Embed Size (px)
Citation preview

©2
01
6 D
r. W
alte
r F.
de
Aze
ve
do
Jr.
1
000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000010000000000000000000000000000000000111111111100000000000000000000000000001111110000000000110000000000000110000001111100000000111110000000000001000011001111111111111111000000000000010001110110001111111000000000000000000011111111111111111111111110000000001111111111111111111111111111110000000000111111111111111111111111110000000000000000111111111111111111111000000000000000000111111111111111110000000000000000000000111111111111111111110000000000000000001111101111111111111111000000000000000001111100111111111111111000000000000000000000111111111111110011100000000000000000000001111111110111111100000000000000000000001110000011101100000000000000000000000000000011111001100000000000000000000000000000000000110000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000
azevedolab.net

Implemente os seguintes códigos:
1) Área acessível ao solvente de proteínas (versão 1). Programa: asa1.py
2) Área acessível ao solvente de proteínas (versão 2). Programa: asa2.py
3) Energia livre de Gibbs para o sistema proteína-ligante. Programa: freeEnergy.py
Data da entrega: Até dia 26/07/2016.
Favor enviar para o e-mail: [email protected]
2
Trabalho 1
www.python.org

Resumo
O programa asa1.py usa uma equação empírica para o cálculo aproximado da
área acessível ao solvente de uma proteína. A entrada é a massa molecular da
proteína em Daltons, variável (molWeight). A saída é área acessível ao solvente
em Å2, variável area. O cálculo é uma aproximação válida para proteínas
monoméricas, publicada em: LESK Arthur M. Introduction to Protein
Architecture. Oxford: Oxford University Press, 2001. página 27. A equação em
Python para o cálculo da ASA é a seguinte: area= 11.1*(molWeight)**(2/3).
Área acessível ao solvente de proteínas (versão 1)
Programa: asa1.py
3
Programa: asa1.py
www.python.org

Diversos programas gráficos permitem a visualização molecular, como a representada
abaixo para o aminoácido alanina. A representação da figura é chamada CPK,
referente aos nomes dos cientistas que a propuseram (Corey, Pauling e Koltun)
(COREY e PAULING, 1953; KOLTUN, 1965). A figura abaixo foi gerada com o
programa VMD (visual molecular dynamics, disponível em:
http://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD
)(HUMPHREY et al, 1996). As esferas em ciano representam os átomos de carbono,
em branco os átomos de hidrogênio, vermelho para oxigênio e azul para o nitrogênio.
Os bastões são ligações covalentes entre os átomos.
VMD é o acrônimo para visual molecular dynamics.4
Programa: asa1.py
www.python.org

Abaixo temos a representação de van der Waals do aminoácido alanina, onde os
átomos são desenhados como esferas com o raio de van der Waals. Esta
representação mostra a superfície de van der Waals, definida pela área contínua das
superfícies esféricas. A tabela abaixo mostra os raios de van der Waals dos principais
átomos encontrados em moléculas biológicas.
Na figura as esferas
representam átomos, onde o
raio de cada esfera é o raio de
van der Waals. O código de
cores é o mesmo usado para
representação CPK.
Átomo Raio de van der Waals (Å)
H 1,20
C 1,85
N 1,54
O 1,40
P 1,90
S 1,85
5
Programa: asa1.py
www.python.org

A superfície molecular é a superfície de van der Waals suavizada nas reentrâncias,
pois consideramos uma molécula de água esférica rolando sobre a superfície de van
der Waals. Esta molécula de água é chamada sonda. Quando aplicada à uma proteína
permite a visualização de cavidades na superfície proteica.
Simplificação da representação
da molécula de água, como uma
esfera de raio de 1,4 Å.
Superfície molecular
A superfície molecular do
aminoácido alanina. O código
de cores é o mesmo usado
para representação CPK.
6
Programa: asa1.py
www.python.org

As proteínas em solução permitem a interação de moléculas de água com sua
estrutura. A área acessível ao solvente (ASA, para accessible surface area) é um
parâmetro geométrico usado para analisar a interação de ligantes e água com a
superfície da proteína. O conceito de tal parâmetro é relativamente simples. Imagine
uma molécula de água deslizando sobre a superfície da proteína, uma molécula
esférica com um raio 1,4 Å é usada na representação, como mostrado abaixo. Esta
forma esférica para a molécula de água não é realista, mas é uma boa aproximação
para nossos propósitos. A superfície acessível ao solvente é traçada pelo
movimento do centro da esfera do solvente, indicada pela linha tracejada.
Simplificação da representação
da molécula de água, como uma
esfera de raio de 1,4 Å.
ASA é o acrônimo em inglês para accessible surface area.
Molécula de água
Superfície acessível ao solvente
Cada átomo está
representado por uma esfera,
onde o raio é o de van der
Waals.
7
Superfície molecular
Programa: asa1.py
www.python.org

Há uma equação simples (LESK, 2001), que relaciona a massa molecular em Daltons
(MM) de uma proteína monomérica, com sua área acessível ao solvente (ASA), esta
equação é uma aproximação, mas pode ser usada para termos uma ideia preliminar
da ASA. O resultado sai em angstroms ao quadrado (Å2).
ASA = 11,1 (MM)2/3
Superfície molecular
Molécula de águaSimplificação da representação
da molécula de água, como uma
esfera de raio de 1,4 Å.
Superfície acessível ao solvente
Referência para a equação da área acessível ao solvente: LESK A. M. Introduction to Protein
Architecture. Oxford University Press, Oxford UK, 2001, página 27.8
Cada átomo está
representado por uma esfera,
onde o raio é o de van der
Waals.
Programa: asa1.py
www.python.org

Vamos preparar o pseudocódigo para o cálculo da área acessível ao solvente de uma
proteína, para a qual sabemos a massa molecular (MM). O algoritmo é bem simples,
temos como variável independente o MM (molWeight) e a saída é a área acessível ao
solvente (area). A equação fica da seguinte forma:
area = 11,1 (molWeight)2/3
O pseudocódigo está no quadro abaixo. O cálculo só é efetuado se a massa molecular
for maior que zero, caso contrário é mostrada uma mensagem de erro.
Início
Leia (molWeight)
Se (molWeight > 0)
area = 11.1*(molWeight)**(2/3)
Escreva "Area: ",area
Senão
Escreva "\nErro! A massa deve ser > 0!"
Fim
9
Programa: asa1.py
www.python.org

Teste o seu programa para os valores de massas moleculares indicados abaixo. Na
tabela temos as massas moleculares em Daltons e a área acessível ao solvente em
Å2. Os números estão em notação inglesa, ou seja, com ponto decimal.
A informação sobre as massas moleculares (MW) das proteínas foi obtida da base de
dados Protein Data Bank (PDB)(http://www.rcsb.org/pdb/home/home.do ).
Proteína Fonte Massa molecular (Daltons)* ASA (Å2)
Mioglobina Physeter catodon 17868.60 7586.649088941961
CDK2 Homo sapiens 34331.25 11725.086527666364
Protease
(monômetro)
HIV-1 11125.23 5531.747024763476
Chiquimato quinase Mycobacterium
tuberculosis
19309.07 7989.088217383464
10
Programa: asa1.py
www.python.org

11
Referências relacionadas ao programa asa1.py
-COREY, RB; PAULING L. Molecular models of amino acids, peptides and proteins.
Review of Scientific Instruments, Nova York, v. 24, n.8, p.621-627, 1953.
-HUMPHREY W; DALKE A; SCHULTEN K. VMD - Visual Molecular Dynamics. Journal
of Molecular Graphics, Amsterdã, v.14, p.33-38, 1996.
-KOLTUN WL. Precision space-filling atomic models. Biopolymers, Hoboken, v.3, n.6,
p.665-79, 1965.
-LESK, Arthur. M. Introduction to Protein Architecture. Oxford: Oxford University
Press, 2001. 347 p.
-Proteopedia. Disponível em: <
http://www.proteopedia.org/wiki/index.php/CPK#cite_note-0 >. Acesso em 25 de
março de 2016.
Programa: asa1.py
www.python.org

Resumo
O programa asa2.py calcula a área acessível ao solvente de uma proteína de
forma similar ao programa asa1.py. A modificação ocorre na entrada, que agora
é o número de resíduos de aminoácidos. A massa molecular da proteína é
calculada multiplicando-se o número de resíduos por 118,89 Daltons, que é a
massa molecular média de cada aminoácido. A saída é área acessível ao
solvente em Å2. O cálculo é uma aproximação válida para proteína
monoméricas, publicada em: LESK Arthur. M. Introduction to Protein
Architecture. Oxford: Oxford University Press, 2001. página 27. A equação em
Python para o cálculo da ASA é a seguinte: area= 11.1*(molWeight)**(2/3).
Área acessível ao solvente de proteínas (versão 2)
Programa: asa2.py
12
Programa: asa2.py
www.python.org

O programa anterior (asa1.py) calcula a
área acessível ao solvente de proteínas, a
partir do conhecimento da massa
molecular. Podemos modificar o programa
asa1.py para que o novo programa
(asa2.py) considere o número de resíduos
de aminoácidos como entrada.
Normalmente usamos a unidade Dalton
(Da) para medir a massa molecular de
aminoácidos e moléculas biológicas em
geral. Um Dalton equivale à massa de
1/12 do átomo de carbono 12. Para
proteínas usamos o kiloDalton (kDa), 1
kDa = 103 Da. Na figura ao lado, temos o
aminoácido alanina, que apresenta uma
massa molecular de 89,09 Da, e uma
proteína chamada lisozima, que
apresenta uma massa molecular de
14331,20 Da. Usaremos o símbolo MM
para representarmos a massa molecular.
Lisozima de ovo de galinha (MM = 14331,20 Da ou
14,331 kDa). Código PDB: 6LYZ.
As figuras não estão na mesma escala.
Alanina (MM= 89,09 Da)
13
Programa: asa2.py
www.python.org

Considerando-se a massa molecular de
cada resíduo de aminoácido, temos uma
massa molecular média de 118,89 Da.
Este valor médio é útil na estimativa da
massa molecular aproximada de
proteínas e peptídeos, a partir do
conhecimento do número de resíduos de
aminoácidos presentes na molécula. A
equação abaixo indica a massa molecular
aproximada (MM),
MM = 118,89 . Naa
onde Naa é o número de resíduos de
aminoácidos presentes na molécula.
No exemplo ao lado temos a CDK2 (cyclin-
dependente kinase)(quinase dependente
de ciclina 2), com massa molecular de
34331,25 Da e 298 resíduos de
aminoácidos.
CDK2 humana (MW = 34331,25 Da )
Código PDB: 2A4L.
A massa molecular calculada pela equação, é de MM =
35429,22 Da, aproximadamente 3,2 % maior que a
massa molecular precisa da proteína. A equação é útil
para aproximações. 14
Programa: asa2.py
www.python.org

A nova versão do programa, usa como entrada o número de resíduos de aminoácidos
da proteína (vale para peptídeos também). Vamos preparar o pseudocódigo para o
cálculo da área acessível ao solvente de uma proteína, para a qual sabemos número
de resíduos de aminoácidos (numRes). Temos como variável independente o numRes
e a saída é a área acessível ao solvente (area). Assim temos que incluir uma linha
adicional de código para calcular a massa molecular (molWeight), como na equação
indicada abaixo:
molWeight = 118.89*numRes
A equação para a área acessível ao solvente é a mesma: area = 11.1 (molWeight)2/3
O pseudocódigo está no quadro abaixo. O cálculo só é efetuado se o número de
resíduos for maior que zero, caso contrário é mostrada uma mensagem de erro.
Início
Leia (numRes)
Se (numRes> 0)
molWeight = 118.89*numRes
area = 11.1*(molWeight)**(2/3)
Escreva "Area: ",area
Senão
Escreva "\nErro! A num. de res. deve ser > 0!"
Fim 15
Programa: asa2.py
www.python.org

Teste o seu programa para os valores de número de resíduos de aminoácidos
mostrados abaixo. Na tabela temos o número de resíduos de aminoácidos para cada
proteína e a área acessível ao solvente em Å2. Os números estão em notação inglesa,
ou seja, com ponto decimal.
A informação sobre os números de resíduos de aminoácidos das proteínas foi obtida
da base de dados Protein Data Bank (PDB)(http://www.rcsb.org/pdb/home/home.do ).
Proteína Fonte Número de resíduos de aa* ASA (Å2)
Mioglobina Physeter catodon 153 7677.399687811302
CDK2 Homo sapiens 298 11973.76433504305
Protease HIV-1 99 5743.500456050333
Chiquimato quinase Mycobacterium
tuberculosis
176 8428.720293276137
16
Programa: asa2.py
www.python.org

