Embed Size (px)
Citation preview

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Capítulo 3 Conceitos Termodinâmicos
3.1 – Sistemas, Estado e Propriedades Básicas:
3.1.1 – Sistemas:
A Termodinâmica utiliza uma classe de sistemas para desenvolver estudos básicos a nível clássico. O sistema em questão será chamado de sistema simples por estar destituído de limites internos rígidos, adiabáticos e impermeáveis e, ainda, não ser afetado por campos de forças externas ou por forças de inércia.
Outra definição importante a ser feita aqui é a de fase. Entende-se por fase a região dentro de um sistema onde todas as propriedades são uniformes. Assim, o sistema mais simples que se pode imaginar será um sistema unifásico. O sistema pode ser multifásico e mesmo assim ser considerado simples se as fases não tiverem limites adiabáticos, rígidos e impermeáveis com relação às outras fases.
Sistemas Compostos são sistemas formados por dois ou mais sistemas simples sem colocar restrições do tipo de limite que separa os diferentes subsistemas simples. Sistemas Simples Compressíveis são sistemas que contém substâncias simples compressíveis, ou seja, substâncias nas quais efeitos de superfícies, magnéticos ou elétrico não são significativos, mas efeitos causados pela mudança de volume, como por exemplo, expansão, compressão de gases, são muito importantes.
Dentro de um sistema pode haver restrições internas, que são barreiras para prevenir mudanças espontâneas ou para simplificar o estudo de sistemas tais como reações químicas, mudanças de fase, e outras que podem ser aplicadas a sistemas simples. Um exemplo interessante seria a reação, em condições ambientes, de hidrogênio e oxigênio para formar água que pode ser evitada se um catalisador apropriado for incorporado ao sistema, portanto, o catalisador é uma restrição interna. Em sistemas compostos, limites adiabáticos rígidos ou impermeáveis são também restrições internas.
A termodinâmica usa, freqüentemente, o sistema simples compressível, tendo a água como substância de trabalho em suas diferentes fases a fim de estudar leis e conceitos básicos. Nesse caso, a água é considerada substância simples compressível e o sistema é chamado de Sistema Hidrostático Simples (SHSC).
Em termos gerais podemos dizer que a termodinâmica não estabelece os limites que podem estar presentes em um sistema, cabe a usuário dos conceitos e métodos termodinâmicos estabelecer esses limites e restrições. A esse respeito devemos considerar os tipos de limites e restrições mais comuns:
19

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
a) Adiabáticos: São limites que não permitem o intercâmbio de energia em forma de calor.
b) Rígidos: São limites que não permitem mudanças de volume (limites fixos).
c) Diatérmicos: São limites que permitem o intercâmbio de energia em forma de calor.
d) Móveis: São limites que permitem mudanças de volume (limite móveis). Um exemplo seria um pistão de um sistema pistão-cilindro.
3.1.2 – Estado:
A metodologia formal para conduzir experimentos e analisar resultados é definido em condições de estado de equilíbrio estável do sistema, utilizando as propriedades adequadas para reproduzir o comportamento do sistema.
Apesar da dificuldade em definir o número de propriedades que caracterizam um determinado estado, a experiência acumulada em várias centenas de anos, através de dados experimentais, permitem determinar quando um número limitado de propriedades define completamente um determinado estado de equilíbrio estável.
3.1.2.1 – Teorema de Dunhem (postulado I):
“Para sistemas simples compressíveis (SSC), de massa fixa (fechados), com restrições internas definidas, existem estados de equilíbrio termodinâmico que podem ser completamente caracterizados ou definidos por duas propriedades independentes, além da massa e das substâncias utilizadas”.
Para aplicar esse postulado é necessário definir corretamente o conceito de propriedades independentes. Podemos afirmar que para um sistema dado, com restrições definidas, as leis da termodinâmica definidas a partir dos postulados básicos têm, necessariamente, que fornecer informações sobre as relações entre as propriedades, sendo, as relações entre propriedades dependentes dos sistemas e de suas restrições. Portanto, poderemos definir quais propriedades não são independentes. Para um sistema fechado de líquido e vapor em equilíbrio estável, temos como requisito básico de equilíbrio de fases, a pressão de vapor que é função única da temperatura logo, a pressão e temperatura não são independentes.
Saber que o equilíbrio estável existe é importante, mas é mais valioso, do ponto de vista informativo, saber quando ele existe. A esse respeito é bom estabelecer o postulado II da Termodinâmica.
3.1.2.2 – Postulado II:
20

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
“Em processos que não exercem qualquer efeito líquido sobre os arredores (ambiente), todos os sistemas (simples ou composto), com restrições internas especificas, tendem a se aproximar a um, e somente um estado de equilíbrio estável. Na condição limite o sistema como um todo estará em equilíbrio”.
Esse postulado junto com o Teorema de Dunhem aplicados a sistemas simples levam a acreditar que, uma vez atingido o equilíbrio estável, são necessárias somente duas propriedades independentes para definir este estado e, como conseqüência, o restante das propriedades do sistema simples são variáveis dependentes.
Esta análise realizada é valida para sistemas simples únicos e não serão totalmente validos para sistemas compostos, onde os diferentes subsistemas podem ter restrições diferentes e onde os limites de cada subsistema podem permitir o intercâmbio de energia e matéria.
3.1.3 – Propriedades Primitivas:
Propriedades primitivas são definidas em termos das medidas realizadas sobre o sistema em algum instante onde, estas medidas são realizadas de forma a não se perturbar o sistema.
Essas propriedades não estão restritas a estados de equilíbrio estável. Todas as propriedades que não podem ser medidas diretamente serão chamadas de propriedades derivadas, ou propriedades dependentes das primitivas.
3.2 – Densidade e volume específico:A densidade é uma propriedade primitiva que especifica a massa de uma
substância por unidade de volume. Do ponto de vista matemático, a densidade é definida como base um volume elementar da substância v ao redor de um ponto p da substância. A relação entre massa m da substância contida no diferencial de volume v é definida como densidade:
Figura 3.1 – Exemplo de volume limite numa porção contínua de massa
21

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
; (3.1)
Onde v’ é o diferencial limite de volume até onde o sistema pode ser considerado contínuo. O volume específico v, é uma propriedade primitiva definida como o inverso da densidade.
; (3.2)
Tanto a densidade quanto o volume específico são propriedades primitivas intensivas.
3.3 – Pressão:
O conceito de pressão como propriedade primitiva de uma subst6ancia pode ser, didaticamente definido, como sendo as forças específicas que atuam através da superfície livre de uma substância. Desta forma é conveniente definir essas forças por unidade de área. A pressão, portanto, seria a força normal Fn por unidade de área, atuando em algum contorno real ou imaginário, sempre e quando o sistema estiver em equilíbrio estável.
As forças normais podem ser chamadas de esforços compressíveis, onde, do ponto de vista mecânico, atuam sempre na direção da área. Estas forças podem ser completamente descritas em termos das dimensões básicas como: massa, comprimento e tempo. Assim podemos dizer que a pressão pode sempre ser considerada como uma propriedade local e positiva. Se considerarmos uma porção de substância em equilíbrio estável (ex.: um fluido estático), a pressão atuando num ponto p da substância deverá ser a mesma em todas as direções, independente da direção e podemos defini-la como o valor limite da razão entre força normal Fn
atuando nos arredores do ponto p e o diferencial da área A ao redor de p.
(3.3)
22

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.2 – Pressão sobre um fluído em repouso de fluido sob ação de um campo gravitacional
Onde A’ é o diferencial limite da área de uma porção elementar da substância considerada contínua. Consideremos uma substância em equilíbrio estável, se fizermos um balanço das forças que atuam numa porção elementar da substância em direção paralela ao campo gravitacional teremos a situação representada na figura 3.2.
Na superfície livre atua a pressão pA e a força que atua sobre a superfície na posição 1, da porção elementar de formato cilíndrico será p1.dA. A força quer atua sobre a área na posição 2 sobre a porção elementar de cilindro será p2.dA. Onde:
(3.4)
Podemos observar que a pressão não é afetada pela forma da porção elementar, pois, as forças provocadas numa superfície devido a pressão, atuam sempre sobre a projeção da superfície na direção da força (princípio de hidrostática), assim fazendo um balanço de forças na direção horizontal (x ou y dos eixo cartesianos), contatamos que as forças opostas se anulam, portanto fazendo um balanço de forças sobre a porção da substância representada pelo cilindro elementar na direção do campo
gravitacional , teremos: (3.5)
Onde: .dA.dz é o peso do cilindro elementar no campo gravitacional, com aceleração gravitacional , onde:
(3.6)
Sendo o peso especifico da substância. Desta forma simplificando a equação 3.5, teremos como resultado:
(3.7)
Onde: dz = altura do cilindro elementar
23

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
p = pressãoIntegrando a equação 3.5 e considerando a substância como sendo
incompressível ( = cte.), teremos:
Onde : :
(3.8)
A equação 3.8 obtida pode ser usada como base para medidas de pressão de um fluído para outro. Para uma dada diferença de pressão temos:
(3.9)
Onde: h1 = coluna de fluído 1.h2 = coluna de fluido 2.
A equação 3.9 é utilizada como principio de medida dos nanômetros, sendo o mais simples deles o manômetro de tubo em U mostrado na figura 3.3. O principio deste manômetro tem como base o fato de que a pressão num ponto x exercida pela coluna da esquerda é igual a pressão exercida pela coluna da direita.
Figura 3.3 – Manômetro de Tubo em U
Assim: (3.10)
24

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Simplificando a equação acima obteremos:
(3.11)
Podemos fazer algumas considerações como as abaixo:1- Quando 1 é bem menor que 2 (Ex.: fluido 1 é um gás e fluido 2 um
liquido), o efeito de 1 pode ser desprezível.2- Quando 1 tende para o valor de 2, para uma dada diferença de pressão
o valor de h tende a ser muito grande, inviabilizando o manômetro.Pelas razões anteriormente expostas é necessário escolher corretamente os dois
fluídos para se Ter um bom grau de sensibilidade e sempre ter em conta que os fluídos não podem ser miscíveis.
Em resumo, pressão é força por unidade de área:
(3.12)
Em hidráulica, a pressão é o peso específico vezes a altura:
(3.13)
Em processos termodinâmicos, que consideram somente intercâmbios de energia por compressão e expansão de uma substância de trabalho, pressão é o trabalho por unidade de volume.
O conceito de trabalho será explicado em capítulos posteriores; podemos porém, adiantar que trabalho é uma função de trajetória (processo), ou seja, um diferencial inexato, e a pressão pode também ser vista como fator de integração que transforma a função de trajetória w na função pontual (propriedade) dv
(3.14)
3.3.1 – Escalas de Medida de Pressão:
A pressão de um fluído, se mede normalmente com respeito a uma pressão de referência. Se a pressão de referência é pressão nula (ausência de matéria) a esta pressão damos o nome de pressão absoluta.Se a pressão de referência é a pressão atmosférica a esta pressão damos o nome de pressão manométrica ou diferencial. A pressão manométrica pode ser positiva ou negativa dependendo da pressão absoluta ser superior ou inferior a pressão atmosférica.
(3.15)
25

