Embed Size (px)
Citation preview

Imunodeficiência Primária: Abordagem Diagnóstica
Susana Lopes da Silva
Centro de Imunodeficiências Primárias Hospital de Santa Maria, CHLN
Faculdade de Medicina da Universidade de LisboaInstituto de Medicina Molecular
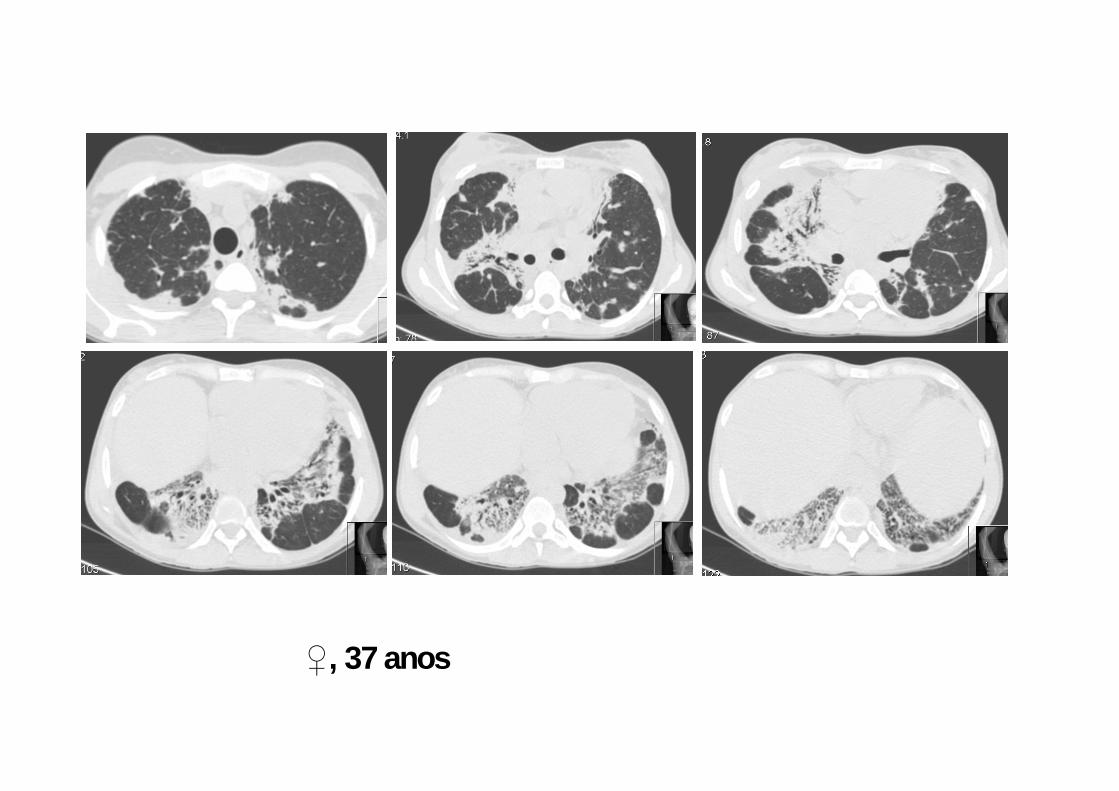
♀, 37 anos
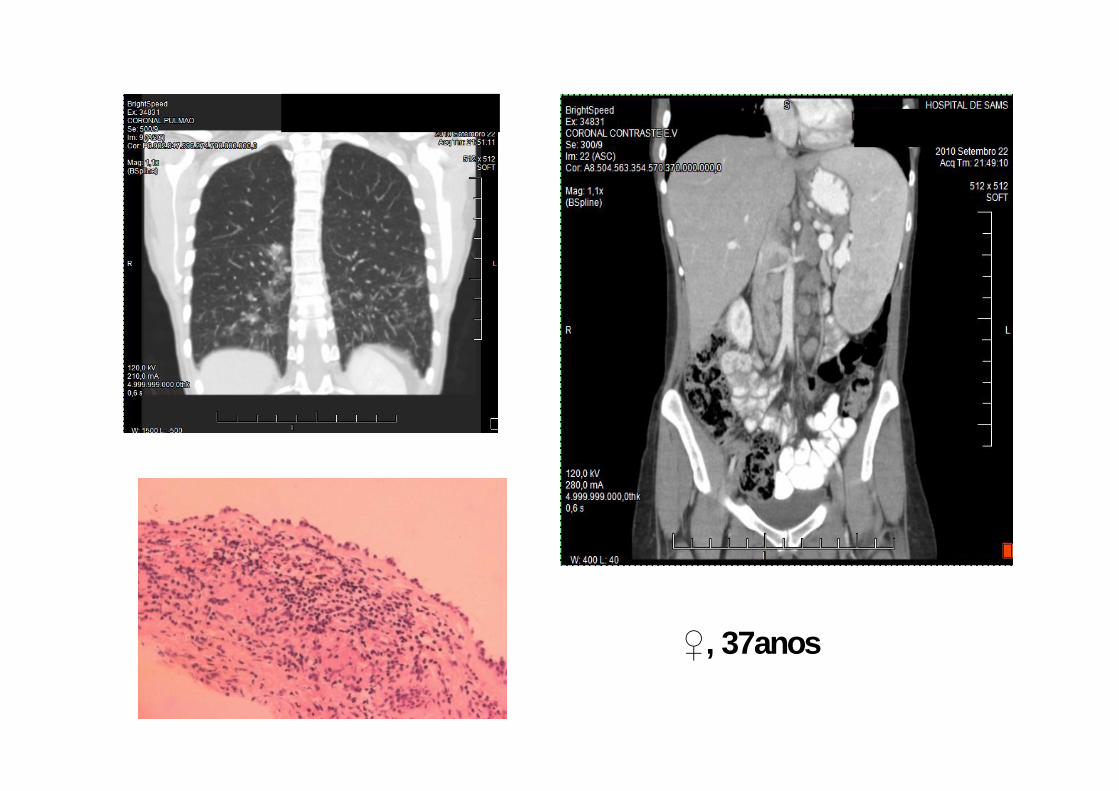
♀, 37anos
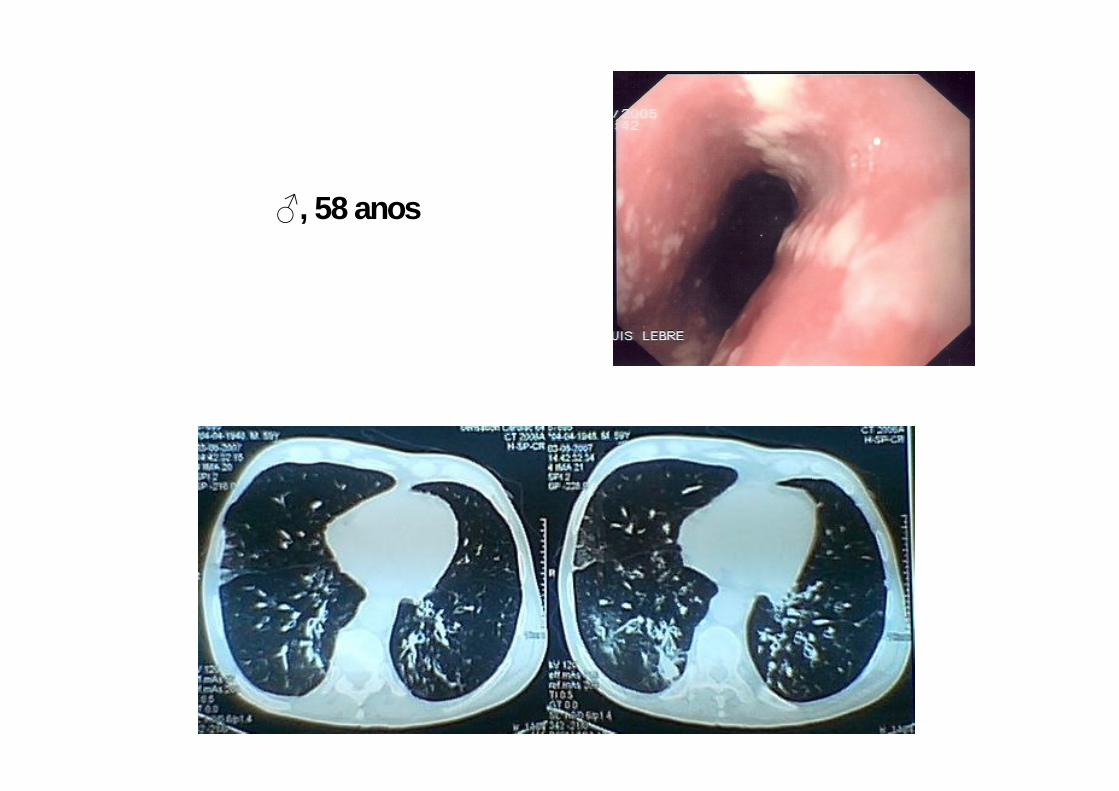
♂, 58 anos
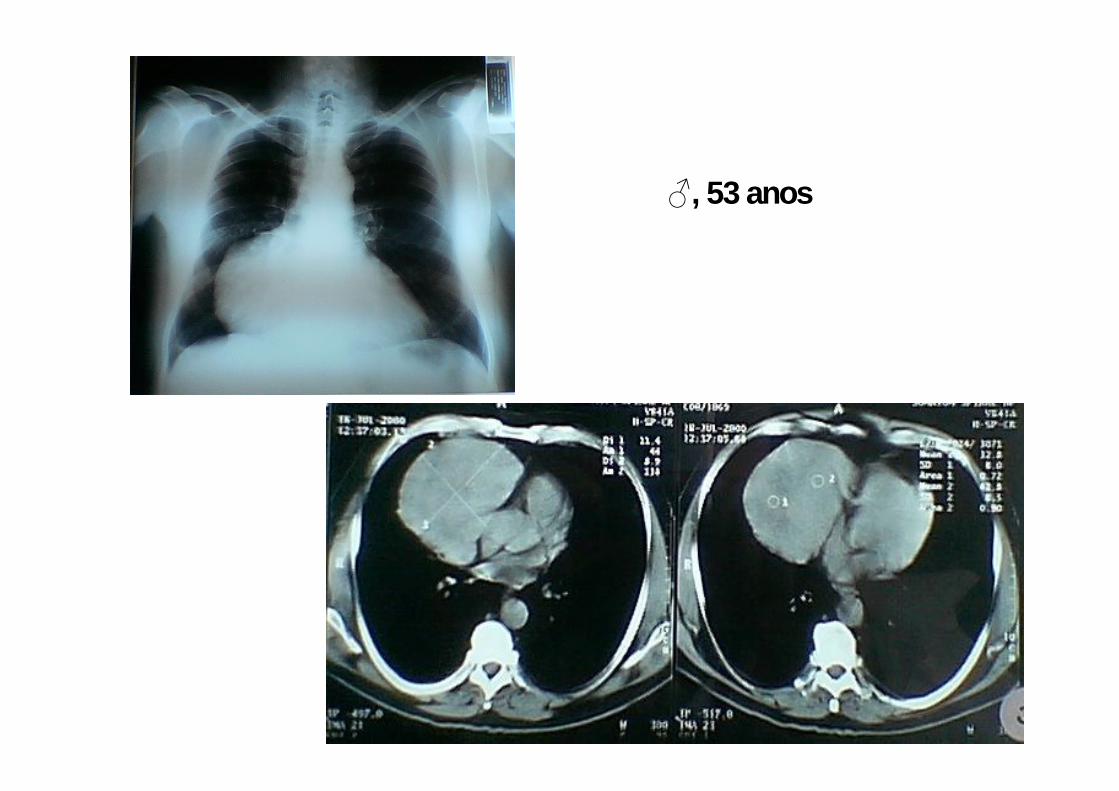
♂, 53 anos
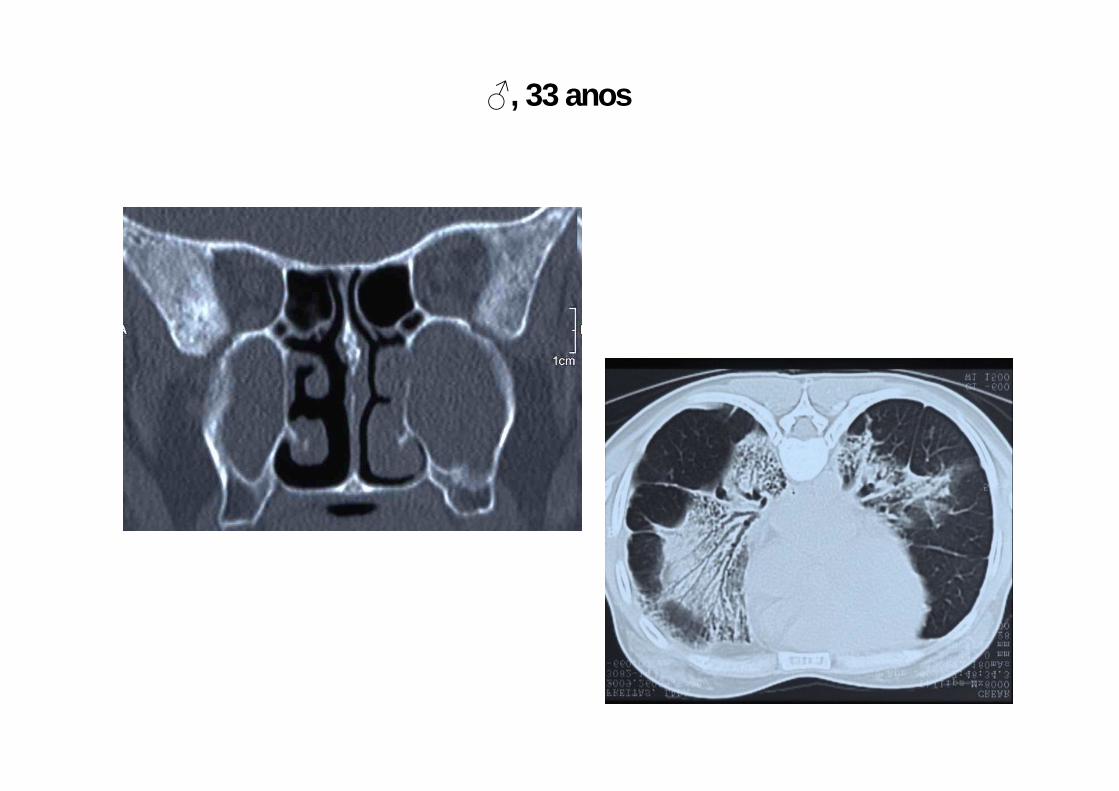
♂, 33 anos

