Embed Size (px)
Citation preview

Avanços regulatórios na área de fármacos e medicamentos
Varley Dias Sousa, Ph.DGerente Geral de Medicamentos e Produtos Biológicos/ GGMED
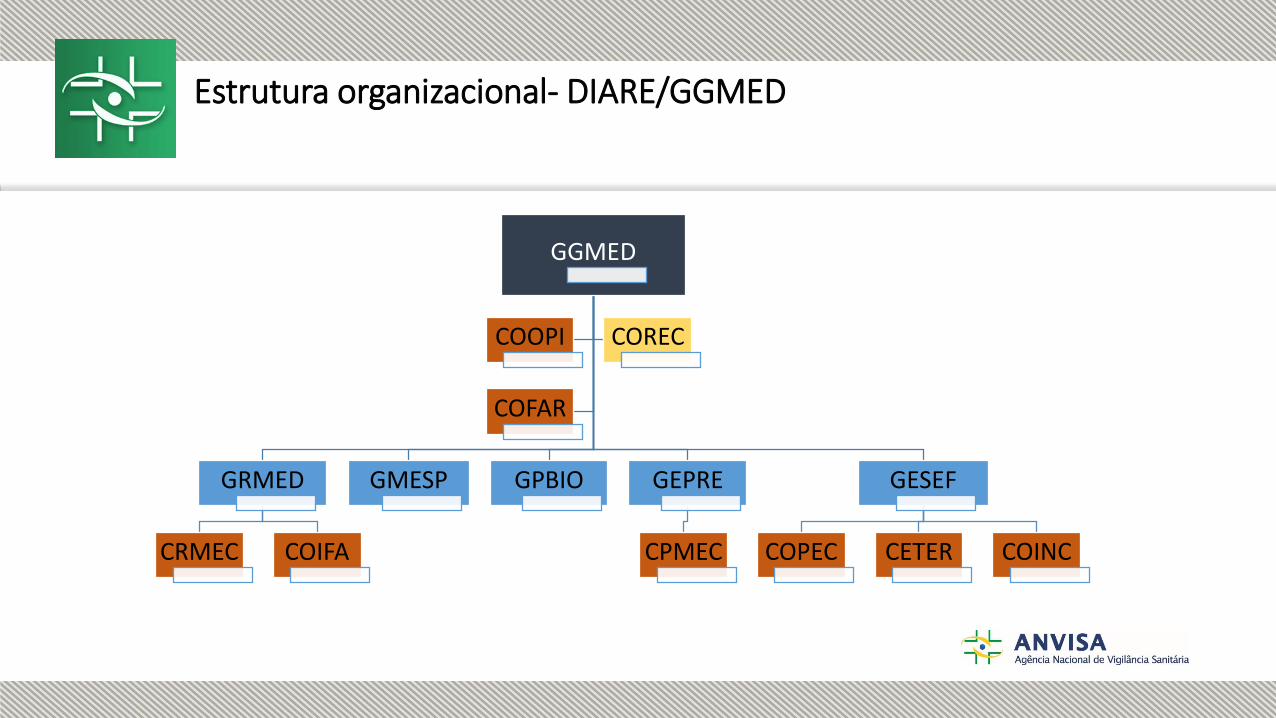
Estrutura organizacional- DIARE/GGMED
GGMED
GRMED
CRMEC COIFA
GMESP GPBIO GEPRE
CPMEC
GESEF
COPEC CETER COINC
COOPI COREC
COFAR

Estrutura organizacional
DIARE/GGMED
Gerência–Geral de Medicamentos e Produtos Biológicos:a) Coordenação de Instrução e Análise de Recursos de Medicamentos e Produtos Biológicos; ***b) Coordenação da Farmacopeia;c) Coordenação de Propriedade Intelectual;d) Gerência de Avaliação de Segurança e Eficácia:1. Coordenação de Pesquisa Clínica em Medicamentos e Produtos Biológicos;2. Coordenação de Equivalência Terapêutica;3. Coordenação de Inovação Incremental.e)Gerência de Avaliação de Tecnologia de Registro de Medicamentos Sintéticos:1. Coordenação de Registro de Medicamentos de Menor Complexidade2. Coordenação de Registro de Insumos Farmacêuticos Ativos;f)Gerência de Avaliação de Tecnologia de Pós–Registro de Medicamentos Sintéticos:1. Coordenação de Pós Registro de Medicamentos de Menor Complexidade;g) Gerência de Medicamentos Específicos, Fitoterápicos, Dinamizados, Notificados e Gases Medicinais;eh) Gerência de Avaliação de Produtos Biológicos.

