Embed Size (px)
Citation preview

Universidade Federal do Rio de Janeiro
Instituto de Química
Programa de Pós Graduação em Química
DESIDRATAÇÃO OXIDATIVA DO GLICEROL SOBRE CATALISADORES METÁLICOS SUPORTADOS EM FOSFATO E ÓXIDO DE NIÓBIO: FORMAÇÃO DE
ACROLEÍNA E ÁCIDO ACRÍLICO
Carolina Fernandes de Miranda Pestana
Professor orientador:
Claudio José de Araujo Mota
Doutor em Ciências
Rio de Janeiro, RJ – Brasil
Setembro de 2014

Universidade Federal do Rio de Janeiro Instituto de Química
Programa de Pós Graduação em Química
DESIDRATAÇÃO OXIDATIVA DO GLICEROL SOBRE CATALISADORES METÁLICOS SUPORTADOS EM FOSFATO E ÓXIDO DE NIÓBIO: FORMAÇÃO DE
ACROLEÍNA E ÁCIDO ACRÍLICO
Carolina Fernandes de Miranda Pestana
Essa tese de Doutorado foi realizada com o apoio financeiro da ANP - Agência Nacional do Petróleo, Gás Natural e Biocombustíveis.
Tese de Doutorado apresentada ao Programa de Pós
Graduação em Química da Universidade Federal do
Rio de Janeiro, como parte integrante dos requisitos
necessários para a obtenção do grau de Doutor em
Ciências, Química

P476 Pestana, Carolina Fernandes de Miranda. Desidratação oxidativa do glicerol sobre catalisadores metálicos suportados em fosfato e óxido de nióbio: formação de acroleína e ácido acrílico / Carolina Fernandes de Miranda Pestana – Rio de Janeiro: UFRJ/IQ, 2014.
128f., il. Tese (Doutorado em Ciências) – Universidade Federal do Rio de
Janeiro, Instituto de Química, Programa de Pós-Graduação em Química, Rio de Janeiro, 2014. Orientador: Claudio José de Araujo Mota. 1. Glicerol. 2. Desidratação Oxidativa. 3. Síntese de Acroleína. 4. Nióbio. 5. Síntese de Ácido Acrílico. I. Mota, Claudio José de Araujo. (Orient.). II. Universidade Federal do Rio de Janeiro. Instituto de Química. Programa de Pós-Graduação em Química. III. Título.
CDD: 547

Carolina Fernandes de Miranda Pestana
DESIDARTAÇÃO OXIDATIVA DO GLICEROL SOBRE CATALISADORES
METÁLICOS SUPORTADOS EM FOSFATO E ÓXIDO DE NIÓBIO: FORMAÇÃO
DE ACROLEÍNA E ÁCIDO ACRÍLICO
Tese de Doutorado apresentada ao Programa de Pós-Graduação em Química,
Universidade Federal do Rio de Janeiro, como requisitos parcial à obtenção do título de
Doutor em Ciências, Química
Aprovada em _______________
________________________________________________________ - Orientador
DSc. Claudio José de Araujo Mota (IQ – UFRJ)
________________________________________________________
DSc. Marco André Fraga (INT)
_________________________________________________________
DSc. Ana Maria Rocco (EQ - UFRJ)
___________________________________________________________
DSc. Elizabeth Roditi Lachter (IQ – UFRJ)
____________________________________________________________
DSc. Jussara Lopes Miranda (IQ – UFRJ)

À minha família, Rafael e Bernardo;
Aos meus pais, Pedro Paulo e Nilcinéa

AGRADECIMENTOS
Agradeço ao meu marido Rafael que, além de companheiro para todas as
horas, é o meu maior incentivador. Ao meu filho Bernardo por ter me dado a honra
de possuir o título mais importante da vida, o de mãe. A vocês agradeço pela
compreensão nas horas que estive ausente, pelo apoio e amor irrestrito em todos
os momentos. Amo vocês acima de tudo.
Aos meus pais, Pedro Paulo e Nilcinéa, que sempre incentivaram e
proporcionaram a minha formação. Vocês são um exemplo a se seguir, sem vocês
nada eu seria. Ao meu irmão, Camilo.
A todos os meus familiares e amigos que sempre estiveram perto de mim,
torcendo e acreditando no meu potencial.
Ao meu orientador Claudio José de Araujo Mota pelos ensinamentos,
confiança no meu trabalho e pela oportunidade.
A amiga Íria Bassan, que além das questões acadêmicas, se tornou uma
grande amiga para todas as horas. Obrigada por me confortar e ajudar no
momento mais difícil de toda esta jornada.
A todos os meus colegas de laboratório: Obrigada pelos momentos de
descontração em nosso ambiente de trabalho. Em especial, a Renata Jorge da
Silva e a Carolina Xavier pela companhia na sala de estudos e também nos
congressos nos quais estivemos juntas. A Luciana Machado pelas análises de
ASAP, além das risadas garantidas.
Aos funcionários do Pólo de Xistoquímica Professor Claudio Costa Neto, em
especial a Brunhilde Henker e Cláudia Caruzo.
A todos que participaram direta ou indiretamente deste trabalho.
A vocês meus agradecimentos!

RESUMO
PESTANA, Carolina Fernandes de Miranda. Desidratação Oxidativa do Glicerol Sobre Catalisadores Metálicos Suportados em Fosfato e Óxido de Nióbio: Formação de Acroleína e Ácido Acrílico. Orientador: Claudio José de Araujo Mota. Rio de Janeiro: UFRJ/IQ, 2014. Tese (Doutorado em Química).
Foi estudada a desidratação do glicerol à acroleína sob catalisadores ácidos,
bem como a desidratação oxidativa do glicerol ao ácido acrílico na presença de
catalisadores preparados por impregnação de sais de vanádio, molibdênio e/ou
cobalto em fosfato e óxido de nióbio. O desempenho desses catalisadores foi
avaliado nestas duas reações num reator de fluxo contínuo com leito fixo de
catalisador, sob atmosfera de nitrogênio e ar sintético, à pressão atmosférica e
temperatura de 300°C. Todos os catalisadores testados na desidratação do glicerol
(Nb2O5 e NbOPO4) produziram acroleína como produto principal e a seletividade
variou entre 70 e 90%. Outros produtos identificados foram acetaldeído,
formaldeído e hidroxiacetona. O acetaldeído e formaldeído são formados pela
decomposição térmica do 3-hidroxipropanal, a seletividade a este produto aumenta
com o aumento da temperatura. A hidroxiacetona é formada pela desidratação com
a hidroxila terminal do glicerol. Para a desidratação oxidativa do glicerol foram
avaliados catalisadores preparados com impregnação de sais metálicos em
suportes de nióbio. Os catalisadores que apresentaram maior seletividade a ácido
acrílico foram Co/V/NbOPO4, com 10,5%, e V/Mo/Nb2O5, com 10,1%. Além dos
produtos de desidratação (acroleína, acetaldeído e formaldeído) foi observado
ácido acético, possivelmente proveniente da oxidação do acetaldeído.
Palavras chave: glicerol, acroleína, ácido acrílico, desidratação, nióbia

ABSTRACT
PESTANA, Carolina Fernandes de Miranda. Desidratação Oxidativa do Glicerol Sobre Catalisadores Metálicos Suportados em Fosfato e Óxido de Nióbio: Formação de Acroleína e Ácido Acrílico. Orientador: Claudio José de Araujo Mota. Rio de Janeiro: UFRJ/IQ, 2014. Tese (Doutorado em Química).
Dehydration of glycerol to acrolein under acid catalysts and oxidative dehydration of
glycerol to acrylic acid in the presence of catalysts prepared by impregnation of
salts of vanadium, molybdenum and / or cobalt species of niobium, niobium oxide
and phosphate, were investigated. The performance of these catalysts was
evaluated in these two reactions in a continuous flow reactor with a fixed catalyst
bed, under nitrogen or synthetic air at atmospheric pressure and 300 °C. All
catalysts tested in the dehydration of glycerol (Nb2O5 and NbOPO4) produced
acrolein as the main product and the selectivity varied between 70 and 90%. Other
products were identified as acetaldehyde, hydroxyacetone and formaldehyde. The
formaldehyde and acetaldehyde are formed by thermal decomposition of 3-
hydroxypropanal, these products selectivity is higher with increasing temperature.
The hydroxyacetone is formed by dehydration of the terminal hydroxyl of glycerol.
Catalysts prepared with impregnating metal salts in niobium supports were
evaluated for oxidative dehydration of glycerol . The catalysts that showed higher
selectivity to acrylic acid were Co/V/NbOPO4, with 10.5%, and V/Mo/Nb2O5, with
10.1%. Aside from dehydration products (acrolein, acetaldehyde and
formaldehyde), acetic acid was observed, possibly from the oxidation of
acetaldehyde.
Keywords: glycerol, acrolein, acrylic acid, dehydration, niobium

Sumário
CAPÍTULO 1 - INTRODUÇÃO ................................................................................ 14
1.1 Objetivos .................................................................................................... 17
CAPÍTULO 2 – REVISÃO BIBLIOGRÁFICA ........................................................... 18
2.1 Glicerol ........................................................................................................... 18
2.2 Desidratação do Glicerol ................................................................................ 28
2.3 Oxidação da Acroleína................................................................................... 35
2.4 Desidratação Oxidativa do Glicerol ................................................................ 41
2.5 Nióbio ............................................................................................................. 48
CAPÍTULO 3 – MATERIAIS E MÉTODOS .............................................................. 54
3.1 Materiais ........................................................................................................ 54
3.2 Preparo dos Catalisadores ............................................................................ 55
3.3 Caracterizações ............................................................................................. 57
3.3.1 Espectrometria de Fluorescência de Raios-X ............................................. 57
3.3.2 Difração de Raios-X .................................................................................... 59
3.3.3 Análise Textural .......................................................................................... 59
3.3.4 Dessorção com Temperatura Programada de n-butilamina ....................... 60
3.4 Condições de Análise .................................................................................... 61
3.5 Testes Catalíticos .......................................................................................... 62
CAPÍTULO 4 – RESULTADOS E DISCUSSÃO ...................................................... 65
4.1 Caracterização dos Catalisadores ................................................................. 65
4.1.1 Espectrometria de Fluorescência de Raios-X ............................................. 65
4.1.2 Difração de Raios-X .................................................................................... 67
4.1.3 Dessorção com Temperatura Programada de n-butilamina ....................... 73
4.1.4 Análise Textural .......................................................................................... 77
4.2 Testes Catalíticos. ......................................................................................... 80
4.2.1 Avaliação dos Suportes .............................................................................. 80
4.2.2 Avaliação dos Catalisadores Bifuncionais .................................................. 85
4.2.2.1 Catalisadores Suportados em Óxido de Nióbio ....................................... 85
4.2.2.2 Catalisadores Suportados em Fosfato de Nióbio ..................................... 90
4.2.2.3 Estimativa da quantidade de CO e CO2 formados ................................... 98
4.2.2.4 Uso do Suporte ...................................................................................... 100

4.2.2.5 Proposta de mecanismo de desidratação/oxidação do glicerol ............. 102
CAPÍTULO 5 – CONCLUSÕES ............................................................................ 110
5.1 Perspectivas Futuras ................................................................................... 111
CAPÍTULO 6 – REFERÊNCIAS BIBLIOGRÁFICAS ............................................. 112
CAPÍTULO 7 - APÊNDICES .................................................................................. 123

LISTA DE TABELAS
Tabela 1 Propriedades Físico-químicas do Glicerol (Adaptada de Perry, 1997) ..... 19
Tabela 2: Massa de sal metálico utilizado em cada catalisador preparado ............. 56
Tabela 3 Condições reacionais utilizadas ............................................................... 62
Tabela 4: Condições cromatográficas utilizadas ..................................................... 64
Tabela 5: Teores dos elementos, em percentual de massa em base seca,
presentes nos catalisadores, obtidos por espectroscopia de fluorescência de Raios-
X .............................................................................................................................. 66
Tabela 6: Teores dos elementos, em mmol, presente em 1 grama de catalisador,
em base seca, onde “T” representa o valor teórico, e “E”, o valor experimental. .... 67
Tabela 7: Acidez dos catalisadores por dessorção com temperatura programada de
n-butilamina ............................................................................................................. 75
Tabela 8: Área Superficial e Volume de poro dos Catalisadores ............................ 78
Tabela 10 Seletividade média (Xm) e desvio padrão (σ) dos produtos formados nas
reações de desidratação com os suportes sem impregnação metálica .................. 84
Tabela 11 Caracterizações dos Catalisadores suportados em óxido de nióbio ...... 88
Tabela 12 Seletividade média (Xm) e desvio padrão (σ) dos produtos de
desidratação oxidativa do glicerol com catalisadores suportados em Nb2O5 .......... 90
Tabela 13 Comparação das caracterizações dos catalisadores suportados em
NbOPO4 .................................................................................................................. 96
Tabela 14 Seletividade média (Xm) e desvio padrão (σ) dos produtos da
desidratação oxidativa do glicerol com catalisadores suportados em NbOPO4 ...... 97
Tabela 15 Rendimento a CO e CO2 (Adaptado de DELAPLANQUE, 2010) ......... 100
Tabela 16 Acidez de Brønsted e Lewis de Nb2O5 e NbOPO4 calcinados a 300ºC
(Adaptado de LACHTER, 2013) ............................................................................ 105

LISTA DE FIGURAS
Figura 1 Distribuição da Matriz Energética Mundial (Adaptado de IEA, 2013) ........ 15
Figura 2: Reação de transesterificação do triglicerídeo com metanol ..................... 20
Figura 3 Distribuição dos usos da glicerina purificada (MOTA, 2009) ..................... 22
Figura 4. Reação de produção do propilenoglicol (1,2 propanodiol) ....................... 23
Figura 5: Reação de hidrogenólise de glicerina com produção de propeno ............ 23
Figura 6: Reação de eterificação da glicerina com etanol ....................................... 24
Figura 7: Esterificação da glicerina com ácido acético ............................................ 24
Figura 8: Reação de glicerina com aldeídos para produção de acetais cíclicos ..... 25
Figura 9: Reação de glicerina com acetona. Produção de solketal ......................... 25
Figura 10: Produtos de oxidação da glicerina ......................................................... 26
Figura 11 Reação de produção do carbonato de glicerina ...................................... 27
Figura 12: Possíveis produtos da reação de desidratação do glicerol .................... 29
Figura 13: Rota industrial de produção do ácido acrílico a partir do propeno ......... 35
Figura 14: Mecanismo da oxidação da acroleína a ácido acrílico
(ANDRUSHKEVICH, 1992) ..................................................................................... 40
Figura 15: Reação de desidratação oxidativa do glicerol com produção de ácido
acrílico ..................................................................................................................... 43
Figura 16: Possíveis produtos da reação de glicerina com catalisador ácido e
oxidante (HUNSOM, 2013)...................................................................................... 48
Figura 17 Estrutura do H8Nb6O19 (TANABE, 1987) ................................................. 51
Figura 18: Unidade de Fluxo Contínuo utilizada para a realização das reações. .... 63
Figura 19 Difratograma de Raios-X do Nb2O5 antes e após a calcinação a 550ºC68
Figura 20 Difratograma de raios-X do fosfato de nióbio antes e após a calcinação, a
550ºC ...................................................................................................................... 69
Figura 21: Difratograma do óxido de nióbio com e sem a impregnação de metal ... 70
Figura 22 Difratograma de Raio-X do fosfato de nióbio com e sem impregnação de
metais. ..................................................................................................................... 71
Figura 23 Difratograma de Raio-X de Fosfato de Nióbio com e sem impregnação de
vanádio (SUN, 2007). (▲) fase cristalina de V2O5. ................................................. 72
Figura 24 Difratograma do catalisador mássico de V e Mo, e dos respectivos óxidos
................................................................................................................................ 73

Figura 25: Curva termogravimétrica e DrTGA de perda de massa em função da
temperatura do catalisador Nb2O5 ........................................................................... 74
Figura 26 Isoterma de adsorção do catalisador V/Mo/Nb2O5 ................................. 80
Figura 27: Conversão do glicerol e seletividade aos produtos para a reação de
desidratação com NbOPO4 ..................................................................................... 82
Figura 28: Conversão do glicerol e seletividade aos produtos para a reação de
desidratação com Nb2O5 ......................................................................................... 82
Figura 29 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação Oxidativa com Mo/V/Nb2O5 .............................................................. 85
Figura 30 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação Oxidativa com Mo/Co/Nb2O5 ............................................................ 86
Figura 31 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação d Oxidativa com V/Co/Nb2O5 ........................................................... 87
Figura 32 Seletividade média dos produtos dos catalisadores suportados em Nb2O5
................................................................................................................................ 89
Figura 33 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação Oxidativa com 5%V/NbOPO4 ............................................................ 91
Figura 34 Conversão do glicerol e seletividade dos produtos da reação de
desidratação oxidativa com 20V/NbOPO4 .............................................................. 92
Figura 35 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação d Oxidativa com V/Mo/NbOPO4 ....................................................... 93
Figura 36 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação d Oxidativa com Co/Mo/NbOPO4 ..................................................... 94
Figura 37 Conversão do Glicerol e Seletividade dos Produtos da Reação de
Desidratação Oxidativa com Co/V/NbOPO4 ........................................................... 95
Figura 38 Seletividades média dos produtos com catalisador suportados em
NbOPO4 .................................................................................................................. 97
Figura 39 Curva de calibração do CO2 ................................................................... 99
Figura 40 Cromatograma do CO e CO2 utilizando DCT .......................................... 99
Figura 41 Seletividades Médias dos catalisadores a base de V/Mo com e sem
suporte .................................................................................................................. 102
Figura 42 Possíveis produtos formados na reação de desidratação oxidativa do
glicerol (Adaptado de DELAPLANQUE, 2010) ...................................................... 103

Figura 43 Possíveis produtos da primeira desidratação do glicerol (Adaptado de
COLL, 2011 ) ......................................................................................................... 104
Figura 44 Mecanismo de desidratação do glicerol a hidroxiacetona (Adaptado de
POSSATO, 2013) .................................................................................................. 106
Figura 45 Regeneração do sítio de Lewis ............................................................. 106
Figura 46 Mecanismo de desidratação do glicerol a acroleína (adaptado de
POSSATO, 2013) .................................................................................................. 107
Figura 47 Regeneração do sítio de Bronsted do catalisador ................................. 108
Figura 48 Produção de acetaldeído (Adaptado de SHEN, 2014) .......................... 108
Figura 49 Curva Termogravimétrica de perda de massa do catalisador
Co/Mo/Nb2O5 ....................................................................................................... 123
Figura 51 Curva Termogravimétrica de perda de massa do catalisador V/Mo/Nb2O5
.............................................................................................................................. 124
Figura 52 Curva Termogravimétrica de perda de massa do catalisador
Co/Mo/NbOPO4 .................................................................................................... 125
Figura 53 Curva Termogravimétrica de perda de massa do catalisador Nb2O5 ... 125
Figura 54 Curva Termogravimétrica de perda de massa do catalisador
V/Mo/NbOPO4 ...................................................................................................... 126
Figura 55 Curva Termogravimétrica de perda de massa do catalisador
Co/V/NbOPO4 ....................................................................................................... 126
Figura 56 Curva Termogravimétrica de perda de massa do catalisador
Co/Mo/NbOPO4 .................................................................................................... 127
Figura 57 Curva Termogravimétrica de perda de massa do catalisador NbOPO4 127
Figura 58 Curva Termogravimétrica de perda de massa do catalisador
20V/NbOPO4 ........................................................................................................ 128
Figura 59 Curva Termogravimétrica de perda de massa do catalisador 5V/NbOPO4
.............................................................................................................................. 128

14
CAPÍTULO 1 - INTRODUÇÃO
A crescente preocupação com o meio ambiente e o aquecimento global
motivam diversas pesquisas que visam minimizar o impacto causado pelas ações
do homem. Neste contexto as novas fontes de energia, principalmente as
renováveis, ganham cada vez mais destaque por emitirem menores quantidades de
gases do efeito estufa e, também, pela possibilidade de os gases produzidos serem
absorvidos pelas próprias plantas cultivadas para a produção dos biocombustíveis.
O mundo contemporâneo ainda é muito dependente dos derivados fósseis.
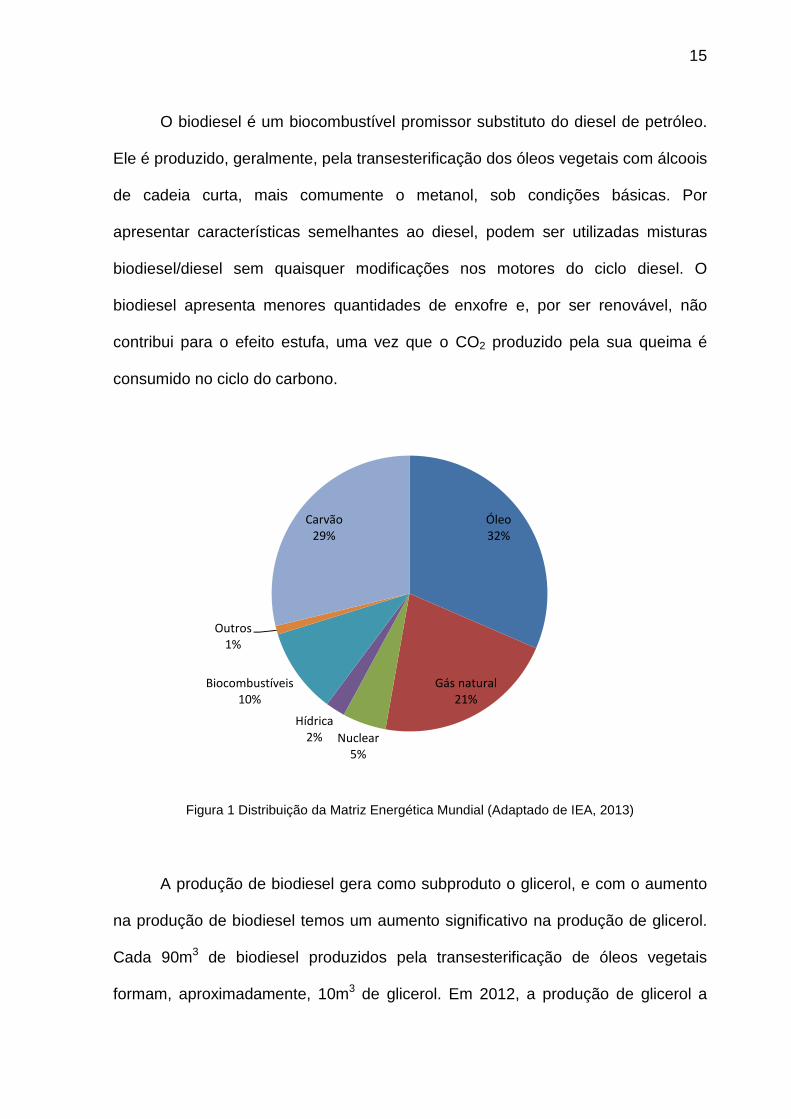
Cerca de 80% da matriz energética mundial é proveniente do petróleo, gás natural
e carvão mineral (Figura 2), que são fontes esgotáveis e não renováveis (IEA,
2013). Segundo dados do Anuário Estatístico Brasileiro do Petróleo, Gás Natural e
Biocombustíveis da ANP, o consumo mundial de petróleo em 2013 foi superior a 89
milhões de barris por dia, o que representa um aumento de 1% ao ano anterior. No
Brasil, o consumo foi pouco mais de 2,8 milhões de barris por dia, sendo esse valor
2,35% maior que em 2012. Na mesma direção do petróleo, a produção de
biocombustíveis também tem aumentado. No Brasil, em 2013, foram produzidos
cerca de 2,7 milhões de metros cúbicos de biodiesel, representando um aumento
de 1,67% do período anterior. (ANP,2013)
15
O biodiesel é um biocombustível promissor substituto do diesel de petróleo.
Ele é produzido, geralmente, pela transesterificação dos óleos vegetais com álcoois
de cadeia curta, mais comumente o metanol, sob condições básicas. Por
apresentar características semelhantes ao diesel, podem ser utilizadas misturas
biodiesel/diesel sem quaisquer modificações nos motores do ciclo diesel. O
biodiesel apresenta menores quantidades de enxofre e, por ser renovável, não
contribui para o efeito estufa, uma vez que o CO2 produzido pela sua queima é
consumido no ciclo do carbono.
Figura 1 Distribuição da Matriz Energética Mundial (Adaptado de IEA, 2013)
A produção de biodiesel gera como subproduto o glicerol, e com o aumento
na produção de biodiesel temos um aumento significativo na produção de glicerol.
Cada 90m3 de biodiesel produzidos pela transesterificação de óleos vegetais
formam, aproximadamente, 10m3 de glicerol. Em 2012, a produção de glicerol a
Óleo32%
Gás natural21%
Nuclear5%
Hídrica2%
Biocombustíveis10%
Outros1%
Carvão29%

16
partir do biodiesel foi de 274mil m3 e esse grande volume de glicerol ofertado
acarreta queda no preço de mercado. (ANP, 2013)
A transformação do glicerol em produtos de maior valor agregado pode ser
um fator fundamental para o sucesso do Programa Nacional de Produção do
Biodiesel (PNPB), pois pode diminuir o custo final do biodiesel ao consumidor,
tornando-o mais atrativo economicamente. Ademais, a utilização econômica do
glicerol evita prejuízos ambientais, com a sua queima direta, ou dejetos em rios e
lagos.

