Embed Size (px)
Citation preview

I
(Actos aprovados ao abrigo dos Tratados CE/Euratom cuja publicação é obrigatória)
REGULAMENTOS
REGULAMENTO (CE) N.o 429/2008 DA COMISSÃO
de 25 de Abril de 2008
relativo às regras de execução do Regulamento (CE) n.o 1831/2003 do Parlamento Europeu e doConselho no que se refere à preparação e apresentação de pedidos e à avaliação e autorização de
aditivos destinados à alimentação animal
(Texto relevante para efeitos do EEE)
A COMISSÃO DAS COMUNIDADES EUROPEIAS,
Tendo em conta o Tratado que institui a Comunidade Europeia,
Tendo em conta o Regulamento (CE) n.o 1831/2003 doParlamento Europeu e do Conselho, de 22 de Setembrode 2003, relativo aos aditivos destinados à alimentaçãoanimal (1), nomeadamente os n.os 4 e 5 do artigo 7.o,
Após consulta da Autoridade Europeia para a Segurança dosAlimentos, nos termos dos n.os 4 e 5 do artigo 7.o doRegulamento (CE) n.o 1831/2003,
Considerando o seguinte:
(1) É necessário estabelecer regras de execução relativas aoprocedimento de autorização de aditivos destinados àalimentação animal ao abrigo do Regulamento (CE)n.o 1831/2003, incluindo regras para a preparação eapresentação de pedidos e para a avaliação e autorização detais aditivos. Estas regras têm por objectivo substituir asdisposições previstas no anexo da Directiva 87/153/CEE doConselho (2), que fixa linhas directrizes para a avaliação dosaditivos na alimentação para animais.
(2) Essas regras devem prever os requisitos a satisfazer peloprocesso que acompanha o pedido. Devem, em particular,definir os dados científicos a apresentar para a identificação
e caracterização do aditivo em causa e os estudos aapresentar para demonstrar a sua eficácia e segurança parao ser humano, para os animais e para o ambiente tendo emvista a verificação e avaliação dos pedidos de autorizaçãopela Autoridade Europeia para a Segurança dos Alimentos(a Autoridade).
(3) A extensão dos estudos necessários para avaliar as suaspropriedades ou efeitos pode variar em função da naturezado aditivo ou das condições de utilização solicitadas. Porconseguinte, deve conceder-se aos operadores certa flexi-bilidade no que diz respeito ao tipo de estudos e de materiala submeter para demonstrar a segurança e eficácia doaditivo em causa. Os operadores que utilizem estaflexibilidade devem ter de justificar no processo a suaescolha.
(4) A Autoridade deve dispor da possibilidade de solicitarinformação suplementar, quando necessário, a fim dedeterminar se o aditivo cumpre as condições de autorizaçãoreferidas no artigo 5.o do Regulamento (CE) n.o 1831/2003.
(5) É indispensável aplicar normas de qualidade adequadas aodesenvolver processos para aditivos destinados a alimentospara animais ou água no sentido de garantir que osresultados dos ensaios laboratoriais não sejam contestados.
(6) Se necessário, devem ser definidos requisitos específicospara cada categoria de aditivos referida no n.o 1 doartigo 6.o do Regulamento (CE) n.o 1831/2003.
(7) Com o objectivo de estimular os esforços para obterautorizações para espécies menores, mantendo todavia onível necessário de segurança, devem ser previstas
22.5.2008 PT Jornal Oficial da União Europeia L 133/1
(1) JO L 268 de 18.10.2003, p. 29. Regulamento alterado peloRegulamento (CE) n.o 378/2005 da Comissão (JO L 59 de 5.3.2005,p. 8).
(2) JO L 64 de 7.3.1987, p. 19. Revogada pelo Regulamento (CE)n.o 1831/2003.

condições específicas para ter em conta a possibilidade dese extrapolarem para as espécies menores os resultados dosestudos efectuados sobre as espécies principais.
(8) As regras de execução relativas a pedidos de autorizaçãodevem ter em conta requisitos diferentes para animaisprodutores de géneros alimentícios e para outros animais,para os quais os aspectos relativos à avaliação de segurançapara o consumidor humano não são relevantes.
(9) Deve-se reduzir ao mínimo o recurso a procedimentos queenvolvam a utilização de animais de laboratório para finsexperimentais ou outros fins científicos e para ensaios emanimais, de acordo com a Directiva 86/609/CEE doConselho, de 24 de Novembro de 1986, relativa àaproximação das disposições legislativas, regulamentares,e administrativas dos Estados-Membros respeitantes àprotecção dos animais utilizados para fins experimentaise outros fins científicos (1).
(10) Para evitar repetir estudos desnecessariamente, devem serprevistos procedimentos simplificados de autorização deaditivos já autorizados para utilização em géneros alimen-tícios.
(11) No que respeita aos aditivos já autorizados por um períodoilimitado ao abrigo da Directiva 70/524/CEE do Conse-lho (2), deve ser prevista a possibilidade, se necessário, de orequerente demonstrar a eficácia, sempre que não estejamdisponíveis estudos, através de qualquer outro materialdisponível para esse fim, em especial material referente aolongo historial de utilização do aditivo em causa.
(12) Devem ser previstas regras para pedidos de alteração deautorizações, em conformidade com o n.o 3 do artigo 13.odo Regulamento (CE) n.o 1831/2003.
(13) Devem também ser previstas regras para pedidos derenovação de autorização, em conformidade com oartigo 14.o do Regulamento (CE) n.o 1831/2003.
(14) No que diz respeito às disposições referentes aos estudos desegurança e eficácia a realizar em apoio ao pedido, énecessário prever um período de transição durante o qualcontinuam a aplicar-se as regras actuais. Os pedidosapresentados antes da entrada em vigor do presenteregulamento devem continuar a ser tratados em conformi-dade com o anexo da Directiva 87/153/CEE. No que dizrespeito aos pedidos apresentados durante um determinadoperíodo após a entrada em vigor, tendo em conta o longoperíodo de tempo necessário para alguns estudos, osrequerentes devem poder optar entre as regras previstas nopresente regulamento e as do anexo da Directiva 87/153//CEE. As regras de execução foram definidas com base nosconhecimentos científicos e técnicos actuais e devem seradaptadas, se for caso disso, a qualquer novo progresso.
(15) As medidas previstas no presente regulamento estão emconformidade com o parecer do Comité Permanente daCadeia Alimentar e da Saúde Animal,
ADOPTOU O PRESENTE REGULAMENTO:
Artigo 1.o
Definições
Para efeitos do presente regulamento, entende-se por:
1. «Animais de companhia e outros animais não produtoresde géneros alimentícios», animais pertencentes a espéciesnormalmente alimentadas, criadas ou mantidas, mas nãoconsumidas por seres humanos, à excepção dos cavalos;
2. «Espécie menor», animais produtores de géneros alimentí-cios à excepção dos bovinos (animais produtores de leite ede carne, incluindo vitelos), ovinos (animais produtores decarne), suínos, galinhas (incluindo galinhas poedeiras),perus e peixe pertencentes aos Salmonidae.
Artigo 2.o
Apresentação de um pedido
1. Um pedido de autorização de um aditivo destinado àalimentação animal, tal como previsto no artigo 7.o doRegulamento (CE) n.o 1831/2003, é apresentado utilizando oformulário definido no anexo I.
É acompanhado de um processo, tal como previsto no artigo 3.o(a seguir designado «o processo»), contendo os dados edocumentos referidos no n.o 3 do artigo 7.o do Regulamento(CE) n.o 1831/2003.
2. Sempre que, em conformidade com o artigo 18.o doRegulamento (CE) n.o 1831/2003, o requerente solicite quedeterminadas partes do processo referido no n.o 1 sejammantidas confidenciais, este apresenta uma justificação verificá-vel para cada documento ou cada parte de um documento cujadivulgação da informação nele contida poderia significativa-mente lesar a sua posição concorrencial. As partes confidenciaissão submetidas separadamente do resto do processo e não sãoincluídas no resumo referido no n.o 3, alínea h), do artigo 7.o doRegulamento (CE) n.o 1831/2003. O requerente envia àComissão uma cópia das partes do processo cujo tratamentofoi solicitado como confidencial e a justificação que as deveacompanhar.
Artigo 3.o
Processo
1. O processo demonstra adequada e suficientemente que oaditivo destinado à alimentação animal satisfaz as condições deautorização prevista no artigo 5.o do Regulamento (CE)n.o 1831/2003.
L 133/2 PT Jornal Oficial da União Europeia 22.5.2008
(1) JO L 358 de 18.12.1986, p. 1; versão rectificada no JO L 117 de5.5.1987, p. 31. Directiva com a redacção que lhe foi dada pelaDirectiva 2003/65/CE do Parlamento Europeu e do Conselho(JO L 230 de 16.9.2003, p. 32).
(2) JO L 270 de 14.12.1970, p. 1. Directiva com a última redacção quelhe foi dada pelo Regulamento (CE) n.o 1800/2004 da Comissão(JO L 317 de 16.10.2004, p. 37).

2. Os requisitos gerais para a preparação e apresentação doprocesso são os estabelecidos no anexo II.
Os requisitos específicos a cumprir pelo processo, conforme ocaso, são os estabelecidos no anexo III.
A duração mínima dos estudos a longo prazo é a estabelecida noanexo IV.
3. Em derrogação ao disposto no n.o 2, o requerente podeapresentar um processo que não cumpra os requisitos previstosno n.o 2, desde que apresente uma justificação para cadaelemento que não obedeça a esses requisitos.
Artigo 4.o
Medidas de transição
1. Continua a aplicar-se a pedidos de autorização apresentadosantes da data de entrada em vigor do presente regulamento oanexo da Directiva 87/153/CEE.
2. Para os pedidos de autorização apresentados antes de11 de Junho de 2009 os requerentes podem optar pelacontinuação da aplicação das secções III e IV das partes I e IIdo anexo da Directiva 87/153/CEE em vez dos pontos 1.3, 1.4,2.1.3, 2.1.4, 2.2.3, 2.2.4, 3.3, 3.4, 4.1.3, 4.1.4, 4.2.3, 4.2.4, 5.3,5.4, 6.3, 6.4, 7.3, 7.4, 8.3 e 8.4 do anexo III e em vez dasdisposições previstas na coluna «Duração mínima dos estudos deeficácia a longo prazo» dos quadros do anexo IV.
Artigo 5.o
Entrada em vigor
O presente regulamento entra em vigor no vigésimo dia seguinteao da sua publicação no Jornal Oficial da União Europeia.
O presente regulamento é obrigatório em todos os seus elementos e directamente aplicável emtodos os Estados-Membros.
Feito em Bruxelas, em 25 de Abril de 2008.
Pela Comissão
Androulla VASSILIOU
Membro da Comissão
22.5.2008 PT Jornal Oficial da União Europeia L 133/3

ANEXO I
FORMULÁRIO DE PEDIDO REFERIDO NO N.o 1 DO ARTIGO 2.o E DADOS ADMINISTRATIVOS
1. FORMULÁRIO DE PEDIDO
COMISSÃO EUROPEIA
DIRECÇÃO-GERAL
SAÚDE E DEFESA DO CONSUMIDOR
(Endereço)
Data: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Assunto: Pedido de autorização de um aditivo destinado à alimentação animal em conformidade com oRegulamento (CE) n.o 1831/2003.
Autorização de um aditivo destinado à alimentação animal ou de uma nova utilização de um aditivo destinadoà alimentação animal [n.o 1 do artigo 4.o do Regulamento (CE) n.o 1831/2003]
Autorização de um produto existente [n.os 2 ou 7 do artigo 10.o do Regulamento (CE) n.o 1831/2003]
Alteração de uma autorização existente [n.o 3 do artigo 13.o do Regulamento (CE) n.o 1831/2003]
Renovação de uma autorização de um aditivo destinado à alimentação animal [artigo 14.o do Regulamento(CE) n.o 1831/2003]
Autorização urgente [artigo 15.o do Regulamento (CE) n.o 1831/2003]
(Por favor indicar claramente seleccionando uma das caixas)
O(s) requerente(s) e/ou o(s) seu(s) representante(s) na Comunidade [n.o 3 do artigo 4.o do Regulamento (CE)n.o 1831/2003], nas condições exigidas no n.o 3, alínea a), do artigo 7.o do Regulamento (CE) n.o 1831/2003 (nome,endereço…)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
apresenta(m) o presente pedido a fim de obter uma autorização para o seguinte produto como um aditivo destinadoà alimentação animal:
1.1. Identificação e caracterização do aditivo
Nome de aditivo [caracterização da(s) substância(s) ou do(s) agente(s) activo(s), tal como definido nassubsecções 2.2.1.1 e 2.2.1.2 do anexo II]:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Designação comercial (se adequado para as autorizações ligadas ao detentor):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
L 133/4 PT Jornal Oficial da União Europeia 22.5.2008

na(s) categoria(s) e no(s) grupo(s) funcional(ais) de aditivos (1) (lista):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Espécie(s)-alvo:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Nome do detentor da autorização: [n.o 6 do artigo 9.o do Regulamento (CE) n.o 1831/2003]
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Este aditivo já está autorizado em legislação relativa à alimentação animal pela Directiva …/…/CE(E) ou peloRegulamento (CE) n.o …/… sob o número … como (categoria do aditivo)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Este aditivo já está autorizado em legislação relativa à alimentação humana pela Directiva …/…/CE(E) ou peloRegulamento (CE) n.o …/… sob o número … como
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
para utilização em
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Se o produto consistir em, contiver ou for produzido a partir de um organismo geneticamente modificado (OGM),por favor faculte a seguinte informação:
Identificador único [Regulamento (CE) n.o 65/2004 da Comissão (2)] (sempre que adequado):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Quer os pormenores sobre qualquer autorização concedida em conformidade com o Regulamento (CE)n.o 1829/2003 do Parlamento Europeu e do Conselho (3):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Quer os pormenores de qualquer pedido de autorização pendente ao abrigo do Regulamento (CE) n.o 1829//2003:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2. Condições de utilização
1.2.1. Utilização em alimentos completos para animais
Espécie ou categoria animal:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
22.5.2008 PT Jornal Oficial da União Europeia L 133/5
(1) Para o grupo funcional «Outros aditivos zootécnicos», na categoria dos aditivos zootécnicos, é necessário definir claramente qual a funçãopretendida para o aditivo.
(2) JO L 10 de 16.1.2004, p. 5.(3) JO L 268 de 18.10.2003, p. 1. Regulamento com a última redacção que lhe foi dada pelo Regulamento (CE) n.o 298/2008 (JO L 97 de
9.4.2008, p. 64).

Idade ou peso máximos:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Dose mínima (se adequado): mg ou unidades de actividade (4) ou unidades formadoras de colónias (UFC) ou ml/kgde alimento completo para animais com um teor de humidade de 12 %
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Dose máxima (se adequado): mg ou unidades de actividade ou UFC ou ml/kg de alimento completo para animaiscom teor de humidade de 12 %
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Para alimentos líquidos para animais as doses mínimas e máximas podem ser expressas por litro.
1.2.2. Utilização em água
Dose mínima (se adequado): mg ou unidades de actividade ou UFC ou ml/l de água
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Dose máxima (se adequado): mg ou unidades de actividade ou UFC ou ml/l de água
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2.3. Condições especiais de utilização (se adequadas)
Espécie ou categoria animal:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Idade máxima:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Dose mínima (se adequado): mg ou unidades de actividade ou UFC/Kg de alimentos complementares para animaiscom teor de humidade de 12 %
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
L 133/6 PT Jornal Oficial da União Europeia 22.5.2008
(4) A definição de «unidade» é apresentada pelo requerente.

Dose máxima (se adequado): mg ou unidades de actividade ou UFC/Kg de alimentos complementares para animaiscom teor de humidade de 12 %
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Para alimentos líquidos para animais as doses mínimas e máximas podem ser expressas por litro.
Condições ou restrições de utilização (se adequado):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Condições ou restrições específicas de manuseamento (se adequado):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Limite máximo de resíduos (se adequado):
Espécie ou categoria animal:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Resíduo marcador:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Tecidos ou produtos-alvo:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Resíduos máximos em tecidos ou produtos (μg/kg):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
22.5.2008 PT Jornal Oficial da União Europeia L 133/7

Intervalo de segurança:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.3. Amostras de referência
Número de amostra do Laboratório Comunitário de Referência (LCR) (se aplicável):
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Número/código do lote:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Data de fabrico:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Data do termo de validade:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Concentração:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Peso:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Descrição física:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Descrição dos contentores:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Requisitos de armazenamento:
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.4. Modificação solicitada (quando necessário)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A cópia deste pedido foi enviada directamente com o processo à Autoridade e ao LCR com as amostras de referência.
Assinatura . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
L 133/8 PT Jornal Oficial da União Europeia 22.5.2008

1.5. Anexos:
Processo completo (apenas para a Autoridade);
Resumo público do processo;
Resumo pormenorizado do processo;
Lista das partes do processo cujo tratamento foi solicitado como confidencial e uma cópia das mesmas (apenaspara a Comissão e para a Autoridade);
Cópia dos dados administrativos do(s) requerente(s);
Três amostras do aditivo destinado à alimentação animal para o LCR, em conformidade com o n.o 3, alínea f),do artigo 7.o do Regulamento (CE) n.o 1831/2003 (apenas para o LCR);
Ficha de segurança para o material (apenas para o LCR);
Certificado de identificação e análise (apenas para o LCR); bem como
Confirmação do pagamento da taxa ao LCR [artigo 4.o do Regulamento (CE) n.o 378/2005 (5)].
Preencher as partes adequadas do formulário e suprimir as que não são relevantes. O formulário de pedido original (com osrestantes anexos solicitados) é enviado directamente para a Comissão Europeia.
2. DADOS ADMINISTRATIVOS DO(S) REQUERENTE(S)
Pormenores de contacto para apresentar um pedido de autorização de um aditivo destinado à alimentação animal aoabrigo do Regulamento (CE) n.o 1831/2003
1. Empresa ou pessoa requerente
a) Nome da empresa ou pessoa requerente
b) Endereço (rua, número, código postal, cidade, país)
c) Telefone
d) Fax
e) Correio electrónico (se disponível)
2. Pessoa de contacto (para toda a correspondência com a Comissão, a Autoridade e o LCR)
a) Nome da pessoa de contacto
b) Cargo
c) Endereço (rua, número, código postal, cidade e país)
d) Telefone
e) Fax
f) Correio electrónico (se disponível)
22.5.2008 PT Jornal Oficial da União Europeia L 133/9
(5) Regulamento (CE) n.o 378/2005 da Comissão, de 4 de Março de 2005, sobre as regras de execução do Regulamento (CE) n.o 1831/2003do Parlamento Europeu e do Conselho relativo às competências e funções do Laboratório Comunitário de Referência no respeitante aospedidos de autorização de aditivos destinados à alimentação animal (JO L 59 de 5.3.2005, p. 8). Regulamento alterado pelo Regulamento(CE) n.o 850/2007 (JO L 188 de 20.7.2007, p. 3).

ANEXO II
REQUISITOS GERAIS A CUMPRIR PELO PROCESSO PREVISTO NO ARTIGO 3.o
ASPECTOS GERAIS
O presente anexo define os requisitos para estabelecer a lista e as características de estudos e informação sobre substâncias,microrganismos e preparados a apresentar com os processos nos termos do artigo 7.o do Regulamento (CE) n.o 1831/2003para:
— uma autorização como novo aditivo destinado à alimentação animal,
— uma autorização para uma nova utilização de um aditivo destinado à alimentação animal,
— uma alteração de uma autorização existente de um aditivo destinado à alimentação animal, ou
— uma renovação da autorização de um aditivo destinado à alimentação animal.
Os processos devem permitir a realização de uma avaliação de aditivos com base no estado actual dos conhecimentos epermitir a verificação do cumprimento por parte destes aditivos dos princípios fundamentais para a autorização,estabelecidos no artigo 5.o do Regulamento (CE) n.o 1831/2003.
Os estudos a apresentar e a respectiva extensão dependerão da natureza do aditivo, da categoria e grupo funcional, do tipode autorização (não específica ao requerente vs específica ao requerente), da própria de substância, dos animais-alvo e dascondições de utilização. O requerente usa como referência o presente anexo e o anexo III a fim de avaliar quais os estudos e ainformação a apresentar com o pedido.
O requerente apresenta claramente as razões para a omissão ou o desvio do processo em relação a quaisquer dados previstosno presente anexo, no anexo III e no anexo IV.
O processo inclui os relatórios pormenorizados de todos os estudos efectuados, apresentados em conformidade com osistema de numeração proposto no presente anexo. O processo inclui referências e cópias de todos os dados científicospublicados mencionados e cópias de quaisquer outros pareceres relevantes que já tenham sido emitidos por qualquerorganismo científico reconhecido. Sempre que estes estudos já tenham sido avaliados por um organismo científico europeuno seguimento da legislação em vigor na Comunidade, é suficiente uma referência ao resultado da avaliação. Os dados dosestudos que foram conduzidos e publicados previamente ou provenientes da avaliação interpares referem-se claramente aomesmo aditivo que o apresentado no pedido de autorização.
Os estudos, incluindo os que foram conduzidos e publicados previamente ou provenientes da avaliação interpares, sãoexecutados e documentados de acordo com normas de qualidade adequadas [por exemplo, as Boas Práticas de Laboratório(BPL)], em conformidade com a Directiva 2004/10/CE do Parlamento Europeu e do Conselho, de 11 de Fevereiro de 2004,relativa à aproximação das disposições legislativas, regulamentares e administrativas respeitantes à aplicação dos princípiosde boas práticas de laboratório e ao controlo da sua aplicação nos ensaios sobre as substâncias químicas (1) ou com aOrganização Internacional de Normalização (ISO).
Quando forem realizados estudos in vivo ou in vitro fora da Comunidade, o requerente demonstra que as instalações emcausa cumprem os princípios da Organização de Cooperação e de Desenvolvimento Económico (OCDE) das Boas Práticasde Laboratório ou de normas ISO.
A determinação das propriedades físico-químicas, toxicológicas e ecotoxicológicas tem de ser efectuada pelos métodosfixados na Directiva 67/548/CEE do Conselho, de 27 de Junho de 1967, relativa à aproximação das disposições legislativas,regulamentares e administrativas respeitantes à classificação, embalagem e rotulagem das substâncias perigosas (2), com aúltima redacção que lhe foi dada pela Directiva 2004/73/CE da Comissão (3), ou por métodos actualizados reconhecidos pororganismos científicos internacionais. A utilização de métodos diferentes dos referidos deve ser justificada.
É incentivada a utilização de métodos in vitro ou de métodos que limitam ou substituem os ensaios habituais com recurso aanimais para ensaio em laboratório ou que reduzem o número de animais utilizados nestes ensaios. Tais métodos devemapresentar a mesma qualidade e fornecer o mesmo nível de garantia que o método que pretendem substituir.
L 133/10 PT Jornal Oficial da União Europeia 22.5.2008
(1) JO L 50 de 20.2.2004, p. 44.(2) JO L 196 de 16.8.1967, p. 1. Directiva com a última redacção que lhe foi dada pela Directiva 2006/121/CE do Parlamento Europeu e do
Conselho (JO L 396 de 30.12.2006, p. 855. Rectificação no JO L 136 de 29.5.2007, p. 281).(3) JO L 152 de 30.4.2004, p. 1. Rectificação no JO L 216 de 16.6.2004, p. 3.

A descrição dos métodos de análise em alimentos para animais ou na água está em conformidade com as regras de BPL talcomo definidas na Directiva 2004/10/CE e/ou na norma EN ISO/IEC 17025. Estes métodos cumprem o disposto noartigo 11.o do Regulamento (CE) n.o 882/2004 do Parlamento Europeu e do Conselho, de 29 de Abril de 2004, relativo aoscontrolos oficiais realizados para assegurar a verificação do cumprimento da legislação relativa aos alimentos para animais eaos géneros alimentícios e das normas relativas à saúde e ao bem-estar dos animais (4).
Cada processo contém um resumo público e um resumo científico pormenorizado, a fim de permitir ao aditivo em causaser identificado e caracterizado.
Cada processo contém uma proposta de monitorização pós-comercialização, sempre que tal seja exigido pelo n.o 3,alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003 e uma proposta de rotulagem, tal como referida no n.o 3,alínea e), do artigo 7.o do Regulamento (CE) n.o 1831/2003.
Avaliação da segurança
Baseia-se em estudos destinados a demonstrar a segurança da utilização do aditivo em relação:
a) Às espécies-alvo ao níveis mais elevados propostos de incorporação nos alimentos para animais ou na água e nummúltiplo desse nível para determinar uma margem de segurança;
b) Aos consumidores que ingerem produtos alimentares obtidos de animais que receberam o aditivo, os seus resíduos ouos seus metabolitos. Neste caso, a segurança será garantida através da definição de Limites Máximos de Resíduos (LMR)e de intervalos de segurança com base numa Dose Diária Admissível (DDA) ou um Nível Máximo de IngestãoTolerável (UL);
c) Às pessoas susceptíveis de serem expostas ao aditivo por contacto respiratório, das mucosas, oftalmológico oucutâneo ao manipular o aditivo ou ao incorporá-lo em pré-misturas ou em alimentos completos para animais ou naágua ou ao utilizar alimentos para animais e água que contenham o aditivo em causa;
d) Aos animais e seres humanos, no que se refere à selecção e propagação de genes com resistência antimicrobiana; e
e) Ao ambiente, em consequência do próprio aditivo ou de produtos dele derivados, directamente e/ou excretados pelosanimais.
Sempre que um aditivo contenha múltiplos constituintes, cada um pode ser separadamente avaliado em termos desegurança para o consumidor e considerar-se depois o efeito cumulativo (sempre que se possa demonstrar que não háinteracções entre os constituintes). Alternativamente, é avaliada a mistura completa.
Avaliação de eficácia
É feita com base em estudos cuja finalidade é demonstrar a eficácia de um aditivo em termos dos objectivos da sua utilizaçãoprevista, tal como definido no n.o 1 do artigo 6.o e no anexo I do Regulamento (CE) n.o 1831/2003.
1. SECÇÃO I: RESUMO DO PROCESSO
1.1. Resumo público em conformidade com o n.o 3, alínea h), do artigo 7.o do Regulamento (CE) n.o 1831//2003
O requerente apresenta um resumo que indica as principais características do aditivo em causa. O resumo nãocontém informação confidencial e é estruturado do seguinte modo:
1.1.1. Conteúdo
a) Nome do(s) requerente(s);
b) Identificação do aditivo;
c) Método de produção e método de análise;
d) Estudos sobre segurança e eficácia do aditivo;
e) Condições propostas de utilização; e
f) Proposta de monitorização pós-comercialização no mercado.
22.5.2008 PT Jornal Oficial da União Europeia L 133/11
(4) JO L 165 de 30.4.2004. Rectificação no JO L 191 de 28.5.2004, p. 1.

