Embed Size (px)
Citation preview

ESTUDIO DE PROPIEDADES TERMOQUÍMICAS DE COMPUESTOS NITRATOS DE
PEROXIACILO (PANs, RC(O)OONO2) DE RELEVANCIA ATMOSFÉRICA
Darcy Hasbleidy Parra Correa
Informe final
Proyecto Trabajo de Grado bajo la Modalidad Pasantía para optar por el título
de Licenciada en Química
Dirigido por:
Cristian Buendía Atencio, PhD.
Universidad Antonio Nariño
Dirigido por:
Miguel Ángel Delgado Gómez M. Sc
Universidad Distrital Francisco José de Caldas
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA
BOGOTÁ D.C.
2020

2
TABLA DE CONTENIDO
RESUMEN ..................................................................................................................................... 3
1. OBJETIVOS ............................................................................................................................... 5
1.1. General ................................................................................................................................. 5
1.2. Específicos ........................................................................................................................... 5
2. METODOLOGÍA ....................................................................................................................... 6
2.1. Estructuras de equilibrio y Frecuencias vibracionales ......................................................... 6
2.2. Orbitales naturales de enlace ............................................................................................... 6
2.3. Termoquímica Computacional ............................................................................................. 7
3. RESULTADOS .......................................................................................................................... 9
3.1. Estructuras de equilibrio y Frecuencias vibracionales ......................................................... 9
3.1.1. Optimización de las estructuras .................................................................................... 9
3.1.2 Parámetros de equilibrio ................................................................................................ 9
3.1.3. Frecuencias vibracionales ........................................................................................... 11
3.2. Orbitales naturales de enlace ............................................................................................. 14
3.2.1 Orbitales moleculares ................................................................................................... 14
3.2.2 Orden de enlace ............................................................................................................ 18
3.3. Termoquímica .................................................................................................................... 19
4. DISCUSIÓN ............................................................................................................................. 20
5. EVALUACIÓN Y CUMPLIMIENTOS DE OBJETIVOS ...................................................... 24
6. CONCLUSIONES .................................................................................................................... 25
7. ANEXOS .................................................................................................................................. 26
BIBLIOGRAFÍA .......................................................................................................................... 39

3
RESUMEN
Los nitratos de peroxiacilo (PANs) son considerados contaminantes secundarios inestables,
producto de la reacción de los radicales peroxi alquilo (procedentes de la degradación oxidativa
de los aldehídos y cetonas) y dióxido de nitrógeno vía fotoltica.
El primer compuesto fue detectado y denominado como sustancia “X” en 1953 utilizando un
espectrofotómetro IR de largo recorrido. Hasta 1960, mediante la técnica de cromatografía de
gases fue nombrado como nitrato de peroxiacetilo (PAN), siendo este el más abundante de la
familia de las PANs encontrado en todo el mundo tanto en la troposfera como en regiones urbanas
contaminadas. PAN ha sido ampliamente estudiado a nivel ambiental como en laboratorio.
Desde los años 70 se han realizado diversas investigaciones empleando técnicas como
espectroscopía infrarroja (IR) de largo recorrido, cromatografía de gases con detección de captura
de electrones, detección quimio-luminiscente, espectrometría de masas, resonancia magnética
nuclear y otros métodos para detectar compuestos derivados de PAN tales como el peroxinitrato
de propionilo (PPN), peroxinitrato de isobutirilo (PiBN), peroxinitrato de metacriloilo (MPAN),
peroxinitrato de benzoilo (PBzN) y peroxycrotonyl nitrate (CPAN).
Esta familia de compuestos se caracteriza por ser estables a bajas temperaturas e inestables a
temperaturas altas aportando grandes cantidades de NOX que contribuyen a la producción
fotoquímica de O3. Son considerados transportadores de óxidos de nitrógenos (NOX) a grandes
distancias, marcadores para la identificación de compuestos orgánicos volátiles (COVS), potentes
irritantes respiratorios y oculares, y agentes nocivos en la vegetación.
Considerando la importancia de esta familia en la atmósfera y la escases de datos fisicoquímicos,
es de gran interés realizar estudios a través de la química computacional y de teorías de alta
precisión química para estimar datos estructurales, vibracionales y termodinámicos que ayudaran
conocer las propiedades químicas de esta familia de PANs.
En este trabajo se estudiaron 8 compuestos pertenecientes a la familia de los nitratos de
peroxiacilo: peroxinitrato de metoxiformilo (MoPAN), peroxinitrato de acriloilo (APAN), (PPN),
peroxinitrato de n-butirilo (PnBN), (CPAN), (PiBN), (MPAN) y peroxy-n-valeryl nitrate (PnVN).
Nuestros cálculos emplearon la teoría funcional de densidad (DFT) a través de los funcionales
híbridos, B3LYP y M06-2X en combinación con el conjunto de base 6-311++G(3df,3pd). Nuestros

4
resultados nos permitieron estimar parámetros geométricos que nos muestran unas estructuras muy
flexibles que tienen una conformación estable en su isomería cis en relación al grupo carbonilo y
el grupo nitro (NO2) de estos compuestos, las frecuencias vibracionales estimadas nos permitieron
caracterizar las bandas del grupo peroxiacil nitrato -C(O)OONO2 las cuales aparecen en las
frecuencias promedio para la banda C=O (1875 cm-1), dos bandas para el grupo nitro NO2
simétrico (1353 cm-1) NO2 asimétrico (1803 cm-1), una banda para el O-O (982 cm-1) y una banda
para C-O (1076 cm-1) que se encuentran en concordancia con lo reportado en la literatura. Se
exploraron los orbitales moleculares y nos permitieron calcular los ordenes de enlace de las
moléculas estudiadas enfocandonos principalmente en el grupo -C(O)OONO2 y los sistemas p de
los derivados PANs. En cuanto a los parámetros termoquímicos estimados, los valores de entalpías
de formación incrementan con la complejidad molecular en un rango de -32,8 kcal/mol hasta -
100,7 kcal/mol y dos de nuestras moléculas están acorde con los datos reportados por otros
investigadores mientras que el resto de valores reportados aquí pueden ser tenidos en cuenta como
valores de referencia.

5
1. OBJETIVOS
1.1. General
Aplicar la teoría funcional de la densidad al estudio de las propiedades estructurales, vibracionales y termoquímicas de compuestos nitratos de peroxiacilo y derivados.
1.2. Específicos
1.2.1. Explorar las estructuras moleculares, las frecuencias vibracionales armónicas y simular los
espectros IR de los PANs y derivados.
1.2.2. Estimar el orden de enlace de los PANs y derivados a partir de cálculos de los orbitales
naturales de enlace.
1.2.3. Estimar las entalpías de formación de los PANs y derivados.

6
2. METODOLOGÍA
2.1. Estructuras de equilibrio y Frecuencias vibracionales
Las estructuras moleculares de equilibrio de los compuestos peroxinitrato de metoxiformilo
CH3OC(O)OONO2 (MoPAN), peroxinitrato de acrioilo CH2CHC(O)OONO2 (APAN),
peroxinitrato de propionilo CH3CH2C(O)OONO2 (PPN), peroxinitrato de n-butirilo
CH3(CH2)2C(O)OONO2 (PnBN), peroxinitrato de crotonilo CH3(CH=CH)C(O)OONO2 (CPAN),
peroxinitrato de isobutirilo (CH3(CH3)2CHC(O)OONO2 (PiBN), peroxinitrato de metacriloilo
CH2=C(CH3)C(O)OONO2 (MPAN) y peroxinitrato de n-valerilo CH3(CH2)3C(O)OONO2 (PnVN)
se optimizaron empleando los funcionales B3LYP [1] y M06-2X [2] de la teoría funcional de la
densidad (DFT, Density Functional of Theory) ver anexo 1. [3] en combinación con el conjunto
de base 6-311++G(3df,3pd) para determinar las conformaciones estructurales más estables. Se
calcularon las frecuencias vibracionales de las estructuras mencionadas y se verificó que cada una
corresponde a un mínimo local en la superficie de energía potencial (SEP). Todos los cálculos se
realizaron con el programa Gaussian 09 [4], el análisis y animación de los modos vibracionales se
realizó a través de la interfaz gráfica GaussView 5.
2.2. Orbitales naturales de enlace
El análisis de los orbitales naturales de enlace (NBO, natural bond orbital) se realizó a través del
programa NBO Versión 3.1 incluido en el paquete Gaussian 09. La visualización y el análisis de
los Orbitales Moleculares Ocupados de más Alta Energía (HOMO) y los Orbitales Moleculares
Desocupados de más Baja Energía (LUMO) se realizó con la interfaz gráfica GaussView 5.0. Este
cálculo nos permitió estimar el orden de enlace a través de la siguiente ecuación.
Ordendeenlace = #e!enlazantes − #e!antienlazantes2 (1)
Para calcular el orden de enlace se identificaron los valores de los orbitales enlazantes BD y
antienlazantes BD*. Lo anteriormente descrito se aplicó para las 8 moléculas estudiadas en este
informe.

