Embed Size (px)
Citation preview

CONCEITOS DEBIOLOGIA MOLECULAR
Jorge Mondego
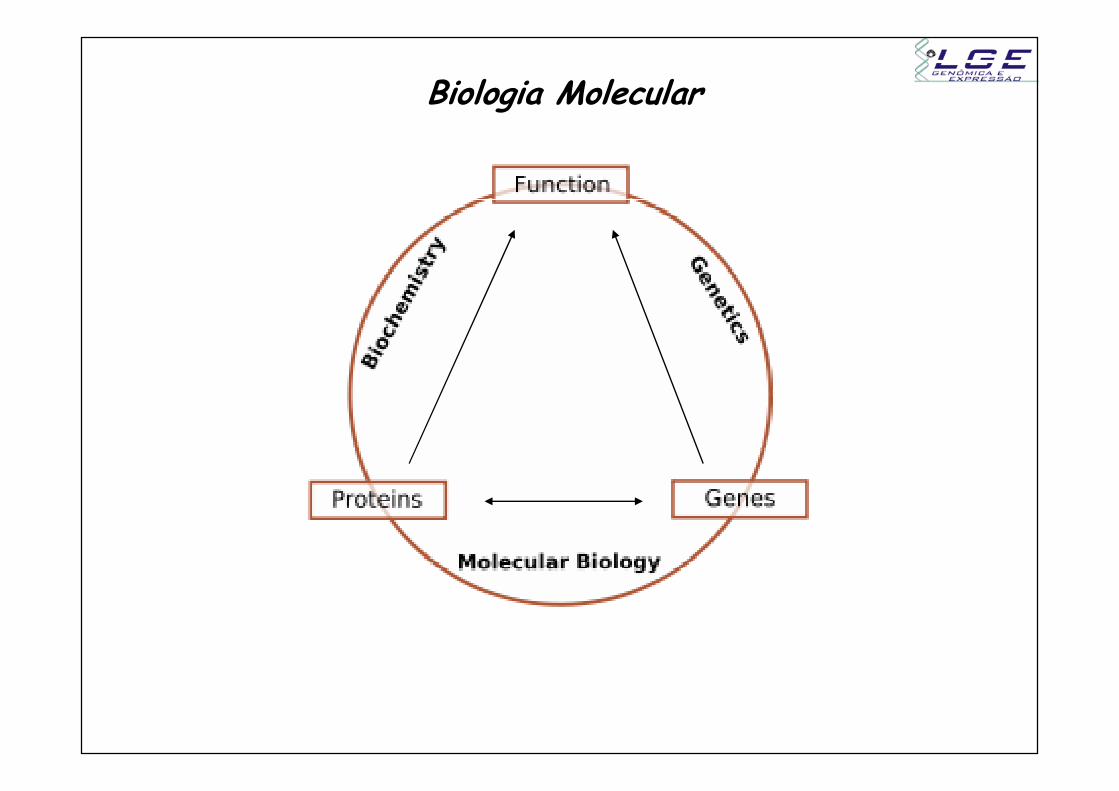
Biologia Molecular

The “OME”-Era
Genome Transcriptome
The “OME”-Era
Proteome Metabolome

- Entendimento da fisiologia e reprodução de microorganismos
- Entendimento dos mecanismos de replicação, transcrição e tradução
- Enzimas de restrição
- Plasmídeos
- Purificação de proteínas e Enzimologia
- Polymerase Chain Reaction (PCR)
- Transcriptase reversa e RT-PCR
- Sequenciamento
- Fluoróforos
- Automação

Cromossomo bacteriano Plasmídeos
Plasmídeos
DNA extracromossômico capaz de se replicar independe ntemente da replicação cromossomal
- Resistência a antibióticos- Produção de toxinas- Conjugação (transmissão de material genético entre as bactérias)- Origem de replicação própria


