Embed Size (px)
Citation preview

T E C N O L O G I A
DE
D N A R E C O M B I N A N T E
Alfredo J. M. Cravador

T E C N O L O G I A
DE
D N A R E C O M B I N A N T E
Alfredo J. M. Cravador
FARO 2001

i
PREÂMBULO
O estudo da base molecular da vida só muito lentamente pôde progredir comparativamente ao das
outras ciências, devido, por um lado, à ausência ou à imperfeição das técnicas de análise e de medida,
mas fundamentalmente à complexidade dos sistemas biológicos. Não será todavia por isso que o
progressivo desvendar dos mecanismos que regem a organização máxima da matéria deixa de ser um dos
capítulos mais apaixonantes da História da Ciência moderna.
Foi há cerca de 50 anos que WATSON e CRICK dobraram um dos cabos mais decisivos da história
da ciência moderna, ao publicarem a estrutura do ácido desoxirribonucleico (DNA). Desde então,
milhares de cientistas e investigadores têm-se interessado pelos diferentes aspectos da estrutura e da
função do DNA, com o fim de compreenderem como a informação genética está contida na molécula de
DNA, e como ela é tratada de maneira tão ordenada e precisa durante os processos biológicos. A
conjugação de todos estes trabalhos de pesquisa levou à acumulação de um volume enorme de
informações e de conhecimentos em genética molecular e ao desenvolvimento de uma metodologia nova,
a engenharia genética.
A engenharia genética ou seja, a pesquisa sobre o DNA recombinante baseia-se num conjunto de
técnicas que conferem a possibilidade, até há pouco apanágio exclusivo da química relativamente a
pequenas moléculas, de romper e formar ligações covalentes de maneira específica e controlada em
moléculas de DNA. O material genético pode hoje ser modificado, mutado, transferido, detectado ou
sintetizado com as finalidades mais diversas tanto aplicadas como fundamentais.
A possibilidade de criar combinações novas, não naturais, a partir de material genético de diferentes
origens e de as introduzir em organismos hospedeiros que as perpetuam e multiplicam, deu uma
dimensão nova à biotecnologia, dotou-a do refinamento próprio à engenharia genética. À exploração
empírica dos fenómenos biológicos contrapõe-se, hoje, a construção racional de sistemas biológicos
artificiais elaborados por manipulação genética no laboratório de investigação, antes de serem explorados
industrialmente ou na produção agrícola. É neste contexto que deve ser considerada a engenharia
genética, não como uma nova disciplina em si, mas antes como um instrumento de investigação poderoso
que tem contribuído para um melhor conhecimento e aprofundamento do conceito de gene.
Esta disciplina não só permite compreender melhor os mecanismos fundamentais da vida, mas
também, pela manipulação dos organismos mais diversos, produzir substâncias úteis para a medicina, a
agricultura e a indústria em geral.
É de salientar que os extraordinários progressos realizados em engenharia genética só foram
possíveis graças aos resultados da investigação fundamental obtidos nos campos da genética, da biologia

ii
celular, da enzimologia, da virologia, da imunologia, da química dos ácidos nucleicos e das proteínas, da
cristalografia e que só a interacção das diferentes competências no seio desta área multidisciplinar
permite abordar nas melhores condições a tecnologia da clonagem de genes.
A racionalidade e os conhecimentos humanos tendo os seus limites, a nova era industrial e agrícola em
que entrámos não é destituída da ameaças e só poderá beneficiar as populações humanas a longo termo se
a exploração dos recursos biológicos respeitar os grandes equilíbrios naturais, os ciclos biológicos e
preservar a riqueza do património biológico terrestre já amplamente enfraquecidos. O mecanismo que o
homem é agora capaz de manipular é o estado superior da complexidade da matéria e é também o mais
frágil. Como corolário, o impacto da acção do homem na natureza passou a ser directo e profundo. As
consequências poderão ser catastróficas ou positivas e só dependem de nós. As ciências da vida ao
desembocarem no domínio das aplicações, a pequena ou a grande escala, tocaram a ecologia e a ética,
dimensões que não podem deixar de estar intimamente associadas à formação científica que têm por
missão ministrar.

iii
ÍNDICE 1.OS INSTRUMENTOS DE BASE 2 1.a.As enzimas, suas actividades e utilizações 2
Endonucleases de restrição e metilases 2 Outras endonucleases 8 Exonucleases e polimerases 9 Fosfatases 12 Ligação de fragmentos de DNA 13
1.b.A sequenciação de DNA 15 1. Método químico ou de Maxam & Gilbert 15 2. Método de terminação de cadeia ou de Sanger 17 3. Utilização da PCR em sequenciação 22 4. Sequenciação "automática" 24 5. Sequenciação multiplex 27 6. Outras tendências 28 7. Sequenciação de oligonucleotídeos 30 8. Chromosome walking 32 1.c.Síntese química de DNA 32
I. Princípios da síntese química de DNA 33 1) Preparação dos blocos de base para a síntese 34 2) Desprotecção selectiva 40 3) Reacções de condensação 42
4) Construção da cadeia oligodesoxinucleotídica 43 5) Desprotecção 46
Rendimentos 48 6) Isolamento, purificação e caracterização 49
2.VECTORES DE CLONAGEM 50 a. Os plasmídeos 50
Concepção de um vector plasmídico 52 b. Bacteriófagos 52
Empacotamento in vitro 54 c. Cosmídeos 55 d. O fago M13 56
Conclusões 57 3. MUTAGÉNESE DIRIGIDA 53 Eliminações 53 Inserções 54 Substituições 54 Mutagénese dirigida com oligonucleotídeos sintéticos 56 Selecção e identificação dos mutantes 59 Aplicações da mutagénese dirigida 60 4. A TRANSFORMAÇÃO BACTERIANA 63 5. BANCO DE CLONES DE cDNA 63 Esquema de construção de um banco de clones de cDNA 64 6. BANCOS GENÓMICOS 66 7. MÉTODOS DE DETECÇÃO E DE ANÁLISE DE CLONES 67
a) Hibridação in situ entre o DNA dos clones recombinantes e uma sonda de DNA radioactiva 67 b) Carta de restrição 67 c) Sequenciação dos nucleotídeos do inserto nos clones positivos por hibridação 68
d) Selecção do mensageiro específico por hibridação com o DNA recombinante e tradução in vitro 68

iv
e) Southern blotting 68
f) Escolha das sondas de DNA sintéticas 68
8. A SÍNTESE QUÍMICA DE GENES 72 9. SISTEMAS DE EXPRESSÃO: ORGANISMOS HOSPEDEIROS - VECTORES 74 a. Expressão em E. coli 74
Vectores de expressão em E. coli 74 1) Construção de vectores de expressão 76
2) Vectores baseados no promotor PL de λ 77
3) λgt11 - vector de expressão derivado do fago λ 79
Identificação das placas produtoras 79 Limitações da produção na bactéria 80 Produção da hormona de crescimento humana (hGH) 81 Hormonas de crescimento animais 85 Produção de insulina humana 86
b. Expressão em Bacillus subtilis 88 c. Expressão na levedura 90
Os vectores de expressão de S. cerevisiae 91 Exemplos de vectores de expressão para produção de proteínas exógenas em leveduras 92 1) A integração dirigida 95 2) O isolamento de alelos mutantes 96 3) Regulação da expressão dos genes na levedura 96 4) Um intrão codifica para uma proteína reguladora 97 5) Estrutura do gene da ferromona de levedura (MFα) 99
d. Expressão nos fungos filamentosos 100 e. Expressão em células de mamíferos 103
Transferência de genes nas células de mamíferos (transfecção) 104 Co-transformação 106 Micro-injecção 106 Isolamento dos genes transferidos 106 Clonagem de oncogenes humanos 107 Vectores virais utilizados para transfectar as células de mamíferos 108 Vector SV40 108 a) Substituição da região tardia de SV40 108
b) Substituição da região precoce de SV40 109
Vector vaivém derivado de SV40 109 Vector BPV (Bovine Papilloma Virus) 110 Vectores derivados de retrovírus 110
f. Introdução de genes estrangeiros em óvulos fertilizados. Animais transgénicos 112 Expressão do DNA estrangeiro nos animais transgénicos 113 A utilização dos retrovírus 115 Conclusões e resumo 115 g. Expressão em células de insectos 116 Construção dum vector de transferência do baculovírus 118 Actividade biológica das proteínas recombinantes 119 10. ENGENHARIA GENÉTICA DE PLANTAS 120 Agrobacterium tumefaciens 120 O plasmídeo Ti como vector 123 Expressão controlada pela luz e específica de órgão, dum gene de trigo em plantas de tabaco 126 Plantas transgénicas 127 a) Resistência a herbicidas 127 b) Resistência a insectos 128 c) Resistência a vírus 130 d) Produtoras de péptidos 133 As plantas monocotiledóneas 134

v
11. O DNA RECOMBINANTE E O DIAGNÓSTICO EM MEDICINA 136 a. Alterações cromossómicas 136 b. Cartografia dos cromossomas humanos 137 c. Mapeamento do genoma humano por RFLP 139 d. Anomalias monogénicas e multifactoriais 141 e. O diagnóstico de defeitos genéticos 142 1) RFLP (Restriction Fragment Length Polymorphism) 143 2) Diagnóstico de mutações identificadas 144 ß-talassemias 144
a) Mutações sem sentido e frame shift 145 b) Mutações que afectam a transcrição 145 c) Mutações que afectam a maturação do RNA 146 d) Utilização dos oligonucleotídeos sintéticos 146 e) Anemia falciforme 148
A deficiência hereditária de αααα1-antitripsina 148 f. Detecção de mutações somáticas 150 g. Amplificação enzimática de DNA (PCR) 152 1) Detecção da anemia falciforme e da hemoglobina C por PCR 154 2) Detecção de doenças infecciosas por PCR 155 Detecção de HPVs 156 Detecção de HIV 158 12.REGULAMENTAÇÃO SOBRE A UTILIZAÇÃO DE ORGANISMOS RECOMBINANTES E A PROTECÇÃO CONTRA
OS RISCOS BIOLÓGICOS 161 ANEXO Directivas do Conselho da CEE 163

1
METODOLOGIA DO DNA RECOMBINANTE
A tecnologia do DNA recombinante representa na realidade, um ramo da engenharia
genética considerada no seu sentido lato. Ela diz respeito à recombinação in vitro do material
genético por meio de enzimas. Estas manipulações in vitro só adquirem realmente um sentido
quando o DNA recombinante é introduzido num hospedeiro biológico, onde pode ser mantido e
propagado indefinidamente. Uma experiência típica de clonagem requer no mínimo
a) um DNA objecto duma pesquisa;
b) um vector de clonagem;
c) enzimas de restrição;
d) DNA ligase;
e) células procariotas ou eucariotas que servem de hospedeiro biológico
(Fig. 1.II.).
Depois da introdução no hospedeiro, as moléculas recombinantes têm que ser isoladas e
caracterizadas. Foi através deste processo de caracterização, que o conhecimento do gene foi
progredindo. O processo de clonagem em si, só fornece um meio de isolar e de identificar
rapidamente os genes de interesse.
Neste capítulo passaremos em revista os diversos métodos utilizados para clonar um gene.

2
1. OS INSTRUMENTOS DE BASE
1.a.As enzimas, suas actividades e utilizações
Endonucleases de restrição e metilases
Sempre que um bacteriófago λ se desenvolve numa estirpe de Escherichia coli C, os
fagos resultantes da lise deste hospedeiro multiplicam-se muito mal noutra estirpe, de tipo K.
Diz-se que o fago é "restrito" pela segunda estirpe bacteriana. Por outro lado, os fagos que
escaparam à restrição são capazes de crescer normalmente se forem utilizados para re-
infectarem a estirpe K. No entanto, se estes mesmos fagos passarem primeiro por E. coli C,
ficam de novo restritos por E. coli K (Fig. 2.II). Portanto, a eficácia com a qual um fago se
multiplica num hospedeiro depende da estirpe na qual ele se multiplicou anteriormente. Esta
modificação não hereditária, conferida ao fago pela segunda estirpe (K) e que lhe permite
voltar a multiplicar-se nesta estirpe sem ser de novo restrito, chama-se modificação.
Os fagos restritos adsorvem-se sobre os hospedeiros restritores e injectam o seu DNA
normalmente. No entanto, este DNA é rapidamente degradado depois da injecção. A enzima
responsável por esta degradação é uma endonuclease, também chamada endonuclease de
restrição ou enzima de restrição. O hospedeiro restritor tem evidentemente que proteger o seu
DNA dos efeitos das enzimas de restrição que ele próprio contém; esta protecção é
assegurada pela modificação do DNA. Mais precisamente, as modificações resultam da
metilação (por metilases) de certas bases num número reduzido de locais do DNA, que
constituem as sequências de reconhecimento para a enzima de restrição. É, pois,
compreensível a razão pela qual um fago que escapa à restrição num dado hospedeiro, pode
em seguida re-infectar eficazmente o mesmo hospedeiro: o DNA do fago ter-se-á replicado
em presença da metilase de modificação e será protegido, assim como o DNA do hospedeiro,
contra os efeitos de restrição.
Os processos de restrição e de modificação existem em todos os sistemas nos quais o
DNA é transferido duma bactéria para outra. A conjugação, a transformação, a transdução e a
transfecção estão todas submetidas ao sistema de restrição-modificação. Os genes que
especificam os sistemas res-mod podem residir no cromossoma do hospedeiro, ou podem
estar localizados num plasmídeo ou num profago.
As endonucleases de restrição classificam-se em 3 categorias. As pertencentes à classe I
são constituídas por 3 subunidades e requerem como cofactores ATP, Mg2+ e S-
adenosilmetionina. Possuem simultaneamente actividades de endonuclease e de metilase
situadas em subunidades diferentes. Os sítios específicos de reconhecimento, longos de 15

3
pares de base, são complexos, situando-se a cerca de 1000 pares de bases de distância do sítio
de clivagem ou de metilação. Esta categoria é pouco interessante em tecnologia genética
porque o local de clivagem não é específico.
As endonucleases de restrição de classe III, constituídas por 2 subunidades têm ATP e
Mg2+ como cofactores obrigatórios, mas não necessariamente a S-adenosilometionina. A
região de clivagem situa-se de 5 a 10 ou de 25 a 27 pares de bases a jusante do sítio de
reconhecimento.
As do primeiro tipo clivam em geral sequências não palindrómicas, dando origem a
populações heterogéneas com extremidades 5' protuberantes, enquanto que as do segundo
tipo clivam o DNA em sítios fixos.
As endonucleases de restrição de classe II são constituídas por uma única subunidade
activa e requerem unicamente Mg2+ como cofactor.
Os locais de clivagem situam-se em geral dentro das sequências de reconhecimento
compostas de 4 a 8 bases. A clivagem dá origem a extremidades 5' ou 3' protuberantes ou a
extremidades rombas ("blunt-end").
Estas enzimas foram isoladas a partir de mais de 200 estirpes bacterianas representando
quase todos os grupos de procariotas. Esta diversidade de enzimas torna indispensável a
utilização duma nomenclatura apropriada. Cada enzima é representada por um código de 3
letras derivado do nome do género e da espécie da bactéria donde a enzima foi isolada. Hae
por exemplo, significa que a enzima foi isolada de Haemophilus aegyptiens.
A maioria das sequências de reconhecimento conhecidas é palindrómica, i.é., possui um
eixo de simetria de rotação. Por exemplo a sequência de reconhecimento de EcoRI
5'-GAATTC-3'
3'-CTTAAG-5'
lê-se da mesma maneira nas duas direcções sobre as cadeias opostas.
O arranjo simétrico dos locais de clivagem tem duas consequências importantes:
1) os cortes em posições desviadas em relação a cada uma das cadeias produzem
extremidades monocatenárias que são complementares em orientação anti-paralela, mas
idênticas em orientação paralela;
2) as extremidades de uma molécula de DNA criadas por uma enzima dada, com uma dada
sequência de reconhecimento, podem formar pares de bases complementares com
qualquer outra molécula de DNA que possua extremidades idênticas:

4
5'...G pApApTpTp C...3 ' EcoRI 5'... GOH3' + 5'pApApTpTpC... 3'
3'...C pTpTpApAp G...5' 3'... CpTpTpApAp5' 3'HOG 5'
fragmento A (corte 1)
5'... GOH
3'... CpTpTpApAp
B
5'... GOHpA p A p T p T p C... 3'
3'... C p T p T p A p Ap HOG....5'
A
pApApTpTpC... 3'
HOG...5'
fragmento B (corte 2)
Esta associação entre bases complementares pode ser completamente estabilizada, pela
formação das ligações covalentes entre as extremidades 3'OH e 5'-fosfato de cada uma das
cadeias de DNA. Esta reacção de ligação é realizada com uma enzima específica, uma ligase,
que tem, como veremos mais adiante, uma utilização importante em metodologia de DNA
recombinante.
Deve ser, no entanto, referido que nem todas as sequências reconhecidas são simétricas.
Devido à existência de ambiguidades nos seus locais de reconhecimento, várias enzimas
reconhecem até 4 sequências alvo diferentes. A digestão duma molécula de DNA com este
tipo de enzima, pode originar extremidades não idênticas que não podem ser recombinadas
ao acaso.
Por exemplo, EcoRII reconhece as sequências seguintes:
CCAGG CCTGG
GGTCC GGACC
e

5
Várias enzimas denominadas isoesquizómeras possuem sítios de reconhecimento
idênticos. HindIII e HsuI por exemplo, partilham as mesmas sequências de reconhecimento e
de clivagem AAGCTT. Outras isoesquizómeras tais como SmaI (CCCGGG) e Xma I
(CCCGGG) possuem a mesma sequência de reconhecimento mas clivam em sítios diferentes
indicados por .Outras enzimas, denominadas isocaudómeras, podem diferir pelo seu sítio de
reconhecimento, mas originar extremidades complementares.
Por exemplo, as digestões por BamHI (GGATCC), BglII (AGATCT), Bc1I (TGATCA) e
Sau3A ( GATC) conduzem às extremidades coesivas 5'-GATC-3'. Todos os fragmentos de
DNA formados por clivagem com qualquer combinação destas enzimas, recombinam-se
entre eles. Assim, os fragmentos formados por digestão BamHI e BglII podem recombinar-se
entre eles, mas neste caso a sequência recombinante 5'-GGATCT-3' é resistente à digestão
por qualquer destas duas enzimas porque as bases externas da sequência hexanucleotídica de
reconhecimento de BamHI e de BglII diferem (G/C e T/A respectivamente).
BamHI Bg II 5'... G GATCT...3' 3'... CCTAG A...5'
5'...GGATCT...3' 3'...CCTAGA...5' não reconhecida por BamHI nem por BglII
mas sim por MboI/Sau3A/DpnI
No entanto, as moléculas originais e as recombinantes podem ser clivadas por outras
enzimas tais como DpnI, Sau3A e MboI e podem ser facilmente distinguidas. Estas enzimas
reconhecem a sequência tetranucleotídica 5'-GATC-3' central e não são influenciadas pelas
bases que a ladeiam.
As endonucleases de restrição têm certas propriedades particulares:
1) Actividade parasita conhecida por "star" que é uma alteração na especificidade da
sequência reconhecida, em condições de pH ou iónicas particulares. Por exemplo, um
aumento do pH, um abaixamento da concentração em NaCl, ou a substituição do Mg por Mn
alteram a especificidade de EcoRI que passa a reconhecer a sequência tetranucleotídica 5'-
AATT-3' que ocorre mais frequentemente. Esta nova especificidade é denominada EcoRI*
(Eco star).

6
2) Sequências na vizinhança de sequências de reconhecimento podem influenciar a
actividade duma enzima, privilegiando a clivagem de certos sítios em relação a outros de
mesma especificidade.
Por exemplo, a actividade de EcoRI pode variar no plasmídeo pBR322 ao ponto de atingir
diferenças de velocidade de 56x, em função da localização do sítio em relação a certas
sequências.
3) A temperatura é outro factor que influencia o desenrolar da clivagem. Certos pequenos
fragmentos oligonucleotídicos desnaturam-se quando a temperatura da reacção enzimática é
superior à sua temperatura de dissociação.
Certas enzimas têm a sua actividade óptima de funcionamento a temperaturas elevadas
(65-79°C), tais como TaqI, Tth111I, Tth111II isoladas de organismos termófilos.
4) Quando o sítio de reconhecimento de certas enzimas se situa perto de uma extremidade
da molécula de DNA a actividade diminui.
Quando dois sítios idênticos ficam demasiados próximos só um deles é clivado, porque
após a primeira clivagem o segundo sítio fica perto duma extremidade da molécula. Esta
propriedade está relacionada com o fenómeno de dissociação mencionado em 3).
5) A probabilidade de uma sequência de restrição estar representada num local com n pares
de bases, numa molécula de DNA, composta de 50 % de AT e de 50 % de CG é
nP4
1=
em que n é o número de bases que comporta o sítio.
Uma sequência tetranucleotídica dada aparecerá num DNA em média cada 44=256 pares
de base e uma hexanucleotídica cada 46=4096 pares de base. Se for conhecida a composição
em GC de um DNA e o seu comprimento, é possível prever o número de vezes que uma
sequência de restrição, de composição e comprimento dados, aparece representada.
Os diferentes fragmentos formados por digestão com uma enzima de restrição podem ser
separados por electroforese em gel de agarose ou de poliacrilamida (Fig. 3.II.). Os
fragmentos correm no gel em função do comprimento e podem ser visualizados por
coloração com o brometo de etídio, que se intercala entre as duas cadeias do DNA.
A utilização de várias enzimas isoladamente, ou em combinação, permite estabelecer a
carta de restrição duma molécula de DNA (Fig.4.II. e 5.II.). A carta indica as posições em
que uma enzima particular cliva o DNA. A distância entre os locais de clivagem expressa-se
em pares de bases (medida em relação a um padrão de referência).

7
As metilases, ou enzimas de modificação, protegem o DNA de um organismo da
actividade endonucleásica ao nível das sequências reconhecidas pela(s) enzima(s) de
restrição própria(s) ao organismo. Estas enzimas, que possuem actividades de metilase e
ausência de actividade de restrição, são a dam metilase, e a dcm metilase que fixam o grupo
CH3 (m de metilo) em posição N6 da adenina e em posição C5 da citosina respectivamente,
no interior dos sítios de restrição.
Dam-DNA adenina metilase GAmTC
Dcm-DNA citosina metilase CCm A/TGG
NHCH3
N
N
N
N
N6-metiladenina
N
N
CH3
NH2
O
C5-metilcitosina
Enquanto que certas enzimas de restrição reconhecem unicamente sequências alvo não
metiladas, os seus isoesquizómeros clivam tanto os sítios metilados como os não metilados.
HpaII (C/CGG), por exemplo, cliva o tetranucleotídeo não metilado, enquanto que o
isoesquizómero MspI cliva a mesma sequência não metilada ou metilada (CCmGG).
As células de eucariotas contêm habitualmente citosinas metiladas em proporção que
pode atingir 10 %. Se bem que de menor importância que no DNA de eucariotas, a metilação
da adenina é altamente relevante no DNA dos procariotas. A metilação dos resíduos adenina
pode ser testada por enzimas, tais como Sau3A ( GATC), MboI ( GATC) e DpnI (GATC).
Enquanto que Sau3A cliva tanto 5'-GATC-3' como 5'-GAmTC-3', MboI cliva unicamente a
sequência não metilada e DpnI cliva exclusivamente 5'-GAmTC-3'. Uma sequência hemi-
metilada, i.é. uma sequência metilada só numa cadeia é resistente à DpnI. Sempre que for
necessário clivar DNA de procariotas em qualquer local possível, este DNA tem que ser
preparado a partir de estirpes de E.coli que são dam-.
Se bem que a maior parte das estirpes de E.coli possua actividades de restrição e de
metilação, certas estirpes utilizadas em engenharia genética possuem uma, ou outra, ou
nenhuma destas actividades.
Ex.: EcoK12 possui actividades de metilase e de restrição

8
MM294 possui modificação mas não restrição
RRI não possui nem uma nem outra
Outras endonucleases [para além das que possuem outra(s) actividade(s)]
α) Desoxi- (Fig. 6.II.)
Desoxirribonuclease I (DNase I) (pâncreas bovino).
Actividade: endonuclease, cadeia dupla ou cadeia simples. Com Mg++ ataca cada cadeia
independentemente e não gera obrigatoriamente extremidades rombas.
Com Mn++ cliva as duas cadeias no mesmo sítio e gera fragmentos com extremidades
rombas ou só com um ou dois nucleotídeos protuberantes.
Utilização: - na introdução de nicks ao acaso em DNA bicatenário a fim de o preparar
para a marcação radioactiva por nick translation;
- na introdução dum nick em DNA circular a fim de o preparar para a mutagénese
mediada por bissulfito (secção 3);
- na criação de clones ao acaso para sequenciação em vectores de bacteriófago M13;
- na análise de complexos proteína-DNA.
ß) Ribonucleases
Ribonuclease A (pâncreas bovino)(Fig. 7.II.).
Actividade: endonuclease - ataca o RNA em 3' de bases pirimidínicas e cliva o fosfato
em 5' do nucleotídeo adjacente. Dá origem à formação de nucleotídeos pirimidínicos-3'P e de
oligonucleotídeos com pirimidinas-3'P.
Utilização: - na remoção de regiões não hibridadas de RNA nos híbridos DNA-RNA;
- no mapeamento de mutações singulares em DNA ou em RNA (desemparelhamentos de
um par de bases entre RNA e DNA são reconhecidos e clivados pela RNase A).
Ribonuclease T1 (Aspergillus oryzae)(Fig. 8.II.)
Actividade: endonuclease - ataca o RNA em 3' da guanosina e cliva o 5'-fosfato do
nucleotídeo adjacente. Dá origem a guanosinas ou a oligonucleotídeos com guanosina-3'P.
Utilização: - na remoção de regiões não hibridadas de RNA em híbridos DNA-RNA.

9
Exonucleases e polimerases
α) DNA polimerase I (E. coli) (Fig. 9.II.)
Actividades: exonuclease 5' → 3' e 3 '→ 5'
polimerase 5' → 3'
reacção de troca
Utilização: - marcação de DNA por nick-translation.
- Na marcação de DNA com extremidades 3' protuberantes.
ß) Fragmento de Klenow da DNA polimerase de E.coli (Fig. 10.II.)
Actividades: exonuclease 3' → 5'
polimerase 5' → 3'
reacção de troca
Utilização: - preenchimento das extremidades 3' retraídas formadas por digestão de DNA
com enzimas de restrição;
- marcação de DNA por preenchimento das extremidades 3' retraídas, com nucleotídeos
trifosfatos 32P;
- marcação terminal de DNA com extremidades 3' protuberantes;
- síntese da segunda cadeia de cDNA em clonagem de cDNA;
- síntese da segunda cadeia de DNA a partir de matrizes monocatenárias em mutagénese in
vitro;
- sequenciação de DNA pelo método dos didesoxinucleotídeos (terminação de cadeia).
γ) T4 DNA polimerase (Fig. 11.II.)
Actividades: exonuclease 3' → 5'
polimerase 5' → 3'
Utilização: - como o fragmento de Klenow, é utilizada na marcação de DNA por
preenchimento de extremidades 3' retraídas;
- na marcação das extremidades de fragmentos de DNA, rombas ou protuberantes em 3'
(reacção de troca);

10
- na marcação de fragmentos de DNA para uso como sondas de hibridação por digestão
parcial de DNA bicatenário utilizando actividade exonuclease 3'→5' seguida por reacção de
preenchimento, utilizando a actividade polimerásica e [α32P]dNTP;
- na conversão de extremidades protuberantes (5' ou 3') de DNA bicatenário, em
extremidades rombas;
- na extensão do iniciador (primer) oligonucletídico mutagénico hibridado à matriz de
DNA monocatenário, em mutagénese in vitro.
δ) Taq polimerase (Thermus aquaticus)(Fig. 12.II)
Actividade: polimerase 5' → 3'
Utilização: - na amplificação in vitro de sequências específicas de DNA por reacção em
cadeia (Polymerase Chain Reaction-PCR);
- na sequenciação de DNA
ε) RNA polimerases dependentes de DNA de bacteriófagos SP6, T3 e T7 (Fig. 13.II)
Actividade : RNA polimerase 5' → 3'
Utilização: - na síntese de RNA monocatenário para utilização como sondas de
hibridação, como mRNAs funcionais em sistemas de tradução in vitro, ou como substratos
em reacções de splicing in vitro. Cada uma destas três polimerases possui um grau elevado de
especificidade para com o seu promotor.
- No caso do sistema de transcrição do bacteriófago T7, para expressão de genes
clonados em bactérias.
ζ) Poli(A) polimerase (Fig. 14.II)
Actividade: polimerisa AMP (derivado de ATP) na extremidade 3'-OH livre do RNA.
Utilização: - preparação de poli (A)- RNA para clonagem;
- marcação da extremidade 3' de RNA com [α-32P]ATP para preparar sondas de
hibridação.
η) Reverse transcriptase (transcritase reversa) (Fig. 15.II.)
(vírus aviano de mieloblastose)
Actividades : polimerase 5' → 3' DNA.
A matriz tanto pode ser o DNA como o RNA.
Exorribonuclease 5' → 3' e 3' → 5' (RNase H)
Degrada especificamente o RNA em híbridos DNA-RNA.

11
Utilização: - síntese de cDNA para clonagem (1a e 2a cadeia) ou para sondas de
hibridação;
- marcação de extremidades de fragmentos de DNA protuberantes em 5' (reacção de
preenchimento).
- Também pode ser utilizada em sequenciação de DNA pelo método de terminação de
cadeia com os didesoxirribonucleotídeos.
θ) Nuclease Bal 31 (Fig. 16.II.)
Actividades: exo e endonuclease 5' → 3' e 3' → 5'
Utilização: - remoção de nucleotídeos das extremidades de DNA bicatenário de maneira controlada. O DNA encurtado pode ser ligado a adaptadores (linkers) ou a vectores com T4
ligase;
- construção de mapas de sítios de restrição;
- mapeamento de estruturas secundárias do DNA, por exemplo junções entre B-DNA e
Z-DNA, ou sítios de modificação em DNA bicatenário;
- remoção de nucleotídeos de RNA bicatenário para preparação de RNAs recombinantes.
ι) Nuclease S1 (Fig. 17.II.)
Actividade: nuclease específica de DNA monocatenário ou RNA;
utilização: - análise de estrutura de híbridos DNA-RNA;
- remoção de extremidades livres (tails) de fragmentos de DNA para produzir
extremidades rombas;
- abertura do gancho formado durante a síntese de cDNA bicatenário;
- na análise da estrutura dos híbridos RNA-DNA.
κ) Mung-bean nuclease
Actividade : nuclease específica de cadeia simples semelhante à nuclease S1 mas mais
suave.
Utilização: - converter extremidades protuberantes de DNA em extremidades rombas.
λ) Exonuclease VII (Fig. 18.II.)
Actividades: exonucleásica sobre cadeia simples de DNA, 3' → 5' e 5' → 3';
dispensa Mg++ e trabalha em presença de EDTA.
Utilização: - recuperação de cDNA inserido em vectores plasmídicos por "tailing" dA-
dT;

12
- construção de mapas dos intrões e dos exões.
µ) Exonuclease III (Fig. 19.II.)
Actividades: exonuclease 3' → 5' de DNA bicatenário com extremidades 3'OH retraídas.
-3' fosfatase.
Utilização: - Preparação de DNA linear como molde para a DNA polimerase (p.ex. na
sequenciação pelo método dos didesoxi; vd. secção sequenciação);
-preparação de sondas específicas de uma cadeia;
-remoção de sequências das extremidades de fragmentos de DNA, criando eliminações
unidireccionais a partir da extremidade 3'OH retraída;
-degradação preferencial da cadeia parental não tiofosfatada, no método de mutagénese
específica pontual que recorre a derivados tiofosfato (vd. secção 3)
ν) Exonuclease λ (Fig. 20.II)
Actividade: exonuclease 5' → 3' em cadeia dupla de DNA com um fosfato em 5'.
Utilização: -na modificação da extremidade 5'-fosfato dos DNAs substratos ulteriores
doutras enzimas (ex.: remoção da extremidade 5'-protuberante do DNA antes do "tailing"
com a terminal transferase).
Fosfatases
Fosfatase alcalina - CAP (intestino de vitela), BAP (bactéria) (Fig. 21.II.)
Actividades: catalisa a remoção dos fosfatos em 5' do DNA bicatenário ou
monocatenário, do RNA e de trifosfatos dos desoxirribonucleotídeos e dos ribonucleotídeos.
Utilização: -eliminação dos grupos fosfato em 5' do DNA ou do RNA para permitir a
marcação com 32P em 5' pela T4 polinucleotídeo kinase e [γ32P]ATP, numa reacção de
troca;
- remoção do fosfato em 5' do DNA a fim de impedir a auto ligação.
T4 polinucleotídeo Kinase (Fig. 22.II.)
Actividades: catalisa a transferência de fosfato γ de ATP para a extremidade 5'OH do
DNA mono ou bicatenário e do RNA por reacção de fosforilação de 5'OH ou de troca com
5'P.
Utilização: - marcação da extremidade 5' do DNA para a sequenciação pela técnica de
Maxam-Gilbert;

13
- fosforilação de oligonucleotídeos sintéticos, ou de outro DNA, para ligação;
- marcação de oligonucleotídeos para serem utilizados como sondas de hibridação;
Ligação de fragmentos de DNA
Existem dois métodos que permitem estabelecer ligações covalentes de DNA in vitro. O
primeiro utiliza uma enzima que intervém na replicação do DNA, a que já nos referimos
anteriormente, a DNA ligase (extraída de E.coli ou de células infectadas pelo fago T4). A
enzima derivada do fago T4 é até capaz de realizar a ligação entre moléculas de DNA com
extremidades rombas, reacção que é estimulada pela RNA ligase de T4.
T4 DNA ligase (Fig. 23.II.)
Actividade: ligação de extremidades coesivas e de extremidades rombas.
Utilização: -catalisa a ligação covalente de moléculas de DNA com extremidades
coesivas complementares;
- ligação covalente de extremidades rombas em moléculas de DNA bicatenário, umas
com as outras, ou com adaptadores ("linkers") sintéticos (observação: a T4 ligase é mais
eficaz nas ligações de extremidades rombas).
T4 RNA ligase (Fig. 24.II)
Actividade: catalisa a ligação covalente de extremidades 5' fosforiladas de DNA
monocatenário ou de RNA, a extremidades 3'OH de DNA monocatenário ou de RNA.
Utilização: -marcação radioactiva das extremidades 3' de RNA (pequenas moléculas tais
como os pNps são substratos eficazes);
- síntese de oligodesoxirribonucleotídeos.
O segundo método utiliza uma enzima que é capaz de adicionar extremidades coesivas
aos fragmentos de DNA. Quando por vezes é necessário ligar fragmentos de DNA
desprovidos de extremidades coesivas complementares, recorre-se a uma enzima que permite
adicionar, in vitro, sequências homopoliméricas complementares, aos fragmentos de DNA. O
método baseia-se na utilização duma enzima isolada de timo de vitelo, a terminal
desoxirribonucleotidiltransferase (terminal transferase). Em presença de um nucleotídeo
trifosfato, a enzima adiciona este nucleotídeo de maneira repetida às extremidades 3' do
DNA.

14
Pode-se, por exemplo, adicionar extremidades poli (A) em 3' dum fragmento de DNA e
extremidades poli (T) em 3' dum outro fragmento de DNA. Ao misturarem-se, estes dois
fragmentos hibridar-se-ão por emparelhamento entre bases complementares A/T e a junção
covalente dos fragmentos emparelhados pode ser efectuada pela enzima DNA ligase.
Terminal desoxirribonucleotidiltransferase (timo de vitelo) (Fig. 25.II.)
Actividade: catalisa a adição de desoxirribonucleotídeos às extremidades 3'-OH das
moléculas de DNA monocatenárias ou 3' protuberantes (Mg++), ou às extremidades rombas
ou 3'OH recessivas (Co++).
Utilização: -adição de caudas (tailing) homopoliméricas complementares a vectores e a
cDNA;
-marcação das extremidades 3'OH de DNA com [α-32P]-3'-desoxirribonucleosídeo ou
com 3'-ribonucleosídeo. A marcação com ribonucleosídeos realiza-se polimerizando com [α-32P]rNTP em presença de Co++ efectuando em seguida um tratamento alcalino.

15
1.b.A sequenciação de DNA
Dois grandes métodos baseados em princípios inteiramente diferentes foram
desenvolvidos para sequenciar longos fragmentos de DNA. Os pequenos fragmentos
oligonucleotídicos são um caso particular que será tratado separadamente.
Um destes métodos é baseado num processo de clivagem química específica e controlada
e o outro na síntese enzimática de fragmentos de DNA interrompidos pela adição controlada
de análogos dos desoxirribonucleotídeos trifosfatos (dNTP), as cordicepinas ou
didesoxirribonucleotídeos. O primeiro método é conhecido pelo nome dos seus autores,
Maxam & Gilbert, ou método químico, e o segundo por método dos didesoxi, ou de
terminação de cadeia, ou ainda de Sanger, nome do seu autor.
1. Método químico ou de Maxam & Gilbert
O princípio do método químico, de Maxam & Gilbert, baseia-se na clivagem duma
cadeia de DNA, induzida por uma reacção química de modificação, específica de cada uma
das 4 bases. Três etapas sucessivas estão implicadas na clivagem específica ao nível de uma,
ou duas das 4 bases (Fig. 26.II.):
1) Modificação da base
2) Remoção da base modificada do ciclo furanôsico
3) Clivagem da cadeia de DNA ao nível deste açúcar.
As etapas 2 e 3 têm a propriedade de serem dependentes da etapa precedente, i.é., o
DNA só será clivado ao nível dum açúcar desprovido da base e somente uma base
modificada de maneira adequada poderá ser removida do ciclo furanôsico que lhe
corresponde. A especificidade reside na primeira reacção. As reacções seguintes têm que ser
quantitativas. Por consequência, os reagentes de modificação que reagem com as bases são
absolutamente fundamentais, porque são eles que determinam o sítio de clivagem do DNA.
O sulfato de dimetilo e a hidrazina são dois reagentes que correspondem aos critérios
exigidos e conhecidos desde há muito tempo pelas suas propriedades de modificação e de
abertura dos ciclos das bases, tornando o DNA vulnerável à clivagem. As Fig. 27.II. e Fig.
28.II. detalham estas reacções. A primeira exemplifica as reacções da guanina: metilação do
azoto 7 com o sulfato de dimetilo, que introduz uma carga positiva na parte N7-C8-N9
imidazólica do ciclo púrico. Quando C8 é atacado por NaOH, a ligação C8-N9 rompe-se. Em
seguida a piperidina remove o ciclo aberto da 7-metilguanina e catalisa a eliminação ß dos
dois fosfatos do açúcar. Um mecanismo semelhante passa-se com a adenina.

16
A segunda figura exemplifica as reacções duma timina. A hidrazina ataca a timina nos
C4 e C6 abrindo o ciclo pirimidínico e ciclisando com C4-C5-C6 para formar um ciclo de 5
membros. A continuação da reacção com a hidrazina remove este novo ciclo pirazolona e o
fragmento ureico N1-C2-N3 da timina, enquanto que os açúcares do esqueleto do DNA
ganham a forma de hidrazona. Em seguida, a piperidina reage com todos estes glicosídeos e
como precedentemente catalisa a eliminação ß dos 2 fosfatos do açúcar. A citosina reage de
maneira semelhante.
Modulando estas reacções pela natureza e concentração da base, o pH, a temperatura, é
possível favorecer a clivagem no sítio de uma ou outra base púrica, ou de uma ou outra base
pirimidínica. Consideremos por exemplo, um fragmento de DNA marcado em 5' com 32P e
submetido às reacções que conduzem à clivagem, parcial mas específica, ao nível das
guaninas. As condições experimentais serão escolhidas de maneira a que cada molécula de
DNA não sofra mais duma clivagem. Neste caso, os fragmentos indicados na Fig. 29.II.,
marcados em 5' com 32P serão formados em proporções idênticas. Se o fragmento de DNA
for submetido separadamente a 4 séries de reacções, cada uma específica de uma base e as 4
séries de fragmentos forem analisadas em paralelo num gel de electroforese e este for em
seguida submetido a uma autorradiografia, a sua sequência pode ser lida seguindo o "padrão"
das bandas na película (Fig. 30.II.).
Uma característica essencial deste método é a necessidade de se dispor de um fragmento
de DNA especificamente marcado numa das suas extremidades, 5' ou 3'. Em geral estes
fragmentos são formados por clivagem com endonucleases de restrição e preenchimento das
extremidades protuberantes com uma polimerase e α32P-ATP. Antes de sequenciar o
fragmento de DNA, é necessário separar as duas extremidades marcadas. Uma maneira de
proceder consiste em digerir o fragmento com uma endonuclease de restrição, que dê origem
à formação de dois fragmentos bicatenários de comprimento diferente que possam ser
separados num gel de electroforese (Fig. 31.II.).
A quantidade de DNA que deve ser utilizada depende do comprimento do fragmento e
do número de reacções que se pretende efectuar, assim que da habilidade do experimentador.
É necessário pelo menos 1 picomole para a marcação. Por exemplo, no caso do pBR322
linearizado, de peso molecular de 2.6106 Da, 1 picomole corresponde a 2,6 µg de DNA. Se a
actividade específica de 32P-dNTP utilizada na marcação for de 1000 Ci/mmol, 1 mci (+ 106
cpm) poderá ser teoricamente incorporada por nucleotídeo adicionado. Depois das etapas de
precipitação sobejarão 105 cpm para colocar no PAGE (PAGE-polyacrilamide gel
eletrophoresis) de separação. A resolução do gel, a sua capacidade para separar fragmentos
de comprimento N dos de comprimento N + 1 depende da natureza do gel, do seu
comprimento e composição. Um gel convencional é composto de 8 % de acrilamida, 8M de

17
ureia, tem 0,2-0,3 mm de espessura e 40 cm de comprimento. Obtém-se uma resolução
satisfatória de 200 nucleotídeos (Fig. 30.II.) Se não se puderem separar 200 nucleotídeos
numa corrida única aplicam-se alíquotas das 4 reacções de clivagem (ou mais) a tempos
diferentes. Na figura, as mesmas amostras aplicadas nos corredores da esquerda (I) foram
aplicadas no gel 2 horas mais tarde, à direita. As moléculas mais pequenas separam-se nos
corredores contendo as últimas alíquotas a serem aplicadas e que correram durante menos
tempo. Começa-se a leitura nos corredores de direita. Na figura, as setas indicam os pontos
que delimitam a zona de sobreposição. Existe equipamento que permite realizar géis mais
compridos e ler um maior número de bases duma vez. A utilização de dNTP marcados com 35S melhora, por sua vez, notavelmente a resolução.
A Fig. 5.3 (Sequenciação de DNA, método de Maxam and Gilbert) ilustra e resume o
princípio do método.
Uma variante importante, que poderia ser potencialmente explorada na sequenciação
automática, é o método em fase sólida. Neste método, o DNA marcado com o 32P é
imobilizado por adsorção num papel de troca iónica (p. ex., DEAE). Todas as reacções e
lavagens são efectuadas em suporte sólido. As vantagens, em relação ao método em solução,
são a rapidez e a simplicidade. As liofilizações e as precipitações são em grande parte
substituídas por simples lavagens rápidas.
2. Método de terminação de cadeia ou de Sanger
Neste método, começa-se por hibridar um iniciador (primer) oligonucleotídico,
complementar do DNA numa região próxima da que se pretende sequenciar. O iniciador é,
em seguida, prolongado (em presença de dNTPs, um dos quais está marcado em posição α
com 32P, ou com 35S) com uma polimerase que utiliza o DNA alvo da sequenciação como
molde.
A cada uma das quatro misturas de reacção realizadas em paralelo, adiciona-se um
didesoxirribonucleotídeo trifosfato correspondente a cada uma das quatro bases, em
concentrações e proporções bem definidas em relação aos dNTP. Obtém-se assim 4
populações de fragmentos, tendo todos a mesma extremidade 5' e terminando com um
didesoxirribonucleotídeo idêntico, no seio de cada população: ddA, ddT, ddC ou ddG (Fig.
32.II.). Os didesoxirribonucleotídeos não possuem os grupos hidroxílicos em 3' e em 2'.

18
Como o grupo 3'OH é necessário para a formação de ligações fosfodiéster, a presença de
ddNTPs provoca a terminação da cadeia. As reacções de síntese são paradas adicionando
formamida que impede as cadeias de DNA de se hibridarem.
Antigamente recorria-se a uma digestão por uma endonuclease que cortasse num sítio
dando origem a fragmentos mais curtos, que serviam de iniciadores. De cerca de 250 em 250
nucleotídeos era necessário recorrer a um novo iniciador. Esta condição obrigatória, e a
necessidade de dispor de DNA monocatenário, eram considerados nos tempos da infância do
método, como inconvenientes sérios. Hoje, se bem que o princípio na base do método não
tenha mudado, a estratégia de clonagem do DNA que se pretende sequenciar, no fago M13,
de DNA circular monocatenário ou em derivados e a possibilidade de recorrer facilmente,
graças aos progressos realizados na síntese química de DNA, a iniciadores oligonucleotídicos
sintéticos, levou à adopção generalizada deste método.
A Fig. 5.4 (Sequenciação de DNA, método d Sanger) ilustra e resume o princípio do
método.
Com a finalidade de o tornar de utilização mais prática, o M13 foi modificado em várias
etapas a partir da sua forma replicativa bicatenária (RF) (Fig. 33.II.). O primeiro derivado é o
mp1, que possui na região intergénica não essencial, representada a preto, uma inserção
composta da região do gene lacI que codifica a porção C-terminal do repressor, a região
reguladora completa promotor-operador e os codões correspondentes aos primeiros 146
aminoácidos da região N-terminal do gene da ß-galactosidase, lac Z.
O derivado mp2 resulta duma simples substituição dum dG em dA, com o fim de criar
um sítio EcoRI (Fig. 34.II.). Esta substituição, que converte o 5o codão do ácido aspártico em
codão de asparagina não tem consequências na actividade do péptido que é assegurada pelos
primeiros 146 aminoácidos da ß-galactosidase. Para tornar o vector mais prático, inseriu-se
no sítio EcoRI um poli adaptador comportando vários sítios de restrição, nas duas orientações
possíveis. Estas construções correspondem aos vectores M13mp8 e M13mp9. A introdução
de aminoácidos suplementares, resultante desta inserção, não interfere com a actividade
complementar que o péptido α assegura a uma ß-galactosidase enzimaticamente inactiva,
O B
H H
O
Estrutura geral dum ddNTP
P O P O P
O O O
OOO
O
19
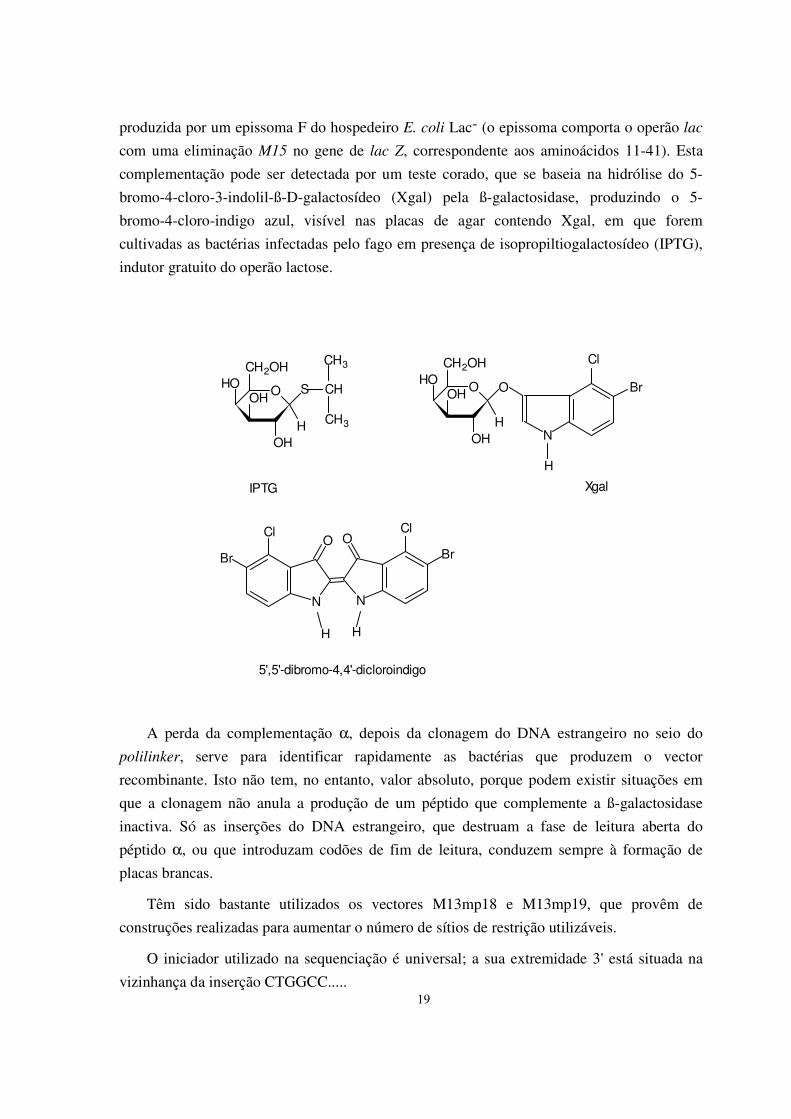
produzida por um epissoma F do hospedeiro E. coli Lac- (o epissoma comporta o operão lac
com uma eliminação M15 no gene de lac Z, correspondente aos aminoácidos 11-41). Esta
complementação pode ser detectada por um teste corado, que se baseia na hidrólise do 5-
bromo-4-cloro-3-indolil-ß-D-galactosídeo (Xgal) pela ß-galactosidase, produzindo o 5-
bromo-4-cloro-indigo azul, visível nas placas de agar contendo Xgal, em que forem
cultivadas as bactérias infectadas pelo fago em presença de isopropiltiogalactosídeo (IPTG),
indutor gratuito do operão lactose.
A perda da complementação α, depois da clonagem do DNA estrangeiro no seio do
polilinker, serve para identificar rapidamente as bactérias que produzem o vector
recombinante. Isto não tem, no entanto, valor absoluto, porque podem existir situações em
que a clonagem não anula a produção de um péptido que complemente a ß-galactosidase
inactiva. Só as inserções do DNA estrangeiro, que destruam a fase de leitura aberta do
péptido α, ou que introduzam codões de fim de leitura, conduzem sempre à formação de
placas brancas.
Têm sido bastante utilizados os vectores M13mp18 e M13mp19, que provêm de
construções realizadas para aumentar o número de sítios de restrição utilizáveis.
O iniciador utilizado na sequenciação é universal; a sua extremidade 3' está situada na
vizinhança da inserção CTGGCC.....
CH3
CH3
IPTG
O
CH2OHHO
OH
OHH
S CH O
CH2OHHO
OH
OHH
O
N
Cl
Br
H
Xgal
N N
Br Br
ClClO O
H H
5',5'-dibromo-4,4'-dicloroindigo

20
As duas cadeias dum inserto, com extremidades coesivas diferentes, podem ser
sequenciadas, uma no mp18 e outra no mp19, pois estas construções contêm o polilinker em
sentidos opostos. Se o fragmento inserido tem extremidades idênticas, obtêm-se as duas
orientações no mesmo vector.
Insertos maiores que 300 a 400 nucleotídeos dificilmente podem ser sequenciados com
um só iniciador. No entanto, a informação obtida sobre a sequência lida o mais longe
possível no inserto, permitirá deduzir a sequência de um segundo iniciador, que servirá para
sequenciar uma nova série de 300 nucleotídeos. Esta estratégia permite a sequenciação de
insertos de mais de 2000 nucleotídeos, graças à síntese química rápida dos iniciadores
oligonucleotídicos. Insertos muito maiores conduzem a um crescimento muito reduzido das
células infectadas.
O DNA de grande comprimento, como por exemplo DNA de vírus de 8000 pares de
bases pode ser sequenciado por este método, utilizando a técnica do shot-gun. Fragmentos de
restrição do DNA são clonados ao acaso nos vectores M13, sem controlo da sua orientação
no genoma. Os fragmentos clonados são sequenciados a partir do iniciador universal e a
informação reunida é em seguida processada num programa computorizado, a fim de
identificar as sequências que se sobrepõem. Para preencher as lacunas é preciso recorrer a
estratégias de sub-clonagem não aleatórias (Fig. 35A.II.). O fragmento de DNA a sub-clonar
para ser sequenciado subsequentemente, é clonado num vector M13mp. O DNA com o
inserto em ambas as orientações é tratado com DNAseI em condições que provocam
linearização em posições ao acaso. A fim de obter uma série de fragmentos contendo o DNA
de M13 intacto (necessário para haver transformação e formar placas de lise), mas
comportando insertos de comprimento variável, estendendo-se do sítio Z até ao sítio Y do
outro lado do inserto, a mistura de fragmentos linearizados é digerida com a enzima X. Após
tratamento com polietilenoglicol (PGE), que precipita unicamente grandes fragmentos de
DNA, estes fragmentos são de novo suspendidos e ligados, para formarem moléculas
circulares de DNA. Entre estas moléculas encontram-se as que foram cortadas com DNAseI
dentro do inserto e que perderam o sítio Y (1, 2 e 3). As que não foram cortadas inicialmente
com DNAseI (esquema de reacção à esquerda na figura 35A.II.), ou que foram cortadas
dentro do segmento de DNA de M13 (molécula 4 na via reaccional da direita), ainda contêm
o sítio Y. Após digestão da mistura de reacção total com a enzima Y, estas últimas moléculas
serão linearizadas, enquanto que as primeiras, com a série de insertos começando no sítio Z,
permanecerão circulares. A eficácia da transformação, ou transfecção, com o DNA linear é
baixa, comparada com a do DNA circular. Assim, unicamente as moléculas de interesse
formarão placas após transformação. Como todos os insertos possuem uma terminação
comum (Z), a sequenciação dos diferentes clones, a partir dum iniciador da síntese
(polimerização) no sítio X, deve eventualmente cobrir todo o inserto.