17
Referências relacionadas ao programa asa2.py
-COREY, RB; PAULING L. Molecular models of amino acids, peptides and proteins.
Review of Scientific Instruments, Nova York, v. 24, n.8, p.621-627, 1953.
-HUMPHREY W; DALKE A; SCHULTEN K. VMD - Visual Molecular Dynamics. Journal
of Molecular Graphics, Amsterdã, v.14, p.33-38, 1996.
-KOLTUN WL. Precision space-filling atomic models. Biopolymers, Hoboken, v.3, n.6,
p.665-79, 1965.
-LESK Arthur. M. Introduction to Protein Architecture. Oxford: Oxford University
Press, 2001. 347 p.
-VOET Donald; VOET Judith G.; PRATT Charlotte W. Fundamentos de Bioquímica.
Porto Alegre: Artmed Editora SA, 2000. 931 p.
Programa: asa2.py
www.python.org

Resumo
Programa para calcular a energia livre de Gibbs de ligação. O foco é no uso para
o estudo da interação proteína-ligante. Os dados de entrada são a temperatura
em Celsius (tempCelsius) e a constante de dissociação (Kd) em molar, chamada
de variável dissConst. A saída é a energia livre de Gibbs em J/mol (deltaG). A
equação da energia livre de Gibbs usa a constante dos gases. O valor foi obtido
da base de dados NIST, e vale gasConst= 8.314 4621 J mol-1 K-1 , disponível
em: < http://physics.nist.gov/cuu/Constants/index.html >. Acesso em 16 de
março de 2015. A energia livre de Gibbs (deltaG) é dada pela seguinte equação:
deltaG= gasConst*tempKelvin*math.log(dissConst) .
Energia livre de Gibbs para o sistema proteína-
ligante
Programa: freeEnergy.py
18
Programa: freeEnergy.py
www.python.org

Consideremos um sistema biológico
formado por uma proteína (P) e um
ligante (L) em solução, sendo que o
ligante é uma pequena molécula que
apresenta afinidade pela proteína, por
exemplo, o fármaco indinavir que inibe a
protease do HIV-1, o agente causador da
AIDS. Tal sistema possibilita a formação
de um complexo binário proteína-ligante
(PL), com constante de reação indicada
por k1 e constante reversa de reação dada
por k-1, ambos descritos na equação
reversível abaixo,
P + L PL
A reação pode ser caracterizada pela
constante de equilíbrio (Keq). Na prática
são usadas as constantes de dissociação
(Kd) e inibição (Ki).
k1
k-1
Formação do complexo proteína (P) com ligante (L),
complexo proteína-ligante (PL).
A estrutura em destaque é o complexo protease do HIV-1
(P) e o fármaco indinavir (L), usado no tratamento contra a
AIDS.19
Programa: freeEnergy.py
www.python.org

As interações entre o inibidor de CDK2
(Cyclin-Dependent Kinase) (roscovitine
mostrado ao lado) e a proteína alvo
(CDK2) são não covalentes. Tais contatos
intermoleculares apresentam uma
interação energética favorável, o que leva
o equilíbrio da reação química para o lado
da formação do complexo binário (PL).
P + L PL
A análise da energia da interação
proteína-ligante, indica a formação de um
composto de transição, onde a energia do
sistema é elevada e o composto formado
instável. Tal estado é chamado estado de
transição, e a reação caminha para uma
menor energia, com a formação do
complexo binário PL, mostrada no
próximo slide.
k1
k-1
Estrutura molecular do inibidor de CDK2 roscovitine. A
figura foi gerada com o programa VMD e a opção CPK. As
coordenadas foram extraídas do arquivo PDB: 2a4l.
20
Programa: freeEnergy.py
www.python.org

+
Proteína + Ligante Estado de transição Complexo proteína-ligante
Energ
ia
Coordenada da interação
Ea
Ed
E
Ea = Energia de ativação para associação
Ed = Energia de ativação para dissociação
E = Variação total da energia
21
Programa: freeEnergy.py
www.python.org

P + L PL
Podemos representar a formação do complexo proteína-ligante (PL), a partir dos
componentes proteína (P) e ligante (L), como esquematizado abaixo. Temos a
proteína (P) e o ligante (L) em solução, no caso do ligante apresentar afinidade pela
proteína, o equilíbrio da reação será favorável à formação do complexo proteína-
ligante (PL).
k1
k-1
22
Programa: freeEnergy.py
www.python.org

No caso do complexo binário proteína-
ligante (PL) apresentar uma energia
menor que as moléculas livres (proteína e
ligante sem interação), a formação do
complexo proteína-ligante ocorrerá de
forma espontânea, ou seja, sem a
necessidade de fornecimento de energia
para que ocorra a formação. Uma forma
de estudar a formação do complexo
binário, é a partir da avaliação da variação
de energia livre de Gibbs (G), definida
pela seguinte equação,
G = H -TS
onde H é a entalpia do sistema proteína-
ligante, T é a temperatura e S é a
variação da entropia do sistema. Na figura acima temos a formação espontânea de um
complexo binário proteína-ligante, pois este apresenta
energia menor que das moléculas livres (proteína e
ligante sem interação)
P
L
PL
23
Programa: freeEnergy.py
www.python.org

PL
G = H -TSCom G > 0 a reação não é favorável à formação do
complexo, como na figura inferior. Com G < 0 temos
uma reação favorável à formação do complexo, como na
figura superior.
L
Se G > 0
O termo H indica a entalpia do sistema,
que representa as forças moleculares
envolvidas nas interações proteína-
ligante. O termo S é a variação da
entropia do sistema, que pode ser
entendida como a quantidade de energia
que não pode ser convertida em trabalho.
Na equação anterior, o produto da
temperatura absoluta (T) pela variação da
entropia (S), indica que um aumento da
entropia favorece a formação do
complexo binário.
Um valor negativo de G indica formação
espontânea do complexo.
Se G < 0
P
24
Programa: freeEnergy.py
www.python.org

Quando o sistema proteína-ligante está
em equilíbrio, a formação do complexo
binário e a dissociação ocorrem, assim a
constante de equilíbrio (Keq) da reação é
dada por,
onde os termos [P], [L], [PL] indicam as
concentrações molares da proteína,
ligante e complexo proteína-ligante,
respectivamente. A partir da análise
dimensional da constante de equilíbrio,
vemos claramente que sua unidade é M-1.
LP
PL
k
kK
1
1eq
Estrutura do complexo entre a proteína quinase
dependente de ciclina 2 (Cyclin-Dependent Kinase 2,
CDK2) e o inibidor roscovitine. 25
Programa: freeEnergy.py
www.python.org

A variação da energia livre de Gibbs da
interação intermolecular entre proteína e
ligante é dada pela seguinte equação,
onde Go é a variação da energia livre de
Gibbs padrão, ou seja, a variação em G
que acompanha a formação do complexo
no estado padrão de equilíbrio
(temperatura de 25oC, pressão de 100
kPa).
)]L][P[
]PL[ln(RTGG o
Estrutura cristalográfica da protease do HIV-1 em
complexo com inibidor.
26
Programa: freeEnergy.py
www.python.org

No equilíbrio termodinâmico temos G = 0 (estado estacionário), assim a equação fica
da seguinte forma,
O termo Go é chamado variação na energia livre de Gibbs da ligação (Gbinding),
assim temos,
)]L][P[
]PL[ln(RTG
)]L][P[
]PL[ln(RTG0
o
o
)]L][P[
]PL[ln(RTGbinding
27
Programa: freeEnergy.py
www.python.org

De uma forma geral, podemos expressar a afinidade proteína-ligante pela variação da
energia livre de Gibbs (Gbinding), quanto menor a energia, maior a afinidade do ligante
pela proteína. Tal grandeza física depende das concentrações do complexo proteína-
ligante [PL], das concentrações da proteína [P] e do ligante [L], bem como da
temperatura. O “R” (R=1,99 cal/mol.K = 8,3144621 J/mol.K)( na equação indica a
constante dos gases).
A constante de dissociação (Kd) é dada por:
Podemos calcular a energia livre de uma interação proteína-ligante, a partir da
constante de dissociação determinada experimentalmente.
][
]][[
PL
LPKd
)ln(
)ln()ln(
)]][[
][ln(
dbinding
dd
binding
binding
K
KK
RTG
RT1
RTG
LP
PLRTG
Fonte da informação sobre as constantes físicas: http://physics.nist.gov/cuu/Constants/index.html
Acesso em 16 de março de 2015. 28
Programa: freeEnergy.py
www.python.org
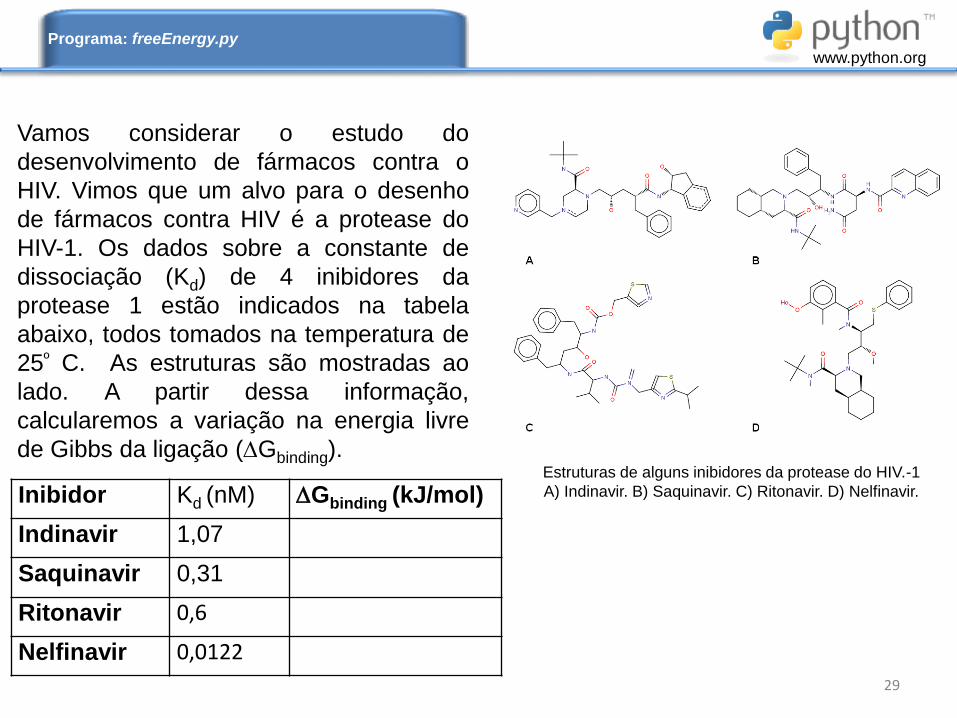
Vamos considerar o estudo do
desenvolvimento de fármacos contra o
HIV. Vimos que um alvo para o desenho
de fármacos contra HIV é a protease do
HIV-1. Os dados sobre a constante de
dissociação (Kd) de 4 inibidores da
protease 1 estão indicados na tabela
abaixo, todos tomados na temperatura de
25º C. As estruturas são mostradas ao
lado. A partir dessa informação,
calcularemos a variação na energia livre
de Gibbs da ligação (Gbinding).Estruturas de alguns inibidores da protease do HIV.-1
A) Indinavir. B) Saquinavir. C) Ritonavir. D) Nelfinavir.Inibidor Kd (nM) Gbinding (kJ/mol)
Indinavir 1,07
Saquinavir 0,31
Ritonavir 0,6
Nelfinavir 0,0122
29
Programa: freeEnergy.py
www.python.org

Estudo do indinavir. As constantes de dissociação foram determinadas a uma
temperatura de 25º C, para converter para Kelvin é só somar 273,15, assim temos T=
298,15 K.
kJ/mol 51,2- G
J/mol 51204,342- (-20,6556) . 2478,9569 G
1,07.10ln . 2478,9569 1,07.10ln . 298,15 . 8,3144621 ln(K R.T. G
binding
binding
-9-9d binding
)
Inibidor Kd (nM) Gbinding (kJ/mol)
Indinavir 1,07 -51,2
Saquinavir 0,31
Ritonavir 0,6
Nelfinavir 0,0122
Estrutura molecular do fármaco indinavir. A
figura foi gerada com o programa VMD e a
opção CPK. As coordenadas foram extraídas
do arquivo PDB: 1hsg.
30
Programa: freeEnergy.py
www.python.org

Repetindo o cálculo para os outros inibidores, chegamos à tabela abaixo. A partir da
análise desta tabela vemos que o inibidor que apresenta menor energia livre de Gibbs
de ligação é o nelfinavir, cujo a estrutura do complexo proteína-ligante está mostrada
abaixo.
Inibidor Kd (nM) Gbinding (kJ/mol)
Indinavir 1,07 -51,2
Saquinavir 0,31 -54,3
Ritonavir 0,6 -52,6
Nelfinavir 0,0122 -62,3
31
Programa: freeEnergy.py
www.python.org

