Embed Size (px)
Citation preview

Diagnóstico das Síndromes Mielodisplásicas
Maria de Lourdes Chauffaille
Unifesp, Grupo Fleury

Diagnóstico das SMD Agenda:
• Definição, fisiopatologia e incidência
• Manifestações clínicas
• Exames diagnósticos: citomorfologia do SP e MO, citogenética e genética molecular
• Diagnóstico diferencial
• Classificação e prognóstico

Diagnóstico das SMD
Definição:
• Doenças clonais de célula tronco hematopoética
• Produção ineficaz de uma ou mais linhagens celulares
• Risco de transformação em LMA

Diagnóstico das SMD
Caracterizadas por:
• Aspectos morfológicos peculiares: displasia
• Anomalias funcionais
• Citopenias irreversíveis
• Medula óssea hipercelular

Diagnóstico das SMD
Heterogeneidade
Clínica, citomorfológica, citogenética e molecular

Diagnóstico das SMD Fisiopatologia:
• Interação entre eventos oncogênicos múltiplos causados por instabilidade genômica intrínseca e por estímulos genotóxicos extrínsecos
• Apoptose aumentada nas fases iniciais
• Lesão genética (cromossômica e molecular) e epigenética contribuem para a patogênese

Diagnóstico das SMD Fisiopatologia:
• Lesão genética: mutações, deleções, modulação epigenética de genes importantes
• Instabilidade genômica
• Transcrição RNAm anormal
• Vias de expressão de genes/RNA desreguladas
• Função ribossômica alterada

Cromotripsis: rearranjo
cromossômico maciço (quebra do cromossomo em
múltiplos pedaços)
Ocorre em 2-3% dos cânceres
Stephens PJ, Cell 2011

Diagnóstico das SMD
Incidência:
3 casos para cada 100.000 hab/ano na pop geral
7 a 35 casos /100.000 hab/ano entre > 60 anos
Mais frequente em homens e idosos
Em jovens, geralmente após exposição a Qt ou Rtx

Diagnóstico das SMD
• SMD primária
• SMD relacionada a terapia

Diagnóstico das SMD
• Idade média ao diagnóstico é 70 anos
• Aumento da incidência por:
– Crescimento da população idosa
– Maior exposição a toxinas ambientais
– Maior sobrevivencia a Qtx e transplantes prévios
– Diagnóstico mais preciso e precoce (Hamblin 2003)

Fatores predisponentes -Idade - Exposição a benzeno -Tabagismo - Radiação (exposição acidental) ou radioterapia - Gases de combustão: diesel e gasolina -Quimioterapia (alquilantes, inibidor de topoisomerase, análogo nucleosideo) - TCTH - G-CSF - Toxina ocupacional/ambiental - Anemia aplástica e HPN

Causas de SMD Herdadas
- Dças genéticas (S. Down, mosaicismo +8, monossomia 7 familial)
- Neurofibomatose
- Tumores germinativos
- Neutropenia congênita (Shwachman-Diamond, Kostmann)
- Reparo de DNA (Anemia de Fanconi)
- Mutações enzimas de desintoxicação (GSTM1 nulo, GSTT1 nulo)

Diagnóstico das SMD
• Período de latência: desconhecido
– 1 a 40 anos para radiação
– 1 a 10 anos para agente alquilante
– 30 anos para benzeno

Diagnóstico das SMD
• Evento inicial
– tempo para aparecimento de anl no SP
– tempo da anl no SP para sintomas


Diagnóstico das SMD
Como se faz o diagnóstico de SMD?