Conjugação
Transmissão de material genéticoentre as bactérias
Transferência horizontal de genes
Transferência de material genético entre reinos
Agrobacterium tumefaciens
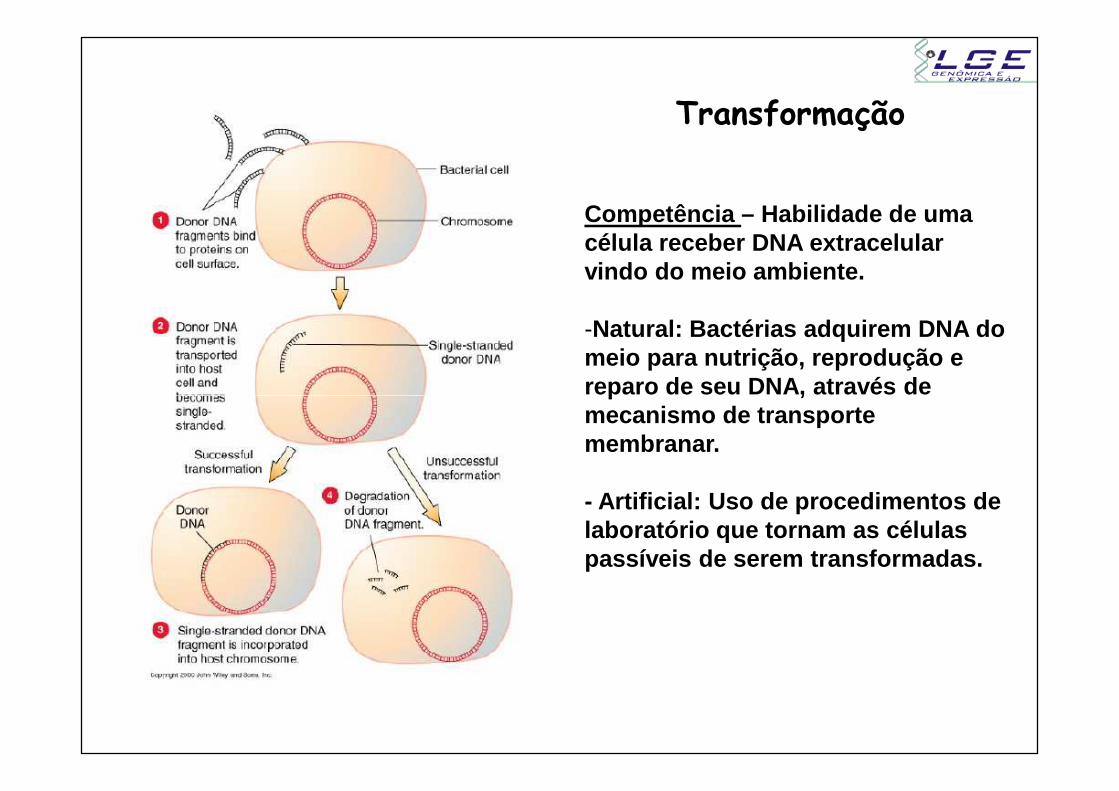
Transformação
Competência – Habilidade de uma célula receber DNA extracelular vindo do meio ambiente.
-Natural: Bactérias adquirem DNA do meio para nutrição, reprodução e reparo de seu DNA, através de reparo de seu DNA, através de mecanismo de transporte membranar.
- Artificial: Uso de procedimentos de laboratório que tornam as células passíveis de serem transformadas.