7
A manera de ejemplo se presenta el cálculo realizado del orden de enlace para la molécula PPN,
más información se encuentra en el anexo 2.
Tabla 1. Orden de enlace calculado para la molécula PPN. Las etiquetas de los correspondientes a los
átomos se pueden verificar en la figura 3.
2.3. Termoquímica Computacional
Para determinar las entalpías (∆"6°) y las energías libres de Gibbs 8∆"9°: de formación de las
especies: MoPAN, APAN, PPN, PnBN, CPAN, PiBN, MPAN y PnVN, se utilizó los valores de
energía estimados a los niveles de teoría B3LYP/6-311++G(3df,3pd) y M06-2X/6-
311++G(3df,3pd), a partir de ello se estimó la ecuación de energía de atomización total como se
muestra en la ecuación (2). Dónde, (M) es molécula y (X) es átomo.
"#!(%) = " (ℇ!(*)á#$%$&
− ℇ!(%) − ℇ'()(%)(2)
Dónde ℇ#(<) es la energía cero puntual de los átomos y ℇ#(=)corresponde a la energía cero
puntual de la molécula yℇ$%&(=)es la corrección de la energía cero puntual. Posteriormente a
partir de las entalpías de formación experimental de cada uno de los átomos involucrados en cada
molécula a 0 K y a 298 K, se estiman las entalpías de formación según las ecuaciones (3) y (4).
∆*.°(%, 02) = " (á#$%$&
∆*.°(*, 02) −"#!(%)(3)
Enlace Cálculo Orden de enlace(@)
H10-C8 (1,97601-0,01194)/2 0,98 C8-H11 (1,97164-0,01148)/2 0,98 C8-C1 (1,97454-0,05467)2 0,96 C8-C9 (1,9705-0,0049)/2 0,98 C1=O2 (1,99637+1,99362-0,01835-0,1561)/2 1,90 C1-O3 (1,99106-0,13748)/2 0,92 O3-O4 (1,98758-0,13748)/2 0,97 O4-N5 (1,98767-0,27582)/2 0,86 N5=O6 (1,99521+1,99348-0,53734-0,10082)/2 1,70 N5-O7 (1,99592-0,05201)/2 0,97 C9-H12 (1,99014-0,00669)/2 1 C9-H14 (1,98673-0,00586)/2 1 C9-H13 (1,99021-0,00586)/2 1

8
∆*.°(%, 2982) = ∆*.°(%, 02) − [.°+(2982) − .°+(02)] − " (á#$%$&
(.°,(2982))(4)
En la ecuación (4), se calcula la diferencia entre las entalpías estándar de la molécula (6°') a
298 K y 0 K, sustrayendo las correcciones térmicas de la sumatoria de los átomos que componen
las moléculas.
Para calcular la energía libre de Gibbs de formación a 298 K se emplea la entalpía de formación a
298 K – TDS como se puede observar en la ecuación (5). Para más información ver el anexo 3
“Thermochemistry in Gaussian”.
∆*9°(2982) = ∆*.°(2982) − :(;°(%, 2982) −∑ ;°(*, 2982)) (5)

9
3. RESULTADOS
3.1. Estructuras de equilibrio y Frecuencias vibracionales
3.1.1. Optimización de las estructuras
Las estructuras de equilibrio (ver Fig.1) para los compuestos MoPAN, APAN, PPN, PnBN, CPAN,
PiBN, MPAN y PnVN fueron optimizadas al nivel de teoría B3LYP y M06-2X en combinación
con el extenso conjunto de base 6-311++G(3df,3pd).
Figura 1. Geometrías moleculares de los compuestos nitratos de peroxiacilo optimizados con la teoría funcional de la densidad.
3.1.2 Parámetros de equilibrio
A continuación, se presenta la figura 2 con las estructuras de equilibrio y sus respectivas etiquetas
necesarias para la discusión de resultados
. Figura 1. Estructuras y etiquetas de los compuestos nitratos de peroxiacilo estudiados.

10
A continuación, en la tabla 2 se presentan todos los parámetros geométricos estudiados, las distancias y ángulos de enlaces están dadas en Angstrom y ángulos, respectivamente. Tabla 1. Parámetros geométricos de los nitratos de perioxiacilo con los niveles de teoría B3LYP y M06-2X en combinación con el conjunto de base 6-311++G(3df,3pd).

11
3.1.3. Frecuencias vibracionales
En la siguiente tabla se presentan las frecuencias vibracionales principales y sus asignaciones de
las moléculas MoPAN, APAN, PPN, PnBN, CPAN, PiBN, MPAN y PnVN.
Tabla 2. Frecuencias vibracionales y sus asignaciones para los nitratos de peroxiacilos MoPAN, APAN, PPN, PnBN, CPAN, PiBN, MPAN y PnVN estudiandos en este trabajo.
Mopan Asignación Frec. (cm-1) Modo O7-N5=O6 811,8 Tijereteo
O3-O4 989,2 Estiramiento O8-C9 1058,3 Estiramiento
H10H11H12-C9 1178,8 Torsión H10H11H12-C9 1212,6 Torsión
C1-O8 1251 Estiramiento O7-N5=O6 1359,7 Estiramiento simétrico
H10H11H12-C9 1478,2 Aleteo H10H11H12-C9 1488,9 Torsión H10H11H12-C9 1498,1 Tijereteo
O7-N5=O6 1815,5 Estiramiento asimétrico C1=O2 1867,2 Estiramiento
H10H11H12-C9 3063,4 Estiramiento simétrico H10H11H12-C9 3142,4 Estiramiento H10H11H12-C9 3174,8 Estiramiento asimétrico
APAN Asignación Frec. (cm-1) Modo
O3-O4 991,9 Estiramiento H12-C9-H11 1020,8 Torsión H12-C9-H11 1028,5 Aleteo
C1-O3 1076,7 Balanceo H12-C9-H11=C8-H10 1091,2 Estiramiento asimétrico
C8-H10 1328 Balanceo O7-N5=O6 1351,5 Estiramiento simétrico H12-C9-H11 1444,6 Tijereteo
H12-C9-H11=C8-H10 1686,9 Estiramiento simétrico O7-N5=O6 1809 Estiramiento asimétrico
C1=O2 1854,4 Estiramiento H12-C9-H11 3152,5 Estiramiento simétrico
C8-H10 3197,9 Estiramiento H12-C9-H11 3246,6 Estiramiento asimétrico
PPN Asignación Frec. (cm-1) Modo
C8-C9 1003,2 Estiramiento H10C8H11 1048 Aleteo H10C8H11 1111,9 Balanceo C1-C8-C9 1116,6 Torsión H10C8H11 1286,4 Torsión O7-N5=O6 1349,9 Estiramiento simétrico H10C8H11 1375,2 Aleteo
H12H13H14C9 1425,8 Aleteo H10C8H11 1460,6 Tijereteo
H12H13H14C9 1496,6 Torsión H12H13H14C9 1503,5 Balanceo
O7-N5=O6 1804,2 Estiramiento asimétrico C1=O2 1875,7 Estiramiento
H10C8H11 3045,5 Estiramiento simétrico H12H13H14C9 3050,4 Estiramiento simétrico
H10C8H11 3074 Estiramiento asimétrico H12H13H14C9 3118 Estiramiento asimétrico
CPAN Asignación Frec. (cm-1) Modo
O3-O4 1000,1 Estiramiento H11C8= C9H12 1007,5 Aleteo
H13H14H15C10-C9H12 1080,1 Aleteo H13H14H15C10-C9H12 1116,3 Torsión
H11C8-C1 1210,4 Tijereteo H11C8 1311,2 Balanceo
H11C8= C9H12 1343,4 Tijereteo O7-N5=O6 1353,7 Estiramiento simétrico
H13H14H15C10 1413,4 Aleteo H13H14H15C10 1476,1 Tijereteo H13H14H15C10 1482,4 Torsión H11C8= C9H12 1699,5 Estiramiento
O7-N5=O6 1806,1 Estiramiento asimétrico C1=O2 1839,7 Estiramiento
H13H14H15C10 3022 Estiramiento simétrico H13H14H15C10 3066 Estiramiento H13H14H15C10 3112,4 Estiramiento asimétrico H11C8= C9H12 3168,2 Estiramiento simétrico H11C8= C9H12 3184,4 Estiramiento asimétrico
PnBN Asignación Frec. (cm-1) Modo
O3-O4 981,2 Estiramiento C1C8C9 1045,2 Estiramiento simétrico C1C8C9 1053,6 Estiramiento simétrico
H15H16C10 1128,7 Torsión H15H16C10 1133,9 Balanceo C8C9C10 1257,5 Torsión H11H12C8 1327,7 Aleteo H11H12C8 1331,2 Torsión O7-N5=O6 1351,5 Estiramiento simétrico
PiBN Asignación Frec. (cm-1) Modo
O3-O4 982,2 Estiramiento H12H13H14C9 1115,6 Balanceo H15H16H17C10 1185,5 Torsión
H11C8 1343,8 Balanceo O7-N5=O6 1345,4 Estiramiento simétrico
H12H13H14C9 1406,5 Aleteo H15H16H17C10 1428,7 Aleteo H15H16H17C10 1492 Balanceo H12H13H14C9 1492,7 Balanceo

