Embed Size (px)
DESCRIPTION
Termodinamica aplicada a metalurgia
Citation preview

197
FACULDADE DE TECNOLOGIA DE PINDAMONHANGABA
FUNDAMENTOS BÁSICOS DE TERMODINÂMICA VOL.III
Prof. César Leandro
18/5/2010
″Saber e não realizar até fazer com competência e exatidão, não se trata de
saber.Mas sim uma falsa ilusão de sua habilidade. ″
Prof.Thiago A.S.Leandro
ESSE TEXTO ABORDA TODOS OS TÓPICOS DA FÍSICO-QUÍMICA METALURGICA QUE AUXILIAM NA COMPREENSÃO DAS REAÇÕES QUÍMICAS E FÍSICAS DESENVOLVIDAS EM PROCESSOS ESSENCIALMENTE METALÚRGICO

198
12 NOÇÕES SOBRE ESCÓRIAS METALÚRGICAS No período de fusão em fornos elétricos a arco (FEA), utiliza-se sempre
a potência máxima, com auxilio do oxigênio para auxiliar na liberação de maior
calor, cortar a sucata através do jato de oxigênio e assim aumentar a troca
térmica, por aumentar a área da sucata, acelerando a fusão. Além disso a
relação H/D (altura/diâmetro) do forno é menor que na panela, ajudando nas
reações entre metal e escória. Portanto é bem claro que o ambiente no forno
elétrico é mais oxidante que na panela.
Essa diferença no teor de oxigênio permite melhores resultados
durante a elaboração do aço, dependendo da etapa do processo a ser
considerada. Aqui exemplifica-se com dois processos bem conhecidos:
Desfosforação e Dessulfuração. A desfosforação é realizada em ambiente com
mais elevado teor de oxigênio FEA (Forno Elétrico a Arco), o contrário é
verdadeiro para a dessulfuração FP (Forno panela). Caso realize um trabalho
muito forte de desfosforação no FEA e durante o vazamento não se faz a
remoção da escória e contínua o trabalho dessulfurando no FP em ambiente
com baixo teor de oxigênio. Tem-se como resultado uma refosforação,
perdendo-se dessa forma todo o trabalho efetuado. Fica bem claro que o
ambiente para desfosforação é o Forno Elétrico. A descarburação também é
realizado em ambiente de alto teor de oxigênio e portanto entende-se realizado
no FEA. O processo de descarburação torna-se importante, pois ajuda a
eliminar impureza para escória, diminuir gases dissolvidos no aço e elevar a
temperatura.
Processos como dessulfuração, desgaseificação, desoxidação e
remoção das inclusões no aço são próprios para o Forno Panela.
12.1 Basicidade O termo basicidade tem sido usado na prática e em geral expressa a
diferença ou a relação entre as concentrações de óxidos denominados “ácidos”
e “básicos” nas escórias. A definição geral do índice de basicidade é

199
ácidosbasesB
%%
= (12.1)
São óxidos básicos – CaO, MgO – base fraca – MnO, FeO – ácidos
fracos – Al2O3, Fe2O3 e ácidos – SiO2
A noção de basicidade é totalmente empírica e arbitrária. A noção de
ácido e base apareceu da observação de que as escórias ricas em sílica
atacam os refratários dolomíticos ou magnesianos e vice-versa. Escórias ricas
em CaO e MgO atacam refratários silicosos. Nenhum outro fundamento teve a
introdução destes termos em metalurgia e nenhuma relação existe com a
noção precisa de ácidos e base de físico-química das soluções aquosas. Trata-
se portanto de noção arbitrária, mas que tem sido útil na metalurgia. O valor do
índice de basicidade, tal como definido, está relacionado com o comportamento
das escórias nos seguintes casos:-
• Ataque das escórias sobre os refratários. A basicidade é um
índice que permite julgar qualitativamente se uma dada escória é
ou não apropriada para uso com um determinado refratário;
• Viscosidade das escórias – Existe uma relação entre a
viscosidade de uma escória e sua basicidade;
• Desfosforação e Dessulfuração – É um fato comprovado que as
escórias básicas são essenciais.
Devido a utilidade desse parâmetro, procurou-se um significado mais
científico. Isso foi conseguido com a noção de ácido e base da físico-química
das soluções aquosas. Uma solução aquosa é um líquido muito diferente das
escórias. Tem-se aí uma dispersão de íons e moléculas, mais ou menos bem
definidas, em um líquido molecular dielétrico. As moléculas desse dielétrico
podem ser rompidas formando íons H+ ou OH-. Quando se predomina o
primeiro tem-se uma solução ácida. Quando o domínio é de OH- tem-se uma
solução básica
Substâncias que se dissolvem em água produzindo grande quantidade
de H+ são ácidos e substâncias que produzem grandes quantidades de OH-
são ditas bases. Naturalmente ácidos tendem a neutralizar as bases.

200
(H+ + Cl-) + (Na+ + OH-) = Na+Cl- + H2O
Ácido + Base = Sal + Água
Nessas soluções aquosas os termos “ácidos” e “base” tem definição
clara e precisa, o que não ocorre com as escórias.
Um significado preciso de basicidade nas escórias foi obtido pelo meio
da atração dos óxidos, que tem encontrado grande aplicação no estudo dos
silicatos fundidos em geral e das escórias em particular.
Segundo essa teoria, os átomos são assimilados a íons tendo uma
carga elétrica igual à valência. Se dois íons a e b de sinais opostos estão
separados por uma distância d, a força de atração entre os mesmos é dado
por:-
2.
dbaFα (12.2)
Os íons oxigênio tem carga 2 e e o cátion metálico tem carga Ze,
sendo z a sua valência. Com d a distância entre os centros do ânion oxigênio e
cátion metálico, a grandeza:
22
2 22d
zdzeI == (12.3)
É utilizada como medida da atração do oxigênio. O termo e2 é omitido
porque é comum a todos os cálculos.
Os raios iônicos são determinados por raio-X. Considera-se o raio do
oxigênio como sendo igual a 1,45 Å . Para um cátion de raio r e valência z, a
atração do oxigênio será:
2)45,1(2+
=r
zI (12.4)
A tabela 12.I classifica os óxidos segundo as atrações crescentes do
cátion para o íon de oxigênio.

201
Tabela 12.I– Mostra os óxidos básicos, anfóteros e ácidos.(LUCIO,A. Apostila de Físico-Quimica, vol.2, 1981, UFMG)
ÓXIDO RAIO CATION ATRAÇÃO O2 BASICIDADE
Na2O 0,98 0,34 Básico
BaO 1,29 0,53 Básico
CaO 0,94 0,70 Básico
MnO 0,80 0,79 Básico
FeO 0,75 0,83 Básico
MgO 0,65 0,91 Básico
BeO 0,30 1,31 Básico
Al2O3 0,45 1,66 Anfótero
TiO2 0,60 1,90 Anfótero
B2O3 0,20 2,20 Anfótero
Cr2O3 0,55 1,50 Anfótero
F2O3 0,53 1,53 Anfótero
SiO2 0,38 2,39 Ácido
P2O5 0,35 3,17 Ácido
A força de atração do íon oxigênio serve para interpretar um grande
número de fenômenos relativos à química das escórias em particular no que se
refere ao caráter mais ou menos ácido ou básico dos óxidos. Com efeito
observando a tabela verifica-se que a série obtida e a série de basicidade
decrescente ou acidez crescente dos óxidos. Do Na2O ao MgO, a basicidade
diminui. No meio da tabela se encontram os óxidos anfóteros, que se
comportam tanto como básico ou ácido.

202
A partir do TiO2 ao P2O5 estão os óxidos ácidos. Vê-se portanto que um
óxido básico é um óxido de fraca atração por oxigênio, portanto sua tendência
é liberar o oxigênio
12.2 Teoria da Estrutura iônica das Escórias
Para se compreender escórias, é necessário dispor de algum conceito
de natureza química das forças inter-atômicas que atuam nas mesmas e o
modo pelo qual estas forças são influenciadas por fatores como composição e
temperatura sempre presentes no controle das operações metalúrgicas.
As primeiras tentativas para se entender as reações metal-escória,
admitia-se que a escória líquida continha compostos moleculares, e que estes
se dissociavam nos óxidos componentes numa extensão que dependiam da
composição e da temperatura. Contudo os resultados encontrados a partir de
medidas de certas propriedades físicas do banho, tais como viscosidade,
condutibilidade elétrica, tensão superficial e números de transporte somente
podiam ser interpretados de modo satisfatório se as escórias forem
consideradas como sendo uma mistura de íons, de modo que o modelo
atualmente proposto para representar a estrutura das escórias líquidas é o de
uma mistura desordenada de ânions e cátions.
Para se entender a estrutura das escórias líquidas, considera-se como
ponto de partida a estrutura da sílica pura que está bem estabelecida e
modelos tem sido propostos para as mudanças estruturais que ocorre quando
óxidos básicos são adicionados a banhos ricos em sílica. Na sílica sólida pura,
cadeias de íons silício compartilham sua carga eletrônica com cada um dos
quatro íons de oxigênio que formam o tetraedo, de modo que cada íon oxigênio
tem uma carga residual negativa, e o grupo tetraédrico tem então quatro cargas
negativas 44−SiO . Neutralidade elétrica é obtida quando cada íon oxigênio é
compartilhado por dois tetraedos, isto é, ligado a dois cátions Si+4 e a relação
cátion/oxigênio é aquela para a sílica, (SiO2). Na coordenação tetraédrica da
sílica sólida, qualquer que seja a forma da mesma, quartzo, tridimita ou
cristobalita, cada átomo de silício está rodeado por quatro átomos de oxigênio,

203
de modo que o silício pode ser considerado como o centro de um tetraedro
com os átomos do oxigênio nos vértices.
Figura 12.1 - Ligação do sílicio com oxigênio, formando grandes células. Eletric Furnace Steelmaking, volume 11,pg 198, 1963, London.
Figura 12.2 - Esquema da célula unitária da sílica, com 1 átomo de silício (cor preta) e 4 átomos de oxigênio, ( cor branca). Eletric Furnace Steelmaking, volume 11,pg 198, 1963, London.

204
Figura 12.3 - Rede de sílica, sendo quebrada por elementos como o Ca e o Fluor. Ref. Alvaro Lúcio, Apostila de Físico-Quimica, vol.2, 1981, UFMG
Cada oxigênio é compartilhado por dois silícios, e a estrutura deve ser
considerada como se estendendo em todas as direções. A figura 12.1 e 12.2 e
12.3 mostram um tetraedro de sílica, mostra como os tetraedros são ligados
entre si de modo a formar uma rede espacial de células hexagonais.
Quando a sílica se funde, os espectros de difração observados tem
sido interpretados como possuindo as mesmas características dos tetraedos de
sílica, com íons oxigênio compartilhados nos vértices, porém o elo de ligação é
mais caótico, de tal modo que os tetraedros não apresentam relações
geométricas entre si. A regularidade da rede sólida é distorcida e pode ser
considerada como uma molécula gigante. Daí a alta viscosidade da sílica
líquida, pois a viscosidade aumenta quando as dimensões da rede de silicatos
aumentam.
No intervalo de temperatura próximo ao ponto de fusão, o líquido é
uma forma desordenada de sólido cristalino, onde subsiste no entanto uma
ordem a curta distância. Observando a figura 12.3 pode se dizer que a
estrutura da sílica é caracterizado pelo fato de que todos os átomos de
oxigênio formarem pontes entre os átomos de sílica, tais como:
- Si – O – Si – O – Si -

205
A estrutura de uma escória liquida rica em sílica é muito análoga
aquela de um vidro. A diferença essencial é que um vidro a desordem é devido
ao fato de que o tempo que seria necessário para formar uma estrutura
ordenada é muito mais longa do que a duração das observações e que a
estrutura tem uma viscosidade muito elevada. No líquido a desordem é devido
às vibrações térmicas dos átomos. As distâncias atômicas são 10% em média
superiores no líquido em relação ao vidro.
Quando um óxido básico como o CaO é adicionado a sílica, cada íon
de oxigênio adicionado penetra na rede e separa os vértices de dois tetraedos,
enquanto o cátion adicionado permanece adjacente à separação e se acomoda
nos vazios da estrutura, figura 12.3. Com adição progressiva de óxidos, a rede
tridimensional é continuamente quebrada pelo rompimento das pontes de
oxigênio, até que eventualmente a sílica está presente como ânions discretos,
o que acontece quando a composição do ortosilicato, 2CaOSiO2 é alcançada.
A figura 12.4 mostra o resultado final de uma adição de óxidos básicos
em uma escória ácida.
Quando este estágio é atingido, novas adições de óxidos básicos não
causam mudanças nas estruturas da sílica.
A incorporação à sílica de uma certa proporção de óxidos básicos, não
destrói os tetraedros de 44−SiO , que permanece sempre na unidade
fundamental. Mas a estrutura é modificada pelo fato de que certos tetraedros
não se tocam mais pelo seus vértices. Em outras palavras um certo número de
pontes de oxigênio são rompidos.
O alumínio se parece com o silício por que tem um íon muito pequeno
e freqüentemente admite ao seu redor quatro íons oxigênio. Nos minerais
naturais o alumínio pode substituir o silício no centro dos tetraedros com a
condição que a diferença entre Al+3 e Si+4 seja compensada por maior número
de cátions da rede. O alumínio forma com a sílica rede mistas de alumino-
silicatos, onde os átomos de alumínio substituem certos átomos de silício e
embora trivalentes, cercam-se de quatro átomos oxigênio. A carga eletrônica
do alumínio sendo igual a três em vez de quatro do silício, é necessário a

206
presença de cátions suplementares. A adição de alumina nas escórias,
contribui para quebrar a rede de silicatos e formar uma rede aluminatos. A
alumina aumenta a viscosidade das escórias básicas e decresce nas escórias
ácidas. Como comprovação experimental, observa-se que escórias de forno
panela com conteúdo acima de 20%, em caso de desgaseificação, pode
ocorrer transbordamento, exatamente por elevar a viscosidade, diminuindo com
isso a permeabilidade dos gases. A figura 12.5 comprova esse efeito do
aumento da viscosidade em escórias básicas com alto teor de alumina.
Figura 12.4 - Representação esquemática do Mg2SO4 cristalino. O silicato fundido seria uma versão desordenada deste esquema. (LÚCIO, A. Apostila de Físico-Quimica, vol.2, 1981, UFMG)
O fósforo é outro íon cujas propriedades químicas são semelhantes às
do silício e pode portanto substituir este último no centro dos tetraedos. O íon
fósforo tem cinco cargas positivas e conseqüentemente quando o fósforo
substitui o silício em coordenação tetraédrica com íons oxigênio a força do
campo elétrico é aumentada e os íons oxigênio são mais fortemente ligados ao
fósforo do que ao silício. Inversamente a substituição do Al enfraquece o
campo elétrico. Estas mudanças refletem nas propriedades termodinâmicas
das escórias. Por exemplo verificou-se que a atividade de um óxido básico é
basicada crescentemente por iguais adição de alumina, sílica e pentóxido de
fósforo.

207
O enxofre na escória provavelmente substitui o oxigênio na rede do
silicato e a estrutura exige a presença do cátion cálcio, mais positivo para se
estabilizar.
Figura 12.5 – Diagrama ternário CaO - 𝐴𝑙2𝑂3 - 𝑆𝑖𝑂2 na temperatura de 1600°C, apresenta as viscosidades em poises de acordo com a relação CaO/Alumina. (COUDURIER,L.; HOPKKINS,D.W.; WILKOMIRSKY,I. Fundamentals of Metallurgical Processes, 2nd Edition, Page 404, 1985).
12.3 Escória Praticada na Fusão dos Fornos Elétricos a Arco Na maioria dos processos de fabricação de aço incl ui a formação da
escória, no qual permanece em contato com o aço, refratário e atmosfera dos
fornos. As escórias podem ser formadas por condições de mistura de óxidos ou
fluxos e freqüentemente promovem reações. A palavra slag em inglês parece
ter origem na Suécia “slagg” significa restos do metal. Conforme definiu
Webster escória é um rejeito do metal, especialmente um produto fundido
contendo muito silicato, que possuem baixa densidade. Em 1934 Herty
caracterizou escórias de fabricação de aço da seguinte forma:- “Escórias são
líquidos complexos consistindo primeiramente de silicato de cálcio com vários
CaO/Al2O3
=0,5
CaO/Al2O3=4,1
CaO/
Al2O3
CaO/SiO2

208
compostos e óxidos em solução ou suspensão.” Escórias básicas são aquelas
no qual CaO e MgO predominam. Escórias ácidas são aquelas com
predominância em sílica. Ocorre sem dúvida uma grande variedade de
composição entre essas escórias básicas e ácidas. Nestas escórias o FeO
pode estar em solução ou combinado com outros elementos e o poder de
oxidação dessas escórias depende da quantidade de óxido de ferro livre
contido.
Tal descrição é inteiramente aceita hoje, nossa atenção pode ser
inteiramente dirigida na formação dessas escórias durante a fabricação do aço
nos fornos elétricos a arco e forno panela.
Durante a etapa de fusão a escória é formada através dos óxidos SiO2,
MnO e FeO. Com a adição de CaO a mistura sólido mais líquido pode ser
representado pelo ponto F no diagrama da figura 12.6. neste instante no
processo de fusão dois fatores tendem a alterar a composição química da
escória que são:-
• Dissolução do CaO;
• Formação de FeO.
O CaO é a componente essencial da escória na fabricação do aço,
provoca uma série de efeitos de significado econômico:-
• Retarda e diminui a oxidação de elementos de liga , cuja
oxidação normalmente formam óxidos básicos;
• Cria condições para eliminação de impurezas, tais como
fósforo e enxofre;
• Retarda e diminui o ataque da escória ao refratário.
Desta forma é interessante que a CaO seja incorporada na escória o
mais rápido possível de tal maneira a manter a trajetória de composição para
se obter alta basicidade, a maior parte do tempo. Em processos rápidos
precisa-se ter essa atenção para que o CaO possa durante o período de fusão
fazer seu papel.

209
Para uma boa dissolução da CaO alguns fatores são de grande
importância como:-
• Viscosidade da escória;
• Temperatura;
• Superfície de contato sólido/líquido;
• Agitação do banho;
• Teor de FeO na escória.
Figura 12.6 - Esquema do diagrama CaO-SiO2-FeO, T = 1600°C mostrando a evolução da composição da escória durante a fusão.(FALCONI, V., Fundamentos de Tecnologia de Fabricação de Aço, vol 1, 2 Ed, 1983, UFMG).
Figura 12.7 - Curva de iso-viscosidade do sistema CaO-SiO2-FeO a 1400ºC (P=poise), segundo Kozakevitch. .(FALCONI, V., Fundamentos de Tecnologia de Fabricação de Aço, vol 1, 2 Ed, 1983, UFMG).

210
A viscosidade da escória constitui um fator importante como mostra a
figura 12.7, a região da flecha é a que procuramos, pois sinaliza o aumento de
CaO na constituição da escória com uma baixa viscosidade, região líquida
conforme mostra a figura 12.6.
Figura 12.8 - Diagrama ternário CaO-SiO2-FeO.Mostra as menos temperaturas para maiores concentrações de FeO e compostos com SiO2 de elevadas temperaturas.(Slag Atlas, 1995)

211
A figura 12.8 mostra que quanto mais FeO o ponto de fusão da escória
decresce, diminuindo com isso a viscosidade da escória.
O aumento da temperatura também favorece o derretimento do CaO e
com isso diminuição da viscosidade com maior fase líquida presente.
É claro que a dissolução da CaO é um processo interfacial e como tal
deve ser diretamente proporcional a superfície de contato escória-metal. Uma
alta superfície de contato pode ser obtida através da utilização de menor
granulometria, ou CaO com alta porosidade.
A agitação do banho também melhora por aumentar a razão A/V (área
/volume) exposta ao calor. Pode-se citar portanto dois tipos diferentes de
mecanismos de dissolução, dependendo do tipo de escória. A figura 12.7
mostra o diagrama ternário CaO-SiO2-FeO, juntamento com os dois tipos de
mecanismos.
Quando a escória inicial é do tipo A (pobre em FeO) a medida que o
CaO dilui forma-se na camada limite o ortosilicato de cálcio (2CaOSiO2), que é
de alto ponto de fusão e prejudica a dissolução do CaO. Quando a escória
incicial é do tipo B, a trajetória de composição da escória não atinge o
ortosilicato (2CaOSiO2), ocorre um equilíbrio entre o líquido e o CaO.
Quando a escória inicial é do tipo A (pobre em FeO) a medida que o
CaO dilui forma-se na camada limite o ortosilicato de cálcio (2CaOSiO2), que é
de alto ponto de fusão e prejudica a dissolução do CaO. Quando a escória
incicial é do tipo B, a trajetória de composição da escória não atinge o
ortosilicato (2CaOSiO2), ocorre um equilíbrio entre o líquido e o CaO.
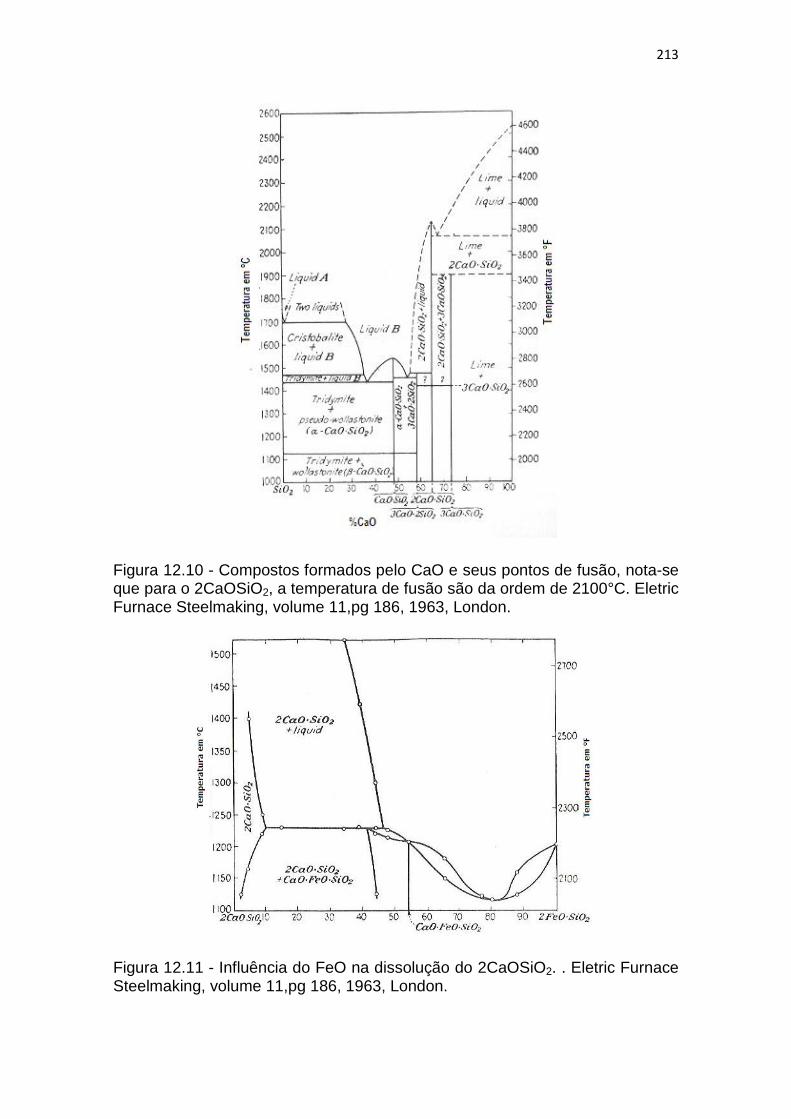
A figura 12.10, mostra em um diagrama binário a temperatura de fus/ao
do 2CaOSiO2, percebe-se o quanto elevada ela é. A figura 12.10, mostra a
influência do FeO na diminuição de temperatura da escória.
A importância do teor de FeO ao definir mecanismos de dissolução do
CaO foi verificado experimentalmente. Demonstrou-se que quanto maior o teor
de FeO da escória, maior será a velocidade de dissolução do CaO.