Diagnóstico das SMD
1. História clínica e exame físico
2. Hemograma e número de reticulócitos
3. Mielograma e morfologia do SP e da MO
4. Histologia da BMO
5. Citogenética: Cariótipo da MO – Genética molecular(FISH e testes moleculares)

Diagnóstico das SMD
6. Testes adjutórios: – Imunofenotipagem (HPN, LGL)
– Citoquímica (peroxidase e esterase)
– Ferritina, ferro sérico, vit B12, folato, etc.
– Eritropoetina
– HLA DR 15

Diagnóstico das SMD
1. Manifestações clínicas:
- Cansaço, fraqueza e sangramento mucoso
História Clínica:
desde indolente a rapidamente progressiva

Diagnóstico das SMD
Necessária anamnese detalhada:
Exposição a mutagênicos, toxinas, uso de medicamentos, álcool, doença infecciosa, dça crônica, insuficiência renal leve, deficiência vitamínica, dça imune, etc.

Diagnóstico das SMD
Citopenias por hematopoese ineficaz
Anemia macrocítica é mais comum (80%)
– Alta morbidade: dependência de transfusões

Diagnóstico das SMD
Leucopenia em 45%
– Mortalidade: por infecção na neutropenia e transformação
Trombocitopenia em 30%
- Sangramentos e dependência de transfusões

Diagnóstico das SMD
Casos sem manifestações clínicas:
Achado incidental de citopenia em hemograma de rotina

SMD
Idosos com anemia defic
vitaminica
an dça crônica, inflam,
IRC
an inexplic
ada
SMD

Diagnóstico das SMD
2.Hemograma e reticulócitos:
- Anemia: Hb < 10g/dL (OMS)
- Leucopenia: < 1800/µL (OMS)
< 1500/µL (IPSS)
< 800/µL (IPSS-R)
-Trombocitopenia: < 150.000/µL (OMS)
< 100.000/µL (IPSS e IPSS-R)

Diagnóstico das SMD
2.Hemograma e reticulócitos:
-Geralmente VCM aumentado, mas pode ser nl
(VCM baixo, raramente é SMD)
-Resposta reticulocitária inadequada

Diagnóstico das SMD
3. Mielograma: geralmente hipercelular
Citomorfologia:
Coloração: Romanowsky, Giemsa, Leishman, azul da Prússia ou Perls para ferro medular
• Estabelece o diagnóstico em 90-95% dos casos

Diagnóstico das SMD
3. Morfologia do SP e da MO
Diseritropoese e sideroblastos em anel

