Embed Size (px)
Citation preview

ALTERACIONS CONGÈNITES
DE LA FUNCIÓ PLAQUETÀRIA
Dr. Ginés Escolar,
Servei d’Hemoteràpia i Hemostàsia
Hospital Clínic,
Barcelona
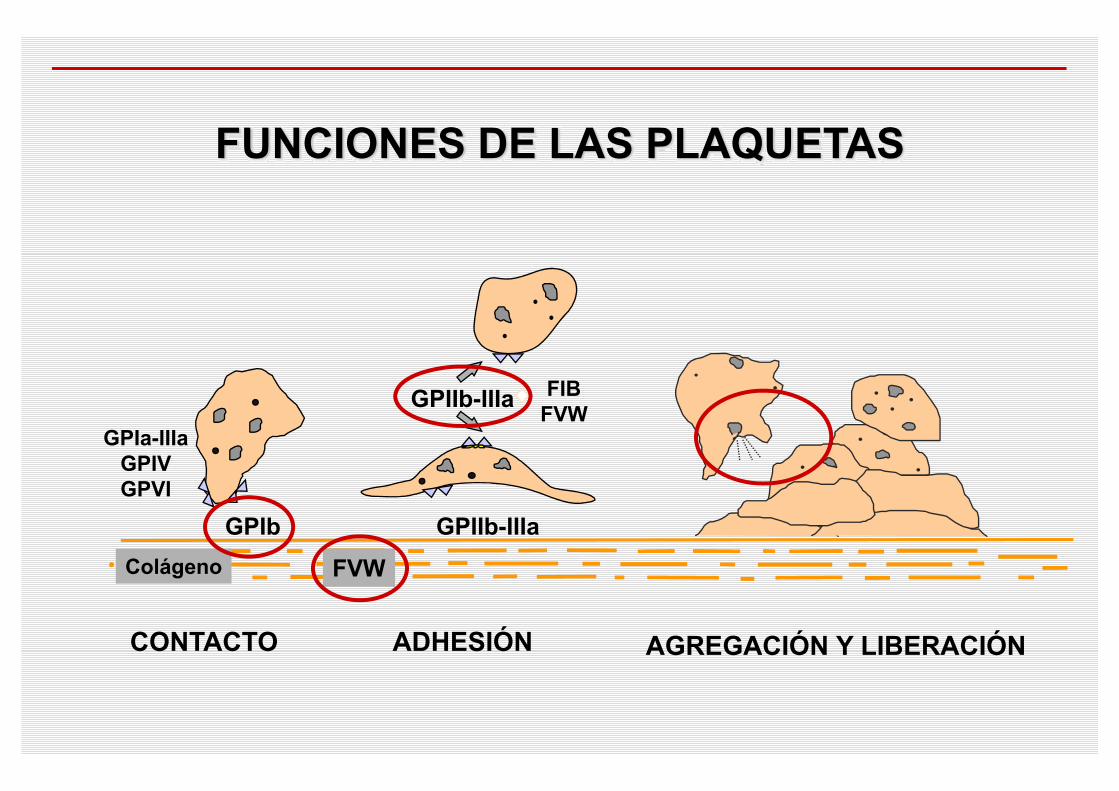
FVW
AGREGACIÓN Y LIBERACIÓN
FUNCIONES DE LAS PLAQUETAS
GPIIb-IIIa FIBFVW
GPIIb-IIIa
ADHESIÓN
GPIb
CONTACTO
GPIa-IIIaGPIVGPVI
Colágeno
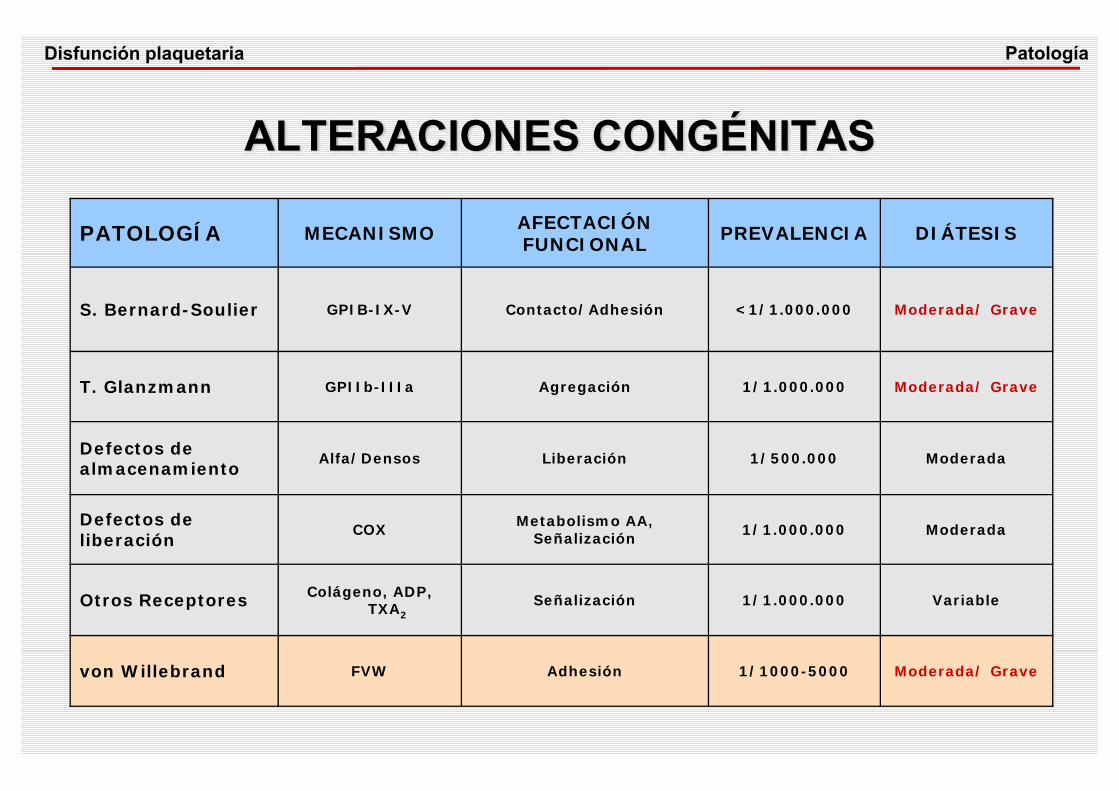
ALTERACIONES CONGÉNITAS
Patología
PATOLOGÍA MECANISMO AFECTACIÓN FUNCIONAL PREVALENCIA DIÁTESIS
S. Bernard-Soulier GPIB-IX-V Contacto/Adhesión <1/1.000.000 Moderada/ Grave
T. Glanzmann GPIIb-IIIa Agregación 1/1.000.000 Moderada/ Grave
Defectos de almacenamiento
Alfa/Densos Liberación 1/500.000 Moderada
Defectos de liberación
COX Metabolismo AA,Señalización 1/1.000.000 Moderada
Otros Receptores Colágeno, ADP, TXA2
Señalización 1/1.000.000 Variable
von Willebrand FVW Adhesión 1/1000-5000 Moderada/ Grave
Disfunción plaquetaria
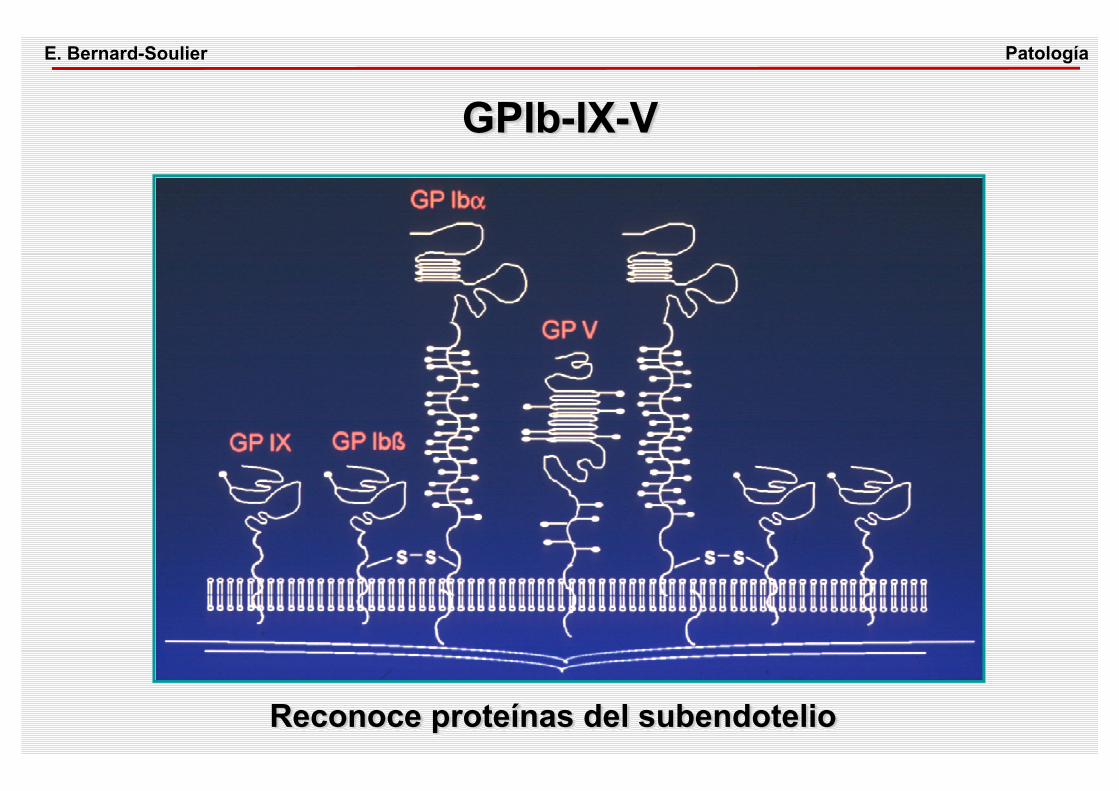
GPIb-IX-VPatologíaE. Bernard-Soulier
Reconoce proteínas del subendotelio

