
UNIVERSIDADE DE MOGI DAS CRUZES
TATIANE FAUSTINO DE MORAES
ESTUDOS TEÓRICOS DA INFLUÊNCIA DA QUIRALIDADE EM
REAÇÕES DE HIDRÓLISE DE COMPOSTOS PALADACICLOS
QUE APRESENTAM ENANTIOSELETIVIDADE EM ATIVIDADES
BIOLÓGICAS
MOGI DAS CRUZES, SP
2008

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

UNIVERSIDADE DE MOGI DAS CRUZES
TATIANE FAUSTINO DE MORAES
ESTUDOS TEÓRICOS DA INFLUÊNCIA DA QUIRALIDADE EM
REAÇÕES DE HIDRÓLISE DE COMPOSTOS PALADACICLOS
QUE APRESENTAM ENANTIOSELETIVIDADE EM ATIVIDADES
BIOLÓGICAS
Dissertação apresentada à
Universidade de Mogi das Cruzes
para a obtenção do Título de
Mestre em Biotecnologia
Área de Concentração: Ciências
Exatas
Orientadora: Profa. Dra. Kaline Rabelo Coutinho
Co-orientador: Prof. Dr. Cláudio Saburo Shida
MOGI DAS CRUZES, SP
2008




Dedico este trabalho primeiramente a
Deus, que nos deu a vida e permite que
façamos dela nossas grandes obras.
Dedico também a meus pais que
juntamente com Deus, me deram à vida e
a razão de viver.

AGRADECIMENTOS
Não posso deixar de aproveitar este espaço e agradecer àqueles que
contribuíram de uma forma ou de outra para a realização desse trabalho, pois,
como diz o slogan de um banco “é impossível chegar lá sozinha”. Assim
gostaria de agradecer:
A professora Kaline Coutinho pela orientação, confiança, amizade,
paciência e oportunidades. À sua maneira, ela soube me incentivar e me
animar nos momentos de dificuldades. Suas críticas e questionamentos
foram fundamentais para o desenvolvimento deste trabalho e para minha
formação como pesquisadora. E a tudo isso eu espero fazer jus. Ao professor Antônio Carlos Fávero Caires pelas boas discussões e por
todo o apoio concedido principalmente para a finalização deste trabalho. Ao professor Sylvio Canuto pela colaboração neste trabalho e em diversos
outros. Ao professor Cláudio Saburo Shida por ceder espaço para utilização do
cluster na realização dos cálculos quânticos e das simulações
computacionais. Aos meus professores da graduação, pela minha formação e que hoje se
tornaram meus colegas de trabalho. Ás pessoas mais importantes da minha vida: meus pais e minha irmã. Por
todo carinho, amor, compreensão, apoio e dedicação incondicional em
todos os momentos. Aos amigos pelo companheirismo, discussões e diversões. Agradeço
especialmente a Fernanda Rezende pelos finais de semana de estudos e
pelos bons e maus momentos que passamos juntas, ao meu grande casal
de amigos Clélia Vieira e Paulo César que sempre tiveram ao meu lado
me apoiando e me incentivando e ao grupo de trabalho da física e biofísica
da USP, em especial ao Herbert de Castro, Cíntia Vequi e Thaciana
Valentina que em nenhum momento mediram esforços para me ajudar. A Universidade de Mogi das Cruzes - UMC pela oportunidade de
realização do curso de Mestrado em Biotecnologia e pelo apoio financeiro. E finalmente a Cleber Leite pelo companheirismo, pela cumplicidade, pela
amizade e pelo amor.

RESUMO
Neste trabalho estudamos as possíveis diferenças nas propriedades conformacionais e eletrônicas de moléculas simplificadas que serviram de modelo para os enantiômeros S e R dos complexos [Pd(C2, N-dmpa)(dppe)]Cl em processos de hidrólise. Essas possíveis diferenças devido a quiralidade podem desempenhar um importante papel no mecanismo de ação anti-tumoral desses complexos, uma vez que a enantioseletividade foi observada experimentalmente e o enantiômero S mostrou-se ativo enquanto que o R inativo. Portanto, o objetivo geral deste trabalho foi conseguir fornecer informações de estudos teóricos, utilizando cálculos quânticos e simulações computacionais, para auxiliar na elucidação da enantioseletividade dos mesmos. Primeiramente, desenvolvemos um estudo dos orbitais moleculares HOMO e LUMO dessas moléculas e foi possível concluir que as quatro ligações do paládio são as mais susceptíveis a um processo de hidrólise. Dessa forma, investigamos duas possibilidades para a quebra de cada uma das 4 ligações: quando o próton da água se liga ao paládio e a hidroxila se liga ao outro átomo da ligação; e quando a hidroxila se liga ao paládio e o próton se liga ao outro átomo da ligação. De forma comparativa, estudamos a precisão e o custo computacional dos métodos ab initio (HF) e o funcional de densidade híbrido com o potencial de troca-correlação (B3LYP) e com diferentes funções de base através de cálculos da variação da entalpia de hidrólise com o objetivo de melhorar a descrição teórica do sistema estudado. E assim foi possível desenvolver, através de cálculos quânticos e simulações computacionais, um estudo completo dos valores da variação da entalpia de 8 rotas diferentes de hidrólise produzindo 16 complexos hidrolisados. Baseados nos resultados que obtivemos, concluímos que a maior diferença da variação da entalpia de hidrólise foi na quebra que envolve a ligação Pd-N e essa diferença foi de aproximadamente 29 kcal/mol, e favor do enantiômero S. Portanto podemos concluir que essa rota de hidrólise (quebrando a ligação Pd-N) apresenta uma diferença significativa das interações desses agentes anti-tumorais com o meio solvente. Acreditamos que essa diferença das interações dos mesmos com meio aquosa pode ser justificada pela localização do átomo de hidrogênio presente no carbono quiral do enantiômero S, que favorece a sua interação com as moléculas de água. Para trabalhos futuros, pretendemos verificar a influência do pH nas rotas de hidrólises estudadas e também realizar cálculos de variação de energia livre para esse processo.

ABSTRACT
In this work we have studied the differences in the conformational and electronic properties of molecules that were proposed as models of the S and R enantiomers of the complex [Pd(C2, N-dmpa)(dppe)]Cl in hydrolysis processes. Due to the chirality these possible differences may play an important role in the antitumoral action mechanism of these complexes, since it was experimentally observed an enantioselectivity and the S enantiomer showed to be an active antitumoral drug whereas the R was inactive. Therefore, the general objective of this work was to supply theoretical information, obtained from quantum calculation and computer simulations, to try to explain the enantioselectivity of these complexes. First, a study of the molecular HOMO and LUMO orbitals of those molecules was made and it was possible to conclude that the four bonds of the palladium are the most susceptible to hydrolysis process. Then, two possibilities of breaking each one of the four bonds were investigated: at first when the proton of the water bonds to the palladium and to hydroxyl bonds to the other atom of the breaking bond; and secondly when to hydroxyl bonds to the palladium and the proton bonds to the other atom of the breaking bond. In a comparative way the precision and the computational cost of the approach of the ab initio method (HF) and the hybrid density functional method with one change-correlation potential (B3LYP) was studied with different basis set through calculations of the hydrolysis variation of enthalpy with the objective of improving the theoretical description of the studied system. Quantum calculations and computer simulations to study the variation of enthalpy of 8 different routes of hydrolysis that produce a total of 16 difference hydrolyzed complexes we used. Based on the results, we have concluded that the larger difference of the enthalpy variation in the hydrolysis process between the enantiomers was in the Pd-N bond breaking. The difference was approximately 29 kcal/mol, favoring the S enantiomer. Therefore, we were able to conclude that this route of hydrolyze (breaking the Pd-N bond) presents a significant difference of the interactions of these antitumoral agents with the solvent environment. We believe that this difference between their interactions with aqueous solution can be due to the location of the hydrogen atom in the chiral carbon of the S enantiomer that favors its interaction with the molecules of water. For future works, we intend to verify the influence of the pH in the studied routes of hydrolysis and also perform the calculation of the variation of free energy in this process.

SUMÁRIO 1 – INTRODUÇÃO ............................................................................................ 15
2 - REVISÃO BIBLIOGRÁFICA ....................................................................... 20
2.1 Complexos a Base de Platina: História de Drogas Antitumorais ............ 20
2.2 Complexos a Base de Paládio: Nova Perspectiva para a Obtenção de
Drogas Antitumorais. ........................................................................................ 23
2.3 Estudos Teóricos de Complexos a Base de Paládio e Platina ............... 27
3 – MÉTODO .................................................................................................... 32
3.1 Introdução .............................................................................................. 32
3.2 Sistemas Estudados .............................................................................. 33
3.3 Cálculos Quânticos ................................................................................ 34
3.3.1 Estudos das Moléculas Isoladas ................................................. 34
3.3.2 Estudos da Entalpia da Reação de Hidrólise .............................. 37
3.4 Simulação Computacional ...................................................................... 40
3.4.1 Estudos das Moléculas em Meio Solvente .................................. 40
3.4.2 Estudos da Entalpia da Reação de Hidrólise em Meio Solvente . 43
4 – RESULTADOS E DISCUSSÕES ............................................................... 46
4.1 Enantiômero Isolado .............................................................................. 46
4.2 Reações de Hidrólise ............................................................................. 48
4.3 Análise das Rotas de Hidrólise em Fase Gasosa ....................................... 51
4.3.1 Quebra da Ligação Pd-P(19) ............................................................. 51
4.3.2 Quebra da Ligação Pd-P(12) ............................................................. 54
4.3.3 Quebra da Ligação Pd-N(20) ............................................................. 56
4.3.4 Quebra da Ligação Pd-C(6) .............................................................. 59
4.3 Sumário e Discussões das Rotas de Hidrólise em Fase Gasosa .......... 61
4.5 Análise das Rotas de Hidrólise em Meio Aquoso ................................... 65
4.5.1 Análise da Energia de Relaxação do Solvente ................................ 66
4.5.2 Análise da Energia de Interação Soluto-Solvente ............................ 69
4.5.3 Análise da Variação de Entalpia de Hidrólise em Solução ............... 71
5 - CONCLUSÕES E SUGESTÕES ................................................................. 75
REFERÊNCIAS ................................................................................................ 79
APÊNDICE ...................................................................................................... 88

LISTA DE ILUSTRAÇÕES
Figura 1-1: Representação esquemática da identificação da nomenclatura
(R/S) retirada da referência (5). ........................................................................ 16
Figura 1-2: Representação esquemática de fármacos com centros quirais (7).
O asterisco identifica o carbono quiral. ............................................................. 17
Figura 1-3: Representação esquemática dos enantiômeros: [Pd(S(-),C2,N-
dmpa)(dppe)]Cl e [Pd(R(+),C2,N-dmpa)(dppe)]Cl que apresentam
enantioseletividade. Ph representa, por simplificação, o grupo fenil................. 18
Figura 2-1: Representação esquemática de complexos de coordenação
inorgânica que apresentam atividade antitumoral............................................. 20
Figura 2-2: Representação esquemática da Oxalinplatina ........................... 21
Figura 2-3: Novos complexos a base de platina com atividade antitumoral. 22
Figura 2-4: Representação esquemática da formação do anel ciclometalada.
Figura retirada da referência (42). .................................................................... 24
Figura 2-5: Representação esquemática do complexo ciclopaladado [Pd2(C2,
N-S(-)dmpa)2(-dppe)Cl2] (9). ........................................................................... 25
Figura 2-6: Representação esquemática dos complexos ciclopaladados
[Pd2(C2, N-S(-)dmpa)2(-dppf)Cl2] (43). ............................................................ 26
Figura 2-7: Representação esquemática íon paladaciclo [Pd(C2,N-S(-)dmpa)
(dppf)]Cl (44). .................................................................................................... 27
Figura 2-8: Representação esquemática dos complexos estudados por
Zeizinger e colaboradores (68). ........................................................................ 29
Figura 2-9: Representação esquemática do mecanismo de substituição
estudada por Jaroslav e colaboradores. X, Y, Z e V são grupos que podem ser
átomos de Cl ou NH3. Figura retirada da referência (70). ................................. 30
Figura 3-1: Representação esquemática das moléculas modelo, na forma
iônica, que foram estudadas neste trabalho. .................................................... 33
Figura 3-2: Representação esquemática das rotas de hidrólise que foram
estudadas neste trabalho. O composto (a) foi chamado de IIR(PdH) e IIS(PdH),
dependendo da quiralidade; (b) IIIR(PdOH) e IIIS(PdOH); (c) IVR(PdH) e
IVS(PdH); (d) VR(PdOH) e VS(PdOH); (e) VIR(PdH) e VIS(PdH); (f)

VIIR(PdOH) e VIIS(PdOH); (g) VIIIR(PdH) e VIIIS(PdH); (h) IXR(PdOH) e
IXS(PdOH). ....................................................................................................... 35
Figura 3-3: Geometrias otimizadas para os enantiômeros: (a) IR (b) IS. As
circunferências indicam o centro quiral. ............................................................ 36
Figura 3-4: Ilustração do processo reativo de hidrólise utilizado no cálculo da
variação de entalpia de hidrólise em solução através de simulações
computacionais. ................................................................................................ 41
Figura 4-1: Geometria otimizada e definição da numeração dos átomos dos
enantiômeros: (a) IR e (b) IS............................................................................. 47
Figura 4-2: Orbitais LUMO dos enantiômeros: (a) IR (b) IS ............................ 48
Figura 4-3: Geometria da molécula (VIIS(PdOH)) formada na quebra da ligação
Pd-N(20) do enantiômero IS, seguindo a rota (f) da Figura 3-2. ......................... 50
Figura 4-4: Valores descritos na Tabela 4-1, da variação da entalpia de
hidrólise do enantiômero IS na rota (f) da Figura 3-2 . Círculo aberto é o valor
da entalpia obtido pelo método HF e os círculos fechados são valores obtidos
pelo método B3LYP. ......................................................................................... 51
Figura 4-5: Geometrias das moléculas formadas na quebra da ligação Pd-P(19)
do enantiômero IR. Em destaque estão as localizações da hidroxila depois do
processo de hidrólise. ....................................................................................... 52
Figura 4-6: Geometrias das moléculas formadas na quebra da ligação Pd-P(19)
do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 53
Figura 4-7: Geometrias das moléculas formadas na quebra da ligação Pd-P(12)
do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 55
Figura 4-8: Geometrias das moléculas formadas na quebra da ligação Pd-P(12)
do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 56
Figura 4-9: Geometrias das moléculas formadas na quebra da ligação Pd-N(20)
do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 57
Figura 4-10: Geometrias das moléculas formadas na quebra da ligação Pd-N(20)
do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 57

Figura 4-11: Geometrias das moléculas formadas na quebra da ligação Pd-N(20)
do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila
depois da hidrólise. A molécula (a) possui uma variação da entalpia de hidrólise
de 73.0 kcal/mol. A molécula (b) possui uma variação da entalpia de hidrólise
de 68.0 kcal/mol. ............................................................................................... 58
Figura 4-12: Geometrias das moléculas formadas na quebra da ligação Pd-C(6)
do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 60
Figura 4-13: Geometrias das moléculas formadas na quebra da ligação Pd-C(6)
do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila
depois do processo de hidrólise. ....................................................................... 60
Figura 4-14: Geometrias das moléculas formadas na quebra da ligação Pd-C(6)
do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila
depois da hidrólise. A molécula (a) possui uma variação da entalpia de hidrólise
de 1.0 kcal/mol. A molécula (b) possui uma variação da entalpia de hidrólise de
-5.7kcal/mol....................................................................................................... 61
Figura 4-15: Variações da energia potencial de interação solvente-solvente em
torno do seu valor médio, em função de ciclos MC. ......................................... 67
Figura 4-16: Gráfico da convergência da variação da energia potencial de
interação soluto-solvente ( WIU ) e da energia potencial de interação solvente
– solvente ( WWU ). ......................................................................................... 68
Figura 4-17: Energia potencial de interação soluto-solvente dos complexos
hidrolisados IR e IS respectivamente, em função dos momentos de dipolo. .... 70

LISTA DE TABELAS
Tabela 4-1: Valores das distâncias de ligações (em Angstron) que envolvem o
átomo de paládio. ............................................................................................. 47
Tabela 4-2: Comparação de métodos/função base para cálculo da variação da
entalpia de hidrólise em fase gasosa (em kcal/mol) (Informar a máquina
utilizada) ........................................................................................................... 50
Tabela 4-3: Valores calculados para variação da entalpia de hidrólise (HF/6-
31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação
Pd – P(19). Rotas (a) e (b) da Figura 3-2. ........................................................... 53
Tabela 4-4: Valores calculados para a variação de entalpia de hidrólise (HF/6-
31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação
Pd – P(12). Rotas (c) e (d) da Figura 3-2. ........................................................... 55
Tabela 4-5: Valores calculados para a variação de entalpia de hidrólise (HF/6-
31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação
Pd –N(20). Rotas (e) e (f) da Figura 3-2. ............................................................ 58
Tabela 4-6: Valores calculados para a variação de entalpia de hidr’ólise (HF/6-
31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação
Pd – C(6). Rotas (g) e (f) da Figura 3-2. ............................................................. 60
Tabela 4-7: Sumário dos valores calculados da variação da entalpia de
hidrólise (HF/6-31G), em fase gasosa para as diferentes rotas mostradas na
Figura 3-2. ........................................................................................................ 63
Tabela 4-8: Sumário dos valores calculados da variação da entalpia de
hidrólise (B3LYP/ 6-31+G(d)), em fase gasosa para as diferentes rotas
mostradas na Figura 3-2. .................................................................................. 64
Tabela 4-9: Valores calculados para a variação da energia potencial de
interação soluto-solvente ( WIU ), e a variação da energia potencial de
interação solvente-solvente ( WWU ). .......................................................... 67
Tabela 4-10: Valores calculados para energia potencial de interação soluto-
solvente dos complexos hidrolisados embebidos em 800 moléculas de água. 70
Tabela 4-11: Valores calculados para a variação da entalpia de hidrólise dos
complexos hidrolisados em solução. A variação da entalpia de hidrólise das
moléculas em fase gasosa foi calculada pelo método HF/6-31G. .................... 72

Tabela 4-12: Valores calculados para a variação da entalpia de hidrólise dos
complexos hidrolisados. A variação da entalpia de hidrólise das moléculas em
fase gasosa foi calculada pelo método B3LYP/ 6-31+G(d)............................... 73

15
1 – INTRODUÇÃO
Atualmente a medicina dispõe de alguns tratamentos com o objetivo de
combater o câncer. O avanço científico alcançado pela quimioterapia tem
propiciado uma estatística crescente no tratamento e na cura de muitos tipos de
câncer [1]. Esse campo de investigação, em constante expansão, que
compreende o entendimento da ação biológica de novas drogas sobre células
normais e cancerígenas, trouxe uma melhoria significativa nas terapias contra o
câncer. A quimioterapia, juntamente com o uso de radiação, é uma das
técnicas mais efetivas no combate ao câncer.
Aproximadamente 50% das drogas, em geral, atualmente
comercializadas são quirais [2], ou seja, tem em sua estrutura um ou mais
átomos de carbono saturado (hibridização sp3), que apresenta quatro radicais
diferentes ligados a ele. Uma molécula com carbono desse tipo pode existir em
dois arranjos espaciais diferentes, que são estereoisômeros um do outro. Esses
tipos de estereoisômeros são chamados de enantiômeros.
Os enantiômeros apresentam a maioria de suas propriedades físicas
idênticas e, quimicamente, eles demonstram comportamentos diferentes. Essa
característica é de extrema importância biológica, uma vez que a maioria das
moléculas que participam de processos bioquímicos, como proteínas de
membranas e enzimas, também são compostos quirais [3].
Se nomearmos dois enantiômeros usando apenas o sistema de
nomenclatura IUPAC (União Internacional de Química Pura e Aplicada), eles
terão o mesmo nome. Portanto com o objetivo de diferenciar esses compostos,
Cahn e colaboradores [4] desenvolveram um método para especificar o
enantiômero em torno do seu centro quiral. Esse método chamado de (R/S) é
amplamente utilizado e faz parte das regras da IUPAC.
Para a aplicação desse método, primeiramente determina-se uma
prioridade (a à d) para os átomos ou grupos diretamente ligados ao centro
quiral. Essa prioridade é definida pelo grau de complexidade do átomo ou grupo
que está ligado ao carbono quiral. Ao grupo com menor complexidade é
atribuído a prioridade mais baixa d.

