Embed Size (px)
Citation preview

11. Descrever o mecanismo de ação e as ações farmacológicas dos anti-
inflamatórios não esteróides. (Documento PDF “Anti-
inflammatory drugs”)
Inibidores da Ciclo-Oxigenase
Este grupo inclui os medicamentos "tradicionais" anti-inflamatórios não-
esteroides (AINEs) assim como os mais recentes coxibes, que são mais seletivos para
COX-2.
Estes medicamentos provocam alívio sintomático da dor e inchaço na doença
articular crónica tal como na osteoartrite e na artrite reumatoide, bem como em
condições inflamatórias agudas, tais com fraturas, entorses, desportos e em outras
lesões dos tecidos moles. Também são úteis no tratamento de dor pós-operatória, dor
dentária e menstrual e, ainda, de dores de cabeça e enxaqueca. Vários AINEs estão
disponíveis ao balcão e são amplamente utilizados para outros tipos de pequenas dores
e sofrimentos. Existem muitas formulações diferentes disponíveis como comprimidos,
injeções e géis. Todos estes medicamentos, em particular os AINEs "tradicionais",
podem ter efeitos indesejados significativos, especialmente nos idosos. Agentes mais
novos têm menos ações adversas. Algumas estruturas representadas na Figura 1 e as
características específicas no Quadro 1.
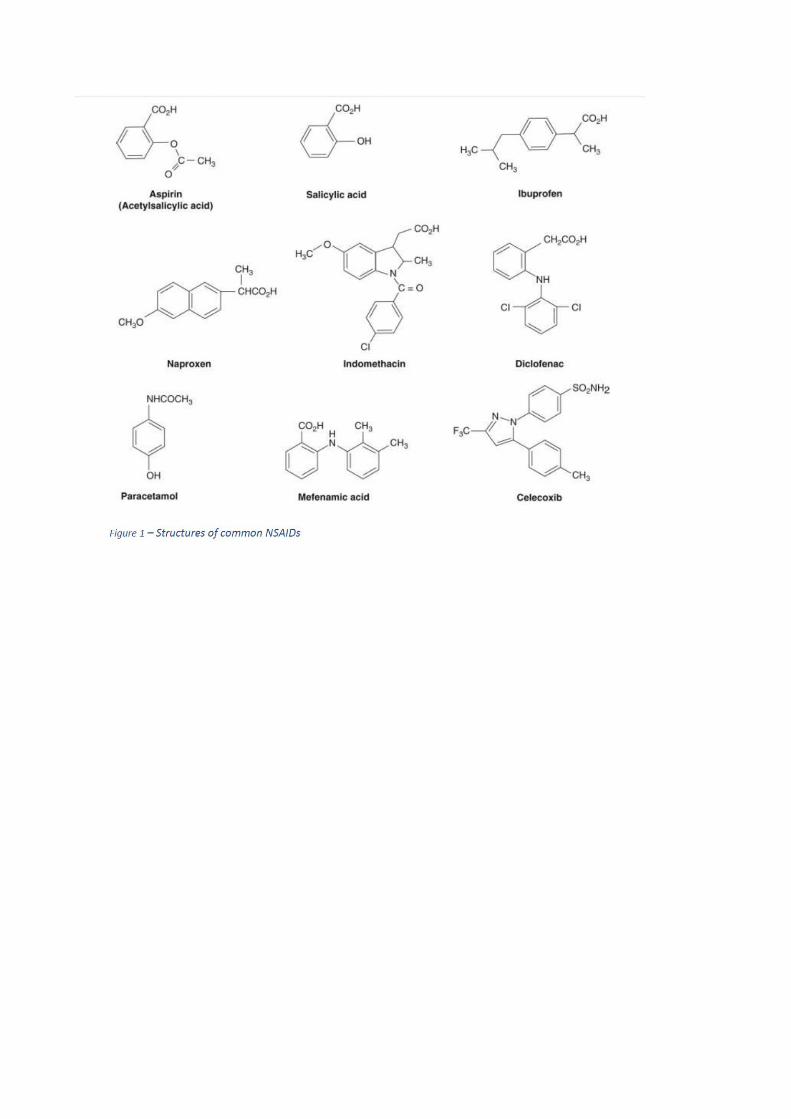

