Embed Size (px)
Citation preview

^ÁlO
MARGARIDA ALICE FERREIRA Assistente da Faculdade de Farmácia da Universidade do Porto
Bolseira da Fundação Calouste Gulbenkian
CONTRIBUIÇÃO PARA O CONHECIMENTO DOS ALCALÓIDES INDÓLICOS
— DE —
BURKEA AFRICANA HOOK
P O R T O 1 9 7 1

M A R G A R I D A A L I C E F E R R E I R A
A s s i s t e n t e da Faculdade de Farmácia
da Universidade do Porto
B o l s e i r a da Fundação Calouste Gulbenkian
CONTRIBUIÇÃO PARA 0 C0ÎHECIMENT0 DOS ALCALÓIDES INDOLICOS
DE
BURKEA AFRICANA HOOK
FACULDA ? ■ ■■ AKIA! A U . " .
B i :■ L . ' ) T ; ;
P O R T O 1971

CONTRIBUIÇÃO PARA O CONHECIMENTO DOS ALCALÓIDES
INDOLICOS
DE
BURKEA AFRICANA HOOK

MARGARIDA ALICE FERREIRA. Assistente da Faculdade de Farmácia
da Universidade do Porto Bolseira da Fundação Calouste Gulbenkian
CONTRIBUIÇÃO PARA 0 CONHECIMENTO DOS ALCALÓIDES INDOLICOS
DE BURKEA AFRICANA HOOX
Dissertação de candidatura ao grau de Doutor, apresenta da à Faculdade de Farmácia da Universidade do Porto
Trabalho realizado no Agrupamento Científico de Farmacognosia
FACULDADE DE FARMÁCIA - UNIVERSIDADE DO PORTO
P O R T O 1971

Aos Excelentíssimos Senhores PROFESSOR DOUTOR ANTONIO CORREIA ALVES PROFESSOR DOUTOR LUÏS VASCO NOGUEIRA PRISTA
Homenagem de gratidão pelos conhecimentos que nos deram de Química Vegetal

AO EXCELENTÍSSIMO SENHOR PROFESSOR DOUTOR JOSE FERREIRA DO VALE SERRANO Ilustre Director da Faculdade de Farmácia
Universidade do Porto
Ao Excelentíssimo Corpo Docente da Faculdade de Farmácia da Universidade do Porto

A M E U S P A I S
A O S M E U S F A M I L I A R E S
A O S M E U S A M I G O S

PREAMBULO
Bastaria a circunstância de sermos assistente de Farmacognosia para justificar a escolha dum tema de índole farmacognósica para a nossa dissertação do doutoramento. Outros motivos, no entanto, concorreram para isso.
Com efeito, terminada a nossa licenciatura em Farmácia, logo nos sentimos atraídas para a investigação,espe_ cialmente no campo da Química. Por isso, quando surgiu a possibilidade de ingressarmos no Agrupamento Científico de Farmacogncsia, um ano apds a conclusão do nosso curso, decidimos aproveitar a oportunidade que nos era oferecida.
Começámos assim a ensaiar, timidamente, os primeiros passos nos complicados meandros da Fitoquímica, ani_ mada do maior desejo de aprender e tendo perante nós a sedutora miragem de virmos a conseguir penetrar nos segredos da química do mundo vegetal.
No entanto, as ilusões que a nossa inexperiência criara depressa se desvaneceram e a dura realidade das no_s sas limitações e das dificuldades que a investigação ofere ce não tardaram a surgir, bem claras, ao nosso espírito.

10
Devemos mesmo confessar que, apesar do entusiasmo com que havíamos principiado o nosso trabalho,certamente não teríamos ultrapassado a fase de iniciação se não f çs sem os incitamentos recebidos dos professores que então orientavam cientificamente o Agrupamento de Farmacognosia.
Contudo, vencido o desânimo momentâneo que nos invadira de começo bem depressa o gosto pela Química Vegetal se foi enraizando em nós à medida que íamos colaborando nos trabalhos realizados no referido Agrupamento.
Foi o capítulo dos alcalóides aquele que desde logo despertou o nosso maior interesse e tal facto explica -se, em parte, pela circunstância de o primeiro trabalho de investigação em que colaborámos ter sido um estudo de uma planta contendo substâncias daquela natureza. Por outro lado, as marcadas e variadas acções farmacológicas de muitos alcalóides constituíram o principal motivo para nos sentirmos atraídas por este grupo de compostos, dado o espírito farmacêutico de que estamos imbuídas.
Esta nossa preferência não passou despercebida ao Exme. Senhor Prof. Doutor Nogueira Prista que nos entusia_s mou a escolher para tema da nossa dissertação de doutoramen to, o estudo de espécies contendo tais princípios.
Em face disso, formulámos o propósito de pesquisarmos alcalóides indólicos nalgumas espécies oriundas da nossa Província de Angola, mas das seis plantas daí provenientes que conseguimos obter para o efeito, somente em

11
Burkea africana HOOK pudemos assinalar a existência dos re feridos compostos.
Mercê desta circunstância, o nosso trabalho teve de restringir-se ao estudo desta espécie.
Demos então início à tarefa que nos propusemos le var a cabo, agora já com plena consciência das limitações e perfeitamente cientes das dificuldades com que iríamos de parar.
Está fora de dúvida que são modestos os resultados obtidos neste trabalho. Pensamos no entanto que valeu a pena realizá-lo porquanto, pudemos isolar e caracterizar algumas bases indólicas existentes numa planta complet amen. te inexplorada neste domínio.
Aos Exm^s. Senhores Profs. Doutores António Correia Alves e Luís Vasco Nogueira Prista que, desde a nossa entrada para o Agrupamento de Farmacognosia, sempre nos distinguiram com a sua amizade e compreensão, e aos quais devemos a possibilidade de termos realizado este trabalho, queremos expressar o nosso mais profundo reconhecimento e gratidão.
Ao colega Dr. Alberto Figueira de Sousa,assisten te do Instituto de Investigação Científica de Angola.agra-decemos a boa vontade e o não se ter furtado a sacrifícios

12
para nos fornecer o material utilizado neste trabalho.
Ao Exme. Senhor Prof. Doutor Alberto Carlos Correia da Silva agradecemos o ter acedido a ensaiar farmacologicamente Burkea africana II00PI, sendo de notar que os resultados desses ensaios contribuiram decisivamente para nos entusiasmar no prosseguimento do seu estudo químico.
Ao Exme. Senhor Prof. Doutor Armando Larose Rocha agradecemos o auxílio que nos prestou na tradução da bibliografia alemã.
Ao Exms. Senhor Prof. Doutor Joaquim António de Barros Polónia o nosso reconhecimento pelos úteis ensinamentos que nos transmitiu quando fomos sua aluna, e ainda pela ajuda e pelas palavras de confiança que tantas vezes nos dirigiu.
Ao ExmQ. Senhor Prof. Doutor Abílio Fernandes, Ilustre Tn rector do Instituto Botânico "Pr. Júlio Henriques", da Universidade de Coimbra, expressamos a nossa profunda gratidão não só por se ter dignado classificar o material utilizado no nosso estudo bem como pela gentileza de nos ter facultado o exame dos exemplares de herbário de Burkea africana HOOK existentes naquele Instituto.
Aos colegas da nossa Faculdade e aos que trabalham no Agrupamento Cientifico de Farmacognosia pela colaboração que nos deram e pela amizade que sempre nos dispen

13
saram, um muito obrigada.
A Fundação Calouste Gulbenkian e ao seu Ilustre Director do Serviço de Bolsas de Estudo, ExmQ. Senhor Engenheiro D. Duarte de Castro, os nossos agradecimentos pela concessão duma bolsa que nos ajudou a minorar as dificuldades de ordem económica.
A todas as pessoas amigas que directa ou indire_c tamente nos ajudaram, quer pelos serviços que nos prestaram quer pelos conselhos e encorajamentos que nos dispensaram, o nosso reconhecido agradecimento.
Aos Exmes. Senhores Albino Teixeira da Silva e Manuel da Rocha Gonçalves o nosso agradecimento pela colaboração que nos deram na dactilografia do nosso trabalho.

P A R T E I

15
1.- NOÇÕES GERAIS SOBRE O GÉNERO BURKEA HOOK
1.1.- SISTEMÁTICA E CARACTERES BOTÂNICOS
O género Burkea foi classificado e estudado em 1843 por HOOKER (l). E um género monoespecífico se bem que OLIVIER (2) admita duas variedades para a única espécie que o compõe.
HIERN (3) e BAKER (4), provavelmente fundamentados nas observações de WELW1TCH e de OLIVIER, são também da mesma opinião.
TORRE e HILCOAT (5) referem* todavia, que em Angola apenas existe a espécie Burkea africana, var. cordata WELW.ex OLIVIER, e põem em dúvida a existência da var. an-dongensis OLIVIER.
Aqueles autores afirmam ter consultado volumoso material de herbário e ter-lhes sido difícil definir os li mites das duas variedades de OLIVIER. Em sua opinião, OLIVIER e WELWITCH teriam sido levados a admitir as duas cit_a das variedades por terem observado espécimes provenientes

16
da floresta densa e dos rochedos áridos, nos quais se observam, de facto, diferenças notórias.
Nos mesmo tivemos oportunidade de verificar que tal assim acontece, ao observar os 32 exemplares de Burkea africana HOOK que constam das colecções do herbário do Instituto Botânico "Pr. Júlio Henriques" da Universidade de Coimbra.
Na opinião do Exme. Senhor Prof. Doutor Abílio Fernandes, deve considerar-se como válida somente a espécie Burkea africana HOOK. Este conceito é, aliás,o da maio ria dos botânicos, como BENTEAM e HOOKER (ô),HUTCHINS0N(7), WHITE (8), etc..
Segundo ENGLER (9), Burkea africana HOOK pertence à família das Leguminoseae, subfamília das Caesalpinoi-deagj tribo Dimorphandreae, sendo composta por árvores ou arbustos altos, inermes e com as extremidades espessadas.
As folhas são bipinadas, corn (l)-3-6(8)pares de pínulas opostas ou sub-opostas; cada pínula tem, geralmente, 6-12 folíolos, alternos, coriáceos, com pecíolo curto ou sub-séssil, de forma ovada ou oblonga e de ápice arredonda do, imarginado, obtuso e apiculado; têm estipulas, filiformes, muito pequenas e caducas.
As flores são pequenas, hermafroditas,espessadas, dispostas em espiga axilar comprida e simples ou ligeiramente ramosa, aparecendo num tufo de folhas nas extremidades dos ramos°, brácteas pequenas.

17
O cálice é campanulado, com os segmentos ligados em tubo, com 5 lobos iguais, arredondados; pétalas em número de 5; subiguais, obovadas, imbricadas em botão; estâmes 10, subiguais( o filete é curto; anteras uniformes, oblongas e oblanceoladas, conectivo estreito e apiculado, com dois lóculos deiscentes longitudinalmente; o ovário é séssil ou subséssil, hirsuto, 12 ovulado; o estilete é curtíssimo e espessado e o estigma côncavo ou obliquamen
te truncado. 0 fruto va^em é de forma oblonga, elíptica,
planocomprimido, levemento coriáceo, indeiscente e, em re_ gra, monospérmico; a semente é suborbiculada, planocompri mida (1,2,4,6).
1.2. DISTRIBUIÇÃO GEOGRÁFICA
As plantas da família das Leguminoseae, ■flubfamilia das Gaesalpinoideae, encontramse largamente distribui_ das pelas regiões tropicais e subtropicais da América e da Africa. Parece que alguns géneros existem, apenas, na Ame rica, ao passo que outros, habitam as terras africanas, en_ contrandose a maioria,simultaneamente, nos dois continentes.
O género Burkea parece estar restrito à Africa(7, 8,10,11). Tem larga dispersão pela África TrópicoAustral e África do Sul, sendo abundante na Rodésia, Niassalândia,

18
Moçambique, Transval, Congo ex-Belga, etc.. Em Angola (5), donde é originária a amostra que
estudámos, existe de Norte a Sul da Província, estando par ticularmente espalhada pela comunidade pirolítica.
Nesta Província portuguesa a planta habita as fio restas decídua,ou mista dos planaltos, atingindo a altura de 5 a 10 metros, e também os terrenos áridos e rochosos, mas nestes atinge, em regra, somente o porte de arbusto.En tre as regiões de Angola onde a sua presença foi assinalada contam-se a Lunda, Benguela, Bié, Moxico,Moçâmedes,Kuí-la, etc.
1.3,- INTERESSE ECONÓMICO (7,9,10,13,14)
As Caesalpinoideae têm interesse particularmente pelas madeiras que fornecem. Algumas delas servem como material tanante e outras, aindajcomo fonte de corantes.
Secundariamente, fornecem óleo-resinas, gomas,em bora de diminuto valor comercial, e, também,fibras utilizáveis no fabrico de cordas e tecidos grosseiros. Burkea a-fricana HOOK tem idênticos empregos.
A madeira é acastanhada, escura ou avermelhada^ Apresenta estriação ondeada com filas concêntricas de poros com goma. Ë uma madeira muito dura, resistente, pesada e duradoira por ser pouco atacada pelos insectos. Ë bastan

19
te grosseira mas adquire polimento razoável. Em Angola e na África Oriental é muito usada no
fabrico de carruagens de caminho de ferro, barcos, pavimen_ tos, na fabricação de estacas para cercas e de mobiliário diverso. Tem largo uso como combustível e, por isso, em aj-gumas regiões Angolanas é conhecida por "árvore do carvão".
A casca tem apreciável quantidade de taninos,seri do, por isso,u?ía curtidura de peles, no norte da Nigéria e no Sudoeste do Tanganica.
A planta fornece também uma goma semitranslúci-da, avermelhada, de qualidade razoável, que parece ter u-tilidade na Africa Oriental.
1.4.- NOMES VERNÁCULOS (12,13,14)
São variadíssimos os nomes pelos quais é conheci da esta espécie entre os nativos. Referiremos apenas alguns.
Assim, na Guiné ex-Francesa é conhecida por Kom-bukuru; na Costa do Ouro por pirimo ou pirimu; no Tojo por Kinkiri, atakplar. tschingli ou tscheseli. esçeresu, opi-rium, etc.; na Nigéria por Kokolu, glaugblongum e dagbomgum.
Em Angola, em dialecto Kinbundum, ehama-se Kabi-langu e musese, sakambua ou sagambua; nas regiões de Huila e Malange os nativos conhecem-na por pgiukalati. etc.

20
Na África do Sul são correntes os nomes de "Wild syringa","Wild lilac" e "Rhodesian ash".
1.5.- USOS DA MEDICINA INDÍGENA (14)
Esta espécie, à semelhança do que acontece com tantas outras, é utilizada com fins medicinais pelos nativos das regiões ..onde cresce, sempre interessados em recor_ rer à forma terapêutica mais acessível e natural, para curarem ou tentarem aliviar os seus males.
Assim, os sul-rodesianos usam a casca mastigada para aplicar em feridas sépticas e as folhas em casos de obstipação e flatulência.
Na África Central, o decocto das folhas aplica-se em fricções, para aliviar dores de cabeça e de dentes.. '
Em Malange, os nativos mastigam a casca para tra tarem anginas e outras inflamações da gargaírta , enquanto que na Huíla, segundo informações particulares, a casca é empregada em casos de indigestão, simplesmente mastigada ou em decocto. Descreve-se também o seu emprego como contra--veneno, especialmente para o gado bovino, em casos de intoxicação com "otyiheké", uma forma de capim tóxico que se desenvolve nos terrenos arenosos.
1,6.- ESTUDOS QUÍMICOS
São incipientes as determinações químicas efectua

cl
das sobre esta planta. MARK (l5), em 1930, fgz a determinação do teor
de taninos nas cascas e frutos duma amostra de Burkes africana HOOK da África Oriental.
0 objectivo da análise foi o de obter indicações acerca do possível aproveitamento económico da piarta, ums vez que as cascas e os frutos pareciam conter elevada pe""> centagem de taninos.
0 material em estudo era proveniente dos arretares da vila Bismark, na antiga Africa Oriental alemã. 0 u tado autor encontrou, na parte mais externa da casca,16,í% de tanino e, nos frutos isentos de sementes, 5,7%. Embora o problema não tivesse ficado bem esclarecido, porquanto i.e impunham mais determinações para verificar qual a época i-deal de colheita da planta e também em que idade apresentai va maior percentagem de taninos, estes ensaios permitirau excluir os frutos para fins tanantes e sugeriram o aprove_i tamento da casca.
HOWES (l6) refere a existência, na planta, duna goma semi-translúcida, amarela ou avermelhada, de razoável qualidade.
ISTAS e col. (17) fizjram, em 1959, uma análise global de "sese", nome vernáculo por que Burkea africana é conhecida no Congo ex-Belga. Procederam à análise de amestras de planta adulta e jovem, determinando nelas a húmida, de, as cinzas, alguns elementos minerais,extracto etéreo,

22
extracto alcoólico-benzénico, e fizeram ainda o doseamento dos taninos.
Aqueles autores procederam também à pesquisa de glucosidos e de alcalóides, nesta planta e não tendo carac_ terizado os primeiros, assinalam no entanto a presença dos segundos, dado que obtiveram reacções positivas com os reja gentes gerais deste grupo de compostos.
1,7.- ESTUDOS FAPAfACODINAMCOS
Alguns povos nativos de Africa servem-se da casca e do fruto de Burkea africana HOOK para matar os peixes por lançamento daqueles produtos nos cursos de água (14),
A acção ictiotóxica da casca poderia ter suscita do o interesse dos farmacologistas para estudar esta planta.
Apesar disso, apenas temos conhecimento de um ú-nico estudo sobre as propriedades farmacodinâmicas desta espe'cie, o qual foi realizado no Laboratório de Farmacodi-namia da Faculdade a que pertencemos, por Correia da Silva e colab. (lQ), a nosso pedido e depois de nós termos averi guado a presença de alcalóides indólicos na planta.
Estes autores ensaiaram o infuso da droga sobre a pressão sanguínea em gatos, cães e coelhos, sobre o mus culo recto abdominal da rã, preparação do frénico-diafrag-ma do rato, intestino isolado do coelho e cobaia,ainda,so-

bre o útero isolado de cobaia e coelha. 0 efeito mais marcado da droga foi observado so
bre a pressão sanguínea. Na realidade, em todos os animais provocou uma forte subida na pressão sanguínea, a qual,por vezes era precedido por uma ligeira hipotensão.
De certo modo, a verificação deste marcado efeito hipertensor da droga constituiu um incentivo para prosseguirmos com o estudo quíaiico dos alcalóides que vínhamos empreendendo.
Como veremos mais tarde, um dos alcalóides isola dos poderá ser o responsável por aquela acção,
Parece-nos, também de assinalar a acção que o in fuso determina, em doses convenientes, sobre o coração is_o lado da rã. De facto, observa-se um aumento ligeiro 'las cori tracções cardíacas, com diminuição da tonicidade e, em doses um pouco mais elevadas acentuada bradicardia e aumente da amplitude das contracções.
BIBLIOGRAFIA
1 -HOOKER, J .B = I o . r i , VI, 393,595 (3943) . 2 - OLIVIER, D-> P i o r a Trop ica l África,, L o n d r e s / T o l . I I .
(1871) . 3 - HIERN, Cat , Afr. P I . Welw. 1. ,304 (1896). 4 - BAKER, E. G.,The Leguminosa© of Trop ica l A f r i c a , d i t a s
P r e s s , 0 s t e n d ( l b 3 0 ) 0 5 - TORRE, A. R. e HILLCOAT, em EXELL. A. W. e MENDOLÇA,F.

24
6 - BENTEAM, G. e HOOKER, J, D,- Genera Plantarum, Vol. I (1892/96).
7 - HUTCHINSON, J., The genera of flowering plants, the Clarendon Press, Oxford, Vol.1.(1964).
8 - WHITE, P., Forest Flora of Northern Rhodesia, Oxford University Press (196!),
9 - ENGLER, A., Syllabus der Pfianzenfamilien,Berlin,Vol. II (1964).
10 - PARKES RILEY, H., Families of flow-ermg plants of Southern Africa (1963.).
11 - WILCZEK, Flora du Congo Belge e du Ruanda-Urandi,Bruxelas, Vol. Ill (1952).
12 - Agronomia Angolana, Luanda, ne. 7.(1953). 13 - DALZIEL, J. Ï/U, The Useful Plants of West Tropical
Africa (en Apendix to the Flora of West tropical Africa de J.HUTCHINSON e J. M. DALZIEL), 2». edição (1955.).
14 - WATT e BEEYER-BEANDWIJK, Medicinal and Poisonous Plants of Southern and Eastern Africa, E. & S. Livingstone Ltd,Edinburgh and London,25 edição (1962).
15 - MARX,T.He; Tropenpflanzer, pag 227-228 (l93o). 16 - HOWES, F. N. - Vegetable gums and resins, Waltham Mass
Chronica Botânica C§ (1949) . 17 - ISTAS, J. R. e RAEKELB00M, F. L. - Bull. Agric. Congo
Beige, 50,697(1959). 18 - CORREIA DA SILVA. A. C., PAIVA, M. Q., COSTA, A.
Anais da Faculdade de Farmácia do Porto XXVIII, 85-112 (1968).

P A R T E I I

27
1 . - ALCALOÏDES INJX3LIC0S
1.1.- GENERALIDADES
A descoberta do indol remonta há. maia de um século. De facto, BAEYER anunciou, em trabalhos publicados em 1866 e 1868 (l), tê-lo descoberto no decurso das inves_ tigações que vinha empreendendo sobre a isatina, tendo a sua fórmula de estrutura sido proposta por este autor e por EMMERLING (2).
2" certo que à data da descoberta do indol já se tinham assinalado alguns alcalóides em plantas, mas só mui to posteriormente se reconheceu serem alguns deles de nat_u reza indólica.
Constituem exemplos deste tipo de compostos a _es~ tricuina e a brucina, descobertas, em 1819, por PELLETIER e CAY-IHTOIJ (3,4)$ a harmalina (5) e a harmina de Peganum harmala L_. « e fisostigmina, t isolada, em 1864, por JCBST e HESSE (7)t das sementes de Physostigma venenosum EALF.
A ocorrência de indol livre nas plantas, se bem

28
que inicialmente contestada, veio a ser provada por ÏÏESSE (8), ao identificá-lo como um dos constituintes dos óleos essenciais de várias espécies de Jasminium e de Citrus, En tretanto, o constituinte mais simples de natureza indólica a ser primeiramente identificado nas plantas superiores pa rece ter sido o escato1 (9),reconhecido na madeira da plan ta javanesa, Ceitis raticulosa MIQ,, em 1899.
Um ano depois seguiu-se a descoberta por GÏÏESÏÏGFF (IO), da primeira "base indólica, a hipaforina,na3 sementes de Erythrina hipaphorus BOERL.
0 triptofano, aminoácido isolado, em 1903 da ca seína (li), não tardou a ser identificado nos vegetais e a ser considerado como o derivado indólico neles predominante.
Dia após dia, e à medida que o progresso técnico se acentuava, novos compostos indólicos foram sendo descobertos» Alguns, revelando-se comuns a animais e vegetais, permitiram que, da comparação dos seus metabolismos,os bio_ químicos e os fisiologistas tirassem proveitos mútuos.
Os efeitos hormonais de alguns compostos indólicos, como o ácido indolilacótico-hormona de crescimento pa ra as plantas, e as propriedades neurohumorals dos hidrox-indois para os mamíferos, constituíram os motivos mais importantes do prosseguimento do estudo destas substâncias.
No reino vegetal, é, sem dúvida, no domínio dos alcalóides indólicos complexos, que se têm feito os maiores

2 y
progressos nos últimos anos. Estes dizem respeito ao isola mento de novas bases indólicas, à elucidação das suas estruturas químicas, ao estudo das suas propriedades farmacjD lógicas e terapêuticas e à sua biogénese.
lia história da investigação dos alcalóides indo licos ficou célebre a data que assinala a descoberta da re-serpina, não só pelo interesse das suas propriedades terapêuticas mas também pelo incentivo que provocou no estudo destes compostos.
De facto, após o isolamento desta substância da planta indiana Rauwolfia serpentina BEIITH^realizadO'em 1952, por MULLER, SCHLITTLER e BEIN, (l2) mas, mais especialmente, depois do seu estudo farmacológico e da demonstração da sua utilidade como droga hipotensora e em clínica psiquiátrica, os químicos lançaram-se na procura deste alcalóide em outras espécies.
Os ensaios de prospecção, desde então levados a cabo sobre o género Rauwolfia, com o fim de descobrir outras espécies igualmente ricas naquele alcalóide, tiveram como consequência a descoberta de numerosas outras bases.
Ass::m, em 1956, isto é, decorridos apenas quatro anos após a d> acoberta da reserpina, conheciam-se 40 alcalóides des Ra; wolf ia,
Vários outros géneros, tais como Vinca,Voacanga, Picralina, Mitragvna, Uncaria, etc., foram objecto de meti culosas investigações e, como resultado dis30,o número de

30
alcalóides indólicos, que em 1952 (13) pouco passava duma escassa meia centena, elevou-se, em 1960, (14), para ce_r ca de 350, havendo já, nessa altura, duas centenas deles cuja constituição química era perfeitamente conhecida.
A descoberta de vincaleucoblastina (l5,16s.l7,18, 19), alcalóide indólico de acção antitumoral, isolado de Gafaharanthus roseus (L«) G. DON, originou novo e poderoso impacto no prosseguimento do estudo destes compostos, para o qual muito tem contribuído a técnica de espectrometria de massa, que nessa altura começou a manifestar-se promete dora na análise destas substâncias. 0 facto é bem patentea, do na obra de HESSE (20) (1964), na qual o autor inscreve 511 alcalóides indólicos.
Hão parou aqui a poderosa actividade das escolas que se dedicam à investigação deste assunto, antes pele con trário ela tem-se intensificado e, como e natural, as espé cies que têm merecido maior atenção são as que manifestam actividade farmacológica.
Assim, a Vinca rosea RBICHB, o Curare de cabaça, a Rauwolfia vomitória AFZEL, a Vallesia dichotomy RUIZ e PAV., Unteria ebúrnea PICHOÎf, Vinca minor L.,Vinca major L., Stryohnos toxifera E. SGIiOi.íB, Pleiocarpa inutica BBHTH.,,P,tubi oina STAPF, etc., apontam-se entre as mais exaustivamen te estudadas. Algumas delas mostraram conter um elevado nu mero de bases indólicas de estruturas diversas e complexas.
Estão descritos hoje mais de 800 alcalóides indo

31
licos, distribuídos por cerca de 80 génaros pertencentes na sua maioria às famílias das Apocy.nacee.9 (subfamília das Plumerj.oideae ), Lo. aniaceae e Rubiaceae (2l).
Embora estas três famílias sejam, fora de dúvida, as mais ricas nestes compostos, elas encontram-se ainda no_u trás famílias filogenèticamente distantes daquelas, como, por exemplo, nas Ânnonaceae (22), Euphorbiaceae (23) Sapo-taceae (24), Biffnoniaceae (25), etc. e recentemente também foram isolados de algumas Alangiaoeae (26-29) e Icacianace-ae (30), etc.
BIBLIOGRAFIA
1 - BAEYER e KNOP. Ann. 140, 1 (1866); BAEYER, Ber.1,17 TÎ868).
2 - BAEYER e EMMERLING, Ber . 2 , 679 . (1869) . 3 - PELLETIER e CAVENTOÏÏ. Ann. Chim. ( P a r i s ) 10, 144
(1819) . 4 - PELLETIER e CAVENTOU, Ann c_Chim.(Paris) 12, 117 (1820). 5 - GOEBEL, P . , Ann. 38, 363 (1841). 6 - PRITZSCHE, J . , Ann. 64, 360 , (1848) . 7 - JOBST, J . e HESSE, 0 . , Aim. 129, 115 (1864) . 8 - HESSE, A. , Ber . 32,261Í~[Í889) 5 i b i d . , 33, 1585 ( l 9 0 0 ) . 9 - DUNSTAN, W.R., Proc.Roy.Soc. (London) 46 , 211 (1889) .
10 - GKSSHOFF, M., Ber . 23 , ~3537 (1890) . 11 - HOPKINS, F.G. e COLE, S.W., J . P h y a i o l . , 2 9 , 451 (190 3). 12 - MTJLLER, G., SCHLITTLER, E. e BEDT, H . J . E z p e r i e n t i a b,
338 (Tãsiy. 13 - MANSKE, e HOLMES, The Alkaloids - Chemistry and Physio-
logy, Academic Pres3 Inc.,New York, Vol. 11(1952),

32
14 - BOIT, KANS.-G,,, Ergebnisse à e r alkaloid-Chemie b i s I960 , Akademie - Verlag - B e r l i n
(1961) . 15 - NOBLE, R.L . , BEER, G,T. e CUTTS, J .M. , Azin.JM.
Acad, S o i . 76, 893 (1958)1 i b i d . , Biochem, Pharmacol. 1, 34? (1958) .
16 - SVOBODA, G.H., J , Am. Pharm. Assoc. S e i . Ed. 47 , 834 (1958) ,
17 - NEUSS, N . , GORMâN, M,, SVOBODA, G „11., MâClAK, G. e BEER, C.T. , J . Am. Cheiru Sçc . 81 , 4754 (1959) .
18 - CUITS, J 5.,BEER, G .T . e NOBLE, R.L . , Cancer Res. 20, 1023 (1960) .
19 - SVOBODA, G .H. , Lloydia 24, 173 ( l 9 6 l ) . 20 - HESSE, M., I n d o l a l k a l o i d e in Tabe l len Spr inger -
-Verlag - B e r l i n , Gõt t ingen , He ide lberg (.1964).
21 - Le MEN, J . e TAYLOR, W . I . , E x p e r i e n t i a 2 1 , 508 (1965) .
22 - BUZAS, A. e EGNELL, C , Ann. Pharm .Franc, 23, 351 (1965) .
23 - LAUTER, W.II, e FOOTE, P.A. , J . Am, Pharm. Assoc. Sei., Ed. 44,561 (1955) .
24 - PARIS, R#) COUTAREL, R., Ann, Pharm, Franc. 16, 15 XÏ958),
25 - FERREIRA. M.A,, ALVES, A.C., PRISTA, L,N., Garcia de Orta 1 1 , 477 (196377"
26 - POPELAK, A. , HAACK, E . , SPINGLER, I I . , Tetrahedron L e t t e r s . 1081 (1966)? i b i d , , 5077 (19667.
27 - BATTERSBY, A.R, , MERCHANT, J . R . , RLVEDA, E.A. , SAL GAR, S . 3 . , Chenu Comm. 315 (1965) .
28 - BATTERSBY, A.R. , FAPIL, R„S.., BHAKTMI, D,S„,POPLI, S.P„, MERCHANT, J 0 R o , SALGAR, S . S . , Te-t rahedron L e t t e r s 4965 (1966) .
29 - PAKASHI, ' 3 . 0 . , Current Science 35, 468 (1966) . 30 - MONTEIRO, H , , BUDZIKIEWIOZ, H . , BJERASSI, C.,ARNDO?,
R.R, e BAARSOHERS, W.H., Chem.Comm. 317 (1965) .

"3
1.2.- DISTRIBUIÇÃO BOTÂNICA DOS ALCALÓIDES INDdLlCOS M S LEGUMINOSAS
A3 Leguminosas, conquanto não muito ricas em alcalóides indólicos, também os possuem, se cem que até ao momento apenas tenham sido isolados destas plantas compostos de constituição relativamente simples.
Na realidade, entre os milhares de espécies que constituem o vastíssimo conjunto das leguminosas, somente cerca de 60 delas, pertencentes a 14 géneros, revelaram, até à data, conter, estes constituintes, todos eles pertencentes a um dos grupos seguintes;
Grupo I - Bases indólicas simples
^Q-nn R2 R3
1 Grupo II - Alcalóides do núcleo dep -caihò-
lina '

34
Grupo III Alcalóides do tipo fisostigminaggnsserina
R 3
^1 /^H'
i k 3 2
No QUADRO I seriámos, por . crdem alfabëtiea as espécies que a literatura refere conterem estes compostos, indicandose ainda a subfamília a que pertencem, os alcalo'ides delas isolados e o grupo químico a que per tericem,
Q U A D R O I LEGUMINOSAS CONTENDO ALALOIDBS INDOLICOS
ESPÉCIE 03
! H PH "i5vi (5 a it.
Abrus precatorius L. (l) Acacia acuminat a BENTÏÏ.(2) â* oardiophylla A, CUM
ex, B J M U (iá)
^ complanata A. CUNN ex. BKMIH*(7)
''♦ confusa MERR, (3)
c u l t r i f o r m i a A.CUM ex . G. DON (A)
P M M
M
o
lã I
I
I I
I
I
ALCALÓIDE ISO
LADO
Abrina Triptamina Triptamina
Leptocladina Eleagnina Dipterina Nigerina Triptamina

35
Acacia floribunda SIEE. (5) A. longifolia WILLD. (4) A. maidenii F.MUELL (6)
A .
A . phlebophyla p.M m&Up)
A.
podalyriaefolia A.
pru inosa A. CUNN ex. BENTfi. (5)
A- ves t i t a KER-Gl^L (2)
Desraodium gangeticum D.C,(12)
Deaaiodiiun pulchellum BENTH, ex. BAKER (9,10,11)
M I M I
M I I
M 1
M I
M I
M I
P I
I
I
I
I I
I I
P I
I
Triptamina Triptamina Oipterina Nigerina Nigerina
Triptamine
Triptamina
Triptamina H. ,K. -Dimetil-b b t r i p t amina
N, ,N. - D i m e t i l -b b
t r i p t a n i n a - N , -oxido 5-Metoxi-N, ,N, -b b - D i m e t i l t r i p t a -mina ;
5-Metoxi-N, ,N, -b ' b - D i m e t i l t r i p c a -
mina-N. -óxido b N. -Met i l t e t r a i -droarmana
Cat ião 6-Metoxi -Nb -met i 1 ft - c a r -bo l ina
Gramina
Niger ina

36
De s ao dl um pui she Hum BENTH. ~ ex . BAKER '
Dicorynia gu ianens i s (13)
Dioclea b i c o l o r BEÎWH.(14)
D. l a s i o c a r p a BETO.Ij(l4)
D. macrocarpa 11116,(14)
T). r e f l e x a HOOK.( 14)
D. v i o l á c e a MART. ex, BEOTH, (14)
E r y t h r i n a a b y s s i n i c a LAl(l5)
E.
E.
acanthocarpa E.MEY. TieT' americana MILL»ou
E. ou
cárnea AIT,
E. be r t eo roana URB. JÏ5)
p I
I I
I
I
M I
P I I I
P I I I
P I I I
P I I I
P I I I
D I
P I
P I
P I
P I
N , K , - D i m e t i l t r J tamina ïï, -óxido b
Bufotenina 5-Metoxl-N^-me-t i l t r i p t a m m a
5-Met oxi-N-b, N-jj-- d i m e t i l t r i p t a m i
na
5-lVIetoxi-Nb5N-b--dimet i l t r i p t ami
na-N-^--óxido
Tr ip tamina
Fioos t igmina
ïïipaforina

E r y t h r i n a c o s t a r i c e n s l á ! fcUMIGHELI (16)
E.
E.
E.
c r i s t a - g a l l i L.(17)
Ej domirtguezii HASSLER (16)
exce l sa BAKER#(22)
E. f a l c a t a BENTS( 18)
E. flabelliformis KEABN (15;
E* folkersii KRU-KOFF e MOLDÏÏNKE
E. fusca LOUR, ( l ? )
E. g lauca WILLD.(lQ)
E. g r i s e b a c h i i ÎJRB. (19)
E" herbácea L. ( l 5 )
E. hipaphorus BOERL. " ex . KOOED.
ou subumbrans MERRÏ.L
ou Hipaphorus subumbras HASSK.
C2Õ) E r y t h r i n a indica_LAM.
ou E. variegai a var.
orientalis L.(24)
' macrophylla D.C.

38
Erythina orophylla GHESQj(22)
E. pallida BRITTON e ROSE (15)
E» poeppigiana O.F. COOK (15) ■
E.
E.
rubrinervia H.B. e K. (16) sandwicensis DEGNER
E. (23)
senegalensis D.C,
E.
E.
Tie) tol loniana HHA. (2l)
velutina WILLD.(l9) Lens esculenta(MOENCH>ŒffH.
T25J ou L. culinare
Lespedesa Taicolor var . japonica HHiïï.(YAMAHAGI) ( 26,27,
28)
Lupinus luteus L. (29) Mimosa h o s t i l i s BEMffi.(31,32)
p I
p I
p I
p I
p I
p I
p I
p I
p I
p I
p I M I
Hipaforina
Triptamina
Uigerina' Nb*NVB±fliot4Utri pt amina1\ - óx idõ Lespedamina Bufotenina 5MetoxiN^, N dimet i l t r ip t ami na 5Met oxiN^, Kjjdimet i l t r i p t ami naH^óxido Gramina Nigerina

ÒS
Mueuna pruriens D.C. (30)
Petalostylis lapicheoides R. BR. (34) P. labicheoides Tar.casseoid.es BEBTa.(33)
Physostigma cylindrospermum HOLMES (35)
P . venenosum BALP. (36,37)"
Piptadenja colu"brina BENTH, (321
P. excelsa LILLO Ç 3 B T -
P . macrocarpa BENÏB-. (38,39)
P
C
M
M
I Serotonina
I I Tetraidroaraiana-
I Triptamina I líigerina I I Tetrt idroarmrna-
III Pisostigmina
III Piacatigniina III Geneserina III Eseramina III Pis over; ins. I l l Eseridir.a I I I N-8-Norfisostig-
mir.a I I I Calabatina I I I Calabacina
I Bufotenina
I '• I Butotenina-N^-
-óxido I iNn-jerina
I Ni^orina I J5-Mexoxi-N-b-me-
jtiltriyUUnltia

40
Piptadenia, mac roc arpa M I Bufotenina BENXH. (38,39) I B tCot enina N,
óxido B tCot enina N,
óxido r • pe reg r ina M I
I Dip te r ina Niger ina BEMIH »(39,41)
M I I
Dip te r ina Niger ina
I NbfNjjDimetiltri, ptcmina N^óxido
I 5Met oxiNjjmet i l t r i p t a m i n a
I 5lVietoxiHb,Nbd i m e t i l t r i p t a m i na
I Bufotenina
Prosopis j u l i f l o r a D.G. (42) M I
I Triptamina
Serotonina
M I
I Triptamina
Serotonina
Pul tenaea a l t í s s i m a F.MDBLL I E s t e r m e t í l i c o do Njjdimeti lL t r i p t o f a n o
e x . BEHUfí.(43) I E s t e r m e t í l i c o
do Njjdimeti lL t r i p t o f a n o
1 ■
G representa subfamília das Gaesalpinoideae M " " " Mimosoideae P '' " " Papilionoideae

41
BIBLIOGRAFIA
1 - GHATAK, N. e KADL, R., J , Indian Ohem.Snp. 9 , 383. (1932).
2 - WHITE, E.P., New Zealand J. Soi. Tohnol. B. 38, 718 (1957) .
3 - LOU, V. , KOO, W. - Y. , e RAMSTAD, E..OoydJLa 28,207 71965).
4 - WHITE, E . P . , New Zealand J . S e i . Technol . B. 33 , 54 (1951) .
5 - WHITE, E . P . , New Zealand J . S e i . Technol . B . 25,157 (1944).
6 - FITZGERALD, J.S. e SIOUMIS, A.A., Aust. J. Chem. 18, 433 (1965).
7 - JOHNS, S.R., LAMBERTON, J.A. e SIOUMIS. A.A..Aust. J . Chem. 19, 1539 (1966) .
8 - ROVELLI, B. e VAUGHAN, G.N., A u s t . J . Chem. 20 ( 6 ) , 1299 (1967) .
9 - GHOSAL, S. , e MUKHERJEE, B . , Chem. and Ind.(London) 793 (1964) .
10 - GHOSAL, S. e MUKHERJEE, B . , J . Org. Chem. 3 1 , 2284 (1966) .
11 - GHOSAL, S. e MUKHERJEE, B . , Chem. and Ind . (London) 1800 (1964) .
12 - BANERJEE, P . K., GHOSAL, S . , Aus t . J . Chem. 22 ( l ) 275 (1969) .
13 - SANDERMANN, W. e LANGE, W., Naturwisaenschaften.54 (10), 249 (1967).
14 - TREISE, Fharm. Z e n t r a l h a l l e 77, 378 (1936) . 15 - FOLKERS, K. e KONIUSZY, F . , J . Am. Chem. Soe. 62,
1677 (1940) . 16 - FOLKERS, K. SHAVEL, J . e KONIUSZY, F . , J.Am.Chem.
S o e . . 63 , 1544 (1941*77"

42
17 DEULOFEU, V., HUG, E . , MA.ZZOCCO, P . , J . Chem. Soc. 1841 (1939).
18 GENTILE, R.A. e LABRIOIA, R., J. Org. Chem. 7,136 (1942).
19 _ POLKEES, K. e KONIUSZY, K., J. Am. Chem. Soc, 62, 436 (1940).
20 GRESHOFF, M., Ber. 23, 3537 (l890). 21 LAPIERE, C , Bull» Soc. Chim. Biol., 31, 862 (1949) 22 FOLKEBS, K., SHAVEL, J., J. Am. Chem. Soc, 64,
1892 (1942). 23 FOLKERS, K., KONIUSZY, F. J. Am. Chem. Soc, 61,
1232 (1939). 24 MARANON, J. e SANTOS, J. K., Philippine "J. ^ k '
48 , 563 (1932) ; C.A. 26, 5609 (1932). 25 PILET, P . E . , Rev. Gen. B o t a n . t 65 , 605 (1958) . 26 GOTO, M., NOGUCHl, T. e WATANABE T . , J.Pharm.Soc
Japan, 78, 464 (1958) . 27 MORIMOTO, H. e OSHIO, H . , Arm. 682, 212 (1965) . 28 MQRIMÛTO, H. e MATSUMOTO, N.,Ann. 692, 194(1966). 29 SPARATORB, P . , Ann. Chim. (Rome) 54, 246 (1964) . 30 BOWDEN, K., BROW, B.G. e BATTY, J . E . , Nature 174,
925 (1954). 31 LIMA. O.G. Arquiv. Inst. Pesquisas Agron.(Pernambu
H o ) T , _ 4 5 (1946) . 32 PACHTER, I . J , , ZACHARIAS, D.E. e RIBEIRO, 0 . , J . O r g .
Chem., 24, 1285 (1959) . 33 JOINS, S.R. , LAMBERTON, J .A . e SIOUMIS, A.A.,
Aust . J . Chem. 19,893 (1966) . 34 BADGER, G.M. e BEECHAM, A.E. , Nature , 168, 517
(1951)., 35 HOLMES, H . L . , Pharm. J . 9 ( 3 ) . , 913 (1879) . 36 ROBINSQN, B . , J . Chem. S o c 1503 (1964) . 37 MANSKE, R.H.F . , Academic P r e s s , Kew York London
Vol. V I I I , pag. 48 (1965) e Vol . X, pag. 383 (1968) .
38 JACOBUCC.I,G.A. e RUVEDA, E.A. , Phyt o chemis t ry 3 , 465 (1964) .
39 FISH, M.S. , JOHNSON, N.M. e HORNING, B . C . ; J . Am. ■■■■; Chem. Spc. 77, 5892 (1955) .

43
40 - LEGLER, G. e TSCHESCHE, R. , Naturwissenschaften 50, 94 (1963) .
41 - STROMBERG, V.L. , J . Am. Chem. SQC. 76, 1707(1954) 42 - PISH, M.S. , t r a b a l h o não publicado (1958) a t r a v é s
de STOWE, F o r t s c h r . Chem. Org., Natu-r s t o f f e 17, 248 (1959) .
43 - FITZGERALD, J . S . , Aus t . J . Chem.- 16, 246 (1963) .

