Embed Size (px)
Citation preview

Deficiencia congénita de fibrinógeno
Dr. Ramiro José Núñez Unidad de Hemofilia.
Hospital Universitario Virgen del Rocío. Sevilla

Fibrinógeno
Proteína compleja y multifuncional.
Importante papel en la hemostasia.
Múltiples procesos fisiológicos.
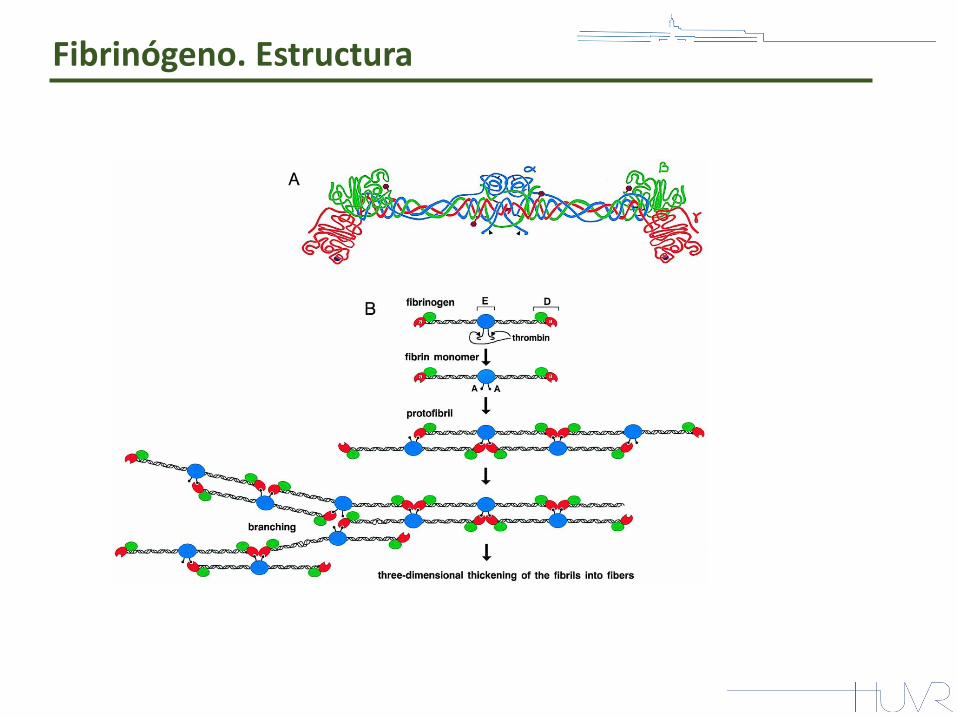
Fibrinógeno. Estructura
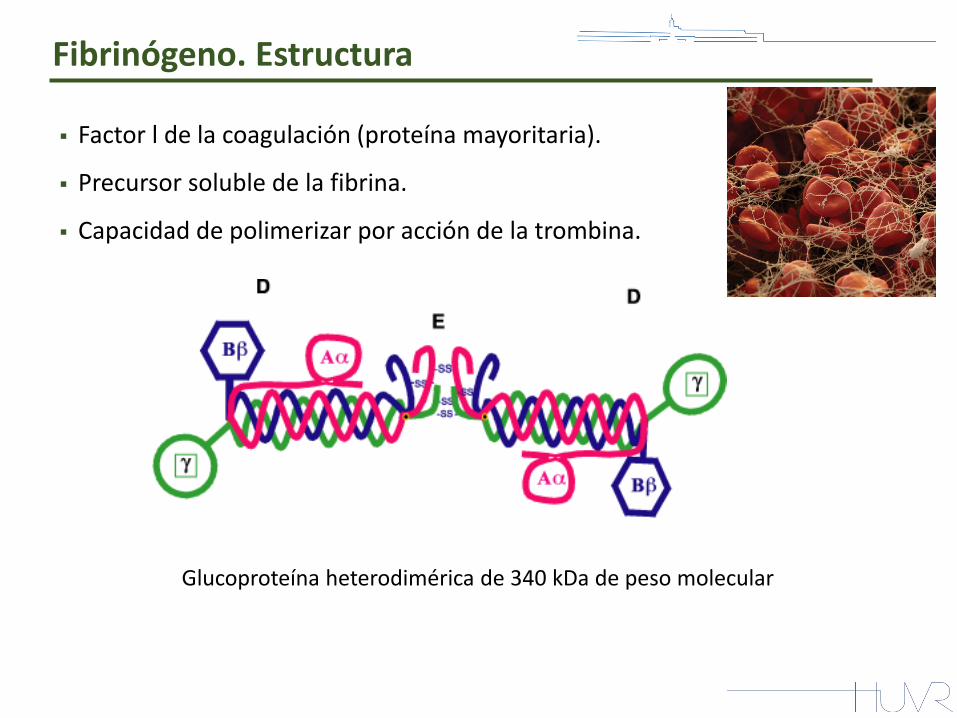
Factor l de la coagulación (proteína mayoritaria).
Precursor soluble de la fibrina.
Capacidad de polimerizar por acción de la trombina.
Glucoproteína heterodimérica de 340 kDa de peso molecular
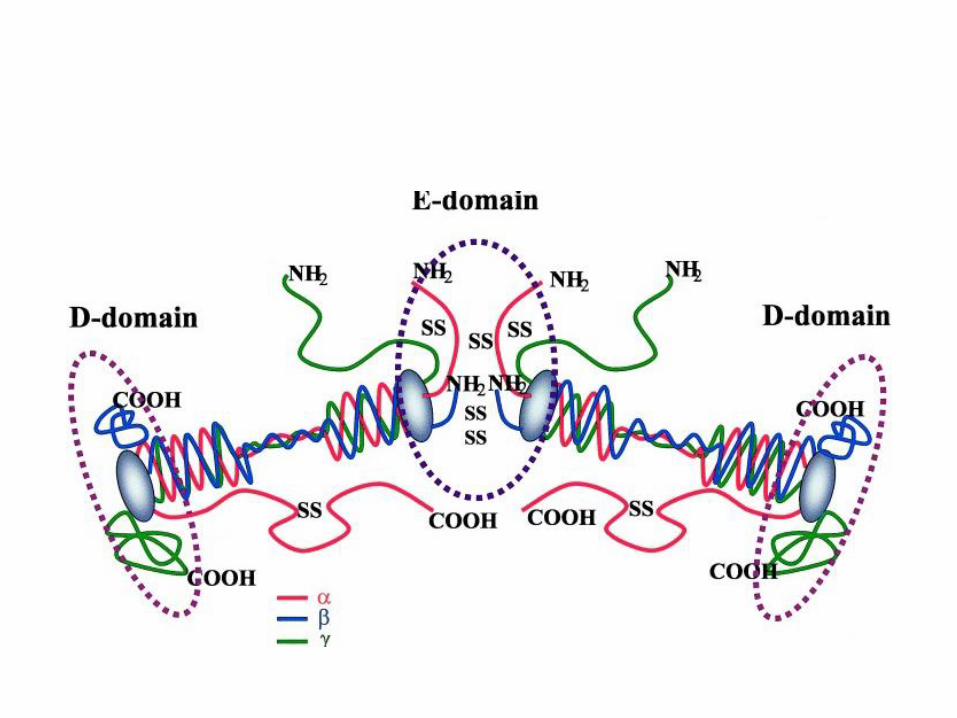
Fibrinógeno. Estructura
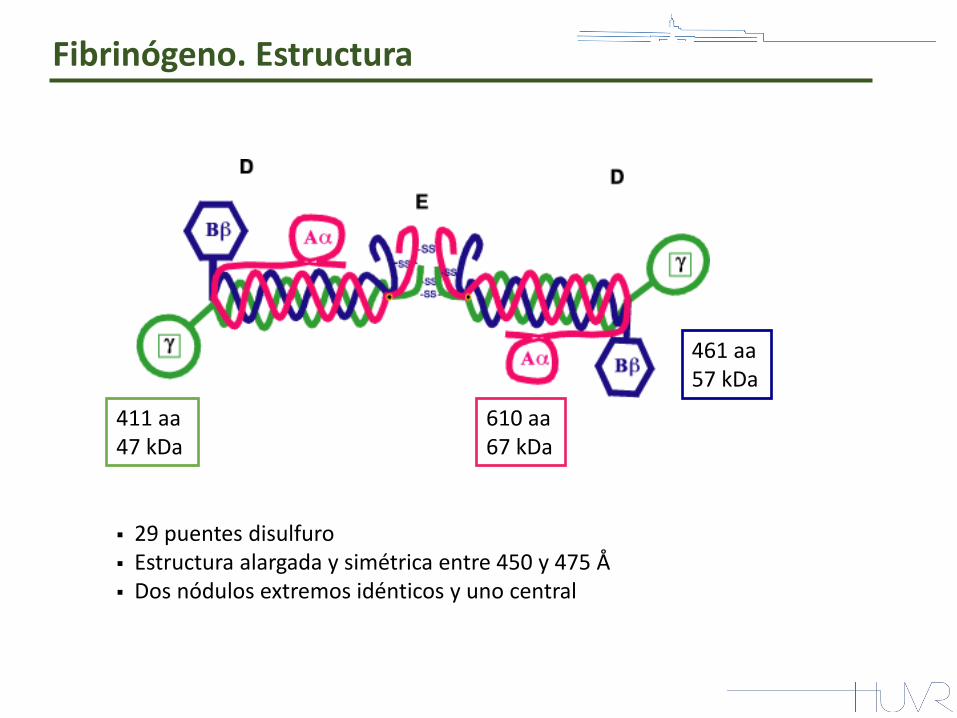
411 aa 47 kDa
610 aa 67 kDa
461 aa 57 kDa