Transdução
Fagos – vírus que infectam bactérias

Daniel Nathans, Werner Arber, Hamilton Smith - 1978
Enzimas de restrição (endonucleases)
Sistema de modificação - restrição
- Enzimas bacterianas que reconhecem e clivam DNA in vasor - Reconhecimento de seqüência palindrômica de DNA exó geno- Seqüências bacterianas equivalentes são metiladas
5'-GTATAC-3'||||||
3'-CATATG-5'Seqüência palindrômica
- Seqüências bacterianas equivalentes são metiladas

Pontas coesivas Pontas “cegas”Pontas coesivasCohesive ends
Pontas “cegas”Blunt ends

Manipulação dos plasmídeos e a tecnologia do DNA recombinante
Sítio de
Resistênciaa antibióticos
Origem de replicação
clonagem

-Plasmídeo é digerido com enzimas
-Gene específico é ligado no plasmídeo,replicado em diversas cópias
“Clonagem” em vetores

Transformação
Choque térmico
Choque osmótico


Shuttle vectors
Plasmídeos que se propagam em duas espécies
-Transformação de plantas, leveduras, fungos filamen tosos e células animais
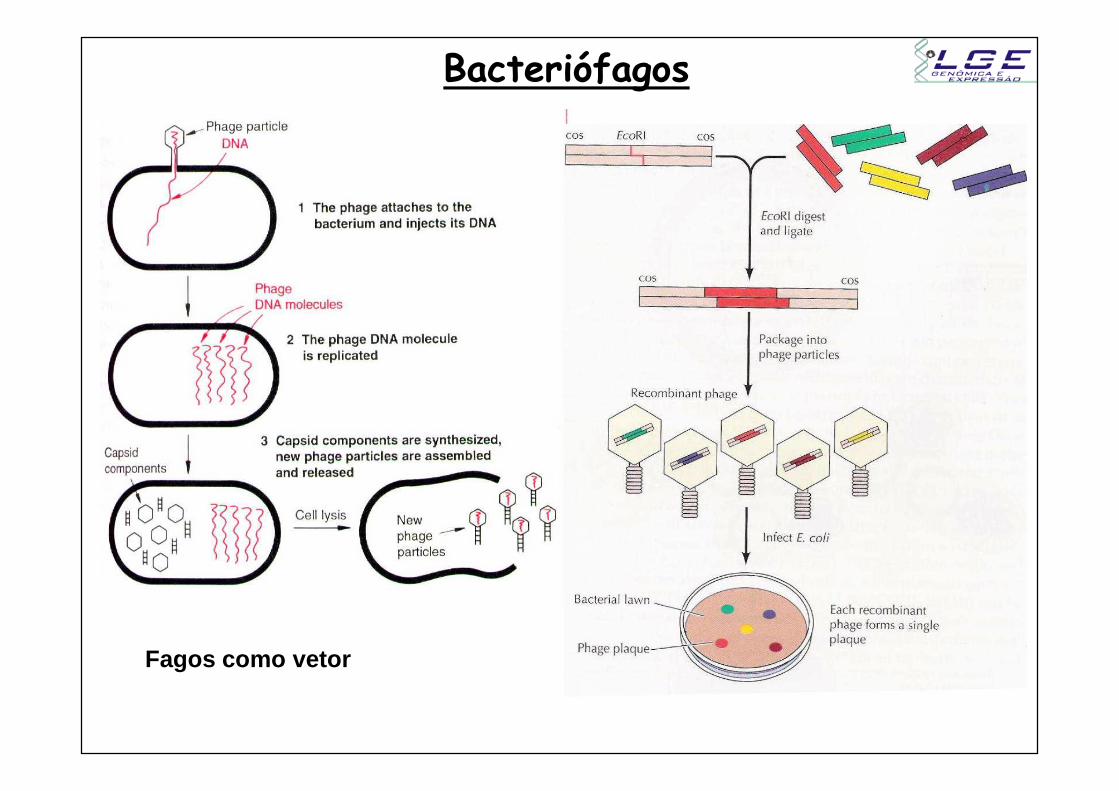
Bacteriófagos
Fagos como vetor

Vetores Tamanho do inserto
Plasmídeos de alta cópia 0–10 kb
Bacteriófago λ (inserção) 0–10 kb
Bacteriophage λ (substituição) 9–23 kb
Cosmídeos 30–44 kb
Bacteriófago P1 70–100 kb
BAC (cromossomos artificiais de bactérias) até 300 k b
YAC (cromossomos artificiais de leveduras) 0.2–2.0 M b