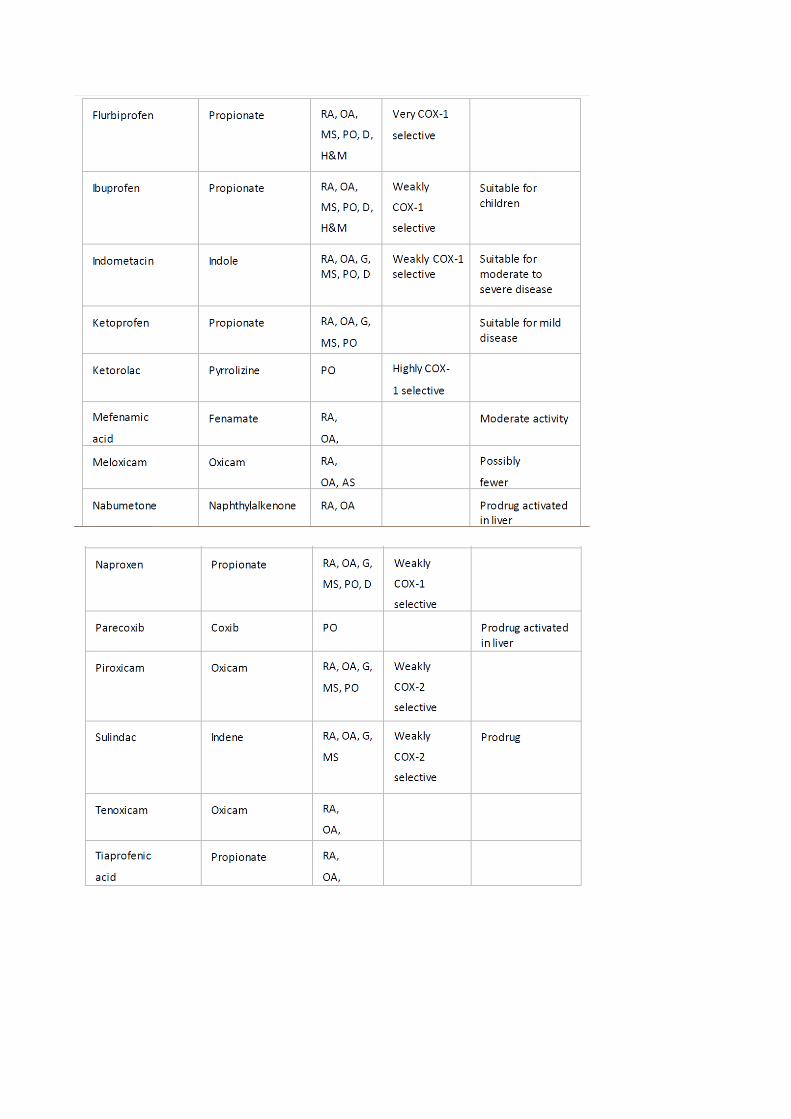


Mecanismo de Ação
Vane e seus colegas estabeleceram, em 1971, que as principais ações de AINEs
eram na inibição da oxidação do ácido araquidónico pelas COXs de ácidos gordos.
Embora hajam diferenças entre os diversos AINEs, a sua ação farmacológica primária
está relacionada com a sua capacidade de inibir a enzima COX do ácido gordo, inibindo
assim a produção de prostaglandinas e tromboxanos. Existem duas isoformas comuns
desta enzima, COX-1 e COX-2. Também pode haver outras enzimas COX que podem
gerar prostaglandinas, mas estes não foram completamente esclarecidos. COX-1 e
COX-2 estão intimamente relacionados (> 60% de identidade de sequência) e
catalisam a mesma reação, é evidente que existem diferenças importantes entre a
expressão e papel destas duas isoformas. COX-1 é uma enzima constitutiva expressa
na maioria dos tecidos, incluindo as plaquetas do sangue; tem um papel
“housekeeping” no corpo, pode estar envolvido na hemóstase dos tecidos e é
responsável pela produção de prostaglandinas (envolvidos na proteção gástrica, na
agregação plaquetar, na autorregulação do fluxo sanguíneo renal e na iniciação do
parto.
COX-2 é induzida em células inflamatórias quando são infetadas, feridas ou
ativadas (por citoquinas inflamatórias - interleucina (IL-1) e factor de necrose tumoral
(TNF)- . Assim, a COX-2 é a principal isoforma responsável pela produção dosα
mediadores prostanóides da inflamação, embora haja algumas exceções significativas.
Por exemplo, existe um considerável conjunto "constitutivo" de COX-2 presente no
sistema nervoso central (SNC) e em alguns outros tecidos, embora a sua função nestes
locais ainda não é completamente clara.
A maioria dos AINEs "tradicionais" inibem tanto a COX-1 como a COX-2, embora
variem no grau ao qual eles inibem cada isoforma. Acredita-se que a ação anti-
inflamatória (e provavelmente a maior parte analgésica e antipirética) dos AINEs esteja
relacionada com a inibição da COX-2, enquanto os seus efeitos indesejados
(particularmente os que afetam o trato gastrointestinal) são, em grande parte, um
resultado da inibição da COX-1. Os compostos que inibem seletivamente a COX-2 são
usados na clínica, mas, enquanto estes fármacos mostrarem efeitos colaterais
gastrointestinais, eles não são tão bem tolerados. Existe uma preocupação sobre os

efeitos cardiovasculares de todos os AINEs quando estes são tomados por um longo
tempo.
Outras ações para além da inibição da COX podem contribuir para os efeitos anti-
inflamatórios de alguns AINEs. Os radicais reativos de oxigénio produzidos pelos
macrófagos e neutrófilos estão implicados nos danos dos tecidos e alguns AINEs
(sulindac) têm o efeito sequestrador dos radicais de oxigénio, bem como atividade
inibidora da COX, o que pode diminuir o dano tecidual. A aspirina inibe também a
expressão do fator de transcrição NFkB, que tem um papel chave na transcrição dos
genes para a mediadores inflamatórios.
Ação Terapêutica
Todos os AINEs têm ações muito similares às da aspirina. Os AINEs padrão foram
introduzidos na medicina clínica na década de 1890.
Efeitos Anti-inflamatórios
Muitos mediadores coordenam reações inflamatórias e alérgicas. Os AINEs
reduzem principalmente os componentes da resposta inflamatória e imune na qual, as
prostaglandinas, derivadas principalmente da COX-2, desempenham um papel
significativo. Esses incluem:
Vasodilatação (redução da síntese de prostaglandinas
vasodilatadoras);
Edema (ação indireta: a vasodilatação facilita e potencia a ação de
mediadores como a histamina, que aumenta a permeabilidade das
vênulas pós-capilares).~
Enquanto os AINEs suprimirem os sinais e sintomas de inflamação, têm pouca ou
nenhuma ação na doença crónica subjacente. Como classe, geralmente, não têm efeito
direto sobre outros aspetos da inflamação, como a citocina/libertação de quimiocina, a
migração de leucócitos, a libertação da enzima lisossomal e a produção de radicais de

oxigénio tóxicos, que contribuem para o dano tecidual em condições inflamatórias
crónicas tais como artrite reumatoide, vasculite e nefrite.
Efeito Antipirético
O hipotálamo controla o equilíbrio entre a perda de calor e a produção de calor,
regulando a temperatura corporal. A febre ocorre quando há um distúrbio do
'termostato' hipotalamico, o que leva ao aumento da temperatura do corpo. AINEs
'reiniciam' este termostato. Uma vez que ocorre um retorno ao estado normal, os
mecanismos de regulação de temperatura (dilatação dos vasos sanguíneos superficiais,
sudorese, etc.) operaram no sentido de reduzir a temperatura. Temperatura normal do
corpo em seres humanos não é afetada pelos AINEs.
Os AINEs exercerem a sua ação antipirética, em grande medida, através da
inibição da produção de prostaglandinas no hipotálamo. Durante uma reação
inflamatória, endotoxinas bacterianas provocam a libertação, por macrófagos, de IL-1, o
qual estimula a gerar, no hipotálamo, prostaglandinas de tipo E- que elevam a
temperatura. COX-2 pode ter um papel aqui, porque a IL-1 induz o endotélio vascular
do hipotálamo. Há algumas evidências que as prostaglandinas não são os únicos
mediadores de febre. Os AINEs podem ter um efeito antipirético adicional por
mecanismos ainda desconhecidos.
Efeito Analgésico
Os AINEs são eficazes contra a dor leve ou moderada, especialmente as
decorrentes da inflamação ou dano tecidual. Dois locais de ação foram identificados:
1. Periférica: diminuem a produção de prostaglandinas que sensibilizam os
nociceptores de mediadores inflamatórios (como a bradicinina) e são
eficazes em casos de artrite, bursite, dores musculares e de origem
vascular, dor de dentes, dismenorreia, dores pós-parto e dores de