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
As unidades de pressão utilizadas em engenharia são comumente referidas à atmosfera física os standard, que seria a pressão produzida por uma coluna de mercúrio de 760 mm a 0oC e atração normal da gravidade (g = 9.809 m/s2).Podemos fazer a seguintes relações para a atmosfera física:
1 atmosfera física = 1 atm = 760 mmHg = 1,01325 x 105 N/m2 = 1.01325 bar = 1.33227 kgf/cm2 = 10.3327 0H2O
A figura 3.4 mostra a relação entre pressão absoluta e manométrica, onde a
pressão na posição A representa pressão nula (zero), o ponto B representa o nível de pressão atmosférica, podemos observar que a única pressão que não muda com o tempo ou posição sobre a terra seria a pressão absoluta, já que a pressão atmosférica é função de muitas variáveis físicas (altura com relação ao nível do mar, fenômenos meteorológicos, temperatura da terra, etc.).
Figura 3.4 – Escalas de pressões normalmente utilizadas
3.3.2 – Manômetro:
Foi utilizado inicialmente (1662) por Boyle e determina de forma precisa pressões de fluídos estáticos. Ele opera nos princípios da hidráulica e normalmente são chamados de tubo em U. Medem pressões desde 1mm de H2O até a altura tecnicamente possível de ser construído. Sua incerteza é normalmente considerada de 0,2% da leitura.
26

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.5 – Manômetro de Tubo e “U”
O Manômetro consiste de um tubo transparente (vidro) constituído na forma de U, e parcialmente cheio com fluido chamado de fluido manométrico.
O fluido manométrico deve Ter como características a definição clara do seu peso específico, deverá ser imiscível com os fluidos em contato com ele.
Assim a pressão desconhecida a ser avaliada PA, mostrada na figura 3.5, é balanceada pelo peso por unidade de área da coluna de líquido manométrico e pela pressão de referência PB
(3.16)
Correções podem ser feitas tendo em conta:
a) Variações do peso específico do fluído manométrico com a temperatura;
b) Correções por efeito de capilaridade;c) Variações da gravidade local;d) Correções dos pesos específicos devido a efeitos hidráulicos
(recomendamos para leitura sobre estas correções o livro: Fundamentals of Temperature, Pressure and Flow Measurements, 3rd
Edition, by Robert P. Benedct, 1984 páginas 300, 301, 302 e 303).
3.3.3 – Micromanômetros
27

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
São manômetros que estendem a capacidade do manômetro convencional de tubo em U para medir pressões muito pequenas, da ordem de 0,005 mmH2O, até 500 mmH2O. O micromanômetro mais conhecido é o tipo Prandtl.O micromanômetro de Prandtl uma vez aplicada uma diferença de pressão, tanto o recipiente quanto o tubo inclinado podem ser movidos de forma precisa até que o menisco esteja em posição nula.
Figura 3.6 – Micromanômetro de Prandtl
3.3.4 – Barômetro
O Barômetro utilizado em seus primórdios por Torricelli (1643) serve para determinar a pressão atmosférica com incertezas de calibração de 0,01 a 0,03% da leitura.O princípio de operação consiste de uma coluna de mercúrio tendo como referência à pressão nula em seu extremo cego imersa num recipiente com grande diâmetro e aberto ao ambiente.O tipo mais comum é mostrado na figura 3.7, onde o nível de mercúrio no recipiente pode ser ajustado para assim fixar o zero de referência. O tubo de metal que contém o tubo de vidro é fixo rigidamente ao recipiente, permitindo observar a leitura da coluna de mercúrio. Para se fazer uma boa leitura e obter com boa precisão para a pressão atmosférica é necessário fazer correções:
a) Correções por temperatura,b) Correções por gravidade,c) Correções por visibilidade (alinhamentos),d) Correções por efeitos capilares.Para maiores informações a respeito das correções, consultar Fundamentals of Temperature, “Pressure and Flow Measurements”, 3o edition, Robert P. Benedict, 1984.
28

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
3.4 – Temperatura
Tomando como base George Sarton, historiador em ciência da origem Americana que diz: “Ninguém deverá ser reconhecido como mestre em qualquer área, se não conhece no mínimo as origens de sua história.”, portanto, para se entender as medidas das propriedades primitivas e especialmente medidas de temperatura que é uma propriedade reconhecida como básica em qualquer ciência (termodinâmica, mecânica dos fluídos, transferência de calor, aeronáutica, ciência do espaço, química e física ) é necessário, para que não se perca nos conceitos, um pouco de história.
Se retornarmos no tempo até onde nossa mente alcança, o ser humano esteve sempre consciente da existência do calor (sol escaldante, água fria, desertos infernais, gelo paralisante), mas os sentidos humanos nunca puderam nem poderão distinguir valores extremos de calor ou frio fora de uma faixa bem limitada, que causa nos seus extremos limitados a sensação de dor.
Figura 3.7 – Barômetro tipo Fortin-type(fonte: ASTM PTC 19.2 [3])
As primeiras tentativas para avaliar (medir) graus de calor pertenceu a Galileo Galilei, nos anos de 1592, quando Galileu era mestre de matemática em Pádua, Itália,
29

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
e inventou o termômetro de vidro contendo ar e água, que era afetado pela pressão atmosférica e que hoje em dia são chamados de “Barotermoscópios”.
A palavra termômetro apareceu pela primeira vez na literatura em 1624, num livro de J. Leurechan (La Récréation Mathématic) o autor descreve um termômetro como “um instrumento de vidro com um pequeno bulbo, um pequeno pescoço e um tubo liso suave e no final um recipiente pequeno cheio de água. Aqueles que desejam determinar mudanças em forma de números tem que fazer uma leitura ao longo do tubo e dividi-la em 8 graus de acordo com o recomendado pelos filósofos”.
Mais tarde Ferdinand, duque de Tuscany (1654), Robert Hooke (1664), Cristian Huygen (1665), Robert Boyle (1665) expressaram suas opiniões a respeito de frio e quente mais ou menos assim: é necessário, de forma universal e padronizada, determinar calor e frio, fazendo uma proporção definida entre a capacidade do bulbo e suas dimensões (termômetro de vidro), descobrindo as origens do frio nas quais a água começa a congelar ou a água começa a ferver (primeira tentativa de definir pontos fixos de temperatura).
Foi Daniel Gabriel Farenheit (1706) que com instrumentos por ele desenvolvidos em Amsterdã, Holanda, fixou a escala de temperatura Farenheit. Onde, foi definido o ponto de fusão e de ebulição da água como sendo 32º F e 212º F, respectivamente.
Mais tarde Anders Celsius (1745), professor de Astronomia na Universidade de Upsala, propôs uma escala fazendo zero o ponto de fusão do gelo e cem o ponto de ebulição da água, esta foi chamada Centígrado ou Celsius.
Ao longo do tempo foi passivamente aceita a definição de um grande número de pontos fixos minuciosamente definidos. No final do século XVIII existiam milhares de instrumentos que permitiam fazer interpolações entre as temperaturas de pontos fixos, assim como milhares de substâncias termométricas e de forma geral era possível notar que os termômetros graduados existentes quando atingiam o equilíbrio térmico, não forneciam a mesma indicação de temperatura.
3.4.1 - Lei Zero da Termodinâmica
Neste ponto se faz necessário definir a temperatura como uma propriedade termométrica, a partir do conceito de equilíbrio térmico, utilizando a lei zero da termodinâmica que diz:
“Quando dois corpos estão em equilíbrio térmico com um terceiro, os três estão em equilíbrio térmico entre si”.
A figura 3.8 ilustra esta lei, da seguinte forma: suponhamos que os corpos A e B estão em equilíbrio térmico; vamos então colocar o corpo A em contato com o outro corpo C, se observarmos que as propriedades primitivas de A e C não mudam com o tempo, podemos afirmar que os três corpos estão em equilíbrio térmico entre si.
30

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.8 – Corpos A, B e C em equilíbrio térmico
A lei zero é a base para se medir temperaturas, que exigem o conceito de Equilíbrio térmico entre dois corpos. Podemos concluir então que quando dois corpos estão em equilíbrio térmico, eles devem compartilhar uma propriedade que indique ou se relacione com este estado de equilíbrio. Esta propriedade é chamada de temperatura. Medir temperaturas requer a definição de escala de temperaturas que fazem atribuição de valores numéricos aos diferentes estados térmicos das substâncias (pontos fixos).
Normalmente as escalas usam como referência pontos fixos ou estados térmicos fixos como nas escalas definidas por Farenheit e Celsius; as temperaturas assim medidas são conhecidas como temperaturas empíricas (t).
Pontos Fixos Valor NuméricoPonto de Oxigênio (temperatura de equilíbrio entre o oxigênio líquido e seu vapor) -182,962Ponto Triplo da Água (temperatura de equilíbrio entre gelo, água líquida e vapor) 0,01Ponto de vapor (temperatura de equilíbrio entre água líquida e seu vapor) 100Ponto do zinco (temperatura de equilíbrio entre o zinco líquido e sólido) 419,58Ponto da prata (temperatura de equilíbrio entre a prata líquida e sólida) 961,93Ponto do ouro (temperatura de equilíbrio entre o ouro líquido e sólido) 961,93
Tabela 3.1 – Pontos fixos definidos em 1968 pelo comitê assessor em termometria da conferência geral de pesos e medidas (IPTS-68). Os valores numéricos correspondem a escala Celsius de temperatura e todos os pontos
foram fixados a pressão atmosférica Standard (760 mmHg), com exceção do ponto tríplice da água.
3.4.2 – Escalas de Temperaturas
As escalas para se medir temperaturas empíricas tem como base qualquer ponto fixo conveniente, mas dependem da escolha da substância termométrica, da propriedade termométrica, e da função que relaciona a temperatura empírica com a mudança da propriedade termométrica.
A escala é arbitrária até o ponto de ser necessário escolher uma constante multiplicadora para poder fixar um e somente um número para cada ponto fixo.
31