SÍNDROME DE BERNARD-SOULIER CUADRO CLÍNICO• Trombopenia, volúmen aumentado, sintomatología
hemorrágica• Herencia autosómica recesivaETIOPATOGENIA• Déficit o ausencia de GPIb DIAGNÓSTICO• Respuesta de agregación plaquetaria disminuida frente
a la ristocetinaFISIOPATOLOGÍA• La adhesión de las plaquetas sobre el subendotelio
mediada a través del GPIb/FVW es defectuosa
PatologíaE. Bernard-Soulier
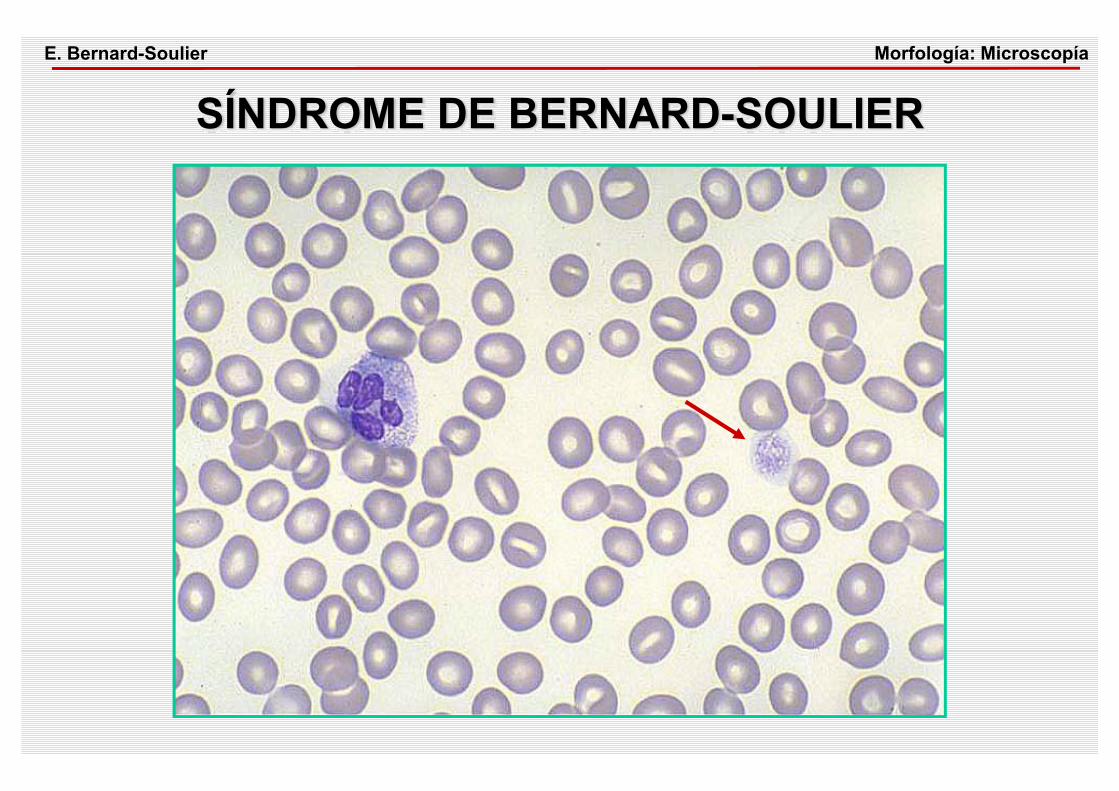
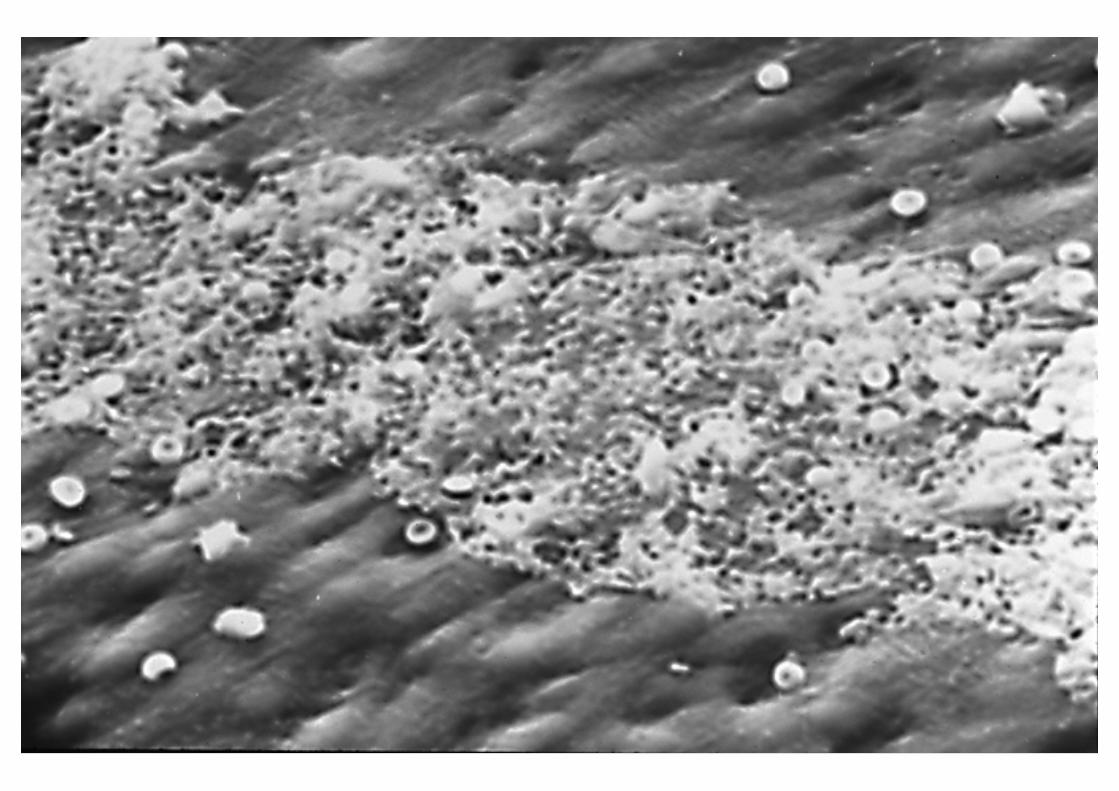
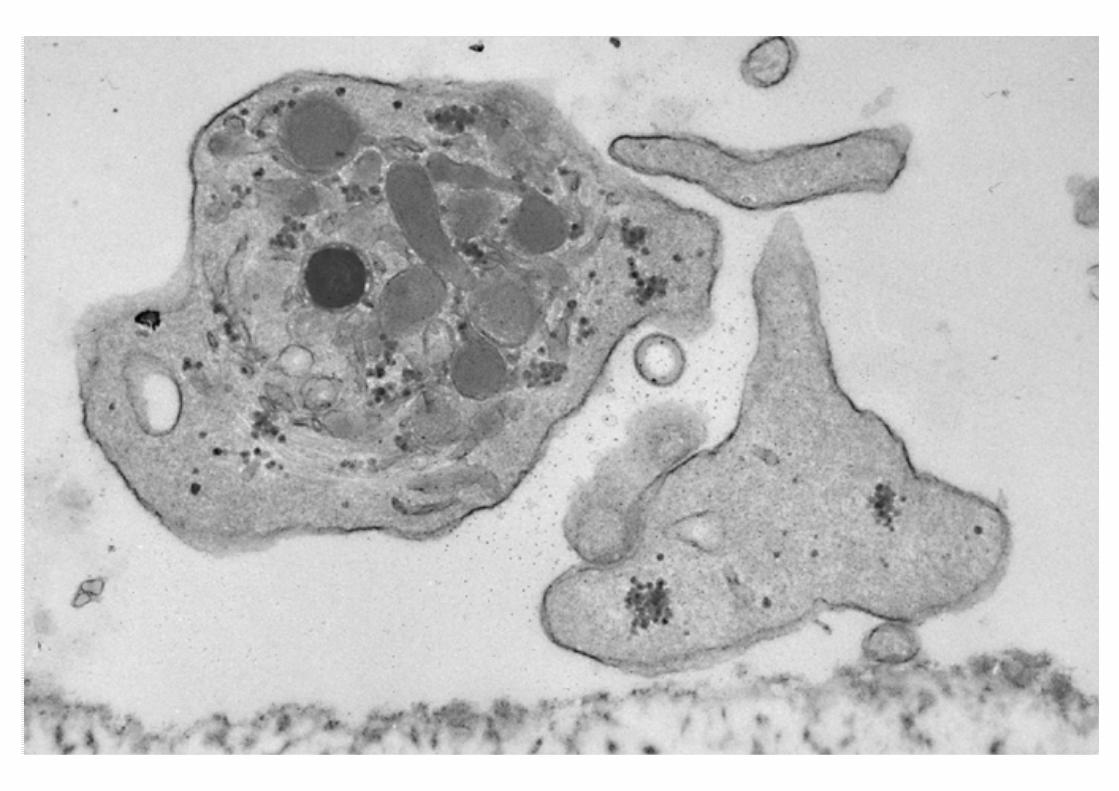
Morfología: MicroscopíaE. Bernard-Soulier
SÍNDROME DE BERNARD-SOULIER
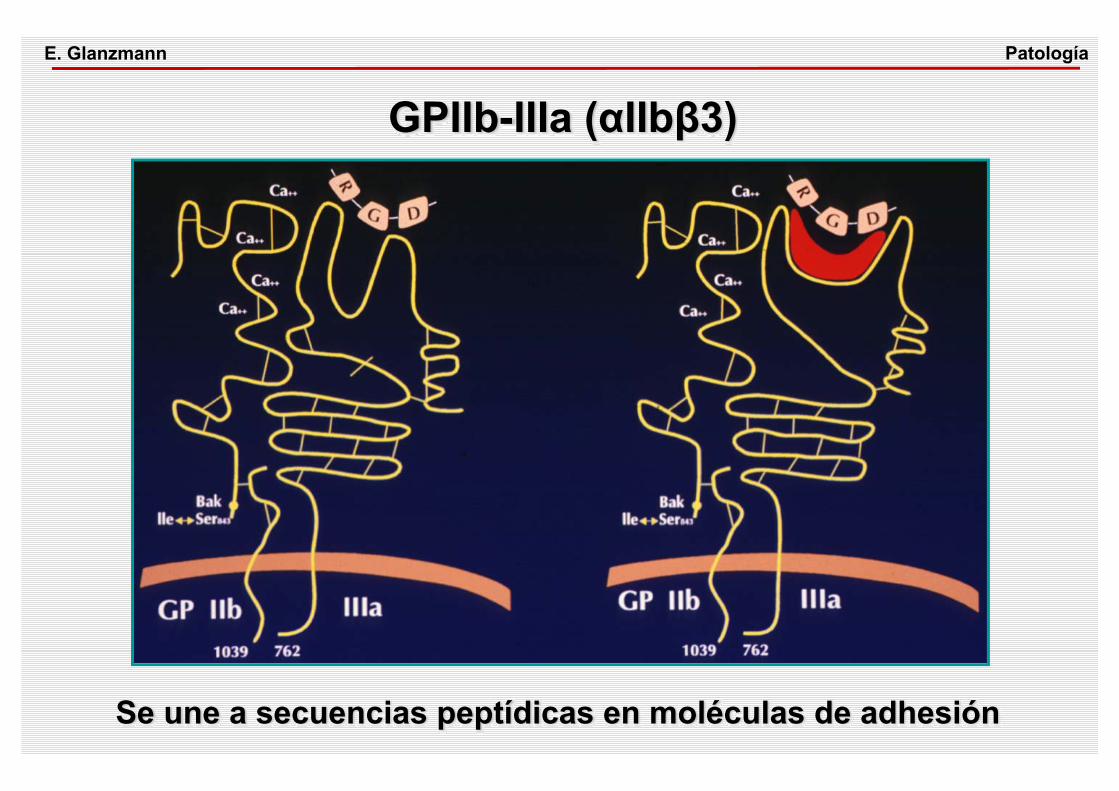
GPIIb-IIIa (αIIbβ3)PatologíaE. Glanzmann
Se une a secuencias peptídicas en moléculas de adhesión

TROMBASTENIA DE GLANZMANN
• Sintomatología hemorrágica• Autosómica recesiva
• GPIIb-IIIa ausente o deficiente
• La agregación plaquetaria mediada a través de la GPIIb-IIIa está disminuida o ausente
CUADRO CLÍNICO
ETIOPATOGENIA
DIAGNÓSTICO
FISIOPATOLOGIA• La formación de agregados que constituyen el
tapón hemostático está inhibida
PatologíaE. Glanzmann

