Embed Size (px)
Citation preview

MIRA WENGERT
ENVOLVIMENTO DE RECEPTOR P2 NA EXCREÇÃO RENAL DE SÓDIO: POSSÍVEL PAPEL PROTETOR CONTRA ESTADOS
ISQUÊMICOS
TESE SUBMETIDA À UNIVERSIDADE FEDERAL DO
RIO DE JANEIRO VISANDO A OBTENÇÃO DO GRAU
DE
DOUTOR EM CIÊNCIAS
Universidade Federal do Rio de Janeiro Centro de Ciências da Saúde Instituto de Biofísica Carlos Chagas Filho 2 0 0 8

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

ii

iii
Esta tese foi desenvolvida entre julho de 2004 e abril de 2008 sob
orientação do Prof. Dr. Celso Caruso Neves e do Prof. Dr. Luiz
Roberto Leão Ferreira no laboratório de Bioquímica Renal do
Instituto de Biofísica Carlos Chagas Filho da Universidade Federal
do Rio de Janeiro. O trabalho foi financiado com auxílios
concedidos pelas seguintes agências de fomento: Coordenação de
Desenvolvimento de Pessoal de Nível Superior (CAPES), Fundação
Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de
Janeiro (FAPERJ) e Conselho Nacional de Desenvolvimento
Científico e Tecnológico (CNPq).

iv
AAAAo meu orientador, Celso, ao o meu orientador, Celso, ao o meu orientador, Celso, ao o meu orientador, Celso, ao meu pai Andreas, ao meu irmão meu pai Andreas, ao meu irmão meu pai Andreas, ao meu irmão meu pai Andreas, ao meu irmão
Maxim, a minha mãe SMaxim, a minha mãe SMaxim, a minha mãe SMaxim, a minha mãe Sigrid (igrid (igrid (igrid (in in in in memorianmemorianmemorianmemorian)))) eeee ao meu ao meu ao meu ao meu
MarquinhosMarquinhosMarquinhosMarquinhos, com todo carinho., com todo carinho., com todo carinho., com todo carinho.

v
Agradecimentos
A Deus por ter concluído mais uma etapa da minha vida, e ter tido condições de concretizar um sonho. Aos meus pais e meu irmão, vocês são muito importantes na minha vida! Nossa união é fundamental para a minha vida. Vocês dois são exemplos de pai, irmão, amigo, família... Obrigada pela preocupação, pela ajuda, pelo amor, pelas discussões, pelas festas e comemorações. Agradeço por ter estas duas pessoas tão maravilhosas na minha vida. Daddy, obrigada pela paciência, pelas conversas, pelo apoio, pelo incentivo, por todo o seu esforço de ter feito tudo e mais um pouco para me ajudar. Daddy e Maxim, vocês são a minha escola de vida. Por isso continuo dizendo: tenho o melhor PAI e o melhor IRMÃO do mundo! A pessoa que eu amo tanto: Marquinhos, obrigada por me ajudar a vencer mais uma fase da minha vida. Sempre do meu lado me ajudando, me animando, e fazendo tudo ser mais agradável. A vida com vc é mais especial! Com você tudo é sempre muito melhor! Isso é AMOR. Sempre agradeço por ser vc o meu Marquinhos pq não teria como ser melhor! Você é muito importante na minha vida. Te amo!!! Ao meu amigo e orientador Celso. Não existem palavras para te agradecer por tudo. Se fosse apenas uma orientação comum seria mais fácil. Mesmo quando a distância era grande, vc sempre esteve presente, sempre me ajudou em tudo que eu precisei, sempre se preocupou com a minha formação. Uma pessoa assim é muito mais que um orientador. Realmente não existem palavras para te agradecer, pq pessoas como vc também não existem. Existem pessoas que passam pela nossa vida e nunca mais são esquecidas, vc é uma dessas. Para isso acontecer eu teria que desaprender muita coisa. Obrigado por tudo que eu aprendi e aprendo com vc!! Digo que é uma honra ter vc como meu orientador e amigo! OBRIGADA POR TUDO MESMO, CELSO! Ao Luiz, meu segundo orientador, sempre com alto astral e aquela super energia positiva! Sempre disposto a ajudar dando aquela força, e realmente vibrando com as minhas conquistas! À professora Doris por ter revisado esta tese tão rápido, obrigada!. À Ingrid, pela sua ajuda, sua presença, sua preocupação, por brigar pelas minhas coisas, estar sempre do meu lado me defendendo e por toda a dedicação que vc sempre teve comigo. Sempre agradeço por vc ser parte da minha família. Você sempre foi e sempre será muito importante na minha vida. Agradecer uma pessoa como vc com palavras é difícil, OBRIGADA POR TUDO MESMO!!! À minha melhor amiga Camille com quem eu sempre posso contar a qualquer momento. Valeu Méu, por sempre me dar força!!

vi
À Belzinha por toda a sua doçura, seu carinho, e por sempre torcer por mim. Vc é muita coisa para mim, cunhada, amiga, irmã... Valeu Belzinha!!! À Carol que sempre vibrou e comemorou bastante as minhas vitórias. À Aloa que me ajudou desde o começo, antes do mestrado. Pessoas como você não se encontra toda hora! Obrigada pela ajuda, pelo apoio, e por tudo que você me ensinou. Valeu Aloinha!!!!!! Minha amigona!! Como ela diz: não joga cordinha, desce para ir buscar uma amiga!!! À Marizete pelo seu amor e carinho, e por sempre rezar por mim para que tudo dê certo. À Ana Acácia, por todo o seu carinho, apoio, por torcer por mim, por sempre passar aquela tranqüilidade e pela sua alegria e alto astral. Valeu Ana !!!! A Jana, minha amiga que eu sempre posso contar, pára até as coisas dela para me ajudar, sempre dando força. Valeu por tudo, Jana!! A Sharon, minha nova amiga! Acho que é a pessoa mais querida que eu conheço! Ao Thiago, meu novo aluno de iniciação científica, que participou deste trabalho sempre com muito carinho, muita dedicação e empolgação. Ao Shan por todas as ajudas e pelo seu alto astral. As outras pessoas do laboratório de Bioquímica Renal, que são todos muito legais! Valeu pela paciência e pelos momentos de descontração.

vii
ABREVIAÇÕES
ADA adenosina desaminase ADE adenina ADO adenosina ADP adenosina 5’-difosfato AMP adenosina 5’-monofosfato AMPc adenosina 3’,5’ monofosfato cíclico ATP adenosina 5’-trifosfato DAG diacilglicerol DIP dipiridamol DMPX 3,7-dimetil-1-propargil xantina DPCPX 8-ciclopentil-1,3-dipropilxantina EDTA ácido etileno diamino tetra acético Gi proteína G inibitória HEPES N-[2-hidroxi etil] piperazina-N’-[2-ácido etano sulfônico] HIPO hipoxantina INO inosina IP3 fosfatidil inositol 1,4,5 trifosfato IP3 inositol 1,4,5 trifosfato LDH lactato desidrogenase MRS1523 (3-propil-6-etil-5-[(etiltio)carboni]-2-fenil-4-propil-3-piridina carboxilato) NBTI 6-[(4-Nitrobenzyl)thio]-9-β-D-ribofuranosylpurine PIP2 fosfatidil inositol 4,5 bifosfato PKA proteína cinase A PKC proteína cinase C PLC fosfolipase C PNP Purina nucleosídeo fosforilase PPDAS 4-[[4-Formyl-5-hydroxy-6-methyl-3-[(phosphonooxy)methyl]-
2-pyridinyl]azo]-1,3-benzenedisulfonic acid tetrasodium salt PRT Purina ribosilfosfotransferase PTN proteína SAH S-adenosil-L-homocisteína SAHH S-adenosil-homocistína hidrolase SCH (7-(2- fenil etil_-5-amino-2(2-furil)-pirazolo-[4,3-e]-1,2,4-
trizolo[1,5-c]pirimidina SDS dodecilsulfato de sódio VEC volume extracelular

viii
RESUMO A adenosina e o ATP extracelulares modulam funções fisiológicas
importantes, tais como excreção de sódio e de água. Além disso, adenosina apresenta um papel protetor contra a injuria renal em eventos fisiopatológicos como a isquemia. Neste trabalho, a linhagem celular LLC-PK1 foi usada como modelo para o estudo do metabolismo de purinas em condições de normóxia e isquemia, a fim de entender a relação estabelecida entre os principais metabolitos purínicos gerados em cada uma das condições e as atividades dos transportadores primários de sódio no túbulo proximal, (Na++K+)ATPase and Na+-ATPase. O metabolismo de purinas foi avaliado através da incubação de células com [3H]-adenosina, fracionamento dos derivados radioativos por TLC e quantificação de cada metabólito por cintilação líquida. Os níveis de equilíbrio de derivados de [3H]-adenosina revelam que os nucleotídeos de adenina, bem como as nucleobases hipoxantina e adenina, são as purinas mais abundantes no meio extracelular,em condição de normóxia. A conversão intracelular de derivados de [3H]adenosina em nuclotideos de adenina foi demonstrada pelo uso do dipiridamol (um inibidor não especifico de transportadores equilibrativos e concentrativos de nucleosideo). A avaliação dos efeitos dos derivados purínicos extracelulares mais abundantes sobre as atividades (Na++K+)ATPase and Na+-ATPase demonstram que adenosina e ATP são potentes estimuladores da Na+-ATPase, enquanto o nucleosideo e provavelmente o nucleotídeo não afetam a atividade (Na++K+)ATPase. O uso de ferramentas farmacológicas tais como agonistas e antagonistas seletivos de receptores P1 e P2 demonstram que os efeitos estimulatórios de adenosina e ATP são mediados por receptor P2. Além disso, nossos resultados demonstram que o efeito estimulatório induzido por adenosina depende da fosforilação intracelular do nucleosideo à ATP e liberação do nucleotídeo por um canal de ânions sensível a DIDS. A via de sinalização ativada por ATP também foi investigada. Nossos resultados demonstram que o ATP extracelular estimula a liberação de cálcio intracelular e ativação de PKC, embora o efeito do ATP sobre a atividade Na+-ATPase pareça ser mediado por uma isoenzima de PKC insensível ao cálcio. Os níveis de equilíbrio observados em condição de isquemia para os derivados de [3H]-adenosina revelam que o nível da nucleobase adenina é significativamente maior que aquele observado em condição de normoxia. A condição isquêmica foi mimetizada pela incubação das células com antimicina A (inibidor da fosforilação oxidativa) e 2-deoxi-glicose (análogo não metabolizável de glicose). Períodos isquêmicos de 10 ou 30 minutos reduziram a atividade Na+-ATPase de modo reversível por meio de reperfusão de 2 horas. Em condição similar a atividade (Na++K+)ATPásica não foi afetada. A pré-incubação com ATP por 30 minutos anteriores ao período isquêmico preveniu o efeito inibitório de uma isquemia de 20 minutos sobre a atividade Na+-ATPásica. Em conjunto, nossos dados indicam que o efeito estimulatório da adenosina sobre a Na+-ATPase de células LLC-PK1 depende da capatação do nucleosídeo, conversão metabólica à ATP e liberação do nucleotídeo pelo canal de anions sensível a DIDS. No meio extracelular, o ATP estimula a atividade Na+-ATPásica através da ativação de PKC mediada pelo receptor P2. Além disso, a inibição da Na+-ATPase induzida pela isquemia é prevenida pela pré-incubação com ATP, sugerindo um papel para o nucleotídeo no pré-condicionamento isquêmico, reduzindo a perda de sódio resultante de isquemia.

ix
ABSTRACT Extracellular adenosine and ATP modulate important physiological
functions in the kidney, such as sodium and water excretion. In addition, adenosine has a protective role against renal injury in pathophysiological events such as ischemia. In this work, LLC-PK1 cell line was used as a model to study purine metabolism in normoxic and ischemic conditions in order to understand the relationship between the major purine metabolites generated in each condition and the sodium primary transporters activities in the proximal tubule, the (Na++K+)ATPase and Na+-ATPase. Purine metabolism was evaluated by incubation of the cells with [3H]-adenosine, fractionation of the radiolabeled derivatives by TLC and quantification of each metabolite by liquid scintillation. The steady-state levels of [3H]-adenosine-derivatives reveals that adenine nucleotides as well as both purine nucleobases hypoxanthine and adenine are the most abundant purines in the extracellular medium, in normoxic condition. The intracellular conversion of [3H]-adenosine-derivatives to [3H]-adenine nucleotides was demonstrated by using dipiridamole (a nonspecific inhibitor of equilibrative and concentrative nucleoside transporters). Evaluation of the effects of the most abundant extracellular purine-derivatives on both (Na++K+)ATPase and Na+-ATPase activities demonstrates that adenosine and ATP are potent stimulators of the Na+-ATPase while the nucleoside and probably the nucleotide does not affect the (Na++K+)ATPase activity. By using pharmacological tools such as selective agonists and antagonists for P1 and P2 receptors we showed that stimulatory effects induced by both adenosine and ATP are mediated by P2 receptor. Furthermore, our results demonstrate that adenosine-induced stimulatory effect depends on intracellular phosphorylation of the nucleoside to ATP and release of the nucleotide by the DIDS-sensitive anion channel. The signaling pathway triggered by ATP was also investigated. Our results demonstrates that extracellular ATP stimulates intracellular calcium release and PKC activation, although the stimulatory effect of ATP on the Na+-ATPase seems to be mediated by a calcium-insensitive PKC isoenzyme. The steady-state levels observed for the [3H]-adenosine-derivatives in the extracellular medium in ischemic condition reveals that the level of the nucleobase adenine is significantly higher in that condition than in mormoxia. The ischemic condition was mimicked by incubation of the cells with antimycin A (an inhibitor of the oxidative phosphorylation) and 2-deoxi-glucose (non-metabolized analog of glucose). Ischemic periods of 10, 30 minutes reduced the Na+-ATPase activity in a reversible manner upon 2 hours of reperfusion while the same condition does not affect the (Na++K+)ATPase activity. Pre-incubation with ATP for 30 minutes before the ischemic period prevented the inhibitory effect of 40 minute-ischemia on the Na+-ATPase activity. Altogether, our data indicate that adenosine induced-stimulatory effect on the Na+-ATPase activity from LLC-PK1 depends on nucleoside uptake, metabolic conversion to ATP and nucleotide release by the DIDS-sensitive anion channel. In the extracellular medium, ATP stimulates the Na+-ATPase activity by P2-mediated PKC activation. Furthermore, the ischemia-induced inhibition of the Na+-ATPase activity is prevented by pre-incubation of the cells with ATP, suggesting a role for the nucleotide in pre-ischemic conditioning reducing the ischemia-related sodium loss.

x
FICHA CATALOGRÁFICA
Wengert, Mira
Envolvimento de receptor P2 na excreção renal de sódio: possível papel protetor contra estados isquêmicos ./ Mira Wengert -- Rio de Janeiro: UFRJ / IBCCF, 2008.
12, 110 f. il.; 29,7 cm.
Orientadores: Prof. Dr. Celso Caruso Neves, Luiz Roberto Leão Ferreira
Tese (doutorado) – Universidade Federal do Rio de Janeiro, IBCCF, Programa de Pós-Graduação em Ciências Biológicas (Fisiologia), 2008.
Referências bibliográficas: f. 111-135.
1. Na+-ATPase 2. (Na++K+)ATPase 3. Adenosina 4. Receptores 5. túbulo proximal 6. Sinalização celular – Tese. I. Caruso-Neves, Celso. II Leão-ferreira, Luiz Roberto. IV. Universidade Federal do Rio de Janeiro, Instituto de Biofísica Carlos Chagas Filho. V. Título

xi
ÍNDICE
1. INTRODUÇÃO __________________________________________________ 1
1.1. ESTRUTURA E FUNÇÃO RENAL______________________________________________________1 1.2.TRANSPORTE TRANSCELULAR DE SÓDIO NO TÚBULO PROXIMAL: PAPEL DA (NA
++K+)ATPASE E NA+-
ATPASE__________________________________________________________________________7 1.2.1 (Na++K+)ATPase e Na+-ATPase _______________________________________________10
1.3. O RIM COMO ALVO E PRODUTOR DE COMPOSTOS MODULADORES DA FUNÇÃO RENAL___________18 1.4. ATP COMO FONTE DE ADENOSINA RENAL: MECANISMO DE SECREÇÃO DE ATP _______________19 1.5.METABOLISMO RENAL DE ADENOSINA_______________________________________________25 1.6. RECEPTORES DE NUCLEOTÍDEOS E NUCLEOSÍDEOS_____________________________________31 1.7. EFEITOS DE ADENOSINA E ATP NO RIM: RECEPTORES E SEUS EFEITOS ACOPLADOS____________35
1.7.1-Efeitos renais da adenosina ___________________________________________________35 1.7.2-Efeitos renais mediados pelo receptor A1 ________________________________________35 1.7.3- Efeitos renais mediados pelo receptor A2 ________________________________________37 1.7.4- Efeitos renais mediados pelo receptor A3 ________________________________________38 1.7.5-Efeitos renais do ATP________________________________________________________39 1.7.6- Efeitos renais mediados pelos Receptores P2X____________________________________39 1.7.7- Efeitos renais mediados pelo Receptor P2Y ______________________________________42
1.8. PAPEL DO ATP E DA ADENOSINA NA ISQUEMIA RENAL __________________________________45 1.9. RESULTADOS ANTERIORES_______________________________________________________48
2. OBJETIVOS___________________________________________________ 52
3. MATERIAIS E MÉTODOS ________________________________________ 53
3.1.MATERIAIS____________________________________________________________________53 3.2. CULTURA DE CÉLULAS LLC-PK1 __________________________________________________53 3.3. MEDIDA DA ATIVIDADE ATPÁSICA _________________________________________________53 3.4. MEDIDA DA ATIVIDADE DE PKA E PKC _____________________________________________54 3.5. MEDIDA DE CÁLCIO INTRACELULAR ________________________________________________54 3.6. CROMATOGRAFIA DE CAMADA FINA PARA ANÁLISE DE METABOLISMO DE PURINAS.____________55 3.7.ENSAIO PARA SUBMETER AS CÉLULAS A ISQUEMIA_____________________________________56 3.8. ANÁLISE ESTATÍSTICA___________________________________________________________56
4. RESULTADOS_________________________________________________ 58
4.1. METABOLISMO DE ADENOSINA EM CÉLULAS LLC-PK1 _________________________________58 4.2. EFEITO DE ADENOSINA E DE SEUS METABÓLITOS SOBRE AS ATIVIDADES NA
+-ATPASE E
(NA++K+)ATPASE EM CÉLULAS LLC-PK1 ______________________________________________64
4.3. ENVOLVIMENTO DE RECEPTORES ESPECÍFICOS PARA ATP E CANAIS DE ÂNIONS NO MECANISMO
ESTIMULATÓRIO DA ADENOSINA SOBRE A NA+-ATPASE EM CÉLULAS LLC-PK1 _________________71
4.4. ESTUDO DA VIA DE SINALIZAÇÃO ENVOLVIDA NO ESTÍMULO DA NA+-ATPASE POR ATP ________80
4.5. ESTUDO DA ISQUEMIA METABÓLICA SOBRE O METABOLISMO CELULAR DE ADENOSINA _________86 4.6. EFEITOS DA ISQUEMIA METABÓLICA SOBRE AS ATIVIDADES DAS ENZIMAS NA
+-ATPASE E
(NA++K+)ATPASE _________________________________________________________________94
5. DISCUSSÃO __________________________________________________ 99
6. REFERÊNCIAS BIBLIOGRÁFICAS ______________________ _________ 111

xii
ÍNDICE DE FIGURAS Figura 1. Estrutura do néfron e a morfologia glomérulo e de cada segmento.... 3 Figura 2. Mecanismos de transporte tubular no túbulo proximal. ....................... 9 Figura 3. Estrutura e topologia da (Na+ + K+) ATPase. .................................... 12 Figura 4. Ciclo catalítico proposto para a Na+-ATPase .................................... 17 Figura 5. Mecanismo de liberação de ATP, papel do receptor P2X na transdução de sinal autócrino e/ou parácrino de ATP...................................... 23 Figura 6 Esquema do metabolismo de adenosina nos espaços intra e extracelular....................................................................................................... 30 Figura 7. Receptores P1 e P2. ......................................................................... 34 Figura 8 Distribuição dos receptors P2 ao longo do nefron.............................. 41 Figura 9. Adenosina estimula Na+-ATPase através da ativação de receptor A2A via AMPc/PKA. ................................................................................................. 50 Figura 10. Efeito da adenosina sobre a atividade Na+-ATPásica via receptores A1 e A2A ............................................................................................................ 51 Figura 11. Metabolismo extracelular de adenosina. ......................................... 59 Figura 12. Metabolismo intracelular de adenosina. .......................................... 61 Figura 13. Metabolismo de adenosina na presença de NBTI e Dipiridamol..... 62 Figura 14. Níveis intra e extracelular de ATP na presença de NBTI e Dipiridamol ....................................................................................................... 63 Figura 15. Dose resposta de adenosina sobre a atividade Na+-ATPásica e (Na+-K+)ATPásica. .................................................................................................... 65 Figura 16. Dose resposta de adenina sobre a atividade Na+-ATPásica........... 66 Figura 17. Dose resposta de hipoxantina sobre a atividade Na+-ATPásica. .... 67 Figura 18. Dose resposta de AMP sobre a atividade Na+-ATPásica................ 68 Figura 19. Dose resposta de ADP sobre a atividade Na+-ATPásica. ............... 69 Figura 20. Dose resposta de ATP sobre a atividade Na+-ATPásica................. 70 Figura 21. Efeito de adenosina sobre a atividade Na+-ATPásica, na presença de DMPX, DPCPX e MRS, antagonistas seletivos de A1, A2 e A3 respectivamente. .............................................................................................. 73 Figura 22. Efeito estimulatório de ATP na presença de PPADS, antagonista de receptor P2....................................................................................................... 74 Figura 23. Efeito estimulatório de adenosina na presença e ausência de PPADS, antagonista de receptor P2. ............................................................... 75 Figura 24. ATP sintetizado a partir de adenosina estimula a atividade Na+-ATPásica. ......................................................................................................... 76 Figura 25. Efeito de ATP e adenosina na presença de iodotubericidina, inibidor de adenosina cinase. ....................................................................................... 77 Figura 26. Efeito de adenosina na presença de DIDS, inibidor de transportador de ânions.......................................................................................................... 79 Figura 27. Efeito de ATP e adenosina sobre a atividade de PKA. ................... 82 Figura 28. Efeito de ATP e adenosina sobre a atividade de PKC. ................... 83 Figura 29. Envolvimento de PKC no efeito de ATP e adenosina sobre a Na+-ATPase. ........................................................................................................... 84 Figura 30. Efeito de ATP sobre o aumento de cálcio citoplasmático................ 85 Figura 31. Níveis intra e extracelular de nucleotídeos de [3H]-adenina em condições de normóxia e hipóxia metabólica. .................................................. 88 Figura 32. Permeabilidade aparente das cálulas a nucleotídeos de [3H]-adenina em condições de normóxia e isquemia. ........................................................... 89

xiii
Figura 33. Níveis intra e extracelular de adenosina e permeabilidade aparente da célula a adenosina em condições de normóxia e isquemia. ....................... 90 Figura 34. Níveis intra e extracelular de hipoxantina formada a partir de adenosina em condições de normóxia e isquemia........................................... 91 Figura 35. Níveis intra e extracelular de inosina formada a partir de adenosina em condições de normóxia e isquemia. ........................................................... 92 Figura 36. Níveis intra e extracelular de adenina em condições de normóxia e......................................................................................................................... 93 isquemia. .......................................................................................................... 93 Figura 37. Hipóxia metabólica inibe a atividade Na+-ATPásica e a reperfusão reverte este efeito............................................................................................. 96 Figura 38. Hipóxia metabólica não modula a atividade (Na++K+)ATPásica...... 97 Figura 39. Pré-condicionamento com ATP protege a atividade Na+-ATPásica contra a inibição promovida por períodos isquêmicos...................................... 98 Figura 40. Modelo proposto para o metabolismo de adenosina em membrana luminal de células LLC-PK1, e seu efeito na reabsorção de sódio................. 105 Figura 41. Esquema do metabolismo de adenosina em membrana luminal de células LLC-PK1 durante isquemia curta, e o pré-condicionamento isquêmico com ATP......................................................................................................... 109

1
1. Introdução
1.1. Estrutura e função renal
O rim é um órgão fundamental na manutenção do meio interno,
desempenhando funções importantes, tais como: controle do balanço hidro-
eletrolítico, regulação do equilíbrio ácido-base, conservação de nutrientes,
excreção de resíduos metabólicos, metabolismo de cálcio e fósforo, entre outras.
Estes processos somente são possíveis devido a uma complexa e integrada rede
de modulação dos mecanismos renais de manipulação de água e íons.
Atualmente, o grande desafio é a determinação dos agentes ativos desta rede e
os possíveis pontos de interação entre eles.
Cada rim é composto por aproximadamente 1,5 milhões de unidades
funcionais: os néfrons. Esses são formados pelo glomérulo, onde ocorre a
ultrafiltração, seguido pelos segmentos tubulares, onde ocorre o processamento
do fluido filtrado, secreção ou reabsorção de solutos e água (Figura 1). A primeira
etapa na formação da urina é a ultrafiltração glomerular, que ocorre nos
glomérulos, levando a formação de um ultrafiltrado glomerular, cuja composição é
similar àquela do plasma, mas com poucas proteínas e macromoléculas de baixo
peso molecular (Mello-Aires, 2008). Isto se deve ao fato da existência de “poros
funcionais” na membrana filtrante que restringem a passagem de moléculas em
função de seu peso, carga e forma (Mello-Aires, 2008). Aproximadamente 180
litros de plasma são filtrados diariamente e ao longo do néfron sua composição e
volume são modificados, através dos mecanismos específicos de secreção e
reabsorção. Apenas 1% do ultrafiltrado glomerular é eliminado na urina, devido a
grande capacidade de reabsorção dos diferentes segmentos tubulares. Igualmente
importtante, o mecanismo de secreção também desempenha papel relevante,
participando na regulação fina da excreção de solutos (Schnermann e Sayegh,
1998).
Na Figura 1 pode-se observar os diferentes segmentos do néfron: túbulo
proximal, alça de Henle, túbulo distal e duto coletor. Vale ressaltar que no ducto
coletor desenbocam vários túbulos distais de diferentes néfrons mas, do ponto de

2
vista funcional, o ducto coletor é considerado parte de um néfron (Mello-Aires,
2008). Cada segmento tubular apresenta características histológicas e funcionais
distintas e, portanto, é regulado por hormônios e autacóides de maneira distinta e
específica.
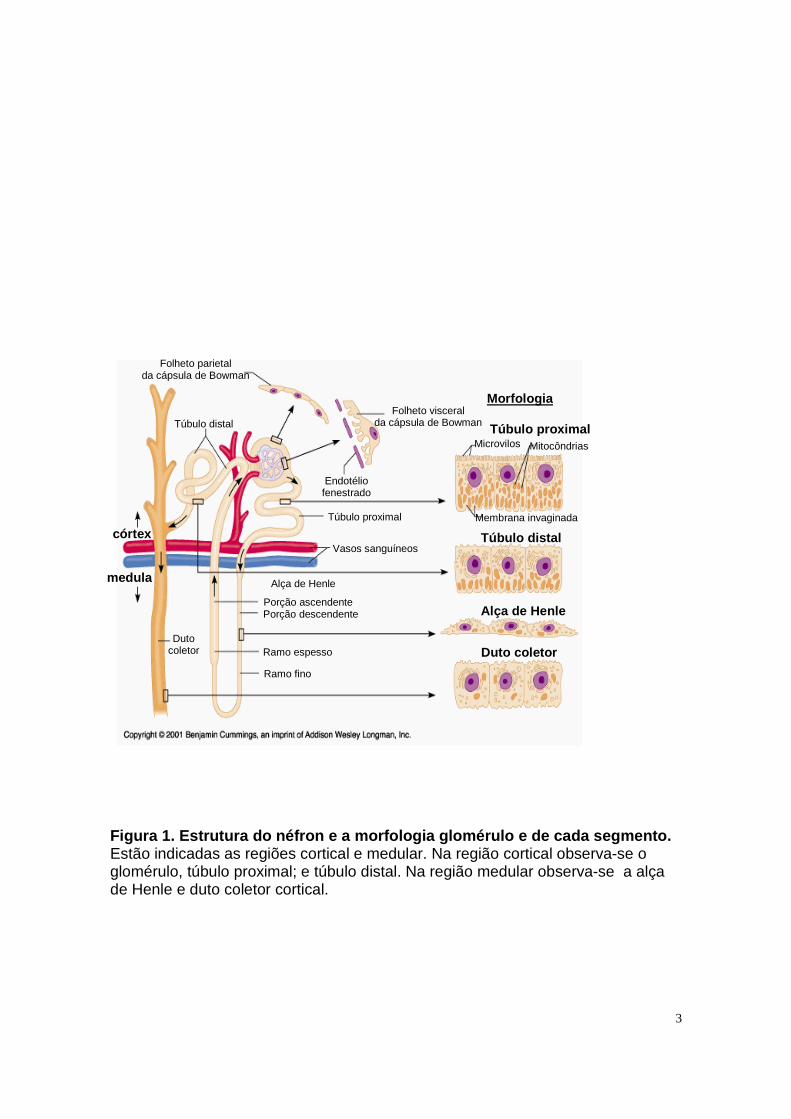
3
Figura 1. Estrutura do néfron e a morfologia glomér ulo e de cada segmento. Estão indicadas as regiões cortical e medular. Na região cortical observa-se o glomérulo, túbulo proximal; e túbulo distal. Na região medular observa-se a alça de Henle e duto coletor cortical.
Folheto parietal da cápsula de Bowman
Folheto visceral da cápsula de Bowman
Endotélio fenestrado
Vasos sanguíneos
Túbulo proximal
Alça de Henle
Porção ascendente Porção descendente
Ramo espesso
Ramo fino
Túbulo distal
Duto coletor
Microvilos Mitocôndrias
Membrana invaginada
Túbulo distal
Alça de Henle
Duto coletor
Morfologia
córtex
medula
Túbulo proximal

4
O túbulo proximal, formado por epitélio cúbico simples, é o primeiro segmento
após o glomérulo e suas células apresentam membranas com diferentes
permeabilidades e características de transporte, são elas: a membrana luminal,
que separa a célula da luz tubular, e a membrana basolateral, que limita a célula
com o interstício (Brenner e Rector, 2000). A membrana luminal é do tipo borda
em escova, o que lhe confere grande superfície de reabsorção. Além disso,
podemos observar inúmeras mitocôndrias adjacentes à membrana basolateral o
que é coerente com sua alta capacidade de transporte ativo (Figura 1). Em
condições normais, os túbulos proximais reabsorvem 126 litros de fluido tubular
isotônico por dia, o que corresponde a aproximadamente 70% do ultrafiltrado
glomerular (Brenner e Rector, 2000). A reabsorção de sódio neste segmento
ocorre tanto pela via paracelular quanto pela transcelular, sendo a última
responsável pela maior parte da reabsorção (Mello-Aires, 2008). Os mecanismos
de transporte de soluto e água deste segmento serão discutidos detalhadamente
no tópico subsequente.
A alça de Henle é formada por uma porção fina descendente, outra fina
ascendente (presente nos néfrons justamedulares) e uma espessa ascendente
(também chamada de túbulo distal reto). A porção fina descendente, constituída
por epitélio pavimentoso, é permeável a água e pouco permeável a solutos (Mello-
Aires, 2008). A porção fina ascendente é impermeável a água e altamente
permeável a solutos (Brenner e Rector, 2000). As diferenças de permeabilidade
características destes segmentos são fundamentais para o mecanismo
multiplicador contra corrente. A alta permeabilidade à água neste segmento é
devida a elevada expressão constitutiva de aquaporinas do tipo I e II (Jeyaseelan
et al., 2006). A porção ascendente espessa, constituída por células epiteliais
cúbicas, é o segmento onde ocorre a reabsorção ativa de NaCl pelo co-
transportador tríplice Na+/K+/2Cl- (NCKK), um transportador ativo secundário
localizado na membrana luminal, cuja atividade depende do gradiente de sódio
gerado pela (Na++K+)ATPase presente na membrana basolateral. O acoplamento
entre transportadores e canais localizados nas membranas basolateral e luminal,

5
respectivamente, constitui o mecanismo primário de concentração do interstício
medular.
O túbulo distal convoluto é constituido de células cúbicas com raros
microvilos na membrana luminal e pregas na superfície basolateral, sendo através
destas ultimas o encaixe entre células vizinhas. Nesta região de contato entre
células são criadas as vias de transporte paracelular, menos pronunciadas que
aquelas observadas no túbulo proximal. O segmento inicial do túbulo distal
convoluto é relativamente impermeável a água. Sua porção final responde ao
hormônio antidiurético, exibindo permeabilidade à água na presença deste
hormônio e impermeabilidade na sua ausência. A condutância iônica é baixa,
desfavorecendo o transporte passivo de íons. Portanto, este segmento possui
baixa capacidade de transporte e alto gradiente de concentração. No túbulo distal
convoluto ocorre uma baixa reabsorção de NaCl, bicarbonato, cálcio, e secreção
de hidrogênio e amônia. O potássio é reabsorvido no início e secretado no final
deste segmento. Tanto a reabsorção de sódio como a secreção de potássio e
hidrogênio são estimuladas pela aldosterona.
Os dutos coletores corticais são compostos de epitélio simples com formato
cúbico a pavimentoso (Figura 1). Os medulares possuem células cúbicas com
transição para colunar conforme os dutos crescem em direção a medula interna. O
epitélio tubular coletor revela essencialmente dois tipos de células: a) as células
principais, em maior número e b) as células intercalares, cuja freqüência diminui à
proporção que o túbulo desce à medula (Mello-Aires, 2008). Nesses segmentos,
ocorre o ajuste fino da composição urinária, sendo esta porção do néfron um sítio
de regulações hormonais como ADH, angiotensina II, prostaglandinas entre outros
(Brenner e Rector, 2000).
Do ponto de vista da funcionalidade, o nefrón é dividido em néfron proximal,
formado pelo túbulo proximal, e néfron distal formado pelos demais segmentos.
Cabe ao néfron proximal a reabsorção de maior parte do ultrafiltrado (cerca de
70%) e ao néfron distal o ajuste fino da composição final da urina (Brenner e
Rector, 2000). Esta observação juntamente com o balanço túbulo-glomerular levou

6
durante muitos anos ao postulado de que os mecanismos regulatórios tivessem no
néfron distal seu sítio de ação.
É interessante ressaltar que os mecanismos de reabsorção e secreção entre
o segmento proximal e os segmentos distais estão correlacionados através do
balanço túbulo-glomerular, mencionado acima. Este balanço envolve uma
estrutura denominada de aparelho justaglomerular formada pela porção espessa
ascendente da alça de henle, formando a células da mácula densa, e pelas
células musculares da arteríola aferente, formando as células justaglomerulares
(Brenner e Rector, 2000). Esta estrutura permite que um aumento do fluxo distal
seja sentido pelas células da mácula densa e, através de sinalizadores específicos
de ação parácrina irá modular a constricção da arteriola aferente, modulando
assim o rítmo de filtração glomerular. Este balanço é de fundamental importância
uma vez que ele evita a perda ou a retenção excessiva de solutos e água pelos
rins. O mecansimo molecular responsável por este balanço ainda não esta
totalmente elucidado, como discutido posteriormente.
Atualmente, o túbulo proximal é considerado um importante sítio de ação
para diversos hormônios e autacóides, tais como angiotensina II, urodilatina,
bradicinina, adenosina, entre outros. Estes, por sua vez, são capazes de regular a
atividade de transportadores de sódio e a reabsorção do fluido tubular neste
segmento (Brenner e Rector, 2000). Esta observação pode ser mais bem
exemplificada com compostos que agem no túbulo proximal e também diminuem a
sensibilidade do balanço tubulo-glomerular. Alguns exemplos de substâncias
natriuréticas que agem no túbulo proximal são: peptídeo atrial natriurético (ANP),
peptídeo natriurético do tipo C (CNP), peptídeo natriurético cerebral (BNP) e
urodilatina (Brenner e Rector, 2000). Um exemplo clássico de composto
antinatriurético é a angiotensina II componente do Sistema Renina Angiotensina
Aldosterona. Os alvos destas substâncias natriuréticas e antinatriuréticas são,
entre outros, os transportadores ativos de sódio. Além dos hormônios
mencionados acima, autacóides moduladores como prostaglandinas, óxido nítrico,
endotelina, dopamina, ATP e adenosina também modulam a reabsorção de sódio
e água no túbulo proximal. Estes efeitos em conjunto levam a uma modificação na

7
capacidade de excreção renal de solutos e água, levando a variações na
composição final da urina (Mello-Aires, 2008). Além disso, em algumas patologias
como na hipertensão arterial primária, ocorrem modificações somente nos
mecanismos transportadores de água e soluto do túbulo proximal. Portanto, nesta
tese detalharemos os estudos sobre os mecanismos de reabsorção de soluto, em
particular o sódio, que ocorrem no túbulo proximal.
1.2.Transporte transcelular de sódio no túbulo proximal: papel da
(Na++K+)ATPase e Na+-ATPase
A reabsorção transcelular de solutos e água é acoplada à reabsorção de
sódio. A etapa limitante deste processo é o transporte de sódio através da
membrana basolateral, realizado por duas bombas de sódio: a clássica
(Na++K+)ATPase e a Na+-ATPase. As duas ATPases geram o gradiente
eletroquímico de sódio que possibilita o transporte de outros solutos por meio de
co-transportadores de sódio presentes na membrana luminal (Figura 2), tais como
sódio/glicose (Na/Gli), sódio/aminoácido (Na/AA) e sódio/proton (NHE).
O transportador Na/Gli pertence à família de transportadores SGLT
constituída por 6 membros (SGLT1-6), todos possuem 12 domínios transmembrana
(Lee e Han, 2007). O túbulo proximal expressa somente as isoformas SGLT1 e
SGLT2, que apresentam um papel importante na glicotoxicidade associada a
patologias renais, como as que ocorrem no diabetes melitus (Lee et al., 2005).
Neste segmento tubular, aproximadamente 90% da glicose filtrada nos glomérulos
é reabsorvida pelo co-transportador SGLT2 com baixa afinidade e alta capacidade
(Lee e Han, 2007).
A isoforma NHE3 do Trocador Na+/H+, localizado na membrana luminal de
células do túbulo proximal, é a principal responsável pela secreção do próton na
luz tubular acoplada à reabsorção de sódio. Existem 9 isoformas do co-
transportador NHE, NHE1-9. As isoformas NHE1,2,3,4,5,8 estão presentes na
membrana plasmática enquanto as isoformas NHE6,7,9 estão presentes em
membranas intracelulares (Slepkov et al., 2007). No túbulo proximal encontra-se
predominantemente a isoforma NHE3 e baixa expressão da isoforma NHE2 (Moe,
1999). Os membros desta família apresentam dois domínios distintos, um

8
transmembrana e um citosólico. Os domínios transmembrana apresentam alta
homologia e contém resíduos responsáveis pela atividade do trocador e pela
sensibilidade farmacológica a inibidores (Orlowski e Kandasamy, 1996; Shrode et
al., 1998). O domínio citosólico C-terminal apresenta menor homologia entre as
isoformas e contém domínios responsáveis pela modulação da atividade do
trocador.
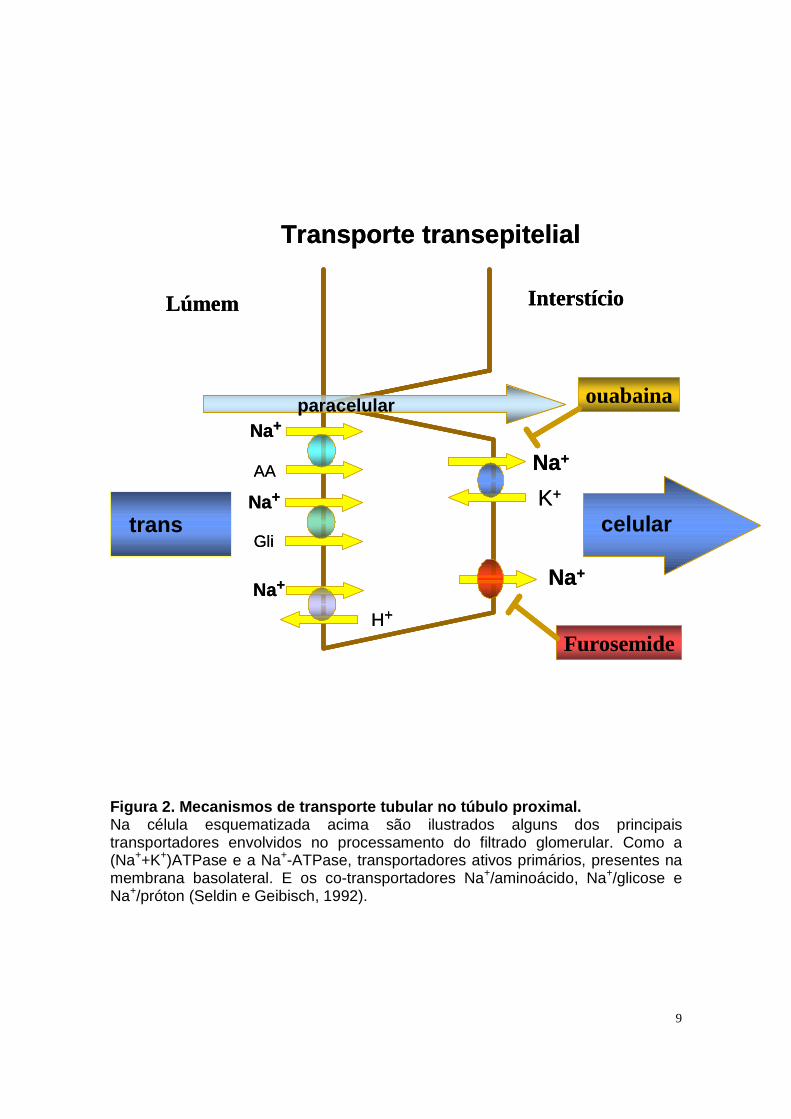
9
Figura 2. Mecanismos de transporte tubular no túbul o proximal. Na célula esquematizada acima são ilustrados alguns dos principais transportadores envolvidos no processamento do filtrado glomerular. Como a (Na++K+)ATPase e a Na+-ATPase, transportadores ativos primários, presentes na membrana basolateral. E os co-transportadores Na+/aminoácido, Na+/glicose e Na+/próton (Seldin e Geibisch, 1992).
trans
Transporte transepitelial
Lúmem Interstício
paracelular
AA
Na+
Na+
H+
Na+
K+
Na+
Furosemide
celularNa+
Gli
ouabaina
trans
Transporte transepitelial
Lúmem Interstício
paracelular
AA
Na+
Na+
H+
Na+
K+
Na+
Furosemide
celularNa+
Gli
ouabaina