1.1.2. Descrição
a) Nome e endereço do(s) requerente(s);
Esta informação é facultada em todos os casos, independentemente do tipo de autorização do aditivodestinado à alimentação animal (específica ao requerente ou não específica ao requerente). Quando umprocesso for apresentado por um grupo de requerentes, é indicado o nome de cada um deles.
b) Identificação do aditivo
A identificação do aditivo contém um resumo da informação exigida de acordo com anexo II ou III, emfunção do tipo de autorização do aditivo destinado à alimentação animal. Designadamente: nome doaditivo, classificação proposta por categoria e grupo funcional, espécies/categorias animais-alvo e doses.
c) Método de produção e método de análise
É descrito o processo de fabrico.
São descritos os procedimentos gerais dos métodos analíticos a utilizar para a análise para efeitos decontrolos oficiais do aditivo como tal, em pré-misturas e em alimentos para animais, tal como definidono presente anexo e no anexo III. Se necessário, com base na informação apresentada de acordo com opresente anexo e o anexo III, é incluído o protocolo do(s) método(s) a utilizar para a análise para efeitosde controlos oficiais dos aditivos ou seus metabolitos em alimentos de origem animal.
d) Estudos sobre segurança e eficácia do aditivo
É fornecida a conclusão em relação à segurança e eficácia do aditivo com base nos diferentes estudosexecutados. Os resultados dos estudos podem ser incluídos em forma de tabela para apoiar a conclusãodo(s) requerente(s). Apenas são indicados no resumo os estudos exigidos de acordo com anexo III.
e) Condições propostas de utilização
O(s) requerente(s) apresenta(m) a proposta de condições de utilização. Em particular, o requerentedescreve o nível de utilização na água ou em alimentos para animais, conjuntamente com as condiçõespormenorizadas de utilização em alimentos complementares para animais. É igualmente exigidainformação quando forem utilizados outros métodos de administração ou incorporação em alimentospara animais ou na água. São descritas quaisquer condições específicas de utilização (por exemplo,incompatibilidades), requisitos específicos de rotulagem e espécies animais às quais se destinado o aditivo.
f) Proposta de monitorização pós-comercialização no mercado
Esta parte refere-se apenas a aditivos que, de acordo com o n.o 3, alínea g), do artigo 7.o do Regulamento(CE) n.o 1831/2003, não pertençam às categorias mencionadas no n.o 1, alíneas a) e b), do artigo 6.o domesmo regulamento e a aditivos abrangidos pelo âmbito de legislação comunitária referente àcomercialização de produtos que consistam em, contenham ou sejam produzidos a partir de OGM.
1.2. Resumo científico do processo
É fornecido um resumo científico incluindo detalhes de cada parte dos documentos apresentados para apoiar opedido, de acordo com o presente anexo e o anexo III. Este resumo inclui as conclusões elaboradas pelo(s)requerente(s).
O resumo tem de seguir a ordem do presente anexo e abordar todos os elementos com referência às páginasrelevantes do processo.
1.3. Lista de documentos e outros pormenores
O requerente tem de identificar o número e os títulos dos volumes de documentação apresentados em apoio dopedido. É incluído um índice pormenorizado com referência a volumes e páginas.
L 133/12 PT Jornal Oficial da União Europeia 22.5.2008

1.4. Lista de partes do processo cujo tratamento foi solicitado como confidencial, se necessário
A lista faz referência aos volumes e às páginas relevantes do processo.
2. SECÇÃO II: IDENTIDADE, CARACTERIZAÇÃO E CONDIÇÕES DE UTILIZAÇÃO DO ADITIVO;MÉTODOS DE ANÁLISE
O aditivo tem de ser plenamente identificado e caracterizado.
2.1. Identidade do aditivo
2.1.1. Designação do aditivo
Se necessário, é feita uma proposta para a designação comercial para aditivos ligados a um detentor deautorização.
2.1.2. Proposta de classificação
É feita uma proposta para a classificação de um aditivo para uma ou mais categorias e grupos funcionais, deacordo com as suas funções principais, nos termos do artigo 6.o e do anexo I do Regulamento (CE) n.o 1831//2003.
Têm de ser fornecidos quaisquer dados relativos a outras utilizações conhecidas de substâncias activas ouagentes idênticos (por exemplo, nos géneros alimentícios, na medicina humana ou veterinária, na agricultura ena indústria). Tem de ser especificada qualquer outra autorização como aditivo destinado à alimentaçãohumana ou animal, medicamento para uso veterinário ou outro tipo de autorização da substância activa.
2.1.3. Composição qualitativa e quantitativa (substância activa/agente, outros constituintes, impurezas, variação entre lotes)
Enumeram-se a(s) substância(s) activa(s)/agente(s) e todos os outros constituintes do aditivo, fornecendo-se aproporção em peso no produto final. É determinada a variação qualitativa e quantitativa da(s) substância(s)activa(s)/agente(s) lote a lote.
Quanto aos microrganismos: É determinado o número das células ou esporos viáveis expressas como UFC porgrama.
Quanto às enzimas: É descrita cada actividade (principal) declarada e é revelado o número de unidades de cadaactividade no produto final. São igualmente mencionadas as actividades colaterais relevantes. As unidades deactividade são definidas preferivelmente em μmoles de produto libertado por minuto a partir do substrato,indicando-se igualmente o pH e a temperatura.
Se o componente activo do aditivo for uma mistura de substâncias activas ou de agentes, sendo cada uma delasmanifestamente definível (qualitativa e quantitativamente), os constituintes da(s) substância(s) activa(s)/agente(s)têm de ser descritos separadamente e fornecidas as respectivas proporções na mistura.
Outras misturas nas quais os componentes não podem ser descritos por uma única fórmula química e/ou ondenem tudo pode ser identificado são caracterizadas pelos componente(s) que contribuem para a sua actividade e//ou componente(s) principal(ais) típico(s).
Sem prejuízo de qualquer pedido de informação suplementar feito pela Autoridade em conformidade com on.o 2 do artigo 8.o do Regulamento (CE) n.o 1831/2003, o requerente pode omitir a descrição de outrosconstituintes que não suscitem problemas de segurança à excepção de substâncias activas ou agentes paraaditivos que não se enquadrem nas categorias dos aditivos zootécnicos, coccidiostáticos e histomonostáticos enão sejam abrangidos pelo âmbito de aplicação do Regulamento (CE) n.o 1829/2003. Em qualquer caso, todosos estudos assinalados no processo devem ser baseados no aditivo próprio cuja autorização se solicita e podemfacultar informação sobre outros preparados diferentes possíveis que poderiam ser efectuados. Pode serpermitido um identificador interno, incluído em documentos de terceiros, e é necessária uma declaração paraenumerar os identificadores e confirmar que o(s) identificador(es) se refere(m) à(s) formulação(ões) para a(s)qual(ais) é apresentado o pedido.
22.5.2008 PT Jornal Oficial da União Europeia L 133/13

2.1.4. Pureza
O requerente identifica e quantifica as impurezas químicas e microbianas, substâncias com propriedades tóxicasou outras propriedades indesejáveis que não são intencionalmente acrescentadas e que não contribuem para aactividade do aditivo. Além disso, para produtos de fermentação, o requerente confirma a ausência deorganismos produtores no aditivo. É descrito o protocolo utilizado para o rastreio de rotina de lotes deprodução para detecção de contaminantes e impurezas.
Todos os dados apresentados têm de apoiar a proposta de especificação do aditivo.
Encontram-se enumerados infra os requisitos específicos em função do processo de produção, emconformidade com a legislação comunitária em vigor.
2.1.4.1. Aditivos cuja autorização está ligada a um detentor de autorização
Para aditivos cuja autorização esteja ligada a um detentor de autorização, é fornecida a informação relevanterelativa ao processo específico utilizado pelo fabricante, com base nas normas existentes utilizadas para outrosfins relacionados. Podem ser utilizadas as especificações do Comité Misto FAO-OMS de Peritos em AditivosAlimentares (CMPAA) ou as especificações de autorizações de aditivos alimentares da Comunidade Europeia.
2.1.4.2. Aditivos cuja autorização não está ligada a um detentor de autorização
Para aditivos destinados à alimentação animal cuja autorização não esteja ligada a um detentor de autorização,podem ser utilizadas as normas existentes utilizadas para outros fins relacionados, ou que possuamespecificações para aditivos alimentares, tal como autorizados na Comunidade Europeia ou provenientes doCMPAA. Sempre que tais normas não estiverem disponíveis, ou sempre que relevante para o processo defabrico, são, pelo menos, descritos os seguintes aspectos e determinadas as suas concentrações:
— quanto aos microrganismos: contaminação microbiológica, micotoxinas, metais pesados,
— para produtos de fermentação (que não contêm microrganismos como agentes activos): seguem osmesmos requisitos que os dos relativos aos produtos de microrganismos (ver supra). É igualmenteindicado até que ponto o meio de crescimento gasto é incorporado no produto final,
— para substâncias derivadas de plantas: contaminação microbiológica e botânica (por exemplo, planta deóleo de rícino, sementes de ervas daninhas e, em especial, cravagem de centeio), micotoxinas,contaminação por pesticidas, valores máximos para solventes e, quando necessário, substâncias quelevantam preocupação a nível toxicológico cuja ocorrência se conhece na planta original,
— para substâncias derivadas de animais: contaminação microbiológica, metais pesados e valores máximospara solventes, quando necessário,
— para substâncias minerais: metais pesados, dioxinas e PCB,
— para produtos produzidos por síntese e processos químicos: são identificados todos os produtosquímicos utilizados nos processos de síntese e quaisquer produtos intermédios que permaneçam noproduto final e são dadas as suas concentrações.
A selecção de micotoxinas para análise é feita de acordo com as diferentes matrizes, quando necessário.
2.1.5. Estado físico de cada forma do produto
No que se refere a preparados sólidos são apresentados dados relativos à distribuição da dimensão daspartículas, forma das partículas, densidade, densidade aparente, potencial de formação de poeiras e à utilizaçãode processos que afectam as propriedades físicas. Para preparados líquidos, são fornecidos dados sobreviscosidade e tensão superficial. Sempre que o aditivo se destine a ser utilizado na água, são demonstradas asolubilidade ou o grau de dispersão.
2.2. Caracterização da(s) substância(s) activa(s)/agente(s)
2.2.1. Descrição
É fornecida uma descrição qualitativa da substância activa ou do agente. Inclui a pureza e a origem dasubstância ou do agente e quaisquer outras características relevantes.
L 133/14 PT Jornal Oficial da União Europeia 22.5.2008

2.2.1.1. Produtos químicos
As substâncias quimicamente bem definidas são descritas pela designação genérica, designação químicasegundo a nomenclatura IUPAC (União Internacional de Química Pura e Aplicada), outras designações eabreviaturas genéricas internacionais e/ou Número CAS (Chemical Abstracts Service). Devem ser incluídos afórmula estrutural e molecular e o peso molecular.
Para compostos quimicamente definidos utilizados como aromatizantes, é incluído o número FLAVIS relativoao grupo químico relevante. Para extractos de plantas, devem ser incluídos os marcadores fitoquímicos.
As misturas nas quais os componentes não podem ser descritos por uma única fórmula química e/ou nemtodos podem ser identificados são caracterizadas pelos componente(s) que contribuem para a sua actividade e//ou componente(s) principal(ais) típico(s). É identificado o composto marcador para permitir avaliar aestabilidade e apresentar uma forma de rastreabilidade.
Para as enzimas e preparados enzimáticos, são fornecidos para cada actividade declarada o número e o nomesistemático propostos pela União Internacional de Bioquímica (UIB) na edição mais recente da «Nomenclaturadas Enzimas». Para as actividades ainda não incluídas, é utilizada uma designação sistemática coerente com asregras de nomenclatura da UIB. São aceitáveis designações triviais desde que sejam inequívocas e utilizadas deforma coerente no processo e que possam ser claramente relacionadas com a designação sistemática e onúmero UIB na sua primeira menção. Deve ser fornecida a origem biológica de cada actividade enzimática.
A origem microbiana de substâncias químicas produzidas por fermentação é igualmente descrita (ver 2.2.1.2Microrganismos).
2.2.1.2. Microrganismos
É apresentada a origem para todos os microrganismos, quer sejam utilizados como produto ou como estirpe deprodução.
Para microrganismos utilizados como produto ou como estirpe de produção, é indicada qualquer história demodificação. A designação e a classificação taxonómica de cada microrganismo são apresentadas, de acordocom a última informação publicada nos Códigos Internacionais de Nomenclatura (CIN). As estirpesmicrobianas são depositadas numa colecção de culturas internacionalmente reconhecida (de preferência naUnião Europeia) e mantidas pela colecção de culturas durante a vida autorizada do aditivo. Tem de serfornecido um certificado de depósito emitido pela colecção, que especifica o número de adesão sob o qual aestirpe é mantida. Além disso, são descritas todas as características morfológicas, fisiológicas e molecularesrelevantes necessárias para a identificação única da estirpe e os meios de confirmar a sua estabilidade genética.Para os OGM é fornecida a descrição das modificações genéticas. É incluído um identificador único a cadaOGM, nos termos do Regulamento (CE) n.o 65/2004 da Comissão, de 14 de Janeiro de 2004, que estabeleceum sistema para criação e atribuição de identificadores únicos aos organismos geneticamente modificados.
2.2.2. Propriedades relevantes
2.2.2.1. Produtos químicos
É fornecida a descrição das propriedades físicas e químicas. São fornecidas, sempre que adequado, a constantede dissociação, pKa, propriedades electrostáticas, ponto de fusão, ponto de ebulição, densidade, pressão devapor, solubilidade em água e em solventes orgânicos, Kow e Kd/Koc, espectro de massa e de absorção, dados deRMN, possíveis isómeros e qualquer outra propriedade física pertinente.
Uma substância produzida através de fermentação está isenta de actividades antimicrobianas relevantes para autilização de antibióticos em seres humanos ou animais.
2.2.2.2. Microrganismos
— Toxinas e factores de virulência
É demonstrada a ausência ou a insignificância de toxinas ou factores de virulência. As estirpes debactérias pertencentes a um grupo taxonómico que inclua membros cuja capacidade para produzirtoxinas ou factores de virulência seja conhecida estão sujeitas a ensaios adequados para demonstrar anível molecular e, se necessário, a nível celular a ausência de motivos de apreensão.
Para as estirpes de microrganismos para as quais não existe historial de utilização segura aparente e cujabiologia permaneça mal conhecida, é necessário um conjunto completo de estudos toxicológicos.
22.5.2008 PT Jornal Oficial da União Europeia L 133/15

— Produção de antibióticos e resistência aos antibióticos
Os microrganismos utilizados como aditivos ou como estirpe de produção estão isentos de actividadeantibiótica ou não são capazes de produzir substâncias antibióticas relevantes enquanto antibióticos emseres humanos e animais.
As estirpes de microrganismos destinadas a utilização como aditivos não contribuem para aumentar oreservatório de genes de resistência aos antibióticos já presentes na flora intestinal dos animais e noambiente. Consequentemente, todas as estirpes de bactérias são testadas à resistência aos antibióticosutilizados em medicamentos para uso humano e veterinário. Sempre que for detectada resistência, sãoestabelecidas a base genética da resistência e a probabilidade de transferência da resistência a outrosorganismos presentes no intestino.
As estirpes de microrganismos que transportam uma resistência adquirida a antimicrobiano(s) não sãoutilizadas como aditivos destinados à alimentação animal, a menos que se possa demonstrar que aresistência é um resultado de mutação(ões) cromossomática(s) e não é transferível.
2.3. Processo de fabrico, incluindo quaisquer tratamentos específicos eventuais
Para definir os pontos críticos do processo que podem ter uma influência na pureza da(s) substância(s) activa(s)//agente(s) ou do aditivo é fornecida uma descrição do processo de fabrico. É fornecida uma ficha de segurançados produtos químicos utilizados no processo de produção.
2.3.1. Substância(s) activa(s)/agente(s)
É apresentada uma descrição do processo de produção (por exemplo, síntese química, fermentação, cultivo,extracção do material orgânico ou destilação) utilizado na preparação da(s) substância(s) activa(s)/agente(s) doaditivo, se adequado, por meio de um fluxograma. É apresentada a composição dos meios de fermentação//cultivo. Os métodos de purificação são pormenorizadamente descritos.
Para microrganismos geneticamente modificados (MGM), utilizados como fonte de aditivos e produzidos emcondições de confinamento, aplica-se a Directiva 90/219/CE do Conselho (5). É incluída uma descrição dosprocessos de fermentação (meio de cultura, condições de fermentação e tratamento a jusante dos produtos dafermentação).
2.3.2. Aditivo
É apresentada uma descrição pormenorizada do processo de fabrico do aditivo. Devem ser apresentadas asfases-chave na preparação do aditivo, incluindo o(s) ponto(s) de introdução da(s) substância(s) activa(s)/agente(s)e outros constituintes, e quaisquer etapas de tratamento subsequentes que afectem o preparado de aditivos, seadequado, por meio de um fluxograma.
2.4. Propriedades físico-químicas e tecnológicas do aditivo
2.4.1. Estabilidade
A estabilidade é, de modo geral, medida pelo acompanhamento analítico da(s) substância(s) activa(s)/agente(s)ou da sua actividade/viabilidade. Para as enzimas, a estabilidade pode ser definida em termos de perda deactividade catalítica; para os microrganismos em termos de perda de viabilidade; para as substânciasaromatizantes em termos de perda de aroma. Para outras misturas/extractos químicos a estabilidade pode seravaliada mediante a monitorização da concentração de uma ou mais substâncias marcadoras adequadas.
Estabilidade do aditivo
É estudada a estabilidade de cada formulação do aditivo à exposição a diferentes condições ambientais (luz,temperatura, pH, humidade, oxigénio e material de embalagem). O prazo de validade esperado para o aditivo nasua forma comercial deve ser baseado em, pelo menos, dois modelos de situação que abranjam a gama provávelde condições de utilização [por exemplo, 25 oC, 60 % humidade relativa do ar (HR) e 40 oC, 75 % HR].
L 133/16 PT Jornal Oficial da União Europeia 22.5.2008
(5) JO L 117 de 8.5.1990, p. 1. Directiva com a última redacção que lhe foi dada pela Decisão 2005/174/CE da Comissão (JO L 59 de5.3.2005, p. 20).

Estabilidade do aditivo utilizado em pré-misturas e alimentos para animais
Para aditivos utilizados em pré-misturas e em alimentos para animais, com excepção de compostos dearomatizantes, é estudada a estabilidade de cada formulação do aditivo em condições de fabrico earmazenamento comuns de pré-misturas e de alimentos para animais. Os estudos de estabilidade em pré--misturas têm, pelo menos, seis meses de duração. A estabilidade é testada preferivelmente com pré-misturascontendo oligoelementos; de outra forma, o aditivo deve ser rotulado como «Não misturar comoligoelementos».
Os estudos de estabilidade em alimentos para animais têm normalmente uma duração de, pelo menos, trêsmeses. A estabilidade é verificada de modo geral em alimentos para animais em pasta e granulados (incluindo ainfluência da granulação ou outras formas de tratamento) para as espécies animais principais do pedido.
Para aditivos destinados a serem utilizados em água, a estabilidade de cada formulação do aditivo tem de serestudada em água em condições que imitem a utilização prática.
Sempre que se verifique uma perda de estabilidade, e sempre que necessário, são caracterizados os produtospotenciais da degradação ou decomposição.
São apresentados os dados de análises que incluam, pelo menos, uma observação no início e uma no final doperíodo de armazenamento.
Se necessário, os estudos contêm a composição quantitativa e qualitativa pormenorizada das pré-misturas oudos alimentos para animais utilizados nas experiências.
2.4.2. Homogeneidade
Tem de ser demonstrada a capacidade para a distribuição homogénea do aditivo destinado à alimentaçãoanimal (com excepção de compostos de aromatizantes) em pré-misturas, alimentos para animais ou água.
2.4.3. Outras características
Têm de ser descritas outras características, como potencial de formação de poeiras, propriedades electrostáticasou dispersibilidade em líquidos.
2.4.4. Incompatibilidades ou interacções físico-químicas
Têm de ser demonstradas as incompatibilidades ou interacções físico-químicas que se possam esperar com osalimentos para animais, excipientes, outros aditivos aprovados ou medicamentos.
2.5. Condições de utilização do aditivo
2.5.1. Modo de utilização proposto na alimentação animal
São indicadas as espécies ou categorias animais, a faixa etária ou a fase de produção de animais, emconformidade com as categorias constantes do anexo IV do presente regulamento. São mencionadas as contra--indicações possíveis. É definida a utilização proposta, em alimentos para animais ou água.
Os pormenores do método de administração proposto e do nível de inclusão devem ser apresentados para pré--misturas, alimentos para animais ou água para abeberamento. Além disso, devem ser fornecidos quandonecessário, a dose proposta nos alimentos completos para animais, a duração de administração proposta e ointervalo de segurança proposto. É exigida uma justificação sempre que uma utilização particular de um aditivoem alimentos complementares para animais seja proposta.
2.5.2. Informação relacionada com a segurança de utilizadores/trabalhadores
2.5.2.1. Produtos químicos
Tem de ser fornecida uma ficha de segurança para o material formatada em conformidade com a Directiva 91//155/CEE da Comissão, de 5 de Março de 1991, que define e estabelece, nos termos do artigo 10.o da Directiva88/379/CEE do Conselho, as modalidades do sistema de informação específico relativo às preparaçõesperigosas (6). Se necessário, são propostas medidas de prevenção dos riscos profissionais e meios de protecçãodurante o fabrico, manipulação, utilização e eliminação.
22.5.2008 PT Jornal Oficial da União Europeia L 133/17
(6) JO L 76 de 22.3.1991, p. 35. Directiva com a última redacção que lhe foi dada pela Directiva 2001/58/CE (JO L 212 de 7.8.2001, p. 24).

2.5.2.2. Microrganismos
É apresentada uma classificação em conformidade com a Directiva 2000/54/CE do Parlamento Europeu e doConselho, de 18 de Setembro de 2000, relativa à protecção dos trabalhadores contra riscos ligados à exposiçãoa agentes biológicos durante o trabalho (Sétima directiva especial nos termos do n.o 1 do artigo 16.o daDirectiva 89/391/CEE) (7). Para microrganismos não classificados no grupo 1 nesta directiva, é facultada aosclientes informação que lhes permita adoptar as medidas de protecção relevantes para os seus trabalhadores, talcomo definido no n.o 2 do artigo 3.o da referida directiva.
2.5.2.3. Requisitos de rotulagem
Sem prejuízo das disposições de rotulagem e embalagem definidas no artigo 16.o do Regulamento (CE)n.o 1831/2003, são indicados quaisquer requisitos específicos de rotulagem e, quando necessário, as condiçõesespecíficas para utilização e manipulação (incluindo incompatibilidades e contra-indicações conhecidas) einstruções para a utilização adequada.
2.6. Métodos de análise e amostras de referência
Os métodos de análise são apresentados no formato normalizado recomendado pela ISO (ou seja, ISO 78-2).
De acordo com o Regulamento (CE) n.o 1831/2003 e com o Regulamento (CE) n.o 378/2005, são avaliadospelo LCR os métodos de análise incluídos nesta secção. O LCR apresenta à Autoridade um relatório deavaliação que indica se estes métodos são apropriados para ser utilizados em controlos oficiais do aditivodestinado à alimentação animal, objecto do pedido. A avaliação do LCR concentra-se nos métodos definidosnas secções 2.6.1 e 2.6.2.
Se o Regulamento (CEE) n.o 2377/90 do Conselho, de 26 de Junho de 1990, que prevê um processocomunitário para o estabelecimento de limites máximos de resíduos de medicamentos veterinários nosalimentos de origem animal (8), tiver estabelecido um LMR para a substância objecto do pedido, a secção 2.6.2não será sujeita a avaliação pelo LCR. O requerente preenche a secção 2.6.2 facultando o mesmo método,informação e pormenores (incluindo actualizações relevantes) para apresentação à Agência Europeia dosMedicamentos (AEAM) em conformidade com o anexo V do Regulamento (CEE) n.o 2377/90 e emconformidade com a «Nota aos requerentes e orientações», volume 8 da série «Regras que regem os produtosfarmacêuticos na Comunidade Europeia».
Podem igualmente ser incluídos na avaliação os métodos analíticos descritos em 2.6.3, se o LCR, a Autoridadeou a Comissão assim considerar necessário.
Em conformidade com o Regulamento (CE) n.o 378/2005, o requerente fornece amostras de referênciadirectamente ao LCR antes da avaliação do dossier técnico e amostras de substituição antes da data de validade.
Os requerentes referem-se à orientação pormenorizada apresentada pelo LCR em conformidade com oartigo 12.o do Regulamento (CE) n.o 378/2005.
2.6.1. Métodos de análise para a substância activa
É fornecida a caracterização pormenorizada do(s) método(s) analítico(s) qualitativo(s) e, sempre que aplicável,quantitativo(s) para determinar o cumprimento dos níveis máximo ou mínimo propostos da(s) substância(s)activa(s)/agente(s) no aditivo, pré-misturas, alimentos para animais e, sempre que adequado, na água.
2.6.1.1. Estes métodos cumprem os mesmos requisitos que os relativos aos métodos de análise utilizados para fins decontrolo oficial em conformidade com o artigo 11.o do Regulamento (CE) n.o 882/2004. Cumprem,nomeadamente, pelo menos um dos seguintes requisitos:
— cumprimento das regras comunitárias relevantes (por exemplo, métodos comunitários de análise) sempreque existirem,
— cumprimento de regras ou protocolos reconhecidos internacionalmente, por exemplo, os que o ComitéEuropeu de Normalização (CEN) aceitou, ou os acordados na legislação nacional (por exemplo, métodos--padrão do CEN),
L 133/18 PT Jornal Oficial da União Europeia 22.5.2008
(7) JO L 262 de 17.10.2000, p. 21.(8) JO L 224 de 18.8.1990, p. 1. Regulamento com a última redacção que lhe foi dada pelo Regulamento (CE) n.o 203/2008 da Comissão
(JO L 60 de 5.3.2008, p. 18).