12
H13H14C9 1401,4 Aleteo H15H16C10 1422 Aleteo H11H12C8 1457,7 Tijereteo H15H16C10 1494,2 Balanceo H15H16C10 1502,9 Torsión H13H14C9 1510,8 Tijereteo O7-N5=O6 1803,3 Estiramiento asimétrico
C1=O2 1875,3 Estiramiento H15H16H17C10 3025,5 Estiramiento simétrico
H11H12C8 3034,6 Estiramiento simétrico H13H14C9 3049,8 Estiramiento simétrico H11H12C8 3074,6 Estiramiento asimétrico H13H14C9 3097,3 Estiramiento asimétrico
H15H16H17C10 3098,5 Estiramiento asimétrico
H15H16H17C10 1504,9 Balanceo H12H13H14C9 1513,8 Torsión
O7-N5=O6 1804 Estiramiento asimétrico C1=O2 1865 Estiramiento
H15H16H17C10 3037,8 Estiramiento simétrico H12H13H14C9 3040,4 Estiramiento simétrico
H11C8 3070,8 Estiramiento H15H16H17C10 3104,2 Estiramiento asimétrico H12H13H14C9 3106,5 Estiramiento asimétrico
C10-C8-C9 3112,7 Torsión C10-C8-C9 3118,9 Tijereta
MPAN Asignación Frec. (cm-1) Modo
O3-O4 995,7 Estiramiento H12C9H11 1017,4 Balanceo
H13H14H15C10 1078,4 Balanceo H13H14H15C10 1078,7 Aleteo
C8-C1=O2 1315,4 Estiramiento O7-N5=O6 1353,5 Estiramiento simétrico
H13H14H15C10 1421,7 Aleteo H12C9H11 1450 Tijereteo
H13H14H15C10 1474,4 Torsión H13H14H15C10 1494,2 Balanceo H11H12C9=C8 1692,3 Estiramiento
O7-N5=O6 1808 Estiramiento asimétrico C1=O2 1837,6 Estiramiento
H13H14H15C10 3041,5 Estiramiento simétrico H13H14H15C10 3125,7 Estiramiento asimétrico
H12C9H11 3155,6 Estiramiento simétrico H12C9H11 3249 Estiramiento asimétrico
PnVN Asignación Frec. (cm-1) Modo
O3-O4 976,7 Estiramiento C10-C11 1015,8 Estiramiento
H12H13C8-H14H15C9-H16H17C10-C11
H18H19H20 1065,5 Balanceo
H16C17C10 1239,1 Balanceo H12H13C8-H14H15C9-
H16H17C10 1296,5 Aleteo
H12H13C8-H14H15C9-H16H17C10-C11
H18H19H20 1315,4 Torsión
H12H13C8-H14H15C9-H16H17C10 1335,2 Torsión
O7-N5=O6 1350,1 Estiramiento simétrico H12H13C8 1375,3 Aleteo
H12H13C8-H14H15C9-H16H17C10-C11
H18H19H20 1404,5 Aleteo
H18H19H20C11 1419,4 Aleteo H12H13C8 1457,6 Tijereteo
H18H19H20C11 1489,7 Balanceo H14H15C9 1498,3 Tijereteo
H18H19H20C11 1500,1 Torsión H14H15C9-H16H17C10 1512,7 Tijereteo
O7-N5=O6 1803,2 Estiramiento asimétrico C1=O2 1875,1 Estiramiento
H16C17C10 3008,9 Estiramiento simétrico H18H19H20C11 3025 Estiramiento simétrico
H12H13C8-H14H15C9 3032,1 Estiramiento simétrico H16H17C10 3033,4 Estiramiento asimétrico
H12H13C8-H14H15C9-H16H17C10 3042,5 Estiramiento simétrico
H12H13C8-H14H15C9 3081,3 Estiramiento asimétrico H12H13C8-H14H15C9-
H16H17C10 3089,3 Estiramiento asimétrico
H18H19H20C11 3093 Estiramiento asimétrico

13
En la siguiente figura se presentan los espectros infrarrojos simulados (ver fig. 3) y la numeración
de las bandas mayores a 900 cm-1 para las moléculas MoPAN, APAN, PPN, PnBN, CPAN, PiBN,
MPAN y PnVN.
Figura 2. Gráfica de los espectros IR, de los compuestos: MoPAN, APAN, PPN, PnVN, CPAN, MPAN, PnBN y PiBN.

14
3.2. Orbitales naturales de enlace
3.2.1 Orbitales moleculares
A continuación, se presentan los orbitales moleculares HOMO-LUMO y sus descripciones de las
moléculas APAN, CPAN y MPAN.
APAN
Orbital Número Figura Descripción
LUMO
38
Orbital s* de los átomos O=C-CH2
37
Orbital !* presente en el fragmento
de la cadena O=C-CH=CH2
36
Orbital !* del grupo funcional NO2
35
Orbital tipo p presente en los O del
carbinilo y el enlace peroxido, en
combinación con un orbital tipo !*
presente en O=C-O
HOMO
34
Orbital tipo n del grupo de peroxido
O-O
33
Orbital d enlazante de O-O

15
APAN
Orbital Número Figura Descripción
32
Orbital tipo par libre (n) del grupo de
nitro NO2
31
Orbital tipo P del grupo de Nitro NO2
30
Orbital tipo ! del grupo de peroxido
O-O
CPAN
Orbital Número Figura Descripción
LUMO
42
Orbital s* presente en O=C-CH2
41
Orbital tipo !* presente en CH2=CH,
combinado con un orbital !* de dos
nodos en O-C=O
40
Orbital tipo !* del grupo funcional
NO2
39
Orbital tipo !* de tres nodos en CH3-
CH=C=O

16
CPAN
Orbital Número Figura Descripción
Orbital tipo P enlazante en el grupro
en CH3-CH=C=O
HOMO
37
Orbital tipo ! enlazante en el grupro
CH2=CH
36
Orbital de tipo par libre (n) del grupo
NO2
35
Orbital tipo P del grupo de nitro NO2
34
Orbital tipo P del grupo de peróxido, O-O, en combinación con un orbital s del grupo C=C
MPAN
Orbital Número
Figura Descripción
LUMO
Orbital s* presente en el gupo peróxido O-O, en combinación con un orbital ! enlazante del grupo C=CH2
41
Está presente un orbital tipo !* presente en CH2=C-CH3, combinado con un orbital pi enlazante O-C=O

17
MPAN
Orbital Número
Figura Descripción
40
Orbital tipo ! * antienlazante presente en el NO2
39
Orbital tipo ! enlazante en el grupo CH2C-
HOMO
38
Orbital de tipo par libre (n) del grupo NO2, en combinación con un orbital !* del grupo O-O
37
Orbital tipo ! enlazante en el grupo CH2=CH
36 Orbital de tipo par libre (n) del grupo NO2
35
Orbital de tipo P del grupo NO2
34
Orbital d enlazante de grupo O-O