10
1.2.1 (Na++K+)ATPase e Na +-ATPase A (Na++K+)ATPase foi descoberta por SKOW em 1957. Posteriormente, a
presença desta enzima foi revelada em diferentes tecidos de animais pertencentes
a diferentes espécies, onde desempenha a função de transporte de íons sódio e
potássio contra seus respectivos gradientes de concentração (Jorgensen e
Pedersen, 2001). Alterações na atividade desta enzima têm sido relacionadas à
gênese ou manutenção de diferentes patologias em mamíferos, como por
exemplo, a hipertensão arterial. No rim, a (Na++K+)ATPase é fundamental no
processo de reabsorção transcelular de sódio e de água. Deste modo, a regulação
da atividade desta enzima está diretamente relacionada à excreção renal dos
mesmos. A (Na++K+)ATPase é inibida por ouabaína, transporta 3 íons sódio para
fora da célula e 2 íons potássio para dentro, as custas da hidrólise do ATP. Esta
diferença estequiométrica entre o sódio e o potássio transportado através da
membrana plasmática confere à enzima uma característica eletrogênica. A
(Na++K+)ATPase é uma enzima do tipo P (P-ATPase), pois apresenta um
intermediário fosforilado em seu ciclo catalítico. Os dois estados conformacionais
da enzima são relacionados ao estado fosforilado e não fosforilado (E1 e E2) e são
caracterizados pelas diferenças de afinidade por sódio, potássio e ATP, além de
apresentarem diferenças de acesso aos sítios catiônicos intra e extracelulares da
membrana (Moller et al., 1996). A conformação E1 apresenta alta afinidade para
sódio e ATP e baixa para potássio; ambos os sítios de ligação de cátions estão
voltados para o lado intracelular. A conformação E2 apresenta baixa afinidade para
sódio e ATP e alta para potássio; ambos os sítios de ligação de cátions estão
voltados para o lado extracelular (Moller et al., 1996).
A (Na++K+)ATPase é composta de duas cadeias polipeptídicas principais,
denominadas subunidades α e β, associadas em uma razão estequiométrica de
1:1 (Blanco e Mercer, 1998). A subunidade α, composta de aproximadamente
1000 aminoácidos e peso molecular em torno de 110 KDa, possui 10 domínios
transmembrana e porções NH2- e COOH- terminal intracelulares, além de uma
grande alça citoplasmática entre os domínios M4-M5 (Figura 3). Esta subunidade é
responsável pelas propriedades catalíticas e de transporte da enzima, contendo

11
sítios de ligação para cátions, ATP e para o inibidor, ouabaína (Blanco e Mercer,
1997). Além disso, a subunidade α possui sítios de fosforilação para proteína
cinase C e tirosina cinase na extremidade NH2, e para proteína cinase A na alça
citosólica M8-M9 (Figura 3) (Lingrel e Kuntzweiler, 1994; Feraille e Doucet, 2001).
Estes dados revelam o papel de proteinas cinases na modulação da
(Na++K+)ATPase por diferentes hormônios e autacóides.
A subunidade β é um polipeptídio de 300 aminoácidos com um único
domínio transmembrana, contendo sítios de glicosilação extracelulares. Esta
subunidade é essencial para a atividade normal da enzima e parece estar
envolvida na oclusão do K+ e na modulação da afinidade da enzima pelo Na+ e
pelo K+ (Chow e Forte, 1995). Além das subunidades α e β, uma terceira
subunidade, denominada γ, um pequeno polipeptídio hidrofóbico (contém 53
aminoácidos) pode estar associado à enzima. Esta subunidade não é encontrada
em todos os tecidos, portanto, ao contrário das subunidades α e β, a subunidade γ
não é necessária para a atividade da (Na++K+)ATPase (Feraille e Doucet, 2001).
Quando é expressa em conjunto com as subunidades α e β, a estequiometria do
complexo α:β:γ é 1:1:1. A subunidade γ parece estar envolvida na estabilidade da
conformação E1 e na passagem de íons através da membrana (Mercer et al.,
1993; Feraille e Doucet, 2001).
As subunidades da (Na++K+)ATPase podem ser formadas a partir de
diferentes isoformas para as subunudades alfa e beta. As quatro variantes para
subunidade α e as três para a subunidade β são codificadas por genes distintos,
enquanto as duas variantes para a subunidade γ são geradas a partir de um único
gene, mas derivam de variantes de “splicing” (Shull et al., 1986; Lemas e
Fambrough, 1993; Shamraj e Lingrel, 1994; Besirli et al., 1997; Feraille e Doucet,
2001). O principal heterodímero, formado no tecido renal entre as subunidades α e
β da (Na++K+)ATPase, é o α1β1, pois correspondente a mais de 90% da enzima
encontrada neste tecido (Chow e Forte, 1995; Feraille e Doucet, 2001; Jorgensen
e Pedersen, 2001).
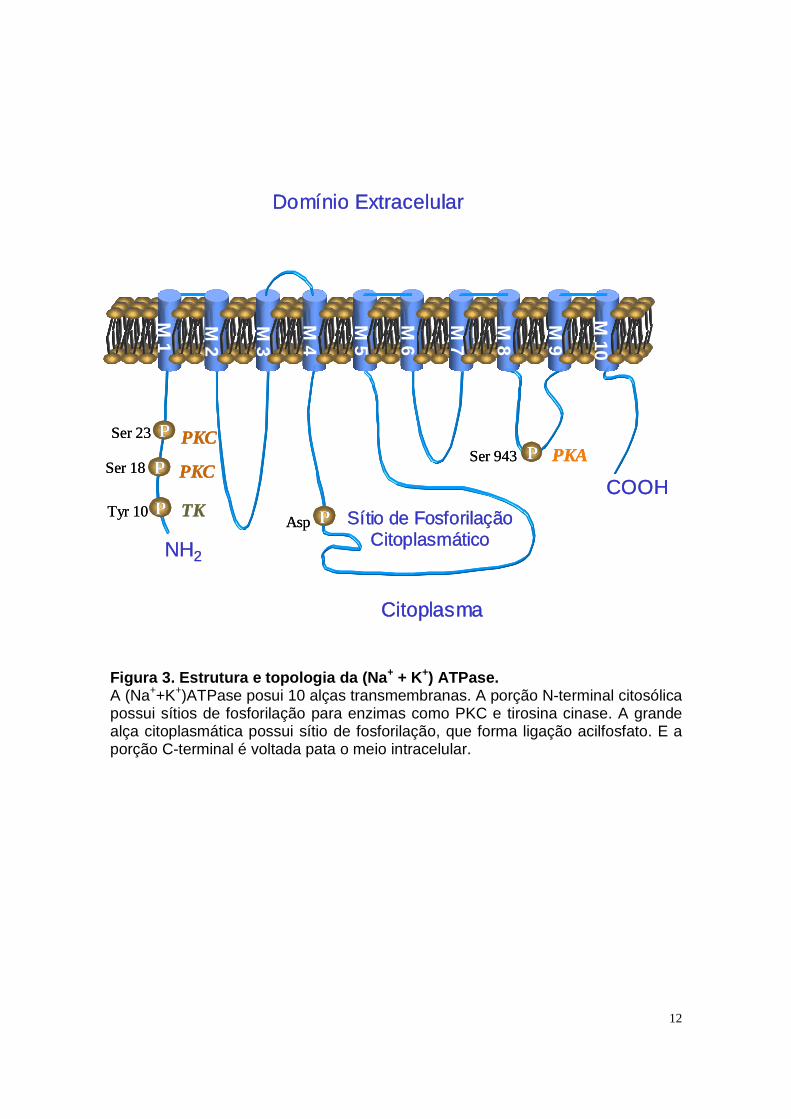
12
Figura 3. Estrutura e topologia da (Na + + K+) ATPase. A (Na++K+)ATPase posui 10 alças transmembranas. A porção N-terminal citosólica possui sítios de fosforilação para enzimas como PKC e tirosina cinase. A grande alça citoplasmática possui sítio de fosforilação, que forma ligação acilfosfato. E a porção C-terminal é voltada pata o meio intracelular.
Domínio ExtracelularDomínio Extracelular
COOHCOOH
NHNH22
Sítio de Sítio de FosforilaçãoFosforilaçãoCitoplasmáticoCitoplasmático
M1 2
M 1 M1 2
M 3 M1 2
M 5 M1 2
M 7 M1 2
M 9
M 2
M 4
M 6
M 8
M 10
CitoplasmaCitoplasma
PPAspTyr 10
Ser 18
Ser 23
PP
PP
PP PKCPKC
PKCPKC
TKTK
Ser 943 PP PKAPKA
Domínio ExtracelularDomínio Extracelular
COOHCOOH
NHNH22
Sítio de Sítio de FosforilaçãoFosforilaçãoCitoplasmáticoCitoplasmático
M1 2
M 1 M1 2
M 3 M1 2
M 5 M1 2
M 7 M1 2
M 9
M 2
M 4
M 6
M 8
M 10
CitoplasmaCitoplasma
PPAspTyr 10
Ser 18
Ser 23
PP
PP
PP PKCPKC
PKCPKC
TKTK
Ser 943 PP PKAPKA

13
Uma segunda atividade ATPásica dependente de sódio foi demonstrada
inicialmente em fatias de córtex renal, por meio de medida da extrusão de sódio e
água na ausência de potássio e na presença de 2 mM de ouabaína, inibidor da
(Na++K+)ATPase (Whittembury e Fishman, 1969; Whittembury e Proverbio, 1970).
Em 1975, PROVERBIO e colaboradores (Proverbio et al., 1975) propuseram o
envolvimento de uma enzima diferente da (Na++K+)ATPase na reabsorção de
sódio em cortex de rim de rato. A proposta baseava-se, inicialmente, na
insensibilidade da enzima à ouabaína e na sensibilidade ao ácido etacrínico e
furosemide (Whittembury e Proverbio, 1970). Posteriormente, PROVERBIO e DEL
CASTILLO (1981) demonstraram que a atividade Na+-ATPásica estava localizada
na membrana basolateral de túbulo proximal. Atualmente, sabe-se que esta
enzima está presente em diferentes tecidos animais e apresenta uma distribuição
similar à da (Na++K+)ATPase.
Os genes codificantes para Na+-ATPase foram clonados em plantas, algas,
protozoários e Saccharomyces cerevisiae (Wada et al., 1992; Ueno et al., 2000).
Contudo, nenhum gene codificante para esta enzima foi identificado em animais.
Apesar da existência em organismos tão distintos, os genes codificantes para a
Na+-ATPase apresentam características básicas similares e por este motivo
pertencem a família ENA (Haro, et al., 1991). Em muitos dos organismos inferiores
que expressam a Na+-ATPase não se observa a expressão da (Na++K+)ATPase,
sugerindo que a primeira apresente uma origem filogenética anterior à
(Na++K+)ATPase (Popova et al., 1998; Suzuki et al., 2005; Iizumi et al., 2006). Tem
sido sugerido que a conservação da Na+-ATPase durante a evolução esteja
associada à capacidade e necessidade de diversos organismos se adaptarem ao
estresse salino. Esta hipótese é confirmada pela observação de que a
Na+-ATPase está envolvida na regulação do volume celular durante estresse
osmótico em diferentes organismos (Proverbio et al., 1989; Iizumi et al., 2006).
A associação entre atividade Na+-ATPásica insensível à ouabaína e
transporte de sódio foi demonstrada em vesículas “inside-out” de células epiteliais
de intestino de cobaia (Del Castillo et al., 1982) e em membrana basolateral de
células de túbulo proximal de rim de porco (Marin et al., 1985). Nesses trabalhos,

14
os autores demonstraram que a atividade ATPásica é insensível a potássio,
inibida por furosemide, e insensível a ouabaína em concentrações de até 7 mM.
Nestas condições, a captação de sódio mediada pela (Na++K+)ATPase é
completamente inibida (Marin et al., 1989).
Diversos estudos têm demonstrado diferenças bioquímicas importantes
entre a clássica (Na++K+)ATPase e Na+-ATPase, tais como, dependência de
cátion; dependência de substrato; sensibilidade a inibidores; pH ótimo;
temperatura ótima; sensibilidade a SDS e tripsina (Proverbio et al., 1986;
Proverbio et al., 1989; Caruso-Neves et al., 1997). Além disso, as enzimas
também apresentam diferenças na regulação por hormônios, autacóides e etanol
em células de mamíferos (Proverbio et al., 1986; Proverbio et al., 1989; Caruso-
Neves et al., 1997; Rangel et al., 2002).
O fato da (Na++K+)ATPase e Na+-ATPase terem distribuição paralela e a
atividade da segunda ser 10 vezes menor que a atividade da primeira, torna difícil
o isolamento da Na+-ATPase. Com o objetivo de caracterizar a Na+-ATPase de
túbulo proximal de rim de porco, nosso grupo realizou um trabalho recentemente,
onde a (Na++K+)ATPase foi imunoprecipitada e as atividades das bombas foram
medidas no sobrenadante. Após a imunoprecipitação da (Na++K+)ATPase, com
anticorpo monoclonal contra isoforma α1 (isoforma presente no tecido renal), a
atividade da Na+-ATPase remanescente no sobrenadante foi maior que 95%,
enquanto a atividade da (Na++K+)ATPase foi menor que 5% (De Souza et al.,
2007). Nestas condições, a adição de 120 mM de NaCl aumentou em duas vezes
o nível do intermediário fosforilado (E-P, ligação acilfosfato), formado pela
fosforilação por ATP, como observado na Figura 4, que ilustra o ciclo catalítico
proposto para a Na+-ATPase (De Souza et al., 2007). A ligação acil-fosfato foi
insensível a 100mM de KCl e inibida por vanadato e hidroxilamina. O peso
molecular do intermediário fosforilado (E-P), determinado por eletroforese em gel
ácido, foi de 100 kDa. Uma característica bem estabelecida das P-ATPases é sua
fosforilação por Pi formando E-P em condições específicas, através de uma
reação denominada “backdoor phosphorylation”. THOMAS e colaboradores (2003)
demonstraram que a Na+-ATPase de membrana de eritrócitos é fosforilada por Pi,

15
apresenta peso molecular de 100 kDa, e os valores de fosfoproteína aumentam na
presença de furosemide, um inibidor desta enzima. O mesmo peso molecular foi
encontrado para a Na+-ATPase de organismos inferiores, tais como: Tetraselmis
viridis (Popova, Balnokin et al., 1998), Exiguobacterium aurantiacum (Suzuki et al.,
2005) e a recentemente clonada enzima de Trypanosoma cruzi (Iizumi et al.,
2006). Estes dados demonstram que a Na+-ATPase é uma entidade enzimática
independente da (Na++K+)ATPase. Esta proposta é reforçada pela observação de
que membranas de glóbulos vermelhos de ovelha Ovies aries, sem atividade
(Na++K+)ATPase, têm uma atividade ATPásica estimulada por sódio e insensível a
ouabaína (Proverbio et al., 1989).
Uma das características da Na+ATPase é sua completa inibição por
furosemide, um inibidor característico do co-transportador Na+/K+/2Cl– (NCKK) e
sua insensibilidade à ouabaína, o clássico inibidor da (Na++K+)ATPase. Uma vez
que tem sido observado que isoformas da (Na++K+)ATPase possuem diferentes
sensibilidades à ouabaína, seria plausível imaginar que a ausência da inibição da
ouabaína seria devida à isoforma estudada. Entretanto, 1 mM de ouabaína é
capaz de inibir completamente todas as isoformas de (Na++K+)ATPase. Estas
observações, associadas ao fato de que a atividade Na+-ATPásica não é inibida
por concentrações de ouabaína até 10 mM descartam a participação da
(Na++K+)ATPase na atividade sensível à furosemide (Blanco e Mercer, 1998).
Acreditamos que a sensibilidade da atividade ATPásica a furosemide não pode ser
relacionada a inibição do co-transportador, pois NCKK: a) não hidrolisa ATP; b)
não forma intermediário fosforilado (E-P) com ligação acil-fosfato sensível a
hidroxilamina; d) seu peso molecular é de 170kDa. Além disso, o possível efeito
de furosemide em outras ATPases pode ser descartado, pois foi observado que a
furosemide não modula as atividades (Na++K+)ATPase, Mg2+-ATPase and Ca2+-
ATPase de diferentes tecidos (Caruso-Neves et al., 2002). Outra possibilidade
seria a sensibilidade de alguma atividade ecto-ATPásica sensível a furosemide.
Entretanto, estudos têm demonstrado que atividades ecto-ATPásicas não são
inibidas por furosemide nem estimuladas por sódio (Junior et al., 2005; De Souza,

16
et al., 2007). Portanto, podemos considerar furosemide um inibidor específico para
a Na+-ATPase.
Duas importantes características que distinguem a Na+-ATPase da
(Na++K+)ATPase são afinidade por sódio e a razão entre as duas atividades em
diferentes tecidos. A afinidade aparente da Na+-ATPase para sódio é cerca de
duas vezes maior do que a da (Na++K+)ATPase, enquanto a atividade da primeira
é um décimo da segunda (Proverbio 1981). Estes dados sugerem um possível
papel para a Na+-ATPase no ajuste fino da reabsorção de sódio, cabendo à
(Na++K+)ATPase o transporte em massa de sódio.
A relação entre a Na+- ATPase e a excreção renal de sódio foi inicialmente
proposta por MALNIC e colaboradores (1969) (Malnic et al., 1969). Eles
observaram que furosemide inibe a reabsorção de sódio no túbulo próximal,
resultando em aumento na excreção renal de sódio. Apesar dos estudos sobre a
Na+-ATPase terem iniciado há mais de 30 anos, apenas na última década alguns
grupos têm se dedicado ao estudo da modulação desta enzima por hormônios e
autacóides, buscando entender seu papel na reabsorção renal de sódio (Moretti et
al., 1991; Thomas e Qian, 2003; Beltowski et al., 2004; Czaplinski et al., 2005).
Nosso grupo tem demonstrado que diferentes hormônios e autacóides modulam a
Na+-ATPase resultando em modificações na excreção renal de sódio (Caruso-
Neves et al., 1997; Caruso-Neves et al., 2004; Caruso-Neves, et al., 1997). Uma
possível relação entre hipertensão e a atividade Na+-ATPase tem sido investigada.
MARIN e colaboradores (1989), observaram aumento da atividade Na+-ATPase e
do volume extracelular em animais alimentados com uma dieta alta de sódio por
quatro meses (Yao et al., 1997). Cabe destacar que não foi detectada alteração da
atividade (Na++K+)ATPásica nesses animais. Nosso grupo demonstrou
recentemente que a atividade Na+-ATPásica em túbulo proximal de animais
espontâneamente hipertensos é significativamente maior do que a observada em
animais normotensos (Silva, 2007; Madeira, 2008), embora nenhuma alteração na
atividade (Na++K+)ATPásica tenha sido detectada. Por este motivo postulamos
que a Na+-ATPase é um alvo importante para a modulação da excreção renal de
sódio por hormônios e autacóides.
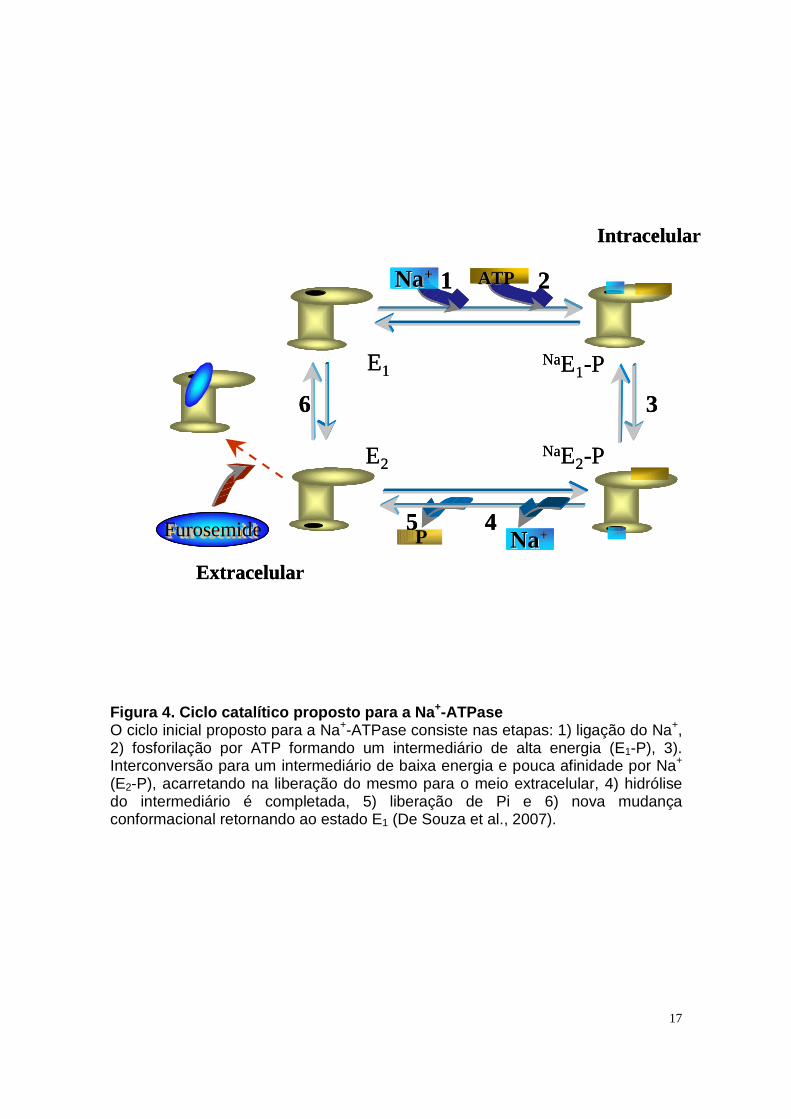
17
Figura 4. Ciclo catalítico proposto para a Na +-ATPase O ciclo inicial proposto para a Na+-ATPase consiste nas etapas: 1) ligação do Na+, 2) fosforilação por ATP formando um intermediário de alta energia (E1-P), 3). Interconversão para um intermediário de baixa energia e pouca afinidade por Na+
(E2-P), acarretando na liberação do mesmo para o meio extracelular, 4) hidrólise do intermediário é completada, 5) liberação de Pi e 6) nova mudança conformacional retornando ao estado E1 (De Souza et al., 2007).
Intracelular
FurosemideFurosemideFurosemide
Na Na + + ATP ATP
Na Na + +
E 1
E 2 Na E 2 - P
Na E 1 - P
Extracelular
1 2
3
4 5
6
Intracelular
FurosemideFurosemideFurosemideFurosemideFurosemideFurosemide
Na Na + + ATP ATP ATP ATP
Na Na + + Na Na + + Pi
E 1
E 2 Na E 2 - P
Na E 1 - P
Extracelular
1 2
3
4 5
6

18
1.3. O rim como alvo e produtor de compostos moduladores da função renal
Existem diversos sistemas capazes de modular a função renal. Como
exemplo temos o sistema renina-angiotensina (SRA), cuja ativação inicia-se pelo
aumento da liberação de renina, principalmente pelas células justaglomerulares
(células modificadas da arteríola aferente). A renina catalisa a clivagem
proteolítica do angiotensinogênio, uma γ-globulina plasmática, gerando o
decapeptídeo angiotensina I (Ang I), principal precursor dos demais peptídeos
derivados do angiotensinogênio. A angiotensina I sofre posterior hidrólise podendo
gerar angiotensina II (Ang II) pela ação da Enzima Conversora de Angiotensinas
(ECA). A Ang II pode ser metabolizada em outros peptídeos com importante
atividade biológica. Estes peptídeos modulam o ritmo de filtração glomerular, a
hemodinâmica renal e a reabsorção de soluto e solvente em diferentes segmentos
do néfron, incluindo o túbulo proximal (Caruso-Neves et al., 2002; Caruso-Neves
et al., 2003; Caruso-Neves et al., 2004). Por outro lado, além de alvo destes
diferentes peptídeos de angiotensina, o rim também é capaz de produzí-los
através de um SRA local. Os peptídeos gerados neste caso terão uma ação
parácrina ou mesmo autócrina.
Além do SRA local, outros sistemas peptídicos também são produzidos no
rim, como o sistema de peptídeos natriuréticos e o sistema calicreína-bradicinina.
A urodilatina é um peptídeo com ação diurética e natriurética, possui estrutura
semelhante ao ANP (peptídeo atrial natriurético), com apenas 4 aminoácidos a
mais que o ANP. Esse peptídeo é produzido no túbulo distal (Ogawa et al., 1999;
Beltowski e Wojcicka, 2002), e tem sido proposto que pode agir como um
composto parácrino modulando o transporte de sódio nos segmentos adjacentes,
como o túbulo proximal. A ligação de urodilatina a receptores de ANP em
membranas luminal e basolateral de túbulo proximal de rato já foi demonstrada e
sua administração aumenta a excreção fracional de sódio (Endlich et al., 1995).
A bradicinina pertence a um grupo de peptídeos chamados cininas. A
infusão de bradicinina na artéria renal induz natriurese sem alterar o rítmo de
filtração glomerular, sugerindo um efeito direto no transporte tubular de sódio

19
(Flamenbaum et al., 1979; Siragy, 1993). Foi demonstrado pelo nosso grupo, que
a bradicinina inibe a Na+-ATPase de túbulo proximal e reverte o efeito
estimulatório promovido por angiotensina 1,7 (Caruso-Neves et al., 2003).
O rim ainda dispõe de uma maquinaria de produção de compostos de
diferente natureza que irão modular sua função de maneira parácrina ou autócrina.
Entre eles podemos citar o sistema purínico, no qual ATP e adenosina aparecem
como os principais compostos ativos.
1.4. ATP como fonte de adenosina renal: mecanismo de secreção de ATP
Os nucleotídeos extracelulares de adenina, em particular o ATP, liberados
por terminações simpáticas renais, células endoteliais renais, células musculares
lisas vasculares e células epiteliais renais, podem ser importantes precursores de
adenosina gerada no ambiente extracelular por ação de ecto-enzimas (Jackson e
Dubey, 2001).
A concentração citosólica de ATP situa-se em torno de 3-5 mM, podendo
chegar a 10 mM (Vallon, 2008). Já a concentração extracelular de nucleotídeos
pode variar de 0,1 a 10 µM o que representa um gradiente de concentração de
ATP favorável à sua saída da célula. No túbulo proximal a concentração
extracelular de ATP em condições basais é de aproximadamente 1µM podendo
atingir a concentração de 5-10 µM em condições de hipotonicidade. Estas
observações sugerem que o ATP poderia ter um papel autócrino e/ou parácrino de
maneira direta neste segmento tubular, através de seus receptores específicos,
ou, mesmo, sendo fonte primária para a formação de adenosina. Apesar do
gradiente de concentração, não ocorre difusão de nucleotídeos através da
membrana plasmática, uma vez que estes compostos apresentam carga negativa
no pH intracelular.
Os mecanismos de transporte descritos para a liberação de ATP celular
são: 1) lise celular 2) exocitose de vesículas contendo ATP; 3) transporte mediado
por transportadores ou canais (Figura 5) (Burnstock, 2006).

20
A liberação de ATP por lise celular é consequência de uma injúria tecidual
que resulta em morte celular por necrose (Sabirov e Okada, 2004). Este
mecanismo não é específico ou modulado diretamente por compostos que
induzem a liberação de ATP. O ATP liberado por este processo pode contribuir,
entre outras coisas, com o processo de inflamação tecidual (Erb et al., 2006).
A excitose de ATP através de vesículas é importante em neurônios, células
neuroendócrinas e plaquetas (Schwiebert e Zsembery, 2003). Células excitáveis
possuem vesículas secretórias contendo ATP e neurotransmissores clássicos
(acetilcolina em neurônios e noradrenalina em neurônios e células cromafins).
Uma vez deflagrado o estímulo, ocorre a exocitose das vesículas, liberando seu
conteúdo no meio extracelular (Pangrsic et al., 2006). Estudos realizados em
astrócitos demonstram que o transporte das vesículas contendo ATP para a
membrana plasmática é dependente de cálcio e proteínas do citoesqueleto
(Potokar et al., 2007). Entretanto, este mecanismo de liberação de ATP não é
descrito para células epiteliais como aquelas presentes no túbulo proximal (Vallon,
2008).
Os canais de ânions são importantes candidatos a mediadores do
transporte de ATP para o meio extracelular em células epiteliais polarizadas. Os
hemicanais de conexina, bem como os transportadores do tipo ABC (do inglês
“ATP-binding cassette”), também podem participar deste processo (Komlosi et al.,
2005). Cada hemicanal é constituído por seis conexinas integrais de membrana,
que são dispostas em arranjo hexagonal formando um canal (Harris e Bevans,
2001). Os hemicanais das junções comunicantes encontram-se fechados em
condições normais e só abrem após dois heminacanais se acoplarem, formando
um canal hidrofílico entre o interior de duas células, estabelecendo assim uma
comunicação intercelular. Entretanto, existem várias isoformas de conexinas, e
algumas delas formam canais que não têm apenas a função de comunicação
intercelular. Neste caso estas isoformas de conexinas (por exemplo, Cx46 e Cx50)
formam hemicanais abertos, que permitem a liberação de íons, NAD+ e ATP para
o meio extracelular (Cotrina et al., 1998; Bruzzone et al., 2001). Além disso, a
abertura dos canais de conexina é dependente de cálcio e eles são importantes na

21
propagação das ondas de cálcio intercelulares (Toyofuku et al., 1998; Scemes et
al., 2000). É descrito na literatura que em astrócitos, conexinas, como a Cx43,
podem formar hemicanais abertos, que, estimulados por estresse mecânico
liberam ATP (Scemes et al., 2000; Yamamoto et al., 2000), assim como células
endoteliais liberam ATP, através de hemicanais, em resposta a estresse causado
por estiramento (Yamamoto et al., 2000). Existem alguns trabalhos, em ratos,
demonstrando a expressão contínua de hemicanais de conexina (Cx30) ao longo
da membrana luminal do segmento fino ascendente da alça de Henle até o duto
coletor, com maior nível de expressão no túbulo distal (Mcculloch et al., 2005). A
expressão em camundongos e coelhos é menor que em ratos e limitada às células
intercalares do duto coletor (Mcculloch et al., 2005). Entretanto, não existem
evidências conclusivas para o papel fisiológico de hemicanais de conexina na
liberação de ATP nestes segmentos (Lazarowski et al., 2003; Vallon, 2008). Não
existem evidências sobre a expressão de hemicanais no túbulo proximal.
Os transportadores ABC compõem uma super família de transportadores
que contêm um domínio de ligação e hidrólise de ATP, altamente conservado, e
transportam ativamente diversos solutos hidrofóbicos e hidrofílicos através da
membrana plasmática (Sabirov e Okada, 2004; Hollenstein et al., 2007). MDR (do
inglês “multidrug resistance transporter”), CFTR (do inglês “cystic fibrosis
transmembrane condutance regulator”) e canal maxi ânion pertencem a esta
família de transportadores. O canal MDR desempenha um papel importante na
defesa do organismo contra xenobióticos, seus metabólitos e outros produtos,
através da excreção destes compostos na bile, lumen intestinal e urina. A
“multidrug resistance 1/P-glycoprotein” (MDR1/Pgp) e as “multidrug resistance
proteins” (MRPs) são ATPases presentes na membrana plasmática, capazes de
transportar solutos com ou sem carga. No intestino, os canais MDR1/Pgp limitam
a bioatividade de drogas e outros compostos através do transporte destas
moléculas dos enterócitos para o lúmen do intestino. Nos hepatócitos e no túbulo
proximal, estes transportadores contribuem para a excreção de drogas através da
bile e urina, respectivamente (Van De Water et al., 2007). Diferentes isoformas de
MRPs (MRD1, MRD2 e MRD4) são expressas na membrana de borda em escova

22
no túbulo proximal, e desempenham um importante papel na excreção urinária de
ânions orgânicos (Van Aubel et al., 2002; Masereeuw et al., 2003). Além disso,
existem evidências demonstrando que o canal MDR1/Pgp é capaz de transportar
ATP em hepatócitos e em células do epitélio intestinal. Entretanto, o papel
fisiológico desta proteína sobre a permeabilidade celular ao ATP ainda deve ser
estudado. Não existem evidências diretas demonstrando que os canais MDR1/Pgp
transportem ATP no túbulo proximal.
O CFTR funciona como canal de cloreto estimulado por AMPc e tem baixa
condutância (Kim et al., 2007). Diversas evidências demonstram o papel de CFTR
na secreção de cloreto pelas células do túbulo distal, células principais do duto
coletor cortical e medular (Schwiebert e Zsembery, 2003). Em relação ao
transporte de ATP, sua função tem sido motivo de controvérsia. De fato, o
aumento da atividade de CFTR aumenta a condução de ATP (Reisin et al., 1994;
Schwiebert e Zsembery, 2003). Entretanto, tem sido proposto que o CFTR não
seja apenas um canal para cloreto e ATP, mas o regulador de um outro canal
condutor de ATP (Sugita et al., 1998; Braunstein, Roman et al., 2001). Não
existem evidências demonstrando o envolvimento de CFTR na permeabilidade de
ATP no túbulo proximal.
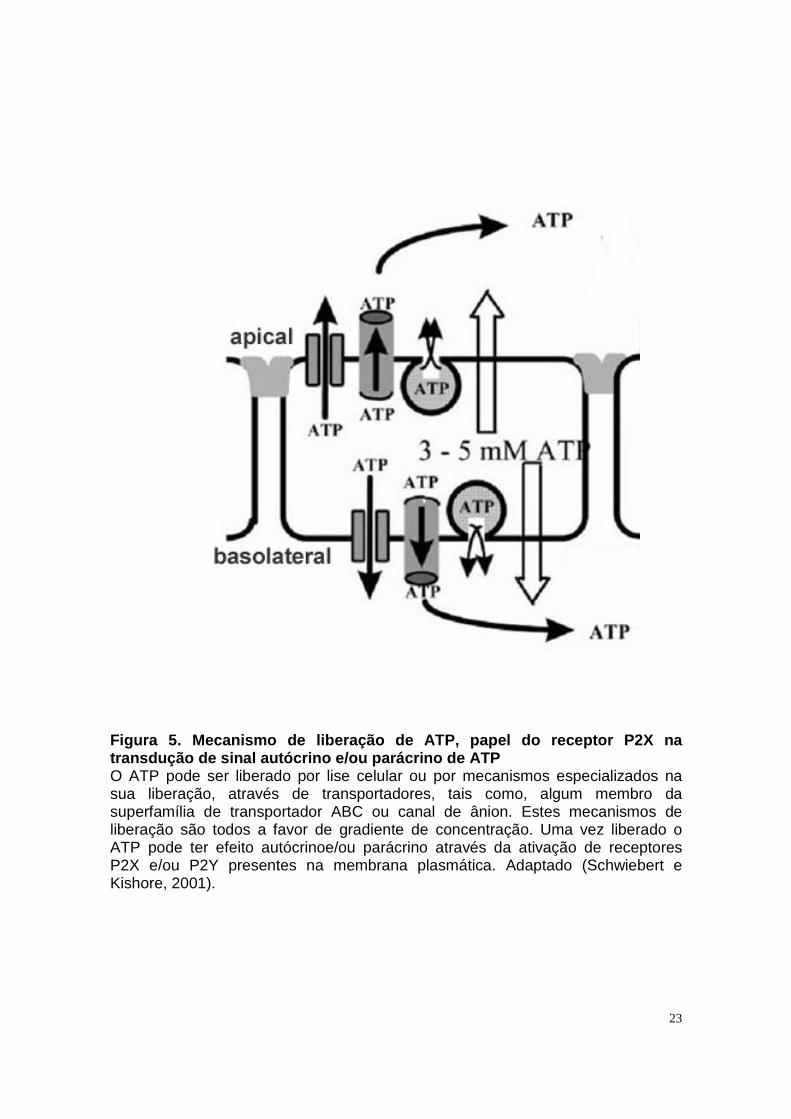
23
Figura 5. Mecanismo de liberação de ATP, papel do r eceptor P2X na transdução de sinal autócrino e/ou parácrino de ATP O ATP pode ser liberado por lise celular ou por mecanismos especializados na sua liberação, através de transportadores, tais como, algum membro da superfamília de transportador ABC ou canal de ânion. Estes mecanismos de liberação são todos a favor de gradiente de concentração. Uma vez liberado o ATP pode ter efeito autócrinoe/ou parácrino através da ativação de receptores P2X e/ou P2Y presentes na membrana plasmática. Adaptado (Schwiebert e Kishore, 2001).

24
O Canal maxi-ânion é uma variação do canal de ânion voltagem
dependente (VADC) presente na membrana externa das mitocôndrias. O canal
VADC é responsável pelo transporte de ATP para o citosol e ADP para a matriz
mitocondrial (Rostovtseva e Colombini, 1996). Estudos recentes de eletrofisiologia
têm sugerido que uma isoforma de VADC, chamado canal maxi ânion, esteja
presente em membrana plasmática de células epiteliais, transportando ATP para o
meio extracelular, (Okada et al., 2004).
Recentemente foi demonstrado por BELL e colaboradores (2003) que
alterações na concentração de NaCl luminal nas células da mácula densa,
promove a liberação de ATP através de canais maxi ânion presentes na
membrana basolateral. Os autores propõem que este mecanismo de liberação de
ATP esteja envolvido no balanço tubuloglomerular (Bell et al., 2003; Komlosi et al.,
2004). O ATP pode ser liberado passivamente, a favor do gradiente de
concentração, por células epiteliais polarizadas, através deste canal de ânion.
Estes canais estão presentes nas membranas luminal e basolateral de células de
túbulo distal e são sensíveis a DIDS, um bloqueador de canal de ânions
(Braunstein et al., 2001; Schwiebert e Zsembery, 2003).
O túbulo proximal é o segmento com maior concentração de ATP luminal,
entretanto não existem estudos sobre o seu mecanismo de liberação. Portanto,
existe uma questão em aberto: será que o ATP presente no lúmen do túbulo
proximal é proveniente do filtrado glomerular ou é secretado pelas células do
túbulo proximal? Para responder esta pergunta, um grupo de pesquisadores
mediu a concentração luminal de ATP na porção S2 do túbulo proximal de ratos
Munich-Wistar e observou que a concentração de ATP era 4 vezes maior que no
espaço da cápsula de Bowman (Vekaria et al., 2006). Como a reabsorção de água
entre estes dois segmentos do néfron não seria suficiente para promover esse
aumento na concentração de ATP, os dados indicam que as células do túbulo
proximal secretam ATP para lúmen. Outra questão em aberto é a seguinte: qual
seria o mecanismo de secreção de ATP nestas células. Apesar de vários
mecanismos de secreção serem descritos para diferentes tipos celulares, como

25
discutido anteriormente, nada é conhecido até o momento sobre a secreção de
ATP pelas células do túbulo proximal.
Diversos estímulos levam à liberação de ATP, como por exemplo:
hipotonicidade (Sabirov e Okada, 2004); aumento de cálcio intracelular
(Boudreault e Grygorczyk, 2004); aumento do nível celular de AMPc (Prat et al.,
1996). Algumas condições fisiopatológicas, também estimulam a liberação de
ATP, tais como hipóxia, estiramento tecidual, perda da viabilidade celular e lise
celular (Leipziger, 2003).
1.5.Metabolismo renal de adenosina
Uma vez no meio extracelular, o ATP pode ligar-se a receptores
específicos, como será discutido na próxima seção, ou ser metabolizado por ecto-
enzimas. No rim, o primeiro dado indicando que túbulos isolados eram capazes de
hidrolisar o ATP extracelular foi publicado em 1972 (Sauer et al., 2000).
Posteriormente, foi observada a presença da atividade ecto-ATPase nas
membranas basolateral e luminal do túbulo proximal (Schulze-Lohoff et al., 1998)
O conceito de ecto-enzimas não é novo. Entretanto, estudos mais
detalhados acerca desta classe de proteínas são recentes. Tem sido descrita a
existência de um número considerável de enzimas de membrana com o sítio
catalítico voltado para o lado extracelular (Vekarian et al., 2006). Essas enzimas
modulam os níveis de purinas extracelulres, modificando seus efeitos sobre suas
funções biológicas (Jackson e Dubey, 2001; Burnstock, 2006).
Os estudos iniciais sobre estas enzimas foram muito lentos por diferentes
razões. Quando a idéia de ecto-enzima foi primeiramente proposta, a avaliação da
orientação das proteínas de membrana era inadequada para definir se o sítio ativo
estava realmente localizado do lado de fora da célula (Zimmermann, 2000). Além
disso, existia grande dificuldade em aceitar a idéia de que uma ecto-enzima
poderia utilizar um substrato muito abundante no meio intracelular como o ATP,
uma molécula envolvida em funções energéticas na célula, mas que poderia ter
funções altamente especializadas e diferentes quando presente no lado de fora da
célula (Abbracchio e Burnstock, 1998).