metástases de cancro no osso. Todas as condições estão associadas com o
aumento da síntese local de prostaglandinas provavelmente como
resultado da indução da COX-2. Sozinha, ou em combinação com opióides,
diminuem a dor de pós-operatório e em alguns casos, pode reduzir a
necessidade de opióides. A sua capacidade para aliviar a dor de cabeça
pode estar relacionada com a redução de prostaglandinas vasodilatadoras
que atuam na vasculatura cerebral.
2. Para além de efeitos periféricos, existe uma segunda ação central (menos
bem definida) possivelmente na medula espinal. Lesões inflamatórias
aumentam a COX-2 e libertação de prostaglandina, causando uma
facilitação da transmissão, pelas fibras de dor aferentes, para retransmitir
neurónios no corno dorsal.
Efeitos Indesejados
No geral, os efeitos indesejáveis são bastantes, provavelmente refletido no facto de
os AINEs serem extensivamente usados na população idosa vulnerável, e frequentemente
por períodos de tempo prolongados. Quando usado para doenças de articulações (o que
geralmente necessita de doses bastante elevadas e uso continuado longo), há uma
incidência elevada de efeitos colaterais, particularmente no trato gastrointestinal mas
também no fígado, rim, baço, o sangue e medula óssea.
Como as prostaglandinas estão envolvidas na proteção gástrica, na agregação
plaquetária, na autorregulação vascular renal e na indução do parto, todos os AINEs
compartilham um perfil semelhante de efeitos secundários dependentes do mecanismo,
embora possam haver outros efeitos indesejados adicionais. Inibidores seletivos da COX-2
têm menos, mas não desprezáveis, toxicidade gastrointestinal.

Distúrbios Gastrointestinais
Efeitos gastrointestinais são os efeitos indesejados mais comuns dos AINEs.
Acredita-se que resultam, principalmente, da inibição de COX-1 gástrica, que é
responsável pela síntese das prostaglandinas que normalmente inibem a secreção de
ácido e protegem a mucosa. Incluem desconforto gástrico, dispepsia, diarreia (mas às
vezes obstipação), náuseas e vómitos, e em alguns casos, sangramento e ulceração
gástrica. Foi estimado que 34-46% dos utilizadores de AINEs irá sustentar algum dano
gastrointestinal que, embora possa ser um sintomático, acarreta um risco de
hemorragia e/ou perfuração grave. O dano é observado quando os fármacos são
administrados oral ou sistemicamente. A Figura 2 apresenta os riscos relativos aos
danos gastrointestinais de alguns AINEs. A administração oral de análogos de
prostaglandinas, como o misoprostol, pode diminuir o dano gástrico produzido por
estes agentes.
Efeitos gastrointestinais dos AINEs diferem da úlcera péptica pela localização
das lesões mais frequentemente no estômago e não no duodeno. Além disso, as lesões
induzidas pelos AINEs ocorrem com mais frequência nos idosos, do que as úlceras
pépticas típicas. Normalmente, o estômago protege-se contra os efeitos nocivos de
ácido clorídrico e da pepsina por uma série de mecanismos protetores referidos como a
barreira da mucosa gástrica, a qual é constituída por células epiteliais, camada de muco
e bicarbonato, e o fluxo de sangue na mucosa. A mucosa gástrica é um gel constituído
por polímeros de glicoproteína, que limitam a difusão de iões de hidrogénio. Estes
polímeros reduzem a taxa com que os iões de hidrogénio (produzida no lúmen) e iões
de bicarbonato (secretada pela mucosa) se misturam; Deste modo, um gradiente de pH
é criado através da camada de muco. Normalmente, as células da mucosa gástrica são
rapidamente reparadas quando são danificados por diversos fatores, como alimentos,
etanol, ou ingestão aguda de AINEs. Entre os mecanismos citoprotetores, está a
capacidade de prostaglandinas das séries PGE, particularmente PGE-1, para aumentar a
secreção de muco e bicarbonato de iões e para manter o fluxo de sangue na mucosa. As
prostaglandinas também diminuem a secreção de ácido, permitindo que a barreira da
mucosa gástrica permaneça intacta. O uso de PGE-1 para reduzir lesões gástricas
induzidas pelos AINEs é limitada pelo facto de não ser eficaz por via oral e degradar-se

rapidamente na administração parentérica, principalmente por oxidação do grupo 15-
hidroxi. Para superar estas limitações, o misoprostol foi sintetizado como um pró-
fármaco análogo de prostaglandina onde a atividade oral foi conseguida por
administração do fármaco como éster metílico, permitindo que o ácido bioativo seja
libertado após a absorção.
Baseado na extensa evidência experimental, era previsto que os agentes seletivos
da COX-2 teriam boas ações anti-inflamatórias e analgésicas com menores danos
gástricos. Dois grandes estudos prospetivos compararam os efeitos colaterais
gastrointestinais do celecoxib e rofecoxib com os AINEs padrão em pacientes com
artrite e mostrou algum benefício, embora os resultados não fossem tão claros como
se esperava.

Reações da Pele
Erupções cutâneas são o efeito indesejável idiossincrático comuns dos AINEs, em
particular com ácido mefenâmico (frequência de 10-15%) e sulindac (frequência de 5-
10%). Variam de leve eritematosa, reações de urticária e fotossensibilidade para
doenças mais graves e potencialmente fatais, incluindo síndrome de Stevens-Johnson
(uma erupção vesicular que se estende para o intestino) e necrólise epidérmica tóxica,
caracterizada por necrose epitelial generalizada (felizmente raro). O mecanismo não é
claro.
Efeitos adversos Renais
Doses terapêuticas de AINEs em indivíduos saudáveis representam pouca
ameaça à função renal mas em pacientes suscetíveis causam insuficiência renal aguda,
que é reversível com a suspensão/interrupção do fármaco. Isto ocorre através da
inibição da biossíntese dos prostanóides (PGE2 e PGI2; prostaciclina) envolvidos na
manutenção do fluxo sanguíneo renal, especificamente no PGE2 - vasodilatação
compensatória mediada que ocorre em resposta à ação de noradrenalina
(norepinefrina) ou à angiotensina II. Os recém-nascidos e os idosos estão
especialmente sobe risco, assim como os pacientes com doença ao nível do coração,
rins ou fígado, ou um reduzido volume de sangue circulante.
O consumo crónico de AINEs, especialmente «abuso» AINEs, pode causar
nefropatia analgésica caracterizada por nefrite crónica e necrose papilar renal. O uso
regular de doses prescritas de AINEs é menos perigoso para o rim que o uso intenso e
prolongado de analgésicos “sobre-o- balcão” no contexto social.