Para implementar a equação para energia livre de Gibbs, usamos como entrada a
temperatura em graus Celsius (tempCelsius) e a constante de dissociação (dissConst)
em molar. Temos que inicialmente converter a temperatura em Celsius para Kelvin,
como segue:
tempKelvin = 273.15 + tempCelsius
A equação para a energia livre de Gibbs (deltaG) fica da seguinte forma:
deltaG= gasConst*tempKelvin*math.log(dissConst)
O resultado será em J/mol.
O valor da constante molar dos gases foi obtido de
http://physics.nist.gov/cuu/index.html (Acesso em 16 de março de 2015).
32
Programa: freeEnergy.py
www.python.org

O pseudocódigo está no quadro abaixo. Escreva o seu código fonte e teste para os 4
valores de constante de dissociação indicados nos slides anteriores. Lembre-se que a
constante de dissociação está em nanomolar, ou seja, para entrar os dados temos que
usar notação científica, por exemplo: 1,07 nM = 1.07e-9. Usaremos o método
math.log(x) para o logaritmo natural. Só efetuamos o cálculo da energia livre de Gibbs,
se a constante de dissociação for maior que zero e a temperatura em Celsius for maior
que -273.15, caso contrário mostramos uma mensagem de erro.
Início
Chama módulo math
Definição da constante: gasConst= 8.3144621
Leia (tempCelsius, dissConst)
Se dissConst>0 and tempCelsius >= -273.15
tempKelvin = 273.15 + tempCelsius
deltaG= gasConst*tempKelvin*math.log(dissConst)
Escreva "Energia = ",deltaG
Senão
Escreva "Erro! t>0 e Kd>0"
Fim 33
Programa: freeEnergy.py
www.python.org

34
Referências relacionadas ao programa freeEnergy.py
-FERSHT, Alan. Structure and Mechanism in Protein Science. A Guide to Enzyme
Catalysis and Protein Folding. Nova York: W. H. Freeman and Company, 1999. 631
p.
-VOET, Donald; VOET, Judith G; PRATT, Charlotte W. Fundamentos de Bioquímica.
Porto Alegre: Artmed Editora SA, 2000. 931 p.
-TINOCO, Ignacio Jr.; SAUER, Kenneth.; WANG, James C. Physical Chemistry.
Principles and Applications in Biological Sciences. 3a Ed. Nova Jersey: Prentice
Hall, Inc., 1995. 761 p.
-VAN HOLDE, Kensal Edward; JOHNSON, W. Curtis; HO P. Shing. Principles of
Physical Biochemistry. Nova Jersey: Prentice Hall, Inc., 1998. 657 p.
Programa: freeEnergy.py
www.python.org

A linguagem de programação Python tem um arsenal de métodos para manipulação
de strings. O conceito de string é simples, uma string é um conjunto de caracteres,
que pode conter números, letras e símbolos especiais. Toda leitura de informação
via teclado, com a função input(), considera que o que está sendo digitado é uma
string. Como vimos, para que o programa considere a informação digitada, como um
número inteiro, temos que usar a função int(), antes da função input(), que converte a
string para inteiro, como indicado abaixo.
Vimos, também, que podemos usar uma entrada de dados de ponto flutuante, a partir
da função float(), que converte de string, ou de inteiro, para float, como indicado
abaixo.
35
Strings em Python
www.python.org
my_var = int(input(“Type an integer =>"))
my_var = float(input(“Type a float =>"))

my_var = input(‘Type a string =>')
Quando não indicamos as funções float() ou int(), antes da função input(), o
interpretador Python assume que a informação sendo digitada é uma string, como
indicado no trecho de código a seguir.
Variáveis em Python fazem referencias às posições na memória, onde estão
armazenados dados, como no exemplo das strings, indicadas abaixo.
Usamos aspas duplas para indicar a string em Python, podemos usar, também,
aspas simples, a única exigência é que, uma vez iniciado com um tipo de aspa,
esta deve está no final, como no exemplo abaixo.
36
Strings em Python
www.python.org
my_var = input(“Type a string =>")
my_var1 = “Here we have a string"
my_var2 = “Here we have a new string with 1 e 2"
my_var3 = "11235813213455"

As variáveis, para as quais foram atribuídas strings, podem ser mostradas na tela
com a função print(), como no exemplo a seguir.
Podemos usar a função print() para mostrar resultados compostos na tela, onde
strings, inteiros, floats e sequências de escape aparecem numa única linha, o código
a seguir, showStrings1.py, ilustra a situação.
Ao executarmos o código showStrings1.py, temos o seguinte resultado.
37
Strings em Python
www.python.org
my_var = “A new string"
print(my_var)
String: My string , Integer: 112358 , Float: 3.14159
my_var1 = "My string"
my_var2 = 112358
my_var3 = 3.14159
print("String: ",my_var1,", Integer: ",my_var2,", Float: ",my_var3,"\n\n")

Vamos olhar alguns detalhes importantes do código showStrings1.py. A primeira linha
traz a atribuição do conteúdo da memória “My string” à variável my_var1. Usamos
aspas duplas, poderíamos ter usado aspas simples, o resultado seria o mesmo (veja
Exercício de programação 1). A segunda linha traz a variável my_var2, veja que
não usamos aspas, o interpretador Python considera a variável my_var2 como inteira,
sem necessidade que explicitemos com a função int(). Para a variável my_var3,
temos situação similar, só que agora a presença do ponto decimal indica que o dado
é do tipo float. A função print() traz conteúdo entre aspas, as variáveis e a sequência
de escape \n. Veja que separamos por vírgulas os conteúdos a serem mostrados na
tela. Se omitirmos a vírgula, que separa, por exemplo, o primeiro conteúdo entre
aspas e a variável my_var1, teremos um erro de execução do programa, também
chamado de erro de sintaxe.
Veja, ainda, que as vírgulas inseridas entre aspas são caracteres de uma string, por
isso são mostradas na tela.
38
Strings em Python
www.python.org
my_var1 = "My string"
my_var2 = 112358
my_var3 = 3.14159
print("String: ",my_var1,", Integer: ",my_var2,", Float: ",my_var3,"\n\n")

Exercício de programação 1. Modifique o código fonte do programa
showStrings1.py, use aspas simples para a variável my_var1. Salve o novo programa
com o nome showStrings2.py. Rode o programa. Houve diferenças nos resultados?
Explique.
Exercício de programação 2. Modifique o código fonte do programa
showStrings1.py, crie um nova variável, a variável my_var4, que recebe o resultadoda divisão da variável my_var2 pela my_var3 (my_var4 = my_var2/my_var3).
Insira a variável my_var4 na função print(), mas antes coloque uma informação que o
resultado mostrado é a divisão (sem acento, por exemplo, Division). Salve o novo
programa com o nome showStrings3.py. Rode o programa. Nota: Não use acentos
nas strings. Abaixo temos o resultado esperado
Exercício de programação 3. Modifique o código fonte do programa
showStrings3.py, e coloque aspas simples nos conteúdos atribuídos às variáveis
my_var2 e my_var3. Mantenha a variável my_var4 inalterada. Salve o novo programa
com o nome showStrings4.py. Rode o programa. O que aconteceu? Explique.
39
Strings em Python
www.python.org
String: My string , Integer: 112358 , Float: 3.14159 Division: 35764.69240098167

Vimos o uso de funções internas em Python (Python’s built-in functions) para
manipulação de strings, como as funções print(), input(), float() e int(). Temos,
também, duas funções adicionais para strings, são elas as funções len() e str(). A
função len() retorna o tamanho de uma string que é dada como argumento, ou seja, o
número de caracteres da string. A função str() converte o argumento para string, por
exemplo, de inteiro para string. Vejamos um exemplo simples onde as duas funções
aparecem. O código showStrings5.py converte um inteiro para string e, depois,
mostra o número de caracteres da string.
Ao executarmos o código acima, temos o resultado mostrado abaixo.
40
Strings em Python
www.python.org
myInt = int(input("Type an integer => ")) # Reads an integer and assigns it to the variable myInt
myString = str(myInt) # Converts to string
countChar = len(myString) # Counts characters in myString
print("Number of characters of ",myString," is ",countChar) # Shows results
Type an integer => 12345
Number of characters of 12345 is 5

Como vimos, há diversos tipos de valores que podem ser usados como argumentos
das funções internas da linguagem Python. Contudo, a maioria das funções, aplica-se
a um tipo específico de dados, por exemplo, às strings. Tais recursos da linguagem
Python, específicos para um tipo de dado, são chamados de métodos. A
chamada de um método em Python, ocorre de forma similar às funções, exceto que o
primeiro argumento aparece antes do nome do método, seguido por um ponto (.).
Esta forma de chamar um método, é denominada de notação dot. Vejamos o
método .count(), que retorna o número de vezes que uma dada string, fornecida
como argumento, aparece na string, myString.
41
Strings em Python
www.python.org
myString.count(“I”)
String sobre a qual será aplicado o método
Método aplicado à string do lado esquerdo
Ponto indicador da notação “dot”
Argumento do método

O programa, showStrings6.py, conta o número de vezes que o caractere “I” aparece
na string atribuída à variável myString.
Ao executarmos o código, temos o resultado abaixo.
Veja que o código é sensível ao tipo de letras, maiúsculas ou minúsculas, dizemos
ser “case sensitive”.
42
Strings em Python
www.python.org
myString = input("Type a string => ") # Reads a string and assigns it to variable myString
# .count("I") method to count the number of times its argument appears in the string
countI = myString.count("I")
print("I appears ",countI, "time(s)") # Shows results
Type a string => BIOINFORMATICS
I appears 3 time(s)

Com a explosão de informações sobre
sequência de genomas completos, as
bases de dados passaram a armazenar
tal informação como strings de bases
nitrogenadas, como a mostrada abaixo,
onde temos 18 bases. Usando-se o
código de uma letra das bases
nitrogenadas, para representar a estrutura
primária do DNA, temos uma forma
condensada de armazenar tal informação.
Adenina = A
Guanina = G
Citosina = C
Timina = T
Representação artística do genoma humano, com uma
molécula de DNA o homem vitruviano. Disponível em: <
http://www.sciencephoto.com/media/408761/enlarge >. .
Acesso em: 25 de março de 2016.
CGATATCGAATTCCGGAT
43
Concatenação de trechos de DNA
www.python.org
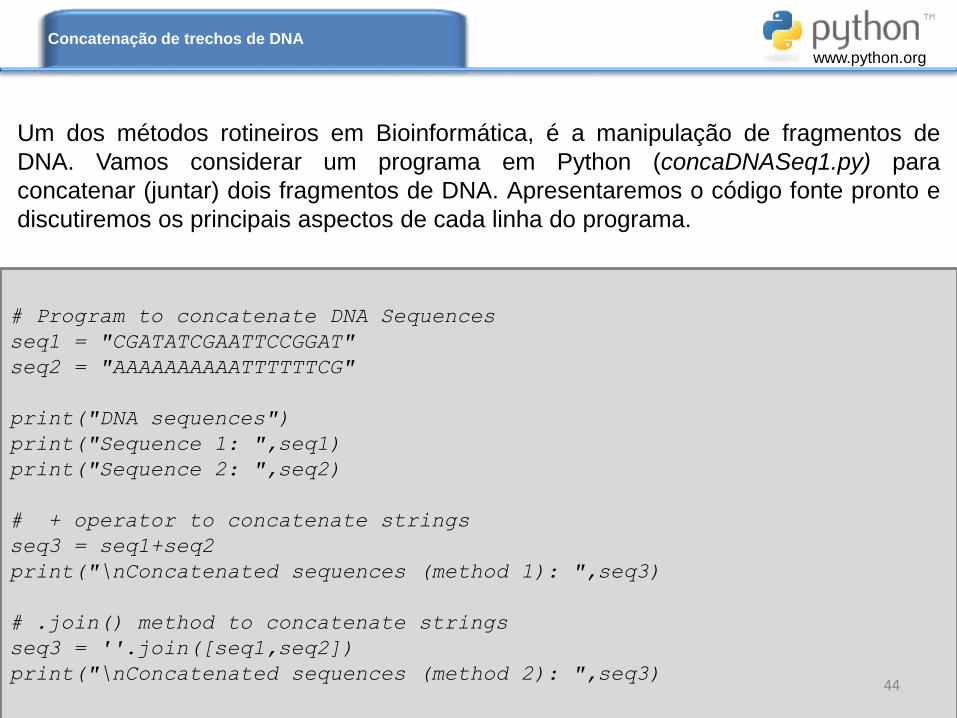
# Program to concatenate DNA Sequences
seq1 = "CGATATCGAATTCCGGAT"
seq2 = "AAAAAAAAAATTTTTTCG"
print("DNA sequences")
print("Sequence 1: ",seq1)
print("Sequence 2: ",seq2)
# + operator to concatenate strings
seq3 = seq1+seq2
print("\nConcatenated sequences (method 1): ",seq3)
# .join() method to concatenate strings
seq3 = ''.join([seq1,seq2])
print("\nConcatenated sequences (method 2): ",seq3)44
Um dos métodos rotineiros em Bioinformática, é a manipulação de fragmentos de
DNA. Vamos considerar um programa em Python (concaDNASeq1.py) para
concatenar (juntar) dois fragmentos de DNA. Apresentaremos o código fonte pronto e
discutiremos os principais aspectos de cada linha do programa.
Concatenação de trechos de DNA
www.python.org