— são aptos para o objectivo pretendido, desenvolvidos em conformidade com protocolos científicos evalidados numa prova do anel em conformidade com um protocolo internacionalmente reconhecido emmatéria de experiências colaborativas (por exemplo, ISO 5725 ou IUPAC), ou
— são validados internamente de acordo com orientações internacionais harmonizadas para a validaçãointerna de métodos de análise (9) no que diz respeito aos parâmetros de caracterização mencionadosem 2.6.1.2.
2.6.1.2. A caracterização pormenorizada do(s) método(s) inclui as características adequadas estabelecidas no anexo IIIdo Regulamento (CE) n.o 882/2004.
2.6.1.3. As características de desempenho dos métodos validados internamente são verificadas mediante o ensaio dométodo num segundo laboratório acreditado e independente. Os resultados de tais ensaios são apresentadosconjuntamente com qualquer outra informação que apoia a transferibilidade do método a um laboratório decontrolo oficial. Por motivos de independência e participação na avaliação da documentação fornecida pelorequerente, sempre que o segundo laboratório seja um laboratório participante no consórcio de laboratóriosnacionais de referência (LNR) assistindo o LCR, em conformidade com o Regulamento (CE) n.o 378/2005, olaboratório envia uma declaração de interesses ao LCR, assim que o pedido seja recebido pelo LCR, descrevendoo trabalho do laboratório no pedido e não participa na sua avaliação.
2.6.1.4. O LCR pode seleccionar características adequadas, tal como mencionado ao abrigo do anexo III doRegulamento (CE) no 882/2004 no seu relatório de avaliação à Autoridade.
2.6.1.5. Os critérios de desempenho para métodos de grupos específicos de substâncias (por exemplo, enzimas) podemser estabelecidos nas orientações pormenorizadas apresentadas pelo LCR em conformidade com o artigo 12.odo Regulamento (CE) n.o 378/2005.
2.6.2. Métodos de análise para a determinação dos resíduos do aditivo ou dos seus metabolitos em géneros alimentícios
É apresentada a caracterização pormenorizada do(s) método(s) analítico(s) qualitativo(s) e quantitativo(s) para adeterminação dos resíduos marcadores e/ou metabolitos do aditivo em tecidos-alvo e em produtos de origemanimal.
2.6.2.1. Estes métodos cumprem os mesmos requisitos que os relativos aos métodos de análise utilizados para fins decontrolo oficial em conformidade com o artigo 11.o do Regulamento (CE) n.o 882/2004. Nomeadamente, osmétodos cumprem, pelo menos, um dos requisitos mencionados em 2.6.1.1.
2.6.2.2. A caracterização pormenorizada do(s) método(s) inclui as características adequadas tal como estabelecidas noanexo III do Regulamento (CE) n.o 882/2004 e tem em conta os requisitos estabelecidos na Decisão 2002/657//CE da Comissão (10). São considerados, quando necessário, os mesmos critérios de desempenho definidos emdecisões da Comissão que estabelecem métodos analíticos a utilizar para detectar certas substâncias e seusresíduos em produtos de origem animal, de acordo com a Directiva 96/23/CE do Conselho.
O limite de quantificação (LOQ) para cada método não deve exceder metade do LMR correspondente e deve servalidado na gama situada entre, pelo menos, metade e duas vezes o valor do LMR.
2.6.2.3. As características de desempenho dos métodos validados internamente são verificadas mediante o ensaio dométodo num segundo laboratório acreditado e independente. São apresentados os resultados de tais ensaios.Por motivos de independência e participação na avaliação da documentação fornecida pelo requerente, sempreque o segundo laboratório seja um laboratório participante no consórcio de laboratórios nacionais dereferência (LNR) assistindo o LCR, em conformidade com o Regulamento (CE) n.o 378/2005, o laboratórioenvia uma declaração de interesses ao LCR, assim que o pedido seja recebido pelo LCR, descrevendo o trabalhodo laboratório no pedido e não participa na sua avaliação.
2.6.2.4. O LCR pode seleccionar as características adequadas das mencionadas no ponto 2.6.2.2 no seu relatório deavaliação à Autoridade.
22.5.2008 PT Jornal Oficial da União Europeia L 133/19
(9) M. Thompson et al.: Harmonized Guidelines For Single Laboratory Validation Of Methods Of Analysis (IUPAC Technical Report) PureAppl. Chem., Vol. 74, No. 5, pp. 835-855, 2002.
(10) JO L 221 de 17.8.2002, p. 8. Decisão com a última redacção que lhe foi dada pela Decisão 2004/25/CE (JO L 6 de 10.1.2004, p. 38).

2.6.2.5. Os critérios de desempenho para métodos de grupos específicos de substâncias (por exemplo, enzimas) podemser estabelecidos nas orientações pormenorizadas apresentadas pelo LCR em conformidade com o artigo 12.odo Regulamento (CE) n.o 378/2005.
2.6.3. Métodos de análise relativo à identidade e caracterização do aditivo
O requerente apresenta uma descrição dos métodos utilizados para a determinação das característicasconstantes sob os números 2.1.3, 2.1.4, 2.1.5, 2.2.2, 2.4.1, 2.4.2, 2.4.3, e 2.4.4.
Em conformidade com anexo II do Regulamento (CE) n.o 1831/2003, com a redacção que lhe foi dada peloRegulamento (CE) n.o 378/2005, os métodos apresentados ao abrigo desta secção podem igualmente seravaliados, se tal for considerado relevante pela Autoridade ou pela Comissão para a avaliação do pedido.
Recomenda-se que os métodos descritos ao abrigo desta secção sejam internacionalmente reconhecidos. Paraos métodos que não sejam internacionalmente reconhecidos, os mesmos têm de ser plenamente descritos.Nesses casos, os estudos são executados por laboratórios acreditados e independentes e documentados deacordo com as normas de qualidade adequadas (por exemplo, BPL em conformidade com a Directiva 2004/10//CE ou normas ISO).
Os métodos de identificação e caracterização do aditivo cumprem os mesmos requisitos que os relativos aosmétodos de análise utilizados para fins de controlo oficial em conformidade com no artigo 11.o doRegulamento (CE) n.o 882/2004, em especial sempre que estejam definidos requisitos legais (por exemploimpurezas, substâncias indesejáveis).
3. SECÇÃO III: ESTUDOS RELATIVOS À SEGURANÇA DO ADITIVO
Os estudos incluídos nesta secção e nos anexos específicos destinam-se a permitir a avaliação:
— da segurança de utilização do aditivo nas espécies-alvo,
— de qualquer risco associado à selecção e/ou transferência de resistência a antimicrobianos e ao aumentoda persistência e disseminação de agentes enteropatogénicos,
— dos riscos para o consumidor de géneros alimentícios derivados de animais alimentados com alimentoscontendo ou tratados com o aditivo ou que poderiam resultar do consumo de géneros alimentícioscontendo resíduos do aditivo ou dos seus metabolitos,
— dos riscos da inalação e do contacto com outro tecido das mucosas, com os olhos ou com a pele, para aspessoas susceptíveis de manipular o aditivo, tal qual ou incorporado nas pré-misturas ou nos alimentospara animais, e
— dos riscos de efeitos nocivos para o ambiente decorrentes do próprio aditivo ou de produtos delederivados, tanto directamente como excretados pelos animais.
3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Os estudos incluídos nesta secção destinam-se a avaliar:
— a segurança de utilização do aditivo nas próprias espécies-alvo, e
— qualquer risco associado à selecção e/ou transferência de resistência a antimicrobianos e ao aumento dapersistência e disseminação de agentes enteropatogénicos.
3.1.1. Estudos de tolerância para as espécies-alvo
O objectivo do ensaio de tolerância é apresentar uma avaliação limitada da toxicidade a curto prazo do aditivopara os animais-alvo. É igualmente utilizado para determinar uma margem de segurança, se o aditivo forconsumido em doses mais elevadas do que as recomendadas. Tais ensaios de tolerância devem ser realizadospara apresentar provas da segurança para cada uma das espécies/categorias animais-alvo para as quais éefectuada um pedido. Em alguns casos é aceitável incluir alguns elementos do ensaio de tolerância numa dasexperiências de eficácia desde que os requisitos para estes ensaios referidos infra sejam cumpridos. Todos osestudos relatados nesta secção devem ser baseados no aditivo descrito na secção II.
L 133/20 PT Jornal Oficial da União Europeia 22.5.2008

3.1.1.1. A concepção de um ensaio de tolerância inclui um mínimo de três grupos:
— um grupo sem suplemento,
— um grupo com a dose recomendada mais elevada, e
— um grupo experimental com o nível múltiplo da dose recomendada mais elevada.
No grupo experimental, o aditivo é dado, de modo geral, a um valor correspondente a dez vezes a doserecomendada mais elevada. Os animais testados são submetidos a uma rotina de monitorização para detecçãode provas visuais dos efeitos clínicos, características do desempenho, qualidade do produto, hematologia eanálises químicas de rotina do sangue e de outros parâmetros susceptíveis de estarem relacionados com aspropriedades biológicas do aditivo. São considerados parâmetros críticos conhecidos dos estudos toxicológicosem animais para ensaio em laboratório. Qualquer efeito nocivo detectado durante os ensaios de eficácia étambém comunicado nesta secção. A mortalidade inexplicada no ensaio de tolerância é investigada pornecropsia e, se necessário, histologia.
Se se puder demonstrar a tolerância a um valor igual a 100 vezes a dose máxima recomendada, não é exigida arealização de hematologia ou análises químicas de rotina do sangue. Se o produto for tolerado apenas a umnível inferior a dez vezes a dose recomendada mais elevada, o estudo é projectado de modo a que possa sercalculada uma margem de segurança para o aditivo e são apresentados parâmetros adicionais (por necropsia ehistologia, se relevante, e outros critérios adequados).
Para alguns aditivos, em função da sua toxicologia e metabolismo ou utilização, pode não ser necessáriorealizar ensaios de tolerância.
A concepção experimental utilizada tem de incluir elementos estatísticos de eficácia adequada.
3.1.1.2. Duração dos ensaios de tolerância
Quadro 1
Duração dos ensaios de tolerância: Suínos
Animais-alvo Duração dos estudos Característica dos animais-alvo
Leitões não desmamados 14 dias Preferivelmente, a partir de 14 dias anteriores aodesmame
Leitões desmamados 42 dias Durante 42 dias após o desmame
Suínos de engorda 42 dias Massa corporal no início do estudo ≤ 35 kg
Porcas reprodutoras 1 ciclo Desde a inseminação até ao fim do período dedesmame
Se se solicitar a aplicação em leitões não desmamados e desmamados, seria considerado suficiente um estudocombinado (14 dias para leitões não desmamados e 28 dias para leitões desmamados). Se a tolerância paraleitões desmamados for demonstrada, não é exigido um estudo separado para suínos de engorda.
Quadro 2
Duração dos ensaios de tolerância: Aves de capoeira
Animais-alvo Duração dos estudos Característica dos animais-alvo
Frangos de engorda/fran-gos para postura
35 dias A partir da incubação
Galinhas poedeiras 56 dias Preferivelmente, durante o primeiro terço do períodode postura
Perus de engorda 42 dias A partir da incubação
Podem ser utilizados dados de tolerância de frangos ou perus de engorda para demonstrar a tolerânciarelativamente a frangos/galinhas ou perus/peruas para postura/criação, respectivamente.
22.5.2008 PT Jornal Oficial da União Europeia L 133/21
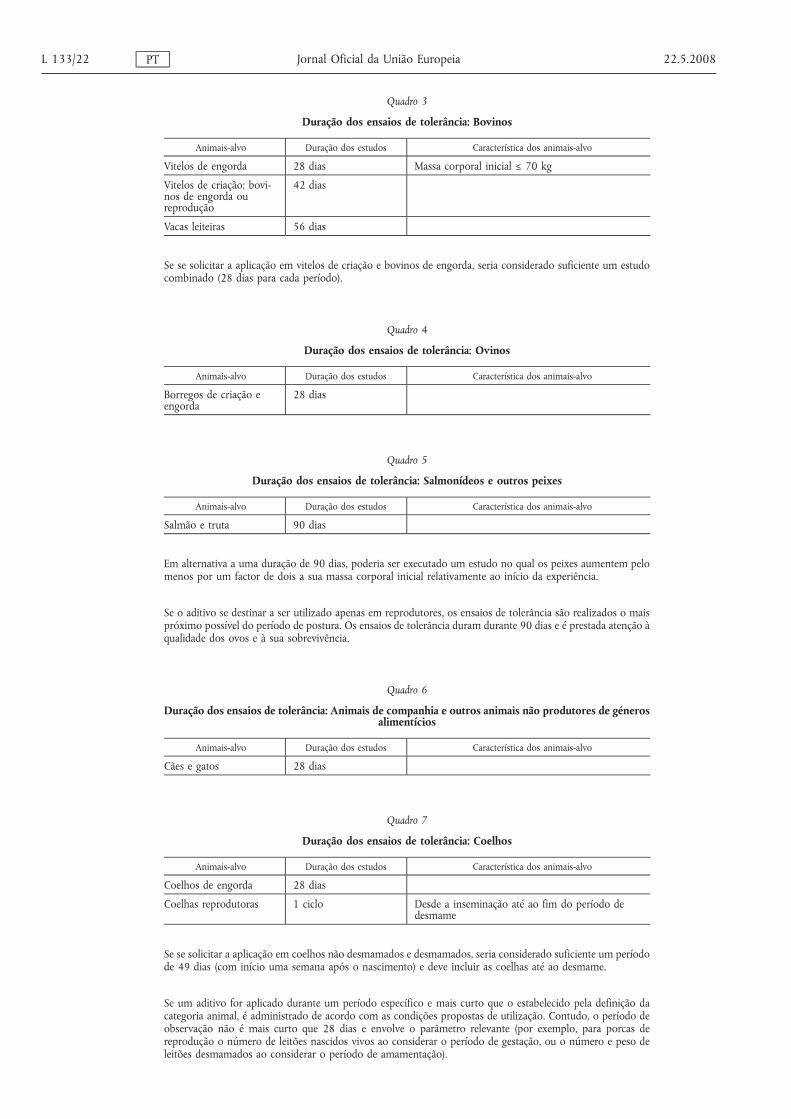
Quadro 3
Duração dos ensaios de tolerância: Bovinos
Animais-alvo Duração dos estudos Característica dos animais-alvo
Vitelos de engorda 28 dias Massa corporal inicial ≤ 70 kg
Vitelos de criação; bovi-nos de engorda oureprodução
42 dias
Vacas leiteiras 56 dias
Se se solicitar a aplicação em vitelos de criação e bovinos de engorda, seria considerado suficiente um estudocombinado (28 dias para cada período).
Quadro 4
Duração dos ensaios de tolerância: Ovinos
Animais-alvo Duração dos estudos Característica dos animais-alvo
Borregos de criação eengorda
28 dias
Quadro 5
Duração dos ensaios de tolerância: Salmonídeos e outros peixes
Animais-alvo Duração dos estudos Característica dos animais-alvo
Salmão e truta 90 dias
Em alternativa a uma duração de 90 dias, poderia ser executado um estudo no qual os peixes aumentem pelomenos por um factor de dois a sua massa corporal inicial relativamente ao início da experiência.
Se o aditivo se destinar a ser utilizado apenas em reprodutores, os ensaios de tolerância são realizados o maispróximo possível do período de postura. Os ensaios de tolerância duram durante 90 dias e é prestada atenção àqualidade dos ovos e à sua sobrevivência.
Quadro 6
Duração dos ensaios de tolerância: Animais de companhia e outros animais não produtores de génerosalimentícios
Animais-alvo Duração dos estudos Característica dos animais-alvo
Cães e gatos 28 dias
Quadro 7
Duração dos ensaios de tolerância: Coelhos
Animais-alvo Duração dos estudos Característica dos animais-alvo
Coelhos de engorda 28 dias
Coelhas reprodutoras 1 ciclo Desde a inseminação até ao fim do período dedesmame
Se se solicitar a aplicação em coelhos não desmamados e desmamados, seria considerado suficiente um períodode 49 dias (com início uma semana após o nascimento) e deve incluir as coelhas até ao desmame.
Se um aditivo for aplicado durante um período específico e mais curto que o estabelecido pela definição dacategoria animal, é administrado de acordo com as condições propostas de utilização. Contudo, o período deobservação não é mais curto que 28 dias e envolve o parâmetro relevante (por exemplo, para porcas dereprodução o número de leitões nascidos vivos ao considerar o período de gestação, ou o número e peso deleitões desmamados ao considerar o período de amamentação).
L 133/22 PT Jornal Oficial da União Europeia 22.5.2008

3.1.1.3. Condições experimentais
Os estudos são relatados individualmente, dando pormenores de todos os grupos experimentais. O protocolodos ensaios é cuidadosamente elaborado em relação aos dados descritivos gerais. Em especial, é registado oseguinte:
1. Efectivo ou bando: localização e tamanho; condições de alimentação e criação, método de alimentação;para espécies aquáticas, dimensão e número de tanques ou recintos na exploração, condições de luz equalidade da água, incluindo temperatura e salinidade;
2. Animais: espécie (para as espécies aquáticas destinadas ao consumo humano, deve referir-se o seu nomecomum seguido, entre parêntesis, do seu binómio latino), raça, idade (tamanho para as espéciesaquáticas), sexo, modo de identificação, fase fisiológica e estado geral de saúde;
3. Datas e duração exacta dos ensaios: Datas e natureza dos exames efectuados;
4. Regimes alimentares: descrição do fabrico e da composição quantitativa do ou dos regimes alimentaresem termos de ingredientes utilizados, nutrientes relevantes (valores analisados) e energia. Registosrelativos à ingestão de alimentos;
5. Estabelece-se a concentração da(s) substância(s) activa(s) ou agente(s) (e, quando for caso disso, de outrasubstâncias utilizadas como termo de comparação) nos alimentos para animais através de uma análise decontrolo, utilizando um método reconhecido adequado: Número(s) de referência dos lotes;
6. Número de grupos de ensaio e de grupos-testemunha, número de animais de cada grupo: O número deanimais envolvidos nos ensaios deve permitir a realização de análises estatísticas. Devem referir-se osmétodos utilizados para as avaliações estatísticas. O relatório inclui todos os animais e/ou unidadesexperimentais envolvidos nos ensaios. Os casos que não puderem ser avaliados devido à falta ou perda dedados são comunicados e é classificada a sua distribuição nos grupos de animais;
7. Deve comunicar-se o momento e a prevalência de quaisquer consequências indesejáveis do tratamentoem indivíduos ou grupos (fornecer pormenores acerca do programa de observações utilizado no estudo);e
8. Os tratamentos terapêuticos/preventivos, se necessário, não interagem com o modo proposto de acçãodo aditivo e são registados individualmente.
3.1.2. Estudos microbianos
São apresentados estudos para determinar a capacidade do aditivo de induzir resistência cruzada a antibióticosutilizados em medicamentos para uso humano ou veterinário, de seleccionar estirpes bacterianas resistentes emcondições reais nas espécies-alvo, de causar efeitos em organismos patogénicos oportunistas presentes notracto digestivo, de causar a disseminação ou excreção de microrganismos zoonóticos.
Se a(s) substância(s) activa(s) possuir(em) actividade antimicrobiana ao nível de concentração no alimento paraanimais, é determinada a concentração inibitória mínima (MIC, Minimum Inhibitory Concentration) em espéciesbacterianas adequadas, em conformidade com os procedimentos normalizados. Sempre que seja demonstradaactividade antimicrobiana relevante, é estabelecida a capacidade do aditivo de seleccionar estirpes bacterianasresistentes in vitro e nas espécies-alvo e de induzir resistência cruzada aos antibióticos relevantes (11).
São apresentados para todos os aditivos microbianos ensaios a nível de utilização recomendada bem como paraaqueles outros aditivos para os quais se possa antecipar um efeito na microflora do intestino. Estes estudosdemonstram que a utilização do aditivo não cria condições conducentes a um crescimento excessivo edisseminação de microrganismos potencialmente patogénicos.
A escolha dos microrganismos a monitorizar depende das espécies-alvo, mas inclui espécies zoonóticasrelevantes, independentemente de se produzirem efectivamente ou não sintomas em animais-alvo.
3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
O objectivo é avaliar a segurança do aditivo para o consumidor e estabelecer resíduos potenciais do aditivo oudos seus metabolitos em alimentos derivados de animais alimentados com alimentos ou água contendo outratados com o aditivo.
22.5.2008 PT Jornal Oficial da União Europeia L 133/23
(11) Uma lista não exaustiva está disponível em: www.efsa.europa.eu/en/science/feedap/feedap_opinion/993.html

3.2.1. Estudos metabólicos e de resíduos
O estabelecimento do destino metabólico do aditivo nas espécies-alvo é uma etapa determinante naidentificação e quantificação dos resíduos nos tecidos ou produtos comestíveis derivados de animaisalimentados com alimentos ou água contendo o aditivo. Têm de ser apresentados estudos no que se refere àabsorção, distribuição, metabolismo e excreção da substância (e dos seus metabolitos).
Têm de ser realizados estudos utilizando métodos de ensaio internacionalmente validados e estes sãoexecutados em conformidade com a legislação europeia em vigor ou as directrizes da OCDE relativas aospormenores metodológicos e de acordo com os princípios de BPL. O estudo respeita as regras relativas ao bem--estar dos animais estabelecidas na legislação comunitária e não são repetidos se tal não for necessário.
Os estudos metabólicos e de resíduos no(s) animal(ais)-alvo são executados com a substância activaincorporada na alimentação (não dada por gavagem a menos que tal seja adequadamente justificado).
É feita a identificação estrutural de metabolitos que representem mais de 10 % dos resíduos totais nos tecidos eprodutos comestíveis e mais de 20 % dos resíduos totais nas excreções. Se os passos metabólicos da substânciaactiva suscitarem quaisquer apreensões a nível toxicológico, são identificados os metabolitos abaixo dos limitessupra.
Os estudos cinéticos dos resíduos constituirão a base para o cálculo da exposição dos consumidores e doestabelecimento de um intervalo de segurança e de LMR, se necessário. É apresentada uma proposta pararesíduo marcador.
Para alguns aditivos, em função da sua natureza ou utilização, pode nem sempre ser necessário realizar estudosmetabólicos e de resíduos.
3.2.1.1. Estudos metabólicos
O objectivo dos estudos metabólicos é avaliar a absorção, distribuição, biotransformação e excreção do aditivonas espécies-alvo.
Os estudos exigidos são:
1. O balanço metabólico no seguimento da administração de uma dose única da substância activa nadosagem proposta para utilização (montante total correspondente à ingestão diária) e possivelmente umadose múltipla (se justificada) para avaliar a taxa e o grau aproximados de absorção, distribuição (plasma//sangue) e excreção (urina, bílis, fezes, leite ou ovos, ar expirado, excreção através das guelras) em machose fêmeas, quando necessário; e
2. O perfil metabólico, a identificação do(s) metabolito(s) em excreções e tecidos e a distribuição nos tecidose produtos são estabelecidos no seguimento da administração repetida da dose do composto marcado aanimais até ao estado estacionário (equilíbrio metabólico) identificado através dos níveis no plasma. Adose aplicada corresponde à dose mais elevada proposta para utilização e é incorporada na alimentação.
3.2.1.2. Estudos de resíduos
Tem-se em consideração a quantidade e natureza dos resíduos não extraíveis em tecidos ou produtoscomestíveis.
São exigidos estudos de resíduos para todas as substâncias para as quais são necessários estudos metabólicos.
Se a substância for um constituinte natural de fluidos do corpo ou tecidos ou se encontrar naturalmentepresente em montantes significativos em géneros alimentícios ou alimentos para animais, a exigência derealização de estudos de resíduos limita-se à comparação dos níveis no tecido/produto num grupo não tratadoe no grupo a quem foi dada em suplemento a dose mais elevada objecto do pedido.
Para as espécies principais, os estudos avaliam simultaneamente os resíduos totais de importância toxicológica eidentificam o resíduo marcador da substância activa no tecido comestível (fígado, rim, músculo, pele, pele +gordura) e em produtos (leite, ovos e mel). O resíduo marcador é o resíduo seleccionado para ensaio cujaconcentração tem uma relação conhecida com o resíduo total nos tecidos que causa apreensão a níveltoxicológico. Os estudos mostram igualmente a permanência dos resíduos nos tecidos ou produtos de forma aestabelecer um intervalo de segurança adequado.
L 133/24 PT Jornal Oficial da União Europeia 22.5.2008