212
Figura 12.9 - a) mostra a velocidade de dissolução em função de cada tipo de scoria A,B,.. b) mostra no diagram ternário a composição de cada tipo de scoria A, B....., segundo Jon e Riboud.(RICHARD,F. Course on Clean Steel techonologies, March 24-25, 2003 BH, MG, Brazil).
A figura 12.9 mostra os resultados de uma experiência, indicando que
a velocidade de dissolução do CaO pode crescer de 0,55 para 5,9 mg/cm2/s
quando o teor de FeO é aumentado. Assim pode-se concluir que para aços que
permitam a utilização de oxigênio no período de fusão, que não é o caso de
aços inoxidáveis ou rápidos, é possível a não utilização de fluorita, pode ser
perfeitamente substituída pelo FeO gerado na oxidação da carga. Também é
importante comentar que esse valor na escória não deve ser elevado, pois uma
oxidação intensa prejudica e descontrola a marcha da corrida, podendo em
uma eventual adição de carbono ocorrer uma forte espumação com danos
prováveis para o equipamento.
213
Figura 12.10 - Compostos formados pelo CaO e seus pontos de fusão, nota-se que para o 2CaOSiO2, a temperatura de fusão são da ordem de 2100°C. Eletric Furnace Steelmaking, volume 11,pg 186, 1963, London.
Figura 12.11 - Influência do FeO na dissolução do 2CaOSiO2. . Eletric Furnace Steelmaking, volume 11,pg 186, 1963, London.

214
12.4 Influência do MgO nas Escórias de Fusão dos Fornos Elétricos a Arco
Até aproximadamente10% de MgO como constituinte das escórias, não
se altera suas constiuições físico-químicas.
No entanto se esse teor de MgO não for propositadamente inserido em
sua composição, a escória para manter seu equilíbrio químico com o sistema,
rouba MgO do refratário. Uma prática comum também utilizado nos fornos
elétricos e fornos panelas é a adição de MgO para neutralizar a reatividade da
escória.
Desta maneira a escória é saturada com MgO, inibindo a erosão do
refratário da linha de escória das panelas ou fornos. Em adições acentuadas,
muito mais que 10% a escória torna-se muito viscosa e não reage mais, ou
seja, não dessulfura e também não retém inclusões, a partir do momento da
adição. Aços que possuem faixas de enxofre, nesse caso passam pelo forno
panela sem nenhuma dessulfuração. Com esse procedimento as escórias
também não atacam os refratários.
12.5 Escória Espumante Praticada nos Fornos Elétricos a Arco A formação da escória espumante consiste na evolução de bolhas de
gás através da escória. A formação dessa escória se faz durante
principalmente no período de fusão, até o vazamento. A grande vantagem da
escória espumante é esconder o arco do forno elétrico, protegendo as paredes
do forno contra a irradiação. Para o forno panela no lugar de escória
espumante, pratica-se apenas um maior volume de escória até a cobertura do
arco, minimizando as perdas térmicas por irradiação e a erosão dos refratários.
A espumação acontece através da injeção de carbono mais oxigênio,
propiciando monóxido de carbano que possui um tempo para passar através da
escória. A figura 12.9 mostra a influência na escória da basicidade e da
temperatura no tempo de espumação, nota-se que diminuindo a basicidade,
eleva-se o tempo de espumação e a elevação da temperatura diminui o tempo
de espumação. A figura 12.10, mostra a influência da fluorita no tempo de
espumação, a medida que o teor de fluorita é elevado, o tempo de espumação
é descrescido, por diminuir muito a viscosidade da escória.

215
Figura12.1 - Apresenta a influência da basicidade em função do tempo de espumação. .(RICHARD,F. Course on Clean Steel techonologies, March 24-25, 2003 BH, MG, Brazil).
Figura 12.10 - Tempo de espumação em segundos em função do teor de fluorita da escória. .(RICHARD,F. Course on Clean Steel techonologies, March 24-25, 2003 BH, MG, Brazil).

216
Figura 12.11 - A altura de escória para forno elétrico a arco em função do sôpro de oxigênio. .(RICHARD,F. Course on Clean Steel techonologies, March 24-25, 2003 BH, MG, Brazil).
Um aumento também da vazão de oxigênio, figura 12.11, eleva o teor
de FeO da escória no período de fusão dos fornos elétricos a arco, diminuindo
com isso a viscosidade. Essa diminuição da viscosidade diminui o tempo de
residência da passagem dos gases através da escória e assim a altura da
escória espumante é descrescido, podendo expor o arco elétrico.
12.6 Escórias Praticadas nos Fornos Panelas A figura 12.12 mostra um diagrama indicando as escórias que mais se
praticam nas Aciarias. As escórias na região da letra S são mais ácidas, com
alto teor de sílica da ordem de 50%, com menor ponto de fusão, são praticadas
em altos fornos, pois escórias com alto ponto de fusão, do tipo básicas, nesses
fornos, podem obstruir a marcha do forno. Essas escórias, localizadas na
região S, possuem muito pouca capacidade para dessulfurar e desfosforar e
podem dependendo do revestimento utilizado, atacar os revestimentos internos
dos fornos. Esse tipo de escória aparece na elaboração de aço em fornos
panelas destinados a trefilação em bitolas minhas pequenas na ordem de 7
mm, aplicados a cordoalha de pneus de carros, ou utilizados nos motores como
mola de válvulas. Com o objetivo de elevar a resistência a fadiga desses aços,

217
as inclusões devem ser deformáveis na temperatura de laminação e para a
ocorrência dessas inclusões, a elaboração de escórias do tipo S são
necessárias. Essas escórias também são praticadas para aços com alto teor de
enxofre na ordem de 0,30%, destinados a usinagem fácil.
Figura 12.12 - Diagrama ternário Al2O3 – CaO - SiO2, e. ( Refino Secundário dos Aços, 15 a 19 de setembro de 2003, ABM).
As escórias na região da letra A predominam os aluminatos de cálcio,
são mais próprias para o trabalho tanto de dessulfuração quanto para
desfosforação e captação de inclusões. São utilizadas também para aços
fabricados pela rota de lingotamento contínuo no qual o refino de grão é
importante (aços com teor de alumínio < 0,050%). Nessa prática as inclusões
sólidas de alumina oriundas da desoxidação do alumínio, são transformadas
para inclusões líquidas de aluminatos de cálcio, através da adição de cálcio.
Assim não ocorre obstrução de válvula por entupimento. Na elaboração
de aço rolamento do tipo 52100, onde o teor de oxigênio solicitado é menor que
10 ppm, também as escórias de aluminato de cálcio são bem utilizadas, com
resultados promissores na utilização de alumina calcinada na escória em lugar
de fluorita. Essa prática diminui a viscosidade da escória sem a utilização de

218
fluorita, que vem ultimamente tornando-se escassa no mercado, além de
resultados mais reprodutivos dos aços produzidos por essa prática.
13 PROPRIEDADES PARCIAL MOLAR – POTENCIAL QUÍMICO A teoria da termodinâmica o qual tem sido desenvolvida é aplicável
somente para sistemas fechados. Isto é, aplica-se portanto para as importantes
relações, apresentadas pelas equações 9.26 e 9.28:
dU = TdS – PdV
dG = -SdT + VdP
São fundamentos entendidos para um sistema que não troca matéria
com o meio. Em geral essas relações não refletem a experiência. Contudo é
possível para a entropia aumentar um simples aumento na quantidade de
reagentes em uma solução. A partir da equação dG = -SdT + PdV, pode-se
concluir que se a temperatura e pressão do sistema são mantidos constantes
não haverá mudança. Isto está correto para sistema sem variação de massa, G
é uma quantidade extensiva e seu valor pode ser aumentado simplesmente por
aumentar a quantidade de matéria no sistema, em temperatura e pressão
constante. Por exemplo se aumentarmos a quantidade de reagentes na célula
galvânica é possível ocorrer maior formação de produtos, devido a esse
aumento de reagentes, espontâneamente.
Essas dificuldades são devidas ao assunto implícito, no qual duas
variáveis são capazes de ser suficiente para fixar um estado no sistema. Isto
pode somente ser verdadeiro para corpos com composição fixa. Devemos
então introduzir dentro das equações as variáveis para a composição e
dimensão do sistema, ou seja, o número de moles. Consideremos uma fase
homogênea no qual existem diferentes substâncias. Sendo n1 (mols), a
quantidade da substância na fase 1, n2 a quantidade em mols da fase 2 e
assim por diante. De acordo com a equação dU = TdS – PdV, se n1,n2..... são
constantes, a energia interna U da fase depende somente de S e V. Contudo
para uma composição variável.
U = U(S,V, n1,n2....)

219
Então para uma diferenciação total
𝑑𝑈 = �𝜕𝑈𝜕𝑆�𝑉,𝑛1,𝑛2..
𝑑𝑆 + �𝜕𝑈𝜕𝑉�𝑆,𝑛1,𝑛2..
𝑑𝑉 + ∑ �𝜕𝑈𝜕𝑛𝑖�𝑉.𝑆𝑑𝑛𝑖𝑘
𝑖=1 (13.1)
Na equação 13.1 o subscrito ni para as primeiras duas derivadas
parciais implica que a quantidade de todas as espécies são constantes. O
último termo na equação é a soma das partículas considerando a entropia e
volume constantes.
Para quantidades constantes é inteiramente válido, as seguintes
equações:
�𝜕𝑈𝜕𝑆�𝑉,𝑛𝑖
= 𝑇 (13.2)
�𝜕𝑈𝜕𝑉�𝑆,𝑛𝑖
= −𝑃 (13.3)
Defini-se então potencial químico como sendo:
𝜇𝑖 = �𝜕𝑈𝜕𝑛𝑖�𝑆,𝑉,𝑈
(13.5)
Então dU pode ser reescrito
𝑑𝑈 = 𝑇𝑑𝑆 − 𝑃𝑑𝑉 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.4)
A quantidade µi é chamado de Potencial Químico, foi introduzido na
termodinâmica por Willard Gibbs
𝜇𝑖 = �𝜕𝑈𝜕𝑛𝑖�𝑆,𝑉,𝑈
(13.6)
, e ele facilita grandemente a discussão de
sistemas abertos ou para alguns sistemas fechados no qual haverá mudança
de composição. Foi definido por Gibbs como segue: Se para alguma massa
homogênea nós supormos uma infinitesimal quantidade de qualquer substância
a ser adicionada, a massa permanecendo homogênea e sua entropia e volume
permanecendo inalterados. O aumento da energia da massa devido a
quantidade de substância adicionada é o potencial para aquela substância na
massa considerada, representado pela equação:

220
O potencial químico tem uma função importante, análogo a
temperatura e pressão. Uma temperatura diferente determina a tendência do
calor passar de um corpo para o outro, a diferença entre pressões determina a
tendência para movimento. Será demonstrado que a diferença em potencial
químico pode causar tendência a reação química ou a difusão.
O potencial químico pode ser introduzido por um método alternativo no
qual algumas vezes encontrou-se ser mais claro. É baseado na equação
dG = -SdT + VdP (13.7)
A equação 13.7, não representa uma reação quando ocorre variação
de produtos ou reagentes. De uma forma geral G deve ser determinado
também pela quantidade dos elementos adicionados, bem como T e P. Então:
G = G(T, P, n1, n2....)
E assim
𝑑𝐺 = �𝜕𝐺𝜕𝑇�𝑃,𝑛1.𝑛𝑖
𝑑𝑇 + �𝜕𝐺𝜕𝑃�𝑇,𝑛1,..𝑛𝑖
𝑑𝑃 + ∑ �𝜕𝐺𝜕𝑛𝑖�𝑇.𝑃,𝑛𝑖
𝑑𝑛𝑖𝑘𝑖=1 (13.8)
Agora a primeira derivada parcial na expressão refere-se a mudança
de G para a pressão e substâncias constantes e a segunda para temperatura e
substâncias constantes. Assim
�𝜕𝐺𝜕𝑇�𝑃,𝑛𝑖
= −𝑆 (13.9)
�𝜕𝐺𝜕𝑃�𝑇,𝑛𝑖
= 𝑉 (13.10)
E portanto
𝑑𝐺 = −𝑆𝑑𝑇 + 𝑉𝑑𝑃 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.11)
Sendo
𝜇𝑖 = �𝜕𝐺𝜕𝑛𝑖�𝑇,𝑃
(13.12)

221
O potencial químico de um componente de uma fase é a capacidade
da fase em realizar trabalho através de infinitésimas quantidade de substância
adicionadas com a temperatura e pressão constantes.
Por exemplo o potencial químico do sulfato de cobre na solução
aquosa na célula de Daniel é igual ao aumento da capacidade da célula
fornecer energia elétrica por quantidade de sulfato adicionado (a adição
atualmente sendo um infinitesimal).
Da mesma forma que se definiu µi para G, defini-se para as outras
funções U, H e A. Então
𝑑𝑈 = 𝑇𝑑𝑆 − 𝑃𝑑𝑉 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.13)
𝑑𝐺 = −𝑆𝑑𝑇 + 𝑉𝑑𝑃 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.14)
𝑑𝐻 = 𝑇𝑑𝑆 + 𝑉𝑑𝑃 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.15)
𝑑𝐴 = −𝑆𝑑𝑇 − 𝑃𝑑𝑉 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.16)
A energia livre parcial molar de um componente é igual então ao
potencial químico do componente na mistura.
13.1 Equilíbrio Térmico e Mecânico
É a propriedade de temperatura no qual dois sistemas A e B estão em
equilíbrio térmico, Tα = Tβ. Para equilíbrio mecânico as pressões entre
interfaces dos fluidos devem ser iguais. Consideraremos dois fluídos A e B, que
não estejam enclausurados em rígidos recipientes, mantendo a temperatura
constante sob tal circunstância, a total energia de Helmoltz dos dois fluidos
deve ser um mínimo junto ao equilíbrio.
dAα + dAβ = 0 (13.17)
Considerando uma possível variação no volume dVα e em dVβ, a
temperatura e composição permanecem inalteradas.
𝑑𝐴 = −𝑆𝑑𝑇 − 𝑃𝑑𝑉 + ∑𝜇𝑖 𝑑𝑛𝑖 (13.18)

222
Para n, T = constante
dAα = - PαdVα (13.19)
dAβ = - PβdVβ (13.20)
Mas dVα= - dVβ desde que o volume total é constante. Então a condição
de equilíbrio reduz para:
(Pα – Pβ)dVβ = 0 (13.21)
No qual pode ser satisfeito a equação para a condição
Pα = Pβ (13.22)
Esta condição naturalmente não é correta se os dois fluidos são
separados por barreiras móveis ou então por superfícies curvas. A variação
dVα no volume implica uma variação interfacial entre os dois fluidos. Em tal
sistema a área e a tensão interfacial (ɤ), tem adicional significância na variável
de estado. Sob tal condição, pode ser mostrado a condição do equilíbrio
mecânico.
�𝑃𝛽 − 𝑃𝛼� = 𝛾 �1𝑟1− 1
𝑟2� (13.23)
Sendo r1 e r2 os raios da s superfícies curvas das interfaces.
13.2 Equilíbrio para Transferência entre Fases
Considera-se um sistema de diversas fases no qual estão em equilíbrio
térmico junto a temperatura constante. Cada fase é um típico sistema aberto,
desde que pode trocar substância com as fases vizinhas. Será mostrado que
para a condição de equilíbrio para todas fases acontece, quando o potencial
químico delas são iguais.
Para uma variação do sistema no qual um dn da substância i passa
através da fase β e entra na fase α. De acordo com a equação dA = -SdT –
PdT + ∑µidni. A variação para o sistema fica:
−𝑆𝑖𝛼𝑑𝑇 − 𝑃𝑖𝛼𝑑𝑉 + ∑ 𝜇𝑖𝛼𝑑𝑛𝑖𝛼 −𝑛𝑖=1 𝑆𝑖
𝛽𝑑𝑇 − 𝑃𝑖𝛽𝑑𝑉 + ∑ 𝜇𝑖
𝛽𝑑𝑛𝑖𝛽 < 0𝑛
𝑖=1 (13.24)

223
Para Temperatura e Volume constantes
∑ 𝜇𝑖𝛼𝑑𝑛𝑖𝛼𝑛𝑖=1 + ∑ 𝜇𝑖
𝛽𝑑𝑛𝑖𝛽 < 0𝑛
𝑖=1 (13.25)
Para transferir i de β para fase α, −𝑑𝑛𝑖𝛽 = 𝑑𝑛𝑖𝛼. Como i é o mesmo
elemento para as duas fases, pode-se entender que ni é igual, mudando
apenas o valor no sinal.
𝜇𝑖𝛼𝑑𝑛𝑖 − 𝜇𝑖𝛽𝑑𝑛𝑖 < 0 (13.26)
Que significa a diferença entre os potenciais
𝜇𝑖𝛼 − 𝜇𝑖𝛽 < 0 (13.27)
Assim a diferença no potencial é o que faz ocorrer a troca entre
substâncias ou a difusão da matéria.
13.3 Princípio da Pressão Parcial de Dalton Considerando-se um volume fixo V, temperatura T, nA moles do gás
perfeito A. A pressão exercida será:
PA.V= nA.RT ⇒PA=nART/V (13.28)
Nesse volume adicionamos nB moles de um gás B, então a pressão total
passa a ser:
P = PA + PB = (nA + nB)RT/V (13.29)
Dividindo as duas equações anteriores tem-se:
(13.30)
Entende-se que a pressão parcial de um elemento A é diretamente
proporcional a sua fração molar na solução.
13.4 A Energia Livre de Mistura dos Gases
Definiu-se µi = Gi/ni, portanto a equação da energia livre para um mol e
para apenas um tipo de gas fica: µi = Gi
AAABA
A
BA
A
BA
AA XPPXnn
n
VRTnn
VRTn
PPP
PP .)( =⇒=
+=
+=
+=

224
Para determinar o valor da energia para um gás perfeito evoluindo de
uma pressão de 1 atm até uma pressão P, temos, conforme equação 9.28:
dG = -SdT + VdP, para temperatura constante
dG = VdP, mas sabe-se que PV = nRT, portanto pode-se fazer:
(13.31)
(13.32)
Em termodinâmica nada pode se dizer em valores absolutos G(P2,T)
ou G(P,T). Sempre os valores são comparativos. Portanto escolhe-se um
estado de referência, no qual as mudanças de energia livre podem ser
medidas. Esse estado de referência é conhecido como o estado padrão e este
foi escolhido como sendo o gás puro em 1 atm de pressão na temperatura de
interesse. A energia livre de n moles do gás no estado padrão G(P=1,T) é
designado por G°(T) e para a energia livre de n mol do gás em qualquer outra
pressão P e temperatura T é dado por:
G(P,T) = G°(T) + nRTLnP, ou simplesmente G = G° + RTLnP (13.33)
Dividindo essa equação pelo número de moles, torna-se portanto o
potencial:
µi = µ° + RTLn(pi) (13.34)
Seja a mistura de gases, indicado na figura 13.1
𝐺𝐼𝑛𝑖𝑐𝑖𝑎𝑙 = 𝜇1° + 𝜇2° + 𝜇3° = 𝑛1𝜇1° + 𝑛2𝜇2° + 𝑛3𝜇3° = ∑ 𝑛𝑖𝜇𝑖°3𝑖=1 (13.35)
𝐺𝐹𝑖𝑛𝑎𝑙 = 𝑛1𝜇1 + 𝑛2𝜇2 + 𝑛3𝜇3 = ∑ 𝑛𝑖𝜇𝑖3𝑖=1 (13.36)
As pressões P1,P2 e P3, representam as pressões das substâncias
n1,n2 e n3 nos seus estados puros, portanto pode-se considerar todas iguais a
pressão P°
(13.37)
( ) ∑∑ ∑ ∑ =−=−=∆= = =
n
i
ii
i i iiiiiiiiMistura P
pLnnRTnnnG
3
1
3
1
3
1µµµµ
[ ]
==−⇒= °
°
°∫∫ οPpiLnnRTLnPnRTGG
PdPnRTdG i
iPP
p
P
G
G0
)(1:; ioi pnRTLnGGatmPSe
Pp
nRTLnGG +=⇔=
+= ο
οο

225
(13.38)
Figura 13.1 - Energia livre na mistura de gases ideais
Como Xi é menor que 1, pode-se afirmar que ∆G mistura é menor que 1,
portanto apresenta uma variação de energia livre negativa (processo
espontâneo). Se houver apenas duas substâncias então se X1 = X e X2 = 1 –
X. A variação da energia livre de mistura pode ser escrita:
(13.39)
Considera-se para sempre 1 mol de solução, portanto a equação da
energia de mistura é representada para nT = 1
(13.40)
A figura 13.2 mostra a variação de energia livre de mistura em função
da fração molar X. A curva é simétrica em torno de X = ½. A maior diminuição
da energia livre no processo de mistura está associado com a formação de
uma mistura tendo igual número de moles dos dois constituintes. Num sistema
ternário a maior diminuição da energia livre corresponde ao processo de
mistura correspondente a fração molecular equivalente a 1/3.
T, P°, n1
T, P°, n2
T, P°, n3
G inicial, antes da mistura
T, Pi para cada
substância e Po
como pressão
total
n1+n2+n3
G final após a mistura
∑∑∑===
===∆3
1
3
1
3
1
.i
iii
ii
i
iiMistura LnXnRT
PPX
LnnRTPp
LnnRTG
[ ]221 LnXXXLnXRTGMistura +=∆
( ) ( )[ ]XLnXXLnXRTnG TMistura −−+=∆ 11

226
Figura 13.2 - Variação da energia livre em uma solução binária
13.5 A Entropia de Mistura dos Gases Ideais Estudou-se que a derivada da Energia Livre em função da temperatura
é igual ao negativo da entropia:
(13.41)
Como:
(13.42)
A forma funcional da entropia para o processo de mistura é a mesma
para a energia livre. Como Xi < 1 , o valor da entropia será > 0, figura 13.3.
Figura 13.3 - Variação da entropia em uma mistura
Ent
ropi
a d
e M
istur
a
0 1
Ener
gia
Livr
e X = 1/2
X=1
[ ] [ ]∑∑ −=
∆∂
∆∂−=∆⇒=∆
n
ili
MisturaMistura
n
iii
Mistura XRLnnT
GSXRTLnnG
MisturaMistura
constP
ST
GSTG
∆−=
∆∂
∆∂⇒−=
∂∂
=
,

227
13.6 Variação de Calor na Mistura de Gases Ideais O calor do processo na mistura de gases, pode ser calculado pela
equação, 9.16. A letra M, indica que é uma mistura de substâncias:
∆GM = ∆HM – T.∆SM (13.43)
Como já foi calculado o valor da variação de energia livre de mistura e
da entropia, basta somente fazer a substituição para encontrar ∆H
RTΣLnXi = ∆HM – (- T.RΣLnXi) (13.44)
Resulta que ∆HM = 0 (13.45)
13.6 Variação do Volume de uma Mistura Ideal Sabe-se que da equação 9.28 ou seja, dG = - SdT + VdP.
Para T constante
(13.46)
Portanto para uma mistura
(13.47)
14 CONSTANTE DE EQUILÍBRIO
Quando se altera a concentração dos reagentes ou produtos para uma
mesma temperatura e pressão. O sistema procura se deslocar para minimizar a
ação externa. Seja a seguinte reação:
OHHCOOCCHOHHCCOOHCH 2523523 +↔+
Ácido Acético(AA) + Alcool Etílico(AE) → Acetato de Etila(AT) + Água(A)
Comprova-se experimentalmente que misturando 1 mol de ácido
acético e 1 mol de álcool etílico (quantidades iniciais) e mantendo-se a mistura
0=∂
∂=
∆∂∆∂
=∆ ∑P
RTLnXiP
GVM
M
VPG
=
∂∂

