Embed Size (px)
Citation preview

INSTITUTO DE PESQUISAS ENERGÉTICAS F NUCLEARESSECRETARIA DA INDÚSTRIA. COMÉRCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA A UNIVERSIDADE DE SAO PAULO
ESTUDO DO COMPORTAMENTO ESTRUTURAL E CINÉTICA DE
OXIDAÇAO DE DIÓXIDO DE URÂNIO, POR DIFRATOMETRIA DE
RAIOS-X EM ALTA-TEMPERATURA
Silvio Rainho Teixeira
Dissertação apresentada aoInstituto de Pesquisas Energéticas e Nucleas-sscomo parte dos requisitos para obtenção
do grau de «Mestre-Área Tecnologia Nuclear"
Orientador: Dr, KENGO IMAKUMA
S&o Paulo1981

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARESSECRETARIA DA INDUSTRIA, COMERCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA A" UNIVERSIDADE DE SBO PAULO
ESTUDO DO COMPORTAMENTO ESTRUTURAL E CINÉTICA DEOXIDAÇÃO DE DIÕXIDO DE URÂNIO, POR DIFRATOMETRIA DE
RAIOS-X EM ALTA-TEMPERATURA
SILVIO RAINHO TEIXEIRA
Dissertação apresentada aoInstituto de Pesquisas Energéticas e Nuclearescomo parte dos requisitos para obtençãodo grau de "Mestre-Ãrea Tecnologia Nuclear"
Orientador: Dr. KENGO IMAKUMA
SÃo PAULO1981

AGRAfECIMEWTOS
PA.. Kenge ~ma.tu.ma On.-icnta.dc K
PA. Cláudio Rcdnigu.e.& Gz.ne.ntt da AFE
. Uo.Kn.an.JL A . I . de. kmon.in Supcfiinttndínte. do 7PEN
Vn.. Claae.fi T. da F-te-í. ai
Vn.. Re.ginaldc
Raimundo C. flune.4
Gzuznto. do Cl'.N
cmCMW
M.Sc. Aníonio
Agn&ó K. ttaaamine.
CPV
CTV
U.Sc. Cibe.lt B. CFW
PA.. Sp?./io P. MoKato
M.Sc. l-.xLzia Caaanlio
hi.Sc. Luiz C. de. P. Rcino
M.Sc. Ma.viii In£4 Coita
Eguit>e,n.to Galego
;.VEAVE
APE
APE
ATE
PRMUCLEAK
A0.6
de.( ime.nto&
peta. c.i.izc dt, incentivo e
cclciboft.ac.ac.

. rc-:.»..'.o. c.Í p c 6 a , 2L' oc
.li \.fji { •'.!. La.'. , / ' . . * : / . ' ; ; a \iv. ,\t'..,o'^ian) c
i A c ; . / ; 1

S U M Á R I O
PAGINA
HESUMO i
ABSTRACT ii
CAPÍTULO I
1. INTRODUÇÃO 1
1.1. Revisão biblicgrãfice 2
1.2. Objetivos 5
CAPITULO II
2. DIFRATOMETRIA DE RAIOS-X 6
2.1. Dif:rat ime-ria de pó 6
2.2. Aná.i e quantitativa 10
CAPÍTULO III
3. si£:?a^ UR?".7ÍIO-O>::G;.-:IO 13
3 . 1 . L'iaçram£ cie f.azss 14
3 . 1 . 1 . ; . Í = Í 2 U, ', 14
3 . 1 . 2 . A füse U.o7 16
3.2. Oxióéçío 17
3.3. Estrutura cr;.rt ! . l ;n;. 19
3 . 3 . 1 . EEt::utv._-a :•:• í".iLj;:ir~r> ei vríi:.;o 21
3.3 .2 . Estrutura 3o.-; ó::idos "âo a cruionvúuri_
cor, 23C/JPlTULO IV
4. P;.Í>TS E;:PDÍ.:;:~KT;.L 26
4.1. r'repi.rrcno òc L.;i3ztrr.z 26
4.2. Instrument! 28

PAGINA
4.3. Condições experimentais 31
4.3.1. Série TA 34
4.3.2. Série TB 35
4.3.3. Temperatura ambiente 36
CAPlTUIiO V
5. DADOS E DISCUSSÃO 37
5.1. Série TA 37
5.2. Sfrie TB 49
5.3. Temperatura ambiente 68
CAPITULO VI
6. CONCLUSÕES 74
REFERÊNCIAS BIBLIOGRÁFICAS 76

ESTUDO DO COWPORTAWEKTO ESTRUTURAL E CINÊTICA DE OXIDAÇAO DE DlOXIDO DE URÂNIO , POR DIFRATOMETRIA DE RAIOS-X DE ALTA TEMPERATURA
R E S U M O
Foi estudado o comportamento estrutural do 00,(placas sinteri
zadas), quando aquecido em atmosfera inerte com baixa concentração
de oxigênio (-140 ppm); utilizando a técnica de difratometria de
raios-X, ••'
. Em temperaturas maiores que 165°C foi determinado o coeficien
te de expansão térmica do Ü0- Q 5: 10,5 x 10 °C~ . Nas temperatu
ras de 170, 235 e 275j foram acompanhadas as transformações estru
turais devido ã oxidaçao. A oxidação isotérmica do U02 até a com
posição D3°7» se processa de forma parabólica e a difusão de oxi
gênio, através da camada produto (U.Og)> é o processo controlando
a velocidade de oxidação. As fases observadas foram: UO2 (cúbico)
— U.OQ (cúbico) — U3O7 (tetragonal). Foram determinadas dife
rentes energias de ativação de oxidação, para diferentes famílias
de planos cristalogrãficos (khl), indicando uma ocupação preferen
ciai dos átomos de^ oxigênio (intersticiais),dentro da estrutura
do 002. (

CRYSTALLOGRAPHIC AND OXIDATION KINETIC STUDY OF URANIUM DIOXIDE BYHIGH TEHPE.WURE X-RAY DIFFRACTOHETRY
A B S T R A C T
The structural behavior of UO, sintered plates was studied
as a function of temperature by X-ray diffractonetry. All the ex
periments were carried out under an inert atmosphere with low
oxygen content (-140 ppn).
. The thermal expansion coefficient of UO2 Q 5 was found to be
10,5 x 10 °C~~ for temperatures above 165°C. Structural trans
formations during oxidation were observed at 170,235 and 275°C. The
isothermal oxidation of UO2 to U3O7 follows a parabolic form and
the diffusion of oxygen through the product layer UjOg is the
mechanism contrclling the oxidation rate. The phases observed were
UOj (cubic) — °4°9 (cubic) — °3°7 (tetragonal). Activation en
ergies of oxidation were found for different crystallographic planes
(hkl). From this one can conclude that there is a preferential
occupation of interstitial oxygen within the VO2 structure.
ii

LISTA DE FIGURAS
PAGINA
FIGURA 2.1. Dedução da equação de Bragg 07
FIGURA 2.2. Geometria do Goniômetro SG-8 08
FIGURA 2.3. Perfil de difração , teórico, para õxidos
não estequiomêtrico (A), ou paticulas com
2 fases (B) 12
FIGURA 3.1. Diagrama de fases do sistema urânio- oxigê18
nio (a) apresentado por Naito e Kamegashira
(1976) e (b) por Gordfunke 1969 15
FIGURA 3.2. Comportamento do parâmetro de rede em fun18 ~"
çao da razão O/U : (1) Suzuki e colabora
dores, (2) Young e colaboradores , (3)
Perio18 e (4) Matsui e Naito16 20
FIGURA 3.3. Estrutura do U02 22
FIGURA 3.4. Estrutura cristalina do UO,,., proposta por6,12,18 2 + x
Willis . Os oxigenios normais em A e
B, no UO^, são substituídos por oxigeniosin
tersticiais O'em C e D e por 0"em E (2:1:2)
ou em E e F (2:2:2) 25
FIGURA 4.1. Porta amostra da câmara de alta temperatura 29
FIGURA 4.2. Goniômetro SG-8, câmara de alta temperatura
instalada e gerador de raios-X 29
FIGURA 4.3. Câmara de alta temperatura desmontada 30

PAGINA
FIGURA 4.4. Controlador de temperatura (ã direita), sis
tema step-scan (em baixo) e diagramas co
mum e step-scan (acima) 30
FIGURA 4.5. Diagrama do controlador de temperatura .... 32
FIGURA 4.6. Câmara de Guinier-Hâgg 33
FIGURA 5.1. Variação do parâmetro de rede, do U02+x,com
a temperatura 40
FIGURA 5.2. Comportamento da reflexão 6-311, durante a
oxidação (série TA-3) 43
FIGURA 5.3. Comportamento da reflexão 0-220, durante a
oxidação (série TA-3) 44
FIGURA 5.4. Amostra-A sem tratamento térmico (simetria
cúbica) 45
FIGURA 5.5. Amostra-A apôs tratamento térmico (série
TA-1) 46
FIGURA 5.6. Amostra-A apôs tratamento térmico (série
TA-2) 47
FIGURA 5.7. Amostra-A após tratamento térmico, apresen
tando simetria tetragonal (série TA-3) 48
FIGURA 5.8. Curvas de taxa de oxidação do UO 2 + X até
Ü4°9 57
FIGURA 5.9. Comportamento da reflexão 6-111, durante a
oxidação a 170°C 61
FIGURA 5.10. Gráficos da constante de taxa (K) e da ener
gia de ativação, para a reflexão 0-311 .... 64
FIGURA 5.11. Gráficos da constante de taxa (K) e da ener

PÁGINA
gia de ativação, para a reflexão a-200 .... 65
FIGURA 5.12. Comportamento da reflexão a-200, durante a
oxidação a 235°C 67
FIGURA 5.13. Comportamento da reflexão 0-311, durante a
oxidação a 235°C 67
FIGURA 5.14. Comportamentos diferentes das reflexões 8-311
e a-200, para a mesma temperatura e tempos
de oxidação 68
FIGURA 5.15. Gráfico da varição da intensidade (cps) das
fases U0- e U.Og, em função do tempo de
oxidação,para as reflexões a-200 e 0-311 .. 70
FIGURA 5.16. Comportamento do parâmetro de rede, das
duas fases, em função do tempo de oxidação
(reflexão B-311) 71
FIGURA 5.17. Estado inicial e final da reflexão B-311, a
temperatura ambiente (série Bi) 72

LISTA DE TABELAS
PÁGINA
TABELA 3.1. Coeficiente de expansão térmica linear paraH» 12
o IX>2 sinterizado 19
TABELA 3.2. Parâmetro de rede do U02, em Angstron, obti
do por vários autores 27
TABELA 4.1. Preparação das amostras 27
TABELA 4.2. Análise química das impurezas presentes nas
amostras de UO-, pela técnica de espectros
copia de emissão 27
TABELA 4.3. Especificações do gás argônio-U 33
TABELA 4.4. Condições experimentais da série TA-1 34
TABELA 4.5. Condições experimentais da série TA-2 35
TABELA 4.6. Condições experimentais da série TB 36
TABELA 5.1. Comportamento de algumas reflexões em fun
ção da temperatura, série TA-1 37
TABELA 5.2. Comportamento de algumas reflexões em fun
cio da temperatura, série TA-2 38
TABELA 5.3. Parâmetros de rede da amostra-1, para dife
rentes temperaturas 38
TABELA 5.4. Parâmetros de rede da amostra-1 e obtidos
por Gronvold1 ° 39

PAGINA
TABELA 5.5. Resultados apresentados pelo programa ANA
CRON: série B-l (235°C) reflexão 0-311
TABELA 5.6. Série B-l (235°C) reflexão a-200
TABELA 5.7. Série B-2 (170°C) reflexão B-311
TABELA 5.8. Série B-2 (170°C) reflexão a-200
TABELA 5.9. Série B-3 (275°C) reflexão B-311
TABELA 5.10. Série B-3 (275°C) reflexão a-200
TABELA 5.11. Dados obtidos na série TB, reflexão B-311..
TABELA 5.12. Dados obtidos na série TB, reflexão a-200..
TABELA 5.13. Constantes de taxa, para os casos conside
rados, (reflexão B-311)
TABELA 5.14. Constantes de taxa, para os casos conside
rados, (reflexão a-200)
TABELA 5.15. Energias de ativação para as reflexões B-311
e a-200
TABELA 5.16. Energias de ativação de oxidação obtidas
por diversos autores
TABELA 5.17. Resultado dos diagramas de Guinier, da amos_
tra B-l (235°C) antes e após a oxidação ...
50
51
52
53
54
55
59
60
62
62
63
66
73

CAPITULO I
1. INTRODUÇÃO
O dióxido de urânio é atualmente muito usado como combust!
vel nuclear. O alto ponto de fusão e estabilidade sob irradiação
desse material cerâmico, favorece sua utilização pára esse fim. As
propriedades e comportamento desses materiais e;n altas temperatu
ras, as quais influenciam fortemente o comportamento de um reator
nuclear de potência, têm sido matéria de muito estudo.
Uma de suas propriedades, guando sujeito a altas temperatu
ras, é tornar-se não estequiomêtricô, alterando significantemente
suas propriedades físicas e químicas, com a extensão do desvio da
estequiometria. Como a estrutura do material varia continuamente
com sua composição, uma forma de estudar a estequiometria e pro
priedades desses óxidos seria observando suas mudanças estrutu
rais quando defeitos, tais como excesso de oxigênio, são adiciona
dos ao material.
0 conhecimento da estrutura atômica das faseB não estequio
métricas é importante na interpretação do comportamento termodinâ
mico desses oxidos e, também, na determinação da relação entre suas
propriedades de transporte (por exemplo a condutividade térmica),
com a razão oxigênio-metal (O/U).
Apesar dos recentes avanços e inúmeros trabalhos realizados,
ainda existem propriedades e comportamentos, desses materiais, que
não são totalmente conhecidos.

1.1. REVISÃO BIBLIOGRAFICA
Os trabalhos encontrados na literatura sobre oxidaçao, este
quiometria e transformação de fases, utilizando a difração de
raio-X como uma das técnicas, apresentam variadas formas de obten
ção da amostra e procedimento experimental. Dentre eles serão de£
tacados alguns que foram considerados como básicos para a realiza
ção deste:10
Gronvold (1955), estudou por difração de raios-X, em
altas temperatura. , os õxiâos de urânio na região de composiçãoÜO2~Ü3°8* S u a s amostras de composição intermediária foram obtidas
misturando-se quantidades definidas, em peso, de UO2 e ü^Og em pó,
que foram cozidos ("annealed") dentro de tubos de quartzo eva
cuados e lacrados, a 800 C.
Em seu trabalho, além das fases Ü O 2 + X e U4°ç>' Gronvold ob
servou uma nova fase tetragonal de composição aproximada UO, 4 0 e
apresentou uma discussão sobre sua composição, que em outros tra
balhos foi definida como existindo na região UO2 20~UO2 30 * F o i
determinado o coeficiente de expansão térmica do UO 2 + X para vã
rias composições. Também foi concluído que relações de fase em a_l
tas temperaturas não podem ser deduzidas de amostras resfriadas
rapidamente ("quenched") e estudadas ã temperatura ambiente.2
Aronson, Roof e Belle (1956), estudaram a cinetica de
oxidaçao do diõxido de urânio na faixa de composição UC^-t^O-, em
ar seco e em oxigênio, nas temperaturas de 160 até 350°C. Os dados
de cinetica foram obtidos analisando-se o ganho em peso da amos
tra durante a oxidaçao, e os dados de difração de raios-X per
mitiram determinar qual o mecanismo de oxidaçao que atuou. Foi ob
servado que a oxidaçao se processa em dois passos, o primeiro até
a formarão de um oxido tetragonal de composição aproximada UO- A
e no r-sgundo esse oxido tetragonal se converte em um oxido ortor
rômbico, U 3 O Q . Eles obtiveram uma expressão para o coeficiente de
difusão e a energia de ativação do processo (26,3+1,5 kcal/mole).
Hoekstra, Santoro e Siegel (1960), analisaram a oxida
çio do UO2e Ü^Og em baixa temperatura (< 300°C). Observaram que
as medidas de difração utilizando filmes poderiam fornecer conclu
soes errôneas, sendo necessário usar o difratômetro para distin
guir algumas linhas múltiplas não observadas nos filmes. Em tempe
raturas menores que 160°C, a região cúbica UO2+X é, na verdade,uma
região difásica composta de UO. original e uma fase tetragonal com
c/a = 0r989, onde c=5,40 Ã. Acima dessa temperatura tem-se uma mi£