16
A molécula pode ser considerada como um modelo físico tridimensional
a qual pode ser desenhada ou, ainda visualizada mentalmente com uma
representação tridimensional de tal modo que o grupo de menor prioridade d
seja posicionado do lado oposto ao do observador como mostra Figura 1-1. O
arranjo dos grupos remanescentes é então considerado para especificar a
configuração R ou S. Um círculo imaginário é traçada partindo-se do grupo de
maior prioridade (a) para o de segunda prioridade e finalmente para o grupo de
terceira prioridade. Se o caminho percorrido for no sentido horário, a
configuração é R e se for no sentido anti-horário a configuração é S [5].
Figura 1-1: Representação esquemática da identificação da nomenclatura (R/S) retirada da referência [5].
Existe também uma outra nomenclatura atribuída aos enantiômeros e
que esta relacioanda à sua capacidade de desviar a luz plano polarizada para a
direita (D ou +) e para a esquerda (L ou -). Esta propriedade que enantiômeros
podem apresentar, depende não somente do arranjo espacial dos seus átomos
ou grupos, mas também do meio em que eles se encontram. Um exemplo são
os enantiômeros do anticoagulantes varfarina, onde o enantiômero S apresenta
rotação positiva (D) e o R rotação negativa (L) em n-hexano-2-propanol,
enquanto que em n-heptano-acetato de etila o desvio é negativo para o
enantiômero S e positivo para R [6]. Dessa forma, pode-se perceber que as
denominações R-S e D-L são independentes e, portanto podemos ter
enantiômeros S(+) ou S(-) e da mesma maneira o enantiômero R(+) ou R(-).
Na Figura 1-2 apresentamos alguns exemplos de substâncias vendidas
nas farmácias como fármacos. Todos os fármacos mencionados nessa figura
são quirais. É sabido que qualquer modificação da orientação espacial dos

17
substituintes ao redor do centro quiral pode mudar completamente o efeito
biológico de um fármaco em nosso corpo. A esse efeito se dá o nome de
enantioseletividade.
Um exemplo da enantioseletividade, muito conhecido e que teve efeitos
muito graves, é o da talidomida (Figura 1-2a) que é um sedativo leve e pode ser
utilizado no tratamento de náuseas, muito comum no período inicial da
gravidez. Quando foi lançado, no final da década de 50, era considerado
seguro para o uso de grávidas, sendo administrado com uma mistura
equivalente dos dois enantiômeros. Entretanto, não se sabia na época é que
um dos enantiômeros apresenta uma atividade teratogênica, ou seja, leva à má
formação congênita, afetando principalmente o desenvolvimento normal dos
braços e pernas de bebês. O uso indiscriminado desse fármaco levou ao
nascimento de milhares de bebês com graves defeitos físicos [7].
Figura 1-2: Representação esquemática de fármacos com centros quirais [8]. O asterisco identifica o carbono quiral.
Esse lamentável acontecimento despertou a atenção da comunidade
científica e das autoridades farmacêuticas sobre a importância da quiralidade
na atividade biológica.
Neste trabalho, utilizamos técnicas de modelagem molecular para
estudar compostos quirais paladaciclos que apresentam enantioseletividade na
atividade antitumoral. De acordo com resultados obtidos por Caires [9] os
Talidomida (a)
C*
N
O
O N
O
O
H
Ketamida (b)
Aspartame (c)
OOH
CH2
*C
NH2
O
NC*
O
O CH3
HHH
C*
N
CH3
H
O

18
enantiômeros do complexo organometálico [Pd(C2, N-dmpa)(dppe)]Cl onde
dmpa representa a N,N-dimetil-1-fenetilamina, e o dppe o ligante 1,2
bis(difenilfosfina)etano, apresentados na Figura 1-3, mostraram uma
significativa enantioseletividade no combate a células do melanoma. O
enantiômero [Pd(S(-) C2, N-dmpa)(dppe)]Cl (Sdmpa), apresenta uma atividade
antitumoral impedindo de forma significativa a formação de tumor in vitro e in
vivo; enquanto o enantiômero [Pd(R(+) C2, N-dmpa)(dppe)]Cl (Rdmpa),
mostrou-se inativo.
Figura 1-3: Representação esquemática dos enantiômeros: [Pd(S(-),C
2,N-dmpa)(dppe)]Cl e
[Pd(R(+),C2,N-dmpa)(dppe)]Cl que apresentam enantioseletividade. Ph representa, por
simplificação, o grupo fenil.
O trabalho de Rodrigues e colaboradores [10] discute, através das
análises dos efeitos biológicos desses complexos, que a atividade antitumoral
desses compostos apresenta claramente uma dependência com a estrutura do
anel ciclo-Pd. Além disso, destaca a importância do carbono quiral, o qual no
complexo Sdmpa uma interação intramolecular, como por exemplo, uma
ligação de hidrogênio poderia estabilizar a molécula e gerar uma forte atividade
antitumoral in vivo. Outro resultado mencionado nesse trabalho é que o
complexo Sdmpa afeta, muito ativamente, o metabolismo respiratório das
células tumorais, causando um colapso no gradiente de próton da cadeia
respiratória da mitocôndria, gerando uma diminuição na taxa de acidificação
extracelular. Como conseqüência dessas observações experimentais,
concluímos que a estrutura molecular desses complexos parece ter um
importante papel no mecanismo de ação biológica e adicionalmente acredita-se
que ao serem expostos ao ambiente biológico, eles sofrem um processo de
hidrólise.
Pd
NH
CH3 CH
3
CH3
P
P
Ph
Ph
Ph
Ph
+
*
Cl
Pd
NCH3
HCH
3
CH3
P
P
Ph
Ph
Ph
Ph
+
*
Cl
(a) (b)

19
Portanto este trabalho teve como objetivo central estudar através de
métodos teóricos as possíveis modificações estruturais desses complexos em
processos de hidrólise, que podem auxiliar na compreensão da atividade
biológica seletiva desses enantiômeros. Além disso, este trabalho teórico teve
também como objetivo, adicionar informações e servir como uma ferramenta de
apoio para interpretações de alguns resultados experimentais ainda pouco
elucidados.
Neste trabalho, estudamos primeiramente os compostos paladaciclos de
forma isolada utilizando cálculos quânticos. Determinamos que as 4 ligações do
paládio são suscetíveis as possíveis rotas de hidrólise e estudamos a quebra
dessas quatro ligações através de duas possibilidades:
O próton (H+) da água se ligando com o paládio (Pd) e a hidroxila
(OH-) se ligando ao restante da molécula, ou;
A hidroxila da água se ligando com o paládio e o próton se ligando
ao restante da molécula.
Dessa forma, estudamos 16 complexos hidrolisados isoladamente, com
o objetivo de identificar possíveis diferenças com relação à quiralidade presente
nos enantiômeros estudados. Em seguida, utilizando simulações
computacionais, incluímos o meio aquoso a fim de estudar os efeitos de
solvente no processo de hidrólise desses compostos.
No capítulo seguinte, discutimos recentes avanços que envolvem
complexos a base de paládio para a terapia do câncer e apresentamos uma
perspectiva de trabalhos teóricos que envolvem o metal paládio (capítulo 2). O
capítulo 3 é destinado à descrição dos procedimentos teóricos utilizados. No
capítulo 4 apresentamos e discutimos os resultados obtidos e por fim, no
capítulo 5 expomos nossas conclusões e perspectivas .

20
PtNH
3
NH3
O
O
C
C
O
O
2 - REVISÃO BIBLIOGRÁFICA
2.1 Complexos a Base de Platina: História de Drogas Antitumorais
Por um longo tempo, as drogas inorgânicas, especialmente as que
possuíam metais, não foram estudadas porque metais eram considerados
poderosos agentes cancerígenos [11]. Porém, em 1965, Rosenberg e Van
Camp e Krigas investigando os efeitos de campos elétricos em processos de
crescimento de colônias de bactérias [12], observaram uma poderosa atividade
antitumoral proveniente do composto de coordenação chamado cisplatina [13].
Essa descoberta originou novos estudos e perspectivas para uma nova classe
de agentes antitumorais chamados de complexos de coordenação inorgânicos.
O sucesso clínico do composto, descoberto por Rosenberg e colaboradores cis-
[diaminodicloroplatina II], cis – [PtCl2(NH3)2] denominado cisplatina (Figura 2-1
a) no combate de tumores em pacientes terminais [14] e depois no tratamento
de tumores localizados como de testículos [15] e de ovário [[16], bem como a
necessidade de diminuição da toxicidade renal em tratamentos clínicos,
motivaram o estudo desses complexos e outros similares [17] por várias
décadas.
Não obstante, o intenso trabalho realizado ao longo desses anos [18],
por pesquisadores do mundo inteiro, avançaram até a fase de testes clínicos só
alguns complexos semelhantes como é o caso da Carboplatina [19], (Figura 2-
1b), que recebeu aprovação do FDA (U.S. Food and Drug Administration) para
comercialização em grande escala.
Figura 2-1: Representação esquemática de complexos de coordenação inorgânica que apresentam atividade antitumoral.
Cisplatina (a) Carboplatina (b)
PtCl
Cl
NH3
NH3

21
A determinação estrutural dos sistemas obtidos pela interação entre
complexos de platina e oligonucleotídeos [20], aumentaram a compreensão
dos mecanismos biológicos desses compostos de coordenação. Os estudos
foram objetivamente focados na síntese de complexos de platina contendo
ligantes que, em hipótese, aumentariam a afinidade dos mesmos pelo DNA por
meio da interação desses complexos com o meio biológico [21],[22],[23]. Esse
é o caso da oxalinplatina (1R, 2R - diaminociclohexano)oxalato de platina II,
(Figura 2-2), que pertence a terceira geração de compostos antitumorais a base
de platina [24]. A combinação desse último complexo com o 5-fluorouracil, foi
recentemente aprovado na Europa, Ásia e América Latina para o tratamento de
câncer de cólon [24].
NH2
NH2
Pt
O
O
O
O
Figura 2-2: Representação esquemática da Oxalinplatina
O entusiasmo inicial causado pela descoberta de novos agentes
antitumorais foi, porém, diminuindo com a observação de que a cisplatina,
agente quimioterápico mais usado, apresentava vários efeitos tóxicos,
realçando a toxicidade renal, gastrointestinal e neurotoxicidade [25]. Por causa
desses efeitos tóxicos, o uso da cisplatina foi suspenso por um tempo. Foi
procurado então, o uso de técnicas, que viabilizassem o uso dessas drogas. A
toxicidade renal é reversível quando ministradas em pequenas doses, mas em
altas dosagens as lesões causadas a esse órgão são severas e irreversíveis
[26]. A toxicidade gastrointestinal manifesta-se através de náuseas e vômitos
intensos tornando difícil o manejo terapêutico. A neurotoxicidade manifesta-se
principalmente através da neuropatia periférica, embora seja geralmente de
pequena intensidade e raramente limita o uso da droga.
Atualmente, as pesquisas têm sido direcionadas para o desenvolvimento
de [27]:
(a) Melhores técnicas de administração de drogas, diminuindo a toxicidade,
sem interferir no efeito citotóxico;
(b) Melhor dosagem terapêutica para cada tipo de tumor;

22
(c) Melhores vias de administração; e
(d) Funcionamento simultâneo de associações terapêuticas.
Embora na maioria das combinações utilizadas, não se saiba a relação
entre a estrutura da substância química e sua atividade biológica, estudos com
novas estratégias foram desenvolvidos com resultados positivos. O mecanismo
biológico de cross-link de drogas a base de cisplatina com bases nitrogenadas
de moléculas de DNA são bem documentadas [20],[21],[22],[23],[24]. Foi
demonstrado que complexos de platina contendo ligantes planares como
[Pt(terpy)(HET)], são capazes de se inserir em dinucleotídeos, por diferentes
caminhos[28) , assim complexos derivados de pirimidina [29],[30] e aminas
[31] em configuração trans apresentam atividade comparável a cisplatina. No
complexo trans – [Pt (Cl2py2)], (Figura 2-3 (a)), o plano formado por moléculas
de piridina permite a inserção dos complexos entre as bases nitrogenadas das
moléculas de DNA. A ligação Pt – Cl perpendicular a esse plano é de certa
forma orientado para favorecer a interação entre a platina e o DNA. Na
geometria cis do mesmo complexo, cis – [Pt (Cl2py2)] apenas uma das
moléculas de piridina pode ser inserida de cada vez [32].
O mecanismo biológico dessas novas drogas ainda não é claro.
Acredita-se que a estrutura planar dos ligantes possa aumentar a
especificidade dos complexos por meio de seqüência de bases nitrogenadas
favorecendo diferentes formações com o DNA e até mesmo por reações com
outras biomoléculas, sendo um importante aspecto para o mecanismo
biológico.
Pt
COCH3
COCH3
NH3
NH2
Cl
Cl
Pt
COCH3
COCH3
NH3
NH2
Cl
Cl
Figura 2-3: Novos complexos a base de platina com atividade antitumoral.
Complexos de platina (IV) são mais solúveis em água que complexos
semelhantes à base de platina (II) e têm sido objeto de intensa investigação e
provavelmente serão usados na quimioterapia no futuro. A Satraplatina, (Figura
(b) (a)
Pt NN
Cl
Cl

23
2-3 b), é o primeiro complexo de platina ministrado oralmente, e apresenta uma
alta atividade biológica contra linhagens de câncer de próstata. Acredita-se que
os complexos a base de platina (IV) manifestem sua atividade biológica depois
de sofrerem redução à platina (II) dentro do organismo e também provoquem
lesões celulares através de interações com o DNA.
Com base no sucesso alcançado pelos complexos de coordenação de
platina como agentes antitumorais, outros estudos enfocando a química dos
complexos a base de paládio têm surgido no meio científico. Porém,
comportamentos químicos e biológicos diferentes e peculiares desses
complexos, quando comparados aos complexos de platina, tornam esses novos
compostos, uma das estratégias mais importantes para o design de novas
drogas antitumorais.
2.2 Complexos a Base de Paládio: Nova Perspectiva para a Obtenção de Drogas Antitumorais.
Inicialmente a grande maioria dos complexos metálicos antitumoras
sintetizados e caracterizados eram estruturalmente análogos à cisplatina [33].
Entretanto, devido a alta toxicidade e labilidade apresentada por esses
complexos, houve uma diminuição no interesse de novos compostos desse tipo
[34],[35]. Assim o trabalho desenvolvido por Khan e colaboradores [36], discute
que complexos de paládio possuem pouca ou nenhuma aplicação como drogas
antitumorais. Esse fato é devido à grande labilidade e a rápidas reações de
hidrólise de complexos a base de paládio no meio biológico quando comparado
aos complexos a base de platina [36]. No esforço de superar esse problema
esse mesmo autor e colaboradores usaram ligantes quelantes na síntese de
complexos a base de paládio. Os resultados demonstraram que o composto
[Pd (meth)(2-merpy)Cl]Cl tem uma significante citotoxidade, podendo agir como
um poderoso agente antitumoral [36]. Assim, trabalhos com outros complexos a
base de paládio começaram a ter outras perspectivas de uso, especialmente à
classe dos complexos ciclopaladados.
Na área médica e bioquímica, alguns trabalhos mencionam os
compostos ciclopaladados como efetivos agentes antitumorais [37], [38]

24
principalmente no combate as células leucêmicas e do câncer mamário . Esse
é o caso do composto contendo ligante N-(4-metoxifenil)- -
benzoilidenoanilina [39].
O trabalho realizado por Navarro-Ranninger e colaboradores [40], e
incluindo referências, demonstrou várias propriedades estruturais interessantes
acerca de compostos ciclometalados a base de platina e paládio na sua função
como agentes antitumorais.
O termo ciclometalado, introduzido por Trofimenko [41], descreve
reações nas quais ligantes orgânicos sofrem uma reação de metalação
intramolecular levando a formação de um anel quelato com uma coordenação
entre o metal M e um átomo doador Y dos grupos V (N, P, As) ou VI(O, S, Se) e
uma ligação covalente metal-carbono, sendo X um halogênio ou grupo alquil
como ilustra a Figura 2-4.
Figura 2-4: Representação esquemática da formação do anel ciclometalada. Figura retirada da referência [42].
Rodrigues e colaboradores [10], estudaram compostos paladaciclos
obtidos por agentes ciclometalantes N,N dimetil-1-fenetilamina (dmpa), fenil-2-
piridinilacetileno e 1-fenil-3-N,N dimetilaminapropeno, respectivamente e
contendo o ligante bifosfínico 1,2 bis (difenilfosfina) etano (dppe). Os complexos
foram testados “in vitro” e “in vivo” contra células de melanoma de ratos do tipo
B16F10-Nex2. Os três complexos foram inibitório “in vitro” em baixas
concentrações (<1,25M) e o complexo [Pd2(C2,N-S(-)dmpa)(-dppe)Cl2]
(Figura 2-5) foi o mais ativo “in vivo” retardando o crescimento do tumor e
prolongando a sobrevivência do animal. “In vitro” o complexo causou um
colapso na atividade respiratória com uma repentina diminuição da acidificação
extra celular em incubação curta (até 100 minutos) acompanhado por
degradação do DNA após 24 horas.

25
O complexo a base de paládio [Pd2(C2, N-S(-)dmpa)(-dppe)Cl2] foi
muito efetivo no combate a células do melanoma murino. Esses resultados
introduzem o complexo de paládio mencionado, como uma droga antitumoral
promissora com excelente especificidade estrutural e ativa “in vivo” e “in vitro”.
Em outro trabalho, envolvendo compostos paladaciclos antitumorais com
ligantes bifosfínicos, Bincoletto e colaboradores [43], descreveram importantes
propriedades exibidas pelos ciclopaladados quirais derivados da N,N dimetil-1-
fenetilamina e o ligante de coordenação 1,1 bis(difenilfosfina)ferroceno(dppf).
Esses complexos (Figura 2-6) foram sintetizados e estudados como inibidores
de catepsina B e como agentes antitumorais contra tumores sólidos.
Figura 2-5: Representação esquemática do complexo ciclopaladado [Pd2(C
2, N-S(-)dmpa)2(-
dppe)Cl2] [10].
O resultado revelou que o complexo de paládio [Pd2(C2, N-S(-)dmpa)2(-
dppf)Cl2] (2) pode inibir a atividade da catepsina B de uma forma reversível. A
aplicação desses complexos em tumores de Walker implantados em ratos,
resultou em 90% de inibição no crescimento de tumores. Estudos toxicológicos
usando camundongos tratados com uma grande dose desse complexo
(100mg/kg) não apresentaram nenhuma alteração na morfologia das células
sanguíneas. Resultados similares foram obtidos com tecidos hepáticos, tecidos
do rim e tecidos do baço.

26
Figura 2-6: Representação esquemática dos complexos ciclopaladados [Pd2(C
2, N-S(-
)dmpa)2(-dppf)Cl2] [43].
Os resultados obtidos por esse trabalho [43], mostra que complexos
ciclopaladados binucleados derivados do enantiômero S(-)dimetil-1-fenetilamina
contendo o ligante 1,1 bis(difenilfosfina)ferrocene é um promissor agente
antitumoral com toxicidade reduzida.
Outro resultado mencionado nesse trabalho, é a verificação do efeito
inibitório dos compostos paladaciclos na atividade da catepsina B. A catepsina
B está envolvida na proliferação de células endoteliais em meio de cultura e
angiogênese. É importante salientar que os efeitos inibidores do S(-)dimetil-1-
fenetilamina contendo o ligante 1,1 bis(difenilfosfina)ferrocene na atividade da
catepsina B foram verificados em tumores de Walker implatados em ratos,
sugerindo, portanto uma relação entre os efeitos antitumorais do S(-)dimetil-1-
fenetilamina com o efeito inibidor do complexo na atividade da catepsina B.
Em outra publicação, ainda envolvendo estudos de compostos
paladaciclos derivados do enantiômero S(-) do N,N dimetil-1-fenetilamina e do
bis (difenilfosfina) ferrocene, Barbosa e colaboradores [44] observaram que o
íon do complexo paladaciclo mononucleado [Pd(C2,N-S(-)dmpa)(dppf)]Cl,
Figura 2-7, tem uma notável ação biológica antitumoral, agindo por via
lisossomal. Além disso, estudos toxicológicos demonstraram que o complexo
paladaciclo apresenta baixo nível de toxicidade para tecidos normais, o que
sugere uma alta seletividade para células tumorais.