228
em ebulição por várias horas, a reação chega a um estado de equilíbrio no qual
estarão presentes as seguintes quantidades (quantidades finais)
0,33 Mol de AA + 0,333 Mol de AE → 0,666 Mol AT + 0,666 Mol de A
𝐾𝐸 = 0,666.0,6660,333.0,333
= 4 (14.1)
Adiciona-se então nesse sistema 1 mol de ácido acético. Então o
sistema ficará com 1,333 mols de ácido acético ( 1 + 0,333). O equilíbrio é
então pertubado. Sendo KR (constante da reação), dessa equação = 0,999;
menor que 4.
(14.2)
Novamente deixamos a mistura reagir até atingir o equilíbrio, utilizando
o mesmo procedimento da mistura inicial. Então recalculamos novamente K,
com as novas concentrações em equilíbrio.
(14.3)
Esse resultado indica que o sistema esta novamente em equilíbrio e o
rendimento da reação aumentou. Assim conclui-se que:
• Adição de reagente – A reação é obrigada a consumir os
reagentes, produzindo mais produtos. Deslocando o equilíbrio
para a direita.
• Adição de produtos – A reação é forçada a produzir maiores
quantidades de reagentes. Deslocando o equilíbrio para
esquerda
• Retirada de reagentes – A reação é forçada a produzir maior
quantidade de reagentes. Deslocando a reação para a
esquerda.
• Retira de produtos – A reação é forçada a produzir maiores
quantidades de produtos. Deslocando o equilíbrio para a direita.
999,0333,0.333,1666,0.666,0
==RK
4154,0.154,0845,0.845,0
==EK

229
Portanto quando se adiciona qualquer participante, o equilíbrio se
desloca no sentido de consumi-lo tendendo a minimizar o efeito da adição. Da
mesma forma retirando qualquer participante, o equilíbrio se desloca no sentido
de recolocá-lo, tendendo a minimizar o efeito da retirada.
14.1 Influência da Pressão Sobre a Constante de Equilíbrio Vamos considerar a síntese do amoníaco (NH3)
N2 + 3H2 → 2NH3 (14.4)
O que acontece quando a pressão é elevada, considerando constantes
a concentração e a temperatura. A tabela 14.I resume alguns dados
experimentais
Tabela 14.I – Mostra a variação da pressão em função do rendimento, ou a produção do NH3.
Pressão(atm) Produção do NH3 (%)
10 2,04
100 13,36
300 25,59
1000 69,40
Como é percebido, o aumento da pressão total sobre o sistema,
aumenta a produção do NH3. Isso significa que o equilíbrio desloca para a
direita, para o lado de formação do NH3.
O aumento da pressão total desloca o equilíbrio para o lado menor em
volume (lado de menor em mols). Lado esquerdo possui 4 mols e lado direito
possui 2 mols.
Então aumento da pressão total faz a reação deslocar o sentido da
direção para o menor volume, pois a redução do volume minimiza o efeito da
pressão aplicada. Assim a redução da pressão, desloca o equilíbrio no sentido
do maior volume, pois o aumento do volume minimiza a redução da pressão

230
14.2 Influência da Temperatura na Constante de Equilíbrio Seja novamente a equação de formação da amônia. A tabela 14.II
mostra dados experimentais em relação ao rendimento da equação versus o
aumento da temperatura para pressão e concentração constantes.
Como essa reação é exotérmica, então nota-se que aumentando a
temperatura a reação desloca-se no sentido de diminuir esse efeito ou seja,
desloca no sentido esquerdo.
Em reações endotérmicas seria ao contrário. O aumento da
temperatura, deslocaria o equilíbrio no sentido direito. Nessa equação como a
reação é exotérmica para direita e portanto é endotérmica para esquerda. Seu
equilíbrio será deslocado para a esquerda.
Tabela 14.II – Relação entre temperatura, rendimento e constante de equilíbrio
Temperatura (°C) Produção NH3(%) Constante – KE
200 82,14 0,65
300 51,36 0,011
400 25,22 6,2.10-4
500 8,75 7,4.10-5
14.3 Influência de Catalisador em uma Reação Química Através do catalisador a Ea (Energia de ativação, é a barreira para uma
reação ocorrer), é decrescida por isso aumenta a velocidade da reação.
Quando a reação é reversível, a influência do catalisador, é percebida tanto na
reação direta, quanto na inversa.
Aumentando por igual as velocidades das reações direta e inversa, o
catalisador diminui o tempo necessário para atingir o equilíbrio, mas não altera
o próprio estado de equilíbrio, isto é, não altera a produção obtido no processo,
nem o valor das constantes de equilíbrio. O catalisador não participa dos
produtos finais na reação, apenas age como uma ponte para aproximar os
reagentes dos produtos, figura 14.1

231
Figura 14.1 - Mostra a influência do catalisador em uma reação química
14.4 A Síntese de Haber-Bosch No mundo atual são produzidos milhões de toneladas de produtos
nitrogenados, adubos agrícolas, explosivos, fibras texteis, corantes,
medicamentos, entre outros. Os mais importantes são sem dúvida os adubos
agrícolas como NH3, NH4NO3, (NH4)2SO4, H2NCONH2 (uréria) e etc. Sem essa
produção de produtos nitrogenados a plantação mundial seria comprometida,
agravando ainda mais o problema da fome.
Até o início do século XX, a principal fonte de produtos nitrogenados
era o NaNO3 (salitre do Chile), que resulta da transformação de excrementos
de aves marinhas em regiões de clima seco. Como aconteceu no Chile. O
salitre natural, não era suficiente para suprir o consumo atual de compostos
nitrogenados. Por outro lado é interessante notar que existe uma quantidade
quase inesgotável de nitrogênio no ar, sob a a forma de N2. A molécula de N2 é
muito estável (inerte) ou seja, é muito difícil rompê-la. Conseguindo-se uma
primeira reação, torna-se mais fácil, daí em diante, produzir outros compostos
nitrogenados. Esse problema foi resolvido em 1909 pelo químico alemão Fritz
Haber (1868 – 1934) como a síntese do amoníaco. N2 + 3H2 ⇔ NH3. O
processo foi aperfeiçoado por Carl Bosch (1874-1940), que deu origem ao
nome síntese de Haber-Bosch. Até hoje é o processo mais importante de
fixação do ar do N2 para obtenção de produtos nitrogenados.
Com
Catalisador
Sem
Catalisador
Ener
gia
Caminho da reação
Reagentes
Produtos

232
14.5 Novo Desenvolvimento Descoberto para a Síntese da Amônia Uma equipe de pesquisadores da Universidade Cornell, chefiada pelo
professor Paul Chirik, (Estados Unidos) conseguiu converter nitrogênio em
amônia utilizando um processo vislumbrado há décadas, mas que até agora
ninguém havia conseguido realizar. O novo processo utiliza um complexo de
zircônio metálico para adicionar átomos de hidrogênio a uma molécula de
nitrogênio e convertê-la para amônia sem a necessidade de altas pressões.
O valor desse trabalho é que foi respondida à questão básica da
química de como pegar essa molécula de nitrogênio inerte e não reativa e
transformá-la em algo útil. O nitrogênio representa 79% da atmosfera terrestre
e, graças a um processo industrial de mais de 90 anos de idade ele pode ser
convertido em fertilizantes a base de amônia, representando a base da
agricultura moderna. O problema com a conversão do nitrogênio em uma forma
industrial e utilizável é que sua molécula é formada por dois átomos com
ligações químicas extremamente fortes. Somente o monóxido de carbono tem
uma ligação mais forte do que a da molécula de nitrogênio. Mas, enquanto o
monóxido de carbono adere facilmente a outras moléculas, o nitrogênio não é
polar e não se liga facilmente a metais. É também muito difícil inserir ou retirar
elétrons das moléculas de nitrogênio.
O método de conversão de nitrogênio em amônia, chamado de
processo Haber-Bosch (em homenagem a seus criadores – Fritz Haber e Carl
Bosch) é responsável pela produção de mais de 100 milhões de toneladas de
amônia anualmente para a agricultura e para a indústria química. O processo
exige altas temperaturas e pressões para que o nitrogênio e o hidrogênio
interajam sobre uma superfície de ferro, que serve como catalisador. Mas a
equipe do Prof. Paul conseguiu quebrar a ligação atômica da molécula de
nitrogênio utilizando zircônio em forma solúvel e apenas 45°C e adicionar
átomos de hidrogênio a esta ponte de di-nitrogênio. A completa conversão para
amônia foi feita a 85°C.
Essa descoberta poderá ter impacto na produção de amônia. Antes
disso porém, precisam transformar o processo de laboratório, que age

233
molécula a molécula, em um processo de larga escala que possa ser utilizado
em ambiente industrial.
Com essa descoberta existe chance de que o processo Haber-Bosch
venha a ser substituído, por esse novo método ser mais útil para a produção de
combustível de foguete ou química fina para sinterização de drogas. Esse novo
processo não utiliza catalisador, apenas átomos de hidrogênio são adicionados
à ponte de di-nitrogênio, formando a amônia, uma molécula por vez. A próxima
etapa dos pesquisadores, são justamente a descoberta de um catalisador para
poder tornar esse processo industrial.
15 Variação da Energia Livre para uma Reação Química Seja a reação química
aA + bB → lL + mM (15.1)
Da equação, 13.34, tem-se:
µi = µ° + RTLn(pi) (15.2)
A variação da energia livre para a reação 15.1, fica
∆G = (lµL + mµM ) - (aµA + bµB) (15.4)
Substituindo cada potencial por 15.2
( ) ( )[ ]−+++=∆ MMlL RTLnpmRTLnplG µµ
( ) ( )[ ]BBAA RTLnpbRTLnpa +++− µµ (15.5)
Separando em duas equações tem-se
( ) ( )BAML bamlG µµµµ +−+=∆ (15.6)
(15.7)
(15.8)
bB
aA
Mm
Ll
E pppp
RTLnRTLnK..
=
[ ] [ ]BAMlE bRTLnpaRTLnpmRTLnplRTLnpRTLnK +−+=

234
Encontramos então a constante de equilbrio da reação química 15.1
(15.9)
Substituindo esses valores se tem
(15.10)
15.1 Constante de Equilíbrio em Função das Concentrações Molares
A equação 14.1, mostra experimentalmente o cálculo da constante de
equilíbrio, que é desenvolvido através do potencial químico de acordo com a
equação 13.34, com origem na referência da pressão igual a 1 atm, P° = 1 atm.
Utilizando esse mesmo raciocínio a constante de equilíbrio para ser
considerada constante é calculada da seguinte maneira:
𝐾𝐸 = ∏𝑃𝑟𝑜𝑑𝑢𝑡𝑜𝑠∏𝑅𝑒𝑎𝑔𝑒𝑛𝑡𝑒𝑠
(15.11)
Para o caso de uma reação gasosa, o cálculo fica melhor abravés das
pressões parciais:
𝑎(𝐴)𝑔 + 𝑏(𝐵)𝑔 → 𝑐(𝐶)𝑔 + 𝑑(𝐷)𝑔
𝐾𝐸 =�𝑃𝐶𝑃° �
𝑐�𝑃𝐷𝑃° �
𝑑
�𝑃𝐴𝑃° �
𝑎�𝑃𝐵𝑃° �
𝑏 (15.12)
Considerando P° = 1 atm, a equação 15.2 torna-se:
𝐾𝐸 = 𝑃𝐶𝑐𝑃𝐷
𝑑
𝑃𝐴𝑎𝑃𝐵
𝑏 (15.13)
A equação 15.3 é praticamente igual a equação 15.9. Também a
constante de equilíbrio pode ser escrita através das concentrações n/V
(mols/volume ou mols/l). Neste caso a referência passa a ser C° = 1 mol/l, para
as concentrações molares, similar a P° = 1 atm.
bB
aA
Mm
Ll
E pppp
K..
=
ERTLnKGG +∆=∆

235
𝐾𝐸 =�𝐶𝐶𝐶°�
𝑐�𝐶𝐷𝐶°�
𝑑
�𝐶𝐴𝐶°�𝑎�𝐶𝐵𝐶°�
𝑏 = �1𝐶°�
𝑐+𝑑−𝑎−𝑏 𝐶𝐶𝑐𝐶𝐷𝑑
𝐶𝐴𝑎𝐶𝐵𝑏
𝑃𝑉 = 𝑛𝑅𝑇 → 𝐶 =𝑛𝑉
=𝑃𝑅𝑇
→ 𝐶° =𝑃°
𝑅𝑇,𝑚𝑎𝑠 𝑃° = 1 → 𝐶° =
1𝑅𝑇
𝐾𝐶 =𝐶𝐶𝑐𝐶𝐷𝑑
𝐶𝐴𝑎𝐶𝐵𝑏
𝐾𝐸 = ��1𝐶°�
(𝑐+𝑑−𝑎−𝑏)
𝐾𝐶� = ��11𝑅𝑇
�
(𝑐+𝑑−𝑎−𝑏)
𝐾𝐶� = (𝑅𝑇)(𝑐+𝑑−𝑎−𝑏)𝐾𝐶
∆𝑛 = 𝑐 + 𝑑 − 𝑎 − 𝑏
𝐾𝐸 = ��1𝐶°�
(𝑐+𝑑−𝑎−𝑏)
𝐾𝐶� = ��11𝑅𝑇
�
(𝑐+𝑑−𝑎−𝑏)
𝐾𝐶� = (𝑅𝑇)∆𝑛𝐾𝐶
𝐾𝐸 = (𝑅𝑇)∆𝑛𝐾𝐶 (15.14)
A constante de equilíbrio além das concentrações, pode ser escrita em
função das frações molares Xi, pois através da lei de Dalton Pi=XiPT.Então:
𝐾𝐸 =𝑃𝐶𝑐𝑃𝐷𝑑
𝑃𝐴𝑎𝑃𝐵𝑏=
(𝑋𝐶𝑃𝑇)𝑐(𝑋𝐷𝑃𝑇)𝑑
(𝑋𝐴𝑃𝑇)𝑎(𝑋𝐵𝑃𝑇)𝑏 =𝑋𝐶𝑐𝑋𝐷𝑑
𝑋𝐴𝑎𝑋𝐵𝑏(𝑃𝑇)∆𝑛
𝐾𝐸 = 𝑋𝐶𝑐𝑋𝐷
𝑑
𝑋𝐴𝑎𝑋𝐵
𝑏 (𝑃𝑇)∆𝑛 (15.15)
Para Δn = 1, KE = KX (KX = concentração em fração molar). No caso de Δn ser
diferente de 1, verifica-se que para valores acima de 1, como KE é um valor
constante KX, deve decrescer, assim favorece a elevação dos reagentes, caso
contrário favorece o acréscimo dos produtos, conforme a lei de Chatelier.
15.2 Efeito da Temperatura na Constante de Equilíbrio de uma Reação Química - Equação de Van’t Hoff Conforme a equação 9.20, temos:

236
G = H – TS
Conforme a equação 9.28:
dG = -SdT + VdP; para P = constante dG = -SdT
𝐺 = 𝐻 + 𝑑𝐺𝑑𝑇
.𝑇 (15.16)
dT.G = dG.T + HdT (15.17)
dT.G - dG.T = H.dT (15.18)
Dividindo a equçào 15.13 por T2 nos dois membros, tem-se
22...
TdTH
TTdGGdT=
− (15.19)
(15.20)
(15.21)
(15.22)
(15.23)
Para o caso da energia livre de helmolthz, as equações são similares à
energia livre, apenas troca-se o H por U: A = U – TS. A o sistema da função A,
é desenvolvido a volume constante e da função H a pressão constante.
(15.24)
Conforme a equação 15.20:
H
T
TG
∆=
∂
∂
1
U
T
TA
∆=
∂
∂
1
22
..T
HdTT
TdGGdTTGd −=
+−=
2
.T
dTHTGd −=
22
01TdT
TdT
Td −=
−=
2TH
TTG
∆−=
∂
∆∂

237
Considerando na equação 15.10, ΔG = 0
(15.25)
Substituindo a equação 15.25 em 15.10
(15.26)
De acordo com a equação 15.22, pode-se escrever:
(15.27)
Essa equação mostra que KE é inversamente proporcional a
temperatura para uma reação exotérmica, equação 15.28. Ou seja para
reações exotèrmicas, KE diminui de valor ou a reação desloca-se para a
esquerda com a elevação da temperatura, figura 15.1A, ou aumenta com a
diminuição da temperatura. Desta forma reações de desfosforação, oxidação
do cromo, do manganês são favorecidas em temperaturas baixas, por serem
exotérmicas..
Aqui se faz necessário um comentário, para o caso da reação
exotérmica do carbono com o oxigênio formando CO, a variação da entropia é
positiva, portanto essas equações devido a entropia positiva possuem grande
valor negativo da energia livre. A variação com a constante de equilíbrio
contínua válida ou seja, diminuindo a temperatura a constante tende a diminuir
por ser reação exotérmica, mas nesse caso devido ao forte valor negativo da
energia livre a variação da constante é menor, ou seja, não se percebe muito,
pois a reação continua ser muito favorável devido a entropia ser positiva.
Exotérmica - ∆H ⇒ (15.28)
( )RH
T
LnKE∆
−=
∂
∂1
ERTLnKG −=∆
( ) ( )RH
T
LnKTH
TRLnK EE
∆−=
∂
∂⇒
∆+=
∂∂
12
( )RH
RH
T
LnK E ∆
+=
∆−−=
∂
∂1

238
Endotérmica +∆H ⇒ (15.29)
Quando os valores de KE são muito grandes, significa que o
deslocamento da reação será para a direita. Também se KE for um número
pequeno, entende-se que a reação estará mais para o lado esquerdo.
Figura 15.1 - Mostra a relação da constante de equilíbrio com variação da entalpia
.
Para o caso de reações endotérmicas, representado pela equação
15.29, mostra um direto relacionamento com a temperatura ou seja,
aumentando a temperatura aumenta também o valor de KE, fazendo o
deslocamento da reação ir para a direita, figura 15.1B.
A constante de equilíbrio expressa o valor no qual a reação está
totalmente equilibrada de acordo com as condições de T e P apresentados.
Portanto se compararmos uma constante fora do equilíbrio, a constante de
equilíbrio mostra em comparação, qual o sentido em que a reação se
deslocará.
Pode-se aqui já se ter um entendimento das reações de desfosforação
e dessulfuração:
LnK
Aumento de T ⇐ 1/T
(A) (B)
Aumento de T ⇐ 1/T
RH ∆
RH ∆
−
LnK
Exotérmica
Endotérmica
( )RH
RH
T
LnKE ∆
−=
∆+−=
∂
∂1

239
15.3 Variação da Energia Livre conforme a Entropia e Constante de Equilíbrio para Reações Exotérmicas e Endotérmicas
Voltando um pouco em entropia estatística, aprende-se que se em um
sistema ocorre um aumento do número de partículas então, também acontece
um aumento no número de configurações e isto resulta em aumento de
entropia. Em caso de diminuição no número de configurações ou diminuição no
número de partículas, traduz-se em diminuição da entropia. Para
reconhecermos em uma reação química um aumento das configurações,
podemos recorrer a variação do número de moles dessa reação. Por exemplo
se ocorre aumento no número de moles, necessariamente pelo elevado
número de partículas, também se elevará as configurações e portanto a
entropia. Como exemplo vamos analisar a oxidação do silício com a formação
da sílica.
�𝑆𝑖�𝑆
+ (𝑂2)𝐺 → (𝑆𝑖𝑂2)𝑆 (15.30)
Considerando apenas a variação do número de moles dessa reação,
referente a fase gasosa, pois em reações metalúrgicas em temperaturas
elevadas, a variação do número de moles da fase gasosa, aumenta em volume
e pressão muito mais expressivamente do que as fases sólidas ou líquidas.
Portanto calcula-se uma variação de -1, pois do lado direito da reação (15.30)
não tem gases, aparecendo apenas do lado esquerdo 1 mol de oxigênio.
portanto Δn = 0 - 1 = -1, ocorre desaparecimento de moles, como
conseqüência, diminui o número de configurações e assim a entropia da
reação é decrescida. Pode-se considerar que para essa reação de oxidação,
houve um decréscimo de entropia, -ΔS. A maioria das reações de oxidação
possui esse comportamento, com algumas exceções que discutiremos.
Considerando a equação da energia livre, equação 9.20
∆𝐺 = ∆𝐻 − 𝑇∆𝑆
Para reação 15.30, por ser uma reação exotérmica, a variação da
entalpia é negativa, - ΔH e a variação da entropia, será – ΔS. Substituindo,
tem-se:
∆𝐺 = −∆𝐻 − 𝑇(−∆𝑆) = ∆𝐺 = −∆𝐻 + 𝑇∆𝑆 (15.31)

240
De acordo com a equação 15.31, observa-se que ao elevarmos a
temperatura o termo TΔS se eleva e assim o valor de ΔG, torna-se menos
negativo, até atingir um valor igual a zero e a partir desse ponto, positivo. Essa
conclusão está de acordo com a equação de Van’t Hoff, 15.27. Mas para o
caso de reações exotérmicas com variação positiva do número de moles e
portanto variação positiva da entropia, acontece uma concorrência entre a
constante de equilíbrio e a entropia. Vamos observar a conseqüência da
variação da energia livre para uma reação exotérmica com variação positiva do
número de moles, por exemplo a queima do carbono por oxigênio. Seja a
reação química:
2𝐶 + (𝑂2)𝐺 → 2(𝐶𝑂)𝐺 (15.32)
A variação do número de moles para essa reação é Δn = 2 -1=1,
levando em consideração apenas a participação dos gases. Dessa forma com
uma variação positiva do número de moles, a entropia também se eleva e
assim +ΔS. Voltando a equação da energia livre de Gibbs:
∆𝐺 = −∆𝐻 − 𝑇(+∆𝑆) = ∆𝐺 = −∆𝐻 − 𝑇∆𝑆 (15.33)
Observa-se, portanto através da equação 15.33 que um aumento da
temperatura, a variação da energia livre torna-se cada vez mais negativa. Esse
fato, faz com que o carbono seja um excelente redutor em determinadas
situações, por possuir em altas temperaturas um valor de energia livre mais
negativa que determinados óxidos, tornando-se com isso um bom redutor de
óxidos, por formar gás como seu produto de reação.
Vamos analisar agora a participação da constante de equilíbrio nessas
circunstâncias, pois pela equação de Van`t Hoff, a equação sendo exotérmica
com o aumento da temperatura faz com que a constante de equilíbrio diminua.
Assim vamos verificar sua influência na variação da energia livre.
De acordo com as equações 9.20 e 15.10, temos:
∆𝐺° = ∆𝐻° − 𝑇∆𝑆° 𝑒 ∆𝐺 = ∆𝐺° + 𝑅𝑇𝐿𝑛𝐾𝐸

241
Consideraremos a condição padrão e que tanto a entropia e a entalpia
são independentes da temperatura, portanto ΔCp = 0. Isso facilita a análise de
uma forma mais clara e objetiva. Calculando ΔG, fica:
∆𝐺 = ∆𝐻° − 𝑇∆𝑆° + 𝑅𝑇𝐿𝑛𝐾𝐸 (15.34)
Para reações exotérmicas, a variação da entalpia é negativa e no caso
de uma variação positiva da entropia, torna-se:
∆𝐺 = −∆𝐻° − 𝑇(+∆𝑆°) + 𝑅𝑇𝐿𝑛𝐾𝐸 = −∆𝐻° − 𝑇∆𝑆° + 𝑅𝑇𝐿𝑛𝐾𝐸 (15.35)
Assim a equação da energia livre torna-se dependente dos termos
−𝑇∆𝑆° e 𝑅𝑇𝐿𝑛𝐾𝐸. Nesse caso o termo da temperatura é muito mais elevado do
que o termo da constante de equilíbrio. Esse fato faz com que ocorra uma
concorrência entre os dois, apesar da constante de equilíbrio ir para esquerda
com a elevação da temperatura, a reação torna-se a ir para direita devido a
entropia e quanto mais elevada for a temperatura menos importância o termo
da constante se confirma. No caso de reações exotérmicas com variação
negativa da entropia, soma-se tanto a entropia quanto a constante de equilíbrio
na diminuição negativa da variação da energia livre, piorando no caso de uma
elevação da temperatura, 15.34.
∆𝐺 = −∆𝐻° − 𝑇(−∆𝑆°) + 𝑅𝑇𝐿𝑛𝐾𝐸 = −∆𝐻° + 𝑇∆𝑆° + 𝑅𝑇𝐿𝑛𝐾𝐸 (15.36)
15.4 Variação da Concentração em relação a Pressão
Seja a seguinte reação química:
𝑎𝐴 + 𝑏𝐵 → 𝑐𝐶 + 𝑑𝐷 (15.37)
A constante de equilíbrio é calculada
𝐾𝐸 =𝑃𝐶𝑐𝑃𝐷𝑑
𝑃𝐴𝑎𝑃𝐵𝑏=𝑋𝐶𝑐𝑃𝑇𝑐𝑋𝐷𝑑𝑃𝑇𝑑
𝑋𝐴𝑎𝑃𝑇𝑎𝑋𝐵𝑏𝑃𝑇𝑏=𝑋𝐶𝑐𝑋𝐷𝑑
𝑋𝐴𝑎𝑋𝐵𝑏𝑃𝑇
(𝑐+𝑑−𝑎−𝑏) = 𝐾𝑋𝑃𝑇(𝑐+𝑑−𝑎−𝑏)
Portanto a constante da concentração será igual:
𝐾𝑋 = 𝐾𝐸𝑃𝑇
(𝑐+𝑑−𝑎−𝑏) (15.38)
No caso de c+d > a+b, a concentração Kx decresce, pois KE contínua
constante e PT se eleva. A constante KX se elevara quando PT diminuir, c+d <
a+b.

