Embed Size (px)
Citation preview

FUNDAÇÃO FACULDADE FEDERAL DE CIÊNCIAS MÉDICAS DE PORTO ALEGREDisciplina de Genética Básica
DOENÇA DE TAY SACHS
Caroline Gregoletto MolinariMaria Julia Machline CarrionMelissa Fernanda Steigleder
Monitor: Rafael Bonfá

A Doença de Tay Sachs é uma gangliosidose na qual ocorre um acúmulo imtralissomal do gangliosídeo GM2 devido à deficiência da enzima hexosamidase A.
As gangliosidoses GM2 possuem 3 formas de apresentação clínica:
-Doença de Sandhoff;-Doença de Tay Sachs;-Deficiência do GM2 ativador.

HISTÓRICO

1881- Warren Tay amaurose infantil idiopática;
1896 - Bernard Sachs amaurose familiar idiopática;
1930 - Ernst Klenk identificação dos gangliosídeos.

1962 - gangliosídio GM2;
1968 - Robinson e Stirling duas formas de hexosaminidase;
1969 - Okada, O’Brien e Sandhoff atividade ausente de um dos componentes da hex A em judeus Ashkenazi;
Susuki e Chen introduzem o termo gangliosidoses GM2.

EPIDEMIOLOGIA

A DTS tem sido observada em crianças de toda as etnias, mas principalmente em populações geneticamente isoladas ( Amish, Franco-canadenses,Cajuns).
Especialmente prevalente entre os Judeus Ashkenazi.
Incidência: • 1 em 3600 judeus Ashkenazi
(f de portadores - 1 em 30)• 1 em 360 000 população em geral
(f de portadores - 1 em 300)

Programas de rastreamento podem a incidência em + de 90%.
No Brasil- estudo da USP concluiu que a f de portadores da DTS entre os judeus brasileiros é similar à de outros países onde o screening é realizado justificativa para implemento do programa de rastreamento.

FISIOPATOLOGIA

Aspectos Bioquímicos
Gangliosídeo GM2:
Ceramida+ glicose +
N-acetilgalactosamina+N-acetilneuramínico

