Embed Size (px)
Citation preview

Análises de sequências
Marcelo Falsarella Carazzolle
Laboratório de Genômica e Proteômica
Unicamp

Resumo
- Revisão
- Processamento das sequências
- DNA
- ESTs
- Instalação e uso do programa phred
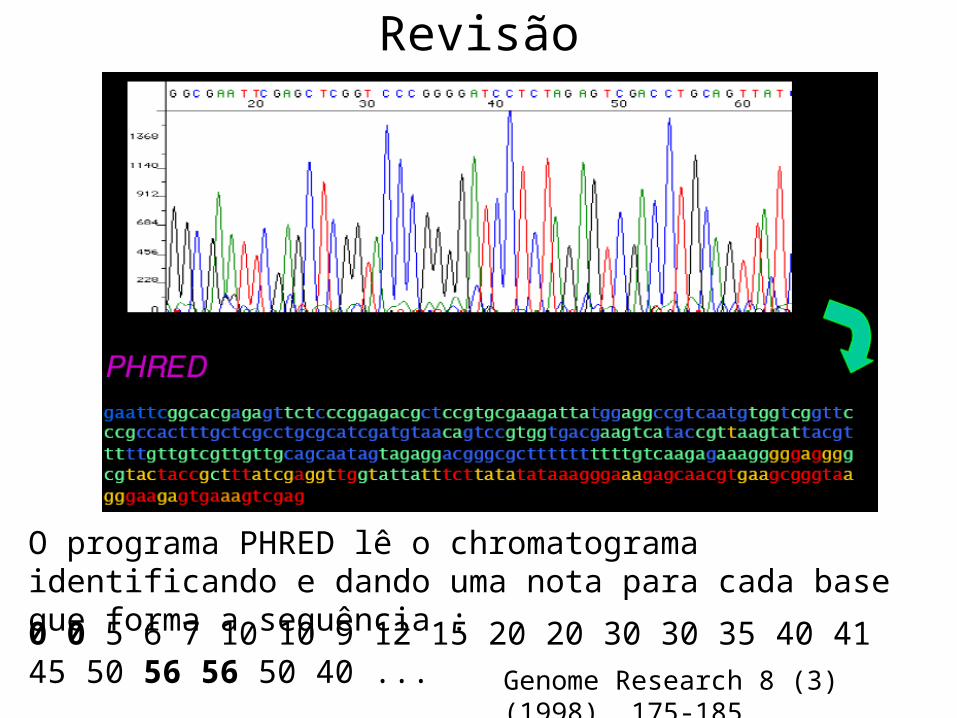
Revisão
0 0 5 6 7 10 10 9 12 15 20 20 30 30 35 40 41 45 50 56 56 50 40 ...
O programa PHRED lê o chromatograma identificando e dando uma nota para cada base que forma a sequência :
Genome Research 8 (3) (1998), 175-185

