfície em função do tempo devido à adsorção da proteína em monocamadas de
DPPC e DPPG, respectivamente. Através das curvas, pode-se verificar que a

104
cinética de adsorção da proteína ocorre em três etapas bem definidas, denominadas
a partir de agora de E1, E2 e E3 (representação na figura 37A).
O estágio E1, denominado tempo de indução (lag time), corresponde a um
progressivo incremento na concentração das moléculas na vizinhança da interface
(ou subsuperfície), geralmente acompanhada por pequena ou praticamente
nenhuma variação na pressão superficial. Uma vez que o número de moléculas na
região da subsuperfície tenha atingido um limite, o processo de adsorção atinge o
estágio E2. Durante este estágio, um valor de área superficial necessita estar
disponível para que ocorra a inserção da molécula na interface. Em outras palavras,
podemos atribuir ao estágio E2 o processo de adsorção/incorporação propriamente
dito. Já o estágio E3 representa alterações conformacionais que a molécula sofre,
para melhor se acomodar na interface, até atingir sua disposição final.

HS100A12 105
A
0 10 20 30 40 50 60 70 80 90
0
1
2
3
4
5
6
7
E1
E3
DPPC + HS100A12 (0,5µg/mL) Apo Ca+2
Zn+2
Ca+2 e Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Tempo (min)
E2
B
0 10 20 30 40 50 60 70 80
0
1
2
3
4
5
6
7
8
9
10
11
12
Apo Ca+2
Zn+2
Ca+2 e Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Tempo (min)
DPPC + HS100A12 (2,5µg/ml)
Figura 37 - Cinética de adsorção da proteína HS100A12 sobre monocamada de DPPC. (A): 0,5 µg/ml de
proteína. (B): 2,5 µg/ml de proteína. HS100A12 apo (preto), HS100A12 + Ca+2 (vermelho), HS100A12 + Zn+2 (azul) e HS100A12 + Ca+2 + Zn+2 (verde).

106
A
0 10 20 30 40 50 60 70 80
0
1
2
3
4
5
6
7
DPPG + HS100A12 (0,5µg/ml) Apo Ca+2
Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Tempo (min)
B
0 10 20 30 40 50 60 70 80
0
1
2
3
4
5
6
7
8
9
10
11
12
DPPG + HS100A12 (2,5µg/ml) Apo Ca+2
Zn+3
Ca+2 e Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Tempo (min)
Figura 38 - Cinética de adsorção da proteína HS100A12 sobre monocamada de DPPG. (A): 0,5 µg/ml de proteína. (B): 2,5 µg/ml de proteína. HS100A12 apo (preto), HS100A12 + Ca+2 (vermelho), HS100A12 + Zn+2 (azul) e HS100A12 + Ca+2 + Zn+2 (verde).
A variação da pressão de superfície para os dois fosfolipídios estudados se
mostrou dependente tanto da concentração da proteína (0,5 e 2,5 µg/ mL) como
esperado, mas também da identidade dos íons ligantes, cálcio e zinco (Tabela 4).
Para a monocamada de DPPC, verificou-se que o estágio E1, relacionado ao
tempo de indução, se mostrou muito maior para a proteína na presença de Ca+2,
diminuindo na presença de Zn+2 e sendo muito menor na presença dos dois íons.
Interessantemente, sabe-se da literatura que a interação de HS100A12 com Ca+2

HS100A12 107 provoca uma mudança na conformação da proteína de mais fechada para uma
conformação mais aberta, tendo como conseqüência maior exposição das regiões
hidrofóbicas.
Na presença do Zn+2, a proteína se encontra com estado de oligomerização
diferente daquele na presença de íons cálcio, pois nessas condições, ela se
encontra na forma tetramérica e não dimérica como quando na presença de cálcio.
O estado de oligomerização parece também afetar na interação com o fosfolipídio,
visto que esta ocorreu a diferentes intervalos de tempo, e provocando diferentes
variações de pressão superficial. Esse resultado corrobora com os estudos
realizados por Moroz et al (102, 104), onde foi observado que o meio de cultura de
células contendo zinco é importante para a oligomerização da proteína HS100A12 e
para sua ligação com moléculas da superfície celular.
Já na presença dos dois íons, a proteína também se encontra em uma
conformação diferente daquela na presença apenas do íon Ca+2, facilitando assim, a
adsorção da mesma na interface.
Os resultados obtidos com o fosfolipídio aniônico, DPPG, apontam que, com
exceção da proteína incubada com íons Ca+2, as variações no valor de pressão
superficial foram maiores (Tabela 4), indicando maior afinidade por lipídios
carregados negativamente. Essa afinidade deve ocorrer justamente pela atração da
HS100A12 pelos grupos da cabeça polar, uma vez que ela se encontra carregada
positivamente no pH 7,5.
Tabela 4 - Valores da pressão de superfície de dois modelos de membrana na presença da proteína HS100A12 à concentração de 0,5 e 2,5 µµµµg/ml, na presença e ausência dos íons cálcio e/ ou zinco.
Componentes
da
Monocamada
Pressão de Superfície (mN/m)
HS100A12-Apo HS100A12-Ca+2 HS100A12-Zn+2 HS100A12-Ca+2 e
Zn+2
0,5 µg/ml 2,5 µg/ml 0,5 µg/ml 2,5 µg/ml 0,5 µg/ml 2,5 µg/ml 0,5 µg/ml 2,5 µg/ml
DPPC 0,86 4,17 2,65 5,40 3,25 7,42 6,37 10,77
DPPG 2,94 4,81 1,07 2,84 6,79 10,14 10,89

108
O estudo da adsorção de proteínas na interface ar-água ou em uma interface
lipídica dificilmente pode ser descrito em termos apenas de um parâmetro, sendo
necessário um estudo individual para cada proteína. Tripp et al (118), por exemplo,
estudaram a adsorção de diversas proteínas à interface líquido-ar e verificaram que
mesmo proteínas com maior grau de hidrofobicidade nem sempre apresentavam
adsorção mais rápida na interface, visto que o tamanho da proteína e estabilidade
conformacional também afetavam diretamente o tempo de indução à interface. Além
disso, na presença de monocamadas lipídicas, mesmo a baixos empacotamentos, a
afinidade da proteína pelo lipídio pode atuar como força motriz para a adsorção
deste (111).
Com isso, conhecendo o mecanismo de ligação de proteínas EF-hand com o
íon Ca+2, em que são expostos os resíduos hidrofóbicos da mesma, podemos inferir
que a cinética de adsorção da HS100A12 em monocamada de DPPC é atribuída à
interação proteína-DPPC. O comportamento da proteína incubada com cálcio se
apresentou diferenciado das demais, visto que a maior interação ocorreu com a
monocamada de DPPC e não com DPPG. Além disso, a adsorção da proteína com
Ca+2 apresentou-se mais lenta. Provavelmente, um estudo mais apropriado
utilizando técnicas espectroscópicas de superfície como PM-IRRAS (“Polarization-
Modulation Infrared Reflection Absorption Spectroscopy”) poderiam responder se o
comportamento observado para a proteína incubada com íons cálcio resulta de
alterações conformacionais desta que não apenas a exposição das regiões
hidrofóbicas induzidas pelo íon.
Os valores de pressão de superfície para a proteína na presença de zinco são
maiores que para a proteína apo e com cálcio, levando-nos a concluir que a
eficiência de interação da proteína com a monocamada é maior na presença deste
íon. As mudanças conformacionais induzidas pelo zinco, discutidas na seção de
dicroísmo circular (v. seção 4.4.4.), provavelmente favorecem a interação da
proteína com a monocamada. Já quando a proteína é incubada com cálcio e zinco,
provavelmente ocorre um processo cooperativo resultando em uma interação maior
do que aquela que ocorre quando a proteína está incubada com os íons
separadamente. Esse resultado está de acordo com alguns estudos que
demonstraram que a ligação do zinco aumenta a afinidade da proteína pelo seu alvo
extracelular, o receptor para produtos de glicosilação (RAGE), presente na
membrana biológica (52, 102).

HS100A12 109
Após as cinéticas de adsorção, os sistemas mistos em equilíbrio foram
comprimidos, e os resultados são apresentados a seguir (Figuras 39 e 40).
Inicialmente, foram medidas as isotermas π-A para as monocamadas de DPPC e
DPPG em subfase de tampão 50 mM Tris, 25mM NaCl pH 7,5, na ausência da
proteína (curvas em cinza das figuras 39 e 40). Diferente da isoterma do DPPG, a
curva para o filme de DPPC apresenta o patamar correspondente à coexistência das
fases líquida-expandida e líquida-condensada (LE-LC) pouco definido. A alteração
deste patamar, que é característico da isoterma desse fosfolipídio, pode ter ocorrido
devido à presença dos sais contidos no tampão utilizado em nosso estudo.
Em seguida, verificamos o efeito da adição da proteína, na ausência e na
presença dos íons. Através das figuras 39 e 40, verificamos efeitos distintos da
proteína sobre as monocamadas de DPPC e DPPG. O deslocamento das curvas
para valores de áreas por molécula maiores quando da presença da proteína indica
a incorporação do polipeptídeo na monocamada lipídica. Essa expansão para
maiores áreas é dependente dos íons que estão ligados à proteína e a amplitude
deste deslocamento na área por molécula corrobora os dados obtidos na cinética de
adsorção.

110
A
0 20 40 60 80 100 120
0
10
20
30
40
50
60
70DPPC + HS100A12 (0,5µg/ml) Tampao
Apo Ca+2
Zn+2
Ca+2 e Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
area molecular (A2)
B
0 20 40 60 80 100 120
0
10
20
30
40
50
60
70
80 DPPC + HS100A12 (2,5µg/ml)
Tampao Apo Ca+2
Zn+2
Ca+2 e Zn+2
Area molecular (A2)
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Figura 39 - Isoterma de pressão de superfície para o DPPC com tampão (cinza), HS100A12 apo (preto), HS100A12 + Ca+2 (vermelho), HS100A12 + Zn+2 (azul) e HS100A12 + Ca+2 + Zn+2 (verde). (A): HS100A12 - 0,5µg/ml. (B): HS100A12 - 2,5µg/ml.

HS100A12 111
A
20 40 60 80 100 120 140
0
10
20
30
40
50
60
70 DPPG + HS100A12 (0,5µg/ml)
Tampao Apo Ca+2
Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Area molecular (A2)
B
20 40 60 80 100 120 140
0
10
20
30
40
50
60
70 DPPG + HS100A12 (2,5µg/ml)
Tampao Apo Ca+2
Zn+2
Ca+2 e Zn+2
Pre
ssao
de
Sup
erfic
ie (
mN
m-1)
Area molecular (A2)
Figura 40 - Isoterma de pressão de superfície para o DPPG com tampão (cinza), HS100A12 apo (preto), HS100A12 + Ca+2 (vermelho), HS100A12 + Zn+2 (azul) e HS100A12 + Ca+2 + Zn+2 (verde). (A): HS100A12 - 0,5µg/ml. (B): HS100A12 - 2,5µg/ml.
Vale ressaltar, que as curvas de π-A não tem início no zero de pressão
superficial porque a compressão da monocamada foi efetuada após a adsorção das
proteínas. Interessantemente, para a isoterma das proteínas com o lipídio DPPC,
notamos o aparecimento de um plateau bem definido ocorrendo em pressões
superficiais entre 25-30 mN m-1. Este fenômeno ocorre para todas as proteínas,
independente da presença e do tipo de íon, sendo que as únicas variações são no
valor exato de pressão em que ocorre, bem como na extensão, em área por
molécula. Vale lembrar que, de acordo com Marsh (119), esta faixa de pressão

112
sueprficial corresponde exatamente ao empacotamento lipídico em uma membrana
real.
Para o caso da proteína na forma apo e na presença do íon cálcio na
concentração de 0,5 µg/ mL, nota-se que após esse plateau, as curvas começam a
coincidir com a do lipídio puro, sugerindo a expulsão da proteína da interface. Já
para a concentração mais elevada, 2,5 µg mL-1, observamos que uma parte da
proteína permanece na monocamada lipídica, provavelmente pelo excesso da
macromolécula adicionado.
Já na presença de íons zinco, bem como de cálcio e zinco essa transição
ocorre, mas a área mínima para cada um dos sistemas (obtida através da tangente à
curva imediatamente antes do colapso) ainda é muito maior que a do lipídio puro,
indicando a permanência das proteínas na interface.
Para o caso do DPPC, os resultados sugerem que na presença dos íons zinco,
a proteína deve expor uma maior região hidrofóbica para o meio, visto que a
incorporação desta foi maior e mais forte, não sendo expulsa da interface. Dessa
forma, a interação com este íon (zinco) deve induzir alterações conformacionais na
proteína que facilitam sua inserção na membrana, desempenhando assim seu papel
biológico.
Para monocamadas de DPPG, nota-se que os mesmos patamares são
observados, entretanto, o efeito para este lipídio parece ser ligeiramente mais
proncunciado, principalmente quando observamos os deslocamentos para maiores
valores de área por molécula.

Lisozima T4


T4L 115 5 As Proteínas Lisozimas
As lisozimas pertencem à classe de enzimas cuja atividade catalítica está
relacionada à hidrólise da ligação glicosídica β (1,4) que liga o carbono C-1 do ácido
N-acetilmurâmico (NAM) e o carbono C-4 do N-acetilglicosamina (NAG),
componentes do peptidoglicano, principal constituinte de parede celular de bactérias.
Em plantas e animais, elas constituem um mecanismo de defesa natural contra
patógenos bacterianos (120, 121, 122). Em bacteriófagos, as lisozimas, chamadas
de endolisinas, hidrolisam o peptidoglicano para facilitar a penetração do aparelho
de injeção do fago ou induzem a lise celular no final do seu ciclo de replicação (123,
124, 125). Por outro lado, as bactérias também produzem lisozimas, chamadas de
autolisinas, que estão envolvidas na síntese de novo da parede celular durante o
crescimento e divisão da célula (126, 127).
As lisozimas são excelentes candidatas à biopreservativos porque existem
naturalmente no corpo humano e em muitos produtos alimentícios, podendo ser
consideradas aditivos alimentícios “naturais”. A primeira enzima a ser utilizada na
preservação de alguns alimentos, cosméticos e produtos farmacêuticos foi a
lisozima da clara de ovo de galinha (Gallus gallus) ou tipo-C. Trata-se de uma
enzima pequena com 129 resíduos de aminoácidos dispostos em cadeia única,
resultando numa massa molecular próxima a 14,5 kDa. A enzima possui um sítio
catalítico formando uma fenda profunda no interior da molécula no qual estão
localizados os resíduos Glu-35 e Asp-52 responsáveis pela hidrólise da ligação
glicosídica citada anteriormente. Acredita-se que o Glu-35 protona o laço acetal do
substrato, enquanto o Asp-52, de carga negativa, estabiliza o íon carbocátion
resultante (128). Novas enzimas com propriedades similares foram encontradas na
lágrima, no leite, em secreções e extratos de vários tecidos animais, bem como na
clara de ovo de inúmeras aves.
Como dito acima, a lisozima além de ser utilizada na indústria alimentícia na
preservação de alimentos e na indústria farmacêutica, como potencializador de
antibióticos, é tambem utilizada nos laboratórios de Genética Molecular, em
experimentos envolvendo tecnologia do DNA recombinante, clonagem e expressão

116
gênica, onde se faz necessária a lise de bactérias hospedeiras dos plasmídeos
recombinantes (129).
5.1 Lisozima do Bacteriófago T4 (T4L)
A lisozima do fago T4 é uma enzima produzida nas células de Escherichia coli
depois da sua infecção com o bacteriófago T4. Apesar da análise da sequência
indicar que a enzima lisozima do fago T4 apresenta baixa identidade sequencial com
a lisozima de clara de ovo de galinha (Figura 41), ambas possuem atividade
catalítica similar sendo, portanto, endoacetilmuramidases (130).
Figura 41 - Alinhamento seqüencial entre Lisozima do bacteriófago T4 (T4Lysozyme) e Lisozima de clara
de ovo de galinha (eggLysozyme). Alinhamento e figura foram feitos utilizando o programa MULTALIN (91).
A enzima lisozima do bacteriófago T4 (T4L) contém 164 resíduos, peso
molecular de 18635,4 Da, pI=9,59 e sua estrutura foi resolvida por Matthews et al
(131). Como pode ser observado na figura 42, a estrutura consiste de dois lóbulos
totalmente distintos, com a parte C-terminal da cadeia polipeptídica exclusivamente
no domínio “superior”, enquanto que no domínio “inferior” está a porção N-terminal
da molécula. A hélice amino-terminal interage extensivamente com o domínio
superior e, com isso, ajuda a manter os dois lóbulos ligados.
Na porção inferior, os resíduos 18-34 formam três fitas β antiparalelas
distorcidas, sendo as únicas estruturas β existentes na molécula. Por outro lado, o
domínio superior possui caráter helicoidal e é constituído de uma combinação de
cinco α hélices, cada uma com aproximadamente 10 resíduos, arranjadas de tal
forma que fazem uma parede de cilindros formando um barril com uma cavidade
hidrofóbica no interior. Essa região hidrofóbica conecta-se através da parte central
da molécula com outra região hidrofóbica na parte inferior.