17
1.1 Objetivos
Objetivo Geral
O trabalho tem como objetivo principal a transformação química do glicerol
em produtos de maior valor agregado, que possam ser utilizados na cadeia de
produção de plásticos e outros produtos. Mais especificamente, a proposta é
converter o glicerol (1,2,3 propanotriol) em acroleína (propenal) e ácido acrílico
(ácido propenóico) através da desidratação e desidratação oxidativa,
respectivamente, sobre catalisadores bifuncionais de metais suportados em
compostos de nióbio.
Objetivos Específicos
• Formular catalisadores metálicos bifuncionais suportados em compostos a
base de nióbio;
• Caracterizar os catalisadores metálicos bifuncionais suportados em
compostos a base de nióbio;
• Avaliar os catalisadores preparados na reação de desidratação oxidativa do
glicerol sob condição de fluxo contínuo para a produção de acroleína e ácido
acrílico

18
CAPÍTULO 2 – REVISÃO BIBLIOGRÁFICA
Neste capítulo é feita a revisão bibliográfica da literatura sobre os temas e
reações abordados neste trabalho.
2.1 Glicerol
Glicerol ou 1,2,3 propanotriol é um álcool, líquido a temperatura ambiente,
viscoso, inodoro e incolor. Também pode ser denominado comercialmente como
glicerina, quando se apresenta com pureza acima de 95%. O glicerol é encontrado
naturalmente em óleos e gorduras de origem vegetal ou animal na forma
combinada, ligado a ácidos graxos.
O glicerol foi descoberto em 1783 pelo químico sueco Carl Wilhelm Scheele,
quando tratava óleos naturais com uma substância alcalina. O glicerol recebeu
esse nome por Michel Eugene Chevreul em 1811, originado da palavra grega
“glycos”, que significa doce. Em 1866 teve seu primeiro uso técnico na produção de
dinamite, nitroglicerina (BEHR, 2008).
Em sua forma pura e anidra a glicerina tem densidade específica de 1,261
g/mL, ponto de fusão de 18,2 ºC e ponto de ebulição de 290 ºC sob pressão
atmosférica normal. Possui uma combinação própria de propriedades físico-
químicas como pode ser observado na Tabela 1 (Perry, 1997).

19
Tabela 1 Propriedades Físico-químicas do Glicerol (Adaptada de Perry, 1997)
Propriedades Valores
Peso Molecular 92,09
Densidade (100% pureza a 25ºC) 1,261 g/mL
Viscosidade (20ºC) 939 cps
Ponto de Ebulição 290ºC
Ponto de Fusão 18ºC
Tensão superficial (20ºC) 63,4 mN/m
Calor específico (99,96% de pureza, 26ºC) 2,435 J/g
Calor de evaporação (55ºC) 88,12 J/mol
Calor de Dissolução 5,8 J/mol
Calor de evaporação 667,8 J/mol
A produção do glicerol foi feita a partir da epicloridrina desde a década de
40. Essa rota caiu em desuso pelo fato da epicloridrina ser proveniente do propeno,
que por sua vez é proveniente de combustíveis fósseis. Antes disso, todo o glicerol
produzido no mundo era proveniente da indústria de sabão. A partir do aumento do
uso do biodiesel, um grande excedente de glicerol tem sido gerado, fazendo com
que diminua cada vez mais as plantas de produção de glicerol com consequente
aumento das plantas que utilizam glicerol como matéria prima para diversos
produtos químicos, inclusive a própria epicloridrina (BEATRIZ, 2011).
Atualmente, a maior parte da glicerina produzida é proveniente do processo
de produção do biodiesel. Esse biocombustível é um éster de ácido graxo obtido
pela transesterificação de óleos ou gorduras, de origem vegetal ou animal, com

20
álcoois de cadeia curta, como o metanol ou etanol, sob condições básicas ou
ácidas, sendo formada glicerina como subproduto (PINTO, 2005). A reação de
transesterificação é mostrada na Figura 2.
R1
O
O
O
R3
OR2
O
O
+ CH3OH
R1 OCH3
O
R3 OCH3
O
R2 OCH3
O
+
+
+
HO
HO
HO
Figura 2: Reação de transesterificação do triglicerídeo com metanol
A reação de transesterificação pode ser catalisada por meio básico ou ácido,
utilizando processo catalítico homogêneo ou heterogêneo. Hidróxidos de sódio e de
potássio são os catalisadores mais comumente utilizados nas indústrias. Têm como
vantagens serem baratos e bastante reativos mas, como desvantagens, podem
produzir sabões pela hidrólise e neutralização dos ácidos graxos livres, assim como
pela saponificação do triglicerídeo (BORGES, 2012). A formação de sabão é uma
reação secundária indesejável porque ela consome o catalisador, diminui o
rendimento de biodiesel e dificulta as etapas de purificação e separação. A
remoção destes catalisadores é tecnicamente difícil e traz custos adicionais ao
produto final. Além disso, a dificuldade para a reciclagem e a grande quantidade de
resíduos gerados faz o tradicional catalisador ser menos favorável (MOTA, 2006).

21
O crescimento da demanda e, consequentemente, da produção de biodiesel
são acompanhados pelo aumento da quantidade de glicerina produzida, seu
principal subproduto; para cada 90m3 de biodiesel produzidos são gerados cerca
de 10m3 de glicerina. A glicerina proveniente da produção do biodiesel é chamada
de bruta ou loira e apresenta em torno de 20% de impurezas, entre elas, o álcool,
água, mono, di e triglicerídeos não reagidos, sais, além de apresentar alta
basicidade por conta do catalisador utilizado. As características da glicerina bruta
dependem do óleo vegetal, do álcool e do catalisador utilizados. Os processos de
purificação da glicerina bruta são complexos e bastante onerosos, o que incentiva
ainda mais a utilização dela com o mínimo de tratamento em reações de
transformação química (BEATRIZ, 2011).
Segundo dados do Anuário Brasileiro de Petróleo, Gás Natural e
Biocombustíveis, em 2012, foram produzidos pouco mais de 274 mil m3 de glicerina
provenientes da transesterificação de ácidos graxos (ANP, 2012). O grande
aumento na oferta de glicerina pressionou a queda dos preços. Em 2005, quando a
produção nacional era muito pequena, cerca de 69 m3, a glicerina custava em
média R$3,00/quilo. Em 2007, com a produção elevada para 36mil m3, o preço não
passava de R$1,70/quilo.
Os setores farmacêutico, de cosméticos, higiene, alimentos e fumo são os
maiores responsáveis pela aplicação da glicerina purificada. Uma pequena
quantidade é utilizada para transformação química, principalmente na produção de
explosivos, como a nitroglicerina, e resinas alquílicas (MOTA, 2009). A Figura 3
mostra os usos mais comuns para a glicerina purificada.

22
Figura 3 Distribuição dos usos da glicerina purificada (MOTA, 2009)
A transformação química da glicerina comercialmente ainda é pouco
utilizada apesar de muitas serem as possibilidades. Diversos estudos mostram a
conversão deste reagente em diversos outros insumos químicos de grande
importância industrial, dentre eles estão a hidrogenólise, eterificação, esterificação,
oxidação, desidratação, acetalisação (MOTA, 2011).
A reação de hidrogenólise do glicerol produz, principalmente, 1,2
propanodiol (Figura 4), que pode ser usado como aditivo anti-congelante para
combustíveis, e 1,3 propanodiol, utilizado na produção de fibras sintéticas. Esta
reação é normalmente realizada na presença de catalisadores metálicos, a base de
Cu, Ni ou Pd, tanto em solução como em fase gasosa (DASARI, 2005).
Papéis1%
Ésteres13%
Poliglicerina12%
Tabaco3%
Filmes e celulose5%
Alimentos e bebidas
8%
Resinas Alquílicas
6%
Cosméticos Saboaria Farmacos
28%
Revenda 14%
Outros10%

23
Figura 4. Reação de produção do propilenoglicol (1,2 propanodiol)
A hidrogenólise mais profunda da glicerina pode produzir n-propanol e
isopropanol, além de propeno e propano (Figura 5). Um processo para produção de
propeno a partir de glicerina foi recentemente desenvolvido e se mostra
interessante como meio de produzir plásticos verdes, produzidos a partir de
matéria-prima renovável, a partir da cadeia do biodiesel (MOTA, 2008).
Figura 5: Reação de hidrogenólise de glicerina com produção de propeno
A eterificação do glicerol produz compostos de menor polaridade e
viscosidade. A produção de éteres de glicerina pode ocorrer pela reação com
álcoois, como metanol, etanol e álcool benzílico, podendo formar mono, di ou tri
éteres (Figura 6). Os éteres etílicos de glicerina são capazes de diminuir o ponto de
congelamento do biodiesel, portanto se apresentam como promissores aditivos
para esta função. (Da Silva, 2009).

24
Figura 6: Reação de eterificação da glicerina com etanol
A produção de mono, di e triacetatos de glicerol, também conhecidos como
mono, di ou triacetina, é obtida pela esterificação, onde ocorre a reação de glicerina
com ácido acético, em presença de catalisadores ácidos, como mostrado na
Figura 7 (GONÇALVES, 2008). Os monoésteres de glicerol podem ser usados
como emulsificantes na indústria alimentícia, cosmética e farmacêutica.
Figura 7: Esterificação da glicerina com ácido acético

25
Reações de acetalisação do glicerol com aldeído formam, em geral, acetais
cíclicos com anéis de 5 ou 6 membros (Figura 8). Tais produtos melhoram as
propriedades de fluxo a frio do biodiesel, como o ponto de névoa e o ponto de
fluidez (MOTA, 2010A). Quando esta mesma reação é realizada com acetona
forma-se como único produto o solketal (Figura 9), que adicionado à gasolina, é
capaz de diminuir a formação de goma e aumentar a octanagem (MOTA, 2010B).
Figura 8: Reação de glicerina com aldeídos para produção de acetais cíclicos
Figura 9: Reação de glicerina com acetona. Produção de solketal
É possível, também, a produção de hidrogênio e gás de síntese (mistura de
CO e H2) a partir da reforma a vapor do glicerol (LIN, 2013). O gás de síntese
apresenta diversas aplicações industriais, como a produção de metanol que pode
ser reintegrado na cadeia do biodiesel, sendo utilizado na reação de
transesterificação de óleos vegetais.
Muitos são os possíveis produtos da reação de oxidação do glicerol (Figura
10). A oxidação catalítica da hidroxila terminal do glicerol forma ácido glicérico e

26
ácido tartrônico. A oxidação da hidroxila central produz di-idroxiacetona, enquanto
a oxidação de ambas as hidroxilas produz ácido hidroxi-pirúvico e ácido
mesoxálico. Além desses produtos, podem ser formados, com quebra de ligação
carbono-carbono, ácido oxálico, hidroxietanóico e fórmico.
Figura 10: Produtos de oxidação da glicerina
A formação de carbonato de glicerina a partir da reação da glicerina com
dióxido de carbono, utilizando catalisadores a base de metais de transição, é uma
rota bastante interessante por utilizar dois rejeitos para a produção de um
composto com valor comercial (DIBENEDETTO, 2006). O carbonato de glicerina
tem aplicações em diversas áreas, por exemplo, como solvente industrial e também
como precursor na preparação de policarbonatos, poliésteres, poliuretanas e
poliamidas (MOTA, 2009).

27
OH
OH
OH + CO2 O
OO
OH
+ H2Ocat
Figura 11 Reação de produção do carbonato de glicerina
Os processos de transformação química da glicerina em produtos de maior
valor agregado representam uma forma de utilização deste insumo e, também,
rotas sustentáveis para a produção da maioria dos produtos citados, que possuem
processos de obtenção caros e dependentes de matéria-prima de origem fóssil.
Além das transformações química já citadas, há a reação de desidratação do
glicerol, que será explanada em um tópico posterior.
Muitos são os estudos da transformação química do glicerol puro, mas
poucos mostram o uso de glicerol bruto, oriundo do processo de produção do
biodiesel. Geralmente, a purificação do glicerol bruto passa pelas seguintes etapas:
neutralização do catalisador remanescente; remoção dos sais; evaporação do
álcool e da água. Etapas de destilação, desodoridação e branqueamento ainda
podem ser feitas para obter uma maior pureza (SKRZYNSKA, 2014). Na reação de
oxidação seletiva do glicerol, por exemplo, a conversão do glicerol puro é de 45%
enquanto a do glicerol bruto não ultrapassa os 5%, em catalisadores de ouro
suportado em titânia (SULLIVAN, 2014).

28
2.2 Desidratação do Glicerol
Desidratação é a reação de eliminação de uma ou mais moléculas de água
de um determinado composto orgânico. A desidratação catalítica do glicerol é uma
rota alternativa para a produção de acroleína. Normalmente, esta reação é
realizada sobre catalisadores com características ácidas e temperaturas até 400ºC.
Esta reação pode ser feita em fase líquida ou em fase gasosa, com utilização de
catalisador homogêneo ou heterogêneo (ZHOU, 2013). Em regra geral, as reações
de hidratação são favorecidas em baixas temperaturas enquanto as de
desidratação em temperaturas elevadas. Foi mostrado que em temperaturas
relativamente baixas (250°C) há uma diminuição da conversão do glicerol enquanto
se tem uma elevada seletividade a acroleína (ARKEMA, 2005).
O glicerol é um álcool com três hidroxilas e, portanto, a desidratação pode
acontecer em duas hidroxilas diferentes. A Figura 12 mostra as possíveis rotas de
desidratação do glicerol. A rota 1 é referente à desidratação pela hidroxila central,
formando 3-hidroxipropanal, seguida de uma segunda desidratação (rota 3) com
formação de acroleína. O intermediário 3-hidroxipropanal ainda pode sofrer uma
clivagem térmica (rota 4) em formaldeído e acetaldeído. A desidratação do glicerol
pode ser, também, através de uma das hidroxilas terminais, rota 2, onde há
formação de hidroxiacetona, também chamado de acetol.
A reação de desidratação do glicerol em acroleína é geralmente
acompanhada de reações secundárias com formação de subprodutos como
hidroxiacetona, propanal, acetaldeído, formaldeído, acetona, e produtos da
policondensação do glicerol e ésteres cíclicos do glicerol (ARKEMA, 2006).

29
Figura 12: Possíveis produtos da reação de desidratação do glicerol
No estudo de Mark R. Nimlos foi investigado, através de cálculos
computacionais, os mecanismos de desidratação do glicerol em fase gasosa. Este
trabalho mostra que, apesar das reações serem endotérmicas, ocorrem com
significativa conversão, uma vez que os estados de transição são muito bem
estabilizados pelas fortes ligações de hidrogênio formadas. Ainda segundo Nimlos
e colaboradores a afinidade das hidroxilas central e terminal ao próton, na reação
de desidratação ácida, são praticamente idênticas, em torno de 195 kcal mol-1
(NIMLOS, 2006).
Sob condições reacionais específicas, a seletividade a acroleína aumenta
com o aumento da acidez total do catalisador, assim como a seletividade a
acetaldeído, propionaldeído e acetona. Já a seletividade a hidroxiacetona e álcool
alílico diminuem. A desativação do catalisador está relacionada com a formação de
depósitos de carbono na sua superfície, consequência das reações consecutivas

30
do glicerol e das reações paralelas entre os produtos formados na desidratação
(SUPRUN, 2009).
A desidratação a acroleína é citada em uma patente (DEGUSSA, 1995),
onde é descrito que a reação ocorre mesmo com o glicerol cru, oriundo da
clivagem de óleos, sem prévia purificação ou concentração. É utilizada uma
solução aquosa de glicerol, e quando a concentração, em peso, é menor que 40%,
a desidratação ainda ocorre. Contudo, o decréscimo dessa concentração diminui
tanto a seletividade quanto a recuperação do catalisador. Vários catalisadores
sólidos, como sulfatos, fosfatos, zeólitas e ácido fosfórico sólido, têm sido testados
para a desidratação do glicerol em fase gasosa ou líquida.
Diversos estudos mostram que o produto principal da desidratação ácida do
glicerol é a acroleína, que é um aldeído insaturado, quimicamente chamado de
propenal, de cor amarelo pálido, odor forte e irritante, inflamável, tóxico e com
grande facilidade de polimerização. Este insumo é usado na medicina, no
tratamento de águas e como biocida na indústria do petróleo. Mas também é
necessário como material de partida para a síntese de produtos químicos finos e
intermediários, como a metionina, fragrâncias e corantes. Além dos usos já citados,
a síntese de ácido acrílico é a principal aplicação da acroleína (HOELDERICH,
2011).
A rota mais usada de produção de acroleína é pela oxidação parcial do
propeno utilizando catalisadores a base de óxidos mistos de metais (HOLBROOK,
1971). Desde o início do século XX (SEBATIER, 1918, apude HOELDERICH,
2011) diversos catalisadores heterogêneos têm sido estudados na reação de

31
desidratação do glicerol, como os a base de sílica, zeólitas, heteropoliácidos e
óxidos mistos.
As zeólitas são materiais constantemente estudados como catalisadores
para a reação de desidratação do glicerol e apresentam altas conversões e
seletividade a acroleína. Segundo a definição clássica, o termo zeólita se refere a
alumino-silicatos cristalinos hidratados de estrutura aberta, constituídos por
tetraedros de SiO4 e AlO4 ligados entre si pelos átomos de oxigênio. Algumas
características das zeólitas são responsáveis pela sua grande eficiência, entre elas:
alta área superficial, alta capacidade de adsorção de moléculas que variam de
altamente hidrofóbicas à altamente hidrofílicas, estruturas que permitem a criação
de sítios ativos, tamanho das cavidades e possibilidade de diferentes tipos de
seletividade de forma. Estudos com H-Beta (QUERINI, 2014), H-ZSM -5 (CORMA
2008; SCHÜTH, 2010; LI, 2012), H-ZSM-11 (LI, 2012), assim como estruturas
análogas às zeólitas como MCM-22 (MASCARENHAS, 2013) mostram altas taxas
de conversão do glicerol (perto de 100%), com acroleína como produto principal
(entre 50 e 80% de seletividade).
Estudo utilizando catalisador de cobre impregnado em zeólita ZSM-5 mostra
que o aumento da temperatura favorece a conversão do glicerol e a seletividade a
acroleína, em detrimento à quantidade de acetol formada. Quando é utilizado
temperatura fixa de 200ºC, o aumento do tempo de reação implica em menores
seletividades a acroleína, com consequente, maiores seletividades a acetol (YUE,
2014).
Os heteropoliácidos são eficientes catalisadores para a reação de
desidratação do glicerol, pois possuem acidez elevada e ajustável para uma melhor

32
atividade. Apesar de apresentar resultados iniciais bastante promissores
catalisadores a base de heteropoliácidos se desativam após algumas horas de
reação (SATO, 2007; MARTIN, 2008; KOZHEVNIKOV, 2010).
Estudos da reação de desidratação do glicerol sobre catalisadores de
tungstênio suportado em zircônia mostra que a conversão do glicerol e a
seletividade a acroleína dependem, majoritariamente, da fração de sítios ácidos
moderados que são formados basicamente pelos sítios ácidos de Brønsted.
Enquanto a seletividade a hidroxiacetona é dependente dos sítios ácidos de Lewis
(GINJUPALLI, 2014).
Zhou e colaboradores estudaram a reação de desidratação do glicerol com a
argila montmorilonita ativada com soluções de H2SO4, em diferentes
concentrações. A reação foi conduzida em fase gasosa, com a utilização de leito
fixo de catalisador. O melhor resultado obtido foi para o catalisador com 10% de
H2SO4, a 320º C, onde houve 54% de conversão do glicerol com 83% de
seletividade a acroleína. Neste estudo foi verificado que o aumento da temperatura
favorece a rota de formação da hidroxiacetona (ZHOU, 2013).
A reação de desidratação do glicerol foi conduzida em catalisadores de
óxido de titânio impregnados com diferentes teores de óxido de tungstênio. Foi
verificado que o aumento da quantidade, em massa, de WO3 no catalisador
aumenta a seletividade a acroleína enquanto diminui a formação de CO e CO2. Na
temperatura de 280ºC o catalisador contendo 17,8% em massa de WO3 em TiO2
apresentou 90% de conversão de glicerol com 72% de seletividade a acroleína
(HOELDERICH, 2011).

