Embed Size (px)
Citation preview

Alinhamentos de sequências e
Busca de Similaridade
Ariane Machado Lima [email protected]
Escola de Artes, Ciências e Humanidades - USP

“Eu não vim para explicar,
eu vim para confundir”
Chacrinha

Alinhamentos veremos em breve
Primeiro: busca de
similaridade

Contexto
http://www.ekac.org/gene.html
http://www.fuzzco.com/news/wp-content/uploads/2007/11/genome.jpg

Contexto

Buscas por sequências (o sentido biológico)
• Busca de identidade:
– SABER o que é, onde está, etc.
• Busca de similaridade:
– INFERIR o que é

Busca de identidade
• Comparar 2 sequências para saber se:
– são iguais
– possuem uma subsequência em comum

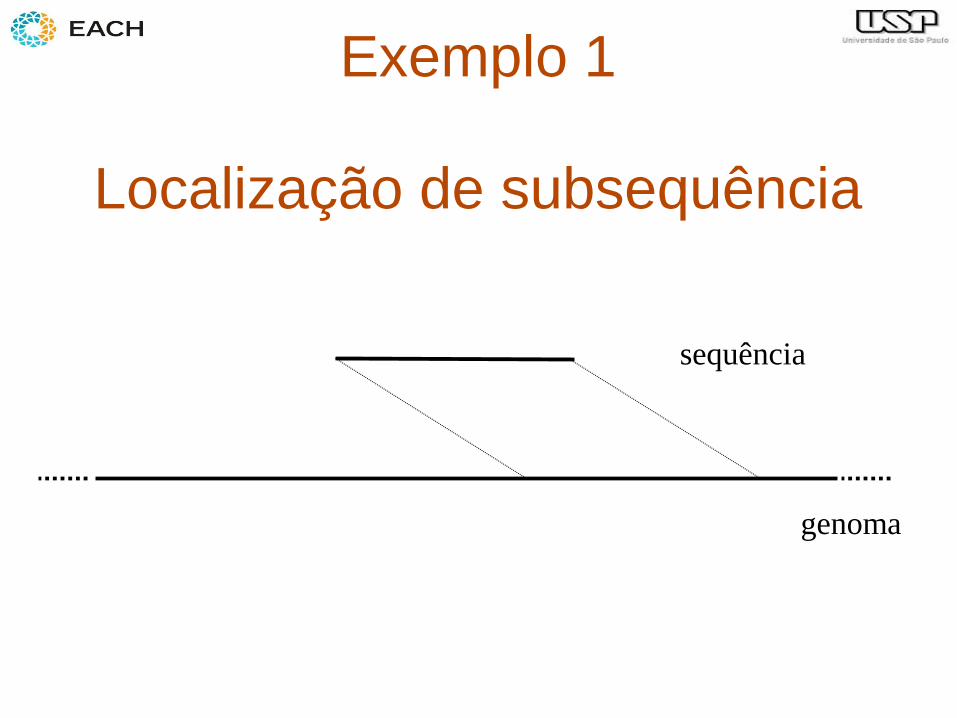
Exemplo 1
Localização de subsequência
sequência
genoma
Exemplo 1
Localização de subsequência
sequência
genoma
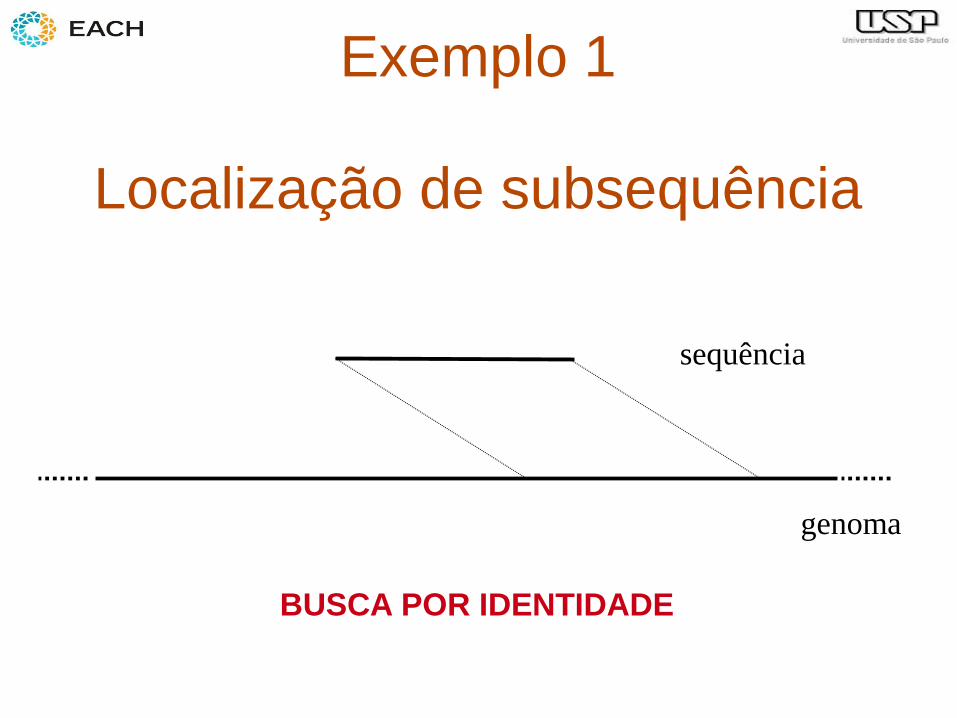
Exemplo 1
Localização de subsequência
sequência
genoma
BUSCA POR IDENTIDADE

