
Linfohistiocitose Hemofagocítica: Estamos diagnosticando?
Monica CyprianoGRAACC/IOP/UNIFESP

Declaração de Conflito de Interesse
Nenhum Conflito de Interesse

Linfohistiocitose Hemofagocítica (HLH)
Síndrome Hemofagocítica Linfohistiocitose eritrofagocítica/
hemofagocítica familar (FEL ou FHL) Síndrome de Ativação Macrofágica (MAS)


Classificação das HistiocitosesIHS, 1987 IHS, 1997 Desordens
Classe I Desordens Relacionadas às Células Dendríticas
Histiocitose de Células de Langerhans Xantogranuloma Juvenil
Classe II Desordens Relacionadas a Macrófagos
Síndromes Hemofagocíticas
Primária Secundária
Rosai-Dorfman
Classe III Histiocitoses Malignas LMA-M4 LMA-M5 LMMC Sarcoma Histiocítico
Favara e cols. MPO 1997;29:157Chu, Lancet 1987;1:208

Histology and somatic mutations of histiocytoses of group L, C, R, M, and H.
Jean-François Emile et al. Blood 2016;127:2672-2681©2016 by American Society of Hematology

Linfohistiocitose Hemofagocítica: Estamos diagnosticando?
Levantamento GRAACC (25 anos de RHC) 11 casos desde 2003 (RHC)
• 5 casos entre 2003 – 20141 vivo
• 6 entre 2015- 20165 vivos
Resposta: Sim• Nos grandes centros/ hospitais universitários• Tratando mais precocemente e melhor

O que é LHF?
Síndrome grave e potencialmente fatal Caracterizadas pela proliferação de macrófagos
ativados associadas e hemofagocitose generalizada
Resposta imune descontrolada e ineficaz • Hiperinflamação importante• Proliferação de macrófagos ativados• Acúmulo de fagócitos no SRE• Hemofagocitose generalizada


Quando ocorre LHF?
Lactentes • < 1 ano em 70-80% dos casos• Geralmente genética• Imunodeficiência• Mutações em genes responsáveis pela função
citotóxica das céls NK e linfócitos T citotóxicos
Crianças mais velhas, adolescentes e adultos• Secundária

