Embed Size (px)
Citation preview

Plano de aula
Profa. Dra Michele Lima Gregório
Data: 18/03/2015
“Neurogenética”
Conteúdo Programático
Genética Humana - avanços
Marcadores Biológicos e Aconselhamento Genético
Farmacogenômica e Terapia Gênica
Doenças neurológicas geneticamente determinadas
Doença de Parkinson
Distonia
Doença de Alzheimer
Doença de Huntington
Ataxia espinocerebelar
Doenças mitocondriais
Doenças mediadas por RNA: distrofia miotônica
Bibliografia
Smeets CJ, Verbeek DS. Cerebellar ataxia and functional genomics: Identifying the
routes to cerebellar neurodegeneration. Biochim Biophys Acta. 2014
Oct;1842(10):2030-2038.
Katsuno M, Banno H, Suzuki K, et al. Molecular genetics and biomarkers of
polyglutamine diseases. Curr Mol Med. 2008 May;8(3):221-34. Review.
Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic
dystrophies. Acta Myol. 2013 Dec;32(3):154-65. Review.
Björklund A, Lindvall O. Cell replacement therapies for central nervous system
disorders. Nat Neurosci. 2000 Jun;3(6):537-44. Review.
Ross CA1, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers
and prospects for therapeutics. Nat Rev Neurol. 2014 Apr;10(4):204-16.
Kumar A, Kumar Singh S, Kumar V, et al. Huntington's disease: an update of
therapeutic strategies. Gene. 2015 Feb 10;556(2):91-7.

Bonifati V. Genetics of Parkinson's disease--state of the art, 2013. Parkinsonism Relat
Disord. 2014 Jan;20 Suppl 1:S23-8.
Eskow Jaunarajs KL, Bonsi P, Chesselet MF, et al. Striatal cholinergic dysfunction as a
unifying theme in the pathophysiology of dystonia. Prog Neurobiol. 2015 Feb 17.
Charlesworth G, Bhatia KP, Wood NW. The genetics of dystonia: new twists in an old
tale. Brain. 2013 Jul;136(Pt 7):2017-37.
Lehmann S, Delaby C, Touchon J, et al. Biomarkers of Alzheimer's disease: the present
and the future. Rev Neurol (Paris). 2013 Oct;169(10):719-23.
American Academy of Neurology, 2014.
SLIDES
Faculdade de Medicina de São José do Rio Preto
FAMERP
Profa. Dra. Michele Lima Gregório
2015

mecanismo de transmissão
dos caracteres de uma
espécie
Avanços da Genética – melhoria na saúde pessoal e
pública
Genética Ciência da hereditariedade
Através das gerações (variabilidade genética)
Evolução
Avanços no conhecimento
da Genética HumanaMedicina
personalizada
Informação clínica, genética e ambiental – melhor
tratamento individualizado
Além do diagnóstico, estabelecer a origem de
determinada doença – risco em outros familiares
Como usar essa informação biológica na
terapia?
Testes genéticos nas decisões terapêuticas
Uso dos genes como agentes ou alvos
terapêuticos
Farmacogenômica
Terapia gênica

Farmacogenômica
Como as variações gênicas influenciam
na resposta ao tratamento
Terapia gênica
O uso de ácidos nucléicos como agente terapêutico
Novas estratégias terapêuticas em doenças neurológicas são
baseadas em expressão gênica
Processo patogênico de doenças neurológicas envolve
desregulação desse processo
Farmacogenômica
Diferença entre pacientes na resposta ao tratamento –
fatores genéticos e ambientais
• Melhorar a abordagem terapêutica para cada paciente –
tratamento individualizado
• Evitar efeitos colaterais
• Otimização de doses

Terapia gênica
Uso de ácido nucléico como agente
farmacêutico no tratamento de doenças
herdadas ou esporádicas.
2011 – mais de 1.700 testes clínicos com uso de terapia gênica.
Nenhum tratamento ainda foi aprovado pelo FDA
• Substituição de genes – inserção de genes que produzem determinada proteína
• Terapia de reposição enzimática
• Silenciamento gênico – suprimir a expressão do gene mutado (miRNA and
RNAi) causadores de doenças.
Integrar informação genética e não-genética sobre um determinado
paciente no delineamento de um tratamento mais adequado
Kumar et al., 2015.