27
Figura 2-7:Representação esquemática íon paladaciclo [Pd(C2,N-S(-)dmpa) (dppf)]Cl [44].
2.3 Estudos Teóricos de Complexos a Base de Paládio e Platina
Com o desenvolvimento da química computacional nas últimas décadas,
a modelagem teórica de metais de transição está cada vez mais evoluída.
Alguns trabalhos de revisão discutem a importância da química teórica na
descrição de propriedades químicas e físicas de sistemas envolvendo metais
de transição [45],[46]. Cálculos de mecanismos de reações através da química
teórica também são bem documentados [47],[48]. Geralmente os métodos
utilizados para otimização de geometria e cálculos de energia de complexos a
base de metais de transição são métodos que envolvem Funcional de
Densidade (DFT) [49],[50],[51] Hartree-Fock (HF) [52] e teoria de perturbação
de Moller-Plesset de segunda ordem (MP2) [53].
O progresso proeminente na química teórica no estudo de metais de
transição estimulou os profissionais dessa área a estudar complexos de paládio
e platina [54]. Isso porque, esses dois metais podem facilmente interagir com
moléculas orgânicas e inorgânicas dando origem a interessantes complexos
[55], [56],[57],[58],[59]. A descoberta da atividade antitumoral na cisplatina é
um grande exemplo disso.
Foram publicados muitos estudos computacionais em cisplatina e
complexos relacionados, que elucidam sua interação com peptídeos e com
moléculas de DNA. O trabalho de Deubel [60] compara a afinidade de

28
interação da cisplatina com vários sítios contendo átomos de enxofre e
nitrogênio em moléculas de aminoácidos e bases purinas de DNA. Nesse
trabalho cálculos quânticos utilizando o método BP86 com um conjunto de base
grande (VTZP) foram usados. Os cálculos mostram que diferenças
consideráveis nas energias de ligação do complexo de cisplatina com
compostos N-heterocíclicos como 1-metilimidazol, 9-metiladenina, e 9-
metilguanina surgem de interações eletrostáticas ao invés de interações
envolvendo orbitais.
Um estudo comparativo entre a estrutura e o espectro vibracional das
moléculas de cisplatina e carboplatina foi desenvolvido por Wysokinski e
Michalska [61]. Oito modelos de DFT (G96LYP, G96PW91, mPWPW,
PW91PW91, mPW1PW, mPW1LYP, B3LYP [62] e B3PW91) , como também o
HF e MP2, foram estudados para determinar as estruturas moleculares,
freqüências vibracionais, intensidades no infra-vermelho e Raman dessas
moléculas. Para todos os átomos foram utilizados pseudopotenciais Lanl2DZ
[63] e SDD [64],[65]. Foi concluído a partir desse estudo que
mPW1PW/Lanl2DZ é um método seguro, que pode ser usado para predizer
estruturas moleculares e espectros vibracionais de compostos de coordenação
que contêm platina (II). Fazendo uma avaliação comparativa dos métodos
utilizados por Wysokinski e Michalska [61], observa-se que o método de cálculo
HF/Lanl2DZ descreve bem a geometria das moléculas estudadas, cerca de
aproximadamente 2% de diferença do melhor resultado obtido. Outro método
de cálculo que também descreve bem a geometria das moléculas de cisplatina
e carboplatina é o B3LYP/Lanl2DZ, menos de 1% de diferença do melhor
resultado obtido. Por esse motivo, os autores concluem que o uso de funções
base mais estendidas e a utilização de outros Funcionais não introduzem
melhorais importantes nos cálculos de otimização de geometria para essas
moléculas.
Em outra publicação, Zhang e colaboradores [66] apresentam um estudo
sobre o mecanismo de hidratação da cisplatina. Como parte dos resultados
obtidos pelos autores, destaca-se o estudo comparativo dos diferentes métodos
computacionais utilizados na otimização da estrutura da molécula de cisplatina.
Os métodos de cálculo utilizados foram HF, MP2, BPL, BVWN, BVWN5, BLYP,
BP86, BPW91, B3LYP, B1LYP, B3PW91, B3P86 e mPW1PW91 com os

29
pseudopotenciais SDD e Lanl2DZ para a platina e função base STO-3G para
todos os outros átomos. Os resultados obtidos por esse trabalho, sugerem que
a estrutura do complexo de platina tenha uma dependência com o
pseudopotencial utilizado. Além disso, concluiu-se que o método mPW1PW91,
utilizando o pseudopotencial SDD [64], [65] foi o nível de cálculo que forneceu
a estrutura molecular da cisplatina, que melhor se aproxima da estrutura
cristalina obtida por raio X [67] . Fazendo uma avaliação comparativa dos
métodos utilizados por Zhang e colaboradores [66], observa-se que o método
de cálculo HF/Lanl2DZ descreve bem a geometria da molécula de cisplatina,
cerca de 2% de diferença do melhor resultado obtido. Outro método de cálculo
que também descreve bem a geometria da molécula é o B3LYP/Lanl2DZ, cerca
de 1% de diferença do melhor resultado obtido.
Figura 2-8: Representação esquemática dos complexos estudados por Zeizinger e colaboradores [68].
Em outro trabalho envolvendo mecanismo de hidratação de metais de
transição, Zeizinger e colaboradores [68], estudaram a energia de solvatação
de quatro complexos quadrado-planar a base de paládio (Figura 2-8). A
estrutura de equilíbrio desses complexos foi obtida utilizando o método de
cálculo MP2, com função base 6-31G* para todos os átomos com exceção do
paládio. O átomo de paládio foi descrito pelo pseudopotencial de Stuttgart [69].
Os resultados foram comparados com dados correspondentes à complexos
PdNH
3
Cl
NH3
Cl
cis - [diaminodicloropaládio II]
PdCl
NH3
NH3
Cl
trans - [diaminodicloropaládio II]
PdNH
3
NH3
NH3
NH3
2+
tetraaminopaládio
PdCl
Cl
Cl
Cl
2-
tetracloropaládio

30
análogos de platina, e observaram que a energia de solvatação dos complexos
de paládio e platina difere em aproximadamente 10 kcal/mol. Considerando a
atividade de hidratação da cisplatina e de complexos análogos de paládio em
processos bioquímicos e farmacológicos, conclui-se que não há diferença
significativa no processo de solvatação entre os complexos de paládio e platina.
Jaroslav e colaboradores [70] também trabalharam com métodos ab
initio para estudar o processo de substituição de dois ligantes das moléculas de
cisplatina, transplatina e análogos de paládio. O método de cálculo utilizado
primeiramente foi HF, em seguida, o método de cálculo MP2 foi utilizado para
obter a estrutura de equilíbrio dessas moléculas. Ambos os cálculos foram
realizados utilizando a função base 6-31+G(d) para todos os átomos com
exceção do átomo de cloro, paládio e platina. Para esses átomos foram
utilizados os seguintes pseudopotenciais: Cl (MWB-10)[65], Pd (MWB-28)[71] e
Pt (MWB-60)[65]. Foram estudadas as características dos reagentes, produtos
e estados de transição no processo de substituição de dois ligantes do metal
por moléculas de água ou íons hidroxila como mostra a Figura 2-9.
Figura 2-9: Representação esquemática do mecanismo de substituição estudada por Jaroslav e colaboradores. X, Y, Z e V são grupos que podem ser átomos de Cl ou NH3. Figura retirada da referência [70].
Os resultados obtidos por esse trabalho [70] mostram que os complexos
de paládio são ligeiramente mais sensíveis a efeitos de substituição trans
comparado com complexos de platina. Além disso, os autores mencionam a
diferença de interação dos dois metais com o átomo de oxigênio do íon OH ,
sendo os complexos de paládio o que apresenta uma maior interação. Também
foi observado que todos os processos de hidratação são endotérmicos, e que
as reações de substituição ocorrem mais rápido em complexos de paládio
comparado com complexos de platina, isto porque as energias dos complexos

31
de paládio no estado de transição são menores comparado com complexos de
platina.
Complexos de paládio também vêm sendo estudados pelo nosso grupo
de pesquisa. Iadoccico [72] estudou os enantiômeros que são objeto de estudo
neste trabalho. Foram feitos à otimização de geometria dos enantiômeros R
dmpa e S dmpa, cálculo da energia, dipolo e cargas atômicas que descrevem
o potencial eletrostático. Nessa etapa, foi concluído que não há diferença
significativa nas estruturas tridimensionais e nas interações eletrostáticas
nessas moléculas devido à quiralidade. Numa segunda etapa desse
trabalho[72], foi feito uma validação do potencial empírico utilizado nas
simulações computacionais em meio aquoso através de comparação com
curvas de potencial de interação dos enantiômeros com uma molécula de água
obtidas de cálculos quânticos. Se concluiu que, os parâmetros do OPLS [73]
para os átomos H, N, C, O, P juntamente com os parâmetros de Lienke [74]
para o átomo de paládio descrevem muito bem a interação desses compostos
paladaciclos com a água. E finalizando o trabalho [72], não foi identificado
nenhuma interação específica (ligações de hidrogênio) desses complexos com
a água.

32
3 – MÉTODO
3.1 Introdução
Este é um trabalho teórico e teve o objetivo de estudar as possíveis
mudanças na estrutura geométrica de moléculas simplificadas que serviram de
modelo para os enantiômeros S(-) e R(+) dos complexos [Pd(C2, N-
dmpa)(dppe)]Cl (Sdmpa e Rdmpa apresentados na Figura 1-3) em processos
de hidrólise.
Como já apresentado no capítulo 1 dessa dissertação, essas possíveis
mudanças estruturais devem desempenhar um importante papel no mecanismo
de ação anti-tumoral desses complexos, uma vez que a enantioseletividade foi
observada experimentalmente [9]-[10] (a saber: o enantiômero S(-) mostrou-se
ativo enquanto que o R(+) inativo).
A alta reatividade desses complexos, Sdmpa e Rdmpa em ambiente
aquoso, que pode existir em várias etapas do mecanismo de ação biológica,
nos levou a estudar o processo de hidrólise desses complexos como sendo um
possível processo de diferenciação entre os enantiômeros que resultaria na
enantioseletividade observada. Sendo assim, para estudar o processo de
hidrólise nas moléculas de interesse dividimos nossos cálculos em duas
etapas: a primeira, onde realizamos cálculos quânticos dos enantiômeros,
utilizando o programa GAUSSIAN [75] e a segunda, onde realizamos
simulações computacionais dos enantiômeros embebidos em água nas
condições normais de temperatura e pressão utilizando o programa DICE [76]
Nas próximas seções, descreveremos os procedimentos utilizados para
ambas as etapas sem entrar em detalhes na descrição dos métodos teóricos
uma vez que existem excelentes livros texto abordando esse tema [50]-[77]-
[78]-[79]-[80]

33
3.2 Sistemas Estudados
Os complexos Sdmpa e Rdmpa são as moléculas de interesse neste
trabalho. Entretanto, devido à complexidade e tamanho dessas moléculas
optamos em simplificá-las, transformando as moléculas aqui estudadas em
sistemas modelos. Duas simplificações foram realizadas. A primeira foi de
retirar o contraíon, Cl-, do sistema e estudar apenas os enantiômeros em sua
forma iônica [Pd(C2, N-dmpa)(dppe)]+. Essa simplificação é bem justificada
considerando que em meio aquoso, onde o processo de hidrólise ocorre, a
interação entre a molécula iônica e o contraíon é enfraquecida devido a forte
interação de ambos com a água. A segunda simplificação é na própria
molécula, onde substituímos os 4 anéis aromáticos do grupo dppe
(representados por Ph na Figura 1-3) por átomos de hidrogênio. Para justificar
essa simplificação fizemos um estudo comparativo das moléculas Sdmpa e
Rdmpa em sua forma iônica (previamente estudada por Iadoccico [72]) e das
formas iônicas substituídas com H (moléculas apresentadas na Figura 3-1 e
denominadas de R (IR) e S (IS)).
Figura 3-1: Representação esquemática das moléculas modelo, na forma iônica, que foram estudadas neste trabalho.
Para estudar as possíveis rotas de hidrólise das moléculas IR e IS,
investigamos a quebra das 4 ligações do átomo de Pd (Pd-X), onde X pode ser
os átomos de N, P e C, através de duas possibilidades:
O próton (H+) da água se ligando com o Pd e a hidroxila (OH-) se ligando
ao outro átomo X da ligação quebrada.
A hidroxila da água se ligando com o Pd e o próton se ligando com X.
Pd
NH
CH3 CH
3
CH3
P
P
+
*
Pd
NCH3
HCH
3
CH3
P
P
+
*

34
Desta forma, geramos e estudamos 8 compostos hidrolisados para cada
enantiômero. Totalizando 16 compostos a partir dos enantiômeros IR e IS. Na
Figura 3-2 apresentamos esses compostos e a nomenclatura que foi utilizada
nesta dissertação.
3.3 Cálculos Quânticos
3.3.1 Estudos das Moléculas Isoladas
As propriedades estruturais e eletrônicas dos enantiômeros IS e IR foram
estudadas através de cálculos quânticos. Inicialmente realizamos a otimização
de geometria de ambas as moléculas. O procedimento de otimização de
geometria busca localizar o mínimo de energia potencial de uma superfície, ou
seja, busca predizer as posições atômicas que geram a estrutura molecular de
equilíbrio. Para gerar as geometrias, inicialmente utilizamos valores
característicos para as distâncias de ligação, ângulos de ligação e ângulos de
torção. A primeira geometria a ser gerada foi IR, composta por 38 átomos (12
carbonos, 22 hidrogênios, 1 nitrogênio, 2 fósforos e 1 paládio = C12H22NP2Pd),
148 elétrons de valência, 39 distâncias de ligação, 72 ângulos de ligação e 109
ângulos de torção. Utilizamos o método HF [52] com função base 6-31G [81]
para todos os átomos, com exceção do paládio, para o qual utilizamos o
pseudopotencial LANL2DZ [74].
Partimos da geometria de equilíbrio do IR para gerar a geometria do IS,
onde invertemos os grupos CH3 e H do carbono quiral. Dessa forma, ao final do
processo de otimização, determinamos as geometrias de equilíbrio para os
enantiômeros IS e IR estudados. As geometrias de equilíbrio dos enantiômeros
IS e IR estão apresentadas na Figura 3-3.
O método HF utiliza o conceito de campo médio, ou seja, baseia-se na
equação de Schrödinger [82] para um elétron movendo-se simultaneamente no
campo criado pelos núcleos e no campo criado por todos os outros elétrons.

35
Figura 3-2: Representação esquemática das rotas de hidrólise que foram estudadas neste trabalho. O composto (a) foi chamado de IIR(PdH) e IIS(PdH), dependendo da quiralidade; (b) IIIR(PdOH) e IIIS(PdOH); (c) IVR(PdH) e IVS(PdH); (d) VR(PdOH) e VS(PdOH); (e) VIR(PdH) e VIS(PdH); (f) VIIR(PdOH) e VIIS(PdOH); (g) VIIIR(PdH) e VIIIS(PdH); (h) IXR(PdOH) e IXS(PdOH).
(a)
(b)
(d)
(c)
(e)
(f)
(g)
(h)

36
É bem estabelecido que o método HF, ao considerar a interação entre as
partículas através de campo médio, negligência a correlação eletrônica
existente em um sistema de muitos corpos, introduzindo um erro da ordem de 1
a 2 por cento na energia interna total obtida. Essa diferença é conhecida como
energia de correlação. Alguns trabalhos na literatura se dedicam a discutir, os
desvios médios de ângulos de ligação e ângulos de torção, obtidos através de
cálculos de otimização de geometria utilizando métodos de cálculo HF e
comparando com os valores experimentais [83],[84]. Esses trabalhos mostram
que o método HF é bom para determinação das geometrias de equilíbrio.
Figura 3-3: Geometrias otimizadas para os enantiômeros: (a) IR (b) IS. As circunferências indicam o centro quiral.
A partir da estrutura molecular de equilíbrio, obtida pelos cálculos de
otimização de geometria, realizamos cálculos quânticos das freqüências
vibracionais harmônicas. Essas freqüências vibracionais foram utilizadas para a
obtenção da energia vibracional do ponto zero e assim, determinamos à
energia interna molecular dos enantiômeros IS e IR.
Após gerarmos as geometrias de equilíbrio dos enantiômeros e
analisarmos as suas respectivas energias internas, determinamos o momento
de dipolo e as cargas atômicas para descrever o potencial eletrostático.
Foram desenvolvidos inúmeros métodos que descrevem a distribuição
eletrônica molecular em termos de cargas atômicas [85], dentre os quais a
análise populacional de Muliken, as cargas CHELPG (Charges from eletrostatic
Potential Grid Based), o modelo dipolar GAPT (Generalized Atomic Polar
Tensor) e a partição molecular AIM (Atoms in Molecules). Embora esses
(a) (b)

37
métodos sejam baseados em aproximações distintas, suas aplicações os
tornaram vastamente difundidos [86],[87],[88], conforme pode ser observado
em revisão detalhada apresentada por Guadagninie Bruns [89].
Neste trabalho, escolhemos o método CHELPG, para determinarmos as
cargas atômicas dos enantiômeros IS e IR. O método CHELPG proposto por
Breneman e Wiberg [90], é um método de obtenção de cargas pontuais que
serve para representar adequadamente o potencial eletrostático Coulombico.
As cargas derivadas do potencial incluem: (i) a determinação de uma função de
onda apropriada; (ii) a determinação do potencial eletrostático em vários pontos
em torno da molécula e; (iii) a determinação das cargas, em posições
predefinidas, por meio de um ajuste de mínimos quadrados dos potenciais
eletrostáticos, calculados classicamente, e valores os potencias calculados
quanticamente [91].
Dessa forma, utilizando a geometria otimizada, realizamos cálculos
quânticos para a determinação do momento de dipolo e das cargas atômicas
para os enantiômeros IS e IR. Esses cálculos quânticos para a determinação
das cargas foram realizados com o mesmo método e funções bases utilizadas
na otimização de geometria.
3.3.2 Estudos da Entalpia da Reação de Hidrólise
Utilizando a estrutura molecular dos enantiômeros obtida anteriormente,
iniciamos os cálculos quânticos para a determinação da variação de entalpia da
reação de hidrólise. A entalpia de hidrólise é o nome dado à variação de
entalpia associado à quebra de uma molécula na presença de água.
Primeiramente, de forma análoga aos cálculos quânticos realizados
anteriormente, estudamos a estrutura eletrônica e geométrica da molécula de
água. Os cálculos quânticos para a otimização de geometria foi no nível HF
com base 6-31G para todos os átomos e essa geometria foi utilizada para
determinar o valor da energia interna da molécula de água.
Em seguida, investigamos as 8 rotas de hidrólise mostradas na Figura 3-
2 para cada enantiômero. Para gerar as geometrias dos 16 complexos
hidrolisados, partimos da geometria otimizada dos enantiômeros IS e IR, onde

38
rompemos uma das ligações do paládio e completamos os átomos
desemparelhados com os íons OH e H provenientes da molécula de água.
Em seguida, realizamos cálculos quânticos para a otimização de geometria
desses complexos hidrolisados. Os cálculos quânticos foram realizados com o
mesmo nível de cálculos e funções bases utilizadas na otimização de geometria
dos enantiômeros IS e IR
Após gerarmos as geometrias de equilíbrio dos complexos hidrolisados,
realizamos cálculos quânticos para obtermos as energias internas. Para isto,
escolhemos o método HF[52], já descrito anteriormente. A partir dessas
energias será obtida a variação da entalpia na reação de hidrólise )(gásH .
Desta forma, é importante analisar se o cálculo da energia pode ser melhorado
para essas geometrias otimizadas.
Com esse objetivo realizamos cálculos quânticos das energias para uma
das possíveis rotas de hidrólise, utilizando DFT [49],[50],[51] com o potencial
híbrido de troca-correlação B3LYP [62], com as seguintes funções bases: 6-
31G, 6-31+G, 6-31G(d), 6-31+G(d), 6-31+G(2d), cc-pvDZ e cc-pvTZ . Assim, foi
possível compararmos os resultados das energias que serão apresentados no
próximo capítulo.
O DFT considera o sistema eletrônico como um todo, e toda a
informação sobre o sistema é então transferida da função de onda para a
densidade eletrônica. A grande vantagem do DFT, é a implementação
computacional eficiente da correlação eletrônica, que possibilita uma boa
descrição de sistemas complexos.
A variação da entalpia de hidrólise é calculada pela diferença entre a
entalpia do estado final e do estado inicial como mostra as equações 1 e 2:
I + W IW (1)
onde, I é o enantiômero, W representa a molécula de água. A interação dessas
duas moléculas, gera o complexo hidrolisado simbolizado por IW. Dessa forma
temos que:
Estado Inicial
Estado Final

39
)gás(inicial)gás(final)gás( HHH (2)
onde, )(gásH é a variação da entalpia de hidrólise das moléculas em fase
gasosa e é obtida pela diferença entre a entalpia do complexo hidrolisada
simbolizado por )(gásHfinal , menos a )(gásHinicial que representa a soma das
entalpias do enantiômero e da água. Dessa forma podemos reescrever os
termos da equação 2 na forma simplificada (equação 3):
)HH(HH WIIW)gás( (3)
Segundos os Princípios da Termodinâmica [92], a entalpia pode ser
definida pela equação 4:
pVKEH (4)
onde H é a entalpia do sistema , E é a energia interna do sistema, K é a energia
cinética (3kT por molécula) e pV é o produto entre a pressão e o volume do
sistema. Dessa forma podemos substituir a Equação 4 em 3:
WWIIIWIW)gás( pVkT3EpVkT3EpVkT3EH (5)
Como as moléculas isoladas têm o comportamento de gás ideal, então
suas propriedades podem ser descritas pela equação 6:
nkTpV (6)
onde pV é o produto entre a pressão e o volume do sistema, n é o número de
moléculas do sistema, k é a constante de Boltzmann e T é a temperatura do
sistema.
Como estamos estudando as moléculas de forma isolada, então n=1,
dessa forma podemos reescrever a equação 6:

40
kTpV (7)
Assim, escrevemos a equação para calcularmos a variação da entalpia
de hidrólise, a partir da substituição da Equação 7 em 5:
kT4EEEH WIIW)gás( (8)
onde )(gásH é a variação da entalpia de hidrólise das moléculas em fase
gasosa e é obtida pela diferença da energia interna do complexo hidrolisado
IWE , da energia interna resultante do enantiômero e da água ( WI EE ) e pela
energia térmica que é o produto da constante de Boltzmann e da temperatura
do sistema 4kT, onde 3 é devido a parte cinética e 1 devido a diferença de
volume.
3.4 Simulação Computacional
3.4.1 Estudos das Moléculas em Meio Solvente
Após termos estudado a variação da entalpia de hidrólise em fase
gasosa. Estudamos a influência do meio aquoso nesse processo. Para isso
realizamos simulações computacionais com método Monte Carlo (MC) [93]-
[94]. O método MC é estocástico e usa uma distribuição no equilíbrio para
gerar configurações de sistemas moleculares que são acessíveis em um
determinado ensemble.
Neste trabalho, optamos em usar o método MC por ser
computacionalmente mais rápido que o método de Dinâmica Molécular (DM)
[93]-[94] e por não estudarmos nenhuma propriedade que necessite da
evolução temporal. O interesse é calcular a variação de entalpia de hidrólise em
solução aquosa, )sol(H .
Realizamos simulações computacionais dos enantiômeros IS e IR e dos
complexos hidrolisados em solução aquosa para analisarmos o efeito do
solvente nas rotas de hidrólise.