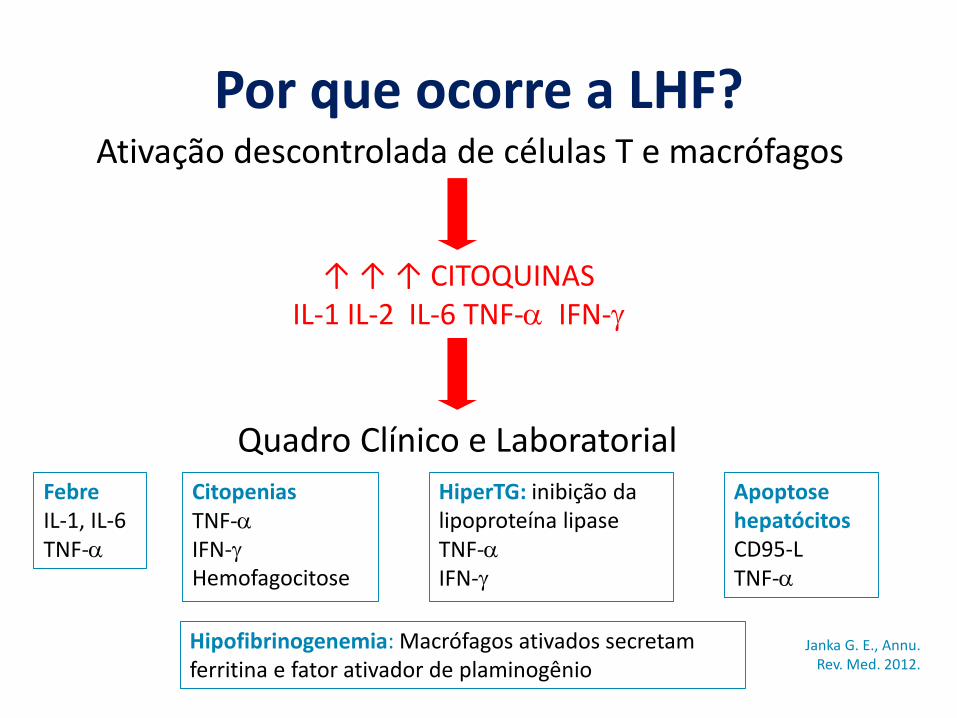
Por que ocorre a LHF?
Primárias (genéticas)• Familial: Herança autossômica recessiva (FEL/FHL)• Imunodeficiências: Sindr Griscelli 2, Chédiak-Higashi,
síndr. linfoproliferativa ligada ao X, sindr. Di George • Doença rara e rapidamente fatal
Secundárias (adquirida, não familial)• Infecção (viral - EBV, bacteriana, parasitária)• Doença autoimune: Síndr de Ativação Macrofágica• Neoplasia (células T)• Imunodeficiência adquirida/ Imunossupressão
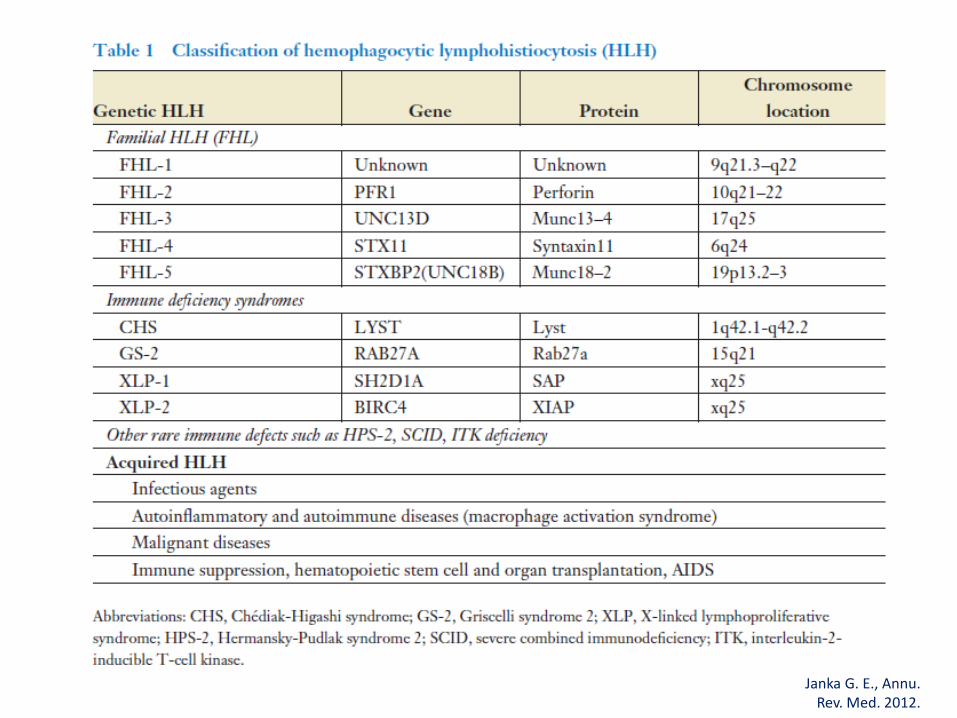
Janka G. E., Annu. Rev. Med. 2012.

Como ocorre a LHF?Caso Clínico
ACTSA, 4 anos, feminina, parda, natural e procedente de São Paulo
1 mês da internação:• Febre intermitente de até 39°C• Dor em MMII, dificuldade para deambular• Perda de 5kg
1 semana da internação:• Aumento do volume abdominal• Palidez cutânea• Icterícia

Caso Clínico – Exame Físico
REG, descorada, ictérica, febril Abdome: distendido, doloroso, fígado a 5 cm
do RCD e baço a 5 cm do RCE
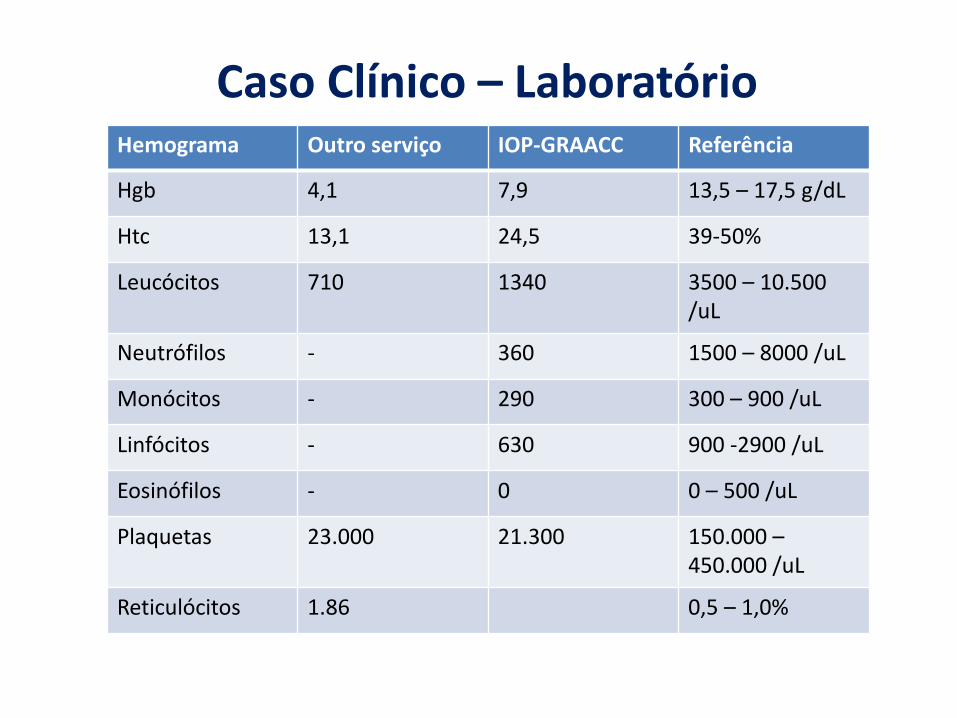
Hemograma Outro serviço IOP-GRAACC Referência
Hgb 4,1 7,9 13,5 – 17,5 g/dL
Htc 13,1 24,5 39-50%
Leucócitos 710 1340 3500 – 10.500 /uL
Neutrófilos - 360 1500 – 8000 /uL
Monócitos - 290 300 – 900 /uL
Linfócitos - 630 900 -2900 /uL
Eosinófilos - 0 0 – 500 /uL
Plaquetas 23.000 21.300 150.000 –450.000 /uL
Reticulócitos 1.86 0,5 – 1,0%
Caso Clínico – Laboratório