21
Outra abordagem para sequenciar um longo fragmento de DNA consiste em criar uma
família de clones M13, contendo cada um, um sub-fragmento do gene. Isto pode ser realizado
preparando eliminações ordenadas, como ilustrado na Fig. 35B.II. A cadeia de DNA
bicatenária, comportando o inserto completo é aberta numa extremidade deste, com uma
enzima de restrição. O inserto é em seguida digerido com uma exonuclease. Porções da
mistura de reacção de digestão são removidas a diferentes tempos, antes da ligação ser
completa, de maneira a parar a reacção. Formam-se populações de moléculas com
comprimentos diminuindo progressivamente. As moléculas com eliminações são de novo
circularizadas com DNA ligase, colocando o sítio de ligação perto do iniciador universal
(USP, Universal Sequencing Primer), que fica adjacente da nova extremidade criada pela
digestão pela exonuclease. Otém-se uma colecção de clones, nos quais a região de hibridação
do iniciador universal está bem situada para sequenciar diferentes porções do inserto. Ao
sequenciar esta colecção de clones com um único iniciador pode-se obter um elevado número
de sequências que se sobrepõem e assim determinar a sequência total do gene.
Existe uma variante em fase sólida do método de sequenciação de Sanger, que se baseia
na imobilização de um plasmídeo contendo o inserto que se quer sequenciar (Fig. 36.II.). Este
é linearizado com uma enzima de restrição A, e as extremidades protuberantes são
preenchidas por uma polimerase com os 4 nucleotídeos trifosfatos, dos quais um é um
derivado contendo biotina, por exemplo a 11-bio-dUTP comercial. Depois de uma segunda
digestão com uma outra enzima de restrição B, a mistura é passada numa coluna de avidina-
agarose. A avidina, ao complexar fortemente a biotina, assegura a imobilização do DNA
biotinilado. As cadeias não biotiniladas são em seguida eliminadas por desnaturação e
lavagem. O DNA biotinilado monocatenário imobilizado servirá de molde para a hibridação
com um iniciador de sequenciação e para as reacções de polimerização. Os oligómeros são
eliminados, depois de desnaturados, deixando as cadeias matrizes disponíveis para a reacção
de sequenciação seguinte.
O método de sequenciação de terminação de cadeia pode ser aplicado à sequenciação de
DNA bicatenário, clonado por exemplo, no pBR322 ou em derivados da família pUC (vd.
secção 9.). Na prática, as moléculas recombinantes na forma circular fechada são
completamente desnaturadas a pH fortemente básico (NaOH). A re-naturação em estrutura
inicial é difícil a este pH (>12); forma-se uma rede de moléculas híbridas que expõem uma
proporção suficiente de DNA monocatenário. A utilização da sequenase (variante genética da
DNA polimerase do bacteriófago T7), em vez do fragmento de Klenow da DNA polimerase
I, foi, sem dúvida, um factor de sucesso na sequenciação de DNA pelo método dos didesoxi,
incluindo o bicatenário. A actividade exonucleásica 3'→5' da forma nativa foi removida por
manipulação genética in vitro. A enzima possui elevada processividade e actividade
específica, e uma boa estabilidade.
22
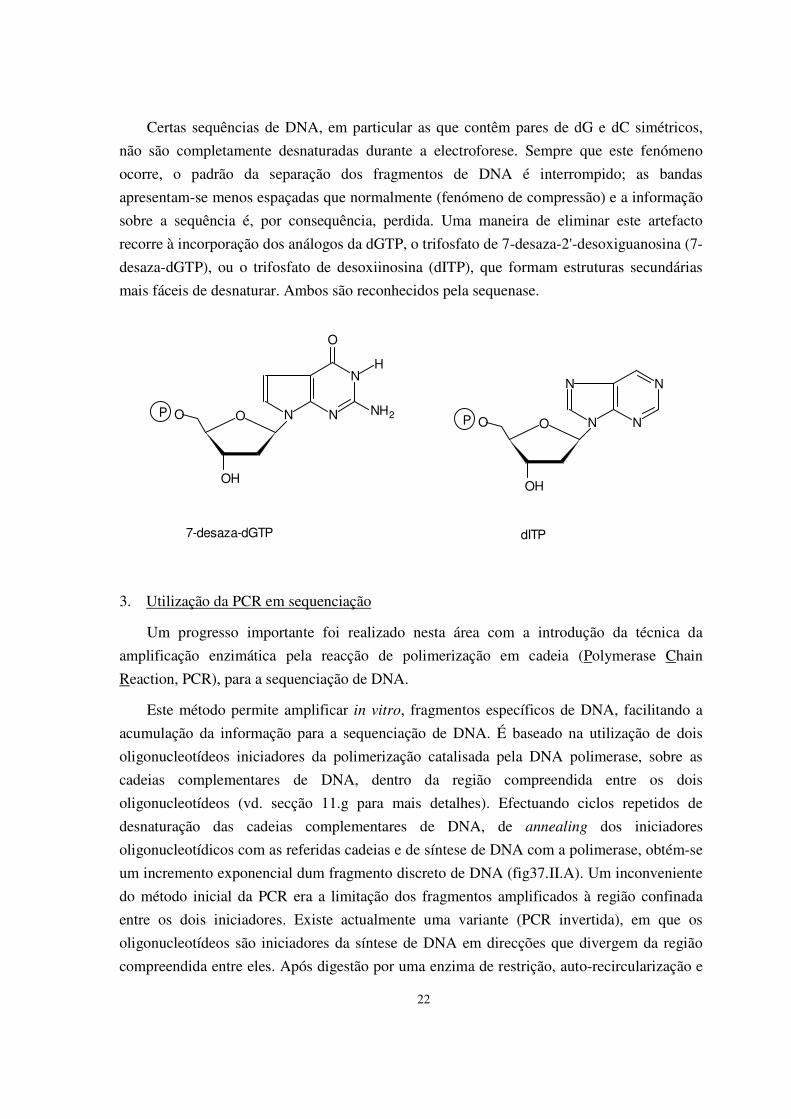
Certas sequências de DNA, em particular as que contêm pares de dG e dC simétricos,
não são completamente desnaturadas durante a electroforese. Sempre que este fenómeno
ocorre, o padrão da separação dos fragmentos de DNA é interrompido; as bandas
apresentam-se menos espaçadas que normalmente (fenómeno de compressão) e a informação
sobre a sequência é, por consequência, perdida. Uma maneira de eliminar este artefacto
recorre à incorporação dos análogos da dGTP, o trifosfato de 7-desaza-2'-desoxiguanosina (7-
desaza-dGTP), ou o trifosfato de desoxiinosina (dITP), que formam estruturas secundárias
mais fáceis de desnaturar. Ambos são reconhecidos pela sequenase.
3. Utilização da PCR em sequenciação
Um progresso importante foi realizado nesta área com a introdução da técnica da
amplificação enzimática pela reacção de polimerização em cadeia (Polymerase Chain
Reaction, PCR), para a sequenciação de DNA.
Este método permite amplificar in vitro, fragmentos específicos de DNA, facilitando a
acumulação da informação para a sequenciação de DNA. É baseado na utilização de dois
oligonucleotídeos iniciadores da polimerização catalisada pela DNA polimerase, sobre as
cadeias complementares de DNA, dentro da região compreendida entre os dois
oligonucleotídeos (vd. secção 11.g para mais detalhes). Efectuando ciclos repetidos de
desnaturação das cadeias complementares de DNA, de annealing dos iniciadores
oligonucleotídicos com as referidas cadeias e de síntese de DNA com a polimerase, obtém-se
um incremento exponencial dum fragmento discreto de DNA (fig37.II.A). Um inconveniente
do método inicial da PCR era a limitação dos fragmentos amplificados à região confinada
entre os dois iniciadores. Existe actualmente uma variante (PCR invertida), em que os
oligonucleotídeos são iniciadores da síntese de DNA em direcções que divergem da região
compreendida entre eles. Após digestão por uma enzima de restrição, auto-recircularização e
O N
N
N
O
H
NH2
OH
OPO N
NN
N
OH
OP
7-desaza-dGTP dITP

23
ligação com uma ligase do fragmento de DNA alvo, a PCR com iniciadores invertidos dará
origem a um fragmento linear contendo regiões de sequências desconhecidas divergindo dos
iniciadores.
A utilização mais evidente da PCR em sequenciação, é na preparação das matrizes de
DNA; existe uma variedade grande de métodos para diferentes aplicações. No entanto, a Taq
polimerase utilizada na reacção de amplificação é uma enzima ideal para sequenciação, que
pode assim ser efectuada directamente subsequentemente à amplificação, sem que sejam
necessárias modificações significativas em relação aos componentes reaccionais.
Habitualmente, a quantidade de matriz de DNA necessária para sequenciação obtém-se
inserindo o DNA alvo em vectores bacterianos ou virais e multiplicando os insertos em
células hospedeiras. Todavia, as matrizes para sequenciação podem-se obter mais
eficazmente que com os métodos dependentes de células, a partir de alvos genómicos ou de
insertos de DNA clonados em vectores bacterianos ou virais, recorrendo à amplificação por
PCR dos insertos clonados. Mesmo a partir de sequências completamente desconhecidas é
possível recorrer à amplificação, utilizando oligonucleotídeos iniciadores, no interior, ou
próximos, do poli-linker do vector de clonagem. Desta maneira não é necessário fazer
culturas repetidas e purificações das matrizes de sequenciação a partir de bactérias ou de
vírus.
Um problema inerente à sequenciação directa pelo método da PCR resulta da re-
associação rápida das duas cadeias de DNA linear amplificadas, após desnaturação, que
bloqueia a extensão do complexo iniciador oligonucleotídeo-matriz de DNA, ou impede o
annealing desta com o oligonucleotídeo. Vários métodos podem ser utilizados para contornar
este inconveniente e obter preferencialmente uma mono cadeia matriz para sequenciação.
1) O DNA bicatenário gerado por PCR é utilizado como matriz de sequenciação
directamente, ou após separação por electroforese e purificação a partir do gel, a fim de
remover o DNA não amplificado, os iniciadores e os dNTPs. Em seguida, um dos
oligonucleotídeos iniciadores é utilizado para iniciar a reacção de sequenciação. O DNA
desnaturado pode ser arrefecido rapidamente em neve carbónica, a fim de retardar a re-
associação das cadeias.
2) Quando os produtos da PCR são de comprimento superior a 500 pb, o DNA
bicatenário pode ser desnaturado e as cadeias monocatenárias separadas por electroforese.
Este método é apropriado para DNA de grande comprimento, cujas cadeias monocatenárias
são difíceis de obter por outros métodos.
3) O DNA monocatenário pode ser produzido directamente por PCR utilizando uma
proporção molar assimétrica dos dois iniciadores oligonucleotídicos, ou ainda recorrendo a
uma segunda reacção de PCR utilizando um excesso dum dos iniciadores e um fragmento

24
alvo, proveniente duma PCR simétrica, separado por electroforese e purificado a partir de gel
electroforético (Fig. 37.II.B).
4) Uma alternativa para produzir DNA monocatenário por PCR sem diminuir a eficácia
da amplificação, (como pode ser o caso do método assimétrico), recorre ao bloqueamento
dum dos iniciadores da PCR. Um excesso de um terceiro oligonucleotídeo complementar de
um dos iniciadores da PCR é adicionado durante a reacção de amplificação. O terceiro
oligonucleotídeo retira da competição as moléculas duma das cadeias acabada de ser
sintetizada, impedindo-as de se complexarem com o iniciador bloqueado. A PCR é assim
transformada a dada altura numa reacção de extensão de mono cadeia (Fig. 37.II.C).
5) Uma abordagem completamente diferente recorre à combinação da PCR com a
transcrição reversa, utilizando uma sequência promotora de fago, específica da enzima,
ligada a um dos iniciadores da PCR. Inicialmente a PCR é efectuada de maneira equilibrada,
simétrica, a fim de produzir DNA bicatenário. A reacção de transcrição é efectuada num
segundo tempo e dará origem a um incremento suplementar do número de cópias da cadeia
monocatenária desejada (RNA), devido ao reconhecimento específico da sequência
promotora pela transcritase do fago. Este transcrito é em seguida sequenciado recorrendo à
transcritase reversa (Fig. 37.II.D).
A Taq polimerase é uma enzima adequada para a sequenciação de DNA porque possui
uma elevada processividade e incorpora em proporção excelente; além disso incorpora o
análogo 7-desazaguanosina, utilizado para resolver a compressão das sequências de DNA
ricas em C-G. Para além destas propriedades, que compartilha com a T7 DNA polimerase,
funciona a 55-75°C, condição que pode ser útil para resolver estruturas secundárias
eventualmente presentes no DNA e que podem dificultar a aquisição da informação de
sequenciação.
4. Sequenciação "automática"
O princípio utilizado nos sistemas ditos "automáticos", actualmente disponíveis no
mercado é o do método de terminação de cadeia de Sanger. As suas características principais
são o sistema de detecção não radioactivo e o processamento da informação, que é
automatizado.
Foram desenvolvidos dois sistemas que se baseiam na derivatização química de
desoxirribonucleotídeos, modificados por grupos fluorescentes que são incorporados nos
fragmentos de DNA sintetizados pela polimerase. Cada um deles recorre a uma série
particular de 4 grupos fluorescentes.

25
Num dos sistemas, os grupos fluorescentes são a fluoresceína, o nitrobenzoxadiazolo, a
tetrametilrodamina e o Texas Red (Fig. 38.II.). Os espectros destes compostos apresentam
máximos de absorvência a comprimentos de onda diferentes. Cada um destes grupos
fluorescentes está ligado quimicamente ao nucleotídeo modificado em 5' do iniciador
universal. Obtêm-se 4 iniciadores que diferem unicamente pela natureza da entidade química
fluorescente fixa na sua extremidade 5'. Se se utilizar cada um destes iniciadores em cada
uma das reacções de polimerização em que intervém um dos quatro
didesoxirribonucleotídeos, associa-se um grupo fluorescente a um terminador de cadeia. A
mistura dos produtos das 4 reacções é separada por electroforese, num corredor único duma
coluna de gel de poliacrilamida, ou num gel com vários corredores. As bandas fluorescentes
são excitadas por um laser e detectadas perto da base do gel durante a electroforese. A
informação é transferida directamente para um computador, que identifica cada uma das
bandas coradas, em tempo real, à medida que elas passam, em função do comprimento de
onda do máximo de absorvência. A sequência temporal dos diferentes grupos fluorescentes
representa a sequência do DNA (Fig. 39.II.).
Os dados sobre os comprimentos de onda são continuamente recolhidos e armazenados
num computador, durante a corrida electroforética e, em seguida, analisados para fornecerem
a sequência final.
No segundo sistema os grupos fluorescentes estão ligados à parte básica dos trifosfatos
dos derivados didesoxirribonucleotídeos (Fig. 40.II.). Os grupos trifosfatos asseguram a
incorporação, sem que o grupo fluorescente impeça aparentemente o reconhecimento da base
complementar e o reconhecimento pela polimerase; a ausência de grupo hidroxílico em 3'
termina a polimerização. Cada um dos quatro didesoxirribonucleotídeos é modificado com
um grupo fluorescente absorvendo a um comprimento de onda diferente, permitindo, como
no primeiro sistema, uma detecção diferencial.
O laser é necessário como fonte de excitação, a fim de se obter uma sensibilidade
máxima a partir de cada banda de DNA.
A figura 41.II mostra a informação bruta, a informação processada e um detalhe
ampliado, entre os nucleotídeos 89 e 110 de um gráfico de análise. Este processamento da
informação, que torna a diagrama mais claro e de mais fácil interpretação, é de momento
indispensável, por 3 razões fundamentais:
1) Os espectros de emissão dos diferentes corantes sobrepõem-se parcialmente. Os picos
correspondendo à presença de um só corante são, por isso, detectados por mais de um canal.
É, portanto, necessário determinar as diferentes proporções dos 4 grupos fluorescentes
presentes no gel a cada instante.

26
2) Uma segunda complicação provém do facto de os grupos fluorescentes influenciarem a
mobilidade dos fragmentos de DNA. Por exemplo, a fluoresceína e a rodamina diminuem a
mobilidade dos fragmentos de DNA de uma base e o Texas Red de 1 base e 1/4. É, portanto,
necessário fazer a correcção.
3) Uma terceira complicação é inerente ao método enzimático, que dá por vezes origem a
variações de intensidade das bandas ou a regiões de leitura difícil, devido à existência de
estruturas secundárias no seio do DNA.
Foram desenvolvidos três outros sistemas que utilizam cada um, um só marcador
fluorescente e quatro corredores electroforéticos. Esta modificação evita os problemas
associados à mobilidade diferencial dos fragmentos, causada pelos 4 diferentes marcadores
químicos e a sobreposição dos espectros de emissão dos múltiplos grupos fluorescentes.
Qualquer deles lê a sequência em bases de DNA de maneira contínua e em tempo real, e
possui uma sensibilidade entre 1 e 50 10-18 moles por banda, próxima da obtida com a
autorradiografia convencional.
Os marcadores químicos fluorescentes encontram-se no iniciador do M13, utilizado na
sequenciação pelo método dos didesoxirribonucleotídeos. Num dos sistemas o marcador é
introduzido na extremidade 5' dum derivado sulfídrico do iniciador, por reacção com a
iodoacetamida fluoresceína de estrutura representada na figura 42.A.II. O feixe de excitação
laser atravessa o interior do gel em linha recta, no sentido da largura (lateralmente),
controlando continuamente os quatro corredores (disposição que elimina a necessidade de
movimentos de "scanner" e aumenta a sensibilidade). Um gel de 20 a 30 cm de comprimento
é suficiente para efectuar a leitura de 300-400 bases em 5 horas.
Nos dois outros sistemas, os iniciadores são marcados com o isotiocianato de
fluoresceína (FITC), introduzido em 5'-OH, ou num fosfato internucleotídico (Fig. 42.B.II),
ou em posição C5 da base desoxiuridina, segundo o caso, por reacção com derivados
oligonucleotídicos alquilaminados nas posições referidas. Num dos sistemas, dois grupos
fluorescentes foram introduzidos no mesmo iniciador utilizando os derivados aminados em
C5 de duas desoxiuridinas diferentes, com a finalidade de aumentar a sensibilidade da
detecção e sem que haja interferências com a actividade biológica (Fig. 42.C.II.).
As Fig. 43.II. e 44.II. ilustram esquematicamente a disposição da aparelhagem de
irradiação de excitação laser, de filtração, de irradiação emitida e de análise computorizada
dos dados. A irradiação de excitação é lateral num dos sistemas (Fig. 43.II.) e orientada num
ângulo, que minimiza os reflexos do vidro do gel e optimiza a energia transmitida às bandas,
no sistema que utiliza a bi-marcação fluorescente (Fig. 44.II.). Este último processo introduz
uma focalização do feixe laser que aumenta a resolução das bandas separadas por espaços
muito curtos e um sistema de scanning horizontal do gel que permite a criação duma imagem

27
bidimensional (Fig. 45.II.). Este sistema permite sequenciar com óptima resolução 500-600
bases duma única aplicação, em gel de 30 cm correndo 4 amostras (16 poços)
simultaneamente, com uma percentagem média de erro de 0,83 %.
5. Sequenciação multiplex
A necessidade crescente da determinação de sequências de DNA e nomeadamente o
projecto de sequenciação da totalidade do genoma humano (3 biliões de bases), levaram ao
desenvolvimento de técnicas mais elaboradas e rápidas. Uma actividade intensa foi sido
desenvolvida na pesquisa dedicada a este tema.
Os princípios básicos da sequenciação, do método enzimático de terminação de cadeia
de Sanger e do método de clivagem química de Maxam & Gilbert, continuaram a ser
aplicados, apesar das variações e aperfeiçoamentos introduzidos. Um exemplo dos
desenvolvimentos realizados nas técnicas de sequenciação de DNA baseia-se em métodos
que recorrem à hibridação de DNA-DNA. O sistema "multiplex" de sequenciação tira partido
da possibilidade de aplicar, num corredor electroforético, uma multiplicidade de amostras
correspondentes a uma clivagem específica, ao nível de uma base (método de Maxam &
Gilbert) efectuada simultaneamente com múltiplos fragmentos de DNA, cuja sequência
individual se deseja determinar. O padrão individual das bandas correspondentes a cada
fragmento é decifrado por hibridação com sondas específicas marcadas radioactivamente.
A Fig. 46.II. esquematiza as diferentes etapas deste método. Uma mistura de fragmentos
de DNA (de 900 a 1500 pb, provenientes por exemplo, do fraccionamento por vibração
ultrassónica, de DNA genómico) é clonada separadamente em 20 vectores, que foram
construídos especialmente para o sistema multiplex. Estes 20 vectores plasmídicos diferem
unicamente pela sequência de dois fragmentos, que flanqueiam o sítio de restrição utilizado
para a clonagem do DNA que se pretende sequenciar. Os 40 fragmentos correspondentes
foram sintetizados quimicamente, com sequências suficientemente diferentes para permitirem
a identificação fácil de cada um dos 20 vectores, por hibridação com uma sonda
complementar (Fig. 47.II.). As 20 misturas de clonagem são levadas a transformar
separadamente uma estirpe de E. coli e os 20 bancos de DNA resultantes são amplificados,
utilizando a resistência à tetraciclina como marcador de selecção. Os plasmídeos que contêm
um inserto único são seleccionados por electroforese em gel de agarose, purificados e levados
a transformar separadamente a mesma estirpe bacteriana. Os transformantes são plaqueados
em 20 placas (1 cultura de transformante por placa) formando cerca de 100 colónias por
placa. São em seguida constituídos grupos de 20 colónias, uma de cada placa, de maneira a
que cada um dos vectores plasmídicos, especificamente rotulado com as sequências
oligonucleotídicas sintéticas, se encontre representado uma vez em cada grupo. Misturando

28
várias colónias desta maneira, obtêm-se várias séries de grupos, compostos cada um de 20
colónias, comportando, portanto, 20 clones de fragmentos de DNA, um por plasmídeo
diferente. Cada um destes grupos de colónias misturadas é cultivado separadamente e os
plasmídeos de cada cultura são purificados e cortados pela enzima de restrição NotI. O DNA
proveniente de cada grupo é submetido separadamente a quatro reacções químicas de
clivagem de base e os fragmentos resultantes são fraccionados electroforeticamente como já
foi descrito antes.
Esta técnica tem a particularidade de recorrer à aplicação de amostras provenientes de 12
séries de reacções, utilizando portanto 48 corredores num só gel (Fig. 48.II.). Os fragmentos
fraccionados são transferidos electricamente do gel para uma membrana de nylon, onde são
fixos por reacção induzida pela luz U.V.. Como cada série de reacção faz intervir um grupo
de 20 fragmentos de DNA, provenientes dos 20 bancos iniciais, é possível ler numa só
membrana, portanto a partir dum único gel, 240 sequências de DNA. Com efeito, cada
oligonucleotídeo marcado radioactivamente com 32P e complementar da sequência que
rotula cada um dos vectores plasmáticos, revela, após hibridação e autorradiografia, o padrão
de clivagem do DNA clonado em cada um destes plasmídeos. Isto é possível porque a
membrana em nylon retém fortemente os fragmentos de DNA, permitindo pelo menos 50
hibridações, intercaladas por 50 operações de desnaturação, sem perca de sensibilidade ou de
resolução, mantendo a especificidade das hibridações.
6. Outras tendências
Para acelerar a velocidade de sequenciação do DNA de várias ordens de grandeza, pode-
se teoricamente recorrer à combinação de várias técnicas existentes: à sequenciação
multiespectral, à fotoquímica dos grupos fluorescentes, à sequenciação multiplex, à
transferência (blotting) directa e automática do resultado da eletroforese para membranas e às
técnicas de figuração electrónica.
A proposta de sequenciação pelo método multiespectral-multiplex ambiciona
desenvolver um sistema capaz de processar um milhão de bases por gel de sequenciação.
A primeira etapa no sentido da automatização do sistema multiplex visa a transferência
directa do gel de sequenciação para uma membrana. Já utilizado para o blotting de proteínas,
este dispositivo compreende uma matriz para imobilização (nitrocelulose ou nylon)
incorporada numa correia transportadora, que se move ao longo do fundo do gel. Os
fragmentos de DNA fraccionados são sequencialmente transferidos para a matriz,
preservando a informação espacial. Uma das vantagens deste sistema é o aumento da
resolução espacial entre os fragmentos de maior comprimento, porque a separação em função

29
do tempo, mediada pela correia móvel, é linear, ao contrário do fraccionamento em função da
distância, cuja relação é logarítmica, o que provoca a compressão das bandas. Este sistema
permite resolver e manter uma resolução linear até aproximadamente 600 nucleotídeos num
gel de 5 % de poliacrilamida.
A proposta multiespectral-multiplex recorre a 50 séries diferentes de sondas, destinadas a
serem hibridadas com um número igual de iniciadores de sequenciação do M13, de
sequências complementares à das sondas. Estas séries de sondas e de iniciadores serão
utilizadas para a determinação de 2000 famílias de sequências (50 ciclos multiplex x 40
corredores electroforéticos) por gel. Não são necessários vectores especiais porque a sonda
fluorescente é dirigida para o iniciador de sequenciação e não para o vector (Fig. 49.II.).
Cada família de bandas de sequenciação será lida a quatro comprimentos de onda específicas,
por um sistema de detecção em que a resolução é função do tempo. Só são necessárias 160
reacções separadas de sequenciação pelo método dos didesoxinucleotídeos para as 2000
famílias de sequências, porque todas as reacções A, G, C e T podem ser agrupadas num
corredor dado (4 bases x 40 corredores = 160 reacções).
As seis etapas esquematizadas na Fig. 50.II. incluem:
1) Preparação para sequenciação de 2000 fragmentos de DNA monocatenário (no M13
mp18, por exemplo). Este DNA é preparado a partir do fago, sem necessidade de vectores ou
métodos especiais.
2) Divisão dos 2000 fragmentos de DNA em 40 grupos de 50 fragmentos cada. Os 50
fragmentos de cada grupo (totalizando 40 x 4 = 160 reacções de terminação de cadeia com os
didesoxinucleotídeos) serão aplicados num só corredor de electroforese. São portanto
necessários 40 corredores (por gel) para a sequenciação multiespectral-multiplex de 2000
cadeias de DNA. Como a sequenciação é tetracromática, cada família de sequências será lida
a partir de um corredor, por ciclo.
3) Quatro iniciadores diferentes de sequenciação serão utilizados para cada fragmento de
DNA (um para cada uma das reacções de terminação com os didesoxinucleotídeos A, C, G e
T). É necessária uma série única de 4 iniciadores oligonucleotídicos para cada grupo de
fragmentos de DNA (4 x 50). São, portanto, necessários 200 oligonucleotídeos únicos. A
mesma série de iniciadores pode ser utilizada para cada um dos grupos. Estes 200
oligonucleotídeos serão as sequências alvo das hibridações da sequenciação multiplex.
4) As reacções dos didesoxinucleotídeos A, C, G e T serão agrupadas para cada grupo, o
que reduz o número total de reacções, de 8000 (2000 x 4) da sequenciação convencional,
para 160 (4 tipos de reacção por cada corredor).

30
5) Durante a electroforese recorrer-se-á em simultâneo à transferência directa para uma
matriz de imobilização.
6) Cada família de sequências será lida hibridando uma série de 4 sondas fluorescentes
diferentes (multiespectrais) por ciclo. No total, há 50 séries de 4 sondas multiespectrais. O
factor 50 corresponde ao número de fragmentos multiplex aplicados em cada corredor e o
factor 4 corresponde às 4 bases do DNA.
Após figuração electrónica de um blot (membrana depois de ter recebido a transferência)
a quatro comprimentos de onda, uma família de sequências será lida em cada corredor. A
membrana será lavada, de novo hibridada e a sua figuração representada electronicamente
num total de 50 vezes, até fornecer a sequência das 2000 famílias.
Como o DNA alvo das sondas fluorescentes é monocatenário, será possível utilizar
condições de hibridação suaves, depois da transferência do gel desnaturante.
Espera-se poder processar rapidamente 1 milhão de bases por gel, após os
desenvolvimentos necessários para a automatização do sistema multiespectral-multiplex.
7. Sequenciação de oligonucleotídeos
O método de Maxam & Gilbert pode-se aplicar à sequenciação de fragmentos de DNA
de pequeno comprimento (4 a 40 nucleotídeos). São no entanto necessárias modificações
experimentais em relação às utilizadas com grandes fragmentos. A percentagem de
modificação e, portanto, de clivagem dos grandes fragmentos de DNA, necessária para a
análise da sequência, é relativamente baixa (1-2 %). Se se utilizarem estas mesmas condições
com os oligonucleotídeos, a maior parte das moléculas de DNA permanecem não
modificadas. Para obter resultados satisfatórios são necessárias de uma maneira geral
condições reaccionais mais vigorosas, prolongando o tempo das reacções ou aumentando a
temperatura, ou ambos. Outras modificações de detalhe incluem o método de separação do
excesso de [γ32P]-ATP do oligonucleotídeo marcado, por cromatografia de Sephadex, ou
papel de DEAE celulose. A variante em fase sólida, na qual o oligonucleotídeo é imobilizado
num suporte apropriado para trocas iónicas, também é eficaz.
Um outro método muito útil e que permite simultaneamente controlar a pureza dos
oligonucleotídeos é o método do Wandering spot. Baseia-se nas diferenças de mobilidade dos
fragmentos de degradação sequencial e parcial do oligonucleotídeo, num cromatograma
bidimensional que se obtém por electroforese de alta voltagem (1a dimensão) e
homocromatografia (2a dimensão). O oligonucleotídeo que se pretende sequenciar é marcado
na sua extremidade 5', com a T4 polinucleotídeo kinase e [γ32]-ATP e em seguida é
hidrolisado com a fosfodiesterase de veneno de serpente, enzima que digere o DNA de 3' em

31
5'. Se a digestão é controlada temporalmente, podem-se obter fragmentos de todos os
comprimentos marcados em 5' em quantidades equivalentes (Fig. 51.II.). A mistura é em
seguida submetida a uma electroforese de alta voltagem, numa tira de acetato de celulose, a
pH 3,5. A este pH, a diferença de protonação das 4 bases heterocíclicas é máxima (Fig.
52.II.). A diferença de mobilidade electroforética de 2 oligonucleotídeos com n e n-1
nucleotídeos depende da natureza do nucleotídeo clivado na extremidade 3' do fragmento.
Para identificar os produtos de degradação sequencial é necessário efectuar, depois da
electroforese, uma homocromatografia que os separa em função do comprimento. O material
é transferido da banda de acetato de celulose para a linha de base duma placa de camada fina
de DEAE celulose. A separação efectua-se em função da carga do fosfato em meio básico,
portanto em função do comprimento, sendo os mais compridos os mais retidos.
O efeito combinado da electroforese e da homocromatografia na mobilidade, aparece na
impressão bidimensional, ilustrada na Fig. 53.II., exemplificada com um octâmero.
Utilizando as regras representadas nas figuras pode-se deduzir a sequência a partir do
fingerprint. pdG corre conjuntamente com o xilenocianol e é identificado sem ambiguidade.
Variações mais ou menos importantes em relação às regras surgem por vezes, pelo que é
aconselhável prudência na interpretação dos resultados. Para além de 20 nucleotídeos é muito
difícil interpretar os resultados, devido à sobreposição das bandas. Pode-se, no entanto, obter
informação sobre sequências de fragmentos maiores, recorrendo à marcação do
oligonucleotídeo em 3' pela desoxirribonucleotidiltransferase com [α32P]-dNTPs, digerindo
o oligonucleotídeo de 5' em 3' pela diesterase de baço de vitela e analisando os fragmentos
pelo método acima descrito. Sobrepondo os resultados das duas análises, de 5' em 3' e de 3
em 5' obter-se-á informação complementar.
Um outro método muito rápido e preciso e que se aplica aos oligonucleotídeos de
pequeno comprimento, de 1 a menos 15 nucleotídeos, é a espectrometria de massa por
bombardeamento atómico rápido, conhecido por FAB (Fast Atomic Bombardment).
Resumidamente, uma amostra de oligonucleotídeo de cerca de 10 nmol é submetida a um
bombardeamento de átomos neutros de xénon, que provoca uma fragmentação do
oligonucleotídeo em partículas de carga negativa. Estes fragmentos, em geral de carga -1, são
acelerados e separados num campo magnético em função da massa, para uma carga idêntica.
Do registo do padrão de fragmentação resulta um espectro de massa como o exemplificado
na Fig. 54.II. As fragmentações ocorrem preferencialmente nos locais indicados. No entanto,
como a ligação entre o átomo de oxigénio em 3' e o átomo de carbono secundário do ciclo
furanôsico é mais lábil que a ligação entre o átomo de oxigénio em 5' ligado ao carbono
primário, o registo dos iões correspondentes aos 5'-fosfatos aparece mais intenso que o dos
3'-fosfatos.

32
Na prática, classificam-se os iões em 5'-fosfato ou em 3'-fosfato, segundo a intensidade a
partir do sinal correspondente a (M-H)-. As diferenças exactas das massas, entre os picos
vizinhos da mesma família de fragmentação, são determinadas por contagem e permitem a
determinação exacta da sequência oligonucleotídica correspondente. A sequência completa
das bases do oligonucleotídeo pode então ser lida 2 vezes, partindo da extremidade 5' ou da
extremidade 3'.
As limitações deste método são o preço do equipamento e o comprimento limitado do
oligonucleotídeo que é possível sequenciar.
8. Chromosome walking
O chromosome walking não é propriamente um método de sequenciação. É antes um
método para fazer uma carta, o mapeamento duma região muita vasta de DNA, em geral mais
longa que os insertos habituais obtidos quando se prepara um banco genómico (20-45 kb).
Um passo desta marcha (walking) consiste no escrutínio do banco com uma sonda conhecida
e na identificação de clones que apresentam uma sobreposição com a sonda. Os insertos que
se sobrepõem são mapeados e os que se localizam nas extremidades são utilizados como
sondas para escrutinar o banco e identificar outros clones, cujo DNA se estende mais para a
esquerda e a direita (Fig. 55.II.). Estas etapas são repetidas até se percorrer todo o gene, ou a
região do cromossoma que se pretende estudar: a sonda 1 identifica os clones λ1 e λ2. A
sonda 2, derivada de λ2, é utilizada para identificar o clone λ3. Uma sonda derivada da
extremidade 3' de λ3 servirá para identificar λ4 e assim de seguida. A mesma estratégia pode
ser utilizada para marchar na direcção 5'.
Em vez de marchar pode ser necessário saltar (jumping), o que acontece sempre que há
rearranjos. A parte B da figura ilustra este género de ocorrência. A parte central do DNA está
invertida em relação à situação em A. A sonda 4 identificará neste caso, um novo clone 5,
que cobre a região A e uma região suplementar do cromossoma, que fica na vizinhança de A
depois do rearranjo. A sonda 5, tirada da extremidade 3' do inserto λ5, pode ser utilizada para
continuar a avançar no estabelecimento do mapa.
1.c.Síntese química de DNA
Há cerca de 25 anos, a síntese química de pequenos fragmentos de DNA
(oligonucleotídeos) era um domínio esotérico, apanágio de alguns químicos especializados.
Calculava-se, em 1975, que seriam necessários 20 anos para sintetizar um gene de 100
nucleotídeos segundo um esquema optimizado por computador. Actualmente, é possível
realizar este trabalho em algumas horas. Esta proeza técnica, só é possível graças ao

33
desenvolvimento acelerado das técnicas de síntese química, catalisado pelo advento das
técnicas da Engenharia Genética. O alargamento contínuo do campo de aplicação dos
fragmentos sintéticos de DNA, assim como o aspecto fundamental do estudo estrutural de
certas sequências particulares, constituíram estímulos poderosos para os químicos orgânicos,
cujos esforços conduziram ao ajustamento e ao aperfeiçoamento das técnicas de síntese.
O domínio das aplicações dos oligonucleotídeos sintéticos, ou de análogos, pode ser
assim resumido:
Síntese total ou parcial de genes
Construções (adaptadores, mutagénese...)
Iniciadores para polimerases e transcritases
DNA (ex., diagnóstico..)
Sondas de hibridação para escrutinar RNA (ex., Northern...)
bancos genómicos
bancos de cDNA
Mutagénese dirigida localisada
Determinação de estruturas de DNA e de RNA
Mecanismos de interacção DNA-proteína
Inibição de tradução
transcrição
Apresentamos neste sub capítulo um resumo histórico geral da química que esteve, ou
está na base da síntese de DNA.
I. Princípios da síntese química de DNA
A síntese química em solução de um oligodesoxinucleotídeo comporta as seguintes
etapas:
1) Preparação dos quatro desoxinucleotídeos: timidina, desoxicitidina, desoxiadenosina e
desoxiguanosina, completamente protegidos. Estes são os blocos fundamentais para a síntese.

34
2) Desprotecção selectiva de dois desoxinucleotídeos a condensar, de maneira a libertar em
cada um dos blocos, unicamente as funções implicadas na formação da ligação
internucleotídica.
3) Condensação dos compostos assim gerados para formar um dímero inteiramente
protegido.
4) Extensão da cadeia de DNA repetindo as reacções das etapas 2 e 3 até à obtenção de um
oligodesoxirribonucleotídeo completamente protegido.
5) Desprotecção sequencial e controlada do oligodesoxirribonucleotídeo.
6) Isolamento, purificação e caracterização.
Quando a extensão da cadeia desoxirribonucleotídica é realizada em fase sólida, são
necessárias algumas etapas suplementares:
i) funcionalização de um polímero insolúvel;
ii) fixação do primeiro desoxirribonucleotídeo protegido da cadeia
oligodesoxirribonucleotídica a sintetizar, sobre a função do suporte sólido;
iii) clivagem da cadeia de DNA do suporte sólido.
1) Preparação dos blocos de base para a síntese
As substâncias de base na síntese de DNA são os quatro desoxirribonucleosídeos:
timidina, desoxicitidina, desoxiadenosina e desoxiguanosina; (obtidos por degradação de
ADN de origem natural) (Fig. 56.II.).
Na molécula desoxinucleosídica podem-se distinguir três centros que vão ser objecto de
três transformações distintas: as bases e as funções hidroxílicas, em posição 5' e em posição
3'.
a) Protecção das funções aminas exocíclicas
As bases que possuem funções aminas primárias (a timidina não possui) têm que ser
protegidas. Esta protecção é permanente, visto que o centro aminado não está implicado nas
reacções de extensão oligomérica. Portanto, os grupos protectores devem ser estáveis ao
longo de todas as etapas de síntese. No entanto, eles devem poder ser retirados no fim da
síntese em condições que preservem a integridade da molécula final. Os grupos que, nas
estratégias de síntese mais utilizadas, melhor satisfizeram estas condições, são o grupo
benzoilo para a desoxicitidina e a desoxiadenosina e o grupo isobutanoilo para a
desoxiguanosina. A sua introdução tem de ser selectiva, a fim de não bloquear os grupos
hidroxílicos do ciclo desoxirribofuranosilo. Estes grupos são objecto de uma protecção
35
transitória, que pode ser especificamente removida após acilação dos grupos aminados (ex.,
Fig. 57.II.).
Uma protecção suplementar original foi introduzida por várias equipas, destinada a
proteger a função lactâmica da desoxiguanosina, fonte de reacções secundárias durante as
reacções de condensação e de fosforilação.
Timidina
1'O N
N
O
O
H
HO
OH
2'3'4'
5'
1'O N
N
OHO
OH
2'3'4'
5'
Desoxicitidina
NH2
NH2
Desoxiadenosina
1'OHO
OH
2'3'4'
5'N
N
N
O
Desoxiguanosina
1'OHO
OH
2'3'4'
5'N
N
N
N
NH2
H
Fig. 56.II
36
NH2
Desoxicitidina
1'O N
N
OHO
OH
2'3'4'
5' ClSi(CH3)3
NH2
1'O N
N
O O
O
2'3'4'
5'Si
Si
O
Cl
O
SiO
Si 5'
4'3' 2'
O N
N
OO1'
NH
NH4OH dil.
O
O
5'
4'
3' 2'
N
N
OO1'
NH
HO
H
Fig. 57.II
b) Transformação dos grupos hidroxílicos
Das funções hidroxílicas em posição 5' e 3', uma tem de ser protegida por um grupo
protector transitório, a outra fosforilada. A escolha do centro a fosforilar depende da
estratégia adoptada. A estratégia que consiste em fosforilar a posição 3' deu os melhores
resultados e tem sido utilizada nos métodos gerais de fosfatotriéster e de fosfitotriéster.
α) A protecção do grupo hidroxílico em posição 5' tem de ser temporária. Ela tem de ser
removida antes de cada etapa de condensação, durante a extensão da cadeia
desoxirribonucleotídica. O grupo protector deve neste caso possuir as propriedades seguintes:
i) poder ser introduzido selectivamente em posição 5' deixando intacta a função
hidroxílica em posição 3';
ii) poder ser retirado em condições suaves que não dêem lugar a nenhuma reacção
secundária;

37
iii) ser estável em todas as etapas de síntese, de desprotecção e de purificação que se seguem
à sua introdução;
iv) eventualmente, facilitar, pela sua introdução, a purificação dos intermediários de
síntese.
Entre outros grupos introduzidos com bons resultados o 4,4'-dimetoxitritilo é um dos
mais correntemente adoptados. É introduzido através do seu cloreto, tirando-se beneficio da
sua selectividade pela posição 5', devido a factores estéricos. (ex., Fig. 58.II.).
Fig. 58.II
O grupo 4,4-dimetoxitritilo pode ser quantitativa e rapidamente retirado em condições de
baixa acidez (ex., ácido dicloroacético, brometo de zinco) sem ruptura, ou com ruptura
insignificante da ligação glicosídica; esta reacção conduz à formação do catião dimetoxitritilo
de cor laranja, cuja absorvência pode ser medida espectrofotometricamente. Esta medida é
particularmente útil na síntese em fase sólida, pois ela é a única indicação da eficácia das
reacções de condensação durante a construção da cadeia oligonucleotídica.
ß) A fosforilação em posição 3' dos nucleosídeos protegidos na bases e no grupo hidroxílico
em 5' pode conduzir, segundo a estratégia escolhida, a um bloco completamente protegido,
ou a uma espécie fosforilada activa, capaz de reagir directamente com outro bloco
desoxinucleotídico desprotegido em posição 5' para formar a ligação internucleotídica.
Numerosos agentes de fosforilação têm sido utilizados em síntese de
oligodesoxirribonucleotídeos, sobretudo na metodologia do fosfatotriéster (Fig. 59.II.).
OCH3
OCH 3
OCH 3
OCH 3
O
Cl
H
+
O
H N
1 ' O O
OH
2 ' 3 ' 4 '
5 ' N
N
N C C
N
Desoxiadenosina protegida na base
1 ' O HO
OH
2 ' 3 ' 4 '
5 ' N
N
N
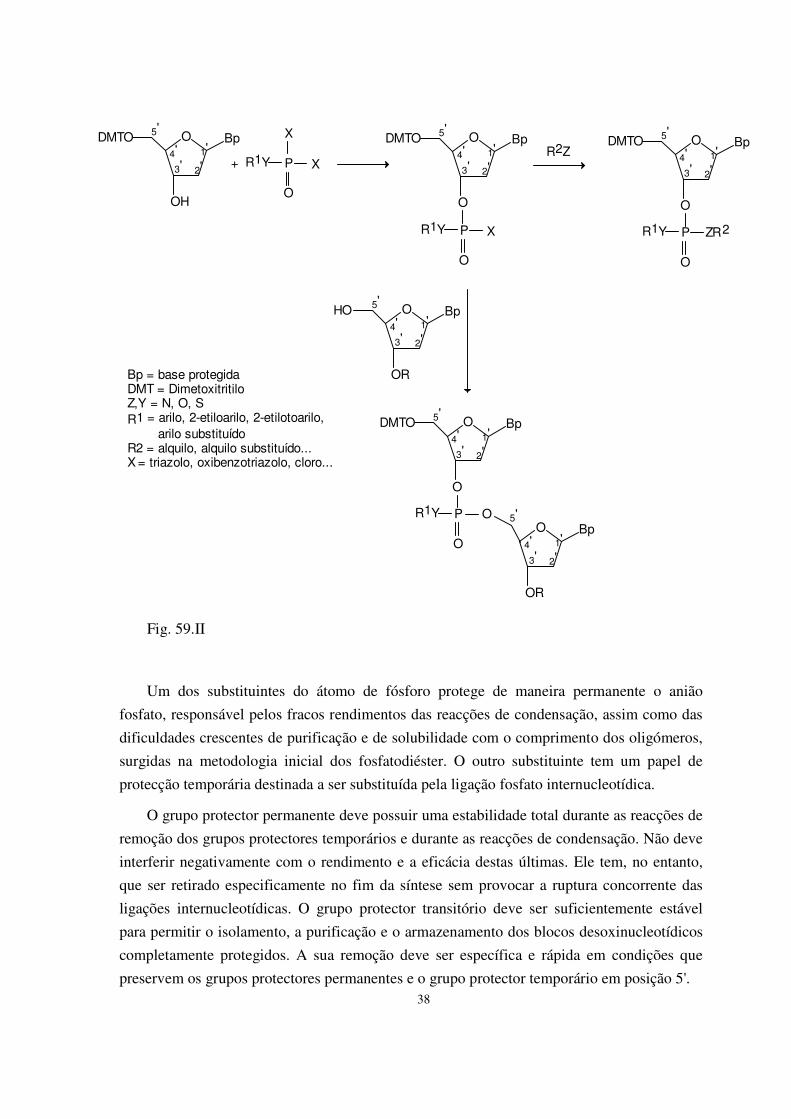
38
+1'
ODMTO
OH
2'3'4'
5'Bp
P
O
X
XR1YR2Z1'
ODMTO
O
2'3'4'
5'Bp
P
O
XR1Y
1'O HO
OR
2'3'4'
5'Bp
1'ODMTO
O
2'3'4'
5'Bp
P
O
ZR2R1Y
1'ODMTO
O
2'3'4'
5'Bp
P
O
R1Y O
1'O
OR
2'3'4'
5'Bp
Bp = base protegidaDMT = DimetoxitritiloZ,Y = N, O, SR1 = arilo, 2-etiloarilo, 2-etilotoarilo, arilo substituídoR2 = alquilo, alquilo substituído...X = triazolo, oxibenzotriazolo, cloro...
Fig. 59.II
Um dos substituintes do átomo de fósforo protege de maneira permanente o anião
fosfato, responsável pelos fracos rendimentos das reacções de condensação, assim como das
dificuldades crescentes de purificação e de solubilidade com o comprimento dos oligómeros,
surgidas na metodologia inicial dos fosfatodiéster. O outro substituinte tem um papel de
protecção temporária destinada a ser substituída pela ligação fosfato internucleotídica.
O grupo protector permanente deve possuir uma estabilidade total durante as reacções de
remoção dos grupos protectores temporários e durante as reacções de condensação. Não deve
interferir negativamente com o rendimento e a eficácia destas últimas. Ele tem, no entanto,
que ser retirado especificamente no fim da síntese sem provocar a ruptura concorrente das
ligações internucleotídicas. O grupo protector transitório deve ser suficientemente estável
para permitir o isolamento, a purificação e o armazenamento dos blocos desoxinucleotídicos
completamente protegidos. A sua remoção deve ser específica e rápida em condições que
preservem os grupos protectores permanentes e o grupo protector temporário em posição 5'.
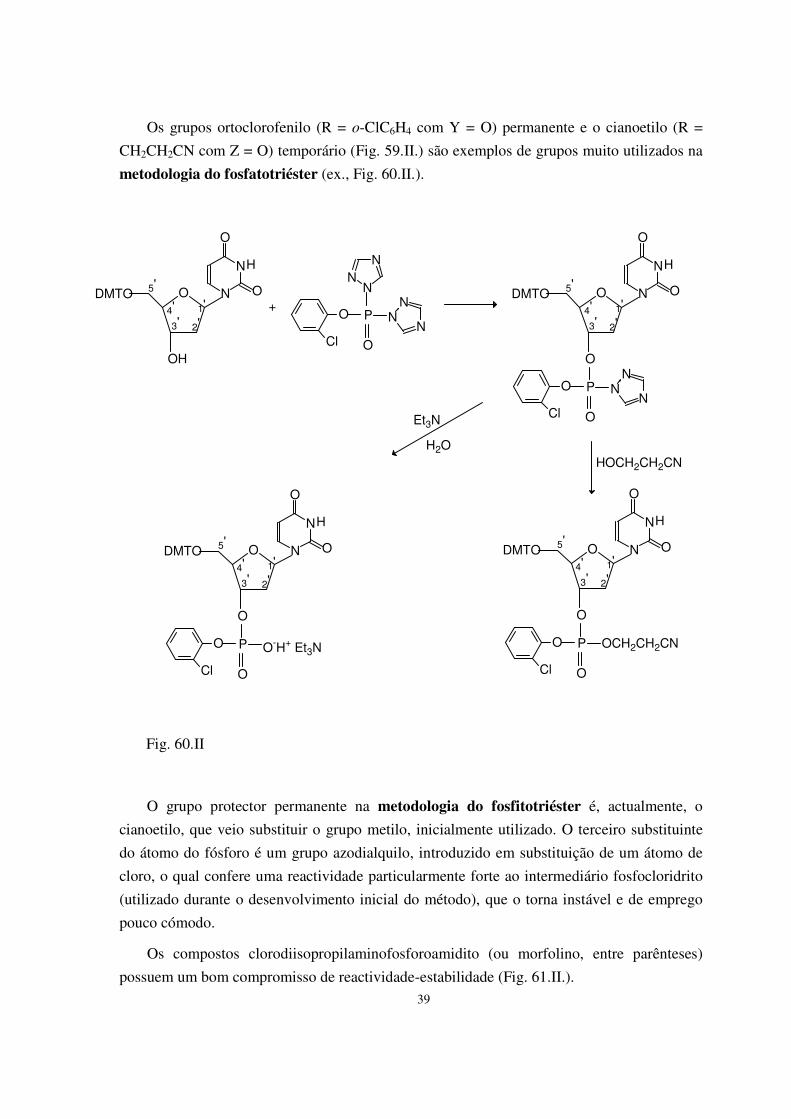
39
Os grupos ortoclorofenilo (R = o-ClC6H4 com Y = O) permanente e o cianoetilo (R =
CH2CH2CN com Z = O) temporário (Fig. 59.II.) são exemplos de grupos muito utilizados na
metodologia do fosfatotriéster (ex., Fig. 60.II.).
+
NN
N
P
O
O
ClN
NN
HOCH2CH2CN
1'O N
N
O
O
H
DMTO
OH
2'3'4'
5'
P
O
O
ClN
NN
DMTO1'
O N
N
O
O
H
O
2'3'4'
5'
Et3N
H2O
P
O
O
Cl
DMTO1'
O N
N
O
O
H
O
2'3'4'
5'
OCH2CH2CNP
O
O
Cl
DMTO1'
O N
N
O
O
H
O
2'3'4'
5'
O-H+ Et3N
Fig. 60.II
O grupo protector permanente na metodologia do fosfitotriéster é, actualmente, o
cianoetilo, que veio substituir o grupo metilo, inicialmente utilizado. O terceiro substituinte
do átomo do fósforo é um grupo azodialquilo, introduzido em substituição de um átomo de
cloro, o qual confere uma reactividade particularmente forte ao intermediário fosfocloridrito
(utilizado durante o desenvolvimento inicial do método), que o torna instável e de emprego
pouco cómodo.
Os compostos clorodiisopropilaminofosforoamidito (ou morfolino, entre parênteses)
possuem um bom compromisso de reactividade-estabilidade (Fig. 61.II.).