41
IW I W
A Figura 3-4 ilustra o processo reativo de hidrólise que foi estudado nas
simulações. O estado inicial do processo é representado pelo enantiômero
solvatado em 800 moléculas de água, adicionado a uma molécula de água
solvatada em 450 e o estado final é representado pelo enantiômero hidrolisado
solvatado em 1250 moléculas de água.
Figura 3-4: Ilustração do processo reativo de hidrólise utilizado no cálculo da variação de entalpia de hidrólise em solução através de simulações computacionais.
Nas simulações computacionais foi utilizado o método MC com
amostragem de Metropolis. O método Monte Carlo Metropolis [93],[96] pode
ser descrito como um método que utiliza seqüências de números aleatórios
para gerar configurações do sistema molecular e aceitá-las com uma
probabilidade mínimo [1, exp (E/kT)], de tal forma a satisfazer à distribuição
de equilíbrio de Boltzmann [97],[98].
As simulações realizadas neste trabalho partem de uma configuração
aleatória, onde a energia inicial é sempre maior que a energia de equilíbrio. Por
essa razão, a evolução dessas simulações, pode ser dividida em dois estágios:
o primeiro é um estágio conhecido como termalização, no qual a energia inicial
vai decrescendo até alcançar o valor de equilíbrio. O segundo estágio,
conhecido como equilíbrio, é iniciado a partir da última configuração gerada
pela termalização, e é responsável por gerar as configurações que são
consideradas nos cálculos das propriedades estruturais e termodinâmicos do
sistema.
As propriedades termodinâmicas de um sistema são definidas a partir de
médias e flutuações da energia gerada durante as simulações e de outras
grandezas que definem a condição termodinâmica do sistema, o conjunto
dessas grandezas recebe o nome de ensemble. Apesar das simulações
levarem mais tempo, a utilização do ensemble NPT se torna mais adequado
quando for estudado a solvatação de moléculas grandes que podem alterar a
+
1250 H2O 800 H2O 450 H2O

42
densidade do sistema. Dessa forma, optamos por utilizar o ensemble NPT em
nossas simulações.
Cada simulação foi gerada a uma temperatura de 36ºC (temperatura do
corpo humano). Em todas as simulações realizadas, as moléculas são rígidas,
ou seja, elas podem transladar e rotacionar, porém não podem vibrar nem
deformar, pois não possuem graus de liberdade interno.
Foi feito essa escolha por falta de parâmetros na literatura para
descrever os movimentos de vibração, ângulos de ligação e parâmetros de
deformação dos ângulos de torção do átomo de paládio. Esses parâmetros
desempenham um importante papel nas rotas de hidrólise estudadas, uma vez
que suas ligações são rompidas. Adicionalmente a isto, acredita-se que essa
restrição não será importante em nosso estudo uma vez que as geometrias dos
enantiômeros são bastante rígidas não apresentando muita possibilidade de
deformação (ver Figura 3-1) e as geometrias dos enantiômeros hidrolisados
(ver ilustração na Figura 3-2) que são flexíveis, nós estudamos a possibilidade
da existência de mais de uma conformação, a saber, conformação aberta e
fechada (ver detalhes no próximo capítulo). As geometrias rígidas utilizadas nas
simulações são as obtidas das otimizações de geometria no caso dos solutos:
enantiômeros e complexos hidrolisados, e no caso do solvente: água,
utilizamos a geometria do modelo SPC [99].
Geramos a configuração inicial da simulação aleatoriamente. A molécula
do soluto é posicionada na origem da caixa, enquanto que as moléculas de
solvente são posicionadas aleatoriamente com orientações também aleatórias.
Utilizamos o potencial de interação entre as moléculas do tipo Lennard-
Jones + Coulomb, como mostra a Equação 9. Os parâmetros e para as
moléculas de soluto que utilizamos neste trabalho, foram obtidos do campo de
força OPLS [73], [100], [101] para todos os átomos com exceção do paládio.
Os parâmetros utilizados para o paládio foram desenvolvidos por LIENKE e
colaboradores [74] e possui os seguintes valores: 0.2 Ǻ e 1.7 kcal/mol
[74]. As cargas parciais utilizadas nas simulações foram extraídas do ajuste do
potencial eletrostático calculado no nível HF/6-31G [52], [82] utilizando a
técnica CHELPG [90].

43
ijr
jqiq
ijr
ijσ
ijr
ijσ
ijε4)ijU(r
612
(9)
Para o solvente (água) utilizamos o potencial de interação do modelo
SPC [99].
Todas as simulações realizadas nesse trabalho, utilizaram o método
Monte Carlo Metropolis e o potencial de interação do tipo Lennard-Jones +
Coulomb, implementados no programa DICE [77].
3.4.2 Estudos da Entalpia da Reação de Hidrólise em Meio Solvente
Através do processo reativo de hidrólise representado pela Figura 3-4,
calcula-se a variação da entalpia de hidrólise através da equação 10:
)sol(
inicial
)sol(
final)sol( HHH (10)
onde )sol(H é a variação da entalpia de hidrólise das moléculas em solução
aquosa. Dessa forma, podemos reescrever os termos da equação 10 na forma:
)HH(HH)sol(
W
)sol(
I
)sol(
IW)sol( (11)
onde,
)X()X(
WWWXXWX)sol(X pVUUKEH (12)
sendo Ex a energia interna molecular do soluto X, KXW a energia cinética (3kt
por molécula) total do sistema, WXU a energia de interação soluto-solvente e
)X(
WWU a energia de interação solvente-solvente na presença do soluto. Então
substituindo a equação 12 em 11, temos:
)W()W(
WWWWW
)I()I(
WWWII
)IW()IW(
WWWIWIW)sol(
pVUUkT3451E
pVUUkT3801(E
pVUU)kT31251(EH
(13)
Desta forma reorganizamos os termos:

44
kTVVVpUUU
)UU(UkT4EEEH
)W()I()IW()w(
WW
)I(
WW
)IW(
WW
WWWIWIWWIIW)sol(
(14)
e reescrevemos na forma simplificada:
kTVpUUHH WWWI)gás()sol( (15)
onde )gás(H é a variação da entalpia de hidrólise das moléculas em fase
gasosa, WIU é a variação da energia potencial de interação soluto-solvente,
WWU é a variação da energia potencial de interação solvente-solvente,
também conhecida como energia potencial de relaxação do solvente, Vp é a
variação do volume multiplicado pela pressão e kT é a diferença da variação da
energia cinética, isto porque o sistema final tem uma molécula a menos que o
sistema inicial.
Desta última equação, o 1º termo à direita, )gás(H , já foi calculado nos
cálculos quânticos descritos na seção 3.3.2. O 2º termo, WIU , é facilmente
calculado das simulações computacionais e apresentam uma rápida
convergência. Em geral, esse valor está convergido a partir de 3x104 ciclos MC,
no equilíbrio. Cada ciclo MC é definido quando todas as moléculas do sistema
sofrem a tentativa de um movimento aleatório. O 3º termo, WWU é uma
quantidade bastante difícil de ser calculada [102],[103],[104],[105], pois esse
termo é a diferença da energia de interação das 1250 moléculas de água na
presença do soluto hidrolisado, )IW(
WWU , do estado final, com a soma das
energias de interação das 800 e 450 moléculas de água na presença do soluto,
)I(WWU , e da água,
)W(WWU , respectivamente, do estado inicial. Pode-se
perceber claramente que esses três termos, )IW(
WWU , )I(
WWU , )W(
WWU são
números bastante grandes (da ordem de 103 – 104 kcal/mol) e apresentam
flutuações também grandes (da ordem 102 kcal/mol).
Dessa forma, o cálculo da relaxação do solvente, devido à hidrólise do
soluto apresenta uma convergência bastante lenta e é necessária uma grande
quantidade de ciclos MC para garantir sua convergência. Em geral, a partir de
106 ciclos MC atinge-se a convergência [104]-[105].

45
Devido a essa dificuldade de calcular esse termo com precisão, é
comum desprezá-lo e assumir que a relaxação do solvente não desempenha
um papel importante no cálculo da variação de entalpia num processo reativo
em solução.
Entretanto, essa suposição pode estar errada. Para analisar a relevância
desse termo, escolhemos uma das possíveis rotas de hidrólise e calculamos de
forma bastante criteriosa esse termo, garantindo sua convergência. Essa
análise está apresentada no próximo capítulo.
O 4º termo da equação 15, Vp , para sistemas líquidos, é calculado
facilmente durante as simulações e, em geral, para soluções aquosas
apresenta valor da ordem de 10-2 kcal/mol. Como esse valor é inferior à
precisão numérica que temos para o cálculo do )sol(H , esse termo pode ser
desprezado sem nenhuma perda de informação do sistema.
O último termo da equação 15, kT, é constante e igual a 0,6 kcal/mol.

46
4 – RESULTADOS E DISCUSSÕES
4.1 Enantiômero Isolado
O estudo das propriedades estruturais e eletrônicas dos enantiômeros
(IS e IR) do complexo N,N-dimetil-1-fenetilamina (mostrados na Figura 1-3) já
foi previamente realizado por Iadoccico [72] através de cálculos quânticos.
Nessa fase as moléculas foram consideradas isoladamente. Nesse estudo o
nível de cálculo utilizado foi HF [52] com base 6-31G [82] para todos os
átomos, com exceção do paládio para o qual foi utilizado o pseudopotencial
LANL2DZ [74]. No total foram 457 funções bases.
As conclusões da autora, após ter feito uma análise sistemática de todas
as distâncias e ângulos de ligação e ângulos de torção, foram que as
diferenças encontradas na geometria da forma iônica dos enantiômeros são
apenas pequenas mudanças conformacionais. As maiores diferenças entre os
enantiômeros foram observadas nas orientações relativas dos grupos fenil e no
anel formado pelo paládio e os átomos de fósforo. Essas mudanças são da
ordem de 17º e não geram diferenças significativas na energia (0,04 kcal/mol).
Com relação à distribuição eletrônica a diferença, no momento de dipolo, obtida
pela autora, foi de 0,03D, o que também não representa uma mudança
significativa.
Considerando que os cálculos de otimização de geometria levaram muito
tempo de processamento (mais de 15 dias), optamos neste trabalho por
simplificar as moléculas estudadas e substituir esses anéis aromáticos por
átomos de hidrogênio como mostra a Figura 4-1.
Fazendo uma análise sistemática da geometria dos enantiômeros IR e
IS, observamos que em ambas as moléculas, o paládio prefere a conformação
quadrado planar. Essa conformação também já foi encontrada em outros
complexos contendo paládio [106]. A diferença de energia encontrada entre as
duas geometrias é de 1kcal/mol, tendo o enantiômero IR energia mais baixa.
Com relação à distribuição eletrônica, temos que o momento de dipolo do

47
N20
Pd11
P19
P12
C6
(a)
enantiômero IR é de 6,4 D e do IS é de 6,4 D, o que também não representa
mudança.
Figura 4-1: Geometria otimizada e definição da numeração dos átomos dos enantiômeros: (a) IR e (b) IS.
Na Tabela 4-1 apresentamos as distâncias de ligação que envolvem o
átomo de paládio dos enantiômeros IR e IS. Ainda nesta tabela, apresentamos
os valores das distâncias de ligação envolvendo o átomo de paládio, obtidos no
trabalho de Iadoccico [72], que utiliza as moléculas em substituição dos grupos
fenis por H, como é o nosso caso.
Comparando todas as distâncias, ângulos de ligação e ângulos de torção
pode-se concluir que obtivemos os mesmos resultados que Iadoccico [72], ou
seja, não existem diferenças significativas entre os enantiômeros IR e IS e nem
entre as moléculas com ou sem os anéis.
Tabela 4-1: Valores das distâncias de ligações (em Angstron) que envolvem o átomo de paládio.
Ligações IR(Å) Rdmpa (Å)
[72] IS(Å)
Sdmpa(Å) [72]
R(C6,Pd11) 2.020 2.032 2.010 2.027
R(Pd11,P12) 2.462 2.470 2.466 2.458
R(Pd11,P19)
R(Pd11,N20)
2.660
2.171
2.625
2.202
2.670
2.173
2.659
2.196
(b)

48
4.2 Reações de Hidrólise
Várias reações podem ser interpretadas fazendo a análise dos orbitais
de fronteira, isto é, o orbital molecular ocupado de maior energia, habitualmente
designado pela sigla HOMO, de “highest occupied molecular orbital” e o orbital
molecular virtual de menor energia, habitualmente designado pela sigla LUMO,
de “lowest unnoccupied molecular orbital”. A importância desta análise foi,
sobretudo salientada por Fukui [107].
Em reações que uma molécula atua como doador de elétrons, é
importante o HOMO dessa molécula, enquanto que, para moléculas as quais
atuam como aceitadores de elétrons é o LUMO que, sobretudo importa
considerar. Como a molécula em estudo é carregada positivamente, ela atua
com um aceitador de elétrons, logo o orbital de interesse para esse sistema é o
LUMO. Na Figura 4-2 ilustra a função de onda que descreve o LUMO das
moléculas IR e IS obtidos após a otimização de geometria de cada uma com o
método de cálculo HF/6-31G.
Figura 4-2: Orbitais LUMO dos enantiômeros: (a) IR (b) IS
Analisando a Figura 4-2 (a) e (b), observamos que o LUMO sobrepõe o
elemento paládio, isto significa que em uma possível captação de elétrons,
ocorrerá um aumento da população eletrônica do LUMO, conseqüentemente
ocorrerá uma ruptura na ligação envolvendo o átomo de paládio originando
uma nova estrutura.
Portanto, através de uma análise dos orbitais moleculares, confirmamos
que as ligações susceptíveis a uma possível quebra são as quatro do paládio,
Em reações que uma molécula atua como doador de elétrons é
especialmente importante o HOMO dessa molécula, enquanto para moléculas que atuam como
aceitadores de elétrons é o LUMO que, sobretudo importa
considerar. Como a nossa molécula de estudo é carregada positivamente ela atua com um
aceitador de elétrons, logo o nosso orbital de interesse é o LUMO, ilustrada na Figura 14
(a) (b)

49
ou seja, os átomos de fósforo (P12 e P19); o átomo de carbono (C6) e o átomo
de nitrogênio (N20).
Como não há indícios fortes quanto à ligação que será quebrada em
uma reação de hidrólise, decidimos estudar a quebra das quatro ligações do
paládio.
Desta forma, estudamos a quebra das 4 ligações do Pd (ver Figura 3-2),
otimizamos os 16 compostos hidrolisados e calculamos as energias para
obtermos as variações de entalpia de hidrólise das moléculas em fase gasosa
)gás(H .
Como a boa descrição teórica de um dado sistema molecular está
intimamente relacionada à escolha adequada dos métodos e das funções
bases, analisamos de uma forma comparativa a precisão e o custo
computacional dos métodos ab initio (HF) e o funcional de densidade híbrido
com o potencial de troca-correlação B3LYP, com as seguintes funções bases:
6-31G, 6-31+G, 6-31G(d), 6-31+G(d), 6-31+G(2d), cc-pvDZ e cc-pvTZ para
todos os átomos com exceção do paládio que usa o pseudopotencial
LANL2DZ, com o objetivo de melhorarmos o cálculo da energia das
geometrias otimizados dos complexos hidrolisados.
O sistema escolhido para desenvolver essa análise, foi à rota de
hidrólise que envolve a quebra da ligação dos átomos Pd–N(20) do enantiômero
IS. Calculamos a geometria otimizada do sistema (Figura 4-3) e em seguida,
calculamos a variação da entalpia de hidrólise da molécula em fase gasosa,
)gás(H , empregando os métodos HF e o DFT, através da Equação 8, para o
complexo hidrolisado.
Na Tabela 4-2 estão descritos os valores das entalpias de hidrólise.
Esses resultados permitem fazer uma análise do efeito do método e do
conjunto de funções base utilizado nos cálculos da energia.
Os valores da variação da entalpia de hidrólise obtidos com a base 6-
31G diminuem em aproximadamente 83% quando modificamos o nível de
cálculo de HF para B3LYP. Isto evidência o efeito da inclusão da correlação
eletrônica.

50
Figura 4-3: Geometria da molécula (VIIS(PdOH)) formada na quebra da ligação Pd-N(20) do enantiômero IS, seguindo a rota (f) da Figura 3-2.
Tabela 4-2: Comparação de métodos/função base para cálculo da variação da entalpia de
hidrólise em fase gasosa (em kcal/mol). Os cálculos foram executados em um Pentium IV com
2Gb de memória RAM.
Método E(IW) E(I) E(W) H CPU Funções
base SW
B3LYP/cc-pvTZ -1411.238 -1334.799 -76.459 10.79 10h59 min 846
B3LYP/6-31+G(2d) -1410.951 -1334.548 -76.428 13.62 2h18min 446
B3LYP/cc-pvDZ -1411.002 -1334.595 -76.418 11.61 1h00 374
B3LYP/6-31+G(d) -1410.945 -1334.544 -76.421 10.92 1h15min 366
B3LYP/6-31G(d) -1410.922 -1334.530 -76.407 7.22 25min 302
B3LYP/6-31+G -1410.750 -1334.359 -76.400 3.82 42min 286
B3LYP/6-31G -1410.727 -1334.345 -76.385 -0.07 12min 222
HF/6-31G -1404.462 -1328.491 -75.985 8.03 10min 222
+

51
Comparando os valores da variação da entalpia de hidrólise obtida pela
base cc-pvTZ e 6-31+G(d), observa-se que a energia obtida pela função base
6-31+G(d) é aproximadamente 1% maior do que o valor obtido pela base cc-
pvTZ. Apesar da base cc-pvTZ ser a mais completa neste nível de cálculo, o
tempo de processamento foi extremamente elevado (10h59 min). Nesse caso,
o aumento do tempo de processamento, em relação à função base 6-31+G(d)
(1h15min), não compensou o ganho no valor da energia (Figura 4-4). Sendo
assim, concluímos que o cálculo da energia pode ser bem descrito utilizando o
método de cálculo B3LYP/6-31+G(d). Na seção 4.3 apresentaremos os
resultados obtidos com HF/6-31G e na seção seguinte 4.4 esses valores serão
comparados com os obtidos com B3LYP/6-31+G(d).
Figura 4-4: Valores descritos na Tabela 4-1 da variação da entalpia de hidrólise do enantiômero IS na rota (f) da Figura 3-2. Círculo aberto é o valor da entalpia obtido pelo método HF e os círculos fechados são valores obtidos pelo método B3LYP.
4.3 Análise das Rotas de Hidrólise em Fase Gasosa
4.3.1 Quebra da Ligação Pd-P(19)
Considerando o procedimento descrito na Seção 3.3.2, estudamos o
processo de hidrólise envolvendo primeiramente a quebra da ligação Pd-P(19)

52
dos enantiômeros IR e IS, rotas (a) e (b) da Figura 3-2, resultando nos
complexos: II(PdH) e III(PdOH), respectivamente.
Os complexos II(PdH) foram obtidos em duas conformações: uma que
chamamos de fechada e que forma uma ligação de hidrogênio intramolecular
entre o hidrogênio ligado ao paládio e a hidroxila ligada ao P(19); e outra que
chamamos de aberta onde não há essa ligação. Essas conformações estão
ilustradas na Figura 4-5 e na Figura 4-6. É importante salientar que para os
complexos III (PdOH) não encontramos nenhuma forma aberta. Os valores
calculados da variação da entalpia de hidrólise de cada complexo estão
apresentados na Tabela 4-3.
Figura 4-5: Geometrias das moléculas formadas na quebra da ligação Pd-P(19) do enantiômero IR. Em destaque estão as localizações da hidroxila depois do processo de hidrólise.
IR
IIR(PdH)aberto
IIIR(PdOH)
IIR(PdH)fechado
+
IR IR

53
Tabela 4-3: Valores calculados para variação da entalpia de hidrólise (HF/6-31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação Pd – P(19). Rotas (a) e (b) da Figura 3-2.
Composto Hgás
(kcal/mol) Composto
Hgás
(kcal/mol)
H (IS-IR)
(kcal/mol)
IIR(PdH)aberto 82.0 IIS(PdH)aberto 82.3 0.3
IIR(PdHfechado 65.0 IIS(PdH)fechado 65.2 0.2
IIIR(PdOH) 31.6 IIIS(PdOH) 31.3 -0.3
Figura 4-6: Geometrias das moléculas formadas na quebra da ligação Pd-P(19) do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
Comparando as variações da entalpia do processo, observamos que, os
complexos IIIR(PdOH) e IIIS(PdOH) são os que necessitam de menos calor
para a quebra da ligação Pd – P(19), uma vez que o grupo hidroxila que está
ligado ao paládio consegue interagir com dois átomos de hidrogênio que estão
ligados ao fósforo formando duas ligações de hidrogênio. Essa interação
+
IS
IIS(PdH)fechado
IIS(PdH)aberto
IIIS(PdOH)

54
intramolecular faz diminuir significativamente (em torno de 60%) a variação da
entalpia de hidrólise dessas moléculas.
Analisando a Tabela 4-3, observamos que as diferenças das entalpias de
hidrólise encontrada para os complexos são inferiores à energia térmica (0.6
kcal/mol), portanto para essa quebra de ligação não foram encontradas
diferenças com relação quiralidade da molécula. Ainda observando a Tabela 4-
3, concluímos que essas rotas de hidrólise são endotérmicas.
4.3.2 Quebra da Ligação Pd-P(12)
Outra ligação que também está sujeita a sofrer um processo de hidrólise,
é a ligação Pd-P(12). De forma análoga ao processo descrito anteriormente,
estudamos também a quebra dessa ligação em fase gasosa, seguindo as rotas
(c) e (d) da Figura 3-2 e resultando nos complexos IV(PdH) e V(PdOH). Esses
complexos estão ilustrados nas Figuras 4-7 e 4-8. Na Tabela 4-4 apresentamos
os valores da variação da entalpia de hidrólise.
Comparando as moléculas obtidas na quebra envolvendo os dois átomos
de fósforo, observamos que esses complexos hidrolizados são muito
semelhantes. Porém quando comparamos a variação da entalpia de hidrólise
notamos que, os complexos IV (PdH) necessitam de menos calor que os
complexos II(PdH) em torno de 6 kcal/mol. É interessante ressaltar também que
os complexos que necessitam de menos calor são os complexos V(PdOH)
devido às interações intramoleculares. Como já comentado anteriormente, para
os complexos III (PdOH) e que para os complexos V(PdOH) não encontramos
nenhuma forma aberta.
Analisando a Tabela 4-4, observamos que as diferenças das variações
da entalpia de hidrólise encontradas para os enantiômeros hidrolisados são
inferiores à energia térmica, portanto, para essa quebra de ligação não foram
encontradas diferenças com relação quiralidade da molécula. Ainda
observando a Tabela 4-4, concluímos que essas rotas de hidrólise são
endotérmicas.

