Embed Size (px)
DESCRIPTION
ESTUDOS EM NEUROLOGIA
Citation preview

Síndrome de West
Síndrome de Ohtahara
Marcus Petindá
Estagiário
Neurologia Infantil
HBDF

Síndrome de West
W. J. West em 1841
Vasquez e Turner, 1951
1ª resposta terapêutica, em 1958, Sorel e Dusaucy-
Bauloye
É uma epilepsia constituída por espasmos
epiléticos em grupos, associados a um traçado
eletroencelagráfico intercrítico do tipo
hipsarrítmico, independente do atraso do DNPM

Conceito
Crise epiléptica do tipo espasmo epiléptico
EEG com traçado hipsarrítmico
• Multifocal - vídeoEEG (Gaily e cols)
Resposta idade-específica de um cérebro
imaturo a uma lesão focal ou generalizada

Espasmos em salvas – 1ª manifestação
clínica
Deterioração psicomotora
• Perda do contato visual
• Hipotonia axial
Outros tipos de crises

Epidemiologia
Incidência – 2,9 a 4,5/100000 nascidos
vivos
Sexo masculino – 60% dos casos
3 e 7 meses – 77%; mas podem surgir até os
6 anos
Mais frequente causa de ADNPM na
infância

Tipos de crises
Breves movimentos axiais, 0,2 a 2 s
Flexão > extensão , ou mistos
Flexão dos MMSS e extensão dos MMII
Seguido de grito
Hipertonia axial
Desvio ocular para cima

Espasmos – 20 a 40 (>100)
2 a 138
Duração – 5 a 30 s
Salvas – 10/dia
3 a 763
Sonolência, manipulação, alimentação, som
inesperado
Mais comum em vigília

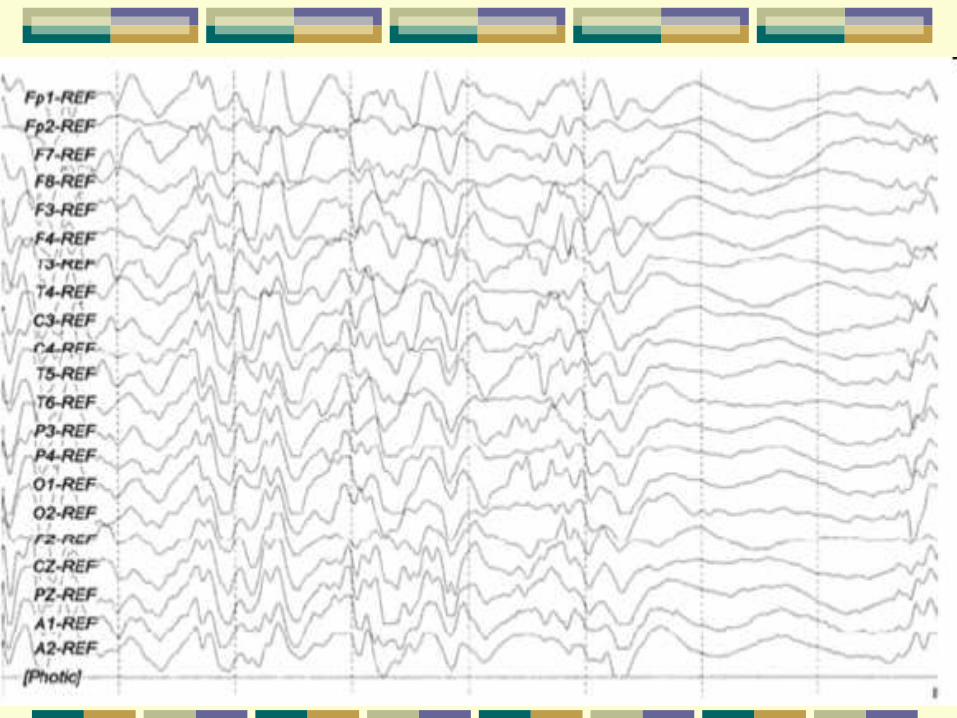
Aspectos eletroencefalográficos
Ictal
• Onda lenta de alta voltagem
• Todos os espasmos
• Região central ou vértex
• Paroxismos de ondas rápidas
• Podem surgir sozinhos ou seguidos de ondas lentas
• Olhar sem expressão
• Crises sutis antes dos espasmos
• Dessincronização/atenuação do traçado
• Seguida de onda lenta
• Espasmos clínicos