33
O efeito do suporte foi investigado em catalisadores preparados pela
impregnação de óxido de tungstênio em diferentes óxidos. Os catalisadores
suportados em alumina ou zircônia apresentam pouca desativação ao longo do
tempo de reação, enquanto os suportados em alumina desativam a partir da
segunda hora de reação. Nos três tipos de catalisadores a seletividade a acroleína
não varia com o tempo, mas é maior nos catalisadores suportados em zircônia
(CHAI, 2014).
A desidratação do glicerol foi realizada em fase líquida utilizando diversos
catalisadores iônicos líquidos com acidez de Brønsted. Nestas reações o
rendimento a acroleína não passou de 58% (SHEN, 2014).
Além dos catalisadores já citados, materiais a base de nióbio também têm se
mostrado bons catalisadores para reações como desidratação, hidratação, hidrólise
por possuir uma boa capacidade de tolerar a água, entre outras características
(TANABE, 1995). O pentóxido de nióbio (Nb2O5) é um sólido branco, estável,
insolúvel em água. Quando hidratado (Nb2O5.nH2O) apresenta forte acidez, com
sítios de Bronsted e de Lewis. O fosfato de nióbio (NbOPO4) possui acidez mais
elevada que o óxido hidratado, pois os grupos P-OH de comportam como sítios de
Brønsted (SUN, 2006).
Xu e colaboradores mostraram a relação entre a temperatura de calcinação
do óxido de nióbio, com suas propriedades e atividade catalítica na reação de
desidratação do glicerol. O óxido de nióbio calcinado a 400ºC apresentou o melhor
resultado catalítico, sendo capaz de converter 100% do glicerol com 50% de
seletividade à acroleína. Além disso, pode-se concluir por esse trabalho que o
aumento da temperatura de calcinação acarreta uma diminuição da área BET,

34
diminuição da acidez total, aumento da cristalinidade e aumento da seletividade a
hidroxiacetona (XU, 2007).
Catalisadores de nióbio suportado em diferentes materiais, em conjunto ou
não com outros metais, foram testados na reação de desidratação do glicerol.
Quando suportado em alumina (Al2O3) ou titânia (TiO2) a conversão do glicerol não
é alterada, mas há um aumento significativo na seletividade a acroleína, de 38%
sem nióbio para 60% com nióbio no caso da alumina, e de 14% para 55% no caso
da titânia. Em ambos os suportes, os catalisadores impregnados com nióbio e
tungstênio, concomitantemente, apresentaram seletividade em torno de 70% a
acroleína. Utilizando sílica (SiO2) como suporte, a impregnação do metal altera
tanto a conversão (de 1,3 % para 82%) quanto a seletividade a acroleína (de 0%
para 57%), sem e com nióbio, respectivamente. Nos catalisadores suportados em
Al2O3 e TiO2 são identificadas fases de niobato e tungstato, enquanto nos
catalisadores de SiO2 são encontrados os óxidos de Nb e W na forma mássica
(ANDERSSON, 2013). Nióbia impregnado em zircônia (ZrO2) também se mostra
um catalisador eficiente para desidratar o glicerol, com conversões perto de 100%
e altas seletividades a acroleína, além de não sofrer desativação ao longo do
tempo de reação (MILLET, 2011).
O óxido de nióbio pode funcionar como suporte para outros metais como,
por exemplo, no estudo de Wang e colaboradores, onde Cs, P e W são
impregnados em Nb2O5. A impregnação dos metais melhora a atividade catalítica,
aumentando a conversão de glicerol e a seletividade a acroleína, além de diminuir
a deposição de coque, em comparação com o Nb2O5 puro. A deposição de coque e

35
reações laterais são relacionadas com a formação de multicamadas do óxido
metálico (WANG, 2013).
Catalisadores preparados pela impregnação de diferentes proporções de
PO4 em Nb2O5 foram testados na reação de desidratação do glicerol. O melhor
resultado apresentado foi com o 50%WPO4/Nb2O5 com 68% de conversão do
glicerol e 72% de seletividade a acroleína. Os autores relacionam à incorporação
do PO4 o aumento da atividade catalítica, apesar da perda de quantidade desta
espécie durante a reação, o que impede a completa regeneração do catalisador
(LEE, 2014).
2.3 Oxidação da Acroleína
A reação de oxidação da acroleína é a principal rota de produção de ácido
acrílico. Na indústria, essa reação é subsequente à reação de oxidação do
propeno, onde a acroleína é formada, como mostrado na Figura 13.
O
CO2H
O2 O2
[cat] [cat]Propeno Acroleína Ácido Acrílico
Figura 13: Rota industrial de produção do ácido acrílico a partir do propeno
O ácido acrílico é importante na indústria química pela sua extraordinária
facilidade para polimerização. Por isso, requer cuidados em seu manuseio,
transporte e estocagem. É considerado um produto prejudicial à saúde e, por ser

36
miscível em água, é contaminante de esgotos, rios e córregos. É inflamável e pode
formar uma mistura explosiva com o ar. Apresenta-se na forma de um líquido
corrosivo incolor e de cheiro penetrante. É usado, principalmente, na indústria para
a produção de ésteres acrílicos, mas também pode ser utilizado na fabricação de
polímeros superabsorventes usados em fraldas descartáveis, detergentes, agentes
para tratamento de água, tintas, adesivos, entre outros. Recentemente, ocorreu um
aumento pronunciado em seu consumo devido, sobretudo, ao aumento da
demanda de polímeros superabsorventes. Pode ser utilizado também para a
produção de acrilonitrila, que é empregada como fibra sintética e em painéis e
interiores de automóveis.
De acordo com dados da Secretaria de Comércio Exterior (SECEX), do
Ministério do Desenvolvimento, Indústria e Comercio (MDIC), o Brasil importou no
ano de 2008 aproximadamente 50 mil toneladas de ácido acrílico, o que representa
algo em torno de 60 milhões de dólares (apud SILVA, 2011).
É sabido que o processo de oxidação da acroleína é favorável com excesso
estequiométrico de oxigênio. A reação é acelerada na presença de vapor d’água e
que a temperatura de reação não deve ultrapassar 300ºC (TICHÝ, 1997). Além
disso, é uma reação bastante exotérmica e suscetível a reações paralelas ou
subsequentes (BASF, 1998).
Do ponto de vista ácido-básico, a reação de oxidação da acroleína pode ser
definida como um sistema de reagente básico se convertendo em um produto
ácido. Por isso um catalisador com características ácidas e sem basicidade seria o
ideal para este tipo de reação pois, o reagente básico é ativado pelo sítio ácido,
enquanto o produto ácido não. Catalisadores com diversas combinações de metais

37
são os mais usados para a oxidação da acroleína. Na maioria deles, o principal
componente é o molibdênio (THE DOW CHEMICAL COMPANY, 1977).
Catalisadores preparados a partir da mistura de óxidos de molibdênio,
vanádio, cromo, cobre, tântalo e nióbio, suportados ou não em sílica, se mostraram
eficientes na oxidação da acroleína, com conversões acima de 90%, e bastante
seletivos a ácido acrílico, com taxas perto de 90%. Os experimentos foram feitos
em temperatura de aproximadamente 300ºC, em fase gasosa. Interessante
ressaltar que, quando o óxido de vanádio é retirado da composição do catalisador,
a conversão de acroleína passa a ser bastante baixa, menos de 5%, e ocorre um
aumento significativo da seletividade a CO e CO2 (THE DOW CHEMICAL
COMPANY, 1977).
Diversos outros documentos de patente descrevem processos com
catalisadores a base de mistura de óxidos metálicos. Molibdênio, vanádio,
tungstênio e cobre são os metais predominantes nestes catalisadores, que ainda
podem conter cobalto, nióbio, zircônia, bismuto, alcalinos, alcalinos terrosos, entre
outros. Conversões acima de 90% de acroleína e ácido acrílico como produto
principal são mostrados em todos estes documentos (BASF, 1981; BASF, 1998;
EUTECO, 1981).
Andrushkevich e colaboradores relacionaram a razão vanádio e molibdênio
com a seletividade e atividade catalítica da oxidação da acroleína, em catalisadores
de óxidos mistos dos dois metais acima. Com o aumento da quantidade de vanádio
na composição, há uma diminuição da seletividade a ácido acrílico com
consequente aumento na seletividade de CO/CO2. Catalisadores com teores de

38
vanádio entre 7 e 14 %mol mostraram-se mais ativos e seletivos a ácido acrílico
(ANDRUSHKEVICH, 1992).
O tungstênio é um dos metais mais utilizados como promotor nos
catalisadores de vanádio e molibdênio para a reação de oxidação da acroleína. O
uso de metais promotores tem como finalidade o aumento da conversão, da
seletividade ao produto desejado ou da estabilidade do catalisador. A oxidação da
acroleína é uma reação a qual é acompanhada por uma rápida desativação do
catalisador mássico tradicionalmente utilizado, misto de vanádio e molibdênio. Isso
ocorre porque os óxidos destes dois metais, após algum tempo nas condições
reacionais, se rearranjam em suas formas mais termodinamicamente estáveis, que
não são ativas para a reação pretendida (ADAMS, 2004).
Experimentos mostram que o tungstênio não somente serve como promotor
estrutural do catalisador, mas influencia na atividade e seletividade. Uma pequena
quantidade de tungstênio em óxidos mistos de V/Mo aumenta a atividade, mas com
o aumento da quantidade do metal promotor a atividade diminui (VOGEL, 2007).
Catalisadores mistos de vanádio e molibdênio preparados por spray drying
contendo tungstênio em sua composição se mostraram mais seletivos a ácido
acrílico do que os que não continham o metal promotor. Os autores do trabalho
justificam essa melhora nos resultados reacionais pela mudança na cristalinidade
do sólido (GIEBELER, 2006). Outro estudo mostra a catálise heterogênea com
óxidos mistos de V-Mo-W para a reação de oxidação seletiva de acroleína a ácido
acrílico, onde os óxidos mistos de V-Mo são mais seletivos e ativos na oxidação de
acroleína a ácido acrílico que os óxidos puros dos mesmos metais mas somente
quando é acrescentado W resulta em um catalisador estável (KAMPE, 2007).

39
Para compostos orgânicos, o termo oxidação é aplicado quando ocorre a
diminuição da densidade eletrônica do carbono, normalmente, com a adição de um
átomo mais eletronegativo a uma ligação do carbono com outro átomo menos
eletronegativo como, por exemplo, a introdução de oxigênio em uma ligação
carbono-hidrogênio. Como o hidrogênio é menos eletronegativo que o carbono,
este cria uma carga parcial negativa, atraindo para si os elétrons da ligação. Com a
introdução do oxigênio, os papeis se invertem e o carbono "perde" parcialmente os
elétrons para o átomo mais eletronegativo, criando uma carga parcial positiva.
A acroleína possui a função aldeído, que é caracterizado pela presença de
uma carboxila ligada a um átomo de hidrogênio. Esse grupo é suscetível ao ataque
nucleofílico de íons oxigênio presentes no meio oxidante, colocando-se exatamente
entre a ligação carbono-hidrogênio e formando um composto do grupo dos ácidos
carboxílicos.
Em 1954, Mars e van Krevelen publicaram o primeiro trabalho onde
mencionavam o mecanismo de oxidação, que hoje recebe o nome dos autores. A
taxa de oxidação era diretamente proporcional à quantidade de íons oxigênios
situados em um plano paralelo ao do íon metálico (MARS, 1954).
Estudos de um grupo de pesquisadores russos mostram um possível
esquema mecanístico para a reação de oxidação da acroleína a ácido acrílico,
mostrado na Figura 14. A primeira etapa, formação da espécie (SI-I), ocorre em
íons de elevado estado de oxidação, como o Mo6+, capazes de fazer a interação σ
com a acroleína. Para a conversão do acrilato (SI-III) em ácido acrílico é necessário
que a superfície contenha elementos anfóteros ou com propriedades básicas fracas

40
como o V4+ (ANDRUSHKEVICH, 1992). O mecanismo envolve a participação do
oxigênio presente na superfície do catalisador (POPOVA, 1979).
Figura 14: Mecanismo da oxidação da acroleína a ácido acrílico (ANDRUSHKEVICH, 1992)
A adição de água pode ser um fator importante no andamento da reação. A
adsorção da água sobre a superfície do catalisador ocorre com posterior
dissociação como na equação 2.1. Nas condições catalíticas da oxidação da
acroleína, a água também pode ser responsável pela dissociação do complexo
entre a espécie acrilato e a superfície do catalisador, equação 2.2 (TICHÝ, 1997).
Vacância + O-2 + H2O(g) = 2OH- Equação 2.1
CH2CHCOO- + H2O(g) = CH2CHCOOH(g) + OH- Equação 2.2

41
Segundo Tichý e colaboradores a oxidação da acroleína em ácido acrílico
segue como uma reação acontecendo em quatro etapas:
1ª etapa: Adsorção da acroleína na superfície do catalisador através de um
dos átomos de O:
C3H4O + Cat(O) = Cat(OC3H4O) Equação 2.3
2ª etapa: Transferência do hidrogênio do grupo aldeído da acroleína para um
átomo de O diferente ao que a molécula está adsorvida e, consequente formação
da ligação entre o C-O com posterior formação da espécie acrilato:
Cat(OC3H4O) = Cat(C3H4O2) Equação 2.4
3ª etapa: Adsorção da água na vacância deixada pela saída do oxigênio.
Dessorção do ácido acrílico:
Cat(C3H4O2) + H2O = Cat(H2O) + C3H4O2 Equação 2.5
4ª etapa: Reconstituição da estrutura do catalisador, utilizando oxigênio do
meio, com dessorção de uma molécula de água:
Cat(H2O) + 0,5 O2 = Cat(O) + H2O Equação 2.6
2.4 Desidratação Oxidativa do Glicerol
O glicerol pode ser facilmente desidratado tendo como produto principal a
acroleína. Esta, por sua vez, pode gerar ácido acrílico por uma reação de oxidação.
Estas duas reações foram explanadas nos tópicos 2.2 e 2.3, respectivamente.

42
Alguns trabalhos mostram estas duas reações ocorrendo
subsequentemente, em dois leitos catalíticos fixos. Num primeiro leito, recheado
com catalisador com propriedades ácidas e usando carga de glicerol puro ou em
solução aquosa, ocorre a desidratação do glicerol a acroleína. No segundo leito,
recheado com catalisador com propriedades oxidantes e tendo como carga os
produtos produzidos no primeiro leito, ocorre a oxidação da acroleína a ácido
acrílico, sob a atmosfera oxidante.
No documento de patente FR2884818A1 é mostrada a produção de ácido
acrílico a partir do glicerol utilizando um leito duplo, com temperatura entre 250 e
350ºC e pressão de 1 a 5 bar. A carga utilizada foi uma solução aquosa de glicerol
entre 15 e 30%. Altas conversões de glicerol são alcançadas, mas a seletividade a
ácido acrílico não ultrapassa 8%. A acroleína é o produto principal encontrado,
mostrando que o catalisador utilizado para a sua oxidação não apresenta alta
eficiência (ARKEMA, 2006). Em um documento mais recente da mesma empresa é
descrita a unidade catalítica utilizada (ARQUEMA, 2012).
Em um dos exemplos mostrados no documento de patente EP1710227A1 é
obtido 65% de seletividade a ácido acrílico, a partir do glicerol, com catalisador de
ácido fosfórico impregnado em sílica e alumina, como função desidratante, e óxido
misto de V/Mo/W/Cu com função oxidante. A carga utilizada foi uma solução
aquosa de 92% em massa de glicerol e temperatura de 292ºC na primeira etapa e
270ºC na segunda (NIPPON SHOKUBAI, 2005).
Sooknoi e colaboradores também estudaram a produção de ácido acrílico a
partir do glicerol, em dois leitos catalíticos subsequentes. A zeólita HZSM-5 foi
utilizada como catalisador ácido para a desidratação e óxido misto de vanádio e

43
molibdênio (contendo 45% em mol de vanádio) como catalisador oxidante. Além
disso, esses mesmos catalisadores foram misturados e o sólido obtido foi utilizado
como leito único. As reações foram feitas a 300ºC com carga de solução aquosa de
glicerol a 10% em massa. Quando utilizados leitos separados obteve-se as
seguintes seletividades: 45% de acroleína, 40% de ácido acrílico, 10% de ácido
acético e 5% de acetaldeído. Com o leito misto obteve-se: 30% de acroleína, 30%
de ácido acrílico, 20% de ácido acético e 20% de acetaldeído (SOOKNOI, 2012).
Segundo os autores, quando os catalisadores são misturados em um único leito
pode ocorrer a oxidação direta do glicerol, se este entrar em contato primeiro com o
catalisador oxidante.
A partir de 2010 começaram a ser publicados os primeiros trabalhos que
mostram a reação direta de glicerol a ácido acrílico, reação de oxi-desidratação ou
desidratação oxidativa (Figura 15) com a utilização de catalisadores bifuncionais.
Figura 15: Reação de desidratação oxidativa do glicerol com produção de ácido acrílico

44
A maioria dos artigos que descrevem a reação de desidratação oxidativa do
glicerol utilizam catalisadores mássico, constituídos por óxidos mistos de diversos
metais. Os metais mais estudados são o vanádio, o molibdênio e o tungstênio.
Os catalisadores MoVTeNbO, Mo3VO e W3VO foram testados para a reação
direta de glicerol a ácido acrílico em fase gasosa. Os resultados mostram que os
produtos de oxidação (ácido acético, ácido acrílico, CO, CO2, ácido fórmico e ácido
propanóico) somam mais de 90% da seletividade, mostrando que estes
catalisadores são bastante ativos para a oxidação, mas pouco seletivos ao ácido
acrílico, com seletividade variando entre 23 e 28% (DELEPLANQUE, 2010).
A variação do estado de oxidação dos metais presentes no catalisador
influencia na distribuição dos produtos. Soriano e colaboradores mostram que o
catalisador onde está presente o tungstênio no estado de oxidação +6, e o vanádio
nos estados de oxidação +5 e +4, em proporção de 1:1,2, é o que apresenta maior
seletividade a ácido acrílico (26%), a 320ºC. O aumento da temperatura diminui a
seletividade a ácido acrílico, aumentando a formação de CO e CO2 (SORIANO,
2011).
A soma da seletividade a acroleína e ácido acrílico é menor no catalisador
de W/V, do que a seletividade a acroleína na reação com o catalisador de óxido de
tungstênio, sem a presença de vanádio, no qual somente ocorre a reação de
desidratação. Isto sugere que a presença do vanádio no catalisador transforma a
acroleína tanto em ácido acrílico quanto em CO e CO2. A melhor razão molar V/W é
entre 0,1 e 0,2. Maiores quantidades de vanádio aumenta a formação das espécies
COx, enquanto menores teores deste metal leva a uma produção menos eficiente
de ácido acrílico e maiores quantidade de compostos pesados (SORIANO, 2011).

45
Em um trabalho posterior do mesmo grupo (SORIANO, 2011) o nióbio é
utilizado como dopante do catalisador de W/V/O, na tentativa de aumentar a
seletividade a ácido acrílico. A adição de Nb+5 diminui a densidade superficial de
sítios ácidos comparativamente com os catalisadores em nióbio (WO3 e W/V/O),
mas apresenta uma maior fração de sítios mais fortes. Em temperatura de 290ºC,
os catalisadores com proporções atômicas de Nb/(V+W+Nb)=0,13 e
V/(V+W+Nb)=0,13 apresentaram 34% de seletividade a ácido acrílico
(CHIEREGATO, 2012).
Experimentos com o mesmo catalisador composto por V/W/Nb utilizado por
(CHIEREGATO, 2012) demonstrou que é necessário um excesso estequiométrico
de oxigênio na mistura reacional. Em quantidade estequiométrica de glicerol:O2
(1:2) somente 50% do glicerol é convertido; em razões acima desta ocorre a
conversão de 100% do glicerol. Isto indica que o parâmetro mais importante que
afeta o comportamento catalítico é a pressão parcial de oxigênio; teores de
oxigênio maiores aumentam a oxidação de acroleína formando ácido acrílico. Isso
limita a formação de condensação indesejada de acroleína em subprodutos. Ao
mesmo tempo, o aumento da pressão parcial de O2 aumenta a quantidade de CO2
formada (NIETO, 2014).
A conversão do glicerol e seletividade a ácido acrílico sobre catalisadores de
óxido misto de vanádio/molibdênio e vanádio/tungstênio foi estudada em função da
proporção de vanádio nestes materiais. Tanto no Mo/V quanto no W/V a tendência
foi a mesma: diminuindo a quantidade de vanádio obtém-se um aumento da
seletividade a ácido acrílico e ácido acético, com menor produção de CO e CO2,

46
em temperatura de 300ºC. Para o catalisador Mo1V0,25 são produzidos 20% de
ácido acrílico e para o W1V0,25, 26% de ácido acrílico (YIN, 2014).
Fosfato ácido de vanádio hemi-idratato (VOHPO4.0,5H2O) e di-idratado
(VOHPO4.2H2O), assim como o pirofosfato (VO)2P2O7 não se mostraram
catalisadores seletivos para a desidratação oxidativa do glicerol. O melhor
resultado foi com o VOHPO4.0,5H2O, que produziu 8% de ácido acrílico em
temperatura de 350ºC (WANG, 2009).
Nb2O5 tratada com H2O2 foi utilizado na reação em batelada de desidratação
oxidativa do glicerol a 200ºC por 3 horas a pH 6. A conversão do glicerol é
analisada por espectroscopia de massa com ionização por eletrospray. A
distribuição dos produtos da reação não é mencionada (OLIVEIRA, 2012).
Os catalisadores mais relatados na literatura para a reação de desidratação
oxidativa do glicerol são na forma mássica, como os apresentados acima. Mas
também existem alguns poucos estudos que mostram catalisadores na forma
suportada, os quais serão apresentados a seguir.
Catalisadores preparados a partir de polioxometalatos suportados em
alumina foram testados para a reação de desidratação oxidativa do glicerol, em
temperatura de 90ºC e na presença de H2O2. Os catalisadores sintetizados são de
ácido fosfomolibdênico (PMo), ácido fosfotunstênico (PW) e ácido silicotungstênico
(SiW), em proporção de 2, 4 e 8% em massa, e suportados em alumina. Os
melhores resultados foram obtidos com os catalisadores com 4% de
polioxometalatos para o PMo/Al2O3 obteve-se 6,3% de seletividade a ácido acrílico;
para o PW/ Al2O3, 16,2%; e para o SiW/Al2O3, 25,1%. A maior conversão de

47
glicerol (83%) foi encontrada no catalisador de menor acidez, SiW/Al2O3, pois este
apresenta melhor estabilidade sob altas temperaturas e em meio aquoso que os
catalisadores com fósforo (HUNSOM, 2013).
Estudo de Mota e colaboradores mostra que a impregnação úmida de
vanádio (na forma de metavanadato de amônio) em zeólita Beta resulta em um
catalisador capaz de produzir até 25% de ácido acrílico a partir do glicerol.
Catalisadores preparados pela mistura física de V2O5 com zeólita Beta, não
produzem ácido acrílico. Isso se deve ao fato de que na impregnação úmida os
átomos de vanádio se encontram, mais dispostos, dentro dos poros da zeólita,
enquanto na mistura física se apresentam em sua quase totalidade na superfície
externa do suporte, conforme observado por XPS (MOTA, 2013).