PCR - A REVOLUÇÃO DA TAQ
Kari Mullis – 1985Premio Nobel de química -1993

PCR 1

95°°°°C
Tm
72°°°°C
PCR 2
n Ciclos de PCR

- Concentração dos reagentesTris-HCl pH 8,8 (20 mM)KCl (10-50 mM)MgCl 2 (1 a 4 mM) – Menos Mg – mais especificidadeGlicerol (menos que 5%)dNTPs (200 a 10 µM – Menos dNTP - mais especificidade)Taq (1 unidade)
PCR – Otimização da reação
Taq (1 unidade)
-Condições dos ciclosEtapa de denaturação inicial (1-3 min a 95°C)Etapa de denaturação (30 seg a 2 min a 95°C)Etapa de anelamento Etapa de extensão (30 seg a 1min e 30 seg a 70-75°C)
- Desenhos dos primers

Características desejáveis em um Primer
● Temperatura de melting (Tm) na faixa de 50 ºC a 65 ºC.
● Tamanho de 15 a 28 pares de bases
● Ausência da capacidade de dimerização.
● Ausência significativa da formação de grampos (>3 bp)
● Inexistência de sítios secundários de anelamento dos primers.
● Temperaturas de anelamento de primers formando um par devem ser próximas

Comprimento
- Quanto maior o comprimento do primer, maior apossibilidade deste ser exclusivo; da mesma forma quemaiores serão as temperaturas de melting e anelamento.
- De uma forma geral, o comprimento do primer não deve serinferior a 15 bases para assegurar a unicidade. inferior a 15 bases para assegurar a unicidade.
- A existência desta faixa está baseada no fato de se buscarunique primers que apresentem temperatura de anelamentodentro da faixa considerada como a mais adequada.

Temperatura de Melting (TM)
- É a temperatura na qual metade das fitas de DNA está na forma de fitas simples e a outra metade naforma de dupla hélice.- Tm é dependente da composição do DNA, de modo queaumento do conteúdo de G+C no DNA gera um incremento na Tm ocasionado pelo maior número de ligações de H.ligações de H.
Temperatura de Anelamento
– É a temperatura na qual os primers se pareiam ao DNA molde. Ela pode ser calculada a partir da Tm .
T anneal = Tm_primer – 4°C

Estringência no Anelamento do Primer
- A estringência determina a especificidade no produto deDNA a ser amplificado.
- T anneal é o fator mais significante que afeta a estringência no anelamento do primer.
-T anneal :
Muito baixa = menor estringência = primer pareia em qualquer lugar.
muito alta = maior estringência = primer pode não parear.

Estrutura interna do primer
- Evitar essas estruturas...
..Pode-se utilizar primers com estas estruturas se estas forem formadas
a uma temperatura em torno de 30°C menor que a Tm

Tm = 4 (G+C) + 2 (A+T)
atgatagaggctctcgaag ctgaggtgaccaggagaacgctcgagtttgacacgtgtaaa·M··I··E··A··L··E··A··E··V··T··R··R··T··L··E··F··D· ·T··C··K·
gtcgt cgctctcccaatggtataa·V··V··A··L··P··M··V··*·
5´- atgatagaggctctcgaag – 3´ 5´- ttataccattgggagagcg – 3 ´
5´- atgatagaggctctcgaag ctgaggtgaccaggagaacgctcgagtttgacacgtgtaaagtcgtcgctc tcccaatg3´- tactatctccgagagcttcgactccactggtcctcttgcgagctcaaa ctgtgcacatttcagcagcgagagggttac
gtataa – 3´catatt – 5´

RNA
DNA
TRANSCRIPTASE REVERSA e A QUEBRA DO PARADIGMA
RNA
Proteína
Quebra do paradigma

