Embed Size (px)
Citation preview

“Caso de Leucemia Aguda agressiva com
características clínico-patológicas distintas de
Leucemia Mielóide Crónica em fase blástica”
Case-report
Mestrado Integrado em Medicina
2009/2010
Diogo Gil Moreira de Sousa
I
INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR

2
RESUMO
A Leucemia Mielóide Crónica (LMC)
é uma neoplasia mieloproliferativa com
evolução trifásica (fase crónica, acelerada e
blástica), sendo rara a sua apresentação em
fase blástica – cerca de 1% dos casos
diagnosticados nos países ocidentais. Esta
apresentação encerra um diagnóstico
diferencial importante com Leucemia
Mielóide Aguda (LMA) Philadelfia (Ph)
positivo, uma entidade ainda com poucos
casos reportados.
O cromossoma Ph além de estar
presente na esmagadora maioria de casos de
LMC (95%), também é detectado em cerca de
20% dos casos de Leucemia Linfóide Aguda
(LLA) e em cerca de 3% dos casos de LMA,
enfatizando a importância de outros critérios
para a distinção destas duas neoplasias
mielóides agressivas – LMC com apresentação
em fase blástica e LMA Ph positivo.
Neste estudo, relata-se um caso de um
doente do sexo masculino, 37 anos de idade,
sem antecedentes patológicos relevantes, que
deu entrada no Centro Hospitalar do Porto
(CHP) com queixas de dores ósseas lombares
e astenia generalizada desde há cinco dias. Ao
exame físico sem alterações de relevo,
nomeadamente ausência de hepato-
esplenomegalia palpável. Investigação
laboratorial revelou hiperleucocitose marcada
no sangue periférico (180,95x103/µL), com
critérios de Leucemia Aguda no mielograma e
coloração citoquímica compatível com
linhagem mielóide, apresentando cariótipo
complexo com t(9;22) e detecção do transcrito
bcr-abl b3a2 (p210) no sangue periférico. O
doente iniciou tratamento com Imatinib e foi
proposto para transplante alogénico de Medula
Óssea (MO) de dador familiar, realizado em
Outubro de 2009. Observou-se rápida e
agressiva recidiva da doença após
alotransplante de MO, com falecimento em
Janeiro de 2010.
ABSTRACT
Chronic myeloid leukemia (CML) is a
myeloproliferative neoplasm with a thriphasic
natural evolution (chronic phase, accelerated
and blast), being rarely presented in blast
phase - around 1% of diagnosed cases in
Western countries. This presentation contains
an important differential diagnosis with Acute
Myeloid Leukemia (AML) Philadelfia (Ph)
positive, an entity with few cases reported.
The Ph chromosome besides being present in
the overwhelming majority of CML cases
(95%) is also detected in about 20% of cases
of Acute Lymphoblastic Leukemia (ALL) and
about 3% of AML cases, emphasizing the
importance of other criteria for distinguishing
these two aggressive myeloid malignancies -
CML blast crisis at presentation and AML Ph
positive.

3
In this study, we report a case of a 37-
year-old men patient without relevant
pathological antecedents received at Oporto’s
Hospital Center (CHP) with complaints of
lumbar bone pain and general malaise for 5
days. On physical examination no significant
alterations, namely there wasn’t palpable liver
and spleen. Laboratory investigations revealed
marked hyperleucocytosis (180.95 x103/μL)
in peripheral blood, with criteria for acute
leukemia in bone marrow examination and
cytochemical staining consistent with myeloid
lineage, with complex karyotype with t(9; 22)
and detection of bcr-abl b3a2 transcript (p210)
in peripheral blood. The patient began
treatment with Imatinib and was proposed for
allogeneic bone marrow (BM) transplantation
from family's donnor, held in October 2009.
We observed rapid and aggressive disease
recurrence after allotransplantation of BM,
with death in January 2010.
INTRODUÇÃO
O cromossoma Filadélfia (Ph) foi a
primeira cromossomopatia consistente
identificada numa neoplasia.1
Em 1973, Rowley descreveu o
cromossoma Ph, ou 22q-, como resultante da
translocação recíproca e equilibrada entre os
cromossomas 9 e 22.2 Esta translocação
origina um gene híbrido bcr-abl que envolve o
proto-oncogene c-abl do cromossoma 9 e a
região bcr do cromossoma 22.3
A formação do cromossoma Ph está
assim associada à formação do gene quimérico
bcr-abl que manifesta actividade tirosina-
quinase constitutivamente elevada, perdendo a
normal capacidade de regulação/feedback.
Esta acção parece exercer papel central na
patogénese da Leucemia Mielóide Crónica
(LMC) e outras leucemias com cromossoma
Ph positivo.4
A translocação t(9;22)(q34;q11),
característica da LMC, é detectada em 95%
dos doentes com este diagnóstico,5 sendo que
nos restantes 5% esta translocação pode estar
sub-diagnosticada, não sendo detectada pelo
cariótipo convencional mas apenas por
técnicas moleculares como hibridação in situ
por fluorescência (FISH) ou reacção em cadeia
da polimerase com transcrição reversa (RT-
PCR).6
No entanto, esta alteração cromossómica
não é exclusiva da LMC, tendo sido estudada
a sua presença em leucemias agudas. A
incidência de Leucemia Linfoblástica Aguda
(LLA) com cromossoma Ph positivo é
estimada em 17-25%, e na Leucemia Mielóide
Aguda (LMA) a positividade desta
translocação, ainda que mais rara, atinge os
3% em algumas séries.7,8 Em geral, estes casos
de LMA Ph positivo estão associados aos
subtipos FAB M1 e M2.9,10