18
3.2.2 Orden de enlace
En la tabla 4 se presentan los orden de enlace de las moléculas MoPAN, APAN, PPN, PnVN, CPAN, MPAN, PnBN y PiBN
Tabla 3. Resultado del Orden de enlace para la moléculas MoPAN, APAN, PPN, CPAN, MPAN, PiBN, PnBN y PnVN. Los valores se aproximaron a la unidad.
MOPAN APAN PPN CPAN MPAN PIBN PnBN PnVN
Enlace Orden
de
enlace
Enlace Orden
de
enlace
Enlace Orden de
enlace Enlace
Orden de
enlace
Enlace Orden de
enlace Enlace
Orden de
enlace
Enlace Orden
de
enlace
Enlace Orden de
enlace
C1=O2 2 C1=O2 1,9 H10-C8 1,0 C1=O2 1,9 C1=O2 1,9 C8-H11 1,0 H1-C2 1,0 H12-C8 1,0
C1-O3 0,9 C1-O3 0,9 C8-H11 1,0 C1-O3 0,9 C1-O3 0,9 C8-C1 1,0 C2-H3 1,0 C13-H8 1,0
C1-O8 1,0 C1-C8 1,0 C8-C1 1,0 C1-C8 1,0 C1-C8 1,0 C8-C10 1,0 C8-C1 1,0 C8-C1 1,0
O3-O4 1,0 O3-O 4 1,0 C8-C9 1,0 O3-O4 1,0 O3-O4 1,0 C8-C9 1,0 C8-C9 1,0 C8-C9 1,0
O4-N5 0,9 O4-N5 0,9 C1=O2 1,9 O4-N5 0,9 O4-N5 0,9 C1=O2 1,9 C1=O2 1,9 C1-O2 1,9
N5=O6 1,7 N5=O6 1,7 C1-O3 0,9 N5=O6 1,7 N5=O6 1,7 C1-O3 0,9 C1-O3 0,9 C1-O3 0,9
N5-O7 1,0 N5-O7 1,0 O3-O4 1,0 N5-O7 1,0 N5-O7 1,0 O3-O4 1,0 O3-O4 1,0 O3-O4 1,0
O8-C9 1,0 C9-H11 1,0 O4-N5 0,9 C8-H11 1,0 C8=C9 1,9 O4-N5 0,9 O4-N5 0,9 O4-N5 0,9
C9-H10 1,0 C9-H12 1,0 N5=O6 1,7 C8=C9 1,9 C8-C10 1,0 N5=O6 1,7 N5=O6 1,7 N5=O6 1,7
C9-H11 1,0 C9-C8 1,9 N5-O7 1,0 C9-H12 1,0 C9-H11 1,0 N5-O7 1,0 N5-O7 1,0 N5-O7 1,0
C9-H12 1,0 C11-H12
1,0 C9-H12 1,0 C9-C10 1,0 C9-H12 1,0 C10-H17 1,0 C9-H14 1,0 C9-H14 1,0
C9-H14 1,0 C10-H14 1,0 C10-H15 1,0 C10-H16 1,0 C9-H13 1,0 C9-H15 1,0
C9-H13 1,0 C10-H13 1,0 C10-H14 1,0 C10-H15 1,0 C9-C10 1,0 C9-C10 1,0
C10-H15 1,0 C10-H13 1,0 C9-H12 1,0 C10-H15 1,0 C10-H17 1,0
C9-H14 1,0 C10-H17 1,0 C10-H16 1,0
C9-H13 1,0 C10-H16 1,0 C10-C11 1,0
C11-H19 1,0
C11-H18 1,0

19
3.3. Termoquímica
Se presentan los valores termoquímicos estimados de las moléculas MoPAN, APAN, PPN, PnVN,
CPAN, MPAN, PnBN y PiBN. Para obtener los resultados expuestos en la tabla 5. Se siguió la
metodología descrita en el anexo 3.
Tabla 4. Valores termoquímicos estimados para las moléculas MoPAN, APAN, PPN, PnVN, CPAN,
MPAN, PnBN y PiBN obtenidos a través de la teoría funcional de la densidad. Las unidades de energía
están dadas en kcal/mol-1
Molécula ΣD0 ∆ f H° (0 K) ∆ f H° (298 K) ∆ f G° (298 K) B3LYP MoPAN
1052,4 -91,0 -95,7 -67,9
M06-2X 1057,2 -95,8 -100,8 -73,8 B3LYP APAN
1101,1 -28,8 -32,8 [-34,8] -5,5
M06-2X 1102,1 -29,8 -34,1[-34,8] -7,3 B3LYP PPN
1232,6 -57,0 -61,6[-62,9] -32,8
M06-2X 1234,4 -58,8 -62,5[-62,9] -36,1 B3LYP PnBN
1506,7 -57,9 -64,9 -34,1
M06-2X 1512,1 -63,3 -71,1 -38,1 B3LYP CPAN
1380,0 -34,4 -39,7 -10,0
M06-2X 1382,8 -37,2 -42,7 -13,5 B3LYP PiBN
1505,7 -56,9 -63,1 -32,3
M06-2X 1512,1 -63,3 -70,5 -40,7 B3LYP MPAN
1378,1 -32,5 -37,9 -8,1
M06-2X 1382,3 -36,7 -42,3 -13,1 B3LYP PnVN
1781,0 -58,9 -66,5 -32,6
M06-2X 1787,9 -65,8 -74,2 -43,3

20
4. DISCUSIÓN
Las estructuras moleculares de equilibrio de una familia de 8 moléculas derivadas del nitrato del
peroxiacilo conocido como PAN fueron estudiadas empleando la teoría funcional de la densidad,
método muy utilizado en el estudio de esta familia. [5][6][7]
Los valores obtenidos de longitudes de enlace, ángulos, y diedros se encuentran consignados en la
tabla 2. Las moléculas estudiadas en este trabajo presentan el grupo -C(O)OONO2 con diversos
grupos R tales como R1= O-CH3; R2= CH=CH2; R3= CH2-CH3; R4= (CH2)2CH3; R5= (CH=CH)-
CH3; R6= CH-(CH3)2CH3); R7= C=CH2-CH3; R8= (CH2)3CH3.
Branko S. Jursic (1996) estudió del PAN la energía de disociación y geométria del enlace entre el
peróxido y el óxido de nitrógeno (IV), comparando métodos ab intio convensionales (ROHP y
MP2), local (SVWN) e híbridos (BhandH, BHandHLYP y B3LYP) con el conjunto de base 6-
31+G(d) y 6-311+G (3df, 3pd). Concluye que los dos primeros métodos no se ajustan para el enlace
O-O-NO2. El método de tipo local podría funcionar en la parte geométrica, pero en cuanto a la
energía de disociación los valores son muy grandes respecto a los experimentales y los métodos
híbridos BhandH, BHandHLYP, aunque sus valores son cercanos a los valores experimentales aun
no son aceptados. Y como método más confiable para la determinación de la energía de disociación
es el modelo teórico B3LYP/6-311+G(3df, 3pd). [8]
Los resultados reportados en la literatura para el PAN estudiado por Byung Jin Mhin y
colaboradores en el año 2000 a través de estimaciones computacionales muestran que las
características estructurales de PAN obtenida a los niveles de teoría B3LYP y MP2 en
combinación con el conjunto de base 6-31G(d) indican una estructura muy flexible destacando dos
conformaciones del tipo cis y trans, la primera presenta un angulo diedro C(O)OO muy pequeño
menor de 10º y la segunda conformación presenta un ángulo de 178º. También destacan que las
energías obtenidas para la conformación más estable es la estructura trans con una diferencias de
3,64 kcal/mol frente a la estructura cis, [9] sin embargo el pobre nivel de teoría pueden dar un
valor dudoso para estas estructuras.
En el 2004, Joseph Francisco [10] estudió la misma estructura PAN empleando un nivel de teoría
más complejo CCSD(T) en combinación con un conjunto de base tipo Dunning cc-pVDZ. En sus
estudios comparó la estructura PAN con los datos experimentales del grupo OONO2 con el
experimental reportado para el ácido pernítrico HOONO2 [11] encontrando excelente concordancia

21
entre estos compuestos. Miller y colaboradores estudiaron la molécula PAN al nivel teórico
B3LYP/6-311++G(3df,3pd).[12] Sus resultados muestran que el nivel de teoría puede estimar las
longitudes de enlace para el O-O que solo difieren en 0,001 Å al igual que el enlace N-O de 1,198
Å con una diferencia de 0,002 Å al compararlos con estructuras análogas.
Wen Mei Wei y colaboradores (2005) estudian las geometrías y energías relativas de 11 isómeros
de PAN, ocasionado por la rotación interna del grupo O-N-O-O, coinciden con autores
anteriormente mensionados que la conformación mas estable es cis-PAN. [13]
Con base a las referencias mostradas anteriormente se concluye que las estructuras más estables
son las que se encuentran en conformación cis y la metodología empleada es correcta para estudiar
otras moléculas análogas a PAN.
En las moléculas estudiadas aquí, el grupo peroxinitrato de acilo fue etiquetado de la siguiente
forma O2C1O3O4N5O6O7, como se observa en la figura 2 estas estructuras presentan conformación
cis, esta característica se debe principalmente a los cinco átomos electronegativos consecutivos
O3O4N5O6O7. [14]
Al comparar los datos de longitudes y ángulos de enlace obtenidos con los funcionales B3LYP y
M06-2X y el conjunto de base 6-311++G(3df,3pd) no se encuentran diferencias muy notables en
estos parámetros con valores de incertidumbre de ± 0,012 Å y ± 0,4º respectivamente. Sin embargo
Hermann y colaboradores indican que la distancia de enlace entre O-N son extremadamente largas,
y se le atribuye la baja estabilidad térmica de las especies de los nitratos de peroxiacilo que oscilan
entre 1,49 Å - 1,53 Å, en nuestro caso, los valores encontrados entre 1,50 Å - 1,52 Å con el nivel
de teoría B3LYP y 1,44 Å - 1,45 Å con M06-2X. B3LYP se ajustan muy a bien a lo reportado. A
esto se le atribuye la baja estabilidad térmica de las especies de los nitratos de peroxiacilo. [15]
Los datos estimados a los niveles de teoría mencionados se presentan en la tabla 2, aquí se
seleccionaron en esta tabla los ángulos diedros más importantes que inciden en la posición de los
grupos complementarios, respecto al grupo funcional -C(O)O2NO2.
Al evaluar el comportamiento sobre el plano de los átomos que conforman cada molécula se
destaca que APAN, MPAN y CPAN son moléculas planas con un doble enlace en C8=C9. Las
moléculas con mayor número de átomos de carbono en este estudio computacional son PiBN,
PnBN y PnVN, en la figura 1 y tabla 2 se puede observar que la distribución en el espacio de los

