Embed Size (px)
Citation preview

UNIVERSIDADE DE SÃO PAULO
INSTITUTO DE FÍSICA DE SÃO CARLOS
ANA LAURA DE LIMA
Caracterização estrutural da proteína spliceosomal de Trypanosoma
brucei U5-15K
São Carlos
2015


ANA LAURA DE LIMA
“Caracterização estrutural da proteína spliceossomal de Trypanosoma
brucei U5-15K”
Dissertação apresentada ao Programa de Pós-
Graduação em Física do Instituto de Física de
São Carlos da Universidade de São Paulo, para
obtenção do título de Mestrado em Ciências.
Área de concentração: Física Aplicada
Opção: Biomolecular
Orientador: Dr. Marco Túlio Alves da Silva
Versão Corrigida (Versão original disponível na Unidade que aloja o Programa)
São Carlos
2015


Aos meus pais, minha irmã e meus amigos, pelo apoio
ao longo do período de elaboração deste trabalho.


AGRADECIMENTOS
Aos meus pais, Maria Lúcia e Marcos César, e à minha irmã, Maria Cecília, pelo apoio
incondicional e por nunca me deixaram desistir, mesmo nos dias mais difíceis.
Ao Dr. Marco Túlio Alves da Silva e ao Prof. Dr. Otavio Henrique Thiemann pela orientação,
ajuda, discussões e ideias que me ajudaram a desenvolver e melhorar esse trabalho.
Ao Ivan pela ajuda com os experimentos, principalmente com a parte computacional.
Ao B2, Matheus, Nath, Paulo, Tereresa, Thomas, pelas discussões construtivas nas reuniões
de grupo e fora delas, pela amizade e por tornar o ambiente de trabalho mais agradável. Aos
ICs, Adriana, Camila, Guilherme, Jéssica e Paola por contribuir com o ambiente agradável no
laboratório e fora dele.
Aos amigos da Biomol que continuaram em São Carlos, Pan, Lok, Ila, Bibi, Fofo, Gringo e
àqueles que mudaram de curso ou cidade, mas que mesmo assim fizeram diferença, MM,
Babi, Rafael, Pikachu, Jiraya, Saia, pela amizade e pelos momentos de alívio quando o
nervosismo estava alto.
Aos professores, técnicos e alunos do Grupo de Biologia Molecular Estrutural e Cristalografia
de Proteínas, e técnicos do Grupo de Biofísica pelo apoio nos experimentos.
À Universidade de São Paulo, ao Instituto de Física de São Carlos, aos professores do IFSC-
USP, IQSC-USP, ICMC-USP, pela participação fundamental na construção do conhecimento.
Ao Grupo de Biologia Molecular Estrutural e Cristalografia de Proteínas, pela oportunidade
de realização desse trabalho.
À CNPq pela ajuda financeira.
Ao LNLS pela colaboração e disponibilização dos equipamentos para a realização desse
trabalho.
Enfim, à todos que contribuíram de alguma forma para a realização desse trabalho.


“Nada é tão poderoso quanto uma ideia cuja hora chegou”
Victor Hugo


RESUMO
DE LIMA, A. L. Caracterização estrutural da proteína spliceosomal de Trypanosoma
brucei U5-15K. 2015. 99 p. Dissertação (Mestrado em Ciências) – Instituto de Física de São
Carlos, Universidade de São Paulo, São Carlos, 2015.
A doença do sono é um dos maiores obstáculos para o desenvolvimento das áreas rurais da
África Subsaariana. O diagnóstico positivo da doença do sono, bem como o estágio em que
ela se encontra, é essencial perante a severidade da doença e toxicidade dos medicamentos
disponíveis para o tratamento. A eficiência do tratamento e diagnóstico depende do
conhecimento do ciclo de vida, biologia do parasito e seu metabolismo. Os
tripanossomatídeos possuem mecanismos conservados entre si como a expressão gênica, neste
contexto o Trypanosoma brucei pode ser considerado um organismo modelo e o estudo do
processamento de RNA mensageiro por splicing neste parasito pode ser extrapolado para
outros tripanosomatídeos. O processo de excisão dos íntrons e junção dos éxons é chamado
splicing, e tanto o cis quanto o trans-splicing são reações de transesterificação realizados pelo
spliceosomo, que consiste de 5 partículas nucleares, as snRNPs (small nuclear
ribonucleoprotein) U1, U2, U4, U5 e U6 bem como proteínas não específicas de snRNPs. As
snRNPs são complexos que consistem de pequenos RNAs ricos em uridina (U snRNAs)
unidos fortemente a proteínas. São conhecidas oito proteínas específicas de U5 snRNP em
humanos 220K, 200K, 116K, 102K, 100K, 52K, 40K e 15K. Foi mostrado que a U5-15K
humana é essencial em diversos pontos da formação do spliceosomo. O objetivo desse
trabalho foi a caracterização estrutural e da atividade de autoclivagem da proteína U5-15K de
T. brucei. Essa proteína pertencente à família Dim1e é formada por 155 resíduos de
aminoácido e massa molecular de 17,7 kDa. A proteína recombinante clonada no vetor de
expressão pET SUMO (Invitrogen) foi expressa em E. coli por indução com IPTG e
purificada por cromatografia de afinidade por íons metálico em resina de Cobalto. O produto
foi usado para testes da atividade de auto-clivagem, contendo ou não inibidores de protease. A
proteína pura também foi usada em estudos de suas propriedades em solução por
experimentos de DLS, que mostrou uma maior homogeneidade da proteína na presença de
MgCl2, DSF, que mostrou a estabilidade da U5-15K em solução com relação ao pH. As
mudanças de conformação da estrutura secundária da U5-15K e U5-15K clivada foi estudada
por experimentos de CD, que mostraram uma redução na porcentagem de folhas-β na
estrutura terciária da U5-15K clivada, e foi construído um modelo tridimensional através da

modelagem por homologia, que foi comparado com os resultados obtidos por experimentos
de SAXS. Foram realizados ensaios de cristalização em diversas condições provenientes de
kits comerciais com a proteína nas formas clivada e não clivada e em diferentes
concentrações. Como perspectivas ficam a definição exata do ponto de clivagem por
espectrometria de massas, proteólise da alça flexível para novos ensaios de cristalização e
estudo das mutações nos possíveis sítios ativos e sítio de clivagem.
Palavras-chave: Dim1. Trans-splicing. Tripanossomatídeos.

ABSTRACT
DE LIMA, A.L. Structural characterization of Trypanosoma brucei’s spliceossomal
protein U5-15K. 2015. 99 p. Dissertação (Mestrado em Ciências) – Instituto de Física de
São Carlos, Universidade de São Paulo, São Carlos, 2015.
Sleep sickness is one the most important public health problem in the Africa and it causative
agent, Trypanosma brucei, is an organism model for the study of different conserved process
among the trypanosomatids. In trypanosomes, mRNAs are processed by trans-splicing, in
which a common spliced leader sequence (SL) is acquired at the 5’ end of the mRNA to yield
a mature transcript. RNA splicing is carried out by the spliceosome, which consists of the U1,
U2, U4, U5 and U6 U snRNPs particles and non-snRNP proteins. The ribonucleoproteins are
complexes that consist of small uridine-rich RNAs (U snRNAs) and interact with common
Sm proteins and proteins that are specific for each snRNP. Seven U5 snRNP specific proteins
are known in trypanosomes, 220K, 200K, 116K, 102K, Cwc21, 40K e 15K. The spliceosomal
protein U5-15K is essential for the parasite viability and various evidences suggest
participation of the member in spliceosome assembly. U5-15K presents a conserved domain
dim1, its molecular weight is estimated of 17,7 kDa and 155 amino acids residues. In this
work, U5-15K was cloned into pET SUMO (Invitrogen), transformed in BL21(DE3)pLysS
and recombinant protein was purified by immobilized ion affinity chromatography. It was
possible observe that U5-15K undergo self-cleavage, process inhibited by serine and cysteine
protease inhibitors. Dynamic Light Scattering (DLS) experiments showed higher protein
homogeneity in the presence of MgCl2 and Differential Scanning Fluorimetry (DSF) data
demonstrated higher stability at neutral pH. Circular dichroism (CD) spectra obtained using
U5-15K native and cleaved suggest a reduction in percentage of β-sheets at cleaved U5-15K
secondary structure. Native and cleaved proteins at different concentrations were used in
crystallization trials, however, no suitable protein crystals were observed. A tridimensional
homology model for U5-15K from Trypanosoma brucei, using as template the human
homologue, present a thioredoxin folding, although it has observed a possible central loop not
present in the template. We intent to determine the exact cleavage point using mass
spectrometry and new crystallization trials will be performed after removal of probable loops
by limited proteolysis.

Keywords: Dim1. Trans-splicing. Trypanossomatids.

Lista de Figuras
Figura 1 - Mapa de risco de infecção por T. b. ganbiense (esquerda) e T. b.
rhodensiense (direita)....................................................................................
23
Figura 2 - Representação do ciclo de vida do Trypanosoma brucei.............................. 23
Figura 3 - Esquema de U1 snRNP de mamíferos. m3G: cap trimetilguanosina; B,
D1, D2, D3, E, F e G: proteínas Sm; asteriscos: proteínas específicas de
U1 snRNP .....................................................................................................
28
Figura 4 - (A)Elementos conservados da sequencia do pré-mRNA de metazoários e
leveduras. Nesse esquema N representa qualquer nucleotídeo, R uma
purina e Y uma pirimidina. (B) Modelo esquemático do cis-splicing..........
29
Figura 5 - Representação esquemática do cis (A) e trans-splicing (B)......................... 30
Figura 6 - Estrutura secundária de SL RNA. Os stem-loops estão indicados por sl I,II
e III e o ponto sombreado na extremidade 5’ do SL RNA é a estrutura do
cap4...............................................................................................................
31
Figura 7 - Modelo do core Sm (esquerda), variação do core Sm de U2 snRNP de T.
brucei (centro), variação do core Sm de U4 snRNP de T. brucei (direita)...
32
Figura 8 - Modelo de interação do core U2 snRNP de T. brucei (parte de cima) em
comparação com o de humano (parte de baixo)...........................................
34
Figura 9 - Estrutura resolvida por RMN da U5-15K humana (esquerda) e estrutura
cristalográfica (direita).................................................................................
37
Figura 10 - Diagrama esquemático do sistema TALON mostrando a ligação entres as
histidinas da cauda N-terminal e o íon Co2+ imobilizado à resina formada
por esferas de sefarose ...............................................................................
40
Figura 11 - (I) As amplitudes da luz polarizada para a esquerda e para a direita são
iguais, o que resulta em um feixe de luz linearmente polarizado.(II) A
diferença nas amplitudes da luz polarizada para a esquerda em relação a
polarizada para a direita resultam em uma polarização elíptica...................
41
Figura 12 - Espectro de CD no UV distante, onde a linha cheia representa a α-hélice,
a linha tracejada representa a folha-β e a linha pontilhada representa uma
estrutura irregular..........................................................................................
42
Figura 13 - Representação esquemática da curva de desenovelamento de uma
proteína, onde Tm (temperatura de melting) é definida como a
temperatura em que metade das proteínas em solução se encontram
desenoveladas................................................................................................
45

Figura 14 - Desenho esquemático da técnica de gota pendurada (hanging drop)
(esquerda) e gora apoiada (sitting drop) (direita).........................................
47
Figura 15 - Representação do domínio da U5-15K de T. brucei................................... 59
Figura 16 - Alinhamento entre U5-15K de T. brucei, T. cruzi, L. major U5-15K
humana (1QGV), Dim1 de Plasmodium yoelli (2AV4) e Dim1de S.
cerevisiae, gerado pelo programa ClustalW. As marcações verdes
representam hélices-α e as amarelas as fitas-β presentes nas estruturas
cristalográficas da U5-15K humana e Dim1 de P. yoelli, e preditas pelo
programa PSIPRED. As letras em vermelho representam o provável
peptídeo resultante da autoclivagem............................................................
60
Figura 17 - Gráfico de Ramachandram do modelo da U5-15K gerado pelo SWISS-
MODEL92
......................................................................................................
61
Figura 18 - Avaliação do Z-score (direita). Avaliação da energia por aminoácido
(esquerda)......................................................................................................
62
Figura 19 - Modelo da U5-15K de T. brucei gerado pelo SWISS-MODEL92
,
visualizado no PYMOL. A estrutura está colorida em arco-íris, de forma
que as cores vão do vermelho (C-terminal) ao azul (N-Terminal). Os
retângulos vermelhos mostram a alça flexível e a região do alinhamento
com a U5-15K humana em que ela se encontra....................................
62
Figura 20- Alinhamento das estruturaras da U5-15K de T. brucei (verde) e Humana
(azul), os pontos em vermelho representam as cisteínas.......................
63
Figura 21 - Diagrama do sitio de clonagem do vetor pET SUMO, mostrando a
sequencia da proteína de fusão SUMO.........................................................
64
Figura 22 - Alinhamento mostrando a ligação da U5-15K no pET SUMO................... 64
Figura 23 - Cromatograma da primeira etapa de purificação da U5-15K, o primeiro
pico corresponde às proteínas que não aderiram a coluna e o pico mais
definido, nas frações 16 a 20, corresponde ao pico de eluição da U5-15K.
A linha verde corresponde a concentração de imidazol no tampão..............
65
Figura 24 - Gel SDS-PAGE 15% da cromatografia de afinidade. (1) Marcador de
Massa Molecular, (2) fração 16, (3) fração 17, (4) fração 18, (5) fração
19, (6) fração 20............................................................................................
66
Figura 25 - Gel SDS-PAGE 15% da segunda parte da purificação. (1) Marcador de
massa molecular, (2) fração contendo SUMO protease e SUMO eluida
com 200mM de imdazol , (3)fração contendo U5-15K................................
66
Figura 26 - Cromatografia de exclusão molecular.................................................. 67
Figura 27 - Gel SDS-PAGE 15% onde 1é o marcador de massa molecular, 2 a fração
42 da cromatografia de exclusão molecular, 3 a fração 43, 4 a fração 44, 5

a fração 45, 6 a fração 46, 7 a fração 47, 8 a fração 48, 9 a fração 49...... 67
Figura 28 - Gel SDS-PAGE 15% do teste de auto-clivagem. (1) Marcador de massa
molecular, (2) amostra logo após a purificação, 0h, (3) amostra 5 dias
após a purificação, 120h................................................................................
69
Figura 29 - Geis SDS-Page 20% do teste de inibição onde (a) controle, (b) leupeptina,
(c)EDTA, (d) quimostatina, (e) PMSF e (f) DTT. Em todos eles (1)é o
marcador de massa molecular, (2) 0h, (3)12h, (4) 24h, (5) 36h, (6) 48h,
(7) 60h, (8) 72h, (9) 84h, (10) 96h, (11)108h...............................................
69
Figura 30 - Gráfico da porcentagem de U5-15K não clivada na amostra versus tempo
(horas). A curva em preto representa o controle (sem inibidor), em
vermelho, amostra contendo 0,1 mM mg leupeptina, em verde, amostra
contendo 1 mM de EDTA, em azul escuro, amostra contendo, 2 mM
PMSF, em rosa, 6 μgmL-1 mg de quimostatina e em azul claro, 2 mM de
DTT...............................................................................................................
70
Figura 31 - Representação esquemática da autoclivagem............................................... 71
Figura 32 - O gel (a) contem amostras encubadas com 2mM de DTT e o gel (b)
amostras encubadas sem DTT. Em ambos os géis (1) é o marcador de
massa molecular, (2) amostra recém purificada (0h), (3) 12h, (4)24h,
(5)36h, (6)48h...............................................................................................
72
Figura 33 - Curva do logaritmo da massa molecular (logMM) pelo logaritmo da
distância percorrida pela banda no gel pelo tamanho do gel (log (d/D)). O
ponto vermelho indica a U5-15K clivada, cuja massa foi calculada a partir
da equação da reta..............................................................................
72
Figura 34 - Representação po ponto de clivagem (vermelho) na estrutura da U5-15K
de T. brucei (verde) alinhada com a estrutura da U5-15K de H. sapiens
(azul).................................................................................................
73
Figura 35 - (Preto) Espectro de Dicroísmo Circular (CD) no UV distante da U5-15K
de T. brucei.(Vermelho) Espectro de Dicroísmo Circular (CD) no UV
distante da U5-15K de T. brucei clivada.............................................
74
Figura 36 - (a) Distribuição de tamanho da amostra de U5-15K. (b) Distribuição de
tamanho da amostra de U5-15K com MgCl2...............................................
75
Figura 37 - O gráfico da esquerda representa os dados experimentais de SAXS da U5-
15K de T. brucei. O gráfico da direita é a junção dos dados experimentais
pelo programa PRIMUS101
...........................................................................
76
Figura 38 - Curva de espalhamento de raio X a baixo ângulo para a U5-15K de T.
brucei. A linha preta representa os dados experimentais e a vermelha a
curva extrapolada para q = 0.........................................................................
77
Figura 39 - Gráfico de Guinier......................................................................................... 77

Figura 40 - Distribuição de distâncias, p(r)..................................................................... 78
Figura 41 - Comparação da curva de espalhamento experimental com a curva teórica
gerada para o modelo construído com o SWISS-MODEL92
........................
78
Figura 42 - Média dos envelopes gerada pelo programa DAMAVER, alinhado pelo
programa SUPCOMB a estrutura tridimensional da U5-15K construído
por modelagem por homologia.....................................................................
79
Figura 43 - Gráfico da temperatura de melting pela concentração de NaCl.................... 80
Figura 44 - Grafico de DSF de ΔTm vs pH. O ΔTm é calculado subtraindo a Tm da
proteína em tampão HEPES 10mM pH7,5 da Tm de cada uma das
condições de pH. Os valores negativos de ΔTm indicam uma redução na
Tm.................................................................................................................
81
Figura 45 - Grafico de DSF de ΔTm vs sal. O ΔTm é calculado subtraindo a Tm da
proteína em tampão HEPES 10mM pH7,5 da Tm de cada uma das
soluções salinas. Os valores negativos de ΔTm indicam uma redução na
Tm.................................................................................................................
82
Figura 46 - Fotos dos cristais gerados nas condições descritas na tabela 6. A
numeração das fotos corresponde à numeração da tabela.............................
83

Lista de Tabelas
Tabela 1 - Composição de aminoácidos na sequência da U5-15K em porcentagem..... 59
Tabela 2 - Candidatos a inibidores da auto-clivagem e suas concentrações.................. 69
Tabela 3 - Deconvolução do espectro experimental dado em porcentagem de
estrutura secundária comparado com o predito (PSIPRED).........................
74
Tabela 4 - Deconvolução do espectro experimental dado em porcentagem de
estrutura secundária comparado com o predito (PSIPRED).........................
75
Tabela 5 - Relação de sais usados no experimento de DSF........................................... 81
Tabela 6 - Contições de cristalização do kit Morpheus HT (Molecular Dimensions)
que resultaram no crescimento de cristais.....................................................
83


Lista de Abreviaturas e Siglas
BPS Branch Point Sequence
CaCl2 cloreto de cálcio
CATT Card Agglutination Trypanosomiasis Test
CD Dicroísmo circular, em inglês Circular Dichroism
Co Cobalto
DLS Espalhamento dinâmico de luz, em ingês Dinamic Light Scattering
DO Densidade Óptica
DSF
Fluorimetria diferencial de varredura, em inglês Differential Scanning
Fluorimetry
DTT Ditiotreitol
EDTA Ácido rtilenodiamino tetra-acético
GMP Guanosina monofosfato
GTP Guanosina trifosfato
HAT
Tripanossomíase Africana Humana, em inglês Human African
Trypanosomiasis
IPTG Isopropil-β-D-tiogalactopiranoside
KCl Cloreto de potássio
KOAc Acetato de potássio
LB Luria-Bertani
LCR Líquido cerebrorraquidiano
m7G 7-metilguanosina
MgSO4 Sulfato de magnésio
MnCl Cloreto de manganês
MOPS Ácido 3-(n-morfolino) propanosulfônico
mRNA RNA mensageiro
NaCl Cloreto de sódio
Nt Nucleotídeos
NTC Nineteen complex
OMS Organização Mundial da Saúde
ORF Open Reading Frame
PCR Reação em cadeia da polimerase, em inglês polymerase chain reaction
PEG Polietilenoglicol
PMSF Fluoreto de fenilmetilsulfonil
RMN Ressonância Magnética Nuclear
RNA Ácido ribonucleico, em inglês ribonucleic acid
RNAi RNA de interferência
rpm Rotações por minuto
SAXS
Espalhamento de raio X em baixo ângulo, em inglês small angle x-ray
scattering
SDS Dodecilsulfato de sódio
SDS-
PAGE Gel de Poliacrilamida com SDS

SMN Survival of Motor Neuron
SL Sliced leader
snRNP Small nuclear ribonucleoprotein
SS Splice site
SUMO Small ubiquitin-like modifier
TAP
Purificação por afinidade em tandem, em inglês tandem affinity
purification
TRIS Tris(hidroximetil)aminometano
WHO World Health Organization

Sumário
1 Introdução ........................................................................................................................... 23
1.1 Tripanossomíase Africana Humana (Doença do Sono) ............................................... 23
1.2 Ciclo do Trypanosoma brucei ........................................................................................... 24
1.3 Diagnóstico e Tratamento ............................................................................................... 25
1.4 Processamento do RNA mensageiro (mRNA) .............................................................. 27
1.5 Cis e Trans-splicing .......................................................................................................... 28
1.6 O SL (spliced leader) RNA .............................................................................................. 31
1.7 Ribonucleoproteínas (snRNPs) e snRNAs ...................................................................... 32
1.8 Proteínas específicas de snRNPs ..................................................................................... 33
1.9 U5-15k ................................................................................................................................ 35
1.10 Técnicas empregadas ...................................................................................................... 39
1.10.1 Modelagem por homologia ......................................................................................... 39
1.10.2 Cromatografia de afinidade ........................................................................................ 39
1.10.3 Cromatografia de exclusão molecular ....................................................................... 40
1.10.4 Dicroísmo circular ....................................................................................................... 40
1.10.5 Espalhamento de luz dinâmico ................................................................................... 42
1.10.6 Espalhamento de raio X a baixo ângulo .................................................................... 44
1.10.7 Fluorimetria diferencial de varredura ...................................................................... 45
1.10.8 Ensaios de cristalização ............................................................................................... 46
2 Objetivos específicos ......................................................................................................... 49
3 Materiais e Métodos ......................................................................................................... 51
3.1 Análise bioinformática da sequência de aminoácidos da proteína U5-15k ................. 51
3.2 Modelagem da estrutura terciária .................................................................................. 51
3.3 Obtenção do plasmídeo .................................................................................................... 51
3.4 Cultura de Bactérias ......................................................................................................... 51
3.4.1 Preparação das bactérias competentes ........................................................................ 51
3.4.2 Reação de transformação .............................................................................................. 52
3.5 Expressão da proteína recombinante ............................................................................. 52
3.5.1 Expressão da U5-15K .................................................................................................... 52
3.5.2 Expressão da SUMO protease ...................................................................................... 53
3.6 Purificação da proteína recombinante ........................................................................... 53
3.6.1 Purificação da U5-15k ................................................................................................... 53