26
Atualmente, conhecem-se quatro famílias de ectonucleotidases conhecidas:
ectonucleotideo pirofosfatase/fosfodiesterase (NPP1-NPP5); ecto-nucleosideo
trifosfato difosfo-hidrolase (NTPDase1-NTPDase8); ecto-5’nucleotidase e
fosfatase alcalina (Zimmermann, 2000; Vekaria et al., 2006). Estas famílias de
enzimas diferem entre si nas suas atividades hidrolíticas e na afinidade por
nucleotídeos.
Os membros da família NPP são capazes de hidrolizar ATP e ADP e
possuem afinidades distintas para os nucleotídeos. Além disso, uma característica
desta família é a capacidade de hidrolizar ligações fosfodiéster de ácidos
nucléicos. Estudos de imunohistoquímica demonstraram alto nível de expressão
de NPP1 na membrana basal de células de túbulo distal de camundongo e baixo
nível de expressão no túbulo proximal (Vekaria et al., 2006). A presença do RNAm
da isoforma NPP2 foi detectada no rim de rato (Vekaria et al., 2006). A isoforma
NPP3 foi demonstrada no glomérulo e no túbulo proximal de rato (Edwards et al.,
1999). As isoformas de proteínas NPP4 e NPP5 foram descritas recentemente e
não existem evidências da sua presença no rim (Gijsbers et al., 2001).
Dos oito membros da família NTPDase, apenas as isoformas 1, 2, 3 e 8
hidrolizam nucleotídeos no meio extracelular. As isoformas 4-7 estão localizadas
nas membranas intracelulares de organelas, como aparelho de Golgi e retículo
endoplasmático (Wang et al., 1998; Trombetta e Helenius, 1999; Braunr et al.,
2000). Apesar de todos os membros da família de NTPDase hidrolizarem ATP e
ADP eles diferem na afinidade e capacidade de hidrólise de seus substratos. A
NTPDase1 tem afinidades similares para ATP e ADP, é expressa
predominantemente nos glomérulos, no espaço peritubular e, em baixo nível, no
duto coletor medular interno (Zimmermann, 2000). A NTPDase2 tem afinidade 30
vezes maior para ATP que para ADP, está presente no segmento espesso da alça
de Henle, no túbulo distal e no duto coletor medular interno (Vekaria et al., 2006).
A NTPDase3 tem afinidade 3 vezes maior para ATP que para ADP, é expressa na
alça de Henle, no túbulo distal e no duto coletor, bem como nas células
intercalares e principais (Kishore et al., 2005).

27
A ecto-5’nucleotidase defosforila AMP gerando adenosina e pode ser
encontrada ancorada nos fosfolipídeos de membrana plasmática. Estudos de
imunohistoquímica demonstraram alto nível de expressão desta enzima na
membrana de borda em escova no túbulo proximal e baixo nível de expressão na
membrana apical e no citoplasma das células intercalares do duto coletor, assim
como no espaço peritubular e nos glomérulos (Gandhir et al., 1990; Le Hir e
Kaissling, 1993). LE HIR e KAISSLING demonstraram maior atividade de ecto-
5’nucleotidase no ínicio do túbulo proximal, segmento S1, que nos segmentos S2
e S3 em rins de rato. Estes dados, em conjunto, indicam que o túbulo proximal
produz adenosina proveniente de ATP.
A fosfatase alcalina é capaz de hidrolizar ATP, ADP e AMP, é encontrada
ancorada nos fosfolipídeos de membrana luminal do túbulo proximal na maioria
dos mamíferos (Burnstock, 2006; Vallon, 2008). Esta característica permite usar a
fosfatase alcalina como um marcador de membrana luminal do túbulo proximal.
Os nucleotídeos formados por ação das ecto-enzimas descritas acima (ADP
e AMP) poderão ser metabolizados à adenosina através das vias descritas abaixo
e esquematizadas na Figura 6. No ambiente extracelular, ADP e AMP poderão
sofrer a ação da 5´ecto-nucleotidase e serem convertidos a adenosina (Vallon et
al., 2006).
Outra fonte renal para a produção de adenosina é o AMPc (adenosina 3’,5’-
monofosfato cíclico) (Inscho, 2001;Schwiebert e Kishore, 2001) (figura 6). Esta via
é independente do estado metabólico do tecido e pode variar dependendo da ação
de hormônios e autacóides que modulem os níveis de AMPc, via adenilato ciclase
ou fosfodiesterase, enzimas responsáveis pela síntese e degradação do
nucleotídeo, respectivamente. Desta forma, esta via poderia ser importante na
interação entre hormônios com o sistema purínico. Esta hipótese é favorecida pela
observação que adenosina pode modular o efeito de Ang II e bradicinina nos rins
(Caruso-Neves et al., 2003; Gomes et al., 2005). Esta via de produção de
adenosina pode ocorrer nos meios intracelular ou extracelular, pois o AMPc
poderia ser liberado da célula por transportador especifico e convertido a AMP por
ação de atividade ecto-fosfodiesterase (Jackson e Dubey, 2001). O papel renal do

28
sistema AMPc-adenosina na concentração de adenosina extracelular ainda não
está bem estabelecido.
Além do metabolismo extracelular de nucleotídeos purínicos, devemos
considerar também o metabolismo intracelular. No meio intracelular, o aumento do
consumo de ATP pode resultar em elevação de AMP que, por ação da enzima
5’nucleotidase, resulta em elevação dos níveis de adenosina. O mesmo efeito
pode ocorrer em decorrência de uma redução na fosforilação oxidativa do ADP.
Uma alternativa metabólica de utilização de adenosina é a desaminação oxidativa,
catalisada pela enzima adenosina desaminase, gerando inosina como produto.
Esta reação pode ser catalisada por enzimas intra ou extracelulares. Uma possível
via de formação de adenosina exclusiva do compartimento intracelular envolve a
reação reversível de condensação com a homocisteína, por ação da S-Adenosil
Homocisteína hidrolase. Esta via tem sido mostrada em cardiomiócitos (Vallon et
al., 2006), porém seu papel renal ainda não foi determinado.
A concentração extracelular de adenosina em todos os fluidos corporais é
constante em condições basais (30- 300nM), mas pode ser elevada a 10 µM ou
mais em condição de hipóxia (Hagberg et al., 1987; Tomanin et al., 1991).
Uma vez formada, a adenosina pode ser transportada do meio extracelular
para o meio intracelular e vice-versa. O transporte de adenosina através da
membrana plasmática de diferentes tipos celulares pode ser classificado como
concentrador ou equilibrador (Cass et al., 1999). O primeiro promove o transporte
do nucleosídeo contra o gradiente de concentração e depende do gradiente de
sódio. O transporte equilibrador transporta o nucleosídeo a favor do gradiente de
concentração, sem gasto energético (Cass et al., 1998). Deste modo, o aumento
do gasto energético ou a redução do suprimento de oxigênio podem resultar em
queda nos níveis de ATP e, conseqüentemente, aumento dos níveis intra e
extracelulares de adenosina. Isto se deve ao processo de transporte equilibrador,
capaz de promover a difusão de adenosina intracelular para o espaço extracelular,
como observado em situações de hipóxia.
Estudos sobre a variação nos níveis de adenosina em rins isquêmicos,
derivada da defosforilação seqüencial do ATP, demonstraram que a concentração

29
tecidual deste nucleosídeo aumenta significativamente durante os 10-30 minutos
iniciais de uma isquemia renal global (Millers et al., 1978; Osswald et al., 1978).
Depois deste período, ocorre uma diminuição do nível de adenosina devido,
provavelmente, à sua desaminação, catalisada pela adenosina deaminase, e não
pela sua fosforilação, catalisada pela adenosina cinase (Spielman e Thompson,
1982; Navar et al., 1999). Esta idéia é reforçada pela observação de PAWELCZYK
e colaboradores (1992) que demonstraram que a atividade de adenosina
desaminase é significativamente maior que a atividade de adenosina cinase em
glomérulo e túbulos corticais renais.
Os mecanismos descritos até aqui permitem concluir que a razão entre os
níveis intra e extracelulares de ATP e adenosina depende de um balanço entre as
atividades de diferentes enzimas e da disponibilidade de oxigênio. Os efeitos
fisiológicos e fisiopatológicos destas purinas são mediados por receptores
específicos.

30
Intracelular Extracelular
HYPO
5` IMP
Gs
AC
ATP
ADPAMPc AMPc
5`AMP 5`AMP
HYPO
ADEADE
INOINO
ADENOSINA
SAH
ADENOSINA
AMPdeaminase
PNP
ADA
PNP
ADOQuinase
PDE
5`NT
PRT
ATP Ecto -ATPase
ADP
Ecto -ADPase
ECTO-5’NT
ECTO-PDE
ECTO-ADA
SAHhidrolase
Intracelular Extracelular
HYPO
5` IMP
Gs
AC
ATP
ADPAMPc AMPc
5`AMP 5`AMP
HYPO
ADEADE
INOINO
ADENOSINA
SAH
ADENOSINA
AMPdeaminase
PNP
ADA
PNP
ADOQuinase
PDE
5`NT
PRT
ATP Ecto -ATPase
ADP
Ecto -ADPase
ECTO-5’NT
ECTO-PDE
ECTO-ADA
SAHhidrolase
Intracelular Extracelular
HIPO
5` IMP
Gs
AC
ATP
ADPAMPc AMPc
5`AMP 5`AMP
HIPO
ADEADE
INOINO
ADENOSINA
SAH
ADENOSINA
AMPdeaminase
PNP
ADA
PNP
ADOQuinase
PDE
5`NT
PRT
ATP Ecto -ATPase
ADP
Ecto -ADPase
ECTO-5’NT
ECTO-PDE
ECTO-ADA
SAHhidrolase
Figura 6 Esquema do metabolismo de adenosina nos es paços intra e extracelular. ADA, adenosina deaminase; ADE, adenina; INO, inosina; HYPO, hipoxantina; 5´NT, 5´nucleotidase; PDE, fosfodiesterase; PNP, Purina nucleosídeo fosforilase; PRT, purina ribosilfosfotransferase; SAH, S-adenosil homocisteína hidrolase.

31
1.6. Receptores de nucleotídeos e nucleosídeos
O papel das purinas como moléculas de sinalização extracelular foi
proposto inicialmente por Drury e Szent-Györgyi em 1929, quando demonstraram
que adenosina e adenosina 5`- monofosfato (AMP), extraídas de músculo
cardíaco, promoviam efeitos biológicos como dilatação arterial, diminuição da
pressão sanguínea e inibição da contração intestinal. GILLESPIE, em 1934,
estudou a relação entre estrutura e atividade da adenosina e de nucleotídeos de
adenina, mostrando que a deaminação reduzia fortemente a atividade
farmacológica da adenosina, enquanto a remoção do fosfato dos nucleotídeos
influenciava não apenas a potência, mas também o tipo de resposta. Os
resultados demonstraram, ainda, que a remoção do fosfato aumentava a
capacidade dos compostos de adenina de induzir hipotensão e vasodilatação. Por
outro lado, o ATP aumentava a pressão sanguínea em ratos e coelhos, efeito que
não havia sido observado anteriormente com AMP ou adenosina. O ATP foi ainda
relacionado a um efeito indutor da contração do útero mais potente que o AMP e a
adenosina. Estes dados, em conjunto indicavam que os efeitos de adenosina e de
nucleotídeos de adenina seriam mediados por receptores distintos. Esta hipótese
foi confirmada após a clonagem dos receptores de purinas (Burnstock, 2007).
Atualmente, estes receptores, denominados purínicos, são classificados em
dois grupos: receptores P1, nos quais a adenosina é o principal ligante endógeno,
e receptores P2, que ligam preferencialmente ATP, ADP e AMP (Abbracchio e
Burnstock, 1994) (Figura 7). A classificação atual dos receptores de purinas é
baseada, principalmente, em critérios moleculares e farmacológicos. Os
receptores P1 são subdivididos em A1, A2 e A3, enquanto os receptores P2 são
subdivididos em P2X e P2Y.
Inicialmente, foi demonstrado que a ativação dos receptores A1 e A2
provocava inibição e ativação da adenilato ciclase, respectivamente, em culturas
de células de cérebro de camundongo (Van Calker et al., 1979). E estes foram os
primeiros receptores de adenosina clonados, inicialmente chamados de RDC7
(Libert et al., 1990a) e RDC8 (Libert et al., 1990b). As propriedades básicas de

32
ligação com seus agentes farmacológicos e seus efeitos sobre a atividade da
adenilato ciclase, mostraram que RDC7 e RDC8 correspondiam aos já
caracterizados receptores A1 e A2, respectivamente.
Mais tarde, o receptor A2 foi subdividido em duas classes, em função da
diferença de afinidade pela adenosina. Os resultados demonstraram a existência
de sítios de alta (0,1-1µM) e de baixa afinidade (≥10µM) para adenosina. Sendo
assim, os dois sítios foram denominados receptores A2A e A2B, respectivamente
(Bruns et al., 1986). Evidências definitivas sobre a existência destes dois
receptores foram obtidas a partir da clonagem dos genes codificantes (Fredholm
et al., 2001). O receptor A3 foi descoberto mais recentemente. A sua
caracterização funcional, expressão e clonagem foram feitas a partir de estriato de
rato em 1992 por ZHOU e colaboradores (Zhou et al., 1992).
Baseado nos resultados acima, os receptores de adenosina podem ser
divididos em quatro subtipos: A1, A2A, A2B, e A3, e pertencem à grande família de
receptores acoplados à proteína G que, caracteristicamente, são constituídos por
sete domínios transmembrana. O domínio NH2 terminal destes receptores está
localizado no lado extracelular da membrana, enquanto o domínio COOH terminal
esta voltado para o lado intracelular (Figura 7). A terceira alça intracelular destes
receptores é o sítio de acoplamento com a proteína G (Olah e Stiles, 1995). Os
quatro subtipos de receptores foram identificados em várias espécies, inclusive em
humanos (Furlong et al., 1992; Townsend-Nicholson e Shine, 1992). Os
receptores humanos A1, A2a, A2b, e A3 são proteínas contendo 326, 412, 332, e
318 aminoácidos respectivamente (Linden, 2001). A análise das seqüências de
aminoácidos dos quatro subtipos de receptores de adenosina humanos revela
30% de identidade entre eles; a identidade chega a 45% ao compararmos os
domínios transmembrana.
Os receptores P2 são subdivididos em duas classes, P2X e P2Y, baseado
nas diferenças estruturais e nas vias de sinalização acopladas. P2X possui sete
subtipos conhecidos, P2X1-7, já foram clonados (Valera et al., 1994; Garcia-
Guzman et al., 1997; Garcia-Guzmanr et al., 1997; Le, Paquet et al., 1997;
Rassendren et al., 1997; Urano et al., 1997; Lynch et al., 1999), e possuem 30-

33
50% de identidade entre eles. São canais iônicos, compostos por dois domínios
transmembrana acoplados a uma grande alça extracelular e a uma pequena alça
intracelular (Benham e Tsien, 1987). Quando o ATP se liga na alça extracelular o
receptor é ativado e o canal é aberto, permitindo a passagem não seletiva de
cálcio e sódio para dentro da célula e potássio para fora.
Os receptores P2Y são subdivididos em oito subtipos P2Y1,2,4,6,11-14, todos
também já foram clonados (Ayyanathan et al., 1996; Communi et al., 1996;
Communi et al., 1997; Otero et al., 2000; Communi et al., 2001; Hollopeter et al.,
2001), e possuem entre 19 e 55% de identidade entre eles. São todos acoplados a
uma proteína G, com sete domínios transmembrana (Figura 7). Podem ser
acoplados a um ou mais subtipos de proteínas G (GS, Gi/0, Gq/11, e G12/13,)
(Abbracchio e Burnstock, 1998; Erb et al., 2006). A estrutura geral para os
receptores P2Y consiste em uma porção N-terminal voltada para o meio
extracelular, com vários sítios de glicosilação, e a porção C-terminal voltada para o
meio intracelular, com vários sítios de ligação e fosforilação por proteínas cinases
(Figura 7) (Abbracchio e Burnstock, 1998; Erb et al., 2006). Em geral, as células
de mamíferos expressam receptores de nucleotídeos junto com ecto-
nucleotidases, enzimas que degradam nucleotídeos extracelulares (Zimmermann,
2000). Este sistema representa um importante mecanismo de controle dos níveis
de ATP e, consequentemente, de seu efeito, como discutido anteriormente.
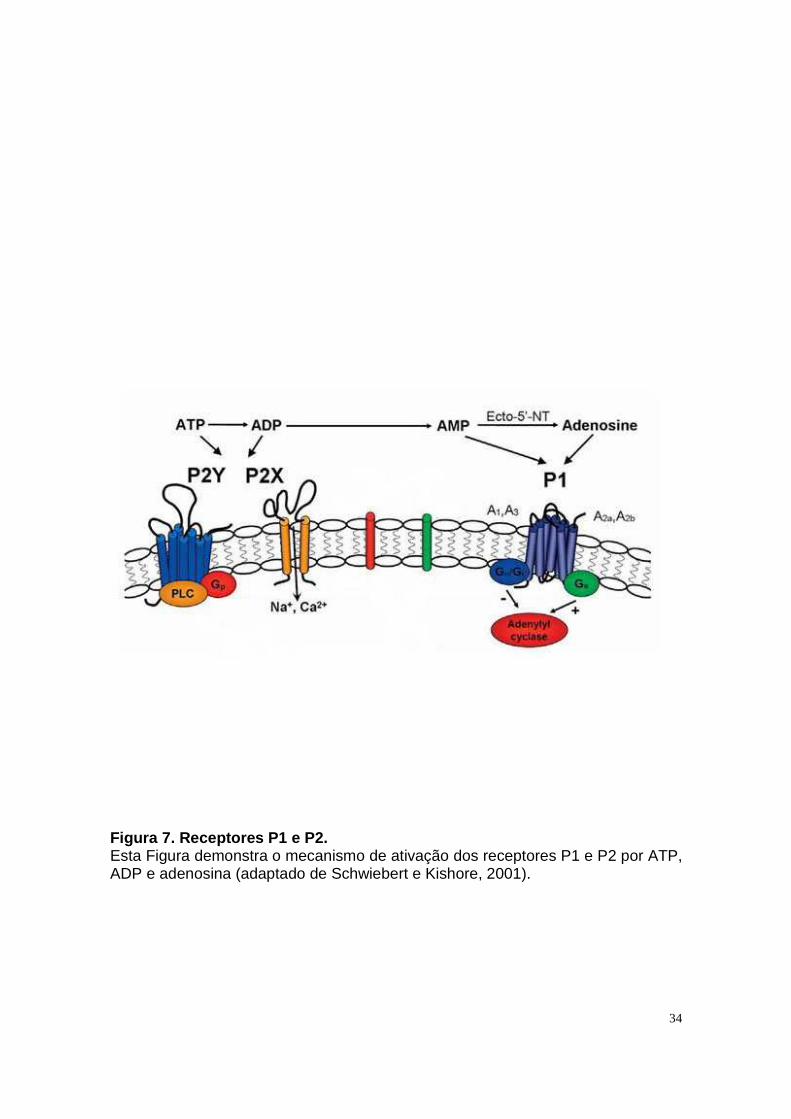
34
Figura 7. Receptores P1 e P2. Esta Figura demonstra o mecanismo de ativação dos receptores P1 e P2 por ATP, ADP e adenosina (adaptado de Schwiebert e Kishore, 2001).

35
1.7. Efeitos de adenosina e ATP no rim: receptores e seus efeitos acoplados
1.7.1-Efeitos renais da adenosina
Os níveis de adenosina dependem do balanço entre sua produção e
metabolização, como discutido anteriormente. Os níveis deste nucleosídeo podem
aumentar significativamente em condições fisiopatológicas tais como isquemia e
inflamação (Vallon et al., 2006). Em condições de hipóxia, a elevação dos níveis
de adenosina no tecido renal pode servir como um sinalizador parácrino, capaz de
promover o restabelecimento do suprimento de oxigênio sanguíneo, através da
dilatação dos capilares peritubulares, aumentando assim a perfusão sanguínea
tecidual (Miller et al., 1978).
Os efeitos da adenosina na função renal são associados a modificações
tanto na hemodinâmica renal quanto nos transportadores de solutos ao longo do
néfron (Okusa, 2002). Os efeito da adenosina sobre função renal estão
correlacionados abaixo com os diferentes subtipos (A1, A2A e A3) de receptores
envolvidos.
1.7.2-Efeitos renais mediados pelo receptor A 1 WEAVER E REPPERT (1992), utilizando técnica de hibridização in situ com
sondas radioativas, determinaram a localização do receptor A1 em rim de rato. O
RNAm de receptor A1 foi detectado no ducto coletor medular interno e nas células
do aparelho justaglomerular. O receptor foi, ainda, detectado imunologicamente
nas arteríolas aferentes e células mesangiais, no túbulo convoluto proximal e
ducto coletor medular, e na superfície do epitélio papilar renal (Smith et al., 1999).
Através da utilização de agonistas seletivos para os receptores de
adenosina, foram determinados seus efeitos na hemodinâmica renal. Na presença
de agonista seletivo do receptor A1 ocorre vasoconstrição da arteríola aferente,
próximo ao glomérulo (Hansen et al., 2005). Este efeito, mediado pelo receptor A1,
está relacionado com o balanço túbulo-glomerular envolvido na auto-regulação
renal, discutido anteriomente. Foi observado que a exposição do túbulo distal, na
região das células da mácula densa, a altas concentrações de NaCl promove a

36
elevação dos níveis de adenosina neste segmento do túbulo (Vallon, 2003;
Komlosi et al., 2005). A adenosina intracelular, formada pela intensa hidrólise de
ATP no processo de intensa reabsorção de sódio pelas ATPases, deixa a célula
através de transporte passivo, e vai atuar de modo parácrino sobre as células da
musculatura lisa vascular pré-glomerular, promovendo vasoconstrição e
diminuição na resistência da arteríola aferente. Este efeito da adenosina tem sido
atribuído à ativação do receptor A1 (Jackson et al., 2002b).
Além do efeito na constricção da arteriola aferente tem sido mostrado o
efeito de adenosina no transporte de sódio tubular (Vallon et al., 2006). Há
evidências de aumento na eficiência do transporte de sódio em células epiteliais
de túbulo proximal quando o receptor A1 é ativado. A ativação desse receptor em
cultura de células epiteliais de túbulo proximal aumenta o cotransporte Na+-glicose
e Na+-fosfato, por transportadores na membrana luminal (Coulson et al., 1991).
Técnicas de microperfusão, aplicadas ao estudo do túbulo convoluto proximal,
demonstraram que a ativação do receptor A1 estimula o cotransporte basolateral
de Na+-3HCO-3 (Takeda et al., 1993). Ainda utilizando esta técnica, foi verificado
que o transporte de fosfato dependente de Na+,é inibido pelo aumento de AMPc,
promovido por um antagonista de receptor A1, em células de túbulo proximal (Cai
et al., 1994). As vias de sinalização induzidas por receptores A1 em células (HK-2)
com características de túbulo proximal podem envolver a proteína G sensível à
toxina pertussis e a ativação da proteína cinase C (PKC) (Lee e Emala, 2002).
Além disso, já foi descrito que a adenosina é capaz de modular a atividade
de ATPases presentes na membrana basolateral do túbulo proximal, como por
exemplo: a bomba de Ca+2 e Mg+2 (Ca+2/Mg+2-ATPase) (Coka-Guevara et al.,
1999) e a bomba de sódio (Na+-ATPase) (Caruso-Neves et al., 1997; Wengert. et
al., 2005), como será discutido posteriormente.
O papel da adenosina na reabsorção distal de sódio ainda não esta
completamente elucidado. MA e colaboradores (1996), usando cultura de células
do túbulo distal (células A6), mostraram que adenosina (30nM) aumenta a
probabilidade de abertura de canais de sódio localizados na membrana luminal
(ENAC). O efeito foi mimetizado por um agonista seletivo para o receptor A1 e

37
bloqueado por um antagonista seletivo deste receptor. Por outro lado, a exposição
luminal a altas concentrações de adenosina (1 ou 10µM) promoveu uma
diminuição na probabilidade de abertura destes canais. Estes dados indicam que a
abertura de canais de sódio no néfron distal é mediada pela ativação luminal do
receptor A1 e que a concentração urinária basal de adenosina (na faixa de nM)
poderia estimular a reabsorção de sódio por estes canais (Ma e Ling, 1996).
Apesar dos resultados descritos acima, pouco se sabe acerca do efeito da
adenosina sobre a excreção renal de sódio. Contudo, os dados descritos acima
indicam que adenosina poderia, através da ação em diferentes sítios do néfron,
modular a excreção renal de sódio e água.
1.7.3- Efeitos renais mediados pelo receptor A 2 O papel do receptor A2 na excreção renal de sódio e água ainda não foi
determinado. Algumas evidências, discutidas abaixo, indicam que ele teria efeito
contrário ao receptor A1 na excreção renal de sódio e de água. O RNAm para o
receptor A2 já foi observado em preparação de rim total, na papila renal, no
glomérulo, na vasa reta descendente medular externa, no segmento fino
ascendente da alça de Henle e no túbulo distal de rim de rato (Jackson et al.,
2002a; Vitzthum et al., 2004) . Dados do nosso laboratório demonstram a
expressão do receptor A2 na membrana basolateral do túbulo proximal (Wengert,
Berto Jr. et al., 2005).
Foi observado que o agonista seletivo de receptor A2 promove
vasodilatação principalmente na arteríola eferente (Vallon et al., 2006). Além
disso, também já foi observado que a adenosina promove diminuição da
resistência vascular da vasa reta, através da ativação de receptor A2, diminuindo a
reabsorção de NaCl e, conseqüentemente, aumentando a excreção urinária deste
sal (Zou et al., 1999). Não existem muitas informações sobre o envolvimento de
receptor A2 na modulação de transportadores de sódio, principalmente no túbulo
proximal. Em 2005 foi demonstrado, pelo nosso grupo, que, em membrana
basolateral isolada de túbulo proximal, na presença de antagonista de receptor A1,
adenosina tem efeito estimulatório, de maneira dose dependente sobre a atividade

38
Na+-ATPásica e este efeito era revertido por um antagonista seletivo de receptor
A2A. Além disso, foi demonstrado que a ativação de receptor do tipo A2A por
agonista seletivo estimula a via AMPc/PKA (Wengert. et al., 2005).
1.7.4- Efeitos renais mediados pelo receptor A 3 Por ter sido descoberto mais recentemente, os efeitos de adenosina
mediados por este receptor são os menos conhecidos. Já foi demonstrada, em
diferentes trabalhos, a expressão do mRNA para este subtipo de receptor em
preparação de rim total (Linden, Taylor et al., 1993). Entretanto, a distribuição
destes receptores, bem como seu papel na fisiologia renal ainda não está muito
bem definida (Vitzthum et al., 2004). Experimentos feitos para examinar a
excreção renal de sódio, através da técnica de microdiálise, em ratos
anestesiados, demonstrou que a infusão de agonista seletivo de receptor A3 na
medula renal não tem efeito no fluxo sanguíneo medular nem na função renal (Zou
et al., 1999). Trabalhos mais recentes demonstraram que o receptor A3 não tem
papel significativo na regulação da excreção renal em condições fisiológicas
(Mozaffari et al., 2000). No entanto, em experimentos realizados in vivo, observou-
se que o pré-condicionamento isquêmico com a infusão de antagonista de
receptor A3 tem efeito protetor contra isquemia renal (Lee e Emala, 2000).
Corroborando estas observações, LEE e colaboradores, observaram que ratos
“knockout” para o receptor A3 apresentam um efeito protetor renal contra
isquemia, quando comparados ao animal selvagem (Lee et al., 2003).
Os efeitos de adenosina descritos até aqui demonstram a sua importância
na hemodinâmica renal e na reabsorção de sódio ao longo do néfron. Seu efeito
final na função renal é, com certeza, uma consequência de sua ação através dos
diferentes receptores que podem ser expressos de maneira diferenciada, de
acordo com as condições fisiológicas e fisiopatológicas, em diferentes segmentos
do nefron. Além disso, pouco se sabe sobre os mecanismos de modulação da
excreção renal de sódio por adenosina no túbulo proximal.

39
1.7.5-Efeitos renais do ATP
Evidências sugerem que as células liberem ATP in vitro e in vivo. Células
epiteliais renais, em particular, liberam ATP em resposta a perturbações de
membrana ou a estímulos específicos como hipotônicidade e hipóxia (Schwiebert
e Zsembery, 2003; Unwin et al., 2003).
Muitas evidências demonstram que o ATP possui ainda outro papel, além
da troca de energia em sistema biológicos. O ATP extracelular, assim como outros
nucleotídeos tem ações autócrinas e parácrinas com um papel importante em
condições fisiológicas e fisiopatológicas através da ativação de receptores de
membrana (Burnstock, 2006). A liberação local de ATP pelas células tubulares
renais modula a função tubular e vascular renal. A função vascular é um
importante processo fisiológico regulado por ATP, uma vez que receptores de
nucleotídeos são expressos na vasculatura renal e no glomérulo, e já foi
demonstrado que dependendo de qual receptor é ativado, o ATP promove tanto
vasodilatação como vasoconstrição (Guan et al., 2007).
1.7.6- Efeitos renais mediados pelos Receptores P2X Utilizando técnicas de imunohistoquímica, CHAN e colaboradores (1998)
demonstraram a expressão do receptor P2X1 em tecido muscular liso vascular de
artéria arqueada e interlobar e em arteríola aferente, mas não no glomérulo, na
arteríola eferente, ou nos túbulos renais. Além disso, demonstraram elevada
expressão de P2X2 nas artérias e veias intrarenais e P2X7 nos glomérulos (Chan
et al., 1998; Turner et al., 2003). O receptor P2X5 é expresso em membrana apical
ao longo do túbulo proximal, P2X4 e P2X6 estão presentes no túbulo proximal, em
pequena quantidade, e duto coletor. Além disso, através da técnica de “western
blotting” foi demonstrada a expressão de P2X2, P2X4 e P2X7 (Turner et al., 2003).
Através da técnica de RT-PCR foi observada a expressão de RNAm de P2X1,
P2X2, P2X3, P2X4, P2X7 em células mesangiais de rato (Turner et al., 2003). A
Figura 8 mostra a distribuição de receptores P2X ao longo do nefron.
Poucos estudos referentes ao efeito dos receptores P2X na função renal
foram realizados. A maioria dos estudos é referente ao papel destes receptores no
balanço tubuloglomerular (Vallon e Schnermann, 2003). Um estudo recente, in

40
vivo em rim de coelho, demonstrou que a infusão de agonistas P2X1 e P2X3 na
artéria renal numa concentração de 7-56 µg x Kg-1 por minuto, que não é capaz de
modular a pressão arterial sistêmica, causa significativa diminuição no fluxo
sanguíneo cortical e medular confirmando a expressão de receptor P2X na
vasculatura renal (Eppel et al., 2006). Outros estudos, in vitro de microperfusão da
arteríola aferente de coelho com o glomérulo anexado, demonstraram que a
perfusão de agonista de P2X1 ou de ATP, leva a uma significativa diminuição no
diametro da arteríola aferente (Weihprecht et al., 1992). Em cultura de células A6,
células com característica de túbulo distal, a adição de agonista de receptor P2X4
estimula o canal de sódio sensível a amiloride (Zhang et al., 2007).

41
Figura 8 Distribuição dos receptors P2 ao longo do nefron (Burnstock, 2006).

42
1.7.7- Efeitos renais mediados pelo Receptor P2Y O grupo de CHAN e colaboradores demonstrou a expressão de receptor
P2Y1 nas arteríolas aferente e eferente (Chan et al., 1998). Os receptores P2Y1 e
P2Y2 são expressos no glomérulo, sendo o P2Y1 principalmente nas células
mesangiais e P2Y2 nos podócitos (Turner et al., 2003; Bailey et al., 2004). Através
da técnica de imunodetecção foi demonstrada a presença de P2Y4 em cultura de
células mesangiais. (Bailey et al., 2004). Os receptores P2Y também são
expressos nas células epiteliais dos túbulos renais. Por exemplo, P2Y4 é expresso
na membrana basolateral ao longo do túbulo proximal, enquanto o P2Y1 encontra-
se na membrana apical deste segmento. (Turner et al., 2003). Outros estudos
também demonstraram RNAm de P2Y1, P2Y2, P2Y6, em túbulo proximal (Bailey et
al., 2000; Bailey et al., 2001; Kishore et al., 2005). Pode-se observar na Figura 8 a
distribuição de receptores P2Y ao longo do néfron.
Utilizando uma preparação de célula muscular lisa vascular pré-glomerular
isolada, INSCHO e colaboradores (Inscho et al., 1996) verificaram que o ATP
promove efeito bifásico no aumento de cálcio citoplasmático. Este aumento de
cálcio citoplasmático é derivado do estoque intracelular e do influxo de cálcio
externo. Na presença de bloqueador de canal de cálcio tipo-L, o influxo de cálcio é
atenuado (Inscho et al., 1999). Estes estudos indicam que a ativação de canal de
cálcio tipo-L é essencial para a vasoconstrição da arteríola aferente mediada por
ATP.
Experimentos feitos em cultura de células do ducto coletor medular interno
(IMCD-K2) demonstraram que a ativação do receptor P2Y2 estimula a secreção de
cloreto e inibe a reabsorção de sódio (Lu et al., 2000), e a secreção de potássio
através de canal ROMK. Em experimentos de perfusão do duto coletor cortical de
rato, foi observado que a ativação de receptor P2Y2 aumenta a concentração de
cálcio intracelular, ativa a secreção de cloreto, e inibe a reabsorção de sódio e
cálcio (Cuffe et al., 2000). Além disso, também já foi demonstrado, neste modelo,
que a ativação de receptor P2Y2 basolateral inibe o transporte de água através de
aquaporina-2 (Kishore et al., 1995). Recentemente, KISHORE publicou um
trabalho que confirma estes dados, o estudo foi feito com ratos “knockout” para

43
receptor P2Y2 e observou-se que estes ratos apresentavam um aumento na
expressão de aquaporina-2 e produziam uma urina concentrada (Kishore et al.,
2005). Portanto, a ativação de receptor P2Y2 modula a reabsorção de sal e água
nestes segmentos.
BAILEY e colaboradores, em 2000, utilizando túbulo proximal isolado de rim
de coelho, demonstraram que a adição de10 µM de ATP na membrana basolateral
promoveu efeito máximo no aumento de cálcio intracelular de maneira transiente,
através da ativação de P2Y1 e P2Y2 (Bailey et al., 2000). Um ano depois, este
mesmo grupo demonstrou que a administração de UTP, agonista de P2Y6, em
membrana basolateral induzia o aumento de cálcio intracelular de maneira dose
dependente, associado com o aumento da produção de inositol fosfato em túbulo
proximal (Baileyl et al., 2001). Utilizando ferramentas farmacológicas, em
experimentos in vivo de microperfusão, foi investigado o efeito de nucleotídeos na
reabsorção de bicarbonato no túbulo proximal. Observou-se que a ativação de
receptor P2Y1 apical promove diminuição na reabsorção de bicarbonato (Bailey,
Turner et al., 2004).
Foi observado, por JIN e colaboradores em 1997, que o ATP inibe a
atividade da (Na++K+)ATPase em cultura primária de células de túbulo proximal
de rim de rato Wistar-Kyoto. (Jin e Hopfer, 1997). Um trabalho recente de
LISTHROP e colaboradores, publicado em 2007, demonstrou que ratos “knockout”
para o receptor P2Y2 apresentaram diminuição na expressão do co-transportador
Na+/Pi-2, entretanto não houve nenhuma alteração significativa na expressão do
trocador NHE3 e da isoforma α da (Na++K+)ATPase, e nenhuma mudança na
função renal foi observada. Portanto, estes dados indicam que a modificação na
atividade da (Na++K+)ATPase em cultura primária de células de túbulo proximal
de rim de rato Wistar-Kyoto não era devida ao aumento da expressão da enzima.
Uma possibilidade poderia ser devido a uma mudança nas propriedades cinéticas
da enzima ou mesmo na translocação de subunidades para a membrana
plasmática.
A interação entre receptores P1 e P2 é bem conhecida no caso de
heterodímeros formados entre P2Y1 e A1. Em célula HEK293, estes dois
44
receptores foram co-expressados e co-imunoprecipitados do lisado de células,
indicando que os dois receptores formam heterodímeros. A co-expressão do
receptor P2Y1 não modulou a expressão de receptor A1 mas, em ensaios de
“binding”, houve inibição na ligação de agonista e antagonista seletivos para o
receptor A1 em preparação de membranas (Yoshioka et al., 2001). Indicando que
a formação de heterodímero entre P2Y1 e A1 diminui a afinidade do receptor A1
por seus ligantes.
As características dos receptores P2 expressos no túbulo proximal estão
listadas na tabela 1. No entanto, pouco se sabe sobre os papeis fisiológicos e
fisiopatológicos destes receptores neste segmento tubular, embora este seja o
sítio que apresenta a maior concentração de ATP luminal (Vallon, 2008).
Tabela 1 – Características de receptores P2 express os no túbulo proximal
(Erb, Liao et al., 2006)
Receptores Agonistas Mecanismo de transdução
P2X4 ATP Intrínseco do canal de íon
(especialmente Ca2+)
P2X5 ATP Intrínseco do canal de íon
P2X6 Baixa afinidade para nucleotídeos Intrínseco do canal de íon
P2Y1 ADP > ATP Gq/G11, ↑ PLCβ, ↑ [Ca2+]i
P2Y2 ATP = UTP Gq/G11, ↑ PLCβ, ↑ [Ca2+]i
P2Y4 ATP = UTP Gq/G11, ↑ PLCβ
P2Y6 UDP > UTP > ADP Gq/G11, ↑ PLCβ

45
1.8. Papel do ATP e da adenosina na isquemia renal
Na isquemia as células sofrem depleção de energia, o que diminui os
transportes ativos e resulta em aumento do volume celular. Subsequentemente, o
ATP é liberado por estas células epiteliais. Este ATP pode ter uma ação autócrina
e/ou parácrina, ativando receptores P2 (Burnstock, 2006). Essa liberação do ATP
pode acontecer em caso de dano à membrana plasmática ou através de canais,
como discutido anteriormente. Além disso, o ATP liberado pode seguir pelo fluido
tubular ao longo do nefron e inibir os processos de transporte que consomem
energia nos segmentos mais distais protegendo-os para diminuir o consumo de
energia. Os receptores P2 basolaterais também podem mediar processos
similares. Os nucleotídeos também podem originar do espaço vascular e entrar no
nefron através da filtração glomerular. É proposto por LEIPZIGER (2003) que o
ATP extracelular luminal age como uma molécula sinalizadora, que confere
proteção ao epitélio tubular em condições isquêmicas.
Na isquemia renal ocorre um aumento na concentração de adenosina na
urina (Osswald et al., 1977). Embora o ATP seja o primeiro metabólito liberado
pelas células isquêmicas, no meio extracelular ele é convertido em adenosina.
Contudo, a possível conversão intracelular de ATP à adenosina, com consequente
liberação para o meio extracelular não pode ser descartada. Diversos estudos
demonstraram que o ATP promove o crescimento e a divisão celular através da
ativação de receptores P2 (Huwiler e Pfeilschifter, 1994; Paller et al., 1998; Harada
et al., 2000). Além disso, foi demonstrado por KISHORE e colaboradores (2001),
que, após isquemia renal, o transporte de íons é inibido enquanto a expressão de
receptor P2Y2 é aumentada (Kishore et al., 2005). Portanto, é sugerido que o ATP
seja capaz de evitar os danos isquêmicos tubulares e promover a sua
regeneração após os danos terem ocorrido.
LEE e colaboradores (Lee e Han, 2005), em estudo com cultura primária de
túbulo proximal de coelho, demonstraram que períodos isquêmicos longos
promovem a liberação de LDH, um marcador de dano celular estrurural, e a
ativação de NF-κβ. Porém, o pré-condicionamento com ATP bloqueia a liberação

46
de LDH induzida pela isquemia, assim como a ativação de NF-κβ. Estes dados
sugerem um efeito protetor para o ATP contra a injúria isquêmica.
Seria interessante considerar que o efeito de ATP poderia ser mediado por
adenosina, uma vez que este nucleosídeo é um dos metabólitos de ATP. Existem
trabalhos demonstrando o efeito protetor de adenosina no tratamento pré-
isquemico. O pré-tratamento com dose única de teofilina, antagonista não seletivo
de receptores de adenosina, atenuou a redução do fluxo sanguíneo renal e do
rítmo de filtração glomerular no período pós-isquemico (1 hora e meia após o
início da reperfusão) em ratos e coelhos (Lin et al., 1986; Gouyon e Guignard,
1988).
Outros estudo, feitos em ratos, demonstraram que a administração
intravenosa de uma dose única de teofilina (20 minutos antes da reperfusão) na
artéria “clampeda“ durante o período de hipóxia (60 minutos), melhorou a
recuperação da função renal após injúria isquêmica, através do aumento do fluxo
urinário, do rítmo de filtração glomerular, da diminuição da obstrução tubular e da
alteração patológica nos glomérulos (Jackson et al., 2003). Além disso, o
tratamento com teofilina após o período isquêmico, em ratos não tratados
previamente, aumentou o fluxo sanguíneo renal e o rítmo de filtração glomerular,
indicando que a adenosina contribui para a supressão do fluxo sanguíneo renal e
do rítmo de filtração glomerular na fase de reperfusão (Lin et al., 1988).
Estudos realizados em humanos demonstraram que a dose única de
teofilina, administrada imediatamente após o nascimento, tem efeitos benéficos
reduzindo a diminuição no rítmo de filtração glomerular e o envolvimento do rim na
asfixia perinatal (Bakr, 2005).
Para verificar o efeito direto da adenosina, foi obervado por LEE e
colaboradores, que o pré-tratamento com o nucleosídeo 1,75 mg x Kg-1 por
minuto, durante 2 minutos, antes de 45 minutos de isquemia confere proteção a
função renal contra isquemia, indicada pela melhora da morfologia renal após 24h
de reperfusão. Foi proposto que o efeito protetor de adenosina seja mediado pela
ativação de receptor A1 (Lee e Emala, 2000), envolva proteína Gi/0, a ativação de
PKC, e a redução na necrose, inflamação e apoptose (Lee et al., 2004).