Sangue Periférico

Medula óssea

Medula óssea

Medula óssea

Diagnóstico das SMD
3. Morfologia do SP e da MO
Disgranulopoese

Sangue Periférico

Sangue Periférico

Sangue Periférico
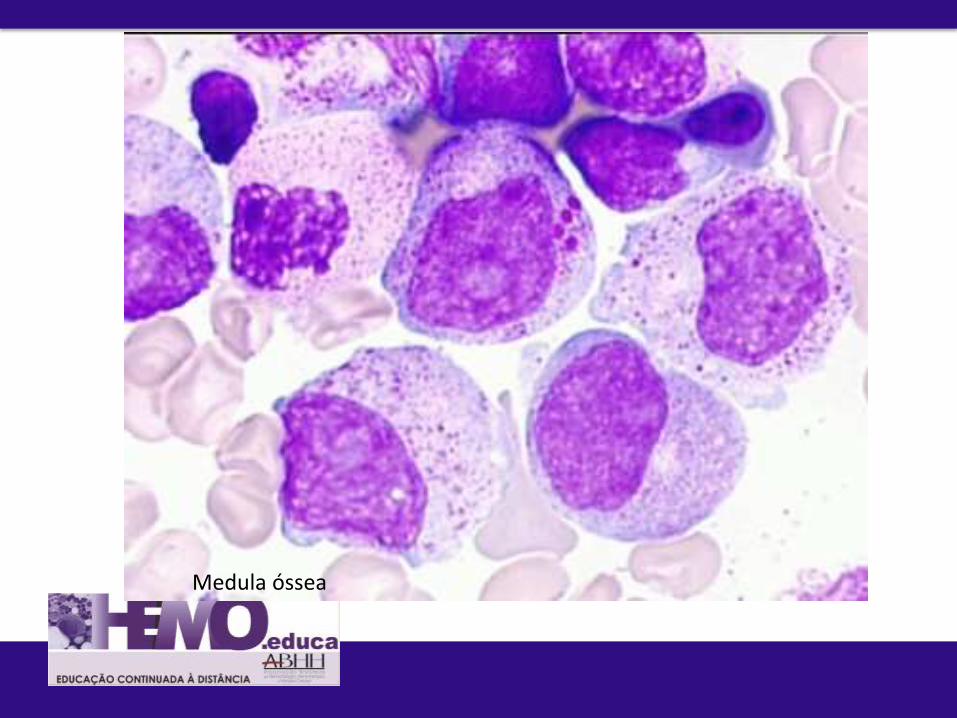
Medula óssea

Medula óssea

Diagnóstico das SMD
3. Morfologia do SP e da MO
Dismegacariopoese

Sangue Periférico de del(5q)

Medula óssea

Medula óssea

Medula óssea

Diagnóstico das SMD
Fundamental importância:
• Porcentagem de blastos
Contagem de 500 células na MO
Contagem 200 células no SP
Avaliação de pelo menos 20 megacariócitos
• Sideroblastos em anel
(contagem de 100 eritroblastos)

Diagnóstico das SMD
4.Histologia da BMO: • Avaliação da celularidade
• Distribuição geográfica de células precursoras
• Grau de fibrose (coloração pela prata de Gömöri)
• Aspecto do estroma
• Colorações imunoistoquímicas: CD34, cél T e B

HE 400x : AR com hiperplasia medular

HE 200x : AREB com hiperplasia medular


Diagnóstico das SMD
5. Citogenética:
• Importância:
– Diagnóstico, classificação,
– Prevê risco de evolução para LMA e sobrevida global
• Classificação:
IPSS (Greenberg et al,1997)
OMS (Jaffe et al, 2001 e Swerdlow 2008)

Citogenética das SMD
• Cariótipo da medula
• 20 metáfases analisadas
• Banda G

Citogenética das SMD
• 30 a 50% de alterações citogenéticas em SMD primária
• 80 a 90% de alterações citogenéticas em SMD secundária

Citogenética das SMD
• Deleções presentes em > 50% casos
• Intersticial mais comum que terminal
• Mais frequentes: 5q-, 7q-, 20q-, 11q-,13q-, 12p-, 17p-
• Deleção geralmente isolada nos casos de baixo risco, mas associada nos de alto risco.


11/2008
Aberrações cromossômicas não equilibradas:
Alteração SMD SMD-t
+8 10%
-7/7q- 10% 50%
-5/5q- 10% 40%
del20q 5-8%
-Y 5%
i(17q) 5%
-13/13q- 3%
11q- 3%
12p-/t12p 3%
9p- 1-2%
idic(X)(q13) 1-2%

11/2008
Equilibrados
Rearranjo SMD SMDt
t(11;16)(q23;p13.3) 3%
t(3;21)(q26.2;q22.1) 2%
t(1;3)(p36.3;q21.2) 1%
t(2;11)(p21;q26.2) 1%
inv(3)(q21q26.2) 1%
t(6;9)(p23;q34) 1%

11/2008
SMD com del(5q) isolada
• Incidência 10%
• Tipo específico de SMD com:
– <5% blastos
– Mais comum em mulheres idosas
– Anemia macrocítica
– GB diminuídos ou nls
– Displasia eritróide e plaquetas nls ou aumentadas
Citogenética em SMD

11/2008
SMD com del(5q) isolada
– Dismegacariocitopoese com elementos monolobulados
– Necessidade transfusional importante
– Necessidade de quelação de ferro
– Evolução favorável
– Transformação ocorre em 2% (ou 25% após 15 anos)
Citogenética em SMD