4
A evolução natural da LMC é trifásica -
evoluindo de fase crónica, para uma fase de
aceleração e, finalmente, fase blástica. Nos
dois primeiros anos após o diagnóstico inicial
de LMC, 5-15% dos doentes não tratados
progridem para fase blástica e nos anos
seguintes a taxa de progressão aumenta 20-
25% por ano, ocorrendo frequentemente entre
os 3 e os 6 anos após os diagnóstico.11
A apresentação de LMC em fase blástica
é rara, representando apenas 0,9% dos doentes
diagnosticados com LMC nos países
Ocidentais. A grande maioria destes doentes é
diagnosticada durante a fase crónica (96,8%) e
uma minoria também na fase de aceleração
(2,2%).12
No maior estudo realizado até à data
envolvendo doentes com LMA Ph positivo
(16), verificaram-se diferenças
estatisticamente significativas em parâmetros
clínicos (esplenomegalia), laboratoriais
(basofilia no sangue periférico e mielograma)
e citogenéticos (alterações cromossómicas
major para além da translocação t(9;22) e tipo
de rearranjo molecular do cromossoma Ph),
que diferenciavam estes casos dos doentes
diagnosticados com LMC em fase blástica,
indiciando que se trata de duas patologias
distintas e cujo diagnóstico diferencial é
relevante em termos de prognóstico e
tratamento.13 No entanto, os autores
interpretam com prudência estes resultados
uma vez que é uma patologia rara e o número
de doentes estudados é reduzido, salientando a
importância de haver mais registos nesta
área.13
O tipo do rearranjo molecular do
cromossoma Ph nas leucemias Ph positivo é
um indicativo da origem da LMA: a presença
do rearranjo do oncogene de fusão bcr-abl
envolvendo o primeiro intrão do gene BCR ou
região m-BCR (minor breakpoint cluster
region), que codifica a proteína p190bcr-abl
, é
característico de LMA de novo, enquanto os
casos que apresentam o rearranjo na região M-
BCR (major breakpoints cluster region), que
codifica a proteína p210bcr-abl
, podem
corresponder à LMA de novo ou à crise
blástica mielóide de LMC, até então não
diagnosticada, não permitindo tirar conclusões
em relação à sua patogénese.4,14
Actualmente, a discussão reside na
diferenciação destes raros casos de LMA Ph
positivo e LMC com apresentação já em fase
blástica, estando a ser estudados diversos
casos de LMC com rápida progressão para
fase blástica ou assintomáticos durante a fase
crónica e os verdadeiros casos de LMA Ph
positiva de novo.13,14
É neste contexto que se insere o
objectivo da dissertação no relato deste caso,
de um doente com provável diagnóstico de
LMA Ph positivo, de forma a permitir
acrescentar mais informação a uma
problemática que reúne um escasso número de
casos nos estudos realizados e que condiciona

5
a validação dos resultados obtidos. Este caso
permite ainda explorar possíveis diferenças
entre o diagnóstico, prognóstico e tratamento
de LMC com apresentação em fase blástica e
os escassos casos estudados na literatura de
LMA Ph positivo.
APRESENTAÇÃO DO CASO
J.R.S.B., sexo masculino, 37 anos,
caucasiano, natural e residente no Porto,
empregado de escritório, sem antecedentes
médicos relevantes; desconhece história
familiar de distúrbios hematológicos.
Transferido do Hospital São Sebastião em 15
de Maio de 2009, com queixas de dores ósseas
lombares e astenia generalizada desde há 5
dias, hiperleucocitose e presença de blastos no
sangue periférico.
À entrada, no Centro Hospitalar do
Porto (CHP), o hemograma demonstrava
hiperleucocitose (180,95x103/µL) com formas
imaturas mielóides (12% blastos, 14%
mielócitos e 9% metamielócitos), com
basofilia (3,62x103/µL; 2,0%), eosinofilia
(1,81x103/µL; 1,0%) e monocitose
(26,43x103/µL; 10,0 %) marcadas; sem
alterações à apresentação das outras linhagens
(Hb 13,4 g/dL; plaquetas 180x103/µL). Ao
exame físico apresentava-se com bom estado
geral, corado, anictérico, apirético, ausência de
adenomegalias periféricas, organomegalias,
nomeadamente hepatoesplenomegalia, ou
sufusões hemorrágicas na pele e mucosas.
Iniciou-se a investigação de provável
neoplasia mieloproliferativa.
O mielograma revelou Medula Óssea
(MO) marcadamente hipercelular,
representando os blastos 46% do total das
células da amostra sem sinais evidentes de
displasia, com reacção mieloperoxidade
positiva, e a alfa naftil butirato esterase era
positiva em 7,0% do total das células da
amostra (MO compatível com LMA-M2).
Na imunofenotipagem de sangue
periférico confirmou-se leucocitose acentuada
(170,4x103/µL), fundamentalmente à custa da
linha granulocítica (78,5%), estando esta
representada por células em diferentes fases de
diferenciação mas com evidência fenotípica
clara de bloqueio maturativo, traduzido pela
acumulação de células nos estadios de
promielócito e mielócito, embora com
morfologia blástica: blastos CD34+ (2%),
promielócitos (35%), mielócitos (12%),
metamielócitos (12%), neutrófilos (18%) e
basófilos (1,5%). A linha monocítica (16,5%)
incluía os estados de promonócito (4,5%) e
monócito maduro (12%) e apresentava um
perfil fenotípico normal. Estes resultados eram
sugestivos de Neoplasia Mieloproliferativa
(LMC) ou (LMMC) em transformação
blástica.
O estudo imunofenotipico da MO
apresentou conclusões sobreponíveis às
observadas no sangue periférico.