Para a determinação de um intervalo de segurança, são os seguintes os números mínimos sugeridos de animaise/ou produtos amostrados em cada ponto temporal:
— Tecidos comestíveis:
— bovinos, ovinos, suínos e espécies menores: 4,
— aves de capoeira: 6,
— salmonídeos e outros peixes: 10.
— Produtos:
— leite: 8 amostras por ponto temporal,
— ovos: 10 ovos por ponto temporal,
— mel: 8 amostras por ponto temporal.
É tida em conta uma distribuição por sexo adequada.
Os resíduos são medidos em intervalo de segurança zero (estado estacionário) e, pelo menos, em três outrospontos temporais de amostragem.
É apresentada uma proposta para resíduo marcador.
Os estudos sobre a absorção, distribuição e excreção, incluindo a identificação dos metabolitos principais têmde ser executados nas espécies animais para ensaio em laboratório nas quais é obtido o NOAEL mais baixo ou,por defeito, no rato (ambos os sexos). Podem ser necessários estudos adicionais relativos a metabolitosespecíficos se estes metabolitos forem produzidos por espécies-alvo e não se formem de forma significativa nasespécies para ensaio em laboratório.
3.2.1.3. Estudos metabólicos e de eliminação
É executado um estudo relativo ao metabolismo, incluindo o balanço, o perfil metabólico e a identificação dosmetabolitos principais na urina e nas fezes. Se outra espécie para ensaio em laboratório mostrar uma diferençaacentuada em relação à sensibilidade do rato, será exigida informação adicional.
3.2.1.4. Biodisponibilidade dos resíduos
A avaliação dos riscos para os consumidores relacionados com resíduos associados a produtos de origemanimal pode ter em consideração um factor de segurança adicional na determinação da sua biodisponibilidadeutilizando animais para ensaio em laboratório adequados bem como métodos reconhecidos.
3.2.2. Estudos toxicológicos
A segurança do aditivo é avaliada com base nos estudos toxicológicos executados in vitro e in vivo em animaispara ensaio em laboratório. De modo geral, incluem medições de:
1. Toxicidade aguda;
2. Genotoxicidade (mutagenicidade, clastogenicidade);
3. Toxicidade oral subcrónica;
4. Toxicidade oral crónica/carcinogénese;
5. Toxicidade na reprodução, nomeadamente teratogenicidade; e
6. Outros estudos.
Caso subsistam motivos de preocupação, realizam-se estudos complementares que forneçam informaçõesadicionais necessárias para a avaliação da segurança da substância activa e dos seus resíduos.
Com base nos resultados destes estudos tem de ser estabelecido um NOAEL toxicológico.
22.5.2008 PT Jornal Oficial da União Europeia L 133/25

Podem ser necessários estudos adicionais relativos a metabolitos específicos se estes metabolitos foremproduzidos por espécies-alvo e não se formem de forma significativa nas espécies para ensaio em laboratório.Se estiverem disponíveis estudos metabólicos em seres humanos, os dados são tomados em consideração aodecidir da natureza de eventuais estudos adicionais.
Têm de ser realizados estudos toxicológicos com a substância activa. Se a substância activa estiver presentenum produto de fermentação, é testado o produto de fermentação. O produto de fermentação testado tem deser idêntico ao que se destina a ser utilizado no produto comercial.
Têm de ser realizados estudos utilizando métodos de ensaio internacionalmente validados e estes sãoexecutados em conformidade com a legislação europeia em vigor ou as directrizes da OCDE relativas ospormenores metodológicos e de acordo com os princípios de BPL. Os estudos envolvendo animais para ensaioem laboratório respeitam as regras relativas ao bem-estar dos animais estabelecidas na legislação comunitária enão são repetidos se tal não for necessário.
3.2.2.1. Toxicidade aguda
São necessários estudos de toxicidade aguda para classificar e apresentar a caracterização limitada da toxicidadedo composto.
Os estudos de toxicidade aguda são efectuados em, pelo menos, duas espécies de mamíferos. Uma espécie deanimais para ensaio em laboratório pode ser substituída, se adequado, por uma espécie-alvo.
Não é necessário identificar o valor DL50 exacto; é normalmente considerada suficiente uma determinaçãoaproximada da dose letal mínima. A dosagem máxima não excede 2 000 mg/kg de massa corporal.
De forma a reduzir o número e o sofrimento dos animais em questão, estão continuamente a ser elaboradosnovos protocolos de ensaios de toxicidade aguda da dose. Os estudos realizados por estes novos procedimentosserão aceites, quando validados adequadamente.
Devem ser seguidas as Directrizes 402 (Toxicidade Dérmica Aguda), 420 (Método da Dose Fixa), 423 (Métododa Classe de Toxicidade Aguda) e 425 (Protocolo Up and Down) da OCDE.
3.2.2.2. Estudos de genotoxicidade incluindo a mutagenicidade
De forma a identificar as substâncias activas e, quando apropriado, os seus metabolitos e produtos dedegradação com propriedades mutagénicas e genotóxicas, deve efectuar-se uma combinação seleccionada dediferentes ensaios de genotoxicidade. Se adequado, os ensaios são realizados sem e com activação metabólicamamífera e é tida em conta a compatibilidade do material de ensaio com o sistema de ensaio.
O grupo principal inclui os seguintes ensaios:
1. Indução de mutações genéticas em bactérias e/ou células mamíferas (preferivelmente, o ensaio TK dolinfoma no rato);
2. Indução de aberrações cromossomáticas em células mamíferas; e
3. Ensaio in vivo em espécies mamíferas.
Pode ser necessária a realização de ensaios complementares dependendo dos resultados obtidos nos ensaiosmencionados supra e tendo em conta o perfil toxicológico da substância bem como a sua utilização prevista.
Os protocolos devem estar em conformidade com as Directrizes 471 (ensaio de mutação reversa de Salmonellatyphimurium), 472 (ensaio de mutação reversa de Escherichia coli), 473 (ensaio de aberração cromossomática demamíferos — in vitro), 474 (ensaio dos micronúcleos em eritrócitos de mamíferos), 475 (ensaio de aberraçãocromossomática da medula óssea de mamíferos), 476 (ensaio de mutação genética em células de mamíferos —in vitro) ou 482 (síntese de ADN não programada em células de mamífero — in vitro) da OCDE, assim comocom outras directrizes da OCDE pertinentes para ensaios in vitro e in vivo.
3.2.2.3. Estudos de toxicidade oral subcrónica por repetição de dose
Para investigar o potencial tóxico subcrónico da substância activa, deve apresentar-se, pelo menos, um estudosobre uma espécie roedora com uma duração mínima de 90 dias. Se considerado necessário, tem ser executadoum segundo estudo com uma espécie não roedora. O item de ensaio deve ser administrado oralmente com ummínimo de três níveis para além de um grupo de controlo para obter uma resposta à dose. Normalmente, deveesperar-se que a dose máxima utilizada revele provas de efeitos nocivos. A dose mínima não deve produzirquaisquer provas de toxicidade.
Os protocolos para estes estudos devem estar em conformidade com as Directrizes 408 (roedores) ou 409 (nãoroedores) da OCDE.
L 133/26 PT Jornal Oficial da União Europeia 22.5.2008

3.2.2.4. Estudos de toxicidade crónica oral (incluindo estudos relativos aos efeitos cancerígenos)
Para investigar o potencial crónico tóxico e cancerígeno, tem de ser realizado um estudo de toxicidade crónicaoral em, pelo menos, uma espécie cuja duração é de no mínimo 12 meses. As espécies escolhidas são as maisadequadas com base em todos os dados científicos disponíveis, incluindo os resultados dos estudos de 90 dias.A espécie por defeito é o rato. Se for solicitado um segundo estudo, são utilizados um roedor ou uma espéciemamífera não roedora. O item de ensaio deve ser administrado oralmente com um mínimo de três níveis paraalém de um grupo de controlo para obter uma resposta à dose.
Se o estudo de toxicidade crónica for combinado com um exame de carcinogenicidade, então a duração éalargada a 18 meses para ratinhos e hamsters e a 24 meses para ratos.
Os estudos dos efeitos cancerígenos podem não ser necessários se a substância activa e os seus metabolitos:
1. Derem resultados constantemente negativos nos ensaios de genotoxicidade;
2. Não estiverem estruturalmente relacionados com agentes cancerígenos conhecidos; e
3. Não produzirem efeitos indicativos de (pré)neoplasia potencial em ensaios de toxicidade crónica.
Os protocolos deveriam estar em conformidade com as Directrizes 452 (estudo de toxicidade crónica) ou 453(estudo combinado de toxicidade crónica/carcinogenicidade) da OCDE.
3.2.2.5. Estudos de toxicidade na reprodução (incluindo toxicidade de desenvolvimento pré-natal)
Para identificar os danos possíveis para a função reprodutora masculina ou feminina ou os efeitos nocivos paraa progenitura resultantes da administração da substância activa, têm de se realizar os seguintes estudos dafunção reprodutora:
1. Estudo da toxicidade para a função reprodutora em duas gerações; e
2. Estudo da toxicidade de desenvolvimento pré-natal (estudo de teratogenicidade).
Para novas experiências, podem ser utilizados métodos alternativos validados que reduzam a utilização deanimais.
3.2.2.5.1. Estudo da toxicidade para a função reprodutora em duas gerações
Os estudos da função reprodutora devem ser efectuados e estender-se por, pelo menos, duas gerações em linhadirecta (F1, F2) em pelo menos uma espécie, normalmente roedora, e podem combinar-se com um estudo deteratogenicidade. A substância a investigar é administrada oralmente aos machos e às fêmeas num momentoadequado antes do acasalamento. A administração prossegue até ao desmame da geração F2.
Têm de se observar e registar cuidadosamente todos os parâmetros relevantes relativos à fertilidade, gestação,parto, comportamento materno, lactação, crescimento e desenvolvimento da geração F1 desde a fertilização atéà maturidade bem como o desenvolvimento da geração F2 até ao desmame. Os protocolos para o estudo detoxicidade na reprodução devem estar em conformidade com a Directriz 416 da OCDE.
3.2.2.5.2. Estudo da toxicidade de desenvolvimento pré-natal (estudo de teratogenicidade)
O objectivo é detectar quaisquer efeitos nocivos na fêmea grávida e no desenvolvimento do embrião e do fetoem consequência da exposição a partir da implantação e durante todo o período de gestação. Tais efeitosincluem o aumento da toxicidade nas fêmeas grávidas, da morte do feto/embrião, a alteração do crescimentofetal e malformações e anomalias estruturais no feto.
O rato é geralmente a espécie de escolha para o primeiro estudo. Se for observado um resultado negativo ouambíguo para a teratogenicidade, é conduzido outro estudo de toxicidade de desenvolvimento numa segundaespécie, preferivelmente o coelho. Se o estudo no rato for positivo para a teratogenicidade, não é necessário umestudo numa segunda espécie, excepto se um exame de todos os estudos principais indicar que a DDA seriabaseada na teratogenicidade do rato. Neste caso, seria exigido um estudo numa segunda espécie paradeterminar a espécie mais sensível a este parâmetro. Os protocolos devem estar em conformidade com aDirectriz 414 da OCDE.
22.5.2008 PT Jornal Oficial da União Europeia L 133/27

3.2.2.6. Outros estudos toxicológicos e farmacológicos específicos
Caso subsistam motivos de preocupação, realizam-se estudos complementares que forneçam informaçõesadicionais úteis para a avaliação da segurança da substância activa e dos seus resíduos. Tais estudos podemincluir o exame dos efeitos farmacológicos, de efeitos em animais juvenis (pré-púberes), imunotoxicidade ouneurotoxicidade.
3.2.2.7. Determinação dos níveis de efeito negativo não observado (NOAEL)
O NOAEL tem por base, regra geral, os efeitos toxicológicos, mas os efeitos farmacológicos poderiam,ocasionalmente, ser mais adequados.
Selecciona-se o NOAEL mais baixo. Na identificação de um NOAEL expresso em mg/kg de massa corporal pordia, têm-se em conta todos os resultados das secções anteriores bem como todos os dados publicadosrelevantes (incluindo qualquer informação adequada relativa aos efeitos da substância activa na espéciehumana) e, se adequado, informações relativas a estruturas químicas próximas.
3.2.3. Avaliação de segurança para o consumidor
A segurança para o consumidor é avaliada por uma comparação da DDA estabelecida (dose diária admissível) eda ingestão teórica calculada do aditivo ou dos seus metabolitos a partir dos géneros alimentícios. No caso devitaminas e oligoelementos, pode ser utilizado o UL (Nível Máximo de Ingestão Tolerável) em vez da DDA.
3.2.3.1. Proposta de Dose Diária Admissível (DDA) para as substâncias activas
A Dose Diária Admissível (DDA) (expressa como mg de aditivo ou de material relacionado com o aditivo porpessoa por dia) obtém-se dividindo o NOAEL mais baixo (mg por kg de massa corporal) por um factor desegurança adequado e multiplicando por uma massa corporal humana média de 60 kg.
Sempre que adequado, propõe-se uma DDA. Uma DDA pode igualmente ser «não especificada» devido a umabaixa toxicidade em ensaios animais. Não é proposta uma DDA se a substância mostrar propriedadesgenotóxicas ou cancerígenas relevantes para o ser humano.
A definição de uma DDA exige normalmente a semelhança do destino metabólico da substância activa emanimais-alvo e em animais para ensaio em laboratório (ver 3.2.1.4 Biodisponibilidade dos resíduos), o queassegura que os consumidores são expostos aos mesmos resíduos que os animais para ensaio em laboratórioutilizados nos estudos toxicológicos. Se não for este o caso, estudos adicionais numa segunda espécie de animalpara ensaio em laboratório ou com os metabolitos específicos às espécies-alvo podem ainda permitir adefinição de uma DDA.
O factor de segurança utilizado para determinar a DDA para um aditivo particular tomará em consideração anatureza dos efeitos biológicos e a qualidade dos dados utilizados para identificar o NOAEL, a importânciadestes efeitos para o ser humano e a sua reversibilidade, bem como qualquer conhecimento do(s) efeito(s)directo(s) dos resíduos no ser humano.
É utilizado um factor de segurança de, pelo menos, 100 no cálculo da DDA (se for apresentado um conjuntotoxicológico completo). Quando estiverem disponíveis dados relativos à substância activa em relação ao serhumano, pode ser aceitável um factor de segurança mais baixo. Poderiam ser aplicados factores de segurançamais elevados para justificar fontes de incerteza adicionais em relação a dados ou sempre que o NOAEL sejadefinido com base num parâmetro crítico particular, como a teratogenicidade.
3.2.3.2. Nível Máximo de Ingestão Tolerável (UL)
Para alguns aditivos pode ser mais adequado basear a avaliação de segurança no UL, que é o nível máximo deingestão diária crónica total de um nutriente (de todas as fontes) considerado (por organismos científicosnacionais ou internacionais) como não sendo susceptível de constituir um risco de efeitos nocivos para a saúdedos consumidores ou de grupos específicos de consumidores.
O processo contém dados para demonstrar que a utilização do aditivo não levaria a uma situação na qual o ULpoderia ser excedido considerando todas as fontes possíveis do nutriente.
Se os níveis de resíduos resultantes do aditivo alimentar ou do(s) seu(s) metabolito(s) em produtos de origemanimal forem mais elevados que o considerado normal ou esperado para estes produtos, este facto é claramenteindicado.
3.2.3.3. Exposição do consumidor
A ingestão total do aditivo e/ou dos seus metabolitos de todas as fontes pelo consumidor é inferior à DDA ouao UL.
L 133/28 PT Jornal Oficial da União Europeia 22.5.2008
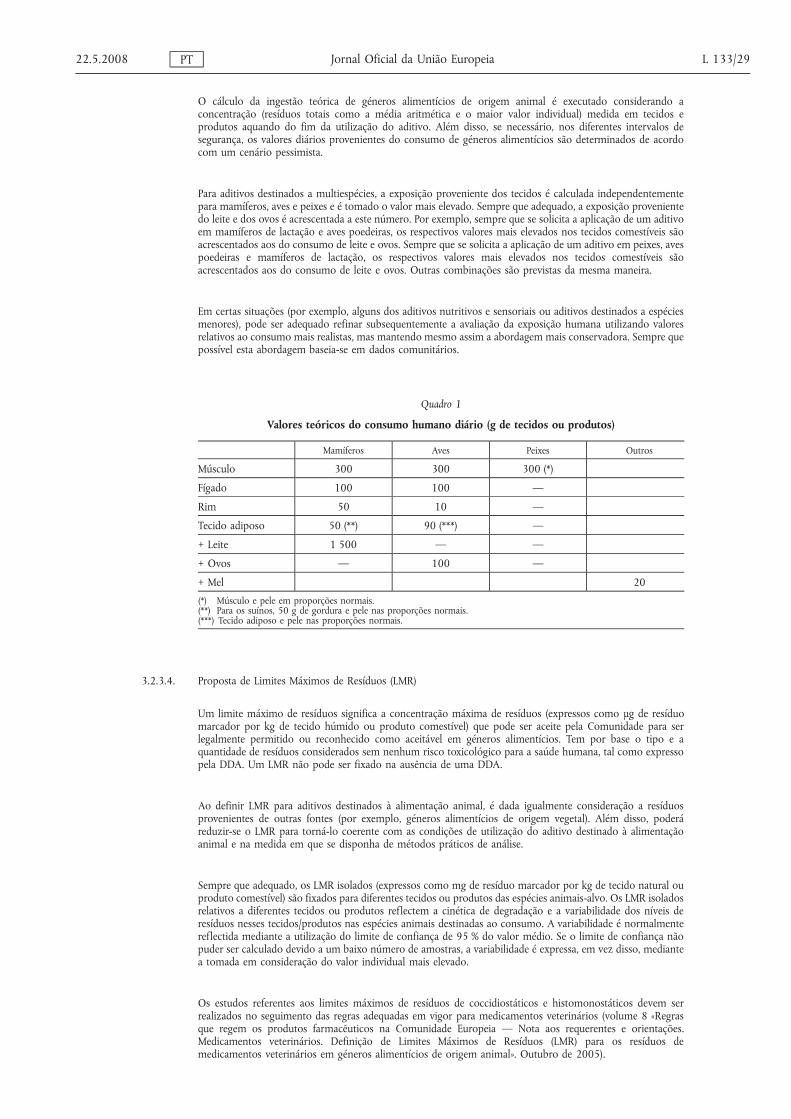
O cálculo da ingestão teórica de géneros alimentícios de origem animal é executado considerando aconcentração (resíduos totais como a média aritmética e o maior valor individual) medida em tecidos eprodutos aquando do fim da utilização do aditivo. Além disso, se necessário, nos diferentes intervalos desegurança, os valores diários provenientes do consumo de géneros alimentícios são determinados de acordocom um cenário pessimista.
Para aditivos destinados a multiespécies, a exposição proveniente dos tecidos é calculada independentementepara mamíferos, aves e peixes e é tomado o valor mais elevado. Sempre que adequado, a exposição provenientedo leite e dos ovos é acrescentada a este número. Por exemplo, sempre que se solicita a aplicação de um aditivoem mamíferos de lactação e aves poedeiras, os respectivos valores mais elevados nos tecidos comestíveis sãoacrescentados aos do consumo de leite e ovos. Sempre que se solicita a aplicação de um aditivo em peixes, avespoedeiras e mamíferos de lactação, os respectivos valores mais elevados nos tecidos comestíveis sãoacrescentados aos do consumo de leite e ovos. Outras combinações são previstas da mesma maneira.
Em certas situações (por exemplo, alguns dos aditivos nutritivos e sensoriais ou aditivos destinados a espéciesmenores), pode ser adequado refinar subsequentemente a avaliação da exposição humana utilizando valoresrelativos ao consumo mais realistas, mas mantendo mesmo assim a abordagem mais conservadora. Sempre quepossível esta abordagem baseia-se em dados comunitários.
Quadro 1
Valores teóricos do consumo humano diário (g de tecidos ou produtos)
Mamíferos Aves Peixes Outros
Músculo 300 300 300 (*)
Fígado 100 100 —
Rim 50 10 —
Tecido adiposo 50 (**) 90 (***) —
+ Leite 1 500 — —
+ Ovos — 100 —
+ Mel 20
(*) Músculo e pele em proporções normais.(**) Para os suínos, 50 g de gordura e pele nas proporções normais.(***) Tecido adiposo e pele nas proporções normais.
3.2.3.4. Proposta de Limites Máximos de Resíduos (LMR)
Um limite máximo de resíduos significa a concentração máxima de resíduos (expressos como μg de resíduomarcador por kg de tecido húmido ou produto comestível) que pode ser aceite pela Comunidade para serlegalmente permitido ou reconhecido como aceitável em géneros alimentícios. Tem por base o tipo e aquantidade de resíduos considerados sem nenhum risco toxicológico para a saúde humana, tal como expressopela DDA. Um LMR não pode ser fixado na ausência de uma DDA.
Ao definir LMR para aditivos destinados à alimentação animal, é dada igualmente consideração a resíduosprovenientes de outras fontes (por exemplo, géneros alimentícios de origem vegetal). Além disso, poderáreduzir-se o LMR para torná-lo coerente com as condições de utilização do aditivo destinado à alimentaçãoanimal e na medida em que se disponha de métodos práticos de análise.
Sempre que adequado, os LMR isolados (expressos como mg de resíduo marcador por kg de tecido natural ouproduto comestível) são fixados para diferentes tecidos ou produtos das espécies animais-alvo. Os LMR isoladosrelativos a diferentes tecidos ou produtos reflectem a cinética de degradação e a variabilidade dos níveis deresíduos nesses tecidos/produtos nas espécies animais destinadas ao consumo. A variabilidade é normalmentereflectida mediante a utilização do limite de confiança de 95 % do valor médio. Se o limite de confiança nãopuder ser calculado devido a um baixo número de amostras, a variabilidade é expressa, em vez disso, mediantea tomada em consideração do valor individual mais elevado.
Os estudos referentes aos limites máximos de resíduos de coccidiostáticos e histomonostáticos devem serrealizados no seguimento das regras adequadas em vigor para medicamentos veterinários (volume 8 «Regrasque regem os produtos farmacêuticos na Comunidade Europeia — Nota aos requerentes e orientações.Medicamentos veterinários. Definição de Limites Máximos de Resíduos (LMR) para os resíduos demedicamentos veterinários em géneros alimentícios de origem animal». Outubro de 2005).
22.5.2008 PT Jornal Oficial da União Europeia L 133/29

Os estudos para estabelecer limites máximos de resíduos para categorias de aditivos que não coccidiostáticos ehistomonostáticos são apresentados, se necessário, de acordo com o presente anexo.
Para determinar a exposição do consumidor aos resíduos totais (tal como calculados em 3.2.3.3), os LMRpropostos para os diferentes tecidos ou produtos têm em conta a razão entre o resíduo marcador e o resíduototal (quadro 2).
Quadro 2
Definições usadas para a determinação de LMR
i-j Tecidos/produtos isolados (fígado, rim, músculo, pele + tecido adiposo, leite, ovos, mel) emmomentos diferentes
LMRi-j Limite máximo de resíduos em tecidos/produtos (mg de substância marcadora kg-1)
Qti-j Consumo humano diário de tecidos/produtos isolados (kg) fixados pelo quadro 1 ou seurefinamento
TRCi-j Concentração de resíduos total em tecidos/produtos isolados (mg kg-1)
MRCi-j Concentração de resíduo marcador em tecidos/produtos isolados (mg kg-1)
RMTRi-j Razão entre MRCi-j e TRCi-j para tecidos/produtos isolados
DITRi-j Ingestão alimentar para tecidos/produtos isolados calculada a partir dos resíduos totais (mg)DITRi-j = Qti-j × TRCi-j
DITRLMRi-j Ingestão alimentar calculada a partir de LMR (mg) dos tecidos/produtos isoladosDITRMRLi-j = Qti-j × MRLi-j × RMTRi-j
-1
Os valores medidos para TRC e MRC inserem-se segundo o caso no modelo constante no quadro 3,calculando-se os outros valores. Sempre que não estiver disponível um conjunto de dados completo porque osvalores são inferiores ao limite de detecção (LD), uma extrapolação de RMTR pode ser aceitável.
A definição de um LMR apenas pode ser executada se a soma dos DITR isolados for inferior à DDA. Se a DDAfor ultrapassada, uma alternativa seria utilizar dados de um intervalo de segurança mais longo ou dosagensinferiores. Uma primeira proposta para um LMR pode ser obtida utilizando o valor MRC como guia e tomandoem consideração o LOQ do método analítico. A soma do DITRLMR obtido a partir dos LMR propostos deve serinferior à DDA e próximo da soma dos DITR isolados. Se a DDA for ultrapassada, é então proposto um LMRinferior e a comparação é repetida.
Em relação a determinados aditivos, os resíduos podem apresentar níveis inferiores ao LMR no leite, nos ovosou na carne, mas não obstante, podem interferir com a qualidade dos géneros alimentícios, nomeadamente emprocedimentos de transformação de géneros alimentícios. Para estes aditivos, pode ser adequado considerar um«nível máximo de resíduos compatível com a transformação (MPCR) (de produtos alimentares)», para além doestabelecimento do LMR.
Quadro 3
Modelo para definir uma proposta de LMR
Fígado Rim MúsculoPele +tecidoadiposo
Leite Ovos Mel Soma
TRC (1) (mg kg-1) —
MRC (2) (mg kg-1) —
RMTR (2) —
DITR (3) (mg)
LMR proposto (mgkg-1)
—
DITRLMR(mg)
(1) Considerando o intervalo de segurança proposto.(2) Estabelecido idealmente ao mesmo tempo que TRC.(3) Calculado a partir de valores TRC.
L 133/30 PT Jornal Oficial da União Europeia 22.5.2008