Bioquímica Resultados Valor de Referência
Bilirrubina Total /BD 7,3/ 6,9 <1/ <0,3
Albumina 2.4 3.5-5.5 g/dL
TGO/ TGP 85/ 43 <32/ <33
Ureia/ Creat 13/ 0.5 10-50/ 0,3-0,5
Na/ K/ P 132/ 4.6/ 2.2 137-148/ 3,5-5/ 4-7
Triglicérides 318 <100
DHL 485 <250
Ferritina 1512 13-150 ng/ml
Fibrinogênio 185 200-400
Caso Clínico – Laboratório

Caso Clínico - Resumo
Menina de 4 anos com febre, dor óssea, perda de peso há 1 mês Há 1 semana, icterícia, palidez cutânea,
hepatoesplenomegalia Exames laboratoriais: pancitopenia
• ↑ BT, BD, TGO, TGP, Triglicérides, DHL, Ferritina• ↓ Albumina, fibrinogênio• MO: figuras de hemofagocitose

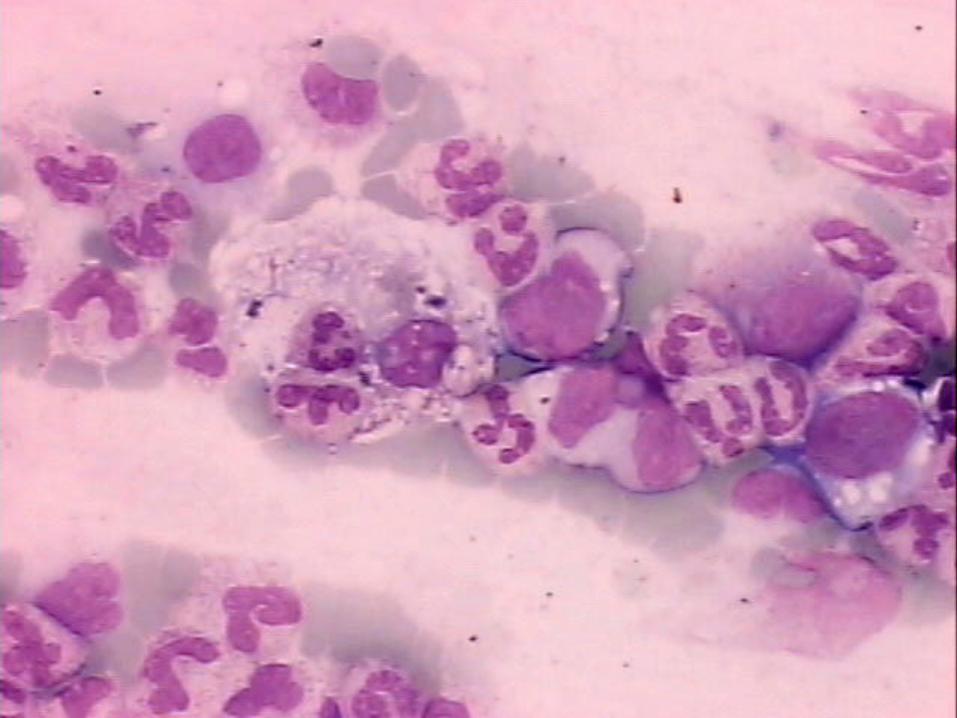

Como Diagnosticar LHF?
Embora todos esses sintomas sejam característicos Nenhum é específico É a magnitude dos sintomas clínicos e
alterações laboratoriais e especialmente sua progressão que sugerem que o paciente não está respondendo adequadamente à uma infecção

Critérios Diagnósticos Revisados Sociedade de Histiocitose
Doença familial/ Defeito genético conhecido Critérios clínicos e laboratoriais (mínimo 5/8)
• 1. Febre: duração > 7 dias, picos > 38,5ºC• 2. Esplenomegalia: > 3cm RCE• 3. Citopenia: bi ou pancitopenia, não associada à
MO hipocelular ou displásicaHb: < 9g/dlPlaquetas < 100.000/mm3
Neutrófilos < 1.000/mm3
Janka G. E., Annu. Rev. Med. 2012.