48
Figura 16: Possíveis produtos da reação de glicerina com catalisador ácido e oxidante (HUNSOM, 2013)
2.5 Nióbio
O elemento 41 foi descoberto na Inglaterra em 1801, por Charles Hatchett, e
na época foi denominado de Colúmbio. Posteriormente, o químico alemão Heinrich
Rose, pensando haver encontrado um novo elemento ao separá-lo do metal
tântalo, deu-lhe o nome de nióbio em homenagem a Níobe, filha do mitológico rei
Tântalo com Dione.
O nióbio é um elemento de transição pertencente ao grupo 5 ou VB da
classificação periódica dos elementos. Em condições normais, é sólido. Sua

49
principal utilização é em ligas de aço para a produção de tubos condutores de
fluidos. A mais antiga utilização do nióbio é de 1925, quando este metal foi usado
na substituição do tungstênio na produção de ferramentas de aço. No início da
década de 1930, o nióbio passou a ser utilizado na prevenção de corrosão em aços
inoxidáveis. Outras aplicações do nióbio são em ligas metálicas com titânio,
zircônia, entre outras, desenvolvidas para utilização nas indústrias espacial e
nuclear, além do uso para fins relacionados à supercondutividade. Ele também está
presente em tomógrafos de ressonância magnética para diagnóstico por imagem,
na forma de magnetos supercondutores feitos com a liga Nb/Ti.
O Brasil se encontra em uma posição favorável, pois detêm
aproximadamente 98% das reservas mundiais exploráveis, seguido do Canadá
com 1,5%. As reservas brasileiras se concentram nas cidades de Araxá (MG),
Catalão (GO) e São Gabriel da Cachoeira (AM).
O óxido de nióbio hidratado, também conhecido como ácido nióbico ou
pentóxido de nióbio, representa 13% do mercado mundial de nióbio e é matéria-
prima para fabricação de produtos especiais como: ferronióbio de alta pureza,
níquel-nióbio, óxidos especiais de nióbio (grau ótico e grau cristal) e nióbio metálico
e suas ligas.
O nióbio apresenta significativa importância para a balança comercial
brasileira do setor mínero-metalúrgico, representando um saldo acima de US$ 240
milhões (1998) ou o equivalente a 43% do faturamento externo de toda indústria
nacional de ferroligas. Porém, este mercado estabilizou-se. Somente na aplicação
em supercondutores (demanda menor que 2% do mercado mundial) o nióbio não
sofre concorrências técnicas de outros metais. Para todos os outros tipos de

50
produtos há a concorrência com o vanádio, titânio, molibdênio, tungstênio e tântalo
que, isoladamente ou combinados em certas proporções, podem conferir
características próximas e altamente substitutivas do nióbio em diversos produtos
(LA CRUZ, 2004).
A produção mundial de nióbio teve um significativo aumento desde 2004,
quando foram produzidas pouco mais de 30 mil toneladas. Em 2007, a produção foi
de mais de 60 mil toneladas. O mercado de nióbio teve um acelerado crescimento
durante a década de 2000, chegando a um pico em 2008. É esperado que a
demanda por nióbio retorne a uma tendência de crescimento de longo prazo (8-
10% ao ano), pois os segmentos de mercado onde o nióbio é bem estabelecido
muitas vezes não têm substituto (GLOBE, METALS AND MINING).
A Companhia Brasileira de Metalurgia e Mineração (CBMM), situada em
Araxá, estado de Minas Gerais, é a maior produtora de nióbio, atendendo a
demanda do mercado nacional, além de exportar para diversos países.
A composição do Nb2O5.nH2O é incerta e a quantidade de água não é
constante. Podem formar diferentes hidratos, estequiométricos ou não-
estequiométricos. A estrutura do hidrato H8Nb6O19 (Figura 17) é ilustrada pela
presença de oito prótons acima de oito fases triangulares do octaedro formado por
seis átomos de nióbio. Cada hidrogênio é envolvido por uma nuvem eletrônica
produzida por três oxigênios em ponte, três oxigênios terminais e um oxigênio
central (TANABE, 1987).
Os prótons do ácido nióbico não estão presente na forma de moléculas de
água e sim, na forma de grupos hidroxila ligados ao átomo de nióbio (SEN, 1981).

51
Figura 17 Estrutura do H8Nb6O19 (TANABE, 1987)
Além das utilizações já mencionadas para o nióbio, outra importante forma
de aproveitamento deste metal é em catalisadores. A presença de compostos de
nióbio em catalisadores pode estar relacionada à função de promover a fase ativa,
servir de suporte a outros metais ou ser o próprio sólido ácido (ZIOLEK, 1999).
Óxidos de nióbio melhoram a atividade catalítica e prolongam a vida útil do
catalisador quando pequenas quantidades são adicionadas a este catalisador
(TANABE, 2003).
Uma das maiores aplicações de catalisadores a base de nióbio é em
catalisadores para reações de oxidação. Eles podem ser de diversas maneiras,
como: nióbia suportada em óxidos, óxidos metálicos suportados em nióbia, óxidos
metálicos mistos contendo nióbio, entre outras configurações.
A elevada atividade de catalisadores de vanádio suportado em nióbia em
reações de oxidação está associada à fácil redução da ponte V-O-Nb e reflete o

52
potencial redox favorável da niobia como suporte em detrimento de outros tipos de
suporte, como sílica e alumina (ZIOLEK, 1999).
Quando calcinado em baixa temperatura (100 a 300ºC) o Nb2O5 apresenta
acidez comparável a cerca de 70% da acidez do H2SO4. Já o fosfato de nióbio, nas
mesmas condições de pré-tratamento, apresenta aproximadamente 90% da acidez
do H2SO4. O aumento da temperatura de calcinação diminui a acidez desses dois
materiais. Acima de 500ºC pouquíssima acidez é observada na superfície
(TANABE, 1987).
Diversos estudos mostram a utilização tanto do óxido de nióbio quanto do
fosfato de nióbio em reações de desidratação. Em um trabalho de Okasaki e Wada
é mostrado que o fosfato e o óxido de nióbio tratados com H3PO4 são os
catalisadores mais seletivos na síntese de MTBE (metil-terc-butil éter). Eles ainda
apresentaram maior conversão na desidratação do terc-butil álcool, dentre os
catalisadores testados (OKAZAKI, 1993).
A desidratação de sacarídeos como a frutose e a sacarose para a formação
de 5-hidroximetil-2-furaldeído (HMF) tem apresentado bons resultados quando o
fosfato de nióbio é utilizado como catalisador. A performance do fosfato de nióbio
nesta reação é superior à do fosfato de zircônia, resinas catiônicas, zeólitas ou
heteropoliácidos, por exemplo (CARLINI, 1999).
A síntese de ácido metacrílico a partir do ácido propiônico e formaldeído
(equação 2.7) apresenta maior atividade e estabilidade quando é utilizado 20% de
óxido de nióbio suportado em sílica (Nb2O5/SiO2), do que em outros catalisadores
como o Ta2O5/SiO2 ou óxido misto de V/Si/P (SPIVEY, 1997).

53
CH3CH2COOH + HCHO CH2=C(CH3)COOH + H2O Equação 2.7
Na seção 2.2 foram mostrados estudos onde o óxido de nióbio é utilizado na
reação de desidratação do glicerol a acroleína.

54
CAPÍTULO 3 – MATERIAIS E MÉTODOS
__________________________________________________________________
3.1 Materiais
• Metavanadato de amônio (NH4VO3) - Vetec - 99% de pureza
• Metamolibdato de amônio ((NH4)6Mo7O24.4H2O) – Vetec –81-83% de MoO3
• Nitrato de Cobalto (Co(NO3)2.6H2O) – Vetec – 98-102% de pureza
• Óxido de Nióbio hidratado (Nb2O5. xH2O) – CBMM – Lote AD4488
• Fosfato de Nióbio (NbOPO4) – CBMM – Lote AD4761
• Glicerol PA - Vetec – 99,5% de pureza
• Gases - AGA:
• Nitrogênio (99,9%)
• Ar Sintético (99,9%)
• Hélio (99,9%)
• Hidrogênio (99,9%)

55
3.2 Preparo dos Catalisadores
Antes de iniciar o processo de impregnação, o óxido de nióbio foi calcinado a
550ºC em mufla, em atmosfera ambiente, por 2 horas, para obter a estrutura
cristalina. Enquanto o fosfato de nióbio não sofreu qualquer pré-tratamento antes
da impregnação.
Os catalisadores foram preparados pela técnica de impregnação úmida.
Nesta técnica, uma massa calculada do sal a ser impregnado foi dissolvida em
água deionizada. A esta solução foi adicionada uma massa conhecida do suporte a
ser utilizado. Em seguida, o sistema ficou em agitação magnética por 24 horas a
temperatura ambiente. Após este período, a água foi evaporada em rotaevaporador
sob pressão reduzida até completa secagem. Em seguida, o catalisador preparado
foi calcinado em mufla, sob pressão atmosférica. Os catalisadores suportados em
Nb2O5 foram calcinados a 400ºC por 2 horas, e os suportados em NbOPO4, a
550ºC também por 2 horas. Quando o catalisador é bimetálico, adicionou-se ao
suporte soluções aquosas dos dois sais metálicos, simultaneamente.
Além dos catalisadores suportados foi preparado um catalisador mássico
misto de óxido de vanádio e molibdênio. Para este catalisador foram preparadas
duas soluções aquosas dos sais metálicos. A primeira solução foi preparada
adicionando 1,17g de metavanadato de amônio em 60 mL de água destilada. A
segunda, adicionando 4,41g de molibdato de amônio em 60 mL de água destilada.
As duas soluções foram mantidas em agitação magnética, à temperatura ambiente
por 10 minutos. Em seguida, as duas soluções foram misturadas e deixadas em
agitação magnética por mais 10 minutos. A água foi evaporada em rotaevaporador

56
a pressão reduzida e calcinado em mufla a 400ºC por 2 horas, resultando em um
sólido de cor preta. Neste catalisador a razão Mo/V teórica é 2,5.
Na Tabela 2: Massa de sal metálico utilizado em cada catalisador
preparadoTabela 2 são apresentadas as quantidades em massa de sal metálico
utilizadas nos catalisadores preparados. Todos os catalisadores suportados foram
preparados utilizando 5g de suporte.
Tabela 2: Massa de sal metálico utilizado em cada catalisador preparado
Catalisador Massa de NH4VO3 Massa de
(NH4)6Mo7O24.4H2O
Massa de
Co(NO3)2.6H2O
V/Co/Nb2O5 0,574g - 1,230g
Mo/Co/Nb2O5 - 0,552g 0,493g
Mo/V/Nb2O5 0,574g 0,459g -
20V/NbOPO4 2,296g - -
5V/NbOPO4 0,574g - -
Mo/NbOPO4 - 0,460g -
Co/V/NbOPO4 0,574g - 1,248g
V/Mo/NbOPO4 0,574g 0,460g -
Co/Mo/NbOPO4 - 0,552g 0,493g
V/Mo/O(1) 1,170g 4,410g -
(1)catalisador mássico, não foi utilizado suporte na preparação deste catalisador

57
Os suportes catalíticos são materiais que sustentam a fase ativa em um
catalisador suportado. As características mais desejadas em um suporte são a alta
área específica, porosidade e a resistência mecânica. O suporte pode afetar o
tempo de vida, a seletividade, sensibilidade ao envenenamento, entre outras
propriedades do catalisador. A utilização de suportes nos catalisadores conduz à
diversas vantagens, entre elas proporcionar maior dispersão da fase ativa,
dependendo do tipo de sistema, o que é particularmente importante no caso de
metais de alto custo, como ouro ou ródio, além de garantir uma distribuição mais
uniforme das espécies ativas. Os suportes também melhoram a estabilidade do
catalisador evitando a sinterização da fase ativa por efeito da alta temperatura. Eles
facilitam a transferência de calor em reações fortemente exotérmicas, como nas
oxidações, evitando a acumulação de calor e o aumento de temperatura. Os
suportes podem ainda melhorar a atividade ou seletividade do catalisador, assim
como a resistência ao envenenamento. Em alguns casos, a interação entre o
suporte e o material ativo pode resultar na formação de compostos superficiais ou
complexos que podem ter efeitos catalíticos mais eficientes que o próprio
componente ativo.
3.3 Caracterizações
3.3.1 Espectrometria de Fluorescência de Raios-X
A análise por fluorescência de raios X é um método baseado na medida das
intensidades (número de fótons detectados por unidade de tempo) dos raios X
característicos emitidos pelos elementos que consistem a amostra. Os raios X
excitam os elementos constituintes, os quais, por sua vez, emitem linhas espectrais

58
com energias características. Esse método é, geralmente, aplicado para a
determinação de elementos com número atômico maior ou igual a 11. Os limites de
detecção em materiais sólidos são, tipicamente, de algumas partes por milhão, mas
a obtenção destes resultados depende de fatores como o elemento em análise e a
composição da matriz de átomos.
A análise de fluorescência de raios X consiste em: excitação dos elementos
que constituem a amostra, dispersão dos raios X característicos emitidos pela
amostra e detecção desses raios X. O objetivo desta técnica é determinar a
composição elementar quantitativa da amostra.
O equipamento utilizado foi um espectrômetro PW2400 da Philips, do
Departamento de Geologia da UFRJ, que é munido de tubo de Rh de 3 KW de
potência, seis cristais analisadores e dois detectores (selado e fluxo).
A perda de fogo (LOI) foi determinada através da obtenção do peso da
amostra antes e depois da mesma ser levada a 950°C por 30 minutos. Os
elementos foram detectados a partir de 1,0 g de pó do material misturados com 7,0
g de tetraborato de lítio. As condições analíticas para a dosagem dos elementos
presentes foram: detectores selado e de fluxo, cristais analisadores PET, Ge, PX1,
PX3 e LIF200 e potência de 24 KV e 90 mA ou 50 KV e 50 mA, dependendo do
elemento químico a ser detectado. Com base nas análises de padrões, o erro
analítico relativo estimando é: Si, Al (<1%), Fe, Mg, Ca (1-2%), Ti, Na,K (3-5%), P e
outros elementos traços (≤ 6%). As curvas de calibração foram obtidas a partir de
análises de padrões de óxidos puros dos elementos.

59
3.3.2 Difração de Raios-X
A difração de raios-X é um método bastante aplicado, principalmente, para
determinar a estrutura cristalina. Ou seja, a maneira na qual os átomos, íons ou
moléculas estão arranjados espacialmente. Os raios-X são ondas eletromagnéticas
com comprimento de onda da ordem de grandeza de 1 Angstrom. Essa é a mesma
ordem de grandeza das ligações entre os átomos em materiais líquidos ou sólidos.
A aplicação da difração de raios-X para caracterizar catalisadores pode ir desde a
simples identificação de fases, por comparação a um banco de dados de padrões
de referência, até a simulação e o refinamento de nanoestruturas (SCHMAL, 2011).
As análises por difração de raios X foram utilizadas para a identificação das
fases presentes nos catalisadores, e executadas em um difratômetro modelo XRD
6000 da marca Shimadzu, utilizando-se radiação CuKα, com 40 kV de voltagem e
corrente de 30 mA. Os espectros foram registrados em ângulos de Bragg (2θ)
crescentes, partindo-se de 5 a 80º, e velocidade de varredura de 4º/min.
3.3.3 Análise Textural
A avaliação textural é feita através das informações obtidas a partir das
isotermas de adsorção-dessorção (fisissorção) do N2 sob a sua temperatura de
ebulição. Fisissorção é um processo em que as moléculas gasosas interagem com
superfícies sólidas apenas por interações físicas. O processo baseia-se
fundamentalmente em interações de Van der Waals. É um processo reversível,
exotérmico e com baixo calor de adsorção. Os gases entram em contato com um
sólido adsorvente e, em função da superfície, apresentam um desbalanceamento

60
de forças moleculares. As moléculas de gás adsorvidas atraem novas moléculas,
formando multicamadas de gás adsorvido na superfície adsorvente.
As análises foram realizadas em um equipamento ASAP 2020 (Surface Area
and Porosity Analyser) da Micromeritics. Cerca de 200 mg de amostra foi pré-
tratada a 300 °C por 10 horas sob pressão reduzida. Após o pré-tratamento, as
isotermas de adsorção de N2 foram obtidas a temperatura de N2 líquido (-196 °C),
com diferentes pressões parciais de N2. O modelo para o estudo de fisissorção
utilizado foi o BET, criado por Brunauer, Emmett e Teller (BRUNAUER, 1940).
3.3.4 Dessorção com Temperatura Programada de n-but ilamina
A técnica de dessorção com temperatura programada, utilizando a
n-butilamina como molécula sonda, é utilizada para medir tanto a quantidade de
sítios ácidos quanto a força ácida de um sólido.
Nesta técnica, a molécula sonda é adsorvida na superfície do catalisador e
depois é dessorvida em equipamento de análise termogravimétrica, pelo aumento
gradativo da temperatura.
A adsorção de n-butilamina consiste nas seguintes etapas: pré-tratamento
do catalisador a 550ºC por 30 minutos sob fluxo de hélio de 40mL/min;
resfriamento até 150ºC; 20 minutos de fluxo de n-butilamina introduzida na corrente
de hélio através de um saturador mantido à temperatura ambiente; 10 minutos de
fluxo de hélio para retirada da n-butilamina fisicamente adsorvida.
A dessorção a temperatura programada foi realizada em analisador
termogravimétrico TGA-50 da Shimadzu. A amostra foi pesada no prato

61
termogravimétrico do equipamento e submetida à análise, operando numa faixa de
temperatura de 20°C a 900°C com taxa de aquecimento de 10°/min, sob fluxo de
nitrogênio (N2). Para a melhor identificação das faixas de temperatura nas quais
ocorrem as perdas de massa, foi utilizada a derivada da curva de ATG (curva de
DTG).
Com esta análise é possível observar a perda de massa no catalisador com
o aumento da temperatura, permitindo assim, a quantificação dos sítios ácidos
totais presentes no material analisado através da dessorção da n-butilamina, de
acordo com determinadas faixas de temperatura. Esta técnica não consegue
distinguir sítios ácidos de Bronsted e Lewis, mas permite se ter uma ideia da força
ácida.
3.4 Condições de Análise
A conversão foi calculada subtraindo a quantidade de glicerol na saída do
reator de 100. Quando não há pico referente ao glicerol a conversão foi
considerada 100%. A seletividade foi calculada dividindo a área do pico de
interesse pela soma das áreas de todos os picos presentes no cromatograma e
multiplicando por 100.
Utilizou-se um reator de vidro em forma de tubo em U com haste lateral para
introdução de reagente líquido. Cerca de 100mg do catalisador já calcinado foram
pré-tratados sob fluxo de N2 a 350°C por 30 minutos. Em seguida, o reator foi
colocado na temperatura de reação e deu-se início a introdução da solução aquosa

62
de glicerol pela haste lateral, com o auxílio de uma bomba seringa, as condições
reacionais estão listadas na Erro! Fonte de referência não encontrada. .
Tabela 3 Condições reacionais utilizadas
Temperatura do Reator Pré-tratamento 350ºC
Reação 300ºC
Temperatura do Corpo
da Unidade
230ºC -
Vazão do Gás de Arraste 40mL/min -
Gás de Arraste Pré-tratamento
Reação
N2
Ar Sintético
Carga de Glicerina Solução aquosa
50% massa
0,50 mL/min
WHSV 313h-1
Razão O2/Glicerina 3:1
3.5 Testes Catalíticos
As reações foram realizadas em uma unidade de fluxo contínuo (Figura 18)
acoplada a um cromatógrafo a gás da marca Thermo, modelo Trace GC Ultra com
detector DIC, para análise em linha dos produtos da reação. Foi utilizada uma
coluna capilar DB-225, com fase estacionária de 50% Cianopropilmetil, 50%

63
Fenilmetilpolisiloxano, com 30 metros de comprimento, 0,25 mm de diâmetro
interno e 0,25 µm de filme.
As análises do efluente do reator foram realizadas por cromatografia em fase
gasosa, utilizando uma válvula pneumática de amostragem de seis vias. Durante
as reações foi mantido fluxo de ar sintético como gás de arraste na unidade. O
corpo da unidade foi mantido a 230ºC, para evitar a condensação dos produtos. A
unidade catalítica utilizada é mostrada na Figura 18.
Figura 18: Unidade de Fluxo Contínuo utilizada para a realização das reações.
Para a identificação dos produtos foram feitas corridas cromatográficas,
seguindo as condições descritas na Tabela 4, com padrões dos possíveis produtos
da desidratação da glicerina. Entre eles estão o acetaldeído, formaldeído,
hidroxiacetona, 1,2 propanodiol, ácido acético, ácido acrílico e glicerina.