3.2.3.5. Proposta de um intervalo de segurança
Este intervalo de segurança inclui o período que é necessário para permitir que os níveis de resíduos se situemabaixo do LMR após a cessação da administração do aditivo.
3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Os trabalhadores podem ser expostos principalmente por inalação ou por exposição tópica durante o fabrico, amanipulação ou a utilização do aditivo. Por exemplo, os trabalhadores de explorações agrícolas sãopotencialmente expostos ao manipular ou misturar o aditivo. É fornecida informação adicional sobre a formade manipular as substâncias.
Inclui-se uma avaliação dos riscos para os trabalhadores. Sempre que disponível, a experiência nas unidades deprodução é frequentemente uma importante fonte de informação na avaliação dos riscos para os trabalhadoresdecorrentes da exposição ao aditivo tanto por via aérea como tópica. Merecem uma especial atenção os aditivosou os alimentos para animais tratados com aditivos ou ainda as excreções animais que se encontram na formapulverulenta ou que a possam originar, bem como os aditivos destinados à alimentação animal que possam terum potencial alergénico.
3.3.1. Avaliação do risco toxicológico em relação à segurança dos utilizadores/trabalhadores
Os riscos para trabalhadores são avaliados numa série de estudos utilizando o aditivo na forma para a qual opedido é apresentado. Realizam-se também estudos de toxicidade relativos à inalação aguda excepto se oproduto não possa formar um pó ou vapor respirável. Efectuam-se estudos relativos à irritação cutânea e casotenham resultados negativos, avalia-se a irritação das mucosas (por exemplo, os olhos). É também avaliado opotencial alergénico/potencial de sensibilização cutânea. Os dados de toxicidade gerados para se responder aoscritérios de segurança dos consumidores (ver o ponto 3.2.2) são utilizados para avaliar a toxicidade sistémicapotencial do aditivo. Todos estes elementos são avaliados, se necessário, por medição directa e estudosespecíficos.
3.3.1.1. Efeitos no sistema respiratório
Fornecem-se provas de que os níveis de poeiras ou de vapor no ar não constituem um perigo para a saúde dosutilizadores/trabalhadores. Estas provas incluem, se necessário:
— ensaios de inalação em animais para ensaio em laboratório,
— dados epidemiológicos publicados e/ou dados do próprio requerente sobre a sua instalação de trabalho e//ou irritação, e
— ensaios de sensibilização do sistema respiratório.
São executados estudos de toxicidade relativos à inalação aguda se mais de 1 % em peso do produto forconstituído por partículas ou gotas com um diâmetro de menos de 50 μm.
Os protocolos para os estudos de toxicidade relativos à inalação aguda devem estar em conformidade com aDirectriz 403 da OCDE. Se forem considerados necessários estudos de toxicidade subcrónica, os mesmosdeveriam seguir a Directriz 412 da OCDE (toxicidade de inalação em doses repetidas: estudo de 28 ou de 14dias) ou 413 (toxicidade subcrónica de inalação: estudo de 90 dias).
3.3.1.2. Efeitos nos olhos e na pele
Quando disponível, fornecem-se provas directas da ausência de irritação e/ou de sensibilização com base emsituações humanas conhecidas. Estes dados são complementados com resultados de ensaios validados emanimais relativos à irritação dos olhos e da pele, bem como ao potencial de sensibilização utilizando o aditivoadequado. É também avaliado o potencial alergénico/potencial de sensibilização cutânea. Os protocolos paraestes estudos deveriam estar em conformidade com as Directrizes 404 da OCDE (irritação/corrosão cutânea),405 (irritação/corrosão ocular), 406 (sensibilização da pele), 429 (sensibilização da pele — ensaio do nódulolinfático local).
Se forem conhecidas propriedades corrosivas, quer dos dados publicados quer dos ensaios in vitro específicos,não são executados outros ensaios in vivo.
A toxicidade cutânea tem de ser considerada se o aditivo for tóxico por inalação. Os estudos têm de estar emconformidade com a Directriz 402 da OCDE (toxicidade cutânea aguda).
22.5.2008 PT Jornal Oficial da União Europeia L 133/31

3.3.1.3. Toxicidade sistémica
Os dados de toxicidade gerados para se responder aos critérios de segurança dos consumidores e outrosrequisitos (incluindo a toxicidade em doses repetidas, a mutagenicidade, o potencial cancerígeno, os ensaios defunção reprodutora e o destino metabólico) são utilizados para avaliar a toxicidade sistémica.
3.3.1.4. Avaliação da exposição
Fornecem-se informações acerca das utilizações do aditivo susceptíveis de originar uma exposição através detodas as vias (inalatória, cutânea ou ingestão). Esta informação inclui uma avaliação quantitativa quando esta seencontrar disponível, tal como a concentração atmosférica típica, a contaminação cutânea ou a ingestão.Quando não se dispuser de informações quantitativas, fornecem-se informações suficientes para permitir umaavaliação adequada da exposição.
3.3.2. Medidas para controlar a exposição
Com recurso à informação da avaliação toxicológica e de exposição, é tirada uma conclusão sobre os riscospara a saúde dos utilizadores/trabalhadores (inalação, irritação, sensibilização e toxicidade sistémica). Podempropor-se medidas cautelares para reduzir ou eliminar a exposição. Contudo, a utilização de equipamentos deprotecção pessoal apenas é encarada como uma medida de última instância para prevenir qualquer riscoresidual após a implementação de medidas de controlo. É preferível, por exemplo, considerar a reformulação doproduto.
3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
A consideração do impacto ambiental dos aditivos é importante uma vez que a administração de aditivosocorre tipicamente durante períodos longos, envolve frequentemente grandes grupos de animais e a(s)substância(s) activa(s) podem ser excretadas em grande medida sob a forma de composto principal ou seusmetabolitos.
É seguida uma abordagem por etapas para determinar o impacto ambiental dos aditivos. Todos os aditivos têmde ser avaliados através da Fase I para identificar quais os aditivos que não precisam de mais ensaios. Para osrestantes aditivos, é necessária uma segunda fase de estudo (Fase II) para fornecer informações adicionais, asquais podem revelar a necessidade de estudos complementares. Estes estudos são efectuados em conformidadecom a Directiva 67/548/CEE.
3.4.1. Fase I da avaliação
O objectivo da fase I da avaliação é determinar se é provável um efeito ambiental significativo do aditivo ou dosseus metabolitos e se é necessária uma fase II da avaliação (ver árvore de decisão).
A isenção da fase II da avaliação pode ser concedida com base num de dois critérios, a menos que haja motivospara preocupação com base científica:
a) A natureza química e o efeito biológico do aditivo e das suas condições de utilização indicam que oimpacto será negligenciável, ou seja, sempre que o aditivo por:
— uma substância fisiológica ou natural que não resultará num aumento substancial da suaconcentração no ambiente, ou
— destinado a animais não produtores de géneros alimentícios;
b) A concentração ambiental previsível (PEC), no caso mais desfavorável, é demasiado baixa para serpreocupante. A PEC é avaliada para cada compartimento que suscita preocupações (ver infra), supondoque 100 % da dose ingerida é excretada enquanto composto principal.
Se o requerente não puder demonstrar que o aditivo é abrangido por uma destas categorias de isenção, seráexigida uma fase II da avaliação.
3.4.1.1. Aditivos para animais terrestres
Quando as excreções do gado forem aplicadas na terra, a utilização de aditivos destinados à alimentação animalpode dar origem à contaminação do solo, da água subterrânea e da água superficial (através de drenagem eescoamento).
Considerando todos os compostos excretados espalhados na terra, verificar-se-ia a PEC no caso maisdesfavorável para o solo (PECsoil). Se a PECsoil (por defeito: 5 cm de profundidade) for inferior a 10 μg/kg, não éexigida outra avaliação.
L 133/32 PT Jornal Oficial da União Europeia 22.5.2008

Se a PEC para a contaminação da água subterrânea (PECgw) for inferior a 0,1 μg/l, não é necessária a fase II daavaliação do impacto ambiental do aditivo na água subterrânea.
3.4.1.2. Aditivos para animais aquáticos
Os aditivos destinados à alimentação animal utilizados em aquicultura podem resultar em contaminação dosedimento e da água. Na avaliação dos riscos ambientais para peixes de viveiro em gaiolas, supõe-se que seja osedimento o compartimento que suscita preocupação. Para peixes de viveiro em sistemas em terra, considera-seque o efluente que flui para a água superficial constitui o risco ambiental principal.
Considerando todos os compostos excretados depositados no sedimento, verificar-se-ia a PEC no caso maisdesfavorável para o sedimento (PECsediment). Se a PECsediment (por defeito: 20 cm de profundidade) é inferior a10 μg/kg de peso húmido, não é exigida outra avaliação.
Se a PEC na água superficial (PECsw) for inferior a 0,1 μg/l, não é exigida outra avaliação.
Fase I — Árvore de decisão
22.5.2008 PT Jornal Oficial da União Europeia L 133/33

3.4.2. Fase II da avaliação
O objectivo da fase II é avaliar o potencial de os aditivos afectarem espécies não alvo no ambiente, incluindoespécies aquáticas e terrestres ou de alcançarem a água subterrânea a níveis inaceitáveis. Não é prático avaliar osefeitos dos aditivos em todas as espécies no ambiente que podem ser expostas ao aditivo no seguimento da suaadministração às espécies-alvo. Os níveis taxonómicos testados destinam-se a servir de substitutos ouindicadores para a gama de espécies presentes no ambiente.
A fase II da avaliação tem por base uma abordagem de quociente de risco, na qual é comparada a PEC calculadae a concentração previsível sem efeitos (PNEC) para cada compartimento. A PNEC é determinada a partir deparâmetros determinados experimentalmente divididos por um factor de avaliação adequado. O valor PNEC écalculado para cada compartimento.
A fase II da avaliação começa, se possível, com um refinamento da PEC e utiliza uma abordagem a dois níveisrelativamente à avaliação dos riscos ambientais.
O primeiro nível, Fase II A, utiliza um número limitado de estudos de destino e efeito para produzir umaavaliação de risco conservadora baseada em exposição e efeitos no compartimento ambiental de preocupação.Se o rácio PEC/PNEC for inferior à unidade, não é exigida mais nenhuma avaliação, a menos que se esperebioacumulação.
Se o rácio PEC/PNEC previr um risco inaceitável (rácio > 1), o requerente progride para a fase IIB para refinar aavaliação dos riscos ambientais.
3.4.2.1. Fase II A
Para além dos compartimentos considerados na Fase I, a PEC para a água superficial tem de ser calculadaconsiderando o escoamento e a drenagem.
Com base em dados não considerados na Fase I, pode calcular-se uma PEC mais refinada para cadacompartimento ambiental em causa. Ao estimar a PEC refinada, tem-se em conta o seguinte:
a) A concentração da(s) substância(s) activa(s)/metabolitos que causam preocupação no estrume/fezes depeixe após a administração do aditivo a animais no nível de dose proposto. Este cálculo inclui aconsideração das taxas de dosagem e da quantidade de excreções produzidas;
b) A degradação potencial da(s) substância(s) activa(s)/metabolitos que causam preocupação excretadosdevido à prática normal de processamento e armazenamento do estrume antes da sua aplicação na terra;
c) A adsorção/dessorção da(s) substância(s) activa(s)/metabolitos que causam preocupação em solo ousedimento para aquicultura, determinada, de preferência, por estudos em solo/sedimento (OCDE 106);
d) Degradação em sistemas de solo e água/sedimento (OCDE 307 e 308, respectivamente); e
e) Outros factores tais como hidrólise, fotólise, evaporação, diluição através de lavra.
Para efeitos da Fase II da avaliação dos riscos, adopta-se o maior valor da PEC obtida através destes cálculos paracada um dos compartimentos ambientais em causa.
Se estiver prevista uma persistência elevada no solo/sedimento (tempo para a degradação de 90 % daconcentração original do composto: DT90 > 1 ano), é considerado o potencial para acumulação.
São determinadas as concentrações de aditivos (ou metabolitos) que produzem efeitos nocivos graves paravários níveis tróficos nos compartimentos ambientais que causam preocupação. Estes ensaios são, em grandemedida, ensaios agudos e deveriam seguir as directrizes da OCDE ou directrizes semelhantes bem estabelecidas.Os estudos para o ambiente terrestre incluem: toxicidade para minhocas; três plantas terrestres; emicrorganismos do solo (por exemplo, efeitos na fixação do azoto). Os estudos para o ambiente da águadoce incluem: toxicidade em peixes; Daphnia magna; algas; e um organismo que habite no sedimento. Em casode gaiolas marinhas, são estudadas três espécies das diferentes famílias de organismos que habitam nosedimento.
Efectua-se o cálculo do valor PNEC para cada compartimento ambiental em estudo. A PNEC é normalmentederivada do valor da toxicidade mais baixa observada nos ensaios supra e da divisão por um factor de segurançade, pelo menos, 100 em função do parâmetro e do número de espécies de ensaio utilizadas.
L 133/34 PT Jornal Oficial da União Europeia 22.5.2008

O potencial de bioacumulação pode ser estimado a partir do valor do coeficiente de partição n-octanol/água,Log Kow. Valores ≥ 3 indicam que a substância pode ser bioacumulável. A fim de avaliar o risco para oenvenenamento secundário, considera-se realizar um estudo do factor de bioconcentração (BCF) na fase II B.
3.4.2.2. Fase II B (estudos toxicológicos mais pormenorizados)
No que respeita aos aditivos para os quais, na sequência da Fase II A da avaliação, não possa ser excluído umrisco ambiental, é necessária mais informação relativa aos efeitos sobre as espécies biológicas no(s)compartimentos(s) ambiental(ais) em que os estudos da Fase II A indicaram possíveis problemas. Nestasituação, são necessários mais ensaios para determinar os efeitos crónicos e mais específicos nas espéciesmicrobianas, vegetais e animais adequadas. Esta informação adicional permitirá a aplicação de um factor desegurança inferior.
Os ensaios adicionais de ecotoxicidade adequados encontram-se descritos em diversas publicações, porexemplo, nas directrizes da OCDE. É necessário escolher adequadamente estes ensaios para garantir que sãoapropriados à situação em que o aditivo e/ou os seus metabolitos podem ser libertados e dispersos noambiente. O refinamento da avaliação do efeito para o solo (PNECsoil) poderia basear-se em estudos sobre osefeitos crónicos nas minhocas, em estudos adicionais sobre a microflora do solo e em algumas espécies vegetaisrelevantes, em estudos dos invertebrados das pastagem (incluindo insectos) e em aves selvagens.
O refinamento da avaliação do efeito para a água/sedimento poderia basear-se em ensaios de toxicidade crónicanos organismos aquáticos/bentónicos mais sensíveis identificados na Fase II A da avaliação.
Se necessário, deveriam ser executados estudos de bioacumulação, de acordo com a Directriz 305 da OCDE.
4. SECÇÃO IV: ESTUDOS RELATIVOS À EFICÁCIA DO ADITIVO
Os estudos demonstram a eficácia para cada utilização proposta e satisfazem pelo menos uma dascaracterísticas definidas no n.o 3 do artigo 5.o do Regulamento (CE) n.o 1831/2003, de acordo com ascategorias e os grupos funcionais de aditivos destinados à alimentação animal da maneira prevista peloartigo 6.o e o anexo I do referido regulamento. Além disso, os estudos têm de permitir a avaliação da eficácia doaditivo de acordo com as práticas comuns de pecuária na União Europeia.
A concepção experimental utilizada tem de ser justificada de acordo com a utilização do aditivo, da espécie e dacategoria animal. Ao utilizar animais, as experiências são conduzidas de modo a que a sua saúde e as condiçõesde criação dos animais não afectem negativamente a interpretação dos resultados. Para cada experiência,descrevem-se os efeitos positivos e negativos, tanto tecnológicos como biológicos. É igualmente demonstrada aausência de efeitos que prejudiquem as características distintivas dos produtos de origem animal. Asexperiências são, idealmente, conformes com os critérios estabelecidos por um regime de garantia de qualidadereconhecido e sujeito a auditoria externa. Na ausência de tal regime, são apresentados os elementos de provapara demonstrar que o trabalho foi feito por pessoal qualificado, utilizando as instalações e o equipamentoadequados e é efectuado sob a responsabilidade de um director de estudo nomeado.
O protocolo experimental é cuidadosamente elaborado pelo director de estudo no que se refere a dadosdescritivos gerais, por exemplo, métodos, aparelhos e materiais utilizados, detalhes das espécies, da raça ou dasestirpe de animais, seu número e condições sob quais foram alojadas e alimentadas. Para todos os estudos queenvolvam animais, as condições experimentais são descritas de acordo com 3.1.1.3. Os relatórios finais, dadosbrutos, planos de estudo, bem como as substâncias de ensaio bem caracterizadas e identificadas são arquivadospara consulta futura.
Os estudos são concebidos para demonstrar a eficácia da mais baixa dose recomendada de aditivo, visandoparâmetros sensíveis em comparação com um grupo de controlo negativo e, opcionalmente, positivo. Taisestudos incluem igualmente a dose máxima recomendada, sempre que seja proposta. Não se recomenda umaconcepção única, dando-se flexibilidade que permita uma liberdade científica na concepção e na condução dosestudos.
É igualmente prestada atenção às interacções biológicas ou químicas conhecidas ou potenciais entre o aditivo,outros aditivos e/ou medicamentos veterinários e/ou constituintes do regime alimentar, onde tal seja relevantepara a eficácia do aditivo em causa (por exemplo, compatibilidade do aditivo microbiano com coccidiostáticose histomonostáticos ou ácido orgânico).
22.5.2008 PT Jornal Oficial da União Europeia L 133/35

4.1. Estudos in vitro
Para todos os aditivos tecnológicos e alguns aditivos sensoriais que afectam as características da alimentação, aeficácia é demonstrada utilizando um estudo baseado em laboratório. O estudo é concebido para abranger umagama representativa de materiais à qual o aditivo é aplicado. Os resultados são avaliados preferivelmente porensaios sem parâmetros, e demonstram mudanças previstas com uma probabilidade de P ≤ 0,05.
Podem ser utilizados estudos in vitro, particularmente os que simulam aspectos do tracto gastrointestinal, paraoutros tipos de aditivos a fim de apoiarem a sua eficácia. Estes estudos deveriam permitir a avaliação estatística.
4.2. Estudos de eficácia a curto prazo com animais
Os estudos de disponibilidade biológica podem ser utilizados para demonstrar até que ponto uma nova forma,fonte de um nutriente ou corante podem substituir um aditivo equivalente já aprovado ou estabelecido.
Os estudos de digestão/balanço podem ser utilizados em apoio dos estudos de desempenho em animais paraapresentar elementos de prova sobre modos de acção. Em alguns casos, particularmente em relação abenefícios ambientais, a eficácia pode ser melhor demonstrada por estudos de balanço que podem serpreferidos a estudos de eficácia a longo prazo. Tais experiências utilizam números e espécies/categorias animaisadequados às condições de utilização propostas.
Conforme o caso, podem ser propostos outros estudos de eficácia a curto prazo com animais e estes podemsubstituir os estudos de eficácia a longo prazo com animais, desde que plenamente justificado.
4.3. Estudos de eficácia a longo prazo com animais
Os estudos devem ser efectuados em, pelo menos, duas localizações diferentes.
A concepção experimental utilizada tem de incluir consideração de poder estatístico adequado e riscos de tipos1 e 2. O protocolo tem de ser suficientemente sensível para detectar quaisquer efeitos do aditivo na dose maisbaixa recomendada (risco de tipo 1 α, P ≤ 0,05 em geral e P ≤ 0,1 para ruminantes, espécies menores, animaisde companhia e animais não destinados à produção de alimentos) e de apresentar poder estatístico suficientepara garantir que o protocolo experimental responde ao objectivo do estudo. O risco do tipo 2 β é inferior ouigual a 20 %, em geral, e 25 % para experiências com ruminantes, espécies menores, animais de companhia eanimais não destinados à produção de alimentos, o que dá origem a um poder (1-β) superior ou igual a 80 %(75 % para ruminantes, espécies menores, animais de companhia e animais não destinados à produção dealimentos).
Reconhece-se que a natureza de alguns aditivos torna difícil definir as condições experimentais nas quaispodem ser alcançados resultados óptimos. Consequentemente, a possibilidade de utilizar a metanálise éconsiderada quando o número de experiências disponíveis for superior a três. Por este motivo, são utilizadasconcepções de protocolo semelhantes para todas as experiências para que os dados possam no fim ser testadosem termos de homogeneidade e agrupados (se os ensaios assim o indicarem) para uma avaliação estatística anível de P ≤ 0,05.
4.4. Duração de estudos de eficácia a longo prazo com animais-alvo
Em geral, a duração de experiências de eficácia corresponde ao período de aplicação solicitado.
As experiências de eficácia são realizadas de acordo com as práticas agrícolas na União Europeia e com aduração mínima, tal como indicada no anexo IV.
Se um aditivo for aplicado durante um período específico e mais curto que o estabelecido pela definição decategoria animal, é administrado de acordo com as condições propostas de utilização. Contudo, o período deobservação não é mais curto que 28 dias e envolve o parâmetro relevante (por exemplo, para porcas dereprodução o número de leitões nascidos vivos ao considerar o período de gestação, ou o número e peso deleitões desmamados ao considerar o período de lactação).
Para outras espécies ou categorias animais para as quais não foi estabelecido no anexo IV um período mínimode duração dos estudos, é tomado em conta um período de administração, de acordo com as condiçõespropostas de utilização.
4.5. Requisitos de eficácia para categorias de aditivos e grupos funcionais
Para todos os aditivos destinados a produzir um efeito em animais, são solicitados estudos in vivo.
L 133/36 PT Jornal Oficial da União Europeia 22.5.2008

Para as categorias de aditivos zootécnicos e coccidiostáticos e histomonostáticos, a eficácia é demonstrada por,pelo menos, três estudos de eficácia a longo prazo. Contudo, para alguns dos aditivos zootécnicos e as restantescategorias de aditivos que produzem efeito em animais, podem ser aceites estudos de eficácia a curto prazo se aeficácia puder ser demonstrada inequivocamente.
Para outras categorias de aditivos sem efeito directo em animais é apresentado, pelo menos, um estudo deeficácia in vitro.
4.6. Estudos sobre a qualidade dos produtos de origem animal sempre que este não seja o efeito alegado
A fim de demonstrar que o aditivo não produz um efeito negativo, ou outro efeito não solicitado (higiénico etecnológico se adequado), nas características organolépticas e nutritivas de géneros alimentícios derivados deanimais alimentados com o aditivo (quando este não for o efeito desejado), são colhidas as amostras adequadasdurante uma das experiências de eficácia. São observados dois grupos: um grupo sem suplemento; e um grupocom a dosagem mais elevada proposta para o aditivo. Os dados permitem a avaliação estatística. A omissãodestes estudos tem de ser adequadamente justificada.
5. SECÇÃO V: PLANO DE MONITORIZAÇÃO PÓS-COMERCIALIZAÇÃO
Em conformidade com o n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003, é apresentada umaproposta de monitorização pós-comercialização no mercado para certas categorias de aditivos, a fim de seguire identificar quaisquer efeitos directos ou indirectos, imediatos, deferidos ou imprevistos resultantes dautilização do aditivo na saúde humana, animal ou no ambiente, em conformidade com as características dosprodutos em causa.
A concepção do plano de monitorização é pormenorizada caso a caso e identifica quem (por exemplo, orequerente, os utilizadores) executará as várias tarefas que o plano de monitorização exige, quem é responsávelpor assegurar que o plano de monitorização é posto em vigor e executado adequadamente e assegura que hajaum itinerário através do qual as autoridades competentes são avisadas de qualquer nova informação relativa àsegurança de utilização do aditivo. A Comissão e a Autoridade serão informadas de quaisquer efeitos nocivosobservados, sem prejuízo das disposições de supervisão estabelecidas no artigo 12.o do Regulamento (CE)n.o 1831/2003.
Em casos onde a substância activa seja igualmente um antibiótico reconhecido e a sua utilização tenha reveladoseleccionar estirpes bacterianas resistentes no seu nível de utilização como alimento para animais, sãoempreendidos estudos de terreno para monitorizar a resistência bacteriana ao aditivo como elemento damonitorização pós-comercialização.
Para os coccidiostáticos e histomonostáticos, a monitorização no terreno da resistência de Eimeria spp. eHistomonas meleagridis, respectivamente, é empreendida, preferivelmente, durante a última parte do período deautorização.
22.5.2008 PT Jornal Oficial da União Europeia L 133/37