Marcadores biológicos
São componentes celulares, estruturais e bioquímicos, que podem definir
alterações celulares e moleculares tanto em células normais e em células
alteradas.
Detectam alterações no DNA, que podem influenciar diferenças fenotípicas
O marcador perfeito deveria ser altamente específico para uma determinada
doença e suficientemente sensível para detectar a presença de pequeno número
de células alteradas, permitindo o diagnóstico precoce e alta especificidade
para excluir falsos positivos.
Ainda de uma forma ideal, o marcador deveria facilmente detectado no
sangue ou fluidos biológicos.
Ross et al., 2014

Processo de comunicação que lida com problemas humanos associados com a
ocorrência, ou risco de ocorrência, de uma doença genética em uma família.
Participação de uma ou mais pessoas treinadas para ajudar o indivíduo ou sua
família a:
1) compreender os fatos médicos: diagnóstico, provável curso da doença e as
condutas disponíveis;
2) apreciar o modo como a hereditariedade contribui para a doença e o risco de
recorrência para parentes específicos;
3) entender as alternativas para lidar com o risco de recorrência;
4) escolher o curso de ação que pareça apropriado em virtude do seu risco,
objetivos familiares, padrões éticos e religiosos, atuando de acordo com essa
decisão;
5) ajustar-se, da melhor maneira possível, à situação imposta pela ocorrência do
distúrbio na família, bem como à perspectiva de recorrência do mesmo.
Aconselhamento genético
Heredograma

Doenças neurológicas geneticamente
determinadas
• Genética das doenças do movimento: doença de Parkinson
e distonia
• Genética da demência: doença de Alzheimer e doença de
Huntington
•Ataxia espinocerebelar (SCA)
• Genética das doenças mitocondriais
•Genética das doenças doenças musculares mediadas por
RNA: distrofia miotônica
Doença de Parkinson
• Degeneração dos neurônios dopaminérgicos da SNc - depleção de dopamina estriatal
(Núcleos da base )
• Sinais motores – rigidez muscular, tremor de repouso, instabilidade postural e lentidão
nos movimentos
•PARK - 10 formas monogênicas da doença – 3 autossômicas dominantes e 7 recessivas.
• PARK1 e 4 – SNCA
• Formas mais comuns da doença: autossômica dominante (AD), início tardio – mutações
LRRK2 e AD de início precoce - mutações no gene Parkin.
• ATP13A2, FBOX07, DNAJC6 e SYNJ1 – início juvenil e padrões atípicos e e
multisistêmicos – demência, movimento anormal do olho, sinais piramidais

Circuito motor
DAUER e PRZEDBORSKI, 2003
PARK 1-4
PARK 8
PARK 2
PARK 6
PARK 7
PARK 9
PARK 15

Distonia
“Síndrome da contração sustentada dos músculos, frequentemente causando
torção e movimentos repetidos do corpo ou posturas anormais”
sintomas - início gradual - com o tempo: os espasmos involuntários podem
continuar mesmo no repouso.
Classificação: idade de início, localização corporal, padrão temporal associação
com outras características.
Modo de herança: autossômico dominante
Isolada: ou primária – sem associação de outras características
Combinada: ou distonia “plus” – associação com outras desordens do
movimento.
Jananarajs et al., 2015
Em condições normais –
Dopamina ativa receptores estriatais D2
nos interneurônios colinérgicos e reduz
influxo de Ca2+ e inibindo liberação de
Ach.
Na distonia causada por mutação DYT1 – ativação de receptores D2 provoca
inibição anormal da dos canais de Ca2+, resultando em um aumento da liberação
de Ach.

Charlesworth et al., 2013
Charlesworth et al., 2013

Doença de Alzheimer
Demências – classificadas como “proteinopatias” – acúmulo anormal de
proteínas específicas. Ex.: amilóidopatias, tauopatias, sinucleinopatias....
• Proteinopatias – podem ser hereditárias (mutações dominantes) ou
esporádicas (vários fatores como idade) – fator de risco genético.
• Demências apresentam influencia genética e ambiental.
• Misto de proteinopatias. Ex.: Doença de Alzheimer – amiloidopatia e tauopatia.
• Início tardio ou precoce: média de idade 65 anos, de progressão lenta,
iniciando com prejuízo cognitivo leve até demência total.
• Atrofia cerebral acompanhada de placas amilóides (β-amilóide) e
emaranhados neurofibrilares de proteína tau.
Doença de Alzheimer
Deterioração cognitiva
Atrofia cerebral:
- Placas senis ß-amilóide
- Emaranhados neurofibrilares proteína tau
Déficit de memória/raciocínio Desorientação
Alois Alzheimer – 1906
Sá et al., Front Neurol. 2012;3:81.
Du Y et al. Brain Res Mol Brain Res 2005; 136: 177-88.