Qualidade boa Qualidade média Qualidade ruim
background

Onde q é a nota phred e P é a probabilidade encontrar uma base errada :
- Nota phred = 20 => 1 base errada a cada 100 (99%)
- Nota phred = 30 => 1 base errada a cada 1000 (99.9%)
- Sequenciamento produz seqüências da ordem de 500 pb
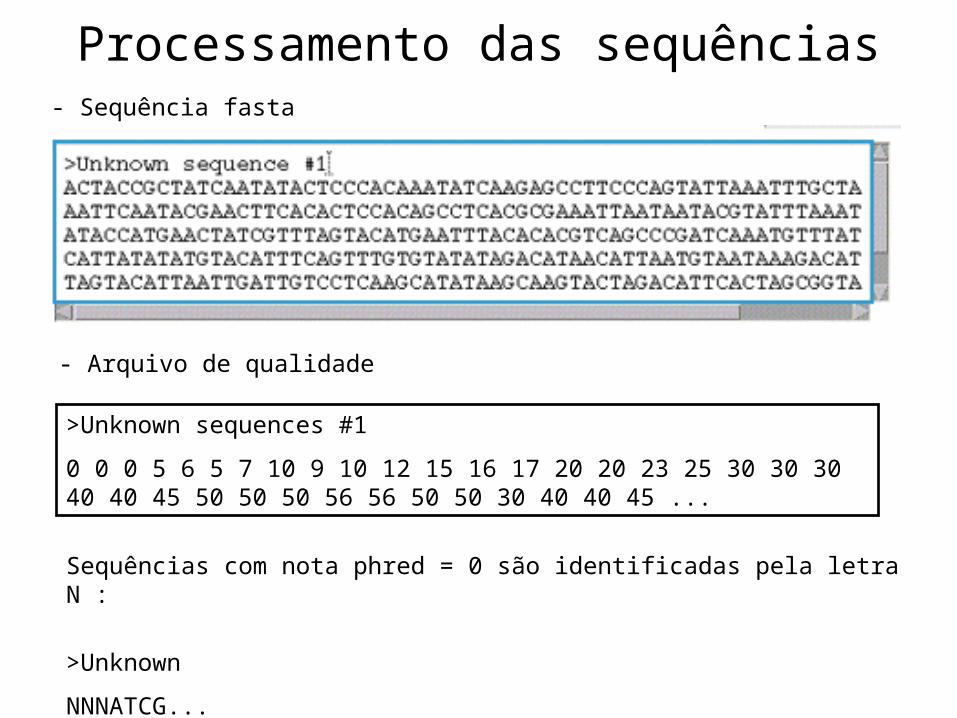
>Unknown sequences #1
0 0 0 5 6 5 7 10 9 10 12 15 16 17 20 20 23 25 30 30 30 40 40 45 50 50 50 56 56 50 50 30 40 40 45 ...
- Sequência fasta
- Arquivo de qualidade
Sequências com nota phred = 0 são identificadas pela letra N :
>Unknown
NNNATCG...
Processamento das sequências

clonar em vetor
sequenciamento
reads
DNA genômico
>Unknown sequence
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCC
Sequência do vetor de clonagem

Como identificar as regiões do vetor ???
- Necessita saber qual o vetor utilizado na clonagem dos fragmentos para pegar a sequência fasta desse vetor (site do fabricante/distribuidor)
http://www.invitrogen.com/content.cfm?pageid=94
- Ou de forma mais geral e automática, criando um arquivo com todas as sequências fastas de todos os vetores utilizados, ou pelo menos os mais utilizados
http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html

A identificação da região do vetor é feita através da comparação da sequência com o banco de vetores e pode ser feita usando vários programas. Exemplo :
- Usando o cross_match :
- Este programa faz uma comparação entre as sequências e mascara a região do vetor na sequência. Isto é, substitui os nucleotídeos vindos do vetor pela letra X,
>Unknown sequence
XXXXXXXXXXXXXXXXXXXAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAXXXXXXXXXXXXXXXXXX
X => Sequência do vetor de clonagem

-Usando o LUCY (Bioinformatics 17 (2001), n. 122001, 1093-1104) :
- Este programa faz uma comparação entre as sequências e coloca no cabeçalho do fasta as coordenadas da região sem vetor na sequência
>Unknown sequence 19 140
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCC
Sequência do vetor de clonagem
Início da região sem o vetor Fim da região sem o vetor

Identificar regiões de baixa qualidade
Identificar regiões de vetores
Cortar regiões de baixa qualidade e vetor
- Como as regiões de vetor e qualidade ruim estão sobrepostas o problema pode ser complicado