22
grupos R son típicos de moléculas orgánicas, conservando distancias y ángulos de enlace
característicos C-C y C-H.
APAN y PPN fueron estudiados por Badenes y colaboradores [16] [17], su analisís muestran que
la estructura más estable presentan la conformación cis al calcular las energías de multiples
esturcturas al variar ángulos y diedros de enlaces empleando el nivel de teoría B3LYP/6-
311++G(3df,3pd). Los datos reportados en estas publicaciones fueron exactamente los mismos que
se reportan en este trabajo, lo anterior es de esperar debido a la reproducibilidad de los métodos
en la química cuántica.
El estudio vibracional para los derivados de nitratos de peroxiacilo estudiados en este trabajo
presentan bandas del grupo - C(O)OONO2 que fueron común en todas las moléculas de la familia
de los 8 compuestos. Las bandas numeradas del 2-5 en la figura 3 de la simulación de los espectros
infrarrojo para MoPAN, APAN, PPN, PnVN, CPAN, MPAN, PnBN y PiBN nos muestran este
detalle lo cual esta acorde con bandas reportadas por compuestos análogos. [18] [19] [20]
En la figura 3, la etiqueta 2 que se encuentra entre 1837 cm-1 y 1878 cm-1 pertenecen a la banda
del grupo C=O asociado a una vibración de estiramiento. Para el grupo funcional O=N-O se
encontraron dos frecuencias que oscilan entre 1803 cm-1 a 1815 cm-1 (etiqueta 3) y 1350 cm-1 a
1359 cm-1(etiqueta 4) correpondientes a un estiramiento asimétrico y simétrico de los átomos de
oxigeno sobre el nitrógeno. [21] En la región de las huellas dactilares del espectro se encuentra
una banda correspondiente al estiramiento del enlace O-O con valores que están entre 972 cm-1 y
1000 cm-1 (etiqueta 5). [22]
Una completa asignación de las frecuencias vibracionales para las moleculas MoPAN, APAN,
PPN, PnVN, CPAN, MPAN, PnBN y PiBN se muestran en la tabla 3, estas son consistentes con
lo reportado por Badenes y colaboradores para el PPN a excepción de la asignación de la banda en
1810 cm-1 que está erroneamente reportada al estar cruzada con la banda en 1854 cm-1. [17]
Los cálculos de orbitales naturales de enlace nos proporcionan información molecular valiosa
basada en la teoría de orbitales moleculares (TOM), una exploración y descripción de los orbitales
más cercanos a la barrera HOMO-LUMO se presentan en la sección de resultados para las
moléculas que tienen sistemas p correspondientes a hibridaciones del tipo sp2 como lo son APAN,
CPAN y MPAN. Se seleccionó un espacio activo correspondiente a 5 orbitales HOMO y 4
orbitales LUMO para un total de 9 orbitales de un espacio activo (EA) en donde se identificaron
orbitales del tipo s, s*, p, p*, d, pares libres (n) y orbitales p tipo P los cuales son muy útiles en

23
cálculos de la teoría funcional de la densidad dependientes del tiempo (TD-DFT) que nos permiten
estimar transiciones electrónicas. Este tipo de estudios son muy complejos y no se realizaron en
este trabajo. Se pudo identificar orbitales en el espacio activo seleccionado que coinciden con
trabajos anteriores reportados en la literatura para moléculas como los peroxinitratos de
alquilo.[23] En la tabla 4 se presentan los resultados para los cálculos del orden de enlace de las
moléculas MoPAN, APAN, PPN, PnVN, CPAN, MPAN, PnBN y PiBN. Estos resultados nos
ayudan a tener una correcta descripción de la naturaleza del enlace químico correspondiente a las
moléculas estudiadas, podemos apreciar que los valores obtenidos están acordes con las estructuras
de equilibrio obtenidas. No se observó ninguna anomalía en los valores reportados aquí.
Las entalpías de formación estimadas para los compuestos MoPAN, APAN, PPN, PnBN, CPAN,
PiBN, MPAN y PnVN se presentan en la Tabla 5. Los valores reportados han sido estimados,
partir de las energías con los métodos híbridos B3LYP/6-311++G(3df,3pd) y M06-2X/6-
311++G(3df,3pd). Todos los datos necesarios para calcular las entalpías y la energía libre de Gibbs
se presentan en el anexo 3.
Se puede apreciar que al comparar los métodos B3LYP y M06-2X los valores de entalpías de
formación a 298K obtenidos no presentan una diferencia significativa de forma general. Sin
embargo, si comparamos los resultados obtenidos de valores de entalpías de formación a 298 K
con los reportados en la literatura para APAN de -34,8 kcal/mol y PPN de -62,9 kcal/mol, el
funcional M06-2X estima valores de -34,1 kcal/mol y -62,5 kcal/mol para el APAN los cuales son
excelentes frente a los reportados por Badenes y colaboradores estimados con niveles teóricos de
alta precisión como lo son los métodos compuestos Gaussian n, G3MP2 (ver tabla 5). En general
los parámetros termoquímicos estimados presentan valores de entalpías de formación que
incrementan con la complejidad molecular en un rango de -32,8 kcal/mol hasta -100,7 kcal/mol
para MoPAN, PnVN, CPAN, MPAN, PnBN y PiBN, los valores reportados aquí para estas
moléculas son nuevos y pueden ser tomados como valores de refencia.

24
5. EVALUACIÓN Y CUMPLIMIENTOS DE OBJETIVOS
Para la evaluación y cumplimiento de los objetivos expuestos en este informe se intentó dar
respuesta a la siguiente pregunta de investigación: ¿Se puede emplear la teoría funcional de la
densidad para estimar las estructuras de equilibrio, las frecuencias vibracionales, explorar los
orbitales molecualres y estimar los parámetros termoquímicos de compuestos derivados de los
nitratos de peroxiacilo?
Fue posible determinar los parámetros estructurales, frecuencias vibracionales, orbitales naturales
de enlace y las propiedades termoquímicas (energías de atomización, entalpías de formación y
energía libre de Gibbs) a través de la teoría funcional de la densidad, empleando los funcionales
B3LYP y M06-2X en combinación con el extenso conjunto de base 6-311++G(3df,3pd). Los
resultados indican que los dos funcionales reproducen buenos valores corroborados con otros
autores que han empleado esta metodología. Los valores obtenidos de las entalpías de formación
y energías libres de Gibbs indican que la formación de las moléculas estudiadas son
energéticamente favorable.
La mayoría de los valores estimados son nuevos y podrían considerarse como referencia de partida
para futuras investigaciones de los nitratos de peroxiacilo.
Según lo propuesto anteriormente se cumple con el objetivo principal de aplicar la teoría funcional
de la densidad al estudio de las propiedades estructurales, vibracionales y termoquímicas de los
compuestos MoPAN, APAN, PPN, PnBN, CPAN, PiBN, MPAN y PnVN.