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Desta forma uma vez um número fixado para um estado termométrico arbitrário (273 K ponto do gelo) e uma vez uma diferença de temperatura e designada entre dois pontos fixos de referência (Ex: 100O C para Tvapor – Tgelo), todo o resto de temperatura nas escalas de temperatura assim definidas podem ser determinadas.
É necessário neste ponto definir escala, de temperaturas absolutas:“A temperatura na escala absoluta não depende de qualquer suposição relativa
à pressão, ou a volume de uma substância (Ex.: pressão a volume zero é zero absoluto de temperatura), não depende da existência do hipotético gás ideal, não envolve qualquer estamento, ou postulado acerca da ausência de movimento molecular a zero absoluto e não pressupõe que a temperatura de zero absoluto seja a menor possível de ser obtida.”
3.4.3 – Escalas Empíricas Normatizadas
As escalas de temperaturas usadas como ‘normas’, foram fixadas a partir de acordos internacionais que são chamadas International Temperature Scales (ITS), seguidos de um número que geralmente é o ano no século XX. Assim, ITS-27 é a escala definida pela conferência internacional de pesos e medidas de 1927. A escala ITS-27 foi definida de forma que fosse facilmente reproduzida e coincidente quanto possível com a escala de temperaturas termodinâmicas. Normalmente estas escalas tem um número mínimo de pontos fixos (ITS-27 tem seis pontos fixos), aos quais foram fixados valores na escala Celsius, sendo a escala a seguir dividida em faixas de temperaturas que tem como base os meios disponíveis de interpolação.
3.4.4 – Escala ITS – 27:
1ª faixa: -190 A 0ºC. Esta faixa foi definida como a resistência medida pelo termômetro de platina normatizado, sendo um polinômio de 4º grau, único, que passa pelos valores de temperatura dos pontos fixos do oxigênio, gelo, vapor de água e enxofre.
2ª faixa: 0 a 660ºC. Esta faixa foi definida pela resistência medida pelo termômetro de platina normatizado sendo a parábola única que passa pelos valores de temperatura dos pontos fixos de gelo, vapor de água e enxofre.
3ª faixa: 660 a 1063ºC. Esta faixa foi definida pela f.e.m. (força eletro-motriz) de um termopar platino (10%)-ródia e platina normalizados, sendo uma parábola única que passa pelos valores de temperatura dos pontos fixos de antimônio, prata e ouro.
4ª faixa: Acima do ponto do ouro. A temperatura foi definida pela radioatividade medida pelo Pirômetro Ótico normatizado. A energia radiante de um corpo negro a uma temperatura desconhecida com comprimento de onda conhecido pode ser comparada com a temperatura de um corpo negro com comprimento de onda similar a emitida no ponto do ouro, ou seja, pela lei de Wein:
32

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
(3.17)
Onde é a radiosidade de um corpo negro, com comprimento de onda a uma temperatura T, esta equação é usada para extrapolar temperaturas nas faixas da tabela 3.4.
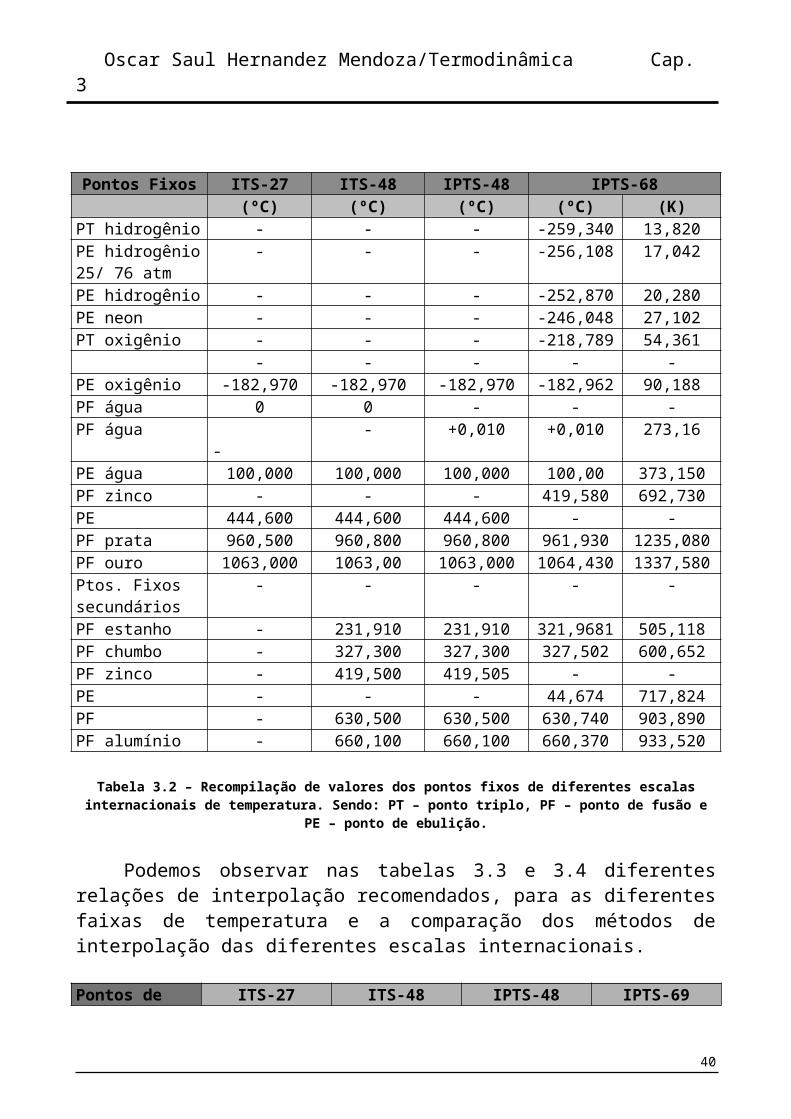
Pontos Fixos ITS-27 ITS-48 IPTS-48 IPTS-68(ºC) (ºC) (ºC) (ºC) (K)
PT hidrogênio - - - -259,340 13,820PE hidrogênio 25/ 76 atm
- - - -256,108 17,042
PE hidrogênio - - - -252,870 20,280PE neon - - - -246,048 27,102PT oxigênio - - - -218,789 54,361
- - - - -PE oxigênio -182,970 -182,970 -182,970 -182,962 90,188PF água 0 0 - - -PF água - - +0,010 +0,010 273,16PE água 100,000 100,000 100,000 100,00 373,150PF zinco - - - 419,580 692,730PE 444,600 444,600 444,600 - -PF prata 960,500 960,800 960,800 961,930 1235,080PF ouro 1063,000 1063,00 1063,000 1064,430 1337,580Ptos. Fixos secundários
- - - - -
PF estanho - 231,910 231,910 321,9681 505,118PF chumbo - 327,300 327,300 327,502 600,652PF zinco - 419,500 419,505 - -PE - - - 44,674 717,824PF - 630,500 630,500 630,740 903,890PF alumínio - 660,100 660,100 660,370 933,520
Tabela 3.2 – Recompilação de valores dos pontos fixos de diferentes escalas internacionais de temperatura. Sendo: PT – ponto triplo, PF – ponto de fusão e PE – ponto de ebulição.
Podemos observar nas tabelas 3.3 e 3.4 diferentes relações de interpolação recomendados, para as diferentes faixas de temperatura e a comparação dos métodos de interpolação das diferentes escalas internacionais.
Pontos de comparação
ITS-27 ITS-48 IPTS-48 IPTS-69
1ª Série – Instrumentos de interpolação: resistência termométrica da platinaLimites de temperatura
-190 a 0ºC -182,970 a 0ºC -182,970 a 0ºC 13,81 a 273,15 K
33

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Relação de interpolação
Callendar-Van Dusen
Callendar-Van Dusen
Callendar-Van Dusen
Função de referência
2ª Série – Instrumentos de interpolação: resistência termométrica da platinaLimites de temperatura
0 a 660ºC 0 a 630,5ºC 0 a 630,5ºC 0 a 630,74ºC
Relação de interpolação
Callendar(parábola)
Callendar(parábola)
Callendar(parábola)
Modified-Callendar
3ª Série – Instrumentos de interpolação: termopar de platina-10%ródio e platinaLimites de temperatura
660 a 1064ºC 630,5 a 1063ºC 630,5 a 1063ºC 630,74 a 1064,43ºC
Relação de interpolação
Parábola Parábola Parábola Parábola
4ª Série – Instrumentos de interpolação: pirômetro ópticoLimites de temperatura
Acima de 1063ºC Acima de 1063ºC Acima de 1063ºC Acima de 1064,43ºC
Relação de interpolação
Lei de Wien Lei de Planck Lei de Planck Lei de Planck
Tabela 3.3 – Comparação de faixas de temperaturas e métodos de interpolação de diferentes escalas internacionais de temperaturas.
Itens de comparação
ITS-27 ITS-48 IPTS-48 IPTS-68
Resistência termométrica standart da platinaR100/R0 1,390 1,3910 1,3920 1,3925
Termopar standart de platina-10% rádio e platinaR100/R0 1,390 1,3910 1,3920 1,3920
EAu 10,300 50 V 10,300 50 V 10,300 50 VEAu-EAg 1185+0,158
x(EAu-10,310)5 V
1185+0,158x(EAu-10,310)
1185+0,158x(EAu-10,310)
EAu-ESb 4776+0,631x(EAu-10,310)
4766+0,631x(EAu-10300)
4766+0,631x(EAu-10,300)
Pirômetro óptico Standart Segunda constante de radiaçãoC2(m-K) 0,01438 0,01438 0,01438 0,014388
Tabela 3.4: Comparação de instrumentos de interpolação proposta pelos diferentes escalas de temperaturas internacionais
As tabelas 3.2, 3.3 e 3.4, mostram os pontos fixos definidos pelas ITS-27, ITS-48, IPTS-48 e IPTS-68 que são escalas de temperaturas internacionais recomendadas pelo Comitê Internacional de Pesos e Medidas (CIPM).Estas escalas têm algumas diferenças que se encontram especificados com mais detalhe no livro “Instruments of Temperature, Pressure and Measuremeants”, de Robert P. Benedict, 1984. Atualmente temos a IPTS-88 que foi elaborada pelo comitê consultivo em termometria do CIPM, que substituiu a IPTS-68.
34

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
A idéia deste comitê de definir uma escala que seja mais coincidente com as temperaturas termodinâmicas surgiu da necessidade de se evitar discrepância em nível de termometria de gases e de termometria óptica. A termometria de gases é utilizada até 500ºC onde as diferenças com as escalas de temperaturas termodinâmicas são da ordem de 0,1ºC. A termometria óptica é utilizada em temperaturas até 800ºC e acusa diferenças da ordem de 0,5ºC. Estes e outros problemas são resolvidos com a IPTS-88.
3.4.4.1 – Métodos de Medidas de Temperatura
Devemos sempre ter em mente que a temperatura de um objeto ou de um fluido não pode ser medida diretamente, comparando seu valor com um valor padrão, como se faz por exemplo com o comprimento. Normalmente, medidas de temperatura são feitas medindo-se a mudança de alguma propriedade física escolhida mude proporcionalmente com a variação de temperatura e que não ocorra histerese, ou seja, que a medida seja sempre bem reduzida. As propriedades físicas normalmente utilizadas são:
1. Expansão térmica2. Efeitos termoelétricos3. Resistência elétrica4. Cores de superfície5. Radiação térmica6. Pontos de fusão, ebulição e congelamento.
3.4.4.2 - Termômetros de expansão volumétrica:
O mais comum é o termômetro de vidro utilizando mercúrio como substância termométrica. Estes termômetros normalmente trabalham na faixa de temperaturas de -30ºC a 350ºC. Se quisermos aumentar a faixa de medida podemos incrementar a pressão acima da coluna de mercúrio introduzindo nitrogênio pressurizado e utilizando vidro temperado para resistir a altas temperaturas. Quando desejamos utilizar termômetros de vidro para medir temperaturas abaixo de –30ºC poderemos utilizar fluidos tais como álcool (-117ºC a 250ºC) ou pentano.
Normalmente estes termômetros são utilizados para medir temperaturas de fluidos em repouso ou fluidos em movimento utilizando fossas termoelétricas (doing de gent) como mostrando na figura 3.9.
35