Imunodeficiências Primárias
O que são
Classificação
Sinais de alerta / manifestações clínicas
Investigação diagnóstica / Quando referenciar
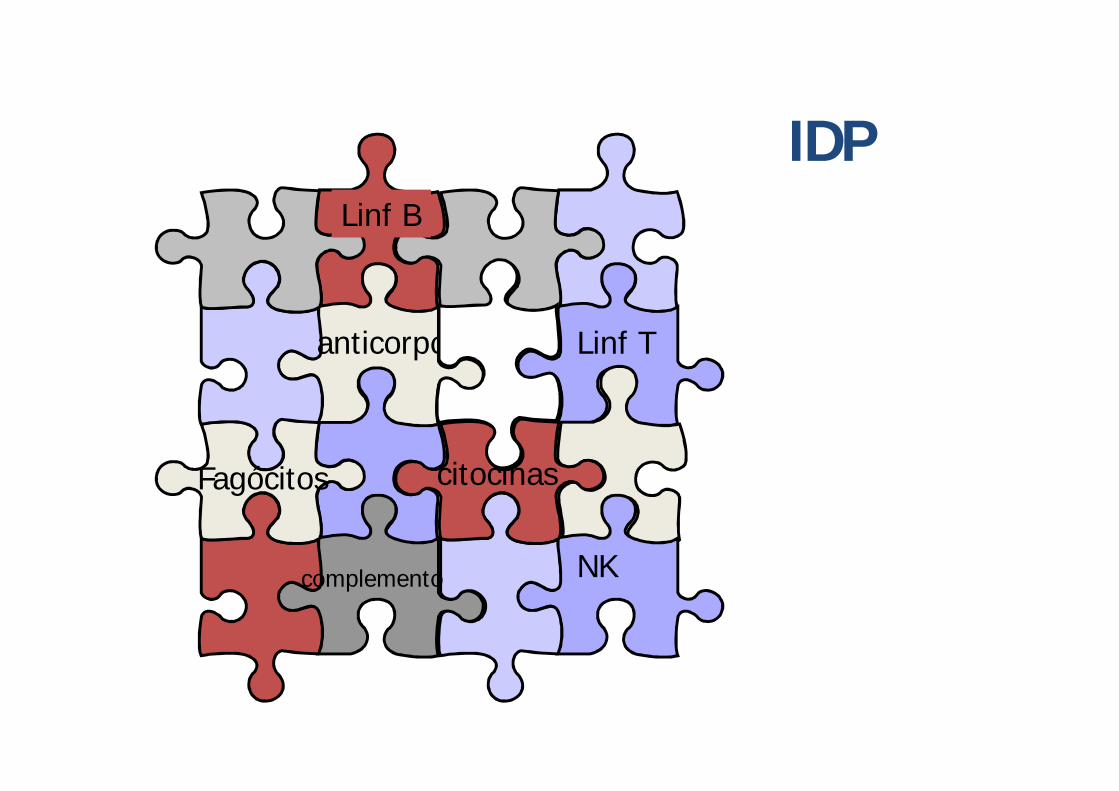
Linf B
Linf T
NKcomplemento
Fagócitos
anticorpos
citocinas
IDP

A. Imunodeficiências combinadas T e B
B. Imunodeficiências por défice predominante de anticorpos
C. Outros síndromes bem definidos com Imunodeficiência
D. Imunodeficiências por défice de regulação imunológica
E. Defeitos congénitos do número e / ou função dos fagócitos
F. Imunodeficiências por défice da imunidade inata
G. Doenças auto-inflamatórias
H. Imunodeficiências por défices do complemento

56% 8%
8%
4%
15%
4%
Predominantly antibody disordersCombined ImmunodeficienciesCongenital defects of phagocyteComplement DeficienciesOther well defined PIDsAutoimmune & immunedysregulation syndromes Autoinflammatory syndromesInate immunity defectsUnclassified PIDs
www.esid.org(n=16547; 30/04/2013)

Considerar a hipótese de IDP!


Infecções bacterianas recorrentes (demonstradas)
>1 Infecção gravee (ex. meningite, osteomielite, pneumonia)
Infecções com apresentação / evolução atípica, sem resposta tratamento habitual
Infecções causadas por agentes oportunistas
Verrugas / molusco contagioso generalizado
Candidíase mucocutânea extensa / persistente
Complicações de vacinas
Abcessos recorrentes / orgãos internos
Diarreia prolongada ou recorrente
Infecção: O paradigma da imunodeficiência
Défice IL12 / IFN
Diagnóstico de IDP

• Alergia respiratória
• Fibrose quística
• Discinésia ciliar
• Eczema
• Drepanocitose
• Cardiopatia congénita
• Nefropatia
• Má higiene
• Desnutrição
• Terapêutica imunossupressora
• VIH
• Esplenectomia
• Diabetes Mellitus
• ...
Será MESMO IDP?Doenças / Factores predisponentes a infecções
Imunodeficiências secundárias
MAIS FREQUENTES no adulto que as IDP!