Exemplo 2 • Como faço para saber que proteína é essa?
MMETERLVLPPPDPLDLPLRAVELGCTGHWELLNLPGAPESSLPHGLPPCAPDLQQEAEQLFLSSPAWLPLHGVEHSARKW
QRKTDPWSLLAVLGAPVPSDLQAQRHPTTGQILGYKEVLLENTNLSATTSLSLRRPPGPASQSLWGNPTRYPFWPGGMDEPTITDLNTREEAEEEIDFEKDLLTIPPGFKKGMDFAPKDCPTPAPGLLSLSCLLEPLDLGGGDEDENEAVGQPGGPRGDTVSASPCSAPLARASSLEDLVLKEASTAVSTPEAPEPPSQEQWAIPVDATSPVGDFYRLIPQPAFQWAFEPDVFQKQAILHLERHDSVFVAAHTSAGKTVVAEYAIALAQKHMTRTIYTSPIKALSNQKFRDFRNTFGDVGLLTGDVQLHPEASCLIMTTEILRSMLYSGSDVIRDLEWVIFDEVHYINDVERGVVWEEVLIMLPDHVSIILLSATVPNALEFADWIGRLKRRQIYVISTVTRPVPLEHYLFTGNSSKTQGELFLLLDSRGAFHTKGYYAAVEAKKERMSKHAQTFGAKQPTHQGGPAQDRGVYLSLLASLRTRAQLPVVVFTFSRGRCDEQASGLTSLDLTTSSEKSEIHLFLQRCLARLRGSDRQLPQVLQMSELLNRGLGVHHSGILPILKEIVEMLFSRGLVKVLFATETFAMGVNMPARTVVFDSMRKHDGSTFRDLLPGEYVQMAGRAGRRGLDPTGTVILLCKGRVPEMADLHRMMMGKPSQLQSQFRLTYTMILNLLRVDALRVEDMMKRSFSEFPSRKDSKAHEQALAELTKRLGALEEPDMTGQLVDLPEYYSWGEELTETQHMIQRRIMESVNGLKSLSAGRVVVVKNQEHHNALGVILQVSSNSTSRVFTTLVLCDKPLSQDPQDRGPATAEVPYPDDLVGFKLFLPEGPCDHTVVKLQPGDMAAITTKVLRVNGEKILEDFSKRQQPKFKKDPPLAAVTTAVQELLRLAQAHPAGPPTLDPVNDLQLKDMSVVEGGLRARKLEELIQGAQCVHSPRFPAQYLKLRERMQIQKEMERLRFLLSDQSLLLFPEYHQRVEVLRTLGYVDEAGTVKLAGRVACAMSSHELLLTELMFDNALSTLRPEEIAALLSGLVCQSPGDAGDQLPNTLKQGIERVRAVAKRIGEVQVACGLNQTVEEFVGELNFGLVEVVYEWARGMPFSELAGLSGTPEGLVVRCIQRLAEMCRSLRGAARLVGEPVLGAKMETAATLLRRDIVFAASLYTQ

Exemplo 2
• Como faço para saber que proteína é essa?
MMETERLVLPPPDPLDLPLRAVELGCTGHWELLNLPGAPESSLPHGLPPCAPDLQQEAEQLFLSSPAWLPLHGVEHSARKWQRKTDPWSLLAVLGAPVPSDLQAQRHPTTGQILGYKEVLLENTNLSATTSLSLRRPPGPASQSLWGNPTRYPFWPGGMDEPTITDLNTREEAEEEIDFEKDLLTIPPGFKKGMDFAPKDCPTPAPGLLSLSCLLEPLDLGGGDEDENEAVGQPGGPRGDTVSASPCSAPLARASSLEDLVLKEASTAVSTPEAPEPPSQEQWAIPVDATSPVGDFYRLIPQPAFQWAFEPDVFQKQAILHLERHDSVFVAAHTSAGKTVVAEYAIALAQKHMTRTIYTSPIKALSNQKFRDFRNTFGDVGLLTGDVQLHPEASCLIMTTEILRSMLYSGSDVIRDLEWVIFDEVHYINDVERGVVWEEVLIMLPDHVSIILLSATVPNALEFADWIGRLKRRQIYVISTVTRPVPLEHYLFTGNSSKTQGELFLLLDSRGAFHTKGYYAAVEAKKERMSKHAQTFGAKQPTHQGGPAQDRGVYLSLLASLRTRAQLPVVVFTFSRGRCDEQASGLTSLDLTTSSEKSEIHLFLQRCLARLRGSDRQLPQVLQMSELLNRGLGVHHSGILPILKEIVEMLFSRGLVKVLFATETFAMGVNMPARTVVFDSMRKHDGSTFRDLLPGEYVQMAGRAGRRGLDPTGTVILLCKGRVPEMADLHRMMMGKPSQLQSQFRLTYTMILNLLRVDALRVEDMMKRSFSEFPSRKDSKAHEQALAELTKRLGALEEPDMTGQLVDLPEYYSWGEELTETQHMIQRRIMESVNGLKSLSAGRVVVVKNQEHHNALGVILQVSSNSTSRVFTTLVLCDKPLSQDPQDRGPATAEVPYPDDLVGFKLFLPEGPCDHTVVKLQPGDMAAITTKVLRVNGEKILEDFSKRQQPKFKKDPPLAAVTTAVQELLRLAQAHPAGPPTLDPVNDLQLKDMSVVEGGLRARKLEELIQGAQCVHSPRFPAQYLKLRERMQIQKEMERLRFLLSDQSLLLFPEYHQRVEVLRTLGYVDEAGTVKLAGRVACAMSSHELLLTELMFDNALSTLRPEEIAALLSGLVCQSPGDAGDQLPNTLKQGIERVRAVAKRIGEVQVACGLNQTVEEFVGELNFGLVEVVYEWARGMPFSELAGLSGTPEGLVVRCIQRLAEMCRSLRGAARLVGEPVLGAKMETAATLLRRDIVFAASLYTQ
Posso procurá-la em bancos de proteínas anotadas (procuro por ela, ou seja, por uma sequência idêntica)

Exemplo 2
• Como faço para saber que proteína é essa?
MMETERLVLPPPDPLDLPLRAVELGCTGHWELLNLPGAPESSLPHGLPPCAPDLQQEAEQLFLSSPAWLPLHGVEHSARKWQRKTDPWSLLAVLGAPVPSDLQAQRHPTTGQILGYKEVLLENTNLSATTSLSLRRPPGPASQSLWGNPTRYPFWPGGMDEPTITDLNTREEAEEEIDFEKDLLTIPPGFKKGMDFAPKDCPTPAPGLLSLSCLLEPLDLGGGDEDENEAVGQPGGPRGDTVSASPCSAPLARASSLEDLVLKEASTAVSTPEAPEPPSQEQWAIPVDATSPVGDFYRLIPQPAFQWAFEPDVFQKQAILHLERHDSVFVAAHTSAGKTVVAEYAIALAQKHMTRTIYTSPIKALSNQKFRDFRNTFGDVGLLTGDVQLHPEASCLIMTTEILRSMLYSGSDVIRDLEWVIFDEVHYINDVERGVVWEEVLIMLPDHVSIILLSATVPNALEFADWIGRLKRRQIYVISTVTRPVPLEHYLFTGNSSKTQGELFLLLDSRGAFHTKGYYAAVEAKKERMSKHAQTFGAKQPTHQGGPAQDRGVYLSLLASLRTRAQLPVVVFTFSRGRCDEQASGLTSLDLTTSSEKSEIHLFLQRCLARLRGSDRQLPQVLQMSELLNRGLGVHHSGILPILKEIVEMLFSRGLVKVLFATETFAMGVNMPARTVVFDSMRKHDGSTFRDLLPGEYVQMAGRAGRRGLDPTGTVILLCKGRVPEMADLHRMMMGKPSQLQSQFRLTYTMILNLLRVDALRVEDMMKRSFSEFPSRKDSKAHEQALAELTKRLGALEEPDMTGQLVDLPEYYSWGEELTETQHMIQRRIMESVNGLKSLSAGRVVVVKNQEHHNALGVILQVSSNSTSRVFTTLVLCDKPLSQDPQDRGPATAEVPYPDDLVGFKLFLPEGPCDHTVVKLQPGDMAAITTKVLRVNGEKILEDFSKRQQPKFKKDPPLAAVTTAVQELLRLAQAHPAGPPTLDPVNDLQLKDMSVVEGGLRARKLEELIQGAQCVHSPRFPAQYLKLRERMQIQKEMERLRFLLSDQSLLLFPEYHQRVEVLRTLGYVDEAGTVKLAGRVACAMSSHELLLTELMFDNALSTLRPEEIAALLSGLVCQSPGDAGDQLPNTLKQGIERVRAVAKRIGEVQVACGLNQTVEEFVGELNFGLVEVVYEWARGMPFSELAGLSGTPEGLVVRCIQRLAEMCRSLRGAARLVGEPVLGAKMETAATLLRRDIVFAASLYTQ
Posso procurá-la em bancos de proteínas anotadas (procuro por ela, ou seja, por uma sequência idêntica)
BUSCA POR IDENTIDADE