Control
Síndrome de Bernard-Soulier
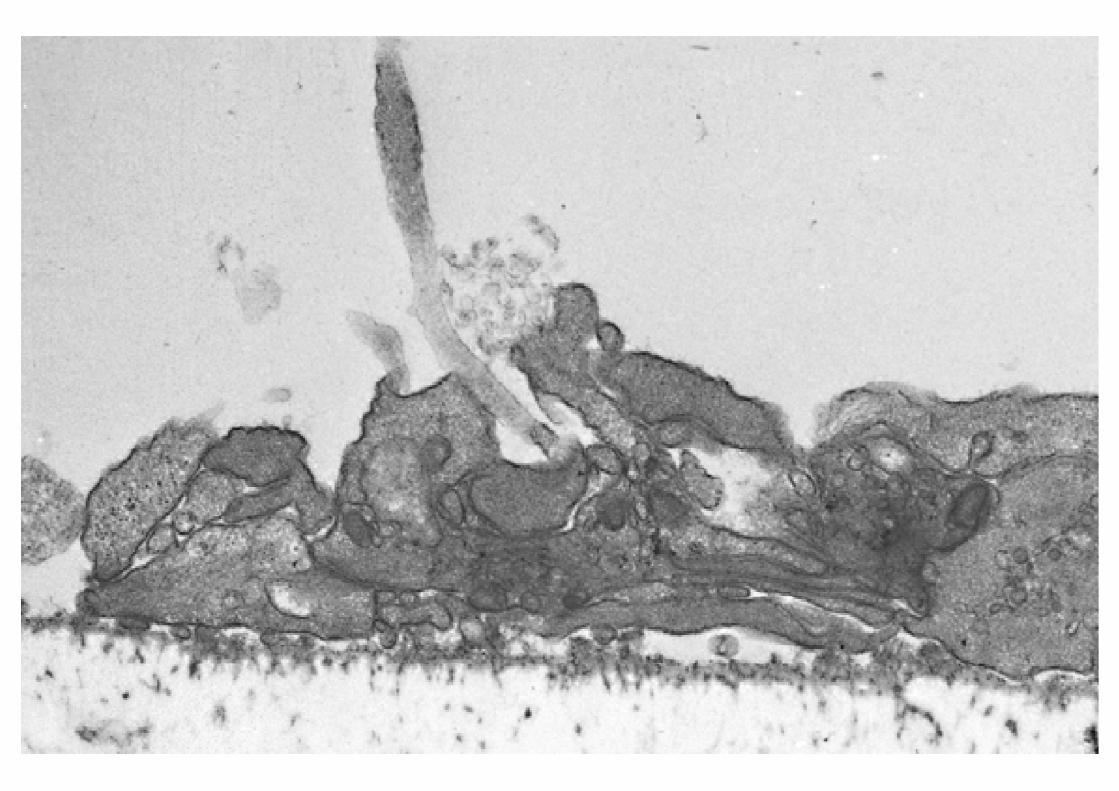
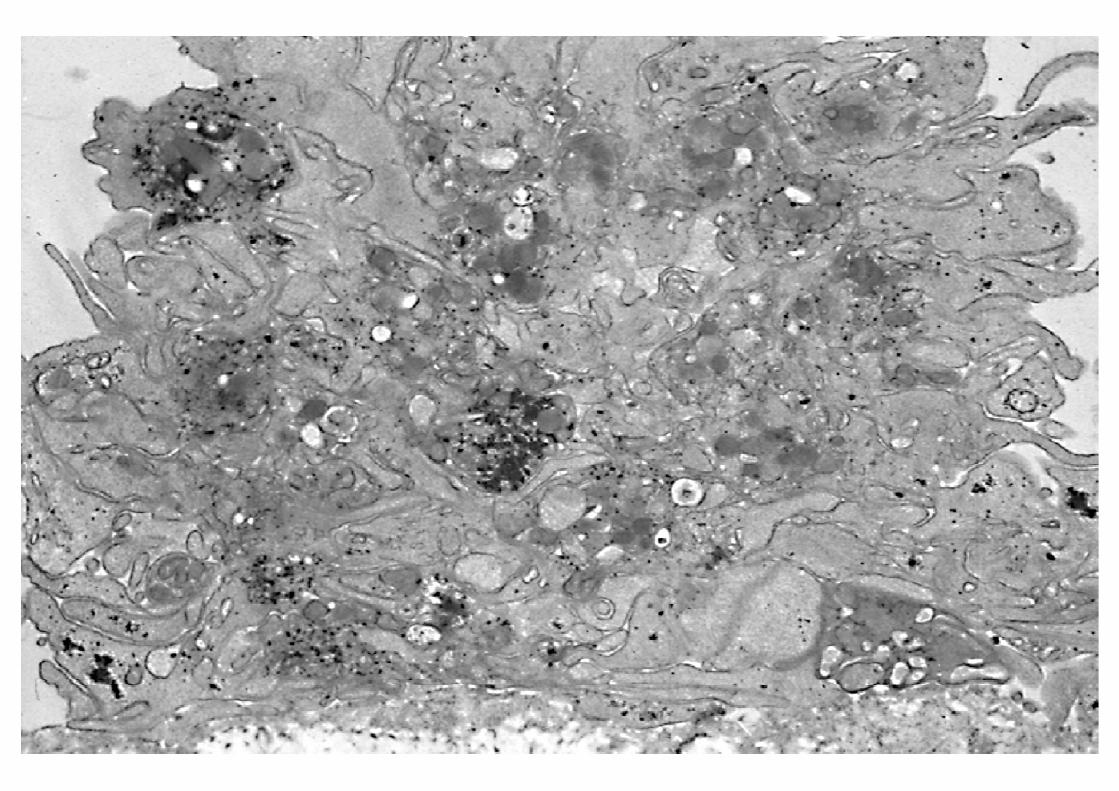
INTERACCIÓN PLAQUETA-SUBENDOTELIO
Thrombastenia deGlanzmann
PatologíaDéficits GPs

DEFECTOS “ASA-LIKE” / ALMACENAMIENTO
CUADRO CLÍNICO• Sangrado moderado
ETIOPATOGENIA• Defectos en CLOX/TXA2• Trastornos en el almacenamiento o liberación de los
gránulos
DIAGNÓSTICO• La agregación mediada a través de la vía del ácido
araquidónico y/o GPIIb-IIIa están reducidas
FISIOPATOLOGÍA• La formación del tapón hemostático está disminuida
PatologíaDefectos liberación/secreción

ENFERMEDAD DE VON WILLEBRAND
CUADRO CLÍNICO• Hemorragia predominantemente de mucosasETIOPATOGENIA• Defecto cuantitativo o funcional del FVWCLASIFICACIÓN• Tipo 1 (disminuido cuantitativamente)• Tipo 2 (variantes funcionales)• Tipo 3 (ausente)DIAGNÓSTICO• Aglutinación reducida frente a la ristocetina• Niveles reducidos de FVW• Tiempo de cefalina discretamente prolongado
PatologíaE. von Willebrand
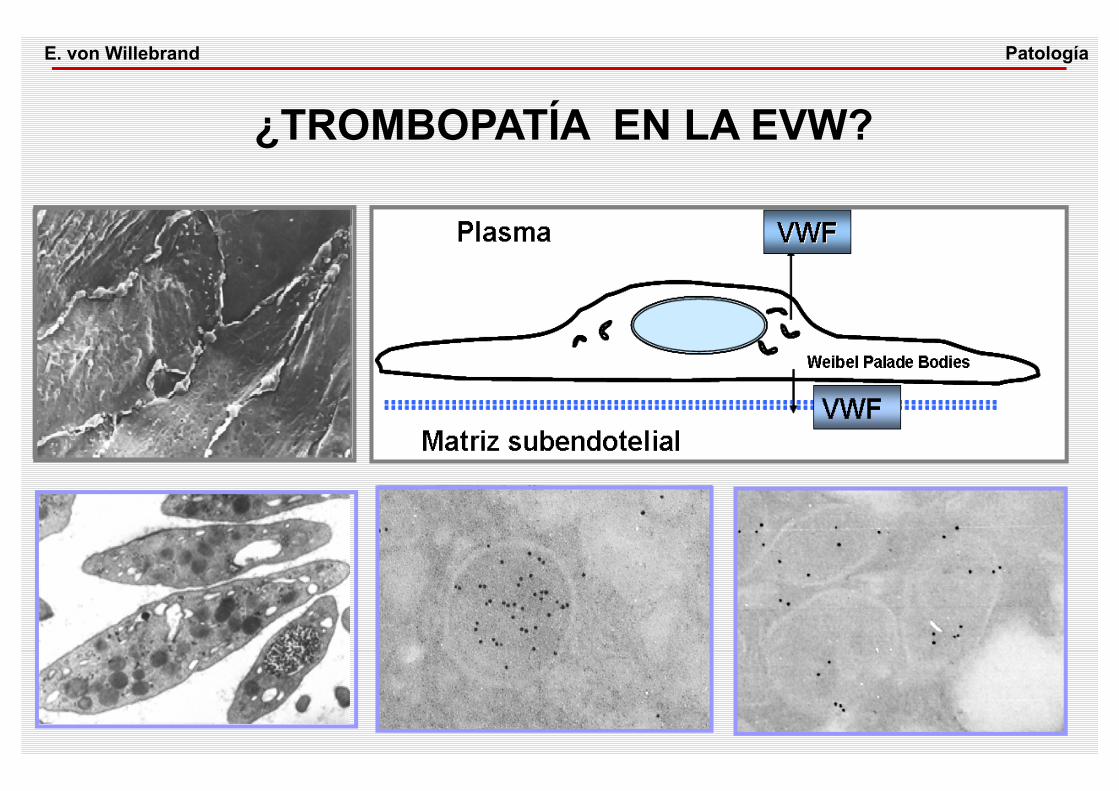
¿TROMBOPATÍA EN LA EVW?