# Program to concatenate DNA Sequences
seq1 = "CGATATCGAATTCCGGAT"
seq2 = "AAAAAAAAAATTTTTTCG"
print("DNA sequences")
print("Sequence 1: ",seq1)
print("Sequence 2: ",seq2)
# + operator to concatenate strings
seq3 = seq1+seq2
print("\nConcatenated sequences (method 1): ",seq3)
# .join() method to concatenate strings
seq3 = ''.join([seq1,seq2])
print("\nConcatenated sequences (method 2): ",seq3)45
Antes de começarmos a descrição linha por linha do programa, gostaria de destacar
alguns aspectos. Veja no código que temos linhas de comentários, iniciadas por #, em
diversas partes, além do início do código. Como já destacado, tal inserção visa
documentar o programa, o que facilita seu entendimento para posteriores
modificações.
Concatenação de trechos de DNA
www.python.org
1# Program to concatenate DNA Sequences
2seq1 = "CGATATCGAATTCCGGAT"
3seq2 = "AAAAAAAAAATTTTTTCG"
4
5print("DNA sequences")
6print("Sequence 1: ",seq1)
7print("Sequence 2: ",seq2)
8
9# + operator to concatenate strings
10seq3 = seq1+seq2
11print("\nConcatenated sequences (method 1): ",seq3)
12
13# .join() method to concatenate strings
14seq3 = ''.join([seq1,seq2])
15print("\nConcatenated sequences (method 2): ",seq3)46
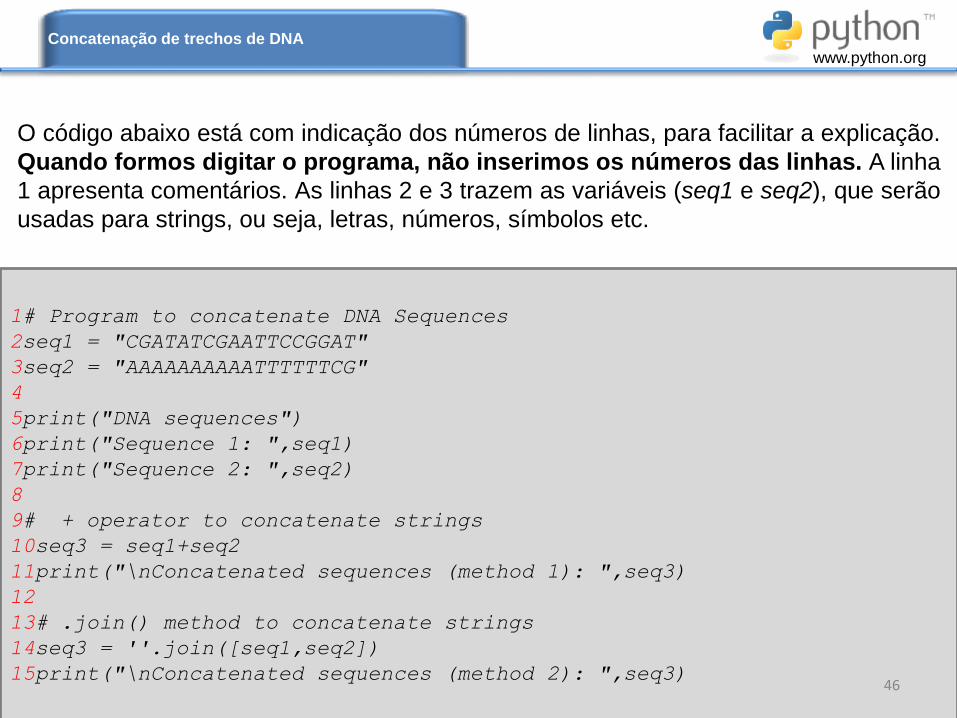
O código abaixo está com indicação dos números de linhas, para facilitar a explicação.
Quando formos digitar o programa, não inserimos os números das linhas. A linha
1 apresenta comentários. As linhas 2 e 3 trazem as variáveis (seq1 e seq2), que serão
usadas para strings, ou seja, letras, números, símbolos etc.
Concatenação de trechos de DNA
www.python.org

1# Program to concatenate DNA Sequences
2seq1 = "CGATATCGAATTCCGGAT"
3seq2 = "AAAAAAAAAATTTTTTCG"
4
5print("DNA sequences")
6print("Sequence 1: ",seq1)
7print("Sequence 2: ",seq2)
8
9# + operator to concatenate strings
10seq3 = seq1+seq2
11print("\nConcatenated sequences (method 1): ",seq3)
12
13# .join() method to concatenate strings
14seq3 = ''.join([seq1,seq2])
15print("\nConcatenated sequences (method 2): ",seq3)47
As aspas duplas “” indicam que o conteúdo entre aspas é uma string a ser atribuída a
uma variável. A string é uma sequência de DNA. A linha 4 está em branco, tais
inserções são usadas para facilitar a leitura do programa por humanos, não são
consideradas pelo interpretador Python.
Concatenação de trechos de DNA
www.python.org

1# Program to concatenate DNA Sequences
2seq1 = "CGATATCGAATTCCGGAT"
3seq2 = "AAAAAAAAAATTTTTTCG"
4
5print("DNA sequences")
6print("Sequence 1: ",seq1)
7print("Sequence 2: ",seq2)
8
9# + operator to concatenate strings
10seq3 = seq1+seq2
11print("\nConcatenated sequences (method 1): ",seq3)
12
13# .join() method to concatenate strings
14seq3 = ''.join([seq1,seq2])
15print("\nConcatenated sequences (method 2): ",seq3)48
As linhas 5, 6 e 7 indicam mensagens a serem mostradas na tela, inclusive as
sequências de DNA. Podemos usar aspas simples ‘ ‘ para indicar a string a ser
mostrada na tela, não há diferença entre o uso das aspas simples e das duplas em
Python, como destacamos anteriormente.
Concatenação de trechos de DNA
www.python.org

1# Program to concatenate DNA Sequences
2seq1 = "CGATATCGAATTCCGGAT"
3seq2 = "AAAAAAAAAATTTTTTCG"
4
5print("DNA sequences")
6print("Sequence 1: ",seq1)
7print("Sequence 2: ",seq2)
8
9# + operator to concatenate strings
10seq3 = seq1+seq2
11print("\nConcatenated sequences (method 1): ",seq3)
12
13# .join() method to concatenate strings
14seq3 = ''.join([seq1,seq2])
15print("\nConcatenated sequences (method 2): ",seq3)49
A linha 8 está em branco. A linha 9 é um comentário. A linha 10 concatena as duas
strings, a partir do operador “+”, que atua colando as duas sequências atribuídas às
variáveis seq1 e seq2. O resultado da concatenação é atribuído à nova variável, seq3.
A linha 11 mostra o resultado na tela. A linha 12 está em branco.
Concatenação de trechos de DNA
www.python.org

1# Program to concatenate DNA Sequences
2seq1 = "CGATATCGAATTCCGGAT"
3seq2 = "AAAAAAAAAATTTTTTCG"
4
5print("DNA sequences")
6print("Sequence 1: ",seq1)
7print("Sequence 2: ",seq2)
8
9# + operator to concatenate strings
10seq3 = seq1+seq2
11print("\nConcatenated sequences (method 1): ",seq3)
12
13# .join() method to concatenate strings
14seq3 = ''.join([seq1,seq2])
15print("\nConcatenated sequences (method 2): ",seq3)50
A linha 13 é comentário. A linha 14 traz o método join, que concatena os conteúdos
atribuídos às variáveis seq1 e seq2, o resultado é atribuído à variável seq3. As aspas
simples, antes do método join, indicam que as strings não apresentarão espaços, ou
qualquer outro símbolo entre elas. A linha 15 mostra o resultado na tela.
Concatenação de trechos de DNA
www.python.org

51
Vamos ao resultado do programa. Chamamos o programa de concaDNASeq1.py.
Vemos que os resultados da concatenação são exatamente idênticos para os 2
métodos.
DNA sequences
Sequence 1: CGATATCGAATTCCGGAT
Sequence 2: AAAAAAAAAATTTTTTCG
Concatenated sequences (method 1):
CGATATCGAATTCCGGATAAAAAAAAAATTTTTTCG
Concatenated sequences (method 2):
CGATATCGAATTCCGGATAAAAAAAAAATTTTTTCG
Concatenação de trechos de DNA
www.python.org

14seq3 = ‘-'.join([seq1,seq2])
52
Se tivéssemos colocado um caractere qualquer entre as aspas, antes do método join,
este seria inserido entre as duas sequências. Como no pequeno trecho de código
mostrado abaixo. Veja a nova linha 14.
Concatenação de trechos de DNA
www.python.org

53
Concatenação de trechos de DNA
www.python.org
Exercício de programação 4. Modifique as duas sequências de DNA e rode o
programa.
Exercício de programação 5. Modifique o programa concaDNASeq1.py. Insira
diferentes símbolos entre as aspas simples da linha 14 e rode o programa para
verificar o resultado.

54
Uma fita isolada de DNA é lida do terminal
5’ para o terminal 3’. Ao formarem
fragmentos de DNA, o terminal 5’ de um
nucleotídeo, liga-se ao terminal 3’ do
fragmento em formação. Quando duas
fitas de DNA unem-se, para formar a
hélice dupla de DNA, uma fita com
sentido de leitura do 5’ para o 3’ liga-se
por meio de ligações de hidrogênio entre
os pares de bases, a outra fita no sentido
oposto, ou seja, do 3’ para o 5’, seguindo
a ordem CG e AT. As sequências ao lado
ilustram tal complementaridade entre as
fitas. Visto que, os pares de bases sempre
combinam A com T e C com G e a
orientação das fitas é invertida, uma com
relação à outra, usamos o termo
complemento reverso (reverse
complement em inglês) para descrever a
relação de bases das duas fitas.
CGATATCGAATTCCGGAT
GCTATAGCTTAAGGCCTA
5’ 3’
3’ 5’
Leitura da esquerda para direita
Leitura da direita para esquerda
Complemento reverso do DNA
www.python.org

55
Na figura abaixo, à esquerda, temos a estrutura 3D de um trecho da molécula de DNA.
A figura da direita é um zoom da parte superior da molécula, onde vemos o início da
fita esquerda com a posição 5’, que faz par com a fita à direita que emparelha com a
posição 3’.
O5’
O3’
Complemento reverso do DNA
www.python.org

56
Abaixo temos a montagem da fita dupla do DNA, com uma fita servindo de molde para
a montagem do complemento reverso. Essa reação é catalisada pela enzima DNA
polimerase.
Figura disponível em: < http://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/nucacids.htm >.
Acesso em: 25 de março de 2016.
Complemento reverso do DNA
www.python.org