T4L 117
A conexão mais óbvia entre os dois domínios é feita por uma longa α-hélice
(hélice C), de 20 resíduos, que se estende quase de uma extremidade a outra da
molécula. Adicionalmente, os dois lóbulos estão também conectados através da
hélice amino-terminal, como já mencionado anteriormente.
H
C
C
N
H
C
C
N
Figura 42 - Estrutura da Lisozima do bacteriófago T4, indicando a região N e C-terminal e as hélices C e
H. Verde: porção N-terminal. Azul: porção C-terminal. Lilás: α-hélice C que se estende de uma porção a outra da molécula.
Na parte central da molécula há uma fenda de aproximadamente 6-8 Å de
diâmetro, que se acreditava fosse a responsável por acomodar o N-acetilmuramil-N-
acetilglucosamil da parede celular (131). Esta hipótese foi confirmada por Tsugita
(132) quando propôs estudar a atividade catalítica da enzima focalizando os
resíduos Asp 20, Glu 22, Glu 105, Trp 138, Asn 140 e Glu 141, cujas cadeias laterais
estão localizadas na fenda da molécula. Tsugita observou que a troca desses
aminoácidos por outros diminuía drasticamente a atividade, concluindo, portanto,
que eles são essencias para a atividade catalítica da enzima. Este e outros estudos
com uma variedade de mutantes da lisozima do fago T4 são feitos rotineiramente a
fim de se obter mais detalhes a respeito da correlação estrutura e função dessa
enzima.
No presente trabalho, abordamos a suposta criação de uma cavidade no “core”
hidrofóbico da porção C-terminal da enzima, quando mutamos a Leu 133 por Ala
e/ou Gly, ou seja, quando trocamos um resíduo grande por um de menor volume.
Acredita-se que a proteína sofra um reajuste estrutural com o intuito de preencher o
espaço vazio criado por essa substituição. Com isso, propusemos estudar por SDSL

118
o movimento da α-hélice H inserindo o marcador de spin na posição 131 ocupada na
forma nativa por um resíduo de valina.
Adicionalmente, analisamos a estrutura da enzima T4L e observamos que a
hélice H possui uma certa “complementariedade” com a hélice que contém os
resíduos 61 ao 81, chamada de hélice C (Figura 43). Essa “complementariedade” é
devida aos seguintes fatores:
• Na hélice H, o resíduo 127 é um Aspartato, enquanto que na hélice C, o
resíduo 68 é uma Asparagina, ou seja, a diferença é o grupo hidroxila (OH)
do Aspartato e o grupamento amina (NH2) da Asparagina.
• O resíduo 128 da hélice H é um Glutamato, enquanto que o resíduo 69 é
uma Glutamina, a diferença entre eles é a mesma que para o resíduo 127 e
68.
• Na hélice H, há dois resíduos de Alanina na mesma posição que os dois
resíduos de Valina na hélice C.
Além dessa “complementariedade” existente entre as duas hélices,
observamos que muitos estudos foram realizados com a hélice C. Mchaourab et al.
(18), por exemplo, realizaram um estudo utilizando SDSL com a hélice C, inserindo o
marcador de spin na posição 72, que é um sítio de superfície de hélice exposto ao
solvente, cujo espectro de RPE reflete um dos movimentos mais imobilizados para
essa classe de sítios. Ao passo que, quando os autores inseriram o marcador de
spin MTSSL na posição 131 da hélice H, um sítio também de superfície de hélice
exposto ao solvente, o espectro de RPE apresentou características de um
movimento anisotrópico simples semelhante ao sítio 72, mas com uma maior
amplitude de movimento para o nitróxido.
Diante da semelhança de ambos os sítios e diferentes espectros de RPE
decidimos estudar a interação entre as cadeias laterais do marcador de spin na
posição 131 e dos resíduos vizinhos da hélice H (resíduo 126-135) da lisozima do
fago T4, mutando os resíduos dessa hélice pelos seus respectivos
“complementares” da hélice C (Figura 43). No total, caracterizamos quatro diferentes
mutantes, primeiramente o resíduo Asp 127 foi mutado por uma Asparagina, sendo
denominado D127N; depois, no lugar do Glu 128 colocamos uma Glutamina,
D127N/E128Q; em seguida, o resíduo Lys 135 foi mutado por uma Arginina,
resultando em um mutante com três substituições, D127N/E128Q/K135R e

T4L 119 finalmente, no lugar da Asn 132 colocamos uma Alanina, obtendo o mutante
D127N/E128Q/K135R/N132A.
72
Arg
Asn Gln
Val Ala
Val
131
Glu
Lys
Asn
Asp
Ala
Ala
72
Arg
Asn Gln
Val Ala
Val
131
Glu
Lys
Asn
Asp
Ala
Ala
131
Glu
Lys
Asn
Asp
Ala
Ala
Figura 43 - Estrutura da Hélice C e da Hélice H indicando seus respectivos resíduos e destacando os
ambientes estruturais em torno da posição 72 e 131 que serão explorados nos estudos por SDSL.
5.2 Metodologia
As etapas nesta seção descrevem as metodologias em biologia molecular
utilizadas para a obtenção e caracterização dos mutantes da lisozima do
bacteriófago T4 (T4L), o seu procedimento de expressão e purificação e
experimentos de ressonância paramagnética eletrônica (RPE).
5.2.1 Produção dos mutantes da proteína T4L
O protocolo para obtenção dos mutantes da enzima T4L foi o mesmo utilizado
para a produção dos mutantes da proteína HS100A12, descrito no capítulo anterior
(seção 4.3). Contudo, as enzimas de restrição utilizadas para a subclonagem do
gene contendo as mutações T4L diferem daquelas utilizadas para as mutações da
proteína HS100A12, devido ao fato do vetor utilizado ser pHSe5 ao invés de pQE60.
A sequência que codifica a proteína T4L utilizada na estratégia de amplificação
para obtenção dos genes de interesse, os oligonucleotídeos forward e reverse e a

120
sequência dos sítios de restrição para a enzima BamHI são apresentados na figura
44. O “template” utilizado como molde na amplificação dos “primers” dos mutantes
da T4L possui três substituições, a primeira delas no resíduo de cisteína 54 que foi
mutado por uma treonina (C54T), na segunda, a cisteína 97 foi mutada por uma
alanina (C97A) e na terceira o resíduo de cisteína foi inserido na posição 131
(V131C).
1
GGAAACAGGATCCATCGATGCATGAATATATTTGAAATGTTACGTATAGATGAAGGTCTTAGACTTAA
AATCTATAAAGACACAGAAGGCTATTACACTATTGGCATCGGTCATTTGCTTACAAAAAGTCCATCAC
TTAATGCTGCTAAATCTGAATTAGATAAAGCTATTGGGCGTAATACTAATGGTGTAATTACAAAAGAT
GAGGCTGAAAAACTCTTTAATCAGGATGTTGATGCTGCTGTTCGCGGAATTCTCAGAAATGCTAAATT
AAAACCGGTTTATGATTCTCTTGATGCGGTTCGTCGCGCTGCATTGATTAATATGGTTTTCCAAATGG
GAGAAACCGGTGTGGCAGGATTTACTAACTCTTTACGTATGCTTCAACAAAAACGCTGGGATGAAGCA
GCAGTTAACTTAGCTAAAAGTAGATGGTATAATCAAACACCTAATCGCGCAAAACGAGTCATTACAAC
GTTTAGAACTGGCACTTGGGACGCGTATAAAAATCTATAAGCCAAGCTTATCGATGATAAG
492
Figura 44 - Seqüência codificadora da Lisozima T4. A seqüência mostra 534 nucleotídeos sendo que 492 nucleotídeos codificam para a proteína Lisozima do bacteriófago T4 (C54T, C97A e V131C). Os nucleotídeos em negrito vermelho representam o início e o fim da seqüência que codifica para a proteína. As seqüências em negrito representam os oligonucleotídeos foward e reverse (5´ e 3’). Os nucleotídeos sublinhados representa o sítio de clivagem para a enzima de restrição BamHI.
Os oligonucleotídeos (“primers”) contendo as mutações de interesse para a
enzima T4L que foram desenhados e sintetizados são listados na Tabela 5. Com
isso, obtemos os mutantes da enzima que serão utilizados para marcação sítio
dirigida.
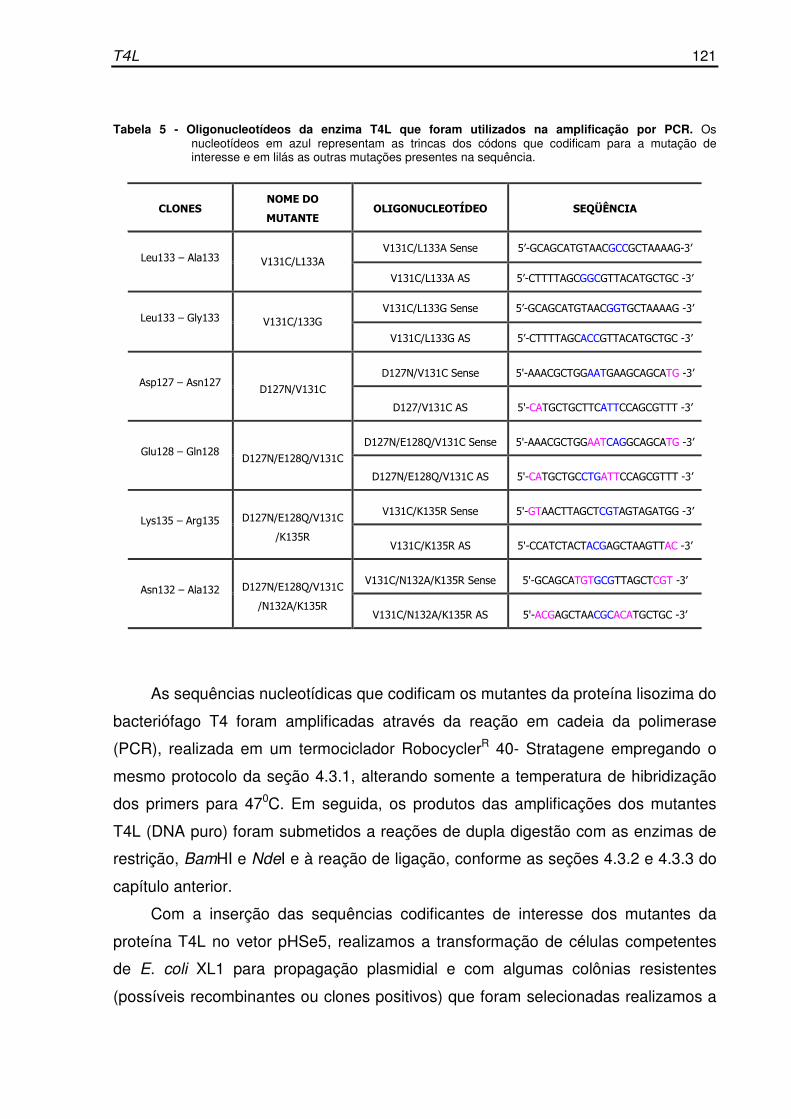
T4L 121 Tabela 5 - Oligonucleotídeos da enzima T4L que foram utilizados na amplificação por PCR. Os
nucleotídeos em azul representam as trincas dos códons que codificam para a mutação de interesse e em lilás as outras mutações presentes na sequência.
CLONES NOME DO
MUTANTE OLIGONUCLEOTÍDEO SEQÜÊNCIA
V131C/L133A Sense 5’-GCAGCATGTAACGCCGCTAAAAG-3’ Leu133 – Ala133 V131C/L133A
V131C/L133A AS 5’-CTTTTAGCGGCGTTACATGCTGC -3’
V131C/L133G Sense 5’-GCAGCATGTAACGGTGCTAAAAG -3’ Leu133 – Gly133 V131C/133G
V131C/L133G AS 5’-CTTTTAGCACCGTTACATGCTGC -3’
D127N/V131C Sense 5'-AAACGCTGGAATGAAGCAGCATG -3’ Asp127 – Asn127
D127N/V131C
D127/V131C AS 5'-CATGCTGCTTCATTCCAGCGTTT -3’
D127N/E128Q/V131C Sense 5'-AAACGCTGGAATCAGGCAGCATG -3’ Glu128 – Gln128
D127N/E128Q/V131C
D127N/E128Q/V131C AS 5'-CATGCTGCCTGATTCCAGCGTTT -3’
V131C/K135R Sense 5'-GTAACTTAGCTCGTAGTAGATGG -3’ Lys135 – Arg135 D127N/E128Q/V131C
/K135R V131C/K135R AS 5'-CCATCTACTACGAGCTAAGTTAC -3’
V131C/N132A/K135R Sense 5'-GCAGCATGTGCGTTAGCTCGT -3’ Asn132 – Ala132 D127N/E128Q/V131C
/N132A/K135R V131C/N132A/K135R AS 5'-ACGAGCTAACGCACATGCTGC -3’
As sequências nucleotídicas que codificam os mutantes da proteína lisozima do
bacteriófago T4 foram amplificadas através da reação em cadeia da polimerase
(PCR), realizada em um termociclador RobocyclerR 40- Stratagene empregando o
mesmo protocolo da seção 4.3.1, alterando somente a temperatura de hibridização
dos primers para 470C. Em seguida, os produtos das amplificações dos mutantes
T4L (DNA puro) foram submetidos a reações de dupla digestão com as enzimas de
restrição, BamHI e NdeI e à reação de ligação, conforme as seções 4.3.2 e 4.3.3 do
capítulo anterior.
Com a inserção das sequências codificantes de interesse dos mutantes da
proteína T4L no vetor pHSe5, realizamos a transformação de células competentes
de E. coli XL1 para propagação plasmidial e com algumas colônias resistentes
(possíveis recombinantes ou clones positivos) que foram selecionadas realizamos a

122
extração dos seus DNAs plasmidiais e, posteriormente, estes foram analisados por
sequenciamento.
Uma vez confirmada a presença dos DNAs plasmidiais que contêm as
sequências codificantes das enzimas mutantes nestas células, os mesmos foram
estocados a -200C para posterior transformação em células de expressão (E. coli
BL21) e obtermos os mutantes da enzima que serão utilizados para marcação sítio
dirigida.
5.2.2 Expressão da Enzima Lisozima do Bacteriófago T4
Os procedimentos utilizados para realizar o teste de expressão e a expressão
da enzima T4L foram os mesmos para todos os diferentes mutantes.
Para realizar os testes de expressão foram feitos pré-inóculos das colônias
resultantes da transformação de células E.coli BL21 com os plasmídeos contendo a
sequência que codifica o mutante da proteína, selecionadas em meio sólido com
antibiótico. As colônias foram inoculadas em 5 mL de meio LB líquido contendo
ampicilina (1 µL a 100 mg/mL) e mantidas “overnight” a 37oC sob 250 rpm de
agitação. Em seguida os inóculos foram diluídos em meio de cultura LB contendo
antibiótico, segundo a razão 1:100. A cultura foi mantida a sob agitação a 37°C até
atingir DO600 nm = 0.6-0.8. Após a indução com 1mM IPTG foram realizados testes de
expressão dos seis mutantes da proteína a 30oC e 37oC por 2, 4 e 16 horas.
As células resultantes das melhores condições de expressão para os mutantes
foram coletadas por centrifugação a 5.000 rpm e congeladas a -20oC até o uso. A
expressão dos mutantes foram analisadas em gel eletroforético (SDS-PAGE),
utilizando os géis PhastGel- Gradiente 8-25 e tampão PhastGel SDS Buffer Strips,
ambos fabricados pela GE Healthcare.