RT “REVOLUTION”
PRIMERS UTILIZADOS
Oligo(dT)15-25-“Somente mRNA”
Hexâmeros randômicosHexâmeros randômicos- Anelamento em regiões aleatórias
Primer específico do gene- Amplificação somente do gene alvo

Bibliotecas

Expressed Sequence Tags (ESTs)
Extrair RNA de diferentes tecidos/condições
cDNA
Bibliotecas
3’ EST5’ EST
Clonar em vetor

Full-Lenght cDNA

Controle Tratado
Extração de RNA e síntese de cDNA
Northern Eletrônico
sequenciamento sequenciamento
clusterizaçãoSequência consenso
controletratado = 2x
Adaptors
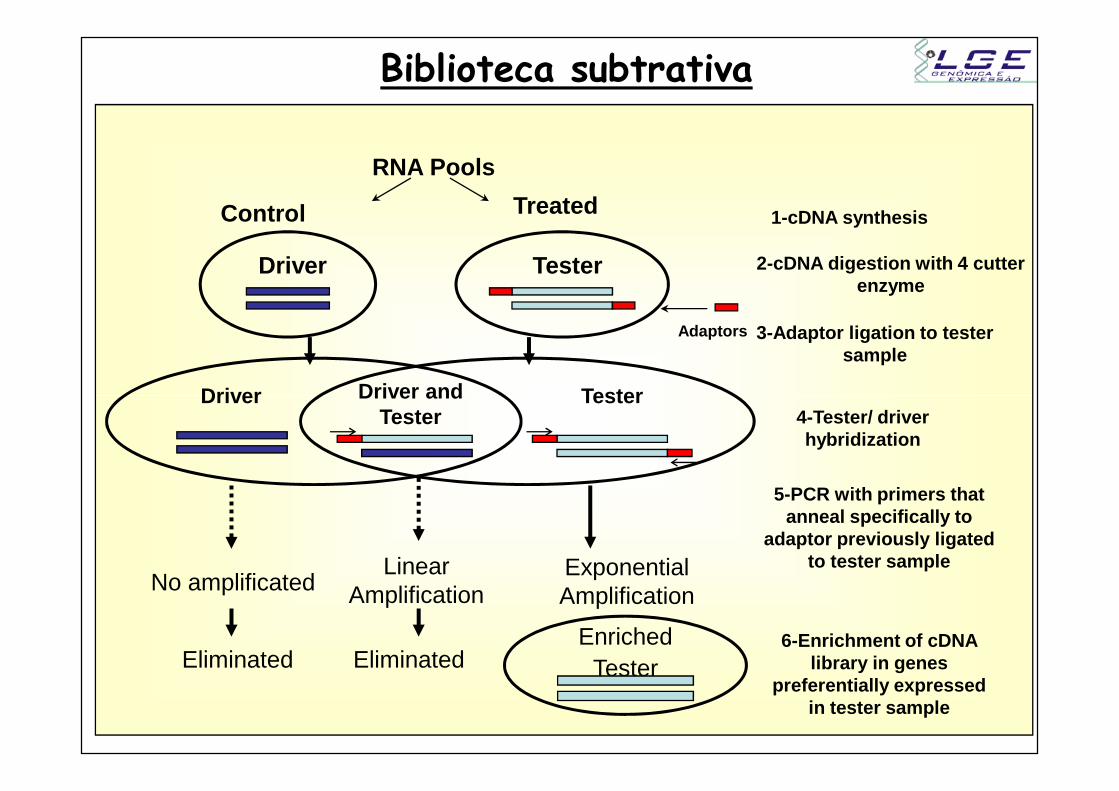
Driver Driver and Tester
Driver Tester
1-cDNA synthesis
2-cDNA digestion with 4 cutter enzyme
3-Adaptor ligation to tester sample
Control Treated
RNA Pools
Biblioteca subtrativa
Driver Driver and Tester
Tester
No amplificatedExponential Amplification
Linear Amplification
EliminatedEliminatedEnriched
Tester
4-Tester/ driver hybridization
5-PCR with primers that anneal specifically to
adaptor previously ligated to tester sample
6-Enrichment of cDNA library in genes
preferentially expressed in tester sample