Vamos ilustrar a transformação da informação codificada numa fita de DNA, para sua
complementar (complementar reverso). Usaremos um pequeno trecho de DNA com 12
nucleotídeos. A informação está armazenada num arquivo no formato FASTA, usado
para guardar informações sobre a estrutura primária de ácidos nucleicos e proteínas.
57
>DNA:A|PDBID|CHAIN|SEQUENCE
CGCGAATTCGCGArquivo no formato FASTA com a sequência de nucleotídeos. A primeira linha inicia com
o símbolo > e traz a identificação do sequência. As bases começam na segunda linha.
>DNA:B|PDBID|CHAIN|SEQUENCE
CGCGAATTCGCG Arquivo no formato FASTA com a sequência de nucleotídeos do complemento reverso,
o sentido de leitura é do terminal 5’ para o 3’, veja que a complementaridade dos pares
de bases ocorre da última base do arquivo original para a primeira base do arquivo do
complemento reverso e assim sucessivamente.
Complemento reverso
Complemento reverso do DNA
www.python.org
# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
58
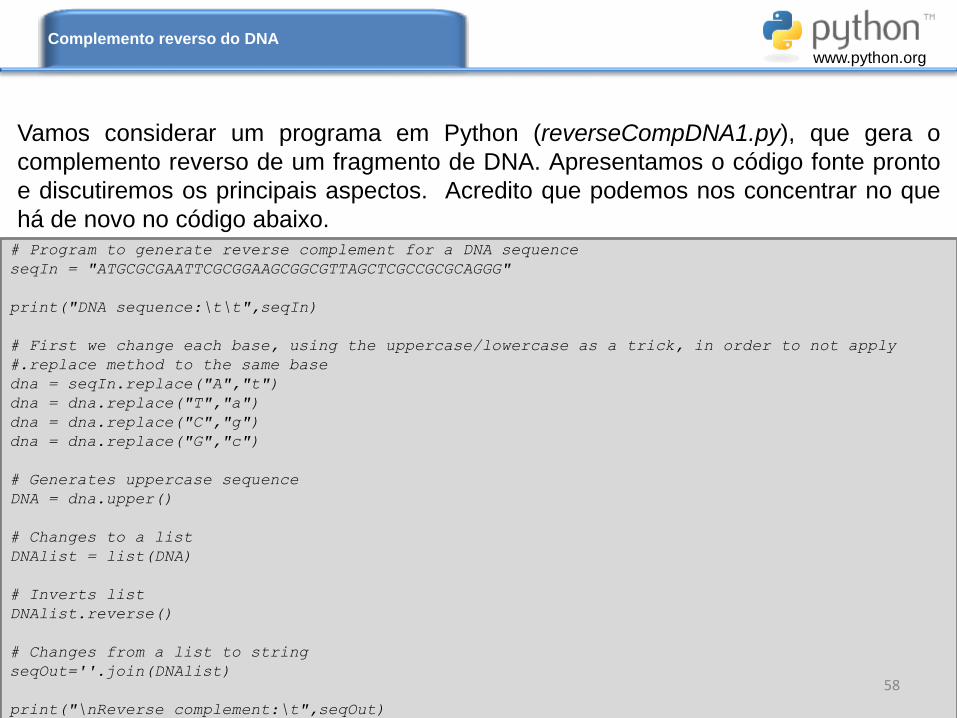
Vamos considerar um programa em Python (reverseCompDNA1.py), que gera o
complemento reverso de um fragmento de DNA. Apresentamos o código fonte pronto
e discutiremos os principais aspectos. Acredito que podemos nos concentrar no que
há de novo no código abaixo.
Complemento reverso do DNA
www.python.org
# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
59
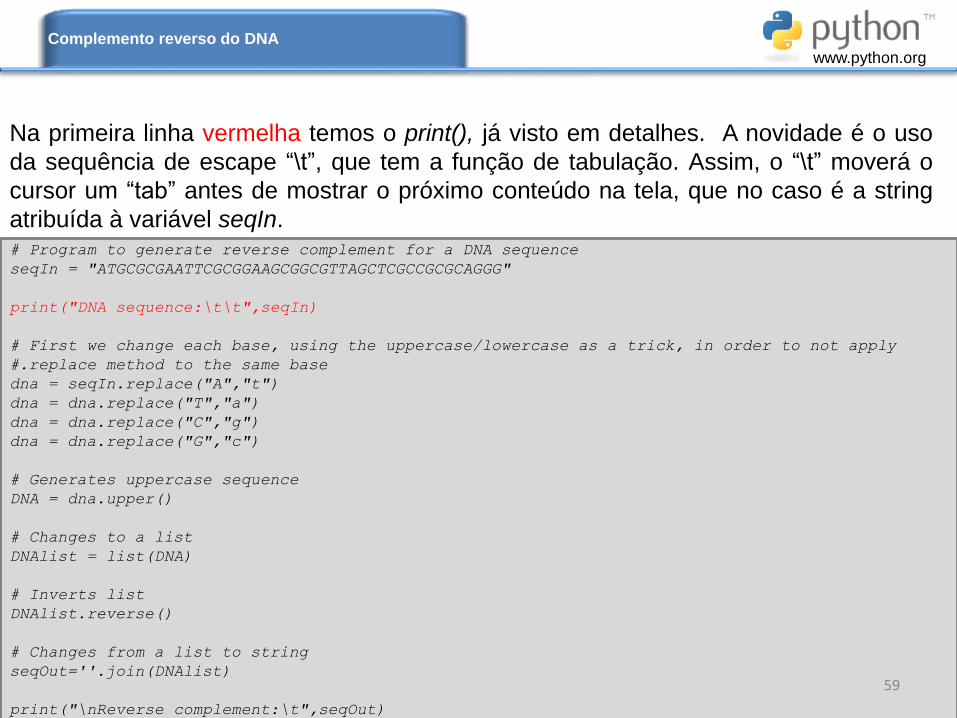
Na primeira linha vermelha temos o print(), já visto em detalhes. A novidade é o uso
da sequência de escape “\t”, que tem a função de tabulação. Assim, o “\t” moverá o
cursor um “tab” antes de mostrar o próximo conteúdo na tela, que no caso é a string
atribuída à variável seqIn.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
60
O método replace(“string 1”,”string 2”) troca a string 1 pela string 2, atribuindo o
resultado à variável à esquerda, que no caso é a variável dna. Veja que trocamos por
uma base com letra minúscula, tal truque evita que troquemos as bases que já foram
trocadas.
Complemento reverso do DNA
www.python.org

61
Abaixo temos um detalhamento da aplicação do método replace(). O recurso de
trocarmos uma base de letra maiúscula, por uma de letra minúscula, evita que o “T”
que foi obtido de “A” volte para “A”. O Python diferencia maiúsculas de minúsculas
(case sensitive).
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
Base Base
A t
T a
C g
G c
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
62
Para voltarmos a ter uma string com letras maiúsculas, usamos o método .upper(). Na
implementação abaixo, atribuímos a string com letras maiúsculas à variável DNA.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
63
Agora convertemos a string atribuída à variável DNA em uma lista, como o método list.
Uma lista apresenta dados, strings ou números, com índices, ou seja, podemos
acessar cada elemento da lista chamando especificamente a posição da lista, no caso
abaixo a string é decomposta e cada letra agora é um elemento da lista.
Complemento reverso do DNA
www.python.org

64
Uma lista apresenta a informação de forma indexada, cada trecho da informação é
chamado elemento da lista. Por exemplo, podemos criar uma lista com os 10 primeiros
elementos da sequência de Fibonacci, como indicado abaixo.
Para nos referirmos diretamente a um elemento da lista, chamamos a ordem do
elemento, tendo em mente que em Python usamos o índice “0” para o primeiro
elemento. Assim, a lista my_list tem a seguinte distribuição.
Complemento reverso do DNA
www.python.org
1 1 2 3 5 8 13 21 34 55
my_list = [1,1,2,3,5,8,13,21,34,55]
my_list
my_list[0] my_list[1] my_list[2] my_list[3] my_list[4] my_list[5] my_list[6] my_list[7] my_list[8] my_list[9]

65
O trecho de código abaixo cria a lista my_list e imprime o elemento 7 da lista, ou seja,
o oitavo elemento da lista.
O resultado das linhas de código acima, cria a lista, e imprime o elemento “7” da lista,
indicado no diagrama abaixo.
Complemento reverso do DNA
www.python.org
1 1 2 3 5 8 13 21 34 55my_list
my_list[0] my_list[1] my_list[2] my_list[3] my_list[4] my_list[5] my_list[6] my_list[7] my_list[8] my_list[9]
my_list = [1,1,2,3,5,8,13,21,34,55]
print(my_list[7])

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
66
A partir da lista podemos usar métodos para sua manipulação, como o método
reverse, que inverte a sequência dos elementos originais. Os métodos para
manipulação de listas, como o .reverse(), são chamados usando-se a notação dot (.),
onde o ponto (.) é colocado após a lista, o resultado é que a lista é invertida.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
67
Podemos voltar a ter uma string, com o método join, que une os elementos da lista
numa string. Por último usamos o print() para mostrar os resultados.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence
seqIn = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
#.replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequence
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)
68
A variável seqOut tem a string obtida da conversão da lista DNAlist, mostramos o
resultado na tela chamando a função print().
Complemento reverso do DNA
www.python.org
69
DNA sequence: ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Reverse complement: CCCTGCGCGGCGAGCTAACGCCGCTTCCGCGAATTCGCGCAT
Complemento reverso do DNA
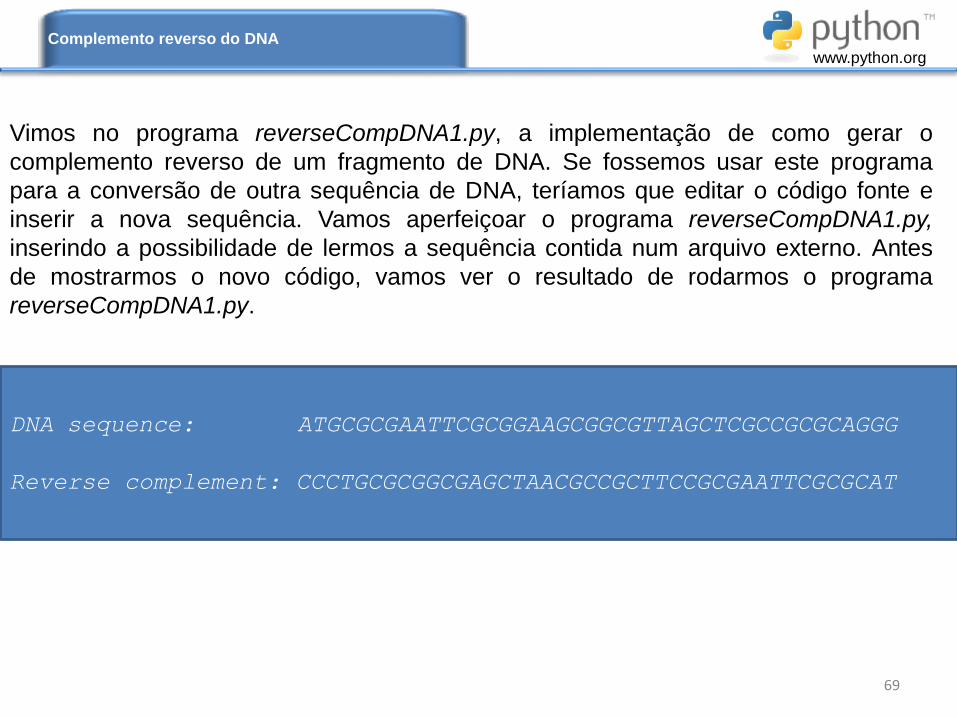
Vimos no programa reverseCompDNA1.py, a implementação de como gerar o
complemento reverso de um fragmento de DNA. Se fossemos usar este programa
para a conversão de outra sequência de DNA, teríamos que editar o código fonte e
inserir a nova sequência. Vamos aperfeiçoar o programa reverseCompDNA1.py,
inserindo a possibilidade de lermos a sequência contida num arquivo externo. Antes
de mostrarmos o novo código, vamos ver o resultado de rodarmos o programa
reverseCompDNA1.py.
www.python.org