64
Tabela 4: Condições cromatográficas utilizadas
Temperatura do Forno 65ºC 2 minutos
65 - 200ºC 10ºC/minuto
200ºC 2 minutos
Temperatura do Detector 200ºC -
Temperatura do Injetor 200ºC -
Gás de arraste Hélio 1,2 mL/min
Razão de Split 1 / 50 -
Foi efetuado para o catalisador V/Mo/Nb2O5 uma estimativa da quantidade
de CO e CO2 formados. Para isso foi coletado na ajusante da unidade catalítica o
gás contendo os produtos de reação com o catalisador citado. Esse gás foi
analisado em um cromatógrafo a gás equipado com detector de condutividade
térmica capaz de quantificar os compostos de interesse.

65
CAPÍTULO 4 – RESULTADOS E DISCUSSÃO
__________________________________________________________________
Neste capítulo, são mostrados os resultados dos testes catalíticos e de
caracterização dos catalisadores. Os resultados são apresentados na forma de
conversão do reagente (glicerol) e seletividade aos produtos (acroleína,
acetaldeído, hidroxiacetona, ácido acético e ácido acrílico).
4.1 Caracterização dos Catalisadores
4.1.1 Espectrometria de Fluorescência de Raios-X
Os teores dos elementos contidos nos catalisadores preparados e dos
suportes foram determinados pela análise química, feita por fluorescência de raios-
X. Os valores encontrados são mostrados na Tabela 5.
Os valores encontrados na análise química coincidem, de maneira
satisfatória, com os valores teóricos calculados para a impregnação.

66
Tabela 5: Teores dos elementos, em percentual de massa em base seca, presentes nos catalisadores, obtidos por espectroscopia de fluorescência de Raios-X
Catalisador
Elementos (%)
Nb2O5 P2O5 V2O5 MoO3 Co3O4
Nb2O5 100 - - - -
V/Co/Nb2O5 87,1 - 5,0 - 5,1
Mo/Co/Nb2O5 89,5 - - 5,7 5,3
Mo/V/Nb2O5 89,3 - 5,8 4,9 -
NbOPO4 54,8 45,1 - - -
20V/NbOPO4 58,8 15,5 25,7 - -
5V/NbOPO4 71,6 18,1 9,5 - -
Co/V/NbOPO4 66,9 18,2 8,4 - 6,4
V/Mo/NbOPO4 67,9 18,4 8,1 5,6 -
Co/Mo/NbOPO4 66,0 26,3 - 5,1 3,3
Para uma melhor comparação da quantidade de metal contida em cada
catalisador é apresentado na Tabela 6 os teores dos elementos, em mmol, contido
em um grama de catalisador.
Nos catalisadores impregnados com vanádio e molibdênio, a quantidade de
vanádio no suporte é bem maior que a quantidade de molibdênio. Essa diferença
não é tão significativa nos catalisadores impregnados com vanádio e cobalto.

67
Tabela 6: Teores dos elementos, em mmol, presente em 1 grama de catalisador, em base seca, onde “T” representa o valor teórico, e “E”, o valor experimental.
Catalisador
Elementos (mmol por grama de catalisador)
Nb P V Mo Co
T E T E T E T E T E
Nb2O5 - 7,5 - - - - - - - -
V/Co/Nb2O5 - 6,5 - - 1,0 0,5 - - 0,8 0,6
Mo/Co/Nb2O5 - 6,7 - - - - 0,5 0,4 0,8 0,7
Mo/V/Nb2O5 - 6,7 - - 1,0 0,6 0,5 0,3 - -
NbOPO4 - 4,1 - 6,3 - - - - - -
20V/NbOPO4 - 4,4 - 2,2 4,0 2,8 - - - -
5V/NbOPO4 - 5,4 - 2,5 1,0 1,0 - - - -
Co/V/NbOPO4 - 5,0 - 2,6 1,0 0,9 - - 0,8 0,8
V/Mo/NbOPO4 - 5,1 - 2,6 1,0 0,9 0,5 0,4 - -
Co/Mo/NbOPO4 - 5,0 - 3,7 - - 0,6 0,4 0,4 0,4
Nos catalisadores suportados em fosfato de nióbio, após a impregnação dos
metais, houve uma grande diminuição na quantidade de P contida no catalisador,
em relação ao fosfato de nióbio sem impregnação. No entanto, a quantidade de Nb
tem um discreto aumento após a impregnação dos metais.
4.1.2 Difração de Raios-X
Pela análise de difração de raios-X dos catalisadores pode-se verificar que
não há mudança estrutural do suporte antes e após a impregnação dos metais.

68
Na figura 19, são mostrados os difratogramas de raios-X do óxido de nióbio
antes e após a calcinação, a 550ºC. O óxido de nióbio apresenta estrutura amorfa
antes e estrutura cristalina após o processo de calcinação. Por isso a etapa de
calcinação antes de se iniciar a impregnação dos sais metálicos se torna
indispensável.
0 10 20 30 40 50 60 70 80
0
50000
100000
150000
200000
250000
300000
350000
Inte
nsid
ade
(u.a
.)
2θ
Nb2O
5 (não calc.)
Nb2O5 (calc.)
Figura 19 Difratograma de Raios-X do Nb2O5 antes e após a calcinação a 550ºC
Na Figura 20, são mostradas os difratogramas de raios-X do fosfato de
nióbio antes e após a calcinação, a 550ºC. O fosfato de nióbio apresenta estrutura
amorfa mesmo após o processo de calcinação. Portanto, esta etapa é
desnecessária antes de iniciar o preparo do catalisador impregnado com metais.

69
0 10 20 30 40 50 60 70 800
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000In
tens
idad
e (u
.a.)
2θ
NbOPO4 (não calc.)
NbOPO4 (calc.)
Figura 20 Difratograma de raios-X do fosfato de nióbio antes e após a calcinação, a 550ºC
Pela Figura 21, onde há a comparação entre os difratogramas do óxido de
nióbio e dos catalisadores preparados pela impregnação metálica neste suporte,
percebe-se que as fases cristalinas são idênticas. Ou seja, mesmo no catalisador
impregnado com o metal, não há picos referentes às fases cristalinas dos óxidos
metálicos impregnados. Este resultado era esperado, pois a quantidade de metal é
pequena (cerca de 5% em massa), possivelmente por estarem abaixo do limite de
detecção da técnica utilizada.
O difratograma do óxido de nióbio é corroborado pela literatura
(AVELLANEDA, 1998) e pelo banco de dados de cristalografia.

70
0 10 20 30 40 50 60 70 80
0
200000
400000
600000
800000
1000000
Inte
nsid
ade
(u.a
.)
2θ
VMoNb2O5 CoVNb2O5 CoMoNb2O5 Nb2O5
Figura 21: Difratograma do óxido de nióbio com e sem a impregnação de metal
A ausência de picos referentes à fase cristalina dos óxidos metálicos sugere
que estes óxidos estejam incorporados no suporte de nióbia formando soluções
sólidas. Em catalisadores de V2O5/Nb2O5 com diferentes teores de V2O5, a fase
cristalina do óxido de vanádio somente é detectada quando o óxido metálico se
encontra em concentração maior que 15%. Nos catalisadores de MoO3/Nb2O5,
somente é detectada fase cristalina referente ao óxido de molibdênio quando este
está presente acima de 5% (JEHNG, 1992).
É sabido na literatura que o fosfato de nióbio é um material amorfo. Ou seja,
não apresenta fase cristalina. Na Figura 22 são mostrados os difratogramas dos

71
catalisadores suportados em fosfato de nióbio, assim como do suporte sem a
impregnação. A característica amorfa do suporte é mantida após a impregnação do
metal.
0 10 20 30 40 50 60 70 800
50000
100000
150000
200000
250000
300000
350000
400000
450000
500000
Inte
nsid
ade
(u.a
.)
2θ
MoCoNbOPO4 VCoNbOPO4 MoVNbOPO4 VNbOPO4 NbOPO4
Figura 22 Difratograma de Raio-X do fosfato de nióbio com e sem impregnação de metais.
É sabido que o fosfato de nióbio só apresenta estrutura cristalina quando
pré-tratado em temperaturas acima de 800ºC, apesar disso, apresenta alta
atividade catalítica em temperatura de pré-tratamento superiores a 500ºC
(TANG, 2010).
Segundo estudo de Sun e colaboradores, nos catalisadores de vanádio
suportados em fosfato de nióbio, a fase referente ao V2O5 somente é detectada no
difratograma quando o teor de vanádio é de 25% em massa, como mostrado na

72
Figura 23. Com vanádio impregnado entre 5 e 15% em massa não são observados
os picos relativos ao V2O5 devido a alta dispersão do metal no suporte (SUN,
2007). Vale ressaltar que no trabalho de Sun os catalisadores impregnados foram
calcinados a 350ºC.
Figura 23 Difratograma de Raio-X de Fosfato de Nióbio com e sem impregnação de vanádio (SUN, 2007). (▲) fase cristalina de V2O5.
Quando calcinado a 1100ºC o fosfato de nióbio apresenta estrutura cristalina
e o padrão de difração de raios-X encontrado é referente à forma β do oxifosfato de
nióbio, NbOPO4 (MARTINS, 1989).
Na Figura 24 são mostrados os difratogramas do catalisador mássico de
óxido de vanádio e molibdênio, assim como os difratogramas dos óxidos
separadamente.

73
0 10 20 30 40 50 60 70 80
0
100000
200000
300000
400000
500000
600000
700000In
tens
idad
e (u
.a.)
2θ
VMoO V2O5 MoO3
Figura 24 Difratograma do catalisador mássico de V e Mo, e dos respectivos óxidos
4.1.3 Dessorção com Temperatura Programada de n-but ilamina
A Figura 25 mostra o gráfico com o perfil de perda de massa contra a
temperatura (curva em azul) e da primeira derivada em mg/min (curva em rosa). A
primeira perda de massa, antes de 100ºC, refere-se à perda de água. Portanto, não
é considerada no cálculo da quantidade de sítios ácidos. Os gráficos de perda de
massa em função da temperatura dos demais catalisadores são mostrados no
Apêndice A.
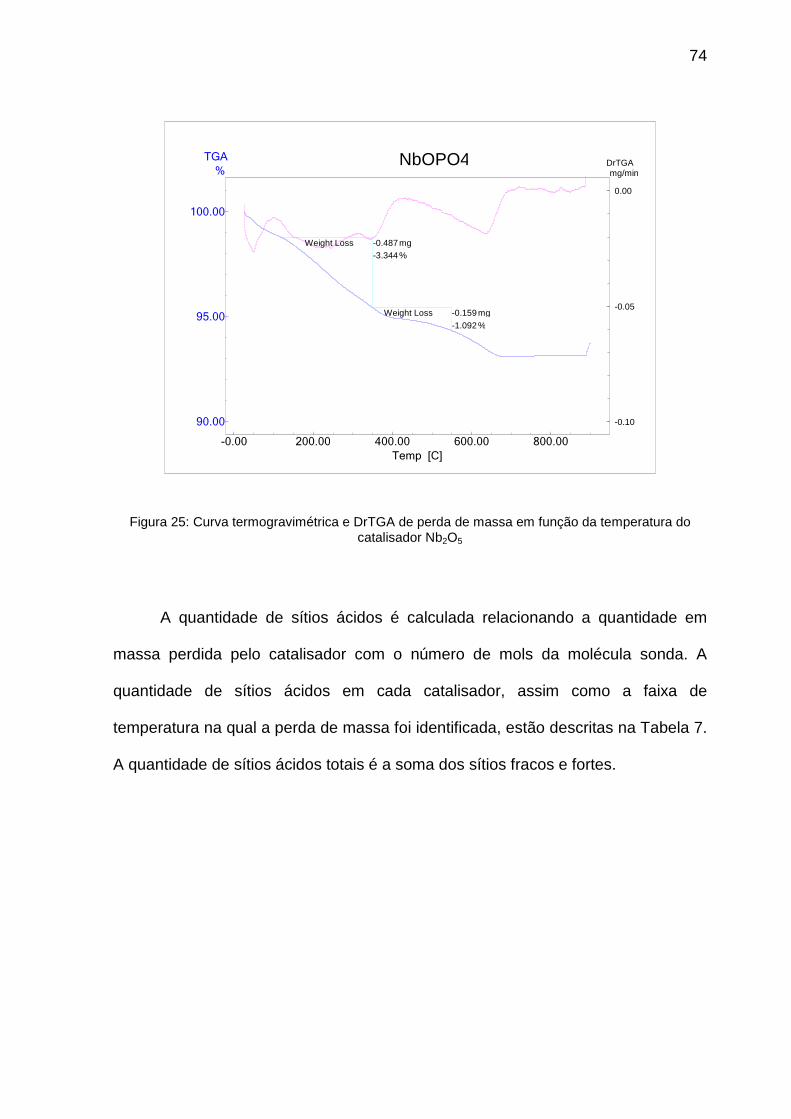
74
-0.00 200.00 400.00 600.00 800.00
Temp [C]
90.00
95.00
100.00
%
TGA
-0.10
-0.05
0.00
mg/minDrTGA
-0.487 mg-3.344 %
Weight Loss
-0.159 mg
-1.092 %
Weight Loss
NbOPO4
Figura 25: Curva termogravimétrica e DrTGA de perda de massa em função da temperatura do catalisador Nb2O5
A quantidade de sítios ácidos é calculada relacionando a quantidade em
massa perdida pelo catalisador com o número de mols da molécula sonda. A
quantidade de sítios ácidos em cada catalisador, assim como a faixa de
temperatura na qual a perda de massa foi identificada, estão descritas na Tabela 7.
A quantidade de sítios ácidos totais é a soma dos sítios fracos e fortes.

75
Tabela 7: Acidez dos catalisadores por dessorção com temperatura programada de n-butilamina
Catalisador
Sítios Fracos
(120–350ºC)
Sítios Fortes
(350- 550ºC)
Sítios
Totais
(µmol/g) Acidez
(µmol g -1)
Força
ácida
Acidez
(µmol g -1)
Força
ácida
Nb2O5 98 57% 74 43% 172
V/Co/Nb2O5 114 67% 56 33% 170
Mo/Co/Nb2O5 51 68% 24 32% 75
Mo/V/Nb2O5 60 87% 9 13% 69
NbOPO4 456 75% 149 25% 605
20V/NbOPO4 35 73% 13 27% 48
5V/NbOPO4 247 68% 114 32% 361
Co/V/NbOPO4 169 76% 53 24% 222
V/Mo/NbOPO4 91 65% 48 35% 139
Co/Mo/NbOPO4 280 76% 88 24% 368
O aumento da quantidade de metal no catalisador de vanádio suportado em
fosfato de nióbio acarretou uma acentuada diminuição na acidez total. Sun e
colaboradores mostram esta mesma tendência, onde a acidez decresce com o
aumento da quantidade de vanádio impregnada no fosfato de nióbio (NbP > 5VNbP
> 20VNbP > 25VNbP) (SUN, 2007).
O aumento da quantidade total de vanádio acarreta, também, em uma
diminuição na área específica. Pode-se, então, afirmar que maior quantidade de

76
espécies V2O5 se encontram na superfície externa do catalisador. Estas espécies
possuem características menos ácidas que o suporte.
Os catalisadores metálicos suportados em fosfato de nióbio apresentam uma
acidez muito inferior ao do suporte sem a impregnação. Isso pode ser devido ao
fato da perda de átomos de P, comprovada pela análise de FRX. O fosfato de
nióbio possui 6,3 mmol de P por grama de catalisador, enquanto após a
impregnação metálica esse valor fica em torno de 3 mmol (valores mostrados na
Tabela 6).
Em estudo de Sun e colaboradores, a acidez total do fosfato de nióbio é
calculada em 0,644 mmol.g-1 e de 5%V/NbOPO4 em 0,641 mmol.g-1, ambas
medidas pela adsorção de NH3 a 150ºC e 67 Pa de pressão, sendo analisada por
espectroscopia na região do infravermelho (SUN, 2007). Apesar de terem sido
utilizadas técnicas diferentes, o valor encontrado para o fosfato de nióbio é
condizente com o da literatura.
Em estudo de Florentino e colaboradores, é verificado que a acidez do óxido
de nióbio é praticamente nula, quando este é calcinado em temperaturas
superiores a 400ºC. A acidez tanto do fosfato de nióbio quanto do óxido de nióbio é
inversamente proporcional à temperatura de calcinação (FLORENTINO, 1992).
Análise da adsorção de piridina em óxido de nióbio mostra que a quantidade
de sítios ácidos de Bronsted diminui com o aumento da temperatura de pré-
tratamento, tendo seu máximo valor quando o material é pré-tratado a 100ºC. Os
sítios ácidos de Lewis apresentam maior quantidade em temperatura intermediária
(300ºC). Quando o óxido de nióbio é pré-tratado a 500ºC apresenta uma

77
quantidade muito pequena de sítios ácidos (TANABE, 1987). Os dados citados
anteriormente corroboram os resultados da análise de acidez feita nos
catalisadores suportados em Nb2O5, calcinados a 550ºC, que apresentam menos
de 0,2 mmol de sítios ácidos por grama de catalisador.
A acidez do óxido de nióbio foi investigada em função da temperatura de
calcinação. Quanto maior a temperatura de pré-tratamento utilizada menor é a
quantidade de sítios ácidos totais encontrados. Para o material calcinado a 500ºC
são encontrados cerca de 0,16 mmol de sítios ácidos por grama de catalisador
(XU, 2007). Este valor é bem semelhante ao mostrado em nossos resultados.
A diminuição da acidez decorrente do aumento da temperatura de pré-
tratamento pode ser atribuída à transformação dos sítios ácidos de Brønsted em
sítios ácidos de Lewis, pela eliminação de água e perda da área específica
(ZIOLEK, 1999). O fosfato de nióbio possui propriedades texturais, ácidas e
catalíticas similares à do óxido de nióbio, mas com a vantagem de conservar estas
propriedades mesmo em altas temperaturas de pré-tratamento.
4.1.4 Análise Textural
As propriedades texturais dos catalisadores (área específica e volume de
poros) foram determinadas por fisissorção de N2. Os valores encontrados para a
área BET dos sólidos são mostrados na Tabela 8. Como era de se esperar,
observa-se uma diminuição da área do sólido após a impregnação dos metais.

78
Tabela 8: Área Superficial e Volume de poro dos Catalisadores
Catalisador
Área BET
(m2/g)
Volume de
Poro (m 3/g)
Nb2O5(1) 40 0,117
V/Co/Nb2O5(1) 18 0,076
Mo/Co/Nb2O5(1) 24 0,087
Mo/V/Nb2O5(1) 25 0,083
NbOPO4(2) 162 0,182
20V/NbOPO4(2) 2 0,006
5V/NbOPO4(2) 82 0,239
Co/V/NbOPO4(2) 63 0,221
V/Mo/NbOPO4(2) 14 0,063
Co/Mo/NbOPO4(2) 29 0,183
(1) Calcinados a 400ºC
(2) Calcinados a 550ºC
Para o fosfato de nióbio impregnado somente com vanádio, quanto maior a
quantidade de metal menor é a área superficial. O fosfato de nióbio puro tem área
específica de 162 m2/g; com 5% de vanádio em massa a área é 82 m2/g enquanto
com 20% de vanádio impregnado a área diminui para apenas 2 m2/g. Isto pode
indicar que o metal está obstruindo os poros do suporte.
Tanto para os catalisadores suportados em óxido quanto para os suportados
em fosfato de nióbio ocorre uma diminuição da área superficial após a impregnação
dos metais. Esse comportamento é devido, provavelmente, à deposição de metais

79
no interior dos poros do suporte, que pode ser corroborado nos valores de volume
de poro mostrados anteriormente.
Nos catalisadores suportados em óxido de nióbio ocorre uma diminuição do
volume de poros em todos os catalisadores impregnados, mas a queda é mais
acentuada no catalisador V/Co/Nb2O5, que tem o volume de poro quatro vezes
menor do que o do material de partida.
Os catalisadores 20V/NbOPO4 e V/Mo/NbOPO4 apresentam volume de poro
bastante inferior ao do suporte sem impregnação. Enquanto os outros catalisadores
impregnados em fosfato de nióbio não apresentam queda, ou até mesmo um ligeiro
aumento do valor de volume de poro.
Em trabalho de Xu e colaboradores, é mostrado que a área superficial do
Nb2O5 calcinado a 500ºC é de 42 m2.g-1 (XU, 2007); valor pouco maior que o
encontrado nos nossos resultados, levando-se em consideração que os
catalisadores utilizados foram calcinados a 550ºC. No mesmo trabalho, é descrito
que quanto maior a temperatura de calcinação menor é a área superficial do óxido
de nióbio. Quando calcinado a 350ºC o óxido de nióbio apresenta 115m2/g; a
400ºC, 99m2/g; a 500ºC, 42m2/g; a 600ºC, 12m2/g e a 700ºC, 7m2/g (XU, 2007).

80
Florentino et al mostra em seu trabalho que a área específica do fosfato de
nióbio também diminui com o aumento da temperatura de calcinação. Para 426ºC
foi obtido um material com cerca de 125 m2/g (FLORENTINO, 1992).
Os catalisadores preparados no presente trabalho apresentam isoterma de
adsorção do tipo IV (Figura 26). Essa isoterma é a mais frequente em alguns tipos
de catalisadores heterogêneos, representando a adsorção em multicamadas e
mesoporosidade.
Figura 26 Isoterma de adsorção do catalisador V/Mo/Nb2O5
4.2 Testes Catalíticos.
4.2.1 Avaliação dos Suportes
Como mostrado nos itens 2.2 e 2.5 do capítulo de revisão bibliográfica, o
óxido e o fosfato de nióbio têm se apresentado como um bom catalisador para
reações de desidratação, incluindo a desidratação do glicerol. Para garantir que
estes materiais seriam efetivos como suporte para os catalisadores bifuncionais a

81
serem inicialmente preparados para a produção de ácido acrílico, eles foram
testados na reação de desidratação do glicerol.
Ambos os materiais foram calcinados a 550ºC em mufla, à pressão
atmosférica, por 2 horas. Antes das reações, 100 mg de catalisador foi pré-tratado
a 350ºC por 30 minutos sob fluxo de N2 a 40 ml/min. Em seguida, o reator foi
resfriado para 300ºC, passando a ter ar sintético (80% N2 e 20% O2) a 40 mL/min
como gás de arraste. Neste momento, é iniciada a introdução de glicerina, pela
bomba seringa, com vazão de 0,5 mL/h. Vinte minutos após o início da introdução
da glicerina é feita uma injeção, via válvula pneumática, do efluente da reação no
cromatógrafo a gás. Esse procedimento é repetido a cada 20 minutos, até o fim da
reação, de forma a se acompanhar a atividade do catalisador.
Nas Figuras 27 e 28, são mostradas a conversão do glicerol e seletividade
aos produtos quando utilizado NbOPO4 e Nb2O5, respectivamente. Em ambos os
catalisadores, a acroleína é observada como produto principal, com seletividade
média de 76% para o fosfato de nióbio e 72% para o óxido de nióbio. Este era o
resultado esperado, pois a acroleína é o produto precursor do ácido acrílico, que
será formado pela oxidação desta. Ambos os catalisadores apresentam 100% de
conversão para o glicerol, não apresentando desativação apreciável durante todo o
período o qual a reação foi monitorada.