Critérios Diagnósticos Revisados Sociedade de Histiocitose
4. Hipertrigliceridemia e/ou hipofibrinogenemia 5. Ferritina > 500 microg/L
• Níveis acima de 10.000 microg/L tem 90% de sensibilidade e 96% de especificidade
6. sCD 25 (receptor IL-2) > 2400 U/ml 7. Diminuição ou ausência na atividade NK 8. Hemofagocitose MO, baço, SNC ou linfonodos
• Suportam o diagnóstico: sintomas neurológicos com pleiocitose moderada e/ou proteína elevada no LCR, ↑ transaminases, bili, DHL
Janka G. E., Annu. Rev. Med. 2012.
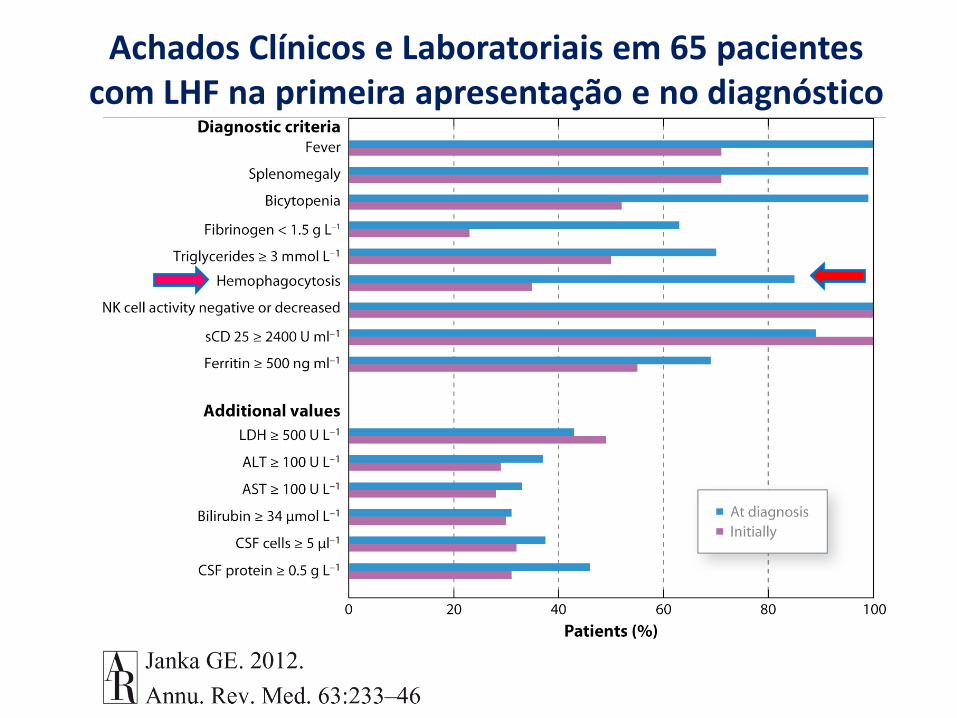
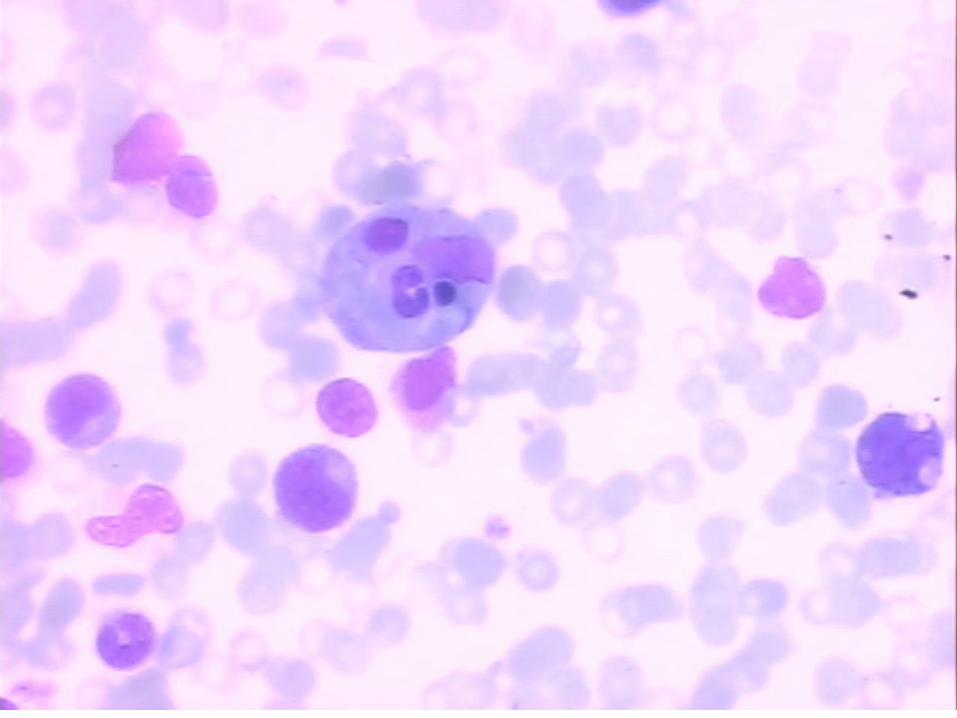
Achados Clínicos e Laboratoriais em 65 pacientes com LHF na primeira apresentação e no diagnóstico
Por que ocorre a LHF?
Janka G. E., Annu. Rev. Med. 2012.
↑ ↑ ↑ CITOQUINASIL-1 IL-2 IL-6 TNF-α IFN-γ
Ativação descontrolada de células T e macrófagos
Quadro Clínico e LaboratorialFebre IL-1, IL-6 TNF-α
CitopeniasTNF-αIFN-γHemofagocitose
HiperTG: inibição da lipoproteína lipaseTNF-αIFN-γ
Hipofibrinogenemia: Macrófagos ativados secretam ferritina e fator ativador de plaminogênio
Apoptose hepatócitosCD95-L TNF-α