Diagnóstico de IDP no adulto

• Outros sinais ou sintomas que podem sugerir IDP
– História clinica• Doenças autoimunes (++ citopenias autoimunes ou lúpus)• Diarreia crónica, má absorção / DII (atípica)• Doença obstrutiva pulmonar difícil de tratar• Bronquiectasias sem causa aparente, pneumatocelos,
doença pulmonar intersticial• Granulomas• Timoma• Neoplasias ( ++ linfoma)
• Angioedema (edema sem etiologia alérgica)
Diagnóstico de IDP no adulto

Exame objectivo– Pele:
• palidez; petéquias; • vitiligo;• verrugas múltiplas e/ou exuberantes; molusco contagioso
– Cavidade oral:• candidíase; • gengivoestomatite; periodontite; • aftas recorrentes
– Tecidos linfoides:• ausência de ggls linfáticos ou amigdalas; • linfadenopatias (++ generalizadas);• organomegálias – hepato e/ou esplenomegalia
Diagnóstico de IDP

Diagnóstico de IDP

Investigação laboratorial sequencial
Exames de 1ª linha / exclusão de DD
Orientar de acordo com apresentação clínica
Colocar / escalonar hipóteses de diagnóstico
Diagnóstico de IDP


Exames de primeira linha
Hemograma e contagem diferencial de leucócitos
Doseamento de IgG, IgA, IgM e IgE total
Quantificação de subpopulações linfocitárias(CD3, CD4, CD8, CD19, NK)
C3, C4, CH50
Diagnóstico de IDP


Protocolo 1: Investigação de deficiência de produção de anticorpos ou de complemento
Protocolo 2: Investigação de deficiência de linfócitos T
Protocolo 3: Investigação de neutropénias e de deficiência de fagócitos
Protocolos específicos

Quantificação de populações linfocitárias – linf B
Produção de anticorpos específicos
Avaliação de subclasses de IgG (IgG1 – IgG4)
Hemograma
Doseamento de IgG, IgA, IgM
Suspeita de ID humoral
Diagnóstico de IDP

Doenças Infecciosas
VIH
Rubéola congénita
Infecção CMV congénita
Toxoplasmose congénita
Vírus Epstein-Barr
DD de hipogamaglobulinémia
Doenças genéticas
Ataxia telangiectasia
ID combinada grave (autossómica / ligada X)
Síndrome hiper IgM
Deficiência de transcobalamina II
Agamaglobulinémia ligada ao X
Sindrome linfoproliferativo ligado ao X
Doenças metabólicas
Síndroma do cromossoma 18q
Monossomia 22
Trissomia 8
Trissomia 21HISTÓRIA FAMILIARSEROLOGIA NÃO CHEGA!!

Doença linfoproliferativa
Leucemia Linfocítica Crónica
Linfoma não Hodgking
Neoplasia de linfócitos B
….
Doenças Sistémicas
ID por hipercatabolismo de Ig´s
ID por perdas de Ig´s
Induzida por fármacos
Anti-maláricos
Captopril
Carbamazepina
Corticosteróides
Sais de Ouro
Penicilamina
Fenitoina
Sulfasalazina
ACTUAL E PASSADA!
DD de hipogamaglobulinémia

Avaliação da produção de anticorpos específicos
isohemaglutininas (IgM anti-A, anti-B)
serologias de acordo com antecedentes e calendário vacinal
(Tétano, hepatite B, pneumococos, H influenzae)
Pneumococo (vacina polissacárida – Pneumo23)
Toxoide tetânico
Naturais
Resposta à vacinação
Diagnóstico de IDP

Suspeita de ID celular / combinada
proliferação celular T em cultura
mitogénios: PHA, PMA + ionomicina, anti-CD3, PWM antigénios: PPD, candidina e toxóide tetânico
quantificação de subpopulações linfocitárias
caracterização imunofenotípica das diferentes populações
Avaliação funcional
Diagnóstico de IDP
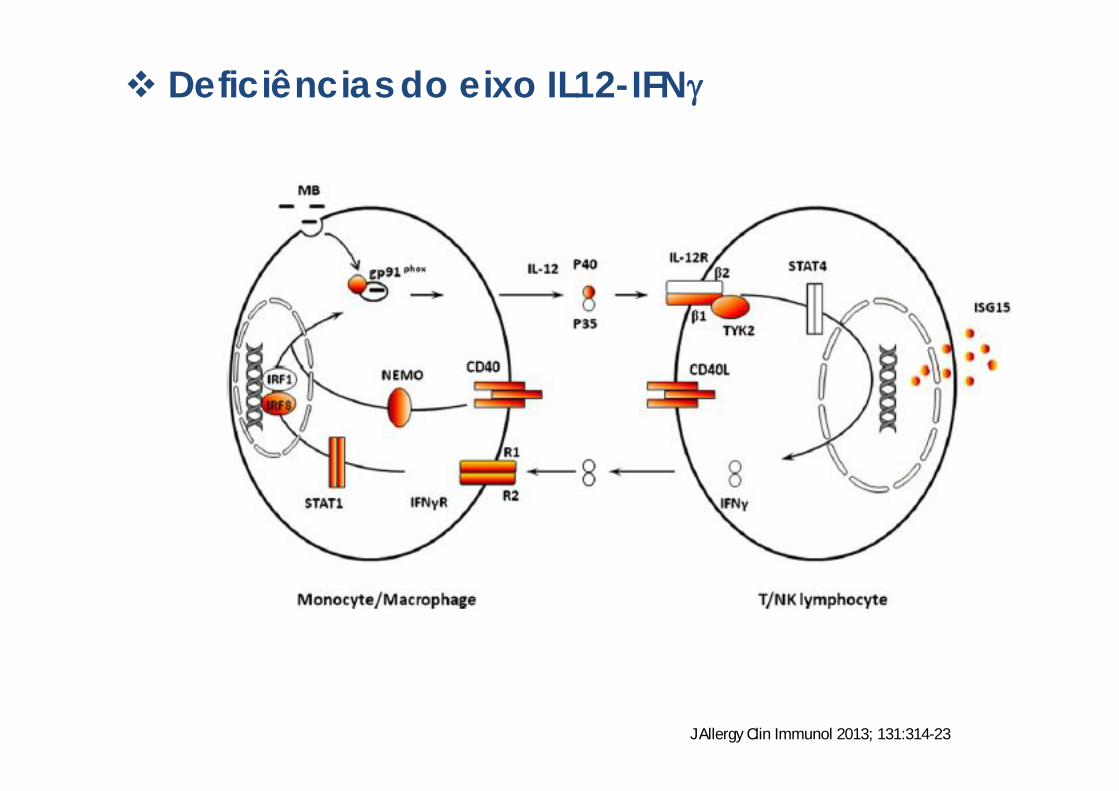
Deficiências do eixo IL12-IFN
J Allergy Clin Immunol 2013; 131:314-23