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.9 – termômetro de vidro instalado em fossa termométrica.
Os problemas apresentados por estes termômetros são a sua fragilidade, condução de calor através das fossas termoelétricas, a grande capacidade térmica deste tipo de termômetros o qual faz deles mais lentos de se medir temperatura, projeto geométrico adequado das fossas termométricas (diâmetro, comprimento, material).
Estes termômetros normalmente são calibrados para imersão total ou parcial. No caso de imersão parcial os termômetros possuem um anel que indica até onde o termômetro deverá ser mergulhado, da mesma forma é necessário Ter cuidado de se saber qual deverá ser a temperatura ambiente onde deverá permanecer a parte não mergulhada do termômetro. Os termômetros de imersão total são calibrados para serem totalmente mergulhados no líquido ou fossa termométrica onde desejam medir a temperatura, como mostrado na figura 3.10.A associação americana de engenheiros mecânicos (ASME) na sua publicação de normas sobre ensaios de imersão total, caso esteja sendo utilizado em imersão parcial, pela seguinte equação (equação válida somente para termômetros com mercúrio como substância termométrica).
(3.18)
Onde em ºF, D é o comprimento indicado na figura 3.9 medido em graus, e t1 e t2 são as temperaturas em ºF do termômetro calibrado para imersão total e o termômetro acoplado para fazer a correção como mostrado até que o resíduo de cálculo seja inferior a 0,5 ºF.
36

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.10 – Utilização de termômetro calibrado para imersão total em caso de imersão parcial
Figura 3.11 – Indicador de temperatura do tipo bobina bimetálica (Westonn electrical instrument corp.)
37

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
3.4.4.3 – Termômetros de expansão fluidos:
O princípio de operação destes termômetros é a expansão de um fluido (fase líquida, vapor ou gás). O termômetro é composto de um bulbo cheio de fluido que é elemento transmissor e um elemento que responde à mudança de pressão do tipo Bourbon na forma de hélice ou espiral. Dependendo da faixa de temperatura desejada, escolhemos o fluido e a fase em que deverá estar dentro do termômetro. O único cuidado recomendado no uso deste termômetro, quando utilizar fluido em fase líquida, é que exige a posição vertical do bulbo e indicador pis a quantidade de líquido no capilar será suficiente para afetar a leitura da hélice. A figura 3.12 mostra diferentes tipos de termômetros de expansão de fluido.
Figura 3.12 – Termômetros de expansão de fluidos (Courtesy foxboro Co.)
3.4.4.4 – Termômetros de resistência:
Este termômetro é o mais efetivo e preciso para se medir temperaturas até 150ºC, tem como vantagem o fato de poder ser mostrado à distância, não requerendo junta de referência, e pela sua origem elétrica é o termômetro mais indicado para medir temperaturas médias de grandes espaços.
O princípio de operação do termômetro é a mudança de resistência elétrica com a variação de temperatura. O termômetro está basicamente composto de um elemento resistente, normalmente um fio de níquel, platina ou cobre, com seus extremos eletricamente ligados a m instrumento de medida (milivoltímetro).
38

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
A figura 3.13 a, b e c, mostram diferentes arranjos de montagem de termômetros de resistência.
Figura 3.13 – arranjos diversos para termômetros de resistência
No arranjo da figura 3.13 a, a resistência do bulbo é obtida substituindo a resistência de bc, da resistência ad; no caso da figura 3.13 b, a resistência do bulbo é obtida substituindo a resistência bc de ac ou ab; no termômetro da figura 3.13 c, supõe-se que a resistência dos fios ab e cd são iguais, depois aplicados o mesmo procedimento dos casos a e b.
Normalmente estes termômetros eram operados pelo método da ponte de Wheastone, como mostrando na figura 3.14.
Figura 3.14 – Diagrama esquelético de um termômetro de resistência de leitura direta.
Na montagem mostrada na figura acima, uma resistência conhecida e fixa C e juntamente com uma outra resistência variável D colocada no circuito da bateria são a base principal de operação do termômetro. O circuito é periodicamente ajustado. Colocando-se o interruptor SW na posição 1 e, ajustando-se a resistência D até que o medidor analise uma leitura de referência previamente fixada. Feito isso o interruptor é colocado na posição 2 e o instrumento estará pronto para ser usado. Se leituras diferentes da previamente fixada são verificadas, é uma indicação de quebra do balanço do circuito causado pela mudança de resistência do bulbo sensor.
39
Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Hoje existem instrumentos em forma de pequenas placas autocolantes em tamanhos minúsculos (5x5mm) ou pequenos semicondutores que podem ser monitorados por placas de aquisição de dados acoplados a computadores pessoais (PC’s).
3.4.4.5 – Pirômetros Termométricos (Termopolar):
Este é o termômetro mais usado devido a sai simplicidade, confiabilidade, forma de operação e de sua construção ser relativamente simples. O efeito foi descoberto por Seebeck em 1821 e ele é provocado quando dois metais diferentes são unidos e aquecidos. A figura 3.15 mostra um Termopar simples que é composto de dois metais diferentes A e B, unidos térmica e eletricamente nos pontos X e Y e tendo como agente intermediário um aparelho de medida de voltagem.
Figura 3.15 – Circuito de um Termopar com um terceiro metal.
Na figura 3.15 a junta X poderá ser colocada no lugar onde desejamos medir a temperatura e a junta Y chamada de junta referência pode ser mantida a uma temperatura constante bem conhecida (ponto do gelo). A força eletromotriz (fem) originais dessas duas juntas em duas diferentes fontes é chamada de Peltier e de efeito Thomson. Podemos dizer que a força eletromotriz de Peltier é a porção de fem total produzida pela diferença de potencial que existe num condutor simples que está submetido a um gradiente de temperatura.Os termopares obedecem à lei dos metais intermediários que diz: “Qualquer terceiro metal pode ser introduzido num circuito tipo Termopar sem afetar a fem gerada se seus pontos de entrada e saída estão à mesma temperatura. A figura 3.16 mostra a lei dos metais intermediários atuando num circuito simples de Termopar”.
40

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.16 – Termopar com ponto do gelo como referência.
No caso da figura 3.16 o metal A e o cobre e o metal B juntamente com o cobre se encontram em contato térmico (localizados numa região de temperatura conhecida), para que a lei dos metais intermediários possa ser aplicada duas conexões deverão estar eletricamente isoladas.
É desejável que o Termopar produza uma fem grande na sua faixa de operação e que não tenha efeitos de histerese e que a relação fem versus temperatura seja linear. A figura 3.17 mostra esta relação para os metais mais comumente utilizados para se fabricar termopares com temperatura com de referência igual a 0O C. A força eletromotriz gerada por qualquer das combinações escolhidas será a diferença de fem do par escolhido quando se encontra a mesma temperatura (distância vertical entre o par de metais escolhidos da figura 3.17).
Existem tabelas de conversão que fornecem a temperatura correspondente aos milivolts gerados pelos vários tipos de juntas termopares, baseados normalmente na junta de referência de 0OC.
41

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.17 – Força eletromotriz gerada por diferentes termopares, relativas ao termopar de platina.
3.5 – Substâncias Puras
3.5.1 – Definição:
Uma substância pura é aquela que tem uma composição química homogênea. Pode existir em mais de uma fase, porém sua composição química é a mesma em todas as fases não sendo afetada em seu comportamento por campos elétricos, magnéticos, ou efeitos de superfície (tensão superficial). O comportamento das substâncias puras que são utilizados amplamente no estudo da termodinâmica clássica será realizado na seguinte ordem:
1- Estudo de fases, propriedades e estados;2- Estudo da água como substância pura modelo, utilizando um sistema
fechado de massa fixa do tipo pistão-cilíndrico, mostrando o seu comportamento em diagramas P-v e p-T;
3- Métodos utilizados para obter informações sobre as relações das propriedades primitivas p,v e T:
a) Tabelas de vapor;b) Equações de estado para gases e vapores;
42

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
c) Diagramas de compressibilidade.A importância do fluido puro no estado da termodinâmica clássica é devido ao
faro de que qualquer fluido considerado como quase puro poder ter seus estados termodinâmicos completamente definidos por suas propriedades primitivas independentes sem, contudo, seu estado ser afetado por efeitos de superfície, gravidade, capilaridade, eletricidade ou magnetismo.
A termodinâmica utiliza a água como fluido modelo quase puro por ser uma substância que é conhecida por todos e pela facilidade em observar o comportamento dela em diferentes fases (sólida, líquida e gasosa). O mais importante é ter em mente que o resto das substâncias existentes na natureza têm comportamento análogo ao da água.
3.5.2 – Fases:
Em geral, uma substância pura pode existir em qualquer uma das três fases conhecidas: fase sólida, fase líquida e fase gasosa ou de vapor. Em condições particulares as três fases podem coexistir não sendo raro a mistura de duas fases.
Existem os seguintes processos de mudança de fase de uma substância pura:- Fusão: mudança da fase sólida para a líquida;- Solidificação: mudança da fase líquida para a sólida;- Vaporização: mudança da fase líquida para a gasosa;- Condensação: mudança da fase gasosa para a fase líquida;- Sublimação: transformação direta da fase sólida para a fase gasosa sem que
passe pela líquida. A transformação inversa também é denominada sublimação.Podemos verificar a existência de fases de uma substância pura, sendo que
estas devem necessariamente, homogêneas. Por exemplo, a fase sólida submetida a diferentes pressões pode apresentar fases distintas.
Foram descobertas oito fases de uma água sólida por Bridgman a aproximadamente 19423,8 bar (282000 lb/pol2 ) , neste caso a água se solidifica a 52OC e é uma fase diferente do gelo que conhecemos.
Vapor, Gás e Gás Ideal;Vapor é o nome que se dá a uma fase gasosa que está em contato com a fase
líquida e esta está na eminência de um estado em que parte do mesmo pode condensar-se. O vapor é um gás imperfeito porque suas moléculas sofrem os efeitos de atração e repulsão por estarem muito perto uma das outras. As propriedades primitivas do vapor não podem relacionar-se simplesmente, como é de se esperar numa equação de estado (f(p,v,T) =0), que não é mais do que a relação matemática entre as propriedades primitivas.
Um gás é um vapor altamente superaquecido a baixas pressões e seu estado de equilíbrio está longe do estado de saturação. Já um gás ideal é um vapor altamente superaquecido em que as interações entre suas moléculas são do tipo colisão elástica.
Definições sobre comportamento das substâncias puras:
43