242
15.5 Reação de Desfosforação O fósforo (P), é interpretado no aço como um residual que traz
fragilidade a frio, ou seja, torna o aço mais quebradiço e eleva a temperatura
de transição no ensaio charpy, ficando o aço mais suscetível à fratura frágil.
A reação de desfosforação é apresentada pela seguinte reação
química
(15.39)
(15.40)
(15.41)
Observa-se que a variação do número de moles é negativa portanto a
entropia tende ser negativa, entende-se que existe uma variação da energia
livre em função da temperatura, ou seja, aumentando a temperatura a reação
se movimenta para esquerda (reação exotérmica), ou diminui o valor negativo
da energia livre. Para diminuir o teor de P, (fósforo), do aço em análise
representado pela equação 15.39, o termo Lp, equação 15.41, indica que
quanto mais alto mais fosfato tem na escória e menos fósforo tem no aço. É
possível observar que é necessário aumentar o numerador e diminuir o
denominador. Para aumentar o denominador então é preciso elevar o teor de
oxigênio do aço, o valor de O-2 (representa escória básica com teores CaO
elevados), e para elevar o valor de KE basta diminuir a temperatura, pois essa
reação é exotérmica. Já no caso do numerador que deve ser elevado, resta
somente aumentar o volume de escória para formar mais íon fosfato na
escória. Fazendo essas observações é possível compreender que esse
processo é melhor desenvolvido no final de fusão dos fornos elétricos a arco.
Nesse período a temperatura é baixa na ordem de 1580°C em um ambiente
bastante oxidante.
( ) ( )34
2 2352 −− ↔++ POOOP
( )( )
( )( )325
2342
3252
234
−
−
−
−
=⇒=OOK
POPOOP
POKE
E
( ) ( ) EKOOP
POLp 3252
34 −−
==

243
15.6 Reação de Dessulfuração O enxôfe (S), promove fragilidade a quente nos aços. Durante o
processo de laminação em aços com teores de enxofre elevados, pode ocorrer
formação de sulfeto de ferro, um composto com ponto de fusão mais baixo que
as temperaturas praticadas de laminação. Assim esse sulfeto funde e fragiliza o
aço durante a laminação. Para evitar esse problema, adiciona-se Manganês
(Mn), em uma relação de pelo menos Mn/S > 4, para substituir o Fe por
manganês, originando o sulfeto de MnS, que possui ponto de fusão muito mais
elevado, Mas mesmo assim em teores de oxigênio baixo nesses aços o sulfeto
de manganês, torna-se alongado e faz com que o material a ser laminado se
divida nas regiões com sulfetos, sendo necessário sucatar a peça laminada.
Para evitar isso, objetiva-se teores de oxigênio na ordem de 120 ppm a 150
ppm, o oxigênio modifica as inclusões de sulfeto de manganês para globular
em vez de laminar, propiciando ao material mais resistência e tenacidade ao ler
laminado.
O processo de dessulfuração é representado pela seguinte equação
química:
(15.42)
(15.43)
(15.44)
Nessa equação 15.42 a variação de moles é praticamente constante,
portanto a variação da energia livre em relação a temperatura é muito pequena.
Na equação 15.44, observa-se que para diminuirmos o teor de enxofre (S), ou
seja elevar o valor de Lp do aço, o numerador deve ser elevado e o
denominado decrescido. No caso do numerador na equação 15.44, o íon O-2
representa a basicidade da escória, portanto quanto mais básica melhor
(valores altos de CaO, Na2O,..). Para o numerador é importante decrescer o
teor de oxigênio. A constante de equilíbrio como não varia muito com a
( ) ( ) OSOS +↔+ −− 22
( )( )
( )( )2
2
2
2
−
−
−
−
=⇒=OK
OSSOS
OSKE
E
OKO
SSLp E.22 −−
==

244
temperatura, não interfere nesse caso, pois é possível notar que a variação do
número de moles é nula, equação 15.42. Mas praticamente se percebe que
aumentando a temperatura melhora o processo de dessulfuração. Esse fato é
entendido devido a temperatura acelerar a cinética das reações e com isso
acelera o processo de dessulfuração.
15.7 Produto de Solubilidade Dado uma solução saturada de um eletrólito pouco solúvel. AxBy, tal que
(15.45)
Chama-se produto de solubilidade PS, ao produto
(15.46)
Por exemplo seja as reações:
(15.47)
(15.48)
(15.49)
(15.50)
Os valores numéricos citados nesses dois exemplos são obtidos
experimentalmente e são sempre números muito pequenos, pois o PS é
definido para substâncias muito pouco solúveis. Além disso o PS varia com a
temperatura. Quanto menor for o PS para uma substância, menos solúvel será.
Para valores < PS não haverá precipitação na solução. Caso contrário ocorrerá
precipitação. Aqui aplica-se um exemplo para lingotamento contínuo de aços:
O CaS possui baixa solubilidade no aço e decresce com a temperatura.
Portanto como a temperatura de lingotamento é menor que a liberação da
corrida a partir do forno panela, a medida que a temperatura diminui poderá
ocorrer precipitação do sulfeto de cálcio, CaS. Esse precipitado adere a
superfície da válvula por ser uma inclusão sólida e bloqueia a continuação do
lingotamento se ocorrer em grande quantidade. Assim deve-se ter em mente
sempre nesses casos que a grande perda de temperatura durante o
lingotamento oferece perigo.
xyyx yBxABA +− +↔
[ ] [ ]yxsy BAPS +−= .
23
23
−+ +↔ COCaCaCO
[ ][ ] )25(10.7,8. 923
2 CTCOCaPS === −−+
( ) −+− +↔ OHFeOHFe 333
( ) [ ] )25(10.1,1. 3633 CTOHFePS === −+−+

245
(15.51)
Objetivando em minimizar esse problema existem siderúrgicas como
por exemplo a Nippon Steel no Japão, desenvolvendo aquecimento por plasma
no tundish, com isso se consegue uma variação de temperatura do aço líquido
no ordem de ≅ 5°C, estabilizando bastante o processo de lingotamento
contínuo.
15.8 Equilíbrio Químico em uma Mistura Considera-se um sistema fechado com temperatura e pressão
constante. O sistema consiste de uma mistura de várias espécies químicas que
podem reagir de acordo com a equação:
αA + βB → γC + δD (15.52)
Onde A, B, C, e D representam as fórmulas químicas das substâncias
enquanto que α,β.γ e δ representam os coeficientes estequiométricos. Depois
da reação ter se completado uma vez como está escrita, isto é, depois de α
moles de A e β moles de B tenham sido consumidos para formar γ moles de C
e δ moles de D, dizemos que ocorreu um avanço na reação. Consideremos um
avanço da reação de ξ unidades. Então o número de moles de forma geral
pode ser:
(15.53)
ln = número iniciais de moles
iυ = coeficiente estequiométrico
Considerando uma quantidade de moles dni em relação a um avanço
(15.54)
(15.55)
(15.56)
No equilíbrio tem:
16,1028300+
−=
TLgKCaS
ξυiii nn +=
ξυ ddn ii =
ξυµµ ddndG iiii ==
∑=
∂∂
iiPT
G υµξ ,

246
(15.57)
Entende-se que o avanço de uma equação química no equilíbrio é igual
a zero. Assim o potencial químico é função de:
µi = f(T,P,ξ) (15.58)
Graficamente, figura 15.2 podemos interpretar a variação da Energia
Livre de uma mistura
(15.59)
Figura 15.2 - Variação da energia livre em uma mistura
O sistema aproxima-se do estado de equilíbrio de energia livre mínima
formando substância de energia intrínseca menor; isto torna G pequeno (G
puro). Também abaixa a energia livre através do processo de mistura dos
reagentes e produtos. Um compromisso é atingido entre um material puro
tendo uma baixa energia livre intrínseca e uma alta energia livre no estado
misturado.
Sejam dois componentes A e B misturados e o Gráfico da Energia Livre
de mistura, figura 15.3:
G dos puros
elementos G dos elementos
misturados
G Total
Valor G Minimo
1 0
G
X = Fração Molar
∑ ∑ ∑∑ −+=−+== )()( iiiiiiiiiii nnnnG µµµµµµµ
0,
==
∂∂ ∑ ii
PT
G υµξ

247
Figura 15.3 - Variação da energia livre parcial molar em uma mistura
(15.60)
(15.61)
(15.62)
XA + XB = 1; XA = 1 - XB; ⇒ dXA = - dXB (15.63)
(15.64)
15.9 Princípio de Le Chatelier Conforme visto, uma mudança na temperatura, pressão e variação de
massa influencia no potencial químico dos elementos.
Portanto pode-se escrever
(15.65)
Então
( )A
M
AMM
A XGXGG∂∂
−+= 1
B
M
B
MA
M
XG
XGGtg
∂∂
=−
=φ
B
M
BMA
M
XGXGG∂∂
=−
B
M
BMM
A XGXGG∂∂
−=
( )ξµ ,, PTfi =

248
(15.66)
(15.67)
(15.68)
(15.69)
Substituindo 15.67; 15.68 e 15.69 em 15.66
(15.70)
Considerando o sistema no equilíbrio
(15.71)
Substituindo em 15.70
(15.72)
Considerando P constante; dP = 0 e substituindo em 15.70
(15.73)
Considerando T = constante; dT = 0 em 15.70
(15.74)
São expressões quantitativas que descrevem a dependência do
avanço da reação no equilíbrio com a temperatura e pressão .
''i
i
T
HdTd
µ
ξ ∆=
''i
iVdPd
µ
ξ ∆−=
ξξµµµµ
ξξ
ddPP
dTT
dPT
i
T
i
P
ii
,,,
∂∂
+
∂∂
+
∂∂
=
iiii
i SnS
TG
nnG
TT∆−=
∂∂
−=
∂∂
∂∂
=
∂∂
∂∂
=
∂∂µ
iiii
i VnV
PG
nnG
PP∆=
∂∂
=
∂∂
∂∂
=
∂∂
∂∂
=
∂∂µ
''i
ii
i Gnn
G µξξξ
µ=
∂∂
∂∂
=
∂∂
∂∂
=
∂∂
ξµµ ddPVdTSd iiii''+∆+∆−=
ξµ ddPVdTTH
iii ''0 +∆+
∆−=
THSd l
ii∆
=∆= ;0µ

249
Para a equação:
(15.75)
Se o segundo termo é positivo, ou a variação da entalpia positivo, a
reação é endotérmica, então a avanço em relação a temperatura também é
positivo. Então um avanço da temperatura avança a reação para manter o
equilíbrio. Para uma reação exotérmica, ou seja, o segundo termo negativo de
acordo com a variação de entalpia negativo, implica em um aumento da
temperatura e uma diminuição no avanço da reação.
No caso da segunda equação:
(15.76)
Se a variação do volume for negativo, a variação do avanço em relação
a pressão torna-se positivo, assim um aumento da pressão, aumenta o avanço
da reação. Correspondentemente se a variação do volume for positivo, o
avanço em relação a pressão será negativo, assim um aumento da pressão,
diminui o avanço da reação.
O efeito global dessas relações é que um aumento na pressão desloca
o equilíbrio para o lado do menor volume, enquanto que uma diminuição da
pressão desloca para o lado de maior volume, figura 15.4. Semelhante um
aumento da temperatura que desloca o equilíbrio para o lado de maior entalpia,
enquanto que uma diminuição desloca para o lado de menor entalpia.
O princípio de Le Chatelier pode ser enunciado do seguinte modo:
“Se os vínculos externos sob os quais se estabelece um equilíbrio
químico forem alterados, o equilíbrio se deslocará de tal modo a moderar o
efeito dessa mudança”
O princípio de Le Chatelier nos ajuda a interpretar que os
desgaseificadores são as maiores fábricas de gelo, pois trabalham com vapor
de água a baixa pressão, portanto sempre esses sistemas tenderão a formar
gelo.
''i
i
P T
HdTd
µ
ξ ∆=
''i
iVdPd
µ
ξ ∆−=
250
Henry Louis Le Chatelier nasceu na França em 1850, foi professor da
Escola de Minas de Paris, faleceu em 1936. Projetou o maçarico a oxi-
acetileno. Sempre procurou mostrar a necessidade da união entre a Química
Aplicada e a Teórica.
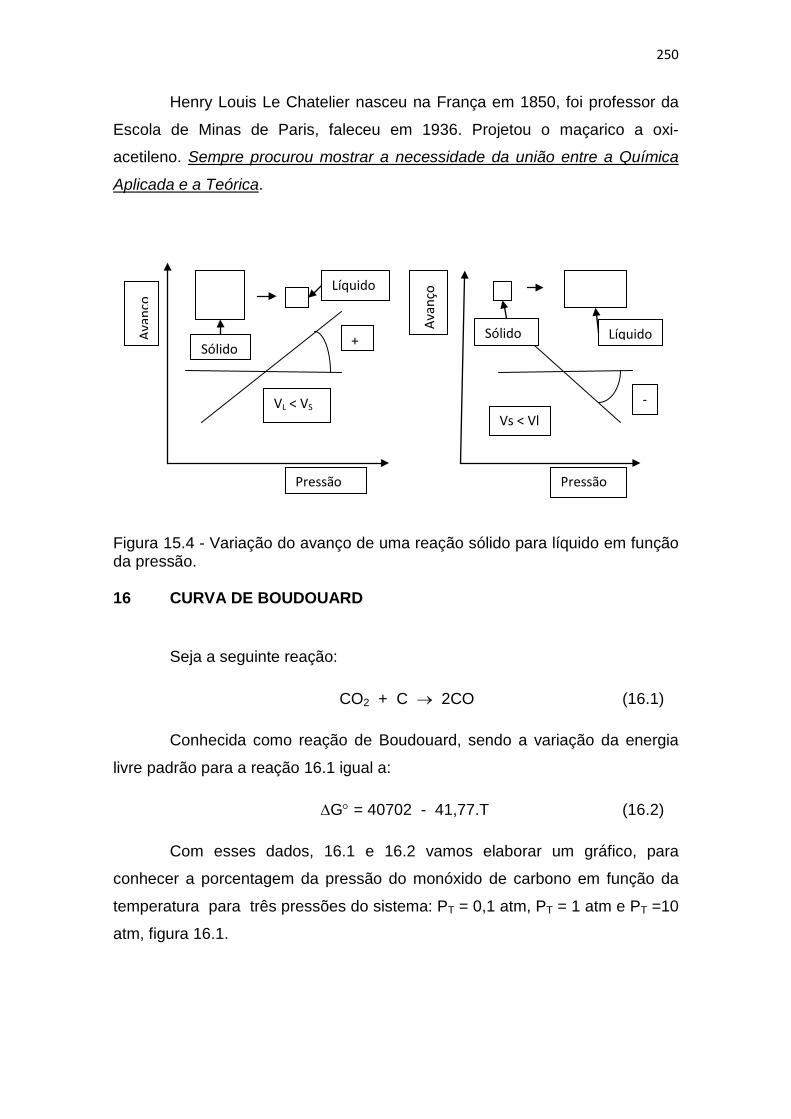
Figura 15.4 - Variação do avanço de uma reação sólido para líquido em função da pressão. 16 CURVA DE BOUDOUARD
Seja a seguinte reação:
CO2 + C → 2CO (16.1)
Conhecida como reação de Boudouard, sendo a variação da energia
livre padrão para a reação 16.1 igual a:
∆G° = 40702 - 41,77.T (16.2)
Com esses dados, 16.1 e 16.2 vamos elaborar um gráfico, para
conhecer a porcentagem da pressão do monóxido de carbono em função da
temperatura para três pressões do sistema: PT = 0,1 atm, PT = 1 atm e PT =10
atm, figura 16.1.
Avan
ço
Avan
ço
Pressão Pressão
+
- Vs < Vl
VL < VS
Sólido
Líquido
Sólido Líquido

251
PCO/(PCO + PCO2) (16.3)
Para construirmos esse gráfico temos que em primeiro lugar
encontrarmos o valor da constante de equilíbrio KE em função da temperatura.
Com o valor de KE e PT, encontra-se os valores de PCO, e em seguida PCO2.
Cálculo do valor de KE
(16.4)
(16.5)
Cálculo de PCO em função da constante KE e a pressão total
Através da reação 16.1, podem ser escritas as seguintes relações
(16.6)
(16.7)
Substituindo a equação 16.7 em 16.6, encontra-se:
(16.8)
(16.9)
(16.10)
A partir dessas equações a tabela 16.I, mostra os valores dos cálculos
efetuados de acordo com as equações. A Pressão Total para ilustração aqui é
de 1 atm. Pode-se repetir esses cálculos para pressões maiores ou menores
que 1 atm.
Precisa-se também prestar atenção nos valores do eixo y, para se
obter resultados comparativos é necessário utilizar a relação Pco/(Pco+Pco2).
Caso contrário o gráfico não se torna de fácil comparação, com valores
máximos variando de acordo com a pressão total.
TRTLnKG E 77,4140702 −=−=∆
096,2156,2055698,1
77,4140702+
−=
+−=
TTTLnK E
TCOCO PPP =+ 2
2
2
CO
COE P
PK =
2.42
TEEECO
PKKKP
+±−=
COTCO PPP −=2
0..2 =−+ TECOECO PKPKP
252
Tabela 16.I – Mostra as relações de temperatura, constante de equilíbrio, pressão do monóxido e dióxido de carbono
Temperatura
(K)
LnK KE Pco/(Pco+
Pco2)
Pco2
373 -34 1,68 E –15 4,1 E –08 1
473 -22,4 1,94 E –10 1,39 E -05 0,999
573 -14,8 3,81 E –07 0,0006 0,999
673 -9,45 7,88 E – 05 0,008 0,991
773 -5,5 0,004 0,062 0,938
873 -2,45 0,086 0,254 0,746
973 -0,031 0,97 0,613 0,387
1073 1,94 6,94 0,887 0,113
1173 3,57 35,6 0,973 0,027
1273 4,95 140,9 0,993 0,00 7
Conforme a figura 16.1, a linha rosa, com símbolo quadrado
representa pressão de 1 atm, a laranja, com símbolo em triângulo de 10 atm e
a azul, com símbolo em losango de 0,1 atm. Pode-se verificar que para
temperaturas elevadas praticamente ocorre apenas a presença de CO, ao
contrário para temperaturas baixas o CO2 é muito presente. Para pressões

253
mais elevadas o teor de CO diminui e para pressões mais baixas aumenta o
teor de CO.
Figura 26.1 - Mostra a curva de Boudouard. A linha azul, com símbolo em losango, Pt = 0,10 atm; linha rosa, com símbolo quadrado, Pt = 1 atm e a linha laranja com símbolo em triângulo, Pt = 10 atm.
Figura 16.2 - Mostra a importância da equação de boudouard na redução do minério de ferro nos altos fornos e as reações envolvidas no processo de redução do minério de ferro. (FELTRE, R. Química Geral, Vol.1, 6ª edição).
A reação de Boudouard é muito importante para redução da hematita
(Fe2O3) para Fe, figura 16.2, pois em um ambiente bastante rico em carbono, a
0
0,5
1
1,5Pc
o/Pc
o+Pc
o2
Temperatura (K)
Curva de Boudouard
Pt =1,0Pt = 10Pt = 0,10

254
reação de Boudouard é uma fonte da geração de CO, composto fundamental
para redução
Além do CO, também o H2 pode ser um agente redutor. A obtenção de
H2 é também CO mais largamente utilizado é através do craqueamento do
CH4, onde:
CH4 + H2O → CO + 3H2 (16.11)
Processos foram desenvolvidos utilizando o gás como redutor, como o
caso da obtenção de ferro esponja, também o processo Midrex é um exemplo
dessa base de raciocínio.
No caso de redução direta (obtenção de gusa através de reação
sólida), passa-se gás através dos minérios fazendo uso de CO e H2 como
resultado do craqueamento do gás. Para minimizar o teor de S, também é
possível misturar esse gás de redução com CaO, fixando o S em CaS, assim o
aumento de S no gusa obtido é reduzido.
Portanto pode-se reduzir o minério de ferro através do CO e H2
Hematita: 3Fe2O3 + H2 (CO) ⇔ 2Fe3O4 + H2O (ou CO2) (16.12)
Magnetita: 2Fe3O4 + 2H2(2CO) ⇔ 6FeO + 2H2O (ou 2CO2) (16.13)
Wustita: 6FeO + 6H2(6CO) ⇔ 6Fe + 6H2O (ou 6CO2)1 (16.14)
Em temperaturas menores que 840 K a magnetita é transformada
diretamente para ferro de acordo com as curvas de Chandron, figura 16.3.
A transfomação direta do Fe3O4 para Fe em temperaturas baixas
menores que 600°C, é dificultada devido a instabilidade do CO, pois nessa
temperatura o gás mais estável é o dióxido de carbono. Observa-se também
que para temperaturas na ordem de 900°C o melhor gás redutor é o CO, pois
para reduzir com hidrogênio é preciso uma menor porcentagem em relação ao
H2.
O hidrogênio é um bom redutor em temperaturas acima de 1000°C. O
diagrama também mostra a redução do zinco. O ponto x indica a região melhor
para a redução, pois nessa região o zinco está na forma de vapor e o FeO na

255
forma líquida, no caso de temperaturas superiores ocorre redução do FeO para
Fe e o Fe nessa temperatura ele se apresenta na forma sólida que pode
obstruir o processo de redução. Também verifica-se que um abaixamento da
pressão, ocorre diminuição da pressão de vapor do zinco e com isso decresce
o rendimento da reação. É importante então tomar cuidado com resfriamento
para não piorar o rendimento, Figura 16.3.
Figura 16.3 - Curva de Chandron, (COUDURIER,L.; HOPKKINS,D.W.; WILKOMIRSKY,I. Fundamentals of Metallurgical Processes, 2nd Edition, Page 404, 1985)
O hidrogênio é um bom redutor em temperaturas acima de 1000°C. O
diagrama também mostra a redução do zinco. O ponto x indica a região melhor
para a redução, figura 16.2, pois nessa região o zinco está na forma de vapor e
o FeO na forma líquida, no caso de temperaturas superiores ocorre redução do
FeO para Fe e o Fe nessa temperatura ele se apresenta na forma sólida que
pode obstruir o processo de redução. Também verifica-se que um abaixamento
da pressão, ocorre diminuição da pressão de vapor do zinco e com isso

256
decresce o rendimento da reação. É importante então tomar cuidado com
resfriamento para não piorar o rendimento.
Observa-se que o monóxido de carbono não é somente um agente
redutor, mas também é um agente carburador na área superior a wustita (FeO)
e a curva de boudouard o monóxido de carbono se dissocia em carbono e
dióxido de carbono, figura 16.4.
O carbono passa então a fazer parte do metal ou dissolver no ferro de
acordo com:
2CO → C + CO2
Figura 16.4 - Mostra o diagrama de redução do ferro em função dos vários teores da wustita em relação ao teor de oxigênio, (COUDURIER,L.; HOPKKINS,D.W.; WILKOMIRSKY,I. Fundamentals of Metallurgical Processes, 2nd Edition, Page 404, 1985)
17 REDUÇÃO DO MOLIBDÊNIO E TUNGSTÊNIO EM ÓXIDOS POR HIDROGÊNIO
Puro tungstênio e molibdênio são obtidos pela redução de seus óxidos
com hidrogênio. No caso do molibdênio o óxido é produzido por ustulação a
partir do molybdnite para MoS2. O principal mineral do tungstênio é o scheelite,
CaWO4 e wolfranite (Fe,Mn)WO4.