Efeitos adversos Cardiovasculares
Embora tivesse sido reconhecido há algum tempo que os AINEs poderiam opor
os efeitos de alguns fármacos hipertensivos, não há atualmente preocupação sobre o
potencial desses fármacos, quando administrado sozinho, para aumentar a pressão
arterial, e, portanto, predispor a eventos cardiovasculares adversos, como acidente
vascular cerebral e enfarte do miocárdio. Este primeiro surgiu durante os ensaios do
inibidor da COX-2 rofecoxib.
Parece agora que a farmacologia cardiovascular adversa, especialmente após uso
prolongado ou em pacientes com risco cardiovascular pré-existente, pode ser um efeito
comum de todos os AINEs, embora alguns (por exemplo, naproxeno) parece ser melhor
tolerado a este respeito do que outros. A explicação mais provável para este efeito é que
a hipertensão é secundária à inibição de COX-2 na secreção de renina na região da
mácula densa do rim. O efeito hipertensivo é dose e tempo-dependente.
Outros efeitos indesejáveis
Aproximadamente 5% dos pacientes expostos aos AINEs podem experimentar
asma sensível à aspirina. O mecanismo exato é desconhecido, mas a inibição da COX
está implicada e a presença de um sensibilizador, pré existência de infeção viral pode
ser culpada. A aspirina é o pior “ofender”, mas não há reação cruzada com os outros
AINEs, exceto inibidores da COX-2 possivelmente. Outros, muito menos comuns, os
efeitos indesejáveis dos AINEs incluem efeitos sobre o SNC, distúrbios da medula
óssea e distúrbios do fígado, sendo o último mais provável se já houver insuficiência
renal.
Overdose de Paracetamol provoca insuficiência hepática. Todos os AINEs (exceto
inibidores COX-2) previnem a agregação de plaquetas e, portanto, podem prolongar
hemorragias. Mais uma vez, a aspirina é o principal problema a este respeito.

PARACETAMOL
Paracetamol (acetaminofeno nos EUA) é um dos analgésicos-antipiréticos não
narcóticos mais vulgarmente utilizados. Em alguns aspetos, o fármaco constitui uma
anomalia: enquanto ele tem excelente atividade analgésica e antipirética, que pode ser
atribuída a inibição da síntese das prostaglandinas no CNS, que tem atividade anti-
inflamatória fraca (exceto em alguns casos específicos) e não compartilha os efeitos
secundários gástricos e plaquetários dos outros AINEs. Por esta razão, o paracetamol
por vezes não é classificado como um AINE.
Uma potencial solução para este problema foi fornecido pela observação de uma
nova isoforma da COX, COX-3 (um produto de splicing alternativo da COX-1) existente,
predominantemente, no SNC de algumas espécies, e que o paracetamol, assim como
outros fármacos com propriedades semelhantes (por exemplo, antipirina e dipirona),
são inibidores seletivos desta isoforma. Esta ideia ainda está sob investigação.
Antipiréticos interferem com os processos em que os fatores pirogénicos
produzem febre, mas não aparecem para baixar a temperatura do corpo em sujeitos
febris. Tinha sido historicamente aceite que os antipiréticos exercem as suas ações no
sistema nervoso central (SNC), principalmente no centro termorregulador do
hipotálamo, mas as evidências mais recentes sugerem que as ações periféricas
também podem contribuir. Pirogénicos leucocitários endógenos podem ser libertados
a partir de células que foram ativadas por vários estímulos.
Antipiréticos podem atuar através da inibição da ativação destas células por
pirogénicos exógenos ou por inibição da libertação de pirogénicos leucocitários
endógenos pelas células que tenham sido ativadas por pirogénicos exógenos. A
evidência substancial sugere um mecanismo central antipirético, um antagonismo que
pode resultar de qualquer uma competição direta de pirogénico e um agente
antipirético nos recetores do SNC, ou uma inibição das prostaglandinas do SNC. O
paracetamol pode inibir impulsos de dor, exercendo um efeito depressor sobre os
recetores periféricos; um efeito antagónico sobre as ações da bradicinina pode ter um
papel. Os efeitos antipiréticos podem não resultar da inibição da libertação de
pirogénicos endógenos a partir de leucócitos, mas sim da inibição da ação de pirogénico
endógeno lançado no centro termorregulador do hipotálamo. O facto de o paracetamol

ser um antipirético/analgésico eficaz, mas um agente anti-inflamatório ineficaz pode
resultar da sua maior inibição da biossíntese de prostaglandina via inibição da isoforma
COX-3 no SNC comparativamente com a na periferia.
Outros estudos têm mostrado que o paracetamol aumenta os níveis de
endocanabinóides nos tecidos traumatizados pela inibição das enzimas
metabolizadoras de endocanabinóides e que isto pode ser um mecanismo importante
para a analgesia. Vários, se não todos, os AINEs exibem a capacidade de inibir a
hidrolase de amida de ácido gordo (FAAH) e elevar os níveis de endocanabinóides. Os
endocanabinóides trouxeram uma maior contribuição para o mecanismo de ação do
paracetamol. É baseado na observação do ibuprofeno e de outros AINEs, que inibem a
FAAH. Outras pesquisas têm sustentado este ponto de vista e podem explicar a
atividade anómala de paracetamol, uma vez que recetores agonistas de canabinóides
são conhecidos por terem ações analgésica e hipotérmica.
Graham e Scott apresentaram uma hipótese onde o paracetamol atua pela
depleção dos stocks de glutationa, que é um co-fator conhecido por PGE sintase. Isto
explicaria a diminuição da produção de PGE e o concomitante efeito analgésico. O
esgotamento da glutationa é a principal causa da toxicidade do paracetamol. A alta
reatividade da benzoquinona-imina, formado pela isoforma CYP2E1, deve ser
conjugada com glutationa antes que possa reagir com outros componentes celulares
cruciais. Em caso de overdose, a falha do mecanismo molecular resulta em sérios danos
no fígado.
Aspetos farmacocinéticos
O paracetamol é administrado por via oral e é bem absorvido, atingindo o pico das
concentrações plasmáticas em 30-60 min. O tempo de semi-vida no plasma das doses
terapêuticas é 2-4h, mas em doses tóxicas, pode ser alargada a 4-8h. O paracetamol é inativado
no fígado, sendo conjugado para dar o glucurónido ou sulfato. Liga-se às proteínas do plasma
(18% a 25%).
Efeitos indesejáveis