29 puentes disulfuro Estructura alargada y simétrica entre 450 y 475 Å Dos nódulos extremos idénticos y uno central
Fibrinógeno. Estructura
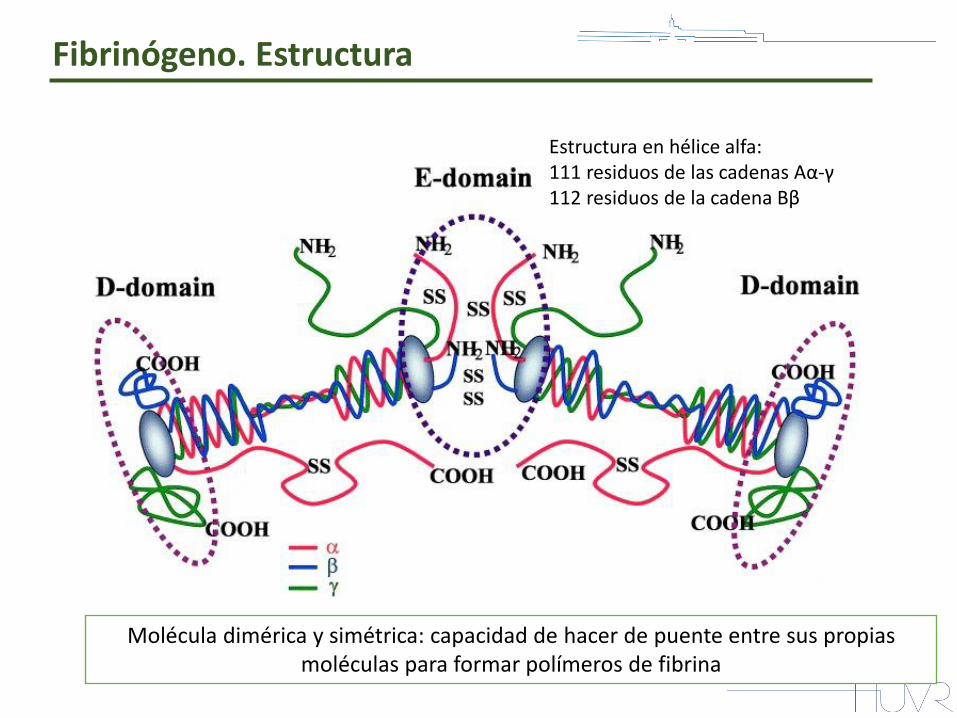
Molécula dimérica y simétrica: capacidad de hacer de puente entre sus propias moléculas para formar polímeros de fibrina
Estructura en hélice alfa: 111 residuos de las cadenas Aα-γ 112 residuos de la cadena Bβ
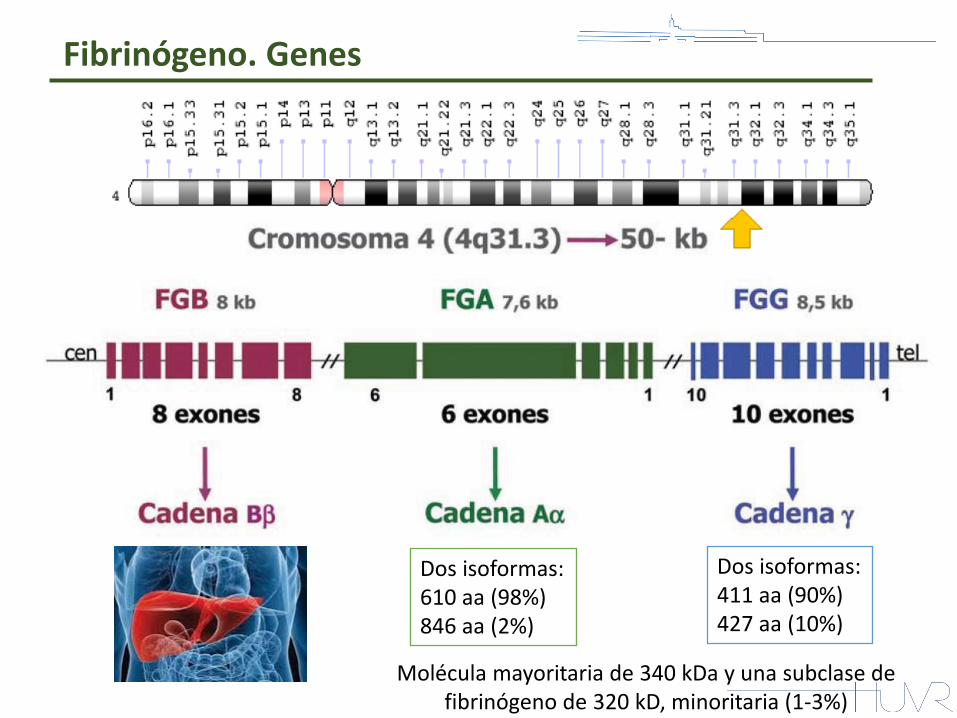
Fibrinógeno. Genes
Dos isoformas: 610 aa (98%) 846 aa (2%)
Dos isoformas: 411 aa (90%) 427 aa (10%)
Molécula mayoritaria de 340 kDa y una subclase de fibrinógeno de 320 kD, minoritaria (1-3%)
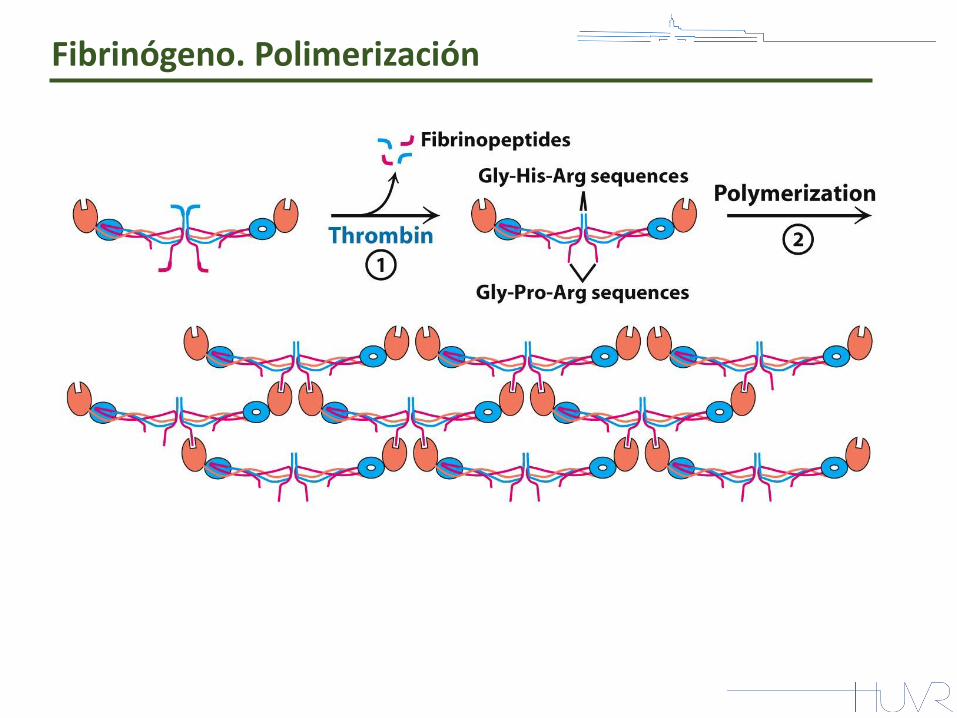
Fibrinógeno. Polimerización
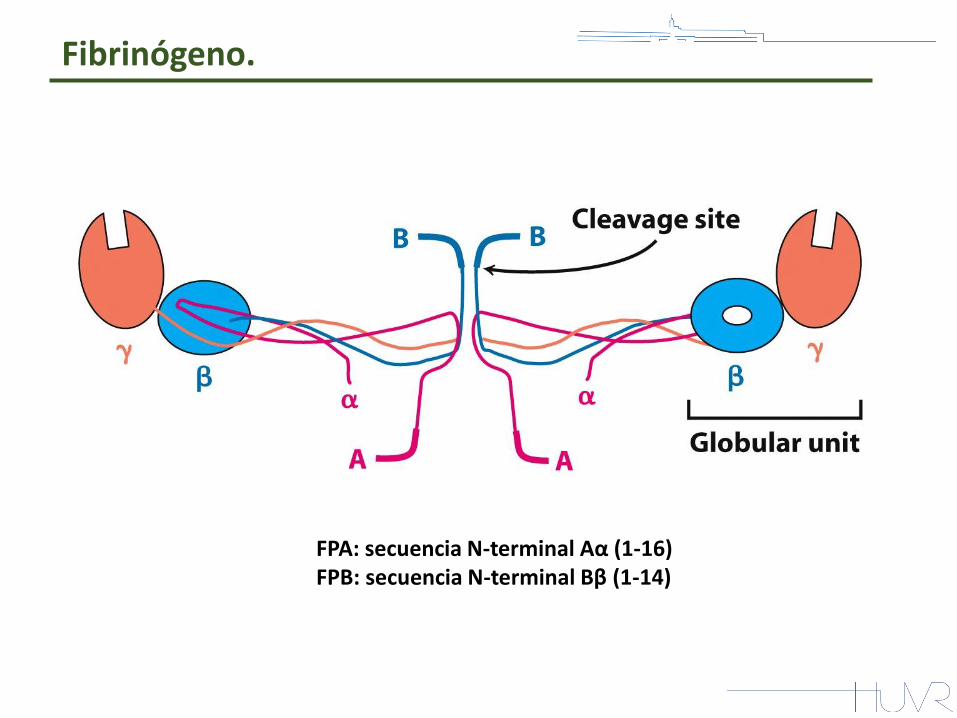
Fibrinógeno.
FPA: secuencia N-terminal Aα (1-16) FPB: secuencia N-terminal Bβ (1-14)