3.6.2 Cromatografia por exclusão de tamanho .................................................................... 54
3.6.3 Purificação da SUMO protease ................................................................................... 55
3.7 Caracterização da auto-clivagem ................................................................................... 55
3.7.1 Teste de auto clivagem .................................................................................................. 55
3.7.2 Inibição da auto clivagem ............................................................................................. 55
3.7.3 Caracterização do ponto de clivagem .......................................................................... 56
3.8 Caracterização biofísica da U5-15k ................................................................................ 56
3.8.1 Dicroísmo circular (CD) ............................................................................................... 56
3.8.2 Espalhamento de luz dinâmico (DLS) ......................................................................... 57
3.8.3 Espalhamento de raio X a baixo ângulo (SAXS) ........................................................ 57
3.8.4 Fluorimetria diferencial de varredura (DSF) ............................................................. 57
3.8.5 Ensaios de Cristalização ............................................................................................... 58
4 Resultados e Discussão........................................................................................................ 59
4.1 Análise bioinformática da sequência de aminoácidos da proteína U5-15K ............... 59
4.2 Modelagem por homologia da U5-15K de T. brucei ..................................................... 61
4.3 Obtenção da proteína U5-15K de T. brucei ................................................................... 63
4.4 Caracterização da auto-clivagem ................................................................................... 67
4.4.1 Teste de auto-clivagem.................................................................................................. 67
4.4.2 Teste de Inibição da auto-clivagem ............................................................................. 68
4.4.3 Caracterização do ponto de clivagem .......................................................................... 71
4.5 Caracterização biofísica da U5-15K de T. brucei .......................................................... 73
4.5.1 Dicroísmo Circular ....................................................................................................... 73
4.5.2 Espalhamento de luz dinâmico (DLS) ......................................................................... 75
4.5.3 Espalhamento de raio X a baixo ângulo (SAXS) ....................................................... 76
4.5.4 Fluorimetria diferencial de varredura (DSF) ............................................................. 79
4.5.5 Ensaios de Cristalização ............................................................................................... 83
5 Conclusões............................................................................................................................ 85
6 Perspectivas ......................................................................................................................... 87
Referências .............................................................................................................................. 89

23
1 Introdução
1.1 Tripanossomíase Africana Humana (Doença do Sono)
A tripanossomíase africana humana (sigla em inglês HAT), também conhecida como
doença do sono, é fatal se não for tratada e, apesar de alguns casos terem sido reportados em
áreas urbanas e periurbanas, afeta principalmente a área rural da África subsaariana, onde o
ambiente é propício para o desenvolvimento do inseto vetor1-2
. Existem duas formas da
doença, uma mais comum, causada pelo Trypanosoma brucei gambiense e encontrada nas
partes oeste e central da África, e uma menos comum, causada pelo Trypanosoma brucei
rhodensiense comum nas regiões sul e leste do continente. A forma da doença causada pelo T.
b. gambiense é crônica e pode permanecer assintomática por meses ou até anos enquanto que
o T. b. rhodensiense é responsável pela forma mais agressiva3.
Figura1- Mapa de risco de infecção por T. b. gambiense (esquerda) e T. b. rhodensiense
(direita).
Fonte: Adaptada de SIMARRO et al4.
O T. brucei, protozoário pertencente à família Trypanisomatidae e gênero
Trypanosoma, é transmitido de um mamífero para outro pela mosca hematófaga Tsetse do

24
gênero Glossina. Tanto o macho quanto a fêmea se alimentam de sangue e, portanto, podem
transmitir o parasito5-6
.
A doença do sono, juntamente com a forma animal da tripanossomíase africana
(Nagana), é um dos maiores obstáculos para o desenvolvimento das áreas rurais da África
Subsaariana, tanto pela redução da mão de obra, decorrente da infecção em humanos, quanto
pela redução da disponibilidade de leite, carne e força animal, que por sua vez minimiza a
produção agrícola3. Acreditando que a doença do sono era responsável pela limitação do
desenvolvimento da região, logo que o agente infeccioso e seu modo de transmissão foram
descobertos, as autoridades coloniais estabeleceram operações de controle7.
Na década de 1960, 60 anos mais tarde, a disseminação da doença do sono foi quase
interrompida em toda a área endêmica, mostrando que a erradicação da tripanossomíase era
possível. A raridade de casos levou a uma perda de interesse na manutenção da vigilância e o
risco do reaparecimento da doença foi negligenciado, esse desinteresse resultou em um
crescimento no número de casos na década de 1980 e na década de 1990 alguns casos
apareceram fora das áreas endêmicas. No fim do século 20, 30000 casos eram reportados
anualmente8-9
.
Desde o pico na disseminação da doença do sono em 1998, quando mais de 37 000
novos casos foram reportados no continente, uma aliança entre governos africanos e
internacionais, liderados pela Organização Mundial da Saúde (OMS ou WHO em inglês)
criaram estratégias visando à eliminação da doença. Como resultado o número de casos caiu
em 82,3% em 2011 e alguns países onde a doença do sono era endêmica deixaram de reportar
novos casos e em 2013 o número de novos casos caiu para 62288, 10
.
Entre 2000 e 2009, 30 dos 36 países listados como endêmicos receberam suporte da
OMS na forma de suporte técnico, acesso a diagnóstico, incluindo equipamentos, reagentes e
suporte financeiro para os centros de pesquisa nacionais, treinamento e acesso a tratamento7.
1.2 Ciclo do Trypanosoma brucei
A transmissão da tripanossomíase africana é feita durante a picada, quando o vetor
contaminado injeta as formas tripomastigota metacíclicas no sangue do hospedeiro mamífero
(1). Essas formas então se diferenciam em tripomastigotas sanguíneas (2), que se multiplicam
na corrente sanguínea e se espalham pelo corpo, podendo atingir o sistema nervoso central, o
que caracteriza o estágio avançado da doença. Essas formas podem então ser ingeridas por
outra mosca (4). No vetor, as formas tripomastigotas sanguíneas se diferenciam em

25
procíclicas (6) e em seguida em epimastigotas (7). A forma epimastigota se multiplica nas
glândulas salivares e se diferencia em tripomastigota metacíclica (8). Essas formas serão
injetadas em um novo hospedeiro dando continuidade ao ciclo121
.
Figura 2- Representação do ciclo de vida do Trypanosoma brucei.
Fonte: Adaptado de KENNY, P. G. E.121
1.3 Diagnóstico e Tratamento
O diagnóstico positivo da doença do sono, bem como o estágio em que ela se
encontra, é essencial perante a severidade da doença e toxicidade dos medicamentos
disponíveis para o tratamento. No caso da forma da doença causada pelo T. b. rhodensiense, o
parasito pode ser identificado no sangue retirado de vasos superficiais, devido à alta
parasitemia11
. Já no caso do T. b. gambiense, a presença do tripanossoma no sangue é cíclica,
o que dificulta o diagnóstico 12
.
Na década de 1970, o CATT (Card Agglutination Trypanosomiasis Test) foi
estabelecido para a triagem sorológica da população com risco de contrair a forma gambiense
da doença do sono. Entretanto há limitações para esse método, incluindo sensibilidade

26
limitada e alta frequência de diagnósticos errados13
. Por esse motivo, formas alternativas de
diagnóstico vêm sendo desenvolvidas. Um exemplo é o uso da técnica de PCR (Reação em
Cadeia da Polimerase) que possui alta sensibilidade e especificidade. Entretanto, a técnica
geralmente não é viável no campo14
.
O método mais utilizado para diagnóstico da doença do sono em estágio avançado é a
presença de tripanossomas no líquido cerebrorraquidiano (LCR) ou a contagem de glóbulos
brancos superior a 5 células por μL de sangue. Entretanto, ainda há discussões sobre qual é o
número adequado de glóbulos brancos15
.
Como consequência de 50 anos de investimentos insuficientes, tantos por parte dos
governos africanos quanto pela indústria farmacêutica, na pesquisa e desenvolvimento de
novos fármacos, o tratamento da doença do sono tem sido insatisfatório. As quatro
substâncias usadas como tratamento não estão disponíveis para administração oral e muitas
vezes são tóxicas e ineficientes 16
.
Para os estágios iniciais da doença do sono causada pelo T. b. gambiense, o tratamento
de primeira linha consiste de injeções intramusculares de pentamidina por uma semana. A
pentamidina está em uso desde 1940 e os efeitos colaterais mais frequentes envolvem dores
locais, inchaço, dores abdominais, problemas gastrointestinais e hipoglicemia12
. Para o
estágio inicial da doença causada pelo T. b. rhodesiense o medicamento usado desde a década
de 1920 é a suramina. Essa substância é administrada de forma intravenosa e o tratamento
dura cerca de 30 dias. Os efeitos colaterais são frequentes, porém brandos e reversíveis12
.
Para o estágio avançado da infecção, quando o parasito ultrapassou a barreira
hematoencefálica e invadiu o sistema nervoso central, o tratamento é mais complicado devido
a maior toxicidade dos medicamentos disponíveis. O único tratamento efetivo contra o T. b.
rhodesiense é o melarsoprol, um composto orgânico de arsênio, usado pela primeira vez em
1949. O tratamento por si só é fatal em 5% dos casos 17
. Em 1981 a eflornitina começou a ser
usada contra o estágio avançado da doença do sono causada por T. b. gambiense, estudos
mostraram que essa molécula possui uma taxa de mortalidade inferior a do melarsoprol apesar
de ainda apresentar efeitos colaterais como anemia, leucopenia, trombocitopenia, problemas
gastrointestinais e, com menor frequência, convulsões. O tratamento possui duração de duas
semanas e, devido a meia-vida curta da molécula, são necessárias 4 infusões por dia. Esse
medicamento só se tornou disponível para a maior parte dos pacientes em 2001, graças à
parceria entre a OMS e companhias farmacêuticas 18-19
.
Recentemente os esforços se voltaram para a otimização da terapia utilizando os
medicamentos já existentes, bem como desenvolvimento de formas de diagnóstico confiáveis

27
e economicamente viáveis, que possam identificar o estágio em que a doença se encontra. A
eficiência do tratamento e diagnóstico depende do conhecimento do ciclo de vida,
metabolismo e biologia do parasito. Os tripanossomatídeos possuem mecanismos conservados
entre si como a expressão gênica, neste contexto o Trypanosoma brucei pode ser considerado
um organismo modelo e o estudo do processamento de RNA mensageiro por splicing neste
parasito pode ser extrapolado para outros tripanosomatídeos125
.
1.4 Processamento do RNA mensageiro (mRNA)
A maioria dos genes eucarióticos apresenta uma estrutura interrompida que alterna
íntrons e éxons. Após a transcrição, os íntros devem ser removidos do transcrito primário
(pré-mRNA) para gerar o mRNA maduro. O processo de excisão dos íntrons e junção dos
éxons é chamado splicing. Quando os éxons unidos fazem parte do mesmo pré-mRNA o
processo é chamado cis-splicing 20
. Uma variação importante desse processo ocorre quando os
éxons são provenientes de moléculas diferentes, esse caso recebe o nome de trans-splicing 21
.
O trans-splincing ocorre em diversos organismos incluindo namátodas 22
,
euglenóides23
, tremátodas24
, dinoflagelados25
, e cordatos26
. Apesar de ser encontrado em
células de mamíferos, o trans-splicing não é essencial, o que o torna um potencial alvo
quimioterápico27
.
Esse processo foi descoberto em 1982 quando Boothroyd e Cross observaram que
diferentes glicoproteínas variantes de superfície de T. brucei possuíam uma sequência comum
de 39 nucleotídeos (nt), denominada spliced leader (SL), sequências similares também foram
encontradas na extremidade 5’ de outros tripanossomatideos, o que mostrou que esse
fenômeno era comum nesses organismos28-29
. Mais tarde, observou-se que o trans-splicing se
estendia para todos os mRNAs de tripanossoma, com exceção da poli(A) polimerase (PAP),
cujo gene é interrompido por sequências intervenientes, estabelecendo que o cis-splicing
também está presente nos tripanossomatideos30-31.
Atualmente sabe-se que além do transcrito
de PAP, o gene de uma helicase putativa também e processado por cis-splicing31
.
Tanto o cis quanto o trans-splicing são reações de transesterificação realizados pelo
spliceosomo, que consiste de 5 partículas nucleares, as snRNPs (small nuclear
ribonucleoprotein) U1, U2, U4, U5 e U6, bem como proteínas não específicas de snRNPs. As
snRNPs são complexos que consistem de pequenos RNAs ricos em uridina (U snRNAs)
unidos fortemente a proteínas. Os snRNAs apresentam diferentes tamanhos e formas,
podendo ser muito abundantes (cerca de 107 cópias por célula de mamífero) e altamente

28
conservadas de leveduras ao homem. Além disso, esses pequenos RNAs possuem um sítio
Sm rico em uridina (AAUUUUUGA) composto por duas regiões conservadas chamadas de
Sm1 e Sm2, separadas por uma região não-conservada32-33
. As proteínas que compõem as
snRNPs podem ser agrupadas em proteínas específicas de snRNP e proteínas comuns, essas
são compostas de sete polipeptídeos chamados proteínas Sm (SmE, SmF, SmG, SmD1,
SmD2, SmD3 e SmB) que formam um anel ao redor dos sítios Sm dos snRNAs34
.
Figura 3 - Esquema de U1 snRNP de mamíferos. m3G: cap trimetilguanosina; B, D1, D2, D3, E, F e G:
proteínas Sm; asteriscos: proteínas específicas de U1 snRNP.
Fonte: Adaptada de WILL; LURHMANN45
1.5 Cis e Trans-splicing
O splicing do pré-mRNA envolve duas reações de transesterificação de tipo SN2 e
grupos funcionais de três regiões reativas do mRNA. Primeiro, a ligação fosfodiéster do 5’
splice site (SS) é atacada pela hidroxila 2’ de uma adenosina da sequência do branch point
(BPS) do íntron. No cis-splicing, essa reação gera um éxon com a extremidade 3’ livre e um
íntron em forma de laço (lariat) ligado a extremidade 3’ do éxon adjacente.
Subsequentemente, a hidroxila 3’ da extremidade 3’ do éxon livre ataca a ligação fosfodiéster
no 3’SS, o que leva à ligação dos dois éxons e excisão do íntron35
.
O cis-splicing tem início com a formação do complexo A, resultado da interação entre
U1 e U2 snRNP por pareamento dos snRNAs com a região 5’ splice site do íntron e com a
BPS, respectivamente. Ao complexo A é adicionado U4/U6 di-snRNP e U5 snRNP como um
tri-snRNP, resultando no complexo B. O complexo U1 então se dissocia da região 5’ splice
site e U6 se liga nesta mesma região e o duplex entre U4/U6 snRNAs, gerado pela união de
U4/U6 snRNPs, é desfeito e permite o pareamento de bases entre U2-U6. Concomitante a
liberação de U1 e U4 snRNP, ocorre a incorporação do complexo associado a Prp19p (Prp19-

29
associated complex, Nineteen complex ou NTC), formando um complexo ativo com
capacidade catalítica, nomeado de Complexo B*. Após este primeiro passo de reação,
ocorrem novos rearranjos, levando a formação do Complexo C, que catalisa o segundo passo
do splicing, e em seguida, o complexo é completamente desorganizado 36
e seus componentes
podem ser reutilizados.
Figura 4 -
(A)Elementos conservados da sequencia do pré-mRNA de metazoários e leveduras. Nesse
esquema N representa qualquer nucleotídeo, R uma purina e Y uma pirimidina. (B) Modelo
esquemático do cis-splicing.
Fonte: Adaptada de WAHL et al37
.
O trans-splicing apresenta as mesmas etapas do cis-splicing (dois passos de reação);
na primeira etapa, ocorre clivagem no sitio doador do SL gerando 5’ SL éxon; o SL
remanescente liga-se à adenosina do íntron no pré-mRNA aceptor para formar o Y-

30
intermediário (correspondente ao lariat do cis-splicing). Na segunda etapa, a clivagem no
sítio 3’ e ligação do éxon geram os éxon ligados e um Y-íntron excisado38-39
.
O SL RNA cumpre duas funções. Primeiramente, essa sequencia atua de maneira
correspondente ao éxon 5’ dos mRNAs processados por cis-splicing, excisando o mRNA do
transcrito primário. Em segundo, a estrutura m7G cap é adicionada a cada mRNA codificante
40. A figura a seguir ilustra as principais diferenças entre o processamento de RNA por cis ou
trans-splicing (Figura 4).
Figura 5 - Representação esquemática do cis (A) e trans- splicing (B).
Fonte: Adaptada de MICHAELI41
.
A maioria dos eventos de SL trsns-splicing descritos em cinetoplastídios mostram a
utilização precisa do sítio aceptor 3’. Entretanto, experimentos mostraram que mais sítios
aceptores pode ser usados para gerar um mRNA maduro que codifique a mesma ORF mas
possuam extensões 5’UTR distintas. O SL trans-splicing alternativo em cinetoplastídeos é um
mecanismo capaz de produzir diferentes transcritos a partir do mesmo pre-mRNA e poderia
contribuir para a expansão da diversidade de proteínas pelo aumento na possibilidade da
combinação de éxons42
.

31
1.6 O SL (spliced leader) RNA
A estrutura secundária do SL RNA de todos os organismos que carregam a maquinaria
de trans-splicing é composta de três stem-loops. Em tripanossomatídeos, a sequência SL
possui de 39 a 41 nucleotídeos, seguida de um íntron de tamanho variado que pode se dobrar
em dois stem-loops (II e III) separados por uma sequência de fita única. Essa região é
chamada sítio Sm-like, já que, em vários tripanossomatídeos, ela é derivada da sequência de
ligação de Sm canônica 43
. O stem-loop I possui duas conformações (forma 1 e 2), embora a
forma 2 pareça ser predominante e proteínas específicas são relevantes para obtenção desta
estrutura 21
. O SL RNA também possui um cap hipermodificado referido como cap4, devido
à modificação no quarto nucleotídeo depois da 7-metilguanosina (m7G)40
.
Figura 6 - Estrutura secundária de SL RNA. Os stem-loops estão indicados por sl I,II e III e
o ponto sombreado na extremidade 5’ do SL RNA é a estrutura do cap4 .
Fonte: LIANG et al
44.
Sequências relevantes estão presentes no SL íntron e ao redor do sítio de ligação a Sm
e são capazes de interagir com U6 snRNA. Esta interação é importante para trazer o SL
5’splice site para o centro catalítico do trans-spliceosomo. Em tripanosomatídeos, entretanto,
não há aparente complementaridade entre regiões do SL e U6 snRNAs e os estudos ainda não
demonstraram esta interação in vivo. Experimentos com células permeáveis de T. brucei
revelaram duas regiões relevantes para SL RNA no trans-splicing. A primeira, do nucleotídeo
7-19, cujo mascaramento inibe a metilação na extremidade 5’ do SL; e a segunda região está
compreendida entre as posições 40-43 downstream do 5’ splice site21
.

32
1.7 Ribonucleoproteínas (snRNPs) e snRNAs
Cada ribonucleoproteína (snRNP – U1, U2, U4/U6, U5) consiste de um pequeno RNA
rico em uridina, ou dois no caso do complexo U4/U6, sete proteínas Sm comuns (B, D3, D2,
D1, E, F, G no caso de mamíferos) e um grande número de proteínas específicas45
. As
snRNPs são nomeadas de acordo com o tipo de RNA associado a elas46
.
Em contraste com as subunidades do ribossomo, nenhuma das snRNPs possui um sítio
ativo pré formado e muitas delas são remodeladas no decorrer do splicing. A montagem do
spliceossomo acontece por interações ordenadas das snRNPs e inúmeros outros fatores 47
.
Experimentos mostraram que o splicing de transcritos, gerados pela RNA polimerase
II in vitro, foram realizados entre 15 e 60 minutos, enquanto que in vivo esse tempo é de
menos de 3 minutos, mostrando que a montagem correta do spliceossomo no pré-mRNA é
mais eficiente no núcleo de uma célula viva do que em um extrato nuclear 48-49
.
Os snRNA U1, U2, U4 e U5 possuem um sítio Sm flanqueado por steem-loops, ao
redor desse sítio as proteínas Sm se unem para formar um anel heptamérico, formando o core
das partículas snRNP. Similarmente, o U6 snRNA adquire um anel heptamérico de LSm (like
Sm). Em tripanossomatídeos, os snRNAs U2 e U4 se associam com proteínas Sm específicas
(SSm) assim como proteínas canônicas. Esperimentos mostraram que no U2 snRNP o
complexo SmB/D3 é substituído pelas proteínas SSm2-1 (U2-15K) e SSm2-2(U2-16,5K). Já
no U4 snRNP, a proteína SmB continua presente mas a SmD3 é substituída pela proteína
única SSm450
.
Figura 7 - Modelo do core Sm (esquerda), variação do core Sm de U2 snRNP de T. brucei (centro), variação
do core Sm de U4 snRNP de T. brucei (direita).
Fonte: Adaptada de TKACZ et al50
.
Os snRNAs recém transcritos são exportados do núcleo para o citoplasma, onde se
associam a sete proteínas Sm para formar o snRNP core. A montagem do Sm core ao redor do

33
snRNA é um passo fundamental na biogênese dos Sn RNPs. Em tripanossomatídeos o
complexo SMN (survival of motor neurons) atua na supressão da montagem do Sm core ao
redor de RNA não relacionados ou snRNA com mutações no sítio Sm. Estudos mostraram
que na ausência do complexo SMN, a maior parte do RNA se liga ao core Sm até certo ponto,
mas na presença do complexo, essas ligações erradas são reduzidas a níveis não detectáveis51
.
Também foi identificado em tripanossomatídeos o complexo PRP19, que é necessário
para a associação dos complexos U5 e U6 com o pré-mRNA. Experimenros com o
silenciamento do PRP19 sugerem que esse complexo seja um fator que se associa ao SL RNA
cedo na biogênese, mas permanece retido durante toda a reação de splicing, e deve funcional
como um fator que ajuda na incorporação do SL RNP no spliceossomo52
.
1.8 Proteínas específicas de snRNPs
Dentre as snRNPs, o complexo U1 é o melhor caracterizado, tanto do ponto de vista
estrutural quanto do seu papel na montagem do spliceosomo53
. As proteínas específicas do U1
snRNP (U1-70K, U1A e U1C) são bem conservadas de leveduras ao homem. As proteínas
U1-70K e U1A associam-se diretamente com o U1 snRNA, enquanto que a U1C interage a
partir de ligações proteína-proteína54
. O U1 snRNA de tripanossomatídeos é bem menor que
os homólogos conhecidos (aproximadamente 75 nucleotídeos contra 164 nucleotídeos em
mamífero), sugerindo que o snRNP opera de maneira mínima ou especializada nesses
organismos. Experimentos de purificação por afinidade em tandem (TAP) e espectrometria de
massas evidenciaram a existência de uma proteína homóloga a 70K (TbU1-70K), a U1C
(TbU1C) e uma nova proteína de 24 kDa (TbU1-24K)55
.
Proteínas específicas de U2 snRNP são conhecidas em mamíferos e leveduras
(denominação entre parênteses para membros de leveduras); são elas, U2-A’ (LEA1), U2-B”
(MSL1), bem como fatores associados a esta partícula: SF3a120 (PRP21), SF3a66 (PRP11),
SF3a60 (PRP9), SF3b155 (HSH155), SF3b145 (CUS1), SF3b130 (RSE1), SF3b125, SF3b49
(HSH49), SF3b14b (RDS3) e SF3b10. Além dessas, são conhecidas proteínas que estão
presentes em purificações de U2 snRNP, mas que apresentam uma ligação fraca, sendo
denominadas de proteínas U2-relacionadas. Algumas delas são Prp5, SR140, CHERP,
PRP43/DDX15, SPF45, SPF31 e SPF3045, 55
. A PRP5, uma proteína DEAD-box e U2-
relacionada, parece estar implicada na aproximação de U1 e U2 snRNPs durante o splicing56
.
Em T. brucei, a proteína U2-40K foi identificada como homóloga à U2-A’ e U2-B”. A U2-