Em doses terapêuticas, os efeitos colaterais são poucos e raros, apesar de
reações alérgicas de pele ocorrerem às vezes. É possível que a ingestão regular de
grandes doses num longo período de tempo possa causar danos renais.
Doses tóxicas (10-15 g) causam hepatotoxicidade potencialmente fatal. Isto
ocorre quando as enzimas hepáticas que catalisam as reações de conjugação estão
saturadas. O metabolito tóxico resultante, N-acetil-p-benzoquinona imina, é inativado
pela conjugação com glutationa. Quando esta se esgota, o intermediário tóxico acumula-
se e provoca necrose no fígado e também nos túbulos renais. Há aumento da toxicidade
do paracetamol entre os alcoólatras uma vez que a ingestão de álcool induz tanto a
CYP2E1 e a CYP3A4 e esgota os stocks de glutationa.
Os sintomas iniciais de intoxicação aguda por paracetamol são náuseas e
vómitos, a hepatotoxicidade pode ser uma manifestação atrasada que ocorre 24-48 h
mais tarde. Se o doente for visto logo após a ingestão, a lesão hepática pode ser
impedida por agentes que aumentam a formação de glutationa no fígado (acetilcisteína
por via intravenosa, por via oral ou metionina). Se passarem mais de 12h após a
ingestão de uma dose elevada, os antídotos, que podem causar efeitos adversos
(náusea, reações alérgicas), têm menos probabilidade de serem úteis. Infelizmente, a
ingestão de grandes quantidades de paracetamol é um método comum de suicídio.
Interações
Às 4 g/dia, o paracetamol foi relatado para potenciar a resposta aos
anticoagulantes orais, o aumento do tempo de protrombina (valores internacionais
razão normalizada) de duas a três vezes. Interações com a varfarina, dicumarol,
anisindione, e difenadiona foram sugeridas. O mecanismo destas interações não foi
totalmente elucidado, mas pode ser associado à competição por sítios de ligação às
proteínas do plasma, porque o paracetamol é um ácido fraco e é fracamente ligado,
mas também pode interferir com as enzimas envolvidas na síntese de fatores de
coagulação dependentes de vitamina K. A absorção do paracetamol é reforçada por
polissorbato e sorbitol e é reduzida por anticolinérgicos e analgésicos narcóticos.

Incompatibilidades químicas também foram relatadas com base na hidrólise por
ácidos ou bases fortes, ou por oxidação de compostos fenólicos na presença de agentes
oxidantes. O paracetamol forma misturas “sticky” com HCl difenidramina e quando
descolorado em condições húmidas na presença de cafeína ou codeína fosfato.
Aspirina
A molécula da aspirina é relativamente insolúvel em água, então é
administrada sob a forma de sal cálcio ou sódio.
A aspirina é utilizada na clínica maioritariamente como um fármaco
cardiovascular, devido à sua capacidade de inibir de forma prolongada as COX-1
e desta forma reduzir a agregação plaquetária.
Todos os AINEs possuem a capacidade de reduzir a agregação, no entanto
é a aspirina que possui uma duração mais prolongada. Isto deve-se a capacidade
de acetilar de forma irreversível as COX. As plaquetas, ao contrário do resto dos
tecidos do corpo, são incapazes de formar novas enzimas (COX).
Pequenas doses de aspirina são extremamente eficazes na inibição da
função plaquetária, durante toda a sua vida (10 dias). Desta forma existe uma
menor probabilidade do doente desenvolver problemas cardiovasculares, como
enfarte.
Este fármaco também é utilizado noutras doenças, tais como:
Cancro Rectal e Colónico;
Doença de Alzheimer;
Farmacocinética
Este fármaco é um ácido fraco, protonado no estômago mas que possui
maior absorção no íleo. É rapidamente hidrolisado por esterases, tanto no
plasma como nos tecidos, em especial relevo no fígado. O metabolito é provido

de ação anti-inflamatória, no entanto o mecanismo pelo qual isso acontece não
está completamente explicado, mas pensa-se que envolva o sistema das COX.
Pode ser eliminada de três formas distintas. Pode ser eliminada sob a
forma inalterada na urina, pode ser conjugada a dar glucuronido ou sulfato ou
pode ser oxidada. A eliminação acontece em maiores quantidades numa urina
alcalina.
O tempo de semivida é dependente das doses, mas possui uma duração de
ação elevada, uma vez que inibe irreversivelmente as COX. Possui ainda Uma
elevada ligação às proteínas plasmáticas.
Efeitos Adversos
Efeitos Gástricos;
Salicilismo (tonturas, surdez e zumbido);
Acidose Metabólica em CRIANÇAS;
Encefalite Pós-viral;
Quando dada concomitantemente com Varfarina, aumenta a probabilidade
de sangramentos;
Síndrome de REYES
Desordem rara que ocorre em crianças, caracterizada por encefalite
hepática seguida de uma doença viral aguda e morte.
ASPIRINA NÃO PODE SER DADA A CRIANÇAS!!
Envenenamento Agudo por Salicilatos
Emergência médica que acontece maioritariamente em crianças e
tentativas de suicido. Ocorre uma perturbação no balanço dos electrólitos e de
ácido-base.