PatologíaE. von Willebrand
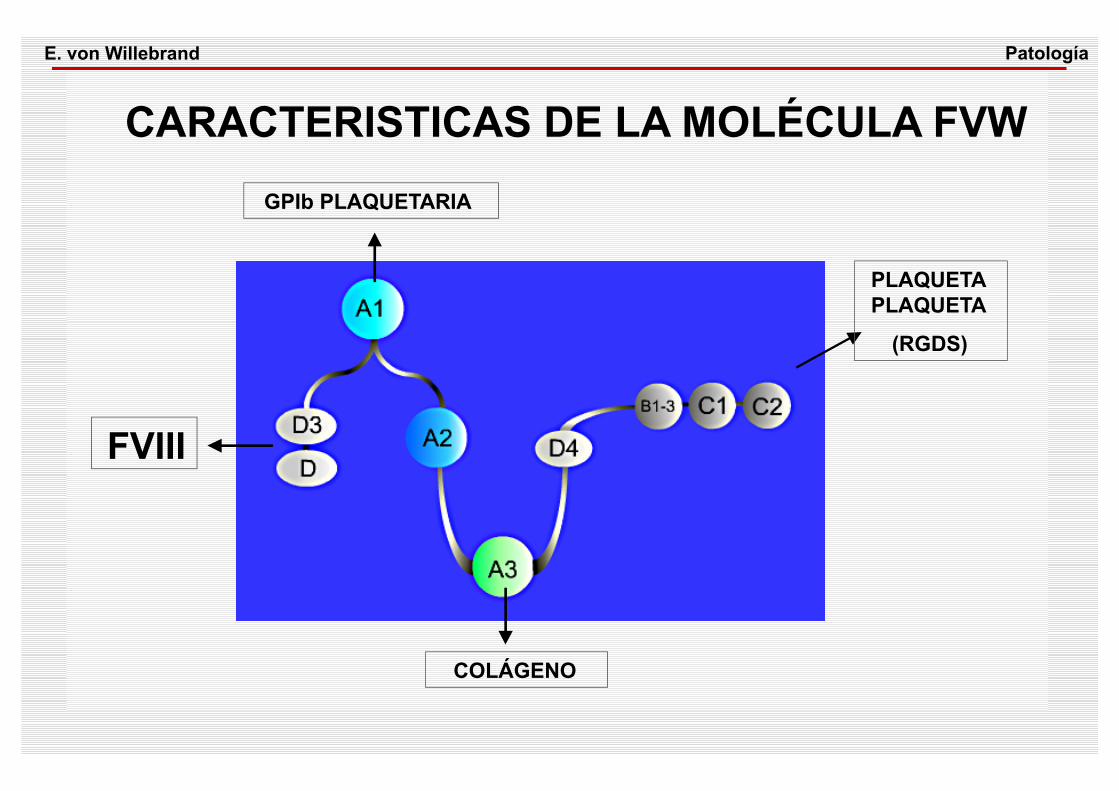
CARACTERISTICAS DE LA MOLÉCULA FVW
PLAQUETAPLAQUETA
(RGDS)
COLÁGENO
GPIb PLAQUETARIA
FVIII
PatologíaE. von Willebrand

• Historia Clínica; Evaluación de Sintomatología Hemorrágica
• Pruebas Básicas de Coagulación
• PFA-100 / Tiempo de Sangría
• Agregometría
• Estudios complementarios
• Estudios Genéticos
DIAGNÓSTICO DE LOS TRASTORNOS DE LA HEMOSTASIA (PRIMARIA)
DiagnósticoDisfunción plaquetaria
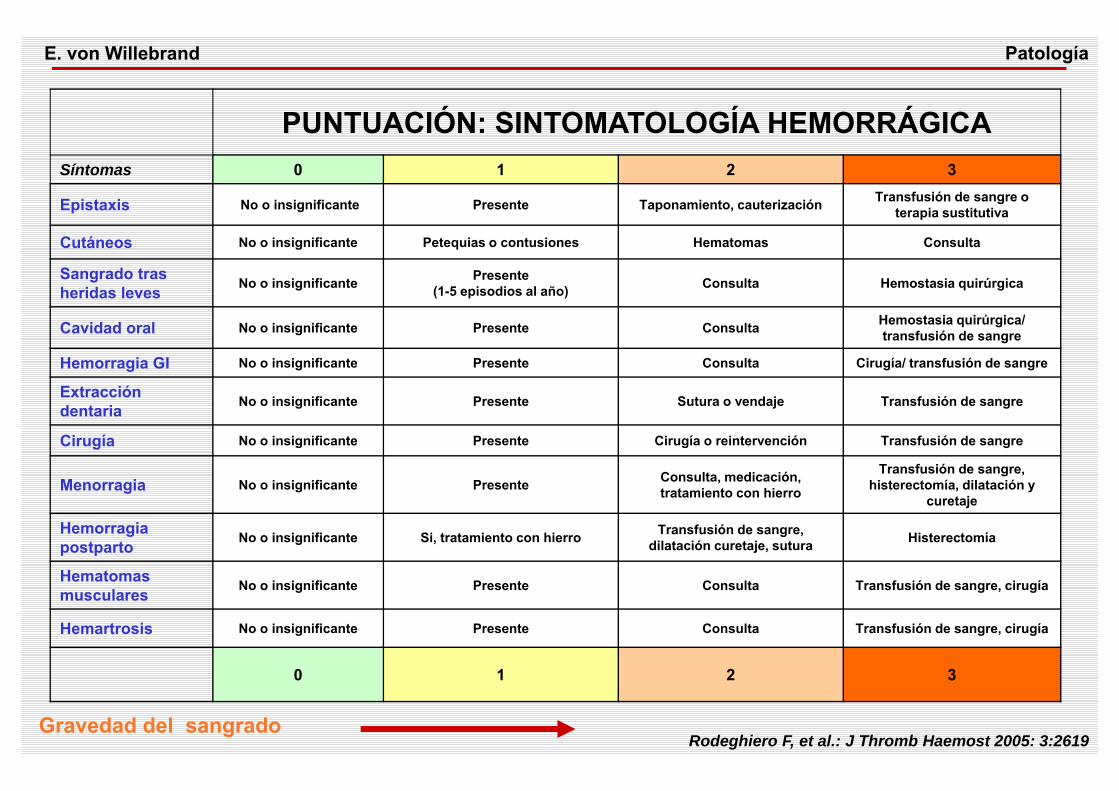
PUNTUACIÓN: SINTOMATOLOGÍA HEMORRÁGICASíntomas 0 1 2 3
Epistaxis No o insignificante Presente Taponamiento, cauterización Transfusión de sangre o terapia sustitutiva
Cutáneos No o insignificante Petequias o contusiones Hematomas Consulta
Sangrado tras heridas leves No o insignificante Presente
(1-5 episodios al año) Consulta Hemostasia quirúrgica
Cavidad oral No o insignificante Presente Consulta Hemostasia quirúrgica/ transfusión de sangre
Hemorragia GI No o insignificante Presente Consulta Cirugía/ transfusión de sangre
Extracción dentaria No o insignificante Presente Sutura o vendaje Transfusión de sangre
Cirugía No o insignificante Presente Cirugía o reintervención Transfusión de sangre
Menorragia No o insignificante Presente Consulta, medicación, tratamiento con hierro
Transfusión de sangre, histerectomía, dilatación y
curetaje