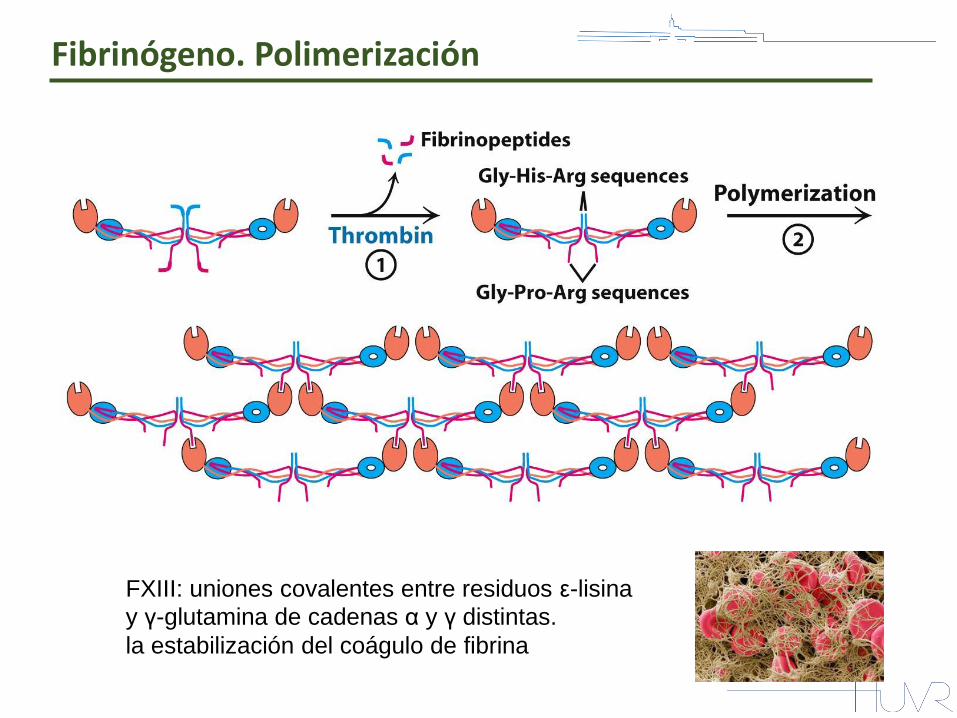
Fibrinógeno. Polimerización
FXIII: uniones covalentes entre residuos ε-lisina
y γ-glutamina de cadenas α y γ distintas. la estabilización del coágulo de fibrina
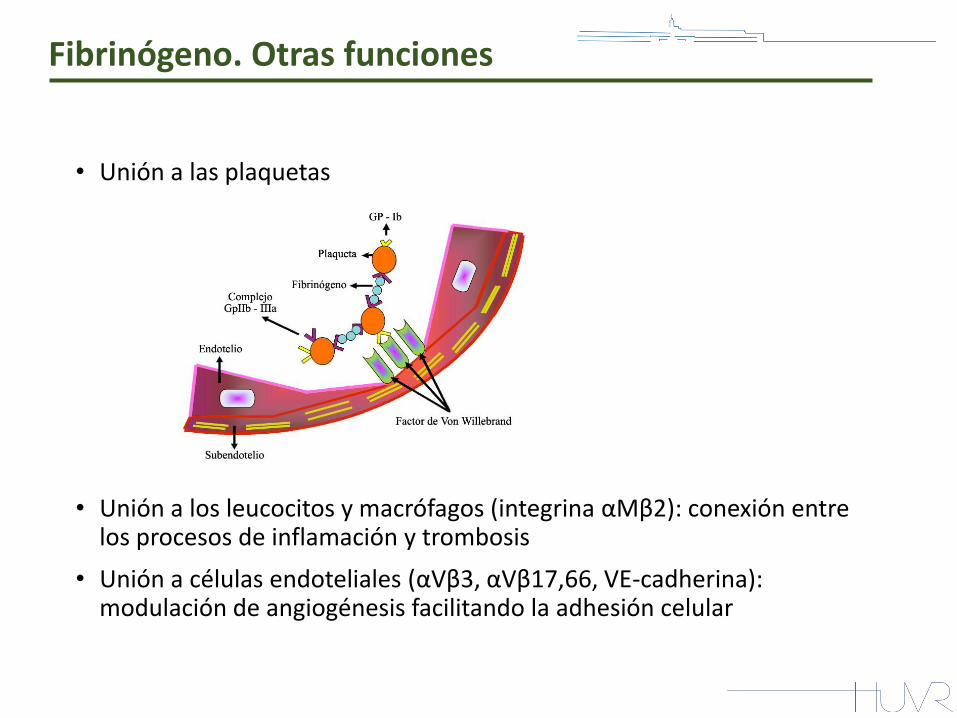
Fibrinógeno. Otras funciones
• Unión a las plaquetas
• Unión a los leucocitos y macrófagos (integrina αMβ2): conexión entre los procesos de inflamación y trombosis
• Unión a células endoteliales (αVβ3, αVβ17,66, VE-cadherina): modulación de angiogénesis facilitando la adhesión celular
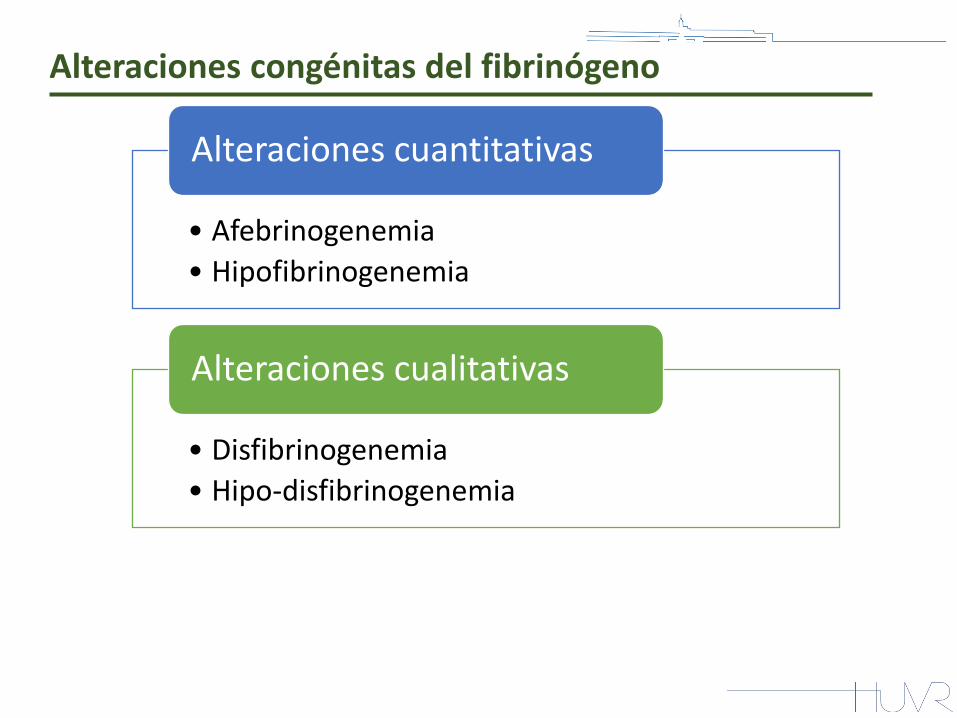
Alteraciones congénitas del fibrinógeno
• Afebrinogenemia
• Hipofibrinogenemia
Alteraciones cuantitativas
• Disfibrinogenemia
• Hipo-disfibrinogenemia
Alteraciones cualitativas