tura de duas fases cúbicas U02+x e U4Og. Os dados de cinética indi
caram que esta reação ê controlada pela difusão, e na oxidação do
UjOg não foi detectada a fase a~U3O7, sendo formada gradualmente
uma fase com c/a = 1,016 . Além da fase a-U,0_, foram identifica
das duas outras fases tetragonais: S-U,O7 com a = 5,38 8 e c=5,55 8
e outro oxido com composição aproximada UO- , com a=5,41 e c=5,49 8.
Young, Lynds, Nohl e Libowitz (1962), obtiveram duas rela
ções entre os parâmetros de rede e a razão O/U, na região UO.-U^Og,
a partir de amostras oxidadas dentro de capilares de quartzo, Ia
crados e resfriados rapidamente ("quenched"). A primeira relação
caracteriza o 1)0, e a outra oü4°9-v' c o m ""^ r e 9 i a o indetermi.
nada na faixa de composição U02 ,,- UO- 17- Também foi observado
que as amostras produzidas desta forma não eram homogêneas,e em ai
guns casos podia ser detectada a presença de duas fases.
Hme. Belbeoch, Mine. Laredo e Perio (1964) ,examinaram, por
difração de raios-X, óxidos de urânio na faixa de composição U0~ -
U02 25' após tratamento em alta temperatura seguido de "quenching".
Os resultados obtidos sugeriram que, em altas temperaturas, o sis
tema pode retornar a uma estrutura de duas fases e que próximo da
composição UO- 2 5 dois tip°s d e difratogramas são obtidos (com ou
sem raias de superestrutura), indicando que o U.Og pode existir em
pelo menos duas modificações polimõrficas.20 —
Ohashí.,Noda e Morozumi (1974), estudaram a oxidação do
dióxido de urânio por termogravimetria (dados de cinética) e difra
ção de raios-X (identificação das fases). Foi notado que quando a
oxidação é lenta, dois estágios distintos de oxidação são observa
dos: no primeiro o U02 oxida-se até a composição U3O7 e depois, no
segundo, vai até U,OC.
Saito (1976), em seu trabalho sobre processos de oxida
ção e reatividade do dióxido de urânio, utilizando as técnicas de
termogravimetria (dados de cinética) e difração de raios-X (identi
ficação de fases), fez as seguintes observações: (1) a oxidação iso
térmica do U02 inativo, na faixa de temperatura 175 a 360°C,se pro
cessa em duas etapas; (a) até a composiação U^O^, de forma parabõ
lica, e (b) até U?Og em forma de "S"; (2) os dados de difração, de
amostras parcialmente oxidadas, identificaram duas fases tetrago
nais, a - U3O7 com a=5,46 8 e c=5,40 8 e & ~Ü3°7 c o m a=5/38 8 e
c«5,55 8; (3) ele sugeriu que a oxidação do U02 obedece a seguinte
seqüência, U02—a-U3O7 B^V^Oy (desordenado) e-UjO^—Ü3°8? *4*
os dados de cinética e de di fração favoreceram um mecanismo de dl.
fusão do oxigênio (através de uma camada tetragonal de oxido for

mada), como senão o processo que controla a taxa, na primeira eta
pa de oxidação; (5) os dados de taxa são bem ajustados pela equa
ção de Jander, no primeiro estágio e (6) a energia de ativação apa
rente para o primeiro estágio de oxidação ê de 24,5 kcal/mol.22
Rand, Achermann, Gronvold, Oetting e Pattoret (1978), fi
zeram uma revisão do estado atual do programa de reavaliação das
propriedades termodinâmicas da fase urânia (UO,), organizado pela
Agência Internacional de Energia Atômica ("IAEA"). Dentre as ob
servações feitas por eles podem ser destacadas as seguintes: (1)
urãnia pode existir sobre a faixa- de composição UO, g_ até UO, 57;
(2) os contornos de fases são bem conhecidos, exceto para a re
gião hipoestequiomêtrica, acima de 2000K; (3) os õxidos U0~, U4^Q»
VJO-J, U2°5' U3°8 e U 03' apresentam apreciável faixas de homogenei.
dade; (4) abaixo de 1400K a fase desordenada UO, 25 pode formar um
composto ordenado Ü4O« ; (5) apesar dos estudos de difração de
neutrons, a posição dos átomos de oxigênio intersticiais, no IX>2+X
e U.Og, não está ainda estabelecida; (6) a faixa de composição
da fase urânia é fortemente dependente da temperatura, existindo
uma estreita faixa de homogeneidade mesmo ã temperatura ambiente ;
(7) medidas em amostras que sofreram "quenching", não são confia
veis devido â extrema mobilidade da sub-rede de ânions neste sis
tema e (8) os estudos, na região hiperestequiométrica (ÜO2+X), es
tão geralmente em concordância dentro de uma pequena incerteza nas
medidas.
Adamson, Schumacher, Navratil e Sundermann (1980), mostra^
ram uma breve revisão do simpósio sobre mudanças na atividade do
oxigênio em combustíveis õxidose um sumário das apresentações fei
tas pelos participantes. Podem ser destacadas nesse trabalho as
seguintes recomendações resultantes do encontro: (1) . obre a neces
sidade de mais estudos sobre as relações entre atividade do oxi_
gênio (Po2) ~ composição - temperatura, para vários tipos de com
bustíveis; (2) ê necessário um maior estudo sobre organização e
energias dos defeitos nos õxidos não-estequiométricos; (3) para
os combustíveis irradiados, concordou-se que técnicas deveriam ser
desenvolvidas ou aperfeiçoadas para medir O/M e Po2; (4) a medida
de parâmetros de rede por difração de raios-X, é uma das técnicas
recomendadas para ser aperfeiçoada na deter nação da razão O/M e
o grau de não estequiometria (2-O/M); e (5) 10Í recomendada uma rea
valiação da literatura sobre a relação entre parâmetros de rede e
razão O/U.
Além dos trabalhos citados, foram consultadas as seguintes re

*• ,6 #12, 18
visões bibliográficas sobre U0 2:
1,2. OBEJTIVOS
Nos trabalhos sobre cinética e processos de oxiâação do2,20,2*)
U0-, encontrados na literatura , os resultados de d.tfração
de raios-X são utilizados como complementação dos dados de cini
tica (obtidos por outras técnicas), na interpretação do mecanismo
de oxidação. Essa utilização cjorre porque, do ponto de vista e£
trutural, a difração ê uma das ferramentas mais poderosas na iden
tificação de fases presentes em materiais cristalinos. Os dados de
cinética somente, não são suficientes para distinguir, entre os
processos de oxidação, que possuem curvas de taxa muito semelhantes.
Considerando esses pontos e o fato que a estrutura e o nume
ro de fases formadas estão associadas ao mecanismo de difusão, â
oxiâação do material e à estabilidade termodinâmica das fases, os
objetivos desse trabalho são:
1) observar as transformações estruturais dos õxidos de ura
nio na faixa de composição UO^-U.O^, em temperaturas entre 100 e
300°C;
2) estudar a cinética de oxidação, na faixa UO 2—U4°g» util^
zando somente os dados de difração de raios-X, e
3) caracterizar algumas vantagens e viabilidade de utiliza
ção da técnica de difração de raios-X em análises de estequiome
tria e cinética em alta temperatura.

CAPITULO II
2. DIFRATOHETRIA DE RAIOS-X
A difratometria de raios-X ê tecnologicamente muito impor
tante, devido a sua aplicação na análise de fases e controla da
qualidade. Ela possui também ampla aplicação dentro da física, no
estudo de transições ordem-desordem e no estudo de imperfeições em
cristais (falhas de empilhamento, tensões elásticas, tamanho de
partículas).
Em altas temperaturas, ela possui a vantagem de poder acom
panhar as transformações de fases (em sólidos) dinamicamente, per
mitindo a obtenção da dependência da intensidade em função da con
centração a ser determinada
2.1. DlFRATOETRIA DE PÓ
Quando um feixe monocromático de raios-X atinge uma amostra
policristalina, ele é espalhado coerentemente pelos elétrons dos
átomos da estrutura . O feixe difratado só é produzido por tal
espalhamento quando certas condições geométricas são satisfeitas,
istoé,ê necessário que muitos grãos orientados ao acaso, apresen
tem uma série particular de planos cristalográficos (hkl) espaça
dos de d.. ,, formando um ângulo apropriado 0 (ângulo de Bragg) com
o feixe incidente, Figura 2.1. Esta reflexão ocorre na direção do
detetor, fazendo um ângulo 29 com o feixe incidente, Figura 2.2.

FIGURA 2.I. DEDUÇÃO DA EQUAÇÃO DE BRAGG.
Cada feixe difratado formara um ângulo ^íkl, com a família deplanos (hkl), o qual será a solução da equação de Bragg, equação2 12.1:
nx = 2d, . .sen Q. , ,TIKI hkl, 2.1
onde ^ é o comprimento de onda da radiação utilizada, d... é a dis
tância interplanar e n ó um número inteiro (geralmente igual a 1).
As relações que determinam as possíveis direções de espalha
mento, podem ser obtidas combinando a equação 2 1 com a equação
interplanar, adequada â estrutura do cristal que está sendo anali
sado. Para as estruturas de alta simetria elas são:
cúbica sí»n2 o - *2 (h2+ k2+ l2) , . . .cuDica sen enJçl ^- l« ' > onde a=b=c,ÕL
tetragonal s e n2 Q ^ = j£ (h2 * k2 + 1^ ) , onde a=b^c,
4 a2 c2
ortorrombica sen2 ehkl - - } ! té * * l2) . ondea b e

fm»
A- Etao «t t
R- M o tf» g68SSmttro*IB5iMii0 - Angule dt BraggI - F M B SoütrlMÜto «o Tabo)O- Fcndo «olterlEnlro*» d» Otttiw)
FIGURA 2.2 GEOMETRIA DO GONIÔMETRO SG-S.

Os valores (a,b,c) são os parâmetros de rede do cristal, (hkl) são
os indices de Miller e representam famílias de planos cristalogrã
ficos paralelos e eqOidistantes.
A intensidade é alterada por qualquer mudança nas posições
ãon átomos da rede. O estabelecimento de uma relação associando as
posições atômicas com intensidade ê um problema complexo, pois en
volve muistas variáveis.
Para a difratometria de pó, existem seis variáveis afetando
a intensidade relativa do feixe dlfratado ' :
- {ato* de Lo*entz,
- {ato* de pota*izaçã.o,
- {ato* de e*t*atu*a { hkt),
- {ato* de multiplicidade [mhkl),
- {ato* de tempe*atu*a.
A equação da intensidade integrada relativa, das reflexões,
fornece a área relativa sob a curva da intensidade pela posição
(2©) do pico. Para uma amostra policristalina ela é representada
por:
-2MEP I 2 , p e - 3rhkl I "hkl LPhkl ~ ^ ~ ' imi
onde v ê o coeficiente de absorção linear do material estudado, K
ê uma constante relacionada com parâmetros eletrônicos, M é o fa
tor de temperatura e I»PhJcl são os fatores de Lorentz e polariza
ção juntos (os dois estão relacionados ã posição 0 do pico).
O fator de estrutura P.. . descreve a eficiência de espalha
mento dos átomos do material, numa dada direção (hkl). Ele express
sa a amplitude e fase da onda resultante e é representado por:
nP u i , i = z í » e x P (2ní ( h x _ + k y + l z ) ) , 7 .
n x i . ^ n n n n & • *
onde n ê o número total de átomos, f é o fator de espalhamen
to atômico do n-ésimo átomo e (x , y , z ) são as coordenadas car
tesianas com valores semi-inteiros (O Í x . y , z # 1).n n n
Na estrutura do U0~ (cfc), a célula cúbica contém quatro ato
mos de urânio e oito átomos de oxigênio, nas seguintes posições:

10
0 , 0 , 0 1/4,1/4,1/4 3/4,3/4,3/4
urânio 1/2.1/2^0 ( O x i g ê n Í £ > 3/4.3/4,1/4 % 1/4,1/4,3/4
1/2, 0 ,1/2 3/4,1/4,3/4 1/4,3/4,1/4
0 ,1/2,1/2 1/4,3/4,3/4 3/4,1/4,1/4
Calculando o seu fator de estrutura tem-se os seguintes resultados:
se h-rk+1 - 4n F2. = 16 (f + 2Í) 2.para (hkl) pares: h K I ü ° 2
se h+k+1 - 2(2n+l) F 2h k l =16 (fy - 2 f Q ) ,
para (hkl) impares: p2hkl = 1 6 fu'
2para (hkl) mistos: F hkl = °*
2.2. AKALISE QUANTITATIVA
Análise guantitiva por difração de raios-X, baseia-se no fa
to que a intensidade difratada de uma fase particular, em uma mis;
tura,depende da sua concentração. A relação entre intensidade e
corcentração não é em geral linear, uma vez que a intensidade di
fratada depende particularmente do coeficiente de absorção da mis;
tura, que por sua vez varia com a concentração
A relação entre intensidade difratada e concentração; para uma
amostra policristalina, é obtida a partir da equação 2.3.
Para analisar uma mistura de duas fases, chamadas de a e B ,
multiplica-se a equação 2.3 pela concentração de cada uma delas.As
equações da intensidade para a e 6 são dadas por :
|2m0Lpa " T «
2.5
T C«m
r
onde Ia e Ig representam as intensidades difratadas de cada fase ,
ca e Cg são as frações de volume de cada fase na mistura e v ê
o coeficiente de absorção linear da mistura.
Como na faixa de composição UO. - U0_ 2 & existem somente duas
fases, com mesma simetria cristalina e parâmetros de rede aproxima
damente iguais, os fatores das equações 2.5 podem ser considera
dos como sendo iguais, exceto as frações de volume das duas fa

11
ses (ca+c =1).
Portanto, a variação relativa da fração de volume (ou concen
tração) da fase em formação na mistura, em função do tempo, dará a
taxa de formação de nova fase para uma dada temperatura e pressão
de oxigênio constantes. A equação 2.6 expressa a variação relati
va da concentração da fase que está sendo formaia.
Ig4°9 - Co 0 . 2.6
1uo2+ au 4o 9
Quando uma análise quantitativa é baseada na variação relati
va da intensidade dos picos, para duas fases, como neste trabalho
(equação 2.6 ),oerro devido â aproximação é minimizado.9
O exemplo a seguir , ilustra o uso de dados de raios-X pa
ra detectar o inicio de formações de novas fases.
A determinação precisa das dimensões da célula geralmente in
dica se uma fase não estequiometrica está sendo formada, sua faixa
de existência e o modelo de incorporação do desequilíbrio este
quiomêtrico. Por outro lado, confiar na detecção de uma nova fase
somente pelo aparecimento de novas linhas, não fornece um valor exa
to da faixa de existência da nova fase, devido ao fato de que os
novos picos aparecerão somente após uma suficiente concentração ter
sido formada.
Em trabalhos mais refinados, é examinado o perfil de difra
ção(de linhas individuais), mudanças de intensidade, largura e a
forma das linhas para anunciar a formação incipiente de uma nova
fase. 0 mesmo método tem sido usado para detectar gradientes de
concentração dentro de uma fase estequiometrica, durante reações qua
micas. Por exemplo, a oxidação de partículas monocristalinas de
U0-, em temperaturas inferiores a 180°C, estabelecem um gradiente
de concentração de oxigênio intersticial, da superfície para o cen
tro de cada partícula, melhor que a formação de uma camada oxí.
dada de uma nova fase sobre o núcleo não reagido.
Os perfis de linha, calculados para o pico K-a (usando o mo
de Io de gradiente de difusão), são mostrados na Figura 2.3.A, a se
guir. Quando x aumenta, a primeira linha desenvolve outro pico em
um ângulo maior que se separa e afina, â medida que a composição se
aproxima do limite de composição (U0_ . . ) . Este comportamento, que
é o observado, é diferente do predito pelo modelo de duas fases,
Figura 2.3.B, que prediz o aparecimento de uma nova linha em alto

12
ângulo, aumentando em intensidade ã medida que a linha original de
saparece.
L1l
«Vi»
FIGURA 2.3. PERFIL DE DIFRAÇÃO .TEÓRICO , PARA ÍXIDOS NÃO ESTEouiojfTRICÔS (A) > OU PARTÍCULAS COM 2 FASES (B)?

13
CAPITULO III
3. >ISTEMA URAKIO-OXIGENIO
Devido ao fato dos átomos de urânio poderem existir em dife
rentes estados de Valencia, o comportamento dos seus õxidos é mais
complexo que o comportaaento dos õxidos dos outros metais. Esses
õxidos apresentam uma larga faixa de cooposição não estequionêtri^
ca, onde a razão oxigênio metal (O/U) difere substancialmente de21
dois, mesmo onde o sistema consiste de uma única fase
No urânio os estados de Valencia , + e , tendem a
ser os mais estáveis. No oxido estequiométrico o Ion metálico pesa
do possui carga +4. Para manter a neutralidade elétrica no cris
tal, quando Ions oxigênio são removidos ou adicionados, é necessá
rio que alguns cãtions mudem de Valencia. Portanto, os Ions urânio
no U09 formarão uma mistura de Ions U e U ou possivelmente 0
e U . Esse desvio da estequiometria é acompanhado pela formação
de defeitos Frenkel na sub-rede dos Ions oxigênio, cujo excesso
é acomodado nos interstícios da estrutura flúorita .
Devido a essa facilidade em formar compostos não estequiomé
tricôs, vários õxidos de urânio já foram caracterizados. A existên
cia de pelo menos quatro fases (U02, U.Og, V^Og e U03) termodina
micamente estáveis foram estabelecidas, e vários õxidos adicionais
(U.jO_, U2°5* f o r a m apresentados CORO fases metaestáveis21*.
Essa capacidade do diôxido de urânio de desviar-se facilmen
te da composição estequiométrica (IX>2 O Q ) , justifica as mudanças
das suas propriedades físicas .

