Embed Size (px)
Citation preview

1
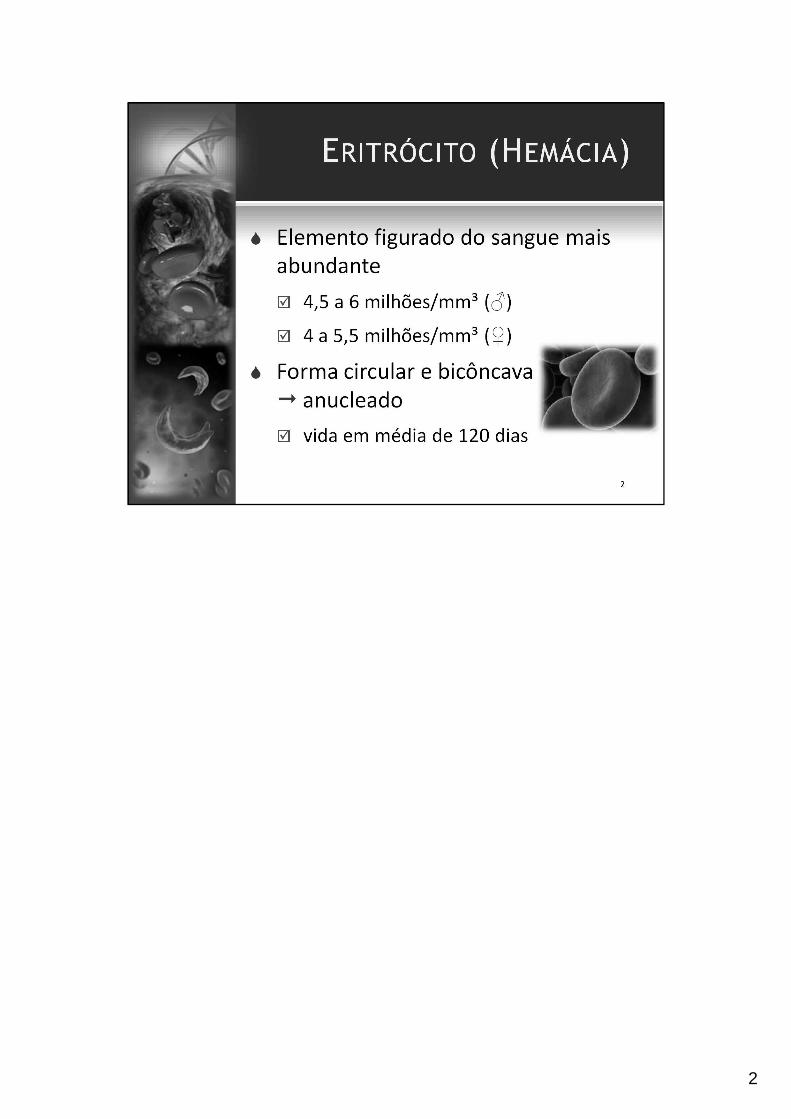
2

As proteínas que compõem a membrana eritrocitária são estruturalmente classificadas emintegrais ou transmembranárias e periféricas ou extramembranárias. Essas proteínas docitoesqueleto membranário formam uma verdadeira malha, que constitui quase uma conchapara o material intracelular. Este esqueleto é responsável pela forma, bicôncava normal ouanormal, em caso de defeitos genéticos, dos glóbulos vermelhos, e representa por si só 60% damassa proteica de toda a membrana. As proteínas integrais penetram ou atravessam a bicamadalipídica e interagem com a porção hidrofóbica das moléculas lipídicas. Fazem parte destas asproteínas de transporte, como a banda 3, denominada proteína transportadora de íons, e asglicoforinas A, B, C e D, que possuem receptores de membrana e antígenos que participam doreconhecimento célula-célula na extremidade externa, e auxiliam na estabilização docitoesqueleto através de ligações com proteínas na face interna da membrana. Das diferentesproteínas da membrana eritrocitária, o domínio citoplasmático da banda 3 se destaca como umgrande centro organizacional que interage com muitas outras proteínas periféricas ou ligantes,como a anquirina (a maior ponte para o citoesqueleto espectrina-actina) e outras proteínas queregulam a interação do citoesqueleto com enzimas glicolíticas.
3

Cerca de 70% do CO2 é transportado no plasma sob a forma de íons bicarbonato; 23% se liga àhemoglobina (Hb), formando a carboemoglobina e o restante é transportado dissolvido noplasma.
4