Fenotipo clínico y de laboratorio
J Thromb Haemost 2012; 10: 1938–43
Pacientes con niveles indetectables: episodios graves espontáneos
Pacientes con valores > 1 g/L: mayoría serán asintomáticos.

Afibrinogenemia
• Ausencia total de fibrinógeno.
• Primer caso descrito en 1920.
• Prevalencia: 1/millón de habitantes.
• Herencia: autosómica recesiva.
• Espectro mutacional:
• Grandes deleciones.
• Mutaciones puntuales (non-sense, missense).
• Mismas mutaciones en heterocigosis: hipofibrinogenemia.
Rabe F. Arch Intern Med. 1920;95:2-14.
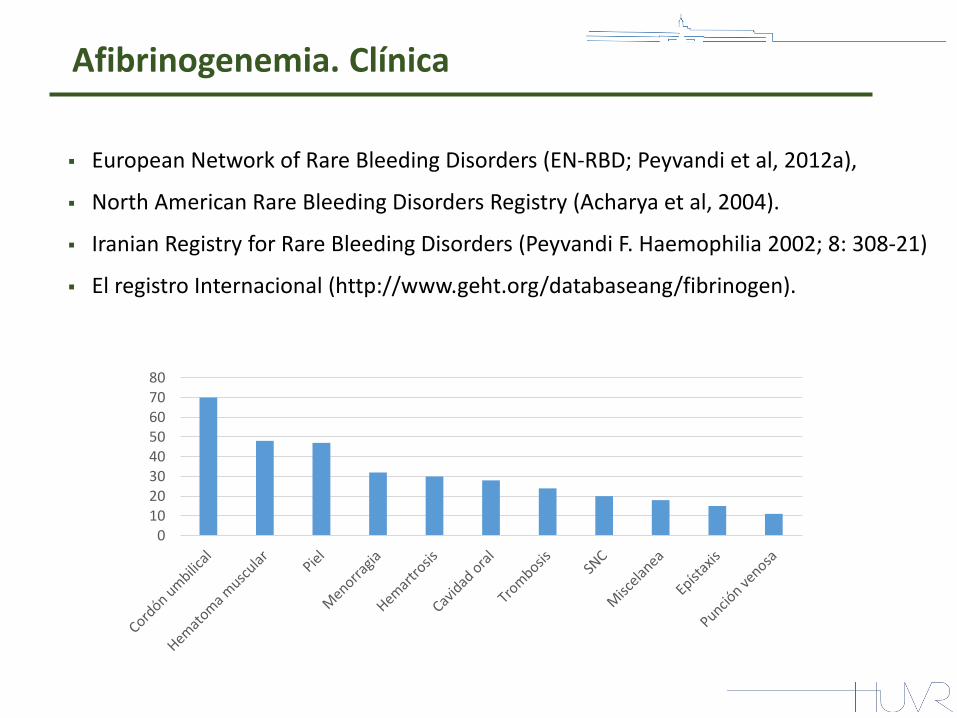
Afibrinogenemia. Clínica
European Network of Rare Bleeding Disorders (EN-RBD; Peyvandi et al, 2012a),
North American Rare Bleeding Disorders Registry (Acharya et al, 2004).
Iranian Registry for Rare Bleeding Disorders (Peyvandi F. Haemophilia 2002; 8: 308-21)
El registro Internacional (http://www.geht.org/databaseang/fibrinogen).
01020304050607080

Afibrinogenemia. Clínica
Peculiaridades
Susceptibilidad aumentada a la rotura espontánea del bazo.
Deficiente cicatrización de heridas.
Formación de quistes óseos dolorosos:
Se desarrollan en la diáfisis de los huesos largos durante la infancia
Parecen beneficiarse del tratamiento sustitutivo con fibrinógeno, sugiriendo una posible etiología hemorrágica durante el remodelado óseo
Aubrey-Bassler.BMC Emerg Med 2012; 12(1):11. Rodriguez-Merchan EC. Haemophilia 2012;18(4):487–490. van Meegeren ME, Haemophilia 2014; 20(2):244–248.
Mujeres: frecuencia aumentadas de complicaciones ginecológicas
Meno-metrorragias.
Hemorragia periparto (21%).
Abortos de repetición (si mutaciones relacionadas con trombosis).
Cetinkaya SE, Acta Obstet Gynecol Scand 2011; 90(2):192–194. Iwaki T,. Am J Pathol 2002; 160(3):1021-1034.

Afibrinogenemia. Trombosis
Generación de trombina aumentada, niveles del complejo
trombina-antitrombina y de los fragmentos 1+2 de la protrombina.
Falta de inhibición de la trombina causada por la activación del
fibrinógeno y la adsorción de la trombina en la red de fibrina.
La trombina se encuentra libre para activar las plaquetas,
proliferación y migración de las células de músculo liso: activación
trombótica.
Trombosis arterial y venosa.
De Bosh NB. Thromb Haemost 2002; 88: 253-8

Afibrinogenemia. Trombosis
Teresa SM. Haemophilia 2015; 21, 88-94.
14 casos de trombosis arterial.
Dos casos deficiencia de la proteína C
Un heterocigoto: mutación del factor V Leiden
Otra paciente: anticonceptivos orales
5 en tratamiento con CF/PFC
No se detectaron factores de riesgo para arterioesclerosis.
Eventos a una edad temprana (mediana 31.5 años).

Hipofibrinogenemia
Peyvandi F. J Thromb Haemost 2012; 10: 615–21.
Concentraciones de fibrinógeno circulante <1,5 g/L.
Mismas mutaciones en estado heterocigoto que las afibrinogenemias.
Mayor prevalencia (no bien definida).
Espectro hemorrágico similar.
Los pacientes son habitualmente asintomáticos
Niveles de fibrinógeno de 1g/L son suficientes.
Episodios más frecuentes en traumatismos, o una segunda anomalía hemostática asociada.
Al igual que en las afibrinogenemias se han descrito casos de hipofibrinogenemias asociados a clínica trombótica.
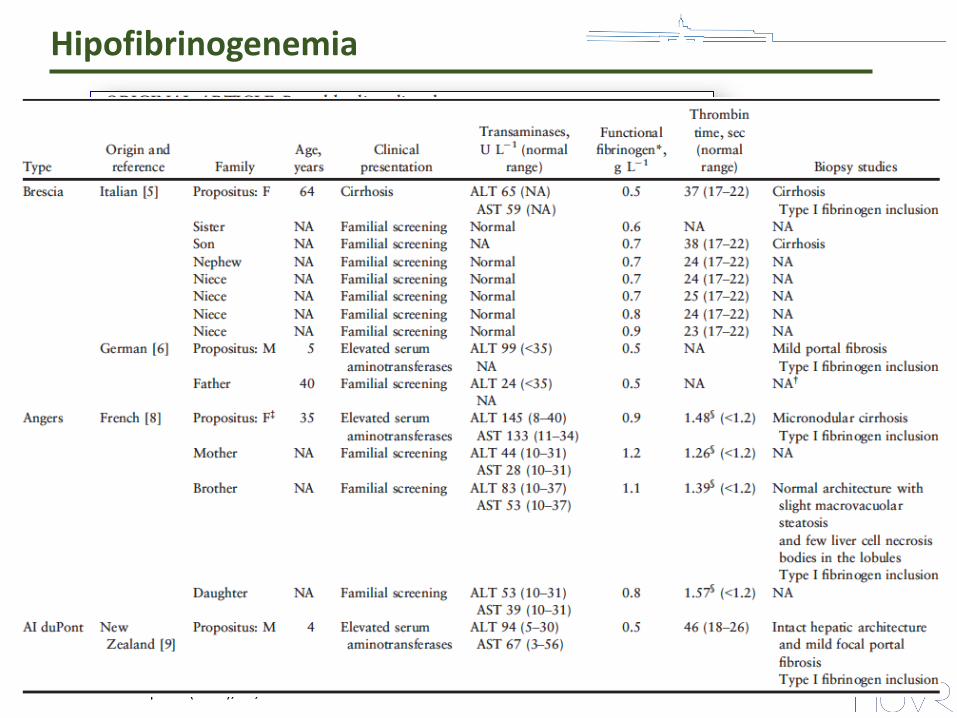
Hipofibrinogenemia
Casini A. Haemophilia (2015), 21, 820-827.
• Hepatopatía producida por una liberación disminuida y acúmulo de fibrinógeno en el retículo endoplásmico: FSD (fibrinogen storage disease).
• Histología: Distinto grado de inflamación y fibrosis. Cirrosis.
• 11 familias genotipadas han sido publicadas con seis variantes heterocigotas: fibrinógenos Brescia, Aguadilla, Angers, AI duPont, Pisa y Beograd.
• Valorar elastografía al diagnóstico.