14
3.1. DIAGRAMA DE FASES
O diagrama de fases do sistema urânio oxigênio ê um dos mais
complicados e interessantes sistemas binãrios oxigênio-metal. Ape
sar de ter sido examinado por ruitos pesquisadores, utilizando ãl
ferentes técnicas, ainda existem regiões que não são bem definidas
ou são inconsistentes; Figura 3.1a .
A solubilidade de oxigênio no urânio metálico é baixa au
mentando de 0,05% no ponto de fusão (113?°C), para 0,4% a 2000°C .
0 oxido em equilíbrio com urânio é o U00, que é a fase mais impor
tante no sistema .
A fase U02 não adquire composição hipoestequiomêtrica (°D-_ ),
em temperaturas moderadas (< 1000°C). Da temperatura ambiente até
aproximadamente 300°C, o oxigênio não entra na estrutura do
UO2» formando solução sólida estável. Entretanto, em temperaturas
maiores ele penetra intersticialmente na rede do UO2 formando um
composto UC>2+ , no qual o valor de x depende da temperatura, da
área superficial da amostra e da pressão parcial de oxigênio. 0 va
lor limite de x,no qual o Ü&2+X e s t* e m equilíbrio com a fase U.Og,
aumenta com a temperatura de 0,17 (a 950°C), para 0,244 (a 1123°C),
que é a maior temperatura em que o U^On pode existir . Acima des
sa temper itura o valor limite de x, dado pelo equilíbrio entre £D_
e Ü3°o_z» aumenta gradualmente.
A fase tetragonal U.Og, tem uma faixa de homogeneidade, cuja
extensão não ê bem definida. Em adição às fp.ses estáveis, existe
uma fase metaestável, de composição aoroximada U^Oy, que pode ser
obtida por oxidação do UO, em temperaturas inferiores a 200°C.
Um oxido, com composição aproximada Ü2°5' f o i o b t i d o e aPre_
sentado como sendo um composto de estrutura hexagonal estável (a =
3,996 R e c=4,117 8), o qual acima de 250°C se decompõe em U4Og e
Ü3°8 * • . o o
O diagrama de fases, Pi. ira 3.1b, na faixa de 500 C até 1500°<;
pode se considerado como bem estabelecido. Entretanto, apesar dos
vários trabalhos realizados ' ' ' ' ' t incerteza ainda exis
te abaixo de 500°C, principalmente quanto ã faixa de homogeneida
de do U02+x e U4O9_y!
3.1.1. A FASE U4Q9
A fase desordenada U02+x adquire uma composição U02 25, for

(a)
15
"2P 2,1
(b)
1000
soo
2J0 220 2.30HatSe 0 / U -
F1GURA3.1. DIAGRAMA DE FASES DO SISTEMA URÂNIO OXIGÊNIO (a) APRESENTADO POR NAITO E KAMAGASHIRA1"(1976) E W PORCORDFUNKE6 (1969).

16
mando uma nova fase U^Og. A transformação envolve ordenação de lon
go alcance dos átomos de oxigênio intersticiais e é caracterizada
por uma perda de entropia. Medidas de intensidade por difração de
neutrons em U.Og, indicaram que os átomos de oxigênio ocupam exa
tamente os mesmos tipos de posições como na célula estatística dou o
2 + x- Ambos os resultados, de difração de neutrons e raios-X, mos
trar. que o U^Og tem grupo espacial I43d com parâmetro de rede de
21,77 8, ou aproximadamente quatro vezes o do U0-. Um modelo re
presentando as posições do oxigênio foi proposto por Belbeoch en
tretanto, ele não concordou com as observações realizadas por
Willis (cap. 3.3.).
Trabalhos sobre U.0g mostraram que essa fase possui três di
ferentes formas cristalogrãficas: a - U.0g que é estável até apro
ximadainente 65°C, B-U^Og que se transforma, em aproximadamente 600°Q
numa fase desordenada Y-UAOQ . Apesar de grande número de traba10, 13, m , 16, 19 9 -
lhos realizados , a faixa de homogeneidade da fase
o - ü*Og_ não é bem estabelecida. A temperatura ambiente, a faixa
de composição ê muito estreita e o valor de % provavelmente não
excede a 0,1.
3.1.2. A FASE U 30 7
A oxidação do UO2 em temperaturas inferiores a 200°C, pode
resultar na formação de fases metaestãveis,tetragonais, na faixa
de composição UO2 3 até U02 4- Um composto U3O7, foi identifica
do empiricamente pela primeira vez em 1947 por Jolibois, sendo po£
teriormente confirmada sua existência por vários pesquisadores '.
Embora várias fases tetragonais tenham sido determinadas e
classificadas em grupos, a relação entre essas fases e grupos ain
da não é reconhecida, havendo discordâncias quanto a sua caracter^
zação precisa, particularmente com relação ãs razões O/U e a/c1 8
(a e c são os parâmetros de rede da fase tetragonal)
A fase o-U307 é geralmente obtida oxidando o U€>2 em tempera
turas inferiores a 135°C. A razão c/a é aproximadamente menor que
1. A fase a é transformada em 6-U3O7 acima de 180°C, uma conversão
que é completa quando se tem: O/U =2,33 e c/a = 1,033 com c*5,556
ft.6
Em maiores temperaturas (> 350°C), uma terceira fase Y - U 3 O 7
foi determinada com c/a=l,017 a 350°C e c/a=l,010 a 650°C.

17
3.2. OXIDAÇÃO
As propriedades químicas do diôxido de urânio têm sido exten
si vãmente estudadas, particularmente com respeito à oxidação . O
oxido estável, em temperaturas inferiores a 500°c, em contato com
oxigênio puro ê o ÜO3 e acima dessa temperatura tem-se U^Og. so
mente pequenas partículas de U02 (-100 8) são oxidadas a UO^.
De forma geral, a oxidação de um sólido, por oxigênio gasoso,
pode ser representada pela equação geral A (s) + B(g) C(s). A
transformação ocorre em dois passos; formação de C sobre a super
flcie de A e o crescimento de C para dentro de A. Dois casos são
considerados: (1) a reação na superfície é muito rápida, compara
da com a taxa de crescimento e (2) a nucleação superficial e cres_
cimento ocorrem em taxas comparáveis. A geometria e estado de
agregação do sólido são fatores importantes controlando a taxa de
reação. 0 sólido A é considerado como sendo um composto policris
talino de partículas esféricas uniformes e ê exposto a um gás B
mantido a pressão constante. De acordo com os objetivos desse tra
balho,só será apresentado o primeiro caso:
0 crescimento controlando a taxa foi discutido por Alberman
e Anderson. A superfície de todas as partículas é coberta com
pletamente com camadas continuas de produto, antes que uma quanti
dade sijnificante de transformação tenha ocorrido. A taxa de rea
ção é controlada pela taxa de crescimento da fase C dentro de A.
Se as fases A e C existem somente como compostos estequiométricôs, a
taxa de crescimento de C pode ser controlada ou pela taxa de rea
ção no contorno C-A, ou pela taxa de difusão de B, possivelmente na
forma iônica, através da camada superficai Ç.
Se a difusão, através da camada produto, controla a veloci
dade de reação, a equação de taxa pode ser desenvolvida utilizando
a aproximação do estado quase estacionãrio. Se os pesos molécula
res e densidade das fases reagentes e produtos são aproximadamente
iguais, a equação pode ser representada por:
2 P H Mo t = 1 - (l-f)2/3 - (2/3) f, 3.1
onde a é o raio das partículas, D é o coeficiente de difusão, f
é a fração de material transformado, t é o tempo de reação , M e
d^ sEoa forma peso e a densidade do reagente e H ê a diferença na

id
concentração de B nas interfaces B-A e A-C.
£ possível que a fase A ou C exista sobre uma larga faixa de
composição. Se a face A pode absorver B dentro de sua rede atê
uma composição limite (correspondente â fase C) ser alcançada, e
se a superfície das partículas de A tornar-se rapidamente satura
da com B, a reação química ê matematicamente análoga â absorção de
calor par um corpo esférico que está a uma temperatura inicial un.i
forme e é exposto a um ambiente numa temperatura constante maior.
Para uma reação química, um gradiente de concentração ocorrerá ao
longo do raio da partícula, e a equação de taxa integrada será:
C = 1 £ — I -1^ exp (-it2m2Dt/a2). 3.21TT 1
As equações 3.1 e 3.2, apresentam curvas de taxas similares.
Ê difícil discriminar entre os dois mecanismos, baseando-se so
mente em dados de taxa; informação adicional , talvez de dados de
raios-X ou análise microscópica, é necessário.
Os dados de cinêtica obtidos nesse trabalho , são bem ajus
tados pelas equações 3.1 e 3.2, indicando que a taxa é controla
da por um processo de difusão. Os dados de difração de raios-X fa
voreceram um mecanismo no qual a oxidaçao é controlada pela difu
são do oxigênio através da fase formada superficialmente.
Trabalhos sobre a oxidaçao do U02 2''*r6i11'2* f mostram que
uma discussão sobre o assunto pode ser convenientemente dividida
em duas ou três faixas de temperatura, nas quais diferentes me ca
nismos de oxidaçao ocorrem (1) a temperatura do nitrogênio líqui
do onde absorção química de oxigênio é observada; (2) até aproxjL
madamente +50°C ocorre oxidaçao superficial e o fator controlando
a taxa de oxidaçao é a difusão dos Ions oxigênio através da camada
superficial. A oxidaçao superficial é responsável pelo caráter não
estequiométrico do UO, , em contato com ar seco e a temperatura
ambiente. Nessas condições a razão O/U aumenta gradualmente com o
tempo de exposição e alcança um valor limite O/U = 2,33 , não sen
do possível, entretanto, se obter uma oxidaçao homogênea; (3) aci
ma de 60°C, tem inicio a oxidaçao total ("bulk oxidation"), que
depende da área superficial, e ocorre em um ou dois estágios. Oxi
dos com uma area superficial maior que lm/g oxidam em dois pas
sos. No primeiro passo de oxidaçao a amostra atinge a composição
U3O7 e é controlado pela difusão de oxigênio. 0 segundo passo, que
ocorre acima de 200°C, atinge a composição U,Og e é controlado

19
por vim processo de nucleação e crescimento.
3,3. ESTRUTURA CRISTALINA
Os ôxidos de urânio com composição até UO, oc, possuem es
tutura cúbica, passando para tetragonal (U-07) e finalmente ortor
rômbica (U~Og) .
O comportamento dos parâmetros de rede depende de três fato
res: conteúdo de oxigênio, grau de aproximação da composição de
equilíbrio e da temperatura . Durante a oxidação, o parâmetro de
rede (a) do UO, diminui, provocando uma contração na rede à medi18 ~
da que o valor de O/U aumenta . Trabalhos anteriores a 1960, so
bre a variação do parâmetro de rede em função da composição O/U ,
mostram uma variação linear, do U02 até a composição U^Og. Entre
tanto, era assumido que a estrutura do U^O. era a mesma do UO.
com um oxigênio adicional na posição (1/2, 1/2, 1/2). Posterior
mente, Belbeoch e colaboradores mostraram que a estrutura do U*Oa25 1 2 1 8 2 5 ^ -*
é mais complexa ". Trabalhos mais recentes ' ' , mostram que es_
sa variação ê melhor representada por duas linhas retas. Figura
3.2, uma característica da região UO2+X e a outra caracterizando o
°4°9_v' existindo uma região intermediária ainda indefinida.
Quando não há oxidação durante um tratamento térmico do U02,
o aumento da temperatura provoca uma expansão da rede. Muitos tra- '•,13,12,18 ~
balhos apresentam resultados de expansão térmica do UO2 -Al.
guns valores são apresentados na Tabela 3.1.TABELA 3.1. COEFICIENTE DE EXPANSÃO TÉRMICA
LINEAR PARA 0 U02 SINTERIZADO "'12
TEMPERATURA COEFICIENTE DE EXPANSÃO
(°c) (°c r 1
20 - 2000 9,4 x 10~6
20 - 950 10,8 x IO"6
20 - 800 9,9 x IO"6
20 - 1000 10,5 x IO"6
Mesmo possuindo estrutura muito próxima da estrutura do UO
° U4°9 aPresenta um parâmetro de rede menor devido â contra

20
ção provocada pelo excesso de ânions na estrutura. Como o parâme
tro de rede é inversamente proporcional ao ângulo de Bragg (26),os
picos de difração caracterizando a fase U*Og surgirão acima das
posições que caracterizam a fase UO2.
Próximo da composição UO2 25 dois tipos de difratogramas são
observados: com ou sem raias de superestrutura, sugerindo a exis
tência de duas estruturas (U,0<,+ ). A estrutura do U0, 2 5 e
desordenada ou ordenada de forma diferente da ordenação do U^Og
As raias de superestrutura caracterizam um arranjo ordenado dos
defeitos da estrutura. Como já mencionado, o equilíbrio de fases do
UjOg tem sido examinado por muitos pesquisadores, mas a concordân
cia entre seus dados é ainda muito pobre
F I G U R A 3 . 2
B4M0-2P0
COMPORTAMENTO DO PARÂMETRO DE REDE EM FUNÇÃO DAo/u1B; O) SUZUKI E COLABORADORES (2) YOUNG E
25 ( 3 ) P 1 8 ( « ) K N
RAZÃOCOLABO
RADORES2 (3) E («) KlATSUI E NAITO
A medida que a oxidação do UO2 se processa, as linhas de
difraçao alargam-se com diminuição da sua intensidade ou dividem-
-se (exceto os picos do tipo (hhh)), sofrendo um pequeno desloca
mento para ângulos maiores . Essa mudança de posição sugere a
formação de solução solida e o alargamento das linhas é provocado pe
Ia tensão produzida pelo excesso de oxigênio. Essas mudanças nos
perfis de difraçao, permitem concluir que a estrutura do ÜO2 é ir.an
tida na amostra até a composição de UO 2 2 0-UO 2 > 2 5. Acima dessa fai
xa de composição, desenvolve-se gradualmente uma fase tetragonal
com razão c/a crescente

21
Como na fase tetragonal os parâmetros de rede sao diferentes
(a=b?«c)7'15, todos os picos, exceto os dos tipo (hhh), se apresen
tarãu na forma de dubletos ou tripletos.
3.3.1. ESTRUTURA DO DlOXIDO DE URÂNIO
A estrutura do U09 foi primeiramente determinada por
Goldschimidt e Thomassen . Esse oxido estequiomêtrico possui uma
estrutura cúbica do tipo fluorita, característica dos õxidos do
tipo MX2> com um cátion tetravalente. Cada átomo do metal H está
coordenado por oito átomos X, cada um dos quais é por sua vez
coordenado por um tetraedro de átomos M.
Em geral, o valor aceito para seu parâmetro de rede, ã tem
pjratura ambiente, é de 5,470 angstron (8) , Tabela 3.2, correspon
dendo a uma densidade teórica de 10,952 g/cm (assumindo quatro uni
dades de UO, por cela unitária).
TABELA 3.2. PARÂMETRO DE REDE DO UO2,EM ANGSTRON,OBTIDO POR viRIOS AUTORES ".
5,4690 - 0,0001
5,4704 - 0,0008
5.4703 - 0,0002
5,4698 - 0,0008
5,4720 - 0,0005
5,4698 - 0,0002
As posições atômicas são aquelas correspondentes ao arranjo
da estrutura fluorita, Figura 3.3
As posições (0,0,0), (1/2,1/2,0), (1/2,0,1/2) e (0,1/2,1/2),
nessa estrutura, são ocupadas por átomos de urânio, sendo que os
átomos de oxigênio ocupam as posições - (1/4,1/4,1/4), mais as po£
slveis translações, Os ions oxigênio estão distribuídos sobre uma
rede cúbica simples, enquanto que os íons U formam uma sub-rede
cúbica de face centrada (cfc).
Um aspecto típico da estrutura fluorita são os grandes va
zios intersticiais, nas posições (1/2, 1/2, 1/2), na sub-rede dos
ions metálicos. Essa característica levou os primeiros pesquisado
res a acreditarem que o excesso de oxigênio no composto não este

22
quionêtrico (hiper-estequioaetrico), ocupava estas posições.
|*-9L4TO4 I—I
FIGURA 3.3. ESTRUTURA DO UO2.
Um cristal estequiométrico ideal sô pode estar em equilíbrio
termodinâmico real na temperatura absoluta, ou seja, com entropia
zero. Em temperaturas acima de 0 K todos os cristais desviam-se do
estado perfeito, devido ao aparecimento de defeitos na rede. Os ti
pos principais de defeitos existentes na estrutura do tipo fluori
ta são os chamados defeitos de equilíbrio atômico: (a) vacâncias
nos sítios da rede, (b) átomos ou Ions intersticiais ou (c) áto
nos de impurezas intersticiais ou substitucionais.Estes defeitos
são os que estão mais diretamente relacionados com as propriedadesq
químicas dos cristais
Os defeitos predominantes nos óxidos do tipo MO-, são os do
tipo (a) e (b), citados acima. Essas imperfeições são classifica
das em duas categorias:
(1) aquelas que são inerentes na termodinâmica do estado só
lido e que ocorrem em todos os cristais;
(2) aquelas que são específicas ao composto cristalino C M
siderado.
Na primeira categoria existem dois tipos de defeitos termodi^
nâmicos inerentes: defeitos Schottky e defeitos Frenkel .
Devido a sua natureza, defeitos Frenkel são geralmente impo£
tantes em cristais com estrutura cristalina apresentando vazios que
possam acomodar íons intersticiais, sem muita distorção. Esse é o
caso de substâncias de baixo número de coordenação, ou quando exi£
te disparidade de tamanho entre os ions M e x". Em estruturas com