Suspeita de ID das células fagocitárias (nº e/ou função)
quantificação e observação morfológica
leucogramas seriados e durante intercorrências infecciosas
doseamento de IgE total, G6PD e mieloperoxidase
Diagnóstico de IDP

Suspeita de ID das células fagocitárias (nº e/ou função)
quantificação e observação morfológica,
leucogramas seriados e durante intercorrências infecciosas.
doseamento de IgE total, G6PD e mieloperoxidase.
Estudo da capacidade oxidativa
Estudo da expressão de moléculas de adesão (CD18/CD11a-c)
Estudo da capacidade fagocítica
Estudo da quimiotaxia
Diagnóstico de IDP

Imunodeficiências Primárias
O que são
Classificação
Sinais de alerta / manifestações clínicas
Investigação diagnóstica
IDPs mais frequentes
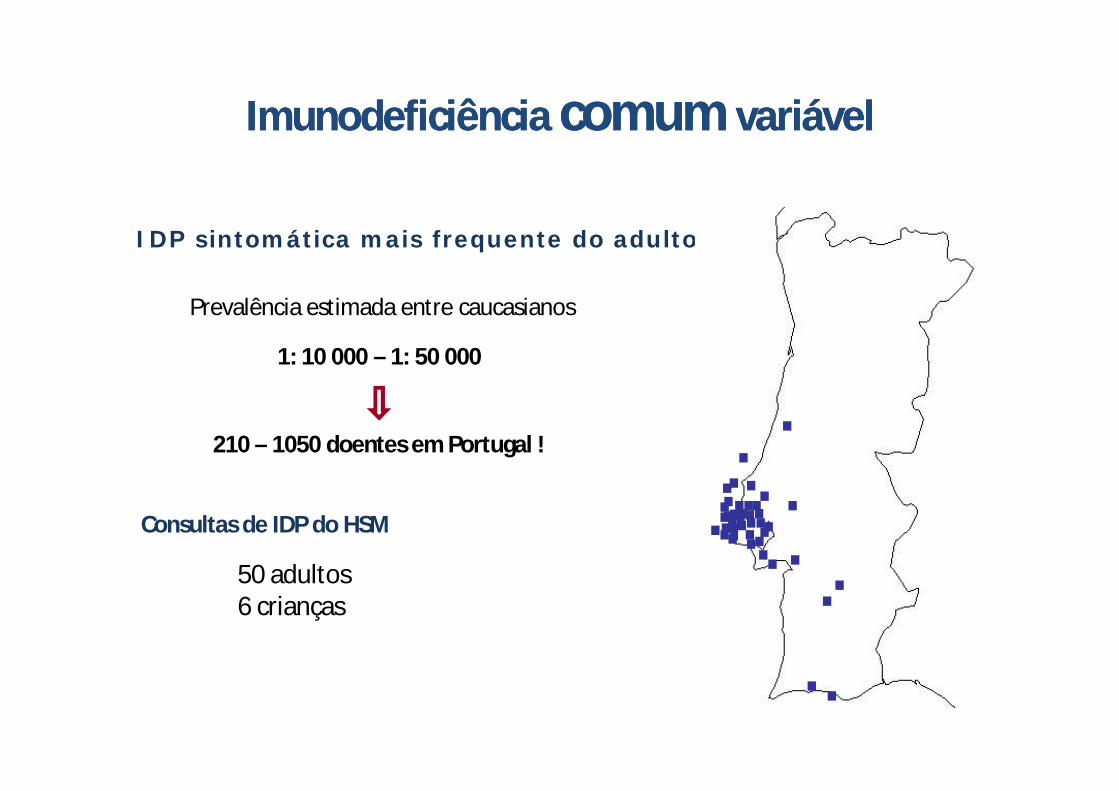
IDP sintomática mais frequente do adulto
Imunodeficiência comum variávelImunodeficiência comum variável
...
..
...
1: 10 000 – 1: 50 000
210 – 1050 doentes em Portugal !
Prevalência estimada entre caucasianos
Consultas de IDP do HSM
50 adultos6 crianças
..
.
............ ..
. .. .... ........
.
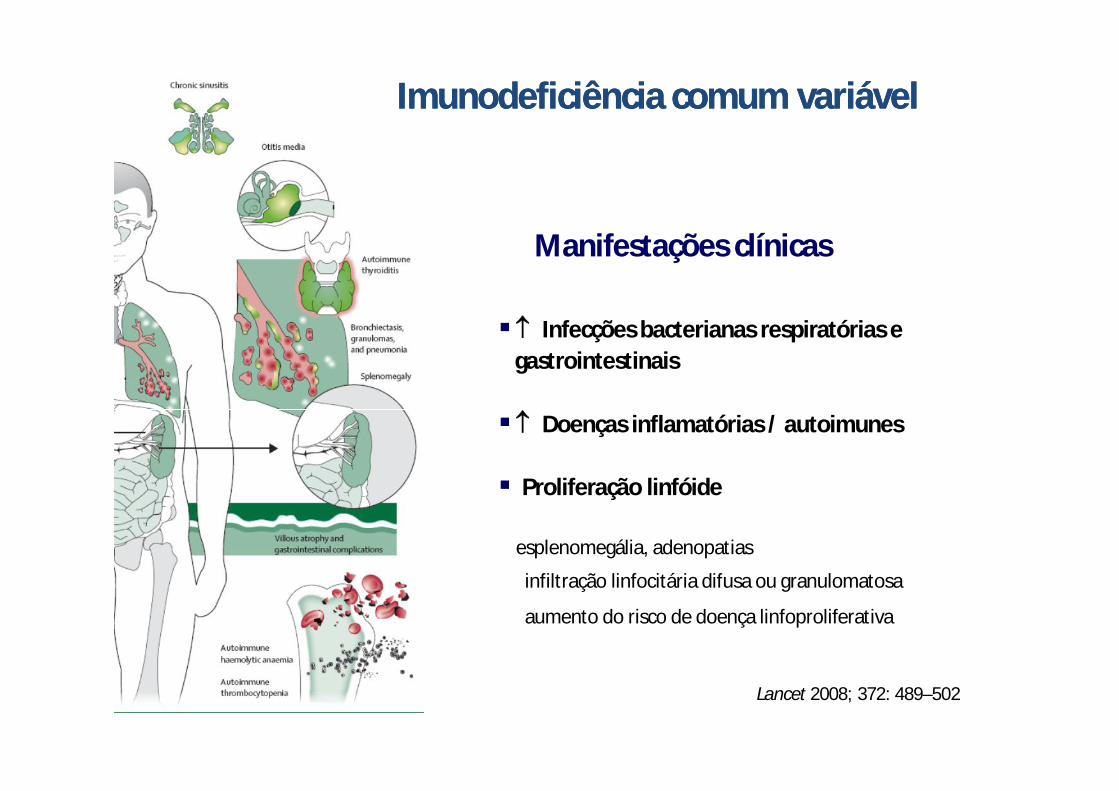
Lancet 2008; 372: 489–502
Infecções bacterianas respiratórias egastrointestinais
Doenças inflamatórias / autoimunes
Proliferação linfóide
esplenomegália, adenopatias
infiltração linfocitária difusa ou granulomatosa
aumento do risco de doença linfoproliferativa
Imunodeficiência comum variávelImunodeficiência comum variável
Manifestações clínicas