45
1.3.- CONSTITUIÇÃO QUÍMICA DOS ALCALÓIDES INDÓLICOS ISOLADOS DE LEGUMINOSAS
1.3.1,- Generalidades
O critério inicialmente seguido para classifi car os alcalóides indólicos consistiu em criar grupos em função das suas origens botânicas.
Assim procederam, por exemplo, os clássicos tratadistas HENRY (l) e MANSKE (2),critério ,que este.úl timo autor ainda mantém no vol. VII (3) da sua valiosa obra sobre alcalóides.
À medida, porém,que se foram elucidando as suas estruturas, a circunstância de um mesmo alcalóide ter si do caracterizado, simultaneamente, em várias espéoies próximas ou não? o facto, assaz frequente da coexistência, numa mesma espécie, dum elevado número de alcalói des indólicos e ainda a circunstância de estes, embora com a mesma proveniência botânica, possuirem estruturas e acções farmacológicas diversas, que os separam entre si e os aproximam mais de outros já conhecidos, têm pro vocado sensíveis alterações no critério primitivamente adoptado de classificação destes compostos.
A tendência actual parece ser a de agruppá-lcs de acordo com as suas semelhanças estruturais, o que é sem dúvida mais lógico e torna possível uma sistematiza

46
ção mais racional do seu estudo (4c-6). No entanto, alguns autores, talvez por conve
niência de exposição, adoptam um critério misto, botâni co e químico (7,8), para agruparem os alcalo'ides indóli cos, enquanto que outros se decidiram, ultimamente, por uma classificação atendendo especialmente à sua nature-aa química (5,6).
Uma vez que na planta que estudámos caracteri zámos uma indolalquilamina e derivados. (3-carbolínico3, serão estas unicamente as bases indólicas que considera remos neste capítulo.
lo3«2-> Bases indólicas simples
Este grupo de compostos, como o seu nome suge_ re, inclui os alcalóides indólicos de estrutura mais sim plificada, os quais possuem, além do núcleo benzopirró-lico, um substituinte alquilamínico em posição _3. Por tal facto, estas bases são correntemente designadas na literatura, de modo genérico, por indolalquilaminas e Por indol-3-alquilaminas, 3-aminoalquilindóis ou mais especificamente por 5-indolalquilaminas.
As indolalquilaminas existentes nas Leguminosas são, sem excepção, derivadas do triptofano ou do 5--hidroxitriptofano.
A abrina, a hipaforina e o ester metílico do
)

47
N-h-dimetil-L-triptofano podem considerar-se derivados di rectos do triptofano, por metilação na cadeia lateral deste aminoácido.
As outras bases deste grupo poderão filiar-se naqueles dois aminoácidos, por descarboxilação, com encurtamento ou não da cadeia etilénica e metilação.
Esta metilação pode ocorrer no hidroxilo liga do a C-5, quando este existe; no azoto do núcleo indóli co, Naf ou no azoto da cadeia lateral, Nb, onde, aliás, é mais frequentemente observada.
Assim, o esqueleto molecular comum a estas su bstâncias é o seguintes
(CH2) r (br^3
n = 1 ou 2
\ - H ou 0CH3
Rg= H ou CH3
R3= H ou CH3
%= H ou OH ou 0CH3
A Êfjj pode l i g a r - s e um átomo de oxigénio (Nv-óxidos)

48
Registe-se, desde j á , que os únicos compostos correspondentes a n = 1 isolados de plantas sãos a g ra -mina e os derivados 3-(aminometil) indol, 5-(metilaminç>-metil) indol e 1T5-dimetoxi-5-(dimetilaminometil) indol. Destes compostos apenas a gramina fo i , a té à presente _da t a , assinalada nas Leguminosas e o último encontrado nu ma Laurácea, Gymnacranthera paniculata var . zippeliana..
(9 ) . Nesta sequência, e se atendermos ao QUADRO I
(pag. 34-40), podemos agrupar as bases indólicas simples existentes nas Leguminosas nos t r ê s subgrupos seguintes;
I - Hipaforina, abrina e és te r metíl ico do ffh-dimet i l -L- t r iptofano
I I - Gramina
I I I - Triptamjna e .derivadojg.
I - Hipaforina. abrina e é s t e r metílico do W-h-dimetil-L-triptofano
A hipaforina ( I I ) foi a primeira base indól i -ca simples a ser descoberta nas Leguminosas. Deve-se a GRESHOFF o seu isolamento, em 1890, a p a r t i r das sement e s de Erythrina hipaphorus BOERL (lO) e principalmente

49
a ROMBURGH (li) o estudo das suas propriedades e consti tuição.
Este investigador, após ter determinado algumas das suas características físicas, formulou a hipóte se da sua estrutura, induzido pelo cheiro fétido manifçe tado pela madeira da planta.
ROMBURGH admitiu que o cheiro da madeira de Erythrina hipaphorus se devia ao indol e à trimetilami-na resultantes da decomposição da hipaforina e daí admitir que a base devia ser a betaina do triptofano,o que ele próprio confirmou, ao realizar a síntese da substância, verificando que a hipaforina, na realidade, por a-quecimento com hidróxido de potássio originava aqueles dois subprodutos.
r^N i H
-CH2-GE-COO aN(GH 3) 3
0
II
Abrina (III) foi o nome dado a uma substância amorfa isolada das sementes do Abrus precatorius L.
M o obstante a crítica feita por MILLER e RCS SON (12) ao nome dado a e3ta substância (abrine, em inglês), pela confusão que pode ocasionar com o da proteí

50
na t ó x i c a a b r i n , que também e x i s t e na p l a n t a , t a l desig_
nação temse conservado.
GHATAK e KA.DL (13) obtiveram, em 1932, aquela
base no estado c r i s t a l i n o e EOSHINO, ( l 4 ) d e c o r r i d o s t r ê s
anos, es t abe leceu a sua fórmula de e s t r u t u r a .
Por desca rbox i l ação , a abr ina or ig inou 3(me
t i l a m i n o e t i l ) indo l opticamente ac t ivos ( ^ )j) + 46 (íICl).
Alem d i s s o , or ig inou um n i t r o s o e um a c e t i l der ivado ,o
que mostrou a e x i s t ê n c i a dum átomo de azoto secundário»
Gomo, por outro l ado , a met i lação do t r i p t o f a
no com i ode to de m e t i l o , em presença do hidróxido de só.
d i o , levou à obtenção de um composto i dên t i co ao que re
s u i t a do t ra tamento daquela base em iguáiJs ■ciréítnstan
c i a s , t a l f a c t o pe rmi t iu e s t a b e l e c e r que a ab r ina co r
respondia ao N b m e t i l t r i p t o f a n o ,
.CH2ÇHC00H feCH3
f H I I I
GORDON e JACKSON (15) obtiveram a forma r a c é
mica da abr ina por reacção do 5 indo la lde ído com 1metil
i d a n t o i n a , seguida de redução e h i d r ó l i s e do produ to .
CAHILL e JACKSON (16) e s t a b e l e c e r a m , p o s t è r i o r
mente, a iden t idade conf igurac iona l da a b r i n a com o L
t r i p t o f a n o , sendo, p o r t a n t o , a ab r ina o N b m e t i l L t r i

51
ptofano.
FITZGERALD ( l7) isolou, em 1963, uma base de Pultenaea alt íssima F. MDELL ex, BENTH., cuja estrutura foi determinada por espectrometria de massa.
Assim, o ião molecular mostrou possuir m/e = =246 e o espectro exibia, ainda, entro outros os picos correspondentes a m/e=187, m/e=130 e M/e=116. Por outro lado, a comparação do espectro de massa desta substân
cia com o co t r iptofano permitiu averiguar da existên
cia de fragmentos semelhantes e r eg i s t a r as respectivas diferenças quanto à localização de alguns picos e no que respe i ta às percentagens re la t ivas entre os mesmos.
Dado o facto de o pico base re la t ivo à subs
tância por s i isolada corresponder a m/e=116 e em face de outros fragmentos registados no espectro , FITZGÏÏML1) admitiu que o composto em causa correspondia ao és te r metíl:ir..q9. ào N^dlmetilLtriptofann (iv),podendo in te r
p r e t a s s e a fragmentação sofrida pela mole'cula do se
guinte modos m/e 187
m /e 150
m /e 116
■CHfCOOCH
U(CH3)j 3
m m/e 246"
7e 116
IV

52
I I - Graining
A gramina (Vi) foi isolada do extracto alcoól ico dos mutantes albinos da cevada, em 1932, por Von EULSR e HELLSTROM ( l ô ) .
Estes autores verificaram t r a t a r - s e duma subs tância de fórmula "bruta C^H^Ng» 1 u e fundia a 134 C e se comportava como uma monobase, contendo dois radicais metilo, alem de apresentar um espectro de absorção no U; T, tipicamente indólico,
OREROV e co l , ( l9) observaram que continha um hidrogénio activo e t r a t a r - s e de um composto inactivo à luz polarizada, o que fez admitir como provável a inexis tencia de carbonos assimétricos na molécula.
A confirmação da presença de um núcleo indóli_ co na molécula da gramina foi conseguida por EULERecoL (20) ao verificarem que a dest i lação desta base com zin co em pó originava esca to l . (V)
Várias hipóteses de es t rutura foram admitidas para es ta substância t a i s comos 5-metil-2-dimetilamino-indol; 2-metiI-Z-dimetilaminoindol, e t c . .

53
Todavia, aquelas duas hipóteses foram postas de lado, porquanto a oxidação da substância pelo método de KTHN-ROTH, não conduzindo à obtenção de ácido a c é t i co, excluia a possibil idade da presença de um grupo CII3
em posição 2 ou_3.
Este facto levou, portanto, a admitir a existência de uma cadeia . l a t e r a l dimetilaminometilénica
(-CB^-N-^.^3)em posição 2 ou 3 . Wig — —
A obtenção do escato l , só por s i , b a s t a r i a para supor como mais provável a subst i tuição na posição _3j de mais, sabe-se que a substi tuição ocorre mais f a c i l mente nesta posição porquanto a carga posi t iva do de r i vado indólico intermediário se e s t ab i l i s a gem des t ru i ção da ressonância do anel benzénico.
A confirmação de que a es t ru tura da gramina correspondia de facto ao 5-(dimetilaminometil)indol(Vl) foi obtida quando WIELAHD e HSING (2l)prepararam , por via s i n t é t i c a , es ta última substância e verificaram serem perfeitamente idênticos os referidos compostos.
SX >, rrCHp-K
N H 3

54
I I I - Tr iptamina e der ivados
WILKINSON (22) e s c r e v i a , em 1958, que das n o ve bases da s é r i e da t r i p t a m i n a (T) que provavelmente £ cor re r iam nas p l a n t a s ( i ; R]_=H$ R2=H ou CHj? R3=H ouCH3
e R4=H ou CE ou OCII3 pag . 4?) j á t inham s ido i s o l a d a s t o d a s , à excepção da 5-metoxi t r ip tamina (5-MeQ-S?)' e da 5-metoxi -N^,N^-dimet i l t r ip t amina ( 5-MeO-MP ) Í
No ano s e g u i n t e , com o isolamento da 5-MeO-DMT do t ronco da árvore b r a s i l e i r a Dictyoloma incanescens D„ C. ( 2 3 ) , da f a m í l i a das Rutáceas,apenas r e s t a v a por de_s c o b r i r a 5-MeO-T.
Acontece que com a de scobe r t a , recentemente
o c o r r i d a , da lespedamina ou Na-metoxi-Nh,Nfr-dimetiltri]>-
tamina e dos Nh-óxidos, as bases t r i p t a m í n i c a s i s o l a d a s
são j á em número de onze . Note -se , no e n t a n t o , que . a l
guns au tores defendem a h i p ó t e s e de que os N^-óxidos cor
respondem a produtos de oxidação espontânea das t r i p t a
minas, durante as manipulações a que é neces sá r io subme
t e r os e x t r a c t o s das p l a n t a s para se conseguir o i s o l a
mento das b a s e s .
Pa rece , no e n t a n t o , que pe lo menos alguns d e
l e s só podem s e r obt idos por oxidação mais enérg ica do
que o simples contacto com o oxigénio do a r à temperatu
r a ambiente. Outros au to res defendem a t é que a forma
ção dos óxidos des t a s aminas pode t e r papel importante
na formação de c e r t o s a l c a l ó i d e s por t r a n s p o s i ç ã o a ca r

55
tinolaminas.
A triptamina (T) foi a primeira indolalqui la
mina deste subgrupo a ser isolada de plantas . Foi WHITE (24) quem, em 1944, a identif icou simultaneamente em duas acácias (Acacia floribunda SIEB ■ o 'Acaoia prui
nosa A. GUKN^se bem que à data t ivesse j á sido s i n t e t i zada e reconhecida como um produto resul tante da putre
facção do t r iptofano por acção bacter iana,
0 alcalóide foi isolado na forma de cloreto e purificado por cristalização de álcoolacetona.
0 carácter indólico da base foi evidenciado pe lo reagente de EHRLICH, pela vanilina clorídrica e pelo reagente HOPKINSCOLE.
A base obtida fundia a 116°G, era óptioàment© inactiva e os seus cloreto e picrato fundiam,respectivamente, a 246° e 244 G, características estas que coin cidiam com as descritas para a triptamina obtida por sín tese.
Depois desta data, tem sido encontrada em várias Leguminosas, como se pode ver no QUADRO I, e ainda em espécies pertencentes a outras famílias botânicas.
A segunda base triptamínica a ser isolada nas Leguminosas foi a serqtonina (5JfiEj), tendo sido a sua presença assinalada por BOWDEN e col. (25) em Queima pruriens D.C.

56
A acção farmacológica da planta e o comportamento do alcalóide dela isolado frente ao reagente de ÍHRLICH, aeido' 3uifàn#ít<í-d, reagente de JEPSON e STEVEN (reagente de ninidr ina em ácido acé t ico) , e t c . , fizeram supor ao citado autor es tar em presença de um derivado t r iptamínico.
A identificação do alcalóide foi conseguida por cromatografia ascendente em papel, no sistema n-bu-tanol-ácido acético ( lOsl ) , comparativamente com a 5-HT s i n t é t i c a e pelo seu espectro de absorção no ïï»v«(/min
=
=247 nm e 300 nm./í =273 nm, o qual é próximo do re ' ' ' max
ferido por HAMLIN e FISHER (26) para a 5-HT a pH = 5,4 ^ m i n = 2 5 0 e 2 " ™>Á max = 2 7 5 m)«
GONÇALVES DE LIMA. (27) publicou em 1946 um in teressante trabalho sobre Mimosa hostilis BSNTH,, relatando os efeitos duma bebida com ela preparada e usada pelos índios do Estado de Pernambuco nas suas cerimónias religiosas. 0 autor refere que a planta o continha um alcalóide a que chamou nigerina, o qual mais tarde foi identificado por PACHTER e col. (23) como sendo a N>,.Nh-dimetlitriptamina (DMT) à custa do respectivo espectro no U.V. e pelos seus picrato e metiliodeto. Aliás ? a DMT já anteriormente tinha sido caracterizada por FISH na Uimosaceae. Piptadenia peregrina.
Em 1964, JACGBUCCI (28), numa re invest Igapo sobre Piptadenia macrocarpa e numa primeira análise so-

57
bre P. excelsa, confirmou a existência de bases j á anteriormente descobertas e caracterizou uma outra, a 5-MeO-T.
GHOSAL e MUKEEJEE (£9) isolaram e iden t i f i ca ram sete indolalquilaminas em Desmodium pulchellum (QUA DRO I ) .
Os alcalóides foram extraídos da planta previamente desengordurada e separados por cromatografia em coluna de alumina de BROCKMAN. Todas as bases reagiram com p-dimetilaminobpnzaldeido, com o reagente vaní-l ico e exibiam os espectros no U.V. carac te r í s t i cos dos 3^1quiJLindjoia\_.
Três destas bases (5-metoxi-^-met i l t r ip tami-na, bufotenina e N^Wfr-dimetiltriptamina) existem na planta em quantidades diminutas e a sua separação só foi possível recorrendo à cromatografia preparat iva em pap e l .
As identif icações foram fe i t as à custa da obtenção de derivados, em função dos valores de Rf em cro matografia em papel, e por espectrofotometria.
A bufotenina ou mapina (5-Œi-DMT) representa, por ordem cronológica, a t e r c e i r a base indólica simples a ser rattcnhecída nas Leguminosas.
A sua identificação nas sementes de Piptadenia peregrina BEMH. por STROMBERG (30) foi conseguida preparando derivados (p icra to , meti l iodeto, oxalato e m-ni

58
trobenzoato), bem como pelos seus espectros no U.V. e no I. V.
A dedução da estrutura da bufotenina foi efe£ tuada por WIELAKD e col. (3l), em 1931. Estes autores, tendo em conta a semelhança que a substância apresentava com a hipaforina, propuseram para ela,iniciaimente ,«.
estrutura (VII) que vieram a regeitar mais tarâe,da da a diferença de comportamento desta substância e o do Fa-met ilt riptofano
De facto, um estudo ulterior permitiu averigu ar a existência de um grupo amínico terciário e possuir propriedades fracamente ácidas, que foram atribuídas a um grupo fenólico. Admitiu-se ainda a presença de um gm po íminico livre, uma vez que a base continha dois hi-drogénios activos e dava um diacetato.
WIELAKD e col. indicaram para o hidroxilo as posições j5 ou _6 como as mais prováveis, pois na natureza não eram conhecidos até à data os derivados substitui dos em 4_ ou _7.
Prepararam, então, os iodetos de amónio qua ternário (IX) da 5-metoxitriptamina e da 6-metoxitripta-mina (VIII) e a comparação daqueles compostos com o me-tiliodeto da O-metilbufotenina mostrou que este correspondia ao derivado com o CE em posição 5, ficando assim determinado que a bufotenina ê a 5-CH-DMT ou 5-(2-dime-tilaminoetil)-5-indolol.

59
CH2-CH2-Î?--Cg3 COCH
VII
+ /CH3 N^GH3 r
vin IX
FISH e co l . (32), analisando amostras de Pip-tadenia macrocarpa e P. peregrina, isolaram delas em 1955, além da j á ci tada MF, t r ê s outras bases s a bufo-tenina, o seu Nb-óxido e o MF-Ny,-óxido.
A identif icação destes compostos foi consegui da por comparação com amostras s i n t é t i c a s , por cromato-

60
grafia em papel e pelos espectros de fluorescência e na região do U.V,
Como os dois óxidos eram desconhecidos naquela altura, os referidos autores preparam-nos, a partir das bases correspondentes, por oxidação com peróxido de hidrogénio.
A Hh-metiltriptamina (MP) foi isolada,em 1963, de Fiptadenia peregrina BENTH. (33). El conhecida vulgar mente pela designação de dipterina, em consequência de ter sido isolada pela primeira vez de Girgensohnia dip-tera BG.(34,35).
A lespedamina foi assinalada por MOEIMOTO e OSHIO (36) em Lespedeza bicolor var. japonica, ao lado de outra base, DMP, que existia em pequena quantidade.
Este alcalóide mostrou ter fórmula molecular G13ÏÏ1QN2O. p.e. = 113 - 114°C, dar com o reagente de EHRLIGH cor vinosa e ser opticamente inactivo.
0 espectro no U.V. (/( - 278 e 291 nm), o espectro no I.V., bem como a reacção com o reagente de EHRLICH tornavam provável uma estrutura indólica para este alcalóide.
Por comparação do espectro de ressonância mag nética nuclear da substância com o da DMP, os autores pu deram concluir pela falta, na molécula, de um protão no N§, típico do indol (2,27-3,13 ltf)mas que em compensação mostra a existência de três protões 'típioes do CH3O

61
(6,03cj). Os valores de J dos outros protões eram idênticos para as duas substâncias. Daí concluiram que a lespedamina possuía um grupo metoxilo em alguns pontos da arquitectura indólica da DMF.
Ora, os derivados metoxilados nas posições 4_; 5; 6 e 7 eram já conhecidos e não correspondiam à lespedamina. Restavam três possibilidades para a localização do OCÏÏ3: em li 2. ou _3. Se o grupo metoxilo estivesse em C-3, a lespedamina seria uma indolenina. Ora o seu espec tro no TJ.V. (/ = 278 e 291 nm) é semelhante ao 1,5-w ^ max -dimetilindol (A = 282,5 e 292 nm ) e não ao da in-max doleninaí/. sensivelmente igual a 260 nm). ' max
A hidrogenação catalítica da lespedamina (x) com paládio e carvão conduziu à obtenção da DMP (X.Ï)
r N OU Fd/G
( LiA]H4
CH3
XI
Além disso, no espectro do I.V. faltava a ban da em 3.490 cm--'-, característica do HH indólico,o que veio justificar a falta do hidrogénio Na indólico tam-

62
têm observada no espec t ro de R.íi.N. Ta is f ac tos depõem a favor da e s t r u t u r a 1-metoxi-N h ,N^-dimeti l t r iptamine, pa
r a a lespedamina. No QUADRO I I indicamos a e s t r u t u r a dos de r iva
dos t r i p t a m í n i c o s i so l ados dte Leguminosas que encon t ra
mos r e f e r i d o s na l i t e r a t u r a .
QUADRO I I
ESTRUTURA QUÍMICA DA TRIITAMINA E DERIVADOS
ISOLADOS DE LEGUMINOSAS
R
^ N-
-CHg-CHgJU
Nome do Composto
Abrevia tura adoptada mais correntemente na l i t e r a t u r a
RADICAIS ; Nome do Composto
Abrevia tura adoptada mais correntemente na l i t e r a t u r a % R2 % R4 ÏÏD
Triptamina
D i p t e r i n a
Niger ina
Sero tonina
T
MT
DMT
5-HT OU5-0ÏÏ-T
H
H
H
H
H
CH3
CH3
ïï
H H
CH3
ïï
ïï
ïï
ïï
Œ

63
Bufotenina 5-CH-DMT H CH3 cn3 CH
Lespedamina 1-MeO-DMF 00¾ CH3 CH3 H
5 - l b t o x i - ^ -meti l t r iptami na
5-MeO-MP H CH3 H 0CH3
-d ime t i l t r i p -mina
5-MeO-DMT H CH3 CH3 0(¾
N^N^-Dimetil triptamina-N^--óxido
DMT-¾-óxido H CH3 GH3 H Rb-0
Bufotenina--N^,-óxido
o-CH-DMr-N^xido H CH3 CH3 kR %r0
5 - M e t o x i - N ^ -- i d i m e t i l t r i p -tamina -
-N-jj-óxido
5-MeO-DMT-N^-óxido H CH3 CH3 00¾ «b-0
1.3.3,- Derivados da Q - cartolina
Foi ao estudar os alcalóides do Peranum har-mala, harmalina, harmina e harmol, que,pela primeira vez, se reconheceu a existência do núcleo da l-carholina.cons tituído pelo núcleo do indol fundido com o da piridina.
A elucidação das estruturas dos alcalóides iso_ lados desta Zigofilacea oriunda da Turquia deve-se, sobretudo, aos trabalhos de FISHER, de PERKIN e ROBINSON, que durante largos anos se dedicaram ao seu estudo.
Assim, a oxidação da harmalina com ácido azó-

64
tico fumante permitiu a FISHER otter o ácido 3-nitro-anísico (XII), o que veio confirmar as suspeitas de que aquele composto continha um anel benzénico ligado em duas posições orto ao resto da molécula e um grupo OCH3.
0 mesmo autor (38) conseguiu, aliás, demonstrar que a harmina continha um núcleo de piridina, ao isolar o ácido isonicotínico (XIIl) como produto da oxi dação do ácido harmínico com ácido azótico a 180-200 C, o que provou ainda que o núcleo piridínico devia estar substituído na posição. Y.
COCE C00H
^°2 CH3
XII Baseados nestes e em muitos outros factos,PER
KEN e ROBINSON (39) foram levados a admitir que os alcalóides de P„ h armai a eram constituídos por um anelben zénico, um anel pirrdlico e um anel piridínico fundidos entre si, ocupando o núcleo pirrdlico a posição central das respectivas moléculas.
Partindo desta hipótese, estes autores propuseram as fórmulas de estrutura seguintes para a harmina (Xiy),harmalina (XV), nor.armina (XVI), ácido harmínico

65
(XVII) a apcarmina (XVIII) .
CH30
XIV XV
XVI
HOOC
H 00'
XVII
/V
l C H 3 II
XVIII
De facto , a harmina e a harmalina t quando a-quecidas com ácido clor ídr ico fumante a 140°G, originaram, respectivamente, as bases fenélicas harmo1 e harma-lo i e cloreto de meti lo, o que indica a presença de um grupo metoxilo na molécula.
A oxidação da harmina com ácido cromico em meio acético originou um ácido - o ácido harmínioo,que, pela circunstância de formar uma f ta le ina com a resor-cina, deixou antever t r a t a r - s e de um ácido o-dicarboxí-

66
lico. Além disso, o ácido harmínico, por pirólise a alta temperatura (300°C), sofreu descarboxilação originan
Por outro lado, a acção do cloreto de zinco,_a mónia e cloreto de amónio sobre o harmol (46) convertem este em amino armaria que, por sua vez, se transformou nqu tra "base - a harmana (XIX).
Mais tarde, os mesmos autores, obtiveram a pro va de que a sequência por que tinham indicado a fusão dos três núcleos estava correcta ao verificariam que a base preparada por HOPKINS e COLE, (42) por oxidação do triptofano com o cloreto férrico em presença do éter era idêntica à harmana »
XIX
Foi então que 1ERKHÍ e ROBINSON (39,43)intro duziram na literatura química os vocábulos carbolina co mo sinónimo de norarmana. e o de pirindol para designar o fragmento constituído pelo núcleo pirrólico fundido com o piridínico.

67
Segundo a nomenclatura inicialmente adoptada por PERKIN e ROBINSON (44), a norannana era designado por 4-carbolina.
Mais tarde, GULAND, ROBINSON, SCOTT e TQHBK-LEY (45), passaram a utilizar as letras gregas , Q, Ve
fj , para indicarem a localização do átomo de azoto no núcleo da piridina nos vários tipos de cartolinas.
Em virtude dos diferentes critérios para a nu meração do núcleo da norarmana^. a literatura apresen-ta-se um pouco confusa encontrando-se como sinónimo da quele composto as designações de 4-carbolina. 3- ou ( ) carbolina e ainda 2- ou ($)carbolina.
Actualmente, porém, segundo as determinações da IM.Q.P.A., o núcleo da norannana ou(3-carbolina e numera-se como se indica na fórmula XX e corresponde-Ihe a nomenclatura ?H-pirido-|Í5,4-b-)-indol.
1 H XX
Se bem que estejam já isolados cerca de três dezenas de alcalóides derivados da 8 -oarbolina.nas Leguminosas apenas foram assinalados que saibamos,reduzi-

68
do número destes compostos. Deles nos vamos ocupar seguidamente.
I - Tirhraidroarmana ou eleagnina
XXI
lio decurso dum estudo sobre alcalóides de Leguminosas, BADGER e BEECHAM (46) tiveram oportunidade de isolar de Petalostylis labichaoides, uma substância cristalina, em forma de prismas e agulhas, com ponto de fusão de 178-180°G. 0 espectro desta substância na região do U.V. apresentava as bandas de absorção seguintes* X =225,5 nm, (log. 6 =4,57) ; £83 nm (log £=3,90) e 291 nm (log 1=3,82) e o seu derivado benzoílico fundia a 165-167°G.
Estas características, bem como a composição centesimal, coadunavam-se com as descritas na literatura para o produto já então sintetizado:atetraidroarma-na (XXI).
Os autores obtiveram prova evidente de que e-

69
fectivamente estavam perante este alcalóide,determinando o ponto de fusão misto do seu produto, sol) a forma de cloreto,com o de uma amostra sintética de cloreto de tetraidroarmana que MANSKE lhes cedera.
Por sua vez JOHNS, LAMBEHTON e SI0UMIS(4?) re ferem terem encontrado vestígios deste alcalo'ide em Pe-talostylis labicheoides var. casseoides B3NEI.
11-^h-metiltetraidroa.rmana ou leptocladina
Ko prosseguimento do estudo dos alcalóides que JCHNS, LAMBEHTON e SIOTMS (48) vinham realizando, isola ram das folhas e do tronco de Acacia complanata A.CUNN, ex. BEHEH, Nh-metiltetraidroarmar- a (XXIl), na percentagem de 0,3 g.
Além deste composto, assinalaram também a e-xistência de tetraidroarmanrv., estando presente apenas em vestígios, tendo-a caracterizado por cromatografia em camada delgada e em fase gasosa»

70
Ë de notar que a leptocladina apenas tinha si do reconhecida até essa data em ArthrQphytum leptocladiua (49), da família das Quenopodiáceas.
A W>-metiltetraidroarmana foi ohtida no estado puro por fraccionamento do extracto "bruto alcalóidi-co da planta em coluna de alumina neutra, fundindo a ha se a 109-110°C. e o picrato a 182-184°C, e apresentando (O()D °° (etanol).
A identificação do alcalóide foi conseguida por comparação com os produtos que os próprios autores prepararam por tratamento da tetraidroarmana com CH^I.Além disso, o espectro de ressonância magnética nuclear mos-trou-se conforme com a suposta estrutura (XXIl),porquan to foi possível assinalar um pico o 7,88, correspondente ao protão do W. indólico, um multipleto «6,85 - 7,55 (protões aromáticos do anel henzénico), um dupleto tri-protónico tf 1,30 e um quarteto monoprotónico a c)3,40 (devido a CH3 -ÇH) e um simpleto triprotónico a cÍ2,42 (N-GH3) e um multipleto estendendo-se de cí 2,60 acÍ3,15 (devido a CH2-CH2-).
Este alcalóide foi de novo isolado, dois anos mais tarde, em Desmodium gangeticum P.C.. por BAMEEJEE e GHOSAL (50). Estes autores fizeram a sua identificação recorrendo, entre outras determinações,ao espectro no U.V. (A m a x 226, 277 e 298 nm) e no I.V. (em KBr),ten do podido localizar uma handa em 2,95 U. (M indólico) , além de outra em 3,55 //(Nb-CHg).

71
I I I - Catião 6-metoxi-2-metil- W-carbolina (XXIII)
CH30
i H
XXIII
Da planta anteriormente referida, os mesmos autores isolaram, a par da leptocladina e de quatro in-dolalquilaminas, uma base quaternária sob a forma de rei neckato.
0 carácter do base quaternária foi detectado pelo seu pKa - 10,8; além disso o espectro no U.V, indicou existir uma certa analogia com o catião da 2--metiltetraidroarmana, pois em meio etanólico exibia os máximos de absorção localizados em 240, 250, 272-275(in flexão), 290 e 342 nm, e em meio etanólico alcalino(0,05 li) observava-se um desvio batocrómico de 10 nm nas bandas de maior absorção.
A confirmação da sua estrutura química foi conseguida por redução do alcalóide com sódio e etanol,não se observando depressão do ponto de fusão quando misturado com uma amostra autêntica de l,2,3,4-tetraidro-6--metoxi-2-metil-fí -carbolina. o que levou a concluir pe

72
la existência de grupo metoxilo, em posição _6 no núcleo benzénico,
BIBLIOGRAFIA
1 - HENRY, T„A.,The Plant Alkaloids, The Blakiston Com pany, Philadelphia-Toronto, 4s. ed. (1949),,
2 - MMJSKE, R.H.F. e HOLMES ,H*L., The Alkaloids-Chemistry e Physiology,Academic Press Inc. New York,vol.II(1952),
3 - MANSKE, RJEt.F,, The Alkaloids-Chemistry e Physiology Academic Press, New York -London, vol. VII (i960).
4 - BOIT. RCG„, Ergebnise der alkaloid - Chemie his 1960, Akademie - Verlag, Berlin - (l96l).
5 - HESSE, Me, Indolalkaloide in Tabellen.Spinger-Verlag, Berlin-Go'ttingen-Heidelberg (1964).
6 - HESSEj M., Indolalkaloide in Tabellen,Springer-Ver-lag, Berlin-Eeidelherg-New York (1968).
7 - MANSKEjR.H.F., The Alkaloids-Chemistry and Physiolo gy, Academic Press - New York-London vol, VIII (1955),
3 - MANSKE,RnH.F., TT,P Alkaloids-Chemistry and Physiolo-gy_, Academic Press - New York-London, vol. X (1968),
9 - JOHNS, S.R,, LAMBERTON, J .A . , 0CC0LCWIÏZ*J.L., Auet . J , ghem. 20 (8) (1890) .
10 - GRESHOFF, 11., Be r . 23> 3 5 3 7 ( 1 8 9 0 ) « 11 -Von RQMBUEG,P.e BARGEE, G., J.Chem.Soc. 99, 2068
(1911) .

73
12 - MILLER, E,J. e ROBSON, W., J.Chem,Soc. 1938 XÎ9IOT
13 - GHATAK, N. e KAUL, R#, J . I n d i a n Chem.Soc, 9, 383 TÏ932).
14 - HOSHINO, T., Proc. Imp. Acad. 11, 227 (1935) Ibid Ann. 520, 31 (1935).
15 - GORDON. W.G., JACKSON, B.W., J, Biol, Chem. 110 151 ( Ï 9 3 5 ) ,
16 - GAHILL, W.M. e JACKSON, R.Wr, J , B i o l . Cheat, 126,
; 2?~Tl9387. 17 - FITZGERALD, J . S . , Aunt. J . Chem. 16, 246 (1963) .
18 - Von EULER, H. e HELLSTROM, H . , Z .Phys io l . Chem. 208, 43 (1932)7
19 - OREKHOV, A.P. e NORKINA, S . S . , Zh.Ohohoh- Khinu 7. 673 (1937)? C.A.'31, 5801 (1937).
20 - Von EULER, H.,HELLSTROM, H., Z. Physiol. Chem. 217, 23 (19337.
21 - WIELAND, T . e USING, C.Y., Ann. 526, 188 (1936) . 22 - WILKINSON, S . , J . Chem. S o c . 2079 (1958)
23 - PACHTER, I . J . , ZACHARIAS, D.E. e RIBEIRO, 0 . J. Org. Chem. 24, 1285 (1959).
24 - WHITE, E.P., New Zealand, J. Sçj. Technol. B, 25, 1 5 7 ^ 1 9 4 4 ) .
25 -BOVSCT, K., BROWN, B.G. e BATTY, J . E . , Nature 174, 925 (1954) .
26 - HAMLIN, K.E. e FISCHER, F.E., J. Am. Chem. Soc.75, 500? (1951/.
27 - LIMA, O.G., Arquiv -, I n s t .Pesquisas Agron. ( Pernambuco) 4 , 45 (1946) .
28 - JACOBUCCI, G.A. e RUVEDA, E.A. , Phytoshem4s'ry 3 , 465 T Î 9 6 4 ) .

74
29 - GHOSAL, S. e MUKHERJEB, B . , J . Org. Ohem. 3 1 , 22847(196677"
30 - STROMBERG, V.L . , J . Am. Chem. Spc. 76, 1707 (1954).
31 - WIELAND, H . , HESS, G.,IS2ETASGH, H . , Be r . 64, 2099 (1931) .
32 - PISH, M.S. , JOHNSON, N.M, e HORNING, E .C . , J . Am. Chem. Soc. 77, 5892 (1955). "
33 - LEGLER, G. e TSHESGHE, R., Naturwissenschaften 50, 941Í963"},
34 - YURASHE7SKII, N.K., STEPANOV, S . I . , Zh. Ofrahch. Khiffi. 9, 2203 (1939) , C.A. 34, 4071 (1940) .
35 - YUPASHE7SKII, N.K., Zh. Obahch. Khim.10, 1781 (1940)? G.A. 35, 4016 (l94l).
36 - MÔRIM0ÍÔ, H. e MàTSUMOTO, N., Ann. 692, 194(1966). 37 - FISHER, 0. e BOESLER, W., Ber. 45, 1930 (1912). 38 - FISHER, 0., .Ber. 47, 99 (1914). 39 - PERKIN, W.ÏÏ. e ROBINSON, R.s J . Chem. S 0 c . 101,
1775~XÎ912)« 40 - FISHER, 0 . e TAUBER, E . , Ber . 18, 400 (1885) . 41 - FISHER, 0 . , Chem. Soc. Abs t . 405 ( l 9 0 l ) . 42 - HOPKINS, F.G. e COLE, S.W., J , P h y s i o l . 29, 451
(1903). 43 - KERMACK, W.O., PERKIN, W.H. e ROBINSON, R.,
J. Chem. Soc. 119, 1602 (192l). 44- - PERKIN, W.H. e ROBINSON, R., J .Chem» Soc. 115,
967 (1919)7 45 - GULIAND, ROBINSON, SCOTT, THORNLEY, J . Chem.Soc.
2924 (1929) .

75
46 - BADGER, G .H. e BEECHAM, A , F . , Nature 168, 517 TÏ95ÏT.
47 - JOHNS, S.R., LAMBERTON, J . A . e SIOUMIS, A.A. .Auet . J , Chem. 19, 893 (1966) .
48 - Ib id . 19, 1539 (1966) . 49 - YURASCHETSKI, N.K., J» A l l g . Chem. (U.D.SS.R.) 9,
595 (1959); Chem. Zen t r . 1,551, (1940) .
50 - BANESJEE, P.K., GHOSAL, S . , Aust . J . Chem. 22 , 275 (1969) .

T:
1.4.- PROPRIEDADES FÍSICAS g QUÍMICAS DAS IHDOIAtaUITA-g j S E DOS DERIVADOS DA ft-CAE30LINA
1.4.1,- Propriedades Gerais (l - li)
Iniciámos este capítulo com uma certa preocupação, porquanto as generalizações são quase sempre difíceis e imprecisas e, muitas vezes, criticáveis,: E isto é tanto mais verdade quanto mais elevado é o número de excepções que a generalização não consegue comportar.
Ora, as indolalquilaminas e os alcalóides Q --carbolínicos constituem dois grupos de substâncias de certo modo numerosos, tendo cada um destes grupos de com postos,como é o'bvio, características físicas e químicas pro'prias que fazem dele uma entidade bem definida e pelas quais se distingue de outros estruturalmente próxi mos.
Não obstante isto, é manifesto que entre os membros de cada série e entre os dois grupos de compostos há semelhanças notórias de còmportamento,as quais se devem, naturalmente, à presença nas suas moléculas do sistema bicíclico comum, o 2,5-benzopirrol.
Acresce, ainda, que cada um destes dois grupos cent So um azoto, N^, separado por dois átomos de car bono da posição _3 do núcleo indólico (exceptua-se a gratina) que lhes confere basicidade. Tal átomo de azoto po

78
de encontrar-se na forma de amina primária, secundária ou terciária (XXIV), ou interoalado num anel hexagonal (C), podendo este apresentar três graus diferentes de hidrogenação (XXV, XXVI, XXVII).
CH2-CH2- K^
XXIV
XXV XXVI
N-R2
XXVII

79
Estes factos fazem com que os dois grupo de substâncias se relacionem na origem, no modo de obtenção, pelas suas propriedades físicas e químicas e de certo modo pelas suas acções farmacológicas.
Embora sejam as propriedades físicas e químicas aquelas de que nos vamos ocupar neste capítulo, diremos que, na realidade, todos eles se filiam geneticamente no triptofano; que os derivados /?-carbolínicoS se obtêm laboratorialmente, em regra, a partir das indolal quilaminas pela clássica condensação de MAMICH e que alguns deles são manifestamente hipertencores e outros alucinogénicos.
Estes alcalo'ides são substâncias sólidas, ex-ceptuando-se a serotonina, a 5-met oxi-Nfr-metiltriptami-na e a lespedamina? de cor variável, sendo uns incolores e outros amarelados, podendo alguns apresentar cor alaranjada ou mesmo vermelha, estando a coloração especialmente dependente da presença, nas respectivas moléculas, de auxdcromos (OH; 0CH3 ou N-»0). Assim, a harma-na (XX?| KL-=CH3; R3=H e a tetraidroarmana (XXVII;Ik=CHa íi2= R3=H) são incolores; a harmina (XXV; %.=CH3^3=0013) e a harmalina (XXVI5 Rj=CH3, R3=0CH"3) são amarelas, enquanto que o harmalol(XXvT ; R:j_=CH3 R3=Œl) é já vermelho e os derivados triptamínicos-Nb-óxidos são acastanhados.
Cristalizam de solventes apropriados com rela tiva facilidade (especialmente OG derivados da /?-carbo-

80
lina) e os cristais fundem a temperaturas bastante distanciadas, muito baixas em alguns casos (DMTjp.f. = 48--49°C) e relativamente elevadas noutros casos (harmol | p.f. = 321°C).
As indolalquilaminas têm, em regra, pontos de fusão mais baixos do que os derivados (3-carbolínicos. Estes fundem a temperaturas moderadamente altas (à volta de 200°C), variando, em regra, na razão inversa do grau de hidrogenação do núcleo piridínico (C).
Assim a tetraidroarmana e a Nh-met iltetraidro-armana fundem a 178-180°C a primeira e a segunda a 109--110°C, enquanto que os correspondentes harmana e %-me-tilannana fundem a 237-238°C e 288°C,respectivamen te.
A introdução de oxigénio na molécula da p-car bolina faz subir como é,aliás, natural a temperatura de fusão, sendo esta máxima quando o radical fenólico se encontra livre. Assim, a harmina (XXV; Í^OCHs) funde a 262°G enquanto que o harmol (XXV; R3=0íl) funde a 321°C«
São em geral sublimáveis à pressão normal, a temperaturas não muito elevadas na sua maioria, são inac tivos à luz polarizada, o que está de acordo com a ausência de carbonos assimétricos nas suas moléculas como aliás, se verifica na maioria destes compostos(XXIV,XXV, XXVI). Mesmo os alcalóides com carbonos assimétricos (XXVII5 C-I assimétrico) não desviam, geralmente,o pia-

81
no de polarização da luz (exceptuam-se a d-tetraidroar-mina, (C/JT) =+ 32 (clorofórmio) e poucos mais .
Conhecem-se duas bases indólicas simples com actividade óptica» a abrina. (0()3=+ 46° (HCl) e a hipa-forina, (çA)D=+114° (água), o que está de acordo com a presença de um carbono assimétrico na cadeia lateral das moléculas destas substâncias.
Tanto as indolalquilaminas como os derivados (3-carbonílieos, apesar de possuirem dois átomos de a-zoto fazendo parte das respectivas moléculas, comportam -se como monobases, embora possuam valores de pkb bastante dispares. Esta basicidade é-lhes conferida quase exclusivamente pelo azoto não indolico.Efectivamente, o par de electrões responsável pela basicidade dos compos, tos de nitrogénio no anel pirrólico faz parte da nuvem 77 deslocalizada do anel aromático e, como tal, não se encontra livre para ser compartilhado pelo protão dos á eidos. Daí ser extremamente baixa a basicidade do núcleo pirrólico (K^ 2,5 x IO"14). (l2).
A piridina tem já um carácter básico apreciável (Kb- 2,3 x 10" ), mas inferior ao da piperidina ou das aminas alifáticas terciárias. Nestas o par de electrões do azoto faz parte de um orbital spg,enquanto que, na 1piridina, participa de um orbital sp2. Considerando o maior carácter _s dos orbitais sp2, o par de electrões de azoto será mais intensamente atraído para o núcleo

82
deste elemento, tornando-se, assim, menos disponível pa ra estabelecer ligações de covalência coordenada com ra dicais de carácter cationóide. Isto explica que a piri-dina apresente um carácter básico inferior aos das ami-nas alifáticas terciárias.
A piperidina tem já uma basicidade apreciável, semelhante à das aminas alifáticas secundárias(1^=2x101
Como consequência deste seu carácter básico , estes alcalóides formam sais, por reacção com ácidos e sais de amo'nio quaternário, por tratamento com halogène tos de alquilos.
1.4.1.1.- Formação.dá sais
A adição de ácidos a estas substâncias (XXIV a XXVII) origina sais solúveis na água, (XXVIII a XXXI ) ao passo que a adição de álcalis às suas soluções aquosas salinas regenera as bases por cedência ao ião hidróxilo do protão por elas fixado, como se indica no ESQUEMA. I

ESQUEMA. I
83
N / \
XXIV
J MI / \
R2R3
XXVII
N N
XXV
H
CE"
:.w.+
N E l
XXIX
1 1 %
XXVI
À/1 IT ^ V l
fe. R 2
NH+ I R2
XXVII XXXI

84
Embora possamos dizer, de uma maneira genérica, que estes compostos têm carácter alcalino,são pouco solúveis na água e solúveis nos solventes orgânicos, há a salientar dois factos que muito perturba este comportamento s
1) a presença de grupos OH no núcleo benzéni-co comunica-lhes carácter ácido e torna-os solúveis nos alcalis fortes °,
2) a função amina forma pontes de hidrogénio com a água, o que dificulta a sua extracção dos meios _a quosos.
Em apoio ao que acabámos de referir lembramos que a serotina (li) (I; R1=%=R3-fí e B^MÉ^t (pag. 47 )é bastante solúvel ria água, solúvel no ácido acético e pou. co solúvel nos solventes orgânicos, acontecendo que a sua solução aquosa 0,01 M tem pH = 3,6.
1.4el.2,- Formação de sais de amónio quaternário
Estes compostos (XXXII.. a XXXV) formam-se por tratamento dos alcalóides (XXIV a XXVII) com os haloge-netos de alquilos (ESQUEMA, II) e são convertidos por a dição de álcalia forces uns em hidróxidos de amónio qua ternário e outros em anidrobases dependendo esse comportamento da substituição ou não em Na,

85
ESQUEMA. I I
N N / \
xxiv
ïf tcH3 r
/ \ R2 %
XXXII
N'
K XXV
CH3 I
N ■CH3 I"
XXXIII
J^f1
XXVI
N / N ^ ^ ^ I "
«1
XXXIV
N' N %
Ri XXVII
H*
X tftCH» I" I ó
R 2 XXXV

86
Os derivados da metilação exaustiva podem ser convertidos pelo hidróxido de prata em hidróxido de amo nio quaternário, os quais se degradam por aquecimento, segundo o esquema de ÏÏOFFMAïW(l3), com abertura do núcleo tetraidropiridínico (C) ESQUEMA. III.
ESQUEMA. I I I
*- N + -, T l £ g £ g / G H 3 I Aquec. VGH3
GEr. CH3
XXXVI
-H 20 r C H 3
1 C2Hc 5
XXXVII
CH3I MT
' t C 2 H 5 X G H 3
CHT. Aquec,
JDÏÏÇ.CH2
CH3
XL XXXIX

07
Esta degradação, frequentemente praticada, é de grande utilidade i,o esclarecimento da estrutura destes compostos, por análise dos fragmentos (XXXIX e XL),
1.4,2,- Reacções de coloração
São numerosas as reacções coradas que a literatura refere para os alcalóides indólicos. Contudo, no âmbito deste trabalho, não nos parece ter utilidade nem cabimento citá-las a telas e, por isse, seleccionámos algumas entre as que é possível praticar»
Para além dns reacções gerais e comuns a todos os alcalóidesj as reacções de condensação com os ai deídos aromáticos e com o xantidrol,, as diazoreacções e a reacção com ninidrina parecem-no? das mais úteis para detectar os compostos de que estamos a tratar, permitiri do algumas delas, inclusivamente, averiguar a existência de certos grupos funcionais e serem aplicadas para fins quantitativos.
1.4.2.1,- Reacções com aldeídos
A maioria dos indois (XLI), reage com os aldeídos aromáticos (XLIl), dando derivados corados(XLIIl). Acontece, porem, que os indo'is 2,3-disubstituidos não são, por vezes, facilmente detectados por este género

de reacções porquanto a formação da cor dá-se,geralmente, por condensação do aldeído com o núcleo indólico,pe las posições 2 ou 3, de acordo com o ESQUEMA. IV (14).
ESQUEMA. IV
^ \ *./R
F i H
XLI
+ OHC-^ y-*i
XLII
R
V " "O^1
XLIII
A reacção mais antiga deste tipo foi descrita por EHRI.IGH, em 1901, e utiliza como reagente o p-dime-tilaminobenzaldeído em solução ácida. Parece não haver uma correlação lógica entre as estruturas do composto indólico e a cor produzida. Entretanto, a maioria reage com formação de tono purpúreos e a existência, na posição _5, de um grupo CM ou de um OCH5 origina o aparecimento de cor azul, enquanto que a localização daqueles

89
grupos em G6 faz coin que a cor mude para azulesverdea da. A reacção de EHRLICH pode ser utilizada não só na detecção como na determinação quantitativa dos compos
tos indólicos. Não é, contudo, exclusiva do núcleo indo. lico e há alguns derivados indólicos que não coram com pdimetilaminobenzaldeído «o que pode induzir em erro um investigador menos precavido. Há, portanto, que a consi_ derar como uma reacção complementar e não como reacção geral dos alcalóides indólicos, mas se se desenvolver cor azul rapidamente a frio é uma boa indicação do núcleo indólico.
Outros aldeídos têm sido propostos em substituição deste. Assim, MÏÏLLER (15) propôs o uso do cina
maldeídoç STRELL e KàLOJANOFF (16) sugeriram o emprego de homólogos vinflicos do pdimetilaminobenzaldeído, op_ tando HARLBYMASON e ARCHER (17) de preferência pelo ^ dimetilaminocinamaldeído na revelação destes compos
tos nos cromatogramas, por ter maior sensibilidade.0 me canismo de reacção é' idêntico com todos os aldeídos entretanto a cor obtida no caso do cinamaldeído ê diferen
te. De facto enquanto que com o pdimetilaminobenzaldeído e os homólogos vinílicos os tons são principalmente purpúreos ou azuis com o cinamaldeído são vermelhos a castanhos s ■ Entretanto ainda hoje o reagente de EHR
LICH ê talvez o mais frequentemente usado.

90
1.4 .2 .2 . - Reacções com xantjdrol
0 xantidrol (XLIV) reage com os derivados do indol dando xan t id r i l indó i s , os quais são produtos intensamente corados e bastante estáveis a pïï ácido (18) . A cor formada depende da es t ru tura do alcalóide e a intensidade de coloração es tá relacionada com a concentra ção do derivado indólico presente, podendo, portanto, ser ut i l izado em avaliações quan t i t a t ivas . Assim, o de rivado xant idr í l ico da triptamina mostra um máximo de absorção localizado em 510 nm, enquanto que o xan t id r i l -5-HT o apresenta em 557 nm, o que pode permitir numa primeira aná l i se , a dist inção destes dois compostos(19).
1 .4 ,2 .3 , - Reacções com o l -n i t roso-2-naf to l (XLY) KO ^ > _
OH

91
Este reagen te fo i proposto por GEKNGROSD. e
c o l . (20) pa ra detecção da t i r o 3 Í n a e t i r a m i n a . !JDE_N
FRIEND e c o l . ( 2 1 , 22) apl icaram-no à pe squ i sa de i n -
dóiso Ensaiaram-no com vá r io s compostos i ndó l i co s e v e
r i f i c a r a m que, em meio c l o r í d r i c o ou s u l f ú r i c o d i l u i d o ,
contendo v e s t í g i o s de n i t r i t o , somente os 5 -h id róx indo is ,
reagiam com produção de cor v i o l e t a purpúrea , sendo deo_
conhecida a e s t r u t u r a do composto formado,
1 , 4 . 2 . 4 , - BeacçSes de d iazo tação
Os compostos de d iazónio mais frequentemente
u t i l i z a d o s na a n á l i s e dos i ndó i s são :
1) Clore to de d iazónio do á í i d o p-benzenosul -
fonico (XLVI) ou reagente de PAULI ou de
VAN DEU BERGc
2) Clore to de p-ni t robenzenodiazónio
(XLVIl)
3) Te t r a f luo robora to de p-ni t robenzenodiazónio
(XLVIII)
4) 2 -naf ta lenosu l fona to do 2 - c l o r o - 4 - n i t r o b e n
zenodiazdnio (XLIX), r eagen te NNCD.