Hemorragia postparto No o insignificante Si, tratamiento con hierro Transfusión de sangre,
dilatación curetaje, sutura Histerectomía
Hematomas musculares No o insignificante Presente Consulta Transfusión de sangre, cirugía
Hemartrosis No o insignificante Presente Consulta Transfusión de sangre, cirugía
0 1 2 3
Gravedad del sangradoRodeghiero F, et al.: J Thromb Haemost 2005: 3:2619
PatologíaE. von Willebrand
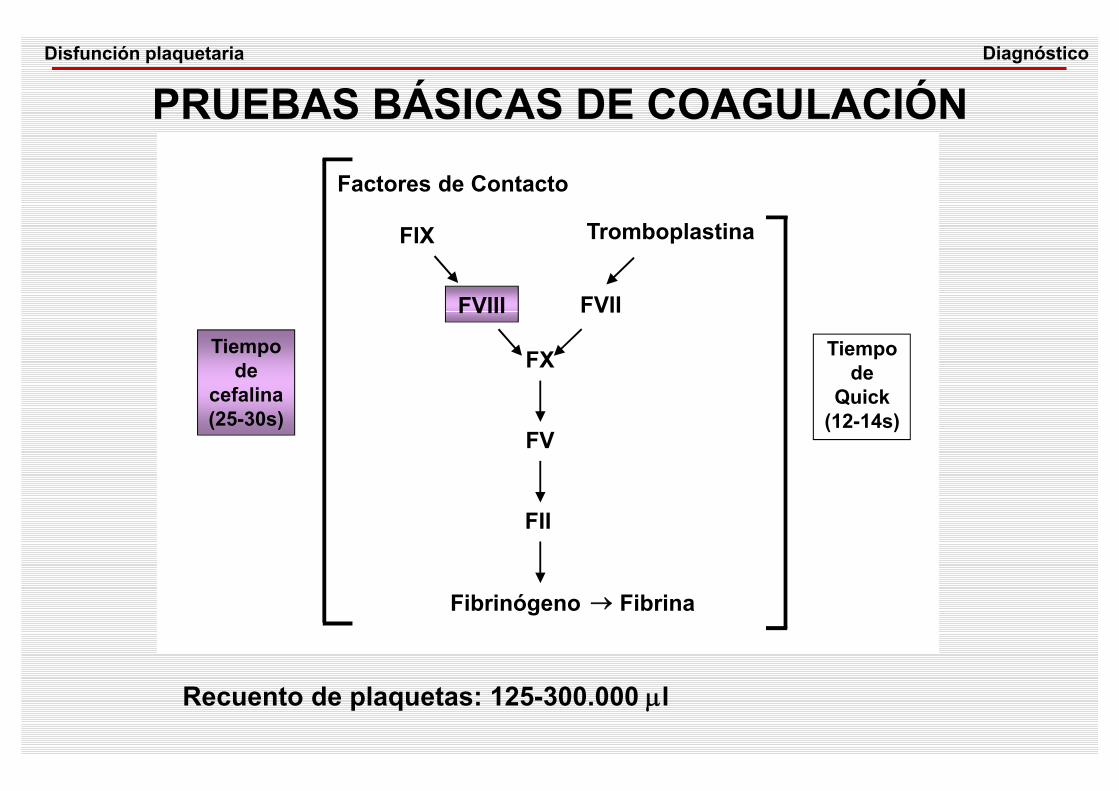
PRUEBAS BÁSICAS DE COAGULACIÓN
Recuento de plaquetas: 125-300.000 μl
Tiempode
cefalina(25-30s)
Tiempode
Quick(12-14s)
FIX
FVIII
FX
FVII
FV
FII
Fibrinógeno → Fibrina
Factores de Contacto
Tromboplastina
DiagnósticoDisfunción plaquetaria

Contenedor de la solución activadora
TecladoPantalla
Impresora
Carrusel
CassetteCartucho
ANALIZADOR DE LA FUNCIÓN PLAQUETARIA (PFA-100TM)
DiagnósticoDisfunción plaquetaria

INTERPRETACIÓN DE ALTERACIONES DE LA FUNCIÓN PLAQUETARIA CON EL PFA-100®
Normal Aspirina EVW Diátesis grave
COL-ADP Normal Normal Prolongado Muy prolongado
COL-EPI Normal Prolongado Prolongado Muy prolongado
0 1 2 3
COL-ADP COL-EPI113 ± 2487 ± 18
137105161123
T. obturación (s)+1 SD+2 SD
Valores de normalidad
Gravedad del sangrado
DiagnósticoDisfunción plaquetaria
AGREGACIÓN PLAQUETARIA
DiagnósticoDisfunción plaquetaria
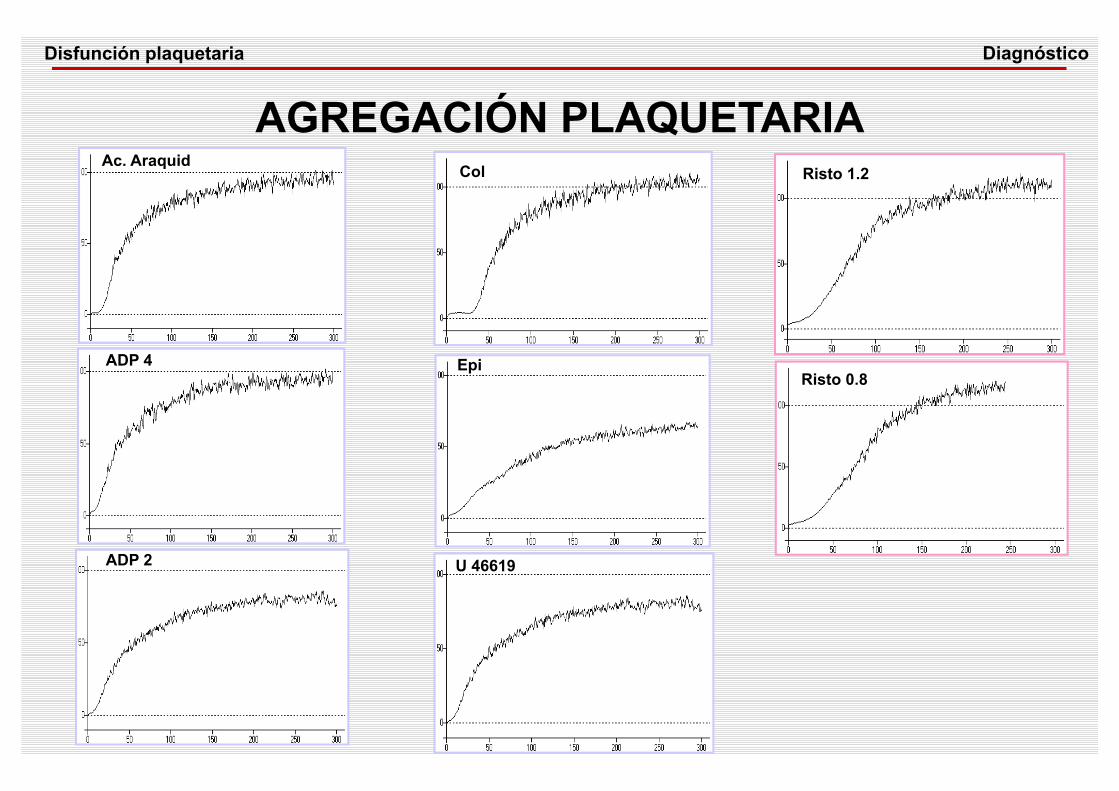
AGREGACIÓN PLAQUETARIAAc. Araquid