40
O
1'O
OH
2'3'4'
5'N
N
N
N
N
H
HC
O
DMTO
O
1'O
O
2'3'4'
5'N
N
N
N
N
H
HC
O
DMTO+
P
Cl
NNCCH2CH2O
NCCH2CH2OP
N
N O
N O
Fig. 61.II
O método do fostitotriéster é o correntemente utilizado em síntese em fase sólida, em
rotina, num processo inteiramente automatizado de elongação da cadeia oligonucleotídica.
É importante sublinhar que o sucesso da síntese química de DNA se baseia sobretudo na
preparação destes blocos de base, isto é, na escolha dos diferentes grupos protectores, no
rendimento das diferentes reacções e na pureza dos desoxinucleotídeos completamente
protegidos.
2) Desprotecção selectiva
Para poder condensar dois desoxinucleotídeos pelo método do fosfatotriéster, é
necessário libertar separadamente, por um lado a função fosfato e por outro o grupo
hidroxílico 5', das suas protecções temporárias. Por exemplo, o grupo dimetoxitritilo é
retirado em condições ácidas suaves e o grupo cianoetilo em condições básicas suaves (Fig.
62.II.).
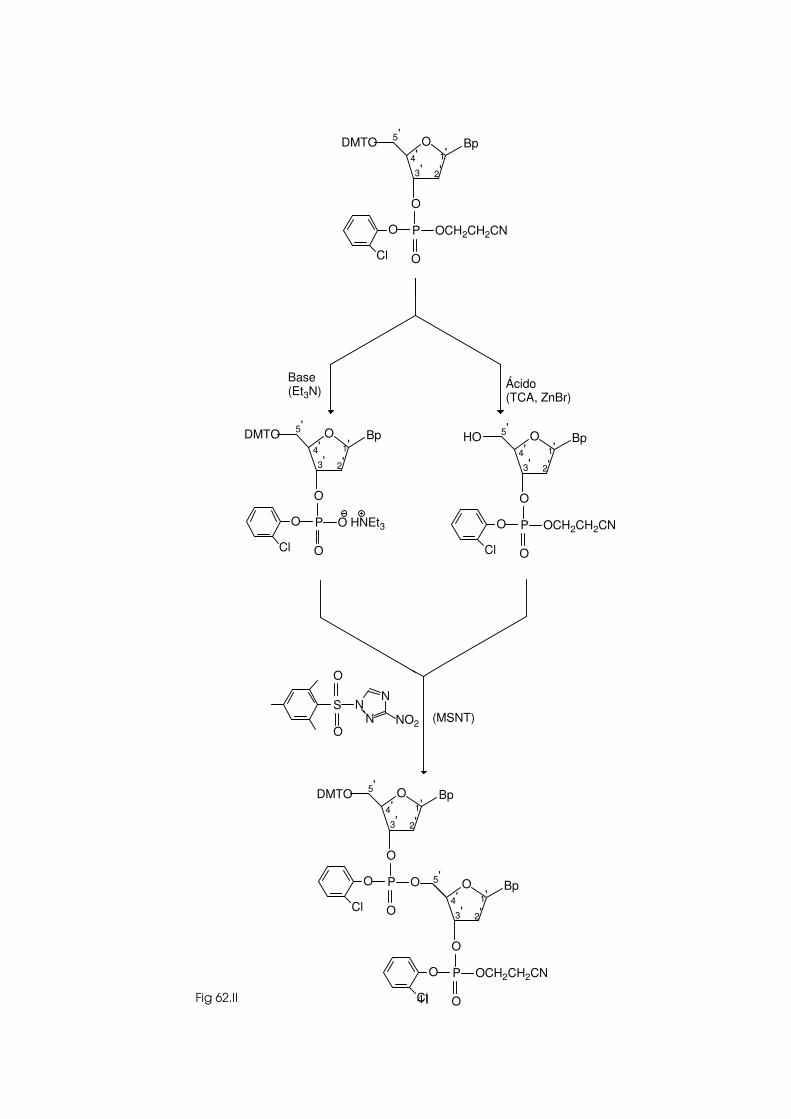
41
Fig 62.II
(MSNT)
Base (Et3N)
Ácido(TCA, ZnBr)
P
O
O
Cl
DMTO1'
O
O
2'3'4'
5'
OCH2CH2CN
Bp
P
O
O
Cl
HO1'
O
O
2'3'4'
5'
OCH2CH2CN
Bp
S
O
O
NN
N NO2
P
O
O
Cl
DMTO1'
O
O
2'3'4'
5'
O HNEt3
Bp
P
O
O
Cl
DMTO1'
O
O
2'3'4'
5'
O
Bp
P
O
O
Cl
1'
O
O
2'3'4'
5'
OCH2CH2CN
Bp
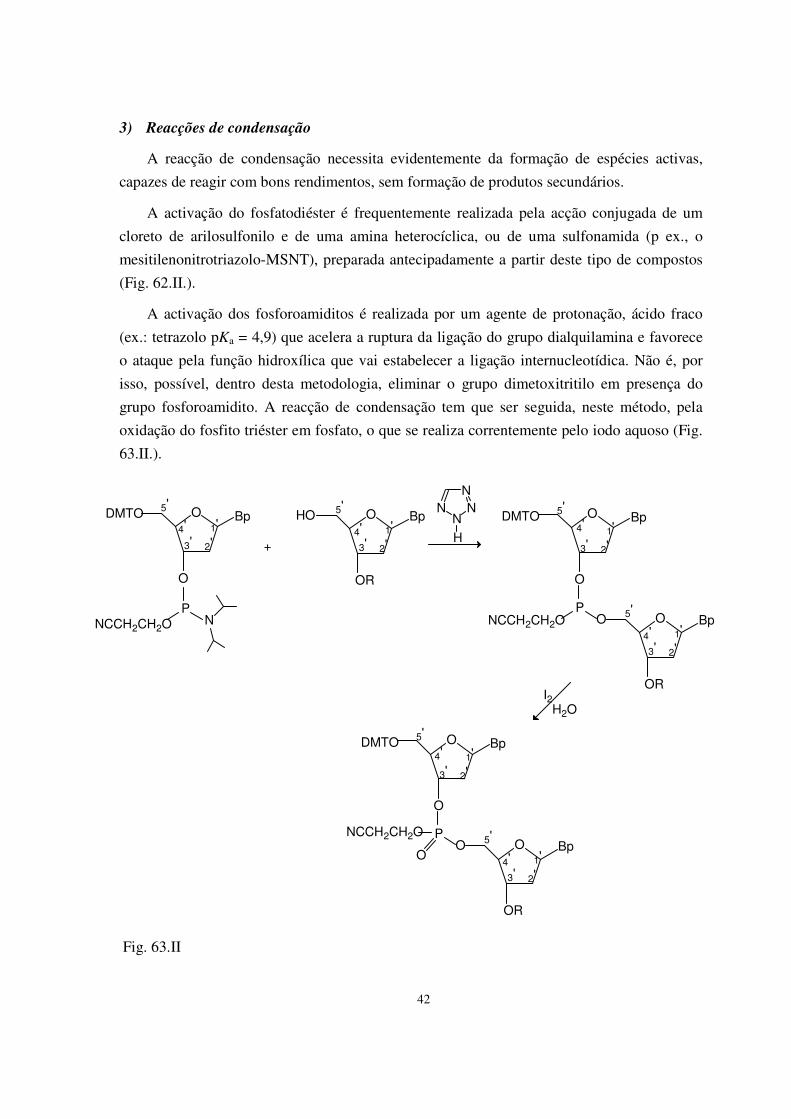
42
3) Reacções de condensação
A reacção de condensação necessita evidentemente da formação de espécies activas,
capazes de reagir com bons rendimentos, sem formação de produtos secundários.
A activação do fosfatodiéster é frequentemente realizada pela acção conjugada de um
cloreto de arilosulfonilo e de uma amina heterocíclica, ou de uma sulfonamida (p ex., o
mesitilenonitrotriazolo-MSNT), preparada antecipadamente a partir deste tipo de compostos
(Fig. 62.II.).
A activação dos fosforoamiditos é realizada por um agente de protonação, ácido fraco
(ex.: tetrazolo pKa = 4,9) que acelera a ruptura da ligação do grupo dialquilamina e favorece
o ataque pela função hidroxílica que vai estabelecer a ligação internucleotídica. Não é, por
isso, possível, dentro desta metodologia, eliminar o grupo dimetoxitritilo em presença do
grupo fosforoamidito. A reacção de condensação tem que ser seguida, neste método, pela
oxidação do fosfito triéster em fosfato, o que se realiza correntemente pelo iodo aquoso (Fig.
63.II.).
NCCH2CH2OP
N
O
1'O HO
OR
2'3'4'
5'Bp
+
NN
NN
H
I2H2O
NCCH2CH2O P
DMTO1'
O
O
2'3'4'
5'Bp
1'O O
OR
2'3'4'
5'Bp
O
DMTO1'
O
2'3'
5'Bp
4'
NCCH2CH2OP
DMTO1'
O
O
2'3'4'
5'Bp
1'O O
OR
2'3'4'
5'Bp
Fig. 63.II

43
4) Construção da cadeia oligodesoxinucleotídica
a) Em solução
Na síntese em solução e pela metodologia do fosfatotriéster a extensão da cadeia
polinucleotídica pode fazer-se de maneira alternada no sentido 3'→ 5' ou 5'→ 3', visto que se
pode desproteger o grupo hidroxílico na extremidade 5', ou o triesterfosfato na posição 3' do
oligómero em construção. Dois oligómeros podem igualmente ser condensados um com o
outro. Devido à sensibilidade dos fosforoamiditos às condições ligeiramente ácidas, esta
flexibilidade própria aos triesterfosfatos, não se aplica ao método do fosfitotriéster, que não
foi desenvolvido em solução.
A síntese em solução permite a detecção de reacções secundárias de maneira mais
directa que a síntese em fase sólida. Ela permite a identificação e a compreensão da origem
dos produtos parasitas e portanto possibilita a intervenção sobre os parâmetros correctores
(Fig. 64.II.).
b) Em fase sólida
A extensão da cadeia de DNA em fase sólida realiza-se numa só direcção (em geral 3'→
5'), a partir de um desoxirribonucleosídeo fixo, por uma das funções hidroxílicas, a um
suporte sólido. A fixação efectua-se em geral através da formação de uma ligação amida entre um 2'-desoxi-3'-succinilorribonucleosídeo e um polímero insolúvel aminado (P-NH2)
(Fig. 65.II.).
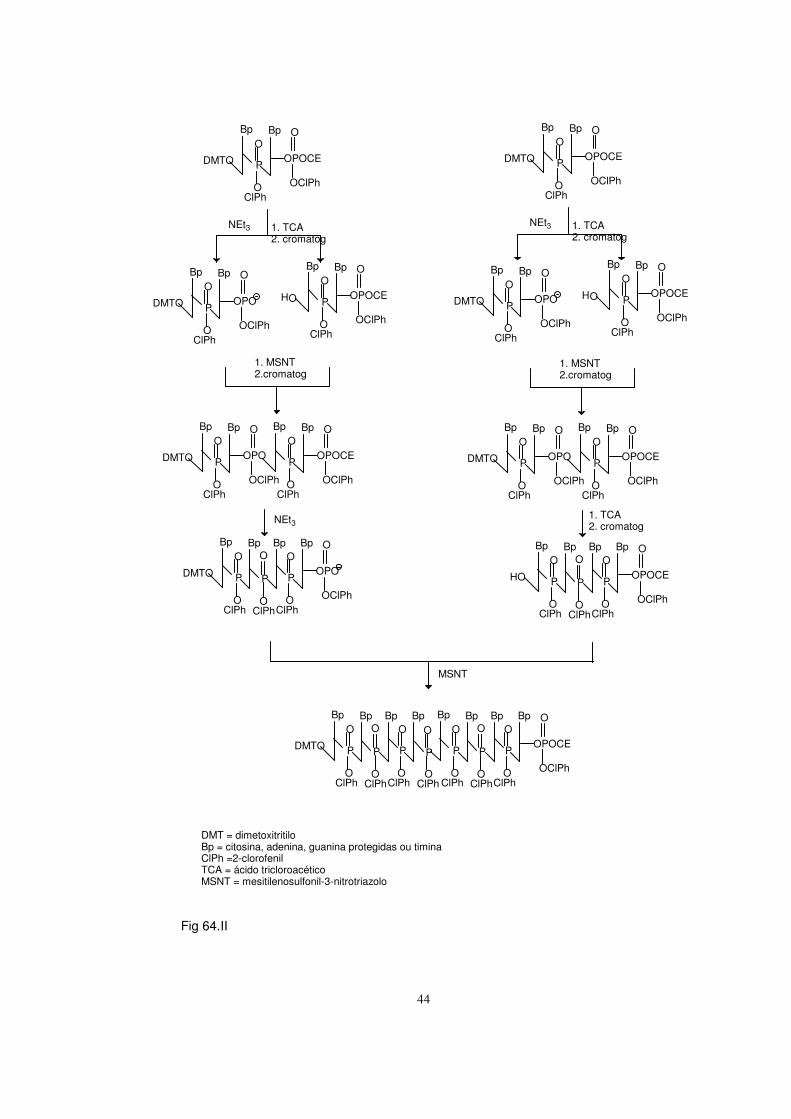
44
ClPh
Bp
DMTO
Bp
P
O
O
O
OPOCE
OClPh
NEt3 1. TCA 2. cromatog
ClPh
Bp
DMTO
Bp
P
O
O
O
OPO
OClPhClPh
Bp
O
Bp
P
O
O
O
OPOCE
OClPh
H
1. MSNT2.cromatog
ClPh
Bp
DMTO
Bp
P
O
O
O
OPO
OClPhClPh
Bp Bp
P
O
O
O
OPOCE
OClPh
1. MSNT2.cromatog
ClPh
Bp
DMTO
Bp
P
O
O
O
OPO
OClPhClPh
Bp Bp
P
O
O
O
OPOCE
OClPh
ClPh
Bp
DMTO
Bp
P
O
O
O
OPOCE
OClPh
ClPh
Bp
DMTO
Bp
P
O
O
O
OPO
OClPh
NEt3 1. TCA 2. cromatog
ClPh
Bp
O
Bp
P
O
O
O
OPOCE
OClPh
H
1. TCA 2. cromatog
ClPh
HO
Bp
P
O
O
O
P
OClPh
Bp Bp
ClPh
Bp
P
O
O
O
OPOCE
OClPh
MSNT
NEt3
ClPh
DMTO
Bp
P
O
O
O
P
OClPh
Bp Bp
ClPh
Bp
P
O
O
O
OPO
OClPh
ClPh
DMTO
Bp
P
O
O
O
P
OClPh
Bp Bp
P
ClPh
Bp
P
O
O
O
OClPh ClPh
Bp
P
O
O
O
P
OClPh
Bp Bp
ClPh
Bp
P
O
O
O
OPOCE
OClPh
DMT = dimetoxitritiloBp = citosina, adenina, guanina protegidas ou timinaClPh =2-clorofenilTCA = ácido tricloroacéticoMSNT = mesitilenosulfonil-3-nitrotriazolo
Fig 64.II

45
O+
O
O
H2N
DCC
ONH
Bp = citosina, adenina, guanina protegidas ou timina
= polímero insolúvel
Fig. 65.II
DMTO O Bp
OH
PDMTO O Bp
O
C O
CH2
CH2
CO2H
DMTO O Bp
O
C O
CH2
CH2
C P
P
O poliestirenodivinilbenzeno, a resina composta polidimetilacrilamida-kieselguhr, a
sílica, as esferas de vidro de porosidade controlada, a celulose, o co-polímero teflon-
poliestireno são exemplos de polímeros insolúveis que podem ser utilizados.
A extensão da cadeia polinucleotídica em fase sólida consiste na repetição de um ciclo que
compreende essencialmente:
a) a reacção de desprotecção do grupo hidroxílico em 5' do primeiro nucleosídeo (ou de
cadeia em crescimento) fixo no polímero insolúvel;
b) a reacção de condensação entre a função hidroxílica libertada e a espécie nucleotídica
introduzida em solução e activada in situ no átomo de fósforo em posição 3'.
Com o método do fosfitotriéster, uma terceira reacção, que consiste na oxidação do
fosfito em fosfato, é, como já acima foi referido, necessária. Uma reacção suplementar é por
vezes efectuada (caping). Ela visa o bloqueamento da ligeira percentagem dos grupos
hidroxílicos que não reagiram na reacção de condensação a fim de os desactivar para as
reacções ulteriores. Os reagentes em excesso, os subprodutos e os solventes das reacções e de
lavagem são eliminados por simples filtração do suporte sólido (Fig. 66.II.).
repetição
n vezes
OO
OC(CH 2)2CNH PP
O
OR
BpBpBp
DMT
Bp
P
O
OR
P
O
OR
POC(CH 2)2CNH
O O
DMT = dimetoxitritiloBp = citosina, adenina, guanina protegidas ou timinaR = grupo protector do fosfatoP = polímero insolúvel
Bp
P
O
OR
Bp
DMT DMT
1) Desprotecção 5'OH
2) Condensação
3) capping4) oxidação
Bp
OO
OC(CH 2)2CNH P
Bp
DMT P
OR
N
Fig. 66.II

46
O isolamento do produto ligado ao polímero insolúvel é, por consequência, simples e
rápido, em relação aos métodos convencionais em solução. O conjunto das operações presta-
se bem à automatização.
Uma desvantagem da fase sólida é a cinética desfavorável. Para conseguir reacções
completas dentro de tempos razoáveis, é indispensável utilizar excessos importantes de
reagentes, cuja pureza é portanto crítica. Vestígios de impurezas reactivas podem inibir
completamente a reacção de condensação.
Em fase sólida a acumulação de produtos indesejáveis é inevitável, visto não haver
purificação em nenhuma etapa da extensão do fragmento de DNA. Uma maneira de
minimizar este inconveniente consiste em utilizar dímeros ou trímeros, preparados em
solução, estratégia que diminui de um factor 2 ou 3 o número de reacções efectuadas no
polímero insolúvel.
O comprimento do fragmento de DNA que é possível obter por síntese química depende
evidentemente do rendimento da etapa de condensação, que tem que ser mantido o mais alto
possível (> 99 %).
Do ponto de vista das vantagens e inconvenientes, o método do cianoetilofosforamidito,
pela alta velocidade da reacção de condensação e sobretudo pela possibilidade de obter
produtos de reacção praticamente isentos de produtos secundários (altos rendimentos) foi
universalmente adoptado nos sintetizadores automáticos. O polímero insolúvel que melhores
resultados tem dado é constituído por esferas de vidro de porosidade controlada e
alquiloaminadas.
5) Desprotecção
A desprotecção do fragmento de DNA é uma operação delicada que deve preservar a
integridade do edifício molecular. Reacções incompletas, ou uma degradação parcial,
conduzem a uma mistura complexa de produtos.
A desprotecção compõe-se de 3 etapas distintas:
I) transformação dos grupos triesterfosfatos em diesterfosfatos;
II) desprotecção das bases;
III) desprotecção da função OH terminal em 5'.
I) A desprotecção dos grupos fosfatos é uma fonte possível de ruptura internucleotídica.
Esta pode ser minimizada utilizando reagentes selectivos antes de desproteger as bases.
Se a extensão da cadeia de DNA for realizada pelo método do fosfatotriéster em fase
sólida, a clivagem do suporte insolúvel efectua-se com o mesmo reagente de desprotecção
dos grupos fosfatos clorofenilados, por exemplo com o 2-nitrofenilcarbaldoximato de

47
tetrametilguanidina. O grupo metilo utilizado no começo do método do fosfitotriéster era
deslocado por ataque nucleofilico pelo anião tiofenalato, antes da clivagem do suporte sólido.
Esta é efectuada em condições suaves. O grupo cianoetilo, utilizado actualmente, é removido
em condições ainda mais suaves e sem necessidade de fazer intervir um reagente
suplementar: por NH4OH à temperatura ambiente.
II) As aminas exocíclicas das bases são desaciladas por amonólise a 50°C. Este tratamento
provoca uma ruptura, que não é desprezível, das ligações fosfatos internucleotídicas quando o
fosfato está na forma de triéster; esta é a razão pela qual se converte antecipadamente e
selectivamente o fosfatotriéster em fosfatodiéster.
III) O grupo protector da função hidroxílica em posição 5' é o último a ser retirado (pelo
ácido acético 80 % no caso do grupo dimetroxitritilo). É conservado até ao fim para impedir
a formação de triésteres fosfóricos cíclicos que conduzem a estruturas
oligodesoxirribonucleotídicas com ligações 5'→ 5, durante as primeiras etapas de
desprotecção. Devido ao seu carácter hidrófobo, o grupo dimetoxitritilo protector da função
5'OH facilita o isolamento do produto por cromatografia de sílica em fase inversa (Fig.
67.II.).
DMT
Bp Bp
P
O
OR
Bp
P
O
OR
POC(CH2)2CNH
O O
n
Desprotecção dosfosfatos eClivagem OH
O O
Desprotecçãodas bases
DMT
Bp Bp
P
O
Bp
P
O
n
B
P
O
B
P
O
B
O
DMTOH
n
Desprotecçãodo grupo 5'OH
B
P
O
B
P
O
B
O
HOOH
n
DMT = dimetoxitritiloBp = citosina, adenina, guanina protegidas ou timinaR = grupo protector do fosfatoP = polímero insolúvel
Fig 67.II
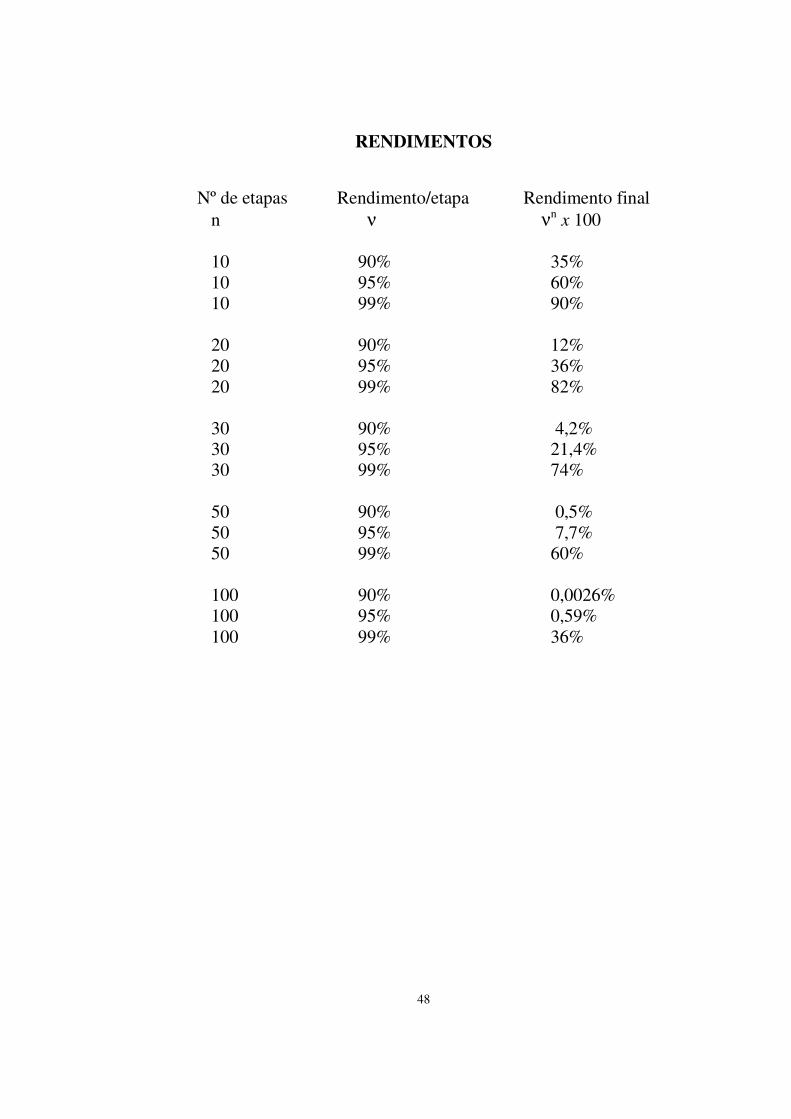
48
RENDIMENTOS
Nº de etapas Rendimento/etapa Rendimento final n ν νn x 100
10 90% 35% 10 95% 60% 10 99% 90% 20 90% 12% 20 95% 36% 20 99% 82% 30 90% 4,2% 30 95% 21,4% 30 99% 74% 50 90% 0,5% 50 95% 7,7% 50 99% 60% 100 90% 0,0026% 100 95% 0,59% 100 99% 36%

49
6) Isolamento, purificação e caracterização
Os métodos de isolamento e de purificação correntemente utilizados são a cromatografia
de DEAE-celulose, de Sefadex, de camada fina de sílica, a cromatografia liquida a alta
pressão (HPLC), com coluna de sílica de fase inversa, ou de troca catiónica e a electroforese
preparativa em gel de poliacrilamida, ou capilar.
A caracterização do produto isolado e marcado numa das extremidades com um
radioisótopo pode ser efectuada pelo método de sequenciação de DNA de Maxam & Gilbert.
É, no entanto, necessário um reajustamento das condições das reacções aos fragmentos de
pequeno comprimento. Um outro método, conhecido pelo nome de Wandering spot, baseia-
se na análise a duas dimensões (electroforese em acetato de celulose, seguida de
cromatografia em camada fina de DEAE-celulose) dos fragmentos de oligonucleotídeo
sintético marcado numa extremidade, obtidos por digestão parcial com uma exonuclease.
Este método restringe-se a fragmentos de comprimento inferior a 20 nucleotídeos (vd. secção
"Sequenciação").
As aplicações dos oligodesoxirribonucleotídeos de sequência definida abrangem quase
todos os aspectos da investigação que implicam a recombinação de DNA, tanto no que diz
respeito à construção, à identificação e à caracterização de clones bacterianos particulares,
como à manipulação do DNA clonado com o fim de modificar a sua estrutura.
Nos campos da amplificação de DNA, da determinação de sequências de DNA, do
estudo das interacções proteína-DNA, ou da análise estrutural do DNA, os
oligodesoxirribonucleotídeos sintéticos têm-se revelado um instrumento extremamente útil e
indispensável.

50
2. VECTORES DE CLONAGEM
A utilização de novas moléculas de DNA formadas por engenharia genética depende da
sua separação e da sua propagação. Para isso é necessário introduzi-las em células
(bacterianas, por exemplo) de tal maneira que elas possam ser replicadas, preservadas e
recuperadas à vontade. Para o realizar recorre-se à utilização de vectores particulares, cujo
DNA pode ser unido por ligação ao DNA que se deseja introduzir no hospedeiro biológico e
que podem depois ser levados a infectar eficazmente esse hospedeiro. Assim que a molécula
recombinante se encontra no hospedeiro, pode-se replicar e propagar de maneira permanente.
Portanto, em princípio, segmentos de DNA de qualquer origem podem ser clonados e
preparados em quantidade a partir de cultura de células, contendo aquele DNA.
Quatro tipos de vectores podem ser utilizados para clonar um fragmento de DNA e
propagá-lo na bactéria E. coli.
a) plasmídeos
b) bacteriófago λ
c) cosmídeos
d) bacteriófago M13
e) sistemas de vectores bacterianos de elevada capacidade
Se bem que diferentes em comprimento e em estrutura, estes 5 tipos de vectores
compartilham um certo número de propriedades:
1) replicam-se de maneira autónoma na bactéria;
2) podem ser facilmente separados do DNA do hospedeiro e purificados;
3) contêm regiões de DNA não essenciais à sua propagação no hospedeiro, nos quais
um DNA estrangeiro pode ser inserido.
a) Os plasmídeos
Os plasmídeos são moléculas de DNA circular bicatenário capazes de se replicar na
bactéria de maneira independente do cromossoma do hospedeiro. Os plasmídeos estão
distribuídos nos procariotas de maneira muito ampla; variam de massa molecular de + 106 a
mais de 200 106 daltons. Conferem um certo número de fenótipos ao hospedeiro que os
contém (Tabela 1.II).
Os plasmídeos podem-se agrupar segundo duas categorias principais, os plasmídeos
conjugantes e os não conjugantes. Estes últimos são destituídos de uma série de genes (tra)

51
que conduzem à conjugação bacteriana. As funções de transferência (tra) incluem um cluster
de 12 genes, pelo menos, que são responsáveis, entre outras coisas, pela síntese de pili e de
outros componentes de superfície celular. Estes pili permitem o contacto físico -um pré-
requisito para a conjugação- entre as células doadoras e as receptoras.
Independentemente destas funções, todos os plasmídeos possuem regiões específicas,
responsáveis pela sua replicação autónoma (rep), que inclui uma origem de replicação (ori).
Dado todos os genes e sequências de DNA num plasmídeo, estarem habitualmente
organizados em clusters funcionais, é possível desenhar uma estrutura geral dos plasmídeos
(Fig. 68. II A).
Os mecanismos que regulam o número de cópias dos plasmídeos são da maior
importância para a sua manutenção na bactéria. O número de cópias é, ou estritamente
controlado por, e correlacionado com, o número de moléculas de DNA cromossómico (i. é., é
dificilmente superior a um) ou relaxado (i. é., uma célula bacteriana pode conter múltiplas
cópias dum plasmídeo). Assim, uma categoria de plasmídeos mantém-se no hospedeiro em
número elevado de cópias (plasmídeo relaxado) e outra em pequeno número de cópias
(plasmídeo restritivo). Com algumas excepções, os plasmídeos restritivos possuem uma
massa molecular relativamente elevada e são conjugativos, enquanto que os relaxados são
normalmente não-conjugativos e têm uma massa molecular baixa.
Os plasmídeos que se replicam sob controlo restrito requerem biossíntese proteica activa,
mas não DNA polimerase I para a sua replicação; os plasmídeos relaxados, contudo,
necessitam duma DNA polimerase I funcional (produto do gene polA), mas replicam-se sem
biossíntese proteica de novo. Muitos dos plasmídeos multicópia apresentam um
comportamento pouco usual: são capazes de continuar a replicação, mesmo na presença dum
inibidor da biossíntese de proteínas. Praticamente, quando se interrompe a síntese proteica do
hospedeiro com um antibiótico (p. ex., cloranfenicol), bloqueia-se também a replicação do
seu DNA. No entanto, a do plasmídeo relaxado continua e podem-se obter vários milhares de
cópias deste plasmídeo por célula (amplificação). Assim, os plasmídeos relaxados podem-se
replicar mesmo se a replicação do DNA do hospedeiro estiver interrompida. Como a
amplificação afecta simultaneamente os insertos de DNA recombinante na molécula de
plasmídeo, os plasmídeos multicópia relaxados desempenham um papel central na tecnologia
do gene. Inversamente, a replicação dos plasmídeos restritivos está ligada de maneira
dependente à do DNA do hospedeiro.
O número de cópias de plasmídeos é controlado geneticamente e pode ser alterado por
mutagénese, de maneira a tornar a replicação do plasmídeo sensível à temperatura e, ou
cessa, após uma elevação da temperatura, ou, pelo contrário, torna o plasmídeo relaxado.

52
Concepção de um vector plasmídico
1. Um vector plasmídico deve ser o mais pequeno possível em relação ao DNA
cromossómico. Esta propriedade facilita a manipulação do vector e aumenta as possibilidades
de transformação eficaz do hospedeiro bacteriano.
2. O vector deve estar bem caracterizado em relação aos genes que contém, no que respeita
à sua carta de restrição e à sua sequência em bases.
3. O vector deve poder replicar-se de maneira autónoma (relaxado) e propagar-se
eficazmente no hospedeiro, de maneira a poder obter-se facilmente grandes quantidades de
DNA do vector e do DNA que nele tiver sido inserido.
4. O vector deve comportar um marcador de selecção que permita distinguir as células
transformadas pelo vector das que não o são.
Habitualmente este marcador é uma resistência a um antibiótico, mediada por um gene
que codifica para uma enzima de modificação de antibiótico (Tcr, Apr, Kanr, Cmr, ...).
5. O vector ideal deve conter um marcador genético adicional, que pode ser inactivado por
inserção do DNA que se quer clonar. A inactivação insercional deste marcador genético
permite distinguir as células que contêm um plasmídeo recombinante, das que contêm o
plasmídeo original (Fig. 68.II.B.).
6. O vector deve conter o maior número possível de sítios de restrição únicos, localizados
num dos marcadores genéticos. Esta propriedade confere um máximo de flexibilidade em
termos de clonagem. Evidentemente que estes sítios de restrição não podem existir nas zonas
do plasmídeo essenciais para a sua replicação.
O plasmídeo pBR322 desenvolvido por Bolivar et al. (1979) tem em conta estas
exigências (Fig. 69.II.).
Neste vector, a replicação é do tipo relaxado e os genes de resistência Tr e Apr podem
servir, um e outro, de marcadores para a selecção ou para a inactivação insercional (Fig. 70.II
e Fig. 71.II).
Os vectores de tipo plasmídico são preciosos, na medida em que não há praticamente
nenhum limite imposto ao comprimento do fragmento de DNA que pode ser inserido neles.
No entanto, em certos casos pode ser mais vantajoso utilizar vectores fágicos.
b) Bacteriófagos
A transformação de E. coli pelo DNA de um fago manifesta-se pela produção de
partículas fágicas que resultam da replicação e da expressão do DNA absorvido. O genoma

53
do fago pode ser utilizado de maneira similar aos plasmídeos como vector para os fragmentos
de DNA.
Uma das vantagens dos vectores fágicos sobre os plasmídeos resulta da grande
experiência bioquímica e genética existente sobre estes fagos. Com os fagos não lisantes
pode-se, nomeadamente, tirar partido dos sistemas de regulação próprios, para optimizar a
expressão dos genes clonados, assim como a acumulação dos seus produtos nas células
infectadas.
Outra vantagem resulta da possibilidade de escrutinar e caracterizar vários milhares de
placas fágicas de lise, p.ex., por hibridação DNA-DNA, numa placa de Petri. Além disso,
existe disponível um sistema de empacotamento in vitro, que permite empacotar o DNA em
cabeças fágicas vazias. A infectividade do DNA recombinante é aumentada várias ordens de
grandeza, quando comparada com a de preparações de DNA puro.
Ao contrário do que acontece com os vectores do tipo plasmídico, o comprimento da
inserção do DNA estrangeiro é limitado no caso da utilização dos vectores fágicos. Pode-se
evidentemente suprimir regiões não essenciais do DNA fágico, mas apesar disso, o
comprimento do DNA tem que permanecer dentro dos limites impostos pelo seu
empacotamento no invólucro do fago. O sistema de empacotamento in vitro permite tirar
partido desta limitação, impondo uma selecção do tamanho do DNA empacotado, de tal
maneira que, em condições apropriadas, só as moléculas de DNA recombinante serão
empacotadas.
Finalmente, milhões de partículas virais clonadas independentemente, constituídas na
forma de banco genómico, representando a informação genética completa dum organismo
complexo, podem ser armazenadas num número reduzido de mililitros de meio de cultura.
O escrutínio dos fagos recombinantes numa população mista, baseia-se essencialmente
na morfologia das placas, ou na sua cor sobre um tapete de bactérias indicadoras e ainda na
hibridação DNA-DNA com uma sonda marcada.
Lembramos que o fago λ contém uma molécula de DNA linear com 45 kbases,
comportando extremidades coesivas. Estas extremidades coesivas naturais associam-se para
formar o sítio cos. Na Fig. 72.II. os genes da esquerda codificam para as proteínas da cabeça
e da cauda do fago; os genes centrais estão implicados na recombinação e na lisogenia e
podem ser considerados como não essenciais ao desenvolvimento lítico do fago. Os genes da
direita conduzem nomeadamente à lise. Um esquema simplificado do desenvolvimento
fágico está representado na Fig. 73.II.
O DNA de λ possui numerosos sítios de clivagem para quase todas as enzimas de
restrição correntes e como tal não é apropriado para vector de DNA. Foi, portanto, preciso

54
construir derivados do DNA selvagem, nos quais existe um só sítio de restrição permitindo a
inserção de um fragmento de DNA exogéneo (vectores de inserção), ou nos quais, um par de
sítios de restrição flanqueia um fragmento de DNA não essencial, que pode ser excisado e
substituído por um fragmento de DNA estrangeiro (vectores de substituição).
O conteúdo em DNA das partículas fágicas está submetido a limites quantitativos. Por
um lado, um conteúdo inferior a 75 % da capacidade normal dos invólucros proteicos (cerca
de 45 kb) não será empacotado correctamente e não conduzirá à formação de partículas
fágicas viáveis. Por outro lado, um excesso de DNA (>105 % do conteúdo normal) levará a
um empacotamento defeituoso. Estes limites são impostos por um sistema de clivagem
específico dos sítios cos, sistema composto de nucleases localizadas na base de cabeça do
fago. Muitos derivados de λ do tipo substituição, contêm também eliminações de algumas
kbases nas regiões não essenciais. Deste modo, sempre que se substitui um fragmento de
DNA do fago, pode-se inserir um fragmento maior de DNA estrangeiro (até 22 kbases em
certos casos) (Fig. 74.II.).
A clonagem que utiliza os bacteriófagos λ requer várias etapas:
1) O DNA do vector é digerido com as enzimas de restrição apropriadas e as extremidades
esquerda e direita do DNA de λ são separadas da região não essencial.
2) As extremidades são ligadas em presença de fragmentos de DNA estrangeiro que
possuam extremidades compatíveis com as das extremidades.
3) O DNA recombinante é empacotado in vitro no invólucro proteico. As partículas fágicas
assim formadas são utilizadas para infectar o hospedeiro bacteriano apropriado.
4) Os fagos recombinantes contendo as sequências de DNA clonadas são a seguir
identificados por diferentes processos.
Empacotamento in vitro
O empacotamento de DNA recombinante λ nos invólucros de fago in vitro, permite que
ele seja introduzido no hospedeiro bacteriano pelo processo normal da infecção fágica com
um rendimento muito elevado (± 106 placas de lise por µg de DNA recombinante).
Uma série de acontecimentos particulares que se desenrolam durante o empacotamento
do DNA no hospedeiro podem ser reproduzidos in vitro (Fig. 75.II.). O DNA do fago, na
forma de concatâmero (forma produzida após replicação), constitui o substrato da reacção de
empacotamento. Na presença de precursores da cabeça (essencialmente produto do gene E) e
do produto do gene A, o DNA concatâmero é clivado em monómeros nos sítios cos e
empacotado. O produto do gene D é em seguida incorporado para formar as cabeças fágicas

55
completas. Finalmente, os produtos doutros genes, tais como W e FII e estruturas da cauda
unidas independentemente, juntam-se às cabeças para formar partículas fágicas maduras.
Para efectuar o empacotamento in vitro, coloca-se o DNA recombinante em presença de
concentrações elevadas de precursores da cabeça, da cauda e de proteínas de empacotamento.
Esta operação é realizada na prática num lisado misto, derivado de duas estirpes bacterianas
lisogénicas, que foram induzidas antecipadamente. Uma das estirpes contém uma mutação
sem sentido no gene D de λ, que bloqueia a etapa de maturação das cabeças dos fagos. A
estirpe acumula pois, os precursores das cabeças fágicas. A segunda estirpe tem uma mutação
sem sentido no gene E de λ, que impede a formação das cabeças fágicas. No lisado misto, a
complementação genética permite a formação de cabeças normais e o empacotamento do
DNA recombinante.
c) Cosmídeos
O sistema de clonagem no DNA de λ possui limitações respeitantes ao tamanho máximo
do fragmento que se quer clonar (de 15 a 25 kbases). Para clonar fragmentos de DNA de
tamanho superior (até 40 kbases), é preciso recorrer aos vectores do tipo cosmídeo. São
plasmídeos que contêm um fragmento de DNA de λ, incluindo o sítio cos. Estes vectores são
utilizados em combinação com o sistema de empacotamento in vitro. Após o empacotamento,
a partícula é levada a infectar um hospedeiro apropriado. O DNA injectado circulariza-se
como o DNA fágico, mas replica-se como um plasmídeo, sem haver expressão das funções
fágicas. As células que contêm os cosmídeos são então seleccionadas com base na resistência
a um antibiótico (Fig. 76.II.).
Os sítios cos foram clonados no gene Amp do plasmídeo pBR322, tendo sido mantido o
gene de resistência à tetraciclina. O DNA que se pretende clonar é primeiro clivado em
fragmentos de + 35 a 40 kbases e ligado aos sítios cos presentes no plasmídeo (o plasmídeo
foi previamente clivado com uma enzima similar à que foi utilizada para cortar o DNA).
Cada um dos fragmentos clonados comportará pois, um sítio cos a cada extremidade, visto
ser a ligação efectuada em condições que dão origem a concatâmeros, DNA-cosmídeo-DNA-
cosmídeo ...
Quando o extracto do empacotamento in vitro é adicionado, os sítios cos são
reconhecidos, o DNA clonado é clivado em fragmentos de + 45 kbases flanqueados de sítios
cos (conteúdo óptimo das cabeças de λ) e estes fragmentos são em seguida empacotados,
originando partículas fágicas. Estas partículas não são evidentemente fagos viáveis, mas
podem ser utilizadas para infectar E. coli e são seleccionadas com base na resistência à
tetraciclina.

56
Assim que se encontra no interior da bactéria, o híbrido DNA-Cos comporta-se como um
plasmídeo. Os cosmídeos combinam as propriedades dos plasmídeos e dos fagos e têm
diversas vantagens sobre estes dois tipos de vectores. A capacidade de um vector cosmídeo é
superior à de um vector λ. A selecção do comprimento (complemento normal da cabeça do
fago) elimina o barulho de fundo próprio aos cosmídeos sem inserto ou aos que contêm
insertos demasiado pequenos (< 75 % do conteúdo normal da cabeça λ). A infecciosidade do
DNA plasmídico empacotado nas cabeças fágicas é pelo menos 3 vezes superior à do DNA
plasmídico puro.
d) O fago M13
O fago M13 é um fago filamentoso que contém uma molécula de DNA circular
monocatenário (cadeia +). O DNA de M13 injectado na bactéria replica-se de maneira a
formar uma molécula bicatenária (+/-), que se amplifica até atingir 300 cópias por célula
(forma replicativa, RF). Esta forma replicativa pode ser isolada e utilizada como vector de
clonagem.
Assim que a forma replicativa se acumula na bactéria, a síntese de M13 torna-se
assimétrica e conduz à síntese em excesso de uma das duas cadeias de DNA (+), que será
finalmente incorporada nas partículas fágicas maduras. Os fagos são produzidos em contínuo
pela célula infectada sem conduzir à sua lise.
A vantagem principal do fago M13, enquanto vector de clonagem, reside na propriedade
que tem de largar no meio de cultura da bactéria infectada, partículas fágicas contendo um
DNA circular monocatenário, homólogo de uma das duas cadeias da forma replicativa. Além
disso, esta molécula monocatenária pode ser utilizada como molde no sistema de
sequenciação de Sanger (método dos didesoxi) (Fig. 77.II.).
A clonagem na forma replicativa de M13 efectua-se como no caso dos plasmídeos. O
fragmento de DNA clonado será inserido na forma replicativa em qualquer das duas
orientações possíveis, visto que as extremidades do DNA são idênticas. Assim, em função da
orientação, quer uma quer outra cadeia do DNA estrangeiro corresponderá à cadeia (+) e será
empacotada no fago.
Para tirar partido do fago M13 na sequenciação do DNA, sintetiza-se um iniciador de
cerca de 17 bases, complementar de uma região do DNA de M13 próxima do sítio de
inserção. Este iniciador serve de substrato à DNA polimerase para fabricar os fragmentos
marcados que serão analisados em gel de poliacrilamida (Fig. 78.II. e 79.II.).
Um certo número de derivados do fago M13 foram construídos para facilitar, por um
lado a identificação dos fagos que contêm o inserto apropriado e para aumentar, por outro
lado, as possibilidades de clonagem em numerosos sítios de restrição (Fig. 80.II.).

57
Em primeiro lugar, um fragmento do DNA de E. coli, contendo as regiões de regulação e
a informação que codifica uma parte da ß-galactosidase (lacZ), foi introduzido numa região
não essencial do DNA de M13. Por outro lado, a estirpe receptora do fago M13 contém um
gene ß-gal-deficiente. Assim, quando o fago M13 infecta a bactéria hospedeira, há
complementação e produção de ß-galactosidase, em presença de um indutor gratuito, o IPTG
(isopropiltiogalactosídeo). As células portadoras do fago são detectadas por um indicador
corado (XGal) que reage com a ß-galactosidase. As colónias produtoras de fago aparecem
azuis sobre o meio sólido.
Além disso, uma sequência de DNA multi-adaptadora (polylinker), contendo diferentes
sítios de restrição únicos, foi inserida no DNA do fago, na parte do gene truncado da região
codificando a extremidade amino-terminal da ß-galactosidase. Esta inserção não afecta a
capacidade de complementação com o gene ß-gal-deficiente do hospedeiro. Em
compensação, qualquer inserção adicional de DNA na região do polylinker destrói a
complementação. Por conseguinte, os fagos contendo insertos dão origem a placas brancas
em presença de indutor IPTG e de XGal (vd. Secção 1.b.2, sequenciação pelo método de
Sanger).
Conclusões
Os quatro tipos de vectores, plasmídeos, bacteriófago λ, cosmídeos e M13, possuem
propriedades biológicas e físicas que os tornam apropriados para diferentes tipos de
clonagem (Tabela 2.II.).
Devido ao grande comprimento dos DNAs estrangeiros que se podem introduzir nos
cosmídeos e nos fagos λ, estes vectores são apropriados para a construção e a propagação de
bancos de DNA de eucariotas. Estes vectores são no entanto, relativamente difíceis de
manipular e são pouco adequados para a análise de pequenos fragmentos de DNA.
Os vectores do tipo M13 são adaptados essencialmente para a sequenciação de DNA
segundo o método de Sanger, ou como fonte de DNA monocatenário utilizável como sonda
de hibridação. Em compensação, os plasmídeos são apropriados para os diferentes processos
de clonagem. Podem aceitar fragmentos de DNA de comprimento reduzido e podem servir de
veículo para a subclonagem de grandes fragmentos, previamente clonados em cosmídeos ou
em fagos λ. São geralmente munidos de numerosos sítios de restrição únicos, permitindo
uma grande flexibilidade em clonagem. Por último, os plasmídeos são particularmente
apropriados para a clonagem de DNAs complementares (cDNA) e podem ser concebidos
para dirigir a síntese abundante e controlada de proteínas (vectores de expressão).

58
3. MUTAGÉNESE DIRIGIDA
A criação de mutantes é uma prática bem conhecida já em genética clássica, sendo
correntemente utilizada no estudo de funções genéticas. A abordagem de genética clássica,
recorre a agentes mutagénicos químicos ou físicos, que induzem mutações ao acaso. Os
mutantes são em seguida seleccionados, com base num fenótipo desejado. As posições
relativas das mutações num cromossoma podem ser determinadas, utilizando técnicas
convencionais de complementação e de análise de recombinantes. As técnicas modernas de
sequenciação permitem conhecer a natureza precisa da mutação.
Alternativamente, pode-se actualmente adoptar um procedimento antitético, i. é., criar
modificações específicas e controladas no DNA e analisar os efeitos destas modificações no
organismo, in vivo, após ter introduzido nele o DNA mutado (Fig. 81.II.). Os métodos
modernos de isolamento e clonagem de genes e, acima de tudo, a síntese química de DNA,
possibilitam a introdução de alterações específicas nos genes tais como eliminações
(deletions), inserções ou substituições. Nesta última classe de mutações estão incluídas as
trocas pontuais de bases.
Eliminações
A maneira mais simples de criar uma eliminação num cromossoma circular, é de cortar o
DNA com uma enzima de restrição que reconhece dois sítios e produz dois fragmentos.
Após separação e eliminação dum dos fragmentos (em geral o menor) o outro pode ser de
novo circularizado, ligando as suas extremidades coesivas intramolecularmente (Fig. 82.II.).
Também se podem obter eliminações por digestão com uma exonuclease, das
extremidades duma molécula de DNA linear, formada a partir dum DNA circular por uma
endonuclease de restrição que reconhece um sítio único. No exemplo da Fig. 83.II., o
fragmento linear, produzido por digestão com a endonuclease BamHI duma molécula de
DNA circular, é digerido de maneira limitada com a exonuclease Bal31, dando origem a
fragmentos de comprimento decrescente, em função do tempo de digestão e da concentração
da enzima. Estas cadeias de DNA de comprimento variável são de novo circularizadas,
adicionando fragmentos adaptadores sintéticos (linkers) que, após ligação com uma ligase,
criam de novo um sítio BamHI.
Para além de Bal31, que possui actividade nucleásica em 3' e em 5', podem ser utilizadas
outras exonucleases, isoladamente ou em associação, tais como a exonuclease λ (do fago λ),
que remove especificamente os nucleotídeos da extremidade 5' do DNA linear, a exonuclease
III (ExoIII de E. coli) que remove os mononucleotídeos em 5' de DNA bicatenário, mas na

59
direcção de 3' para 5', ou a nuclease S1 que cliva e digere o DNA monocatenário (ganchos ou
extremidades protuberantes).
Inserções
Oligonucleotídeos sintéticos podem ser utilizados como adaptadores e introduzidos em
sítios particulares do DNA clonado. A sequência original deste DNA é assim interrompida e
alterada, podendo-se, por exemplo criar novos sítios de restrição utilizáveis em mutagénese.
A Fig. 84.II. ilustra esta abordagem. Plasmídeos recombinantes são cortados pela
endonuclease DNaseI pancreática, uma vez em média, ao acaso. A adição de adaptadores
sintéticos Xho I, seguida de clivagem com a enzima Xho I e ligação com a T4 DNA ligase
para recircularizar o DNA, cria uma família de plasmídeos portadores de insertos de 8 bases.
Um outro método importante de mutagénese por inserção recorre à propriedade que têm
os transposões de saltar de um loco de DNA cromossómico para outro, por um mecanismo de
recombinação homóloga, que faz intervir sequências nucleotídicas idênticas orientadas em
direcção oposta (vd. I.8., os genes móveis). O transposão Tn5 que contém 5.7 Kb e codifica
para a resistência à kanamicina e/ou à neomicina, é um exemplo, de elemento móvel utilizado
para inserir genes num cromossoma. Esta técnica consiste em inserir o Tn5 numa molécula
de DNA clonada num vector, que é em seguida reintroduzida num DNA cromossómico a fim
de obter mutantes cromossómicos. A Fig. 85.II. ilustra as diferentes etapas deste processo.
O plasmídeo pRK290-nif::Tn5 de 30 Kb contém um gene clonado (nif de Rhizobium
melioti), no qual foi inserido o transposão Tn5. Quando este vector é introduzido por
conjugação em células de R. melioti transfere a região nif para o DNA cromossómico por um
mecanismo de recombinação homóloga. O gene nif cromossómico é substituído pelo gene nif
portador da inserção Tn5, que confere resistência à neomicina ao genoma de R. melioti.
Substituições
Antes de estudar em detalhe mutações pontuais únicas, é por vezes aconselhável examinar as
consequências biológicas de várias mutações numa região dada dum cromossoma. O método
de desaminação química de citidina modificando-a em uridina permite realizar este
objectivo.
Modificação de C em U por desaminação química
Esta técnica tira partido da propriedade que possui o bissulfito de sódio de desaminar
resíduos de citidina em fragmentos de DNA monocatenário. Estes podem ser criados por
corte com enzimas de restrição, seguido de digestão com uma polimerase possuindo

60
actividade exonucleásica. No exemplo da Fig. 86.II. um plasmídeo recombinante é cortado
por SmaI (CCCGGG) e as cadeias do DNA são digeridas de 3' em 5' pelo fragmento de
Klenow da DNA polimerase, em presença de ATP, unicamente. A enzima deter-se-á na
primeira adenosina que encontra.
As citosinas desemparelhadas, situadas nas extremidades protuberantes, são convertidas em
uridinas, por desaminação com o bissulfito de sódio. O preenchimento das extremidades
protuberantes, utilizando agora a actividade polimerásica da DNA polimerase de Klenow,
em presença dos 4 nucleotídeos trifosfato, seguido de ligação das extremidades rombas para
recircularizar o plasmídeo, gera uma molécula contendo, neste exemplo, duas mutações G-C
em A-U.
Existe um outro método para gerar DNA monocatenário, que recorre a fagos
monocatenários, tais como o M13. Qualquer DNA clonado neste vector pode ser obtido na
forma de molécula monocatenária e mutagenisado com bissulfito. A cadeia complementar (-)
é em seguida sintetizada, a partir da cadeia mutagenisada utilizando iniciadores específicos.
A região mutada de interesse, excisada da forma replicativa bicatenária, poderá ser reclonada
num vector não mutagenisado (Fig. 87.II).
Substituição por incorporação incorrecta
Em presença de brometo de etídeo, certas endonucleases de restrição (por exemplo, ClaI,
EcoRI) clivam a ligação fosfodiéster do DNA no sítio de reconhecimento, unicamente numa
das cadeias. Este corte pode ser alargado, sem ruptura da cadeia intacta, por uma polimerase
com actividade exonucleásica. O preenchimento da região monocatenária pela polimerase,
em presença de só 3 dos 4 nucleotídeos trifosfatos, resultará ocasionalmente, na introdução
incorrecta de um nucleotídeo em face da base cujo par complementar foi omitido. No
exemplo da Fig. 88.II. foi omitido o dCTP. Se o mecanismo corrector natural de excisão,
próprio à DNA polimerase não funcionar, o resultado final será uma molécula de DNA
contendo uma substituição de base. O desemparelhamento criado in vitro será reparado na
bactéria, durante a replicação, após transformação pelo plasmídeo. Os plasmídeos não
mutados podem ser identificados pela presença do sítio de restrição alvo do corte inicial
(ClaI, neste exemplo). Os mutados não serão reconhecidos pela endonuclease.
Uma alternativa para superar o mecanismo corrector da DNA polimerase, recorre a
análogos dos nucleotídeos, os nucleotídeos α-tiofosfatos, (Fig. 94.II.) que são substratos da
DNA polimerase mas não da sua actividade exonucleásica 3' → 5'. Uma vez incorporado, o
análogo não pode ser excisado pela enzima, mas a sua extremidade 3' é reconhecida como
iniciador para prolongar a síntese do DNA.