Agentes mais frequentes na IDCV
Haemophilus influenzae Streptococcus pneumoniaeMoraxella catarrhalis
Giardia lamblia Campylobacter jejuni Salmonella Shigella
Mycoplasma
Enterovirus CNS (echoviruses)
HSV 1 VZV HHV8
Pseudomonas Aspergillus fumigatus Pneumocystis jiroveciiMycobacterium tuberculosis Atypical mycobacteria
Imunodeficiência comum variávelImunodeficiência comum variável
IDP

www.esid.org
Probable Diagnosis
Male or female with a marked decrease of IgG (at least 2 SD below the mean for age) and a marked decrease in at least one isotype among IgM or IgA
Possible Diagnosis
Male or female with a marked decrease (at least 2 SD below the mean for age) in at least one isotype among IgM, IgG and IgA
1) Onset of immunodeficiency at greater than 2 years of age2) Absent isohemagglutinins and/or poor response to vaccines3) Defined causes of hypogammaglobulinemia have been excluded
Fulfilling all of the following criteria:
Imunodeficiência comum variávelImunodeficiência comum variável
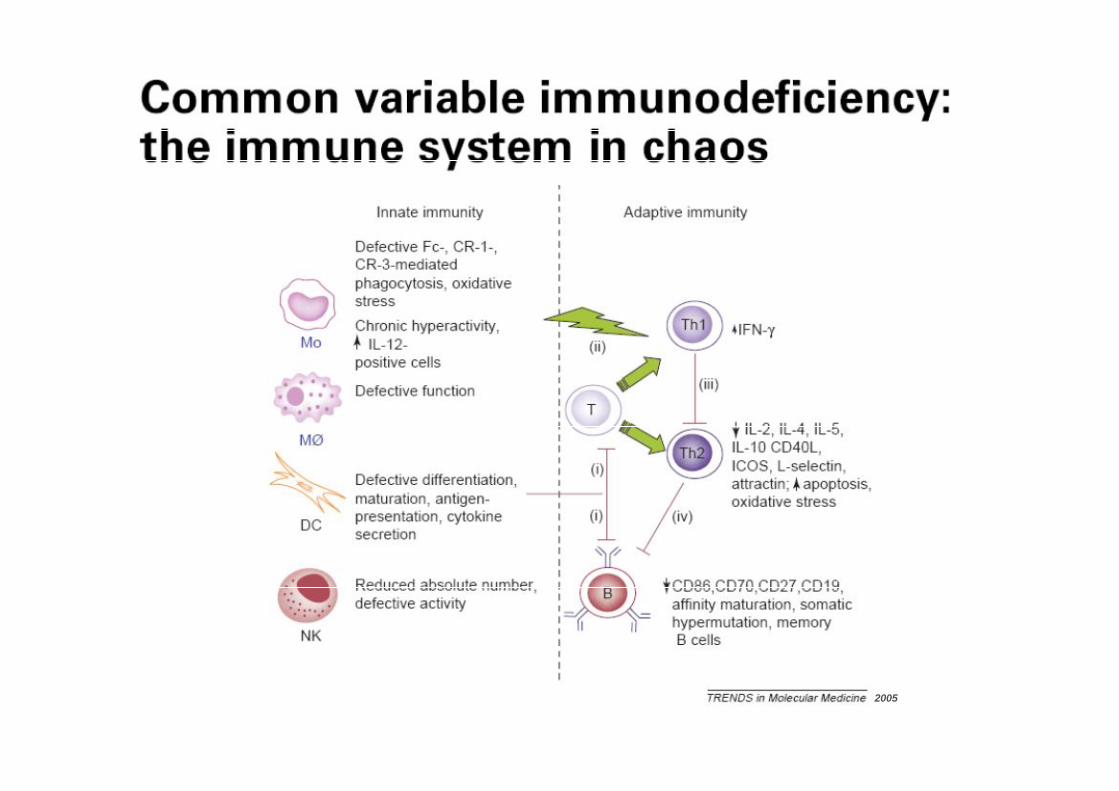
Common Variable ImmunodeficiencyCommon Variable Immunodeficiency
2005
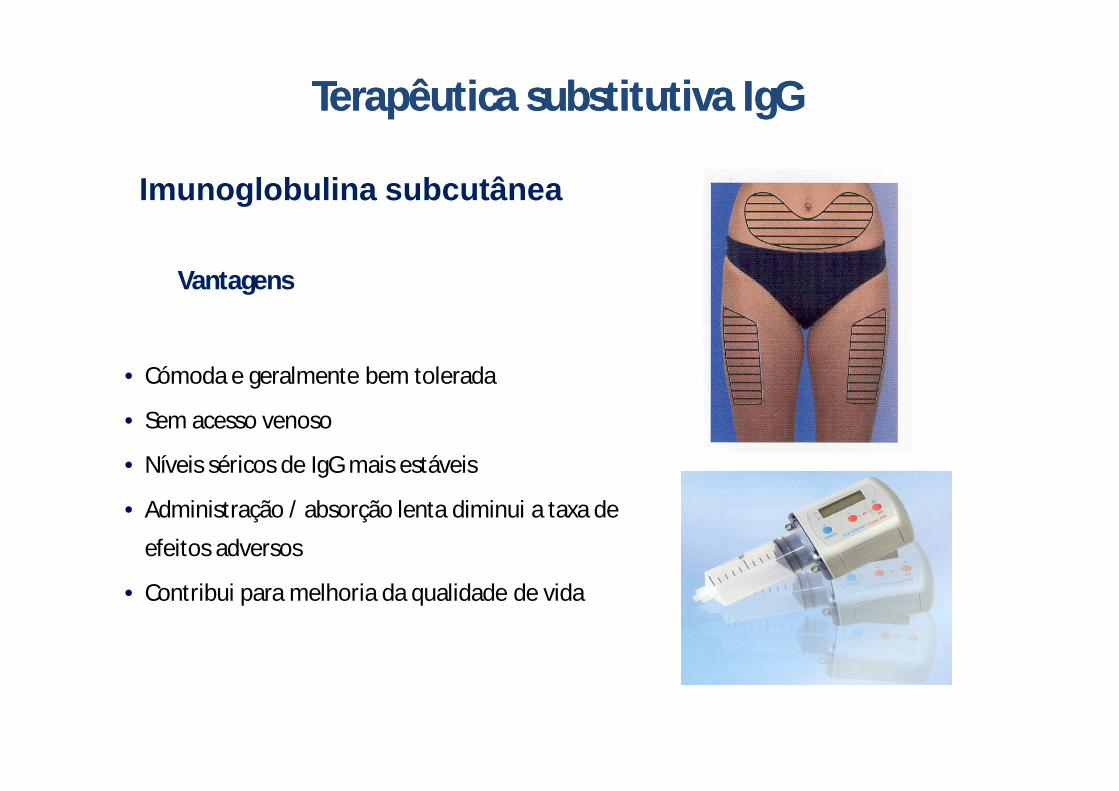
Imunoglobulina subcutânea
Terapêutica substitutiva Terapêutica substitutiva IgGIgG
• Cómoda e geralmente bem tolerada
• Sem acesso venoso
• Níveis séricos de IgG mais estáveis
• Administração / absorção lenta diminui a taxa de
efeitos adversos
• Contribui para melhoria da qualidade de vida
Vantagens