T4L 123 5.2.3 Purificação da Enzima Lisozima do Bacteriófago T4
A purificação dos mutantes da proteína T4L iniciou com o descongelamento
das células que foram ressuspensas em 25 mM Tris, 25 mM MOPS, 0,1 mM EDTA,
pH 7,6, e lisadas por sonicação em 2 ciclos de 4 minutos e intervalo de 5 minutos.
Os resíduos celulares e o material insolúvel foram removidos por centrifugação a
15.000 rpm por 30 minutos. O sobrenadante foi coletado e filtrado, utilizando uma
membrana de 0,2 µm e então, aplicado na coluna HiTrap SP HP (GE Healthcare)
(interação iônica-aniônica) previamente equilibrada com o tampão 25 mM Tris, 25
mM MOPS, 0,1 mM EDTA, 5 mM DTT, pH7,6, e eluída com gradiente de 0 a 1 M de
NaCl, em que geralmente a proteína elui entre 15-20% de NaCl. A purificação foi
realizada utilizando o sistema ÄKTA Purifier (GE Healthcare).
5.2.4 Determinação da Concentração da Proteína T4L
A concentração da proteína recombinante purificada foi determinada medindo-
se sua absorbância em espectrofotômetro UV-Vis a 280 nm, sendo esta relacionada
ao seu coeficiente de extinção molar, 24750 M-1cm-1.
5.2.5 Medidas de RPE
O procedimento para a marcação de spin sítio dirigida dos mutantes da
proteína T4L é o mesmo descrito no capítulo anterior (seção 4.3.12) para os
mutantes da proteína HS100A12, alterando somente o tampão de marcação para 50
mM MOPS, 25 mM NaCl pH 6,8.
Os mutantes marcados com MTSSL foram medidos no espectrômetro de RPE
Bruker ESP300, equipado com uma cavidade de alta sensibilidade, operando em

124
banda X e utilizando uma potência de microondas de 20 mW. Os espectros de RPE
foram obtidos variando-se a temperatura de 273 a 313 K, em intervalos de 5 K. A
temperatura foi controlada através de um sistema de fluxo de nitrogênio (Bruker,
Rheinstetten, Germany).
Para os experimentos de RPE foram utilizadas alíquotas de 4 µL de soluções
das proteínas marcadas em um intervalo de concentração de 300 a 600 µM, em
30% de sacarose para aumentar a viscosidade da solução e, com isso, minimizar a
contribuição da rotação da proteína na forma de linha do espectro. As amostras
foram colocadas em capilares de vidro com 0,84 o.d. e 0,60 i.d. (VitroCom Inc.,
Mountain Lakes, NJ).
5.3 Resultados e Discussões
5.3.1 Expressão dos Mutantes da Enzima T4L
A fim de produzir os mutantes da enzima T4L em quantidade suficiente para
análises espectroscópicas, foram realizados testes de expressão sob as condições
descritas na seção 5.2.2.
Nas figuras 45, 46 e 47 como ilustração, são apresentados os géis
eletroforéticos (SDS-PAGE) do teste de expressão de três mutantes da enzima T4L,
todos evidenciando a existência de frações induzidas com o tamanho esperado para
a proteína, aproximadamente 18,6 kDa. No gel do mutante V131C/L133A (Figura
45), a fração induzida e mantida a 370C por 2 horas revela a existência de maior
quantidade da enzima, com isso, esse mutante foi expresso em 2 L de meio LB,
nessas condições. O mutante D127N/E128Q/V131C, como é apresentado na figura
46, revelou sucesso na metodologia de expressão a 370C, “overnight”; enquanto que
o mutante D127N/E128Q/V131C/K135R apresentou melhor resultado, quando foi
induzido e mantido a 300C, “overnight”. Entretanto, como pode ser observado na
figura 47, esse mutante também revela uma boa expressão a 300C por 2 horas,
então optamos por expressá-lo a 300C por 4 horas.

T4L 125
As condições de expressão para os seis mutantes da enzima T4L são listadas
na Tabela 6. Com isso, obtivemos todos os mutantes da enzima em quantidade e
estáveis para posterior purificação.
kDa
97.4
66.245.0
31.0
21.514.4
1 2 3 4 5 6
kDa
97.4
66.245.0
31.0
21.514.4
1 2 3 4 5 6
Figura 45 - Expressão do mutante V131C/ L133A. Proteínas visualizadas em gel SDS-PAGE, gradiente
8-25. Poços: 1- marcadores de massa molecular; 2- amostra nao induzida; 3- amostra induzida a 370C por 2 horas; 4- amostra induzida a 300C por 2 horas; 5- amostra induzida a 370C “overnight” e 6- amostra induzida a 300C “overnight”. A posição da T4L está indicada pela seta vermelha.
1 2 3 4 5 6
kDa
97.466.245.031.021.514.4
1 2 3 4 5 6
kDa
97.466.245.031.021.514.4
Figura 46 - Expressão do mutante D127N/E128Q/V131C. Proteínas visualizadas em gel SDS-PAGE,
gradiente 8-25. Poços: 1- marcadores de massa molecular; 2- amostra nao induzida; 3- amostra induzida a 300C por 2 horas; 4- amostra induzida a 370C por 2 horas; 5- amostra induzida a 300C “overnight” e 6- amostra induzida a 370C “overnight”. A posição da T4L está indicada pela seta vermelha.

126
kDa
97.4
66.245.0
31.021.514.4
1 2 3 4 5 6
kDa
97.4
66.245.0
31.021.514.4
1 2 3 4 5 6
Figura 47 - Expressão do mutante D127N/E128Q/V131C/K135R. Proteínas visualizadas em gel SDS-PAGE,
gradiente 8-25. Poços: 1- marcadores de massa molecular; 3- amostra induzida a 300C por 2 horas; 4- amostra induzida a 370C por 2 horas; 5- amostra induzida a 300C “overnight” e 6- amostra induzida a 370C “overnight”. A posição da T4L está indicada pela seta vermelha.
Tabela 6 - Condições de expressão para os mutantes da proteína T4L
MUTANTE TEMPERATURA (0C) TEMPO DE INDUÇÃO
(horas)
V131C/L133A 37 2
V131C/L133G 30 4
D127N/V131C 37 2
D127N/E128Q/V131C 37 4
D127N/E128Q/V131C/K135R 30 4
D127N/E128Q/V131C/N132A/K135R 37 2
5.3.2 Purificação dos Mutantes da Enzima T4L
O resultado do protocolo descrito na seção de Métodos é a obtenção dos
mutantes da enzima T4L em quantidade e grau de pureza suficientes para as
medidas espectroscópicas. Na figura 48 é apresentado o gel eletroforético (SDS-
PAGE, gradiente 8-25) das várias frações eluídas do mutante V131C, para efeito de
ilustração, pois todos os mutantes da enzima apresentam o mesmo perfil de
purificação. A existência da enzima T4L é confirmada pela banda em
aproximadamente 18 kDa.

T4L 127
1 2 3 4 5 6 7
kDa
97.466.245.0
31.021.514.4
Figura 48 - Purificação do mutante da proteína T4L V131C por cromatografia de interação iônica.
Proteínas visualizadas em SDS-PAGE, gradiente 8-25. Poços: 1- marcadores de massa molecular; 2- void (lixo); 3- amostra eluida com 12% de NaCl; 4- amostra eluida entre 12-15% de NaCl; 5- amostra eluida entre 15.5 – 18% de NaCl; 6- amostra eluida entre 18-19.5% de NaCl e 7- amostra eluida com porcentagem de NaCl maior que 22%. A posição da T4L está indicada pela seta vermelha.
5.3.3 Resposta Estrutural da Lisozima T4 a Substituições de Aminoácidos Grandes por Pequenos no Interior da Proteína
Um dos objetivos desse trabalho é utilizar a técnica de “Site-Directed Spin
Labeling” (SDSL) para estudar mudanças conformacionais da proteína T4L quando
ocorre a formação da cavidade pela substituição de um resíduo grande por um
pequeno (leucina por alanina e/ou glicina). Para isso, dois mutantes da T4L foram
construídos com o marcador de spin inserido sempre no resíduo de cisteína 131. No
primeiro deles, substituímos a leucina 133 (Leu-133) por alanina e no segundo o
mesmo resíduo de leucina foi substituído por glicina. Assim, tratamos com os
seguintes mutantes da T4L nativa: uma mutação da valina 131 (trocada por cisteína
e depois marcada com MTSSL) mais a mutação da leucina 133 (trocada ou por
alanina L133A ou por glicina L133G).
Na figura 49 apresentamos os espectros de RPE do marcador de spin na
posição 131 da enzima T4L (V131R1 – espectro usado como controle) e do
marcador de spin na posição 131 dos mutantes L133A e L133G. O espectro da
enzima T4L marcada (V131R1) apresenta uma única componente associada a uma
população de nitróxidos em movimento anisotrópico característico de R1 (cadeia
lateral do marcador nitróxido) localizado em sítios expostos ao solvente de hélices
ordenadas, onde o nitróxido não tem interação com resíduos vizinhos. A substituição
do resíduo de leucina por alanina resultou em um espectro de RPE com poucas

128
mudanças em relação ao espectro da proteína “nativa” (V131R1), apresentando o
aparecimento de uma discreta segunda componente espectral. Por outro lado, a
substituição do resíduo de leucina por glicina resultou em um espectro de RPE com
duas componentes claras, uma livre e outra relativamente imobilizada,
característicos de sítios com contato terciário, onde interações com resíduos
próximos restringem o movimento do grupo N-O.
Esse resultado concorda com os estudos realizados por Eriksson et al. (133)
quando da resolução da estrutura cristalográfica do mutante em que a leucina 133 é
substituída por alanina. Neste trabalho, é observado que o C-β do resíduo mutado
(133) se deslocou 0,8 Å, que os átomos da cadeia principal da α-hélice que inclui os
resíduos 108 ao 113 se moveram por volta de 0,6 Å e, por fim, que os átomos da
cadeia lateral dos resíduos de Met-106, Phe-114, Leu-118 e Leu-121 se moveram
em torno de 0,5 Å, assim ocupando o espaço que ficou vago pela substituição da
leucina por alanina. Com isso, podemos inferir que a hélice H, onde se encontra o
resíduo 131 marcado e o resíduo 133 mutado, sofreu uma mudança conformacional,
permitindo que o radical nitróxido estabeleça uma interação com resíduos vizinhos,
interação esta que ainda não sabemos identificar, mas possivelmente ligações de
hidrogênio ou interações eletrostáticas, que são as responsáveis pelo aparecimento
da componente imobilizada no espectro de RPE do mutante L133G.

T4L 129
Figura 49 - Espectros de RPE em banda X da proteína T4L com o marcador de spin MTSSL na posição
131 (preto) e dos mutantes da enzima T4L L133A e L133G (vermelho) a 298K.
5.3.4 SDSL na determinação de diferenças estruturais entre sítios topologicamente equivalentes da Enzima T4L
O espectro de RPE da cadeia lateral do marcador de spin (R1) em uma
posição específica da proteína nos dá informação sobre a dinâmica do nitróxido.
Essas informações são fundamentais e de grande interesse porque o movimento do
nitróxido reflete uma variedade de fatores estruturais e dinâmicos da proteína:
difusão rotacional da proteína, dinâmica da cadeia principal (“backbone”) e
isomerização rotacional das ligações que conectam o nitróxido à cadeia principal da
proteína (134).
Para proteínas de membranas ou globulares com massa molecular acima de
50 kDa a difusão rotacional das mesmas é muito lenta e por isso não contribue com
o espectro de RPE. Para proteínas globulares menores, como a lisozima T4 (massa
molecular igual a 17 kDa), o efeito da difusão rotacional pode ser reduzido através
do artifício de se fazer as medidas de RPE em solvente contendo, por exemplo, 30%
T 4L V 131R 1 T 4L V 131R 1-L133A
T 4L V 131R 1 T 4L V 131R 1-L133G

130
de sacarose, aumentando assim a viscosidade do meio ou usando RPE de alta-
frequência. Com isso, é possível em ressonância paramagnética eletrônica se
estudar os efeitos das flutuações da cadeia principal e movimentos internos da
cadeia lateral sem a interferência da difusão rotacional da proteína.
Alguns progressos foram e estão sendo obtidos em termos de estrutura
protéica quando os movimentos internos da cadeia lateral são analisados. Para este
propósito, uma descrição semiquantitativa acabou por tornar-se comum para análise
dos espectros obtidos, permitindo-nos interpretar os dados experimentais em termos
da relação mobilidade da cadeia lateral e estrutura protéica (18). Essa descrição
semiquantitativa envolve uma análise da dinâmica da cadeia lateral do marcador de
spin nos sítios de interesse da proteína e se reflete na forma da linha espectral de
uma maneira dependente da estrutura local experimentada pela sonda
paramagnética. A relação entre a estrutura local e a dinâmica pode ser resumida da
seguinte forma: os resíduos R1 são fortemente imobilizados em sítios enterrados, ou
seja, em sítios no interior da proteína; são imobilizados ou possuem espectros
complexos com multicomponentes em sítios com contato terciário; possuem uma
mobilidade intermediária em sítios de superfície de hélice e são movéis em regiões
de loop (Figura 3).
Enquanto os estudos citados acima fornecem uma relação geral entre a
mobilidade do nitróxido e a topografia da proteína, eles também revelam os efeitos
da mobilidade que podem não ser esclarecidos pela diferença na localização
topográfica. Por exemplo, os espectros de RPE para R1 em diferentes sítios de
superfície de hélice expostos ao solvente refletem graus de mobilidade que os
distinguem claramente dos sítios enterrados ou de contato terciário (18). Contudo, a
mobilidade de R1 em cada um desses sítios expostos ao solvente é única em
detalhes quantitativos e é uma “impressão digital” para cada posição do resíduo (18,
28).
O objetivo desta parte do trabalho é identificar a origem de tal “impressão
digital” para sítios situados em regiões topologicamente equivalentes da T4L. Neste
caso, escolhemos dois sítios de superfície de hélice expostos ao solvente (posição
72 e 131) e que apresentam espectros de RPE do marcador R1 diferentes (os
espectros do resíduo 72 foram reportados na referência 134). Para tanto, realizamos
um experimento de “transmutação” no qual os resíduos vizinhos ao sítio 72 na hélice
C são mutados de forma a, após realizadas as mutações, termos um ambiente

T4L 131 estrutural em torno do R1 introduzido no sítio 131 da hélice H similar àquele
experimentado pelo R1 na posição 72 (Figura 43). Para isso, foram construídos
quatro mutantes: D127N-V131R1, D127N/E128Q–V131R1, D127N/E128Q/K135R–
V131R1 e D127N/E128Q/K135R/N132A–V131R1. Os espectros de RPE de todos os
mutantes foram obtidos a 273, 298 e 313K.
O espectro da proteína T4L com o marcador de spin inserido na posição 72
apresenta um movimento anisotrópico (134), um dos movimentos mais ordenados
para essa classe de sítios (Figura 50). Nesse sítio, a flexilbilidade da cadeia principal
é mínima, pois a estrutura do “backbone” é relativamente rígida, e não há contatos
terciários, nem interações com vizinhos na própria hélice. Ao passo que no sítio 131
da T4L, acreditamos que haja um modo dinâmico adicional para a flexibilidade da
cadeia principal, refletido no espectro de RPE do mesmo que, apesar de similar ao
do sítio 72, apresenta menor separação hiperfina máxima e com menor resolução
(Figura 50).
T4L 72R1
T4L 131R1
T4L 72R1
T4L 131R1
Figura 50 - Espectros de RPE em banda X da proteína T4L com o marcador de spin R1 nas posições 72 e
131. As setas marcam a extrema hiperfina, característica de espectro anisotrópico. Figura adaptada de Columbus et al (134).
A Figura 51 mostra os espectros de RPE do mutante T4L D127N – V131R1 em
diferentes temperaturas e compara com os espectros da proteína “nativa” marcada
(T4L V131R1). Como pode ser observado, a substituição do resíduo aspartato pelo
resíduo asparagina não causou nenhum efeito nos espectros. Em todos os valores
de temperatura os espectros da proteína mutante são idênticos aos da proteína que
apresenta esses resíduos nativos.

132
T 4 L V 1 3 1 R 1 2 7 3 K
T 4 L 1 2 7 N - 1 3 1 R 1 2 7 3 K
T 4 L V 1 3 1 R 1 2 9 8 K T 4 L 1 2 7 N - 1 3 1 R 1 2 9 8 K
T 4 L V 1 3 1 R 1 3 1 3 K T 4 L 1 2 7 N - 1 3 1 R 1 3 1 3 K
Figura 51 - Espectros de RPE em banda X da proteína T4L com o marcador de spin MTSSL na posição
131 (preto) e do mutante da enzima T4L D127N (vermelho) a 273K, 298K e 313K, respectivamente.
Quando mutamos também o resíduo i–3 ao longo da cadeia principal, neste
caso o resíduo de glutamato por glutamina, a forma anisotrópica da linha não se
altera significativamente a 298K e 313K, refletindo a geometria básica do movimento
do N-O, com mudanças sutis nos espectros (Figura 52). Por outro lado, no espectro
obtido a 273K (Figura 52) observamos duas componentes e podemos, com isso,
especular que o nitróxido que antes não interagia com COO- do glutamato, agora
interage com NH2+ da glutamina, pois este possui um volume maior. A maior
mudança no espectro é observada a 273K, pois a baixas temperaturas a
contribuição entálpica é dominante e as cadeias laterais, tanto do marcador de spin
quanto do resíduo de glutamina, fazem um número máximo de contatos enquanto
que a temperatura ambiente (298K) a contribuição entrópica torna-se significativa,
aumentando com isso os graus de liberdade de ambas as cadeias laterais.