82
0 50 100 150 200 250 300 350 400-10
0
10
20
30
40
50
60
70
80
90
100
110%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína Hidroxiacetona
NbOPO4
Figura 27: Conversão do glicerol e seletividade aos produtos para a reação de desidratação com NbOPO4
0 100 200 300 400 5000
10
20
30
40
50
60
70
80
90
100
110
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína
Nb2O
5
Figura 28: Conversão do glicerol e seletividade aos produtos para a reação de desidratação com Nb2O5

83
Somente quando o fosfato de nióbio é utilizado como catalisador é formada
hidroxiacetona entre os produtos da reação. Este é um produto da desidratação em
uma das hidroxilas terminais do glicerol.
Na Tabela 9 são mostrados os valores de seletividade média e desvio
padrão para os produtos da reação de desidratação realizadas com o fosfato e
óxidos de nióbio.
Em estudo anterior constatamos que sobre zeólitas ácidas (HBeta, HY e
HZSM-5) ocorre a formação de hidroxiacetona. Na zeólita Beta é observado o
aumento da seletividade à hidroxiacetona com o aumento da temperatura de
reação. Quando a reação é mantida a 250ºC, cerca de 5% de hidroxiacetona são
formados. Já em temperatura de 300ºC, há formação de aproximadamente 20% do
mesmo produto (PESTANA, 2010). Estas zeólitas na forma ácida apresentam
quantidade muito maior de sítios ácidos totais do que as espécies de nióbio
estudadas. Além da acidez a temperatura de reação também influencia na
seletividade a hidroxiacetona.
Catalisadores com forte acidez tendem a formar mais coque que
catalisadores com uma acidez fraca. Isso poderia insinuar que as moléculas de
acroleína formadas sobre os catalisadores com forte acidez são facilmente sujeitas
a reações secundárias conduzidas na superfície, levando a formação de depósitos
de carbono (CHAI, 2007). Por isso, a utilização de suportes com acidez mais baixa,
porém suficiente para realizar a desidratação do glicerol, parece ser mais
interessante, como é o caso do óxido e fosfato de nióbio.

84
Tabela 9 Seletividade média (Xm) e desvio padrão (σ) dos produtos formados nas reações de desidratação com os suportes sem impregnação metálica
Nb2O5 NbOPO4
Formaldeído
Xm (%) 5,0 2,4
σ 5,1 1,3
Acetaldeído
Xm (%) 22,0 7,4
σ 1,9 2,3
Acroleína
Xm (%) 72,7 77,9
σ 5,3 7,9
Xm (%) 0 12,3
Hidroxiacetona σ 0 6,0
Em trabalho anterior foi mostrado que a zeólita Beta ácida sofre
desativação na reação de desidratação do glicerol. A 250ºC a conversão inicial é
de 75% e, após 120 minutos, a conversão é em torno de 10%. Com o aumento da
temperatura para 300ºC, a conversão inicial é mantida e, após 180 minutos de
reação a conversão diminui menos que com a temperatura mais baixa, mas ainda
sim cai pra 50% (PESTANA, 2010). Esse dado pode indicar que a não desativação
do catalisador não esteja somente ligada ao fato de o gás de arraste conter
oxigênio, que auxilia na eventual queima do coque formado, mas também com a
natureza do catalisador.

85
4.2.2 Avaliação dos Catalisadores Bifuncionais
Os resultados obtidos nas reações com os catalisadores suportados em
fosfato e óxido de nióbio são apresentados relacionando a distribuição dos
produtos e a conversão da glicerina com o tempo de reação. As reações foram
realizadas em temperatura de 300ºC, velocidade espacial horária mássica (WHSV)
de 313h-1 e razão molar de O2/glicerina de 3:1.
4.2.2.1 Catalisadores Suportados em Óxido de Nióbio
Todos os catalisadores suportados em óxido de nióbio apresentaram
conversões de glicerol igual a 100%, mantendo esse valor durante todo o período
em que a reação foi monitorada. A não desativação dos catalisadores pode estar
associada com o excesso de oxigênio utilizado no meio reacional, que auxilia na
eventual queima do coque na superfície do catalisador.
0 50 100 150 200 250 300 350 400 4500
10
20
30
40
50
95
100
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína Ác. Acético Ác. Acrílico
V/Mo/Nb2O5
Figura 29 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação Oxidativa com Mo/V/Nb2O5

86
É mostrada na Figura 29 a convFigura 29 Conversão do Glicerol e
Seletividade dos Produtos da Reação de Desidratação Oxidativa com
Mo/V/Nb2O5ersão do glicerol e seletividade dos produtos com o catalisador
V/Mo/Nb2O5. Os valores de seletividade não variam de forma significativa em
função do tempo reacional. Ele também apresentou as maiores seletividade a ácido
acrílico, dentre os catalisadores suportados em óxido de nióbio, com média de
10%. Em contrapartida, o catalisador apresentou alta seletividade ao ácido acético,
mais de 35%. Mesmo com a grande formação de ácido acético a acroleína foi o
produto principal, com cerca de 45% de seletividade. Pouca quantidade de
acetaldeído e formaldeído são formados.
0 200 400 600 800 10000
5
10
15
20
60
80
100
120
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína AcAcético AcAcrílico
Mo/Co/Nb2O5
Figura 30 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação Oxidativa com Mo/Co/Nb2O5

87
É mostrada na Figura 30 a conversão do glicerol e seletividade dos produtos
com o catalisador Mo/Co/Nb2O5, sendo este o que apresentou maior variação entre
os valores de seletividade dos produtos em função do tempo. A acroleína é o
produto principal com mais de 75% de seletividade. Os produtos de oxidação, ácido
acético e ácido acrílico, são formados em pequenas quantidades, 7 e 3% de
seletividade, respectivamente. O acetaldeído é formado com quase 11% de
seletividade média.
0 200 400 600 800 1000 1200 1400 1600
0
20
40
60
80
100
120
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína AcAcético
V/Co/Nb2O
5
Figura 31 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação d Oxidativa com V/Co/Nb2O5
É mostrada na Figura 31 a conversão do glicerol e a seletividade dos
produtos com o catalisador V/Co/Nb2O5 que foi o único dentre os suportados em
óxido de nióbio que não foi capaz de produzir ácido acrílico. Mesmo assim, tem-se
uma pequena seletividade a ácido acético, cerca de 8% de média com picos de até
quase 35% de seletividade a este produto de oxidação.

88
Na Tabela 10 são comparados os resultados das caracterizações feitas nos
catalisadores suportados em óxido de nióbio. A razão metal/suporte foi calculada
dividindo a soma da quantidade de metal encontrada no catalisador pela
quantidade de nióbio, em mmol.
Pelos resultados de caracterização destes catalisadores podemos relacionar
a baixa seletividade aos produtos de oxidação com a elevada acidez do catalisador
Co/V/Nb2O5, além da ausência de molibdênio. Na caracterização textural, este
catalisador apresentou os menores valores de área superficial e volume de poro,
apesar de a diferença entre os três catalisadores suportados em óxido de nióbio
ser pequena, o que pode indicar que no processo de preparação deste catalisador
os poros tenham sido bloqueados.
Tabela 10 Caracterizações dos Catalisadores suportados em óxido de nióbio
Catalisador Razão
Metal/Suporte
Área
Superficial
(m2/g)
Volume de
Poro (m 3/g)
Acidez Total
(µmol/g)
V/Mo/Nb2O5 0,15 25 0,083 69
Co/Mo/Nb 2O5 0,16 24 0,087 75
Co/V/Nb2O5 0,17 18 0,076 170
Na Figura 32, são comparadas a distribuição das seletividades aos produtos
dos catalisadores suportados em óxido de nióbio. O catalisador V/Mo/Nb2O5 foi o
que apresentou menor quantidade de acroleína e maior soma das seletividades

89
dos produtos de oxidação, ácido acético e ácido acrílico. Nos outros dois
catalisadores, a distribuição entre os produtos de oxidação, produto de
desidratação e produtos de clivagem térmica se mantêm estável.
Na Tabela 11, são mostrados os valores de seletividade média e desvio
padrão dos produtos formados pelas reações de desidratação oxidativa com os
catalisadores suportados em óxido de nióbio. Quanto menor o valor do desvio
padrão, mais os valores de seletividade estão próximos do valor da média.
Figura 32 Seletividade média dos produtos dos catalisadores suportados em Nb2O5
0
10
20
30
40
50
60
70
80
90
100
V/Mo/Nb2O5 Co/Mo/Nb2O5 V/Co/Nb2O5
Ác. Acrílico
Ác. Acético
Acroleína
Acetaldeído
Formaldeído

90
Tabela 11 Seletividade média (Xm) e desvio padrão (σ) dos produtos de desidratação oxidativa do glicerol com catalisadores suportados em Nb2O5
V/Mo/Nb2O5 Co/Mo/Nb 2O5 V/Co/Nb2O5
Formaldeído
Xm (%) 4,7 2,9 5,1
σ 0,5 1,2 2,6
Acetaldeído
Xm (%) 4,3 10,9 14,0
σ 0,3 1,7 3,7
Acroleína
Xm (%) 45,1 75,2 73,1
σ 3,5 4,9 7,9
Ác. Acético
Xm (%) 35,7 7,8 7,8
σ 2,5 4,1 5,8
Ác. Acrílico
Xm (%) 10,1 3,1 0
σ 2,4 1,5 0
4.2.2.2 Catalisadores Suportados em Fosfato de Niób io
Os catalisadores suportados em fosfato de nióbio apresentam conversão de
glicerol igual a 100%, assim como os suportados em óxido de nióbio.

91
0 50 100 150 200 250 300 350 4000
5
10
15
60
70
80
90
100
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína Ac.Acético Ac.Acrílico
5V/NbOPO4
Figura 33 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação Oxidativa com 5%V/NbOPO4
É mostrada na Figura 33 a conversão do glicerol e a seletividade dos
produtos com o catalisador monometálico de 5% de V impregnado em fosfato de
nióbio. Este catalisador apresenta boa seletividade a ácido acrílico, cerca de 10%,
Com a vantagem de produzir pouco ácido acético. O produto principal formado é a
acroleína, com seletividade superior a 75%. Formaldeído e acetaldeído são
formados em pouca quantidade.

92
20 40 60 80 100 120
0
20
40
60
80
100
120
%
Tempo (minutos)
Conversão Formaldeído Acetaldeído Acroleína Ác. Acético
20V/NbOPO4
Figura 34 Conversão do glicerol e seletividade dos produtos da reação de desidratação oxidativa com 20V/NbOPO4
É mostrada na Figura 34 a conversão do glicerol e a seletividade dos
produtos com o catalisador monometálico de 20% de V impregnado em fosfato de
nióbio. O aumento da quantidade mássica de vanádio suportado em fosfato de
nióbio não melhora os resultados, pois este catalisador não é capaz de formar
ácido acrílico, além de formar cerca de 20% de ácido acético.

93
0 50 100 150 200 250 300
0
20
40
60
80
100
120
%
Tempo (minutos)
Conversão Acetaldeído Acroleína Ac.Acético Ac.Acrílico
V/Mo/NbOPO3
Figura 35 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação d Oxidativa com V/Mo/NbOPO4
É mostrada na Figura 35 a conversão do glicerol e seletividade dos produtos
com o catalisador V/Mo/NbOPO4. Este catalisador se mostrou capaz de produzir
ácido acrílico somente durante a primeira metade do tempo reacional monitorado.
Pelos resultados fica bem claro que a seletividade a acroleína diminui quando a
seletividade a ácido acético aumenta, enquanto a seletividade a acetaldeído
permanece inalterada. Isso pode ser um indicativo de que nem todo o ácido acético
produzido seja proveniente da oxidação do acetaldeído formado pela clivagem
térmica do 3-hidroxipropanal, que é uma rota que compete com a rota de formação
da acroleína.

94
0 50 100 150 200 250 300 350 400-5
0
5
10
15
60
70
80
90
100
110
%
Tempo (minutos)
Conversão Acetaldeído Acroleína Ac.Acético Ac.Acrílico
Co/Mo/NbOPO4
Figura 36 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação d Oxidativa com Co/Mo/NbOPO4
É mostrada na Figura 36 a conversão do glicerol e seletividade dos produtos
com o catalisador Co/Mo/NbOPO4, que apresenta valores de seletividade dos
produtos bastante estáveis em função do tempo de reação. É o catalisador que
produz maior quantidade de acetaldeído, dentre os suportados em fosfato de
nióbio. A seletividade a ácido acético e ácido acrílico tem praticamente o mesmo
valor, cerca de 7%.

95
0 50 100 150 200 250 300
0
20
40
60
80
100
120
%
Tempo (minutos)
Conversão Acetaldeído Acroleína Ac.Acético Ac.Acrílico
Co/V/NbOPO4
Figura 37 Conversão do Glicerol e Seletividade dos Produtos da Reação de Desidratação Oxidativa com Co/V/NbOPO4
É mostrada na Figura 37 a conversão do glicerol e seletividade dos produtos
com o catalisador Co/V/NbOPO4. Este catalisador foi capaz de produzir cerca de
11% de seletividade a ácido acrílico, sendo o que mais produziu este produto
dentre os catalisadores suportados em fosfato de nióbio, com a vantagem de
produzir pouca quantidade de ácido acético e acetaldeído.
Comparando os resultados mostrados nos gráficos de conversão e
seletividades das reações realizadas com catalisadores suportados em fosfato de
nióbio com as caracterizações destes catalisadores, pode-se relacionar a
seletividade a ácido acrílico com as caracterizações texturais. Nos catalisadores
que possuem menores valores de área superficial e volume de poros (20V/NbOPO4
e V/Mo/NbOPO4), não é observada a produção de ácido acrílico. Nos catalisadores
que apresentam as maiores áreas superficiais e volume de poros (5V/NbOPO4 e

96
Co/V/NbOPO4) são obtidas as maiores seletividades a ácido acrílico. Os resultados
das caracterizações dos catalisadores suportados em fosfato de nióbio estão
descritos na Tabela 12.
Tabela 12 Comparação das caracterizações dos catalisadores suportados em NbOPO4
Catalisador Razão
metal/suporte
Área
superficial
(m2/g)
Volume de
Poro
(m3/g)
Acidez
(µmol/g)
V/Mo/NbOPO4 0,17 14 0,063 139
Co/Mo/NbOPO4 0,09 29 0,183 368
Co/V/NbOPO4 0,23 63 0,221 222
5V/NbOPO4 0,13 82 0,239 361
20V/NbOPO4 0,43 2 0,006 48
Na Figura 38 são comparadas a distribuição das seletividades aos produtos
dos catalisadores suportados em fosfato de nióbio. Eles apresentam pouca
variação na seletividade a acroleína, produto principal da reação. Os produtos de
oxidação, ácido acético e ácido acrílico, somam menos de 20% da seletividade
total.
Na Tabela 13 são mostrados os valores de seletividade média (Xm) e desvio
padrão (σ) para os catalisadores suportados em fosfato de nióbio.

97
Figura 38 Seletividades média dos produtos com catalisador suportados em NbOPO4
Tabela 13 Seletividade média (Xm) e desvio padrão (σ) dos produtos da desidratação oxidativa do glicerol com catalisadores suportados em NbOPO4
V/Mo/ NbOPO4
Co/Mo/ NbOPO4
Co/V/ NbOPO4
5V/ NbOPO4
20V/ NbOPO4
Formaldeído
Xm (%) 0 0 0 3,1 2,5
σ 0 0 0 1,2 1,1
Acetaldeído
Xm (%) 8,5 12,6 5,8 5,2 7,0
σ 2,9 0,7 2,2 1,1 1,4
Acroleína
Xm (%) 67,9 70,5 71,7 75,1 74,3
σ 15,1 2,1 8,7 3,4 1,2
Ác. Acético
Xm (%) 21,7 7,7 8,4 7,1 16,1
σ 12,4 0,8 3,8 2,7 1,2
Ác. Acrílico
Xm (%) 1,8 7,2 10,5 9,5 0
σ 3,6 0,9 3,2 2,3 0
0
10
20
30
40
50
60
70
80
90
100
Ác. Acrílico
Ác. Acético
Acroleína
Acetaldeído
Formaldeído

98
4.2.2.3 Estimativa da quantidade de CO e CO 2 formados
As reações de oxidação de compostos orgânicos têm como subprodutos
uma mistura de CO, CO2 e H2O. Como os catalisadores utilizados têm a função
oxidante é importante que seja investigado a quantidade de CO2 e CO que,
possivelmente, estejam sendo formados durante a reação de desidratação
oxidativa do glicerol.
O analisador presente no equipamento de cromatografia utilizado é um
detector por ionização em chama (DIC). O princípio deste tipo de detector é que o
efluente da coluna ao ser misturado com hidrogênio é queimado ao ar produzindo
uma chama que tem energia o suficiente para ionizar as moléculas do soluto desde
que tenham potencial de ionização baixos. As espécies iônicas produzidas são
coletadas, amplificadas e enviadas para o sistema de registro. Possui grande
aplicabilidade, mas somente detecta espécies que possuem a ligações C-H.
Portanto, CO e CO2 não podem ser detectadas por este tipo de analisador, sendo
necessário o uso de um detector por condutividade térmica (DCT).
Para se obter uma estimativa da quantidade de CO e CO2 formados, o
efluente da reação foi coletado em uma bolsa de coleta de gás, utilizando o
catalisador V/Mo/Nb2O5. Durante 10 minutos a bolsa de coleta de gás ficou
acoplada à jusante da unidade catalítica. Com o auxílio de uma seringa de gás
0,5mL do gás contido na bolsa foi injetado em um equipamento de cromatografia
gasosa com detector por condutividade térmica. A partir da curva de calibração
feita para o CO2 (Figura 39) e da média dos valores das áreas do pico referente ao
CO2 do cromatograma, foi calculado uma concentração de CO2 de 3,75% em mol.

99
Figura 39 Curva de calibração do CO2
Com isso tem-se um rendimento de 18% a CO2. Indicando que 18% da
glicerina injetada como reagente da reação é convertida a CO2.
Infelizmente, não foi possível fazer a mesma análise para o CO, pois a
coluna utilizada no cromatógrafo não era específica para este gas e havia co-
eluição entre o CO e a mistura N2/O2 do gás de arraste da reação, como mostrado
na Figura 40, onde o primeiro pico é referente ao CO, juntamente com o N2/O2, e o
segundo pico ao CO2. O terceiro pico corresponde a água.
Figura 40 Cromatograma do CO e CO2 utilizando DCT
y = 26,153x + 0,9405R² = 0,9945
0,00
20,00
40,00
60,00
80,00
100,00
120,00
140,00
160,00
180,00
200,00
0 1 2 3 4 5 6 7

100
A reação de desidratação oxidativa do glicerol foi avaliada em três diferentes
óxidos mistos (DELAPLANQUE, 2010). O rendimento a CO e CO2 em cada um dos
catalisadores é mostrado na Tabela 14.
Tabela 14 Rendimento a CO e CO2 (Adaptado de DELAPLANQUE, 2010)
Catalisador Rendimento CO (%) Rendimento CO 2 (%)
Mo3VO 18,1 17,0
MoVTeNbO 29,7 15,1
W3VO 28,3 26,4
Nieto e colaboradores mostram em seu trabalho que a seletividade a
espécies COx aumentam com o aumento da quantidade de oxigênio disponível no
meio reacional, nas reações de desidratação oxidativa do glicerol. Para o
catalisador de óxido misto de W-V-Nb, a 265ºC, são formadas as espécies COx
com seletividade em torno de 50% para a proporção molar de O2/glicerina igual a
3:1(NIETO, 2014).
O aumento da quantidade de vanádio em óxido misto de W-V-Nb provoca
um aumento na seletividade a espécies COx como produto da reação de
desidratação oxidativa do glicerol. Em temperatura de 300ºC, a seletividade a COx
varia entre 38 e 76% (CHIREGATO, 2012).
4.2.2.4 Uso do Suporte
Além dos catalisadores de vanádio e molibdênio suportados nas espécies de
nióbio, foi preparado um catalisador mássico de óxido misto destes dois metais, a

101
fim de comparar o uso de suportes para a reação de desidratação oxidativa do
glicerol.
O catalisador mássico foi preparado com a razão Mo/V igual a 2,5, ou seja,
cerca de 71% de Mo e 29 % de V. Esta proporção foi escolhida devido a estudos
da literatura mostrarem que catalisadores com 7 a 30% de vanádio representam os
que possuem maior atividade e seletividade a ácido acrílico na reação de oxidação
da acroleína (ANSDRUSHKEVICH, 1979) (KUSNETSOVA, 1979)
(ANSDRUSHKEVICH, 1992).
Na Figura 41 são mostradas as seletividades médias aos produtos da
reação de desidratação oxidativa com os catalisadores preparados com vanádio e
molibdênio, com e sem suporte. O catalisador mássico apresentou a maior
seletividade a ácido acrílico, aproximadamente 23%. A quantidade de ácido acético
produzida no catalisador mássico e no suportado em Nb2O5 são equivalentes,
enquanto a seletividade a acroleína diminui quando utilizado o catalisador não-
suportado.
Possivelmente, a maior disponibilidade dos sítios metálicos é responsável
pelo aumento da seletividade ao ácido acrílico no catalisador mássico pois ele
possui uma maior densidade metálica que os catalisadores suportados. O
catalisador V/Mo/Nb2O5 possui 10,1 mmol de metais (soma da quantidade de
vanádio e molibdênio), V/Mo/NbOPO4, 13,7 mmol de metais e o V/Mo mássico
possui 35 mmol de metais por grama de catalisador. Além da quantidade total de
metais, existe a diferença entre a razão Mo/V. O catalisador mássico apresenta
razão Mo/V 5 vezes maior que nos catalisadores suportados.