11/2008
SMD com del(5q) isolada

Diagnóstico das SMD
5. Citogenética
Cariótipo da MO: não informativo, pode ser feita a hibridação in situ por fluorescência (FISH) nas interfases, para pesquisar as anomalias citogenéticas mais frequentes em SMD:
-5/5q-, -7/7q-; +8, 11q, 20q-,-Y,

FISH
FISH é técnica adjutória e deve ser reservada para situações nas quais:
• Cariótipo sem resultado
• Cariótipo não analisável
• Cariótipo complexo
• Cariótipo com cr 5 envolvido em translocação



11/2008
5. Testes Moleculares:
– JAK2 = ARSA
– FLT3, família gene RAS (N, H, K), NPM1, AML1, KIT
– P53 (17p), EVI1c [t(3q26)] e 11q23(MLL)= SMDt
– TET2 (4q24)
– outros
Diagnóstico das SMD

Diagnóstico das SMD
6. Testes adjutórios: – Imunofenotipagem (HPN, LGL)
– [Citoquímica (peroxidase e esterase)]
– Ferritina, ferro sérico, vit B12, folato, etc.
– Eritropoetina
– HLA DR 15

Papel a IMF por citometria de fluxo:
1. Caracterização da diferenciação mieloide Normal /Reacional Neoplásico
2. Quantificação de células progenitoras
3. Classificação
4. Prognóstico
5. Avaliação de efetividade de tratamento

Normal Caso SMD
IMF
Figura cedida por Alex Sandes, Grupo Fleury
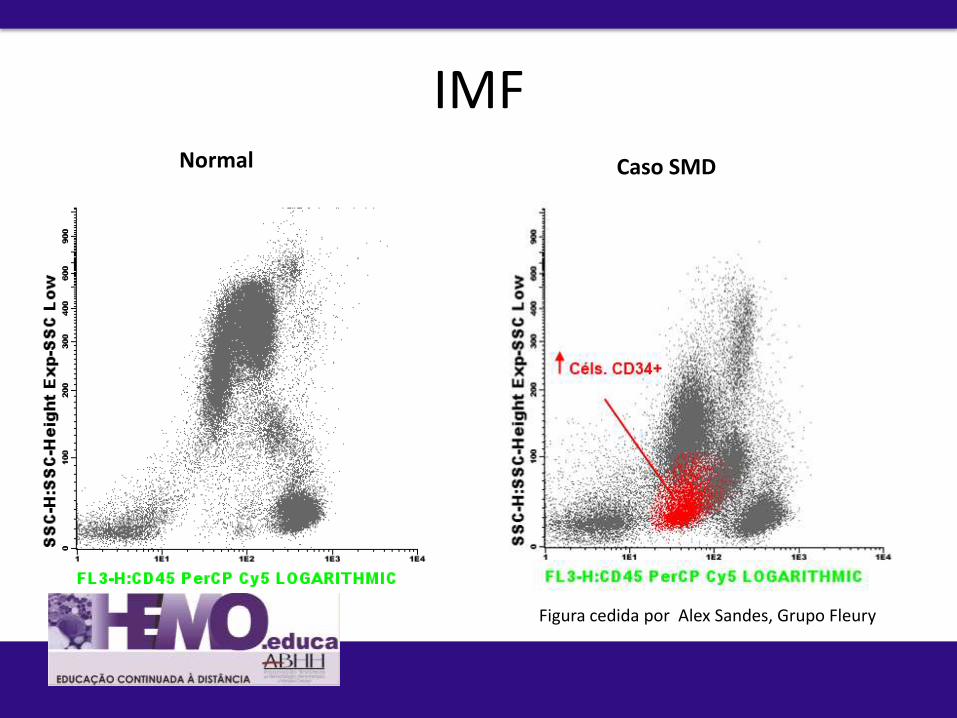
IMF Normal Caso SMD
Figura cedida por Alex Sandes, Grupo Fleury