Disfibrinogenemia
Casini A. Haemophilia (2015), 21, 820-827 Casini A. Blood 2015; 125: 553-61.
• Defecto funcional del fibrinógeno.
• Herencia autosómica dominante.
• La identificación de la primera disfibrinogenemia: 1958.
• Manifestaciones clínicas muy heterogéneas.
• Penetrancia del fenotipo clínico a menudo incompleta.
• Episodios hemorrágicos leves en la mayoría de los casos. • Pacientes con fenotipo grave y hemorragias con
compromiso vital. • Mujeres presentan una alta prevalencia de menorragias y
sangrados obstétricos.
• Existen dos mutaciones frecuentes (74%): • Mutación en FGA Arg35, exón 2 • Mutación FGG Arg301, exón 8.
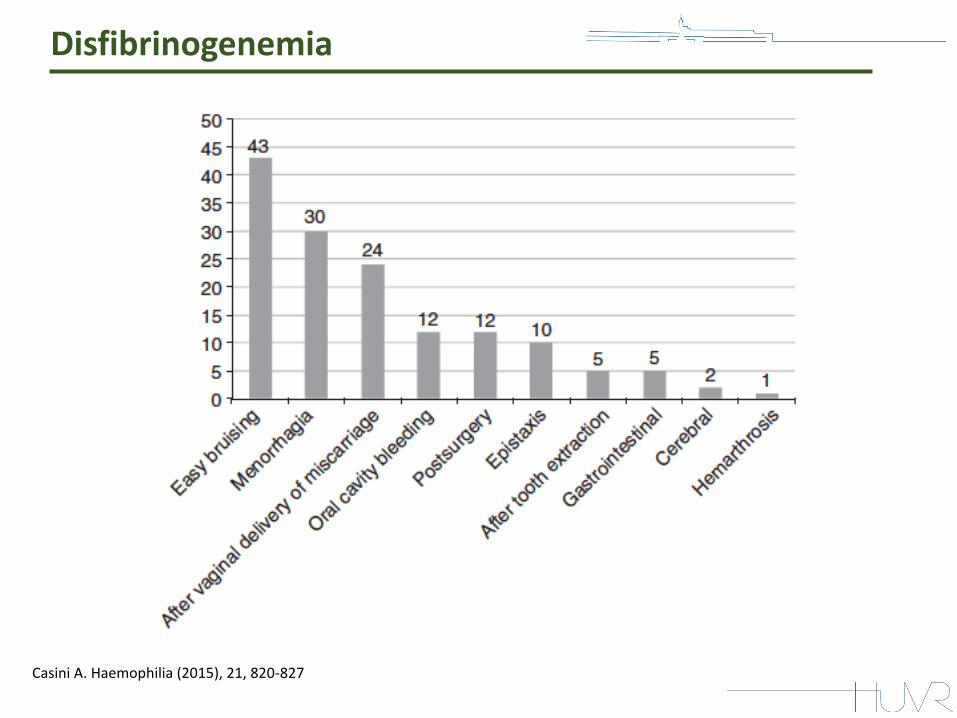
Disfibrinogenemia
Casini A. Haemophilia (2015), 21, 820-827
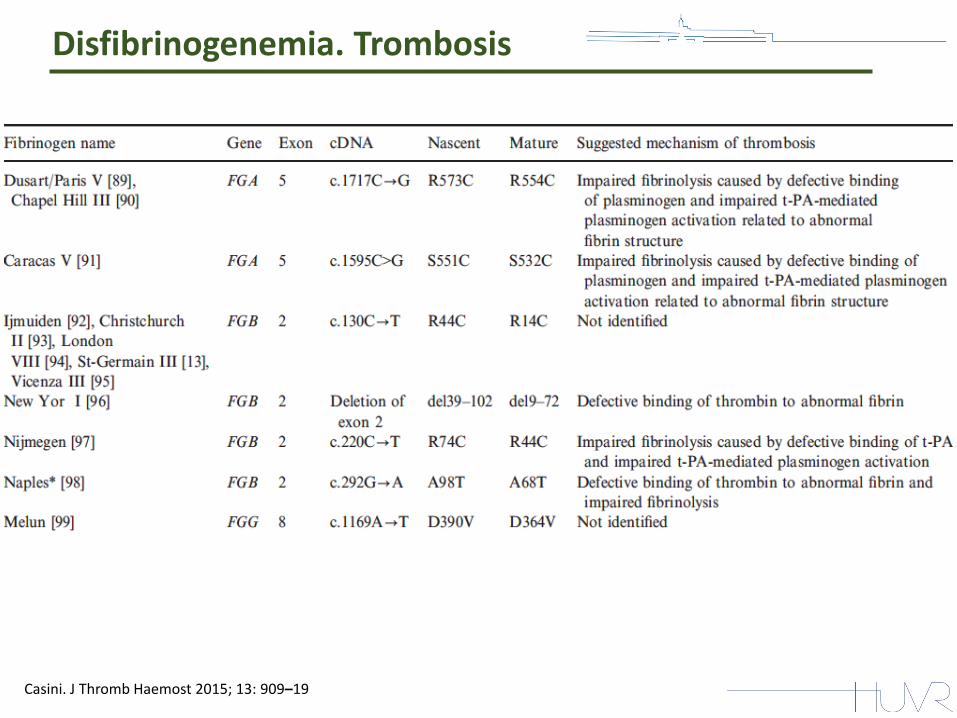
Disfibrinogenemia. Trombosis
Se han implicado diversos mecanismos:
Niveles elevados de trombina circulante.
Red de fibrina con una estructura, fuerza y estabilidad alteradas.
Fibrinólisis disminuida por una unión defectuosa del plasminógeno o del activador tisular del plasminógeno en relación con una estructura anormal de la fibrina.
187 familiares:
20/99: disfibrinogenemia + trombosis.
vs 88 sin el defecto y sin trombosis.
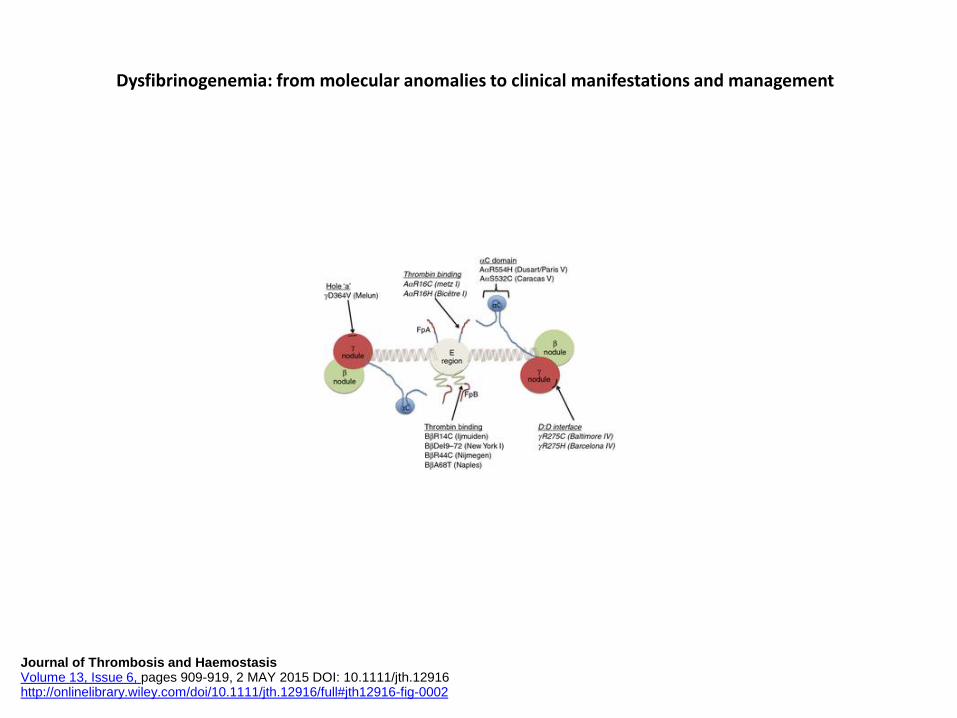
Se encontró asociación para 5 tipos de fibrinógenos anormales: Caracas V, Vlissingen, Melun, Naples, and Dusart .