Terapêutica de intercorrências
Tratamento PrecoceProlongadoAgressivo (doses elevadas, espectro alargado, >1 antimicrobiano)Drenagem de abcessos
Procura exaustiva do(s) agente(s) etiológico(s)Culturas, PCR, biópsiasSerologias não valorizáveis
Infecções
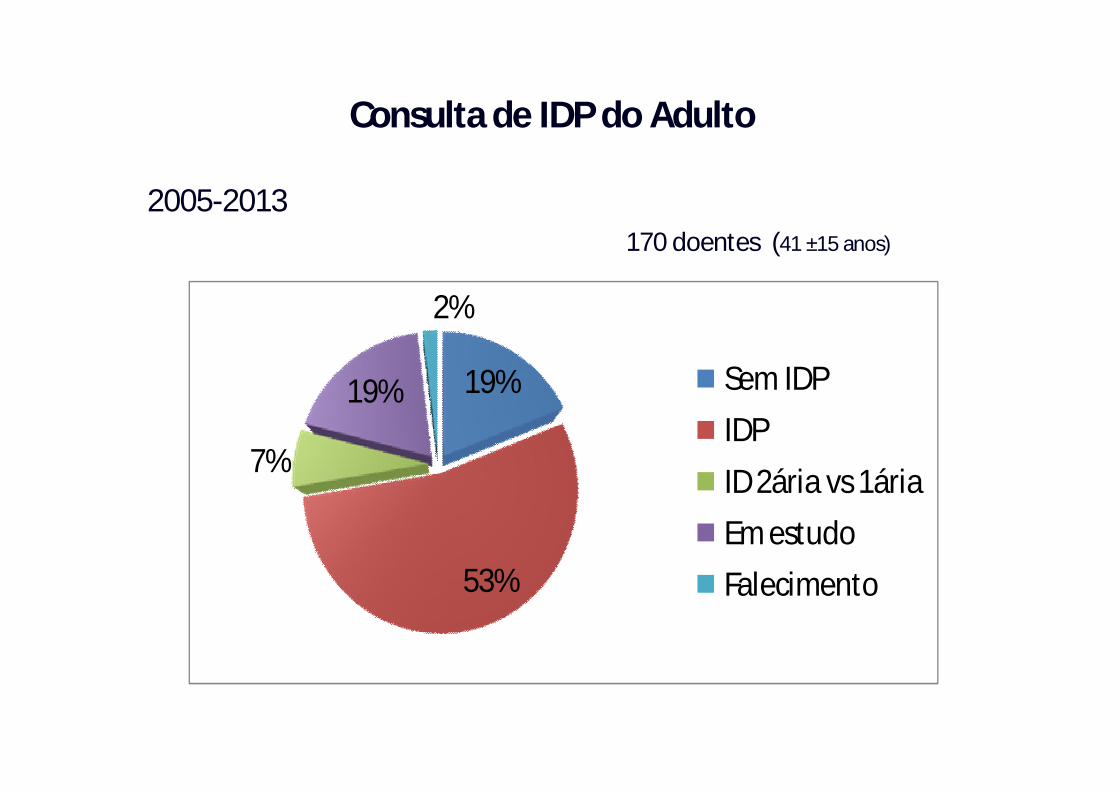
2005-2013170 doentes (41 ±15 anos)
19%
53%
7%
19%
2%
Sem IDPIDPID 2ária vs 1áriaEm estudoFalecimento
Consulta de IDP do Adulto
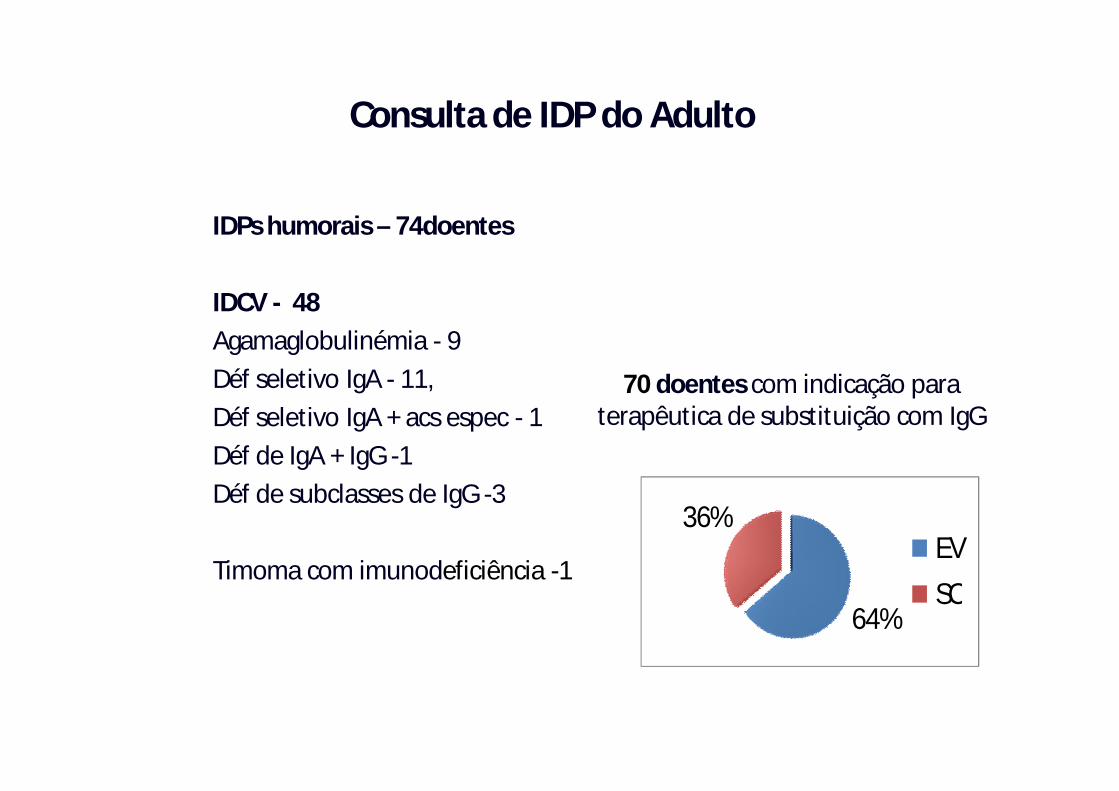
IDPs humorais – 74doentes
IDCV - 48Agamaglobulinémia - 9 Déf seletivo IgA - 11, Déf seletivo IgA + acs espec - 1Déf de IgA + IgG -1Déf de subclasses de IgG -3
Timoma com imunodeficiência -1
70 doentes com indicação para terapêutica de substituição com IgG
64%
36%EVSC
Consulta de IDP do Adulto