A molécula de hemoglobina é um tetrâmero com peso molecular de 64.458 dáltons (64kD), formado por quatro subunidades, iguais duas a duas. Cada subunidade é compostade duas partes: a globina, cadeia polipeptídica que varia muito geneticamente, e aheme, grupo prostético que consiste em um átomo de ferro situado no centro de umanel de porfirina, sendo semelhante em todas as formas geneticamente diferentes dehemoglobina.
5

Essa molécula tem estrutura aproximadamente esférica, com as cadeias de globinadobradas, de modo a que os quatro grupos heme se localizem em fendas superficiaisequidistantes umas das outras. Esse tetrâmero é mantido junto por ligações entre asquatro cadeias de globina, e sua estrutura quaternária muda à medida que o oxigênio écaptado pela oxigenação de cada grupo heme. As duas cadeias ( 2) são iguais,possuindo cada uma 141 aminoácidos; as duas cadeias ( 2) são também iguais entresi, compreendendo cada uma 146 aminoácidos.Na Figura está representada a molécula da hemoglobina humana adulta normal.
6

Assim, a hemoglobina normal do adulto (Hb A) tem a seguinte fórmula: 2 2. As cadeiase são quase iguais em comprimento, estrutura primária (sequência de aminoácidos)
e estrutura terciária configuração tridimensional). Elas também se assemelham àmioglobina (proteína transportadora de oxigênio no músculo), mas essa possui apenasuma cadeia polipeptídica. As semelhanças na sequência de aminoácidos e na estruturaterciária sugerem que as moléculas da hemoglobina e da mioglobina evoluíram a partirde um polipeptídeo ancestral comum.
7

O grupo heme é tão importante como a globina, por dois motivos: é o agente quedisponibiliza o oxigênio para a célula, e é um pigmento corado que possibilita o estudoda diferenciação e maturação dos precursores eritrocitários. Portanto, é o ferro,componente do heme que se combina com o oxigênio, conferindo à molécula dehemoglobina sua capacidade de transporte de O2.
8

Existem pelo menos oito locos bem conhecidos comandando a síntese da globina: alfa 1( 1), alfa 2 ( 2), beta ( ), delta ( ), gama A ( A), gama G ( G), épsilon ( ) e zeta ( ). Cadaloco é responsável pela estrutura de um tipo de cadeia polipeptídica. Existe ainda o locodo gene teta ( ), de função ainda não bem conhecida e atividade no saco vitelínico efígado fetal.
9

Os genes das globinas e fazem parte das famílias multigênicas, que são grupamentosde muitos genes, alguns deles não transcritos.
10

Os genes do grupamento da -globina são genes muito ligados, situados no braço curtodo cromossomo 16 (16p13.3). São eles: zeta ( ), alfa 2 ( 2), alfa 1 ( 1) e teta ( )(Figura). Os três primeiros genes são ativados e transcritos nessa mesma ordem duranteo desenvolvimento. Esse complexo engloba cerca de 30 kb ou 30 mil pares de basesnitrogenadas. Na literatura genética consultada, também se encontram valores quevariam de 28 a 40 kb para o tamanho desse grupamento gênico humano. Cada gene éformado por três éxons e dois íntrons. Entre os genes e 2, existem três pseudogenes( , 1 e ). Os pseudogenes (representados pela letra grega psi ( ) e o símbolo dogene a que mais se assemelham) são remanescentes de genes que outrorafuncionavam, mas sofreram tantas mutações, que se tornaram incapazes de sertranscritos e produzir proteínas. São relíquias evolutivas, que conservam grandehomologia de sequências com os genes funcionais.
11

Os genes do grupamento da -globina são genes ligados, situados no braço curto docromossomo 11 (11p15.5). São eles: épsilon ( ), gama G ( G), gama A ( A), delta ( ) ebeta ( ), sendo expressos nessa mesma ordem durante o desenvolvimento (Figura).Cada gene está formado também por três éxons e dois íntrons. Os dois genes gama G( G) e gama A ( A) codificam cadeias que diferem em apenas um aminoácido naposição 136: gama G ( G) possui glicina; gama A ( A), alanina. A expressão desses genesno desenvolvimento fetal é evolutivamente recente, tendo surgido nos primatas.Especula-se que a hemoglobina fetal (HbF), da qual participam as cadeias , sejavantajosa para esses organismos, devido ao seu tempo maior de gestação, que exige ummeio mais eficiente de assegurar o transporte adequado de O2 no feto. Essahemoglobina tem uma afinidade mais alta pelo oxigênio do que a hemoglobina adulta,sendo, portanto, capaz de captar oxigênio da circulação materna com mais eficiência,através da placenta. O gene delta ( ) assemelha-se ao beta ( ), mas adquiriu muitasalterações em seu promotor, que o tornam relativamente ineficiente.Esse complexo gênico é mais extenso do que o grupamento , compreendendo cerca de60 kb ou 60 mil pares de bases nitrogenadas. A literatura genética consultada tambémmenciona valores que variam de 50 kb a 65 kb para esse complexo gênico. Nogrupamento da -globina, há um pseudogene ( ), localizado entre os genes A e .
12