ADP 4
ADP 2
Col
U 46619
Epi
Risto 1.2
Risto 0.8
DiagnósticoDisfunción plaquetaria
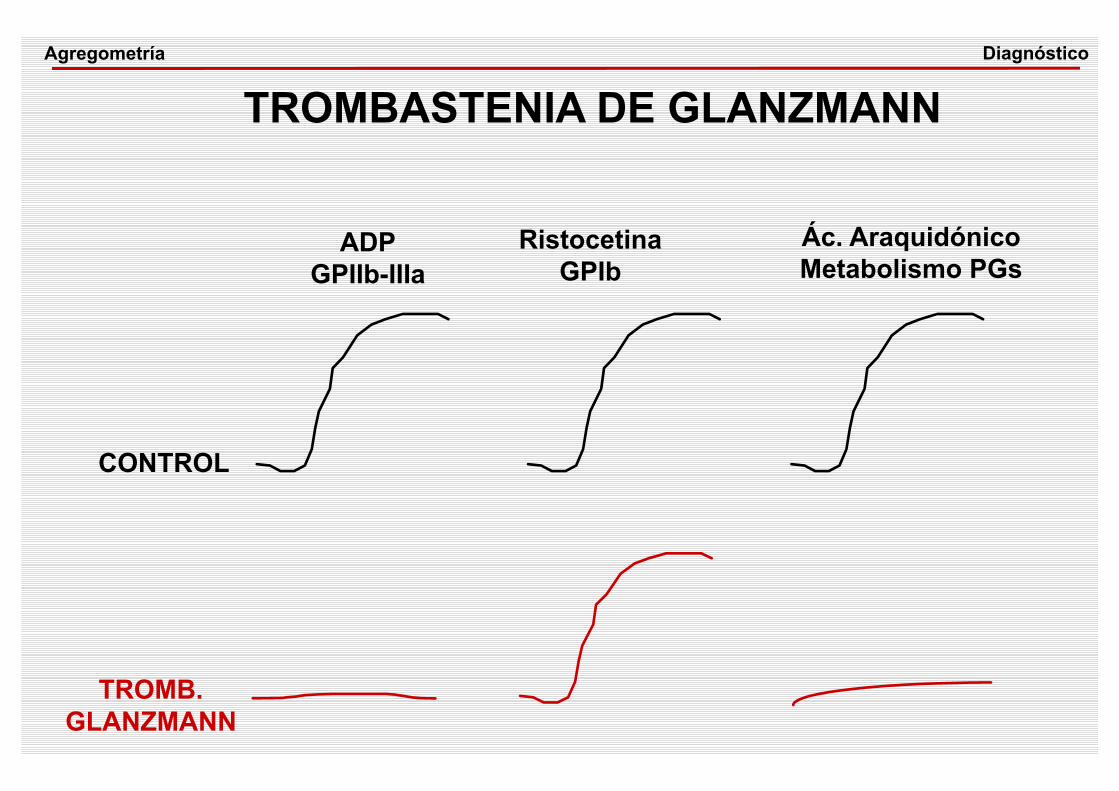
ADPGPIIb-IIIa
RistocetinaGPIb
Ác. AraquidónicoMetabolismo PGs
CONTROL
TROMB.GLANZMANN
TROMBASTENIA DE GLANZMANNDiagnósticoAgregometría
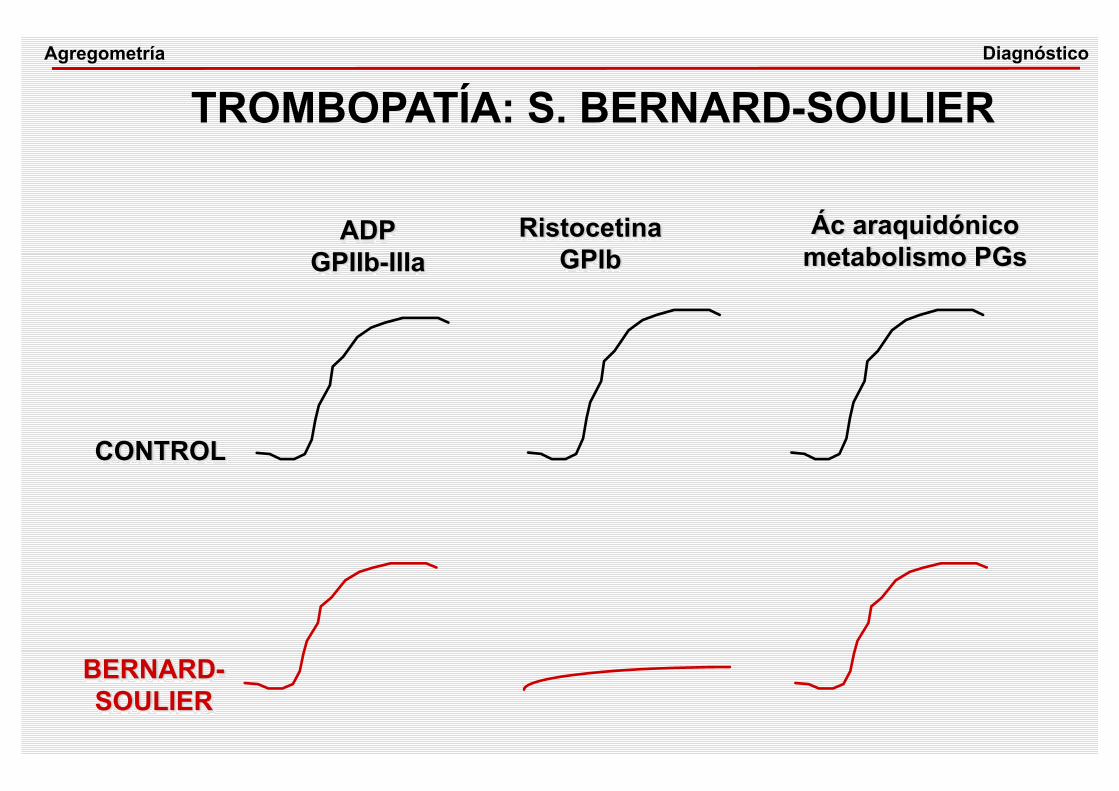
ADPGPIIb-IIIa
RistocetinaGPIb
Ác araquidónico metabolismo PGs
CONTROL
BERNARD-SOULIER
TROMBOPATÍA: S. BERNARD-SOULIERDiagnósticoAgregometría
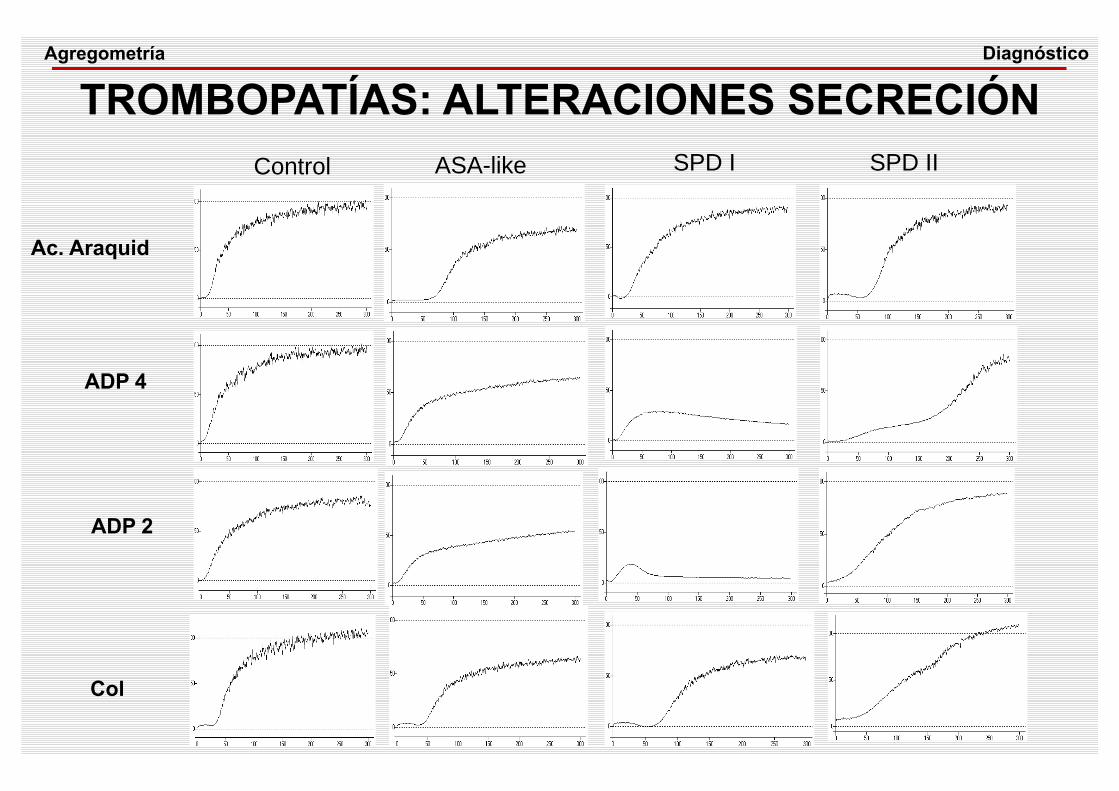
TROMBOPATÍAS: ALTERACIONES SECRECIÓN
Ac. Araquid
ADP 4
ADP 2
ASA-like
Col
SPD I SPD II
ADP 4
ADP 2
Control
Col
DiagnósticoAgregometría

PRUEBAS ADICIONALES
• Contenido y liberación ATP/ADP
• Glicoproteínas de membrana (FACS o PAGE)
• Estudios genéticos
• Ultraestructura plaquetaria
• Estudios de perfusión
• Transducción de señal (Fosforilación)
• Actividad FVW, antígeno y RiCof /Multímeros
Diagnóstico
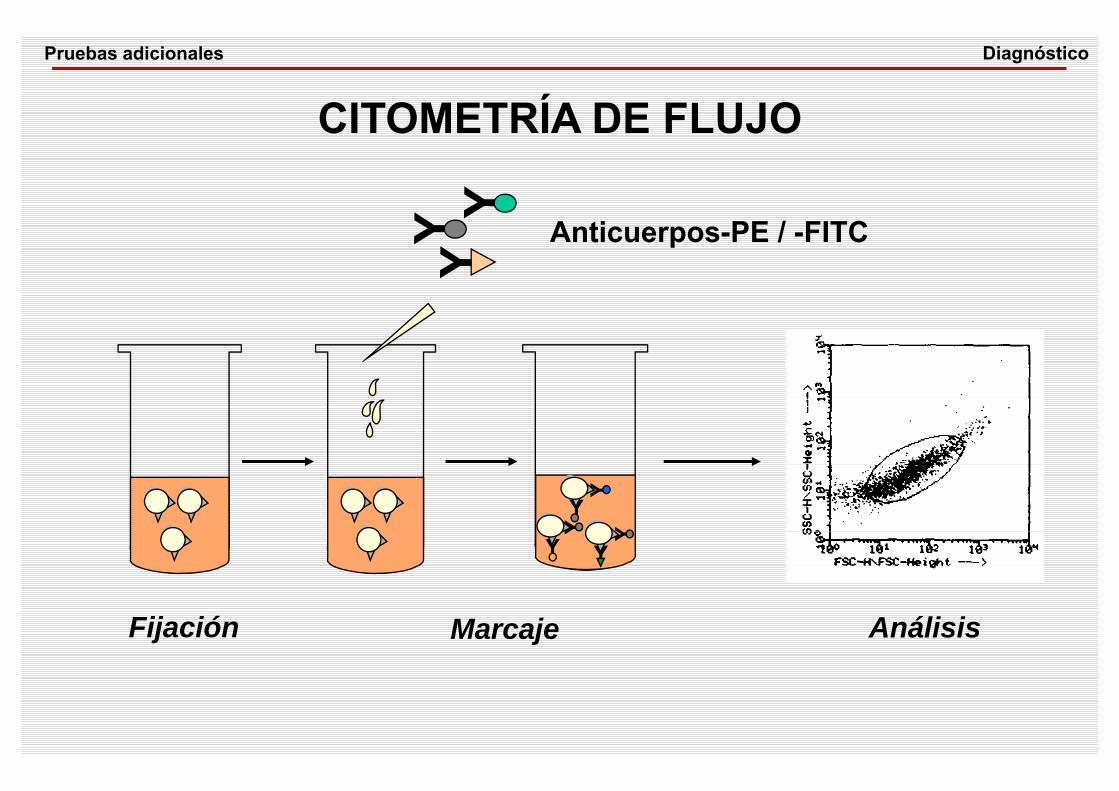
Y
YYY
Y
Y
Fijación Marcaje Análisis
Anticuerpos-PE / -FITC
CITOMETRÍA DE FLUJODiagnósticoPruebas adicionales
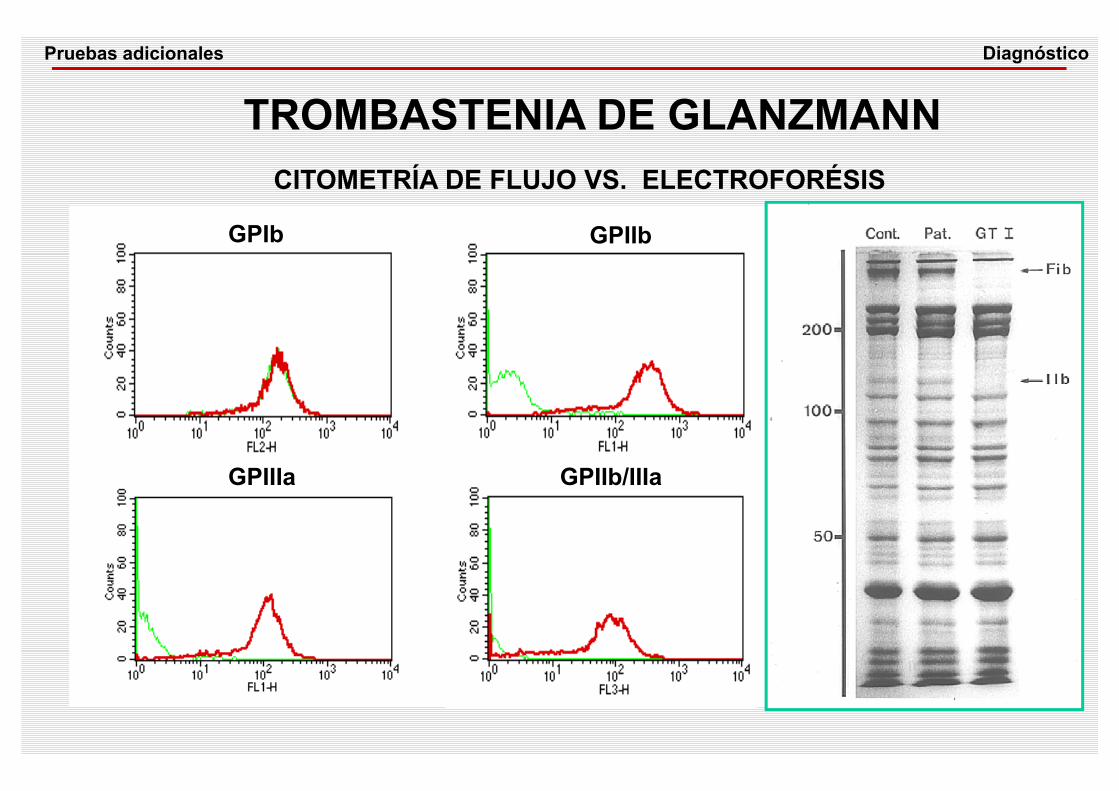
GPIb GPIIb
GPIIb/IIIaGPIIIa
CONTROLTROMBASTENIA GLANZMANN
TROMBASTENIA DE GLANZMANNCITOMETRÍA DE FLUJO VS. ELECTROFORÉSIS
DiagnósticoPruebas adicionales
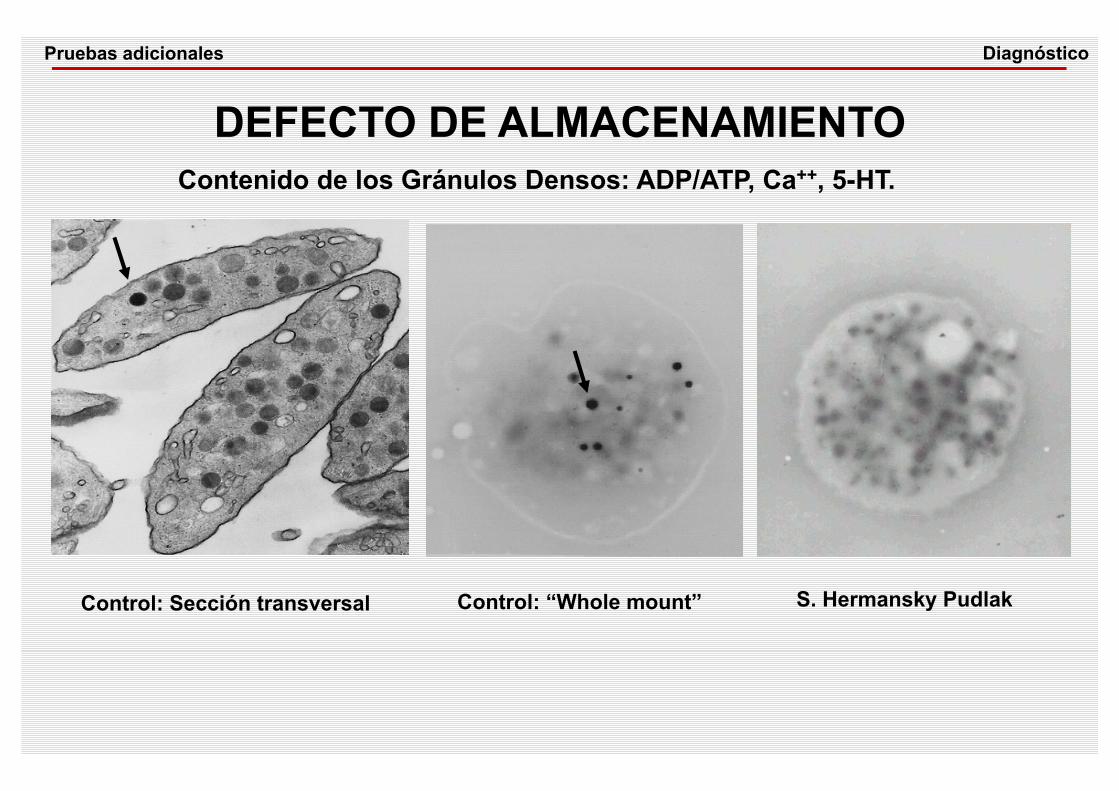
DEFECTO DE ALMACENAMIENTOContenido de los Gránulos Densos: ADP/ATP, Ca++, 5-HT.