Locais: terminações nervosas; células gliais.
Síntese: RE e Golgi; adição seqüencial de monômeros glicosila
transferidos de açúcares.
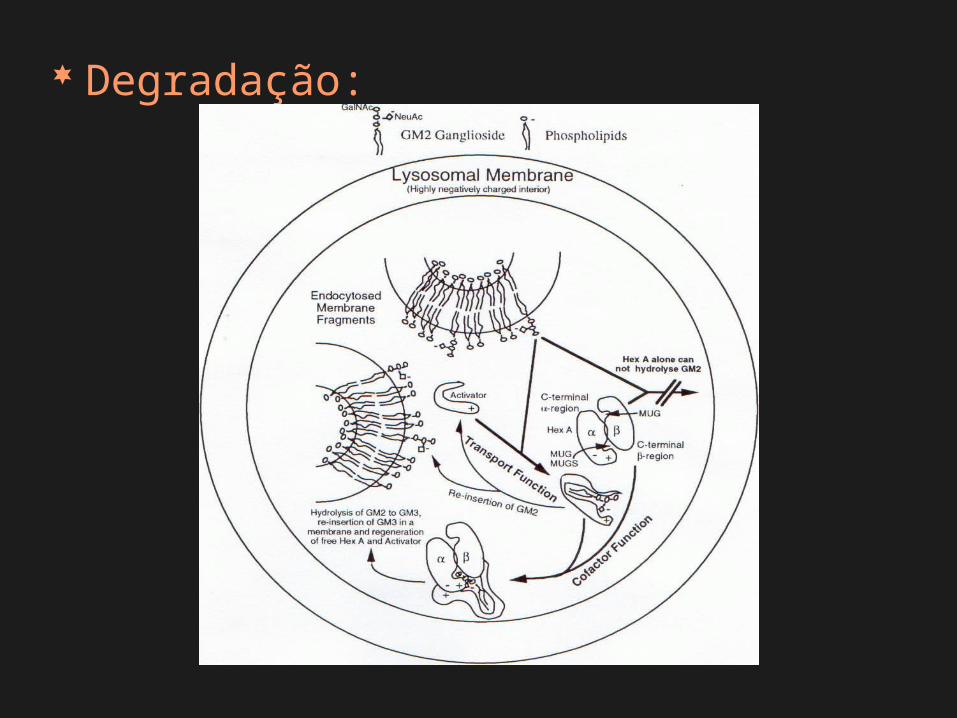
Degradação:
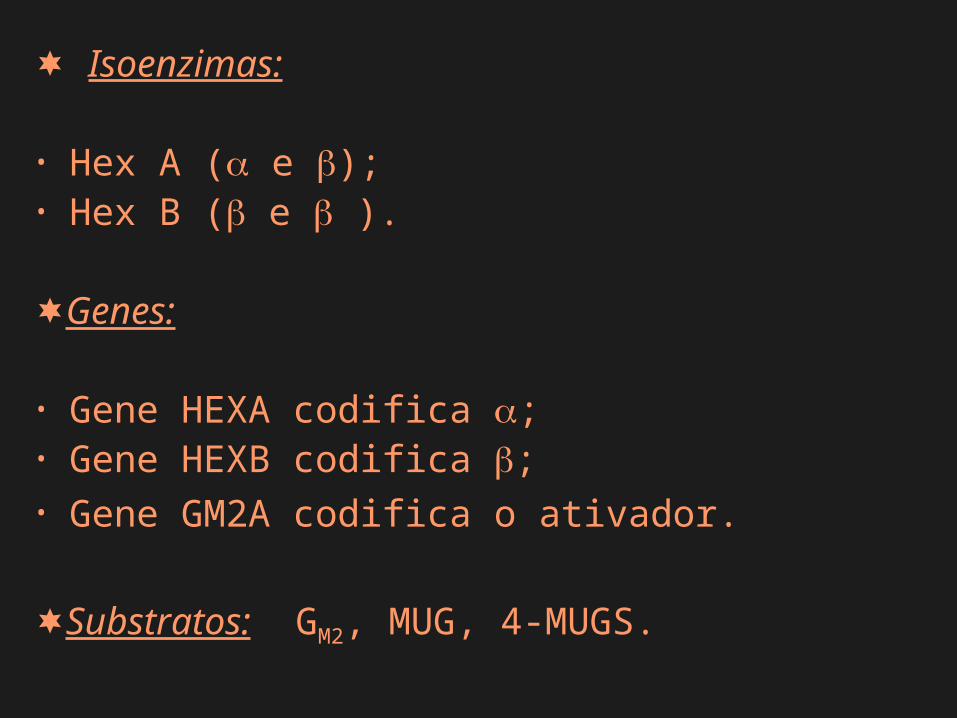
Isoenzimas:
• Hex A ( e );• Hex B ( e ).
Genes:
• Gene HEXA codifica ;• Gene HEXB codifica ;• Gene GM2A codifica o ativador.
Substratos: GM2, MUG, 4-MUGS.

Deficiência enzimática
catabolismo incompleto acúmulo
intralisossomal
funcionamento anormal das células
distúrbios do armazenamento lisossômico

Aspectos Patológicos
Similaridade: Sandhoff DTSdef. GM2 ativador.
Achados patológicos:
- entumescimento neuronal;
- gliose;
-mancha vermelho cereja.
Microscopia Eletrônica: MCB.


MANIFESTAÇÕES CLÍNICAS

Forma Aguda ou Infantil
Ao nascimento: aparentemente normal

3º ao 6º m: - fraqueza motora moderada + contrações mioclônica
- “startle reaction”


6 ao 10ºm:- perda de habilidades; - não adquire novas habilidades;- fraqueza progressiva;- hipotonia;- dificuldade para engatinhar e sentar.

Acuidade visual + movimentos oculares não usuais + cherry-red spot

Após 8º m: progressão rápida
- mov.voluntária;
- responsividade;
- visão deteriora. 12ºm: convulsões (parciais e crises de
ausência)


18º m: macrocefalia – gliose
2º ano: deterioração
- postura de decerebração;
- dificuldade para engolir;
- convulsões;
- estado vegetativo.
Óbito broncopneumonia.


Forma Subaguda ou Juvenil
Entre 2º e 10º ano: ataxia e incoordenação.
Regressão do desenvolvimento, demência, declínio da cognição
marcantes dessa variante. Final da 1ª déc.: espasticidade e
convulsões.

Perda da visão. Cherry-red spot: ñ tão f. Atrofia ótica e retinite pigmentosa. Entre 10º e 15º a: estado vegetativo c/
rigidez de decerebração morte em poucos anos.

Forma Crônica ou Adulta
Neurodegeneração lenta + baixos níveis de atividade residual da Hex A.
Idade de início: desde 1ª infância até final da 1ª déc.
Fenótipos variam sobreposição.