Diagnóstico das SMD
Heterogeneidade diagnóstica:
• Raros casos com MO hipoplásica e nesses a discrepancia diagnóstica chega a 20%
• Diagnóstico é óbvio em paciente com excesso de blastos, mas difícil em citopenia isolada com displasia

Diagnóstico das SMD
Heterogeneidade diagnóstica:
• Se cariótipo anormal, fecha diagnóstico
• Se não, pode ser difícil conclui-lo
• Necessária a ampliação da investigação ou repetição dos testes em alguns meses

Diagnóstico diferencial
A. Exclusão de causas de anemia:
Perda sanguínea, processo inflamatório ou infeccioso
- avaliação do TGI e ginecológica
- doenças reumáticas

Diagnóstico diferencial
B. Presença de sideroblastos em anel:
Intoxicação por chumbo, etilismo, deficiência de piridoxina, deficiencia de cobre, hipotermia, uso de medicamentos como isoniazida, pirazinamida, cloranfenicol, d- penicilamina e progesterona

Diagnóstico diferencial C. Deficiência de cobre: causa habitualmente não
reconhecida de anemia e neutropenia reversível e confundida com
SMD, ainda que rara.
- Gastrectomisados, nutrição parental de longa duração, uso de suplementos de zinco
- Vacuolização de citopolasma mieloide e eritroide, sideroblasto em anel

MO de paciente com deficiência de cobre: maturação granulocítica prejudicada, vacuolização de precursores eritroides (seta vermelha) e inclusão de ferro em plasmócito (seta preta). Hafdanarson, 2007
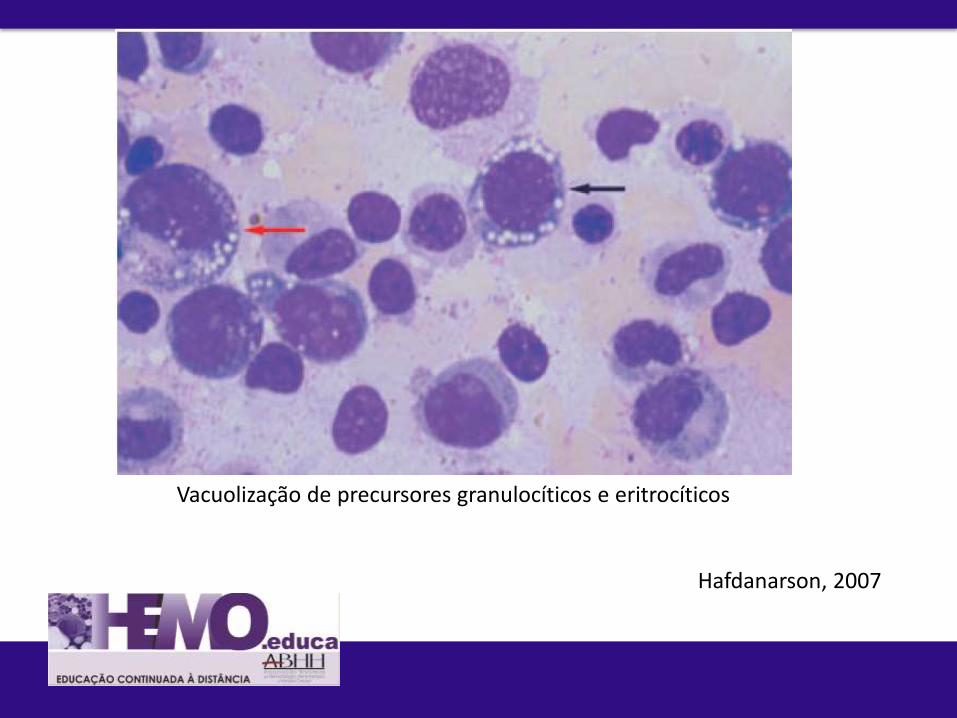
Hafdanarson, 2007
Vacuolização de precursores granulocíticos e eritrocíticos