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
1)Temperatura de saturação: é a temperatura na qual se efetua a mudança de fase: líquido-vapor ou sólido-líquido.
2)Pressão de saturação: é a pressão na qual se efetua a mudança de fase: líquido-vapor ou sólido-líquido.
3)Ponto tríplice (PT): é o estado no qual coexistem as três fases em equilíbrio. As substâncias puras simples tem dependência definida entre pressão e temperatura de saturação e as curvas geradas num diagrama PsxTs são chamadas de curvas limites entre os domínios de fases simples (como exemplo podemos citar a curva entre as fases líquida e de vapor que é chamada de pressão de vapor).
4)Ponto crítico (C): acima deste ponto a substância não consegue mudar de fase líquida para vapor e vice-versa.
5)Líquido saturado: é a substância simples pura em fase líquida a pressão e temperatura de saturação.
6)Líquido subesfriado: é a substância simples pura em fase líquida a temperatura inferior à de saturação a uma dada pressão.
7)Líquido comprimido: substância simples pura em fase líquida a temperatura inferior a de saturação a uma dada pressão.
8)Título de vapor (x): é a razão entre massa de vapor e massa de líquido, que existe quando uma substância simples pura tem as duas fases (líquido e vapor) coexistindo em equilíbrio termodinâmico em estado de saturação.
9)Vapor de saturação: é a substância simples pura em fase de vapor a pressão e temperatura de saturação.
10)Vapor superaquecido: é a substância simples pura em fase de vapor a temperatura superior à temperatura de saturação a uma pressão dada.
3.5.3 – Diagrama p -T :
O diagrama de pressão (p) versus temperatura (T) tem muita importância em termodinâmica, pois, explica as fronteiras existentes entre fases e quando é possível mudar de fases, assim como os estados de equilíbrio que permitem a presença de mais de uma fase. Imaginemos um processo a pressão constante de ma substância pura, representado pela linha horizontal ituzn, (veja figura 3.18), partimos de um estado i na fase sólida e fornecemos calor à substância até chegar ao estado t onde começa a fusão (se a substância se dilata ao congelar-se, como a água, o ponto de fusão dar-se-á no estado u). Se continuarmos fornecendo calor à substância a pressão e temperatura permanecem constantes enquanto houver a presença das duas fases (sólida-líquida). Este calor fornecido neste período é chamado calor latente de fusão. Uma vez a fase sólida tenha desaparecido a substância estará em fase líquida. Se continuarmos fornecendo calor à substância observaremos que a temperatura começa novamente a aumentar, até chegar ao estado de equilíbrio z. Neste ponto observamos que começa a aparecer a fase vapor e novamente a pressão e temperatura permanecem constantes enquanto as duas fases coexistirem. O
44

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
calor fornecido neste período é chamado calor latente de vaporização. Uma vez que a fase líquida desaparece totalmente a substância estará na fase de vapor. Se continuarmos fornecendo calor a substância não mudará mais de fase e passará pelos estados de vapor superaquecido, gás real e, finalmente, seu comportamento poderá se aproximar do gás ideal, que será definido na seção de equações de equações de estado.
Figura 3.18 – diagrama p-T da água
É conveniente notar que as mudanças sólido-líquido e líquido-vapor tem limites. Os limites mais conhecidos são o do estado crítico (limite superior da mudança líquido-vapor) e o ponto tríplice (limite inferior desta mesma mudança). Podemos afirmar que acima do ponto crítico a substância não comporta mudanças líquido-vapor e abaixo do ponto tríplice a substância faz o processo de sublimação. Observamos que a linha compreendida entre o ponto tríplice e o ponto crítico é chamado de vaporização, pois ela comporta a substância no estado de líquido saturado, vapor saturado ( ou vapor saturado seco), e a mistura dos dois. O mesmo podemos dizer da linha de fusão, que contém a substância como líquido saturado, sólido saturado ou a mistura dos dois. A linha de sublimação, localizada abaixo do ponto tríplice, divide a fase sólida e a fase de vapor além de conter a substância na fase sólida saturada, vapor saturado ou a mistura das duas fases.
Do ponto de vista do engenheiro mecânico, interessam os estados da substância na forma fluida (fase líquida, fase vapor), por várias razões:
a) A substância na fase líquida pode ser conduzida através de condutos e não apresenta resistência à deformação (fluido). Isso representa facilidade nos processos de troca de energia em forma de calor, processos de transformação
45

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
de energia potencial em cinética e vice-versa (trocadores de calor, turbinas hidráulicas, bobas).
b) a substância na fase vapor apresenta baixa densidade, grande volume específico e pode conter altos níveis de acumulação de energia térmica, o que permite o seu uso em máquinas térmicas geradoras de trabalho mecânico (turbinas de vapor, sistema pistão-cilíndrico) e processos alimentícios e industriais que tenham limites em seus níveis de temperaturas.
3.5.4 – Regras das Fases:
O professor J. Willard Gibbs da Universidade de Yale desenvolveu a regra das fases, que é aplicável a substância que mudam de fase sem realizar reações químicas.
(3.19)Onde:
= número de fases presentes em equilíbrio,= grau de liberdade, ou seja, o número de propriedades intensivas
independentes que fixam o estado de equilíbrio, = número de componentes presentes na substância.
Seja uma substância pura ( = 1,0) que se encontra na fase líquida ( =1) e portanto, =2 então, deveremos ter duas propriedades intensivas ou apenas uma , respectivamente, for necessária para determinar o estado de equilíbrio do sistema. Caso apliquemos a regra de fases ao ponto tríplice observaremos que
=0, ou seja, a mudança de qualquer propriedade faz a substância sair do ponto tríplice.
46

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Figura 3.19: diagrama de uma substancia pura mostrando processos isobáricos e isotérmicos
Para observar melhor o que acontece durante o processo de mudança de fase utilizarem um sistema pistão-cilindro, no qual existe água e a mesma é considerada substância quase pura. Vamos seguir o mesmo processo relatados anteriormente (processo isobárico); faremos a representação desses processos no diagrama pressão versus volume específico. No diagrama da figura 3.19 observamos que do estado i até t (estado onde começa a fusão) um volume específico muda ligeiramente e, como tínhamos observado anteriormente, a temperatura também muda, o que significa serem as propriedades p,v e T (fase líquida). Durante o processo de fusão t'- t" é uma linha de pressão e temperatura constantes (isobárica e isotérmica). O processo t"- z" é o caracterizado, novamente, pela independência da propriedade p,v e T (fase líquida). De novo, podemos observar que quando começa o processo de vaporização (z') a pressão e temperatura são dependentes até o estado z" onde a substância chega à fase de vapor.
Observamos na figura 3.19 a isobárica it' t" z' z" e as isotermas T1,T2, T3 e T4 que são caracterizadas por terem pontos de inflexão quando a substância começa ou termina as mudanças ( co t' t" z' z" ). O diagrama P-v representa todos os estados da substância quando ela realiza processos de fusão, vaporização e sublimação. Podemos então definir os seguintes lugares geométricos:
a) O lugar geométrico acima do ponto de todos os estados no qual o sólido começa a fusão ou termina o processo de solidificação (similares ao estado t"), será chamado de linha de sólido saturado relativo à fusão ou solidificação.
b) O lugar geométrico acima do ponto tríplice de todos os estados no qual o sólido termina o processo de fusão começa o processo de solidificação (similares ao estado “t”), será chamado de linha de líquido saturado relativo à fusão ou solidificação.
47

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
c) O lugar geométrico acima do ponto tríplice até o ponto crítico de todos os estados no qual o líquido começa vaporização ou termina o processo de condensação (similar ao estado “z”), será chamado de linha de líquido saturado relativa a vaporização ou condensação.
d) O lugar geométrico acima do ponto tríplice até o ponto crítico de todos os estados no qual o líquido termina a vaporização ou começa o processo de condensação (similares ao estado “z”), será chamado de linha de vapor saturado relativa a vaporização o que ou condensação.
e) O lugar geométrico abaixo do ponto tríplice de todos os estados no qual o sólido inicia o processo de sublimação é chamado de linha de sólido saturado relativa a sublimação.
f) O lugar geométrico abaixo do ponto tríplice de todos os estados nos quais o sólido termina o processo de sublimação é chamado de linha de sólido saturado relativa a sublimação.
Observemos que entre o sólido saturado e líquido (fusão) duas fases coexistem em equilíbrio térmico-mecânico, ou seja, termodinâmico. Neste processo de fusão, pressão e temperatura são propriedades dependentes, assim para definir o estado da substância é necessário definir outra propriedade que seja indicativa da quantidade mássica de cada uma das fases coexistindo em forma de mistura.
Levando em conta que numa mistura sólido-líquido saturado o volume total (propriedade extensiva) da mistura é a soma do volume ocupado pelo sólido saturado mais o volume ocupado pelo líquido saturado, logo:
(3.20) Mas:
(3.21)
Da mesma forma que foi definido o volume da mistura sólido - líquido do () saturado no processo de fusão, podemos definir o volume específico da mistura líquido-vapor saturado no processo de vaporização. Definindo:
(3.22)
Nesse caso é definido o título do vapor (x) como se segue:
Assim: (3.23)
O mesmo procedimento pode ser aplicado nós processos de sublimação. Para que ressaltar neste ponto que as fases sólidas, líquida ou gasosas em processo de
48

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
mudança de fase conservam suas propriedades dos respectivos estados de saturação representados pelas linhas de sólido saturado, linha de líquido saturado (fusão ou vaporização), que linha de vapor saturado, respectivamente. Fica, também, claro que no ponto crítico deixa de ter sentido o título do vapor, portanto este seria o ponto de origem das linhas de título constantes. Cabe também aqui, esclarecer que qualquer propriedade das substâncias puras intensiva, em mistura de fases, pode ser calculada seguindo o mesmo procedimento empregado para calcular o volume específico, tendo como resultado final equações similares a equação 3.24, em que o valor total da propriedade intensiva é uma função dos valores da propriedade nos estados de saturação e do título, respectivamente.
Figura 3.20: superfícies termodinâmicas representando mudanças de fase de uma substância pura.
Para melhor entendermos o os diagramas p-T e p-v representaremos estes diagramas na figura 3.20 de forma tridimensional. Dessa figura podemos concluir que quando uma substância pura muda de fase ela passa por uma superfície plana atípica onde duas fases saturadas podem coexistir em equilíbrio. Podemos também observar que a substância tem limite duplo na mudança líquido-vapor a (ponto crítico e ponto tríplice) e limite o único (ponto tríplice) na mudança gás -sólido (fusão ou sublimação), ou seja poderíamos afirmar sem medo que gases têm mais tendência a serem sólidos do que líquidos.Isto poderia explicar o porquê da existência de planetas sólidos com condições ambientais extremas ao seu redor.
Com base nos diagramas p-T e p-v foi possível examinar o comportamento pagamento das propriedades primitivas (p, v e T) e representar o comportamento básico da matéria, em forma geral, quando muda de .
Observamos que as fases sólida, líquida e gasosa aparecem como superfícies de limitadas pelas superfícies planas de mudança de fases (fusão, vaporização,
49