6
O estudo citogenético revelou um
cariótipo complexo com t(9;22): 50xy, +4, +6,
t(9;22)(q34;q41), +17, +der(22). Com a
utilização de sondas específicas dos loci
22q11.2 e 9q34 para detecção da fusão dos
genes bcr-abl, por FISH, foram observados
200 núcleos, tendo sido detectada a respectiva
fusão em 100% dos núcleos. A pesquisa do
rearranjo bcr-abl no sangue periférico detectou
o transcrito bcr-abl b3a2 (p210). Restante
estudo molecular para factores de prognóstico
de LMA - detecção do transcrito pml/rar e
pesquisa de mutações no gene FLT3 - foi
negativo.
Análise bioquímica geral mostrou
função renal e hepáticas normais e uma
desidrogenase do lactato (DHL)
marcadamente aumentada – 1590 U/L. A
ecografia abdominal superior mostrou
esplenomegalia ligeira (15,2cm), com fígado
com padrão de esteatose, sem lesões ocupantes
de espaço e de tamanho normal.
O estudo da coagulação realizado à
entrada era normal.
No exame virológico para pesquisa de
vírus da hepatite B (VHB), vírus da hepatite C
(VHC), vírus da imunodeficiência humana 1 e
2 (HIV 1/2), vírus Epstein Barr (EBV), vírus
Herpes Simplex 1 e 2 (HSV 1 e 2), vírus
Herpes Zooster (VZV), Citomegalovírus e
Parvovírus, destacava-se a positividade para
EBV VCA IgG (87,1 RU/mL), EBV EBNA
IgG (40,9 RU/mL), HSV 1 IgG (>200,0
U/mL), VZV IgG (4077,0 mU/mL).
Perante um quadro mieloproliferativo
com hiperleucocitose, decidiu-se introduzir
terapêutica citorredutora com Hidroxiureia
(HU) 2g/dia, tendo-se observado uma
citorredução rápida com normalização dos
dados hematológicos e o doente teve alta
mantendo terapêutica citorredutora.
Dois dias após alta foi reinternado, por
dores ósseas intensas que não cediam aos
analgésicos convencionais e um quadro de
dispneia e insuficiência respiratória. Por
suspeita clínica de tromboembolismo
pulmonar, foi pedido angio-TAC que não
confirmou a hipótese proposta e revelou áreas
de consolidação parenquimatosa
bilateralmente, no lobo médio, segmento
lingular e em ambas as bases, em provável
relação com alterações de natureza
inflamatória/infecciosa. Iniciou antibioterapia
de largo espectro, sem agente isolado mas com
boa resposta e com resolução do quadro
respiratório.
No dia 2 de Junho, e por se tratar de uma
neoplasia mieloproliferativa bcr-abl positiva
iniciou-se tratamento com inibidores da
tirosina-cinase - Imatinib - numa dose diária
de 400mg/dia. Pelo facto de ser um doente
jovem com um quadro agressivo complexo,
com numerosas células imaturas no sangue
periférico, decidiu-se iniciar terapêutica de
indução com Citosina-Arabinosídeo (Ara-C)