ANEXO III
REQUISITOS ESPECÍFICOS A SATISFAZER PELO PROCESSO PREVISTO NO ARTIGO 3.o NO QUE DIZRESPEITO A CERTAS CATEGORIAS DE ADITIVOS OU CERTAS SITUAÇÕES PARTICULARES, TAL COMO
PREVISTO NO N.o 5 DO ARTIGO 7.o DO REGULAMENTO (CE) N.o 1831/2003
O Regulamento (CE) n.o 1831/2003 prevê o auxílio adicional para a preparação de processos, se necessário, para cada dascategorias de aditivos ou para outras situações particulares, em conformidade com o n.o 5 do artigo 7.o do Regulamento(CE) n.o 1831/2003.
Lista dos requisitos específicos para estabelecer processos para:
1. Aditivos tecnológicos
2. Aditivos sensoriais
3. Aditivos nutritivos
4. Aditivos zootécnicos
5. Coccidiostáticos e histomonostáticos
6. Extrapolação de espécies principais para espécies menores
7. Animais de companhia e outros animais não produtores de géneros alimentícios
8. Aditivos já autorizados para utilização em géneros alimentícios
9. Alteração das autorizações
10. Renovação das autorizações
11. Reavaliação de certos aditivos já autorizados ao abrigo da Directiva 70/524/CEE.
Quaisquer pedidos podem ser apresentados seguindo um ou mais dos requisitos específicos mencionados supra.
Condições gerais
É justificada a ausência, no processo, de qualquer dado previsto nestas secções.
1. ADITIVOS TECNOLÓGICOS
1.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
1.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos não sujeitos a um detentor de autorização específico aplicam-se os números 2.1.2, 2.1.3,2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 e 2.6,
— para outros aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II.
L 133/38 PT Jornal Oficial da União Europeia 22.5.2008

1.3. Secção III: Estudos referentes à segurança do aditivo
As subsecções 3.1, 3.2 e 3.4 do anexo II não se aplicam a aditivos de silagem sempre que se puder demonstrarque:
— nenhuma quantidade detectável da(s) substância(s) activa(s) ou metabolitos relevantes ou o(s) agente(s)activo(s) sobrevivem no alimento para animais final, ou
— a(s) substância(s) activa(s) e agente(s) ocorrem como componente de silagem normais e a utilização doaditivo não aumenta substancialmente a sua concentração comparada com a silagem preparada semutilização do aditivo (ou seja, sempre que não existir alteração substancial em termos da exposição).
Nos restantes casos, aplica-se toda a secção 3 do anexo II.
1.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Para substâncias xenobióticas (1): aplica-se toda a subsecção 3.1 do anexo II.
1.3.1.1. Estudos de tolerância para as espécies-alvo
Para aditivos de silagem:
— O produto é acrescentado a um regime alimentar básico e os resultados são comparados a um controlonegativo com o mesmo regime. O regime alimentar básico pode conter uma única fonte de silagempreparada sem a utilização de um aditivo.
— A dose seleccionada para os estudos de tolerância é um múltiplo da concentração presente no materialensilado na altura da utilização normal sempre que este valor possa ser estabelecido conclusivamente.Consideração particular é dada a um produto contendo microrganismos viáveis e a sua capacidade desobrevivência e multiplicação durante a ensilagem.
Os estudos de tolerância podem geralmente ser limitados a uma espécie ruminante, normalmente vacas leiteiras.Apenas são exigidos estudos que envolvam outras espécies quando a natureza do material ensilado o tornar maisadequado para utilização com não ruminantes.
Outras substâncias:
Para as outras substâncias para as quais se solicita autorização como aditivos tecnológicos ainda não autorizadospara utilização na alimentação animal, é demonstrada a ausência de consequências nocivas para os animais aonível mais elevado proposto. Esta demonstração pode ser limitada a uma experiência numa das espécies-alvo maissensíveis ou numa espécie animal para ensaio em laboratório.
1.3.1.2. Estudos microbianos
Aplica-se a totalidade da subsecção 3.1.2 do anexo II.
1.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
1.3.2.1. Estudos metabólicos e de resíduos
Não são exigidos estudos metabólicos e de resíduos se:
1. A substância ou os seus metabolitos não estiverem presentes no alimento para animais aquando daalimentação; ou
2. A substância for excretada inalterada, ou se puder demonstrar que os seus metabolitos não são absorvidosem grande medida; ou
22.5.2008 PT Jornal Oficial da União Europeia L 133/39
(1) Um xenobiótico é uma substância química que não é um componente natural do organismo a ela exposto. Pode igualmente abrangersubstâncias presentes em concentrações muito mais elevadas que o habitual.

3. A substância for absorvida sob a forma de compostos fisiológicos; ou
4. O(s) componente(s) activo(s) do aditivo consiste(m) apenas em microrganismos ou enzimas.
Também não são exigidos estudos metabólicos se a substância estiver naturalmente presente em montantessignificativos em géneros alimentícios ou alimentos para animais ou a substância for um constituinte normal defluidos ou tecidos corporais. Contudo, nestes casos, há uma exigência de estudos de resíduos que podem serlimitados à comparação dos níveis nos tecidos/produtos num grupo não tratado e no grupo a quem foi dada emsuplemento a dose mais elevada recomendada.
1.3.2.2. Estudos toxicológicos
Não são exigidos estudos toxicológicos se:
1. A substância ou os seus metabolitos não estiverem presentes no alimento para animais aquando daalimentação; ou
2. A substância for absorvida sob a forma de composto(s) fisiológico(s); ou
3. O produto consistir em microrganismos encontrados geralmente em materiais ensilados ou aqueles jáutilizados em géneros alimentícios; ou
4. O produto consistir em enzimas com um grau de pureza elevado provenientes de microrganismos com umhistorial de utilização segura documentado.
Para microrganismos e enzimas não excluídos supra, são exigidos estudos de genotoxicidade (incluindomutagenicidade) e um estudo de toxicidade oral subcrónica. Os estudos de genotoxicidade não são feitos napresença de células vivas.
Para substâncias xenobióticas, não isentas anteriormente, aplica-se a totalidade da subsecção 3.2.2 do anexo II.
Para outras substâncias, é tomada uma abordagem caso a caso, tendo em conta o nível e os meios de exposição.
1.3.2.3. Avaliação de segurança para o consumidor
Aplica-se a totalidade da subsecção 3.2.3 do anexo II para aditivos cuja utilização se solicita para animaisprodutores de géneros alimentícios.
1.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II. Presume-se que os aditivos contendo enzimas emicrorganismos sejam sensibilizadores respiratórios, a menos que sejam apresentados elementos convincentes deprova em contrário.
1.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Aplica-se a totalidade da subsecção 3.4 do anexo II. Para os aditivos de ensilagem, são considerados os efeitos doaditivo na produção de efluente proveniente do silo fossa ou silo durante a ensilagem.
1.4. Secção IV: Estudos referentes à eficácia do aditivo
Os aditivos tecnológicos têm por objectivo melhorar ou estabilizar as características dos alimentos para animaismas não produzem, em geral, um efeito biológico directo na produção animal. Têm de se fornecer provas daeficácia do aditivo mediante critérios adequados, indicados em métodos reconhecidos e aceites, nas condições deutilização prática e em comparação com alimentos para animais de controlo adequados.
L 133/40 PT Jornal Oficial da União Europeia 22.5.2008

A eficácia será avaliada por estudos in vitro, com excepção de substâncias para controlo de contaminação porradionuclídeo. O seguinte quadro indica os parâmetros adequados para os vários grupos funcionais.
Valores-limite para diferentes aditivos tecnológicos
Grupo funcional Parâmetros para demonstração de eficácia
a) Conservantes Inibição de crescimento microbiano, particularmente o de bióticos e deorganismos de deteriorações. É demonstrado o período para o qual éalegado um efeito de preservação.
b) Antioxidantes Protecção contra dano oxidativo dos nutrientes/componentes-chavedurante transformação e/ou armazenamento do alimento para animais.É demonstrado o período para o qual é alegado um efeito de protecção.
c) Emulsionantes Formação/manutenção de emulsões estáveis dos ingredientes dosalimentos para animais, de outra maneira imiscíveis ou insuficiente-mente miscíveis.
d) Estabilizantes Manutenção do estado físico-químico dos alimentos para animais.
e) Espessantes Viscosidade das matérias-primas para a alimentação animal ou dosalimentos para animais.
f) Gelificantes Formação de um gel resultante numa mudança na textura do alimentopara animais.
g) Aglutinantes Durabilidade do granulado ou desempenho da formação de grânulos.
h) Substâncias para o controlo deradionuclídeos
Provas de contaminação reduzida de alimentos de origem animal.
i) Antiaglomerantes Capacidade de fluxo. É demonstrado o período para o qual é alegadoum efeito antiaglomerante.
j) Reguladores de acidez pH e/ou capacidade-tampão em alimentos para animais.
k) Aditivos de silagem — Melhoria da produção de silagem;— Inibição de microrganismos indesejáveis;— Redução de efluentes;— Melhoria da estabilidade aeróbia.
l) Desnaturantes Identificação indelével de matérias-primas para a alimentação animal.
Aditivos de silagem
São efectuados ensaios separados para demonstrar o efeito solicitado no processo de silagem (2). As experiênciassão realizadas com um exemplo de cada uma das seguintes categorias (sempre que estejam envolvidas todas asforragens ou algumas forragens não especificadas):
— forragem fácil de ensilar: > 3 % de hidratos de carbono solúveis no material fresco (por exemplo, milho deplanta inteira, azevém, bromus ou polpa de beterraba sacarina),
— forragem moderadamente difícil de ensilar: 1,5-3 % de hidratos de carbono solúveis no material fresco (porexemplo, erva de febra, festuca ou luzerna murcha),
— forragem difícil de ensilar: < 1,5 % de hidratos de carbono solúveis no material fresco (por exemplo,panasco ou leguminosas).
Quando os pedidos são restritos a subcategorias de forragem descritas em termos de matéria seca (MS), éexplicitamente indicada a gama de matéria seca. São então realizados três ensaios com material representativo dagama utilizando, sempre que possível, exemplos de origem botânica diferente.
São exigidos ensaios específicos para alimentos especiais para animais.
22.5.2008 PT Jornal Oficial da União Europeia L 133/41
(2) Para efeitos do presente regulamento, entende-se por «Processo de ensilagem» o processo pelo qual a deterioração natural da matériaorgânica é controlada por acidificação em meio anaeróbico resultando da fermentação natural e/ou da adição de aditivos de silagem.

A duração do estudo é normalmente de 90 dias ou mais a uma temperatura constante (intervalo recomendado15-25 oC). A utilização de uma duração mais curta tem de ser justificada.
Regra geral, são apresentadas as medições dos seguintes parâmetros em comparação com o controlo negativo:
— matéria seca e perdas calculadas de matéria seca (corrigidas para voláteis),
— diminuição do pH,
— concentração dos ácidos gordos voláteis (por exemplo, ácido acético, butírico e propiónico) e ácido láctico,
— concentração de álcoois (etanol),
— concentração de amoníaco (g/kg de azoto total), e
— teor de hidratos de carbono hidrossolúveis.
Além disso, são incluídos outros parâmetros microbiológicos e químicos, segundo o caso, para fundamentar aalegação específica feita (por exemplo, número de leveduras assimiladoras de lactato, número de Clostridia, deListeria e aminas biogénicas).
Um efeito procurado para a redução de efluente será avaliado em função do volume total de efluente produzidodurante todo o período experimental, tendo em conta o efeito provável no ambiente (por exemplo, ecotoxicidadedo efluente ou da procura biológica de oxigénio). A redução de produção de efluente é demonstradadirectamente. A capacidade do silo é suficiente para permitir a libertação de efluente com a aplicação de pressão.A duração do estudo é normalmente de 50 dias. Se for utilizado um período diferente, tal decisão é justificada.
A melhoria da estabilidade aeróbia é demonstrada em comparação com um controlo negativo. Os estudos deestabilidade têm uma duração de, pelo menos, sete dias após a exposição ao ar e o aditivo apresenta provas deestabilidade durante, pelo menos, mais dois dias em relação à do grupo de controlo não tratado. Recomenda-seque a experiência seja feita a uma temperatura ambiente de 20 oC e que seja utilizado como indicador deinstabilidade um aumento de temperatura de 3 oC ou mais em relação à temperatura de base. A medição datemperatura pode ser substituída pela medição da produção de CO2.
1.5. Secção V: Monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.Ou seja, é exigido um plano de monitorização pós-comercialização no mercado apenas para aditivos que sejamOGM ou que sejam produzidos a partir de OGM.
2. ADITIVOS SENSORIAIS
2.1. Corantes
2.1.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
2.1.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos não sujeitos a um detentor de autorização específico aplicam-se os números 2.1.2, 2.1.3,2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 e 2.6,
— para outros aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II.
L 133/42 PT Jornal Oficial da União Europeia 22.5.2008

2.1.3. Secção III: Estudos relativos à segurança de utilização do aditivo
Aplica-se na íntegra para cada aditivo a subsecção 3.3 do anexo II.
1. Para substâncias que, quando dadas a animais, acrescentam cores a géneros alimentícios de origem animalaplicam-se na íntegra as subsecções 3.1, 3.2 e 3.4 da secção III do anexo II;
2. Para substâncias que acrescentam ou restituem a cor em alimentos para animais, os estudos referidos nasubsecção 3.1 da secção III são executados em animais alimentados com o aditivo na dose recomendada. Oselementos de prova podem igualmente ser apresentados por referência à literatura científica existente.Aplicam-se as subsecções 3.2 e 3.4 da secção III do anexo II;
3. Para substâncias que afectam favoravelmente a cor de peixes ou aves ornamentais, são exigidos os estudosreferidos na subsecção 3.1 da secção III do anexo II e executados em animais alimentados com o aditivo nadose recomendada. Os elementos de prova podem igualmente ser apresentados por referência à literaturacientífica existente. Contudo, não é exigido o cumprimento das subsecções 3.2 e 3.4.
2.1.4. Secção IV: Estudos referentes à eficácia do aditivo
Aplica-se a totalidade da secção IV do anexo II.
a) Para substâncias que, quando administradas aos animais, conferem cor aos géneros alimentícios de origemanimal:
As mudanças da cor dos produtos obtidos de animais alimentados com o aditivo nas condiçõesrecomendadas de utilização são medidas utilizando a metodologia adequada. É demonstrado que autilização do aditivo não afecta adversamente a estabilidade dos produtos ou as qualidades organolépticas enutritivas dos géneros alimentícios. Em princípio, se os efeitos de uma substância particular na composição//características de produtos de origem animal estiverem bem documentados, então outros estudos (porexemplo, estudos de disponibilidade biológica) podem apresentar elementos adequados de prova da eficácia;
b) Para substâncias que conferem ou restituem a cor dos alimentos para animais:
São apresentadas provas de eficácia pelos estudos de laboratório adequados que reflictam as condições deutilização pretendidas em comparação com alimentos para animais de controlo;
c) Para substâncias que afectam favoravelmente a cor de peixes e pássaros ornamentais:
São executados estudos que demonstram o(s) efeito(s) em animais alimentados com o aditivo nos níveisrecomendados de utilização. As mudanças de cor são medidas utilizando a metodologia adequada. Asprovas de eficácia podem igualmente ser apresentadas por outros estudos experimentais (por exemplo,disponibilidade biológica) ou por referência à literatura científica.
2.1.5. Secção V: Plano de monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.Ou seja, é exigido um plano de monitorização pós-comercialização apenas para aditivos que sejam OGM ou quesejam produzidos a partir de OGM.
2.2. Compostos aromatizantes
2.2.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
2.2.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Em geral, no caso do grupo «produtos naturais», as plantas inteiras, os animais e outros organismos e partes destesou os produtos deles derivados resultantes da transformação muito limitada, tal como o esmagamento, a moagemou a secagem (por exemplo, várias plantas aromáticas e especiarias), não são considerados como abrangidos poreste grupo funcional da categoria dos aditivos sensoriais.
22.5.2008 PT Jornal Oficial da União Europeia L 133/43

Para efeitos da avaliação de pedidos relativos a estes produtos, os aromatizantes são classificados do seguintemodo:
1. Produtos naturais:
1.1. Produtos naturais — definidos botanicamente.
1.2. Produtos naturais — de origem não vegetal.
2. Aromatizantes naturais ou correspondentes sintéticos quimicamente definidos
3. Substâncias artificiais.
É indicado o grupo em causa, ao qual pertence o produto objecto do pedido. Se o produto não se enquadrarem nenhum dos grupos mencionados supra, tal facto é mencionado e justificado.
2.2.2.1. Caracterização da(s) substância(s) activa(s)/agente(s)
Aplica-se a totalidade da subsecção 2.2 do anexo II.
Além disso:
Para todos os grupos de aromatizantes, são sempre fornecidos, quando disponíveis, o(s) número(s) deidentificação em causa (como o do FLAVIS (3), o do Conselho da Europa (4), o do CMPAA, o do CAS (5) ouqualquer outro sistema de numeração internacionalmente aceite) utilizado(s) especificamente para a identificaçãodos produtos aromatizantes nos alimentos para animais e nos géneros alimentícios.
1. Produtos naturais — definidos botanicamente
A caracterização dos produtos naturais definidos botanicamente inclui o nome científico da planta deorigem, a sua classificação botânica (família, género, espécie, subespécie e variedade, se adequado) e osnomes e sinónimos comuns em tantas línguas europeias quanto possível ou noutra(s) língua(s) (tal como a(s) do(s) lugar(es) de cultivo ou origem), quando disponível. São indicadas as partes da planta utilizadas(folhas, flores, sementes, frutos, tubérculos, etc.) e, para plantas menos conhecidas, o lugar de cultivo, oscritérios de identificação e outros aspectos relevantes destas plantas. São identificados e quantificados oscomponentes principais do extracto e é apresentada a sua gama ou variabilidade. É dada atenção especial aimpurezas, tal como mencionado na subsecção 2.1.4 do anexo II. São igualmente relatadas asconcentrações de substâncias que levantam preocupação a nível toxicológico (6) para seres humanos ouanimais, que podem ocorrer na planta a partir da qual o extracto é produzido.
As propriedades farmacológicas ou conexas da planta de origem, das suas partes ou dos produtos deladerivados são plenamente investigadas e relatadas.
2. Produtos naturais — de origem não vegetal
Pode ser utilizada uma abordagem equivalente à mencionada supra.
3. Aromatizantes naturais ou correspondentes sintéticos quimicamente definidos
Além dos requisitos gerais da subsecção 2.2.1.1 do anexo II, é especificada a origem do aromatizante.
L 133/44 PT Jornal Oficial da União Europeia 22.5.2008
(3) Número de identificação para substâncias aromatizantes quimicamente definidas utilizadas no FLAVIS, o sistema de informação da UEsobre aromatizantes, a base de dados utilizada no âmbito do Regulamento (CE) n.o 1565/2000 da Comissão, de 18 de Julho de 2000,que estabelece as medidas necessárias para a adopção de um programa de avaliação em aplicação do Regulamento (CE) n.o 2232/96 doParlamento Europeu e do Conselho (JO L 180 de 19.7.2000, p. 8).
(4) N.o COE: Número do Conselho da Europa para os produtos aromatizantes botanicamente definidos no Relatório n.o 1 do Conselho daEuropa sobre «Fontes de aromas naturais», volume I, Estrasburgo, 2000 e volumes subsequentes.
(5) N.o CAS: Número de registo do «Chemical Abstracts Service» (CAS), identificador único para substâncias químicas amplamente utilizadoem inventários químicos.
(6) Para efeitos desta secção do presente regulamento, entende-se por «substância que levanta preocupação a nível toxicológico» umasubstância com uma dose diária ou semanal tolerável (DDT ou DST), uma DDA, ou com uma restrição na sua utilização, ou um princípioactivo tal como definido na Directiva 88/388/CEE do Conselho relativa à aproximação das legislações dos Estados-Membros no domíniodos aromas destinados a serem utilizados nos géneros alimentícios e dos materiais de base para a respectiva produção, ou uma substânciaindesejável.

2.2.2.2. Método de produção e fabrico
Aplica-se a totalidade da subsecção 2.3 do anexo II.
No caso dos produtos naturais não quimicamente bem definidos, de modo geral misturas complexas de muitoscompostos obtidos por um processo de extracção, é apresentada uma descrição pormenorizada do processo deextracção. Recomenda-se que a descrição utilize a terminologia relevante tal como óleo essencial, absoluto,tintura, extracto e termos relacionados (7), utilizados amplamente para produtos aromatizantes botanicamentedefinidos para descrever o processo de extracção. São especificados os solventes de extracção utilizados, asprecauções tomadas para evitar resíduos de solventes e os níveis de resíduos sempre que estes levantem umapreocupação a nível toxicológicos e se a sua presença for inevitável. Os termos utilizados para caracterizar oextracto podem incluir uma referência ao método de extracção.
2.2.2.3. Métodos de análise
1. Para produtos naturais (definidos botanicamente ou de origem não vegetal) que não contêm substâncias quelevantem preocupação a nível toxicológico para seres humanos ou animais, o requisito normalizado paramétodos de análise da subsecção 2.6 do anexo II pode ser substituído por um método de análise qualitativomais simples adaptado ao objectivo para os componentes principais ou característicos do produto.
2. Para aromatizantes naturais ou correspondentes sintéticos quimicamente definidos que não são substânciasque levantam preocupação a nível toxicológico para seres humanos ou animais, o requisito normalizadopara métodos de análise da subsecção 2.6 do anexo II pode ser substituído por um método de análisequalitativo mais simples adaptado ao objectivo.
Aplica-se a totalidade da subsecção 2.6 do anexo II para todos os outros aromatizantes, tais como os extractosnaturais que contêm substâncias que levantam preocupação a nível toxicológico, aromatizantes naturais oucorrespondentes sintéticos quimicamente definidos que são, eles próprios, substâncias que levantam preocupaçãoa nível toxicológico e aromatizantes artificiais.
2.2.3. Secção III: Estudos referentes à segurança do aditivo
Para todos os aromatizantes, são apresentados os cálculos da exposição animal e da ingestão a partir da exposiçãonatural e no seguimento da adição do aromatizante aos alimentos para animais.
Para aromatizantes pertencentes ao grupo das substâncias artificiais, aplica-se a totalidade da secção III doanexo II.
2.2.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
1. Produtos naturais (definidos botanicamente ou de origem não vegetal)
A segurança destes produtos pode ser avaliada com base nos seus componentes principais e característicos etendo igualmente em consideração substâncias conhecidas que levantam preocupação a nível toxicológico.Se os componentes principais ou característicos não estiverem ainda autorizados como aromatizantesquimicamente definidos ou como aditivos destinados à alimentação animal, tem de se verificar se sãosubstâncias que levantam preocupação a nível toxicológico para seres humanos ou animais e as suaspropriedades toxicológicas têm de ser apresentadas em conformidade com a subsecção 3.1 do anexo II.
2. Aromatizantes naturais ou correspondentes sintéticos quimicamente definidos
Se estas substâncias forem aromatizantes autorizados para seres humanos, a segurança para espécies-alvopode ser avaliada tendo em conta a comparação entre o nível de ingestão pelas espécies-alvo a partir dosalimentos para animais proposto pelo requerente com o dos seres humanos através dos génerosalimentícios. São apresentados os dados metabólicos e toxicológicos utilizados para a realização daavaliação para uso humano.
Em todos os outros casos diferentes do caso em que ambos os níveis de ingestão sejam semelhantes, talcomo no caso em que o nível de ingestão pelo animal-alvo proposto pelo requerente seja substancialmentemais alto que o do ser humano através dos géneros alimentícios, ou em que a substância não estejaautorizada para géneros alimentícios, a segurança para os animais-alvo pode ser avaliada tendo em conta osseguintes dados: o princípio do limiar de preocupação a nível toxicológico (8), dados metabólicos etoxicológicos disponíveis para compostos conexos, e consideração do alerta de estrutura química (seguindo,por analogia, o Regulamento (CE) n.o 1565/2000 da Comissão, de 18 de Julho de 2000, que estabelece asmedidas necessárias para a adopção de um programa de avaliação em aplicação do Regulamento (CE)n.o 2232/96 do Parlamento Europeu e do Conselho (9).
São necessários estudos de tolerância apenas quando os limiares são excedidos ou não podem serdeterminados.
22.5.2008 PT Jornal Oficial da União Europeia L 133/45
(7) Definidos no apêndice 4 do relatório n.o 1 do Conselho da Europa sobre «Fontes de aromas naturais», volume I, Estrasburgo, 2000.(8) CMPAA (FAO/OMS, 1996, Aditivos alimentares, série 35, IPCS, OMS, Genebra), o limiar correspondente para o animal-alvo deveria ser
ajustado para ter em conta o peso do animal e a ingestão através dos alimentos para animais.(9) JO L 180 de 19.7.2000, p. 8.

