Embed Size (px)
Citation preview

INSTITUTO POLITÉCNICO NACIONAL
Escuela Nacional de Ciencias Biológicas Departamento de Fisiología
“Dr. Mauricio Russek Berman”
Laboratorio de metabolismo I
Efecto neuroprotector del extracto etanólico de
Buddleja cordata kunth en un modelo de epilepsia generado
por la administración de pentilentetrazol
Tesis
Para obtener el título de
Químico Farmacéutico Industrial
Presenta Director de tesis
Erick García Pliego Dra. Margarita Franco Colín
Co-director
Dr. Edgar Cano Europa
CIUDAD DE MÉXICO, 2018

aa

I
Índice
Relación de figuras………………………………………………………………III
Relación de tablas………………………………………………….…………….IV
Abreviaturas………………………………………………………..……………..V
Resumen………………………………………………………………………….VI
I.- Introducción……………………………………………………………………1
I.I Anatomía y fisiología del Sistema Nervioso Central (SNC)……………...………………...1
I.II Antecedentes de la epilepsia…………………………………………………………….....17
I.III Epilepsia………………………………………………..………………………………….18
I. IV Epidemiología………………………………………….……………………….................19
I.V Etiología………………………………………………….……………………………...…..19
I.VI Clasificación de la epilepsia…………………………………………..…...……...............21
I.VII Fisiopatología de la epilepsia………………………….…………………………………23
I.VIII Tratamiento de la epilepsia………………………………………………………….….23
I.IX Uso de plantas medicinales para el tratamiento de la epilepsia………………………...26
I.X Buddleja cordata kunth………………..……………………………………………………26
I.XI Modelos para el desarrollo de crisis epiléptica………………………………..................27
II.- Justificación………………………………………………………………….31

II
III.- Hipótesis...………………………………………………………………..…31
III.- Objetivo general…...……………………………………………………..…31
IV.- Objetivos particulares……………………………………………………...31
V.- Metodología…...……………………………………………………………...32
V.I Material vegetal…………………………………………………………………………….32
V.II Preparación del extracto etanólico de Buddleja cordata kunth..………..………………32
V.III Análisis fitoquímico preliminar…………………………………………………………32
V.IV Dosis letal 50…..…………………………………………………………………………..33
V.V Efecto anticonvulsivo.……………………………………………………………………..33
V.VI Marcadores de función hepática y renal……………………………………...………...35
V.VII Análisis histológico………………………………………………………………………35
V.VIII Análisis estadístico……………………………………………………………………...35
VI.- Resultados……...……………………………………………………………36
VII.- Discusión.…...……………………………………………………………...45
VIII.- Conclusión………………………………………………………………...49
IX.- Referencias Bibliográficas……………...…………………………………..50

III
Relación de figuras
Figura 1.- Representación celular de la neurona……….…………………………………………1
Figura 2.- Núcleo y nucléolo de una neurona…………...………………………………………..2
Figura 3.- Clasificación anatómica y funcional de las neuronas…………………………………3
Figura 4.- Diferentes tipos de neuronas…………………………………………………………..4
Figura 5.- Células de la glía en el sistema nervioso.………………...……………………………5
Figura 6.- Células de Schwann en el SNP………………………………………………………..6
Figura 7.- Etapas del potencial de acción………………………………………………...………8
Figura 8.- Representación de la unión GAP…………………………………………………….10
Figura 9.- Representación de la fusión de membranas catalizada por proteínas SNARE………12
Figura 10.- Estructura química del ácido-γ-aminobutírico (GABA)...………………….…........13
Figura 11.- Biosíntesis y metabolismo del GABA………………………...………………........14
Figura 12.- Vista de un modelo homólogo del receptor GABAA……………………………….15
Figura 13.- Representación de los canales iónicos regulados por ligando tipo cys-loop……….16
Figura 14.- Estructura química del glutamato……..……………………………………………17
Figura 15.- Clasificación de crisis generalizadas, focales y desconocidas de acuerdo con la
ILAE……………………………………………………………………………………………..23
Figura 16.- Mecanismo de acción de los fármacos antiepilépticos……………………………..24
Figura 17.- Estructura química del ácido kaínico……………………………………………….29
Figura 18.- Estructura química del pentilentetrazol……………………………………………..29
Figura 19.- Efecto del extracto etanólico de Buddleja cordata kunth sobre la duración de las
convulsiones (min) en ratas Fischer 344 tratadas con PTZ……………………………………...38
Figura 20.- Efecto del extracto etanólico de Buddleja cordata kunth sobre el tiempo de latencia
de las convulsiones (min) en ratas Fischer 344 tratadas con PTZ……….....................................38
Figura 21.- Efecto del extracto etanólico de Buddleja cordata kunth sobre los niveles de BUN,
bilirrubina total, bilirrubina directa y bilirrubina indirecta en los grupos estudiados………...….39
Figura 22.- Efecto del extracto etanólico de Buddleja cordata kunth sobre la morfología de
neuronas en el núcleo basolateral de amígdala, zona M1 de corteza motora, estría terminal y zona
CA3 de hipocampo……………………...…………………………………………………….…40

IV
Figura 23.- Fotomicrografías del núcleo basolateral de amígdala de los grupos estudiados…...41
Figura 24.- Fotomicrografías de la zona M1 de corteza motora de los grupos estudiados…......42
Figura 25.- Fotomicrografías de estría terminal de los grupos estudiados……...………………43
Figura 26.- Fotomicrografías de la zona CA3 de hipocampo de los grupos estudiados…..…….44
Relación de tablas
Tabla 1.- Etiología de la epilepsia respecto a la edad de los pacientes………………………….20
Tabla 2.- Cambio de la terminología etiológica de la epilepsia…………………………………20
Tabla 3.- Sitio de acción de fármacos antiepilépticos de primera, segunda y tercera
generación………………………………………………………………………………..………25
Tabla 4.- Agentes para la generación de kindling químico…………………………………...…28
Tabla 5.- Grupos formados para realizar las pruebas de DL50……..…………………………....33
Tabla 6.- Grupos formados para realizar las pruebas del efecto anticonvulsivo a dosis crecientes
del extracto etanólico de Buddleja cordata kunth………………………………………………..34
Tabla 7.- Metabolitos secundarios presentes en el extracto ácido, acuoso y etanólico de hojas de
Buddleja cordata kunth…………………………………………………………………………..36
Tabla 8.- Manifestaciones presentes en los grupos experimentados a diferentes dosis del extracto
etanólico de Buddleja cordata kunth……………………………………………………….…….37 Tabla 9.- Dosis letal media (DL50) y nivel de toxicidad del extracto etanólico de hojas de
Buddleja cordata kunth…………………………………………………………………………..37

V
Lista de abreviaturas
AMPA
DL50
BDZ
EE
ELT
FAE
GABA
Glu
I.G.
ILAE
I.P.
KA
MES
OMS
PA
PTZ
REA
SNARE
SNC
SNP
V.O.
Ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico
Dosis letal 50
Benzodiazepina
Estado epiléptico
Epilepsia de lóbulo temporal
Fármacos antiepilépticos
Ácido γ-aminobutírico
Glutamato
Intra gástrico
International League Against Epilepsy
Intra peritoneal
Ácido kaínico
Maximal Electroshock Seizure
Organización Mundial de la Salud
Potencial de acción
Pentilentetrazol
Relación estructura actividad
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor
Sistema Nervioso Central
Sistema Nervioso Periférico
Vía oral

VI
Resumen
La epilepsia es un trastorno que afecta a millones de personas en el mundo que se presenta como
crisis epilépticas espontaneas por una sobre excitación de una población neuronal. El objetivo del
presente trabajo fue demostrar que el extracto etanólico de Buddleja cordata kunth presenta un
efecto neuroprotector ante los procesos que engloban la epilepsia.
Para este propósito se procedió en un comienzo a realizar el extracto etanólico de hojas de Buddleja
cordata kunth, se procedió a determinar la DL50 del extracto mediante el método modificado de
Lorke así como la determinación de la presencia de metabolitos secundarios en las hojas de
Buddleja cordata kunth.
Se administraron por vía oral a grupos de experimentación con dosis de 75, 150 y 300 mg/kg del
extracto 30 minutos previos a la generación del estado epiléptico inducido por pentilentetrazol
(PTZ), posteriormente se determinó el efecto anticonvulsivo así como la cuantificación de
marcadores de función hepática y renal junto a estudios histológicos para cuantificar la densidad
de neuronas normales y anormales en amígdala, corteza motora, estría terminal e hipocampo. Los
resultados del presente estudio demostraron que el uso del extracto etanólico de Buddleja cordata
kunth además de disminuir los efectos convulsivos también aminora el daño y la muerte neuronal
producido por el PTZ en las distintas zonas del cerebro estudiadas, por lo cual se podría utilizar
como un agente terapéutico en personas que sufren procesos epilépticos.
Palabras clave:
Epilepsia del lóbulo temporal, amígdala, corteza motora, estría terminal, hipocampo,
pentilentetrazol, neuro protección, Buddleja cordata kunth

1
Introducción
Anatomía y fisiología del Sistema Nervioso Central (SNC)
Para conocer y poder comprender la fisiopatología de la epilepsia y al mismo tiempo entender la
forma en que actúan los fármacos antiepilépticos es necesario explicar algunos conceptos básicos
que engloben la función y conformación del sistema nervioso central (SNC) así como las
moléculas que son esenciales para la correcta homeostasis del SNC.
Estructura de las neuronas
El SNC está conformado por neuronas (figura 1) que producen y conducen impulsos eléctricos, y
por células de sostén (Ira, 2011), las cuales su población varía entre 100 000 millones de neuronas
y de 10 a 50 mil millones son células neurogliales (Barrett et al., 2013).
Figura 1. Representación celular de la neurona, se observan las estructuras que conforman la neurona así como
la presencia de vaina de mielina que no se presenta en todas las neuronas (Joel y Sabyasachi, 2012).

2
La unidad básica del SNC es la neurona y de su buen funcionamiento depende todas las actividades
de los seres vivos. La membrana de las neuronas recibe el nombre de plasmolema que al igual que
otro tipo de células representan una barrera impermeable a sustancias hidrosolubles (Bustamante,
2007). Las neuronas están conformadas por las siguientes estructuras:
• Soma o cuerpo celular, lleva a cabo el metabolismo de la célula por medio de los cuerpos de
Nissl (pilas de retículo endoplásmico rugoso) y aparato de Golgi, además en el soma también se
encuentra el citosol que contiene proteínas necesarias para el metabolismo, mitocondrias para la
producción de energía, neurofilamentos, citoesqueleto y microtúbulos. El núcleo (figura 2), cuya
estructura presentan forma esférica con dimensiones de 3 y 18 µm; y el nucléolo, se encarga de la
producción de ácido nucleico y proteínas citoplasmáticas (Bustamante, 2007; Barrett et al., 2013).
• Dendritas, son prolongaciones delgadas y ramificadas procedentes del soma que se encargan
de proporcionar un área receptiva que transmite impulsos eléctricos hacia el soma (Ira, 2011).
Presentan proyecciones diminutas llamadas espinas, que tienen la capacidad de formar sinapsis,
cuando se produce la transmisión sináptica, la concentración de Ca2+ se eleva a las espinas y
contribuyen al incremento de la duración del potencial postsináptico (Joel y Sabyasachi, 2012).
• Axón o fibra nerviosa, llevan a cabo la conducción de señales lejos del soma, se pueden
encuentran rodeados por una vaina aislante, denominada vaina de mielina y formada por células
de Schwann o por la oligodendroglía, cuya función es aumentar la conducción de información a
alta velocidad.
• Terminales axónicas o sinápticas, son ramificaciones provenientes de la terminación del axón
que permiten enviar señales simultaneas a otras neuronas, glándulas o fibras musculares (Randall
et al., 1998; Cardinali, 2007).
Figura 2. Núcleo y nucléolo de una neurona, el aspecto pálido del núcleo se debe a la presencia de ácido
desoxirribonucleico (ADN) mientras que el nucléolo presenta una gran cantidad de ácido ribonucleico (ARN)
como ADN (Bustamante, 2007).
Núcleo
Nucléolo

3
Clasificación morfológica de las neuronas
Las neuronas se clasifican según su ubicación en el SNC, sus neurotransmisores o el modelo de
sus conexiones sinápticas y morfológicamente con base en el número, la longitud y la forma de
sus dendritas (figura 3):
• Neurona unipolar o pseudounipolar, presenta una única dendrita con una corta distancia al
soma. En invertebrados se denominan unipolares y su función es dar origen a varias ramas,
mientras que las neuronas pseudounipolar da origen a dos ramas funcionales, una periférica o
dendrítica y otra central (Cardinali, 2007; Snell, 2007).
• Neurona bipolar, presenta 2 axones que se encuentran en los extremos opuestos del soma y
que se extienden hacia afuera de este, se hallan en el ganglio espiral (de Corti) de la cóclea del
oído interior o neuronas de la retina (Silverthorn, 2008).
• Neurona multipolar, de su soma emerge una gran cantidad de dendritas y solo un axón, se
encuentra en el encéfalo, corteza cerebral, motoneuronas espinales, células de Purkinje del cerebro
y medula espinal. (Cardinali, 2007; Snell,2007).
• Neurona anaxónica, se caracteriza por ausencia de axón, suele encontrarse en el bulbo olfatorio
y en la retina (Welsch y Sobotta, 2008).
Si del tamaño se trata las neuronas se pueden clasificar de la siguiente manera:
• Neuronas de Golgi de tipo I o neuronas de proyección, son neuronas más grandes con axones
de hasta 1 metro o más de longitud, ejemplos son las neuronas piramidales de la corteza cerebral,
células de Purkinje de la corteza cerebelosa y células motoras de la médula espinal.
Figura 3.- Clasificación anatómica y funcional de las neuronas, se presenta los tipos de neuronas, su estructura
celular que las caracteriza y su localización dentro del sistema nervioso (Silverthorn, 2008).

4
• Neuronas de Golgi de tipo II o neuronas de circuito local, no presentan axón y si lo hay es
con una longitud corta que finaliza cerca del soma observándose de manera estrellada, se halla en
corteza cerebral y cerebelosa (Joel y Sabyasachi, 2012; Snell, 2007).
De acuerdo con Snell (2007) y Welsch y Sobotta (2008), existe otra manera de nombrar a las
neuronas multipolares (figura 4):
• Neuronas piramidales, presentan un cuerpo triangular y de gran tamaño, una dendrita apical
larga, así como varías dendritas basales y se encuentran en el cerebro (Berne y Levi, 2009).
• Neuronas mitrales, son neuronas que se encuentran en el bulbo olfatorio (Welsch y Sobotta,
2008).
• Neuronas estrelladas, llamadas también neuronas granulares, son interneuronas que poseen un
cuerpo pequeño y numerosas dendritas ramificadas, se presentan en la capa IV de la corteza y
tienen la función de inhibir usando el neurotransmisor GABA (Berne y Levi, 2009).
• Neuronas de Purkinje, se encuentran en el cerebelo que se encarga de enviar señales a otros
sitios del cerebro para llevar a cabo funciones de coordinación motora, así como efectos
inhibitorios con otras neuronas (Ira, 2011).
Con base a esto se puede comprender la organización de las neuronas; cuando un cúmulo de somas
se encuentran dentro del SNC se denomina “núcleo”, mientras que si ese cúmulo se encuentra
fuera del SNC se denomina “ganglio”. Por otra parte, un haz de axones dentro del SNC se
denomina “tracto” mientras que si ese haz de axones se localiza fuera del SNC se denomina
“nervio” (Joel y Sabyasachi, 2012).
Figura 4.- Diferentes tipos de neuronas, se aprecia la forma que toma el cuerpo de la neurona que puede ser
de forma estrellada, piramidal u ovoidea (Snell, 2007).

5
Células de sostén
Las células nerviosas a diferencia de las demás células del cuerpo, no se encuentran recubiertas
por tejido conjuntivo, si no por un tipo diferente de células, llamadas células de la glía (significa
pegamento nervioso) que son el tejido conectivo del cerebro (figura 5), dichas células derivan de
la misma capa de tejido embrionario (ectodermo) que produce las neuronas (Ira, 2011). A pesar de
no participar en la transmisión de señales eléctricas proporcionan un soporte físico, bioquímico y
metabólico que mantienen la homeostasis del líquido extracelular del encéfalo ya que captan el
exceso de metabolitos y de iones (Silverthorn, 2008), de acuerdo con Cardinali (2007) presentan
las siguientes funciones:
• Soporte para las neuronas.
• Eliminar productos de desecho del metabolismo neuronal o restos celulares.
• Provisionar vaina de mielina.
• Funciona como buffer espacial de K+.
• Guía para la migración neuronal durante el desarrollo.
• Captación de neurotransmisores.
• Generar señales de tipo paracrino, como distintas citocinas.
• Capacidad de regeneración neuronal.
Figura 5.- Células de la glía en el sistema nervioso, el tipo de célula va a depender del lugar en que se
encuentra, el SNP presenta células de Schwann y células satélite; mientras que en el SNC se presentan cuatro
tipos de células: oligodendrocitos, microglía, astrocitos y células ependimarias (Silverthorn, 2008).

6
Las células de la glía se encuentran tanto en el SNC (reciben el nombre de neuroglia) como en el
SNP. Las células de sostén presentes en el SNP son:
• Células de Schwann, este tipo de células se encarga de rodear los axones amielínicos de las
neuronas, estos se rodean sola por una célula de Schwann (figura 6).
• Células satélite o gliocitos ganglionares, son células de Schwann amielínicas, forman cápsulas
de sostén alrededor de los cuerpos de las neuronas ganglionares de la raíz dorsal y de los pares
craneales (Silverthorn, 2008; Berne y Levi, 2009; Ira, 2011).
La glía presente en el SNC se compone de:
• Oligodendrocitos, tienen como papel facilitar la comunicación eléctrica entre las neuronas, ya
que se encargan de la producción de mielina que envuelve a los axones, existen tres tipos de
oligodendroglía en el SCN que son: perineuronales, perivasculares e interfasciculares (Reyes et
al., 2014).
• Microglía, semejantes a los oligodendrocitos pero en menor tamaño, son células inmunitarias
especializadas que residen y migran a través del SNC y se activan cuando existe una lesión central,
un proceso inflamatorio o cualquier otro evento que pueda perturbar la homeostasis del SNC, se
activan por quimiocinas y sufren una transformación para tener la capacidad de fagocitar desechos
celulares, así como la secreción de sustancias que permitan la supervivencia celular (Cerdón y
Troncoso, 2016; Joel y Sabyasachi, 2012).
Figura 6.- Células de Schwann en el SNP, las células de Schwann se caracterizan por envolver únicamente a un
solo axón, dicho axón puede presentar hasta 500 células de Schwann (Silverthorn, 2008).