Control: Sección transversal Control: “Whole mount” S. Hermansky Pudlak
DiagnósticoPruebas adicionales
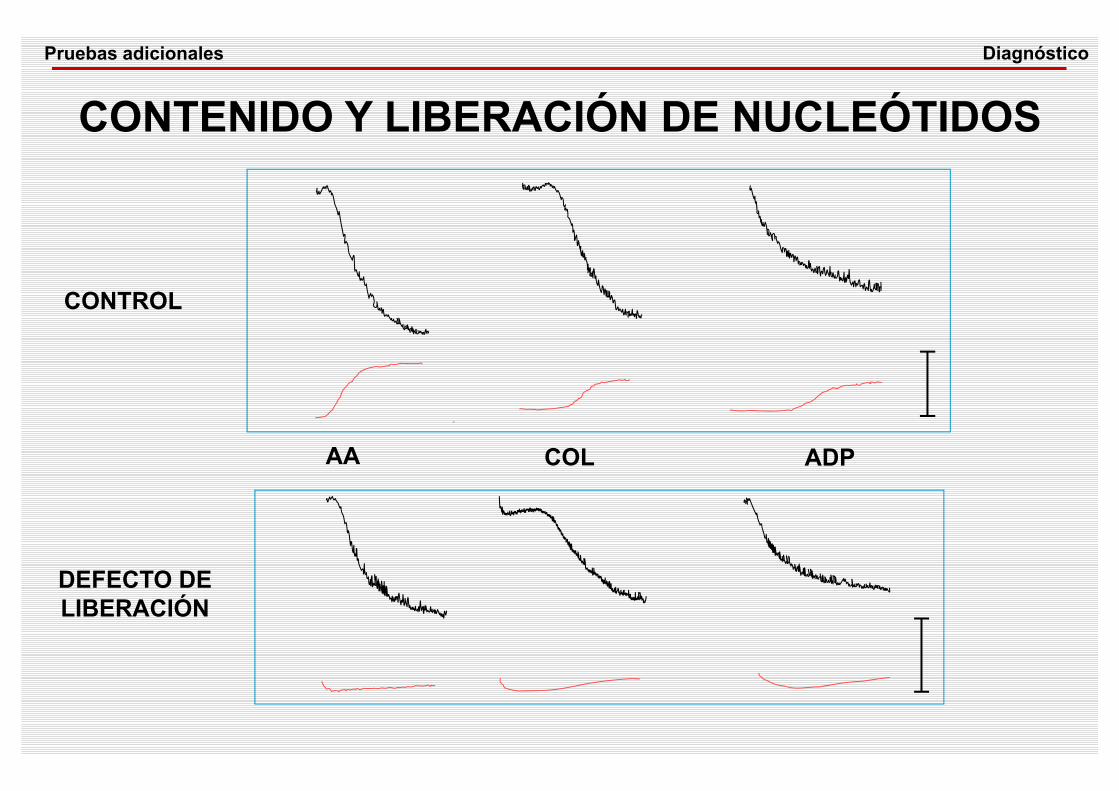
AA COL ADP
CONTENIDO Y LIBERACIÓN DE NUCLEÓTIDOS
CONTROL
DEFECTO DE LIBERACIÓN
DiagnósticoPruebas adicionales
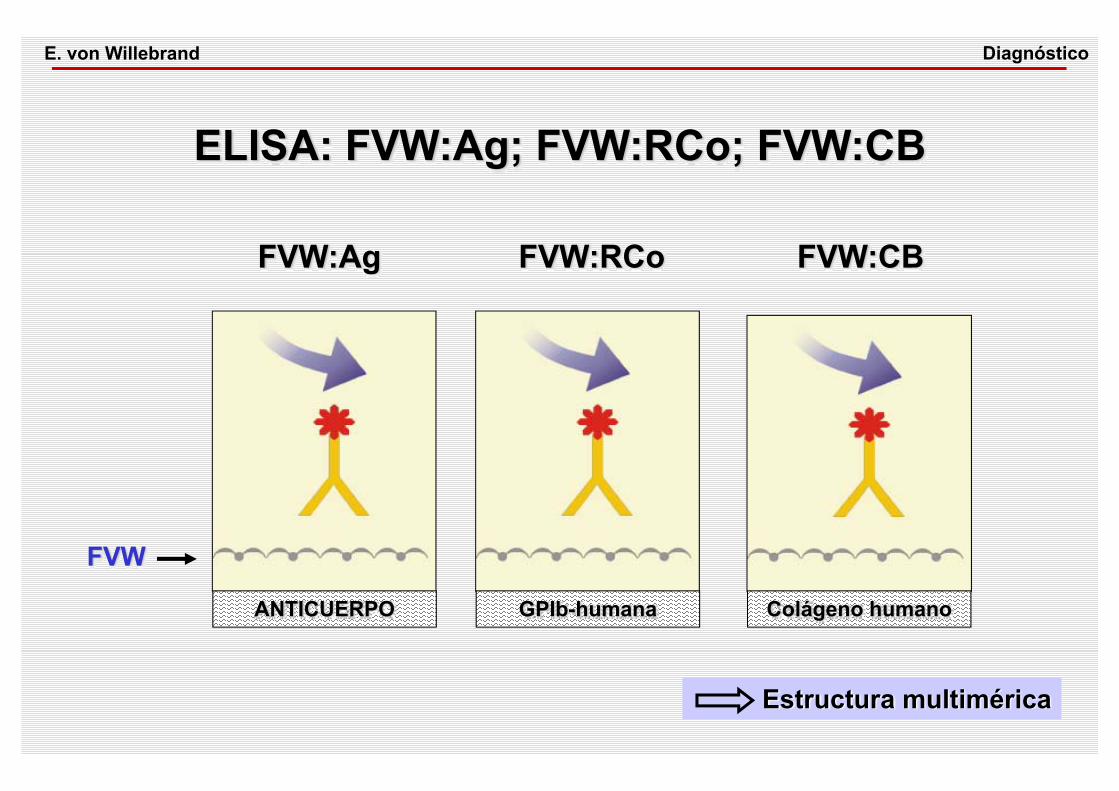
ELISA: FVW:Ag; FVW:RCo; FVW:CB
ANTICUERPO GPIb-humana Colágeno humano
FVW:Ag FVW:RCo FVW:CB
FVW
Estructura multimérica
DiagnósticoE. von Willebrand
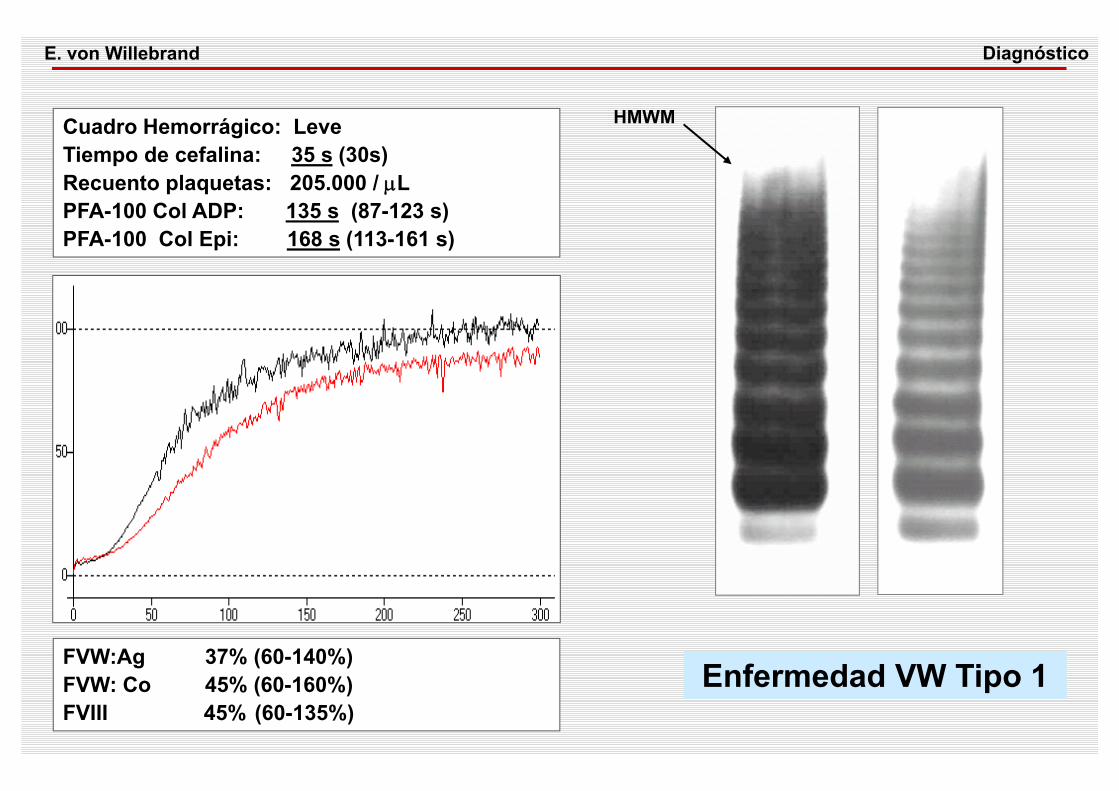
Cuadro Hemorrágico: LeveTiempo de cefalina: 35 s (30s) Recuento plaquetas: 205.000 / μLPFA-100 Col ADP: 135 s (87-123 s)PFA-100 Col Epi: 168 s (113-161 s)
FVW:Ag 37% (60-140%) FVW: Co 45% (60-160%)FVIII 45% (60-135%)
Enfermedad VW Tipo 1
HMWM
DiagnósticoE. von Willebrand

TRATAMIENTO
Desmopresina (DDAVP)
Antifibrinolíticos
Concentrados Factor von Willebrand
FVIIar
Atención a las transfusiones de plaquetas en pacientes Bernard-Soulier o con trombastenia de Glanzmman
Disfunción plaquetaria

• Los trastornos congénitos de la función plaquetaria queproducen sintomatología hemorrágica grave son raros.