Bioinformatics 17 (2001), n. 122001, 1093-1104
- Possíveis combinações de regiões com qualidade ruim e vetores
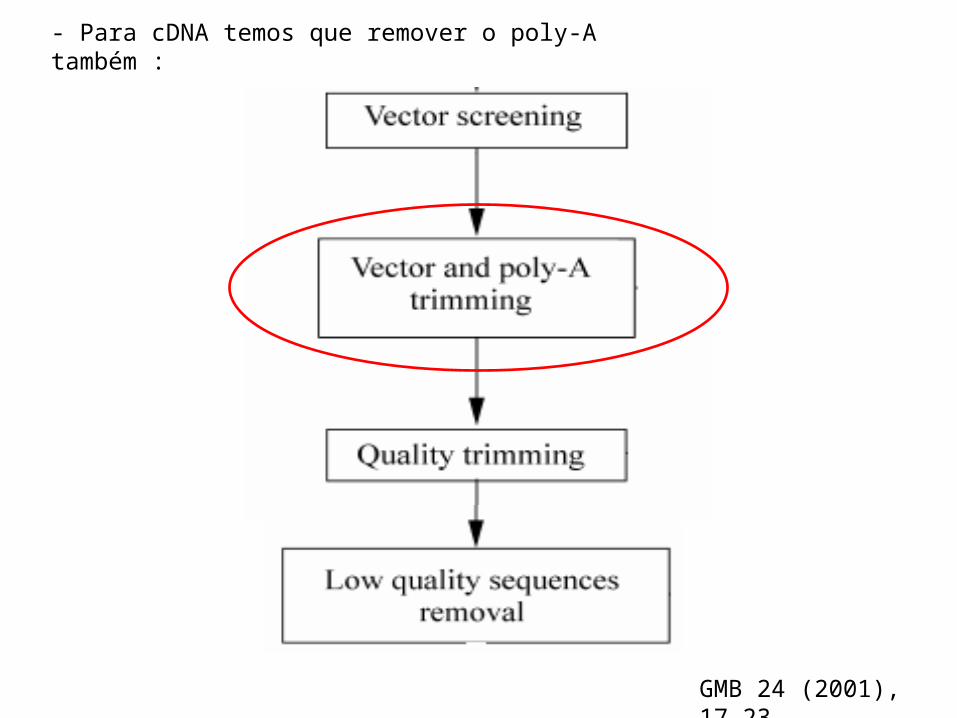
- Para cDNA temos que remover o poly-A também :
GMB 24 (2001), 17-23
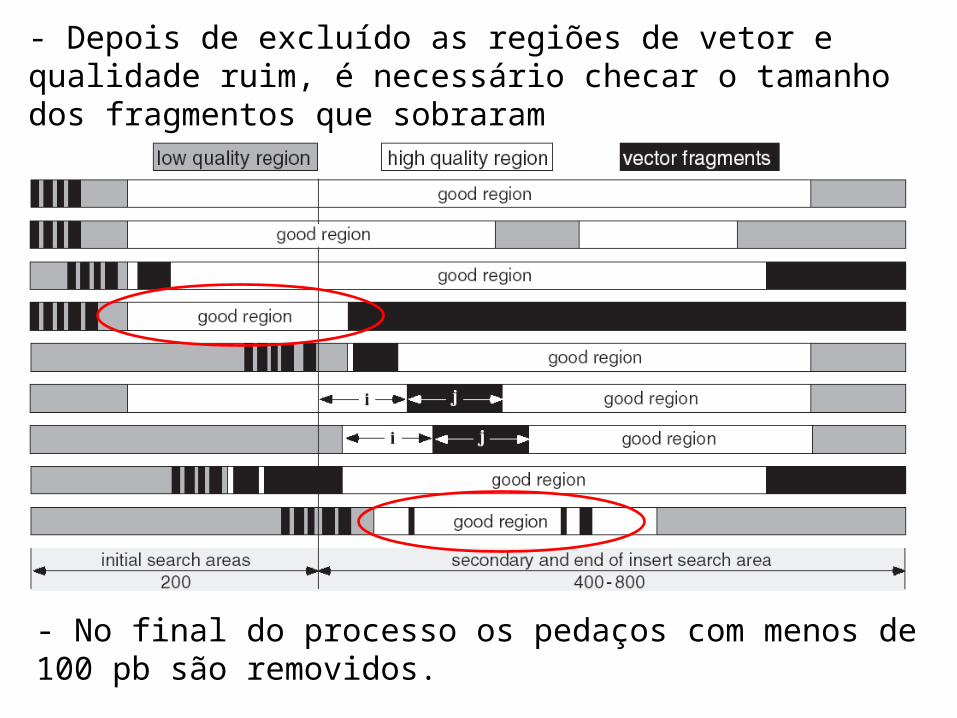
- Depois de excluído as regiões de vetor e qualidade ruim, é necessário checar o tamanho dos fragmentos que sobraram
- No final do processo os pedaços com menos de 100 pb são removidos.