92
XLIX
Estes reagentes dão com os 4,5 ou 5-hidrb.xin-dóis, produtos de copulação fortemente corados. São muito sensíveis para a pesquisa dos hidroxindóis,permitindo detectá-los em quantidades diminutas,enquanto que os não hidroxilados ou que tenham este radical bloqueado não dão cor ou coram muito ligeiramente.
A diazotação-copulação processa-se melhor em meio ligeiramente alcalino e a cor resultante é "bastante variável conforme o pH do meio e, portanto, obtêm-se

93
dados úteis, praticando-a a diferentes valores de pïï. Por isso, os cromatogramas são revelados com a solução ácida do sal de diazónio e observa-se a cor formada, após o que se faz uma aspersão com uma solução de carbonato de so'dio, de modo a criar um meio alcalino, ob-servando-se a cor originada nestas condições (23-26).
1.4,2.5.- Reacções com a ninidrina
Gomo se sabe, o mecanismo da reacção que con. duz à formação de cor quando se faz actuar a ninidrina (L) sobre as aminas e os aminoácidos é complicado devido à ocorrência de reacções parciais, e a cor resultante ê devida não a uma simples substância ma3 uma mistu ra de vários produtos e necessita de ser feita a quente (14). Na análise cromatográfica os cromatogramas são as pergidos com solução de ninidrina e aquecidos durante 5 a 10 minutos, a 100-105°C.
Ultimamente HUTZINGER (2?) experimentou na re

94
velação dos cromatogramas reagentes de complexação ace_i tadores de pares de electrões-Das substâncias experimentadas pareceu ser mais útil o 2.4.7-trinitro--9--f luoreiío-na. Segundo o autor, estes reagentes são menos sensíveis do que os já referidos, mas têm a vantagem de se po der regenerar o derivado indólico a partir do complexo formado. Além disso, reagem com os indóis 2,3-dissubsti tuidos e parece fornecerem algumas indicações quanto ao tipo de substituição no núcleo.
1.4.-3,- Reacções de fluorescência
A fluorescência do núcleo indólico transmite--se a muitos produtos dele derivados.
Assim, os alcalóides da -carbolina e as bases indólicas simples exibem, em regra,fluorescência de intensidade e cor variáveis desde esbranquiçada, a azul pálida, a azul violácea, verde, etc. (2,4),
Nas t?> -carbolinas. a fusão do núcleo piridíni-co com o indólico foz exacerbar a fluorescência compara tivamente à das indolalquilaminas. Entretanto, os compostos com o núcleo piridínico completamente hidrogenado, as tetraidro fí> -carbolinas,não são naturalmente flu orescentes mas podem adquirir esta propriedade em virtu de da sua oxidação ao ar.
Esta propriedade é de grande utilidade na de-

95
tecção destes compostos em produtos biológicos sobretudo para a sua localização por técnica histológica, e é de extrema comodidade para os evidenciar nos cromatogra mas. Além disso, o espectro de fluorescência é um elemento que muito pode contribuir para a identificação des tes compostos.
Os indóis simples e sem substituintes no anel benzénico, em solução neutra ou fracamente ácida, apresentam, quando excitados com luz de comprimento de onda da ordem dos 275 nm, o máximo de emissão próximo de 350 nm.
Os 5-hidróxindóis e 5-metoxindóis necessitam de menor energia de excitação, localizando-se o máximo de emissão por volta dos 350 nm, quando excitados com radiações de comprimento de onda da ordem de 295 nm(28)L
A título de exemplo, apresentam-se 03 resulta, dos obtidos por FISH e col. (29) para quatro compostos, sendo dois de cada um dos grupos mencionados QUADRO IIIA
QUADRO III FLUORESCÊNCIA DE ALGTMàS IITOQLALQUILAMEMS
Composto máx„, em nm
1¾,H^-Dimetiltriptamina N-jj jK- -Dimet iltriptamina-N-^-óxi
do
Excitação Emissão 1¾,H^-Dimetiltriptamina N-jj jK- -Dimet iltriptamina-N-^-óxi
do
283 283
350 349
Bufotenina Bufotenina-Nb-óxido
300 308
340 330

96
Segundo UDEHFRIEKD e col. (30) e QUAY (3l) é ainda possível, operando em meio fortemente ácido(HC13M), distinguir os 5-hidroxindóis e os 5-metoxindóis dos in-dóis simples não substituídos»Em tais condições1 dê pti estes perdem a fluorescência, enquanto que aqueles mostram um deslocamento do máximo de fluorescência, locali zando~se esta a 550 nm por excitação com radiações de A próximo de 295 nm.
Em certos casos, os ensaios de fluorescência entre os compostos do mesmo grupo permitem uma melhor discriminação do que os ensaios polarimétricos ou mesmo a espectrofotometria no ultravioleta. Assim, por exemplo, os 5-hidroxindóis dão a mesma cor com o reagente es pecífico deste grupo, o l-nitroso-2-naftol,os espectros no U,V, têm sensivelmente o mesmo traçado gráfico e tam DÓm apresentam espectro de fluorescência idênticos em meio ácido ou neutro. Contudo, dentro de certa medida é possível distinguí-los determinando os respectivos espectros de fluorescência a vários e convenientes valores de pH.
As indolalquilaminas, além da flúoresÔência própria que possuem, originam com determinados reagentes produtos mais intensamente fluorescentes. Assim, a trip-tamina (32), quando oxidada com iodato em meio acético, origina, triptocromo (Li) intensamente fluorescente.

97
LI
A maioria das reacções que se praticam com o fim de obter compostos mais fluorescentes ba3eiam-se na formação de derivados carbolínicos e foram inicialmente propostas por JEPSON e STEVENS (33) para a detecção da3 triptaminas em cromatogramas.
HESS e uDEEFRIEffl) (34) aplicaram, com grande êxito, uma reacção de fluorescência deste tipo na pesquisa e doseamento da triptamina nos tecidos.
Esta amina (LIl) reage com formaldeído para dar tetraidronorarmana (LUI), que não é fluorescente, mas que, por desidrogenação por tratamento com peróxido de hidrogénio, origina norarmana (LIV), com fluorescência azul violácea intensa.

ECHO N-H
LIV
Outra reacção também baseada na formação . de um derivado (Hidrogenado fc-oarbolinioo, com intensa flu orescência verde foi proposta por JEPSON (35) para detectar a 5-HT e indolalquilaminas primárias em cromato-gramas, usando ninidrina em solução acética.
PROCHAZKA (36) usou, para a detecção em croma togramas dos indóis substituídos em C-3, uma mistura de formaldeído e ácido clorídrico aplicado por pulveriza ção. Nestas condições, estes derivados mostram cores a-laranjadas e acastanhadas e fluorescência amarela ou a-laranjada. Mas para que este comportamento se observe

99
é necessário que o núcleo indoXico se encontre separado do substituinte (R) por um CH2 (LV), O que, aliás, se observa nas indolalquilaminas.
r^N J CHo—R
LV
Se E é um grupo hidroxilo ou um grupo dimetil amínico (como acontsoe na gramina) observa-se cor verme lha a violácea.
SHEKEED e col. (37) usaram para revelar cro-matogramasformaldeído e dicromato de potássio. Nestas condições, a 5-HT e a bufotenina produzem fluorescência amarela dourada.
QUAY (38) efectuou em 1968 um estudo tendente a averiguar da especificidade da determinação fluoromá-trica da 5-HT com a ninidrina. Com este objectivo teve oportunidade de estudar 121 compostos contando-se entre eles 72 indolaminas e mais de uma vintena, de derivados Q -carbolínicos.
0 espectro de fluorescência destes últimos com postos foi determinado antes e após aquecimento, em pre sença da ninidrina. Na sua maioria, quando em meio tam-

100
ponado a pll=7,0 e irradiados com luz de /\ = 380-385 nm emitem radiações de fluorescência de A - 490 nm.
0 autor verificou também o que no início deste capítulo havíamos afirmado, isto é, que os compostos, carecendo de insaturação no núcleo piridínico/ não emitem fluorescência e que os compostos mais intensamente fluorescentes eram os que possuíam duas duplas ligações em 1 e 3. Além disso, segundo afirmam McISAAC e col. , os compostos 6-hidroxilados e metoxilados têm fluorescência rósea, enquanto que os hidroxilados ou metoxilados em C-7 exibem fluorescência azul,
0 aquecimento dos derivados(^-oarbblíriicos opm ninidrina, durante uma hora, a 60 C, não determinou,sobre a fluorescência destes compostos, um efeito uniforme. Alguns deles, apresentando, naturalmente, forte, mo derada ou mesmo fraca fluorescência, quando tratados com ninidrina, tornam-se menos intensamente fluorescentes,
Comportaram-se assim a maioria dos derivados analisados, e entre eles, podemos destacar a harmalina, o harmalol, a harmana, a harmina, a leptoflorina,etc.
Noutro grupo destes compostos, mas menos nume roso que o anterior, embora apresentem inicialmente ligeira fluorescência, após aquecimento com ninidrina, re gista-se um aumento da sua intensidade de fluorescência. Comportaram-se assim a 6-metoxiarmalana e a 6-hidroxite-traidroarmana,

101
Outros derivados, taJSo&mo- á totraidroarmaia-na, não exibem fluorescência em 490 nm, quer antes,quer após aquecimento com o hidrato de 1,213-idantriona.
1.4.4,- Comportamento cromatográfico
0 emprego da técnica cromatográfica encontra -se largamente generalizado no estudo dos produtos natu rais.
No domínio dos alcalóides indólicos todas as modalidades da análise cromatográfica têm aplicação, sendo cada uma das técnicas escolhida conforme o objectivo em vistaí isolamento ou identificação dos compostos em causa.
A excepcional importância da cromatografia em coluna reside na possibilidade de permitir purificar e isolar estes compostos, pois que o fraccionamento dos extractos se processa de acordo com a afinidade dos cons tituintes do extracto para o adsorvente e com a respectiva solubilidade nos eluentes.
Esta técnica é quase imprescindível para o iso lamento dos alcalóides indólicos, sendo mais eficaz do que os processos clássicos (cristalização fraccionada , precipitação com reagentes, sublimação, etc.).
A cromatografia em coluna é menos selectiva do que a de placa e a de papel, mas tem sobre estas grande

102
vantagem de permitir separações em grande escala. Basta apenas escolher a coluna com tamanho apropriado,e enche -la com adsorvente em quantidade proporcional ao extracto, em geral uma parte deste..para 30 partes de adsorvente. Apresenta, no entanto» alguns inconvenientes relativamente às outras duas técnicas mencionadas,E ba_s tante morosa, pois, enquanto que o desenvolvimento dum cromatograma em papel se processa em algumas horas e em placa em alguns minutos, a elução da coluna demora bas tante tempo. Este tempo está, como é evidente, dependen te do tamanho da coluna e do escalonamento dos eluentes que se tenha que utilizar para obter a separação preteri dida.
Dos adsorventes a que é possível recorrer, os mais vulgarizados são a alumina e a sílica, os quais o-riginam, neste caso, boas separações e só episòdicamen te é necessário recorrer a outros (celulose, magnesol, etc.).
A cromatografia em papel conheceu uma larga di vulgação tanto para isolar como para identificar alcalói tes indólicos e cremos, até, que no campo das indolalqui-laminas ainda não cedeu o lugar conquistado. Pratica-se sobretudo em duas modalidades - ascendente e descendente - qualquer que seja o propósito a que se destine,isto é, com fins separativos (técnica cromatográfica preparativa), com o fim de identificar os compostos pelos

103
v a l o r e s dos seus Rf ou por comparação com amostras pa
d r õ e s .
0 papel que se usa é , em r e g r a , o WHATMAN n ° .
1, o de SCHLEICHER e o de SCHULL n ° . 2043 b , recorrendo,
- s e , muitas v e z e s , ao WHATMAN n». 3 ou 3 MM, os q u a i s ,
por serem mais e spes sos , absorvem maior quant idade de
e x t r a c t o , sempre que se pre tende fazer uma .. cromatogra
f i a p r e p a r a t i v a . São v á r i o s os s i s temas u t i l i z a d o s , s e n d o em ge
r a l b a s t a n t e p o l a r e s e de na tu r eza ác ida ou a l c a l i n a , c o n s t i t u i d o s por mis tu ras de á l coo i s com ácidos ou com amónia, p i r i d i n a ou aminas a l i f á t i c a s .
Assim, referem-se frequentemente, e n t r e out r o s , o bu tano l -ác ido a c é t i c o - água (4s 1; 5 ) , o i s o -propanol-amónia-água ( 8 : Is l ) , o b u t a n o l - p i r i d i n a - á g u a ( i s Is l ) , bu tanol sa turado de HC1 N e butano 1-metilamina a 25 $> (8s 3) ( 3 9 ) .
A cromatograf ia em camada delgada tem sobre
a do papel a vantagem de s e r mais r áp ida execução e p e r m i t i r , em r e g r a , uma melhor separação dos compostos.
No estudo dos der ivados ^ - c a r b o l i n i c o s , se bem que a t é c n i c a cromatográf ica em papel cont inue em voga, a c romatograf ia em camada f i n a é francamente mais p r a t i c a d a .
Os adsorventes mais v u l g a r i z a d o s , n e s t e caso , são também a s í l i c a e a alumina.

104
Dos vários desenvolventes que têm sido preconizados podemos destacar s butanol-ácido acético-água(4s Is l), propanol-hidróxido de amónio (8s 2) e clorofc'r-mio-metanol (em proporções variadas), etc.
No desenvolvimento dos cromatogramas de papel os sistemas são bastante idênticos aos que foram indica dos para o desenvolvimento das indolalquilaminas.
Para detecção dos alcalóides nos cromatogra-mas, como a maioria destes compostos são fluorescentes, basta a simples exposição daqueles à luz de uma lâmpada de WOOD.
Por vezes, a observação da fluorescência pode ajudar-nos a formular uma hipótese, pois que os deri vados tetraidro(3-carbolínicos não são fluorescentes,os
diidro ^ -carbolínicos apresentam fluorescência verde enquanto que os -carbolínicos mostram intensa fluo rescência azul.
Os reagentes químicos mais vulgarizados para a sua revelação sãos reagente de DRAGSNDORFF, iodoplatí nico, de EHRLICH, xantidrol, l-nitroso-2-naftol, ácido fosfovanílico, ácido sulfanílico diazotado, ninidrina, etc.
Deve-se a McISAAC e col. (40) um interessante trabalho de análise cromatográfica de derivados^-carbolínicos naturais e sintéticos, Submeteram a análise cro matrográfica em papel e em camada delgada de sílica se-

105
tenta e três desses derivados, tendo usado no desenvolvimento os seguintes solventes s
Para cromatografia em papel; butanol-ácido _a cético-água (l20s 30s 50); butanol--isopropanol-hidrdxi_ do de amo'nio-água (3s Is Is 1)5 melanol-butanol-benzeno água(£í 1: Is l).
Para cromatografia em camada delgada; butanqi -ácido acético-água (4s Is l); propanol-hidróxido de a mdnio (8; 2)§ cloroférmio-metanol (9; l).
Para revelação usaram vários reagentes de coloração s xantidrol 1% em álcool a 95°, reagente de EHR-LICH, azul de brentamina em carbonato-bicarbonato (pH= =9,2), ácido sulfanílico diazotado, ninidrina a Vfo em butanol, mistura oxidante -ácido sulfúrico a 30%, solução aquosa de cloreto férrico a 5%, ácido perclórico a 36% (9,8; 0,2s 10 volumes), ácido cítrico a 2% em ani-drido acético.
0 comportamento destas bases, quando cromato-grafadas em camada delgada de sílica, no sistema clorofórmio-metanol (9s l), permitiu-lhes estabelecer certas relações entre o comportamento observado e as respectivas estruturas moleculares.
Na realidade, todos os derivados tetraidro -í-carbolínicqs têm Rf inferiores aos das @> -carbolinas a-romátiças. Poderemos exemplificar com dois pares destes compostos ;

106
Tetra idro\ i -carbol ina , Rf = 0,05 (¾-carbolina, Rf = 0,57
Tetraidroarmaria, Rf = 0,06 Harmana, Rf = 0,54
Por outro lado, sempre que as t e t r a id ro (¾ -car bolinas se encontrem alquiladas nas posições 2 ou 9 migram nas placas de s í l i c a para níveis mais elevados do que as respectivas bases não subst i tuídas nessas posições como podemos observar no seguinte exemplos
1 - Metil tetraidroÇ^-carbolina, Rf = 0,06 l ,2 -Mmet i l t e t ra id ro(Vcarbo l ina , Rf = 0,19 1,9-Mmet i l t et raidro ^ -ca rbo l ina , Rf = 0,25 1,2,9-Trimeti l tetraidro ($-carbolina,Rf = 0,40
Estas observações sugerem uma atracção entre os grupos N-H dos núcleos p i r ró l i co (B) e t e t r a i d r o p i r i dínico ( c ) , e a s í l i c a , provavelmente através de pontes de hidrogénio» Sempre que este hidrogénio do Na ou do Su se encontre substi tuído por um alquilo (normalmente é um radical metilo nos produtos naturais) é de prever uma menor atracção para a s í l i c a , v i s to que a ponte de hidrogénio j á se não pode estabelecer ,
A maior migração dos derivadosft -carbolínicos aromáticos relativamente aos derivados t e t r a id ro Q -car bolínicos poderá também ser interpretada pela sua menor

107
basicidade, As 6-carbolinas hidroxiladas cromatografadas
em sílica apresentam comparativamente aos corresponde^ tes derivados metoxilados valores de B£ inferiores, ex-plicando-se o facto pela forte adsorçâo daqueles à si li ca, por uma ligação de hidrogénio do grupo ÇH.Embora os derivados metoxilados, em virtude da sua menor adsorçâo à sílica, apresentem valores de Rf mais elevados estes são, no entanto, inferiores aos dos derivados correspondentes sem substituintes no núcleo benzénico.
Os compostos com um grupo carboxílico em posj. ção j. migram nos solventes básicos anidros para níveis pouco elevados, reduzindo-se ainda essa migração se es tiver presente outro grupo do mesmo tipo e isso devido à formação de sais.
Os compostos quaternários, quando aplicados em placa de sílica e estas desenvolvidas em cloroférmio--metanol (9s l) ou em sistemas anidros ácidos ou alcali nos, localizam-se em zonas mais baixas do que as bases ternárias.
A cromatografia em fase gasosa tem sido prati cada ultimamente, porquanto esta técnica permite mais cò modamente não só uma separação das bases como também a sua identificação e a determinação quantitativa das mes_ mas.
HOLMSTEDT e col. (4l), aplicando esta técnica à separação de uma mistura de indolalquilaminas, conse-

108
guiram separar, entre outras, as bases seguintes; DMT, 5-MeO-T,. 5-MeO-DMT, 5-Hî, 5-CB-DMT.
1.4.5,- Comportamento espectral
Nas três últimas décadas os químicos orgânicos têm vindo a fazer cada vez maior uso dos métodos físicos e instrumentais em substituição parcial ou qua se total dos laboriosos e complicados processes puramen. te químicos, até então classicamente utilizados, A razão deve-se ao facto de estes últimos, além de serem,par veze3, de realização difícil e morosa, fornecerem resultados quantas vezes de interpretação ambígua e exigi_ rem, em regra, apreciável quantidade de.:'- matéria*.-prima para poderem ser praticados.
Ora a obtenção de um produto em quantidade su ficiente e em perfeito estado de pureza são duas das grandes dificuldades que os químicos que se dedicam ao estudo das substâncias orgânicas naturais, têm de enfrentar com frequência e que nem sempre vencem fàcilmen te,
Dado que os processos instrumentais de análise (espectrofotometria no U,V„, no I,V, e de fluorescein cia, espectrometria de massa, ressonância magnética nuclear, etc.) apenas exigem diminutas quantidades de substância, mostraram-se extremamente úteis no estudo dos alcalóides indolicos, pelo que ganharam plena aceitação

109
neste domínio. Note-se, no entanto, que a interpretação dos
resultados a que estas técnicas conduzem por vezes não é fácil e nem sempre aqueles são totalmente esclarecedo, res quanto à estrutura dos compostos em causa. Por isso em alguns casos, não obstante o incontestável mérito e as apreciáveis informações fornecidas por estas técnicas, a estrutura de um composto por vezes só fica esclarecida após aturado trabalho, tomando-se necessária a preparação de derivados ou de compostos marcados para serem analisados pelos métodos referidos. Além disso, a síntese do alcalóide em estudo é imprescindível pa ra a confirmação plena da estrutura que a análise permi tiu antever e estabelecer.
1.4.5.1,- Espectrofotometria na região do ultravioleta
0 comportamento de uma substância na região e lectromagnética do espectro, entre 200 a 400 nm,raramen te permite, só por si, a dedução da estrutura global dum composto, pois que nesta região apenas se evidencia a porção molecular responsável pelas transições electron! cas CA-7» e n-*|). Contudo, algumas das estruturas ini cialmente formuladas para certos alcalóides indólicos fo ram quase exclusivamente fundamentadas em observações es pectrofotométricas nesta região, Mas, na maioria dos ca

110
sos, o espectro no JJ<|V, du™ alcalóide indclico permite, deduzir com certa segurança o cromóforo presente e este representa, em regra, uma imagem incipiente da sua mole cuia.
De facto,pelo espectro no U.V.é possível iden tificar nestes alcalóides os cromóforos seguintes s indol, indolina, fi carbolina, ■ ÓLmetilenoindolina,oxJridol,in
dolenina e U/ indoxilo» Destes, o indólico é predominan te neste grupo de alcalóides seguindose o diidroindóli co? e o (¾ carbolínico, etc.
Cada um destes cromóforos origina na região do U.V. bandas de absorção localizadas em comprimentos de onda bem caracterizados como se pode ver no QUADRO IT, onde inscrevemos as estruturas daqueles cromóforos, os comprimentos de onda aproximados de absorção máxima bem como a frequência com que cada cromóforo aparece nes te grupo de alcalóides (9, 10 e 42). As indicações £&« das englobam os resultados 'publicados até 1968.

I l l
Q U A D R O rv
Cromdforo
> s c
^
Indol
Atsorção no U.V. Á max,em nm(va-lores aproximados)
225 230-292
I Indolina
(diidroindol)
245 295
-Cartolina
^ -Metilenoindolina
234 287 347
252 298 361
Frequência do cromcforc nos alcalóides in-dólicos isolado f!
39,67$
Z&fo
7,5/o
5,6%

112
s£^ t « 210 210 252 280(inflexão)
5,6$
Oxindol
^ \ 218 2,6$ I 218 2,6$ 1
^ 257
Indolenina
^ \ ^0 235 400
1,1$ O I
235 400
1,1$
^ Indoxilo
A indicação que a espectrofotometria no U. V, proporciona acerca dos alcalóides indólicos é de extraordinária valia como ajuda na interpretação dos espectros no I.V. e de massa,sem dúvida mais válidos na elucidação da estrutura de um determinado composto.
As indo1alqui1aminas apresentam espectros de absorção típicos dos 5-indóis substituídos. As que não contêm substituintes no anel benze'nico apresentam um má ximo de absorção na vizinhança de 220 nm e outro em 280

113
nm e um pequeno pico subsidiário em 288291 nm, em moio ácido neutro ou alcalino (l0<> 42, 43).
■A3 5hidroxi e 5metoxindolalquilaminas apre
tam em meio ácido ou neutro um máximo próximo de 275 nm e também um pico subsidiário para além de 295 nm. A pre sença de um grupo OH ou 0CH3 determina ligeiro efeito batocrómico neste último pico.
E" notáriaa influência do meio alcalino sobre os 5hidroxindóis. De facto, o efeito ionizante do meio sobre o alcalóide, em virtude de este possuir um grupo fenolico, determina apreciável efeito batocrómico,sendo o pico desviadopara 322 nm.
Tanto a bufotenina como a serotonina exemplificam claramente o que acabamos de referir. A bufotenina (44), que tem um máximo em 277 nm e uma inflexão cm 295 nm (A ^ 249 nm), em solução de hidro'xido de sódio 0,1 N, sofre um deslocamento da inflexão a qaal se transforma num máximo em 322 nm.
A serotonina, segundo referem ESPAMER e ASERO (45), apresenta a pH 56 um máximo em 275 nm e uma in
flexão em 293295 nm. Esta banda sofre desvio similar ao da bufotenina a pH alcalino.
As 5metoxindolalquilaminag possuem um espectro semelhante às 5hidroxindolalquilaminas mas, pelo facto de o grupo fenolico se encontrar bloqueado por me tilação, não apresentam aquele desvio em meio alcalino.
Parecenos, também, de referir que o comprimen

114
to da cadeia alquilamínica ou a circunstância da função amina ser primária, secundária ou terciária não a-fectam, essencialmente, a posição das bandas referidas, o mesmo podendo dizer-se relativamente aos N^-óxidos (QUADRO V).
As características espectrais das indolalqui*-laminas Na substituídas (46) são mais aproximadas das correspondentes às do 1,3-dimeti1indo1 do que das que correspondem ao 5-metilindo1 e em meio alcalino a localização dos máximos não é afectada.
Os espectros de absorção dos derivados tetrai_ dro p -carbolínicos são ainda tipicamente indólicos(Fig, 13 na parte experimental - PARTE III). Na realidade, se o grupo indólico não apresenta qualquer conjugação com duplas ligações ou com qualquer outro cromdforo, não so_ fre apreciável variação na sua energia de ressonância . E, sendo assim, não surpreende que estes alcalóides pos suam um espectro idêntico ao do 2,3,-dimetilindol, com uma banda de absorção intensa perto de 226 nm e outra de menor intensidade, próxima de 280 nm, e um pico em 290.292 na (43),
Os derivados com um substituinte no núcleo ben zénico(OH ou dOH3) apresentam um espectro com a banda de absorção mais longínqua deslocada no sentido dos maio res comprimento de onda.

115
Q U A D R O V
Espectros de absorção no U,V. de algumas indolalquilaminas extraídas de Leguminosas
a) Indolalquilaminas não substituídas no núcleo benzéni co
Alca ló ides max, em nm Referência Bible
Gramina 2175280 43 Hipa fo r ina , ÏÏC1 219?280;289 43 T r i pt amina,, HC1 221;275;281;2S3 11 N-jj, N^-Dimet i l t r i p -
tamina 274;283;291 29 \ jN^ -Dime t i l t r i p -
tamina-Kjj--óxido 274;282;290~291 29
Lespedamina 278;291 46 Bufotenina 277;295 44
223;277;300 43 279;301;314 29
Buf ot enina-K-,0-dxi-do 278;302;314 29
Sero tonina 275;293-295 45
5-Met oxi t r i p t amina, HC1 221 ; 2.74; 307 43
5-Metoxi- í í^-meti l -t r i p t amina . HC1 222;274;295 43
Uriptamina 222;276;382;291 11

116
A tetraidroarmina,que segundo BERNAUER (4?)ab sorve em 225 (log £,-4,52), 269 (log £ = 3,69) e 296 (log £= 3,75) nui)concretiza o que afirmamos.
0 meio ácido provoca, nos alcalóides tetrai-droft -cartolínicos, um ligeiríssimo efeito hipsocrómi* co, na banda de maior intensidade.
No núcleo da fi -carbolina, o cromóforo funda mental, o indol, encontra-se conjugado com um segundo sistema insaturado, no qual as duplas ligações são tem bem conjugadas. A molécula apresenta, por consequência, uma maior energia de ressonânica e, portanto, o seu espectro é já bem diferente do do indol, sendo de esperar um deslocamento das bandas para os maiores comprimentos de onda e também de prever o aparecimento dum maior número de bandas. Assim acontece, de facto, apresentando estes derivados três máximos bem evidentes, que se loca lizam para a harmana (43) em 234, 287 e 347 nm (Fig. 16 PARTE III) e para a norarmana respectivamente em 235, 288 e 349 nm (48), além de outros menos maroadoBj
0 meio ácido (49) determina nestes compostos um notável efeito batocr'mieo com perda das bandas finas (Fig. 16 - PARTE III).
A transformação do núcleo^-carbolínico em com posto derivado do amónio quaternário(iodometilato) faz modificar o espectro de maneira bastante semelhante ao resultante da transformação em cloreto, sendo mais in-

117
tenso o efeito b at ocrónico, mas igualmente nítido o esbatimento das "bandas finas.
Se o hidrogénio de Na se encontra substituído por um alquilo, o meio alcalino forte não tem efeito apparente nos espectros destes derivados quaternários. Em caso contrário, a alcalinidade provoca a formação de a-nidro-base de cor amarela ou alaranjada e os três máximos sofrem apreciável deslocação batocrómica (10 e 5l) (Fig. 22 - PARTE III).
A formação destas anidro-bases, (LVIII-LIX) , que são híbridos de ressonância,pode explicar-se pela deslocação do protão do átomo Na pela presença activado hidroxildã© do mAo,seguida de uma mobilização electróni ca como a seguir se esquematiza.
LIX LVIII

118
O estudo destas bases anidrónicas no U.V.e no I.V. apresenta enorme interesse no esclarecimento da es trutura dos compostos que lhe deram origem. Na realidade o espectro no ÏÏ.V. em meio ácido ou neutro dos derivados iodometilados apresenta o mesmo aspecto, mas orneio alcalino provoca um típico efeito batocrómico nas bandas de absorção, ao mesmo, t empo ,que no I.*V. se nota ode saparecimento da banda característica do' M indólico.
1,4.5.2.- Espectrofotometria na região do infravermelho
A espectrofotometria no I.V. tem sido largamen te utilizada na análise dos alcalo'ides indólicos e permite confirmar, com segurança, a natureza indólica de um determinado composto.
Na realidade, o estudo das propriedades espectrais de 23 alcalóides indólicos da mais variada estrutura química na região do I.V. longínquo permitiu a WAR BEN, EISDORFER, THOMPSON e ZAREMBO (52)verifiçarem que tais compostos apresentam duas bandas de absorção caraç.
_1 terísticas, na zona compreendida entre 700 e 400 cm . -1 A primeira delas situa-se em 620 + 20 cm , sendo atribuída pelos referidos investigadores ao núcleo indo'lico. Nalguns dos compostos que figuram na série estudada ohserva-se mais do que uma banda nesta região, facto esse, devido ao desdobramento de uma banda

119
simples e larga. Conforme WARREN e col. põem em destaque,a fre
quência desta banda aumenta ligeiramente em relação ao indol nalguns dos compostos examinados. Isso verifica--se nos metil e metoxindóis substituídos nas posições 4, 5., 6. ou j nãor-se "registando .qualquer efeito-perceptível sobre a frequência desta banda quando a substituição se faz nas posições 2_ ou 3.
A segunda banda de absorção característica de_s tes compostos localiza-se em 575 + 25 cm~ ,sendo também devida ao núcleo indólico.
Tal absorção, como, aliás, a anterior, é notavelmente persistente e foi observada em todos os membros da série estudada.
Uma vez que a posição dos substituintes apenas provoca ligeiros deslocamentos na frequência de absorção, a qual, porém, é independente da natureza dos mesmos, tais bandas podem ser tomadas como característi cas dos compostos possuindo um núcleo indólico.
KâNOAKA e col. (S5) descrevem uma banda, nas proximidades de 810 cm , que parece ser característica dos 5-indóis substituídos e que julgam 3er devida a uma vibração £[ do C-H em posição _2 do núcleo indólico.
Por seu turno, MARION, RAMSAY e JONES (54) as_ sinalam que um grupo imino,quando introduzido em núcleo indólico, origina uma banda nítida na região de

120
3.480 - 3 460'qm" ,. (Pigs. 14» 20, :39,'-.:49, 90 e 51rPAR~ TE III) a- quai tém iîm coeficiente de extinção^molecular aparente de 140-210. (QUADRO VI).
Q U A D R O VI Bandas (ff-ff alongamento) em alguns alcalóides indólicos
Composto f\ , em cm" g N-acet ilarmina 3460 210 Evodiamina 3470 170 Gramina 3480 170 Harmalina 3470 140 Harmana 3475 150 Isoevodiamina 3480 210 Ruteacarpina 3465 190 Yoimbina 3480 180
Para a distinção das indolalquilaminas primárias, secundárias e terciárias interessa considerar as oandas no espectro respeitantes às funções -Mo N-H e
As regiões do espectro entre 3.500-3.300 cm e na vizinhança de 1650 cm" \ assinalam-se como as mais úteis para distinguir os três tipos de aminassprimárias, secundárias e terciárias (55 a 63).(Fig. 39 - PARTE III),

121
A absorção na zona de maior frequência ë devi da à vibração de ligação N-H, enquanto que a absorção nas proximidades de 1650 cm" se deve a vibrações de deformação no NB?«
Assim., as aminas primárias e secundárias ab-sorvem em 3,500-3.300 cm" , enquanto que ás aminae t e r c i á r i a s , por carecerem da função NH, não apresentam, em pr inc íp io , absorção nesta zona.
As aminas primárias mostram duas bandas de ab sorção nesta região e as secundárias uma BO, mas ambas podem ainda apresentar uma outra,, Esta, que é, em regra, mais larga que as r e s t an te s , s i tua-se em frequências l i geiramente mais baixas e parece ser devida a as so ciar -_.es moleculares por formação de pontes de hidrogénio (64).A dist inção entre as diferentes aminas nem sempre é f ác i l e isso deve-se a vários factores., segundo DU7AL,tais co mo associações moleculares,intcr&o^õèa cem os solventes dispersantes , à presença de outras funções na molécula, e t c .
Por seu turno os espectros dos sais dos a l calóides R - l r / , R-KHo e R-NH nos quais o par de elec t roes do azoto da cadeia l a t e r a l j á não pode funcionar como aceitador de protões, mostram em geral um pico bem evidente entre 3,400 e 3.100 (ver espectro do cloreto de de triptamina , ; Fig„ 32 - PARTE I I I ) , estando a l o c a l i zação da banda dependente do grau de substi tuição do n i

122
trogénio. Assim, segundo referem MESLEY e EVANS (57),os
sais das indolalquilaminas primárias absorvem em 3*230 -3,250 cm e os das aminas secundárias em 3.400 cm " , enquanto que os saia &03 àarrsaa larciáriaa.absorvem -em 3,320-3,,125. cm"1.
1.4,5.3,- Espectrometria de massa
A espectrometria de massa foi na sua origem aplicada quase exclusivamente na análise quantitati va de gases. Vencidas, porém, dificuldades técnicas de vária ordem e aperfeiçoada a aparelhagem necessária à sua pratica;e3tendëu~se gradualmente, à análise qualitativa dos compostos orgânicos, sector em que, nesta úl tima década, tem vindo a ser cada vez mais utilizada.
Vários são os atributos desta técnica que a tornam tão atractiva na análise de produtos naturais,Is to, em primeiro lugar, pelo facto de, para a obtenção dum espectro, ser necessária uma quantidade pequeníssima de produto,depois, o pico correspondente ao ião molecular fornece a indicação da massa molecular do composto com um extremo rigor, não atingido nos processos clássi. cos para a sua determinação. Por outro lado, além de cons tituir um bom processo de identificação, por comparação do espectro da substância problema com o de um padrão ,

123
oferece possibilidades enormes na análise de produtos de estrutura não completamente esclarecida, permitindo, como nenhuma outra técnica até agora uoada sobretudo quandr apoiada pela R.M.N., estabelecer estruturas desconhecidas. Por último, é também útil e odmodo para a-companhar um processo de síntese e de purificaçãoesendo ainda de considerar que o espectro de um compcsto,rcs mo quando impuro, pode fornecer dados de inestimável va lor.
Esta técnica foi inicialmente aplicada à determinação de estruturas dos alcalóides do grupo indôli co pelas escolas de BIEMANN (65-66) e de DJBMSSI (67 --68). E de tal modo foi espectacular a sua aceitação nos te campo da química que nc curto espaço de tempo (10 a-nos) que medeia entre as suas primeiras aplicações até hoje, há talvez mais estruturas de bases indolicas determinadas por este processo do que as estabelecidas a-té àquela data pelas técnicas convencionais. Em todo o caso, a determinação de uma nova estrutura necessita mui tas vezes de apoiar-se também em reacções químicas, nos processos espectrofotométricos clássicos (ultravioleta, e infravermelho) e em dados de ressonância magnética nuclear, porque convém reunir o maior número possível de informações a fim de que fique determinada com segurança.

124
O notável sucesso registado pelo uso da espeç_ trofotonietria de massa neste campo está indubitavelmente ligado a factores inerentes à estrutura destes compostos.
De facto, após o impacto electrónico originam em regra um ião molecular bastante estável e por isso bem reconhecível no gráfico em virtude de estes compostos conterem dois centros (um sistema heteroaromático e um nitrogénio básico) cnde a carga positiva se pode estabilizar. Além disso, no seu esqueleto há ligações particularmente propensas à ruptura, originando estes compostos, por isso, um certo número de fragmentos comuns e bem característicos.
Assim, as indolalquilaminas sofrem cisão ao ní vel da cadeia lateral e os derivados \S -carbolínicos em regra no núcleo piridínico.
A interpretação do espectro de massa de uma indolalquilamina é, em regra, formulada por cisões do ião molecular progenitor formado por remoção de um eleç_ trão pertencente ao Na da molécula.
Em alguns casos, porém, a explicação para o aparecimento de alguns picos só é viável ou pelo menos mais fácil se, se admite a fragmentação do ião formado por remoção do electrão de N],, 0 ESQUEMA V permite explicar segundo o primeiro critério a formação de 3 frag mentos mais comuns resultantes da cisão destas substân-

12b
c i a s , ( a , b , c ) .
ESQUEMA. V
CH2CH2 N
CHoN
R l
m/e = 130
+ / R i
CHo
m/e ■ 144 0 i ã o m/e=130 corresponde em r eg ra ao pico "ba
se e r e s u l t a da cisão do i ão molecular ao n í v e l do
Cl—rC2 da cade ia l a t e r a l , enquanto que o ião m/e 144

126
se forma por cisão ao nível da ligação CH<? — ; — ^^^ daquela cadeia e é, em geral, menos abundante.
BIEMATOÍ observou que este esquema geral nafrag mentação das indolalquilaminas não é afectado pela presença de substituintes no anel benzénico. Como esses substituintes são, nas indolalquilaminas das Leguminosas, grupos ÇH ou OCH3, os ,picos base correspondem aos fragmentos referidos mas acrescidos de 16 ou 30 unidades massa, conforme na molécula se encontra o primeiro ou o segundo dos substituintes citados. Assim é que AHLBORG, HOLMSTEDT e LTNDGHEK (70) indicam para o DM?, 5-CE-DMFe 5-MeO-DMT os picos base respectivamente de m/e 130, 146 e 160. Os picos m/e 130 e 144 podem também observar--se por eliminação daqueles substituintes.
A detecção no espectrograma do fragmento (o) é de enorme utilidade e através dele se pode averiguar se a indolalquilamina em análise é uma amina primária, secundária ou terciária.
0 fragmento mais abundante nos alcalóides te-traidro fó -carbolínicos corresponde, em regra, ao ião de m/e = M-l, o qual se forma, a partir do ião molecular (ivl), por perda de um hidrogénio do C-l. Naquele ião a carga positiva fica perfeitamente estabilizada por efe_i to conjunto do sistema indólico e do par de electrões de H|JI sendo por isso o predominante.
Além destes picos podem observar-se outros de

127
m/e = 184; m/e - 170 e m/e = 169 cuja formação se pode facilmente conceber a partir do ião M-l.
^X <^X
LXIII ou
m/e - i69
LXVI
m/e = 176
LXVII
Tanto nos espectros de massa dos alcalóides te-traidro- |3 -carbolínicos como nos (¾ -carbolínicos podem detectar-se, além de muitos outros, os fragmentos seguintes!
^ \ ^
m/e = 168 LXVI
m/e =182 LXVII

128
El também característico o ião m/e = 156, cuja formação se pode explicar a partir do ião molecular,por fragmentação do anel tetraidropiridínico (C),estando en volvido neste mecanismo primeiramente uma fragmentação retro Diels-Alder seguida de uma cisão homolítica activada alilicamente.
^H^s CHr
LKVIII LXIX

129
BIBLIOGRAFIA
1 - HENRY. T.A., The Plant Alkaloids, The Blakiston Com pany-Philadelphia-Toronto, 4 s ed.(l949j.
2 - MANSKE, R.H.F. e HOLMES, H.L., The Alkaloids -Chemistry and Physiology, Academic Press Inc., Pub., New York, yol.II (1952).
3 - MANSKE, R.H.F., The Alkaloids - Chemistry and Physiology,Academic Press, New York -Lon don, Vol. VII (i960).
4 - RODD, E.H., Chemistry of Carbon Compounds, Elsevier Publishing Company, Vol. IV, Parte C (i960).
5 - BOIT H.-G., Ergebnisse der Alkaloid Chemie bis 1960, Akademie - Verlag-Berlin (l96l).
6 - HESSE, M., Indolalkaloide in Tabellen,Springer-Ver-lag-Berlin-Gõttingen-Heidelberg (1964)c
7 - MANSKE, R.H.F., The Alkaloids - Chemistry and Pny-siology, Academic Press - New York--London, Vol, VIII (1965).
8 - MANSKE, R.H.F., The Alkaloids - Chemistry and Pny-siology - Academic Press, New York--London, Vol. X (1968).
9 - HESSE, M„, Indolalkaloide in Tabellen,Springer-Verlag-Berlin- New York (1968).
10 - BERNAUER, K., Forschr. Chenu Org, Naturstoffe 17 , 192 (1959).
11 - The Merck Index of Chemicals and Drugs, 7s ed„Merck & C2.,Inc.,Rahwag,N.I.,U.S.A. (i960).

130
12 - MORRISON, R. e BOYD, R., Química Orgânica ( tradução de SILVA, M.A., Fundação Calouste Gulbenkian -Lisboa (1967).
13 - HOFMANN, A.W., Ber. 14, 659 (l88l)j ref. em GILMAN, H., Org. Chemistry II, p. 1172 New York (1943).
14 - FEIGL, P., Sppt tests in organic analysis, Elsevier Publishing Company. (1956)
15 - MULLER, R., Beitr, Biol, Pflanzen 30, 1 (1953). 16 - STRELL, 1., e KALOJANOFF, A., Chem. Ber. 87, 1025
("1954)7" 17 - EAELEY-MASON, J , e ARCHER, A.A.P.G., Biochem. J .
69, 60 (1958). " 18 - DICKMAN, S.R., WESCOTT, W.L., J. Biol. Chem. 210,
481 0-954*). 19 - DICKMAN, S.R. e CROCKETT, A.L., J. Biol. Chem.220.
957 (1956). 20 - GERNGR03S, 0 . , VOSS, K. e HERFELD, H. B e r . d t s c h .
Chem. Ges.66. 435 ( 193377"^ 21 - TTDENFRIEND, S . , WEISSBACH, S . e CLARK, C.T. ,
J . B i o l . Chem. 215, 337 (1955) . 22 - IIDËNFRIEND, S . , e WEISSBACH, H.-, em COLOWIGK S. P .
e KAPLAN, N.O., Method in Enzymolo-gy, Vol. 6, 598-605^.New York(l963).
23 - ERSPAMER, V., BORETTI, G0, Arch, int.. Pharmacodyn, 88, 296 (1951).
24 - ERSPAMER, V. , R.C. S o i . F a r m i t a l i a 1, 1 (1954) . 25 - ERSPAMER, V. , J j f t g s i o l . 127, 118 (1955) . 26 - JEPSON, J . B . e ZALTZMAN, P . , DDENFRIEND, Biochim.
Biophys. Acta 62, 91 (1962) . 27 - HDTZINGER, 0 . , J . Chromâtog. 40, 117 (1969) .

151
28 -BOWMAN, R .L . , CAULFIELD, P.A. e UDENFRIEND, S . , Science 122. 32 (1955).
29 - FISH, M.S., JOHNSON, M.M, e HORNING, E.C., J. Am. Chem. Soo. 77, 5892 (1955).
30 - UDENFRIEND, S,, Science 122, 972 (.1955). 31 - QUAY, W.B., Analyt. Biochem. 5, 51 (1963). 32 - FEARON, W.R. e BOGGUST, W.A., Biochem, J. 46, 62
TTo"5Õ). " 33 - JEPSON, J . B . e STEVENS, B . J . , Nature 172, 772
XÏ953). 34 - HESS, S.M. e UDENFRIEND, S., J Pharmacol, exp.
Ther. 127, 175 (1959). 35 - JEPSON, J . B . , em SMITH, I . , Chromatographic and
e l e c t r o p h o r e t i c t e c h n i q u e s , V o l . 1 , p . 183-211 - London ( i 9 6 0 ) .
36 - PROCHAZKA, 2 . , em HAIS, I .M. e MAGEK, K., Handbuch der Papierchromatographie , Vol. I , 585-592. Jena (1958) .
37 - SHEPHERD, D.M., WEST, G.B. e ERSPAMER, V. , Nature 172, 357 (1953).
38 - QUAY, W.B., J. Pharm. Soi. 57, 1568 (1968). 39 - BLOCK, R.J.,DURRUM, E.L., ZHEIG, G. A manual of
paper Chromatography and paper electro phoresis - Acad-eniic PreE3 Inc, New York ( 195*8).
40 - McISAAC, W.M., HO, B.T., ESTEVEZ, V.C., POWERS. D., J . Chromât,og. 3 1 , 444 (1967) .
41 - HOLMSTEDT, Be e VANDENHEUVEL, V.J .A. e GARDINER,W. L. e HORNING, E .C . , Analyt.Biochem. 8, 151 (1964) .
42 - SANGTER, A.W. e STUART, K.L., Chem. Rev. 65, 69 (1965) .

132
43 - KBÍJSS, N . , Phys ica l Dat a of Indole e Dihydro.indolè. A l k a l o i d s , Ely L i l l y & C2. , I r id ianopol i s 6, Ind iana , U.S.A. Ed. 1954, 1956,1960, 1961,1962,1964,1966, 1968.
44 - STEOMBERG,.: V.L. , J . Am. Chem. Spc. 76, 1707 (1954).
45 - ERSPAMER, V. e ASERO, B . , Nature 169, 800 (1952) .
46 - M0RIM0T0, H . , OSHIO, H . , Ann.de r Ghem. 682, 212 Tl965 )•
47 - BERNATJER, K., Helv . Chim. Acta 47 , 1075 (1964) .
48 - KEUFER, J » , Tese dout . , Travaux de Labora to i res de Matière Médicale e t de Pharmacie Galé-nique (1954) .
49 - RAYMOND-HAMET, Comp. Rend. 229, 1165 ( 1 9 4 9 ) , i b i d . 1359 TÎ949J .
50 - RáYMOND-HAMET, Comp. Rend. 232, 507 ( l 9 5 l ) . 51 - SPENSER, I . D . , J„ Chem. Soc. 3659 (1956) . 52 - WARREN, R . J . , EISDORFER, I . B . , THOMPSON, W.E., e
ZAREMBO, J . E . , J . Pharm. So i , 54, 1806 (1965) , 53 - KANAOKA, Y., BANY, Y . , OISHI, I . , YONEMETSU, 0 . ,
TEPASHlMà, M., KEMÏÏRATT e NAKâGAWA, M., Chenu Pharm. B u l l . Tokyo 8,294^1960)
54 - MARION, L . , RAMSAY, D.A., e JONES, R.N., J . Chem, Soc. 305, (l95Ï77~
55 - THOMPSON, W.E., WARREN, R.J., EISDORFER, I.B. e ZâREMBO, J.E., J. Pharm. Soi. 54, 1819 (1965),
56 - EISDORFER, I.B., WARREN, R.J. e ZâREMBO, J.E., J . Pharm. Soi 57, 195 (1968) .
57 - MESLEY, R . J . , e EVANS, ïï.II., J . Pharm. Pharmac.22, 321 (1970) .
58 - RAO, C.N.R., Chemical Applications of Infrared Spe'ctro'sas.opy,Academic PTPSS Ire..
New York U963).