Primeiros sintomas: desde fraqueza muscular até manifestações:• Extrapiramidais (distonia, coreoatetose,
ataxia);• Cerebelares (disartria, descoordenação
motora e anormalidades posturais entre 2º e 10º a);
• Fala e pensamento envolvidos tardiamente.
Diferenciar c/ decebração espinocerebelar,
ataxia de Friedriech e Esclerose lateral amiotrófica (ALS).

Fraqueza muscular, fasciculações e disartria: indistingüíveis da forma progressiva da D. de Kugelberg- Welander ou início precoce da ALS.
40% dos pctes: manifestações psiquiátricas (sem demência).

ASPECTOS GENÉTICOS

Padrão de Herança
Doença Autossômica Recessiva.

Estrutura do Gene HEXA
Cromossomo 15q23-q24:
Subunidade da enzima Hex A; 26Kb-35Kb; 14 éxons e 13 íntrons; Vários alelos grau variável de deficiência
enzimática manifestações clínicas diversas.

Cadeias e 57% de homologia. Genes HEXA e HEXB impressionante
grau de homologia entre os números e a localização das junções éxon-íntron.
Genes HEXA e HEXB diferentes cromossomos ancestral comum.

Subunidades e áreas idênticas entre as estruturas primárias domínios funcionais comuns.
Arg178/Arg211 atividade catalítica das duas subunidades.

Mutações
91Total
1Grandes deleções
4Pequenas inserções
13Pequenas deleções
19Subs. nucleotídeos (splicing)
54Subst.nucleotídeos (missense/nonsense)
Nº de mutaçõesTipos de mutações

Mutações que causam completa da atividade da Hex A rápido acúmulo de gangliosídeo GM2
manifestações clínicas mais precoces forma aguda.
Mutações que permitem um nível residual de atividade a Hex A acúmulo mais lento do gangliosídeo GM2 manifestações clínicas mais tardias forma de início tardio.

Correlação entre a atividade residual da enzima Hex A e a severidade da doença resultante:
• aguda 0,1% do controle normal;• subaguda 0,5% do controle normal;• crônica 2-4% do controle normal.

Dois probandos clinicamente saudáveis com baixa atividade da Hex A foram identificados como possuindo uma atividade da enzima entre 11 e 20%.
HIPÓTESE DO LIMIAR CRÍTICO

Forma aguda infantil dois alelos nulos sem atividade enzimática da Hex A.
Formas juvenil e crônica heterozigotos compostos para um alelo nulo e um alelo expressivo baixa atividade residual da Hex A.

Mutações associadas com a DTS Infantil
1ª mutação identificada deleção de 7,6 Kb na extremidade 5’ do gene HEXA.
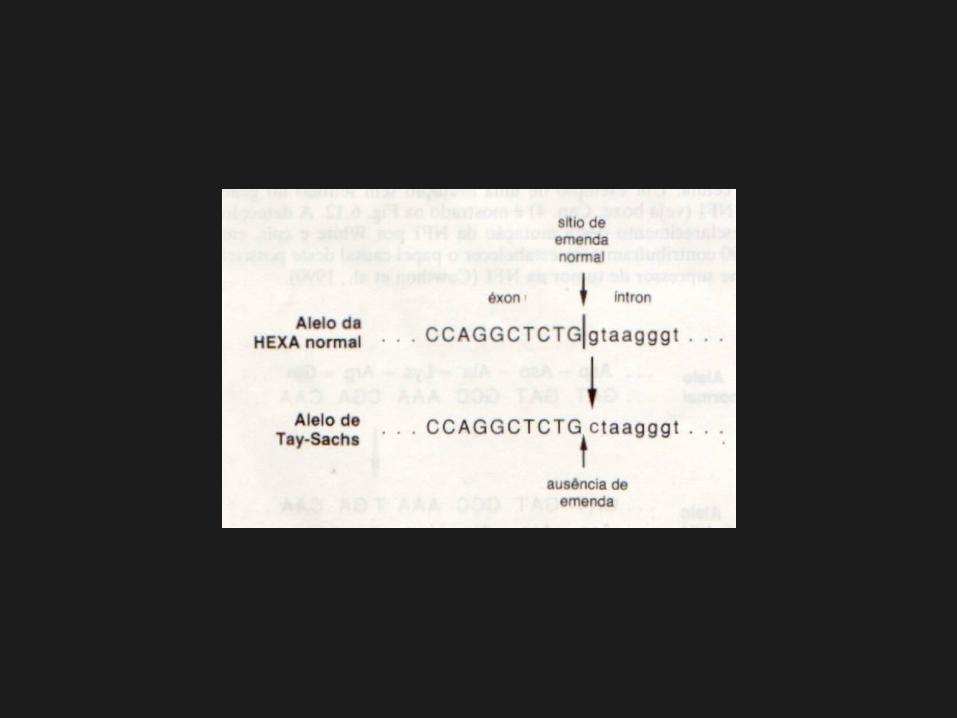
Mutação mais freqüente inserção de 4pb, +TATC1278, no éxon 11.
2ª mutação mais freqüente GC (íntron 12).