Fluoróforos e “molecular beacons”
Cianinas
Emissão fluorescente - 570 nm (região do verde do espectro de luz)
Emissão fluorescente - 670 nm Emissão fluorescente - 670 nm (região do verde do espectro de luz)
SYBR GREEN
Emissão fluorescente - 522 nm (região do verde do espectro de luz)

“molecular beacons”
Loop – complementar a seqüência alvo

Análise Qualitativa Visa detectar a presença ou não do gene
Análise QuantitativaVisa detectar a quantidade (expressão) do gene na
amostra
PCR – Quantidade X Qualidade
amostra
Difícil diferenciar 10 cópias de 50 cópias em gel de agarose

Detecção do PCR tradicional
Detecção do Real Time PCR

SYBR green assay
SYBR se liga a DNA dupla fita e fluoresce. Durante o processo
de amplificação, a fluorescência vai aumentando, tornando mais fácil aumentando, tornando mais fácil
a quantificação de DNA

Taq Man®
Sonda anela no gene alvoPolimerase desloca repórter
liberação de fluorescência verde
Polimerase desloca quencher
www.appliedbiosystems.com

TTGTTCGAAGACTGGAAaCAACAGGTCGTCAGGCAGCATAACGAGTACAGGGCCCGTTATGGTGCACCCAACCTGTCCTGGAGCGATGCTCTGTACCCGGATACTGCTCGATATGCCGGACAGTGCAA GTTCcAACACAGGTATGACACGTCGTTGGTTCGTCGACATGTA AGGGTACTGACGACACATTCAAAG
Desenho de primers real time
Junção éxon - íntron
GTTCGTCGACATGTA AGGGTACTGACGACACATTCAAAGCAACAGTGGCGGCAAGTACGGCGAAAACTTGGCTGCTGGTACTGGAAACGCCTATGGTTTCTCGAGCGGCTTGAAGTCGTGGATGGATGAAGCTTGTATGTCTAC
http://www.genscript.com/cgi-bin/tools/primer_gensc ript.cgi


Quebrar em pedaços aleatórios ~2000pb
(shotgun)
DNA genômico
reads
Clonagem de genomas
clonar em vetor
sequenciamento

reads
Sequência consenso(DNA original)
Reconstrução do DNA original a partir do fragmentos (clusterização)
A reconstrução é feita a partir de sobreposição dos fragmentos

Quebrar em pedaços Quebrar em pedaços Quebrar em pedaços Quebrar em pedaços aleatoriamente desde aleatoriamente desde aleatoriamente desde aleatoriamente desde
50Kpb até 300Kpb50Kpb até 300Kpb50Kpb até 300Kpb50Kpb até 300Kpb
DNA genômicoDNA genômicoDNA genômicoDNA genômico
Clonar em BAC’s e
Shotgun de pedaços do genoma
~800 bp ~800 bpQuebrar em pedaços de 2000pb
Clonar em BAC’s e sequenciar apenas as
pontas de cada fragmento
clonar em vetor e sequenciar os fragmentos

Primer Walking
Clone to sequenceVector
Primer Sequence
New Primer
Sequence
Repeat
Sempre desenhar o primer de forma que a sequência amplificada tenha sobreposição com a anterior (tipicamente 100 pb de sobreposição)