70
Complemento reverso do DNA
A função para abertura de arquivos em Python é open(), vamos destacar abaixo, as
principais características da função open() para leitura de arquivos pré-existentes.
Usamos a linha de comando abaixo, para leitura do arquivo my_file, como segue:
my_file é uma variável para a qual foi atribuída o nome do arquivo, a informação ‘r’, é
chamada de modo de acesso ao arquivo, no caso indica que será realizada uma
operação de leitura do conteúdo do arquivo my_file. A informação, contida no arquivo
de entrada, será referenciada à variável my_info. Resumindo, usaremos a variável
my_info para manipularmos o conteúdo lido do arquivo my_file. Por exemplo,
consideremos que a informação do nosso arquivo de entrada está contida numa linha
de texto, por exemplo a sequência de um DNA. Para lermos essa informação, usamos
o comando,
.readline() é um método que lerá uma linha da variável my_info e atribuirá à variável
my_line. O método .readline() é aplicado à variável my_info.
www.python.org
my_info = open(my_file,’r’)
my_line = my_info.readline()
# Program to demonstrate open() function in Python
my_file = input("Enter file name = >")
# Opens input file
my_info = open(my_file,'r')
# Reads one line of the input file
my_line = my_info.readline()
# Closes file
my_info.close()
# Shows file content on screen
print(my_line)71
Complemento reverso do DNA
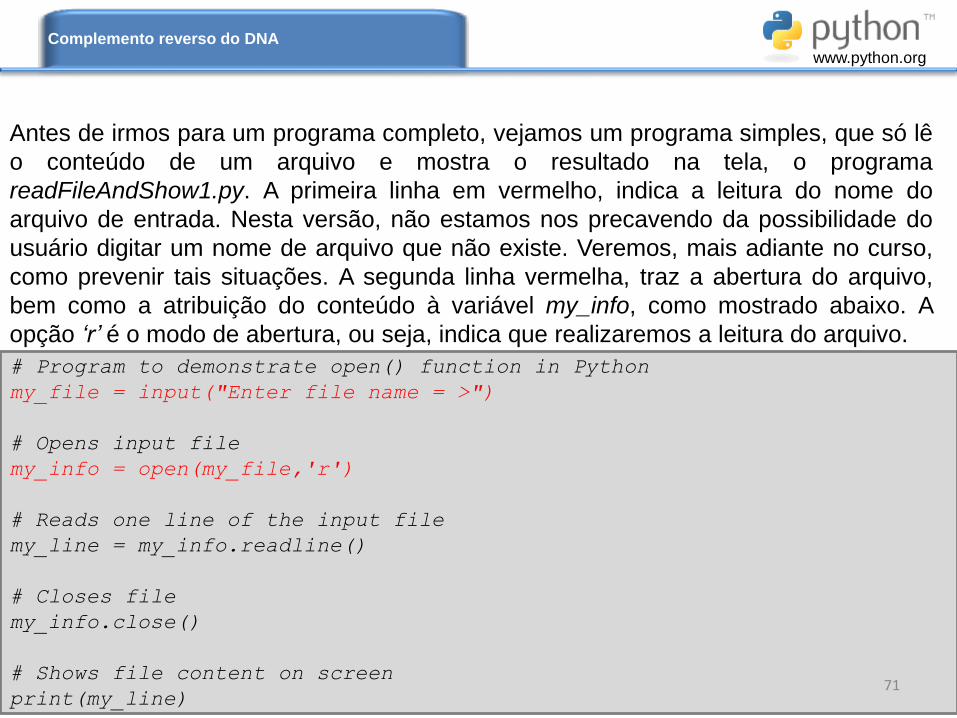
Antes de irmos para um programa completo, vejamos um programa simples, que só lê
o conteúdo de um arquivo e mostra o resultado na tela, o programa
readFileAndShow1.py. A primeira linha em vermelho, indica a leitura do nome do
arquivo de entrada. Nesta versão, não estamos nos precavendo da possibilidade do
usuário digitar um nome de arquivo que não existe. Veremos, mais adiante no curso,
como prevenir tais situações. A segunda linha vermelha, traz a abertura do arquivo,
bem como a atribuição do conteúdo à variável my_info, como mostrado abaixo. A
opção ‘r’ é o modo de abertura, ou seja, indica que realizaremos a leitura do arquivo.
www.python.org

# Program to demonstrate open() function in Python
my_file = input("Enter file name = >")
# Opens input file
my_info = open(my_file,'r')
# Reads one line of the input file
my_line = my_info.readline()
# Closes file
my_info.close()
# Shows file content on screen
print(my_line)72
Complemento reverso do DNA
A terceira linha em vermelho traz a atribuição do conteúdo à variável my_line. Tal
passagem pode parecer desnecessária, mas veja que à variável my_info foi
referenciado o conteúdo do arquivo, mas para manipularmos o conteúdo, temos que
atribuí-lo a uma nova variável, no caso a my_line. O método usado é o .readline(), que
é aplicado à variável my_info. Uma vez que o conteúdo foi atribuído a uma variável,
podemos fechar o arquivo, com o método .close(), aplicado ao my_info. Por último,
mostramos o conteúdo atribuído à variável my_line na tela, com a função print().
Algumas linguagens, como Perl, chamam variáveis como my_info de filehandle.
www.python.org

73
Complemento reverso do DNA
A partir do filehandle my_info, podemos
manipular o arquivo de entrada, contudo,
para acessar seu conteúdo de forma
específica, temos que atribuí-lo a uma
variável, como a my_line do programa. O
fluxograma ao lado ilustra os principais
conceitos na abertura e leitura de um
arquivo com Python. Inicialmente temos o
arquivo my_file, que será aberto com a
função open() e seu conteúdo
referenciado ao filehandle my_info. A linha
de comando para a tarefa, está indicada
abaixo:
my_info = open(my_file, ‘r’)
A seguir, a primeira linha do arquivo, via
filehandle é atribuída à variável my_line,
com a aplicação do método .readline(),
my_line = my_info.readline()
Podemos fechar o arquivo como
my_info.close().
www.python.org
Arquivo my_file
Abertura do arquivo
my_file, conteúdo
referenciado ao
filehandle my_info.
Primeira linha é
atribuída à variável
my_line, com o
método .readline()
Arquivo de entrada
é fechado com o
método close()
my_info = open(my_file,’r’)
my_line = my_info.readline()
my_info.close()

74
Enter file name => dnafile1.fasta
ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Complemento reverso do DNA
Rodaremos o programa readFileAndShow1.py e usaremos o arquivo dnafile1.fasta
como entrada. O resultado está mostrado abaixo.
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input file
dnaFileIn = input("Enter DNA sequence file => ")
fh = open(dnaFileIn,'r') # Opens input file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply .replace
# method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequences
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)75
A abertura de um arquivo em Python usa a função open(), que tem como argumento o
nome do arquivo a ser aberto, ou uma variável com o nome. Assim, antes de abrirmos
o arquivo, o programa tem que perguntar o nome do arquivo. Abaixo temos o código
reverseCompDNA2.py .
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input file
dnaFileIn = input("Enter DNA sequence file => ")
fh = open(dnaFileIn,'r') # Opens input file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply .replace
# method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequences
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)76
A primeira linha vermelha lê o nome do arquivo de entrada, que será atribuído à
variável dnaFileIn. Esta variável será o argumento da função open(). O open() cria um
filehandle, que é usado para fazer referência ao arquivo aberto.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input file
dnaFileIn = input("Enter DNA sequence file => ")
fh = open(dnaFileIn,'r') # Opens input file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply .replace
# method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequences
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)77
No código abaixo, o filehandle é o fh, assim qualquer operação com o arquivo aberto,
deve ser realizada sobre o filehandle fh.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input file
dnaFileIn = input("Enter DNA sequence file => ")
fh = open(dnaFileIn,'r') # Opens input file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply .replace
# method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequences
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)78
O conteúdo arquivo é lido fazendo-se referência ao filehandle fh, usando-se o método
.readline(). O conteúdo do arquivo, no caso uma linha somente, é atribuído à variável
seqIn.
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input file
dnaFileIn = input("Enter DNA sequence file => ")
fh = open(dnaFileIn,'r') # Opens input file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn)
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply .replace
# method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
# Generates uppercase sequences
DNA = dna.upper()
# Changes to a list
DNAlist = list(DNA)
# Inverts list
DNAlist.reverse()
# Changes from a list to string
seqOut=''.join(DNAlist)
print("\nReverse complement:\t",seqOut)79
Depois fechamos o arquivo, com close(), a ser aplicado ao filehandle fh. O restante do
código é idêntico ao programa reverseCompDNA1.py, visto que já temos a sequência
de DNA na variável seqIn.
Complemento reverso do DNA
www.python.org
80
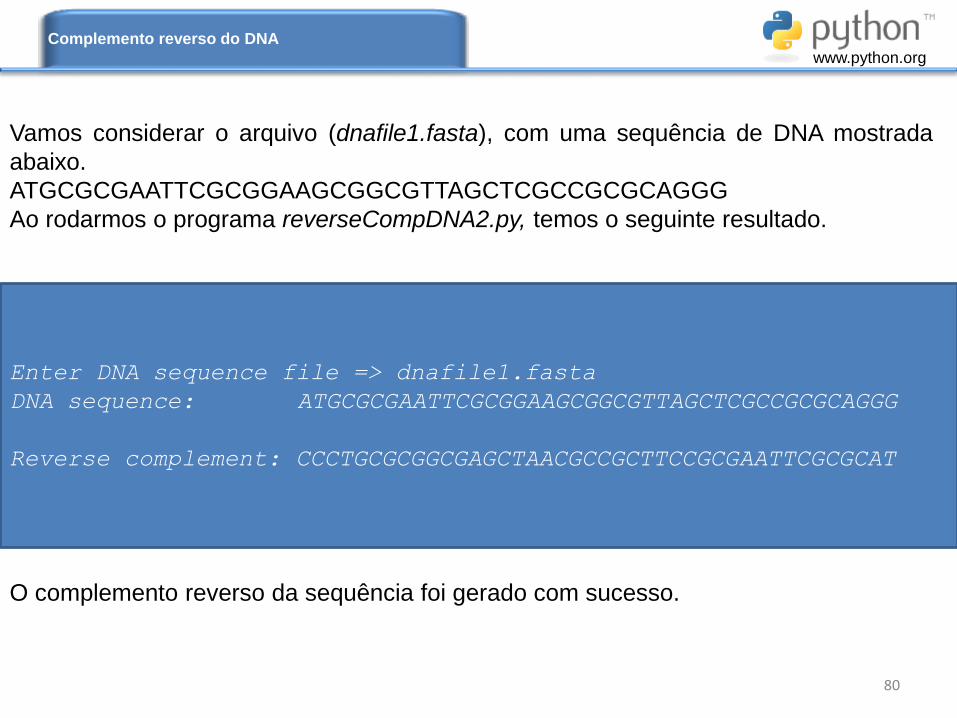
Vamos considerar o arquivo (dnafile1.fasta), com uma sequência de DNA mostrada
abaixo.
ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Ao rodarmos o programa reverseCompDNA2.py, temos o seguinte resultado.
O complemento reverso da sequência foi gerado com sucesso.
Enter DNA sequence file => dnafile1.fasta
DNA sequence: ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Reverse complement: CCCTGCGCGGCGAGCTAACGCCGCTTCCGCGAATTCGCGCAT
Complemento reverso do DNA
www.python.org

# Program to generate reverse complement for a DNA sequence, using a sequence read from an input
# file.
# Reverse complement will be written in an output file
dnaFileIn = input("Enter DNA sequence file => ") # Reads input file name
dnaFileOut = input("Enter output file => ") # Reads output file name
fh = open(dnaFileIn,'r') # Opens input file
fw = open(dnaFileOut,'w') # Opens output file
seqIn = fh.readline() # Assigns file content to seqIn
fh.close() # Closes file
print("DNA sequence:\t\t",seqIn) # Shows sequence on screen
# First we change each base, using the uppercase/lowercase as a trick, in order to not apply
# .replace method to the same base
dna = seqIn.replace("A","t")
dna = dna.replace("T","a")
dna = dna.replace("C","g")
dna = dna.replace("G","c")
81
Vamos criar mais um aperfeiçoamento. Tal tarefa ilustra uma abordagem comum em
programação, criamos um protótipo e vamos aperfeiçoando. Vamos escrever o
complemento reverso num arquivo de saída. O código vai ficar mais extenso, de forma
que não caberá num slide. O novo programa chama-se reverseCompDNA3.py. Neste
programa lemos, também, o nome do arquivo de saída (primeira linha vermelha), que
será atribuído à variável dnaFileOut. O filehandle fw será usado para fazer referência
ao arquivo de saída, com o open(dnaFileOut,’w’) indicando que é um arquivo para
escrita. O ‘w’ é o parâmetro indicativo que será um arquivo para escrita.
Complemento reverso do DNA
www.python.org

DNA = dna.upper() # Generates uppercase sequences
DNAlist = list(DNA) # Changes to a list
DNAlist.reverse() # Inverts list
seqOut=''.join(DNAlist) # Changes from a list to string
print("\nReverse complement:\t",seqOut) # Shows reverse complement sequence
fw.write(seqOut) # writes sequence into output file
fw.close() # Closes file
82
Destacando-se só as novidades do código, temos que escrever o complemento
reverso no arquivo de saída. Para isto usamos o filehandle fw e o método write(). Por
último fechamos o arquivo de saída com close().
Complemento reverso do DNA
www.python.org

83
Vamos considerar o arquivo (dnafile1.fasta), mostrado abaixo.
ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Ao rodarmos o programa reverseCompDNA3.py temos o seguinte resultado.
O complemento reverso da sequência foi gerado com sucesso e escrito no arquivo
test.fasta.
Enter DNA sequence file => dnafile1.fasta
Enter output file => test.fasta
DNA sequence: ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG
Reverse complement: CCCTGCGCGGCGAGCTAACGCCGCTTCCGCGAATTCGCGCAT
Complemento reverso do DNA
www.python.org