34
40K se liga eficientemente à U2-B” na ausência de U2 snRNA e aumenta a afinidade de
ligação entre U2-B” e o U2 snRNA57-58
.
Figura 8 - Modelo de interação do core U2 snRNP de T. brucei (parte de cima) em comparação com o de
humano (parte de baixo).
Fonte: PEUßER et al58
.
In vivo, U4 e U6 snRNPs não existem na forma de mono-snRNP, mas formam um
complexo U4/U6 pelo pareamento de seus snRNAs e adição de proteínas específicas dessas
di-snRNP. Além das proteínas Sm e Sm like que associadas ao complexo U4 e U6, cinco
proteínas foram identificadas como específica da U4/U6 di-snRNP, o complexo heteromérico
20/60/90K, uma proteína que se liga especificamente ao U4 snRNA, 15,5K, e a proteína 61K,
que se mostrou necessária para a formação da tri snRNP U4/U6.U559-61
. A formação do di-
snRNP é hierárquica, de forma que a ligação da 15,5K à extremidade 5’ do stem-loop do U4
snRNA é necessária para que tanto a 61K quanto o complexo 20/60/90K interajam com o
duplex U4/U6 snRNA 62
.
Com exceção da proteína 20K, as proteínas específicas de U4/U6 snRNP são
evolutivamente conservadas e seus ortólogos em Saccharomyces cerevisiae são nomeados
Snu13p (15,5K), Prp4p (60K), Prp3p (90K), Prp31p (61K) 63
. Em tripanossomatídeos foram
descritos quatro membros de U4 snRNP; Prp31, Prp4, Prp3 e Snu13 64
.
São conhecidas oito proteínas específicas de U5 snRNP em humanos, e suas ortológos
em S. cerevisiae (entre parênteses): 220K (PRP8), 200K (BRR2), 116K (SNU114), 102K
(PRP6), 100K (PRP28), 52K (LIN1 ou SNU40), 40K e 15K (DIB1) 55
.
Em um dos maiores e mais conservados fatores de splicing, PRP8, tem sido observado
interação física e funcional com o dinucleotídeo GU do sítio de splicing 5´ e com sítio de

35
splicing 3´65-68
. Além disso, PRP8 também faz contato com branch point e o trato de
polipirimidina65, 69
, estando também relacionada ao reconhecimento do sítio de splicing 5´,
parecendo ter uma posição específica neste sítio durante a primeira reação catalítica. Acredita-
se que após esse passo, PRP8 alinha os sítios 5´ e 3´ para a segunda reação de catálise70
e
pode auxiliar no reconhecimento do sítio de splicing 3´. A região de PRP8 em levedura
relacionada com o reconhecimento do sítio de splicing 3´ apresenta-se conservada na proteína
de tripanosomas 71
.
U5-116K está relacionada com a separação do duplex U4/U6 durante a ativação do
spliceosomo72
. As proteínas U5-220K, -200K, -116K e -40K formam um complexo
heteromérico na ausência do RNA, sugerindo um complexo pré-formado antes da montagem
da partícula de U5 snRNP73
.
A proteína 52K foi identificada em vertebrados, insetos, nemátodes, fungos e plantas,
mas não em tripanosomatídeos. Essa proteína é encontrada na partícula 20S U5 snRNP, mas
não na partícula 35S U4/U6.U5 tri-snRNP, sugerindo que essa proteína esteja associada a
formação do tri-snRNP, mas que não tenha função na reação de splicing propriamente dita. A
deleção da proteína homóloga a 52K em levedura (Lin1) não se mostrou letal ao organismo, o
que sugere que a 52K tenha uma função auxiliar ou redundante para o processo de splicing.
Também foi visto que a 52K interage com as proteínas específicas de U5 snRNP, 102K e
15K, sendo que cada uma dessas interações ocorre em um domínio diferente, domínio N-
terminal no caso da 102K e domínio GYF (glicina-tirosina-fenilalanina) do C-terminal no
caso da 15K124
.
A última proteína específica de U5 snRNP de tripanosomatídeo descrita foi U5-
Cwc21, e é considerada exclusiva deste organismos64
. Trata-se de proteína essencial para o
parasito, como demonstrado por experimentos de RNAi, e com alto grau de ligação ao U5
snRNA. Destaca-se ainda por possui localização tanto nuclear quanto citoplasmática, fato
inesperado para proteína com participação na reação de splicing74
. Técnicas de purificação de
complexos utilizando U5-Cwc21 como isca demonstraram a copurificação deste membro com
quatro proteínas específicas de U5 snRNP (U5-220K, -200K, 116K e 40K), além do anel de
proteínas Sm, sugerindo a participação de U5-Cwc21 no 35S U5 snRNP 75
.
1.9 U5-15k
U5-15K em humano e tripanossomatídeos, Dibp em S. cerevisae ou Dim1p em
Schizosaccharomyces pombe, é um membro altamente conservado entre os eucariotos. O gene

36
dim1 foi inicialmente isolado em S. pombe em condições que indicavam seu papel no
progresso do ciclo celular, sendo que o mutante dim1-35 apresenta fenótipo sensível à
temperatura, com perda da viabilidade e defeitos na mitose que incluem problemas na
separação das cromátides irmãs e síntese de agentes desestabilizantes de microtúbulos. Já a
deleção do dim1 resulta em letalidade na fase G2 com células alongadas. Esses dois fenótipos
sugerem que a proteína Dim1p é necessária para que a célula entre na fase M da mitose 76
.
Entretanto, experimentos cujo objetivo era encontrar novos componentes de
U4/U6.U5 tri-snRNP em S. cerevisae levaram à identificação da Dibp, sugerindo provável
função no processamento de RNA mensageiros, mais especificamente na reação de splicing.
Este achado sugere que o efeito observado no ciclo celular seria indireto77
. Essa segunda
proposta de função da Dim1p foi apoiada por experimentos que mostraram que a deleção da
Dibp resultava em falha no splicing de pequenos RNAs78
.
O domínio conservado Dim1 (Mitosis protein DIM1) foi identificado entre os
aminoácidos 53-157 na seqüência de T. cruzi e T. brucei. Presente em tiorredoxinas, este
domínio é altamente conservado e está presente em organismos de diversos reinos.
Tiorredoxinas são conhecidas como doadoras de hidrogênio para ribonucleotídeo redutase,
uma enzima essencial para produção de deoxiribonucleotídeos para replicação do DNA.
Tiorredoxinas e glutaredoxinas são pequenas proteínas que contem um sítio ativo com
atividade redutora, responsável pela transferência de elétrons a partir de duas proteínas com
grupos SH para formação de pontes de dissulfeto79
.
Experimentos de RNAi de U5-15K em C. elegans levaram a letalidade embrionária,
sugerindo que esta proteínas é essencial para o processamento de mRNA no zigoto.
Experimentos em S. cerevisae, S. pombe e T.brucei também demonstraram a essencialidade
desta proteína na reação de splicing, bem como para viabilidade celular. Entretanto, novos
experimentos são necessários para demonstrar essencialidade em células de mamífero75, 80
.
A análise das propriedades de interação da Dim1 mostrou uma conexão direta dessa
proteína com elementos da maquinaria de splicing. Observou-se que U5-15K humana interage
com hnRNP F e hnRNP H’, proteínas tecido-específicas, envolvidas no aumento da atividade
da reação de splicing, e que apresentam motivos de reconhecimento de RNA (RRM-like) que
se ligam diretamente com sítios de poli-glutamina (rG). U5-15K humana também interage
com Npw38/PQBP1, que possuem capacidade de se ligar a poli(rG) e colocaliza com os
coativadores de splicing SRm160/SRm300 quando coexpressa com U5-15K. Em algumas
circunstâncias esta interação pode regular a transcrição dependente de RNA polimerase II. Por
fim, U5-15K interage fortemente com U5-102K, uma proteína importante para acúmulo do

37
U4/U6.U5 tri-snRNP, sendo esta interação observada em humano, leveduras e T. brucei.
Juntos, estes dados sugerem que a U5-15K pode estar envolvida em múltiplos pontos de
controle do processamento de mRNA por splicing 63, 75, 80-81.
A estrutura tridimensional de U5-15K foi analisada tanto por Ressonância Magnética
Nuclear (RMN) 82
quanto por cristalografia78
. Sendo que a estrutura cristalográfica da U5-
15K humana foi a primeira estrutura tridimensional a ser resolvida dentre as proteínas
específicas de U5 snRNP Sua estrutura apresenta um enovelamento semelhante a uma
tiorredoxima, que é caracterizado por uma folha β formada for 4 fitas β com pareamento
paralelo e anti-paralelo flanqueadas por 3 hélices α. As estruturas de U5-15K e de uma
tioredoxina humana se sobrepõem, sendo que a primeira proteína possui 37 resíduos
adicionais, concentrados principalmente na região N-terminal 78
.
Figura 9- Estrutura resolvida por RMN da U5 -15K humana (esquerda) e estrutura
cristalográfica (direita) .
Fonte: Adaptada de REUTER et al78
; ZHANG et al82
.
A estrutura calculada por RMN confirma que a hDim11-128 (U5-15K humana sem 14
resíduos do C-terminal) assume um enovelamento semelhante a uma tiorredixina, como foi
visto na estrutura cristalográfica da U5-15K. Apesar da semelhança, há algumas diferenças
entre essas estruturas, como a presença de uma pequena folha β formada pelos resíduos 91-93
e 129-131 na estrutura cristalográfica, que não está presente na estrutura resolvida por RMN,
onde os resíduos 91-93 assumem conformações que não são registradas como de folha β, o
que provavelmente é resultado da deleção dos 14 aiminoácidos do C-terminal 82
.
Outra diferença está relacionada ao motivo Cys-X-X-Cys que em tiorredoxinas forma
uma ligação dissulfeto funcional no N-terminal da hélice α2. Na U5-15K apenas a Cys38,

38
correspondente a segunda cisteína do motivo Cys-X-X-Cys, é conservada, sendo que na
estrutura cristalográfica essa cisteína forma uma ligação dissulfeto com a Cys79 da fita β3. Os
dados de RMN não mostram essa ligação, sugerindo que esta ponte não seja essencial para
estrutura ou função da proteína. Soma-se a isto o fato da cisteína 79 não ser evolutivamente
conservada 82
.
A habilidade da forma truncada da U5-15K, hDim11-128, agir como um inibidor
negativo dominante da proteína selvagem em levedura, juntamente com o fato da sequência
C-terminal ser conservada, indica que esses 14 resíduos possuem um papel importante para a
função da proteína83
. A contribuição desta região para estabilidade foi analisada por
Dicroismo Circular (CD) na presença de agentes desnaturantes. Embora tanto a forma
selvagem quanto a proteína truncada sofram desnaturação em concentrações semelhantes de
agentes desnaturantes, alterações no espectro de CD sugerem uma região de desnaturação
local na referida região C-terminal82
.
Comparações entre o espectro de CD entre U5-15K nativa e a forma trucada mostra
significativas alterações de estrutura secundária, sendo possível observar significativo
decréscimo de estrutura helical na forma truncada. Associado aos dados da estrutura
cristalográfica é possível que os resíduos de 129-131 estejam envolvidos na interação com
interações com folhas β (β4) e que o restante da sequência de 14 aminoácidos possua
conformação helical82
.
Demonstrou-se que a perda da região C-terminal ocorre pela ação de uma peptidase,
sendo reportado que a U5-15K retém atividade de autoclivagem. Atividade essa que é inibida
por diferentes inibidores de protease, como EDTA, ácido 6-aminohexanóico e quimostatina84
.
Experimentos usando a sequência de 20 aminoácidos no C-terminal da U5-15K como ligação
entre VHb (Vitreoscilla haemoglobin) e GST (Glutathione S-Transferase) mostram que essas
duas proteínas eram separadas na presença da U5-15K truncada (hDim11-128), o que sugere
que a U5-15K é capaz de clivar o mesmo sítio em diferentes contextos e que o core
semelhante a tiorredoxina retém a atividade de peptidase84
.
Durante os primeiros experimentos de expressão de U5-15K de T. brucei foi possível
observar a presença a superexpressão de um banda de aproximadamente 24 kDa, entretanto,
ora observava-se um banca única em torno de 24 kDa ora uma banda extra mais baixa,
levantando a possibilidade da presença de contaminantes ou degradação da proteína.
Entretanto, baseado na literatura, é possível que U5-15K de T. brucei esteja sofrendo
autoclivagem como descrito para os homólogos em humano e leveduras78, 81-82
.

39
Neste trabalho a estrutura da proteína U5-15K de T. brucei foi caracterizada em
solução e foi mostrado que, assim como as homólogas humana e de levedura, essa proteína
também apresenta atividade de autoclivagem.
1.10 Técnicas empregadas
1.10.1 Modelagem por homologia
A estrutura tridimensional de uma proteína pode fornecer informações valiosas quanto
sua função, o que permite o design efetivo de experimentos. Apesar do grande progresso no
campo da solução experimental da estrutura terciária com as técnicas de cristalografia de raio
X e RMN, o processo ainda é demorado e possui limitações, como a necessidade de alta
concentração proteica e condições experimentais difíceis de ser alcançadas com amostras
proteicas. O número de estruturas depositadas em bases de dados como o PDB (Protein Data
Bank) é muito inferior ao número de sequências proteicas conhecidas, para minimizar essa
lacuna no conhecimento, métodos computacionais de predição de estrutura tridimensional
vem sendo desenvolvidos 90
.
Evolutivamente, a estrutura terciária de proteínas homólogas é mais estritamente
conservada que sua sequência de aminoácidos. Aliada ao fato de que existe um número
limitado de enovelamentos existentes na natureza, essa afirmação permitiu que métodos como
a modelagem comparativa fossem desenvolvidos 91
.
A modelagem comparativa, ou modelagem por homologia, é capaz de gerar um
modelo tridimensional confiável da proteína a partir de sua sequência de aminoácidos, usando
como base ao menos uma estrutura experimentalmente resolvida. Esse método segue quatro
passos básicos: seleção da proteína molde, alinhamento entre as sequências da proteína alvo e
molde, construção do modelo e avaliação 90
.
1.10.2 Cromatografia de afinidade
Os sistemas de expressão pET SUMO e pET28a(+) possuem uma cauda N-terminal de
6 histidinas, que interage reversivelmente com os íons Co2+
imobilizados em uma resina de
sefarose, permitindo separação da proteínas recombinantes das demais. Para a eluição da
proteína foi usado o composto imidazol, uma vez que este compete pela ligação com o íon
Co2+ com o anel imidazólico presente na cadeia lateral da histidina.

40
Figura 10 - Diagrama esquemático do sistema TALON mostrando a ligação entres as histidinas da cauda N-
terminal e o íon Co2+ imobilizado à resina formada por esferas de sefarose.
Fonte: CLONTECH ...1 15
.
1.10.3 Cromatografia de exclusão molecular
Na cromatografia de exclusão molecular, os componentes da amostra são separados de
acordo com o seu tamanho e massa molecular. A retenção das partículas depende da
penetração relativa da amostra nos poros da matriz que constitui a fase estacionária da coluna.
As moléculas que percorrem a coluna, fase móvel, podem entrar nos poros caso esses sejam
maiores que a molécula122
.
A teoria mais simples da cromatografia de exclusão molecular assume que uma
molécula de um determinado tamanho entra e sai de um poro em média n vezes durante a
migração pela coluna. Cada molécula possui um caminho individual, sendo que moléculas do
mesmo tamanho se comportam de maneira semelhante. Moléculas menores podem penetrar
em mais poros, ficando retidas na coluna por mais tempo que moléculas maiores, que passam
pela matriz sem penetrar os poros122
.
1.10.4 Dicroísmo circular
Muitas moléculas apresentam assimetria, ou seja, não é possível sobrepor a sua
estrutura com a imagem especular. Esse fenômeno é chamado quiralidade e cada um dos
enantiômeros é capaz de rotacionar o plano da luz polarizada para a direita ou para a
esquerda. A quiralidade de uma molécula pode ser estudada através da técnica de
espectroscopia de dicroísmo circular94
.
A espectrometria de dicroísmo circular usa uma fonte de luz circularmente polarizada,
na qual o vetor oscila rotativamente para a direita ou para a esquerda, formando uma hélice

41
em relação ao eixo de propagação. Essa técnica consiste em incidir um feixe de luz,
polarizado para a direita quanto para a esquerda em uma amostra e medir a diferença de
absorção em relação aos dois componentes. Uma amostra opticamente ativa absorve as duas
componentes de maneira diferente, o que resulta em uma polarização elíptica da luz 94
.
Figura 11 - (I) As amplitudes da luz polarizada para a esquerda e para a direita são iguais, o que resulta em
um feixe de luz linearmente polarizado.(II) A diferença nas amplitudes da luz polarizada para a
esquerda em relação a polarizada para a direita resultam em uma polarização elíptica.
Fonte: KELLY et al
95.
A elipticidade (θ) do feixe resultante está relacionada com a absorção da luz pela
amostra como mostrado na equação 2, onde AL e AR são as absorbâncias do feixe polarizado a
direita (R) e a esquerda (L), obedecendo à lei de Beer-Lambert:
𝐴(𝜆) = log(𝐼 𝐼𝑜)⁄ = 𝐶𝜀(𝜆)𝑙 (1)
Onde A é a absorbância a um determinado comprimento de onda, λ, I0 é a intensidade
da luz incidente, I é a intensidade da luz após passar pela amostra, C é a concentração, ε é o
coeficiente de extinção molar e l é o caminho óptico percorrido95
.
𝜃 =2,303(𝐴𝐿−𝐴𝑅)
4𝑙 (2)
A assimetria das ligações peptídicas permite que a estrutura secundária das proteínas
seja estudada por espectrofotometria de CD e os resultados são geralmente expressos em
termos da elipticidade residual molar média, [θ], dada em deg.cm2.dmol-1:
[𝜃] =𝜃.100.𝑀𝑀
𝐶.𝑙.𝑛 (3)

42
Onde θ é a elipticidade em graus, MM é a massa molecular, C é a concentração em
mgmL-1, l o caminho óptico em centímetros e n é o número de resíduos na proteína95
.
O espectro de CD da proteína, abaixo de 250nm, é dominado pelo cromóforo amida. A
amida possui duas transições eletrônicas de baixa energia bem caracterizadas,
n→π*,responsável pelas bandas negativas próximas a 222 nm características de α-hélice e
216-218 nm características de folha-β, e π0→π*, responsável pelas bandas positivas a 190 nm
e negativas a 208 nm características de α-hélice e bandas positivas em 198 nm características
de folha-β95
.
Figura 12 - Espectro de CD no UV distante, onde a linha cheia representa a α-hélice, a linha tracejada
representa a folha-β e a linha pontilhada representa uma estrutura irregular.
Fonte: KELLY et al95
.
A estimativa da porcentagem de cada estrutura secundária presente em uma proteína, a
partir do espectro de CD, é feita pela combinação linear dos espectros característicos de cada
estrutura secundária.
1.10.5 Espalhamento de luz dinâmico
O espalhamento de luz dinâmico, também conhecido como espectroscopia de
correlação de fótons, é um dos métodos usados na determinação do tamanho de uma proteína
e seu estado oligomérico em solução96
.
Para partículas menores que o comprimento de onda, o campo elétrico do feixe de luz
incidente em uma amostra induz um dipolo elétrico oscilante nas moléculas, que agem como
uma fonte de luz secundária, ou seja, luz espalhada. Esse fenômeno é chamado espalhamento

43
de Rayleigh. A intensidade da luz espalhada varia com o tempo e a razão com que essas
flutuações ocorrem depende do tamanho da partícula. De forma que as partículas menores
provocam uma variação mais rápida na intensidade se comparada a partículas maiores 96
.
Uma forma de extrair informação a partir da variação de intensidade da luz espalhada
é a função de correlação. Para um grande número de partículas monodispersas em movimento
Browniano, a função de correlação é uma exponencial que decai com τ, tempo de correlação.
A soma dos decaimentos exponenciais contidos na função de decaimento é frequentemente
chamado de função de espalhamento intermitente medido, F, e é dada por 96
:
𝐹(𝑞,𝜏) = exp(−𝑞2⟨∆𝑟2(𝜏)⟩ 6⁄ ) (4)
Onde ⟨∆𝑟2(𝜏)⟩ é o deslocamento quadrático médio de uma partícula no tempo τ e q é
o vetor espalhamento.
A velocidade de difusão para partículas em movimento Browniano é definida pelo
coeficiente translacional de difusão, D0, o que resulta em um deslocamento quadrático médio
dado por96
:
⟨∆𝑟2(𝜏)⟩ = 6𝐷0𝜏 (5)
Usando a relação de Einstein, o coeficiente translacional de difusão pode ser escrito da
seguinte forma.
𝐷0 = 𝑘𝐵𝑇 𝑓⁄ (6)
Onde kB é a constante de Boltzmann, T é a temperatura e f é a constante de fricção.
Para partículas esféricas, a aproximação de Stokes é válida, o que resulta em f = 6πηRH, onde
η é a viscosidade do solvente e RH o raio hidrodinâmico da partícula96
.
Portanto, o tamanho de uma partícula em solução pode ser calculado a partir do
coeficiente de difusão usando a equação de Stokes-Einstein.
𝑅𝐻 = 𝑘𝐵𝑇 6𝜋𝜂𝐷0⁄ (7)
O resultado de experimento de DLS é o valor de τ através da função de correlação e o
calculo de D0, e consequentemente de RH. Considerando a proteína esférica, também é

44
possível calcular sua massa e densidade, e dessa forma estimar seu estado oligomérico em
solução96
.
1.10.6 Espalhamento de raio X a baixo ângulo
O SAXS é um método analítico de determinação da estrutura de um sistema de
partículas (no caso desse trabalho, de uma proteína) em termos do seu tamanho e forma
médios. O espalhamento a baixo ângulo aparece quando um feixe monocromático de raio X é
espalhado pelos átomos de uma molécula. Diferente da difração de raio X, o SAXS pode ser
observado em moléculas em solução e, apesar de ser descrita como uma técnica de baixa
resolução, o SAXS é capaz de fornecer informação de alta precisão quanto ao tamanho e
forma das moléculas97
.
Por razões de conveniência matemática, o padrão de espalhamento é descrito pela
intensidade (I) em função da amplitude do vetor de espalhamento, q:
𝑞 =4𝜋
𝜆𝑠𝑖𝑛𝜃 (8)
Onde λ é o comprimento de onda do feixe incidente e 2θ é o ângulo de espalhamento.
Para sistemas diluídos, I(q) versus q está relacionado com a forma da molécula em
solução, mas seu perfil não é intuitivo e para ser interpretado em termos da estrutura é
necessário fazer a transformada de Fourier do perfil de espalhamento para obter a função de
distribuição das distâncias interatômicas, P(r), cujo perfil é sensível à simetria e à estrutura
dos domínios da molécula, e dessa forma pode fornecer informação sobre a sua forma e o
volume que ocupa 98
.
Os dados de espalhamento só podem ser medidos em um range finito de valores de q,
dessa forma, o cálculo de P(r) depende de suposições como P(r) ser zero quando r = 0 e Dmax,
onde Dmax é dimensão máxima da molécula e sua incerteza é altamente dependente da
qualidade dos dados coletados99
.
De um perfil de espalhamento de alta qualidade, dois parâmetros podem ser calculados
com precisão, I(0) e Rg. I(0) está relacionada ao número de partículas que espalham raio X
por unidade de volume (N) e ao volume da partícula (V) ao quadrado. Rg é definido como a
distância quadrática média dos elétrons em relação ao centro de massa da molécula. Uma
forma rápida de determinar esses valores é através da aproximação de Guinier100
, dada por:

45
𝐼(𝑞) = 𝐼(0)𝑒𝑥𝑝 (−𝑞2𝑅𝑔
2
3) (9)
1.10.7 Fluorimetria diferencial de varredura
A cristalização de uma proteína depende da sua homogeneidade, estabilidade e
solubilidade em uma determinada solução, essas características dependem de uma grande
variedade de fatores, como pH, força iônica, presença de aditivos, precipitantes e
concentração de proteína. Dessa forma, a otimização do tampão é um passo importante para
aumentar as chances de sucesso em ensaios de cristalização 106
.
O conceito central da técnica de fluorimetria diferencial de varredura é a possibilidade
de diferenciação de uma proteína enovelada e uma desenovelada pela exposição de uma sonda
fluorescente hidrofóbica. Essa sonda se liga preferencialmente ao interior hidrofóbico exposto
da proteína quando essa está se desenovelando. Conforme a proteína perde sua estrutura
terciária, a sonda perde a fluorescência de modo que essa emissão pode ser detectada em
função da temperatura 107
.
Figura 13 - Representação esquemática da curva de desenovelamento de uma proteína, onde Tm
(temperatura de melting) é definida como a temperatura em que metade das proteínas em
solução se encontram desenoveladas.
Fonte: Adaptado de NIESEN et al
1 08.
Estudos mostraram que a probabilidade de sucesso na cristalização está relacionada
com a forma da curva de desenovelamento (melting), de forma que curvas com a transição

46
entre o estado enovelado e desenovelado bem definida, combinada com uma baixa
fluorescência inicial resultam em maiores chances da proteína formar cristais 109
.
1.10.8 Ensaios de cristalização
A cristalização de proteínas apareceu pela primeira vez no século 19, com a
publicação de 1840 sobre a observação de cristais durante a precipitação de sangue, que se
tratava de cristais de hemoglobina. Nessa época a cristalização de proteínas tinha como
propósito a sua purificação e as proteínas cristalizadas eram basicamente de origem vegetal,
com poucas de origem animal como hemoglobina e albumina 110
.
O foco da cristalização de proteínas mudou quando, em 1934, foi tirada a primeira
fotografia de raio x de um cristal de proteína. Desde então houve grandes melhoramentos nos
processos de obtenção de cristais adequados para a determinação estrutural da proteína, bem
como avanços nas técnicas de cristalografia de raio X 110
.
A cristalização é um fenômeno multiparamétrico, ou seja, depende de uma diversidade
de aspectos como solubilidade e concentração da proteína, tipo de sal usado para induzir
supersaturação, temperatura e necessidade de íons metálicos. A função primordial dos
experimentos é escolher quais desses parâmetros levam a melhores condições de crescimento
de cristais 110
.
Os métodos de difusão de vapor começaram a ser usados em 1968 e significaram um
grande avanço na produção de cristais. O método se baseia no equilíbrio entre uma gota
contendo a proteína a ser cristalizada e uma solução de cristalização em um reservatório
contendo a mesma solução em concentração maior. O equilíbrio ocorre por difusão dos
componentes mais voláteis da solução até que a pressão de vapor seja a mesma na gota e no
reservatório. Esse processo leva a uma redução do volume da gota e consequente aumento da
concentração da proteína, que pode entrar na fase de supersaturação onde a cristalização pode
ocorrer. Entre os métodos que usam esse princípio estão as técnicas da gota pendurada
(hanging drop) e da gota apoiada (sitting drop)110
.

47
Figura 14 – Desenho esquemático da técnica de gota pendurada (hanging drop) (esquerda) e gora apoiada
(sitting drop) (direita).
Fonte: Elaborada pelo autor.

48

49
2 Objetivos específicos
Otimizar a purificação de U5-15K recombinante de T. brucei;
Realizar ensaios de rastreamento das condições de cristalização da proteína U5-15K de T.
brucei nas diferentes formas de clivagem;
Realizar medidas de SAXS de U5-15K nas diferentes formas de clivagem;
Caracterizar a autoclivagem da proteína, identificando potenciais inibidores da atividade
peptidase.

50

51
3 Materiais e Métodos
3.1 Análise bioinformática da sequência de aminoácidos da proteína U5-15k
A análise dos parâmetros físico-químicos teóricos da proteína U5-15K de T. brucei,
bem como de sua forma clivada, foi feita a partir da sua sequência de aminoácidos usando a
ferramenta PROTPARAM 85
do servidor ExPASy.
A predição da estrutura secundária foi feita através do servidor PSIPRED86
e a análise
de aminoácidos conservados foi feita através do alinhamento múltiplo com sequências
homólogas, gerado pelo programa ClustalW87
e a análise dos domínios foi feita pelos
servidores CDD88
e SMART89
.
3.2 Modelagem da estrutura terciária
Para a modelagem por homologia da U5-15K a estrutura cristalográfica da homóloga
humana foi usada como molde (código PDB 1QGV) e o modelo foi gerado pelo servidor
SWISS-MODEL92
. A avaliação foi feita através do servidor SAVES93
e a visualização foi
feita com o programa PYMOL (DeLano Scientific LLC).
3.3 Obtenção do plasmídeo
O gene da U5-15K de T. brucei clonado em pET28a(+) foi cedido pelo Dr. Marco
Túlio Alves da Silva, que os obteve durante seu doutorado. A clonagem no vetor pET SUMO
foi realizada pelo então aluno de mestrado Pedro Neuber sob a orientação da Prof. Dra.
Claudia Elisabeth Munte no Instituto de Física de São Carlos da Universidade de São Paulo.
3.4 Cultura de Bactérias
3.4.1 Preparação das bactérias competentes
Um pré-inóculo de 5 mL de meio TYM (2% de triptona; 0,5% de extrato de levedura;
0,58% de NaCl e 0,25% de MgSO4.7H2O) contendo bactérias BL21(DE3)pLysS, após
crescimento por 16 horas, foi transferido para 10 mL de meio TYM, em uma diluição de

52
1:100 e incubado a 37ºC até que a densidade óptica (D.O.) medida a 600 nm atingisse entre
0,2 e 0,6. Em seguida 10 mL da cultura foram diluídos em 40 mL de meio TYM e incubado,
nas mesmas condições, até que a D.O. 600 nm atingisse entre 0,5 e 0,9. Os 50 mL foram
então diluídos em 200 mL de meio TYM e novamente incubados até que a D.O. 600 nm
atingisse 0,6 e então a cultura foi resfriada rapidamente em gelo.
Em seguida a cultura foi centrifugada a 4000 rpm por 15 minutos a 4ºC em tubos de
50 mL previamente gelados. As células foram ressuspendidas gentilmente em 100 mL de
TBF1 (30 mM de KOAc; 50 mM de MnCl2; 100 mM de KCl; 10 mM CaCl2 e 15% de
glicerol) gelado. As células foram novamente centrifugadas a 4000 rpm e 4ºC por 8 minutos e
ressuspendidas em 10 mL de TFB2 (10 mM de MOPS pH 7; 75 mM de CaCL2.2H2O; 10
mM de KCl e 15% de glicerol) gelado. As células foram mantidas em gelo e uma alíquota de
100 μL foi transferida para um microtubo de 1,5 mL previamente gelado. As células foram
então congeladas em nitrogênio líquido e estocadas à -80ºC.
3.4.2 Reação de transformação
Para a transformação das bactérias competentes, 20 μL do vetor pET SUMO contendo
o gene da proteína U5-15Kde T.brucei foram misturados a 80 μL de tampão de transferência
(100 mM de KCl; 30 mM de CaCl2; 50 mM de MgCl2 e 1% de PEG 6000 ou PEG 4000),
adicionados a 100 μL da bactéria. A mistura foi mantida em gelo por 30 minutos e em seguida
10 minutos a temperatura ambiente. Em seguida foi adicionado 1 mL de meio Luria Bertani,
LB (1% de triptona; 0,5% de extrato de levedura e 1% de NaCl) e incubados a 37ºC por 50
minutos. Por fim, 200 μL da cultura foram plaqueados em meio LB sólido (1% de triptona;
0,5% de extrato de levedura; 1% de NaCl e 1,5% de ágar) contendo 30 μgmL-1 do antibiótico
canamicina e incubados a 37ºC por 16 horas.
3.5 Expressão da proteína recombinante
3.5.1 Expressão da U5-15K
O sistema de expressão de proteína pET SUMO (InvitrogenTM) utiliza um pequeno
modificador semelhante a uma ubiquitina (small ubiquitin-like modifier, SUMO). A fusão
com esse modificador de 11 kDa pode aumentar a expressão e solubilidade da proteína

53
recombinante. A SUMO fusionada ao N-terminal da proteína de interesse é facilmente
reconhecida pela SUMO protease e a sua clivagem resulta na proteína nativa.
As bactérias da cepa BL21(DE3)pLysS, transformadas com a construção U5-
15K/pETSUMO, foram inoculadas em 5 mL de meio LB contendo Kanamicina (50 μg mL-1)
e incubadas sob agitação de 250 rpm a 37ºC por 16 horas. Após o crescimento das células,
foram usados 10 mL desse pré-inóculo em 1 L de meio LB líquido. Esse meio foi mantido sob
agitação de 150 rpm a 37ºC por uma hora, em seguida a temperatura foi reduzido para 20ºC.
Quando a D.O a 600 nm da cultura era de aproximadamente 0,6 a expressão da proteína foi
induzida com 0,6 mM de IPTG por 16 horas.
Após a expressão cada litro de cultura foi centrifugado a 4500 rpm, 4ºC por 40
minutos e o sedimento foi congelado (-20ºC). O sedimento descongelado foi ressuspendido
em 50 mL de Tampão de lise (150 mM de NaCl e 20 mM de Tris HCl pH 7,8) e submetido a
ultrassonicação por 10 ciclos (pulsos de 30s com intervalo de 45s) no equipamento . O lisado
foi então centrifugado a 13000 rpm, 4ºC por 30 minutos e o sobrenadante foi separado para a
purificação.
3.5.2 Expressão da SUMO protease
A expressão da SUMO protease clonada em pET 28a(+) seguiu a metodologia descrita
no item 3.5.1 com algumas modificações. A indução foi feita com 0,1 mM de IPTG por
aproximadamente 24h. Após a expressão, as culturas foram centrifugadas e congeladas como
descrito no item anterior. O sedimento descongelado foi ressuspenso em 50 mL de tampão de
lise (150 mM de NaCl e 20 mM de Tris HCl pH 7.5 ) e submetido a ultrassonicação. O lisado
foi centrifugado para a separação das proteínas solúveis e insolúveis, como descrito no item
anterior.
3.6 Purificação da proteína recombinante
3.6.1 Purificação da U5-15k
A purificação da U5-15K foi realizada em duas etapas. Na primeira etapa foi realizada
a cromatografia de afinidade acoplada ao sistema de cromatografia ÄKTA explorer10 (GE),
utilizando uma coluna TALON™ Metal Affinity Resin com íons Co2+
imobilizados. Nessa
etapa a proteína fusionada com a SUMO e a cauda de histidina é separada das demais

54
proteínas da bactéria. Em seguida foi usada a SUMO protease para clivar a SUMO da
proteína de interesse, e procedeu-se com uma nova cromatografia de afinidade. Nessa etapa,
tanto a SUMO quanto a SUMO protease, que possuem cauda de histidina, continuam
interagindo com a resina, enquanto que a proteína recombinante de interesse passa livremente
pela coluna.
Para a primeira etapa a coluna foi lavada com 5 volumes de água mili-Q e equilibrada
com 5 volumes do tampão A (100 mM NaCl; 20 mM Tris HCl pH7,5). O sobrenadante (50
mL) resultante da lise de 1 L de cultura bacteriana descrita no item 3.4.1 foi lavado com
aproximadamente 8 volumes do tampão A, seguido por 4 volumes de tampão A com 25 mM
de imidazol, 4 volumes de tampão A contendo 200 mM de imidazol e 4 volumes de tampão A
contendo 500 mM de imidazol. Para a confirmação da purificação as frações das eluições de
25 mM e 200 mM de imidazol forma analisas através de eletroforese em gel de poliacrilamida
15% com SDS (SDS-PAGE). As alíquotas analisadas foram fervidas com tempão de amostra
(188 mM de Tris HCl pH 6,8; 6% de SDS; 30% de glicerol; 15% de β-mercaptoetanol; 0,01%
de azul de bromofenol) e, para a visualização das bandas, o gel foi corado com solução
corante (0,2% de coomassie brilliant blue R-250; 50% de metanol; 10% de ácido acético) e
descorado com ácido acético (10%) e metanol (45%).
Para os experimentos de clivagem, o imidazol foi retirado com uma coluna HiTrap
Desalting (GE Healthcare) acoplada ao Äkta e a solução foi mantida em gelo por 2 horas com
0,5 mg de SUMO protease. Para os outros experimentos o imidazol foi retirado por diálise a
4ºC, com duas trocas de tampão (1 L de tampão A). A solução de proteína foi incubada com
0,5 mg de SUMO protease durante o processo de diálise, e em seguida foi feita uma nova
cromatografia de afinidade para retirada da proteína de fusão SUMO, bem com da SUMO
protease.
Para a concentração, a amostra de interesse foi submetida à ultra-filtração em
membrana Milipore, sob rotação de 2500 g a 4ºC. A concentração de proteína foi determinda
utilizando espectrofotômetro NanodropTM 1000 (Thermo Scientific), que lê a absorbância, A,
a 280 nm e calcula a concentração, C, dado o coeficiente de extinção molar da proteína, ε, e o
caminho óptico percorrido, l, que nesse caso é 1 mm, usando a lei de Beer-Lambert (equação
1).
3.6.2 Cromatografia por exclusão de tamanho

55
Para confirmar se a pureza da amostra de U5-15K, foi feita a cromatografia de
exclusão molecular utilizando uma coluna HiLoad™ 16/60 Superdex 200 (GE) acoplada ao
sistema de cromatografia ÄKTA Explorer 10 (GE). A amostra de proteína com concentração
aproximada de 2mgmL-1
, foi aplicada em uma coluna previamente equilibrada com tampão
20mM Tris-HCl pH 7,5, 100mM NaCl e 2mM DTT.
3.6.3 Purificação da SUMO protease
Assim como o pET SUMO, o vetor de expressão pET 28a(+) (Novagen) também
fornece a produção da proteína fusionada com uma cauda de histidina. Dessa forma, a
purificação da SUMO protease também foi realizada por cromatografia de afinidade. A
coluna com íons Co2+
foi lavada com 4 volumes de água mili-Q e equilibrada com tampão B
(150 mM de NaCl, 20 mM de Tris HCl pH 7,5), em seguida aplicou-se o sobrenadante
proveniente da lise de 2 L da cultura bacteriana descrita no item 3.4.2. Lavou-se a coluna com
5 volumes do tampão B, 4 volumes de tampão B com 10 mM de imidazol e por fim a SUMO
protease foi eluída com 2 volumes de tampão B com 150 mM de imidazol.
Após a purificação, a amostra da proteína passou por uma diálise a 4ºC para a retirada
do imidazol, o tampão B foi trocado duas vezes de 6 em 6 horas. Em seguida adicionou-se 2
mM DTT; composto que impede a formação de ligações dissulfeto, evitando a agregação de
proteínas, além de ser essencial para a atividade da SUMO protease. A amostra foi
concentrada como descrito no item 3.6.1.
3.7 Caracterização da auto-clivagem
3.7.1 Teste de auto clivagem
Para demostrar que a U5-15K possui atividade de autoclivagem, como descrito para os
homólogos (1.8), a proteína pura foi incubada à temperatura ambiente por 5 dias. Duas
alíquotas foram retiradas para aplicação em SDS-PAGE 15%, uma logo após a purificação e
outra após diferente períodos de incubação.
3.7.2 Inibição da auto clivagem

56
Para os testes de inibição da autoclivagem, alíquotas da proteína recém purificada
(item 3.5.1) foram incubados com os seguintes inibidores: DTT (2 mM), EDTA (1 mM),
PMSF (2 mM), quimostatina (6 μgmL-1), leupeptina (10 μM), e uma alíquota controle sem
nenhum inibidor. Alíquotas foram retiradas de 12 em 12 horas para aplicação em SDS PAGE
20%. A cinética qualitativa da clivagem foi feita usando o software IMAGEJ para a
quantificação das bandas.
3.7.3 Caracterização do ponto de clivagem
Para determinar se o ponto de clivagem acontece na porção C ou N terminal, realizou-
se apenas a primeira parte da purificação (sem clivagem com SUMO protease). A amostra de
proteína foi então separada em dois microtubos de 1,5 mL. Em um deles adicionou-se 2 mM
de DTT. Alíquotas foram retiradas de 12 em 12 horas e analisadas em um gel SDS-PAGE
15%.
A predição do ponto de clivagem foi feita através da relação existente entre a massa da
proteína e a distância que a banda percorre no gel SDS-PAGE. As distâncias percorridas pelas
bandas do marcador de massa molecular do gel SDS-PAGE 20% (Figura 25b), normalizadas
pelo comprimento do gel, e o valor das massas, foram usados para construir uma curva de
logMM vs log (d/D), onde MM é a massa molecular, d a distância percorrida pela banda e D o
comprimento do gel. A massa da proteína clivada foi calculada a partir da equação dessa
curva.
3.8 Caracterização biofísica da U5-15k
3.8.1 Dicroísmo circular (CD)
O espectro de CD da U5-15K e sua variante clivada foram obtidos utilizando um
espectropolarímetro JASCO J-810 (JASCO International Co Ltd). O espectro foi coletado
entre 190 e 280 nm, com resolução de 0,2 nm e tempo de resposta de 0,5 segundo, em uma
cubeta de quartzo de caminho óptico de 0,1 cm. A concentração das proteínas eram de 0,1
mgmL-1 e 0,09 mgmL-1 para a U5-15K nativa e clivada, respectivamente. O programa usado
para registro dos dados foi o Spectra Manager (JASCO) e a deconvolução do espectro foi feita
usando o programa CONTIN111
.

57
3.8.2 Espalhamento de luz dinâmico (DLS)
Para esse experimento, foi utilizado o equipamento Zetasizer UV (Malvern), a amostra
de proteína concentrada a 4,5 mgmL-1
, purificada como descrito no item 3.5.1, concentrada
por ultrafiltração e contrifugada a 10000 g por 10 minutos, foi aplicada em uma cubeta de
quartzo para a realização das medidas em diferentes tampões. A seleção das medidas mais
representativas e cálculo do seu valor médio foi realizada pelo software ZETASIZER.
3.8.3 Espalhamento de raio X a baixo ângulo (SAXS)
Nesse trabalho, os dados experimentais de SAXS das amostras de U5-15K nas
concentrações 2mgmL-1 e 7mgmL-1 em tampão 20 mM de TRIS-HCl pH 7,5; 100 mM de
NaCl, 2mM de MgCl2, na presença de 2mM de DTT, foram coletados na linha SAXS2 do
Laboratório Nacional de Luz Síncrotron (LNLS, Campinas-SP) utilizando um comprimento
de onda de 1,488 Ӑ em um detector bidimensional a 1968,97 mm da amostra.
A análise sistemática dos resultados experimentais foi feita, inicialmente, através do
programa FIT2D, onde os dados foram normalizados pela intensidade do feixe e
multiplicados pela absorção da amostra. O espalhamento da amostra contendo apenas tampão
foi subtraído da curva de espalhamento da solução de proteína. As curvas de espalhamento
das amostras de concentrações diferentes foram fundidas usando o programa PRIMUS101
.
Dessa curva foram calculados o raio de giro da proteína, através da aproximação de Guinier,
com o programa AUTORG102
. O valor de I(0) foi utilizado pelo programa SAXS MoW103
para estimar a massa molecular da proteína. A função de distribuição de distâncias foi
calculada pelo método de transformada inversa de Fourier implementado pelo programa
GNOM104
. O CRYSOL105
foi usado para comparar os resultados experimentais com os da
estrutura tridimensional da U5-15K obtido no item 3.2. Por fim, 10 modelos independentes de
baixa resolução foram gerados pelo método ab initio através do programa DAMMIN118
e
foram premediados com o programa DAMAVER119
.
3.8.4 Fluorimetria diferencial de varredura (DSF)
Para a fluorimetria diferencial de varredura foi usada uma amostra de 0,1 mgmL-1 de
U5-15K em 10 mM de HEPES, pH 7,5. A sonda utilizada foi o Sypro Orange. Foram testados

58
diferentes sais e condições de pH, descritas nos kits StockOptionsTMSalt e
StockOptionsTMpH, Hampton Research. A varredura dessas condições foi realizada em
triplicata, em uma placa de 96 poços no aparelho CFX96™ Real-Time System (Bio-Rad) e a
análise das curvas foi feita com o software Bio-Rad CFX manager. Com as curvas de
desenovelamento foram feitos gráficos de ΔTm (Temperatura de desenovelamento da
condição menos a temperatura de desenovelamento da amostra em tampão HEPES 10mM pH
7,5) pela condição estudada (pH ou sal).
3.8.5 Ensaios de Cristalização
A triagem inicial das condições de cristalização da U5-15K de T. brucei nas
concentrações 4,5 mgmL-1, 7,5 mgmL-1 e 10 mgmL-1, bem como da variação clivada na
concentração 5mgmL-1 foi feita em placas de cristalização de 96 poços através do método de
difusão de vapor. Foram utilizados os kits comerciais Salt RT, Crystal Screen HT, Index HT
(Hampton Research), PEGII Suite (Qiagen), MORPHEUS HT, MIDAS, JCSG-plus HT
(Molecular Dimensions). O método usado foi o da gota apoiada, feitas com o robô Honeybee
939 (Molecular Dynamics), contendo 0,7μL de solução de proteína (20mM de Tris HCl pH
7,5; 100 mM NaCl; 2mM de DTT; 2 mM de MgCl2) e 0,7μL de solução precipitante.