Diagnóstico das SMD
Diagnóstico diferencial:
D. Exclusão de causas de trombocitopenia:
-PTI
-Infecções
-Medicamentos

Diagnóstico das SMD
Diagnóstico diferencial
E. Presença de monocitose:
Peristente (há mais de 3 meses) ou isolada,
infecção, doença autoimune, inflamatória e neoplásica podem causar monocitose

Diagnóstico das SMD
Diagnóstico diferencial:
F. Exclusão de outras doenças:
Hepatites, EBV, HIV, insuficiencia renal,
HPN
Parvovírus

Diagnótico diferencial
Hemoglobinúria paroxística noturna

Parvovirus

Diagnóstico diferencial
AA
HPN
LGL
DIS ñ
SMD
MPC
LMA
Eritroleucemia/
Leuc eritroide pura
SMD

Exames gerais
• Perfil bioquímico, eletrólitos, creatinina, DHL, enzimas hepáticas, albumina, imunoglobulinas, ferritina , fibrinogenio, proteina C reativa, sorologia para virus, dosagem de vitamina B12, ácido fólico, eritropoetina sérica,
• Triptase

Classsificação
< 1970: Pré-leucemia
1982: Classificação FAB
1999: Classificação OMS
2008: Classificação OMS revista

Classsificação
< 1970: Pré-leucemia
1982: Classificação FAB
1999: Classificação OMS
2008: Classificação OMS revista
1997: Classificação IPSS
2005: Classificação WPSS
2012 : IPSS - R

Diagnóstico das SMD
Classificação - OMS 2008 (Swerdlow et al)
A classificação substitui a FAB e a OMS 1999 e incorpora histologia da medula, porcentagem de blastos e achados citogenéticos

Classificação da OMS 2008
Citopenia refratária com displasia uni linhagem:
– Anemia refratária
– Neutropenia refratária
– Trombocitopenia refratária
MO: Uni ou bicitopenia, displasia >10% das células, <5% de blastos, < 15% de sideroblastos em anel

Classificação OMS 2008
Anemia refratária com sideroblastos em anel:
– Anemia
–15% de sideroblastos em anel
–Apenas displasia eritroide
–<5% de blastos

Classificação OMS 2008
Citopenia refratária com displasia multilinhagem:
SP: citopenias, <1% blastos, sem bastão de Auer, <1000/μL monócitos
MO: Displasia >10% em duas linhagens, <5% de blastos, sem Auer pode ou não ter sideroblastos em anel

Classificação OMS 2008
Anemia refratária com excesso de blastos I:
SP: citopenias, < 5% blastos, sem bastão de Auer, <1000/μL monócitos
MO: Displasia >10% em uma ou mais linhagens, sem bastão de Auer, 5-9% de blastos

Classificação OMS 2008
Anemia refratária com excesso de blastos II:
SP: citopenias, 5-19% blastos, com ou sem Auer, <1000/μL monócitos
MO: Displasia >10% em uma ou mais linhagens, com ou sem Auer, 10-19% de blastos

Classificação OMS 2008
SMD inclassificável:
SP: citopenias, <1% blastos
MO: Displasia inequívoca em <10% das células de uma ou mais linhagens acompanhada de alteração citogenética considerada presuntiva de SMD, <5% de blastos

Classificação OMS 2008
• SMD associada a del(5q) isolada:
SP: anemia, <1% blastos, plaquetas normais ou aumentadas
MO: megacariócitos hipolobulados ou monolobulados, <5% de blastos, del(5q) isolada, sem Auer

Estratificação de risco / Prognóstico
Prognóstico de SMD é muito variável:
desde expectativa de vida quase normal a poucos meses de sobrevida
Escores de prognóstico tentam identificar a estimativa de sobrevida e o risco de transformação em leucemia

Estratificação de risco / Prognóstico
Escores de prognóstico levam em conta:
Idade, sexo, subtipo, porcentagem de blastos na MO, número de citopenias, alterações citogenéticas, dependencia transfusional, etc.