Exemplo 3
• Como faço para saber que proteína é essa?
MMETERLVLPPPDPLDLPLRAVELGCTGHWELLNLPGAPESSLPHGLPPCAPDLQQEAEQLFLSSPAWLPLHGVEHSARKWQRKTDPWSLLAVLGAPVPSDLQAQRHPTTGQILGYKEVLLENTNLSATTSLSLRRPPGPASQSLWGNPTRYPFWPGGMDEPTITDLNTREEAEEEIDFEKDLLTIPPGFKKGMDFAPKDCPTPAPGLLSLSCLLEPLDLGGGDEDENEAVGQPGGPRGDTVSASPCSAPLARASSLEDLVLKEASTAVSTPEAPEPPSQEQWAIPVDATSPVGDFYRLIPQPAFQWAFEPDVFQKQAILHLERHDSVFVAAHTSAGKTVVAEYAIALAQKHMTRTIYTSPIKALSNQKFRDFRNTFGDVGLLTGDVQLHPEASCLIMTTEILRSMLYSGSDVIRDLEWVIFDEVHYINDVERGVVWEEVLIMLPDHVSIILLSATVPNALEFADWIGRLKRRQIYVISTVTRPVPLEHYLFTGNSSKTQGELFLLLDSRGAFHTKGYYAAVEAKKERMSKHAQTFGAKQPTHQGGPAQDRGVYLSLLASLRTRAQLPVVVFTFSRGRCDEQASGLTSLDLTTSSEKSEIHLFLQRCLARLRGSDRQLPQVLQMSELLNRGLGVHHSGILPILKEIVEMLFSRGLVKVLFATETFAMGVNMPARTVVFDSMRKHDGSTFRDLLPGEYVQMAGRAGRRGLDPTGTVILLCKGRVPEMADLHRMMMGKPSQLQSQFRLTYTMILNLLRVDALRVEDMMKRSFSEFPSRKDSKAHEQALAELTKRLGALEEPDMTGQLVDLPEYYSWGEELTETQHMIQRRIMESVNGLKSLSAGRVVVVKNQEHHNALGVILQVSSNSTSRVFTTLVLCDKPLSQDPQDRGPATAEVPYPDDLVGFKLFLPEGPCDHTVVKLQPGDMAAITTKVLRVNGEKILEDFSKRQQPKFKKDPPLAAVTTAVQELLRLAQAHPAGPPTLDPVNDLQLKDMSVVEGGLRARKLEELIQGAQCVHSPRFPAQYLKLRERMQIQKEMERLRFLLSDQSLLLFPEYHQRVEVLRTLGYVDEAGTVKLAGRVACAMSSHELLLTELMFDNALSTLRPEEIAALLSGLVCQSPGDAGDQLPNTLKQGIERVRAVAKRIGEVQVACGLNQTVEEFVGELNFGLVEVVYEWARGMPFSELAGLSGTPEGLVVRCIQRLAEMCRSLRGAARLVGEPVLGAKMETAATLLRRDIVFAASLYTQ
Posso procurá-la em bancos de proteínas anotadas (procuro por ela, ou seja, por uma sequência idêntica)
E SE EU NÃO ENCONTRASSE UMA IDÊNTICA, MAS UMA SIMILAR?

Inferência de função a partir de similaridade

Inferência de função a partir de similaridade

Inferência de função a partir de similaridade

Nem sempre funciona...

2 sequências
cacttttaactctctttccaaagtccttttcatctttccttcacagtacttgttcactat
cacttttaactctctttccaaagaacttttcatctttccctcacggtacttgtttgctat


Processo evolutivo

Homologia, paralogia e ortologia
• Homologia: 2 sequências são homólogas se elas possuem uma sequência ancestral comum
• Ortologia
• Paralogia

Ortologia: homologia por especiação
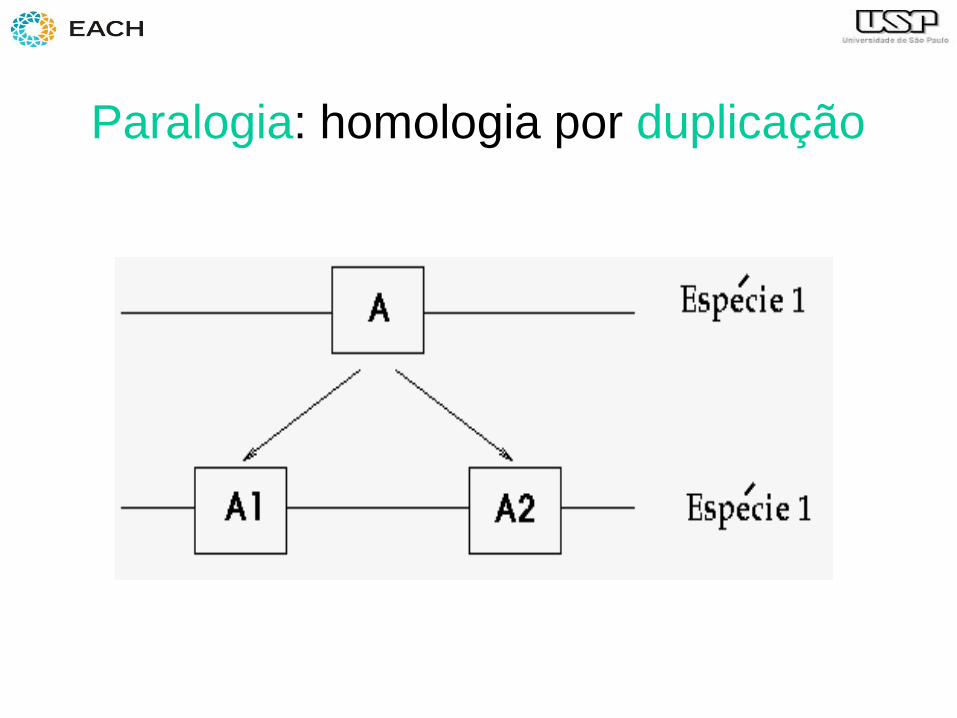
Paralogia: homologia por duplicação

Homologia, paralogia e ortologia
Paralogia
Ortologia

Aplicações de busca de similaridade
• ?