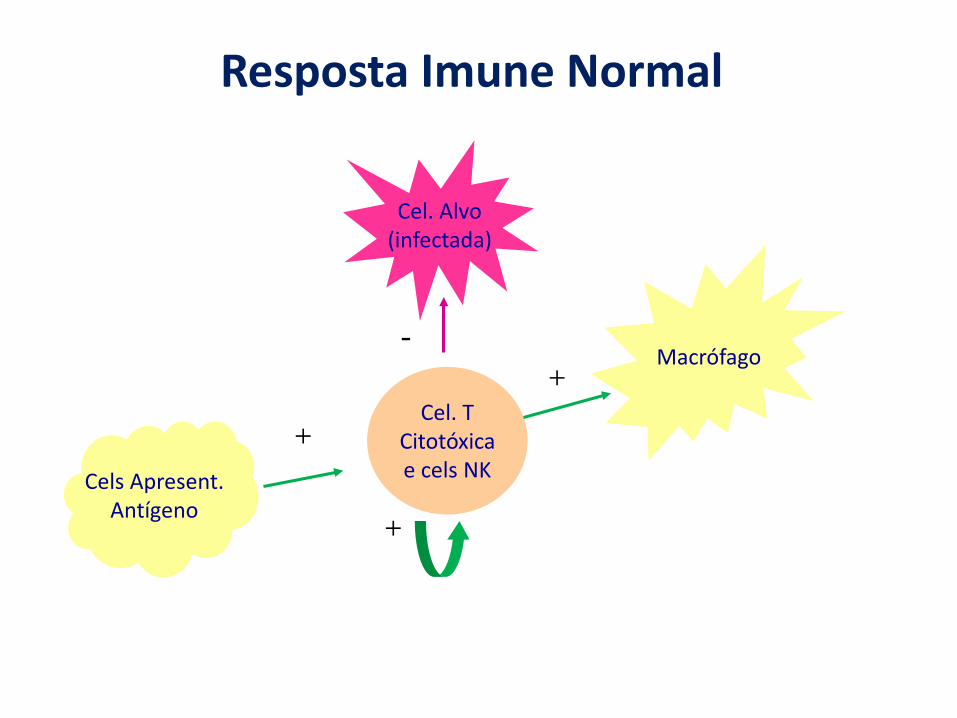
Resposta Imune Normal
Macrófago
Cel. TCitotóxicae cels NK
Cel. Alvo(infectada)
Cels Apresent.Antígeno
+
+-
+