Sistemas de escores propostos: • Bournemouth (Mufti et al), 1985
• FAB (Varella et al), 1985 • Espanhol (Sanz et al), 1989 • Cassano et al, 1990 • Rubio-Feliz et al, 1991 • Lille (Morel et al), 1993 • Unifesp (Souto et al), 1996 • IPSS (Greenberg et al,) 1997 • Dusseldorf (Aul et al, 1998) • Nordic (Hellstrom-Lindberg et al, 2003) • WPSS (Malcovatti et al, 2007) • MDACC (Kantarjian et al, 2008) • IPSS- R (Greenberg et al, 2012)

Estratificação de risco / Prognóstico
• IPSS
• WPSS - dinâmico
• IPSS-R

IPSS Blastos na MO:
< 5% = 0
5 - 10% = 0,5
11 - 20% = 1,5
21 - 30% = 2
Cariótipo:
nl, -Y, del(5q), del(20q) = 0
3 anls ou alts 7 = 1
outras anls = 0,5
Citopenias periféricas:
nenhuma ou única = 0
2 a 3 citopenias = 0,5
Escore: BR = 0
Int1 = 0,5-1
Int2 = 1,5-2
AR = >2,5
Greenberg et al, 1997

IPSS
Sobrevida livre de LMA variando de <4a para AR até >17a para BR
Evolução para LMA variando de <3a para AR até indeterminada para BR

Escore de prognóstico dinâmico
• Maioria dos sistemas de escore são estáticos, isto é, avaliação feita ao diagnóstico e daí é estratificado o risco
• Avaliação dinâmica aparece com WPSS, no qual é possível realocar o paciente conforme a doença evolui e estabelecer nova expectativa.

Variável 0 1 2 3
Categoria
OMS
AR, ARSA
5q-
CRDM
CRDM-SA
AREB1 AREB2
Cariótipo Bom
nl, -Y, del(5q), del(20q)
Interm
≥ 3 anls ou alts 7
Desfav
≥ outras anls
-
Neces.
Transf*
Não Regular - -
*1 transfusão a cada 8 sem em 4 meses
Malcovati et al, 2007
WPSS

WPSS
• Muito baixo risco = escore 0
• Baixo risco = escore 1
• Intermediário = escore 2
• Alto = escore 3 a 4
• Muito alto = escore 5 a 6

Sobrevida global WPSS
Malcovati et al, 2007

Risco de trasnformação WPSS Malcovati et al, 2007

IPSS-R
• Revisado com 7012 pacientes
• Incluidas novas anomalias citogenéticas
• Definiu cinco categorias
Greenberg et al, 2012

IPSS-R
• Muito bom: del(11q),-Y
• Bom: nl,del(20q),del(5q),del(12p),só ou em dupla
• Intermediário: +8, 7q-, i(17q),+19,+21, ou qlq outra isolada ou não, e clones independentes
• Pobre: der(3)q21/q26,-7, duplas incluindo 7q-,
complexo (3 anl)
• Muito pobre : complexo (>3 anl)
Schanz et al, J Clin Oncol, 2012


Prognóstico
Decisão terapêutica é baseada no risco:
• Baixo risco e intermediário baixo = medidas para diminuir a necessidade transfusional
• Intermediário alto e alto risco = opções de medicamentos hipometilantes ou quimioterápicos

Referências • Alcindor & Bridges. Br J Haematol 2002,116:733-43
• Garcia-Manero G. Am J Haematol 2011,86:491-8
• Halfdanarson et al. Eur J Heamatol 2008,80:523-31
• Nagoshi et al, Curr Mol Med 2011,11:678-85
• Swerdlow et al, 2008
• Malcovati e t al, JCO 2007,25:3503-10
• Malcovati e t al , Haematologica 2011; 96:1433-40
• Greenberg et al, Blood , 2007
• Greenberg et al, Blood 2012, 120: 2454
