55
Figura 4-7: Geometrias das moléculas formadas na quebra da ligação Pd-P(12) do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
Tabela 4-4: Valores calculados para a variação de entalpia de hidrólise (HF/6-31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação Pd – P(12). Rotas (c) e (d) da Figura 3-2.
Composto Hgás
(kcal/mol) Composto
Hgás
(kcal/mol)
H (IS-IR)
(kcal/mol)
IVR(PdH)aberto 76.6 IV(PdH)Saberto 76.6 0.0
IVR(PdH)fechado 59.1 IVS(PdH)fechado 59.2 0.1
VR(PdOH) 31.4 VS(PdOH) 31.2 -0.2
+
IR
IVR (PdH) aberto
IVR(PdH) fechado
VR(PdOH)
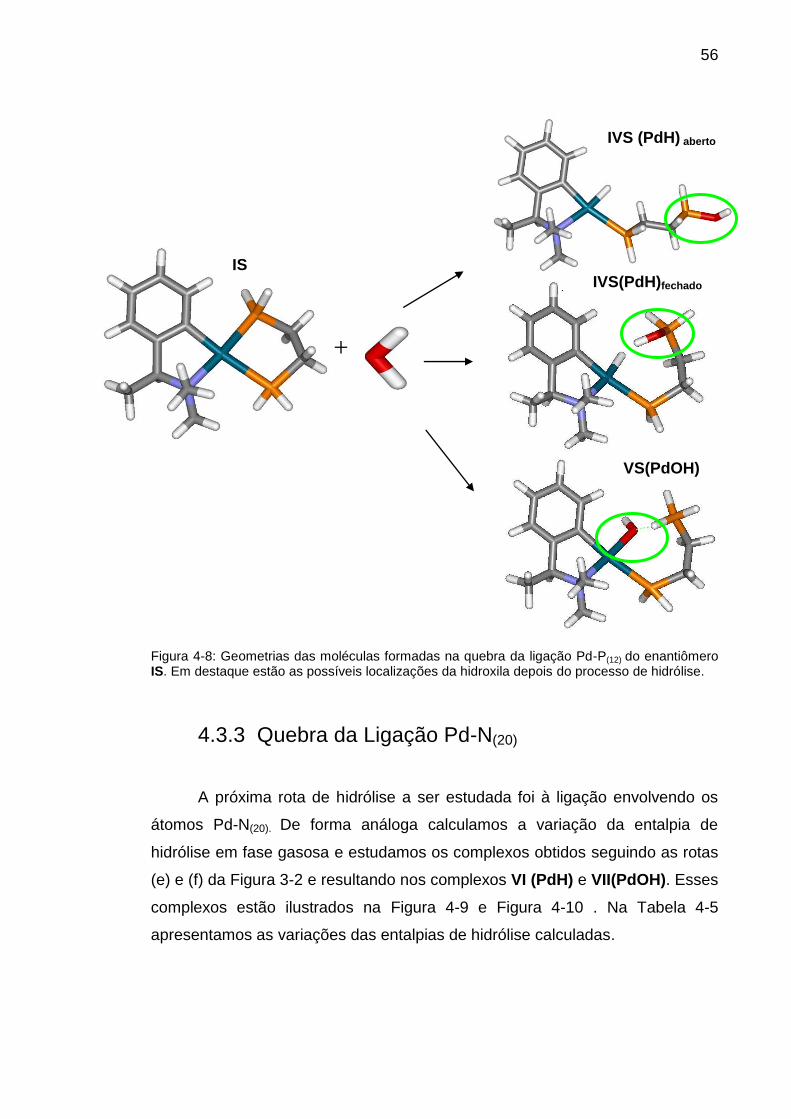
56
Figura 4-8: Geometrias das moléculas formadas na quebra da ligação Pd-P(12) do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
4.3.3 Quebra da Ligação Pd-N(20)
A próxima rota de hidrólise a ser estudada foi à ligação envolvendo os
átomos Pd-N(20). De forma análoga calculamos a variação da entalpia de
hidrólise em fase gasosa e estudamos os complexos obtidos seguindo as rotas
(e) e (f) da Figura 3-2 e resultando nos complexos VI (PdH) e VII(PdOH). Esses
complexos estão ilustrados na Figura 4-9 e Figura 4-10 . Na Tabela 4-5
apresentamos as variações das entalpias de hidrólise calculadas.
IS
IVS (PdH) aberto
IVS(PdH)fechado
VS(PdOH)
+
IVS(PdH)fechado
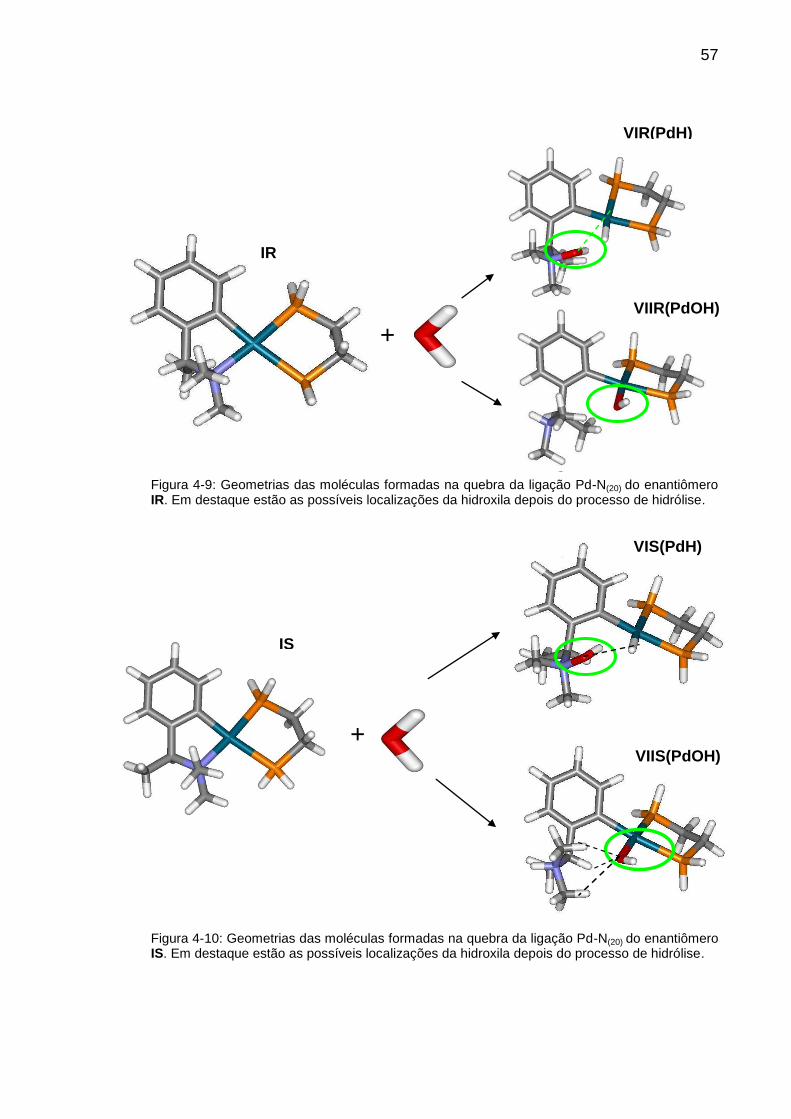
57
Figura 4-9: Geometrias das moléculas formadas na quebra da ligação Pd-N(20) do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
Figura 4-10: Geometrias das moléculas formadas na quebra da ligação Pd-N(20) do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
+
IR
VIR(PdH)
VIIR(PdOH)
+
IS
VIIS(PdOH)
VIS(PdH)

58
Tabela 4-5: Valores calculados para a variação de entalpia de hidrólise (HF/6-31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação Pd –N(20). Rotas (e) e (f) da Figura 3-2.
Ao fazer a análise da quebra da ligação Pd-N(20), observamos que os
grupos ligados ao átomo N(20) possuem maior grau de flexibilidade ao sofrer o
processo de hidrólise, comparado com os átomos de fósforo. E essa
flexibilidade (observado no ângulo torcional C1-C29-N20-O39) é determinante na
obtenção da variação da entalpia de hidrólise, principalmente quando a
hidroxila se liga ao átomo de N(20) formando interações intramoleculares com o
próton da água que se ligou ao paládio como ilustra a Figura 4-11.
Ainda analisando a Figura 4-11 observamos que ao rotacionarmos os
grupos que estão ligados ao átomo N(20), observamos que a energia do
complexo diminui em torno de 5 kcal/mol devido às interações intramoleculares.
Figura 4-11: Geometrias das moléculas formadas na quebra da ligação Pd-N(20) do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila depois da hidrólise. A molécula (a) possui uma variação da entalpia de hidrólise de 71.2 kcal/mol. A molécula (b) possui uma variação da entalpia de hidrólise de 66.2 kcal/mol.
É interessante salientar que, o complexo com mais baixa variação da
entalpia de hidrólise foi o VIIS(PdOH) no valor de 8.03 kcal/mol. O motivo
dessa baixa variação da entalpia de hidrólise é devido à formação de três
Composto Hgás
(kcal/mol) Composto
Hgás
(kcal/mol)
H (IS-IR)
(kcal/mol)
VIR(PdH) 75.8 VIS(PdH) 71.2 -4.6
VIIR(PdOH) 18.8 VIIS(PdOH) 8.03 -10.8
(b)
(a)

59
ligações de hidrogênio que se formam entre a hidroxila e os hidrogênios dos
grupos metil do átomo N(20).
De acordo com a Tabela 4-5, observamos que as diferenças da entalpia
(H(IS-IR)) encontradas para os enantiômeros hidrolisados são superiores à
energia térmica, portanto para essa quebra de ligação encontramos diferenças
com relação à quiralidade das moléculas. Ainda observando a Tabela 4-5,
concluímos que essas rotas de hidrólise são endotérmicas.
4.3.4 Quebra da Ligação Pd-C(6)
Para finalizarmos nossa análise na determinação das possíveis rotas de
hidrólise a serem estudadas, quebramos a ligação Pd-C(6). De forma análoga
às análises anteriores seguimos as rotas (g) e (h) da Figura 3-2 e geramos os
complexos VIII(PdH) e IX(PdOH). Esses complexos são ilustrados na Figura 4-
12 e a Figura 4-13. Na Tabela 4-6 apresentamos os valores da variação da
entalpia de hidrólise calculados.
Figura 4-12: Geometrias das moléculas formadas na quebra da ligação Pd-C(6) do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
+
IR
VIIIR(PdH)
IXR(PdOH)

60
Tabela 4-6: Valores calculados para a variação de entalpia de hidrólise (HF/6-31G), em fase gasosa dos complexos hidrolisados obtidos na quebra da ligação Pd – C(6). Rotas (g) e (f) da Figura 3-2.
Figura 4-13: Geometrias das moléculas formadas na quebra da ligação Pd-C(6) do enantiômero IS. Em destaque estão as possíveis localizações da hidroxila depois do processo de hidrólise.
Analisando a quebra da ligação Pd-C(6) observamos que o ciclo que
contém os átomos de fósforo ligados ao paládio, possui uma significativa
flexibilidade ao sofrer o processo de hidrólise. E essa flexibilidade (observadas
nos ângulos C6-C29-N20-Pd11; C25-N20-Pd11-O39 e C6-C1-C29-C31) também é
determinante na obtenção da variação da entalpia de hidrólise, principalmente
quando a hidroxila se liga ao paládio como ilustra a Figura 4-14.
Ainda analisando a Figura 4-14, observamos que ao rotacionarmos o
ciclo que contém os átomos de fósforo, observa-se que a energia do complexo
diminuiu em torno de 6.7 kcal/mol devido à mudança na localização da hidroxila
Composto Hinterna
(kcal/mol) Composto
Hinterna
(kcal/mol)
H (IS-IR)
(kcal/mol)
VIIIR(PdH) 5.33 VIIIS(PdH) 3.03 -2.3
IXR(PdOH) -4.17 IXS(PdOH) -7.47 -3.3
+
IS
VIIIS(PdH)
IXS(PdOH)

61
que interage através de ligações de hidrogênio com os átomos de hidrogênio
do grupo metil ligado ao carbono quiral.
Analisando a Tabela 4-6, observamos que as diferenças das variações
da entalpia de hidrólise (H (IS-IR)) encontradas para os complexos
hidrolisados, são superiores à energia térmica, portanto para essa quebra de
ligação encontramos diferenças com relação à quiralidade das moléculas.
Figura 4-14: Geometrias das moléculas formadas na quebra da ligação Pd-C(6) do enantiômero IR. Em destaque estão as possíveis localizações da hidroxila depois da hidrólise. A molécula (a) possui uma variação da entalpia de hidrólise de -0.77 kcal/mol. A molécula (b) possui uma variação da entalpia de hidrólise de -7.47 kcal/mol.
Ainda analisando a Tabela 4-6 e a Figura 4-14 podemos concluir que, a
estrutura que necessita de menos calor encontrado para essa rota de hidrólise
foi a IXS(PdOH), isto porque o próton da água que se ligou ao carbono do anel,
interage com o paládio através de ligações de hidrogênio. A variação da
entalpia de hidrólise encontrada para o complexo IXS(PdOH) é de -7.47
kcal/mol sendo, portanto uma quebra exotérmica.
4.3 Sumário e Discussões das Rotas de Hidrólise em
Fase Gasosa
A investigação completa dos complexos hidrolisados envolve uma
grande quantidade de cálculos quânticos; por isso organizamos todos os
valores da variação da entalpia de hidrólise de todas as rotas na Tabela 4-7. Na
(b) (a)

62
última coluna dessa tabela temos as diferenças da entalpia de hidrólise para os
enantiômeros IR e IS.
É importante destacar que encontramos valores positivos para a
variação da entalpia de hidrólise, ou seja, (hidrólise endotérmica) para as
ligações Pd-P(19); Pd-P(12) e Pd-N(20); e valores negativos para a variação da
entalpia de hidrólise (hidrólise exotérmica) apenas para a ligação Pd-C(6).
Fazendo uma análise dos valores da Tabela 4-7, observamos que as
diferenças da entalpia entre os enantiômeros hidrolisados foram baixas, ou
seja, diferenças inferiores à energia térmica para todos os complexos
hidrolisados, com exceção dos complexos que envolvem a hidrólise na ligação
Pd-N(20) e Pd-C(6). Na rota de hidrólise envolvendo a ligação Pd-N(20), na qual
ocorre a protonação do grupo amina terciário presente no complexo,
observamos a maior diferença energética, em torno de 10.8 kcal/mol. Portanto
para essa quebra de ligação encontramos uma diferença significativa com
relação à quiralidade das moléculas.
Como já descrito anteriormente, na seção 4.2, melhorando o método de
cálculo quântico e aumentando o conjunto de funções base, espera-se
melhorar a qualidade dos resultados. Desta forma, calculamos os valores da
variação da entalpia de hidrólise das diferentes rotas utilizando o método de
cálculo B3LYP/ 6-31+G(d) e seus valores estão apresentados na Tabela 4-8.
Na última coluna dessa tabela temos as diferenças da entalpia de hidrólise dos
enantiômeros. É importante salientar que esses novos cálculos foram
realizados apenas para as entalpias, mantendo as mesmas geometrias das
moléculas.
A partir dos dados da Tabela 4-7 e da Tabela 4-8 observa-se que com o
método de cálculo B3LYP/6-31+G(d) os resultados obtidos apresentam o
mesmo comportamento descrito anteriormente para o método de cálculo HF/6-
31G, porém os valores numéricos alteraram.
Fazendo uma análise da Tabela 4-8, observamos que para todas as rotas
de hidrólise, a maior diferença entre os enantiômeros se manteve nos
complexos hidrolisados VII (PdOH), no valor de 26.7 kcal/mol, em favor do
enantiômero IS. Essa rota é a (f) da Figura 3-2 que representa a quebra da
ligação Pd-N(20) com protonação no N gerando o grupo NH+.