59
4 Resultados e Discussão
4.1 Análise bioinformática da sequência de aminoácidos da proteína U5-15K
A U5-15K de T. brucei possui 155 aminoácidos, massa molecular de 17,71 kDa e
ponto isoelétrico teórico, calculado pela ferramenta PROTPARAM85
, de 6,07. A porcentagem
de aminoácidos na sequência está descrita na tabela abaixo (TABELA 1).
Tabela 1: Composição de aminoácidos na sequência da U5 -15K em porcentagem.
Resíduo Porcentagem
(%)
Resíduo Porcentagem
(%)
Resíduo Porcentagem
(%)
Ala (A) 9,0 Gly (G) 4,5 Pro (P) 1,9
Arg (R) 7,7 His (H) 3,9 Sec (U) 0,0
Asn (N) 3,9 Ile (I) 5,2 Ser (S) 5,8
Asp (D) 4,5 Leu (L) 7,7 Thr (T) 7,1
Cys (C) 0,6 Lys (K) 4,5 Trp (W) 0,6
Gln (Q) 1,3 Met (M) 2,6 Tyr (Y) 4,5
Glu (E) 9,7 Phe (F) 4,5 Val (V) 10,3
Fonte: Elaborada pelo autor.
O coeficiente de extinção molar calculado considerando todas as cisteínas reduzidas é
de 15930 M-1
cm-1
, valor usado para calcular a concentração da proteína em solução (3.6.1).
A análise de domínios feita pelo servidor SMART89
mostra a presença do domínio
Dim1 (PFAM PF02966), presente em proteínas da família Dim1. O domínio se estende do
aminoácido na posição 3 à 148 com e-value de 1.2e-36.
Figura 15 - Representação do domínio da U5 -15K de T. brucei
Fonte: SMART
89.
A análise com o servidor CDD88
mostra dois sítios críticos conservados dentro do
domínio Dim1, o primeiro composto pelos aminoácidos nas posições 12,14, 15 e 91-93 e o
segundo entre as posições 42 e 45.

60
Figura 16 - Alinhamento entre U5-15K de T. brucei, T. cruzi, L. major U5-15K humana (1QGV), Dim1 de
Plasmodium yoelli (2AV4) e Dim1de S. cerevisiae, gerado pelo programa ClustalW. As
marcações verdes representam hélices-α e as amarelas as fitas-β presentes nas estruturas
cristalográficas da U5-15K humana e Dim1 de P. yoelli, e preditas pelo programa PSIPRED.
As letras em vermelho representam o provável peptídeo resultante da autoclivagem.
Fonte: Elaborado pelo autor.
A U5-15K de T. brucei apresenta 73 % de identidade com a proteína de T. cruzi e
46% com a de L. major, o que mostra uma alta conservação da U5-15K entre os
trypanossomatídeos, e 41% de identidade com a U5-15K humana. Como as proteínas
pertencentes a família Dim1, a U5-15K não possui as cisteínas no motivo Cys-X-X-Cys típico
das tiorredoxinas, apesar de sua estrutura terciária (item 4.2) ser semelhante a uma
tiorredoxina.

61
4.2 Modelagem por homologia da U5-15K de T. brucei
O modelo para a proteína U5-15K de T. brucei foi obtido através do servidor SWISS-
MODEL92
, usando a estrutura cristalográfica da homóloga humana, U5-15K (identificação no
PDB 1QGV). O gráfico de Ramachandram (Figura 17) gerado pelo servidor SAVES mostra
85,8% dos resíduos em posições favoráveis, 12,7% em regiões adicionalmente permitidas e
1,5% em regiões generosamente permitidas. Esses resíduos são a Glu 123 e a Ser 30 e se
encontram em regiões de conexão entre duas estruturas secundárias e que não apresentam um
bom alinhamento com a sequência da U5-15K humana.
Figura 17 – Gráfico de Ramachandram do modelo da U5 -15K gerado pelo SWISS-MODEL92
.
Fonte: SAVES...93
.
O Z-score, que mede a qualidade do modelo de estrutura tridimensional de uma
proteína por meio da medida da energia total da estrutura em relação a uma função
distribuição de energia de conformações aleatórias, fornecendo uma faixa de valores
característicos para proteínas nativas comparadas às proteínas com estruturas conhecidas por
cristalografia ou RMN, foi calculado pelo servidor ProSa. O Z-score calculado foi de -4,87,
que está dentro dos valores típicos para proteínas com o mesmo número de resíduos na
sequência123
(Figura 18).
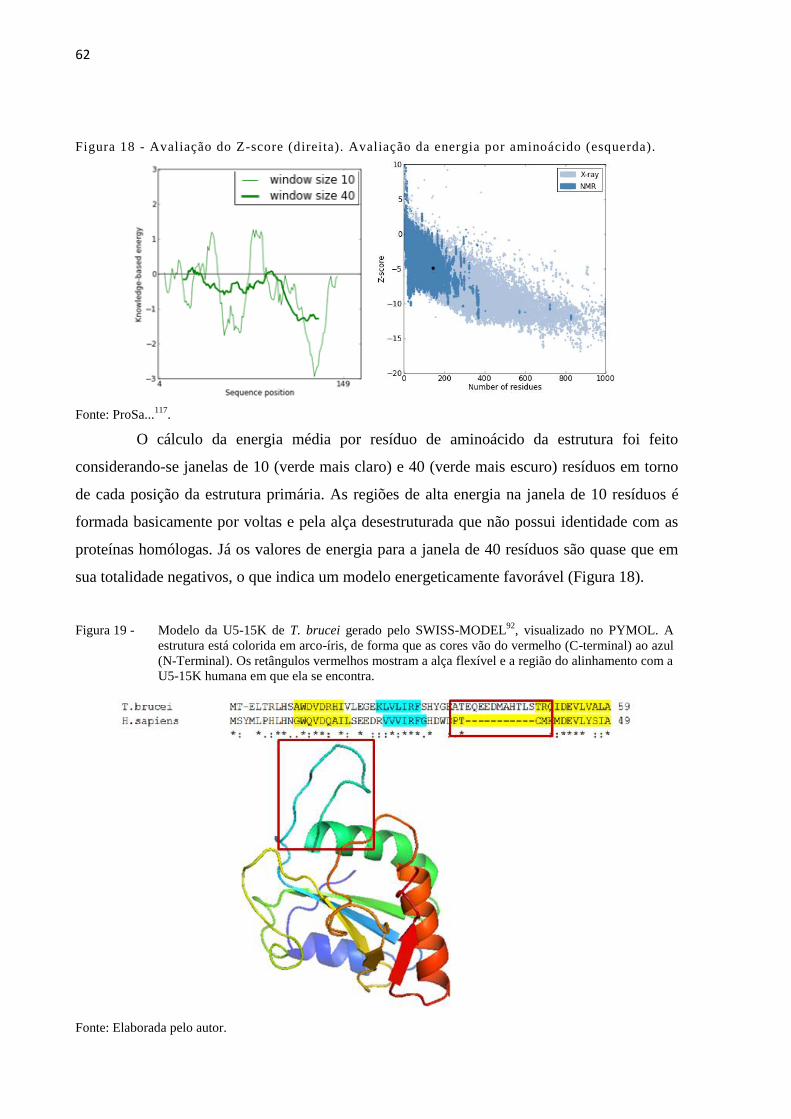
62
Figura 18 - Avaliação do Z-score (direita). Avaliação da energia por aminoácido (esquerda).
Fonte: ProSa...117
.
O cálculo da energia média por resíduo de aminoácido da estrutura foi feito
considerando-se janelas de 10 (verde mais claro) e 40 (verde mais escuro) resíduos em torno
de cada posição da estrutura primária. As regiões de alta energia na janela de 10 resíduos é
formada basicamente por voltas e pela alça desestruturada que não possui identidade com as
proteínas homólogas. Já os valores de energia para a janela de 40 resíduos são quase que em
sua totalidade negativos, o que indica um modelo energeticamente favorável (Figura 18).
Figura 19 - Modelo da U5-15K de T. brucei gerado pelo SWISS-MODEL92
, visualizado no PYMOL. A
estrutura está colorida em arco-íris, de forma que as cores vão do vermelho (C-terminal) ao azul
(N-Terminal). Os retângulos vermelhos mostram a alça flexível e a região do alinhamento com a
U5-15K humana em que ela se encontra.
Fonte: Elaborada pelo autor.

63
Figura 20 - Alinhamento das estruturaras da U5-15K de T. brucei (verde) e Humana (azul), os pontos em
vermelho representam as cisteínas.
Fonte: Elaborada pelo autor.
Como as outras proteínas da família Dim1, é possível ver o core bem modelado,
semelhante a uma tiorredoxina78
, formado por uma folha β, flanqueada por três hélices α, mas
diferentemente das homólogas que possuem estrutura cristalográfica, a proteína de T. brucei
apresenta uma alça flexível, na região de baixa identidade, que pode atrapalhar os ensaios de
cristalização (item 4.5.5). Também e possível ver uma fita β na porção C-terminal interagindo
com uma das fitas β mais externas do core, que nesse modelo aparece com um tamanho
reduzido se comparada à da homóloga humana. Outra diferença entre as estruturas é a posição
das cisteínas, que aparecem em regiões opostas (Figura 20).
4.3 Obtenção da proteína U5-15K de T. brucei
Devido a grande quantidade de proteína insolúvel obtida com a expressão no vetor
pET28a(+), a U5-15K foi clonada no vetor de expressão pET SUMO (item 3.5.1), as enzimas
de restrição usadas foram XhoI e BamHI.

64
Figura 21 - Diagrama do sitio de clonagem do vetor pET SUMO, mostrando a sequencia da proteína de
fusão SUMO.
Fonte: INVITROGEN™...116
.
O sequenciamento foi realizado pelo método Sanger e as sequências obtidas foram
analisadas usando a ferramenta BLAST102
do NCBI (National Center for Biothecnology
Information). O alinhamento da sequência gerada pelo sequenciamento com a sequência da
U5-15K de T. brucei (linha vermelha) e com a parte C-terminal da proteína SUMO (linha
rosa) estão representadas na figura abaixo.
Figura 22 - Alinhamento mostrando a ligação da U5 -15K no pET SUMO.
Fonte: Elaborado pelo autor.
As células da cepa BL21(DE3) pLysS, transformadas com a construção U5-15K/pET
SUMO (item 3.4.2) foi inoculada em 1L de meio de cultura LB e a expressão da proteína
recombinante foi induzida com 0,6 mM de IPTG, como descrito no item 3.5.1.

65
A primeira etapa da purificação da U5-15K foi feita por cromatografia de afinidade
em coluna de Co+2
, como descrito no item 3.6.1. Abaixo são mostrados o cromatograma e o
gel SDS-PAGE 15% obtidos nesta etapa (figuras 23 e 24).
Figura 23 - Cromatograma da primeira etapa de purificação da U5-15K, o primeiro pico corresponde às
proteínas que não aderiram a coluna e o pico mais definido, nas frações 16 a 20, corresponde
ao pico de eluição da U5-15K. A linha verde corresponde a concentração de imidazol no
tampão.
Fonte: Elaborada pelo autor.
Alíquotas das frações 16, 17, 18, 19 e 20 foram analisadas em gel SDS-PAGE 15%
para confirmação da purificação. A banda acima de 31kDa corresponde a U5-15K fusionada
com à SUMO e à cauda de histidinas (His-tag), como demonstrado na figura 24.

66
Figura 24 - Gel SDS-PAGE 15% corado com Comassie Blue contendo amostras após cromatografia de
afinidade. (1) Marcador de Massa Molecular, (2) fração 16, (3) fração 17, (4) fração 18, (5)
fração 19, (6) fração 20.
Fonte: Elaborada pelo autor.
Em seguida foi feita a diálise para a retirada do imidazol e clivagem com a SUMO
protease. A segunda parte da purificação também foi realizada utilizando cromatografia de
afinidade contendo íons cobalto imobilizado (item 3.6.1).
Figura 25 - Gel SDS-PAGE 15% corado com Comassie Blue contendo amostras da segunda parte da
purificação. (1) Marcador de massa molecular, (2) fração contendo SUMO protease e SUMO
eluída com 200mM de imdazol , (3) fração contendo U5-15K.
Fonte: Elaborada pelo autor.
A concentração da amostra de U5-15K após o segundo passo da purificação, medida
com o equipamento NanodropTM 1000 (Thermo Scientifc) foi de aproximadamente 1mgmL-1
em 10 mL de amostra.
Para avaliar a pureza da amostra (item 3.6.2) foi feita a cromatografia de exclusão
molecular, o cromatograma está representado abaixo (Figura 26).
67
Figura 26 - Cromatografia de exclusão molecular.
Fonte: Elaborada pelo autor.
Alíquotas das frações de 42 a 49 foram aplicadas em gel SDS-PAGE 15% (Figura 27).
Figura 27 - Gel SDS-PAGE 15% onde 1é o marcador de massa molecular, 2 a fração 42 da cromatografia
de exclusão molecular, 3 a fração 43, 4 a fração 44, 5 a fração 45, 6 a fração 46, 7 a fração 47, 8
a fração 48, 9 a fração 49.
Fonte: Elaborada pelo autor.
Tanto pelo gel quanto pelo cromatograma é possível confirmar que a U5-15K se
encontra pura na amostra.
4.4 Caracterização da auto-clivagem
4.4.1 Teste de auto-clivagem

68
Para verificar se a U5-15K de T. brucei apresenta atividade de auto-clivagem, como
observado na homóloga humana (item 1.8) uma alíquota da U5-15K recém purificada, na
concentração de aproximadamente 1mg mL-1, foi mantida a temperatura ambiente por cinco
dias. Para certificação da auto-clivagem amostras retiradas logo após a purificação e outra
incubada a temperatura ambiente foram analisadas em gel SDS-PAGE 15%. É possível ver
que a banda da amostra incubada é mais grossa do que a banda da amostra recém purificada, e
em uma posição inferior do gel (Figura 28).
Figura 28 - Gel SDS-PAGE 15% do teste de auto-clivagem. (1) Marcador de massa molecular, (2)
amostra logo após a purificação, 0h, (3) amostra 5 dias após a purificação, 120h.
Fonte: Elaborada pelo autor.
Pela diferença na posição das duas bandas e pelo arraste na amostra incubada, é
possível afirmar que há uma redução na massa da proteína ao longo do tempo, que concorda
com a afirmação de a U5-15K de T. brucei também apresenta atividade de auto-clivagem.
4.4.2 Teste de Inibição da auto-clivagem
Visto que a U5-15k de T. brucei apresenta atividade de autoclivagem, foram testadas
algumas moléculas para rastrear a inibição da autoclivagem, entre elas EDTA e quimostatina,
que, como visto no item 1.8, foram descritos como inibidores da autoclivagem na U5-15K
humana. Na tabela abaixo encontram-se as moléculas usadas no teste de inibição e suas
concentrações.

69
Tabela 2 - Candidatos a inibidores da auto -clivagem e suas concentrações.
Inibidor Concentração
EDTA 1 mM Quimostatina 6 µgmL
-1
PMSF 2 mM DTT 2 mM
Leupeptina 0,1 mM Fonte: Elaborada pelo autor.
A amostra de proteína recém purificada foi separada em seis microtubos de 1,5mL,
cada um contendo 500 μL da amostra na concentração de 1mg mL-1, como descrito no item
1.7.2. Após 112h, as alíquotas foram aplicadas em gel SDS-PAGE 20%. O aumento na
concentração de poliacridamida no gel permitiu melhor resolução, permitindo uma melhor
visualização das proteínas antes e depois da autoclivagem.
Figura 29 - Geis SDS-Page 20% do teste de inibição onde (a) controle, (b) leupeptina, (c)EDTA, (d)
quimostatina, (e) PMSF e (f) DTT. Em todos eles (1) é o marcador de massa molecular, (2) 0h,
(3)12h, (4) 24h, (5) 36h, (6) 48h, (7) 60h, (8) 72h, (9) 84h, (10) 96h, (11)108h.
Continua
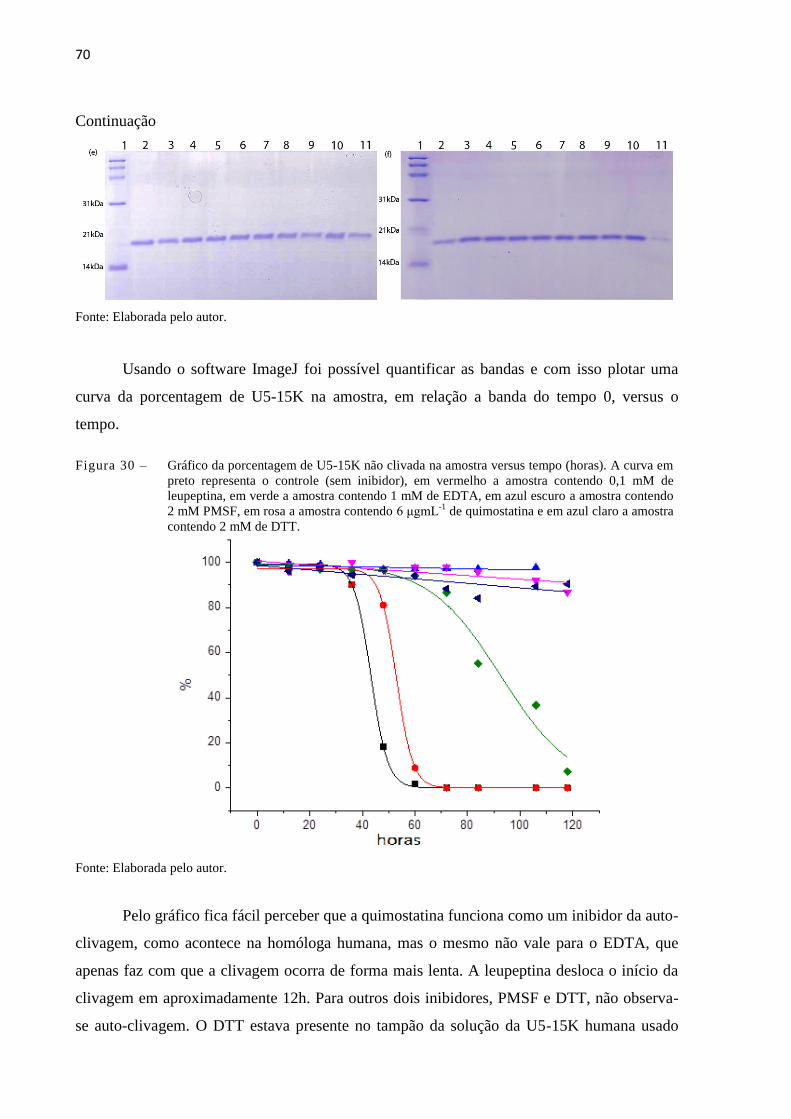
70
Continuação
Fonte: Elaborada pelo autor.
Usando o software ImageJ foi possível quantificar as bandas e com isso plotar uma
curva da porcentagem de U5-15K na amostra, em relação a banda do tempo 0, versus o
tempo.
Figura 30 –
Gráfico da porcentagem de U5-15K não clivada na amostra versus tempo (horas). A curva em
preto representa o controle (sem inibidor), em vermelho a amostra contendo 0,1 mM de
leupeptina, em verde a amostra contendo 1 mM de EDTA, em azul escuro a amostra contendo
2 mM PMSF, em rosa a amostra contendo 6 μgmL-1
de quimostatina e em azul claro a amostra
contendo 2 mM de DTT.
Fonte: Elaborada pelo autor.
Pelo gráfico fica fácil perceber que a quimostatina funciona como um inibidor da auto-
clivagem, como acontece na homóloga humana, mas o mesmo não vale para o EDTA, que
apenas faz com que a clivagem ocorra de forma mais lenta. A leupeptina desloca o início da
clivagem em aproximadamente 12h. Para outros dois inibidores, PMSF e DTT, não observa-
se auto-clivagem. O DTT estava presente no tampão da solução da U5-15K humana usado
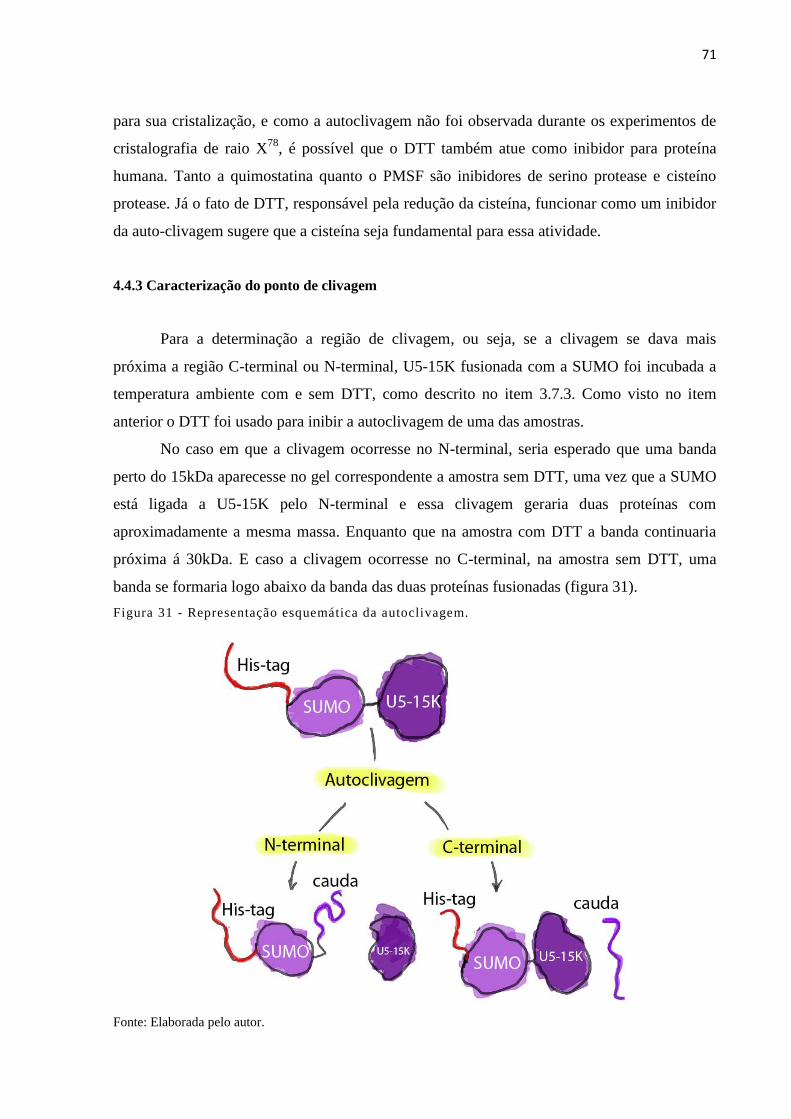
71
para sua cristalização, e como a autoclivagem não foi observada durante os experimentos de
cristalografia de raio X78
, é possível que o DTT também atue como inibidor para proteína
humana. Tanto a quimostatina quanto o PMSF são inibidores de serino protease e cisteíno
protease. Já o fato de DTT, responsável pela redução da cisteína, funcionar como um inibidor
da auto-clivagem sugere que a cisteína seja fundamental para essa atividade.
4.4.3 Caracterização do ponto de clivagem
Para a determinação a região de clivagem, ou seja, se a clivagem se dava mais
próxima a região C-terminal ou N-terminal, U5-15K fusionada com a SUMO foi incubada a
temperatura ambiente com e sem DTT, como descrito no item 3.7.3. Como visto no item
anterior o DTT foi usado para inibir a autoclivagem de uma das amostras.
No caso em que a clivagem ocorresse no N-terminal, seria esperado que uma banda
perto do 15kDa aparecesse no gel correspondente a amostra sem DTT, uma vez que a SUMO
está ligada a U5-15K pelo N-terminal e essa clivagem geraria duas proteínas com
aproximadamente a mesma massa. Enquanto que na amostra com DTT a banda continuaria
próxima á 30kDa. E caso a clivagem ocorresse no C-terminal, na amostra sem DTT, uma
banda se formaria logo abaixo da banda das duas proteínas fusionadas (figura 31).
Figura 31 - Representação esquemática da autoclivagem.
Fonte: Elaborada pelo autor.