25
6. CONCLUSIONES
Se han empleado los funcionales B3LYP y M06-2X para estudiar 8 miembros de la familia de
nitratos de peroxiacilo, peroxinitrato de metoxiformilo (MoPAN), peroxinitrato de acriloilo
(APAN), (PPN), peroxinitrato de n-butirilo (PnBN), (CPAN), (PiBN), (MPAN) y peroxy-n-
valeryl nitrate (PnVN).Las estructuras de equilibrio y las frecuencias vibracionales obtenidas son
consistentes con los datos experimentales disponibles de compuestos relacionados y los estimados
teoricamente encontrados en la literatura. Nuestros resultados muestran que el grupo sustituyente
no afecta la conformación cis caracteristica de estos compuestos C(O)OONO2. Los espectros
infrarrojo simulados muestran bandas caracteristicas en esta familia y pueden ser usados para su
identificación experimental.
Una exploración de los orbitales moleculares fueron realizadas para las moleculas estudiadas aquí,
sin emabrgo consideramos de especial importancia reportar las moleculas con sistemas p de
hidración sp2, de igual forma los ordenes de enlaces calculados son consistentes con las estructuras
moleculares estudiadas. Los valores reportados en este trabajo pueden ser empleados como
referencia ante la ausencia de valores experimentales.
Los valores para las entalpías de formación a 298 K han sido calculados usando la teoría funcional
de la densidad, nuestros valores obtenidos para el funcional M06-2X en tan preciso como los
niveles de teoría Gaussian-n reportados para APAN y PPN.

26
7. ANEXOS
Anexo 1. Descripción de la teoría funcional de la densidad.
Teoría del funcional de la densidad
A finales de 1920 Llewellyn Thomas y Enrico Fermi desarrollaron un modelo de la teoría del
funcional de la densidad electrónica. Cuatro décadas más tarde Pierre Hohenberg, Walter Kohn y
Lu Sham fueron quienes establecieron teóricamente el método. Finalmente en 1998, Walter Kohn
fue premiado con el Nobel de Química por sus contribuciones al desarrollo de esta teoría.
La teoría funcional de la densidad (DFT), está definida como un método diseñado para dar solución
a la ecuación de Schrödinger. No obstante hay otros métodos tradicionales incluidos en las teorías
de las estructura electronica de la materia, como por ejemplo: la teoría de Hartree Fock que tienen
como base la función de onda multielectrónica, este permite obtener información del
comportamiento de sistemas con un tamaño reducido. Sin embargo el limitante mas grande para
su uso, es el alto grado de complejidad para resolver de sus ecuaciones. En relación a la
problemática expuesta la DFT reemplaza el uso de la función de onda por el funcional de la
densidad electrónica, siendo esta mas sencilla de calcular.
Born-Oppenheimer propone que la energía electrónica, Ε! [$(&)], se puede reescribir como una
función de la densidad electrónica, representada en la ecuación 1.
Ε! [$(&)] = Τ[$(&)] + -!"[$(&)] + .[$(&)] + /[$(&)] (1)
Siendo Τ[$(&)] la energía cinética de los e-, -!"[$(&)] la energía de atracción de los e- nucleares,
.[$(&)] es la energía clásica de repulsión e-- e- y /[$(&)] es la energía de inteacción e-- e- no
clásica. Para los miembros 2 y 3 de la ecuación 1. existe solución aplicando las ecuaciones (2) y
(3). De la misma forma la DFT tiene como finalidad efectuar funciones que den solución a los
términos 1 y 4 de la ecuación 1.

27
-!"[$(0)] = −∑ ∫ $!%&'!
$ (0)d0()*+ (2)
.[$(0)] = +,∬
-(%")-(%#)0"#
d0+d0, (3)
Se considera que la energía cinética del electrón es la magnitud mas dificil de calcular, por lo tanto,
el funcional de la energía cinética debe ser muy preciso al momento de ser reemplazado. Para ello
Thomas- Fermi en 1930 describe en la ecuación (ver ecuación 4.) una forma sencilla de calcular
la Τ[$(0)] para un gas con la característica de ser uniformemente infinito.
6[$(0)] = 1+2 (38
,)#$ ∫$(0)
%$ d0 (4)
Se plantea entonces el problema que la ecuación se ajusta sólo para sistemas con densidades
electrónicas uniformes, y no logra incluir de forma correcta los enlaces químicos. Se concluye que
plantear funciones de energía cinética con precisión para aplicaciones moleculares tiene un grado
de dificultad alto. Kohn y Sham revelan que la energía cinética se puede aproximar con tal
precisión por medio de el determinante de Slater (ver ecuación 5. y 6.), considera el sistema como
ficticio, dónde los electrones no interactuan y su densidad electrónica es similar a la función de
onda electrónica. Lo anteriormente descrito fue nombrado como KS-DFT, siendo esta una junto
con DFT una teoría exacta. [3]
63[{∅4}] = − +, ∑ ∫ ∅4∗"
4*+ (0)∇,∅4(0)d0 (5)
Ε67 [$(&)] = Τ[$(&)] − 63[{∅4}] + /[$(&)] (6)

28
Anexo 2. Matriz de orbitales de enlaces naturales (orbitales enlazantes BD y orbitales
antienlazantes BD*) correspondiente a la molécula PPN.

29
Anexo 3. Material instructivo para estimar las entalpías de formación, energías de atomización
totales y energía libre de Gibbs, A partir de los valores obtenidos por Gaussian 09.
Termoquímica en Gaussian
Ejemplo
En el siguiente apartado se mostrará cómo se debe dar uso a los valores termiquímicos obtenidos
por el programa Gaussian 09. Se tomó como ejemplo la molécula de etileno, dónde se extrae un
átomo de hidrógeno (H), del hidrógeno diatómico (H2). Se realizaron cálculos utilizando los
términos, reactivos y productos de la reacción. Ver ecuación 7.
C2H5 +H2 → C2H6 +H (7)
El archivo de salida de Gaussian proporciona datos de todas las especies (Tabla 6). Usted puede
seleccionar los valores de su interés para calcular las propiedades que desee. Las entalpías están
calculadas usando 298K ≈ 298.15K.
Tabla 6. Valores termoquímicos de la reacción del etileno obtenidos de Gaussian 09. Las unidades
están dadas en Hartrees.
Energías libres y entalpías de reacción
Para determinar las entalpías de reacción es necesario calcular los calores de formación, realizando
operaciones de suma y diferencias. Generalmente se usa la ecuación 8.

30
(8)
El programa Gaussian calcula las sumas de las entalpías térmicas y electrónicas. Proporciona de
manera directa la diferencia de las sumas entre los valores correspondientes a los reactivos y los
productos. Esto se debe a que hay una igualdad de átomos en ambos lados de la reacción, y por
con siguiente se puede cancelar la información atómica, y se utiliza los datos moleculares. La
entalpía de reacción se puede calcular utilizando la información de la tabla 6. de la siguiente
manera:
De igual manera se determina la energía libre de Gibbs.
Velocidad de reacción
Para calcular las velocidades de reacción dados por el archivo de salida de Gaussian, se debe
utilizar los valores derivados de la teoría del estado de transición, que se puede ver en la sección
28-8 del libro “Química físíca un enfoque molecular" de D. A. McQuarrie y J. D. Simon.

31
Tabla 7. Energías electrónicas totales HF / STO-3G y suma de energías libres electrónicas y
térmicas para moléculas de átomos y complejo en estado de transición de la reacción FH + Cl →
F + HCl y el análogo sustituido con deuterio.
La ecuación que se debe utilizar corresponde a número 28.72 en el texto (ver ecuación 9).
(9)
Se usará cº =1 para la concentración. Para reacciones simples, se debe conectar los números. Para
ello se debe realizar un cálculo de frecuencias HF/STO-3G, para la reacción FH + Cl → F + HCl
y el deuterio sustituido por el hidrógeno. Como se puede ver la tabla 7. está en resumen las energías
electrónicas totales de cada compuesto. El punto de la geometría final, la energía electrónica,
constantes de fuerza cartesiana son independientes de la masa de los átomos. Sólo el análisis
vibracional y las cantidades derivadas de él dependen de la masa.
Lo primero que se debe hacer para calcular las velocidades de reacción es calcular la energía libre
de activación, ∆‡G º. H corresponde a la reacción de hidrógeno y D para reacción de deuterio).
Ahora se puede calcular las velocidades de reacción, para ellos es necesario tener las constantes,
aquí se utilizó c º=1.k (298, H).

32
La reacción de hidrógeno es más rápida, respecto a la del deuterio, como se esperaría. Los cálculos
realizados se obtuvieron aplicando un nivel de teoría HF/STO-3G.
Estos cálculos se realizaron con fines ilustrativos, puesto que para reacciones mas complejas es
necesario aplicar análisis mas sofisticados, que valore con detalle los efectos de los modos de baja
frecuencia en los estados de transición y el efecto túnel.
• Entalpías y energías libres de formación
Para determinar las entalpías de formación se debe evaluar en dos rangos de temperatura: 0K y
298K. En la energía libre de Gibbs se debe utilizar la entropía.
Para estimar estas propiedades, se debe tener en cuenta algunos componentes. Se utilizará X, M y
yx, siendo: elemento, molécula y número de átomos de X en M, respectivamente.
• Energía de atomización de la molécula S D0 (M):
Esta energía se calcula a partír de la energías totales de la molécula (ℇ0 (M)), la energía zero puntual
de la molécula (ℇZPE (M)) y los átomos que la constituyen.