O fármaco contribui para uma diminuição da fosforilação oxidativa, que
por sua vez leva a um aumento do consumo de oxigénio pelo músculo
esquelético e desta forma ocorre um aumento da quantidade de dióxido de
carbono produzido. Isto leva a um aumento da frequência respiratória que pode
resultar em situações de hiperventilação. Tudo isto provoca uma alcalose
respiratória que normalmente é compensada pela libertação de bicarbonato de
sódio pelos rins.
Grandes quantidades libertadas de bicarbonato de sódio podem levar a
uma depressão do sistema respiratório, e desta forma vai ocorrer uma retenção
de dióxido de carbono no plasma sanguíneo. Sendo assim ocorre uma acidose
metabólica que leva a acumulação de metabolitos de ácido pirúvico, láctico e
acetoacético.
Tudo isto leva a hiperpirexia secundaria, desidratação e vómitos. Ocorre a
excitação do SNC, seguida de coma e depressão respiratória.
Hipersensibilidade aos Salicilatos
Pouco comum no caso da aspirina, no entanto quando acontece, é fatal. A
hipersensibilidade é composta por vários sintomas, desde secreções, urticária,
edema, broncoconstrição, choque anafilático, etc...
Asmáticos apresentam uma maior risco de hipersensibilidade.
Interações Medicamentosas
Uma vez que é um fármaco com elevada ligação às proteínas plasmáticas,
vai competir pela ligação com outros fármacos (p.e. Metotrexato).
Pode ocorrer um aumento do efeito da Varfarina.
Pode interferir com o efeito de alguns hipertensivos e outros agentes
como o probenicide. Pode aumentar a toxicidade das sulfonamidas.

Ácidos Arilalcanóicos
Tal como os salicilatos, estes ácidos têm a propriedade de inibir a
biossíntese de prostaglandinas através da inibição da COX-1 e COX-2, com vários
graus de seletividade.
Todos os compostos que fazem parte dos ácidos arilalcanóicos são
extensamente metabolizados. Ocorre primariamente através de sistemas
enzimáticos hepáticos microssomais e pode levar a desactivação ou bioactivação
de moléculas parentais.
Possuem uma elevada ligação às proteínas plasmáticas e podem afectar a
ligação de outros, resultando num aumento de atividade e toxicidade. A
Indometacina não apresenta este efeito.
A interação mais comum é observada entre os ácidos arilalcanóicos e
anticoagulantes orais, nomeadamente a Varfarina. A coadministração pode
prolongar o tempo de protrombina.
Potenciais interações com outros fármacos acídicos, tais como
hidantoínas, sulfonamidas e as sulfonilureias, devem ser monitorizadas.
A administração concomitante de aspirina diminui os níveis plasmáticos
de ácidos arilalcanóicos. O Probenecid, por outro lado, tende a aumentar estes
níveis.
Interações com fármacos capazes de induzir os sistemas enzimáticos
microssomais hepáticos, tal como o fenobarbital, podem aumentar ou diminuir a
atividade anti-inflamatória, dependendo se o ácido arilalcanóico é
metabolicamente bioactivado ou inativado pelo sistema enzimático.
Alguns diuréticos, tal como a Furosemida, inibem o metabolismo das
prostaglandinas e o consequente aumento dos níveis de PGE2 induz a actividade
da renina do plasma. Visto que os ácidos arilalcanóicos bloqueiam a biossíntese

de prostaglandinas, os efeitos da furosemida podem ser antagonizados, com uma
interação medicamentosa potencialmente significativa.
Derivados do Ácido Acético
Indometacina
É um dos NSAIDs mais potentes em uso. É
mais potente como antipirético do que a aspirina
ou o acetaminofeno e possui cerca de 10 vezes a
potência analgésica da aspirina. No entanto, o
efeito analgésico é desvalorizado devido à
frequência de efeitos secundários.
As soluções aquosas de indometacina não
são estáveis.
A sua absorção ocorre rapidamente através de administração oral.
Forte ligação às proteínas plasmáticas.
É convertida em metabolitos inactivos, 5-O desmetilados pelas CYP2C9.
A sua capacidade para inibir potencialmente a biossíntese de
prostaglandinas pode explicar a sua acção anti-inflamatória, antipirética e
analgésica.
Efeitos secundários são frequentemente observados em doses anti-
reumáticas. Indivíduos com mais de 70 anos de idade experienciam efeitos
indesejáveis ao nível do TGI (náuseas, dispepsia, diarreia, erosão/úlceras nas
paredes estomacais), do SNC (dores de cabeça, tonturas e vertigens) e das
orelhas (zumbidos). Posto isto, poderá ser necessário interromper seu uso. Tal
como acontece com outros ácidos arilalcanóicos, a administração de
indometacina com alimentos ou leite diminui os efeitos colaterais a nível
gastrointestinal.

É usada no tratamento a curto prazo da artrite gotosa aguda, dor aguda da
espondilite anquilosante e osteoartrite. A sua forma injectável está disponível
como sal sódico tri-hidratado para uso IV em prematuros com ‘patent ductus
arteriosus’. Devido à sua capacidade para suprimir a actividade uterina através
da inibição da biossíntese de prostaglandinas, também é utilizada para evitar o
parto prematuro.
Diclofenac
Possui propriedades anti-inflamatórias,
analgésicas e antipiréticas.
É único entre os AINES na medida em que
possui três possíveis mecanismos de ação:
1) inibição do sistema COX do ácido
araquidónico, tendo como resultado a
diminuição da produção de prostaglandinas e
tromboxanos ;
2) inibição da via da lipoxigenase, o que resulta na diminuição da
produção de leucotrienos, particularmente do LKB4 pró-inflamatório;
3) inibição da libertação do ácido araquidónico e estimulação da sua
recaptação, reduzindo o ácido araquidónico disponível;
É rápida e completamente absorvido por administração oral.
Tem Forte ligação às proteínas plasmáticas.
Apresenta um extenso metabolismo hepático.
Foram identificados 4 metabolitos resultantes da hidroxilação aromática.
O principal, metabolizado pela CYP3A4, é o derivado 4'-hidroxi.
Apesar do metabolito maioritário ser menos ativo que o composto inicial,
pode exibir atividade biológica.