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
sublimação). Também concluímos que qualquer estado que esteja sobre os limites separadores das fases únicas dos regimes bifásico são chamados de estados de saturação. É bom ressaltar a descontinuidade nas isotermas quando ocorrem mudança de fase mostrando, claramente, que não ocorre mudança de fase se temperatura e pressão forem independentes.
Podemos, de forma quantitativa e baseados em experiências, dizer que na maioria das substâncias o espaçamento entre partículas (volume) muda só ligeiramente na mudança de fase sólido - líquido. Logo, em um diagrama p-v o plano de fusão deverá ser uma fita muito fina, comparada como plano de vaporização, devido ao fato de na mudança de fase e líquido-vapor as alterações (no caso, aumento), no volume da mistura são muito maiores (104 vezes) que as mudanças ocorridas na fusão. Porém, a medida que nos movimentamos da linha tríplice para o estado crítico a mudança de volume causada pelo processo de vaporização se torna cada vez menor, até chegar ao estado crítico onde não existe mudança de volume.
Acima do estado crítico o processo de vaporização ou condensação não existem. O estado crítico caracterizado pelos valores de pressão (pc), volumes (vc) e temperatura (Tc) é bem conhecido dos termodinâmicos. Alguns desses valores são mostrados na tabela 3.5. Tabela mais completa se encontra no Anexo 1.
Pontos críticos para as fases sólida - líquida ou sólido - gasosa não tem sido observados experimentalmente. O nome da fase da substância que existe acima do estado crítico não é importante, e devido ao fato de não existir diferença entre a fase líquida e gasosa a substância comumente é chamada de fluido.
Da tabela 3.5 podemos observar que normalmente as pressões críticas são superiores a pressão atmosférica, o que não é regra para as temperaturas críticas.
Substância Temperatura PressãoºK ºR bar Psia
Amoniaco (NH3) 405,5 724,8 112,8 1636Dióxido de Carbono (CO2) 304,2 547,5 73,9 1071Monóxido de Carbono(CO) 133 240 35,0 507
Hélio(He) 5,3 9,5 2,29 33,2Hidrogênio (H2) 33,3 59,9 13,0 188,1Nitrogênio (N2) 126,2 227,1 33,9 492
Àgua (H2O) 647,3 1165,2 220,9 3204
Tabela 3.5 – Propriedades críticas de algumas substânciasO ponto tríplice é caracterizado pelos valores de temperatura( ), volume( ) e
pressão( ). São também conhecidos alguns desses valores que estão mostrados na tabela 3.6. Mais dados a reais a esse respeito serão encontrados no Anexo1.
Substância T(K) T(*C) P(bar)Hélio 4 (ponto ) 2,17 -271,11 0,05066Hidrogênio (H2) 13,84 -223,88 0,07092Oxigênio (O2) 54,36 -218,88 0,001519
Nitrogênio (N2) 63,18 -210,00 0,12564Amônia (NH3) 195,40 -77,77 0,06180
50

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Dióxido de Carbono (CO2) 216,55 -56,66 5,16757Àgua(H2O) 273,16 0,00 0,00607
Tabela 3.6 - Dados de estado tríplice
Devemos, nesse ponto, ressaltar que o ponto tríplice da água é o utilizado o como referência para estabelecer a escala de temperatura absoluta chamada escala Kelvin, também ressaltaremos, como observação, que a água é uma substância anormal na solidificação, pois ela se expande a congelar, ou seja, o volume específico da fase sólida é maior que o da fase líquida.
Se compararmos o comportamento p, v e T de um vapor ou gás real, observaremos que os limites representativos da isotermas tendem a ser hipérboles equiláteras. A medida que nos afastamos da limite de vapor saturado vai-se para uma região de vapor superaquecido a baixa pressão (pressão quase nula) e temperatura superior à temperatura de saturação. Isso pode ser representado como função matemática do tipo:
pv = cte. (hipérbole eqüilátera) (3.24)
Quando esta relação se cumprem dizemos que o gás se encontra em estado ideal. A figura 3.21 mostra o diagrama b-ter e P-veio dar a de gás e ideal onde observamos que as curvas de pressão e volume constantes são linhas retas.
Figura 3.21-superfície p, v e T e diagramas p-T e P-v para gás com comportamento ideal3.6 - Tabelas de propriedades da substância pura
Depois da introdução dada sobre o comportamento de p,v e t de substâncias puras é necessário conhecer valores numéricos das propriedades das diferentes substâncias mais utilizadas na engenharia para poder realizar cálculos quantitativos.
51

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Atualmente, existem tabelas para quase todas as substâncias conhecidas acessíveis aos estudiosos da termodinâmica e as mesmas estão organizadas de forma similar em toda a literatura termodinâmica.
As tabelas foram, inicialmente, obtidas de forma experimental e logo expandidas utilizando relações termodinâmicas que serão devidamente estudadas em capítulos posteriores.
Para nosso estudo inicial das tabelas, as seguintes as propriedades intensivas serão de interesse primário: pressão, volume e temperatura.Outras propriedades como energia interna (u), entalpia(h) e entropia(s) serão apresentadas no decorrer deste livro.
3.6.1-Tabelas de saturação
Para poder utilizar as que ações de mistura de fases é necessário conhecer valores dados das propriedades nos estados de saturação das duas fases envolvidas assim como o título das mistura. Somente uma propriedade intensiva é necessária para identificar o estado das duas fases em equilíbrio numa mistura saturada. Assim as propriedades intensivas de cada fase podem ser tabuladas em função seja da pressão ou temperatura de saturação. Nestas tabelas, a variável independente que utilizada (expressão de saturação) normalmente aparece com valores inteiros.
A tabela 3.7, incompleta, mostra as propriedades da água saturada como função da temperatura em unidades do sistema internacional (SI). Tabelas mais completas se encontram no anexo 1. Algumas dessas tabelas mostram colunas de dados organizadas de forma similar a da tabela 3.7,e diferenças entre o valor das propriedades do estado de vapor saturado e líquido saturado.
TemperaturaTºC
PressãoP (bar)
Volume EspecíficoV (cm³/Kg)
Entalpiah (KJ/Kg)
Entropias (KJ/Kg.K)
Líq.Saturado
(vl)
Vapor Saturado
(vg)
Líq.Saturado
(hl)
Vapor Saturado
(hg)
Líq.Saturado
(sl)
Vapor Saturado
(sg)20 0,02339 1,0018 57791 83,96 2538,1 0,2966 8,667240 0,07384 1,0078 19523 167,57 2574,3 0,5725 8,257060 0,19940 1,0172 7671 251,13 2609,6 0,8312 7,909680 0,47390 1,0291 3407 334,91 2643,7 1,0753 7,6122100 1,01400 1,0435 1673 419,04 2676,1 1,3069 7,3549
Tabela 3.7 - Propriedades de líquido e vapor saturado para a água(Fonte:Keenan J.F; F.G.Keyes; P.G.Hill, and J.G.Moore, "SteamTables", New York,1969.)
3.6.2 - A tabela de líquido comprimido o subresfriado
A literatura especializada não apresenta muitos dados nesta fase, é certo para água% fluindo muito utilizado. A primeira linha de cada conjunto de dados apresentados na tabela 3.8 mostra os dados de líquido saturado na respectiva temperatura de saturação; observamos que a mudança das propriedades na fase
52

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
líquida com a pressão (liquido comprimido fecha parênteses é muito pequena. Podemos, então, em caso de ausência de dados experimentais, dizer que os dados de líquido comprimido são mais dependentes da temperatura do que da pressão.
p (bar) v (cm(3)/g) u (Kj/kg) h (Kj/kg) s (Kj/kg)0,474(saturação) 1,0291 334,86 334,91 1,0753
50 1,0268 333,72 338,85 10720100 1,0245 332,54 342,83 1,0688
Tabela 3.8 - Propriedades da água na fase líquida vida (líquido comprimido) e o(Fonte:Keenan J.F; F.G.Keyes; P.G.Hill, and J.G.Moore, "SteamTables", New York,1969.)
3.6.3 - Tabela de vapor superaquecido
Na região de fases únicas (sólido, líquido, vapor) tal como região superaquecida duas propriedades intensivas são necessárias para fixar os estados de equilíbrio. Normalmente, as propriedades v,u,h e s são tabeladas em função de pressão e temperatura.
O formato destas tabelas é fácil de entender se nos referirmos ao diagramas p-T. Os valores das propriedades v, u,h e s. são arbitrários com base em dados fixados para o estado de referência (exemplo: u = 0 para tref = 0 ºC)
A tabela 3.9 mostra o formato da organização dos dados. Observamos que os dados começam com estado de saturação (vapor saturado)e continuam mantendo a pressão constante e mudando a temperatura.
Quando, em problemas de engenharia, nos deparamos com estados de equilíbrio que não se encontram exatamente definidos na tabela é possível interpolar de forma linear e sem perder muita precisão.
Temperatura (*C) v (cm(3)/Kg u (kj/kg) h (kj/kg) s (kj/kgk)1,0bar(99,63*C)
sat100120160200
1694,01696,01793,01984,02172,0
2506,12506,72537,32597,82658,1
2675,52676,22716,62796,22875,3
7,35947,36147,46687,65977,8343
10 bar(179,91*sat.200240280320
194,4206,0227,5248,0267,8
2583,62621,92692,92760,22826,1
2778,12827,92920,43008,23093,9
6,58656,69406,88177,04657,1962
Tabela 3.9 – Propriedades de Vapor de água Superaquecida(Fonte:Keenan J.F; F.G.Keyes; P.G.Hill, and J.G.Moore, "SteamTables", New York,1969.)
53

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
É conveniente neste ponto explicar como fazer a consulta às tabelas oferecidas pela literatura. Quando a temperatura, pressão ou outro valor de uma propriedade é dada, o melhor método de encontrar a tabela apropriada a consultar é procurar primeiro a tabela de saturação. Seja dada uma pressão (p) e uma temperatura (T). Observando o diagrama p-v podemos comparar os valores de p e T e dados com os valores de ps e Ts (temperatura de saturação), exatamente como mostrado na figura 3.23. Podemos fazer as seguintes comparações:
Figura 3.22- Diagrama p x v de qualquer substância
Figura 3.23 - Diagrama p x v da água pura. Gráfico feito pelo EES.
1- T<Ts: a substância está na região de líquido comprimido 2- T>Ts: a substância está na região de vapor superaquecido3- T=Ts: a substância está no estado saturação e devemos obter o título.
Hoje os trabalhos de termodinâmica que a transferência de calor podem ser facilmente realizados sem a necessidade de consultar tabela sistema de mantas,
54

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
existem códigos constitucionais que proporciona as propriedades da substância de forma direta, fornecendo dos propriedades independentes como propriedade e o título.
O Código computacional EES - Engineering Equation Solver, desenvolvido o pela companhia F-Chart Software, Winsconsin,U.S.A é um dos códigos mais versáteis, fáceis e práticos para resolver o problemas termodinâmicos, a função básica fornecida por este programa é a solução de conjuntos de Algébricas ou problemas de Equações Diferenciais de valor inicial. O programa fornece muitas funções matemáticas e funções termofísicas prontas para serem usadas. Para um conjunto de substâncias que cobrem praticamente todo o campo aplicativo da termodinâmica são também fornecidas propriedades de transporte como a competitividade, viscosidade, etc. O programa permite:
Facilidades para entrar com dados tabulados que podem ser usados na solução de conjunto de equações,
a) A linguagem computacional pelo EES permite o uso de funções e procedimentos desenvolvidos pelo usuário os quais são armazenados em bibliotecas que são automaticamente lidas quando o EES começa a operar,
b) Funções e procedimento compilados e escritos em linguagem de alto nível como Pascal ou C podem ser dinamicamente vinculados ao EES, utilizando uma biblioteca incorporada ao sistema de operação do Windows.Este programa permite ao estudante resolver inúmeros problemas de
termodinâmica ou transferência de calor, sem se preocupar com a busca de propriedades, nem com métodos algébricos de solução de conjuntos de equações, permitindo ao estudante se concentrar mais no projeto ou na física do problema.
Estudos paramétricos são facilitados pelo uso de tabelas paramétricas que são similares a speedsheets, o usuário identifica as variáveis que são independentes, fornecendo seus valores no espaço da tabela. O EES calculará a partir do conjunto e de equações fornecido na janela de equações, todas as variáveis dependentes e as indicará na tabela se assim o usuário quiser.
Uma opção de gráfico é fornecida, que poderá indicar a relação entre quaisquer pares de variáveis contidos na tabela.
Os comandos do EES são intuitivos e conscientes com a interface Macintosh, que um novato aprende rapidamente. Este programa está idealmente projetado para o auxílio em cursos de engenharia mecânica e para o auxílio a engenheiros formados na solução de problemas do seu dia-a-dia.3.7 - Problemas Ilustrativos
Problema 3.1:
Comprimimos 10 Kg de ar de acordo com relação pv1,50=cte, desde uma pressão de 1 bar e 15 graus até uma pressão de 10 bar, qual será a temperatura final ?
Dados:
55