33
• Calores de formación de los átomos a 0K, ∆fHº (X,0K):
En la tabla 8 se encuentran los valores tabulados recomendados para loa calores de formación de
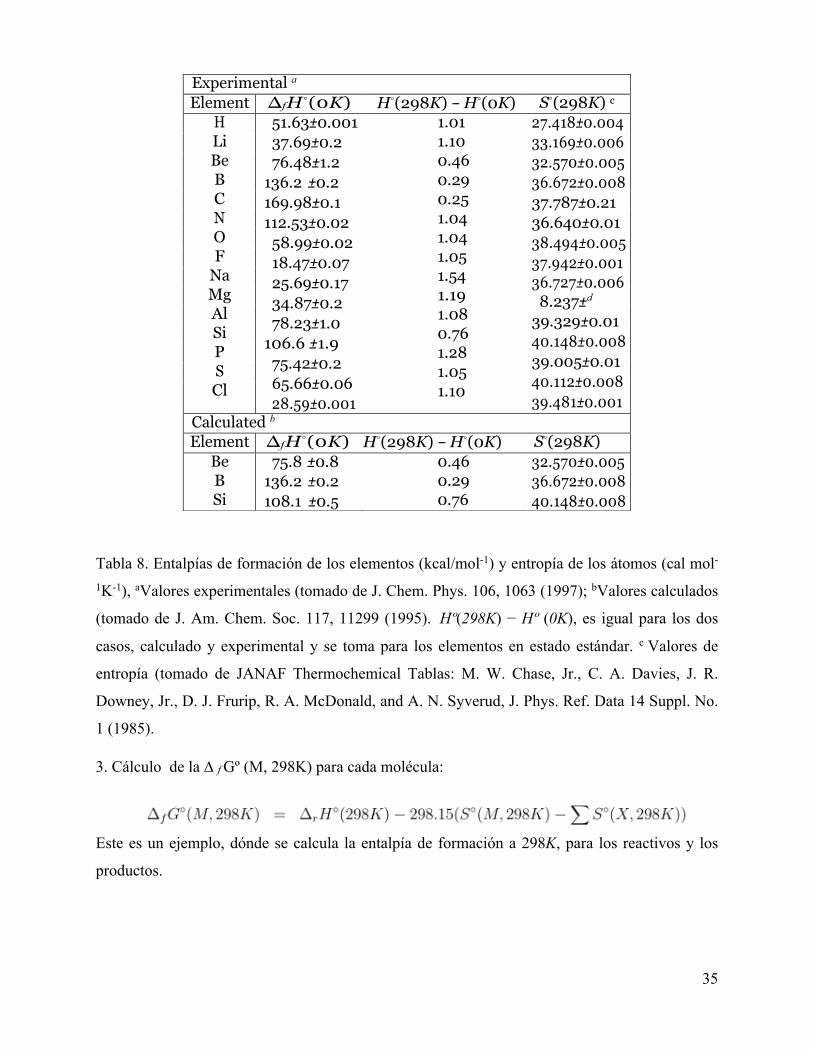
los elementos atómicos a 0K (Primera y segunda fila). Hay por lo menos dos escuelas que
proporcionan valores de calores de atómicos de fomación con información confiable. Hay autores
que prefieren utilizar datos de tipo experimental para los calores de formación, como: “Curtiss, et.
Al., J. Chem. Phys. 106, 1063 (1997)). Otros utilizan valores combinados entre lo experimental y
calculado para lograr una mayor precisión “Ochterski, y col., J. Am. Chem. Soc. 117, 11299.
1995”. En la parte superior de la tabla 8 se encuentran los valores esperimentales y los valores
computacionales en la parte inferior, como es el caso de berilio, boro y silicio con un alto grado
de incertidumbre.
• Correcciones de entalpía de los elementos atómicos, HºX (298K) – HºX (0K)
En las filas 1 y 2 de la tabla 8 se encuentran las correcciones de las entalpías de los elementos
atómicos. Se utilizan para convertir los calores atómicos de formación en 0K a 298,15K, esto se
da para los elementos en su forma estándar. Esto se debe a que no depende de la precisión del calor
de formación del átomo, los valores calculados y experimentales son los mismos.
En general, lo anteriormente descrito no pasa en un archivo de salida de Gaussian en un cálculo
para un átomo aislado en fase gaseosa. Los valores obtenidos se refieren a los estados estándares
de los elementos. En el caso del átomo de hidógeno, su valor es 1,01 kcal/mol. Esto es: (?°8#
(298K) – ?°8# (0K))/2, y no (?°8 (298K) – ?°8 (0K)), que es lo que calcula Gaussian.

34
• Correción de la entalpía para la molécula HºM (298K) – HºM (0K)
Para una molécula, Hcorr - ℇZPE (M), dónde Hcorr es el valor de la “Corrección térmica de la entalpía”
en el archivo de salida de Gaussian. Es necesario realizar la conversión de Hartrees/particula a
kcal/mol. Se debe aplicar el siguiente factor de conversión: 1 Hartree = 627,5095 kcal/mol.
• Entropía de los átomos SºX (298K)
Los valores ubicados en la columna 3 de la tabla 8, fueron tomados de las tablas de JANAF (MW
Chase, Jr., CA Davies, JR Downey, Jr., DJ Frurip, RA McDonald y AN Syverud, J. Phys. Ref.
Data 14 Suppl. No. 1 (1985)). Estos valores se refieren a los estados estándar de los elementos y
no concuerdan con los valores calculados por Gaussian para los átomos aislados en fase gaseosa.
• Entropía de las moléculas SºM (298K)
Gaussian calcula G = H-TS, este valor se situa en la linea denominada, “Corrección térmica de la
energía libre de Gibbs”. La entropía se puede estimar usando S = (H-G)/T.
Uniendo todo lo anteriormente descrito, podemos resumirlo de la siguinete manera:
1. Cálculo de la ∆ f Hº (M, 0K) para cada molécula:
2. Cálculo de la ∆ f Hº (M, 298K) para cada molécula
35
Tabla 8. Entalpías de formación de los elementos (kcal/mol-1) y entropía de los átomos (cal mol-
1K-1), aValores experimentales (tomado de J. Chem. Phys. 106, 1063 (1997); bValores calculados
(tomado de J. Am. Chem. Soc. 117, 11299 (1995).Hº(298K) − Hº (0K), es igual para los dos
casos, calculado y experimental y se toma para los elementos en estado estándar. c Valores de
entropía (tomado de JANAF Thermochemical Tablas: M. W. Chase, Jr., C. A. Davies, J. R.
Downey, Jr., D. J. Frurip, R. A. McDonald, and A. N. Syverud, J. Phys. Ref. Data 14 Suppl. No.
1 (1985).
3. Cálculo de la ∆ f Gº (M, 298K) para cada molécula:
Este es un ejemplo, dónde se calcula la entalpía de formación a 298K, para los reactivos y los
productos.
Experimental a Element ∆f H◦(0K) H◦(298K) − H◦(0K) S◦(298K) c
H 51.63±0.001 37.69±0.2 76.48±1.2
136.2 ±0.2 169.98±0.1 112.53±0.02
58.99±0.02 18.47±0.07 25.69±0.17 34.87±0.2 78.23±1.0
106.6 ±1.9 75.42±0.2 65.66±0.06 28.59±0.001
1.01 1.10 0.46 0.29 0.25 1.04 1.04 1.05 1.54 1.19 1.08 0.76 1.28 1.05 1.10
27.418±0.004 33.169±0.006 32.570±0.005 36.672±0.008 37.787±0.21 36.640±0.01 38.494±0.005 37.942±0.001 36.727±0.006
8.237±d
39.329±0.01 40.148±0.008 39.005±0.01 40.112±0.008 39.481±0.001
Li Be B C N O F
Na Mg Al Si P S Cl
Calculated b Element ∆f H◦(0K) H◦(298K) − H◦(0K) S◦(298K)
Be B Si
75.8 ±0.8 136.2 ±0.2 108.1 ±0.5
0.46 0.29 0.76
32.570±0.005 36.672±0.008 40.148±0.008

36
Inicialmente se debe calcular ∆fHº (0K) para cada una de las especies:
El siguiente paso es calcular el ∆ f Hº (298K):
Para calcular la energía libre de formación de Gibbs, tenemos (los factores de 1000 son para
realizar la conversión)