Aplicações de busca de similaridade
• Predição de genes
• Predição de estrutura
– de proteínas
– de RNA/DNA
• Inferência de árvores filogenéticas
• Busca de polimorfismos / marcadores

• CUIDADO: Se duas (ou mais) sequências são parecidas:
– elas podem ser homólogas
– elas podem ter funções similares
– elas podem ter a mesma estrutura
Identidade, similaridade e homologia

Como encontrar identidade e similaridade?

ALINHAMENTOS!
Como encontrar identidade e similaridade?

Alinhamentos de 2 sequências
• “Deixar2sequênciasomaisparecidaspossível”
ROSAVERMELHA
AMOROSOVERME
• Ajustando as posições de suas letras, se necessário usando espaços:

Alinhamentos de 2 sequências
• “Deixar2sequênciasomaisparecidaspossível”
ROSAVERMELHA
AMOROSOVERME
---ROSAVERMELHA
AMOROSOVERME---
• Ajustando as posições de suas letras, se necessário usando espaços:

• Alinhamentos permitem comparações entre as sequências
– Identidade
– Similaridade
ROSAVERMELHA
AMOROSOVERME
---ROSAVERMELHA
AMOROSOVERME---

ROSAVERMELHA
|
AMOROSOVERME
Identidade: 8% (1/12)
---ROSAVERMELHA
||| |||||
AMOROSOVERME---
Identidade: 53% (8/15)

Sistema de scores
• Pontos para match (ex: +2)
• Penalidades para mismatch (ex: -1)
• Penalidades para gap
– abertura (ex: -3)
– extensão (ex: -1)

ROSAVERMELHA
|
AMOROSOVERME
Identidade: 8% (1/12)
SCORE: ???
---ROSAVERMELHA
||| |||||
AMOROSOVERME---
Identidade: 53% (8/15)
SCORE: ???

ROSAVERMELHA
|
AMOROSOVERME
Identidade: 8% (1/12)
SCORE: -9
---ROSAVERMELHA
||| |||||
AMOROSOVERME---
Identidade: 53% (8/15)
SCORE: ???

ROSAVERMELHA
|
AMOROSOVERME
Identidade: 8% (1/12)
SCORE: -9
---ROSAVERMELHA
||| |||||
AMOROSOVERME---
Identidade: 53% (8/15)
SCORE: +3

ROSAVERMELHA
|
AMOROSOVERME
Identidade: 8% (1/12)
SCORE: -9
---ROSAVERMELHA
||| |||||
AMOROSOVERME---
Identidade: 53% (8/15)
SCORE: +3
Para um dado sistema de score, calculo o alinhamento de maior score (alinhamento ótimo)
PROBLEMA DE OTIMIZAÇÃO

Similaridade entre os aminoácidos

Identidade, similaridade e homologia
Identidade
Similaridade
Homologia
Tipo de Medida
Quantitativa
Quantitativa
QUALITATIVA
Sentido
quantos idênticos
quantos parecidos
TEM ou NÃO TEM
um ancestral comum

• Matrizes 20x20
• Algumas matrizes:
– PAMs
– BLOSUMs
Matrizes de score (matrizes de substituição de aa)

Reference: Henikoff, S. and Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89: 10915-10919.
A R N D C Q E G H I L K M F P S T W Y V B Z X *
A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4
R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4
N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4
D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4
C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4
Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4
E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4
H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4
I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4
L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4
K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4
M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4
F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4
P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4
S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4
T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4
W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4
Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4
V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4
B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4
Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4
* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1

• Matrizes 20x20
• Algumas matrizes:
– PAMs
– BLOSUMs
Matrizes de score (matrizes de substitição de aa)
Também pode usar matrizes de nucleotídeos....

• Matrizes 20x20
• Algumas matrizes:
– PAMs
– BLOSUMs
Matrizes de score (matrizes de substitição de aa)
Também pode usar matrizes de nucleotídeos....
Veremos sobre essas matrizes mais adiante....

Alinhamentos
• Pairwise: 2 sequências
• Múltiplo: mais de 2 sequências

Tipos de alinhamentos
• Global
• Semi-global
• Local

Alinhamento global
QUERIDA---ROSAVERMELHA
|||| ||| |||||
QUEROUMAMOROSOVERME---

Alinhamento global
• Aplicação: – comparar 2 proteínas (ex. para inferir
estrutura secundária)

Estrutura 3D de proteínas

Alinhamento global
• Aplicação: – comparar 2 proteínas (ex. para inferir
estrutura secundária)
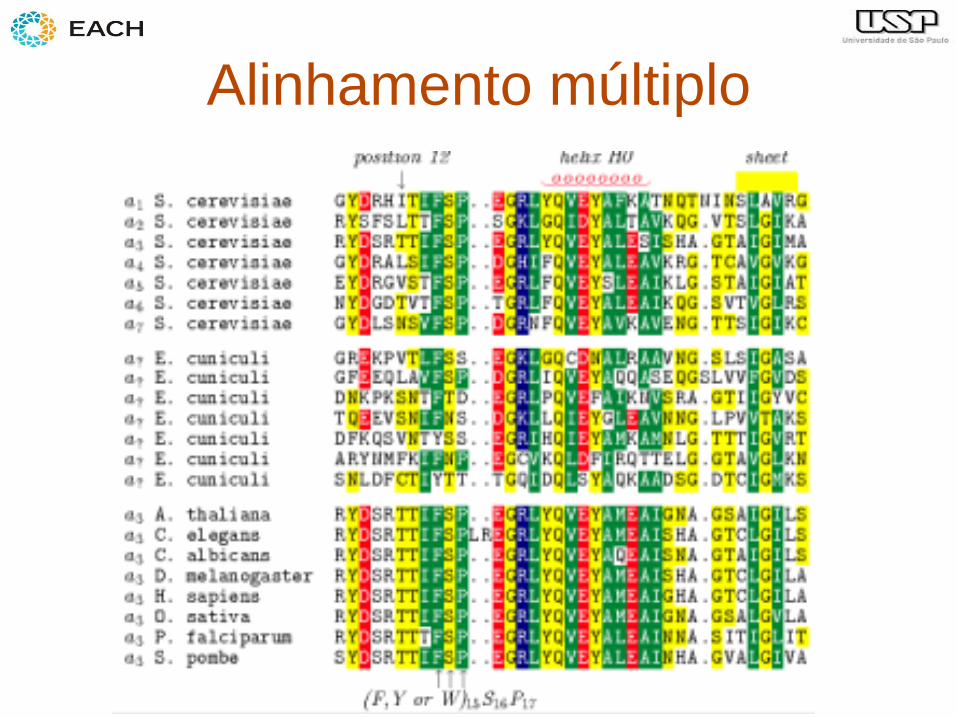
Alinhamento múltiplo

Alinhamento global
• Outras aplicações
– Identificação de SNPs (single nucleotide polimorphism) e outros polimorfismos
– Identificação de domínios proteicos mais conservados
– Identificação de isoformas
– Construção de árvores filogenéticas