• Con la excepción de los déficits congénitos de glicoproteínas,la correspondencia entre alteración de las pruebas ysignificación clínica es irregular.
• La historia clínica y una buena anamnesis son de gran utilidaden el diagnóstico de las alteraciones de la hemostasia.
• Es aconsejable la aplicación de sistemáticas para unagradación de la sintomatología hemorrágica.
• El PFA-100 permite la determinación de tiempos de sangrado“in vitro” y facilita la orientación diagnóstica de lasalteraciones de la hemostasia primaria.
• Son precisas varias tecnologías para alcanzar un diagnósticoespecífico de la patología.
CONSIDERACIONES DiagnósticoE. von Willebrand

• Carmen Altisent • Maribel Díaz-Ricart• Marc Pino• Juan Monteagudo• Juan Carlos Reverter• Dolors Tassies
AGRADECIMIENTOS
http://www.platelet-research.org/
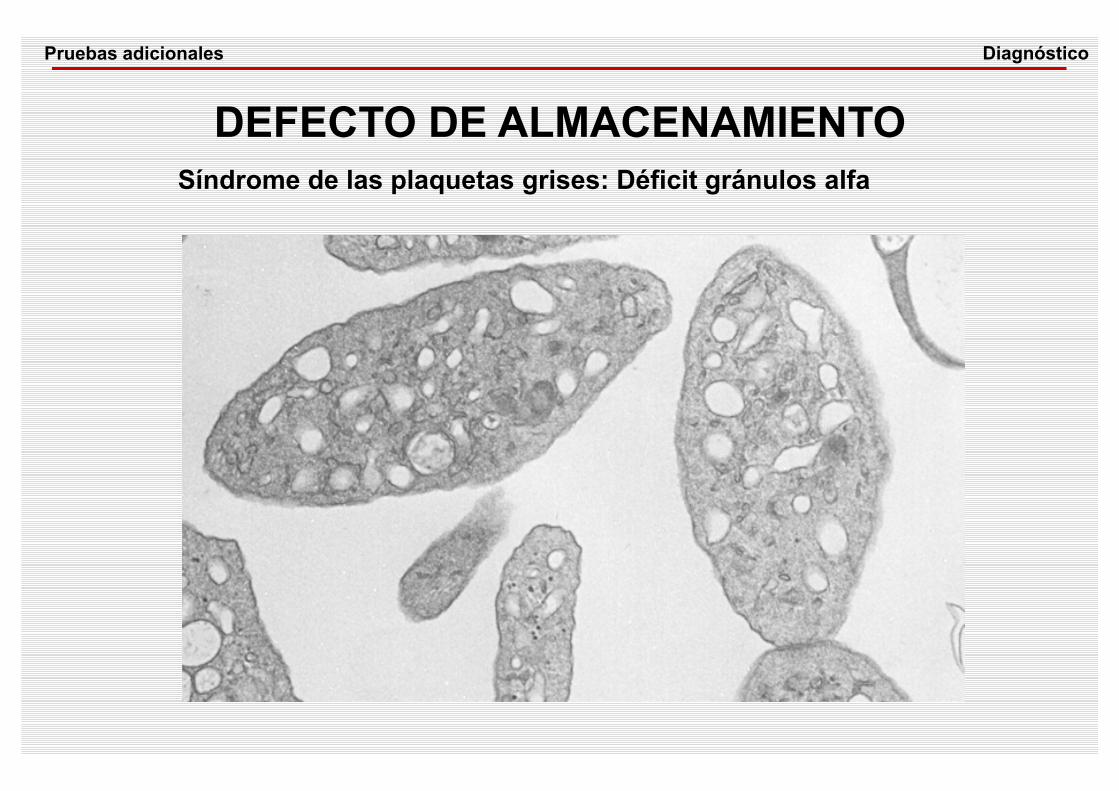
DEFECTO DE ALMACENAMIENTOSíndrome de las plaquetas grises: Déficit gránulos alfa
DiagnósticoPruebas adicionales
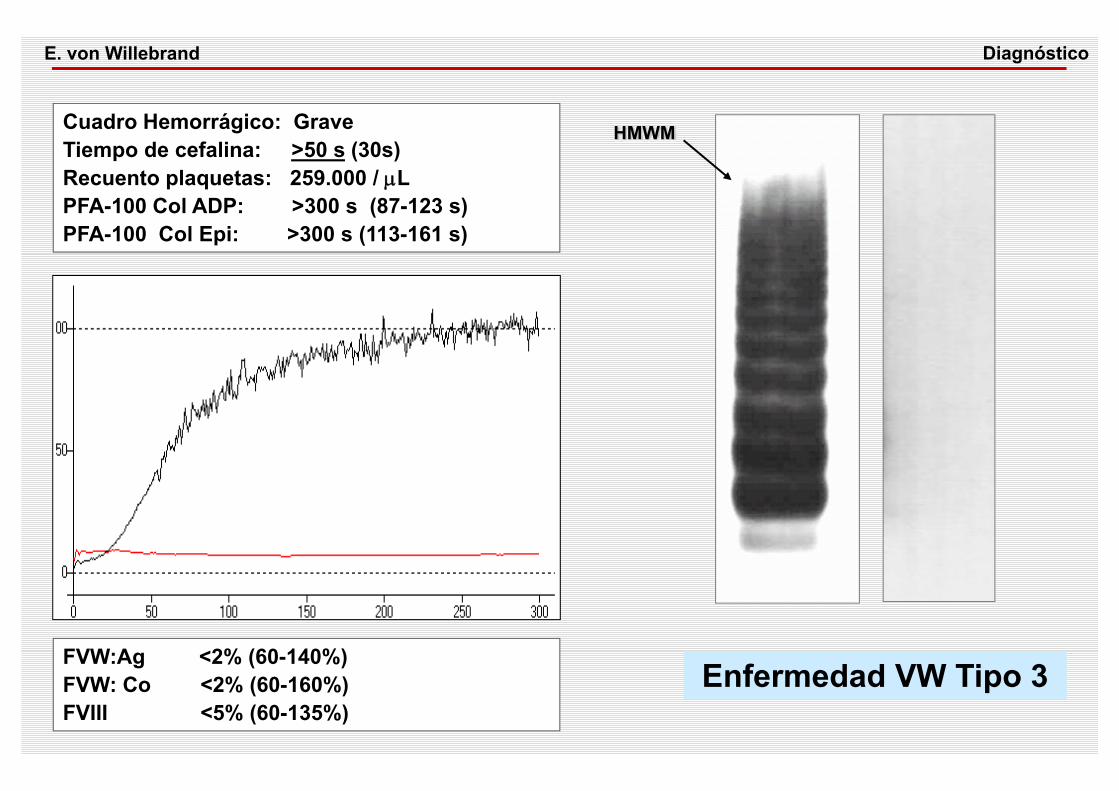
Cuadro Hemorrágico: Grave Tiempo de cefalina: >50 s (30s) Recuento plaquetas: 259.000 / μLPFA-100 Col ADP: >300 s (87-123 s)PFA-100 Col Epi: >300 s (113-161 s)
FVW:Ag <2% (60-140%) FVW: Co <2% (60-160%)FVIII <5% (60-135%)
Enfermedad VW Tipo 3
HMWM
DiagnósticoE. von Willebrand
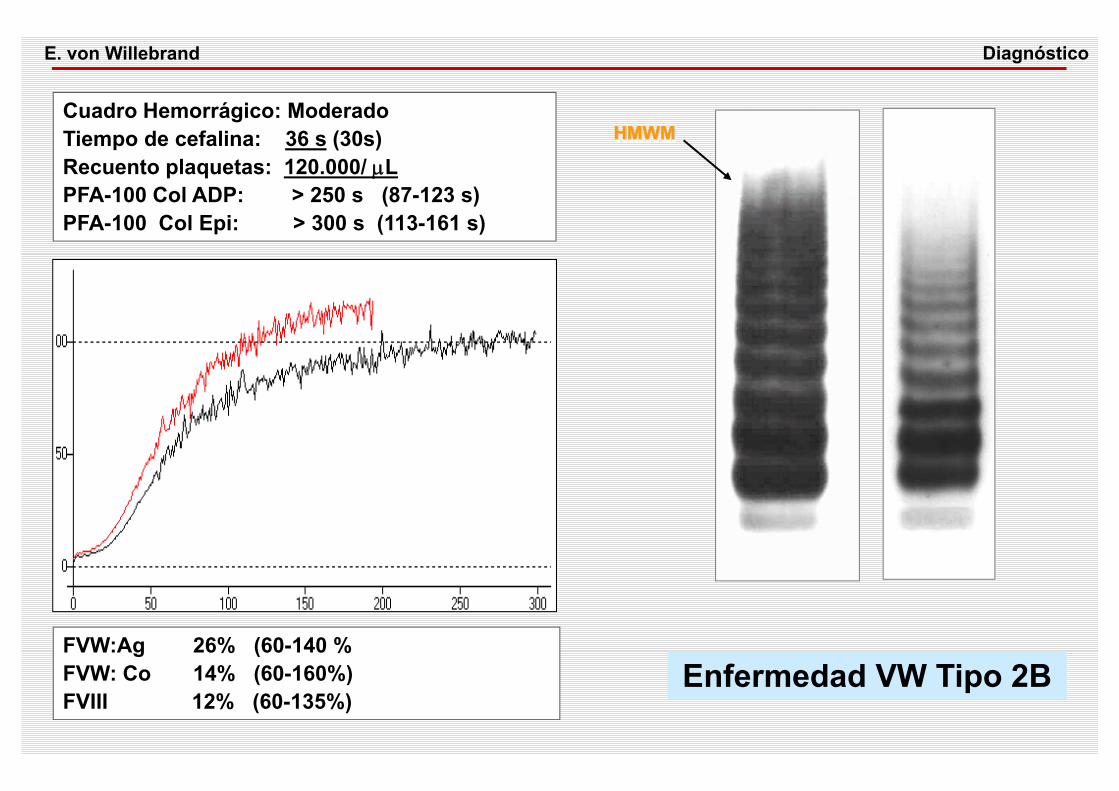
Cuadro Hemorrágico: ModeradoTiempo de cefalina: 36 s (30s) Recuento plaquetas: 120.000/ μLPFA-100 Col ADP: > 250 s (87-123 s)PFA-100 Col Epi: > 300 s (113-161 s)
FVW:Ag 26% (60-140 %FVW: Co 14% (60-160%)FVIII 12% (60-135%)
Enfermedad VW Tipo 2B
HMWM
DiagnósticoE. von Willebrand





