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
a)Constante Universal dos Gases: R=8,316Kj x Kmol -1 x K-1.b)Massa molecular: M=28,96kg x Kmol-1.c)Volume ocupado por 1Kmol de ar a p0=1,01355bar e t0=0*C: 22,414m3/Kmol.
Solução:
Deve-se primeiramente verificar se o ar nas condições se encontra num estado como g que ás ideal para compra ou vapor superaquecido. A forma mais rápida e segura de se saber é obtendo o fator de compressibilidade Z, onde:
Do diagrama de Nelson e Obert, 'Generalized compressibility charts', Chem Eng. Vol.61, pg.203:
Z 1,0
Podemos então considerar o ar no estado inicial como gás quase ideal é. A prática nos mostra que este problema não apresenta erros consideráveis utilizando a equação de estado para gases e ideais
A constantes particulares de ar serão:
Sabendo que o ar realiza um processo que segue a relação p.v1,5 = cte
56

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Assim poderemos dizer:
Onde:
Portanto:
Problema 3.2
1 kg de vapor de água está confinado numa esfera elástica que suporta sua pressão de forma proporcional ao seu diâmetro. Inicialmente o vapor se encontrava em estado de saturação a 105*C. Se fornecermos calor ao vapor até que se atinja uma pressão de 1,5 bar, qual será sua temperatura final?
Solução : Das tabelas de vapor de água, obteremos para vapor saturado a pressão de
saturação ps = 1,207bar e o seu volume específico vg = 1,42m3.Kg-1. Portanto:
Vi = M.vg = 1Kg.1,42m3.Kg-1 = 1,42m3
O diâmetro inicial da esfera será:
Sabendo que: . Portanto:
Assim podemos obter o diâmetro que terá a esfera quando sua pressão final for de 1,5 bar.
57

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Seu volume final será:
Para obtermos a temperatura final utilizaremos a mudança as tabelas de vapor superaquecido, sabendo que o estado do vapor procurado tem como propriedades vf = 2,7218m3 . Kg-1 e pf = 1,5bar a temperatura encontrada é:
Sabendo que o ar realiza um processo que segue a relação p.v1,5 = cte
Assim poderemos dizer:
Onde:
Portanto:
58

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Problema 3.2
1 kg de vapor de água está confinado numa esfera elástica que suporta sua pressão de forma proporcional ao seu diâmetro. Inicialmente o vapor se encontrava em estado de saturação a 105*C.Se fornecermos calor ao vapor até que se atinja uma pressão de 1,5 bar , qual será sua temperatura final?
Solução:Das tabelas de vapor de água, obteremos para vapor saturado a pressão de
saturação ps = 1,207bar e o seu volume específico vg = 1,42m3.Kg-1. Portanto:Vi = M.vg = 1Kg.1,42m3.Kg-1 = 1,42m3
O diâmetro inicial da esfera será:
Sabendo que: . Portanto:
Assim podemos obter o diâmetro que terá a esfera quando sua pressão final for de 1,5 bar.
Seu volume final será:
Para obtermos a temperatura final utilizaremos a mudança as tabelas de vapor superaquecido, sabendo que o estado do vapor procurado tem como propriedades vf = 2,7218m3 . Kg-1 e pf = 1,5bar a temperatura encontrada é:
Problema 3.3
Um sistema cilindro-pistão como mostrado na figura, contém 3 litros de água em estado líquido a uma pressão manométrica de 0,2 bar, o resto do volume é
59
Tf =

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
ocupado por vapor saturado em equilíbrio térmico e mecânico com a água líquida. Fornecendo-se calor ao sistema até que o pistão equilibre seu peso com a força exercida pelo vapor sobre o pistão.
Pergunta-se:a) Qual será o título de mistura vapor-líquido ao iniciar o processo?b) Qual será o título de mistura vapor-líquido no instante em que a força que realiza
o vapor sobre o pistão seja igual ao peso do pistão?c) Qual será o volume ocupado pelo vapor quando só exista vapor saturado?d) Qual será amassa de vapor remanescente no sistema se num instante antes de se
iniciar o movimento do pistão extrairmos toda água em fase líquida?
Dados: Volume de água líquida = 3 litros. Massa do pistão = 200 kg.
Solução : a) O título da mistura no início do processo pode ser obtido da seguinte forma:
A mistura se encontra saturada à pressão nanométrica Pim = 0,2 bar A pressão atmosférica dada é a correspondente a pressão exercida por uma coluna de mercúrio de 760mmHg. Para se calcular o título, precisamos conhecer, inicialmente, a pressão absoluta em que se encontra a mistura.
Pi = Pim + Patm
60

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Para se calcular o peso específico do mercúrio faz-se da seguinte forma:
Portanto:
Das tabelas de vapor de água saturado obteremos para P=l,2062 bar os seguintes valores:
Calculando as massas de líquido e vapor presentes no estado inicial:
A massa total contida no sistema vale:
Sendo a massa total de mistura e o volume total da mistura valem:
Podemos conhecer o volume especifico inicial da mistura:
61

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
O título da mistura será:
Este título pode ser igualmente calculado da forma proposta inicialmente:
Onde:
A pequena diferença observada é devido ao número de termos decimais utilizados nos dois casos.
b) Cálculo da pressão devida somente ao pistão (P):
Deve-se calcular a pressão absoluta que deverá ter a mistura para movimentar o pistão:
Das tabelas de vapor saturado procuramos o volume de líquido e vapor para P2= 162691,10 N1m2 e volume especifico igual ao volume inicial porque o pistão não foi movimentado v4.3737 x 103rn3 Kg’.
c) A pressão em que o vapor saturado estará será a necessária para
62

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
movimentar o pistão, ou seja, Pí=l,6269 bar. O valor do volume específico do vapor saturado a esta pressão foi anteriormente obtido (Vg2=4,lO8 m3 Kg5.
Como a massa deverá ser a mesma, então:
d) Podemos considerar que o vapor remanescente permaneça saturado à pressão P=1,6269 bar, assim saberíamos que o volume especifico deste vapor seria v = v g2 = 1,108m Kg’, portanto, como conhecemos o volume total quando o pistão ainda está sobre os topes iniciais:
Problema 3.4:
Determinar a mudança de volume quando 11 grama de água, em estado de liquido aturado, é vaporizada completamente a 100GC e a 3000C.
SoluçãoA mudança de volume na vaporização será:Teremos das tabelas _____ no anexo _____ ,
v100º = 1673 – 1,04 = 1672 cm3 / g
Este problema ilustra que a mudança de volume diminui à medida que nos aproximamos do ponto critico (3740C). Isto também indica que a linha indicativa de vapor saturado deve mudar lentamente de tangente, isso acontece com a maior parte das substâncias.
Problema 3.5:
Determinar a energia interna de vapor de água superaquecido a 1,0 bar e 1100C e 6,0 bar a 2200C.
Solução:
63

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
• valor da energia interna u a 1,0 bar e 1100C pode ser obtido fazendo-se o seguinte teste: em um diagrama p-v, para a pressão de 1 bar, a temperatura de saturação é 99,620C, portanto, podemos fazer um teste de localização da região na qual se encontra o estado procurado.
O nosso estado se localiza na região de vapor superaquecido, consultando a tabela teremos os seguintes valores de energia interna:
Fazendo-se uma interpolação entre estes dois estados, teremos:
2506,7 KJ / Kg u 2537,3 KJ / Kg100 ºC 110 ºC 120 ºC
64

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
A diferença de energia interna deve ser diminuída da energia interna a 1 200C porque a energia interna aumenta com a temperatura e, portanto, a energia interna a 11 00C será menor que a energia interna a 1 200C.
Para se obter a energia interna a 6 bar e 2200C, observamos que temos dados para 5 bar e 7 bar a 2000C e 2400C. Neste caso, teremos que fazer uma interpolação dupla da forma apresentada abaixo na tabela.
1ª InterpolaçãoT [ºC] U5,T[KJ/Kg] U7,T[KJ/Kg]
200 U5,200ºC(tabela)
U7,200ºC(tabela)
220 U5,220ºC(interp.)
U7,220ºC(interp.)
240 U5,240ºC(tabela)
U7,240ºC(tabela)
Fazendo-se a 1a interpolação teremos como resultado:a) Para uma pressão de 5 bar e temperatura de 2200C z~’ us.22o=2675,2
kJ/kgb) Para uma pressão de 7 bar e temperatura de 2200C ~ u7~22o=2668,3
kJ/kg.Na 2a interpolação, realizada da mesma maneira que a anterior, teremos como
resultado para uma pressão de 6 bar e temperatura de 2200C uma energia interna igual a:
Problema 3.6:
2ª InterpolaçãoUP,,220ºC[KJ/
Kg]P [bar]
2657,2 5X 6
2668,3 7
65

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
Determinar a pressão do fluido refrigerante Ri 2 no estado a 40~C e com entalpia de 214,44 kJ/kg e 214,70 kJ/kg.
Solução:Este problema ilustra o caso de como qualquer par de propriedades
independentes fixa o estado de uma substância pura simples compressível. Neste caso procuramos p = f(T.h).
Observamos que fazendo o teste de localização através das tabelas de saturação, que o estado procurado está na região de vapor superaquecido, nestas tabelas a temperatura muda de cima para baixo a qualquer pressão. Começando com 400C e à pressão de 6 bar notamos que a entalpia começa com 216,77 kJ/kg e decresce à medida que a pressão se incrementa para a mesma temperatura. Se continuarmos a ler pressões superiores e a temperatura de 400C, finalmente obteremos h=2 14,44 kJ/kg a 2,4 bar.
O valor de h~2 14,70 kJ/kg para a entalpia se encontra entre 2,0 e2,4 bar, neste caso, faremos uma interpolação linear para estimar a pressão correspondente a entalpia h=2 14,70 kJ/kg, e obteremos com resultado uma pressão igual a 2,2 bar.
Problema 3.7:
Obter a mudança na energia interna específica da água, para a mudança de estado de 400C e 25 bar até 80ºC e 75 bar:
a) Utilizar a tabela de líquido comprimido.b) Fazer cálculo aproximado com dados de saturação.
Solução:
a) Da tabela _____ do anexo _____ para liquido comprimido encontramos,
u1 – u2 = 331,15 – 167,25 = 165,90 KJ / Kg
b) Pelo método aproximado, os valores de u2, saturado, são utilizadas as temperaturas especificadas e o dado de pressão é ignorado:
u1 – u2 = 334,86 – 167,56 = 167,30 KJ / Kg
O erro encontrado é da ordem de 0,84% o qual mostra que no caso de ausência de tabelas de líquido comprimido este método aproximado pode se utilizado.
Problema 3.8:
Desejamos saber a pressão do C2H~ que se encontra a 293 K e que tem uma
66