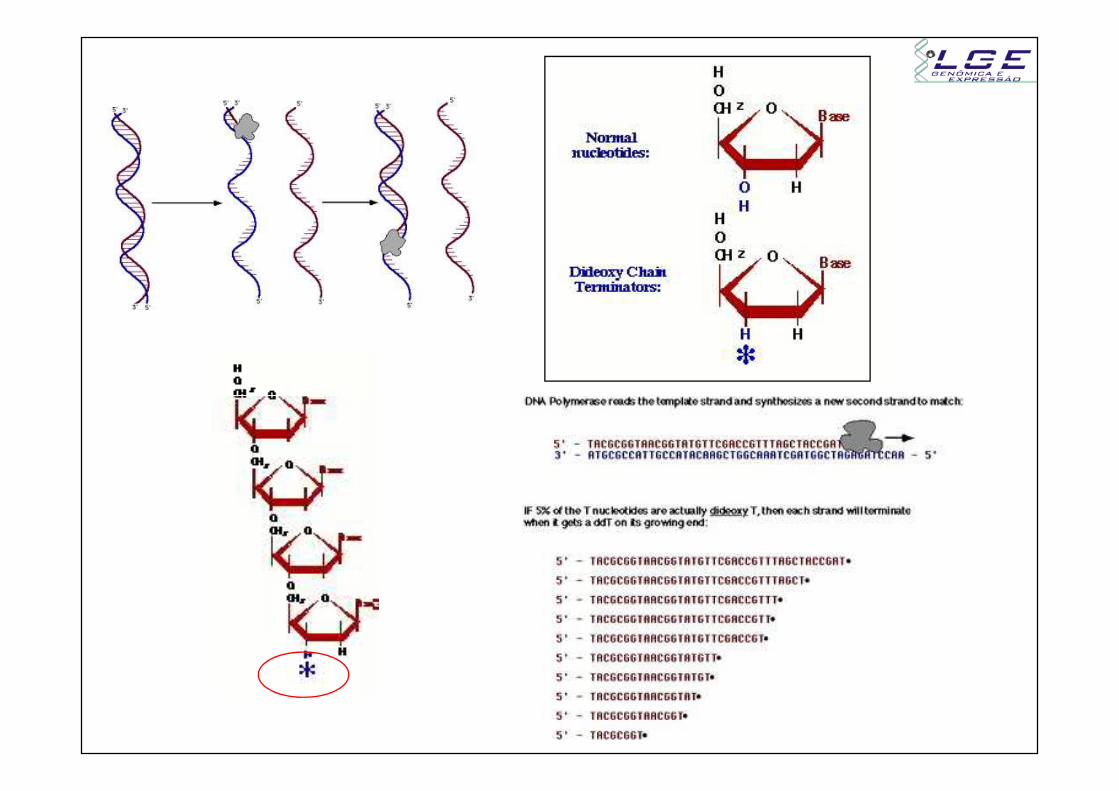

anelamento dos primers
denaturação


TTGGCGTAATCATGGTCATAGCTGTTTCCTGTGTGAAATTGTTATCC
0 0 5 6 7 10 10 9 12 15 20 20 30 30 35 40 41 45 50 56 56 50 40 ...
O programa PHRED lê o chromatograma identificando e dando uma nota para cada base que forma a sequência :

- A identificação dos picos é feita através de uma transformada de fourier do sinal
- A nota é ligada com a resolução entre os picos vizinhos e a altura do background

Região de qualidade alta
Analisando o cromatograma
• Picos bem definidos e grandes.
• Linha de base boa.
• Distância entre picos anterior e posterior constante.

Região de qualidade média – poucas ambigüidades
• Picos razoavelmente bem definidos e de tamanho médio.
• Linha de base boa a razoável.
• Distância entre picos anterior e posterior razoável.