7
• Astrocitos, son las células más abundantes en el SNC y su nombre se debe a su forma de
estrella, proporcionan soporte estructural que define límites celulares además de contribuir a la
formación de la barrera hematoencefálica (BHE), es decir, la regulación del ambiente externo de
las neuronas en el SNC, otras de sus funciones es el control de la formación y función sináptica,
neurogénesis y la regulación del tono vascular del cerebro (Martínez, 2014; Ira, 2011).
• Células ependimarias, recubren los ventrículos encefálicos y el conducto central de la médula
espinal, separándolo del líquido cefalorraquídeo, creando así una capa epitelial con permeabilidad
selectiva (Silverthorn, 2008; Berne y Levi, 2009).
Función de la neurona
La neurona es una célula excitable ya que tiene la capacidad de enviar información en forma de
impulsos eléctricos denominados “potenciales de acción (PA)” o “impulsos nerviosos” que llevan
a cabo diferentes tipos de información motora y sensitiva (Joel y Sabyasachi, 2012). Dicha
información de acuerdo con Hall (2011) puede quedar bloqueada en su transmisión de una neurona
a otra, convertirse en una cadena repetitiva a partir de un solo impulso o integrarse con los
procedentes de otras células para originar patrones muy intrincados en las neuronas sucesivas.
Las neuronas tienen la característica de poseer propiedades eléctricas pasivas (capacitancia y
resistencia) y propiedades eléctricas activas (conducir señales eléctricas sin decremento), la
propiedad eléctrica activa depende de proteínas especializadas en la membrana celular
denominadas canales iónicos dependientes de voltaje que permiten a los iones moverse a través de
la membrana por cambios en el campo eléctrico de la membrana (Randall et al., 1998). El PA se
produce cuando se abren canales iónicos regulados por voltaje que altera la permeabilidad de la
membrana hacia los iones Na+ y K+, el PA se divide en las siguientes fases (figura 7):
• Despolarización: cuando se produce un estímulo hay un aumento de la permeabilidad de la
neurona para el ion Na+, el PA comienza de manera lenta despolarizando a la membrana hasta
alcanzar el umbral (-55 mV), la membrana conforme se despolariza abre más canales de Na+
haciendo que esta sea aún más permeable al ion Na+.
• Pico máximo: es el valor máximo de despolarización, es decir, el potencial de membrana se
vuelve positivo (+55 mV), cuando se alcanza este punto se detiene la entrada de Na+ y se trata de
llevar a un potencial de equilibrio del Na+ (ENa) de +60 mV, pero antes de alcanzar dicho equilibrio
los canales de Na+ en el axón se inactivan, al suceder esto el PA alcanza un pico de +30 mV.

8
• Repolarización: una vez que los canales de Na+ se inactivan, los canales de K+ dependientes
de voltaje se abren haciendo permeable a la membrana, de tal manera que el gradiente de
concentración produce la salida de iones K+ hacia el exterior de la célula generando un potencial
de membrana negativo, es decir, la repolarización de la membrana.
• Hiperpolarización o postpotencial: cuando han salido los iones K+ y el potencial de
membrana alcanza un valor de -70 mV, el K+ sigue saliendo a través de los canales regulados por
voltaje y los canales permeables de K+ y la membrana se hiperpolariza aproximadamente a -90 mV
(denominada subestimulación). Cuando los canales de K+ regulados por voltaje se cierran y se
detiene la salida de K+ se restaura el potencial de membrana a -70 mV (Córdova, 2003; Silverthorn,
2008; Rosenzweig et al., 2005).
Cuando el PA ha terminado las neuronas requieren un tiempo para que los canales de Na+
dependientes de voltaje regresen a su configuración original, a este tiempo se le denomina periodo
refractario y es necesario para que las neuronas puedan generar nuevamente un PA. El periodo
refractario está dado en dos fases: periodo refractario absoluto que corresponde con generar el PA
(1-2 ms) y se caracteriza porque la membrana es incapaz de responder a un estímulo sin importar
su magnitud; y periodo refractario relativo (10-15 ms) que corresponde con el aumento de la
permeabilidad de los iones K+ pero con la ventaja de poder generar un segundo PA siempre y
cuando se aplique un estímulo de mayor intensidad al umbral (Córdova, 2003).
Figura 7.- Etapas del potencial de acción, se muestran los cambios de voltaje y de permeabilidad a los iones
que ocurren en una sección de la membrana durante las distintas etapas del PA, se aprecian 3 etapas del PA:
fase de crecimiento del potencial de acción, fase de caída y fase de poshiperpolarización. (Silverthorn, 2008).

9
Las neuronas en reposo presentan un potencial eléctrico llamado “potencial de reposo” de la
membrana, la diferencia de potencial de una neurona típica es de 65 mV, como la carga neta fuera
de la célula es 0 se dice que el potencial de reposo de la membrana de una neurona es de -65 mV.
La diferencia en el potencial de reposo se debe a la desigual distribución de iones positivos (Na+
y K+) en el exterior de la membrana así como a los aminoácidos y proteínas en el interior de la
célula con carga negativa y a la permeabilidad por parte de la membrana por iones (Bustamante,
2007). Los PA se caracterizan porque:
• Se producen por estímulos despolarizadores.
• La despolarización (disminución del potencial de membrana) alcanza el umbral para
desencadenar el estímulo.
• Responden a la ley del “todo o nada”, se entiende que si no alcanza el umbral el estímulo no se
genera (nada) y en caso de que alcance el umbral se produce el potencial de acción completo (todo),
despreciando la intensidad o duración del estímulo.
• La propagación del potencial de acción se produce sin decremento.
• Después de que se produce un potencial de acción existe un tiempo en el cual la neurona no es
capaz de producir un segundo potencial de acción, denominado “período refractario” (Córdova,
2003; Rosenzweig et al., 2005).
Tipos de sinapsis
La sinapsis es un sitio especializado de interacción funcional entre neuronas mediante el axón,
dendritas o cuerpo celular (Bennet, 1997; Coaquira et al., 2012). Se puede clasificar a la sinapsis
como química o eléctrica y estas pueden excitar o inhibir a las células postsinápticas.
Sinapsis eléctrica
Una sinapsis eléctrica es el paso rápido de información entre células por acoplamiento iónico
directo, en la cual, dicha transferencia de información tiene lugar por medios únicamente
eléctricos, sin la participación de un transmisor químico (Randall et al., 1998). Para que las células
tengan la posibilidad de intercambiar información se necesita de uniones específicas denominadas
uniones gap, nexo, hendidura o interconexión (Dvorkin et al., 2010). Son una serie de proteínas
integrales que permiten el paso de iones y moléculas de bajo peso molecular, están formadas por
la oposición de conexones de células adyacentes y cada conexón está formado a su vez por seis
proteínas de conexina (figura 8).

10
Son esenciales en varios procesos fisiológicos como la propagación de señales eléctricas y
coordinación de señalización celular por transferencia de segundos mensajeros, encontrándose
expresados en todos los tejidos excepto en músculo esquelético, eritrocitos y células espermáticas
maduras (Nielsen et al., 2012).
Actualmente se han identificado un segundo grupo de proteínas que forman uniones gap, dichas
proteínas reciben el nombre de panexinas, estas uniones gap se han implicado en diversas
patologías incluyendo enfermedades cardiovasculares, cáncer y neuropatías, sin embargo,
investigaciones recientes hechas en ratones han demostrado que las panexinas también han
contribuido a la protección de la lesión por accidente cardiovascular isquémico, modulación de
excitabilidad neuronal y aprendizaje (Beyer y Berthoud, 2017).
Sinapsis química
En este tipo de sinapsis existe un espacio entre la membrana celular presináptica y la postsináptica
denominada hendidura sináptica por la cual es transmitida la información a través de sustancias
químicas que reciben el nombre de neurotransmisores (Costanzo, 2011). De acuerdo con Palacios
et al (2005) las características de una sinapsis química son:
• Unidireccional: su estructura determina que sólo se pueda transmitir en un sentido.
• Retarda la conducción de impulsos nerviosos: produce fatiga sináptica, es decir, que una
estimulación repetitiva de las terminales presinápticas produce que la actividad postsináptica se
detenga con el tiempo, debido al agotamiento del neurotransmisor y como protección contra el
exceso de excitabilidad.
• Integración neuronal: es decir a la suma temporal y espacial de sinapsis en el soma que
procede de diferentes señales sinápticas.
Figura 8.- Representación de la unión GAP. La ubicación del poro del canal GAP se ha indicado con un
circulo amarillo, se aprecia la conformación de conexón formado a su vez por 6 conexinas. Modificado de
Nielsen et al., 2012.
Citoplasma
Espacio
extracelular
Conexón
(célula A)
Conexón
(célula B)
Conexina
Citoplasma

11
La mayoría de las transmisiones en el SNC son de naturaleza química, en donde los
neurotransmisores son captados por proteínas receptoras presentes en la membrana postsináptica
ya sea para excitarla, inhibirla o modificar su sensibilidad. En la actualidad se conocen más de 40
neurotransmisores y entre los más importantes se citan la acetilcolina, noradrenalina, adrenalina,
histamina, ácido γ-aminobutírico (GABA), glicina, serotonina y glutamato (Hall, 2011).
De acuerdo con Cardinali (2007) para que una sustancia sea considerada un neurotransmisor
tiene que cumplir con las siguientes especificaciones:
• Tiene que ser sintetizado por la neurona presináptica y almacenarse en vesículas sinápticas (a
excepciones de algunos neurotransmisores como gases o lípidos).
• Se tiene que liberar por el estímulo neural fisiológico.
• Tiene que actuar en la postsinapsis.
• Tienen que existir mecanismos efectivos para que su acción finalice (recaptación en la terminal
neural, difusión al espacio extrasináptico, metabolismo) y así pueda producir una rápida acción y
difusión del neurotransmisor.
La comunicación entre las neuronas es a través de la liberación de neurotransmisores, esto
involucra la formación de una vesícula desde la neurona precursora seguido del transporte de la
vesícula a la membrana de la célula y finalmente la fusión de la vesícula con la membrana neuronal,
en dicho proceso participan las proteínas SNARE (figura 9) que permiten la comunicación
mediante la liberación del neurotransmisor a través de vesículas (Jahn y Scheller, 2006). Las
SNARE (Soluble N-ethylmaleimide-sensitive factor attachment protein receptor) son una
superfamilia de proteínas pequeñas que tienen un motivo conservado de aproximadamente 60-70
aminoácidos (motivo SNARE) en su dominio citoplásmico. Los procesos en los que participan
estas proteínas son el crecimiento celular, la citocinesis y la transmisión sináptica (Van den
Bogaart et al., 2013). Los motivos SNARE no presentan una estructura definida, pero cuando estos
entran en contacto entre sí y adoptan una conformación de hélice alfa formado por 4 hélices alfas
entrelazadas y en el que cada hélice corresponde a un motivo SNARE diferente, dentro de las 4
hélices se encuentra 16 anillos que pueden presentar 3 residuos de glutamina (Q) y 1 de arginina
(R). De acuerdo con la presencia de alguno de estos aminoácidos las proteínas SNARE se
clasifican en Q y R además de esto las SNARE Q se subdivide en Qa, Qb, Qc y Qb (Merino et al.,
2009; Jahn y Scheller, 2006).

12
Varios procesos de fusión en la membrana se encuentran catalizados por distintos conjuntos de
proteínas SNARE, encontrándose las proteínas asociadas a la liberación de neurotransmisores
dependientes de Ca2+ llamadas (vesicular) v-SNARE sinaptobrevina-2 (llamada también VAMP2
o R-SNARE) en la membrana de la vesícula, y las proteínas (target) t-SNARE sintaxina-1 (Qa) y
SNAP25 (Qb y Qc) en el plasma de la membrana neuronal (Jahn y Scheller, 2006).
Otra familia de proteínas a considerar es el de las proteínas SM (Sec/Munc) también conocidas
como nSec1, Munc18-a y rbSec1, poco se sabe de la función de dichas proteínas, sin embargo se
tiene conocimiento de que dichas proteínas se encuentran implicadas en la regulación del complejo
SNARE dado a que las proteínas SM interactúan de manera específica y con alta afinidad a la
sintaxina de las proteínas SNARE (Voets et al., 2001).
Munc18-1 interactúa con SNARE de dos formas diferentes al unirse con alta afinidad al dominio
Habc de la sintaxina. La primera forma Munc18 interactúa con el segmento helicoidal N-terminal
de la sintaxina llamado dominio Habc formando el complejo Habc/sintaxina. El segundo modo
Munc18 se une al núcleo de 4 hélices de SNARE formado por sintaxina, VAMP y SNAP25. Ante
esto las proteínas SM son importantes para mantener un efecto estabilizante de la conformación
cerrada de la sintaxina durante el transporte de la vesícula en la membrana (Diao et al., 2010).
Las sinaptotagminas se tratan de una pequeña familia de proteínas membranales expresadas en
neuronas y células endócrinas que actúan como sensores de calcio en los procesos de exocitosis
Figura 9.- Representación de la fusión de membranas catalizada por proteínas SNARE. (A) Dominio topológico
neuronal SNARE: sinaptobrevina-2 y proteínas SNARE de membrana neuronal SNAP25 y sintaxina-1, se indica
el dominio Habc de la sintaxina-1. (B) Proceso de exocitosis donde las proteínas SNARE de la vesícula
interactúan con un complejo SNARE complementario aceptor en la membrana neuronal, dando la formación de
un dominio SNARE de espiral enrollada alfa-helicoidal estrecha que supera la barrera de energía para la fusión
de la membrana. (C) Estructura del complejo neuronal SNARE. Modificado de Van den Bogaart et al., 2013.
SNAP25
Sinaptobrevina-2
Sintaxina-1
Sintaxina-1
Sinaptobrevina-2
SNAP25
Dominio
Habc

13
de neurotransmisores. Estas proteínas se pueden encontrar en varias isoformas en la membrana
vesicular (Syt1, Syt2 y Syt9) como en la membrana plasmática (Syt3 y Syt7) pero siendo
principalmente la Syt1 y Syt2 las principales isoformas que funcionan como sensor sináptico de
Ca2+ (sensor que acopla Ca2+) con la liberación rápida del transmisor. Adicionalmente las
sinaptotagminas interactúa también con otras proteínas que forman parte de la neuroexocitosis,
como la sintaxina 1, SNAP-25, complejo SNARE, RIM, SV2 o canales de Ca2+ de tipo N (Lang y
Jahn, 2008: Merino et al., 2009).
Sistema gabaérgico
El ácido γ-aminobutírico o denominado como GABA (figura 10) es el neurotransmisor inhibitorio
más abundante en el SNC ya que se encuentra cerca del 50% en las sinapsis centrales inhibitorias
(Dvorkin et al, 2010). La función del GABA es la de inhibición a través de un potencial
postsináptico inhibitorio (PPSI) al incrementar la conductancia del ion Cl-, produciendo una
hiperpolarización de la membrana celular (Mendoza, 2008).
Biosíntesis del GABA
Se realiza en el SNC (Figura 11) ya que el GABA es incapaz de atravesar la barrera
hematoencefálica, el GABA proviene cuando el glutamato es descarbolixado por la enzima
glutamato descarboxilasa (GAD). Además, el GABA se liga al ciclo de Krebs por medio del ácido
2-oxoglutárico y del semialdehído succínico que es precursor del ácido succínico a través de la
enzima semialdehído succínico deshidrogenasa. Otra enzima de importancia en el metabolismo
del GABA es la GABA-transaminasa (GABA-T) ya que por un lado convierte el ácido 2-
oxaglutárico a glutamato, así como la conversión del GABA a semialdehído succínico (Delgado
et al., 2004).
Figura 10.- Estructura química del ácido γ-aminobutírico (GABA).

14
Almacenamiento, liberación, recaptación e inactivación del GABA
Cuando el GABA ha sido sintetizado este es almacenado en vesículas presinápticas y es liberado
por exocitosis de manera Ca2+ dependiente. Cuando es liberado al espacio sináptico, el GABA que
no interactúa con su receptor postsináptico es recapturado por una proteína transportadora para ser
utilizado nuevamente, en caso de no ser recapturado es degradado por la enzima GABA-T, cuando
una molécula de GABA es degradada, una molécula de α-cetoglutarato es metabolizada a
glutamato (Mendoza, 2008; Redolar et al., 2010).
Receptores GABA
Actualmente se conocen 3 tipos diferentes de receptores GABA: receptor ionotrópico GABAA,
receptor metabotrópico GABAB y el recién descubierto receptor GABAC, todos estos con
propiedades farmacológicas, electrofisiológicas y bioquímicas diferentes (Wu y Sun, 2014).
El receptor GABAA (figura 12) se trata de una proteína oligomérica transmembranal formada por
cinco subunidades unidas por enlaces covalentes formando un pentámero que en su centro forman
un canal iónico, las subunidades presentes en el receptor GABAA son: α1-6, β1-3, γ1-3, δ, ε, π y ρ1-3
(Burón et al, 2008; Jacob et al., 2008).
Figura 11.- Biosíntesis y metabolismo del GABA, la síntesis comienza con la descarboxilación del glutamato
por la glutamato descarboxilasa (GAD), la GABA-T es una enzima clave en la biosíntesis del GABA ya que
cataliza tanto la desaminación oxidante del GABA a semialdehído succínico como la conversión del ácido 2-
oxoglutárico a ácido L-glutámico (precursor inmediato del GABA), siendo así la enzima idónea para la
regularizar indirecta de los niveles de GABA en el SNC (Delgado et al., 2004).

15
Los receptores GABAA se pueden localizar también en regiones que no conforman el SNC como
en las células granulares cerebelares, retina, hígado, músculo liso de vías respiratorias y diversas
células inmunes (Sigel y Steinmann, 2012).
Además los receptores GABAA son parte de la superfamilia del canal iónico del ligando
pentamérico Cys-loop (figura 13). Todos estos canales presentan funciones similares para
establecer la topología de la membrana, estructura del canal iónico, sitios de unión del agonista así
como sitios de unión para otro tipo de moduladores alostéricos, cada subunidad del receptor Cys-
loop está conformado por una larga región hidrofóbica extracelular N-terminal, seguido por cuatro
α-hélices transmembranales (TM) con un largo lazo intracelular entre TM3 y TM4, finalizando
con un dominio extracelular corto C-terminal (Olsen y Sieghart, 2008). Se ha determinado que
varias proteínas interactúan con el bucle intracelular entre TM3 y TM4, estas proteínas son
relevantes ya que se encuentran implicadas en el movimiento de receptores y en el anclaje de los
receptores en el citoesqueleto y en la membrana postsináptica (Sigel y Steinmann, 2012).
Figura 12.- Vista de un modelo homólogo del receptor GABAA, el canal GABA se encuentra formado por cinco
subunidades que forman un pentámero (α1, β1, γ1, δ, ε, π y ρ) que se ensamblan para formar un canal permeable
al ion Cl-. Debido a su heterogeneidad puede ser blanco de varias entidades químicas las cuales se unen a las
distintas subunidades del canal que desencadenan la apertura o cierre de dicho canal lo que permite el paso o no
de iones Cl- provocando distintos efectos sobre la neurona entre ellos convulsiones, efectos sedantes, etc. (Jacob
et al., 2008).