23
alto número de coordenação, onde não existe espaço para ions in
tersticiais, a energia de formação para defeitos Frenkel atinge ai
tos valores. Nesse caso a foraação de defeitos Schottky é mais
provável .
O diõxido de urânio que apresenta estas características dis
solve facilmente grandes quantidades de oxigênio intersticialmente,
adquirindo composição ÜO2+X e formando vacâncias nas posições nor
mais dos átomos de oxigênio, sem que a sub-rede de átomos de ura
nio seja alterada2S. X medida que o composto se afasta da estequio
metria, maior é o número de defeitos na estrutura.
3.3.2. ESTRUTURA DOS OXIDOS MO-ESTEQUIOMETRICOS
Os óxidos do sistema urânio-oxigênio, na faixa de composição
UO, 75~üO- -Q, apresentam estrutura cúbica de face centrada com
arranjo atômico do tipo CaF_ (fluorita) .
Os átomos de urânio e oxigênio, na estrutura do U0~, vibram
isotopricamente mesmo em baixas temperaturas. Em altas temperatu
ras os átomos de oxigênio podem sofrer um relaxamento ao longo das
quatro direções <111> . No U02 desordenado a rede fluorita ace_i
ta Ions oxigênio nas posições intersticiais, sem mudar a sime
tria dos cãtions, enquanto que na fase ordenada U.Og algumas ato
mos de urânio sofrem pequenos deslocamentos nas posições fcc. Essa
alteração na estrutura é caracterizada pelo aparecimento de raias
de superestrutura nos difratogramas de raios-X e neutrons,com a
formação de uma célula cúbica de corpo centrado quatro vezes maior
que a célula fluorita.
Para o UO- hiper-estequiométrico, dois modelos de defeitos
são possíveis: excesso de oxigênio nos interstícios e vacai rias na
sub-rede de urânio. Numerosos dados, entretanto, mostram que os
efeitos principais no U0»+x são produzidos pela excesso de oxigênio
intersticial 12'JIYoung e colaboradores25,concluíram, baseados em
resultados experimentais, que o desvio estequiométrico seria me
lhor descrito pelo excesso de oxigênio do que pela ausência de áto
mos de urânio, provocando uma pequena contração na rede cristalina.
Segundo o relatório da Agência Internacional de Energia Ato
mica (IAEA), publicado em 1965 12, quando o U02 é oxidado não
aparecem linhas extras nos difratogramas de raios-X. Estudos em
monocristais, utilizando difração de neutrons, confirmaram o não
aparecimento de reflexões adicionais, sendo portanto o grupo espa
ciai do U02+x o mesmo do U02 (Fm3m). Isso evidencia a possibilidade

24
de ocorrência de ordenação de curto alcance entre pequenos grupos
de átomos, excluindo a possibilidade de formação de ordenação de
longo alcance que resultaria em um grupo espacial diferente 12'17.
Em altas temperaturas o diõxido de urânio»com excesso de oxi
gênio, apresenta duas fases com faixa de composição sobrepostas:
uma fase U.Og deficiente de oxigênio (U02 25-v* e u m a f a s e n a o e£
tequiomêtrica desordenada üO2+x" E m a m b a s existem complexos de defei
tos ou aglomerados, consistindo basicamente de dois ãnions inters
ticiais, uma vacância e ions D que mantém a neutralidade ele
trica do composto. Um complexo deste tipo provém da introdução de
um ânion intersticial na estrutura do U0-, provocando o desiocamen
to de um ânion vizinho, da sua posição regular na rede.
Na estrutura do UO2+X, os complexos são distribuídos e orien
tados ao acaso quando x é pequeno, mas quando seu valor aumenta hã
uma tendência para a ordenação com a formação da estrutura do ti
po ü^Og , dentro da fase ÜO2+ .
Para explicá-los foram criados dois modelos de defeitos :8
Kofstad apresentou um modelo que consiste em dois anions desloca
dos, duas vacâncias correspondentes e um ou dois ânions intersti
ciais. O modelo 2:1:2, tem somente um dos sítios 0* ocupado, des
locando dois ânions da rede para as posições 0",Figura 3.4. Quando
ambos os sítios O* são ocupados, dois dos complexos, representados
por I (0.~ ) Q I , podem descrever a estrutura que corresponde ao
modelo designado 2:2:2 (dois ânions deslocados, dois intersticiais. - . . 12, 17, 26
e duas vacâncias)26
Analise estrutural do U0 2 + x realizada por Willis , utilizan
do difração de neutrons, o levou a propor uma estrutura para oU O2 12 * F i9 u r a 3-4'' a qual postula a existência de três tipos de
átomos de oxigênio na estrutura: 0,0* e 0". 0 sitio £ corresponde
â posição normal dos oxigênios na rede, 0_ e 0^ são ocupados por
oxigênio intersticiais e 0_ está localizado no interstício cen
trai (1/2, 1/2, 1/2), deslocando em 0,86 8 na direção <110>, e os
átomos 0" ocupam as mesmas posições mas deslocados em 1,05 8 na
direção <111> . Por exemplo ; o U02 1 2, b a s e a do n o modelo 2:2:2,
apresenta a composição seguinte: UO^ g 7 O'Q 1 30" Q 1 2
9 23 26 ' ' '
Kosfstad , Saito e Willis , baseados em dados obtidos utl.lizando as técnicas de condutividade elétrica e difração de nêutrons, consideram que o modelo 2:2:2 explica melhor a estrutura do

25
Átomo de oxigânio
Átomo interstíciol O*
Átomo interstício! O
• Átomo de Urânio
• \toz\o no sub-rede de oxigfinio
FIGURA 3.4. ESTRUTURA CRISTALINA DO UO Z + X PROPOSTA POR WILLIS '
OS.OXIGÊNIOS NORMAIS EM A E B, NO U02> SÃO SUBSTITUÍDOS POR OXIGÊNIOS INTERSTICIAIS O1 EM C E D E POR 0"EM E (2:1:2) OU EM E E F (2:2;2).

26
CAPÍTULO IV
4. PARTE EXPERIMENTAL
Os experimentos foram realizados em duas etapas: altas tempe
raturas e temperatura ambiente. O primeiro caso foi divido em sé
ries (TA e TB). Na série TA foi determinado o coeficiente de expan
são térmica do U02 e em TB foi acompanhada a transformação de fa
ses, durante a oxidaçlo, determinando-se parâmetros estruturais e
de cinética.
4,1, PREPARAÇÃO DE AMOSTRAS
As amostra foram fornecidas pelo Centro de Metalurgia Nu
clear (CMN) do Instituto de Pesquisas Energéticas e Nucleares
(IPEN). Elas foram produzidas em duas etapas, serie-A e série-B, e
suas condições de preparação são especificadas na Tabela 4.1.
Os métodos utilizados na determinação das densidades, nessa
Tabela, foram:
Série-A - picnometria de mercúrio,
Série-B - picnometria de tolueno.
0 resultado da análise química de impurezas presentes na
amostra é apresentado na Tabela 4.2.
As amostras da série-A foram compactadas e sinterizadas em
forma de placas finas (-1,2 mm). Após terem sido cortadas nas di
mensoes do porta amostras, Figura 4.1., suas superfícies d^ estu
do foram polidas com lixas 220, 400 e 600, respectivamente, fabr;L
cas pela Scotch 3M do Brasil. Após o polimento elas eram lavadas
com acetona e guardadas dentro de um dessecador.

27
TABELA 4.1. PREPARAÇÃO DAS AMOSTRAS
Material te Partida: Diuranato de Amõnia (DOA)
Calcinação: Temperatura: 700°CAtmosfera: arTempo: 3:00 hsEn bote de inconel
Reduçio: Temperatura: 700°CAtmosfera: HidrogênioTempo: 0:30 hs
Compactação: Prensa manualEsforço: 40 ton.
Sinterização: Temperatura: 1700°CAtmosfera: Hidrogênio
1:30 hs
Densidade: Série-A -10,56 g/cm3-Série-B - 9,593 g/cmJ
TABELA 4.2. ANALISE QUÍMICA DAS IMPUREZAS PRESENTES NAS AMOSTRASDE U02 PELA TÉCNICA DE ESPECTROSCOPIA DE EMISSÃO
ELEMENTO CONCENTRAÇÃO (PP«)
Si 82
Al 200
Cr 12
Mi 6
Mn 3,6
Cu 0,9
B < 0,1
Pb < 1
Sn < 1
Bi" < 1
V < 3
Mg < 2,4
Cd < 0,1
P <55
Mo < 2
Zn <10
' Fe -20
Os sinais "-" e "<" indicam "aproximadamente" e "menor" respectiva
mente.

28
As amostras da sêríe-B foram compactadas e sinterizadas em
forma de pastilhas, com 4 cm de diâmetro por 1 cm de altura, sendo
posteriormente cortadas no tamanho do porta amostras. Apôs o
mento elas foram
rante 5h 30 min.
mento elas foram reduzidas em atmosfera de hidrogênio a 500°C du
4,2. INSTRUMENTAL
Para obtenção dos raios-X foi utilizado um conjunto gera
dor-estabilizador de tensão, marca Rigaku Denki Co. Ltda., equipa
do com um tubo com alvo de cobre, de 2 kw. A potência média de
trabalho foi de 1,35 kW (45 kV/30mA).
Foram utilizados dois métodos de difração: método fotogrãfi
co e difratometria. Os acessórios utilizados são os seguintes:
- goniõmetro SG-8, Figura 4.2.,
- câmara de alta temperatura modelo A-4, Figura 4.2. e 4.3.,
- controlador de temperatura modelo MTC-3, Figura 4.4.,
- sistema "step-sca*»", Figura 4.4.,
todos fabricados pela Rigaku.
O passo fundamental para a obtenção de bons resultados é o
alinhamento da amostra e câmara de alta temperatura, que deve ser
ajustada no centro (A) do goniõmetro (Figura 2.2.).
A câmara de alta temperatura possui um conjunto de blinda
gens térmicas, um sistema para controle de atmosfera,um sistema de
refrigeração a água e um 'ermopar de platina-rõdio (Pt-PtRh 13%).0
termopar fica posicionado dentro do porta amostras (ao lado da
amostra), de modo que reproduza mais fielmente o valor da sua tem
peratura. 0 forno resistivo e o porta amostra são de platina e
foram projetados para que o calor se distribua homogeneamente so
bre a amostra.
0 sistema de alinhamento da câmara, possui parafusos para o
ajuste fino dos movimentos de translação longitudinal e inclina
ção e uma alavanca para o movimento de rotação da câmara.
0 ajuste horizontal visa colocar a superfície da amostra pa
ralela do feixe de raios-X, ou seja, tangente ao circulo de foca
gem do difratômetro (Figura 2.2.).
0 ajuste na inclinação deve colocar a superfície da amostra
paralela ao plano vertical central, do feixe direto.

29
1 •I
FIGURA 4.1. PORTA AMOSTRA DA CÂMARA DE ALTA TEMPERATURA.
FIGURA 4.2. ÓONIÔMETRO SG-8, CÂMARA DE ALTA TEMPERATURAINSTALADA E GERADOR DE RAIOS-X.

30
FIGURA 4.3. CÂMARA DE ALTA TEMPERATURA DESMONTADA-
FIGURA 4.4. CONTROLADOR DE TEMPERATURA (À DIREITA), SI£TEMA STEP-SCAN (EM BAIXO) E DIAGRAMAS CQ.MUM E STEP-SCAN (ACIMA),

31
O ajuste longitudinal fará com que o centro da amostra cor
responda ao plano vertical central do feixe, isto é, o centro do
feixe incidente atingirá o centro da amostra quando esta estiver
girando em torno do eixo de rotação (A),(Figura 2.2.).
Esse alinhamento ê um processo lento, envolvenão o uso de
fenâas especiais e muita prática. Deve-se tomar muito cuidado,pois
durante o alinhamento a câmara permanece aberta sem o sistema de
blindagem. Todo esse procedimento deverá ser repetido sempre que
a amostra for substituída.
0 termopar está ligado diretamente ao controlador de tempera
tura que é composto de duas unidades: de controle e de indicação
da temperatura. Figura 4.5.
0 sistema "step-scan" é um acessório muito importante, prin
cipalmente no estudo de transformação de fases e análises quantita
tiva das fases presentes. Ele é um sistema automático de varredura,
que faz contagens acumuladas ponto a ponto. 0 sistema possui os
seguintes recursos:
- controle de largura de passo,
- programa para repetir as medidas na mesma faixa de ângulos
(26) ou em outra, várias vezes,
- controle de tempo das contagens acumuladas,
- os resultados são impressos na forma de histoçramas e os
valores das contagens são registrados em fita de papel.
As medidas realizadas utilizando esse sistema foram obtidas
sob <s seguintes condições: tamanho de passo 0,02 , tempo de con
tagem 20 segundos.
Na Figura 4.4., pode-se observar um difratograma normal e
um "step-scan" do pico 6-311.
A temperatura ambiente foi utilizada uma câmara de Guinier-
Hâgg modelo XDC-700, fabricada pela Jungner Instrument AB, Figu
ra 4.6.-Essa câmara foi adaptada a um gerador de mesa, de 1 kW de
potência, fabricado pela Rigaku Denki. Ela é uma câmara de alta
precisão, cujas medidas dos parâmetros de rede podem ser compara
das com os dados da literatura ou associadas â composição da amos_
tra obtida por outra técnica (TGA), para uma estimativa da compo
sição inicial e final da amostra.
4.3, CONDIÇÕES EXPERIMENTAIS
A instalação e alinhamento da câmara de alta temperatura, no
goniõmetro SG-8, obedeceu as especificações do catálogo fornecido

A- AmpwiiMtro, 0 o I2AA.-T—Auto-TKm.
nmvi
P.U- Lonpoda piloto
V-VtNUMtro
t) !
A-T
Ô
UNdadt 4 t Indteoçôo d»
FIGURA 4.5. DIAGRAMA DO CONTROLADOR DE TEMPERATURA.

33
FIGURA 4.6. CÂMARA DE GUINIER-HAGG-
pelo fabricante.
Antes do inicio de cada série de medidas a câmara era evacua^
da (-10 torr.), utilizando-se uma bomba mecânica, após o qual era
preenchida com a atmosfera de trabalho. Quando o fluxo de gás de
sejado era obtido, iniciava-se o aumento da temperatura. O fluxo
foi controlado através da válvula de regulagem de pressão, do ci
lindro, e por um medidor de fluxo de alta precisão, fabricado pe
Ia Union Carbide.
Os gases foram fornecidos pela Oxigênio do Brasil S.A., com
as seguintes especificações, Tabela 4.3.
TABELA 4.3. ARGÔNIO ULTRA"PURO <Ar~U)
Pureza - 99,995%O2 < 5 vpmH-0 < 5 vpm
53< 5 vpm- 15,3 vpm

34
A pressão parcial de oxigênio, desses gases, foi determinada
pelo CMN/IPEN, utilizando um elétrolito sólido de ZrO2 + 10% Y 2 03>
Os valores obtidos foram:
Ar-Ü (sêrie-A) - 1 5 7 + 4 3 ppm
Ar-U (série-B) - 1 4 4 + 3 2 ppm
Este aumento na concentração de oxigênio, no Ar-U, ê prova
velmente devido a utilização de tubos plásticos e válvulas não ade
quadas para esse tipo de gás.
4.3.1. Série TA
Com as amostras-A, foram realizadas três séries de experi
ntentos: TA-1, TA-2 e TA-3. As Tabelas 4.4. e 4.5. apresentam as
condições experimentais de cada difratograma das séries TA-1 e
TA-2.
DIFRATOGRAMA
Tfl-l-l1R-1- 1
TÍ-1-?1n-1-C
TA-1-3
TA-1-4
TA-1-5
TA-1-6
TA-1-7Í
TA-1 -7Ü
TA-1-8
TA-1-9
ATMOSFERA
ai
ar
Ar-UAr-U
Ar-U
Ar-U
Ar-U
Ar-U
AR-U
ar
TABELA 4 . 4 .
TEMPERATURA
Cs L
165°C
350°C
460°C
620°C
800°C
810°C
950°C
25°C
SERIE T A - 1
TEMPO P/ EQUILÍBRIO
16:00 hs
10:30 hs
16:30 hs
16:00 hs
ZERD
25:00 hs
ZERO
30 dias
FLUXO GAS
-200 cc/min
210 cc/min
180 cc/min
180 cc/min
170 cc/min
180 cc/min
180 cc/min
Além dos difratogramas normais, foram estudados, utilizando
-se o sistema "step-scan", os seguintes picos:
Série TA-1 - Ficos 0 (511), (531) e (600)
Série TA--2 - Picos B (220), (531) e (600)
A série TA-3 foi realizada sob condições diferentes das duas
anteriores. Seu principal objetivo foi observar as transformações
que ocorrem durante a oxidacão, para o estabelecimento das condi
ções e parâmetros para a série TB.

35
TABELA 4.5. SÉRIE TA-2
DIFRATOGRAMA ATMOSFERA TEMPERATURA TEMPO P/ EQUILÍBRIO FLUXO GÍS
TA-2-1
TA-2-2
TA-2-3
TA-2-4
TA-2-5
TA-2-6
TA-2-7
TA-2-8
TA-2-9
TA-2-10
TA-2-11
TA-2-12
Ar-U
Ar-U
Ar-U
Ar-U
Ar-U
Ar-U
Ar-U
Ar-U
AR-U
ar
ar
ar
530°C835°C1070°C1080°C1070°C995°C908°C908°C20°C
23°C
17:45 hs
16:25 hs
14:15 hs
38:45 hs
62:00 hs
14:50 hs
14:00 hs
38:00 hs
150 cc/min
200 cc/min
150 cc/min
270 cc/min
220 cc/min
170 cc/min
150 cc/min
120 cc/min
120 cc/min
Na série TA-3, a amostra foi aquecida em argõnio-U até-o.
a
temperatura de 145"C, sendo posteriormente aumentada por duas ve
zes (para 280°C e 330°C), durante o experimento. Também foi inje
tada uma mistura de gases. Ar (80%) e O- (20%), por nove vezes no
interior da câmara, sem interrupação do fluxo de gás. 0 objetivo
dessas mudanças nos parâmetros (Po_ e T), foi acelerar a oxidação
e observar as modificações no difratograma.
4.3.2. SERIE TB
Com as amostras B, foram realizadas três séries de experimen
tos (TB-1, TB-2 e TB-3). Cada uma dessas séries foi realizada em
temperaturas constantes (oxidação isotérmica): 235 - 5°C, 170 -5°C
e 275 - 5°C, respectivamente.
O comportamento dos picos foi acompanhado através de difra
togramas normais, que eram tirados geralmente de 24 em 24 horas. Al
guns picos foram estudados utilizando-se o "step-scan". As carac
teristicas da série TB são apresentadas na Tabela 4.6., a seguir:
Os resultados do "step-scan" eram impressos em fita de pa
pel e enviados ao Centro de Processamento de Dados do IPEN, para
serem analisados pelo programa ANACRON, desenvolvido por Antonio
Gouveia e Carlos H. Mesquita. 0 programa foi adaptado a esse tipo
de análise. Ele ajusta os dados a gaussianas simples ou modifica
das â direita e/ou â esquerda.