Determinadas mutaciones asociadas
Casini. J Thromb Haemost 2015; 13: 909–19
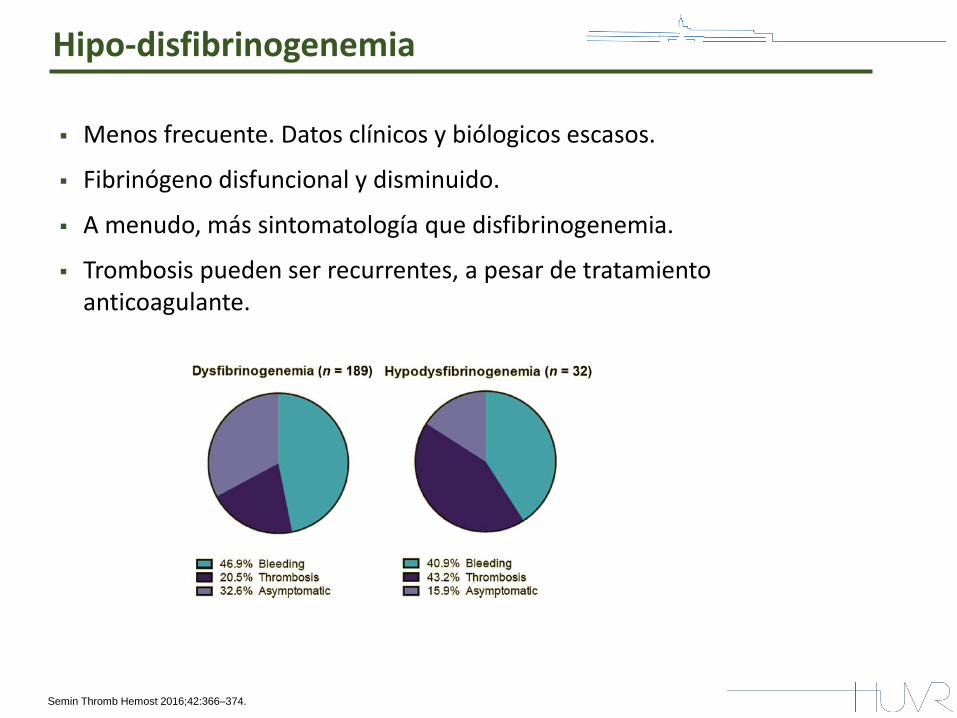
Hipo-disfibrinogenemia
Menos frecuente. Datos clínicos y biólogicos escasos.
Fibrinógeno disfuncional y disminuido.
A menudo, más sintomatología que disfibrinogenemia.
Trombosis pueden ser recurrentes, a pesar de tratamiento anticoagulante.
Semin Thromb Hemost 2016;42:366–374.
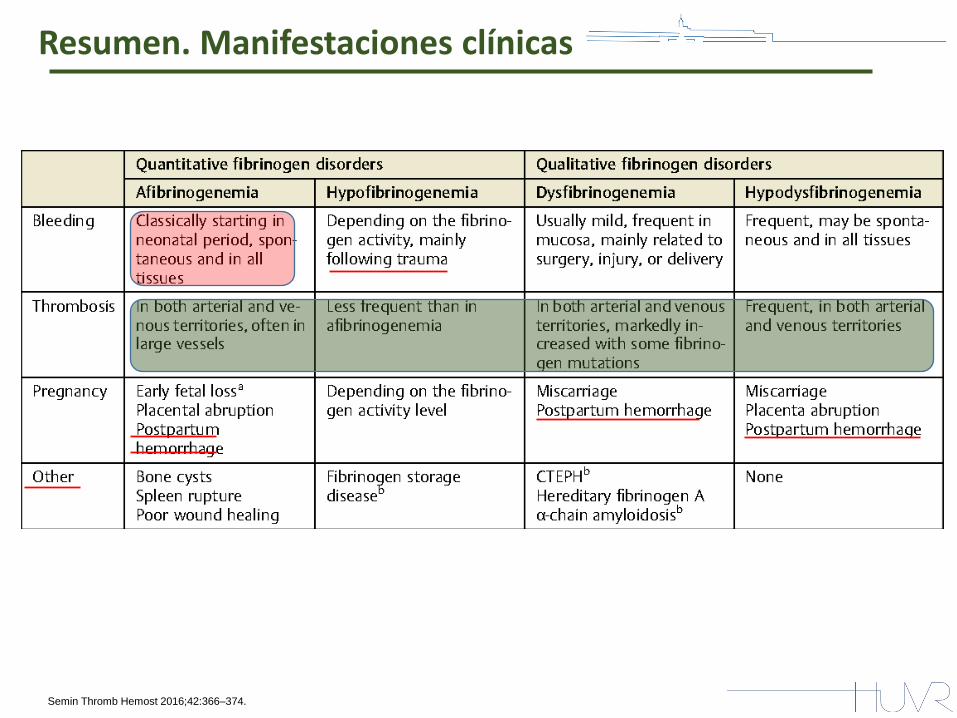
Resumen. Manifestaciones clínicas
Semin Thromb Hemost 2016;42:366–374.
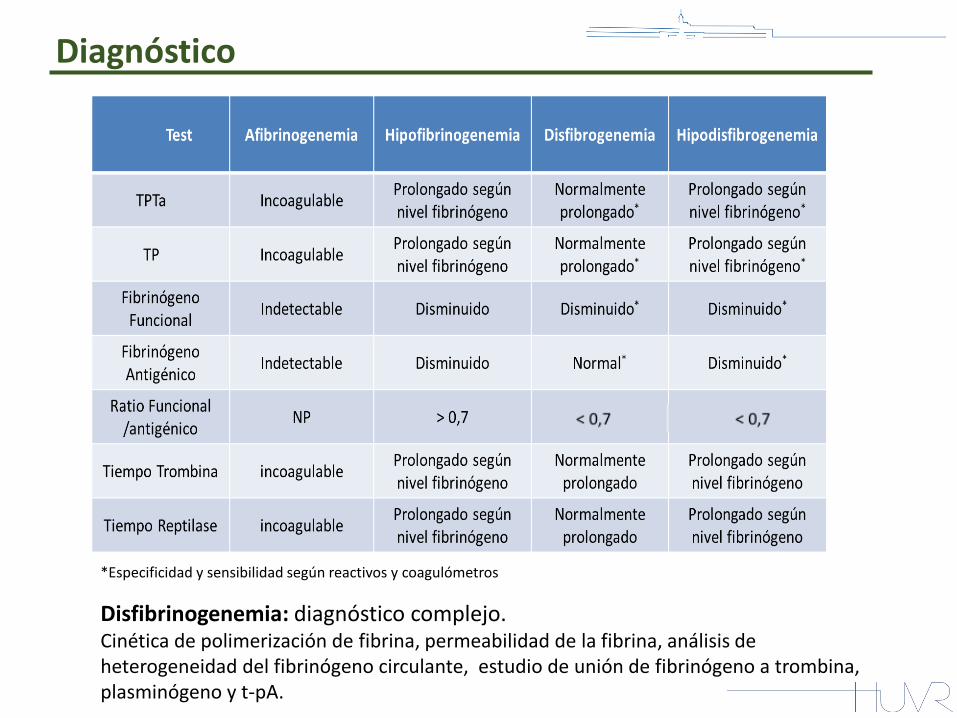
Diagnóstico
*Especificidad y sensibilidad según reactivos y coagulómetros
Disfibrinogenemia: diagnóstico complejo. Cinética de polimerización de fibrina, permeabilidad de la fibrina, análisis de heterogeneidad del fibrinógeno circulante, estudio de unión de fibrinógeno a trombina, plasminógeno y t-pA.
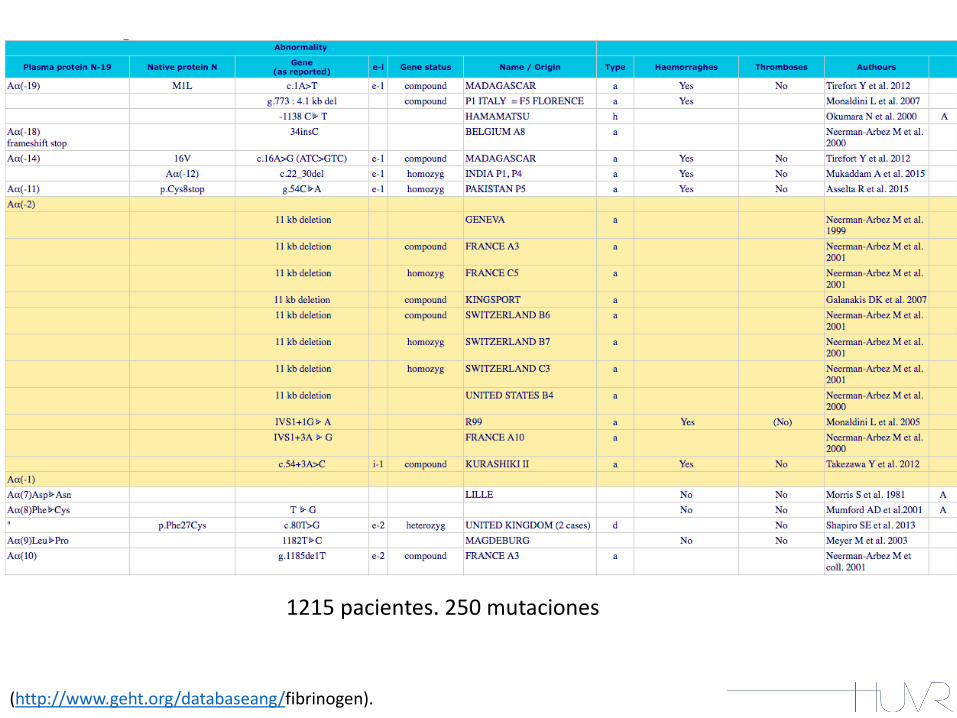
(http://www.geht.org/databaseang/fibrinogen).
1215 pacientes. 250 mutaciones
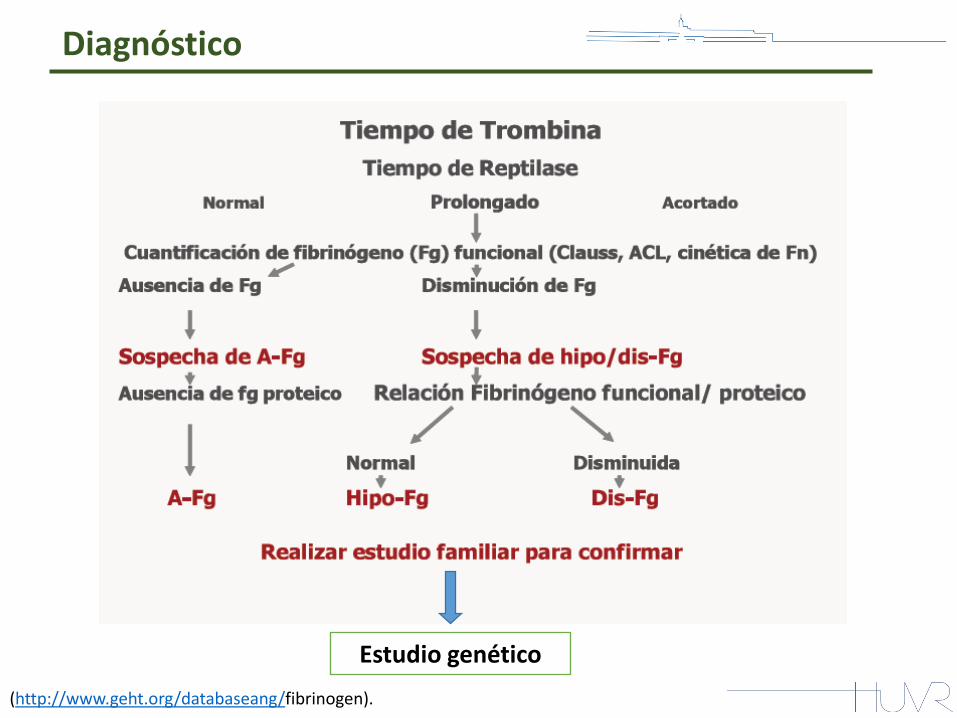
Diagnóstico
(http://www.geht.org/databaseang/fibrinogen).
Estudio genético
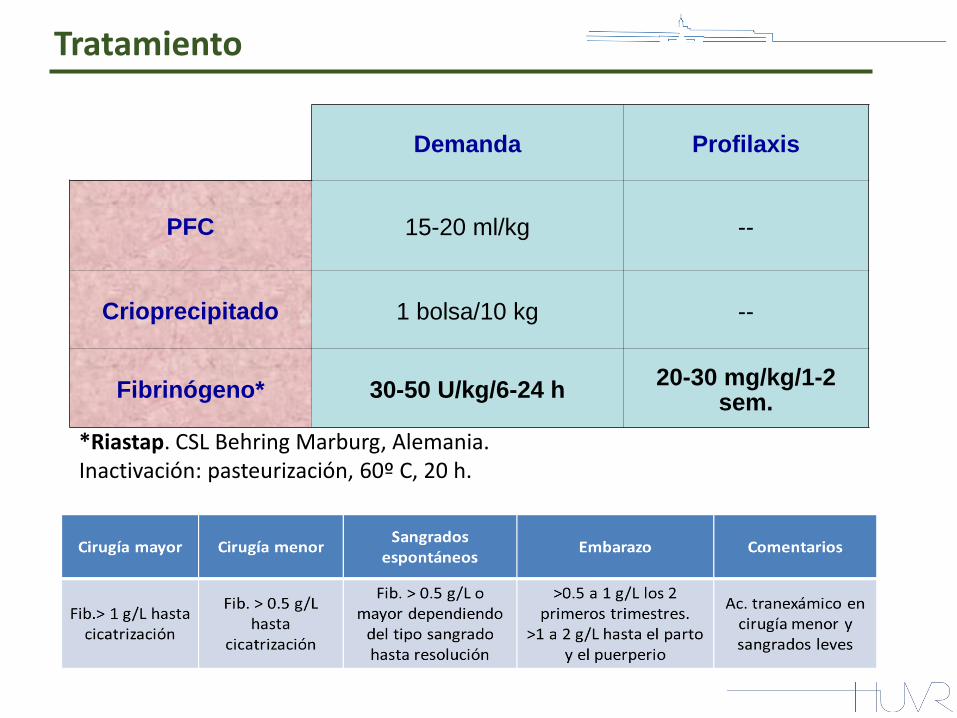
Tratamiento
Demanda Profilaxis
PFC 15-20 ml/kg --
Crioprecipitado 1 bolsa/10 kg --
Fibrinógeno* 30-50 U/kg/6-24 h 20-30 mg/kg/1-2
sem.
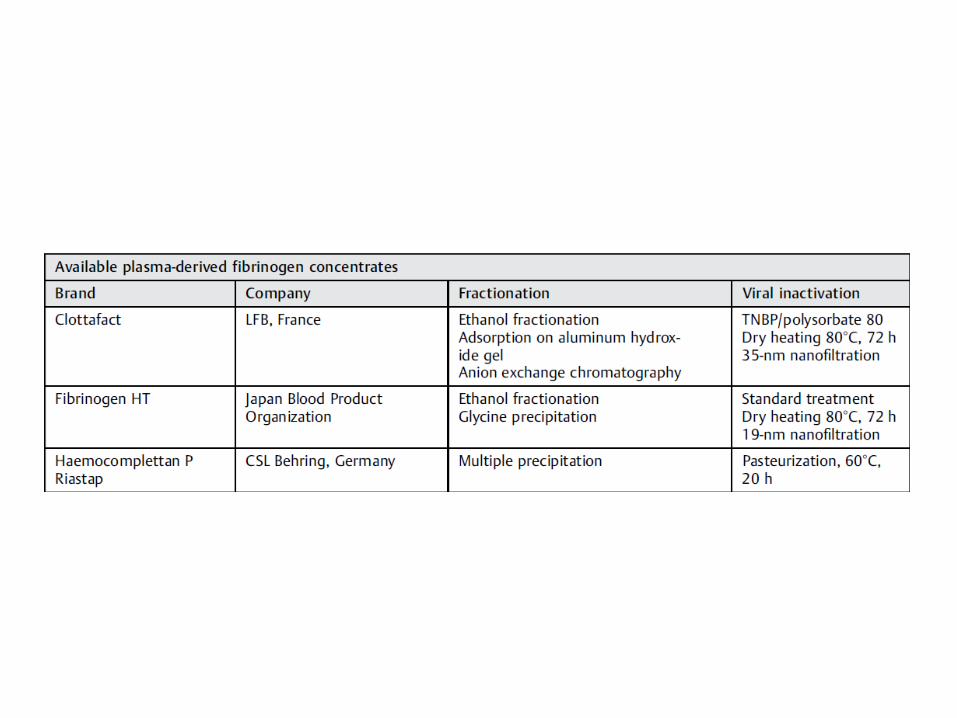
*Riastap. CSL Behring Marburg, Alemania. Inactivación: pasteurización, 60º C, 20 h.