257
Esses minerais são dissolvidos em solução alcalina (NaOH e NH4OH)
formando o molybdate ou tungstate. As impurezas são retiradas por meio de
um filtro, os óxidos são precipitados pela mudança do pH da solução. Depois
de seco, o material é levado para um forno. No primeiro estágio são reduzidos
para MoO2 e WO2 em temperaturas de 870K e 970K respectivamente. No
segundo estágio os óxidos são reduzidos em temperatura de 1270K e 1370K
para o tungstênio. O hidrogênio circula em contra corrente e o excesso é
queimado na saída do forno. O molibdênio e tungstênio em pó é obtido e
compactado embarras por pressão e sinterização por hidrogênio em 2300K,
através de uma corrente elétrica.
18 EXERCÍCIOS RESOLVIDOS
1 – Tomando por base o princípio de Le Chatelier, analise os efeitos de
temperatura e da pressão total no equilíbrio das seguintes reações
a) 2COg + (O2)g ⇔ 2(CO2)g
b) H2O g + COg ⇔ (H2)g + (CO2)g
c) (CO2)g + Cs ⇔ 2COg
d) (FeO)s + COg ⇔ Fes + (CO2)g
a) A variação de entalpia dessa reação é de ∆H298 = -67,634 kcal. Como a
reação é exotérmica, a redução da temperatura, favorece a formação de
CO2. Para a variação da pressão tem-se: que a variação do número de
moles é de 3 para 2 de reagentes para produtos. Portanto um aumento da
pressão favorece a produção de CO2, pois os produtos favorecem por
possuir menos moles.
b) A variação de entalpia para essa reação na temperatura de 298 K é de –9,8
kcal, portanto é uma reação também exotérmica, assim uma redução da
temperatura desloca a reação para o lado direito na produção de H2 e CO2.

258
A variação do número de moles é igual a zero, portanto variação na pressão
não favorece a reação.
c) A variação da entalpia na temperatura de 298 K é de 4102 kcal, portanto
reação endotérmica, um aumento da temperatura favorece a reação para o
lado direito. A variação do número de moles é de +1, portanto um aumento
de pressão desfavorece a reação para o lado direito.
d) A entalpia padrão para essa reação é de – 4,4 kcal. A reação é portanto
exotérmica, aumento de temperatura desfavorece a reação para o lado
direito. A variação do número de moles é igual a Zero, assim a pressão não
interfere no resultado da reação.
2 – Calcular a variação da Energia Livre ∆G° para a reação abaixo na
temperatura de 298 K e 1000 K.
(CO2)g + Cs ⇔ 2COg
A tabela abaixo apresenta os valores para a variação da entalpia e entropia na
temperatura de 298 K e 1000 K
Tabela 18.I – Dados termodinâmicos para o Carbono, Dióxido de Carbono (CO2) e Monóxido de Carbono (CO).
Substâncias
Químicas ∆H para 298 K
(kcal/mol)
∆S para 298 K
(cal/K.mol)
C0g -26,42 47,21
Cs 0 1,361
(CO2)g -94,054 51,07
Para ∆G° (temperatura de 298 K); ∆G° = ∆H° - T. ∆S°
∆H° = 2∆H°CO - ∆H°CO2 - ∆H°Cs = 2(-26,42) – (- 94,054) – 0 = 41,214 kcal
∆S°=2∆S°CO - ∆S°CO2 - ∆S°Cs = 2.47,21 – 51,07 – 1,361 = 41,989 cal/mol.K
calTSTHG 989,41.214.41000 −=∆−∆=∆
Para temperatura T = 298 K
kcalSTHG 7,28989,41.298214.41000 =−=∆−∆=∆

259
Para temperatura T = 1000 K
kcalSTHG 775,0989,41.1000214.41000 −=−=∆−∆=∆
Uma outra forma mais fácil de resolver esse problema é a utilização das
tabelas termodinâmicas através da equação do ∆G°
2(C)s + O2 = 2CO ∆G° = -53680 – T41,7 cal/mol
CO2 = (C)s + O2 ∆G° = 94074 + T.0,49 cal/mol
Somando as duas equações encontramos
(C)s + CO2 = 2CO ∆G° = 40394 – T. 41,21
Para T = 1000 K, ∆G = - 0,816 kcal/mol
Para T = 298 K
(C)s + CO2 = 2CO ∆G° = 40702 – 298. 41,77 = 28,254 kcal
Comparando os dois modos de calcular a variação da energia livre, percebe-
se, que os dois métodos fornecem resultados praticamente iguais. Portanto, as
duas formas de calculo são válidas.
Conclui-se portanto que a reação é na direção de formação do CO2 na
temperatura de 298 K e na formação do CO quando estiver com 1000 K.
3 – Utilizando as expressões para cálculo do ∆G°, determinar se a reação
abaixo são espontâneas ou não?
(FeO)s + COg = (Fe)s + (CO2)g
Considerar as reações abaixo
(C)s + ½O2 = CO ∆G° = -26840 - 20,85.T
(C)s + O2 = CO2 ∆G° = - 94074 - 0,49.T
(Fe)s + ½O2 = (FeO)s ∆G° = -64490 + 15,02.T
Rearranjando

260
(FeO)s = (Fe)s + ½Og ∆G° = 64490 - 15,02.T
CO = (C)s + ½O2 ∆G° = 26840 + 20,85.T
(C)s + O2 = CO2 ∆G° = - 94074 - 0,49.T
Somando tem-se
(FeO)s + CO = (Fe)s + CO2 ∆G° = -2744 + 5,34.T
Para T = 900 K ⇒ ∆G° = -2744 + 5,34.900 = 2062 cal/mol
A reação no sentido da esquerda para direita não é espontânea
4 – Utilizando as relações para ∆G°, determinar a temperatura em que se tem o
equilíbrio termodinâmico na seguinte reação:
3Fe2O3 → 2Fe3O4 + ½O2
3(Fe2O3 → 2Fe + 3/2O2 ) ∆G° = 586350 - 185,4.T
2(3Fe + 2O2 → Fe3O4) ∆G° = - 531320 + 153,6.T
Somando
3Fe2O3 → 2Fe3O4 + ½O2 ∆G° = 55030 - 31,8.T
Fazendo ∆G° = 0, o valor de T = 1730,50 K
5 – Determinar a temperatura a partir do qual torna-se viável a redução do FeO
pelo carbono, seguindo a reação abaixo.
(FeO)s + (C)s → (Fe)s + CO
(FeO)s → (Fe)s + ½O2 ∆G° = 63051 - 15,39.T
(C)s + ½O2 → CO ∆G° = - 26816 - 21,03.T

261
Somando as reações tem-se
(FeO)s + (C)s → (Fe)s + CO ∆G° = 36235 - 36,42.T
Para ∆G° = 0 ⇒ T = 994,92 K Significa que para temperaturas maiores que
está o valor de ∆G° passa a ser negativo.
6 – Considerando as reações abaixo a 1000°C. Determinar em que sentido vai
ocorrer a transformação quando os gases tiverem as seguintes composições:
(Considerar Ptotal = 1 atm)
%H2 = 25; %CO = 40; %CO2 = 25; %H2O = 10
CO2 + H2 → CO + H2O
Dados
C + ½O2 → CO ∆G° = -26840 – 20,85.T
C + O2 → CO2 ∆G° = - 94074 + 0,49.T
H2 + ½O2 → H2O ∆G° = -58850 + 13,12.T
Fazendo o seguinte rearranjo nessas equações
C + ½O2 → CO ∆G° = -26840 – 20,85.T
H2 + ½O2 → H2O ∆G° = -58850 + 13,12.T T
CO2 → C + O2 ∆G° = 94074 - 0,49.T
Somando
CO2 + H2 → CO + H2O ∆G° = 8384 - 8,22.T
( ) ( )( ) ( )
( ) ( )( ) ( )
64,025,0.25,040,0.10,0
...
.. 22
2
21
21
12
1.
12
12
112. =====
HCO
COOH
HTCOT
COTOHT
HTCOT
COTOHTE XX
XXXPXPXPXP
XPXPXPXPK

262
Caso os coeficientes dos elementos químicos da reação fossem diferentes de
1, então em vez de utilizarmos as frações molares, utilizaríamos as pressões
parciais.
Substituindo T = 1273 K na equação ∆G° = 8384 - 8,22.T; ∆G° = - 2080,06cal
No equilíbrio
( ) 9,112464,01273.98,1 −== LnRTLnKE
Como a equação está fora do equilíbrio temos que considerar a seguinte
equação
calRTLnKGG E 94,32049,112406,2080 −=−−=+∆=∆
Como a variação da energia livre é negativa, a reação é espontânea da
esquerda para a direita. No sentido em que se apresenta.
7 – Considerando o exercício anterior, determinar a composição química dos
gases em equilíbrio a 1000°C e 1 atm. Sabendo-se que a composição inicial é
a mesma do exercício anterior.
Considerando que os reagentes no equilíbrio estarão menor x% e os produtos
+ x%. No equilíbrio ∆G = 0; então ∆G° = - RTLnKE
Assim a equação fica:
3,21273.98,152,832
06,20801273;22,88384
=⇒=−−
−=∆⇒=−=−=∆
EE
E
KLnK
GTTRTLnKG
( )( )( )( ) 010,0.65,1.3,139,1
25,025,010,040,0 2 =+−⇒=
−−++
= xxxxxxK E
Resolvendo está equação encontra-se x = 1,2 e x = 0,073
Assim a resposta será:

263
CO = 0,40 + 0,073 = 0,4073
H2O = 0,10 + 0,073 = 0,173
CO2 = 0,25 – 0,073 = 0,177
H2 = 0,25 – 0,073 = 0,177
Para X = 1,296 é inconsistente pois os valores de H2 e CO2 seriam negativos,
portanto não são corretos.
8) Determinar a variação da energia livre do sistema, através do número de
moles dos gases nA, nB formando dois moles de nC e também para uma
variação mínima da energia livre do sistema, calcular a variação do número de
moles de nA em função da constante de equilíbrio e temperatura?
𝐴𝑔 + 𝐵𝑔 → 2𝐶𝑔
A energia livre do sistema é dado por:
𝐺 = 𝑛𝐴𝜇𝐴 + 𝑛𝐵𝜇𝐵 + 𝑛𝐶𝜇𝐶
𝑛𝐴 = 𝑛𝐵
𝑛𝐶 = 2 − 𝑛𝐴 − 𝑛𝐵 = 2 − 2𝑛𝐴 = 2(1 − 𝑛𝐴)
𝑛𝑇 = 𝑛𝐴 + 𝑛𝐵 + 𝑛𝐶 = 2𝑛𝐴 + 2(1 − 𝑛𝐴) = 2
𝜇𝑖 = 𝜇𝑖0 + 𝑅𝑇𝐿𝑛𝑝𝑖 = 𝜇𝑖0 + 𝑅𝑇𝐿𝑛(𝑋𝑖.𝑃)
𝜇𝑖 = 𝜇𝑖0 + 𝑅𝑇𝐿𝑛𝑋𝑖 + 𝐿𝑛𝑃
𝑋𝐴 =𝑛𝐴2
,𝑋𝐵 =𝑛𝐵2
,𝑋𝐶 =2(1 − 𝑛𝐴)
2= 1 − 𝑛𝐴
Para calcular o G do sistema, fazemos:
𝐺 = 𝑛𝐴 �𝐺𝐴0 + 𝑅𝑇𝐿𝑛 �𝑛𝐴2� + 𝑅𝑇𝐿𝑛𝑃� + 𝑛𝐴 �𝐺𝐵0 + 𝑅𝑇𝐿𝑛 �
𝑛𝐵2� + 𝑅𝑇𝐿𝑛𝑃�
+ (2 − 2𝑛𝐴)(𝐺𝐶0 + 𝑅𝑇𝐿𝑛(1 − 𝑛𝐴) + 𝑅𝑇𝐿𝑛𝑃)
𝐺 = 𝑛𝐴(𝐺𝐴0 + 𝐺𝐵0 − 2𝐺𝐶0) + 2𝐺𝐶0 + 2𝑅𝑇𝐿𝑛𝑃 +
+2𝑅𝑇 �𝑛𝐴𝐿𝑛 �𝑛𝐴2� + (1 − 𝑛𝐴)𝐿𝑛(1 − 𝑛𝐴)�

264
Fazendo
∆𝐺0 = −(𝐺𝐴0 + 𝐺𝐵0 − 2𝐺𝐶0)
Considerando pressão igual a 1 atm
𝐺 − 2𝐺𝐶0 = 𝑛𝐴(−∆𝐺0) + 2𝑅𝑇 �𝑛𝐴𝐿𝑛 �𝑛𝐴2� + (1 − 𝑛𝐴)𝐿𝑛(1 − 𝑛𝐴)�
O termo do lado esquerdo mostra a variação quando se forma 2 moles
de nC a partir de 1 mol de nA e 1 mol de nB. O primeiro termo da equação a
direita do sinal de igual, representa a energia padrão, início da reação que
desaparece reagentes e aparece produtos. O segundo termo da equação a
direita do sinal de igual mostra a energia livre de mistura dos constituintes da
reação.
Para entendermos melhor essas reações, vamos dar valores para que
possamos representar essas equações graficamente. Assim o valor de
𝐺𝐶0 = −500𝑐𝑎𝑙 e vamos considerar a soma de 𝐺𝐴0 + 𝐺𝐵0 = 0 𝑒 𝑇 = 500 𝐾. Desta
maneira podemos efetuar os seguintes cálculos para nA = 1.
∆𝐺0 = −(𝐺𝐴0 + 𝐺𝐵0 − 2𝐺𝐶0) = ∆𝐺0 = −(0 + 2.500) = −1000 𝑐𝑎𝑙
𝐺 − 2𝐺𝐶0 = 𝑛𝐴(−∆𝐺0) + 2𝑅𝑇 �𝑛𝐴𝐿𝑛 �𝑛𝐴2� + (1 − 𝑛𝐴)𝐿𝑛(1 − 𝑛𝐴)�
Utilizando o valor de ΔG° = - 1000 cal, T = 500 K e variando os valores
de nA de 1 até zero na figura 18.1, é calculado o valor de ΔG do sistema.
A Linha cinza representa os elementos ainda não misturados
𝑛𝐴(−∆𝐺0), a linha vermelha representa os elementos químicos
misturados, 2𝑅𝑇 �𝑛𝐴𝐿𝑛 �𝑛𝐴2� + (1 − 𝑛𝐴)𝐿𝑛(1 − 𝑛𝐴)� e a linha azul é a soma das
duas anteriores.
A figura 18.2, mostra o mesmo cálculo da variação da energia livre do
sistema, utilizado na figura 18.1, para temperatura de 1000 K.
Em seguida será calculado o valor do número de moles para um
mínimo valor de G, da seguinte maneira:

265
𝐺 = 𝑛𝐴𝜇𝐴 + 𝑛𝐵𝜇𝐵 + 𝑛𝐶𝜇𝐶 = 𝑛𝐴𝜇𝐴 + 𝑛𝐴𝜇𝐵 + 2(1 − 𝑛𝐴)𝜇𝐵
Figura 18.1 – Variação da energia de mistura para temperatura de 500 K, ΔG° = - 1000, variando o número de moles de A de, 0 a 1.
Figura 18.2 – Variação da energia livre do sistema para temperatura de 1000 K, ΔG° = - 1000, variando o número de moles de A, de 0 a 1.
𝜕𝐺𝜕𝑛𝐴
= 𝜇𝐴 + 𝜇𝐵 + 2𝜇𝐶 = 0

266
𝜇𝐴 = 𝜇𝐴0 + 𝑅𝑇𝐿𝑛𝑝𝐴, 𝜇𝐵 = 𝜇𝐵0 + 𝑅𝑇𝐿𝑛𝑝𝐵, 𝜇𝐶 = 𝜇𝐶0 + 𝑅𝑇𝐿𝑛𝑝𝐶
𝑝𝐴 = 𝑝𝐵 =𝑛𝐴2
, 𝑝𝐶 =2(1 − 𝑛𝐴)
2= 1 − 𝑛𝐴
𝜕𝐺𝜕𝑛𝐴
= 𝜇𝐴0 + 𝑅𝑇𝐿𝑛 �𝑛𝐴2� + 𝜇𝐵0 + 𝑅𝑇𝐿𝑛 �
𝑛𝐴2� − 2�𝜇𝐶0 + 𝑅𝑇𝐿𝑛(1 − 𝑛𝐴)� = 0
𝜕𝐺𝜕𝑛𝐴
= (𝜇𝐴0 + 𝜇𝐵0 − 2𝜇𝐶0) + 𝑅𝑇𝐿𝑛𝑝𝐴 + 𝑅𝑇𝐿𝑛𝑝𝐵 − 2𝑅𝑇𝐿𝑛𝑝𝐶 = 0
∆𝐺0 = −𝑅𝑇𝐿𝑛𝑝𝐶2
𝑝𝐴𝑝𝐵= −𝑅𝑇𝐿𝑛𝐾𝑝
Chega-se portanto em um resultado igual a equação 15.10, considerando
o sistema no equilíbrio e portanto a variação da energia livre igual a zero.
∆𝐺 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝐾𝑝 = 0
Considerando então ΔG° = - 1000 cal e T = 500 K, pode-se calcular o
valor no número de moles de A para um valor mínimo de variação de energia
de mistura.
−10001,98.500
= −𝐿𝑛�𝑝𝐶2
𝑝𝐴𝑝𝐵� = −1,01 →
𝑝𝐶2
𝑝𝐴𝑝𝐵= 2,7457
2,74 =(1 − 𝑛𝐴)2
𝑛𝐴24
→ 𝑛𝐴 = 0,547
𝑛𝐵 = 0,547 𝑒 𝑛𝐶 = 0,906
9) Utilizando os dados do problema anterior, mostrar a variação da energia livre
de mistura para uma reação exotérmica? Considerar ∆S = 1 e ∆𝐻0= - 500 cal
para T = 500 K, 1000 K e 1500 K, para valores mínimos de ΔG, ou seja
considerando-o igual a zero.
Utilizamos a seguinte equação: ∆𝐺0 = ∆𝐻0 − 𝑇∆𝑆0 e substituindo-os
em:

267
∆𝐺0 = −𝑅𝑇𝐿𝑛𝑝𝐶2
𝑝𝐴𝑝𝐵= −𝑅𝑇𝐿𝑛𝐾𝑝 =
�(1 − 𝑛𝐴)�2
𝑛𝐴24
Pode-se determinar o valor de Kp em função de ΔG° e também o valor
do número de moles de A para cada caso e também calculá-los com a variação
da temperatura e da energia livre padrão. Os resultados são mostrados na
tabela 8.1 e graficamente na figura 18.3.
Tabela 18.1 – Resultados dos cálculos do número de moles e constante de equilíbrio em função da energia livre padrão como constante e também em função da temperatura e energia livre padrão como variáveis.
ΔG°(cal) Temp K na(Gcons.) Kp(1000) KpF(G) na(F(G,T) -1000 500 0,768 2,746 2,746 0,547 -1500 1000 0,720 1,657 2,133 0,578 -2000 1500 0,703 1,400 1,961 0,588 -2500 2000 0,694 1,287 1,880 0,593 -3000 2500 0,689 1,224 1,833 0,596 -3500 3000 0,685 1,183 1,803 0,598 -4000 3500 0,683 1,155 1,781 0,600
Figura 18.3 – Valores da constante de equilíbrio e número de moles para um valor fixo de energia livre padrão e para valores onde tanto a energia quanto a temperatura tornam-se variáveis.
Conclui-se que para uma reação exotérmica (∆𝐻0= - 500 cal), a medida
que a temperatura se eleva a constante de equilíbrio tende a diminuir, tanto

268
para valores fixos da energia livre padrão de ΔG°= - 1000 cal, quanto para
valores variáveis de temperatura e energia livre padrão. Observa-se quando a
constante é calculado em função de energia livre padrão temperatura, decresce
menos. Esse fato é possível devido a variação da entropia no sistema. O
mesmo se repete para o número de moles.
Em reações onde ∆S é maior que zero, apesar de Kp se deslocar para
a esquerda em reações exotérmicas, a reação desloca-se menos para
esquerda, em determinadas situações pode chegar a anular o efeito da
temperatura, devido ao efeito da entropia na equação da variação da energia
livre, direcionando a reação ainda mais para a direita.
10) Mostrar que para uma reação química quando os coeficientes dos
elementos tanto dos reagentes quanto dos produtos forem iguais a 1, a
constante de equilíbrio, pode independer da pressão total do sistema.
Seja a reação:
𝑎𝐴 + 𝑏𝐵 → 𝑐𝐶 + 𝑑𝐷
Utilizando a equação da pressão parcial
𝑝𝐴 = 𝑃.𝑋𝐴 → (𝑝𝐴)𝑎 = (𝑃.𝑋𝐴)𝑎 = 𝑃𝑎.𝑋𝐴𝑎
𝐾𝐸 =𝑝𝐷𝑑𝑝𝐶𝑐
𝑝𝐴𝑎𝑝𝐵𝑏=𝑋𝐷𝑑𝑋𝐶𝑐
𝑋𝐴𝑎𝑋𝐵𝑏=𝑋𝐷𝑑𝑃𝑑𝑋𝐶𝑐𝑃𝑐
𝑋𝐴𝑎𝑃𝑎𝑋𝐵𝑏𝑃𝑏= 𝐾𝑋 .𝑃(𝑐+𝑑−𝑎−𝑏)
Portanto se tivermos os coeficientes iguais a 1 a soma c+d-a-b = 0 e P0
é igual a 1. Assim a constante de equilíbrio torna-se apenas função das frações
molares.
𝐾𝐸 = 𝐾𝑋
11) Calcular ∆𝐺10000 para a equação 𝐶𝐻4 + 𝐶𝑂2 → 2𝐶𝑂 + 2𝐻2
a) Em qual temperatura KE=1
b) A temperatura no qual os produtos aumentam
c) Se a pressão total é decrescida qual o sentido da reação

269
a)
(𝐶𝐻4)𝑔 → 𝐶𝑠 + (𝐻2)𝑔 ∆𝐺0 = 22100 − 26,45𝑇
(𝐶𝑂2)𝑔 + 𝐶𝑠 → 2𝐶𝑂 ∆𝐺0 = 38499 − 40,4𝑇
Somando as duas equações encontramos
𝐶𝐻4 + 𝐶𝑂2 → 2𝐶𝑂 + 2𝐻2 ∆𝐺0 = 60500 − 66,85𝑇
Para K = 1 ∆𝐺0 = 0 e ∆𝐺0 = −𝑅𝑇𝐿𝑛𝐾𝐸 Então 60500 − 66,85𝑇 = 0
Assim T = 905 K
b) ↑ 𝑇,∆𝐺0 fica mais negativo, portanto mais produtos são produzidos
c) Pela lei de Chatelier se ↑ 𝑃 o equilíbrio é deslocado para a esquerda, por
apresentar menor número de moles.
12) Para a reação de oxidação do Mn calcular o valor da variação da energia
livre para pressão de oxigênio igual a 1 atm, 10-4atm e 102atm. na temperatura
de 1600°C.
𝑀𝑛𝑠 +12𝑂2 → 𝑀𝑛𝑂𝑠
∆𝐺 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝐾𝐸
Esse sistema não é um sistema que se encontra no estado padrão
portanto o valor da energia livre precisa ser calculado pela equação geral.
∆𝐺 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝐾𝐸 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝑃𝑀𝑛𝑂
𝑃𝑀𝑛.�𝑃𝑂2= ∆𝐺0 + 𝑅𝑇𝐿𝑛1 = ∆𝐺0
∆𝐺0 = −91980 + 17,48.1873 = −59,2 𝑐𝑎𝑙
Para pressão de oxigênio igual a 10-4atm
∆𝐺 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝐾𝐸 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝑃𝑀𝑛𝑂
𝑃𝑀𝑛.�𝑃𝑂2= −59,2 + 1,98.1873𝐿𝑛
1√10−4
∆𝐺 = −42122 𝑐𝑎𝑙