Regulação da Resposta Imune
Macrófago
Cel. TCitotóxicae cels NK
Cel. Alvo(infectada)
Cels Apresent.Antígeno
+
+
+-
Cel. TCitotóxicae cels NK
_
_ _
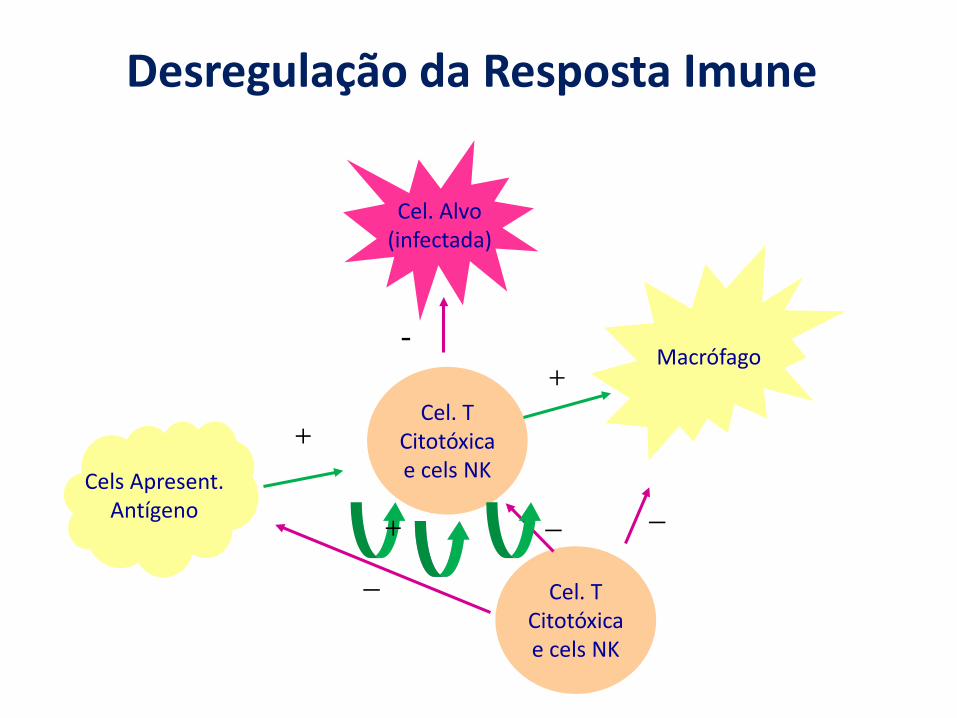
Desregulação da Resposta Imune
Macrófago
Cel. TCitotóxicae cels NKCels Apresent.
Antígeno
+
+-
Cel. TCitotóxicae cels NK
_
_ _+
Cel. Alvo(infectada)

Janka G. E., Annu. Rev. Med. 2012.
FHL - 2
FHL - 3
FHL – 4/5
CHS
GS-2

Por que tratar a LHF?
Sem tratamento é uma doença rapidamente fatal• Infecção bacteriana ou fúngica: neutropenia
prolongada• Falência de múltiplos órgãos• Disfunção SNC

Como tratar a LHF?
1° Objetivo (imediato): suprimir a inflamação exacerbada que é responsável pela sintomatologia• Tratamento da infecção quando identificável
Leishmaniose: suficienteCasos leves
2° Objetivo: interromper a ativação mútua de céls T citotóxica e macrófagos• Destruição de macrófagos infectados• Destruição de céls T citotóxicas
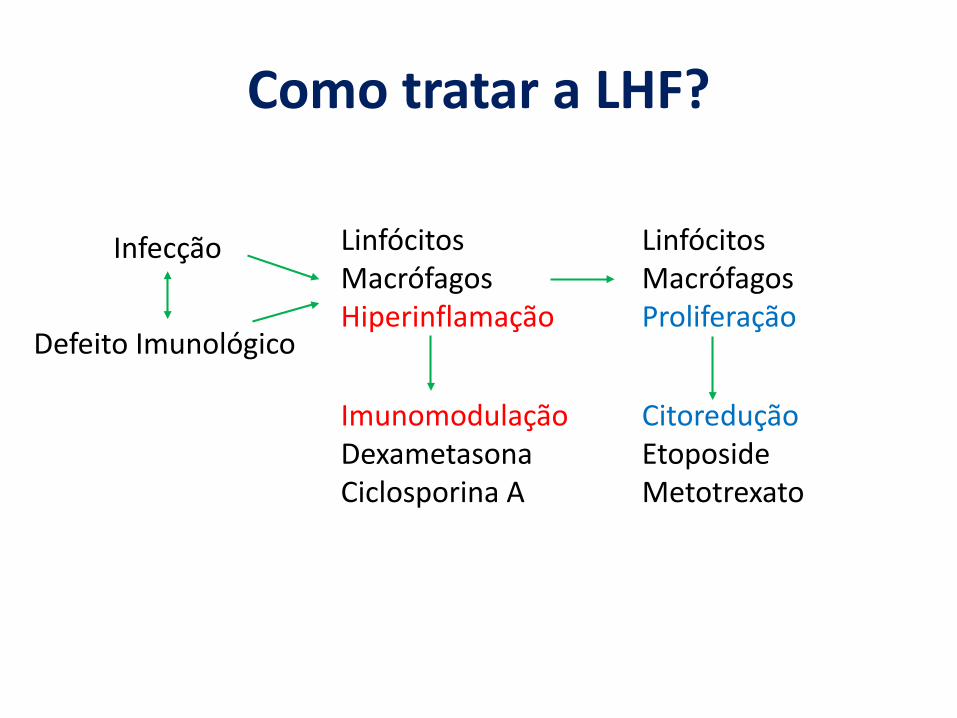
Como tratar a LHF?
LinfócitosMacrófagosHiperinflamação
LinfócitosMacrófagosProliferação
Infecção
Defeito Imunológico
ImunomodulaçãoDexametasonaCiclosporina A
CitoreduçãoEtoposideMetotrexato