13

14

15

16

17

18

19

20

21

22

As anormalidades de membrana que resultam em anemia hemolítica são devido a alterações de umaestruturais. Os lipídeos da membrana, bem como o sistema de transporte de cátions, podem estaralterados e, consequentemente, produzir defeitos no funcionamento da membrana. Todas essas trêsanormalidades (proteínas, lipídeos e sistema de transporte de cátions) alteram a superfície da área damembrana, promovem aumento de sua rigidez, fragmentação ou deformação. Os eritrócitos anormaisapresentam dificuldades no trânsito vascular do baço. As doenças causadas por anormalidades demembrana inclem esferocitose hereditária, eliptocitose hereditária, piropoiquilocitose hereditária,estomatocitose hereditária e xenocitose.Hemoglobinas anormais: As hemoglobinas anormais costumam abranger as que são consideradasvariantes, bem como as hemoglobinas normais com alterações quantitativas, por exemplo: Hb A2 elevada,Hb F elevada, HbA2 diminuída. Há centenas de variantes já descritas, porém nem todas estão associadas amanifestações clínicas e alterações hematológicas. Essas variantes podem ser classificadas em duascategorias principais: as variantes estruturais e os defeitos de síntese das hemoglobinas.As variantes estruturais abrangem cinco classes: (1) hemoglobinas de agregação, que formam cristais,com repercussões clínicas e laboratoriais variáveis, como as hemoglobinas S e C; (2) hemoglobinas comfenótipos talassêmicos, devidas a falhas na regulação da síntese da globina por adição de aminoácidos àextremidade C-terminal das globinas e , e por fusão de cadeias devido ao crossing-over desigualdurante a meiose, sendo exemplos, respectivamente, a Hb Cranston e as Hbs Lepore/anti-Lepore; (3)hemoglobinas instáveis, com graus variáveis de manifestações clínicas e hematológicas, bem comoexpressão laboratorial diversificada entre os tipos já descritos, cujo exemplo é a Hb Niterói; (4)hemoglobinas com alterações funcionais que causam metemoglobinemias por HbM, cianose e alteraçãoda afinidade hemoglobínica pelo oxigênio; e (5) hemoglobinas sem alterações funcionais, que consistemna maioria das variantes estruturais e, embora apresentem importância bioquímica, genética eantropológica, não produzem efeitos clínicos e laboratoriais significativos, como a Hb Kenya.Os defeitos de síntese das hemoglobinas englobam as -talassemias e as -talassemias, as síndromes depersistência hereditária da hemoglobina fetal e as metemoglonemias.
23

As talassemias consistem em um conjunto de síndromes motivadas principalmente poralterações quantitativas da síntese de globinas e , causando desequilíbrio entre elas e grausvariáveis de anemias hemolíticas, além de outras consequências patológicas.
24

As talassemias são também chamadas de anemia do mediterrâneo, entre outros nomes.
25

26

Levam a um quadro clinico variável, dependendo do número de locos alfa deletados.
27

28

A perda de um gene constitui um estado de portador silencioso, sem importância clínica.
29

Quando dois dos genes estão inativos, há duas situações possíveis: na primeira, frequentementea causa de -talassemia heterozigota no sudeste asiático, ambos os genes deletados localizam-se no mesmo cromossomo ( ); na outra, frequente entre os afrodescendentes com -talassemia heterozigota, onde em cada cromossomo há um gene deletado ( / ). Essefenótipo é relativamente benigno, resultando em anemia leve com microcitose.
30

A perda de três genes resulta em problemas clínicos graves. As cadeias de -globinapredominam e formam homotetrâmeros ( 4), resultando a hemoglobina H (HbH), de reduzidacapacidade para o transporte de oxigênio e visualizada como corpos de inclusão nas hemáciasde indivíduos com -talassemia.
31


Hidropsia fetal de Bart (Hb de Bart): quando houve a perda dos quatro genes alfas devidodeleção. É um tipo incompatível com a vida. Provoca hepatomegalia severa e morte fetal.
33