72
Figura 32 – O gel (a) contem amostras incubadas com 2mM de DTT e o gel (b) amostras incubadas sem
DTT. Em ambos os géis (1) é o marcador de massa molecular, (2) amostra recém purificada
(0h), (3) 12h, (4)24h, (5)36h, (6)48h.
Fonte: Elaborada pelo autor.
Comparando os dois géis é possível ver que em ambos há um aumento na banda em
torno de 17kDa, o que pode ter sido causado pela clivagem no sitio de clivagem da SUMO
protease resultante de contaminação com SUMO protease. Olhando para as bandas próximas
a 30kDa é possível ver que na amostra do gel (b) (Figura 32), sem DTT, uma segunda banda
se forma logo abaixo da banda da U5-15K fusionada com a SUMO, o que não acontece com a
amostra do gel (a). Comparando esses resultados com o esquema da figura 27 é possível dizer
que a autoclivagem acontece no C-terminal, como foi observado para a homóloga humana.
A predição do ponto de clivagem da U5-15K de T. brucei foi feita através da relação
existente entre massa da proteína e a distância em que a banda aparece no gel SDS-PAGE
(item 3.7.3).
Figura 33- Curva do logaritmo da massa molecular (logMM) pelo logaritmo da distância percorrida pela
banda no gel pelo tamanho do gel (log (d/D)). O ponto vermelho indica a U5-15K clivada,
cuja massa foi calculada a partir da equação da reta.
Fonte: Elaborada pelo autor.

73
Da curva da figura 29 foi calculada sua equação de reta e aplicando a distância
percorrida pela banda da proteína clivada na equação, obteve-se que a massa da proteína
clivada é de 15,8 ± 0,3 kDa. Subtraindo esse valor da massa molecular da proteína inteira
(17,7 kDa) e dividindo pela massa média de um aminoácido (120Da), tem-se que o tamanho
do peptídeo clivado é de 15 ± 2 resíduos, esse resultado preliminar será confirmado por
espectrometria de massas.
Figura 34 – Representação po ponto de clivagem (vermelho) na estrutura da U5-15K de T. brucei (verde)
alinhada com a estrutura da U5-15K de H. sapiens (azul).
Fonte: Elaborada pelo autor.
4.5 Caracterização biofísica da U5-15K de T. brucei
4.5.1 Dicroísmo Circular
A análise da composição da estrutura secundárias das formas clivada e não clivada da
U5-15K de T. brucei foi feita através da espectroscopia de dicroísmo circular, como descrito

74
no item 3.8.1. Após o predição do ponto de clivagem, foi feita uma nova predição coeficiente
de extinção para a proteína clivada usando o PROTPARAM85
.
O espectro da amostra de 0,1 mgmL-1
de U5-15K está mostrado na figura 35, os picos
positivo em torno de 190 nm e negativo em torno de 210, são típicos de proteínas com
predominância de hélice-α (item 3.8.1).
Figura 35 – (Preto) Espectro de Dicroísmo Circular (CD) no UV distante da U5-15K de T.
brucei.(Vermelho) Espectro de Dicroísmo Circular (CD) no UV distante da U5-15K de T.
brucei clivada.
Fonte: Elaborada pelo autor.
A deconvolução do espectro experimental foi feita com o programa CONTIN do
servidor DichroWeb, os resultados estão apresentados na tabela 3, junto com uma comparação
da estrutura secundária predita pelo Psipred.
O espectro experimental de dicroísmo circular da amostra de 0,09 mgmL-1
da U5-15K
clivada é mostrado na figura 32. Esse espectro apresenta os picos próximos a 190nm e 210nm
ainda mais acentuados que o espectro da proteína não clivada.
A deconvolução do espectro de CS da U5-15K clivada tembém for realizada com o
programa CONTIN e os resultados estão apresentados na tabela 4.
Tabela 3 –
Deconvolução do espectro experimental dado em porcentagem de
estrutura secundária comparado com o predito (PSIPRED)
Hélice-α(%) Folha-β(%) Região desordenada (%)
CD 29 25 46 PsiPred 32 36 32
Fonte: Elaborada pelo autor.
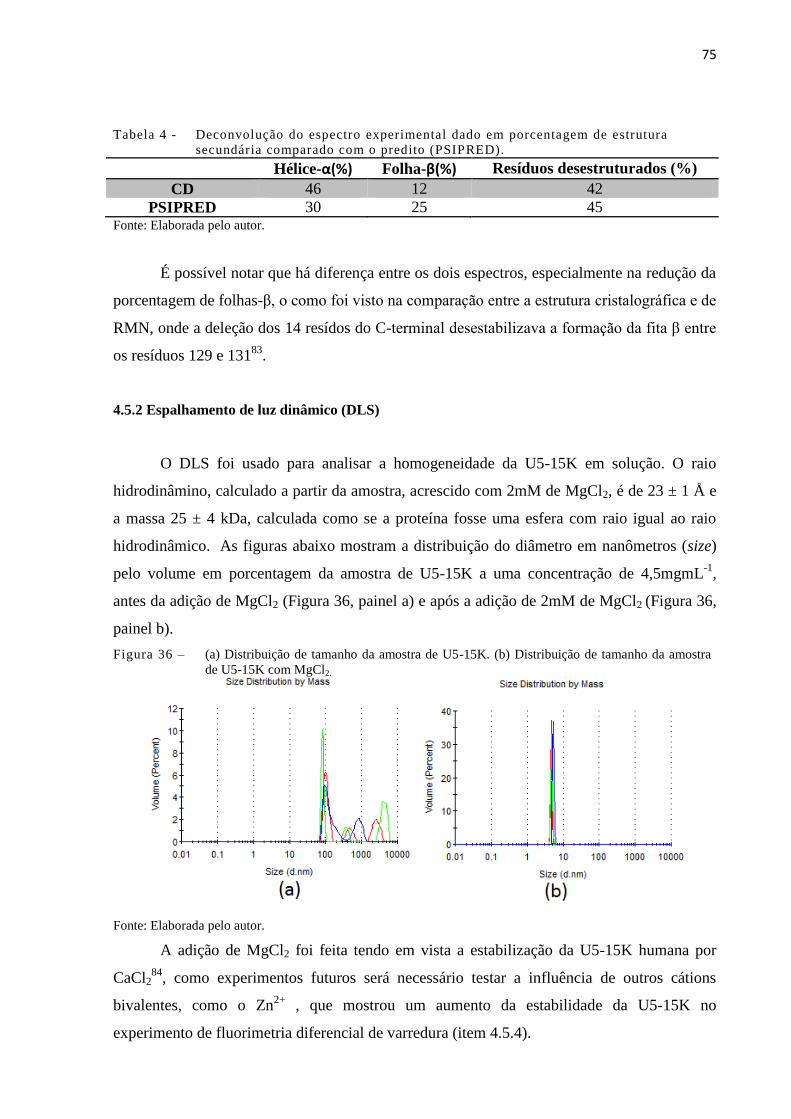
75
Tabela 4 - Deconvolução do espectro experimental dado em porcentagem de estrutura
secundária comparado com o predito (PSIPRED).
Hélice-α(%) Folha-β(%) Resíduos desestruturados (%)
CD 46 12 42
PSIPRED 30 25 45 Fonte: Elaborada pelo autor.
É possível notar que há diferença entre os dois espectros, especialmente na redução da
porcentagem de folhas-β, o como foi visto na comparação entre a estrutura cristalográfica e de
RMN, onde a deleção dos 14 resídos do C-terminal desestabilizava a formação da fita β entre
os resíduos 129 e 13183
.
4.5.2 Espalhamento de luz dinâmico (DLS)
O DLS foi usado para analisar a homogeneidade da U5-15K em solução. O raio
hidrodinâmino, calculado a partir da amostra, acrescido com 2mM de MgCl2, é de 23 ± 1 Å e
a massa 25 ± 4 kDa, calculada como se a proteína fosse uma esfera com raio igual ao raio
hidrodinâmico. As figuras abaixo mostram a distribuição do diâmetro em nanômetros (size)
pelo volume em porcentagem da amostra de U5-15K a uma concentração de 4,5mgmL-1
,
antes da adição de MgCl2 (Figura 36, painel a) e após a adição de 2mM de MgCl2 (Figura 36,
painel b).
Figura 36 – (a) Distribuição de tamanho da amostra de U5-15K. (b) Distribuição de tamanho da amostra
de U5-15K com MgCl2.
Fonte: Elaborada pelo autor.
A adição de MgCl2 foi feita tendo em vista a estabilização da U5-15K humana por
CaCl284
, como experimentos futuros será necessário testar a influência de outros cátions
bivalentes, como o Zn2+
, que mostrou um aumento da estabilidade da U5-15K no
experimento de fluorimetria diferencial de varredura (item 4.5.4).

76
A massa calculada, relativamente próxima a massa teórica da U5-15K, indica presença
de monômeros na solução, apesar do coeficiente de polidispersão ser próximo a 50%
Comparando as curvas geradas para as duas amostras é possível ver que a adição de MgCl2
diminui a agregação das moléculas, dessa forma é possível conseguir amostras mais
concentradas de U5-15K, necessárias para os experimentos de SAXS e ensaios de
cristalização.
4.5.3 Espalhamento de raio X a baixo ângulo (SAXS)
A técnica de SAXS foi empregada para duas concentrações diferentes de U5-15K não
clivada, 2mgmL-1
e 7mgmL-1
, as curvas experimentais são mostradas a seguir, bem como a
junção das duas curvas, realizada pelo programa PRIMUS101
.
Figura 37 – O gráfico da esquerda representa os dados experimentais de SAXS d e
U5-15K de T. brucei . O gráfico da direita é a junção dos dados
experimentais pelo programa PRIMUS101
.
Fonte: Elaborada pelo autor.
Com esses dados foram construídas a curva extrapolada para q = 0 (programa
GNOM102
, Figura 38) e o gráfico de Guinier (programa AutoRG, Figura 38), mostrados na
figura abaixo.
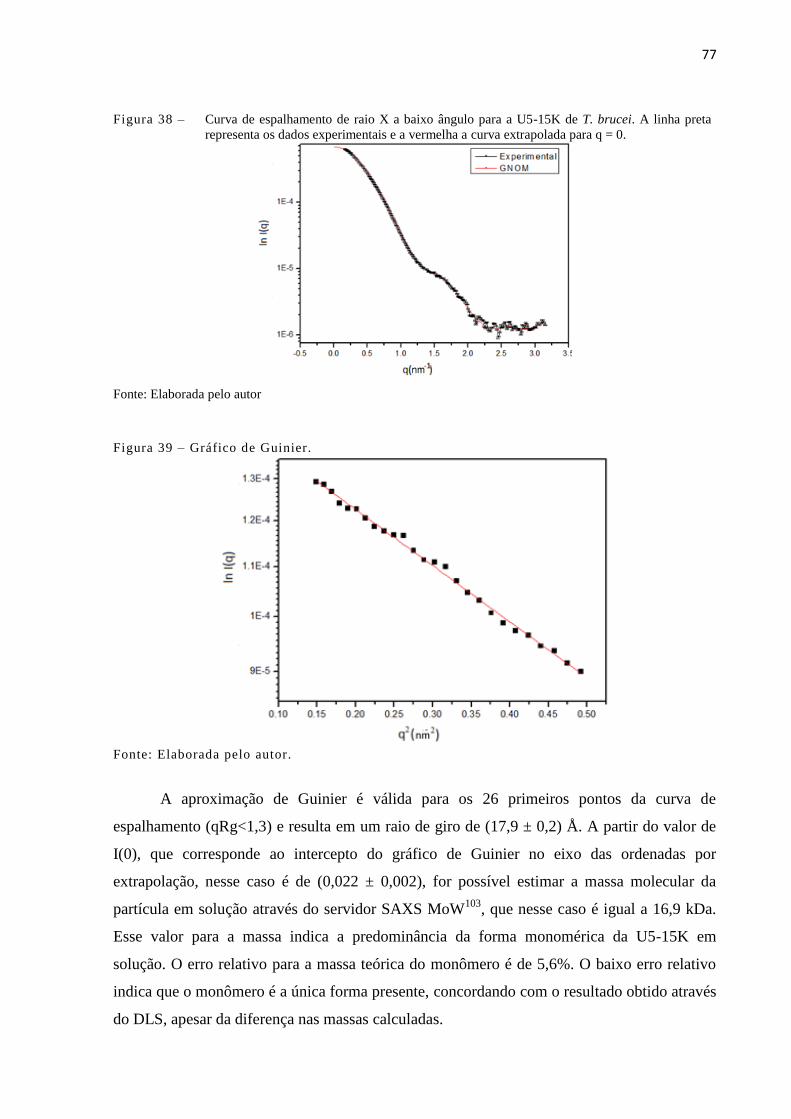
77
Figura 38 – Curva de espalhamento de raio X a baixo ângulo para a U5-15K de T. brucei. A linha preta
representa os dados experimentais e a vermelha a curva extrapolada para q = 0.
Fonte: Elaborada pelo autor
Figura 39 – Gráfico de Guinier.
Fonte: Elaborada pelo autor.
A aproximação de Guinier é válida para os 26 primeiros pontos da curva de
espalhamento (qRg<1,3) e resulta em um raio de giro de (17,9 ± 0,2) Å. A partir do valor de
I(0), que corresponde ao intercepto do gráfico de Guinier no eixo das ordenadas por
extrapolação, nesse caso é de (0,022 ± 0,002), for possível estimar a massa molecular da
partícula em solução através do servidor SAXS MoW103
, que nesse caso é igual a 16,9 kDa.
Esse valor para a massa indica a predominância da forma monomérica da U5-15K em
solução. O erro relativo para a massa teórica do monômero é de 5,6%. O baixo erro relativo
indica que o monômero é a única forma presente, concordando com o resultado obtido através
do DLS, apesar da diferença nas massas calculadas.
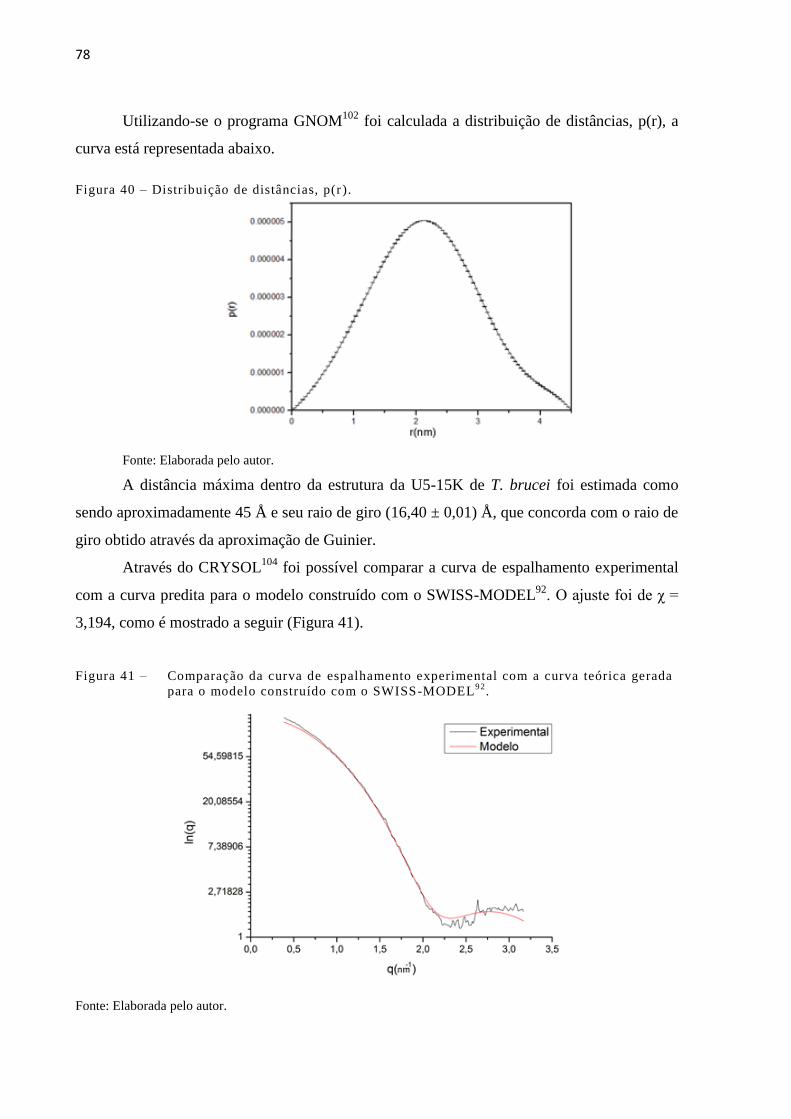
78
Utilizando-se o programa GNOM102
foi calculada a distribuição de distâncias, p(r), a
curva está representada abaixo.
Figura 40 – Distribuição de distâncias, p(r ).
Fonte: Elaborada pelo autor.
A distância máxima dentro da estrutura da U5-15K de T. brucei foi estimada como
sendo aproximadamente 45 Å e seu raio de giro (16,40 ± 0,01) Å, que concorda com o raio de
giro obtido através da aproximação de Guinier.
Através do CRYSOL104
foi possível comparar a curva de espalhamento experimental
com a curva predita para o modelo construído com o SWISS-MODEL92
. O ajuste foi de χ =
3,194, como é mostrado a seguir (Figura 41).
Figura 41 – Comparação da curva de espalhamento experimental com a curva teórica gerada
para o modelo construído com o SWISS-MODEL92
.
Fonte: Elaborada pelo autor.
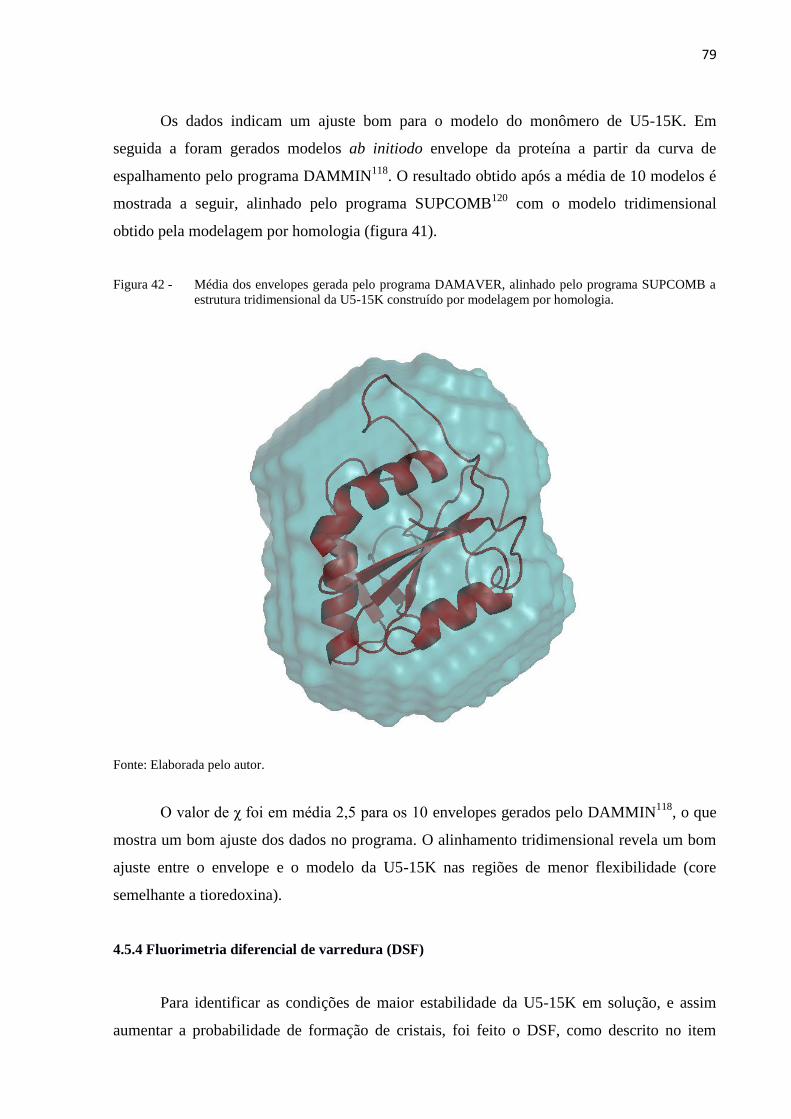
79
Os dados indicam um ajuste bom para o modelo do monômero de U5-15K. Em
seguida a foram gerados modelos ab initiodo envelope da proteína a partir da curva de
espalhamento pelo programa DAMMIN118
. O resultado obtido após a média de 10 modelos é
mostrada a seguir, alinhado pelo programa SUPCOMB120
com o modelo tridimensional
obtido pela modelagem por homologia (figura 41).
Figura 42 - Média dos envelopes gerada pelo programa DAMAVER, alinhado pelo programa SUPCOMB a
estrutura tridimensional da U5-15K construído por modelagem por homologia.
Fonte: Elaborada pelo autor.
O valor de χ foi em média 2,5 para os 10 envelopes gerados pelo DAMMIN118
, o que
mostra um bom ajuste dos dados no programa. O alinhamento tridimensional revela um bom
ajuste entre o envelope e o modelo da U5-15K nas regiões de menor flexibilidade (core
semelhante a tioredoxina).
4.5.4 Fluorimetria diferencial de varredura (DSF)
Para identificar as condições de maior estabilidade da U5-15K em solução, e assim
aumentar a probabilidade de formação de cristais, foi feito o DSF, como descrito no item