47
Concordando com este resultado, ratos “knockout” para receptor A1 apresentaram
um aumento significativo na creatinina plasmática e no dano tecidual renal,
comparado com os ratos controles, 24h após 30 minutos de isquemia (Lee et al.,
2004). Ainda nesse trabalho, foi observado que ratos controles, pré-tratados com
antagonista ou agonista de receptor A1 apresentaram significativa piora e melhora,
respectivamente, após injúria de isquemia/reperfusão, associadas com aumento e
diminuição de marcadores da inflamação renal (Lee et al., 2004).
A infusão contínua de agonista de receptor A2A no período de reperfusão
também protege o rim contra injúria de isquemia/reperfusão. Análise histológica
demonstrou melhora na necrose tubular e na congestão vascular nos ratos
tratados farmacologicamente, quando comparados com os ratos tratados com
veículo (Okusa, 2002). Por outro lado, foi demonstrado, em ratos, que a ativação
do receptor A3 potencializa a injúria de isquemia/reperfusão, enquanto a inibição
deste receptor protege a função renal (Lee e Emala, 2000). Ratos “knockout” para
receptor A3 apresentaram significativa proteção, funcional e morfológica, contra
isquemia renal (Lee et al., 2003). Entretanto, a via de sinalização ativada por
receptor A3 ainda não foi estabelecida. Uma possibilidade é que a ausência de
receptor A3 seja antiapoptótica, pois a ativação de receptor A3 promove apoptose
em diversa linhagens celulares, inclusive em células renais (Vallon et al., 2006).
Em conjunto, estes dados indicam que o efeito protetor da função renal,
contra a injúria decorrente de isquemia/reperfusão, pode ser induzido por: 1) pré-
tratamento com adenosina; 2) pré-tratamento com agonista de receptor A1; 3)
tratamento durante a reperfusão com agonista de receptor A2A; 4) deficiência de
receptor A3, e 5) pré-tratamento com teofilina, antagonistas dos receptores A1 e A2
de adenosina. Uma vez que a teofilina antagoniza somente os efeitos de
adenosina mediados por receptores A1 e A2, seria possível supor que, na
presença de teofilina, a adenosina seja um agonista seletivo para receptor A3.
Desta forma, o efeito de adenosina observado seria somente devido à sua
interação com A3 e não uma resultante da ação de adenosina nos receptores A1 e
A2.

48
O efeito final da adenosina depende do subtipo de receptor ativado e a
ativação de cada subtipo de receptor de adenosina depende da concentração de
adenosina no meio, uma vez que estes receptores apresentam diferentes
afinidades para o nucleosídeo, e do nível de expressão de cada receptor. Além
disso, o nível extracelular de adenosina depende do seu metabolismo e
transporte. Considerando a relevância clínica da injúria renal de isquemia e
reperfusão, muito ainda deve ser estudado sobre a proteção da função renal.
1.9. Resultados Anteriores
Em 1997, demonstramos que a adenosina é capaz de modular a atividade
Na+-ATPásica em membrana basolateral isolada de túbulo proximal. A modulação
ocorre de maneira bifásica, promovendo efeito inibitório através da ativação de
receptor A1, em baixas concentrações (10-3-10-8 M) do nucleosideo, e reversão do
efeito em concentrações mais elevadas de adenosina (Caruso-Neves et al., 1997).
Foi demonstrado, ainda, que a inibição da Na+-ATPase envolve diminuição dos
níveis de AMPc. A questão, naquele momento, era determinar se a reversão do
efeito inibitório seria devida a ligação de adenosina a outro receptor ou se
ocorreria a desensibilização do receptor A1. Este questionamento deu origem à
minha tese de mestrado (Wengert et al., 2005). Nós observamos que a reversão
da inibição do efeito da adenosina era devido à interação de adenosina com o
receptor A2A. Demonstramos que adenosina estimulava a Na+-ATPase de maneira
dose dependente na presença de antagonista de A1 e que o efeito máximo era
obtido na concentração de 10-6 M (Figura 9). O estudo do mecanismo de ativação
da Na+-ATPase por adenosina revelou envolvimento da ativação da via
AMPc/PKA (Figura 9). Este resultados indicam que o balanço entre a expressão
de A1 e A2 poderia ser um importante fator de modulação da Na+-ATPase e,
consequentemente, da excreção renal de sódio (Figura 10). Contudo, o resultado
que mais nos chamou a atenção foi o metabolismo de adenosina, na membrana
basolateral isolada de túbulo proximal, gerando principalmente inosina.
Posteriormente, foi observado, em nosso laboratório, que a inosina inibe a Na+-
ATPase de maneira similar e não aditiva à adenosina. A idéia de que adenosina

49
poderia ser metabolizada e seus metabólitos pudessem ter ação renal nos levou a
pensar sobre o metabolismo de adenosina em células intactas. Esta foi a pergunta
inicial do projeto que resultou no desenvolvimento desta tese de doutorado.
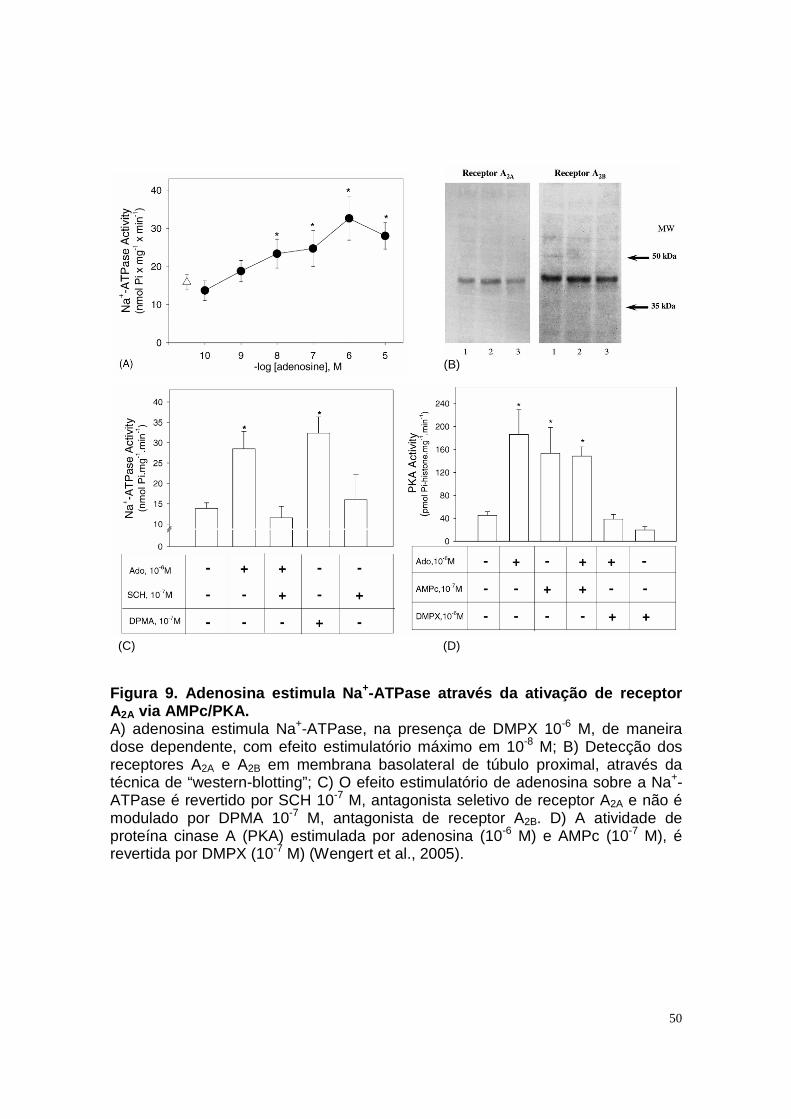
50
Figura 9. Adenosina estimula Na +-ATPase através da ativação de receptor A2A via AMPc/PKA. A) adenosina estimula Na+-ATPase, na presença de DMPX 10-6 M, de maneira dose dependente, com efeito estimulatório máximo em 10-8 M; B) Detecção dos receptores A2A e A2B em membrana basolateral de túbulo proximal, através da técnica de “western-blotting”; C) O efeito estimulatório de adenosina sobre a Na+-ATPase é revertido por SCH 10-7 M, antagonista seletivo de receptor A2A e não é modulado por DPMA 10-7 M, antagonista de receptor A2B. D) A atividade de proteína cinase A (PKA) estimulada por adenosina (10-6 M) e AMPc (10-7 M), é revertida por DMPX (10-7 M) (Wengert et al., 2005).
(B)
(C) (D)
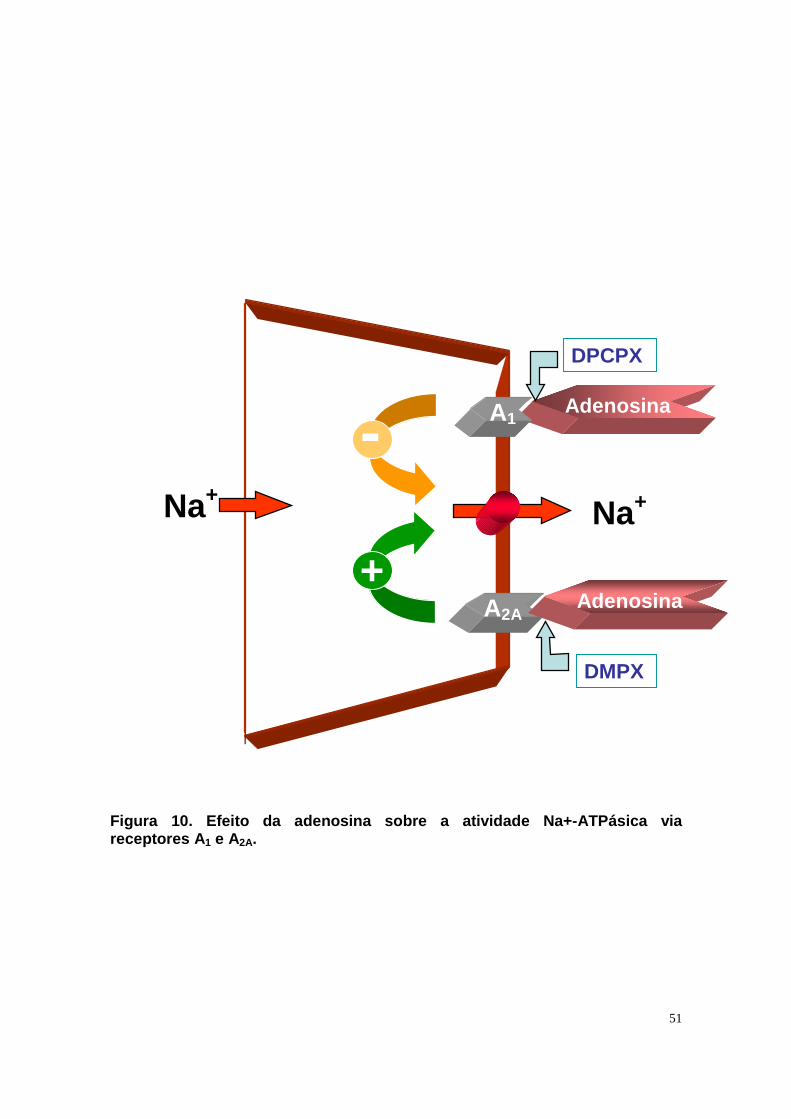
51
Figura 10. Efeito da adenosina sobre a atividade Na +-ATPásica via receptores A 1 e A2A.
Na+ Na+
A1 Adenosina
DPCPX
A2A
+
-
DMPX
Adenosina

52
2. Objetivos
O objetivo do presente trabalho foi verificar o metabolismo de adenosina na
membrana luminal de células LLC-PK1, uma bem estabelecida linhagem de
células do túbulo proximal de rim de porco, e seu possível papel na reabsorção de
sódio no túbulo proximal. Para isto as seguintes etapas serão desenvolvidas:
1) estudar o metabolismo de adenosina quando adicionada ao lado luminal;
2) identificar os efeitos de adenosina sobre as atividades da Na+-ATPase e
(Na++K+)ATPase;
3) identificar os receptores e as vias de sinalização envolvidos na modulação das
bombas de sódio por metabólitos de adenosina;
4) verificar o efeito da isquemia no metabolismo de adenosina;
5) verificar a possível modulação das bombas de sódio durante a isquemia;
6) estudar o efeito do pré-condicionamento isquêmico com ATP na modulação da
atividade das bombas de sódio durante a isquemia.

53
3. Materiais e métodos
3.1.Materiais
ATP, ouabaína, EGTA, HEPES, Tris, adenosina, furosemide, calfostina C,
peptídeo inibidor de PKA de coelho (Thr-Thr-Tyr-Ala-Asp-Phe-Ile-Ala-Ser-Gly-Arg-
Thr-Gly-Arg-Arg-Asn-Ala-Ile-His-Asp) (Cheng et al., 1986) foram comprados da
Sigma Chemical Co., St. Louis, MO, USA. DPCPX, MRS e DMPX foram
comprados da Research Biochemicals International, Natick, MA, USA. Percol foi
comprado da Pharmacia Biotech, Uppsala, Sweden. Todos os outros reagentes
foram da mais alta pureza disponível. Todas as soluções foram preparadas com
água destilada e desionizada. O ATP[γ-32P] foi preparado como descrito por MAIA
e colaboradores (1993).
3.2. Cultura de células LLC-PK1
Células LLC-PK1, célula epitelial renal de porco, são cultivadas em meio
DMEM com 10% de soro fetal bovino, 1g/L de bicarbonato de sódio e 1% de
antibiótico/antimicótico (10000 unidades/ml de penicelina G sódica, 10000 µg/ml
de sulfato de streptomicina e 25 µg/ml de anfotericina B com fungisona a 85 %),
pH 7,4, em atmosfera de 95% ar e 5% CO2. São utilizadas células entre as
passagens 20 e 60, com três dias de crescimento, e 90-100% de confluência.
3.3. Medida da atividade ATPásica
A atividade ATPásica foi medida pelo método descrito por Grubmeyer e
Penefsky (1981) (Grubmeyer e Penefsky, 1981). A reação foi iniciada com a
adição do homogenato de células (concentração final de proteína: 0,2 mg/ml). A
reação ocorreu a 370C por 20 minutos, foi parada pela adição de 0,5ml de carvão
ativado por HCl (0,1N). O [32P]Pi liberado foi medido em alíquotas do
sobrenadante, obtidas após a centrifugação da suspensão de carvão a 2000 rpm,

54
por 5 minutos, em centrífuga clínica. A radioatividade foi determinada por
cintilação líquida (Packard Tri-Carb 2100 TR).
O cálculo da atividade (Na++K+)ATPásica foi obtido pela diferença de [32P]Pi
liberado na ausência e na presença de 0,1mM de ouabaína, e a atividade Na+-
ATPásica foi obtida pela diferença de [32P]Pi liberado na ausência e na presença
de furosemide 2mM (inibidor específico da Na+-ATPase), ambos na presença de
0,1mM de ouabaína (inibidor específico da Na+/K+-ATPase) (Osswald et al., 1978;
Proverbio et al., 1986; Vallon, 2003). A concentração de proteína foi determinada
pelo método de Folin fenol (Lowry et al., 1951), usando albumina sérica bovina
como padrão.
3.4. Medida da atividade de PKA e PKC
A atividade de PKA no homogenato de células foi medida pela incorporação
de 32Pi à histona sensível ao peptídeo inibidor de PKA (PKAi). A reação, com
volume final de 0,1ml, foi iniciada pela adição de 10µM de [γ-32P]ATP (sal de
magnésio, 7µCi x µmol-1). A composição do meio reacional foi: 4mM de MgCl2,
20mM HEPES-Tris (pH 7,0), 1,5 mg/ml de histona, e 0,7 mg/ml de homogenato de
células. Após 20 minutos, a reação foi parada pela adição de TCA 20% e
imediatamente incubada em gelo até a filtração em filtros milipore (0,45 µm). Os
filtros foram lavados com solução gelada de TCA 20% e tampão fosfato 2mM (pH
7,0).
A radioatividade incorporada foi quantificada por cintilação líquida (Packard
Tri-Carb 2100 TR). A atividade especifica de PKA e PKC foi calculada pela
diferença de incorporação de 32Pi na presença e na ausência de inibidores
específicos para cada uma das enzimas, peptídeo inibidor de PKA (10-8M) e
calfostina C (10-8M).
3.5. Medida de Cálcio intracelular
As células foram plaqueadas em lamínulas de 40mm por 48 horas DMEM
com 10% de soro fetal bovino, 1g/L de bicarbonato de sódio e 1% de
antibiótico/antimicótico, pH 7,4, em atmosfera de 95% ar e 5% CO2 a 37oC em

55
atmosfera umidificada e 5% de CO2. Para a medida de cálcio, as células foram
incubadas com o fluorocromo FURA-2-AM a 4µM (Molecular Probes) e
Probenicide (Sigma) a 5mM em meio de cultura, à temperatura ambiente e ao
abrigo da luz, por 40 minutos. As lamínulas foram, então, lavadas por duas vezes
com PBS e montadas em câmara de perfusão de três compartimentos no
microscópio NIKON Diaphot 300. A câmara central, contendo as células,
permanece com volume de 200µL, sendo sua velocidade de vazão de 1mL x min-
1. A perfusão foi feita com solução de PBS suplementada com CaCl2 (Sigma) a
1mM, pH 7,4, a 25 oC. O ATP (Sigma) (diluídos em PBS/Ca2+ 1mM) foi aplicado
em pulsos em diferentes concentrações. O ATP foi aplicado cinco vezes mais
concentrado que a concentração final desejada na pré-câmara de perfusão. Para
a realização de curva dose-resposta, concentrações crescentes do nucleotídeo
foram aplicadas em pulsos seqüenciais com intervalos fixos de 5 minutos.
A captação da alteração de cálcio foi feita através do sistema de aquisição de
um fotômetro de fluorescência Photon Technology, Princetown, NJ. O fluorocromo
de escolha, FURA-2 AM (Molecular Probes) é excitado alternadamente a 340-
380nm e sua emissão a 510nm é medida. A taxa de medida que é proporcional à
variação da concentração de cálcio intracelular é determinada a cada 100ms. A
escala de variação de emissão é arbitrária.
3.6. Cromatografia de camada fina para análise de metabolismo de purinas.
As culturas de células foram lavadas com PBS e incubadas com [3H]-
adenosina por diferentes períodos a 37ºC. A reação foi interrompida pela adição
de TCA 10%. No final de cada período retirou-se o sobrenadante, e uma alíquota
deste (3 x 30µl) foi aplicada em placa cromatográfica (sílica gel 60 F254), medindo
14 x 4 cm, para quantificar os metabólitos no meio extracelular. As monocamadas
celulares foram raspadas e submetidas a centrifugação, 15000rpm por 15 minutos,
reservou-se o sobrenadante, e uma alíquota deste (3 x 30µL) foi aplicada em
placa cromatográfica (sílica gel 60 F254), medindo 14 x 4 cm. Em todas as placas
cromatográficas foi aplicada uma solução padrão de adenosina, inosina,
hipoxantina e adenina (5mM), para intensificar as bandas no momento da

56
revelação. Como eluente utilizamos a seguinte mistura de solventes: metanol
1,7mL, acetato de etila 2,2mL, butanol 3,8mL e hidróxido de amônio 2,2mL. Após
a corrida, a marcação das manchas correspondentes ao padrão de adenosina e
de seus metabólitos foi feita por meio de luz ultravioleta. As áreas marcadas foram
raspadas e transferidas para um recipiente e a radioatividade de cada mancha foi
medida por meio de cintilação líquida.
3.7.Ensaio para submeter as células a isquemia
Estes ensaios foram realizados de acordo com o método descrito por
DOCTOR e colaboradores (1994). As culturas de células foram lavadas com PBS
e incubadas por 3 horas a 37ºC em meio de pré-incubação sem glicose (125 mmol
NaCl, 5mmol KH2PO4, 2 mmol MgSO4, 25 mmol NaHCO3, 1,5 mmol CaCl2, 2
mmol de glutamina), para reduzir os substratos glicolíticos endógenos. Logo após
as células foram incubadas com meio de depleção de ATP (a mesma composição
do meio de pré-incubação, mas com 10 µmol antimicina A, 10 mmol 2-deoxi-
glicose, sem glutamina). A antimicina A é um inibidor do complexo III envolvido na
fosforilação oxidativa, portanto na presença deste inibidor não há a síntese de
ATP derivado da fosforilação oxidativa mitocondrial, e a 2-deoxi-glicose é um
análogo não hidrolisável da glicose, impedindo a formação de ATP através da via
glicolítica.
3.8. Análise estatística
Inicialmente, as médias das atividades ATPásicas foram comparadas
através da análise de variância (ANOVA), considerando os fatores de tratamento
reservados aos grupos experimentais. A magnitude das diferenças foi
posteriormente verificada através do teste de comparação múltipla de Bonferroni.
Os dados são apresentados como média ± erro padrão (número de
experimentos em cada condição testada). Nos ensaios, cada condição
experimental foi realizada em triplicata, sendo computadas nos resultados as
médias aritméticas assim obtidas. Cada dado experimental (n) corresponde a

57
resultados obtidos em diferentes placas, sendo que em cada experimento eram
testadas todas as condições experimentais. As diferenças foram consideradas
significativas quando p < 0,05.

58
4. RESULTADOS
4.1. Metabolismo de adenosina em células LLC-PK1
O primeiro grupo experimental foi realizado com o objetivo de verificar o
metabolismo de adenosina na membrana luminal de células LLC-PK1. Este
processo foi investigado através da incubação das células LLCPK-1 com [3H]-
adenosina no meio de cultura sem soro, seguida de posterior determinação dos
níveis de produtos metabólicos gerados (adenina, inosina, hipoxantina e
nucleotídeos de adenina) no homogeneizado celular (meio intracelular) e em meio
de cultura (meio extracelular). Os derivados metabólicos da [3H]-adenosina foram
fracionados por TLC e quantificados por cintilação líquida.
Nossos resultados demonstram uma queda considerável no nível
extracelular de [3H]-adenosina, de modo tempo dependente, que atinge o valor
mínimo aos 30 minutos e, então, se mantém por até 4h de reação (Figura 11A).
Nesta condição os níveis de adenosina caem de 1000 pmol x mg-1 ptn (2 minutos)
para 180 ± 16 pmol x mg-1 ptn (30 minutos de reação), correspondendo a uma
diminuição de 82 %. Seguindo o mesmo curso temporal, podemos observar uma
intensa e rápida elevação de [3H]-adenina (nos dois minutos iniciais de
incubação), além de uma elevação mais lenta de [3H]-hipoxantina e nucleotídeos
de [3H]-adenina. A elevação da [3H]-adenina é revertida após 2 minutos de reação,
enquanto o nível de [3H]-hipoxantina e nucleotídeos de [3H]-adenina se eleva até
30 minutos de reação e se mantém elevado por até 4h. Vale ressaltar que, após
30 minutos de reação, pode-se observar um equilíbrio dinâmico entre o
metabolismo de [3H]-adenosina e a formação de nucleotídeos de [3H]-adenina,
[3H]-inosina e [3H]-hipoxantina. A Figura 11B resume os resultados do
metabolismo de [3H]-adenosina em 30 minutos de reação no meio extracelular.
Neste tempo os principais metabólitos formados a partir de [3H]-adenosina são
[3H]-hipoxantina, [3H]-adenina e nucleotídeos de [3H]-adenina.

59
Figura 11. Metabolismo extracelular de adenosina. A) Curso temporal do metabolismo de adenosina adicionada à cultura de células LLC-PK1. As células foram incubadas com [3H]-adenosina 10-7 M por diferentes períodos (2, 5, 10, 30, 60 e 240 minutos) e processadas como descrito em materiais e métodos. Os metabólitos extracelulares presentes no meio de cultivo foram separados por cromatografia em camada fina e quantificados por cintilação líquida. B) Quantificação dos metabólitos presentes no meio extracelular em 30 minutos de incubação, período em que o metabolismo atinge o estado de equilíbrio (n=10).
0 10 20 30 40 50 60 240
pmol
/mg
ptn
0
200
400
600
800
1000
1200
1400
0
200
400
600
30 minutos
pmol
/mg
ptn
A
B
minutos
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
aden
ina
aden
osin
a
hipo
xant
ina
inos
ina
Nuc
leot
ídeo
s de
ade
nina
0 10 20 30 40 50 60 240
pmol
/mg
ptn
0
200
400
600
800
1000
1200
1400
0
200
400
600
30 minutos
pmol
/mg
ptn
A
B
minutos
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
aden
ina
aden
osin
a
hipo
xant
ina
inos
ina
Nuc
leot
ídeo
s de
ade
nina

60
No meio intracelular pode-se observar a formação majoritária de
[3H]nucleotídeos de adenina de maneira tempo dependente, sendo o nível máximo
alcançado aos 30 minutos de reação e mantido até 4 h (Figura 12A). A Figura 12B
resume os resultados do metabolismo de [3H]adenosina em 30 minutos de reação
no meio intracelular. Neste tempo o principal metabólito de [3H]adenosina formado
são os nucleotídeos de [3H]adenina.
Estes dados nos permitem propor que a [3H]adenosina adicionada à cultura
seja rapidamente convertida em [3H]adenina e [3H]hipoxantina ao nível
extracelular. Além disso, estes resultados indicam que possivelmente uma fração
considerável de [3H]adenosina seja convertida em nucleotídeos de [3H]adenina
(provavelmente ATP) no interior das células, para serem, em seguida,
transportados para o meio extracelular a favor do gradiente de concentração.
Nucleosídeos e bases nitrogenadas são transportados através da
membrana plasmática por meio de transportadores específicos, classificados
como equilibrativos (denominados ENT1 e ENT2) e concentrativos (denominados
CNT1,2,3). Com o objetivo de verificar se [3H]adenosina estaria sendo convertida
em nucleotídeos de [3H]adenina (ATP) e este estaria sendo secretado para o meio
extracelular, realizamos experiências na presença de dipiridamole, um bloqueador
específico de transportadores concentrativos e equilibrativos, ou NBTI, um
bloqueador da isoforma ENT1 de transportadores equilibrativos (Figura 13). A
presença de dipiridamole reduziu a captação de [3H]adenosina, quando
comparada ao controle (Figura 13A), apesar deste efeito não ter como
consequência uma elevação resultante do nível extracelular de [3H]adenosina,
quando comparada ao controle (Figura 13B). A presença de NBTI não promoveu
alterações nos níveis intra ou extracelulares de [3H]adenosina. Por outro lado, a
incubação com dipiridamol resultou em diminuição nos níveis intra e extracelulares
de nucleotídeos de [3H]adenina (Figuras 14A e 14B), confirmando a origem
intracelular dos nucleotídeos de [3H]adenina .
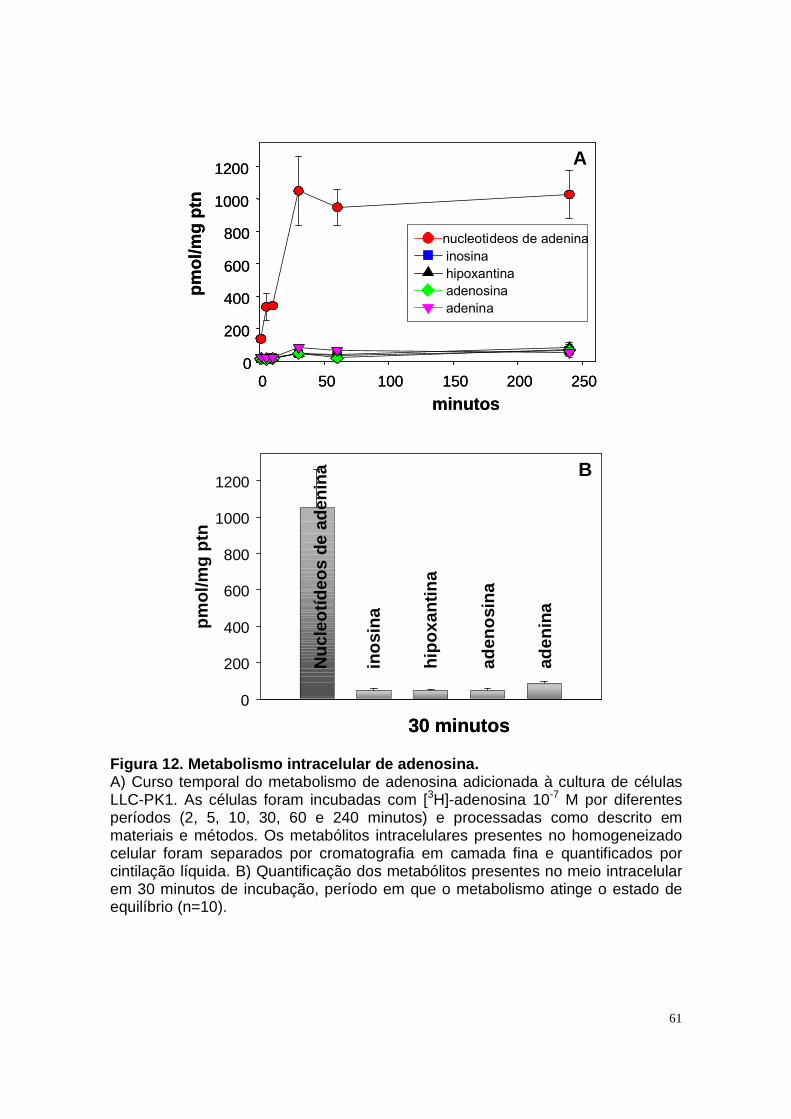
61
Figura 12. Metabolismo intracelular de adenosina. A) Curso temporal do metabolismo de adenosina adicionada à cultura de células LLC-PK1. As células foram incubadas com [3H]-adenosina 10-7 M por diferentes períodos (2, 5, 10, 30, 60 e 240 minutos) e processadas como descrito em materiais e métodos. Os metabólitos intracelulares presentes no homogeneizado celular foram separados por cromatografia em camada fina e quantificados por cintilação líquida. B) Quantificação dos metabólitos presentes no meio intracelular em 30 minutos de incubação, período em que o metabolismo atinge o estado de equilíbrio (n=10).
0
200
400
600
800
1000
1200
minutos0 50 100 150 200 250
0
200
400
600
800
1000
1200
pmol
/mg
ptn
pmol
/mg
ptn
30 minutos
aden
ina
aden
osin
a
hipo
xant
ina
inos
ina
Nuc
leot
ídeo
s de
ade
nina
A
B
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
0
200
400
600
800
1000
1200
minutos0 50 100 150 200 250
0
200
400
600
800
1000
1200
pmol
/mg
ptn
pmol
/mg
ptn
30 minutos
aden
ina
aden
osin
a
hipo
xant
ina
inos
ina
Nuc
leot
ídeo
s de
ade
nina
A
B
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
nucleotideos de adeninainosinahipoxantinaadenosinaadenina
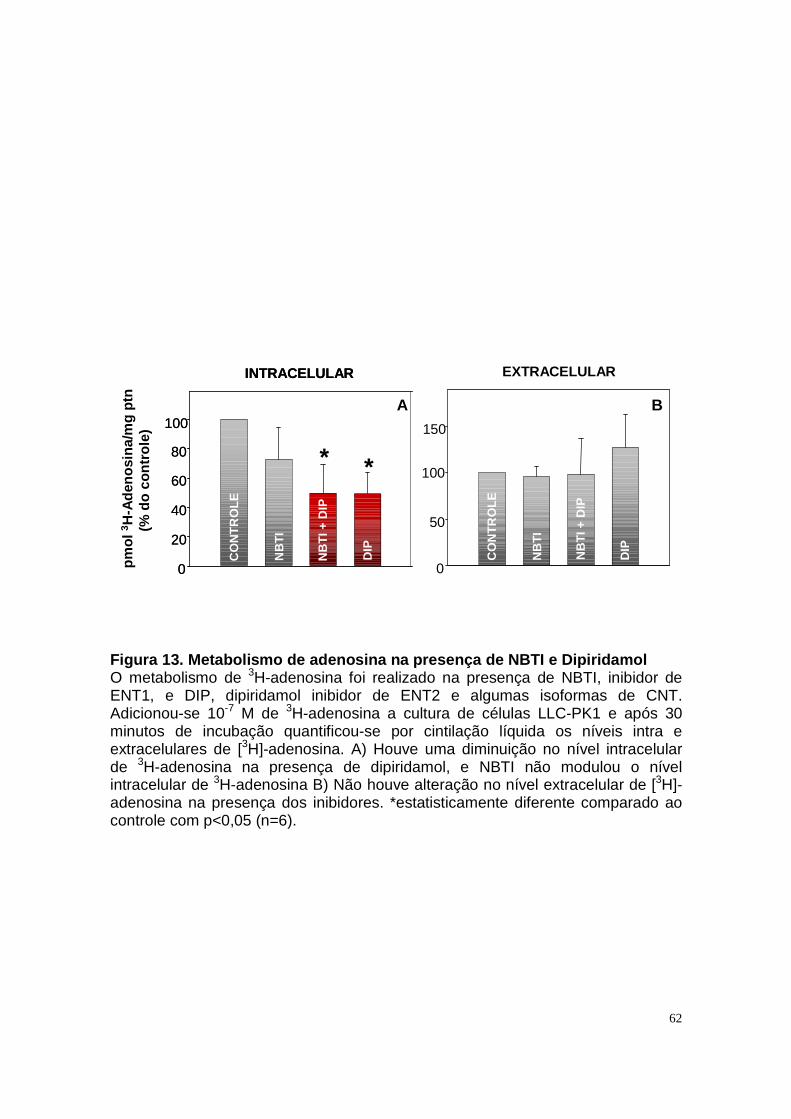
62
Figura 13. Metabolismo de adenosina na presença de NBTI e Dipiridamol O metabolismo de 3H-adenosina foi realizado na presença de NBTI, inibidor de ENT1, e DIP, dipiridamol inibidor de ENT2 e algumas isoformas de CNT. Adicionou-se 10-7 M de 3H-adenosina a cultura de células LLC-PK1 e após 30 minutos de incubação quantificou-se por cintilação líquida os níveis intra e extracelulares de [3H]-adenosina. A) Houve uma diminuição no nível intracelular de 3H-adenosina na presença de dipiridamol, e NBTI não modulou o nível intracelular de 3H-adenosina B) Não houve alteração no nível extracelular de [3H]-adenosina na presença dos inibidores. *estatisticamente diferente comparado ao controle com p<0,05 (n=6).
100
150
EXTRACELULAR
0
50
pmol
3 H-A
deno
sina
/mg
ptn
(% d
o co
ntro
le)
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
100
INTRACELULAR
0
20
40
60
80
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
* *
A B
100
150
EXTRACELULAR
0
50
pmol
3 H-A
deno
sina
/mg
ptn
(% d
o co
ntro
le)
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
100
INTRACELULAR
0
20
40
60
80
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
* * 100
150
EXTRACELULAR
0
50
pmol
3 H-A
deno
sina
/mg
ptn
(% d
o co
ntro
le)
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
100
INTRACELULAR
0
20
40
60
80
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
* *
100
INTRACELULAR
0
20
40
60
80
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
* *
A B
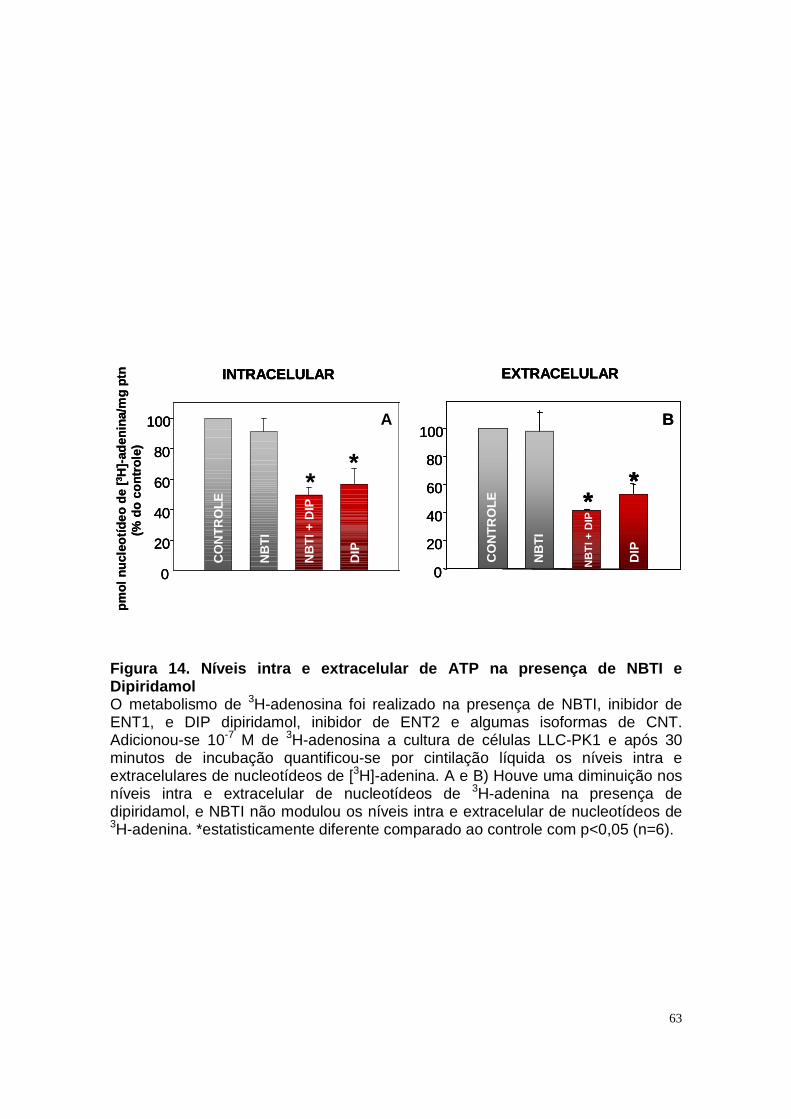
63
Figura 14. Níveis intra e extracelular de ATP na pr esença de NBTI e Dipiridamol O metabolismo de 3H-adenosina foi realizado na presença de NBTI, inibidor de ENT1, e DIP dipiridamol, inibidor de ENT2 e algumas isoformas de CNT. Adicionou-se 10-7 M de 3H-adenosina a cultura de células LLC-PK1 e após 30 minutos de incubação quantificou-se por cintilação líquida os níveis intra e extracelulares de nucleotídeos de [3H]-adenina. A e B) Houve uma diminuição nos níveis intra e extracelular de nucleotídeos de 3H-adenina na presença de dipiridamol, e NBTI não modulou os níveis intra e extracelular de nucleotídeos de 3H-adenina. *estatisticamente diferente comparado ao controle com p<0,05 (n=6).
pmol
nucl
eotíd
eo d
e [
3 H]-
aden
ina/
mg
ptn
(% d
o co
ntro
le)
INTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
EXTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
A B
pmol
nucl
eotíd
eo d
e [
3 H]-
aden
ina/
mg
ptn
(% d
o co
ntro
le)
INTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
INTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
EXTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
EXTRACELULAR
0
20
40
60
80
100
DIP
CO
NT
RO
LE
NB
TI +
DIP
NB
TI
**
A B