2.2.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
São apresentados os elementos de prova de que os metabolitos do aromatizante não resultam numa acumulaçãono animal de produtos que levantam preocupação a nível toxicológico para os seres humanos. No caso em que autilização do produto aromatizante solicitado, em consequência da sua adição a alimentos para animais, resulteem resíduos em géneros alimentícios de origem animal, é apresentado o cálculo pormenorizado da exposição dosconsumidores.
a) Estudos metabólicos e resíduos
1. Produtos naturais (definidos botanicamente ou de origem não vegetal)
A segurança destes produtos para seres humanos quando utilizados como aromatizantes emalimentos para animais, no que respeita ao seu metabolismo, pode ser baseada no metabolismo (noanimal-alvo), em estudos de resíduos dos seus componentes principais e característicos e na ausênciano extracto de substâncias que levantam preocupação a nível toxicológico.
Se os componentes principais ou característicos não estiverem ainda autorizados como aromatizantesquimicamente definidos ou se o nível de ingestão pelos animais-alvo a partir da respectivaalimentação for substancialmente mais elevado que o dos seres humanos a partir dos génerosalimentícios, é exigida a aplicação da totalidade da subsecção 3.2.1 do anexo II.
2. Aromatizantes naturais ou correspondentes sintéticos quimicamente definidos
Se estes produtos não estiverem autorizados como aromatizantes para seres humanos ou se o nível deingestão pelos animais-alvo a partir da respectiva alimentação, como proposto pelo requerente forsubstancialmente mais elevado que o dos seres humanos a partir dos géneros alimentícios, sãofornecidos os dados disponíveis sobre o destino metabólico e os mesmos são utilizados para avaliar aacumulação potencial em tecidos e produtos comestíveis, de acordo com subsecção 3.2.1 do anexo II.
b) Estudos toxicológicos
1. Produtos naturais (definidos botanicamente ou de origem não vegetal)
A segurança destes produtos para os seres humanos quando utilizados como aromatizantes emalimentos para animais pode ser baseada nos dados toxicológicos dos seus componentes principais oucaracterísticos e na ausência no extracto de substâncias que levantam preocupação a níveltoxicológico.
É exigido um conjunto toxicológico quando os estudos metabólicos dos compostos principais oucaracterísticos revelarem que existe acumulação em tecidos ou produtos de origem animal e forexcedido limiar de preocupação a nível toxicológico para o animal-alvo. Este conjunto toxicológicoinclui estudos de genotoxicidade, incluindo mutagenicidade e um estudo de toxicidade oralsubcrónica, de acordo com subsecção 3.2.1 do anexo II.
2. Aromatizantes naturais ou correspondentes sintéticos quimicamente definidos
É exigido um conjunto toxicológico que inclui estudos de genotoxicidade, incluindo mutagenicidade eum estudo de toxicidade oral subcrónica, de acordo com a subsecção 3.2.1 do anexo II, quando osestudos metabólicos destes produtos mostrarem que existe acumulação em tecidos ou produtos deorigem animal e for excedido o limiar de preocupação a nível toxicológico para o animal-alvo.
2.2.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
2.2.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Aplica-se a totalidade da subsecção 3.4 do anexo II.
2.2.4. Secção IV: Estudos referentes à eficácia do aditivo
São apresentados os elementos de prova das propriedades aromatizantes, geralmente com base na literaturapublicada. As mesmas podem igualmente ser demonstradas por experiência derivada da utilização prática, sempreque disponível, ou, em caso contrário, podem ser exigidos estudos em animais.
Tem de ser plenamente investigado e relatado se o produto objecto do pedido exerce outras funções naalimentação animal, no animal ou nos géneros alimentícios de origem animal para além das referidas na definiçãode compostos aromatizantes constante do anexo I do Regulamento (CE) n.o 1831/2003.
L 133/46 PT Jornal Oficial da União Europeia 22.5.2008

2.2.5. Secção V: Plano de monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.Ou seja, é exigido um plano de monitorização de pós-comercialização no mercado apenas para aditivos que sejamOGM ou que sejam produzidos a partir de OGM.
3. ADITIVOS NUTRITIVOS
3.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
3.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos não sujeitos a um detentor de autorização específico aplicam-se os números 2.1.2, 2.1.3,2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 e 2.6,
— para outros aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II.
3.3. Secção III: Estudos referentes à segurança dos aditivos
3.3.1. Estudos relativos à segurança de utilização do aditivo em espécies-alvo
3.3.1.1. Testes de tolerância nas espécies-alvo
1. Não são exigidos estudos para a ureia e aminoácidos, seus sais e análogos autorizados pela Directiva 82//471/CEE nem compostos de oligoelementos e vitaminas, provitaminas e substâncias quimicamente bemdefinidas que produzem um efeito semelhante que não possuem um potencial de acumulação jáautorizados como aditivos destinados à alimentação animal ao abrigo da Directiva 70/524/CEE.
2. Para os aditivos abrangidos pelo grupo funcional «Vitaminas, provitaminas e substâncias quimicamente bemdefinidas que produzem um efeito semelhante» e que tenham um potencial de acumulação, será apenasnecessário demonstrar tolerância para compostos para os quais se preveja ou tenha sido demonstrado que apotência seja diferente da da(s) vitamina(s) bem estabelecida. Em certos casos, os elementos do ensaio detolerância (concepção ou critérios) poderiam ser combinados com uma das experiências de eficácia.
3. Será demonstrada a tolerância para os derivados da ureia, análogos de aminoácidos e compostos deoligoelementos não previamente autorizados. Será necessária uma demonstração de tolerância para osprodutos de fermentação, a menos que a substância activa esteja separada do produto de fermentação brutoe seja altamente purificada, ou o organismo de produção tenha um historial de utilização segura aparente eseja bem conhecido no que se refere à sua biologia, no sentido de excluir um potencial para a produção dosmetabolitos tóxicos.
4. Quando o pedido for solicitado para todas as espécies/categorias animais, é suficiente um estudo detolerância na espécie mais sensível (ou mesmo um animal adequado para ensaio em laboratório) segundo osconhecimentos mais recentes.
3.3.1.2. Estudos microbianos
Aplica-se a totalidade da subsecção 3.1.2 do anexo II.
3.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
3.3.2.1. Estudos metabólicos e resíduos
Normalmente, não são exigidos estudos metabólicos. Para derivados de ureia, é estudado o metabolismoruminante nas experiências de eficácia.
22.5.2008 PT Jornal Oficial da União Europeia L 133/47

Apenas são exigidos estudos de resíduos ou de deposição para os aditivos abrangidos pelo grupo funcional«Vitaminas, provitaminas e substâncias quimicamente bem definidas que produzam um efeito semelhante» quetenham um potencial de acumulação no corpo e para o grupo funcional dos compostos de oligoelementossempre que tenha sido aumentada a biodisponibilidade. Nesse caso, não se aplica o procedimento descrito nasubsecção 3.2.1 do anexo II. O requisito está limitado à comparação dos níveis nos tecidos ou produtos entre ogrupo a quem foi dada em suplemento a dose mais elevada da substância objecto de pedido e um controlopositivo (composto de referência).
3.3.2.2. Estudos toxicológicos
Estes são exigidos para produtos de fermentação e aditivos ainda não autorizados. Para produtos de fermentação,têm de se apresentar estudos de genotoxicidade e de toxicidade subcrónica, a menos que:
1. A substância activa esteja separada do produto de fermentação bruto e seja altamente purificada; ou
2. O organismo de produção tenha um historial de utilização segura aparente e haja um conhecimentosuficiente da sua biologia de forma a excluir um potencial de produção de metabolitos tóxicos.
Sempre que o organismo de produção pertença a um grupo no qual algumas estirpes sejam conhecidas porproduzir toxinas, a sua presença é especificamente excluída.
3.3.2.3. Avaliação de segurança para o consumidor
Aplica-se a totalidade da subsecção 3.2.3 do anexo II.
3.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
3.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Aplica-se a totalidade da subsecção 3.4 do anexo II às novas substâncias activas pertencentes ao grupo«Compostos de oligoelementos».
3.4. Secção IV: Estudos referentes à eficácia do aditivo
Não são exigidos estudos de eficácia para a ureia, os aminoácidos, sais de aminoácido e análogos já autorizadoscomo aditivos destinados à alimentação animal, compostos de oligoelementos já autorizados como aditivosdestinados à alimentação animal e vitaminas, provitaminas e substâncias quimicamente bem definidas queproduzam um efeito semelhante já autorizadas como aditivos destinados à alimentação animal.
É exigido um estudo de curta duração para apoiar a eficácia dos derivados da ureia, aminoácidos, sais deaminoácido e análogos ainda não autorizados como aditivos destinados à alimentação animal, compostos deoligoelementos ainda não autorizados como aditivos destinados à alimentação animal e vitaminas, provitaminas esubstâncias quimicamente bem definidas que produzam um efeito semelhante ainda não autorizadas comoaditivos destinados à alimentação animal.
Para outras substâncias para as quais seja alegado um efeito nutritivo é solicitado, pelo menos, um estudo deeficácia a longo prazo, nos termos de secção 4 do anexo II.
Quando tal seja necessário, os estudos demonstram que o aditivo pode responder às necessidades nutritivas dosanimais. Os ensaios incluem um grupo de ensaio com um regime alimentar contendo o nutriente emconcentrações inferiores às necessidades dos animais. Contudo, evitam-se as experiências que utilizam um grupode controlo severamente deficiente. Em geral, será suficiente demonstrar a eficácia numa única espécie oucategoria animal, incluindo animais para ensaio em laboratório.
3.5. Secção V: Monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.
L 133/48 PT Jornal Oficial da União Europeia 22.5.2008

4. ADITIVOS ZOOTÉCNICOS
4.1. Aditivos zootécnicos que não enzimas e microrganismos
4.1.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
4.1.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a totalidade da secção II do anexo II.
4.1.3. Secção III: Estudos referentes à segurança dos aditivos
4.1.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Aplica-se a totalidade da subsecção 3.1 do anexo II.
4.1.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
1. Estudos metabólicos e de resíduos
Estes estudos não são exigidos se:
— se puder demonstrar que a substância ou os seus metabolitos são excretados inalterados e não sãoabsorvidos em grande medida, ou
— a substância for absorvida sob a forma fisiológica e ao nível fisiológico do(s) composto(s).
Não são necessários estudos metabólicos se a substância estiver naturalmente presente em montantessignificativos em géneros alimentícios ou alimentos para animais ou se a substância for um constituintenormal de fluidos ou tecidos corporais. Contudo, nestes casos, há uma exigência de estudos de resíduos quepodem ser limitados à comparação dos níveis nos tecidos ou produtos num grupo não tratado com osníveis constatados no grupo a quem foi dada em suplemento a dose mais elevada recomendada.
Nos restantes casos, aplica-se toda a subsecção 3.2.1 do anexo II.
2. Estudos toxicológicos
Não são exigidos estudos toxicológicos se a substância for absorvida sob a forma do(s) composto(s)fisiológico(s).
Para substâncias xenobióticas, aplica-se a totalidade da subsecção 3.2.2 do anexo II.
Para outras substâncias, é utilizada uma abordagem caso a caso, tendo em conta o nível e os meios deexposição e qualquer omissão de dados prescritos nesta secção tem de ser plenamente justificada.
3. Avaliação de segurança para o consumidor
Aplica-se a totalidade da subsecção 3.2.3 do anexo II para animais produtores de géneros alimentícios.
4.1.3.3. Estudos relativos à segurança do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
4.1.3.4. Estudos relativos à segurança do aditivo para o ambiente
Aplica-se a totalidade da subsecção 3.4 do anexo II.
22.5.2008 PT Jornal Oficial da União Europeia L 133/49

4.1.4. Secção IV: Estudos referentes à eficácia do aditivo
Aplica-se a totalidade da secção IV do anexo II.
1. Aditivos que afectam favoravelmente a produção, o desempenho ou o bem-estar animal e o grupo funcional«Outros aditivos zootécnicos».
Os efeitos podem apenas ser demonstrados em relação a cada espécie ou categoria animal-alvo. Em funçãodas propriedades do aditivo, as medidas resultantes podem ser baseadas em características de desempenho(por exemplo, eficiência da alimentação, ganho diário médio, aumento de produtos de origem animal), nacomposição da carcaça, no desempenho do rebanho, nos parâmetros de reprodução ou de bem-estar dosanimais. Os elementos de prova do modo de acção podem ser apresentados por estudos de eficácia a curtoprazo ou por estudos de laboratório medindo o parâmetro relevante.
2. Aditivos que influenciam favoravelmente as consequências da produção animal sobre o ambiente
Para estes aditivos que afectam favoravelmente o ambiente (por exemplo, redução da excreção de azoto oude fósforo ou produção reduzida de metano, odores anormais), as provas de eficácia para as espécies-alvopodem ser dadas por três estudos de eficácia a curto prazo com animais que revelem efeitos benéficossignificativos. Os estudos tomam em consideração a possibilidade de uma resposta adaptável ao aditivo.
4.1.5. Secção V: Monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.
4.2. Aditivos zootécnicos: enzimas e microrganismos
4.2.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
4.2.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a totalidade da secção II do anexo II.
4.2.3. Secção III: Estudos referentes à segurança dos aditivos
4.2.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Aplica-se a totalidade da subsecção 3.1.1 do anexo II.
Os candidatos são incentivados a utilizar, na medida do possível, pelo menos, uma dose 100 vezes superior nogrupo experimental e reduzir consequentemente o número de parâmetros exigidos. Pode ser utilizada para estefim uma forma concentrada do aditivo. A concentração é ajustada mediante a redução do montante de excipientepresente mas a razão agente(s) substância(s) activa(s) para os outros produtos de fermentação deve permanecer amesma que no produto final. Para as enzimas, o regime alimentar fornece o(s) substrato(s) adequado(s).
Aplica-se a totalidade da subsecção 3.1.2 do anexo II a todos os microrganismos e enzimas com um efeitocatalítico directo em elementos da microbiota ou para os quais, de outra forma, se alegue afectarem a microbiotado intestino.
Quando haja uma nova exposição ou um aumento substancial da extensão de exposição a microrganismos,podem ser necessários estudos adicionais para demonstrar a ausência de efeitos nocivos na microbiota comensaldo tracto digestivo. Para ruminantes, apenas serão necessárias contagens directas da microbiota se tal for indiciadopor provas de uma mudança negativa de função do rúmen (medida in vitro como uma mudança em concentraçõesvoláteis de ácido gordo, redução da concentração de propionato ou celulólise reduzida).
4.2.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
1. Não são exigidos estudos metabólicos e de resíduos.
2. Estudos toxicológicos, de acordo com subsecção 3.2.2 do anexo II.
L 133/50 PT Jornal Oficial da União Europeia 22.5.2008

As enzimas e os microrganismos constituem apenas uma parte do aditivo completo que, na maioria dos casos,pode incluir outros componentes originados a partir do processo de fermentação. Consequentemente, énecessário testar o aditivo para assegurar que não contém material mutagénico ou outro que possa lesar osconsumidores humanos de géneros alimentícios derivados de animais alimentados com alimentos para animaisou água tratados com estes aditivos.
Contudo, a maioria das bactérias viáveis destinadas à ingestão directa ou indirecta por mamíferos (incluindo sereshumanos) são seleccionadas de grupos de organismos com um historial de utilização segura aparente ou degrupos onde os riscos tóxicos são bem definidos. Do mesmo modo, os riscos associados aos microrganismosutilizados actualmente para a produção de enzimas são bem conhecidos de modo geral e substancialmentereduzidos pelos métodos de produção modernos. Por conseguinte, para enzimas de fontes microbianas e paramicrorganismos com um historial de utilização segura aparente e sempre que os componentes do processo defermentação sejam bem definidos e conhecidos, não são considerados necessários ensaios de toxicidade (porexemplo, ensaios de toxicidade oral ou genotoxicidade). Contudo, para organismos vivos e aqueles utilizados paraa produção de enzimas, são sempre abordadas as preocupações específicas na secção 2.2.2.2 do anexo II.
Quando o organismo ou a sua aplicação forem novos e a biologia do organismo (de produção) insuficientementeconhecida de forma a excluir um potencial de produção de metabolitos tóxicos, são introduzidos estudos degenotoxicidade e toxicidade oral feitos com os aditivos que contêm microrganismos viáveis ou enzimas. Nestecaso, assumem a forma de estudos de genotoxicidade incluindo mutagenicidade e um estudo de toxicidade oralsubcrónica. Recomenda-se que tais estudos sejam executados com o caldo de fermentação sem células ou, no casode uma fermentação em estado sólido, um extracto adequado.
4.2.3.3. Estudos relativos à segurança do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II, excepto quando:
— se presume que as enzimas e os microrganismos, enquanto substâncias proteináceas, sejam sensibilizadoresrespiratórios, a menos que sejam apresentados elementos convincentes de prova em contrário. Porconseguinte, não é exigido um ensaio directo,
— a formulação do produto (por exemplo, microcapsulação) pode eliminar a necessidade de alguns ou detodos os ensaios. Em tais casos, é apresentada uma justificação adequada.
4.2.3.4. Estudos relativos à segurança do aditivo para o ambiente
Aplica-se plenamente a totalidade da subsecção 3.4 do anexo II a microrganismos que não tenham origem nointestino ou que não sejam ubíquos no ambiente.
4.2.4. Secção IV: Estudos referentes à eficácia dos aditivos
Aplica-se a totalidade da secção IV do anexo II.
1. Aditivos que afectam favoravelmente a produção, o desempenho ou o bem-estar animal e para o grupofuncional «Outros aditivos zootécnicos»
Os efeitos podem apenas ser demonstrados em relação a cada espécie ou categoria animal-alvo. Em funçãodas propriedades do aditivo, as medidas resultantes podem ser baseadas em características de desempenho(por exemplo, eficiência da alimentação, ganho diário médio, aumento de produtos de origem animal), nacomposição da carcaça, no desempenho do efectivo, nos parâmetros de reprodução ou de bem-estar dosanimais. Os elementos de prova do modo de acção podem ser apresentados por estudos de eficácia a curtoprazo ou por estudos de laboratório medindo o parâmetro relevante.
2. Aditivos que influenciam favoravelmente as consequências da produção animal sobre o ambiente
Para estes aditivos que afectam favoravelmente o ambiente (por exemplo, redução da excreção de fósforo ouazoto ou redução da produção de metano, odores anormais), as provas de eficácia para as espécies-alvopodem ser dadas por três estudos de eficácia a curto prazo com animais que revelem efeitos benéficossignificativos. Os estudos tomam em consideração a possibilidade de uma resposta adaptável ao aditivo.
22.5.2008 PT Jornal Oficial da União Europeia L 133/51

4.2.5. Secção V: Plano de monitorização pós-comercialização
Esta secção aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE) n.o 1831/2003.
5. COCCIDIOSTÁTICOS E HISTOMONOSTÁTICOS
5.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
5.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a totalidade da secção II do anexo II.
5.3. Secção III: estudos referentes à segurança dos aditivos
5.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Aplica-se a totalidade da subsecção 3.1 do anexo II.
5.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
Aplica-se a totalidade da subsecção 3.2 do anexo II.
5.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
5.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Aplica-se a totalidade da subsecção 3.4 do anexo II.
5.4. Secção IV: Estudos referentes à eficácia do aditivo
Estes aditivos protegem os animais dos resultados de uma invasão de Eimeria spp. ou Histomonas meleagridis. É dadaimportância aos elementos de prova dos efeitos específicos do aditivo (por exemplo, espécies controladas) e dassuas propriedades profilácticas (por exemplo, redução da morbilidade, mortalidade, contagem de oocistos e índicede lesões). São facultadas, se necessário, as informações sobre o efeito no crescimento e na conversão alimentar(aves de engorda, poedeiras de substituição e coelhos), os efeitos na eclosibilidade (aves de reprodução).
Os dados de eficácia exigidos derivam a partir de três tipos de experiências com animais-alvo:
— infecções artificiais únicas e mistas,
— infecção natural/artificial para simular condições de utilização,
— condições de utilização reais em experiências de terreno.
As experiências com infecções artificiais únicas e mistas (por exemplo, gaiolas em bateria para aves de capoeira)têm por objectivo demonstrar a eficácia relativa contra os parasitas e não exigem réplica. São exigidos trêsresultados significativos para estudos simulando condições de utilização (por exemplo, estudos de criação no soloem recinto fechado com aves de capoeira, estudos de gaiolas em bateria com coelhos). São igualmente exigidostrês estudos de terreno nos quais um grau de infecção natural esteja presente.
5.5. Secção V: Plano de monitorização pós-comercialização
Esta secção do anexo II aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE)n.o 1831/2003.
L 133/52 PT Jornal Oficial da União Europeia 22.5.2008

6. EXTRAPOLAÇÃO DE ESPÉCIES PRINCIPAIS PARA ESPÉCIES MENORES
As espécies menores estão definidas no n.o 2 do artigo 1.o do presente regulamento.
Normalmente, aceitar-se-á uma candidatura mais reduzida para uma proposta de extensão da autorização do usonuma espécie próxima, do ponto de vista fisiológico, de outra espécie para a qual a utilização do aditivo já tenhasido concedida.
Os seguintes requisitos aplicam-se apenas às autorizações solicitadas para espécies menores de aditivos jáautorizados para espécies principais. Para as autorizações solicitadas para novos aditivos destinados à alimentaçãoanimal solicitadas apenas para espécies menores, aplicam-se plenamente todas as secções, em função da categoria//grupo funcional do aditivo (ver requisitos específicos correspondentes do anexo III).
6.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
6.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II,
— para outros aditivos aplicam-se os números 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4,2.5, 2.6.
6.3. Secção III: Estudos relativos à segurança de utilização do aditivo
6.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
6.3.1.1. Estudos de tolerância nas espécies-alvo
Aplicam-se os requisitos para as diferentes categorias/grupos funcionais de aditivos.
Em princípio, não são exigidos estudos de tolerância para as espécies menores se o aditivo revelar uma amplamargem de segurança (pelo menos, um factor de dez) nas espécies principais fisiologicamente semelhantes.
Se três espécies-alvo principais (incluindo mamíferos monogástricos e ruminantes e aves de capoeira) revelaremuma margem de segurança semelhante e ampla, não seria exigido um estudo de tolerância adicional para espéciesmenores não semelhantes fisiologicamente (por exemplo, cavalos ou coelhos). Sempre que for exigida tolerância, aduração dos estudos para as espécies menores (excepto coelhos) é de, pelo menos, 28 dias para animais emcrescimento e 42 dias para animais adultos. Para coelhos, aplicam-se as seguintes durações: Coelhos de engorda:28 dias; Coelhas reprodutoras: Um ciclo (desde a inseminação até ao fim do período de desmame). Se se solicitar aaplicação em coelhos não desmamados e desmamados, seria considerado suficiente um período de 49 dias (cominício uma semana após o nascimento) e deve incluir as coelhas até ao desmame. Para peixes (com excepção desalmonídeos) é exigido um período de 90 dias.
6.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores humanos
6.3.2.1. Estudos metabólicos
Aplicam-se os requisitos para as diferentes categorias e grupos funcionais de aditivos.
Além disso, não são exigidos estudos metabólicos se o aditivo já estiver autorizado para utilização numa espéciefisiologicamente comparável às espécies menores para as quais a autorização é solicitada. Na ausência desemelhança fisiológica, é considerada suficiente uma comparação do perfil metabólico baseada em estudos in vitro(por exemplo, executado em hepatócitos, utilizando o composto marcado) para avaliar a proximidade metabólica.
Se a espécie menor não for fisiologicamente semelhante a uma espécie principal, é obtida então uma indicação dodestino metabólico do aditivo nas espécies menores.
22.5.2008 PT Jornal Oficial da União Europeia L 133/53

6.3.2.2. Estudos de resíduos
Apenas é necessária a quantificação do resíduo marcador em tecidos e produtos comestíveis quando aproximidade metabólica for dada ou demonstrada. Em todos os restantes casos, aplica-se plenamente asubsecção 3.2.1.2 do anexo II.
6.3.2.3. Avaliação de segurança para o consumidor
Proposta de Limites Máximos de Resíduos (LMR)
A definição de LMR pode ser feita supondo-se que não se verifica nenhuma diferença significativa no teor deresíduos nos tecidos comestíveis de espécies menores em comparação com uma espécie principal semelhante.
Os LMR podem ser extrapolados em classes de animais do seguinte modo:
— de ruminantes principais em crescimento para todos os ruminantes em crescimento,
— de leite de vacas leiteiras para leite de outros ruminantes leiteiros,
— de suínos para todos os mamíferos monogástricos, com exclusão dos cavalos,
— de galinhas ou perus para outras aves de capoeira,
— de galinhas poedeiras para outras aves poedeiras, e
— de salmonídeos para todas as espécies de pescado.
Os LMR para cavalos poderiam ser extrapolados quando existem LMR para um ruminante principal e ummamífero monogástrico principal.
Se forem derivados LMR idênticos em gado bovino (ou ovino), suínos e galinhas (ou aves de capoeira), querepresentem espécies principais com capacidades metabólicas e composição de tecido diferentes, pode igualmenteser definido o mesmo LMR para ovinos, equídeos e coelhos, o que significa que é considerada possível umaextrapolação para todos os animais produtores de géneros alimentícios, excepto peixes. Considerando aorientação do Comité dos Medicamentos para Uso Veterinário (CMUV) (10), na definição de LMR parasalmonídeos e outro pescado, que já permite uma extrapolação de LMR no músculo de uma espécie principal parasalmonídeos e outro pescado desde que as substâncias de origem sejam aceitáveis como resíduo marcador para oLMR no músculo e na pele, os LMR podem ser extrapolados para todos os animais produtores de génerosalimentícios.
Estão disponíveis métodos analíticos para monitorizar resíduos no tecido e produtos comestíveis de todos osanimais produtores de géneros alimentícios.
6.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
6.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Pode ser extrapolada uma avaliação dos riscos ambientais a partir da avaliação executada para as espéciesprincipais fisiologicamente comparáveis. Para os aditivos destinados a serem utilizados em coelhos, aplica-se todaa secção, tendo em consideração os requisitos para cada categoria/grupo funcional de aditivos.
6.4. Secção IV: Estudos referentes à eficácia do aditivo
Quando o aditivo já estiver aprovado para uma espécie principal fisiologicamente comparável para a mesmafunção e sempre que o modo de acção do aditivo seja conhecido ou demonstrado, os elementos de prova domesmo modo de acção nas espécies menores podem ser consideradas como provas de eficácia. Quando tal ligaçãonão puder ser feita, a eficácia é demonstrada de acordo com as regras gerais da secção IV do anexo II. Em algunscasos pode ser adequado combinar espécies animais na mesma fase produtiva (por exemplo, caprinos e ovinosutilizados para a produção de leite). A significância deveria ser demonstrada em cada estudo (P ≤ 0,1) ou, sepossível, por metanálise (P ≤ 0,05).
L 133/54 PT Jornal Oficial da União Europeia 22.5.2008
(10) Nota de orientação relativa à definição de limites máximos de resíduos para salmonídeos e outro pescado. Agência Europeia de Avaliaçãodos Medicamentos. Unidade de avaliação de medicamentos veterinários. EMEA/CVMP/153b/97-FINAL.