61
Os métodos descritos até agora dependem da presença dum sítio de restrição apropriado,
mais ou menos perto do sítio da mutação. Esta limitação pode ser superada, recorrendo a um
método universal e extremamente específico, que faz intervir oligonucleotídeos sintéticos
como agentes mutagénicos.
Mutagénese dirigida com oligonucleotídeos sintéticos
Este método oferece a possibilidade de mutar uma ou várias bases perfeitamente
definidas e localizadas, substituindo cada uma delas por qualquer outra antecipadamente
escolhida. O oligonucleotídeo sintético de sequência definida é complementar de uma das
cadeias do DNA que se pretende mutar, excepto numa ou várias posições alvo das
substituições mutagénicas. Em condições escolhidas judiciosamente, visando
nomeadamente, estabilizar a hibridação, o híbrido formado entre o oligonucleotídeo e o
DNA circular monocatenário, clonado num vector M13, se bem que imperfeitamente
emparelhado, será suficientemente estável para servir de iniciador numa reacção de
polimerização que levará à síntese da cadeia complementar do fago. A molécula bicatenária
resultante será portadora de um ou vários desemparelhamentos. Uma vez introduzida em
células E. coli competentes, os "erros" serão reparados e dois tipos de moléculas
descendentes serão obtidas: umas possuem a sequência do tipo selvagem, outras contêm as
modificações desejadas. Os fagos cuja cadeia (-) foi só parcialmente sintetizada darão
origem, após transfecção ao tipo selvagem (Fig. 89.II.).
Para distinguir as moléculas modificadas das do tipo selvagem, pode-se recorrer à
clivagem dos sítios de restrição, eventualmente criados, ou suprimidos pelas mutações, ou se
esse não for o caso, à hibridação do oligonucleotídeo sintético marcado radioactivamente
(com 32P por exemplo) com os dois tipos de fagos. A optimização das condições de
hibridação selectiva, nomeadamente a temperatura, permitirá distinguir o DNA mutado do
selvagem.
Os rendimentos geralmente obtidos em fago mutado são baixíssimos. Esta fraca
eficiência de mutação tem várias causas. A eficácia variável da reacção de polimerização é
certamente uma. Como o DNA monocatenário do fago também é infeccioso, se a síntese do
DNA complementar não for quantitativa as moléculas parcialmente monocatenárias, cuja
cadeia intacta é a não mutada, replicar-se-ão, dando origem ao tipo selvagem.
Um outro factor que favorece a ocorrência de clones do tipo selvagem, é a correcção dos
desemparelhamentos na forma replicativa (RF) antes da replicação in vivo. Este fenómeno
explica-se pela actividade 3'-exonucleásica ainda presente no fragmento de Klenow da DNA
polimerase I, que teoricamente pode digerir o iniciador antes do começo da síntese do DNA
in vitro. Mesmo um heterodúplex completo, contendo desemparelhamentos, pode ser
reparado por um mecanismo de excisão dos nucleotídeos desemparelhados. Ora, a cadeia

62
sintetizada in vitro é o alvo privilegiado destas reparações, porque não é metilado, ao
contrário do DNA parental, visto a metilação ocorrer in vivo após a replicação. Com efeito, o
mecanismo de reparação in vivo baseia-se, pelo menos em parte no reconhecimento do nível
de submetilação. A cadeia não metilada é portanto um bom substrato para o mecanismo de
reparação.
Numerosas estratégias foram desenvolvidas para superar estes problemas e elevar o
rendimento em moléculas mutadas, diminuindo de preferência a necessidade de escrutinar ou
purificar laboriosamente os mutantes. Este objectivo é tanto mais importante que a relação
entre o número de clones que é necessário escrutinar para ter uma probabilidade de 90 % de
obter um mutante, e a ineficácia da mutação, é logarítmica (Fig. 90.II.).
Uma das estratégias adoptadas recorre à utilização de DNA parental não metilado que
pode ser facilmente obtido através de estirpes de E. coli mutantes dam- que são deficientes no
mecanismo de metilação. Um inconveniente deste sistema é a elevada frequência de
mutações e de recombinações in vivo.
Foi desenvolvido um outro método que recorre a dois iniciadores oligonucleotídicos. O
segundo iniciador, que não introduz mutações, é dirigido para uma região da cadeia de DNA
monocatenário do fago em 5' do oligonucleotídeo mutagénico. O primeiro iniciador é
prolongado em presença do fragmento de Klenow da DNA polimerase, de DNA ligase e de
nucleotídeos trifosfatos. Este método visa proteger o oligonucleotídeo mutagénico da
actividade exonucleásica 5'→ 3' contaminante, da polimerase. Esta estratégia não impede no
entanto completamente o deslocamento do segundo oligonucleotídeo pela polimerase.
A fim de remediar o problema de reparação dos desemparelhamentos, foi estudado o
sistema do dúplex interrompido (gapped duplex). Este pode ser obtido por hibridação com o
DNA monocatenário dum vector que não possui a sequência clonada. A Fig. 91.II.
esquematiza as diferentes etapas deste método. O vector fágico M13 contém uma mutação
âmbar sem sentido e por consequência, só pode multiplicar-se num hospedeiro supressor de
âmbar. O DNA alvo da mutagénese é clonado neste fago e obtido na sua forma
monocatenária. Ao hibridar este DNA circular, com o DNA linearizado e desnaturado da
forma RF, não âmbar, do mesmo vector sem DNA estrangeiro, forma-se um dúplex parcial
na região do DNA clonado. O oligonucleotídeo mutagénico é dirigido para esta região
monocatenária e prolongado por uma polimerase, em presença dos 4 nucleotídeos trifosfatos.
Após ligação, a cadeia mutante poderá ser seleccionada num hospedeiro não supressor,
porque o vector selvagem não possui a mutação sem sentido. Na prática, a frequência de
mutantes é muito variável e frequentemente baixa. O método tem ainda a desvantagem de
produzir uma descendência não portadora do marcador sem sentido (âmbar), o que obriga a

63
recorrer de novo à clonagem no vector âmbar, se outras mutações forem necessárias no DNA
clonado.
Um sistema engenhoso de selecção, baseado na semelhança das sequências entre os
sítios de restrição EcoK e EcoB, possibilita a execução de vários ciclos de mutagénese com
elevada eficiência. EcoK e EcoB são enzimas de restrição do tipo I, que reconhecem um sítio
com cerca de 15 pb e cortam a uma distância de 1000 a 5000 bases dele:
EcoB 5'...... TGA ..... N8 ..…….TGCT......3'
EcoK 5'.…. GAAC .... N6 .….GTGCT.........3'
Um vector especial foi construído, que contém um sítio único EcoK num inserto
polylinker de 30 pb (vector M13K19 derivado de M13m19, Fig. 92.II.). São utilizados dois
iniciadores; um chamado iniciador de selecção modifica a sequência EcoK em EcoB, e o
outro, o mutagénico modifica a sequência alvo. Os iniciadores são prolongados pelo
fragmento de Klenow em presença dos 4 nucleotídeos trifosfatos e de DNA ligase e a
molécula bicatenária é levada a transformar uma estirpe bacteriana restritiva para EcoK. A
descendência com o sítio EcoK não sobreviverá e a mutada que contém o sítio EcoB e a
mutação de interesse será seleccionada. Esta selecção pode ser repetida com um iniciador de
selecção que modifica o sítio EcoB em EcoK e uma estirpe bacteriana restritiva para EcoB
(Fig. 93.II.). A eficácia pode atingir 70 % de rendimento.
Uma outra estratégia visa a destruição da cadeia (+) não interessante. Ela tira partido de
um hospedeiro especializado, para a multiplicação do fago antes da mutagénese. O vector
fágico monocatenário é multiplicado numa estirpe de E. coli Dut-Ung-. A deficiência no gene
dut, que codifica para a dUTPase leva a uma superprodução de dUTP que será incorporada
no DNA no lugar de dTTP. A deficiência em uraciloglicosidase impede a eliminação dos
resíduos de U do DNA (papel próprio a esta enzima). É assim possível obter um DNA fágico
monocatenário contendo 20 a 30 resíduos de uracilo. Este fago será inactivado por um
hospedeiro Ung+. Utilizada como matriz em mutagénese dirigida, em presença de dTTP, a
cadeia parental contendo os resíduos uracilo será copiada em cadeia mutada, contendo
timidina em vez de uracilo. Multiplicado num hospedeiro Ung+, o fago dará lugar a uma
descendência predominantemente mutada. Devido à multiplicação fraca das estirpes Dut-
Ung- o rendimento em fago é baixo apesar da frequência de mutação ser elevada (+ 80 %).

64
Uma das estratégias mais eficazes baseia-se na utilização e nas propriedades dum
isómero óptico, de um análogo de nucleotídeo trifosfato, o isómero Sp do dCTPαS que
contém um átomo de enxofre, em substituição dum átomo de oxigénio, ligado ao fósforo α
(Fig. 94.II.). Neste método, a cadeia não mutada é eliminada in vitro, sendo seleccionado um
puro homodúplex de DNA mutado. O oligonucleotídeo mutagénico é utilizado como
iniciador para a polimerização com o fragmento de Klenow, em presença de T4 DNA ligase.
O análogo dCTPαS é utilizado no lugar do dCTP e incorporado normalmente, o que permite
a eliminação selectiva da cadeia não mutada (Fig. 95.II.). Com efeito, certas enzimas de
restrição não clivam o DNA nos sítios onde um fosforotioato foi incorporado, mas criam
cortes simples na cadeia nativa. Estes cortes são o ponto de partida para a degradação
enzimática parcial, pela exonuclease III, do DNA não mutado. Este pode em seguida ser
utilizado como matriz para a reconstrução duma molécula bicatenária circular, que será por
consequência um homodúplex mutado. A eficácia da mutação é frequentemente superior a 80
%.
Selecção e identificação dos mutantes
A técnica mais directa para escrutinar os clones interessantes é a sequenciação do DNA
monocatenário purificado de cada clone, utilizando uma única reacção, com o
didesoxinucleotídeo que diferenciará o tipo selvagem dos mutantes. Se a mutação estiver
situada longe no sítio de clonagem pode ser necessário sintetizar um oligonucleotídeo
apropriado para ser utilizado como iniciador.
Se a mutação desejada criar ou destruir um sítio de restrição, a análise electroforética da
forma RF do DNA, preparado in vitro a partir do DNA monocatenário, purificado e cortado
pela enzima que reconhece o sítio, permitirá a identificação dos clones mutados.
O método de hibridação é o mais versátil e possibilita o escrutínio de um grande número
de mutantes potenciais. Utiliza-se directamente o iniciador mutagénico marcado com 32P,
como sonda de hibridação, que pode ser aplicada tanto com colónias bacterianas, como com
lisados de fagos. Um desemparelhamento entre o oligonucleotídeo mutagénico e o DNA
fágico monocatenário destabilizará o híbrido. Um aumento progressivo da temperatura
dissociará selectivamente a sonda marcada dos clones não mutados, mas permite que a sonda
permaneça hibridada com os mutantes de sequência perfeitamente complementar. A reacção
de hibridação e as etapas de lavagem a temperaturas crescentes são realizadas com o DNA
alvo fixo a um filtro de nitrocelulose ou de nylon.

65
Aplicações da mutagénese dirigida
A mutagénese dirigida é um instrumento extremamente poderoso para estudar
fenómenos bioquímicos e biofísicos, tais como o sítio de ligação com os receptores de
proteínas biologicamente activas, o sítio activo de enzimas (identificando os domínios
implicados na interacção com os receptores), porque permite modificar a especificidade das
ligações mediante alterações pontuais específicas dos genes.
O estudo da estrutura e da função das proteínas, assim como a engenharia de proteínas
com o fim de aperfeiçoar as suas propriedades, são sem dúvida áreas de grande aplicação
desta técnica.
Um exemplo é a mutagénese específica pontual do gene do interferão ß humano (IFN-ß).
Este gene foi clonado e abundantemente expresso em E. coli, mas a actividade específica
antiviral da proteína é apenas 1/10 da da proteína nativa glicosilada. O IFN-ß contém três
resíduos de cisteína, localizados nos aminoácidos 17, 31 e 141. Estas cisteínas podem formar
pontes dissulfureto intermoleculares, dando origem a dímeros ou oligómeros inactivos. Por
outro lado, podem reagir ao acaso intramolecularmente levando à formação de três tipos de
moléculas, correspondentes às três ligações dissulfureto intramoleculares possíveis. Só uma
destas formas possuirá a conformação nativa e poderá ter actividade biológica. A substituição
duma destas cisteínas por um outro aminoácido, é uma solução possível para eliminar a
formação de pontes dissulfuretos indesejáveis. Para manter as propriedades biológicas da
proteína é necessário, por um lado, que o aminoácido que substitui uma das cisteínas tenha
um comportamento próximo do do aminoácido original, para não provocar distorções
relativamente à conformação nativa e por outro lado, que a cisteína substituída não esteja
implicada numa ponte dissulfureto na proteína natural. A primeira condição tem grandes
probabilidades de ser satisfeita, escolhendo a serina para aminoácido de substituição, visto só
diferir da cisteína pela natureza dum átomo (um átomo de oxigénio no lugar de um átomo de
enxofre). A decisão de escolher a cisteína 17 para alvo da substituição é baseada na analogia
com o IFN-α que possui quatro cisteínas implicadas em duas pontes dissulfureto, entre as
posições Cys29 - Cys138 e Cys1 - Cys98. Sabe-se por outro lado, que a Cys-141 é essencial
para a actividade do IFN-ß, porque a sua substituição por uma tirosina dá origem a uma
modificação conformacional importante da proteína, que perde a especificidade de se ligar a
anticorpos dirigidos contra o interferão natural e de competir com os receptores do IFN-ß
nativo nas células alvo. A ligação dissulfureto mais provável no IFN-ß é pois entre a Cys-31
e a Cys-141.
A figura 96.II. ilustra a estratégia adoptada para mutagenisar especificamente o gene do
IFN-ß. A substituição pontual foi induzida por um decaheptanucleotídeo G-C-A-A-T-T-T-T-
C-A-G-A-G-T-C-A-G complementar de 16 nucleotídeos da cadeia não codificante do gene.

66
Um simples desemparelhamento na posição 12 do iniciador (um A no lugar dum T) induz a
mutação de cisteína em serina. Este gene clonado em E. coli, produz uma proteína mutada
IFN-ß Ser-17, que possui actividades biológicas, incluindo actividade antiviral, em tudo
comparáveis às da proteína natural, em contraste com a proteína IFN-ß Cis-17 produzida pelo
mesmo hospedeiro. A proteína mutada neutraliza o anti-soro dirigido contra o IFN-ß com a
mesma eficácia que a proteína natural, o que indica que as duas proteínas possuem
determinantes antigénicos comuns que são reconhecidos de maneira idêntica pelo anti-soro.
Os dois interferões são pois imunologicamente semelhantes. A substituição do aminoácido
Cis-17 por uma serina não causa alterações importantes na estrutura da proteína, que forma a
ponte dissulfureto e se enrola correctamente, ao contrário do IFN-ß Cis-17 expresso no
mesmo hospedeiro.
Para além da recuperação da actividade específica, o INF-ß Ser-17 possui uma estabilidade
muito superior à do IFN-ß Cis-17 recombinante produzido pela bactéria, o qual perde
rapidamente a actividade específica.
Este caso concreto aplicado a uma proteína de grande interesse terapêutico ilustra o
potencialidade da mutagénese dirigida em engenharia das proteínas. As aplicações desta
metodologia de mutagénese perfeitamente definida e controlada são numerosas, tanto no
plano teórico de investigação de mecanismos fundamentais, como no plano prático da
produção. Nesta área da investigação aplicada, descrevemos, como exemplo, a mutação no
gene do interferão-ß que modifica uma cisteína em serina. Esta alteração tem como resultado
melhorar o rendimento de produção da proteína activa por E. coli.
A mutagénese dirigida por oligonucleotídeos tem também, sido amplamente utilizada na
análise funcional de genes, no estudo de regiões reguladoras, de operões, de promotores ou
outras sequências de controlo (como por exemplo a elucidação do papel das sequências
consenso presentes nas junções intrão-exão), na elucidação da influência de sequências
específicas na biossíntese de tRNA, no estudo de alterações das propriedades catalíticas de
enzimas, na avaliação da influência da natureza da carga dos aminoácidos das sequências
sinal, no transporte das proteínas através da membrana celular. A aplicação desta técnica à
elucidação dos mecanismos de acção das enzimas e das relações existentes entre a estrutura
primária e as funções biológicas desenvolveu-se rapidamente e tem tido importantes
repercussões.

67
4. A TRANSFORMAÇÃO BACTERIANA
As moléculas de DNA recombinante formadas in vitro por ligação só começam a ter
interesse depois de introduzidas num hospedeiro biológico. De facto, a utilização das novas
moléculas de DNA formadas por engenharia genética depende da sua separação e
propagação. Por isso, é necessário introduzi-las em células (bacterianas, por exemplo) de
maneira a poderem replicar-se e serem preservadas e recuperadas facilmente. Um meio de o
conseguir é recorrer, como vimos, a vectores particulares recombinados com o gene que nos
interessa, para o introduzir num hospedeiro biológico em geral um colibacilo.
Uma série de métodos foi desenvolvida para assegurar a introdução eficaz de DNA no
hospedeiro. Sabe-se que o DNA exógeno pode entrar nas bactérias naturalmente. Estas
passam por um estado de competência que as torna aptas para aceitar o DNA exógeno,
fixando o DNA numa forma resistente às nucleases. O estado de competência pode ser
reproduzido artificialmente pela exposição das bactérias ao cloreto de cálcio antes da adição
do DNA exógeno (plasmídeo recombinante) (Fig. 97.II.)
Para manter o DNA recombinante nas células transformadas e evitar problemas de
degradação pelo sistema hospedeiro, as bactérias receptoras são geralmente tornadas
deficientes para o sistema de restrição (hsdR-) e por vezes também para o sistema de
modificação (hsdM-). A proliferação permite a obtenção de biliões de cópias idênticas do
fragmento clonado.
5. BANCO DE CLONES DE CDNA
Para construir um clone contendo sequências derivadas de um mensageiro de eucariota é
necessário obter, primeiramente, esta sequência na forma de DNA. A cópia DNA de um
RNA mensageiro chama-se DNA complementar (cDNA). O termo clone de cDNA descreve
uma bactéria portadora de um plasmídeo que contém a cópia DNA de uma molécula de
RNA.
Uma célula de eucariota comum contém vários milhares de sequências de RNA
mensageiros diferentes. Um banco de clones de cDNA, para ser representativo, deve conter
um número suficiente de transformantes distintos, para que cada RNA mensageiro esteja
representado pelo menos uma vez na população bacteriana. O número de transformantes
distintos a obter para ter uma elevada probabilidade de identificar o clone que se procura
varia entre +5.000 e +100.000, segundo a abundância relativa dum mensageiro na célula.

68
Esquema de construção de um banco de clones de cDNA
a) Preparação do DNA complementar bicatenário a partir de RNA poli (A)+ (Fig. 98.II.)
A partir de células pertencentes a um órgão apropriado isolam-se os RNA totais. Estes
são passados numa coluna de oligo-dT celulose de maneira a reter especificamente os RNA
poli (A)+ (hibridação A-T). Um iniciador oligo-dT é hibridado com os RNA poli (A)
+ para
iniciar a transcrição reversa em DNA complementar monocatenário, utilizando uma
transcritase reversa isolada do vírus aviano de mieloblastose. Para a síntese da segunda
cadeia do cDNA, pode utilizar-se a transcritase reversa, o fragmento de Klenow da
polimerase I de E. coli (enzima de Kornberg) ou a T4DNA polimerase. Estas últimas duas
enzimas não possuem actividade exonucleásica 5'→3' própria da DNA polimerase I e não
degradam o DNA bicatenário à medida que ele é sintetizado. Tanto a enzima de Kornberg
como a DNA polimerase de T4 requerem um iniciador para a extremidade hidroxílica em 3'.
Este iniciador pode criar-se espontaneamente, após a terminação da cadeia monocatenária do
cDNA, em cuja extremidade se forma um gancho (hairpin), que serve de iniciador para a
síntese da segunda cadeia de DNA pelo fragmento de Klenow da DNA polimerase I (ou a
trancritase reversa ou a T4 DNA polimerase). Este gancho pode ser depois clivado pela
nuclease S1, de maneira a produzir um DNA complementar bicatenário linear.
Também é possível prolongar a extremidade 3' do cDNA com uma cauda
homopolimérica. Neste caso, a síntese da segunda cadeia pode ser iniciada utilizando um
iniciador homopolimérico.
Uma etapa crítica na síntese do cDNA é o tratamento com a S1 nuclease. Existe um
processo alternativo, pelo qual o RNA do híbrido RNA-DNA obtido após a síntese da
primeira cadeia é retirado, não por um tratamento alcalino, mas antes por um tratamento
combinado RNAase H/DNA polimerase I de E. coli. A RNAase H (H de híbrido), que ataca
especificamente a cadeia de RNA do híbrido RNA-DNA, introduz um certo número de cortes
(nicks), i. é., extremidades 3'-hidroxílicas na cadeia de RNA, que servem de iniciadores para
a reacção de nick translation catalisada pela DNA polimerase I.
Observações:
1) A origem da região do gancho em 3' da primeira cadeia do cDNA não está bem
elucidada. É possível que muitos mensageiros tenham em 5' uma região complementar que
conduza à formação deste gancho.

69
2) Se o RNA não possui a região poli(A)+, esta pode ser adicionada in vitro à
extremidade 3' pela enzima poli(A) polimerase, para ser depois utilizada com o iniciador
oligo-dT.
3) Se a sequência nucleotídica do RNA mensageiro for conhecida, pode-se utilizar,
para a síntese do DNA complementar, um iniciador oligonucleotídico sintético complementar
da extremidade 3'do RNA mensageiro.
b) Inserção do cDNA num plasmídeo - Transformação das bactérias
Quando se dispõe dum DNA complementar bicatenário, é preciso em seguida inseri-lo
num vector destinado a transformar o hospedeiro bacteriano. Para isso, adicionam-se
extremidades coesivas às moléculas de cDNA bicatenário, segundo um dos esquemas
seguintes :
1) Adição de adaptadores sintéticos que geram sítios de restrição (Fig. 99.II.).
2) Adição de extremidades homopoliméricas complementares (Fig. 100.II.).
No termo destas manipulações dispõe-se de um banco de clones de cDNA.

70
6. BANCOS GENÓMICOS
Um banco de cDNA de um órgão, ou de uma dada população de células, é representativo
da população de genes expressos nestas células. Na célula, só alguns milhares, entre centenas
de milhares de genes dos cromossomas, são transcritos em mRNA. Um banco representativo
do mRNA destas células contém forçosamente menos clones diferentes que um banco
representativo da totalidade dos genes (banco genómico), o que simplifica a procura de um
gene.
Um banco genómico é uma colecção de clones plasmídicos ou de lisados fágicos, que
contém moléculas de DNA recombinante. A totalidade dos insertos individuais de DNA
representa a informação genética completa dum organismo.
A estratégia geral para a construção de bancos genómicos requer primeiro o isolamento
de fragmentos de DNA do organismo dado. Estes fragmentos têm, evidentemente, que
representar o genoma inteiro e são derivados do DNA genómico total por digestão enzimática
ou ruptura mecânica suave. Estes fragmentos são em seguida introduzidos num vector ad hoc
do tipo λ, ou cosmídeo, afim de assegurar a sua propagação (Fig. 101.II.).
O número mínimo de clones necessários para se obter a representação do genoma inteiro
dum organismo pode ser calculado. A probabilidade P de se obter um gene específico numa
colecção de clones depende do comprimento dos insertos de DNA e da complexidade do
genoma. Quanto maior e mais complexo for o genoma, maior é o número de clones de um
tamanho específico necessário para se obter uma representação completa da informação
genética. Este número N de clones pode ser determinado utilizando a fórmula :
)1ln(
)1ln(
f
PN
−
−=
em que P é a probabilidade de se obter uma sequência particular e f é a relação entre o
comprimento do inserto e a totalidade do genoma.
A tabela 3.II. fornece as valores de N para um inserto de 20 kb e para as probabilidades
de 90 % e de 99 % de se obter um clone particular. Estes valores devem ser considerados
como uma estimativa por defeito, porque foram obtidos fazendo a hipótese que cada
molécula híbrida têm uma capacidade de transformação semelhante e que cada colónia
representa uma ocorrência de transformação independente.
A tabela 4.II. fornece o número de clones teoricamente necessários para que o genoma
apareça representado pelo menos uma vez, em função dos tamanhos dos fragmentos clonados
e da origem do genoma. No entanto, este número mínimo, só representa estatisticamente uma

71
possibilidade em duas de se encontrar o clone alvo. Em geral, é necessário escrutinar de 3 a
10 vezes o número mínimo de clones, para se ter uma probabilidade razoável de identificar
um gene particular.
O esquema de clonagem no DNA de fago λ e o empacotamento nos invólucros fágicos
está ilustrado na Figura 102.II. A eficácia deste tipo de sistema é muito elevada; obtêm-se da
ordem de 108 pfu/µg de DNA (pfu = plaque forming units).
Em princípio, qualquer gene para o qual existe um sistema de detecção apropriado, por
exemplo, uma sonda de hibridação, pode ser isolado de um banco genómico, contanto que o
banco satisfaça os critérios mencionados e seja realmente representativo. Se o banco
genómico contiver fragmentos de DNA obtidos por digestão parcial, estes fragmentos
apresentarão sobreposições e o banco oferecerá a possibilidade de examinar a estrutura, não
apenas de genes individuais, mas também de sequências vizinhas. Este tipo de análise é
conhecido por chromosome walking (Secção 1.1.b.8).
7. MÉTODOS DE DETECÇÃO E DE ANÁLISE DE CLONES
Várias técnicas são utilizadas para detectar, entre os clones dum banco, os que
comportam a sequência de DNA desejada.
a) Hibridação in situ entre o DNA dos clones recombinantes e uma sonda de DNA
radioactiva (Fig. 103.II.)
A sonda radioactiva pode ser um cDNA purificado, um RNA mensageiro purificado ou
um oligonucleotídeo sintético. Estas sondas são marcadas in vitro com 32P por nick
translation (cDNA) ou por quinação (oligonucleotídeo de síntese, RNA poli (A)+).
b) Carta de restrição
Os clones positivos por hibridação podem ser analisados por restrição. A carta de
restrição é em seguida comparada com uma carta conhecida ou teórica (derivada da
sequência em aminoácidos da proteína cujo DNA foi clonado).
c) Sequenciação dos nucleotídeos do inserto nos clones positivos por hibridação
d) Selecção do mensageiro específico por hibridação com o DNA recombinante e tradução
in vitro
Neste método, o DNA plasmídico dos clones é isolado e em seguida fixo num filtro de
nitrocelulose. Em condições apropriadas de temperatura e de força iónica, hibrida-se com o
RNA mensageiro total que serviu para a síntese do DNA complementar. Só o RNA
mensageiro, que possui sequências complementares das do DNA imobilizado no suporte,

72
permanecerá associado a este. Pode-se assim isolar este RNA mensageiro, extrai-lo do
suporte e traduzi-lo num sistema de tradução in vitro. O produto codificado pelo RNA
mensageiro é em seguida identificado por imunoprecipitação com um anticorpo dirigido
contra a proteína cujo mensageiro foi clonado (Fig. 104.II.).
e) Southern blotting
Para localizar sequências particulares de DNA nos fragmentos de restrição, utiliza-se o
método desenvolvido por Southern (Fig. 105.II.). O DNA é clivado em fragmentos, que são
separados em gel de agarose e em seguida transferidos para uma folha de nitrato de celulose.
O filtro é depois hibridado com uma sonda radioactiva apropriada. A autorradiografia da
folha de nitrocelulose, depois de lavada, revela fragmentos contendo sequências
complementares das da sonda utilizada.
Uma técnica análoga existe para a caracterização do RNA (chamada neste caso Northern
blotting): os RNA totais ou mensageiros são separados em gel de agarose e depois
transferidos para um suporte apropriado (nitrocelulose, papel activado, ...). A hibridação com
uma sonda marcada permite em seguida conhecer o número e o comprimento dos RNA
mensageiros complementares da sonda.
f) Escolha das sondas de DNA sintéticas
Oligonucleotídeos sintéticos, escolhidos judiciosamente podem ser utilizados para escrutinar
um banco de clones por hibridação, ou para isolar um DNA complementar pouco abundante,
contanto que se conheça, pelo menos, uma parte da sequência dos aminoácidos da proteína,
cujo gene se deseja clonar. Uma sequência de alguns aminoácidos é suficiente para deduzir
todas as sequências do RNA que podem codificar para este péptido. Com efeito, se só a
sequência proteica for acessível, a estratégia na escolha dos oligonucleotídeos a utilizar para
escrutinar os bancos é menos directa. O conhecimento da sequência da proteína não permite
deduzir uma sequência de DNA única, devido à degenerescência do código genético. É, no
entanto, possível sintetizar todas as sequências nucleotídicas correspondentes.
Uma das estratégias consiste em escolher, como sequência alvo da sonda
oligonucleotídica, uma região correspondente a uma sequência de aminoácidos pertencendo
aos menos degenerados. A Fig. 106.II. exemplifica a aplicação desta estratégia à detecção de
clones de cDNA da uroquinase humana.
Não é só a complementaridade perfeita da sonda que é crítica para obter uma boa
hibridação; o seu comprimento é igualmente importante. Quanto maior for a complexidade
do DNA alvo, maior é a probabilidade de nele existir uma sequência qualquer dada, escolhida
ao acaso. 4n é o número total de sequências diferentes possíveis, constituídas pelos 4
nucleotídeos e com o comprimento de n nucleotídeos. A frequência com que uma sequência

73
dada de n nucleotídeos, considerada ao acaso, é susceptível de se encontrar num genoma de
3.5 109pb, como é o genoma humano, é dada pela equação:
n4
510.3 9
Esta frequência é pois, de 1/5 para uma sequência definida considerada ao acaso de 17
nucleotídeos. Para maior segurança e eliminar problemas de barulho de fundo, escolheu-se no
exemplo dado n = 23.
É corrente utilizar uma mistura de fragmentos sintéticos marcados, formada de todas as
combinações correspondentes à degenerescência, para escrutinar um banco de clones. Em
condições adequadas, só um oligómero correspondente a uma sequência clonada se hibridará
com esta e dará um sinal após autorradiografia. No exemplo da figura foi sintetizada uma
mistura de 16 oligonucleotídeos, desprezando os codões menos prováveis, afim de manter
uma concentração elevada do oligonucleotídeo perfeitamente complementar da sequência
alvo. Com efeito, quanto maior for a ambiguidade ao nível da utilização dos codões, mais
difícil é a utilização da mistura dos oligonucleotídeos de grande comprimento, porque o
número de possibilidades aumenta rapidamente e a concentração do oligonucleotídeo único,
que corresponde à verdadeira sequência clonada alvo, diminui proporcionalmente.
Neste contexto, uma outra estratégia, para evitar a síntese de misturas complexas e um
grande número de desemparelhamentos entre a sonda sintética e o DNA clonado, consiste em
utilizar a desoxiinosina para substituir todas as bases ambíguas. Este análogo tem um
comportamento aproximadamente neutro no emparelhamento entre as moléculas de DNA e
não destabiliza, ou destabiliza pouco, os híbridos.
desoxiinosina
O N
N
NNHO
OH
O
H
A especificidade de uma pequena sonda sintética de cerca de 20 nucleotídeos é notável.
Um único desemparelhamento no interior da região de emparelhamento, entre a sonda e a
sequência alvo, é suficientemente destabilizador do híbrido para provocar a sua dissociação,
em condições de temperatura e concentração salina que mantêm o híbrido perfeito estável.
Uma série de regras, relativamente empíricas, determinam a escolha e as condições de
utilização óptimas duma sonda oligonucleotídica. Nomeadamente, a temperatura de

74
hibridação deve ser determinada judiciosamente. Quanto mais baixa for a temperatura maior
será a probabilidade de o oligonucleotídeo se hibridar, não só com a sequência pretendida,
mas também com sequências próximas; por outro lado, a temperatura não pode ser
incrementada indefinidamente, dado que a eficiência da hibridação diminui com o aumento
da temperatura e só é de 50% à temperatura de dissociação (Td) do DNA. A regra empírica
seguinte permite calcular a temperatura de dissociação de híbridos formados com
oligonucleotídeos de comprimento não superior a 18 nucleotídeos:
CxCGxTATd º4)(2)( +++=
Esta temperatura aumenta 4°C por cada par de bases G:C e 2°C por cada par T:A hibridados
entre o oligonucleotídeo e o DNA alvo, em condições de concentração salina correntes
(6xSSC: NaCl 0.9 M, citrato de Na 50 mM, pH 7).
Para sequências mais compridas é conveniente utilizar a fórmula seguinte, válida para
oligonucleotídeos de comprimento compreendido entre 14 e 60-70 nucleotídeos:
[ ] )/600()(%41.0(log6.165.81 NCGNaTd −++−= +
em que N é o número de nucleotídeos. Na prática, quando existem desemparelhamentos
prováveis subtrai-se 1°C à temperatura calculada, por cada 1% de desemparelhamento e
efectuam-se as primeiras lavagens pós-hibridação a 1-10°C abaixo da Td calculada.
É todavia possível adoptar condições em que a temperatura de dissociação é
independente da proporção de G:C. Este efeito obtém-se utilizando uma concentração 3M de
cloreto de tetrametilamónio (ou de tetraetilamónio) em vez de NaCl, o qual se liga
selectivamente às sequências AT, aumentando a Td do DNA bicatenário (a temperatura de
dissociação de A:T passa a ser igual à de G:C). A Tabela 5.II fornece a Td dos híbridos de
DNA em função do comprimento e da composição das sondas, nas condições mencionadas.

75
8. A SÍNTESE QUÍMICA DE GENES
Uma alternativa à clonagem de cDNA, ou de um gene a partir de fragmentos do DNA
genómico, é a síntese total do DNA correspondente a uma proteína cuja sequência é
conhecida. Esta abordagem é viável graças ao refinamento que atingiu a química de síntese
dos oligonucleotídeos, tanto no que respeita aos rendimentos, como à pureza dos produtos e
à rapidez da sua obtenção, devido nomeadamente aos progressos realizados no campo do
automatismo (vd. Secção 1.1.c.).
No entanto, dada a impossibilidade de as reacções químicas atingirem repetidamente
rendimentos de 100 %, existe um limite ao comprimento dos fragmentos de DNA que é
possível sintetizar quimicamente. A síntese total de um gene realiza-se, por consequência,
por complementação de meios químicos e de meios enzimáticos.
Actualmente, é possível obter, em condições optimizadas de síntese, fragmentos de uma
centena de desoxinucleotídeos, com um grau de pureza aceitável. O exemplo das Fig. 107.II,
108.II e 109.II ilustra a síntese do gene do hGRF (human Growth Hormone Releasing Factor,
factor de libertação da hormona humana de crescimento), descrita em 1985 e realizada por
métodos manuais. O princípio da síntese do gene permanece válido.
O hGRF é uma pequena proteína de 44 aminoácidos, de cuja sequência foi deduzida a
sequência nucleotídica das duas cadeias do gene. Foram incluídos nos fragmentos sintéticos,
o codão de fim de tradução e um sítio Sal I, que servirá para a clonagem do gene num
plasmídeo linearizado pela mesma enzima.
Os fragmentos a sintetizar foram definidos de maneira a que cada um deles seja
complementado por hibridação com dois outros, conduzindo, após a sua mistura em solução,
a uma estrutura correspondente à sequência do gene, mas apresentando ainda nicks entre 2
fragmentos consecutivos. O fecho destes nicks realiza-se com uma ligase, que estabelece
enzimáticamente a ligação covalente entre o fosfato em 5' e o HO em 3', de dois
oligonucleotídeos consecutivos, mantidos nas suas posições correctas por hibridação entre
bases complementares (Fig. 108.II.). A figura ilustra esquematicamente as diferentes etapas
no sentido antitético, i.é., por ordem inversa à da realização, na prática, da síntese do gene.
No sentido sintético, cada um dos fragmentos purificados, depois de síntese, (excepto os
que correspondem às extremidades 5' da construção final) é fosforilado enzimáticamente. Os
fragmentos são em seguida misturados em condições que favorecem a sua hibridação
correcta (annealing), e ligados. O fragmento A31 na extremidade 5' do gene possui uma
extremidade coesiva própria para entrar num sitio de restrição NcoI (Fig. 109.II). É uma
variante da construção indicada na Fig. 107.II (comparar com A27), que ilustra a

76
flexibilidade introduzida pela possibilidade de sintetizar fragmentos de DNA de sequência
definida. Os oligonucleotídeos nas extremidades da construção contêm, não só as sequências
próprias ao gene, mas também sequências correspondentes a sítios de restrição úteis,
funcionando, portanto, igualmente como adaptadores, na construção.
A clonagem deste gene exemplifica outras aplicações dos oligonucleotídeos sintéticos.
Afim de expressar este gene sintético numa bactéria, este é inserido num plasmídeo, construído a partir do vector pCQV2 (Fig. 110.II), no qual o promotor PR de λ foi substituído
pelo promotor PL de λ, mais eficiente. Este foi introduzido por ligação de fragmentos
sintéticos, contendo a sequência do PLλ, com o plasmídeo em que foi suprimida a sequência PRλ. Os fragmentos sintéticos comportam, além das sequências do PLλ, sequências
correspondentes ao sítio de fixação do ribossoma (RBS-"Ribosoma Binding Site") e a sítios
múltiplos de restrição, potencialmente úteis para outras construções eventuais. A linearização
do plasmídeo, digerido pelas enzimas NcoI e SalI, expõe as extremidades coesivas às
extremidades complementares do gene sintético. Em presença de um excesso deste último, o
vector readquire a sua forma circular, ao hibridar as suas extremidades complementares e
enfim, a acção enzimática da ligase estabelece as ligações covalentes. Após transformação
bacteriana com a mistura de ligação, as colónias transformadas com o plasmídeo que contém
o inserto sintético, podem ser seleccionadas por hibridação com um dos oligonucleotídeos
marcados que serviram para a construção do gene. A visualização, como já vimos, é baseada
na marcação radioactiva do oligonucleotídeo, por fosforilação enzimática com uma quinase e
γ32P-ATP. O oligonucleotídeo marcado hibridar-se-á com a sua sequência complementar, no
inserto e só as colónias transformadas pelo plasmídeo contendo o hGRF e imobilizadas num
filtro de nylon ou de nitrocelulose emitirão um sinal radioactivo.

77
9. SISTEMAS DE EXPRESSÃO: ORGANISMOS HOSPEDEIROS - VECTORES
Um certo número de produtos biológicos não pode ser obtido em quantidade suficiente
para permitir um estudo pormenorizado da sua estrutura e da sua função. Clonar o DNA
correspondente a estes produtos não basta, é também preciso poder expressá-los em
abundância. Como vimos, um gene não é unicamente uma sequência de DNA codificando
para uma sequência de aminoácidos, mas comporta ainda um conjunto de sinais moleculares,
que diferem de um organismo para outro (idênticos ou parecidos, por exemplo, entre
mamíferos, mas diferentes na levedura ou no colibacilo). Estas diferenças têm que ser
consideradas para optimizar a expressão, tanto para fins de produção como de investigação.
Além disso, os vectores possuem a sua própria coerência genética e não são capazes de
se multiplicar em qualquer hospedeiro (são-no em geral só no hospedeiro de origem).
a. Expressão em E. coli
Vectores de expressão em E. coli
O colibacilo é o hospedeiro mais utilizado, tirando a sua supremacia das vantagens
inerentes à sua genética relativamente bem conhecida, à facilidade de ser cultivado,
recuperado e lisado; tem sido utilizado para a produção em massa de algumas proteínas.
Exemplos de vectores de expressão muito utilizados em E. coli são os plasmídeos
multicópias, comportando um promotor forte de origem bacteriana ou fágica (trp, lac, lpp, PR
ou PL de λ). A existência deste promotor forte assegura a transcrição eficaz do gene clonado
a jusante, mas não garante necessariamente a sua expressão eficaz. É, evidentemente,
necessário que os sinais moleculares apropriados estejam presentes: o promotor que indica o
local onde a cópia do gene em mRNA começa, a sequência de Shine-Dalgarno, sítio de
fixação reconhecido pelos ribossomas, o local da iniciação da tradução do mRNA em
proteína, os sinais de fim de transcrição e de tradução.
Além disso, é preciso conceber o sistema de tal maneira que o produto resultante da
expressão do DNA clonado seja autêntico, e não na forma de fusão com um fragmento
proteico codificado pelo plasmídeo.
É igualmente necessário evitar a presença e o efeito de sinais de terminação fortuitos da
transcrição (efeitos polares), assegurar-se da estabilidade do RNA mensageiro resultante da
transcrição do gene clonado, assim como da proteína sintetizada e enfim, do efeito letal
potencial do produto superexpresso, no crescimento do organismo hospedeiro.
O vector de expressão ideal deve teoricamente possuir as seguintes propriedades:

78
1. Ser estável, para não se perder nem modificar o gene estrangeiro.
2. Estar representado em número elevado e constante em cada célula, para aumentar o
número de cópias do gene, susceptíveis de serem traduzidas em proteínas.
3. Poder funcionar como vector vaivém (shuttle), quer dizer poder multiplicar-se em mais
de um tipo de hospedeiro.
4. Possuir genes marcadores de selecção, como por exemplo de resistência aos antibióticos.
5. Possuir um sistema de expressão potente (promotor forte), que permita uma produção
abundante de proteína.
6. Não possuir sequências terminadoras que interrompam prematuramente a transcrição.
7. Ser concebido de maneira que se possa inserir nele, facilmente e correctamente (em fase
de leitura), o cDNA ou o gene que se deseja expressar.
8. Possuir as instruções moleculares, as sequências de DNA que permitam a síntese de uma
proteína que seja segregada para fora da célula, para facilitar o seu isolamento e purificação.
Esta última propriedade é proporcionada por uma curta sequência de aminoácidos, que
precede a sequência da proteína propriamente dita, a sequência sinal. Ela orienta a síntese da
proteína nascente para a secreção para fora da célula. Esta sequência é clivada, perdida,
durante a passagem da proteína através da membrana.
A obtenção da proteína no meio extracelular têm vantagens na produção industrial,
porque facilita a sua purificação, já que não fica perdida no meio da massa de proteínas
celulares. Por outro lado, uma toxicidade eventual da proteína em relação ao hospedeiro será
reduzida. Enfim, mesmo pequenos péptidos que seriam normalmente destruídos no seio da
célula, podem ser produzidos.
O vector de expressão ideal não existe. Para optimizar a expressão de um gene e o
rendimento de obtenção duma proteína particular, é necessário dispor de um bom sistema
expressão-hospedeiro que não é forçosamente universal.
1) Construção de vectores de expressãobaseados no promotor lac
O DNA que se pretende expressar (um cDNA por exemplo) é removido do plasmídeo de
clonagem original por digestão enzimática (por exemplo, PstI, Fig 111.II). Um local de
restrição é em seguida colocado na proximidade do codão de iniciação ATG. O DNA é
primeiramente submetido à acção combinada das enzimas ExoIII e S1, ou à de Bal31. As
condições de digestão dependem da distância entre o local de clivagem inicial (neste exemplo

79
PstI) e o codão ATG, e devem ser ajustadas separadamente para cada caso particular. O
fragmento de DNA é em seguida ligado com linkers, neste exemplo EcoRI, de maneira a
poder ser inserido no local EcoRI de um vector adequado. Antes da clonagem, o fragmento é
digerido por EcoRI e outra enzima de restrição que cliva no local X, no interior do gene.
Obtêm-se assim fragmentos com uma extremidade direita definida, que pode ser utilizada
para mais tarde reconstruir o gene inteiro. Se bem que a extremidade esquerda seja definida
pelo local EcoRI, a distância entre este local e o codão de iniciação ATG varia duma
molécula para outra. A ligação a um vector adequado dará origem a uma gama larga de
clones diferentes, nos quais a distância entre o local EcoRI e ATG varia. Certos insertos
poderão não estar na fase de leitura correcta. Os clones na fase correcta de leitura poderão,
todavia, ser identificados, explorando o fenómeno da complementação α (vd. Secção
1.1.b.2). Para isso, como no caso da clonagem em M13, foram construídos vários plasmídeos
da série pUC (Fig. 112.II). Estes plasmídeos possuem a região de regulação lac e uma parte
do gene lac Z que codifica os 59 aminoácidos N-terminais da ß-galactosidase. A estirpe
hospedeira (JM 83) possui a eliminação M15 do operão lac, correspondente à remoção dos
aminoácidos 11-44 da ß-galactosidase, mas conserva a parte C-terminal completa da enzima.
Todos os genes lac Z incompletos expressarão um polipéptido inactivo. Todavia, estes
polipéptidos poderão complementar a parte C terminal da enzima produzida pela bactéria, na
forma de agregados. A actividade enzimática resultante poderá ser detectada em placas com
indicador X-gal. Os plasmídeos pUC 7, 8, 9, 12, 13, 18 e 19 contêm polilinkers no interior da
região do gene lac Z, correspondente aos 59 aminoácidos N-terminais. Estes polilinkers
possibilitam a clonagem de fragmentos de DNA comportando diferentes extremidades. Os
polilinkers não interferem com a complementação α, se a fase de leitura correcta for
preservada.
Quando os fragmentos de cDNA de diferentes comprimentos, descritos acima (Fig.
111.II), são inseridos no polilinker dum plasmídeo pUC, unicamente os clones possuindo
inserções na fase de leitura correcta, relativamente à ß-galactosidase, originarão colónias
azuis. O comprimento máximo dum inserto, que dá ainda lugar à complementação α, não é
conhecido; é, pois, preferível clonar fragmentos de DNA relativamente curtos. A intensidade
do azul dos diferentes clones é bastante variável; os de coloração mais intensa apresentam um
nível de expressão mais elevado e podem ser seleccionados para receberem a parte do gene
que falta, para a produção da proteína completa fusionada. As proteínas fusionadas, com
longas sequências peptídicas que incluem a parte N-terminal da ß-galactosidase (≈ 600
aminoácidos), são, em geral, insolúveis no interior da célula bacteriana, o que as protege da
degradação proteolítica e pode facilitar a purificação. É, geralmente, possível detectar
determinantes antigénicos na proteína fusionada. No entanto, a proteína pretendida deve
poder ser separada da componente bacteriana, do produto quimérico fusionado. A clivagem

80
com o brometo de cianogénio (vd. mais abaixo) é limitada às proteínas que, tais como a
proinsulina, não possuem metioninas internas. Caso contrário, é necessário recorrer à
clivagem enzimática, que exige construções comportando codões codificando sequências de
aminoácidos específicos (p. ex., Arg e Lys, típicos da clivagem tríptica, i. é., pela tripsina),
sensíveis a proteases, que evidentemente não podem estar presentes na sequência da proteína
fusionada. Duma maneira geral, este sistema de expressão de proteína fusionada é útil para a
produção de pequenas proteínas e de péptidos.
Para produzir directamente proteínas não fusionadas, a síntese proteica deve ser iniciada,
não na primeira metionina do péptido guia procariótico, tal como lac Z ou trp E, mas na
primeira metionina do próprio péptido pretendido. As construções biologicamente activas
possuem pois, em geral, promotores procarióticos indutíveis e um sítio de fixação
ribossómico híbrido, constituído pela sequência bacteriana de Shine-Dalgarno e por um
codão ATG, correctamente distanciado, que não tem que ser necessariamente de origem
bacteriana. No caso duma proteína eucariótica, este ATG pode constituir o próprio codão de
iniciação do gene eucariótico.
2) Vectores baseados no promotor PL de λ
O plasmídeo pAS1, representado na Fig. 113.II, deriva do plasmídeo pBR322 e comporta
sinais de regulação derivados do fago λ. O promotor PLλ foi escolhido em particular, porque
a eficácia com a qual ele permite a transcrição parece superior à da maioria dos promotores
bacterianos (lac, por exemplo).
A fim de poderem controlar a transcrição iniciada a partir de PLλ, os inventores do vector
introduziram um sistema de repressão na bactéria hospedeira, cujo genoma comporta um
profago λ contendo um repressor cI termo-lábil, cI857.
Nas bactérias lisogénicas, a transcrição a partir de PLλ é inibida pelo repressor cI, em
contínuo, mas unicamente a 30°C, i. é., a temperaturas em que o repressor é termo-estável.
Um simples aumento da temperatura de crescimento das bactérias (32°C→ 42°C) desencadeia a
transcrição a partir de PLλ, visto que, nestas condições, o repressor é desactivado e deixa de
bloquear o promotor.
Na prática, as bactérias portadoras do vector pAS1 podem ser cultivadas em massa a
30°C, sem que haja expressão do gene clonado a jusante do promotor, e serem em seguida
induzidas a 42°C, para sintetizar o produto do gene. Esta possibilidade de controlar a
expressão do gene clonado no vector, e de o poder induzir rapidamente, permite obter uma
super-expressão dos produtos clonados num prazo curtíssimo. Estas propriedades são
particularmente importantes para a expressão de produtos que poderiam ser tóxicos para o

81
hospedeiro no qual eles são sintetizados, e/ou cuja estabilidade no hospedeiro é pouco
elevada.
Além disso, o vector pAS1 é concebido de maneira a assegurar uma transcrição eficaz,
por intermédio de PLλ, através de qualquer sequência de DNA clonada a jusante. Para isso, o
vector comporta, a jusante de PLλ, os sítios nut (utilização de N, sítio de reconhecimento
necessário para a acção de N) e a bactéria lisogénica fornece o produto N (proteína
antiterminadora).
A expressão de N, no hospedeiro lisogénico, suprime a polaridade transcricional,
permitindo à RNA polimerase ler através de tL e tR (terminadores esquerdo e direito) e
impede, por consequência, qualquer paragem fortuita da transcrição, na unidade de
transcrição PLλ e na sequência de DNA clonada a jusante. É de notar, que um terminador
natural tR1 de λ, derivado da construção, existe no vector e que o seu efeito é contrariado por
N e nutR.
Por fim, o vector pAS1 comporta os sinais de regulação da tradução, necessários à
expressão de sequências de DNA que não possuam estas informações. Para este efeito, no
gene cII de λ, gene expresso eficazmente, foi suprimida a sua sequência codificante, de
maneira a manter unicamente a região de Shine-Dalgarno e o triplet de iniciação.
Imediatamente adjacente ao triplet ATG, foi introduzido um local de restrição único, prático
(BamH1), que permite a fusão directa de qualquer sequência codificante, a jusante do ATG, e
a obtenção duma junção na fase correcta de leitura, mediante adaptações para cada caso
particular.
Em certos casos, a utilização do vector de expressão pAS1 conduz à produção de 10 a 20
% da proteína desejada, em relação às proteínas totais.
3) λgt11 - vector de expressão derivado do fago λ
Este vector, representado na Fig. 114.II., comporta um local de restrição único, EcoRI,
que permite a inserção de DNA exógeno (possuindo extremidades EcoRI naturais ou
adicionadas) no gene da ß-galactosidase (lacZ). Esta inserção conduz à síntese de proteínas
híbridas (em fusão com a ß-galactosidase), sob a acção do promotor lac, em presença do
indutor IPTG (indutor gratuito que desactiva o repressor, impedindo-o de se fixar ao operador
de Z-ßlac, Y-permease e A-transacetilase, ao mesmo título que o indutor natural, a lactose,
mas sem ser consumido pela enzima; vd. Secção 1.1.b.2). Como a proteína fusionada perde a
actividade ß-galactosidásica, é possível, em presença de Xgal, distinguir os fagos
recombinantes (placas brancas) dos fagos não recombinantes (placas azuis com indicador
Xgal).