16
En pacientes que presentan convulsiones parciales así como en modelos experimentales destinados
a la generación de Epilepsia de lóbulo temporal (ELT) se tiene documentado un aumento o
disminución de las subunidades específicas del receptor GABAA pero suele aparecer un
incremento de las subunidades del receptor en las regiones afectadas por la neurodegeneración del
cual no se sabe si este aumento conduce a una hipersincronización anormal de las redes neuronales
o es parte de una manera compensatoria para proteger contra las convulsiones recurrentes
(Fritschy, 2009).
Por otro lado, los receptores GABAB se encuentran constituidos por dos subunidades de siete
subunidades transmembranales, GABAB1 y GABAB2 que se encuentran unidos por medio de una
interacción espiral/espiral entre sus colas C terminales (Kubo y Tateyama, 2005), estos son
receptores pre y postsinápticos, además de que dichos receptores se encuentran acoplados a canales
de K+ y Ca2+ vía proteína Gi. Los receptores GABAB funcionan como autorreceptores inhibiendo
la liberación de GABA ya que inhiben la adenilatociclasa y activan los conductos de K+
hiperpolarizando la membrana reduciendo así la conductancia de Ca2+ (Wu y Sun, 2014), también
pueden disminuir el flujo de iones Ca2+ disminuyendo la producción de inositoltrifosfato (IP3) y/o
inhibiendo la producción de AMPciclico (Olsen y Sieghart, 2008).
Figura 13.- Representación de los canales iónicos regulados por ligando cys-loop. Las subunidades de los
receptores GABAA están formados por 4 dominios transmembranales (TM1-4). La terminación extracelular
amino es el sitio de unión de GABA así como drogas psicoactivas como las benzodiazepinas, también se
representan los dominios intracelulares (TM3 y TM4) sitios de unión de proteínas. (Jacob et al., 2008).

17
Sistema glutamatérgico
El ácido glutámico (figura 14) o glutamato (Glu) es el principal neurotransmisor excitatorio del
SNC, el Glu se encuentra implicado en procesos fisiológicos como la proliferación celular,
apoptosis, supervivencia celular, proliferación de células nerviosas, aprendizaje y memoria, así
mismo se encuentra implicado en procesos patológicos como la epilepsia (Lujan, 2004).
Receptores de glutamato (Glu)
El glutamato tiene la posibilidad de unirse a dos tipos de receptores:
Receptores metabotrópicos (mGluRs) que se dividen en mGluRs del grupo I (mGluR1 y mGluR5)
que activan a la fosfolipasa C (PLC) ubicados en membranas postsinápticas, mGluRs del grupo II
(mGluR2 y mGluR3) y mGluRs del grupo III (mGluR4, mGluR6, mGluR7 y mGluR8) se hallan
en la presinapsis y se activan mediante la inhibición de la adenil ciclasa (Rivera y López, 2005).
Receptores ionotrópicos, estos últimos se dividen de acuerdo con sus agonistas específicos: N-
metil-D-aspartato (NMDA), ácido α-amino-3-hidroxi-5-metil-4-isoxazol (AMPA) y ácido kaínico
(AK) (Flores et al, 2012).
Antecedentes de la epilepsia
La epilepsia data de años muy antiguos, incluso la presencia de este trastorno va más allá de la
existencia del hombre mismo, ya que como lo menciona Medina et al. (2001) los propios
dinosaurios padecían de este trastorno, un claro ejemplo de esto es el Apatosaurus al que se le
calcula que vivió hace unos 150 millones de años en América del norte.
En tiempo arcaicos, la epilepsia solo se lograba explicar por pensamiento mágicos-religiosos
puesto que “el epiléptico ha incumplido una norma divida y ha sido castigado por ello”, la epilepsia
se convierte así en la enfermedad diabólica por excelencia (García et al., 2007). Sin embargo,
Hipócrates en el siglo 400 AC, explicaba que la epilepsia se debía a una alteración de carácter
natural, hereditario, producto de un desequilibrio de humores, que debía tratarse con dietas y
drogas (Acevedo et al., 2007).
Figura 14.- Estructura química del glutamato.

18
En la actualidad el avance científico ha llevado al desarrollo de fármacos antiepilépticos que
permitan controlar las crisis convulsivas, entre estos fármacos se encuentran los bromuros (1857),
los barbitúricos (1864), la difenilhidantoina (1937), la carbamacepina (1954) y ácido valproico
(1973) como principales tratamientos antes pacientes epilépticos (García et al., 2007).
Epilepsia
De acuerdo con la Liga Internacional Contra la Epilepsia (ILAE, 2005) la epilepsia puede definirse
de manera conceptual y para fines diagnósticos. Conceptualmente se denomina “crisis epiléptica”
a la ocurrencia transitoria de signos y/o síntomas debidos a actividad neuronal excesiva o
sincrónica en el cerebro. Por otra parte, de acuerdo con Fisher et al. (2014) la epilepsia es un
trastorno cerebral que se caracteriza por una predisposición continuada a la aparición de crisis
epilépticas y por las consecuencias neurobiológicas, cognitivas, psicológicas y sociales de
enfermedad, la definición de epilepsia requiere la presencia de al menos una crisis epiléptica.
Con respecto a la definición diagnostica o clínica operativa la ILAE (2005) menciona que la
epilepsia es una enfermedad cerebral que se define por cualquiera de las siguientes circunstancias:
1.- Al menos dos crisis no provocadas (o reflejas) con al menos 24 h de separación.
2.- Una crisis no provocada (o refleja) y una probabilidad de presentar nuevas crisis durante los 10
años siguientes similar al riesgo general de recurrencia (al menos el 60 %) tras la aparición de dos
crisis no provocadas.
3.- Diagnóstico de un síndrome de epilepsia (Fisher et al., 2014).
En el caso de la Organización Mundial de la Salud (OMS, 2017) “la epilepsia es una afección
crónica de etiología diversa, caracterizada por crisis recurrentes debido a una descarga excesiva
de neuronas cerebrales, con pérdida o no de la conciencia, asociadas a diversas manifestaciones
clínicas y para clínicas, que puede ocurrir a cualquier edad”.
El síndrome epiléptico se caracteriza por un conjunto de signos y síntomas que incluyen el tipo de
crisis, localización anatómica, factores desencadenantes, edad de comienzo del síndrome
epiléptico, gravedad, cronicidad, carácter diurno o nocturno y en algunos casos su pronóstico. Es
importante conocer el tipo de epilepsia que presentan los pacientes ya que esto definirá el tipo de
fármaco antiepiléptico a utilizar (Armijo y Herranz, 2014). En cambio, el término convulsión está
referido como un trastorno transitorio del comportamiento que se debe a la activación desordenada,
sincrónica y rítmica de poblaciones enteras de neuronas cerebrales, dichas convulsiones se
originan en la corteza cerebral y en otras estructuras del sistema nervioso central como lo es el

19
tálamo, tallo encefálico o cerebelo (McNamara, 2012). Otro termino importante a conocer es el de
epileptogénesis, el cual es el proceso dinámico en el que el cerebro se convierte en epiléptico y
comienza a generar crisis epilépticas espontáneas recurrentes (López et al., 2009).
Epidemiología
La OMS (2017) destaca los siguientes datos y cifras con respecto a la prevalencia de la epilepsia:
• La epilepsia es un trastorno crónico que afecta a personas de todas las edades.
• En todo el mundo, unos 50 millones de personas padecen epilepsia, lo que la convierte en uno
de los trastornos neurológicos más comunes.
• Cerca del 80 % de los pacientes viven en países de ingresos bajos y medianos.
• Las personas con epilepsia responden al tratamiento en aproximadamente un 70 % de los casos.
Corroborando estos datos, en el documento técnico basado en las presentaciones del taller
internacional efectuado en Santiago de Chile de la ILAE (Kestel et al., 2013) menciona que:
• A nivel mundial, cerca de 2.4 millones de personas son diagnosticadas con epilepsia cada año.
• La proporción estimada de la población con epilepsia activa (convulsiones continuas o
necesidad de tratamiento) está entre 4 y 10 por cada 1000 personas.
Con respecto al territorio mexicano, el Instituto Nacional de Neurología y Neurocirugía reportó
una prevalencia de epilepsia de 18 casos por cada 1000 habitantes; con respecto a lo publicado por
la Secretaria de Salud que calculó 12 casos por cada 1000 habitantes durante el año 2001, además,
este trastorno presenta una mortalidad del 4% en México (Epilepsia en México, 2017).
Etiología
Generalmente el origen que produce una crisis epiléptica no tiene en sí una etiología especifica si
no que el inicio de dicha crisis está más allá de lesiones o anormalidades congénitas o genéticas
cerebrales que generen explicación de la crisis convulsiva. Palacios et al. (2017) establece que
estudios de cohorte en centros de referencia han sugerido que las epilepsias parciales al menos el
25% son estructurales, el 18% de etiología indeterminada y el 0.1% de origen genético. También
se tiene la propuesta de que las crisis convulsivas van dadas de acuerdo con la edad del paciente,
dicha propuesta viene dada por Waaler et al. (2000), la epilepsia genética es la que se presenta en
un mayor porcentaje en todas las edades de acuerdo con la siguiente tabla:
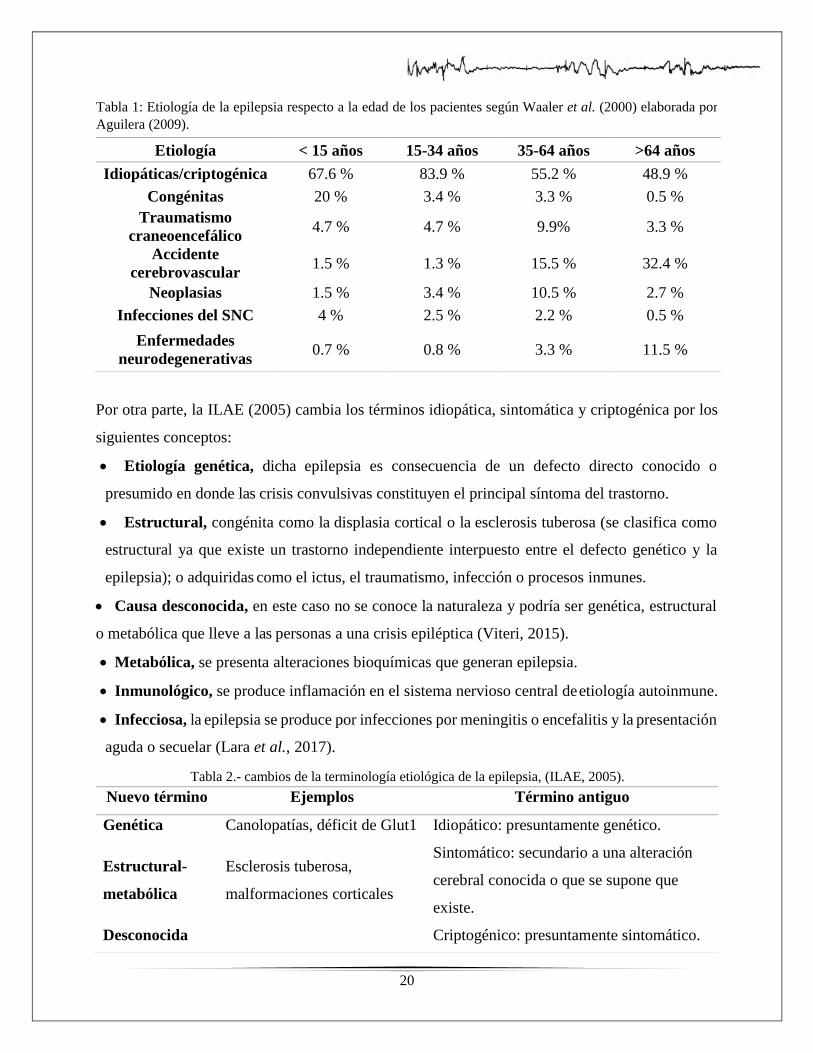
20
Por otra parte, la ILAE (2005) cambia los términos idiopática, sintomática y criptogénica por los
siguientes conceptos:
• Etiología genética, dicha epilepsia es consecuencia de un defecto directo conocido o
presumido en donde las crisis convulsivas constituyen el principal síntoma del trastorno.
• Estructural, congénita como la displasia cortical o la esclerosis tuberosa (se clasifica como
estructural ya que existe un trastorno independiente interpuesto entre el defecto genético y la
epilepsia); o adquiridas como el ictus, el traumatismo, infección o procesos inmunes.
• Causa desconocida, en este caso no se conoce la naturaleza y podría ser genética, estructural
o metabólica que lleve a las personas a una crisis epiléptica (Viteri, 2015).
• Metabólica, se presenta alteraciones bioquímicas que generan epilepsia.
• Inmunológico, se produce inflamación en el sistema nervioso central de etiología autoinmune.
• Infecciosa, la epilepsia se produce por infecciones por meningitis o encefalitis y la presentación
aguda o secuelar (Lara et al., 2017).
Etiología < 15 años 15-34 años 35-64 años >64 años
Idiopáticas/criptogénica 67.6 % 83.9 % 55.2 % 48.9 %
Congénitas 20 % 3.4 % 3.3 % 0.5 %
Traumatismo
craneoencefálico 4.7 % 4.7 % 9.9% 3.3 %
Accidente
cerebrovascular 1.5 % 1.3 % 15.5 % 32.4 %
Neoplasias 1.5 % 3.4 % 10.5 % 2.7 %
Infecciones del SNC 4 % 2.5 % 2.2 % 0.5 %
Enfermedades
neurodegenerativas 0.7 % 0.8 % 3.3 % 11.5 %
Nuevo término Ejemplos Término antiguo
Genética Canolopatías, déficit de Glut1 Idiopático: presuntamente genético.
Estructural-
metabólica
Esclerosis tuberosa,
malformaciones corticales
Sintomático: secundario a una alteración
cerebral conocida o que se supone que
existe.
Desconocida Criptogénico: presuntamente sintomático.
Tabla 1: Etiología de la epilepsia respecto a la edad de los pacientes según Waaler et al. (2000) elaborada por
Aguilera (2009).
Tabla 2.- cambios de la terminología etiológica de la epilepsia, (ILAE, 2005).

21
Se consideran 2 tipos de epilepsias, epilepsias de etiología sintomática que se produce al detectarse
una lesión cerebral (estructuras o metabólica) acompañándose de otras manifestaciones
neurológicas además de la crisis convulsiva (Ochoa et al., 2015). Así como crisis precipitadas por
situaciones especiales como la lectura, el pensamiento, la memoria, los estímulos táctiles y
sonoros, etc (Targas et al., 2014).
Clasificación de la epilepsia
En los últimos años la ILAE ha facilitado la clasificación de las epilepsias, eliminando algunos
términos para facilitar y comprender mejor los tipos de epilepsia que se pueden producir. La
clasificación más reciente considera tanto los síntomas que se producen durante la crisis, así como
la localización de la descarga eléctrica que se registra en el electroencefalograma, se considera que
de acuerdo con el lugar en donde se origina la descarga eléctrica será la manifestación clínica,
como ejemplo la zona del hemisferio cerebral que rige los movimientos, la crisis epiléptica se
presenciará por cambios motores en el lado opuesto del cuerpo (Acevedo et al., 2007).
De acuerdo con la ILAE (2005), las crisis epilépticas se clasifican en crisis de comienzo
generalizado, focal o desconocido:
Crisis Generalizadas
En dichas crisis, las manifestaciones se caracterizan por implicar a los dos hemisferios cerebrales
desde el inicio de la crisis, las descargas electroencefalográficas son bilaterales (Mulantinho et al.,
2011). Las redes bilaterales pueden incluir o no estructuras corticales y subcorticales, pero no
incluyen necesariamente la corteza (ILAE, 2005) así mismo cabe mencionar que las crisis
generalizadas, puede provenir de una lesión focal.
Crisis focales
Este tipo de crisis se desarrolla únicamente en redes neuronales limitadas a un hemisferio, así como
estructuras subcorticales o del neocórtex (Viteri, 2015). Además, las crisis focales pueden
evolucionar a una crisis tónico-clónica generalizada (llamada también como crisis focal con
generalización secundaria) (Mulantinho et al., 2011).
Crisis desconocidas
En este tipo se clasifican de origen desconocido ya que no se sabe si el comienzo es focal,
generalizado o ambas (Viteri, 2015).

22
Fig
ura
15.-
Cla
sifi
caci
ón d
e cr
isis
gen
eral
izad
as,
foca
les
y d
esco
noci
das
de
acuer
do c
on l
a IL
AE
(2005).
Se
clas
ific
an l
os
tip
os
de
cris
is d
e ac
uer
do
co
n
sus
man
ifes
taci
ones
que
se p
rese
nta
n e
n l
os
indiv
iduos
y s
i se
tra
ta d
e una
cris
is f
oca
l o g
ener
aliz
ada.
Las
cri
sis
des
con
oci
das
son
deb
ido
a i
nad
ecu
ada
info
rmac
ión
o i
mposi
bil
idad
de
ubic
arla
s en
las
otr
as c
ateg
orí
as d
e ti
pos
de
cris
is.