Se for exigida uma demonstração da eficácia, a duração dos estudos de eficácia é análoga às fases de produçãocomparáveis das espécies principais fisiologicamente comparáveis. Noutros casos, a duração mínima do estudosegue as disposições relevantes da subsecção 4.4 do anexo II e do anexo IV.
6.5. Secção V: Plano de monitorização pós-comercialização
Esta secção do anexo II aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE)n.o 1831/2003.
7. ANIMAIS DE COMPANHIA E OUTROS ANIMAIS NÃO PRODUTORES DE GÉNEROS ALIMENTÍCIOS
Os animais de companhia e outros animais não produtores de alimento são definidos no n.o 1 do artigo 1.o dopresente regulamento.
7.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
7.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II,
— para outros aditivos aplicam-se os números 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4,2.5, 2.6.
7.3. Secção III: Estudos referentes à segurança do aditivo
7.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Aplicam-se os requisitos para as diferentes categorias/grupos funcionais de aditivos. Quando for exigido umestudo de tolerância, a sua duração é de, pelo menos, 28 dias.
Não é exigido um estudo de tolerância se o aditivo revelar uma margem de segurança comparável e ampla em trêsespécies principais (incluindo mamíferos monogástricos e ruminantes e aves de capoeira).
7.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
Esta subsecção não é geralmente exigida. É tida em consideração a segurança do proprietário.
7.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
7.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
Não é exigida a aplicação da subsecção 3.4 do anexo II.
7.4. Secção IV: Estudos referentes à eficácia do aditivo
Aplicam-se os requisitos para as diferentes categorias/grupos funcionais de aditivos.
Quando o aditivo, para o qual são exigidos estudos animais, for previamente autorizado para outras espéciesfisiologicamente semelhantes, não é exigida mais nenhuma demonstração de eficácia, desde que o efeito solicitadoe o modo de acção sejam o mesmo. Se o aditivo não for previamente autorizado ou o efeito solicitado ou o modode acção forem diferentes da autorização anterior, a eficácia é demonstrada no seguimento das regras gerais dasecção IV do anexo II.
A duração das experiências de eficácia a longo prazo é de, pelo menos, 28 dias.
22.5.2008 PT Jornal Oficial da União Europeia L 133/55

7.5. Secção V: Plano de monitorização pós-comercialização
Esta secção do anexo II aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE)n.o 1831/2003.
8. ADITIVOS JÁ AUTORIZADOS PARA UTILIZAÇÃO EM GÉNEROS ALIMENTÍCIOS
8.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
8.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II,
— para outros aditivos aplicam-se os números 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4,2.5, 2.6.
8.3. Secção III: Estudos referentes à segurança dos aditivos
São incluídas as avaliações formais de segurança mais recentes do aditivo alimentar e completadas com quaisquerdados produzidos subsequentemente.
Para os aditivos que são autorizados como aditivos alimentares ou aprovados como constituintes para produtosalimentares na União Europeia sem nenhuma restrição, não são, em geral, necessários estudos referentes àsegurança para o consumidor e trabalhadores.
São apresentados os dados referidos nas subsecções 3.1, 3.2 e 3.3 do anexo II, tendo em conta o conhecimentoactual sobre a segurança destas substâncias quando utilizadas em géneros alimentícios. Deste modo, taissubstâncias utilizadas igualmente em alimentos podem classificar-se como:
— DDA não especificada (sem uma indicação explícita do limite superior de ingestão, atribuído a substânciasde toxicidade muito baixa),
— DDA ou UL definidos, ou
— DDA não atribuída (aplicável a substâncias para as quais a informação disponível não é suficiente paraestabelecer a sua segurança).
8.3.1. Estudos relativos à segurança de utilização do aditivo em animais-alvo
Se o nível de utilização no que diz respeito ao aditivo destinado à alimentação animal for semelhante ao dosgéneros alimentícios, a segurança para espécies-alvo pode ser avaliada com base nos dados toxicológicos in vivodisponíveis, na consideração da estrutura química e na capacidade metabólica das espécies-alvo. Se o nível deutilização em alimentos para animais for consideravelmente mais elevado que a correspondente nos génerosalimentícios, pode ser exigido um estudo de tolerância no animal-alvo, em função da natureza da substância.
8.3.2. Estudos relativos à segurança de utilização do aditivo para os consumidores
Se a utilização como aditivo destinado à alimentação animal resultar numa exposição mais elevada dosconsumidores, ou num padrão de metabolitos diferente do resultante da utilização em géneros alimentícios, serãoentão exigidos outros dados toxicológicos e de resíduos.
8.3.2.1. Aditivos alimentares para os quais não está especificada uma DDA
Não é exigida a avaliação de segurança para os consumidores, excepto quando a utilização do aditivo emalimentos para animais resultar num padrão de metabolitos diferente do resultante da sua utilização em génerosalimentícios.
L 133/56 PT Jornal Oficial da União Europeia 22.5.2008

8.3.2.2. Aditivos alimentares com DDA ou UL definidos
A segurança dos consumidores tem de ser avaliada tomando em consideração a exposição adicional decorrente dautilização em alimentos para animais, ou na exposição específica relacionada com os metabolitos com origem naespécie-alvo. Esta avaliação pode ser feita mediante a extrapolação de dados de resíduos a partir da literatura.
Quando são necessários estudos de resíduos, o requisito está limitado a uma comparação dos níveis em tecido ouprodutos num grupo não tratado com o grupo a quem foi dada em suplemento a dose solicitada mais elevada.
8.3.2.3. Aditivos alimentares para os quais não está atribuída uma DDA
São claramente especificadas as razões pelas quais não foi atribuída uma DDA. Se tal facto levantar preocupação ea utilização do aditivo em alimentos para animais contribua para um aumento significativo da exposição dosconsumidores, é exigida uma avaliação toxicológica completa.
A exposição adicional decorrente da utilização em alimentos para animais pode ser extrapolada de dados deresíduos a partir da literatura.
Quando são necessários estudos de resíduos, o requisito está limitado a uma comparação dos níveis em tecido ouprodutos num grupo não tratado com o grupo a quem foi dada em suplemento a dose solicitada mais elevada.
8.3.3. Estudos relativos à segurança de utilização do aditivo para os utilizadores/trabalhadores
Aplica-se a totalidade da subsecção 3.3 do anexo II.
São tidas em conta as medidas cautelares estabelecidas para manipular estas substâncias utilizadas em génerosalimentícios, ao considerar a segurança para o utilizador do aditivo destinado à alimentação animal.
8.3.4. Estudos relativos à segurança de utilização do aditivo para o ambiente
É exigida a aplicação da subsecção 3.4 do anexo II.
8.4. Secção IV: Estudos referentes à eficácia do aditivo
Sempre que a função solicitada para a alimentação animal for a mesma que a dos géneros alimentícios, poderianão ser necessária mais nenhuma demonstração de eficácia. De outra forma, os requisitos para eficácia são comoos referidos na secção IV do anexo II.
8.5. Secção V: Plano de monitorização pós-comercialização
Esta secção do anexo II aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE)n.o 1831/2003.
9. ALTERAÇÃO DAS AUTORIZAÇÕES
Uma vez que se pode confiar na avaliação dos dados fornecidos para autorizações precedentes, um processopreparado para um pedido nos termos do n.o 3 do artigo 13.o do Regulamento (CE) n.o 1831/2003 tem deobedecer apenas aos requisitos mencionados infra.
Um pedido de alteração dos termos incluídos num regulamento de autorização em vigor, como a identificação,caracterização ou condições de utilização do aditivo, demonstra que a alteração não produz um efeito nocivo nasespécies-alvo, no consumidor, no utilizador ou no ambiente. Um aditivo pode ser, para este fim, consideradocomo idêntico se a(s) substância(s) activa(s) ou o(s)agente(s) e as condições de utilização forem as mesmas, a suapureza for essencialmente semelhante e não tenha sido introduzido nenhum constituinte que levantepreocupação. Para tais produtos pode ser apresentado um pedido abreviado visto que, normalmente, não seránecessário repetir estudos para demonstrar a segurança para as espécies-alvo, o consumidor, o ambiente e aeficácia.
O pedido aborda os seguintes requisitos:
1. Aplica-se a totalidade do anexo I — incluem-se pormenores da alteração solicitada;
2. Aplica-se a totalidade da secção II do anexo II;
22.5.2008 PT Jornal Oficial da União Europeia L 133/57

3. Os dados têm de ser apresentados indicando que as características químicas ou biológicas do aditivo sãoessencialmente as mesmas que as do produto estabelecido;
4. Sempre que adequado, os elementos de prova para a bioequivalência são apresentados por especificação,pela literatura publicada ou através de estudos específicos. Quando a bioequivalência não for inteiramentedemonstrada, tem de ser demonstrada a conformidade do intervalo de segurança com o LMR;
5. Devem apresentar-se provas de que, à luz dos conhecimentos científicos actuais, o aditivo continua a serseguro ao abrigo das condições aprovadas para as espécies-alvo, os consumidores, os trabalhadores e oambiente;
6. É apresentado um relatório sobre os resultados da monitorização pós-comercialização, caso a autorizaçãoespecifique requisitos nesta matéria; e
7. Têm de ser apresentados os dados específicos que apoiam o pedido de alteração, em conformidade com aspartes relevantes de secções III, IV e V do anexo II.
10. RENOVAÇÃO DE AUTORIZAÇÕES
Os pedidos de renovação de autorização nos termos do artigo 14.o do Regulamento (CE) n.o 1831/2003obedecem aos seguintes requisitos:
10.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II. É apresentada uma cópia da autorização original da Comunidadepara colocar o aditivo destinado à alimentação animal no mercado, ou a última renovação de autorização. Épreparado um processo actualizado de acordo com os requisitos mais recentes e uma lista que apresente todas asvariações desde a autorização original ou é apresentada a última renovação de autorização. O requerente tem deapresentar um resumo do processo, pormenorizando o âmbito do pedido e qualquer nova informação que ficoudisponível desde a autorização/renovação anterior em termos de identidade e segurança.
10.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II,
— Para outros aditivos aplicam-se os números 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4,2.5, 2.6.
São apresentadas provas demonstrativas de que o aditivo não foi significativamente alterado nem se modificou asua composição, pureza ou actividade em relação ao aditivo que foi autorizado. É comunicada qualquer alteraçãono processo de fabrico.
10.3. Secção III: Estudos referentes à segurança dos aditivos
São apresentados elementos de prova de que, à luz dos conhecimentos actuais, o aditivo continua a ser seguro aoabrigo das condições aprovadas para as espécies-alvo, os consumidores, os trabalhadores e o ambiente. Éapresentada uma actualização em matéria de segurança desde a autorização original ou desde a última renovaçãocom informações relativas às seguintes áreas:
— relatórios sobre os efeitos nocivos incluindo os acidentes (efeitos anteriormente desconhecidos, efeitosgraves de qualquer tipo, aumento da incidência dos efeitos conhecidos) para os animais-alvo, osconsumidores, os utilizadores e o ambiente. O relatório relativo aos efeitos nocivos inclui a natureza doefeito, o número de indivíduos/organismos afectados, o resultado, as condições de utilização e a avaliaçãodo nexo de causalidade,
— relatórios relativos a interacções e a contaminações cruzadas anteriormente desconhecidas,
— dados relativos ao controlo de resíduos, sempre que adequado,
L 133/58 PT Jornal Oficial da União Europeia 22.5.2008

— dados de estudos epidemiológicos e/ou toxicológicos,
— qualquer outra informação referente à segurança do aditivo e aos riscos do aditivo para os animais, os sereshumanos, e o ambiente.
Caso não se forneçam informações complementares relativas a qualquer destas áreas, as razões para este facto sãoclaramente identificadas.
É apresentado um relatório sobre os resultados do programa de monitorização de pós-comercialização nomercado, se tal requisito de monitorização estiver incluído na autorização anterior.
Sempre que, tal como previsto no n.o 2, alínea d), do artigo 14.o do Regulamento (CE) n.o 1831/2003, o pedidode renovação da autorização incluir uma proposta para alterar ou completar as condições da autorização original,nomeadamente as condições referentes à monitorização futura, têm de ser apresentados os dados específicos queapoiam a proposta de alteração, em conformidade com as partes relevantes de secções III, IV e V do anexo II.
11. REAVALIAÇÃO DE CERTOS ADITIVOS JÁ AUTORIZADOS AO ABRIGO DA DIRECTIVA 70/524/CEE
Os aditivos abrangidos por este ponto 11 são aditivos que foram autorizados ao abrigo da Directiva 70/524/CEE,que devem ser reavaliados em conformidade com o n.o 2 do artigo 10.o do Regulamento (CE) n.o 1831/2003 eque pertencem aos seguintes grupos:
— substâncias antioxidantes,
— substâncias aromatizantes e apetentes,
— substâncias emulsionantes e estabilizantes, espessantes e agentes gelificantes,
— corantes, incluindo os pigmentos,
— conservantes,
— vitaminas, provitaminas e substâncias com efeito análogo quimicamente bem definidas,
— oligoelementos,
— qgentes aglutinantes, antiaglomerantes e coagulantes,
— reguladores de acidez, e
— aglomerantes de radionuclídeos.
O nível e a qualidade da avaliação do risco para estes aditivos são semelhantes a outros aditivos. Contudo, devidoao seu longo historial de utilização segura, podem ser utilizados os dados de estudos já publicados, ao abrigo dedisposições estabelecidas pelo presente regulamento, para demonstrar que o aditivo permanece seguro nascondições aprovadas para as espécies-alvo, os consumidores, os utilizadores e o ambiente.
11.1. Secção I: Resumo do processo
Aplica-se a totalidade da secção I do anexo II.
11.2. Secção II: Identidade, características e condições de utilização do aditivo; métodos de análise
Aplica-se a secção II do anexo II do seguinte modo:
— para aditivos não sujeitos a um detentor de autorização específico aplicam-se os pontos 2.1.2, 2.1.3, 2.1.4,2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 e 2.6,
— para outros aditivos sujeitos a um detentor de autorização específico, aplica-se toda a secção II.
22.5.2008 PT Jornal Oficial da União Europeia L 133/59

11.3. Secção III: Estudos referentes à segurança dos aditivos
Sempre que um aditivo for avaliado em termos de segurança para as espécies-alvo, os consumidores, osutilizadores/trabalhadores e o ambiente, é apresentado um resumo dos estudos de segurança apresentados para aautorização anterior, juntamente com qualquer nova informação que tenha surgido desde essa autorizaçãoanterior. Quando não tenha sido efectuada uma avaliação formal de segurança para a utilização da substânciacomo aditivo destinado à alimentação animal, podem ser utilizados estudos e dados da literatura científica desdeque seja equivalente aos que seriam exigidos para um novo pedido. Caso contrário, é apresentado um conjuntocompleto de estudos de segurança.
11.4. Secção IV: Estudos referentes à eficácia do aditivo
Sempre que adequado, o cumprimento do requisito de eficácia previsto no n.o 3 do artigo 5.o do Regulamento(CE) n.o 1831/2003 pode ser demonstrado por apresentação de material, com excepção de estudos, em especialreferentes ao longo historial de utilização.
11.5. Secção V: Plano de monitorização pós-comercialização
Esta secção do anexo II aplica-se ao abrigo do disposto no n.o 3, alínea g), do artigo 7.o do Regulamento (CE)n.o 1831/2003.
L 133/60 PT Jornal Oficial da União Europeia 22.5.2008

ANEXO IV
Categorias e definições de animais-alvo e indicação da duração mínima dos estudos de eficácia
1. Quadro. Categoria animais: Suínos
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período/idade Idade Peso
Leitões (não desmama-dos)
Suínos jovens que obtêm leite de porcas Desde o nascimento Até 21-42 dias Até 6-11 kg 14 dias
Leitões (desmamados) Suínos jovens que terminaram o período delactação e criados para reprodução ou produção decarne
A partir de 21-42dias
Até 120 dias Até 35 kg 42 dias
Leitões (não desmama-dos e desmamados)
Suínos jovens para fins de reprodução ou produçãode carne desde o nascimento
Desde o nascimento Até 120 dias Até 35 kg 58 dias
Suínos de engorda Suínos que terminaram o período de desmame edestinados à produção de carne até ao dia dotransporte para o matadouro
A partir de 60-120dias
Até 120-250 dias (oude acordo com ocostume local)
80-150 kg (ou deacordo com o cos-tume local)
Até ao peso de abate, mas não menos que 70 dias
Porcas reprodutoras Suínos fêmeas fecundados/acasalados pelo menosuma vez
A partir da primeirainseminação
Desde a inseminação até ao fim do segundo períodode desmame (dois ciclos)
Porcas, a fim de bene-ficiar os leitões
Suínos fêmeas fecundados/acasalados pelo menosuma vez
Pelo menos, duas semanas antes do parto até ao fimde período de desmame
2. Quadro. Categoria animais: Aves de capoeira
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Frangos de engorda Aves criadas para engorda A partir da incubação Até 35 dias Até ~1 600 g (até2 kg)
35 dias
Frangas para postura Aves fêmeas criadas para produção de ovos de mesaou para fins de reprodução
A partir da incubação Até ~16 semanas(até 20 semanas)
— 112 dias (se não estiverem disponíveis dados deeficácia para frangos de engorda)
22.5.2008PT
JornalOficialda
União
EuropeiaL133/61
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Galinhas poedeiras Aves fêmeas produtivas mantidas para fins deprodução de ovos
A partir de 16-21semanas
Até ~13 meses(até 18 meses)
A partir de 1 200 g(brancas) 1 400 g(castanhas)
168 dias
Perus de engorda Aves criadas para engorda A partir da incubação Até ~14 semanas(até 20 semanas)Até ~16 semanas(até 24 semanas)
Peruas: até ~7 000 g(até 10 000 g)Perus: até ~12 000 g(até 20 000 g)
84 dias
Perus reprodutores Aves fêmeas e machos mantidas para fins dereprodução
Todo o período Desde 30 a ~60semanas
Peruas: Desde~15 000 gPerus: Desde~30 000 g
Pelo menos seis meses
Perus criados parareprodução
Aves fêmeas e machos jovens criadas para fins dereprodução
A partir da incubação Até 30 semanas Peruas: até ~15 000 gPerus: até ~30 000 g
Todo o período (se não estiverem disponíveis dadosde eficácia para perus de engorda)
3. Quadro. Categoria animais: Bovinos (bovinos domésticos, incluindo espécies de Bubalus e Bisonte)
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Vitelos de criação Vitelos que são criados para reprodução ou paraprodução de carne de bovino
Desde o nascimento Até 4 meses Até 60-80 kg (até145 kg)
56 dias
Vitelos de engorda Vitelos para produção de carne de vitela Desde o nascimento Até 6 meses Até 180 kg(até 250 kg)
Até ao abate, mas não menos que 84 dias
Bovinos de engorda Animais bovinos que tenham terminado o períodode desmame e destinados à produção de carne atéao dia do transporte para o matadouro
A partir do desenvol-vimento completo daruminação
Até 10-36 meses Até 350-700 kg 168 dias
L133/62
PTJornalO
ficialdaUnião
Europeia22.5.2008

Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Vacas leiteiras paraprodução de leite
Bovinos fêmeas que produziram pelo menos umvitelo
84 dias (é relatado o período total de lactação)
Vacas para reprodução Bovinos fêmeas fecundados/acasalados pelo menosuma vez
Desde a primeirainseminação até aofim do segundoperíodo de desmame
Dois ciclos (se forem solicitados os parâmetros dereprodução)
4. Quadro. Categoria animais: Ovinos
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Borregos de criação Borregos criados para reprodução futura Desde o nascimento Até 3 meses 15-20 kg 56 dias
Borregos de engorda Borregos que são criados para produção de carne deborrego
Desde o nascimento Até 6 meses (ou mais) Até 55 kg Até ao peso de abate, mas não menos que 56 dias
Ovinos leiteiros (paraprodução de leite)
Ovinos que tenham produzido pelo menos umborrego
84 dias (é relatado o período total de lactação)
Ovelhas para reprodu-ção
Ovinos fêmeas fecundados/acasalados pelo menosuma vez
Desde a primeirainseminação até aofim do segundoperíodo de desmame
Dois ciclos (se forem solicitados os parâmetros dereprodução)
5. Quadro. Categoria animais: Caprinos
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Cabritos de criação Caprinos jovens criados para reprodução futura Desde o nascimento Até 3 meses 15-20 kg Pelo menos 56 dias
22.5.2008PT
JornalOficialda
União
EuropeiaL133/63
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Cabritos de engorda Caprinos jovens que são criados para produção decarne de caprino
Desde o nascimento Até 6 meses Pelo menos 56 dias
Caprinos leiteiros (paraprodução de leite)
Caprinos que tenham produzido pelo menos umcabrito
84 dias (é relatado o período total de lactação)
Cabras para reprodução Caprinos fêmeas fecundados/acasalados pelo menosuma vez
Desde a primeirainseminação até aofim do segundoperíodo de desmame
Dois ciclos (se forem solicitados os parâmetros dereprodução)
6. Quadro. Categoria animais: Peixes
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Salmão e truta 200-300 g 90 dias ou até a massa corporal inicial ser duplicados.
Salmão e truta Reprodutores O mais próximopossível do períodode postura
90 dias
7. Quadro. Categoria animais: Coelhos
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Coelhos não desmama-dos e desmamados
Início uma semanaapós o nascimento
56 dias
Coelhos de engorda Coelhos que são criados para produção de carne Após o período dedesmame
Até 8-11 semanas 42 dias
L133/64
PTJornalO
ficialdaUnião
Europeia22.5.2008
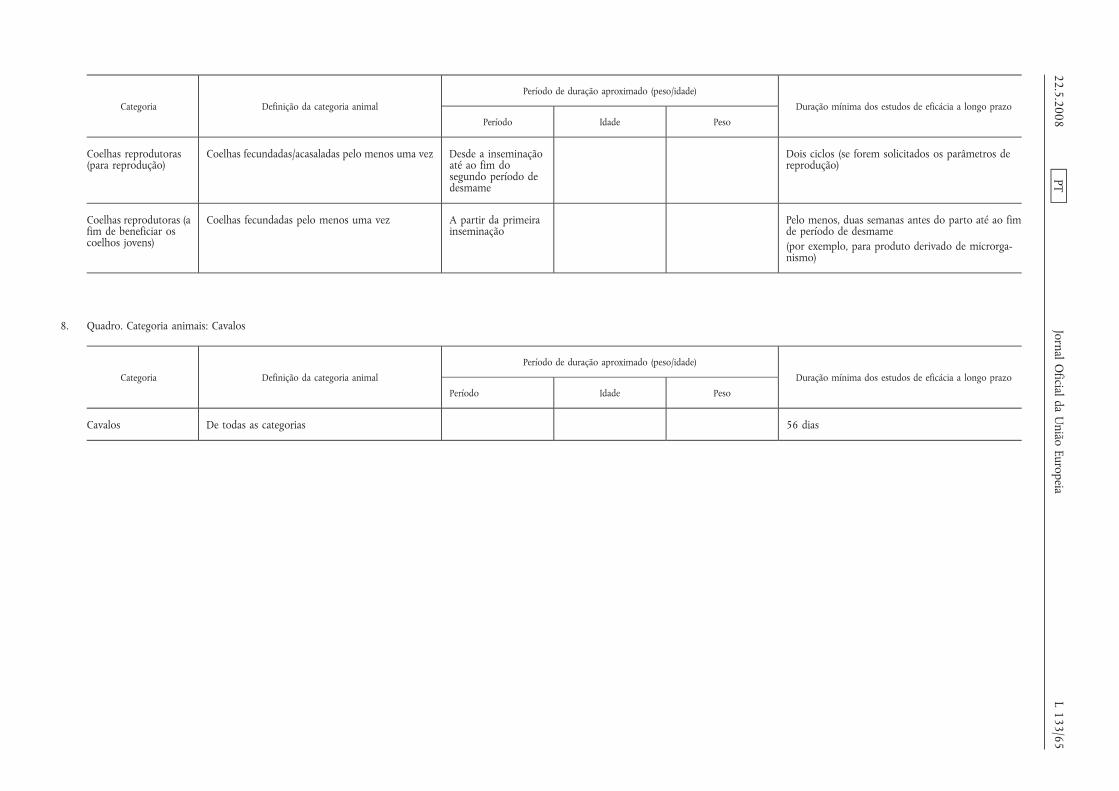
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Coelhas reprodutoras(para reprodução)
Coelhas fecundadas/acasaladas pelo menos uma vez Desde a inseminaçãoaté ao fim dosegundo período dedesmame
Dois ciclos (se forem solicitados os parâmetros dereprodução)
Coelhas reprodutoras (afim de beneficiar oscoelhos jovens)
Coelhas fecundadas pelo menos uma vez A partir da primeirainseminação
Pelo menos, duas semanas antes do parto até ao fimde período de desmame(por exemplo, para produto derivado de microrga-nismo)
8. Quadro. Categoria animais: Cavalos
Categoria Definição da categoria animal
Período de duração aproximado (peso/idade)
Duração mínima dos estudos de eficácia a longo prazo
Período Idade Peso
Cavalos De todas as categorias 56 dias
22.5.2008PT
JornalOficialda
União
EuropeiaL133/65





