36
O programa ANACftON fornece as seguintes informações sobre os
dados (para cada pico observado).
- número de picos observado,
- altura dos picos, corrigida com relação ao B6,
- área real e porcentual,
- posição (26),
- largura ã meia altura,
- grafico do ajuste, com os pontos experimentais e calculados,
- gráfico dos resíduos e
- gráfico probabilistico.
Todos os valores calculados eram fornecidos com o erro pa
drão assintótico.
TABELA 4.6. SÉRIE TB
TB-1 TB-2 TB-3
Picos estudados ^zando o Step-Scanning
Temperatura de trabalho em °C
Fluxo de gás
Início do experimento
Fim do experimento
Tempo total de tratamento térmico ~
* - 311a - 200
235 í5°C
153cc/min
04/09/80
16/09/80
17375 min.
e - 311B - ina - 200
170±5°C
-144cc/min
25/09/80
07/10/80
17191 min.
B - 311 e 111a - 200 e 220
275 - 5°C
-169cc/min
13/10/80
27/10/80
20043 min.
4.3.3. TEMPERATURA AMBIENTE
As amostras foram analisadas antes e após o tratamento tér
mico, utilizando uma câmara de alta precisão (diagrama de põ),
Guinier-HSgg.
A amostra era triturada, manualmente, em um almofariz de ága
ta. Além de ser possível observar quantas fases estão presentes pô
de-se estimar a composição (O/U) das amostras, comparando o valor
do parâmetro de rede medido oom tabelas,encontradas na literatu
ra ' , associando o parâmetro de rede com a composição esteguio
métrica.

37
CAPITULO V
5. DADOS E DISCUSSÃO
5 . 1 . SÉRIE TA
O comportamento dos picos de difração para as séries TA-1 eTA-2, em função da temperatura, é apresentado nas Tabelas 5 .1 . e5 . 2 . , respectivamente.
TABELA 5 . 1 . S É R I E T A - 1
DIFRATO TEMPERA TEMPO/ PICO fr-511GRAMA NQ TURA EQUIL. 26 ALTURA
TA-1-3
TA-1-4
TA-1-5
TA-1-6
TA-1-7
PICO p-531
26 ALTURA
PICO 0-600
26 ALTURA
165°C 16:00hs 82.6° 1808cps 97.44° 1680cps 99.32°
350°C 10:30hs 82.44° 2160cps
460°C 16:30hs 82.34° 2500cps
625°C 16:00hs 82.12° 2640cps
810°C 25:00hs 81.92° 2968cps
1050cps
1300cps
1300cpS
1460cps
96.56° 2550cps 98.38 1650cps
97.24 1950cps 99.10
97.06° 2000cps 98.96
95.86° 2280cps 97.72
Os parâmetros de rede para a s é r i e TA-1, Tabela 5 . 3 . , s ã o com1 o
parados com os valores obtidos por Gronvold , para o UO, no'00? 05e UO- | g , Tabela 5 . 4 .
Pode-se observar nos dados apresentados na Tabelas 5.1. e
5.2., que houve um aumento sistemático das intensidades dos picos
com a temperatura.
Nas Tabelas 5.3, e 5.4., observa-se a variação do parâmetro

TABELA 5.2. sERIE TA-2
38
DIFRATO TEMPERA TEMPO/
GRAMA N9 TURA EQUIL.
TA-2-2
TA-2-3
TA-2-4
TA-2-5
TA-2-6
TA-2-7
TA-2-8
TA-2-9
525°C
330°C
1080°C
1080°C
1070°C
993°C
91O°C
910°C
17:45hs
16:25hs
14:15hs
38:45hs
62:OOhs
14:50hs
14:OOhs
38:00hs
PICO B-22O
26 ALTURA
PICO P-531
26 ALTURA
97.12° 2014cps
97.70° 2511cps
41.77" 8200cps 96.32° 2584cps
41.73° 8351cps 96.28° 2682cps
42.10"
41.95° 7579cps
41.74° 8281cps
41.74°
41.77°
41.77°
7872cps
7512cps
7513cps
96.30 24%cps96.38° 2503cps
O96.5296.50°
2369cps2459cps
PICO P-60026 ALTURA
98.98"98.5498.14
98.12
98.12
98.2298.3298.32
1407cps
1731cps
1894cps
1751cps
1691cps
1767cps
1699cps
1718cps
TABELA 5 . 3 . PARÂMETROS DE REDE DA AMOSTRA-1 ( S É R I E T A - 1 ) .
TEMPERATURA
°C
25
165
350
460625
610
PARÂMETRO
6-511
5.464
5.472
5.480
5.486
5.496
5.509
S DE REDE EM
6-531
5.4635.471
5.480
5.487
5.496
5.509
ANGSTRON (
6-600
5.464
5.471
5,480
5.486
5.496
5.509

TABELA 5.4. PARÂMETROS DE REDE DA AMOSTRA-1 E OBTIDOS PORGRONVOLD10, EM (*).
39
TEMPERATURAft
°C
2025
138165250260350
397
456460
520522
536599607625661
724778785810946
951969
ü02,00
5.4704
-
--
5.4839
--
-
--
-
5.4988
---
-
5.5087
-
5.5153
-
-
5.5246
-
GRONVOLD
U02,05
-
-
--
---
-
5.4907
-
5.4943
-
--
5.4980
--
5.5052
--
--
5.5194
U02,10
5.4696
-
5.4769
-
-
5.4841
-
5.4896
--
--
5.4936
5.4936
-
---
-
5.5038
--
-
5.5148
AMOSTRA-1
-
5.464
-
5.472
-
-
5.480
-
-
5.486
--
---
5.496--
-
5.509
-
-

40
de rede em função da temperatura. A partir dos dados da Tabela 5.4,
foram calculados os coeficientes de expansão térmica para o üO_
das seguintes amostras: amostra da série TA-1 e amostras estudadas10
por Gronvold (UO2 f 0 0, Figura 5.1.
&930-
9.499
S/MO1
900 1000itAptfOfiro («C)-
F1GURA 5.1. VARIAÇÃO DO PARÂMETRO DE REDE, DO U0 2 + x, COMA TEMPERATURA.
10Segundo Gronvold , o UO- O Q constitui uma única fase na fai_
xa de temperatura 20 a 946°C, apresentando um coeficiente de ex
pansão térmica linear da ordem de 10,8 x 10~6 °c" , que representa
muito bem a dependência da constante da rede em função da tempera
tura. Por outro lado, o composto UO~ nt. consiste áe duas fases abai
xo e uma acima de 460°C, o composto UO2 1 Q possui também duas fa
ses abaixo de 550 C, sendo homogênea acima dessa temperatura.
O ajuste dos pontos para essas três fases forneceu as se
guintes equações lineares:
UO2 0 0 para T*20°C, a = 5,468 (1+ aT) onde a = 10,7 x10"6 °c"1,
U02 0 5 para T»456°C, a = 5,464 (1+ aT) onde o = 10,5 x l O ^ V 1 ,
UO2'10 para T>536°C, a = 5,459 (1+ aT) onde a - 10,5 x IO"6 °c"1t
O ajuste dos dados da série TA-1 fornece as seguintes equa
ções:

41
para T * 25°C - a = 5.462 (1 + « T> onde a = 10,2 x 10~6 °C~1,
para T * 165°C - a = 5.461 (1 + « T) onde a « 10,5 x 10~6 °C~1,
para T j 350°C - a * 5.457 (1 + a T) onde a = U . 5 x 10~6 °C~1.
A Figura 5.1. apresenta a variação do parânetro de rede en
função da temperatura. Pode-se observar que as amostras estudadas
por Gronvold10apresentam una região (400 - 600°C), onde o parâme
tro de rede não varia com a temperatura, caracterizando una homo
geneização da amostra. A amostra-1 não apresenta un gráfico total_
mente linear, indicando una pequena oxidaçao para T<350°C e una
pequena redução en temperaturas maiores que 700 C; estando a con
posição final da amostra entre 2,05 e 2,10. Essas observações são
válidas, uma vez que na série-B estudou-se a oxidaçao do IK>2 en
temperaturas inferiores a 300°C. Apôs a oxidaçao a 165°C a compo
sicão da amostra era de aproximadamente 2,10. Cono o gás possui
baixa pressão de oxigênio, e está fluindo continuamente através
da câmara, em temperaturas maiores rrue 600°C o equilíbrio do sis
tema favoreceu a liberação de oxigênio pela amostra,provocando uma
pequena redução da mesma. Nas amostras estudadas por Gronvold10 não
ocorre esse fato, provavelmente porque as amostras estavam lacra
das dentro de capilares de quartzo com quantidades de oxigênio cons
tantes.
Os valores obtidos para o coeficiente de expansão térmica
estão dentro da faixa apresentada na literatura, por vários auto
res (Tabela 3.1.). Levando em consideração os comentários acima e
os resultados de Gronvold10 ,o valor considerado para a amostra-1 foi
de 10,5 x 10~6 °C"1.
A série TA-3 objetivou mostrar a ordem das fases formadas
durante a oxidaçao e fornecer parâmetros para a próxima série de
medidas. 0 comportamento dos picos de difração forneceu as seguin
tes informações. Figuras 5.2. e 5.3.:
- decréscimo na intensidade dos picos U02,
- aparecimento das linhas de unia nova fase cúbica (provável^
mente U.0~),
- a altura, do pico 6-311, da nova fase ultrapassa a altura
do U02
- desaparecimento quase que total do pico 6-311 do UO2 en
quanto que em B-220 as duas fases se confundem,
- formação de um novo pico na posição do U02, sem alterar
o pico do U4O9 , formando um dubleto ((311 mais 131) e 113)
que caracteriza a formação de outra fase que ê tetragonal
(provavelmente U,0 7);

42
- afastamento e aumento da intensidade dos picos do dubleto,
com o tempo de tratamento,
- observa-se que a velocidade e comportamento de transforma
ção é diferente para diferentes picos.
As Figuras 5.4., 5.5., 5.6. e 5.7., apresentam os perfis de
difração das seguintes amostras: sêrie-A sem tratamento, séries
TA-1, TA-2 e TA-3 apôs tratamentos térmicos.
O perfil de difração da série TA-3 foi indexado e calculados
os parâmetros de rede da composição final:
a = 5,401 - 0,004 8C = 5,506 - 0,004 ft
Comparando esse resultado com os apresentados por Aronson ,2 „
Roof e Belle , a composição final dessa amostra foi estimada como
sendo aproximadamente 2,30.
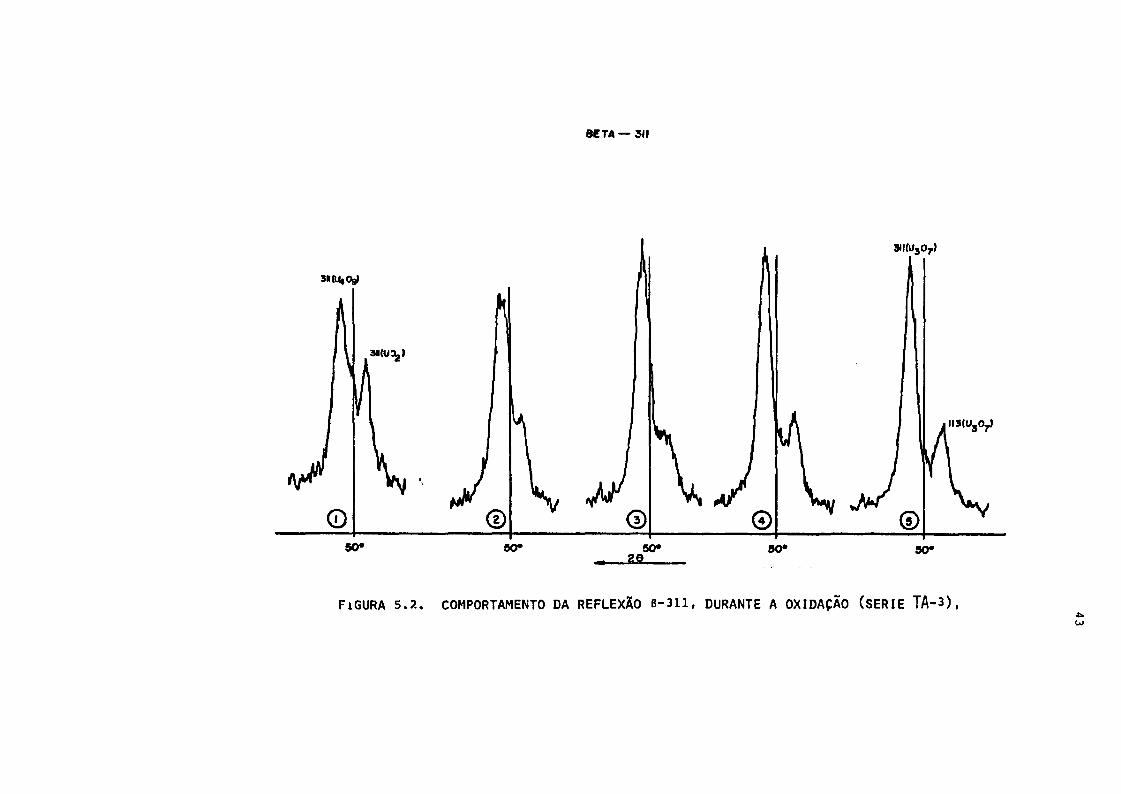
BETA — 311
3ll(U307)
50» 60" 50» 50# SO»
FiGURA 5.?.. COMPORTAMENTO DA REFLEXÃO B-311, DURANTE A OXIDAÇAO (SERIE TA-3),

BETA— 220
42»
FIGURA 5 . 3 . COMPORTAMENTO DA REFLEXÃO S-220,DURANTE A OXIDAÇAO (SERIE T A - 3 ) .

oo
ujWuTravtfi^^
FIGURA 5.4. AMOSTRA-A SEM TRATAMENTO TÉRMICO (SIMETRIA COBIÇA).

Ü J
FIGURA 5.5. AMOSTRA-A APÓS TRATAMENTO TERMICü (SÉRIE TA-1). 0%

*xjnrr ^
FIGURA 5.6. AMOSTRA-A APÔS TRATAMENTO TÉRMICO (SERIE TA-2).

~>-« O N
FIGURA 5.7. AMOSTRA-A APÓS TRATAMENTO TÉRMICO, APRESENTANDO SIMETRIA TETRAGONAL (SÉRIE TA-3) 00

49
5.2. SÉRIE TB
Os dados apresentados nas Tabelas 5.5. a 5.10., com exceção
do parâmetro de rede, foram calculados utilizando-se os pontos ex
perimentais, pelo programa ANACRON.
Devido ao pequeno número de pontos obtidos e ã falta de reso
lução no cálculo da área e largura ã meia altura dos picos, guando
estes eram muito pequenos, tornou-se impossível fazer os cálculos
utilizando a área dos picos e, também, fazer maiores considera
ções sobre o aumento do tamanho médio dos cristalitos (da fase em
formação). O tamanho médio dos cristalitos está associado ã largu
ra ã meia altura dos picos de difração.
Além do programa ANACRON foi utilizado o SAS (Statistical
Analysis System), na análise dos dados. Foram fornecidas as altu
ras das linhas para as duas fases (ANACRON), I, (UO_) e I- (U^O^),
e seus respectivos tempos de tratamento. Os cálculos efetuados pe
Io SAS foram:
- valores de f = (I-/(Ii + I?)) = concentração do U,Og, que
ê a fração de conversão do UO2 em U^O.;
- valores de Kt, utilizando as equações 3.1. e 5.1., onde
K é a constante de taxa;
- as energias de ativação, obtidas a partir dos gráficos
lnKxl/T. .
As análises quantitativas, utilizando-se a difração de raios-X,
devem basear-se na intensidade integrada (área) dos picos em lugar
da intensidade da linha (altura), porque essa última diminui quan
do tamanho médio dos cristalitos são menores que aproximadamente
0,1 u (1000 8 ) 1 5 .
Quando a nova fase começa a ser formada os cristalitos são
muito peguenos, não sendo portanto possível determinar o inicio da
nova fase. Pelos dados das Tabelas 5.5. a 5.10., pode-se observar
que o limite de detecção é da ordem de 20%. No entanto, é obser
vado um decréscimo na altura dos picos da UO-, cujo tamanho médio
dos cristalitos permanece invariável devido ao tratamento térmico
durante a sinterização e redução do material.
Somente a área do pico 3-311, a 235 C, apresentou um compor
tamento regular íurante a transformação. Na tentativa de estimar o
erro, devido ã utilização da altura em lugar da área dos picos,nos
cálculos da K (constante de taxa), ela foi calculada utilizando a
área . 0 resultado foi:

04/69
05/09
05/09
06/09
07/09
08/09
09/09
10/09
U/09
12/09
13/09
15/09
16/09
TEMPODE
TRATAMENTÜ(min)
44
1180
1425
2760
4085
5810
6973
8405
9831
11267
1Z933
15606
17040
TEMPERA
TURA
<°0230
238
235
240
230
233
235
235
232
231
235
230
235
TABELA 5
ALTURA
(cont/seg)
4732 ± 49
3662 ± 22
3557 ± 27
2934 ± 20
2531 ± 20
2226 ± 26
2019 ± 22
1727 ± 25
1489 ± 27
1318 ± 28
1172 ± 21
989 ± 23
846 ± 16
.5. SÉRIE B-l
uo2
CENTRO RREA29
GRAUS %
( 2 3 5 ° ) REFLEXÃO 6 - 3 1 1 .
LARG. PARAM.MEIA DE REDEALTURA (ft)
49,914±0,00149,924±0,00149,923±0,00149,925±0,001
49,930±0,001
49,931-0,001
49,930±0,00149,927±0,00249,932±0,00249,900±0,003
49,946±0,00349,947"O,004
49, « 2±0,003
100
8:1§1,9±0,8
$6,3±0,840,7±1,037,7±0,8
34,1±0,9
J9.4-1.0
28.6-1.12,6,1±0,8
23,3? l , 022,8*0,8
.0,247Í0.003+0,235Í0.002.0,238Í0.002
+0,246Í0.002
.0,249*0,003
.0,252*0,004
+0,256Í0.004
.0,261Í0.005
+0,269Í0.006.0,285Í0.008.0,304*0,007
.0,317*0,010,0,362-0,009
5,471
5,470
5,470
5,470
5,469
5,469
5,469
5,470
5,469
5,472
5,468
5,467
5,467
ALTURA
(cont/seg)
1010 í 15
1070 í 24
1477 í 16
1669 Í 17
1930 i 21
2045 t 18
2141 í 20
2282 - 24
2424 - 26
2605 - 18
2741 - 22
2809 - 16
Ü4°9CENTRO AREA LARG.
29 MEIAGRAUS X ALTURA
PARÍM.DE REDE
50,349Í0.004
50,359Í0,005
50,425Í0.002
50,453Í0.00250,475Í0.00250,480Í0.002
50,482Í0.00250,515*0,002
50,469*0,002
50,518Í0.00150,516±0,00150,523Í0.001
25.9Í1.3}6.9J1.9J8.1fl.l§3.7J1.Z
59,3Í1.362,3=1.255,9±1.370,6±1.771.4±1.8J3.9±1,2
76,7±1.4
17,2±1.1
.0,477Í0.014
.0,463±0,020
.0,454±0,007
+0,439±0,007
.0.423±0,008
.0.418±0,005
.0.408±0,005
.0,422±0,006.0,387±0,006
.0,388±0,003.0.377±0,004
0,369±0,003
5,427
5,426
5,419
5,416
5,414
5,414
5,413
5,410
5,415
5,410
5,410
5,409o

TABELA 5 . 6 . S É R I E B - l ( 2 3 5 ° C ) REFLEXÃO a - 2 0 0 .
D TEMPOA DE
T TRATAA MENTÜ
(min)
06/09 2888
07/09 4210
08/09 5490
09/09 7346
10/09 8517
11/09 9945
TEMPERA
TURA ALTURA
(°C) (cont/seg)
240 2922 - 31
235 2454 ± 26
232 2169 ± 24
230 1847 - 23
235 1628 ± 23
230 1413 ± 24
12/09 11398 235 1714 ± 27
15/09 15718 240 1494 ± 26
uo2
CENTRO AREA26
GRAUS %
32.771 34,8±0,003 Í0.8
32.772 30,5±0,003 Í0,7
32,784 28,1±0,004 Í0.7
32,788 ?6,2±0,005 ±0,8
32,803 26,0±0,007 ?l,0
U4°9
32,822±0,009
,2l
16/09 17158 235 1377 ± 28
32,971 43,0±0,005 ±1 ,0
32.968 34,0±0,005 Í0.8
32.969 35,6±0,006 Í0.9
LAR6.MEIA
ALTURA
.0,294±0,005
.0,309±0,006
.0,323±0,007
.0,355±0,009
.0,389±0,013
+0,431±0,016
.0,637±0,006
.0,657*0,007
.0,677±0,008
PARÍM.DEGREDE(8)
5,466
5,466
5,464
5,463
5,461
5,458
5,434
5,434
5,434
ALTURA
(cont/seg)
4750 í 23
5262 - 24
5600 - 24
5912 - 29
5936 - 39
6049 í 57
5306 - 35
6015 - 33
6042 - 35
CENTRO29
GRAUS %
33,115 65,2"0,002 Í1.0
23,132 69,5Í0.002 í l ,0
J3.148 71,9-0,001 . Í l , 0
3.3,150 73,8ÍO.OOl II,0
33,155 74,0Í0.001 11,4
33,167 74,8^0,001 Í1.3
LARG.MEIA
ALTURA
.0,338Í0.004
.0,328±0,003
.0,320±0,003
.0,313±0,002
x0,303±0,002
x0,299±0,002
33,172 57,0 .0,273±0,000 ÍO, 7 ±0,002
33,172 63,0±0,000 30,7
|3,174 64,4±0,000 ±0,8
+0,278±0,001
.0,280±0,002
PARÍM.DEGREDE8
5,411
5,408
5,406
5,405
5,405
5,403
5,402
5,402
5,402

D TEMPOA DE
T TRATAA MENTTJ
(min)
24/09 O
25/09 147
26/09 1224
28/09 4443
29/09 5593
30/09 7014
01/10 8566
03/10 11281
06/10 15624
07/10 17052
08/10
TEMPERA
TURA
20
169
170
170
170
170
170
168
170
165
20
TABELA S . 7 .
UOo
SÉRIE B - 2 ( 1 7 0 ° C ) REFLEXÃO < * - 3 1 1 .
ALTURA
(cont/seg)
3319 - 32
4966 • 47
4777 "- 49
4471 ± 34
4125 ± 15
3939 ± 20
4029 ± 19
3794 ± 25
3675 í 35
3620 ± 30
2906 Í 25
CENTRO20
GRAUS
50,000±0,001
49,913±0,001
49,924±0,001
49,929±0,008
AREA
%
100
100
100
100
49,945 60,3±0,003 ±0,7
49,948 55,8±0,005 ±1,1
49.940 57,2±0,000 ±0,9
49,962 54,3±0,000 ±2,5
49,942 66,2±0,001 ±1,8
49.941 67,2±0,001 ±1,6
49,979 68,5±0,001 ±1,7
LARG.MEIA
ALTURA
.0,249±0,003
0,270±0,003
.0,252±0,003
.0,229±0,002
+0,217±0,001
.0,219±0,001
.0,218±0,001
.0,213±0,002
JJ.230±0,003
.0,235±0,002
+0,Z59±0,003
PARAM.DE REDE(*)
5,462
5,471
5,470
5,469
5,468
5,467
5,468
5,466
5,468
5,468
5,464
u4o9
ALTURA CENTRO AREA29
(cont /seg) GRAUS %
629 Í 11
629 - 16
660 - 13
661 ± 45
698 ± 19
731 ± 1 9
581 ± 16
LARG.MEIA
ALTURA
50,107 39,7±0,006 =1,1
§0,139 44,2±0,011 ±1,1
50,180 42,8±0,010 ±1,5
J0.249 45,7±0,024 ±4,9
50,421 33,8±0,011 ±2,2
50,434 32,8±0,010 ±1,8
50,508 31,5±0,010 ±1,9
.0,938±0,017
.1,088±0,033
J>,996±0,024
.1,029±0,072
.0,617±0,033
+0,568±0,025
.0 ,597±0,030
PARAM.DEGREDE
5,451
5,448
5,444
5,437
5,419
5,418
5,411

TABELA 5 . 8 . SERIE B-2 (170°C) REFLEXÃO a - 2 0 0 .
n TEMPO TEMPERAA 0E
T TRATA TURA ALTURAA MENTO
(min) (°C) (cont/seg)
25/09 66 170 6807 - 35
26/09 1338 170 6427 - 73
29/09 5722 168 4892 ± 26
30/09 7378 168 4506 ± 45
01/10 8686 170 5390 ± 43
07/10 17166 '70 4250 ± 32
CENTRO28
GRAUS
32,783±0,001
32,780±0,001
32,781±0,000
32,785±0,001
32,783±0,002
03/10 11402 168 5244 ± 47 2 2 ,780
06/10 15738 168 4311 ± 30
22,780
ío,oo2
08/10 20 2898 - 19
32,773Í0.001
32,774Í0.001
32,79430,002
AREA
100
100
§0,13
$3,63
+1,71±1,56
$7,22- 1 . 5 2
$6,88±1,00
J6.47
58,83±1,01
LARG.MEIAALTURA
0,257±0,002
+0,272±0,004
+0,225*0,001
+0,217Í0.002
+0,269±0,003
+0,265±0,003
+0,253Í0.003
,0,254±0,003
.0,306Í0.003
PARÍM.DE REDE(*>
5,464
5,465
5,465
5,464
5,464
5,465
5,466
5,466
5,462
ALTURA
(cont/seg)
1681 - 23
1899 í 37
1581 í 30
1847 t 31
2259 Í ?.3
^349 í 25
1704 í 16
U4°9ÍENTRO 'ÍREA LARG. PARAM.
20 MEIA DE.REDEGRAUS X ALTURA (A)
32,935*0,003
32,947Í0.005
33,137Í0.007
33,139Í0.006
33,142Í0.003
Í0.003
33.191*0,003
$9,87Í0.95
56,37Í1.62
28,29Í1.39
32,78*1,44
$3.12=1.15
43,53-1,19
41.17Í1.00
+0,651+0,006
0,665Í0.010
+0,316Í0.015
.0,367Í0.014
+0,367+0,008
+0,354Í0.008
0,365Í0.007
5 '

TABELA 5 . 9 . SERIE B-3 ( 2 7 5 ° C ) REFLEXÃO B - 3 1 1 .
D TEMPO TEMPERAA DE
T TRATA TURA ALTURAA MENTTT
1O> 10
(min) (ÜC) (cont/seg)
21 3557 - 31
13/10 63 275 3905 ± 31
14/10 1281 280 1589 ± 16
14/10 1691 275 1319 ± 14
15/10 2743
15/10 3234
16/10 4185
17/10 5637
272
275
272
270
777 t 14
714 - 12
519 - 12
433 - 14
uo2
CENTRO AREA26
GRAUS %
$ 9 ' 9 5 6 100±0,001 10°49,863 65,53±0,001 ±1,49
49,89' 31,57±0,002 -0,62
49,893 26,73±0,002 ±0,55
49,918 20,78±0,004 ±0,75
49,931 21,05±0,005 ±0,74
49,960 20,53±0,013 ±1,26
49,994 21,24±0,027 **,02
LARG.MEIAALTURA
0,283±0,003
.0,240±0,002
0,255Í0.004
.0,272Í0.004
+0,344ÍO.010
.0,391Í0.012
+0,515*0,028
.0,638*0,005
PARAM.DEnREDE(8)
5,647
5,476
5,473
5,473
5,470
5,469
5,466
5,463
ALTURA
(cont/seg)
692 - 29
2063 - 13
2332 • 12
2664 - 14
2815 - 12
2964 t 21
3067 Í 41
CENTRO29
GRAUS
4o«AFREA LARG. PARAM.
MEIA DE REDEALTURA (A)
49,935Í0.007
50,359Í0.002
50,380Í0.001
50,410Í0.001
§0,417Í0.001
50,424Í0.002
50,433*0,002
34,47Í1.97
68,43íl .01
73,2710,93
19,22-1.14
78,95-1,04
79,47-1.47
78,76-2,30
0,712Í0.024
.0.425-0,004
.0,422-0,003
.0,383Í0003
.0,372±0,003
+0,349*0,003
0,334IO,003
5,469
5,426
5,424
5,421
5,420
5,419
5,418

TABELA 5 . 1 0 . SÉRIE B-3 (275°C) REFLEXÃO a - 2 0 0 .
TEMPO TEMPERADE
TRATA TURA ALTURAMENTÜ(min) (°C) (cont/seg)
10/10
13/10 154
14/10 1397
14/10 1817
15/10 2974
16/10 4294
17/10 5749
21
275
278
277
275
275
275
5044 - 23
6787 - 189
1726 í 105
2108 - 64
1334 3 45
1164 í 32
1101 - 32
uo2
CENTRO AREA29
GRAUS %
LARG. PARAM.MEIA DE REDE
ALTURA (%)
n-JSSt - 4 :32,728 81,37 .0,30,003 Í3.73 ±0,
iO.020 12,48 -0,023
32,919 45,19 .0 ,Í0.007 Í1.68 -O,
32,885 31,94 .0 ,Í0.010 31,32 -o ,
32,898 29,31 0,665 -Í0.010 31,00 *0,012 5 t
|2,885 27,86 +0,686 - 4 4 8
30,011 Í1.06 30,015 5 > 4 4 B
u4o9
ALTURA CENTRO AREA I.ARG. PARAM.29 MEIA DEGREDE
(cont/seg) GRAUS % ALTURA (?)
1 3 1 4 í l 2 8
32,792 28,18 0,421 - . f i , . . . . + m
Í0.020 32.48 30.023 5«^6lJ S M S ]W
4780 i 78
18,63 .0,345•2,51 -0,030
71,82 x0,33333,50 30,004
54,81 0,300*1,43 30,003
« l f i + u 33»0 8 1 S8»06 +0»3 0 25 9 1 6 " 5 9 Í0.001 31J30 Í0.002
6323 i 41 70,69 0,29530,98 30,002
72,14 J),29431,06 -0,002
5,434
5,418
5,416
5,412
5,412
5,411
in

56
K = 8,7 x 10~ (min. ) utilizando a área,
K = 8,8 x 10~6 (min."1) utilizando a altura,
indicando que nesse caso deve ter ocorrido somente uma translação
no gráfico Kt x t. O mesmo argumento não pode ser extrapolado pa
ra as outras temperaturas, pois pode ser que o tamanho dos crista
litos tenha tido maior influência nos resultados, principalmente
em 170°C, onde eles são muito pequenos, < 100 8 (para uma largura
â meia altura maior que -0,79, tem-se cristalitos < 100 8).Também
deve-se considerar que a 235°C os.resultados obtidos para a refle
xão 6-311, foram excelentes em todos os aspectos (Tabela 5.5.).
A variação relativa na intensidade dos picos difratados (f =
Ij/dy + IjJKcom o tempo, Figura 5.8., para uma dada temperatura
e pressão de oxigênio constantes, permitiu acompanhar a transfo£
inação de fases UO2—U.Og, bem como determinar alguns parâmetros de
cinética de oxidação para o UC^.
Pode-se observar na Figura 5.8., que as curvas de taxa apre
sentam forma parabólica atê a composição estável menor que U00 ,,.21» 2 *»*:>
(f<1). Segundo Saito e Aronson e colaboradores , essa tran£
formação parabólica implica em um estágio de oxidação controlado
pela difusão. Inicialmente, se a difusão de oxigênio ocorre na
estrutura do VOJI formando solução sólido UO,. numa reação mono
fãsica, uma equação análoga â equação utilizada na condução de
calor (equação 3.2.), pode ser usada. Por outro lado, se a difu
são através de uma fase tetragonal U3O7*, numa reação difásica, é
c processo que controla a taxa de oxidação, a equação 3.1. ou a
equação relacionada 5.1. conhecida como equação de Jander , pode
ser usada:
Kt « 1 - (2/3) f - (l-f) 2 / 3, 3.1
Kt - (1 - (1 - f ) 1 / 3 ) 2 , 5.1
onde f_ é a fração de conversão de U(>2 em U.O7, t é o tempo de rea
ção e K é a constante de taxa e está relacionada com o coeficiente
* Saito sugeriu que a oxidação do UO~ obedece a seguinte ordem
de transformação:
uo 2— a-u3o? — e-u3o7 — B'-u3o7 — u3og.
No entanto Andressen , utilizando difração de neutrons, apontou
a similaridade entre as lases u-U,o7 e U4O9.