270
Para pressão de oxigênio igual a 100 atm
∆𝐺 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝐾𝐸 = ∆𝐺0 + 𝑅𝑇𝐿𝑛𝑃𝑀𝑛𝑂
𝑃𝑀𝑛.�𝑃𝑂2= −59,2 + 1,98.1873𝐿𝑛
1√102
∆𝐺 = −67739 𝑐𝑎𝑙
Observa-se portanto que aumentando a pressão de oxigênio, tornamos a
reação possível. Aumentamos com isso a quantidade de reagentes conforme a
reação de Chatelier confirma.
19 O ESTADO PADRÃO O processamento na fabricação de aço desde a redução do minério até
o lingotamento, envolve operações em que o teor de oxigênio presente no
sistema é abaixado ou elevado, a partir do processo de redução do minério de
ferro. Desta forma o oxigênio é manuseado até a composição química final.
Para se efetuar cálculos termodinâmicos com o objetivo de verificar os
processos, um estado de referência ou estado padrão é necessário. Esse tema
será abordado nos próximos capítulos, tem fundamental importância no
entendimento dos processos metalúrgicos. Caso os estados padrões não forem
considerados corretamente nos cálculos, o resultado final pode não ser o
esperado. Considera-se estado padrão, quando determinamos a pressão do
sistema igual a 1 atm para os gases, em uma temperatura desejada. Esse
estado é deixado evoluir e comparado com os outros.
19.1 Estado Padrão e Estado de Equilíbrio Imagina-se uma caixa ideal completamente fechada e no qual não
exista nada. Submeta-se essa caixa a uma temperatura T e coloque dentro
dela sílica sólida pura, silício sólido puro e oxigênio, a pressão de 1 atm.
Este sistema está por definição no estado padrão. Portanto um sistema
qualquer encontra-se no estado padrão se a pressão do gás reagente ou
produto for de 1 atm e se os componentes sólidos ou líquidos, estiverem puros.
Nesse exemplo foi escolhido o estado sólido do silício puro e da sílica
como estado padrão. No entanto poderia ter sido escolhido o estado líquido ou

271
gasoso dos dois ou líquido de um e sólido do outro. A escolha do estado físico
dos componentes (sólido, líquido ou gás) é livre, mas deve ser mencionado.
O estado padrão é um estado imaginário, é apenas um ponto de
referência, a partir do qual se pode fazer medidas comparativas. Por exemplo,
para o caso do Si – O ao fim de um certo tempo ele entrará em equilíbrio às
custas da reação do silício com o oxigênio, até que o Si não tenha mais força
para reagir, então o sistema torna-se estável.
Assim o estado padrão é imaginário não está necessariamente no
equilíbrio e o oxigênio tem pressão de 1 atm. O estado de equilíbrio se
caracteriza por uma pressão de oxigênio específica que é função da
temperatura e da força dos outros componentes do sistema que estarão puros
se forem sólidos ou líquidos e a 1 atm para cada elemento gasoso se forem
gases.
Verifica-se que para passarem do estado (1) para o estado (2), de
acordo com a figura 18.1, o oxigênio passou de 1 atm para uma pressão y
(atm).
(19.1)
A variação da energia livre para a condição de referente a reação 19.1
fica:
(19.2)
Figura 19.1 - Evolução de um sistema do estado padrão ao de equilíbrio
Analisando a equação 19.2, ou seja, substituindo a pressão de oxigênio
por 1 atm, tem-se a energia livre padrão ∆G° = 0. Isto significa que a reação
química está equilibrada. Se a pressão de oxigênio for menor que 1 atm, a
Pressão de Oxig = 1 atm Pressão de Oxig = y atm
Si puro
SiO2 puro Si Puro SiO2 puro
22 SiOOSi ↔+
222
11O
OO
RTLnPP
RTLnGP
RTLnGG =−=∆⇒+∆=∆

272
sílica será reduzida enriquecendo o sistema com oxigênio. Para o caso de
pressão de oxigênio maior que 1 atm, a reação ocorre com diminuição do teor
de silício e aumento da sílica.
Para o caso de T = 1673 K (1400°C) e P = 1,79,10-18 atm
∆G° = 1673.1,987.Ln(1,79.10-18) = - 135843,24 cal/mol
A denominação de caloria por mol de oxigênio é conhecido como
Potencial do Oxigênio µi , sendo R = constante dos gases ideais = 1,987
cal/mol.K e PO2 = pressão parcial do oxigênio em equilíbrio (atm) .
A variação do potencial do oxigênio de vários sistemas a partir do
estado padrão (P = 1 atm) até o estado de equilíbrio, pode levar as seguintes
informações se for realizado em função da temperatura:
• Potencial do oxigênio em função da temperatura
• Comparação de vários sistemas
Para o caso do silício pode ser comparado o potencial de acordo com a
temperatura e pressão. Utilizando os dados da tabela termodinâmica
(www.fatecpindamonhangaba.edu.br), para cálculo do ΔG°, variando o valor da
temperatura, conforme a equação 19.2, chega-se nos resultados da tabela 19.I.
Observando os resultados da tabela 19.I, é possível verificar que a
medida que a temperatura se eleva, o potencial se torna menor negativamente,
ou seja diminui a força da reação para o lado direito.
Tabela 19.1 – Relação entre potencial, temperatura e pressão e variação de energia livre padrão para o silício
Temperatura (K) Pressão do oxigênio (atm)
∆G°(cal/mol)
773 9,03.10-51 -176367
1273 2,14.10-27 -154787
1773 3,33.10-17 -133208
. Chega-se a um momento que a reação é totalmente interrompida em
uma determinada temperatura. Também é possível perceber o caráter
exotérmico da reação, pois a medida que a temperatura sobe a constante de
equilíbrio torna-se menor.

273
Quando os reagentes e produtos com excessão do oxigênio puros,
tendo a pressão do oxigênio igual a 1 atm. e portanto no estado padrão em
uma determinada temperatura T, a partir das equações 9.16 e 19.1, pode-se
escrever:
STHG ∆−∆=∆ (19.3)
∆G° = Energia livre padrão de formação
∆H° = Entalpia padrão de formação
∆S° = Entropia padrão de formação
O símbolo (°), quando utilizado, refere-se a equação de um sistema no
estado padrão.
Esta equação de ∆G° é apresentada em tabelas termodinâmicas para
várias reações de oxidação ou outras, variando para cada uma a entalpia e a
entropia. Usualmente chama-se de A a entalpia e B a entropia na seguinte
forma:
∆G° = A + B.T (19.4)
Então para uma determinada reação encontra-se A = -209730 cal/mol e
B = 46,16 cal/molK. Substituindo em 19.4.
∆G° = -209730 + 46,16.T (cal/mol) (19.5)
Para T = 1282 K ⇒ ∆G° = -150552,88 cal/mol
Caso não se encontre os dados termodinâmicos para o estado padrão,
para determinados compostos na forma de líquidos ou gases, equação 19.9.
Pode-se compor os resultados da seguinte maneira:
SiS + O2G → SiO2S ∆G° = - 209730 + 43,16T (19.6)
SiL→ SiS ∆G° = - 13100 + 7,19T (19.7)
SiO2S → SiO2L ∆G° = 3600 - 1,8T (19.8)
Somando as equações 19.6 + 19.7 + 19.8, tem-se a equação 19.9:
SiL + O2G → SiO2L ∆G° = - 219230 + 48,55T (19.9)

274
Obtendo os valores de ∆H° de fusão do silício de 12100 cal/ mol na
temperatura de fusão 1410°C(1683K) e da sílica de 3600 cal/mol na
temperatura de fusão de 1723°C(1996K) é possível calcular a variação da
energia livre padrão de fusão, por exemplo para o silício, equação 19.7 e para
a sílica 19.8:
(19.10)
19.2 Energia Livre como Critério de Equilíbrio
Conforme a equação 15.10, A variação da energia livre realizada desde
um estado qualquer até o estado de equilíbrio é dado por:
∆G é o trabalho do oxigênio na pressão do sistema até o estado
considerado.
∆G° é o trabalho do estado padrão até o estado de equilíbrio.
RTLnKE é o trabalho do estado padrão até o estado considerado.
A equação ∆G = ∆G° + RTLnK E permite saber se o processo está ou
não em equilíbrio e se a reação é possível ou não, no sentido considerado.
Se tem um sistema de silício líquido puro, sílica líquida pura e oxigênio
na pressão de oxigênio de 10-30 atm na temperatura de 1409°C. O sistema
estará em equilíbrio?
É necessário para isso calcularmos a equação 15.10. Temos os
seguintes dados, para reação de oxidação da sílica:
∆G° = -218230 + 48,55T; (19.11)
Para 𝑃𝑂2 = 10−30𝑎𝑡𝑚; como a pressão parcial da sílica e do silício são muito
baixas, pode–se considerar: 𝑃𝑆𝑖𝑂2𝑃𝑆𝑖
= 1, e T = 1682 K
Substituindo então temos:
(19.12)
RTLnKGG +∆=∆
TTHTHSTHG
F
FFFFF 19,712100+−=
∆−∆=∆−∆=∆
)/(9429898,155,482182302
2 molcalPP
PTLnTG
OSi
SiO =
++−=∆

275
Nesse caso por ser a variação de energia livre positiva, a tendência é da
dissociação da sílica.
Vamos considerar a oxidação do silício para sílica em um forno em uma
determinada temperatura e aplicar a esse cálculo as equações 15.10 e 19.1.
∆𝐺 = ∆𝐺° + 𝑅𝑇𝐿𝑛 � 𝑃𝑆𝑖𝑂2𝑃𝑆𝑖𝑃𝑂2
�𝐹𝑜𝑟𝑛𝑜
(19.13)
Por se tratar de material sólido, a pressão parcial dos dois se cancela, 𝑃𝑆𝑖𝑂2𝑃𝑆𝑖
= 1
∆𝐺 = ∆𝐺° + 𝑅𝑇𝐿𝑛 � 1𝑃𝑂2
�𝐹𝑜𝑟𝑛𝑜
(19.14)
Substituindo na equação o valor de ∆G° pelo valor da pressão de oxigênio em
equilíbrio, ou seja, para ∆G = 0, temos o seguinte valor para ∆G°
(19.15)
(19.16)
(19.17)
Portanto conclui-se que:
�𝑃𝑂2�𝐸𝑞𝑢𝑖𝑙í𝑏𝑟𝑖𝑜 < �𝑃𝑂2�𝑆𝑖𝑠𝑡𝑒𝑚𝑎⇒ ∆G < 0 (19.18)
e se
�𝑃𝑂2�𝐸𝑞𝑢𝑖𝑙í𝑏𝑟𝑖𝑜 > �𝑃𝑂2�𝑆𝑖𝑠𝑡𝑒𝑚𝑎⇒ ∆G > 0 (19.19)
Assim pode-se entender que se o teor de oxigênio do sistema é menor que o
da equação química, haverá dissociação do óxido de modo a elevar o teor de
oxigênio do sistema. Podemos dizer que o óxido nesse caso é instável. Mas
caso contrário se o teor de oxigênio do sistema for maior que o teor de oxigênio
em equilíbrio da reação envolvida, a tendência é a de o óxido ficar estável, sem
o perigo da dissociação. Para melhor compreensão a figura 19.2 mostra que
ERTLnKGG +∆=∆
EquilOPRTLnG
−=°∆
2
1
FornoOEquilOFornoO PRTLn
PRTLn
PRTLnGG
+
−=
+°∆=∆
222
111
( )
=
+=∆
FornoO
EquilbrioO
FornoOO P
PRTLn
PRTLnPRTLnG
)()(1
2
2
22

276
quando um sistema possuir potencial de oxigênio menor que a do equilíbrio de
uma determinada reação, a tendência é a reação fornecer oxigênio para o
sistema e assim ocorre também da forma inversa, ou seja, se o potencial do
sistema for maior que a do equilíbrio, a reação tenderá anular esse potencial
maior eliminando do sistema o oxigênio através da oxidação do metal.
Figura 19.2 – Mostra as regiões de estabilidade e instabilidade dos óxidos em função do potencial de oxigênio de um determinado sistema. Para o caso de dois tipos de óxidos de acordo com o potencial de cada um, o
óxido mais estável deve reduzir o menos estável, ou seja, aquele que
apresenta o menor potencial de oxigênio em equilíbrio é capaz de reduzir o
outro que apresenta maior potencial. Então para a seguinte equação química:
OMMOMM 2112 +→+
Nesse caso o M2 é o redutor do M1O, pois o potencial do oxigênio do M2O é
menor que do M1O.
Figura 19.3 – Mostra o potencial do oxigênio para dois diferentes óxidos
A figura 19.3 mostra o potencial do oxigênio para dois diferentes
óxidos, o M2O, possui um potencial menor que o M1O, assim o M2 passa a ser
o redutor do óxido M1O.
M1 + O ⇐ M1O (1)
M2 + O ⇒ M2O (2)
POxigênio(M2) < POxigênio(M1)
PO2 > PO2 equilíbrio
M + O2 ⇒ MO2
PO2 < PO2 equilíbrio
M + O2 ⇐ MO2

277
Desta forma os metais posicionados em locais onde o potencial do
oxigênio são os mais baixos, podem ser utilizados como redutores daqueles
óxidos com potencial de oxigênio mais elevados. Com esse entendimento
pode-se concluir que:
• O alumínio pode ser utilizado para produzir titânio metálico;
• O silício produz Mn metálico, reduzindo o MnO;
• A 1400°C o C pode ser usado para produzir Fe;
• A 1600°C o carbono reduz o óxido de ZnO, mas não reduz o MgO.
20 SOLUÇÕES
Até agora foi considerado o puro componente ou seja um único
elemento e seu óxido. Mas isso não ocorre na prática ou seja, os óxidos não
aparecem unicamente, mas sim misturados e para determinar a pressão de
vapor em equilíbrio é necessário conhecer a influência de todos esses
elementos na solução.
20.1 Princípio de Raoult e Henry
Seja um recipiente previamente evacuado com uma substância líquida
A em seu interior. Assim que a substância A entra no recipiente, inicia-se a
evaporação até chegar a uma pressão de equilíbrio P°A. A partir desse
momento se estabelece o equilíbrio e(A) = c(A), evaporação(A) =
condensação(A). Portanto para uma temperatura fixa pode-se escrever.
e(A) = c(A) = kR P°A (20.1)
(°) esse símbolo representa que a substância é pura, então no caso de P°A,
mostra que A está puro em uma determinada temperatura T. Sem esse
símbolo significa que a substância encontra-se em solução, portanto uma
mistura de dois ou mais substâncias.
kR, kR1, kR2... são as constantes de proporcionalidade de cada elemento
químico, ou substância que relaciona a velocidade de vaporização ou
condensação com a pressão parcial em equilíbrio em uma determinada
temperatura e solução.

278
Considerando agora o efeito de uma pequena quantidade de B
adicionado no líquido A, obtendo portanto a liga A-B. Assumindo os diâmetros
atômicos de A e B equivalentes e a composição da solução a mesma tanto
internamente quanto na superfície do líquido. Isto significa que A somente
vaporize da superfície nos lugares ocupados por A, então a razão de
evaporação de A decresce pelo fator de XA e a pressão diminui de P°A para PA.
e(A) = c(A) = kR1 P°A e e(A).XA = kR2 .PA ⇒ kR1 . P°A.XA = kR2 PA
𝑃𝐴 = 𝐾𝑅1𝐾𝑅2
𝑃𝐴𝑜𝑋𝐴 (20.2)
Considerando soluções ideais kR1 = kR2, caso contrário kR1 ≠ kR2, ou
seja ocorre interações entre as substâncias da solução. Define-se atividade de
um elemento, a razão entre a pressão parcial na solução e a sua pressão
parcial como elemento puro na mesma temperatura da solução.
𝑘𝑅1𝑘𝑅2
= 𝛾 = 1 → 𝑘𝑅1 = 𝑘𝑅2
𝑃𝐴 = 𝑃𝐴𝑜𝑋𝐴 𝑒 𝑎𝐴 = PA𝑃𝐴𝑜 → 𝑎𝐴 = 𝑋𝐴 (20.3)
𝑃𝐵 = 𝑃𝐵𝑜𝑋𝐵 𝑒 𝑎𝐵 = PB𝑃𝐵𝑜 → 𝑎𝐵 = 𝑋𝐵 (20.4)
Então a equação 20.3 é para a substância A e a equação 20.4 para a
substância B.
As substâncias que obedecem as equações 20.3 e 20.4 são aquelas
que seguem a lei de Raoult, expressadas na figura 20.2
O símbolo 𝛾 (gama), significa o desvio da idealidade, para 𝛾 = 1 trata-
se de uma solução ideal, se for diferente de um, trata-se de uma substância
que apresenta desvio da idealidade, ou interações entre os componentes da
solução. A figura 20.1, mostra para uma solução, as relações entre pressões
parciais para um situação ideal. Quando os elementos puros, representados
pelos eixos verticais, as pressões são dos componentes puros. Quando
misturados, a medida que se misturam as pressões parciais ou se elevam ou
decrescem, dependendo da temperatura de vaporização de cada elemento
químico.

279
Se considerarmos que tenhamos na liga pouco A, ou seja A passa ser
uma substância diluída. Nestas circunstâncias a razão de evaporação de A
será decrescida proporcionalmente a pressão parcial de A, de maneira
constante, pois existe grande separação entre os átomos do soluto, fazendo
com que a razão de evaporação seja independente da composição.
Figura 20.1 – Mostra a relação das pressão parciais de A e B de acordo com a fração molar de A.
XA.e(A) = γºPA (substância diluída) e e(A) = γP°A (substância pura)
γº = coeficiente de atividade raoultano do soluto em condições diluídas
Substituindo o valor de e(A), obtém-se
(20.5)
(20.6)
Pelo fato de f ser uma constante e oAP também, pode-se considerar o
seguinte:
XB
PA PB
PA + PB
P°A
P°B
XA
oAAoA PXP
γγ
=
fo =γγ
AAAo
AoA XkPPk =⇒=γγ

280
De uma maneira mais geral
(20.7)
𝑘𝐴,𝑘𝑖 = constante de proporcionalidade para substâncias diluídas, que
relacionam diretamente a pressão parcial com a fração molar.
A pressão parcial para concentrações de solutos muito pequenos é
proporcional a sua pressão parcial. Esse fato verifica-se por exemplos em
concentração de gases na água. Quanto maior a pressão na superfície, mais
os gases se encontram dissolvidos na água. No caso da temperatura é o
oposto, quanto maior for a temperatura menor sua concentração, pois ocorre
dilatação do líquido, deixando menos espaço para os gases. Esse é um fato
muito preocupante para a elevação da temperatura nos mares, pode ocasionar
a falta de oxigênio para uma grande espécies de peixes, por exemplo a truta
vive em ambiente de temperatura igual a 15°C, a Carpa 32°C e o Bragre em
34°C.
Portanto a partir das equações 20.5 e 20.6:
(20.8)
E como estamos considerando uma solução diluída, a atividade
raoultiana passa a ser conhecida por henriana, troca-se a por h, para indicar
que estamos trabalhando com soluções diluídas de acordo com o princípio de
Henry.
ℎ = 𝑓.𝑋𝐴 (20.9)
Para soluções diluídas ideais, o coeficiente de atividade f passa a ser
1, considerando A e B como solutos de uma solução
ℎ𝐴 = 𝑋𝐴 𝑒 ℎ𝐵 = 𝑋𝐵 (20.10)
As equações 20.10 são equações válidas somente para uma variação
de soluto e conhecidas como o princípio de Henry. O coeficiente f, representa a
relação entre os coeficientes de uma substância com comportamento
raoultiano e henriano quando não são ideais. Para soluções ideais ou diluídas:
(20.11)
AAAoA
AA
oAA XfaXf
PPXPfP .... =⇒=⇒=
1== ofγγ
iii XkP =

281
Quando não houver desvios f torna-se igual a 1, portanto para f = 1, na
região onde a solução apresenta tal comportamento, a solução obedece a lei
de Henry. A figura 20.2 mostra a relação das atividades graficamente.
Figura 20.2 – Quando a atração entre A – B é maior que entre A-A ou B-B, então ocorre um desvio negativo de γ. Do modo contrário quando A – B, for menor que A-A e B-B, o desvio é positivo de γ. 20.2 Diagrama Pressão Total Versus Fração Molar de Gases e Líquidos
Considerando o gráfico 20.1 onde Pt = PA + PB. Aqui precisa-se fazer
uma distinção entre fração molar da fase gasosa e fração da fase líquida.
Quando utiliza-se a lei de Raoult, equações 20.3 e 20.4 estamos relacionando
pressão de vapor da fase gasosa com a pressão parcial de uma solução
líquida, portanto a fração molar representativa é da fase líquida. Para a
equação das pressões parciais calculadas à partir da equação 13.30 relaciona
a fração molar apenas da fase vapor. Então vamos considerar Y a fase vapor e
X a fase líquida através das deduções de Dalton e Raoult, podemos fazer uma
relação entre essas duas frações molares, ou seja, para uma mesma pressão
total quanto teríamos Y moles na fase vapor e quanto seria X moles na fase
líquida da solução.
Seja então uma solução binária, figura 20.3, para dois elementos 1 e 2
misturados na fase líquida com uma fase gasosa em equilíbrio com essa
solução.
Princípio
de Raoult
Princípio de
Henry
γ < 1, aA<XA
γ >1, aA>XA
ai
0 1 Fração Molar 0 1 Fração Molar
h
γ°
γ=1,
aA=XA
f = γ/γ°

282
Figura 20.3 – Solução líquida em equilíbrio com sua fase gasosa, sendo X representante do número de moles da fase líquida e Y representante do número de moles da fase gasosa.
Considerando a lei de Raoult tem-se que 𝑃1 = 𝑋1.𝑃1° e a equação de
Dalton fornece 𝑌1 = 𝑃1𝑃𝑡
. Vamos entender que X1 representa a fração molar na
fase líquida e Y1 representa a fração molar na fase gasosa em equilíbrio com a
solução líquida. Portanto relacionando a lei de Raoult com a de Dalton concluí-
se que:
𝑌1 = 𝑃1𝑃𝑡
= 𝑋1.𝑃1°
𝑃𝑡 (20.12)
Mas
𝑃𝑡 = 𝑃1 + 𝑃2 = 𝑋1.𝑃1° + 𝑋2.𝑃2° = 𝑋1.𝑃1° + (1 − 𝑋1).𝑃2° = 𝑋1.𝑃1° + 𝑃2° − 𝑋1.𝑃2°
𝑃𝑡 = 𝑃2° + 𝑋1(𝑃1° − 𝑃2°) (20.13)
Substituindo em 20.12
𝑌1 =𝑋1.𝑃1°
𝑃𝑡=
𝑋1.𝑃1°
𝑃2° + 𝑋1(𝑃1° − 𝑃2°)⇒
⇒ 𝑋1 = 𝑌1.𝑃2°
𝑃1°−𝑌1(𝑃1°−𝑃2°) (20.14)
Cálculo de Pt em função de Y1, a partir de 20.12
𝑃𝑡 = 𝑋1.𝑃1°
𝑌1 (20.15)
Fase líquida, solução
misturada de X1 e X2 em
equilíbrio com a fase vapor
Y1, Y2.
Fase vapor Y1, Y2 em
equilíbrio com fase líquida
X1,X2 número de
moles da fase
líquida e Y1, Y2
número de
moles da fase
gasosa.