23
Fisiopatología de la epilepsia
Las regiones del cerebro que se suelen estudiar debido a que se consideran focos epilépticos son:
la neurocorteza, el hipocampo, la amígdala, así como los bulbos olfatorios y el colículo inferior de
la vía auditiva, estas regiones son áreas de relevo y/o de procesamiento de información de otros
núcleos del cerebro las cuales se encuentran interconectadas y con una citoarquitectura compleja
(Cabo et al, 2006).
La fisiopatología de las crisis epilépticas se ha clasificado en tres factores (Medinilla et al., 2005):
1.- Modificación de la permeabilidad de la membrana debida a un fallo de la ATPasa-Na+/K+ que
podría disminuir la conductancia de iones K+ y aumentar la conductancia de iones Ca2+ y Na+.
2.- Alteración de los mecanismos inhibitorios y excitatorios: se ha demostrado que sustancias
bloqueadoras gabaérgicas producen convulsiones, considerando así que el GABA contribuye en
los mecanismos implicados con el tratamiento de la epilepsia (Germán, 2010). Otro mecanismo es
sobre la alteración de Ca2+/calmodulina dependiente de la proteína cinasa II (CaMKII) ya que su
activación incrementa la fosforilación del receptor AMPA provocando un aumento de su función
que produce la formación de potenciación a largo plazo (Torres et al., 2010).
3.- Acoplamiento sináptico excitador entre neuronas de la región epileptógena, produciendo una
sincronización no fisiológica entre neuronas y teniendo la posibilidad de propagarse en zonas
vecinas así como de generalizarse a toda la corteza cerebral, explicando la existencia de
manifestaciones localizadas, propagadas a regiones vecinas o secundariamente generalizadas
(Medinilla et al., 2005).
Tratamiento de la epilepsia
En la actualidad existe un gran número de fármacos antiepilépticos (FAE) (tabla 3) disponibles en
el mercado, sin embargo no todos son satisfactorios puesto que presentan desventajas en su eficacia
clínica, tolerancia, en sus propiedades farmacocinéticas y en su toxicidad (López et al., 2005).
De acuerdo con Armijo y Herranz (2014) los mecanismos por los que actúan los fármacos
antiepilépticos son los siguientes (figura 16):
• Modulación de canales iónicos, inhibición de los canales de Na+ dependientes de voltaje y de
canales de Ca2+ tipo T y canales P/Q (Medinilla et al., 2005; Armijo y Herranz, 2014).
• Sistema GABAérgico, los fármacos que actúan sobre este sistema pueden actuar en diferentes
fases del ciclo GABA, se pueden unir selectivamente al receptor GABAA inhibiendo el flujo de

24
iones Cl-2; inhibir a la enzima GABA transaminasa, bloqueo del transportador GAT-1 o evitar la
recaptación de GABA en la hendidura sináptica (Sankaraneni y Lachhwani, 2015).
• Sistema glutamatérgico, al ser el principal sistema excitatorio es blanco de diferentes fármacos
antiepilépticos, ya que se unen a receptores AMPA y NMDA (Armijo y Herranz, 2014).
• Nuevos mecanismos, los fármacos de tercera generación presentan la novedad de actuar sobre
proteínas SV2A en vesículas presinápticas, en las que se han propuestos distintos mecanismos que
incluyen atrapar las moléculas de neurotransmisores, modificar la exocitosis de vesículas
sinápticas y regular la forma y el tráfico de vesículas (Sankaraneni y Lachhwani, 2015).
A pesar del esfuerzo en la investigación de nuevos FAE en la cual se presenta un mecanismo de
acción conocido permitiendo una estrategia terapéutica más apropiada, propiedades
farmacocinéticas más atractivas como una fácil absorción, eliminación sin metabolizarse
presentando una interacción mínima, ninguno cumple todas las características del antiepiléptico
ideal aunado a esto un elevado costo (Medinilla et al., 2005).
Figura 16.- Mecanismos de acción de los fármacos antiepilépticos. Se indica la acción inhibidora (-) o
estimuladora (+) de los fármacos antiepilépticos sobre receptores GABA, NMDA, AMPA, canales de Na+,
K+ y Ca2+ dependientes de voltaje así como actividad sobre la proteína SV2A (Armijo y Herranz 2014).
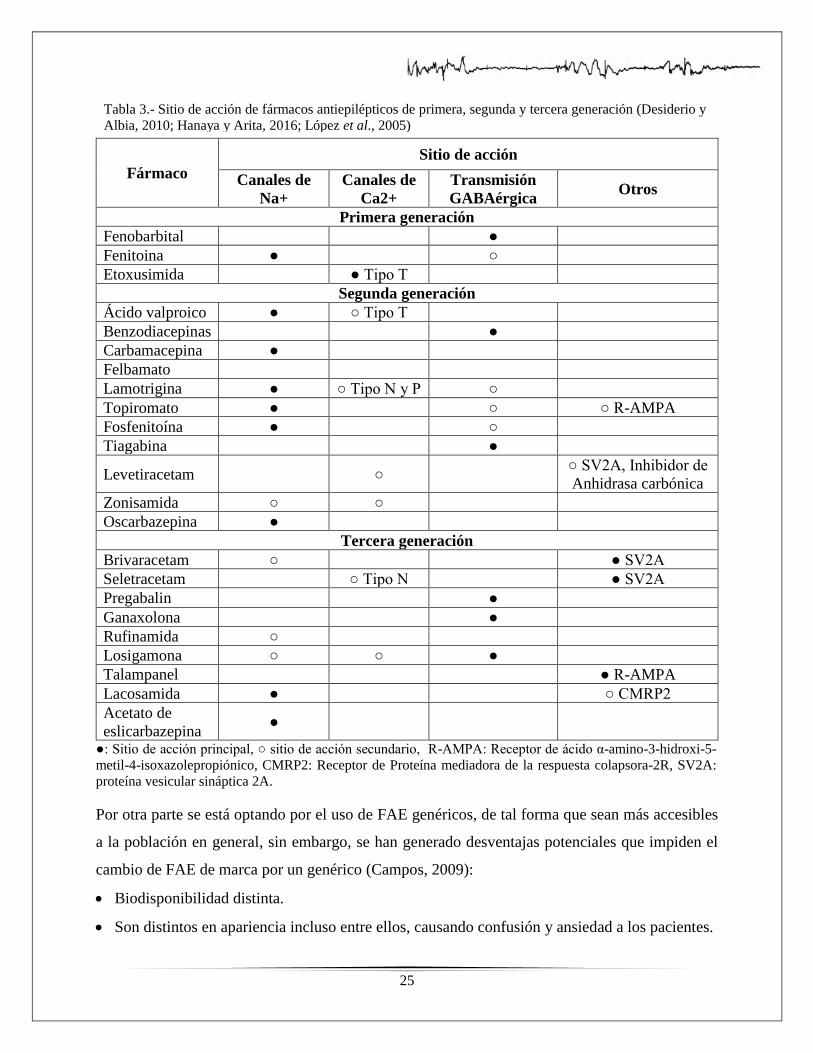
25
Fármaco
Sitio de acción
Canales de
Na+
Canales de
Ca2+
Transmisión
GABAérgica Otros
Primera generación
Fenobarbital ●
Fenitoina ● ○
Etoxusimida ● Tipo T
Segunda generación
Ácido valproico ● ○ Tipo T
Benzodiacepinas ●
Carbamacepina ●
Felbamato
Lamotrigina ● ○ Tipo N y P ○
Topiromato ● ○ ○ R-AMPA
Fosfenitoína ● ○
Tiagabina ●
Levetiracetam ○ ○ SV2A, Inhibidor de
Anhidrasa carbónica
Zonisamida ○ ○
Oscarbazepina ●
Tercera generación
Brivaracetam ○ ● SV2A
Seletracetam ○ Tipo N ● SV2A
Pregabalin ●
Ganaxolona ●
Rufinamida ○
Losigamona ○ ○ ●
Talampanel ● R-AMPA
Lacosamida ● ○ CMRP2
Acetato de
eslicarbazepina ●
●: Sitio de acción principal, ○ sitio de acción secundario, R-AMPA: Receptor de ácido α-amino-3-hidroxi-5-
metil-4-isoxazolepropiónico, CMRP2: Receptor de Proteína mediadora de la respuesta colapsora-2R, SV2A:
proteína vesicular sináptica 2A.
Por otra parte se está optando por el uso de FAE genéricos, de tal forma que sean más accesibles
a la población en general, sin embargo, se han generado desventajas potenciales que impiden el
cambio de FAE de marca por un genérico (Campos, 2009):
• Biodisponibilidad distinta.
• Son distintos en apariencia incluso entre ellos, causando confusión y ansiedad a los pacientes.
Tabla 3.- Sitio de acción de fármacos antiepilépticos de primera, segunda y tercera generación (Desiderio y
Albia, 2010; Hanaya y Arita, 2016; López et al., 2005)

26
• Los pacientes pueden ser no inertes.
• El escalado de introducción y politerapias pueden alterarse en algunos pacientes.
• El estrecho índice terapéutico, la baja solubilidad y parámetros farmacocinéticos de algunos
FAE genéricos impiden resultados de eficacia y tolerancia sean similares a los FAE de marca.
• Reaparición de crisis en pacientes con EE controlado.
• La bioequivalencia no asegura una terapia equilibrada, puesto que, la variabilidad permitida de
genéricos en el que se evalúa una baja cantidad de voluntarios sanos y la variabilidad individual
de la respuesta de los FAE en el paciente epiléptico no permite predecir para un caso determinado.
Uso de plantas medicinales para el tratamiento de la epilepsia
A partir del siglo XX las personas epilépticas tienen la oportunidad de emplear un gran número de
fármacos para tratar este trastorno, sin embargo, cuando estos fármacos no generan el efecto
deseado o produce efectos adversos se implementa el uso de otras terapias, como el uso de plantas
medicinales teniendo además la ventaja de ser menos costoso (Sucher y Carles, 2015).
Las plantas medicinales con actividad antiepiléptica se emplean en distintas partes del mundo, en
Iran se emplea un gran número de plantas como Paeonia officinales L., Bryonia alba L., Lavandula
stoechas L. así como distintas especies de la familia Ferula (Sahranavard et al., 2013); por otro
lado en china se emplean otras plantas tradicionales como Acorus tatarinowii Schott, Buplerum
chinese, Ligusticum chuanxiong Hort, Paeonia suffruticosa y Salvia miltiorrhiza (Ling et al.,
2014); en Europa se han identificado 221 plantas de 53 familias con actividad antiepiléptica
(Adams et al., 2012). En México también se ha reportado una gran variedad de plantas utilizadas
en la medicina tradicional como la Indigofera suffruticosa Millar (añil), Tillandsia usneoides L.
(Heno), Citrus aurantium L. (Naranja agria), Amanita muscaria (Mosquero), etc (Medicinal
tradicional mexicana-UNAM, 2018).
Buddleja cordata kunth
Buddleja cordata kunth es un arbusto o árbol que pertenece a la familia Loganiaceae llamada
tradicionalmente en México como tepozán, es originario de México y Guatemala y habita en
climas semisecos y templados entre los 2200 y 3000 msnm, tiene la ventaja de colonizar con
facilidad sitios abiertos gracias a que sus semillas son fácilmente dispersadas por el viento
(Mendoza, 2003).

27
Esta planta tradicionalmente se utiliza para lesiones de la piel como heridas, llagas, ulceras o para
madurar abscesos y granos, también es utilizada para tratar diarrea, afecciones renales,
propiedades analgésicas, diuréticas y eupépticas además se emplea para tratas reumas, tos,
calambres tumores y úlceras (Medicinal tradicional mexicana-UNAM, 2018; Rangel et al., 2003).
De manera experimental se ha aislado componentes químicos y se les ha demostrado efectos
terapéuticos como lo son sesquiterpenos con actividad pesticida, el iridoide aucubina con actividad
antiséptica (Houghton, 1984), el flavonoide linarina con propiedades diuréticas (Martínez et al.,
1995), el fenilpropanoide verbasido con efecto fotoprotector (González et al., 2016; Avila et al.,
2014) y fenoles con actividad antioxidante (Estrada et al., 2016); sin embargo también se le ha
demostrado otras propiedades terapéuticas como efectos bactericida y bacteriostático (Avila et al.,
1999) antifúngicos (Houghton et al., 2003), antiparasitario (Díaz et al., 2000) y amebicida
(Rodríguez et al., 1999), efecto analgésico y antipirético (Martínez et al., 1996), efecto
antiinflamatorio (Martínez et al., 1998) y recientemente se le ha asociado actividad
neuroprotectora (Pérez et al., 2015).
Modelos para el desarrollo de crisis epiléptica
Para llevar a cabo el estudio de la epilepsia y la propuesta de nuevos tratamientos es necesario el
uso de modelos experimentales que generen estados epilépticos en animales de laboratorio para
promover las herramientas necesarias para el estudio celular y mecanismos moleculares de esta
patología (Villalpando y Medina, 2015). Los modelos más usados para estudiar la epilepsia de
lóbulo temporal (ELT) son el método de Kindling y el método de estado epiléptico (EE).
Método kindling
Este método fue descubierto por Goddard (1969), demostró que las convulsiones conductuales se
podían generar con estimulaciones subcortical de baja intensidad en la amígala, pero esto solo
después de la aplicación continua por días o semanas. Este modelo en la actualidad también se
puede aplicar con estímulos subumbrales de manera eléctrica o química en diversas áreas del
sistema límbico (amígdala, corteza e hipocampo) provocando una excitabilidad de las neuronas al
paso del tiempo promoviendo la patología de las neuronas y generar crisis epilépticas. Para la
aplicación del kindling químico se utilizan diversas sustancias con actividad convulsivante con
dosis subumbrales, para la generación de una crisis epiléptica con este método depende del tipo de
fármaco convulsivante utilizado, el tiempo determinado entre cada administración, duración del

28
tratamiento, vía de administración y la dosis del fármaco a emplear (Delgado et al., 2014).
Al aplicar este modelo de crisis epiléptica se presentan los siguientes cambios:
• Liberación de glutamato que activa receptores NMDA.
• Incremento de Ca2+ que activa la proteincinasa II dependiente de Ca2+/calmodulina.
• Apoptosis y muerte neuronal de las áreas CA1 y CA3 del hipocampo.
• Proliferación de neurogénesis en el giro dentado, las neuronas desarrolladas en este sitio
presentan propiedades hiperexcitables (García et al., 2009).
Método de Estado Epiléptico
Se utilizan diversas sustancias con actividad convulsiva como el PTZ, ácido kaínico y pilocarpina,
este método consta de dos fases, la primera es un EE caracterizado por crisis tónico-clónicas de
origen límbico que no ceden (fase aguda), seguido de un periodo libre de crisis (etapa latente) para
finalizar con crisis recurrentes espontáneas (fase crónica) semanas más tardes (Pereno, 2010).
Crisis inducidas por ácido kaínico
El ácido kaínico (AK) (figura 17) es un neurotóxico análogo del glutamato capaz de activar dichos
receptores, su mecanismo de acción consiste en que al unirse a receptores de glutamato aumenta
la entrada de calcio intracelular y activa enzimas dependientes de este ion incrementando así las
especies reactivas de oxígeno, disfunción mitocondrial, condensación y fragmentación del núcleo
Agente Zona Animal por estudiar Intervalo (horas)
Aminoácidos:
Glutamato/aspartato, NMDA Amígdala Rata 48
Opiáceos:
β-endorfina, met-encefalina
Amígdala,
hipocampo
y ventrículo
Rata 48
Agonistas colinérgicos: Carbacol,
acetil- β-metilcolina, muscarina,
pilocarpina, fisostigmina
Amígdala Rata 24-48
Nucleótidos cíclicos:
AMPc Amígdala Rata 48
Antagonistas GABAérgicos:
Pentilentetrazol, picrotoxina,
bicuculina, β-carbolina
Amígdala Rata 48-96
Tabla 4.- Agentes para la generación de kindling químico. Modificado de Delgado et al., 2014.

29
y la apoptosis celular (Amador et al., sf). De acuerdo con Rao et al. (2006) citado por Concepción
(2009) se observa perdida neuronal en las áreas CA1 y CA3 del hipocampo y efectos neurogénicos.
Crisis inducida por pentilentetrazol (PTZ)
Everett y Richards (1944) presentaron resultados positivos sobre el PTZ al demostrar que este
agente químico es eficaz para la generación de convulsiones dando así la importancia de
implementarlo como un modelo para inducir y estudiar la epilepsia.
El PTZ (figura 18) es ampliamente utilizado para generar EE, el efecto que produce está de acuerdo
con la dosis que se administra, dosis bajas induce ausencias epilépticas, dosis moderas conlleva a
crisis de tipo clónicas y a dosis altas genera crisis tónico-clónica y eventualmente un estado
epiléptico generalizado (Pereno, 2010).
Se trata de un antagonista no competitivo de receptores GABAA (subunidad β) a los sitios de unión
de picrotoxica bloqueando el canal de Cl-, sirve para evaluar convulsiones tónico-clónicas (Ramos
et al., 2012; Huang et al., 2001). Además de que dicha sustancia genera desordenes cognitivos,
cambios en el comportamiento emocional y perdida de neuronas en la zona CA3 del hipocampo
(Pavlova et al., 2006; Szyndler et al., 2008).
El uso de PTZ presenta ventajas en el estudio de la epilepsia ya que además de ayudar en detectar
fármacos con mayor con eficacia antiepiléptica y en el estudio de genes relacionados con la
epilepsia también nos proporciona información sobre el daño neuronal después de crisis epilépticas
puesto que los daños histológicos que se observan en los pacientes que sufren epilepsia también
se aprecian en los cerebros de animales tratados con este sustancia entre las que se encuentra la
Figura 18.- Estructura química del pentilentetrazol.
Figura 17.- Estructura química del ácido kaínico.

30
migración de neuronas de los gránulos, astrogliosis, muerte de neurona en hipocampo y esclerosis
de hipocampo (Shimada y Yamagata, 2018), presentando además la ventaja de ser un método
económico y reproducible.
Crisis inducida por pilocarpina-cloruro de litio
El uso de la pilocarpina como modelo para estudiar la epilepsia es utilizado para desarrollar EE y
ELT (Römermann et al., 2015). Este modelo fue descrito por primera vez por Turski et al. (1989)
y es ampliamente utilizado por que tiene la capacidad de producir un EE altamente isomórfico con
la epilepsia humana. Se trata de un agente químico que se une a receptores muscarínicos del tipo
1 (M1) del hipocampo (Hamilton et al., 1997) que al administrar genera un EE que es procedida
por inmovilidad, cabeceo, temblores, masticación y ataques tónico-clónico de tipo 4 y 5 en la
escala de Racine (Römermann et al., 2015).
Adicionalmente el modelo puede ser utilizado es conjunto con litio, el cual se administra 24 horas
antes del EE producido por pilocarpina, al realizar este proceso la dosis de pilocarpina disminuye
(30 mg/kg), sin embargo los efectos metabólicos, electrográficos, conductivos y neuropatológicos
son similares, la única diferencia es que el uso previo de litio las ratas tienden a ser más sensibles
a la pilocarpina (Clifford et al., 1987; Honchar et al., 1983).
Maximal Electroshock Seizure (MES)
Este modelo fue desarrollado por Toman et al. (1946), es un modelo de convulsiones tónico-
clónicas generalizadas y de la estimulación de estructuras límbicas como modelos de convulsiones
parciales complejas (Mares y Kubová, 2006). El modelo MES se considera como el mejor modelo
para el estudio de fármacos efectivos con convulsiones tónico-clónico (gran mal), además tiene la
ventaja de que fármacos como fenitoína, carbamazepina, fenobarbital y primidona son muy
efectivos en este modelo pero tiene la desventaja de que al producir estímulos muy fuertes de
convulsión puede generar falsos positivos en aquellas moléculas que tienen etapas tempranas de
estudios (Löscher y Schmidt, 1988).