84
Além de mostrar na tela o resultado do complemento reverso, o programa armazena a
sequência de DNA no arquivo test.fasta, mostrado abaixo.
CCCTGCGCGGCGAGCTAACGCCGCTTCCGCGAATTCGCGCAT
Temos agora os comandos básicos para ler arquivos existentes e criarmos novos
arquivos. Os procedimentos de manipulação de arquivos serão usados no curso para
leitura de arquivos com dados biológicos, tais como sequência de nucleotídeos (DNA e
RNA) e sequência de aminoácidos (proteínas). Usaremos, também, para
manipulações de coordenadas atômicas de macromoléculas biológicas, contidos em
bases de dados, como o Protein Data Bank (http://rcsb.org/pdb ).
Complemento reverso do DNA
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
85
Há diversos métodos em Python para manipulação de strings, o código
methods4Strings1.py traz alguns deles aplicados à string “GATTACA”. Os próximos
slides destacam em vermelho o trecho de código discutido.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
86
O método s.lower() transforma a string s para letras minúsculas e o s.upper() para
letras maiúsculas. Ambos resultados são mostrados na tela com a função print().
Método associados à manipulação de strings
www.python.org
# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
87
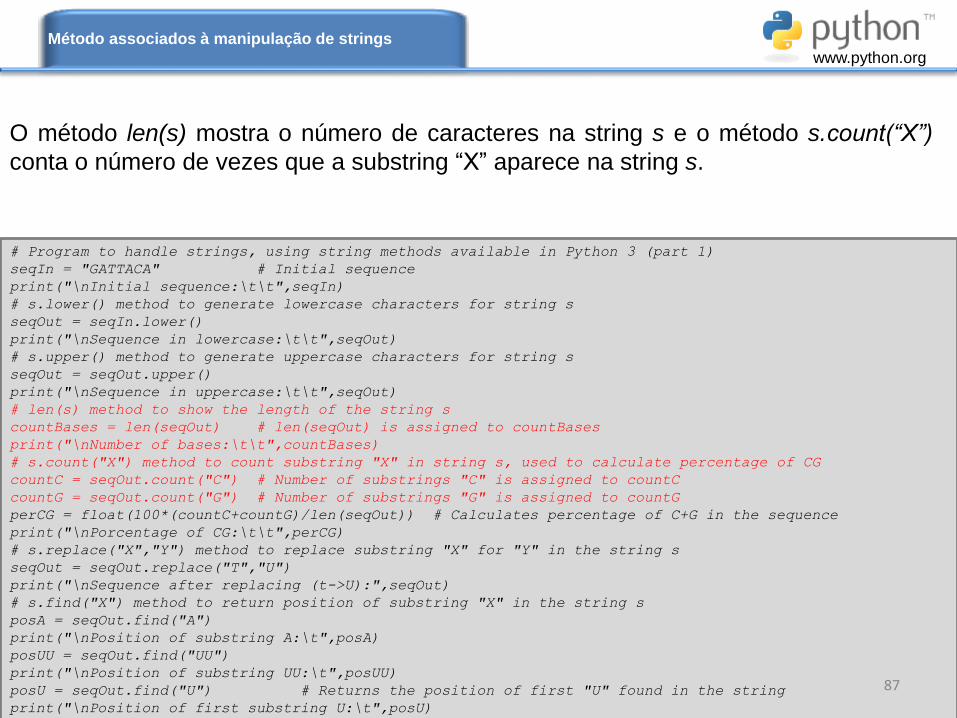
O método len(s) mostra o número de caracteres na string s e o método s.count(“X”)
conta o número de vezes que a substring “X” aparece na string s.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
88
A partir do conteúdo atribuído às variáveis countC e countG, é calculada a
porcentagem de C + G na sequência.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
89
O método replace(“X”,”Y”) troca a substring “X” pela substring “Y”.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 1)
seqIn = "GATTACA" # Initial sequence
print("\nInitial sequence:\t\t",seqIn)
# s.lower() method to generate lowercase characters for string s
seqOut = seqIn.lower()
print("\nSequence in lowercase:\t\t",seqOut)
# s.upper() method to generate uppercase characters for string s
seqOut = seqOut.upper()
print("\nSequence in uppercase:\t\t",seqOut)
# len(s) method to show the length of the string s
countBases = len(seqOut) # len(seqOut) is assigned to countBases
print("\nNumber of bases:\t\t",countBases)
# s.count("X") method to count substring "X" in string s, used to calculate percentage of CG
countC = seqOut.count("C") # Number of substrings "C" is assigned to countC
countG = seqOut.count("G") # Number of substrings "G" is assigned to countG
perCG = float(100*(countC+countG)/len(seqOut)) # Calculates percentage of C+G in the sequence
print("\nPorcentage of CG:\t\t",perCG)
# s.replace("X","Y") method to replace substring "X" for "Y" in the string s
seqOut = seqOut.replace("T","U")
print("\nSequence after replacing (t->U):",seqOut)
# s.find("X") method to return position of substring "X" in the string s
posA = seqOut.find("A")
print("\nPosition of substring A:\t",posA)
posUU = seqOut.find("UU")
print("\nPosition of substring UU:\t",posUU)
posU = seqOut.find("U") # Returns the position of first "U" found in the string
print("\nPosition of first substring U:\t",posU)
90
O método s.find(“X”) retorna a posição da substring “X”. O primeiro caractere da string
tem posição zero “0”, o segundo caractere tem posição “1”, e assim sucessivamente.
Método associados à manipulação de strings
www.python.org

Initial sequence: GATTACA
Sequence in lowercase: gattaca
Sequence in uppercase: GATTACA
Number of bases: 7
Porcentage of CG: 28.571428571428573
Sequence after replacing (t->U): GAUUACA
Position of substring A: 1
Position of substring UU: 2
Position of first substring U: 2 91
Ao executarmos o código methods4Strings1.py, temos os resultados mostrados
abaixo.
Método associados à manipulação de strings
www.python.org

Exercício de programação 6. Modifique o código fonte do programa
methods4Strings1.py, de forma que a nova versão leia a sequência a partir de um
arquivo de entrada. Salve o novo programa com o nome methods4Strings2.py. Rode
o programa.
92
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
93
O código methods4Strings3.py traz outros métodos aplicados à string “GaTTaCa”,
como segue.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
94
O método s.swapcase() troca de maiúscula para minúscula e vice-versa.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
95
O método s.strip() elimina espaços em branco, tabs e newlines no início e final da
string.
Método associados à manipulação de strings
www.python.org
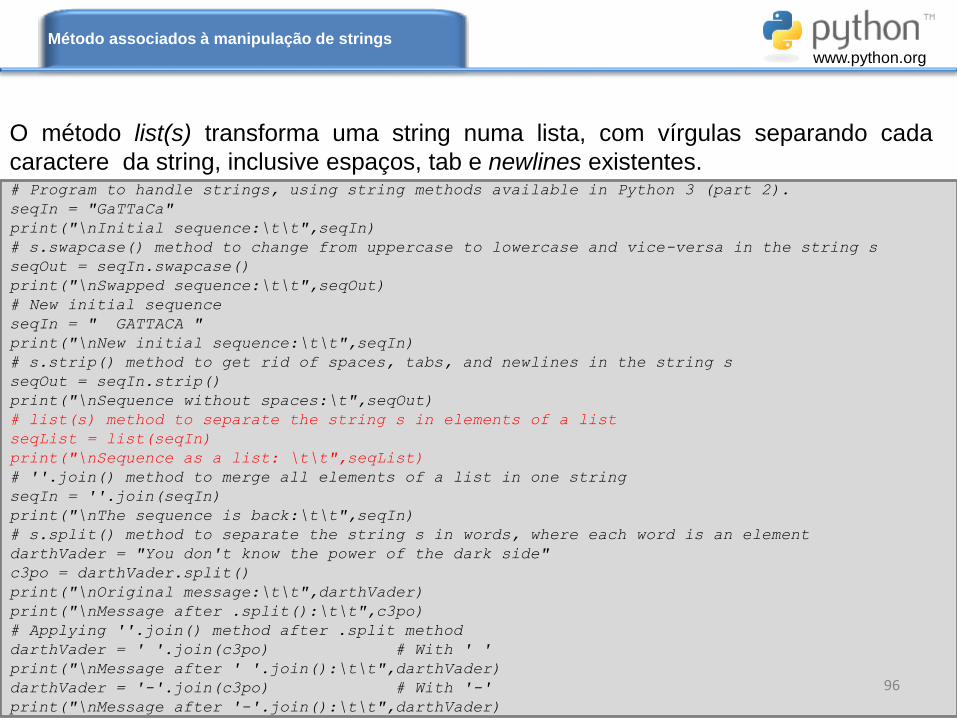
# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
96
O método list(s) transforma uma string numa lista, com vírgulas separando cada
caractere da string, inclusive espaços, tab e newlines existentes.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
97
O método ‘X’.join(ls) transforma uma lista ls numa string, inserindo o argumento X
entre os elementos da lista na composição da string.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
98
O método s.split() divide uma string s em palavras, formando uma lista.
Método associados à manipulação de strings
www.python.org

# Program to handle strings, using string methods available in Python 3 (part 2).
seqIn = "GaTTaCa"
print("\nInitial sequence:\t\t",seqIn)
# s.swapcase() method to change from uppercase to lowercase and vice-versa in the string s
seqOut = seqIn.swapcase()
print("\nSwapped sequence:\t\t",seqOut)
# New initial sequence
seqIn = " GATTACA "
print("\nNew initial sequence:\t\t",seqIn)
# s.strip() method to get rid of spaces, tabs, and newlines in the string s
seqOut = seqIn.strip()
print("\nSequence without spaces:\t",seqOut)
# list(s) method to separate the string s in elements of a list
seqList = list(seqIn)
print("\nSequence as a list: \t\t",seqList)
# ''.join() method to merge all elements of a list in one string
seqIn = ''.join(seqIn)
print("\nThe sequence is back:\t\t",seqIn)
# s.split() method to separate the string s in words, where each word is an element
darthVader = "You don't know the power of the dark side"
c3po = darthVader.split()
print("\nOriginal message:\t\t",darthVader)
print("\nMessage after .split():\t\t",c3po)
# Applying ''.join() method after .split method
darthVader = ' '.join(c3po) # With ' '
print("\nMessage after ' '.join():\t\t",darthVader)
darthVader = '-'.join(c3po) # With '-'
print("\nMessage after '-'.join():\t\t",darthVader)
99
No final do programa temos a inserção de espaço ‘ ‘ e traço ‘-’ entre os elementos da
lista ls, com os métodos ‘ ‘.join(ls) e ‘-’.join(ls), respectivamente.
Método associados à manipulação de strings
www.python.org

Initial sequence: GaTTaCa
Swapped sequence: gAttAcA
New initial sequence: GATTACA
Sequence without spaces: GATTACA
Sequence as a list: [' ', ' ', 'G', 'A', 'T', 'T', 'A', 'C', 'A', ' ']
The sequence is back: GATTACA
Original message: You don't know the power of the dark side
Message after .split(): ['You', "don't", 'know', 'the', 'power', 'of', 'the',
'dark', 'side']
Message after ' '.join(): You don't know the power of the dark side
Message after '-'.join(): You-don't-know-the-power-of-the-dark-side100
Ao executarmos o código methods4Strings3.py, temos os resultados mostrados
abaixo.
Método associados à manipulação de strings
www.python.org

101
Uma lista completa dos métodos usados para manipulação de strings em Python pode
ser encontrada em:
http://www.tutorialspoint.com/python/python_strings.htm
Acesso em: 25 de março de 2016.
Método associados à manipulação de strings
www.python.org

Na figura ao lado temos a pedra de Rosetta,
que traz a descrição de um culto executado
em honra ao faraó Ptolomeu V do Egito. A
descrição está em hieróglifos na parte
superior, em demótico cursivo na parte
central e, em grego, na parte final. Antes da
descoberta da pedra de Rosetta, em 1799,
o entendimento dos hieróglifos estava
perdido. Ou seja, toda riqueza de
informações contidas nas pirâmides e nos
templos egípcios não era entendida. Como
o trecho final da pedra de Rosetta está em
grego, os estudiosos usaram tal informação
para traduzir as informações dos hieróglifos,
o que permite hoje a leitura dos
documentos do Egito antigo. A pedra de
Rosetta está em exposição no British
Museum em Londres.
Foto feita por Linus S. Azevedo (5/2/2011).
British Museum, London-UK102
Dogma central da Biologia Molecular

Toda vida na Terra é baseada num conjunto
de moléculas (DNA, RNA e proteínas). O
DNA é uma hélice dupla de nucleotídeos,
que codificam um organismo. Cada tripleto
de nucleotídeos forma um códon, específico
para um aminoácido numa cadeia proteica.
Em outras palavras, três nucleotídeos (um
códon) especificam um aminoácido na
cadeia proteica.
A lista completa, relacionando cada códon
com um aminoácido, recebe o nome de
“código genético”. O RNA também é um
ácido nucleico, sua função é transferir a
informação do DNA para ser traduzida em
proteína.
F. H. C. Crick, "The Origin of the Genetic Code," Journal of Molecular Biology 38(3), 1968 pp. 367-379
DNA
Pares de bases
103
Códon
Dogma central da Biologia Molecular