63
Tabela 4-7: Sumário dos valores calculados da variação da entalpia de hidrólise (HF/6-31G),
em fase gasosa para as diferentes rotas mostradas na Figura 3-2.
No trabalho de Barbosa e colaboradores [44], foi observado que
complexos ciclopaladados bifosfínicos (BPC), têm uma notável ação biológica
antitumoral, agindo por via lisossomal, e que o enantiômero IS apresenta uma
maior seletividade comparada ao enantiômero IR. É sabido que, as enzimas
lisossomais podem ser liberadas do lisossomo para dentro do citosol, as quais
contribuem para a ativação do processo de apoptose [108]. A presença de
grupos hidrofóbicos, bem como a protonação do grupo amina presente na
estrutura da BPC, sugere que essa molécula ativa a permeabilização da
membrana lisossomal, permitindo a liberação de enzimas lisossomais.
Ainda no trabalho de Barbosa e colaboradores [44] foi realizado um
ensaio que usou a comparação de espectros vibracionais na região do
infravermelho dos compostos em uma escala de pH (variando de 1 a 12). Os
Composto Hgás
(kcal/mol) Composto
Hgás
(kcal/mol)
H (IS-IR)
(kcal/mol)
Quebra Pd-P
IIR(PdH)aberto 82.1 IIS(PdH)aberto 82.3 0.2
IIR(PdH)fechado 65.0 IIS(PdH)fechado 65.2 0.2
IIIR(PdOH) 31.6 IIIS(PdOH) 31.3 -0.3
IVR(PdH)aberto 76.6 IVS(PdH)aberto 76.6 0.0
IVR(PdH)fechado 59.1 IVS(PdH)fechado 59.3 0.2
VR(PdOH) 31.4 VS(PdOH)fechado 31.2 -0.2
Quebra Pd-N
VIR(PdH) 75.8 VIS(PdH) 71.2 -4.6
VIIR(PdOH) 18.83 VIIS(PdOH) 8.03 -10.8
Quebra Pd-C
VIIIR(PdH) 5.33 VIIIS(PdH) 3.03 -2.3
IXR(PdOH) -4.17 IXS(PdOH) -7.47 -3.3

64
resultados indicam que, podem ser obtidas bandas de absorção entre os
comprimentos de onda 2750 a 2300 cm-1, que envolvem modos de vibração
normais do grupo –NH+. Esse resultado pode ser atribuído na quebra da
ligação de coordenação Pd-N e uma protonação do grupo amina terciário
presente no complexo de paládio.
Tabela 4-8: Sumário dos valores calculados da variação da entalpia de hidrólise (B3LYP/ 6-31+G(d)), em fase gasosa para as diferentes rotas mostradas na Figura 3-2.
Considerando a hidrólise dos enantiômeros que resultam nos complexos
VII(PdOH), ambos apresentam um processo endotérmico como mostrado nas
Tabelas 4-7 e 4-8, porém o trabalho de Barbosa e colaboradores [44], sugere
um possível processo de protonação do grupo amina terciário presente na
molécula BPC em meio ácido, com pH variando de 5.1 a 6.5. Como os cálculos
realizados neste trabalho não envolve o efeito do pH é possível que em meio
ácido essa rota de hidrólise seria favorável energéticamente ocorrendo a
Composto Hgás
(kcal/mol) Composto
Hgás
(kcal/mol)
H (IS-IR)
(kcal/mol)
Quebra Pd-P
IIR(PdH)aberto 57.1 IIS(PdH)aberto 57.4 0.3
IIR(PdH)fechado 38.3 IIS(PdH)fechado 32.6 -5.7
IIIR(PdOH) 24.0 IIIS(PdOH) 24.2 0.2
IVR(PdH)aberto 50.7 IVS(PdH)aberto 50.9 0.2
IVR(PdH)fechado 32.3 IVS(PdH)fechado 32.6 0.3
VR(PdOH) 28.0 VS(PdOH) 27.8 -0.2
Quebra Pd-N
VIR(PdH) 63.3 VIS(PdH) 58.8 -4.5
VIIR(PdOH) 38.3 VIIS(PdOH) 11.6 -26.7
Quebra Pd-C
VIIIR(PdH) 5.83 VIIIS(PdH) 2.43 -3.4
IXR(PdOH) 3.23 IXS(PdOH) 0.63 -2.6

65
protonação do grupo amina terciário presente na molécula de BPC e ativando
proteínas lisossomais, desencadeando, portanto, um processo de apoptose.
Identificamos ainda que, nas ligações Pd-N e Pd-C há diferenças
significativas (em ambos os métodos) com relação à quiralidade presente nos
enantiômeros estudados. Nas quebras das ligações Pd-N e Pd-C do
enantiômero IS, observamos que a hidroxila ligada ao paládio pode formar mais
ligações de hidrogênio com o grupo metil (ligados ao átomo de N(20)) e também
com o átomo de hidrogênio ligado ao carbono quiral. Isto não ocorre com o
enantiômero IR, pois esses átomos de hidrogênio estão do lado oposto à
hidroxila como mostra a Figura 4-9, Figura 4-10, Figura 4-12 e Figura 4-13.
Portanto, numa possível quebra dessas ligações, no enantiômero IS existe uma
possibilidade maior de ocorrer mais interações intramoleculares, comparado ao
enantiômero IR, o que nos indica a possibilidade de explicarmos a
enantioseletividade desses compostos.
Porém é sabido que, não podemos extrapolar os resultados obtidos
através das análises realizadas de moléculas isoladas por cálculos quânticos
para o meio líquido, pois o efeito do solvente nas reações que ocorrem hidrólise
é de grande importância, e esse efeito não é incluído nesse tipo de análise
devido a limitações que existem com relação aos números de átomos
envolvidos nos cálculos quânticos. Dessa forma, realizamos estudos das rotas
de hidrólise em meio aquoso com o objetivo de analisar o efeito do solvente
nessas rotas de hidrólise.
4.5 Análise das Rotas de Hidrólise em Meio Aquoso
Utilizando o procedimento descrito na seção 3.4, realizamos simulações
de cada um dos enantiômeros IR e IS, simulações de uma molécula de água e
simulações para cada um dos 16 complexos hidrolizados, todos embebidos em
solução aquosa. Nosso objetivo com essas simulações foi estimar a variação
da entalpia de hidrólise através da variação da energia potencial de interação
soluto-solvente ( WIU ), e a variação da energia potencial de interação
solvente-solvente ( WWU ), também conhecida como energia potencial de
relaxação do solvente.

66
É sabido que, o cálculo da relaxação do solvente apresenta uma
convergência bastante lenta e é necessário uma grande quantidade de passos
de simulação para garantir sua convergência.
Devido à dificuldade de calcular esse termo com precisão, escolhemos a
rota de hidrólise que envolve a quebra da ligação Pd-C(6), IXR(PdOH), para a
análise da relevância desse termo e calculamos a variação da energia potencial
de interação solvente-solvente de forma bastante criteriosa, garantindo sua
convergência.
4.5.1 Análise da Energia de Relaxação do Solvente
Utilizando o procedimento descrito pela Figura 3-4, realizamos 71
simulações do IR solvatado em 800 moléculas de água, 71 simulações para
uma molécula de água solvatado em mais 450 moléculas de água e 71
simulações para o complexo hidrolisado IXR(PdOH) solvatado em 1250
moléculas de água , todos embebidos em solução aquosa.
Cada simulação passou por um processo de termalização de 3x104
ciclos MC. Em seguida, foram realizados 24 x103 ciclos MC do sistema em
equilíbrio térmico. No total das 71 simulações, analisamos a variação da
energia potencial de interação soluto-solvente ( WIU ), e a variação da
energia potencial de interação solvente-solvente )U( WW com um total de
17x 105 ciclos MC. Esses valores foram calculados e são mostrados na Tabela
4-9.
Na Figura 4-15 mostra os valores da variação da energia potencial de
interação solvente-solvente em torno do seu valor médio, em função dos ciclos
MC. Fica evidente através dessa figura, que a relaxação do solvente apresenta
grandes variações em torno do seu valor médio. Portanto, é de grande
importância confirmar a convergência desse valor para garantir a confiabilidade
dos nossos resultados.
Na Figura 4-16, analisamos a convergência dos termos WIU e WWU .
Do lado direito temos a média acumulada das médias da variação da energia
potencial de interação soluto-solvente ( WIU ), e do lado esquerdo temos a
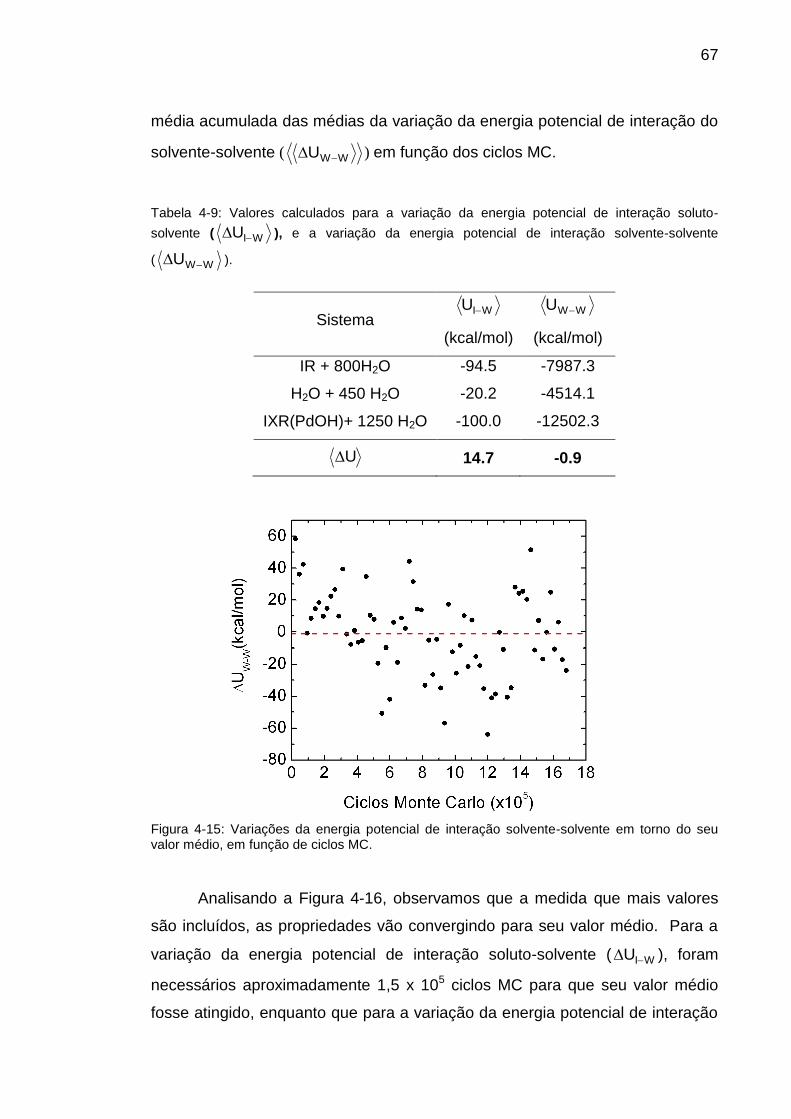
67
média acumulada das médias da variação da energia potencial de interação do
solvente-solvente ( WWU ) em função dos ciclos MC.
Tabela 4-9: Valores calculados para a variação da energia potencial de interação soluto-
solvente ( WIU ), e a variação da energia potencial de interação solvente-solvente
( WWU ).
Sistema WIU
(kcal/mol)
WWU
(kcal/mol)
IR + 800H2O -94.5 -7987.3
H2O + 450 H2O -20.2 -4514.1
IXR(PdOH)+ 1250 H2O -100.0 -12502.3
U 14.7 -0.9
Figura 4-15: Variações da energia potencial de interação solvente-solvente em torno do seu valor médio, em função de ciclos MC.
Analisando a Figura 4-16, observamos que a medida que mais valores
são incluídos, as propriedades vão convergindo para seu valor médio. Para a
variação da energia potencial de interação soluto-solvente ( WIU ), foram
necessários aproximadamente 1,5 x 105 ciclos MC para que seu valor médio
fosse atingido, enquanto que para a variação da energia potencial de interação

68
solvente-solvente ( WWU ) foram necessários 14 x 105 ciclos MC. Ou seja,
para a estimativa da propriedade WWU , foi necessário um grande número de
ciclos MC para obtermos um valor confiável, enquanto para a estimativa do
termo WIU bem menos ciclos são necessários para sua convergência.
Figura 4-16: Gráfico da convergência da variação da energia potencial de interação soluto-
solvente ( WIU ) e da energia potencial de interação solvente – solvente ( WWU ).
Através da Tabela 4-9, verificamos que o valor médio calculado da
variação da energia potencial de interação solvente-solvente WWU foi de -0,9
kcal/mol, ou seja, a contribuição energética da relaxação do solvente para esse
sistema estudado é pequena e podemos, portanto, despreza-la em nossos
cálculos sem muita perda de informação do sistema.
Dessa forma, podemos reescrever a Equação 15 descrita na seção
3.4.2. Excluindo o termo WWU :
kTUHH WI)gás()sol( (16)
onde )gás(H é a variação da entalpia de hidrólise das moléculas em fase
gasosa, WIU é a variação da energia potencial de interação soluto-solvente,
e kT é a diferença de variação da energia cinética.
Dessa última equação, o 1º termo à direita, )gás(H , já foi calculado e
está mostrada nas Tabelas 4-7 e 4-8. O 2º termo, UI-W, pode ser facilmente
calculado realizando uma única simulação computacional com

69
aproximadamente 1,5x105 ciclos MC e o último termo dessa equação, kT, é
constante e igual a 0.6 kcal/mol.
4.5.2 Análise da Energia de Interação Soluto-Solvente
Realizamos simulações dos enantiômeros IS e IR e dos complexos
hidrolisados em solução aquosa utilizando o procedimento descrito no capítulo
anterior. Nosso objetivo com essas simulações é estimar a variação da energia
potencial de interação soluto-solvente (UI-W).
Cada simulação passou por um processo de termalização com 21 x103
ciclos MC. Em seguida, foram realizados 1,5 x 105 ciclos MC no equilíbrio. Na
Tabela 4-10 pode-se observar os valores das energias potenciais de interação
soluto-solvente, obtidos em cada simulação.
Um fator importante na análise da energia potencial de interação soluto -
solvente que deve ser levado em consideração é o momento de dipolo () do
soluto envolvido no meio solvente. Normalmente é esperado que a
estabilização em um solvente polar, seja dominada pelo momento de dipolo
com o soluto [109]. Na realidade, esse princípio, dá origem aos tratamentos de
campo de reação de efeitos solventes [110],[111].
Na Figura 4-17 analisamos as energias potencias de interação soluto-
solvente dos complexos hidrolisados em função dos seus momentos de dipolo.
Do lado esquerdo temos a energia potencial de interação soluto-solvente dos
complexos hidrolisados do enantiômero IR e do lado direito temos a energia
potencial de interação do soluto-solvente dos complexos hidrolisados do
enantiômero IS.
Através da análise da Figura 4-17 observamos que os complexos com o
momento de dipolo maior apresentam conseqüentemente, um valor de energia
potencial de interação soluto-solvente mais forte em meio aquoso.
Verificamos ainda, a partir da Tabela 4-10, que a diferença das energias
potenciais de interação soluto-solvente entre os complexos hidrolisados IR e IS
foram baixas, com exceção dos complexos que envolvem a quebra da ligação
Pd-P(12). Na rota de hidrólise envolvendo a ligação Pd-P(12), na qual ocorre a
protonação do átomo de paládio e o anel bifosfínico permanece próximo ao

70
centro metálico formando uma estrutura fechada como ilustrado na Figura 4-7 e
na Figura 4-8, obtemos a maior diferença energética, em torno de 10.97
kcal/mol, sendo o complexo hidrolisado IVS(PdH)fechado o que interage mais
fortemente com o meio solvente.
Tabela 4-10: Valores calculados para energia potencial de interação soluto-solvente dos complexos hidrolisados embebidos em 800 moléculas de água.
Figura 4-17: Energia potencial de interação soluto-solvente dos complexos hidrolisados IR e IS respectivamente, em função dos momentos de dipolo.
Composto
Debye)
UI-W
(kcal/mol) Composto
Debye)
UI-W
(kcal/mol)
IIR(PdH)aberto 25.45 -175.88 II(PdH)Saberto 25.54 -177.27
IIR(PdH)fechado 12.76 -120.10 IIS(PdH)fechado 12.78 -122.72
IIIR(PdOH) 10.78 -107.61
IIIS(PdOH) 10.97 -105.19
IVR(PdH)aberto 23.28 -178.11 IVS(PdH)aberto 23.11 -183.41
IVR(PdH)fechado 14.58 -133.01 IVS(PdH)fechado 11.43 -122.04
VR(PdOH) 9.91 -106.16 VS(PdOH) 9.82 -107.68
VIR(PdH) 4.55 -95.98 VIS(PdH) 11.64 -99.41
VIIR(PdOH) 5.12 -103.69 VIIS(PdOH) 3.68 -105.71
VIIIR(PdH) 5.66 -94.21 VIIIS(PdH) 5.12 -93.91
IXR(PdOH) 7.39 -104.99 IXS(PdOH) 7.81 -104.82

71
Comparando os momentos de dipolo dos complexos hidrolisados
IVR(PdH)aberto e IVS(PdH)aberto encontramos respectivamente os valores de
14.58 D e 11.43 D, ou seja uma diferença de 3.15 D. Se analisarmos de forma
análoga, os complexos hidrolisados VIIR(PdH) e VIIS(PdH) encontra-se
respectivamente os valores do momento de dipolo de 4.55 D e 11.64 D, ou
seja, uma diferença de 7.17 D. Apenas para essas duas rotas de hidrólise foi
verificada uma diferença significativa entre os valores dos momentos de dipolo
dos complexos hidrolisados IR e IS.
4.5.3 Análise da Variação de Entalpia de Hidrólise em Solução
Finalmente podemos fazer uma análise do processo de hidrólise desses
enantiômeros, através do cálculo da variação da entalpia de hidrólise em
solução, a fim de propormos uma explicação da enantioseletividade desses
compostos.
A variação da entalpia de hidrólise pode ser calculada pela Equação 16
descrita na seção 4.5.1. Os valores calculados da variação da entalpia de
hidrólise de cada complexo são mostrados na Tabela 4-11. Os valores das
variações das entalpias apresentados nesta tabela foram calculados utilizando
a variação da entalpia de hidrólise das moléculas em fase gasosa (H(gás)),
obtida através do método de cálculo HF/6-31G. Na última coluna dessa tabela
temos as diferenças das variações da entalpia entre os enantiômeros IR e IS
hidrolisados.
Observamos através da Tabela 4-11, que para todas as possíveis rotas
de hidrólise estudadas, dos enantiômeros IR e IS, os valores das variações de
entalpia de hidrólise foram endotérmicas, ou seja, do ponto de vista energético
estas rotas necessitam de calor para romper as ligações. Verificamos ainda
que, as maiores diferenças energéticas se concentram nas rotas de hidrólise
que envolve a quebra das ligações Pd-P(12) no valor de 11.07 kcal/mol e a rota
que envolve a quebra da ligação Pd-N(20) no valor de -12.91 kcal/mol.
Considerando os valores obtidos na Tabela 4-11, decidimos investigar a
variação da entalpia de hidrólise considerando a variação da entalpia de
hidrólise das moléculas em fase gasosa (H(gás)) calculada pelo método B3LYP/

72
6-31+G(d). Os valores das novas variações das entalpias de hidrólise estão
apresentados na Tabela 4-12. Na última coluna dessa tabela temos as
diferenças das variações das entalpias entre os enantiômeros IR e IS
hidrolisados.
Tabela 4-11: Valores calculados para a variação da entalpia de hidrólise dos complexos hidrolisados em solução. A variação da entalpia de hidrólise das moléculas em fase gasosa foi calculada pelo método HF/6-31G.
Analisando os valores da Tabela 4-12, nota-se que duas possíveis rotas
de hidrólise são exotérmicas, e são elas as rotas que envolvem a quebra da
ligação Pd-P(19) (II(PdH)aberto) e a quebra da ligação Pd-P(12) (IV(PdH)aberto),
sendo o enantiômero hidrolisado IS o que libera maior calor na quebra das
ligações, em ambas as rotas.
Através de testes experimentais desenvolvidos por Caires e
colaboradores, observa–se que quando esses enantiômeros são expostos
Composto H(sol)
(kcal/mol) Composto
H(sol)
(kcal/mol)
H (IS-IR)
(kcal/mol)
Quebra Pd-P
IIR(PdH)aberto 21.38 IIS(PdH)aberto 20.18 -1.20
IIR(PdH)fechado 60.15 IIS(PdH)fechado 57.66 -2.49
IIIR(PdOH) 39.20
IIIS(PdOH) 41.26 2.06
IVR(PdH)aberto 13.66 IVS(PdH)aberto 8.36 -5.30
IVR(PdH)fechado 41.30 IVS(PdH)fechado 52.37
11.07
VR(PdOH) 40.44 VS(PdOH) 38.68 -1.76
Quebra Pd-N
VIR(PdH) 85.44 VIS(PdH) 81.78 -3.66
VIIR(PdOH) 30.38 VIIS(PdOH) 17.47 -12.91
Quebra Pd-C
VIIIR(PdH) 26.25 VIIIS(PdH) 24.21 -2.04
IXR(PdOH) 6.01 IXS(PdOH) 2.84
-3.17

73
muito tempo em solução aquosa com pH neutro (aproximadamente 7), ocorre
um desligamento do grupo bifosfínico, o qual se precipita no meio formando um
sólido branco.
Tabela 4-12: Valores calculados para a variação da entalpia de hidrólise dos complexos hidrolisados. A variação da entalpia de hidrólise das moléculas em fase gasosa foi calculada pelo método B3LYP/ 6-31+G(d).
Considerando esses resultados, acredita-se que a hidrólise desses
agentes anti-tumorais possa ocorrer no enantiômero IS, na quebra da ligação
Pd-P sendo a ligação com o fósforo 12 a mais provável devido à maior
liberação de calor na quebra da ligação.
Verificamos ainda através da Tabela 4-12 que a maior diferença da
variação da entalpia de hidrólise foi encontrada entre os enantiômeros IR e IS
hidrolisados envolvendo a quebra da ligação Pd-N(20). A diferença entre os
enantiômeros IR e IS hidrolisados, foi de aproximadamente 29 kcal/mol, ou
Composto H(sol)
(kcal/mol) Composto
H(sol)
(kcal/mol)
H (IS-IR)
(kcal/mol)
Quebra Pd-P
IIR(PdH)aberto -3.51 IIS(PdH)aberto -4.74 -1.23
IIR(PdH)fechado 33.46 IIS(PdH)fechado 25.03 -8.43
IIIR(PdOH) 32.31
IIIS(PdOH) 34.15 1.84
IVR(PdH)aberto -12.20 IVS(PdH)aberto -17.32 5.12
IVR(PdH)fechado 14.49 IVS(PdH)fechado 25.71
11.22
VR(PdOH) 37.08 VS(PdOH) 35.28 -1.80
Quebra Pd-N
VIR(PdH) 73.08 VIS(PdH) 70.02 -3.06
VIIR(PdOH) 49.87 VIIS(PdOH) 21.03 -28.84
Quebra Pd-C
VIIIR(PdH) 26.87 VIIIS(PdH) 23.69 -3.18
IXR(PdOH) 13.44 IXS(PdOH) 10.92
-2.52

74
seja, essa rota de hidrólise apresenta uma diferença significativa das
interações desses agentes anti-tumorais quirais com o meio solvente.
Analisando os valores das variações da entalpia de hidrólise dos
complexos VII(PdOH), observamos que o mesmo precisa de menos calor para
romper a ligação . Acredita-se, portanto, que a localização do átomo de
hidrogênio presente no carbono quiral do enantiômero IS, favoreça a sua
interação com as moléculas de água.
Como descrito anteriormente, na seção 4.4, acredita-se através de
espectroscopia vibracional na região do infravermelho que ocorra a quebra da
ligação Pd-N com a formação de uma espécie capaz de romper os lisossomos
quando expostos ao pH ácido (aproximadamente o pH dos lisossomos, ou
seja, 5).
Na tentativa de verificar a influência do pH nessas rotas de hidrólise,
realizamos uma nova simulação dos enantiômeros IR e IS nas mesmas
condições termodinâmicas descritas anteriormente, porém incluindo agora
moléculas de água dissociadas, buscando simular um pH ácido para o meio
solvente, e de forma análoga, calculamos a variação da entalpia de hidrólise
para os complexos estudados.
Quando simulamos os enantiômeros IR e IS em meio solvente na
presença dos íons H+ e OH-, observamos que todas as rotas de hidrólise são
exotérmicas. Verificamos ainda que, as maiores diferenças energéticas
permanecem nas mesmas rotas de hidrólise descrita anteriormente, ou seja,
nas quebras das ligações Pd-P12 e Pd-N.
Acredita-se, portanto que quando os enantiômeros IR e IS forem
expostos a um pH ácido por um longo período de tempo, todas as ligações
envolvendo o metal paládio serão rompidas. O mais adequado para esse tipo
de análise é identificar o efeito temporal da interação dos enantiômeros IR e IS
com o meio ácido, priorizando a identificação das rupturas das ligações mais
exotérmicas em um curto tempo de exposição.