Possíveis cortes de qualidade
- Nota de corte phred :
- Igual a 16 para corte processamento em larga escala
- Igual a 30 quando tem a necessidade de trabalhar com sequências de alta qualidade (Exemplo : SNPs)
- Igual a 0 quando se está interessado no máximo de informação possível sobre a sequência estudada. Possivelmente uma região de qualidade ruim pode continuar dando similaridade com a proteína de interesse

Query: 469 TTAGGAGGATCGTTTTTAGAATCCCCTGCAACGTTACCACGGTGGATTTCACTGACTGCG 528 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||Sbjct: 1038 ttaggaggatcgtttttagaatcccctgcaacgttaccacggtggatttcactgactgcg 979
Query: 529 ACGTTCTTAACGTTGAATCCAACGTTGCTACCAgggagagcctcagtaagtgcttcatga 588 ||||||||||||||||| || |||||||||||||||||| ||||||||||||||||||||Sbjct: 978 acgttcttaacgttgaagcccacgttgctaccagggagaccctcagtaagtgcttcatga 919
Query: 589 tgcatttcgacagaattgacttcagtcgacaaaccttgcggagcaaaagtgacgaccata 648 |||||||||||||| |||||||||| |||| ||||||||||| |||||||||||||||||Sbjct: 918 tgcatttcgacagacttgacttcagccgaccaaccttgcggaccaaaagtgacgaccata 859
Query: 649 ccaggcttgatgataccagtttcaacgc 676 ||||||||||||||||||||||||||||Sbjct: 858 ccaggcttgatgataccagtttcaacgc 831
.TGAAGCTTTCAGCTTCTTTAGGAGGATCGTTTTTAGAATCCCCTGCAACGTTACCACGGTGGATTTCACTGACTGCGACGTTCTTAACGTTGAATCCAACGttGCTACCAgggagagcctcagtaagtgcttcatgatgcatttcgacagaattgacttcagtcgacaaaccttgcggagcaaaagtgacgaccataccaggcttgatgataccagtttcaacgcctcggggccaggctggcgtgaacagggcctagcgggtccgcgggggaagggtcccggctcaatccaccaatagagcggagctaaagtgacgggggcgcca
Phred 15

Instalação e uso do phred
- Download (www.phrap.com)
- “you must email David Gordon the information requested in the academic user agreement including which platform(s) you want and your ip address”
- U$10.000,00 para uso não acadêmico
- O programa phred faz parte do pacote : phred / phrap / cross_match / consed
- Escrito em linguagem C
- Roda em sistema operacional linux