283
Substituindo em 20.15, a equação 20.14
𝑃𝑡 =𝑋1.𝑃1°
𝑌1=𝑃1° �
𝑌1.𝑃1°𝑃1° − 𝑌1(𝑃1° − 𝑃2°)
�
𝑌11
=𝑃1°.𝑃2°
𝑃1° − 𝑌1(𝑃1° − 𝑃2°)
𝑃𝑡 = 𝑃1° .𝑃2°
𝑃1°−𝑌1(𝑃1°−𝑃2°) (20.16)
Assim as duas equações que representam a pressão total em função
de X1 (fração molar de X1 na solução líquida) e Y1 (fração molar de Y1 na fase
gasosa), equações 20.13 e 20.16, são:
𝑃𝑡 = 𝑃2° + 𝑋1(𝑃1° − 𝑃2°)
𝑃𝑡 = 𝑃1° .𝑃2°
𝑃1°−𝑌1(𝑃1°−𝑃2°)
Com as equações 20.13 e 20.16 é possível construir um gráfico que
mostra a pressão total Pt em função das frações molares na fase líquida X1 e a
fração molar na fase gasosa Y1, figura 20.4
O figura 20.4 mostra então a fração molar para um mesma composição
da solução na fase líquida o correspondente que se teria na fase gasosa. A
figura 20.5 faz essa correlação de uma forma mais esclarecedora. A reta
vertical mostra a composição de uma solução, o encontro com essa reta na
primeira reta que liga os eixos de X1 e X2 encontra-se a fração molar na fase
líquida. Fazendo uma paralela com o eixo dos x (abcissa), encontra-se a fração
molar da fase gasosa em equilíbrio com a solução líquida, na mesma pressão.
Percebe-se assim que a fração molar para um determinada substância
é maior na fase gasosa que na fase líquida. A substância que possui maior
pressão de vapor, (menor temperatura de vaporização), apresenta maior
número de moles na fase gasosa.
Essa substância deve ser mais volátil. A parte superior do diagrama
indica a fase líquida, que corresponde ao aumento da pressão. A parte inferior
corresponde a fase gasosa que aparece quando a pressão está baixa. A
porção intermediária entre a reta e a curva que correspondem o ponto de
orvalho e o borbulhamento da fase líquida indicam uma região onde existe

284
equilíbrio entre a fase líquida e a fase vapor. Esse diagrama é conhecido como
diagrama de fase pressão-composição.
Figura 20.4 – Diagrama da Pressão Total em função das frações molares na fase líquida e fase gasosa.
Através desses gráficos é possível entender como é o mecanismo da
destilação fracionada. Seja então a figura 20.6 onde mostra a pressão em
função da composição para uma mistura na solução líquida X1,X2. Nessa
solução ocorrerá uma fase gasosa em equilíbrio com a fase líquida. A fração
molar para uma substância mais volátil corresponderá uma maior fração molar
na fase gasosa.
A fração molar na fase líquida (X1) é representada pelo ponto B, que
corresponderá ao ponto A, a fração molar em equilíbrio com a fase gasosa
(Y1), figura 20.6.
Agora isolando essa fase gasosa e deixando-a em contato com um
meio refrigerante, a solução passará de gasosa para líquida e novamente
constituirá uma fase gasosa em equilíbrio, enriquecendo cada vez mais a
fração molar de Y1.
Repetindo esse processo por várias vezes, chega-se a substância pura
X1. Esse processo não se aplica somente a soluções que apresentam pontos
azeotrópicos.
100% X1 100% X2
Fase vapor - V
Fase líquida - L
L + V
Linha que representa o ponto de
borbulhamento Ptf(X1)
Linha que representa o ponto
de orvalho Ptf(Y1)
P1°
P2°

285
Figura 20.5 – Diagrama que mostra a fração molar da fase líquida equivalente a fase gasosa para uma dada solução líquida.
Figura 20.6 – Representa o mecanismo de uma destilação fracionada
Essas soluções possuem um ponto característico no qual as duas
curvas se encontram, passando a pressão de vapor tanto do vapor quanto a do
líquido serem as mesmas, figura 20.7.
Também é possível construir esse gráfico substituindo a pressão pela
temperatura, o gráfico torna-se bem similar de acordo com a figura 20.8.
Apenas as fases são invertidas e apresentam maior TEB (Temperatura
de ebulição) a substância com pressão de vapor menor e TEB menor para a
pressão de vapor maior.
B
A
Fase gasosa
Fase líquida P°1
P°2
100% X1 100% X2
L + V
Mistura na
solução X1;X2
Fração da solução na
fase líquida X1
Fração molar da solução
na fase gasosa Y1
100% X1 100% X2
P°1
P°2

286
Figura 20.7 – Mostra um diagrama pressão versus fração molar com um ponto azeotrópico.
Figura 20.8 – Mostra o diagrama de equilíbrio, temperatura versus composição.
Analisando o diagrama 20.8 pode-se observar que também ocorre
diferença entre a fração molar da fase gasosa para a fase líquida. No caso de
sólido e líquido o fenômeno também se repete. Assim pode-se entender que
utilizando o processo de fusão localizada para barras de silício por exemplo é
possível direcionar as impurezas toda para o líquido, purificando esse material
para valores elevados em silício (altas purezas). Também entende-se que ao
solidificar um lingote de aço, ocorre diferenças de composição entre a parte
sólida e líquida, de maneira que elementos com alta mobilidade na fase líquida,
da mesma forma que elementos voláteis segregam para a última porção a se
solidificar (similar a destilação fracionada). Esse fenômeno em metalurgia
Fase gasosa
Fase líquida
Temperatura
A B
Fase Líquida + fase Vapor
Fase gasosa
Fase líquida P°1
P°2
Ponto azeotrópico 100% X1 100% X2

287
chama-se segregação. Durante o processo de laminação ou forjamento são
analisados a superfície e o centro dos materiais processados que devem
obedecer uma especificação mínima de diferenças conforme as normas pré-
estabelecidas de acordo com a aplicação. O C (carbono), S (enxôfre) e H2
(Hidrogênio), são os que mais segregam apresentando-se sempre em maior
porcentagem no centro que na superfície. É claro que esse fenômeno tem a
grande influência da temperatura e das ligas produzidas. Quanto maior for o
intervalo de solidificação e maior for a temperatura maior será a segregação.
Além de C e S também os carbonetos podem segregar e dependendo do
processo não mais dissolver e prejudicar posteriormente as propriedades
mecânicas dos materiais como por exemplo a resistência a fadiga. Esse é um
dos problemas apresentados pelos fabricantes de rolamentos para o tipo de
aço SAE 52100, que por apresentar Cr em sua composição, pode ocorrer
formação de carbonetos grandes primários, (oriundos do processo de
solidificação), que prejudicará a resistência a fadiga no produto final.
Um exemplo da destilação fracionada é a redução do óxido de zinco
ZnO, obtido através das seguinte reação química:
(𝑍𝑛𝑂)𝑆 + 𝐶𝐺𝑟𝑎𝑓𝑖𝑡𝑒 = 𝑍𝑛𝑔 + 𝐶𝑂𝑔 (20.17)
Também a redução pode ser representada pelas seguintes equações:
(𝑍𝑛𝑂)𝑆 + 𝐶𝑂𝑔 → 𝑍𝑛𝑔 + (𝐶𝑂2)𝑔 (20.18)
𝐶𝑂2 + 𝐶 = 2𝐶𝑂 (20.19)
As reação do (ZnO)S com COg , como a reação de Boudourad são
endotérmicas portanto temperaturas elevadas são necessárias. Assim a
utilização de 1000°C para redução do (ZnO)s é razoável, fazendo com que o
zinco torna-se vapor, pois seu ponto de vaporização é de 906°C e portanto a
sua recuperação deve ser pela condensação do vapor de zinco para líquido. O
cádmio vem associado com o zinco, é mais volátil, pois sua temperatura de
vaporização é de 765°C, portanto também é separado por condensação Outro
metal que também é extraído através de destilação é o mercúrio. Através da
reação do sulfeto de mercúrio com oxigênio, por apresentar temperatura de
evaporação de 357°C.

288
(𝐻𝑔𝑆)𝑆 + 𝑂2 → (𝐻𝑔)𝑔 + 𝑆𝑂2 (20.20)
Na redução do sulfeto de mercúrio também pode ser utilizado CaO ou Fe:
4(𝐻𝑔𝑆)𝑆 + 4(𝐶𝑎𝑂)𝑆 → 4(𝐻𝑔)𝑔 + 3(𝐶𝑎𝑆)𝑆 + (𝐶𝑎𝑆𝑂4)𝑆 (20,21)
(𝐻𝑔𝑆)𝑆 + (𝐹𝑒)𝑆 → (𝐻𝑔)𝑔 + (𝐹𝑒𝑆)𝑆 (20.22)
20.3 Regra da Alavanca
Para um diagrama onde existe um intervalo de solidificação entre a
linha de liquidus e linha de solidus, durante o processo de solidificação, ocorre
uma certa quantidade de líquido e sólido. Para quantificar essas quantidades
em forma de porcentagens utiliza-se a regra da alavanca.
Figura 20.9 - Diagrama isomorfo, sendo XA – fração molar de A, nA - número de moles de A, nS
A – número de moles de A na fase sólida, nAL – número de
moles de A na fase líquida, XAS – fração molar da fase sólida, XA
L – fração molar da fase líquida.
A quantidade de moles de A na liga é igual a quantidade de moles na
fase sólida de A, mais a quantidade de moles de A na fase líquida, assim:
𝑛𝐴𝑋𝐴 = 𝑛𝐴𝐿𝑋𝐴𝐿 + 𝑛𝐴𝑆𝑋𝐴𝑆 (20.23)
Líquido
Sólido
Líquido + Sólido
XA=0
XA=1
XA XAS
XAL
T
fS fL

289
nA - número de moles de A, nSA – número de moles de A na fase sólida, nA
L –
número de moles de A na fase líquida, XAS – fração molar da fase sólida, XA
L –
fração molar da fase líquida.
Dividindo a equação 20.23 por nA
𝑋𝐴 = 𝑛𝐴𝐿𝑋𝐴
𝐿
𝑛𝐴+ 𝑛𝐴
𝑆𝑋𝐴𝑆
𝑛𝐴 (20.24)
𝑓𝑆 = 𝑛𝐴𝑆
𝑛𝐴; 𝑓𝐿 = 𝑛𝐴
𝐿
𝑛𝐴 (20.25)
𝑋𝐴 = 𝑓𝑆𝑋𝐴𝑆 + 𝑓𝐿𝑋𝐴𝐿 = (1 − 𝑓𝐿)𝑋𝐴𝑆 + 𝑓𝐿𝑋𝐴𝐿 (20.26)
𝑓𝐿 = 𝑋𝐴−𝑋𝐴𝑆
𝑋𝐴𝐿−𝑋𝐴
𝑆 ; 𝑓𝑆 = 𝑋𝐴−𝑋𝐴𝐿
𝑋𝐴𝑆−𝑋𝐴
𝑆 (20.27)
fL representa a fração de líquido e fS a fração de sólidos
20.3 Volatilidade Relativa
Para que ocorra a destilação entre duas substâncias, é preciso entre as
duas existir uma diferença nas pressões de vapores para uma determinada
temperatura. A relação das pressões parciais pode ser desenvolvida, a partir
da equação 20.2:
𝑎 = 𝛾𝑋 =𝑃𝑃° ⇒ 𝑃 = 𝛾𝑋𝑃°
A relação entre duas pressões parciais pode ser desenvolvida como:
𝑃1𝑃2
= 𝛾1𝑋1𝑃1°
𝛾2𝑋2𝑃2° (20.28)
Entende-se que 𝑃1,𝑃2, .. são pressões parciais; 𝑋1,𝑋2, .. são frações
molares e 𝑃1°,𝑃2°, …são pressões de vapores dos componentes puros em uma
determinada temperatura e 𝛾1,𝛾2 são coeficientes de atividades. O critério para
o sucesso da destilação, para um determinado componente 1 é a fração molar
desse componente da solução gasosa (Y1), ser maior que na solução
liquidam,(X1).
Igualando as equações 20.13 e 20.16

290
𝑃1°𝑃2°
𝑃1° − 𝑌1(𝑃1° − 𝑃2°)= 𝑃2° + 𝑋1(𝑃1° − 𝑃2°)
𝑃1°
𝑃1° − 𝑃2°−
𝑃1°𝑃2°
�𝑃1° − 𝑃2°�[𝑃2° + 𝑋1(𝑃1° − 𝑃2°)]= 𝑌1
1𝑃1° − 𝑃2°
�𝑃1° −𝑃1°𝑃2°
𝑃2° + 𝑋1(𝑃1° − 𝑃2°)� = 𝑌1
𝑃1°𝑃2°
𝑃1° − 𝑃2°�
1𝑃2°−
1𝑃2° + 𝑋1(𝑃1° − 𝑃2°)
� = 𝑌1
𝑃1°𝑃2°
𝑃1° − 𝑃2° .
1𝑃2°�1 −
𝑃2°
𝑃2° + 𝑋1(𝑃1° − 𝑃2°)� = 𝑌1
𝑃1°
𝑃1° − 𝑃2°�𝑃2° + 𝑋1�𝑃1° − 𝑃2°� − 𝑃2°
𝑃2° + 𝑋1(𝑃1° − 𝑃2°)� = 𝑌1
𝑃1°
𝑃1° − 𝑃2°�
𝑋1�𝑃1° − 𝑃2°�𝑃2° + 𝑋1(𝑃1° − 𝑃2°)
� = 𝑌1
�𝑃1°𝑋1
𝑃2° + 𝑋1(𝑃1° − 𝑃2°)� = 𝑌1
Dividindo o numerador e o denominador por 𝑃2° e chamando 𝛼 = 𝛾1𝑃1°
𝛾2𝑃2°
(Esse termo é conhecido como volatilidade relativa) e considerando os
coeficientes de atividades iguais a 1, ou seja 𝛾1𝛾2
= 1. Então teremos 𝛼 = 𝑃1°
𝑃2° .
𝑃1°𝑋1𝑃2°
𝑃2°𝑃2°
+𝑋1�𝑃1° − 𝑃2°�
𝑃2°
=𝛼𝑋1
1 + 𝑋1(𝛼 − 1)= 𝑌1
𝑌1 = 𝛼𝑋11+𝑌1(𝛼−1)
(20.29)
Para 𝛼 = 1 teremos 𝑥1 = 𝑦1, sem condições de ocorrer destilação na
solução, pois não haverá separação na fase gasosa com maior fração molar
que na fase líquida. Essa solução pode ser identificada como apenas um
componente puro.

291
𝛼 = 𝛾1𝑃1°
𝛾2𝑃2° (20.30)
Para um processo de separação das substâncias na fase vapor ter
sucesso, precisa-se de um 𝛼 maior que 1. Portanto é importante verificar em
qual temperatura principalmente esse fato ocorre na solução.
Para exemplificar esse processo, vamos calcular a pressão parcial e o
coeficiente 𝛼 para o equilíbrio entre três componentes: Cadmium (Cd), Bismuto
(Bi) e Chumbo (Pb), contendo as seguintes porcentagens em fração molar:
60% de Cd, 20% de Bi e 20% de Pb. Comentar a possível destilação do Cd na
temperatura de 500°C, com os seguintes coeficientes de atividades: 𝛾𝐶𝑑 =
1,2; 𝛾𝐵𝑖 = 0,9 𝑒 𝛾𝑃𝑏 = 0,2 .
Em primeiro lugar vamos Calcular a pressão de valor do Cd, Bi e Pb na
temperatura de 723 K. Para isso utilizamos os dados da tabela termodinâmica
na aba da planilha Excel a pressão de vapor no site www.fatecpinda.edu.br e
encontramos:
𝑃𝐶𝑑° = 4,41 𝑚𝑚𝐻𝑔 ; 𝑃𝐵𝑖° = 2,36. 10−6𝑚𝑚𝐻𝑔 ; 𝑃𝑃𝑏° = 2,15. 10−6𝑚𝑚𝐻𝑔
Cálculo das pressões parciais:
𝑃𝐶𝑑 = 𝑋𝐶𝑑𝛾𝐶𝑑𝑃𝐶𝑑° = 0,6.1,2.4,41 = 3,17 𝑚𝑚𝐻𝑔
𝑃𝐵𝑖 = 𝑋𝐵𝑖𝛾𝐵𝑖𝑃𝐵𝑖° = 0,2.0,9.2,36. 10−6 = 0,42. 10−6𝑚𝑚𝐻𝑔
𝑃𝑃𝑏 = 𝑋𝑃𝑏𝛾𝑃𝑏𝑃𝑃𝑏° = 0,2.0,2.2,15. 10−6 = 0,086. 10−6𝑚𝑚𝐻𝑔
Percebe-se através das pressões parciais que o Cd entre os três é o
mais apropriado para ser separado. Calculando agora a volatilidade relativa,
utilizando a equação 20.25:
Primeiro entre o Cd e o Pb:
𝑃𝐶𝑑𝑃𝑃𝑏
=3,17
0,086. 10−6= 3,7. 107
Entre Cd e Bi

292
𝑃𝐶𝑑𝑃𝐵𝑖
=3,7
0,42. 10−6= 7,54. 106
Finalmente entre Pb e Bi
𝑃𝑃𝑏𝑃𝐵𝑖
=0,086. 10−6
0,42. 10−6= 0,2
Nesse caso o cádmio tem mais chance de ser separado da liga do que
os outros elementos.
A pressão parcial de um determinado elemento em uma determinda
condição de trabalho torna-se importante durante o processo de fabricação de
aço. Por exemplo um típico aço como o utilizado para nitretação onde o teor de
alumínio é da ordem de 1%. Nesse caso esse aço por possuír como liga
cromo, níquel e molibdênio deve ser desgaseificados. Mas adicionar o alumínio
antes ou após o processo de desgaseificação. Se adicionar após para um
banho contendo 90 ton de aço, seria necessário aproximadamente 900 kg para
uma rendimento de 100%, portanto recomenda-se adicionar um pouco mais.
Nesse caso a adição após a desgaseificação, haverá alguma exposição para
homogeneização e poderá ter elevação do teor de hidrogênio. Poderia então
fazer uma adição parcial antes do processo de vácuo. Nesse caso seria
interessante fazer alguns cálculos e verificar se ocorrerá evaporação do
alumínio adicionado durante a desgaseificação. Devido ao teor de alumínio
elevado, esse aço deverá ter inclusões de alumina, aços com inclusões não
são tão sensíveis a trincas por hidrogênio, dependendo das condições de
resfriamento e de conformação. Seria então interessante nesse caso elevar o
teor de enxofre ao máximo permitido pelo cliente e fazer alguns testes, talvez
nem seria necessário o processo de desgaseificação. Como cálculo podemos
fazer o seguinte: a temperatura de início de desgaseificação é na ordem de
1630°C, conhecendo a pressão de vapor do ferro de 0,060 mmHg e para o
alumínio puro de 2,3 mmHg. Os coeficientes de atividades são para o ferro 1 e
para o alumínio 0,031. Assim a volatilidade relativa pode ser calculada:
𝛼 =𝛾𝐴𝑙𝑃𝐴𝑙°
𝛾𝐹𝑒𝑃𝐹𝑒°=
0,031.2,31.0,060
= 1,19

293
Através desse resultado, entende-se que pode ocorrer vaporização do
alumínio durante o processo de desgaseificação. Portanto se for desgaseificar,
deve-se adicionar um mínimo antes e completar depois, ou então não utilizar o
processo de desgaseificação e sim resfriamento controlado após a laminação
ou forjamento.
20.5 Fugacidade para Gases Reais
Para um gás não ideal a equação G = G° + RTLnP que é uma função
linear do logarítmico de P, não é mais linear. Assim para se conhecer a
variação de G em função de P para um gás real, deve-se saber a dependência
de V em relação a P, com o objetivo de que a relação dG = RTLnP possa ser
integrada e como essa característica é individual para cada gás, o resultado
seria um dG para cada gás. Para continuar com a mesma simplicidade e
assegurar a linearidade de G em função de P, foi determinado uma outra
função que substitui P, a função fugacidade f. Assim a equação da atividade
(𝑎𝑖) passa a ser representada por:
𝑎𝑖 = 𝑓𝑖𝑓° (20.31)
fi é conhecido como fugacidade. O componente i na solução na temperatura T
e fo a fugacidade do puro i (estado padrão), na temperatura T. Se o vapor
considerado for uma solução ideal,então fi = pi. Em qualquer caso.
(20.32)
(20.33)
Z = medida dos desvios para gases reais
f = medida da pressão real dos gases
A atividade de i na solução com respeito ao puro i, é representado pela
razão da pressão de vapor i exercido pela solução para a pressão de vapor do
puro i, na mesma temperatura. Se o componente i conduz idealmente na
solução, então a equação fica:
ai = Xi (20.34)
°=PPiai
∫−
+=P
dPP
ZLnPLnf0
1

294
20.5 Atividade de Fases Condensadas
Vamos considerar a reação de oxidação do cobre, onde existem um
equilíbrio entre um sólido e um gás
4(𝐶𝑢)𝑠 + (𝑂2)𝑔 → 2(𝐶𝑢2𝑂) (20.35)
De acordo com as equações: 15.10 e 19.1.
∆𝐺 = ∆𝐺° + 𝑅𝑇𝐿𝑛 � �𝑎𝐶𝑢2𝑂�2
(𝑎𝐶𝑢)4𝑃𝑂2� (20.36)
Considerando a equação no equilíbrio, a equação 20.36 fica:
∆𝐺° = −𝑅𝑇𝐿𝑛� �𝑎𝐶𝑢2𝑂�2
(𝑎𝐶𝑢)4𝑃𝑂2� (20.37)
Como o óxido de cobre é uma fase condensada, considera-se como se esta
ocorre em seu estado puro como estado padrão e portanto de atividade igual a
1, também essa condição e estendida para o cobre, assim a equação 20.37
torna-se:
∆𝐺° = −𝑅𝑇𝐿𝑛 � 1𝑃𝑂2
� (20.38)
Integrando
𝐾𝐸 = 1𝑃𝑂2
(20.39)
Essa prática é sempre realizada para quando principalmente estiver em
equilíbrio, fases sólidas e fases gasosas, onde a atividade das fases
condensadas são consideradas desprezíveis em relação as atividades das
fases gasosas.
20.7 Conceito de Desgaseificação em Função da Pressão Parcial
Quanto menor for a pressão parcial de um determinado elemento,
menor será a sua fração molar na solução de acordo com a lei de Raoult, ou
menor será sua porcentagem na solução. Pode-se aplicar essa definição, por
exemplo no processo de desgaseificação, especificamente para retirada do
hidrogênio. Quando ocorre o processo de desgaseificação, o aço está
ERTLnKGG +∆=∆

295
submetido a um local estanque (presume-se que seja o mais estanque
possível) sob uma pressão muito baixa na ordem de 1 torr ou até menos.
Dessa forma todas as pressões parciais devem entrar em equilíbrio com a
pressão total do sistema. Como a difusão do hidrogênio é maior que todos os
outros elementos, provavelmente ele será eliminado com maior facilidade pela
diferença de pressão parcial.
Seja a pressão total em um desgaseificador em equilíbrio com os
gases presentes na fase liquido do aço a 1600°C:
P = PCO2 + PN2 + PCO + PH2 + PSiO + PAlO + ..... (20.40)
Se diminuirmos a pressão total P do sistema, pode-se rearranjar as
outras pressões de maneira que também serão diminuídas. Aqueles elementos
com maior energia cinética, escaparão primeiro, que será o caso do hidrogênio.
Também pensando nesse sistema em pressão de 1 atm, uma forma de
diminuirmos a pressão parcial dos gases, é introduzir um gás inerte adicional
em pressão muito maior que os demais, por exemplo Argônio. Então a equação
(20.34), ficaria:
P = PCO2 + PN2 + PCO + PH2 + PSiO + PAlO + PAr + .... (20.41)
Assim quanto maior for a pressão do argônio menor será as outras
pressões parciais. Pensando em melhorar o processo, também é possível
diminuir a pressão total P do sistema e adicionar o Argônio (aumentando PAr),
tem-se então nesse caso uma otimização, pois estamos promovendo a retirada
de elementos cineticamente mais rápidos por dois motivos, diminuição da
pressão total do sistema por vácuo e por introdução de mais um elemento
gasoso.
O argônio também auxilia na renovação de superfície. A partir de uma
certa profundidade a pressão é muito alta devido a coluna de aço. Essa coluna
de aço, dificulta a saída do hidrogênio, devido a alta pressão exercida. Nesse
caso o argônio sendo injetado pela parte inferior da panela de aço líquido, faz o
aço circular, forçando o hidrogênio contido no metal ir para superfície e pela
diferença de pressão ser retirado.