102
Figura 41 Seletividades Médias dos catalisadores a base de V/Mo com e sem suporte
Os óxidos metálicos são depositados no suporte através da reação
com os grupos hidroxila da superfície das espécies de nióbio para formar uma nova
fase de óxido metálico na superfície (JEHNG, 1992).
4.2.2.5 Proposta de mecanismo de desidratação/oxida ção do glicerol
A conversão do glicerol a ácido acrílico ocorre em duas etapas: primeiro a
desidratação do glicerol a acroleína, e posteriormente oxidação da acroleína a
ácido acrílico. Mesmo o processo ocorrendo em um catalisador bifuncional, em um
único leito catalítico, as duas etapas ocorrem sucessivamente para que seja
formado o ácido acrílico.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
V/Mo/NbOPO4 V/Mo/Nb2O5 V/Mo
Ác.Acrílico
Ác. Acético
Acroleína
Acetaldeído
Formaldeído

103
Muitos são os possíveis produtos quando glicerol é reagido com um
catalisador com função ácido e oxidante. Na Figura 42 são mostrados os possíveis
produtos de desidratação e também os produtos de oxidação.
OH OHOH
O
CH3OH
OH OCH2 O + CH2
OH CH3
O
CH2O
CH2O
OH
O OH
CH3 O
OH
COx
- H2O
- H2O
- H2O
+ 1/2 O2
+ 1/2 O2
+ 1/2 O2
Glicerol
Hidroxiacetona
3 hidroxi-propanal
Acroleína
Ácido Acrílico
Ácido Acético
AcetaldeídoFormaldeído
Ácido Fórmico
Álcool Vinílico
Figura 42 Possíveis produtos formados na reação de desidratação oxidativa do glicerol (Adaptado de DELAPLANQUE, 2010)

104
Dependendo do grupo hidroxila que for protonado, até seis produtos
intermediários podem ser formados na primeira etapa de desidratação (Figura 43).
Destes 6, somente 2 são estáveis: o 3-hidroxipropanal e a hidroxiacetona.
Figura 43 Possíveis produtos da primeira desidratação do glicerol (Adaptado de COLL, 2011 )
A hidroxiacetona (também chamada de acetol) é formada pela desidratação
a partir de uma das hidroxilas laterais do glicerol. Não há a possibilidade de uma
segunda desidratação da hidroxiacetona devido à presença de carbonila adjacente
à hidroxila. A hidroxiacetona é formada com seletividade média igual a 12%,
quando utilizado o fosfato de nióbio como catalisador da reação de desidratação do
glicerol. Esse produto não é observado nas reações realizadas com o óxido de
nióbio. Possivelmente isso ocorre devido a diferença na quantidade de sítios ácidos
totais, 605 µmol.g-1 no fosfato e 172 µmol.g-1 no óxido de nióbio e também pela
presença de maior quantidade de sítios ácidos fortes no fosfato de nióbio.

105
O fosfato de nióbio apresenta maior acidez de Brønsted, enquanto o óxido
de nióbio, maior acidez de Lewis quando calcinados a 300ºC, conforme mostrado
na Tabela 15 (LATCHER, 2013). Mas o perfil muda com o aumento da temperatura
de pré-tratamento devido à transformação dos sítios ácidos protônicos em sítios
ácidos de Lewis pela eliminação de água e perda da área superficial (ZIOLEK,
1999). Como os suportes utilizados nos catalisadores deste trabalho foram
calcinados a 550ºC, possivelmente eles apresentam maior quantidade de sítios de
Lewis.
Tabela 15 Acidez de Brønsted e Lewis de Nb2O5 e NbOPO4 calcinados a 300ºC (Adaptado de LACHTER, 2013)
Catalisador Sítio Ácido de Br ønsted
µmol g -1
Sítio Ácido de Lewis
µmol g -1
Nb2O5.nH2O 41 86
NbOPO4 70 63
Segundo Possato e colaboradores, o mecanismo da desidratação do glicerol
a hidroxiacetona se dá nos sítios ácidos de Lewis (Figura 44). A reação ocorre
através da interação de uma hidroxila terminal com o sítio metálico e, com a
transferência de um próton para um átomo de oxigênio da superfície catalítica. Há
a formação do 2,3 di-hidroxipropeno que, por rearranjo ceto-enólico, permite a
produção de hidroxiacetona (POSSATO, 2013). Após a reação de desidratação
ocorre a regeneração do sítio ativo com a liberação de uma molécula de água
(Figura 45).
Segundo o mecanismo proposto por Martinuzzi e colaboradores, uma outra
rota reacional para a hidroxiacetona é formar acetaldeído pela perda de uma

106
espécie COx (MARTINUZZI, 2014). Essa poderia ser uma das possibilidades da
alta seletividade a ácido acético, produto da oxidação do acetaldeído, em alguns
dos catalisadores testados.
M M
O
OHOH
OH
M M
OOH
OH
OHH
CH2
OH
OHCH3
O
OH
Hidroxiacetona
Glicerol
Figura 44 Mecanismo de desidratação do glicerol a hidroxiacetona (Adaptado de POSSATO, 2013)
M M
O
M M
OH2
+OH OH
Figura 45 Regeneração do sítio de Lewis
A hidroxiacetona é o produto principal da desidratação do glicerol quando a
reação é realizada em baixa temperatura (260ºC), e utilizando um catalisador com
características básicas, como La2CuO4 (VELASQUEZ, 2014) e hidroxiapatita
trocada com cobre (OLIVEIRA, 2014).
Diferente da hidroxiacetona, o 3-hidroxipropanal pode sofrer uma segunda
desidratação formando acroleína; ou ainda ser clivado termicamente a formaldeído
e acetaldeído.

107
Segundo Possato e colaboradores, a desidratação na hidroxila central do
glicerol ocorre através dos sítios ácidos de Brønsted (Figura 46). O mecanismo se
inicia com a protonação da hidroxila central pelo sítio protônico, ocorrendo a
formação do intermediário 1,3-di-hidroxipropeno que, por rearranjo ceto-enólico,
forma o 3-hidroxiapropanal. A segunda desidratação ocorre através da protonação
do grupo hidroxila do 3-hidroxipropanal, produzindo a acroleína (POSSATO, 2013).
Outro possível caminho reacional do 3-hidroxipropanal é a degradação
térmica formando acetaldeído e formaldeído.
OH
OHOH
OH
O-
OHOH
OH2+
OHOH OHO
OH O-
OHO OH2+
OCH2
O
Hidroxipropanal
Acroleína
Glicerol
Figura 46 Mecanismo de desidratação do glicerol a acroleína (adaptado de POSSATO, 2013)
Após as reações subsequentes de desidratação do glicerol e do
3-hidroxipropanal, ocorre a regeneração do sítio ativo com liberação de água
(Figura 47).

108
O-OH
OH3+
OH2
+
Figura 47 Regeneração do sítio de Bronsted do catalisador
A produção do acetaldeído se dá, em sua maioria, pela decomposição
térmica do 3-hidroxipropanal. Mas este produto também pode ser produzido,
quando a reação é realizada em fase líquida, pela protonação de uma das
hidroxilas laterais do glicerol pelo sítio ácido de Brønsted, seguida de
craquemaneto da espécie formada (Figura 48) (SHEN, 2014).
OH OHOH
+ H+ OH OH2+
OH
- H2OCH3
OH
OH
+ - H+
craqueamentoOCH3
Figura 48 Produção de acetaldeído (Adaptado de SHEN, 2014)
A oxidação da acroleína a ácido acrílico segue o mecanismo de Mars van
Krevelen, no qual o oxigênio adicionado ao reagente é proveniente da estrutura do
catalisador, e a vacância deixada pela saída desse átomo de oxigênio é preenchida
pelo oxigênio molecular do meio reacional.
As etapas do mecanismo de oxidação compreendem em:
1. Adsorção da acroleína na superfície do catalisador através de um dos
átomos de O do suporte;

109
2. Transferência do hidrogênio do grupo aldeído da acroleína para um
átomo de O diferente ao que a molécula está adsorvida e, consequente, formação
da ligação C-O com posterior formação da espécie acrilato;
3. Adsorção da água na vacância deixada pela saída do oxigênio e
dessorção do ácido acrílico
4. Reconstituição da estrutura do catalisador, utilizando oxigênio do meio
reacional, com dessorção de uma molécula de água.
A confirmação que a formação de ácido acrílico a partir da acroleína
acontece através do complexo de acrilato na superfície do catalisador é feita por
estudo de infravermelho. As bandas de absorção em 1445 e 1570 cm-1 podem ser
relacionadas, respectivamente, com as vibrações simétrica e assimétrica do grupo
COO-, enquanto a banda em 1620 cm-1 indica a existência de dupla ligação na
estrutura (TICHY, 1976 apud TICHY, 1997).
A oxidação do acetaldeído a ácido acético possivelmente ocorre pelo mesmo
mecanismo da oxidação da acroleína. Portanto, essas duas reações competem
pelos mesmos sítios metálicos. Por ter menor tamanho, o acetaldeído pode ter
melhor permeabilidade pelos poros do suporte tendo mais acesso aos sítios ativos
para a oxidação. Isso explicaria a maior seletividade a ácido acético que a ácido
acrílico na maioria dos catalisadores testados.

110
CAPÍTULO 5 – CONCLUSÕES
__________________________________________________________________
No presente trabalho foi estudada a reação de desidratação oxidativa do
glicerol, visando a formação de ácido acrílico e acroleína, com a utilização de
catalisadores preparados a partir da impregnação úmida de sais metálicos de
vanádio, molibdênio e cobalto em espécies de nióbio, fosfato e óxidos de nióbio.
O objetivo proposto foi alcançado com êxito, pois seis dos oito catalisadores
preparados foram capazes de produzir ácido acrílico dentre os produtos da reação.
O catalisador Co/V/NbOPO4 produziu uma média de 10,5% de seletividade a ácido
acrílico, e o catalisador V/Mo/Nb2O5, 10,1% de seletividade ao mesmo produto. O
produto principal de todas as reações foi a acroleína.
Todas as reações com os catalisadores utilizados manteve 100% de
conversão do glicerol em todo o tempo na qual a reação foi monitorada, mostrando
que não houve desativação dos catalisadores.
Após a impregnação dos metais os catalisadores não apresentaram
modificações na estrutura em relação à do suporte. Os catalisadores suportados
em óxido de nióbio mantiveram a estrutura cristalina do suporte e não
apresentaram picos referentes aos sítios metálicos, pela baixa concentração
desses elementos. Os catalisadores suportados em fosfato de nióbio apresentam
estrutura amorfa, assim como o suporte.
Os catalisadores impregnados apresentam quantidade total de sítios ácidos
menores do que os respectivos suportes, mas a distribuição de força ácida é pouco

111
alterada. A análise textural mostra que há uma diminuição na área superficial dos
catalisadores após a impregnação metálica.
5.1 Perspectivas Futuras
A titulo de sugestão para continuação da presente pesquisa, recomenda-se:
1. Realizar testes catalíticos com soluções de diferentes
concentrações de glicerol, a fim de verificar se a quantidade de água interfere na
conversão e seletividade, e também a sua influencia na desativação do catalisador;
2. Preparar catalisadores impregnando, além dos metais aqui
mencionados, sais de tungstênio, uma vez que este metal é descrito com bastante
frequência na literatura em catalisadores de oxidação de acroleína a ácido acrílico;
3. Variar a quantidade e a razão entre os metais nos catalisadores
suportados, a fim de se obter um catalisador com quantidade de molibdênio
superior a do segundo metal;
4. Mensurar com mais preciosismo a quantidade de CO e CO2
possivelmente formados na reação, com a utilização de equipamentos específico
para esta finalidade.

112
CAPÍTULO 6 – REFERÊNCIAS
BIBLIOGRÁFICAS
__________________________________________________________________
ADAMS, A. H.; HAAB, F.; BURHMESTER, T.; KUNERT, J.; OTT, J.; VOGEL, H.; FUESS, H.; Structure and Reaction Studies on Vanadium Molybdenum Mixed Oxides, Journal of Molecular Catalysis A: Chemical, v. 216, p. 67-74, 2004
ANDERSSON, A.; MASSA, M.; FINOCCHIO, E.; BUSCA, G.; Gas-phase Dehydration of Glycerol to Acrolein Over Al2O3-, SiO2-, and TiO2-supported Nb- and W-oxides Catalysis, Journal of Catalysis , v. 307, p. 170-184, 2013
ANDRUSHKEVICH, T. V.; PLYASOVA, L. M.; KUSNETSOVA, G. G.; BONDAVERA, V. M.; GORSHKOVA, T. P.; OLENKOVA, I. P.; LEBEDEVA, N. I.; Catalytic Properties of the Vanadium-Molybdenium Oxide System for Acrolein Oxidation, Reaction, Kinetcs, Mechanism and Catalysis, v. 12, p. 463-467, 1979
ANDRUSHKEVICH, T. V.; BONDAREVA, V. M.; POPOVA, G. Ya.; PLYASOVA, L. M.; New Compounds of Vanadium-molybdenum Oxide System. In Situ Investigation of the Mechanism of Acrolein Oxidation to Acrylic Acid. The Role of the Structure and Bond Energy of the Intermediate Compounds. Studies in Surface Science and Catalysts, v. 72, p. 91-100, 1992
ANP (Agência Nacional do Petróleo, Gás Natural e Biocombustíveis), Estatístico Brasileiro do Petróleo, Gás Natural e Biocombustíveis, 2013
ARKEMA, Jean-Luc Dubois; Christopher Duquenne; Wolfgang Holderich, Procede de Deshydratation du Glycerol en Acroleine , FR 2 882 052 A1, 2005
ARKEMA, Jean-Luc Dubois; Christopher Duquenne; Wolfgang Holderich, Procede de Preparation D’Acide Acrylique a partir de Glycer ol , FR 2 884 818 A1, 2006
ARKEMA, Jean-Luc Dubois; Greègory Patience; Method for Preparing Acrylic Acid from Glycerol, US8212070B2, 2012

113
AVELLANEDA, C. O.; AEGERTER, M. A.; PAWLICKA, A.; Caracterização de Filmes Finos de Nb2O5 com Propriedades Eletrocrômicas, Química Nova, v. 21, p. 365-367, 1998
BASF Akteingesellschaft; Richard Krabetz, Wlater Herrmann, Norbert Scholz, Heinz Engelbach, Gerd-Juergen Engert, Carl-Heinz Willersinn, Gerd Duembgen, Fritz Thiessen; Catalysts for Oxidation of Acrolein and MethaAcrole in to acrylic Acid and Methaacrylic Acid, Respectively, U.S Pat nº 4,259,211; 31 de março de 1981
BASF Akteingesellschaft; Wilhelm Ruppel, Ulrike Wegerle, Andreas Tenten, Ulrich Hammon; Catalytic Gas-phase Oxidation of Acrolein to Acryli c Acid, U.S. Pat nº 5,739,391, 14 de abril de 1998
BEATRIZ, A.; ARAÚLO, Y.J.K.; LIMA, D.P.; Glicerol: Um breve Histórico e aplicação em Sínteses Estereoseletivas, Química Nova, v.34, p. 306-319, 2011
BEHR, A.; EILTING, J.; IRAWADI, K.; LESCHINSKI, J.; LINDR, F. Improved utilization of renewable resources: New important derivates of glycerol, Green Chemistry , v.10, p. 13-30, 2008
BORGES, M.E.; DÍAZ, L.; Recent Developments on Heterogeneous Catalysts for Biodiesel Production by Oil Esterification and Transesterification Reactions: A Review, Renewable and Sustainable Energy Reviews , v. 16, p. 2839-2849, 2012
Brunauer, S.; Deming, L. S.; Demming, W. S.; Teller, E.; Journal of American Chemical Society , v.62, p. 1723, 1940
CARLINI, C.; GIUTTARI, M.; GALLETTI, A. M. R.; SBRANA, G.; ARMAROLI, T.; BUSCA, G.; Selective Saccharides Dehydration to 5-hydroxymethyl-2-furaldehyde by Heterogeneous Niobium Catalysts, Applied Catalysis A: General, v. 183, p. 295-302, 1999
CHAI, S., WANG, H., LIANG, Y., XU, B. Sustainable production of acrolein: investigation of solid acid-base catalysis for gas-phase dehydration of glycerol. Green Chemistry , v. 9, p. 1130-1136, 2007

114
CHAI, S. H.; YAN, B.; TAO, L. Z.; LIANG, Y.; XU, B. Q.; Sustainable Production of Acrolein: Catalytic Gas-phase Dehydration of Glycerol over Dispersed Tungsten Oxides on Alumina, Zirconia and Silica, Catalysis Today, v. 234, p. 215-222, 2014
CHIEREGATO, A.; BASILE, F.; CONCEPCION, P.; GUIDETTI, S.; LIOSI, G.; SORIANO, M. D.; TREVISANUT, C.; CAVANI, F.; LÓPEZ NIETO, J. M.; Glycerol Oxidehydration into Acrolein and Acrylic Acid over W-V-Nb-O Bronzes with Hexagonal Structure, Catalysis Today, v. 197, p. 58-65, 2012
CORMA, A.; HUBER, G. W.; SAUVANAUD, L.; O’CONNOR, P.; Biomass to Chemicals: Catalytic Conversion of Glycerol/water Mixtures into Acrolein, Reaction Network, Journal of Catalysis, v. 257, p. 163-171, 2008
Da SILVA, C. R. B.; GONÇALVES, V. L. C.; LATCHER, E. R.; MOTA, C. J. A.; Etherification of Glycerol with Benzyl Alcohol Catalyzed by Solid Acids, Journal of Brazilian Chemical Society, v. 20, n. 2, p. 201, 2009
DASARI, M. A.; KIATSIMKUL, P.; SUTTERLIN, W. R.; SUPPES, G. J.; Low Pressure Hydrogenolysis og flycerol to propylene glycol, Applied Catalysis A, v, 281, p. 225, 2005
DEGUSSA AKTIENGESELLSCHAFT; Armim Neher; Thomas Hass, Diertrich Arntz; Hebert Klenk; Walter Girke; Process for the production of acrolein , 5,387,720; 1995
DELEPLANQUE, J.; DUBOIS, J. L.; DEVAUX, J. F.; UEDA, W.; Production of Acrolein and Acrylic Acid Through Dehydration and Oxydehydration of Glycerol over Mixed Oxide Catalysts, Catalysis Today, v. 157, p. 351-358, 2010
DIBENEDETTO, A.; ARESTA, M.; NOCITO, F.; PASTORE, C.; A Study on the Carboxilation of Glycerol to Glycerol Carbonate with Carbon Dioxide: The Role of the Catalyst, Solvent and Reactions Conditions, Journal of Molecular Catalysis A: Chemical , v. 257, p. 149, 2006
EUTECO IMPIANTI S.p.A.; Natale Bertoloni, Natale Ferlazzo; Supported Catalysts for the Oxidation of Acrolein to Acrylic Acid , U.S. Pat nº 4,289,654; 15 de setembro de 1981

115
FLORENTINO, A.; CARTRAUD, P.; MAGNOUX, P.; GUISNET, M.; Textural, Acid and Catalytic Properties of Niobium Phosphate and of Niobium Oxide, Applied Catalysis A: General, v. 89, p. 143-153, 1992
GIEBELER, L.; KAMPE, P.; WIRTH, A.; ADAMS, A. H.; KUNERT, J.; FUESS, H.; VOGEL H.; Structural Changes of Vanadium-molybdenum-tungsten Mixed Oxide Catalysts During the Selective Oxidation of Acrolein to Acrylic Acid, Journal of Molecular Catalysis A: Chemical, v. 259, p. 309-318, 2006
GINJUPALLI, R. S.; MUGAWAR, S.; RAJAN, P.; BALLA, P. K.; KOMANDUR, V. R. C.; Vapour Phase Dehydration of Glycerol to Acrolein over Tungstated Zirconia Catalysis, Applied Surface Science, v. 309, p. 153-159, 2014
GLOBE, METALS AND MINING, http://www.globemetalsandmining.com.au, acessado em 05/05/2014
GONÇALVES, V. L.C.; PINTO, B. P.; SILCA, J.C.; MOTA, C. J. A.; Acetylation of Glycerol Catalyzed by Different Solid Acids, Catalysis Today, v. 133, p. 673, 2008
HOELDERICH, W. F.; ALGEN, A.; Conversion of Glycerol to Acrolein in Presence of WO3/TiO2 Catalysts; Applied Catalysis A: General , v. 400, p. 34-38, 2011
HOLBROOK, L. L.; WISE, I.; Mechamism of Promoter Action in Catalytic Oxidation of Propilene to Acrolein, Journal of Catalysis, v. 20, p. 367-373, 1971
HUNSOM, M.; THANASILP, S.; SCHWANK, J. W.; MEEYOO, V.; PENGPANICH, S.; Preparation of Supported POM Catalyst for Liquid Phase Oxydehydration of Glycerol to Acrylic Acid, Journal of Molecular Catalysis A: Chemical, v. 380, p. 49-56, 2013
IEA, International Energy Agency, Key World Energy Statistics, 2013
JEHNG, J. M.; TUREK, A. M.; WACHS, I. E.; Surface Modified Niobium Oxides: Synthesis, Characterization and Catalysis, Applied Catalysis A: General, v. 83, p. 179-200, 1992
KAMPE, P., GIELER, L., SAMUELIS, D., KUNERT, J., DROCHNER, A., HAAB, F., ADAMS, A.H., OTT, J., ENDRES, S., SCHIMANKE, G., BUHRMESTER, T.,