64
4.2. Efeito de adenosina e de seus metabólitos sobre as atividades Na+-ATPase e
(Na++K+)ATPase em células LLC-PK1
Considerando nosso objetivo inicial de avaliar o papel da adenosina e de
seus metabolitos na reabsorção de sódio, continuamos nosso trabalho avaliando
os efeitos de cada purina sobre as atividades Na+-ATPásica e (Na++K+)ATPásica
em células LLC-PK1ATPásicas.
Culturas de células confluentes foram incubadas com doses crescentes de
adenosina ou de seus derivados metabólicos por período de 30 minutos. Em
seguida, as células foram raspadas das placas, homogeneizadas e centrifugadas.
As atividades ATPásicas foram determinadas nos sobrenadantes, conforme
descrito em Materiais e métodos. A adenosina promoveu um efeito estimulatório
dose-dependente sobre a atividade Na+-ATPásica, atingindo intensidade máxima
(58 %) em concentrações superiores a 10-13 M. Nenhum efeito foi observado sobre
a atividade (Na+/K+)ATPásica (Figura 15). Considerando que a adenosina foi
capaz de promover efeito somente sobre a atividade Na+-ATPásica, os efeitos dos
outros metabolitos foram avaliados somente em relação a esta enzima. As bases
purínicas adenina e hipoxantina não induziram qualquer alteração sobre a
atividade da enzima (Figuras 16 e 17).
Apesar de nossos dados relacionados ao metabolismo de [3H]adenosina
demonstrarem uma elevação significativa nos níveis intra e extracelulares de
nucleotídeos de [3H]adenina, a metodologia de TLC utilizada para fracionar os
derivados metabólicos da [3H]adenosina não permitiu resolver os nucleotídeos
mono-, di- e trifosfatados de adenina. Por este motivo, decidimos testar o efeito de
AMP, ADP e ATP sobre a atividade da Na+-ATPase. O aumento da concentração
de AMP e ADP (de 10-11 para 10-7 M) não promoveu nenhum efeito na atividade
da enzima (Figuras 18 e 19). Por outro lado, o aumento da concentração de ATP
de 10-12M para 10-8 M promoveu efeito estimulatório sobre a atividade Na+-
ATPásica, atingindo o efeito máximo (42 %) na concentração de 10-9M (Figura 20).
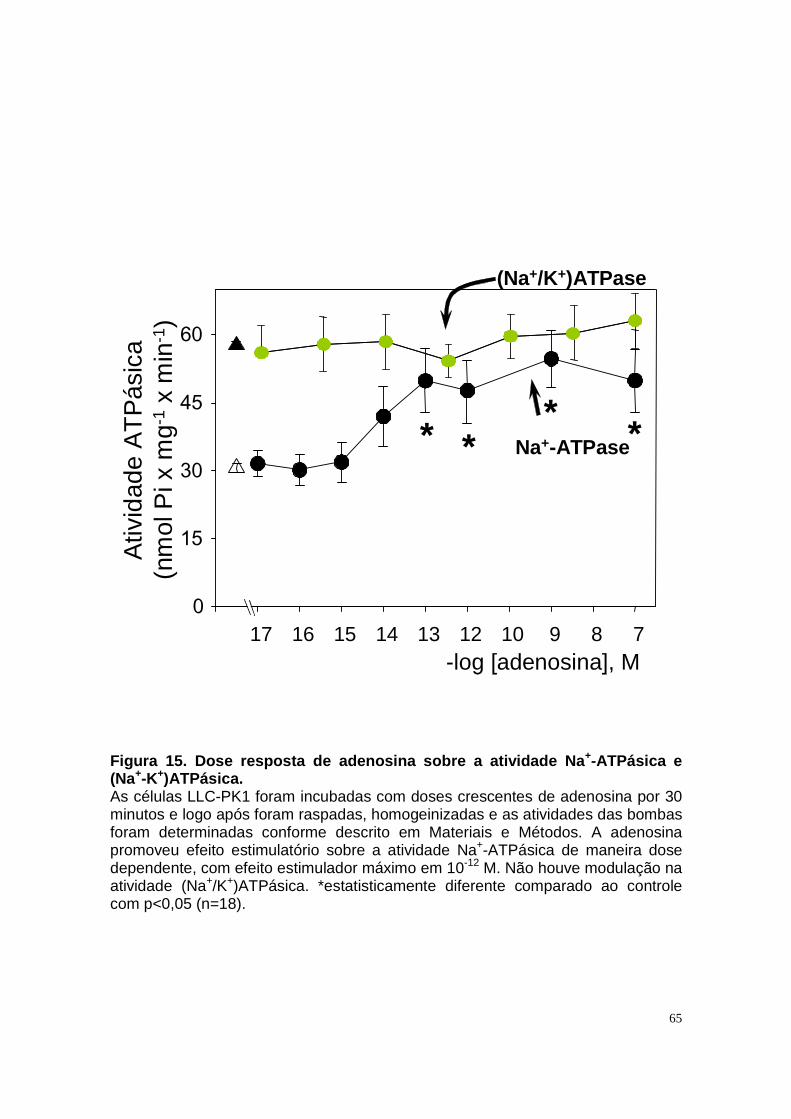
65
Figura 15. Dose resposta de adenosina sobre a ativi dade Na +-ATPásica e (Na+-K+)ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de adenosina por 30 minutos e logo após foram raspadas, homogeinizadas e as atividades das bombas foram determinadas conforme descrito em Materiais e Métodos. A adenosina promoveu efeito estimulatório sobre a atividade Na+-ATPásica de maneira dose dependente, com efeito estimulador máximo em 10-12 M. Não houve modulação na atividade (Na+/K+)ATPásica. *estatisticamente diferente comparado ao controle com p<0,05 (n=18).
-log [adenosina], M17 16 15 14 13 12 10 9 8 7
0
15
30
45
60
(Na+/K+)ATPase
Na+-ATPase
Ativ
idad
e A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*** *
-log [adenosina], M17 16 15 14 13 12 10 9 8 7
0
15
30
45
60
(Na+/K+)ATPase
Na+-ATPase
Ativ
idad
e A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*** *

66
Figura 16. Dose resposta de adenina sobre a ativida de Na+-ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de adenina por 30 minutos e logo após foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O quadrado representa a condição controle, e os cículos representam concentrações crescentes de adenina. Adenina não modulou a atividade Na+-ATPásica, nas concentrações de 10-13 até 10-5 M. (n=6).
13 11 9 7 50
10
20
30
40
-log [adenina], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
13 11 9 7 50
10
20
30
40
-log [adenina], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
13 11 9 7 50
10
20
30
40
-log [adenina], M13 11 9 7 5
0
10
20
30
40
-log [adenina], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)

67
Figura 17. Dose resposta de hipoxantina sobre a ati vidade Na +-ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de hipoxantina por 30 minutos e logo após foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O quadrado representa a condição controle, e os cículos representam concentrações crescentes de hipoxantina. Hipoxantina não modulou a atividade Na+-ATPásica, nas concentrações de 10-12 até 10-6 M. (n=7).
-log [hipoxantina], M12 10 9 8 6
0
10
20
30
40
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
-log [hipoxantina], M12 10 9 8 6
0
10
20
30
40
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
-log [hipoxantina], M12 10 9 8 6
0
10
20
30
40
-log [hipoxantina], M12 10 9 8 6
0
10
20
30
40
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
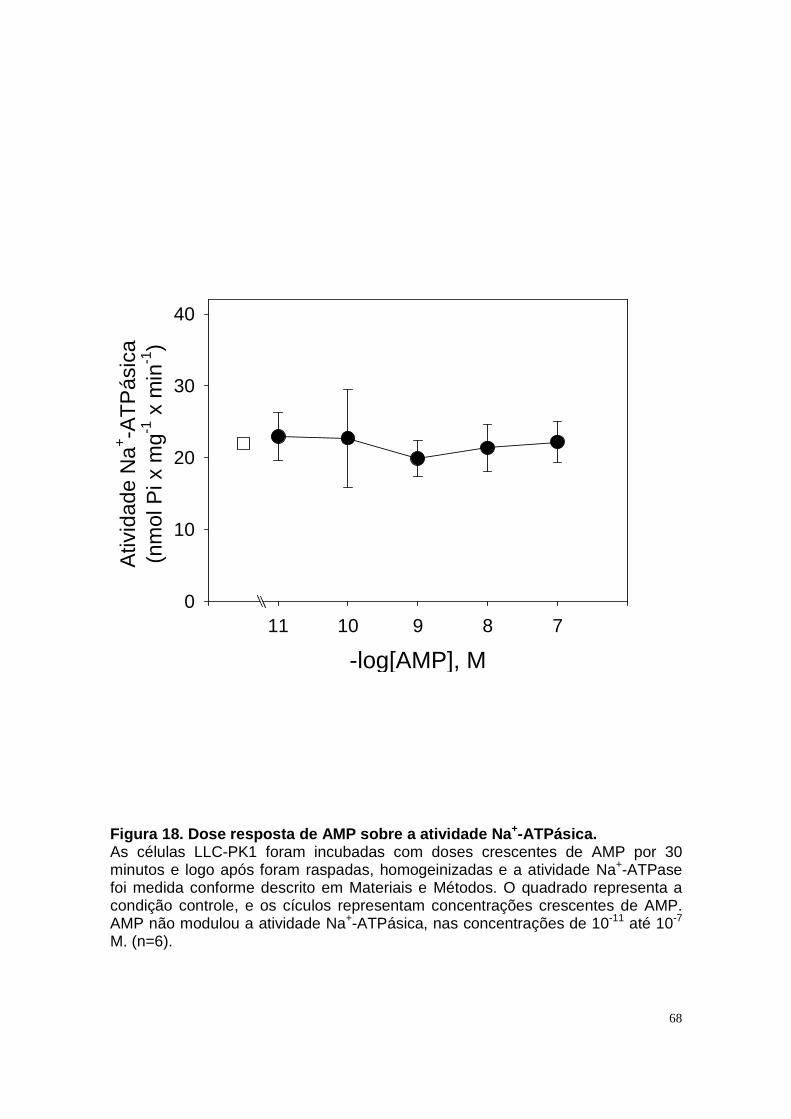
68
Figura 18. Dose resposta de AMP sobre a atividade N a+-ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de AMP por 30 minutos e logo após foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O quadrado representa a condição controle, e os cículos representam concentrações crescentes de AMP. AMP não modulou a atividade Na+-ATPásica, nas concentrações de 10-11 até 10-7 M. (n=6).
-log[AMP], M
11 10 9 8 7
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
0
10
20
30
40

69
Figura 19. Dose resposta de ADP sobre a atividade N a+-ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de ADP por 30 minutos e logo após foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O quadrado representa a condição controle, e os cículos representam concentrações crescentes de ADP. ADP não modulou a atividade Na+-ATPásica, nas concentrações de 10-11 até 10-7 M. (n=8).
-log [ADP], M
11 10 9 8 7
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
0
10
20
30
40
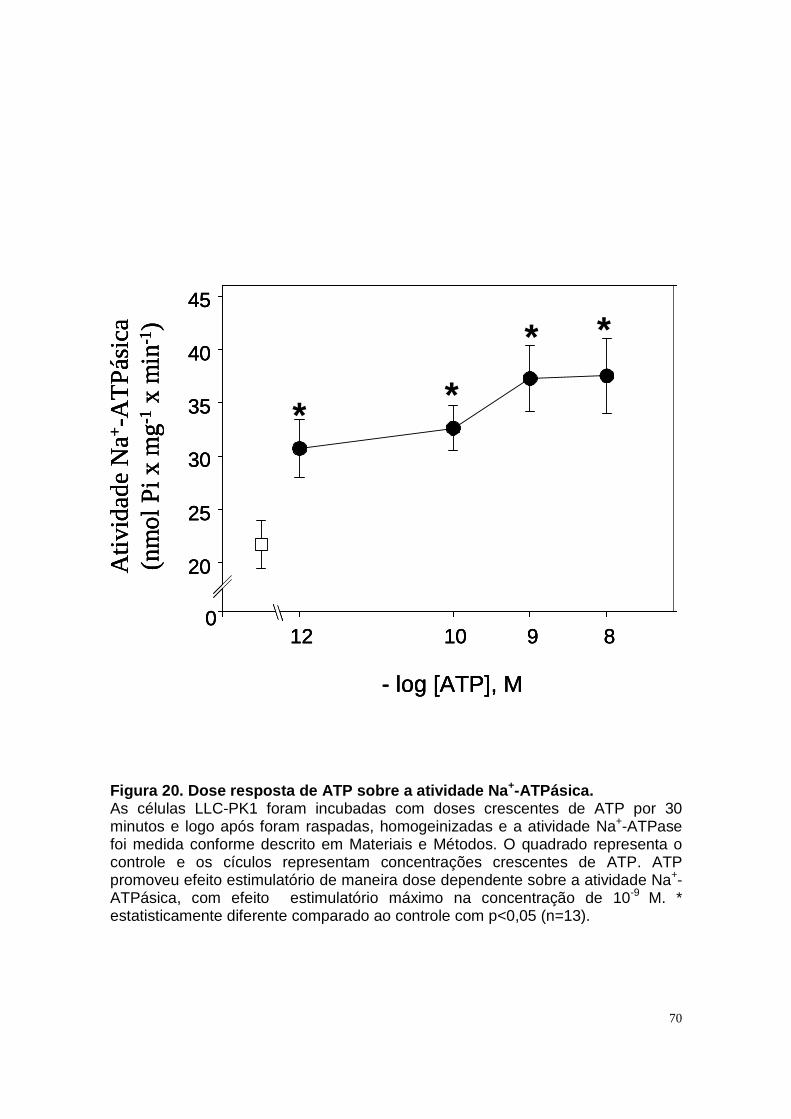
70
Figura 20. Dose resposta de ATP sobre a atividade N a+-ATPásica. As células LLC-PK1 foram incubadas com doses crescentes de ATP por 30 minutos e logo após foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O quadrado representa o controle e os cículos representam concentrações crescentes de ATP. ATP promoveu efeito estimulatório de maneira dose dependente sobre a atividade Na+-ATPásica, com efeito estimulatório máximo na concentração de 10-9 M. * estatisticamente diferente comparado ao controle com p<0,05 (n=13).
12 10 9 80
20
25
30
35
40
45
*
*
*
*
- log [ATP], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mo
lPi x
mg-1
x m
in-1)
12 10 9 80
20
25
30
35
40
45
*
*
*
*
- log [ATP], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mo
lPi x
mg-1
x m
in-1)
12 10 9 80
20
25
30
35
40
45
*
*
*
*
- log [ATP], M
Ativ
idad
e N
a+-A
TP
ásic
a(n
mo
lPi x
mg-1
x m
in-1)

71
Considerando a ausência de efeito da adenosina sobre a (Na++K+)ATPase
e que ATP pode ser produzido a partir de adenosina, não testamos o efeito do
ATP sobre a (Na++K+)ATPase. Baseado nas observações de que adenosina é
convertida em nucleotídeos de adenina e que o efeito de ATP na atividade da Na+-
ATPase é similar aquele observado para adenosina, decidimos investigar a
possibilidade do efeito de adenosina sobre a enzima ser mediado por ATP.
4.3. Envolvimento de receptores específicos para ATP e canais de ânions no
mecanismo estimulatório da adenosina sobre a Na+-ATPase em células LLC-PK1
Com o objetivo de determinar os receptores envolvidos na modulação da
atividade Na+-ATPásica por adenosina utilizamos antagonistas seletivos para os
diferentes subtipos de receptores P1 e P2. Os resultados obtidos demonstram que
o efeito de adenosina não é revertido por DPCPX 10-7 M, DMPX 10-6 M ou MRS
10-9 M, antagonistas seletivos de receptores A1, A2 e A3, respectivamente (Figura
21), indicando que o efeito da adenosina não é mediado por receptores do tipo P1.
Isoladamente, os antagonistas de receptores de adenosina não foram capazes de
promover alteração na atividade Na+-ATPásica (dados não mostrados). Por outro
lado, o efeito estimulatório do ATP sobre a atividade Na+-ATPásica foi revertido
por PPADS (50 e 10 µM), antagonista dos receptores P2 (Figura 22).
Dados da literatura demonstram que a conversão metabólica de
sinalizadores extracelulares purínicos pode resultar em inicio ou termino de efeitos
biológicos específicos (Jacksoni et al., 2003; Vallon et al., 2006). Ao observarmos
que o efeito induzido por adenosina não é mediado por seus receptores
específicos, decidimos investigar se este poderia depender de sua conversão em
ATP, que, como demonstrado acima, seria mediado por receptor P2. Deste modo,
determinamos a atividade Na+-ATPásica em homogeneizados de células
incubadas com adenosina, na presença do antagonista de receptor P2, PPADS. O
bloqueio do efeito da adenosina por PPADS 50 µM (Figura 23) foi a primeira
evidência de que o efeito induzido por adenosina depende de sua conversão em
ATP.
Para confirmar esta hipótese foram realizadas duas experiências descritas
a seguir. Na primeira delas, incubamos uma cultura de células confluentes com

72
adenosina 10-8 M, por 30 minutos. Recolhemos o meio de cultura, considerado por
nós como “meio condicionado”, e o adicionamos a duas outras culturas de células,
uma sem e outra com PPADS 50 µM. As culturas foram incubadas nestas
condições por 30 minutos. Em seguida, as células foram raspadas das placas,
homogeinizadas e a atividade Na+-ATPásica foi determinada em cada uma das
condições. Podemos observar que o “meio condicionado” promoveu efeito
estimulatório de 58 % sobre a atividade Na+-ATPásica (Figura 24), em intensidade
similar àquela induzida por adenosina 10-8 M e ATP 10-9 M (Figura 15 e 20). De
modo semelhante ao observado em células incubadas com adenosina, o efeito
induzido pelo “meio condicionado” gerado na presença de adenosina, também foi
bloqueado por PPADS. Em um segundo experimento, avaliamos os efeitos da
adenosina (10-8 M) e do ATP (10-9 M) na ausência e na presença de
iodotubericidina (Ido), inibidor de enzima adenosina cinase (envolvida na
fosforilação da adenosina a AMP, um importante precursor do ATP). Nossos
dados revelam que o efeito estimulatório de adenosina, mas não do ATP, foi
revertido por iodotubericidina (Figura25).
Em conclusão, os dados apresentados acima confirmam a hipótese de que
o efeito estimulatório induzido por adenosina sobre a atvidade Na+-ATPásica
depende de sua captação pela célula, e sua conversão em ATP, e que seria
mediado por receptores do tipo P2. Considerando a localização intracelular para a
síntese ATP e seu efeito mediado pela ligação a um sitio extracelular de um
receptor P2, tornou-se necessária a elucidação do mecanismo de liberação do
nucleotídeo.
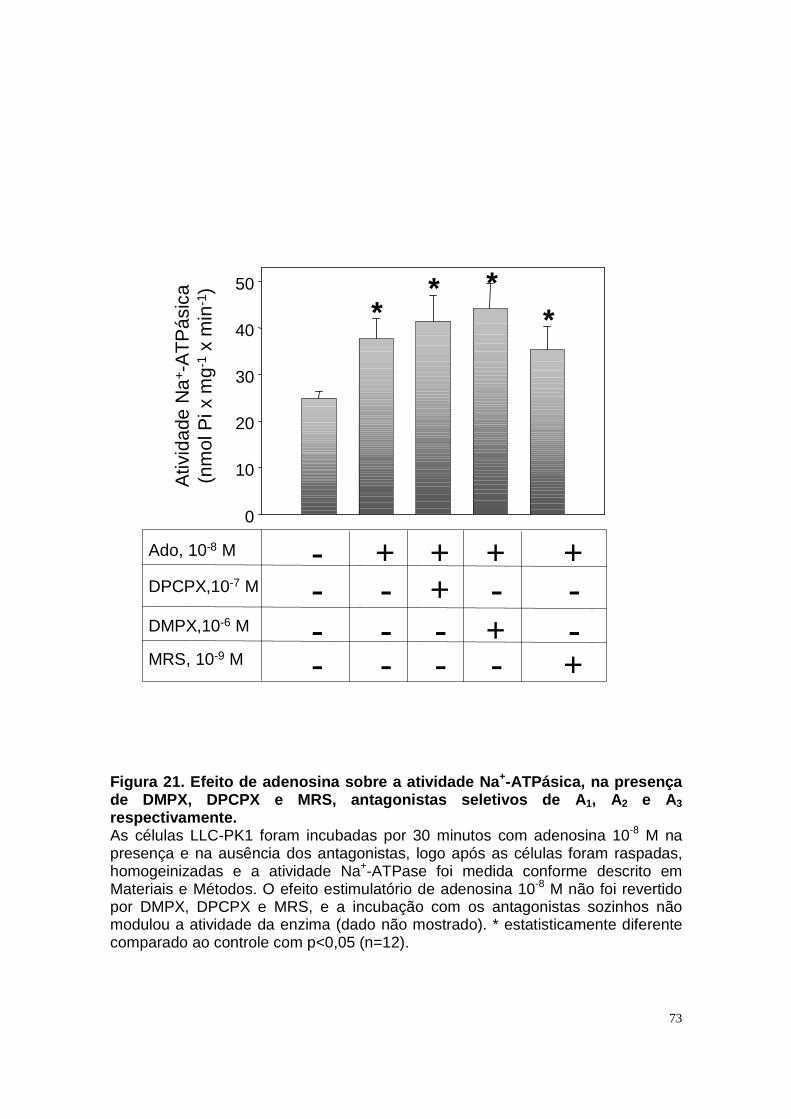
73
Figura 21. Efeito de adenosina sobre a atividade Na +-ATPásica, na presença de DMPX, DPCPX e MRS, antagonistas seletivos de A 1, A2 e A3 respectivamente. As células LLC-PK1 foram incubadas por 30 minutos com adenosina 10-8 M na presença e na ausência dos antagonistas, logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O efeito estimulatório de adenosina 10-8 M não foi revertido por DMPX, DPCPX e MRS, e a incubação com os antagonistas sozinhos não modulou a atividade da enzima (dado não mostrado). * estatisticamente diferente comparado ao controle com p<0,05 (n=12).
+- + +
+
++
+--
---
--
----
-
10
20
30
40
50
Ado, 10-8 M
DPCPX,10-7 M
DMPX,10-6 M
MRS, 10-9 M
0
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-
1x
min
-1)
***
*
+- + +
+
++
+--
---
--
----
-
10
20
30
40
50
Ado, 10-8 M
DPCPX,10-7 M
DMPX,10-6 M
MRS, 10-9 M
0
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-
1x
min
-1)
***
*
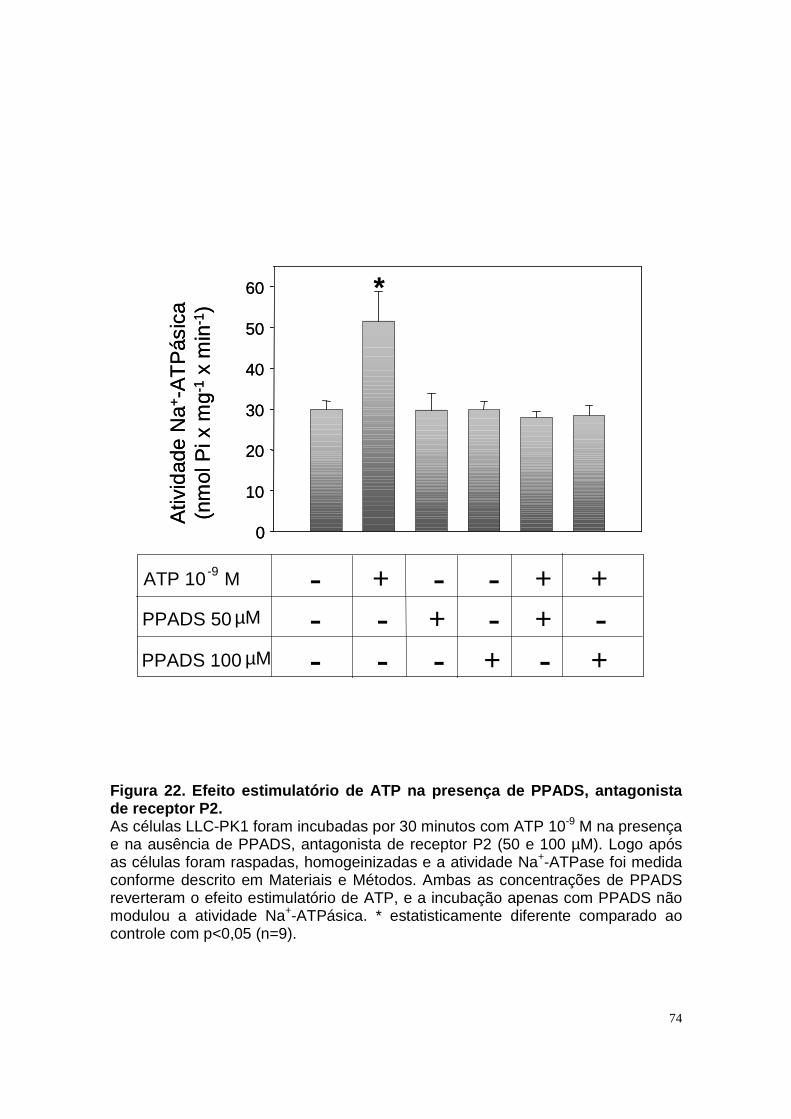
74
Figura 22. Efeito estimulatório de ATP na presença de PPADS, antagonista de receptor P2. As células LLC-PK1 foram incubadas por 30 minutos com ATP 10-9 M na presença e na ausência de PPADS, antagonista de receptor P2 (50 e 100 µM). Logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. Ambas as concentrações de PPADS reverteram o efeito estimulatório de ATP, e a incubação apenas com PPADS não modulou a atividade Na+-ATPásica. * estatisticamente diferente comparado ao controle com p<0,05 (n=9).
ATP 10 -9 M
PPADS 50 µΜ
PPADS 100 µΜ
---
--
-
-
--
--
++
+++
+
+
0
10
20
30
40
50
60
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*
ATP 10 -9 M
PPADS 50 µΜ
PPADS 100 µΜ
---
--
-
-
--
--
++
+++
+
+
0
10
20
30
40
50
60
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*
ATP 10 -9 M
PPADS 50 µΜ
PPADS 100 µΜ
---
--
-
-
--
--
++
+++
+
+
0
10
20
30
40
50
60
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*
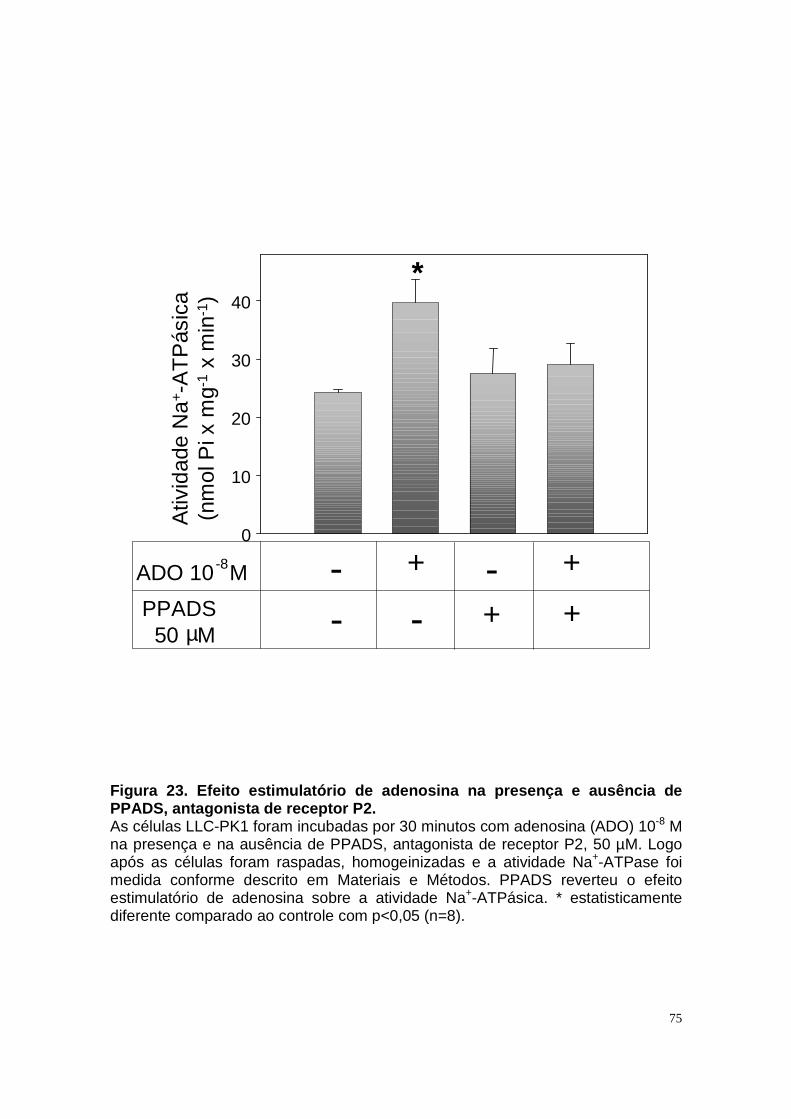
75
Figura 23. Efeito estimulatório de adenosina na pre sença e ausência de PPADS, antagonista de receptor P2. As células LLC-PK1 foram incubadas por 30 minutos com adenosina (ADO) 10-8 M na presença e na ausência de PPADS, antagonista de receptor P2, 50 µM. Logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. PPADS reverteu o efeito estimulatório de adenosina sobre a atividade Na+-ATPásica. * estatisticamente diferente comparado ao controle com p<0,05 (n=8).
0
10
20
30
40
ADO 10 -8M
PPADS 50 µM
+--
--
+
+ +
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*
0
10
20
30
40
ADO 10 -8M
PPADS 50 µM
+--
--
+
+ +
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-1
x m
in-1
)
*
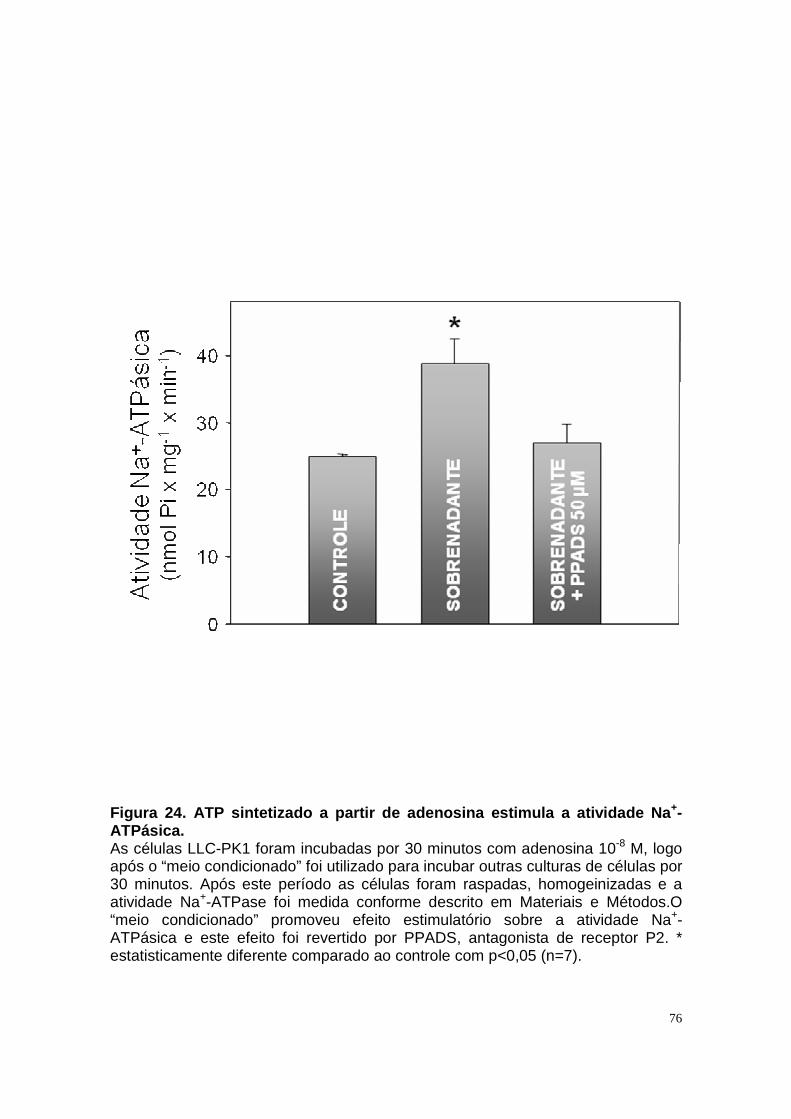
76
Figura 24. ATP sintetizado a partir de adenosina es timula a atividade Na +-ATPásica. As células LLC-PK1 foram incubadas por 30 minutos com adenosina 10-8 M, logo após o “meio condicionado” foi utilizado para incubar outras culturas de células por 30 minutos. Após este período as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos.O “meio condicionado” promoveu efeito estimulatório sobre a atividade Na+-ATPásica e este efeito foi revertido por PPADS, antagonista de receptor P2. * estatisticamente diferente comparado ao controle com p<0,05 (n=7).
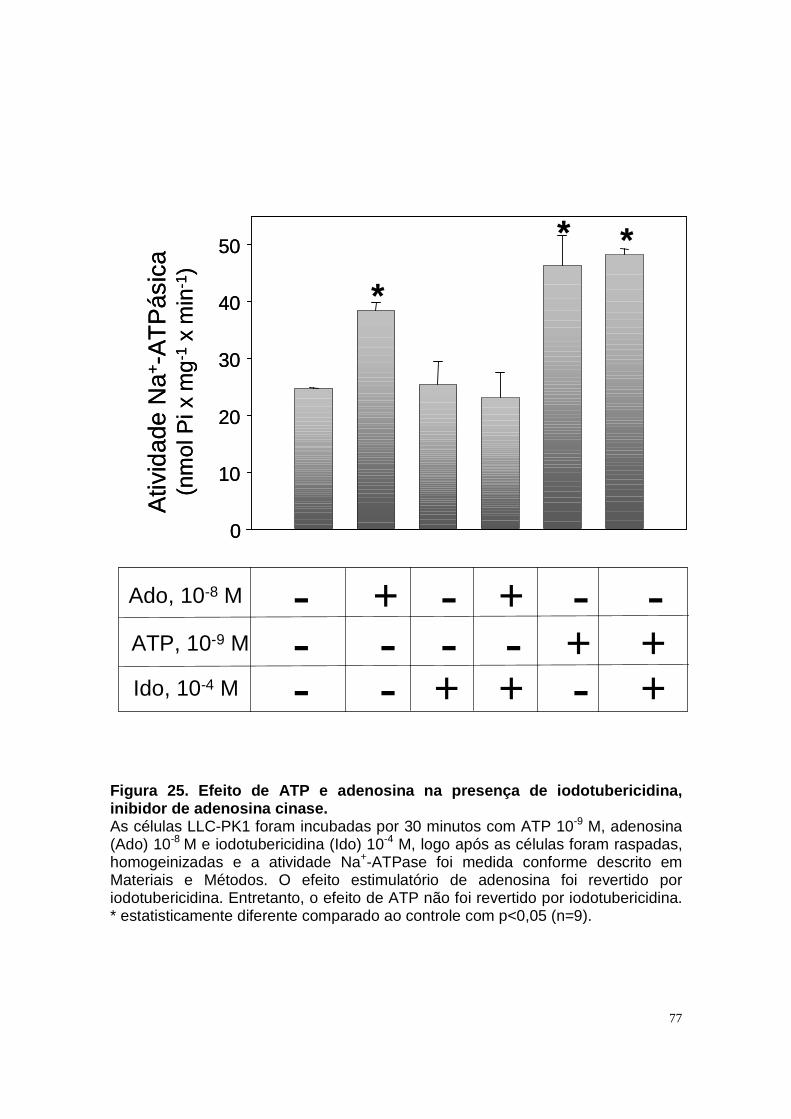
77
Figura 25. Efeito de ATP e adenosina na presença de iodotubericidina, inibidor de adenosina cinase. As células LLC-PK1 foram incubadas por 30 minutos com ATP 10-9 M, adenosina (Ado) 10-8 M e iodotubericidina (Ido) 10-4 M, logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. O efeito estimulatório de adenosina foi revertido por iodotubericidina. Entretanto, o efeito de ATP não foi revertido por iodotubericidina. * estatisticamente diferente comparado ao controle com p<0,05 (n=9).
0
10
20
30
40
50
Ado, 10-8 M
Ido, 10-4 M
ATP, 10-9 M
-++
+
+
+
+
+---
-
---
-
--
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-
1x
min
-1)
* *
*
0
10
20
30
40
50
Ado, 10-8 M
Ido, 10-4 M
ATP, 10-9 M
-++
+
+
+
+
+---
-
---
-
--
Ativ
idad
e N
a+-A
TP
ásic
a(n
mol
Pi x
mg-
1x
min
-1)
* *
*

78
O ATP é transportado para o meio extracelular por transporte vesicular ou
liberação mediada por canal (Sabirov e Okada, 2005). Como o ATP é carregado
negativamente, em pH fisiológico, os canais de ânions presentes na membrana
plasmática passaram a ser considerados como fortes candidatos a mediadores do
transporte de ATP para o meio extracelular. Esta função tem sido atribuída a
diferentes proteínas, tais como hemicanais de conexina, transportadores ABC
“ATP- Binding Cassete transporter” (tais como as proteínas MDR – Multi Drug
Resistance - e CFRT – “Cystic Fibrosis Transmembrane Conductance Regulator”),
e canais de ânion voltagem-dependente (Komlosi et al., 2005).
Considerando a existência de canais de ânions voltagem-dependentes
sensíveis a DIDS em células de túbulo proximal (Millar e Robson, 2001) decidimos
iniciar nossa investigação sobre o mecanismo de extrusão de ATP avaliando o
papel destes canais no processo. Ao avaliarmos a atividade Na+-ATPásica de
homogeneizados de células incubadas com adenosina (10-8 M), na presença e na
ausência de DIDS (10-4 e 10-5 M), observamos que o inibidor seletivo do canal de
ânions bloqueou o efeito da adenosina (Figura26), indicando a participação deste
canal no mecanismo de extrusão de ATP, uma etapa fundamental no mecanismo
estimulatório induzido por adenosina.
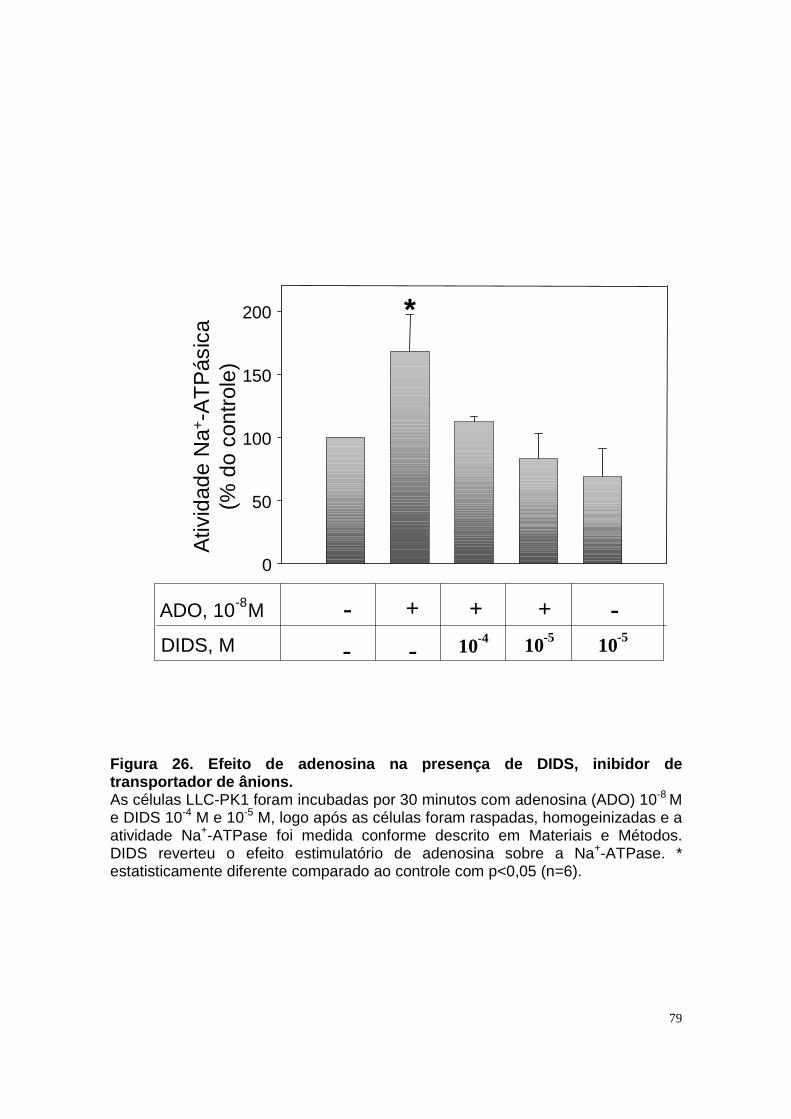
79
Figura 26. Efeito de adenosina na presença de DIDS, inibidor de transportador de ânions. As células LLC-PK1 foram incubadas por 30 minutos com adenosina (ADO) 10-8 M e DIDS 10-4 M e 10-5 M, logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. DIDS reverteu o efeito estimulatório de adenosina sobre a Na+-ATPase. * estatisticamente diferente comparado ao controle com p<0,05 (n=6).
0
50
100
150
200
ADO, 10-8M
DIDS, M
- + + + -
- 10-4 10-5 10-5-
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
*
0
50
100
150
200
ADO, 10-8M
DIDS, M
- + + + -
- 10-4 10-5 10-5-
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
*

80
4.4. Estudo da via de sinalização envolvida no estímulo da Na+-ATPase por ATP
Tem sido mostrado que a Na+-ATPase é modulada por proteínas cinases;
entre elas podemos destacar PKA e PKC (Caruso-Neves et al., 1997; Caruso-
Neves et al., 2000). A ativação destas ezimas isoladamente promove o aumento
da atividade Na+-ATPase (Wengert et al., 2005; Wengert et al., 2007). A próxima
etapa teve como objetivo verificar o papel de PKA e PKC na modulação da Na+-
ATPase por ATP. Portanto, para avaliarmos a possibilidade de envolvimento
destas vias no mecanismo de estimulação da Na+-ATPase por ATP e adenosina,
medimos as atividades de PKA e PKC em resposta a ambos. Com este objetivo,
incubamos culturas confluentes de células LLC-PK1 com ATP 10-9 M ou
adenosina 10-8 M, na presença ou ausência de PPADS 5x10-5 M, por período de
30 minutos. Nossos dados revelam que ATP (10-9 M) e adenosina (10-8 M) não
foram capazes de modular a atividade de PKA (Figura 27). Por outro lado,
adenosina e ATP estimularam a atividade de PKC de maneira similar, sendo estes
efeitos revertidos por PPADS,.50 µM, antagonista de receptor P2 (Figura 28). Para
confirmar que a PKC estava envolvida no efeito estimulatório de ATP sobre a Na+-
ATPase, a atividade Na+-ATPásica foi medida na presença de calfostina C 10-8 M,
inibidor de PKC. Os efeitos estimulatórios de adenosina 10-8 M e ATP10-9 M foram
revertidos pelo inibidor específico de PKC, confirmando o envolvimento de PKC
nos estímulos de adenosina e ATP sobre a bomba de sódio (Figura 29).
A hidrólise de fosfatilinositol 4,5 bisfosfato (PIP2) por PLC resulta em
produção de diacilglicerol (DAG) e inositol-3P (IP3). A ativação de PKC pode
resultar da sua interação com DAG e o íon cálcio. No caso da isoforma sensível
ao calcio, a ativação de PKC não dependeria exclusivamente da atividade de PLC,
mas também da liberação do cátion a partir de estoque intracelular, no reticulo
endoplasmático, promovida pelo IP3. Uma vez que adenosina e ATP promoveram
efeito estimulatório sobre a atividade de PKC, o próximo passo foi investigar a
participação do cálcio intracelular no mecanismo de sinalização disparado por
ATP. Os níveis intracelulares de cálcio foram determinados por fluorescência,
usando um derivado, permeável à membrana, do fluoróforo denominado Fura-2.