31
Justificación
En la actualidad la epilepsia presenta una prevalencia de 18 casos por cada 1000 habitantes
presentando así una mortalidad del 4% en México, de tal manera que la implementación de nuevas
terapias como el uso de plantas medicinales contribuirán a mejorar la calidad de vida de los
pacientes epilépticos.
Hipótesis
Si el extracto etanólico de tepozán (Buddleja cordata kunth) ejerce actividad neuroprotectora
disminuirá la duración de las convulsiones provocadas por la administración de pentilentetrazol,
así como la muerte neuronal en amígdala, corteza motora, estria terminal e hipocampo de ratas.
Objetivo General
• Determinar el efecto neuroprotector del extracto etanólico de Buddleja cordata kunth en ratas
administradas con pentilentetrazol.
Objetivos particulares
Del extracto etanólico de Buddleja cordata kunth:
• Determinar los metabolitos secundarios.
• Cuantificar la dosis letal 50 (DL50.)
En ratas:
• Determinar el efecto anticonvulsivo producido por el extracto etanólico de Buddleja cordata
kunth.
• Cuantificar el nivel de bilirrubina y urea en sangre.
• Cuantificar el número y morfología de neuronas presentes en amígdala, corteza motora, estría
terminal e hipocampo.

32
Metodología
Material vegetal
Una parte aérea de la planta fue preparada para su identificada taxonómica, la cual fue identificada
como Buddleja cordata kunth perteneciente a la familia Scrophulariaceaea por la Dr. Raquel
Galván Villanueva del departamento de botánica de la Escuela Nacional de Ciencias Biológicas
del Instituto Politécnico Nacional.
Preparación del extracto etanólico de Buddleja cordata kunth
Se pesaron 100 g de hojas secas trituradas de la planta de Buddleja cordata kunth y se transfirió a
un frasco de vidrio color ámbar, al cual se le agregó 1 L de etanol; dicho macerado se dejó reposar
por un periodo de 15 días a temperatura ambiente en oscuridad con agitación de una vez al día.
Después de los 15 días el macerado se filtró para obtener el etanol, el etanol se evaporó con ayuda
de un evaporador (rotavapor) el cual se configuró para alcanzar una presión reducida de 10 pg. con
una temperatura de 60°C a 30 rpm/hora, al término, el extracto se concentró mediante la aplicación
de nitrógeno gaseoso y se guardó a una temperatura de 4°C hasta realizar los estudios
correspondientes.
Análisis fitoquímico preliminar
Para la identificación de los metabolitos secundarios presentes en las partes aéreas de la planta se
procedió a preparar 3 diferentes extractos:
Extracto ácido
En un vaso de precipitado de 250 mL se pesó 1 g de hojas trituradas de Buddleja cordata kunth y
se agregaron 20 mL de HCl al 10%, se calentó hasta ebullición y se dejó enfriar para la
determinación de alcaloides.
Extracto acuoso
Se pesaron 3 g de hojas trituradas de Buddleja cordata kunth en un vaso de precipitado de 250 mL
y se agregó 50 mL de agua, se calentó hasta ebullición y se dejó enfriar para determinar la presencia
de azúcares reductores, taninos y glucósidos cianogénicos.

33
Extracto etanólico
En un vaso de precipitado de 250 mL se colocaron 5 g de hojas trituradas de Buddleja cordata
kunth y se agregó 50 mL de etanol, se llevó a ebullición, se filtró y dejó enfriar para determinar la
presencia de flavonoides, cumarinas, glucósidos cardiotónicos, quinonas y sesquiterpenlactonas.
Dosis letal media (DL50)
Para la determinación de la DL50 se utilizó el método modificado de Lorke (1983), para lo cual se
distribuyeron de manera aleatoria 12 ratones de la cepa NHI con un peso promedio de 300 g en 4
grupos, cada grupo constituido por 3 ratones. Los animales se alojaron en una cámara con una
temperatura de 21 ± 1 °C y humedad relativa de 40–60 %, con ciclos de luz/ oscuridad de 12/12
horas. Las ratas fueron administradas con el extracto a diferentes dosis por vía oral (v.o.) de
acuerdo con la siguiente tabla:
Grupo Tratamiento
1 Control (S.S 0.9 %)
2 Extracto etanólico 2 mg/kg
3 Extracto etanólico 200 mg/kg
4 Extracto etanólico 2000 mg/kg
Los grupos de experimentación se mantuvieron en observación durante un periodo de 10 días en
los cuales se examinaron que no presentaran anormalidades como alteraciones físicas y motoras,
diarrea, piloerección, irritación ocular, temblores, miosis, midriasis, convulsiones o muerte.
Efecto anticonvulsivo
Para realizar la determinación del efecto anticonvulsivo del extracto etanólico de Buddleja cordata
kunth se utilizaron 48 ratas Fisher 344 con un peso promedio de 150-200 g que se distribuyeron
de manera aleatoria en 6 grupos, cada grupo constituido por 8 ratas, los grupos de experimentación
fueron tratados de acuerdo con la siguiente tabla:
Tabla 5.- Grupos formados para realizar las pruebas de DL50. Se preparó 1 grupo control y 3 grupos
administrando dosis crecientes del extracto etanólico de Buddleja cordata kunth.

34
El extracto etanólico de Buddleja cordata kunth de los grupos 3, 4, 5 y 6 fue administrado por vía
intragástrica (i.g.) 30 minutos previos de la administración con pentilentetrazol. Al cabo de 30
minutos se les administro por vía intraperitoneal (i.p.) a los grupos 2, 4, 5, y 6 pentilentetrazol a
una dosis de 50 mg/kg, posteriormente se colocaron en jaulas individuales y se procedió a observar
las convulsiones que durante 10 minutos. La intensidad de las convulsiones se calificó de acuerdo
con la escala propuesta por Racine (1972) de la siguiente forma:
0: sin respuesta.
1: espasmos faciales y del oído.
2: sacudidas mioclónicas sin alzarse.
3: tirones mioclónicos.
4: vueltas en posición lateral y convulsiones tónico-clónicas no generalizadas.
5: vueltas en posición hacia atrás y convulsiones tónico-clónicas generalizadas.
A estas ratas se les midió el tiempo de latencia (tiempo en que tarda en surgir efecto el PTZ) así
como la duración de las convulsiones de fase 5, 24 horas después se administró nuevamente el
extracto a los grupos 3, 4, 5 y 6 a su dosis correspondiente, 48 horas posteriores se administró de
nuevo el extracto para finalmente sacrificar a las ratas por decapitación. Una vez sacrificados los
animales de experimentación se obtuvieron muestras de sangre y se diseco el cerebro para realizar
el análisis histológico.
Grupo Tratamiento
1 Control (S.S 0.9%)
2 Pentilentetrazol (50 mg/kg)
3 Extracto etanólico (300 mg/kg)
4 Extracto etanólico (75 mg/kg) + PTZ (50 mg/kg)
5 Extracto etanólico (150 mg/kg) + PTZ (50 mg/kg)
6 Extracto etanólico (300 mg/kg) + PTZ (50 mg/kg)
Tabla 6.- Grupos formados para realizar las pruebas del efecto anticonvulsivo a dosis crecientes del extracto
etanólico de Buddleja cordata kunth administrados 30 min previos al PTZ.

35
Marcadores de función hepática y renal
Con las muestras de sangre recolectadas de los diferentes grupos de experimentación se procedió
a procesarlas para la cuantificación de bilirrubina y BUN de acuerdo con los métodos establecidos
en los manuales de los kits RANDOX de los respectivos metabolitos.
Análisis histológico
Se colocaron los cerebros en formalina al 10% durante 48 horas y se lavaron con agua durante 24
horas, posteriormente los cerebros se colocaron en alcohol al 60%, 70%, 80%, 96%, 100% y
alcohol 100%-xilol cada uno por 1 hora; xilol 30 minutos, xilol-parafina 15 minutos y parafina I,
II y III cada uno por una hora. Se incluyeron los cerebros en parafina III y se realizaron los cortes
histológicos con un grosor de 6 μm desde la posición de -0.80 a 1.70 mm de Bregma.
Para la tinción de los cortes histológicos, primero se hidrataron colocándolos en cubas que
contenían xilol (durante 5 min), xilol-alcohol 100%, alcohol 100%, 96%, 80%, 70% y 60% (1
minuto cada uno), posteriormente las muestras se colocaron en azul de toluidina por 10 min y
nuevamente se deshidrataron pasando los cortes histológicos en soluciones de alcohol al 60%,
70%, 80%, 96%, 100%, alcohol 100%-xilol (1 minuto cada uno) y xilol (1 hora); y se fijaron con
resina.
Finalmente se procedió a contar la cantidad de neuronas normales y anormales presentes en el
núcleo basolateral de la amígdala, zona M1 de la corteza motora, estría terminal y zona CA3 del
hipocampo.
Análisis estadístico
Todos resultados obtenidos en los diferentes estudios se expresaron como la media ± error
estándar, en el caso de la actividad anticonvulsiva y cuantificación de bilirrubina y BUN se empleó
una prueba ANOVA unifactorial, mientras que para los datos de la cuantificación neuronal
obtenidos de los cortes histológicos se empleó una prueba ANOVA bifactorial (neuronas normales
y anormales como factores) y Tukey post hoc. En caso de que los valores presentaran *P < 0.05
se consideraron estadísticamente diferentes. Dichos análisis se realizaron utilizando el programa
estadístico SIGMA STAT.

36
Resultados
Análisis fitoquímico preliminar
En la tabla 7 se reportan los resultados del análisis fitoquímico para evidenciar los tipos de
metabolitos presentes en las hojas de Buddleja cordata kunth en cada tipo de extracto.
Extracto Metabolito
secundario Evidencia del metabolito Resultado
Extracto
ácido Alcaloides
Precipitado color
verdoso
Extracto
acuoso
Azúcares
reductores
Precipitado naranja
Extracto
etanólico
Antraquinonas
Coloración roja
Derivados de
catecol
Coloración verde
Flavonoides
Coloración amarilla-
marron
Glucósidos
cardiotónicos
Coloración roja poco
estable (izquierda)
Coloación que varia
del naranja a rojo
oscuro (derecha)
Tabla 7.- Metabolitos secundarios presentes en el extracto ácido, acuoso y etanólico de las hojas de Buddleja
cordata kunth.

37
Dosis letal media (DL50)
En la tabla 8 se reportan que al emplear diferentes dosis del extracto etanólico no se observa ningún
tipo de alteraciones físicas, motoras, diarrea, piloerección, irritación ocular, temblores, miosis,
midriasis, convulsiones o incluso la muerte en los grupos estudiados.
Mientras que en la tabla 9 se determina la dosis letal media (DL50) así como el grado de toxicidad
al que se encuentra el extracto de acuerdo con la clasificación establecida por la OMS (2009) y
Repetto (2009).
Grupo Tratamiento Alteraciones observadas
1 Control (S.S 0.9 %) Sin alteraciones
2 Extracto etanólico 2 mg/kg Sin alteraciones
3 Extracto etanólico 200 mg/kg Sin alteraciones
4 Extracto etanólico 2000 mg/kg Sin alteraciones
Dosis letal media DL50 Nivel de toxicidad
Mayor a 2000 mg/kg Ligeramente tóxico
Tabla 8.- Manifestaciones presentes en los grupos experimentados a diferentes dosis del extracto etanólico
de Buddleja cordata kunth.
Tabla 9.- Dosis letal media (DL50) y nivel de toxicidad que presenta el extracto etanólico de hojas de Buddleja cordata kunth.

38
Efecto anticonvulsivo
En la figura 19 se presentan los datos de la duración de las convulsiones de tipo 5, los grupos que
fueron tratados con el extracto etanólico a diferentes dosis presentaron una disminución de la
duración de las convulsiones, mientras que la figura 20 se demuestra que los grupos tratados con
el extracto etanólico presentaron un aumento del tiempo de latencia, protegiendo así del efecto
convulsivo generado por PTZ.
C o n tr o l P T Z 3 0 0 m g /k g 7 5 m g /k g 1 5 0 m g /k g 3 0 0 m g /k g
0 .0
0 .5
1 .0
1 .5
2 .0
Du
rac
ión
de
la
s c
on
vu
lsio
ne
s (
min
)
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
P T Z 5 0 m g /kg
S .S 0 .9 %
Tie
mp
o d
e l
ate
nc
ia (
min
)
0 .0
0 .5
1 .0
1 .5
2 .0
2 .5
3 .0
3 .5
4 .0
C o n tro l 3 0 0 m g /K g 1 5 0 m g /K g 3 0 0 m g /K gP T Z 7 5 m g /K g
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
S .S 0 .9 %
P T Z 5 0 m g /kg
Figura 19.- Efecto del extracto etanólico de Buddleja cordata kunth sobre la duración de las convulsiones
(min) en ratas Fischer 344 tratadas con PTZ. Los datos representan la media ± error estándar.
*P < 0.05 vs PTZ (50 mg/kg) ANOVA unifactorial y Dunn post hoc.
* * *
Figura 20.- Efecto del extracto etanólico de Buddleja cordata kunth sobre el tiempo de latencia de la
convulsión (min) en ratas Fischer 344 tratadas con PTZ. Los datos representan la media ± error estándar.
*P < 0.05 vs PTZ, ANOVA unifactorial y Newman-Keuls post hoc.
*
*
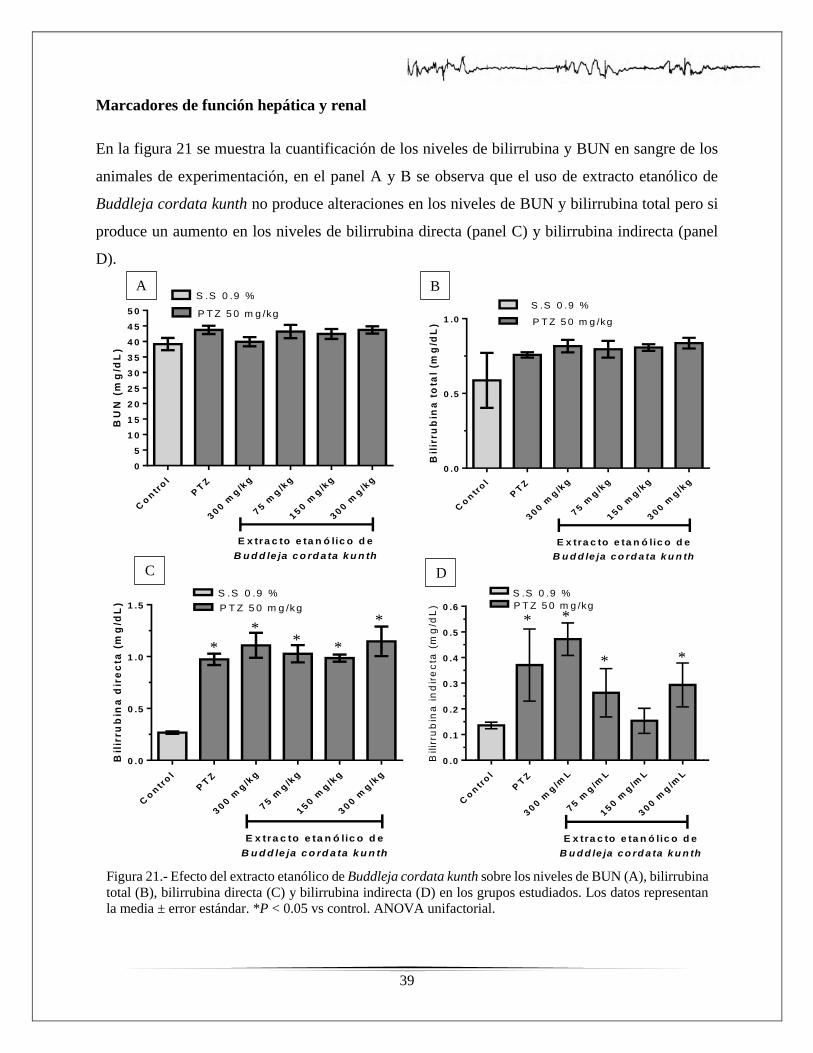
39
Marcadores de función hepática y renal
En la figura 21 se muestra la cuantificación de los niveles de bilirrubina y BUN en sangre de los
animales de experimentación, en el panel A y B se observa que el uso de extracto etanólico de
Buddleja cordata kunth no produce alteraciones en los niveles de BUN y bilirrubina total pero si
produce un aumento en los niveles de bilirrubina directa (panel C) y bilirrubina indirecta (panel
D).
Co
ntr
ol
PT
Z
300 m
g/k
g
75 m
g/k
g
150 m
g/k
g
300 m
g/k
g
0
5
1 0
1 5
2 0
2 5
3 0
3 5
4 0
4 5
5 0
BU
N (
mg
/dL
)
P T Z 5 0 m g /kg
S .S 0 .9 %
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
C o n tro l
PTZ
Co
ntr
ol
PT
Z
300 m
g/k
g
75 m
g/k
g
150 m
g/k
g
300 m
g/k
g
0 .0
0 .5
1 .0
Bil
irru
bin
a t
ota
l (m
g/d
L) P T Z 5 0 m g /kg
S .S 0 .9 %
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
C o n tro l
Co
ntr
ol
PT
Z
300 m
g/k
g
75 m
g/k
g
150 m
g/k
g
300 m
g/k
g
0 .0
0 .5
1 .0
1 .5
Bil
irru
bin
a d
ire
cta
(m
g/d
L)
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
P T Z 5 0 m g /kg
S .S 0 .9 %
Co
ntr
ol
PT
Z
300 m
g/m
L
75 m
g/m
L
150 m
g/m
L
300 m
g/m
L
0 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
Bil
irru
bin
a i
nd
ire
cta
(m
g/d
L)
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
S .S 0 .9 %
P T Z 5 0 m g /kg
Figura 21.- Efecto del extracto etanólico de Buddleja cordata kunth sobre los niveles de BUN (A), bilirrubina
total (B), bilirrubina directa (C) y bilirrubina indirecta (D) en los grupos estudiados. Los datos representan
la media ± error estándar. *P < 0.05 vs control. ANOVA unifactorial.
*
*
*
* *
A B
C D
* *
* *