82
O vector comporta ainda o repressor cI termo-lábil (cI857) e uma mutação sem sentido
no gene S de λ, que torna a lise defectiva (na ausência de supressão pelo hospedeiro, supF).
Por consequência, as bactérias lisogénicas podem ser induzidas a 42°C, para acumularem
grandes quantidades de produtos fágicos, sem que haja lise. Esta pode ser efectuada expondo
as colónias a vapores de clorofórmio.
Por outro lado, o hospedeiro bacteriano possui o repressor do gene lac Z, que impede a
expressão da proteína fusionada, durante as primeiras horas do crescimento. É só no
momento apropriado, que se desactiva este repressor com o indutor IPTG, levando à
produção maciça da proteína fusionada.
A eficácia de clonagem do sistema λgt11 é de + 107 pfu/µg de DNA.
O escrutínio dos fagos recombinantes pode realizar-se de duas maneiras:
1) por hibridação com uma sonda de DNA marcada;
2) por detecção imunológica.
A expressão correcta do DNA clonado no λgt11, depende da orientação e da fase de
leitura do inserto em relação às do gene lac Z.
Identificação das placas produtoras
Um dos métodos de detecção das proteínas produzidas pelo fago recombinante baseia-se
na interacção dessas proteínas com anticorpos dirigidos contra elas. Na prática, reveste-se um
suporte sólido, inerte, com esses anticorpos e em seguida expõe-se esta matriz às colónias
(placas), lisadas in situ. Os sítios de fixação do antigénio podem ser detectados por incubação
com o anticorpo marcado (com 125I ou com peroxidase, por exemplo). A autorradiografia do
suporte sólido, ou a reacção corada com um substrato cromogénico da peroxidase, revela as
colónias produtoras do antigénio que se procura (Fig. 115.II.).
Alternativamente, o método de western blotting permite identificar o antigénio produzido
no seio duma população de proteínas. Este método baseia-se na separação dos componentes
proteicos dum extracto total, derivado de bactérias, por exemplo, por electroforese em
poliacrilamida e em seguida, na transferência eléctrica destas proteínas para uma folha de
nitrocelulose, onde elas aderam fortemente. A folha de nitrocelulose é embebida num
anticorpo, específico da proteína que se procura identificar e em seguida, após lavagem, é
tratada com um segundo anticorpo, dirigido contra a parte constante do primeiro anticorpo e
marcado com a peroxidase. Por fim, um substrato cromogénico permite revelar os complexos
imunoespecíficos (Fig. 116.II.).

83
Limitações da produção na bactéria
E. coli têm, no entanto, vários inconvenientes, derivados da sua incapacidade em
processar as proteínas, realizar modificações pós-traducionais, que são muitas vezes
necessárias para a sua actividade biológica (p. ex., glicosilação, fosforilação, carboxilação), e
em segregar certas proteínas para fora do citoplasma, que por vezes se acumulam em grandes
quantidades no citoplasma, de forma insolúvel e inactivas, cuja renaturação é extremamente
difícil. Estes agregados têm o nome de corpos de inclusão (inclusion bodies), partículas
constituídas essencialmente de proteína recombinante ou polímeros derivados, não
reductíveis e de proteínas endógenas, tais como RNA polimerase, proteínas de membrana e
de rRNA e de DNA plasmídico.
Para libertar a proteína é usual recorrer a agentes caotrópicos fortes, tais como a ureia 6
M ou o cloridrato de guanidina 8 M. O enrolamento correcto da proteína depois de remover o
agente desnaturante, é frequentemente um passo bastante difícil, por vezes impossível,
nomeadamente com proteínas de elevado peso molecular e numerosas pontes dissulfureto.
Por consequência, proteínas, como o activador tissular de plasminogéneo (t-PA) ou o factor
VIII da coagulação sanguínea, são produzidas de preferência em cultura de células de
mamíferos.
Certas proteínas, tais como o interferão-α2 (IFN-α2), o interferão γ (IFN-γ) ou a proteína
MX (induzida pelo interferão), que formam agregados insolúveis e são inactivas, quando
produzidas em culturas de E. coli a 37°C, podem ser obtidas na forma solúvel e activa,
quando a temperatura da cultura é de 30°C ou abaixo. Uma medida possível para obter
proteínas solúveis é, portanto, diminuir a temperatura de crescimento das culturas e evitar
vectores plasmídicos indutíveis termicamente.
A proteína exógena, formada no citoplasma da célula bacteriana, que é um meio redutor
menos estável que o citoplasma das células de mamífero e sujeito a variações de composição,
para manter o pH e as propriedades osmóticas, apresenta-se na sua forma reduzida. Só depois
da lise bacteriana, a molécula pode formar as pontes dissulfureto, o que pode ser uma
dificuldade suplementar na aquisição da conformação correcta, tanto menos provável quanto
o número de átomos de S for maior. Além disso, pequenos péptidos de E. coli, como por
exemplo o glutatião, podem fixar-se nos átomos de enxofre livres e perturbar a formação das
pontes S-S, conduzindo a proteínas anormalmente enroladas. Sabendo-se que, a menor
anomalia em relação à molécula nativa pode influir nas suas propriedades imunogénicas e
que uma proteína produzida, para ser utilizada com fins terapêuticos, não pode provocar o
aparecimento de anticorpos, podem-se prever impasses frequentes.
Assim, apesar dos sucessos obtidos na síntese de algumas proteínas de mamífero, na
forma solúvel, em E. coli, outras vias têm sido exploradas para forçar a sua secreção para

84
fora do citoplasma. E. coli e, em geral as bactérias gram-negativas, possuem uma membrana,
que se pode considerar dupla, constituída por uma parede interna rígida, coberta por uma
camada exterior menos espessa, composta por proteínas, lipoproteínas e lipopolissacáridos.
As proteínas segregadas para fora do citoplasma acumulam-se, em geral, neste espaço
periplasmático que constitui um meio mais favorável ao seu enrolamento correcto.
Existem exemplos que demonstram que esta abordagem é uma alternativa interessante. A
proteína, para ser segregada para o periplasma, deve possuir uma sequência sinal, que dirija a
cadeia polipeptídica através da parede interna da membrana. O péptido sinal é, normalmente,
clivado, durante a travessia para o espaço periplasmático, por uma protease que reconhece
uma sequência particular de aminoácidos.
Produção da hormona de crescimento humana (hGH)
Um exemplo de produção em E. coli, à escala industrial, de uma proteína segregada no
espaço periplasmático, é o da hGH. Esta hormona é um polipéptido de 191 aminoácidos,
segregada pela hipófise. É utilizada para estimular o crescimento de crianças, cuja deficiência
parcial ou total em hGH se traduzirá na idade adulta por uma altura abaixo do mínimo, ou por
uma estatura de anão.
A acção da hormona de crescimento não se limita à estimulação do crescimento do
esqueleto. Ela permanece pela vida inteira, mesmo após a interrupção do crescimento. A
hGH está, com efeito, implicada no crescimento da massa de todos os tecidos do corpo. Ela
parece também poder ser utilizada eficazmente no tratamento de fracturas, de queimaduras e
de úlceras. Esta hormona é específica de espécie (a hormona de crescimento isolada de
diferentes espécies é sem acção na espécie humana), razão pela qual, a sua única fonte, antes
do advento das técnicas de engenharia genética, provinha da hipófise extraída de cadáveres
humanos. Para além da insuficiência do material assim obtido, para curar todos os casos de
deficiência, existe ainda o risco de contaminações, provenientes de purificações insuficientes
de hGH, a partir de glândulas infectadas.
A cadeia polipeptídica, no momento da sua síntese na hipófise, na forma de precursor,
comporta 217 aminoácidos. Os 26 aminoácidos suplementares em relação à proteína madura,
correspondem à sequência sinal. O enrolamento no espaço, preciso e complexo, cuja
estabilização é, em parte, assegurada por duas pontes S-S, está representado na Fig. 117.II, de
maneira muito rudimentar.
Uma primeira abordagem, para a clonagem e a expressão do DNA que codifica a hGH,
ignora a sequência sinal e visa a produção no citoplasma da proteína madura. A clonagem
recorre à combinação de dois métodos. Por um lado, um DNA complementar foi obtido por
cópia do RNA mensageiro da hGH, derivado da hipófise; por outro, um fragmento de DNA

85
correspondente aos aminoácidos 1 a 24, provido com um triplet de iniciação, foi sintetizado
quimicamente (Fig. 118.II.). Os dois fragmentos foram introduzidos, após ligação, num
vector de expressão, a jusante dum promotor forte (lac). O plasmídeo recombinante foi em
seguida utilizado para transformar um hospedeiro bacteriano. Os clones obtidos foram
seleccionados pela resistência a um antibiótico e caracterizados por hibridação com sondas
radioactivas apropriadas. Os clones que contêm a informação genética da hGH produzem a
hormona de crescimento humana, cuja actividade biológica foi demonstrada por imunoensaio
e por testes in vivo, em ratos desprovidos de hipófise. A actividade biológica manifesta-se por
um aumento de peso e um crescimento, nos ratos, da cartilagem das tíbias.
A natureza do produto clonado é idêntica à do produto natural, no que respeita à actividade biológica. No entanto, a metionina presente na extremidade NH2 da proteína
(derivada do codão de iniciação ATG e que, durante o processo natural, é eliminada com o
péptido sinal) não é clivada na bactéria. A proteína expressa não pode, por consequência, ser
considerada rigorosamente autêntica, visto possuir um aminoácido a mais (met-hGH). A
percentagem de expressão varia de 2 a 30 % de proteínas solúveis, segundo o tipo de
promotor utilizado. Neste exemplo, a hGH é produzida no citoplasma e não é segregada. No
entanto, ao contrário doutras proteínas super-expressas na bactéria, a hGH permanece solúvel
e é produzida por fermentação a grande escala e purificação em grandes quantidades (Tabelas
6.II. e 7.II.). A quantidade obtida, a partir dum fermentador de 500 litros, é equivalente à
obtida por extracção de 12.000 hipófises de cadáveres. A met-hGH é clinicamente activa, se
bem que, a longo termo, a experiência clínica revele o aparecimento de anticorpos não
neutralizantes, em quantidade reduzida. O seu efeito, ao longo de vários anos de tratamento, é
uma incógnita. O aparecimento de anticorpos pode estar correlacionado com um perfil
antigénico particular da hGH produzida por engenharia genética. A presença da metionina
excedentária poderia, por exemplo, conferir uma estrutura terciária particular à hormona e
criar um sítio antigénico novo. O enrolamento diferente da met-hGH poderia ser, igualmente,
causado pelo facto de as pontes dissulfureto, S-S (que estabilizam normalmente a molécula
enrolada), se formarem unicamente no momento da lise da bactéria (devido ao meio redutor
do citoplasma). No momento da lise, pequenos péptidos de E. coli podem-se fixar ao nível
dos átomos de enxofre das cisteínas e perturbar a formação das pontes S-S internas,
conduzindo a moléculas anormalmente enroladas. Uma contaminação, mesmo fraca, da
hormona, por produtos bacterianos, pode também ser responsável pela resposta imunitária.
Uma segunda abordagem recorre à clonagem do DNA completo, incluindo a sequência
que codifica para o péptido sinal da hGH (Fig. 119.II.). Esta sequência sinal de mamífero é
reconhecida pelos mecanismos celulares bacterianos, que presidem à expulsão das proteínas
para fora do citoplasma. A hGH acumula-se no espaço periplásmico, libertada da sequência
sinal, clivada durante a travessia da parede interna da membrana citoplásmica (Fig. 120.II.).

86
Ela comporta, por consequência 191 aminoácidos, como a molécula natural. É possível
extrair a hormona acumulada no periplasma, sem ter que recorrer à lise das células
bacterianas (Fig. 121.II.). Utiliza-se a técnica do choque osmótico, que consiste num primeiro
tempo, em mergulhar as bactérias numa solução muito concentrada de soluto. Nesta solução
hipertónica, a célula retrai o citoplasma, aumentando por consequência o espaço
periplásmico, onde se acumulam as moléculas de hGH. Num segundo tempo, as bactérias são
transferidas bruscamente para um meio muito pouco concentrado de soluto. Nesta solução
hipotónica o citoplasma aumenta brutalmente de volume, reduzindo o espaço periplásmico ao
mínimo e forçando as moléculas de hGH a atravessar os poros da parede exterior da
membrana. A proteína recuperada do meio extracelular é muito pouco contaminada por
proteínas bacterianas.
As análises físicas e químicas da hGH, obtida por esta metodologia, revelaram
propriedades idênticas às da proteína natural. As análises clínicas tendem a comprovar que
não há desenvolvimento de anticorpos nas crianças tratadas com este produto.
A secreção da hGH para o meio de cultura de E. coli é a terceira via possível. Um
exemplo desta abordagem recorre à transformação de E. coli com dois vectores, um dos
quais, pOmpA-hGH2, expressa a proteína híbrida, constituída pela sequência sinal de OmpA
(Outer membrane protein A, proteína cuja expressão é regulada pela osmolaridade do meio) e
da cadeia polipeptídica da hGH (a Fig. 122.II. esquematiza os passos essenciais da
construção deste vector) e o outro, pJL3 expressa a BRP (Bacteriocin Release Protein),
proteína que activa a fosfolipase A da membrana externa, permeabilizando as membranas
interna e externa. A sequência sinal de OmpA guia a cadeia polipeptídica através da
membrana interna e a BRP leva à formação de zonas permeáveis nas membranas celulares,
através das quais a hGH pode passar e ser libertada no meio de cultura.
A Fig. 123.II. ilustra alguns elementos essenciais dos vectores utilizados. O vector de
secreção, pOmpA-hGH2, possui o sistema promotor-operador lpp-lac, que é regulado pelo
repressor lac. A indução parcial da síntese de hGH, em E. coli, é provocada, cultivando as
células em meio rico, (TY), que contém lactose, ou em presença de níveis fracos ou
moderados de IPTG. O vector contém ainda a sequência ompA que codifica o péptido sinal, o
qual, fusionado ao DNA da hGH, orienta a sua secreção. Este plasmídeo, pOmpA-hGH2,
comporta o gene de resistência à ampicilina.
O plasmídeo pJL3 possui o mesmo híbrido, composto do promotor de lipoproteína (lppp)
e do promotor-operador lac (lacpo) de E. coli, que o vector de secreção, e que dirige a
expressão do gene de BRP clonado a jusante. A expressão desta proteína é também regulada
pelo repressor lac, codificado pelo gene lacI. O cloranfenicol é utilizado como marcador de

87
resistência. Os dois plasmídeos são compatíveis, e após co-transformação de E. coli, os
transformantes são seleccionados com base na resistência dupla aos antibióticos.
Os dois genes, de BRP e de hGH, são pois regulados pelo repressor lac. Em presença de uma
concentração baixa de isopropil-1-tio-ß-D-galactopiranosídeo (IPTG, 20 mM), tanto a BRP,
como a proteína híbrida OmpA-hGH se expressam e a hormona de crescimento madura é
libertada no meio de cultura, processada correctamente. Com efeito, a sequência de
aminoácidos N-terminal é correcta, o que indica que o péptido sinal foi clivado como se
esperava (Fig. 124.II.).
O nível de produção situa-se na ordem de 4,5 mg/ml. Apesar de ser mais baixo que o
rendimento da secreção periplásmica (10-15 mg/ml), a purificação, numa única etapa, por
HPLC, em coluna de fase reversa, atinge 98 %.
Este sistema têm a vantagem de não requerer, nem a lise das células bacterianas, nem
operações suplementares para permeabilizar ou romper a membrana exterior, necessárias no
caso da produção periplásmica. Um dos interesses maiores desta técnica é a possibilidade
teórica de poder, no futuro, assegurar uma produção em contínuo a partir de células de E.
coli, imobilizadas num suporte sólido. É necessário, evidentemente, avaliar previamente o
comportamento imunogénico da proteína in vivo.
Outros sistemas de secreção no meio de cultura de E. coli têm sido estudados, que
favorecem a permeabilização da membrana exterior, recorrendo a mutações específicas ou à
hiper-expressão de outras proteínas que alteram a permeabilidade da membrana, tais como o
péptido kil. Este péptido induz a secreção de proteínas heterólogas ou homólogas, em duas
etapas, primeiro actuando sobre a membrana citoplásmica, em seguida permeabilizando a
membrana externa.
Hormonas de crescimento animais
Para além da hGH, outras hormonas de crescimento de origem animal (porco, boi...)
foram objecto de trabalhos de clonagem. Visto ser a hormona de crescimento específica de
espécie, a produção de hormona de crescimento de porco e de boi, com fins veterinários,
impunha-se. A estratégia adoptada é paralela à que conduziu à produção da hGH. O RNA
mensageiro foi extraído a partir de hipófise de porco e de boi, e o DNA complementar foi
sintetizado e clonado num vector plasmídico. A análise das sequências nucleotídicas mostrou
que os dois cDNA são muito homólogos (+ 90 %). As hormonas diferem apenas em 18
aminoácidos. As duas sequências contêm uma sequência sinal, que codifica um péptido (27 e
20 resíduos) implicado na secreção da hormona pelas células somatotrópicas.

88
Para expressar os cDNA em E. coli, recorreu-se, como acima descrito com a hGH, a uma
construção que comporta adaptadores sintéticos, contendo um codão ATG em 5' e a
sequência de bases correspondente aos primeiros 24 aminoácidos da hormona. Estes
fragmentos sintéticos foram, depois de associados e ligados, fusionados num vector de
expressão. A expressão, obtida em elevado nível, em E. coli leva à produção da hormona sem
sequência sinal.
Produção de insulina humana
A insulina é uma hormona peptídica segregada pelas células pancreáticas e presente na
circulação sanguínea. A insuficiência em insulina conduz ao diabete, perturbação grave do
metabolismo dos glúcidos, dos lípidos e dos prótidos. Esta hormona, antes da aplicação das
técnicas da engenharia genética, era isolada a partir das fontes convencionais, para ser
utilizada no tratamento do diabete. Ela comporta um péptido sinal, que permite o transporte
da molécula através das membranas intracelulares. Durante o transporte, este péptido sinal é
clivado. O resto da molécula é, em seguida, armazenado nas vesículas ligadas à membrana
das células pancreáticas.
A forma acumulada nas vesículas, chamada proinsulina, difere da insulina
fisiologicamente activa (Fig. 125.II.). A proinsulina é um polipéptido simples, enrolado numa
forma particular, que é estabilizado por 3 pontes dissulfureto. A insulina madura é, pelo
contrário, constituída por duas cadeias separadas, uma de 21 aminoácidos (cadeia A) e a
outra de 30 aminoácidos (cadeia B), mantidas associadas pelas mesmas pontes S-S. A
proinsulina é convertida em insulina, no seio das vesículas pancreáticas, por maturação
enzimática do precursor e libertação dum polipéptido excedentário, de 33 aminoácidos
(péptido C). A insulina madura é, em seguida, acumulada na forma de complexo com iões de
zinco, pronta para ser exportada.
Resumindo, a insulina existe em 3 formas: a pré-proinsulina (produto de tradução
original), a proinsulina (destituída de péptido sinal) e a insulina activa.
A insulina é produzida actualmente em E. coli, programada por técnicas de DNA
recombinante, numa forma idêntica à da insulina natural. Conhecendo a sequência de
aminoácidos das cadeias A e B da insulina humana, é possível reconstituir, por síntese, os
fragmentos de DNA correspondentes (cadeia A: 63 pares de bases e cadeia B: 90 pares de
bases). Cada fragmento de DNA é provido, em 5', com um codão especificando a metionina,
e em 3', com um codão de fim de cadeia (TGA). Cada um dos genes sintéticos, A e B, é em
seguida fusionado em fase com o gene da ß-galactosidase (ou pelo menos com uma parte
deste), contido num plasmídeo de expressão. Os plasmídeos recombinantes são, em seguida,

89
utilizados na transformação de E. coli, replicam-se, e o RNA mensageiro, sob controlo dos
sinais de regulação do gene ß-gal (promotor, sequência Shine-Dalgarno), é traduzido em
proteína híbrida, comportando uma parte da ß-galactosidase e a cadeia da insulina, A ou B.
Como a insulina não contém metioninas, com excepção da que foi deliberadamente
introduzida em 5', durante a síntese das cadeias A e B, é possível clivar a proteína fusionada,
com brometo de cianogénio (que cliva especificamente ao nível das metioninas), de maneira
a libertar a insulina dos fragmentos de ß-galactosidase. Depois da purificação, as duas
cadeias A e B, ao serem misturadas, associam-se correctamente, formando as três pontes
dissulfureto. O resultado é uma insulina pura e biologicamente activa (Fig. 126.II.).
Alternativamente, a insulina humana pode ser produzida a partir dum clone comportando
o DNA complementar, derivado do mRNA da insulina (proinsulina) (Fig. 127.II.). O RNA
mensageiro que codifica a proinsulina é isolado e copiado em cDNA. Imediatamente a
montante do codão correspondente ao primeiro aminoácido da pro-insulina, introduz-se um
triplet ATG que codifica a metionina. Este DNA complementar é em seguida ligado ao gene
ß-gal, contido num plasmídeo de expressão. As bactérias transformadas pelo plasmídeo
recombinante produzem uma proteína híbrida, que contém um fragmento de ß-galactosidase
e a proinsulina. Esta é libertada da fusão, por clivagem com o brometo de cianogénio (que
destrói a metionina), é purificada e obtida na forma enrolada natural, com 3 pontes S-S
correctas. É em seguida submetida a uma clivagem enzimática, in vitro, para originar a
insulina biologicamente activa.

90
b. Expressão em Bacillus subtilis
A secreção das proteínas que se deseja produzir para o meio extracelular é, como já
salientámos, vantajoso porque simplifica a recuperação e a purificação, favorece o
enrolamento correcto e abre perspectivas para o desenvolvimento de biorreactores de células
imobilizadas mais eficazes. É, pois, natural que bactérias gram-positivas, como Bacillus
subtilis, Staphylococcus aureus ou Streptomycetes, que não possuem membrana externa e
que são excelentes excretoras, tenham suscitado elevado interesse enquanto organismos
potencialmente produtores de proteínas recombinantes. Com efeito, ao contrário do
colibacilo, B. subtilis secreta enzimas naturalmente e várias estirpes de Bacillus são utilizadas
na produção industrial de amilases e de proteases, enzimas que intervêm nas indústrias do
amido e dos detergentes, e Streptomycetes, na produção de antibióticos naturais.
As propriedades de B. subtilis, i) secretor de proteínas no meio de cultura, ii) bem
conhecido e aceite industrialmente na fabricação de proteínas, iii) não patogénico (não
produz endoxinas), tornam este organismo extremamente atractivo para produzir proteínas
humanas e animais pela tecnologia do DNA recombinante. No entanto, a expressão eficaz de
genes exógenos e a secreção das proteínas correspondentes têm sido dificultadas em razão de
vários problemas. Os mecanismos de secreção dos bacilos gram-positivos não estão bem
caracterizados, e em particular, os mecanismos de controlo são mal conhecidos. Têm faltado,
por outro lado vectores apropriados para a expressão e a secreção dos produtos de genes
heterólogos. A maiora das estirpes de colecção de Bacilli contém unicamente plasmídeos
crípticos, destituídos de marcadores de selecção. Foram, por isso, desenvolvidos vários
plasmídeos, sobretudo derivados de S. aureus, capazes de transformar B. subtilis, possuindo
genes de resistência a antibióticos, para serem utilizados em clonagem. Alguns foram, ainda,
manipulados para serem utilizados como vectores de inactivação insercional e como vectores
vaivém (shuttle) para E. coli e B. subtilis. Foram introduzidos marcadores adicionais de
resistência, derivados de outros plasmídeos de S. aureus, do DNA cromossómico de outras
estirpes de Bacilli ou do plasmídeo pBR322 de E. coli (Fig. 128.II.).
A partir destes plasmídeos foram construídas séries de vectores de secreção para
clonagem em B. subtilis, na esperança de obter expressão e secreção eficazes de produtos de
genes estrangeiros. Como a experiência têm mostrado que a expressão de genes heterólogos
em B. subtilis requer aparentemente sinais de transcrição e de tradução de Bacillus ou doutras
bactérias gram-positivas, é frequente recorrer a sequências promotoras, ao local de ligação do
ribossoma e à sequência sinal do gene da α-amilase (que é secretada em grande quantidade
por alguns Bacilli no meio de cultura), para orientarem a síntese e a secreção de genes
estrangeiros clonados em pUB110, por exemplo. Um exemplo é a clonagem do gene da ß-
lactamase de E. coli a jusante de sequências reguladoras do gene da α-amilase de Bacillus

91
amyloliquefaciens. Estas foram previamente introduzidas no plasmídeo pUB110 para
construir o vector pKTH38 (Fig. 129.II.), a partir do qual foram realizadas várias construções
que diferem pela junção entre o linker sintético - gene da ß-lactamase (destituído da sua
sequência sinal) e a sequência sinal do gene da α-amilase que se apresenta quase completa
(pKTH78), cortada (pKTH86), ou aumentada com sequências codificantes de diferentes
comprimentos (pKT16, pKT83 e pKT84) (Fig. 130.II.).
Uma estirpe de B. subtilis mutante deficiente em protease, transformada com as
construções que apresentam a sequência sinal quase completa (-1) ou contendo 11
núcleotídeos do gene da α-amilase (+4) segrega 95 % da actividade de ß-lactamase no meio
de cultura, à razão de 20 mg/ml. As outras construções são muito menos produtivas e a (-7),
correspondente ao péptido sinal amputado dos 6 aminoácidos carboxílicos terminais não
segrega a enzima.
Apesar destes resultados encorajadores, as tentativas de secreção de interferões humanos
(IFN-α2 e IFN-γ, por exemplo) em sistemas semelhantes têm-se revelado infrutíferas. O
nível de secreção é apenas de 1 a 3 % do da α-amilase produzido por B. subtilis transformado
com o mesmo vector (1-2 mg/l contra 250-400 mg/l). As razões desta ineficiência são em
parte a degradação proteolítica, mas, sobretudo, o transporte e/ou o processamento deficiente
do precursor do IFN. O IFN-γ é, por outro lado, aparentemente tóxico para as células
produtoras. Com efeito, os níveis de RNAs mensageiros da α-amilase e do IFN não são
significativamente diferentes, como foi demonstrado por análise do RNA, isolado de culturas
produtoras de IFN e de α-amilase, por eletroforese e hibridação com sondas radioactivas de
DNA (Fig. 131.II., tabela 8.II.). Na realidade, o precursor do IFN acumula-se sob forma
insolúvel nas células por razões ainda não bem esclarecidas.
Outros sistemas têm sido utilizados para expressar diferentes genes correspondentes a
proteínas e enzimas humanas, mas sempre com fracos rendimentos de secreção.
Em conclusão, e apesar dos progressos realizados na elucidação da bioquímica e da
genética de B. subtilis, a caracterização de promotores eficazes e de sinais de tradução
apropriados neste organismo está muito aquém do que é conhecido no colibacilo: não se
conhecem promotores com uma força comparável à dos lac, trp de E. coli ou PL de λ. Por
outro lado, os vectores recombinantes introduzidos em B. subtilis são frequentemente
instáveis e perdem ou transformam o gene heterólogo, faltando por isso bons sistemas
hospedeiro-vector. Apesar de B. subtilis possuir potentes sistemas de secreção autóctones,
como por exemplo o da α-amilase, as construções que recorrem à sequência sinal desta
proteína, para guiar genes estrangeiros codificando proteínas de mamíferos clonados a
jusante, têm-se revelado ineficazes, com baixos níveis de produção e de secreção, e as

92
proteínas são mal processadas e inactivas e, por vezes, degradadas por proteínas
extracelulares endógenas.
c. Expressão na levedura
A levedura Saccharomyces cerevisiae é um dos organismos eucariotas mais úteis para
estudar a regulação da expressão dos genes. Devido ao seu genoma relativamente pequeno (4
vezes o de E. coli) e do seu tempo de geração relativamente curto, a levedura pode ser
manipulada tão comodamente como a maioria dos procariotas. A genética da levedura é
relativamente bem conhecida; foram isoladas, caracterizadas e mapeadas centenas de
mutações por métodos genéticos convencionais e, actualmente, a sequência do seu genoma
(de S. cerevisiae) está totalmente determinada. Além disso, a levedura possui a propriedade
de se propagar de maneira estável do estado haplóide ao estado diplóide, o que permite a
complementação entre marcadores genéticos por conjugação de estirpes haplóides,
portadoras cada uma de um dos dois marcadores que se pretende analisar. O diplóide
resultante da conjugação pode ser depois induzido afim de regenerar haplóides, por meiose
(tetrada), os quais podem ser manipulados e tratados isoladamente.
No que respeita à clonagem, um dos meios originais de identificação dos genes de
levedura consiste em complementar mutações nos genes correspondentes de E. coli. O
genoma inteiro da levedura pode ser clonado em plasmídeos (banco genómico), os quais,
nesta forma recombinante, podem ser utilizados para transformar estirpes bactérianas
portadoras de diferentes mutações. Vários genes de levedura que codificam enzimas do
metabolismo dos aminoácidos ou dos ácidos nucleicos foram assim clonados directamente (p.
ex., LEU2, URA3).
É evidente que, se bem que a RNA polimerase de E. coli transcreva fragmentos de DNA,
seja qual for a sua origem, o RNA correspondente a um gene de levedura só será traduzido
correctamente se o gene em questão não comportar intrões.
Este método foi, no entanto, substituído pela clonagem de genes de levedura por
complementação directa de mutantes. Esta abordagem foi implementada graças ao
desenvolvimento de métodos de introdução de DNA na própria levedura. Um DNA exógeno
introduz-se relativamente facilmente na levedura, eliminando a parede celular para formar
esferoplastos (digestão dos polissacáridos por enzimas específicas). Em presença de iões de
cálcio e de polietilenoglicol (PEG) os esferoplastos absorvem o DNA.
Após regeneração da parede celular o crescimento das leveduras transformadas pode ser
iniciado. Este método baseia-se igualmente na utilização de plasmídeos vaivém que contêm,

93
simultaneamente, sequências necessárias à replicação do DNA em E. coli e à replicação na
levedura. Os fragmentos de DNA de levedura podem ser assim introduzidos por ligação
nestes plasmídeos vaivém e propagados em E. coli. A população heterogénea dos plasmídeos
recombinantes é em seguida introduzida nos esferoplastos de levedura. Qualquer plasmídeo
recombinante que complemente uma mutação no hospedeiro, em condições apropriadas de
selecção, pode ser identificado, reintroduzido em E. coli e multiplicado à vontade (Fig.
132.II.).
Os vectores de expressão de S. cerevisiae
A maioria das estirpes de levedura contém naturalmente um plasmídeo chamado 2µ,
constituído por uma molécula de DNA bicatenário, circular, com ≈6300 pb, localizado no
núcleoplasma à razão de 60 a 100 cópias por célula, e dotado de replicação autónoma. 2µ
comporta uma origem de replicação e codifica três funções REP que determinam a
amplificação quando o número de cópias baixa. Em condições normais, o círculo 2µ replica-
se a velocidade idêntica à do resto do genoma; quando o número de cópias diminui, as
proteínas REP dissociam a replicação do plasmídeo da do genoma e iniciam ciclos repetidos
de replicação independente do 2µ, até que a quantidade de cópias atinja de novo o número de
+ 60 a 100 por célula. Este sistema de regulação assegura a propagação do plasmídeo 2µ em
número elevado de cópias durante o ciclo celular da levedura (Fig. 133.II.). Na figura estão
indicados a origem de replicação e os três genes conhecidos, codificados por 2µ-REP1, REP2
e REP3 que são necessários à conservação dum número elevado de cópias. FLP determina a
passagem da forma (A) à forma (B) do círculo 2µ.
O DNA transformante pode-se estabelecer na levedura, quer por integração no genoma,
quer por replicação autónoma, na qualidade de epissoma. A presença ou a ausência de
sequências particulares, chamadas ARS (Autonomously Replicating Sequence), determina o
destino do DNA transformante. Um vector plasmídico para clonagem na levedura replicar-
se-á de maneira autónoma, contanto que possua as sequências ARS. Estas sequências têm um
comprimento de 60 pb, são ricas em AT (80 %) e possuem uma sequência consenso
AAATATAAA
C Normalmente, durante a divisão celular, os plasmídeos que se encontram na levedura não
segregam regular e uniformemente. Na ausência de pressão de selecção, as leveduras perdem
o plasmídeo durante o crescimento. Para remediar esta situação tira-se partido de sequências
de DNA derivadas da região centromérica dos cromossomas de levedura. As sequências CEN
asseguram naturalmente a ligação dos cromossomas à rede fibrosa do aparelho mitótico e
contribuem para a repartição igual dos cromossomas quando a levedura se divide. Clonadas

94
num plasmídeo, estas sequências CEN asseguram a sua permanência na célula durante a
divisão.
O conjunto das condições descritas está resumido na Fig. 134.II, dentro do contexto da
construção dum vector plasmídico ideal para a clonagem do DNA na levedura. Este vector
ideal deveria portanto comportar:
1°) uma sequência derivada do plasmídeo pBR322 que permita a replicação e a
selecção na bactéria (AmpR);
2°) uma sequência ARS que permita a replicação autónoma na levedura;
3°) uma sequência CEN que assegure a conservação do plasmídeo durante a divisão da
levedura;
4°) um marcador de selecção para a levedura (por exemplo o marcador nutricional
LEU-2, que complementa uma mutação auxotrófica no hospedeiro);
5°) um ou vários locais únicos de restrição para a introdução de DNA exógeno.
Exemplos de vectores de expressão para produção de proteínas exógenas em leveduras
Uma série de vectores plasmídicos destinados a clonar e a expressar um DNA heterólogo
na levedura foram construídos. Um exemplo está representado na Fig. 135.II.). Este vector
vaivém possui o gene amp utilizado para selecção na bactéria, a origem de replicação
específica de E. coli, um marcador de selecção (LEU2) adequado para a levedura, a origem
de replicação própria da levedura (2µ parcial ou 2µ completo) e uma cassete de expressão
ARG3. Esta cassete contém o promotor ARG3, derivado do gene de levedura ornitina
carbamóil transferase e a sequência terminadora, derivada do mesmo gene.
A actividade do promotor ARG3 é regulável; em presença de concentração elevada de
arginina este promotor é reprimido; quando a concentração baixa é desbloqueado.
Estas duas sequências, promotora e terminadora estão flanqueadas por locais de restrição
que permitem remover os sinais promotor ou terminador ARG3 e substitui-los por outros. Por
outro lado, no vector pRIT10774 um local BamH1, situado entre o promotor e o terminador,
permite a inserção e a expressão dum DNA heterólogo possuindo o seu próprio sinal de
iniciação ATG. A versão pRIT10779 comporta ainda um local NcoI entre o promotor e o
terminador que permite a introdução de DNA desprovido do sinal de iniciação ATG.
Um exemplo de aplicação prática de S. cerevisiae como organismo hospedeiro de
expressão de DNA exógeno é a produção industrial dum antigénio de superfície, HBsAg, do
vírus da hepatite B, capaz de induzir a produção de anticorpos protectores e comercializado
recentemente como vacina. A expressão deste antigénio sob controlo do promotor

95
cromossómico, altamente eficiente, de levedura, TDH3 (triosefosfato desidrogenase), é
constitutiva durante a multiplicação celular. Em 3' um sinal eficaz de terminação de
transcrição de ARG3 assegura um nível elevado de mRNA estável. LEU-2 é utilizado como
marcador de selecção (Fig. 136.II.).
Várias outras proteínas de mamíferos foram expressas na levedura (Tabela 9.II.).
Algumas são produzidas em quantidade suficiente e possuem actividade biológica suficiente
para serem comercialmente viáveis.
A levedura pode efectuar as modificações postraducionais de acetilação, fosforilação e
glicosilação, frequentemente necessárias para obter completa actividade biológica das
proteínas. O grau de glicosilação é, no entanto, bastante variável, diferente da realizada nas
células de mamífero e é por vezes observada hiperglicosilação (sobretudo N-glicosilação com
manose pesado), o que pode originar instabilidade e antigenicidade.
Muitas das proteínas produzidas pela levedura possuem alguma actividade biológica, o
que pressupõe uma conformação tridimensional correcta. Nalguns casos a formação de
pontes dissulfureto faz-se ao acaso, como acontece por exemplo com o t-PA (tissue
plasminogen activator), expresso em quantidade elevada mas pouco activo biologicamente e
não segregado para fora da célula.
No que se refere à secreção das proteínas pela levedura foram construídos vários
vectores que contêm sinais de secreção de ferromonas, tais como o factor sexual α, que induz
o acoplamento entre o dois tipos sexuais α e a e é naturalmente segregado pela estirpe
haplóide. O gene da proteína que se deseja fazer segregar para meio é inserido naquele
vector, imediatamente a seguir ao sinal de secreção. Algumas proteínas activas maduras
foram assim recuperadas no meio de cultura, se bem que os rendimentos sejam em geral
modestos.
A maioria das proteínas estrangeiras não é produzida em S. cerevisiae com níveis de
expressão elevados. A proteína endógena correspondente da levedura é expressa por vezes
neste organismo, sob controlo dos seus próprios promotores, de 15 a 50 vezes mais
intensamente. Com efeito, se bem que cassetes de expressão, comportando os sinais de
começo e de fim de transcrição dos genes (da glicólise da levedura, por exemplo), sejam
extremamente eficazes num sistema homólogo (para expressar um gene de levedura), elas
funcionam em geral com um rendimento muito menor para expressar um gene estrangeiro.
Qualquer "coisa" na região codificante do gene de levedura facilita a sua transcrição ou
aumenta a estabilidade dos mensageiros. Estes sinais, ausentes nos genes estrangeiros são
objecto de pesquisas intensivas.

96
A levedura Pichia pastoris metilotrófica (metabolisadora de metanol) é outro sistema de
produção de proteínas com grandes potencialidades. As regiões reguladoras de genes
implicados na via de utilização do metanol foram isoladas. Por exemplo, sequências que
regulam a expressão das enzimas álcool desidrogenase e álcool oxidase, foram utilizadas para
controlar a expressão de várias proteínas exógenas importantes. A modulação pela fonte de
carbono permite regular os níveis de expressão.
Existem vectores de expressão replicativos (de expressão autónoma) e integrativos. A
utilização dos integrativos, capazes de induzir a incorporação de cópias múltiplas de um gene
seleccionado no DNA cromossómico do hospedeiro, e a selecção de estirpes recombinantes
com capacidade para se multiplicarem mais rapidamente, são desenvolvimentos que levaram
em certos casos à obtenção de níveis de expressão elevadíssimos. Exemplos da aplicação
deste sistema à produção de proteínas de interesse em medicina são a expressão do antigénio
de superfície do vírus da hepatite B (HBsAg), o factor de necrose tumoral (TNF), a
estreptoquinase e o t-PA.
Assim, o gene do HBsAg, controlado pelo promotor regulado pelo metanol, da álcool
oxidase I (AOX1) e flanqueado em 3' pelas sequências terminadoras do mesmo gene AOX1,
constitui uma cassete de expressão que foi inserida no plasmídeo pBSAGI51 (Fig. 137.A.II.).
O gene his4 de P. pastoris em 3' desta cassete funciona como marcador de selecção na
transformação duma estirpe auxotrófica que requer histidina por ser deficiente em histidinol
desidrogenase (HIS4). Para inserir a cassete no genoma de P. pastoris, recorre-se ao método
de substituição de gene, já anteriormente utilizado em S. cerevisia, pelo qual os fragmentos
de DNA clonados vão substituir sequências genómicas nativas por um mecanismo que
implica uma recombinação estimulada por sequências flanqueantes, homólogas de regiões
genómicas (Fig. 137.B.II.). Neste exemplo, o gene AOX1 genómico foi o alvo da
substituição pela construção cassete de expressão - HIS4.
A inserção da cassete no genoma hospedeiro elimina problemas potenciais de
instabilidade dos plasmídeos. A cultura do P. pastoris recom(-HAUT-) utilizando fontes de
carbono repressivas, tais como glucose ou glicerol, leva à acumulação duma massa ilimitada
de células, sem selecção significativa de mutantes deficientes em expressão genética
heteróloga.
Quando se adiciona metanol, depois de deixar esgotar a fonte de carbono repressor,
induz-se a expressão do gene heterólogo controlado pelo promotor AOX1. A limitação do
período de expressão de HBsAg, graças à possibilidade de regulação pelo metanol, minimiza
as probabilidades de selecção de mutantes não expressores. O nível de expressão em
produção à escala industrial atinge 0,4 g de HBsAg por litro de cultura (3 a 4 % da massa
proteica total). Esta proteína parece ter as mesmas propriedades antigénicas in vitro e in vivo

97
(induz anticorpos neutralizantes no rato) que a proteína nativa e constitui uma concorrente
potencial à produzida por S. cerevisiae e já utilizada como vacina humana.
A aplicação das técnicas da engenharia genética às leveduras não têm unicamente a
finalidade de clonar e expressar DNA heterólogos. Muitas informações fundamentais têm
sido obtidas sobre a estrutura dos cromossomas dos eucariotas e sobre a organização e a
regulação dos genes de levedura.
Exemplos:
1) A integração dirigida
O DNA introduzido nos esferoplastos, pode-se integrar nos cromossomas de levedura.
Este processo efectua-se por recombinação entre sequências homólogas presentes no DNA
transformante e no cromossoma. Um plasmídeo de levedura pode-se integrar nos
cromossomas, contanto que seja desprovido da sequência ARS. No entanto, a frequência da
integração de moléculas de DNA circulares é fraca, ao contrário dum plasmídeo previamente
linearizado que se integrará com uma frequência 100 vezes superior à forma circular. A
integração do plasmídeo no cromossoma da levedura pode ser dirigida pelo local do corte de
linearização com a enzima (Fig. 138.II.). Esta propriedade de dirigir a integração do DNA
num sítio específico pode servir para substituir um gene normal por um gene mutado. Este
processo, chamado substituição alélica, permite estudar os efeitos duma mutação específica
num gene, realizada in vitro, na expressão deste gene na levedura. Pode-se, por exemplo,
modificar in vitro por mutagénese dirigida, a sequência do DNA que codifica para a actina.
Este DNA "mutante", transportado por um plasmídeo de integração e linearizado por corte no
gene da actina, num sítio apropriado, será reintroduzido na levedura e integrar-se-á no lugar
da actina. Os efeitos da mutação na expressão do gene da actina podem assim ser medidos in
vivo. No caso deste exemplo, os efeitos da modificação introduzida por mutagénese dirigida
são recessivos e letais, indicando que a levedura não sobrevive se a actina não for funcional.
2) O isolamento de alelos mutantes
Pode-se tirar partido do processo de integração do DNA nos cromossomas de levedura
para isolar alelos mutantes, contanto que o gene selvagem clivado esteja disponível. Toda a
região codificante do gene, flanqueado pelas sequências não codificantes em 5' e em 3', é
excisado do plasmídeo portador de um marcador de selecção e de uma ARS. O plasmídeo
amputado é em seguida introduzido na levedura; para se replicar ele têm que ser reparado e
recircularizado, o que só acontece por recombinação das sequências flanqueantes, presentes
no plasmídeo, com as sequências homólogas que delimitam o gene mutado presente no
cromossoma. Assim, durante a duplicação, o gene mutado será introduzido no plasmídeo.

98
Este contém, pois, uma cópia do alelo cromossómico que poderá replicar-se e propagar-se
(Fig. 139.II.).
3) Regulação da expressão dos genes na levedura
Ao contrário do que acontece com as bactérias, os genes cujo controlo se efectua de
maneira coordenada não são obrigatoriamente contíguos e podem, até, estar localizados em
cromossomas diferentes. Assim, por exemplo, três genes de levedura que intervêm na
biossíntese da histidina encontram-se em cromossomas diferentes. As regiões que flanqueiam
estes três genes em 5' têm muito pouco homologia e há pouco tempo, pelo menos,
desconhecia-se como se efectua a coordenação da expressão. Cada gene contém, no entanto,
sequências de controlo essenciais para haver expressão normal. Um tipo de sequência
assegura um nível de transcrição baixo (nível basal) e um outro tipo de sequência
desencadeia uma transcrição abundante (regulação positiva em condições de carência) (Fig.
140.II.).
O fenómeno de regulação a longa distância da expressão dos genes de levedura já foi
evocado. Por exemplo, a expressão do tipo "mating" na levedura faz intervir mecanismos de
inserção de gene afastados num determinado locus de expressão.
4) Um intrão codifica para uma proteína reguladora
Certos genes de mitocôndria de levedura possuem propriedades genéticas complexas,
que só puderam ser elucidadas por análise molecular de DNA. O gene box que codifica para
o citocroma b existe em duas versões :
a) um gene "longo", de 1155 pb codificantes, distribuído ao longo de 6400 pb em 6
exões (B1 a B6) e 5 intrões (I1 a I5);
b) noutras estirpes, este gene é "curto"; as sequências correspondentes aos 4 primeiros
exões do gene "longo" são contíguas no gene "curto".
As duas formas do gene box expressam-se de maneira idêntica.
O gene box "longo" (Fig. 141.II.).
Os RNAs correspondentes a este gene existem em vários tamanhos distintos. O mais
curto têm 3300 pb e corresponde ao RNA mensageiro (incluindo as sequências flanqueantes
em 3' e 5'). Os RNAs de comprimento superior contêm uma ou várias sequências
correspondentes aos intrões. O transcrito primário têm um comprimento de 8500 pb.
Um certo número de mutações no gene box foram isoladas e caracterizadas. Distribuem-
se por grupos separados e por distâncias bastante grandes. Uma primeira classe de mutações
afecta a proteína directamente; o RNA mensageiro é normal mas durante a tradução há

99
interrupção prematura, sem sentido (nonsens), ou incorporação errada (missence). Estas
mutações situam-se nos exões B1, B3, B4 e B6.
Um outro grupo de mutações, box9 e box2, possui características particulares,
provocando a síntese de RNA anormais. Estas mutações situam-se todas no intrão 4. As
mutações box9 estão situadas a +350 pb a jusante da extremidade 5' do intrão e as mutações
box2, a 25 pb a montante da extremidade 3' do intrão. Os dois grupos de mutações impedem
o splicing e a junção dos exões B4 e B5. Mutações em sítios específicos podem impedir o
reconhecimento das junções de splicing, mesmo estando relativamente afastadas destas
junções.
Por outro lado, as mutações de tipo box3, 10 e 7 estão localizadas nos intrões I2, I3 e I4.
Nenhuma pode complementar outra mutação do mesmo tipo, mas podem, no entanto,
complementar mutações noutros clusters. Isto significa que os grupos box3, box10 e box7
codificam para um produto difusível, distinto do cromossoma b, que actua em trans e é
necessário para a síntese do citocroma b.
Cada um dos três intrões I2, I3 e I4 comporta sequências que codificam para um produto
cuja existência é independente e que desempenha um papel regulador na produção do
citocroma b. Com efeito, estas mutações bloqueiam a maturaçãodo RNA mensageiro do
citocroma b o que provoca a acumulação dos seus precursores. Cada grupo de mutações
bloqueia a maturação numa fase particular do processo desactivando um produto difusível
necessário ao splicing dum intrão. O produto difusível é uma RNA maturase. É provável que
esta proteína reconheça as extremidades intrónicas e assegure a execução dum splicing
correcto.
A análise da sequência núcleotídica do gene box dá peso a esta hipótese. Com efeito, o
primeiro exão (B1) codifica para os primeiros 139 aminoácidos do citocroma b (417 pb);
segue-se um intrão (I1) de 765 pb sem fases de leitura aberta, a seguir um segundo exão (B2,
15 pb ou seja 5 codões) e, finalmente, o segundo intrão (I2) cujas 840 primeiras bases estão
em fase em relação às do exão precedente (Fig. 142.II.). As mutações box3 criam codões sem
sentido na fase de leitura aberta do intrão I2. Por consequência, o produto codificado por esta
fase de leitura não é sintetizado e o splicing deixa de ser correcto, o que provoca a
acumulação do RNA precursor.
Na ausência da mutação box3, supõe-se que a tradução que conduz à formação da RNA
maturase começa no exão B1 e continua através de B2 e do intrão I2, contanto que o primeiro
intrão tenho tido um splicing correcto. A RNA maturase seria assim constituída por 424
aminoácidos [139 (B1) + 5 (B2) + 279 (I2)].