7
na dose de 1,5g/m2, 12/12 horas e efectuar
estudo HLA para pesquisa de dador com vista
a transplante alogénico com dador familiar.
Após ter completado 3 ciclos de Ara-C
mantendo o Imatinib, atingiu Remissão
Hematológica e Remissão Citogenética Parcial
(3/18 mitoses Ph positivo).
Nessa altura verificou-se existir um
dador familiar compatível pelo que o doente
foi proposto para transplante.
Em Outubro de 2009 o doente foi
internado para realização de alotransplante de
progenitores hematopoiéticos periféricos de
irmão HLA idêntico e isogrupal. À entrada na
unidade de transplante apresentava critérios de
Remissão Hematológica e Remissão
Citogenética Parcial (5/20 mitoses Ph
positivo),
Submetido a regime de condicionamento
com Bussulfano e Ciclofosfamida (BuCy2) e
profilaxia da doença do enxerto contra
hospedeiro (DECH) com Metotrexato e
Ciclosporina, foi realizado o alo-transplante
em 15/10/09 com infusão de 5,68x106/Kg de
células CD34.
Na reavaliação do 1º mês pós alo-
transplante apresentava MO normocelular,
sem alterações morfológicas aparentes, o
cariótipo de MO era complexo e a pesquisa de
transcripto bcr-abl era positiva por RT-PCR.
Evidenciou quimerismo completo, havendo
apenas um quimerismo misto para linfócitos T
com predomínio de linfócitos do dador.
Na consulta de rotina pós-transplante, a
23-11-09, o doente referiu queixas de
lombalgia e apresentava parâmetros
hematológicos compatíveis com progressão da
doença: hiperleucocitose (64,45x103/µL) com
26% de blastos, anemia normocrómica
normocítica (Hb-11,3 g/dL; VGM-94,9 fL;
HGM-31,9 pg) e trombocitopenia (plaquetas-
114x103/µL). Iniciou-se citorredução com HU
(500+500mg/dia) e Imatinib 400mg/dia, e
desmame progressivo de ciclosporina. O
estudo do cariótipo revelava cariotipo
complexo com persistência da t(9;22): 50xy,
+4, +6, +8, t(9;22), t(9;22) [18]/46xy[2].
Dois dias depois foi internado no CHP
em crise blástica pós-transplante,
evidenciando no hemograma hiperleucocitose
(Leucócitos – 72,36x103/µL) com aumento
das formas blásticas (36%), agravamento da
anemia (Hb-11,0 g/dL) e trombocitopenia
(plaquetas-104x103/µL). Apresentava ainda
critérios de síndrome de lise tumoral:
insuficiência renal aguda (IRA) (Creatinina:
2,21 mg/dL, Ureia: 135 mg/dL), hiperuricemia
(ácido úrico 13mg/dL) e elevação marcada de
DHL (1580 UI/L); mantendo dorsalgia
bilateral difusa, que agrava com a mobilização
e não resolve com anti-inflamatórios não
esteróides (AINEs), tendo havido necessidade
de recorrer morfina endovenosa para controlo.
8
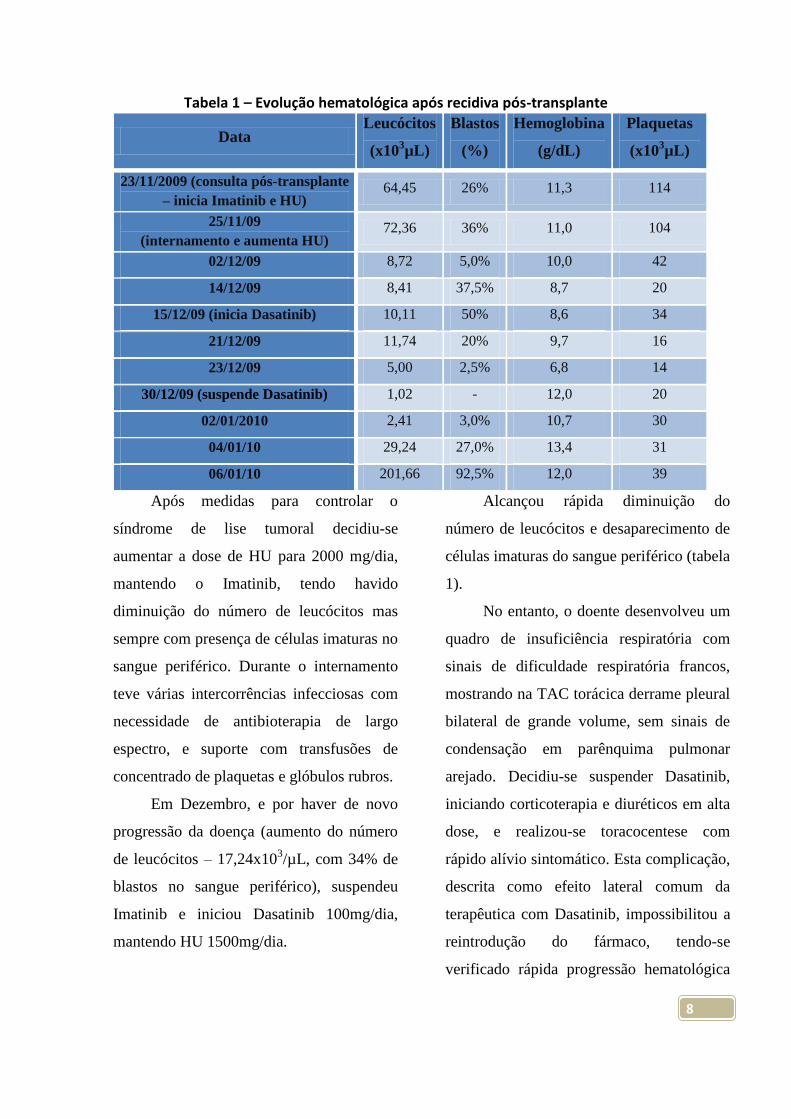
Tabela 1 – Evolução hematológica após recidiva pós-transplante
Data Leucócitos
(x103µL)
Blastos
(%)
Hemoglobina
(g/dL)
Plaquetas
(x103µL)
23/11/2009 (consulta pós-transplante
– inicia Imatinib e HU) 64,45 26% 11,3 114
25/11/09
(internamento e aumenta HU) 72,36 36% 11,0 104
02/12/09 8,72 5,0% 10,0 42
14/12/09 8,41 37,5% 8,7 20
15/12/09 (inicia Dasatinib) 10,11 50% 8,6 34
21/12/09 11,74 20% 9,7 16
23/12/09 5,00 2,5% 6,8 14
30/12/09 (suspende Dasatinib) 1,02 - 12,0 20
02/01/2010 2,41 3,0% 10,7 30
04/01/10 29,24 27,0% 13,4 31
06/01/10 201,66 92,5% 12,0 39
Após medidas para controlar o
síndrome de lise tumoral decidiu-se
aumentar a dose de HU para 2000 mg/dia,
mantendo o Imatinib, tendo havido
diminuição do número de leucócitos mas
sempre com presença de células imaturas no
sangue periférico. Durante o internamento
teve várias intercorrências infecciosas com
necessidade de antibioterapia de largo
espectro, e suporte com transfusões de
concentrado de plaquetas e glóbulos rubros.
Em Dezembro, e por haver de novo
progressão da doença (aumento do número
de leucócitos – 17,24x103/µL, com 34% de
blastos no sangue periférico), suspendeu
Imatinib e iniciou Dasatinib 100mg/dia,
mantendo HU 1500mg/dia.
Alcançou rápida diminuição do
número de leucócitos e desaparecimento de
células imaturas do sangue periférico (tabela
1).
No entanto, o doente desenvolveu um
quadro de insuficiência respiratória com
sinais de dificuldade respiratória francos,
mostrando na TAC torácica derrame pleural
bilateral de grande volume, sem sinais de
condensação em parênquima pulmonar
arejado. Decidiu-se suspender Dasatinib,
iniciando corticoterapia e diuréticos em alta
dose, e realizou-se toracocentese com
rápido alívio sintomático. Esta complicação,
descrita como efeito lateral comum da
terapêutica com Dasatinib, impossibilitou a
reintrodução do fármaco, tendo-se
verificado rápida progressão hematológica