Doença de Alzheimer
Início precoce – mutações – formas herdadas e familiais da DA
(~2% de todas as DA) – 3 diferentes genes
• Gene da proteína precursora amilóide (APP) – codifica APP através da qual
a β-amilóide é liberada.
• Presenilinas 1 e 2 - componentes do complexo da γ-secretase (clivagem da
APP) – mutações da presenilina 1 – forma mais comum de DA familial.
Proteína Precursora da β Amilóide
http://www.nia.nih.gov/alzheimers/scientific-images. National Institute on Aging
α γ β secretases
βA-42 aa.
Insolúvel
21q21.1

Doença de Alzheimer
Início tardio – maioria da DA - as causas podem ou não serem genéticas –
embora fatores genéticos sejam importantes.
• Apolipoproteína E (ApoE) – principal fator de risco da forma tardia.
•Alelo E4 – risco aumentado de DA e E2 – risco reduzido (proteção).
- não usado clinicamente para diagnóstico.
Determinantes
(2% casos)
PS1
PS2
APP
Herança autossômica dominante
Apolipoproteína E
APOE*4
Du Y et al. Brain Res Mol Brain Res 2005; 136: 177-88.
PRÉ SENIL (<65 anos de idade)
TARDIO (>65 anos de idade)
Patogênese da DA – aumento da produção ou acúmulo de amilóide

Lehmann et al., 2013
Doença de Huntington
George Huntington (1872) – doença hereditária – pai e avô afetados.
Doença neurodegenerativa progressiva – movimentos involuntários, com
desordem intelectual e psiquiátrica
Média de idade – 25 a 70 anos
Graus variados de atrofia estriatal – lesão dos neurônios estriatais (inibitórios –
GABA) que se projetam a substância negra e globo pálido (DA) - níveis
GABA
DA Sinais motores da doença
Drogas antidopaminérgicas - tratamento
Kumar et al., 2015.

Björklund et al., 2000; Ross et al., 2014
Doença de Huntington
Doença autossômica dominante – 5/100.000 indivíduos
Região 4p16.3
Kumar et al., 2015.
Mutação gene IT15 –proteína huntigtina (htt) – função
desconhecida – essencial ao desenvolvimento –
ausência é letal em ratos
Importante no funcionamento normal dos núcleos da
base –
Proteína normal – regula expressão do BDNF

Doença de Huntington
Doença poliglutamina – mais de 39 repetições CAG
(glutamina)
Repetição promove agregação – toxicidade: apoptose,
excitotoxicidade, disfunção mitocondrial, desregulação
transcricional – características neuropatológicas da doença
Ataxia espinocerebelar
Grupo heterogêneo de doenças neurodegenerativas geneticamente heterogênea –
atrofia do cerebelo e perda das células de Purkinje –
- ataxia dos membros, tronco e marcha (e outras específicas de cada doença)
Desafio – sobreposição fenotípica entre as etiologias adquiridas, idiopáticas e
herdadas
Doença autôssomica dominante – SCAs – ações motoras descoordenadas –
disartria, disfagia, alterações no movimento dos olhos, desequilíbrio postural.
SCAs – repetição CAG (glutamina) – ataxias poliglutaminas (SCA1, 2, 3, 6 e 7)
Doença autôssomica recessiva– tb apresentam sinais cerebelares, entretanto
possuem neuropatia periférica associada.
Repetição intron GAA no gene frataxin –
ataxia de Friedreich
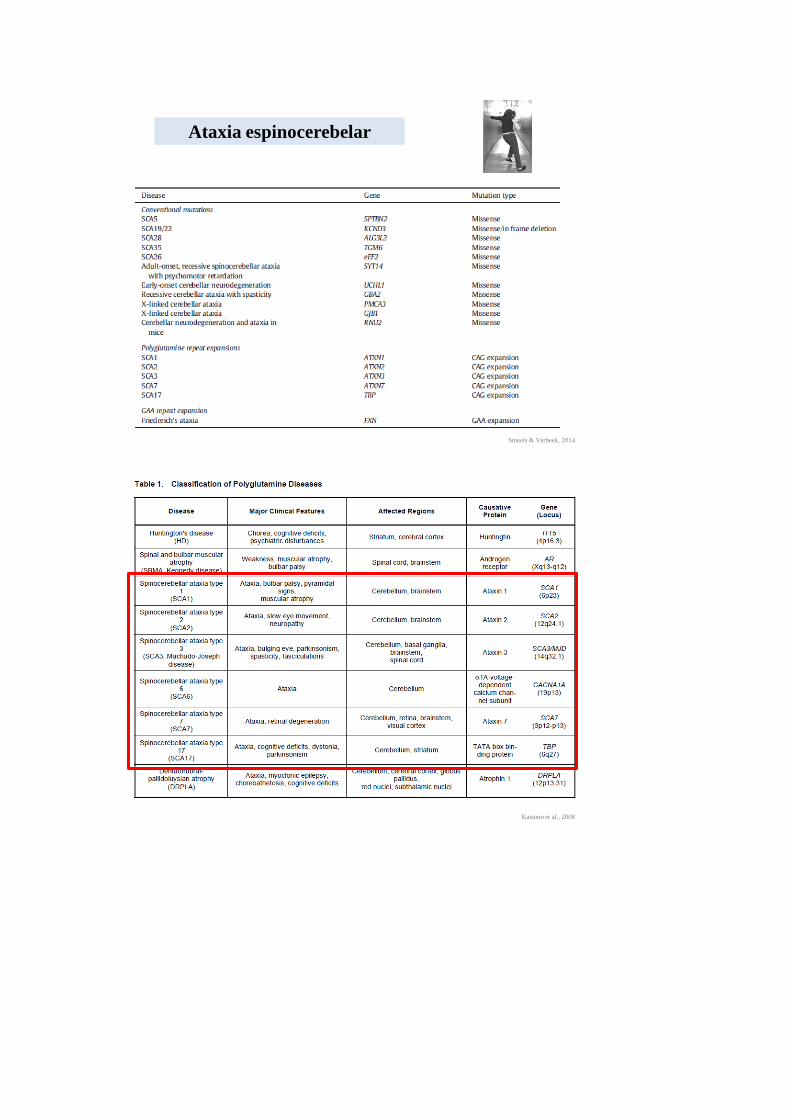
Ataxia espinocerebelar
Smeets & Verbeek, 2014
Katsuno et al., 2008