Helicases humanas (SNPs)
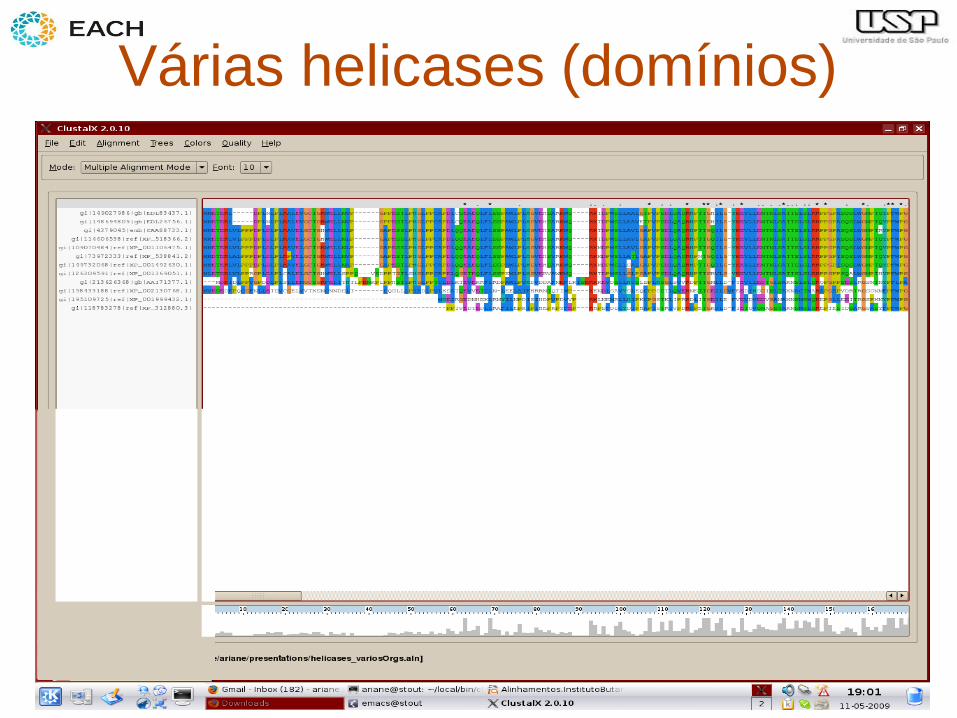
Várias helicases (domínios)

Várias helicases (domínios)
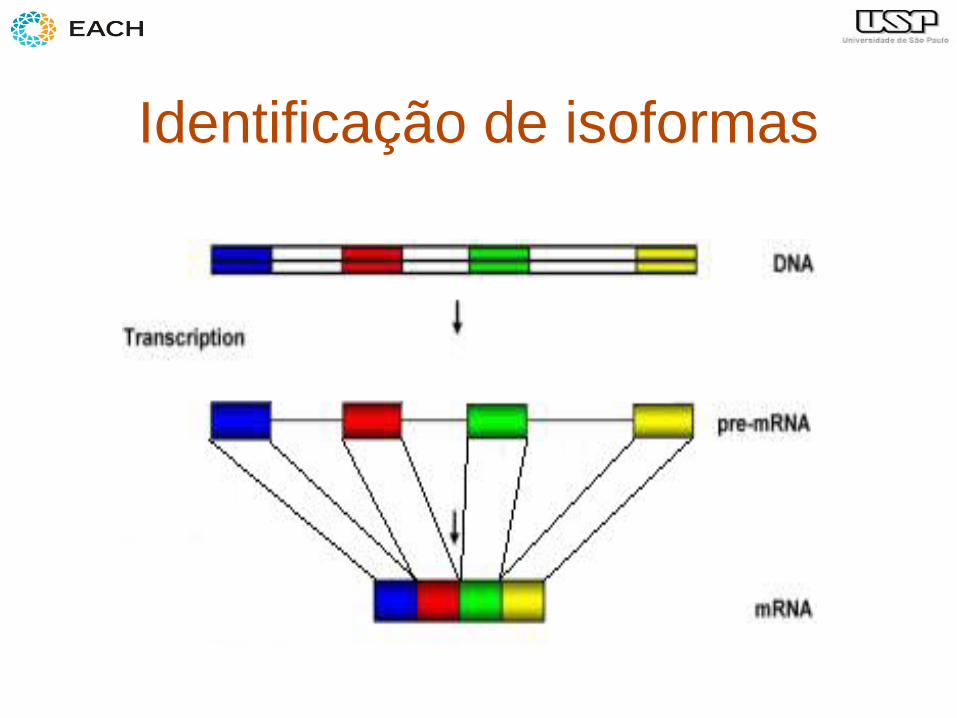
Identificação de isoformas

Identificação de isoformas

Alinhamento pairwise global
• Algoritmo Exato: Needleman-Wunsch
(pairwise)
• Programas: – needle (EMBOSS)
– stretcher (EMBOSS) (demora mais, mas economiza memória)
– FASTA

Alinhamento múltiplo (global)
• Ferramentas normalmente usadas NÃO SÃO EXATAS!
• Necessita alguma edição manual
• Parece não haver um consistentemente melhor que todos

Alinhamento múltiplo (global)
• Algumas ferramentas:
–ClustalW / ClustalX
–T-Coffee
–Muscle


Outra aplicação
• Criação de modelos e identificação de
RNAs não codificantes (ou outros
elementos) com estrutura secundária
• Ex: microRNAs
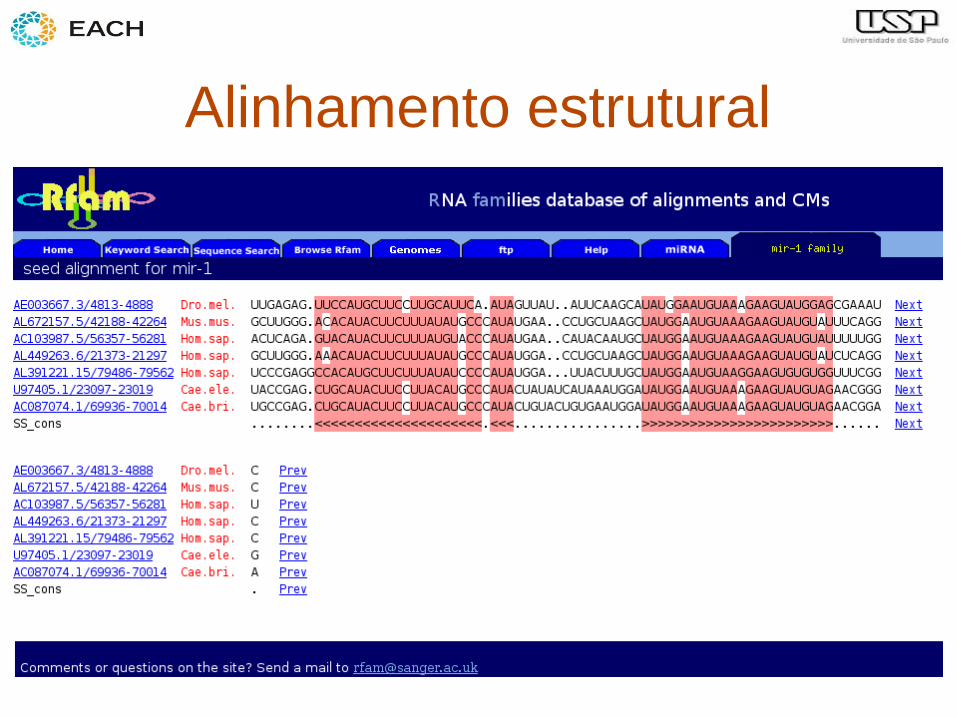
Alinhamento estrutural