80
3.8.4. Primeiro foi testada a concentração salina mais adequada para a realização do
experimento. A temperatura de desenovelamento (Temperatura de Melting, Tm) está
relacionada com a estabilidade de proteína em solução, de forma que quanto mais estável a
proteína maior a Tm. O gráfico de Tm pela concentração salina mostra uma tendência
crescente da Tm com a redução da concentração salina. Isso indica que a U5-15K apresenta
maior estabilidade em menores concentrações de NaCl (Figura 43).
Figura 43 – Gráfico de DSF da temperatura de melting pela concentração de NaCl .
Fonte: Elaborada pelo autor.
A partir desse primeiro resultado, foram testadas condições de pH de 2,4 a 10,8. Pelo
gráfico é possível dizer que a proteína é mais estável (tem maior temperatura de melting) entre
6,4 e 6,8 e entre 7,4 e 7,6. Esse poder ser explicado pelo fato do splicing ocorrer no núcleo
celular, cujo pH está dentro dessa faixa.
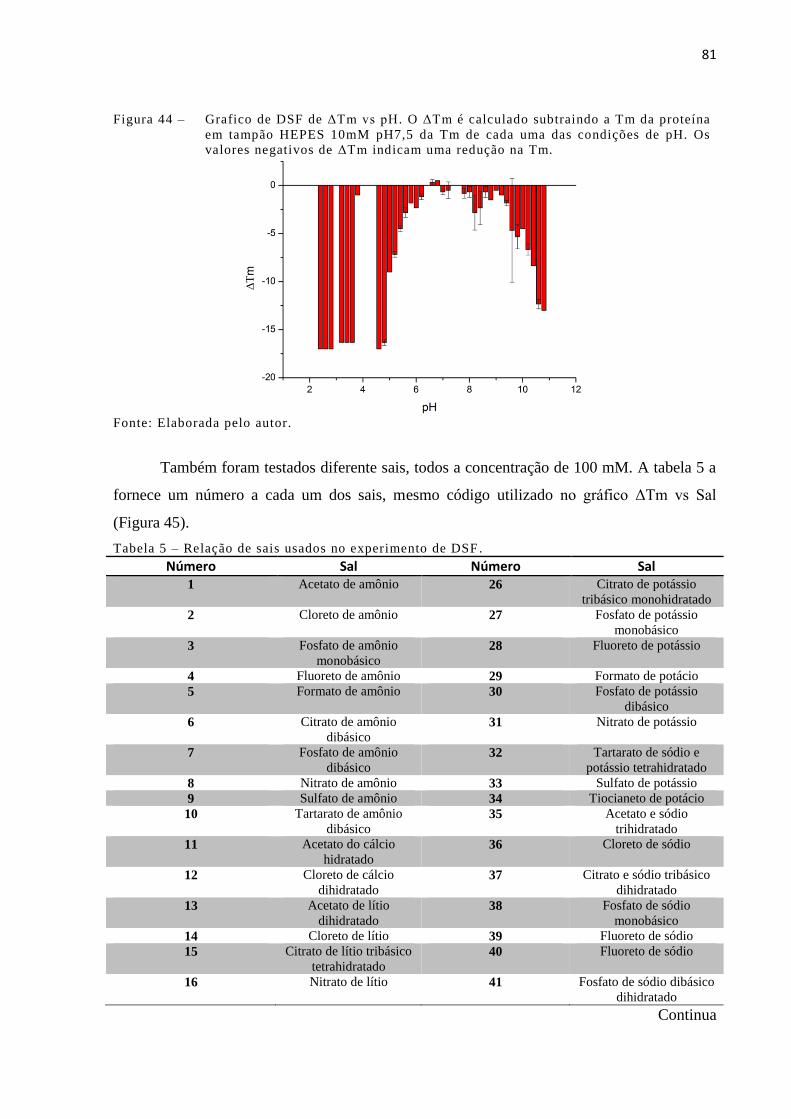
81
Figura 44 – Grafico de DSF de ΔTm vs pH . O ΔTm é calculado subtraindo a Tm da proteína
em tampão HEPES 10mM pH7,5 da Tm de cada uma das condições de pH. Os
valores negativos de ΔTm indicam uma redução na Tm.
Fonte: Elaborada pelo autor.
Também foram testados diferente sais, todos a concentração de 100 mM. A tabela 5 a
fornece um número a cada um dos sais, mesmo código utilizado no gráfico ΔTm vs Sal
(Figura 45).
Tabela 5 – Relação de sais usados no experimento de DSF .
Número Sal Número Sal 1 Acetato de amônio 26 Citrato de potássio
tribásico monohidratado
2 Cloreto de amônio 27 Fosfato de potássio
monobásico
3 Fosfato de amônio
monobásico 28 Fluoreto de potássio
4 Fluoreto de amônio 29 Formato de potácio
5 Formato de amônio 30 Fosfato de potássio
dibásico
6 Citrato de amônio
dibásico 31 Nitrato de potássio
7 Fosfato de amônio
dibásico 32 Tartarato de sódio e
potássio tetrahidratado
8 Nitrato de amônio 33 Sulfato de potássio
9 Sulfato de amônio 34 Tiocianeto de potácio
10 Tartarato de amônio
dibásico 35 Acetato e sódio
trihidratado
11 Acetato do cálcio
hidratado 36 Cloreto de sódio
12 Cloreto de cálcio
dihidratado 37 Citrato e sódio tribásico
dihidratado
13 Acetato de lítio
dihidratado 38 Fosfato de sódio
monobásico
14 Cloreto de lítio 39 Fluoreto de sódio
15 Citrato de lítio tribásico
tetrahidratado 40 Fluoreto de sódio
16 Nitrato de lítio 41 Fosfato de sódio dibásico
dihidratado
Continua

82
Continuação
17 Sulfato de lítio
monobásico 42 Malonato de sódio pH 7,0
18 Acetato de magnésio
tetrahidratado 43 Nitrato de sódio
19 Cloreto de magnésio
hexahidratado 44 Sulfato de sódio
decahidratado
20 Formato de magnésio
dihidratado 45 Tartarato de sódio
dibásico dihidratado
21 Nitrato de magnésio
hexahidratado 46 Tiocianato de sódio
22 Sulfato de magnésio
hidratado 47 Ácido succinico pH 7.0
23 Cloreto de níquel (II)
hexahidratado 48 Acetato de zinco
dihidratado
24 Acetato de potássio 49 Sulfato de zinco
heptahidratado
25 Cloreto de potássio 50 e 51 água
Fonte: Elaborada pelo autor.
Figura 45 – Grafico de DSF de ΔTm vs sal. O ΔTm é calculado subtraindo a Tm da proteína
em tampão HEPES 10mM pH7,5 da Tm de cada uma das soluções salinas. Os
valores negativos de ΔTm indicam uma redução na Tm.
.
Fonte: Elaborada pelo autor.
O gráfico da relação entre os diferentes sais e o ΔTm mostra que praticamente todos os
sais, incluindo o MgCl2, resultaram em uma diferença de temperatura de desenovelamento
negativa, ou seja, tem efeito negativo na estabilidade da U5-15K, embora o DLS tenha
mostrado que a adição de MgCl2 resulta em uma maior homogeneidade da solução. A
diferença nos resultados se deve à diferença de concentração da proteína usada no
experimento de DLS (4,5 mg mL-1
) e DSF (0,1 mg mL-1
). No DSF, pela baixa concentração
de proteína, o efeito da aglomeração dessas moléculas não é significativo. Antes de considerar
algum dos sais testados como um bom aditivo para a solução de cristalização seria necessário

83
estudar o comportamento da proteína em altas concentrações nas soluções salinas descritas na
tabela 5 utilizando o DLS.
4.5.5 Ensaios de Cristalização
Com o objetivo de obter informação de alta resolução da estrutura tridimensional da
U5-15K de T. brucei, foram realizadas triagens iniciais das condições de cristalização, como
descrito no item 3.8.5. As condições que resultaram em cristais estão descritas na tabela
abaixo.
Tabela 6 – Condições de cristalização do kit Morpheus HT (Molecular Dimensions) que resultaram no
crescimento de cristais.
1 2 3 4
10 % w/v PEG
20000, 20 % v/v
PEGMME 550 0.1 M
MES/Imidazol pH
6.5 0.03 M nitrato de
sódio, 0.03 M di-
hidrogenofosfato
de sódio, 0.03 M sulfato de
amônio
10 % w/v PEG
8000, 20 % v/v
etilenoglicol 0.1 M
MOPS/HEPES-Na
pH 7.5 0.03 M nitrato de
sódio, 0.03 M di-
hidrogenofosfato
de sódio, 0.03 M sulfato de
amônio
10 % w/v PEG
8000, 20 % v/v
etilenoglicol 0.1 M
MES/Imidazol pH
6.5 0.03 M nitrato de
sódio, 0.03 M di-
hidrogenofosfato
de sódio, 0.03 M sulfato de
amônio
12.5 % w/v PEG
1000, 12.5% w/v
PEG 3350, 12.5%
v/v MPD 0.1 M
MES/Imidazol pH
6.5 0.03 M nitrato de
sódio, 0.03 M di-
hidrogenofosfato
de sódio, 0.03 M sulfato de
amônio
Fonte: Elaborada pelo autor.
Figura 46 – Fotos dos cristais gerados nas condições descritas na tabela 6. A numeração das fotos
corresponde à numeração da tabela.
Continua

84
Continuação
Fonte: Elaborada pelo autor.
Apenas algumas condições do kit comercial Morpheus resultaram em cristais para a
U5-15K inteira, entretanto, para a proteína clivada nenhum cristal foi observado. A partir do
padrão de difração apresentado por esses cristais ao serem testados no equipamento de
difração de raio X, concluiu-se que na verdade se tratam de cristais de sal. Para solucionar
esse problema será necessário verificar a influência de outros sais com espalhamento
dinâmico de luz (DLS).
Entre os possíveis motivos da dificuldade de cristalização da U5-15K de T. brucei
encontra-se a grande flexibilidade conferida à molécula pela alça desestruturada vista no
modelo por homologia. Uma solução para esse problema seria uma proteólise limitada da
alça, seguida de uma nova purificação por cromatografia de exclusão molecular. A proteólise
limitada consiste no uso de proteases para a retirada das regiões flexíveis, permitindo que
apenas o domínio cristalize113
. Essa técnica já foi usada com sucesso no laboratório pela Dra.
Lívia Faim114
.

85
5 Conclusões
Nesse trabalho foram apresentados os resultados da purificação da proteína
recombinante U5-15K de Trypanosoma brucei, sua caracterização estrutural e caracterização
da atividade de auto-clivagem, bem como quais inibidores de protease interferem nesta
atividade.
O estudo através de ferramentas de bioinformática revela que a proteína faz parte da
família Dim1, que dentre outras funções estão presente proteínas spliceossomais com
estrutura terciária semelhante a tioredoxina. Observou-se a conservação de possíveis sítios
ativos e identidade de 73% com a homóloga de T. cruzi, 46% com a homóloga de L. major,
mostrando que há uma grande conservação entre as proteínas dos tripanossomatídeos. A U5-
15K de T. brucei também apresenta identidade de 41% com a homóloga humana (Figura 15),
cuja estrutura já foi elucidada por cristalografia de raio X e RMN, permitindo que um modelo
por homologia fosse construído para a proteína de T. brucei. A partir do modelo foi visto que
não há conservação da posição da cisteínas na estrutura e que a U5-15K de T. brucei possui
uma região de alça grande onde a sequência não se alinha com as duas proteínas que possuem
estrutura resolvida por cristalografia de raio X.
Os experimentos de clivagem mostraram que, como a homóloga humana e de S.
cerevisiae, a U5-15K possui atividade de auto-clivagem no C-terminal, aproximadamente na
mesma região, apesar da sequência da porção C-terminal não se conservar. Esses
experimentos também mostraram relativa semelhança entre os inibidores dessa atividade.
Os espectros de CD mostram uma diminuição na porcentagem de folha-β na estrutura
secundária da U5-15K clivada em comparação com a nativa, já que a fita- β mais próxima a
porção C-terminal é perdida com a clivagem, assim como a interação com outra fita- β mais
interna, o que pode ter levado a sua desestruturação, como foi visto no modelo de RMN da
homóloga humana. Foi possível construir um envelope de baixa resolução para a U5-15K a
partir dos dados de SAXS, que se ajustaram bem ao modelo construído por homologia.
Os experimentos de DLS e DSF mostraram melhores condições para manter U5-15K
em solução, homogênea e em concentrações mais elevadas; fundamentais para os
experimentos de SAXS e ensaios de cristalização. Apesar das diversas condições de
concentração e soluções de cristalização, os ensaios de cristalização não resultaram em
cristais.

86

87
6 Perspectivas
Determinação exata do ponto de clivagem através de experimentos de
Espectrometria de Massas.
Estudo das mutações nos possíveis sítios ativos indicados pelo alinhamento
com outras proteínas da família Dim1, bem como no sítio de clivagem.
Proteólise limitada das regiões flexíveis da estrutura terciária e novos ensaios
de cristalização.
Estudo da interação entre as proteínas U5-15K e U5-102K e sua importância
para a formação do spliceosomo.

88

89
Referências
1 BRUN, A.; BLUM, J.; CHAPPUIS, F.; BURRI, C. Human African Trypanosomiasis.
Lancet, v. 375, n. 9709, p148-59, 2010.
2 ROBAYS, J.; EBEJA KADIMA, A.; LUTUMBA, P.; MIAKA MIA BILENGE, C.;
KANDE BETU KU MESU, V.; DE DEKEN R.; MAKABUZA, J.; DEGUERRY M.; VAN
DER STUYFT, P.; BOELAERT, M. Human African trypanosomiasis amongst urban
residents in Kinshasa: a case-control study. Tropical Medicine and International Health, v.
9, n. 8, p. 869–875, 2004.
3 SIMARRO, P. P.; JANNIN, J.; CATTAND, P. Eliminating human african trypanosomiasis:
Where do we stand and what comes next. PLoS Medicine, v. 5, n. 2, p.55. 2008.
4 SIMARRO, P. P.; CECCHI, G.; FRANCO J. R.; PAONE, M.; DIARRA, A.; RUIZ-
POSTIGO J. A.; FÈVRE, E. M.; MATTIOLI, R. C.; JANNIN, J. G. Estimating and mapping
the population at risk of sleeping sickness. PLoS Neglected Tropical Diseases, v. 6, n. 10 , p.
1859, 2012.
5 JORDAN, A.M. Tsetse-flies (Glossinidae). In: LANE, R. P.; CROSSKEY, R. W.; (Ed).
Medical insects and arachnids. London: Chapman and Hall, 1993: 333–88.
6 STEVENS, J. R.; BRISSE, S. Systematics of trypanosomes of medical and veterinary
importance. In: MAUDLIN, I.; HOLMES, P. H.; MILES, M. A.; (Ed). The
trypanosomiases. Wallingford: CABI Publishing, 2004. p.1–23.
7 SIMARRO, P. P.; CECCHI, G.; FRANCO, J. R.; PAONE, M.; FÈVRE, E.M.; DIARRA,
A.; POSTIGO, J. A.; MATTIOLI, R. C.; JANNIN, J. G. Risk for human African
trypanosomiasis, Central Africa, 2000-2009. Emerging Infectious Diseases, v. 17, n. 12, p.
2322-2324, 2011.
8 FRANCO, J. R.; SIMARRO, P. P.; DIARRA, A.; RUIY-POSTIGO, J. A.; JANNIN, J. G.;
The journey towards elimination of gambiense human African trypanosomiasis: not far, nor
easy. Parasitology, v. 141, n. 6, p. 748–760, 2014.
9 WORLD HEALTH ORGANIZATION. Accelerating work to overcome neglected
tropical diseases: a roadmap for implementation. 2012. Disponivel em: <
http://whqlibdoc.who.int/hq/2012/WHO_HTM_NTD_2012.1_eng.pdf> Acesso em: 21
jan.2015.
10 SIMARRO, P. P.; CECCHI, G.; PAONE, M.; FRANCO, J. R.; DIARRA, A.; RUIZ, J. A.;
FÈVRE, E. M.; COURTIN, F.; MATTIOLI, R. C.; JANNIN, J. G. The Atlas of human
African trypanosomiasis: a contribution to global mapping of neglected tropical diseases.
International Journal of Health Geographics, v.9, p.57, 2010. doi: 10.1186/1476-072X-9-
57
11 KENNEDY, P. G. E.; Diagnostic and neuropathogenesis issues in human African
trypanosomiasis. International Journal of Parasitology, v. 36, n. 5, p. 505-512, 2006.

90
12 BRUN, R.; BLUM, J.; CHAPPUIS, F.; BURRI, C. Human african trypanosomiasis.
Lancet, v. 375, n. 9709, p. 148-159, 2010.
13 TRUC, P.; LEJON, V.; MAGNUS, E.; JAMONNEAU, V.; NANGOUMA, A.; VERLOO,
D.; PENCHENIER, L.; BÜSCHER, P. Evaluation of the micro-CATT, CATT/Trypanosoma
brucei gambiense, and LATEX/T b gambiense methods for serodiagnosis and surveillance of
human African trypanosomiasis in west and central Africa. Bulletin of World Health
Organization, v. 80, n. 11, p. 882–886, 2002.
14 MUGASA, C. M.; ADAMS, E. R.; BOER, K. R.; DYSERINCK, H. C.; BÜSCHER, P.;
SCHALLING, H. D.; LEEFLANG, M. M. Diagnostic accuracy of molecular amplifi cation
tests for human African trypanosomiasis—systematic review. PLoS Neglected Tropical
Diseases, v. 6, n.1, e1438, 2012. doi: 10.1371/journal.pntd.0001438.
15 WORLD HEALTH ORGANIZATION. Control and surveillance of African
trypanosomiasis. 1998. Disponível em: < http://whqlibdoc.who.int/trs/WHO_TRS_881.pdf>
Acesso em: 21 jan.2015. 16 FAIRLAMB, A. H. Chemotherapy of human African trypanosomiasis: current and future
prospects. Trends in Parasitology, v. 19, n. 11, p. 488-494, 2003.
17 FAIRLAMB, A. H.; HENDERSON, G. B.; CERAMI, A. Trypanothione is the primary
target for arsenical drugs against African trypanosomes. Proceedings of the National
Academy of Sciences, v. 86, n. 8, p. 2607-2611, 1989.
18 KENNEDY, P. G. E. Diagnosing central nervous system trypanosomiasis: two stage or not
to stage? Transactions of the Royal Society of Tropical Medicine and Hygiene, v. 102, n.
4, p. 306-307, 2008.
19 BALASEGARAM, M.; HARRIS, S.; CHECCHI, F.; GHORAHSOAN, S.; HAMEL, C.;
KARUNAKARA, U. Treatment outcomes and risk factors for relapse in patients with early-
stage human African trypanosomiasis (HAT) in the Republic of the Congo. Bulletin of
World Health Organization, v. 84, n. 10, p. 777-782, 2006.
20 MOORE, M.; QUERY, C. C.; SHARP, P. A. Splicing of precursors to mRNA by the
spliceosome. In GESTELAND, R. F.; ATKINS J. F. (Ed), The RNA world. New York :
Cold Spring Harbor Press, 1993. p. 303-357.
21 ULLU, E.; TSCHUDI, C.; GUNZL, A. Trans-splicing in trypanosomaid protozoa. In:
SMITH, D. F.; PARSONS, M. (Ed), Molecular biology of parasitic protozoa. Oxford : IRL
Press, 1996. p.115-133.
22 KRAUSE, M.; HIRSH, D. A trans-spliced leader sequence on actin mRNA in C. elegans.
Cell, v. 49, n. 6, p. 753-761, 1987.
23 TESSIER, L. H.; KELLER, M.; CHAN, R. L.; FOURNIER, R.; WEIL, J. H.; IMBAULT,
P. Short leader sequences may be transferred from small RNAs to pre-mature mRNAs by
trans-splicing in Euglena. EMBO Journal, v. 10, n. 9, p. 2621-2625, 1991.

91
24 RAJKOVIC, A.; DAVIS, R. E.; SIMONSEN, J. N.; ROTTMAN, F. M. A spliced leader is
present on a subset of mRNAs from the human parasite Schistosoma mansoni. Proceedings
of the National Academy of Sciences, v. 87, n. 22, p. 8879-8883, 1990.
25 ZHANG, H.; HOU, Y.; MIRANDA, L.; CAMPBELL, D. A.; STURM, N. R.;
GAASTERLAND, T.; LIN, S. Spliced leader RNA trans-splicing in dinoflagellates.
Proceedings of the National Academy of Sciences, v.104, n. 11, p. 4618-4623, 2007.
26 VANDENBERGHE, A. E.; MEEDEL, T. H.; HASTINGS, K. E. mRNA 5'- leader trans-
splicing in the chordates. Genes & Development, v. 15, n. 3, p. 294- 303, 2001.
27 DENKER, J. A.; ZUCKERMAN, D. M.; MARONEY, P. A.; NILSEN, T. W. New
components of the spliced leader RNP required for nematode trans-splicing. Nature, v. 417,
n. 6889, p. 667-670, 2002.
28 BOOTHROYD, J. C.; CROSS, G. A. Transcripts coding for variant surface glycoproteins
of Trypanosoma brucei have a short, identical exon at their 5' end. Gene, v. 20, n. 2, p. 281-
289, 1982.
29 PARSONS, M.; NELSON, R. G.; WATKINS, K. P.; AGABIAN, N. Trypanosome
mRNAs share a common 5' spliced leader sequence. Cell, v. 38, n. 1, p. 309-316, 1984.
30 AGABIAN, N. Trans splicing of nuclear pre-mRNAs. Cell, v. 61, n.7, p. 1157–1160,
1990.
31 MAIR, G.; SHI, H.; LI, H.; DJIKENG, A.; AVILES, H. O.; BISHOP, J. R.; FALCONE, F.
H.; GAVRILESCU, C.; MONTGOMERY, J. L.; SANTORI, M. I.; STERN, L. S.; WANG,
Z.; ULLU, E.; TSCHUDI, C. A new twist in trypanosome RNA metabolism: cis-splicing of
pre-mRNA. RNA, v. 6, n. 2, p. 163–169, 2000.
32 HERMANN, H.; FABRIZIO, P.; RAKER, V. A.; FOULAKI, K.; HORNIG, H.;
BRAHMS, H.; LÜRHMANN, R. snRNP Sm proteins share two evolutionarily conserved
sequence motifs which are involved in Sm protein interactions. EMBO Journal, v. 14, n. 9,
p. 2076-2088, 1995.
33 SÉRAPHIN, B. Sm and Sm-like proteins belong to a large family: identification of
proteins of the U6 as well as the U1, U2, U4 and U5 snRNPs. EMBO Journal, v. 14, n.9, p.
2089-2098, 1995.
34 KAMBACH, C.; WALKE, S.; YOUNG, R.; AVIS, J. M.; LA FORTELLE, E.; RAKER,
V. A.; LÜHRMANN, R.; LI, J.; NAGAI, K. Crystal structures of two Sm protein complexes
and their implications for the assembly of the spliceosomal snRNPs. Cell, v. 96, n. 3, p. 375-
387, 1999.
35 TOOR, N.; KEATING, K. S.; TAYLOR, S. D.; PYLE, A. M. Crystal structure of
a self-spliced group II intron. Science, v. 320, n. 5872, p. 77–82, 2008.
36 LAGGERBAUER, B.; LIU, S.; MAKAROV, E.; VORNLOCHER, H. P.; MAKAROVA,
O.; INGELFINGER, D.; ACHSEL, T.; LÜHRMAN, R. The human U5 snRNP 52K protein

92
(CD2BP2) 92 interacts with U5-102K (hPrp6), a U4/U6.U5 tri-snRNP bridging protein, but
dissociates upon trisnRNP formation. RNA, v.11, n. 5, p. 598-608, 2005.
37 WAHL, M. C.; WILL, C. L.; LUHRMANN, R. The Spliceosome: design principles
of a dynamic RNP machine. Cell, v. 136, n. 4, p. 701–718, 2009.
38 ULLU, E.; TSCHUDI, C. Accurate modification of the Trypanosome spliced leader cap
structure in a homologous cell-free system. Journal of Biological Chemistry, v. 270, n. 35,
p. 20365-10369 , 1995.
39 VIANNA, V. F.; AMBRÓSIO, D. L.; CICARELLI, R. M. B. Mapeamento de bandas de
trans-splicing in vitro com extratos nucleares de Trypanosoma cruzi. Revista de Ciências
Farmacêuticas, v. 22, n. 2, p. 295-306, 2001.
40 BANGS, J. D.; CRAIN, P. F.; HASHIZUME, T.; MCCLOSKEY, J. A.; BOOTHROYD,
J. C. Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides.
Journal of Biological Chemistry, v. 267, n.14, p. 9805-9815, 1992.
41 MICHAELI, S. Trans-splicing in trypanosomes: machinery and its impact on the parasite
transcriptome. Future Microbiology, v. 6 ,n. 4, p. 459–474, 2011.
42 DA SILVA, E. R.; CASTILHO, T. M.; PIOKER, F. C.; TOMICH DE PAULA SILVA, C.
H.; FLOETER-WINTER, L. M. Genomic organisation and transcription characterisation of
the gene encoding Leishmania (Leishmania) amazonensis arginase and its protein structure
prediction. International Journal of Parasitology, v. 32, n. 6, p. 727-737, 2002.
43 BRUZIK, J. P.; VAN DOREN, K.; HIRSH, D.; STEITZ, J. A. Trans splicing involves a
novel form of small nuclear ribonucleoprotein particles. Nature, v. 335, n. 6190, p. 559–562,
1988
44 LIANG, X. H.; HARITAN, A.; ULIEL, L.; MICHAELI. S. Trans and cis splicing in
trypanosomatids: mechanism, factors, and regulation. Cell, v. 2, n. 5, p. 830–840, 2003.
45 WILL, C. L.; LÜRHMANN, R. Spliceosome structure and function. Cold Spring Harbor
Perspectives in Biology, v. 3, n. 7, p. 369-400, 2006.
46 BURGE, C. B.; TUSCHL, T.; SHARP, P. A. Splicing of precursors to mRNAs by the
spliceosomes. In: GESTERLAND, R. F.; CECH, T. R. (Ed): The RNA world. 2nd ed. New
York: Cold Spring Harbor, 1999. p. 525–560.
47 MATLIN, A. J.; MOORE, M. J. Spliceosome assembly and composition. Advances in
Experimental Medicine and Biology. v. 623, p.14-35, 2007. PMID:18380338.
48 DAS, R.; DUFU, K.; ROMNEY, B.; FELDT, M.; ELENKO, M.; REED, R. Functional
coupling of RNAP II transcription to spliceosome assembly. Genes, v. 20, n. 9, p.1100-1109,
2006.