American Academy of Neurology, 2014
Katsuno et al., 2008

Doenças mitocondriais
Doenças heterogêneas – clínico, bioquímico e genético.
Mitocôndria: diversas funções
Produção de ATP e controle genético (DNAnuclear e
DNAmitocondrial)
Definição – defeitos na via energética final do metabolismo
mitocondrial, ou seja, na fosforilação oxidativa.
Doenças mitocondriais
Sob controle do DNAn e DNAmt
Das 90 proteínas, 13 são codificadas por DNAmt –
mutações nesse DNAmt ocasionam as doenças
Fosforilação oxidativa

Tipos de Mutação
Pequena escala de mutação
Substituição de nucleotídeos
ATCGAATCGA
ATGGAATCGA
Doenças mitocondriais
Mutações no DNAmt – Deleções e mutações pontuais
Síndrome Kearns-Sayre (KSS) –deleções em larga escala
Epilepsia mioclonica (MERRF) – m.8344A>G no gene RNAtLys
Síndrome Kearns-Sayre (KSS) – m.11778A>G gene ND4
MELAS – m.3243A>G no gene RNAtLeu(UUR)
Defeitos na síntese de proteína mitocondrial

Doenças mitocondriais
Mutações no DNAn – defeitos na cadeia respiratória
mitocondrial
Síndrome de Leigh – mutações heterozigotas na subunidade
flavoproteína do complexo II, e outras mutações no complexo I, III,
IV e V
Doenças mitocondriais
Mutação POLG – codifica polimerase γ do DNAmt – doença
autossômica dominante
MNGIE – encefalopatia neurogastrointestinal mitocondrial –
depleção, deleções multiplas e mutações pontuais no DNAmt
(autossômica recessiva)
TYMP – codifica a timidina
fosforilase – importante na
replicação do DNAmt

Doenças musculares
Distrofia miotônica – doença mustissistêmica autossômica
dominante.
Miotonia, distrofia muscular, defeitos na condução cardíaca, envolvimento cerebral
e endócrino.
Consórcio Intern. Distrofia Muscular – nomenclatura com base no
DNA
Distrofia muscular tipo 1 (DM1) – expansão instável da repetição
de trinucleotídeos CTG no cromossomo 19.
Distrofia muscular tipo 2 (DM2) – expansão instável da repetição
de tetranucleotídeos CCTG no cromossomo 3.
Meola et al., 2013

Mutação
DM1
Repetição instável do trinucleotídeo
CTG na região 3’UTR do gene
DMPK
Repetição 150-12.000 pb
19q13.3
Mutação
DM2 Repetição instável do
tetranucleotídeo CCTG no intron 1
do gene CNBP
Repetição <30 em indivíduos
normais
3q21

Mutação
DM1
Mutação
DM2
Em cromossomos diferentes, mas com padrões
semelhantes da doença
RNA ocasiona a patogênese da doença
Expansão é transcrita e o
RNA mutante contendo as
expansões acumula-se no
núcleo
Inclusões
ribonucleares
Padrões
patológicos
das doenças

“Existem muitas hipóteses em ciências que estão erradas.
Isso é perfeitamente aceitável, elas são a abertura
para achar as que estão certas”
(Carl Sagan)Obrigada !!!
Núcleo de Pesquisa em Bioquímica e Biologia
Molecular – NPBIM/FAMERP
Ramal 5864