9
da doença em poucos dias (Leucócitos –
201,66x103/µL; blastos – 92,5%). Iniciou
perfusão de Ara-C 100mg/m2 para controlo
da doença, mas o doente manteve-se em
progressão e foi decidido manter apenas
medidas de suporte e vigilância de sinais de
sofrimento. Em 10-01-2010, ocorreu
agravamento súbito da insuficiência
respiratória e falecimento do doente.
DISCUSSÃO
A LMC é uma neoplasia
mieloproliferativa caracterizada geralmente
por uma evolução insidiosa, sendo a maioria
dos doentes detectados na fase crónica. A
apresentação de LMC em fase blástica é
uma entidade rara, representando apenas
0,9% dos doentes com LMC ao diagnóstico
nos países ocidentais.12
Ao diagnóstico cerca de 40% dos
doentes com LMC são assintomáticos,
sendo os sintomas mais comuns de
apresentação a fadiga, letargia e
esplenomegalia.
A esplenomegalia está presente ao
diagnóstico na esmagadora maioria dos
doentes com LMC (cerca de 95%), variando
de apenas ponta palpável até uma massa
ocupando grande parte da cavidade
abdominal; cerca de 50% dos doentes
apresentam baço palpável 10 cm abaixo da
margem costal, sendo que o tamanho
esplénico mostrou em diversos estudos uma
boa relação com o número de leucócitos.15 A
hepatomegalia é menos comum (cerca de
50% ao diagnóstico).
O caso em estudo, de um doente com
hiperleucocitose com cromossoma Ph
positivo em apresentação blástica (MO
hipercelular com 46% de blastos ao
diagnóstico, com coloração citoquímica
compatível com linhagem mielóide) com
dores ósseas, apresenta características
clínicas incomuns, com manifestações
agressivas e atípicas para LMC à
apresentação. A ausência de esplenomegalia
palpável ao diagnóstico (apenas detecção
ecográfica – 15,2 cm) é rara em doentes
com LMC, e torna-se ainda mais incomum
perante o número de leucócitos verificado à
apresentação (180,95x103/µL), contrariando
a literatura existente que relaciona a
leucometria com o aumento do tamanho
esplénico.15
Devido à apresentação blástica
mieloide, em indivíduo de 37 anos, sem
sintomas prévios e sem evidência de
alterações hematológicas anteriores, a
primeira suspeita diagnóstica foi LMA de
novo. No entanto, a evidência de t(9;22) nas
200 células observadas nos estudo
citogenético de sangue periférico,
colocaram problemas de diagnóstico
diferencial entre uma apresentação em fase
blástica de LMC ou Leucemia Aguda Ph
positiva.

10
A t(9;22)(q34;q11), encontrada neste
doente, resultou na formação do
cromossoma Ph, com geração de tirosina-
quinase bcr/abl activa. Esta anomalia
cromossómica está comummente associada
à LMC e LLA. No entanto, estão descritos
casos de LMA com positividade para o
cromossoma Ph, constituindo cerca de 2%
de novos casos de LMA diagnosticados.16,17
A controvérsia mantém-se acerca da
origem desta entidade, discutindo-se se
representa uma verdadeira leucemia aguda
ou apresentação em crise blástica mielóide
de LMC. No entanto esta distinção é muito
difícil, se não mesmo impossível à luz do
conhecimento actual, devido à semelhança
de apresentação destas duas entidades e ao
escasso número de casos estudados na
literatura, normalmente envolvendo estudos
com número reduzido de doentes que
dificultam a sua validação estatística. Existe
actualmente nesta área intensa discussão e
investigação.
Critérios clínicos estudados que
sugerem a diferenciação entre LMA Ph
positivo e crise blástica de LMC incluem
ausência de história prévia de distúrbio
hematológico, ausência de evidência de fase
crónica ou acelerada de LMC após
quimioterapia de indução e inexistência de
características clínicas e laboratoriais de
LMC, como esplenomegalia e basofilia.18
Na análise destes relatos da literatura
encontrou-se dados clínicos conflituantes
com o caso abordado, uma vez que apesar
de se apresentar sem história prévia de
distúrbios hematológicos, sem
esplenomegalia palpável e basofilia ligeira
(2%) ao diagnóstico - características
sugestivas de LMA Ph positiva de novo -,
por outro lado, evidenciou resposta
hematológica e remissão citogenética
parcial (17% de metáfases da MO com
t(9;22)) com regressão para uma fase
crónica após início de terapêutica de
indução com Imatinib e 3 ciclos de
quimioterapia com ARA-C. No entanto esta
resposta foi de curta duração, com rápida
recidiva pós transplante, e resistência
secundária ao Imatinib, que motivou a
introdução de inibidor alternativo de
tirosina-quinase (Dasatinib).
No estudo de Chad et al (2007),13
envolvendo doentes com LMA Ph positivo,
através de uma análise retrospectiva multi-
institucional, estudaram-se características
clínicas, analíticas, imunofenotípicas e
citogenéticas possivelmente diferenciadoras
de doentes com diagnóstico documentado
de LMC em fase blástica e LMA Ph
positivo.1 Entre os resultados
estatisticamente significativos, observou-se
que os doentes com LMA Ph positivo
apresentavam menos comummente
esplenomegalia (25% vs 65%), menor

11
contagem absoluta e percentagem de
basófilos no mielograma (0,2% vs 6%) e
hemograma (0,6% vs 4,6%), menor rácio
mieloide/eritroide no mielograma (mediana
2,0 vs 4,8). Outras alterações citogenéticas
major, para além da translocação t(9;22),
foram também menos observadas na LMA
Ph positivo (70% vs 25%) e em 17% destes
doentes com LMA Ph positivo foi expressa
a proteína p190bcr-abl
, raramente expressa
nos doentes com LMC(<1%).13, 19
No caso em estudo, a pesquisa do
rearranjo bcr/abl no sangue periférico
detectou o transcrito bcr/abl b3a2 (p210),
não ajudando portanto na distinção entre
uma possível LMA de novo e LMC em crise
blástica mielóide ao diagnóstico.
Os rearranjos que codificam para a
proteína p210bcr-abl
tornam a LMA
indistinguível da LMC em crise blástica
mielóide, enquanto a expressão da p190bcr-
abl identifica com elevada fiabilidade a
verdadeira LMA, também chamada LMA
de novo.8,19 Esta premissa resultou de vários
estudos que identificaram diferenças
significativas nos rearranjos do gene bcr-abl
e na consequente expressão proteica nas
diferentes leucemias: a proteína p210bcr-abl
,
identificada no caso em estudo, é
encontrada em 99% dos casos de LMC, 50-
80% dos adultos com LLA Ph positiva e
50% das LMA Ph positivas; por sua vez a
proteína p190bcr-abl
é encontrada em menos
de 1% dos casos de LMC e nos restantes
casos de leucemia aguda Ph positivo.6 A
expressão conjunta de ambas as proteínas
(p210bcr-abl
e p190bcr-abl
) na LMC é muito
rara.19,20
Evolução clonal na LMC, definida
como anormalidades cromossómicas
adicionais ao cromossoma Philadelphia,
ocorre em 60 a 80% das crises blásticas de
LMC,21-23 sendo que as mais frequentemente
observadas são a trissomia do cromossoma
8, cromossoma Philadelphia adicional,
trissomia do cromossoma 19 e
isocromossoma 17q.21,24 Estas alterações
citogenéticas revelaram-se em diversos
estudos significativamente menos
frequentes nos doentes com LMA Ph
positivo (apenas 25% dos casos), sendo
assim mais um dado a relevar no
diagnóstico diferencial.21,24
Estes dados são a favor de uma
apresentação blástica mielóide de LMC no
caso em estudo, uma vez que o doente
evidenciou regressão para uma fase crónica
após terapêutica de indução com Imatinib e
ciclos de quimioterapia com ARA-C (ainda
que de curta duração e sem nunca atingir
resposta citogenética completa – à data do
internamento para alotransplante para MO
apresentava resposta citogenética parcial
com 25% de metáfases na MO com t(9;22)).
A aquisição de anormalidades
cromossómicas adicionais também foi