Alinhamento semi-global
---ROSAVERMELHA
||| |||||
AMOROSOVERME---

Alinhamento semi-global
• Aplicação: montagem de genomas!
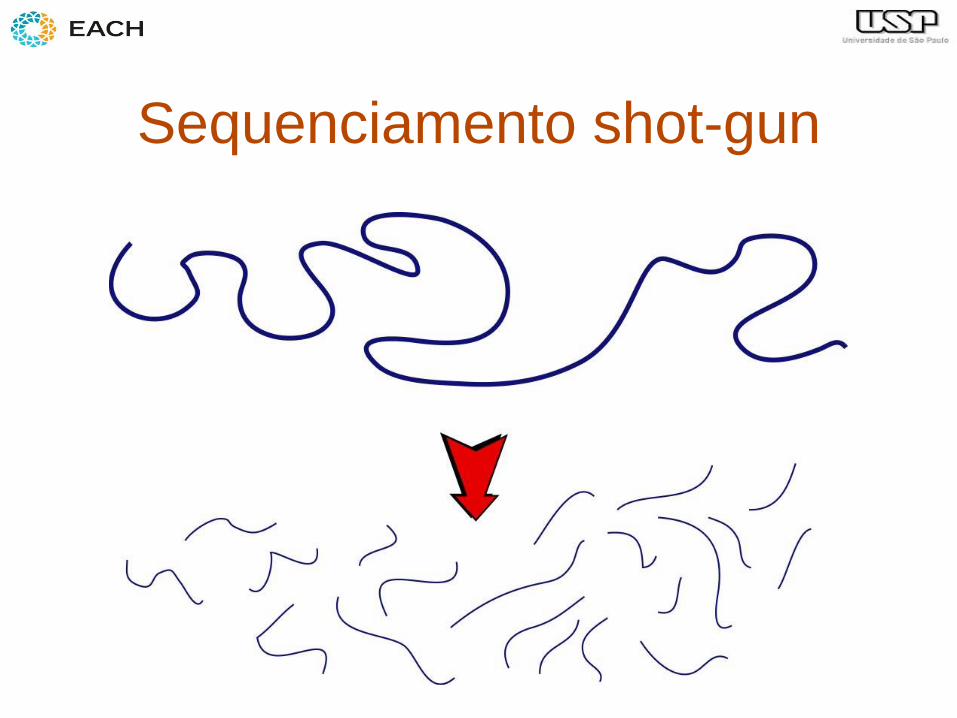
Sequenciamento shot-gun

Alinhamento semi-global
• Aplicação: montagem de genomas!

Alinhamento local
QUERIDA---ROSAVERMELHA
|||| ||| |||||
QUEROUMAMOROSOVERME---
QUER
||||
QUER
ROSAVERME
||| |||||
ROSOVERME
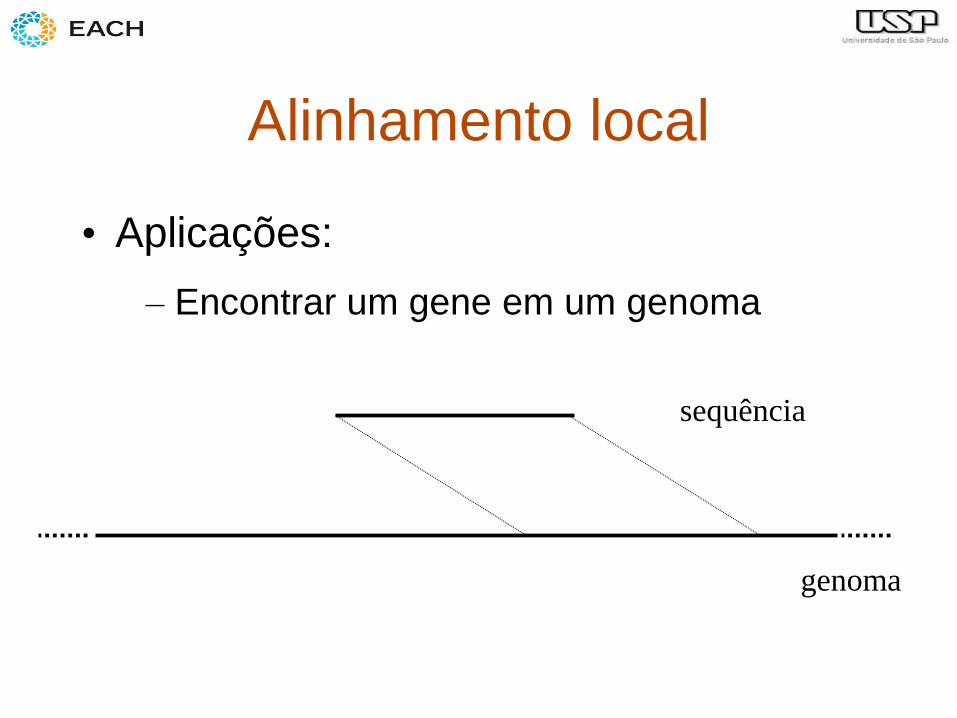
Alinhamento local
• Aplicações:
– Encontrar um gene em um genoma
sequência
genoma