81
Inicialmente, as células foram incubadas com Fura-2 por 40 minutos na presença
de probenecide, para evitar que o cromóforo fosse capturado por organelas
celulares. Em seguida, as células foram perfundidas com salina para remoção do
Fura-2 extracelular e perfundidas (1 ml/min) com concentrações crescentes de
ATP (0,1 nM – 500 µM). Nossos resultados demonstram que concentrações de
ATP entre 0,1 nM e 20 µM não foram capazes de promover alterações nos níveis
de cálcio citoplasmático (Figura 30A). Por outro lado, concentrações entre 100 e
500 µM induziram aumento significativo (Figua 30B). Ao final de cada dose
resposta as células foram perfundidas com saponina 1% como controle da
permeabilidade celular. Apesar da demonstração do efeito estimulatorio do ATP
sobre a liberação de cálcio vesicular, nossos dados indicam que este efeito parece
não fazer parte do mecanismo envolvido na ativação da isoforma de PKC
responsável pela estimulação da atividade da Na+-ATPase por ATP, pois a
concentração do nucleotídeo necessária para promover estimulação máxima da
PKC e da ATPase foi igual a 10-9 M, uma concentração incapaz de promover
alterações nos níveis de cálcio citoplasmático.
Este dados indicam que a modulação da Na+-ATPase em células LLC-PK1
por adenosina é mediada por ATP e envolve a ativação de uma PKC insensível a
Ca2+.

82
Figura 27. Efeito de ATP e adenosina sobre a ativid ade de PKA. As células foram incubadas por 30 minutos com adenosina, ATP, na presença e na ausência de PPADS, antagonista de receptor P2. Logo após as células foram raspadas, homogeinizadas e a atividade de PKA foi medida conforme descrito em Materiais e Métodos. Adenosina e ATP não foram capazes de modular a atividade de PKA (n=4).
0
1
2
3
4
ATP 10 -9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
+++
---
--+--
Ativ
idad
e P
KA
(pm
olde
his
tona
-P x
mg-1
x m
in-1
)
0
1
2
3
4
ATP 10 -9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
+++
---
--+--
0
1
2
3
4
ATP 10 -9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
+++
---
--+--
Ativ
idad
e P
KA
(pm
olde
his
tona
-P x
mg-1
x m
in-1
)

83
Figura 28. Efeito de ATP e adenosina sobre a ativid ade de PKC. As células foram incubadas por 30 minutos com adenosina, ATP, na presença e na ausência de PPADS, logo após as células foram raspadas, homogeinizadas e a atividade de PKC foi medida conforme descrito em Materiais e Métodos. Adenosina (ADO) e ATP estimularam a atividade de PKC e PPADS, antagonista de receptor P2, reverteu o efeito de ambos. * estatisticamente diferente comparado ao controle com p<0,05 (n=8).
Ativ
idad
e P
KC
(pm
olde
his
tona
-P x
mg-1
x m
in-1
)
0
1
2
3
4
5
6
ATP 10-9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
---
--+--
* *
Ativ
idad
e P
KC
(pm
olde
his
tona
-P x
mg-1
x m
in-1
)
0
1
2
3
4
5
6
ATP 10-9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
---
--+--
* *
0
1
2
3
4
5
6
ATP 10-9 M
PPADS 50 µM
ADO 10 -8 M
- ++ -
+++
---
--+--
* *
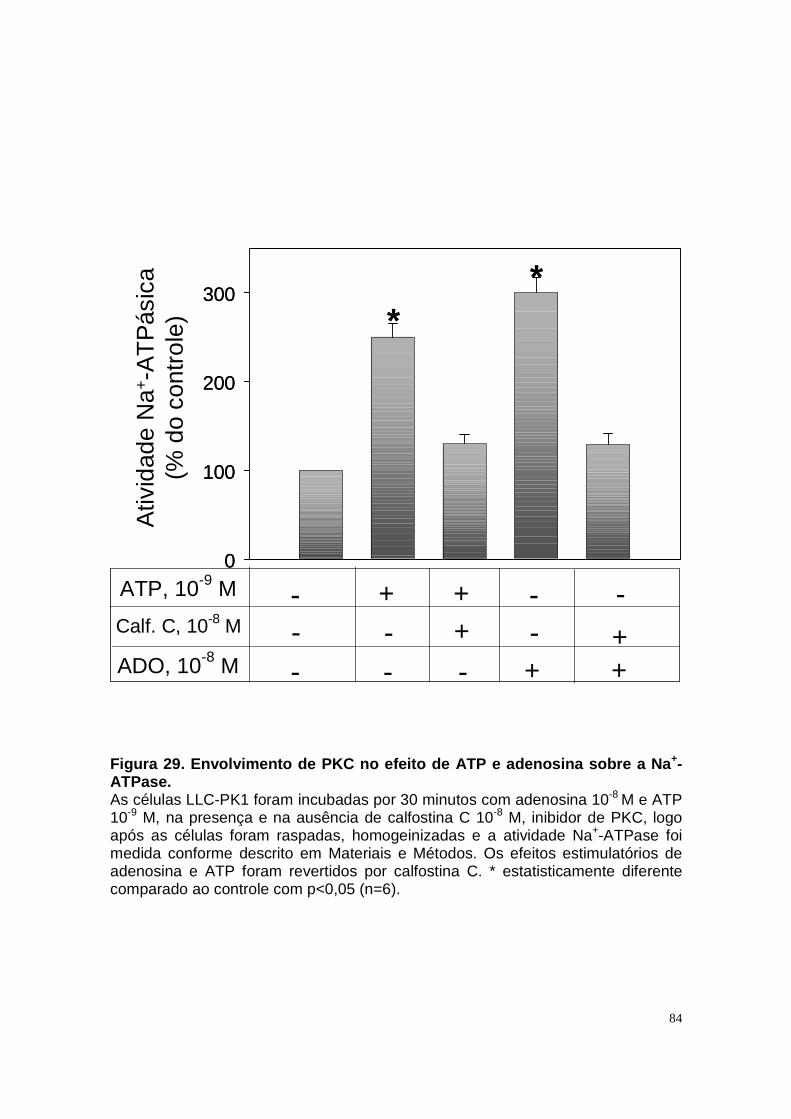
84
Figura 29. Envolvimento de PKC no efeito de ATP e a denosina sobre a Na +-ATPase. As células LLC-PK1 foram incubadas por 30 minutos com adenosina 10-8 M e ATP 10-9 M, na presença e na ausência de calfostina C 10-8 M, inibidor de PKC, logo após as células foram raspadas, homogeinizadas e a atividade Na+-ATPase foi medida conforme descrito em Materiais e Métodos. Os efeitos estimulatórios de adenosina e ATP foram revertidos por calfostina C. * estatisticamente diferente comparado ao controle com p<0,05 (n=6).
0
100
200
300
Col 1
ATP, 10-9 M
Calf. C, 10-8 M
ADO, 10-8 M
+
-
+
+
+
++- -
-
--- -
-
**
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
0
100
200
300
Col 1
ATP, 10-9 M
Calf. C, 10-8 M
ADO, 10-8 M
+
-
+
+
+
++- -
-
--- -
-
**
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
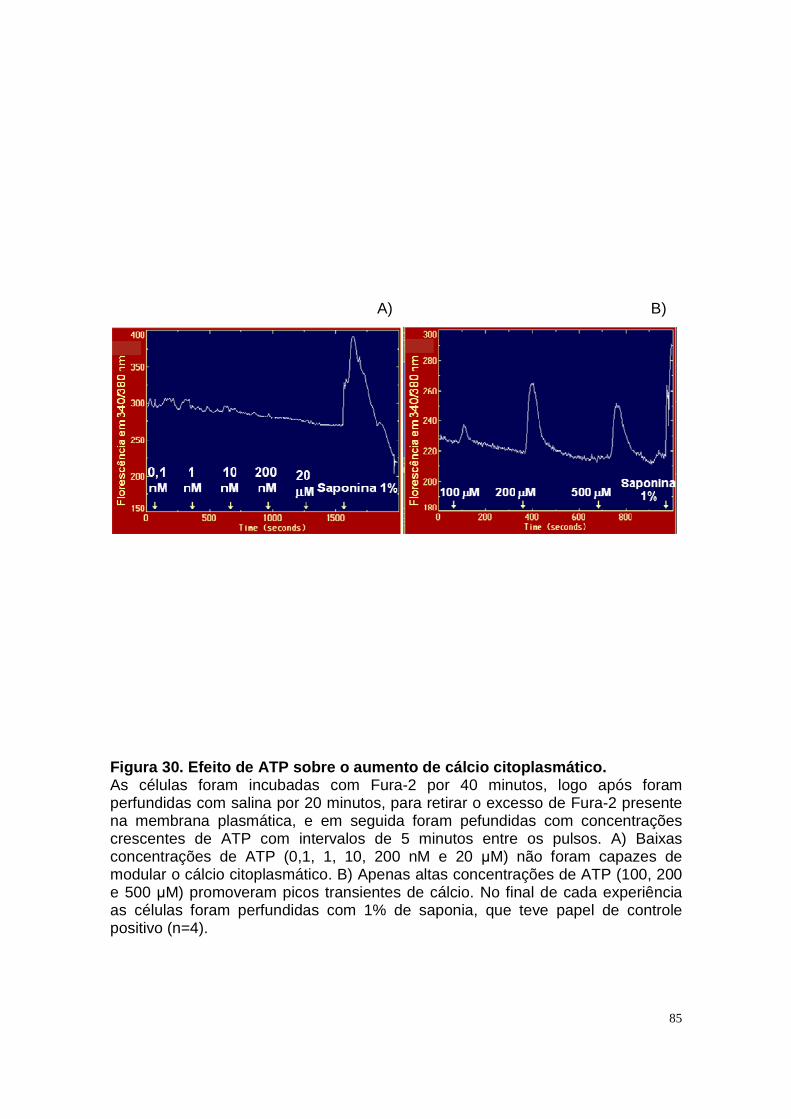
85
A) B)
Figura 30. Efeito de ATP sobre o aumento de cálcio citoplasmático. As células foram incubadas com Fura-2 por 40 minutos, logo após foram perfundidas com salina por 20 minutos, para retirar o excesso de Fura-2 presente na membrana plasmática, e em seguida foram pefundidas com concentrações crescentes de ATP com intervalos de 5 minutos entre os pulsos. A) Baixas concentrações de ATP (0,1, 1, 10, 200 nM e 20 µM) não foram capazes de modular o cálcio citoplasmático. B) Apenas altas concentrações de ATP (100, 200 e 500 µM) promoveram picos transientes de cálcio. No final de cada experiência as células foram perfundidas com 1% de saponia, que teve papel de controle positivo (n=4).

86
4.5. Estudo da isquemia metabólica sobre o metabolismo celular de adenosina
A condição de isquemia renal pode ser observada em situações de
transplante renal ou de estenose da artéria renal, sendo caracterizada pela perda
da circulação vascular e uma conseqüente depleção de oxigênio e nutrientes.
Dados descritos na literatura demonstram a ocorrência de depleção de ATP e
aumento dos níveis de seus metabólitos (ADP, AMP e adenosina), durante
episodio isquêmico renal (Vallon et al., 2006).
O conjunto de resultados apresentado acima demonstra que a adição de
adenosina ao meio de cultura de células LLC-PK1, numa condição metabólica
normal (normóxia), resulta em elevação dos níveis extracelulares de ATP,
hipoxantina e adenina. Considerando a elevação do nível de adenosina em uma
situação de hipóxia e a necessidade do oxigênio como aceptor final de elétrons no
processo de fosforilaçao oxidativa do ADP para geração do ATP, decidimos
avaliar os níveis nucleotídeos de [3H]-adenina, bem como dos outros derivados,
produzidos a partir do metabolismo de [3H]-adenosina adicionada ao meio de
cultura em uma condição de isquemia. Mimetizando uma condição de isquemia
metabólica, culturas de células LLC-PK1 foram submetidas a um tratamento com
antimicina A e 2-deoxiglicose com o objetivo de reduzir a síntese de ATP a partir
da glicolise e da fosforilaçao oxidativa.
Nossos resultados demonstram que os níveis intra e extracelulares de
nucleotídeos de [3H]-adenina produzidos a partir de [3H]-adenosina adicionada ao
meio de cultura foram significantemente reduzidos em condição de hipóxia,
quando comparada à condição de normóxia (Figura 31). Verificamos, ainda, que a
redução do nível extracelular de nucleotídeos de [3H]-adenina observada na
condição de isquemia não se deve a alterações na fração liberada, pois cerca de
30% dos nucleotídeos de [3H]-adenina produzidos a partir da [3H]-adenosina são
liberados para o meio extracelular em ambas as condições metabólicas, normóxia
e hipóxia (Figura 32). Este resultado demonstra que a permeabilidade aparente da
célula ao nucleotídeo não foi afetada pela condição de isquemia metabólica.

87
Embora tenhamos demonstrado que a estimulação da Na+-ATPase por
adenosina tem um mecanismo envolvendo a liberação de nucleotídeos de adenina
por um canal de ânions sensível a DIDS (Figura 26), podemos observar que este
inibidor reduz apenas parcialmente (mais de 50%) a liberação de nucleotídeos de
[3H]-adenina em condição de normóxia (Figura 32). Por outro lado, em condição
de isquemia DIDS não modifica a liberação de nucleotídeos de [3H]-adenina
(Figura 32). Estes resultados indicam que apesar da liberação de nucleotídeos de
[3H]-adenina seguir a mesma proporção em normóxia e isquemia, elas acontecem
por canais diferentes. Em isquemia esta liberação de nucleotídeos de [3H]-adenina
ocorre através de um mecanismo insensível ao DIDS, enquanto que em normóxia
este mecanismo envolve tanto canais sensíveis a DIDS quanto canais insensíveis
a DIDS.
Ao avaliarmos os níveis intra e extracelulares de [3H]-adenosina verificamos
que a isquemia não foi capaz de promover qualquer alteração significativa dos
níveis absolutos ou relativos (%) de [3H]-adenosina observados em normóxia
(Figura 33 A e B, respectivamente). Considerando que a condição de isquemia
reduziu a produção de nucleotídeos, mas não resultou no esperado aumento do
nível intracelular de [3H]-adenosina, avaliamos os níveis dos outros produtos
metabólicos gerados nesta condição. Nenhuma diferença foi observada entre os
níveis intra e extracelulares de hipoxantina ou inosina (Figuras 34 e 35,
respectivamente). Por outro lado, os níveis de adenina extracelular, mas não
intracelular, foram significativamente mais elevados na condição de hipoxia que
em normoxia (Figura 36). Este acúmulo mais elevado de [3H]-adenina no meio
extracelular na condição de isquemia poderia resultar da menor demanda de
precursores de nucleotídeos de [3H]-adenina no meio intracelular.

88
Figura 31. Níveis intra e extracelular de nucleotíd eos de [ 3H]-adenina em condições de normóxia e hipóxia metabólica. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilação oxidativa e análogo não metabolizável de glicose, respectivamente) por período de 10 minutos. Logo após, a solução salina contendo antimicina A e 2 desoxiglicose foi substituida por outra contendo apenas [3H]-adenosina 10-7 M e as células foram incubadas por mais 30 minutos. Os níveis de nucleotídeos de [3H]-adenina foram determinados por TLC conforme descrito em Materiais e Métodos. Ocorreu uma diminuição nos níveis intra e extracelular de nucleotídeos de [3H]-adenina na condição de isquemia comparada à condição de normóxia. * estatisticamente diferente comparado a condição de normóxia com p<0,05 (n=5).
pmol
de
nucl
eotíd
eo d
e [3 H
]-ad
enin
a x
mg-1
x m
in-1
0
400
800
1200
1600 normóxiaisquemia
intracelular extracelular
*
*
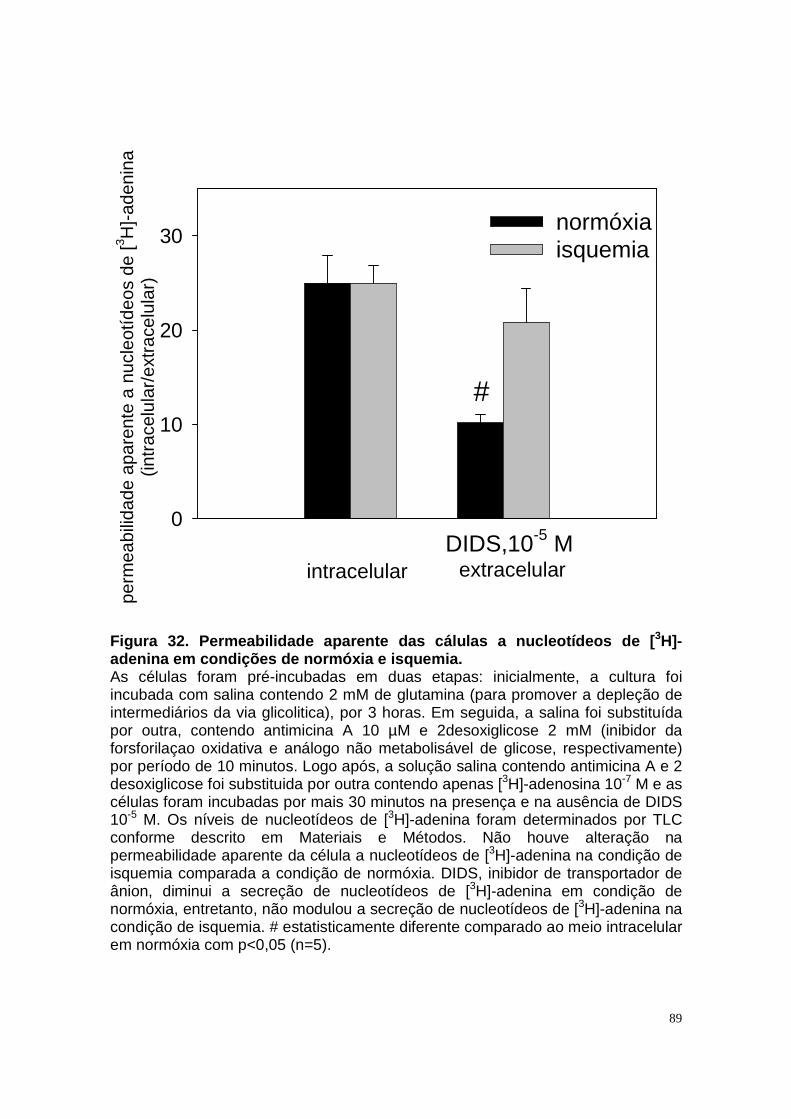
89
perm
eabi
lidad
e ap
aren
te a
nuc
leot
ídeo
s de
[3 H]-
aden
ina
(intr
acel
ular
/ext
race
lula
r)
0
10
20
30normóxiaisquemia
DIDS,10-5 M
#
intracelular extracelular
Figura 32. Permeabilidade aparente das cálulas a nu cleotídeos de [ 3H]-adenina em condições de normóxia e isquemia. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por período de 10 minutos. Logo após, a solução salina contendo antimicina A e 2 desoxiglicose foi substituida por outra contendo apenas [3H]-adenosina 10-7 M e as células foram incubadas por mais 30 minutos na presença e na ausência de DIDS 10-5 M. Os níveis de nucleotídeos de [3H]-adenina foram determinados por TLC conforme descrito em Materiais e Métodos. Não houve alteração na permeabilidade aparente da célula a nucleotídeos de [3H]-adenina na condição de isquemia comparada a condição de normóxia. DIDS, inibidor de transportador de ânion, diminui a secreção de nucleotídeos de [3H]-adenina em condição de normóxia, entretanto, não modulou a secreção de nucleotídeos de [3H]-adenina na condição de isquemia. # estatisticamente diferente comparado ao meio intracelular em normóxia com p<0,05 (n=5).
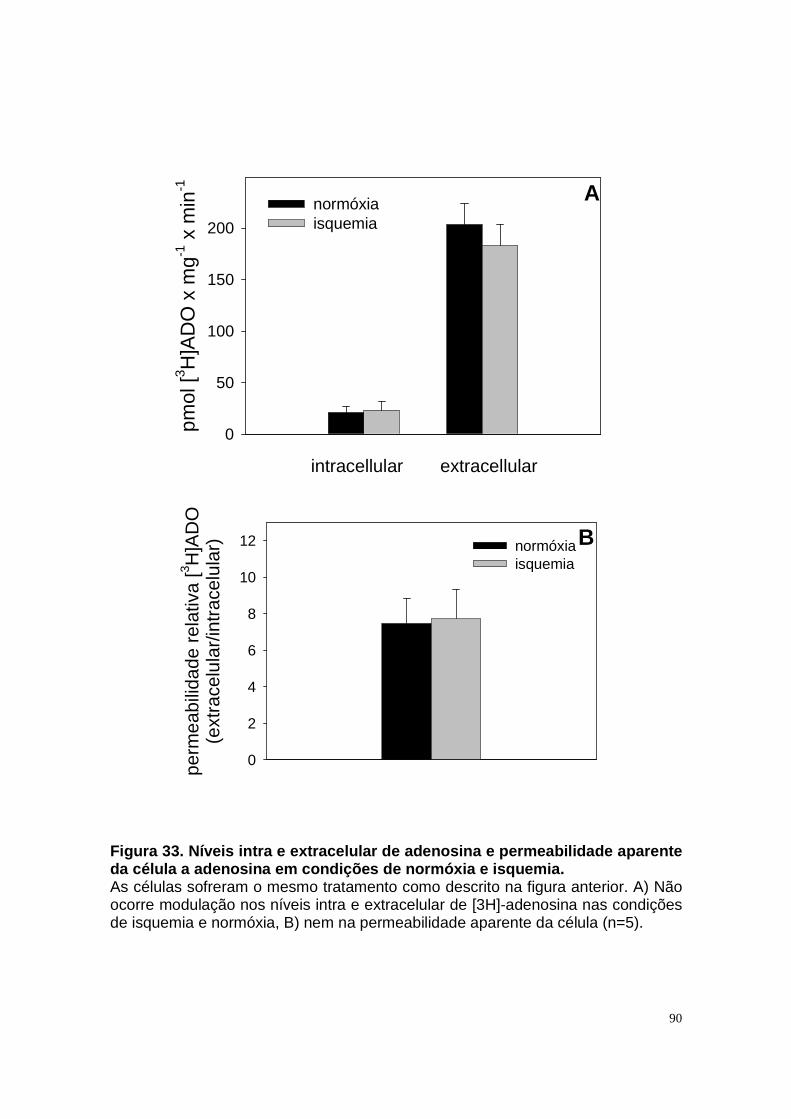
90
pmol
[3 H]A
DO
x m
g-1 x
min
-1
0
50
100
150
200
normóxiaisquemia
intracellular extracellular
A
perm
eabi
lidad
e re
lativ
a [3 H
]AD
O(e
xtra
celu
lar/
intr
acel
ular
)
0
2
4
6
8
10
12 normóxiaisquemia
B
Figura 33. Níveis intra e extracelular de adenosina e permeabilidade aparente da célula a adenosina em condições de normóxia e is quemia. As células sofreram o mesmo tratamento como descrito na figura anterior. A) Não ocorre modulação nos níveis intra e extracelular de [3H]-adenosina nas condições de isquemia e normóxia, B) nem na permeabilidade aparente da célula (n=5).

91
Figura 34. Níveis intra e extracelular de hipoxanti na formada a partir de adenosina em condições de normóxia e isquemia. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por período de 10 minutos. Logo após, a solução salina contendo antimicina A e 2 desoxiglicose foi substituida por outra contendo apenas [3H]-adenosina 10-7 M e as células foram incubadas por mais 30 minutos. Os níveis de [3H]-hipoxantina foram determinados por TLC conforme descrito em Materiais e Métodos. Não ocorre modulação nos níveis intra e extracelular de [3H]-hipoxantina formada a partir de adenosina nas condições de isquemia e normóxia. (n=5).
pmol
[3 H]-
hipo
xant
ina
x m
g-1 x
min
-1
0
200
400
600normóxiaisquemia
intracellular extracellular

92
Figura 35. Níveis intra e extracelular de inosina f ormada a partir de adenosina em condições de normóxia e isquemia. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por período de 10 minutos. Logo após, a solução salina contendo antimicina A e 2 desoxiglicose foi substituida por outra contendo apenas [3H]-adenosina 10-7 M e as células foram incubadas por mais 30 minutos. Os níveis de [3H]-inosina foram determinados por TLC conforme descrito em Materiais e Métodos. Não ocorre modulação nos níveis intra e extracelular de [3H]-inosina formada a partir de adenosina nas condições de isquemia e normóxia. (n=5).
pmol
[3 H] i
nosi
na x
mg-1
x m
in-1
0
50
100
150
normóxiaisquemia
intracellular extracellular

93
Figura 36. Níveis intra e extracelular de adenina e m condições de normóxia e isquemia. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por período de 10 minutos. Logo após, a solução salina contendo antimicina A e 2 desoxiglicose foi substituida por outra contendo apenas [3H]-adenosina 10-7 M e as células foram incubadas por mais 30 minutos. Os níveis de [3H]-adenina foram determinados por TLC conforme descrito em Materiais e Métodos. Não ocorre modulação no nível intracelular de [3H]-adenina na condição de isquemia comparada à condição de normóxia. Porém, ocorre um aumento no nível extracelular de [3H]-adenina na condição de isquemia comparada à condição de normóxia. * estatisticamente diferente comparado ao meio extracelular em normóxia com p<0,05 (n=5).
pmol
[3 H]-
aden
ina
x m
g-1 x
min
-1
0
200
400
600
800
1000
1200
1400normóxiaisquemia
intracellular extracellular
*

94
4.6. Efeitos da isquemia metabólica sobre as atividades das enzimas Na+-ATPase
e (Na++K+)ATPase
Estudos realizados em camundongos demonstraram que um aumento na
excreção fracionária de sódio após um período de 30 minutos de isquemia é
revertido por reperfusão (Park et al., 2001). Por este motivo, decidimos avaliar a
possibilidade de participação de elementos tubulares envolvidos na reabsorção de
sódio no túbulo proximal em uma condição de isquemia e reperfusão, usando
nosso modelo celular para estudo dos efeitos da isquemia metabólica. Com este
objetivo, avaliamos os efeitos da isquemia e reperfusão sobre as atividades Na+-
ATPase e (Na++K+)ATPase.
Culturas de células LLC-PK1 confluentes foram submetidas a dois períodos
distintos de isquemia metabólica (incubação na presença de Antimicina A e 2-
desoxi-glicose), 10 e 30. Em seguida, as células foram raspadas, homogeneizadas
e as atividades das enzimas Na+-ATPase e (Na++K+)ATPase foram determinadas.
Nossos resultados demonstram que a isquemia metabólica resultou em redução
significativa da atividade Na+-ATPásica nos dois períodos de tempo analisados,
quando comparadas às culturas de células que foram submetidas à condição de
normóxia (incubação em meio de cultura sem soro) (Figura 37). A lavagem das
células com PBS e subsequente reperfusão com meio sem antimicina e 2-
deoxiglicose reverteu o efeito inibitório da isquemia nos dois intervalos de tempo
analisados em relação a atividade da Na+-ATPase. Porém, com relação à
(Na++K+)ATPásica, nossos resultados demonstram que não houve modulação da
atividade da enzima nos tempos de 10 e 30 minutos de isquemia, demonstrando
sua maior resistência (Figura 38).
Considerando que a condição de isquemia reduz os níveis de nucleotídeos
extracelulares e que o ATP é um estimulador da atividade Na+-ATPásica,
podemos acreditar que a redução da atividade da enzima observada em condição
de isquemia poderia estar relacionada à queda nos níveis de ATP. Com a
finalidade de avaliarmos esta hipótese realizamos um experimento para verificar
se a pré-incubação com ATP poderia interferir no efeito da isquemia sobre a Na+-

95
ATPase. As culturas de células foram pré-incubadas com ATP (10-9 M), por 30
minutos, antes do inicio da isquemia metabólica. Ao final de um período de 10 ou
30 minutos em isquemia, as células foram raspadas e a atividade Na+-ATPásica
foi determinada. O tratamento (pré-condicionamento) com ATP bloqueou
totalmente o efeito inibitório da hipóxia de 10 minutos, e parcialmente a de 30
minutos (Figura 39). Esta reversão foi completamente abolida quando utilizamos
antagonista de receptor P2, PPADS 100µ M.
Os dados descritos acima demosntram que a isquemia metabólica em
tempos curtos (10 e 30 minutos) exerce um efeito inibitório específico sobre a Na+-
ATPase. Acreditamos que a inibição das atividades destas enzimas em condição
de isquemia possa contribuir para o aumento da excreção fracional de sódio
observada em episódios de isquemia renal. Além disso, mostramos que ATP
exerce um papel fundamental na proteção contra curtos eventos de isquemia.

96
Figura 37. Hipóxia metabólica inibe a atividade Na +-ATPásica e a reperfusão reverte este efeito. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por períodos de 10 e 30 minutos. Nas condições em que havia reperfusão a salina era trocada por meio de cultura sem soro. Logo após, as células foram raspadas, homogeinizadas e mediu-se a atividade Na+-ATPásica, conforme descrito em Materiais e Métodos. A isquemia inibiu a atividade Na+-ATPásica e a reperfusão por 2 horas reverteu a inibição. * estatisticamente diferente comparado ao controle com p<0,05 (n=9).
0
20
40
60
80
100
120
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
* *
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60-- - +
10
-30 10
+
30Tempo de isquemia
(min)
2h de reperfusão
0
20
40
60
80
100
120
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
* *
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60-- - +
10
-30 10
+
30Tempo de isquemia
(min)
2h de reperfusão
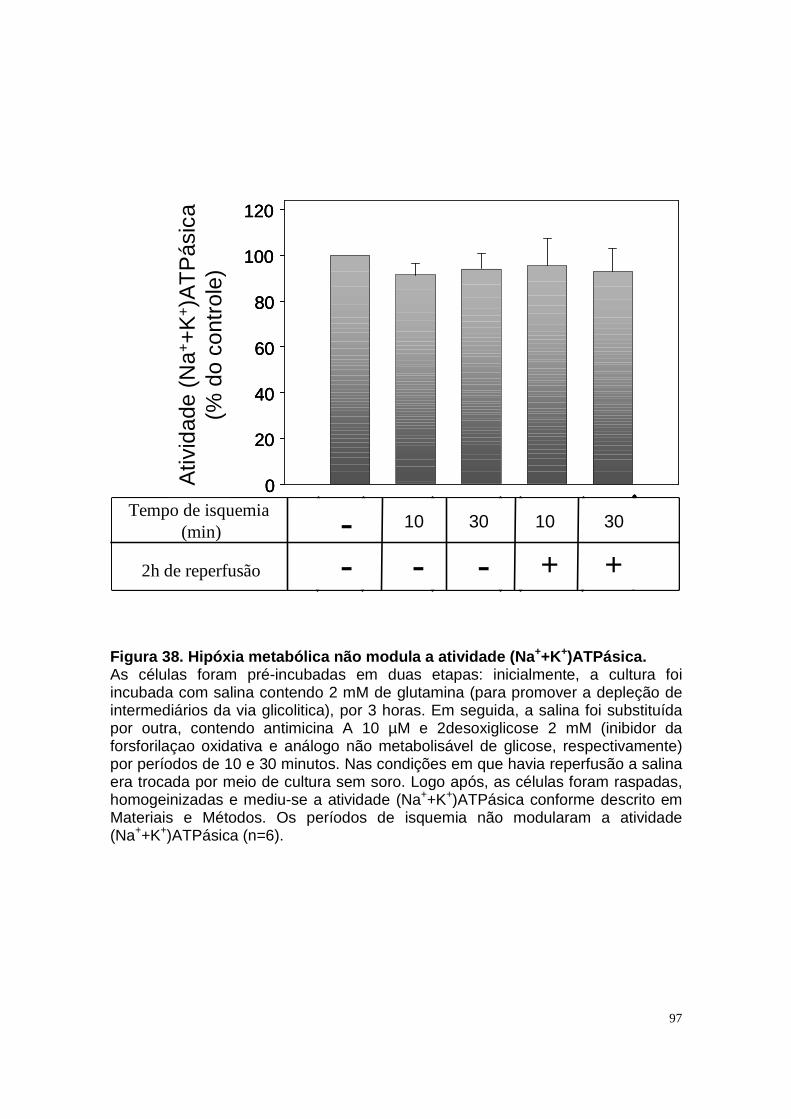
97
Figura 38. Hipóxia metabólica não modula a atividad e (Na++K+)ATPásica. As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por períodos de 10 e 30 minutos. Nas condições em que havia reperfusão a salina era trocada por meio de cultura sem soro. Logo após, as células foram raspadas, homogeinizadas e mediu-se a atividade (Na++K+)ATPásica conforme descrito em Materiais e Métodos. Os períodos de isquemia não modularam a atividade (Na++K+)ATPásica (n=6).
Ativ
idad
e (N
a++K
+)A
TP
ásic
a(%
do
cont
role
)
0
20
40
60
80
100
120
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60-- - +
10
-30 10
+
30Tempo de isquemia
(min)
2h de reperfusão
Ativ
idad
e (N
a++K
+)A
TP
ásic
a(%
do
cont
role
)
0
20
40
60
80
100
120
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60-- - +
10
-30 10
+
30Tempo de isquemia
(min)
2h de reperfusão
0
20
40
60
80
100
120
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60
2 h of reperfusion
ischemic time (min) -- - +
10
-30
-60 10
+
30
+
60-- - +
10
-30 10
+
30Tempo de isquemia
(min)
2h de reperfusão
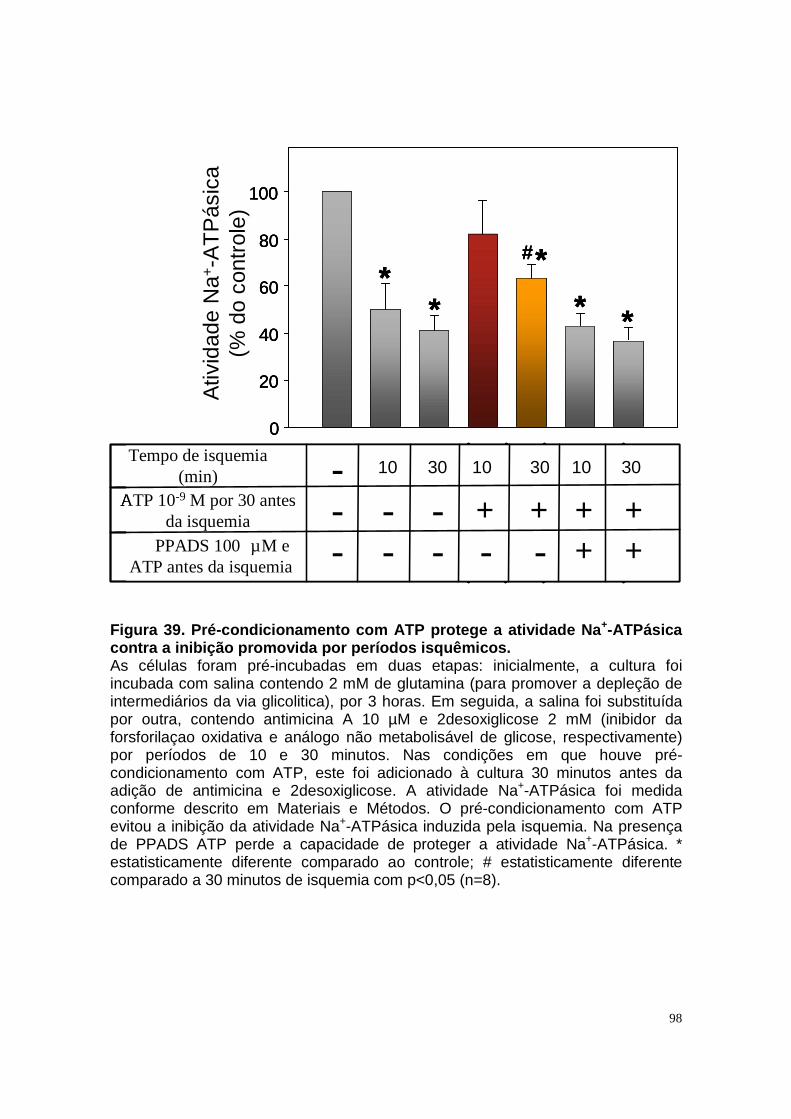
98
Figura 39. Pré-condicionamento com ATP protege a at ividade Na +-ATPásica contra a inibição promovida por períodos isquêmicos . As células foram pré-incubadas em duas etapas: inicialmente, a cultura foi incubada com salina contendo 2 mM de glutamina (para promover a depleção de intermediários da via glicolitica), por 3 horas. Em seguida, a salina foi substituída por outra, contendo antimicina A 10 µM e 2desoxiglicose 2 mM (inibidor da forsforilaçao oxidativa e análogo não metabolisável de glicose, respectivamente) por períodos de 10 e 30 minutos. Nas condições em que houve pré-condicionamento com ATP, este foi adicionado à cultura 30 minutos antes da adição de antimicina e 2desoxiglicose. A atividade Na+-ATPásica foi medida conforme descrito em Materiais e Métodos. O pré-condicionamento com ATP evitou a inibição da atividade Na+-ATPásica induzida pela isquemia. Na presença de PPADS ATP perde a capacidade de proteger a atividade Na+-ATPásica. * estatisticamente diferente comparado ao controle; # estatisticamente diferente comparado a 30 minutos de isquemia com p<0,05 (n=8).
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
0
20
40
60
80
100
--
30
--
60 10
+
30
-
10 30
+ ++ +PPADS 100 µM e
ATP antes da isquemia
---
-- -
+
10
--
30 10
+
30
-
10 30
+ ++ +
Tempo de isquemia (min)
ATP 10-9 M por 30 antes da isquemia
** * *
#*
Ativ
idad
e N
a+-A
TP
ásic
a(%
do
cont
role
)
0
20
40
60
80
100
--
30
--
60 10
+
30
-
10 30
+ ++ +PPADS 100 µM e
ATP antes da isquemia
---
-- -
+
10
--
30 10
+
30
-
10 30
+ ++ +
Tempo de isquemia (min)
ATP 10-9 M por 30 antes da isquemia
** * *
#*
0
20
40
60
80
100
--
30
--
60 10
+
30
-
10 30
+ ++ +PPADS 100 µM e
ATP antes da isquemia
---
-- -
+
10
--
30 10
+
30
-
10 30
+ ++ +
Tempo de isquemia (min)
ATP 10-9 M por 30 antes da isquemia
** * *
#*