Boas Práticas Regulatórias
Transparência
Convergência
regulatóriaResponsabilidade
compartilhada
ANVISAPrevisibilidadeRacional técnico
e científico

Publicação do Parecer Público de Avaliação do Medicamento de registro (desde março de 2015) http://www.anvisa.gov.br/datavisa/Fila_de_analise/index.asp
Divulgação no site da Anvisa dos estudos clínicos aprovadoshttp://portal.anvisa.gov.br/consulta-de-ensaios-clinicos-autorizados
Disponível, no site da Anvisa, o Relatório de Atividades 2016http://portal.anvisa.gov.br/documents/281258/2742545/Relat%C3%B3rio+de+Atividades+2016/d1556cef-8c1f-4b21-ae78-58ad65713d61
Transparência

Novas Substâncias Ativas
(NSA) AprovadasNSA Biológicas NSA Sintéticos
Total Prazo Total Prazo Total Prazo
Japão 52 306 12 300 40 321
EUA 45 343 15 334 30 362
União Europeia 30 418 9 420 21 415
Fonte: The Centre for Innovation in Regulatory Science (CIRS), 2016
Prazos médios (em dias) de aprovação em outros países

Número de novas moléculas registradas (sintéticas e biológicas), em 2015

Simplificação de procedimentos
Resolução RDC nº 73/2016 (pós-registro)
análise de acordo com a complexidadepermite implementações imediatas
Resolução RDC nº 107/2016 (notificação simplificada)
37 novos princípios ativos na categoria “baixo risco” (aumento de 50% na lista)
Otimização do fluxo de análise de medicamentos novos
novo processo de trabalho reduziu o tempo de 14 meses em 2014 para 3 meses em 2016
Avaliação de registro e pós-registro de medicamentos de menor complexidade (CRMEC e CPMEC)
Registro de medicamentos: iniciativas de aprimoramento

Convergência regulatória internacional
Participação em vários fóruns internacionais para promover a convergência regulatória na área de medicamentosInternational Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals
for Human Use (ICH)
Diversos grupos técnicos de especialistas da Organização Mundial da Saúde (OMS)
Rede Pan-Americana da Harmonização de Regulamentação Farmacêutica (Rede PARF) coordenada pela Organização Pan-Americana da Saúde (Opas/OMS)
International Generic Drug Regulators Programme (IGDRP)
Nov/16: Avaliação pelo Centre for Innovation in Regulatory Science (CIRS)Identificação das melhores práticas e das fragilidades dos procedimentos de registro
Comparação com dados internacionais
Recomendações para melhoria do processo de registro de medicamentos no Brasil
Registro de medicamentos: iniciativas de aprimoramento

Planejamento Estratégico da Anvisa
Projetos EstratégicosProjeto P1: “Aperfeiçoamento dos procedimentos de registro de produtos em alinhamento com as
melhores práticas internacionais”: maior celeridade e transparência às solicitações de registro de produtos
Projeto P6: “Ampliação da consistência e transparência de exigências técnicas”: eficiência dos processos de trabalho e a agilidade dos processos de registro.
Resultados esperadosRedução do tempo de fila
Padronização do processo de registro entre as diferentes áreas
Indicadores e metas para os processos de registro para tomada de decisão
Redução do volume, do esforço e da variabilidade de exigências técnicas
Melhorias nas práticas de emissão de exigências técnicas
Registro de medicamentos: iniciativas de aprimoramento

Guia de análise de IFADisponível em inglês e português, no link: https://www20.anvisa.gov.br/coifa/
Transparência ao procedimento de análise de registro de insumos farmacêuticos ativos (RDC no
57/2009)
Portaria conjunta INPI-AnvisaAgilidade para concessão de patentes
Reestruturação das atividades do INPI e da Anvisa
Análise de resíduos de agrotóxicos em fitoterápicosPerguntas e respostas disponível em:
http://portal.anvisa.gov.br/documents/33836/2501251/FAQ_Agrotoxicos_Fito.pdf/c2bcd8cb-a9d4-47ff-aeaa-29ad5203afae
Guia para análise de estudos não clínicos em fitoterápicos (em fase de elaboração)
Outras iniciativas