Mutações missense dois grupos de proteínas mutantes:
o precursor RE proteína não fosforilada não há combinação com subunidade não há formação da Hex Anão há secreção nem transporte para o lisossomo.
• G1444A (no éxon 13) Glu482Lis aguda;

precursor é fosforilado, mas não se associa com a subunidade a enzima não é processada na forma madura.
• C1510T (no éxon 13) Arg504Cis.

“Sistema de controle de qualidade” chaperones sugere que subunidades com mutações missense, não necessariamente são incapazes de formar uma Hex A parcialmente funcional, pois isso pode ser prevenido pela afinidade aumentada dessas subunidades por uma ou mais chaperones.

Diferente grau de afinidade pelas chaperones diferentes formas de manifestação da doença (aguda, subaguda e crônica).

Mutações da emenda:• GC (+1 IVS-12);• GA (+1 IVS-9).

Mutações associadas a DTS Subaguda
Mutação silenciosa G570A (éxon 5) Leu190Leu;
• G1496A (éxon 13) Arg499His;• G1511A (éxon13) Arg504His; - substituição mais conservativa da histidina
permite a algumas cadeias mutantes tornarem-se fosforiladas, associarem-se a cadeias e serem levadas ao lisossomo.

Mutações associadas com a Variante B1
Indivíduos afetados por essa variante produzem Hex A ativa para o substrato sintético 4MUG, mas não para o substrato sintético 4MUGS ou para o gangliosídeo GM2. Isso porque a atividade
da enzima Hex A sobre o substrato 4MUG deve-se à subunidade , que permanece ativa nessa variante, enquanto a subunidade apresenta-se deficiente, provavelmente por uma mutação no sítio ativo dessa subunidade.

• G533A (éxon 5) Arg178His;
- homozigotos apresentam fenótipo subagudo;
- heterozigotos apresentam um segundo alelo agudo associado fenótipo mais severo.

• C532T (éxon 5) Arg178Cis; • G533T (éxon 5) Arg178Leu;
- essas mutações acarretam fenótipos agudos mais severos.

Outro estudo: heterozigoto para um alelo não expressivo
e um alelo causador da variante B1 fenótipo juvenil;
homozigoto para a mutação causadora da variante B1 possui o dobro da atividade enzimática em relação ao heterozigoto composto fenótipo crônico moderado.

Mutações associadas com a DTS Crônica
• G805A (éxon 7) Gli269Ser;• A590C (éxon 6) Lis197Thr;
- identificada num paciente cujo segundo alelo tinha uma mutação Arg499His.

Mutações associadas com a Pseudodeficiência da Hex A
Uma ou duas mutações puntiformes que levam a atividade reduzida, mas variável da enzima sobre os substratos sintéticos (4MUG e 4MUGS), mas com atividade funcional sobre o gangliosídeo GM2.

• C739T (éxon 7) Arg247Trp;
- heterozigotos compostos nos quais o alelo mutado era “acoplado” com outro alelo portador de uma inserção + TATC1278
ou de uma mutação da emenda +1 IVS -12.
• C745T (éxon 7) Arg249Trp.

Percentagem das Mutações encontradas
População de judeus Ashkenazi:• duas mutações forma aguda infantil
90 a 95% de todos os alelos;• mutação Gli269Ser forma crônica
3%;• mutação para a pseudodeficiência
(Arg247Trp) 2%.

População geral não judaica:• 35% dos alelos duas mutações associadas
com o fenótipo da doença aguda;• 5% dos alelos mutações associadas com as
formas juvenil e crônica; • 35% dos heterozigotos não judeus portadores
de um dos dois alelos para a pseudodeficiência (Arg247Trp ou Arg249Trp).