75
5 - CONCLUSÕES E SUGESTÕES
Neste trabalho estudamos as possíveis mudanças nas propriedades
conformacionais e eletrônicas de moléculas simplificadas que serviram de
modelo para os enantiômeros S(-) e R(+) dos complexos [Pd(C2, N-
dmpa)(dppe)]Cl (Sdmpa e Rdmpa apresentados na Figura 1-3) em processos
de hidrólise. Utilizamos técnicas de modelagem molecular, cálculos quânticos
e simulações computacionais, com o objetivo de fornecer subsídios para
elucidar a enantioseletividade desses compostos e consequentemente um
melhor entendimento no mecanismo de ação anti-tumoral desses complexos.
Numa primeira abordagem optamos em simplificar os complexos Sdmpa
e Rdmpa em sistemas modelos que nomeamos de IR e IS. Para justificar essa
simplificação realizamos uma análise comparativa das geometrias dos
inibidores IR e IS, e observamos que as regiões centrais das moléculas são
muito semelhantes. Com relação à distribuição eletrônica também não foi
observado nenhuma mudança significativa entre os enantiômeros. Comparando
esses resultados com os resultados obtidos pelo estudo dos complexos Sdmpa
e Rdmpa realizado por Iadoccico [72], concluímos que não existem diferenças
significativas entre os enantiômeros IR e IS.
O estudo sobre as possíveis mudanças geométricas desses
enantiômeros em um processo de hidrólise iniciou-se através da análise dos
orbitais de fronteira HOMO e LUMO. Como a molécula de estudo é carregada
positivamente ela atua como um aceitador de elétrons, logo o orbital de
interesse é o LUMO. Portanto, através de uma análise dos orbitais moleculares,
podemos concluir que as ligações susceptíveis a uma possível quebra são as
quatro ligações do paládio, ou seja, os átomos de fósforo (P12 e P19); o átomo
de carbono (C6) e o átomo de nitrogênio (N20).
Foi realizado portanto, um estudo comparativo da quebra das quatro
ligações do Pd e foram investigadas duas possibilidades para cada quebra de
ligação:
O próton (H+) da água se ligando com o paládio (Pd) e a hidroxila
(OH-) se ligando ao outro átomo da ligação quebrada, ou;
A hidroxila (OH-) da água se ligando com o (Pd) e o próton se ligando
com o outro átomo da ligação.

76
Desta forma foi estudado 8 compostos hidrolisados para cada
enantiômero (IR e IS) totalizando 16 compostos.
Em seguida, analisamos de forma comparativa a precisão e o custo
computacional dos métodos ab initio (HF) e o Funcional de Densidade Híbrido
com o potencial de troca-correlação B3LYP através de cálculos da variação da
entalpia de hidrólise, utilizando diferentes funções bases, com o objetivo de
melhorar a descrição teórica do sistema estudado. Dessa forma, escolhemos a
rota de hidrólise que envolve a quebra da ligação Pd-N(20) do enantiômero IS
para desenvolver essa análise.
De acordo com os resultados obtidos, observamos que utilizando o
método de cálculo B3LYP com funções base 6-31+G(d), a qual pussuí funções
de polarização e difusas, os resultados da variação da entalpia de hidrólise
apresentam uma melhora significativa comparada com o melhor cálculo
(B3LYP/cc-pVTZ). Sendo assim, concluímos, considerando precisão e tempo
de processamento de cada método, que nos cálculos da energia dos sistemas
estudados, também utilizaríamos o nível de cálculo B3LYP/6-31+G(d).
Uma vez definido o nível de cálculo a ser utilizado, iniciamos uma
investigação completa dos valores da variação da entalpia dos complexos
hidrolisados.
É importante destacar que encontramos valores positivos para a
variação da entalpia de hidrólise, ou seja, (hidrólise endotérmica) para as
ligações Pd-P(19); Pd-P(12) e Pd-N(20); e valores negativos (hidrólise exotérmica)
apenas para a ligação Pd-C(6).
Fazendo uma análise das rotas de hidrólise estudadas cujos valores da
variação da entalpia de hidrólise em fase gasosa estão descritos na Tabela 4-7,
observamos que para todas as rotas de hidrólise que envolvem a quebra da
ligação Pd-P, a rota que obteve uma diferença significativa na variação da
entalpia de hidrólise, ou seja, uma diferença superior à energia térmica, foram
os complexos hidrolisados II(PdH)fechado. Além disso, a maior diferença da
entalpia se manteve nos complexos hidrolisados VI(PdOH), no valor de 26.7
kcal/mol.
Identificamos ainda que nas ligações Pd-N e Pd-C há diferenças
marcantes com relação à quiralidade presente nos enantiômeros estudados.
Nas quebras das ligações Pd-N e Pd-C do enantiômero IS, observamos que a

77
hidroxila da água pode interagir com os átomos de hidrogênio ligados ao grupo
metil (ligado ao átomo de N20) e também ao átomo de hidrogênio ligado ao
carbono quiral, o qual no enantiômero IR esse átomo está do lado oposto à
hidroxila como mostra a Figura 4-9, Figura 4-10, Figura 4-12 e a Figura 4-13.
Portanto, numa possível quebra dessas ligações, o enantiômero IS existe uma
possibilidade maior de ocorrer mais interações intramoleculares, comparado ao
enantiômero IR, o que indica a possibilidade de explicarmos a
enantioseletividade desses compostos.
Porém é sabido que, não pode-se extrapolar os resultados obtidos
através das análises de moléculas isoladas para o meio líquido, pois o efeito do
solvente na análise de reações que ocorrem hidrólise é de grande importância.
Dessa forma, realizamos uma análise das rotas de hidrólise em meio aquoso
com o objetivo de analisar o efeito do solvente nessas rotas de hidrólise.
Portanto, realizamos simulações dos enantiômeros IR e IS, de água e
dos complexos hidrolisados, todos embebidos em solução aquosa. O objetivo
com essas simulações foi estimar a variação da entalpia de hidrólise através da
variação da energia potencial de interação soluto-solvente ( WIU ), e a
variação da energia potencial de interação solvente-solvente ( WWU ), também
conhecida como energia potencial de relaxação do solvente.
Como resultado dessas simulações pode-se concluir que a variação da
energia potencial de relaxação do solvente para esse sistema estudado é
aproximadamente zero e podemos, portanto, desprezá-los em nossos cálculos.
Além disso, observa-se que os complexos hidrolisados com o momento de
dipolo maior apresentam conseqüentemente, um valor da variação da energia
potencial de interação soluto-solvente ( WIU ), mais baixo em meio aquoso.
Foi possível analisar através da Tabela 4-11 que duas possíveis rotas de
hidrólise são exotérmicas, e são elas as rotas que envolvem a quebra da
ligação Pd-P(19) ((PdH)aberto) e a quebra da ligação Pd-P(12) (IV(PdH)aberto),
sendo o complexo hidrolisado IS o que libera maior calor na quebra das
ligações, em ambas as rotas.
Esses resultados teóricos apresentam concordância com testes
experimentais desenvolvidos por Caires e colaboradores. Esses resultados
indicam um possível desligamento do grupo bifosfínico, o qual se precipita no

78
meio formando um sólido branco, quando esses enantiômeros são expostos
muito tempo em solução aquosa com pH neutro. Dessa forma, acredita-se que
a hidrólise desses agentes anti-tumorais possa ocorrer no enantiômero IS na
quebra da ligação Pd-P sendo a ligação com o fósforo 12 a mais provável
devido à maior liberação de calor na quebra da ligação.
Observamos ainda através da Tabela 4-11 que a maior diferença da
variação da entalpia de hidrólise foi encontrada entre os enantiômeros IR e IS
hidrolisados envolvendo a quebra da ligação Pd-N(20). A diferença entre esses
complexos hidrolisados, foi de aproximadamente 29 kcal/mol, sendo que o
complexo VIIS(PdOH) precisa de menos calor para romper a ligação.
Portanto, pode-se concluir que essa rota de hidrólise apresenta uma diferença
significativa das interações desses agentes anti-tumorais com o meio solvente.
Acredita-se que essa diferença das interações dos enantiômeros com meio
aquosa possa ser justificada pela localização do átomo de hidrogênio presente
no carbono quiral do enantiômero IS, que teoricamente favorece a sua
interação com as moléculas de água.
No trabalho de Barbosa e colaboradores [45], foi observado através de
espectroscopia vibracional na região do infravermelho, que ocorre a quebra da
ligação Pd-N com a formação de uma espécie capaz de romper os lisossomos
quando expostos ao pH ácido (aproximadamente o pH dos lisossomos).
Dessa forma, para trabalhos futuros, pretendemos estender o projeto
para o estudo destes complexos em meio ácido, na tentativa de verificar a
influência do pH nessas rotas de hidrólise e realizar cálculos da variação da
energia livre que nos informará sobre a espontaneidade do processo de
hidrólise.

79
REFERÊNCIAS
[1] GUPTA; S. P.; Quantitative Structure – Activity Relationship Studies on anticancer Drugs. Chem. Rev.; 94, 1507, 1994.
[2] MCCONATHY, J.; OWENS, M.J.; Stereochemistry in Drug Action. J. Clin. Psychiatry.; 5, 70, 2003.
[3] SOLOMONS, T.; GRAHAM, W.; Química Orgânica. 7. ed. Rio de Janeiro: LTC Editora, 2000. 165 p.
[4] CAHN, R. S.; INGOLD, C.; PRELOG, V.; Specification of Molecular Chirality. Chem. Int. Ed. Engl.; 5, 385, 1966.
[5] SILVA, L.F.; Esterioquímica.2008. Disponível em: http://www.dqi.ufms.br/~lp4/QFL305Aula008.pdf. Acessado em 02.nov.2008.16h15min. [6] FRANCOTTE, E. R. Enantioselective chromatography as a powerful alternative for the preparation of drug enantiomers. Journal of Chromatography A.; 906, 379, 2001. [7] ERIKSSON, T.; BJORKMAN, S.; HOGLUND, P.; Clinical Pharmacology of Thalidomide. Eur. J. Clin. Pharmacol.; 57, 365, 2001.
[8] SHELDON, R. A. Chirotechnology- Industrial Synthesis of Optically Active Compounds. Nova Iorque: Marcel Dekker Inc, 1993.
[9] CAIRES, A. C. F.; Recent Advances Involving Palladium (II) Complexes for Cancer Therapy. Anticancer Agents Med. Chem.; 7, 484, 2007.
[10] RODRIGUES, E.G.; SILVA, L.; FAUSTO, D. M.; HAYASHI, M.S.; DREHER, S. D.; SANTOS, E.L.; PESQUEIRO, J. B.; TRAVASSOS, L.R.; CAIRES, A. C. F.; Cyclopalladated Compounds as Chemotherapeutic Agents: Antitumor Activity Againsts a Murine Melanoma Cell Line. Int. J. Cancer.; 107, 498, 2003.
[11] HAIDUC; I., SILVESTRE. C.; Metal compounds in cancer chemotherapy. Chem. Rev.; 99, 253, 1990.
[12] ROSENBERG; B.; VAN CAMP; L.; KRIGAS, T.; Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature; 205, 698, 1965. [13] ROSENBERG; B.; VAN CAMP.; TROSKO, J. E.; MANSOUR; V. H.; Platinum Compounds: a New Class of Potent Antitumour Agents. Nature; 222, 385, 1969.

80
[14] HIGBY.; D.J.; WALLACE.; H.; HOLLAND.; J.F.; Cis diamminedichloroplatinum (NSC-119875): a phase I study. Cancer. Chemother. Rep.; 57, 459, 1973.
[15] WALLACE, H.J.; HIGBY, D.J.; Platinum Coordination Complexes in Cancer Chemotherapy; CONNORS, T.A.; ROBERTS, J.J., Eds.; Springer-Verlag, Berlin, 1974, p. 167. [16] WILTSHAW, E.; CARR, B.; Platinum Coordination Complexes in Cancer Chemotherapy; CONNORS, T.A.; ROBERTS, J.J., Eds.; Springer-Verlag, New York, 1974, Vol 48, p. 178.
[17] HOWELL, B.; Platinum and other Metal Coordination Compounds in Cancer Chemotherapy, Plenum Press, New York, 1991.
[18] BARNARD, C.J.F.; CLEARE, M.J.; HYDES, P. C.; Second generation anticancer platinum compounds. Chem. Brit.; 22, 1001, 1986.
[19] BARNARD, C.J.F.; Platinum Anti-Cancer Agents. Platinum. Met. Rev.; 33,162,1989.
[20] BELLON; S. F.; LIPPARD; S.J.; Bending studies of DNA site-specifically modified by cisplatin, trans-diamminedichloroplatinum(II) and cis-[Pt(NH3) 2 (N3-cytosine) Cl]+. Biophys. Chem.; 35, 179,1990.
[21] REEDIJK, J.; FICHTINGER-SCHEPMAN, A.M.J.; VAN OOSTEROM, A.T.; VAN DER PUTTE, P.; Platinum amine coordination compounds as anti-tumor drugs. Molecular aspects of the mechanism of action. Struct. Bond; 67,53,1987.
[22] LIPPARD, S.J.; Chemistry and molecular biology of platinum anticancer drugs. Pure and Appl. Chem.; 59, 731, 1987. [23] ROBERTS, J.J.; KNOX, R.J.; FRIEDLOS, F.; LYDALL, D.A. In Biochemical Mechanism of Platinum Antitumor Drugs; Mc Brain D. C. H.; Slater, T. F.; Eds.; IRL Press Oxford, 1987. [24] SPINGLER, B.; WHITTINGTON, D.A.; LIPPARD, S.J.; Å Crystal Structure of an Oxaliplatin 1,2-d(GpG) Intrastrand Cross-Link in a DNA Dodecamer Duplex. Inorg .Chem.; 40, 5596, 2001.
[25] STRUM, S.B.; MCDERMED, J. E.; LIPONI, D.F.; High-dose intravenous metoclopramide versus combination high-dose metoclopramide and intravenous dexamethasone in preventing cisplatin- induced nausea and emesis: a single-blind crossover comparison of antiemetic efficacy. Clin. Oncol.; 3, 245,1985. [26] ROSENBERG, B.; Fundamental studies with cisplatin. Cancer; 55, 2203, 1985.

81
[27] Sixth International Symposium on Platinum and Other Metal Coordination Compounds in cancer Chemotherapy, Plenum Press, San Diego, 1991, p147-183. [28] WEBSTER, L.K.; DEACON, G. B.; BUXTON, D. P.; HILCOAT, B.L.; JAMES, A. M.; ROSS, I.A. G.; THOMSON, R. J.; WAKELIN, L. P.G.; WILLIANS, T. L.; Cis-Bis(pyridine)platinum(II) organoamides with unexpected growth inhibition properties and antitumor activity. J. Med. Chem; 31, 634, 1992.
[29] VAN BENSICHEM, M.; FARREL, N.; Activation of the trans geometry in platinum antitumor complexes. Synthesis, characterization, and biological activity of complexes with the planar ligands pyridine, N-methylimidazole, thiazole, and quinoline. Crystal and molecular structure of trans-dichlorobis(thiazole)platinum(II). Inorg. Chem.; 31, 634,1992.
[30] GUO, Z.; SADLER, P. In Advances in Inorganic Chemistry; A.G. Sykes, Ed.; Academic Press: New York, 2000, Vol. 49, p.87.
[31] COLLUCCIA, M.; NASSI, A.; LOBETO, F.; BOCCARELLI, A.; MARIGGIO, M.A.; GIORDANO. D.; INTINI, F. P.; CAPUTO, P.; NATILI, G.; A trans-platinum complex showing higher antitumor activity than the cis congeners. J. Med. Chem.; 36, 510, 1993.
[32] FONTES, A. P. S.; ALMEIDA, S. G.; NADER, L. A.; Compostos de Platina em Quimioterapia do Câncer. Química Nova; 20, 398, 1997.
[33] PADHYE, S.B.; KAUFFMAN, G.B.; Transition metal complexes of semicarbazones and thiosemicarbazones. Coord. Chem. Rev.; 63,127,1985.
[34] MATESANZ, A.; PÉREZ, J.M.; NAVARRO, P.; MORENO, J.M.; COLACIO, E.; SOUZA, P.; Synthesis and characterization of novel palladium(II) complexes of bis(thiosemicarbazone). Structure, cytotoxic activity and DNA binding of Pd(II)-benzyl bis(thiosemicarbazonate). J.Inorg. Biochem.; 76, 29, 1999.
[35] KELLAND, L.R.; New platinum antitumor complexes. Crit. Rev. Oncol. Hematol.; 15, 191, 1993.
[36] KHAN, B.T.; BHATT, J.; NAJMUDDIN, K.; SHAMSUDDIN, S.; ANNAPOORNA, K.; Synthesis, antimicrobial, and antitumor activity of a series of palladium(II) mixed ligand complexes. J. Inorg. Biochem.; 44, 55,1991. [37] NAVARRO-RANNINGER, C.; LÓPEZ-SOLERA, I.; PÉREZ, J. M.; MASAGUER, J. R.; ALONSO, C.; In vitro antitumour activity of two isomeric cyclopalladiated compounds derived from benzoylbenzylidenimines. Appl. Organometal. Chem.; 7, 57, 1993.
[38] HIGGINS II, J. D.; FRICKER, S.; Synthesis and cytotoxicity of some cyclometallated palladium complexes. J. Inorg. Biochem.; 49, 149, 1993.

82
[39] NAVARRO-RANNINGER, C.; LÓPEZ-SOLERA, I.; PÉREZ, J. M.; RODRIGUES, J.; GARCIA-RUANO, J. L.; RAITBY, P.R.; MASAGUER, J. R.; ALONSO, C.; Analysis of two cycloplatinated compounds derived from N-(4-methoxyphenyl)-.alpha.-benzoylbenzylidenamine. Comparison of the activity of these compounds with other isostructural cyclopalladated compounds. J. Med. Chem.; 36, 3795, 1993.
[40] NAVARRO-RANNINGER, C.; LÓPEZ-SOLERA, I.; GONZÁLES, V. M.; PÉREZ, J. M.; ALVAREZ-VALDÉS, A.; MARTIN, A.; RAITHBY, P.R.; MASAGUER, J. R.; ALONSO, C. Cyclometalated Complexes of Platinum and Palladium with N-(4-Chlorophenyl)- -benzoylbenzylideneamine. In Vitro Cytostatic Activity, DNA Modification, and Interstrand Cross-Link Studies. Inorg. Chem.; 35, 5181, 1996.
[41] TROFIMENKO. S.; Cyclopalladation reaction, Inorg. Chem.; 12,1215,1973.
[42] ANANIAS; S. R.; SANTANA, A. M.; MAURO, A. E.; Reações de Bis-inserção de 1,2 difenilacetileno na ligação Pd-C de ciclometalados. Química Nova; 26, 53, 2003.
[43] BINCOLETTO, C.; TERSARIOL, I.L.S.; OLIVEIRA, C.R.; DREHER, S.; FAUSTO, D. M.; SOUFEN, M.A.; NASCIMENTO, F.D.; CAIRES, A.C.F.; Chiral cyclopalladated complexes derived from N,N-dimethy-1-phenethylamine with bridging bis(diphenyphosphine)ferrocene ligand as inhibitors of the cathepsin B activity and as antitumoral agents. Bioorg. Med. Chem.; 13, 3047, 2005.
[44] BARBOSA, C.M.V.; OLIVEIRA, R.C.; NASCIMENTO, F.D.; SMITH, M.C.M.; FAUSTO, D.M.; SOUFEN, M.A.; SENA, E.; ARAÚJO, R.C.; TERSARIOL, I.L.S.; BINCOLETTO, C.; CAIRES, A.C.F.; Biphosphinie palladacycle complex mediates lysosomal-membrane permeabilization and cell death in K562 leukaemia cells. Eur. J.Pharmacol.; 542, 37, 2006.
[45] SIEGBAHN, P.E.M.; BLOMBERG, M. R. A.; Transition-Metal Systems in Biochemistry Studied by High-Accuracy Quantum Chemical Methods. Chem. Rev.; 100, 421, 2000.
[46] VANQUICKENBORNE, L.G.; PIERLOOT, K.; HEYLEN, G. A.; CHIBOTARU, L.F.; CEULEMANS, A.; Theoretical Models of Exchange Interactions in Dimeric Transition-Metal Complexes. Chem. Rev.; 100, 787, 2000.
[47] NIU, S.; Hall, M. B.; Theoretical Studies on Reactions of Transition-Metal Complexes. Chem. Rev.; 100, 353, 2000. [48] FRENKING, G.; SOLÀ, M.; TORRENT, M.; Theoretical Studies of Some Transition-Metal-Mediated Reactions of Industrial and Synthetic Importance.
Chem. Rev.; 100, 439, 2000
[49] KOHN, W.; Nobel Lecture. Eletronic Structure of matter-wave functions and density functionals. Rev. Mod. Phys.; 71, 1253, 1998.