T4L 133
T 4 L V 1 3 1 R 1 2 7 3 K T 4 L 1 2 7 N / 1 2 8 Q - 1 3 1 R 1 2 7 3 K
T 4 L V 1 3 1 R 1 2 9 8 K T 4 L 1 2 7 N /1 2 8 Q - 1 3 1 R 1 2 9 8 K
T 4 L V 1 3 1 R 1 3 1 3 K
T 4 L 1 2 7 N / 1 2 8 Q - 1 3 1 R 1 3 1 3 K
Figura 52 - Espectros de RPE em banda X da proteína T4L com o marcador de spin MTSSL na posição
131 (preto) e do mutante da enzima T4L D127N/ E128Q (vermelho) a 273K, 298K e 313K, respectivamente.
Nos espectros do mutante da proteína T4L com três substituições, aquelas
mencionadas acima mais a substituição da lisina por um resíduo de arginina,
D127N/E128Q/K135R, não há nenhuma mudança significativa comparada com o
espectro da proteína que possui os resíduos nativos nessas posições,T4L V131R1
(Figura 53). A mudança observada no espectro do mutante D127N/E128Q/K135R a
273K se deve à presença da mutação E128Q (Figura 52).

134
T 4 L V 1 3 1 R 1 2 9 8 K
T 4 L 1 2 7 N / 1 2 8 Q / 1 3 5 R 2 9 8 K
T 4 L V 1 3 1 R 1 2 7 3 K
T 4 L 1 2 7 N / 1 2 8 Q / 1 3 5 R - 1 3 1 R 1 2 7 3 K
T 4 L V 1 3 1 R 1 3 1 3 K T 4 L 1 2 7 N / 1 2 8 Q / 1 3 5 R 3 1 3 K
Figura 53 - Espectros de RPE em banda X da proteína T4L com o marcador de spin MTSSL na posição
131 (preto) e do mutante da enzima T4L D127N/ E128Q/K135R (vermelho) a 273K, 298K e 313K, respectivamente.
A figura 54 apresenta os espectros do mutante da proteína T4L que contém
quatro substituições: o aspartato 127 foi substituído por asparagina, glutamato 128
por glutamina, a lisina 135 por arginina e por fim a asparagina 132 foi mutada por
alanina (D127N/E128Q/K135R/N132A). Como podemos observar, os espectros do
mutante em todas as temperaturas estudadas são similares aos espectros da
enzima “nativa”. A razão para a forma da linha espectral a 273K voltar a ser
essencialmente idêntica à da enzima “nativa”, mesmo este mutante possuindo a
mutação E128Q, é a possibilidade de haver uma rede de interações terciárias entre
as cadeias laterais do marcador de spin e dos resíduos vizinhos. A substituição da
asparagina 132 por alanina aumentou os graus de liberdade da cadeia lateral do
marcador de spin, pois a cadeia lateral da alanina é menor que da asparagina. Com
isso, a segunda componente que observamos no espectro a 273K da figura 52
desapareceu, resultando em um espectro com apenas uma componente

T4L 135 anisotrópica, semelhante ao espectro da enzima T4L “nativa” (Figura 54, espectro a
273K).
Os resultados acima nos levam a concluir que a diferença na mobilidade dos
dois sítios expostos ao solvente comparados em nosso estudo, 72 e 131,
aparentemente não é determinada pela diferença na interação com os vizinhos na
mesma hélice, pois se assim fosse, quando “transmutamos” os quatro resíduos da
hélice C para a hélice H, observaríamos mudanças na mobilidade do espectro de
RPE do sítio 131 para se tornar similar ao espectro do sítio 72. As mudanças
observadas são interações suficientemente fracas entre o anel nitróxido na posição
131 e grupos vizinhos, pois são detectadas no espectro de RPE somente em
temperaturas reduzidas.
A proposta mais atrativa é que a diferença da mobilidade reflete as diferenças
dinâmicas da cadeia principal (“backbone”) da hélice, surgindo de diferenças
estruturais e do meio entre as duas hélices. Por exemplo, a hélice H consiste em
somente duas voltas e meia, o suficiente para que seja um pouco flexível. Por outro
lado, o sítio 72 está situado no centro de uma hélice longa de 5 voltas. A hélice C
(sítio 72) tem uma treonina estabilizando o N-cap (início da hélice), enquanto que a
hélice H (sítio 131) possui dois resíduos carregados positivamente no N-cap, que
são fracos estabilizadores da hélice. Talvez mais importante ainda, o segmento da
hélice que contém o sítio 72 está localizado próximo ao núcleo hidrofóbico
firmemente empacotado. Estas características nos conduzem a concluir que a
mobilidade mais elevada de 131R1 pode ter sua origem em “flutuações” da cadeia
principal de maior amplitude próximo ao sítio 131 devido a uma estabilidade
termodinâmica menor da estrutura secundária.

136
T 4 L V 1 3 1 R 1 2 7 3 K
T 4 L 1 2 7 N /1 2 8 Q /1 3 2 A /1 3 5 R -1 3 1 R 1 2 7 3 K
T 4 L V 1 3 1 R 1 2 9 8 K T 4 L 1 2 7 N /1 2 8 Q /1 3 2 A /1 3 5 R -1 3 1 R 1 2 9 8 K
T 4 L V 1 3 1 R 1 3 1 3 K
T 4 L 1 2 7 N /1 2 8 Q /1 3 2 A /1 3 5 R -1 3 1 R 1 3 1 3 K
Figura 54 - Espectros de RPE em banda X da proteína T4L com o marcador de spin MTSSL na posição
131 (preto) e do mutante da enzima T4L D127N/E128Q/K135R/N132A (vermelho) a 273K, 298K e 313K, respectivamente.

Conclusões


Conclusões 139
6 Conclusões
Findado o período de doutoramento junto ao IFSC/USP, podemos ressaltar
alguns aspectos e objetivos mais generalistas e outros mais específicos do presente
trabalho. Há alguns anos, um dos interesses centrais de nosso grupo de pesquisa
está na implementação da técnica de marcação de spin sítio dirigida e sua aplicação
em problemas de Biofísica Molecular. Após alguns projetos iniciais, tivemos o
desafio de proceder com uma investigação sistemática e extensiva que envolvesse a
metodologia de SDSL. A proteína ligante de cálcio HS100A12 se encaixou
perfeitamente nos requisitos para um bom objeto de estudo por SDSL: baixa massa
molecular, bastante solúvel, bom rendimento nos protocolos de expressão e
purificação e sem resíduos de cisteína nativos. Além disso, estudos anteriores
mostravam a existência de mudanças conformacionais na estrutura protéica na
presença de alguns ligantes.
Após o início do desenvolvimento do projeto, surgiu a oportunidade de
realizarmos um estágio junto ao grupo liderado pelo Prof. Dr. Wayne Hubbell
(Universidade da Califórnia – Los Angeles). O grupo do Prof. Hubbell não é apenas
um laboratório de excelência na aplicação de métodos modernos de RPE em
biologia estrutural, mas é também o local de onde emergiu a metodologia de SDSL e
suas aplicações em estrutura e dinâmica de proteínas. Durante tal estágio,
trabalhamos em um projeto estudando mudanças conformacionais da proteína
Lisozima do bacteriófago T4. Em seguida, já no Grupo de Biofísica Molecular- IFSC,
buscamos informações estruturais e funcionais da proteína S100A12 humana
utilizando diferentes técnicas, como: dicroísmo circular, monocamadas de Langmuir
e SDSL.
Uma avaliação em termos dos objetivos gerais nos leva a crer que estes foram
alcançados com êxito e temos hoje à disposição em nosso grupo não somente uma
série de mutantes simples e duplos da proteína HS100A12 submetidos a análises de
SDSL apresentadas neste texto, mas também várias outras proteínas que vieram
como desdobramentos naturais do conhecimento e experiência adquiridos ao longo
dos anos de trabalho. Vale ressaltar, ainda, que além das medidas e análises
tradicionais de forma de linha e mudanças espectrais, implementamos as medidas

140
de distância entre sondas introduzidas na proteína HS100A12, o que teve como
consequência natural a implantação, não neste trabalho mas em outro projeto de
nosso grupo, de técnicas de RPE resolvidas no tempo para medida de distâncias.
Em relação aos objetivos específicos, vimos que, através da técnica de
dicroísmo circular, monitoramos mudanças no conteúdo de estrutura secundária da
HS100A12 quando submetida a um processo de desnaturação térmica na ausência
e na presença dos íons Ca+2 e Zn+2. Os efeitos dos íons no enovelamento e
estabilidade da proteína HS100A12 contribuem para entendermos a regulação
dependente de Ca+2 e Zn+2 da proteína. Nossos resultados mostraram que a
desestabilização da proteína na presença do Ca+2 é devido a uma mudança
conformacional induzida pelo metal e que a ligação com o Zn+2 confere uma
estabilidade térmica maior para a proteína. Adicionalmente, quando a proteína está
ligada com os dois íons, a estabilidade térmica é ainda maior, sugerindo que o efeito
do metal é dominado pela contribuição do íon zinco. Utilizamos um modelo
termodinâmico de transição irreversível entre dois estados conformacionais e, assim,
determinamos os valores das temperaturas de transição e das energias de ativação
para cada caso estudado.
O efeito antagônico dos íons cálcio e zinco no controle da estabilidade da
proteína HS100A12 fornece uma razão molecular para a ação dos dois metais. A
conformação da proteína é sensível à presença dos diferentes metais e o rearranjo
resultante da ligação com os íons preserva o enovelamento como um todo da
proteína.
Uma combinação bastante interessante entre métodos experimentais de
investigação, engloba a união de técnicas de medidas globais, como o CD e a
calorimetria, e técnicas de medidas pontuais, como fluorescência e RPE. Com as
primeiras, monitoramos mudanças que advêm da estrutura como um todo e com as
segundas podemos nos focar em uma dada parte da estrutura, onde normalmente
existe uma sonda (intrínseca ou extrínseca) específica. Sendo assim, procedemos
com os experimentos de SDSL-RPE e, ao longo do caminho, fomos capazes de
produzir os mutantes simples e duplos da HS100A12: K33C, I53C, D54C, F57C,
L60C, D61C, A80C, K33C/A80C, I53C/A80C, D54C/A80C, F57C/A80C, L60C/A80C
e D61C/A80C. Os mutantes, apesar de terem protocolos de clonagem, expressão e
purificação similares, requereram otimizações específicas. Ao final, tivemos bom
rendimento para os experimentos de RPE em todos os casos.

Conclusões 141
Os experimentos de RPE focaram principalmente a detecção de mudanças
conformacionais em duas hélices que formam o motivo EF-hand ligante de cálcio na
proteína HS100A12. Esses estudos se dividiram em medidas de RPE-CW realizadas
a temperatura ambiente e a -100oC. Nas primeiras, o objetivo foi averiguarmos
alterações na forma de linha dos espectros, resultado de uma maior ou menor
restrição de movimento das sondas ou de sua aproximação/afastamento, após
adição de íons cálcio ou zinco. Em vários dos duplos mutantes claras mudanças
espectrais foram observadas, principalmente quando da presença do íon zinco.
As medidas de RPE em baixa temperatura serviram para, ao “congelarmos” a
dinâmica da cadeia polipeptídica, podermos determinar as distâncias entre os vários
pares de resíduos contidos nos duplos mutantes produzidos. Esta foi mais uma
contribuição metodológica de nosso trabalho, visto que pudemos estabelecer
protocolos não só para produção dos duplos mutantes, mas também para
realizarmos as medidas de distância e a análise dos dados segundo o método de
desconvolução de Fourier. Uma análise mais sistemática dos dados aqui
apresentados, em que se calculem as distâncias em todos os casos estudados, está
colocada como perspectiva futura do trabalho, juntamente com a realização das
mesmas medidas de distância, desta feita se valendo da técnica de RPE pulsada
conhecida como “Double electron-electron resonance” (DEER).
Por fim, com os experimentos utilizando monocamadas de Langmuir
concluimos que a eficiência de interação da proteína HS100A12 com monocamadas
constituídas pelos fosfolipídios DPPC e DPPG é maior na presença de zinco que na
proteína apo ou com cálcio. Provavelmente, as mudanças conformacionais
induzidas pelo zinco, discutidas no estudo utilizando dicroísmo circular, favorecem a
interação da proteína com a monocamada. Já quando a proteína é incubada com
cálcio e zinco, provavelmente ocorre um processo cooperativo resultando em uma
interação maior do que aquela que ocorre quando a proteína está incubada com os
íons separadamente.
Com relação aos estudos realizados com a enzima T4L, os dados obtidos
neste trabalho são uma pequena parte do projeto coordenado pelo Prof. Dr. Wayne
Hubbell. São resultados bastante promissores e que complementarão outros
resultados obtidos pelo grupo do Prof. Hubbell e que devem fazer parte de uma
biblioteca ampla de espectros de RPE oriundos de resíduos estruturalmente distintos
na lisozima T4L. Com isso e varrendo um número de resíduos suficientemente

142
diversos, pretendemos tentar estabelecer uma correlação posição na estrutura (se
em α-hélice exposta ou enterrado ou folha β) e forma de linha dos espectros de
RPE. Inicialmente, realizamos um estudo com a suposta cavidade criada no “core”
hidrofóbico da porção C-terminal da enzima T4L, quando mutamos a Leu 133 por
Ala e/ou Gly, ou seja, quando substituímos um resíduo de aminoácido grande por
um de menor volume. Como resultados, obtivemos para a enzima T4L “nativa” na
presença do marcador de spin na posição 131 um espectro com uma única
componente espectral anisotrópica, que permanece inalterado quando mutamos o
resíduo de leucina por alanina, enquanto que para o mutante da proteína T4L L133G
(leucina por glicina) o espectro de RPE passa a apresentar duas componentes, uma
livre e outra relativamente imobilizada, indicando que a hélice H sofreu alguma
mudança conformacional, permitindo que o radical nitróxido interaja com resíduos
vizinhos, possivelmente por ligações de hidrogênio ou interações eletrostáticas. Com
isso, podemos confirmar a formação da cavidade no interior da proteína T4L e
propor futuros estudos na presença de ligantes da enzima T4L.
Adicionalmente, na tentativa de investigar a natureza das contribuições para os
diferentes modos dinâmicos experimentados pelo marcador de spin quando
introduzido em sítios topologicamente semelhantes, realizamos um experimento de
“transmutação”, no qual os resíduos vizinhos ao sítio 72 da hélice C são mutados em
locais similares em torno do R1 presente no sítio 131 da hélice H. A fim de realizar
tal experimento, os mutantes da enzima T4L foram: D127N-V131R1,
D127N/E128Q–V131R1, D127N/E128Q/K135R – V131R1 e
D127N/E128Q/K135R/N132A – V131R1. A 298K e 313K, os espectros dos quatro
mutantes manteram a forma da linha anisotrópica encontrada no espectro controle
da enzima “nativa” (T4L V131R1). No entanto, no espectro a 273K do mutante
D127N/E128Q observamos o aparecimento de uma segunda componente, o que
indica que, quando substituímos o glutamato 128 pela glutamina, a cadeia lateral
dessa interage de alguma forma com o anel nitróxido, restringindo o movimento do
mesmo. O mesmo resultado é observado para o mutante D127N/E128Q/K135R, já
que esse também possui a mutação E128Q. Embora o mutante
D127N/E128Q/K135R/N132A também possua a substituição E128Q, o seu espectro
a 273K é similar ao espectro da proteína “nativa”, levando-nos a acreditar que há
uma rede de interações entre as cadeias laterais do marcador de spin e dos
resíduos vizinhos. Essas interações podem ser devido à natureza hidrofóbica do

Conclusões 143
anel nitróxido, a capacidade de formação de ligações de hidrogênio ou interações
eletrostáticas envolvendo o momento de dipolo do N-O, mas suficientemente fracas
pois são detectadas nos espectros de RPE somente a baixas temperaturas. Posto
isto, concluímos que a diferença na mobilidade do marcador R1 em sítios de
superfície de hélice expostos ao solvente se dá pelo movimento do “backbone” e
não pelas interações entre cadeias laterais de resíduos vizinhos.