Porque ela surge quando cada genitor apresenta um cromossomo com deleção de ambos osgenes ( ), que é mais comum em populações de origem asiática. Nos afrodescendentes, osalelos geralmente têm configuração / ( e não ).
34

35

Ao contrário das -talassemias, as -talassemias geralmente não são devidas a uma deleção,mas à redução ou à supressão da síntese das cadeias como resultado de mais de cemdiferentes mutações pontuais, com predomínio das substituições de bases.
36



39


41

42

Tanto na hemoglobina desoxigenada (desoxiemoglobina) quanto na hemoglobinaoxigenada (oxiemoglobina), o ferro constituinte do grupo heme está presente naforma ferrosa (Fe2+) e não há variação na sua valência durante todo o processo deoxigenação e desoxigenação. No entanto, se íon ferroso (Fe2+) for oxidado a íon férrico(Fe3+) por meio de um agente oxidante como o ferrocianeto, forma-se metemoglobina,molécula que não consegue combinar com o oxigênio.
43

44

Glu = ácido glutâmicoVal = valinaLys = lisina
45

46

Essa mutação afeta a solubilidade e causa a cristalização dessa hemoglobina em condições dehipóxia. Com um grau relativamente baixo de hipóxia, a HbS polimeriza, formando feixes defibras. Esses cristais de hemoglobina anormal torcem a membrana da hemácia, dando-lhe umaforma característica de foice (falciforme). Algumas células permanecem irreversivelmentefalciformes após episódios repetidos de hipóxia e reoxigenação, sendo destruídasprematuramente em crises hemolíticas.
47

48

49

Condições de baixa tensão de O2: hipóxia, acidose, desidratação, vasoconstrição, anestesia geral,voo em avião despressurizado, regiões de altitude elevada, mergulhos em profundidade,excesso de esforço físico etc.
50

As áreas africanas onde a malária é endêmica e a área onde é alta a incidência do alelocausador da anemia falciforme coincidem porque os indivíduos heterozigotos HbA/HbS(traço falcêmico) têm maior chance de sobreviver quando infectados pelo Plasmodium(agente causador da malária) e transmitem o alelo mutante (HbS) aos seusdescendentes. Assim, o Plasmodium atua como agente da seleção natural, visto queseleciona indivíduos resistentes à infecção (heterozigotos HbA/HbS), pois os indivíduoshomozigotos HbA/HbA (normais) têm menores chances de sobrevivência em áreasendêmicas de malária (morrem de malária).
51

Palidez, cansaço fácil, por causa da redução do oxigênio circulante.Icterícia: cor amarelada mais visível na esclera (branco dos olhos), por causa do excesso debilirrubina no sistema circulatório, resultante da destruição rápida dos glóbulos vermelhos.
52

53

A viscosidade aumentada e a rigidez da membrana, diminuem a capacidade das célulasfalciformes para atravessarem os capilares. A hipóxia resultante leva a mais falcização, comepisódios característicos de crises de falcização.
54

A oclusão vascular (vaso-oclusão) provoca inchaço doloroso nas mãos e pés.
55

O sequestro esplênico é uma complicação aguda da maior gravidade, sendo causa de grandemorbidade e mortalidade em pacientes com doença falciforme. Instala-se subitamente, havendoqueda progressiva nos valores sanguíneos de hemoglobina e, não raramente, evoluindo aochoque hipovolêmico. É potencialmente fatal se não tratado rapidamente. Durante esta crise, oindivíduo apresenta anemia aguda e choque, sem as características de uma crise hemolítica. Anecrópsia mostra vasos periféricos vazios, isquemia generalizada e grande quantidade dehemácias falciformes sequestradas pelo baço.
56

A asplenia é um termo usado para indica a ausência completa da função esplênica.
57

As crises aplásticas não são muito frequentes e geralmente ocorrem após processosinfecciosos, mesmo após infecções relativamente insignificantes. Clinicamente seapresentam por sintomas de anemia aguda sem aumento esplênico, podendo, emsituações mais severas, estarem presentes sinais de choque hipovolêmico.A principal diferença laboratorial entre estas crises e as de sequestro esplênico é apresença de reticulocitopenia na crise aplástica e reticulocitose na de sequestro. Otratamento é sintomático e transfusões de concentrado de hemácias devem seradministradas se necessário. A monitorização do estado hemodinâmico é quepossibilitará a indicação precisa de hemotransfusão.
58