40
Determinación de la densidad neuronal
La figura 22 presenta el número de neuronas normales y anormales de los grupos de
experimentación en amígdala (panel A), corteza motora (panel B) estría terminal (panel C) e
hipocampo (panel D), en los cuales los grupos que fueron administrados con el extracto
presentaron una disminución del número de neuronas anormales con respecto al grupo
administrado con PTZ.
Co
ntr
ol
PT
Z (
5 0 mg
/kg
)
3 0 0 mg
/kg
PT
Z +
75 m
g/k
g
PT
Z +
15 0 m
g/k
g
PT
Z +
30 0 m
g/k
g
0
5
1 0
1 5
2 0
2 5
Ne
uro
na
s t
ota
les
en
10
,00
0
m2
N e u ro n a s n o rm a le sN e u ro n a s a n o rm a le s
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
Co
ntr
ol
PT
Z (
5 0 mg
/kg
)
3 0 0 mg
/kg
PT
Z +
75 m
g/k
g
PT
Z +
15 0 m
g/k
g
PT
Z +
30 0 m
g/k
g
0
5
1 0
1 5
2 0
2 5
Ne
uro
na
s t
ota
les
en
10
,00
0
m2
N e u ro n a s n o rm a le sN e u ro n a s a n o rm a le s
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
Co
ntr
ol
PT
Z (
5 0 mg
/kg
)
3 0 0 mg
/kg
PT
Z +
75 m
g/k
g
PT
Z +
15 0 m
g/k
g
PT
Z +
30 0 m
g/k
g
0
5
1 0
1 5
2 0
2 5
3 0
3 5
4 0
Ne
uro
na
s t
ota
les
en
10
,00
0
m2 N e u ro n a s n o rm a le s
N e u ro n a s a n o rm a le s
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
Co
ntr
ol
PT
Z (
5 0 mg
/kg
)
3 0 0 mg
/kg
PT
Z +
75 m
g/k
g
PT
Z +
15 0 m
g/k
g
PT
Z +
30 0 m
g/k
g
0
5
1 0
1 5
2 0
2 5
3 0
Ne
uro
na
s t
ota
les
en
10
,00
0
m2
N e u ro n a s n o rm a le s
N e u ro n a s a n o rm a le s
E x tra c to e ta n ó lic o d e
B u d d le ja c o rd a ta k u n th
Figura 22.- Efecto del extracto etanólico de Buddleja cordata kunth sobre la morfología de neuronas en el
núcleo basolateral de amígdala (A), zona M1 de corteza motora (B), estría terminal (C) y zona CA3 de
hipocampo de los grupos estudiados. Los datos representan la media ± error estándar. *P < 0.05 vs PTZ neuronas anormales. ANOVA bifactorial y Tukey post hoc.
A B
C D
* * *
*
* *
* * *
* * * * * * *

41
Se muestran las fotomicrografías de los cortes histológicos del núcleo basolateral de la amígdala de los
diferentes grupos de experimentación en la figura 23. En el panel B se observa como el PTZ ocasiona una
disminución del volumen neuronal así como una disminución de neuronas normales y un aumento de la
microglía por plano, además de alteración en la morfología de las neuronas como lo es la perdida de la
relación núcleo-citoplasma, fragmentación parcial o completa del nucléolo así como prolongaciones en
relación con los grupos tratados con el extracto etanólico que evita la disminución del número total de
neuronas y de las anormalidades morfológicas ocasionadas por el PTZ.
Figura 23.- Fotomicrografías de neuronas del núcleo basolateral de amígdala en los grupos estudiados:
control (panel A), PTZ 50 mg/kg (panel B), 300 mg/kg del extracto etanólico de Buddleja cordata kunth
(panel C), PTZ + extracto 75 mg/kg (panel D), PTZ + extracto 150 mg/kg (panel E) y PTZ + extracto 300 mg/kg (panel F). Técnica azul de toluidina, observado con objetivo 40x.
D
F
B A
C
50 μm 50 μm
50 μm 50 μm
50 μm 50 μm E

42
La figura 24 muestra las fotomicrografías de los cortes histológicos de la zona M1 de la corteza motora de
los diferentes grupos de experimentación. En el panel B se observa como el PTZ ocasiona una disminución
del tanto del número de neuronas totales y neuronas normales así como un aumento de la microglía por
plano, además se observa un aumento del número de neuronas irregulares con una disminución de la
relación núcleo-citoplasma, fragmentación parcial o completa del nucléolo así como prolongaciones en
relación con los grupos tratados con el extracto etanólico que evita la disminución del número total de
neuronas y de las anormalidades morfológicas ocasionadas por el PTZ.
Figura 24.- Fotomicrografías de neuronas de la zona M1 de corteza motora en los grupos estudiados: control
(panel A), PTZ 50 mg/kg (panel B), 300 mg/kg del extracto etanólico de Buddleja cordata kunth (panel C),
PTZ + extracto 75 mg/kg (panel D), PTZ + extracto 150 mg/kg (panel E) y PTZ + extracto 300 mg/kg
(panel F). Técnica azul de toluidina, observado con objetivo 40x.
D
E F
B A 50 μm 50 μm
50 μm 50 μm
50 μm 50 μm
C D
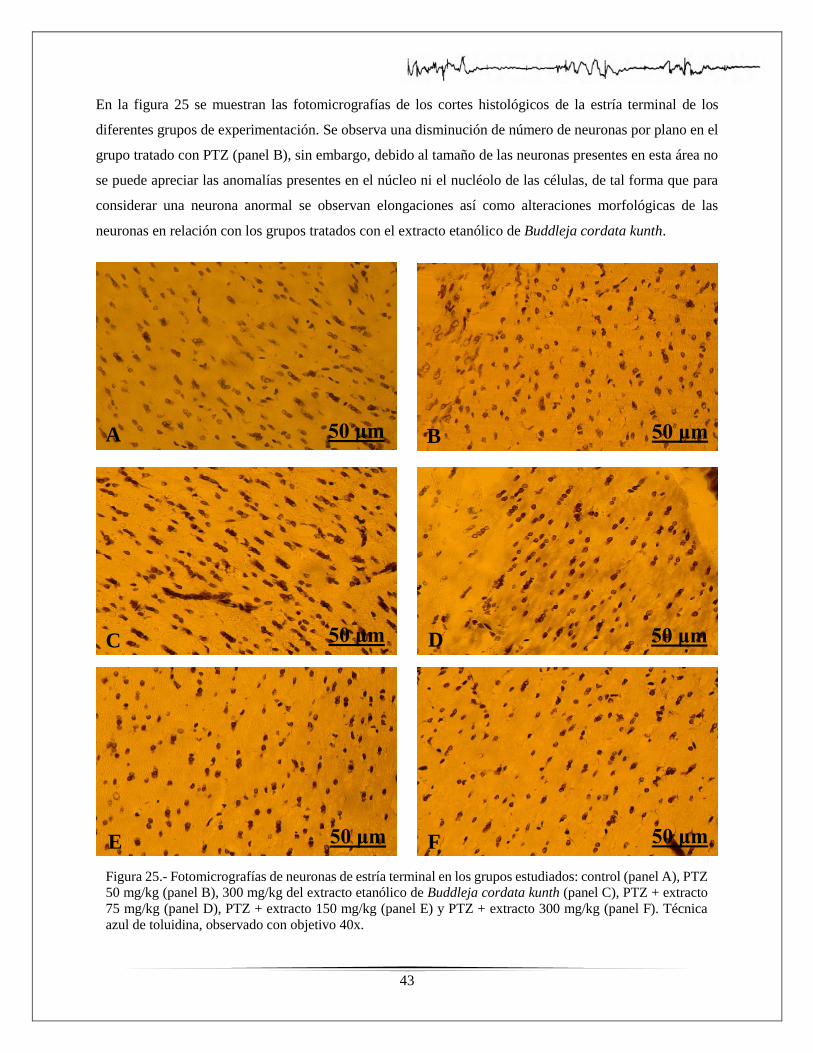
43
En la figura 25 se muestran las fotomicrografías de los cortes histológicos de la estría terminal de los
diferentes grupos de experimentación. Se observa una disminución de número de neuronas por plano en el
grupo tratado con PTZ (panel B), sin embargo, debido al tamaño de las neuronas presentes en esta área no
se puede apreciar las anomalías presentes en el núcleo ni el nucléolo de las células, de tal forma que para
considerar una neurona anormal se observan elongaciones así como alteraciones morfológicas de las
neuronas en relación con los grupos tratados con el extracto etanólico de Buddleja cordata kunth.
Figura 25.- Fotomicrografías de neuronas de estría terminal en los grupos estudiados: control (panel A), PTZ
50 mg/kg (panel B), 300 mg/kg del extracto etanólico de Buddleja cordata kunth (panel C), PTZ + extracto
75 mg/kg (panel D), PTZ + extracto 150 mg/kg (panel E) y PTZ + extracto 300 mg/kg (panel F). Técnica
azul de toluidina, observado con objetivo 40x.
C D
E
B A 50 μm 50 μm
50 μm 50 μm
50 μm 50 μm F

44
La figura 26 muestra las fotomicrografías de los cortes histológicos de la zona CA3 del hipocampo de los
diferentes grupos de experimentación. En el panel C se observa como el PTZ que al igual que las demás
zonas estudiadas ocasiona alteración en la citoarquitectura de la zona, una disminución del número de
neuronas normales y aumento de la microglía por plano, disminución de la relación núcleo-citoplasma,
fragmentación parcial o completa del nucléolo en relación con los grupos tratados con el extracto etanólico
que evita la disminución de las anormalidades morfológicas ocasionadas por el PTZ.
A
Figura 26.- Fotomicrografías de neuronas de la zona CA3 de hipocampo en los grupos estudiados: control
(panel A), PTZ 50 mg/kg (panel B), 300 mg/kg del extracto etanólico de Buddleja cordata kunth (panel C),
PTZ + extracto 75 mg/kg (panel D), PTZ + extracto 150 mg/kg (panel E) y PTZ + extracto 300 mg/kg (panel F). Técnica azul de toluidina, observado con objetivo 40x.
C D
E F
50 μm
50 μm 50 μm
50 μm 50 μm
50 μm B

45
Discusión
El uso de plantas medicinales a lo largo del tiempo ha sido una práctica muy común en las
comunidades rurales de nuestro país; en el presente trabajo estudiamos el efecto neuroprotector de
la planta Buddleja cordata kunth.
Se realizo el análisis fitoquímico preliminar al extracto etanólico de Buddleja cordata kunth, en
dicho análisis se encontró la presencia de diversos metabolitos a los cuales se les puede asociar a
diversas funciones tanto en beneficio para las propias plantas como para su uso medicinal:
alcaloides con función antioxidante (Ibarra et al., 2011), actúan de diversas maneras como
protección en plantas como efecto antiparasitario, antifúngico y como insecticida (Sepúlveda et
al., 2003); azúcares reductores para la obtención de energía (Partearroyo et al., 2013);
antraquinonas con actividad antifúngica, antiviral, antiparasitaria (Saavedra et al., 2012),
anticancerígena y antioxidante (Locatelli et al., 2009); taninos derivados de catecol con actividad
antimicrobiana, antitumoral, antioxidante y antiinflamatoria (Aguirre et al., 2015); flavonoides con
propiedades antioxidantes, antifúngicas y antivirales (Estrada et al., 2012); y glucósidos
cardiotónicos utilizados para el tratamiento de la congestión cardiaca ya que incrementa la fuerza
y la velocidad de las contracciones cardiacas (Casamitjana, 2002).
Además, para asegurar que el extracto es seguro, se determinó la dosis letal 50 (DL50) mediante el
método modificado de Lorke (1983), en dicho estudio en ninguno de los grupos de
experimentación se presentaron signos de toxicidad o muerte, con una supervivencia del 100 % de
los animales, es decir que el extracto presenta un DL50 calculada mayor a 2000 mg/kg que de
acuerdo a la clasificación propuesta por Repetto (2009) y la OMS (2009) el extracto se clasifica
como ligeramente toxico siendo seguro para su consumo.
También se realizó la evaluación de la actividad anticonvulsivante del extracto mediante un
modelo de epilepsia generada por la administración de PTZ, antagonista del receptor GABAA
(Ramírez et al., 2014) que específicamente se une al sitio para picrotoxina produciendo daño en el
canal para iones cloruro bloqueando la actividad inhibitoria mediada por GABA (Pereno et al.,
2010) generando así la excitación neuronal y manifestándose mediante convulsiones en las ratas.
Al evaluar la actividad anticonvulsivante del extracto de Buddleja cordata kunth se encontró una
disminución significativa en la duración de las convulsiones en los tres grupos tratados con el
extracto con las diferentes dosis, respecto al tiempo de latencia la dosis de 150 mg/kg del extracto
mostro un aumento significativo, lo que significa que se presentó un retardo en el inicio de las

46
convulsiones. El efecto anticonvulsivo se asocia con los metabolitos secundarios presentes en el
extracto, autores como Hiller y Zetler (1996) y Pérez de Alejo (1998) asocian la actividad
anticonvulsiva a la presencia de flavonoides en las plantas Valeriana officinalis L. y Indigofera
suffruticosa mil respectivamente, así mismo Celis et al. (2007) en sus estudios farmacológicos
sobre la planta Valeriana pavonii no solo le atribuye el efecto terapéutico ante las crisis tónico-
clónicas a los flavonoides sino también por la presencia de alcaloides, mismo metabolitos que
fueron encontrados en el estudio fitoquímico preliminar de la planta de estudio. Ante estos
antecedentes sobre los metabolitos presentes en plantas medicinales se asocia a fármacos con
actividad antiepiléptica como el levetiracetam y la etosuximida, fármacos que en su estructura
química presentan como grupo funcional al pirrol, un alcaloide que podría conformar varios de los
metabolitos secundarios de la planta Buddleja cordata kunth. El levetiracetam es un fármaco
antiepiléptico de gran efectividad que actualmente se desconoce su mecanismo de acción, pero se
ha demostrado que dicho fármaco tiene efectos sobre la liberación de neurotransmisores
excitatorios e inhibidores, está disminución de la liberación de neurotransmisores se da durante
periodos de activación de alta frecuencia, además de que se une a la proteína vesicular sináptica
2A (SV2A) (Meehan et al., 2012), esta proteína presenta una función desconocida, pero se sabe
que dicha proteína se encuentra en todas la terminales nerviosas (Lynch et al., 2004). Por parte de
la etosuximida, este fármaco bloquea los canales de Ca2+ de tipo T disminuyendo las corrientes de
dicho ion (Huguenard, 2002).
Por otra parte, los marcadores de función de hepática y renal nos sirven como referencia para
mostrar si el extracto genera alguna alteración en el hígado o el riñón, ya que estos órganos son
los encargados de realizar el metabolismo y eliminación de sustancias químicas exógenas (del
Arco, 2001). La bilirrubina se produce a partir de la degradación del grupo hemo de la hemoglobina
procedente de eritrocitos envejecidos y nos indica un mal funcionamiento en hígado (Moreira et
al., 2015; Rincón, 2012), en el panel B de la figura 20 los resultados de bilirrubina total no
muestran anormalidades, pero en el panel C, los resultados para bilirrubina directa muestran
diferencia significativa del grupo control con respecto a los grupos restantes, esto podría deberse
a la presencia de alcaloides que presenten una estructura similar a la bilirrubina, es decir moléculas
con grupo(s) pirrol (Kaplan et al., 1982) produciendo así un aumento de la concentración de
bilirrubina directa. En el caso del BUN, alteraciones en los niveles de ésta puede ser por defectos
en el transporte de metabolitos intermediarios del ciclo de urea o por la acumulación de metabolitos

47
anómalos que interfieren en la ureagénesis (Dimski, 2008), en el panel A de la figura 21 se muestra
que el grupo control no es diferente con relación a las concentraciones de BUN presente en la de
los grupos problemas destacando así que el extracto no provoca alteraciones que pudiera producir
algún daño en la función renal.
La administración de PTZ además de provocar convulsiones en las ratas se ha demostrado que
genera daño y muerte en neuronas piramidales del giro dentado en la zona CA1 y CA3 del
hipocampo; así como de amígdala del cual se desconoce dicho proceso (Pohle, 1997; Pavlova
2004), sin embargo, se han atribuido a otros procesos la neurotoxicidad provocada por PTZ, puesto
que el EE desencadena la activación de fosfolipasas, proteasas y nucleasas de la membrana, dando
como resultado la liberación de peróxidos lipídicos y radicales libres no solo en cerebro sino
también en eritrocitos e hígado (Obay et al., 2008; Akbas et al., 2005; Ilhan et al., 2005), además
de un incremento de lípidos, daño a proteínas, incremento de óxido nítrico (NO) y disminución de
la actividad enzimática antioxidante de la catalasa (CAT) y superóxido dismutasa (SOD)
(Rodriguez et al., 2013). Otro mecanismo propuesto por el cual se produce neurodegeneración es
mediante la activación de la caspasa-3 en hipocampo debida a la exposición al PTZ (Pavlova,
2006).
Las zonas donde se estudiaron la densidad neuronal fue en el núcleo basolateral de amígdala, en
la zona M1 de corteza motora, en estría terminal y en la zona CA3 hipocampo ya que estas zonas
son consideradas como focos epilépticos (Cabo et al., 2006). Al estudiar dichas regiones
encontramos que el número de neuronas normales disminuye significativamente en todos los
grupos tratados con PTZ y al administrar el extracto etanólico de Buddleja cordata kunth se
presentó una disminución significativa del número de neuronas anormales en cada una de las
regiones estudiadas con la dosis de 150 mg/kg y de 300 mg/kg, en tanto la dosis de 75 mg/kg no
fue suficiente para realizar tal efecto. Con base a estos resultados obtenidos de acuerdo con el
número total de neuronas, se confirma que el extracto etanólico de Buddleja cordata kunth produce
una disminución del daño y muerte neuronal en el núcleo basolateral de amígdala, zona M1 de
corteza motora, estría terminal y la región CA3 del hipocampo.
De acuerdo con este resultado se espera que los metabolitos presentes en el extracto sean los que
afecta el PA en las neuronas con una alta excitabilidad, en una primera instancia se le asocia este
efecto a los alcaloides puesto que se le ha atribuido actividad antiepiléptica (Ameri, 1998), se han
realizado experimentos en los que se determina que una gran variedad de alcaloides como

48
alcaloides indólicos (Chen et al., 1996), alcaloides piperidínicos (da Cruz et al., 2012) y alcaloides
isoquinolínicos (Bhutada et al., 2010; da Silva et al., 2006;) presentan efectos anticonvulsivos, el
mecanismo en que generan dicho efecto es bloqueando los canales de Na+ dependientes de voltaje
disminuyendo la conductancia de iones Na+ en las neuronas epileptogénicas (Ameri et al., 1996;
Ameri, 1997; Ameri, 1998), sin embargo a estas entidades químicas se les han atribuido otros
mecanismos de modulación de la epilepsia como unión a canales GABA (da Cruz et al., 2012),
receptores NMDA (Chen et al., 1996) tanto en hipocampo como en amígdala (Lin et al., 2002).
Los otros metabolitos secundarios encontrados en el análisis fitoquímico preliminar son los
flavonoides, diversos estudios indican que los flavonoides son capaces de unirse a los sitios de
unión de las benzodiazepinas (BDZ) en los receptores GABAA de acuerdo con Medina et al.
(1998), los flavonoides además de tener una alta afinidad hacia GABAA podrían incluso presentar
propiedades farmacológicas, se ha considerado esta actividad no solo por estudios de relación
estructura actividad (REA) de flavonoides a receptores GABAA (Huang et al., 2001; Wang et al.,
2005) si no que se han reportado en estudios realizados en plantas medicinales de la familia
Valerianoidea que los flavonoides son capaces de unirse de manera selectiva a receptores GABAA
(Wasowski et al., 2002; Hölzl y Godau, 1989) pero presentando afinidad variable hacia dichos
receptores GABA. Así mismo se tienen la posibilidad de que los flavonoides al entrar al organismo
son metabolizados rápidamente y puede que algunos de esos metabolitos secundarios sean capaces
de unirse con alta afinidad a sitios de unión de BDZ en receptores GABAA (Fernández et al., 2006).
Todo esto en relación de la afinidad de los flavonoides hacia los receptores GABAA con subunidad
α3 acopladas a β3γ2 (Medel et al., 2011) presentes en las neuronas piramidales del hipocampo,
amígala y corteza motora y en la cual esta afinidad hacia dicho receptor es base de la estructura
química de los flavonoides.
Otro mecanismo por considerar en el que podría estar implicada la actividad neuroprotectora de
los flavonoides es el efecto antioxidante, ya que presenta una alta actividad quelante de hierro
(Mira, et al., 2002; Mladěnka et al., 2011), también se les han atribuido efectos inhibidores sobre
oxidasas ya que han demostrado inhibir la formación de productos de lipooxigenasa (LOX)
(Ribeiro et al., 2014; Ferrándiz y Alcaraz, 1991), xantina oxidasa (XO) (Lin et al., 2015; Lin et
al., 2014) y NADPH oxidasa (Bohmont et al., 1987; Hodnick et al.,1987) disminuyendo la
generación de especies reactivas de oxígeno, todos esto dependiendo de la estructura del
flavonoide; se les atribuye además como inhibidores de enzimas presentes en procesos oxidativos

49
como la fosfolipasa A2 (Lindahl y Tagesson, 1997; Lättig et al., 2007). También se les atribuye
en participar en la actividad de la enzima con propiedades antioxidantes como la catalasa (CAT)
(Sudheesh et al., 1999).
Sin embargo para elucidar el mecanismo por el cual los metabolitos presentes en el extracto
etanólico de Buddleja cordata kunth generan efecto neuroprotector es necesario realizar la
extracción de los compuestos mediante solventes con diversas polaridades y de esta manera
identificar la estructura del agente químico responsable por medio de análisis espectroscópicos
como RMN o espectrometría de masas y así estudiar más detalladamente el efecto terapéutico que
genera.
Conclusión
El extracto etanólico de hojas de Buddleja cordata kunth presenta:
• Alcaloides, azúcares reductores, antraquinonas, taninos derivados de catecol, flavonoides y
glucósidos cardiotónicos.
• Dosis letal media mayor de 2000 mg/kg.
Las ratas tratadas con extracto etanólico de Buddleja cordata kunth presentaron:
• Disminución de la duración de convulsiones.
• Aumento del tiempo de latencia de convulsiones.
• Aumento de los niveles de bilirrubina directa.
• Disminución de neuronas anormales en amígdala, corteza motora, estría terminal e hipocampo.