144

145
*De acordo com: ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS NBR6023: Informação e documentação: referências: elaboração. Rio de janeiro, 2002.
Referências*
1 LEHNINGER, A. L.; NELSON, D. L.; COX, M. M. Lehninger- principles of biochemistry. 4th ed. New York: W. H. Freeman, 2005. 2 FRAUENFELDER, H.; PARAK, F.; YOUNG, R. D. Conformational substates in proteins. Annual Review of Biophysics and Biophysical Chemistry, v.17, p.451-479, 1988. 3 FRAUENFELDER, H.; SLIGAR, S.G.; WOLYNES, P.G. The energy landscapes and motions of proteins. Science, v.254, n.5038, p.1598-1603,1991. 4 LÓPEZ, C. J. et al. Osmolyte pertubation reveals conformational equilibria in spin- labeled proteins. Protein Science, v.18, n.8, p.1637-1652, 2009. 5 HODSDON, M.E.; CISTOLA, D.P. Discrete backbone disorder in the nuclear magnetic resonance structure of the apo intestinal fatty acid-binding protein: implications for the mechanism of ligand entry. Biochemistry, v.36, n.6, p.1450-1460, 1997. 6 ALTENBACH, C. et al. Structural studies on transmembrane proteins. Biochemistry, v.28, n.19, p.7806-7812, 1989. 7 KUO, W. et al. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. Journal of Molecular Biology, v.387, n.2, p.284-294, 2009. 8 HUANG, H.; CAFISO, D. S. Conformation and membrane position of the region linking the two C2 domains in synaptotagmin 1 by site- directed spin labeling. Biochemistry, v. 47, n.47, p.12380-12388, 2008. 9 DONG, J. H. et al. Structural basis of energy transduction in the transport cycle of Msb. Science, v.308, n.5724, p.1023-1028, 2005. 10 UEKI, S. et al. Site-directed spin labeling electron paramagnetic resonance study of the calcium- induced structural transition in the N- domain of human cardiac troponin C complexed with troponin I. Biochemistry, v.44, n.1, p.411-416, 2005. 11 BELLE, V. et al. Probing the opening of the pancreatic lipase lid using site-directed spin labeling and EPR spectroscopy. Biochemistry, v.46, n.8, p.2205-2214, 2007. 12 KAVALENKA, A. et al. Conformational analysis of the partially disordered measles virus NTAIL-XD complex explored by SDSL EPR spectroscopy. Biophysical Journal, v.8, n.6, p.1055-1064, 2010. 13 OHNISHI, S.; McCONNEL, H. M. Interaction of radical ion of Chlorpromazine with deoxyribonucleic acid. Journal of the American Chemical Society, v. 87, n. 10, p. 2293, 1965.

146
14 HUBBELL, W. L.; McCONNELL H. M. Molecular motion in spin-labeled phospholipids and membranes. Journal of the American Chemical Society, v.93, n.2, p.314-326, 1971. 15 BERLINER, L. J. Spin labeling theory and applications II. New York: Academic Press, 1979. p. 357. 16 HUBBELL, W. L. et al. Watching proteins move using site-directed spin labeling. Structure, v.4, n.7, p.779-783, 1996. 17 HUBBELL, W. L.; ALTENBACH.C. Investigation of structure and dynamics im membrane proteins using site-directed spin labeling. Current Opinion in Structural Biology, v.4, n.4, p.566-573, 1994. 18 MCHAOURAB, H. S. et al. Motion of spin-labeled side chains in T4 lysozyme. correlation with protein structure and dynamics. Biochemistry, v. 35, n.24, p. 7692-7704, 1996. 19 OH, K. J. et al. Organization of diphtheria toxin t domain in bilayers: a site-directed spin labeling study. Science, v.273, n.9, p.810-812, 1996. 20 PEROZO, E. et al. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nature Structural & Molecular Biology, v.5, n.6, p.459-469, 1998. 21 ADDONA, G. H. et al. Estimating the electrostatic potential at the acetylcholine receptor agonist site using power saturation EPR. Biochimica et Biophysica Acta, v.1329, n.1, p.74-84, 1997. 22 KLUG, C. et al. Mapping of the residues involved in a proposed β-strand located in the ferric enterobactin receptor FepA using site-directed spin-labeling. Biochemistry, v.36, n.42, p.13027-13034, 1997. 23 LIN, Y. et al. Docking phospholipase a2 on membranes using electrostatic potential-modulated spin relaxation magnetic resonance. Science, v. 279, n.5358, p. 1925-1929, 1998. 24 MCHAOURAB, H.S. et al. Conformation of T4 lysozyme in solution. hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry, v. 36, n.2, p. 307-316, 1997. 25 FRAZIER, A. A. et al. Membrane orientation and position of the C2 domain from cPLA2 by site-directed spin labeling. Biochemistry, v.41, n.20, p.6282-6292, 2002. 26 COUTO-CARVALHO, S. G. Estudos estruturais e funcionais de diidroorotato desidrogenases.106p.2008. Tese (Doutorado em Ciências) - Instituto de Física de São Carlos, Universidade de São Paulo,São Carlos, 2008. 27 PISTOLESI, S. et al. Molecular motion of spin labeled side chains in the C-terminal domain of RGL2 protein: A SDSL-EPR and MD study. Biophysical Chemistry, v.123, n.1, p.49-57, 2006.

147
28 MCHAOURAB, H. S. et al. Motion of spin-labeled side chains in T4 lysozyme: effect of side chain structure. Biochemistry, v.38, n.10, p.2947-2955, 1999. 29 BHATTACHARYA, S. et al. Target selectivity in EF-hand calcium binding proteins. Biochimica et Biophysica Acta, v.1742, n.1-3, p.69-79, 2004. 30 KRETSINGER, R. H.; NOCKOLDS, C. E. Carp muscle calcium-binding protein. structure determination and general description. Journal of Biological Chemistry, v. 248, n.9, p. 3313-3326, 1973. 31 NAKAYAMA, N.; KAWASAKI, H.; KRETSINGER, R. Evolution of EF-hand proteins. Topics in Biological Inorganic Chemistry, v. 3, p. 29-58, 2000. 32 HOUDUSSE, A.; COHEN, C. Structure of the regulatory domain of scallop myosin at 2 Å resolution: implications for regulation. Structure, v.4, n.1, p.21-32, 1996. 33 DECLERCQ, J. P. et al. Ionic interactions with parvalbumins. crystal structure determination of pike 4.10 parvalbumin in four different ionic environments. Journal of Molecular Biology, v.220, n.4, p.1017-1039, 1991. 34 MALMENDAL, A. et al. Battle for the EF-hands: magnesium-calcium interference in calmodulin. Biochemistry, v.38, n.36, p.11844-11850, 1999. 35 SKELTON, N. J. et al. Signal transduction versus buffering activity in Ca2+-binding proteins. Nature Structural & Molecular Biology, v.1, n.4, p.239-245, 1994. 36 HOLMES, K. C. et al. Muscle proteins — their actions and interactions. Current Opnion in Structural Biology, v.6, n.6, p.781-789, 1996. 37 YAP, K. L. et al. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins: structure, function, bioinformatics, v.37, n.3, p.499-507, 1999. 38 VAN ASSELT, E. J. et al. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme-like catalytic domain with an EF-hand. Structure, v.7, n.11, p.1167-1180, 1999. 39 LEWIT- BENTLEY, A.; RÉTY, S. EF-hand calcium-binding proteins. Current Opnion in Structural Biology, v.10, n.6, p.637-643, 2000. 40 NELSON, M. R.; CHAZIN, W. J. Structures of EF-hand Ca(2+)-binding proteins: diversity in the organization, packing and response to Ca2+ binding. BioMetals, v.11, n.4, p.297– 318, 1998. 41 STRYNADKA, N. C.; JAMES, M. N. Crystal structures of the helix-loop-helix calcium-binding proteins. Annual Review of Biochemistry, v.58, p.951–998, 1989.

148
42 LINSE, S.; FORSEN, S. Determinants that govern high-affinity calcium binding. In: Advances in Second Messenger and Phosphoprotein Research, v. 30, p. 89–151, 1995. 43 BROKX, R. D. et al. Energetics of target peptide binding by calmodulin reveals different modes of binding. Journal of Biological Chemistry, v. 276, n.17, p. 14083–14091, 2001. 44 MOORE, B. W. et al. A soluble protein characteristic of the nervous system. Biochemical Biophysical Research Communications, v. 19, p. 739–744, 1965. 45 ISOBE, T.; OKUYAMA, T. The amino acid sequence of the S100 protein (PAP I-b protein) and its relation to the calcium binding proteins. European Journal of Biochemistry, v. 89, n.2, p. 379–388, 1978. 46 ISOBE, T.; OKUYAMA, T. The amino acid sequence of the α-subunit in bovine brain S100a protein. European Journal of Biochemistry, v.116, n.1, p. 79–86, 1981. 47 COCCHIA, D.; MICHETTI, F.; DONATO, R. S100 antigen in normal human skin. Nature, v. 294, n. 5836, p. 85–87, 1981. 48 KRETSINGER, R. H. et al. The EF-hand, homologs and analogs. In: HEIZMANN, C. W.(Ed.). Novel calcium binding proteins. fundamentals and clinical applications. New York: Springer-Verlag, 1991. p. 17–37. 49 KISIEL, L., S.; RINTALA-DEMPSEY, A. C.; SHAW, G. S. Calcium-dependent and –independent interactions of the S100 protein family. Biochemistry Journal, v. 396, n.2, p. 201-214, 2006. 50 KREBS, J.; QUADRONI, M.; VAN ELDIK, L. J. Dance of the dimers. Nature Structural & Molecular Biology, v. 2, n.9, p. 711-714, 1995. 51 DONATO, R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochimica et Biophysica Acta, v. 1450, n.3, p. 191-231, 1999. 52 RÉTY, S. et al. Structural basis of the Ca(2+)-dependent association between S100C (S100A11) and its target, the N-terminal part of annexin I. Structure, v. 8, n.2, p. 175-184, 2000. 53 SZEBENY, D. M. E.; MOFFAT, K. The refined structure of vitamin D-dependent calcium-binding protein from bovine intestine. molecular details, ion binding, and implications for the structure of other calcium-binding proteins. Journal of Biological Chemistry, v. 261, n.19, p. 8761-8777, 1986. 54 MIWA, N. et al. Purification and molecular cloning of a novel calcium-binding protein, p26olf, in the frog olfactory epithelium. Biochemical Biophysical Research Communications, v. 251, n.3, p. 860-867, 1998. 55 TANAKA, T. et al. Molecular modeling of single polypeptide chain of calcium-binding protein p26olf from dimeric S100B (ββ).Protein Engineering Design & Selection, v. 12, n.5, p. 395-405, 1999.

149
56 MARKOVA, N. G. et al. Profilaggrin Is a major epidermal calcium-binding protein. Molecular and Cellular Biology, v. 13, n. 1, p. 613-625, 1993. 57 LEE, S. C. et al. The structure of human trichohyalin. potential multiple roles as a functional EF-hand-like calcium-binding protein, a cornified cell envelope precursor, and an intermediate filament-associated (cross-linking) protein. Journal of Biological Chemistry, v. 268, n.16, p. 12164-12176, 1993. 58 ISOBE, T.; ISHIOKA, N.; OKUYAMA, T. Structural relation of two S-100 proteins in bovine brain; subunit composition of S-100a protein. European Journal of Biochemistry, v. 115, n.3, p. 469-474, 1981. 59 SVENSSON, L. A.; THULIN, E.; FORSEN S. Proline cis-trans isomers in calbindin D9k observed by X-ray crystallography. Journal of Molecular Biology, v. 223, n.3, p. 601-606, 1992. 60 MALER, L.; POTTS, B. C.; CHAZIN, W. J. High resolution solution structure of apo calcyclin and structural variations in the S100 family of calcium-binding proteins. Journal of Biomolecular NMR, v. 13, n.3, p. 233-247, 1999. 61 LEE, Y. C. et al. NMR structure of the Apo-S100P protein. Journal of Biomolecular NMR, v. 29, n.3, p. 399-402, 2004. 62 MATSUMURA, C. H. et al. A novel mode of target recognition suggested by the 2.0A structure of holo S100B from bovine brain. Structure, v. 6, n.2, p. 233-241, 1998. 63 OSTENDORP, T. et al. Structural and functional insights into RAGE activation by multimeric S100B. Embo Journal, v. 26, p. 3868-3878, 2007. 64 WRIGHT, N. T. et al. The three-dimensional solution structure of Ca(2+)-bound S100A1 as determined by NMR spectroscopy. Journal of Molecular Biology, v. 353, n.2, p. 410-426, 2005. 65 KOCH, M.; DIEZ, J.; FRITZ, G. Crystal Structure of Ca+2- free S100A2 at 1.6 A resolution. Journal of Molecular Biology, v. 378, n.4, p. 931-940, 2008. 66 MITTL, P.R. et al. Metal-free MIRAS phasing: structure of apo-S100A3. Acta Crystallographica D, v.58, n.8, p. 1255-1261, 2002. 67 MALASHKEVICH, V. N. et al. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proceedings of the National Academy of Science of the United States of America, v. 107, n.19, p. 8605-8610, 2010. 68 BERTINI, I. et al. Solution structure and dynamics of S100A5 in the apo and Ca+2 bound states. Journal of Biological Inorganic Chemistry, v. 14, n.7, p. 1097-1107, 2009.

150
69 OTTERBEIN, L. R. et al. Crystal structures of S100A6 in the Ca(2+)-free and Ca(2+)-bound states: the calcium sensor mechanism of S100 proteins revealed at atomic resolution. Structure, v. 10, n.4, p. 557-567, 2002. 70 BRODERSEN, D. E.; NYBORG, J.; KJELDGAARD, M. Zinc-binding site of an S100 protein revealed. two crystal structures of Ca2+-bound human psoriasin (S100A7) in the Zn2+-loaded and Zn2+-free states. Biochemistry, v. 38, n.6, p. 1695-1704, 1999. 71 ISHIKAWA, K. et al. The structure of human MRP8, a member of the S100 calcium-binding protein family, by MAD phasing at 1.9 Å resolution. Acta Crystallographica D, v.56, n.5, p. 559-566, 2000. 72 ITOU, H. et al. The crystal structure of human MRP14 (S100A9), a Ca+2- dependent regulator protein in inflammatory process. Journal of Molecular Biology, v. 316, n.2, p. 265-276, 2002. 73 RÉTY, S. et al. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nature Structural & Molecular Biology, v. 6, n.1, p. 89-95, 1999. 74 MOROZ, O. V. et al. The three-dimensional structure of human S100A12. Acta Crystallographica D, v.57, n.1, p. 20-29, 2001. 75 MOROZ, O. V. et al. The crystal structures of human S100A12 in apo form and in complex with zinc: new insights into S100A12 oligomerisation. Journal of Molecular Biology, v. 391, n.3, p. 536-551, 2009. 76 BABINI, E.; BERTINI, I.; BORSI, V. Structural characterization of human S100A16, a low- affinity calcium binder. Journal of Biol Inorganic Chemistry, v.16, n. 2, p. 243-256, 2010. 77 ARNESANO, F. et al. Structural Interplay between Calcium(II) and Copper(II) binding to S100A13. Protein Angewandte Chemie International ed in English, v. 44, p. 6341-6344, 2005. 78 DONATO, R. et al. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. The international Journal of Biochemistry & Cell Biology, v. 33, n.7, p. 637-668, 2001. 79 NAKAYAMA, S.; MONCRIEF, N. D.; KRETSINGER, R. H. Evolution of EF-hand calcium-modulated proteins. II. domains of several subfamilies have diverse evolutionary histories. Journal of Molecular Evololution, v. 34, n.5, p. 416-448, 1992. 80 KLIGMAN, D.; HILT, D. C. The S100 protein family. Trends in Biochemistry Science, v. 13, n.11, p. 437-442, 1988. 81 BABU, Y. S. et al. The three dimensional structure of calmodulin. Nature, v. 315, n.6014, p. 37-40, 1985.