99
5. Discussão Os efeitos renais mediados por adenosina parecem variar em função da
distribuição heterogênea de seus receptores nas diferentes regiões do rim (Vallon
et al., 2006). Outro fator determinante para o efeito de adenosina é seu
metabolismo uma vez que adenosina pode ser metabolizada no meio intra e
extracelular em diversos metabólitos ativos (Vitzthum et al., 2004). Os resultados
obtidos nesta tese mostram que adenosina adicionada ao lado luminal de células
LLC-PK1 é transportada para o meio intracelular, onde é convertida em ATP e
este é secretado para o lado luminal. No meio extracelular ATP se liga ao receptor
do tipo P2 ativando PKC o que resulta no estímulo da Na+-ATPase.
Nesta tese foi observado o efeito de adenosina sobre a atividade de duas
bombas de sódio: Na+-ATPase e (Na++K+)ATPase. Uma questão relevante é a
possível interferência da atividade (Na++K+)ATPásica na atividade Na+-ATPásica
uma vez que as enzimas apresentam características comuns como seu estímulo
por sódio. A possibilidade que o estímulo da Na+-ATPase por adenosina possa ser
devido à ação sobre a (Na++K+)ATPase pode ser descartada uma vez que: 1) a
adenosina 10-17-10-7M não modula a atividade (Na++K+)ATPásica; 2) a atividade
Na+-ATPásica foi medida sempre na presença de 1 mM de ouabaína, um inibidor
específico da (Na++K+)ATPase, e na ausência de K+ (Blanco et al., 1995). Na
maioria dos tecidos foi observado que a afinidade da (Na++K+)ATPase por
ouabaína está na faixa micromolar, contudo algumas preparações como túbulo
proximal de rato apresentam afinidade menor (Sweadner, 1989; Blanco et al.,
1995). A sensibilidade da (Na++K+)ATPase para ouabaína depende da isoforma
expressa em cada célula e é, também, tecido específica (Akera et al., 1985;
Blanco et al., 1995). Contudo, 1 mM de ouabaína inibe completamente todas as
isoformas da (Na++K+)ATPase.
Jin e colaboradores (Jin e Hopfer, 1997).(1997) mostraram que a incubação
por 5 minutos com ATP inibe a (Na++K+)ATPase em cultura primária de células de
túbulo proximal de rato Wistar-Kyoto. Por outro lado, nós observamos que
adenosina, em células LLC-PK1, não modifica a atividade desta bomba. Uma vez
que o efeito de adenosina é mediado por ATP, é possível propor que ATP não

100
modula a atividade da (Na++K+)ATPase em células LLC-PK1. Uma possível
explicação para esta possível controvérsia seria o tipo de preparação usada.
Dados de nosso laboratório mostraram que vários compostos, entre eles a
adenosina, não modulam a atividade da (Na++K+)ATPase em membrana
basolateral isolada de túbulo proximal de rim de porco (Caruso-Neves et al., 1997;
Wengert et al., 2005). Naquele momento foi proposto que a não modulação da
(Na++K+)ATPase era devido a falta de integridade celular (Caruso-Neves et al.,
1999; Caruso-Neves et al., 2000; Caruso-Neves et al., 2004). Nesta tese nós
observamos que adenosina adicionada ao lado luminal das céulas LLC-PK1 não
modifica a atividade da (Na++K+)ATPase apesar da integridade celular mantida.
Em aorta isolada de rato, foi observado que a adenosina promove relaxamento
independente de endotélio, o qual é, provavelmente, dependente da ativação da
(Na++K+)ATPase de músculo liso, e este efeito é mediado pela ativação de
receptor A2A (Grbovic e Radenkovic, 2003). Estes resultados podem sugerir que a
(Na++K+)ATPase não seja o alvo de modulação por adenosina em tecido renal.
Contudo, não podemos descartar a possibilidade que a modulação da
(Na++K+)ATPase apenas sede em períodos longos, uma vez que o efeito de
adenosina estudado na presente tese foi aquele induzido por adenosina após
apenas 30 minutos de incubação.
Nucleosídeos e bases nitrogenadas são transportados através da
membrana plasmática por meio de transportadores específicos, classificados
como equilibrativos (denominados ENT1 e ENT2) e concentrativos (denominados
CNT1,2,3) (Damaraju et al., 2007). Os transportadores do tipo equilibrativo estão
presentes nas membranas luminal e basolateral, enquanto que os concentrativos
estão presentes apenas na membrana luminal. NBTI inibe apenas a isoforma
ENT1 enquanto dipiridamol é um inibidor inespecífico, inibe tanto os
transportadores equilibrativos como concentrativos. Nós observamos que a
captação de adenosina pelas células LLC-PK1 não ocorre através de ENT1, uma
vez que na presença de NBTI não houve alteração na captação do nucleosídeo.
Entretanto, na presença de dipiridamol, inibidor inespecífico de ENT e CNT, a
captação de adenosina foi reduzida indicando a participação do transportador

101
ENT2 ou de alguma isoforma de CNT. Estes dados estão de acordo com o
observado para o transporte de adenosina em túbulo proximal (Damaraju et al.,
2007). A observação que o transporte de adenosina não é completamente inibido
por dipiridamiol indica: 1) a participação de outro transportador de adenosina
insensível a dipiridamol e NTBI ; 2) que a adenina formada pelo metabolismo de
adenosina no meio extracelular entra na célula através de transportadores de base
purínica e esta seja convertida à adenosina.
É interessante a observação que na presença de dipiridamol não houve
modulação no nível extracelular de 3H-adenosina, indicando que adenosina deve
ser metabolizada no meio extracelular. Esta hipótese é fortalecida pelas seguintes
observações: 1) adenosina no meio extracelular é convertida à inosina,
hipoxantina e adenina; 2) durante eventos de isquemia ocorre um aumento na
conversão de adenosina a adenina no meio extracelular.
Os nucleotídeos de adenina são gerados no meio intracelular a partir da
adenosina captada pela célula e após algum tempo são transportados para o meio
extracelular a favor de gradiente. Esta proposta é confirmada pela observação de
que o aumento do nível intracelular de nucleotídeos de adenina precede seu
aumento no meio extracelular. Atualmente, é descrito que a secreção de ATP
pode ocorrer por 3 formas: exocitose de vesículas, através do transportador ABC
ou por canal de ânions, como discutido na introdução. A exocitose de vesículas
não é descrita em células epiteliais, apenas em células da glia e astrócitos
(Potokar et al., 2007). Entretanto, o transporte através de canal de ânions ou
transportador ABC é descrito em células epiteliais. É descrito que os canais de
ânions também estão presentes tanto na membrana luminal como basolateral em
células de túbulo distal, e são sensíveis a DIDS, (Braunstein et al., 2001;
Schwiebert e Zsembery, 2003). Entretanto, não existem muitos estudos sobre a
secreção de ATP pelas células do túbulo proximal.
Nossos resultados indicam que a secreção de nucleotídoes de adenina no
túbulo proximal é mediada, em parte, por um canal de ânion sensível a DIDS.
Além disso, observamos que DIDS bloqueia completamente o efeito estimulatório
de adenosina sobre a Na+-ATPase. O fato de DIDS inibir completamente o efeito

102
de adenosina sobre a atividade da enzima e reverter parcialmente a secreção de
nucleotídeos de adenina, sugere que parte dos nucleotídeos de adenina formados
a partir de adenosina e secretados são formados por AMP e ADP. Esta hipótese é
favorecida pela observação que estes nucleotídeos, AMP e ADP, não modulam a
atividade Na+-ATPásica, como discutido abaixo.
Inicialmente, acreditava-se que os nucleotídeos de adenina possuiam
efeitos biológicos enquanto a hipoxantina, inosina e adenina, provenientes da
degradação de adenosina, não apresentam atividade biológica (Nishiyama et al.,
2001). Contudo, alguns trabalhos, incluindo de nosso grupo, têm mostrado que
adenina, hipoxantina e inosina podem ter efeito biológico importante (Wengert et
al., 2007). BLANCO e colaboradores (1993) observaram que o ATP adicionado à
membrana luminal de células de túbulo proximal é rapidamente degradado, e o
principal metabólito formado é a inosina, devido à alta atividade ecto-nucleotidase
e ecto-adenosina deaminase presente nesta membrana. (Blanco et al., 1993).
Contudo, o único metabólito formado a partir da adenosina capaz de modular a
atividade Na+-ATPásica é o ATP (Figura 20). Além disso, o efeito estimulatório de
adenosina sobre a Na+-ATPase não foi revertido por nenhum antagonista de
receptor de adenosina, porém foi totalmente revertido por PPADS, antagonista de
receptor P2. Estes resultados indicam que o efeito de adenosina sobre a Na+-
ATPase é via ATP. Esta hipótese é confirmada pelas seguintes observações: 1)
iodotubericidina, inibidor de adenosina cinase, reverte o efeito de adenosina sobre
a Na+-ATPase; 2) a adição do meio condicionado, obtido pela incubação das
células com adenosina, aumenta a atividade Na+-ATPásica e este efeito é
revertido por PPADS.
Em geral os efeitos de ATP mediados por receptores metabotrópicos (P2Y)
envolve a ativação tanto da via PLC/Ca2+/PKC como AMPc/PKA (Erb et al., 2006).
Os resultados obtidos mostraram que nem adenosina 10-8M nem ATP 10-9M
modificaram a atividade de PKA, porém aumentaram a atividade de PKC. Este
efeito foi completamente revertido por PPADS e calfostina C. Três classes de PKC
têm sido descritas: clássica (α,β,γ), “novel” (δ,θ) e atípica (ζ). A primeira classe é
sensível a éster de forbol e Ca2+, a segunda é insensível a Ca2+ e sensível a éster

103
de forbol, enquanto que a última classe é insensível a ambos. A presença destas
três classes de PKC tem sido mostrada em túbulo proximal, contudo seus efeitos
na reabsorção de Na+ neste segmento são diferentes.
O papel da PKC sobre a modulação da reabsorção de sódio no túbulo
proximal não estão muito claro. LIU e colaboradores mostraram que ativação de
PKC aumenta a reabsorção de bicarbonato e água nos segmentos S1 e S2 do
túbulo proximal (Liu e Cogan, 1990). Outro trabalho de microperfusão de túbulo
proximal de ratos demonstrou que o forbol éster promove efeito bifásico sobre a
reabsorção de fluido: inicialmente o forbol éster estimula a reabsorção de fluido e,
logo após, a inibe (Wang e Chan, 1990). Por outro lado, BAUM e HAYS (1988)
demonstraram que o forbol éster inibe o transporte de íons em túblo proximal de
rato (Baum e Hays, 1988). EFENDIEV e colaboradores (Efendiev et al., 1999)
mostraram que PKCβ (clássica) e PKCζ (atípica) apresentam efeitos opostos
sobre a atividade da (Na++K+)-ATPase em túbulo proximal de ratos. Estes
resultados indicam que o efeito de PKC sobre a reabsorção de Na+ depende da
isoforma ativada e de sua localização celular.
A possível participação de Ca2+ intracelular nas ações promovidas por ATP
no túbulo proximal foi mostrado por BAILEY e colaboradores em 2000. Os autores
utilizando túbulo proximal, isolado de rim de coelho, demonstraram que 10-5M de
ATP na membrana basolateral promovia o aumento de cálcio intracelular de
maneira transiente, e este efeito foi mediado pela ativação de P2Y1 e P2Y2 (Bailey
et al., 2000). Nossos experimentos demonstraram que ATP, nas concentrações
(10-12-10-8M) que promovem o aumento da atividade Na+-ATPásica, não modifica
os níveis intracelulares de Ca2+ nas células LLC-PK1. Somente concentrações
acima de 10-4 M foram capazes de aumentar o Ca2+ intracelular. Estes resultados,
em conjunto, indicam que a PKC envolvida na ativação da Na+-ATPase por ATP
pertence a classe “novel”, ou seja, é insensível ao Ca2+ e sensível a éster de forbol
PMA. Estes resultados estão de acordo com as observações anteriores que a Na+-
ATPase é estimulada pela PKC pertencente a classe “novel”
Em resumo os resultados discutidos até o presente momento mostram um
importante mecanismo de balanço entre os diferentes metabólitos de adenosina e

104
seus papeis na modulação dos transportadadores ativos de sódio, Na+-ATPase e
(Na++K+)ATPase (Figura 40). Estes resultados em conjunto com aqueles obtidos
na literatura, nos permitem postular a hipótese de que em condições de normóxia
as células do túbulo proximal secretam ATP constantemente (Vekaria et al., 2006),
e este, por sua vez, deve ter fundamental importância na manutenção da função
do túbulo proximal, uma vez que este segmento é responsável pela reabsorção
em massa do sódio filtrado nos glomérulos.
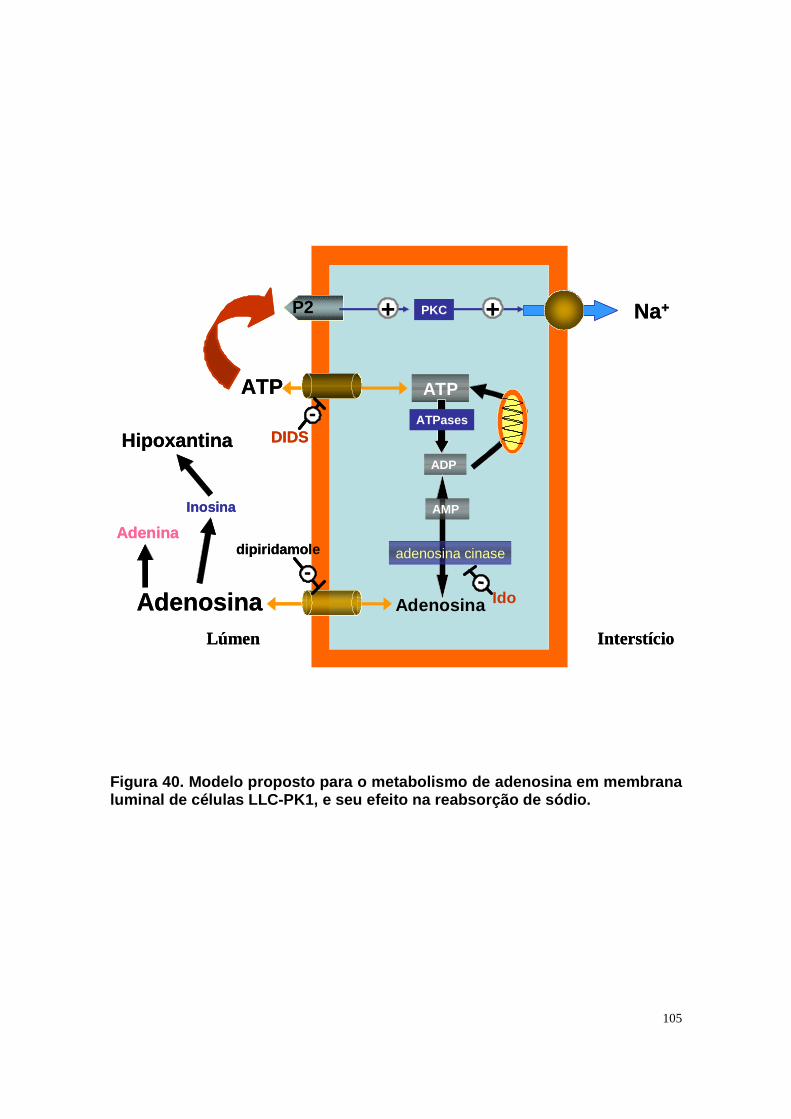
105
Figura 40. Modelo proposto para o metabolismo de ad enosina em membrana luminal de células LLC-PK1, e seu efeito na reabsor ção de sódio.
Lúmen Interstício
Adenosina
ATP
ADP
ATPases
ATP
Na+P2
Adenosina
PKC+ +
DIDS
dipiridamole
-
-
AMP
adenosina cinase
Ido-
Adenina
Hipoxantina
Inosina
Lúmen Interstício
Adenosina
ATP
ADP
ATPases
ATP
Na+P2P2
Adenosina
PKC++ ++
DIDS
dipiridamole
--
--
AMP
adenosina cinase
Ido--
Adenina
Hipoxantina
Inosina

106
Em uma segunda etapa foi estudado o possível papel do metabolismo de
adenosina durante eventos de isquemia. Como mencionado na introdução, a
adenosina pode ter efeito tanto protetor quanto deletério durante eventos de
isquemia renal. Seu efeito final depende do receptor com o qual ela irá interagir e
dos metabólitos formados (Vallon, 2008). Nesta tese, avaliamos o metabolismo da
adenosina em células LLC-PK1 durante isquemia. Observamos que, durante a
isquemia, ocorre diminuição nos níveis intra e extracelulares de nucleotídeos de
adenina. Entretanto, a permeabilidade aparente da célula aos nucleotídeos de
adenina não foi alterada, mesmo na presença de DIDS. Neste momento havia
uma questão em aberto: será que os níveis de nucleotídeos de adenina durante a
isquemia metabólica estavam diminuídos devido à diminuição da conversão de
adenosina ou será que a captação de adenosina pela célula estava diminuída?
A permeabilidade aparente da célula à adenosina não foi modificada e foi
observado que a diminuição na conversão de adenosina a nucleotídeos de
adenina não era devida a uma diminuição na captação de adenosina pela célula.
Contudo, a observação que os níveis de adenosina intra e extracelular não
mudam indica que adenosina deveria estar sendo metabolizada no meio intra ou
extracelular. Trabalhos publicados anteriormente mostraram que durante períodos
longos de isquemia renal (180 minutos) o principal metabólito formado a partir de
adenosina era a hipoxantina (Perico et al., 2004). Nossos resultados mostram que
o principal metabólito formado a paritr de adenosina na membrana luminal das
células LLC-PK1 em eventos de isquemia de 40 minutos é a adenina. Foi
observado, ainda, que os níveis de inosina e hipoxantina não foram modificados. A
aparente contradição entre os resultados obtidos em outros trabalhos e os nossos
poderia ser explicada devido ao tempo da isquemia metabólica. Desta forma,
estes resultados revelam um importante mecanismo pelo qual, dependendo do
tempo de isquemia, ocorre a ativação de diferentes vias metabólicas envolvidas no
metabolismo de adenosina. Este mecanismo poderia determinar o efeito final de
adenosina durante estes eventos de isquemia.
PARK e colaboradores (Park et al., 2001) (2002) utilizando modelos in vivo,
demonstraram que a isquemia renal aguda em ratos promovia um aumento na

107
excreção fracionária de sódio, que era revertida após sete dias de reperfusão.
Este dado indica que durante a isquemia deve haver modulação nos
transportadores de sódio ao longo do néfron e, provavelmente, a região mais
afetada deve ser a região cortical, uma vez que esta região possui 90-95% do
fluxo sanguíneo renal total, enquanto a medula possui apenas 5-10% do fluxo
sanguíneo renal total. Além disso, dentre os segmentos localizados no córtex, o
mais afetado deve ser o túbulo proximal, pois é responsável pela reabsorção de
70% do sódio filtrado nos glomérulos; portanto, pequenas alterações na
reabsorção de Na+ neste segmento resultam em grandes alterações na excreção
renal do íon. Nós observamos que períodos curtos de isquemia, de 10 e 30
minutos, em células LLC-PK1 inibem a atividade Na+-ATPase, porém não inibem a
atividade da (Na++K+)ATPase, sendo este efeito inibitório revertido após 2 horas
de reperfusão. Estes dados estão de acordo com os dados da literatura, os quais
citam que durante isquemia ocorre aumento da excreção fracionária de sódio e
que a reperfusão reverte este efeito. Portanto, a inibição da atividade Na+-ATPase
pode ser em parte responsável pelo aumento da excreção fracionária de sódio
observada em modelos in vivo (Park et al., 2001).
Outra questão importante, ainda em aberto, se refere ao mecanismo de
inibição da Na+-ATPase durante eventos de isquemia de 40 minutos. Alguns
mecanismos poderiam ser sugeridos: 1) a adenina através da interação com
receptores poderia promover a inibição da atividade enzimática; 2) a depleção de
ATP do meio intracelular levaria a uma diminuição na atividade dos
transportadores ativos primários; 3) a não secreção de ATP pelas células levaria a
uma inibição da enzima.
Em um trabalho recente, nós mostramos que adenina se liga ao receptor A1
e promove a inibição da Na+-ATPase em membrana basolateral. Contudo, o
aumento da adenina na membrana luminal das células LLC-PK1, durante eventos
de isquemia, parece não estar correlacionado à inibição da Na+-ATPase uma vez
que a adição de adenina nesta região não promoveu nenhum efeito sobre a
atividade desta enzima, muito embora as experiências de adição de adenina

108
tenham sido realizadas durante períodos não isquêmicos. Seria necessário a
realização de experiências durante eventos de isquemia para testar esta hipótese.
Outra possibilidade seria a depleção de ATP intracelular. Neste caso a
baixa do substrato energético iria levar a uma diminuição da atividade de todos os
transportadores ativos, não apresentando nenhum efeito específico. Este parece
não ser o caso uma vez que a atividade da (Na++K+)ATPase não foi modificada
em períodos de isquemia de 40 minutos. A atividade desta enzima somente é
modificada em períodos de isquemia superior a 180 minutos (dados não
mostrados), o que confirma os dados da literatura, nos quais foi observada a
inibição somente em períodos longos de isquemia (240 minutos) tanto em células
LLC-PK1 e MDCK1 (Wiegele, et al., 1998).
A última possibilidade seria a diminuição da secreção de ATP pelas células
que levaria a uma inibição da Na+-ATPase (Figura 41). Esta hipótese é favorecida
pela observação de que o pré-condicionamento das células LLC-PK1 com ATP
impede o efeito inibitório sobre a Na+-ATPase promovido pela isquemia. Além
disso, nós observamos que o efeito de ATP é revertido por PPADS de maneira
similar aquela observada quando verificamos o efeito do ATP em condições não
isquêmicas (normóxia). Estes resultados estão de acordo com o observado que o
pré-condicionamento de células com ATP previne o processo inflamatório que
sucede ao período de isquemia (Lee e Han, 2005). LEE e colaboradores (Lee e
Han, 2005), em estudo com cultura primária de túbulo proximal de coelho,
demonstraram que períodos isquêmicos longos promovem a liberação de LDH,
um marcador de dano celular estrutural, e a ativação de NF-κβ. Porém, o pré-
condicionamento com ATP bloqueia a liberação de LDH induzida pela isquemia
assim como a ativação de NF-κβ. Estes dados sugerem um efeito protetor para o
ATP contra injúria isquêmica.
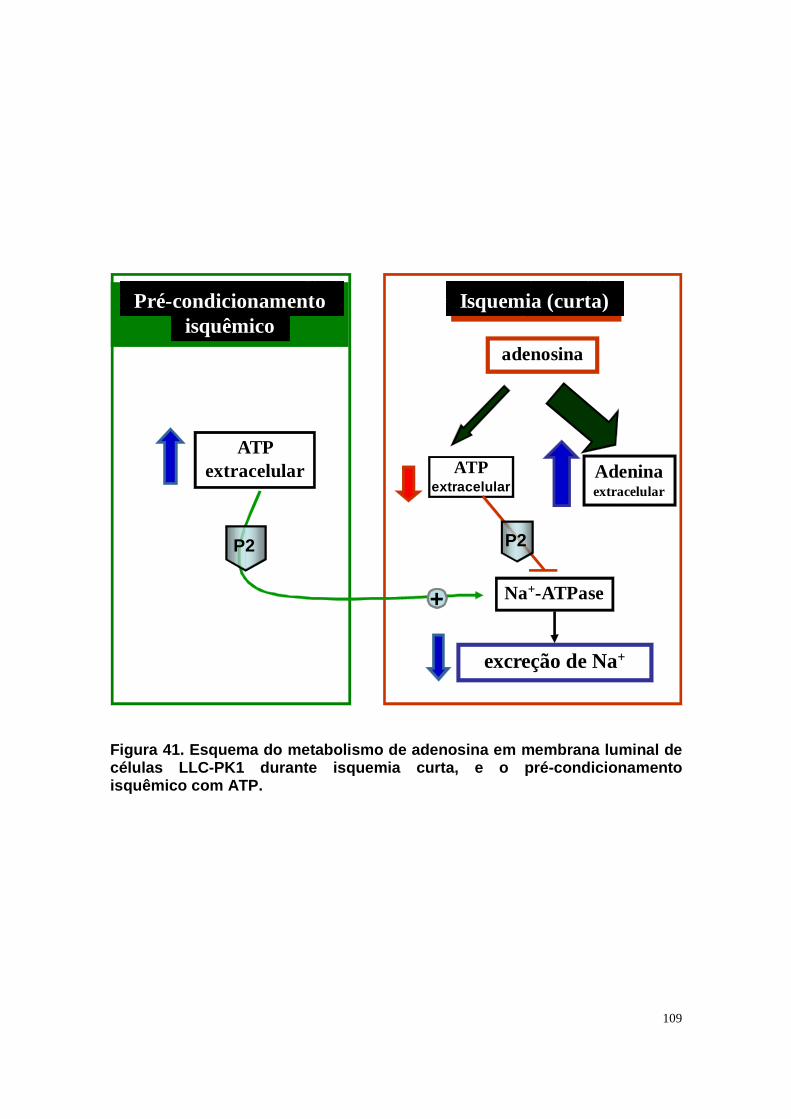
109
excreção de Na+
Na+-ATPase
Isquemia (curta)
adenosina
ATPextracelular
Pré-condicionamento isquêmico
ATP extracelular
P2
+
P2
Adenina extracelular
Figura 41. Esquema do metabolismo de adenosina em m embrana luminal de células LLC-PK1 durante isquemia curta, e o pré-con dicionamento isquêmico com ATP.

110
Quando comparamos os resultados obtidos na presente tese com aqueles
obtidos previamente (Wengert. et al., 2005; Wengert et al., 2007) observamos que
o metabolismo de adenosina na membrana luminal é totalmente diferente daquele
observado na membrana basolateral onde somente a inosina é formada a partir de
adenosina. Esta diferença sugere que o papel de adenosina seja diferente nos
dois lados da membrana e, desta forma, seu efeito final dependeria não somente
da distribuição de receptores mas de sua metabolização. Além disso, deve ser
considerado também o importante papel do metabolimso sobre a adenosina
filtrada no glomérulo. Uma vez que observamos efeitos de metabólitos de
adenosina, como o ATP, em concentrações baixas tais como 10-12 M, é possível
sugerir que pequenas concentrações de adenosina filtrada já possam exercer um
papel relevante na regulação da reabsorção de sal e água no túbulo proximal,
tanto em situações fisiológicas quanto patológicas. Em resumo nossos resultados
permitem um melhor conhecimento do metabolismo luminal de adenosina e do
efeito de seus metabólitos na função do túbulo proximal e, consequentemente, na
excreção renal de sal e água.

111
6. Referências Bibliográficas
Abbracchio, M. P. e G. Burnstock. Purinergic signalling: pathophysiological roles.
Jpn J Pharmacol, v.78, n.2, Oct, p.113-45. 1998.
Abbracchio, M. P. e G. Burnstock. Purinoceptors: are there families of P2X and
P2Y purinoceptors? Pharmacol Ther, v.64, n.3, p.445-75. 1994.
Akera, T., Y. C. Ng, I. S. Shieh, E. Bero, T. M. Brody e W. E. Braselton. Effects of
K+ on the interaction between cardiac glycosides and Na,K-ATPase. Eur J
Pharmacol, v.111, n.2, May 8, p.147-57. 1985.
Ayyanathan, K., T. E. Webbs, A. K. Sandhu, R. S. Athwal, E. A. Barnard e S. P.
Kunapuli. Cloning and chromosomal localization of the human P2Y1
purinoceptor. Biochem Biophys Res Commun, v.218, n.3, Jan 26, p.783-8.
1996.
Bagnasco, S., D. Good, R. Balaban e M. Burg. Lactate production in isolated
segments of the rat nephron. Am J Physiol, v.248, n.4 Pt 2, Apr, p.F522-6.
1985.
Bailey, M. A., C. M. Turner, A. Hus-Citharel, J. Marchetti, M. Imbert-Teboul, P.
Milner, G. Burnstock e R. J. Unwin. P2Y receptors present in the native and
isolated rat glomerulus. Nephron Physiol, v.96, n.3, p.p79-90. 2004.
Bailey, M. A., M. Imbert-Teboul, C. Turner, S. K. Srai, G. Burnstock e R. J. Unwin.
Evidence for basolateral P2Y(6) receptors along the rat proximal tubule:
functional and molecular characterization. J Am Soc Nephrol, v.12, n.8, Aug,
p.1640-7. 2001.
Bailey, M. A., M. Imbert-Teboul, C. Turner, S. Marsy, K. Srai, G. Burnstock e R. J.
Unwin. Axial distribution and characterization of basolateral P2Y receptors
along the rat renal tubule. Kidney Int, v.58, n.5, Nov, p.1893-901. 2000.

112
Bakr, A. F. Prophylactic theophylline to prevent renal dysfunction in newborns
exposed to perinatal asphyxia--a study in a developing country. Pediatr
Nephrol, v.20, n.9, Sep, p.1249-52. 2005.
Baum, M. e S. R. Hays. Phorbol myristate acetate and dioctanoylglycerol inhibit
transport in rabbit proximal convoluted tubule. Am J Physiol, v.254, n.1 Pt 2,
Jan, p.F9-14. 1988.
Bell, P. D., J. Y. Lapointe, R. Sabirov, S. Hayashi, J. Peti-Peterdi, K. Manabe, G.
Kovacs e Y. Okada. Macula densa cell signaling involves ATP release
through a maxi anion channel. Proc Natl Acad Sci U S A, v.100, n.7, Apr 1,
p.4322-7. 2003.
Beltowski, J. e G. Wojcicka. Regulation of renal tubular sodium transport by
cardiac natriuretic peptides: two decades of research. Med Sci Monit, v.8, n.2,
Feb, p.RA39-52. 2002.
Beltowski, J., A. Jamroz-Wisniewska, J. Nazar e G. Wojcicka. Spectrophotometric
assay of renal ouabain-resistant Na(+)-ATPase and its regulation by leptin
and dietary-induced obesity. Acta Biochim Pol, v.51, n.4, p.1003-14. 2004.
Benham, C. D. e R. W. Tsien. A novel receptor-operated Ca2+-permeable channel
activated by ATP in smooth muscle. Nature, v.328, n.6127, Jul 16-22, p.275-
8. 1987.
Besirli, C. G., T. W. Gong e M. I. Lomax. Novel beta 3 isoform of the Na,K-ATPase
beta subunit from mouse retina. Biochim Biophys Acta, v.1350, n.1, Jan 3,
p.21-6. 1997.
Blanco, G. e R. W. Mercer. Isozymes of the Na-K-ATPase: heterogeneity in
structure, diversity in function. Am J Physiol, v.275, n.5 Pt 2, Nov, p.F633-50.
1998.
Blanco, G. e R. W. Mercer. Regulation of the alpha 2 beta 1 and alpha 3 beta 1
isozymes of the Na,K-ATPase by Ca2+, PKA, and PKC. Ann N Y Acad Sci,
v.834, Nov 3, p.572-5. 1997.

113
Blanco, G., J. C. Koster, G. Sanchez e R. W. Mercer. Kinetic properties of the
alpha 2 beta 1 and alpha 2 beta 2 isozymes of the Na,K-ATPase.
Biochemistry, v.34, n.1, Jan 10, p.319-25. 1995.
Blanco, J., E. I. Canela, J. Sayos, J. Mallol, C. Lluis e R. Franco. Adenine
nucleotides and adenosine metabolism in pig kidney proximal tubule
membranes. J Cell Physiol, v.157, n.1, p.77-83. 1993.
Boudreault, F. e R. Grygorczyk. Cell swelling-induced ATP release is tightly
dependent on intracellular calcium elevations. J Physiol, v.561, n.Pt 2, Dec 1,
p.499-513. 2004.
Braun, N., S. Fengler, C. Ebeling, J. Servos e H. Zimmermann. Sequencing,
functional expression and characterization of rat NTPDase6, a nucleoside
diphosphatase and novel member of the ecto-nucleoside triphosphate
diphosphohydrolase family. Biochem J, v.351 Pt 3, Nov 1, p.639-47. 2000.
Braunstein, G. M., R. M. Roman, J. P. Clancy, B. A. Kudlow, A. L. Taylor, V. G.
Shylonsky, B. Jovov, K. Peter, T. Jilling, Ismailov, Ii, D. J. Benos, L. M.
Schwiebert, J. G. Fitz e E. M. Schwiebert. Cystic fibrosis transmembrane
conductance regulator facilitates ATP release by stimulating a separate ATP
release channel for autocrine control of cell volume regulation. J Biol Chem,
v.276, n.9, Mar 2, p.6621-30. 2001.
Brenner, B. M. e F. C. Rector. The kidney: W B Saunders Co. 2000
Bruns, R. F., G. H. Lu e T. A. Pugsley. Characterization of the A2 adenosine
receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol,
v.29, n.4, Apr, p.331-46. 1986.
Bruzzone, S., L. Guida, E. Zocchi, L. Franco e A. De Flora. Connexin 43 hemi
channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells.
FASEB J, v.15, n.1, Jan, p.10-12. 2001.
Burnstock, G. Purinergic signalling--an overview. Novartis Found Symp, v.276,
p.26-48; discussion 48-57, 275-81. 2006.

114
Cai, H., V. Batuman, D. B. Puschett e J. B. Puschett. Effect of KW-3902, a novel
adenosine A1 receptor antagonist, on sodium-dependent phosphate and
glucose transport by the rat renal proximal tubular cell. Life Sci, v.55, n.10,
p.839-45. 1994.
Caruso-Neves, C., A. S. Siqueira, G. Iso-Cohen e A. G. Lopes. Bradykinin
modulates the ouabain-insensitive Na+-ATPase activity from basolateral
membrane of the proximal tubule. Biochim. Biophys. Acta, v.1431, n.2, p.483-
91. 1999.
Caruso-Neves, C., A. T. Malaquias, F. F. Loss, V. M. Correa Da Costa, V. O.
Gomes e A. G. Lopes. Bradykinin B1 receptor stimulates the proximal tubule
Na(+)-ATPase activity through protein kinase C pathway. Regul Pept, v.115,
n.3, p.195-201. 2003.
Caruso-Neves, C., D. Vives, C. Dantas, C. M. Albino, L. M. Fonseca, L. S. Lara, M.
Iso e A. G. Lopes. Ouabain-insensitive Na+-ATPase of proximal tubules is an
effector for urodilatin and atrial natriuretic peptide. Biochim Biophys Acta,
v.1660, n.1-2, p.93-8. 2004.
Caruso-Neves, C., K. Provenzano, F. F. Luz, F. M. Santos, M. S. Fernandes, L. R.
Leao-Ferreira e A. G. Lopes. Bradykinin counteracts the stimulatory effect of
angiotensin-(1-7) on the proximal tubule Na+ -ATPase activity through B2
receptor. Regul Pept, v.110, n.3, p.207-12. 2003.
Caruso-Neves, C., L. B. Rangel, D. Vives, A. Vieyra, S. Coka-Guevara e A. G.
Lopes. Ouabain-insensitive Na(+)-ATPase activity is an effector protein for
cAMP regulation in basolateral membranes of the proximal tubule. Biochim.
Biophys. Acta, v.1468, n.1-2, p.107-14. 2000.
Caruso-Neves, C., L. G. Francisco-Pedro, L. P. Souza, C. Chagas e A. G. Lopes.
Effect of adenosine on the ouabain-insensitive Na+-ATPase activity from
basolateral membrane of the proximal tubule. Biochim. Biophys. Acta, v.1329,
n.2, p.336-44. 1997.

115
Caruso-Neves, C., S. A. Coelho-Souza, D. Vives, G. Goes, L. S. Lara e A. G.
Lopes. Modulation of ouabain-insensitive Na(+)-ATPase activity in the renal
proximal tubule by Mg(2+), MgATP and furosemide. Int. J. Biochem. Cell
Biol., v.34, n.12, p.1586-93. 2002.
Cass, C. E., J. D. Young e S. A. Baldwin. Recent advances in the molecular
biology of nucleoside transporters of mammalian cells. Biochem Cell Biol,
v.76, n.5, p.761-70. 1998.
Cass, C. E., J. D. Young, S. A. Baldwin, M. A. Cabrita, K. A. Graham, M. Griffiths,
L. L. Jennings, J. R. Mackey, A. M. Ng, M. W. Ritzel, M. F. Vickers e S. Y.
Yao. Nucleoside transporters of mammalian cells. Pharm. Biotechnol., v.12,
p.313-52. 1999.
Chan, C. M., R. J. Unwin, M. Bardini, I. B. Oglesby, A. P. Ford, A. Townsend-
Nicholson e G. Burnstock. Localization of P2X1 purinoceptors by
autoradiography and immunohistochemistry in rat kidneys. Am J Physiol,
v.274, n.4 Pt 2, Apr, p.F799-804. 1998.
Chorna, N. E., L. I. Santiago-Perez, L. Erb, C. I. Seye, J. T. Neary, G. Y. Sun, G. A.
Weisman e F. A. Gonzalez. P2Y receptors activate neuroprotective
mechanisms in astrocytic cells. J Neurochem, v.91, n.1, Oct, p.119-32. 2004.
Chow, D. C. e J. G. Forte. Functional significance of the beta-subunit for
heterodimeric P-type ATPases. J Exp Biol, v.198, n.Pt 1, Jan, p.1-17. 1995.
Coka-Guevara, S., R. P. Markus, C. Caruso-Neves, A. G. Lopes e A. Vieyra.
Adenosine inhibits the renal plasma-membrane (Ca2+ + Mg2+)-ATPase
through a pathway sensitive to cholera toxin and sphingosine. Eur. J.
Biochem., v.263, n.1, p.71-8. 1999.
Communi, D., C. Govaerts, M. Parmentier e J. M. Boeynaems. Cloning of a human
purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J
Biol Chem, v.272, n.51, Dec 19, p.31969-73. 1997.

116
Communi, D., M. Parmentier e J. M. Boeynaems. Cloning, functional expression
and tissue distribution of the human P2Y6 receptor. Biochem Biophys Res
Commun, v.222, n.2, May 15, p.303-8. 1996.
Communi, D., N. S. Gonzalez, M. Detheux, S. Brezillon, V. Lannoy, M. Parmentier
e J. M. Boeynaems. Identification of a novel human ADP receptor coupled to
G(i). J Biol Chem, v.276, n.44, Nov 2, p.41479-85. 2001.
Cotrina, M. L., J. H. Lin, A. Alves-Rodrigues, S. Liu, J. Li, H. Azmi-Ghadimi, J.
Kang, C. C. Naus e M. Nedergaard. Connexins regulate calcium signaling by
controlling ATP release. Proc Natl Acad Sci U S A, v.95, n.26, Dec 22,
p.15735-40. 1998.
Coulson, R., R. A. Johnson, R. A. Olsson, D. R. Cooper e S. J. Scheinman.
Adenosine stimulates phosphate and glucose transport in opossum kidney
epithelial cells. Am. J. Physiol., v.260, n.6 Pt 2, p.F921-8. 1991.
Cuffe, J. E., A. Bielfeld-Ackermann, J. Thomas, J. Leipziger e C. Korbmacher. ATP
stimulates Cl- secretion and reduces amiloride-sensitive Na+ absorption in M-
1 mouse cortical collecting duct cells. J Physiol, v.524 Pt 1, Apr 1, p.77-90.
2000.
Czaplinski, M., C. Abad e A. Eblen-Zajjur. Normal expression and inflammation-
induced changes of Na and Na/K ATPase activity in spinal dorsal horn of the
rat. Neurosci Lett, v.374, n.2, Feb 10, p.147-51. 2005.
Damaraju, V. L., A. N. Elwi, C. Hunter, P. Carpenter, C. Santos, G. M. Barron, X.
Sun, S. A. Baldwin, J. D. Young, J. R. Mackey, M. B. Sawyer e C. E. Cass.
Localization of broadly selective equilibrative and concentrative nucleoside
transporters, hENT1 and hCNT3, in human kidney. Am J Physiol Renal
Physiol, v.293, n.1, Jul, p.F200-11. 2007.
De Souza, A. M., T. L. Carvalho, P. M. Sabino, D. Vives, C. F. Fontes, A. G. Lopes
e C. Caruso-Neves. Characterization and partial isolation of ouabain-
insensitive Na(+) -ATPase in MDCK I cells. Biochimie, v.89, n.11, Nov,
p.1425-32. 2007.

117
Del Castillo, J. R., R. Marin, T. Proverbio e F. Proverbio. Partial characterization of
the ouabain-insensitive, Na+-stimulated ATPase activity of kidney basal-
lateral plasma membranes. Biochim Biophys Acta, v.692, n.1, p.61-8. 1982.
Doctor RB, Bacallao R, Mandel LJ. Method for recovering ATP content and
mitochondrial function after chemical anoxia in renal cell cultures. Am J
Physiol. 266:1803-11. 1994.
Drury, A. N. e A. Szent-Gyórgyi. The physiological activity of adenine compounds
with special reference to their action upon the mammalian heart. J. Physiol.,
v.68, p.213-237. 1929.
Edwards, R. M., M. Pullen e P. Nambi. Distribution of neutral endopeptidase
activity along the rat and rabbit nephron. Pharmacology, v.59, n.1, Jul, p.45-
50. 1999.
Efendiev, R., A. M. Bertorello e C. H. Pedemonte. PKC-beta and PKC-zeta
mediate opposing effects on proximal tubule Na+,K+-ATPase activity. FEBS
Lett, v.456, n.1, p.45-8. 1999.
Endlich, K., W. G. Forssmann e M. Steinhausen. Effects of urodilatin in the rat
kidney: comparison with ANF and interaction with vasoactive substances.
Kidney Int, v.47, n.6, Jun, p.1558-68. 1995.
Eppel, G. A., S. Ventura, K. M. Denton e R. G. Evans. Lack of contribution of P2X
receptors to neurally mediated vasoconstriction in the rabbit kidney in vivo.
Acta Physiol (Oxf), v.186, n.3, Mar, p.197-207. 2006.
Erb, L., Z. Liao, C. I. Seye e G. A. Weisman. P2 receptors: intracellular signaling.
Pflugers Arch, v.452, n.5, Aug, p.552-62. 2006.
Feraille, E. e A. Doucet. Sodium-potassium-adenosinetriphosphatase-dependent
sodium transport in the kidney: hormonal control. Physiol. Rev., v.81, n.1,
p.345-418. 2001.
Flamenbaum, W., J. Gagnon e P. Ramwell. Bradykinin-induced renal
hemodynamic alterations: renin and prostaglandin relationships. Am J
Physiol, v.237, n.6, Dec, p.F433-40. 1979.