50
Referencias bibliográficas
Acevedo C., Campos M., Mesa T. y Núñez L. (2007). Epilepsias: todo lo que usted desea saber.
2° edición. Chile: BHA impresores.
Adams M., Vanessa S.S., Luge M.., Kessler M. y Hamburger M. (2012). Epilepsy in the
Renaissance: A survey of remedies from 16th and 17th century German herbals. Journal of
Ethnopharmacology. 143 (1): 1-13.
Aguilera L.P. (2009). Estudio prospectivo de la respuesta al tratamiento farmacológico en la
epilepsia infantil. Epilepsia refractaria y factores pronósticos. (Tesis doctoral, Universidad
de Granada).
Aguirre O.F., Wall M.A., González A.G. López D.J., Álvarez P.E., de la Rosa L.A. y Ramos J.A.
(2015). Taninos hidrolizables; bioquímica, aspectos nutricionales y analíticos y efectos en
la salud. Nutrición Hospitalaria. 31 (1): 55-66.
Akbas S.H., Yegin A. y Ozben T. (2005). Effect of pentylenetetrazol-induced epileptic seizure on
the antioxidant enzyme activities, glutathione and lipid peroxidation levels in rat
erythrocytes and liver tissues. Clinical biochemistry. 38 (11): 1009-1014.
Amador J.M., Pacheco L.J. y Giordano N.M. (sf). Caracterización de la respuesta antioxidante al
estrés inducido por ácido kaínico en estriado de rata. Instituto de neurobiología.
Ameri A. (1997). Inhibition of rat hippocampal excitability by the plant alkaloid 3-acetylaconitine
mediated by interaction with voltage-dependent sodium channels. Arch Pharmacology.
355: 273-280.
Ameri A. (1998). Structure-dependent inhibitory action of the Aconitum alkaloids 14-
benzoyltalitasamine and talitasamine in rat hippocampal slices. Arch Pharmacology. 357:
585-592.
Ameri A. (1998). The effects of aconitum alkaloids on the central nervous system. Progress in
Neurobiology. 56: 211-235.
Ameri A., Gleitz J. y Peters T. (1996). Aconitine inhibits epileptiform activity in rat hippocampal
slices. Arch Pharmacology. 354: 80-85.
Armijo J.A. y Herranz J.L. Fármacos antiepilépticos y anticonvulsivantes. (2014). Flórez J, Armijo
J.A. y Mediavilla A. Farmacología humana. 6° edición. España: Elsevier Masson.
Avila A.J., Espinosa G.A., De Maria y Campos D.M., Benitez F.J., Hernández D.T., Flores M.S.,
Campos C.J., Muñoz L.J. y García B.A. (2014). Photoprotection

51
of Buddleja cordata extract against UVB-induced skin damage in SKH-1 hairless mice.
BMC Complement Altern Med. 14: 281.
Avila J.G., de Liverant J.G., Martínez A., Martínez G., Muñoz J.L., Arciniegas A. y Romo de
Vivar A. (1999). Mode of action of Buddleja cordata verbascoside against Staphylococcus
aureus. Journal Ethopharmacol. 66 (1): 75-78.
Barrett E., Barman M., Boitano S., Heddwen L. y Brooks L. (2013). Ganong Fisiología médica.
24ª edición. México: Mc Graw Hill.
Bennett M.V. (2000). Electrical synapses, a personal perspective (or history). Brain Res Rev. 32
(1): 16-28.
Berne y Levy. (2009). Fisiología. 6ª edición. España: Elsevier.
Beyer C.E. y Berthoud M.V. (2017). Gap junction gene and protein families: Connexins, innexins,
and pannexins. BBA Biomembranes. 1860 (1): 5-8.
Bhutada P., Mundhada Y., Bansod K., Dixit P., Umathe S. y Mundhada D. (2010). Anticonvulsant
activity of berberine, an isoquinoline alkaloid in mice. Epilepsy & behavior. 18: 207-210.
Biblioteca Digital de la Medicina Tradicional Mexicana (2018). En Medicina Tradicional
Mexicana-Universidad Autónoma Nacional de México.
Bohmont C., Aaronson L.M., Mann K. y Pardini R.S. (1987). Inhibition of mitochondrial NADH
oxidase, succinoxidase, and ATPase by naturally occurring flavonoids. Journal of nature
products. 50 (3): 427-433.
Buron E., López M.M. y Navarro F.J. (2008). Farmacología y aspectos conductuales del receptor
α5/GABA-A. 15 (1): 11-15.
Bustamante Z. E. (2007). El sistema nervioso: desde las neuronas hasta el cerebro humano.
Colombia: universidad de Antioquia.
Cabo V.C., Villanueva H.P. y Prieto M.A. (2006). Neuroquímica de la epilepsia, neurotransmisión
inhibitoria y modelos experimentales: nuevas perspectivas. Revista de neurociencia. 42 (3):
159-168.
Campos C.J. (2009). Papel de los fármacos antiepilépticos genéricos en el tratamiento de la
epilepsia infantil. Neurología infantil II. 69 (1): 109-113.
Cardinali P.D. (2007). Neurociencia aplicada: sus fundamentos. Argentina: Médica Panamericana.
Casamitjana C.N. (2002). Glucósidos cardiotónicos. Acción y usos. Centro de información del
medicamento. 16 (4): 5-94.

52
Celis T.C., Rincón J. y Guerrero F.M. (2007). Actividad farmacológica sobre el sistema nervioso
central del extracto etanólico y de la fracción alcaloidal de Valeriana pavonii. Revista
colombiana de ciencias químico-farmacéuticas. 36 (1): 11-22.
Cerdón J. y Troncoso J. (2016). Alteraciones de las células de la microglía del sistema nervioso
central provocadas por lesiones del nervio facial. Biomédica. 36: 619-631.
Chen K., Kokate G.T., Donevan D.S., Carroll I.F. y Rogawski A.M. (1996). Ibogaine Block of the
NMDA Receptor: In Vitro and In Viva Studies. Neuropharmacology. 35 (4): 423-431.
Clifford B.D., Olney W., Maniotis A., Collins C.R. y Zorumski F.C. (1987). The functional
anatomy and pathology of lithium-pilocarpine and high-dose pilocarpine seizures. 23 (3):
953-968.
Coaquira P.S., Condori Q.A. y Fuentes B. P. (2012). Neurofisiología de la conducción nerviosa.
Revista de actualización clínica. 27: 1301-1306.
Concepción R.A. (2009). Uso de neurotoxinas marinas en la experimentación animal. e- Gnosis.
7 (2).
Córdova A. (2003). Fisiología dinámica. España: Masson.
Costanzo S.L. (2011). Fisiología. 4° edición. España: Elsevier.
da Cruz P.G., Felipe B.C., Scorza F.A., da Costa C.M., Tavares A.F., Menezes F.M., de Andrade
M.G., Leal A.M., Brtio C.G., Mazzacoratti N.M., Cavalheiro A.E. y Viana D.G. (2012).
Piperine decreases pilocarpine-induced convulsions by GABAergic mechanisms.
Pharacology biochemistry and behavior. 104: 144-153.
da Silva B.F., de Andrade J.P., Bevilaqua L.R., de Souza M.M., Izquierdo I., Henriques T.A. y
Zuanazzi S.J. (2006). Pharmacology biochemistry and behavior. 85: 148-154.
del Arco C. (2001). Metabolismo de los fármacos. Florez J. Farmacología humana. 3a edición. Ed.
España: Masson.
Delgado C.A., Minguillón L.C. y Joglar T.J. (2004). Introducción a la química terapéutica. 2°
edición. España: Díaz de Santos.
Delgado J.M. (2014). Modelos experimentales de Kindling para el estudio de la epilepsia. Trabajo
de grado. Universidad Complutense de Madrid. España.
Desiderio P.L. y Albia P.A. (2005). Antiepilépticos de tercera generación. Revistas Médicas
Cubanas.
Diao J., Su Z., Lu X., Yoon Y.T., Shin K.Y. y Ha T. (2010). Single-Vesicle Fusion Assay Reveals

53
Munc18-1 Binding to the SNARE Core Is Sufficient for Stimulating Membrane Fusion.
ACS Chem Neuroscience. 1 (3): 168-174.
Díaz S. B., Jiménez E.M. y Auro O.A. (2000). Evaluación del efecto parasiticida de los extractos
acuoso y metabólico de Buddleja cordata HBK (tepozán) sobre Costia necatrix en tilapia
(Oreochromis sp). E-jouernal-UNAM. 1-9.
Dimski S.D. (2008). Ammonia metabolism and the urea cycle: function and Clinical implications.
Journal of veterinary internal medicine. 8 (2): 73-78.
Dvorkin A.M., Cardinali P.D. y Iermoli H.R. (2010). Best & Taylor: Bases Fisiológicas de la
Práctica Médica. 14° edición. Argentina: Médica Panamericana.
Epilepsia en México. (2017).
Estrada R.R., Ubaldo S.D. y Araujo E.G. (2012). Los flavonoides y el sistema nervioso central.
Salud Mental. 35: 375-384.
Estrada Z.M., Aarland R.C., Rivera C.F., Barnabé A.A., Buendía G.L. y Cruz S.F. (2016).
Mcropropagation of buddleja cordata and the content of verbascoside and total phenols
with antioxidant activity of the regenerated plantlets. 15 (2): 333-346.
Everett G.M. y Richards R.K. (1944). Comparative anticonvulsive action of 3,5,5-
trimethyloxazolidine2,4-dione (tridione), dilantin, and phenobarbital. J. Pharmacol. Exp.
Ther. 81: 402-407.
Fernández P.S., Wasowski C., Loscalzo M.L., Granger E.R., Johnston A.R., Paladini C. y Marder
M. (2006). Central nervous system depressant action of flavonoid glycosides. European
journal of pharmacology. 539 (3): 168-176.
Ferrándiz M.L. y Alcaraz M.J. (1991). Anti-inflammatory activity and inhibition of arachidonic
acid metabolism by flavonoids. Agents and actions. 32 (3-4): 283-288.
Fisher S.R., Acevedo C., Arzimanoglou A., Bogacs A., Cross H., Elger E.C., Engel J.Jr., Forsgren
L., French A.J., Glynn M., Hesdorffer C.D., Lee I.D., Mathem W.G., Moshé L.S., Perucca
E., Scheffer E.I., Tomson T., Watanabe M. y Wiebe S. (2014). Definición clínica práctica
de la epilepsia. Informe oficial de la ILEA. 55 (4): 475-482.
Flores S.M., Chaparro H.V., Escoto D.M., Vázquez V.E., González C.R. y Beas Z.C. (2012).
Estructura y función de las subunidades del receptor a glutamato tipo NMDA. Neurología.
27 (5): 301-310.
Fritschy M.J. (2009). Differential alterations of GABAA receptors in epilepsy. 273-277.

54
García E. y Ristol A. (2007). Epilepsia, historia, concepto, síndromes epilépticos, crisis epiléptica,
clasificación, epidemiología, valoración socioeconómica. Medicine. 9 (75): 4801-4805.
García G.M., García M.I. y Matías G.J. (2009) Modelos experimentales en epilepsia. Neurología.
25: 181-188.
Germán L.P. (2010). Fisiopatología de la epilepsia del lóbulo temporal: revisión del proceso de
muerte neuronal a la neuroplasticidad. Revista Argentina de ciencias del comportamiento.
2 (1): 46-57.
Goddard V.G. (1969). Development of Epileptic Seizures through Brain Stimulation at Low
Intensity. Nature Publishing Group. 214: 1020-1021.
González E.A., García B.A., Benítez F.J., Sandocal P.C., González V.M., Céspedes C.L. y Avila
A.J. (2016). Photoprotective effect of verbascoside from Buddleja cordata in SKH-1 mice
exposed to acute and chronic UV-B radiation. Boletin latinoamericano y del Caribe de
plantas medicinales y aromáticas. 15 (5): 288-300.
Hall E. J. (2011). Guyton y Hall: tratado fisiología médica. 12° edición. España: Elsevier.
Hamilton E.S., Loose D.M., Qi M., Levey I.A., Hille B., Mcknight S.G., Idzerda L.R. y Nathanson
M.N. (1997). Disruption of the ml receptor gene ablates muscarinic receptor-dependent M
current regulation and seizure activity in mice. Neurobiology. 94 (24): 13311-13316.
Hanaya R. y Arita K. (2016). The New Antiepileptic Drugs: Their Neuropharmacology and
Clinical Indications. Neurol Med Chir. 56: 205-220.
Hiller O.K. y Zetler G. (1996). Neuropharmacological Studies on Ethanol Extracts of Valeriana
officinalis L.: Behavioural and Anticonvulsant Properties. Phytotherapy research. 10: 145-
151.
Hodnick W.F., Bohmont C.W., Capps C. y Pardini R.S. (1987). Inhibition of the mitochondrial
NADH-oxidase (NADH-coenzyme Q oxido-reductase) enzyme system by flavonoids: a
structure-activity study. Biochemical pharmacology. 36 (17): 2873-2874.
Hölzl J. y Godau P. (1989). Receptor binding studies with Valeriana officinalis on the
benzodiazepine receptor. Planta Med. 55: 642.
Honchar P.M., Olney W.J. y Sherman R.W. (1983). Systemic Cholinergic Agents Induce Seizures
and Brain Damage in Lithium-Treated Rats. Science. 220 (4594): 323-325.
Houghton P.J. (1984). Ethnopharmacology of some Buddleja species. Journal of
Ethnopharmacology. 11: 293-308.

55
Houghton P.J., Mansah A.Y., Lessa N. y Hong L.Y. (2003). Terpenoids in Buddleja: relevance to
chemosystematics, chemical ecology and biological activity. Phytochemistry. 64 (2): 385-
393.
Huang R., Horner B.C., Dibas I.M., Covey F.D., Drewe A.J. y Dillon H.G. (2001).
Pentylenetetrazole-induced inhibition of recombinant ᵞ-aminobutyric acid type A
(GABAA) receptors: mechanism and site of action. The journal of pharmacology and
experimental therapeutics. 298 (3): 986-995.
Huang X., Liu T., Gu J., Luo X., Ji R., Cao Y., Xue H., Wong J.T., Wong B.L., Pei G., Jiang H. y
Chen K. (2001). 3D-QSAR model of flavonoids binding at benzodiazepine site in GABAA
receptors. Journal of medicinal chemistry. 44 (12): 1883-1891.
Huguenard R. J. (2002). Block of T-Type Ca2+ channels is an important action of succinimide
antiabsence drugs. American epilepsy society 2 (2): 49-52.
Ibarra E.E., Pacheco S.M., García M.R., San Miguel C.R., Ramírez V.G. y Soto H.M. (2011).
Actividad antioxidante de alcaloides de Erythrina americana Miller. Rev. Fitotec. 34 (4):
241-246.
Ilhan A., Arif A.M., Kocer A., Boluk A., Gurel A. y Armutcu A. (2005). Erdosteine ameliorates
PTZ-induced oxidative stress in mice seizure model. Brain research bulletin. 65 (6): 495-
499.
Ira F.S. (2011). Fisiología humana. 12ª edición. México: McGraw Hill.
Jacob C.T., Moss J.S. y Jurd R. (2008). GABAA receptor trafficking and its role in the dynamic
modulation of neuronal inhibition. Nature Reviews Neuroscience. 9 (5): 331-343.
Jahn R. y Scheller H.R. (2006). SNAREs – engines for membrane fusion. Nature Reviews
Molecular Cell Biology. 7 (9): 631-643.
Joel M. y Sabyasachi S. (2012). Fisiología humana. México: Manual moderno.
Kaplan D. y Navon G. (1982). Studies of the conformation of bilirubin and its dimethyl ester in
dimethyl sulphoxide solutions by nuclear magnetic resonance. Biochemical journal. 201
(3): 605-613.
Kestel D., Acevedo C., Medina T.M., Mesa T. y Rodríguez J. (2013). Epilepsia en Latinoamérica:
experiencias, documento técnico basado en las presentaciones del taller internacional
efectuado en Santiago de Chile, en agosto de 2013, y otras contribuciones posteriores.
Asociación Internacional Contra la Epilepsia.