93
49 WETTERBERG, I; ZHAO, J.; MASICH, S.; WIESLANDER, L.; SKOGLUND, U. In
situ transcription and splicing in the Balbiani ring 3 gene. EMBO Journal, v. 20, n. 10 p.
2564–2574, 2001
50 TKACZ, I. D.; LUSTING, Y.; STERN, M. Z.; BITON, M.; SALMON- DIVON, M.;
DAS, A.; BELLOFATTO, V.; MICHAELI, S. Identification of novel snRNA-specific Sm
proteins that bind selectively to U2 and U4 snRNAs in Trypanosoma brucei. RNA, v. 13,n. 1,
p. 30–43, 2007.
51 PALFI, Z.; JAÉ, N.; PREUßER, C.; KAMINSKA, K. H.; BUJNICKI, J. M.; LEE, J. H.;
GÜNZL, A.; KAMBACH, C.; URLAUB, H.; BINDEREIF, A. SMN-assisted assembly of
snRNP-specific Sm cores in trypanosomes. Genes & Development, v. 23, n. 14, p. 1650–
1664, 2009.
52 TKACZ, I. D.; GUPTA, S. K.; VOLKOV, V.; ROMANO, M.; HAHAM, T.; TULINSKI,
P.; LEBENTHAL, I.; MICHAELI, S. Analysis of spliceosomal proteins in Trypanosomatids
reveals novel functions in mRNA processing. Journal of Biological Chemistry, v. 285, n.
36, p. 27982–27999, 2010.
53 STARK, H.; DUBE, P.; LÜRMANN, R.; KASTNER, B. Arrangement of RNA and
proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature, v. 409, n.
6819, p. 539–542, 2001.
54 NELISSEN, R. L.; WILL, C. L.; VAN VENROOIJ,W. J.; LÜHRMANN, R. The
association of the U1-specific 70K and C proteins with U1 snRNPs is mediated in part by
common U snRNP proteins. EMBO Journal, v.13, n. 17, p. 4113–4125, 1994.
55 CHEN, Y. I.; MOORE, R. E.; GE, H. Y.; YOUNG, M. K.; LEE, T. D.; STEVENS, S. W.
Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog
of participating factors. Nucleic Acids Research, v. 35, n. 12, p. 3928-3944, 2007.
56 XU, Y. Z.; NEWNHAM, C. M.; KAMEOKA, S.; HUANG, T.; KONARSKA, M. M.;
QUERY, C. C. Prp5 bridges U1 and U2 snRNPs and enables stable U2 snRNP association
with intron RNA. EMBO Journal, v. 23, n. 2, p. 376–385, 2004.
57 CROSS, M.; WIELAND, B.; PALFI, Z.; GÜNZL, A.; RÖTHLISBERGER, U.; LAHM,
H. W.; BINDEREIF, A. The trans-spliceosomal U2 snRNP protein 40K of Trypanosoma
brucei: cloning and analysis of function domains reveals homology to a mammalian snRNP
protein. EMBO Journal, v. 12, n. 3, p. 1239–1248, 1993.
68 PREUßER, C.; PALFI,Z.; BINDEREIF, A. Special Sm core complex functions in
assembly of the U2 small nuclear ribonucleoprotein of Trypanosoma brucei Eukaryotic Cell,
v. 8, n. 8, p. 1228–1234, 2009.
59 HOROWITZ, D. S.; KOBAYASHI, R.; KRAINER, A. R. A new cyclophilin and the
human homologues of yeast Prp3 and Prp4 form a complex associated with U4/U6 snRNPs.
RNA, v. 3, n. 12, p. 1374-1387, 1997.
60 NOTTROTT, S.; HARTMUTH, K.; FABRIZIO, P.; URLAUB, H.; VIDOVIC, I.;
FICNER, R; LÜHRMANN, R. Functional interaction of a novel 15.5Kd [U4/U6´U5] tri-

94
snRNP protein with the 5’ stem-loop of U4 snRNA. EMBO Journal, v. 18, n. 21, p. 6119-
6133, 1999.
61 MAKAROVA, O. V.; MAKAROV, E. M.; LIU, S.; VORNLOCHERr, H. P.;
LÜHRMANN, R. Protein 61K, encoded by a gene (PRPRF31) linked to autosomal dominant
retinitis pigmentosa, is required for [U4/U6´U5] tri-snRNP formation and pre-mRNA
splicing. EMBO Journal, v. 21, n. 5, p. 1148-1157, 2002.
62 NOTTROTT, S.; URLAUB, H.; LÜHRMANN, R. Hierarchical clustered protein
interactions with U4/U6 snRNA: a biochemical role for U4/U6 proteins. EMBO Jorunal, v.
21,n. 20, p. 5527-5538, 2002.
63 LIU, S.; RAUHUT, R.; VORNLOCHER, H. P.; LÜHRMANN, R. The network of protein-
protein interactions within the human U4/U6.U5 tri-snRNP. RNA, v. 12, n. 7, p. 1418-1430,
2006.
64 GÜNZL A. The pre-mRNA splicing machinery of trypanosomes: complex or simplified?
Eukaryotic Cell, v. 9 n.8, p. 1159-1170, 2010
65 TEIGELKAMP, S.; NEWMAN, A. J.; BEGGS, J. D. Extensive interactions of PRP8
protein with the 5' and 3' splice sites during splicing suggest a role in stabilization of exon
alignment by U5 snRNA. EMBO Journal, v. 14, n. 11, p. 2602-2612, 1995.
66 REYES, J. L.; KOIS, P.; KONFORTI, B. B.; KONARSKA, M. M. The canonical GU
dinucleotide at the 5' splice site is recognized by p220 of the U5 snRNP within the
spliceosome. RNA, v. 2, n. 3, p. 213-225, 1996.
67 SIATECKA, M.; REYES, J. L.; KONARSKA, M. M. Functional interactions of Prp8 with
both splice sites at the spliceosomal catalytic center. Genes & Development, v. 13, n. 15, p.
1983-1993, 1999.
68 MARONEY, P. A.; ROMFO, C. M.; NILSEN, T. W. Functional recognition of 5' splice
site by U4/U6.U5 tri-snRNP defines a novel ATP-dependent step in early spliceosome
assembly. Molecular Cell, v. 6, n. 2, p. 317-328, 2000.
69 MacMILLAN, A. M.; QUERY, C. C.; ALLERSON, C. R.; CHEN, S.; VERDINE, G. L.;
SHARP, P. A. Dynamic association of proteins with the pre-mRNA branch region. Genes &
Development, v. 8, n. 24, p. 3008-3020, 1994.
70 COLLINS, C. A.; GUTHRIE, C. Allele-specific genetic interactions between Prp8 and
RNA active site residues suggest a function for Prp8 at the catalytic core of the spliceosome.
Genes & Development, v. 13, n. 15, p. 1970-1982, 1999.
71 LÜCKE, S.; KLÖCKNER, T.; PALFI, Z.; BOSHART, M.; BINDEREIF, A. Trans mRNA
splicing in trypanosomes: cloning and analysis of a PRP8-homologous gene from
Trypanosoma brucei provides evidence for a U5-analogous RNP. EMBO Journal, v. 16, n.
14, p. 4433-40, 1997.

95
72 BARTELS, C.; KLATT, C.; LÜHRMANN, R.; FABRIZIO, P. The ribosomal translocase
homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the
spliceosome. EMBO Reports, v. 3, n. 9, p. 875-880, 2002.
73 ACHSEL,T.; BRAHMS, H.; KASTNER, B.; BACHI, A.; WILM, M.; LÜHRMANN, R. A
doughnut-shaped heteromer of human Sm-like proteins binds to the 3’-end of U6 snRNA,
thereby facilitating U4/U6 duplex formation in vitro. EMBO Journal, v. 18, n. 20, p. 5789-
5802, 1999.
74 AMBRÓSIO, D. L.; LEE, J. H.; PANIGRAHI, A. K.; NGUYEN, T. N.; CICARELLI, R.
M. B.; GÜNZL, A. Spliceosomal proteomics in Trypanosoma brucei reveal new RNA
splicing factors. Eukaryotic Cell, v. 8, n. 7, p. 990-1000, 2009.
75 DA SILVA, M. T. A.; AMBRÓSIO, D. L.; TREVELIN, C. C.; WATANABE, T. F.;
LAURE, H. J.; GREENE, L. J.; ROSA, J. C., VALENTINI, S. L., CICARELLI, R. M. B.
New insights into trypanosomatid U5 small nuclear ribonucleoproteins. Memórias do
Instituto Oswaldo Cruz, v.106, n. 2, p. 130-138, 2011.
76 BERRY, L. D.; GOULD, K. L. Fission yeast dim1(+) encodes a functionally conserved
polypeptide essential for mitosis. Journal of Cell Biology, v. 137, n. 6, p. 1337-54, 1997.
77 STEVENS, S. W.; ABELSON, J. Purification of the yeast U4/U6.U5 small nuclear
ribonucleoprotein particle and identification of its proteins (pre-mRNA splicing & mass
spectrometry). Proceedings of the National Academy of Sciences of the United States of
America, v. 96, n. 13, p. 7226-7231, 1999.
78 REUTER, K.; NOTTROTT, S.; FABRIZIO, P.; LÜHRMANN, R.; FICNER, R.
Identification, characterization and crystal structure analysis of the human spliceosomal U5
snRNP specific15 Kd Protein. Jounal of Molecular Biology, v. 294, n. 2, p. 515-525, 1999.
79 HOLMGREN, A. Thioredoxin and glutaredoxin systems, review. Journal of Biological
Chemistry, v. 264, n. 24, p.13963-13966, 1989.
80 SIMEONI, F.; DIVITA, G. The pre-mRNA splicing machinery of trypanosomes: complex
or simplified? Cellular and Molecular Life Sciences, v. 64, n. 16, p. 2079-2089. 2007.
81 ZHANG, Y. Z.; LINDBLOM, T.; CHANG, A.; SUDOL, M.; SLUDER, A. E.;
GOLEMIS, E. A. Evidence that Dim1 associates with proteins involved in pre-Mrna splicing,
and delineation of residues essential for Dim1 interactions with hnRNP F and Npw38/PQBP-
1. Gene, v. 257, n.1, p. 33–43, 2000.
82 ZHANG, Y. Z.; GOULD, K. L.; DUNBRACK JUNIOR, R. L.; CHENG, H.; RODER, H.;
GOLEMIS, E. A. The evolutionarily conserved Dim1 protein defines a novel branch of the
thioredoxin fold superfamily. Physiological. Genomics, v. 1, n.3, p. 109–118, 1999.
83 ZHANG, Y.; LI, N.; CARON, C.; MATTHIAS, G.; HESS, D.; KHOCHBIN, S.;
MATTHIAS, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo.
Embo Journal, v. 22, n. 5, p. 1168-79, 2003.

96
84 JIN, T.; GUO, F.; WANG, Y.; ZHANG, Y. Z. Identification of Human dim1 as a
Peptidase with Autocleavage Activity. Chemical Biology & Drug Design, v. 68, n. 5, p.
266–272, 2006.
85 GASTEIGER, E.; HOOGLAND, C.; GATTIKER, A.; DUVAUD, S.; WILKINS, M. R.;
APPEL, R. D.; BAIROCH, A. Protein identification and analysis tools on the ExPASy server.
In: WALKER, J. M. (Ed): The proteomics protocols handbook, New York: Humana Press.
p. 571-607. 2005
86 JONES, D. T. Protein secondary structure prediction based on position-specific scoring
matrices. Journal of Molecular Biology, v. 292, n.2, p. 195-202, 1999.
87 LARKIN, M. A.; BLACKSHIELDS, G.; BROWN, N. P.; CHENNA, R.; MCGETTGAN,
P. A.; MCWILLIAM, H.; VALENTINI, F.; WALLACE, I. M.; WILM, A.; LOPEZ,
R.; THOMPSON, J. D.; GIBSON, T. J.; HIGGINS, D. G. Clustal W and Clustal X version
2.0. Bioinformatics, v. 23, n. 21, p. 2947-2948, 2007.
88 MARCHLER-BAUER, A.; ZHENG, C.; CHITSAZ, F.; DERBYSHIRE, M. K.; GEER,
L. Y.; GEER, R. C.; GONZALES, N. R.; GWADZ, M.; HURWITZ, D. I.; LANCZYCKI,
C. J.; LU, F.; LU, S.; MARCHLER, G. H.; SONG, J. S.; THANKI, N.; YAMASHITA, R. A.;
ZHANG, D.; BRYANT, S. H.; CDD: conserved domains and protein three-dimensional
structure. Nucleic Acids Research, v. 41 p. 348-52, 2013. doi: 10.1093/nar/gks1243.
89 SIMPLE modular architecture research tool. Disponível em: < http://smart.embl-
heidelberg.de/ > Acesso em: 23 jan.2015.
90 SCHWEDEL, T.; KOPP, J.; GUEX, N.; PEITSCH, M. C. SWISS-MODEL: an automated
protein homology-modeling server. Nucleic Acids Research, v. 31, n. 13, p. 3381–3385,
2003.
91 KACZANOWSKIi, S.; ZIELENKIEWICZ, P. Why similar protein sequences encode
similar three-dimensional structures? Theoretical Chemistry Accounts, v. 125, p. 643–650,
2010. doi: 10.1007/s00214-009-0656-3.
92 BIASINI, M.; BIENERT, S.; WATERHOUSE, A.; ARNOLD, K.; STUDER, G.;
SCHMIDT, T.; KIEFER, F.; CASSARINO, T. G.; BERTONI, M.; BORDOLI, L.;
SCHWEDE, T. SWISS-MODEL: modelling protein tertiary and quaternary structure using
evolutionary information. Nucleic Acids Research, v. 42, p. 252–258, 2014.
doi: 10.1093/nar/gku340.
93 THE STRUCTURE Analysis and Verification Server. Disponível em:
<http://services.mbi.ucla.edu/SAVES/> Acesso em: 23 jan.2015.
94 SCHELLMAN, J. Circular dichroism and optical rotation. Chemical Reviews, v. 75, n. 3,
p. 323-331, 1975.
95 KELLY, S. M.; JESS, T. J.; PRICE, N. C. How to study proteins by circular dichroism.
Biochimica Biophysica Acta, v. 1751, p. 119-139, 2005. doi:10.1016/j.bbapap.2005.06.005.

97
96 JOHNSON, W. C. Circular dichroism and its empirical application to biopolymers.
Methods of Biochemical Analysis, v. 31, p. 62-163, 1985. PMID:3894885
97 NEYLON, C. Small angle neutron and X-ray scattering in structural biology: recent
examples from the literature. European Biophysics Journal, v. 37, n. 5, p. 531–541, 2008.
98 SVERGUN, D. I.; KOCH, M. H. J. Small-angle scattering studies of biological
macromolecules in solution. Reports on Progress in Physics. v. 66, n. 13, p. 1735–1782,
2003.
99 MOORE, P. B. Small-angle scattering. Information content and error analysis. Journal of
Applied Crystallography, v. 13, n. 2, p. 168–175. 1980.
100 HJELM, R. P. The small-angle approximation of X-ray and neutron scatter from rigid
rods of non-uniform cross-section and finite length. Journal of Applied Crystallography, v.
18, n. 6, p. 452–460. 1985.
101 KONAREV, P. V.; VOLKOV, V. V.; .SOKOLOVA, A. V.; KOCH, N. W. J.;
SVERGUN, D. I. PRIMUS - a Windows-PC based system for small-angle scattering data
analysis. Journal of Applied Crystallography, v. 36, p. 1277-1282, 2003.
doi:10.1107/S0021889803012779.
102 PETOUKHOV, M. V.; SVERGUN, D. I. Analysis of X-ray and neutron scattering from
biomacromolecular solutions. Current Opinion in Structural Biology, v. 17, n.5 ,p. 562-71,
2007.
103 FISCHER, H.; DE OLIVEIRA NETO, M.; NAPOLITANO, H. B.; CRAIEVICH, A. F.;
POLIKARPOV, I. The molecular weight of proteins in solution can be determined from a
single SAXS measurement on a relative scale. Journal of Applied Crystallography, v. 43,
n.1, p. 101-109, 2010.
104 SVERGUN, D. I. Determination of the regularization parameter in indirect-transform
methods using perceptual criteria. Journal of Applied Crystallography, v. 25, n. 4, p. 495-
503, 1992.
105 SVERGUN, D. I.; BARBERATO, C.; KOCH, M. H. J. CRYSOL - a program to evaluate
x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of
Applied Crystallography, v. 28, n. 6, p. 768-773, 1995.
106 SEGELKE, B. W. Efficiency analysis of sampling protocols used in protein
crystallization screening, Journal of Crystal Growth, v. 232, n. 1, p. 553–562, 2001.
107 PANTOLIANO, M. W.; PETRELLA, E. C.; KWASNOSKI, J. D.; LOBANOV, V. S.;
MYSLIK, J.; GRAF, E.; CARVER, T.; ASEL, E.; SPRINGER, B. A.; LANE, P.;
SALEMME, F. R. High-density miniaturized thermal shift assays as a general strategy for
drug discovery. Journal of Biomolecular Screen, v. 6, n. 6, p. 429–440, 2001.

98
108 NIESEN, F. H.; BERGLUND, H.; VEDADI, M. The use of differential scanning
fluorimetry to detect ligand interactions that promote protein stability. Nature, v. 2, n. 9, p.
2212-2221, 2007.
109 ERICSSON, U. B.; HALLBERG, B. M.; DETITTA, G. T.; DEKKER. N.; NORDLUND,
P. Thermofluor-based high-throughput stability optimization of proteins for structural studies.
Analytical Biochemistry, v. 357, n. 2, p. 289–298, 2006.
110 GIEGÉ, R. A historical perspective on protein crystallization from 1840 to the present
day. FEBS Journal, v. 280, n. 24, p. 6456-97, 2013.
111 VAN STOKKUM, I. H.; SPOELDER, H. J.; BLOEMENDAL, M.; VAN GRONDELLE
R.; GROEN, F. C. Estimation of protein secondary structure and error analysis from circular
dichroism spectra. Analytical Biochemistry, v. 191, n. 1, p. 110-118, 1990.
112 THE BASIC local alignment search tool. Disponível em <
http://blast.ncbi.nlm.nih.gov/Blast.cgi> Acesso em: 21 jan.2015.
113 WERNIMONT, A.; EDWARDS, A. In situ proteolysis to generate crystals for structure
determination: an update. PLoS One, v.4, n. 4, e5094, 2009. doi:
10.1371/journal.pone.0005094.
114 FAIM, L. M.; ROSA E SILVA, I.; BERTACINE DIAS, M. V.; D'MUNIZ PEREIRA,
H.; BRANDAO-NETO, J.; ALVES DA SILVA, M. T.; THIEMANN, O. H. Crystallization
and preliminary X-ray diffraction analysis of selenophosphate synthetases from Trypanosoma
brucei and Leishmania major. Acta Crystallografica Section F: structural biolology and
crystallization communications, v. 69, n. 8, p. 864-867, 2013.
115 CLONTECH LABORATORIES. TALO® metal affinity resins user manual. 2007.
Disponivel em: <http://wolfson.huji.ac.il/expression/local/Talon_Products.pdf> Acesso em:
23 jan.2015.
116 INVITROGEM™. Champion™ pET SUMO protein espression system user manual.
2010. Disponível em <
https://classes.soe.ucsc.edu/bme220l/Spring11/Reading/petsumo_man.pdf> Acesso em: 23
jan.2015.
117 PROTEIN structure analysis. Disponível em
<https://prosa.services.came.sbg.ac.at/prosa.php> Acesso em: 23 jan.2015.
118 SVERGUN, D. I. Restoring low resolution structure of biological macromolecules from
solution scattering using simulated annealing. Biophysical Journal, v. 76, n. 6, p. 2879-2886,
1999.
119 VOLKOV, V. V.; SVERGUN, D. I. Uniqueness of ab-initio shape determination in
small-angle scattering. Journal of Applied Crystallography, v. 36, p. 860-864, 2003.
doi:10.1107/S0021889803000268.

99
120 KOZIN, M. ;SVERGUN, D. Automated matching of high- and low-resolution structural
models. Journal of Applied Crystallography, v. 34, p. 33-41,
2001. doi:10.1107/S0021889800014126.
121 KENNEDY, P. G. E. Human African trypanosomiasis of the CNS: current issues and
challenges. Journal of Clinical Investigation, v. 113, p. 496-504. 2004. doi:
10.1172/JCI200421052.
122 SEPSEY, A.; BACSKAY,I.; FELINGER, A. Molecular theory of size exclusion
chromatography for wide pore size distributions. Journal of Chromatography A, v. 1331, p.
52-60. 2014. doi: 10.1016/j.chroma.2014.01.017.
123 SIPPL, M. J. Recognition of errors in three-dimensional structures of proteins. Proteins,
v. 17, n.4, p. 355-362. 1993.
124 LAGGERBAUER, B.; LIU, S.; MAKAROV,E.; VORNLOCHER,H. E.; MAKAROVA,
O.; INGELFINGER, D.; ACHSEL, T.; LÜHRMANN, R. The human U5 snRNP 52K protein
(CD2BP2) interacts with U5-102K (hPrp6), a U4/U6.U5 tri-snRNP bridging protein, but
dissociates upon tri-snRNP formation. RNA Journal, v.11, p. 598-608. 2005. doi:
10.1261/rna.2300805.
125 MARTINEZ-CALVILLO, S.; VIZUET-DE-RUEDA,J. C.; FLORENCIO-MARTINEZ,
L. E.; MANNING-CELA,R. G.; FIGUEROA-ÂNGULO, E. E. Gene expression in
trypanosomatid parasites. Journal of Biomedicine and Biotechnology. 2010.
doi:10.1155/2010/525241.





