Objetivo: conferir maior transparência e previsibilidade ao processo de registrodivulgação do status da análiseprazo para a decisão final fundamentos técnicos das decisões nos processos
Altera a Lei nº 6.360/1976 (registro de medicamentos)Classifica em “prioritárias” e “ordinárias” as petições Critérios de complexidade técnica e benefícios clínicos, econômicos ou sociais Estabelece prazos para a deliberação final processos de registro e pós-registroAprovação condicional (pós-registro), pela não manifestação contrária da AnvisaCria regra de transição (1 ano) para o passivo de petições
Altera a Lei nº 9.782/1999 (Lei da Anvisa)Normas com justificativa e avaliação de impacto econômico e técnico Amplia os prazos para interposição e julgamento dos recursos Contrato de Gestão: descumprimento injustificado exonera a Diretoria Colegiada
Lei 13.411/2016

Guia de estudos não clínicos – pesquisa clínica e produtos de degradação
Guia para a condução de estudos não clínicos de toxicologia e segurança farmacológica necessários ao desenvolvimento de medicamentos (v.2, 2013)
Harmonização internacional (ICH, OECD, NCI, WHO)
Documentação solicitada semelhante a de outras agências regulatórias (FDA, EMA)
Foco nos estudos de segurança (não clínicos)Fortalecimento da pesquisa no Brasil - informações disponíveis em formato de guia: apresenta as
diretrizes, mas não é mandatório (RDC)
Obtenção de informações necessárias e relevantes para realização da pesquisa clínicaProdutos de degradação (RDC 53/2015 e RDC 171/2017)RDC 53/2015: “adequação” dos medicamentos registrados
Entretanto: entrada da Anvisa no ICH em nov/2016
Necessidade de alinhamento com o ICH e mudança das regras e dos prazos

Priorização de análise
Resolução de Diretoria Colegiada (RDC) nº 37, de 16 de junho de 2014 – REVOGADA!
• Priorização da análise técnica de petições de registro, pós-registro e anuência prévia em pesquisa clínica demedicamentos
• Revogada em virtude de Lei nº 13.411, de 28 de dezembro de 2016: os critérios da RDC não atendiam aosnovos critérios estabelecidos pela Lei (complexidade técnica e benefícios clínicos, econômicos e sociais dautilização do medicamento objeto do requerimento)
• Proposta de iniciativa regulatória foi publicada no DOU nº 61, de 29/04/2017 com o objetivo deregulamentar o tema – Consulta Pública no 372, de 02/08/2017: prazo para contribuição até 09/10/2017(http://portal.anvisa.gov.br/consultas-publicas#/visualizar/354234)

Proposta – RDC – DOENÇAS RARAS
Proposta de regulamentação especial de anuência em pesquisa clínica e registro de medicamentos paradoenças raras
• Publicada proposta de iniciativa no DOU nº 232, de 05/12/2016
• 14ª Reunião Ordinária Pública de 2017 (06/06/2017): alterado o regime de tramitação da proposta(inicialmente tramitou em regime especial)
• Consulta Pública nº 355, de 19 de junho de 2017, publicada no DOU nº 116, de 20/06/2017 – 30contribuições recebidas. Período de contribuições encerrado em 26/07/2017. Em fase final de consolidaçãodas contribuições.

RESOLUCAO DA DIRETORIA COLEGIADA - RDC No 172, DE 8 DE SETEMBRO DE 2017
Dispoe sobre os procedimentos para a importacao e a exportacao de bens e produtos destinados a pesquisa cientifica ou
tecnologica e a pesquisa envolvendo seres humanos, e da outras providencias.
A importação de bens e produtos sob vigilancia sanitária, destinados à pesquisa cientifica ou tecnologica, realizada por pesquisadores ou Instituições Cientifica, Tecnologica e de Inovação devidamente credenciados pelo CNPq, nos termos
da Lei no 8.010/90 e suas alterações, terá o deferimento automatico do licenciamento de importação no
SISCOMEX.
Não credenciados pelo CNPq, Envolvendo Seres Humanos, Amostras Biologicas Humanas e Produtos Sujeitos ao Controle Especial.
Em até 48 (quarenta e oito) horas apos a chegada do produto em territorio nacional e o cumprimento dos
requisitos legais pertinentes.
Importacao e a exportacao

Obrigado!
Contatos
Agência Nacional de Vigilância Sanitária - Anvisa
SIA Trecho 5 - Área especial 57 - Lote 200
CEP: 71205-050
Brasília - DF
www.anvisa.gov.br
www.twitter.com/anvisa_oficial
Anvisa Atende: 0800-642-9782