57
OiO
lUJ
I-
ODOOol<o<atoai
XoLU
a
00
m
<
o

58
de difusão do oxigênio e o raio médio das partículas,assumindo que
exas são esferas uniformes ' '
Analisando seus resultados de difração de raios-X e cinética*• 2 *•
(utilizando a técnica de termogravimetria), Saito concluiu gue
o processo gue controla a taxa de oxidação, até a composição U,07,
é a difusão de oxigênio através de uma camada superficial de oxido
da nova fase. Portanto a eguação 5.1. foi utilizada para analisar
seus dados.
As Tabelas 5.11. e 5.12., apresentam os dados obtidos nessa
série (série-TB). Como c seu comportamento é semelhante ao obser2,10,11,20,2t -
vado em trabalhos sobre oxidação , foram utilizadas as
equações 3.1. e 5.1., para interpretá-los.
Os dados referentes ao pico 6-111, não puderam ser aproveita
dos porque sua transformação foi muito rápida e principalmente por
gue em baixos ângulos (26), os picos das duas fases estão muito
próximos e não foi possível separá-los. Figura 5.9.
Como inicialmente (t = 0), a concentração da fase U4Og é nu
Ia (Io = 0 ) , os gráficos f x t e Kt x t deveriam passar na origem.
Entretanto, como as amostras foram aquecidas, até atingirem as tem
peraturas de trabalho, em argõnio-U (atmosfera oxidante) e consi
derando que a oxidação é rápida, principalme»-ce em 235 e 275°C; no
tempo t=0 tem-se I-j* O. Isso poderia ter sido evitado se a amos
tra tivesse sido aquecida em uma atmosfera não oxidante.
Considerando esses fatos, os dados foram analisados, utili
zando-se as equações 3.1. e 5.1., de duas formas: com intercepto
e passando na orige*.i. Os resultados são apresentados nas Tabelas
5.13. (0-311) e 5.14. (a-200).
Vários fatores contribuíram para gue nem todos os dados obti
dos fossem considerados nos cálculos:
- segundo Saito , para f > 0,7 os dados começam a se des_
viarem do ajuste linear. Foi observado que f apresenta diferen
tes valores dependendo da reflexão estudada e da temperatura;
- no cálculo do erro em f e em Kt não foi levado em conta o
erro na temperatura e no tempo, contribuindo para gue algumas medi^
das não estivessem dentro do desvio médio;
- no pico a-200, principalmente, para valores de f>0,7 foi
dificil medir a altura do pico do UO2 gue já havia se transformado
quase gue totalmente em
- or pontos que apresentaram desvios maior que a média, de
acordo com os gráficos dos ajustes e resíduos e testes estatísti

TABELA 5 . 1 1 . PICO 6-311
59
a) T = 170 ! 5°C
TEMPO EM INTENSIDADEMIN. UO2(cps)=I1
INTENSIDADEU4O9(cps)=I2
f=Kt Kt
EQUAÇÃO 3.1. EQUAÇÃO 5.1
O147
12244443559370148566112811562417052
4966 í 474777 l 494471 í 344125 i 153939 * 204029 * 193794 * 253675 i 353620 * 30
629 j 11629 * 16660 * 13661 * 45698 * 19731 - 19
0,132*0,0020,138*0,0030,141*0,0020,148*0,0090,160*0,0040,168*0,004
0,0021*0,00010,0022x0,00010,0024*0,00010,00?6*0,00030,0031x0,00020,0034*0,0002
O,0021ÍO,00010,0023*0,00010,0024*0,00010,0027*0,00030,0032*0,00020,0035*0,0002
b) T = 235 * 5°C
TEMPO EM INTENSIDADEMIN. U02(cps)=I1
O44
11801425276040855810697384059831
11267129331560617040
4782 í 493662 * 223557 i 272934 * 202531 i 202226 * 262019 í 221727 i 251489 i 271318 * 281172 v 21989 2 23846 - 16
INTENSIDADE
1010 2 151070 l 241477 í 161669 2 171930 í 212045 i 182141 2 202282 2 242424 2 262605 2 182741 í 222809 - 16
KtEQUAÇÍO 3 .1
KtEQUAÇÃO 5 . 1 .
0,23320,0030,23.120,0040,33520,0030,39720,0030,46420,0040,50320,0040,55420,0040,60520,0050,64820,0050,690x0,0040,73520,0050.769Í0.004
0,006720,00020,0067^0,00030,014810,00030,021620,00040,030920,00060,037320,00060,046820,00090,058410,00120,069420,00150,081920,00130,097420,00180,1107^0,0015
0,0070*0,00020,0070*0,00030,0161x0,00030,024120,00040,035320,0008O,0433jO,00080,055620,00120,071020,00190,086320,00210,104320,00190,1279x0,00280,1490*0,0024
c ) T = 275 * 5°C
TEMPO EMMIN.
INTENSIDADEU02(cps)=I1
INTENSIDADEU409(cps) I
Kt KtEQUAÇÃO 3.1 . EQUAÇÃO 5.1
063
128116912743323441855637
3905 * 311589 * 161319 * 14777 * 14714 * 12519 * 12
O692 * 292068 * 132332 ± 122664 * 142815 * 122964 * 21
433 * 14 3067 * 41
0,151*0,0060,566*0,0030,639*0,0030,774*0,0030,798*0,0030,851*0,0030,876*0,004
0,0027*0,00020,0493*0,00060,0669*0,00070,1131*0,00140,1236*0,00130,1561*0,00180,1673*0,0025
0,0028*0,00020,0588*0,00080,0828*0,00100,1529*0,00230,1705*0,00220,2208*0,00350,2513*0,0051
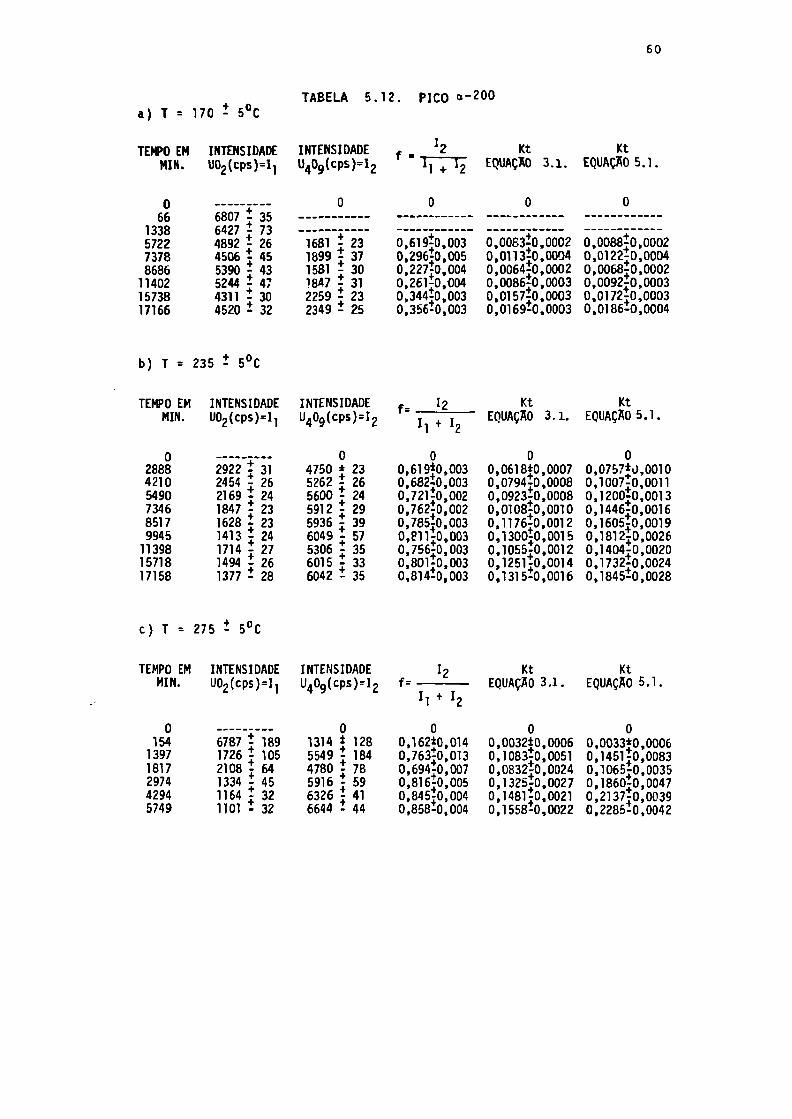
60
a) T = 170 - 5°CTABELA 5 . 1 2 . PICO «~200
TEMPO EMMIN.
u661338
572273788686
114021573817166
b) T =
TEMPO EMMIN.
g288842105490734685179945
113981571817158
INTENSIDADEU02(cps)
6807 *6427 *4892 *4506 *5390 *5244 *4311 *4520 *
235 * 5°C
= I1
3573264543473032
INTENSIDADEUO2(cps)
2922 *2454 *2169 *1847 *1628 *1413 *1714 *1494 *1377 *
= I1
312624232324272628
INTENSIDADEU 4 0 9 ( cps )= I 2
168118991581184722592349
o
* 2 3í 3?* 30* 3 1* 23* 25
INTENSIDADE
ty>9(cpsH2
475052625600591259366049530660156042
D* 23* 26* 24* 29* 39* 57* 35* 33* 35
f - Z
o
0,619*0,0,296*00,227*0,
,003,005.004
0,261*0,0040,344*0,0,356*0
f= I :
o0,619*00,682*00,721*00,762*00,785*00,811*00,756*00,801*00,814*0
,003,003
l
h
,003,003,002,002,003,003,003,003,003
KtEQUAÇÃO
000000
o
,0083*0.,0113*0,,0064*0,,0086*0,,0157*0,,0169*0,
KtEQUAÇÃO
000000000
o,0618±0,0794*0,0923*0,0108*0,1176*0,1300*0,1055*0,1251*0,1315*0
3 . 1 .
,0002,0004,0002,0003,0003.0003
3 . 1 .
,0007,0008,0008,0010,0012,0015,0012,0014,0016
KtEQUAÇÃO 5
0
0,0088*0,0 ,0122*0,0,0068*0,0,0092*0,0 ,0172*0,0,0186*0,
KtEQUAÇÃO 5
o0,0757*ü.0,1007*0,0,1200*0,0,1446*0,0,1605*0,
. 1 .
000200040002000300030004
. 1 .
,0010,0011,0013,0016,0019
0,1812*0,00260,1404*0,0,1732*0;0,1845*0.
,0020,0024,0028
c) T = 275 - 5°C
TEMPO EM INTENSIDADEMIN. UO2(cps)=I1
INTENSIDADEU40g(cps)=I2 f=
KtEQUAÇÃO 3.1.
KtEQUAÇÃO 5.1.
O15413971817297442945749
6787 x 1891726 * 1052108 * 641334 * 451164 * 321101 + 32
1314 ± 1285549 * 1844780 * 785916 * 596326 * 416644 - 44
0,162*0,0140,763x0,0130,694x0,0070,816x0,0050,845x0,0040,858*0,004
0,0032*0,00060,1083x0,00510,0832*0,00240,1325*0,00270,1481*0,00210,1558*0,0022
0,0033*0,00060,1451*0,00830,1065*0,00350,1860*0,00470,2137*0,00390,2285*0,0042
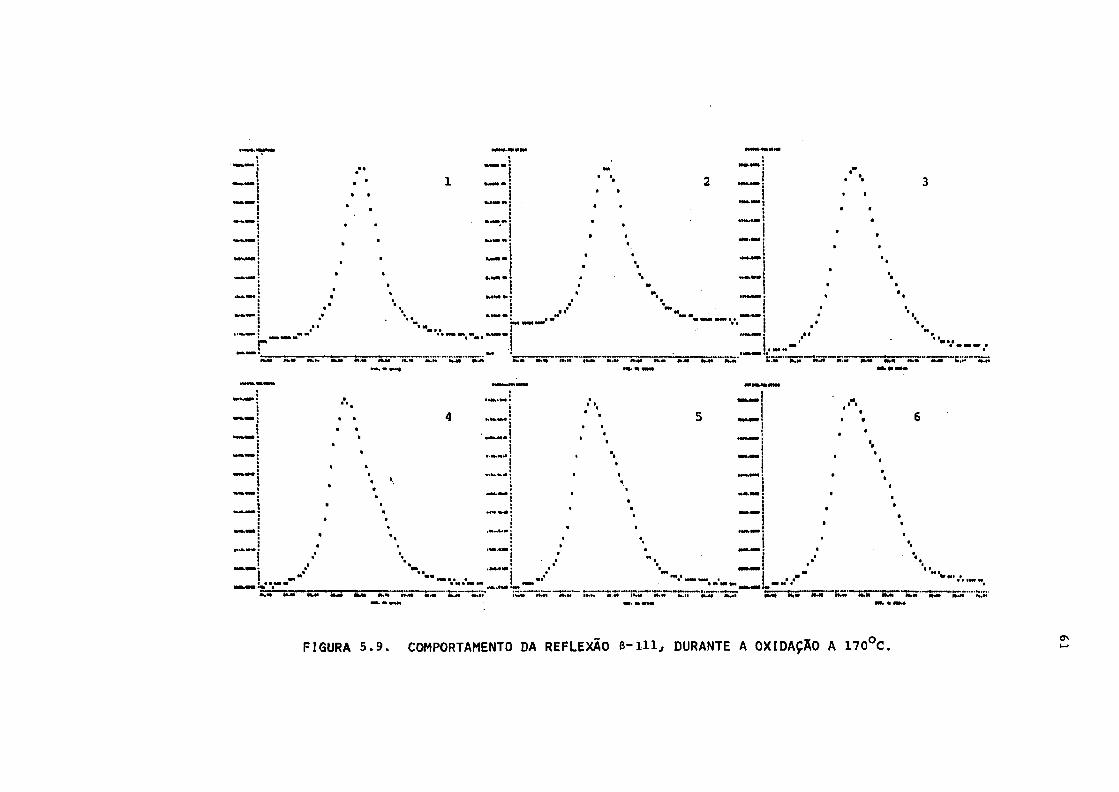
• •
H?m tit «I'M WrW
* •
• •
•%•-.•-.- I -•,- *'•»•
FIGURA 5.9. COMPORTAMENTO DA REFLEXÃO P-lll, DURANTE A OXIDAÇÃO A 170°C.

TABELA 5 . 1 3 . PICO B-311
62
CONSTANTE TEMPERATURADE TAXA( m i n - 1 )
*1
K2
°C
170
235
275
EQUAÇÃO 5 . 1 .COM NA
INTERCEPTO ORIGEM«-7
EQUAÇÃO 3.1.COM NA
INTERCEPTO ORIGEM
. 1,1 x IO"7 2,3 x IO"7 1,1 x IO"7 2,2 x IO"7.± 0,1 x 10"' ± 0,2 x 10 ' ±0,1 x 10 ' ± 0,2 x 10"'^8,83x10"? . 7,86 x 10"f . 6,56 xlO"| 6,15x10"*±0,45x10"° ± 0,25 xlO"° ±0,15 xlO"° ±0,11x10"°
. 5,43x10"! . 5,28xlO"c ^3,69x10"^ . 3,80x10"^±0,19xl0"5 ± 0,10xl0"D -O,17xlO"D ±0,08xl0"s
N9 DE PONTOS AJUS"TADOS ~
6
12
6
TABELA 5 . 1 4 . PICO a-200
CONSTANTE TEMPERATURADE TAXA or(min"1) L
K. 170
235
275
EQUAÇÃO 5.1. EQUAÇÃO 3.1.COM NA COM NA
INTERCEPTO ORIGEM INTERCEPTO ORIGEM
N9 DE PONTOS AJUS
TADOS
,-77. 8,5 x IO"7 .11,13 x 10"? 7,5 x 10"! J 0 . 1 6 x 10"
* 0,2 x 10'7 í 0,70 x 10"' -0,1 x 10"' - 0,70 x 10"
.14.79x10"® .20,42 x 10"5 .9,70x10"? .15,40 x 10~fÍ 0 , 6 4 x l 0 " 6 í 1,03 x 10"° ÍD.5 x 10"° í 1,04x10"°
11,41 xlO"5 10,29 x 10"| 8,46 x IO"5 M 7,68 x IO"5.
* O í 0,90 x IO"* - O Í 0,68 x IO"5

63
cos fornecidos pelo SAS, não foram considerados.
As Figuras 5.10. e 5.11., mostram os gráficos dos ajustes,
de acordo com as Tabelas 5.11. e 5.12., e os gráficos de Arrhenius
com os valores das energias de ativação de oxidação, para as re
flexões B-311 e a-200, respectivamente. As energias de ativação ,
para os casos considerados, são apresentados na Tabela 5.15.
TABELA 5.15. ENERGIAS DE ATIVAÇÃO EM KCAL/MOL.
PICO EQUAÇÍO 5.1. EQUAÇflO 3.1.
com 28,520 t 1,179 26,980 - 1,071intercepto
e-311na 24,812 - 0,500 23,464 - 0,592
origem
com 22,087 ± 2,124 21,095 ± 2,689intercpeto
«-200na 20,607 - 0,607 19,695 ± 0,967
origem
Os resultados obtidos a 170°C mostram que â medida que a
intensidade do pico UO2 diminui a do U.Og, quando começa a ser
detectado, sofre pequenos aumentos (Tabelas 5.11. e 5.12.). Esse
efeito pode ser explicado considerando que nessa temperatura pre
domina a formação de solução sólida ^O^+x'' P r o v o c a n^ 0 ° decrés_
cimo na intensidade e uma mudança na posição (26), dos picos do
UO,. Outros fatores, já mencionados, que também contribuem para
esse comportamento são: o limite de detecção, da fase que se for
ma, que é grande -20% e o tamanho médio dos cristalitos, que pro
duzem picos bem largos quando seu valor é muito pequeno. A peque
na variação na altura dos picos do U4Og, pode estar associada ao
fato que nessa temperatura a camada de oxido formada superficial
mente deve ser muito fina, sendo portanto rapidamente atingido o
equilíbrio do sistema.
Os valores da energia de ativação para a oxidação do IK>2, en
contrados na literatura, são apresentados na Tabela 5.16. Apesar
dos resultados encontrados estarem compatíveis com esses valores,
foram considerados os valores cujo ajuste não passa na origem como
sendo os mais representativos.

64
cvso
OH»
npso
opt»
EOÜAÇÃO3.1-
/ • EQUAÇÃO 5 . 1 -
• PONTOS COINC'OENTES
I7O*C
9000 10000 6000 T»mpoimin}—
FIGURA 5 . 1 0 . GRÁFICOS DA CONSTANTE DE TAXA (K) E DA ENERGIA
DE ATIVAÇÃO, PARA A REFLEXÃO B-311 .

65
4290
0*00
o;so
0,100
0,500
/ • Equocfe 3 1 -
/ « Equocfe 5 . 1 -
Ponto*
FIGURA 5.11. GRÁFICOS DA CONSTANTE DE TAXA (K) E DA ENERGIADE ATIVAÇÃO^ PARA A REFLEXÃO o-200.