59

Os pulmões são um dos principais alvos de complicações agudas e crônicas na anemiafalciforme. Os fenômenos vaso-oclusivos característicos das doenças falciformes podem ocorrerem qualquer órgão, incluindo o coração e os pulmões. Com o aumento da sobrevida dospacientes, aumentou a incidência de falência crônica de órgãos e, apesar do acometimentocardíaco ser comum, ainda é pouco diagnosticado. Já as complicações pulmonares estão entre asprincipais causas de morbidade e mortalidade destas doenças, sendo a segunda causa deadmissão hospitalar e a primeira causa de morte em adultos.
60

O acometimento pulmonar pode ser de natureza aguda ou crônica. As complicações agudas sãorepresentadas pela hiper-reatividade brônquica, pelo tromboembolismo pulmonar e pelasíndrome torácica aguda (STA). As complicações crônicas levam a alterações da função pulmonar(doença restritiva, doença obstrutiva e capacidade anormal de difusão) e à hipertensãopulmonar (HP). A STA e a HP estão entre as causas mais importantes de morbimortalidade nestapopulação. A síndrome torácica aguda é a segunda causa de internação hospitalar apresentandoconsiderável morbimortalidade.
61

As complicações neurológicas da anemia falciforme são causadas pelo acidente vascular cerebral(AVC), ataques isquêmicos transitórios (AIT), infartos cerebrais silenciosos e diminuição dodesempenho neuropsicológico. A consequência do efeito cumulativo destas complicações é ofuncionamento intelectual rebaixado, a diminuição do rendimento acadêmico, ou abandonoescolar, que repercutirão na possibilidade destes indivíduos obterem melhor inserção nomercado de trabalho, além do resultante sofrimento psicoafetivo (Rev. Bras. Hematol. Hemoter.2010;32(2):181-185).
62

Doentes falciformes podem apresentar alterações hepáticas agudas ou crônicas.A colestase intra-hepática resulta de um processo de falcização intra-sinusoidal, hipoxemia eisquemia dos hepatócitos com consequente edema dos hepatócitos e colestase intracanalicular.As manifestações clínicas são semelhantes à crise aguda de falcização hepática, com dor noquadrante superior direito, náusea, vômito, hepatomegalia dolorosa, febre e leucocitose.Entretanto, ocorre um aumento acentuado da bilirrubina, acompanhado de insuficiência renal,coagulopatia e encefalopatia hepática. (Rev. Bras. Hematol. Hemoter. 2007; 29(3):299-303)Hemocromatose é um termo utilizado para se referir à sobrecarga de ferro que é associada àdisfunção pancreática, hepática e cardiovascular. A hemocromatose primária é um problemahereditário (erro inato do metabolismo) que resulta da mutação de um ou mais genesenvolvidos na absorção e transporte de ferro. A hemocromatose secundária é quase semprecausada por distúrbio hereditário ou adquirido da eritropoiese e/ou tratamentos de doençascom transfusão sanguínea, apesar de também poder surgir por ingestão excessiva de ferro oualcoolismo.Hemossiderose: acúmulo anormal de hemossiderina nos tecidos, especialmente nos macrófagosda derme, do fígado, do baço, da medula óssea, linfonodos e pulmões. Hemossiderina: Pigmentobrilhante, amarelo ouro ou castanho escuro, resultante da degradação da Hb.
63



A transfusão tem um papel estabelecido na gestão de complicações agudas e crônicas da anemiafalciforme. Corrige-se a anemia, mantendo a concentração de HbS abaixo de 30%, aumentandoa oxigenação e diminuindo complicações vasculares e reduzindo a hemólise. Além disso, astransfusões previnem acidentes vasculares cerebrais primários e secundários e diminuem afrequência de internações, eventos vaso-oclusivos, síndrome torácica aguda e retardo decrescimento.
66

Os heterozigotos HbA/HbC apresentam de 25-40% de HbC, sendo clinicamente assintomáticos.
67

Quando combinado ao traço falcêmico, um defeito no gene determinante dabetatalassemia produz uma doença bastante semelhante à anemia falciforme. O genedeterminante da betatalassemia diminui a taxa de síntese da cadeia beta-A, resultando napredominância de beta-S em pacientes com traço falciforme. Dependendo de o paciente tertalassemia beta-0 ou beta+, as hemácias contêm quantidades variáveis de HbS, HbA, HbA2 eHbF. Pacientes com talassemia beta-0 não possuem HbA, mas apenas HbS, HbF e HbA2. Por isso,estes pacientes desenvolvem uma doença severa. O diagnóstico é baseado na elevação dosníveis de HbA2, HbF ou ambas detectada por eletroforese de hemoglobina, bem comopela história familiar positiva de talassemia e presença de gene falciforme.
68

69

70





