116
MARTIN, M., FUESS, H., VOGEL, H., Heterogeneously catalysed partial oxidation of acrolein to acrylic acid – structure, function and dynamics of the V-Mo-W mixed oxides. Physical Chemistry Chemical Physics, v. 9, p. 3577-3589, 2007
KOZHEVNIKOV, I. V.; ALHANASH, A.; KOZHVNIKOVA, E. F.; Gas-phase dehydration of Glycerol to Acrolein Catalysed by Caesium Heteropoly Salt, Applied Catalysis A: General , v. 378, p. 11-18, 2010
KUSNETSOVA, T. G.; BORESKOV, G. K.; ANDRUSHKEVICH, T. V.; PLYASOVA, L. M.; MAKSIMOV, N. G.; OLENKOVA, I. P.; Formation of the Active Component of a Vanadium-molybdenium Oxide Catalyst in Acrolein Oxidation, Reaction, Kinetcs, Mechanism and Catalysis, v. 12, p. 531-536, 1979
LACHTER, E. R.; BASSAN, I. A. L.; NSCIMENTO, D. R.; GIL, R. A. S. S.; SILVA, M. I. P.; MOREIRA, C. R.; GONZALEZ, W. A.; FARO JR, A. C.; ONFROY, T.; Esterification of Fatty Acids with Alcohols Over Niobium Phosphate, Fuel Processing Technology, v. 106, p. 619-624, 2013
LEE, Y. Y.; LEE, K. A.; PARK, N. C.; KIM, Y. C.; The Effect of PO4 to Nb2O5 Catalysts on the Dehydration of Glycerol, Catalysis Today, v.232, p. 114-118, 2014
LI, C.; GU, Y.; CUI, N.; YU, Q.; CUI, Q.; Study on the Influence of Channel Structure Properties in the Dehydration of Glycerol to Acrolein over H-zeolites Catalysts, Applied Catalysis A: General , v. 419-430, p. 9-16, 2012
LIN, Y.; Catalytic Valorization of Glycerol to Hydrogen and Syngas, International Journal of Hydrogen Energy, v. 38, p. 2678, 2013
MARS, P.; VAN KREVELEN, D.W.; Oxidations Carried Out by Means of Vanadium Oxide Catalysts, Special Supplement to Chemical Engineering Science, v. 3, p. 41-59, 1954
MARTIN, A.; ATIA, H.; ARMBRUSTER, U.; Dehydration of Glycerol in Gas Phase Using Heteropolyacid Catalists as Active Compound, Journal of Catalysis , v. 258, p. 71-82, 2008

117
MARTINS, R. L.; SCHITINE, W. J.; CASTRO, F. R.; Texture, Surface Acidic and Catalytic Propertiesof Niobium Phosphate, Catalysis Today, v. 5, p. 483-491, 1989
MARTINUZZI, I.; AZIZI, Y.; DEVAUX, JF.; TRETJAK, S.; ZAHRAA, O.; LECLERC, JP.; Reaction Mechanism of Glycerol Dehydration in the Gas Phase over Solid Acid Catalysts Determined with On-line Gas Chomatography, Chemical Engineering Science, v. 116, p. 118-127, 2014
MASCARENHAS, A. J. S.; CARRIÇO, C. A.; CRUZ, F. T.; SANTOS, M. B.; PASTORE, H. O.; ANDRADE, H. M. C.; Efficiency of Zeolite MCM-22 with Different SiO2/Al2O3 Molar Ratios in Gas Phase Glycerol Dehydration to Acrolein, Microporous and Mesoporous Materials, v. 181, p. 74-82, 2013
MILLET, J. M. M.; LAURIOL-GARBAY, P.; LORIDANT, S.; BELLIERE-BACA, V.; REY, P.; New Efficient and Long-life Catalysts for Gas-phase Glycerol Dehydration to Acrolein, Journal of Catalysis , v. 280, p. 68-76, 2011
MOTA, C.J.A Gliceroquímica: A Petroquímica Renovável. Periódico Tchê Química , v. 3, n. 6, Porto Alegre, 2006.
MOTA, C. J. A.; Gonçalves, V. L. C.; Gambetta, R.; Fadigas, J. C.; Preparação de Catalisadores Heterogêneos Utilizados na Hidrogenação Seletiva de Glicerina a Propeno, BR PI 0806337-0, 2008.
MOTA, C.J.A.; SILVA, C.X.A.; GOLÇALVES, V.L.C Gliceroquímica: Novos Produtos e Processos a partir da Glicerina de Produção de Biodiesel. Química Nova , v. 32, p. 639-648, 2009.
MOTA, C. J. A.; GONÇALVES, V. L. C.; SILVA, P. H. R.; Glycerol Acetal as Anti-freezing Additives for Biodiesel, Bioresource Technology, v. 101, 2010A
MOTA, C. J. A.; Da SILVA, C. X. A.; ROSENBACH, N.; COSTA, J.; Da SILVA, F.; Glycerin Derivatives as Fuel Additives: The Adition of Glycrol/Acetone Ketal (Solketal) in Gasolines; Energy & Fuels, v. 24, p. 2733, 2010B
MOTA, C. J. A.; PESTANA, C. F. M.; Co-produtos da Produção de Biosiesel, Revista Virtual de Química, v. 3, n. 5, p. 416, 2011

118
MOTA, C. J. A.; PESTANA, C. F. M.; GUERRA, A. C. O.; FERREIRA, G. B.; TURCI, C. C.; Oxidative Dehydrtaion of Glycerol to Acrylic Acid over Vanadium-Impregnated Zeolite Beta, Journal of Brazilian Chemical Society, v. 24, p. 100-105, 2013
NIETO, J. L. M.; CHIEREGATO, A.; SORIANO, M. D.; BASILE, F.; LIOSE, G.; ZAMORA, S.; CONCEPCION, P.; CAVANI, F.; One-pot Glycerol Oxidehydration to Acrylic Acid on Multifunctional Catalysts: Focus on the Influence of the Reactions Parametersin Respect to the Catalytic Performance, Applied Catalysis B: Environmental , v. 150-151, p. 37-46, 2014
NIMLOS, M.R., BLANKSBY, S.J., QIAN, X., HIMMEL, M.E., JOHNSON, D.K., Mechanisms of glycerol dehydration. Journal of Physical Chemistry A , v. 110, p. 6145-6156, 2006
NIPPON SHOKUBAI, Masahide Shima, Tsukasa Takahashi, Method for Production Acrylic Acid, EP 1 710 227 A1, 2005
OLIVEIRA, L. C. A.; PORTILHO, M. F.; SILVA, A. C.; TAROCO, H. A.; SOUZA, P. P.; Modified Niobia as a Funcional Catalysts for Simultaneos Dehydration and Oxidation of Glycerol, Applied Catalysis B: Environmental , v. 117-118, p. 29-35, 2012
OLIVEIRA, A. C.; CARVALHO, D. C.; PINHEIRO, L. G.; CAMPOS, A.; MILLER, E. R. C.; SOUZA, F. F.; FILHO, J. M.; SARAIVA G. D.; FILHO, A. C. S.; FONSECA, M. G.; Characterization and Catalytic Performances of Cooper and Cobalt-exchange Hydroxyapatite in Glycerol Conversion for 1-Hydroxyacetone Production, Applied Catalysis A: General, v. 471, p. 39-49, 2014
OKAZAKI, S.; WADA, N.; Surface Properties and Catalytic Activities of Amorphous Niobium Phosphate and a Comparison with Those of H3PO4 Treated Niobium Oxide, Catalysis Today, v. 16, p. 349-359, 1993
PERRY, R. H., GREEN, D.W., MALONEY, J.O.H. . Perry’s chemical engineers’ handbook . 7th ed. McGraw-Hill; , 1997.
PESTANA, C. F. M.; Desidratação do Glicerol sobre Catalisadores Ácido Heterogêneos: Formação de Acroléina e Ácido Acrílic o, 2010, 110 f.

119
Dissertação (Mestrado em Ciências, Química) – Universidade Federal do Rio de Janeiro, 2010
POPOVA, G. Ya.; ANDRUSHKEVICH, T. V.; METALKOVA, G. A.; Study of the Steps of Acrolein Oxidation Over a vanadium-molybdenum Catalysts, Reactions Kinects and Catalysis Letters, v. 12, p. 469-473, 1979
QUERINI, C.A.; DALLA COSTA, B. O.; PERALTA, M. A.; Gas Phase Dehydration of Glycerol over Lanthanun-modified Beta-zeolite, Applied Catalysis A: General , v. 472, p. 53-63, 2014
SATO, S.; TSUKUDA, E.; TAKAHASHI, R.; SODESAWA, T.; Production of Acrolein from Glycerol over Silica-supported Heteropoly Acids, Catalysis Communications , v. 8, p. 1349-1353, 2007
SCHAML, MARTIN; Catálise Heterogênea, Editora Synergia, 2011
SCHÜTH, F.; JIA, C.; LIU, Y.; SCHMIDT, W.; LU, A.; Small-sized HZSM-5 Zeolite as Highly Active Catalysts for Gas Phase Dehydration of Glycerol to Acrolein, Journal of Catalysis , v. 269, p. 71-79, 2010
SEBATIER, P.; GAUDION, G.; Compt. Rend. , v. 166, p. 1033-1039, 1918
SEN, B. K.; SAHA, A. V.; CHATTERJEE, N.; On the Nature and Structure of Niobic Acid and Its Pyrolytics Products: 1H NMR, IR, Conductivity and Ion Exchange Studies, Materials Research Bulletin , v. 16, p. 923-932, 1981
SHEN, L.; YIN, H.; WANG, A. LU, X.; ZHANG, C.; CHEN, F.; WANG, Y.; CHEN, H.; Liquid Phase Catalytic Dehydration of Glycerol to Acrolein over Bronsted Acid Ionic Liquid Catalyst, Journal of Industrial and Engineering Chemistry, v. 20, p. 759-766, 2014
SILVA, A. C.; OLIVEIRA, L. C. A.; NOGUEIRA, F. G. E.; NOGUEIRA, A. E.; Processo de Transferência de Tecnologia da Universidade para a Indústria: Estudo de Caso Envolvendo a Conversão de Glicerol, Química Nova , v. 34, p. 1852-1855, 2011

120
SKRZYNSKA, E.; GRABOWSKA, A.; CAPRON, M.; DUMEIGNIL, F.; Crude Glycerol as a Raw Material for the Liquid Phase Oxidation Reaction, Applied Catalysis A: General, v. 482, p. 245-257, 2014
SOOKNOI, T.; WITSUTHAMMAKUL, A.; Direct Conversion of Glycerol to Acrylic Acid via Integrated Dehtdration-Oxidation Bed System, Applied Catalysis A: General, v. 413-414, p. 109-116, 2012
SORIANO, M. D.; CONCEPCION, C.; LÓPEZ NIETO, J. M.; CAVANI, F.; GUIDETTI, S.; TREVISANUT, C.; Tungsten-Vanadium Mixed Oxide for the Oxidehydration of Glycerol into Acrylic Acid, Green Chemistry, v. 13, p. 2954-2962, 2011
SPIVEY, J. J.; GOGATE, M. R.; ZOELLE, J. R.; COLBERG, R. D.; Novel Catalysts for the Envirommentally Friendly Synthesis of Methyl Methacrylate, Industrial & Engineering Chemistry Reserch, v. 32, p. 4600, 1997
SULLIVAN, J. A.; BURNHAM, S.; The Selective oxidation of Glycerol over Model Au/TiO2 Catalysts – The Influence of Glycerol Purity on Conversion and Product Selectivity, Catalysis Communications, v. 56, p. 72-75, 2014
SUN, Q.; AUROUX, A.; SHEN, J.; Surface Acidity of Niobium Phosphate and Steam Reforming of Dimethoxymethane over Cu/ZnO/Al2O3 – NbP Complex Catalysts, Journal of Catalysis, v. 244, p 1-9, 2006
SUN, Q.; FANG, D.; WANG, S.; SHEN, J.; AUROUX, A.; Structural, Acidic and Redox Properties of V2O5/Nb2O5 Catalysts, Applied Catalysis A: General, v. 327, p. 218-225, 2007
SUPRUN, W.; LUTECKI, M.; HABER, T.; PAPP, H.; Acidic Catalysts for the Dehydration of Glycerol: Activity and Dactivation, Journal of Molecular Catalysis A: Chemical , v. 309¸p. 71-78, 2009
TANABE, K.; Niobic Acid as an Unusual Acidic Solid Material, Material Chemistry and Physics, v. 17, p. 217-225, 1987
TANABE, K.; OKASAKI, S.; Various Reactions Catalyzed by Niobium Compounds and Materials, Applied Catalysis A: General , v. 133, p. 191-218, 1995

121
TANABE, K.; Catalytic Application of Niobium Compounds, Catalysis Today, v. 78, p. 65-77, 2003
TANG, Z. C.; YU, D. H.; SUN, P.; LI, H.; HUANG, H.; Phosphoric Acid Modified Nb2O5: A selective and Reusable Catalysts for Dehydration of Sorbitol to Isosorbide, Bull Korean Chemical Society, v. 31, p. 3679-3683, 2010
THE DOW CHEMICAL COMPANY; David L. Childress, William V. Hayes, Richard L. Poppe; Acrolein Oxidation Catalysis , U.S. Pat nº 4,065,262; 12 de julho de 1977
TICHÝ, J.; DAVYDOV, A. A.; Interaction of Acrolein with Vanadium-molybdenium Oxide Catalysts Surface, Collection of Czechoslovak Chemical Communication, v. 41, p. 834-838, 1976
TICHÝ, J.; Oxidation of Acrolein to Acrylic Acid over vanadium-molibdenum Oxide Catalysts, Applied Catalysis A: General , v. 157, p. 363-385, 1997
VELASQUEZ, M.; SANTAMARIA, A.; DUPEYRAT, C. B.; Selective Conversion of Glycerol to Hydroxyacetone in gas Phase over La2CuO4, Applied Catalysis B: Environmental, v. 160-161, p. 606-613, 2014
VOGUEL, H.; ENDRES, S.; KAMPE, P.; KUNERT, J.; DROCHNER, A.; The for Acrolein Oxidation, Applied Catalysis A: General, v. 325, p. 237-243, 2007
XU, B-Q; CHAI, S. H.; WANG, H. P.; LIANG, Y.; Sustainable Production of Acrolein: Gas-phase Dehydration of Glycerol over Nb2O5 Catalysts, Journal of Catalysis , v. 250, p. 342-349, 2007
ZIOLEK, M.; NOWAK, I.; Niobium Compounds: Preparation, Characterization and Application in Heterogeneous Catalysis, Chemical Reviews, v. 99, p. 3606-3624, 1999
ZHOU, C. H.; ZHAO, H.; WU, L. M.; LOU, J. Y.; LI, N.; YANG, H. M.; DONG, D. S.; YU, W. H.; Catalytic Dehydration of Glycerol to Acrolein over Sulfuric Acid-activated Montmorillonite Catalysis, Applied Clay Science , v. 74, p. 154, 2013

122
WANG, F.; DUBOIS, J. L.; UEDA, W.; Catalytic Dehydration of Glycerol over Vanadium Phosphate Oxides in Presence of Molecular Oxygen, Journal of Catalysis, v. 268, p. 260-267, 2009
WANG, T.; LIU, R.; LIU, C.; JIN, Y.; High Selective and Stable CsPW/Nb2O5 Catalysis for Dehydration of Glycerol to Acrolein, Chinese Journal of Catalysis , v. 34, p. 2174-2182, 2013
YIN, H.; SHEN,L.; WANG, A.; LU, X.; ZHANG, C.; Gas Phase Oxidehydration of Glycerol to Acrylic Acid over Mo/V and W/V Oxide Catalysts, Chemical Engineering Journal, v. 244, p. 168-177, 2014
YUE, J. Y.; GAN, M. M.; GU, L. P.; ZHUANG, Y. F.; In Situ Synthesized Nano-cooper over ZSM-5 for the Catalytic Dehydration of Glycerol under Mild Conditions, Journal of the Taiwan Institute of Chemicals Engine ers, v. 45, p. 1443-1443, 2014
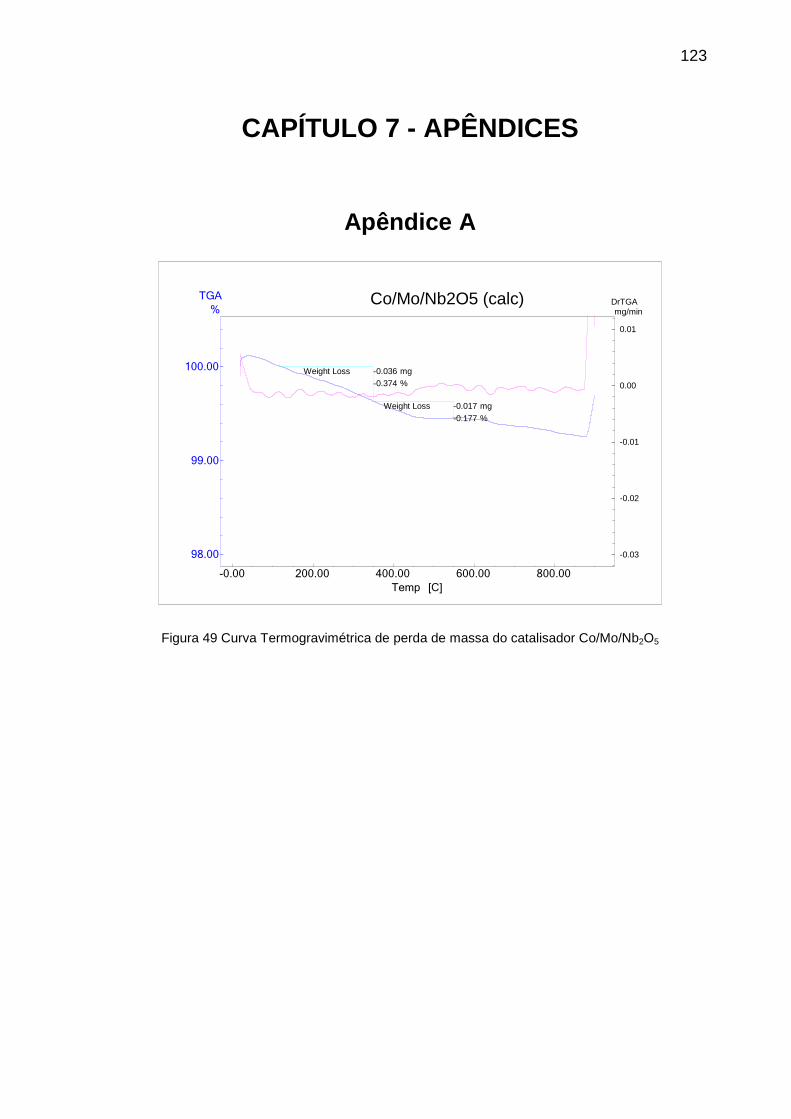
123
CAPÍTULO 7 - APÊNDICES
Apêndice A
-0.00 200.00 400.00 600.00 800.00
Temp [C]
98.00
99.00
100.00
%
TGA
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.036 mg-0.374 %
Weight Loss
-0.017 mg
-0.177 %
Weight Loss
Co/Mo/Nb2O5 (calc)
Figura 49 Curva Termogravimétrica de perda de massa do catalisador Co/Mo/Nb2O5

124
-0.00 200.00 400.00 600.00 800.00
Temp [C]
97.00
98.00
99.00
100.00
101.00
%
TGA
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.101 mg-0.836 %
Weight Loss
-0.050 mg-0.414 %
Weight Loss
Co/V/Nb2O5 (calc)
Figura 50 Curva Termogravimétrica de perda de massa do catalisador Co/V/Nb2O5
-0.00 200.00 400.00 600.00 800.00
Temp [C]
99.00
99.50
100.00
%
TGA
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.066 mg-0.444 %
Weight Loss
-0.010 mg
-0.067 %
Weight Loss
V/Mo/Nb2O5 (calc)
Figura 51 Curva Termogravimétrica de perda de massa do catalisador V/Mo/Nb2O5

125
-0.00 200.00 400.00 600.00 800.00
Temp [C]
96.00
98.00
100.00
%
TGA
-0.04
-0.02
0.00
mg/minDrTGA
-0.072 mg-0.774 %
Weight Loss
-0.067 mg
-0.720 %
Weight Loss
Co/Mo/NbOPO4
Figura 52 Curva Termogravimétrica de perda de massa do catalisador Co/Mo/NbOPO4
-0.00 200.00 400.00 600.00 800.00
Temp [C]
98.00
99.00
100.00
101.00
%
TGA
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.113 mg-0.718 %
Weight Loss
-0.086 mg-0.546 %
Weight Loss
Nb2O5
Figura 53 Curva Termogravimétrica de perda de massa do catalisador Nb2O5

126
-0.00 200.00 400.00 600.00 800.00
Temp [C]
97.00
98.00
99.00
100.00
101.00
%
TGA
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.066 mg-0.668 %
Weight Loss
-0.035 mg
-0.354 %
Weight Loss
V/Mo/NbOPO4
Figura 54 Curva Termogravimétrica de perda de massa do catalisador V/Mo/NbOPO4
-0.00 200.00 400.00 600.00 800.00
Temp [C]
96.00
97.00
98.00
99.00
100.00
101.00
%
TGA
-0.04
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.191 mg-1.240 %
Weight Loss
-0.060 mg
-0.390 %
Weight Loss
Co/V/NbOPO4
Figura 55 Curva Termogravimétrica de perda de massa do catalisador Co/V/NbOPO4

127
-0.00 200.00 400.00 600.00 800.00
Temp [C]
96.00
97.00
98.00
99.00
100.00
101.00
%
TGA
-0.04
-0.03
-0.02
-0.01
0.00
0.01
mg/minDrTGA
-0.191 mg
-1.240 %
Weight Loss
-0.060 mg
-0.390 %
Weight Loss
Co/Mo/NbOPO4
Figura 56 Curva Termogravimétrica de perda de massa do catalisador Co/Mo/NbOPO4
-0.00 200.00 400.00 600.00 800.00
Temp [C]
90.00
95.00
100.00
%
TGA
-0.10
-0.05
0.00
mg/minDrTGA
-0.487 mg-3.344 %
Weight Loss
-0.159 mg-1.092 %
Weight Loss
NbOPO4
Figura 57 Curva Termogravimétrica de perda de massa do catalisador NbOPO4

128
-0.00 200.00 400.00 600.00 800.00
Temp [C]
98.00
99.00
100.00
101.00
%
TGA
-0.02
-0.01
0.00
0.01
0.02
mg/minDrTGA
-0.027 mg-0.262 %
Weight Loss
-0.010 mg
-0.097 %
Weight Loss
20%V/NbOPO4
Figura 58 Curva Termogravimétrica de perda de massa do catalisador 20V/NbOPO4
-0.00 200.00 400.00 600.00 800.00
Temp [C]
96.00
98.00
100.00
%
TGA
-0.04
-0.02
0.00
0.02
mg/minDrTGA
-0.151 mg-1.809 %
Weight Loss
-0.070 mg-0.839 %
Weight Loss
5%V/NbOPO4
Figura 59 Curva Termogravimétrica de perda de massa do catalisador 5V/NbOPO4





