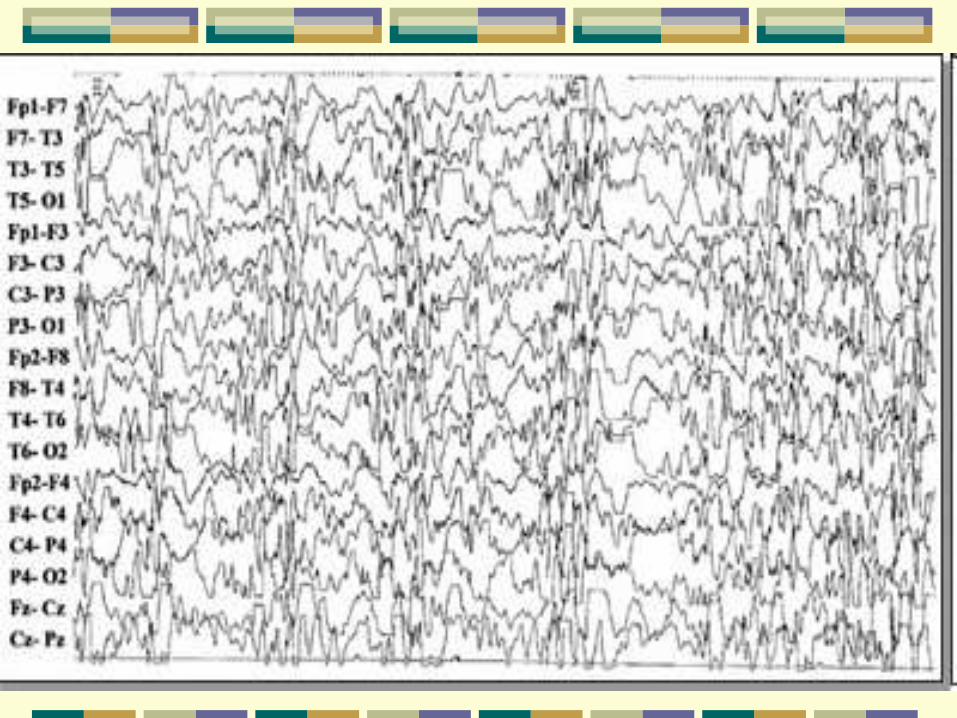
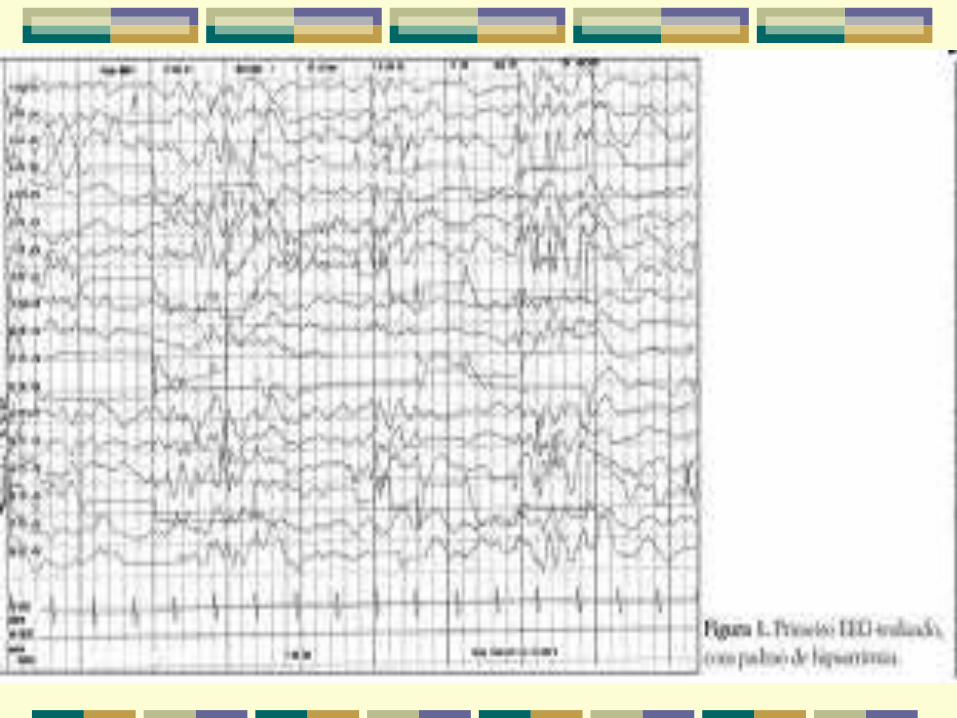
Aspectos eletroencefalográficos
Interictal - Hipsarritmia
Padrão interictal mais característico é uma mistura
de espículas e ondas lentas de alta amplitude, com
duração e localizações variáveis que ocorrem
continuamente ou em paroxismos. Manifesta-se
mais claramente nos estágios 2 e 3 do sono, sem
completa sincronização.
Pode se modificar pela idade, estado sono-vigília e
lesões cerebrais