TratamentoFaca de dois gumes
Tanto a hiperinflamação quanto Destruição da função imune remanescente Podem ser letal para o paciente

Grupo de Estudo da Sociedade Internacional - HLH-94
1° protocolo internacional 1994• Dexametasona e etoposide• Ciclosporina A após 8 semanas• MTX intratecal para pts com persistência de
sintomas neurológicos
Pts com forma familial => TCTH
Henter et al, Blood. 2002;100(7):2367-73

Grupo de Estudo da Sociedade Internacional - HLH-94
N= 113 pacientes (Jul/94 - Jun/98)
• Mortes precoces: 25 (20/25 doença)• TMO: 65• Fora de tratamento, sem TMO: 20• Sobrevida global em 3.1 anos: 55%
Casos familiares: 51%Crianças >1 ano: 72%Crianças <1 ano: 42% (p<0.005)
Henter et al, Blood. 2002;100(7):2367-73

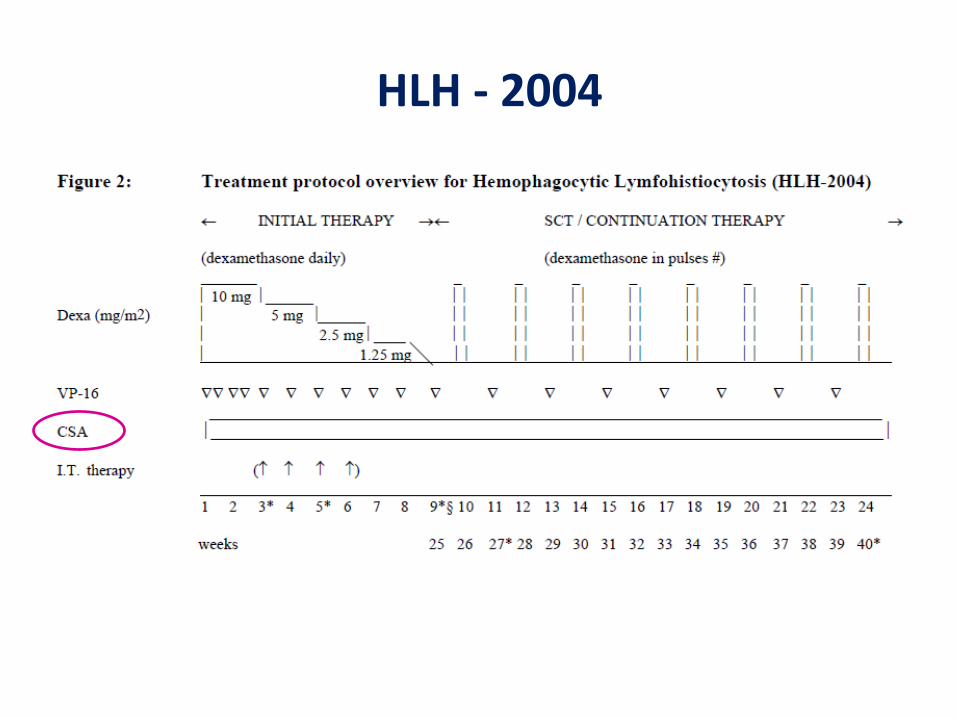
HLH - 2004
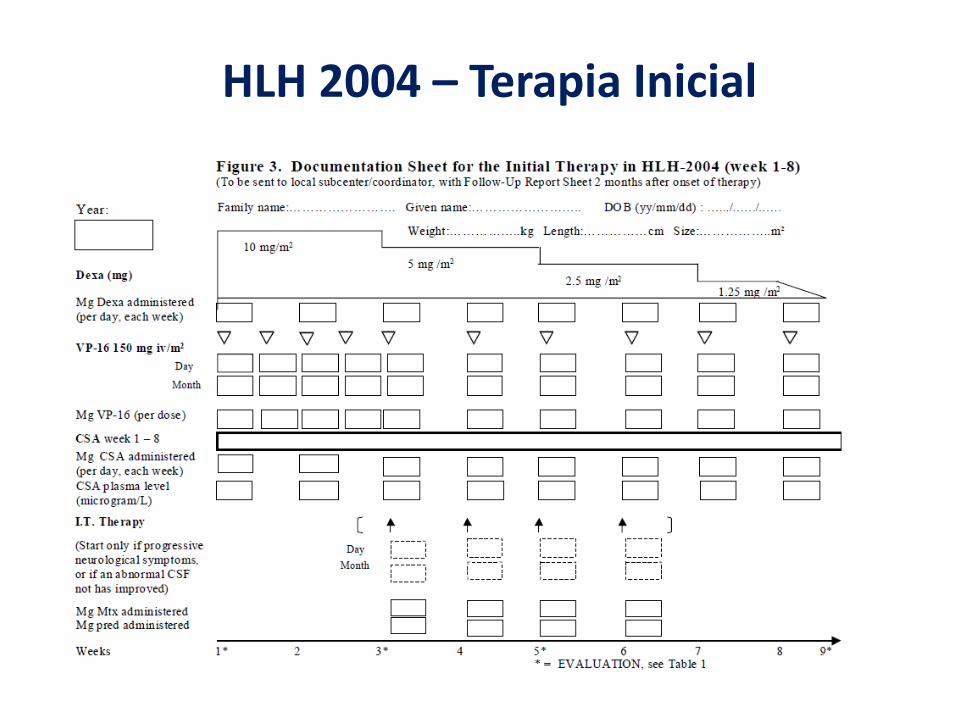