151
82 HERZBERG, O.; JAMES, M. N. G. Structure of the calcium regulatory muscle protein troponin-c at 2.8 resolution. Nature, v. 313, n.6004, p. 653-659, 1985. MOEWS, P. C.; KRETSINGER, R. H. Refinement of the structure of carp muscle calcium-binding parvalbumin by model building and difference Fourier analysis.Journal of Molecular Biology, v. 91, n.2, p. 201-228, 1975. 84 MONCRIEF, N. D.; KRETSINGER, R. H.; GOODMAN, M. Evolution of EF-hand calcium-modulated proteins. I. relationships based on amino acid sequences. Journal of Molecular Evolution, v. 30, p. 522-562, 1990. 85 MALER, L.; SASTRY, M.; CHAZIN, W. J. A structural basis for S100 protein specificity derived from comparative analysis of apo and Ca(2+)- calcyclin. Journal of Molecular Biology, v. 317, n.2, p. 279-290, 2002. 86 MOROZ, O. V. et al. Structure of the human S100A12- copper complex: implications for host- parasite defence. Acta Crystallographica D, v.59, n.5, p. 859-867, 2003. 87 HEIZMANN, C. W.; COX, J. A. New perspectives on S100 proteins: a multi-functional Ca(II)-, Zn(II)- and Cu(II)- binding protein family. Biometals, v. 11, n.4, p. 383-397, 1998. 88 GUIGNARD, F.; MAUEL, J.; MARKETT, M. Identification and characterization of a novel human neutrophil protein related to the S100 family. Biochemistry Journal, v. 309, n.2, p. 395-401, 1995. 89 DELL’ ANGELICA, E. C.; SCHLEICHER, C. H.; SANTOME, J. A. Primary structure and binding properties of calgranulin C, a novel S100-like calcium-binding protein from pig granulocytes. Journal of Biological Chemistry, v. 269, n.46, p. 28929-28936, 1994. 90 NONATO, M. C. et al. Crystallization and preliminary crystallographic studies of calgranulin C, an S-100 like calcium binding protein from pig granulocytes. Acta Crystallographica D, v.53, n.2, p. 200-202, 1997. 91 CORPET, S. Multiple sequence alignment with hierarchical clustering. Disponível em: <http://bioinfo.genopole-toulouse.prd.fr/multalin/multalin.html>. Acesso em: janeiro/2007. 92 KERKHOFF, C.; KLEMPT, M.; SORG, C. Novel insights into structure and function of MRP8 (S100A8) and MRP14 (S100A9). Biochimica Biophysica Acta, v. 1448, n.2, p. 200-211, 1998. 93 VOGL, T. et al. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. Journal of Biological Chemistry, v. 274, n.36, p. 25291-25296, 1999. 94 HOFMANN, M. A. et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell, v. 97, n.7, p. 889-901, 1999.

152
95 HSIEH, H. L. et al. Expression analysis of S100 proteins and RAGE in human tumors using tissue microarrays. Biochemical and Biophysical Research Communications, v. 307, n.2, p. 375-381, 2003. 96 SHISHIBORI, T. et al. Three distinct anti-allergic drugs, amlexanox, cromolyn and tranilast, bind to S100A12 and S100A13 of the S100 protein family. Biochemistry Journal, v. 338, n.3, p. 583-589, 1999. 97 MARTI, T.; ERTTMANN, K.D.; GALLIN, M. Y. Host-Parasite interaction in human onchocerciasis: Identification and sequence analysis of a novel human calgranulin. Biochemical and Biophysical Research Communications, v. 221, n.5, p. 454-458, 1996. 98 NOVITSKAYA, V. et al. Oligomeric forms of the metastasis-related Mts1 (S100A4) protein stimulate neuronal differentiationin cultures of rat hippocampal neurons. Journal of Biological Chemistry, v. 275, n.52, p. 41278–41286, 2000. 99 KLINGELHOFER, J. et al. Up-regulation of metastasis-promoting S100A4 (Mts-1) in rheumatoid arthritis: putative involvement in the pathogenesis ofrheumatoid arthritis. Arthritis and Rheumatism, v. 56, n.3, p. 779–789, 2007. 100 VOGL, T. et al. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochimica Biophysica Acta, v. 1763, n.11, p. 1298–1306, 2006. 101 MOROZ, O. V. et al. The structure of S100A12 in a hexameric form and its proposed role in receptor signalling. Acta Crystallographica D, v.58, n.3, p. 407–413, 2002. 102 MOROZ, O. V. et al. Both Ca2+ and Zn2+ are essential for S100A12 protein oligomerization and function. BMC Biochemistry, v. 10, n.11, p. 11, 2009. 103 FENG, S. S. et al. Interpretation of mechanochemical properties of lipid bilayer vesicles from the equation of state or pressure-area measurement of the monolayer at the air-water or oil-water interface. Langmuir, v. 15, n. 4, p. 998-1010, 1999. 104 BROCKMAN, H. et al. Lipid monolayers: why use half a membrane to characterize protein-membrane interactions? Current Opinion in Structural Biology, v. 9, n. 4, p. 438-443, 1999. 105 CANTOR, C. R.; SCHIMMEL, P. R. Other optical techniques. In: Biophysical chemistry. Part II: techniques for the study of biological structure and function. San Francisco: Ed.Freeman,1980. cap.8. 106 WOODY, R.; BEROVA, N.; NAKANISHI, K. Circular dichroism: principles and applications. New York: VCH Publishers, 1994. 570 p. 107 KELLY, M. et al. How to study proteins by circular dichroism. Biochimica et Biophysica Acta, v. 1751, n.2, p. 119-139, 2005.

153
108 WOODY, R.W. Circular dichroism of protein-folding intermediates. In:Methods in enzymology, v. 380, p. 242-285, 2004. 109 ARANTES, A. R. Caracterização dos estados de spin e formação de agregados das microperoxidases-8 e –9 em função do pH e estudos do citocromo –c (S=1/2) por RPE pulsada.2005. 107p.Tese (Doutorado em Ciências) - Instituto de Física de São Carlos,Universidade de São Paulo, São Carlos, 2005. 110 SCHREIER, S. Apostila de técnicas espectroscópicas- Ciências Moleculares. Disponível em: <http://www.iq.usp.br/schreier/apostila.PDF>. Acesso em: fevereiro/2006. 111 NOBRE, T. et al. The specificity of frutalin lectin using biomembrane models. Biochimica et Biophysica Acta, v.1798, n.8, p. 1547-1555, 2010. 112 SILVA-LUCCA, R. A. et al. Structural and thermodynamic studies of KM+, a D-mannose binding lectin from Artocarpus integrifolia seeds. Biophysical Chemistry, v.79, n.2, p. 81-93, 1999. 113 CHEN, Y. H.; YANG, J. T.; MARTINEZ, H. M. Determination of secondary structures of proteins by circular-dichroism and optical rotatory dispersion. Biochemistry, v. 11, n.22, p. 4120-4131, 1972. 114 SÁNCHEZ-RUIZ, J. M. et al. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry, v. 27, n.5, p. 1648-1652, 1988. 115 DELANO SCIENTIFIC LLC. Pymol. Disponível em: <http://pymol.sourceforge.net>. Acesso em: fevereiro/2010. 116 BANHAM, J.E. et al. Distance measurements in the borderline region of applicability of CW EPR and DEER: A model study on a homologous series of spin-labelled peptides. Journal of Magnetic Resonance, v.191, n.2, p. 202-218, 2008. 117 XIE, J. et al. Hexameric calgranulin (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. Journal of Biological Chemistry, v. 282, n.6, p. 4218-4231, 2007. 118 TRIPP, B. C.; MAGDA, J. J.; ANDRADE, J. D. Adsorption of globular–proteins at the air/water interface as measured via dynamic surface-tension—concentration-dependence, mass-transfer considerations, and adsorption-kinetics. Journal of Colloid and Interface Science, v.173, n.1, p.16–27, 1995. 119 MARSH, D. Lateral pressure in membranes. Biochimica et Biophysica Acta, v.1286, n.3, p.183–223, 1996. 120 BEINTEMA, J. J.; VAN SCHELTINGA, A. C. Plant lysozymes. In: JOLES, P.(Ed.) Lysozymes: model enzyme in biochemistry and biology. Basel: Birkhauser, 1996. p.75–86.

154
121 HANCOCK, R. E. W.; SCOTT, M. G. The role of antimicrobial peptides in animal defense. Proceedings of the National Academy of Science of the United States of America, v. 97, n.16, p. 8856–8861, 2000. 122 YAN, H.; HANCOCK, R. E. W. Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrobial Agents Chemotherapy, v. 45, n.5, p.1558–1560, 2001. 123 YOUNG, R. Bacteriophage lysis: mechanism and regulation. Microbiological Reviews, v. 56, n.3, p. 430–481, 1992. 124 KENNY, J.G. et al. Bacteriophage Tuc2009 encodes a tail-associated cell wall-degrading activity. Journal of Bacteriology, v. 186, n.11, p. 3480–3491, 2004. 125 TAKÁC, M.; BLASI, U. Phage P68 virion-associated protein 17 displays activity against clinical isolates of Staphylococcus aureus. Antimicrobial Agents and Chemotherapy, v. 49, n.7, p. 2934–2940, 2005. 126 HOLTJE J. V. From growth to autolysis: the murein hydrolases of Escherichia coli. Archives of Microbiology, v. 164, n.4, p. 243–254, 1995. 127 NAKIMBUGWE, D. et al. Cell wall substrate specificity of six different lysozymes and lysozyme inhibitory activity of bacterial extracts. FEMS Microbiology Letters, v. 259, p. 41-46, n.1, 2006. 128 QASBA, P. K.; KUMAR, S. Molecular divergence of lysozymes and alfalactalbumin. Critical Reviews in Biochemistry and Molecular Biology, v. 32, n. 4, p. 255-306, 1997. 129 SAMBROOK, J.; FRITSCH, E. F.; MANIATIS, T. Molecular cloning. a laboratory manual. 2nd . ed. New York: Cold Spring Harber Laboratory Press, 1989, 1234p. 130 TSUGITA, A. et al. Purification of bacteriophage T4 Lysozyme. Journal of Biological Chemistry, v. 243, n.2, p. 391-397, 1968. 131 MATTHEWS, B. W.; REMINGTON, S. J. The three Dimensional Structure of the Lysozyme from Bacteriophage T4. Proceedings of the National Academy of Science of the United States of America, v. 71, n.10, p. 4178-4182, 1974. 132 TSUGITA, A. Phage lysozyme and other lytic enzymes. In: BOYER, P.D The Enzymes.3rd ed. New York: Academic Press,1971.v.5.p. 343-411. 133 ERIKSSON, A. E. et al. The response of a protein structure to cavity-creating mutations and its relationship to the hydrophobic effect. Science, v. 255, n.5041, p. 178-183, 1992. 134 COLUMBUS, L. et al. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry, v. 40, n.13, p. 3828-3846, 2001.

155
Apêndice A
Tutorial para mutação sítio dirigida

156

157
1 Tecnologia do DNA Recombinante
Para a aplicação da técnica SDSL é necessária a produção de inúmeros
mutantes em que resíduos de interesse são substituídos por resíduos de cisteína.
Como realizamos um trabalho intenso de produção de mutantes através de técnicas
de DNA recombinante, achamos conveniente apresentar uma introdução sobre os
referidos métodos. Assim, a partir de 1970, foram desenvolvidas novas tecnologias
provenientes da Microbiologia, Bioquímica, Imunologia e Genética Microbiana,
permitindo o isolamento e a purificação de genes específicos num processo
chamado clonagem gênica (1, 2). Com a clonagem gênica, a molécula de DNA
tornou-se uma das moléculas de mais amplo uso e manuseio, com metodologias
que permitem isolar regiões específicas, obtê-las em grande quantidade e
determinar a sua sequência numa velocidade de milhares de nucleotídeos por dia.
Como consequência do desenvolvimento destas tecnologias, a tecnologia do
DNA recombinante, como se convencionou denominar este conjunto de técnicas,
teve ampla aplicação tanto na área biológica como em diversas outras áreas, tais
como: médica, forense, agrícola e química. Atualmente é possível realizar
investigações de paternidade, diagnósticos de doenças genéticas e infecciosas,
estudos dos mecanismos de replicação e expressão, estudos do desenvolvimento
de culturas microbianas capazes de produzir substâncias úteis tais como a insulina
humana, hormônio de crescimento e vacinas, e ainda, produzir enzimas de interesse
industrial aplicáveis no processo de fabricação de pesticidas e fármacos, entre
outros. Portanto, sua aplicação comercial ou biotecnológica parece ter um potencial
inesgotável.
Diante da diversidade de aplicações, a tecnologia do DNA recombinante foi de
extrema importância neste trabalho tanto na obtenção dos genes de interesse como
na confirmação de suas sequências e, consequentemente, na obtenção de proteínas
e seus mutantes.

158
1.1 Clonagem Molecular
A técnica central da metodologia do DNA recombinante é a clonagem
molecular, que consiste no isolamento e propagação de moléculas de DNA
idênticas. Em geral, a clonagem molecular compreende duas etapas importantes:
1. a ligação de um fragmento de DNA de interesse, denominado inserto, a uma
outra molécula de DNA, denominada vetor, a fim de formar uma terceira molécula, o
DNA recombinante;
2. a molécula do DNA recombinante é introduzida numa célula hospedeira
compatível, num processo chamado de transformação. A célula hospedeira que
adquiriu a molécula do DNA recombinante é agora chamada de transformante ou
célula transformada. Um único transformante, em condições ideais, sofre muitos
ciclos de divisão celular, produzindo uma colônia que contém milhares de cópias do
DNA recombinante.
1.2 Reação em Cadeia da Polimerase (PCR)
Partindo do ponto que é necessária grande quantidade do fragmento de DNA
de interesse para iniciar-se a primeira etapa do processo de clonagem molecular, a
amplificação deste via uma reação em cadeia (PCR) torna-se primordial.
A tecnologia da reação em cadeia da polimerase (“Polymerase Chain
Reaction”) consiste da possibilidade de se amplificar uma determinada sequência de
DNA através da síntese enzimática in vitro de milhões de cópias de um segmento
específico na presença da enzima DNA polimerase. A condição básica para a
aplicação dessa técnica é a construção de um par de oligonucleotídeos (“primers” ou
iniciadores) que são complementares às duas fitas opostas do DNA e que delimitam
a sequência alvo da amplificação.
A PCR envolve basicamente 3 etapas: desnaturação, hibridização e extensão,
que constituem um ciclo da reação de amplificação. A figura 1 ilustra a
representação de um ciclo básico da PCR.

159
Figura 1- Ciclo da Reação em Cadeia da Polimerase. O processo envolvendo estas três etapas pode ser
repetido de 25 a 32 ciclos, sendo possível aumentar em cada ciclo, duas vezes a concentração de DNA pré- existente. Figura adaptade de < http://portais.ufg.br/this2/uploads/files/242/image/pcr.jpg>.
Na primeira etapa, a fita dupla do DNA alvo sofre desnaturação a temperaturas
em torno de 950C durante 5 min. Nesta temperatura, as duas fitas do DNA alvo se
separam. Na etapa de anelamento, a temperatura é rapidamente reduzida para 350C
a 600C permitindo que os oligonucleotídeos (“primers”) sejam hibridizados
especificamente na região do DNA molde em que há complementariedade das
bases. Finalmente, a temperatura é elevada para aproximadamente 720C para que a
DNA polimerase realize a extensão a partir das extremidades 3’ dos “primers”.
1.2.A Inserção da Mutação por PCR- “Overlap Extension”
A reação em cadeia da polimerase utilizada para extensões sobrepostas
(“Insertation Mutagenesis by PCR- Overlap Extension”) é uma variante da PCR que
pode tanto inserir mutações em pontos específicos de uma sequência quanto
produzir polinucleotídeos de fragmentos menores (3).
No presente trabalho, utilizamos esta estratégia para inserir mutações em uma
sequência. Para isso, a sequência que está sendo modificada é utilizada para fazer
duas fitas de DNA modificadas, com as modificações no final de cada uma delas.
Primeiramente, produzimos um “primer” que contém a mutação (“primer
Sense”) que será adicionado na extremidade 5’ do “template” (DNA molde), ao
passo, que na extremidade 3’ é adicionado um segundo “primer”, complementar ao
“template”, ou seja sem mutações, de modo que a DNA polimerase possa fazer a
cópia do “primer Sense”. A mesma reação é feita para a extremidade 3’ do DNA
molde, produzindo, portanto, a segunda fita de DNA com a mutação na extremidade
3’. Em seguida, essas duas fitas são misturadas e servirão como moldes para a

160
segunda reação em cadeia da polimerase (PCR) juntamente com os “primers” 5’ e
3’, “primers” sem mutação, utilizados anteriormente (Figura 2).
Figura 2 - Estratégia para mutação sítio-dirigida utilizando o método de “Overlap Extension”. Figura
adaptada de <http://www.medigenomix.de> (4).
1.3 Construção do DNA Recombinante
Como descrito na seção 1.1., o início do processo de clonagem envolve a
construção de vetores contendo os genes de interesse, denominado construção do
DNA recombinante. Na construção do DNA recombinante a primeira etapa envolve a
produção de fragmentos de DNA de origens diferentes com o mesmo conjunto de
extremidades fitas simples, ou seja, a digestão das moléculas de DNA com a mesma
enzima de restrição. Numa segunda etapa, estes dois fragmentos são ligados por
renaturação das regiões de fita simples. Se a ligação for feita com uma enzima, a
DNA ligase após o pareamento das bases, estes fragmentos serão ligados
permanentemente.