DNPM
Normal ou anormal antes do início do
quadro
Anormal
• Controle cefálico
• Pegar objetos
• Rastreamento ocular
• Diplegia, tetraplegia, ataxia, atetose

Etiologia - Classificação
Sintomáticos
• As crises resultam de uma ou mais lesões cerebrais identificáveis
• Grupo mais frequente – 45,7%
• Fatores predisponentes
• Esclerose tuberosa, holoprosencefalia, HIV, cisto de plexo coróide , hidrocefalia congênita, CMV, sofrimento fetal crônico, gangliosidose, síndrome de Down, hipóxia perinatal, infartos hemorrágicos, crises epilépticas

Etiologia - Classificação
Criptogênicos
• São sintomáticos
• Causa oculta

Etiologia - Classificação
Idiopático
• Evolução favorável com desaparecimento das
crises e DNPM normal
• 24,3%
• Características
• Ausência de regressão mental significativa
• Preservação da função visual
• Ausência de alterações no EEG

Fisiopatologia
Disfunção monoaminérgica ou colinérgica ao nível do tronco cerebral que envolve o controle do ciclo sono-vigília
Teoria maturativa
Defeito no sistema imunológico
• Anticorpos contra o SNC
• Aumento de céls Te B no sangue periférico
• Antígeno HLA-DRw52

Diagnóstico
História clínica
Manifestações clínicas associadas ao EEG
EEG normal
• vídeoEEG
• Neuroimagem – espasmos assimétricos e
assíncronos associados a crises parciais
• Pesquisa de EIM

Diagnóstico diferencial
Alterações não epilépticas
• Cólicas
• Reflexo de Moro
• Resposta exacerbada de adaptação ao espaço ou a um estímulo sonoro(crianças espásticas)
• Abalos hipnagógicos
• Abalos mioclônicos
• Movimentos anormais em crianças espásticas
• RGE
• Distúrbios da deglutição

Diagnóstico diferencial
Fenômenos epilépticos
• Epilepsia mioclônica benigna do lactente
• Epilepsia mioclônica do lactente precoce
• Encefalopatia mioclônica neonatal
• Síndrome de Ohtahara

Diagnóstico diferencial
Síndromes genéticas – herança ligada ao X
• Sd de Aicardi
• Doença de Menkes
• Espasmo infantil ligado ao X
• Complexo piruvato-desidrogenase

Diagnóstico diferencial
Autossômica dominante
• Esclerose tuberosa
• Lissencefalia tipo Miller-Dieker

Diagnóstico diferencial
Autossômica recessiva
• Deficiência de biotinidase
• Síndrome de PEHO
• Transtorno congênito da glicosilação, tipo VII
• Doença de Tay-Sachs variante AB
• Agenesia de corpo caloso com neuroniopatia
• Deficiência mitocondrial do complexo IV

Tratamento
ACTH/corticosteróides – 70%; recidiva de 35 a 50%
NTZ – 25 a 50%
Piridoxina – 11 a 25%
TPM – 45%
VPA – 50%
VGB – 40 a 90%
Zonisamida – 25%
Dieta cetogênica
Tratamento cirúrgico

ACTH e corticosteróides
ACTH – não disponível no Brasil
• 0,4 a 150 UI/m²/dia IM
Prednisona
• 2 – 5 mg/kg/dia VO
Efeitos colaterais
• Infecções, HÁ, cardiomiopatia hipertrófica e
insuficiência adrenal

Vigabatrina
25 – 250 mg/kg/dia
• HC-FMUSP
• Dose inicial de 125 mg ao dia até 100 mg/kg/dia,
máximo de 150 mg/kg/dia
Efeitos colaterais
• Trremores, aumento das crises, < horas de sono,
irritabilidade, sonolência e constricção do
campo visual