133
59 MASON, S.E., J. Chem, Soc. 3619 (1958). 60 WITKOP, B, e PATRICK, J.B., J. Am. Chem. Spc. 73,
713, e 1558 (1951) . ol WHETSEL, K.B., ROBERTSON, W.E, e KRELL, N.W.,
Anal . Chem, 32, 1281 ( i 9 6 0 ) .
62 NAKANISHI, K., I n f r a r e d absorp t ion Spectroscopy, HoldenDayIno. , San F r a n c i s c o C a l l f o r n i a (1962) ,
63 SADTLER Standard Spec t r a , publ i shed S a d t l e r Research L a b o r a t o r i e s , I n c . Phi lade lph ia ,Pa .
64 DUVAL, C , Chim, Anal . 42, 117 ( i 9 6 0 ) .
65 BIEMANN, K,, Tetrahedron L e t t e r s 15, 9 ( i 9 6 0 ) .
66 BIEMAM, K., Mass Spetrometry, McGRAWHILL, New York (1962) .
67 BJERASSI, C , GILBERT, B . , SH00LERY, J .N.,JOHNSON, L.F . e BIEMANN, K., E x p e r i e n t i a 17, 162 (1961) .
68 BUDZIKIEWICZ, H.,BJERASSI, C. e WILLIAMS, D.H., In t e r p r e t a t i o n of mans spect ra ' , of Orga, n ic Compounds, HoldenDay, San Franc i s co (1964) .
69 BUDZIKIEWICZ, H.,DJERASSI, C. , WILLIAMS, D .11,, S t r u c t u r e e l u c i d a t i o n of n a t u r a l pro ducts by Mass Spectrometry, Vol. I : A l k a l o i d s , HoldenDay, San Francisco, London Amsterdam (1964) .
70 AHLB0RG,U., ÏÏOLMSTEDT, B. LINDGREN , ■!■■}& 9 Adv., in Pharmacol. 6, ,Parte £,213^1968).

135
1.5-ALGimS ASPECTOS DA FARMACOLOGIA PAS DJDQIAIQ,UI-
LAMINAS E PAS Q) -CARBOLIMS
1.5.1,- Indolalquilaminas
Ao abordar, ainda que vagamente, a fármacolo gia das indolalquilaminas, assunto a respeito do qual se encontra publicada uma extensa bibliografia,tomámos, como substância tipo, a 5-hidroxitriptamina. Na verdade, embora os diversos compostos deste grupo mostrem d_i ferenças na actividade farmacodinâmica,relacionadas com modificações de estrutura química, umas ao nível do a-nel indólico e outras na cadeia lateral, é, contudo, a serotonina o representante mais qualificado. A importai cia deste composto deve-se não só à sua larga distribui ção nos reinos animal e vegetal, mas, sobretudo, ao relevante papel por ele desempenhado na fisiologia animal.
As investigações de EKSPAftïER e col. (l),reali aadas em células enterocromafins, no propósito de isolarem a substância responsável pelas propriedades histo, químicas particulares por elas evidenciadas,levaram à descoberta da entcramina, Os estudos sistemáticos desde então efectuados foram esclarecendo,progressivamente,as propriedades farmacodinâmicas desta substância e a sua constituição química, tendo-lhe atribuído aqueles auto res uma natureza indólica (2, 3).

136
Preocupava-se, por esta mesma data, a equipa de RAPPORT (4, 5 e 6) com o isolamento do princípio res ponsável pelas propriedades vasoconstrictoras e hiperten soras do soro sanguíneo, conhecidas desde há muitos ancs.
Os trabalhos de RAPPORT e col. culminaram com a separação duma substância que designaram s?rotôàJ.na }
tendo ;sugèayidaeque a mesma.corresponde a 5-hidroxitrip-tamina.
Os longos estudos efectuados acabaram por demonstrar que a enteramina e a serotonina eram, na realidade, uma mesma substância.
HAMLIN e FISCHER (7) obtiveram, em 1951, por síntese, a 5-HT, composto que evidenciava as mesmas pro priedades da serotonina natural,
A designação 5-hidroxitriptamina foi a que prevaleceu, adoptando-se, correntemente, a abreviatura proposta por BACQ (8) 5-HT.
São muito numerosos os trabalhos que provam a larga distribuição da 5-HT nos seres vivos e a alguns de les tivemos já oportunidade de aludir na PARTE II, Cap„ 1-2. Diversos autores defendem até que o papel fisiológico desempenhado por esta substância nos animais e vegetais seja de certo modo idêntico, mas tal facto não está esclarecido» Quem, pela primeira vez, chamou a a-tenção para a estreita semelhança entre o ácido 5-hidro-xindolilacetioo, a auxina, hormona de crescimento nas

137
plantas, e a amina animal, serotonina, foi WOOLEY (?.0), em 1957. De facto, ambas se formam partindo do triptofa-no, estando envolvidos, nos processos enzimáticos que estão na sua origem, enzimas comuns. Nos vegetais, contudo, há uma etapa suplementar que conduz ao ácido 5-hi droxindolíacético e nos animais este composto rn^ulta, entre outros processos, do catabolismo e inactivação da sero.tom.na.
IABORÍT (li) atribui à serotonina função no crescimento dos seres vivos, animais e vegetais, NIAU3-3AT, LABORIT e DUBOIS (12) verificaram, em experiências com aveia, uma acção favorecedora do crescimento tauto com a auxina como com a serotonina.
Em relação aos animais, LABOFJT (l3) formula a hipótese de que a somatotrofina hipofisiária daria lu gar à formação de serotonina que seria um factor essencial na actividade da citada hormona. A verdade, porém, é que é grande o número de interrogações que persistem em muitos aspectos da fisiologia, bioquímica e farmacologia da 5-HT, sendo ainda extenso o caminho a trilhar para se chegar ao seu completo esclarecimento. Podemos dizer que é vago e confuso, ainda hoje, o que se sabe quanto ao papel desempenhado pela 5-HT. Parece estar im plicada em diversos mecanismos fisiológicos e na determinação de certos quadros patológicos. Assim,refere-se, entre outros, a sua influência na pressão sanguínea, o

138
seu papel no funcionamento renal, a sua participação na tumefacção mitocondrial, na fagocitose, no colagénio e vários efeitos no S.N.C.
A 5-HT encontra-se no organismo humano, principalmente no intestino e no cérebro, mostrando-se sobretudo ricos o hipotálamo e as plaquetas. Enquanto que as plaquetas captam activamente este autacóide . do sangue, no cérebro e noutros tecidos ele é elaborado a par tir do triptofano que sofre uma hidroxilação em que intervém a triptofanoidroxilaae sendo depois o 5-hidroxi-triptofano descarboxilado por acção da 5-HT.P descarboxJr-lase, como referiremos - PARTE II Gap. 1-6,quando abqr darmo3 o problema da biogénese das indolalquilaminas.
No homem e na maioria dos mamíferos a principal via de destruição da serotonina é por desaminação oxidativa.
Após administração de serotonina (LXXl)marca-da, por via oral, esta é, algumas horas mais tarde, excretada na proporção de 80% sob a forma de ácido ,5~hi-droxindolilacético»
Esta transformação produz--se em 2 tempos(l4). Primeiramente, há formação de ]>-hidroxindqlj.laceta3daí» do (LXXII) sob a influência da monoaminoxidase, e,a per tir deste, por oxidação catalizada pelo DPN é que se for ma o ácido 5-hidroxindolilacético (LXXIIl),segundo o ES_ QUEM. VI.

139
ESQUEMA. VI
CHO
LXXI
! B
LXXII (
DRToxid, \ ic
HO N ^ S CGOÍI
i LXX-».J-J.
BULBRIWG, CREMA (l5) e BOULIN (IG) entre outros, atribuem-lhe uma função reguladora do peristaltiB mo intestinal.
A sua presença nas plaquetas pode srighifioar que estes elementos figurados captam do sangue a amina, com o fim de manter baixa a sua concentração»
Ainda que alguns autores tenham defendido a intervenção deste composto na hemostasû, o corto 6 que hoje não se lhe atribui qualquer papel de importância na coagulação sanguínea ou na hémostase.
Sabemos do notável desenvolvimento já atingido pela psicofarmacologia. 0 estudo de substâncias que

140
provocam modificações fisiológicas que se repercutem no psiquismo, ao mesmo tempo que se perspectiva benefícios terapêuticos destes novos fármacos no campo da psiquiatria, trazem valiosa contribuição para o esclarecimento da psicofisiologia e da etiopatogenia das perturbações mentais,
Nesta experimentação psicofarmacológica tem--se lançado mão de substâncias que interferem com os e-feitos da 5-HT. Admite-se que esta substância é um mediador do sistema nervoso e a experimentação prova que estimula as terminações nervosas sensitivas.
Diversos autores, entre os quais W00LEY e S HAW (l?), contam-se entre os primeiros que sugerem que em certas perturbações mentais, nomeadamente psicoses es-quizóide3; esteja implicado um transtorno no metabolismo da 5-HT.
1.5.1,,1.3- Actividade farmacodinâmica da 5-HT
1,5.1,1*1,- Aocão sobre o músculo liso
A actividade farmacodinâmica fundamental veri ficada na 5-HT é a sua acção estimulante sobre a musculatura lisa.
A contracção do músculo liso é, na realidade, de modo genérico o efeito observado na experimentação realizada "in vitro" e "in vivo". Numerosos ensaios pra

141
ticados com fragmentos de vasos sanguíneos mostram a aç ção constritora desta substância sobre os referidos or_ gãos. Do mesmo modo, em trabalhos realizados com brônquios, isolados ou em experiências "in vivo", se verifi cou que este composto determina broncoconstrição, Re-velam-se particularmente sensíveis as preparações do ú-tero, cólon e estômago de rato que, por esta razão, se encontram propostas para os ensaios biológicos da 5-HT.
No homem, a 5-HT estimula a motilidade intestinal, sobretudo do intestino delgado, já que em certas circunstâncias se tem verificado um efeito distinto sobre o cólon,
Admite-se que este efeito é consecutivo à acção directa sobre a fibra muscular lisa e ainda à excitação de células glanglionares intramurais, Descrevem--se dois tipos de receptores designados por D e M,de a-cordo com as substâncias capazes de promoverem o respe_c tivo bloqueio. A dibenzilina ou fenoxizamina e morfina<,
0 mecanismo de acção não está esclarecido, admitindo-3e 2+
que a 5-HT dá lugar a tun aumento de influxo do ião Ca .
1.5.1.1.2.- Acção 3obi-3 o aparelho cárdi o-vascular
0 fármaco actua sobre a musculatura lisa dos vasos sanguíneos determinando vasoconstrição,efeito so bretudo nítido se se usam nos ensaios preparações ener-

142
vadas, A sensibilidade manifestada é, contudo,diferente, conforme as diversas regiões orgânicas dcnde provêm as preparações, sendo muito intensa a resposta observada com vasos renais. Os vasos sanguíneos dos músculos estriados dilatam-se sobre a acção da 5-HT, sobretudo se esta se aplica em pequena dose. De igual modo, verifica -se dilatação dos vasos coronários em resposta à adminis tração da 5-HT, Conclui-se, pois, que o efeito depende da região vascular considerada, mas há ainda que contar com outros factores, entre eles, a tonicidade inicial e a dose da substância administrada.
As experiências realizadas com músculo papi-lar e coração isolados revelam que a 5-HT tem acção ino trópica e cronotrópica positivas. Esta actividade pode pôr-se em evidência em ensaios praticados com corações de mamíferos, mas é* mais sensível o coração de • Lalgun,? molúsculcs, Compreende-se, assim, que o coração isolado da Venus mercenária tenha sido largamente utilizado para a aferição biológica da 5-HT.
Este efeito estimulante sobre o coração veri-fica-se igualmente quando a substância é administrada ao animal intacto e no homem e é resultante de uma acção directa sobre aquele órgão. Há, no entanto, que atender a que, nesta última condição experimental, são postos em jogo diversos outros mecanismos que influenciam o e-feito final observado.

143
A repercussão verificada na pressão sanguínea determinada pela 5-HT e outras indolalquilaminas depende da dose, da via de administração, do estado anterior da pressão sanguínea, da espécie animal utilizada na ex perimentação, da presença ou não de anestésico, etc,,ha vendo que atender a que dá lugar a taquifilaxia. Dâecre ve-se uma resposta trifásica, em que a substância determina hipertensão precedida e seguida por fases de hi potensão. A "breve queda da pressão sanguínea, observada logo a seguir à injecção do fármaco, atribui-se à esti mulação de quimioreceptores e, possivelmente, de baro-receptores. A hipertensão explica-se pelo aumento da re sistência periférica do afluxo venoso e do volume sistó lico. A hipotensão consecutiva à hipertensão interpreta -se como sendo devida à vasodilatação, sobretudo dos va sos dos músculos esqueléticos.Determinam, mais regularmente, hipertensão a triptamina e substâncias afins sem hidróxilos no anel indólico e a bufotenidina.
1.5.1.1.3,- Acção sobre o aparelho respiratório
A 5-HT também influi na respiração, mas de um modo um tanto variável, sendo, por via de regra, estimu lante. A análise dos resultados experimentais referidos por diversos investigadores mostra que a resposta varia não só com as condições experimentais mas ainda com a

144
dose de fármaco e a espécie animal em ensaio. No homem determina um aumento de volume minuto por mecanismo re flexo, graças à estimulação de químioreceptores aórticos e carotídeos, devendo intervir um efeito central,se aão aplicadas doses elevadas de 5-HT,
1.5.1.1.4,- Acção sobre o sistema nervoso central
A 5-HT atravessa com dificuldade a barreira hemoencefálica. A aplicação parentérica da substância só em dose elevada dá lugar a efeitos centrais.
Não se sabe se os efeitos estimulantes e depressores observados na experimentação animal são o resultado de uma acção directa ou reflexa. Aceita-se que seja consequência, não só das acções vasculares mas tam bem da estimulação de fibras nervosas aferentes. As experiências de FELDBERG e SHERWOOD (19), em que injectaram o fármaco no ventrículo do gato, demonstram que dá lugar a um estado letárgico. Alguns investigadores obti veram respostas de células cerebrais, aplicando a 5-ET com auxílio de micropipet as.
BROME (20) admite que a acção sedativa, veri_ ficada quando se usa na terapêutica a reserpina,seja re sultante de uma libertação de serotonina.
Também FELDBERG (21) atribui à 5-HT hipotaiâ-mica uma função no mecanismo de regularização da temperatura corporal.

145
A experimentação histológica efc- tteurofármacoló gica permite admitir que a 5-HT esteja envolvida no mecanismo do sono.
1.5.2.- Alcalóides (3 -carbolínicos
São pelo contrário pouco numerosas as referências bibliográficas que encontrámos a respeito da far macologia das (¾ -carbolinas naturais.
Num trabalho publicado sobre alcalo'ides indo'-licos pelo departamento de Farmacodinamia da Faculdade de Farmácia do Porto (22), aludem os autores à escassez de dados bibliográficos sobre a harmana ,
Recorremos sobretudo ao trabalho de GUNN (23) de que retirámos algumas notas sobre a actividade farma codinâmica de alguns destes compostos.
1.5.2.1,- Acção sobre o músculo
Refere este autor que a harmina e a harmalina têm actividade inibidora sobre o músculo liso, isolado. São contudo, segundo o referido autor, estimulantes do músculo uterino. Alguns trabalhos levam aconsiderar que esta contracção do útero se verifica sobretudo quando estes alcalo'ides são injectados no animal intacto.
0 músculo esquelético de rã,isolado, contrai--se por acção destes compostos.

146
1.5.3.2.- Acção sobre o coração
Em ensaios de perfusão de corações isolados de rã, coelho e gato, verificou-se um efeito depressor, ao mesmo tempo que um aumento do débito coronário.
1.5.2.3,- Acção sobre a pressão sanguínea
Quando se injectam pequenas doses desta substância em animais preparados para registo da pressão sanguínea, observa-se uma pequena elevação de pressão, enquanto que as doses elevadas têm um efeito hipotensor.
1.5,2.4,- Acgad ãúWéd ãi§tema nervoso eenfral
Estes alcalóides são estimulantes do S.N.C. . Desde há muito que se sabe que a sua adminis
tração a animais de laboratório dá lugar ao aparecimen to de tremor.
A aplicação em doses elevadas origina apareci mento de convulsões epileptiformes, mostrando-se mais aç tiva a harmalina do que a harmana.
Alguns destes derivados têm acção antibacteri ana e actividade em relação a certos protozoários e hel_ mintas.

147
1.5»3.- Acção alucinogénica das indolalquilaminas e dos derivados B-carbolínicos
Entre as tribos de índios do América Central e do Sul, data desde há séculos, nas suas práticas reli giosas com fins proféticos e de divinização, o uso de plantas, de "Snuffs" e beberragens de acção psicomimét? ca.
0 "epena" e o "cohoba" constituem dois desses preparados "Snuffs" que os índios da América do Sul uti lizavam por inalação.
Embora seja grande o mistério envolvido na pre paração destes produtos e a sua composição varie de tri bo para tribo, sabe-se hoje figurarem nela espécies de Virola (.Myristicaceae) e de Piptadenia (jLegyjninoB&Q)f
Parece que fazem parte das beberragens conhecidas por "natem", "caapi", "yahusoa" e "yagé" certas espécies de Banisteriopsis (B. caapi, B. inebrians, B_. rusbyana) da família das Malpighiaceae, quer isoladamente, quer em mistura com outras espécies.
Também às sementes de Poganum harmala, da família das Zygophylaceae, são atiúbuídas na índia efeitos psicotrópicos, bem como a certos cogumelos maxioa-nos dos géneros s Conocybo, Psilocybe e Stropharia.
A análise química destas drogas, dita3 aluci-noffénicas, eidéticas, psicomiméticas, etc., revelou, em

148
Peranum harmala e nas espéc ies de E a n i s t e r i o p s i s , d e r i
vado s \ 3 - c a r b o l í n i c o s , e em P i p t a d e n i a , V i r o l a e nos .co
gumelos mexicanos, i ndo l a lqu i l aminas .
DELAY (24) propo3 para e s t a s drogas mágicas a
designação de p s i c o d i s l é p t i c a s , sendo o grupo encimado
pe lo LSD25.
En t re e s t e preparado de s í n t e s e e as subs tân
c i a s n a t u r a i s i s o l a d a s de p l a n t a s com acção a luc inogéni
ca (harmàtia, harmal ina , t e t r a i d r o a r m i n a t p s i l o c i n a , p s i -
l o c i b i n a , D M T , bufo ten ina , os N-^-óxidos d e s t e s dois
ú l t imos de r ivados , a 5 -metoxi - -HbA,-d imet i l t r ip tamina ,
e t c 3 ) há uma semelhança e s t r u t u r a l f lagrante ,como a l i á s ,
o fazem n o t a r HOFFMAM (35) , VALCUROME e CEERI (26)e
como poderemos observai" nas moléculas i n s c r i t a s no QUA
DRO VI I . Nelas sal ientamos por t r açado mais espesso a
p a r t e molecular comum. Q U A D R O VII
Fórmula A Composto
CH9CH~ u | VH2C5H3
Diet i l amida do ácido l i s é r g i c o LSDgj-
<^" ^ J S « C H 3
Diet i l amida do ácido l i s é r g i c o LSDgj-
^ ^ !r
Diet i l amida do ácido l i s é r g i c o LSDgj-
1 H
Die t i l amida do ácido l i s é r g i c o LSDgj-
1 1

149
f ^ ^ ,., - > " ^ s Harmalina
H3Cr Harmalina
DMT (R=Rx=H)
Bufotenina(R=H5 R-,=OH
5-MeO - Di\ÏT(R=HjR1=ŒH^
P s i l o c i n a (R=OH$ B,«fl)
Psilocibina(R=OPO(OH)25 Ct7 i H
DMT (R=Rx=H)
Bufotenina(R=H5 R-,=OH
5-MeO - Di\ÏT(R=HjR1=ŒH^
P s i l o c i n a (R=OH$ B,«fl)
Psilocibina(R=OPO(OH)25
As drogas psicodislépticas produzem efeitos profundos no psiquismo, caracterizando-se, essencialmen te, por uma acção despersonalizaste, uma alteração da noção do tempo, do espaço e do esquema corporal, e, em doses elevadas, provocam visões e alucinações.
As doses necessárias para que estas substâncias produzam efeitos alucinogénicos no homem são muito variáveis. Enquanto que para o LSDpts bastam doses de 0,08 mg, de psilocina e psiocibina são necessários 10 -- 30 mg, e de dimeti.ltriptamina 50 - 100 mg. A DMT e a 5-MeO-DMT são mais activas que a 5-QH-DMT,explicando-se o facto pela maior lipossolubilidade daqueles compostos,

150
daí resultando uma maior facilidade de penetração no sis tema nervoso.
Parece, pois5 que, para que uma indolalquila-mina seja activa, ê indispensável a alquilação da função amina da cadeia lateral, exacerbando-se essa acção pela presença de grupos OH na posição 4 do núcleo indó-lico (psilocina e psilocibina),
A harmalina parece ser a (\ -carbolina mais a_c tiva, sendo a sua acção cerca de duas vezes superior à da t et raidro armina -,
Muitas experiências tendentes a esclarecer os efeitos destas substâncias seriam dignas de citação,en-contrando-se uma descrição "bastante completa da etnofar macologia dos efeitos psicomiméticos das drogas,na obra Ethnopharmacologie Search for Psychoactive Drugs (27).No entanto, os mecanismos pelos quais os psicodislépticos exercem a sua acção sobre as funções mentais estão ainda longe de ser perfeitamente esclarecidos, sendo de considerar a sua interferência no metabolismo da seroto-nina. Os psicodislépticos mostram alguns efeitos comuns resultantes da estimulação de certas zonas do diencéfa-lOc traduzindo-se nos animais de laboratório por determinada sintomatologia s hipertermia, hiperglicemia , mi-dnase, piloerição e taquipneia. Os centros nervosos ficam sensibilizados à acção dos estímulos exteriores (o'pticos e acústicos) e verifica-se um encurtamento do

151
tempo ao estímulo nervoso. Estas substâncias não têm só a propriedade de
produzir estados de excitação e transtornos sensoriais como, provocam também um verdadeiro ressurgimento do sub consciente e a elas se tem recorrido não só na terapêutica de certas doenças mentais como na investigação far macológica, em "busca do esclarecimento das causas bioquímicas daquelas doenças.
BIBLIOGRAFIA
1 - Através de ERSPAMER, V,, Pharm. Rev, 6, 425 (1954). 2 - ERSPAMER s V., Arch. Sei. Biol. 31, 86 (1946). 3 - ERSPAMER, V., Arch. Internt. Pharmacodyn. e Thér,
"76, 308 (1948), 4 - RAPPORT, M.M., GREEN, A.A., e PAGE, I.H., Science
108, 329 (1948). 5 - I b i d , J . B i o l . Chem. 176, 1243 (1948) .
6 - RAPPORT, M.M., J . B i o l . Chem. 180, 961 (1949) .
7 - HAMLIN, K,E. e FISCHER, F . E . , J . Am. Chem. S Q C 73, 5007 (1951) .
8 - BACQ, Z.M,, II- Congres Intern, de Biochemie,Paris. pag. 59 (1952).
9 - DEYSSON G*, Produi t s Pharmaceutiques 17, 62(1952) 10 - WOOLLEY, D.W., Nature 180, 630 (1957) . 11 - LABORTT, H . , NIAUSSAT, P . , BROUSSOLLE, B . , JOUANY ,
J .M. , Press Med. 67, 927 (1959) .

NIAUSSAT, P . , LABORIT, H . , NIAUStíAT, M. e DUBOIS,C., C R , Soc. B i o l , 152, 945 (1958) .
NIAUSSAT, P . e LABORIT, H . , Med, exp . 1, 207 (1959).
WHITE, A. , HANDLER, P . , SMITH, E. L . , P r i n c i p l e s of Biochemis t ry , Mcgraw-Hill- Book Company, pag . 589 - 4 » . edição (1968) .
BÎJLBRING, E. e CREMA, A . , J . Phva io l . 146, 29(1959).
BOULIN, D . J . , B r . J . Pharmac. Chemother. 23 , 14 -(1954) .
WOOLEY, D.W. e SHAW, E , , Science 119, 587 (1954) .
DOUGLAS, W.W. em GOODMAN, L .S . e GILMA.NN, A, , The Pharmacological b a s i s of Therapeutics , Macmillan Company, New York, 3 a . e d i ção pag. 644 (1966) .
PELDBERG, W. e SHERWOOD, S-.L/. J . P h y s i o l . 123,148 (1954) .
BRODIE, B .B . , PLETSCHER, A. e SHORE, P .A. .Sc ience 122, 968 (1955).
FELDBERG, W. e SMITH, A.N..Br. J. Pharmc. Chemo-"-ttfer. 8, 406 (1953).
CORREIA DA SILVA, A., COSTA, A., PAIVA, M.Q., Garcia de Orta 14 (n^. 1),91 (1966).
GUNN, J.A,, The harmine group of alkaloids,Hahbuch der experimentalen Pharmacologie V band (1937).
DELAY, J., PICHOT, P., LEMPERIERE, T. Press Med. 67, 1731 (1959).
HOFMANN, A., Colloques Internationaux du Centre Na tional de la Recherche Scientifique 144, 223 (1966).
VALCURONE, G. e CERRI, 0., Boll. Chim. Farm. 10s. 340 (1966).
Etnopharmacologic search for Psychoactive Drugs U. S. Public, Health. Serv» Publ. n^.1645,Government Printing Office, Washington, D.C. (196?).

153
1.6. - BIOGËXESS DAS INDOLALQUILAMMS E DOS DERIVADOS (¾ -CAKBOLIECCOS
Os alcalóides indólicos são, desde há muito, considerados biogenèticamente derivado3 do triplofano (l,2). ultimamente, esta origem ficou perfeitamente demonstrada por experiências conduzidas em várias plantas, em que se conseguiu a incorporação daquele am.iiHoáiido marcado na molécula de muitas bases inddlicas (3,4 e 5),
1.6.1,- Origem biogenótica das indolalquilaminas
0 estudo da origem, acção e metabolismo das indolalquilaminas é assunto de grande actualidade e tem sido objecto de intensas especulações, sobretudo no que diz respeito ao reino animal, dado o interesse de que se revestem estas substâncias nos mamíferos.
A primeira referência, quanto à capacidade dos tecidos dos mamíferos catalizarem a síntese da3 indolal_ quilaminao, deve-se, provavelmente, a WERLE e MEMICKEN (6). Estes investigadores, incubando triptofano com extracto de rim de cobaio, obtiveram triptamina cuja iden tificação estabeleceram em função do seu efeito vasopressor.
A hipótese quanto à existência, naquele tecido, de uma triptofanodescarboxilase não teve aceitapo

154
unânime entre os investigadores e concorreram, para que a dúvida surgisse, o modo impreciso de que aqueles auto res se serviram para identificar a triptamina, o facto de ser insignificante a quantidade de substância produzida no decurso da experiência e ainda a possibilidade de esta poder surgir como produto do metabolismo putre-factivo das bactérias sobre o triptofano, durante aquela incubação.
Decorreram mais de dez anos entre as observações de WERLS e MElíNICKM e a descoberta da serotonina, A esta indolalquilamina e em virtude das acções fármaco lógicas que revelou, desde logo foi atribuído um importante papel na actividade mental do homem, e daí todo o interesse manifestado pelos investigadores em esclarecer a sua origem.
BLASGHKO (7), apoiando-se na3 experiências de WERLS e MI3NNICKEM, encontrou a resposta a tal questão> Segundo este autor, o substracto directo da descarboxi-lase referida por WERLE e MENHICKEN seria o 5-hidroxi-triptofano em vez do triptofano_, facto que explicava o baixo rendimento em triptamina que aqueles autores con seguiram nas sua3 experiências,
BLASGHKO admitiu, pois, a formação de serotonina (LXXVl) por descarboxilação enzimática do 5-hidro-xitriptofano (LXXY), O qual, por sua vez, resultaria, possivelmente, da hidroxilação do triptofano (LXXIV)„

155
Esta sugestão que hoje se ace i ta , encontrou a poio em várias experiências e em descobertas porteriores (8) , além de que nenhuma experiência se mostrou convincente quanto à possível hidroxilaçâb se poder ve r i f i ca r sobre a t r iptamina. em vez de se observar inicialmente sobre o t r ip tofano. como BLASGHKO previ ra .
De facto, o isolamento de uma "descarbaiiliaijo do rim de mamíferos, act iva sobre o 5-h idroxi t r i t r ip to-fano e inact iva sobre o tr iptofano e sobre a 7-hidroxi-tr iptofano (9, 10), veio apoiar a hipo'tese a t rás r e fe r i da.
Hoje es tá bem demonstrado que es ta 5-hidroxi-triptofanodescarboxilase é a mesma enzima que H0HTZ,HEI SE e LUDTKE ( l i ) haviam descoberto, em 1938, no rim de cobaio, a dopadescarboxilase.
A TOENFRIEKD e co l . (12, 13) coube a demonstração evidente da hidroxilação do t r ip tofano . Estes au tores alimentaram animais da espe'cie Bufo marinus com I 14 \ ~ ————— (2 - C) L-triptofano e do seu veneno glandular i so la ram, alam de 5-hidroxitriptofano f outros compostos ra dioactivos dele derivados. Além disso , MITOMâ, ViTEIStíBACH e TÍDEEPRIEKD (14) observaram ainda a conversão do t r i p tofano naquele aminoácido pela Chromobacterium - v io la-ceum.
Nessa mesma a l tura DALGLIESH (15) identifioou o 5-hidroxitriptofano na urina de um paciente sofrendo

156
de neoplasia enterocromafínico. Posterior evidência de que o 5-hidroxitripto-
fano é um intermediário na síntese da serotonina foi da da por TTOENFRIEttD e WEISSBACH (lô), os quais administra ram a animais triptofano e 5-hidroxitriptofano. marcados, tendo a administração deste último produzido um aumento de 5-HT no cérebro e nas plaquetas, com subsquente aparecimento desta indolalquilamina noutros tecidos e aumento da excreção urinária de ácido 5-hidroxindolilace-tico.
Estudos efetuados por DONALDSON, GRA.Y e LETtíOU (17) sobre doentes com carcinoma (argentofinoma),a quem fizeram a administração oral de (14C) triptofano, leva ram ao isolamento de (14C)-5-hidroxitriptofano da urina desses pacientes.
Em 1959, restava por esclarecer qual o conjun to enzimático que estaria envolvido nesta hidroxilação. PEBEDLAHD, WADZINSKI e WAISMAN (l8) e ainda BENSON, WEISSBACH e UDENFRIEND (19), entre outros investigadores, conseguiram demonstrar a conversão do triptofano (LXXIV) em 5-hidroxitriptofano, (LXXV), pela fenilalani-naidroxilaae... do fígado dos mamíferos. Apesar disso,crê -se que, embora aquela enzima desempenhe certo papel de hidroxilação do triptofano, esta parece ser efectuada por outra enzima diferente e mais activa (20). Admite--se, pois,o ESQUEMA. VII, para a formação do 5-HT.
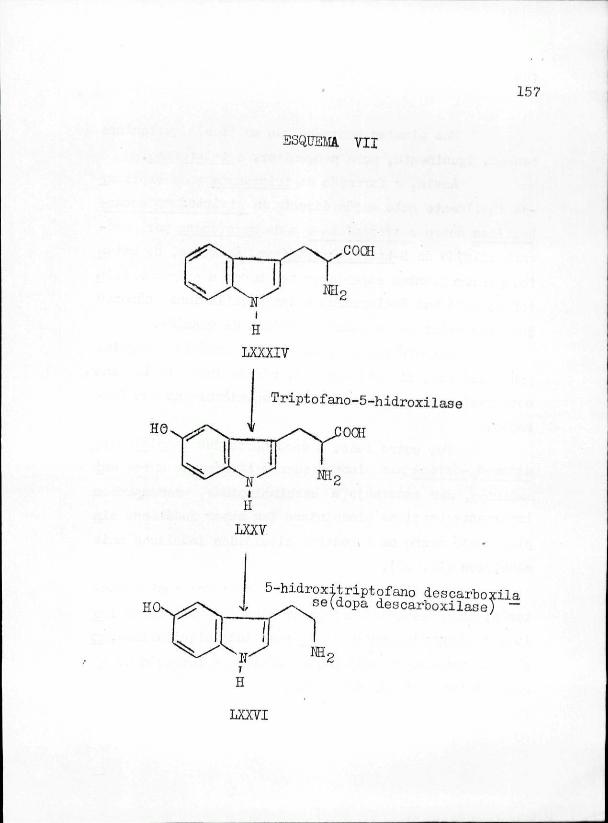
ESQUEMA. V I I
157
HO N ^
coce
LXXXIV
T r i p t o f a n o - 5 - h i d r o x i l a s e
ÎOCH
» H
LXXV
m.
5-hidroxitriptofano descarboxila se(dopa descarboxilase) -
m.
LXXVI

158
Nas plantas pensa-se que as indolalquilaminas tenham, igualmente, como progenitor, o trlptofano.
Assim, a formação da triptamina pode explicar -se facilmente pela acção directa da triptofanodesoar-boxilase sobre o triptofano e a da serotonina por des-carboxilação do 5-hidroxitriptofano. Acontece, no enta» to, que nas plantas superiores nem mesmo a origem do tri£ tofano está cem esclarecida e das alquilaminas somente julgamos estar bem estudada a origem da gramina.
Entretanto, como numa mesma espécie vegetal podem existir, simultaneamente, várias indolalquilaminas, este facto pressupõe certa interdependência na sua formação.
Por outro lado, a descoberta das indplalquila, minas-Nh-óxidos nas plantas sugere também que estes ami nô'oxitosj por rearranjo a carbinolamiBas, desempenhem importante papel na biossíntese das bases indólicas sim pies e- até mesmo na de outros alcalóides indólicos mais complexos (21, 22).
G-HOSAL e MOKHERJEE (23), apoiados nestes factos e, muito especialmente, por eles próprios terem iso_ lado <3c Desmodium pulchellum sete indolalquilaminas,pro põem um esquema segundo o qual admitem a formação de ai gumas delas a partir de outras.

159
ESQJJEMA VIII
Triptofano > Abrina
l amina NbfNb-.Diraetiltriptaml [na
Bufotenina «ufotenina-N^-drido ^Nb ,Nb~Dimetil tr iptami na-Nb-óxido
5-Metoxi-Nb,Nb~])imetiltriptamina-Nb-(5xido
/ \ 5-Metoxi-Nb,Nb-dimetiltriptamina 5-Metoxi-Nb-metirtrip_
tamina
Este esquema, que nos parece bastante incipiente, carece de confirmação experimental,
Aliás, que saibamos e como já dissemos,noa ve getais, somente a origem da gramina está esclarecida.De facto, foi um dos primeiros alcalo'ides a ser estudado com base em progenitores radioactivos,devendo-se a BOW-DEN e MARION (24) a demonstração de que deriva do triptofano, não obstante esta se encontrar desfalcada em um CII2 na cadeia lateral, relativamenbe às outras indolal-quilaminas e ao aminoácido donde provenu
Estes investigadores, apo's administração de (\?~ c) triptofano (LXXVIl) à cevada em germinação,extraíram das folhas da planta gramina radio act i va (LXM.HÍ)

160
a qual, por degradação, mostrou estar exclusivamente mar cada no carbono do grupo metilénico (assinalado com asterisco) da cadeia lateral. De facto, a fusão alcalina do alcalóide com o hidróxido de potássio conduziu à obtenção do indol-5-carboxilato de potássio. radiactivo (LXXIV), o qual, por descarboxilação, originou indol i-nactivo (LXXX) e anidrido carbónico marcado (LXXXl).
Além disso, do tratamento da gramina rádioECtò va com iodeto de etilo e etóxido de potássio resultou 5-etoximetilindol radiactivo (LXXXII) e dimetiletilami-na inactiva (LXXXIIl),como se indica no ESQUEMA. IX
ESQUEMA. IX .COCE
H LXXVTI CsHgl
KDGgHg :C00K
LXXXII
N'-y y G%3 -CHg -CE*
LXXXÏÙ LXXX LXXXI

161
A demonstração inequívoca de que a ligação C-3-C-.jijL da moléoulo do triptofano não sofre ruptura du rante o processo biossintético e que, portanto,a porção a,, assinalada na fórmula do triptofano (LXXVIl),transita para a molécula da gramina (LXXVIII) deve-se a LEETE e MARION (25).
De facto, a administração à cevada duma mistu ra de ((¾ -14C e de 2-14C) DL triptofano levou à formação de gramina marcada no Gr2 do núcleo indólico e no grupo metilénico da cadeia lateral, sendo a relação das radioactividades 0 - c/2 - C na molécula a mesma que se registava no triptofano progenitor.
BEECCIÂ e MARION (26), por um lado e WIGHTMAN e col. (27) por outro, experimentaram vários precursores radioactivos com o fim de melhor conhecerem o meça nismo da formação daquela base, mas de todos os compostos experimentados nenhum se mostrou tão eficiente como
. 14 N o (¾- C) triptofano. Posteriormente, as experiências de 0'DONOVAN
e LEETE (28) mostraram ainda que o grupo metilénico da cadeia lateral também transita intacto da molécula do triptofano (LXXIV) para a gramina (XCl), porquanto a ad ministração do aminoácido, simultaneamente marcado em posição ('icoitt-toâ- isótopos C e TE, originou o alcalóide exclusivamente radioactivo no grupo assinalado (XCl)sen do a relação (3- C/~^»Tl a mesma nos dois composto.

162
WMKEKT (29)5que muito se tem dedicado aos pro
blemas b i o g e n é t i c o s , t i n h a j á formulado n e s t a a l t u r a , a
sua h i p ó t e s e acerca do mecanismo pelo qual o c o r r e r i a es_ t a h io t rans formação . Assim, em sua op in i ão ,o t r i p t o f a n o
(iiXXXIY) condensa r - se - i a com o fosfa to de . p i r i d o x a l
(LXXXV), formando uma hase de SCHIFP (LXXXVl), a qual
s o f r e r i a uma degradação por um mecanismo inverso ao da
reacção de MICHAEL, o r ig inando-se um in t e rmed iá r io ele_c
t r o f í l i c o , a 3 -met i lenoindolen ina p r o t o n a d a (LXXXVTIl).
Este i n t e r m e d i á r i o , por l i g a ç ã o com r a d i c a i s cedidos p_e"
l a met ionina , o r i g i n a r i a , sucess ivamente , 3~(aminometil)
indo l (LXXXÏX) , 3 - (met i l aminomet i l ) indo l (XC) e fí.
nalmente gramina (XCl),de acordo com o ESQUEM X,
ESQUEMA X ,CHO HO. / engOPOgH
H 3 C > ^
LXXXV * H H
H3C
20P0#
LXXXVl

163
1
I H HC,
coce
:H20H)^I ^N—^H
H
LXXXVTI
i H
LXXXVIII
* / H
* H
LXXXIX
* ? ^ N •*? \ « / i' ce.
Till xc
H ^X
ii x r a 3
Foi talvez o facto de na s ín tese microbiológica do triptofano (30) es ta r envolvida a vitEiuina-Bg que sugeriu a WENKEKT tão interessante hipótese. Esta

164
encontrou apoio nos t r a b a l h o s de MUDD ( 3 l ) , pois e s to
au to r conseguiu não só i s o l a r da cevada o 3-(aminometil)
indol e o 3-(met i laminometi l ) índoJL mas a inda conver
t e r aquele derivado em gramina, por incubação com(-)-S
-adenos i l -L-met ion ina e uma enzima e x t r a í d a dos reben
t o s da cevada. Mas foram sobretudo as expe r i ênc ia s com
p recur so res marcados, r e a l i z a d o s por O'DONOVAN e LEETE
(28 ) , GKWER e LEETE ( 3 2 ) , e , ù l t imamente ,por DIGElS^33)
que confirmaram aquela h i p ó t e s e .
Aos t r a b a l h o s de 0'DONOVAN e LESTE j á nos r e
ferimos an t e r io rmen te . As exper iênc ias de GOWER e LESTE
demonstraram que, na r e a l i d a d e , o 5 - (aminomet i l ) indo le
o 3"(met i laminomet i l ) indol são p recu r so re s mais d i r e c t o s
que o t r i p t o f a n o , , j á que a adminis t ração des t e aminoáci
do conduz à formação de gramina apenas com 0,5/6 de i n
corporação r a d i o a c t i v a , enquanto que, aque les do is d e r i
vados conduzem à gramina com 14,2 a 24,5% de incorpora
ção , r e spec t ivamen te .
As r e c e n t e s exper i ênc ias de DIGESTS ( 3 $ não
só vieram apo ia r a h ipó t e se de WEMERT como também ex
p l i c a r o desaparecimento da gramina que se observa nas
fo lhas da cevada, decor r ido um c e r t o per íodo após a sua
germinação. 0 a u t o r u t i l i z o u , para as suas e x p e r i ê n c i a s ,
fo lhas de cevada com se s sen t a e cento e v i n t e d i a s de
i dade , po r t an to numa a l t u r a em que a gramina j á não é de
t e c t á v e l , e admin i s t rou - lhes e s t e a l c a l ó i d e marcado na

165
cadeia metilénica com isótopos G e n» Após sete dias de metabolização do alcalóide pôde extrair triptofano. com 0,84$ de incorporação radioactiva, na qual a relação C/H era a mesma da do alcalóide adnri.nistrado.Iso_ lou ainda 5-hidroximetilindo1 e 5-carborindol contendo, respectivamente, 3.0,1% e 6,25¾ da radioactividade inicial da gramina,
Idênticas experiências, mas conduzidas na ausência da luz, permit iram-lhe verificar que, nestas con_ dições, a planta além de sintetizar triptofano radioactivo, eliminava anidrido carbónico também radioactivo.
Perante estas observações, o autor admitiu que esta biodegradação da graminàa triptofano, deve ocor rer por um mecanismo análogo, embora inverso,dó da trans formação do aminoácido no alcalóide, de acordo com a hi pótese formulada por YffiNKEíff.
Comparando esta degradação com a da seroteni-na no homem, o autor admite que na degradação da cadeia lateral das indolalquilaminas devem estar envolvidas e_n zimas comuns aos dois reinos de seres vivos, pois que a gramina se degrada a 5-indo 1 i 1 carbino 1 e a serotonina a ácido 5-hidroy.indol -:ia.,:.441co o
1,6.2.- Origem biogenética dos alcalóides derivado?, da -carbolina ^ -.s Apesar da simplicidade estrutural destes alça

166
Idides, as experiências directas para comprovar a sua origem são muito recentes. No entanto, há algumas hipóteses formuladas desde longa data acerca desta matéria,,
Assim, PERKIN e ROBINSON (34) sugeriram, em 1919, a sua formação por descarboxilação do triptofano (XCVII) a triptamina (XGVIIl) a qual, por combinação com o aldeído acético, originava uma hase de SCHlF-f (XCIX) de cuja ciclização, resultava tetraidroarmana (C),e esta, desidrogenando-se daria, sucessivamente, harmalana (Cl) e harmana (CIl). Estes autores explicam, portanto, que os alcalóides deste grupo possam apresentar o núcleo hexagonal heterocíclico em três graus diferentes da hidrogenação, como aliás se verifica (ESQUEMA. XII).
fOIKDEXTER e CARPENTER (35) estudaram no fumo do tabaco a formação da harmana e da norarmana,por trans formação do triptofano marcado.
GROEGER e SIMON (36), por administração de ((T)_14C) triptofano a Peganum harmala L,, obtiveram uma mistura radioactiva de três alcalóides,constituída por hariirina, harmalina e harmalol, mas não localizaram a pa_r te radioactiva das moléculas.
A hipótese de PERKIN e ROBINSON parece ser a-poiada pelas experiências de 0'DONOVAN e KENNEALLY(37). Estes autores utilizaram nas suas experiências dois lotes de plantas pertencentes à espécie Elaeagnus angusti-folia L* com a idade de 2 anos. As plantas de um dos lo

167
t e s foram alimentadas com ( ( ^ - C) t r i p t o f a n o e as do out ro com ( l - " ^ ) a c e t a t o de sódio e , decor r idos v i n t e e t r ê s d i a s e v i n t e e s e t e d i a s , r e spec t ivamente , foram co lh idas e o seu a l c a l ó i d e - e laeagnina i s o l a d o .
0 a l c a l ó i d e (XCII) obt ido do pr imei ro l o t e , quando degradado segundo a t é c n i c a de HOFFMAiíN,forneceu a lde ído fórmico (XCV) r a d i o a c t i v o , mostrando,por consequência , que e s t ava marcado no carbono 3 ,
A oxidação de KŒN-ROTH p r a t i c a d a sobre a t e t r a d droarmana (XCII) i s o l a d a do segundo l o t e , pe rmi t iu ob t e r ácido acé t i co (XCVI) r a d i o a c t i v o que fo i t r a n s f o r mado em an id r ido carbónico r a d i o a c t i v o p e l a reacção de SCHMID, provando-se , nes t e caso , que a r a d i o a c t i v i d a d e do a l c a l ó i d e se encontrava cen t rada no carbono 1_ (ESQUE
m X l ) * ESQUEMA XI Oxidação de KIMN-ROTH Degradação de HOFFLIAUff
Xr
CH3C0ŒÏ f-
XCVI
HCHO+
XGV
çQe^Op ^te?