Alinhamento local
• Aplicações:
– Identificar possíveis homólogos em um banco
de dados
MMETERLVLPPPDPLDLPLRAVELGCTGHWELLNLPGAPESSLPHGLPPCAPDLQQEAEQLFLSSPAWLPLHGVEHSARKWQRKTDPWSLLAVLGAPVPSDLQAQRHPTTGQILGYKEVLLENTNLSATTSLSLRRPPGPASQSLWGNPTRYPFWPGGMDEPTITDLNTREEAEEEIDFEKDLLTIPPGFKKGMDFAPKDCPTPAPGLLSLSCLLEPLDLGGGDEDENEAVGQPGGPRGDTVSASPCSAPLARASSLEDLVLKEASTAVSTPEAPEPPSQEQWAIPVDATSPVGDFYRLIPQPAFQWAFEPDVFQKQAILHLERHDSVFVAAHTSAGKTVVAEYAIALAQKHMTRTIYTSPIKALSNQKFRDFRNTFGDVGLLTGDVQLHPEASCLIMTTEILRSMLYSGSDVIRDLEWVIFDEVHYINDVERGVVWEEVLIMLPDHVSIILLSATVPNALEFADWIGRLKRRQIYVISTVTRPVPLEHYLFTGNSSKTQGELFLLLDSRGAFHTKGYYAAVEAKKERMSKHAQTFGAKQPTHQGGPAQDRGVYLSLLASLRTRAQLPVVVFTFSRGRCDEQASGLTSLDLTTSSEKSEIHLFLQRCLARLRGSDRQLPQVLQMSELLNRGLGVHHSGILPILKEIVEMLFSRGLVKVLFATETFAMGVNMPARTVVFDSMRKHDGSTFRDLLPGEYVQMAGRAGRRGLDPTGTVILLCKGRVPEMADLHRMMMGKPSQLQSQFRLTYTMILNLLRVDALRVEDMMKRSFSEFPSRKDSKAHEQALAELTKRLGALEEPDMTGQLVDLPEYYSWGEELTETQHMIQRRIMESVNGLKSLSAGRVVVVKNQEHHNALGVILQVSSNSTSRVFTTLVLCDKPLSQDPQDRGPATAEVPYPDDLVGFKLFLPEGPCDHTVVKLQPGDMAAITTKVLRVNGEKILEDFSKRQQPKFKKDPPLAAVTTAVQELLRLAQAHPAGPPTLDPVNDLQLKDMSVVEGGLRARKLEELIQGAQCVHSPRFPAQYLKLRERMQIQKEMERLRFLLSDQSLLLFPEYHQRVEVLRTLGYVDEAGTVKLAGRVACAMSSHELLLTELMFDNALSTLRPEEIAALLSGLVCQSPGDAGDQLPNTLKQGIERVRAVAKRIGEVQVACGLNQTVEEFVGELNFGLVEVVYEWARGMPFSELAGLSGTPEGLVVRCIQRLAEMCRSLRGAARLVGEPVLGAKMETAATLLRRDIVFAASLYTQ

Alinhamento Local
• Algoritmo Smith-Waterman
• Programas
– BLAST (NCBI / WU)
– BLAT (mais preciso – bom para localização)
– water (EMBOSS - exato)
– matcher (demora mais, mas economiza memória - exato)
– cross_match (swat) – bom para mascaramento
– FASTA

BLAST
Basic Local Alignment Search Tool
• NCBI BLAST ou WU-BLAST
• Heurísticas

“Palavras”doBLAST(W)
MLI LII
MLIIKRDELVISWASHERE sequência query
IIK IKR KRD RDE DEL ELV LVI VIS ISW SWA WAS ASH SHE HER ERE
todas as palavras de tamanho 3 com sobreposição

“Palavras”doBLAST(W)
• Valores default para aminoácidos e para nucleotídeos
• CUIDADO!!!!! Veja se isso não é muito para o seu caso!


Formato FASTA
>Identificador da sequência
GCCCCCGGCCCCGCCCCGGCCCCGCCCCCGGCCCCGCCCCGCAAGGGTC
ACAGGTCACGGGGCGGGGCCGAGGCGGAAGCGCCCGCAGCCCGGTACCG
GCTCCTCCTGGGCTCCCTCTAGCGCCTTCCCCCCGGCCCGACTCCGCTG
GTCAGCGCCAAGTGACTTACGCCCCCGACCTCTGAGCCCGGACCGCTAG
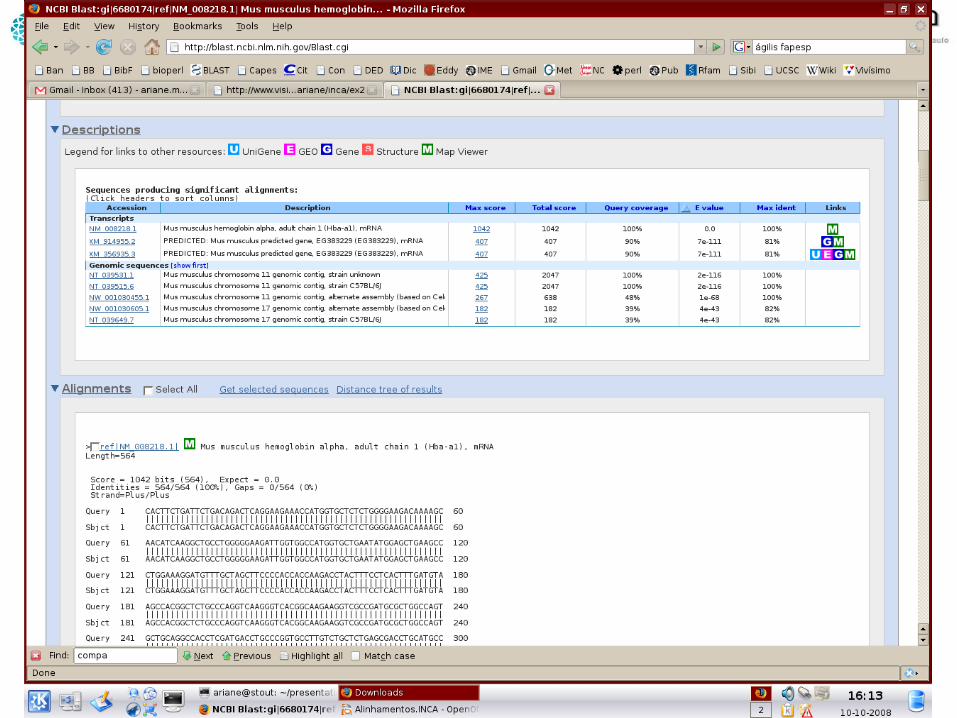

Significância de scores
• E-value (s): número de hits com score tão bom ou melhor que s puramente por chance em um banco de dados aleatório, do mesmo tamanho e com a mesma composição de bases
• Quanto menor...

Significância de scores
• E-value (s): número de hits com score tão bom ou melhor que s puramente por chance em um banco de dados aleatório, do mesmo tamanho e com a mesma composição de bases
• Quanto menor...
... melhor!!!!

Significância de scores
• P-value (s): probabilidade de obter um score tão bom ou melhor que s puramente por chance em um banco de dados aleatório, do mesmo tamanho e com a mesma composição de bases
• E-value (s): número de hits com score tão bom ou melhor que s puramente por chance em um banco de dados aleatório, do mesmo tamanho e com a mesma composição de bases

Significância de scores
• E-value é um número real não negativo
• Quanto menor...
... melhor!!!!
• E-value depende de...
E(S) = Kmne-S
... por isso não existe número mágico

Programas standalone
• Programas como Blast, BLAT e muuuuitos outros: – via web server
– standalone (linha de comando) – Perl scripts!!!!
• NCBI x WU BLAST
• netblast: linha de comando, mas executa remotamente

BLAT – Blast Like Alignment Tool
• Mais rápido e mais preciso (para sequências altamente similares)
• Aplicação: mapeamento de sequências (ex: transcritos)
• Mantém um índice de todo o banco em memória (non-overlapping k-mers)

SIM4 e outros
• Para alinhar regiões sequências em
nucleotídeos de regiões codificantes
(alinhamento de códons)

Cuidado com anotações erradas!!!
• Cuidadocombancosnão“curados”