O diclofenac de sódio é indicado para o tratamento de artrite reumatoide,
osteoartrite e espondilite anquilosante.
Ibuprofeno
É comercializado como uma
mistura racémica, embora a
atividade biológica resida quase
exclusivamente no isómero S-(+).
O ibuprofeno é mais potente do que a aspirina, mas menos potente do que
a indometacina, relativamente à inibição da biossíntese de prostaglandinas e à
sua acção anti-inflamatória. Produz irritação gástrica moderada.
É rapidamente absorvido por administração oral.
Forte ligação às proteínas plasmáticas.
O seu metabolismo ocorre rapidamente. É quase totalmente excretado na
urina na sua forma inalterada e como metabolitos oxidativos.
Todos os metabolitos são inativo.
É indicado para alívio dos sinais e sintomas da artrite reumatoide e
osteoartrite, alívio de dor leva a moderada, redução da febre e o tratamento da
dismenorreia moderada.
Naproxeno

Características gerais
Classe de ácidos enólicos de AINESCompostos acídicos, com valores de pKa na gama de 4 a 6
Mecanismo de ação (caso do Piroxicam)
Inibe a migração de células polimorfonucleares em locais inflamados e inibe a libertação de enzimas lisossomais destas célulasInibe a agregação de plaquetas induzida por colagénioInibidor eficaz da COX – conformação assemelha-se ao precursor do radical peroxi de PGG e inibidores da COX
É comercializado como enantiómero S - (+), mas o sal de sódio do isómero
(-), também se encontra no mercado como Anaprox.
É mais potente do que a aspirina e o ibuprofeno, como inibidor da
biossíntese de prostaglandinas, mas é menos potente do que a indometacina.
É absorvido praticamente na sua totalidade após a administração oral.
É eliminado na sua forma inalterada ou como conjugados do fármaco
inalterado. O restante é convertido no metabolito 6-O-desmetil.
O metabolito 6-O-desmetil é desprovido de atividade anti-inflamatória.
O efeito secundário mais comum é a irritação do TGI. Outras reações
adversas encontram-se associadas com distúrbios ao nível do SNC (náuseas e
tonturas).
O naproxeno é usado no tratamento de artrite reumatoide, osteoartrite,
artrite reumatoide juvenil, espondilite anquilosante, tendinite, bursite, artrite
gotosa aguda, e dismenorreia primária e no alívio de dores ligeiras a moderadas.
Oxicams

Piroxicam
Absorvido por administração oral, atingindo os
níveis máximo plasmáticos em 2 horas.
Os níveis plasmáticos máximos parecem ser menores quando
administrado com alimentos, a doses baixas. No entanto, os alimentos não
afetam significativamente a biodisponibilidade.
Elevada ligação às proteínas plasmáticas.
Possui um tempo de meia-vida de 38 horas, tornando possível a
administração única diária.
É indicado para a utilização a longo prazo da artrite reumatoide e
osteoartrite.
É extensivamente metabolizado em humanos, em que menos de 5% da
dose administrada é excretada inalterada na urina. Os principais
metabolitos resultam da hidroxilação do anel piridina catalizada pela
CYP2C9 e subsequente glucuronidação.
Mais potente que a Aspirina e tão potente como a indometacina.
Aproximadamente 20% dos indivíduos que tomam piroxicam relatam
reacções adversas, sendo os distúrbios gastrointestinais os mais
relatados. Menos de 1% reporta a incidência de úlceras gástricas.
Administração concomitante com aspirina tem demonstrado uma
redução de aproximadamente 20% nos níveis de piroxicam no plasma, ao
contrário do efeito concomitante com o anticoagulante acenocoumarin,
sendo este potenciado.
Meloxicam
Em Abril de 2000, o meloxicam foi aprovado
para o tratamento da osteoartrite nos Estados Unidos. Quando foi inicialmente

introduzido foi promovido como um inibidor selectivo da COX-2. O meloxicam
no entanto é menos selectivo para a COX-2 do que o celecoxib.
Bem absorvido por administração oral.
Liga-se fortemente às proteínas plasmáticas,
É extensivamente metabolizado no fígado, principalmente pela CYP2C9, e
em menor extensão pela CYP3A4.
Gastroenteropatia induzida pelos AINEs não seletivos da COX
A eficácia dos AINEs fazem desta classe uma das classes mais
comummente usada na terapêutica. Até à introdução dos inibidores seletivos da
COX-2, a maior parte destes fármacos produziam efeitos prejudiciais para a
mucosa gástrica e intestinal, resultando em erosão, úlceras e hemorragias. Este
tipo de lesões podem ser agudas ou crónicas e são denominadas de gastropatia
dos AINEs e diferem das úlceras pelas localização, uma vez que acontecem em
maior quantidade no estômago.
Estes efeitos adversos levam muitas vezes ao abandono da terapêutica por
parte do doente.
Entre os mecanismos citoprotetores, temos a capacidade das
prostaglandinas aumentarem a secreção de iões bicarbonato, muco e manterem
o fluxo sanguíneo na mucosa. As prostaglandinas também diminuem a secreção
de ácido, permitindo que a barreira da mucosa gástrica permaneça intacta.
O uso de PGE para reduzir os danos gástricos induzidos pelas AINEs é
limitado, uma vez que são ineficazes por via oral e degradam-se rapidamente
quando administrados por via parentérica. Para superar estas limitações, o
misoprostol foi sintetizado como um análogo de um pró-fármaco da
prostaglandina em que a atividade oral é conseguida através da sua
administração como éster metílico, permitindo que o ácido seja bioactivado após
a absorção.

Ácidos fracos não ionizados
permanecem no estômago
Acumulação nas células da
mucosa gástrica
O elevado pH no meio intracelular faz com que haja a dissociação dos ácidos e estes se
tornem "aprisionados" no interior das
células.
Alteração da membrana celular da mucosa e
acumulação de iões de H
Danos nas células das
mucosas
O misoprostol foi introduzido como uma mistura de estereoisómeros,
denominado Cytotec, para a prevenção de úlceras gástricas induzidas pelos
AINEs (mas não úlceras no duodeno) em doente que apresentem maior
sensibilidade a estes efeitos, como por exemplo os idosos.
A aspirina e os AINEs são substâncias ácidas que podem danificar o TGI,
mesmo na ausência de ácido clorídrico, alterando a permeabilidade das
membranas celulares, permitindo uma difusão de iões de hidrogénio.
Como já foi dito anteriormente, o mecanismo primário de ação dos AINEs
é a inibição da biossíntese das prostaglandinas no passo da ciclooxigenase. A
resultante inibição não seletiva da biossíntese de prostaglandinas no TGI impede
que as estas exerçam o seu mecanismo protetor na mucosa gástrica.
Assim, através deste mecanismo, os AINEs induzem lesões gástricas.