168
Esquema i d ê n t i c o ao de PERKEN e ROBINSON fo i
proposto por ANTONACCIO e BUDZIKEEWICZ (38) para a b i o -
génese dum alcalo ' ide g l i c o s í d i o o , em que a ag l í cona 4 a
5-carboxiarmajia.
Recentemente, SLAYTOR e McFARIANE (39) e s tuda
ram o mecanismo da b i o s í n t e s e da harmana em Passiflora,,,,,
e d u l i s L. Ut i l i za ram para o e f e i t o v á r i o s p recu r so re s ra
d ioac t ivos s ( Ç ) - M C ) t r i p t o f a n o , ( 2 - M C ) a e e t i l t r i p t o f a -
no , c l o r e t o de (dv- 1 4C) t r i p t a m i n a e ( 2 - 1 4 C ) - t e t r a i d r o -
armana e c l o r e t o de ( X C) harmalana.
Das suas expe r i ênc ia s puderam conc lu i r que se
forma N h - a c e t i l t r i p t a m i n a ( C I I l ) na fase i n i c i a l d e s t a
b i o s í n t e s e e que e s t a , provavelmente, r e s u l t a da a c e t i -
lação da t r i p t a m i n a (XCVIIl) pe l a ace t i l -CoA.
Segundo e s t e s a u t o r e s , o ane l daí3 - c a r b o l i n a ,
fo rmar - se - í a por de s id r a t ação e desidrogenação da a c e t i l -
t r ip ta ro ina dando harmalana (Cl) e e s t a , por sua v e z , p e r
dendo h id rogén io , o r i g i n a r i a harmana ( C I l ) ,
0 esquema proposto por SLAYTOR e McFARIANE d_i
f e r e , e s senc ia lmente , do de PERKEN e ROBINSON no queres
p e i t a ao in t e rmed iá r io t r i c í c l i c o que pr imeiro se f o r
ma, além de que se admi te , na h i p ó t e s e mais r e c e n t e e s
t a r em jogo o equipamento enzimático da p l a n t a (ESQUEMA
X I I ) . 03 au tores não excluem, no e n t a n t o , que a t e -

169
traidroarmana (CIV) se possa formar por hidrogenação da harmalana (Cl) ou esta por desidrogenação daquela.
ESQUEMA XII
Hipótese de PERKIN e ROBINSON
I l N cá è '
,CH^CI
CHr XGIX
^
JCOCH
O L
^
Hipótese de SLAYTOR e
McFARIÂNE
l COç
F H
XCVIII
lift MÎ2
? -20.
■i ãT H CHj.
GI
2H
/A
GIL
f c6 H I
GHr, CHI
H
ÍY H CH3
HH
PIV
C i l

170
BIBLIOGRAFIA
1 - PICTET, A, , Arch.Pharm. 244, 389 (1906) . 2 - LEETE, E . , Biogenes is of Na tu ra l P rodu t s ,Be rn f i e ld ,
Pergamon P r e s s , Oxford (1963) . 3 - LEETE, E . , Chem.and Ind . 692 ( i 9 6 0 ) . 4 - YAMASAKI, M,, e LEETE, E«, Tetrahedron Le t te r s . 23 ,
1499 (1964) . 5 - LEETE, B . , AHMAD, A. e KOMPIS, I . , J.Am.Ghem.Soc.
87, 4168 (1965). 6 - WERLE, E. e MENNICKEN, G., Biochem. Z. 291, 325
" (1937) . 7 - BLASCHKO, H . , Biochem. J . 52 , 10 (1952)5 i b i d . P h a r
macol. Rev,. 4 , 415 (1952) . " 8 - UDENFRIEND, S , , C2EVELING, C.R., POSNER, H.,REDFI-
ELD , B.G., DALY, J . e WITKOP, B . , Arch. Biochem, 83,501 (1959) .
9 - UDENFRIEND, S . , CLARK, C.T. e TITUS, E . , J.Am.Ghem. Soc. 75, 501 (1953) ,
10 - CLARK, C.T., WEISSBACH, H, e UDENFRIEND S., J.Biol. Chem. 210, 139 (1954) .
11 - HOLTZ, P . , HEISE, R. e LÍ3DTKE, K„, Arch .exp. Pa th , Pharmak. 191, 87 (19387.
12 - UDENFRIEND, S , , CLARK,~C,T. e TITUS, E . , E x p e r i e n t i a 8, 379 (1952),
13 - UDENFRIEND, s>, TITUS, E„, WEISSBACH,H,, FETTERSON, R.f J. Biol. Chem. 219, 335 (1956).
14 - MITO1.1A, 0., WEISSBACH, H., e UDEHFRIEND, S,, Arch. Biochem. 63, 122 (1956).
15 - DALGLIESH, C.E.t Biochem. J. 64, 431 (1956) 16 - UDENFRIEND, S. e WEISSBACH, H., Proc.Soc.exp.Biol.
97, 748 (1958). 17 - DONALDSON Jr., R..M,, GRAY, S,J, e LETSOU, V.C.,
Lancet II 1002 (1959). 18 - FREEDLAND, R.A., WADZINSKI, IeM. e WAISMAN, H.A.,
Biochem. biophys.Res.Commun.5, 94

171
19 RENSON, J,, WEISSBACH, H. e UDENFRIEND, S..J.BI0I Chem. 237, 2261 (1962).
20 WHITE, A., HANDLER, P., SMITH, E., Principles of Biochemistry, . McGrawRili Bool:. Company 4» ed. (¢068).
21 FISH, M.S., JOHNSON, M.M., HORNING, E.C., J. Am. Chem. SQQ. 77, 5892 (1955) .
22 WENKERT, E . , E x p e r i e n t l a 10, 346 (1954) . 23 GHOSAL, S.,, e MUKHEIÏJKE, B . , J.Org.Chem. 31,2284
(1966) . 24 BOWDEN, K. e MARION. L. , Ca n . J . Chem. 29, 1037
(1951) i b i d 29, 1043 (1951) . 25 LEETE, E . , MARION, L. , C<ân. J , Chem. 3 1 , 1195
(1953) . 26 BRECCIA, A., MARION, L., Can, J. Chem. 37, 1066
TÏ959). 27 WIGHTMAN, P . , CHISHOLM, M.D. e NEISH, A.C. , Phyto
chemitry 1, 30 (1961) , 28 O'DONOVAN, D. e LEETE, E . , J.Am.Chem.S0c.85, 461
(1963) . 29 WENKERT, E . , J . Am. Cheytu SQC. 84, 98 (1962) . 30 DOY, C.H., Pure a p p l . Chem, 10, 185 ( i 9 6 0 ) . 31 MUDD, S.H., Nature 189, 498 ( l 9 6 l ) . 32 GOWER, B.G. e LEETE, E . , J Am. Chem. Soc. 85,3683
(1963) . 33 DIGENIS, G.A,, J. Pharm. Sci. 58, 39 (1969) IMd 58,
42(1969) 34 PERKIN, Jr., W.H. e ROBINSON, R., J. QheauSqQ.. 115.
933 (1919) . 35 POINDEXDER, E.H. e CARPENTER, R.D. , Çhem. e Ind ,
176 (1962). 36 GRÕGER, D. e SIMON, H., Abhandl dent. Akad. Wiss■
Berlin, 343 (1963)". 37 O'DONOVAN, D.G, e KENNEALLY, M.F., J. Chem. SQC.
1109 (1967). 38 ANTONACCIO, L.D. e BUDZIKIEWICZ, Monatshefte Chem.
93, 962~(l962). 39 SLAYTOR, M. e McPARLANE, I.J., Phytochemestry 7,
605 (1968).

P A R T E E X P E R I M E N T A L
P A R T E I I I

175
1. II 0 T A S
1,1.- Os pontos de fusão foram determinados em microscd pio de platina de aquecimento (microscópico de pon to de fusão LEITZ) e não foram corrigidos.
1.2,- As determinações dos poderes rotatórios reâliza-ram-se em polarímetro da marca BELLINGEAM & STANLEY munido de dispositivo analisador LIPPIGH, à luz de sódio.
1.3,- Os pH foram ajustados usando um PHOTOVOLT pH METER da marca PHOT0V0LT CORPORATION L,da,
1.4.- Os espectros na região do U.V. foram realizados u tilizando os solventes s metanol, álcool etílico p.a. MERCK, solução metanólica de hidróxido de só. dio N/20, solução metanólica de ácido clorídrico N/20, em cuvetes de quartzo de 1 cm de espessura e operando em espectrofotómetro de registo automá tico SPECTRONIC 505 BAUSCH & LOMB.

176
1.5,- Os espectros na região do I.V-. foram rea l i za dos- com a substância dispersa em KBr e usando um aparelho Grating Infrared Spectrophotometer PERKffl ELMER 237.
1,6o- Nas cromatografias em coluna usamos os adsorven-tess Alumina"Aluminniumoxid Standardisiez aktivitãts-sufe - (I) (Alg03) MERCK"»
Acido sâlícicó 'MALLINCKRODT silicic acid 100 mesh
(Si02XH2°)"»
1.7,- Na preparação das placas para cromatografia em ca mada fina usamos como adsorventes a alumina ea s_í liça e preparámo-las do modo seguintes
Placas de sílica - 30 g de pò* de "Kieselgel G nach STAHL MERCK" foram agitados em matrás com 60 ml de água destilada durante cerca de 2 minutos. A suspensão foi espalhada sohre placas de vidro de 20x xlO cm numa espessura de 0,25 mm e, depois de a-bandonadas 12 horas à temperatura do laboratório, foram colocadas na estufa durante 2 horas a 100°C.
Placas de Alumina - foram preparadas de modo idên tico às de sílica, mas partindo de 30 g de "Alumi niumoxid G nach STAHL MERCK" suspensos em 45 ml de água.

177
1 . 8 . - Reagentes;
1 . 8 . 1 . - Reagentes de p r e c i p i t a ç ã o
Solução de ácido s i l i c o t ú n g s t i c c
Acido s i l i c o t ú n g s t i c o . . . . 10 g Agua d e s t i l a d a q.b 100 ml
Reagente de VALSER-MAYER
Iodeto de potássio puro cristalino. 25 g Cloreto de mercúrio puro „. 6,67 g Agua destilada q.b, , 500 ml
0 cloreto de mercúrio e o iod_e to de potássio foram dissolvidos em 25 ml de água destilada, completan-do-se depois o volume de 500 ml.
Reagente de DRAGEEDORFF. fórmula de MONIER e MACHBBOEIFs
Solução A S ú b - n i t r a t o de bismuto 1,70 g
Agua destilada „ 80 ml Acido acético cristalizado ........ 20 ml

178
Solução B Iodeto de potássio » 16 S Agua destilada 40 m l
Solução mãe M a t u r a das so luções A e B .
1 . 8 , 2 , Reagente de Qoloração
Para a l c a l ó i d e s s
Beagente da DBAGENDOHFF, formula de MUNIER e
MA.CHEBOEUF "
Solução rQY,a rfivfilaçgo de cromatogramag.
Solução mãe 1 0 m l
Acido acé t i co 20 ml
Agua d e s t i l a d a q./b. »• »• 1 0 ° m l
Reagente foafovan í l i ço
V a n i l i n a . . . , , « . * • • • • » » • *■ S Acido fosfórico •
10 8 Etanol a 96° q . b . • 1 0 ° &
Reagente de pdimeti1aminob enzaldeído pdimet i laminobenzaldeído . . . . . . . . . 0,5 g
Acido c l o r í d r i c o • 1 ml Alcool absolu to q.b 100 ml

179
Reagente de sulfato oéfrioo Sulfato cerico amoniacal \ g
Acido fosfo'rico a 85 fo 99 g
Reagente de njjiidrina Ninidrina 0 ? 2 g
A c e t o n a •••• 100 ml
Reagente l -n i t rosn-2-naf tol l -ni t roso-2-naf tol o 1 g Alcool a 95° 1 0 0 m l
Reagente de ácido ni t roso Solução de ácido sulfúrico 2 N . . . . 5 m l Solução de n i t r i t o de so'dio a 2,5%. 0,2 ml
Para es teróides ; Ãl0001 80 ml Anidrido acético 10 rui Acido sulfúrico , , ]_Q mi
As cr i s ta l izações para purificação dos produ tos i so lados de Burkea cvfrioaniy. .B3ng Q d o s
s i n t e t i z a d o s foram p r a t i c a d a s usando solven t e s p . a .

181
2 ENSAIOS PRELIMINABSS ■
Como já tivemos oportunidade de referir no pre âmbulo deste trabalho, o nosso propo'sitc consistiu em fazer o estudo de alcalóides indóliccs de espécies "botânicas originárias da Província de Angola e que vies
sem a revelar tais compostos. Para o efeito, conseguimos obter pequenas a
mostras das seguintes plantas:
1 BaiC^pa.WaSfhorstii SCHINZ folhas e raízes,
2 Garissa edulis VAÏÏL casca das raízes.
3 Landolphia parvifolia K.SCHUM, casca das raízes,
4 Burkea africana HOOK casca do tronco e folhas.
5 Erythrophloeum africanum HARMS casca do tronco.
6 Pterocarpus angolensis D.C, casca do tronco.
Como se pode ver no QUADRO I, estas plantas pertencem apenas a duas famílias : Apocynaceae e Legume nosae.

182
Q U A D R O I
ESPÉCIE TRIBO SUBFAMILIA FAULIA.
Baissea W&1-fhorstii
Ichnocarpeae Echitioideae Apocynaceae
Burkea africana
Dimorphan-dreae
Caesalpinioi deae
Leguminosae
Carissa edulis
Cariseae Plumerio 1-cleae
Apocynaceae
Erythrcphlo-eum africa-num
Dimorphan-dreae
Caesalpinioi_ deae
Leguminosae
Landolphia parvifólia
Carisseae Plumerioideae Leguminosae Carisseae Plumerioideae Leguminosae
Pt ero carpus, anffoler.sÍ3
Dalbergiae Fahoideae ou Papilionoi-deae
Leguminosae
2 , 1 , - PESQUISA DE ALCALÓIDES
Procedemos à pesquisa de a l c a l ó i d e s em cada
uma das amostras das p l a n t a s c i t a d a s depois de prèviamen
t e reduzidas a pó„
Desengordurámos 100 g de cada droga,,pelo ét 'ar
de p e t r ó l e o (p.e„ 5 0 ° - 7 0 ° ) , em d i s p o s i t i v o de SOXHLETjdu
r a n t e 15 h o r a s . Seguidamente, o pó f o i r e t i r a d o do SOX-
HLET e exposto ao a r para e l imina r o é t e r de p e t r ó l e o , o
que se reconheceu p e l a ausência de cheiro a e s t e solven
t e .

183
O pó foi colocado em balão de 500 ml de capacidade, embebido com 50 ml de amónia a 50$ e adicionado de 500 ml duma mistura éter-clorofórmio (3:1),, Deixámos macerar em balão rolhado durante 48 horas,tendo-se agitado frequentes vezes.
Filtrou-se e D resíduo foi ainda esgotado por uma nova porção de 200 ml da mistura éter-clorofórmio. Os líquidos extractivos foram reunidos e o seu volume re duzido a 100 ml por evaporação dos solventes a 'baixa temperatura (inferior a 50°C) e sob pressão reduzida, A solução foi transvasada para uma ampola de decantação e extraída por três vezes com 25 ml de solução de ácido sulfúrico a 3$, de cada vez. As soluções ácidas foram reunidas e ensaiadas com os reagentes de BERTRAND e com o reagente de MA.YER,
A restante solução ácida (cerca de 50 ml) foi alcalinizada com hidróxido de amónio a pH cerca de 10 e extraída com 25 ml de clorofórmio. Separámos a solução clorofórmica e repetimos a extracção por mais duas vezes.
As soluções clorofórmicas reunidas foram evaporadas a baixa temperatura e pressão, até* reduzir o vo lume a cerca de 5 ml,
Nesta solução pesquisámos os alcalóides pelo reagente de DRAGENDORFF, usando a técnica de toque em papel de filtro.
0 resíduo da evaporação completa do clorofór-

184
mio foi ensaiado com o reagente de EHRLICH para pesquisar alcalóides inddlicos.
Os resultados obtidos inscrevem-se no QUADRO
Q U A D R O II
ESPÉCIE r , « 1 9 Ed fe H S ^ a tai S PH H
M « á sã PH
H M M Ed S
iSi N Q
P H H
1 a Baissea Folhas
r a í z e s i --
--
Wul fho r s t i i Folhas r a í z e s i -
--
-
Burkea Folhas e casca do t ronco
+
+
+
+
+
+
+
+ a f r i cana
Folhas e casca do t ronco
+
+
+
+
+
+
+
+
Folhas e casca do t ronco
+
+
+
+
+
+
+
+
Car i s sa Casca das r a í z e s
- - .«., -e d u l i s
Casca das r a í z e s
- - .«., -Casca das r a í z e s
- - .«., -
Erythrophloeum Casca do t ronco
+ + + + africanum
Casca do t ronco
+ + + + Casca do t ronco
+ + + +
Landolphia Casca das r a í z e s
- - - -c á r v i f õ l i â
Casca das r a í z e s
- - - -
Pterocarpus Casca do t ronco
- - + -angolens i s
Casca do t ronco
- - + -Casca do t ronco
- - + -
A nossa atenção dirigiu-se de início para as plantas da família das Apocynaceae e, mais especialmente, para as duas espécies pertencentes à subfamflia das Plumerioideae,uma vez que nas plantas desta subfamília

185
oa alcalóides têm sido encontrados com certa frequência. Todavia, a nossa expectativa em breve se desvaneceu^após a realização destes ensaios preliminares. De facto, estas espécies ou não contêm alcalóides ou se os possuem trata-se de quantidades tão reduzidas que não puderam ser detectadas, pela técnica seguida, na quantidade de pó utilizado no ensaio.
Uma nova tentativa de extracção dos alcalóides com metanol ácido sobre todo o po que nos restava destas duas espécies (150 g) conduziu a idênticos resulta-tos.
^31 - ffythropbloeuia africanum pudemos assinalar alcalóides. Uma rápida consulta bibliográfica permitiu--nos verificar que esta planta se encontra largamente estudada no que respeita a estes constituintes, sendo, no entanto, os vários alcalóides até agora dela isolados de estrutura não indólica.
Para dar seguimento ao plano traçado reótou-nos Burkea africana HOOK e esta desde logo nos pareceu prometedora, porquanto o resíduo alcaloídico bruto obti do no ensaio prévio fora bem significativo.

187
3 , - ESTUDO QUÍMICO DE BURKSA AFRICANA HOOK
3 , 1 . - MATERIAL UTILIZADO
O material que utilizámos no nosso trabalho foi colhido em Huíla, na nossa Província de Angola, em Outubro de 1968.
A nossa investigação incidiu especialmente so_ bre a casca do tronco e ramos da planta Burkea africana HOOK, se bem que as folhas também tivessem sido ensaia das.
A fotografia (Pig, l) que a seguir incluímos reproduz a planta e dá-nos ideia do seu aspecto e porte, mostrando a (Fig. 2) em mais pormenor um ramo florido da mesma.

Fig. 1 — Burkea africana HOOK

Fig. 2 — Ramo de Burkea africana HOOK

193
• 3.2.- CARACTERES MACROSCÓPICOS DO MATERIAL UTILIZADO
Cascas
Como a fotografia mostra (Fig. 3), as cascas utilizadas correspondem a várias zonas da planta, umas do tronco e outras dos ramos.
As mais velhas, as do tronco, apresentam acor tiça desenvolvida, muito rugosa e fendida e cor avermelhada escura.
As cascas dos ramos têm aspecto rugoso, não fendido e externamerite são mais claras que as do tronco.
0 suber encontra-se perfeitamente aderente à zona cortical, que tem cor amarela avermelhada,tornan-do-se difícil a sua separação. Por isso a nossa investi gação abrangeu a casca composta de suber e entrecasco.

8 o c o u
s CÛ
<u " D 1/) o u IA
ô m en

197
3,3.- COMPOSIÇÃO QUÍMICA GLOBAL DE BURKEA AFRICANA HOOK
3.3,1,- Agua
Foi determinada por perda de peso por aquecimento em estufa a 100-105 , até peso constante e operan do sobre cerca de 5 g de amostra.
Média dos resultados: 10,2 g fo
3,3.2,- Cinzas
Foram determinadas partindo de amostras de ce_r ca de 5 g, por incineração em mufla eléctrica, à temperatura de 500°C, até obtenção de cinzas brancas.
Média dos resultados; 3,40 g fo
3.3,3,- Glúcidos totais, por inversão
Utilizámos a técnica seguintes SubEietemos 3 g de pó desengordurado pelo éter
de petróleo à hidrólise ácida. 0 pó foi lançado em balão e adicionado de uma mistura ácida constituída por 16 ml de ácido clorídrico concentrado e 200 ml de água. Ao balão adaptámos um refrigerante e aqueceu-se a banho maria à ebulição, durante 4 horas.
Após arrefecimento e neutralização com hidróxido de sódio a 30^ em presença da fenolftaleína,proce-

198
demos à sua defecação por adição de excesso de solução de subace ta to de chumbo. 0 excesso fo i eliminado por a
dição de solução sa tu r ada de s u l f a t o de s d d i o . A suspensão fo i f i l t r a d a para balão de 500 ml
e o p r e c i p i t a d o lavado v á r i a s vezes com água, a t é com
p l e t a r o volume de 500 ml. Homogeneizada a so lução , pro cedemos ao doseamento dos açúcares r edu to re s pe l a t é c n i ca grav imé t r i c a de MTJUSON e WALKER, d e s c r i t a por ISIDO
RO KETO ( l ) .
Média dos r e s u l t a d o s s 16,80 f°
3 , 3 , 4 . Prê t idos t o t a i s
Procedemos à sua determinação pelo método de KJELDAHL, pa r t i ndo de amostras de 2 g, aproximadamente.
Média dos r e s u l t a d o s s Azoto = 1,70 g % Pró t idos t o t a i s ■ l ,70x x 6,25 = 10,63 g fo.
3 . 3 . 5 , Lípidos
Foram determinados por extracção pelo éter de petróleo (ponto de ebulição = 5070°C) em aparelho de SOXHLET, partindo de amostras de 20 g„ 0 resíduo, após eliminação do éter de petróleo por destilação, foi seco a 100 , em estufa, até peso constante.
Média dos resultados; 0,65 g fo.

199
3.3.6,- Celulose
A determinação da celulose foi efectuada pelo método de WEEÏÏDE, descrito por ISIDORO KSTO (l)e
Partimos de 2 g do pó da planta desengordurado. Este foi introduzido em balão de 750 ml e adicionado de 200 ml de solução fervente de ácido sulfúrico a 1,25 °/o (p/v). Adaptámos um refrigerante e aquecemos à ebulição durante 30 minutos. Decorrido este tempo,decan támos o líquido, fazendo-o passar através de uma placa filtrante e lavámos o resíduo com água em ebulição, até o líquido deixar de dar reacção ácida ao papel de torne sol.
A fracção do resíduo que passou para a placa filtrante foi de novo arrastada para o balão com 200 ml de solução aquosa fervente de hidróxido de sódio a 1,25 1o (PA),
Ligámos o condensador ao balão e procedemos a um novo aquecimento. Após 30 minutos de ebulição, o líquido foi decantado como anteriormente se descreveu e o resíduo lavado com água, até o líquido filtrado não acu sar reacção alcalina ao tornesolfl
0 resíduo foi quantitativamente transferido , por arrastamento com água destilada, para uma cápsula de platina previamente calcinada.
A água foi evaporada a banho maria e o resí-

200
duo seco em estufa, a 105°C, durante 2 horas.Depois de arrefecida em excicador pesou-se a cápsula. Em seguida foi calcinada em mufla eléctrica a 500 G, deixada arrefecer em exsicador e de novo pesada.
Celulose bruta em gfo =(p-p' ) x [lQQ-(h + g)| •p
h - representa a humidade em g %
g - representa a gordura em g %
p - representa o peso da cápsula de platina com resíduo antes da calcinação, expresso em g.
p'- representa o peso da cápsula de platina com o resíduo depois da calcinação, expre_s_ so em g.
P ~ representa o peso da droga tomada para o ensaio,
iíèdia dos resultados - 26,0 fo,
3.,4.- ANALISE GEPAL DE BÏÏRKEA AFRICAM HOOK
3,4.1,- Pesquisa de compostos polifenólicos
As pesquisas a que seguidamente nos referimos efectuaram-se no infuso a 10 fo. Este tinha aspecto turvo e apresentava cor vermelha acastanhada,

201
3.4.1.1.- Pesquisa de flavondides
A pesquisa destes compostos foi efectuada pela reacção de SHIBATA ou reacção de cianídíj?^.
A 5 ml de infuso adicionámos volume igual do álcool clorídrico a 20 fo e alguns fragmentos de fita de magnésio. Não se observou alteração da cor inicial e,por agitação com álcool amílico, este não corou. S pois de admitir que a planta não contenha flavondides.
3.4.1,2,- Pesquisa de compostos antociânicos
Por adição de ácido clorídrico ao infuso não se observou apreciável alteração da cor e a adição de amónia não provocou variação de cor para tons asuiá esverdeados, como é vulgar acontecer quando estão presentes estes compostos.
3,4.1.3,- Pesquisa de fendis livres
Cerca de 50 ml de infuso foram esgotados várias vezes, com porções de 20 ml de éter sulfúrico de ca da vez.
As soluções etéreas reunidas foram evaporadas à secura a baixa temperatura, sob pressão reduzida, e o resíduo retomado com álcool a 96°.

202
A solução alcoólica, adicionada de solução de cloreto férrico, originou cor verde azulada. A planta contém pois, fenóis livres.
3.4.1,4,- Pesquisa de taninos
A adição de algumas gotas de cloreto férrico ao infuso a ifo originou aparecimento de coloração azul intensa.
Por adição de gelatina salina (solução de gelatina a Vfo em solução aquosa de cloreto de sódio a 10/¾) formou-se precipitado esbranquiçado (taninos gálhicos e catéquicos).
0 infuso, adicionado de umas gotas de ácido clorídrico e aquecido à ebulição, originou precipitado (vermelho catéquico ou flabafeno).
A adição de solução de subacetato de chumbo originou um abundante precipitado amarelo esbranquiçado.
Por adição ao infuso de reagente de STIASNY , (formol-ácido clorídrico 3sl) seguida de aquecimento a banho maria, formou-se precipitado de coloração rósea (taninos catéquicos).
Após repouso e filtração da suspensão anterior, o filtrado corou de azul intenso, por adição de umas gotas de solução de cloreto férrico a Vfo (taninos pirogálhioos).

203
3.4.1.5.- Dosagem de taninos
Usámos para o efeito, a técnica descrita nos métodos oficiais da Sociedade Internacional dos Q'iicicoo da Indústria de Couros (2),
3.4.1,5.1.- Preparação da solução extractiva do tanino
Segundo os referidos métodos, a solução de ta nino pode ser obtida pela água em extractor de KOCH, ou em extractor de PROCTER.
A quantidade de droga a utilizar é condiciona da pelo teor desta em tanino. Deve dispor-se de uma quantidade de droga, tal que dois litros da solução extractiva contenham, tanto quanto possível, cerca de 4gA da matéria tânica adsorvida pelo pó de pele. Por isso, o processo definitivo carece sempre de um ou mais ensaios prévios. No nosso caso utilizámos 75 g de pó e pa ra a extracção do tanino empregámos o extractor de KOCH.
Este, como a Fig. 4 mostra, compô'e-se dum fras co com a capacidade de 200 a 300 ml, onde se coloca a droga.
No fundo do referido frasco dispõe-se uma camada de areia de dois centímetros de espessura e, sobre esta, o pó da droga que, por sua vez, se cobre com outra camada de areia de igual espessura.

O)
u
6)

207
0 frasco é fechado hermeticamente com uma rolha atravessada por dois tubos. Um deles dá entrada à água destilada contida num frasco colocado em plano superior e termina a cerca de um centímetro abaixo da rolha? o outro dá saída à solução extractiva, penetra no frasco até próximo do fundo e no outro extremo tem um tampão de gase.
0 frasco assim preparado é aquecido num "banho maria e o tubo que dá saída à solução extractivo lança--a'num "balão de 2000 ml de capacidade, sendo a velocida de de extracção regulada de modo a obterem--se 2000 ml da solução tânica, no espaço de 4 horas.
Recolhem-se os primeiros 150 ml e, nesta altu ra, eleva-se a temperatura do banho para .50 . A esta temperatura recolhem-se mais 750 ml. Depois aquece-se o banho à ebulição e recolhe-se a quantidade necessária até perfazer o volume atrás referido,
3.4.1.5.2,- Determinação dos solides totais
Os sólidos totais foram determinados evaporãn do à secura aparente 50 ml da solução extractiva,em cáp_ sula previamente tarada. 0 resíduo da cápsula é depois seco, na estufa, a 98,5-100°G. Após arrefecimento,pesag e com a precisão de 0,2 mg.
Média dos resultados = 22,34 g %.

208
3.4.1,5,3,- Determinação dos não taninos
A solução extractiva foi destanizada com pó de pele cromado, que foi preparado no momento de emprego.
Tomámos uma quantidade de pó de pele cromado húmido (com humidade não inferior a 72$ nem superior a 74$), correspondente a 6,25 g de pó de pele cromado seco, e adicionámo-la a 100 ml da solução analítica.Á sus pensão foi agitada em máquina própria, à velocidade de 50 a 60 rotações por minuto, durante 10 minutos.Coou-se através dum pano e à solução adicionámos 1 g de caulino de pureza própria. Filtrámos e repetimos a filtração o número de vezes necessário ate a solução ficar límpida.
Pipetámos 50 ml do filtrado para uma cápsula tarada, evaporámos à secura, secámos o resíduo e pesámos a cápsula arrefecida. Depois de corrigido o peso do resíduo para eliminar o erro causado pela diluição da so. lução pela água do pó de pele, calculámos a percentagem dos não taninos.
Média dos resultados: 11,20 $*
3.4.1.5.4,- Determinação dos taninos adsorvidos pelo pó de pele
Os taninos adsorvidos pelo pó de pele são de-

209
terminados p e l a d i f e r e n ç a en t re os s ó l i d o s t o t a i s e os
não t a n i n o s . Média dos r e s u l t a d o s : 11,14 g %
RESULTADOS
Humidade, expressa em g % 10,20 Não t a n i n o s , e x p r e s s o s em g $ . . . . 11,20
Taninos , expressos em g /» 11,14 Matér ias i n so lúve i s expressos em
g <fo 67,46
Segundo informações colhidas numa unidade industrial de curtumes desta cidade, onde realizámos o do seamento dos taninos, a casca de carvalho do norte de Portugal possui,em regra,características prckinias das seguintes:
Humidade, expressa em g $ 10,8 Não taninos,expressos em g $ .... 6,3 Taninog expressos em g $ . . . . . . . . 11,20 Matérias insolúveis expressas em
g $ 71,70
Se bem que hoje se utilizem como fontes de ta nino espécies mais ricas do que o Quercus robur, tais como, Chizophora mucrunata (mangal)-2l a 48$ (cascas) , Quebrachia lorentzii,(quebracho)-20 a 30$ (tronco), Re-sus estinoides (sumagre) 21$ (folhas)} Burkea africana

210
HOOK de Angola poderá, em princípio, ser aproveitada cp_ mo fonte de matérias tânicas,
3.4.1.6,- Dosagem dos taninos catéquioos
A dosagem efectuada por precipitação dos tani_ nos catéquicos com o reagente de STIASNY (formol-ácido clorídrico (2si)).
2 g de pó desengordurado foram deixados em con tacto, durante 2 horas, com 150 ml de água a 80°G 5 decorrido este tempo filtrou-se e lavou-se o resíduo com água quente, até à obtenção do volume final de 200 ml,
A 100 ml do filtrado adicionámos 50 ml de rea. gente de STIASNY^ apés repouso de 24 horas, o precipita, do foi recolhido em filtro de papel previamente pesadoe lavado até reacção neutra do filtrado. 0 filtro foi seco em estufa a 100 G, até peso constante.
Média dos resultados5 8,3 g fo.
3.4.1.7,- Pesquisa de derivados quinonicos
3.4.1,7,1.- Quinonas livres
20 g de pó de droga foram humedecidos com 10 ml de ácido clorídrico a Stfo, Adicionou-se 100 ml de clorofórmio e, após algumas horas de contacto,filtrou-se. A solução foi reduzida, por evaporação, a 10 ml e adicio-

211
nou-se-lhe 5 ml de amónia diluída; não se observou aparecimento de qualquer coloração.
3.4,1,7.2,- Heterósidos antraquinómcos
20 g de pó de droga foram lançados em balão de rolha esmerilada e adicionados de 100 ml de solução clorídrica a 5$, Adaptou-se ao balão um refrigerante e submeteu-se a aquecimento a banho maria durante uma hora.
A solução ácida foi separada e esgotada pelo clorofórmio? a solução clorofórmica, depois de reduzida a pequeno volume, foi adicionada de amónia diluída.Esta não corou.
Perante estes resultados parece-nos legítimo afirmar da inexistência de compostos quinónicos quer li vres quer sobre a forma heterosídica.
3.4.1.8,- Pesquisa de heterósidos cianogenéticos
20 g de pó da droga foram introduzidos em ma-trás de 250 ml de capacidade e adicionados de 10 ml de solução sulfúrica a. 10$, Rolhámos o matrás tendo previa mente suspendido da rolha uma tira de papel picrossódi-co. Após aquecimento a banho maria, durante duas horas, não observámos alteração da cor amarela do papel picroe

212
sódico, pelo que parece podermos afirmar que a casca da planta não contém estes compostos *
3,4.1.9, Pesquisa de heterosídos cardenol.id_i.oos_
20 g de pó foram tratados com 100 ml de.metanol. 0 solvente foi eliminado, soo a pressão reduzida e a baixa temperatura, e o extracto obtido ensaiado com o reagente de KEDDE por deposição de gotas do extracto em papel de filtro seguida de imersão no reagente citado.
Não foi possível tirar qualquer conclusão des. te ensaio porquanto como o extracto apresentava cor ver melha acastanhada intensa e a aparição da cor azuladavip lácea9 que porventura se produziria_se.este^_noompofetes existissem, era mascarada pela propria cor do extracto,
3.4,1.10, Pesquisa de saponosidos.
0 infuso da droga» quando agitado,espuma a
bundantemente, mas esta é pouco espessa embora ■..pérsia tente,
A circunstância de na literatura encontrarmos referência ao uso de Burkea africana. IIOOK como ictioté.xica levounos a pesquisar saponosidos e, para isso,pro cedemos à determinação do respectivo índice de espuma.A técnica utilizada foi a indicada na Farmacopeia France

213
sa 1965 - VIII edição, usando um digesto a 1%.
Numa série de tubos de ensaio de 16 cm de altura e 16 mm de diâmetro introduzimos quantidades crescentes do digesto (1, 2, 3 10 ml) e oomplòtá-mos o volume de 10 ml em cada tubo com água destilada. Após agitação de --cada um dos tubos durante 15 segundos, seguida de repouso durante 15 minutos,verificámos que no tubo n2. 4 se observava espuma com altura de 1 cm.
0 índice de espuma d, portanto igual a 250. 0 índice é muito baixo e, por isso, pomo3 certas reservas quanto à existência destes compostos na planta.
(

215
P.5,- ESTUDO DO EXTRACTO ÉTER DE PETRÓLEO
Extracto A
Como veremos, a extracção dos alcalóides foi precedida de desengorduramento do pó com éter de petróleo, obtendo-se uma solução que apresentava cor amarela intensa, à luz natural e fluorescência esverdeada. Por sua vez, o resíduo, R^, resultante da eliminação do so_l vente por evaporação a baixa temperatura e pressão ,apre_ sentava cor amarela alaranjada e consistência butirosa.
Como o óter de petróleo pode extrair,além de gordura, outros constituintes, tais como compostos es-terdcLicos, terpénicos, carotenóides, flavonóides, compostos quinónicos e certos alcalóides, etc.,procurámos, seguidamente, pesquisar esses compostos no extracto obtido.
S.5.I.- Ensaios prévios
Para pesquisar as substâncias atráa referidas praticámos as reacções seguintes:
Alcalóides - reacção de DRAGENDORFF (técnica da mancha em papel de filtro).
Esteróides e triterpenos - reacção de LIEBER MAM - BUECHARD e reacção de SAL E&7SKT.

216
Carotenóides - reacção de CAKR-PRICE. Flavonóides - reacção de SHIBATA. Compostos quindnicos - reacção de BORNTRKGER.
Os resultados destas pesquisas inscrevem-se no QUADRO III e, em face deles, parece de excluir a hipóte se de que o éter de petróleo dissolva os alcalóides e-xistentes na planta.
Q U A D R O III
COMPOSTOS REACÇÃO COR Alcalo'ides DRAGENDORFF M o se observou cor verme
lha nem alaranjada
triterpenos
LIEBERM&NN--BURGHARD
Anel vermelho violáceo
triterpenos SALKOWSKE Vermelha violácea
Carotenóides CARR-PRICE Azul violáceo
Flavonóides SHIBATA Negativa, sem alteração
Quinonas BORNTMGER Negativa, sem alteração
Por outro lado, a negatividade da reacção de SHIBATA e de BOTNTMGER confirma os ensaios já anterior mente realizados para a pesquisa de flavonas e quinonas.

217
Pelo contrário, é de admitir a existência de caroteno'i-des e de estero'ides ou triterpenos no extracto éter de petróleo.
A intensa reacção de LIE3EIMA1IN-B1MJIIAED deixou antever que os estero'ides ou triterpenos possivelmen te existentes se encontrariam em razoável quantidade.
A presença de um produto branco,cristalino,no seio da massa gordurosa amarela-alaranjada,que conferia ao extracto aspecto nacarado e um tanto friável, suscitou a nossa curiosidade. Admitimos que tal produto fos se responsável pelas reacções fortemente positivas de LIEBEHMAM e decidimos, por isso, estudar estes compostos.
Começámos por averiguar se no extracto existia apenas um ou vários componentes deste tipo. Para isso, um pouco de resíduo,, RA foi dissolvido em pequenaquan tidade de clorofo'rmio e cromatografado em placa de alumina. Esta foi desenvolvida no sistema constituído por heptano-benzeno-álcool (50s50s0,5).
A revelação da placa, apo's eliminação do solvente desenvolvente por exposição ao ar, foi feita com a mistura de ácido sulfúrico-anidrido acético-álcool(lO; 10s80), nos moldes descritos por IKA.N e col. (3), e já utilizado por nós em trabalhos anteriores (4, 5).As pia cas depois de aspergidas com o referido revelador,foram colocadas na estufa a 120°C durante 5 minutos.

218
O revelador permitiu-nos individualizar várias manchas, coradas umas de violáceo., outras de tons azulados e ainda outras de castanho acinzentado.
Uma outra placa preparada nos mesmos moldes, mas revelada por exposição aos vapores de iodo, mostrou várias outras manchas além das que aquele revelador per mitira assinalar.
Perante a complexa composição do resíduo, RA, procedemos ao seu fraccionamento em coluna de alumina, Gol. I, com eluentes de polaridades sucessivamente cre_s centes, desde o éter de petróleo até ao clorofórmio. Ás quinze fracções eluidas apresentavam aspecto gorduroso e, depois de cromatografadas em camada delgada de alumi na, nos moldes atrás citados, mostraram composição com plexa e bastante similar, se bem que algumas mais enriquecidas num ou noutro componente.
Pof'i improfícua esta primeira tentativa de separação e, por isso, decidimos reunir todas as fracções e submetê-las a uma saponificação prévia para estudar o insaponificável assim obtido,
3.5.2.- Saponificação do resíduo ffy
9,5 g do resíduo A foram saponificados com 500 ml de potassa alcoólica aproximadamente 2 N, por aqueci mento à ebulição, durante 4 horas.

219
Após a saponificação, o álcool foi eliminado sob pressão reduzida a baixa temperatura, adicionando--se ao resíduo 500 ml de água destilada. A suspensão ob tida foi extraída em ampola de decantação com éter de petróleo, num total de 5 veze3, utilizando-se 100 ml der te solvente de cada vez. As soluções etéreas foram reunidas e o resíduo, R.., da evaporação do éter de petróleo apresentava cor amarela alaranjada e pesava,após se_ cagem, 2,92 g.
A solução aquosa remanescente foi acidificada com ácido clorídrico e agitada 5 vezes com 100 ml de é-ter de petróleo de cada vez. Após eliminação do solvente e secagem, o resíduo apresentava cor amarelada acastanhada e pesava 5,51 g.
3.5.3,- Separação dos constituintes do resíduo R.-
0 resíduo RA,, submetido à análise croinatográ fica em camada fina de alumina, corno atrás referimos mos trou vários componentes, sendo de destacar como predomi nantes os componentes A e B, corados respectivamente de rosa violáceo e de acinzentado azulado.
Partindo de 2,85 g do resíduo procedemos ao seu fraccionamento em coluna de alumina (84 g), e eluin do esta com fracções de 280 ml de solventes de polaridades sucessivamente crescentes. 0 resultado deste fraccji onamento regista-se no QUADRO IV.

220
Q U A D R O IV
CROMATOGRAFIA EM ALUMINA DO RESÍDUO, RA 1
C O L U N A I I
Número da
fracção Eluente
Peso do
Resíduo
Cor do
Resíduo
Reacção de
LIEBERMAM
1 Éter de petróleo
0,1878 amarela-ãLaranjada +
2 Éter de petróleo
0.4496 amarela-alaranja- +
3 petróleo
0,2813 amarela-alaranja-da
+
4 E-« pet, 5 Benz, 5
0,3005 amarela +
5 E,pGu. 5 Benz, 5
0,4545 amarela +
6 E,, pet, 5 Benz, 5
0,174 5 amarela clara +
7 Benzeno 0,2909 amarela paj_a.cí.a
+
8 Benzeno 0,1245 amarela paiida -+
9 Benz, 5 Clorof.5
0,1715 amarela pálida +
10 Benz, 5 Clorof.5
0,1385 amarela pálida +

221
11 Benz. 5 Clorof.5
0,1130 amarela pálida +
12 Clorof. 0,0152 amarela +
13 Clorof. 0,0763 amarela +
14 Clorof.5 Metan, 5
0,0712 amarelada -
15 Clorof.5 Metan, 5
0,0000 s/resíduo -
2,8493
Todas as fracções da COLUNA I I foram anal isadas cromatogràficamente em placa de alumina. As f racções menos complexas foram II7 a I I ; Q mas apesar disso ainda mostravam após revelação com a mistura ácido su l -fúrico-anidrido acético - á lcool , t r ê s manchas: A,B e C, A mancha A apresentava cor acastanhada, a mancha B cor azulada violácea e a mancha C cor rósea violácea.
Reunimos es tas fracções (Col. I I 7 _ n i ) , de peso igual a 0,8384, e tentámos a si; i purificação por cris t a l i zaçâb , primeiro em acetona e depois em álcool .
0 resíduo destas duas cr i s ta l izações continua va a apresentar duas manchas B e C e ao microscópio mos t rava aspecto c r i s t a l i no , com c r i s t a i s prismáticos bastante compridos e outros muito pequenos, em agulhas.

222
Em microscópio de KOEFLER observámos que o pro duto começava a fundir a cerca de 135 C e continuando o aquecimento sublimava à volta de 180 C sot a forma de agulhas isoladas e curtas. Todo o resíduo estava fundido a cerca de 200 C.
M o restava dúvida que o resíduo carecia de no vo fraccionamento. Procedemos à separação das substâncias B e C, usando novamente a cromatografia em coluna de alumina (Col. III).
No QUADRO V indica-se este fraccionamento.
Q U A D R O V
CROMATOGRAFIA EM ALUMINA. DAS FRACÇÕES, Col . 117-11
C O L U N A I I I
Número da f raç. Ção
4
Eluente
É t e r de p e t r ó l e o
E t , p e t . 8 Benz. 2
E t « p e t . 6 Benz. 4
E t . p e t . 5 Benz. 5
Peso do res íduo
H resíduo
H resíduo
s/ resíduo
H r e s íduo
P . f . do r e s íduo
Cor do res íduo
Reacção de LIEBEPMNN

223
5 Et.pet.4 Benz, 6
s/ resíduo
- -
1 6 Et.pet.2 Benz, 8
0-,0075 65-70° amarela da +
7 1
Benz. 0,0135 110--115°
Branca +
8 Benz, 0,0160 120--128°
E:;'anca +
9
! Benz. 0,0470 130-
-135° Branca +
1 1 10 1
Benz, 0,0510 130--135°
Branca +
11 Benz, 9 Clorof.l
0,1795 180--183°
Branca +
12 Benz.. 9 Clorof.l
0,0750 180--198°
Branca +
13 Benz. 9 Clorof.l
0,0065 185--200°
Branca +
14 Clorof. 0,0050 185--200°
Branca +
15 Clorof„ 0,0050 amarela da
-
0,4060 As fracções Illgelllig foram cristalizadas em
álcool e depois duas vezes em acetona.

224
3 .5 .4 , - Identificação dorò-s i tos tero l nas fracções
Sa aSiû
Após as três cristalizações citadas o ponto de fusão do resíduo fixou-se em 141-142 G. Desde logo pensamos estarmos em presença do /^-sitosterol (l),este róide muito difundido no reino vegetal que segundo KÃ.R-EEE (6) e Index Merck (?) funde a 139-140°C e 140°C res. pectivamente. Aliás, a temperatura de fusão do nosso pro duto misturada com uma amostra autêntica de fò<~sitoqte rol Fluka, não sofreu alteração.
Cromatografáàoem alumina, usando como desenvol vente o sistema heptano-benzeno-álcool (50s50í0,5) apre sentava o mesmo Rf (0,07) e cor idêntica à da amostra de /3-sitosterol padrão, após revelação, com o reagente á
eido sulfúrico-anidrido acético e álcool (Fig. 5)» Para confirmar que na realidade se tratava do
^-sitosterol determinámos o seu espectro na região do I.V. mas a comparação do espectro do produto como o de uma amostra autêntica de <3-sitosterol revelou certas dis crepâncias, embora ligeiras.
Persistimos, contudo, na ideia de que devíamos ter isolado este composto, embora admitíssimos que se encontrasse ainda impurifiçado, mas uma nova recris-talização do produto de álcool não conduziu a uma perfeita sobreposição dos espectros no I.V. da nossa subs-

225
tância e do padrão.
Resolvemos, por isso, preparar os derivados acetilado e benzoilado do produto I isolado de Burkea africana, na esperança de que a cristalização daqueles ésteres nos permitisse libertarmo-nos da substância im-purificadora.
3.5.5.- Preparação do acetato de^-sitosterol e do acetato do produto I
50 mg do produto I foram adicionados de 3 ml de piridina e 1 ml de anidrido acético. A mistura contactou durante 24 horas à temperatura ambiente. 0 resíduo resultante da eliminação dos solventes foi lavado, várias vezes, com água, até não se notar cheiro a piridina. 0 resíduo foi cristalizado de acetona e recrista-lizado de áloool.
0 derivado acetilado mostrou estar puro quan-

226
do cromatografado em placa de alumina e deslocou-se à mesma altura, Rf = 0,63, que um produto que preparámos de modo idêntico ao citado a partir de uma amostra autêntica de (¾ -sitosterol (Fig 5),
0 p,f. do produto em análise foi de 128-129 C e não sofreu alteração quando misturado com acetato de ft -sitosterol padrão.
Os espectros no I.V, do acetato do produto por nós isolado e do acetato da amostra de A -sitosterol são perfeitamente sobreponíveis (Fig. 6).
3.5.6.- Preparação do benzoato de (^-sitosterol e do benzoato do produto I
Preparámos igualmente o benzoato do produto I isolado de Burkea africana e do ft -sitosterol autêntico pela técnica seguintes
50 mg de produto foram adicionadas de 2 ml de piridina e 0,2 ml de cloreto de benzoílo,deixando-se 24 horas em contacto à temperatura ambiente. Após elimina ção do excesso dos solventes, lavagem do resíduo e cris, talização repetida em éter e acetona, obtivemos um produto cristalino que, depois de seco, fundia a 147-148 G, sendo de notar que a temperatura indicada na literatura (?) para o derivado benzoilado do9>-sitosterol é de 14jS -147°C.
São também coincidentes para os dois produtos

227
o Hf «0 ,85 em placa de alumina (Fig. 5) e os espectros na região do I.V. (Fig. 7) .
Já r&ovw&mm àfrj&asde que o produto i so la do era na realidade o /3-si tosterol mas quisemos no entanto mais uma confirmação e para isso extraímo-lo dos e'steres preparados, apds h id ró l i s e .
3 . 5 .7 , - Hidrólise do acetato e benzoato do produto I isolado de Burkea africana
Estes ésteres resul tantes do produto isolado de Burkea afr icana, depois de cr is ta l izados do é te r e de acetona foram submetidos à h idró l i se durante 4 hor a s , com solução alcoólica de hidróxido de potássio N/2» Após eliminação do álcool por evaporação e adição de á-gua des t i lada , a suspensão foi extraída por agitação com é t e r . 0 resíduo da evaporação do é te r sulfúrico,mostrou t e r um espectro perfeitamente coincidente com o do ft-ai to s t e ro l que cristalizámos de igual modo (Fig. 8).Foram também coincidentes os seus poderes ro ta tór ios específ i cos quando determinados em solução c l o r o f ó r m i c a ^ - S õ 0
(C=2 em clorofórmio).
3 . 5 . 8 . - Estudo da3 fracções I l l i g a I I I15
As fracções 12 a 15 da Col. I l l mostraram

228
placa de alumina no sistema heptano-benzeno-álcool (50s 50s0,5), duas manchas, uma de Rf idêntico ao do\£-sitos, terol (mancha C) e outra de Rf ligeiramente mais baixo (mancha B).
Tentámos a separação dos dois produtos por cromatografia em coluna de ácido silícico (Col. IV),iniciai do a eluição com benzeno.
As fracções seguintes, de polaridades sempre crescentes eram constituidas por misturas de benzeno e clorofórmio. As fracções 4 e 5 correspondentes à eluição com benzeno-clorofo'rmio (5:5), continham ^-sitosterol. As fracções 6 e 7, quando observadas ao microscopio,não se apresentavam cristalinas. Entretanto, procedendo ao seu aquecimento, verificámos que a cerca de 180-185 Cse iniciava a sublimação de substância, depôsitando-se na lamela superior em agulhas agrupadas em ouriços. Continuando o aquecimento à temperatura de 218-220 C esses ouriços converteram-se em longas agulhas que acabaram por fundir a 251°. Embora tivéssemos tentado estudar es te composto, produto II,não nos foi possível,conseguir a sua identificação.