12
observada, apresentando ao diagnóstico
cariótipo complexo 50xy, +4, +6,
t(9;22)(q34;q41), +17, +der(22), que
evoluiu após terapêutica de indução com
Imatinib e transplante alogénico de MO,
com aquisição de trissomia 8, mantendo no
entanto a t(9;22) – 50xy, +4, +6, +8, t(9;22),
t(9;22) [18]/46xy[2]. Por outro lado,
coexistência de metáfases normais e
metáfases com t(9;22) ao diagnóstico, e
retorno ao cariótipo normal após
quimioterapia de indução, foram sugestivas
de LMA Ph positiva,18,25 que não se verificou
no caso em estudo, tendo sido detectada a
respectiva fusão em 100% dos núcleos no
estudo citogenético do sangue periférico.
Devido aos resultados decepcionantes
e prognóstico limitado é difícil estabelecer
uma terapêutica padrão para a crise blástica
da LMC. A sobrevivência média em crise
blástica linfoide após tratamento para
leucemia linfoblástica aguda varia de 9 a 12
meses, 26 e o resultado para crise blástica
após esquemas de tratamento baseados na
Citarabina para LMA é ainda mais curto,
com sobrevivências médias entre 3 e 5
meses.27
O único tratamento com possibilidade
curativa é o alotransplante de células
estaminais hematopoiéticas, no entanto, a
cura é rara, com menos de 10% dos doentes
a alcançar remissão durável.28
A associação de Imatinib com ARA-C
como terapêutica de indução utilizada no
doente em estudo não alcançou resposta
citogenética completa, mas permitiu atingir
resposta citogenética parcial antecedendo o
transplante alogénico de MO. No entanto,
verificou-se rápida recidiva pós-transplante,
mantendo cariótipo complexo e
apresentação em crise blástica um mês após
transplante, com rápida resistência
secundária ao Imatinib reiniciado após
transplante.
A combinação de quimioterapia com
Citarabina e Imatinib demonstrou em vários
estudos um efeito sinérgico no tratamento
de leucemia com cromossoma Philadelphia
positivo, com maiores reduções das células
leucémicas e com potencial para atrasar a
selecção clonal de células leucémicas
resistentes.29,30
Estes resultados foram ainda mais
evidentes em doentes diagnosticados em
fase blástica de LMC sem tratamento prévio
com Imatinib, como é o caso do doente em
estudo. No entanto, mesmo em casos com
resistência parcial, o tratamento combinado
com Citarabina e Imatinib pode ser
considerado, devido o efeito sinérgico do
tratamento combinado.31
Devido a mutações no domínio
tirosina-quinase bcr/abl – causa comum de
resistência ao imatinib e progressão da
doença – foram desenvolvidos novos

13
inibidores alternativos e mais potentes de
tirosina-quinase, como o nilotinib e
dasatinib para o tratamento de indução na
crise blástica.32
A resistência ao imatinib é incomum
em doentes em fase crónica inicial,
enquanto a incidência estimada de
resistência em dois anos é de 10%-20% em
LMC em fase crónica, 40%-50% em fases
acelerada e 70%-80% em crise blástica ou
LLA Ph positiva.33 Alguns doentes falham o
tratamento inicialmente (resistência
primária), enquanto outros perdem uma
resposta previamente adquirida (resistência
secundária), sendo esta última a mais
comum e associada ao desenvolvimento de
mutações no domínio bcr-abl.34,35
Em estudo fase II de doentes com
LMC em crise blástica
resistentes/intolerantes ao Imatinib tratados
com Dasatinib confirmou-se uma boa
resposta a esta terapêutica de salvação,
tendo atingido resposta citogenética
completa 26% dos doentes e 33% resposta
citogenética parcial. Uma resposta
hematológica major foi verificada em 34%
dos doentes em crise blástica. O tempo
médio para a progressão da doença após
início da terapêutica com Dasatinib neste
coorte de doentes foi 6-7 meses.36
A comparação destes resultados com
o doente em estudo foi dificultada pela curta
duração do tratamento (15 dias) devido ao
início de derrame pleural de grande volume
que motivou a sua suspensão. No entanto,
verificou-se uma rápida melhoria
hematológica após a introdução deste
fármaco (com rápida e intensa redução das
formas blásticas) e recidiva agressiva e
fulminante após a sua retirada, o que pesa a
favor da boa resposta do doente ao fármaco.
A presença de derrame pleural é uma
complicação importante no tratamento com
Dasatinib, sendo reportada em várias séries
frequências até 36% nos doentes em fase
blástica mielóide (este grupo de doentes
apresentou maior risco de complicações
com Dasatinib).36 Contrariamente ao descrito
em vários relatos na literatura, que referem
melhoria e ausência de recidiva do derrame
pleural após interrupção ou redução da dose
do fármaco,36,37 o doente em estudo
apresentou recidiva de derrame pleural de
grande volume após a suspensão do
Dasatinib que motivou toracocentese
terapêutica e impediu a reintrodução do
fármaco.
No doente em estudo foi utilizado
Dasatinib na dose de 100mg/dia,
procurando um bom compromisso entre os
possíveis efeitos laterais e controlo da
progressão da doença.
Na avaliação da dose mais eficaz para
o tratamento de doentes com LMC
resistentes ou intolerantes ao Imatinib,
estudo conduzido em 139 centros do