Inibidores selectivos da COX2
Os AINEs tradicionais inibem a COX-1, COX-2, e a tromboxano sintetase
em diferentes graus de seletividade.
Estudos levaram ao desenvolvimento de isoformas de inibidores seletivos
da COX-2, com o objectivo de desenvolver o AINE ideal – um que iniba
seletivamente a COX-2, reduzindo assim a resposta inflamatória, mas que não
interfira com as funções protetoras da COX-1. Desta forma foram desenvolvidos
inicialmente dois compostos de chumbo, o NS-398 e o DuP-697. Tanto o NS-398
como a nimesulida são protótipos conhecidos como “sulides”, enquanto que o
DuP-697 pertence a uma classe de inibidores da COX-2, denominados “coxibes”.
Hoje em dia apenas o celecoxib existe no mercado, uma vez que todos os
fármacos pertencentes aos “coxibes” possuíam muitos riscos associados,
incluindo problemas graves cardiovasculares.
Inibição não-seletiva da COX Diminuição da proteção da mucosa
gástrica
“Stress” induzido pela perfusão renal
Inibição da síntese do tromboxano
Aumento da síntese de
prostaglandinas
Redução da agregação plaquetária
AINEs com maior seletividade para a COX1, por regra, causam maior sangramento gastrointestinal e toxicidade renal do que os que tem maior seletividade para a COX-2 .

Vários estudos tem indicado que os pacientes que tomam AINEs têm uma
incidência mais baixa e uma diminuição da velocidade de progressão da doença
de Alzheimer. Estudos epidemiológicos sugerem uma redução significativa no
risco de cancro do cólon em pacientes que tomam aspirina regularmente. Além
disso, os AINEs têm sido relatados para reduzir a taxa de crescimento de pólipos
no cólon em seres humanos.
Comparação entre a seletividade de vários AINEs e inibidores seletivos da COX-2.
Comparação entre os inibidores seletivos e não seletivos da COX

Celecoxib e Etoricoxib
Primeira NSAID a ser comercializada como inibidor seletivo da
COX-2;
Administração oral com uma boa absorção pelo TGI, atingindo boas
concentrações plasmáticas 1-3 horas após a administração;
Elevada ligação às proteínas plasmáticas;
Extensamente metabolizado pelo fígado e excretado na urina sob a forma
de metabolitos inativos;
Inibe a CYP2D6 e altera os perfis farmacocinéticos de outros fármacos
inibidos por esta isoenzima;
Efeitos secundários
Aumento de riscos cardiovasculares, enfarte de miocárdio e AVC;
Aumento de perturbações gastrointestinais graves, como sangramento,
ulceração e perfuração do estômago ou intestino;
É atualmente indicado para o alívio dos sinais e sintomas de osteoartrite,
artrite reumatoide, entre outras doenças;
A administração concomitante de aspirina e celecoxib pode aumentar a
incidência de efeitos secundários gastrointestinais;
Nimesulide
Indicações terapêuticas
Tratamento da dor aguda;
Tratamento sintomático da osteoartrose dolorosa;
Dismenorreia primária;

Posologia e método de administração
Deve ser usado durante o menor período de tempo possível, como exigido
pela situação clínica;
Contraindicado para crianças com menos de 12 anos;
Deve ser feito um ajuste posológico em adolescentes (12 a 18 anos) e
adultos;
Contraindicado em doentes com insuficiência hepática;
Contraindicações
Hipersensibilidade conhecida ao nimesulide ou a qualquer um dos
excipientes do medicamento;
Histórico de reações de hipersensibilidade (p.ex. broncoespasmo, rinite,
urticária) em resposta ao ácido acetilsalicílico ou outros fármacos não-
esteroides anti-inflamatórios);
Histórico de reações de hepatotoxicidade ao nimesulide:
Histórico de ulceração recorrente ou hemorragia gastrointestinal,
hemorragia cerebrovascular ou outras hemorragias ativas ou doenças
hemorrágicas;
Distúrbios de coagulação severos;
Insuficiência cardíaca grave;
Insuficiência renal grave;
Insuficiência hepática;
Crianças menores de 12 anos;
Terceiro trimestre da gravidez e aleitamento materno;
Advertências e precauções especiais de utilização

Tanto a administração concomitante de fármacos hepatotóxicos , como o
abuso de álcool devem ser evitados durante o tratamento, já que pode
aumentar o risco de reações hepáticas;
Durante a terapia os doentes devem ser aconselhados a não tomarem
outros analgésicos. Não é recomendada a utilização simultânea de outros
AINEs;
Se ocorrer sangramento ou ulceração gastrointestinal, o nimesulide deve
ser interrompido;
Os doentes idosos são particularmente suscetíveis aos efeitos adversos
dos AINEs por isso deve ser realizada monitorização clínica;
O uso de nimesulide pode comprometer a fertilidade feminina e não é
recomendado em mulheres que pretendam engravidar;
Interações medicamentosas
Pacientes que tomaram agentes anticoagulantes, como a Varfarina ou
ácido acetilsalicílico têm um risco aumentado de complicações
hemorrágicas, quando tratados com nimesulide;
A coadministração do nimesulide e furosemida resulta numa diminuição
(cerca de 20%) da AUC e da excreção cumulativa da furosemida, sem
afectar a sua depuração renal;
As concentrações plasmáticas de fármacos que são substratos da CYP2D9
podem ser aumentadas quando o nimesulide é usado concomitantemente;
É necessário ter cuidado se o nimesulide é usado menos de 24 horas antes
ou após o tratamento com metotrexato porque os níveis séricos do
metotrexato podem aumentar e, portanto, a toxicidade do fármaco pode
aumentar;
Devido ao seu efeito sobre as prostaglandinas renais, o nimesulide, pode
levar ao aumento da nefrotoxicidade de ciclosporinas.

Efeitos adversos

Propriedades farmacocinéticas
Bem absorvida quando administrada por via oral;
É atingida a concentração máxima plasmática após 2-3h da
administração;
Liga-se fortemente às proteínas plasmáticas (97,5%);
É extensivamente metabolizado no fígado;
Principalmente excretado pela urina (aproximadamente 50% da dose
administrada). Apenas 1-3% é excretada de forma inalterada. Cerca de
29% da dose é excretada, após metabolização, nas fezes. Cerca de 29% da
dose é excretada, após metabolização, nas fezes;
Overdoses causadas pelos AINEs, geralmente causam apenas letargia,
náuseas, tonturas e vómitos. Estes efeitos são reversíveis, ao parar a toma
do fármaco, estes efeitos desaparecem;




![Apresentação Tintas Inap [Modo de Compatibilidade] · ADITIVOS - modificam as propriedades das tintas – secantes, anti-espumantes, anti-sedimentantes, anti-pele, anti-flotante,](https://img.document.onl/doc/110x75/5c45d0c093f3c34c46595ca6/apresentacao-tintas-inap-modo-de-compatibilidade-aditivos-modificam-as.jpg)
