229
O O °»85
o O °'63
O O0»07
Fig. 5 - Cromatografia em placa de sílica 1 2 3 4 5 6
-A - s i t o s t e r o l Pluka -Produto I. isolado de Burkea africana flOOK - BoîEoato de Ç'j-sitosterol - Benzoato do m-oduto I - Acetato de (*s - s i t o s t e r o l - Acetato do produto I
< 3 DO
E
0) ■o
O ■o
o ■D
o o _CD
8 ' (0 O O) O 09
0) <->
z (0 c
V)
o O
o U -i
Z
o <C0
o
(A 1
"D O a
(0 a> o > - Q T3 "D
_ < (D g í 0 o
0) ** "O ro a +-1
O ai a> O o o
O < < 0) a 1 1 w LU 1 - CM
(O
d) %) 3DNV11IWSNVM1

y. CD
E 0)
o 0)
(Õ, <u
■u o O
O '2 O) (U
o c
,8 .o o
U ai Q.
O l/)
O a) « O o
'5> X C2 LI.
a> O 0 T3 O O
8 O o N N
C t: <u <u m CO
— rsi
d) |%) 3DNV11IWSNVH1

Cû
E 0)
o -o o !2 o> o c o O -
JS a 0)
"D
U ai Q.
O 3
O o I "a c o u
o O)
- y
CO
<u
O -a o L i o
in ei
HH a> 0 o -1
m "H <lí CL
— (N
[/o) 3DNV11IWSNVMX
CO
O) i-L.

237
BIBLIOGRAFIA
1 - NETO, I., Análise de géneros alimentícios - Métodos físicos e químicos, Lisboa (1959).
2 - OFFICIAL METHODS of ANALISIS of the International Society of leather trades chemists,A.Harvey Publ., London (1938),
3 - IKM, R,, HAREL, E., KASHMAN, J,- BERGMANN, E.D., J. Chromatog. 14, 504 (1964).
4 - FERREIRAy M.A,, CORREIA ALVES, A., NOGUEIRA PRISTA, L., CRUZ COSTA, M.A., Garcia de Orta 16 (ne.l), 31 (1968). "
5 - FERREIRA, l£.A.f CRUZ COSTA, M.A,, Garcia de Orta 16, (ne.4) 417 (1968),
6 - KARRER,, W,, Konstituition und Vorkomen dor orga-nischen Fflantzenstoffe, Birkhauser--Verlag Basel (1958).
7 - The Merck Index of Chemicals and Drugs, 76,edição MERCK & CO., Inc., Rahway, N.J,,U,S,A, (i960).

239
3..6. - ESTUDO QUÍMICO DOS ALCALÓIDES DE BURKEA AFRICAM HOOK
3.6.1.- Extracção dos alcalóides totais de Burkea africana HOOK
A primeira extracção de alcalóides que reali zámos foi praticada sobre 1,5 Kg de pó das cascas do tronco e ramos da planta, nos moldes já" descritos: nos. ensaios preliminares. Antes, porem, o pó foi desengordu rado em extractor de SOXHLET, com 5 1 de éter de petróleo puro (p.e.=50-70°C), durante 16 horas. A solução re sultante desta extracção foi destilada a 50 C, sob prés. são reduzida, até ficar reduzida a 250 ml, e nela pesquisámos alcalóides, embora saibamos que,, o éter de petróleo, dada a sua baixa polaridade, não tem, por via de regra, poder dissolvente para a maioria daquelas substâncias. M o obstante ter sido negativa a reacção de_s tes compostos frente ao reagente de DRAGENDORFF modificado, usando a técnica de deposição de gotas da solução em papel de filtro, já observada nos ensaios preliminares, por uma questão de precaução,submetemos a solução a uma extracção com 250 ml de solução de ácido sulfúrico a 3$. A solução aquosa, depois de separada, foi alcalini-zada a pH = 10 com amónia e agitada com três porções de 100 ml de clorofórmio. Após eliminação deste solvente.,
/

240
obtivemos um resíduo, Rç, insignificante, que não reagiu com os reagentes gerais dos alcalóides,excluindo-se assim, a hipótese aliás já remota, do éter de petróleo ter extraído alcalóides existentes na planta. Pelo contrário, o resíduo, R«, resultante da completa eliminação do óter de petróleo,após a referida extracção pelo ácido sulfúrico foi bastante abundante (9,75 g) e ao seu estudo já nos referimos anteriormente,
0 pó resultante do desengorduramento pelo é-ter de petróleo foi exposto ao ar, até eliminação deste solvente. Em seguida, foi lançado em balão de capacidade apropriada e humedecido com 500 ml de solução de amo nia a 50% (preparada a partir de solução com 25% de amo níaco), com a qual esteve em contacto durante 24 horas. Passado este tempo adicionaram-se 5 litros de uma mistu ra de éter-clorofórmio (3si) e deixado em contacto por igual período de tempo com frequentes agitações manuais.
A solução etéreo-clorofórmica foi separada por filtração e o pó, depois de bem espremido, foi de novo lançado no balão e tratado com igual volume de mistura de solventes. 0 resíduo sofreu ainda um terceiro esgota mento com 4 litros desta mistura dissolvente.
As soluções etéreo-clorofórmicas foram reunidas e os solventes recuperados, em parte, por destilação a 50°C e a pressão reduzida. A solução, concentrada a 500 ml e apresentando cor amarela acastanhada,foi lan

241
cada em ampola de decantação e extraída com 1,5 litros de solução sulfúrica a 3$, usando-se fracções de 300 ml em cada extracção. A primeira adição de ácido à solução provocou o aparecimento de cor avermelhada intensa na _fk se aquosa.
As soluções ácidas reunidas foram agitadas por duas vezes com 500 ml de éter de petróleo de cada vez, com o fim de retirar da solução aquosa ácida impurezas lipossolúveis que porventura pudessem ainda existir nela.
A solução ácida, que apresentava cor amarela -avermelhada, exibia à luz ultra violeta intensa fluo rescência azul-violácea,
A adição de amónia a esta solução,até pH =10, provocou forte turvação, aparecimento de cor verde arroxeada e fluorescência verde amarelada intensa à lampa da de WOOD.
As bases foram extraídas com 1,5 litros de clo_ rofórmio, tendo-se usado 300 ml em cada esgotamento. A-pós eliminação do solvente, a baixa pressão e temperatu ra, obtivemos um resíduo, R», que depois de seco pesava 1,1450 g, de cor branco-amarelada, muito leve e de aspecto esponjoso(ESQTJEMA I).

242
ESQUEMA. I
Extracção dos alcalóides de Burkea africana
Pó de casca do tronco
Extr. com éter de petróleo
Sol. éter de petróleo
Ext. c/íí2S04 a 3$
< r ~ Sol- âi-ér de ptí-crõleo
Evaporação
Resíduo, R^ (gordura e outra3 substâncias)
1 Pó desengordurao
Ext.c/é-ter>-clo-rofórmio (3sl) em meio amo niacal
Sol.aquosa ácida Sol. etéreo cloro formica
Amónia Extr. c/clorofórmio
Sol. clorofórmica
Evaporação
Resíduo, Rn (não contém alcalóides)
H 2 S 0 4 a $
Sol.aquosa ácida
Amónia Ext fc/cio rofórmio
Sol. clorofórmica
Evaporação
Resíduo, Rg (alcalóides to ta is)

243
3.6,2.- Reacções de precipitação e de coloração do resíduo, Rp
1 - Uma. pequena porção de resíduo, dissolvida em ácido sulfúrico a 3$, precipitou pelo reagente de VALSER-MA.YER.
2 - Uma poquena porção do resíduo, dissolvida em ácido sulfúrico a 3$, precipitou pelo reagente de DRAGENDORFF.
3 - Uma pequena porção do resíduo, dissolvida em ácido sulfúrico a 3$, precipitou pelo reagente de BERTRAND.
4 - 0 resíduo tratado com reagente fosfovaní-lico originou coloração azul esverdeada.
5 - Um pouco de resíduo, adicionado de reagen te de EHRLICH aquecido a banho maria,originou coloração azul esverdeada.
6 - Um pouco de resíduo, dissolvido numa gota de ácido acético e adicionado de ácido sul fúrico contendo 0,5$ de nitrito de so'dio, provocou aparecimento de cor azul escura,
7 - 0 resíduo, adicionado de ácido sulfúrico, corou de vermelho intenso.
8 - 0 resíduo, adicionado de solução de sulfa.

244
to cérico e aquecido a banho maria duran te 10 minutos, não origiïPUcoloração.
9 - 0 resíduo, dissolvido em 2 ml de ácido clo_ rídrico 0,2 N, foi adicionado de 1 ml de l-nitroso-2-naftol e de 1 ml de reagente de ácido nitroso e aquecido a banho maria a 55 G durante 5 minutos °, não se observou coloração.
Estas reacções são suficientemente elucidativas quanto à existência de alcalóides e depõem a favor da presença do núcleo indólico nas suas estruturas.
A negatividade da reacção com o reagente l-nitroso-2-naftol excluiu, dentro de certos limites, a existência de derivados do 5-hidroxindol. (l,2,3).
3.6c3,- Ensaios cromatográficos praticados com o resíduo RR
Numa tentativa de averiguar do número de alça, lo'ides existentes no extracto alcaloídico bruto fizemos vários ensaios cromatográficos em papel,pela técnica as_ cendente e descendente, e em camada delgada de sílica e de alumina

245
3.6.3,1.- Ensaios cromatográficos em papel
Utilizámos, como suporte para estes ensaios,o papel WHATMAN n2. 1 e como desenvolventes os sistemas: butanol-ácido acético-água (4:1 :5), butanol saturado de água e ainda um terceiro, constituído por tetracloreto de carbono-benzeno-metanol (4:l:l), Com este último sol. vente praticámos o desenvolvimento nas duas modalidades ascendente e descendente, enquanto que com os dois primeiros apenas experimentámos a técnica cromatográfica as cendente.
Os cromatogramas foram revelados por exposição à luz ultra violeta, e depois por imersão em reagen te de DRAGENDOKFF, segundo a formula de MUNIER e MACHE-BOEUF. (4).
A revelação conjunta com os reveladores referidos deixoa-nos antever a presença de mais que um alça lóide , mas, dada a má individualização das manchas,não nos permitiu verificar qual o número existente. De facto, observaram-se em todos os casos manchas muito alongadas que se estendiam do meio do cromatograma até próximo da linha de frente, exibindo fluorescência verde a parte mais próxima da linha de partida e forte fluorescência azul violácea a zona mais próxima da linha de frente, notando-se, além destas duas manchas fluorescen tes, a sobreposição de várias outras zonas com fluorescência amarelada. Dos três sistemas ensaiados o que per

246
mitiu melhor individualização das manchas foi o tetracloreto de carbono-benzeno-metanol (4slsl), por técnica descendente, mas, mesmo com este desenvolvente, a separação estava longe de ser aceitável.
3.6.3.2.- Ensaios cromatográficos em camada delgada
Informadas, através da cromatografia em papel, da complexidade do extracto alcaloídico bruto,resolvemos abandonar .esta técnica e ensaiar o extracto em camada delgada de sílica e de alumina, uma vez que as separações por cromatografia em camada fina são normalmente mais eficientes.
Foram ensaiados vários solventes e os melhores resultados foram conseguidos com combinações,em va-riadasproporçõeB/P.eclorofórmio-metanol sempre que utili závainos as placas de sílica. Assim, os sistemas? clorofórmio-metanol (9,6s0,4), clorofórmio-metanol (9,5s0,5), clorofórmio-metanol (9sl), clorofórmio-metanol (8s2),clo rofórmio-metanol (3s2) pareceram-nos nesta primeira fase os mais eficientes e por isso os utilizámos repetid_a mente no decurso do nosso trabalho.
Com a alumina, o acetato de etilo puro, aceta to de etilo-metanol (3sl e l°l), acetona-etanol (8,5 : s 0,5), também se mostraram úteis na separação dos alça. lóides do extracto bruto.

247
Entretanto, como a revelação com o reagente de DRAGENDORFF com este adsorvente só originava aparecimen to das manchas vermelhas ou alaranjadas em alguns casos após certo tempo de espera, optámos, no decorrer do no_s so trabalho, pelo uso de placas de sílica por esse fao, to e ainda por serem de mais fácil preparação.
Cromatografando o extracto em placas de síli ca e desenvolvendo-aa em clorofórmio-metanol (8s2) e clorofórmio-metanol (9!l),ob8exvavamMse r.pís revelação pelo reagente de DRAGENDOEFF, entre outras, duas manchas intensamente coradas de avermelhado a que chamaremos A e F.
As placas, observadas à luz de U.V.,antes da revelação com o reagente de DRAGENDOEFF, mostravam variadíssimas manchas fluorescentes, sendo a mancha A não fluorescente ou com ligeira fluorescência esverdeada,en cimada por uma outra, com fluorescência verde,e que tam bem reagia com aquele revelador. A mancha F apresentava fluorescência azul violácea muito intensa.
Perante a existência de um tão grande número de manchas fluorescentes, reagindo umas fortemente, com formações de cor rosa avermelhada, com reagente de DRA GENDOEFF, e outras apenas com cor amarela alaranjada,de_ cidimos proceder a um primeiro fraccionamento dos constituintes do extracto com o objectivo de isolar os alça lóides correspondentes às manchas A e F.

248
3,6.4»- Separação dos alcalóides por cromatografia em coluna
Com v i s t a ao isolamento dos produtos A e F ten
támos o fraccionamento do e x t r a c t o b ru to em coluna de a
lumina ,par t indo de 1,53 S ào res íduo % e de 45 g des t e
adso rven te , processando-se a sua e lução com fracções de
100 ml de so lven te escalonados segundo po la r idades su
cessivamente c r e s c e n t e s , desde o I t e r de petro ' leo ao me
tano lc
Os r e s u l t a d o s des te p r imei ro fraccionamen
to inscrevem-se no QUADRO VI.
Q U A D R O VI
CROMATOGRAFIA EM ALUMINA DO RESÍDUO,R^
C O L U N A V
Número de fracção
Eluente Peso do resíduo
Cor da solução
Reacção de DRâGENDORFF(cor)
1 . petróleo
- incolor -
2 Eçpet. 8 Benz, 2
- - -
3 Eopet. 6 Benz, 4
0,0015 incolor (Duvidosa) amarelada

249
4 Eppet, 6 Benz, 4
0,0040 incolor (Duvidosa) amarelada
5 E.pet, 6 Benz. 4
0,0025 amarelada
(Duvidosa) amarelada
6 E.pet, 5 Benz, 5
0,0015 amarelada
(Duvidosa) amarelada
7 E.pet. 5 Benz. 5
0,0015 amarelada
(Duvidosa) amarelada
8 E.pet. 4 Benz. 6
0,0015 amarelada
alaranjada
9 E.pet. 2
Benz. 8
0,0010 amarela esverdeei da
alaranjada.
10 Benz» 0,0010 esverdea da
alaranjada
11 Benz.9,5 Clorof.0,5
0,0020 acastanhada
alaranjada
12 Benz. 8 Clorof.2
0,0040 acastanhada
alaranjada
13 Benz. 6 Clorof.4
0,0030 cl C cio X cl"" nhada
alaranjada
14 Benz. 6 Clorof.4
0,0030 CVCOÍSX cl-" nhada
alaranjada
15 Benz, 6 Clorof.4
0,0065 amarelada
alaranjada
16 Benz. 6 Clorof.4
0,0040 amarelada
alaranj ada

250
17 Benz, 5 Glorof.5
0,0065 amarela da
alaranjada
18 Benz, 5 Clorof»5
0,0080 amarelada
avermelhada
19 Benz p 5 Clorof.5
0,0175 amarelada
avermelhada
20 Benz, 4 Clorof.6
0,0140 amarelada
avermelhada
21 Benz. 4 Clorof.6
0,0550 amarelada
avermelhada
22 Benz. 2 Clorof.8
0,0695 amarelada
avermelhada
23 Benz. $ Clorof.8
0,0145 amarelada
avermelhada
24 V.^TOf , 0,0475 amarelada
avermelhada
25 Clorofo 0,0950 amarelada
avermelhada
26 Clorof, 0,0435 amarelada
avermelhada
27 Clorof .9,5 MeCH 0,5
0j1530 amarelada
avermelhada
28 Clorof .9,5 MeŒÎ 0,5
Of0825 amarelada
avermelhada
29 Clorof. 9 MeŒI 1
0,1040 amarelada
avermelhada

251
30 C lo ro f .9 MeCH 1
0,2855 amarelada
avermelhada
31 Clorof .9 MeCH 1
0,0345 amarela-da
avermelhada
32 Clorof .9 MeCH 1
0,0255 amarelada
avermelhada
33 C lg ro f . 8 M3CÍI 2
0,0485 amarelada
avermelhada
34 C lo ro f . 8 MeOH 2
0,0340 amarelada
avermelhada
35 C l o r o f . 8 MeŒI 2
0,0195 amarelada
avermelhada
36
I
Clorof .6 MeOH 4
0,0250 amarelada
avermelhada
37 Clorofc6 MeCH 4
0,0010 amarelada
avermelhada
38 C lo ro f .5 MeCH 5
0,0115 amarelada
avermelhada
39 Clorof -5 MeCH 5
0 ,0145 amarelada
avermelhada
40 C lo ro f ,5 MeOH 5
0,0250 amarelada
avermelhada
41 Clorof„6 MeCH 4
0,0160 amarelada
avermelhada
42 C lo ro f .S MeŒI 4
0,0295 amarelada
avermelhada

252
43 Clorof . 8 MeOî 2
0,0090 amarela da
avermelhada
44 MèCH 0,03 80 a c a s t a nhada
-
45 MeCH - l i C ci O Ti cl™*
nhada -
1.3445
3,6.5,- Análise das fracções da COLUNA Y
No QUADRO VI podemos ver que as fracções V, g e V não reagiram com o reagente de DRAGENDORFF e, por essa circunstância, desprezámo-las.
As fracções V fl reagiram com o reagente de DRAGENDORFF com cor amarelada e cada uma delas apresentava aspecto gorduroso e resídiuo insignificante.
Todas as fracções desde V a V _ eram constituídas por um resíduo amarelado acastanhado, de aspecto gorduroso; entretanto, quando observadas ao microscópio, mostravam conter cristais aciculares muito pequenos,dis persos no seio de uma massa gordurosa acastanhada esuhli mavam próximo de 120°C à pressão normal. Tal como acontecera com as fracções anteriores, a cor obtida por tra tamento destas com reagente de DRAGENDORFF,quando ensaiadas pela técnica de toque em papel de filtro, não era também suficientemente elucidativa para podermos a-

253
firmar, com segurança, da presença ou da ausência de al_ calóides nestas fracções. Como a dúvida se instalou no nosso espírito, não nos mereceram de início grande aten. ção. Pelo contrário, as fracções a partir de V?r) reagiram fortemente com o reagente citado, com formação -do cor avermelhada, e, por esse facto, foi destas que primeiramente nos ocupámos.
Como ensaio orientador, para averiguação da composição de cada uma destas fracções, cromátografamó--las em placa de sílica e escolhemos para fase móvel a mistura clorofórmio-metanol (8Í2), em consequência de ter sido este um dos sistemas experimentados que nos pa receu fornecer melhores resultados quando ensaiámos o extracto alcamoídico bruto donde provieram as fracções que agora analisámos.
A localização das manchas nas placas foi conseguida por exposição destas à luz de urna lâmpada de WO OD, seguida de aspersão com reagente de DRA.GENDOEFF. Em toda3 as fracções se observaram variadíssimas manchas fluorescentes mas a revelação com o último reagente citado permitiu-nos verificar certa similaridade na compo_ sição de algumas delas,o que nos levou a fazer algumas associações. Assim, em função dessa similitude reunimos as fracções V3 a V8? 7 g a V ^ ^ a Y ^ V ^ a V ^ ; 7 2 8 a 1^ e V 5 8 a V ^ .
A Pig. 9 mostra o aspecto duma placa de síli-

254
ca sobre a qual aplicámos estas fracções,desenvolvida e revelada nos moldes atrás descritos. No QUADRO VII indi cam-se as fluorescências das manchas que o reagente de de DRAGENDORFF permitiu detectar.

255
Fig. 9 - Cromatografia em placa de sílica das fracções da COLUNA V.

256
Q U A D R O VI I
Fracção Mancha Fluorescência no U0V,
Reacção c/reag* DRAGENDORFF(cor)
y 3-8 X amarela-azulada alaranjada
V 9-18 X amarela-azulada alaranjada
V19-21 E
F G X
amarela-alaranjada azul-violácea azul-"brilhante amarela-azulada
alaranjada
vermelha vermelha alaranjada
V22-26 A B C D E F G Y
n/fluorescente verde-hrilhante amarela esverdeada amarela amarela alaranjada azul violácea azul "brilhante rosa violácea à luz natural
vermelha ro s a- avermelha Sosa-avermelha da alaranjada alaranjada vermelha rosada rosa
V 28-36 A B C F
n/fluorescência verde "brilhante Amarela esverdeada azul violácea
vermelha rosa avermelha da rosa avermelha da avermelhada té nue i

257
v 28-36
X amarela asulada alaranjada
v 38-42
A n/fluorescòfi t e - vermelha
B verde "brilhante rosa avermelhada
F azul violácea vermelha
X amarela azulada alaranjada
Em face destes resultados tivemos de proceder à purificação das fracções agora reunidas, visto que ne nhuma se encontrava pura.
3,6.5.1,- Estudo das fracções Vg8-42
Começámos por estudar estas fracções por nos parecerem entre todas as mais limpas. Mesmo assim, como vimos no QUADKO "VII, o reagente de DRAGEKDOKFF detectou as manchas A, B_, P, e X» sendo no entanto a mancha A francamente predominante. A cristalização fraccionada deste resíduo em éter sulfúrico e em metanol permitiu--nos obter um resíduo cada vez menos amarelado.
Tal facto levou-nos a admitir que o resíduo continuava impuro. Uma cromatografia em placa de sílica nos sistemas clorofórmio-metanol (8:2) e clorofo'rmio-me tanol (3:2) mostrou-nos as manchas A, B_ e F_,embora es-

258
tas duas últimas fossem quase imperceptíveis» Além dis. so pelas cristalizações sucessivas a que procedemos eli minámos o produto X. Por outro lado, o ponto de fusão do resíduo obtido baixou consideravelmente em relação ao valor inicial, fixando-se a temperatura de fusão em limites bem mais estreitos. De facto, o resíduo destas cris talizações estava completamente fundido a 180 C,enquanto que com o produto inicial V38..42» embora apresentasse aspecto cristalino, a fusão iniciava-se à volta de 170°, com sublimação parecendo-nos que apenas terminava a 23Q°C.
Embora conscientes de que o alcalóide A não estava perfeitamente isolado, não resistimos a fazer a sua análise espectral na região do U.V. e visível como ensaio de orientação sugerindo o espectro da substância que o alcalóide predominante devia ser um alcalóide tipicamente indclico, provavelmente 2,3-dissubstituído.,da-da a semelhança espectral deste composto com o 2,3-dime-tilindol.
De facto, o produto isolado de Eurkea africa-na_ HOOK aprèsentavanáxinosde absorção em 225-26, 282--83, 290-91 nm, referindo a literatura (5) para o 29-3 --dimetilindol os máxdaos seguintes s 226, 282 e 290 nm.
Prosseguimos então na purificação do produto isolado. Uma das tentativas consistiu em tratar um pouco do produto dissolvido em metanol, com carvão activa-

259
do. 0 resíduo, apds filtração e eliminação do metanol,,, continuava com aspecto amarelado mas a quantidade obtida foi muito inferior à ensaiada. Rejeitámos este preces so visto que o carvão adsorvia fortemente o alcalóide e não permitiu que nos desembaraçássemos dos produtos B e P. Como tínhamos verificado que o produto sublimava, tentamos purificá-lo por este processo.
O sublimado ensaiado em camada fina continuava a apresentar as manchas A, B e F, sendo, no entanto, mais rico em alcalóides B e F do que o resíduo inicial. Este facto surpreendeu-nos desagradàvelmente,mas ao mej_ mo tempo despertou a nossa atenção deixando-nos antever certa semelhança de estrutura entre A, B e P_, já que A, por sublimação, nos pareceu originar B e F. Fomos no eja tanto mais felizes procedendo a um segundo fraccionamen to do resíduo em coluna de alumina (COLUNA VI) sensivej. mente eluida nos moldes descritos para a COLUNA V.Neste caso obtivemos apena3 30 fracções, verificando-se que as fracções eluidas com clorof<5rmio-metanol (6s4)s depois de analisadas em camada fina, apenas continham o alcalóide A.
3.6.5.1.1.- Identificação do alcalóide das fracções VTlO-20 com a tetraidroarmana ou eleag-
nina
0 resíduo conjunto das fracções VI,n a VIo~

260
depois de cristalizado em éter, apresentava cor ligeira mente amarelada e era formado por cristais prismáticosv Estes,depois de perfeitamente secos em exsicador de vazio durante alguns dias, fundiam a 179-180 C.
A cromatografia do produto isolado em placa de sílica nos sistemas clorofórmio-metanol (8s2) e cloro for mio -metanol (3s2) e metano1-amónia (97s3) apresentava uma só mancha de Rf respectivamente igual a 0,19 ; 0,29 e 0,48, a qual reagia com o reagente de EHRLICH , com formação de cor azulada, com o reagente fosfovaníli co, com cor azulada esverdeada, mas não reagia com orea gente de sulfato cérico quando as placas foram aspergidas com estes reveladores e aquecidas na estufa a 100 , durante 5 minutos. Também uma outra placa colocada em atmosfera de iodo permitiu observar apenas a mancha A intensamente corada de castanho.
0 espectro na região do ÏÏ.V. manteve-se perfeitamente coincidente com o que anteriormente tínhamos ob tido.
0 espectro obtido em solução metanólica de h i
dróxido de sódio N/ 2 0 n ã o sofreu alteração relativamen
t e ao determinado em meio neutro neste mesmo solvente.
0 meio ácido (solução metanólica de ácido cio
r íd r ico N/20) provocou um l ige i ro deslocamento hipsocro
mico nas bandas de absorção. Perante a temperatura de fusão e as caracterfe

261
ticas espectrais apresentadas pela substância, suspeita mos que tínhamos isolado o alcalóide tetraidroarmana , pois que elas estavam de acordo com as referidas na li teratura para este composto.
Preparámos seguidamente o cloreto e o iodome-tilato do alcalóide, e os pontos de fusão para este3 dois derivados foram os seguintes s
Gloridrato de alcalóide vT10_20 p.f.=2G8-69°C Iodometilato do alcalóide 1^10-20 P*f*=220 -
—21 C, Na literatura (6) indica-se para o cloridrato
o de teti-aidi^oamana o- p«.f<, de 267 G. Para confirmamos a identificação da substân
cia por nós isolada comparámo-la com a t et raidroarmana que preparámos por síntese por não a termos podido adquirir.
Síntese da tetraidroarmana (?)
1 g de harmana foi dissolvida em 100 ml de ai cool isoamílico em balão de colo esmerilado, ao qual se adaptou um refrigerante, aquecemos a solução à ebulição, sob refluxo, e fomos adicionando 6 g de sódio, em peque nas fracções. Apcís arrefecimento, vertemos o produto s_o bre 70 ml de ácido clorídrico a 1/2, colocado numa amp_q la de decantação, adicionámos éter, de modo a baixar a solubilidade do cloreto de tetraidroarmana formado no álcool isoamílico e esgotámos a solução amílica, várias

262
vezes, com água. Reunidas as soluções aquosas ácidas fo ram estas várias vezes lavadas, primeiro com éter de pe_ tróleo, e, depois, com éter etílico, com o fim de as i-sentar de álcool isoamílico, o que se reconheceu pela au sência de cheiro a este solvente. A solução aquosa foi alcalinizada com amónia e o alcalóide extraído com éter sulfúrico, o ÇuUal, depois de bem lavado com água, e seco pelo sulfato de sódio anidro foi eliminado no vazio a baixa temperatura. 0 resíduo de várias cristalizações em metanol e éter, depois de bem seco, fundia a 180-181°.
0 p. nto de fusão do produto preparado por siri tese,quando misturado com a nossa substância não sofreu alteração «
0 comportamento em placa dos dois alcalóides era também semelhante, assim como as cores que se regis_ taram quando as placas foram reveladas com os reagentes atrás citados, (Fig, 10, 11 e 12).
Tínhamos nesta altura já fortes razões para admitir que o alcalóide imolado era, na verdade, a te-traidr o armaria de 1=186,22. No entanto determinámos ainda o espectro de absorção quantitativo dos dois alcaiójl des operando sobre uma solução metanólica com a concen-tração de 5,36x1.0 gfo. A Fig. 3 mostra os espectros do alcalóide obtido por síntese e do isolado de Burkea a-fricana, registando-se no QUADRO VIII os valores de £"e log. c indicados na literatura para a tetraidroarmana e

263
Fig.10 - Cromatografia em placa de sílica no sistema clorofórmio—metanol (8:2) l-Tetraidroarmanó 2-Alcal6ide A
0,29 (J V J
Fig,11 - Cromatografia em placa de silica no sistema clorofór-mio-::ietanol(3:2)
l~Tetraidroarmana 2~Alcal6ide A
265
Pig. 12- Cromatografia em placa de sílica no sistema me tanol-amónia (97 s3).

266
e os determinados por nÓ3.
Q U A D R O VIII
Alca ló ide X max., nm
Á m i a , , nm l Log £
Alcaló ide A i so lado de Burkea a f r i cana
22526
28283 291
248 31.433
2,400 7.520 6.200
4,50 3,38 3,87 3,79
22526
28283 291
248 31.433
2,400 7.520 6.200
4,50 3,38 3,87 3,79
T et r a id ro arman? t 225
28283 29091
24650 32.136 2.000 7.122 6.080
4,50 3,30 3,85 3,78
s i n t é t i c a prepa :*ada por nós
t 225
28283 29091
24650 32.136 2.000 7.122 6.080
4,50 3,30 3,85 3,78
Tet r a id ro armaria 225,5 283
- 4,57 « 3 , 9 0 i s o l a d a de Pe ta
225,5 283
- 4,57 « 3 , 9 0
l o s t y l i s l a b i
225,5 283
- 4,57 « 3 , 9 0
cheoides (8)
225,5 283
- 4,57 « 3 , 9 0
225,5 283
- 4,57 « 3 , 9 0
Tetraidroarmana 225
280 4 , 5 1
3,85 . 5 so lada de Lep
t a c t i n a densi■■■■*
225
280 4 , 5 1
3,85 f l o r a (9)
225
280 4 , 5 1
3,85
225
280 4 , 5 1
3,85
Tetraidroarmana 225 291
32,100 5.610 s i n t é t i c a (5)
L 225 291
32,100 5.610
A ctîWittOSÇ^o dos espectros na região do I . V, do produto sintet izado e do alcalóide isolado não nos deixaram dúvidas quanto à sua identidade,porquanto são perfeitamente sobreponíveis, como mostra a Fig.14,iden

...,. 2 6 7
■íâ 3&
., 2$
sew
10
9"
r 1 ! 1 1 1 r 200 10 20 30 40 50 60 70
— T 1 ~] "t— 80 90 300 Arm
Fig. 13 1 Espectro de absorção no U.V. de tetraidroarmana,em
alcool. 2 Idem, do a l c a l ó i d e A i s o l a d o de Burkea a f r i cana
HOOK

7o) 3DNV111WSNVÍJ1

o Q>
Dû
-a CÛ o 'O V c n
u w F b n <u
8 O
> "O O
1-H u TJ O
0 a) i - *" a j
o a) E 0
"8 IO
o u o
E 0
"8 I f l a o 0
QJ o
"D ■o E
o
U >
8 | Q o V Qi IA
d)
O O I o c o u
— rM
%>) aDNVinwsNvyi

273
t i f icando-se também com o espectro n° . 21685,incluso na colecção SADTLER (lO). São ainda sobreponíveis pico por pico os espectros dp derivado iodometilado do nosso com posto e o da tetraidroarmana também preparado por nés (Fig. 15) . Podemos, pois afirmar que o alcalóide i so la do das fracções VI 0 ?Q alcalóide A, é, efectivamente,a tetraidroarmana. Al iás , este composto foi j á isolado em Leguminosas como vimos na PARTE I I .
3 .6 .5 .2 . - Estudo das fracções Vi 9.21 e Vg2_9fi
ITo res íduo Vnn A-j_predominava o alcalóide F e em V22-26 a substância A era a mais abundante,se bem que ex i s t i s se também apreciável quantidade de F que nos pareceu de in teresse aprovei tar .
Como o primeiro resíduo a que aludimos estava mais puro do que o segundo, tentámos a purificação dest e último, e, para i s so , fizemo-lo passar através de u-ma nova coluna de alumina (COLUNA V i l ) , depois de dissol vido em benzeno, procedendo à sua elução primeiramente com este solvente e depois com mistura de benzeno-cloro fórmio e clorofórmio-metanol. 03 eluatos VI IQ e V H ^ Q t i nham composição similar a qual , por sua vez, se assemelhava à das fracções Vig_2l Os resíduos resul tantes da eliminação dos solventes destas fracções foram adiciona dos ao resíduo Vig_2l e, para faci l idade de exposição

274
continuaremos a chamar ao resíduo desta segunda combina ção V1 9_2i.
Duas impurezas, E e G, acompanhavam o a lca ló i de F que projectámos estudar, mas a semelhança de polaridade das t r ê s substâncias criou-nos um impasse no i -solamento desta última, que só vencemos com cer ta d i f i culdade .
Foram vár ias as t en ta t ivas empreendidas na sua purificação s c r i s ta l ização fraccionada em é te r su l fú r i co e em acetona, passagem por nova coluna de alumina,s_u blimação no vazio, e t c . Dado o pouco êxito de que se re vestiram t a i s . t e n t a t i v a s não nos parece com interesse o re la to pormenorizado destes ensaios . A dada a l tu ra pare ceu-nos que à única solução ser ia a separação por croma tograf ia preparativa em camada fina de s í l i c a . A aplica, ção desta última técnica util izandocono desaavolsrenixvo. clorofórmio-metanol ( 8 J 2 ) permitiu-nos separar o produto F que depois de eluido para metanol e analisado " e s -pectrofotomètricamente na região de 200 a 500 nm nos le vou a concluir, sem sombra de dúvida, t r a t a r - s e de um aL calóideW -carbolínico t í p i c o . De facto, o traçado gráfico que obtivemos era idêntico à imagem que guardávamos na nossa rot ina de um outro que tínhamos isolado an te r i ormente, de um espécie diferente e que continha este nu
cleo ( i l ) . Aproveitámos ainda a oportunidade para da mes

275
ma placa retiramos E e G, A mancha G depois de eluida para o metanol,a-
presentou um espectro no U.V,scnclhante ao anterior, Isto levou-nos a supor que, dada a contiguidade das duas manchas duas manchas, não tivéssemos feito uma per feita separação.
0 produto E mostrou um espectro de absorção distinto dos dois produtos precedentes. Limitámo-nos a registá-lo (Fig. 30) sem nesta altura procurarmos inte_r pretá-lo.
Já que a cromatografia em placa' de sílica nos permitia uma razoável individualização dos três produ tos fomos tentada a proceder à sua separação em coluna de ácido silícico (COLUNA VIII) nos moldes que o QUADRO IX indica.
Separámos trinta e uma fracções, sendo cada uma delas obtida por elução da coluna com 25 ml dos sol_ ventes que o mesmo quadro indica. Os eluatos VlIIg --VIII5t, bem como VIII8a - VIII^, VIII9a - VIII9b tot a lizaram igualmente aquele volume de solvente, mas foram recolhidos em separado em consequência de nesse altura se destacarem da coluna anéis com colorações diferentes.

276
Q U A D R O IX
CROMATOGRAFIA EM COLUNA DE ACIDO SILIGICO DO RESÍDUO Vlfr-
- 2 1 C O L U N A V I I I
Número da
.fracção
Eluente Cor do eluato
Reacção de DRAGENDORPF (cor)
1 Éter de petróleo
Incolor -
2 E.pet, 5 Benz, 5
Incolor -
3 Benz» Incolor -
4 Benz. 20 E.sulf. 5
Incolor -
5a
5b
Benz. 20 E.sulf. 5
amarelada amarelada 5a
5b
Benz. 20 E.sulf. 5
alaranjada rosada
6 Benz. 15 E.sulf.10
amarelapai ida
amarelada
7 Benz. 15 E.sulf.10
arroxeada rosa intensa
8a
8TD
Benz. 10 E.sulf.15
amarelada pálida
amarelada 8a
8TD
Benz. 10 E.sulf.15
alaranjada ai arau* r/lis.
1 "1

9a
9b
Benz. 5 E.sulf .20
avermelhada! avermelhada 9a
9b
Benz. 5 E.sulf .20
incolor averemlhada
10 Benz. 5 E.sulf .20
incolor avermelhada
11 Benz. 5 E.sulf .20
incolor avermelhada
12 Benz. 5 E.sulf .20
incolor avermelhada
13 Benz, 5 E.sulf .20
incolor avermelhada
14 1 Bènz. 5 E.sulf .20
incolor avermelhada
15 E.sulf . incolor avermelhada
16 E.sulf . incolor avermelhada
1? E.sulf, incolor avermelhada
18 E.sulf . 20 Clorof. 5
incolor avermelhada
19 E.sulf . 20 Clorof. 5
incolor avermelhada
20 E.sulf . 20 Clorof. 5
incolor avermelhada

278
21 Clorof. incolor avermelhada
22 Clorof, incolor avermelhada
23 Clorof, incolor avermelhada
24 Clorof, incolor avermelhada
25 Clorof. 5 Ac.et i lo 5
amarelada avermelhada
26 Clorof. 5 Ac.et i lo 5
amarelada avermelhada
27 Clorof* 5 Ac.et i lo 5
amarelada avermelhada
28 Ac.eti lo amarelada avermelhada
29 Ac.et i lo amarelada avermelhada
30 A c e t i l o 5 Metanol 5
amarelada avermelhada
31 Metanol amarelada avermelhada
As fracções VlIIg a VIII30 foram cromatografia das em placa de sílica no sistema clorof <5rmiometanol( 8: s2) e as manchas detectadas por exposição das , placas aos raios U.V. e por pulverização com reagente de DPA-

279
GENDOEFF. A fracção VlIIga apresentava uma mancha junto
àa linha de solvente, com cor amarela azulada, que reagia com o reagente de DRAGE1ID0EFF,
A fracção VIII5¾ mostrava tr§3 manchas flúores centes muito próximas da linha do solvente,reapectivarnai te amarelada, azulada e acastanhada.
As fracçSes VHIg 9 VIIIQ^ apresentavam uma mancha de fluorescência azulada e coravam pelo reagente de DRAGEEDORFF.
A fracção ?IIIga apresentava quatro manchas fluorescentes? duas de Rf mais baixo, de cor amarelada rosada, a terceira, correspondente ao alcalóide F, com fluorescência azul violácea intensa, e uma quarta mancha com fluorescência azul e contígua à anterior.
As fracções VlIIg^ a VIII24 continham apenas o alcalóide F, de fluorescência aa>ul violácea,
A partir de VlIIgg, embora ainda predominasse als&lolde F, observavam-se várias manchas fluorescentes e por isso as respectivas fracções não foram aproveitadas.
3,6.5.2.1,- Estudo das fracções VIII 9 ..24
Os resíduos destas fracções apresentavam-se cristaMnoss, sendo os cristais de forma acicular. Fo-

280
ram no seu conjunto cristalizados ainda duas vezes em éter sulfúrico p.a., determinando-se algumas das características do produto cristalino isolado.
Assim, o alcalóide não reagiu com o reagente de EHRLICH, mesmo apÓ3 aquecimento a b*m, durante 5 minutos, e dissolvido em ácido acético e adicionado de u-ma gota de ácido sulfúrico concentrado contendo 2 o de nítrito originou cor rósea violácea,
A determinação do ponto de fusão em microscópio LEITZ tornou-se-nos um pouco difícil porque o produ to sublimava, mas pareceu-nos ter fundido completamente a 239°G, Esta mesma determinação em tubo capilar condu-ziu-nos ao valor de 236-237°C, observando-se forte ene grecimento da substância, próximo do início da temperatura de fusão (ô).
0 espectro do produto, dissolvido em metanol, na região do U,V, identifica-se com o da h ar mana inclui do na colecção de espectros do Laboratório Lilly (5),sen do de notar que os valores das extinções moleculares nos vários comprimentos de onda, (C=0,00432 8fo, M=182,22)de terminados para o composto em estudo são próximos,embora inferiores, aos que sa referem para aquele alcalóide.
0 espectro de absorção do nosso produto, deter, minado em solução metanólica de hidróxido de sódio K/20, não sofreu alteração digna de registo, pelo contrário,em solução metanólica de ácido clorídrico ïï/20 as bandas

281
de maior absorção sofreram um notável desvio no sentido dos maiores comprimentos de onda, com desaparecimento de algumas delas como ê característico dos alcalóides (* --carbonílicos típicos (Fig, 16).
Como possuíamos uma amostra de harmana que tínhamos preparado para ser utilizada como intermediário, na síntese de tetraidroarmaria a que já aludimos,foi-nos fácil garantir : a perfeita identidade do nosso composto com a harmana sintética.
A técnica utilizada na preparação deste composto é a descrita por KEUFER (7) e não é mais que uma condensação de MANICH em que o triptofano reage com o acetaldeído em meio sulfúrico. 0 ácido 5-carboxitetrai-droarmana intermediário é descarboxilado e desidrogena* do a harmana por aquecimento e oxidação com solução de dicromato de potássio.
Síntese da harmana
' Dissolveram-se 2 g de triptofano en l^nl do ácido sulfúrico normal e adicionou-se 5 ml de .acetaldeído. A solução foi aquecida sob refluxo a 40°,durante meio hora e, depois durante hora e meia,à ebulição.Após arrefecimento, alcalinizou-se pela amo'nia e deixouese em repouso durante a noite. 0 excesso de amo'nia foi eli minado por evaporação no vazio e adicionou-se ],300 ml de água fervente. Sobre esta solução também aquecida à

282
fervura verteram-se 150 ml de uma solução aquosa dei di-cromato de potássio a 10$ e 35 ml de ácido acético,man-tendo-se em ebulição durante dois minutos. Após arrefecimento, adicionou-se uma quantidade suficiente de hi-drosulfito de so'dio para reduzir o excesso de dicromato. A solução foi alcalinizada pelo carbonato de sódio e ej3 gotada pelo éter. A solução etérea, depois de bem lavada com água, apresentava cor amarelada. 0 resíduo da e-vaporação do éter, submetido a várias cristalizações,pri. meiramente em álcool e depois em éter, apresentava-se for mado por cristais incolores e aciculares.
Este produto, misturado com o alcalóide F,iso lado de_Burkga, não sofreu depressão no seu ponto de fusão.
0 espectro quantitativo do alcalóide sintetizado aproximou-se mais do determinado para o nosso produto do que o referido na obra atrás citada.
Julgamos que as diferenças registadas para os valores de è(QUADR0 X) entre as que nós determinámos e as referidas na literatura sejam devidas ao facto do nos so produto conter água de cristalização e termos feito os cálculos considerando o produto húmido e muito provavelmente o espectro incluido na colecção dos Laborató nos Lilly ter sido determinado com a harmana anidra.

283
Q U A D R O X
CARACTERÍSTICAS ESPECTRAIS DO ALCALOÏDE F E DA HARMA-
NA NA REGIÃO DO IJ.V. s
Alca ló ide A max., nm Âmín^nm r é Log £ F , i so l ado de Burkea , so lu
212
234
259
2 8 0 ( i n f . ) , 286
335
347
217
256
266-68
298-300
340-42
17,176 ' 16.400
34.500 10;500
7.592 4.429 9.070
15.608 1.687 4.640 4,218 4.640
4 ,24 4 ,21 4,54 4,02 3,88 3 , '65 3,96 4,19. 3,23 3,67 3,63 3,67
ção em metanol
212
234
259
2 8 0 ( i n f . ) , 286
335
347
217
256
266-68
298-300
340-42
17,176 ' 16.400
34.500 10;500
7.592 4.429 9.070
15.608 1.687 4.640 4,218 4.640
4 ,24 4 ,21 4,54 4,02 3,88 3 , '65 3,96 4,19. 3,23 3,67 3,63 3,67
F , i s o l a d o de Burkea , so lu
205
246
299
364-66
221
273
318-22
18.982 12.444 29.105
4.429 15.326
1.,265 4 ,851
4 ,28 4 ,09 4 ,46 3,65 4 ,19 3,10 3,69
ção em metano l acido
205
246
299
364-66
221
273
318-22
18.982 12.444 29.105
4.429 15.326
1.,265 4 ,851
4 ,28 4 ,09 4 ,46 3,65 4 ,19 3,10 3,69
F , i so l ado do Burkea , so lu
234
259
2 8 0 ( i n f . )
256
264-68
321.000 8.624 8,625 3,750 7.874
4 ,51 3,94 3,94 3,57 3,90
ção em metanol a l c a l i n o
234
259
2 8 0 ( i n f . )
256
264-68
321.000 8.624 8,625 3,750 7.874
4 ,51 3,94 3,94 3,57 3,90
-

284
F , i so l ado de Burkea em
286
335
347
300
342
14.248 375
4.125 3.562 4 .125
4,16 2,57 3,61 3,55 3,61
meio a l c a l i no
286
335
347
300
342
14.248 375
4.125 3.562 4 .125
4,16 2,57 3,61 3,55 3,61
Harmana se-^ gundo KEDFER (7)
234
289
339
351
237-18
266-67
303
345
4,25 4 ,56 3,64 4,16 2,79 3,59 3,57 3,59
Harmana se— 234 287 347
37.400 16.300 4,550
4 ,57 4 ,21 3,66
gundo c o l e c ção L i l l y ( 5 )
234 287 347
37.400 16.300 4,550
4 ,57 4 ,21 3,66

2ÕÕ7T 30 45" 60 7s "To" 305 20 l o & 65 380 m F i g . 16 1 - Espectro do alcalóide F,em metanol , o n » » F.em Bol.metanolica de NaCH N/^U | - -, „ .. F » " " "HC1 N/20

287
3 . 6 . 5 , 2 , 1 , 1 , - Comportamento cromatográfico do alcalóide F e da harmana
Os valores de Rf dos dois produtos,quando cro-matografados em placa de s í l i c a nos sistemas clorofórmio. -metanol(9,5sO,5),Rf=0,27; clorofórmio-metancl(9sl),Rf -=0,50$ Clorofórmio~metanol(822), Rf=0,77, são idênt icos, não se notando qualquer separação quando em prova cruzada como mostram as Figs . 17, 18 e 19.
A revelação das placas foi f e i t a por exposição destas à luz U,V., observando-se nos t r ê s cases uma única mancha de fluorescência azul violácea.A revelação com o reagente de M1RLICH, após aquecimento a 100 em estufa, durante 10 minutos, provocou -aparecimento de cor rooa violácea sendo no entanto inconstante este comportamento. Com o reagente de DRâGEKDORFF obtinha-se cor avermelhada

288
0 0 0,27
0
1 2 3
F i g . 17 Cromatografia em p laca 1 - Harmana 2 - Harmana e a l c a l ó i d e F 3 - Alca ló ide F .