66
TABELA 5.16,
18
25
24
27aa26
ENERGIAS DE ATIVAÇÃO DE
EM
,5
27
30
,3
KCAL/MOL.
referencia
referência
referencia
referência
referência
0
24
24
24
24
2
Pode-se observar nas Figuras 5.12, 5.13. e 5.14, que o coin
portamento dos picos das duas fases são diferentes para cada faml
lia de planos cristalogfaficos (hkl), isto ê, o valor de f é dife
rente para diferentes picos, para um mesmo tempo e temperatura de
tratamento; implicando em diferentes energias de ativação para ca
da reflexão. Esse comportamento pode estar associado ãs posições
de ocupação e direções de difusão dos átomos de oxigênio inters
ticial, dentro da estrutura, ou seja, os átomos tendem a ocupar
primeiramente as posições de menor energia de ativação. Esta trans;
formação na estrutura do material faz com que os átomos de oxi_
gênio passem a ter uma participação maior, construtiva ou destru
tiva, na intensidade dos pico:: difratados. Este modelo foi est<a- 2t
belecido em trabalhos de duração de neutrons por Willis e no
trabalho teórico de Catlow".
A Figura 5.15., apresenta o comportamento dos picos 6-311 e
a-200, para cada fase, em função do tempo.
Nos resultados obtidos foi observado que para uma melhor in
terpretação do comportamento da transformação UO-—ü4Oq» o s grãfi^
cos de taxa deveriam ser divididos em regiões ou estágios e ana
lisados separadamente. Embora nõo tenha sido possível delimitá-los,
os gráficos foram considerado» COTÜO sendo melhor representados
em três estágios, com as seguintes características:
ÍÇ Eitãgio: Ab&oição de. vxigznio pilo U0£, ^oKmando iolução
iõlida Ü02 + x, até. a íoKmação de uma camada t> up in. facial da nova í£
Ac. Ei tu ê. um iitagio muito lapido, de.pe.nde.ndo da te.mpe.ia tula., e
pode. ÒÍK obi invado atn.ave.i do dccié-icímo na intzntidade. doi pico*
do U(?2« E£e &oi me.lh.on. oba^adu na tzmpzn.atun.a dl 170°C.
Io. Titãqioi t caA.c.ct<zi.izado pzla dífiuião do oxigê.nio attiavib
da camadí. mpe.núíciat da nova ^aiz. Eaa c a Kígiao que deve. dai
o vaiou \e.o.i da zneAgia de. ativação paia a oxidação e. de difiuião
do oxigênio.

67
FIGURA 5.12. COMPORTAMENTO DA REFLEXÃO a-20ü, DURANTEA OXIDAÇAO A 235°C.
.BETA-»
FIGURA 5.13. COMPORTAMENTO DA REFLEXÃO 6-311, DURANTEA OXIDAÇAO A 235°C.

68
OGflD
FIGURA 5.14. COMPORTAMENTOS DIFERENTES DAS REFLEXÕES
6-311 E a-200, PARA A MESMA TEMPERATURA
E TEMPOS DE OXIDAÇÃO-
3? E&tãgio: HcAto. íòtágio a amoòtia titã pn.Oxi.ma da
•&ição di e.quilZbni.0, paia ai condiçõzò e,6tabe.lzcida&. A
mação z muito tinta nzi&a A.e.gião.
compo_
Deve ser observado que, para um maior número de pontos, para
cada temperatura, distribuídos uniformemente sobre os gráficos fxt;
não seria necessário utilizar as equações 3.1. e 5.1. para a
obtenção dos valores de K. A obtenção direta de K, das curvas de
taxa, forneceria a delimitação das regiões discutidas acima.
A variação do parâmetro de rede, em função do tempo, para o
pico B-311, Figura 5.16., mostra que as duas fases apresentam um
decréscimo sistemático, no parâmetro de rede, â medida que a oxi
dação se processa.
A Figura 5.17., ilustra o estado inicial e final do pico
B-311, tratado a 235 C, que apresentou os melhores resultados.
5,3, TEMPERATURA AMBIENTE
Após o resfriamento das amostras, foram tirados diagramas
de Guinier que confirmaram a presença de duas fases. A Tabela 5.17.
apresenta os resultados obtidos para a amostra B-l, antes e após
o tratamento térmico.

69
Difratogramas tirados quatro meses após o resfriamento, apre
sentara» modificações nos perfis das amostras, caracterizando um
rearranjo estrutural dos oxigênios. Com a mudança nas condições ex
ternas, o sistema provavelmente adquiriu outra configuração de
equilíbrio.

70
2000
COO
mfo-200
7OOO
.6000
a o o o .
woo
3C00
zooo
1000 I
3000 10000
beta-311
15000 Téwpo (min)
u4o9
• I7O«C* 23S*C
•VOt
3000 10000 aooo Ttmpotmin)
FIGURA 5 .15 . GRÁFICO DA VARIAÇÃO DA INTENSIDADE (CPS) DASPASES U02 E U4O9, EM FUNÇÃO DO TEMPO DE OXIDAÇÃO, PARA AS REFLEXÕES a-200 E B-311.

5080
5(460
3|»4O
5<»20
5JÍ0O
5000 10000 15000Tampo (min) —~
FIGURA 5.16. COMPORTAMENTO DO PARÂMETRO DE REDE, DAS DUAS FASES,EM FUNÇÃO DO TEMPO DE OXIDAÇÃO (REFLEXÃO B-31l).

72
SCTA-SII.INICIAL
T-ei'c
Sfi*
FIGURA 5.17. ESTADO INICIAL E FINAL DA REFLEXÃO (3-311, ATEMPERATURA AMBIENTE (SÉRIE Bi),
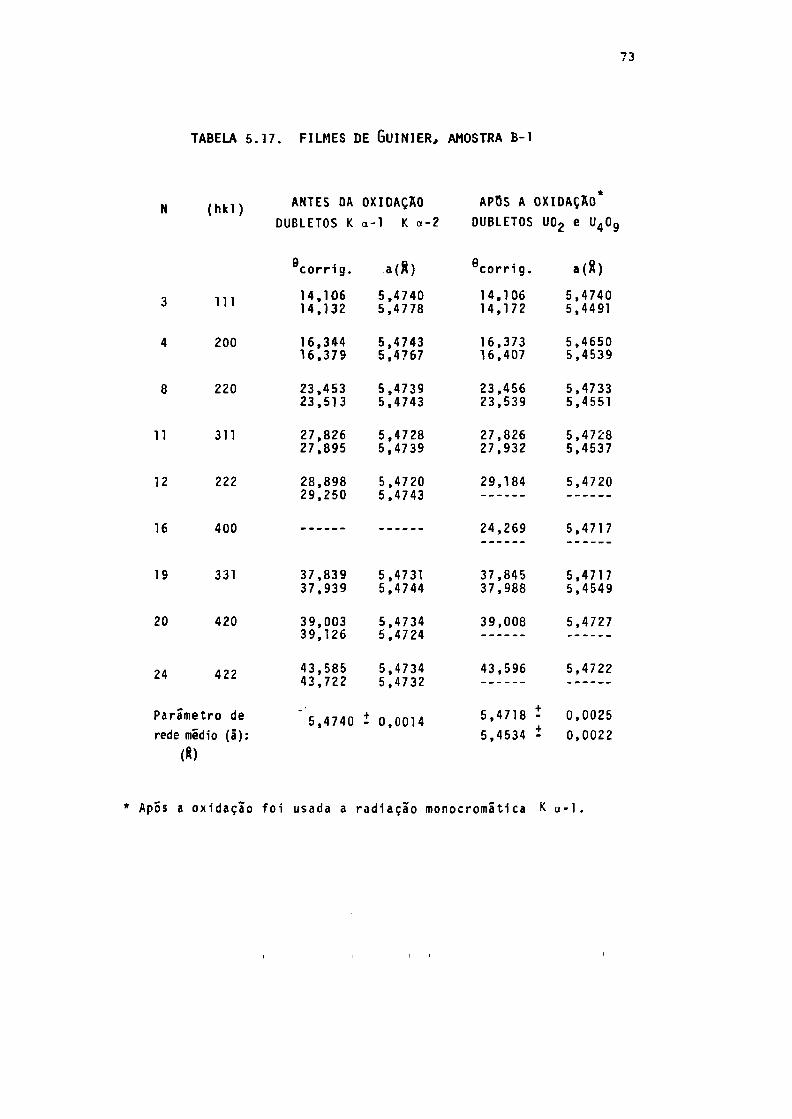
73
TABELA 5.17. FILMES DE GuiNIER, AMOSTRA B-l
(hkl) ANTES DA OXIDAÇÃO
DUBLETOS K a-1 K a-2
APUS A OXIDAÇKODUBLETOS U02 e U
0corrig. a(8) ecorrig. a(8)
11
12
16
24
111
200
220
311
222
400
19 331
20 420
422
Parâmetro de
rede médio (ã):
14,10614,132
16,34416,379
23,45323,513
27,82627,895
28,89829,250
37,83937,939
39,00339,126
43,58543,722
5,47405,4778
5,47435,4767
5,47395,4743
5,47285,4739
5,47205,4743
5,47315,4744
5,47345,4724
5,47345,4732
5,4740 í 0,0014
14.10614,172
16,37316,407
23,45623,539
27,82627,932
29,184
24,269
37,84537,988
39,008
43,596
5,4718 -
5,4534 í
5,47405,4491
5,46505,4539
5,47335,4551
5,47285,4537
5,4720
5,4717
5,47175,4549
5,4727
5,4722
0,0025
0,0022
* Após a oxidação foi usada a radiação monocromática K a-l.

74
CAPITULO VI
6. CONCLUSÕES
Os resultados desse trabalho podem ser sumarizados da seguin
te forma:
(J) 0 aumento na altura dai Unhai de di^ração, com a tempe
ratura, não <L divido ao aumento do tamanho médio doi criitalitoi,
e ilm a outro £ator não determinado nzite trabalho.
(Z) A expamão t~e.Kmi.ca do U0g ê ZoganZtmica, mai divido ao
pzquo.no valoi do i&u coe.iicie.ntz zta pode 6ti apioximada a uma &x
pamão íiníaK. 0 valoi obtido pana U02 QC t de 10,5 x 10~ °C~ ,que
atã em boa concordância com oi valonti enconttadoi na liteiatufia.
(3) Vatia umít atmo&ieia iníKtz com concentração de oxigênio me
qu
da.
not que 140 ppm c te.mpe.natuH.ai iupen.ion.ei a 350°C o HO* não ie oxí
(4) A compoiição inicial do ma.tzn.iat e&tudado faoi eitimada
como iendo de aproximadamente 2,0b.
(5) A ierie TA-3 moitra que a ordem dai &aiei ^ormadai duran
te a oxidação, nai condiçõei eipeci^icadai, ião: Uõ» [cúbico)
Ujfl,, (cúb-tco) "3^7 {tetragonal); com aumento na razão c/a {£a
ií tetragonal),com o tempo de tratamento.
16) Oi comportamentoi di£erentei, nai Unhai de di^ração, du
rante a oxidação, forneceram diferente* valorei de energiai de ati
vação, implicando em poiiçõei pre^erenciaii dt ocupação e direçõei
preierenciaii de di^uião para oi ãtoma de oxigênio intenticiaii.
(7) Eitabzlecendo-ie boai condiçõei de traniformação (prei

75
ião de oxigtnio e tempeiatuia], podt-*e acompanhai a oxidação do
U0», utilizando a di^iatomttiia de. iaio*-X de. alta. tzmpeiatuia.
[6) O* dado* de cinetica,mo*tiam que a oxidaq.Ro £ paiabõlica
[na iaixa de composição UO2-U,O7) e o pn.ime.ino oxido ^olmado e o
UjOq, com e*tiutuia cúbica. PÔde-se ob*eivai com ba&tante. nitidez,
e**a $a*e no diagiama de Guiniei da amo&txa BJ [UO^+ U.Og), apõi
a oxidação.
(9) Um gtã{ico {xt, com maio*, númtfio de pontoò, faoinecenia
diKítamínti OÒ valoitò dai conAtante.6 de taxa (K) e a delimitação
da di^eAentei tbtaaioò de oxidação, onde. di^eitntei mecanibmot>
atuam.
I/O) Ai znexgiai de ativação paia o pico B-3IJ [26,9t- 1,07
kcal/mol, 2S,52 - 1,1» kcal/mol) e paia a-ZOO [21,10 - 2,69 kcal/
mol, 22,09 - 2,12 kcal/mol], e.itão dtntxo da &aixa de. valoieó apie
izntadoò na liteJiatuia, obtido* utilizando-ie outiaò técnica*.
[11) finalmente, cabeKiam alguma* obieivaçõei ciZticai ao
tnabalho n.e.alizadoi
(a) o pe.que.no nãme.10 dl ponto* utilizado* no* calculo* e giá
faio.0*, não ^oinzcenam bon* ie*ultado* z*tatZ*tico*;
(b) paia mtlhoie* valoií* da* e.nzi§ia* de ativação, a axi
dação dzveiia tei *ido e.*tudada em um número maioi de tempeiatuia*,
entie. 17 0 e 275°C;
[c] um maioi nwtneio de plano* cii*talogiã^ico* [hkl) £
lia tei iido eitudado, paia a obt&nção de um maioi númzio de dado*
de cinítica, que con^iimaliam a* poiiçõe* piefieienciai* de ocupa_
ção doi átomo* de oxigênio;
[d] paia a obtenção de melkoie.* iziultado* *obie e*ttquiomeLtiia IO/U) c teimo dinâmica do WC^t de.veiia.rn *ei utilizada* paiale
lamente outia* ttcnica*, como poi exemplo teimogiavimetiia;
(el a utilização de um difaiatometio automático de iai*-X, um
contioladoi de tempeiatuia com alta *zn*ibilidade. e uma camaia de
alta tempeiatuia com maioi fiaixa de atuação, peimitiiia uma o ti
mização no* valoie* e no tempo de tiabalho do* iz&ultado*
tadoi, bem como em tiabalho* na* legiõz* Üí^ e ^2-x' ^°
ma de fia&e*.

76
REFERENCIAS BIBLIOGRÁFICAS
1 A D A M S O N , M . G . ; SCHUMACHER» G . ; N A V R A T I L , J . D . ; S U N D E R M A N N , H .
Oxygen activity changes in oxide fuels. Atom. Energy Rev.,
1£ (1): 247-57, 1980.
2 ARONbCN, S.; ROOF, R.B.; BELLE, J. Kinetic study of the
oxidation of uranium dioxide. J. chem. Phys., 2J_ (1): 137-
44, 1957.
3 BELBEOCH, B.; LAREDO, E.; PERIO, P. Examen par rayons X,apres
trempe , d1 oxydes d'uranium de type UO2. J. nucl. Mater.,
13:100-6, 1964.
4 BELLE, J. Uranium dioxide. Properties and nuclear applications.
USi'VEC, 1961.
5 CATLON, C.R.A. Point defect and eletronic properties of uranium
dioxide. Proc. R. Soc. London; A353: >33-61, 1977.
6 CORDFUNKE, E.H.P. The chemistry of uranium. Amsterdam,Elsevier,
1969.
7 CULLITY, B.D. Elements of X-ray diffraction. Reading, Mass.
Addison-Wesley, 1967.
8 FREITAS, C.T. Uranium dioxide sintering kinetics and mechanisms
under controlled oxygen potentials. Illinois, Univ. of
Illinois, July, 1977. (Ph. D. Thesis). (IEA-DT-141).
9 GREENWOOD, N.N. Ionic crystals, lattice deffects and nonstoi
chiometry. London Butterworths, 1970.
10 GRONVOLD, F. Hihg temperature x-ray study of uranium oxides in
the UO2-U3Og region., J. inorg. nucl.chem., 1:357-70, 19S5.
11 HOEKSTRA, H.R.; SANTORO, A.; SIEGEL, S. The low temperature
oxidation of UO2 and U^Og. J. inorg. nucl. Chem., 16:166-79»
1961.

77
12 INTERNATIONAL ATOMIC ENERGY AGENCY. Thermodynamic and transport
properties of uranium dioxide and related phases. IAEA,1965.
(Technical Report Series, 39).
13 ISHII, T.; NAITO, K.; OSHIMA, K. An X-ray and neutron diffrac
tion study on a phase transition in the U.Og phase.J. Phys.
Chem. Solids.,32:235-41, 1971.
14 ISHII, T.; NAITO, K.; OSHIMA, K. X-ray study on a phase tran
sition and a phase diagram in the U4Og phase. Solid St.Cannon.,
8:677-83; 1970.
15 KLUG, H.P.; ALEXANDER, L.E. X-ray diffraction procedures for
polycrystalline and amorphous materials. New York, Wiley &
Sons, 1974.
16 MATSUI, T.; NAITO, K. Phase relation and defect structures
of nonstoichiometric U.Og+ and UO,. at high temperatures.
J. nucl. Mater., 50:327-35, 1975.
17 MATZKE, H. Lattice defects and irradiation damage in Tho-jUO-
and (U, Pu)O,. IN: Plutonium and other actinides, Proceed
of the 5 international conference on ..., held in
Baden-Baden, September 10-13, 1975. p. 801-31.
1G NAITO, K.; KAMEGASHIRA, N. High temperature chemistry of cera
mie nuclear fuels with emphasis on nonstoichiometry. Adv.
nucl. Sci. Technol., £:99-180, 1976.
19 NAITO, K.; TSUJI, T.; MATSUI, T. An electrical conductivity
and x-ray study of a high-temperature transition in V.On
J.nucl. Mater., 4jl:58-66, 1973.
20 OKASHI, H.; NODA, E.; MOROZUMI, T. Oxidation of uranium dioxi.
de. J. nucl. Sci. Technol., U_(10):445-51, 1974.
21 CLANDER, D.R. Fundamental aspects of nuclear reactor fuel ele
nents. Springfield,Wa., ERDA, 1976.
22 RAl*D, M.H.; ACHERMANN, R.J.; GRONVOLD, F.; GETTING, F.L. ;

76
PATTORET, A. The thermodynamic properties of the uranium
phase. Revue int. Hautes Temper. Refract. Fr., 15_:355- 65 ,
1978.
23 SAITO, Y. Nonstoichiometry in urani?i7ti dioxide. J. nucl.Mater.,
5^:112-25, 1974.
24 SAITO, Y. Oxidation process and reactivity of uranium dioxide.
In: U.S. - JAPAN joint seminar on defects and diffusion in
solids, Tokyo, Oct.4-6, 1976. p. 33-44.
25 YOUNG, W.A.; LYNDS, L.; MOHL, J.S.; LIBOWITZ, G.G. An x-ray
and density study of nonstoichiometry in uranium oxides
Canoga Park, Calif., North American Aviation, 1962. (KAA-
SR-6765).
26 WILLIS, B.T. The defect structure of hyper-stoichiometric ura
nium dioxide. Acta Crystallogr., A34:88-90, 1978.