37
Apéndice
Símbolos
Ce = Contribución a la capacidad calorífica debido al movimiento electrónico
Cr = Contribución a la capacidad calorífica debido al movimiento de rotación
Ctot = Capacidad calorífica total (Ct +Cr +Cv +Ce)
Ct = Contribución a la capacidad calorífica debido a la traslación
Cv = Contribución a la capacidad calorífica debido al movimiento vibratorio
Ee = Energía interna debido al movimiento electrónico
Er = Energía interna debido al movimiento de rotación
Etot = Energía interna total (Et +Er +Ev +Ee)
Et = Energía interna debido a la traslación
Ev = Energía interna debido al movimiento vibratorio
Gcorr = Corrección de la energía libre de Gibbs debido a la energía interna.
Hcorr = Corrección de la entalpía debido a la energía interna
I = Momento de inercia
K = índice de modos vibracionales
N = Número de moles
NA = Número de Avogadro
P = Presión (el valor predeterminado es 1 atmósfera)
R = Constante de gas = 8.31441 J/(mol K) = 1.987 kcal/(mol K)
Se = Entropía debido al movimiento electrónico
Sr = Entropía debido al movimiento rotacional
Stot = Entropía total (St + Sr + Sv + Se)

38
St = Entropía debido a la traslación
Sv = Entropía debido al movimiento vibracional
T = Temperatura (predeterminado es 298.15) V = volumen
qr, qr, (xyz) = Temperatura característica de rotación (en el plano x, y or z)
qv, K = Temperatura característica de vibración K
kB = Constante de Boltzmann = 1.380662 × 10−23 J/K
@n = La energía en ωn = la degradación del n-cimo nivel de energía
An = La degradación del n-cimo nivel de energía Br = symmetry number for rotation
h = Constante de Planck = 6.626176 × 10−34 J s
m = Masa de una molécula
n = Número de partículas (Siempre 1)
qe = Función de partición electronica
qr = Función de partición rotacional
qt = Función de partición traslacional
qv = Función de partición vibratoria
ℰZPE = Energía zero puntual de la molécula
ℰ0 = Energía electronica total, por ejemplo la energía MP2
Hº Sº, Gº = Entalpía estándar, entropía y energía libre de Gibbs – (cada compuesto en estado
estandar a temperatura dada).
∆‡G◦ = Energía libre de activación
c◦ = Concentración (es:1)
k(T) = Rango de reacción de la temperatura

39
BIBLIOGRAFÍA
[1] P. Taylor, A. J. Cohen, and N. C. Handy, “Molecular Physics : An International Journal at
the Interface Between Chemistry and Physics Dynamic correlation Dynamic correlation,”
no. November 2012, pp. 37–41, 2009.
[2] Y. Zhao and D. G. Truhlar, “The M06 suite of density functionals for main group
thermochemistry , thermochemical kinetics , noncovalent interactions , excited states , and
transition elements : two new functionals and systematic testing of four M06-class
functionals and 12 other fun,” pp. 215–241, 2008.
[3] N. Mardirossian and M. Head-Gordon, “Thirty yeras of density functional theory in
computational chemistry: an averview and extensive assessment of 200 density funcionals,”
Mol. Phys., 2017.
[4] M. J. Frisch et al., “Gaussian 09,” Inc., Wallingford CT. 2016.
[5] Xiao-Xia and L. Feng-Ling, “Mechanism for the Gas-Phase Hydrogen Fluoride-Mediated
Decomposition of Peroxyacetyl Nitrate (PAN) Studied by DFT Method,” Int. J. Quantum
Chem., vol. 111, pp. 4020–4029, 2011.
[6] M. P. Badenes, L. L. B. Bracco, and C. J. Cobos, “Theoretical study of the equilibrium
structure, vibrational spectrum, and thermochemistry of the peroxynitrate
CF2BrCFBrOONO2,” J. Phys. Chem. A, vol. 115, no. 26, pp. 7744–7752, 2011.
[7] A. G. Bossolasco, J. A. Vila, M. A. Burgos Paci, F. E. Malanca, and G. A. Argüello, “A
new perfluorinated peroxynitrate, CF3CF2CF 2CF2OONO2. Synthesis, characterization
and atmospheric implications,” Chem. Phys., vol. 441, no. 2, pp. 11–16, 2014.
[8] B. S. Jursic, “Density functional theory and ab initio study of bond dissociation energy for
peroxonitrous acid and peroxyacetyl nitrate,” J. Mol. Struct. THEOCHEM, vol. 370, no. 1,
pp. 65–69, 1996.
[9] B. J. Mhin, W. Y. Chang, J. Y. Lee, and K. S. Kim, “Ab Initio Study of Peroxyacetic Nitric
Anhydride and Peroxyacetyl Radical: Characteristic Infrared Band of Peroxyacetyl
Radical,” J. Phys. Chem. A, vol. 104, no. 12, pp. 2613–2617, 2000.
[10] J. S. Francisco and Y. Li, “Ab initio study of the electronic spectrum of peroxyacetyl

40
nitrate,” J. Chem. Phys., vol. 121, no. 13, pp. 6298–6301, 2004.
[11] E. H. Appelman and D. J. Gosztola, “Aqueous Peroxynitric Acid (HOONO2): A Novel
Synthesis and Some Chemical and Spectroscopic Properties,” Inorg. Chem., vol. 34, no. 4,
pp. 787–791, 1995.
[12] C. E. Miller, J. I. Lynton, D. M. Keevil, and J. S. Francisco, “Dissociation pathways of
peroxyacetyl nitrate (PAN),” J. Phys. Chem. A, vol. 103, no. 51, pp. 11451–11459, 1999.
[13] W. M. Wei, W. Tan, R. H. Zheng, T. J. He, D. M. Chen, and F. C. Liu, “Ab initio studies
of isomerization and dissociation reactions of peroxyacetyl nitrate (PAN),” Chem. Phys.,
vol. 312, no. 1–3, pp. 241–259, 2005.
[14] S. Von Ahsen, H. Willner, and J. S. Francisco, “Thermal decomposition of peroxy acetyl
nitrate CH 3C(O)OONO 2,” J. Chem. Phys., vol. 121, no. 5, pp. 2048–2057, 2004.
[15] A. Hermann et al., “Gas-phase structures of acetyl peroxynitrate and trifluoroacetyl
peroxynitrate,” Inorg. Chem., vol. 40, no. 7, pp. 1672–1676, 2001.
[16] M. P. Badenes and C. J. Cobos, “Ab initio and DFT study of the molecular conformations
and the thermochemistry of the CH2{double bond, long}CHC(O)OONO2 (APAN)
atmospheric molecule and of the CH2{double bond, long}CHC(O)OO and CH2{double
bond, long}CHC(O)O radicals,” J. Mol. Struct. THEOCHEM, vol. 814, no. 1–3, pp. 51–60,
2007.
[17] M. P. Badenes and C. J. Cobos, “Quantum chemical study of the atmospheric
C2H5C(O)OONO2 (PPN) molecule and of the C2H5C(O)OO and C2H5C(O)O radicals,”
J. Mol. Struct. THEOCHEM, vol. 856, no. 1–3, pp. 59–70, 2008.
[18] E. R. Stephens and M. A. Price, “Analysis of an important air pollutant: peroxyacetyl
nitrate.,” J. Chem. Educ., vol. 50, no. 5, pp. 351–354, 1973.
[19] E. Monedero, M. S. Salgado, F. Villanueva, P. Martín, I. Barnes, and B. Cabañas, “Infrared
absorption cross-sections for peroxyacyl nitrates (nPANs),” Chem. Phys. Lett., vol. 465, no.
4–6, pp. 207–211, 2008.
[20] A. G. Bossolasco, F. E. Malanca, and G. A. Argüello, “Peroxy ethoxyformyl nitrate,

41
CH3CH2OC(O)OONO 2. Spectroscopic and thermal characterization,” J. Photochem.
Photobiol. A Chem., vol. 221, no. 1, pp. 58–63, 2011.
[21] J. M. Roberts, “The atmospheric chemistry of organic nitrates,” Atmos. Environ. Part A,
Gen. Top., vol. 24, no. 2, pp. 243–287, 1990.
[22] A. Ramírez-Hernández, C. Aguilar-Flores, and A. Aparicio-Saguilán, “Fingerprint analysis
of ftir spectra of polymers containing vinyl acetate,” DYNA, vol. 86, no. 209, pp. 198–205,
2019.
[23] C. Buendía-Atencio, V. Leyva, and L. González, “Thermochemistry and UV spectroscopy
of alkyl peroxynitrates,” J. Phys. Chem. A, vol. 114, no. 35, pp. 9537–9544, 2010.





