289
Pig. 18 Cromatografia em placa 1 - H armaria 2 - Harmana e alcalóide F 3 - Alcalóide F
Fig. 19 Idem.

290
3.6,5,2.1, ,2,- Comportamento do alcalóide F e da harmana
na região do I,V,
Como a Fig , 20 mostra os espectros na região do I.V. do alcalóide F i so l ado do Burkea -er'da harmana são. idên t icos»
3,6.5o2«l,3„- Preparação de derivados do alcalóide F e d a
harmana
Cloridrato
0 produto dissolvido em éter foi adicionado de solução metanólica clorídrica a 1:1 e deixado na geleira durante alguns dias. Os cristais formados foram separados por centrifugação e recristalizados em metanol. Após secagem sublimavam, a cerca de 200°C. 0 espectro no I.V, do cloridrato de harmana também preparado segundo a técnica referida e o cloridrato do alcalóide F são perfeitamente scbreponíveis (Fig0 2l),
Iodometilato
50 mg de alcalóide F foram dissolvidos em 225 ml de etanol absoluto, adicionou-*se 0,30 ml de iodeto de metilo e abandonou-se a solução durante 5 dias à temperatura ambiente, Obtiveram-se cristais aciculares macroscópicos que depois de recristalizados duas vezes em

CO
E ai
O 10 a i
O O I o c o u
o a>
,.2 CÛ
u T> O
"O O
Q U .
a <u
■o
0)
o u
• ó
;%) 3DNV11IWSNV*U
O c o E
1x1 — <N
O
d)

E
O "D O
IO '5) <D
o c o
IO m o
o 0)
O CD CL
LU
O r -Ï 0 •O | o .c
O
o
"D O O
O
cu ■o 2 o
■v o
f - U U
— <N
íN
cn
;%) 3DNVJ1IWSNVM1

295
metanol e secos fundiam a 275-276 5 Esta era igualmente a temperatura de fusão do iodomtilato de harmana que tam bém preparámos»
Os espectros destes dois produtos são idênticos quer na região do U.V. quer na região do I .V. (Figs . 22 e 23) .
0 espectro no U.V. em solução metanólica de h l dróxido de sódio N/lO deixou antever a formaç~ao de uma anidrobase, porquanto a solução corou de amarelo intenso, notando-se um apreciável efeito bactocrómico (Fig.22) .
Benzalarmana (12, 13, 14)
102 mg de alcalóide F, adicionados de 2 ml de benzaldeído, foram aquecidos, durante 3 horas , a 150 C.
A solução foi evaporada à secura, e o resíduo dissolvido em 20 ml de é t e r ; com a adição de 10 ml de á-cido c lor ídr ico a 20$ obteve-se um precipitado amarelo que" foi recolhido e lavado várias vezes com é te r e re -cr is ta l izado de álcool a 95 . _
Suspendeu-se em água , a lcal inizou-se pela amd n i a e extraiu-se com clorofórmio. Este foi evaporado e o resíduo obtido cr i s ta l izado de acetona. 0 produto,quando cromatografado em camada fina apresentava, vár ias manchas flúores cent es qœ reagiam com DRAGENDOEFF, cas depois do puii ficado por cromatografia em placa fundia a 199-202°C.
0 espectro da substância no U.V. que se reproduz na Fig.24 é i d ê n t i c o ao da JLsnzalarmana que tam-preparámos pela t écn ica acima d e s c r i t a pa r t i ndo duma amostra de harmana.

297
200 20 h0 60 80 300 20 kO 60 30 ^ 0 0 ^ Fig..22 1 - Espectro do iodometilado do alcalóide F
" H II II II -ci F em solução
metanolica de NaOH N/10 2 -
OÙ V E
-S
LU —
3 O)
3
■a
0) ■g :§ o u
o 10 O) 0)
O c o
a o
fc 5 u 9 u c X \£
,8 -s F LU
i n
o 0
3 o •£ o
(U
E 8 >
O 1 û
rM
8 I
1%) 3DNVÍ1IMSNVM1

301
0,7-
0,6-
0,5-
0,4i
0,3-1
0,2-
-i , , — , — y - r ,.
200 20 40 60 80 300 20 40 60 8 0 4 0 0 / ( ^ Pig.24 1 - Espectro do derivado "benailidénico do alcalóide F,
em metanol. 2 - Idem, em metanol alcalino. 3 - Idem, em metanol ácido.

303
3.6.5.3,- Estudo das fracções v"28-56 e HÍ2]-35
Os resíduos V28-36 e VII21»35 (resíduo da elu-ção da COLMA VII com solventes de polaridade crescente desde benzeno-clorofórmio (4:6) até metanol) aps-SeenD ;--vam composição aproximada quando cromatografados em placa de sílica e eram particularmente ricos nos alcalóides A e B, embora também contivessem, além de outros, um po_u co de alcalóide F.
Destas fracções projectámos isolar o alcalóide B, que mostrava, na placa, intensa fluorescência verde e Rf ligeiramente superior ao alcalóide A-tetraidroarma-na.
As tentativas levadas a efeito para o seu isolamento em coluna de alumina ou de sílica foram improfícuas, parecendo-nos até que,a cada tentativa e por serem demoradas as eluções das colunas, o"btinhamos fracções de aspecto cada vez mais escurecido. Atribuímos tal facto , em princípio, a uma alteração do alcalóide A, mas, como veremos mais tarde, julgamos que tal fenómeno não era e-fectivamente, devido à alteração da tetraidroarmana ou alcalóide A,mas, sim à de um outro alcalóide,quo só mais' tarde identificámos, a triptamina.
Como não encontrámos processo mais rendoso para isolar o alcalóide B, decidimo-nos pela sua separação por cromatografia preparativa em camada delgada de síli-

304
ca no sistema clorofórmio-metanol (3Í2).
0 espectro de absorção no U.V. deste produto em solução no metanol mostrou as bandas de absorção loca lizadas nos comprimentos de onda seguintes s
/\max, 209, 240 (inflexão), 320, 350 (inflexão) e um mínimo em 272-276 nm.(Fig. 25).
Em meio ácido (ácido clorídrico N/20 em solução metanólica) observou-se um deslocamento bactocrómico das bandas com um mais perfeito delineamento da banda em 240 nm. Assim, observaram-se no espectro os seguintes má ximos s 209, 245 e 350 nm e mínimos em 238-40 e 290-300 nm. (?ig, 25),
Em solução alcalina (líaOH N/20 em metanol) a substância apresentava os máximos de absorção localizados em, 209, 240 (inflexão) e 315 e um mínimo em 272 nm.(Fig. 25),
A fluorescência verde do produto bem como o e_s pectro de absorção no U.V. indicavam tratar-se de um derivado diidrogenado-fb-carbonílico. De facto estes compostos possuem, em regra, fluorescência verde como acontece com os dois compostos diidrogenados naturais har-malol e harmalina e possuem dois máximos de absorção típicos como se pode ver na Pig, 32 e 31 bem como noutros compostos de síntese com este núcleo e que a ' literatura (15) refere.

305
-, , , ! ,. 200 20 h0 60 80 300 20 kO 60 80 / Fig» 25 1 - Espectro do alcalóide B, em metanol 2 - Idem, em solução metanólica de ÏÏC1 N/20 3 - Idem, em solução metanólica de NaOH N/20

307
Demais, o comportamento anteriormente verifica do de o alcalóide A. se decompor por sublimação em alcalóide B e F, acrescido do facto, que neste momento re-registámos, de ao tentar purificar o composto B em placa de sílica, este se decompor, originando um outro produto de fluorescência azul violácea, que verificámos corresponder à harmana, ao mesmo tempo que escurecia, são ele mentos que, a nosso ver, apoiam a hipótese acima referida.
Estes factos levaram-nos a admitir que o cpm-_ posto B seria um alcalóide intermediário entre A e F,que se formaria por oxidação de A e que, sofrendo uma oxidação subsequente, originaria F, Ora,.A_é um. composto - 0 --carbonílico tetraidrogenado no núcleo C e;F tem o mesmo núcleo completamente desidrogenado, daí admitirmos que,a tratar-se de um composto deste tipo mas diidrcgenado, B deveria corresponder à hamalana. Não temos conhecimento de que a harmalana tenha sido isolada de fonte natural $§n tretanto ela é um produto considerado na linha biogenéti ca dos derivados Q>-carboíínicos e, por isso, não é" de admirar nem nos cueton a admitir que tal substância estivesse presente em Burkea africana, da qual, aliás. . j-á isoláramos a tetraidroarmana e a h armaria. Na altura em que obtivemos o composto B desconhecíamos a publicação do espectro de absorção da harmalana e, por isso,para poder mos compará-lo com o do nosso produto B vimo-nos na necessidade de sintetizar aquela base.

308
Síntese da harmalana (16)
Partimos de 0,01 g de -frriptamina, ALDRICH a que adicionámos 1 ml de anidrido acético e deixámos em con tacto durante a noite, à temperatura ambiente. A acetil-triptamina resultante desta reacção foi extraída pelo é-ter sulfúrico, depois da eliminação do excesso do anidri do acético e alcalinização da solução aquosa com soda a &fo, 0 produto resultante da eliminação do éter foi cristalizado de éter de petro'leo e a partir dele preparámos a harmalana. A acetiltriptamina foi dissolvida em 10 ml de xilol e esta solução foi mantida em ebulição suave du rante três quartos de hora, adicionando-se-lhe 2 g de pai tóxido de fósforo em pequenas porções. Prolongámos o a-quecimento até perfazer uma hora. Filtrámos e eliminámos o xilol por evaporação. 0 resíduo foi retomado com éter, adicionado de água e ácido clorídrico diluído e aquecido até à obtenção de uma solução límpida? separámos então a solução ácida, a qual foi extraída várias vezes, com é-ter, para eliminar impurezas, e em seguida foi alcalini-zada com hidróxido de sódio a 3^ e extraída várias vezes com éter sulfúrico. 0 resíduo da eliminação do éter foi cristalizado de álcool metílico. Obtivemos harmalana com ponto de fusão 179-180°. Não conseguimos obter cristais brancos comotSPATH e LEDERER (l2), referem, nem atingimos o ponto de fusão de 182°C que MOSKE, PERKEN e ROBINSON©

309
encontraram para este composto. 0 ponto de fusão do alcalóide sintetizado mis
turado com o alcalóide B isolado de Burkea manteve-se em 179-180°C.
Em placa de sílica nos sistemas clorofórmio-me tanol (9,30,5); clorofórmio-metanol (8s2); clorofórmio--metanol (3s2) e em metanol, os dois produtos apresenta. vam igual migração (Fig. 26, 27 e 28).
0 espectro de absorção no U.V.desta:substèônia era similar ao atrás referido para o produto isolado de Burkea, em solução metanólica ácida, neutra e alcalina (Fig. 29).
Entretanto os espectros no I.V. das duas substâncias não eram perfeitamente sobreponíveis mas julgamos que o facto se deva à fácil alteração da substância, a cuja labilidade já nos referimos,pelo que não o reproduzimos no nosso trabalho por temermos que nenhum deles corresponda ao produto puro.

310
Fig . 26 Cromatografia em placa 1 - Harmalana de síntese 2 - Alcalo'ide B

311
F i g . 27 F i g . 28 Cromatografia em p laca Idem. 1 - Harmalana de s í n t e s e 2 - Alcalo'ide B

313 A
0,9-
0,8-
0,7H
0,6-
0,5-
0*4*
0,2-
0,1-
1 1 1 1 1 1 r 1 [ ~ 200 20 40 60 80 300 20 40 60 80 / am Fig. 29 1 - Espectro da harmalana sintética, em metanol. 2 - Idem, em solução metanólica de HC1 N/20. 3 - Idem, em solução metanólica de NaOH N/20.

315
3,605.4.- Isolamento do alcalóide E das fracções V22-26
Quando tratámos do estudo das fracções V]_9_2ie ^22-26 "ti'7'611103 oportunidade de referir que nos foi posai, vel isolar, por cromatografia em placa de sílica, no sis_ tema clorofo'rmio-metanol (8:2), um alcalo'ide a que chama mos alcalóide.E.Este produto, que apresentava Rf ligeira mente inferior ao da harmana embora muito próximo, apresentava cor amarela acastanhada na placa e não era fluorescente à luz U«V,
0 espectro de absorção deste alcalóide eluido pelo metanol apresentava o aspecto que a Fig. 30 indica, sendo de assinalar bandas com /\ máx em 213, 252 e 350 nm e Amfn em 248 e 288 nm. Comparando este espectro com os dos três alcalóides já isolados, tetraidroarmana, harmalana e harmana, ele apresenta mais semelhanças com o espectro do segundo destes compostos, sendo o seu pon to de fusão superior ao de qualquer dos três alcalóides citados. Na realidade, o produto sublima em pequenas agu lhas a 217-218°, quando aquecido em microscópio de'LEITZ, parecendo estar completamente fundido a 243°, Além disso, a banda de absorção de maior comprimento de onda é mais intensa do que na harmalana,e localiza-se num comprimento de onda superior ao desta, o que nos fez suspeitar da presença de um auxócromo na molécula. 0 espectro apro xima-se, de facto, bastante dos dois derivados -carbolí

316
nico diidrogenados naturais -harmalina e harmalol (Fig. 31 e 32). E" flagrante a semelhança com a harmalina, que apresenta em metanol bandas localizadas em 218(log. £ = = 4,27) 260 (iog£= 3,90), 346 (log. fc- 4,08), 276 (log. £ » 4,02)(inflexão) (5), A literatura referecomo ponto de fusão da harmalina 229-?31°C (5) e ainda 242-244°C (18). Apesar das semelhanças observadas entre o alcalóide E e a harmalina, as duas substâncias não se identificam nem pela fluorescência (pois que esta apresenta intensa fluo rescência verde e o nosso produto apresenta apenas cor amarela acastanhada), nem migram em placa de sílica para o mesmo nível. Entretanto, o facto de o alcalóide ter um ponto de fusão superior ao da harmalana e apresentar a banda nos maiores comprimentos de onda mais intensa (com parar os espectros nss. 25 e 30) fez-nos suspeitar da e-xistenoia de oxigénio na molécula.
Sabemos da importância dos N-óxidos na biogéne se dos alcalóides indólicos e, como nesta planta nos foi possível encontrar três alcalóides tetraidroarmana, har-•malana e harmana que se encontram na linha biogenéticapro posta por PERKEÏÏ (l3) e SLârTOR ( 14) f suspeitámos que o aL calóide em causa pudesse corresponder a um N^-óxido de qualquer de um dos três compostos já isolados. Estão des eritos a harmanina ou harmana-..'h-oxido (19) e aindan te^ traidroarmana--Nb-óxido (20). As características determinadas para o nosso produto não se condunam, porém,com as

517
A It5-
M-1,3
1,2
Ml
1-
o,H 0,8-,
0,7H
0,6
0,5-
0,4H
0,3
0,2-
0,1-
200 40 -i r 80 300 ' lb" r "8Õ~T; \nm
Fig, 30 1 - Espectro do alcaloïde "E en metanol. 2 - Idem, do um produto de oxidação &a totraidroarmara..
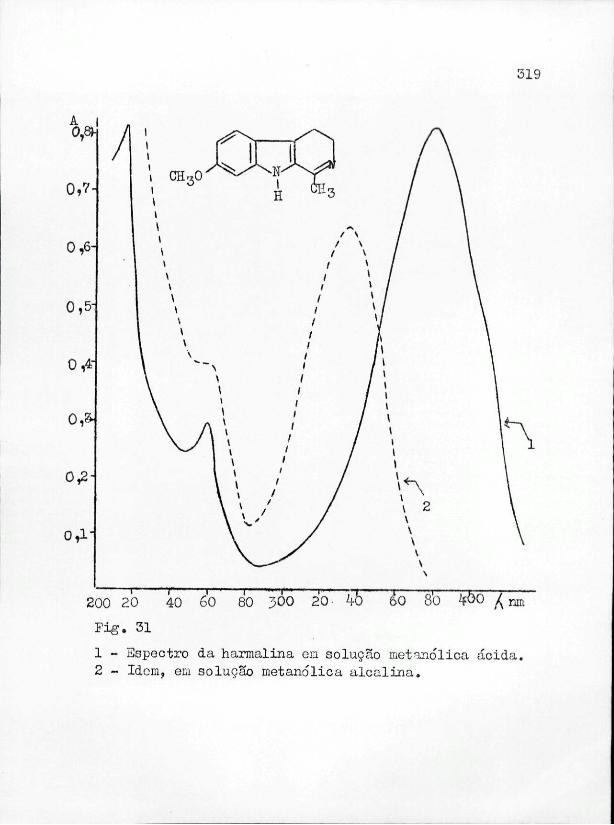
319
A I 0,8)-
0,7-
0,6-
0,5"
0,*
0,¾
0,2-
0,1
200 20 40 30 80 300 20' kb 25 80 IfÔO Fig. 31
run
1 - Espectro da harmalina em solução met ano'li ca ácida. 2 - Idem, em solução metanólica a l ca l ina .

321
0,5
0,3-
0,2-
0,1-
^
I H
Q Au
f \
\ i
\ f *-*\
200 -, , ( 1 1 1 r— ~~\
40 80 300 40. 80 I am.
F i g . 32 1 - Espectro de absorção do harmalol ,em solução me tand l i -
ca á c i d a . 2 - Idem, em solução metandl ica a l c a l i n a .

323
referidas para estes dois alcalóides. Tentámos a prepara ção dos N^-óxidos de cada um dos alcalóides já isolados por oxidação com peróxido de hidrogénio naa concentrações de bio e 30$ quer em meio acético ou neutro operando à tem peratura ambiente durante 24 horas ou por aquecimento a 70° durante 2 horas.
Só com a tetraidroarmana conseguimos a obtenção de um produto com Rf igual ao do nosso alcalóide E. Uma das técnicas usada foi a seguinte:
100 mg de tetraidroarmana foram dissolvidos em 2 ml de metanol e adicionado de 20 ml de água oxigenada a 30$ sendo a solução aquecida a banho maria durante duas horas a 70°. Após este tempo de aquecimento a solução foi alcalinizada com amónia e extraída com clorofórmio.
0 resíduo da evaporação mostrou em placa de s_í liça quando desenvolvida no sistema clorofórmio-metanol (8:2) quatro manchas DRAGENDORPP positivas (Pi. 33).
A de menos: migração mostrava Rf idêntico -, *, tetraarmana(A)pn+ ra imediatamente a seguir apresentava fluo.
rescência verde e migração semelhante à da harmalana,(B) outra ainda de cor amarela acastanhada (E), de Rf inferior mas contígua à mancha predominante com fluorescência azul violácea e de Rf idêntico ao da h armaria (p). Verifi cámos, portanto, que a oxidação da tetraidroarmana com peróxido de hidrogénio conduziu à obtenção de quatro alcalóides. 0 facto não surpreendo se noa lembrarmos

324
de que a estabilidade máxima se atinge com a total aroma tização da molécula e, sendo assim, os derivados tetrai-drogenados e diidrogenadosC>-carbolínicos terão tendência a adquirir o estado de máxima estabilidade por desidrogenação, perdendo, respectivamente, quatro e dois hi-drogénios. Entretanto, como o núcleo Ç_ dos alcalóides do grupo da (^-carbolina pode ser um núcleo piridínico, te-traidropiridínicojo.u-ãiidropiridínico em que, se encontra presente o heteroátomo (¾) que contém um par de elec troes não compartilhado que, não intervindo na aromati-zação do núcleo, torna consequentemente possível,como já vimos anteriormente ( PARTE II), que se formam derivados por protonização, por combinação com ácidos de LEWIS e por oxidação, originando neste último caso ííb-óxidos por tratamento com peróxidos.
A substância correspondente à mancha amarela a castanhada (ïï) contígua à mancha de fluorescência azul violácea por cromatografia preparativa em gel de sílica, utilizando como desenvolvente o sistema cloroformio-meta nol (8Î2) e eluida com metanol, mostrou um espectro no TI, V. idêntico ao de alcalóide E isolado de Burkea africana (Pig. 30).
Também verificamos que sublimava a temperatura idêntica à que registamos para o nosso produto. Dada a instabilidade deste alcalóide e o pouco rendimento obtido, não nos foi possível o seu estudo na região do I«VJf

325
o o, 6 6 D !
O *0 o
1 2
Fig.33 - 'Cromatografia en placa de s í l i ca , 1 - Tetraidroarnanas 2 - Produto dè oxidação da tetráidroannana,

3 26
nem a realização de qualquer outro ensaio além dos- rcfo^ r idos .
Inclinámo-nos, embora com reserva, para que o alcalóide E e o produto de oxidação obtido da t e t r a i d r o -armana correspondam à harmalana-W-h-óxido,

327
3.6,6t- Isolamento de triptamina
Quando procedemos ao isolamento do alcalóide B, como anteriormente relatámos, apercebemo-nos da existência de um outro alcalóide que, em placa de sílica e nos sistemas clorofórmio-metanol (9:l), e (8s2) mostrava Rf semelhante ao do alcalóide A,
Entretanto, cromatografando o resíduo Vg„ ,,- e VII0, „_ em clorofórmio-metano1 (3s2) e em metano1-amónia (97s3), esta mancha separava-se em duas, A de Rf mais e-levado correspondia ao alcalóide A já identificado como sendo a tetraidroarmana, ao passo que a de migração inferior reagia mal com o reagente de DRAGEKDORFF,mostrando apenas cor alaranjada. A separação por cromatografia preparativa deste produto em clorofórmio-metanol(3s2)e a sua análise espectral após elução com metanol indicaram--nos tratar-se de um derivado tipicamente indólico, próxi mo do 3-metilindol. Não nos foi possível, nessa altura, identificar este composto mas ficamos7no entanto, com a ideia de que se tratava de uma substância facilmente alterável, adquirindo cor cada vez mais intensamente verme lha acastanhada a cada tentativa de purificação. DecidjL mos por isso,fazer uma nova extracção dos alcalóides totais da planta para o conseguir o menos alterado possível. Como nos pareceu existir em quantidade relativamente a-bundante partimos, j?ara o -seu isolamento de 200 g do pó

328
da planta desengordurado e submetemo-lo a maceração oom metanol acético a 5$, fazendo três extracções com 600 ml de cada vez. Os filtrados destes três macerados foramreu nidos e por evaporação em vazio, concentrados até 200 ml. Adicionámos 250 ml de ácido sulfúrico N/2 e extraímos a suspensão com 500 ml de éter de petróleo (duas extracções com 250 ml de cada vez). 0 resíduo da evaporação do éter de petróleo pesava 0,6383 g e não continha alcalóidas. 0 líquido aquoso foi depois extraído com 500 ml de clorofór mio e o resíduo da eliminação deste solvente pesava 0,6294 g. 0 pll da solução foi acertado para 6,5 com amónia, e _ex traiu-se com 500 ml de clorofórmio (lOO ml de cada vez). 0 resíduo pesava 0,1671 e corava fortemente com o reagei te de DRAGENDORFF.
Adicionámos à solução aquosa amónia até pH = = 10 e esgotámo-lw novamente com 500 ml de clorofórmio „ 0 peso do resíduo, após evaporação do solvente, era de 0,1886 g. Este resíduo reagia, fortemente, tal como o an terior, com o reagente de DRAGENDOHFF e a sua análise por cromatografia em placa de sílica no sistema metanol-amó nia (97:3), mostrou-nos conter as manchas correspondentes a Aj, A, B e F (esta em pequena quantidade), 0 resíduo foi dissolvido na menor porção de clorofórmio p.a. (cerca de 5 ml) e adicionado de 1 gota de metanol-ácido clorídrico (lsl),observando-se que o líquido tinha cor vermelha acastanhada e à superfície deste se separava um pro duto cristalino amarelo esbranquiçado. Com o auxílio de

329
pipeta Pasteur retirámos o líquido e os cristais sobrena dantes, foram lavados várias vezes com clorofórmio e depois com éter sulfúrico. Obtivemos 0,102o g deste produto após secagem no vazio
Estes cristais apresentavam-se em escamas bri lhantes de cor amarelada, e, observados ao microscópio tinham aspecto de placas transparentes e o seu ponto de fusãoxera de 247-48°C.(6).
A substância, quando cromabografada em placa de sílica nos sistemas clorofórmio-metanol (8:2)(Fig.34), clorofórmio-metanol (3:2)(Fig. 35) e metanol-amónia(97:3) (Fig. 36), migrava para os mesmos níveis que uma amostra padrão de cloreto de triptamina Aldrich , reagindo por pulverização das placas com os reagentes dimetilaminoben zaldeído, reagente fosfovanilico e ninidrina, originando coloração de tons violáceos azulados, e reagindo mal com o reagente de DRAGEMDOHFF.
0 espectro no U.V. do nosso produto e o da a-mostra padrão são muito semelhantes (Fig. 37),localizan-do-se as bandas nos seguintes comprimentos de ondas(2l)
Clnridrato de triptamina padrão.em metanol
/vmax 221 (£ = 33.127) 274 (£ = 6o 147) 281 (£ = 6.318) 290 (i = 6.489)

330
Fig. 34 Cromatografia en placa. 1 - Cloreto de triptamina Aldrich» 2 - Cloreto do alcalóide A »
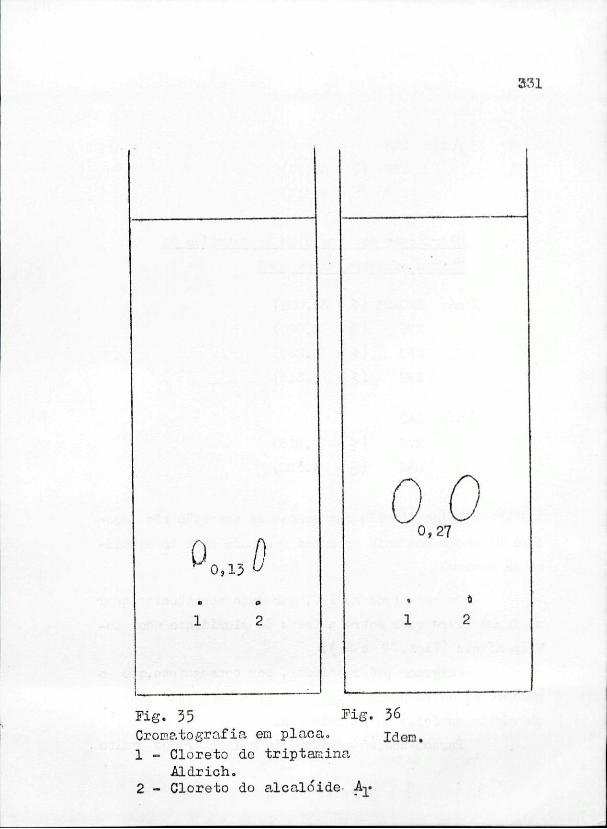
331
Fig. 35 F i s - 36 Cromatografia em placa» Idem. 1 - Cloreto de t r i p t amina
Aldr ich . 2 - Cloreto do a lca ló ide- A-j.

332
/\ min 242 276 (£ = 5.977) 287 (f = 4,610)
Cloridrato do alcalóide Aj extraído de Burkea africana,em metanol
/\ max
Xm£n 242 276 286
A localização das bandas de absorção não :<s-çp-freu alteração nos dois produtos operando em meio alcalino ou neutro.
Os espectros no I»V. das duas substâncias,quer na forma livre quer sobre a forma de cloridrato são so-breponíveis (Figs ,39 e 38- ),
Julgamos poder afirmar, por conseguinte,que o produto Aj corresponde à triptamina, a qual foi isolada em várias espécies de Leguminosas,
Parece--nos,no entanto, de salientar que muito
220-21 (i = 32.000) 275 ("?.= 6.020) 280 (£= 6.230) 289 (£= 6.318)
(<£ = 5.815) (£.= 4.520)

333
embora já canham sido identificados em várias espdbiea triptamina, tetraidroarmana e harmana julgámos ser a pri meira vez que se consegue isolar de uma mesma espécie 08« constituintes que de uma maneira tão completa confirmam a linha biogenética concebida teoricamente por PERKIN e SLAYTOR.
Por outro lado a ocorrência de triptamina om quantidade significativamente mais elevada do que a dos restantes alcalóides justificará, em nossa opinião a acção hipertensora que CORREIA M SILVA e Col. (22) registaram para o infuso de Eurkea africana,

535
»o 3©-X
25-
20-
15
10
i H
If.HCl i \
5 H
200 10 20 30 ^ 40 5() 25 95 §5 90~ÕÕ0/Cnm
F i g . 37 1 Espectro do c l o r e t o de t r i p t a m i n a Aldrich,em meta _, _ . „ „ do a l c a l ó i d e A, i so lado de 2 - Espectro '■ " Bur leá a i r r i c a n a HOOK, em metanol .

CÛ
E CD
O
O c o
o <u
8.
o o I o c o u
o
ca ai
■o o
"D a o
o c < E o
<D E o V n ■O
u u n
l !) T ) o
u 0 o 0) 0)
o o U U
— (N
(7o) 3DNV11IHSNVM1
00 m
ix.

V
CÛ
E
O ■o O '2 O) 0) o c o
10
o 4)
D CO
<D
"O o I o
a < c CU
E 13 o -O a. u
u h- <
o 0)
"O
o •; ±>
Ï í I LU — <N
ON IT)
O)
3/o) 3DNV11IWSNVMÍ

341
3 .6 .7 . - Extracção dos alcalóides do resíduo RB a d i fe
rentes vã
A elução da COLUNA. VIII deixou-nos antever a e. x is tência de outros alcalóides além dos j á a t rás assinalados. Na real idade, as fracções VIII5 a a VIII9¾ mostravam, quando cromatografadas em placa de s í l i c a e desenrol
vidas nos sistemas clorofórmio e clorofórmio-metanol(9,f>; : 0 , 5 ) , manchas que reagiam com o revelador de DRAGENDORFF, com formação de cores alaranjada e avermelhada. Dada a exígua quantidade dessas fracções não nos foi possível nessa a l t u r a fazer o estudo dos alcalóides que as const i tu íam.
Partindo de 2,48 g de extracto alcaloídico bru t o , % , ohtido nos moldes que se indicaram nas páginas 239 a 242 procurámos ex t ra i r estes alcalóides a pH - 6,5. Para i s so , dissolvemos o extracto em 250 ml de ácido cio r íd r ico a 3$ e agitámos a solução com clorofórmio.0 resí, duo obtido das evaporações deste solvente, que e ra , a l i á s , muito ins igni f icauie , não continha a lca ló ides ,
Ao<3ï*ÊBKÎS- o pH da solução aquosa com amónia' para 6,5 e extraímos os alcalóides com 500 ml de clorofórmio, uti l izando 100 ml de cada vez. 0 resíduo da evapora ção do clorofórmio, % x , depois de seco pesava,0,25 g. A solução aquosa foi de novo esgotada com clorofórmio depois de termos ajustado com amónia o pH a 10. 0 resíduo,

542
Rgg» obtido pesava cerca de 2,22 g. Uma nova extracção a que tivemos de proceder
para conseguirmos produtos em quantidade necessária para os ensaios que empreendemos conduziu aos seguintes re sultadoss De 2,30 g de resíduo,%,extraímos a pH = 6,5 0,24 g de R ^ .
3 , 6 . 7 . 1 . - Estudo dos alcalóides do resíduo RRT
Submetendo os resíduos Rg]_ e R-g à anál i se cro matográfica em placa de s í l i c a em vários s is temas, já c i tados anteoriormente(clorofórmio, clorofórmio-metanol (9 ,6 :0 ,4 ) , clorofórmio-metanol(9,5:0,5),clorofórmio-met a n o l ^ ^ ) , clorofórmio-metanol(3:2) e metanol-amónia (9,7:0,3) verificámos que o resíduo %2 continha os a l calóides j á c i tados , Al, A, B e F e por isso não procedemos ao seu estudo. Pelo contrário ,o resíduo % i most rava , além do alcalóide F, j á identificado como sendo a harmana , <• outros de Rf superior a este alcalóide .Ten támos fazer o estudo dos alcalóides deste resíduo.Começámos por submetê-lo a um fraccionamento em coluna de ácido s i l í c i c o . Partimos de 0,25 g de Rg-, e de 8 g de adsorvente. As eluções foram fe i tas com solventes de po]a
ridade® crescentes uti l izando porções de 25 ml de sojL vente em cada uma delas , conforme se indica no QUADRO XI*

343
Q U A D R O XI
CROMATOGRAFIA EM COLUNA DE AGIDO SILlGICO DO RESÍDUO BR-,
C O L U N A IX
Número da fracção
Eluente C o r d o eluáto Reacção de PRkGENDORFF (cor)
• I Éter de petróleo
incolor -
2 E, pet. 9 Benz. 1
incolor -
3 E. pet. 8 Benz. 2
incolor -
4 E. pet. 8 Benz. 2
incolor -
5 E. pet. 8 Benz, 2
incolor -
6 E. pet. 8 Benz. 2
inçolor -
7 E. pet. 7 Benz. 3
incolor -
8 E. pet. 6 Benz„ 4
&aarela rosada avermelhada
9 E. pet. 6 Benz. 4
amarelada avermelhada
10 E. pet. 5 Benz. 5
amarelada alaranjada 1 1 i

344
11 E . . ; )Ví , ,5 Benz. 5
amarelada a l a r an jada
12 E. p e t . 4 Benz. 6
amarelada a l a r an jada
13 E. p e t , 3 Benz. 7
amarelada a l a r a n j a d a
14 E. p e t . 2 Benz. 8
arcarelada avermelhada
15 Boaa. . amarelada avermelhada
16 Benz. 9,5 Glorof.0,5
amarelada avermelhada
17 Benz. 9 Clorofi 1
amarelada avermelhada
18 Benz. 8 Cloro& 2
amarelada avermelhada
19 Benz. 7 Clorcf. »
amarelada avermelhada
20 Benz. 7 Clorof, 3
amarelada avermelhada
21 Benz. 6 Clorofi 4
amarelada avermelhada
22 Benz. 5 Clorof. 5
amarelada n
23 Benz. 5 Clorcfi 5
amarelada

345
24 Benz. 5 Clorof, 5
amarelada ,-
25 1 Benz. 5 Gloro£ 5
i
amarelada .-'
26 Benz. 3 Clorofi 7
amarela--acastanhada
aver me liada
27 Jenz. 2 Glorof 8
amarela--acastanhada avermelhada
28 Benz. .1 Clorof -9
amarela--acastanhada
avermelhada
29 Clorof. amarela--acastanhada
avermelhada
30 Clorof. amarela--acastarihada
avermelhada
31 Clorof, amarela--acastanhada
avermelhada
32 Clorof. 9 Metanol 1
amarela--acastanhada
avermelhada
33 Clorof. 9 Metanol 1
amarela--acastanhada
avermelhada
34 Clorof. 8 Metanol 2
amarela--acastanhada
avermelhada
35 Metanol amarela--acastanhada

34<5
As fracções IXg a IX34 foram cromatografadas em placa de sílica no sistema clorofórmio. A partir da fraç ção IXx9 tivemos que utilizar no desenvolvimento das pia cas um solvente de maior polaridade, clorofórmio-metanol (9,5:0,5) porquanto no anterior sistema as fracções que
reagiam com o reagente de BRA.GEKDORFF praticamente não se deslocavam do local onde eram depositadas.
Nas fracções IX8 e IXg predominava o alcalóide L; nas fracções IX 1 0_ 1 2 o alcalóide J5 em IXT*II5 ' ° .# calóide I; e em IX 1 6. 1 8 o alcalóide H.
Nas fracções IXgQ_2i predominava o alcalóide H mas observam-se ainda outras manchas de maior Rf,que tam bem reagiam com o reagente de DRAGENDORFF,e as ^fracções 1X22-25 não continham alcalóides.
As fracções 1X26-28 continham o alcalóide F(har-mana) e um outro, G, de fluorescência azul contíguo a F. As fracções IX29-52 mostravam três manchas que nos pareceram corresponder aos alcalóides E, F e B já estudados, A fracção IX33 apresentava várias manchas parecendo de-tectar-se os alcalóides A, B, D, E, è' F, '
3.6.7.1.1,- Isolamento do alcalóide L
A fracção IXQ (0,0080 g)foi recromatografado em coluna de ácido silícico (2 g) e eluida com fracções de 10 ml de eluente, segundo o QUADRO XII.

347
Q TI A D E 0 H I
CROMATOGRAFIA EM COLUNA DE ACIDO SILlCICO DO RESÍDUO IX f l
C O L U N A X
Número da
fracção
Eluen-t e
Cor de e lua ; to j
Re acção de DEAGENDOHFF(cor)
1 E. p e t . i nco lo r -
2 E. p e t . 9 Benz. 1
i n c o l o r -
3 E. p e t , 8 Benz. 2
i n c o l o r -
4 E. p e t , 7 Benz. 3
i nco lo r -
5 EP p e t . 6 Benz, 4
i n c o l o r -
6 E. p e t . 5 Benz, 5
amarelada avermelhada
7 E, p e t . 5 Benza 5
rosada avermelhada
8 E, pet , 5 Sanz . 5
rosada avermelhada
9 E. p e t . 4 Benz. 6
rosada avermelhada
10 E. p e t . 3 Benz. 7
amarelada a l a ran jada

34-3
11 E, pet. 3 Benz. 7
amarelada alaranjada
12 E. pet. 2 Benz. 8
amarelada alaranjada
13 Benz. amareiada -
14 Benz. 8 Clorof. 2
amarelada -
^s fracções Xg_io foram reunidas e o resíduo da evaporação espontânea dos respectivos eluentes apresen-tava-se cristalino e com cor rosa alaranjada. Cromatogra fado em placa de sílica ,,pareceu.-mos estar puro, observar;. do~se uma mancha à luz U.V., de cor rosa alaranjada, com Rf = 0,93 e Rf = 0,54 respectivamente nos sistemas cloro fórmio-mecanol(48s2) e clorofórmio (Figs. 40 e 4l).
3,6^7*1,2,,- Isolamento do alcalóide J
Isolámos 6 alcalóifte^T -das fracções IX-|_Q_I2 CLO se guinte modos o resídvxo foi dissolvido em clorofórmio e a plicado em traço contínuo em placas de sílica,sendo estas desenvolvidas no sistema clorofórmio-metanol(48sl)„ Destacámos uma zona amarela que reagia com o reagente de DRAGEEDORFF com cor alaranjada, A sílica contendo o alcalóide J foi lançada numa coluna de ácido silíoico (5 g)e

34V
eluida com éter de petróleo, depois cora éter de petróleo. -"benzeno(5s5) e finalmente "benzeno. As fracções da elu-ção com o "benzeno apresentavam cor amarela e oontirl.am exclusivamente o alcalóide Jt como comprovámos por croma tografia em placa de sílica. Os Rf deste alcalóide nos sistemas citado3 são 0,72 e 0,35 (Figs. 40 e 41).
3,6.7.1,3,- Isolamento do alcalóide I
0 alcalóide I foi isolado das fracções IX14_15
do mesmo modo que se indicou para o alcalóide J. 0 alcalóide I apresentava fluorescência azul e
cristalizou espontaneamente em agulhas de tamanho reduzj. do. Em placa de sílica nos sistemas clorofórmio--me"cand (48s2) o clorofórmio migrou para uma altura com Rf respec. tivamente 0,83 e 0,29 (Figs. 40 e 4l).
3.6.7,1.4.- Isolamento do alcal_óide_H
As fracções IX16-I8 (0,0095 g) apresentavam cor amarelada e ligeira fluorescência azulada «Cor.-» nmtra.-ajL várias outras manchas fluorescentes foram de novo croma» tografadas em coluna de ácido silícico (5 g) e eluidas com fracções de 15 ml de solvente, segunde se indica no QUADRO XIII.

350
Q U A D R O X I I I
CROMATOGRAFIA EM COLIMA DE AGIDO SILlCICO DO RESÍDUO I X 1 6 - 1 8
C O L U N A XI
Número da
fracção
Eluente Cor do eluato Reacção de DRAGENDORPF (cor
1 E. pet, incolor -
2 E, pet. 9 Benz, 1
incolor -
3 E, pet. 8 Benz. 2
incolor -
4 E. pet. 7 Benz„ 3
incolor -
5 E, pet. 6 Benz c 4
incolor -
6 E = pet. 5 Benz. 5
incolor -
7 E„ pet. 4 Benz. 6
incolor -
8 E. pet, 3 Benz. 7
incolor -
9 E. pet. 2 Benz* 8
incolor -
r H

3C1
10 Benz. incolor i
11 Benz, 9 Clorof. 1
incolor -
12 Benz, 8 Clorof, 2
incolor -
13 Benz. 5 Clorof. 5
incolor -
14 Benz » 2 Clorof. 8
incolor -
15 Benz. 9 Clorof. 1
amarelada alaranjada
16 Benz» 9 Clorof. 1
amarelada alaranjada
17 Clorof. incolor -
As fracções XI 1 5_ 1 6 continham o alcalóide U que, V cromatografado em placa de sílica (Figs,40 e 41) nos mql des indicados para os compostos L, J e I, apresentava Rf = 0,54 e 0,16 respectivamente.

352
Fig-. 40 Cromatografia em placa dos alcalóides J, I, H e L no sis tema olorofdrmio-metanol(48:2).

353
O 0,54
0 0,35 0
0,29
0 0,16
j i .H L
Fig, 41 Cromatografia em placa dos alcalóides J, I, H e L no sis tema clorofórmio.

355
3'6,8»- Análise dog alcaloïdes H. IT J o T,
3.6.8.1.- Características do alcalóide H
Este alcalóide tem cor amarelada e em solução metanólica apresenta fluorescência muito ligeiramente a-zulada. Em solução alcalina exibe idêntica fluorescência mas em meio clorídrico ou sulfúrico cora de amarelo e à luz U.V. exibe fluorescência azul bastante mais intensa. Cristaliza do éter sob a forma de agulhas finas e curtas. Os cristais, depois de secos,subiIxaem a cerca de 130-135° e fundem a 198°C com decomposição. Dissolvido em cloro
formio e cromatografado em placa de sílica nos sistemas clorofórmio e cloroformio-metanol(48i2) apresenta ^ os Rf = 0,16 e Rf = 0,54, respectivamente. Cora de avenue lhado, pulverizando as placas com o reagente de DRAGEN DOEPF. Com o reagente p-dimetilaminobenzaldeído cora de amarelo e não reage com a ninidrina. Os espectros no U/V. em soluções em metanol, em metanol alcalino e em metanol ácido estão reproduzidas na Fig. 42, referindo o QUA DRO XIV a localização dos máximos e dos mínimos de absor ção dos respectivos espectros.

357
\ 2
\ "i « r 1 — — i -T 1 , , , .
200 20 40 60 80 300 20 40 60 80 400 20 / ran Fig. 42 1 - Espectro de absorção do alcalóide H,em metanol. 2 - Idem, em metanol ácido. 3 - Idem, em metanol alcalino»

358
Q U A D R O XIV
ESPECTRO DE ABSORÇÃO NO ÏÏ.V. DO AÍÇAL^DEH
Em metanol
X max.nin A mín,nm 238(inf) 228 243 234 252( in f ) 260 271 283 298-302 322 364
Em sol .metanoli jEm s o l . m e t a n ó l i ca de NaCB N/20jca de HC1 H/20
/ máx. 238( inf) 244 252(Lnf) 272 298-300 364
X minerai 228 234 260-62 284 322-26
^ máx 267 318 392-94
/ ,mín,nm 238-40 293 358
3 . 6 . 8 , 1 . 1 , - Rgdugjo do a l c a l ó i d e H.
Dissolvemos cerca de 1 mg do a l c a l ó i d e H,em 5ml
de I t e r , adicionámos 0 , 1 g de h i d r a t o de alumínio e l í
t i o e deixámos em contacto duran te 24 h o r a s . A solução
e t é r e a r e s u l t a n t e da f i l t r a ç ã o da mi s tu r a , f o i evaporada
e c r o m a t o g r a f i a em p laca de s í l i c a no s i s tema c l o r o f ó r -
mio-metanol(48s2) . Obtiveram-se fundamentalmente duas
manchas que coraram com o reagente de DPAGENDORPF. Uma
mancha es t ava l o c a l i z a d a muito próximo do ponto de a p l i
cação da subs t ânc i a e a o u t r a ao n í v e l do a l c a l ó i d e i n i
c i a l . A separação das duas zonas por cromatograf ia prepa
r a t i v a em p l aca de s í l i c a naquele mesmo s i s tema p e r m i t i u
-nos v e r i f i c a r , pelo espec t ro de absorção no U.V.^que a

359
mancha de Rf maior correspondia ao alcalóide inicial que não sofrera total redução, enquanto que a mancha A,de Rf menor, de fluorescência azul violácea, apresentava um es. pectro típico dos compostos fo-carbolínicos (Fig. 43).
3.6.8.1.2,- Metilação do alcalóide H
0 alcalóide H dissolvido em metanol e adicionado de iodeto de metilo não sofreu metilação à temperatura ambiente após 5 dias de contacto. Passado este tempo adicionámos, uma gota de hidróxido de sódio metanólico. N foi aquecido a 70° durante duas ' horas, e o produto resultante destes tratamentos, quando cromatografado empla ca de sílica no sistema clorofórmio-metanol(48s2),originava uma mancha com o mesmo Rf do alcalóide inicial,sendo também coincidentes os espectros no U.V. dos dois pro dutos antes e depois da metilação.
Além desta mancha, observou-se ainda uma outra, de maior Rf e de fluorescência azul, cujo espectro no U, V. é semelhante ao do alcalóide I (Fig. 44).

301
0,7
0,6
0,5-
0,4-
0,3.
0,2-
0,1»
\v
200 20 40 60 80 300 20 40 60 80 X nm Fig, 43 1 - Produto de redução do alcalóide II, em metanol. 2 - Produto de redução do alcalóide-!!, em metano]. 3 - Produto de redução do ^ ' â M e l , em metanol. 4 - Produto de redução dp_aTcaÍ£^s~L, em metanol.

363
A
0,7-
0,6-
0,5
0,4-
0,3-
0,2-
o,i-
T r - i r 200 20 40 60 80 300 20 40 60 80 / Fig. 44 Produtos de iodometilação do alcalóide H. 1- Zona correspondente ao alcalóide H inicial. 2- Zona de Rf superior ao alcalóide H.
nm

365
3 , 6 . 8 , 1 . 3 . - Hidgdllse e descarboxilaçfio do alcalóide H
Cerca de 1 mg do alcalo'ide H foi adicionado de 10 ml de solução de KOH 2N e aquecido a b.m. durante 2 horas. Apo's este tratamento foi adicionado de 15 ml de H2SO4 211 e aquecido à ebulição durante 15 minutos. Alca-linizámos, depois a solução com amónia e extra£mo<-la com clorofo'rmio.
0 produto resul tante da evaporação do clorofo'rmio apresentava fluorescência azul e o espectro no U„V. era tipicamente o de um derivado(¾ -carbol ínico(Pig.45) .