83
[50] MORGON, N.H.; CUSTODIO, R.; Teoria do Funcional de Densidade. Química Nova; 18, 44, 1995.
[51] KOCH, W.; HOLTHAUSEN, M.C.; A Chemist’s Guide to Density Functional Theory, 2nd ed.; Willey-VHC: Weinheim, 2001. [52] HARTREE, D. R. The calculation of atomic structures. New York: John Wiley, 1957, p-181.
[53] MOLLER, C.; PLESSET, M.S.; Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev.; 46, 618,1934.
[54] DEDIEU; A.; Theoretical Studies in Palladium and Platinum Molecular Chemistry. Chem. Rev.; 100, 543, 2000. [55] FIETZEK, C.; MACK, H.G.; Influence of different transition metals in phthalocyanines on their interaction energies with volatile organic compounds: an experimental and computational study. J. Mol. Model.; 13, 11, 2007.
[56] TSUBOMURA, T.; ITO, Y.; INOUE, S.; TANAKA, Y.; MATSUMOTO, K.; TSUKUDA, T.; Strongly luminescent palladium(0) and platinum(0) diphosphine complexes. Inorg. Chem.; 47, 481, 2008.
[57] HEBBEN, N.; HIMMEL, H.J.; EICKERLING, G.; HERRMANN, C.; REIHER, M.; HERZ ,V.; PRESNITZ, M.; SCHERER, W.; The Electronic Structure of the Tris(ethylene) Complexes [M(C(2)H(4))(3)] (M=Ni, Pd, and Pt): A Combined Experimental and Theoretical Study. Chemistry; 13,36,2007.
[58] AUVRAY, N.; BASU BAUL,T.S.; BRAUNSTEIN, P.; CROIZAT, P.; ENGLERT, U.;HERBERICH, G.E.; WELTER, R.; Organometallic building blocks with amino-substituted cyclopentadienyl and boratabenzene ligands for the synthesis of heterometallic complexes and clusters. Dalton Trans., 24, 2950, 2006.
[59] DENUAULT, G.; MILHANO, C.; PLETCHER, D.; Mesoporous palladium the surface electrochemistry of palladium I aqueous sodium hydroxide and the cathodic reduction of nitrite. Phys. Chem.; 7, 3545, 2005.
[60] DEUBEL, D.V.; On the Competition of the Purine Bases, Functionalities of Peptide Side Chains, and Protecting Agents for the Coordination Sites of Dicationic Cisplatin Derivatives. J. Am. Chem. Soc.; 124, 5834, 2002.
[61] WYSOKINSKI, R.; MICHALSKA, D.; The performance of different density functional methods in the calculation of molecular structures and vibrational spectra of platinum(II) antitumor drugs: cisplatin and carboplatin. J. Comput. Chem.; 22, 901, 2001.
[62] BECKE, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys.; 98, 5648, 1993.

84
[63] HAY, P.J.; WADT, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys.; 82, 270, 1985. ibid 284, ibid 299. [64] LEININGER, T.; NICKLASS, A.; STOLL, H.; DOLG, M.; SCHWERDTFEGER, P. J. Chem. Phys.;105, 1052, 1996.
[65] ANDRAE, D.; HAEUSSERMANN, U.; DOLG, M.; STOLL, H.; PREUSS, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13-17. Theor.Chim.Acta.; 77, 123, 1990.
[66] ZHANG, Y.; GUO, Z.; YOU, X. Z.; Hydrolysis Theory for Cisplatin and Its Analogues Based on Density Functional Studies. J. Am. Chem. Soc.; 123, 9378, 2001.
[67] MILBURN, G. H. W.; TRUTER, M. R.; The crystal structures of cis- and trans-dichlorodiammineplatinum(II). J. Chem. Soc. A.; 1609, 1966.
[68] ZEIZINGER, M., BURDA, J.V., SPONER, J., KAPSA, V., LESZCZYNSKI, J. A Systematic ab Initio Study of the Hydration of Selected Palladium Square-Planar Complexes. A Comparison with Platinum Analogues. J. Phys. Chem. A.;105, 8086, 2001. [69] DOLG, M.; WEDIG, U.; STOLL, H.; PREUSS, H. Energy adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys.; 86, 866, 1987. [70] JAROSLAV, B.; ZEIZINGER, M.; LESZCZYNSKI, J; Activation barriers and rate constants for hydration of platinum and palladium square-planar complexes: An ab inito study. J. Phys. Chem.; 120, 1253, 2004.
[71] DOLG, M.; STOLL, H.; PREUSS, H.; RUSSEL, M.P. Relavistic and Correlation Effects for Element 105 (Hahnium, Ha). A Comparative study if M and MO (M = Nb, Ta, Ha) Using Energy-Adjusted ab-inito Pseudopotentials. J.Chem. Phys.; 97, 5852, 1993.
[72] IADOCICCO, K. Estudos Teóricos de Propriedades estruturais e Eletrônicas de Complexos Paladaciclos Derivados dos Enantiômeros R(+) e S(-) N,N-dimetil-1fenetilamina que Apresentam Atividade Antitumoral Diferenciada, Dissertação de Mestrado, Universidade de Mogi das Cruzes, Mogi das Cruzes, (2005). [73] PRANATA, J.; WIERSCHKE, S.G.; JORGENSEN, W.L. OPLS potential functions for nucleotide bases-relative association constants of hydrogen-bonded base-pairs in chloroform. J.Am.Chem.Soc.; 113, 2810, 1991.
[74] LIENKE, A.; KLATT, G.; ROBINSON, D. J.; KOCH, K.R.; NAIDOO, K.J. Modeling platinum group metal complexes in aqueous solution. Inorg. Chem.; 40, 2352, 2001.

85
[75] FRISCH, M. J.; TRUSCKS, G. W.; SCHLEGEL, H. B.; SCUSERIA, G. E.; ROBB, M. A.; CHEESEMAN, J.R.; ZAKRZEWSKI, V. G.; MONTGOMERY, J.A, JR.; STRATMAN, R. E.; BURANT, J. C.; DAPPRICH, S.; MILLAM, J. M.; DANIELS, A. D.; KUDIN, K. N.; STRAIN, M. C.; FARKAS, O.; TOMASI, J.; BARONE, V.; COSSI, M.; CAMMI, R.; MENNUCCI, B.; POMELLI, C.; ADAMO, C.; CLIFFORD, S.; OCHTERSKI, J.; PETERSSON, G. A.; AYALA, P. Y.; CUI, Q.; MOROKUMA, K.; MALICK, D. K.; RABUCK, A.D.; RAGHAVACHARI, K.; FORESMAN, J.B.; CIOSLOWKI, J.; ORTIZ, J.V.; STEFANOV, B. B.; LIU, G.; LIASHENKO, A.; PISKORZ, P.; KOMAROMI, I.; GOMPERTS, R.; MARTIN, R.L.; FOX, D.J.; KEITH, T.; AL-LAHAM, M. A.; PENG, C. Y.; NANAYAKKARA, A.; GONZALEZ, C.; CHALLACOMBE, M.; GILL, P.M. W.; JOHSON, B.G.; CHEN, W.; WONG, M. W.; ANDRES, J. L.; HEAD-GORDON, M.; REPLOGLE, E. S.; POPLE, J. A. Gaussian 98; Gaussian, Inc., PITTSBURGH PA, 1998. [76] COUTINHO, K.; CANUTO, S. DICE (v2,8): A Monte Carlo program for molecular liquid simulation. University of São Paulo, Brazil, 2000
[77] MCWEENY, R.; Methods of Molecular Quantum Mechanics, 2nd Ed., Academic Press, New York, 1992, p.573.
[78] JENSEN, F.; Introduction to Computational Chemistry, 2nd Ed., John Wiley & Sons Ltd, New York, 2006.
[79] CRAMER, C. J.; Essentials of Computacional Chemistry: Theories and Models, 2nd Ed., John Wiley & Sons Ltd, New York, 2004.
[80] SZABO, A.; OSTLUND, N. S.; Modern quantum Chemistry: Introduction to Advanced Eletronic Structure Theory, Dover Publications, New York, 1996. [81] HEHRE, W. J.; DITCHFIELD, R.; POPLE, J. A.; Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian – Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys.; 56, 2257, 1972. [82] ATKINS, P.W.; FRIEDMAN, R.S. Molecular Quantum Mechanic. 3th. Oxford University Press., New York. 1997. [83] JOHNSON, B. G.; GILL, P. M. W.; POPLE, J. A.; The performance of a family of density functional methods. J. Chem. Phys.; 98, 5612, 1993. [84] CURTISS, L. A.; RAGHAVACHARI, K.; TRUCKS, G. W.; POPLE, J. A.; Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys.; 94, 7221, 1991.
[85] WILLIAMS, D.E.; LIPKOWITZ, K.B.; BOYD, D.B. Net atomic charge and multipole models for the ab initio molecular electricpotential. editors. Reviews in Computational Chemistry, New York: Wiley VCH, 1991 Vol. 2, p. 219–271.

86
[86] WIBERG, K. B.; BRENEMAN, The Reactions of 1,4-Dihalocubanes with Organolithiums. The Case for 1,4-Cubadiyl. J. Am. Chem. Soc.; 112, 876, 1990. [87] DIXON, S. L.; LURS, P. C.; Estimation of pKa for organic oxyacids using calculated atomic charges. J. Comput. Chem.; 14, 1460, 1993.
[88] TUPPER, K. J.; GAJEWSKI, J. J.; COUNTS, R. W.; Empirical method for the estimation of electron affinities. J. Mol. Struct. (Theochem).; 235, 263, 1991.
[89] GUADAGNINI, P. H.; BRUNS, R. E.; Cargas Atômicas em Moléculas. Química Nova; 19, 148, 1996.
[90] BRENEMAN, C. M.; WIBERG, K. B.; Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem.; 11, 361,1990. [91] BARLETTE, V.E. FREITAS, L.C.G.; Termodinâmica Estatística de Líquidos com o Método de Monte Carlo. I. Metodologia. Química Nova; 22, 254, 1999. [92] CASTELLAN, G. Fundamentos de Físico-Química. LTC Editora, Rio de Janeiro, primeira edição, 1986. [93] COUTINHO, K.; MORGON, N.H.; Métodos de Química Teórica e Modelagem Molecular. São Paulo: Livraria Física, 2007. [94] ALLEN, M. P.; TILDESLEY, D.J.; Computer Simulation of Liquids, Clarendon Press, Oxford, 1989.
[95] METROPOLIS, N.; ULAM, S.; The Monte Carlo Method, J. Am, Stat. Ass.; 44, 335,1949. [96] METROPOLIS, N.; ROSENBLUTH, W.A.; ROSENBLUTH, M.N.; TELLER, A. H.; TELLER, E.; Equation of state calculations by fast computing machines. J. Chem. Phys.; 21, 1087, 1953. [97] PATHRIA, R.K.; Statistical Mechanics, Pergamon Press, Oxford, 1965. [98] KUBO, R.; Statistical Mechanics, North-Holand, Amsterdam, 1965.
[99] BERENDSEN, H.J.C.; POSTMA, J.P.M.; VAN GUNSTEREN, W.F.; HERMANS, J.; Intermolecular Forces, ED. PULLMAM, B., Redel, Dordrecht, 1981, p.331. [100] JORGENSEN, W.L.; MAXWELL, D.S.; TIRADO-RIVES, J. Development and Testing of the OPLS All-Atom Force Field on Corformational Energetics and Properties of Organic Liquids. J.Am.Chem.Soc.;118, 11225, 1996.

87
[101] PRANATA, J.; WIERSCHKE, S.G.; JORGENSEN, W.L. OPLS potential functions for nucleotide bases-relative association constants of hydrogen-bonded base-pairs in chloroform. J.Am.Chem.Soc.; 113, 2810, 1991.
[102] LEVCHUK, V. N.; SHEYKHET, I.I.; SIMKIN, B.Ya.; Calculation the hydration energy of etanol by the Monte Carlo. Chem. Phys. Lett.; 185, 339,1991.
[103] LAZARIDIS, T.; Solvent Reorganization Energy and Entropy Hidrophobic Hydration. J. Solution. Chem.; 104, 4964, 2000. [104] GUEDES, R.C.; COUTINHO, K.; CABRAL, B.J.C.; CANUTO, S. Differential hydration of phenol and phenoxy radical and the energetics of the phenol OH bond in solution. J. Phys. Chem.B.; 107, 4304, 2003. [105] GUEDES, R.C.; COUTINHO, K.; CABRAL, B.J.C.; CANUTO, S.; CORREIA, C.F.; SANTOS, R. M.B.; SIMÔES, J. A. M.; Solvent Effects on the energetics of the phenol OH bond: Differential Solvation of Phenol and Phenoxy Radical in Benzene and Acetonitile. J. Phys. Chem. A.; 107, 9197, 2003.
[106] FETTOUHI, M.; ELALI, B.; MORSY, M.; GOLHEN, S.; OUAHAB, L.; LE GUENNIC, B.; SAILLARD, J.Y.; DARO, N.; SUTTER, J.P.; AMOUYAL, E.; Metal-dependent ferro-versus antiferromagnetic interactions in molecular crystals of square planar (M (II) imino-nitroxide radical) complexes (M= Pt, Pd). Inorg. Chem.; 42, 1316, 2003. [107] GIL, VICTOR. M.S. Orbitais em átomos e moléculas, Fundação Calouste Gulenkian, Lisboa (1996).
[108] FEHRENBACHER, N.; JAATTELA, M.; Lysosomes as targets for câncer therapy. Cancer Res., 65, 2993, 2005.
[109] COUTINHO, K.; CABRAL, B. J. C.; CANUTO, S.; "Can larger dipoles solvate less? Solute-solvent hydrogen bond and the differential solvation of phenol and phenoxy". Chem. Phys. Lett., 399, 534, 2004.
[110] TAPIA, O.; GOSCINSKI. O.; Self-consistent reaction field theory of solvent effects. Mol. Phys., 29, 1653, 1975.
[111] RIVAIL. J.L.; RINALDI, D.; A quantum chemical approach to dielectric solvent effects in molecular liquids. Chem. Phys., 18, 233, 1976.

88
APÊNDICE A
Neste apêndice mostramos os parâmetros usados nas simulações de
Monte Carlo.
Tabela A.1: Valores das coordenadas cartesianas dos enantiômeros IS e IR, e valores dos
parâmetros do potencial de interação: cargas (q), epsilon () e sigma () de cada átomo.
Parâmetros x y Z carga
(q) x y z
carga (q)
epsilon (ε)
sigma (σ)
Átomo IS IR
C1 -2.34596 0.24094 -0.31260 0.093 -2.42162 0.02043 0.16483 0.086 0,007 3,550
C2 -3.59858 0.84159 -0.32888 -0.196 -3.73267 -0.43567 0.15127 -0.218 0,007 3,550
C3 -3.74517 2.17883 0.01727 -0.101 -4.00682 -1.76912 -0.11916 -0.097 0,007 3,550
C4 -2.63956 2.91873 0.39562 -0.150 -2.96674 -2.64315 -0.38948 -0.154 0,007 3,550
C5 -1.38103 2.32609 0.41237 -0.126 -1.65203 -2.18875 -0.38202 -0.151 0,007 3,550
C6 -1.22090 0.99446 0.04811 -0.177 -1.36435 -0.85771 -0.09720 -0.130 0,007 3,550
H7 -4.46933 0.28120 -0.60463 0.156 -4.54150 0.24481 0.34485 0.156 0,030 2,420
H8 -4.71749 2.63041 -0.00052 0.149 -5.02094 -2.11712 -0.12876 0.148 0,030 2,420
H9 -2.74907 3.94747 0.67946 0.155 -3.17292 -3.67215 -0.61218 0.156 0,030 2,420
H10 -0.54644 2.92067 0.72843 0.124 -0.87428 -2.88887 -0.61722 0.134 0,030 2,420
Pd11 0.48357 -0.07640 0.05573 -0.180 0.43307 0.06306 -0.11125 -0.150 0,200 1,700
P12 1.955.97 1.88280 -0.21693 0.318 1.72895 -1.99748 0.26002 0.281 0,200 3,740
C13 3.72454 1.31778 -0.60902 -0.134 3.53165 -1.55922 0.66055 -0.072 0,066 3,500
H14 3.75384 1.07650 -1.66423 0.102 3.56567 -1.28455 1.70744 0.088 0,030 2,500
H15 4.41126 2.13714 -0.44607 0.089 4.15373 -2.43506 0.53629 0.075 0,030 2,500
C16 4.12608 0.10942 0.24189 -0.146 4.03942 -0.41647 -0.22364 -0.179 0,066 3,500
H17 4.17771 0.37305 1.29084 0.102 4.08680 -0.72048 -1.26181 0.106 0,030 2,500
H18 5.10426 -0.24493 -0.05151 0.090 5.03761 -0.12893 0.07549 0.093 0,030 2,500
P19 2.85558 -1.30064 0.08561 0.295 2.88120 1.09349 -0.14065 0.321 0,200 3,740
N20 -0.94389 -1.71259 0.13557 0.215 -0.84438 1.80582 -0.31858 0.086 0,170 3,250
C21 -1.29707 -1.94846 1.56284 -0.273 -1.19896 1.94732 -1.76182 -0.288 0,066 3,500
H22 -0.39486 -2.14072 2.12522 0.098 -0.29901 2.08752 -2.34222 0.125 0,030 2,500
H23 -1.95197 -2.80501 1.66342 0.109 -1.84395 2.80830 -1.90388 0.109 0,030 2,500
H24 -1.78387 -1.07763 1.96942 0.151 -1.70770 1.06453 -2.10810 0.142 0,030 2,500
C25 -0.37536 -2.95219 -0.44783 -0.286 -0.21579 3.07254 0.12971 -0.277 0,066 3,500
H26 0.47691 -3.26462 0.13570 0.108 0.57847 3.33218 -0.55362 0.110 0,030 2,500
H27 -0.06276 -2.77085 -1.46555 0.108 0.19436 2.97016 1.11910 0.120 0,030 2,500
H28 -1.09573 -3.75991 -0.44108 0.120 -0.94094 3.87997 0.12950 0.128 0,030 2,500
C29 -2.12173 -1.21077 -0.68321 0.022 -2.08422 1.45781 0.47873 0.072 0,066 3,500
H30 -1.76860 -1.25061 -1.70831 0.095 -2.88101 2.11772 0.14370 0.073 0,030 2,500
C31 -3.36723 2.09596 -0.58047 -0.360 -1.89527 1.65807 1.98705 -0.268 0,066 3,500
H32 -3.85444 -2.00488 0.38076 0.101 -1.77686 2.69845 2.25740 0.086 0,030 2,500
H33 -3.13742 -3.13818 -0.75408 0.112 -1.04401 1.09634 2.35330 0.082 0,030 2,500
H34 -4.07907 -1.80542 -1.34159 0.132 -2.77393 1.28618 2.49679 0.112 0,030 2,500
H35 1.65320 2.81366 -1.22704 0.035 3.39146 1.93749 -1.15069 0.007 0,000 0,000
H36 3.28129 -1.97309 -1.08358 0.009 3.33964 1.77507 1.01040 0.007 0,000 0,000
H37 3.28279 -2.21885 1.06952 0.012 1.33993 -2.86375 1.29763 0.041 0,000 0,000
H38 2.11137 2.72158 0.90594 0.035 1.83075 -2.88584 -0.83020 0.038 0,000 0,000

89
Água
O 0,00000 -2,00000 0,00000 -0,820 0,155 3,165
H 0,81650 -2,57740 0,00000 0,420 0,000 0,000
H -0,81650 -2,57740 0,00000 0,420 0,000 0,000
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo
Recommended

