Valproato de sódio
20 – 70 mg/kg/dia
Efeitos colaterais
• < 2 anos - falência hepática, pancreatite
• Plaquetopenia
• Tremores

Benzodiazepínicos
NTZ
• Comprimidos de 5mg – iniciar com ¼ cp à noite até a dose alvo – 0,8 mg/kg/dia
Efeitos colaterais
• Hipotonia
• Sedação
• Diminuição do peristaltismo
• Dificuldade de deglutição
• Salivação
• Estertoração alta

Topiramato
8 a 26/mg/kg/dia – média de 11 mg/kg/dia)
• Iniciar com 25 mg /dia e aumentar 25 mg a
cada 2 ou 3 dias
Efeitos colaterais
• Irritabilidade, distúrbio do sono, constipação,
letargia, respiração

Piridoxina
20 – 50 mg/kg/dia, em 3 tomadas
Efeitos colaterais
• Perda do apetite, vômitos, diarréia, flatulência,
disfunção hepática, apatia, polineuropatia
periférica, rabdomiólise

Outras drogas
Não comercializadas no Brasil
• Zonisamida – 4 – 8 mg/kg/dia
• Sultiamo – 10 mg/kg/dia
• Ganaxolona

Prognóstico
Etiologia – melhor fator preditivo
Taxa de remissão
• 28% no 1º ano de vida
• 49% aos 2 anos
• 65% aos 3 anos
• 74% aos 4 anos
Outros tipos de crises – 50 a 70%
Epilepsia intratável em 50%
Evolução para sd de Lennox-Gastaut – 20 a 50%

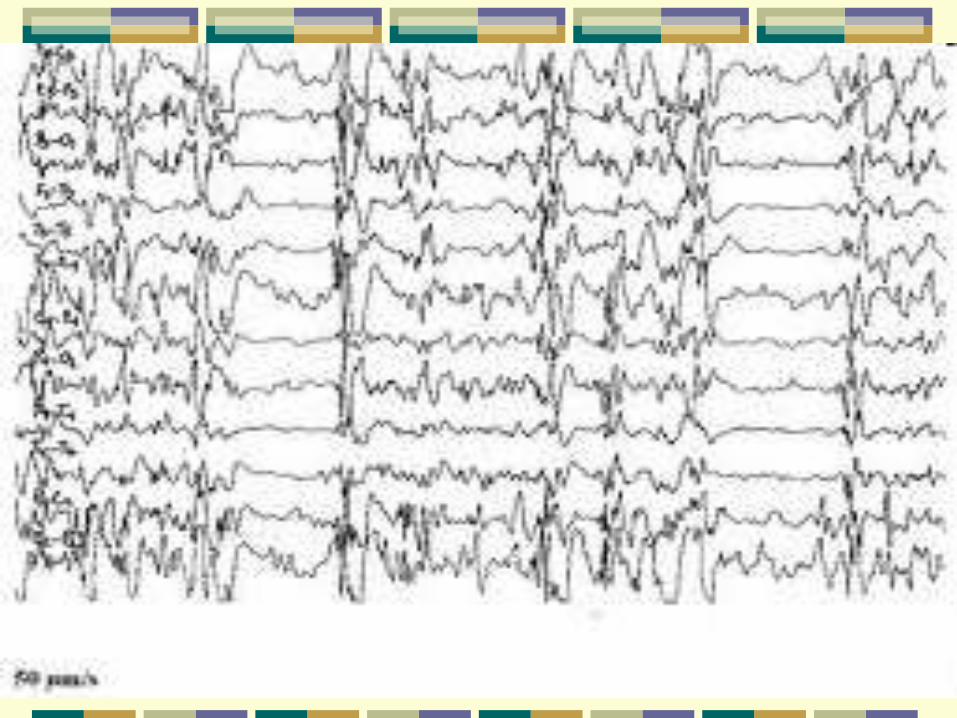
Síndrome de Ohtahara
Início neonatal ou no 1º mês de vida
Deterioração progressiva
Crises tônicas breves, em grupos – principal
Crises clônicas, mioclônicas, focais, TCG
EEG: padrão de surto-supressão, frequentemente assíncrono ou assimétrico, maids evidente em sono
Lesões estruturais focais, pp malformações do SNC
Entre 3 e 6 meses de idade evolui para sd de West
Tratamento: PB




























![Encruzilhada - Kasie West[1]](https://img.document.onl/doc/110x75/56d6c6db1a28ab30169c6c80/encruzilhada-kasie-west1.jpg)