367
A
0*6
-T 1 1 1 1 r 1 i ~r 200 15 30 45 60 75 90 305 20 35 /, nm Fig. 45 1 - Produto da hidrólise e descarhoxilação do alcalói
de Ht em metanol. 2 - Idem, em metanol ácido. 3 - Idem, em metanol alcalino.

369
3,6.8,2. - Características do alcalóide I
0 alcalóide I apresenta cor amarelada e em solu ção metanólica neutra, ácida e alcalina possui fluorescên cia azul, cristalizando sot a forma de agulhas. Os cristais depois de secos stiKO&Mto a cerca de 130-135 C e £-¾ dem a 166-167°G. 0 alcalóide, dissolvido em clorofórmioe cromatografado em placa de sílica nos sistemas clorofórmio e clorofórmio-metanol(48s2), migra com os Hf seguintes! Rf = 0,29 e Rf = 0,83. Reage com o reagente de DRA-GEHDORFF com cor avermelhada e não reage com o reagente p-metilaminohenzaldeído nem com a ninidrina. 0 espectro da substância (Fig. 46) apresenta as seguintes bandas de absorção s
Em solução metanól ica n e u t r a e a l c a l i n a
/ .máx . 245, 258, 274, 300 e 368-370 nm / . m i n . 232-34, 251 , 264, 287 e 328 nm Em solução metanól ica ácida / m á x , 270, 323-24 e 402-404 nm / m i n , 240-42, 295 e 360 nm
3 , 6 . 8 . 2 , 1 , - Redução do a l c a l ó i d e I
A redução do a l c a l ó i d e I f o i p r a t i c a d a nos mol
des que se ind icou pa ra H. 0 produto mais abundante d e s
t a reacção possui o espec t ro da ft-carbolina ( F i g . 4 3 ) ,

371
°,31
0,7-
0,6
0,5
0,4-
0,3-
0,2-
0 , u
^
é
* X 1
C\J<JjÀÍn
W> \ x ■ T
T 1 1 1 1 r 200 20 40 60 80 300 20 40 60 80 ^m
Fig. 46 1 Espectro de absorção no U.V.do alcalo'ide I,em metanol. 2 Idem, em metanol 4lcali.no. 3 Idem, em metanol acido»

373
3.6.8,2,2,- Metilação do alcalóide I
0 alcalóide I não sofreu metilação quando esta foi praticada nos moldes já descritos para o alcalóide H, pois que o produto resultante apresentava espectro no U, V. idêntico ao alcalóide inicial e não sofreu qualquer desvio quando determinado em meio alcalino.
3,6.8.2.3,- Hidrólise e descarboxilaçao do alcalóide I
Foi praticada nos moldes descritos para o alcalóide H, conduzindo o ensaio tambóm ao mesmo resultado que se referiu para este alcalóide.
*

375
3.6.8.3.- Características do alcalóide J
Este alcalóide tem cor amarela, é* pouco flúores, oente em meio neutro e alcalino mas em meio clorídrico ço ra de amarelo com fluorescência azul nítida. Funde a 240° C. 0 espectro no U. V. apresenta os seguintes máximos e mínimos s em solução metanólica neutra e alcalinas /N máx: 282(inflexão), 291, 312 e 390 e A mins 272.304--6 e 354-56.
Em solução metanólica ácidas/máx. 282, 292( inflexão), 317 e 380-82: A mins 270, 302-306, 364 e 370) (Fig. 47) .
3 . 6 . 8 . 3 . 1 , - Redução do alcalóide J
A redução do alcalóide J com o hidreto de alumí nio e l í t i o conduziu-nos a obtenção dum produto com fort e fluorescência azul Violácea com espectro idêntico ao da rt-carbolina (Fig. 43) .
5 .6.8.5.2«- Metilação do alcalóide J
A t en ta t iva da metilação deste alcalóide nos mol des descri tos para o alcalóide H não conduziu ao derivado metilado deste a lca ló ide .

377
0,7-
0,6"
0,5-
0,4-
0,3-
0,2-
o,-i
1 e 2
, , f , , , , -, , , r_
200 20 40 60 80 300 2G 40 60 80 400 / ni:. Fig. 47 1 - Espectro de absorção no TJ.V. do alcalo'ide J, em metano^ 2 - Idem, em meio alcalino. 3 - Idem, em meio ácido.

379
3 , 6 . 8 . 4 , - C a r a c t e r í s t i c a s do a l c a l ó i d e L
Es te a l c a l ó i d e possui cor amarela em solução al_
coó l i ca e não é f l u o r e s c e n t e , enquanto que em meio ácido
exibe f l uo re scênc i a a z u l . C r i s t a l i z a do é t e r sendo os
seus c r i s t a i s de aspecto a c i c u l a r e sublimando a cerca da
110° a p ressão normal . Analisado em cromatografia ,em p i a
ca d e â í l i c a , a p r e s e n t a os Rf s egu in t e s s no s i s tema clo^
rofórmio, Rf = 0,54$ no s i s tema clorofórmio-metanol(48 s
2) Rf - 0 ,93 (Fig . 41 e 4 0 ) .
A pulver ização das p l aca s com o reagente de DRA-
GENDORFF permi t iu -nos l o c a l i z a r o a l ca ló ide , co rado de
avermelftado? com p-dimet i laminobenzaldeído obtivemos co
loração amarelada e não r e a g i u com a n i n i d r i n a . A d e t e r
minação do espec t ro de absorção da s u b s t â n c i a em s o l u
ção metanól ica n e u t r a , a l c a l i n a e ác ida reve lou a e x i s
t ê n c i a das s e g u i n t e s bandas s ( F i g . 4 8 ) .
Em metanol ; / m á x ; 245, 253, 2 6 3 , 2 8 6 , 307 e
380 nm e Amíns 242, 248, 259, 260, 300 e 340-42 nm.
Em solução metanól ica de HC1 W/20 { maxs253(pa-
t amar ) , 264, 286, 308 e 381 e A min s 242, 272,300 e 346-
-52 nm. Em solução metanól ica de NaOH N/20 /{máxs 245
( i n f l e x ã o ) , 253, 263, 286, 307 e 382 e X mins 242, 259,
272, 300 e 344-46.

381
200 20 40 60 80 300 20 40 60 80 400 / m F i g . 49 - 1- Espectro de absorção no U.V.do a l c a l ó i d e L,fcm solução metanol ica . '
2- Idem, em solução metanol ica a l c a l i n a . 3 - Icem, em solução met ano'li ca á c i d a .

333
3, .6 ,8 ,4 ,1 , - Redução do alcalóide L
Praticámos esta^aduçSo com hidreto de alumínio e l í t i o nos moldes que se indicaram para o alcalo'ide H, A análise do produto resul tante desta redução permitiu— -nos ve r i f i ca r que a redução conduziu a um derivado t i p i camente (3-carbolínico (Fig, 43) ,
3.6.8.4. .2,- Met ilação do ale a l ó i d e j .
Foi prat icada nos moldes que se indicaram para o alcalóide H. A análise cromatográfioa e espectrofotomé t r i c a no UUV, do produto resul tante deste t r a t a n ê n t o per mitiu-noa concluir que o alcalóide não metilou.

385
3e6„9. - Análise dos resultados
A análise das ca rac te r í s t i cas dos alcalóides H, I , J e L leva-nos a t i r a r algumas conclusses.
Assim, podemos dizer que se t r a t a de alcalóides com o núcleo fundamental da (3-carbolina, porquanto t o dos e les , por redução com hidreto de alumínio e l í t iopon duziram a um alcalóide cujo espectro é ca rac te r í s t i co do núcleo refer ido . Como é sabido, este redutor não a t inge, em regra, duplas ligações mas reduz funções e'ster, carbo x í l i c a s , cetónicas e a lde íd icas .
Os espectros no U,V£ indicam-nos que todos eles possuem maior conjugação do que a que existe na (J-carbo-l ina e na harmana, porquanto a banda de absorção mais longínqua se encontra neles deslocada para comprimentos de onda bastante maiores do que naqueles dois a lcalóides .
Os alcalóides H e I , por h id ró l i se seguida de aquecimento, originaram noranaana. Este facto fez-nos ad ml t i r a existência duma função és te r na molécula. Eaciol cinando em termos b i o g e n e t i c s , parece lógico admitir-se a possibil idade de esta função poder local izar -se no car bono 3 ou 1 do núcleo da (3-carbolina. Encontrasse i solados de plantas a p -carbolina-1-carboxilato de metilo ( I I ) e o 3-carboxiarmano ou ácido-1-meti l-G-carbolina-3 -carboxilico ( i l l ) . ^

38Ô
COO H
COOH,
I I H I O primeiro foi isolado de Pleiocarpa muticaBENTH
(23) e o segundo como genina dum alcalóide heterosídico L solado HP Anpidosperma polyneurqn IvIULL.ARG. (24)
Os alcalóides H e i assemelham-se mais no que res pe i ta ao espectro no U.V,, forma de cr i s ta l ização e fluo rescÔncia oom a (3-carbolina-l-carboxilato de metilo do
que com o 3-carboxiarmana,.. De facto o 3-carboxiarmamaf ou l-metil~3-carbo
x i - Ç - c a r t o l i n a em etanol ácido tem os seguintes máximos de absorção* A máx. 244( logí= 4 ,48) , 277(log Ç- 4,77), 300(log£ = 4,21) e 365(logi = 4,0?) nm (25),enquanto que KÏÏMP (14) indica para o primeiro composto as bandas de absorção no U.V. (álcool a 96$) seguintes s
A max... 246( log£ 4 ,23) , 250'(log.â 4 ,23) , 274 ( l o g . ^ 4 , 2 8 ) , 300(log. l4,0l) e 372(log.S 3,77) nm.
Al t ín . , 230( log .^3 ,98) , 250 ( log .S4 ,2 l ) , 264
( log^ 4,20), 286( log .^3 ,8ô) , 330(log.<g 3 , l l ) nm.

3.07
ACHEEBACH e BIEMâUN (23) referem as seguintes bandas de absorção?
/max., 245(log.£4,17^ 257(log.14,16), 274 (log. £4,21), 300(log.^3,96), 368(log.£ 3,69) am.
/min., 228(log."£3,96, 25l(log.¢4,14), 263 (log.14,13), 286(log.^3,80), 327(log.£ 3,ll) nm.
Es tes ú l t imos au to re s ass inalam para e s t e compos to as bandas no I .V. em 3440 ( s ) , 1700 (s),1630(s)om~J" bandas que se podem r e p u t a r a p r imei ra ao grupo M indó*-l i c o e as duas ú l t imas ao grupo é s t e r .
Quer no a l c a l ó i d e H quer no .ç.loalo*i&e_I encontrámos bandas próximas das c i t a d a s . Assim o a l c a l ó i d e I a p r e s e n t a , como poderemos v e r na P i g , 49, bandas em 3,410, 1.740 e 1.640 cm" , mas encontram-se o u t r a s também dignas de r e g i s t o dada a sua f o r t e in tens idade? 2 .920 , ' 1,450; 1.290 e 1.150 cm"1 .
No a l c a l ó i d e H destacsun-se as bandas s egu in t e s s 3 ^ 1 0 , 2 .910 . 1.710, 1,640., 1,430, 1,325, 1,295. 1.265, 1.230, 1,120, 800, 730 e 715 cm"1 ( F i g . B0).
Apesar das d i fe renças r e g i s t a d a s , incl inámo-no* pa ra a h i p ó t e s e de que o a l c a l ó i d e I corresponda ao á c i i c - (3 - c a r b o l i n a - 1 - c a r b o x í l i e o , p o i s , como vimos, o ponto' de fusão determinado p a r a o nosso composto corresponde ao daquele , o espec t ro no U . 7 . , o fac to de o produto de dee carboxi lação o r i g i n a r norarmana, o facto de^pca? r edução ,

388
originar um derivado y -carbolínico que supomos ser a M -droximeti lQ-carbol ina, e, ainda, o facto de nos ser pos. s ível loca l iza r no espectro do I.V. as bandas caracter ís t i cas da função és t e r depõem a favor desta h ipótese .
0 alcalóide H muito semelhante nas suas caract e r í s t i c a s espectrais ao alcalóide I , supomos ser o (a --carbolina-1-carboxilato de meti lo . Parece-nos favorável a es ta hipótese o facto de na metilação oom iodeto de me t i l o termos obtido dois produtos, sendo um deles semelhante ao alcalóide de que partimos, e outro semelhante ao alcalóide I também no que respei ta às ca rac te r í s t i cas referidas.(Comparar Figs . 42, 44 e 46). Entretanto a trans formação da função carboxi em és te r metílico ou carboxi_ lato de metilo não é normalmente conseguida por tratamen to com I CE3, mas como neste caso a função se encontra j é na forma deocarboxilato de sódio e assenta no núcleo ben zénico, é de admitir t a l poss ib i l idade .
0 alcalóide L, embora possua o núcleo fundamen t a l da Çí-carbolina, tem um comportamento anormal na região do U.V. relativamente aos derivados -\J-carbol3nioos simples quando o espectro é realizado em meio metanólico ácido (Fig. 43) . Este facto faz-nos supor que a não pro-tonização do composto se deva a qualquer impedimento do átomo de azoto p i r id ín ico , o que j u s t i f i c a r i a também a impossibilidade de iodometilação observada bem como o carácter de base fraca que manifesta. Aliás,qualquer des.

P u 0) "O
Œ •o z o o D
l ã >
0 o < 0
Tl
00
E <U
z
O -a o
IO
8 I
O) o <u c o o u r o o lO o u V « -*
X I - I u ca
X I
u a.
T3 O
XI a o
d) 3DNVl±IWSNVai

'/o) 3DNVlllNSNVai

ZO'â
tes quatro alcalóides são substâncias de reduzida basiqi. dade como comprova o modo como podem ser extraídas do ex. tracto bruto alcaloídico. Na realidade, estes produtos po dem ser extraídos das soluções aquosas a pH entre 4e6,5 como nós tivemos ocasião de verificar. 0 comportamento do alcalóide L em placa de sílica migrando para níveis bastante altos no sistema clorofórmio indica possuir pequena polaridade e também ligeira basicidade. Apenas a títu lo de hipótese poderemos admitir que tal composto possa corresponder a um N^-óxido pois que podemos assinalar u-ma banda muito intensa em 1.390 cm" , que poderá corresponder ao grupo N -» 0 (Fig, 5l). Além disso, é de supor a existência de outros radicais possivelmente carboxi ou carboxilato de metilo, visto que o produto se reduz pelo hidreto de alumínio e lítio podendo aqueles radicais serem responsáveis pelo efeito batocrómico que se observa neste alcalóide relativamente à Q-carbolina.
Quanto ao alcalóide J parece-nos que igualmente haverá impedimento no azoto piridínico visto que não pro toniza nem se iodometila. Não podemos, no entanto, aven tar qualquer hipótese de estrutura para este alcalóide , por falta de elementos o

O £ 0 ™ O T3
O S O E IA
o D
it _l > Q> s •o
•o o o
1 o a
g a.. S UJ Z D z > < 3
-8
_; ca 0 V
T ) o
IO b
O) ai V u O o O c X o IO 0 m t o l/>
o o 0 <D
T ) g U) O v (D n ifl (1)
LU "U
l i (%) 33NVlllWSNVai
LO
O)

397
3.6.10,,- Síntese do e s t e r a -carbolina-1-carboxila.to de metilo
Gomo suspeitámos t e r isolado de Burkea africana o és te r metílico da 1-carboxi- Q - ca r to l ina , tentámos prepar^í.jo para o comparar com o alcalóide por nos isola do. A técnica usada é uma adaptação das u t i l izadas por KERMA.CK e co l . (19), SNIDER e co l . (13) e KDMP e ool.( l4) e fundamenta-se na oxidação da benzilidenoarmana com per_ manganato de potáss io , dando a 1-carboxi-!^-carbolina, a qual, por ester i f icação com metanol em meio clor ídr ico , dá a G -carbolina-1-carboxilato de meti lo. Por sua vez, o derivado benzaiarmana é obtido a p a r t i r da harmana por aquecimento desta com aldeído benzóico. Como vimos a t rás , a harmana foi obtida pela reacção do tr iptofano com ace-ta ldeído, seguida de oxidação com descarboxilação. 0 e s quema Ç.UG indicámos na, página segu in te r e f e r e os pas sos p r i n c i p a i s n e s t a s í n t e s e .
Partimos de 1 g de harmana adicionada de 20 ml de bensaldeído, que aquecemos em banho de s i l icone a 150° C, durante 3 horas . Após es te aquecimento, a cor da solução era amarela acastanhada. Foi, em seguida, evaporada no vácuo e adicionada de 20 ml de é te r e ácido c l o r í drico a 10% (25 ml). Formou-se um abundante , precipitado amarelo que foi separado por f i l t r a ção . 0 precipitado foi lavado várias vezes com é ter e com água, a té deixar de cheirar a benzaldeído. Foi c r i s ta l izado várias vezes em

398
,^>> ■CCOH
'Mr CHOCHO'
! H
GOOH
en* .EH
:X Ozid» Mn04"
Met.
Oxid.
! H
1 ,^
N MeOH e HOI GOOH
■ CO,
E <* OOCH,

odd
álcool a 95 . O precipitado foi suspenso em etanol e neu tralizado com amónia a 20$; a base livre foi separada por centrifugação e o precipitado cristalino obtido,fundia a 200-202 possuindo cor amarela intensa e forte fluorescência azul. 0 produto obtido correspondia à benzalarma-na. .
0 produto foi depois oxidado pelo permanganato de potássio. Para isso, dissolvemo-lo em 10 ml de piridjL na sendo a solução arrefecida em gelo e adicionada de so_ lução aquosa saturada de permanganato de potássio em quan tidade suficiente até que a cor violácea do permanganato se manteve ao fim de 1 hora. 0 excesso de permanganato foi eliminado por tratamento com álcool a banho maria. A suspensão obtida foi filtrada e o filtrado evaporado à secura e o resíduo retomado por ácido clorídrico diluído. Recolhemos um precipitado amarelo. Este foi dissolvido em hidróxido de potássio a 5$, a solução foi filtrada e precipitada com ácido acético. 0 precipitado foi separado por centrifugação, seco no vazio e dissolvido em meta nol saturado de ácido clorídrico e aquecido a banho maria durante 15 horas, após o que se evaporou à secura sob pressão reduzida. Foi depois adicionado de água e amónia e extraído pelo éter sulfúrico. 0 resíduo da evaporação do éter, quando cromatografado em placa de sílica nos sis t.emas clorofórmio e clorofórmio-metanol(48s2) apresentava várias zonas fluorescentes. Procedemos à separação des

400
tas diferentes zonas por cromatografia preparativa em placa de sílica. Separamos 4 zonas que reagiram com o re velador de DRAGENDORFF.
A zona de menor Rf, possuia fluorescência azul Violácea e localizava-se muito próxima do ponto de aplicação. 0 espectro desta zona eluida pelo metanol mostrou espectro tipicamente Q»-carbolínico, A zona B apresentava cor amarelada e fluorescência azulada sendo o seu espectro no U.y, muito semelhante ao alcalóide H isolado de Burkea.
A zona C, de fluorescência azul brilhante, migrou para um nível superior a D e corresponde ao alcalói de I, quer pelo Rf quer pelo espectro no U.V",
A zona D tem cor amarelada e apresenta um espeç. tro bastante semelhante ao do alcalóide L isolado de Burkea, mas os dois compostos não se identificam quer pelos seus Rf em placa de sílica quer pelo espectro no U.V. em meio ácido.
Tentámos a separação destas substâncias em colu na de ácido silícico e conseguimos obter os produtos A, B, 0 e D; entretanto o rendimento foi bastante insignifi cante, só nos sendo possível fazer o estudo cromatográfi co em placa de sílica no desenvolvente clorofórmio-meta-nol(48:2) e determinar o espectro no ÏÏ.V, dos citados pro dutos (Fig. 52),
0 alcalóide A corresponde possivelmente à harma

401
60 80 300 20 40 60 80 /N am -, 1 , — - — p
200 20 40 Pig. 52 Espectro de absorção en solução metano'lica de alguns produtos isolados na síntese daÇ-oarbolina-l-carbo-xilato de metilo.

402
na i n i c i a l , que não reagiu com o benzaldeído. São de prever na oxidação da dupla ligação do
benzilidenoharmana os produtos 1-formil-\-3-carbolina, 1--carboxi- \J-carbol ina e por ester if icação deste o(^-car-bolina-1-carboxilato de meti lo . Supomos que B corresponde à l -carboxi-(3-car tol ina ou formil\3- car to l ina . Eepa-re-se na semelhança do espectro com o alcalóide H i so l a do de Burkea (Fig. 42) . A substância G deve corresponder a^-carbol ina-d-carboxi la to de metilo que pretendíamos preparar e é muito idênt ica ao alcalóide I também iso lado de Burkea.
£ é bastante semelhante ao alcalóide L de Burkea, no entanto as bandas experimentam em relação a L um l ige i ro desvio batocrómico» notando-se que para D o meio ácido provoca também desvio batocrómico, enquanto que para o alcalóide L esse fenómeno se não ver i f i ca em idêntiças condições„
Finalizaremos o nosso trabalho com as conclusões que este nos susci tou. Antes, porém, mencionamos em quadro resumo algumas das ca rac te r í s t i cas dos alcalój. des isolados de Burkea africana, HOOK,

403
, !>
CO O O Ï=J* t O N O 1 <tf LO CV 1 o q o 1 tí rH ÎN CO
to 5 •3 si 11 II II VNJ vt..' W
03 03 r-i r H
• H O
O
-p s § S "»03
O
O j CO CO rH § S 03
W C O O ) i ! J. in cv o
•H -3 Crt O O O O ^ C\2 25
f en o m en o m w (M CD en
•H -3 Crt O O O O ^ C\2 25 O «# rH O ^ rH
N W N 0 Î C\2 CO CO C\J OJ CO CNJ N CO
.¾ O
3 <co o O 03 ca Q)
3 rH 1
£ .ÍH P O > 0 to
rH •Pu
03 - P 3
î> i n t o C- to m <o H O
en a> en cr> rH I N O r H CV2 «* a CM -* à à o o O o o o
CD TJ •H o VD o rH r H
CÔ 03 C\J
rH 3 o°d> n3 03 cr> o j
o <0 w o O 03
"D o H 03
CO . (O HJ
o
g r H CJ 03 H fr 3 U O
1 CJ1
P4 CD o -ri C-r-f rH O
O H rH
03 I Õ i ) Ai •H cô rw 1 \ rH c3
o -a) fi
-d rw « | Lf O r H <
•H cô r i
- P 03
EH
i / o rH
3 C/

404
>
o •
BC o cu Pi CO W O
H t o m c\a c\2 to
i
<CU o co (D
8 rH
Q)
CU
rH O
CO
8
Ítí
0) -d •ri H3
m
(fl m rH r i •H fi> r-l tri rH
a CO CQ
o T i tu
r-i 1 PM r~
rH PM C\i
O !c0 o> Ctí S3
•ri
«M
O O CO
CX! <* CT> O <-0 m ! > 14 t o i n co co co <* C\J C\î W CA2 C\! CO CO
CO i
ID CO CT> ■& o «sh en is CM OJ C\2 Sf>
•a
O I f J • * CO LO LO O CM P- <tf CM CM O <0 CO CM r H t-i
■sjt ^ <* rH . II II II
CT> O «3 LO C-
i n co co t o ^ CM CM CM CM CO CO
CM CO JN to
II I! II
ci-3! co
HOI
> rH :3 ta a
cô CD O
to -^ in
CM o in
U <S>
<+H CD
O
S a CU
o
° to
I CO
O
a 8 CM
a r l C7> ■rl o 05 r H co r-4 ,£! r-3 i crt CQ m
r- a n CM
< t CM - P d rv CI) rH
é erj fn o N o T i « rH O cu
O H M

405
o
o v
cd rf a
-p . C U N ^
ra _W
<cu' o 03 O ÍH Q 3 Cd
H -H
(D
CU
rH o
CO
£
t i •H
N o rH Cd O 03
r-1 . H Cd Cd
03 O
TH OJ
Et
cu Tá ■H ' •O H cd o
rH
cu
O
o o o co O CM CO r H O O CM CO
CM <£> t£> IO CO II II II II
CM
O LO O cn CM £ N CO 0 0 CM CM CM CM
O
H
LO CO
CM O
CO rH CM
O O
¾ ■tf
t i tf C M
u o
rH o
,8 o X M H 0
- p l r H CU
3 3 'ti <H ■H CU
R 0
• H
R •H sã 3(
M CO CO CM rH 00 r\? •*H M 2S 00 CM P J t O tf LO JN 0¾ o «0
W t O H 01 CM CM CM CM CM t o CO CM CO CO
O
3 ícd i8 r i g « H cu cu cfl H r H o H oj
«H «H Q § R O
■ri -H CO
I j 00 xil CM CM a i t f LO tf LO S- CT><£> r 00 xil CM CM a i t f LO tf LO S- CT><£> W m CM CM CMCO
I •H rH Hd ft
to cd cd TJ
CM
«3
o o
03
O O LO
to o LO r H
g' O cu
< ~ o o
?cd o o
ecd -H CO 03 3 o
o o 00 CT> rH
W
\\ f

406
\=> o ti •
P4 u a •p „ CD ^ Pt ra
i CD Q) cri
§H r- l «D Fm o
Cl) o ti
o • r i í-l rH -p cri 0 o fl) rH ti cri
Hi
O I
m co <<# o tu »# m r o to w t\j N B n
o • H O
Vri CM ^ „ I I O W N t - CM O CM to <*
pi tsi cri
c»
f! 'D
,¾ O
ca
• H vO
H cri CQ O -H H cri cri m
■■d
FM
CM
Cl CM
CO CD
O
o
•H ' o ' t H «ri
(D ri •H rH o cri
,a co
LO i> CO U3 J o CO H
tO
NA CE!
/ /
H m—w
0) o o-9 H IH ■g tti
O a; r-í R c3 W Ti
O íctf S « M CD 0
H r-l «H t H q § •H -H
O -d •ri
4 CM ao 03
H N Q W (Tl r-
1 o i p. CM co co
<§ M 0)
rH %} R
•H
CM CM co cr>
CM CO
o ô rH CO
CM CM CO CO
4 ft H cri 3 t i (S H
H CM
m CM CO £>
o o
O o o <* CM

407
tS o a «
si $ ^ CD ft m
o r i
■s cu
O T i •ri
m to co to c*- o a •rf m <o co o co TS CV2 C\2 C\2 OJ CO CO
M - r i (U r
r- l Cí
P! (- '
5 HO <* tO CD rH |Tj l i ) tO 00 O 00 * a
s M cu
r H
5 m to to ia JS ci ■è
1 IO tO 00 O 00 Oi CM C\! 0J CO t" ,
Fluores
cência o
p
r H
03
0)
CU >
rH O
02
H C\2
»
>st< CO
O O
CU TH •H %
o r H cd to O T j r-H cd cd CQ
o o
o ?cd o> ã •ri H
CO
O o o rH rH
ai
■H sO
rH cd O H
t-H

408
c\j 00
u I o
•H
<H <H
in o m
o u o r-i O
O U o
r-H O
02
(J) ffi Œl
rH O
-P I I
O
O
i I t
H W t O
r-H O
I 1
O •H S
. H
O fi O
r-H O
rH Q
-P
I O
•H a l+H <+H
O fi o r-H O
I I ^ m
'S a
o
1 <H O ÍH O
rH O
I
« t o
•d
-P J3
1
(D
0) !> rH O
CO

409
BIBLIOGRAFIA
1 - GERNGROSS, 0 . , VOSS, K. e HERFELT, T . , Ber,Chem.Gefl-66, 435 (1933) .
2 - UDENFRIEND, S. e COOPER.J.R,, J .Biol .Chem. 196, 237 "(195277"
3 - UDENFRIEND, S., WEISSBACH. R. e CLARK, C.T., J. BioL Chem. 215, 337 (1955) .
4 - MUNIER, R. e MASiïEBOEUF, M., C.R, Acád. So. 230, l i 7 7 (1950) . '
5 - NEUSS, N , , Phys ica l Data of Indole asd" Dihydrnindole A l k a l o i d s , Ely L i l l y & C ° , I n d i a n a p o l i s 6, Ind iana , U.S.A. Ed. 1954, 19G, 1960,1961, 1962, 1964, 1966 e 1968.
6 - BOIT, H - G., Ergebnisse de r Alkaloid-Chemie "bis 1960, Akademie-Verlag, B e r l i n ( l 9 6 l ) .
7 - KEUPER,J., Tese Dout . , Travaux des Labora to i res de Matière Médicale e de Pharmac i e Galénique. XXXIX, P a r i s , éd . Vigot F rè r e s (1954)*
8 - BADGER, G.M. e BEECHAM, A . F . , Nature 168.517 (1951). 9 - PARIS, R.R., PERCHERON, F . , MAINIL, J . e GOUTAREL,
R,, B u l l . SOGo Chim. France 780 (1957) , 10 - The SADTLER, Standard Spec t ra , Pulai. SADTLER, Re
search Labora tor ies Ph i l ade lph i a P.A, 11 - FERREIRA, M.A,,CORREIA /.LVES, A . , NOGUEIRA PRISTA L„,
Garcia de Orta 11 ( 3 ) , 477 (1963) . 12 - KERMACK, W.0-, PÊRKCN J r . , W.H. e ROBINSON, N.R. ,
J, Chem. Soc. 119, 1602 (l92l). 13 - SNYDER, H.R., WÀLKER, H*&, WERBER, F.Xa, J,Am,Chem.
Soc. 7 1 , 527 (1949, 14 - KUMP, C . , SEIBL, J . e SCHMID, D.H., Helv.Chlm.Aota
XLVI, 498 (1963). 15 - SPENSER, I.D., Can, J, Chem. 37, 1851 (1959).

410
16 - SPATH, E. e LEDERER, E., Ber. 63, 120 (1930). 17 - MANSKE, R. H. F., PERKIN, W.H. e ROBINSON, N.R.,
' J . Chenu-. Soo, l (1927) . 18 - HESSE, M., I n d o l a l k a l o i d e in Tábe l l en , Spr inger -
-Ver lag-Ber l in -Got t ingen-Heide lberg--New York, (1964) e (1968) .
19 - ABDUSALAMOV, B.A. , SADYKOV, A.S . e ASLANOV, K.A., Nauphn, T r . Tashkentsk.Gos.Univ. 263, 3 (1964) .
20 - ABDUSALAMOV, B.A. e SADYKOV. A . S . , Uzhekak. Khim. Hi . 6., 79 (1962) . ~
21 - THE ïvîERCK IHDEX OF CHEMICALS and DRUGS, Pub l . MERCK & C°, I n c . Rahway,N.3. ,U.S.A., 7 a ed . (1960) .
22 - CORREIA DA SILVA, A.C. , PAIVA, M.Qe, COSTA, A. An» Fac . de Farmácia do Porto,XXVIII, 85 (1968) .
23 - ACHENBACH, H. e BIEMANN, K. J. ,Am, Chem. S o c . 87, 4177 (1965) . .
24 - ANTONACCIO, L.D, e BUDZIKIEWICZ, H . , Monatschefte Chem. 93, '962(1962) ,
25 - UHÎAUS, R,A0 , Science 129, 641 " ( l959) .
26 - PERKIN J r . , W.H., ROBINSON, R., J.Chem-Soc, 115, 933 (1919) .
27 - SLAYTOR, M. e McFARLANE, I . J . , PtiytochemiBtry 7, 605 (1968) .
* * *

411
CONCLUSÕES
1 Na prospecção de alcalóides inclólicos que levámos a efeito em algumas espécies botânicas originárias de Angola pudemos assinalar estes compostos nas cascas do tronco e dos ramos da Leguminosa Burkea africana HOOK.
2 Dos ensaios preliminares executados sobre as cascas desta planta apurámos a existência de lípidos, prdtidos, glúcidos, celulose, fenóis livres,taninos hi drolisáveis e condensados.
3 Não conseguimos detectar pelas técnicas utilizadas: flavonóides, compostos antociânicos, quinonas, hete rosidos cianogenéticoanemcardenólidos e pomos em dúvida a existência de sapononc.dos ■
4 A análise quantitativa global das cascas da planta conduziunos aos resultados seguintes?
Agua 10,2 8¾ Cinzas 3,40 g# Glúcidos totais por inversão .. 16580 g/o Prótidos totais 10,63 gf> Lípidos 0,65 gg Celulose 26,0 g/o Taninos pirocatéquicos . . . . . . . . 8,3 g/o Taninos absorvíveis pelo pó de
pele .... 11,14 g/o Não taninos 11,20 g% Matéria insolúvel 67,46 g/o

412
5 - 0 t e o r em t an inos abso rv íve i s pe lo pó de pe le c r o
mado (11,14 g%) j u s t i f i c a de c e r t o modo o uso que
a lguns povos fazem das cascas de Burkea a f r i c a n a 110-
OK na i n d ú s t r i a de curtumes.
6 - Do e x t r a c t o do é t e r de p e t r ó l e o , após s apon i f i cação ,
isolámos por cromatograf ia em coluna de alumina dois
produtos com f o r t e reacção de LIEBEEMâOT-BUEÍÍHAED e
du SALKOWSKC.
Um de le s fo i i d e n t i f i c a d o com o ^i - s i s t o s t e r o l
comparativamente com uma amostra a u t ê n t i c a de ^ ' - s i
t o s t e r o l F luka , tendo a i d e n t i f i c a ç ã o assentado em
determinações dos pontos de fusão, desv ios polarimé
t r i c ô s , Rf em p l aca de s í l i c a e e s p e c t r o s no I .V , ,
quer do produto i s o l a d o , quer dos r e s p e c t i v o s d e r i
vados ace t i l ado e benzo i l ado .
7 - Do estudo químico que efectuámos do e x t r a c t o a l c a l o í
dico b ru to averiguámos a presença de v á r i o s alcalói_
des 0
8 - Dos a l c a l ó i d e s a s s ina l ados conseguimos i s o l a r nove,
e i d e n t i f i c a r qua t ro de les com segurança, 0 alcalój l
de Aj. fo i i d e n t i f i c a d o com a t r i p t aminas alcalóide_A
com a te t ra idroarmaria t, o a l c a l ó i d e F com a harmana
e o a l c a l ó i d e I a m ^ - c a r p o l i n a - l - c a r b o x i l a t o de me-
t i l o .

413
9 - A identificação da indolalquilamina Aj com a tripta-mina foi fundamentada em determinações de ponto de fusão, comportamento cromatográfico em camada delga da de sílica com revelação com 03 reagentes fosfa-vanílico, de EHRLICH e ninidrina, pelo espectro no no U.V. e I.V. da amina e do derivado sob a forma de cloreto, comparativamente com uma amostra de cloreto de triptamina Aldrich. Esta amina foi já isolada de variadíssimas espécies de Leguminosas.
1 0 - 0 alcalóide A, tetraidroarmaria. foi identificado pe la determinação do ponto de fusão, pelo comportamen to cromatográfico em placa de sílica, espectros no U.V, e I„V„, quer do alcalóide, quer dos respectivos cloridrato e iodometilado, Julgámos ser a terceira vez que esta base foi isolada de uma Leguminosa.
11 -0 alua-lo ide F mostrou-ae peias, características deter minadas, idêntico à harmana,quer no que respeita à fluorescência, ao Rf em placa de sílica em vários de senvolventes, características espectrais da base de alguns derivados s cloridrato, iodometilato e benzil ideno. Julgámos ser a primeira vez que se relata o isolamento de 1-metil-^-carbolina em Leguminosas,
1 2 - 0 alcalóide I mostrou-se idêntico à^b-carbolina-1--carboxilato de metilo, no ponto de fusão, Rf por cromatografia em camada delgada, espectro no U.V.em metanol e metanol ácido e alcalino. A saponificação

414
e descarboxilação deste alcalóide conduziu à obteri ção de ^ j -carbol ina . 0 espectro no I.V,permitiu-.nos, iden t i f i ca r no alcalóide as bandas r e l a t ivas à fun ção é s t e r . Julgámos ser a segunda vez que es te a l calóide foi imolado de vegeta is .
13 - A identidade dos alcalóides B, E, H, I e L não foi estabelecida em bases seguras devendo as hipóteses formuladas para as suas es t ruturas ser encaradas com reserva, Entretanto, o alcalóide B mostrou-se semelhante a uma amostra de ê:\?â3SQívÍv^ °lue s i n t e t i zámos, no que se refere à fluorescência, ao compor, tamento cromatográfico em placa de s í l i c a , ao ponto de fusão e espectros de absorção no TJ.V.em meio metanclico neutro, ácido e a l ca l ino .
1 4 - 0 espectro no UfcY. do alcalóide 13 corresponde ao de um derivado diidrogenado-p-carbolínico e como este se apresenta idêntico a um derivado de oxidação do tetraidroarmana com peróxido de hidrogénio,
julgámos t r a t a r - s e da harmalana-N^-óxido,
15 - A coexistência dos alcalóides tr iptamina, t e t ra id roarmana, harmalana e harmana nesta planta constituem a demonstração inequívoca dos passos comuns a quaJL quer das hipóteses formuladas por PERMÎT e SLAYTOR na biogénese dos alcalóides h -carbol ín icos .

415
16 J\..harra .la a~Ifh-óxido que julgámos existir na planta desempenhará também provavelmente papel importante na biogénese de3te grupo de compostos.
1 7 - 0 comportamento cromatográfico dos alcalóides H ; J, e L em placa de sílica^ os espectros no U.V. em meio metanólico neutro, ácido e alcalino, redução destes compostos com hidreto de alumínio e lítio permitiram-nos concluir tratar-se de bases ternárias com um núcleo fundamental da^i -carbolina mas com maior conjugação do que este núcleo.,
18 - Admitimos para o alcalóide H, embora com muitas re servas, a estrutura l-carboxi-(i-carbolina.
19 - A existência dos alcalóides referidos e particular mente a presença de triptamina podem justificar as acções que COSBEIA DA SILVA e col. averiguaram no infuso da planta.
oooOooo

417
Í N D I C E
pág . PREAMBULO . . ' . ' . '.. ,\"".'. '.'. ..'.,. . 6
P A R T E I
1.NOÇÕES GERAIS SOBRE O GÉNERO BÏÏRKEA HOOK . . . . . 15
1 . 1 . S i s t e m á t i c a e c a r a c t e r e s botân icos 15
1 ,2 , D i s t r i b u i ç ã o geográ f i ca 17
1 . 3 . I n t e r e s s e económico 18
1 ,4 . Nomes vwernáculos i *•>',.. •■ , 19
1 . 5 , Usos na medicina indígena 20
1 ,6 , Estudos químicos 20
1 .7 , Estudos farmacodinâmicos 22
BIBLIOGRAFIA . . . . . , , 23
P A R T E I I
1 , ALCALÓIDES INDÕLICOS 27 1,1, Generalidades 27
BIBLIOGRAFIA 31 1,2, Distribuição botânica dos alcalóides in
dólicos na3 Leguminosas 33 BIBLIOGRAFIA 41
1.3, Constituição química dos alcalóides indólicos isolados de Leguminosas 45
1.3.1, Generalidades 45 1.3.2, Base indólicas simples 46 1.3.3, Derivados da ft.oarbolina.v* .4 • 6 3

418
Page BIBLIOGRAFIA ..., , ,. 72
1.4.- Propriedades físicas e químicas das in dolalquilaminas e dos derivados da Çà^Sjp bolina . .„ , . . " . . , 77
1,4.,1,- Propriedades gerais . , , . . . . 77 1.4« 1»1, - Formação de sais • 82 1 .4 .1 .2 , - Formação de sa is de amónio quaterná
r io • •. 84 1,4.2,,- Reacções de coloração « 87 1,4.2.1,- Reacções com aldeídos 87 1.4,2.2,- Reacções com xantidrol 90 1.4,2.3.- Reacções com l-nitroso-2-naftol ... 90 1,4.,2.4,- Reacções de diazotação 91 1.4,2.5.- Reacções com a ninidrina 93 1,4,3,- Reacções de fluorescência .... 94 1.4.4a- Comportamento cromatográfico ........ 101 1 4,5,- Comportamento espectral ,. « 108 1,4,5,1,- Espectrofotometria na região do ul
travioleta 109 1.4,5.2,- Espectrofotometria na região do in
fra vermelho >,>•.« * 118 1.4,5,3,- Espectrometria de massa 122
BIBLIOGRAFIA ... « , ....,« 129 1,5-.- Alguns aspectos da farmacologia das in
do lalquilaminas e das Ç? -carbolinas ... 135 1,5,1,- Indolalquilaminas ,. 135 1.5.1,1.,- Actividade farmacodinâmica da 5-HT. 140 1,5,1.1.1.- Acção sobre o músculo liso 140 1»5,1.1«20- Acção sobre o aparelho cardiovas
cular 141

419
pág. 1.5,1,1,3.- Acção sobre o aparelho respira-t ório 143
1,5,1.1.4,- Acção sobre o sistema nervoso central g 144
1.5.2. - Alcalo'ides Q -carbolínicos 145 1.5.2,1,- Acção sobre o músculo 145 1,5.2.2,- Acção sobre o coração 146 1.5.2.3,- Acção sobre a pressão sanguínea. '* * 146 1.5,2,4,- Acção sobre o sistema nervoso cen-
t r a l .Vi,.. 146 1.5.3,- Acção alucinogénica dasindolàlqúila
minas e dps, ierivados ft-carbolínicos .,"" 147 BIBLIOGRAFIA 1 5 1
1.6,- Biogénese das indolalquilaminas e dos derivados Çb-carbolínicos , 153
1.6.1,- Origem biogenética das indolalquila minas , I53
1.6,2,- Origem biogenética dos alcalo'ides derivados da Ç>-carbolina 165
BIBLIOGRAFIA 1 7 0
P A R T E I I I
1 . - NOTAS , , 175
2 , - ENSAIOS PRELIMINARES 181
2 . 1 , - Pesquisa de a l c a l ó i d e s , 182
3 . - ESTUDO QUÍMICO DE BURKEA AFRICANA HOOK 187
3 . 1 . - Ma te r i a l v e g e t a l u t i l i z a d o 167
3 , 2 . - Carac teres macroscópicos do m a t e r i a l u t i l izado , 193

420
Pág. 3,3.-— Composição química g loba l da Burkea
africana HOOK 197 3.3.1.- Água 197 3.3.2.- Cinzas 197 3-,.3,,3-- Glúcidos totais por inversão 197 .3:,3,4-,- Pro'tidos totais . 198 3.3.5,- Lípidos r M 198 333„6,- Celulose 199 3.4»- Análise geral de Burkea africana HOOK . 200 3.4.1.- Pesquisa de compostos polifenólicos. 200 3.4,1,1.- Pesquisa de flav^nóides «... 201 3.4.1,2.- Pesquisa de compostos antociânicos. 201 3.4,1*3,- Pesquisa de fenóis livres... 201 3,4,1.4o- Pesquisa de taninos ........ 202 3,4,1.,5,,- Dosagem de taninos 203 3.4.1.,5,1,- Preparação da solução extractiva
do tanino • • • > 203 3,4,1.5,2,- Determinação de sólidos totais .. 207 3-,4,1,5.3,- Determinação dos não taninos .... 208 3,4.1,5.4,- Determinação dos taninos absorvi
dos pelo pó de pele o 208 3.4,.,1.6,- Dosagem dos taninos catéquicos .... 210 3,4.1.7,- Pesquisa de derivados quinónicos .. 210 3.4,1.7,1,- Quinonas livres 210 3.4.1.7.2,-Heteróaid.os antraquincnicos ..... 211 3.4.1.8,- Pesquisa dehe-teró.ai"dos cianogené-
t i c o s . . . » 211 3 , 4 . 1 . 9 , - Pesquisa de heterósidoBS c a r d e n ó l i -
do§ 212

3,4,10,- Pesquisa de saponósidos ..,., 212 3,5,- Estudo do extracto de éter de petróleo, 215 3.5.1,- Ensaios prévios ., 215 3,5,2,- Saponificação do resíduo R^ ., „ 218 3,5.3,- Separação dos constituintes do resí
duo % ] _ , 219 3.5.4,- Identificação do fi -sitosterol nas
fracções III9 a IH10 .... 224 3.5,5,- Preparação do acetato de Çb -sitosterol
e do acetato de produto I 225 3.5.6,- Preparação do benzoato de li-sitosterol
e do benzoato do produto I 226 3,5.7,- Hidrolise do acetato e benzoato do
produto I isolado de Burkea africana . 227 3,5«8,- Estudo das fracções III12 a 11^15 ••• 227
BIBLIOGRAFIA 237 3,6,- Estudo químico dos alcalóides de Burkea
africana HOOK 239 3.6,1,- Extracção dos alcalóides totais de
Burteaafricana HOOK 239 3,6,2,- Reacções de precipitação e de colora
ção do resíduo % • • • • 243 3,6.3,- Ensaios cromatográficos praticados
com o resíduo RJJ , 244 3.6.3,1,- Ensaios cromatográficos em papel ,. 245 3„6,3,2,- Ensaios cromatográficos em camada
delgada ....* , 246 3,,6.4,- Separação dos alcalóides por cromato
grafia em coluna ,,. 248 3.6.5.- Análise das fracções da. coluna.Y.«... 252 3,6.5.1,- Estudo das fracções ^Zd~42 257

422 pág.
3.6.5.1.1.- Identificação do alcalóide das frac ções VI10-20 com a tetraidroarmana ou eleagnina • 259
3.6.5.2.- Estudo das fracções 719-21 e V22-26. 273 3.6.5,2.1,- Estudo das fracções VIII 9 ,-24 *••* 279 3.6.5.2.1.1«- Comportamento cromatográfico do
alcalóide F e da harmana 287 3.6.5.2.1.2,- Comportamento do alcalóide F e
da harmana na região do infravermelho »... 290
3.6.5,2.1,3,- Preparação dos derivados do alcalóide F e da harmana ......... 290
3,6.5.3,- Estudo das fracções V2a.36 e V I I21- „n„ -35 •••••• 3 0°
3.6,5.4,- Isolamento do alcalóide E das fracções V22-26 • ••••#••*•« 315
3.6,6.- Isolamento da triptamina 327 3.6.7.- Extracção dos alcalóides do resíduo
Rg a diferentes pH »• 3 4 1
3.6,7,1,- Estudo dos alcalóides do resíduo 342
*B1 ••• * ' 5,6,7,L1.- Isolamento do alcalóide L 346 3.6.7.1.2,- Isolamento do alcalóide J ....... 348 3,6.7.1.3,- Isolamento do alcalóide I 349 3.6,7.1.4,- Isolamento do alcalóide H 349 3.6.8.- Análise dos alcalóides H,I,J, e L ... 355 3.6.8.1.- Características do alcalóide H .... 355 3,6.8.1.1,- Redução do alcalóide H 358 3.6.8.1,2,- Metilação do alcalóide H 359 3.6,8.1.3.- Hidrólise e descarboxilação do
alcalóide H 365

423
3.6.8.2,- Características do alcalóide I .... 369 3.6.8.2,1,- Redução do alcalóide I 369 3.6.8.2,2,- Metilação do alcalóide I 373 3.6,8.2,3,- Hidrólise e descarboxilaçSo do
alcalóide I 373 3.6.8.3,- Características do alcalóide J .... 375 3.6.8.3.1.-Redução do alcalóide J 375 3.6.8,3,2,- Metilação do alcalóide J 375 3.6.8,4,- Características do alcalóide L .... 379 3.6.8.4,1,- Redução do alcalóide L 383 3.6,8.4,2,- Metilação do alcalóide L 383 3.6.9c- Análise dos resultados 385 3,6.10^- Síntese do éster Ç>-carbolina-l-car-
"boxilato de metilo 392 BIBLIOGRAFIA 409
CONCLUSÕES 411





