296
Continuando com esse raciocínio, existem aços que devem ser
oxidados em ambientes a pressão de 1 atm (sem vácuo), e para melhorar a
eficácia da descarburação, sem oxidar outros elementos. É importante
decrescer a pressão parcial do CO, forçando a reação para a direita no sentido
da formação dos produtos, ou seja CO. Assim se estamos pensando em
oxidação, então pode-se adicionar mais um elemento que será o Oxigênio. A
equação (20.32), então ficaria:
P = PCO2 + PN2 + PCO + PH2 + PSiO + PAlO + PAr + PO2 + .....(20.42)
Assim acabamos de entender o processo AOD (Descarburação com
auxílio de Oxidação e borbulhamento de Argônio sem diminuição da pressão
total). Também nessa linha é o processo VOD (Nesse caso em vez de utilizar o
Argônio, utiliza-se vácuo – diminuição da pressão do sistema – com
borbulhamento de oxigênio por lança).
Como a pressão parcial é diretamente relacionada com a fração molar,
ou porcentagem do elemento na solução. Quanto menor a pressão parcial
menor será a participação de tal elemento na solução.
Mais tarde estudaremos o caso do nitrogênio, sua retirada do aço
ocorre em certas situações, melhor explicadas pelo entendimento da tensão
superficial.
20.8 Lei de Sievert
Considerando a dissolução de gases diatômicos: O2, N2 e H2 e ainda
supondo que esses gases encontram-se na forma atômica dissolvida na fase
metálica. A exemplo do hidrogênio escreve-se
𝐻2 ↔ 2𝐻 (20.43)
Assim o equilíbrio entre o hidrogênio (ou os outros gases diatômicos) e
o metal é representado pela lei de Sievert:
Onde a equação torna-se:
%𝐻 = 𝑘𝐻�𝑃𝐻2 (20.44)
kH – constante de Sievert

297
A constante do hidrogênio é representada por
𝑙𝑔𝑘𝐻 = −1670𝑇
− 1,68 (20.45)
Portanto para temperatura de 1600°C (1873 K), o valor de kH para o
hidrogênio é de kH =10-2,57. Assim através na equação de Sierverts encontra-se
o valor da pressão necessária para se obter um teor de 1 ppm de H2 no aço.
%𝐻 = 𝑘𝐻�𝑃𝐻2 → 𝑃𝐻2 = �%𝐻𝑘𝐻
�2
= �0,000110−2,57�
2
= 0,0014 𝑎𝑡𝑚
Para transformar atm em mmHg ⇒ P = 0,0014.760 = 1,05 mmHg. Essa
equação mostra que para teores de hidrogênio muito baixo, a pressão de
desgaseificação também deve ser, para o caso de 0,5 ppm de H2 a pressão
deve ser de 0,262 torr pelo menos na câmara de vácuo.
20.9 Aspectos Cinéticos da Desgaseificação para o caso do Hidrogênio
Considerando em uma interação onde a diferença de concentração é a
força driving e que o limitante é a difusão através da camada limite, podemos
considerar o seguinte modelo.
Figura 20.10 – Mostra o esquema da espessura da camada limite entre fases imissíveis
A partir da primeira lei de Fick e considerando a espessura da camada
limite como sendo a espessura entre o metal líquido e a fase gasosa em um
reator onde se tem vácuo em baixas pressões, pode-se escrever a seguinte
equação:
Ceoncentração na
fase gasosa
Fase Gasosa
Fase líquida
Concentração no
aço líquido, C

298
𝑑𝑛𝑑𝑡
= −𝐷𝑆 𝑑𝐶𝑑𝑋
= 𝐷𝑆 𝐶−𝐶𝑒𝛿
(20.46)
𝑛 =𝑛𝑉𝑉 ⟹ 𝑑𝑛 = �𝑑 �
𝑛𝑉
= 𝜌�� 𝑉 = 𝑉𝑑𝜌; 𝑐 =𝑛𝑉⟹ 𝑑𝑛 = 𝑉𝑑𝐶
𝑑𝑛𝑑𝑡
=𝑉𝑑𝐶𝑑𝑡
= −𝐷𝑆𝐶 − 𝐶𝑒𝛿
⟹𝑑𝐶
𝐶 − 𝐶𝑒= −
𝐷𝑆𝑉𝛿
𝑑𝑡
�𝑑𝐶
𝐶 − 𝐶𝑒
𝐶𝑓
𝐶𝑖= −
𝐷𝑆𝑉𝛿
� 𝑑𝑡𝑡
0
[𝐿𝑛(𝐶 − 𝐶𝑒)]𝐶𝑖𝐶𝑓 = −
𝐷𝑆𝑡𝑉𝛿
𝐿𝑛 �𝐶𝑓−𝐶𝑒𝐶𝑖−𝐶𝑒
� = −𝐷𝑆𝑡𝑉𝛿
= 𝑘 ҆𝑡 (20.47 )
Essa equação define a teoria da camada limite de Nerst e tem como
qualidade principal, a simplicidade.
Figura 20.11- Mostra o teor de hidrogênio em função do tempo de desgaseificação. Na figura da esquerda mostra a influência da turbulência e na figura da direita mostra a influência do teor inicial de hidrogênio antes da desgaseificação.
Para o caso do hidrogênio, considerando 𝐷𝐻 = 3,2. 10−3. 𝑒−1670𝑇(𝐾) ,k ҆ pode
ser definido como regime lamelar com valor aproximadamente de 19.10-2 cm/s
até 1,5.10-2 cm/s. Em caso de regime turbulento k ҆ é aproximadamente 19.10-2
299
cm/s. O hidrogênio em equilíbrio é de 1 ppm. Aqui vale a lei de Sieverts,
%𝐻 = �𝑃𝐻2
Figura 20.11 – Tempo de desgaseificação em função do tempo. Medição feita com o aparelho Hidrys.
Figura 20.12 – Constante k ҆ (min-1). Medição de hidrogênio feita através do laboratório químico.
Assim utilizando a equação 20.42:

300
𝐻𝑓 = 𝐻𝑒 + (𝐻𝑖 − 𝐻𝑒)𝑒−𝐷𝑆𝑡𝑉𝛿 (20.48)
20.10 Exercícios Resolvidos
1)Vinte gramas de um soluto são adicionados a 100g de água a 25°C. A
pressão do vapor de água pura é de 23,76 mmHg, a pressão do vapor na
solução é de 22,41 mmHg. Dados PM(H2O) = 18 g/mol
a)Calcule a massa molar do soluto, PM ?
𝑀𝑆 = 20𝑔;𝑇 = 298𝐾; 𝑀𝑆𝑉 = 100𝑔; 𝑃𝐻2𝑂0 = 23,76 𝑚𝑚𝐻𝑔;𝑃𝐻2𝑂 = 22,41 𝑚𝑚𝐻𝑔
𝑃𝐻2𝑂 = 𝑋 𝑃𝐻2𝑂0 ⇒ 22,41 = 𝑋. 23,76 ⇒ 𝑋 = 0,94
0,94 =10018
10018 + 20
𝑀𝑆
⇒ 𝑀𝑆 = 57 𝑔/𝑚𝑜𝑙
b)Qual é a massa desse soluto que se deve juntar a 100g de água para reduzir
sua pressão de vapor à metade da pressão de vapor da água pura?
Assim a nova pressão de vapor da solução será igual a 23,762
= 11,88 𝑚𝑚𝐻𝑔
𝑋 = 11,8823,76
= 0,50
0,50 =10018
10018 + 𝑀𝑆
57 ⇒ 𝑀𝑆 = 226𝑔
2)Quantas gramas de sacarose C12H22O23 devem ser dissolvidos em 90g de
água para produzir uma solução, sobre o qual a umidade relativa seja 80%?
Assuma solução ideal. PM(sacarose)= 342,3 g/mol.
Nesse caso a sacarose passa ser o soluto e o gás em equilíbrio o vapor de
água. Assim a fração molar deve ser igual a relação entre a pressão parcial do

301
vapor na solução e a pressão de vapor da água pura na mesma temperatura,
que é a umidade.
𝑋 =𝑃𝐻20𝑃𝐻2𝑂0 = 0,8
0,80 =9018
9018 + 𝑀𝑆𝑎𝑐𝑎𝑟𝑜𝑠𝑒
342,3 ⇒ 𝑀𝑆𝑎𝑐𝑎𝑟𝑜𝑠𝑒 = 427𝑔
3)Suponha que uma série de soluções preparada utilizando-se 180g de H2O
como solvente e 10g de um soluto não volátil. Qual será o abaixamento relativo
da pressão de vapor se a massa moldar do soluto for 100g/mol?
O abaixamento relativo que o problema se refere é a fração molar, pois:
𝑃 = 𝑋𝑃0
𝑃𝐴 = 𝑋𝐴𝑃𝐴0 = (1 − 𝑋𝐵)𝑃𝐴0 = 𝑃𝐴0 − 𝑋𝐵𝑃𝐴0 ⟹ 𝑃𝐴 − 𝑃𝐴0 = −𝑋𝐵𝑃𝐴0
𝑃𝐴0 − 𝑃𝐴 = 𝑋𝐵𝑃𝐴0 ⇒𝑃𝐴0 − 𝑃𝐴𝑃𝐴0
= 𝑋𝐵
𝑋𝐵 =10
10010
100 + 18018
= 0,0099
4)O benzeno e o tolueno formam soluções bem próximas da idealidade. A
300K, 𝑃𝑇0 = 32 𝑚𝑚𝐻𝑔 e 𝑃𝐵0 = 103 𝑚𝑚𝐻𝑔.
a)Uma mistura líquida é composta de 3 moles de tolueno e 2 moles de
benzeno. Se a pressão sobre a mistura a 300 K for reduzida, a que pressão se
formará o primeiro vapor?
𝑃𝑇0 = 32 𝑚𝑚𝐻𝑔 𝑒 𝑛𝑇 = 3
𝑃𝐵0 = 103 𝑚𝑚𝐻𝑔 𝑒 𝑛𝐵 = 2
𝑋𝑇 =35
= 0,60 𝑒 𝑋𝐵 =25
= 0,40
𝑃𝑇 = 𝑋𝑇𝑃𝑇0 = 0,60.32 = 19,2 𝑚𝑚𝐻𝑔 𝑒 𝑃𝐵 = 𝑋𝐵𝑃𝐵0 = 0,40.103 = 41,2 𝑚𝑚𝐻𝑔
O primeiro vapor se formará em 41,2 mmHg.

302
b)Qual a composição dos primeiros traços de vapor?
𝑋𝑇 =19,2
60,44= 0,32 𝑋𝐵 =
41,260,4
= 0,68
c)Se a pressão for reduzida ainda mais, a que pressão desaparecerá o último
traço do líquido?
Desaparecerá na pressão de 19,2 mmHg.
d)Qual será a pressão, a composição do líquido e a composição do vapor
quando 1 mol da mistura for vaporizada?(utilizar a regra da alavanca).
𝑛𝑇𝑋𝑇 = 𝑛𝐿𝑋𝐿𝑇 + 𝑛𝑉𝑋𝑉𝑇 = 𝑛𝐿(1 − 𝑋𝑉𝑇) + 𝑛𝑉𝑋𝑉𝑇
5.0,60 = 4(1 − 𝑋𝑉𝑇) + 1𝑋𝑉𝑇 ⇒ 𝑋𝑉𝑇 = 0,33
𝑃𝑇 = 𝑃𝑇0𝑋𝑉 = 32.0,33 = 10,56 𝑚𝑚𝐻𝑔
𝑛𝑇𝑋𝑏 = 𝑛𝐿𝑋𝐿𝐵 + 𝑛𝑉𝑋𝑉𝐵 = 𝑛𝐿(1 − 𝑋𝑉𝐵) + 𝑛𝑉𝑋𝑉𝐵
0.0,40 = 4(1 − 𝑋𝑉𝐵) + 1𝑋𝑉𝐵 ⇒ 𝑋𝑉𝐵 = 0,67
𝑃𝐵 = 𝑃𝐵0𝑋𝑉𝐵 = 0,67.103 = 69 𝑚𝑚𝐻𝑔
𝑃 = 𝑋𝑉𝑇𝑃𝑇0 + 𝑋𝑉𝐵𝑃𝑉0 = 10,56 + 69 = 79,56 𝑚𝑚𝐻𝑔
5)Dois líquidos A e B formam uma solução ideal. A uma determinada
temperatura, a pressão de vapor de A puro é 200 mmHg enquanto que a de B
puro é de 75 mmHg. Se o vapor sobre a mistura consistir de 50%A, qual é a
porcentagem de A no líquido?
Líquido + vapor
Vapor
Líquido
Tol
Benz

303
Y = fase vapor e X = fase líquida
𝑦𝐴 =𝑃𝐴𝑃𝑇
𝑒 𝑃𝐴 = 𝑋𝐴𝑃𝐴0
𝑦𝐴 =𝑋𝐴𝑃𝐴0
𝑋𝐴(𝑃𝐴0 − 𝑃𝐴0) + 𝑃𝐵0⇒ 0,5 =
𝑋𝐴. 200𝑋𝐴. 125 + 75
⇒ 𝑋𝐴 = 0,27
6)A temperatura de -31,2º C temos os seguintes dados:
Composto: Propano: P0 = 1200 mmHg e n-butano: P0 = 200 mmHg
a)Calcule a fração molar de propano na mistura líquida em entra em ebulição a
-31,2º C sob uma pressão de 760 mmHg?
𝑃𝑇 = 𝑋𝑃𝑃𝑃0 + 𝑋𝐵𝑃𝐵0 = 𝑋𝑃𝑃𝑃0 + (1 − 𝑋𝑃)𝑃𝐵0
760 = 𝑋𝑃. 1200 + (1 − 𝑋𝑃)200
𝑋𝑃 = 0,56
b)Calcule a fração molar de propano no vapor em equilíbrio com o líquido em
a)?
𝑦𝑃 =𝑃𝑃𝑃𝑇
=0,56.1200
760= 0,82
7)A -47º C a pressão de vapor do brometo de etila é 10 mmHg, enquanto a do
cloreto de etila é 40 mmHg. Assuma uma mistura ideal. Se existir apenas um
traço de líquido e se a fração molar do coloreto de etila no vapor for 0,80.
a)Qual será a pressão total e a fração molar do cloreto de etila no líquido?
YA=0,5
Fase gasosa
XA + XB
Fase líquida

304
𝑋𝐶𝑙 =𝑃𝐶𝑙𝑃𝐶𝑙0
𝑒 𝑦𝐶𝑙 =𝑋𝐶𝑙𝑃𝐶𝑙0
𝑋𝐶𝑙(𝑃𝐶𝑙𝑜 − 𝑃𝐵𝑜) + 𝑃𝐵𝑜
0,8 =𝑋𝐶𝑙40
𝑋𝐶𝑙30 + 10⇒ 𝑋𝐶𝑙 = 0,50
𝑋𝐶𝑙 =𝑃𝐶𝑙𝑃𝐶𝑙0
= 0,50 ⇒ 𝑃𝐶𝑙 = 0,5.40 = 20
𝑋𝐵 =𝑃𝐵𝑃𝐵𝑜
= 0,50 ⇒ 𝑃𝐵 = 0,50.10 = 5
𝑃𝑇 = 𝑃𝐵 + 𝑃𝐶𝑙 = 5 + 20 = 25 𝑚𝑚𝐻𝑔
b)Se existirem 5 moles de líquido e 3 moles de vapor presentes na mesma
pressão de a), qual a composição global do sistema?
𝑛𝐿 = 5 𝑚𝑜𝑙𝑒𝑠; 𝑛𝑉 = 3 𝑚𝑜𝑙𝑒𝑠; 𝑃𝐵 = 5 𝑚𝑚𝐻𝑔; 𝑃𝐶𝑙 = 20 𝑚𝑚𝐻𝑔; 𝑃𝑇 = 25 𝑚𝑚𝐻𝑔
X representa a porcentagem na solução líquida e Y na fase gasosa
𝑛𝐶𝑙 = 𝑋𝐶𝑙𝑛𝐿 + 𝑌𝐶𝑙𝑛𝑉 =𝑃𝐶𝑙𝑃𝐶𝑙0
𝑛𝐿 +𝑃𝐶𝑙𝑃𝑇
𝑛𝑉 = 0,5.5 + 0,8.3 = 4,9
𝑋𝐶𝑙 =𝑛𝐶𝑙
𝑛𝐿 + 𝑛𝑉=
4,98
= 0,61
8)Uma mistura gasosa de duas substâncias sob uma pressão total de 0,8 atm,
está em equilíbrio com uma solução ideal líquida. A fração molar da substância
A é 0,5 na fase vapor e 0,2 na fase líquida. Quais são as pressões de vapor
dos dois líquidos puros?
𝑃𝑇 = 0,8 𝑎𝑡𝑚; 𝑋𝐴𝐿 = 0,2 ; 𝑋𝐴𝑉 = 0,5
𝑃𝐴 = 𝑋𝐴𝑉𝑃𝑇 = 0,5.0,8 = 0,4
𝑋𝐴𝐿 =𝑃𝐴𝑃𝐴0
= 0,2 =0,4𝑃𝐴0
⇒ 𝑃𝐴0 = 2 𝑎𝑡𝑚
𝑃𝑇 = 𝑋𝐴𝑃𝐴0 + 𝑋𝐵𝑃𝐵0 ⇒ 0,8 = 0,2.2 + 0,8.𝑃𝐵0 ⇒ 𝑃𝐵0 = 0,5 𝑎𝑡𝑚
9)A composição do vapor em equilíbrio com uma solução binária ideal é
determinada pela composição do líquido. Se X1 e Y1 são as frações molares de
1 no líquido e no vapor, respectivamente, ache o valor de X1 para o qual Y1 - X1
apresenta um máximo. Qual é a pressão nessa composição?

305
𝑓 = 𝑌1 − 𝑋1 =𝑋1𝑃10
𝑃20 + (𝑃10 − 𝑃20)𝑋1− 𝑋1
Derivando e chamando a derivada de f ’, a equação fica
𝑓 , =𝑃10[𝑃20 + (𝑃10 − 𝑃20)𝑋1] − (𝑃10 − 𝑃20)𝑋1𝑃10
[𝑃20 + (𝑃10 − 𝑃20)𝑋1]2− 1 = 0
𝑃10(𝑃20 + 𝑃10𝑋1 − 𝑃20𝑋1)− (𝑃10𝑃10𝑋1 − 𝑃20𝑃10𝑋1) = (𝑃20)2 + 𝑋12(𝑃10 − 𝑃20)2 + 2𝑃20(𝑃10 − 𝑃20)𝑋1
(𝑋1)2[(𝑃10)2 + (𝑃20)2 − 2𝑃10𝑃20] + 𝑋1[2𝑃20𝑃10 − 2(𝑃20)2] + (𝑃20)2 − 𝑃10𝑃20 = 0
(𝑋1)2(𝑃10 − 𝑃20)2 + 2𝑃20𝑋1(𝑃10 − 𝑃20) − 𝑃20(𝑃10 − 𝑃20) = 0
(𝑋1)2�𝑃10 −𝑃2
0� + 2𝑃20𝑋1 − 𝑃20 = 0
𝑋1 =−2𝑃20 ± 2��𝑃10𝑃20�
2(𝑃10 − 𝑃20)
Desprezando o termo negativo
𝑋1 =−𝑃20 + �𝑃10𝑃20
(𝑃10 − 𝑃20)
Substituindo na equação da pressão total fica:
𝑃𝑇 = 𝑋1𝑃10 + 𝑋2𝑃20 = 𝑋1𝑃10 + (1 − 𝑋1)𝑃20
𝑃𝑇 = �−𝑃2
0 + �𝑃10𝑃2
0
(𝑃10 − 𝑃2
0)�𝑃10 + �1 −
−𝑃20 + �𝑃1
0𝑃20
(𝑃10 − 𝑃2
0)�𝑃20
Resolvendo encontramos o valor da pressão igual a:
𝑃𝑇 = �𝑃10𝑃2
0
10)Suponhamos que o vapor acima de uma solução ideal contenha n1 moles
de 1 e n2 moles de 2 e ocupe o volume V sob pressão P = P1 + P2. Se
definirmos 𝑉10 = 𝑅𝑇𝑃10 𝑒 𝑉20 = 𝑅𝑇
𝑃20 , mostre que a lei de Raoult implica que 𝑉 =
𝑛1𝑉10 + 𝑛2 𝑉20 .
1 = 𝑋1 + 𝑋2 =𝑃1𝑃10
+𝑃2𝑃20
=𝑃1𝑅𝑇𝑉10
+𝑃2𝑅𝑇 𝑉20
=𝑃1𝑉10
𝑅𝑇+𝑃2𝑉20
𝑅𝑇

306
1 =𝑃1𝑉10
𝑅𝑇+𝑃2𝑉20
𝑅𝑇
Multiplicando ambos os termos por V
𝑉 =𝑉𝑃1𝑉10
𝑅𝑇+𝑉𝑃2𝑉20
𝑅𝑇= 𝑛1𝑉10 + 𝑛2𝑉20
11)Mostre que enquanto a pressão de vapor numa solução linear da fração
molar de cada componente no líquido, a recíproca da pressão é uma função
linear da fração molar de qualquer um dos componentes no vapor.
𝑃1 = 𝑋1𝑃𝑇
𝑌1 =𝑃1𝑃𝑇
=𝑋1𝑃𝑇𝑃𝑇
= 𝑋1
12)Os pontos de ebulição do benzeno puro e do tolueno são 80,1º C e 110,6º C
sob 1 atm. Admitindo que as entropias de vaporização nos pontos de ebulição
são iguais a 90 J/Kmol e aplicando a eqjuação de Clausius-Clapeyron a cada
substância deduza uma expressão implícita para o ponto de ebulição da
mistura dos dois líquidos em função da fração molar, XB do benzeno.
𝑇𝐵0 = 353,1 𝐾; 𝑇𝑇0 = 383,6 𝐾; 𝑃 = 1 𝑎𝑡𝑚; ∆𝑆𝑉0𝐵 = ∆𝑆𝑉0𝑇 = ∆𝑆𝑉0 = 90𝐽𝐾𝑚𝑜𝑙
Achar a seguinte função:𝑇𝑀𝑒𝑏 = 𝑓(𝑋𝐵)
𝑃 = 𝑃𝐵 + 𝑃𝑇
𝑑𝑃𝑑𝑇
=∆𝑆∆𝑉
=∆𝑆1𝑅𝑇𝑃
=𝑃.∆𝑆𝑅𝑇
�𝑑𝑃𝑃
𝑃𝐵0
𝑃 =∆𝑆𝑅�
𝑑𝑇𝑇⇒ [𝐿𝑛𝑃]𝑃
𝑃𝐵0𝑇
𝑇𝐵0
= −∆𝑆𝑅
[𝐿𝑛𝑇]𝑇𝑇𝐵0
𝐿𝑛𝑃𝐵0 − 𝐿𝑛𝑃 = −∆𝑆𝑅
[𝐿𝑛𝑇𝐵0 − 𝐿𝑛𝑇] = −∆𝑆𝑅𝐿𝑛 �
𝑇𝐵0
𝑇�
𝐿𝑛 �𝑃𝐵0
𝑃� = −
∆𝑆𝑅𝐿𝑛 �
𝑇𝐵0
𝑇� ⇒
𝑃𝐵0
𝑃= �
𝑇𝐵0
𝑇�−∆𝑆𝑅
Então

307
𝑃𝑇0
𝑃= �
𝑇𝑇0
𝑇�−∆𝑆𝑅
𝑃 = 𝑋𝐵𝑃𝐵0 + (1 − 𝑋𝐵)𝑃𝑇0
1 = 𝑋𝐵𝑃𝐵0
𝑃+ (1 − 𝑋𝐵)
𝑃𝑇0
𝑃= 𝑋𝐵 �
𝑇𝐵0
𝑇�−∆𝑆𝑅
+ (1 − 𝑋𝐵)�𝑇𝑇0
𝑇�−∆𝑆𝑅
1 = 𝑋𝐵 �𝑇𝐵0
𝑇�−∆𝑆𝑅
+ (1 − 𝑋𝐵)�𝑇𝑇0
𝑇�−∆𝑆𝑅
b)Qual é a composição do líquido que ferve a 95º C (368 K).
1 = 𝑋𝐵 �353,1368
�−908,3
+ (1 − 𝑋𝐵) �383,6368
�−908,3
1 = 1,57𝑋𝐵 + (1 − 𝑋𝐵)0,64
𝑋𝐵 = 0,39





