-Para usar basta criar 3 pastas :
- chromat_dir
- edit_dir
- phd_dir
- Copiar os chromatogramas na pasta chromat_dir
- Entrar na pasta edit_dir e digitar :
- phred -id chromat_dir -pd phd_dir
- Esta linha de comando informa ao phred que os chromatogramas estão no chromat_dir e os arquivos phds devem ser gravados no phd_dir
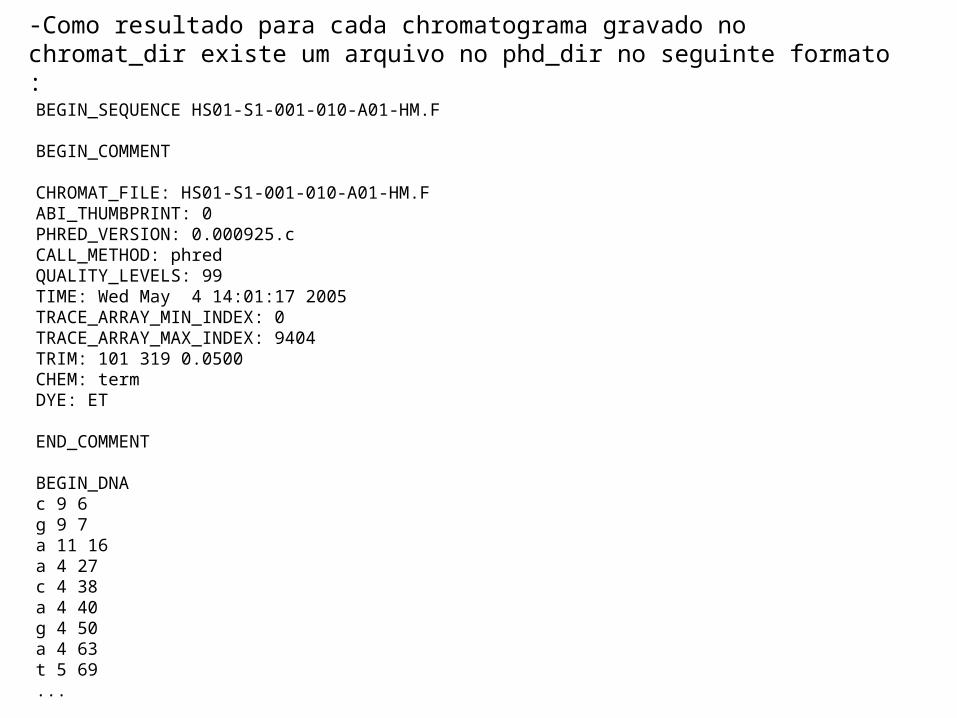
-Como resultado para cada chromatograma gravado no chromat_dir existe um arquivo no phd_dir no seguinte formato :
BEGIN_SEQUENCE HS01-S1-001-010-A01-HM.F
BEGIN_COMMENT
CHROMAT_FILE: HS01-S1-001-010-A01-HM.FABI_THUMBPRINT: 0PHRED_VERSION: 0.000925.cCALL_METHOD: phredQUALITY_LEVELS: 99TIME: Wed May 4 14:01:17 2005TRACE_ARRAY_MIN_INDEX: 0TRACE_ARRAY_MAX_INDEX: 9404TRIM: 101 319 0.0500CHEM: termDYE: ET
END_COMMENT
BEGIN_DNAc 9 6g 9 7a 11 16a 4 27c 4 38a 4 40g 4 50a 4 63t 5 69...

- Na sequência roda-se o phd2fasta :
- phd2fasta -id phd_dir -os seqs_fasta -oq seqs_fasta.qual
- Gerando na pasta do edit_dir :
- Arquivo com todas as sequências fasta de todos os chromatogramas lidos :
- seqs_fasta :
>chromatograma 1
ATCGCGC...
>chromatograma 2
TGCGCCA...
- Arquivo com todas as notas phred de cada base para todas os chromatogramas lidos :
- seqs_fasta.qual :
>chromatograma 1
0 10 12 15 12 20 ...
>chromatograma 2
0 12 13 5 10 10 ...

-Para mascarar o vetor roda-se o cross_match :
- cross_match seqs_fasta vector.seq -minmatch 12 -minscore 20 -screen > screen.out
- No qual gerar os arquivos :
- screen.out => grava as mensagens de saída do programa
- seqs_fasta.screen => arquivo fasta igual ao seqs_fasta mas com a letra X substituindo os nucletídeos vindos do vetor

END