ORIGEM DAS MUTAÇÕES

Mutação inicial na população de judeus Ashkenazi 70-1100 D.C. centro-leste europeu.
Isolados genéticos grupos pequenos nos quais a freqüência de certos genes recessivos raros é bem diferente daquela na população em geral.

Caráter recessivo freqüência numa determinada população consangüinidade não é uma característica marcante.

Efeito do fundador freqüência elevada de um gene mutante numa população fundada por um pequeno grupo ancestral quando um ou mais dos fundadores eram portadores do gene mutante.
Seletividade ambiental vantagem biológica aos heterozigotos.

DIAGNÓSTICO

Métodos Diagnósticos
Historicamente: história clínica, exame físico e determinações histopatológicas e bioquímicas;
Ensaios Enzimáticos; Diagnóstico Molecular.

Ensaios Enzimáticos
Demonstração específica da deficiência da atividade da Hex A na presença de atividade normal ou elevada da Hex B.

Métodos de separação das enzimas
Substratos artificiais cromogênicos ou flurogênicos são hidrolizados pela Hex A e Hex B;
Substrato GM2 + proteína ativador mais específico diferencia entre as variantes infantil, crônica e variante B1;

O substrato 4MUGS, exclusivamente, hidrolizado pela Hex A é útil principalmente na variante B1, não distingue heterozigotos pseudodeficientes, nem é tão sensível como os substratos padrões.

Enzimas são separadas por cromatografia ou por eletroforese.
Programas de rastreamento baseados na estabilidade térmica e no pH das enzimas: ph 4,4- Hex B estável até 55°C e Hex A até 50°C; atividade total é mensurada antes e depois da desnaturação seletiva da Hex A e a atividade é calculada pela diferença entre essas medidas.

Diagnóstico Molecular
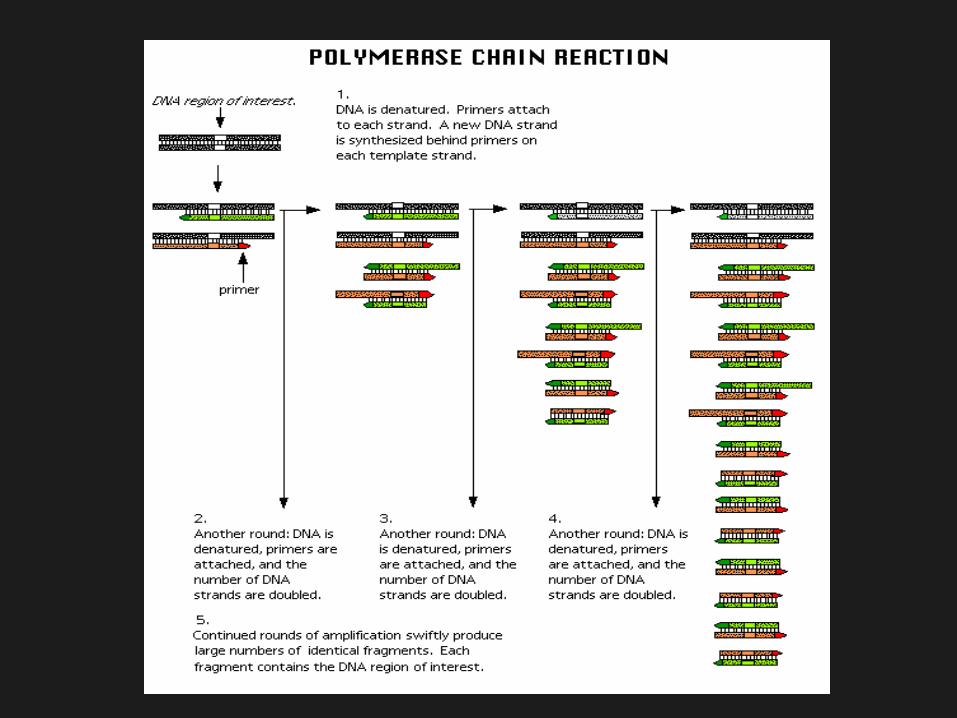
Baseado na análise do DNA; Identifica mutações específicas no gene
HEXA causadoras da DTS; Distingue alelos da pseudodeficiência; Análise através enzimas de restrição,
Southern blotting, técnicas de PCR e MALDI-MS.