Tratamiento. Profilaxis
En los casos de hemorragias vitales se recomienda mantener una profilaxis secundaria.
No existen estudios comparando tratamiento a demanda y profiláctico.
Un estudio retrospectivo: en 100 pacientes con afibrinogenemia y disfibribrinogenemia la incidencia anual de sangrados fue de 0.5 y 0.7 en pacientes en profilaxis y a demanda, respectivamente.
Estudio proRBDD ha mostrado que el riesgo de tener un sangrado mayor no difiere entre los 75 pacientes tratados a demanda de los 12 pacientes en profilaxis.
Peyvandi F. J Thromb Haemost 2006; 4(7):1634–1637. Peyvandi F. XXV Congress of the International Society on Thrombosis and Haemostasis. Toronto, Canada; 2015.

Tratamiento. Profilaxis
Haemophilia (2016), 1–8

Tratamiento. Disfibrinogenemia
Gran heterogeneidad clínica: tratamiento debería ser individualizado en función de los antecedentes personales y familiares, así como del genotipo
Si clínica hemorrágica: recomendaciones similares.

Tratamiento. Trombosis
Si fenotipo trombótico, se deben valorar los riesgos-beneficios de la administración de fibrinógeno.
El manejo de la trombosis a menudo es un reto puesto que es necesario combinar tratamiento antitrombótico y fibrinógeno simultáneamente.
La duración del tratamiento no ha sido establecido y se podrían aplicar las mismas recomendaciones que para la población general.
HBPM son preferidas a AVK. Los inhibidores directos de la trombina o Xa pueden ser más efectivos teniendo en cuenta la elevada trombina circulante.
Tromboprofilaxis debería considerarse en todos los pacientes con antecedentes trombóticos que deban recibir terapia sustitutiva con fibrinógeno, especialmente en situaciones de alto riesgo trombótico, y en aquellos pacientes con disfibrinogenemia y mutaciones asociadas a un aumento de los fenómenos trombóticos.
Haemophilia (2016), 1–8 Semin Thromb Hemost 2016;42:366–374.


Muchas gracias

Comentarios
Peyvandi F. J Thromb Haemost 2006; 4(7):1634–1637. Peyvandi F. XXV Congress of the International Society on Thrombosis and Haemostasis. Toronto, Canada; 2015.
Disfibrinogenemia Any considered treatment should first be based on the personal and family history and discussed in a specialized center In case of bleeding, fibrinogen concentrates are the first choice; dosage and schedule should be adapted to the severity of bleeding, with the aim to target a trough fibrinogen activity level greater than 1 g/L Surgery in patients with a bleeding phenotype should be managed following recommendation for patients with quantitative fibrinogen disorders Surgery in patients with a thrombotic phenotype should be managed if possible without or with a minimum fibrinogen substitution. An accurate thromboprophylaxis is necessary In case of venous thrombosis, low-molecular-weight heparin is the first choice. The length of treatment should be established according to the personal and familial history, the thrombophilia screening, and the genotype Patients with a thrombotic phenotype without anticoagulant treatment should receive an accurate thromboprophylaxis in high-risk situations Asymptomatic patients should benefit from a regular follow-up and, when possible, from functional assays in research laboratory to better establish the clinical phenotype

Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management
Journal of Thrombosis and Haemostasis Volume 13, Issue 6, pages 909-919, 2 MAY 2015 DOI: 10.1111/jth.12916 http://onlinelibrary.wiley.com/doi/10.1111/jth.12916/full#jth12916-fig-0001