161
1.3.A Vetores de Clonagem
Os vetores de clonagem molecular são moléculas de DNA capazes de
amplificar uma informação genética de interesse em centenas de cópias.
Atualmente, os tipos básicos de vetores usados na metodologia do DNA
recombinante apresentam características especiais que os tornam excelentes
veículos de clonagem em diferentes situações: são pequenas moléculas de DNA
dupla fita, que possuem os elementos necessários para a sua replicação e pelo
menos um gene que confere resistência a antibiótico. Estas moléculas variam de 5 a
400 quilobases e comumente estão presentes em duas ou mais cópias por célula.
Os vetores de clonagem mais utilizados normalmente são os plasmídeos de
bactérias, que são moléculas de DNA circular existentes nesses microorganismos,
fora dos seus cromossomos e capazes de se replicar de modo independente.
Um plasmídeo é considerado um bom vetor de clonagem se: possuir uma
origem de replicação (O), ou seja, uma sequência de DNA que permita que o vetor
seja replicado na célula hospedeira; apresentar dois ou mais sítios únicos de
clivagem para endonucleases de restrição onde o inserto é incorporado e possuir um
gene que codifica um produto que distingue a célula transformada da célula não
transformada (Figura 3).
Figura 3- Estrutura molecular de um plasmídeo típico usado em clonagem molecular. O é a origem de
replicação, Amp é gene que confere resistência ao antibiótico ampicilina, por exemplo, e MSC é o sítio de múltipla clonagem.
O conjunto dos sítios de clivagem é denominado de múltiplos sítios de
clonagem (MSC) que permite sua abertura por várias enzimas, a escolha
dependendo do DNA exógeno. Muitos vetores de clonagem carregam genes que
conferem resistência a diversos antibióticos tais como ampicilina (AmpR), tetraciclina
e canamicina. As células transformadas com tais vetores são capazes de crescer

162
num meio contendo o antibiótico, enquanto que as células não tranformadas acabam
morrendo.
1.3.B Enzimas de Restrição
A descoberta das enzimas de restrição revolucionou a biologia molecular, pois
possibilitou montar moléculas de DNA recombinante, tornando possível a
manipulação do DNA com extrema precisão. Estas enzimas são proteínas
bacterianas que reconhecem sequências nucleotídicas específicas de 4 a 8 pares de
base (pb) em uma molécula de DNA, clivando-a em dois lugares da fita dupla que a
constitui. A sequência de reconhecimento é chamada de sítio de restrição.
Na tecnologia do DNA recombinante, o sítio de reconhecimento das enzimas é
normalmente uma sequência denominada palindrômica, ou seja, ela tem um eixo de
simetria e a sequência de bases de uma fita é a mesma da fita complementar,
quando lida na direção oposta. A Tabela 1 mostra o organismo de origem, a
sequência palindrômica e o sítio de clivagem das enzimas de restrição utilizadas
neste trabalho.
Tabela 1 - Endonucleases ou enzimas de restrição. As setas verdes dentro das sequências de reconhecimento indicam o local de clivagem em uma fita dupla de DNA.
ORGANISMO DENOMINAÇÃO DA
ENZIMA SEQÜÊNCIA DE
RECONHECIMENTO
Bacillus amyloliquefaciens BamHI 5’ G↓↓↓↓GATCC 3’ 3’ CCTAG↑↑↑↑G 5’
Haemophilus influenzae Rd HindIII 5’ A↓↓↓↓AGCTT 3’ 3’ TTCGA↑↑↑↑A 5’
Neisseria denitrificans NdeI 5’ CA↓↓↓↓TATG 3’ 3’ GTAT↑↑↑↑AC 5’
Nocardia corallina NcoI 5’ C↓↓↓↓CATGG 3’ 3’ GGTAC↑↑↑↑C 5’
A digestão de DNA por enzimas de restrição é um processo bastante simples.
Basta colocar o DNA em contato com a enzima a uma temperatura ideal, geralmente
37 0C, e a enzima inicia imediatamente o processo de digestão, cortando o DNA em
diversos fragmentos. O número de fragmentos produzido é estabelecido pelo

163
número de sítios de restrição reconhecidos pela enzima utilizada. Uma importante
consequência desta especificidade é que o número de clivagens feito por cada uma
delas no DNA de qualquer organismo é definido e permite o isolamento de
fragmentos deste DNA. Portanto, cada enzima de restrição gera um conjunto único
de fragmentos quando cliva uma molécula de DNA específica. O conjunto de
fragmentos gerados por digestão com enzimas de restrição é geralmente detectado
pela separação destes fragmentos por eletroforese em gel de agarose.
Com isso, as enzimas de restrição constituem a principal ferramenta da
tecnologia do DNA recombinante, permitindo cortar genes específicos de interesse e
colocá-los dentro de outros organismos, tornando possível recombinar ou criar
organismos transgênicos, tão conhecidos popularmente.
1.3.C Enzimas DNA Ligases
Com as duas moléculas de DNA que se pretende recombinar in vitro isoladas e
preparadas, segue-se para o processo de ligação. Se o vetor de clonagem e o
fragmento de DNA a clonar forem tratados com a mesma enzima de restrição, suas
extremidades serão compatíveis e a reação de ligação poderá ocorrer na presença
de uma DNA ligase, que são enzimas codificadas por algumas bactérias e vírus,
como a E. coli e o fago T4.
A ligação é promovida pela ligação covalente (uma ligação fosfodiéster) entre
os dois nucleotídeos das extremidades das duas moléculas. A enzima DNA ligase
catalisa a ligação entre o grupo fosfato ligado ao carbono 5’ e o grupo hidroxila, OH
livre, ligado ao carbono 3’ das moléculas de desoxirribose presentes nos
nucleotídeos que constituem estas moléculas.
Enfim, ocorrendo a ligação entre as duas moléculas, uma nova molécula de
DNA plasmidial contendo o DNA de interesse é gerada, sendo denominado,
plasmídeo híbrido (Figura 4). Este é, então, inserido em uma bactéria por
transformação, para ser replicado, como se fosse parte integrante do plasmídeo.

164
Figura 4 - Construção de um DNA recombinante. Uma molécula de DNA híbrido é formada a partir de
fragmentos de diferentes organismos obtidos com o uso de uma mesma enzima de restrição. Figura adaptada de < http://users.med.up.pt/med05009/bcm/ligase_frame_files/image004.jpg>.
1.4 Sequenciamento de DNA
O sequenciamento de DNA é um método de análise dos clones que se faz
necessário para se obter a sequência completa, ou pelo menos uma porção, do
gene de interesse. Sua estratégia envolve a PCR consistindo na capacidade da DNA
polimerase em adicionar um nucleotídeo na extremidade de alongamento de um
oligonucleotídeo copiando uma fita de DNA molde.
Desde a década de 70, o método descrito por Sanger é o mais utilizado para
análise de sequências de DNA e é conhecido também como método da terminação
da cadeia (1, 2). No início dos anos 70, o processo era manual e eram necessários
dois anos para sequenciar 20 pares de bases de DNA. Atualmente, com o
desenvolvimento das técnicas de sequenciamento automático, um aparelho de
sequenciamento é capaz de sequenciar meio milhão de pares de bases por dia (1,
2).
Esta técnica utiliza dideoxinucleotídeos (ddNTPs) marcados com compostos
fluorescentes e um equipamento provido de um laser. Primeiramente, realizamos a
reação de sequenciamento, que é uma reação em cadeia da polimerase (PCR) com
os genes de interesse e os ddNTPs marcados com os fluoróforos, obtendo assim,
um produto de PCR contendo os ddNTPs fluorescentes. Então, os produtos da

165
reação de sequenciamento, marcados com os fluorocromos, são submetidos à
eletroferese, que os separa pela diferença de tamanho, passando por um feixe de
laser, que promove a excitação dos fluorocromos. A luz emitida pelos fluoróforos é
detectada por um fotomultiplicador e a informação é processada através de um
computador.

166
Referências
1 ZAHA, A.; SCHRANK, A.; FERREIRA, H. B.; SCHRANK, I. S.; RODRIGUES, J. J. S.; REGNER, L. P.; PASSAGLIA, L. M. P.; ROSSETTI, M. L. R.; RAUPP, R. M.; SILVA, S. C.; GAIESKY, V. L.V. Biologia molecular básica, Porto Alegre: Mercado Aberto, 1996. p. 336. 2 STRYER, L. Biochemistry. New York: W. H. Freeman and Company, 1988. p. 881. 3 HIGUCHI R.; KRUMMEL B.; SAIKI R. A general method of in vitro preparation and specific mutagenesis DNA fragments: study of protein and DNA interactions. Nucleic Acids Research, v. 16, n. 15, p. 7351–7367, 1988. 4 EUROFINS MEDIGENOMIX. Disponível em: <http://www.medigenomix.de>. Acesso em: junho/2008

167
Apêndice B
Diidroorotato Desidrogenase de Leishmania major

168

169
1 INTRODUÇÃO
1.1 A enzima Diidroorotato Desidrogenase (DHODH)
Os nucleotídeos não atuam somente como unidades fundamentais que
constituem os ácidos nucléicos DNA e RNA. Atuam também como componentes de
coenzimas envolvidas no metabolismo oxidativo da célula e, ainda, são ativadores e
inibidores em várias vias do metabolismo intermediário da célula. Portanto, todos os
organismos vivos dependem da sua síntese. Embora algumas células sejam
capazes de produzir a quantidade necessária de nucleotídeos, reciclando
nucleosídeos e bases de purina através da via de salvação, outras, especialmente
durante o período de proliferação celular, não conseguem reciclar a quantidade
necessária ou não possuem a via de salvação, passando a depender, portanto, da
síntese de nucleotídeos através de outra via metabólica chamada síntese de novo
de nucleotídeos (1,2).Inibidores da síntese de novo de nucleotídeos formam um
importante grupo de agentes quimoterapêuticos e células que dependem desta via
metabólica são susceptíveis a tais inibidores. Como exemplo, células de linfócitos T
e células cancerígenas dependem da síntese de novo de nucleotídeos, e inibidores
deste processo metabólico têm sido utilizados no tratamento de câncer e rejeição de
órgãos transplantados (1).
No quarto passo da síntese de novo de nucleotídeos atua a enzima
diidroorotato desidrogenase (DHODH), responsável pela conversão de diidroorotato
em orotato (3). Esta enzima foi identificada como sendo o alvo farmacológico de
uma série de compostos químicos e naturais (4),os quais interferem em reações
descontroladas do sistema imune, auxiliam no combate de infecções parasitárias
como malária e em terapias antivirais através da diminuição da concentração
intracelular de nucleotídeos de pirimidina (5). De acordo com a estrutura primária, as
enzimas DHODH dos mais diferentes organismos podem ser divididas em duas
famílias: as enzimas da família 1 são encontradas no citosol, enquanto que
proteínas da família 2 estão localizadas na região interna da membrana mitocondrial
(eucariotos) ou citosólica (procariotos). Todos os membros da família 2 apresentam

170
uma extensão na região N-terminal que permite a interação dessas enzimas com a
membrana (6,7).
No desempenho de suas funções biológicas, DHODH utiliza o
mononucleotídeo flavina (FMN, C17H21N4O9P1) como cofator, para promover a
oxidação do substrato, diidroorotato, enquanto FMN é reduzido. Na segunda parte
da reação, FMN é reoxidado (FMNH2 é convertido em FMN) através do auxílio de
uma terceira molécula cuja natureza varia entre as enzimas de diferentes origens
biológicas (Figura 1) (8). No caso da enzima humana, HsDHODH, ubiquinona é o
agente oxidante (9), enquanto que na enzima do parasita Trypanosoma cruzi,
fumarato é o agente oxidante (10).
Figura 1- Reação de oxi-redução catalisada pela enzima DHODH. O hidrogênio H* é transferido para FMN
como um híbrido, e o hidrogênio H' é perdido como um próton. Figura adaptada de Takashima, et al, 2002 (10).
Até o presente momento, estruturas cristalográficas de quatro enzimas
DHODHs foram determinadas: DHODH de Lactococcus lactis (LlDHODH) e DHODH
de Trypanosoma cruzi (TcDHODH) (11, 12, 13) pertencentes à família 1 das
DHODHs (14); DHODH humana (HsDHODH) (15) e de E. coli (EcDHODH) (16),
membros da família 2. Em termos gerais, as enzimas apresentam uma estrutura
terciária similar, se enovelando em um motivo barril α/β (domínio catalítico).
HsDHODH e EcDHODH contêm na região N-terminal um motivo adicional composto
Orotato Diidroorotato (DHO)
FMN FMNH2
Succinato Fumarato
Oxidação
Redução

171
de duas hélices, responsável pela interação com a membrana e que se encontra
ausente na estrutura de LlDHODH (6).
1.2 Diidroorotato Desidrogenase de Leishmania major
Os protozoários parasitas do genêro Leishmania são agentes etiológicos de
uma série de doenças denominadas leishmanioses que afetam 88 países, inclusive
o Brasil, onde, segundo dados do Ministério da Saúde, a leishmaniose cutânea ou
tegumentar, conhecida popularmente como ferida brava, atinge de 30 a 35 mil
pessoas anualmente, em geral, moradores de áreas rurais ou das proximidades das
matas. No caso da leishmaniose visceral, três mil novos casos são divulgados todos
os anos e se encontram espalhados em 19 estados.
A enzima diidroorato desidrogenase de Leishmania major (LmDHODH) contém
320 resíduos, peso molecular de 34661,8 Da, pI=5,7 e, segundo a sua estrutura
primária, é uma proteína citosólica, membro da família 1 das DHODHs. LmDHODH
apresenta 73% de identidade sequencial com DHODH de Trypanosoma cruzi, 52%
de identidade seqüencial com DHODH de Lactococcus lactis (LlDHODH) e 22,8 %
com a enzima DHODH humana (Figura 2).
Figura 2 - Alinhamento seqüencial entre DHODH de Leishmania major (LmDHODH), Trypanosoma cruzi
(TcDHODH), Lactococcus lactis (LlDHODH) e Homo sapiens (HsDHODH). Alinhamento e figura foram feitos utilizando o programa MULTALIN (17).

172
A estrutura do complexo entre LlDHODH e o produto da reação, orotato, revela
os resíduos envolvidos na função enzimática: Asn67, Asn127, Asn132, Asn193,
Lys43, Ser194, Met69, Gly70, Leu71 (4). Um alinhamento entre as estruturas
primárias da LlDHODH e LmDHODH mostra que todos os resíduos que participam
na ligação com o cofator FMN e com o produto da catálise são conservados na
estrutura de Leishmania major, sugerindo similaridades no modo de atuação de
ambas as enzimas. Além disso, a alta identidade sequencial entre TcDHODH e
LmDHODH (72%) sugere que as duas enzimas utilizam o mesmo substrato para a
reciclagem de FMN, ou seja, ambas têm uma molécula de fumarato no papel de
aceitador de elétrons na segunda etapa da catálise enzimáticas.
Esta parte do trabalho visa a caracterização estrutural da enzima LmDHODH
através da técnica dicroísmo circular que, corroborados com outras abordagens
como estudos bioquímicos, cristalográficos e biofísicos, acreditamos que possa
auxiliar no entendimento do papel desta enzima e desta via metabólica no
mecanismo de funcionamento e patogenicidade em Leishmania major.
2 METODOLOGIA
2.1 Expressão e Purificação da enzima LmDHODH
O gene que codifica a enzima Diidroorotato desidrogenase de Leishmania
major foi clonado no vetor de expressão pET28a (NOVAGEN, Madison, WI) pela
Prof(a). Dr(a). Maria Cristina Nonato do grupo de Cristalografia de Proteínas da
Faculdade de Ciências Farmacêuticas de Ribeirão Preto (FCFRP-USP). Na
construção utilizada a proteína se encontra fusionada a uma sequência de 6
histidinas (cauda de histidina) para facilitar os experimentos de purificação.
O pré-inóculo foi produzido utilizando a cepa BL21(DE3) de Escherichia coli,
transformadas com o plasmídeo que contém o gene que codifica LmDHODH em
meio de cultura LB (Luria-Bertani) (18) sob agitação (225 rpm), durante
aproximadamente 16 horas, a 37°C e na presença de 30 µg/mL de Kanamicina, pois
o gene codificador da enzima LmDHODH confere resistência a esse antibiótico.