Programas de Rastreamento 1ª condição genética com rastreamento
de heterozigotos; Método de escolha: ensaios
enzimáticos • vantagens: econômico, larga escala• desvantagens: sobreposição nas
distribuições da atividade enzimática entre portadores e não portadores = dados inconclusivos; não distingue tipo de mutação do gene.

Testes de DNA são limitados pela sua especificidade singular para cada mutação.

Mesmo com anormalidade da atividade enzimática, a análise das mutações em todos os portadores é essencial e decisiva para definir a forma da DTS para qual a prole teria risco.

Diagnóstico Pré-Natal
Envolve casais identificados como de risco, ou seja, heterozigotos;
A quantificação das enzimas pode ser feita na 1ª metade da gestação;
Material: líquido amniótico, cultura de células do líquido, vilosidades coriônicas e derivados de sua cultura.

Detecção pré-natal análise molecular do DNA = maior especificidade.
Mutações em ambos os genitores, específicas e conhecidas = identificação de homozigose ou heterozigose composta sem dificuldades; do contrário deve-se fazer testes enzimáticos e moleculares combinados para excluir alelos para pseudodeficiência em heterozigotos compostos.

Indicação dos Testes Pré-Natais
Ensaios enzimáticos revelarem genitores heterozigotos;
Testes moleculares um dos genitores com alelo para pseudodeficiência ou mutação no gene HEXA;
Um dos pais heterozigoto e outro com atividade enzimática inconclusiva;
Mutação causadora da doença não identificada.

Casal Heterozigoto:• 25% criança afetada• 50% criança normal portadora • 25% criança normal não portadora; Irmandade heterozigoto = 50% chance
de ser heterozigota; Irmandade afetado = 2/3 chance de ser
heterozigoto; Indivíduo heterozigoto para alelo da
pseudodeficiência = sem risco aumentado de prole com DTS.

Significativo número de gestações monitoradas = freqüência < 25% = testes de rastreamento inconclusivos.

Aconselhamento Genético
Monitoração pré-natal; Interrupção da gestação - quando
permitido; Adoção; Inseminação artificial com gameta doado; Diagnóstico pré-implantação.

Rastreamento de Portadores
+
Aconselhamento Genético
+
Diagnóstico Pré-Natal
Incidência DTS reduzida em 90%

TRATAMENTO

Inespecífico; Tratamento suporte das manifestações
clínicas e manejo de intercorrências; nutrição e hidratação adequada, controle convulsões e infecções;
Diversas técnicas têm sido desenvolvidas com intuito de encontrar terapia viável e efetiva.

Reposição Enzimática
Requer que uma significativa quantidade da Hex A alcance o SNC na forma catalítica ativa e que as células defeituosas liberem o material de armazenamento em excesso;
Efetiva in vitro; In vivo, barreira hematoencefálica não
permite penetração de quantidades suficientes de enzimas no SNC.

Privação de Substrato
Inibidor específico da biossíntese do glicolipídio para reduzir parcialmente a quantidade de glicolipídio sintetizado pelas células = catabolismo glicolipídio restante pela enzima residual do indivíduo;
Funciona em modelos animais, previnindo o acúmulo de gangliosídeo no SNC;
Não se tem certeza dos resultados em humanos.

Transplante de Medula Óssea
Transferência de células hematopoiéticas normais doadoras para células hospedeiras defeituosas;
Corrige defeito enzimático no fígado e outros tecidos afetados;
Correção enzimática no SNC interrogada devido a barreira hematoencefálica excluir a enzima circulante.

Terapia Gênica mediada por Vetor Retroviral
Sistema de liberação de genes para células proliferativas
Vetores retrovirais recombinantes integração estável dos genes terapêuticos com o DNA das células hospedeiras expressão do gene transferido sem gerar resposta imune contra os agentes virais;
Vetores não infectam células estáveis = neurônio = sem efeito na DTS.

Liberação de genes diretamente no SNC
Tentativa de superar a barreira hematoencefálica;
2 métodos : a) vetor viral com tropismo pelo SNC herpes simplex vírus ou adenovírus
b) implante cerebral de células geneticamente modificadas que superexpressam o gene terapêutico.

Infelizmente não permitem uma expressão contínua do gene nas células hospedeiras, pois não se integram no genoma.

Uma nova abordagem que promete grandes mudanças no tratamento das lesões amplas do SNC é o transplante de células neurais progenitoras multipotenciais que podem diferenciar-se em neurônios, astrócitos e oligodendrócitos = possíveis correções, a longo prazo, dos distúrbios do armazenamento.