• Base de datos

Fibrinógeno. Estructura
Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management
Journal of Thrombosis and Haemostasis Volume 13, Issue 6, pages 909-919, 2 MAY 2015 DOI: 10.1111/jth.12916 http://onlinelibrary.wiley.com/doi/10.1111/jth.12916/full#jth12916-fig-0002
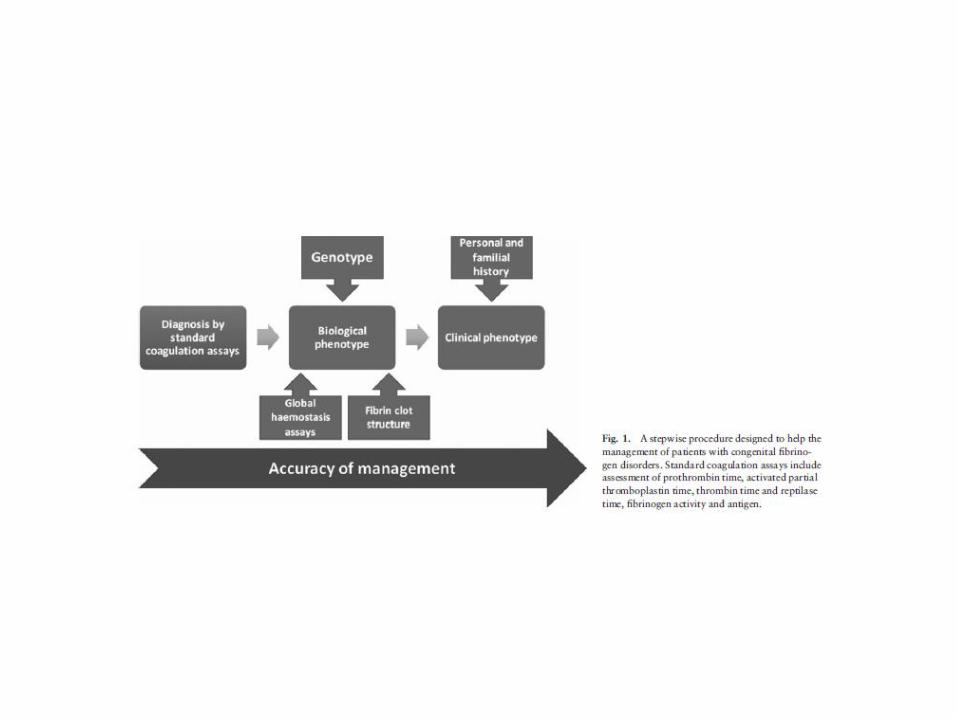
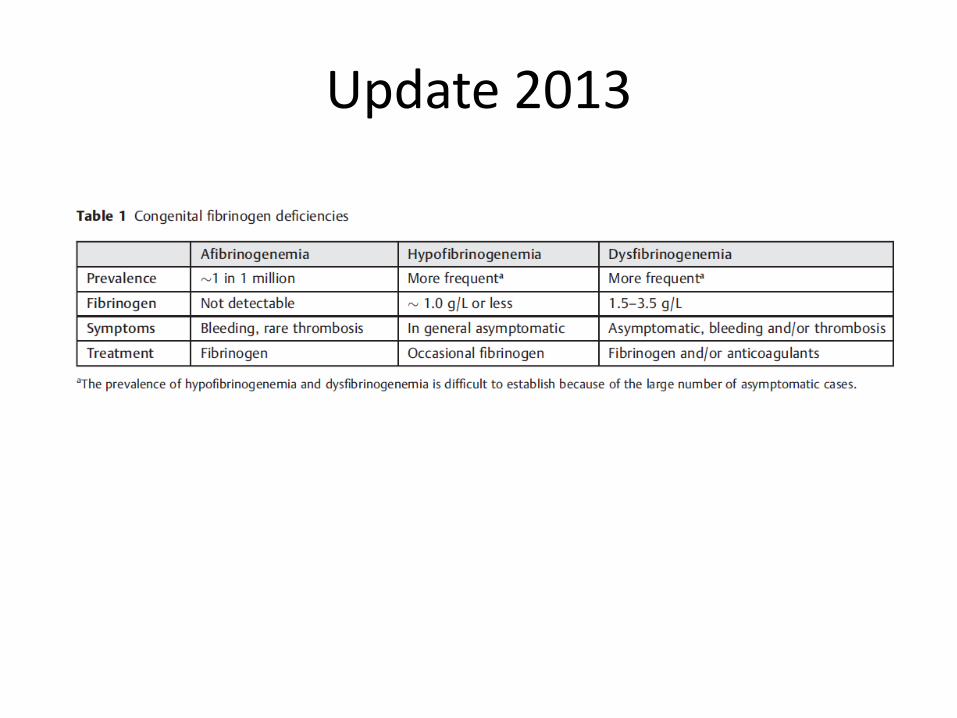
Update 2013
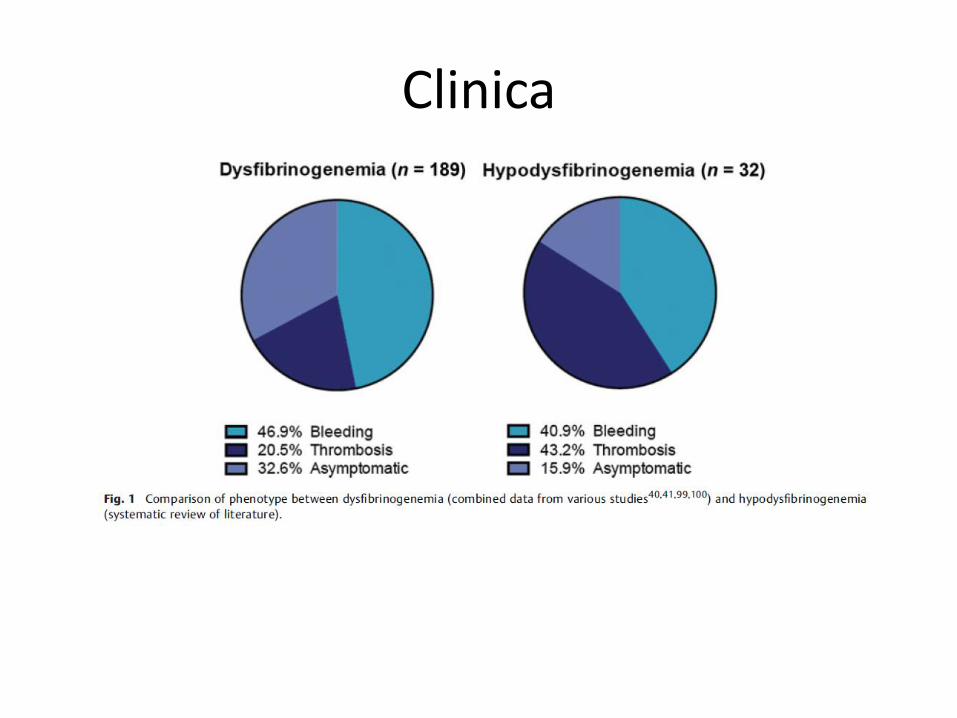
Clinica
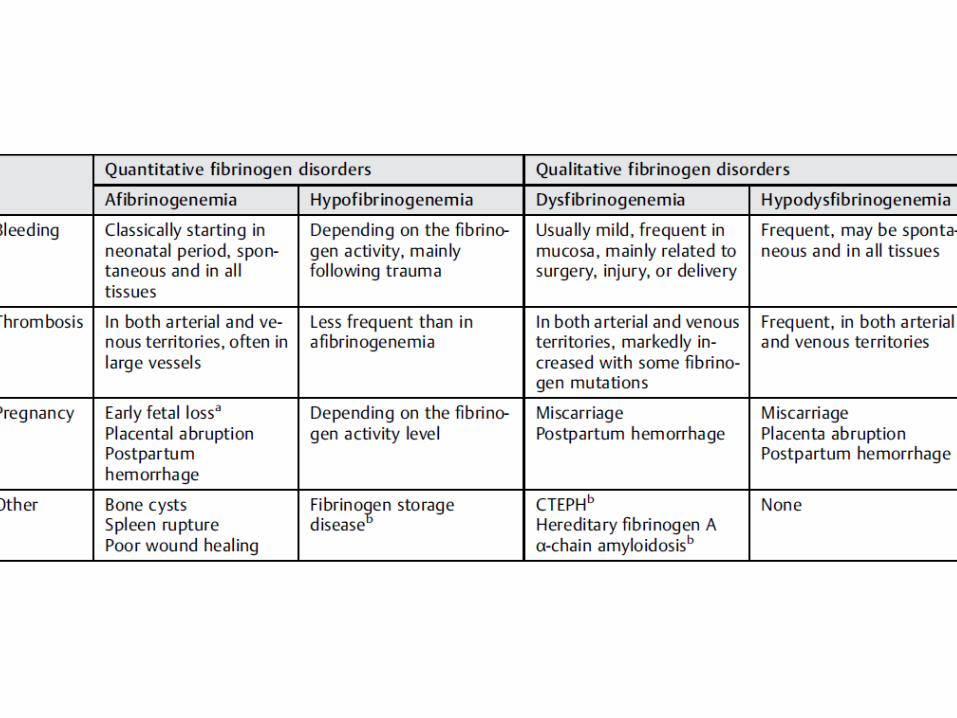

Trombosis (a-hipofibrinogenemia) other with recurrent thrombosis concomitant to life-threatening hemorrhages.81
The management of thrombosis is often challenging, as it is necessary to give both
antithrombotic drugs and fibrinogen concentrate at the same time. It has been reported
that the increased circulating levels of prothrombin fragment 1 þ 2 and thrombin–
antithrombin complexes, abnormally elevated in afibrinogenemia, can be normalized by
fibrinogen infusion.54,82–84 Regarding venous thrombosis, the optimal length of treatment
has not been established. Therefore, the same recommendations as for the general
population could be empirically applied. Low-molecularweight heparin is preferred to
vitamin K antagonists, given the impossibility to use the prothrombin time to monitor
treatment. Direct Xa or thrombin inhibitors may be more effective in this setting of
elevated circulating thrombin.
The successful use of lepirudin has been reported in a afibrinogenemic patient who
suffered recurrent arterial thrombosis,85 and more recently rivaroxaban has been used for
the management of a cerebral venous thrombosis in an afibrinogenemic woman.86
Antiplatelet therapy has safely been used in patients with arterial thrombosis.29 In case of
myocardial infarction, successful medical therapy with dual antiplatelet therapy87 or even
angioplasty has been reported.88 Thromboprophylaxis with low-molecular- weight heparin
should be considered in all patients with thrombotic history receiving fibrinogen
substitution, especially in high-risk thrombotic situations.59

Trombosis (hipo-disfibrinogenemia)
recommendations as those previously mentioned for the quantitative fibrinogen
disorders in case of surgery.4 In case of thrombosis, recommendations for general
population are valuable, although patients with fibrinogen mutations associated with
an increased susceptibility to thrombosis (mentioned earlier) should be treated with
long-term anticoagulation. 90,91 Most patients receive vitamin K antagonists (despite the
fact that the international normalized ratio is not an appropriate indicator in case of a
prolonged baseline PT) or low-weight-molecular heparin while a few have been
successfully treated with new oral anticoagulants.92 It is important to note that patients
still asymptomatic at the time of diagnosis may develop symptoms during the natural
course of the disease.41 Consequently, an accurate and regular follow-up must be
provided to all patients. In addition, functional analyses, such as turbidimetry assaying
fibrin polymerization and lysis and evaluation of the fibrin clot structure (viscoelastic
properties, permeability, fiber density, and architecture), could be performed in
research laboratories to better try to assess the patient phenotype.39,93