118
Fredholm, B. B., A. P. Ijzerman, K. A. Jacobson, K. N. Klotz e J. Linden.
International Union of Pharmacology. XXV. Nomenclature and classification of
adenosine receptors. Pharmacol. Rev., v.53, n.4, p.527-52. 2001.
Furlong, T. J., K. D. Pierce, L. A. Selbie e J. Shine. Molecular characterization of a
human brain adenosine A2 receptor. Brain Res Mol Brain Res, v.15, n.1-2,
Sep, p.62-6. 1992.
Gandhi, R., M. Le Hir e B. Kaissling. Immunolocalization of ecto-5'-nucleotidase in
the kidney by a monoclonal antibody. Histochemistry, v.95, n.2, p.165-74.
1990.
Garcia-Guzman, M., F. Soto, J. M. Gomez-Hernandez, P. E. Lund e W. Stuhmer.
Characterization of recombinant human P2X4 receptor reveals
pharmacological differences to the rat homologue. Mol Pharmacol, v.51, n.1,
Jan, p.109-18. 1997.
Garcia-Guzman, M., W. Stuhmer e F. Soto. Molecular characterization and
pharmacological properties of the human P2X3 purinoceptor. Brain Res Mol
Brain Res, v.47, n.1-2, Jul, p.59-66. 1997.
Gijsbers, R., H. Ceulemans, W. Stalmans e M. Bollen. Structural and catalytic
similarities between nucleotide pyrophosphatases/phosphodiesterases and
alkaline phosphatases. J Biol Chem, v.276, n.2, Jan 12, p.1361-8. 2001.
Gillespie, J. H. The biological significance of the linkages in adenosine
triphosphoric acid. J Physiol, v.80, n.4, Feb 28, p.345-59. 1934.
Gomes, C. P., L. R. Leao-Ferreira, C. Caruso-Neves e A. G. Lopes. Adenosine
reverses the stimulatory effect of angiotensin II on the renal Na+-ATPase
activity through the A2 receptor. Regul Pept, v.129, n.1-3, Jul 15, p.9-15.
2005.
Gouyon, J. B. e J. P. Guignard. Theophylline prevents the hypoxemia-induced
renal hemodynamic changes in rabbits. Kidney Int, v.33, n.6, Jun, p.1078-83.
1988.

119
Grbovic, L. e M. Radenkovic. Analysis of adenosine vascular effect in isolated rat
aorta: possible role of Na+/K+-ATPase. Pharmacol Toxicol, v.92, n.6, Jun,
p.265-71. 2003.
Grubmeyer, C. e H. S. Penefsky. The presence of two hydrolytic sites on beef
heart mitochondrial adenosine triphosphatase. J. Biol. Chem., v.256, n.8,
p.3718-27. 1981.
Guan, Z., D. A. Osmond e E. W. Inscho. P2X receptors as regulators of the renal
microvasculature. Trends Pharmacol Sci, v.28, n.12, Dec, p.646-52. 2007.
Hagberg, H., P. Andersson, J. Lacarewicz, I. Jacobson, S. Butcher e M. Sandberg.
Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to
tissue nucleotides and purines in rat striatum during transient ischemia. J
Neurochem, v.49, n.1, Jul, p.227-31. 1987.
Hansen, P. B., S. Hashimoto, M. Oppermann, Y. Huang, J. P. Briggs e J.
Schnermann. Vasoconstrictor and vasodilator effects of adenosine in the
mouse kidney due to preferential activation of A1 or A2 adenosine receptors.
J Pharmacol Exp Ther, v.315, n.3, Dec, p.1150-7. 2005.
Harada, H., C. M. Chan, A. Loesch, R. Unwin e G. Burnstock. Induction of
proliferation and apoptotic cell death via P2Y and P2X receptors, respectively,
in rat glomerular mesangial cells. Kidney Int, v.57, n.3, Mar, p.949-58. 2000.
Haro, R., B. Garciadeblas e A. Rodriguez-Navarro. A novel P-type ATPase from
yeast involved in sodium transport. FEBS Lett, v.291, n.2, Oct 21, p.189-91.
1991.
Harris, A. L. e C. G. Bevans. Exploring hemichannel permeability in vitro. Methods
Mol Biol, v.154, p.357-77. 2001.
Hollenstein, K., D. C. Frei e K. P. Locher. Structure of an ABC transporter in
complex with its binding protein. Nature, v.446, n.7132, Mar 8, p.213-6. 2007.
Hollopeter, G., H. M. Jantzen, D. Vincent, G. Li, L. England, V. Ramakrishnan, R.
B. Yang, P. Nurden, A. Nurden, D. Julius e P. B. Conley. Identification of the

120
platelet ADP receptor targeted by antithrombotic drugs. Nature, v.409,
n.6817, Jan 11, p.202-7. 2001.
Huwiler, A. e J. Pfeilschifter. Stimulation by extracellular ATP and UTP of the
mitogen-activated protein kinase cascade and proliferation of rat renal
mesangial cells. Br J Pharmacol, v.113, n.4, Dec, p.1455-63. 1994.
Iizumi, K., Y. Mikami, M. Hashimoto, T. Nara, Y. Hara e T. Aoki. Molecular cloning
and characterization of ouabain-insensitive Na(+)-ATPase in the parasitic
protist, Trypanosoma cruzi. Biochim Biophys Acta, v.1758, n.6, Jun, p.738-46.
2006.
Inscho, E. W., A. C. Schroeder, P. C. Deichmann e J. D. Imig. ATP-mediated Ca2+
signaling in preglomerular smooth muscle cells. Am J Physiol, v.276, n.3 Pt 2,
Mar, p.F450-6. 1999.
Inscho, E. W., T. P. Belott, M. J. Mason, J. B. Smith e L. G. Navar. Extracellular
ATP increases cytosolic calcium in cultured rat renal arterial smooth muscle
cells. Clin Exp Pharmacol Physiol, v.23, n.6-7, Jun-Jul, p.503-7. 1996.
Jackson, E. K. e R. K. Dubey. Role of the extracellular cAMP-adenosine pathway
in renal physiology. Am. J. Physiol. Renal Physiol., v.281, n.4, p.F597-612.
2001.
Jackson, E. K., C. Zhu e S. P. Tofovic. Expression of adenosine receptors in the
preglomerular microcirculation. Am J Physiol Renal Physiol, v.283, n.1, Jul,
p.F41-51. 2002a.
Jackson, E. K., C. Zhu e S. P. Tofovic. Expression of adenosine receptors in the
preglomerular microcirculation. Am. J. Physiol. Renal Physiol., v.283, n.1,
p.F41-51. 2002b.
Jackson, E. K., Z. Mi, C. Zhu e R. K. Dubey. Adenosine biosynthesis in the
collecting duct. J Pharmacol Exp Ther, v.307, n.3, Dec, p.888-96. 2003.
Jeyaseelan, K., S. Sepramaniam, A. Armugam e E. M. Wintour. Aquaporins: a
promising target for drug development. Expert Opin Ther Targets, v.10, n.6,
Dec, p.889-909. 2006.

121
Jin, W. e U. Hopfer. Purinergic-mediated inhibition of Na+-K+-ATPase in proximal
tubule cells: elevated cytosolic Ca2+ is not required. Am J Physiol, v.272, n.4
Pt 1, Apr, p.C1169-77. 1997.
Jorgensen, P. L. e P. A. Pedersen. Structure-function relationships of Na(+), K(+),
ATP, or Mg(2+) binding and energy transduction in Na,K-ATPase. Biochim
Biophys Acta, v.1505, n.1, May 1, p.57-74. 2001.
Junior, I. C., M. L. Rodrigues, C. S. Alviano, L. R. Travassos e J. R. Meyer-
Fernandes. Characterization of an ecto-ATPase activity in Cryptococcus
neoformans. FEMS Yeast Res, v.5, n.10, Jul, p.899-907. 2005.
Kim, J. K., H. Y. Yoo, S. J. Kim, Y. S. Hwang, J. Han, J. A. Kim, C. S. Kim e H. S.
Cho. Effects of sevoflurane on the cAMP-induced short-circuit current in
mouse tracheal epithelium and recombinant Cl- (CFTR) and K+ (KCNQ1)
channels. Br J Anaesth, v.99, n.2, Aug, p.245-51. 2007.
Kishore, B. K., C. L. Chou e M. A. Knepper. Extracellular nucleotide receptor
inhibits AVP-stimulated water permeability in inner medullary collecting duct.
Am J Physiol, v.269, n.6 Pt 2, Dec, p.F863-9. 1995.
Kishore, B. K., C. M. Krane, R. L. Miller, H. Shi, P. Zhang, A. Hemmert, R. Sun e
R. D. Nelson. P2Y2 receptor mRNA and protein expression is altered in inner
medullas of hydrated and dehydrated rats: relevance to AVP-independent
regulation of IMCD function. Am J Physiol Renal Physiol, v.288, n.6, Jun,
p.F1164-72. 2005.
Kishore, B. K., J. Isaac, M. Fausther, S. R. Tripp, H. Shi, P. S. Gill, N. Braun, H.
Zimmermann, J. Sevigny e S. C. Robson. Expression of NTPDase1 and
NTPDase2 in murine kidney: relevance to regulation of P2 receptor signaling.
Am J Physiol Renal Physiol, v.288, n.5, May, p.F1032-43. 2005.
Komlosi, P., A. Fintha e P. D. Bell. Renal cell-to-cell communication via
extracellular ATP. Physiology (Bethesda), v.20, Apr, p.86-90. 2005.

122
Komlosi, P., J. Peti-Peterdi, A. L. Fuson, A. Fintha, L. Rosivall e P. D. Bell. Macula
densa basolateral ATP release is regulated by luminal [NaCl] and dietary salt
intake. Am J Physiol Renal Physiol, v.286, n.6, Jun, p.F1054-8. 2004.
Lazarowski, E. R., R. C. Boucher e T. K. Harden. Mechanisms of release of
nucleotides and integration of their action as P2X- and P2Y-receptor
activating molecules. Mol Pharmacol, v.64, n.4, Oct, p.785-95. 2003.
Le Hir, M. e B. Kaissling. Distribution and regulation of renal ecto-5'-nucleotidase:
implications for physiological functions of adenosine. Am J Physiol, v.264, n.3
Pt 2, Mar, p.F377-87. 1993.
Le, K. T., M. Paquet, D. Nouel, K. Babinski e P. Seguela. Primary structure and
expression of a naturally truncated human P2X ATP receptor subunit from
brain and immune system. FEBS Lett, v.418, n.1-2, Nov 24, p.195-9. 1997.
Lee, H. T. e C. W. Emala. Characterization of adenosine receptors in human
kidney proximal tubule (HK-2) cells. Exp. Nephrol., v.10, n.5-6, p.383-92.
2002.
Lee, H. T. e C. W. Emala. Protective effects of renal ischemic preconditioning and
adenosine pretreatment: role of A(1) and A(3) receptors. Am J Physiol Renal
Physiol, v.278, n.3, Mar, p.F380-7. 2000.
Lee, H. T., A. Ota-Setlik, H. Xu, V. D. D'agati, M. A. Jacobson e C. W. Emala. A3
adenosine receptor knockout mice are protected against ischemia- and
myoglobinuria-induced renal failure. Am J Physiol Renal Physiol, v.284, n.2,
Feb, p.F267-73. 2003.
Lee, H. T., G. Gallos, S. H. Nasr e C. W. Emala. A1 adenosine receptor activation
inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion
injury in mice. J Am Soc Nephrol, v.15, n.1, Jan, p.102-11. 2004.
Lee, H. T., H. Xu, S. H. Nasr, J. Schnermann e C. W. Emala. A1 adenosine
receptor knockout mice exhibit increased renal injury following ischemia and
reperfusion. Am J Physiol Renal Physiol, v.286, n.2, Feb, p.F298-306. 2004.

123
Lee, Y. J. e H. J. Han. Effect of adenosine triphosphate in renal ischemic injury:
involvement of NF-kappaB. J Cell Physiol, v.204, n.3, Sep, p.792-9. 2005.
Lee, Y. J. e H. J. Han. Regulatory mechanisms of Na(+)/glucose cotransporters in
renal proximal tubule cells. Kidney Int Suppl, n.106, Aug, p.S27-35. 2007.
Lee, Y. J., S. H. Park e H. J. Han. ATP stimulates Na+-glucose cotransporter
activity via cAMP and p38 MAPK in renal proximal tubule cells. Am J Physiol
Cell Physiol, v.289, n.5, Nov, p.C1268-76. 2005.
Leipziger, J. Control of epithelial transport via luminal P2 receptors. Am J Physiol
Renal Physiol, v.284, n.3, Mar, p.F419-32. 2003.
Lemas, M. V. e D. M. Fambrough. Sequence analysis of DNA encoding an avian
Na+,K(+)-ATPase beta 2-subunit. Biochim Biophys Acta, v.1149, n.2, Jul 4,
p.339-42. 1993.
Libert, F., M. Parmentier, A. Lefort, J. E. Dumont e G. Vassart. Complete
nucleotide sequence of a putative G protein coupled receptor: RDC7. Nucleic
Acids Res, v.18, n.7, Apr 11, p.1915. 1990a.
Libert, F., M. Parmentier, A. Lefort, J. E. Dumont e G. Vassart. Complete
nucleotide sequence of a putative G protein coupled receptor: RDC8. Nucleic
Acids Res, v.18, n.7, Apr 11, p.1914. 1990b.
Lin, J. J., P. C. Churchill e A. K. Bidani. Effect of theophylline on the initiation
phase of postischemic acute renal failure in rats. J Lab Clin Med, v.108, n.2,
Aug, p.150-4. 1986.
Lin, J. J., P. C. Churchill e A. K. Bidani. Theophylline in rats during maintenance
phase of post-ischemic acute renal failure. Kidney Int, v.33, n.1, Jan, p.24-8.
1988.
Linden, J. Molecular approach to adenosine receptors: receptor-mediated
mechanisms of tissue protection. Annu Rev Pharmacol Toxicol, v.41, p.775-
87. 2001.

124
Linden, J., H. E. Taylor, A. S. Robeva, A. L. Tucker, J. H. Stehle, S. A. Rivkees, J.
S. Fink e S. M. Reppert. Molecular cloning and functional expression of a
sheep A3 adenosine receptor with widespread tissue distribution. Mol
Pharmacol, v.44, n.3, p.524-32. 1993.
Lingrel, J. B. e T. Kuntzweiler. Na+,K(+)-ATPase. J Biol Chem, v.269, n.31, Aug 5,
p.19659-62. 1994.
Liu, F. Y. e M. G. Cogan. Role of protein kinase C in proximal bicarbonate
absorption and angiotensin signaling. Am J Physiol, v.258, n.4 Pt 2, Apr,
p.F927-33. 1990.
Listhrop, R; Nelson, R; Ecelbarger, C.A.; Kohan, D.E; Kishore, B. Genetic deletion
of P2Y2 receptor (P2Y2-R) alters the protein abundances of renal sodium
transporters and channels. FASEB J. 21:937.4. 2007.
Lowry, O. H., N. J. Rosebrough, A. L. Farr e R. J. Randall. Protein measurement
with the Folin phenol reagent. J. Biol. Chem., v.193, p.265-275. 1951.
Lu, M., G. G. Macgregor, W. Wang e G. Giebisch. Extracellular ATP inhibits the
small-conductance K channel on the apical membrane of the cortical
collecting duct from mouse kidney. J Gen Physiol, v.116, n.2, Aug, p.299-310.
2000.
Lynch, K. J., E. Touma, W. Niforatos, K. L. Kage, E. C. Burgard, T. Van Biesen, E.
A. Kowaluk e M. F. Jarvis. Molecular and functional characterization of human
P2X(2) receptors. Mol Pharmacol, v.56, n.6, Dec, p.1171-81. 1999.
Ma, H. e B. N. Ling. Luminal adenosine receptors regulate amiloride-sensitive Na+
channels in A6 distal nephron cells. Am J Physiol, v.270, n.5 Pt 2, May,
p.F798-805. 1996.
Madeira, E. P. Q. Modulação da Na-ATPase insensível à ouabaína em túbulo
proximal de córtex de ratos SHR . Tese de Doutorado em Ciências
Biológicas (Fisiologia) - Universidade Federal do Rio de Janeiro. 2008.

125
Maia, J. C., S. L. Gomes e M. H. Juliani. Genes of antigens of parasites. In: C. M.
Morel (Ed.). A laboratory manual. Rio de Janeiro: Fundação Oswaldo Cruz,
1993. Genes of antigens of parasites., p.146-157
Malnic, G., H. Enokibara, M. M. Aires e F. L. Vieira. [Effect of furosemid and NaCl-
loading on chloride excretion in single nephrons of rat kidneys]. Pflugers.
Arch., v.309, n.1, p.21-37. 1969.
Marin, R., G. Ayes, T. Proverbio e F. Proverbio. The effect of cell K+ on the volume
regulation in rat kidney cortex cells. Biomed Biochim Acta, v.48, n.4, p.303-15.
1989.
Marin, R., T. Proverbio e F. Proverbio. Active sodium transport in basolateral
plasma membrane vesicles from rat kidney proximal tubular cells. Biochim.
Biophys. Acta, v.814, n.2, p.363-73. 1985.
Masereeuw, R., S. Notenboom, P. H. Smeets, A. C. Wouterse e F. G. Russel.
Impaired renal secretion of substrates for the multidrug resistance protein 2 in
mutant transport-deficient (TR-) rats. J Am Soc Nephrol, v.14, n.11, Nov,
p.2741-9. 2003.
Mcculloch, F., R. Chambrey, D. Eladari e J. Peti-Peterdi. Localization of connexin
30 in the luminal membrane of cells in the distal nephron. Am J Physiol Renal
Physiol, v.289, n.6, Dec, p.F1304-12. 2005.
Mello-Aires, M. Fisiologia: Guanabara Koogan. 2008
Mercer, R. W., D. Biemesderfer, D. P. Bliss, Jr., J. H. Collins e B. Forbush, 3rd.
Molecular cloning and immunological characterization of the gamma
polypeptide, a small protein associated with the Na,K-ATPase. J Cell Biol,
v.121, n.3, May, p.579-86. 1993.
Millar, I. D. e L. Robson. Na+-alanine uptake activates a Cl- conductance in frog
renal proximal tubule cells via nonconventional PKC. Am J Physiol Renal
Physiol, v.280, n.5, May, p.F758-67. 2001.
Miller, W. L., R. A. Thomas, R. M. Berne e R. Rubio. Adenosine production in the
ischemic kidney. Circ Res, v.43, n.3, Sep, p.390-7. 1978.

126
Moe, O. W. Acute regulation of proximal tubule apical membrane Na/H exchanger
NHE-3: role of phosphorylation, protein trafficking, and regulatory factors. J
Am Soc Nephrol, v.10, n.11, Nov, p.2412-25. 1999.
Moller, J. V., B. Juul e M. Le Maire. Structural organization, ion transport, and
energy transduction of P-type ATPases. Biochim Biophys Acta, v.1286, n.1,
May 6, p.1-51. 1996.
Moretti, R., M. Martin, T. Proverbio, F. Proverbio e R. Marin. Ouabain-insensitive
Na-ATPase activity in homogenates from different animal tissues. Comp.
Biochem. Physiol. B., v.98, n.4, p.623-6. 1991.
Mozaffari, M. S., W. Abebe e B. K. Warren. Renal adenosine A3 receptors in the
rat: assessment of functional role. Can J Physiol Pharmacol, v.78, n.5, May,
p.428-32. 2000.
Navar, L. G., L. M. Harrison-Bernard, C. T. Wang, L. Cervenka e K. D. Mitchell.
Concentrations and actions of intraluminal angiotensin II. J Am Soc Nephrol,
v.10, n.Suppl 11, p.S189-95. 1999.
Nishiyama, A., S. Kimura, H. He, K. Miura, M. Rahman, Y. Fujisawa, T. Fukui e Y.
Abe. Renal interstitial adenosine metabolism during ischemia in dogs. Am J
Physiol Renal Physiol, v.280, n.2, Feb, p.F231-8. 2001.
Ogawa, T., M. Vatta, B. G. Bruneau e A. J. De Bold. Characterization of natriuretic
peptide production by adult heart atria. Am J Physiol, v.276, n.6 Pt 2, Jun,
p.H1977-86. 1999.
Okada, S. F., W. K. O'neal, P. Huang, R. A. Nicholas, L. E. Ostrowski, W. J.
Craigen, E. R. Lazarowski e R. C. Boucher. Voltage-dependent anion
channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in
murine cells. J Gen Physiol, v.124, n.5, Nov, p.513-26. 2004.
Okusa, M. D. A(2A) adenosine receptor: a novel therapeutic target in renal
disease. Am. J. Physiol. Renal Physiol., v.282, n.1, p.F10-8. 2002.
Olah, M. E. e G. L. Stiles. Adenosine receptor subtypes: characterization and
therapeutic regulation. Annu Rev Pharmacol Toxicol, v.35, p.581-606. 1995.

127
Orlowski, J. e R. A. Kandasamy. Delineation of transmembrane domains of the
Na+/H+ exchanger that confer sensitivity to pharmacological antagonists. J
Biol Chem, v.271, n.33, Aug 16, p.19922-7. 1996.
Osswald, H., H. J. Schmitz e R. Kemper. Tissue content of adenosine, inosine and
hypoxanthine in the rat kidney after ischemia and postischemic recirculation.
Pflugers Arch, v.371, n.1-2, Oct 19, p.45-9. 1977.
Osswald, H., W. S. Spielman e F. G. Knox. Mechanism of adenosine-mediated
decreases in glomerular filtration rate in dogs. Circ Res, v.43, n.3, Sep, p.465-
9. 1978.
Otero, M., R. C. Garrad, B. Velazquez, M. G. Hernandez-Perez, J. M. Camden, L.
Erb, L. L. Clarke, J. T. Turner, G. A. Weisman e F. A. Gonzalez. Mechanisms
of agonist-dependent and -independent desensitization of a recombinant
P2Y2 nucleotide receptor. Mol Cell Biochem, v.205, n.1-2, Feb, p.115-23.
2000.
Paller, M. S., E. J. Schnaith e M. E. Rosenberg. Purinergic receptors mediate cell
proliferation and enhanced recovery from renal ischemia by adenosine
triphosphate. J Lab Clin Med, v.131, n.2, Feb, p.174-83. 1998.
Pangrsic, T., M. Potokar, P. G. Haydon, R. Zorec e M. Kreft. Astrocyte swelling
leads to membrane unfolding, not membrane insertion. J Neurochem, v.99,
n.2, Oct, p.514-23. 2006.
Park, K. M., A. Chen e J. V. Bonventre. Prevention of kidney ischemia/reperfusion-
induced functional injury and JNK, p38, and MAPK kinase activation by
remote ischemic pretreatment. J Biol Chem, v.276, n.15, Apr 13, p.11870-6.
2001.
Pawelczyk, T., D. Bizon e S. Angielski. The distribution of enzymes involved in
purine metabolism in rat kidney. Biochim Biophys Acta, v.1116, n.3, Jun 12,
p.309-14. 1992.
Perico, N., D. Cattaneo, M. H. Sayegh e G. Remuzzi. Delayed graft function in
kidney transplantation. Lancet, v.364, n.9447, Nov 13-19, p.1814-27. 2004.

128
Popova, L., Y. Balnokin, K. J. Dietz e H. Gimmler. Na+-ATPase from the plasma
membrane of the marine alga Tetraselmis (Platymonas) viridis forms a
phosphorylated intermediate. FEBS Lett, v.426, n.2, Apr 17, p.161-4. 1998.
Potokar, M., M. Kreft, L. Li, J. Daniel Andersson, T. Pangrsic, H. H. Chowdhury, M.
Pekny e R. Zorec. Cytoskeleton and vesicle mobility in astrocytes. Traffic, v.8,
n.1, Jan, p.12-20. 2007.
Prat, A. G., I. L. Reisin, D. A. Ausiello e H. F. Cantiello. Cellular ATP release by the
cystic fibrosis transmembrane conductance regulator. Am J Physiol, v.270,
n.2 Pt 1, Feb, p.C538-45. 1996.
Proverbio, F. e J. R. Del Castillo. Na+-stimulated ATPase activities in kidney basal-
lateral plasma membranes. Biochim. Biophys. Acta, v.646, n.1, p.99-108.
1981.
Proverbio, F., M. Condrescu-Guidi e G. Whittembury. Ouabain-insensitive Na+
stimulation of an Mg-2+ -dependent ATPase in kidney tissue. Biochim.
Biophys. Acta, v.394, n.2, p.281-92. 1975.
Proverbio, F., R. Marin e T. Proverbio. The 'second' sodium pump and cell volume.
Curr Top Membr Transp, v.34, p.105-120. 1989.
Proverbio, F., T. Proverbio e R. Marin. Na+-ATPase is a different entity from the
(Na+ + K+)-ATPase in rat kidney basolateral plasma membranes. Biochim.
Biophys. Acta, v.858, n.1, p.202-5. 1986.
Rangel, L. B., C. Caruso-Neves, L. S. Lara e A. G. Lopes. Angiotensin II stimulates
renal proximal tubule Na(+)-ATPase activity through the activation of protein
kinase C. Biochim Biophys Acta, v.1564, n.2, p.310-6. 2002.
Rassendren, F., G. N. Buell, C. Virginio, G. Collo, R. A. North e A. Surprenant. The
permeabilizing ATP receptor, P2X7. Cloning and expression of a human
cDNA. J Biol Chem, v.272, n.9, Feb 28, p.5482-6. 1997.
Reisin, I. L., A. G. Prat, E. H. Abraham, J. F. Amara, R. J. Gregory, D. A. Ausiello e
H. F. Cantiello. The cystic fibrosis transmembrane conductance regulator is a

129
dual ATP and chloride channel. J Biol Chem, v.269, n.32, Aug 12, p.20584-
91. 1994.
Rostovtseva, T. e M. Colombini. ATP flux is controlled by a voltage-gated channel
from the mitochondrial outer membrane. J Biol Chem, v.271, n.45, Nov 8,
p.28006-8. 1996.
Sabirov, R. Z. e Y. Okada. ATP-conducting maxi-anion channel: a new player in
stress-sensory transduction. Jpn J Physiol, v.54, n.1, Feb, p.7-14. 2004.
Sauer, H., J. Hescheler e M. Wartenberg. Mechanical strain-induced Ca(2+) waves
are propagated via ATP release and purinergic receptor activation. Am J
Physiol Cell Physiol, v.279, n.2, Aug, p.C295-307. 2000.
Scemes, E., S. O. Suadicani e D. C. Spray. Intercellular communication in spinal
cord astrocytes: fine tuning between gap junctions and P2 nucleotide
receptors in calcium wave propagation. J Neurosci, v.20, n.4, Feb 15, p.1435-
45. 2000.
Schnermann, J. B. e S. I. Sayegh. Kidney Physiology: Lippincott-Raven. 1998
Schulze-Lohoff, E., C. Hugo, S. Rost, S. Arnold, A. Gruber, B. Brune e R. B.
Sterzel. Extracellular ATP causes apoptosis and necrosis of cultured
mesangial cells via P2Z/P2X7 receptors. Am J Physiol, v.275, n.6 Pt 2, Dec,
p.F962-71. 1998.
Schwiebert, E. M. ATP release mechanisms, ATP receptors and purinergic
signalling along the nephron. Clin Exp Pharmacol Physiol, v.28, n.4, Apr,
p.340-50. 2001.
Schwiebert, E. M. e A. Zsembery. Extracellular ATP as a signaling molecule for
epithelial cells. Biochim Biophys Acta, v.1615, n.1-2, Sep 2, p.7-32. 2003.
Schwiebert, E. M. e B. K. Kishore. Extracellular nucleotide signaling along the renal
epithelium. Am J Physiol Renal Physiol, v.280, n.6, Jun, p.F945-63. 2001.
Seldin, D. W. e G. Geibisch. The Kidney: Physiology and Pathophysiology: Raven
Press. 1992

130
Shamraj, O. I. e J. B. Lingrel. A putative fourth Na+,K(+)-ATPase alpha-subunit
gene is expressed in testis. Proc Natl Acad Sci U S A, v.91, n.26, Dec 20,
p.12952-6. 1994.
Shrode, L. D., B. S. Gan, S. J. D'souza, J. Orlowski e S. Grinstein. Topological
analysis of NHE1, the ubiquitous Na+/H+ exchanger using chymotryptic
cleavage. Am J Physiol, v.275, n.2 Pt 1, Aug, p.C431-9. 1998.
Shull, G. E., J. Greeb e J. B. Lingrel. Molecular cloning of three distinct forms of the
Na+,K+-ATPase alpha-subunit from rat brain. Biochemistry, v.25, n.25, Dec
16, p.8125-32. 1986.
Silva, P. A. D. Alterações nos transportadores renais de sódio e na sua regulação
por angiotensina II estão envolvidas na gênese de hipertensão arterial na
desnutrição crônica. Dissertação de Mestrado em Ciências Biológicas
(Biofísica) - Universidade Federal do Rio de Janeiro. 2007.
Siragy, H. M. Evidence that intrarenal bradykinin plays a role in regulation of renal
function. Am J Physiol, v.265, n.4 Pt 1, Oct, p.E648-54. 1993.
Skou, J. C. The influence of some cations on an adenosine triphosphatase from
peripheral nerves. Biochim Biophys Acta, v.23, n.2, Feb, p.394-401. 1957.
Slepkov, E. R., J. K. Rainey, B. D. Sykes e L. Fliegel. Structural and functional
analysis of the Na+/H+ exchanger. Biochem J, v.401, n.3, Feb 1, p.623-33.
2007.
Smith, J. A., E. M. Whitaker, N. Yaktubay, M. J. Morton, C. J. Bowmer e M. S.
Yates. Regulation of renal adenosine A(1) receptors: effect of dietary sodium
chloride. Eur J Pharmacol, v.384, n.1, p.71-9. 1999.
Spielman, W. S. e C. I. Thompson. A proposed role for adenosine in the regulation
of renal hemodynamics and renin release. Am. J. Physiol., v.242, n.5, p.F423-
35. 1982.
Sugita, M., Y. Yue e J. K. Foskett. CFTR Cl- channel and CFTR-associated ATP
channel: distinct pores regulated by common gates. EMBO J, v.17, n.4, Feb
16, p.898-908. 1998.

131
Sun, R., R. L. Miller, A. C. Hemmert, P. Zhang, H. Shi, R. D. Nelson e B. K.
Kishore. Chronic dDAVP infusion in rats decreases the expression of P2Y2
receptor in inner medulla and P2Y2 receptor-mediated PGE2 release by
IMCD. Am J Physiol Renal Physiol, v.289, n.4, Oct, p.F768-76. 2005.
Suzuki, Y., S. Ueno, R. Ohnuma e N. Koyama. Cloning, sequencing and functional
expression in Escherichia coli of the gene for a P-type Na(+)-ATPase of a
facultatively anaerobic alkaliphile, Exiguobacterium aurantiacum. Biochim
Biophys Acta, v.1727, n.3, Mar 10, p.162-8. 2005.
Sweadner, K. J. Isozymes of the Na+/K+-ATPase. Biochim Biophys Acta, v.988,
n.2, May 9, p.185-220. 1989.
Takeda, M., K. Yoshitomi e M. Imai. Regulation of Na(+)-3HCO3- cotransport in
rabbit proximal convoluted tubule via adenosine A1 receptor. Am. J. Physiol.,
v.265, n.4 Pt 2, p.F511-9. 1993.
Thomas, W. G. e H. Qian. Arresting angiotensin type 1 receptors. Trends
Endocrinol Metab, v.14, n.3, p.130-6. 2003.
Tomanin, R., C. Ballarin, B. Nardini, G. Mastrangelo e F. Sarto. Influence of
smoking habit on the frequency of micronuclei in human lymphocytes by the
cytokinesis block method. Mutagenesis, v.6, n.2, Mar, p.123-6. 1991.
Townsend-Nicholson, A. e J. Shine. Molecular cloning and characterisation of a
human brain A1 adenosine receptor cDNA. Brain Res Mol Brain Res, v.16,
n.3-4, Dec, p.365-70. 1992.
Toyofuku, T., M. Yabuki, K. Otsu, T. Kuzuya, M. Hori e M. Tada. Intercellular
calcium signaling via gap junction in connexin-43-transfected cells. J Biol
Chem, v.273, n.3, Jan 16, p.1519-28. 1998.
Trombetta, E. S. e A. Helenius. Glycoprotein reglucosylation and nucleotide sugar
utilization in the secretory pathway: identification of a nucleoside
diphosphatase in the endoplasmic reticulum. EMBO J, v.18, n.12, Jun 15,
p.3282-92. 1999.

132
Turner, C. M., O. Vonend, C. Chan, G. Burnstock e R. J. Unwin. The pattern of
distribution of selected ATP-sensitive P2 receptor subtypes in normal rat
kidney: an immunohistological study. Cells Tissues Organs, v.175, n.2, p.105-
17. 2003.
Ueno, S., N. Kaieda e N. Koyama. Characterization of a P-type Na+-ATPase of a
facultatively anaerobic alkaliphile, Exiguobacterium aurantiacum. J Biol Chem,
v.275, n.19, May 12, p.14537-40. 2000.
Unwin, R. J., M. A. Bailey e G. Burnstock. Purinergic signaling along the renal
tubule: the current state of play. News Physiol Sci, v.18, Dec, p.237-41. 2003.
Urano, T., H. Nishimori, H. Han, T. Furuhata, Y. Kimura, Y. Nakamura e T. Tokino.
Cloning of P2XM, a novel human P2X receptor gene regulated by p53.
Cancer Res, v.57, n.15, Aug 1, p.3281-7. 1997.
Valera, S., N. Hussy, R. J. Evans, N. Adami, R. A. North, A. Surprenant e G. Buell.
A new class of ligand-gated ion channel defined by P2x receptor for
extracellular ATP. Nature, v.371, n.6497, Oct 6, p.516-9. 1994.
Vallon, V. e J. Schnermann. Tubuloglomerular feedback. Methods Mol Med, v.86,
p.429-41. 2003.
Vallon, V. P2 receptors in the regulation of renal transport mechanisms. Am J
Physiol Renal Physiol, v.294, n.1, Jan, p.F10-27. 2008.
Vallon, V. Tubuloglomerular feedback and the control of glomerular filtration rate.
News Physiol. Sci., v.18, p.169-74. 2003.
Vallon, V., B. Muhlbauer e H. Osswald. Adenosine and kidney function. Physiol
Rev, v.86, n.3, Jul, p.901-40. 2006.
Van Aubel, R. A., P. H. Smeets, J. G. Peters, R. J. Bindels e F. G. Russel. The
MRP4/ABCC4 gene encodes a novel apical organic anion transporter in
human kidney proximal tubules: putative efflux pump for urinary cAMP and
cGMP. J Am Soc Nephrol, v.13, n.3, Mar, p.595-603. 2002.

133
Van Calker, D., M. Muller e B. Hamprecht. Adenosine regulates via two different
types of receptors, the accumulation of cyclic AMP in cultured brain cells. J
Neurochem, v.33, n.5, Nov, p.999-1005. 1979.
Van De Water, F. M., J. M. Boleij, J. G. Peters, F. G. Russel e R. Masereeuw.
Characterization of P-glycoprotein and multidrug resistance proteins in rat
kidney and intestinal cell lines. Eur J Pharm Sci, v.30, n.1, Jan, p.36-44. 2007.
Vekaria, R. M., R. J. Unwin e D. G. Shirley. Intraluminal ATP concentrations in rat
renal tubules. J Am Soc Nephrol, v.17, n.7, Jul, p.1841-7. 2006.
Vitzthum, H., B. Weiss, W. Bachleitner, B. K. Kramer e A. Kurtz. Gene expression
of adenosine receptors along the nephron. Kidney Int, v.65, n.4, Apr, p.1180-
90. 2004.
Wada, M., O. Urayama, S. Satoh, Y. Hara, Y. Ikawa e T. Fujii. A marine algal
Na(+)-activated ATPase possesses an immunologically identical epitope to
Na+,K(+)-ATPase. FEBS Lett, v.309, n.3, Sep 14, p.272-4. 1992.
Wang, T. e Y. L. Chan. Time- and dose-dependent effects of protein kinase C on
proximal bicarbonate transport. J Membr Biol, v.117, n.2, Aug, p.131-9. 1990.
Wang, T. F., Y. Ou e G. Guidotti. The transmembrane domains of ectoapyrase
(CD39) affect its enzymatic activity and quaternary structure. J Biol Chem,
v.273, n.38, Sep 18, p.24814-21. 1998.
Weihprecht, H., J. N. Lorenz, J. P. Briggs e J. Schnermann. Vasomotor effects of
purinergic agonists in isolated rabbit afferent arterioles. Am J Physiol, v.263,
n.6 Pt 2, Dec, p.F1026-33. 1992.
Wengert, M., C. Berto Jr., J. Kaufman, L. R. Leão-Ferreira, R. Paes-De-Carvalho,
A. G. Lopes e C. Caruso-Neves. Stimulation of the proximal tubule Na+-
ATPase activity by adenosine A2A receptor. Int J Biochem Cell Biol, v.37, n.1,
p.155-165. 2005.
Wengert, M., J. Adao-Novaes, N. Assaife-Lopes, L. R. Leao-Ferreira e C. Caruso-
Neves. Adenine-induced inhibition of Na(+)-ATPase activity: Evidence for

134
involvement of the Gi protein-coupled receptor in the cAMP signaling
pathway. Arch Biochem Biophys, v.467, n.2, Nov 15, p.261-7. 2007.
Whittembury, G. e F. Proverbio. Two modes of Na extrusion in cells from guinea
pig kidney cortex slices. Pflugers. Arch., v.316, n.1, p.1-25. 1970.
Whittembury, G. e J. Fishman. Relation between cell Na extrusion and transtubular
absorption in the perfused toad kidney: the effect of K, ouabain and
ethacrynic acid. Pflugers Arch, v.307, n.3, p.138-53. 1969.
Wiegele, G., M. Brandis e L. B. Zimmerhackl. Apoptosis and necrosis during
ischaemia in renal tubular cells (LLC-PK1 and MDCK). Nephrol Dial
Transplant, v.13, n.5, May, p.1158-67. 1998.
Yamamoto, K., R. Korenaga, A. Kamiya e J. Ando. Fluid shear stress activates
Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res,
v.87, n.5, Sep 1, p.385-91. 2000.
Yao, S. Y., A. M. Ng, W. R. Muzyka, M. Griffiths, C. E. Cass, S. A. Baldwin e J. D.
Young. Molecular cloning and functional characterization of
nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei)
equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat
tissues. J Biol Chem, v.272, n.45, Nov 7, p.28423-30. 1997.
Yoshioka, K., O. Saitoh e H. Nakata. Heteromeric association creates a P2Y-like
adenosine receptor. Proc Natl Acad Sci U S A, v.98, n.13, Jun 19, p.7617-22.
2001.
Zhang, Y., D. Sanchez, J. Gorelik, D. Klenerman, M. Lab, C. Edwards e Y.
Korchev. Basolateral P2X4-like receptors regulate the extracellular ATP-
stimulated epithelial Na+ channel activity in renal epithelia. Am J Physiol
Renal Physiol, v.292, n.6, Jun, p.F1734-40. 2007.
Zhou, Q. Y., C. Li, M. E. Olah, R. A. Johnson, G. L. Stiles e O. Civelli. Molecular
cloning and characterization of an adenosine receptor: the A3 adenosine
receptor. Proc Natl Acad Sci U S A, v.89, n.16, Aug 15, p.7432-6. 1992.

135
Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn
Schmiedebergs Arch Pharmacol, v.362, n.4-5, Nov, p.299-309. 2000.
Zou, A. P., F. Wu, P. L. Li e A. W. Cowley, Jr. Effect of chronic salt loading on
adenosine metabolism and receptor expression in renal cortex and medulla in
rats. Hypertension, v.33, n.1 Pt 2, Jan, p.511-6. 1999.
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo










![Filo Platyhelminthes [Modo de Compatibilidade] · Sistema Nervoso Possuem sistema nervoso ganglionar difuso. 5 Prof. Gilmar Marques. Excreção Nos platelmintos a excreção é feita](https://img.document.onl/doc/110x75/5be6f7e709d3f23a518db985/filo-platyhelminthes-modo-de-compatibilidade-sistema-nervoso-possuem-sistema.jpg)


