56
Kubo Y. y Tateyama M. (2005). Towards a view of functioning dimeric metabotropic receptors.
Neurobiology. 15: 289-295.
Lang T. y Jahn R. (2008). Core proteins of the secretory machinery. Handbook of Experimental
Pharmacology of Neurotransmitter Release. 184: 107-127.
Lara L.S., Abrahin J.M., Colla M.C., Clement M.E., Coffey P., Furnari A., Marquez F. y Romano
M.L. (2017). Clasificación etiológica de la epilepsia de recién diagnóstico en adultos
mayores: ¿es útil la nueva propuesta? Neurología Argentina. 9 (2): 125- 130.
Lättig J., Böhl M., Fischer P., Tischer S., Tietböhl C., Menschikowski M., Gutzeit H.O., Metz P.
y Pisabarro M.T. (2007). Mechanism of inhibition of human secretory phospholipase A2
by flavonoids: rationale for lead design. Journal of computer-aided molecular design. 21
(8): 473-483.
Liga Internacional Contra la Epilepsia. (2005). En ILAE.
Lin M.T., Wang J.J. y Young M.S. (2002). The protective effect of dl-tetrahydropalmatine against
the development of amygdala kindling seizures in rats. Neuroscience Letters. 320 :113-
116.
Lin S., Zhang G., Liao Y. y Pan J. (2015). Inhibition of chrysin on xanthine oxidase activity and
its inhibition mechanism. International journal of biological macromolecules. 81: 274-282.
Lin W.Q., Xie J.X., Wu X.M., Yang L. y Wang H.D. (2014). Inhibition of xanthine oxidase activity
by gnaphalium affine extract. Chinese medical sciences journal. 29 (4): 225-230.
Lindahl M. y Tagesson C. (1997). Flavonoids as phospholipase A2 inhibitors: importance of their
structure for selective inhibition of group II phospholipase A2. Inflammation. 21 (3): 347-
356.
Ling Z.H., Bo W.J., Tao W.Y., Cai L.B., Xiang C., He J. y Li P. (2014). Medicinal compounds
with antiepileptic/anticonvulsant activities. Epilepsia. 55 (1): 3-16.
Locatelli M., Tammaro F., Menghini L., Carlucci G., Epifano F. y Genovese S. (2009).
Anthraquinone profile and chemical fingerprint of Rhamnus saxatilis L. from Italy.
Phytochemistry Letters. 2 (4): 223-226.
López H.E., Solis H. y Bravo S.J. (2005). Epilepsia y antiepilépticos de primera y segunda
generación. Aspectos básicos útiles en la práctica clínica. Universidad Nacional Autónoma
de México (UNAM). 48 (5): 1-8.
López M.M., Rocha L., Miquel M., Hernández M., Cárdenas T.R., Coria A.G., García I.L., Pérez

57
E.C., Aranda A.G. y Manzo J. (2009). Conceptos básicos de la epilepsia. Revista Médica
UV. 9 (2): 31-37.
Lorke D. (1983). A new approach to practical acute toxicity testing. Archive of Toxicology. 54:
275-287.
Löscher W. y Schmidt D. (1988). Which animal models should be used in the search for new
antiepileptic drugs? A proposal based on experimental and clinical considerations.
Epilepsy Res. 2: 145-181.
Lujan R. (2004). Bases moleculares de la señalización neuronal. Ciencia al Día Internacional. 5
(2): 1-19.
Lynch A.B., Lambeng N., Nocka K., Hammes K.P., Bajjalieh M.S., Matagne A. y Fuks B. (2004).
The synaptic vesicle protein SV2A is the site for the antiepileptic drug levetiracetam. 101
(26): 9861-9866.
Mares P. y Kubová H. Electrical stimulation-induced models of seizures. In: Models of Seizures.
Pitkänen, A., Schwartzkroin, P.A., Moshé, S.L. (Eds.). Elsevier Academic Press and
Epilepsy, Chapter 12: 153-159.
Martínez G.A. (2014). Comunicación entre células gliales y neuronas I. Astrocitos, células de
Schwann que no forman mielina y células de Schwann presinápticas. Medicina e
investigación. 2 (2): 75-84.
Martínez V.M., Ramírez A.T., Aguilar H. y Bye R. (1996). Analgesic and antipyretic activities of
an aqueous extract and of the flavone linarin of Buddleia cordata. Planta Medica. 62 (2):
137-140.
Martínez V.M., Ramírez A.T., Aguilar M.H. y Bye R. (1995). Analgesic and Antipyretic Activities
of an Aqueous Extract and of the Flavone Linarin of Buddleia cordata. Planta Medica. 62:
137-140.
Martínez V.M., Ramírez A.T., Lastra A.L. y Bye R. (1998). A comparative study of the analgesic
and anti-inflammatory activities of pectolinarin isolated from Cirsium subcoriaceum and
linarin isolated from Buddleia cordata. Planta medica. 64 (2): 134-137.
McNamara O.J. Farmacoterapia de las epilepsias. (2012). En Brunton L.L, Chabner A.B. y
Knollman C.B. Goodman & Gilman las bases farmacológicas de la terapéutica. 12° edición.
Pág. 583-608. México: McGraw Hill.
Medel M.J., Cortijo P.L., Gasca P.E., Susan T.P., Pérez P.A. y Ramos M.F. (2011). Receptor

58
GABAA: implicaciones farmacológicas a nivel central. Arch neurociencia. 18 (1): 40-45.
Medina J.H., Viola H., Wolfman C., Marder M., Wasowski C., Calvo D. y Paladini A.C. (1998).
Neuroactive flavonoids: new ligands for the Benzodiazepine receptors. Phytomedicina. 5
(3): 235-243.
Medina T. M., Chaves S. F., Chinchilla C. N. y Gracia G. F. (2001). Las epilepsias en
Centroamérica. Honduras: Tegucigalpa.
Medinilla T.M., Barajas V.G., Ramírez A.E., Barbosa C.O. y Sánchez R.S. (2005) Anatomía de la
epilepsia y fármacos utilizados en su tratamiento. Archivos de medicina. 1 (2): 1-18
Meehan L.A., Yang X., McAdams D.B., Yuan L. y Rothman S.M. (2012). A new mechanism for
antiepileptic drug action: vesicular entry may mediate the effects of levetiracetam. 106 (3):
1227-1239.
Mendoza H.P. (2003). El tepozán. Revista Ciencias. 70: 32-33.
Mendoza P.N. (2008). Farmacología médica. México: Médica Panamericana.
Merino B.D., Martín G.Y. y Rodríguez A.A. (2009). La maquinaria molecular de la exocitosis:
¿un nuevo marcador en las enfermedades neurodegenerativas? Monografías de la real
academia nacional de farmacia. 71-94.
Mira L., Fernandez M.T., Santos M., Rocha R., Florêncio M.H. y Jennings K.R. (2002).
Interactions of flavonoids with iron and copper ions: a mechanism for their antioxidant
activity. Free radical research. 36 (11): 1199-1208.
Mladěnka P., Macáková K., Filipský T., Zatloukalová L., Jahodář L., Bovicelli P., Silvestri IP.,
Hrdina R. y Saso L. (2011). In vitro analysis of iron chelating activity of flavonoids. Journal
of inorganic biochemistry. 105 (5): 693-701.
Moreira V.F. y Garrido E. (2015). Pruebas de función hepática: B, AST, ALT, FA y GGT. Revista
española de enfermedades digestivas. 107 (10): 1.
Mulantinho V.M, Araújo G.E. y Evaristo C.P. (2011). Epilepsia y Anestesia. Revista Brasileña de
anestesiología. 61 (2): 124-136.
Nielsen S.M., Axelsen N.L., Sorgen L.P., Verma V., Delmar M. y Rathlou H.H. (2012). Gap
junctions. Comprehensive Physiology. 2: 1981-2035.
Obay B.D., Taşdemir E., Tümer C., Bilgin H. y Atmaca M. (2008). Dose dependent effects of
ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides. 29
(3): 448-455.

59
Ochoa G.A., López P.J., Fuertes C.R., Martínez R.F., Samper V.P., Monge G.L., Peña S.J. y García
J.M. (2015). Estudio descriptivo de las epilepsias sintomáticas según edad de inicio
controladas durante 3 años en una unidad de neuropediatría de referencia regional.
Neurología. 32 (7): 417-486.
Olsen W.R. y Sieghart W. (2008). Subtypes of gamma-aminobutyric acid(A) receptors:
classification on the basis of subunit composition, pharmacology, and function.
Pharmacological Reviews. 60 (3): 243-260.
Organización Mundial de la Salud (2017).
Palacios E., Bello L., Maldonado D. y Martínez F. (2017). Epilepsia occipital. Repertorio de
medicina y cirugía. 26 (1): 3-8.
Palacios R.L., Blasco M.J. y Pagés C.T. (2005). Fisiología animal. Vol. 1. España: Universitat
Barcelona
Partearroyo T., Sánchez C.E. y Varela M.G. (2013). El azúcar en los distintos ciclos de la vida:
desde la infancia hasta la vejez. Nutr Hosp. 28 (4): 40-47.
Pavlova T.V., Stepanichev M. y Gulyeava N. (2006). Pentylenetetrazole kindling induces neuronal
cyclin B1 expression in rat hippocampus. Neuroscience Letters. 392 (2006): 154-158.
Pavlova T.V., Yakovlev A.A., Stepanichev Y.M., Mendzheritskii A.M. y Gulyaeva N.V. (2004).
Pentylenetetrazole kindling induces activation of caspase-3 in the rat brain. Neuroscience
and behavioral physiology. 34 (1): 45-47.
Pereno G.L. (2010). Fisiopatología de la Epilepsia del Lóbulo Temporal: Revisión del Proceso de
Muerte Neuronal a la Neuroplasticidad. Revista Argentina de Ciencias del
Comportamiento. 2 (1): 46-57.
Pereno G.L. y Beltramino C. (2010). Descifrando la fisiopatología de la epilepsia en un modelo
animal: el pentilentetrazol induce la activación pero no la muerte de las neuronas de la
amígdala extendida medial. Neurología. 25 (3): 148-155.
Pérez B.G., Ávila A.J., García B.A., Montes S., García J.S., León R.I., Rubio O.M. y Monroy N.A.
(2015). Neuroprotective effect of Buddleja cordata methanolic extract in the 1-methyl-4-
phenylpyridinium Parkinson’s disease rat model. J. Nat Med. 69: 86-96.
Pérez de Alejo L.J., Rodríguez R.G. y Miranda F.R. (1998). Actividad anticonvulsivante de las
fracciones butanólica y acetato de etilo de la Indigofera suffruticosa mill (Añil cimarrón).
Revista cubana de plantas medicinales. 3 (3): 7-11.

60
Pohle W., Becker A., Grecksch G., Juhre A. y Willenberg A. (1997). Piracetam prevents
pentylenetetrazol kindling-induced neuronal los and learnig deficits. 6 (6): 467-474.
Racine J.R. (1972). Modification of seizure activity by electrical stimulation: I. after-discharge
threshold. Electroencephalography and Clinical Neurophysiology. 32 (3): 269-279.
Ramírez S.J., Soriano U.M., Espinosa R.J., Correa B.J., Trujillo F.G., Miranda R.R., Delgado R.F.
y Gómez P.R. (2014). Anticonvulsant effects of bis-1,4-dihydropyridines and the probable
role of L-type calcium channels suggested by docking simulations. Medicinal Chemistry
Research. 23 (12): 5149-5159.
Ramos M.F., Correa B.J., Saavedra V.M., Acosta H.M., Gasca P.E., Pérez P.A. y Trujillo F.J.
(2012). Modelo PTZ: un screening primario para el desarrollo de nuevas moléculas con
actividad anticonvulsionante. Neurocien. 17 (1): 45-48.
Randall D., Burggren W. y French K. (1998). Fisiología animal. 4a edición. España: McGraw Hill.
pp. 188-192.
Rangel R.S., Aguilar R.S. y Rojas Z.E. (2003). Buddleja cordata H.B.K. ssp. cordata
(buddlejaceae): propagación y anatomía de la madera. Polibotánica. 16: 63-78
Rao M.S., Hattiangady B., Reddy D.S. y Shetty A.K. (2006). Hippocampal neurodegeneration,
spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe
epilepsy. J neurosci Res. 83 (6): 1088-1095.
Redolar R.D., Moreno A.A., Robles M.N., Torras G.M. y Vale M.A. (2010). Fundamentos de
psicobilogía. España: UOC.
Repetto J.M. y Repetto K.G. (2009). Toxicología fundamental. 4° edición. España: Díaz de Santos.
Reyes H.D., Bulavina L. y Pivneva T. (2014). La glía, el pegamento de las ideas. Revista ciencia.
Ribeiro D., Freitas M., Tomé S.M., Silva A.M., Porto G., Cabrita E.J., Marques M.M. y Fernandes
E. (2014). Inhibition of LOX by flavonoids: a structure-activity relationship study.
European journal of medicinal chemistry. 72: 137-145.
Rincon R.D. (2012). Interpretación diagnóstica y pronóstica de las pruebas de función hepática.
Medicine. 11 (12): 733-739.
Rivera L.I. y López C.A. (2005). Relación de las estructuras de los receptors NMDA con su
función en la retina. Revista especializada en ciencias químico-biológicas. 8 (2): 71-81.
Rodrigues A.D., Scheffel T.B., Scola G., Dos Santos M.T., Fank B., Dani C., Vanderlinde R.,
Henriques J.A., Coitinho A.S. y Salvador M. (2013). Purple grape juices prevent

61
pentylenetetrazol-induced oxidative damage in the liver and serum of Wistar rats. Nutrition
research. 33 (2): 120-125.
Rodríguez Z.S., Ordaz C.A., Muñoz J.L., Arciniegas A. y Romo de Vivar A. (1999). In vitro
evaluation of the amebicidal activity of Buddleia cordata (Loganiaceae, H.B.K.) on
several strains of Acanthamoeba. Journal Ethnopharmacol. 66 (3): 327-334.
Römermann K., Bankstahl P.J., Loscher W. y Bankstahl M. (2015). Pilocarpine-induced
convulsive activity is limited by multidrug transporters at the rodent blood-brain barrier.
Journal Pharmacology of Experimental Therapeutics. 353 (2): 351-359.
Rosenzweig R.M., Breedlove S.M. y Watson V.N. (2005). Psicobiología: una introducción a la
neurociencia conductual, cognitiva y clínica. 2° edición. España: Ariel.
Saavedra E.F., López C.B., Yrei J.V., Gallardo C.T., Ale N. Y Gordillo C.G. (2012). Actividad
antibacteriana y fungicida de las antraquinonas de Aloe vera L. combinadas con cationes
cobre, hierro, plata y bismuto. Ciencia e investigación. 15 (1): 30-35.
Sahranavard S., Ghafari S. y Mosaddegh M. (2013). Medicinal plants used in Iranian traditional
medicine to treat epilepsy. Seizure. 23 (5): 328-332.
Sankaraneni R. y Lachhwani D. (2015). Antiepileptic Drugs-A Review. Pediatric Annals. 44 (2):
36-42.
Sepúlveda J.G., Porta D.H. y Rocha S.M. (2003). La Participación de los Metabolitos Secundarios
en la Defensa de las Plantas. Revista Mexicana de Fitopatología. 21 (3): 355-363.
Shimada T. y Yamagata K. (2018). Pentylenetetrazole-Induced Kindling Mouse Model. Journal
of visualized experiments. 136: 1-10.
Sigel E. y Steinmann E.M. (2012). Structure, function, and modulation of GABAA receptors. The
journal of biological chemistry. 287 (48): 40224-40231.
Silverthorn U.D. (2008). Fisiología humana: un enfoque integrado. 4° edición. Argentina: Médica
Panamericana.
Snell S.R. (2007). Neuroanatomía clínica. 6° edición. Argentina: Médica Panamericana.
Sucher J.N. y Carles C.M. (2015). A pharmacological basis of herbal medicines for epilepsy.
Epilepsy & Behavior. 52: 308-318.
Sudheesh S., Sandhya C., Sarah Koshy A. y Vijayalakshmi N.R. (1999). Antioxidant activity of
flavonoids from Solanum melongena. Phytotherapy research. 13 (5): 393-396.
Szyndler J., Maciejak P., Turzynska D., Sobolewska A., Lehner M., Taracha E., Walkowiak J.,

62
Skorzewska A., Wislowska S. A., Hamed A., Bidzinski A. y Plaznik A. (2008). Changes
in the concentration of amino acids in the hippocampus of pentylenetetrazole-kindled rats.
Neuroscience Letters. 438 (3): 245-249.
Targas Y.E., Contreras C.G. y Ríos P.L. (2014). Tratamiento farmacológico de las epilepsias.
Brasil: Leitura Médica.
Toman E.P., Swinyard A.E. y Goodman S.L. (1946). Properties of maximal seizures, and their
alteration by anticonvulsant drugs and other agents. J. Neuorophysiol. 9 (3): 231-239.
Torres Z.M., Bustos S. J. y Granados L.F. (2010). Fisiopatología del estatus epiléptico. Asociación
colombiana de neurología. 27 (1): 12-20.
Turski L., Ikonomidou C., Turski A.W., Bortolotto A.Z. y Cavelheiro A.E. (1989). Review:
cholinergic mechanisms and epileptogénesis. The seizures induced by pilocarpine: a novel
experimental model of intractable epilepsy. Synapse. 3 (2): 154-171.
Van den Bogaart G., Lang T. y Jahn R. (2013). Microdomains of SNARE proteins in the plasma
membrane. 72: 193-230.
Villalpando V.F. y Medina C.L. (2015). Effect of sparteine on status epilepticus induced in rats by
pentylenetetrazole, pilocarpine and kainic acid. Brain research. 1624: 59-70.
Viteri T.C. (2015). Epilepsia. Medicine. 11 (73): 4364-4373.
Voets T., Toonen F.R., Brian C.E., De wit H., Moser T., Retting J., Sudhof C.T., Neher E. y
Verhage M. (2001). Munc18-1 promotes large dense-core vesicle docking. Neuron. 31 (4):
581-591.
Waaler E.P., Blom H.B., Skeidsvoll H. y Mykletun A. (2000). Prevalence, classification, and
severity of epilepsy in children in Western Norway. Clinical Research. 41 (7): 802-810.
Wang F., Shing M., Huen Y., Tsang S.Y. y Xue H. (2005). Neuroactive flavonoids interacting
with GABAA receptor complex. Current drug targets. CNS and neurological disorders. 4
(5): 575-585.
Wasowski C., Marder M., Viola H., Medina J.H. y Paladini AC. (2002). Isolation and
identification of 6-methylapigenin, a competitive ligand for the brain GABA(A) receptors,
from Valeriana wallichii. Planta médica. 68 (10): 934-936.
Welsch U. y Sobotta J. (2008). Histología. 2° edición. Argentina: Médica panamericana.
Wu C. y Sun D. (2014). GABA receptors in brain development, function, and injury. 30 (2): 367-
379.





















![[Recensão a] BATAILLON, Marcel, MARGOLIN, J. C., MACEDO, …€¦ · cordata e criteriosamente traz os cinco temas à barra judicativa, acaba por concor dar ser esta oração sobretudo](https://img.document.onl/doc/110x75/60804582c098cd50c45b902c/recenso-a-bataillon-marcel-margolin-j-c-macedo-cordata-e-criteriosamente.jpg)