100
Se efectivamente a RNA maturase é necessária para o splicing do intrão I2, a acção desta
enzima implica um mecanismo de retro-controlo negativo bastante sofisticado (Fig. 143.II.).
A eliminação do intrão 2 pela RNA maturase suprime a sua própria síntese, visto que
parte do RNA que codifica para ela neste intrão desaparece. É necessário que se estabeleça
um equilíbrio entre as duas formas de RNA que estão sendo alvo do splicing e a RNA
maturase.
Aparentemente este mecanismo não é único e poderia existir noutros genes tais como o
oxy3 (subunidade 1 da citocroma-oxidase).
5) Estrutura do gene da ferromona de levedura (MFα)
A conjugação na levedura parece facilitada por factores peptídicos chamados
ferromonas, que bloqueiam as células de tipo conjugativo oposto, numa fase particular da
divisão celular. As células haplóides α produzem o factor α com 12 a 13 aminoácidos de
comprimento (2 formas). As células haplóides a produzem o factor a, com 11 aminoácidos de
comprimento (2 formas). As células diplóides α/a não produzem ferromonas e não são
sensíveis a elas.
Devido ao seu tamanho pequeno, e por analogia com outras situações, era de admitir que
estas ferromonas derivassem de precursores muito maiores cujo processamento conduzisse à
forma biologicamente activa (analogia com precursores de hormonas). A clonagem do gene
da ferromona α confirmou esta hipótese.
A estratégia desenvolvida para clonar este gene foi a seguinte:
certas estirpes de levedura mutadas produzem o factor α, mas não o segregam porque ele é
degradado muito rapidamente na célula. Todavia, o gene α clonado num plasmídeo
multicópia deveria expressar-se abundantemente e em quantidade suficiente para saturar a
actividade de degradação e conferir um fenótipo produtor de α que fosse facilmente
identificado. Por conseguinte, o DNA plasmídico isolado dum banco genómico de levedura
foi utilizado para transformar as leveduras não secretoras de α (matα2, leu2). Os
transformantes, seleccionados para leu+, foram escrutinados para a secreção do factor α por
um processo que faz intervir células a sensíveis ao factor α (produção de um halo ao contacto
dos transformantes secretores de α com as células a). Um plasmídeo recombinante
superprodutor de α foi identificado e caracterizado. O gene de estrutura do factor α codifica
para um precursor de 165 aminoácidos (prepropoli α F) (Fig. 144.II. e 145.II.). A
extremidade N-terminal do precursor contém uma sequência sinal hidrófoba e comporta três
sítios de glicosilação. A extremidade C-terminal contém quatro cópias do factor α maturado
unidas por péptidos relativamente homólogos.

101
Maturação do precursor de α
O processamento do precursor α requer a glicosilação dos três sítios na região guia, entre
o sinal e a primeira unidade α e em seguida a proteólise entre as quatro unidades α tornando-
as biologicamente activas (Fig. 146.II.). A proteólise realiza-se por etapas; corte a jusante da
região Lys-Arg por uma endopeptidase (KEX2), eliminação das sequências C-terminais por
uma carboxipeptidase e, enfim, eliminação dos aminoácidos excedentes Glu-Ala no termino NH2 por uma dipeptidilaminopeptidase.
d. Expressão nos fungos filamentosos
A genética dos fungos é ainda pouco conhecida apesar de alguns serem utilizados
industrialmente, nomeadamente na produção de antibióticos. Por exemplo, certos Aspergillus
são utilizados e cultivados industrialmente para a produção de penicilinas e doutros
antibióticos. Outros são utilizados na produção à escala industrial de proteínas segregadas no
meio, como por exemplo Aspergillus niger que pode segregar até 20 g/l de enzima endógena
glucoamilase.
Os fungos são pois candidatos importantes para a produção de proteínas estrangeiras
segregadas. No entanto, as dificuldades são ainda grandes porque não existem bons vectores
de expressão. Os promotores destes fungos não foram bem caracterizados e os vectores têm
bastante inconvenientes: não existem na forma autónoma e têm tendência a integrar-se nos
cromossomas, o que diminui a sua capacidade de expressão. As construções recorrem a
diversos outros elementos derivados de fungos clássicos e, em geral, da levedura S.
cerevisiae.
Tem havido progressos na estabilização destes vectores, mas o nível de expressão e de
secreção de proteínas estrangeiras permanece baixo.
A integração dos vectores no genoma pode ser, no entanto, uma vantagem se o nível de
expressão for melhorado. Foi observado, nalguns casos, a aquisição duma grande
estabilidade, sem pressão de selecção, como se a estirpe não tivesse sido modificada, o que é
importante nas aplicações industriais.
Os fungos filamentosos têm sido utilizados para produzir enzimas industriais. Já
referimos a glucoamilase produzida por A .niger. Outro exemplo é a renina (ou quimosina),
utilizada no fabrico de queijo, que pode ser preparada em larga escala, tanto em A. niger
como em Trichoderma reesei. A renina na sua conformação activa possui 3 ligações
dissulfureto intramoleculares e é difícil de expressar correctamente noutro sistema de
expressão. Existem exemplos de secreção de proteínas de mamíferos na forma activa, que
requerem a formação correcta de pontes dissulfureto para serem activas biologicamente. Não

102
existem, no entanto, dados suficientes para generalizar a capacidade dos fungos em relação às
modificações pos-tradução de proteínas expressas heterologamente.
Um exemplo de expressão de uma proteína de mamífero, aparentemente com a
conformação correcta, é o do interferão humano α-2 cujo cDNA foi clonado num vector de
expressão e colocado sob controlo do promotor indutível pelo etanol, da alcool
desidrogenase, derivado de Aspergillus nidulans. O vector integra-se de maneira estável no
genoma do hospedeiro. Foi ainda desenvolvido um outro vector de expressão comportando o
promotor da glucoamilase de A. niger para expressar aquele interferão humano.
Uma área importante de aplicação dos fungos filamentosos é o da utilização dos seus
genes para produzir enzimas de degradação da celulose e da lenhina. Por exemplo, o fungo T.
reesei segrega um complexo de enzimas que degradam a celulose em glucose. Alguns dos
genes que codificam para estas enzimas foram caracterizados e clonados em vectores de
levedura (nomeadamente de S. cerevisiae), sob controlo de promotores e de terminadores do
hospedeiro, afim de aumentar o nível de expressão, caracteristicamente baixo no organismo
nativo. Existem projectos para produzir, a partir de cassetes de expressão o conjunto do
complexo das celulases.
A expressão do complexo de genes que codificam para as lenhinases é igualmente
objecto de grande interesse, dada a importância que representa, a nível mundia,l o
processamento da pasta de papel.
A tecnologia do DNA recombinante pode também ser aplicada ao melhoramento da
produção de antibióticos em estirpes já altamente produtivas. Obtidas classicamente por
mutagénese ao acaso, com agentes mutagénicos químicos ou físicos, e selecção de mutantes
superprodutores dos precursores dos metabolitos primários, estas estirpes fizeram as suas
provas como organismos de produção industrial. A engenharia genética abre novas
perspectivas de desenvolvimento racional da capacidade produtora dos fungos. A criação de
mutações dirigidas, utilizando métodos de recombinação genética, pode reduzir
drasticamente o número de colónias que é preciso escrutinar para obter uma estirpe
optimizada. É, evidentemente, importante conhecer o pathway de biossíntese do antibiótico e
o complexo de enzimas que nele participa. Um exemplo de aplicação bem sucedida desta
abordagem é a modificação do Cephalosporium acremonium, fungo produtor do antibiótico
cefalosporina C, cujo pathway biossintético está representado na Fig. 147.II.
A primeira etapa consistiu em identificar um intermediário que se acumulasse de
maneira significativa na estipe altamente produtora. Isto foi realizado por análises química e
cromatográfica (HPLC) do meio de fermentação, que mostraram ser a penicilina N o
intermediário procurado. Estes resultados sugerem que a actividade da enzima

103
desacetoxicefalosporina C sintetase (DAOCS), que expande o ciclo tiazolidina da penicilina
N (etapa 4 do pathway biossintético), é limitante na síntese de cefalosporina C.
A segunda etapa é a clonagem do gene cefEF, que codifica para esta enzima, afim de
introduzir cópias suplementares do gene na estirpe produtora. Para isso foi construído um
plasmídeo que, para poder ser obtido em quantidades suficientes, têm que ser amplificado em
E. coli e necessita por consequência da origem da replicação colE1 e dum marcador de
selecção, CAT (cloranfenicol acetiltransferase). A escolha do marcador é importante porque,
se o gene que produz a enzima que inactiva o antibiótico de selecção dos transformantes em
E. coli se expressar gratuitamente nos transformantes de C. acremonium, os antibióticos beta
lactâmicos intracelulares poderão ser destruídos. Por isso foi escolhido CAT, que têm como
substrato um antibiótico que não possui a estrutura ß-lactâmica.
Para seleccionar os transformantes fungais é necessário um outro marcador de resistência
antibiótica, por exemplo a higromicina B fosfotransferase (HPT), conhecido como sendo um
marcador dominante de selecção, útil nas transformações genéticas de eucariotas inferiores.
A transformação de C. acremonium e a expressão de HPT foram conseguidas com
plasmídeos em que foi clonado o gene híbrido, composto pela fase de leitura aberta do gene
que codifica para HPT, derivado de E. coli (HPTorf), e o promotor (IPNSp) do gene pcbC, do
fungo, que codifica para a isopenicilina N sintetase.
O plasmídeo pPS55, cuja construção está esquematizada na Fig. 148.II., é um exemplo
de vector utilizado com o híbrido IPNSp/HPTorf para transformar eficazmente C.
acremonium, levando à integração estável no DNA genómico. O gene cefEF e as suas
sequências flanqueantes reguladoras foram introduzidos em 5' do fragmento IPNSp de pPS55
e o plasmídeo resultante, pPS56, foi utilizado para criar transformantes de C. acremonium,
seleccionados pela resistência à higromicina B. Uma selecção suplementar baseada na análise
da capacidade produtora de cefalosporina C, levou ao isolamento duma estirpe transformante
que produz industrialmente 15 % mais antibiótico que a melhor estirpe não transformada. A
presença, no genoma de C. acremonium, duma cópia suplementar do gene cefEF foi
correlacionada com um aumento duplicado da actividade específica da enzima DAOCS, o
qual se correlaciona por sua vez com uma diminuição substancial da quantidade de penicilina
N sedregada e com o aumento da cefalosporina produzida relativamente à estirpe parental.
e. Expressão em células de mamíferos
Os níveis de expressão e secreção variam de um organismo para outro, segundo a
proteína; esta é uma dificuldade maior que está por resolver em todos os sistemas hospedeiro-
vector conhecidos presentemente.

104
Muitos dos produtos farmacologicamente relevantes, recentemente descobertos e
identificados não podem ser produzidos na sua forma fisiologicamente activa pelas bactérias
ou até pelas leveduras. A capacidade dos organismos para sintetizarem proteínas com a
conformação correcta natural, para as glicosilarem correctamente, processarem com todas as
modificações secundárias necessárias para a sua função e estabilidade, a sua inocuidade e
actividade biológica, é limitada. Em muitos casos, as proteínas produzidas por micróbios têm
que ser enroladas correctamente in vitro e as processadas incorrectamente têm que ser
removidas. Esta purificação não é muitas vezes possível, ou é insuficiente e, além disso,
substâncias contaminantes pirogénicas têm que ser eliminadas, em particular das proteínas
produzidas por E. coli.
As células de mamíferos efectuam um processamento pós-traducional eficaz durante a
biossíntese de proteínas e de péptidos, o que pode favorecer fortemente a estrutura natural do
produto derivado do DNArec, influenciando, por consequência, a sua homogeneidade,
estabilidade e eficácia. Os produtos perfeitamente homólogos da proteína natural humana são
sempre preferíveis, afim de garantirem uma actividade farmacológica, específica e uma
compatibilidade clínica óptima durante as aplicações.
A expressão de DNA recombinante em cultura de células de mamíferos é, em princípio,
o melhor método para produzir quantidades suficientes de substâncias complexas
glicosiladas, com a condição de não estarem contaminadas com, por exemplo, vírus latentes,
ou outras substâncias potencialmente perigosas. A cultura de células animais têm vindo a
impor-se como uma tecnologia das mais importantes para investigação e desenvolvimento
em biologia celular, virologia e imunologia, assim como em investigação na área tumoral.
São utilizadas, por exemplo, linhagens celulares de macaco e de hamster que não têm
grandes exigências de meio de cultura e crescem em grande quantidade. Qualquer gene de
mamífero, tanto na forma genómica como na de cDNA, pode ser colocado num vector de
expressão adaptado às células de mamíferos. Pode ser flanqueado dos seus próprios sinais de
regulação, ou doutros mais potentes e complementados eventualmente com um sinal de
secreção. Estes vectores funcionam bem nas células em que são introduzidos, e os genes de
que são portadores expressam-se normalmente. As proteínas produzidas são segregadas no
meio de cultura e são correctamente processadas, diferindo pouco do produto segregado pela
célula de origem. Assim, por exemplo, a glicosilação efectuada pela célula de rato difere um
pouco da realizada por células humanas, mas esta diferença é sem significado nas
propriedades fisiológicas e imunológicas do produto final. Actualmente, a expressão nas
células de mamíferos em cultura constitui a única maneira de produzir, numa escala
relativamente grande, proteínas de organismos superiores que exigem, para serem activas,

105
modificações pós-traducionais. É o caso de certos factores de coagulação do sangue, do
activador tissular do plasminogéneo, de hormonas, de interleucinas, etc.
Os problemas inerentes à expressão nas células de mamíferos decorrem, primeiramente,
dos vectores próprios deste tipo de células. Como se integram quase sempre nos
cromossomas, o número de cópias activas nas células é limitado. Existem excepções, tais
como a dos vectores derivados do vírus do papiloma bovino (Fig. 149.II.). Existem, no
entanto, métodos para amplificar o número de cópias do gene que se deseja expressar. A
segunda dificuldade é de ordem económica, porque a cultura das células de mamíferos nem
sempre é fácil. Apesar dos enormes progressos realizados na simplificação dos meios de
cultura, no aumento da massa de células, graças ao crescimento em suportes sólidos, no
melhoramento das técnicas de fermentação etc..., a produção industrial de células animais é
ainda relativamente dispendiosa e reservada a produtos de alto valor acrescentado.
É de assinalar, todavia, que a cultura in vitro das células animais têm progredido a ponto
de se poder utilizar células normais não transfectadas para produzir substâncias interessantes,
tais como HBsAg segregada por células hepáticas ou ainda factores imunológicos, como a
interleucina 2 ou os interferões. É possível que, graças a progressos futuros nas técnicas de
cultura das células de mamíferos, as células normais não manipuladas possam vir a suplantar
as linhagens celulares transfectadas com vectores de expressão recombinados.
Transferência de genes nas células de mamíferos (transfecção)
O estudo da expressão dos genes de eucariotas evoluídos não teria sido possível sem o
desenvolvimento de técnicas que permitissem a introdução do DNA nas células. O processo
natural de absorção de DNA viral pelas células é muito pouco eficaz, e só foi possível
melhorá-lo recorrendo ao método de co-precipitação DNA-cálcio. O DNA purificado é
precipitado em presença de iões de cálcio e os grânulos assim formados são fagocitados pelas
células em cultura; uma pequena fracção deste DNA é susceptível de se integrar no DNA
cromossómico das células. Afim de identificar as células que integraram este DNA foi
preciso desenvolver técnicas de selecção apropriadas.
Um sistema muito utilizado recorre ao isolamento de linhagens celulares deficientes em
timidina kinase, enzima que intervém na biossíntese das pirimidinas. Esta enzima fosforila a
timidina proveniente da degradação do DNA celular; a TMP formada é em seguida
transformada em TTP e reincorporada no DNA. Esta via metabólica não é, no entanto,
essencial para a sobrevivência da célula que utiliza habitualmente a dCDP. É portanto
possível obter células tk- perfeitamente viáveis. O isolamento de mutantes tk- efectua-se
segundo o esquema da Fig. 150.II. Em presença de bromodesoxiuridina, as células cuja tk é
funcional são inviáveis porque esta enzima fosforila o composto análogo, que é incorporado

106
no DNA, tornando-o hipersensível à radiação ultravioleta. As raras células deficientes em tk
sobrevivem a este tratamento.
No entanto, as células tk- morrem no meio HAT (meio que contém hipoxantina,
aminopterina e timidina), porque a aminopterina bloqueia a utilização normal de dCDP
obrigando a célula a recorrer à acção da timidina kinase, única via possível para sintetizar a
TTP. Existe, portanto, um sistema de selecção de células tk+ (Fig. 151.II.). O gene tk pode,
portanto, ser utilizado como marcador de selecção para a integração, no DNA de células de
mamíferos, doutros genes que lhe estão ligados.
O sistema de selecção baseado no gene tk apresenta certos inconvenientes. É preciso,
primeiramente, tornar a linhagem tk- e, por outro lado, obter uma mutação tk-, cuja
frequência de reversão seja muito baixa. Foram, por isso, desenvolvidos sistemas melhores,
que se baseiam na utilização de genes de procariotas associados aos sinais de controlo dos
eucariotas (Fig. 152.II.).
Um dos vectores é portador dum gene bacteriano XGPRT (utilização de xantina como
fonte de purinas, xantina guanina fosforribosil transferase). Este gene foi inserido entre o
promotor e o sítio de adição de poli A do gene de SV40, que codifica para o grande antigénio
T (a célula contém o gene HGPRT, utilização de hipoxantina e praticamente de nenhuma
xantina). Uma grande quantidade de XGPRT bacteriana é produzida sob acção do promotor
muito eficaz. É pois possível seleccionar o fenótipo HGPRT+ em meio HAT nas linhagens
celulares HGPRT-; mesmo nas linhagens selvagens, HGPRT+, é possível seleccionar o
fenótipo HGPRT+, em presença de xantina e de ácido micofenólico que bloqueia a enzima
celular HGPRT, porque as células portadoras do vector são capazes de utilizar a xantina,
graças à enzima codificada pelo vector.
O segundo vector contém o gene procariota que confere resistência à neomicina
(inactivação da neomicina por fosforilação enzimática). Este gene é também inserido entre o
promotor e o sítio de adição poli A de SV40. As células eucariotas são sensíveis a um
análogo da neomicina, G418; este análogo é também desactivado pelo produto do gene
NeoR. Este vector SVneo pode, portanto, ser utilizado na transformação de células de
eucariotas, seleccionadas pela resistência ao G418.
Co-transformação
Na prática, não é necessário ligar, por ligação in vitro, o fragmento de DNA que se
deseja introduzir nas células com o marcador de selecção. Basta co-precipitá-los com iões de
cálcio e transfectar as células. O DNA exógeno encontra-se na forma de longo concatâmero
que se integra ao acaso e em bloco no cromossoma (Fig. 153.II.).

107
Micro-injecção
Outra alternativa é a introdução do DNA em células em cultura por microinjecção no
núcleo celular. Este método, se bem que sofisticado e delicado, é muito eficaz, pois 50 % das
células microinjectadas integram e expressam os genes injectados. Além disso, este método
não requer nenhuma pressão de selecção para manter o gene transfectado e permite em
princípio injectar qualquer DNA em qualquer célula.
Isolamento dos genes transferidos
A possibilidade de transferir genes para as células de mamíferos fornece, ao mesmo
tempo, os meios de os isolar. Uma abordagem possível está ilustrada na Fig. 154.II. O DNA
genómico é cortado em fragmentos por uma ou várias enzimas que se sabe não afectarem o
gene que se pretende transferir. Este DNA é em seguida ligado a um DNA heterólogo (por
exemplo pBR322), de maneira a marcar todos os fragmentos de DNA genómico com um
DNA procariota. Após tranfecção e selecção, o gene que se pretende isolar é integrado no
cromossoma das células receptoras na vizinhança imediata deste marcador. Isola-se então o
DNA das células transformadas e prepara-se um banco genómico, num fago λ ou num
cosmídeo. Este banco é em seguida escrutinado com uma sonda característica do marcador
utilizado. É de esperar que o clone positivo para o marcador, contenha igualmente o gene
seleccionado (no exemplo da figura trata-se da adenosina fosforribosil transferase).
Uma outra abordagem possível, chamada rescue, baseia-se na adição dum DNA
funcional ao DNA que se pretende transferir, por exemplo, uma resistência a um antibiótico
ou um supressor procariota como supF (tRNA) (Fig. 155.II.). Depois da transfecção, o DNA
das células seleccionadas é cortado por uma enzima que não afecta o gene que se pretende
isolar, recircularizado e utilizado na transformação de bactérias. A selecção de resistência a
um antibiótico (ampicilina, por exemplo) fornece os clones portadores do gene pretendido.
As técnicas de transfecção e de isolamento dos genes transferidos permitiram identificar,
nomeadamente, sequências de DNA responsáveis pelo controlo da expressão de certos genes
de eucariotas. No caso preciso do vírus MMTV (Mouse Mammary Tumor Vírus), a
expressão do DNA é controlada pelas hormonas glucocorticóides. O provírus clonado,
introduzido nas células de rato em cultura, contendo o receptor hormonal ad hoc continua a
responder à hormona. A transcrição do DNA proviral transferido nas células aumenta de 5 a
10 vezes em presença da hormona. O sítio sensível ao efector foi identificado e localizado no
long terminal repeat (LTR) do provírus (sequências que intervêm na inserção viral nos
cromossomas celulares) (Fig. 156.II.).
Existem outros genes eucariotas, activáveis ou desactiváveis por diferentes factores; são exemplos o gene da α2-globulina de rato, sensível à insulina, o gene da metalotionéina,

108
induzido pelo cadmium. Em todos estes casos, os elementos de regulação estão localizados
na proximidade do próprio gene.
As técnicas de transferência de genes permitiram identificar e clonar genes implicados na
formação de cancros. Estas experiências começaram a desenvolver-se quando se observou
que o DNA extraído de tumores pode transformar células normais e torná-las malignas (Fig.
157.II.).
Estas células transformadas, injectadas em ratos, levam à formação de tumores. A
conclusão é 1) que, com estes tumores pelo menos, o fenótipo é dominante, isto é, devido à
presença e não à ausência dum produto específico; 2) que o fenótipo transformado das células
é induzido pela expressão dum só gene. É, com efeito, pouco provável que a transformação
das células se pudesse realizar se ela exigisse a transferência e a expressão simultânea de dois
genes.
Clonagem de oncogenes humanos
Vários oncogenes humanos foram clonados por uma das técnicas já referidas
anteriormente e que estão ilustradas nas Fig. 158.II. e 159.II. Assim, os oncogenes presentes
nas linhagens cancerosas derivadas de tumores da bexiga, do neuroblastoma, de tumores do
cólon ou do pulmão foram clonados e caracterizados. Estes genes têm comprimentos
compreendidos entre + 5 e 45 kb. Apresentam todos um arranjo intrão-exão idêntico: quatro
exões codificam para proteínas similares mas distintas, de massas moleculares de cerca de
21.000 daltons.
Estas proteínas, chamadas p21, geralmente associadas à membrana plasmática das
células cancerosas são assaz homólogas dos produtos dos oncogenes já identificados nos
retrovírus. O gene do cancro da bexiga, por exemplo, é muito semelhante ao do oncogene ras
encontrado no genoma do vírus HSV (Harvey Sarcoma Vírus), enquanto que o oncogene do
cancro do pulmão é muito parecido com o oncogene ras do vírus KSV (Kirston Sarcoma
Vírus).
Estes oncogenes humanos, assim como os oncogenes virais, têm os seus homólogos
celulares normais de que eles derivam aparentemente por mutação. O oncogene do carcinoma
da bexiga, por exemplo, difere do seu equivalente celular normal numa única mutação pontual (Gly12→Val12 na p21). Está, no entanto, por demonstrar que esta mutação pontual
seja a causa única do cancro de bexiga. Também não se conhece o papel da proteína p21,
nem no estado normal, nem no estado mutado.

109
Vectores virais utilizados para transfectar as células de mamíferos
A infecção viral é um processo natural muito eficaz que conduz à introdução de várias
cópias dum mesmo gene em cada célula receptora. O genoma viral comporta promotores
muito potentes, susceptíveis de expressar eficazmente os genes que dependem deles e de
produzir RNA e as proteínas correspondentes em abundância. Estas propriedades conduziram
naturalmente à construção de vectores derivados de vírus para introduzir DNAs heterólogos
nas células animais.
Vector SV40
Foram adoptadas duas vias para tirar partido do DNA viral como vector. A primeira
consiste em construir recombinantes entre o DNA de SV40 e um fragmento de DNA
heterólogo, e introduzir em seguida as moléculas híbridas nas células receptoras afim de
sintetizar as partículas virais que contêm o DNA recombinante.
a) Substituição da região tardia de SV40
Quando se substitui o DNA de SV40, correspondente às funções tardias (implicadas na
síntese da cápsula viral), por um fragmento de DNA heterólogo é necessário complementar
estas funções perdidas pelas fornecidas por um vírus amputado das funções precoces
(implicadas na replicação do DNA viral). Transfectam-se, simultaneamente, as células pelo
DNA de SV40 recombinante e o DNA helper; um fornece as proteínas da cápsula, o outro, as
funções de replicação. Resulta uma população viral mista, difícil de separar. Utiliza-se, então,
a mistura de partículas virais, para re-infectar outras linhagens celulares e acumular o produto
do gene clonado (Fig. 160.II.).
b) Substituição da região precoce de SV40
Quando se substitui o DNA de SV40, correspondente às funções precoces por um DNA
heterólogo, a produção de partículas virais intactas depende da complementação pelas
proteínas precoces, os antigénios T. É possível evitar a utilização dum vírus helper
introduzindo o DNA de SV40 recombinante nas células COS de macaco. Estas células já
foram transformadas por um vírus SV40 portador de DNA codificando para as funções
precoces, mas deficiente em relação à origem de replicação viral. Por consequência, nestas
células, o vírus SV40 fornece os antigénios T mas não se pode replicar independentemente
dos cromossomas da célula (Fig. 161.a.II.). Quando um DNA de SV40 recombinante,
deficiente em funções precoces, é introduzido nas células COS, a origem de replicação que
ele contém será reconhecida pelos antigénios T celulares e o vírus poderá replicar-se e
fornecer uma descendência viral normal (Fig. 161.b.II.)
A vantagem deste método é que todas as partículas virais produzidas contêm o DNA
recombinante e não ha contaminação pelo vírus helper.

110
É possível, por outro lado, utilizar unicamente a origem de replicação do SV40 para
transfectar células. Constrói-se um DNA recombinante portador desta origem e dum
fragmento de DNA heterólogo. Nas células COS, os antigénios T celulares induzem a
replicação da molécula recombinante na forma plasmídica, até um número de cópias muito
elevado. O DNA heterólogo replicar-se-á pois, pelo menos de maneira transitória,
independentemente do DNA celular, produzindo uma alta percentagem de mensageiro
correctamente processado (Fig. 162.II.).
Vector vaivém derivado de SV40
Um plasmídeo, portador da origem de replicação SV40 ligada a um marcador de
selecção de eucariota, é levado a integrar-se no DNA cromossómico de células de rato.
Quando se fusionam estas células com células COS, os antigéneos T que estas produzem
difundem-se no núcleo das células de rato e induzem a replicação na origem de replicação de
SV40 presente nos cromossomas. Esta replicação bidireccional conduz à produção de
moléculas de DNA circulares que se soltam dos cromossomas e podem ser isoladas
facilmente. Se a origem de replicação procariótica e um marcador de selecção para procariota
estiverem presentes, na vizinhança da origem de SV40, estas moléculas circulares podem ser
utilizadas para transformar bactérias e recuperar o plasmídeo de partida (Fig. 163.II.). Este é
um exemplo de vector vaivém capaz de se replicar, tanto em células animais, como em
bactérias.
Habitualmente, os vectores derivados de SV40 possuem, para além da origem de
replicação SV40, sequências virais estimuladoras (enhancers). Estas sequências estão
localizadas a montante do promotor do gene T de SV40, elas estimulam a expressão de
qualquer promotor contido num plasmídeo no qual elas foram clonadas.
Vector BPV (Bovine Papilloma Virus)
O vírus BPV responsável pelas verrugas do gado bovino tem um genoma de 8 kilobases.
O DNA circular de BPV é capaz de transformar certas linhagens celulares de rato em
fenótipo maligno. Nestas células, o DNA do BPV permanece circular e extra-cromossómico,
à razão de 30 a 100 cópias por célula (situação análoga à dos plasmídeos). Pode-se clonar
DNAs heterólogos num genoma de BPV e introduzi-los desta maneira em células de rato em
cultura (Fig. 164.II.).
Se sequências apropriadas de pBR322 forem clonadas no DNA de BPV, obtém-se um
vector vaivém capaz de se manter na forma plasmídica nas bactérias e nas células de rato. Na
prática, não se utiliza o DNA completo do BPV, mas antes um fragmento correspondente a
69 % do genoma, fragmento dotado da capacidade de transformação das células e de
replicação autónoma extra-cromossómica. Afim de facilitar a identificação das células

111
transformadas pelo vector BPV, introduz-se um marcador de selecção dominante, como por
exemplo, a resistência à neomicina (na realidade resistência a um análogo G418).
Um dos vectores derivados do BPV, correntemente utilizado para expressar DNAs
heterólogos está ilustrado na Fig. 165.II.
Vectores derivados de retrovírus
Vimos que o genoma dos retrovírus é constituído por uma molécula de RNA semelhante
a um RNA mensageiro, possuindo a estrutura cap em 5' e uma cauda poli A em 3'. Um
retrovírus têm um número restrito de genes, para além do gene pol, responsável pela síntese
da enzima característica dos retrovírus, possui um gene gag que governa a síntese de
proteínas estreitamente associadas ao genoma viral e um gene env, que determina a síntese
das proteínas do invólucro; por fim as sequências ditas LTR, nas duas extremidades do
genoma, que intervêm na inserção do vírus nos cromossomas das células.
O RNA do retrovírus é copiado em DNA bicatenário, que se integra no genoma das
células infectadas. Uma vez integrado, o provírus é transcrito como um gene celular. O
provírus integrado possui em cada uma das suas extremidades uma sequência LTR, onde fica
localizado o único promotor conhecido para a expressão dos genes virais. A produção dos
diferentes RNAs que codificam para as diferentes proteínas virais é regulada ao nível do
splicing do transcrito primário. Os provírus clonados podem ser utilizados para introduzir
DNAs heterólogos nas células de mamíferos, se se substituir um dos genes virais por um
fragmento de DNA estrangeiro (Fig. 166.II.).
As células infectadas por um retrovírus recombinante produzirão grandes quantidades do
novo RNA mensageiro cuja transcrição começa no LTR. Com a complementação dum vírus
helper, são fornecidas as funções necessárias ao encapsulamento do RNA do retrovírus
recombinante e por consequência à produção de partículas virais.
Na Fig. 167.II. estão representadas duas maneiras de produzir retrovírus. Na parte A da
figura está representado um primeiro método, segundo o qual se faz penetrar (transfecção),
por um método fisico-químico, o DNA dum retrovírus no qual se introduziu um gene
estrangeiro (este ocupa, por exemplo, o lugar do gene pol), numa célula dita assistente que é,
por outro lado, infectada por um vírus selvagem. Este poderá dirigir a síntese de novas
partículas virais infecciosas selvagens e, além disso, fornecer as moléculas de transcritase
reversa às partículas virais, que incorporaram o RNA que veicula o gene estrangeiro, o qual
foi sintetizado graças ao DNA integrado nos cromossomas da célula. Estas últimas partículas
virais poderão ser utilizadas, conjuntamente com as selvagens, para infectar células alvo, tais
como células de embrião de mamífero.

112
O método representado na parte B da figura é uma versão aperfeiçoada da precedente.
Visa produzir unicamente partículas virais contendo o genoma viral que veicula o gene
estrangeiro. Para isso, prepara-se um DNA mutado, a partir do selvagem, amputado duma
sequência nucleotídica ψ que intervém no empacotamento do RNA viral pelas partículas do
invólucro. O método consiste em fazer penetrar numa célula assistente, uma cópia de DNA
viral amputado da sequência ψ e uma cópia de DNA viral que veicula o gene estrangeiro
(contendo a sequência ψ). As cópias do DNA vão-se alojar nos cromossomas da célula e
dirigir a síntese dos diferentes elementos das partículas virais (RNA e proteínas).
As partículas infecciosas, finalmente formadas, comportarão unicamente o RNA que
veicula o gene estrangeiro, porque só este foi empacotado.
É frequente que os retrovírus, uma vez inseridos no cromossoma, activem genes
vizinhos. Estes genes, que eram silenciosos antes da inserção do retrovírus, podem revelar-se
proto-oncogenes e induzir a transformação cancerosa da célula infectada. Um certo número
de cancros animais são o resultado deste género de mecanismo.
Os retrovírus são relativamente instáveis e, mesmo quando foram tornados não
infecciosos pela eliminação do gene da transcritase reversa, podem, uma vez alojados nos
cromossomas duma célula, produzir de novo um vírus infeccioso recombinando-se a vírus
latentes presentes nas células hospedeiras. Por estas razões, outras vias têm sido estudadas,
tais como o recurso aos AAV (Adeno-Associated Virus) que parecem inserir-se de maneira
estável e sem inconvenientes no genoma das células que ele infecta.
f. Introdução de genes estrangeiros em óvulos fertilizados. Animais transgénicos
A transfecção de células em cultura fornece um instrumento indispensável ao estudo da
regulação dos genes de eucariotas. Esta abordagem é, no entanto, limitada a células muito
diferenciadas e não fornece nenhuma indicação sobre os mecanismos moleculares
responsáveis pelo controlo da expressão dos genes num dado tecido celular, nem sobre a
evolução deste controlo durante o desenvolvimento embrionário.
Uma maneira de abordar estes problemas baseia-se na introdução de genes estrangeiros,
facilmente identificáveis, em zigotos ou embriões precoces e na análise da sua expressão nos
tecidos destes embriões ou dos animais deles nascidos. O método, simples no seu princípio,
consiste na deposição duma solução dos genes clonados no núcleo dum ovo (de rato por
exemplo) recentemente fecundado (a deposição no citoplasma conduz frequentemente à
degradação rápida do DNA por enzimas). Mais precisamente, a solução de DNA é micro-
injectada num dos pro-núcleos pertencentes às células sexuais, antes que elas se associem no
ovo fecundado. O pro-núcleo macho, pertencente ao esperma, é habitualmente escolhido para

113
a micro-injecção, porque é de maiores dimensões e permanece durante algumas horas perto
da superfície do óvulo fecundado (Fig. 168.II.).
Os ovos de mamífero (rato, porco, etc.) assim injectados são, ou reintroduzidos no
oviducto duma fêmea portadora, preparada hormonalmente para o efeito, ou desenvolvidos in
vitro, em cultura, até ao estado blastocito antes de serem implantados no útero (Fig. 169.II.).
Este género de experiências foi realizado com vários genes clonados, correspondentes à
hormona de crescimento, ao interferão, à insulina, à globina, à timidina kinase, etc. De 200 a
30.000 moléculas de DNA, de comprimento variável entre 5 e 50 kbases são injectadas por
ovo. A percentagem de sobrevivência dos ovos injectados de rata é de 10 a 30 % e a
proporção de ratas que integram o DNA injectado nos seus cromossomas é de cerca de 40 %.
A percentagem de sucesso parece, no entanto, depender da estirpe de rato e da espécie
utilizada. A técnica é muito menos eficaz quando aplicada ao carneiro, à vaca, ao porco e ao
coelho.
Na maioria dos casos analisados, várias cópias de DNA micro-injectado encontram-se
integradas num dos cromossomas dum par de cromossomas, no rato adulto. Duma
experiência para outra a integração ocorre num cromossoma qualquer, o que mostra que o
processo de integração não é específico. O DNA integrado encontra-se habitualmente tanto
nas células somáticas como nas células germinais (óvulos e esperma). Ele é, portanto,
transmitido por intermédio destas como um carácter mendeliano (Fig. 170.II.), o que implica
que o acontecimento de integração se passa muito cedo durante o desenvolvimento
embrionário, antes de a população de células germinais se separar das células somáticas de
origem. O DNA estrangeiro pode-se transmitir durante várias gerações; integra-se, portanto,
de maneira estável, no genoma do rato, sem no entanto lhe conferir nenhuma vantagem
selectiva.
Os pontos fracos deste método de micro-injecção são a impossibilidade de prever qual o
número de cópias de um dado gene que se vai inserir no património genético do animal e a
localização da inserção, que se faz ao acaso, o que têm, por vezes, como efeito, desactivar
certos genes da célula hospedeira e provocar anomalias morfológicas mais em menos graves.
Além disso, o nível de funcionamento do gene estrangeiro é totalmente imprevisível.
Expressão do DNA estrangeiro nos animais transgénicos
Os óvulos micro-injectados que chegam ao seu termo normal de desenvolvimento são
unicamente aqueles cujo desenvolvimento embrionário não é perturbado pela integração do
DNA estrangeiro. É, pois, necessário que o DNA integrado o seja de forma passiva, seja
geneticamente silencioso e não expresso, ou controlado normalmente na sua expressão.

114
O exemplo seguinte mostra que a expressão do DNA integrado pode ser específica de
tecidos celulares. Quando um plasmídeo, portador do promotor da metalotioneína (MT) a
montante da sequência que codifica para a timidina kinase, é microinjectado em embriões de
rato, prevê-se que a expressão do gene da timidina kinase seja controlada por sinais
moleculares que desencadeiam a acção do promotor da metalotioneína, por iões Cd, por
exemplo (o papel fisiológico de metalotioneína é de fixar iões de metais pesados, sendo o
promotor MT induzido por estes iões) (Fig. 171.II.). A exposição dos ovos micro-injectados a
estes iões metálicos conduz, com efeito, à síntese de timidina kinase.
Por outro lado, os ovos micro-injectados, após reimplantação em fêmeas portadoras, dão
origem a uma descendência que possui o gene MT-tK integrado no cromossoma numa
proporção de 10 a 15 %. A injecção de sulfato de cádmio nestes ratos conduz à síntese de
timidina kinase, mas a expressão do gene integrado é específica de um tecido dado, neste
caso, o fígado, o que corresponde à localização natural da metalotioneína no rato. Durante as
gerações seguintes, uma parte da descendência perde o gene MT-tK. Entre os ratos que
herdaram o gene, certos deixam de o expressar e outros expressam-no com abundância
superior à dos pais. Aparentemente, durante a passagem pelo óvulo e pelo esperma e durante
o desenvolvimento embrionário, acontecimentos importantes levaram à extinção ou à
amplificação da expressão do gene integrado.
Numa outra abordagem experimental, um plasmídeo, portador do promotor da
metalotioneína de rato e do DNA genómico que codifica para a hormona de crescimento
(GH) de rato, foi micro-injectado em óvulos de rata (a hormona de crescimento, sintetizada
normalmente na hipófise encontra-se no fígado onde desencadeia a síntese de somatomedinas
que estimulam o crescimento muscular, ósseo e cartilaginoso) (Fig. 172.II.). Este plasmídeo
contém os sinais de regulação MT e o DNA genómico que codifica para a GH, como o sinal
de iniciação AUG, os exões e os intrões correspondentes e o sinal de poliadenilação. A
transcrição deste DNA fusionado conduz à síntese de um RNA que codifica para a hormona
de crescimento do rato. Na micro-injecção utiliza-se o plasmídeo linearizado, que se integra
melhor que a molécula circular, à razão de 600 cópias por pro-núcleo macho. Foram
injectados e implantados 170 ovos no útero de ratas portadoras, das quais 21 chegaram ao seu
termo. Sete ratos em 21 tinham o DNA integrado em várias cópias num cromossoma. Seis
destes 7 ratos desenvolveram-se mais rapidamente que os outros, mesmo na ausência de
indução do promotor MT com iões de cádmio. Como previsto, a hormona de crescimento
nestes 6 ratos expressa-se 100 a 800 vezes mais que em ratos de controlo. A aceleração do
crescimento nestes ratos transgénicos resulta, por consequência, da integração e da expressão
do gene fusionado MT-GH (Fig. 173.II.).

115
No domínio da micro-injecção de genes, os trabalhos que se desenvolvem actualmente
visam essencialmente definir as mudanças moleculares que ocorrem durante a diferenciação
e correlacionar estas modificações com as alterações da expressão dos genes integrados.
A utilização dos retrovírus
Um outro tipo de experiências tira partido das propriedades dos retrovírus, afim de os
utilizar como vectores que veiculam genes estrangeiros nos animais. Por exemplo, o DNA
proviral do vírus MLV (vírus da leucémia de rato) pode ser integrado em embriões de rato de
duas maneiras diferentes:
1) infectando os embriões com o vírus para desencadear a síntese de DNA proviral
bicatenário que é seguida de integração num cromossoma;
2) micro-injectando directamente o DNA proviral clonado nos embriões.
No primeiro caso, os resultados obtidos diferem completamente segundo o estado de
desenvolvimento do embrião (32 células ou embrião de 8 a 10 dias) (Fig. 174.II.). A análise
da descendência resultante da integração no embrião de 32 células mostra que o DNA
proviral está integrado em algumas células pertencentes a diferentes tecidos e, sobretudo, que
este DNA proviral está intensamente metilado e não expresso. Na descendência derivada da
infecção de embriões mais velhos, o DNA proviral encontra-se em todos os tecidos sob
forma não metilada e muito expresso. Estes resultados sugerem que a metilação do DNA
integrado determina, pelo menos parcialmente, a ausência de expressão do gene. Parece, pois,
evidente que os embriões tardios não possuem mais a capacidade para metilar e, portanto,
suprimir geneticamente, a expressão do DNA proviral.
Numa segunda experiência, o DNA proviral, clonado a partir de células de um dos ratos
da experiência precedente, foi injectado, 1°) nos núcleos de óvulos fertilizados, ou 2°) no
citoplasma destes óvulos. A descendência resultante da micro-injecção no núcleo contém
várias cópias do DNA proviral em todas as células, altamente metiladas e não expressas. A
descendência derivada da micro-injecção no citoplasma, em compensação, contém uma só
cópia de DNA proviral por célula e em todas as células. Além disso, 10 % dos ratos
produzem RNA mensageiro correspondente ao vírus MuLV, localizado em quantidade muito
variável em todos os tecidos. O sítio de integração no cromossoma parece, pois, influenciar a
expressão do DNA viral duma maneira específica de tecido.
Conclusões e resumo
1) É possível introduzir genes por micro-injecção em células somáticas e germinais.
2) O sítio cromossómico no qual se integra o DNA e o grau de metilação deste DNA
influenciam a activação do gene durante o desenvolvimento.

116
A ausência de metilação pode ser correlacionada com a expressão do gene.
3) O grau de expressão do gene integrado é também influenciado pela diferenciação celular
e pode ser específico do tecido.
4) Genes estrangeiros podem ser colocados sob controlo regulatório normal das células
hospedeiras.
g. Expressão em células de insectos
As células de insectos poderão ser uma alternativa às células animais em cultura. Vários
grupos mostraram que vectores derivados de vírus de insectos podem ser utilizados para a
produção notável de substâncias, como interleucinas, interferões, várias proteínas derivadas
dos vírus HIV, da hepatite B, da influenza, etc.
O sistema que têm sido utilizado com sucesso baseia-se na utilização de vectores de
Autographa californica, vírus das poliedroses nucleares (AcMNPV), protótipo da família
Baculoviridae, que possui um largo espectro de hospedeiros, infectando mais de 30 espécies
de lepidópteros, assim como, tricópteros, himenópteros, dípteros e crustáceos.
A. californica, assim como os outros membros do subgrupo A dos baculovírus, formam
oclusões constituídas por proteínas virais, com a configuração de cristais ou corpos oclusivos,
chamados corpos poliedrais ou poliedros, encerrando cada um, um número elevado de
partículas virais. A proteína principal dos poliedros é a poliedrina de MM de 29.000 daltons,
que é produzida intensamente, acumulando-se, por exemplo, nas culturas celulares de
Spodoptera frugiperda, correntemente utilizadas, a níveis elevadíssimos de 1 mg/ml por 1-2
106 células infectadas, representando, pelo menos, 50 % das proteínas da célula detectáveis
em gel de poliacrilamida-SDS. O ciclo de vida do baculovírus está representado na Fig.
175.II.
Os baculovírus são o grupo mais relevante de vírus conhecido que infecta insectos.
Constituem aparentemente o único grupo que possui os artrópodes como único hospedeiro.
Devido a esta especificidade, à sua natureza contagiosa e à sua estabilidade relativa no meio
ambiente, têm um enorme potencial na agricultura como agentes extremamente úteis, seguros
e selectivos de controlo de pestes.
A aplicação mais atraente do baculovírus, enquanto sistema de vector em tecnologia do
DNA recombinante, é o seu potencial para expressar grandes quantidades de produtos de
genes estrangeiros. A poliedrina expressa-se em abundância superior à de qualquer outra
proteína em células de eucariotas infectadas por vírus. Mais de 15 % da massa total de
proteína encontrada em células de larvas infectadas por NPV é constituída por poliedrina.

117
Para além de sequências promotoras fortes, que dão a vantagem aos vectores de
expressão deste vírus de expressar proteínas recombinantes a elevados níveis, comparados
com os sistemas bacterianos, de levedura ou de mamíferos, os baculovírus possuem várias
outras características que os torna interessantes para a clonagem e expressão de DNA
estrangeiros:
1) Não são patogénicos para os vertebrados ou as plantas porque não se replicam nestes
tipos de células, não sendo necessário recorrer a células transformadas ou a elementos
transformantes, como é o caso com os sistemas de expressão das células dos mamíferos.
2) Possuem um espectro de hospedeiros limitado a certos invertebrados.
3) O genoma foi mapeado relativamente a muitas enzimas de restrição e caracterizado
em relação à actividade transcricional.
4) Os vectores do baculovírus utilizam muitos sistemas de modificação, processamento e
transporte de proteínas que ocorrem nas células superiores de eucariotas, que podem ser
essenciais para as funções biológicas completas das proteínas recombinantes. Estas são,
consequentemente, com frequência, antigenicamente, imunologicamente e funcionalmente
semelhantes às suas homólogas autênticas.
5) O invólucro do baculovírus pode empacotar pelo menos dois comprimentos
genómicos, o que sugere que é capaz de inserir famílias de genes, ou um grande número de
genes.
O genoma do AcMNPV consiste num DNA circular, super-enrolado, bicatenário, com
um comprimento de cerca de 128 kilobases. O gene da poliedrina do AcMNPV foi mapeado
e sequenciado. Foi demonstrado que não é essencial para a replicação ou a produção de vírus
extracelular em células em cultura. A eliminação ou a desactivação insercional do gene da
poliedrina leva à produção de vírus de oclusão negativa (Occ-), que formam placas de lise
distintas das formadas pelos vírus selvagem de oclusão positiva (Occ+). Estas morfologias
distintas das placas fornecem um meio de escrutinar visualmente os vírus recombinantes, nos
quais o gene da poliedrina do tipo selvagem AcMVPV foi substituído por um outro gene, ou
por um híbrido.
A natureza não essencial e os elevados níveis de expressão do gene da poliedrina, aliados
à facilidade com a qual os vírus recombinantes Occ- podem ser detectados, tornam o
promotor deste gene particularmente apropriado para a construção dum vector de expressão.
Construção dum vector de transferência do baculovírus
A maioria dos vectores de transferência contém sequências do AcMNPV que incluem o
promotor do gene da poliedrina e várias percentagens do DNA viral, que flanqueia em 5' e

118
em 3' o gene da poliedrina, clonado num plasmídeo bacteriano de número elevado de cópias.
As sequências do gene estrangeiro no plasmídeo recombinante podem ser transferidas para o
AcMNPV por recombinação homóloga, no interior de uma célula transfectada com os DNAs
do plasmídeo e do vírus selvagem.
A Fig. 176.II. esquematiza a sequência de etapas de clonagem de um gene estrangeiro e
o escrutínio de placas recombinantes. Um fragmento apropriado de gene estrangeiro é
inserido num vector de transferência na orientação correcta a jusante do promotor da
poliedrina. Células de insecto (S. furgiperda) em cultura são co-transfectadas com uma
mixtura de DNA viral e de plasmídeo e a progenitura viral é plaqueada. Os recombinantes
virais Occ- são identificados visualmente ou por hibridação com uma sonda da DNA e
purificados a partir da placa, até atingirem a homogeneidade.
O baculovírus recombinante independente de helpers produz uma proteína recombinante
48-72 horas após a infecção, a qual é essencialmente lítica após 4-5 dias.
O vector de transferência mais correntemente utilizado para a introdução de genes
estrangeiros no AcMNPV é o pAC373 (Fig. 177.II.). Deriva dum plasmídeo constituído por
um fragmento EcoRI de 7kb contendo o gene da poliedrina de AcMNPV clonado no
fragmento EcoRI-HindIII de pVC8. Após mutagénese com Bal31 e adição de linkers BamHI
(CGGATCCG), obtém-se o plasmídeo pAC373 que contém a eliminação duma sequência
entre a posição -8 (8 bases a montante do ATG do gene da poliedrina e aproximadamente 40
bases a jusante do sítio de iniciação de transcrição) e os locais naturais BamHI no núcleotídeo
+121.
Uma boa expressão de proteínas estrangeiras não fusionadas requer que os genes
estrangeiros tenham uma sequência condutora curta. Os comprimentos das sequências não
codificantes em 3' e 5' (cuja influência em relação aos níveis de expressão se desconhece) dos
genes expressos em vectores dos baculovírus têm variado de 3 a 400 bases.
Como a sequência condutora da poliedrina é muito rica em AT, recomenda-se,
geralmente, que sequências guias ricas em G-C ou demasiado compridas, sejam truncadas,
tanto quanto possível, antes do gene estrangeiro ser inserido no vector de transferência.
Todos os vectores de transferência têm o sinal de poliadenilação da poliedrina intacto. Bons
níveis de expressão foram obtidos com genes que possuem os seus próprios sítios de
poliadenilação adicionados aos do gene da poliedrina. Todos os genes expressos com níveis
elevados foram derivados de cDNA ou de clones de genomas que não contêm intrões.
Foram construídos vários outros vectores de transferência apropriados para a produção
de proteínas fusionadas ou não fusionadas .

119
Actividade biológica das proteínas recombinantes
As proteínas recombinantes produzidas nas células de insecto, em vectores de
baculovírus, são biologicamente activas e a maioria é processada pós-tradução, produzindo
produtos recombinantes muito semelhantes às proteínas autênticas. As proteínas
recombinantes podem ser segregadas, dirigidas para o núcleo, para a superfície da célula,
associadas em complexos oligoméricos e em dímeros ligados por pontes dissulfureto,
clivadas proteoliticamente, fosforiladas, N-glicosiladas, O-glicosiladas, miristiladas ou
palmitiladas. Estes produtos são antigenicamente, imunologicamente e funcionalmente
semelhantes aos seus análogos naturais.
Todavia, muito está ainda por elucidar acerca da natureza da glicosilação nas células dos
insectos. A capacidade destas células para efectuarem a N-glicosilação implica a adição de
oligossacarídeos do tipo manose pesado. Estudos efectuados com linhagens celulares de
Aedes aegypti e Aedes albopictus (mosquitos), mostraram que os oligossacarídeos ligados à
Asparagina são deficientes em ácido siálico, galactose e fucose. Assim, a conversão de
manose pesado em complexos oligossacarídicos ligados a N parece não se efectuar naquelas
células de insectos. Nas células de mosquitos a ausência de complexos N-glicanos foi
confirmada pelos baixos níveis de N-acetilglucosaminil-, galactosil- e sialitransferases,
responsáveis pela maturação dos manose-N-glicanos pesados das glicoproteínas dos
mamíferos.
Está ainda por elucidar se esta diferença aparente ou potencial na via do processamento
oligossacarídico entre insectos e vertebrados é um factor importante na produção de
glicoproteínas nas células de insectos, ou se diferentes linhagens de células de insectos
apresentam capacidades diferentes para glicosilar proteínas estrangeiras. Alguma
investigação é ainda necessária para elucidar os detalhes moleculares do processamento,
transporte e condução das proteínas recombinantes produzidas por células de insectos.