HLH 2004 – Terapia Inicial
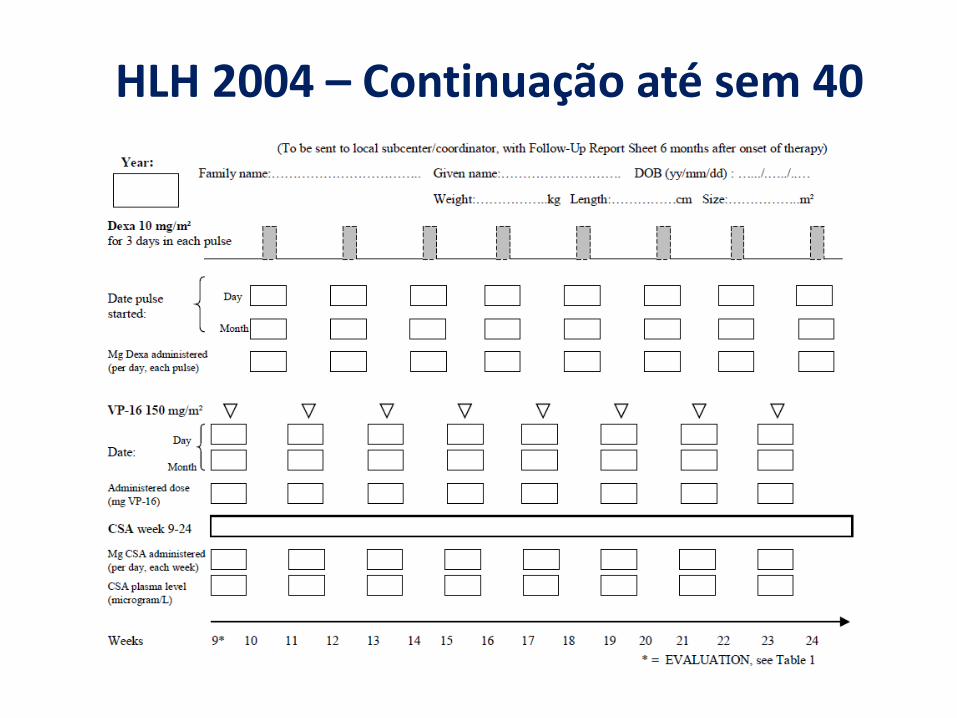
HLH 2004 – Continuação até sem 40

Outras Terapias
ATG e corticosteroides => TSCH (SG=55%) Sindr. de Ativação Macrofágica: metilprednisolona
em altas doses por 3 dias (30mg/kg/dia) Anakinra (Kineret®) antagonista de receptor de IL-1 Casos leves: só costicosteróides Imunoglobulina Rituximab (anti-CD20) : EBV +

Resumo

Resumo
Síndrome clínica grave, ameaçadora de vida Caracterizada por resposta imune
descontrolada e hiperinflamação Difícil diagnóstico e tratamento Na ausência de qualquer marcador específico
de LHF, a terapia muitas vezes é iniciada baseada em forte suspeita clínica, antes que a atividade descontrolada da doença produza danos irreversíveis à órgãos e sistemas

Obrigada!
Recommended

