Visto que temos 3 nucleotídeos possíveis para cada posição no códon, teremos um
total de 4x4x4 = 64 códons possíveis. Na natureza observamos somente 20
aminoácidos, assim podemos dizer que o código genético é degenerado. Cada códon
especifica um aminoácido em particular. Por exemplo, no código genético o
aminoácido glicina tem quatro códons (GGA, GGC, GGU e GGG). Já o aminoácido
triptofano, tem somente um códon (UGG). Além disso, há códons relacionados ao
processo de informação, por exemplo, os códons de parada (stop codons) (UAA, UAG
e UGA).
GGA, GGC, GGU, and GGG codificam a Glicina (Gly)
UGG codificam o triptofano (Trp)
UAA, UAG, and UGA são códons de parada.
Gly
Trp
104F. H. C. Crick, "The Origin of the Genetic Code," Journal of Molecular Biology 38(3), 1968 pp. 367-379
Dogma central da Biologia Molecular

Glicina GGA GGC GGG GGU
Alanina GCA GCC GCG GCU
Serina AGC AGU UCA UCC UCG UCU
Treonina ACA ACC ACG ACU
Cisteina UGC UGU
Valina GUA GUC GUG GUU
Isoleucina AUA AUC AUU
Leucina UUA UUG CUA CUC CUG CUU
Prolina CCA CCC CCG CCU
Fenilalanina UUC UUU
Tirosina UAC UAU
Metionina AUG
Triptofano UGG
Asparagina AAC AAU
Glutamina CAA CAG
Histidina CAC CAU
Aspartato GAC GAU
Glutamato GAA GAG
Lisina AAA AAG
Arginina AGA AGG CGA CGC CGG CGU
Código genético
A tabela abaixo mostra os nomes dos 20 aminoácidos naturais e seus respectivos
códons. Os códons são representados por nucleotídeos presentes no RNA, ou seja,
timina (T) foi substituído por Uracila (U).
105
Dogma central da Biologia Molecular
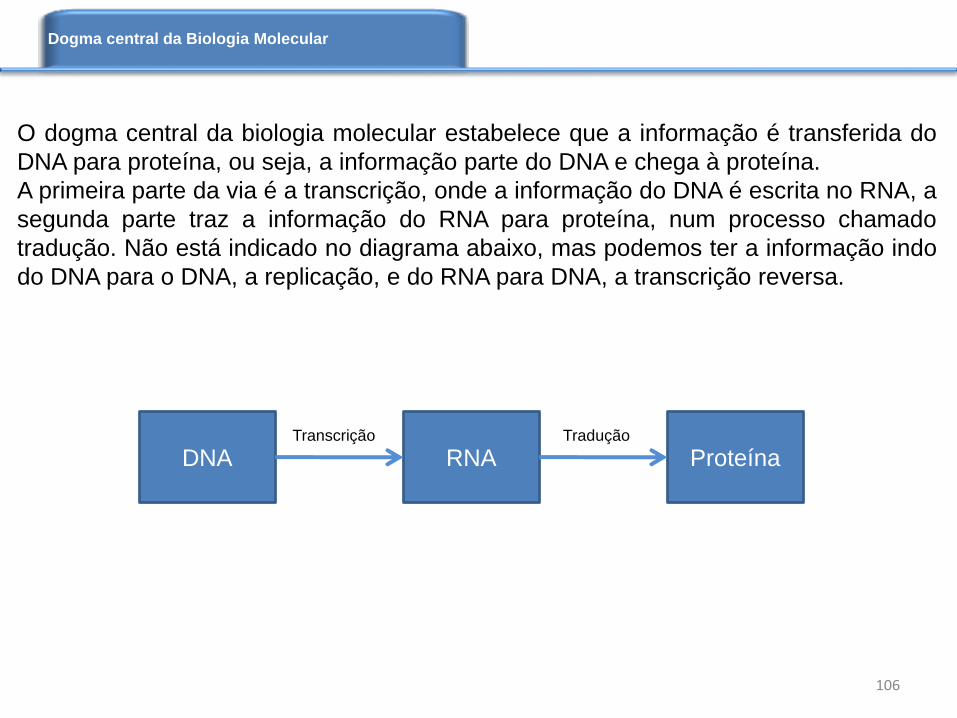
O dogma central da biologia molecular estabelece que a informação é transferida do
DNA para proteína, ou seja, a informação parte do DNA e chega à proteína.
A primeira parte da via é a transcrição, onde a informação do DNA é escrita no RNA, a
segunda parte traz a informação do RNA para proteína, num processo chamado
tradução. Não está indicado no diagrama abaixo, mas podemos ter a informação indo
do DNA para o DNA, a replicação, e do RNA para DNA, a transcrição reversa.
DNA RNA ProteínaTranscrição Tradução
106
Dogma central da Biologia Molecular

O código genético pode ser
considerado como uma “pedra de
Rosetta molecular”, onde temos a
tradução da informação contida no
DNA para proteínas. A primeira parte
dessa via é a transcrição, onde a
informação do DNA é escrita no RNA,
a segunda parte traz a informação do
RNA para proteína, num processo
chamado tradução. Usando a analogia
com a pedra de Rosetta, temos que a
informação dos códigos escritos em
hieróglifos (parte superior da pedra) é
equivalente à molécula de DNA. A
informação passa para o demótico
cursivo (no meio da pedra), que é
equivalente ao RNA e, por último, em
grego (na parte de baixo da pedra),
equivalente às proteínas.
Proteína
DNA
RNA
107
Dogma central da Biologia Molecular
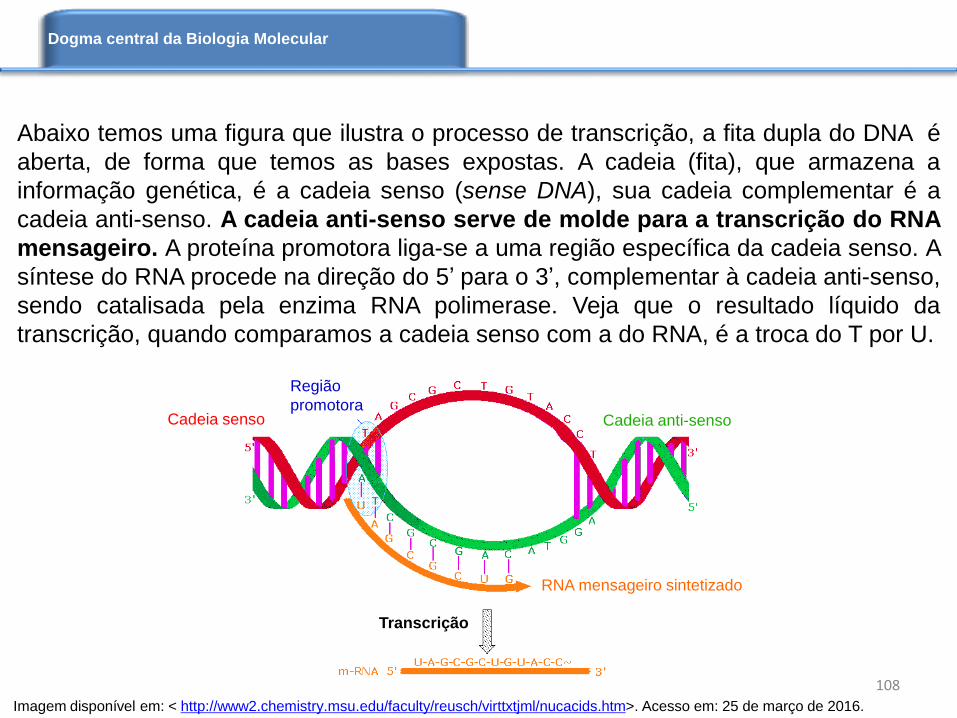
Abaixo temos uma figura que ilustra o processo de transcrição, a fita dupla do DNA é
aberta, de forma que temos as bases expostas. A cadeia (fita), que armazena a
informação genética, é a cadeia senso (sense DNA), sua cadeia complementar é a
cadeia anti-senso. A cadeia anti-senso serve de molde para a transcrição do RNA
mensageiro. A proteína promotora liga-se a uma região específica da cadeia senso. A
síntese do RNA procede na direção do 5’ para o 3’, complementar à cadeia anti-senso,
sendo catalisada pela enzima RNA polimerase. Veja que o resultado líquido da
transcrição, quando comparamos a cadeia senso com a do RNA, é a troca do T por U.
108
Imagem disponível em: < http://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/nucacids.htm>. Acesso em: 25 de março de 2016.
Cadeia senso Cadeia anti-senso
Transcrição
RNA mensageiro sintetizado
Região
promotora
Dogma central da Biologia Molecular

Vamos ilustrar a transformação da informação codificada no DNA para RNA
(transcrição) e, depois, para proteína (tradução). A informação está armazenada num
arquivo no formato FASTA. Inicialmente faremos transcrição do trecho de DNA para
RNA (trocar T por U) e, por último, traduziremos o RNA para uma sequência de
aminoácidos.
109
>DNA:A|PDBID|CHAIN|SEQUENCE
CGCGAATTCGCGArquivo no formato FASTA com a sequência de nucleotídeos. A primeira linha inicia com
o símbolo > e traz a identificação do sequência. As bases começam na segunda linha.
>RNA:A|PDBID|CHAIN|SEQUENCE
CGCGAAUUCGCG Arquivo no formato FASTA com a sequência de nucleotídeos do RNA
Transcrição
>PEP:A|PDBID|CHAIN|SEQUENCE
REFA
Tradução
Arquivo no formato FASTA com a sequência de aminoácidos do trecho traduzido a
partir do RNA.
CGC Arginina (Arg) (R)
GAA Glutamato (Glu) (E)
UUC Fenilalanina (Phe) (F)
GCG Alanina (Ala) (A)
Dogma central da Biologia Molecular

# Program to carry out transcription
dna = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence")
print(dna)
# Transcription
rna = dna.replace("T","U")
print("\nTranscription from DNA to RNA")
print("DNA => ",dna)
print("RNA => ",rna)
110
Vamos considerar o programa abaixo, que transcreve um fragmento de DNA para um
de RNA, a partir de do método .replace(“T",“U") . O programa chama-se
transcription1.py . O código é relativamente simples, o conteúdo da string com
nucleotídeos é atribuído à variável dna, como mostrado na primeira linha vermelha
abaixo. O método dna.replace(“T",“U") faz a troca de T->U e atribui à variável rna,
mostrado na segunda linha vermelha. No final, as duas sequências são mostradas na
tela, com a função print().
Dogma central da Biologia Molecular

111
A rodarmos o programa transcription1.py, temos o resultado mostrado abaixo.
Dogma central da Biologia Molecular
# Program to carry out transcription
dna = "ATGCGCGAATTCGCGGAAGCGGCGTTAGCTCGCCGCGCAGGG"
print("DNA sequence")
print(dna)
# Transcription
rna = dna.replace("T","U")
print("\nTranscription from DNA to RNA")
print("DNA => ",dna)
print("RNA => ",rna)

ALBERTS, B. et al. Biologia Molecular da Célula. 4a edição. Porto Alegre: Artmed editora, Porto Alegre, 2004.
-BRESSERT, Eli. SciPy and NumPy. Sebastopol: O’Reilly Media, Inc., 2013. 56 p.
-DAWSON, Michael. Python Programming, for the absolute beginner. 3ed. Boston: Course Technology, 2010. 455 p.
-HETLAND, Magnus Lie. Python Algorithms. Mastering Basic Algorithms in the Python Language. Nova York: Springer
Science+Business Media LLC, 2010. 316 p.
-IDRIS, Ivan. NumPy 1.5. An action-packed guide dor the easy-to-use, high performance, Python based free open source
NumPy mathematical library using real-world examples. Beginner’s Guide. Birmingham: Packt Publishing Ltd., 2011. 212 p.
-KIUSALAAS, Jaan. Numerical Methods in Engineering with Python. 2ed. Nova York: Cambridge University Press, 2010. 422
p.
-LANDAU, Rubin H. A First Course in Scientific Computing: Symbolic, Graphic, and Numeric Modeling Using Maple, Java,
Mathematica, and Fortran90. Princeton: Princeton University Press, 2005. 481p.
-LANDAU, Rubin H., PÁEZ, Manuel José, BORDEIANU, Cristian C. A Survey of Computational Physics. Introductory
Computational Physics. Princeton: Princeton University Press, 2008. 658 p.
-LUTZ, Mark. Programming Python. 4ed. Sebastopol: O’Reilly Media, Inc., 2010. 1584 p.
-MODEL, Mitchell L. Bioinformatics Programming Using Python. Sebastopol: O’Reilly Media, Inc., 2011. 1584 p.
-TOSI, Sandro. Matplotlib for Python Developers. Birmingham: Packt Publishing Ltd., 2009. 293 p.
Última atualização: 19 de julho de 2016
112
Referências
www.python.org