14
mundo, avaliou quatro possibilidades de
doses do dasatinib: 100 mg 1x/dia, 50 mg 2
x/dia, 140 mg 1 x/dia ou 70 mg 2 x/dia. O
estudo mostrou que, fundamentalmente, em
termos de resposta, tanto hematológica
quanto citogenética, os três grupos
obtiveram resultados muito parecidos. A
principal diferença observada foi que, na
dose de 100 mg 1 x/dia, o perfil de
toxicidade foi menor que nas outras
modalidades de apresentação, com valor
estatístico para anemia, neutropenia e
trombocitopenia.38
No entanto, estudos mais recentes
identificaram vantagens em termos de
resposta hematológica e citogenética no
subgrupo de doentes em crise blástica, com
a utilização da dose de 70mg 2x/dia,39
realçando a importância de avançar na
investigação da dose ideal para este grupo
de doentes de alto risco.
Em conclusão, o estudo e discussão
deste doente com quadro leucémico
cromossoma Ph positivo, que se manifestou
de forma agressiva e atípica, com elevado
número de formas imaturas mielóides no
sangue periférico e resistência a todos os
esquemas terapêuticos propostos (inclusive
rápida recidiva após transplante alogénico
de MO), visa fornecer mais informações a
um diagnóstico diferencial entre duas
neoplasias mielóides raras, de mau
prognóstico e má resposta à terapêutica
actualmente proposta: LMA com
cromossoma Ph positivo e LMC com
apresentação em fase blástica.
AGRADECIMENTOS
À Dra. Luciana Pinho, pela
disponibilidade e colaboração
disponibilizada.
BIBLIOGRAFIA
1. Nowell PC, Hungerford DA. A minute chromosome in
human chronic granulocytic leukemia. Science.
1960;132:1497.
2.Rowley JD. A new consistent chromosomal abnormality
in chronic myelogenous leukaemia identified by quinacrine
fluorescence and Giemsa staining. Nature 1973; 243: 290-
3.
3. de Klein A, van Kessel AG, Grosveld G et al. A cellular
oncogene is translocated to the Philadelphia chromosome
in chronic myelocytic leukemia. Nature 1982; 300: 765-7.
4. Razelle Kurzrock, MD; Hagop M. Kantarjian, MD;
Brian J. Druker, MD; and Moshe Talpaz, MD.
Philadelphia Chromosome–Positive Leukemias: From
Basic Mechanisms to Molecular Therapeutics; Ann Intern
Med. 2003;138:819-830.
5. Shepherd P, Suffolk R, Halsey J, Allan N: Analysis of
molecular breakpoint and m-RNA transcripts in a
prospective randomized trial of interferon in chronic
myeloid leukaemia: No correlation with clinical features,
cytogenetic response, duration of chronic phase, or
survival. Br J Haematol 89546, 1995
6. Melo JV. The molecular biology of chronic myeloid
leukaemia. Leukemia 1996; 10:751–756. 7.Keung YK,
Beaty M, Powell BL, et al. Philadelphia chromosome
positive myelodysplastic syndrome and acute myeloid
leukemia: retrospective study and review of literature.
Leuk Res. 2004;28:579-586.
7. Faderl, S. et al. The biology of chronic myeloid
leukemia. N Engl J Med, v. 341, n. 3, p. 164-72, 1999.
8. Kurzrock R, Gutterman JU, Talpaz M. The molecular
genetics of Philadelphia chromosome-positive leukemias.
N Engl J Med 1988; 319: 990-8.
9. Bennett JM, Catovsky D, Daniel MT et al. Proposed
revised criteria for the classification of acute myeloid