Região de qualidade baixa – baixa confiabilidade
• Picos mal definidos e de tamanho pequeno.
• Linha de base confusa.
• Distância entre picos anterior e posterior inconstante.

http://www.454.com/
PirosequenciamentoRoche (454) GS FLX sequencer

Fita simples
Câmera de CCD
Reação de degradação

Quebrar em pedaços aleatórios ~2000pb
(shotgun)
DNA genômico
Shotgun do genoma inteiro
Ligação do adaptador e
separação em fita simples

- O adaptador permite que o DNA se ligue em grânulosminúsculos (diâmetro de 28 mm). Apenas um DNA éligado em cada grânulo
- Os grânulos são envolvidos em gotas de óleo quecontêm todos os reagentes necessários para amplificar oDNA
- Cada gota contendo o grânulo é mantida isolada para- Cada gota contendo o grânulo é mantida isolada paraevitar contaminação e consegue produzir 10 milhões decópias numa reação de pirosequenciamento
- Um pmol de DNA numa reação de pirosequenciamentoproduz 1011 moléculas de ATP gerando mais de 109
fótons, num comprimento de onda de 560 nm, e numperíodo de 3-4 segundos. Facilmente detectado por umacâmera de CCD



O sequenciador 454
Câmera de CCD
Bombeamento de fluídos
Câmara de fluxo contendo as amostras e as fibras ópticas(1,6 milhões/slide)
Computador

Pirograma
Linearidade é mantida até homopolímeros de 8 nt

Illumina/Solexa Genome Analyzer Sequenciamento por síntese + clustering PCR
http://www.illumina.com/

- O adaptador permite que o DNA se ligue a uma placa nasuperfície dos canais de fluxo
- PCR em fase sólida permite que as moléculasresultantes de uma PCR fiquem próximas. Ciclo érepetido várias vezes
- Adição de polimerase, primers e de nucleotídeos,- Adição de polimerase, primers e de nucleotídeos,marcados por fluoróforos, com o 3’OH inativado – adiçãode um nucleotídeo por vez. Após incorporação, há adetecção do fluoróforo, reverte-se a inativação do 3’OH ee retira-se o fluoróforo. Ciclo se repete.



Applied Biosystems SOLiD TM
Sequencer
http://www.appliedbiosystems.com.br/site/abhome.jsp

- O adaptador ligado ao DNA e a grânulos magnéticos.Ocorre PCR em emulsão e as fitas de DNA sãodepositadas numa placa
- Ligação de primer universal n ao adaptador e de óligosdegenerados (7 bases) marcados contendo duasprimeiras bases fixas.
- Ocorre detecção do sinal, clivagem das duas últimasbases e adição de novos óligos
- Ao fim de n rounds, a fita resultante é liberada e há aligação de um novo primer universal, (agora n-1). Ciclo serepete mais três vezes (até n-4).





SANGER Novas tecnologias
• Depende de clonagem em bactéria (2 semanas de trabalho)
• Não há clonagem
• Reads de ~700 bp • Reads de 200 a 25 pb
• Clones de fita dupla
Sanger vs Novas tecnologias
• Milhões de bp em menos de 4 horas
• 1 milhão de pb em 24 horas
• Clones de fita dupla permitem seqüenciamento em ambas direções (facilita orientação e montagem)
• Fragmentos fita simples não permitem seqüenciamento em ambas direções. Aplicação da técnica de paired-end
• 6 meses de sequenciamento, 24 horas por dia, para sequenciar o genoma de um fungo
• 24 horas para sequenciar o genoma de um fungo
Conclusão : a união faz a força

454 Solexa SoliDSequencing chemistry
PyrosequencingPolymerase-based sequencing-by-synthesis
Ligation-based sequencing
Amplification approach
Emulsion PCR Bridge amplification Emulsion PCR
Paired ends/separation
Yes/3 kb Yes/200 bp Yes/3 kb
Novas Tecnologias
ends/separationYes/3 kb Yes/200 bp Yes/3 kb
Mb/run 100 Mb 1300 Mb 3000 Mb
Time/run (paired ends)
7 h 4 days 5 days
Read length 250 bp 32–40 bp 35 bp
Cost per run (total direct a)
$8439 $8950 $17 447
Cost per Mb $84.39 $5.97 $5.81





