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
constante particular R=276,64 N m/kg K, utilizando:a) A equação reduzida de Van der Waals.b) A equação de estado de Van der Waals com suas constantes calculadas da
seguinte forma:
;
c)Equação de Estado de Beattie – Bridgmann com as constantes
A0 = 659,124 N.m4 / Kg2
B0 = 0,003126 m3 / Kga = 0,001949 m3 / Kgb = 0,000637 m3 / Kgc = 29930 m3.K3 / Kg
d) Lei modificada dos estados correspondentes aplicada à equação de Van der Waals, onde:
; ;
e) Equação de estado de gás ideal:
f) O Utilizar o fator de compressibilidade:
g) Utilizar dados de tabelas, obtidos utilizando o Código computacional EES.
Solução:
67

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
a) Obtendo os valores dos parâmetros críticos das tabelas _______ no anexo___, tem-se:
; ;
Calculando os parâmetros reduzidos:
;
Substituindo seus valores na equação de Van der Waals reduzida teremos:
b) Utilizando a equação de estado de Van der Waals:
c) Utilizando a equação de estado de Beattie-Bridgemann:
d) Utilizando a lei modificada dos estados correspondentes aplicada à equação de Van der Waals, calcula-se o volume ideal:
A pressão reduzida ideal será:
68

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
e) Utilizando a equação de estado de gás ideal:
f) Utilizando o fator de compressibilidade, com os valores de Pr e Tr entramos no diagrama de compressibilidade e obtemos o fator Z=0,735.
g) Utilizando o programa EES, obtem-se:
Programa do ESS
ESS – Equation Window
{Exemplo de uso de equações de estado e lei dos estados correspondentes}v = 0.03T = 293 V1 = 0.03 T1 = 293{LEI DOS ESTADOS CORRESPÓNDENTES}Pr = 8*Tr*(3*Vr-1)-3/Vr**2Vc = 0.00492Pc = 49.3*1 e 4*9.81Tc = 305.27Tr = T / TcVr = V / VcPr = P / Pc{EQUAÇÕES DE VAN DER WALLS}a = (27 / 64)*R**2*Tc**2 / Pcb = R*Tc / 8*PcR = 28.2*9.81P1 = R*T / (V-b) – a / V**2{EQUAÇÕES DE BEATTLE BRIDGMANN}P2 = R*T*(1-C2 / (V*T**3) / V**2)*(V+B0*(1-B2/V))-A0*(1-A2/V)/V**2A0 = 67.189*9.81B0 = 0.003126A2 = 0.001949B2 = 0.000637
69
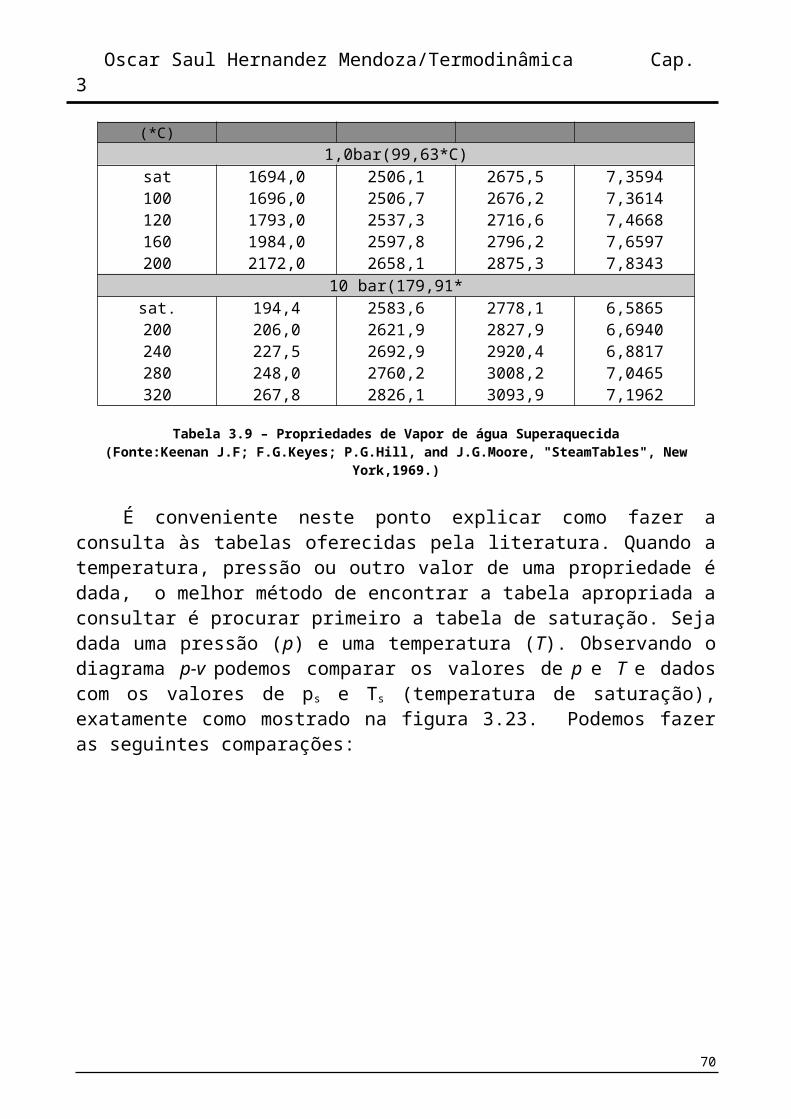
Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
C2 = 29930{LEI MODIFICADA DOS ESTADOS CORRESPONDENTES}Pri = 8*Tr/(8*Vri-1)-0.422/(Vri**2)Vri = V*Pc/(R*Tc)P3 = Pri*Pc{EQUAÇÕES DE ESTADO DE GÁS IDEAL}P4 = R*T/V{UTILIZANDO FUNÇÕES DO EES}PS*PRESSURE(C2H6,T=T1,V=V1)*1e3{FATOR DE COMPRESSIBILIDADE}Z = P3*V/(R*T)
ESS –Solution
Unit Settings: [C] / [Kpa] / [Kg] / [degrees]a=622.1 A0=659.1 A2=0.001949 b=0.002183 B0=0.003126 B2=0.00063
7P=1.757E+0
6P1=2.223E+0
6P2=2.175E+0
6P3=2.222E+0
6P4=2.702E+0
6P5=5.218E+
06Pr=03633 Pri=0.4595 R=276.6 T=293 T1=293 Tc=305.3V=0.03 V1=0.03 Vc=0.00492 Vr=6.098 Vri=1.718 Z=0.8226
Das análises feitas acima podemos tirar algumas conclusões:1 Podemos observar que o gás considerado, se encontrava perto da
temperatura critica (Tr 0,96) e tem um volume reduzido (6,098) o que indica ser um vapor superaquecido.
2. Nas condições estabelecidas, o gás está mais para vapor superaquecido do que para gás ideal, portanto, o código computacional EES, que trata o C2H6 como gás ideal, não é o método mais correto para determinar a pressão.
3 Comparando o resultado obtido, com fator de compressibilidade, com o resultado utilizando a equação de Van der Waals, observamos que sua diferença (D) é muito pequena.
O qual nos leva a concluir que é um gás de Van der Waals.
Resumo
Neste capitulo definimos os conceitos e linguagens básicas utilizadas no desenvolvimento da ciência chamada termodinâmica clássica. E um capitulo aparentemente simples, mas extremamente importante, pois define, por assim dizer, as normas do jogo, que serão utilizadas ao longo de todo o desenvolvimento das leis básicas da termodinâmica e da lógica que deverá ser aplicada na resolução de
70

Oscar Saul Hernandez Mendoza/Termodinâmica Cap. 3
problemas físicos que tratam de relações energéticas.É muito importante entender o que é sistema, estado, limites do sistema e os
postulados básicos da termodinâmica. Assim como nos sistemas de unidades estudados no capitulo 2, que se baseia em suas unidades básicas, a termodinâmica tem como base real seus parâmetros básicos (propriedades primitivas), que pelo fato de serem mensuráveis, constituem o contato entre a teoria clássica utilizada pela termodinâmica e o mundo real.
Este capitulo define como propriedades primitivas mensuráveis: densidade (massa / unidade de volume), pressão (força / unidade de área) e temperatura (nível de energia interno das substâncias). Também relatam, de forma resumida, as técnicas mais conhecidas para avaliar a ordem de grandeza destas propriedades e as escalas utilizadas para fixar seus valores numéricos.
Outro trecho que merece destaque neste capitulo é a definição de “substância pura”, que não é mais que a descrição de uma substância ideal livre de imperfeições que permite a quem está estudando termodinâmica concentrar a sua atenção nos trocas de energia entre sistemas ou nos processos realizados pelo sistema, sem se preocupar com desvios que poderiam ser causados pelo comportamento não ideal das substâncias.
Deveremos observar que é utilizada a “água” como substância modelo para explicar o comportamento de todas as substâncias puras existentes na natureza nas diferentes fases. Por ser uma substância bem conhecida pelo homem, e pelo fato desta substância apresentar suas fases (sólida, líquida e gasosa) perto das condições ambientes presentes na Terra, permite conhecer suas fases visualmente.
Os diagramas p-v e p-T são estudados devido à importância do seu uso. Exemplificando: área abaixo de um processo num diagrama p-v, representa energia em forma de trabalho intercambiada pelo sistema que realiza o processo. O diagrama p-T indica claramente a dependência da pressão e temperatura nas transições de fase, mostrando visualmente a chamada “pressão de vapor”, expressão comumente utilizada para definir a pressão de saturação de uma substância à temperatura conhecida na qual podem ser realizados processos de vaporização ou condensação. De forma similar, poderíamos definir temperatura de fusão ou sublimação para uma determinada pressão.
Finalmente, o capítulo fecha mostrando a forma como, universalmente, é organizada a informação sobre a mudança de propriedades primitivas e de outras não menos importantes (energia interna, entalpia, entropia) quando a substância muda de fase. Neste ponto, convém ressaltar que as fases que mais interessam ao engenheiro mecânico são: liquido, liquido comprimido, líquido-vapor, vapor saturado e vapor superaquecido, por serem estas as fases em que se realizam as maiores mudanças de volume específico quando há mudança de fase. Isto do ponto de vista mecânico, pode ser entendido como processos potenciais de realização de trabalho mecânico.
71





