Voltando ao sistema de score...
• Match/mismatch pode ser substituído por
– uma matriz 4x4 (nucleotídeos)
– uma matriz 20x20 (aminoácidos)
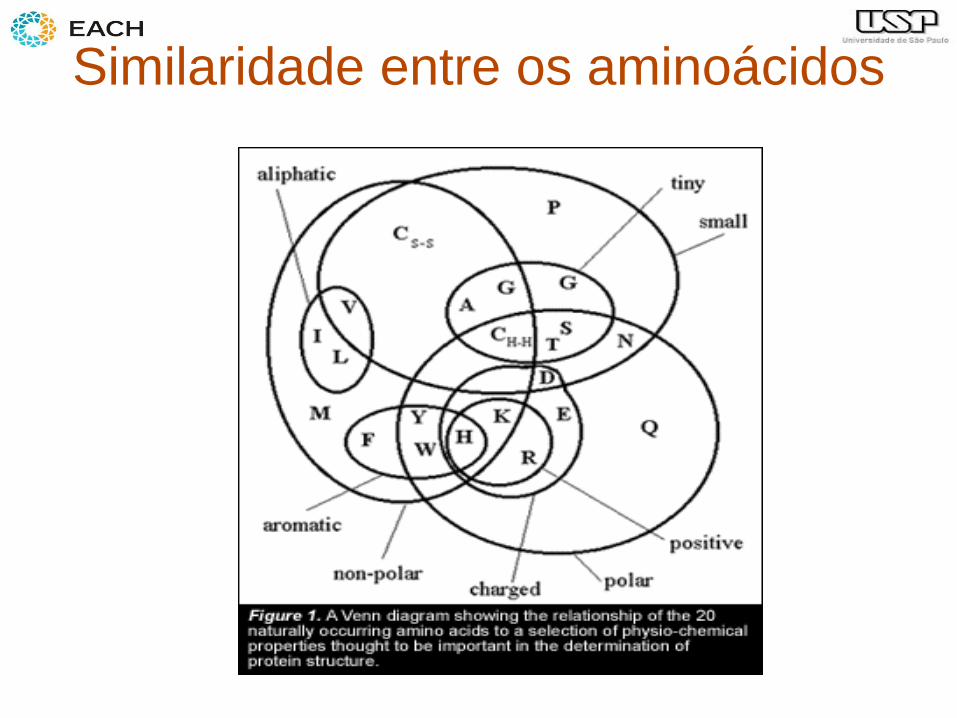
Similaridade entre os aminoácidos

Matrizes de score (matrizes de substituição)

Reference: Henikoff, S. and Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89: 10915-10919.
A R N D C Q E G H I L K M F P S T W Y V B Z X *
A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4
R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4
N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4
D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4
C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4
Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4
E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4
H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4
I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4
L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4
K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4
M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4
F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4
P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4
S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4
T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4
W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4
Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4
V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4
B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4
Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4
* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1

Matrizes de score (matrizes de substituição)
• qij: probabilidade do aminoácido i ser
substituído pelo aminoácido j
• pi: probabilidade do aminoácido i
mij = log (qij / pi pj) = mij

Matrizes de score (matrizes de substituição)
• qij: probabilidade do aminoácido i ser
substituído pelo aminoácido j
• pi: probabilidade do aminoácido i
mij = log (qij / pi pj) = mij

Matrizes de score (matrizes de substituição)
• qij: probabilidade do aminoácido i ser
substituído pelo aminoácido j
• pi: probabilidade do aminoácido i
mij = 1/ log (qij / pi pj) = mij

• Como achar qij, pi e pj?
• Algumas matrizes:
– PAMs
– BLOSUMs
Matrizes de score (matrizes de substitição)

Matrizes PAM de aminoácidos – Point Accepted Mutation
• Dayhoff, 1978
• Processo:
– Alinhamento de conjuntos de sequências relacionadas (85% id)
– Construção de árvores filogenéticas
– Cálculo da frequência de substituição de cada par de aminoácido
– Normalização das frequências: 1% de mudança ~ 50 milhões de anos (PAM1)

Matrizes PAM de aminoácidos – Point Accepted Mutation
• Em um período de 2 PAMs, pode ter havido A ?, e então ? D
• Extrapolação: PAM2 = PAM1 x PAM1
PAMy = PAM1 x PAM1 x .... x PAM1
• PAM120: 40% de identidade
• PAM250: 20% de identidade

PAM250 Diagonal
Hidrofóbicos
Hidrofílicos

Problemas das PAMs
• Inferida por um conjunto restrito de proteínas
• Extrapolação
• Muitas novas proteínas foram sequenciadas desde 78...

Matrizes BLOSUM de aminoácidos
• Henikoff & Henikoff, 1992
• Alinhamentos de blocos de vários grupos de proteínas relacionadas (banco de dados BLOCKS)
• Cálculo de frequência de substituição de cada par de aminoácido
• BLOSUMx: blocos de sequências com no máximo x% de identidade
• Ex: BLOSUM62 e BLOSUM85

BLOSUM62 Reference: Henikoff, S. and Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89: 10915-10919.
A R N D C Q E G H I L K M F P S T W Y V B Z X *
A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4
R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4
N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4
D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4
C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4
Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4
E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4
H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4
I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4
L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4
K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4
M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4
F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4
P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4
S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4
T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4
W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4
Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4
V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4
B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4
Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4
X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4
* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1

PAMs e BLOSUMs
• Para encontrar alinhamentos mais curtos e com maior similaridade:
– PAMs
– BLOSUMs
• Para encontrar alinhamentos mais longos e com menor similaridade:
– PAMs
– BLOSUMs

PAMs e BLOSUMs
• Para encontrar alinhamentos mais curtos e com maior similaridade:
– PAMs
– BLOSUMs
• Para encontrar alinhamentos mais longos e com menor similaridade:
– PAMs
– BLOSUMs
mais baixas
mais altas

PAMs e BLOSUMs
• Para encontrar alinhamentos mais curtos e com maior similaridade:
– PAMs
– BLOSUMs
• Para encontrar alinhamentos mais longos e com menor similaridade:
– PAMs
– BLOSUMs
mais baixas
mais baixas
mais altas
mais altas

Papel dos gaps
• Inserções / deleções
GLOBAL LOCAL
MUITO
ALTAS
Inibir trechos de gap →
alinhamentos ruins (muitos
mismatches)
Inibir trechos de gap → poucos
blocos alinhados
MUITO
BAIXAS
Muitos gaps espalhados pelo
alinhamento (alinhamento
ruim)
Muitos gaps espalhados pelo
alinhamento (alinhamento
ruim e possivelmente maior do
que deveria)



Referências
Caprichado (geral): Mount - http://www.bioinformaticsonline.org/
Básico: O'Reilly - http://www.oreilly.com/catalog/bioskills/
BLAST: http://www.oreilly.com/catalog/blast/
Durbin R, Eddy S, Krogh A, Mitchison G. (1998). Biological sequence analysis: probabilistic models of proteins and nucleic acids, Cambridge University Press, 1998.





