Referenciação
Consulta de IDP do Adulto
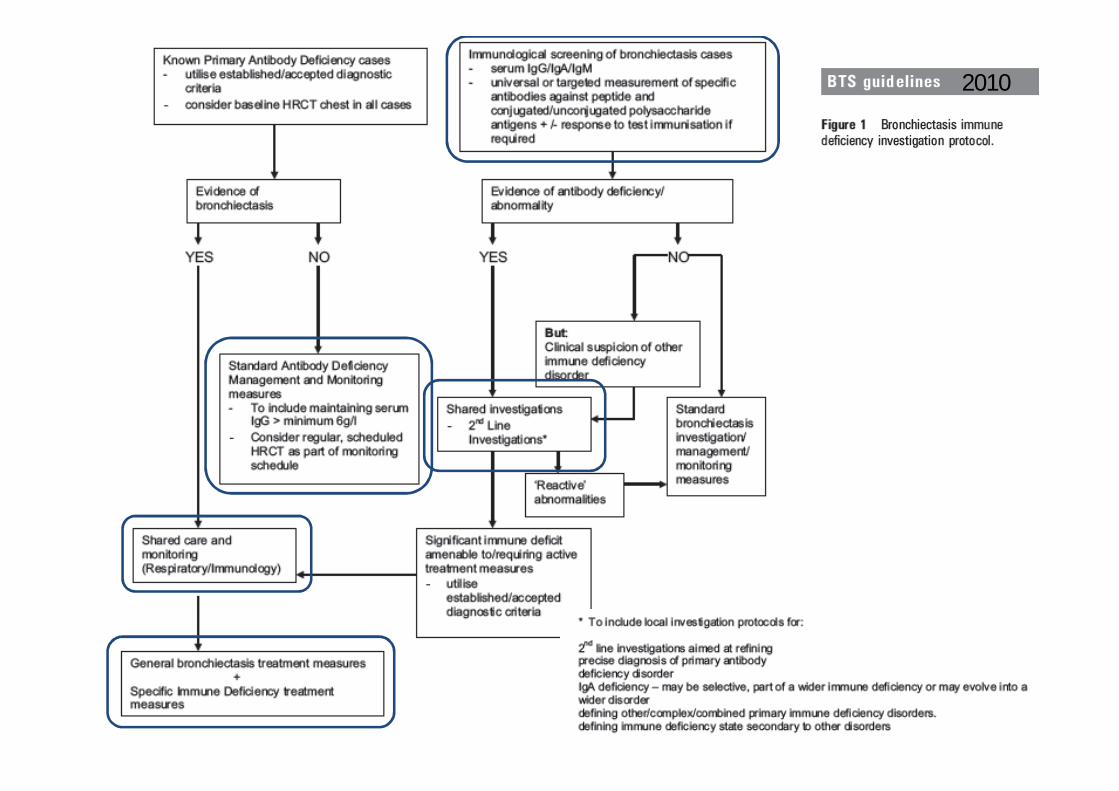
2010

Bronquiectasias : Quando referenciar a Consulta de IDP
Doentes com diagnóstico de IDP
Suspeita clínica de IDP mantida, apesar de avaliação laboratorial sem alterações
Manifestações clínicas associadas / manif. sindromáticas
História familiar
Infecções graves / persistentes / recorrentes / oportunistas
/ múltiplas localizações

Centro de Imunodeficiências Primárias Hospital de Santa Maria, CHLN
Faculdade de Medicina da Universidade de LisboaInstituto de Medicina Molecular
www.spi.pt
Muito Obrigada





