120
10. ENGENHARIA GENÉTICA DE PLANTAS
O melhoramento genético das espécies vegetais é uma prática milenária, cujos princípios
só começaram a ser racionalizados com Mendel. No entanto, uma intervenção racional no
património genético de plantas, só nestes últimos anos têm sido objecto de experiências
laboratoriais, com um impacto, actualmente, já bastante importante nas aplicações de campo
e com um futuro altamente promissor.
Com efeito, os numerosos métodos desenvolvidos nestes últimos 20 anos, que visam
regenerar plantas a partir de células vegetais em cultura, conjugados com as técnicas de DNA
recombinante, abrem perspectivas novas para o melhoramento de plantas, nomeadamente no
que respeita ao seu conteúdo em aminoácidos essenciais, e à resistência a doenças, a pragas, a
herbicidas e a condições climatéricas.
No âmbito desta disciplina serão abordados unicamente os grandes princípios da
manipulação do DNA vegetal.
Agrobacterium tumefaciens
Agrobacterium tumefaciens é uma bactéria que infecta um certo número de plantas
causando a formação de tumores, sobretudo no colo das dicotiledóneas. A excrescência, ou
galha, que se desenvolve no colo da planta têm o nome corrente inglês de crown gall (galha
do colo). Esta bactéria vive habitualmente no solo e penetra na planta por uma ferida, em
geral durante um Inverno rigoroso, exercendo o seu poder patogénico nos tecidos que
apresentam lesões provocadas pela geada. O crescimento do tumor, em geral, só se termina
com a morte da planta ou do órgão atingido (Fig. 178.II.). As células destes tumores
assemelham-se fortemente às células cancerosas do tipo animal, tendo adquirido a
propriedade de se multiplicarem de maneira independente e incontrolada.
Em cultura, as células crown gall aglomeram-se formando um calo, isto é, uma massa
não organizada e pouco diferenciada de células, mesmo na ausência de fito-hormonas
(auxina, citoquinina) que são indispensáveis à cultura in vitro de células vegetais normais.
Uma das primeiras contribuições importantes da cultura in vitro dos tecidos vegetais foi
a de demonstrar que as células atingidas de crown gall contêm substâncias específicas que
não existem nas células normais: as opinas, derivadas da arginina.
Por um lado, estes compostos nunca se encontram nas plantas sãs, eles são utilizados
pela bactéria como fonte de carbono e de azoto. Por outro, o carácter tumoral das células de
crown gall é permanente, e independente da presença das bactérias que provocaram o
aparecimento do tumor. Na realidade, o agente tumorígeno na bactéria é um plasmídeo, que

121
funciona integrando uma parte do seu DNA nos cromossomas das células vegetais
hospedeiras e que expressa as funções necessárias para a indução e a metabolização das
opinas.
As células da planta invadida por A. tumefaciens são incapazes de utilizar as opinas. Por
consequência, a infecção bacteriana não só induz um tumor na planta, mas ainda desvia o seu
metabolismo para fabricar aminoácidos que só podem ser consumidos pela bactéria.
À descoberta, nas estirpes patogénicas de A. tumefaciens, dum plasmídeo dum
comprimento nunca antes observado noutro organismo (circular, 200 kbases, + 112 108
daltons, 3 a 5 % do cromossoma bacteriano) (Fig. 179.II.), seguiu-se rapidamente a
demonstração de que era ele que conferia o carácter patogénico à bactéria; daí o seu nome Ti,
indutor de tumor (tumor inducing).
Existem essencialmente dois tipos de plasmídeos Ti, caracterizados pela natureza da
opina que induzem: plasmídeo octopina e plasmídeo nopalina. A bactéria A. tumefaciens só
pode conter um tipo de plasmídeo Ti.
A sequência em bases do DNA dos plasmídeos Ti é pouco conservada, com excepção de
quatro regiões de elevada homologia, das quais uma, como foi demonstrado, se integra no
genoma das células de crown gall. Ela corresponde a um fragmento de Ti, chamado T-DNA
(DNA transferido). É este T-DNA que, integrado no genoma da célula vegetal, confere à
célula, ao expressar-se, o carácter tumoral e a propriedade de sintetizar opinas.
Várias funções foram identificadas e estudadas no plasmídeo Ti. A função Vir de
virulência, responsável pela transferência do T-DNA para a célula vegetal, a função
oncogénica, Onc, responsável pela proliferação tumoral, a função ou as funções que
especificam a síntese das opinas, Ops, e as funções catabólicas (Opc: opine catabolism) que
conferem à estirpe bacteriana a possibilidade de degradar as opinas produzidas pelo tumor.
Convém distinguir bem dois tipos de funções: as funções situadas fora do T-DNA (Vir,
Opc, etc.), que se expressam na bactéria e não são propriamente patogénicas, e as funções
contidas no T-DNA, que se expressam na célula vegetal depois da transferência deste
fragmento (Onc, Ops) (Fig. 180.II.).
A bactéria, em si, não têm efeito patogénico, não secreta toxinas, nem enzimas que
dissolvam as paredes celulares, como outras bactérias fitopatogénicas. A sua acção limita-se
à transferência de um fragmento de DNA, o T-DNA, cuja expressão na célula vegetal é
responsável pela doença. A eliminação, no plasmídeo Ti do segmento correspondente à
função Onc, ou a desactivação, por mutação, dos seus genes, torna a bactéria que o contém
perfeitamente inofensiva, sem no entanto suprimir a propriedade de poder transferir o DNA
para uma célula de planta.

122
Assim que se transfere para a célula vegetal o T-DNA expressa a suas funções. A função
oncogénica (Onc) é responsável pela proliferação celular que conduz ao desenvolvimento do
tumor. Esta proliferação é a consequência da síntese de auxina e de citoquinina, dois factores
fito-hormonais que são em geral necessários para a multiplicação e a diferenciação das
células vegetais. Aqui se encontra a explicação do facto de as células crown gall em cultura
não terem necessidade de fito-hormonas no meio. Estas hormonas determinam, uma, o
aparecimento dos rebentos e, a outra, a formação das raízes. Nas células normais, o equilíbrio
entre auxinas e citoquininas controla a via de diferenciação. Nas células de crown gall estas
duas hormonas são produzidas em grande excesso, mantendo as células num estado
indiferenciado.
As funções Ops estão implicadas na síntese das opinas; não são patogénicas, visto que o
tumor se desenvolve perfeitamente quando elas são desactivadas por mutação e, por outro
lado, a sua expressão na ausência da função Onc é perfeitamente compatível com o
desenvolvimento normal das células ou das plantas que as comportam.
O significado biológico da correlação entre a síntese de opinas nos tumores e a sua
degradação pelas bactérias é fornecido pelo carácter parasitário da bactéria, que realiza uma
manipulação genética natural para criar o seu nicho ecológico, fazendo a planta produzir
substâncias que lhe servem de alimento. De facto, a síntese das opinas constitui um desvio
duma parte da actividade fotossintética da planta em proveito da bactéria (Fig. 181.II.).
O plasmídeo Ti como vector
O processamento e a transferência do T-DNA são mediados por um sistema bacteriano,
que reconhece as sequências flanqueantes de T-DNA, de 23 pb cada. É a região
compreendida entre estes limites que é transferida e se integra no genoma da planta. Foi
demonstrado que nenhum dos genes do T-DNA é necessário para o desenrolar deste processo
e que qualquer gene estrangeiro colocado entre aquelas sequências é transferido com o T-
DNA para a célula vegetal.
Para fazer funcionar, numa célula vegetal, um gene estrangeiro introduzido pelo T-DNA
e pôr em evidência a sua expressão, foi efectuada uma primeira experiência fazendo intervir
o crown gall de tabaco e um gene bacteriano (de E. coli) que confere resistência ao
antibiótico cloranfenicol. Para obter a transcrição do gene na célula do tabaco, o promotor de
E. coli foi substituído por um promotor reconhecido pela polimerase do tabaco, neste caso, o
do gene da nopalina sintetase, proveniente do T-DNA de A. tumefaciens. O gene quimérico
introduzido no plasmídeo Ti expressa, após inoculação duma estirpe de Agrobacterium em
plântulas de tabaco, a actividade de cloranfenicol transacetilase em extractos dos tumores

123
formados. Esta experiência demonstrou, pela primeira vez, que a expressão dum gene
estrangeiro numa célula vegetal era possível.
Neste exemplo, as células transformadas eram tumorais e eram incapazes de regenerar
um indivíduo inteiro. A experiência fundamental seguinte utiliza um T-DNA "desarmado"
dos genes tumorais, para veicular um gene de resistência à canamicina (Fig. 182.II.). O gene
quimérico foi construído associando in vitro a sequência que codifica para a neomicina
fosfotransferase (NPT), que confere a resistência à canamicina, uma sequência promotora e
uma sequência terminadora do gene da nopalina sintetase (NoS) do T-DNA de A.
tumefaciens. O gene assim construído foi introduzido no plasmídeo Ti desarmado das
funções Onc, o qual foi por sua vez integrado em A. tumefaciens. A bactéria é, em seguida,
levada a infectar protoplastas de tabaco (é, geralmente, mais prático infectar células vegetais
em cultura que induzir tumores numa planta inteira). O método consiste em converter células
derivadas de folhas de plantas em protoplastas, (células destituídas de membrana) que são
infectados com A. tumefaciens portador do plasmídeo Ti recombinante e que, em cultura,
regeneram a parede celular e se dividem. Após algumas horas, as bactérias de A. tumefaciens
são mortas adicionando um antibiótico e as células, depois de algumas semanas de cultura em
meio contendo fito-hormonas, formam amontoados celulares, ou calos. Neste momento, o
meio de cultura é substituído por um meio sem hormonas, em presença de um agente
selectivo que permita seleccionar as células que receberam o plasmídeo Ti. Estas sobrevivem,
multiplicam-se e sintetizam opinas. Em certos casos regeneram espontaneamente rebentos e
plantas inteiras.
Algumas devem ser resistentes à canamicina; sendo férteis, a transmissão do carácter de
resistência à descendência pode ser estudada, semeando as sementes resultantes duma auto-
fecundação, num meio contendo canamicina. A observação do comportamento selectivo é
particularmente simples porque a canamicina interfere com o desenvolvimento dos
cloroplastas (lugar de fotossíntese). As plântulas sensíveis aos antibióticos permanecem
brancas. As plantas resistentes possuem a NPT que desactiva a canamicina e assegura o
funcionamento do sistema cloroplástico, apresentam uma cor verde e continuam a
desenvolver-se, enquanto que as plântulas brancas, incapazes de efectuar a fotossíntese,
morrerão. Cerca de um quarto das plântulas não possui o gene de resistência. Esta é a
proporção esperada para a segregação dum gene qualquer segundo as leis de Mendel. O gene
introduzido comporta-se como os próprios genes de planta, fazendo parte do seu património
genético.
O plasmídeo Ti possui duas características ideais para promover a introdução de DNA
heterólogo em células de plantas.

124
1°) A. tumefaciens pode infectar praticamente todas as plantas dicotiledóneas e em
certas condições monocotiledóneas, tais como o trigo e o milho.
2°) O T-DNA integrado nos cromossomas de plantas transmite-se de maneira
mendeliana. Os genes contidos no T-DNA são, ainda, portadores dos seus próprios
promotores aos quais se podem associar genes estrangeiros e que podem dirigir a expressão
destes.
A maneira mais simples de introduzir o T-DNA nas células vegetais consiste em infectá-
las com A. tumefaciens portador do plasmídeo apropriado e deixar agir a natureza. Bastaria,
como vimos, inserir os genes de interesse na região T do plasmídeo Ti. No entanto, isto não é
muito cómodo, devido ao tamanho enorme deste plasmídeo. Com a descoberta da
propriedade, que possuem as funções de virulência, de agir à distância, foram desenvolvidos
sistemas binários de transferência de genes que, como o nome indica, fazem intervir dois
plasmídeos. Um dos plasmídeos é o Ti sem T-DNA. O outro é um pequeno plasmídeo que
serve de vector aos genes a introduzir. Este segundo plasmídeo têm, além disso, a
propriedade de se poder multiplicar, tanto em E. coli, como em A. tumefaciens. Esta
propriedade, assim como a seu tamanho relativamente pequeno, simplifica
consideravelmente as manipulações para a realização de construções de genes a introduzir
nas plantas.
A abordagem utilizada está representada na Fig. 183.II. O T-DNA é primeiramente
excisado do plasmídeo Ti por digestão com enzimas de restrição e, em seguida, é introduzido
num vector de procariota, como o pBR322. O T-DNA clonado pode assim ser amplificado e
isolado. Na etapa seguinte, um gene dado é inserido no T-DNA, sem afectar as funções
essenciais deste DNA. O híbrido é, em seguida, introduzido nas bactérias de A. tumefaciens
contendo o plasmídeo Ti intacto. Por recombinação genética homóloga entre o T-DNA
híbrido e o T-DNA do plasmídeo Ti, obtêm-se plasmídeos Ti completos portadores do T-
DNA híbrido. Alternativamente, basta fornecer um Ti contendo unicamente a região vir, para
assegurar a transformação das plantas; neste caso, deixa de haver recombinação homóloga
mas existe somente complementação de função.
Também é possível transformar protoplastas vegetais com o DNA do plasmídeo Ti
recombinante; este método é, no entanto, muito menos eficaz que o precedente, mas mostra
que a bactéria A. tumefaciens não é essencial para a transformação e o seu papel é só de
facilitar a introdução do plasmídeo Ti nas células.
A regeneração de plantas inteiras e férteis, a partir de células tumorais, não é, como
vimos, geralmente, possível se se utilizar o plasmídeo Ti intacto. No entanto, certos mutantes
Ti contêm eliminações na região T, que suprimem a capacidade para formar tumores mas

125
mantêm a síntese de opinas. As células vegetais transformadas por este plasmídeo Ti mutante
comportam-se, em cultura, como células normais e podem ser regeneradas em plantas férteis.
A título de exemplo, a Fig. 184.II. descreve um sistema de vectores correntemente
utilizado que consiste, por um lado, num vector vaivém do tipo pBR322 e por outro lado,
num vector Ti aceitador não oncogénico. Este vector é excisado dos genes que impedem a
diferenciação das células transformadas (funções responsáveis, como vimos, pela síntese de
fito-hormonas). Os genes removidos do T-DNA são substituídos pelo plasmídeo pBR322. A
extremidade direita do T-DNA residual codifica a enzima nopalina sintetase. Esta actividade
enzimática serve de marcador para a selecção das células transformadas.
O gene heterólogo que se pretende introduzir na planta é, primeiro, clonado no vector
vaivém derivado do pBR322 e, em seguida, inserido no plasmídeo Ti onc-, aceitador, por
recombinação homóloga. Um simples crossing-over conduz à co-integração do vector
vaivém no Ti onc-, entre as regiões flanqueantes do T-DNA. Se um marcador de selecção
apropriado (resistência a um antibiótico) for utilizado no vector vaivém, poder-se-á
seleccionar para este acontecimento de recombinação em A. tumefaciens (pBR322 não se
replica em A. tumefaciens).
Pode-se, por exemplo, introduzir no vector vaivém, o promotor vegetal (NOS) associado
a um gene de resistência à canamicina, nas plantas, seguido das regiões (NOS) que codificam
para o splicing e a poliadenilação dos mensageiros. Por consequência, esta unidade de
transcrição funcionará na planta e permitirá seleccionar para a resistência à canamicina, i. é.,
para os transformantes.
Expressão controlada pela luz e específica de órgão, dum gene de trigo em plantas de tabaco
A transformação das monocotiledóneas com A. tumefaciens não é corrente.
Aparentemente, o processo de transformação pelo plasmídeo Ti falha ao nível do colo. Ora,
uma grande parte dos vegetais de consumo corrente pertencem à família das
monocotiledóneas, tais como os cereais milho, trigo, arroz. É, no entanto, possível expressar
e controlar normalmente genes de monocotiledóneas em plantas dicotiledóneas. A introdução
do gene de trigo que codifica para uma proteína do complexo de assimilação da luz (cab) no
genoma do tabaco, pelo sistema Ti, e a regeneração de plantas inteiras, é um exemplo.
Na primeira etapa, deriva-se do genoma de trigo o DNA genómico que codifica para a
proteína Cab e clona-se num vector vaivém, do tipo pBR322, contendo um sistema de
selecção para procariotas (SpcR / StrR), um sistema de selecção para células vegetais
(NOS/NPTII) (resistência à canamicina), uma região de homologia com o plasmídeo Ti e a
região do T-DNA (Fig. 185.II). Este plasmídeo recombinante é, em seguida, mobilizado na

126
bactéria A. tumefaciens "desarmada", i. é., contendo um Ti Onc-. Os protoplastas de tabaco são
transformados e as células resistentes à canamicina seleccionadas. Após a obtenção de calos,
as plantas completas são regeneradas em condições apropriadas. Pode-se em, seguida,
verificar que o gene cab, integrado nas plantas transgénicas, é expresso e controlado pela luz
de maneira idêntica à da planta de origem. Para isso, expõem-se as plantas transgénicas a
períodos variáveis de iluminação ou de obscuridade, em seguida, extrai-se o RNA
mensageiro das folhas e doseia-se por hibridação com um fragmento de DNA radioactivo
(Northern blotting), derivado do gene cab. Os resultados mostram que o gene cab do trigo se
expressa, pois o RNA mensageiro é detectado correctamente transcrito, processado e
poliadenilado. Além disso, a percentagem do mensageiro cab é claramente função do tempo
de exposição à luz; decresce quando as plantas permanecem na obscuridade e aumenta
quando são iluminadas.
Constata-se, por outro lado, que a síntese do produto codificado pelo gene cab nas
plantas transgénicas é especifico de órgão; encontra-se uma grande quantidade de
mensageiros nas folhas e praticamente nenhum nos caules, raízes e pétalas (Fig. 186.II.).
Estes resultados mostram que o gene cab do trigo é transcrito correctamente e se
expressa selectivamente nas plantas transgénicas e que, portanto, o sistema de transcrição
nuclear das dicotiledóneas reconhece as regiões reguladoras do gene cab derivado duma
monocotiledónea.
Para que este tipo de manipulação apresente um interesse agronómico é preciso
identificar, isolar e clonar genes susceptíveis de melhorar plantas cultivadas. Se os caracteres
tiverem uma base genética complicada isto pode constituir uma tarefa difícil. O rendimento
de produção ou a resistência duma planta ao frio, por exemplo, são provavelmente caracteres
governados por numerosos genes que não serão, sem dúvida, durante bastante tempo, objecto
de manipulações. Em compensação, quando se trata dum gene microbiano ou mesmo dum
gene vegetal, cujo produto (uma enzima ou uma proteína de estrutura) é conhecido e, de
preferência, abundante ou fácil de purificar, o objectivo é mais fácil de atingir: o gene pode
ser isolado, a sua sequência determinada, pode ser manipulado in vitro, sinais apropriados de
expressão podem ser-lhe associados e a introdução numa planta, efectuada nas condições
definidas acima.
Plantas transgénicas
a) Resistência a herbicidas
O milho é naturalmente resistente a uma família de herbicidas, as triazinas. As condições
de produção deste cereal foram altamente melhoradas pela possibilidade de utilizar estes
herbicidas para matar as ervas daninhas nos milheirais. A resistência do milho às triazinas é o

127
resultado duma desintoxicação enzimática da molécula herbicida. O gene responsável por
esta desintoxicação foi recentemente clonado e é de prever que seja introduzido noutras
plantas que, como o soja, estão frequentemente associadas ao milho no afolhamento.
O estudo e a utilização de genes quiméricos que conferem resistência aos herbicidas, são
objecto de importantes projectos industriais. Um dos resultados mais espectaculares foi
obtido pela introdução no tabaco, na batata, no tomate, no colza, na beterraba, por exemplo,
de um gene de resistência a um herbicida não selectivo correntemente utilizado, a
fosfinotricina. O gene que confere a resistência a este herbicida foi isolado a partir de
Streptomyces hygroscopicus. Ele codifica para a fosfinotricina acetiltransferase que acetila os
grupos NH2 livres da fosfinotricina e, consequentemente, impede naturalmente a auto-
toxicidade da fosfinotricina em Streptomyces. As plantas transgénicas sintetizam a enzima
codificada pelo gene transferido e suportam o herbicida pulverizado em doses dez vezes
superiores às normalmente utilizadas na agricultura. As plantas assim manipuladas
transmitem à descendência o gene nelas expresso (Fig. 187.II.C).
As consequências agro-económicas derivadas deste género de manipulação são fáceis de
imaginar. As previsões são, aliás, pouco ecológicas, se elas se traduzirem unicamente pela
generalização da utilização de herbicidas. Em compensação, as perspectivas parecem mais
favoráveis na luta contra insectos nocivos ou contra vírus, fungos ou bactérias.
b) Resistência a insectos
Preparações comerciais de esporos da bactéria Bacillus thuringiensis têm desempenhado
um papel importante no controlo biológico de insectos nos campos. Elas associam uma
elevada toxicidade para com os insectos e uma segurança biológica em relação ao meio
ambiente. B. thuringiensis não afecta todos os insectos e é completamente não tóxico para os
vertebrados. As inclusões cristalinas produzidas durante a esporulação contêm proteínas
insecticidas que afectam o epitélio do tracto intestinal de certos insectos, nomeadamente as
larvas dos lepidópteros. Os cristais dissolvem-se nas condições alcalinas do tracto intestinal
do insecto e libertam proteínas que são processadas proteoliticamente pelas proteases do
insecto, produzindo fragmentos polipeptídicos tóxicos, de massa molecular compreendida
entre 65.000 e 160.000 daltons.
O gene bt2 de B. thuringiensis codifica uma proteína Bt2, que é uma toxina potente para
as larvas de vários lepidópteros, tais como Manducta sexta, que é uma praga da planta do
tabaco. Bt2 é uma protoxina que gera um polipéptido de menor tamanho (60.000), que
conserva ainda uma plena actividade tóxica.
Nas experiências de transformação da planta do tabaco foram utilizados vários genes
quiméricos contendo a sequência completa de bt2, assim como genes excisados.

128
As construções no T-DNA incluem, para além das sequências de bt2, o gene da
neomicina fosfotransferase (neo), marcador de selecção de resistência à canamicina (NPTII),
um promotor constitutivo do gene 2, que dirige a expressão da manopina sintetase no TR
DNA do plasmídeo pTiA6 (Fig. 188.II.) e sinais de terminação e de poliadenilação da
extremidade 3' do gene.
Os plasmídeos resultantes destas construções no T-DNA dos vectores de expressão em
plantas, pGSH160 ou pGSH150 (Fig. 189.II), foram mobilizados em A. tumefaciens, receptor
que contém um plasmídeo Ti octopina (pGV2260), do qual a região T-DNA foi excisada e
substituída por pBR322. Recombinações entre o pGV2260 e o vector de expressão, através
das sequências homólogas de pBR322, produziram plasmídeos Ti contendo as várias
construções incluindo o gene bt2 e seus derivados excisados (Fig. 190.II.).
As plantas de tabaco transgénicas foram obtidas por infecção de discos de folhas de
Nicotiana tabacum. Rebentos resistentes à canamicina foram seleccionados em todas as
experiências de transformação, indicando que os genes fusionados conferem, com efeito,
actividade NPTII às células das plantas transformadas.
As plantas transformadas foram cultivadas individualmente e controladas,
subsequentemente, em relação à resistência à canamicina, testando a sua capacidade para
produzirem calos a partir de discos de folhas em concentrações crescentes de canamicina.
As folhas das plantas transgénicas, que contêm os vários tipos de construção do gene de
B. thuringiensis, foram dadas como alimento às larvas de M. sexta, a fim de avaliar se os
níveis de toxina expressos na planta tinham efeitos insecticidas. Observou-se 75-100 % de
mortalidade das larvas, num quarto das plantas que expressavam a proteína fusionada Bt-
NPT23 mais longa e em dois terços das que expressam a fusão menor Bt-NPT860 (Fig.
190.II). A toxicidade para com os insectos está directamente correlacionada com o nível de
resistência à canamicina. Constatou-se, ainda, que só os genes bt2 incompletos expressam
níveis de proteína fortemente insecticidas nas plantas transgénicas de tabaco, sendo a fusão
de menor comprimento a que produz os maiores níveis de proteína biologicamente activa.
Os resultados de experiências visando a determinação da real eficácia de protecção
contra os estragos causados pelos insectos estão exemplificados nas Fig. 187.II.A e B e
191.II. Eles mostram que é possível proteger plantas transgénicas programadas para
produzirem toxinas contra pragas de origem larvar.
Para verificar se esta propriedade é hereditária a descendência F1 destas plantas
transgénicas foi testada, tanto em relação à resistência à canamicina, como à toxicidade para
com as larvas de M. sexta. A actividade insecticida revelou-se idêntica à observada nas
plantas parentais, confirmando o carácter hereditário da protecção.

129
O exemplo da construção de plantas de tabaco resistentes às larvas de M. sexta, descrito
com certo pormenor, ilustra as potencialidades ofertas pelas manipulações genéticas das
plantas. Outros estudos sobre a protecção contra doenças fúngicas, bacterianas ou virais têm
sido desenvolvidos. Assim, por exemplo, resultados obtidos com o vírus do mosaico do
tomate, o vírus do mosaico da luzerna, o vírus do mosaico do pepino ou o PVX de batata
mostram que, quando um gene codificando uma proteína da cápsula do vírus se expressa nas
plantas transgénicas, estas adquirem uma protecção considerável contra a infecção viral.
c) Resistência a vírus
O princípio de protecção viral de plantas através da inoculação de estirpes atenuadas de
vírus ou de viróides, a fim de impedir a infecção por estirpes mais virulentas, é conhecido dos
agricultores, que o têm aplicado desde há muito tempo. É uma prática que foi utilizada para
reduzir os prejuízos provocados em culturas de tomates, de batatas ou em citrinos, por
exemplo e causados pelos vírus do mosaico do tomate (TMV), pelo viróide fusiforme do
tubérculo e o vírus da tristeza dos citrinos.
Esta prática, denominada protecção cruzada, têm vários inconvenientes e insuficiências,
tais como o risco de mutação da estirpe atenuada numa estirpe mais virulenta, ou o
desenvolvimento duma condição patogénica derivada de uma eventual sinergia entre a estirpe
atenuada do vírus e um outro vírus presente na cultura, que tenha características mais severas
que a causada por cada um dos vírus ou, ainda, o risco de o vírus protector duma planta poder
ser um patógeno severo para outra. A estirpe protectora pode ainda causar um prejuízo
significativo, mesmo reduzido.
Fazer expressar na planta o princípio activo da protecção, inscrito definitivamente no seu
genoma e, consequentemente transmissível hereditariamente, resolveria os problemas
inerentes à protecção cruzada, com a vantagem suplementar de propagar definitivamente às
gerações futuras as características protectoras.
O desenvolvimento da engenharia genética aplicada às plantas permitiu realizar
progressos notáveis, também nesta área. Foi, com efeito, demonstrado que a expressão
constitutiva, numa planta transformada geneticamente, da proteína do invólucro dum vírus,
ou proteína capsídica (CP), assegura a imunização da planta contra a infecção por esse vírus.
Este fenómeno, ainda mal compreendido, parece ser geral e foi posto em evidência em várias
espécies vegetais, tais como a planta do tabaco, a luzerna, o pepino, a batata.
Analisemos em mais detalhe um exemplo de transformação duma variedade de batata
(Russet Burbank), que confere à planta transgénica resultante a resistência dupla aos vírus
PVX e PVY. Estes vírus causam baixas importantes de rendimento de produção da batata,
que podem atingir 80 %, sobretudo o PVY.

130
O PVY possui um RNA único de cerca de 10.000 nucleotídeos de comprimento. Possui
uma pequena proteína (Vpg) ligada covalentemente à sua extremidade 5' e a extremidade 3' é
poliadenilada. O genoma expressa-se na forma de precursor de poli-proteína que é
processada pós-tradução em proteínas singulares. A proteína CP do invólucro é gerada por
clivagem da poli-proteína por uma protease viral. Ao contrário do PVX, que só se transmite
mecanicamente, o PVY é transmitido também por afídeos.
O PVX possui um RNA monocatenário único, de 6.4 kbases, com estrutura chapéu em
5', uma sequência poli A em 3' e cinco fases abertas de leitura. A sua sequência nucleotídica
era conhecida antes da do PVY. Esta foi determinada a partir do cDNA obtido num banco
construído em λgt10, preparado utilizando um iniciador oligo T e escrutinado com um
oligonucleotídeo sintético degenerado. A sequência desta sonda foi deduzida da sequência
dos aminoácidos N-terminais da proteína do invólucro. Os clones de cDNA identificados
com esta sonda foram sub-clonados no mp19 e as suas sequências nucleotídicas
determinadas.
A sequência do RNA da proteína CP de PVY constituída por 801 nucleotídeos é
traduzida em 267 aminoácidos que constituem a proteína. Para expressar o cDNA da CP do
PVY nas plantas transgénicas, a sequência codificante foi alterada por mutagénese dirigida a
fim de criar uma sequência de começo de tradução, no início da sequência codificante, e um
sítio de restrição para clonar o cDNA em vectores de expressão de plantas. Um sítio NcoI foi
criado a montante do codão do aminoácido N-terminal, alanina e um sítio BglII foi
introduzido 11 pares de bases a montante do sítio NcoI (Fig. 192.II.). A criação dum sítio
NcoI introduz um codão de metionina na extremidade N da CP de PVY. O DNA
mutagenizado, comportando 326 pb da sequência não traduzida em 3', foi inserido no vector
pMON906, entre a sequência estimulada do promotor 35S do CaMV (vírus do mosaico da
couve-flor) e a sequência 3' terminal E9 do gene da RuBP carboxilase (pequena subunidade
da ribulose-1,5-bifosfato carboxilase de ervilha -rbcS), dando origem ao vector pMON9892.
A sequência estimulada do promotor 35S do CaMV é uma construção obtida por duplicação
de 250 pb a montante do promotor, as quais actuam como sequências estimuladoras
(enhancers), aumentando de 10 vezes a actividade da transcrição natural do promotor.
A sequência que codifica para a CP do PVX situa-se na região 3' do genoma que é
traduzida, in vivo, a partir dum RNA sub-genómico. A sequência nucleotídica deste gene
comporta 711 pares de bases e codifica para uma proteína de 237 aminoácidos. Para construir
o vector pMON9898 que contém as sequências codificantes da CP de ambos os vírus, o gene
da proteína do invólucro de PVX foi removido do vector 9809 e ligado no pMON893 que é
um derivado do pMON200, que contém a extremidade 3' da proteína de reserva 7S do soja e
é desprovido do gene da nopalina sintetase. Um fragmento HindIII de DNA, que contém o

131
promotor estimulado 35S do CaMV, o gene da CP do PVY e a extremidade 3' da rbcS E9, foi
excisado de pMON9892 e ligado ao pMON9896. O vector resultante, pMON9898, foi
utilizado para transformar a batata Russet Burbank, recorrendo a um sistema de
transformação mediado por A. tumefaciens cujo princípio descrevemos acima.
A Fig. 193.II. mostra os resultados das análises, por Northern blotting, dos transcritos de
PVX e de PVY obtidos por purificação do RNA total extraído de folhas de plantas
transgénicas e a Fig. 194.II., as análises, por Western blotting, da expressão das CP de PVX e
de PVY separadas da mistura de proteínas extraídas das folhas das mesmas plantas. Os níveis
de expressão das proteínas de invólucro de PVX e de PVY situam-se entre 0.05-0.2 % e 0.01-
0.05 % do total das proteínas das folhas, respectivamente.
Estas proteínas exogénicas asseguram a protecção das plantas transgénicas contra as
infecções por PVX e por PVY. Foi, com efeito, demonstrado em experiências de challenge
nas quais, concentrações crescentes de vírus foram inoculadas em folhas de batata, que as
plantas transgénicas possuem uma excelente resistência à infecção (Fig. 195.II).
No entanto, a maior parte das linhagens transgénicas é sensível à infecção simultânea
pelos dois vírus, o que confirma observações anteriores de que a infecção mista da batata, por
PVX e PVY, causa um acréscimo sinergético da severidade da doença. Só uma linhagem se
revelou resistente à infecção simultânea pelos dois tipos de vírus, tanto por via mecânica,
como por intermédio de afídeos. Não parece existir correlação entre o nível de expressão das
proteínas do invólucro e a capacidade de resistência. Esta observação sugere que factores,
tais como expressão específica de um tipo de tecido ou de célula, ou a localização sub-celular
da CP, podem ser importantes para determinar o nível de resistência.
Como foi demonstrado com outros exemplos, que as características de resistência viral
são herdadas de maneira estável através de várias gerações, esta abordagem poderá ter um
desenvolvimento geral importante na construção de plantas transgénicas resistentes a vírus.
Qual é o princípio que preside a este fenómeno de resistência concedida pela expressão
nos tecidos das plantas, da proteína do invólucro viral?. Uma das hipóteses avançada para
explicar a protecção mediada pela CP afirma que o despojamento do invólucro do vírus é
impedido pela proteína do invólucro. Algumas observações, demonstrando a perda da
resistência à infecção por inoculação do RNA viral, são compatíveis com este mecanismo
mas não excluem outras hipóteses, tais como o impedimento pela CP da penetração do vírus,
ou a possibilidade de que, o impedimento do vírus perder o invólucro diminua a sua
capacidade de disseminação de célula em célula, no caso da deslocação se fazer na forma de
virião.

132
Observações feitas com outros tipos de vírus levaram no entanto a resultados opostos,
tendo sido posta em evidência a protecção contra a infecção pelo RNA. Pode-se, neste caso,
fazer a suposição de que a CP se liga ao RNA do vírus impedindo a tradução da replicase ou
interferindo com a replicação. É, pois, provável, que os mecanismos de protecção variem
com o tipo de vírus.
d) Produtoras de péptidos
Um dos resultados mais espectaculares obtidos com a construção de plantas transgénicas
demonstra a possibilidade de fazer produzir péptídos de aplicação farmacêutica por sementes.
A experiência diz respeito ao neuropéptido Leu-encefalina, pentapéptido de sequência
TyrGlyGlyPheLeu, detentor de actividade opiácea. A estratégia adoptada consiste em
substituir uma parte do gene da proteína de reserva albumina 2S de Arabidopsis thaliana,
pelos codões correspondentes à Leu-encefalina, numa região menos conservada entre as
diferentes espécies, a fim de tirar partido da presença abundante das albuminas 2S (de 20 % a
60 % do total das proteínas de sementes em várias espécies), para explorar a possibilidade de
produzir quantidades grandes de péptidos na forma de proteínas quiméricas de reserva de
plantas.
A região escolhida de inserção na albumina 2S é bastante variável em comprimento e
sequência, portanto, pouco susceptível de afectar a estrutura secundária e a estabilidade da
proteína. A substituição foi realizada de maneira a fazer corresponder os resíduos 35-41 da
proteína resultante à encefalina flanqueada por resíduos lisina, necessários para realizar a
clivagem do péptido do restante da albumina 2S.
A construção do gene híbrido foi efectuada substituindo parte do gene da 2S, clonado
num plasmídeo, por mutagénese dirigida utilizando um oligonucleotídeo sintético. A
construção final do gene quimérico no vector T-DNA, utilizado para transformar a planta,
compreende, nomeadamente, o gene quimérico, os genes neo e hph, marcadores de selecção
de resistência à canamicina e à higromicina, as extremidades 3' do gene da octopina sintetase
e do gene 7 do T-DNA, o promotor bi-direccional, PTR, do T-DNA (Fig. 196.II.). O
plasmídeo pGSATE1, construído, foi mobilizado em A. tumefaciens e as plantas transgénicas
Arabidopsis foram obtidas por infecção em disco de folha e selecção, subsequente, com
canamicina. As sementes obtidas a partir das plantas regeneradas foram germinadas em
presença de canamicina. A proteína foi extraída a partir das sementes provenientes desta
segunda geração, purificada e tratada enzimaticamente para extrair a Leu-encefalina. O
rendimento obtido com este exemplo é de 200 nmol/g de semente.

133
As plantas monocotiledóneas
Até há pouco tempo pensava-se que só a classe das dicotiledóneas podia ser
transformada por A. tumefaciens, sendo as monocotiledóneas resistentes ao crown gall.
Mostrou-se, no entanto, que esta limitação não é absoluta, pois certas monocotiledóneas, como
o espargo ou o narciso, são permeáveis à transformação por A. tumefaciens. No que respeita
aos cereais, começou a poder-se realizar transformações sem recorrer a A. tumefaciens,
adicionando o DNA não integrado num organismo ao meio de cultura de protoplastas de
cereais. Células transformadas em cultura, resistentes à canamicina, podem ser assim obtidas.
O método utilizado, ao começo, com as dicotiledóneas, consiste em fazer penetrar o DNA
purificado nos protoplastas. Diferentes técnicas têm sido utilizadas, entre as quais a
electroporação parece particularmente eficaz.
O princípio consiste em submeter os protoplastas a uma forte descarga eléctrica de curta
duração, que cria na membrana poros minúsculos, pelos quais o DNA pode penetrar. Para
além da electroporação, outros métodos, que não são limitados pela espécie vegetal, e que
podem ser utilizados com tecidos de plantas de raízes, caules ou folhas, são por exemplo a
micro-injecção, o bombardeamento com microprojécteis e a precipitação com o fosfato de
cálcio.
Por outro lado, o número de espécies susceptíveis de serem regeneradas com sucesso, a
partir de células em cultura, aumenta constantemente graças aos progressos da cultura in
vitro. Ao mesmo tempo desenvolvem-se as técnicas que permitem transformar, tanto células
sexuais, como os tecidos embrionários que são normalmente programados para regenerar um
indivíduo completo.
Em paralelo com as aplicações de finalidades mais imediatas, a investigação
fundamental continua a desenvolver-se, tendo por objectivo a compreensão dos mecanismos
que regulam a expressão dos genes. Estes mecanismos, ainda pouco conhecidos, controlam
propriedades importantes, como a síntese de um pigmento, a de uma proteína de reserva na
semente, ou o desenvolvimento dos tecidos ou dos órgãos.
É evidente que, compreender estes mecanismos significa adquirir os meios para
manipular as propriedades que deles emanam.

134
11. O DNA RECOMBINANTE E O DIAGNÓSTICO EM MEDICINA
A tecnologia do DNA recombinante têm também aplicações importantes no campo do
diagnóstico. O princípio geral que preside a esta nova metodologia do diagnóstico é a
detecção de sequências, de fragmentos de DNA resultantes de rearranjos ou de mutações, de
uma maneira geral (inserções, substituições ou eliminações), responsáveis por anomalias
genéticas, ou a detecção, em tecidos ou em fluídos biológicos, de sequências típicas do
genoma de organismos infecciosos de origem viral, bacteriana, fungal ou de parasitas.
Existem pelo menos três categorias de alterações genéticas: cromossómicas,
monogénicas e multifactoriais.
a. ALTERAÇÕES CROMOSSÓMICAS
A maioria dos defeitos cromossómicos é incompatível com a vida, asserção que é
confirmada pela observação da correlação, que excede 50 %, entre a frequência deste tipo de
anomalias e o número de abortos espontâneos ocorridos no primeiro trimestre da gravidez.
Alguns tipos de defeitos cromossómicos menos deletérios, são caracterizados por
aberrações numéricas dos autossomas ou dos cromossomas sexuais e resultam de falhas de
disjunção durante a divisão meiótica. Pertencem a esta categoria de anomalias a síndrome de
Down (trisomia 21), a síndrome de Klinefelter (47, XXY) e a síndrome de Turner (45, XO).
Os pacientes com o cariótipo XXY são fenotipicamente machos, apresentando digénese
testicular e infertilidade. Os indivíduos com o cariótipo XO são fêmeas estéreis deficientes no
seu desenvolvimento sexual secundário.
Uma outra forma de anomalias cromossómicas observadas em certas doenças são as
translocações: durante o fenómeno natural de translocação recíproca, os segmentos de dois
cromossomas diferentes permutam. Bastante frequente no homem, este fenómeno manifesta-
se por uma percentagem elevada de abortos e anomalias congénitas. Foram registadas mais
de 300 translocações recíprocas no homem e as células dos indivíduos afectados existem em
colecção. Várias leucémias e linfomas estão associados a este género de ocorrência. Um
exemplo é a leucémia mielogénica crónica, resultante da translocação do braço comprido do
cromossoma 22 para o braço comprido do cromossoma 9. Esta anomalia estrutural é
conhecida por cromossoma de Filadélfia.
Em certos casos, parece existir uma relação directa, entre uma translocação
cromossómica específica e um carácter maligno. A hipótese supõe que a translocação leva ao
deslocamento de oncogenes não expressos de um cromossoma dado para locais de um outro
cromossoma, onde eles seriam activados e expressos. Esta hipótese foi confirmada com um

135
linfoma humano, o linfoma de Burkitt, que conduz ao desenvolvimento de tumores dos
linfócitos B. O gene c-myc, não expresso em situação normal, é transposto do seu sítio
normal, o cromossoma 8, para uma região de cromossoma 14 que codifica para as cadeias
pesadas das imunoglobulinas (Fig. 197.II.). Nas células B tumorais, a região do DNA que
codifica para as cadeias pesadas das imunoglobulinas está sujeita a recombinações
frequentes. É bastante provável que uma recombinação aberrante, ocorrendo com baixa
frequência, leve à colocação dum promotor activo das cadeias pesadas a montante do gene c-
myc e conduza à sua expressão e à transformação maligna.
b. CARTOGRAFIA DOS CROMOSSOMAS HUMANOS
A obtenção de cartas genéticas detalhadas de cada cromossoma humano poderá em breve
fornecer um instrumento essencial para o diagnóstico das doenças genéticas. Vários genes
foram localizados em cromossomas humanos utilizando a genética mendeliana clássica.
Depois do aparecimento das técnicas de fusão celular entre espécies diferentes e das técnicas
de DNA recombinante, o estabelecimento de cartas genéticas ao nível dos cromossomas
acelerou-se consideravelmente.
Existem actualmente vários métodos que permitem fusionar células humanas em cultura
com células de outras espécies. Por outro lado, é possível transferir em células humanas e
animais, em cultura, fragmentos de DNA do tamanho dum cromossoma ou limitados a um
gene particular. A exploração destes métodos de trocas genéticas artificiais permite
posicionar um gene humano num cromossoma particular ou num fragmento de cromossoma
particular.
Quando se fusionam células humanas com células de rato, os híbridos resultantes
sobrevivem e dividem-se mas perdem, no entanto, progressivamente, os cromossomas
humanos. É possível correlacionar a presença duma proteína humana, com base em testes que
recorrem à expressão do gene nas células híbridas, com a presença dum cromossoma humano
particular (Fig. 198.II.).
Na realidade, este método sofre dum condicionamento importante, visto ser necessário
dosear uma actividade enzimática ou proceder a uma identificação imunológica para
identificar o produto do gene que se quer mapear. Ele foi, no entanto, amplamente utilizado e
permitiu localizar mais de 100 genes humanos em diferentes cromossomas. Quando um gene
foi cartografado num cromossoma dado, qualquer outro gene ligado geneticamente ao
primeiro (com base em estudos genealógicos) pode ser igualmente atribuído a esse
cromossoma.
A combinação dos métodos de fusão celular e das técnicas de DNA recombinante
oferece possibilidades bem mais amplas, pois é baseada na utilização dum gene ou dum

136
fragmento de DNA clonado, como sonda, para procurar sequências homólogas nas células
híbridas. Neste caso, deixa de haver dependência em relação à expressão do gene. O primeiro
exemplo deste tipo de abordagem é a localização do gene da globina ß no cromossoma 11
(Fig. 199.II.). Depois deste primeiro sucesso, numerosos outros genes humanos foram
atribuídos por este método a um ou outro cromossoma (Fig. 200.II., Tabela 10.II.).
Depois de atribuir um gene a um cromossoma, é necessário posicioná-lo em relação a
outros genes situados no mesmo cromossoma. Em genética clássica, a localização determina-
se pela medida das frequências de recombinação, durante a meiose, entre pares ou grupos de
genes. Quanto mais próximos estiverem dois genes, menor é a frequência com que eles se
recombinam. Em genética das células somáticas recorre-se a outros métodos tais como a
indução de eliminações, de translocações recíprocas ou de fragmentação de cromossoma.
Assinalámos, mais acima, a existência das translocações recíprocas espontâneas e a
correlação estabelecida entre este fenómeno e o aparecimento dum carácter maligno
veiculado por um oncogene, posicionado em situação propícia para ser activamente expresso.
A posição das translocações recíprocas pode ser identificada devido à alteração observável ao
microscópio, provocada na distribuição das bandas dos cromossomas, corados por um
processo apropriado. É, assim, possível atribuir um gene a um fragmento particular dum
cromossoma (Fig. 201.II.). O exemplo ilustrado na figura refere-se ao posicionamento do
gene da insulina na parte distal do segmento curto do cromossoma 11.
Alternativamente, pode-se induzir uma fragmentação aleatória dos cromossomas nas
células somáticas por irradiação aos raios X e, em seguida, fusionar estas células com células
receptoras, acompanhando, no híbrido celular, a retenção de dois genes mapeados
previamente num mesmo cromossoma (mede-se, neste caso, uma actividade enzimática típica
destes dois genes). Quanto mais próximos estiverem estes genes no cromossoma, mais
frequentemente permanecerão retidos juntos nos híbridos, visto estarem situados no mesmo
fragmento (Fig. 202.II.). No exemplo da figura foi demonstrado que os genes humanos tk e
gk estão separados por cerca de 1,2 106 pares de bases no cromossoma X.
A cartografia fina dos genes num cromossoma dado pode ser estabelecida directamente,
in situ. Para isso, utiliza-se um DNA radioactivo clonado, para marcar, por hibridação, a
posição dum gene num cromossoma. Praticamente, é preciso bloquear as células humanas no
estado mitótico, de maneira a que cada cromossoma da célula seja claramente identificável
por microscopia. O DNA clonado radioactivo é então hibridado in situ com os cromossomas
e a posição do gene é determinada por autorradiografia e por quantificação da granulometria
da autorradiografia. A localização faz-se em relação a um cliché dos cromossomas expostos
em série e corados. O exemplo da Fig. 203.II. ilustra a atribuição dos genes do interferão α
ao cromossoma 9.

137
Graças ao desenvolvimento das técnicas de DNA-recombinante, nomeadamente de
sequenciação, foi possível , recentemente, concluir a sequenciação total do genoma humano o
que permitirá, em breve obter uma carta genética detalhada completa de cada cromossoma
c. MAPEAMENTO DO GENOMA HUMANO POR RFLP
A organização estrutural dos genes em genomas de grande comprimento, como o
humano, pode ser elucidado utilizando o polimorfismo dos fragmentos de restrição
(Restriction Fragment Length Polymorphisms - RFLP). O polimorfismo é um fenómeno
conhecido há bastante tempo na área das proteínas, sendo um dos exemplos mais conhecidos
o dos grupos sanguíneos de histocompatibilidade. Quando existem modificações ligeiras na
estrutura primária de proteínas originárias de diferentes indivíduos da mesma espécie, que
têm funções e localizações cromossómicas idênticas, estas proteínas apresentam um carácter
polimórfico. As diferenças nas estruturas primárias das proteínas manifestam-se no nível do
DNA e podem ocasionalmente originar novos locais de restrição.
A Fig. 204.II. esquematiza duas moléculas de DNA diferentes, codificando para um
mesmo gene hipotético X, flanqueado por locais de clivagem para as enzimas A e B. Os dois
alelos são idênticos em relação à posição dos locais de clivagem da enzima A, mas diferem
em relação à posição dos locais de clivagem da enzima B. O padrão de separação por
electroforese dos fragmentos de DNA, observado após hibridação com uma sonda do gene X,
marcada radioactivamente, mostra uma banda quando a enzima A é utilizada para clivar o
DNA celular e duas bandas com a enzima B. No segundo caso, as duas bandas diferem no
comprimento e correspondem a duas formas alélicas diferentes. O gene X é dito polimórfico
em relação ao local alvo da enzima B. Na maioria dos casos, estes polimorfismos, também
chamados variações alélicas, são causados por simples substituições de bases que destroem
ou criam um local de restrição, mas, ocasionalmente, os RFLPs podem ser ocasionados por
eliminações ou inserções.
Os polimorfismos de DNA possuem um certo número de vantagens para o mapeamento
do genoma. O número de marcadores conhecidos de DNA excede o das proteínas e além
disso a sequência de DNA não necessita de expressar obrigatoriamente uma proteína para ser
identificada por locais de clivagem polimórficos. Os polimorfismos de DNA podem,
evidentemente, ocorrer em qualquer sequência de DNA e em particular em intrões.
Se os marcadores de polimorfismo estão muito próximos dum gene, isto é, são herdados
juntos, o RFLP pode também ser utilizado para mapear genes e portanto para caracterizar
anomalias genéticas, mesmo quando o gene em questão é completamente desconhecido.
A Fig. 205.II. mostra dois marcadores de polimorfismo, A e B, localizados perto dum
gene. Neste exemplo, o alelo normal está situado no cromossoma do macho e o alelo mutado

138
no da fêmea. A mãe é heterozigótica e o pai homozigótico em relação à variante normal.
Segundo as regras de Mendel, estes quatro alelos (dois paternos e dois maternos) dão origem
a quatro combinações possíveis na descendência. Se o marcador A e o gene defeituoso estão
a curta distância um do outro, serão co-transmitidos, isto é, todos os filhos que herdarem o
defeito genético herdam igualmente o alelo A do marcador. A presença do marcador de
polimorfismo A não implica forçosamente a presença da anomalia genética. Esta correlação
só se aplica a situações em que o gene e o marcador de polimorfismo estão situados muito
perto um do outro. Se estão bastante separados, podem-se realizar recombinações com mais
frequência relativa, colocando o gene defeituoso no cromossoma que contém o marcador B.
A probabilidade de existirem trocas genéticas aumenta com a distância entre o marcador e o
gene em questão. Quanto mais perto estiverem os marcadores mais conclusivas serão as
correlações observadas.
É, portanto, possível seguir a transmissão de qualquer marcador de polimorfismo por
análise genealógica e correlacionar a sua presença com a expressão fenotípica dum defeito
genético, se o marcador estiver situado suficientemente perto do gene mutado.
Na prática, o DNA dum grande número de indivíduos, pertencentes a uma linhagem, é
clivado com uma enzima de restrição que reconhece um polimorfismo na região da sonda de
DNA que será utilizada ulteriormente. Serão obtidos aproximadamente um milhão de
fragmentos diferentes de DNA, que podem ser separados por electroforese em gel de agarose
e submetidos a análise por Southern blotting. Várias centenas de sondas RFLP foram já
identificadas e utilizadas para mapeamento.
d. ANOMALIAS MONOGÉNICAS E MULTIFACTORIAIS
As anomalias monogénicas são causadas por mutações em genes únicos. As suas
características mendelianas de transmissão hereditária são autossómicas dominantes,
autossómicas recessivas ou ligadas ao cromossoma X. As aberrações autossómicas
dominantes manifestam-se mesmo quando um só dos dois alelos dum gene particular é
afectado. No caso das doenças autossómicas recessivas, pelo contrário, é necessário que
ambos os alelos dum gene sejam afectados, para haver manifestação clínica de doença. Os
efeitos das anomalias ligadas ao cromossoma X são diferentes nas fêmeas e nos machos,
visto estes últimos possuírem um só cromossoma X. Uma fêmea com dois cromossomas X,
pode ser homozigótica ou heterozigótica em relação a um gene mutado, ligado ao X, e a
expressão pode ser dominante ou recessiva. Qualquer que seja o modo de expressão nas
fêmeas, um carácter ligado ao X será sempre expresso de maneira dominante nos machos
(que possuam um só cromossoma X).