173
Após 16 h, o pré-inóculo foi utilizado para inocular 1 L de meio de cultura LB
fresco contendo 30 µg/mL de kanamicina em uma diluição 1(pré-
inóculo):100(inóculo). As células foram mantidas sob 225 rpm de agitação, a 37oC,
até que a absorbância a 600 nm (DO600nm) atingisse valores entre 0,5- 0,6. A
expressão da proteína foi, então, induzida pela adição de 0,5 mM de IPTG (isopropil-
β-D-tiogalactopiranosídeo). A temperatura foi imediatamente diminuída para 250C e
a cultura mantida sob a mesma agitação por 3 h. Para o isolamento das células, o
material foi centrifugado a 6.000 rpm, 4 °C, por 10 minutos e o “pellet” congelado.
A purificação da LmDHODH foi feita utilizando cromatografia por afinidade, na
qual nos valemos da afinidade da seqüência de 6 resíduos de histidinas fusionadas
à proteína por íons de níquel encontrados na resina. A purificação por cromatografia
de afinidade constitui um dos métodos mais eficientes para a purificação de
proteína. Para a utilização desta metodologia, as células foram descongeladas e
ressuspensas em 50 mM NaH2PO4, 300 mM NaCl e 1 mM fenilmetilsulfonil fluoreto
(PMSF) (pH 8,0) e lisadas por sonicação em 7 ciclos de 30 s. Os resíduos celulares
e o material insolúvel foram removidos por centrifugação a 12.000 rpm por 30
minutos. O sobrenadante é aplicado em uma coluna com a resina de Níquel-Ácido
Nitriloacético (Ni-NTA Qiagen) previamente equilibrada com o tampão usado para a
ressuspensão das células sem PMSF. Para otimizar a interação da cauda de
histidina (fusionada à nossa proteína) com os íons de níquel da resina, a mistura foi
mantida sob agitação (70 rpm) por 30 min à 40C. Para eliminar contaminantes que
interagem com a resina através de interações não-específicas, a mesma foi lavada
com aproximadamente 50 mL de tampão de ressuspensão sem PMSF. Em seguida,
a eluição da proteína foi feita através de um gradiente crescente de imidazol (25
mM, 50 mM e 100 mM), cuja interação com os íons Ni(II) presentes na resina
compete com a interação entre esses íons e os resíduos de histidina fusionados à
proteína. O gradiente de imidazol é aplicado de maneira que a resina é lavada com
50 mM NaH2PO4, 300 mM NaCl, 25 mM Imidazol pH 8,0; em seguida, passa-se o
tampão com 50 mM Imidazol e, por fim, a proteína é eluída com 100 mM Imidazol.

174
2.2 Dicroísmo Circular
As medidas de dicroísmo circular foram realizadas no espectropolarímetro
Jasco J-720, em um intervalo de 195 a 250nm, utilizando cubetas de quartzo de 1
mm de caminho óptico e 16 varreduras. Para experimentos de desnaturação térmica
foram utilizadas alíquotas de 360 µL da enzima na concentração de 5,8 µM em
tampão fosfato de sódio pH 7,2 e da enzima, na mesma concentração, adicionada
ao orotato na concentração de 58 µM. A estabilidade da proteína LmDHODH e da
mesma na presença de orotato (em concentração dez vezes maior do que a de
LmDHODH) foi estudada variando-se a temperatura de 20o a 80oC, em intervalos de
20C e no final da varredura, resfriada para 200C.
Os dados de CD em função do comprimento de onda foram convertidos em
elipticidade molar média por aminoácido [θ]λ (grau.cm2.dmol-1), de acordo com as
equações da seção 4.3.11. da tese. A análise do processo de desnaturação da
enzima na presença e ausência do orotato foi realizada segundo tratamento
termodinâmico apresentado na seção 4.4.4.B. da tese.
3 RESULTADOS E DISCUSSÕES
3.1 Purificação da enzima LmDHODH
O processo de purificação da enzima LmDHODH por cromatografia de
afinidade (v. seção 2.1.) foi satisfatório e o resultado é a obtenção da enzima em
quantidade e com grau de pureza suficiente para as medidas espectroscópicas. Na
Figura 3 é apresentado o gel eletroforético (SDS-PAGE 15%) das várias frações
eluídas, evidenciando a existência da enzima LmDHODH, com massa molecular de
34,6 kDa, em quantidade (banda larga) e pura (ausência de outras bandas de
contaminantes na coluna).

175
1 2 3 4 5 6 7 8 9 10kDa
66
45
30
20
12
1 2 3 4 5 6 7 8 9 10kDa
66
45
30
20
12
Figura 3 - Frações eluídas a partir de cromatografia por afinidade em coluna de níquel. Proteínas
visualizadas em SDS-PAGE 15%. Poços: 1- marcadores de massa molecular; 2- void (lixo); 3- pellet; 4 e 5- amostra obtida durante a lavagem com 50mM de imidazol; 6 a 10- amostras obtidas durante a eluição com 100mM de imidazol. A posição da LmDHODH está indicada pela seta vermelha.
3.2 Estudo da Estabilidade Térmica da Enzima LmDHODH
O espectro de CD da LmDHODH em tampão 50 mM fosfato de sódio pH 7,2, a
200C é mostrado na Figura 4 e é caracterizado por dois mínimos negativos a 220 e
210 nm e um máximo positivo em torno de 197 nm. O mínimo em 220 nm é atribuído
a transições encontradas em estruturas helicoidais (19).O mínimo em 210 nm é
resultado de uma mistura do mínimo em 208 nm, característico de estrutura em α-
hélice, com o mínimo em 215 nm, característico de folhas β, o que sugere uma
estrutura α/β para a proteína.
Para estudar a influência do ligante quando ligado à enzima, também
realizamos experimentos de dicroísmo circular da LmDHODH na presença do
produto da reação enzimática, orotato. Essa molécula é conhecida por atuar como
inibidora das DHODHs em concentrações saturadas. Por isso, também mostrado na
Figura 4, está o espectro de CD da enzima LmDHODH na presença do orotato a
uma concentração 10 vezes maior que a concentração da proteína. Características
espectrais similares são observadas nesse caso quando comparada ao espectro de
CD da LmDHODH pura indicando portanto, que a composição estrutural secundária
da proteína não é significativamente alterada pela ligação do orotato. Entretanto,
algumas diferenças espectrais podem ser detectadas, como por exemplo, a variação
entre as intensidades das curvas, que é devido à um rearranjo estrutural da proteína
quando da presença do orotato.

176
200 210 220 230 240 250
-15
-10
-5
0
5
10
15
LmDHODH LmDHODH com orotato
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
Figura 4 - Espectros de CD da LmDHODH e LmDHODH na presença de orotato a 200C. A linha sólida
representa o espectro de CD da LmDHODH e a linha tracejada representa o espectro da LmDHODH com orotato, ambas à concentração de 5,8 µM em tampão 50 mM Na2HPO4, pH 7.2.
Os espectros de CD da LmDHODH na ausência e na presença de orotato em
função da variação de temperatura são mostrados nas Figuras 5 e 6,
respectivamente. Quando a temperatura aumenta gradativamente de 20 para 800C,
a mudança no espectro de CD da LmDHODH, com e sem orotato, revela uma
diminuição na elipticidade e a presença de um ponto isodicróico em 203 nm,
apontando ambos, para uma perda cooperativa da estrutura em α− hélice da
LmDHODH e para uma transição de dois estados (nativo e desnaturado). Além
disso, a desnaturação térmica da enzima LmDHODH é um processo de
desenovelamento irreversível, pois a amostra aquecida até 800C não retornou ao
seu estado original depois de ser resfriada rapidamente para 200C (Figura 5 e 6).

177
200 210 220 230 240 250-15
-10
-5
0
5
10
15
200C 200C volta 240C 280C
320C 360C 400C 440C 480C 520C
560C 600C 640C 680C 720C 760C
800C
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
Figura 5 - Espectros de CD, para diferentes valores de temperatura, da proteína LmDHODH à
concentração de 5,8 µµµµM.
200 210 220 230 240 250
-15
-10
-5
0
5
10
15
200C 200C volta
240C 280C
320C 360C 400C
440C 480C
520C 560C 600C
640C 680C
720C 760C 800C
[θ] (
103 de
g. c
m2 . d
mol
-1)
Comprimento de onda (nm)
Figura 6- Espectros de CD, para diferentes valores de temperatura, da proteína LmDHODH (5,8 µµµµM) na
presença de um excesso de orotato.
Resultados similiares são observados quando a elipticidade de ambas as
amostras é monitorada em 220 nm (Figura 7), comprimento de onda considerado um
indicativo prático de estrutura secundária da proteína (20).A desnaturação térmica
da LmDHODH exibe um comportamento do tipo sigmoidal (cooperativo), que
começa em 400C e é completado próximo a 540C, para LmDHODH, e se inicia em
400C e se completa em 580C para a enzima na presença de um excesso de orotato.

178
20 30 40 50 60 70 80-12
-10
-8
-6
-4
-2
0
θ22
0(103 de
g. c
m2 . d
mol
-1)
Temperatura (0C)
20 30 40 50 60 70 80
-16
-14
-12
-10
-8
-6
-4
-2
0
θ2
20(1
03 deg.
cm
2 . dm
ol-1)
Temperatura (0C)
(A) (B)
Figura 7- Transição da desnaturação térmica com a elipticidade monitorada a 220 nm. (A): LmDHODH. (B): LmDHODH na presença de orotato. A diminuição do módulo da elipticidade está associada com a perda de estrutura secundária. As linhas sólidas são ajustes obtidos com funções do tipo sigmóide.
Portanto, a desnaturação térmica da LmDHODH na presença e ausência de
orotato pode ser descrita como um processo irreversível de dois estados, então
tratado como descrito na seção 4.4.4.B. da tese. Os valores da elipticidade foram
transformados em frações de proteína nativa, de acordo com a Eq. 6 e a variação
com a temperatura da fN é apresentada na Figura 8. Esses valores foram então
usados para construir os gráficos de ln[ln(1/fN)] versus 1/T, que foram ajustados para
uma função linear (Eq. 7) pelo método dos mínimos quadrados. Os ajustes
resultaram em uma boa relação linear (dados não mostrados) e, a partir da
inclinação desses gráficos, a energia de ativação média foi calculada para
LmDHODH (257± 9 kJ/mol) e para LmDHODH na presença do orotato (212± 6
kJ/mol).
20 30 40 50 60 70 80
0,0
0,2
0,4
0,6
0,8
1,0
LmDHODH LmDHODH com orotato
FN
Temperatura (0C)
Figura 8- Curva de fração nativa em função da temperatura. A fração nativa de proteína (fN) foi calculada
como indicado na seção 4.4.4.B. da tese.

179
Adicionalmente, a partir do coeficiente linear dos ajustes, determinamos a
temperatura média de transição (Tm) que apresenta pequena variação para maior
temperatura quando o orotato está ligado à enzima (Tm=323,2 K para LmDHODH e
Tm=325,5 K para LmDHODH na presença de orotato em excesso).
Os dois parâmetros, energia de ativação e temperatura de transição, indicam
que a ocupação dos sítios ligantes na estrutura da LmDHODH pelo orotato confere
ao complexo enzima-orotato uma estabilidade maior do que aquela observada
apenas para a LmDHODH. Isto é compatível com uma regulação enzimática do tipo
“feedback”, muito comum em cadeias de reações enzimática como aquela em que
as DHODHs estão inseridas. Podemos aventar a hipótese de que a LmDHODH
permaneça em sua forma inativa (com o orotato ligado) até que a concentração
celular de tal ligante seja diminuída a níveis que promovam o deslocamento do
equilíbrio químico de ligação, levando à liberação do orotato em solução e,
conseqüentemente, à ativação da enzima. Assim, o mecanismo de regulação não
pode estar baseado na existência de um complexo enzima-orotato muito mais
estável do que a enzima, já que o complexo tem que ser desfeito quando o
organismo precisar que a via de novo de síntese de nucleotídeos seja ativada.
4 CONCLUSÕES
Na tentativa de caracterizar a enzima LmDHODH, fizemos medidas de
dicroísmo circular com variação de temperatura da enzima isolada e da enzima na
presença de orotato, nas quais este último foi adicionado em concentração tal que
inibisse a atividade enzimática. Em ambos os casos, observamos que a partir de 40
°C o espectro começa a perder o mínimo em 220 nm característico de estrutura em
α-hélice, que desaparece completamente em temperaturas superiores a 54 °C, para
a enzima isolada e 58 °C para a enzima na presença de orotato. Observamos ainda,
que no final da varredura, ao resfriarmos o sistema novamente para 200C obtivemos
espectros diferentes daqueles obtidos a 20oC no início do processo, indicando assim
um processo de desenovelamento irreversível. Assumindo o processo de
desnaturação como irreversível de dois estados foi possível determinar o valor da
energia de ativação para LmDHODH (256± 9 kJ/mol) e para LmDHODH na presença

180
de orotato (212± 6 kJ/mol). Em adição, determinamos a temperatura média de
transição (Tm) para a enzima isolada, Tm=323,2 K, e para a enzima na presença de
orotato, Tm=325,5 K, indicando que a ligação do orotato confere uma estabilidade
térmica um pouco maior para a enzima.
Podemos, entretanto, especular um pouco em termos do papel do orotato
como promotor da estabilidade do complexo enzima+orotato, o que teria influência
significativa no processo de ativação/desativação da enzima quando em condições
celulares.

181
Referências
1 FAIRBANKS, L.D. et al. Importance of Ribonucleotide availability to proliferating T-lynphocytes from healthy humans. Journal of Biological Chemistry, v. 270, n.50, p. 29682-29689, 1995. 2 STRYER, L. Biochemistry. 4th Edition. New York: WH Freeman and Company, 1995. 3 BALDWIN, J. et al. Malarial Dihydroorotate dehydrogenase. Journal of Biological Chemistry, v. 44, n. 277, p. 41827-41834, 2002. 4 ROWLAND, P. et al. The crystal structure of Lactococcus lactis dihydroorotate dehydrogenase. A complexed with the enzyme reaction product throws light on its enzymatic function. Protein Science, v. 7, n.6, p. 1269-1279, 1998. 5 KNECHT, W.; HENSELING, J.; LOFFLER, M. Kinetics of inhibition of human and rat dihydroorotate dehydrogenase by atovaquone, lawsone derivatives, brequinar sodium and polyporic acid. Chemico-Biological Interactions, v. 124, n.1, p. 61-76, 2000. 6 LIU, S. et al. Structures of human dihydroorotate dehydrogenase in complex with antiploriferative agents. Structure, v. 8, n.1, p. 25-33, 2000. 7 NORAGER, S.; JENSEN, K. F.; BJÖMBERG, O.; LARSEN, S. E. Coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenase. Structure, v. 10, n.9, p. 1211-1223, 2002. 8 NAGY, M.; LACROUTE, F.; THOMAS, D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proceedings of the National Academy of Science of the United States of America, v. 89, n.19, p. 8966-8970, 1992. 9 JONES, M.E. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annual Review of Biochemistry, v. 49, p. 253-279, 1980. 10 TAKASHIMA, E. et al. Characterization of the dihydroorotate dehydrogenase as a soluble fumarate reductase in Trypanosoma cruzi. Molecular and Biochemical Parasitology, v. 122, n.2, p. 189-200, 2002. 11 INAOKA, D. K. et al. Crystal structure of native Trypanosoma cruzi dihydroorotate dehydrogenase. Disponível em: <http://www.rcsb.org/pdb/home/home.doI>. Acesso em: outubro/2005. 12 PINHEIRO, M. P. Estudos cristalográficos da enzima diidroorotato desidrogenase de Trypanosoma cruzi.2008.106p. Dissertação (Mestrado em Farmácia) – Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, Ribeirão Preto, 2008. 13 PINHEIRO, M. P.; IULEK, J.; NONATO, M. C. Crystal structure of T. cruzi DHODH from Y strain. Biochemical Biophysical Research Communications, v. 369, n. 3, p. 812-817, 2008.

182
14 ROWLAND, P. et al. The crystal structure of the flavin containing enzyme dihydroorotate dehydrogenase A from Lactococcus lactis. Structure, v. 5, n.2, p. 239-252, 1997. 15 MCHAOURAB, H.S. et al. Conformation of T4 Lysozyme in solution. hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry, v. 36, n.2, p. 307-316, 1997. 16 ROWLAND, P. et al. Crystalization and preliminary X-ray studies of membrane- associated Escherichia coli dihydroorotate dehydrogenase. Acta Crystallographica D, v.56, n.5, p. 659-661, 2000. 17 CORPET, S. Multiple sequence alignment with hierarchical clustering. Disponível em: <http://bioinfo.genopole-toulouse.prd.fr/multalin/multalin.html>. Acesso em: março/2005. 18 SAMBROOK, J.; FRITSCH, E. F.; MANIATIS, T. Molecular cloning:. a laboratory manual. 2nd . ed. New York: Cold Spring Harber Laboratory Press, U.S.A., 1989. Appendix A, pp. A.1 19 WOODY, R.; BEROVA, N.; NAKANISHI, K. Circular dichroism: principles and applications. New York: VCH Publishers, 1994. 570 p. 20 CHEN, Y. H.; YANG, J. T.; MARTINEZ, H. M. Determination of secondary structures of proteins by circular-dichroism and optical rotatory dispersion. Biochemistry, v. 11, n.22, p. 4120-4131, 1972.





