15
leukemia: a report of the French-American-British
Cooperative Group. Ann Intern Med 1985; 103: 626-9.
10. Cigudosa JC, Almeida MTA, Carrasco V et al. BCR-
ABLrearrangement and “variant” Philadelphia
chromosome in de novo acute myelogenous leukemia FAB
subtype M1. Br J Haematol 1995; 91: 932-4.
11. Ahmed R, Naqi N, Hussain I, Khattak BK, Nadeem M,
Iqbal J. Presentating phases of chronic myeloid leukaemia.
J Coll hysicians Surg Pak. 2009 Aug;19(8):469-72.
12. Tardieu S, Brun-Strang C, Berthaud P, Michallet M,
Guilhot F, Rousselot P, et al. Management of chronic
myeloid leukemia in France: a multi-centered cross-
sectional study on 538 patients. Pharmacoepidemiol Drug
Saf 2005; 14:545-53.
13. Chad P. Soupir, Jo-Anne Vergilio, Paola Dal Cin,
Alona Muzikansky, Hagop Kantarjian, Dan Jones, and
Robert P. Hasserjian Philadelphia Chromosome–Positive
Acute Myeloid LeukemiaAJCP 2007 127:642-650
14. G.Colleoni, M. Satake , C.L. Borovik, J. Kerbauy, M.
Yamamoto. Leucemia mielóide aguda Ph1-positivo de
novo ou criseblástica de leucemia mielóide crônica?
Análise molecular e evolução clínica de um caso. Rev Ass
Med Brasil 1998; 44(3): 253-5
15. Savage DG, Szydlo RM, Goldman JM. Clinical
features at diagnosis in 430 patients with chronic myeloid
leukemia seen at a referral centre over a 16-year period. Br
J Haematolol 1997;96:111-116.
16. Keung YK, Beaty M, Powell BL, et al. Philadelphia
chromosome positive myelodysplastic syndrome and acute
myeloid leukemia: retrospective study and review of
literature. Leuk Res. 2004;28:579-586.
17. Paietta E, Racevskis J, Bennett JM, et al. Biologic
heterogeneity in Philadelphia chromosome–positive acute
leukemia with myeloid morphology: the Eastern
Cooperative Oncology Group experience. Leukemia.
1998;12:1881-1885.
18. Cuneo A, Ferrant A, Michaux JL, et al. Philadelphia
chromosome–positive acute myeloid leukemia:
cytoimmunologic and cytogenetic features.
Haematologica. 1996;81:423-427.
19. Razelle Kurzrock, MD; Hagop M. Kantarjian, MD;
Brian J. Druker, MD; and Moshe Talpaz MD. Philadelphia
Chromosome–Positive Leukemias: From Basic
Mechanisms to Molecular Therapeutics Ann Intern Med.
2003;138:825-830.
20. Dhingra K, Talpaz M, Kantarjian H, Ku S, Rothberg J,
Gutterman JU, et al. Appearance of acute leukemia-
associated P190BCR-ABL in chronic myelogenous
leukemia may correlate with disease progression.
Leukemia. 1991;5:191-5.
21. Cortes J, O’Dwyer ME. Clonal evolution in chronic
myelogenous leukemia. Hematol Oncol Clin North Am
2004;18:671-684, x.
22. Przepiorka D, Thomas ED. Prognostic significance of
cytogenetic abnormalities in patients with chronic
myelogenous leukemia. Bone Marrow Transplant.
1988;3:113-119.
23. Rowley JD. Ph1-positive leukaemia, including chronic
myelogenous leukaemia. Clin Haematol. 1980;9:55-86.
24. Goldman JM, Melo JV. Chronic myeloid leukemia:
advances in biology and new approaches to treatment. N
Engl J Med. 2003;349:1451-1464.
25. Berger R. Differences between blastic chronic myeloid
leukemia and Ph-positive acute leukemia. Leuk
Lymphoma 1993;11(suppl 1):235-237.
26. Derderian PM, Kantarjian HM, Talpaz M, et al.
Chronic myelogenous leukemia in the lymphoid blastic
phase: characteristics, treatment response, and prognosis.
Am J Med. 1993;4:69–74.
27. Sacchi S, Kantarjian HM, O’Brien S, et al. Chronic
myelogenous leukemia in nonlymphoid blastic phase:
analysis of the results of first salvage therapy with three
different treatment approaches for 162 patients. Cancer.
1999;6:2632– 2641.
28. Gratwohl A, Hermans J, Niederwieser D, et al. Bone
marrow transplantation for chronic myeloid leukemia:
longterm results. Chronic Leukemia Working Party of the
European Group for Bone Marrow Transplantation. Bone
MarrowTransplant. 1993;2:509–516.
29. Topaly J, Zeller WJ, Fruehauf S. Synergistic activity of
the new ABL-specific tyrosine kinase inhibitor STI571 and
chemotherapeutic drugs on BCR-ABL-positive chronic
myelogenous leukemia cells. Leukemia. 2001;5:342–347.
30. Topaly J, Zeller WJ, Fruehauf S. Combination therapy
with imatinib mesylate (STI571): synopsis of in vitro
studies. Br J Haematol. 2002;19:3–14.
31. Fruehauf S, Topaly J, Buss EC, Fischer T, Ottmann
OG, Emmerich B, Müller MC, Schuld P, Balleisen L,
Hehlmann R, Ho AD, Hochhaus A. Imatinib combined
with mitoxantrone/etoposide and cytarabine is an effective
induction therapy for patients with chronic myeloid
leukemia in myeloid blast crisis. Cancer. 2007 Apr
15;109(8):1543-9.
32. Delamain MT, Conchon, M. Os inibidores de tirosino
quinase de segunda geração. Rev Bras Hematol Hemoter.
2008;30(1):37-40
33. Brave M, Goodman J, Kaminskas E. Sprycel for
chronic myeloid leukemia and Philadelphia chromosome-
positive acute lymphoblastic leukemia resistent to or
intolerant of imatinib mesylate. Clin Câncer Res.
2008;14(2):352-9.
34. Keam S. Dasatinib. In chronic myeloid leukemia and
Philadelphia chromosome-positive acute lymphoblastic
leukemia. Adis Drug Profile. 2008;22(1):59-69.

16
35. Golemovic M, Verstovsek S, Giles F, et al. AMN107, a
novel aminopyrimidine inhibitor of Bcr-Abl, has in vitro
activity against imatinib-resistant chronic myeloid
leukemia. Clin Cancer Res. 2005;11;4941-7.
36. J Cortes, D-W Kim, E Raffoux, G Martinelli, E
Ritchie, L Roy, S Coutre, S Corm, N Hamerschlak, J-L
Tang, A Hochhaus, H J Khoury, T H Brümmendorf, M
Michallet, G Rege-Cambrin, C Gambacorti-Passerini, J P
Radich, T Ernst, C Zhu, J M A Van Tornout and M Talpaz
Efficacy and safety of dasatinib in imatinib-resistant or -
intolerant patients with chronic myeloid leukemia in blast
phase Dasatinib is effective in blast-phase CML Leukemia,
December 2008; 22, 2176-2183.
37. Bergeron A, Réa D, Levy V, Picard C, Meignin V,
Tamburini J, Bruzzoni-Giovanelli H, Calvo F, Tazi A,
Rousselot P. Lung abnormalities after dasatinib treatment
for chronic myeloid leukemia: a case series. Am J Respir
Crit Care Med. 2007 Oct 15;176(8):814-8.
38. Hochhaus A, Kim DW, Rousselot P, et al. Dasatinib
(Sprycel) 50 mg or 70 mg BID vs 100 mg or 140 mg QD
in patients with chronic myeloid leukemia in chronic phase
(CML-CP) resistant or intolerant to imatinib: results of the
CA180-034 study. Blood. 2006;108:53a.
39. Lindauer M, Hochhaus A. Dasatinib. Recent Results
Cancer Res. 2010;184:83-102.





