139
Cerca de 1400 doenças genéticas conhecidas são causadas por mutações em genes
únicos. Em geral, a prevalência é fraca (como ilustrado nos exemplos da Tabela 11.II.), se
bem que correspondam de 5 % a 10 % de todos os casos registados em pediatria. As lesões
bioquímicas de mais de 250 destas doenças são conhecidas. Elas afectam sobretudo enzimas,
mas existe igualmente uma variedade de outras proteínas implicadas, tais como factores de
coagulação do sangue, proteínas de transporte, hormonas peptídicas, receptores.
As doenças monogénicas podem não ser herdadas, mas serem antes o resultado de uma
nova mutação no indivíduo afectado. A frequência da ocorrência deste género de mutações
foi calculada como sendo de 5 x 10-6 mutação por gene e por geração. Aproximadamente um
em cada 100.000 recém-nascidos deveria, portanto, possuir uma nova mutação num qualquer
locus genético. Mas, visto que muitas destas mutações são ou recessivas, ou geneticamente
silenciosas, porque não afectam a função da proteína correspondente, as novas mutações com
manifestação clínica devem ser extremamente raras. Pensa-se, por exemplo, que todos os
casos de Huntington's chorea (várias dezenas de milhar) tiveram origem num pequeno
número de indivíduos afectados (provavelmente com mutações independentes), que puderam
ser rasteados até ao século XVIII.
Casos assinalados de doenças genéticas aparecendo subitamente na descendência de pais
não afectados podem-se explicar, na maioria dos casos, mais provavelmente, por paternidade
extra-conjugal do que por aquisição duma nova mutação.
Nas perturbações multifactorias estão incluídas uma grande variedade de doenças, tais
como o cancro, diabetes mellitus, hipertensão etc. A predisposição para ser afectado por este
tipo de doenças parece ser geneticamente transmissível. São enfermidades poligénicas; visto
que o número de genes implicados é frequentemente desconhecido, o risco individual é difícil
de avaliar. Contudo, este risco é, em geral, consideravelmente menor (1 a 5 %) que o de
aquisição de doenças monogenéticas a partir de pais afectados (25 % ou 50 %), porque é
necessário que vários genes sejam simultaneamente afectados. Só algumas destas
predisposições podem ser actualmente diagnosticadas.
Entre o pequeno número de loci associados de maneira relevante com predisposições
para várias doenças figuram os do complexo de histocompatibilidade, HLA (Human
Leukocyte Antigen). Consiste em quatro loci genéticos distintos comportando, cada um,
alelos múltiplos que codificam para proteínas diferentes. Um conjunto particular é, por
exemplo, o HLA-B27, associado com um maior risco de contrair a espondilite ancilosante. O
risco de contrair esta doença grave é 90 vezes mais elevado para os indivíduos HLA-B27
positivos do que para os HLA-B27 negativos. Se bem que esta predisposição possa ser
diagnosticada, não existe terapia para a doença, propriamente dita, devido à sua natureza
genética multifactorial.

140
e. O DIAGNÓSTICO DE DEFEITOS GENÉTICOS
O diagnóstico e a prevenção de doenças genéticas podem ser realizados a diferentes
níveis. Se a base molecular de um defeito genético não for conhecida, os dados disponíveis
só podem servir para avaliar estatisticamente o risco de vir a ter uma criança com a doença
familiar.
Um conhecimento profundo da doença ao nível molecular, conjugado com técnicas
apropriadas de detecção do defeito molecular, permite pelo contrário, identificar de maneira
segura uma lesão num dado indivíduo. Isto pode ser realizado ao nível das proteínas,
analisando, por exemplo, espécimes de sangue fetal. As técnicas de clonagem permitem
actualmente a detecção dum defeito genético até ao nível do gene. Como estas análises
podem ser efectuadas com quantidades ínfimas de tecidos, obtidas, por exemplo, por
amniocentese ou biopsia coriónica, as doenças genéticas podem ser diagnosticadas numa fase
muito precoce da gravidez (Fig. 206.II.). Em função do resultado dos testes, a interrupção
voluntária da gravidez pode ser realizada com um mínimo de risco.
Se um defeito genético que se pretende diagnosticar conduz à não expressão do gene,
será evidentemente impossível detectar o produto deste. A identificação duma proteína
mutada não permite definir a natureza da mutação dum gene, não indica se o defeito afecta
um sinal de regulação, ou o transporte duma proteína, ou a sequência codificante.
1) RFLP (Restriction Fragment Length Polymorphism)
Os RFLPs são particularmente úteis para identificar defeitos genéticos no homem e
podem ser aplicados com fins de diagnóstico, contanto que as alterações no DNA implicado
não ocorram várias vezes e que estejam associadas unicamente a genes únicos. A maioria dos
RFLPs conhecidos ocorreram ao acaso e não têm relação com os genes vizinhos.
Um exemplo importante da aplicação desta metodologia é a detecção dum marcador da
Huntington's chorea, doença caracterizada pela degenerescência irreversível das células
nervosas, que ocorre com uma frequência de 1 em 10.000 indivíduos. O DNA proveniente de
duas linhagens genealógicas portadoras desta anomalia genética foi analisado por hibridação
de sondas radioactivas com fragmentos de restrição originados por clivagem em locais
polimórficos (chamados marcadores polimórficos) conhecidos, sem se saber à partida se um
qualquer destes marcadores polimórficos se situava na vizinhança do gene de Huntington e
segregava com ele.
Observou-se, no entanto, que uma das sondas reconhecia um polimorfismo que obedecia
àquelas condições, causado por dois locais HindIII, cuja presença ou ausência dá origem ao
padrão de hibridação representado na Fig. 207.II. A presença ou a ausência do primeiro local
HindIII cria fragmentos de comprimento de 15 ou 17.5 kb, respectivamente, enquanto que a

141
presença ou ausência do segundo local HindIII gera fragmentos de 3.7 e de 4.9 kb,
respectivamente. Existem, portanto, 4 padrões de fragmentação possíveis, chamados
haplotipos A, B, C e D. Como há sempre dois alelos num gene dado duma célula somática,
os quatro haplotipos geram dez combinações duplas diferentes.
A frequência destas combinações duplas numa população normal é variável. A análise da
árvore, representada na Fig. 208.II, revelou que o haplotipo C (presença dos dois locais
HindIII) segrega sempre com a doença. Como este haplotipo também ocorre na população
normal, a única conclusão plausível é que um membro da família, que não herdou o haplotipo
C dos pais, tem uma grande probabilidade de não ser afectado pela doença; por outro lado,
um indivíduo com o haplotipo C não poderá ter a certeza de que a doença se manifestará. Só
aproximadamente 25 % da população normal tem o haplotipo C; portanto, a probabilidade de
um indivíduo de haplotipo C, pertencente a uma família de risco, desenvolver a doença é
bastante alta.
Destas observações pode-se tirar a conclusão de que predições conclusivas só podem ser
feitas após uma análise genealógica. Como o haplotipo C também ocorre na população
normal, o facto de ser identificado num indivíduo pertencente a uma família sem
predisposição, não têm grande sentido.
A associação observada entre o locus marcador G8 e a doença não significa que o gene
responsável pela doença tenha sido identificado. Foi observado um único recombinante na
árvore genealógica analisada, que provém duma translocação do gene de Huntington para um
cromossoma de haplotipo A. A partir desta ocorrência única, conclui-se que a distância entre
o gene responsável pela doença e o marcador é de ordem de pelo menos 2000 kb.
As aplicações do RFLP não se restringem ao mapeamento de defeitos genéticos mas
intervêm também, como vimos, na construção dum mapa genético de cromossomas humanos.
A descoberta dos RFLPs e a generalização das suas aplicações têm consequências práticas
importantes. A determinação do polimorfismo de dadores de orgãos para transplantações,
realizada actualmente por técnicas serológicas, poderá ser ultrapassada por determinações
mais rigorosas e rápidas de polimorfismos de DNA. Será, também, possível detectar
correlações entre a predisposição de alguns indivíduos para certas doenças e a sua
constituição genética.
2) Diagnóstico de mutações identificadas
Se o gene responsável duma dada doença genética estiver identificado pode-se, a partir
dum clone correspondente ao gene normal, determinar a sua sequência nucleotídica e
estabelecer uma carta de restrição detalhada. Por outro lado, o gene normal pode ser utilizado
como sonda para identificar e isolar o gene correspondente, presente no DNA das células

142
fetais. Comparando as sequências e as cartas de restrição é possível identificar as mutações
que afectam o gene fetal.
O exemplo mais estudado é o das ß-talassemias.
ß-talassemias
As pertubações genéticas da síntese de hemoglobina, chamadas talassemias, foram
estudadas em detalhe ao nível do DNA. As hemoglobinas humanas são codificadas por duas
séries de genes: os genes de tipo α, localizados no cromossoma 10 e os genes de tipo ß,
localizados no cromossoma 11 (Fig. 209.II.) Estas regiões foram clonadas e sequenciadas.
A hemoglobina normal dum indivíduo adulto é constituída por duas cadeias α e duas
cadeias ß. As talassemias são patalogias caracterizadas pela ausência total ou parcial de
síntese de um tipo de globina. O resultado é um desequilíbrio entre as cadeias α e ß e uma
anemia, mais ou menos pronunciada, segundo o tipo de talassemia. Estes estados têm origem
numa deficiência de transcrição do DNA, ou num defeito de maturação do RNA, ou numa
instabilidade do RNA ou, ainda, numa incorrecção na tradução do RNA. Estes defeitos
podem resultar de mutações pontuais que conduzem a um desfasamento da tradução ou ao
aparecimento prematuro de sinais de fim de cadeia ou, ainda, derivar de eliminações do gene,
no seu todo ou parciais.
As ß-talassemias homozigóticas são caracterizadas por anemias bastante severas e
ausência total ou quase total de globina ß. A doença é fatal nos primeiros anos de vida porque
as cadeias de globina ß só são activas depois do nascimento. Normalmente, o feto não é
afectado porque utiliza as cadeias de globina γ fetais. Em certos casos, os indivíduos
talassémicos adultos (privados de globina ß) podem permanecer de boa saúde porque
conservam uma percentagem de hemoglobina fetal elevada (persistência hereditária de
hemoglobina fetal).
a) Mutações sem sentido e frame shift (Tabela 12.II)
A ß-talassemia, caracterizada pela ausência total da síntese da globina ß, resulta duma
mutação pontual no codão correspondente ao aminoácido 17 ou no correspondente ao
aminoácido 39. Nos dois casos, a mutação dá origem a um codão sem sentido. Para além de
serem intraduzíveis, os RNA mensageiros correspondentes são muito instáveis in vivo.
Outras talassemias de tipo ß resultam de eliminações ou de inserções na sequência que
codifica para a globina ß; o resultado é uma deslocação da fase de leitura do RNA
mensageiro.

143
b) Mutações que afectam a transcrição
Num dos casos, uma substituição do nucleotídeo em posição -87 em relação ao local de
chapéu do mensageiro conduz a uma percentagem muito fraca de mensageiro de ß globina;
no segundo caso, uma mutação na TATA box em posição -30 bloqueia a iniciação da
transcrição.
c) Mutações que afectam a maturação do RNA
Um certo número de talassemias resultam dum splicing anormal dos transcritos primários
da ß globina.
O gene ß contém dois intrões cujo splicing é necessário para a maturação do mensageiro
(Fig. 210.II.).
Nestes tipos de talassemias, a sequência GT que se encontra sempre presente em 5' da
junção do splicing é mutada em AT, no primeiro ou no segundo intrão. O splicing correcto
não se realiza; no seu lugar são utilizadas outras sequências semelhantes à normalmente
implicada no splicing. O resultado é um RNA mensageiro intraduzível e de estrutura
incorrecta.
Por outro lado, certas talassemias resultam de mutações no interior de um intrão, que
criam uma sequência de splicing 3' (aceitador) que entra em competição com a sequência
natural e provoca um splicing incorrecto.
Outras mutações aparecem na região codificante do gene e criam locais de splicing do
tipo 5' que entram em competição com o local normal. O resultado é, evidentemente, mais
uma vez, a formação de RNAs mensageiros que não podem ser traduzidos correctamente.
O diagnóstico pré-natal das talassemias pode ser, actualmente, efectuado por análise de
Southern blotting do DNA amniótico. Um padrão anormal de fragmentos de DNA aparece
quando genes de globina são suprimidos, como no caso de α-talassemia. A detecção pré-natal
da ß-talassemia é mais complexa, porque muitas mutações diferentes podem dar origem à
síntese da cadeia de ß-globina defeituosa. Algumas mutações alteram os locais de
reconhecimento de enzimas de restrição, directamente, e podem ser detectadas por análise de
restrição (vd. exemplo da anemia falciforme mais adiante), outras não. Para fazer um
diagnóstico, nestes casos, pode-se recorrer à identificação dos cromossomas que contêm as
mutações talassémicas, estabelecendo relações com locais de restrição polimórficos que se
situam ao longo do cluster do gene da ß-globina (vd. mais acima RFLP). Foi, no entanto,
desenvolvida uma abordagem mais directa que recorre à detecção de mutações pontuais que
provocam ß-talassemias, por hibridação com oligonucleotídeos sintéticos.

144
d) Utilização dos oligonucleotídeos sintéticos
Uma das aplicações mais interessantes dos oligonucleotídeos sintéticos de sequência
definida é a sua utilização como sondas para diagnosticar doenças genéticas em fase pré-
natal. Esta área de aplicação só pôde, evidentemente, ser desenvolvida graças à tecnologia do
DNA recombinante.
A especificidade duma pequena sonda sintética de cerca de vinte nucleotídeos é notável.
Um único desemparelhamento (mismatch), no interior da região de associação entre a sonda e
o DNA alvo, é suficientemente destabilizador do híbrido para poder ser posto em evidência.
É este o princípio básico da detecção e do diagnóstico, mesmo em fase pré-natal, de
mutações genéticas pontuais (Fig. 211.II).
Esta técnica foi aplicada para detectar, no DNA, substituições de um nucleotídeo que
provocam a talassemia de tipo +ß. Um exemplo está ilustrado na Fig. 212.II que mostra a
transição de G em A na posição 110 da região IV S-1 do gene. Uma sonda oligonucleotídica
de sequência perfeitamente complementar do DNA normal, marcada por quinação com um
radioisótopo (32P), hibridar-se-á com o fragmento de 1,8 kb produzido por digestão BamHI
do genoma e a hibridação será revelada por emissão radioactiva. A sonda formará um
desemparelhamento C-A com a sequência mutada e, em condições de temperatura de
hibridação selectivas, o híbrido dissociar-se-á, libertando a sonda. O resultado prático será
ausência de sinal radioactivo na autorradiografia. Pelo contrário, uma sonda de sequência
homóloga da região mutada do gene dará um resultado simetricamente oposto, com o gene
normal e o gene mutado. O conjunto da informação permitirá diagnosticar rigorosamente se
um indivíduo é homozigótico normal ou mutado, ou se é heterozigótico.
A fotografia dum gel electroforético do DNA de indivíduos duma família de risco,
digerido por BamHI e hibridado com as duas sondas oligonucleotídicas, ilustra esta análise
(Fig. 213.II). A banda de 1.8 kb do DNA do pai hibrida-se com a sonda normal e com a
mutada. A da mãe responde de maneira semelhante. Os pais são, portanto, os dois
heterozigóticos para a mutação. O DNA do filho, pelo contrário, só se hibrida com a sonda
mutada. É, pois, homozigótico para esta mutação.
Outro exemplo é o da detecção da talassemia ß, que resulta duma mutação sem sentido
no aminoácido 39ß; a glutamina do codão CAG é mutada em codão de fim de cadeia TAG,
causando a terminação prematura da tradução da ß-globina. Esta mutação é a causa
predominante da ß-talassemia na Sardenha e está espalhada por toda a região mediterrânica.
A sequência de DNA correspondente ao codão mutado não comporta locais de
reconhecimento por enzimas de restrição.

145
Num estudo modelo, dois nonadecâmeros sintéticos foram utilizados (sublinhados na
Fig. 214.II.) para diagnosticar a presença de mutação em casos de gravidez, em que os fetos
apresentavam de risco de ß-talassemia. 20 ml de fluído amniótico foram tirados na 18ª
semana de gestação e o DNA isolado e digerido por BamHI. Após separação por
electroforese e hibridação do gel, sequencialmente, com as duas sondas marcadas, o híbrido,
ou a ausência de híbrido radioactivo formado com a banda de 1,8 kb, permite fazer um
diagnóstico. Num caso, ambas as sondas se hibridam com o DNA (Fig. 215.II.), o que
implica um diagnóstico de ß-talassemia heterozigótica (Aβ). Nos outros dois casos, só a
sonda ßA se hibrida com o DNA, indicando que os fetos eram normais (AA).
e) Anemia falciforme
A anemia falciforme é uma doença hereditária que resulta duma mutação que muda um
resíduo de ácido glutâmico (GAG) em valina (GTG) na posição 6 da cadeia ß-globina da
hemoglobina. A consequência desta modificação é a síntese duma hemoglobina que tem
tendência para cristalizar nos glóbulos vermelhos; as células tornam-se menos flexíveis e são
eliminadas do sangue ao nível do baço, o que conduz à anemia.
A mutação de bases A → T suprime um local de restrição no DNA do gene da globina ß.
É, pois, fácil detectar a mutação por análise de restrição do DNA fetal. Um DNA normal
contém o local de restrição, enquanto que no DNA mutado este local está ausente. Na prática,
esta modificação manifesta-se por uma distribuição anormal dos fragmentos de restrição (Fig.
216.II).
Praticamente digere-se o DNA, extraído de células derivadas dum doador normal e dum
indivíduo anémico, pela enzima de restrição DdeI (sequência reconhecida CCTGAG) ou,
melhor, por MstII (sequência reconhecida CCTNAGG), em seguida separam-se os
fragmentos em gel de agarose e transferem-se para uma membrana de nitrocelulose (Southern
blotting). O comprimento dos fragmentos formados por digestão é detectado por hibridação
com o DNA complementar de globina marcado com 32P.
A deficiência hereditária de αααα1-antitripsina
Os indivíduos deficientes em α1-antitripsina têm uma forte predisposição para
desenvolver enfisema pulmonar ou cirrose infantil do fígado. A α1-antitripsina é uma
proteína sintetizada no fígado e presente no plasma. O seu papel é inibir a elastase, proteína
implicada na perca da elasticidade dos tecidos pulmonares. Nos indivíduos normais o equilíbrio entre α1-antitripsina e elastase é controlado; nos deficientes hereditários, pelo
contrário, a elastase não é inibida e destrói os tecidos pulmonares.
A análise dos genes normais (M) e mutados (Z) mostra que eles só diferem numa única
base, G → A. Esta transição provoca a substituição dum resíduo glutâmico por um resíduo

146
lisina, em posição 342 da proteína. Esta substituição não cria nem destrói locais de restrição,
de maneira que é impossível utilizar o polimorfismo dos fragmentos de restrição na região
codificante para revelar a mutação do gene. No entanto, é possível tirar partido de sondas de
DNA sintéticas para diagnosticar a doença (Tabela 13.II), sintetizando, por exemplo, um
oligómero de 19 nucleotídeos, no qual a base G ou a base A é flanqueada de um lado e de outro pelas 9 bases correspondentes à sequência de DNA da α1-antitripsina. Estas sondas
marcadas com 32P por quinaçao foram testadas por hibridação com o DNA clonado da α1-
antitripsina. Uma escolha judiciosa das condições de hibridação permite obter um sinal diferencial (Fig. 217.II.). As duas sondas reconhecem o DNA normal da α1-antitripsina a
45°C; todavia, a 55°C só a sonda totalmente homóloga ao DNA da α1-antitripsina dá um
sinal de hibridação. Em função destas condições, é possível utilizar o oligómero correspondente ao DNA da α1-antitripsina normal, para testar a hibridação com o DNA
derivado de indivíduos deficientes em α1-antitripsina. O DNA genómico derivado das
células de indivíduos MM, MZ e ZZ, digerido pelas enzimas de restrição HindIII/XbaI ou
BamHI/XbaII, e transferido para uma folha de nitrocelulose é em seguida hibridado com o
oligómero sintético marcado, em condições previamente determinadas (55°C para a
hibridação e a lavagem) (Fig. 218.II.).
Os fragmentos do DNA MM normal, com o qual a sonda é completamente
complementar, e que contêm a região correspondente ao aminoácido 342 (fragmento de 2,4
ou de 2,6 kbases), dão um sinal positivo forte. O DNA MZ heterozigótico dá um sinal
reduzido e o DNA ZZ homozigótico não dá sinal. Este método é, pois, capaz de distinguir os
indivíduos ZZ dos indivíduos MM ou MZ e pode ser utilizado para o diagnóstico pré-natal da deficiência em α1-antitripsina.
Também neste caso existe a possibilidade de diagnosticar a deficiência em α1-
antitripsina pela análise do polimorfismo dos fragmentos de restrição, se se estudar o gene
inteiro incluindo os intrões. Com efeito, existe um polimorfismo de restrição para três
enzimas SstI, MspI e AvaII, que pode ser posto em evidência por hibridação com sondas de
fragmentos de DNA genómico clonados, segundo o princípio e técnicas previamente
descritos.
Os métodos de diagnóstico que recorrem à análise do DNA por hibridação com sondas
moleculares são de aplicação geral e tanto mais precisos quanto mais completo for o
conhecimento ao nível molecular dos defeitos do DNA geneticamente transmissíveis.
f. DETECÇÃO DE MUTAÇÕES SOMÁTICAS
A detecção de mutações com sondas oligonucleotídicas não esgota as suas
potencialidades com as doenças genéticas. É também possível detectar mutações somáticas

147
que conduzem a transformações oncogénicas. A família dos genes humanos ras constitui um
exemplo. Estes genes em número de 3, H-ras, K-ras e N-ras codificam proteínas de 21
kdaltons, localizadas na parte interna da membrana celular, que parecem estar implicadas na
transdução de sinais de receptores celulares de superfície para os seus alvos intracelulares.
Uma percentagem importante de linhagens celulares tumorais e de tecido tumoral possui um
gene ras activado. Estes genes caracterizam-se pela propriedade de induzirem uma
transformação oncogénica em células em cultura. Em quase todos os casos analisados a
activação foi correlacionada com mutações pontuais dos codões 13 ou 61 dos genes ras, as
quais levam à substituição de um aminoácido na proteína.
A sua natureza pontual e fixa torna possível a utilização de oligonucleotídeos sintéticos
para escrutinar, directamente, no DNA genómico, a presença das mutações responsáveis pela
activação destes genes. Por exemplo, uma abordagem utilizada para escrutinar mutações no
codão 61 do gene humano N-ras recorre à síntese de quatro grupos diferentes de icosâmeros
(Fig. 219.II.). O primeiro oligonucleotídeo (N61-I) tem uma sequência idêntica a um
segmento de 20 nucleotídeos que inclui o codão 61 (CAA) do gene N-ras normal. N61-II é
uma mistura de 3 oligonucleotídeos que diferem de N61-I pela natureza do primeiro
nucleotídeo do codão 61, em que o C do tipo selvagem foi substituído por A, por G ou por T.
Este grupo de oligonucleotídeos formará sempre um híbrido imperfeito com o gene N-ras
normal. Contudo, se o codão 61 apresenta uma mutação na primeira posição, um dos
oligonucleotídeos do grupo N61-II formará um emparelhamento perfeito com o gene mutado.
De maneira análoga, no grupo N61-III, o segundo nucleotídeo G, T ou C substitui o A na
posição 2 do codão. Por fim, os dois icosâmeros do grupo N61-IV possuem um T, ou um C,
no lugar do A natural na terceira posição do codão (a substituição por G origina uma mutação
silenciosa). As sondas oligonucleotídicas foram marcadas com elevada actividade específica,
incorporando vários nucleotídeos marcados com o radioisótopo 32P. O método utilizado
recorre à síntese química prévia de oligonucleotídeos anti-sentido (i. é., complementares e
orientados em sentido oposto) em relação às sequências das sondas e de um octâmero
complementar da extremidade 3' dos icosâmeros. O radioisótopo é introduzido prolongando o
octâmero, que funciona como primer, com a DNA polimerase I e (α-32P)dNTPs, utilizando
os icosâmeros anti-sentido como matrizes da polimerização.
Polim I5'
3' HO- 5'ATGAGAAGAACATACTCTTCTTGTCCAGCTGT- GGTCGACA-p 5'
3'
α-32PdNTPs
TACTCTTCTTGTCCAGCTGT-
5' 3'
GGTCGACA p
* * * *** ***** *
OH OH
Como o octâmero é portador dum fosfato 5' terminal, o icosâmero marcado
radioactivamente pode ser separado da matriz não fosforilada, num gel electroforético de
poliacrilamida-ureia.

148
Para escrutinar e determinar a natureza das mutações, o DNA genómico é digerido por
uma enzima de restrição e fraccionado num gel de agarose 0.5 %, desnaturado in situ e seco.
O gel seco é humidificado em água destilada e posto directamente em hibridação com os
oligonucleotídeos marcados. O gel é resistente às diferentes manipulações de hibridação e
lavagem e permite a detecção directa de um gene cópia única, a partir de uma quantidade de
DNA genómico de 1 a 5 µg, eliminando a necessidade de transferência para uma membrana
de nylon ou de nitrocelulose (Southern blotting). Em condições salinas apropriadas, a
temperatura de dissociação observada de um híbrido perfeitamente emparelhado, formado
entre os icosâmeros e o DNA genómico, é de 61-62°C, e a de um híbrido contendo um
desemparelhamento é de 55-56°C. Realizando a hibridação a 50°C e as lavagens a 59°C, é
possível detectar especificamente um híbrido perfeito, enquanto que os híbridos com um
desemparelhamento se dissociam. A Fig. 220.II. mostra o resultado das hibridações dos
oligonucleotídeos marcados N61-I (painel I), N61-II (painel II), N61-III (painel III) e N61-IV
(painel IV), com o DNA genómico (10µg), digerido por PstI e fraccionado
electroforeticamente, isolado das linhagens celulares MOLT-4 (corredor-1), HL60 (corredor-
2), Rc-2a (corredor 3), K562 (corredor-4), HT1080 (corredor-5) e RD301 (corredor 6), ou
dos plasmídeos pAT8.8 e pSVN-ras (corredores 7 e 8).
Forma-se um fragmento de 3.6 kb a partir do DNA genómico e fragmentos de 4.4 kb e de
2.8 kb a partir do DNA clonado, respectivamente no pAT8.8 e no pSVN-ras, contendo todos
o codão 61 do gene N-ras. Todas as linhagens celulares contêm um alelo normal N-ras,
dando um sinal positivo por hibridação com o oligonucleotídeo perfeitamente complementar
N61-I, após lavagem a 59°C (painel I). O painel II mostra que só o DNA das células HT1080
se hibrida com a mistura N6-II e o painel III, que só o DNA das células HL60 se hibrida
especificamente com a mistura N61-III. O painel IV mostra por seu lado, que a mistura N61-
IV se hibrida exclusivamente com o DNA de RD301. Este resultado implica que, na proteína
N-ras de RD301, a glutamina natural (CAA) foi mutada em histidina (CAC ou CAT).
Pode-se ainda concluir que, tanto HT1080, como HL60, possuem a mesma mutação
pontual no codão 61 que os genes isolados dos transfectantes celulares e clonados nos
plasmídeos. Com efeito, os DNAs isolados de HT1080 e de HL60 hibridam-se com N61-II e
N61-III, respectivamente, assim como com o DNA dos plasmídeos pAT8.8 e pSVN-ras, os
quais contêm partes dos genes N-ras activados isolados de transfectantes, respectivamente com as mutações conhecidas CAA61 → AAA e CAA61 → CTA.
Conclui-se, ainda, que estas três linhagens celulares contêm, para além do gene N-ras
mutado, um alelo N-ras normal.

149
Não foram detectadas mutações nas linhagens MOLT-4 e Rc-2a com os
oligonucleotídeos utilizados, o que significa que, provavelmente, as mutações podem estar
localizadas no codão 13 ou numa outra posição.
Um problema geral, associado com este método de detecção de mutações no DNA
genómico com oligonucleotídeos sintéticos, decorre da fraca sensibilidade (cf. o complicado
método de marcação para obter sondas de alta actividade específica, descrito mais acima) e o
elevado barulho de fundo devido a hibridações inespecíficas (Figs. 213.II, 215.II e 218.II).
g. AMPLIFICAÇÃO ENZIMÁTICA DE DNA (PCR)
As limitações de sensibilidade e de barulho de fundo, inerentes à necessidade de detectar
modificações pontuais com pequenas sondas cuja marcação é forçosamente reduzida, em
presença de um número enorme de sequências de DNA genómico, foram ultrapassadas com o
desenvolvimento duma técnica extremamente poderosa (denominada PCR: Polymerase
Chain Reaction) que permite amplificar enzimaticamente fragmentos de comprimento bem
definido, a partir de quantidades ínfimas de DNA. A reacção de polimerização em cadeia é
um método enzimático de amplificação de fragmentos específicos de DNA, que revolucionou
o diagnóstico molecular e abriu numerosos campos da biologia molecular, anteriormente
inacessíveis devido à inexistência de métodos analíticos suficientemente sensíveis.
Se a sequência nucleotídica de pequenos segmentos flanqueando a região alvo da
detecção for conhecida, pode-se sintetizar dois oligonucleotídeos, de cerca de 20 bases,
complementares de cada uma das cadeias do DNA, de um lado e de outro da região alvo da
amplificação, e que podem ser prolongados por polimerização enzimática em sentidos
opostos e convergentes.
A amplificação comporta vários ciclos (Fig. 221.II.) compostos, cada um, das seguintes
etapas: 1) desnaturação do DNA, 2) hibridação com os oligonucleotídeos sintéticos e 3)
polimerização enzimática com uma polimerase e dNTPs. Os oligonucleotídeos orientados
com a extremidade 3' na direcção do alvo da amplificação servem de iniciadores da
polimerização a partir das cadeias opostas do DNA. No fim do segundo ciclo obtêm-se
moléculas de DNA com uma extremidade bem definida. No fim do 3° ciclo formam-se
fragmentos de DNA com as duas extremidades definidas.
Para além do DNA ter sido amplificado 8 vezes, a sequência alvo foi extraída do DNA
total. Como os fragmentos sintetizados por polimerização contêm as sequências
complementares dos iniciadores, a utilização dum excesso destes e de dNTPs conduz a uma
amplificação unicamente limitada pelo número de ciclos e pela actividade da polimerase. Em
cada ciclo sucessivo a quantidade do DNA sintetizado duplica, o que leva à acumulação
exponencial do fragmento alvo segundo a lei aproximada 2n, em que n é o número de ciclos.

150
A eficácia da amplificação aumenta com a utilização duma polimerase termo-estável, a
Taq polimerase, isolada de Thermus aquaticus, organismo que vive em condições de
temperatura elevada. O facto de se poder efectuar a polimerização a temperatura elevada (p.
ex., a 70°C) aumenta a especificidade da hibridação dos oligonucleotídeos, eliminando
associações parasitas, e permite a realização dum grande número de ciclos de amplificação
(20 a 60, por exemplo), sem necessidade de voltar a adicionar a enzima em fases intermédias,
como é o caso com a polimerase I (fragmento de Klenow) que vai perdendo
progressivamente a actividade. Este desenvolvimento importante têm como consequência um
aumento considerável da especificidade, do rendimento, da sensibilidade e do comprimento
dos fragmentos alvos da amplificação.
Segmentos de DNA de comprimento que pode variar de uma centena (ou menos) de
pares de bases até mais de 2000 pb, podem assim ser amplificados mais de 10 milhões de
vezes com grande especificidade, a partir de quantidades extremamente pequenas de DNA.
A técnica do PCR têm sido utilizada na análise de variações de sequências nucleotídicas,
nomeadamente na detecção de mutações e de rearranjos cromossómicos, em clonagem de
sequências genómicas com alta eficácia, na sequenciação directa de DNA mitocôndrico e
genómico, na detecção de organismos patogénicos ou contaminantes (virais, bacterianos,
incluindo micloplasmas, fúngicos e parasitas).
A título de exemplo descrevemos, a seguir, a aplicação da técnica do PCR à análise de 2
mutações da ß-globina humana.
1) Detecção da anemia falciforme e da hemoglobina C por PCR
As mutações correspondentes à anemia falciforme e à hemoglobina C situam-se, ambas,
no codão 6 do gene da ß-globulina. A Fig. 222.II. representa uma região do gene de 110 pb,
no centro do qual se situa o codão 6 alvo da detecção por hibridação com sondas
oligonucleotídicas sintéticas marcadas radioactivamente, 19A, 19S e 19C (complementares
do alelo normal, do alelo da anemia falciforme e do alelo da hemoglobina C,
respectivamente). Esta região é amplificada a partir de dois iniciadores, PC03 e PC04, de 20
nucleotídeos cada, que flanqueiam a sequência alvo da amplificação e que estão orientados
em sentidos opostos e convergentes na direcção da polimerização (5' → 3').
O DNA extraído de seis amostras de sangue de indivíduos potencialmente mutados no
codão 6, de um indivíduo normal e da linhagem celular GMZ2064 (que contém uma
eliminação homozigótica do gene) é submetido a 25 ciclos de amplificação. Em seguida, 33
ng (1/30) dos produtos das amplificações são aplicados, na forma de dot blot, num filtro de
nylon, a partir do qual foram preparadas três réplicas. Cada um destes três filtros é hibridado,
em condições de temperatura selectivas (que dissociam os híbridos não perfeitamente

151
emparelhados), com uma das três sondas marcadas com 32P. O autorradiograma (Fig.
223.II.) mostra claramente a existência de 6 combinações diploídicas ßAßA (homozigótico
normal, AA), ßAßS (heterozigótico para a mutação da anemia falciforme, AS), ßSßS
(homozigótico para a anemia falciforme, SS), ßSßC (simultaneamente heterozigótico para as
mutações da anemia falciforme e da hemoglobina C, SC), ßCßC (homozigótico para a
mutação da hemoglobina C, CC), ßAßC (heterozigótico para a hemoglobina C, AC) e
confirma a eliminação do gene nas células GMZ2064 (XX).
Por análise dos produtos da amplificação, clonados, pode-se avaliar a 1 % a frequência
do fragmento de ß-globina amplificado, o que representa um enriquecimento superior a 105
em relação ao DNA genómico não amplificado. É esta redução substancial da complexidade
que permite a aplicação de sondas de 19 nucleotídeos, marcadas por simples fosforilação em
5' com uma quinase, à detecção de mutações utilizando a técnica extremamente simples do
dot blot.
Desenvolvimentos mais recentes da PCR mostram que é possível explorar a elevada
sensibilidade deste método para detectar colorimetricamente mutações genéticas. Assim, o
genótipo relativo à anemia falciforme pode ser determinado utilizando 3 tipos de iniciadores
para a amplificação do mesmo DNA. Dois destes iniciadores (decapentâmeros) são
conjugados de maneira covalente a grupos fluorescentes e são dirigidos para a região do
codão 6 do gene da ß-globina. Um, perfeitamente complementar da sequência normal, está
ligado à fluoresceína e o outro, conjugado à rodamina, difere do primeiro num nucleotídeo e
é complementar da mutação da anemia falciforme. O terceiro iniciador, correspondente à
sequência da cadeia oposta e situado a montante do codão 6, é utilizado como iniciador desta
cadeia, sendo o seu par na reacção de amplificação, ora o iniciador marcado com a
fluoresceína, ora o marcado com a rodamina.
Este método é baseado no princípio duma maior eficácia da PCR quando um dos
iniciadores é completamente complementar da sequência com que se hibrida do que quando
apresenta um desemparelhamento. Quando o DNA normal é amplificado, o iniciador
complementar da sequência é prolongado. Como este iniciador está marcado com a
fluoresceína, o DNA amplificado emite uma cor verde à luz ultravioleta. Quando o DNA
amplificado provém dum feto ou dum paciente com anemia falciforme, só o iniciador
marcado com a rodamina, complementar da mutação, é prolongado, e o DNA amplificado
fluoresce com cor vermelha. Quando o DNA provém dum indivíduo heterozigótico, ambos
os iniciadores, marcados cada um com o seu próprio grupo fluorescente são prolongados e o
DNA amplificado apresenta uma cor amarela resultante da fluorescência complementar dos
dois corantes. A cor do produto amplificado pode ser analisada, após electroforese em gel de
agarose (Fig. 224.II.), por laser de comprimento de onda dupla (vd. secção sobre a

152
sequenciação automática) ou por filtração num sistema de microfiltração selectivo que
permite eliminar o excesso dos iniciadores do meio reaccional e reter o DNA amplificado.
Este é em seguida analisado por laser ou à luz UV.
Estes desenvolvimentos permitem a automatização do diagnóstico, em rotina, de doenças
genéticas, em fase pré-natal, de maneira rápida precisa, sensível e simples.
2) Detecção de doenças infecciosas por PCR
Entre as numerosas aplicações da técnica do PCR, a detecção de infecções numa fase
precoce, têm uma importância particular. Por exemplo, o diagnóstico de infecções
microbacterianas ou microplásmicas é por vezes longo e laborioso, necessitando a cultura do
microorganismo, frequentemente difícil e longa (várias semanas), ou um aumento
significativo do título de anticorpos para fazer um teste serológico.
Foi demonstrado que a introdução da PCR ao diagnóstico da tuberculose, amplificando
um fragmento de 383 pb que é em seguida hibridado com oligonucleotídeos específicos,
permite a detecção rápida de Mycobacterium tuberculosis.
A mesma abordagem, aplicada à detecção de Mycoplasma pneumonia, responsável de
várias doenças respiratórias, revelou que é possível detectar até 102 organismos, após
lavagens bronco-alveolares, com grande especificidade.
O diagnóstico de infecções virais assume um relevo especial pela importância, por vezes
crítica, em detectar a presença de vírus o mais cedo possível no organismo supostamente
exposto, afim de poder aplicar mais eficazmente uma terapia ou tomar medidas preventivas
contra a disseminação e conter um risco epidemiológico.
Dois exemplos característicos são o do HPV (Human Papilloma Vírus, vírus humano do
papiloma) e o do HIV (Human Immunodeficiency Vírus, vírus humano da imunodeficiência).
Detecção de HPVs
Os vírus humanos do papiloma apresentam-se na forma de viriões icosaédricos contendo
um DNA de cerca de 8 kb. Cerca de 60 tipos distintos de HPV foram descritos, estando
muitos associados a sintomas clínicos particulares ou a manifestações patológicas bem
definidas. Por exemplo, cerca de 20 tipos de HPV foram identificados em lesões benignas em
pacientes com epidermodisplasia verruciforme ou com simples verrugas comuns. Dentro
deste grupo foram particularmente identificados 2 tipos, HPV5 e HPV8, presentes em cerca
de 90 % dos casos de cancro da pele.
Dentro do grupo dos HPVs que ocorrem em lesões genitais (pelo menos 7) dois tipos,
HPV16 e HPV18, foram encontrados associados a uma percentagem elevada de cancros do
cérvix uterino ou a outros cancros anogenitais (HPV16 a mais de 50 % e HPV18 a cerca de

153
20 %). Sendo, actualmente, correntemente, admitido que estes tipos de vírus são um cofactor
etiológico importante na patogénese destes cancros e, HPV16 e HPV18, nomeadamente, na
dos cancros anogenitais, torna-se evidente a importância da sua detecção precoce. Não
existindo, actualmente, sistemas de cultura apropriados para replicar os HPV, nem testes
serológicos disponíveis, numerosos esforços foram desenvolvidos para detectar a presença do
vírus por hibridação molecular e, em particular, mais recentemente, por PCR, nos tecidos de
risco.
Um dos factores importantes a ter em consideração para a escolha dos segmentos de
DNA viral, alvos da amplificação, é o da integridade dessa região, no decorrer do processo
infeccioso. Sabe-se, com efeito, que no caso das lesões malignas o DNA viral se integra
frequentemente no DNA genómico das células hospedeiras, conduzindo a eliminações ou a
rupturas de sequências virais. Contudo, duas regiões do vírus, correspondentes aos genes que
codificam para as proteínas E6 e E7 são invariavelmente conservadas e constituem portanto
um alvo privilegiado da amplificação. Uma escolha judiciosa dos iniciadores de
amplificação, dirigidos para sequências pouco conservadas dos diferentes tipos de vírus, e
iniciadores da amplificação de fragmentos de DNA de comprimento facilmente separáveis
por análise electroforética e de sequências diferenciáveis com sondas oligonucleotídicas
específicas, permite detectar e identificar especificamente a presença e o tipo de vírus
infectando potencialmente tecidos de risco.
Exemplo desta abordagem é a detecção dos HPV11 e 16 no DNA celular obtido a partir
de esfregaços cervicais de mulheres suspeitas de possuírem anomalias citológicas. Os
iniciadores sintéticos utilizados para a amplificação enzimática obedecem aos critérios
definidos acima (Fig. 225.II.). São dirigidos para uma região do gene E6 dos vírus HPV11,
16 e 18, são específicos de cada um dos vírus e amplificam segmentos de comprimento
diferente (90, 120 e 100 pb, respectivamente). A Fig. 226.II. atesta a especificidade destes
iniciadores utilizados na aplicação da PCR ao DNA isolado de linhagens celulares que
contêm DNA do HPV16 (CaSki), do HPV18 (HeLa) ou aos DNAs de HPV11, 16 e 18
clonados em plasmídeos.
Os iniciadores dirigidos para o HPV16 iniciam a amplificação do DNA de CaSki e do
DNA de HPV16 clonado (corredor 1 e 2), mas não o do HPV11 ou o do das células HeLa
(corredores 3 e 4), os iniciadores dirigidos para o DNA de HPV11 amplificam um segmento
deste DNA clonado (corredor 5) mas não o das células CaSki (corredor 6) e os iniciadores
específicos de HPV18 iniciam a amplificação do DNA de HeLa e do DNA de HPV18
clonado, (corredores 7 e 8), mas não do DNA de CaSki (corredor 9). Os fragmentos
amplificados, analisados por electroforese em gel de poliacrilamida e visualizados com o
brometo de etídeo, separam-se nitidamente, correndo em função do comprimento respectivo

154
(90, 100 e 120 pb). Este resultado é confirmado por análise por Southern blotting hibridando
os fragmentos amplificados com sondas oligonucleotídicas específicas, marcadas com 32P.
A Fig. 227.II. ilustra o resultado do diagnóstico da infecção por HPV em esfregaços do
cérvix uterino. O espécimen 47 é negativo para HPV16 (Fig. 227.II.A) e positivo para
HPV11 (Fig. 227.II.B) e os espécimes, 76, e 84 são positivos para HPV16, apresentando
bandas amplificadas de intensidade variável. Os espécimes 38, 43 e 47 são positivos para
HPV11, que está normalmente associado com lesões benignas (condilomas). Os espécimes 1,
7 e 87, submetidos à amplificação por PCR com uma mistura de pares de iniciadores
específicos de HPV11 e de HPV16 revelam-se infectados por ambos os vírus. Três controlos
figuram nos corredores centrais (Fig. 227.II.B). Podem, pois, ser efectuadas reacções com
iniciadores múltiplos para a detecção simultânea de diferentes tipos de HPV na mesma
amostra clínica.
Análises com uma amostragem mais ampla mostram que todas os mulheres que
apresentam uma citologia cervical anormal estão infectadas, unicamente com o HPV16 ou
simultaneamente com o HPV16 e o HPV11, mas nunca só com o HPV11.
A enorme sensibilidade (1 molécula de DNA de HPV em 105células), a especificidade
elevada, a possibilidade de utilizar quantidades ínfimas de material (< 0,5 µg de DNA
celular) para as análises de rotina e de detectar simultaneamente mais de um tipo de HPV,
são propriedades que fazem desta técnica, não só um poderoso meio de diagnóstico, mas
igualmente um instrumento de investigação epidemiológica sobre o potencial oncogénico dos
HPVs.
Detecção de HIV
Uma outra aplicação de grande actualidade e enorme interesse é a detecção dos vírus
HIV. Foi, com efeito, demonstrado que a técnica PCR permite identificar indivíduos
infectados com HIV antes de estes terem desenvolvido anticorpos. Sendo a detecção de
anticorpos a base actual do diagnóstico de rotina, é compreensível que numerosos
desenvolvimentos sejam realizados para permitirem a adopção em rotina de análise clínica
por PCR. Estando bem documentada a existência frequente de casos de pessoas infectadas
com HIV que não desenvolvem anticorpos antivirais por períodos para cima de um ano, não
deve ser excepcional que dadores de sangue infectados escapem ao diagnóstico. Um outro
grupo importante são as crianças de mães seropositivas para o HIV e cujo diagnóstico
serológico não é possível durante vários meses antes do desenvolvimento do sistema
imunológico.
O HIV é um vírus de RNA do tipo dos retrovírus. Como vimos, o genoma destes vírus é
copiado em DNA complementar por uma transcritase reversa e eventualmente integrado, na

155
forma de DNA, no DNA cromossómico das células infectadas e, em particular, no caso dos
HIVs, nos linfócitos T, nos macrófagos e nas células microgliais do cérebro.
O vírus pode ser identificado por PCR, nomeadamente no sangue de indivíduos de risco.
Dada a heterogeneidade do genoma viral, bem caracterizada por sequenciação de vírus
isolados de vários indivíduos ou do mesmo indivíduo, a tempos diferentes do processo
infeccioso, é importante escolher para alvo da amplificação uma região do genoma cuja
sequência seja perfeitamente conservada. É este o caso do gene gag de HIV1, dentro do qual
foram seleccionadas duas sequências em cadeias opostas para a hibridação com dois
oligonucleotídeos sintéticos, iniciadores de amplificação de um fragmento de 213 pb.
A amplificação aplicada à detecção directa do HIV em amostras de sangue, para ser
eficaz, têm que ser altamente sensível porque, em geral, só uma pequena percentagem de
linfócitos T está infectada. A Fig. 228.II.A ilustra a sensibilidade da amplificação aplicada ao
segmento de 213 pb, a partir de 2 picogramas de DNA de HIV clonado num plasmídeo, em
função do número de ciclos. A detecção é efectuada por hibridação de uma amostra diluída
de 1/70 de meio reaccional, aplicada numa membrana de nitrocelulose, com uma sonda
oligonucleotídica de 19 nucleotídeos, marcada com 32P (dot blot).
A verificação do comprimento do segmento amplificado efectua-se por electroforese
num gel de agarose 2 % de uma amostra do meio reaccional, seguida de autorradiografia. A
radioactividade provém da incorporação de um nucleosídeo trifosfato (por exemplo 32P-
dCTP), adicionado à mistura dos restantes 3 nucleosídeos trifosfatos durante a reacção de
polimerização (Fig. 228.II.B).
Uma quantificação do número de moléculas de DNA detectáveis dá a medida da
sensibilidade deste método. A Fig. 228.II.C. mostra que é possível detectar até 4 moléculas
de DNA. Este resultado foi obtido amplificando uma mistura composta de uma quantidade
fixa do DNA do HIV plasmídico (4 x 105 moléculas) e de quantidades crescentes (a partir de
1µg) de DNA humano de controlo que serve de meio de diluição serial. Esta sensibilidade de
4 moléculas corresponde à possibilidade de detectar 1 molécula de DNA de HIV em cerca de
106 linfócitos.
Aplicada à identificação de HIV em indivíduos seropositivos, a amplificação enzimática
confirma inequivocamente a infecção. A Fig. 229.II. representa o resultado da hibridação de
DNA, proveniente de sangue de três indivíduos seropositivos, amplificado e aplicado numa
membrana, com a sonda oligonucleotídica. O número de ciclos de amplificação necessários
para obter um sinal é variável de indivíduo para indivíduo e reflecte provavelmente a
abundância de linfócitos T no sangue destes pacientes.

156
Os resultados mais importantes deste estudo aparecem na Fig. 230.II. Entre um grupo de
risco constituído por 16 pessoas seronegativas, parceiras de indivíduos seropositivos, 5
revelaram-se inequivocamente infectadas por HIV. Uma análise electroforetica do DNA
amplificado destes indivíduos confirma plenamente estes resultados (Fig. 231.II.).
A detecção directa do DNA viral pode, pois, antecipar-se ao desenvolvimento de uma
reacção serológica positiva. Este método foi, também, aplicado com sucesso à identificação
do DNA de HIV no sangue de crianças com menos de 18 meses nascidos de mães
seropositivas.
Utilizado a grande escala este teste constituirá, sem duvida, um progresso importante
para conter a disseminação, controlar a epidemia e determinar estratégias terapêuticas.

157
12. REGULAMENTAÇÃO SOBRE A UTILIZAÇÃO DE ORGANISMOS
GENETICAMENTE MODIFICADOS E A PROTECÇÃO CONTRA OS RISCOS
BIOLÓGICOS
As aplicações dos organismos recombinantes que podem beneficiar as nossas sociedades
são, como vimos, numerosas e abrangem importantes sectores económicos e sociais. No
entanto, como qualquer outra actividade humana, a que recorre à utilização dos organismos
geneticamente modificados não é isenta de riscos. Estes riscos potenciais de futuras
aplicações foram, pela primeira vez, objecto de preocupações expressas publicamente pelos
próprios cientistas, em 1974, em carta dirigida à Academia das Ciências dos Estados Unidos
da América. Foi primeiramente pedido que fosse observado uma moratória, aplicando-se às
experiências de clonagem que envolvessem os genomas de certos vírus animais, de
determinantes de resistência a antibióticos, de toxinas e de oncogenes. Foram, em seguida,
elaboradas, pela primeira vez, em 1976, pelo National Institutes of Health (NIH), uma série
de directivas que foram actualizadas em 1986, que definem categorias de confinamento
biológico e físico a que os investigadores desta área se devem submeter. Estes diferentes
níveis de confinamento são definidos em função do tipo de vector e da origem do DNA
objecto da recombinação.
O novo conceito de confinamento biológico preconiza a utilização de sistemas vector-
hospedeiro "desarmados", que só podem sobreviver em condições laboratoriais especiais.
A noção de confinamento físico estabelece quatro níveis de segurança, que vão das
práticas laboratoriais microbiológicas de base (BL-1 = Biosafety Level-1) até às mais
sofisticadas medidas e condições de protecção do pessoal e do meio ambiente (BL-4), por
imposição de progressivas barreiras físicas (pressão negativa, portas duplas e herméticas,
autoclaves, filtração do ar de saída, descontaminação rigorosa de todo o material e dos
efluentes, etc.)
Estas recomendações levaram ao desenvolvimento de sistemas vector-hospedeiro sem
risco para o tipo de actividade em vista (ensino, investigação académica ou industrial,
produção...). Um certo número de estirpes bacterianas foram modificadas de maneira a torná-
las não viáveis fora do laboratório. Por exemplo, a estirpe X1776, derivada de E. coli K12,
possui uma exigência metabólica (biossíntese da lisina), que não pode ser compensada pelas
substâncias presentes no intestino humano (acido diaminopimélico).
Além disso, esta estirpe possui uma membrana celular frágil que se desagrega em
presença de detergente ou de força iónica fraca. A esta estirpe seguiram-se outras derivadas
de K12, de utilização mais cómoda, mas também incapazes de sobreviver fora do laboratório.

158
Os plasmídeos também foram modificados para serem incapazes de se transferirem de
uma estirpe para outra e não poderem disseminar a resistência aos antibióticos.
Quanto aos fagos, uma série de mutações sem sentido nos genes de associação, torna-os
incapazes de se multiplicarem fora dum hospedeiro de laboratório apropriado.
Os riscos potenciais do DNA recombinante continuam a pertencer ao domínio da
especulação, na medida em que os milhões de experiências realizadas nos últimos 25 anos
não permitiram nunca pôr em evidência riscos biológicos causados por uma bactéria, vírus,
fungo, ou célula portadores de DNA recombinante, para além dos perigos inerentes à
natureza da molécula de DNA recombinante. Isto é, a recombinação genética não levou até
hoje ao desenvolvimento de novas propriedades inesperadas nos organismos recombinantes.
Como não é, no entanto, possível demonstrar a segurança absoluta duma construção genética
particular em todas as circunstâncias que se possam imaginar, é normal e aconselhável, que
as experiências envolvendo organismos geneticamente modificados se efectuem adoptando
medidas de precaução definidas por directivas compulsórias, em particular as experiências de
maior risco, pelo impacto que podem vir a ter no meio ambiente, ou as que levantam
problemas de ordem ética, tais como:
-fermentações a grande escala de organismos contendo DNA recombinante,
-a libertação deliberada no ambiente de organismos geneticamente modificados,
-a transferência de genes em células embrionárias de mamíferos e criação de animais
transgénicos,
-a terapia genética e a clonagem de seres humanos.
Em 1990, o Parlamento Europeu aprovou duas directivas, uma relativa à utilização
confinada de microorganismos geneticamente modificados e outra relativa à libertação
deliberada no ambiente de organismos geneticamente modificados, propostas pelo Conselho
das Comunidades Económicas Europeias, que definem a legislação que os Estados Membros
deverão implementar.
Esta legislação é apresentada, na sua forma integral, em anexo.

159
ANEXO
Directivas do Conselho da CEE





























